Настоящее изобретение относится к тетраазафенален-3-оновым соединениям, которые ингибируют поли(АДФ-рибоза)полимеразу (PARP).
Настоящее изобретение относится к ингибиторам ядерного фермента поли(аденозин-5'-дифосфорибоза)полимеразы ["поли(АДФ-рибоза)полимераза" или "PARP", который также называют ADPRT (NAD:белок (АДФ-рибозилтрансфераза (полимеризующая)), и PARS (поли(АДФ-рибоза)синтетаза) и предлагает соединения и композиции, содержащие раскрытые соединения. Кроме того, настоящее изобретение относится к способам применения раскрытых ингибиторов PARP для лечения рака.
Большое внимание уделяется разработке ингибиторов PARP в качестве химиосенсибилизирующих средств, для применения в противораковой терапии и для ограничения повреждения клеток после ишемии или эндотоксического стресса. В частности, усиление цитотоксичности темозоломида, наблюдаемое в доклинических исследованиях с использованием активных ингибиторов PARP-1, связано с ингибированием восстановления удаленных оснований, и последующая цитотоксичность обусловлена неполным процессингом N7-метилгуанина и N3-метиладенина. Существующая в настоящее время совокупность результатов доклинических исследований демонстрирует, что цитотоксичность темозоломида усиливается при совместном введении ингибитора PARP как in vitro, так и in vivo. Plummer, et al, Clin. Cancer Res., 11(9), 3402 (2005).
Темозоломид, ДНК-метилирующее средство, вызывает повреждение ДНК, которое исправляется O6-алкилгуаниналкилтрансферазой (ATазой) и путем поли(АДФ-рибоза)полимераза-1 (PARP-1)-зависимой репарации удаленных оснований. Темозоломид представляет собой монофункциональное ДНК-алкилирующее средство, которое может усваиваться после перорального введения и используется для лечения глиом и злокачественной меланомы. Темозоломид быстро абсорбируется и подвергается спонтанному разрушению с образованием активного монометилтриазена, 5-(3-метил-1-триазено)имидазол-4-карбоксамида. Монометилтриазен дает несколько продуктов метилирования ДНК, среди которых преобладают N7-метилгуанин (70%), N3-метиладенин (9%) и O6-метилгуанин (5%). Если O6-метилгуанин не подвергается репарации под действием O6-алкилгуаниналкилтрансферазы, он является цитотоксичным вследствие неспособности спариваться с тимином в процессе репликации ДНК. Данное нарушение комплементарности распознается на дочерней цепи белками, осуществляющими репарацию ошибочно спаренных оснований, затем вырезается тимин. Однако если исходный нуклеотид O6-метилгуанин в родительской цепи не подвергается репарации в результате опосредованного AT-азой удаления метил-содержащего аддукта, может осуществляться повторная вставка тимина. Повторяющиеся безрезультатные циклы вырезания тимина и вставки на противоположной цепи нерепарированного нуклеотида O6-метилгуанина вызывают состояние непрерывно возобновляющегося разрыва цепи, и ветвь MutS системы репарации ошибочно спаренных оснований сигнализирует о прекращении клеточного цикла на стадии G2-M и инициации апоптоза. Количественно более значимые продукты алкилирования нуклеотидов N7-метилгуанин и N3-метиладенин, образующиеся под действием темозоломида, подвергаются быстрой репарацией путем вырезания оснований. Plummer, et al, Clin. Cancer Res., 11(9), 3402 (2005).
Химиосенсибилизация под действием ингибиторов PARP не ограничивается темозоломидом. Цитотоксичные лекарственные средства или облучение, как правило, могут вызывать активацию PARP-1, и ингибиторы PARP-1, как показано, могут усиливать разрушение ДНК и цитотоксические эффекты химиотерапии и облучения. Kock, et al, 45 J. Med. Chem. 4961 (2002). PARP-1-опосредованная репарация ДНК в ответ на средства, повреждающие ДНК, олицетворяет механизм устойчивости опухолей к лекарственным средствам, и показано, что ингибирование данного фермента повышает активность ионизирующего облучения и некоторых цитотоксичных противоопухолевых средств, включая темозоломид и топотекан. Suto et al, в патенте США 5177075, раскрывают некоторые изохинолины, используемые для усиления летального действия ионизирующего облучения или химиотерапевтических средств на опухолевые клетки. Weltin et al, "Effect of 6(5H)-Phenanthridinone, an Inhibitor of Poly(ADP-ribose) Polymerase, on Cultured Tumor Cells", Oncol. Res., 6:9, 399-403 (1994), описывают ингибирование активности PARP, уменьшение пролиферации опухолевых клеток и значительный синергический эффект при совместной обработке опухолевых клеток алкилирующим средством. Таким образом, PARP-1 является потенциально важной терапевтической мишенью для повышения эффективности ДНК-повреждающих противораковых терапий.
Ингибиторы PARP также могут ингибировать рост клеток, имеющих дефекты в пути репарации двухцепочечной ДНК посредством гомологичной рекомбинации (HR). См. Bryant et al, "Specific killing of BRCA2-deficient tumours with inhibitors of Poly(ADP-ribose) Polymerase," Nature 434, 913 (2005); Farmer et al, "Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy," Nature 434, 917 (2005). Данный эффект реализуется в отсутствии химиосенсибилизирующих средств. Id. Известные состояния, связанные с дефектами HR, включают дефекты BRCA-1, дефекты BRCA-2 и раковые заболевания, ассоциированные с анемией Фанкони. McCabe et al., "Deficiency in the Repair of DNA Damage by Homologous Recombination and Sensitivity to Poly(ADP-ribose) Polymerase Inhibition," Cancer Res. 66. 8109 (2006). Белки, которые были идентифицированы как ассоциированные с анемией Фанкони, включают FANCA, FANCB, FANCC, FANCD2, FANCE, FANCF, FANCG, FANCL и FANCM. Id. Обзор можно найти у Zaremba et al., в "PARP Inhibitor Development for Systemic Cancer Targeting," Anti-Cancer Agents in Medicinal Chemistry 7, 515 (2007) и Lewis et al., "Clinical Poly(ADP-Ribose) Polymerase inhibitors for the treatment of cancer," Curr. Opin. Investigational Drugs 8, 1061 (2007).
Большое число известных ингибиторов PARP описано у Banasik et al., в "Specific Inhibitors of Poly(ADP-Ribose) Synthetase and Mono(ADP-Ribosyl)-Transferase", J. Biol. Chem., 267:3, 1569-75 (1992), а также в Banasik et al., "Inhibitors and Activators of ADP-Ribosylation Reactions", Molec. Cell. Biochem., 138, 185-97 (1994). Однако эффективное применение указанных ингибиторов PARP в описанных выше способах ограничено вследствие присутствия нежелательных побочных эффектов. См. Milam et al., "Inhibitors of Poly(Adenosine Diphosphate-Ribose) Synthesis; Effect on Other Metabolic Processes," Science, 223, 589-91 (1984).
В дополнение к вышесказанному, ингибиторы PARP были раскрыты и описаны в следующих международных патентных заявках: WO 00/42040; WO 00/39070; WO 00/39104; WO 99/11623; WO 99/11628; WO 99/11622; WO 99/59975; WO 99/11644; WO 99/11945; WO 99/11649 и WO 99/59973. Исчерпывающий обзор состояния данной области был опубликован Li and Zhang в IDrugs 2001, 4(7): 804-812 (PharmaPress Ltd ISSN 1369-7056).
Способность ингибиторов PARP усиливать летальное действие цитотоксических средств путем химиосенсибилизации опухолевых клеток к цитотоксическим эффектам химиотерапевтических средств была описана, в числе прочего, в US 2002/0028815; US 2003/0134843; US 2004/0067949; White AW, et al., 14 Bioorg. и Med. Chem Letts. 2433 (2004); Canon Koch SS, et al., 45 J. Med. Chem. 4961 (2002); Skalitsky DJ, et al, 46 J. Med. Chem. 210 (2003); Farmer H, et al, 434 Nature 917 (14 April 2005); Plummer ER, et al., 11(9) Clin. Cancer Res. 3402 (2005); Tikhe JG, et al., 47 J. Med. Chem. 5467 (2004); Griffin R.J., et al, WO 98/33802; и Helleday T, et al, WO 2005/012305.
Индукция периферической невропатии является основной причиной ограничения использования химиотерапевтических средств. Quasthoff and Hartung, J. Neurology, 249, 9-17 (2002). Индуцированная химиотерапией невропатия представляет собой побочный эффект, наблюдающийся после применения многих традиционных (таких как таксол, винкритин, цисплатин) и новых химиотерапевтических средств (таких как велкад, эпотилон). В зависимости от используемого вещества, может развиваться чистая сенсорная и болезненная невропатия (при использовании цисплатина, оксалиплатина, карбоплатина) или смешанная сенсомоторная невропатия, необязательно с участием автономной нервной системы (при использовании винкристина, таксола, сурамина). Нейротоксичность зависит от суммарной дозы и типа используемого лекарственного средства. В отдельных случаях невропатия может развиваться даже после однократного применения препарата. Излечение симптомов часто бывает неполным и для восстановления функции требуется длительный период регенерации. До настоящего времени существовало только небольшое число лекарственных средств, позволяющих надежно предотвращать или лечить невропатию, индуцированную химиотерапией.
Таким образом, остается потребность в эффективных и сильнодействующих ингибиторах PARP, которые могут усиливать летальное действие химиотерапевтических средств на опухолевые клетки, вызывая при этом минимальные побочные эффекты.
В дополнение, сообщалось, что ингибиторы PARP эффективно сенсибилизируют гипоксические опухолевые клетки к облучению и эффективно предотвращают восстановление опухолевых клеток после потенциально летального повреждения ДНК под действием лучевой терапии, предположительно, благодаря их способности предотвращать репарацию ДНК. Патенты США 5032617; 5215738 и 5041653.
Последние публикации позволяют предположить, что ингибиторы PARP убивают раковые клетки, дефицитные по ассоциированным с раком молочной железы генам 1 и 2 (BRCA1/2). Данные исследования позволяют предположить, что с помощью ингибиторов PARP можно эффективно лечить BRCA1/2-ассоциированный рак молочной железы. [Farmer et al., Nature 2005, 434, 917; DeSoto and Deng, Intl. J. Med. Sci. 2006, 3, 117; Bryant et al., Nature, 2005, 434, 913.]
Таким образом, остается потребность в эффективных и сильнодействующих ингибиторах PARP, которые могут усиливать летальное действие ионизирующего облучения и/или химиотерапевтических средств на опухолевые клетки, или ингибировать рост клеток, имеющих дефекты пути репарации двухцепочечной ДНК посредством гомологичной рекомбинации (HR), вызывая при этом минимальные побочные эффекты.
Настоящее изобретение относится к описанным в данном описании соединениям, их производным и их применению для ингибирования поли(АДФ-рибоза)полимеразы ("PARP"), композициям, содержащим данные соединения, а также способам получения указанных ингибиторов PARP и способам их применения для лечения последствий описанных выше состояний.
Настоящее изобретение также относится к тетраазафенален-3-оновому соединению формулы (I) или его фармацевтически приемлемой соли:
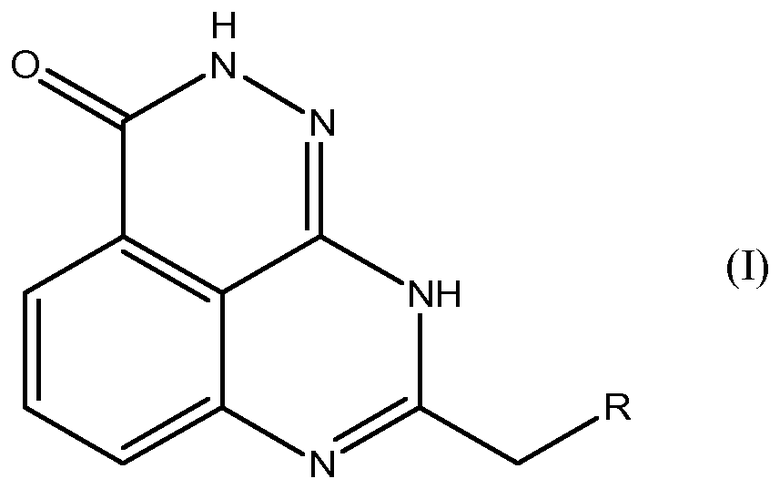
где R означает
(a) NR1R2, где R1 выбирают из группы, включающей водород, C1-C6алкил с линейной или разветвленной цепью, C2-C6алкенил с линейной или разветвленной цепью, C3-C8циклоалкил, C1-C6алкокси, C2-C6алкенилокси, фенил, фенокси, бензилокси, NRARB(C1-C6алкил с линейной или разветвленной цепью), NRARB(C2-C6алкенил с линейной или разветвленной цепью), (C1-C6алкил с линейной или разветвленной цепью)карбонил, (C2-C6алкенил с линейной или разветвленной цепью)карбонил, (C3-C8циклоалкил)карбонил, (C1-C6алкил с линейной или разветвленной цепью)оксикарбонил, (C2-C6алкенил с линейной или разветвленной цепью)оксикарбонил, (C3-C8циклоалкил)оксикарбонил, арилкарбонил, сульфонил, арилсульфонил, арил(C1-C6алкил с линейной или разветвленной цепью), арил(C2-C6алкенил с линейной или разветвленной цепью), арил(C3-C8циклоалкил), (C1-C6алкил с линейной или разветвленной цепью)арил, (C2-C6алкенил с линейной или разветвленной цепью)арил, (C3-C8циклоалкил)арил, арил, гетероциклил, гетероциклил(C1-C6алкил с линейной или разветвленной цепью) и гетероциклил(C2-C6алкенил с линейной или разветвленной цепью); где каждый гетероциклил содержит от 1 до 7 гетероатомов, независимо выбранных из O, N или S, и где каждый из RA и RB независимо выбирают из группы, включающей водород, C1-C6алкил с линейной или разветвленной цепью, C2-C6алкенил с линейной или разветвленной цепью и C3-C8циклоалкил;
и R2 выбирают из группы, включающей C1-C6алкил с линейной или разветвленной цепью, C2-C6алкенил с линейной или разветвленной цепью, C3-C8циклоалкил, C1-C6алкокси, C2-C6алкенилокси, фенил, фенокси, бензилокси, NRXRY(C1-C6алкил с линейной или разветвленной цепью), NRXRY(C2-C6алкенил с линейной или разветвленной цепью), (C1-C6алкил с линейной или разветвленной цепью)карбонил, (C2-C6алкенил с линейной или разветвленной цепью)карбонил, (C3-C8циклоалкил)карбонил, (C1-C6алкил с линейной или разветвленной цепью)оксикарбонил, (C2-C6алкенил с линейной или разветвленной цепью)оксикарбонил, (C3-C8циклоалкил)оксикарбонил, арилкарбонил, сульфонил, арилсульфонил, арил(C1-C6алкил с линейной или разветвленной цепью), арил(C2-C6алкенил с линейной или разветвленной цепью), арил(C3-C8циклоалкил), (C1-C6алкил с линейной или разветвленной цепью)арил, (C2-C6алкенил с линейной или разветвленной цепью)арил, (C3-C8циклоалкил)арил, арил, гетероциклил, гетероциклил(C1-C6алкил с линейной или разветвленной цепью) и гетероциклил(C2-C6алкенил с линейной или разветвленной цепью); где каждый гетероциклил содержит от 1 до 7 гетероатомов, независимо выбранных из O, N или S, и где каждый из RX и RY независимо выбирают из группы, включающей водород, C1-C6алкил с линейной или разветвленной цепью, C2-C6алкенил с линейной или разветвленной цепью и C3-C8циклоалкил;
где R1 и R2 независимо замещены 0-4 заместителями, каждый из которых независимо выбирают из галогена, C1-C6алкила с линейной или разветвленной цепью, C2-C6алкенила с линейной или разветвленной цепью, C1-C6алкокси, трифторметила, трифторэтила и амино; при условии, что R1 и R2, оба не являются метилом, и R2 не является (фенил)проп-1-илом, если R1 означает водород; или
(b) арилокси, замещенный 0-4 заместителями, каждый из которых независимо выбирают из группы, включающей галоген, C1-C6алкокси, трифторметил, трифторэтил, C1-C6алкил с линейной или разветвленной цепью, C2-C6алкенил с линейной или разветвленной цепью, C3-C8циклоалкил, NRCRD, NRCRD(C1-C6алкил с линейной или разветвленной цепью) и NRCRD(C2-C6алкенил с линейной или разветвленной цепью), где каждый из RC и RD независимо выбирают из группы, включающей водород, C1-C6алкил с линейной или разветвленной цепью, C2-C6алкенил с линейной или разветвленной цепью и C3-C8циклоалкил; и если более чем один заместитель представляет собой NRCRD, каждый из RC и RD независимо выбирают из группы, включающей водород, C1-C6алкил с линейной или разветвленной цепью, C2-C6алкенил с линейной или разветвленной цепью и C3-C8циклоалкил; или
(c) гетероциклил, содержащий от 1 до 7 гетероатомов, независимо выбранных из O, N или S; и от 0 до 4 заместителей, независимо выбранных из группы, включающей галоген, галогеналкил, гидроксил, нитро, трифторметил, трифторэтил, C1-C6алкил с линейной или разветвленной цепью, C2-C6алкенил с линейной или разветвленной цепью, C1-C6алкокси, C2-C6алкенилокси, фенил, фенокси, бензилокси, амино, тиокарбонил, циано, имино, NRERF(C1-C6алкил с линейной или разветвленной цепью), NRERF(C2-C6алкенил с линейной или разветвленной цепью)сульфгидрил, тиоалкил, диоксаспироэтил, (C1-C6алкил с линейной или разветвленной цепью)карбонил, (C2-C6алкенил с линейной или разветвленной цепью)карбонил, (C1-C6алкил с линейной или разветвленной цепью)оксикарбонил, (C2-C6алкенил с линейной или разветвленной цепью)оксикарбонил, арилкарбонил, сульфонил, арилсульфонил, арил(C1-C6алкил с линейной или разветвленной цепью), арил(C2-C6алкенил с линейной или разветвленной цепью), арил(C3-C8циклоалкил), (C1-C6алкил с линейной или разветвленной цепью)арил, (C2-C6алкенил с линейной или разветвленной цепью)арил, (C3-C8циклоалкил)арил, арил, гетероциклил, гетероциклил(C1-C6алкил с линейной или разветвленной цепью) и гетероциклил(C2-C6алкенил с линейной или разветвленной цепью), где каждый гетероциклил содержит от 1 до 7 гетероатомов, независимо выбранных из O, N или S, где каждый из RE и RF независимо выбирают из группы, включающей водород, C1-C6алкил с линейной или разветвленной цепью, C2-C6алкенил с линейной или разветвленной цепью и C3-C8циклоалкил; и если более чем один заместитель представляет собой NRERF, каждый из RE и RF независимо выбирают из группы, включающей водород, C1-C6алкил с линейной или разветвленной цепью, C2-C6алкенил с линейной или разветвленной цепью и C3-C8циклоалкил; где каждый из указанных 0-4 заместителей независимо замещен 0-4 дополнительными заместителями, и каждый из указанных дополнительных заместителей независимо выбирают из галогена, C1-C6алкила с линейной или разветвленной цепью, C2-C6алкенила с линейной или разветвленной цепью, C3-C8циклоалкила, C1-C6алкокси, трифторметила, трифторэтила и амино; при условии, что R содержит по меньшей мере один заместитель, если R означает N-пиперидинильную, N-пирролидинильную или N-морфолинильную группу.
В некоторых вариантах осуществления размер каждого кольца всех гетероциклилов формулы (I) независимо составляет 5-7 атомов.
Некоторые варианты осуществления содержат один, два или три атома азота по меньшей мере в одном кольце гетероциклила формулы (I).
В некоторых вариантах осуществления гетероциклил формулы (I) содержит 1-3 кольца. В некоторых вариантах осуществления гетероциклил содержит 1-7 гетероатомов, независимо выбранных из O, N и S. В некоторых вариантах осуществления гетероциклил содержит 1-2 кольца. В некоторых вариантах осуществления гетероциклил содержит одно кольцо. В некоторых вариантах осуществления разные варианты гетероциклила формулы (I) независимо содержат 1-3 кольца. В некоторых вариантах осуществления разные варианты гетероциклила формулы (I) независимо содержат 1-2 кольца. В некоторых вариантах осуществления разные варианты гетероциклила формулы (I) независимо содержат одно кольцо.
В некоторых вариантах осуществления гетероциклил формулы (I) выбирают из группы, включающей пиперидинил, пиперазинил, пиредазинил, дигидропиридил, тетрагидропиридил, пиридинил, пиримидинил, дигидропиримидинил, тетрагидропиримидинил, гексагидропиримидинил, дигидропиразинил, тетрагидропиразинил, пирролидинил, имидазолидинил, пиразолидинил, пирролил, дигидропирролил, имидазолил, дигидроимидазолил, пиразолил, дигидропиразолил, азепанил, [1,2]диазепанил, [1,3]диазепанил, [1,4]диазепанил, индолил, дигидроиндолил, изоиндолил, дигидроизоиндолил, дигидрохинолил, тетрагидрохинолил, дигидроизохинолил и тетрагидроизохинолил; или их подгруппы.
Настоящее изобретение также относится к фармацевтической композиции, содержащей (i) терапевтически активное количество соединения формулы (I) и (ii) фармацевтически приемлемый носитель.
Настоящее изобретение относится к соединениям, которые ингибируют полимеразную активность поли(АДФ-рибоза)полимеразы (PARP) in vitro и/или in vivo, а также композициям, содержащим раскрытые соединения.
Настоящее изобретение относится к способам ингибирования, ограничения и/или подавления полимеразной активности поли(АДФ-рибоза)полимеразы (PARP) in vitro и/или in vivo в растворах, клетках, тканях, органах или системах органов. В одном варианте осуществления настоящее изобретение предлагает местные или системные способы ограничения или ингибирования активности PARP у млекопитающего, такого как человек.
В одном варианте осуществления изобретение относится к способу лечения рака посредством химиосенсибилизации, включающему приведение раковых клеток в контакт с усиливающим цитотоксичность соединением формулы (I), тетраазафенален-3-оном, или его фармацевтически приемлемой солью, а также приведение опухолевых или раковых клеток в контакт с противораковым средством.
Вариант осуществления настоящего изобретения предлагает способ химиосенсибилизации, где, для обеспечения эффективного уровня химиосенсибилизации, нуждающемуся в этом пациенту однократно или многократно вводят первую дозу по меньшей мере одного соединения формулы (I) или его фармацевтически приемлемой соли, и через некоторый промежуток времени указанному пациенту однократно или многократно вводят вторую дозу по меньшей мере одного химиотерапевтического средства.
Аспект настоящего изобретения предлагает фармацевтическую композицию, содержащую соединение формулы (I) в форме, выбранной из группы, состоящей из перечисленных ниже неограничивающих примеров таких химиотерапевтических средств, их фармацевтически приемлемых свободных оснований, солей, гидратов, сложных эфиров, сольватов, стереоизомеров и их смесей. В соответствии с другим аспектом, фармацевтическая композиция дополнительно содержит фармацевтически приемлемый носитель и, необязательно, химиотерапевтическое средство. Следующие варианты осуществления служат только для иллюстрации и не предназначены для какого-либо ограничения объема настоящего изобретения. В одном варианте осуществления фармацевтическая композиция настоящего изобретения содержит соединение настоящего изобретения и фармацевтически приемлемый носитель. В другом варианте осуществления фармацевтическая композиция настоящего изобретения содержит фармацевтически приемлемую соль соединения настоящего изобретения и фармацевтически приемлемый носитель. В следующем варианте осуществления фармацевтическая композиция настоящего изобретения содержит соединение настоящего изобретения, одно или несколько химиотерапевтических средств и фармацевтически приемлемый носитель. В другом варианте осуществления фармацевтическая композиция настоящего изобретения содержит фармацевтически приемлемую соль соединения настоящего изобретения, одно или несколько химиотерапевтических средств и фармацевтически приемлемый носитель. Неограничивающие примеры таких химиотерапевтических средств приведены ниже.
В соответствии с дополнительными аспектами настоящего изобретения химиосенсибилизирующее соединение и химиотерапевтическое средство вводят практически одновременно.
В соответствии с одним аспектом настоящего изобретения химиотерапевтическое средство выбирают из группы, включающей темозоломид, адриамицин, камптотецин, карбоплатин, цисплатин, даунорубицин, доцетаксел, доксорубицин, интерферон-альфа, интерферон-бета, интерферон-гамма, интерлейкин 2, иринотекан, паклитаксел, таксоид, дактиномицин, даунорубицин, 4'-дезоксидоксорубицин, блеомицин, пилкамицин, митомицин, неомицин и гентамицин, этопозид, 4-OH циклофосфамид, координационный комплекс платины, топотекан, терапевтически эффективные аналоги и производные указанных средств, а также их смеси. В соответствии с конкретным аспектом, химиотерапевтическое средство представляет собой темозоломид.
В другом варианте осуществления настоящее изобретение относится к способам лечения последствий рака и/или радиосенсибилизации раковых клеток, с целью повышения чувствительности раковых клеток к лучевой терапии и, следовательно, предотвращения восстановления опухолевых клеток после потенциально летального повреждения ДНК под действием лучевой терапии, включающим введение субъекту эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли. Способ данного варианта осуществления направлен на специфическую и предпочтительную радиосенсибилизацию раковых клеток, в результате чего раковые клетки становятся более чувствительными к лучевой терапии, чем неопухолевые клетки.
Настоящее изобретение также относится к способу лечения рака у субъекта, нуждающегося в этом, который включает введение субъекту терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, где раковые клетки имеют дефект репарации расщепленной двухцепочечной ДНК. В одном варианте осуществления дефект репарации расщепленной двухцепочечной ДНК представляет собой дефект гомологичной рекомбинации. В одном варианте осуществления раковые клетки имеют фенотип, выбранный из группы, состоящей из дефекта BRCA-1, дефекта BRCA-2, дефекта BRCA-1 и BRCA-2, и анемии Фанкони.
В другом варианте осуществления настоящее изобретение относится к способам лечения BRCA1/2-ассоциированного рака молочной железы, которые включают введение соединения формулы (I) или его фармацевтически приемлемой соли.
В соответствии с одним вариантом осуществления настоящего изобретения, соединение, предназначенное для использования в способе химиосенсибилизации настоящего изобретения, способе радиосенсибилизации настоящего изобретения или способе лечения рака настоящего изобретения, где раковые клетки имеют дефект репарации расщепленной двухцепочечной ДНК, представляет собой соединение формулы (I) или его фармацевтически приемлемую соль. В другом аспекте соединение выбирают из группы, включающей
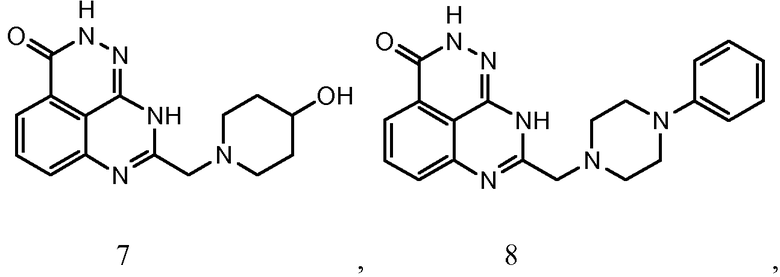
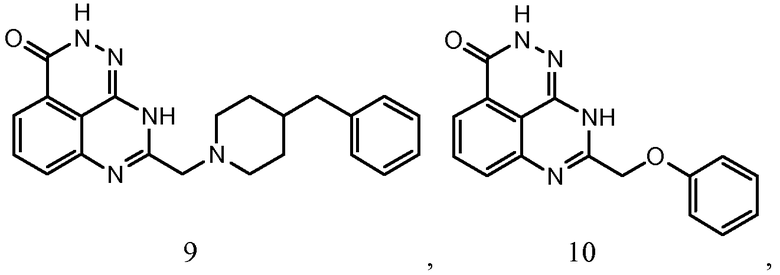
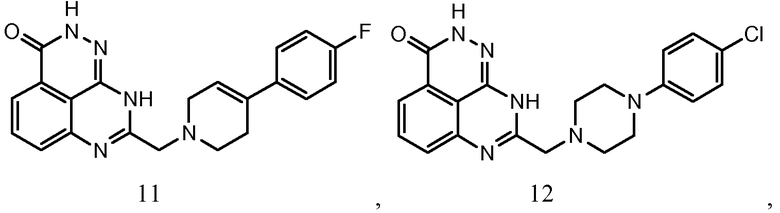
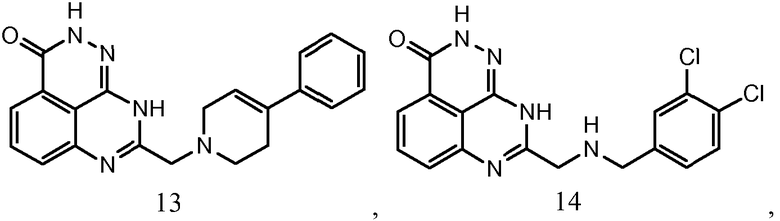

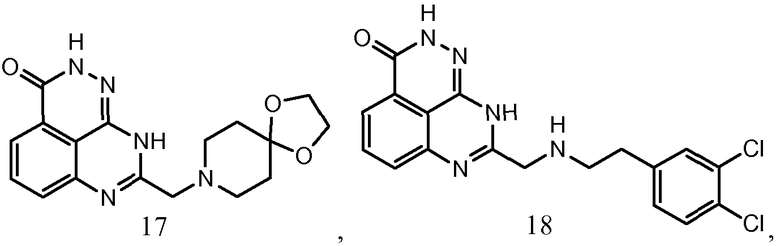
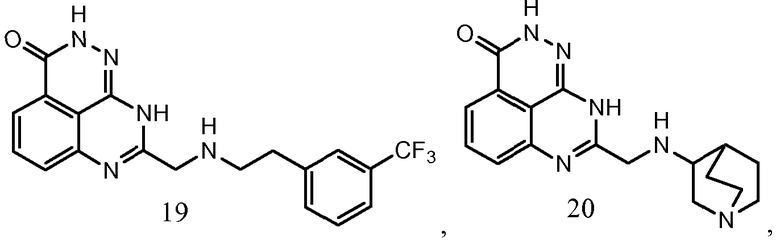
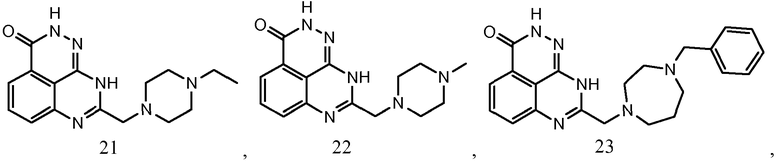
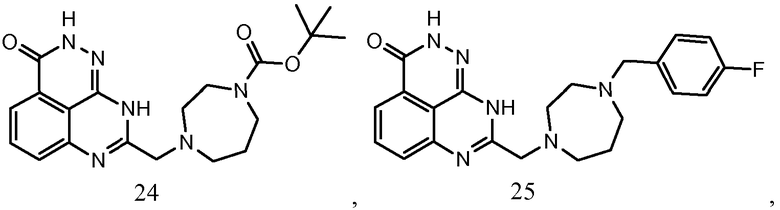

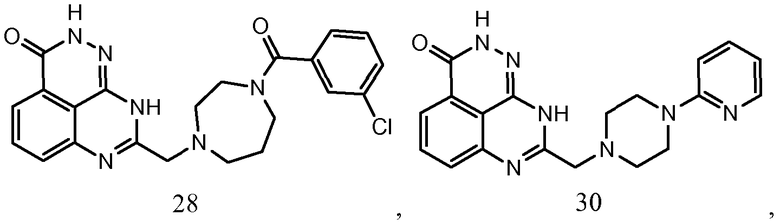

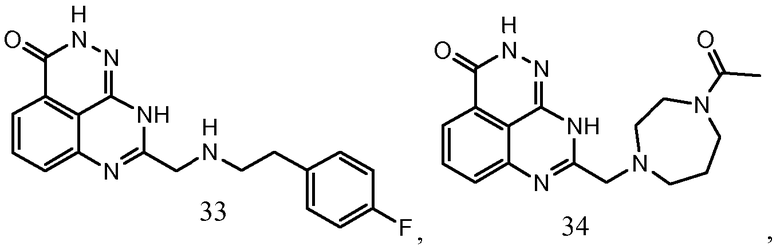
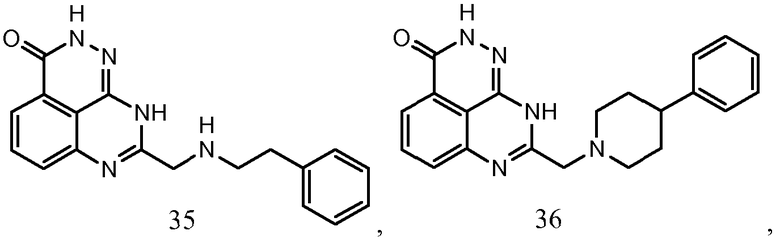
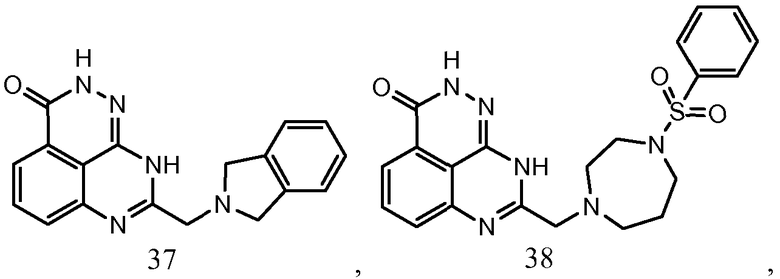
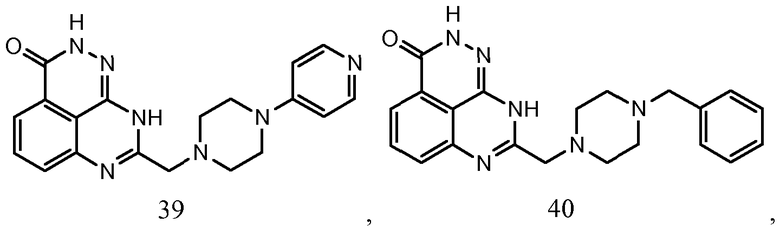
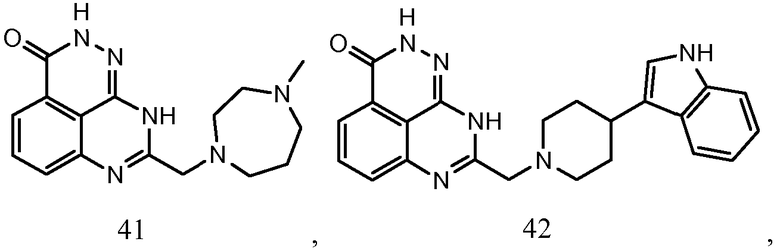
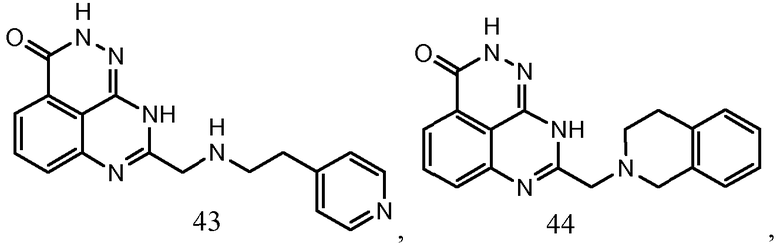
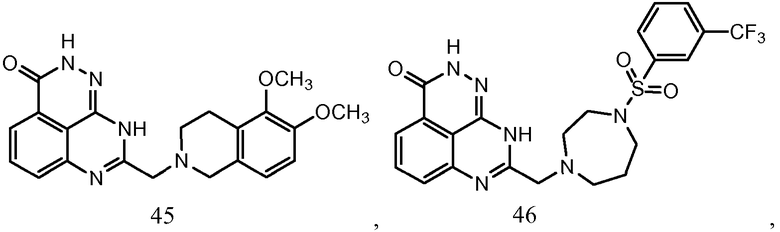
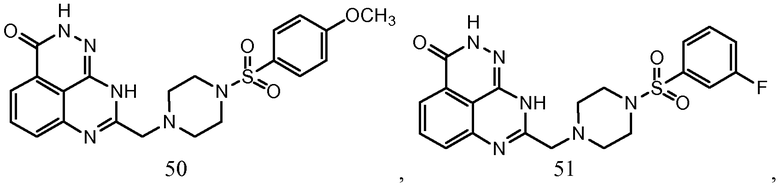
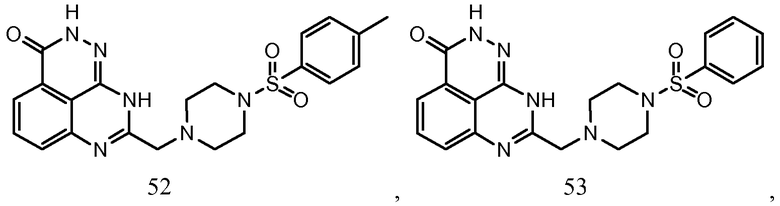
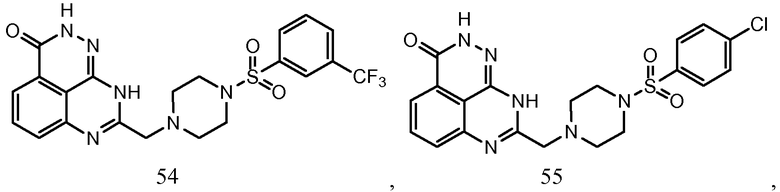
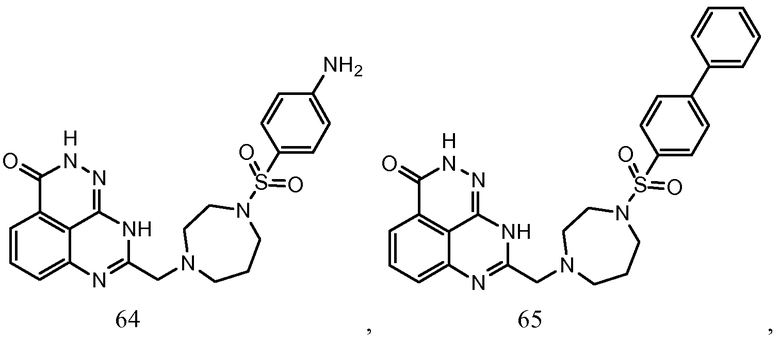
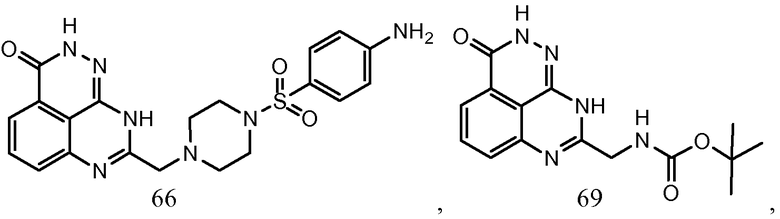
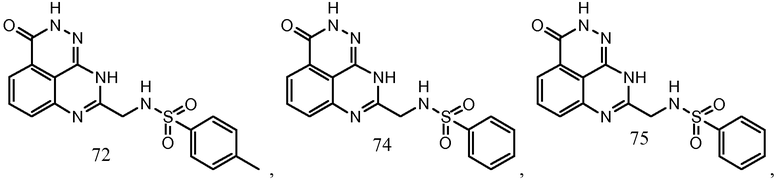
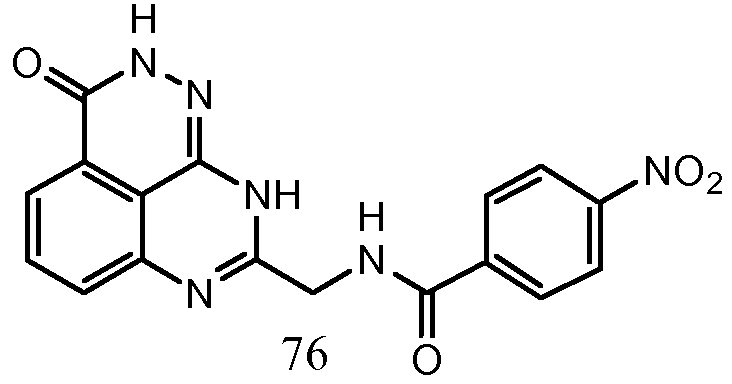
и их фармацевтически приемлемые соли.
Настоящее изобретение также относится к способам лечения периферической невропатии, индуцированной химиотерапией. В соответствии с одним аспектом настоящего изобретения, для предотвращения развития симптомов невропатии или уменьшения тяжести таких симптомов, соединения настоящего изобретения вводят до введения по меньшей мере одного химиотерапевтического средства или одновременно с ним. В соответствии с другим аспектом, соединения настоящего изобретения вводят после введения по меньшей мере одного химиотерапевтического средства для лечения симптомов невропатии у пациента или для уменьшения тяжести таких симптомов. В другом аспекте настоящее изобретение относится к способу замедления, отсрочки или прекращения роста раковых клеток у млекопитающего, включающий введение химиотерапевтического средства и дополнительное введение соединения формулы (I) или его фармацевтически приемлемой соли в количестве, достаточном для усиления противораковой активности указанного химиотерапевтического средства.
Специалисты в данной области могут легко определить другие аспекты и преимущества настоящего изобретения из приведенного ниже подробного описания, в котором продемонстрированы и описаны предпочтительные варианты осуществления настоящего изобретения для иллюстрации наилучшего способа осуществления изобретения. При реализации изобретения могут существовать другие и отличные варианты осуществления, и некоторые детали можно модифицировать в разных очевидных аспектах, не отступая от сущности изобретения. Соответственно, описание следует считать иллюстративным, но не ограничивающим.
Краткое описание чертежей
Фиг.1. - Пероральное введение ингибитора PARP-1 соединения 13+TMZ приводит к повышению выживания мышей, несущих модель меланомы B16.
Фиг.2. - Пероральное введение ингибитора PARP-1 соединения 13+TMZ, демонстрирующее повышение выживания моделей внутричерепной глиомы SJGBM.
Фиг.3. - Пероральное введение ингибитора PARP-1 соединения 37+TMZ, демонстрирующее повышение выживания мышей, несущих модель меланомы B16.
Фиг.4. - Пероральное введение ингибитора PARP-1 соединения 37+TMZ, демонстрирующее повышение выживания моделей внутричерепной глиомы SJGBM.
Фиг.5. - Пероральное введение ингибитора PARP-1 соединения 37+облучение, демонстрирующее ингибирование роста опухоли у модели рака головы и шеи.
Фиг.6. - Пероральное введение ингибитора PARP-1 соединения 37, демонстрирующее ингибирование роста мутантных опухолей BRCA1
Настоящее изобретение относится к описанным в данном описании соединениям, их производным и их применению для ингибирования поли(АДФ-рибоза)полимеразы ("PARP"), композициям, содержащим данные соединения, а также способам получения указанных соединений и способам их применения для лечения, профилактики и/или ослабления последствий раковых заболеваний путем усиления цитотоксического действия ионизирующего облучения на опухолевые клетки.
Настоящее изобретение относится к описанным в данном описании соединениям, их производным и их применению для ингибирования поли(АДФ-рибоза)полимеразы ("PARP"), композициям, содержащим данные соединения, а также способам получения указанных соединений и способам их применения для лечения последствий раковых заболеваний путем усиления цитотоксического действия химиотерапевтических средств на опухолевые клетки.
Настоящее изобретение относится к способу химиосенсибилизации при лечении опухолевых и/или раковых клеток, включающему приведение указанных раковых клеток в контакт с соединением формулы (I) и дополнительно приведение указанных раковых клеток в контакт с противораковым средством.
Настоящее изобретение относится к описанным в данном описании соединениям, их производным и их применению для ингибирования поли(АДФ-рибоза)полимеразы ("PARP"), композициям, содержащим данные соединения, а также способам получения указанных соединений и способам их применения для ингибирования роста клеток, имеющих дефекты в пути репарации двухцепочечной ДНК посредством гомологичной рекомбинации (HR).
Соединения и композиции настоящего изобретения можно использовать при лечении рака в присутствии или отсутствии радио- или химиосенсибилизирующих средств. Соединения и композиции предпочтительно используют в отсутствии радио- или химиосенсибилизирующих средств, где рак связан с дефектом в пути репарации двухцепочечной ДНК посредством гомологичной рекомбинации (HR). Такие дефекты включают фенотипы дефектов BRCA-1, дефектов BRCA-2, двойных дефектов BRCA-1/BRCA-2 и анемии Фанкони.
Анемия Фанкони представляет собой генетически гетерогенное заболевание, и пациенты с анемией Фанкони имеют сильно повышенный риск развития ракового заболевания. Одиннадцать белков ассоциированы с анемией Фанкони. FANCA, FANCB, FANCC, FANCE, FANCF, FANCG и FANCM образуют ядерный комплекс. Комплекс взаимодействует с FANCL с включением убихинона FANCD2. Модифицированный FANCD2 необходим для репарации поперечных связей ДНК. FANCd2 накапливается в участках повреждения ДНК и связан с BRCA-1 и BRCA-2.
Примеры раковых заболеваний, которые могут быть связаны с дефектами HR, включают рак молочной железы и рак яичника. Рак молочной железы, который можно лечить с помощью способов настоящего изобретения, включает все типы рака молочной железы, предпочтительно, инвазивный протоковый рак и инвазивный дольковый рак. Рак яичника, который можно лечить с помощью способов настоящего изобретения, включает все типы рака яичника, предпочтительно, эпителиальные опухоли яичника, герминогенные опухоли яичников и стромальные опухоли зародышевых тяжей.
Соединения настоящего изобретения можно синтезировать с использованием исходных веществ и способов, раскрытых в заявке США 10/853714, которая полностью включена в данное описание посредством ссылки.
Как правило, соединения, используемые в композициях настоящего изобретения, такие как соединения формулы (I), ингибируют поли(АДФ-рибоза)полимеразу in vitro с IC50, составляющей примерно 20 мкМ или менее, предпочтительно, менее чем примерно 10 мкМ, более предпочтительно, менее чем примерно 1 мкМ, или, предпочтительно, менее чем примерно 0,1 мкМ, наиболее предпочтительно, менее чем примерно 0,01 мкМ.
Подходящим способом определения IC50 соединения, ингибирующего PARP, является описанный ниже анализ PARP с использованием очищенного рекомбинантного человеческого PARP от Trevigan (Gaithersburg, Md.): ферментный анализ PARP проводят на льду в объеме 100 микролитров, который содержит 100 мМ Tris-HCl (pH 8,0), 1 мМ MgCl2, 28 мМ KCl, 28 мМ NaCl, 3,0 мкг/мл ДНКаза I-активированной ДНК спермы сельди (Sigma, Mo.), 30 микромолярный [3H]никотинамидадениндинуклеотид (62,5 мКи/ммоль), 15 микрограмм/мл фермента PARP и тестируемые соединения в разных концентрациях. Реакцию инициируют путем добавления фермента, затем смесь инкубируют при 25°C. После 2 минут инкубации реакцию останавливают добавлением 500 микролитров охлажденной на льду 30% (масс/об) трихлоруксусной кислоты. Образовавшийся осадок переносят на фильтр из стекловолокна (Packard Unifilter-GF/C) и промывают три раза 70% этанолом. После сушки фильтра определяют радиоактивность путем подсчета сцинтилляций. С помощью описанного анализа ингибирования обнаружено, что соединения настоящего изобретения обладают высокой ферментативной активностью с IC50 в интервале от нескольких наномолей до 20 микромолей.
В данном описании термин "алкил" означает разветвленную или линейную насыщенную углеводородную цепь, содержащую указанное число атомов углерода. Например, если не указано иное, линейный или разветвленный C1-C6алкил представляет собой углеводородную цепь, которая содержит от 1 до 6 атомов углерода, и включает, но без ограничения, такие заместители, как метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, н-пентил, н-гексил и т.п. В некоторых вариантах осуществления алкильная цепь представляет собой C1-C6 разветвленную или линейную углеродную цепь. В некоторых вариантах осуществления алкильная цепь представляет собой C2-C5 разветвленную или линейную углеродную цепь. В некоторых вариантах осуществления алкильная цепь представляет собой C1-C4 разветвленную или линейную углеродную цепь. В некоторых вариантах осуществления алкильная цепь представляет собой C2-C4 разветвленную или линейную углеродную цепь. В некоторых вариантах осуществления алкильная цепь представляет собой C3-C5 разветвленную или линейную углеродную цепь. В некоторых вариантах осуществления алкильная цепь представляет собой C1-C2 разветвленную или линейную углеродную цепь. В некоторых вариантах осуществления алкильная цепь представляет собой C2-C3 разветвленную или линейную углеродную цепь.
Термин "алкенил" означает разветвленную или линейную ненасыщенную углеводородную цепь, содержащую указанное число атомов углерода. Например, если не указано иное, линейный или разветвленный C2-C6алкенил представляет собой углеводородную цепь, которая содержит от 2 до 6 атомов углерода и по меньшей мере одну двойную связь, и включает, но без ограничения, такие заместители, как этенил, пропенил, изопропенил, бутенил, изобутенил, трет-бутенил, н-пентенил, н-гексенил и т.п. В некоторых вариантах осуществления алкенильная цепь представляет собой C2-C6 разветвленную или линейную углеродную цепь. В некоторых вариантах осуществления алкенильная цепь представляет собой C2-C5 разветвленную или линейную углеродную цепь. В некоторых вариантах осуществления алкенильная цепь представляет собой C2-C4 разветвленную или линейную углеродную цепь. В некоторых вариантах осуществления алкенильная цепь представляет собой C3-C5 разветвленную или линейную углеродную цепь.
Термин "алкокси" означает группу -OZ, где Z представляет собой алкил, определенный в данном описании. Z также может представлять разветвленную или линейную, насыщенную углеводородную цепь, содержащую от 1 до 6 атомов углерода.
Термин "цикло", используемый в данном описании в качестве префикса, относится к структуре, характеризующейся как замкнутое кольцо.
Если не указано иное, термин "галоген" означает по меньшей мере один атом фтора, хлора, брома или йода.
Каждый из описанных в данном описании "NRARB", "NRXRY", "NRCRD" и "NRERF" независимо означает амино (NH2), а также замещенный амино. Например, NRARB может означать -NH(CH3), -NH(циклогексил) и -N(CH2CH3)(CH3). Если более чем один заместитель представляет собой "NRARB", "NRXRY", "NRCRD" или "NRERF", каждый из RA, RB, RC, RD, RX или RY независимо выбирают из группы, включающей водород, C1-C6алкил с линейной или разветвленной цепью, C2-C6алкенил с линейной или разветвленной цепью и C3-C8циклоалкил. Такие примеры приводятся только для иллюстрации и не предназначаются для какого-либо ограничения.
Термин "арилкарбонил" относится к карбонильному радикалу, замещенному арилом, описанным в данном описании. Неограничивающие примеры включают фенилкарбонил и нафтилкарбонил.
Термин "алкилкарбонил" относится к карбонильному радикалу, замещенному алкилом, описанным в данном описании. Неограничивающие примеры включают ацил и пропилкарбонил.
Термин "алкоксикарбонил" относится к карбонильному радикалу, замещенному алкокси, описанным в данном описании. Неограничивающие примеры включают метоксикарбонил и трет-бутилоксикарбонил.
Термин "Ar" или "арил" относится к ароматическому карбоциклическому фрагменту, содержащему одно или несколько замкнутых колец. Примеры включают, но без ограничения, фенил, нафтил, антраценил, фенантраценил, бифенил и пиренил.
Термин "гетероциклил" относится к циклическому фрагменту, содержащему одно или несколько замкнутых колец и один или несколько гетероатомов (например, атом кислорода, азота или серы) по меньшей мере в одном из колец, где кольцо или кольца независимо могут быть ароматическими, неароматическими, сопряженными и/или соединенными мостиком. Примеры включают, но без ограничения, пиперидинил, пиперазинил, пиредазинил, дигидропиридил, тетрагидропиридил, пиридинил, пиримидинил, дигидропиримидинил, тетрагидропиримидинил, гексагидропиримидинил, дигидропиразинил, тетрагидропиразинил, пирролидинил, имидазолидинил, пиразолидинил, пирролил, дигидропирролил, имидазолил, дигидроимидазолил, пиразолил, дигидропиразолил, азепанил, [1,2]диазепанил, [1,3]диазепанил, [1,4]диазепанил, индолил, дигидроиндолил, изоиндолил, дигидроиндолил, дигидрохинолил, тетрагидрохинолил, дигидроизохинолил и тетрагидроизохинолил.
Термин "арилалкил" относится к алкильному радикалу, замещенному арилом. Неограничивающие примеры включают бензил, фенилэтил и фенилпропил.
Термин "алкиларил" относится к арильному радикалу, замещенному алкилом. Неограничивающие примеры включают толил и диметилфенил.
Термин "циклоалкил" относится к углеводородному циклическому фрагменту, который не является ароматическим. Примеры включают, но без ограничения, циклопропан, циклобутан, циклопентан, циклогексан, циклогептан, циклооктан, циклопентен, циклогексен, циклогептен и циклооктен.
Термин "нервное повреждение" относится к любому повреждению нервной ткани с последующим нарушением функционирования или гибелью. Причина нервного повреждения может носить метаболический, токсический, нейротоксический, ятрогенный, термальный или химический характер, и включает, но без ограничения, ишемию, гипоксию, удар, травму, хирургическое вмешательство, давление, избыточный вес, кровоизлияние, облучение, вазоспазм, нейродегенеративное заболевание, инфекцию, болезнь Паркинсона, боковой амиотрофический склероз (ALS), процесс миелинизации/демиелинизации, эпилепсию, когнитивное расстройство, глутаматное расстройство, а также их вторичные эффекты.
Термин "нейропротективный" относится к способности уменьшать, останавливать или облегчать нервное повреждение, а также защищать, восстанавливать или оживлять поврежденную нервную ткань.
Термин "предотвращение нейродегенерации" включает способность предотвращать развитие нейродегенеративного заболевания или предотвращать дополнительную нейродегенерацию у пациентов, уже страдающих от нейродегенеративного заболевания или имеющих его симптомы.
Термин "лечение" относится к:
(i) предотвращению развития заболевания, нарушения или состояния у животного, которое может быть предрасположено к заболеванию, нарушению и/или состоянию, но еще не имеет его симптомов; и/или
(ii) ингибированию заболевания, нарушения или состояния, т.е. прекращения его развития; и/или
(iii) облегчению заболевания, нарушения или состояния, т.е. инициации регрессии заболевания, нарушения и/или состояния.
Термин "химиосенсибилизирующее средство", как использовано в данном описании, относится к молекуле, такой как молекула низкомолекулярного соединения, которая после введения животным в терапевтически эффективных количествах может усиливать противоопухолевую активность химиотерапевтических средств. Такие химиосенсибилизирующие средства можно использовать, например, для усиления эффекта замедления или прекращения роста опухоли, обеспечиваемого конкретной дозой химиотерапевтического средства, или для улучшения профиля побочных эффектов химиотерапевтического средства в результате снижения его дозы при сохранении его противоопухолевой активности.
Термин "радиосенсибилизирующее средство", как использовано в данном описании, относится к молекуле, такой как молекула низкомолекулярного соединения, которая после введения животным в терапевтически эффективных количествах может повышать чувствительность клеток к электромагнитному облучению и/или способствовать лечению заболеваний с помощью электромагнитного облучения. Заболевания, которые можно лечить с помощью электромагнитного облучения, включают неопластические заболевания, доброкачественные и злокачественные опухоли и раковые клетки. С помощью электромагнитного облучения можно лечить и другие заболевания, не приведенные в данном описании.
Термин "эффективное количество" относится к количеству, необходимому для достижения желаемого эффекта.
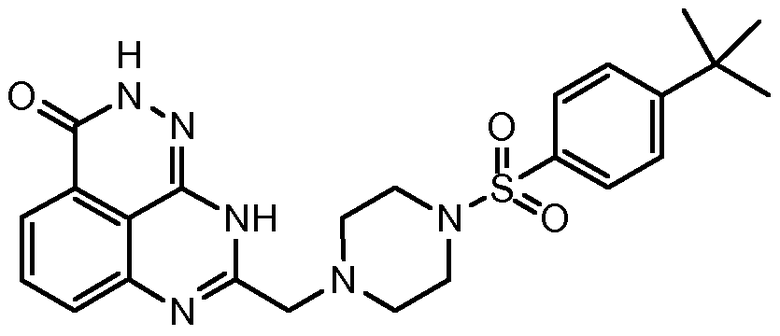
Термин "замещенный" означает, что по меньшей мере один атом водорода на указанной группе заменен другим радикалом с учетом нормальной валентности данной группы. Любая группа может содержать один или несколько заместителей, при условии, что все замены являются стерически возможными, синтетически осуществимыми, и/или полученные в результате структуры являются стабильными. Как описано в данном описании, в некоторых вариантах осуществления настоящего изобретения заместитель может замещать радикал, который сам по себе является заместителем. Например, в соединении, приведенном ниже только для иллюстрации, пиперазинильное кольцо представляет собой гетероциклил, который может быть замещен 0-4 заместителями, как описано в данном описании. В примере соединения пиперазинильное кольцо замещено арилсульфонилом, где арил представляет собой фенил, и где арилсульфонил может быть дополнительно замещен 0-4 заместителями, как описано в данном описании. В примере соединения фенилсульфонильный фрагмент дополнительно замещен трет-бутилом. Такой пример приведен только для иллюстрации и не предназначен для какого-либо ограничения.
Термин "субъект" относится к клетке или ткани in vitro или in vivo, к животному или человеку. Животное или человека также можно называть "пациентом".
Термин "животное" относится к живому организму, обладающему чувствами и способностью к сознательному движению, для жизнедеятельности которого требуется кислород и органическая пища. Примеры включают, но без ограничения, виды человекообразных, млекопитающих и приматов.
Ориентировочно, соединения и композиции настоящего изобретения можно использовать при лечении или профилактике повреждения или гибели клеток в результате некроза или апоптоза, ишемии и реперфузионного повреждения головного мозга, или нейродегенеративных заболеваний у животного, такого как человек. Соединения и композиции настоящего изобретения можно использовать для увеличения продолжительности жизни и пролиферативной способности клеток и, как следствие, их можно использовать при лечении или профилактике заболеваний, связанных с продолжительностью жизни и пролиферативной способностью; они изменяют экспрессию генов в стареющих клетках; кроме того, они обеспечивают радиосенсибилизацию гипоксических опухолевых клеток. Предпочтительно, соединения и композиции настоящего изобретения можно использовать при лечении или профилактике повреждения ткани после повреждения или гибели клеток вследствие некроза или апоптоза, и/или эффекта нейронной активности, опосредованного или не опосредованного токсичностью NMDA. Применение соединений настоящего изобретения не ограничивается лечением глутамат-опосредованной нейротоксичности и/или NO-опосредованных биологических путей. Кроме того, соединения настоящего изобретения можно использовать при лечении или профилактике других повреждений тканей, связанных с активацией PARP, как описано в данном описании.
Настоящее изобретение предлагает соединения, которые ингибируют полимеразную активность поли(АДФ-рибоза)полимеразы (PARP) in vitro и/или in vivo, а также композиции, содержащие раскрытые соединения.
Настоящее изобретение предлагает способы ингибирования, ограничения и/или контролирования полимеразной активности поли(АДФ-рибоза)полимеразы (PARP) in vitro и/или in vivo в любых растворах, клетках, тканях, органах или системах органов. В одном варианте осуществления настоящее изобретение предлагает местные или системные способы ограничения или ингибирования активности PARP у млекопитающего, такого как человек.
Соединения настоящего изобретения, действующие в качестве ингибиторов PARP, можно использовать при лечении или профилактике рака путем химиопотенциирования цитотоксических эффектов химиотерапевтических средств. Соединения настоящего изобретения, действующие в качестве ингибиторов PARP, можно использовать при лечении или профилактике рака путем сенсибилизации клеток к цитотоксическим эффектам облучения. Соединения настоящего изобретения, действующие в качестве ингибиторов PARP, можно использовать при лечении или профилактике BRCA1/2-ассоциированного рака молочной железы.
Соединения настоящего изобретения могут содержать один или несколько асимметричных центров и, следовательно, могут быть получены в виде смесей (рацемических и нерацемических) стереоизомеров, или в виде индивидуальных энантиомеров или диастереомеров. Индивидуальные стереоизомеры можно получить путем использования оптически активного исходного вещества, путем разделения рацемической или нерацемической смеси промежуточного соединения на подходящей стадии синтеза, или путем разделения соединения формулы (I). Следует понимать, что в объем настоящего изобретения входят как отдельные стереоизомеры, так и смеси (рацемические и нерацемические) стереоизомеров.
Соединения настоящего изобретения можно использовать в виде свободных оснований, в виде фармацевтически приемлемых солей, фармацевтически приемлемых гидратов, фармацевтически приемлемых сложных эфиров, фармацевтически приемлемых сольватов, фармацевтически приемлемых пролекарств, фармацевтически приемлемых метаболитов и в виде фармацевтически приемлемых стереоизомеров. Все указанные формы входят в объем настоящего описания.
Термин "фармацевтически приемлемые соль", "гидрат", "сложный эфир" или "сольват" относится к соли, гидрату, сложному эфиру или сольвату соединения настоящего изобретения, которые обладают желаемой фармакологической активностью и не оказывают неблагоприятного биологического или иного действия. Органические кислоты можно использовать для получения солей, гидратов, сложных эфиров или сольватов, таких как ацетат, адипат, альгинат, аспартат, бензоат, бензолсульфонат, п-толуолсульфонат, бисульфат, сульфамат, сульфат, нафтилат, бутират, цитрат, камфорат, камфорсульфонат, циклопентанпропионат, диглюконат, додецилсульфат, этансульфонат, фумарат, глюкогептаноат, глицерофосфат, гемисульфат гептаноат, гексаноат, 2-гидроксиэтансульфонат, лактат, малеат, метансульфонат, 2-нафталинсульфонат, никотинат, оксалат, тозилат и ундеканоат. Неорганические кислоты можно использовать для получения солей, гидратов, сложных эфиров или сольватов, таких как гидрохлорид, гидробромид, гидроиодид и тиоцианат.
Примеры оснований, подходящих для получения солей, гидратов, сложных эфиров или сольватов, включают гидроксиды, карбонаты и бикарбонаты аммония, соли щелочных металлов, такие как соли натрия, лития и калия, соли щелочноземельных металлов, такие как соли кальция и магния, соли алюминия и соли цинка.
Соли, гидраты, сложные эфиры или сольваты также можно получить с использованием органических оснований. Органические основания, подходящие для получения фармацевтически приемлемых основно-аддитивных солей, гидратов, сложных эфиров или сольватов соединений настоящего изобретения, включают нетоксичные основания, достаточно сильные для получения таких солей, гидратов, сложных эфиров или сольватов. В качестве иллюстрации класс таких органических оснований может включать моно-, ди- и триалкиламины, такие как метиламин, диметиламин, триэтиламин и дициклогексиламин; моно-, ди- или тригидроксиалкиламины, такие как моно-, ди- и триэтаноламин; аминокислоты, такие как аргинин и лизин; гуанидин; N-метилглюкозамин; N-метилглюкамин; L-глютамин; N-метилпиперазин; морфолин; этилендиамин; N-бензилфенетиламин; (тригидроксиметил)аминоэтан; и т.п.. См., например, "Pharmaceutical Salts," J. Pharm. Sci., 66: 1, 1-19 (1977). Соответственно, основные азотсодержащие группы можно кватернизировать с помощью средств, включающих: низшие алкилгалогениды, такие как метил-, этил-, пропил- и бутилхлориды, -бромиды и -иодиды; диалкилсульфаты, такие как диметил-, диэтил-, дибутил- и диамилсульфаты; длинноцепочечные галогениды, такие как децил-, лаурил-, миристил- и стеарилхлориды, -бромиды и -иодиды; а также аралкилгалогениды, такие как бензил- и фенетилбромиды.
Кислотно-аддитивные соли, гидраты, сложные эфиры или сольваты основных соединений можно получить путем растворения соединения настоящего изобретения в виде свободного основания в водном или водно-спиртовом растворе, или в другом подходящем растворителе, содержащем подходящую кислоту или основание, с последующим выделением соли путем упаривания раствора. Альтернативно, соединение настоящего изобретения в виде свободного основания можно подвергнуть взаимодействию с кислотой, и соединение настоящего изобретения, несущее кислотную группу, можно подвергнуть взаимодействию с основанием, в органическом растворителе, в котором соль сразу выделяется, или ее можно получить путем концентрирования раствора.
Термин "фармацевтически приемлемое пролекарство" относится к производному соединения настоящего изобретения, которое оказывает фармакологическое действие после биотрансформации. Пролекарство используют для повышения химической стабильности, облегчения одобрения и согласия пациента, улучшения биодоступности, увеличения продолжительности действия, повышения органоселективности, улучшения характеристик композиции (например, повышения растворимости в воде) и/или уменьшения побочных эффектов (например, токсичности). Пролекарство можно легко получить из соединения настоящего изобретения с помощью известных в данной области способов, таких как описано Burgers Medicinal Chemistry and Drug Chemistry, Fifth Ed, Vol. 1, pp. 172-178, 949-982 (1995). Например, соединения настоящего изобретения можно преобразовать в пролекарства путем превращения одной или нескольких гидроксильных или карбоксильных групп в сложные эфиры.
Термин "фармацевтически приемлемый метаболит" относится к лекарственному средству, претерпевшему метаболическую трансформацию. После попадания в организм большая часть лекарственных средств может принимать участие в химических реакциях в качестве субстратов, в результате чего может происходить изменение физических свойств и биологических эффектов. Такие метаболические преобразования, которые обычно изменяют полярность соединения, могут влиять на распределение лекарственных средств в организме и их выведение из организма. Однако в некоторых случаях метаболическое преобразование лекарственного средства необходимо для проявления терапевтического эффекта. Например, противораковые средства, относящиеся к классу антиметаболитов, превращаются в активные формы после их транспортировки в раковые клетки. Поскольку большинство лекарственных средств могут подвергаться метаболической трансформации любого рода, в метаболизме лекарственного средства участвуют многочисленные и разнообразные биохимические реакции. Основным участком метаболизма лекарственных средств является печень, хотя другие ткани также могут участвовать в метаболизме.
Более того, способы настоящего изобретения можно использовать при лечении рака, а также для химиосенсибилизации и радиосенсибилизации раковых и/или опухолевых клеток. Термин "рак" в данном описании используется в широком смысле. Соединения настоящего изобретения могут усиливать эффекты "противораковых средств", причем данный термин также охватывает "средства против роста опухолевых клеток", "химиотерапевтические средства", "цитостатические средства", "цитотоксические средства" и "антинеопластичные средства". Термин "BRCA1/2-ассоциированный рак молочной железы" относится к раку молочной железы, клетки которого являются дефицитными по генам-супрессорам раковой опухоли молочной железы BRCA1 и/или BRCA2.
Например, способы настоящего изобретения можно использовать при лечении раковых заболеваний, таких как АКТГ-продуцирующие опухоли, острый лимфоцитарный лейкоз, острый нелимфоцитарный лейкоз, рак коры надпочечника, рак мочевого пузыря, рак мозга, рак молочной железы, рак шейки матки, хронический лимфоцитарный лейкоз, хронический миелоцитарный лейкоз, рак ободочной и прямой кишки, Т-клеточная лимфома кожи, рак эндометрия, рак пищевода, саркома Юинга, рак желчного пузыря, лейкоз ворсистых клеток, рак головы и шеи, лимфома Ходжкина, саркома Капоши, рак почки, рак печени, рак легкого (мелкоклеточный и немелкоклеточный), злокачественный перитонеальный выпот, злокачественный плевральный выпот, меланома, мезотелиома, множественная миелома, нейробластома, неходжкинская лимфома, остеосаркома, рак яичника, рак яичников (герминогенный рак), рак простаты, рак поджелудочной железы, рак полового члена, ретинобластома, рак кожи, саркома мягкой ткани, плоскоклеточная карцинома, рак желудка, рак яичка, рак щитовидной железы, трофобластические опухоли, рак матки, рак влагалища, рак наружных половых органов и опухоль Вильмса.
В некоторых неограничивающих вариантах осуществления раковые и/или опухолевые клетки выбирают из группы, включающей клетки рака мозга, меланомы, рака головы и шеи, немелкоклеточного рака легкого, рака яичка, рака яичника, рака толстой кишки и рака прямой кишки.
Настоящее изобретение также относится к фармацевтической композиции, содержащей (i) терапевтически эффективное количество соединения формулы (I) и (ii) фармацевтически приемлемый носитель.
Приведенное выше обсуждение использования предпочтительных вариантов осуществления и введения соединений настоящего изобретения также применимо к фармацевтическим композициям настоящего изобретения.
Термин "фармацевтически приемлемый носитель", как использовано в данном описании, относится к любым носителям, разбавителям, эксципиентам, суспендирующим средствам, смазывающим средствам, адъювантам, наполнителям, системам доставки, эмульгаторам, дезинтегрантам, абсорбентам, консервантам, поверхностно-активным веществам, красителям, ароматизаторам или подсластителям.
Для достижения указанных целей композицию настоящего изобретения можно вводить перорально, парентерально, с помощью спрея для ингалиции, путем адсорбции или абсорбции, местно, ректально, назально, буккально, вагинально, интравентрикулярно, посредством имплантируемого резервуара, в виде дозированных форм, содержащих традиционные нетоксичные фармацевтически приемлемые носители, или в виде любых других традиционных дозированных форм. Термин парентеральный в данном описании относится к таким способам введения, как подкожное, внутривенное, внутримышечное, внутрибрюшинное, интратекальное интравентрикулярное, надчревное, а также внутричерепные инъекции или инфузии.
Композиция для парентерального введения обычно находится в виде единичной дозированной формы, стерильной формы для инъекции (раствор, суспензия или эмульсия), которая предпочтительно является изотоничной по отношению к крови реципиента и содержит фармацевтически приемлемый носитель. Примерами таких стерильных форм для инъекций являются стерильные инъецируемые водные или масляные суспензии. Такие суспензии могут быть получены с помощью известных в данной области способов, используя подходящие диспергирующие или увлажняющие средства и суспендирующие средства. Стерильные формы для инъекций также могут представлять собой стерильные инъецируемые растворы или суспензии в нетоксичных парентерально приемлемых разбавителях или растворителях, например, растворы в 1,3-бутандиоле. К подходящим приемлемым средам и растворителям относятся вода, физиологический раствор, раствор Рингера, раствор декстрозы, изотонический раствор хлорида натрия и раствор Хэнкса. Кроме того, в качестве растворителей или суспендирующих сред часто используют стерильные жирные масла. Для данной цели можно использовать любое легкое жирное масло, включая синтетические моно- или диглицериды, кукурузное, хлопковое, арахисовое и кунжутное масло. Для получения инъецируемых препаратов можно использовать жирные кислоты, такие как этилолеат, изопропилмиристат и олеиновая кислота, а также их глицеридные производные, включая оливковое масло и касторовое масло, особенно их полиоксиэтилированные варианты. Указанные масляные растворы или суспензии также могут содержать длинноцепочечные спирты в качестве разбавителей или диспергирующих средств.
Стерильный физиологический раствор представляет собой предпочтительный носитель, и соединения зачастую достаточно хорошо растворяются в воде, позволяя использовать их в виде раствора. Носитель может содержать в небольших количествах вспомогательные средства, такие как вещества, повышающие растворимость, изотоничность и химическую стабильность, например, антиоксиданты, буферы и консерванты.
Композиции, подходящие для назального или буккального введения (такие как распыляемые композиции с самопропеллирующим порошком), могут содержать примерно от 0,1% до 5% масс/масс, например 1% масс/масс активного ингредиента. Композиции настоящего изобретения, предназначенные для лечения человека, содержат активный ингредиент в сочетании с фармацевтически приемлемым носителем и необязательно другой терапевтический ингредиент (ингредиенты).
Композиции для перорального введения обычно получают в виде единичных дозированных форм, таких как таблетки, облатки, порошки, гранулы, шарики, жевательные пастилки, капсулы, жидкости, водные суспензии или растворы, или подобные лекарственные формы, с использованием традиционного оборудования и известных в данной области способов. Такие композиции обычно содержат твердый, полутвердый или жидкий носитель. Примеры носителей включают лактозу, декстрозу, сахарозу, сорбит, маннит, крахмалы, гуммиарабик, фосфат кальция, минеральное масло, кокосовое масло, масло какао, альгинаты, трагакант, желатин, сироп, метилцеллюлозу, полиоксиэтиленсорбитана монолаурат, метилгидроксибензоат, пропилгидроксибензоат, тальк, стеарат магния и т.п.
Композицию настоящего изобретения предпочтительно вводят в виде капсулы или таблетки, содержащей однократную или разделенную дозу соединения формулы (I) или его фармацевтически приемлемой соли. Композицию можно вводить в виде стерильных растворов, суспензий или эмульсий, содержащих однократную или разделенную общую дозу. Таблетки могут содержать носители, такие как лактоза и кукурузный крахмал, и/или смазывающие средства, такие как стеарат магния. Капсулы могут содержать разбавители, такие как лактоза и сухой кукурузный крахмал.
Таблетку можно получить путем прессования или формования активного ингредиента, необязательно вместе с одним или несколькими вспомогательными ингредиентами. Прессованные таблетки можно получить путем прессования в подходящей машине активного ингредиента, находящегося в свободнотекучем виде, например, в виде порошка или гранул, необязательно в смеси со связующим средством, лубрикантом, инертным разбавителем, поверхностно-активным веществом или диспергирующим средством. Формованные таблетки можно получить путем формования в подходящей машине смеси порошкообразного активного ингредиента и подходящего носителя, увлажненной инертным жидким разбавителем.
Соединения настоящего изобретения также можно вводить ректально в виде суппозиториев. Такие композиции можно получить путем смешивания лекарственного средства с подходящим нераздражающим эксципиентом, который является твердым при комнатной температуре и жидким при ректальной температуре, и, следовательно, плавится в прямой кишке, высвобождая лекарственное средство. Такие вещества включают масло какао, пчелиный воск и полиэтиленгликоли.
При применении композиций и способов настоящего изобретения также можно использовать технологию контролируемого высвобождения. Так, например, раскрытые соединения можно внедрить в гидрофобную полимерную матрицу, обеспечивающую контролируемое высвобождение в течение нескольких дней. Затем композицию настоящего изобретения можно ввести в состав твердого имплантата или наружного пластыря, обеспечивающих высвобождение эффективных концентраций ингибиторов PARP в течение длительного периода времени без необходимости частого многократного введения дозы. Пленки с таким контролируемым высвобождением хорошо известны в данной области. Особенно предпочтительны системы чрезкожной доставки. Другие примеры полимеров, традиционно используемых для данной цели и подходящих для применения в настоящем изобретении, включают неразрушаемый этиленвинилацетатный сополимер, разрушаемые сополимеры молочной кислоты и гликолевой кислоты, которые можно использовать наружно или внутренне. Также можно использовать некоторые гидрогели, такие как поли(гидроксиэтилметакрилат) или поли(виниловый спирт), однако они обеспечивают более краткосрочные циклы высвобождения, чем другие полимерные системы высвобождения, такие, как указано выше.
В одном варианте осуществления носитель представляет собой твердый биоразрушаемый полимер или смесь биоразрушаемых полимеров с подходящими временными характеристиками высвобождения и кинетикой высвобождения. Затем композицию настоящего изобретения можно ввести в состав твердого имплантата, подходящего для обеспечения эффективных концентраций соединения настоящего изобретения в течение длительного периода времени без необходимости частого многократного введения дозы. Композицию настоящего изобретения можно внедрить в биоразрушаемый полимер или смесь биоразрушаемых полимеров любым подходящим способом, известным специалисту в данной области, с получением гомогенной матрицы на основе биоразрушаемого полимера, или ее можно заключить в капсулу из полимера, или ввести в состав твердого имплантата.
В одном варианте осуществления биоразрушаемый полимер или смесь биоразрушаемых полимеров используют для получения мягкого "депо", содержащего фармацевтическую композицию настоящего изобретения, которое можно вводить в виде текучей жидкости, например, путем инъекции, но которое остается достаточно вязким, чтобы удерживать фармацевтическую композицию в локализованном участке вблизи места введения. Время разрушения полученного таким образом депо может варьировать от нескольких дней до нескольких лет, в зависимости от типа выбранного полимера и его молекулярной массы. Применение полимерной композиции в инъецируемой форме даже позволяет устранить необходимость делать разрез. В любом случае, пластичное или текучее "депо" доставки принимает форму пространства, которое оно занимает в организме, сводя к минимуму травмирование окружающих тканей. Фармацевтическую композицию настоящего изобретения используют в количестве, которое является терапевтически эффективным и может зависеть от желаемого профиля высвобождения, концентрации фармацевтической композиции, необходимой для достижения эффекта сенсибилизации, и продолжительности времени, в течение которого фармацевтическая композиция должна высвобождаться, обеспечивая лечение.
Соединения настоящего изобретения входят в состав композиции в терапевтических количествах. Композиции могут быть стерильными и/или они могут содержать адъюванты, такие как консерванты, стабилизирующие средства, увлажняющие или эмульгирующие средства, средства, способствующие растворению, соли, регулирующие осмотическое давление, и/или буферы. Кроме того, они могут содержать другие терапевтически полезные вещества, включающие, но без ограничения, конкретные химиотерапевтические средства, описанные в данном описании. Композиции, которые получают традиционными методами смешивания, гранулирования или нанесения покрытия, содержат примерно 0,1-75% по массе, предпочтительно примерно 1-50% по массе соединения настоящего изобретения.
Чтобы оказывать эффективное терапевтическое действие на мишени центральной нервной системы после периферического введения, соединения настоящего изобретения должны легко проникать через гематоэнцефалический барьер. Соединения, которые не могут проникать через гематоэнцефалический барьер, можно эффективно вводить интравентрикулярно или с использованием системы доставки, подходящей для введения в мозг.
При применении в медицине количество активного ингредиента, необходимое для достижения терапевтического эффекта, варьирует в зависимости от конкретного соединения, способа введения, вида млекопитающего, подлежащего лечению, а также конкретного нарушения или заболевания, подлежащего лечению. Подходящая систематическая доза соединения настоящего изобретения или его фармакологически приемлемой соли, вводимая млекопитающему, страдающему от или которое может страдать от любого состояния, описанного выше в данном описании, находится в интервале примерно от 0,1 мг/кг до 100 мг/кг, предпочтительно, примерно от 1 до 10 мг/кг.
Однако следует понимать, что конкретный уровень дозы для любого конкретного пациента зависит от ряда факторов, включающих активность конкретного используемого соединения, возраст, массу тела, общее состояние здоровья, пол, диету, время введения, скорость выведения, сочетание лекарственных средств, тяжесть конкретного заболевания, подлежащего лечению, и способ введения.
Очевидно, что рядовой врач или ветеринар может легко определить и назначить эффективное количество соединения для профилактики или лечения состояния. На практике врач или ветеринар может, например, использовать внутривенную болюсную инъекцию с последующим внутривенным вливанием и многократным введением, парентеральным или пероральным, в зависимости от необходимости. Хотя активный ингредиент можно вводить сам по себе, его предпочтительно использовать в составе композиции.
При получении дозированной формы, содержащей композиции настоящего изобретения, соединения также можно смешивать с традиционными эксципиентами, такими как связующие средства, включающие желатин, желатинированный крахмал и т.п.; лубриканты, такие как гидрированное растительное масло, стеариновая кислота и т.п.; разбавители, такие как лактоза, манноза и сахароза; дезинтегранты, такие как карбоксиметилцеллюлоза и гликолят натрийкрахмала; суспендирующие средства, такие как повидон, поливиниловый спирт и т.п.; абсорбенты, такие как диоксид кремния; консерванты, такие как метилпарабен, пропилпарабен и бензоат натрия; поверхностно-активные вещества, такие как лаурилсульфат натрия, полисорбат 80 и т.п.; красители; ароматизаторы; и подсластители. Фармацевтически приемлемые эксципиенты хорошо известны в области фармацевтики и описаны, например, в Remington's Pharmaceutical Sciences, Mack Publishing Co., Easton, Pa (например, 20th Ed., 2000), и Handbook of Pharmaceutical Excipients, American Pharmaceutical Association, Washington, D.C, (например, 1-ое, 2-ое и 3-ье издания, 1986, 1994 и 2000, соответственно).
Настоящее изобретение относится к применению соединения формулы (I) при получении лекарственного средства для лечения у животного любого описанного в данном описании заболевания или нарушения. В одном варианте осуществления соединения настоящего изобретения используют при лечении рака. В предпочтительном варианте осуществления соединения настоящего изобретения используют для усиления цитотоксических эффектов ионизирующего облучения. В данном варианте осуществления соединения настоящего изобретения действуют как радиосенсибилизаторы. В альтернативном предпочтительном варианте осуществления соединения настоящего изобретения используют для усиления цитотоксических эффектов химиотерапевтических средств. В данном варианте осуществления соединения настоящего изобретения действуют как химиосенсибилизаторы. В другом предпочтительном варианте осуществления соединения настоящего изобретения используют для ингибирования роста клеток, имеющих дефекты в пути репарации двухцепочечной ДНК посредством гомологичной рекомбинации (HR).
В качестве химиотерапевтического средства настоящего изобретения можно использовать любое фармакологически приемлемое химиотерапевтическое средство, которое действует путем повреждения ДНК. В частности, настоящее изобретение охватывает применение химиотерапевтически эффективного количества по меньшей мере одного химиотерапевтического средства, включающего, но без ограничения: темозоломид, адриамицин, камптотецин, карбоплатин, цисплатин, даунорубицин, доцетаксел, доксорубицин, интерферон-альфа, интерферон-бета, интерферон-гамма, интерлейкин 2, иринотекан, паклитаксел, топотекан, таксоид, дактиномицин, даунорубицин, 4'-дезоксидоксорубицин, блеомицин, пилкамицин, митомицин, неомицин, гентамицин, этопозид 4-OH циклофосфамид, координационный комплекс платины, топотекан, и их смеси. В соответствии с предпочтительным аспектом, химиотерапевтическое средство представляет собой темозоломид.
Изобретение, описанное в данном описании, демонстрирует применение соединений и композиций настоящего изобретения при лечении и/или профилактике рака, например, путем радиосенсибилизации и/или химиосенсибилизации опухолевых и/или раковых клеток к химиотерапевтическим средствам, а также для ингибирования роста клеток, имеющих дефекты в пути репарации двухцепочечной ДНК посредством гомологичной рекомбинации (HR).
Следующие примеры приведены только с целью иллюстрации и не предназначены для ограничения объема изобретения.
В одном варианте осуществления настоящее изобретение относится к тетраазафенален-3-оновому соединению формулы (I) или его фармацевтически приемлемой соли:
где R означает
(a) NR1R2, где R1 выбирают из группы, включающей водород, C1-C6алкил с линейной или разветвленной цепью, C2-C6алкенил с линейной или разветвленной цепью, C3-C8циклоалкил, C1-C6алкокси, C2-C6алкенилокси, фенил, фенокси, бензилокси, NRARB(C1-C6алкил с линейной или разветвленной цепью), NRARB(C2-C6алкенил с линейной или разветвленной цепью), (C1-C6алкил с линейной или разветвленной цепью)карбонил, (C2-C6алкенил с линейной или разветвленной цепью)карбонил, (C3-C8циклоалкил)карбонил, (C1-C6алкил с линейной или разветвленной цепью)оксикарбонил, (C2-C6алкенил с линейной или разветвленной цепью)оксикарбонил, (C3-C8циклоалкил)оксикарбонил, арилкарбонил, сульфонил, арилсульфонил, арил(C1-C6алкил с линейной или разветвленной цепью), арил(C2-C6алкенил с линейной или разветвленной цепью), арил(C3-C8циклоалкил), (C1-C6алкил с линейной или разветвленной цепью)арил, (C2-C6алкенил с линейной или разветвленной цепью)арил, (C3-C8циклоалкил)арил, арил, гетероциклил, гетероциклил(C1-C6алкил с линейной или разветвленной цепью) и гетероциклил(C2-C6алкенил с линейной или разветвленной цепью); где каждый гетероциклил содержит от 1 до 7 гетероатомов, независимо выбранных из O, N или S, и где каждый из RA и RB независимо выбирают из группы, включающей водород, C1-C6алкил с линейной или разветвленной цепью, C2-C6алкенил с линейной или разветвленной цепью и C3-C8циклоалкил;
и R2 выбирают из группы, включающей C1-C6алкил с линейной или разветвленной цепью, C2-C6алкенил с линейной или разветвленной цепью, C3-C8циклоалкил, C1-C6алкокси, C2-C6алкенилокси, фенил, фенокси, бензилокси, NRXRY(C1-C6алкил с линейной или разветвленной цепью), NRXRY(C2-C6алкенил с линейной или разветвленной цепью), (C1-C6алкил с линейной или разветвленной цепью)карбонил, (C2-C6алкенил с линейной или разветвленной цепью)карбонил, (C3-C8циклоалкил)карбонил, (C1-C6алкил с линейной или разветвленной цепью)оксикарбонил, (C2-C6алкенил с линейной или разветвленной цепью)оксикарбонил, (C3-C8циклоалкил)оксикарбонил, арилкарбонил, сульфонил, арилсульфонил, арил(C1-C6алкил с линейной или разветвленной цепью), арил(C2-C6алкенил с линейной или разветвленной цепью), арил(C3-C8циклоалкил), (C1-C6алкил с линейной или разветвленной цепью)арил, (C2-C6алкенил с линейной или разветвленной цепью)арил, (C3-C8циклоалкил)арил, арил, гетероциклил, гетероциклил(C1-C6алкил с линейной или разветвленной цепью) и гетероциклил(C2-C6алкенил с линейной или разветвленной цепью); где каждый гетероциклил содержит от 1 до 7 гетероатомов, независимо выбранных из O, N или S, и где каждый из RX и RY независимо выбирают из группы, включающей водород, C1-C6алкил с линейной или разветвленной цепью, C2-C6алкенил с линейной или разветвленной цепью и C3-C8циклоалкил;
где R1 и R2 независимо замещены 0-4 заместителями, каждый из которых независимо выбирают из галогена, C1-C6алкила с линейной или разветвленной цепью, C2-C6алкенила с линейной или разветвленной цепью, C1-C6алкокси, трифторметила, трифторэтила и амино; при условии, что R1 и R2, оба не являются метилом, и R2 не является (фенил)проп-1-илом, если R1 означает водород; или
(b) арилокси, замещенный 0-4 заместителями, каждый из которых независимо выбирают из группы, включающей галоген, C1-C6алкокси, трифторметил, трифторэтил, C1-C6алкил с линейной или разветвленной цепью, C2-C6алкенил с линейной или разветвленной цепью, C3-C8циклоалкил, NRCRD, NRCRD(C1-C6алкил с линейной или разветвленной цепью) и NRCRD(C2-C6алкенил с линейной или разветвленной цепью), где каждый из RC и RD независимо выбирают из группы, включающей водород, C1-C6алкил с линейной или разветвленной цепью, C2-C6алкенил с линейной или разветвленной цепью и C3-C8циклоалкил; и если более чем один заместитель представляет собой NRCRD, каждый из RC и RD независимо выбирают из группы, включающей водород, C1-C6алкил с линейной или разветвленной цепью, C2-C6алкенил с линейной или разветвленной цепью и C3-C8циклоалкил; или
(c) гетероциклил, содержащий от 1 до 7 гетероатомов, независимо выбранных из O, N или S; и от 0 до 4 заместителей, независимо выбранных из группы, включающей галоген, галогеналкил, гидроксил, нитро, трифторметил, трифторэтил, C1-C6алкил с линейной или разветвленной цепью, C2-C6алкенил с линейной или разветвленной цепью, C1-C6алкокси, C2-C6алкенилокси, фенил, фенокси, бензилокси, амино, тиокарбонил, циано, имино, NRERF(C1-C6алкил с линейной или разветвленной цепью), NRERF(C2-C6алкенил с линейной или разветвленной цепью)сульфгидрил, тиоалкил, диоксаспироэтил, (C1-C6алкил с линейной или разветвленной цепью)карбонил, (C2-C6алкенил с линейной или разветвленной цепью)карбонил, (C1-C6алкил с линейной или разветвленной цепью)оксикарбонил, (C2-C6алкенил с линейной или разветвленной цепью)оксикарбонил, арилкарбонил, сульфонил, арилсульфонил, арил(C1-C6алкил с линейной или разветвленной цепью), арил(C2-C6алкенил с линейной или разветвленной цепью), арил(C3-C8циклоалкил), (C1-C6алкил с линейной или разветвленной цепью)арил, (C2-C6алкенил с линейной или разветвленной цепью)арил, (C3-C8циклоалкил)арил, арил, гетероциклил, гетероциклил(C1-C6алкил с линейной или разветвленной цепью) и гетероциклил(C2-C6алкенил с линейной или разветвленной цепью), где каждый гетероциклил содержит от 1 до 7 гетероатомов, независимо выбранных из O, N или S, где каждый из RE и RF независимо выбирают из группы, включающей водород, C1-C6алкил с линейной или разветвленной цепью, C2-C6алкенил с линейной или разветвленной цепью и C3-C8циклоалкил; и если более чем один заместитель представляет собой NRERF, каждый из RE и RF независимо выбирают из группы, включающей водород, C1-C6алкил с линейной или разветвленной цепью, C2-C6алкенил с линейной или разветвленной цепью и C3-C8циклоалкил; где каждый из указанных 0-4 заместителей независимо замещен 0-4 дополнительными заместителями, и каждый из указанных дополнительных заместителей независимо выбирают из галогена, C1-C6алкила с линейной или разветвленной цепью, C2-C6алкенила с линейной или разветвленной цепью, C3-C8циклоалкила, C1-C6алкокси, трифторметила, трифторэтила и амино; при условии, что R содержит по меньшей мере один заместитель, если R означает N-пиперидинильную, N-пирролидинильную или N-морфолинильную группу.
В некоторых вариантах осуществления размер каждого кольца всех гетероциклилов формулы (I) независимо составляет 5-7 атомов.
Некоторые варианты осуществления содержат один, два или три атома азота по меньшей мере в одном кольце гетероциклила формулы (I).
В некоторых вариантах осуществления гетероциклил формулы (I) содержит 1-3 кольца. В некоторых вариантах осуществления гетероциклил содержит 1-7 гетероатомов, независимо выбранных из O, N и S.
В некоторых вариантах осуществления гетероциклил формулы (I) выбирают из группы, включающей пиперидинил, пиперазинил, пиридазинил, дигидропиридил, тетрагидропиридил, пиридинил, пиримидинил, дигидропиримидинил, тетрагидропиримидинил, гексагидропиримидинил, дигидропиразинил, тетрагидропиразинил, пирролидинил, имидазолидинил, пиразолидинил, пирролил, дигидропирролил, имидазолил, дигидроимидазолил, пиразолил, дигидропиразолил, азепанил, [1,2]диазепанил, [1,3]диазепанил, [1,4]диазепанил, индолил, дигидроиндолил, изоиндолил, дигидроизоиндолил, дигидрохинолил, тетрагидрохинолил, дигидроизохинолил и тетрагидроизохинолил.
В другом варианте осуществления настоящее изобретение относится к соединению, выбранному из группы, включающей
и его фармацевтически приемлемым солям.
В некоторых вариантах осуществления изобретение относится к соединению, имеющему структуру
или его фармацевтически приемлемой соли.
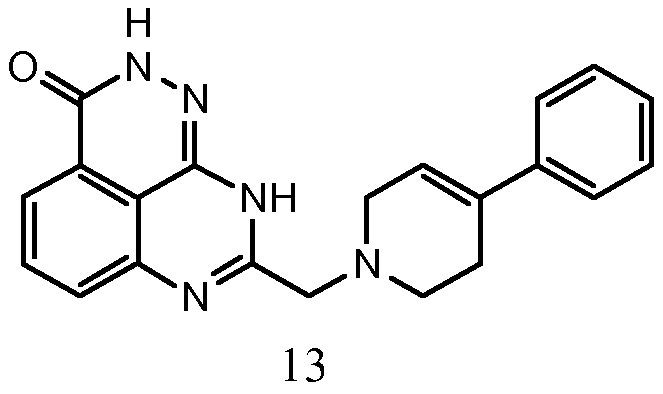
В некоторых вариантах осуществления изобретение относится к соединению, имеющему структуру
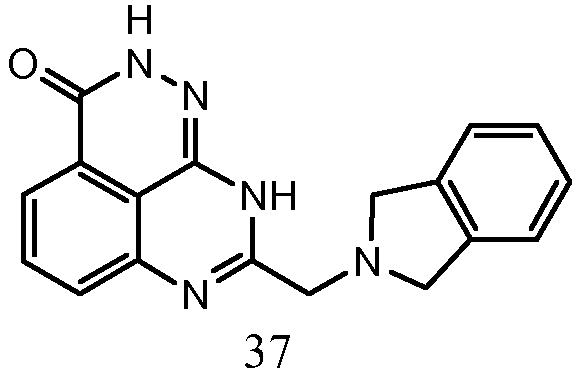
или его фармацевтически приемлемой соли.
В некоторых вариантах осуществления настоящее изобретение относится к способу химиосенсибилизации раковых клеток у млекопитающего, нуждающегося в химиотерапии, включающему введение указанному млекопитающему соединения формулы (I), описанного в данном описании, или его фармацевтически приемлемой соли. В некоторых вариантах осуществления указанным млекопитающим является человек. В некоторых вариантах осуществления указанное введение представляет собой введение фармацевтической композиции, содержащей указанное соединение и фармацевтически приемлемый носитель. В некоторых вариантах осуществления способ химиосенсибилизации дополнительно включает введение указанному млекопитающему химиотерапевтического средства. В некоторых вариантах осуществления указанное химиосенсибилизирующее соединение и указанное химиотерапевтическое средство вводят практически одновременно.
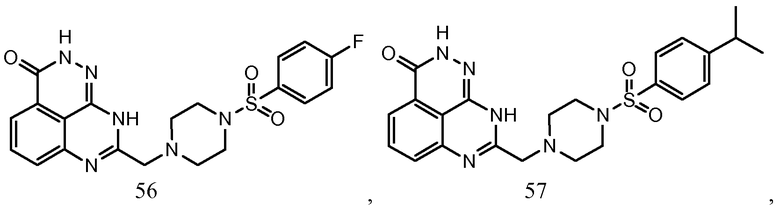
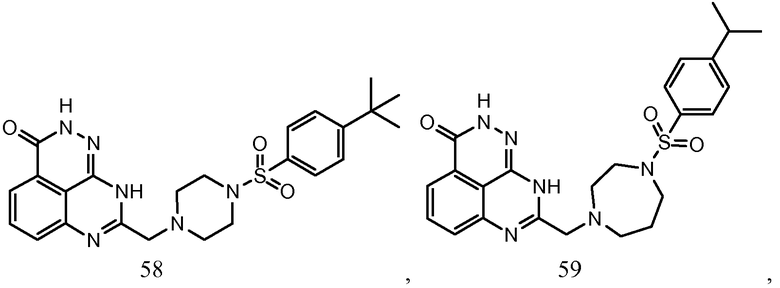
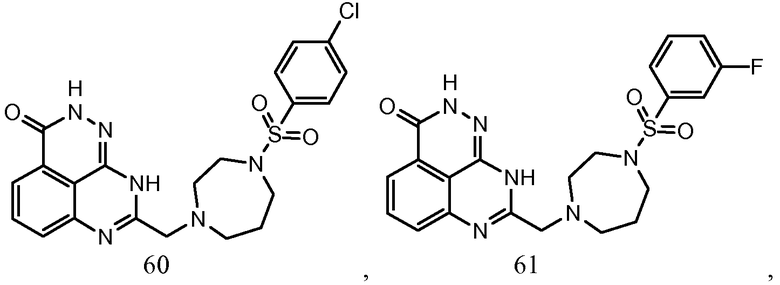
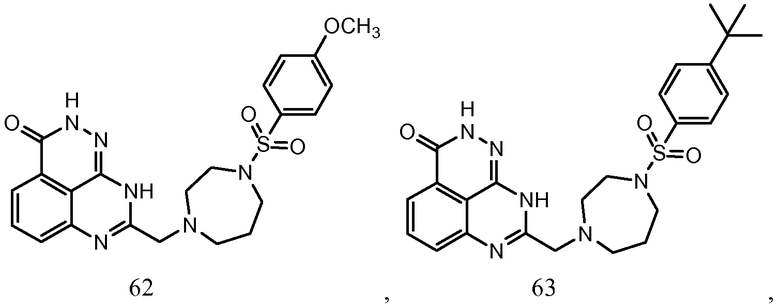
В некоторых вариантах осуществления настоящее изобретение относится к способу химиосенсибилизации раковых клеток у млекопитающего, нуждающегося в химиотерапии, включающему введение указанному млекопитающему соединения, выбранного из группы, состоящей из соединений 7-28, 30-46, 50-66, 69, 72, 74-76 и их фармацевтически приемлемых солей, описанных в данном описании. В некоторых вариантах осуществления указанным млекопитающим является человек. В некоторых вариантах осуществления указанное введение представляет собой введение фармацевтической композиции, содержащей указанное соединение и фармацевтически приемлемый носитель. В некоторых вариантах осуществления способ химиосенсибилизации дополнительно включает введение указанному млекопитающему химиотерапевтического средства. В некоторых вариантах осуществления указанное химиосенсибилизирующее соединение и указанное химиотерапевтическое средство вводят практически одновременно.
В некоторых вариантах осуществления химиотерапевтическое средство настоящего изобретения выбирают из группы, включающей темозоломид, адриамицин, камптотецин, карбоплатин, цисплатин, даунорубицин, доцетаксел, доксорубицин, интерферон-альфа, интерферон-бета, интерферон-гамма, интерлейкин 2, иринотекан, паклитаксел, топотекан, таксоид, дактиномицин, даунорубицин, 4'-дезоксидоксорубициндезоксидоксорубицин, блеомицин, пилкамицин, митомицин, неомицин, гентамицин, этопозид, 4-OH-циклофосфамид, координационный комплекс платины и их смеси. В некоторых вариантах осуществления химиотерапевтическое средство представляет собой темозоломид или его соль.
В некоторых вариантах осуществления настоящее изобретение относится к способу радиосенсибилизации раковых клеток у млекопитающего, нуждающегося в лучевой терапии, который включает введение указанному млекопитающему соединения формулы (I), описанного в данном описании, или его фармацевтически приемлемой соли. В некоторых вариантах осуществления указанным млекопитающим является человек. В некоторых вариантах осуществления указанное введение представляет собой введение фармацевтической композиции, содержащей указанное соединение и фармацевтически приемлемый носитель.
В некоторых вариантах осуществления настоящее изобретение относится к способу радиосенсибилизации раковых клеток у млекопитающего, нуждающегося в лучевой терапии, который включает введение указанному млекопитающему соединения, выбранного из группы, состоящей из соединений 7-28, 30-46, 50-66, 69, 72, 74-76 и их фармацевтически приемлемых солей, описанных в данном описании. В некоторых вариантах осуществления указанным млекопитающим является человек. В некоторых вариантах осуществления указанное введение представляет собой введение фармацевтической композиции, содержащей указанное соединение и фармацевтически приемлемый носитель.
В некоторых вариантах осуществления изобретение относится к фармацевтической композиции, содержащей соединение формулы (I), описанное в данном описании, или его фармацевтически приемлемую соль, и фармацевтически приемлемый носитель. В некоторых вариантах осуществления фармацевтическая композиция дополнительно содержит химиотерапевтическое средство, описанное в данном описании.
В некоторых вариантах осуществления изобретение относится к фармацевтической композиции, содержащей соединение, выбранное из группы, состоящей из соединений 7-28, 30-46, 50-66, 69, 72, 74-76 и их фармацевтически приемлемых солей, описанных в данном описании. В некоторых вариантах осуществления фармацевтическая композиция дополнительно содержит химиотерапевтическое средство, описанное в данном описании.
В некоторых вариантах осуществления раковые клетки, которые можно лечить с помощью способов химиосенсибилизации и/или радиосенсибилизации настоящего изобретения, выбирают из группы, включающей клетки таких опухолей, как АКТГ-продуцирующие опухоли, острый лимфоцитарный лейкоз, острый нелимфоцитарный лейкоз, рак коры надпочечника, рак мочевого пузыря, рак мозга, рак молочной железы, рак шейки матки, хронический лимфоцитарный лейкоз, хронический миелоцитарный лейкоз, рак ободочной и прямой кишки, Т-клеточная лимфома кожи, рак эндометрия, рак пищевода, саркома Юинга, рак желчного пузыря, лейкоз ворсистых клеток, рак головы и шеи, лимфома Ходжкина, саркома Капоши, рак почки, рак печени, рак легкого (мелкоклеточный и немелкоклеточный), злокачественный перитонеальный выпот, злокачественный плевральный выпот, меланома, мезотелиома, множественная миелома, нейробластома, неходжкинская лимфома, остеосаркома, рак яичника, рак яичников (герминогенный рак), рак простаты, рак поджелудочной железы, рак полового члена, ретинобластома, рак кожи, саркома мягкой ткани, плоскоклеточная карцинома, рак желудка, рак яичка, рак щитовидной железы, трофобластические опухоли, рак матки, рак влагалища, рак наружных половых органов и опухоль Вильмса. В некоторых вариантах осуществления раковые клетки, которые можно лечить с помощью способов химиосенсибилизации и/или радиосенсибилизации настоящего изобретения, выбирают из группы, включающей рак мозга, меланома, рак головы и шеи, рак яичка, рак яичника, рак молочной железы, немелкоклеточный рак легкого и рак прямой кишки.
В некоторых вариантах осуществления изобретение относится к способу лечения млекопитающего, страдающего от ракового заболевания, характеризующегося дефектом в пути репарации двухцепочечной ДНК посредством гомологичной рекомбинации (HR), который включает введение указанному млекопитающему соединения формулы (I), описанного в данном описании, или его фармацевтически приемлемой соли. В некоторых вариантах осуществления указанным млекопитающим является человек. В некоторых вариантах осуществления указанное введение представляет собой введение фармацевтической композиции, содержащей указанное соединение и фармацевтически приемлемый носитель. В некоторых вариантах осуществления раковые клетки имеют фенотип, выбранный из группы, включающей i) дефект BRCA-1, ii) дефект BRCA-2, iii) дефект BRCA-1 и BRC A-2 и iv) анемию Фанкони. В некоторых вариантах осуществления раковые клетки выбирают из клеток рака молочной железы или рака яичника.
В некоторых вариантах осуществления изобретение относится к способу лечения млекопитающего, страдающего от ракового заболевания, характеризующегося дефектом в пути репарации двухцепочечной ДНК посредством гомологичной рекомбинации (HR), который включает введение указанному млекопитающему соединения, выбранного из группы, состоящей из соединений 7-28, 30-46, 50-66, 69, 72, 74-76 и их фармацевтически приемлемых солей, описанных в данном описании. В некоторых вариантах осуществления указанным млекопитающим является человек. В некоторых вариантах осуществления указанное введение представляет собой введение фармацевтической композиции, содержащей указанное соединение и фармацевтически приемлемый носитель. В некоторых вариантах осуществления раковые клетки имеют фенотип, выбранный из группы, включающей i) дефект BRCA-1, ii) дефект BRCA-2, iii) дефект BRCA-1 и BRC A-2 и iv) анемию Фанкони. В некоторых вариантах осуществления раковые клетки выбирают из клеток рака молочной железы или рака яичника.
Способы синтеза раскрытых соединений
Схема 1
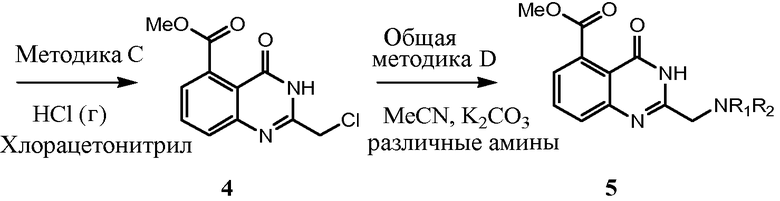
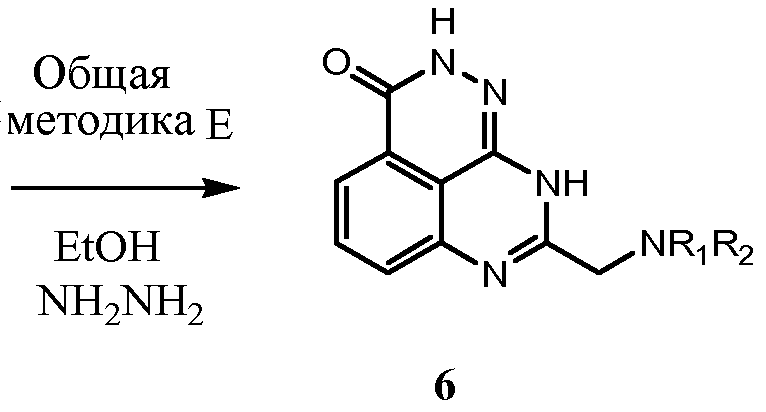
Методика A: Получение диметилового эфира 3-нитрофталевой кислоты, 2
К перемешиваемому раствору 4-нитроизобензофуран-1,3-диона (150 г, 0,78 моль), 1, в 2 л MeOH добавляют 50 мл концентрированной серной кислоты. Реакционную смесь нагревают при кипячении с обратным холодильником в течение 16 часов. Раствор смеси охлаждают до комнатной температуры и затем выливают в воду со льдом объемом 3 л, что приводит к выпадению густого белого осадка. Полученный осадок растирают в течение 15 минут, затем фильтруют, твердое вещество тщательно промывают водой и сушат, получая 120 г диметилового эфира 3-нитрофталевой кислоты, 2, в виде белого твердого вещества (65%).
1H ЯМР (300 МГц, ДМСО-d6): 8,54 (д, J=7,25 Гц, 1H), 8,42 (д, J=7,82 Гц, 1H), 7,98 (т, J=8,20 Гц, 1H), 3,99 (с, 3H), 3,98 (с, 3H); 13C ЯМР: 52,03, 52,29, 111,02, 115,67, 119,08, 131,80, 133,68, 148,80, 167,64, 168,63.
Методика B: Получение диметилового эфира 3-аминофталевой кислоты, 3
Соединение 2 (205 г, 1,0 моль) растворяют в 2 л MeOH. Добавляют каталитическое количество 10% Pd/C, затем раствор гидрируют в атмосфере H2 (45 фунт/кв. дюйм) на аппарате Пара при комнатной температуре в течение ночи. Фильтруют через целит и упаривают, получая диметиловый эфир 3-аминофталевой кислоты, 3, с количественным выходом.
1H ЯМР (300 МГц, ДМСО-d6): 7,26 (т, J=7,33 Гц, 1H), 6,94 (д, J=8,34 Гц, 1H), 6,77 (д, J=8,33 Гц, 1H), 6,12 (с, 2H), 3,77 (с, 3H), 3,76 (с, 3H); 13C ЯМР: 51,51, 51,77, 110,50, 115,16, 118,56, 131,26, 133,16, 148,28, 167,12, 168,11.
Методика C: Получение метилового эфира 2-хлорметил-4-оксо-3,4-дигидрохиназолин-5-карбоновой кислоты, 4
100 мл хлорацетонитрила перемешивают в 130 мл 1,4 диоксана при комнатной температуре. Сухой газообразный HCl барботируют через раствор в течение тридцати минут, затем добавляют 30 г диметилового эфира 3-амино-1,2-фталевой кислоты, 3. Реакционную смесь нагревают при кипячении с обратным холодильником в течение примерно трех часов, с получением густого белого осадка. Суспензию охлаждают на бане со льдом, фильтруют и промывают пентаном, для удаления всех оставшихся растворителей. Выделяют 30 г (83%) химически чистого белого твердого вещества, 4.
1H ЯМР (300 МГц, ДМСО-d6): 7,88 (т, J=8,33 Гц, 1H), 7,79 (д, J=7,08 Гц, 1H), 7,52 (д, J=7,33 Гц, 1H), 4,60 (с, 2H), 3,84 (с, 3H); 13C ЯМР: 42,21, 54,86, 119,95, 127,77, 130,86, 135,71, 136,78, 150,59, 155,70, 162,49, 171,24.
Общая методика D: Получение соединений 5
Замену хлора в соединении 4 нуклеофилами, такими как амин, с использованием общего способа D дает соединения 5. К раствору хлорсодержащего соединения 4 в сухом ДМФА или MeCN добавляют карбонат калия и нуклеофил, такой как амин. Реакционную смесь нагревают при 70°C в течение 12 часов, затем охлаждают до комнатной температуры. К реакционной смеси добавляют воду и затем этилацетат. Органический слой отделяют, промывают водой, насыщенным раствором соли и сушат над сульфатом натрия. Растворители удаляют в вакууме. Остаток очищают колоночной хроматографией на силикагеле, используя смесь этилацетат/гексаны в качестве элюента, с получением продуктов 5 с выходом 50-95%. Примером является получение соединения 7.
Общая методика E: Получение соединений 6
2,9-Дигидро-1,2,7,9-тетраазафенален-3-оновое кольцо может быть образовано путем конденсации соединений 6 с гидразином. К раствору соединений 6 в абсолютном этаноле добавляют избыток безводного гидразина при комнатной температуре. Раствор нагревают при кипячении с обратным холодильником в течение ночи и затем охлаждают до комнатной температуры. Добавляют охлажденную на льду воду и отделяют белое твердое вещество. Твердое вещество отделяют вакуумным фильтрованием, промывают водой и небольшим количеством метанола, с получением продуктов 6 в виде белого твердого вещества с выходом 40-90%. Примером является получение соединения 7.
Пример 1
Получение 8-(4-гидроксипиперидин-1-илметил)-2,9-дигидро-1,2,7,9-тетраазафенален-3-она, 7
Следуя общей методике D: раствор MeCN (25 мл), 4-гидроксипиперидина (0,46 мг, 4,5 ммоль), соединения 4 (1,0 г, 3,9 ммоль) и карбоната калия (1 г, 7 ммоль) нагревают при кипячении с обратным холодильником в атмосфере азота и перемешивают в течение ночи. Реакционную смесь упаривают досуха и экстрагируют дихлорметаном. Очищают на колонке с диоксидом кремния, используя смесь 9:1 дихлорметан/MeOH, с получением 1,05 г (84%) не совсем белого твердого вещества, метиловый эфир 2-(4-гидроксипиперидин-1-илметил)-4-оксо-3,4-дигидрохиназолин-5-карбоновой кислоты, 7a.
Следуя общей методике E: к раствору соединения 7a (1,0 г, 3,1 ммоль) в EtOH (20 мл) при нагревании с обратным холодильником добавляют моногидрат гидразина (7 мл, большой избыток) и нагревают в течение ночи. Реакционную смесь охлаждают до КТ и добавляют H2O (15 мл), получая густой белый осадок. Фильтруют, промывают смесью 1:1 EtOH/H2O, с получением 0,6 г (64%) химически чистого белого твердого вещества 7.
Т.пл.: 168-171°C; МС (ES+): 300; 1H ЯМР (300 МГц, CD3OD): 1,46-1,55 (м, 2H), 1,71-1,75 (м, 2H), 2,15-2,23 (м, 2H), 2,70-2,75 (м, 2H), 3,16-3,18 (м, 1H), 3,25 (с, 2H), 3,47-3,55 (м, 1H), 7,30-7,33 (м, 1H), 7,60-7,64 (м, 2H). Анализ. Вычислено для C15H17N5O2•1,7H2O: C, 56,45; H, 6,06; N, 21,94; найдено: C, 56,10; H, 6,00; N, 22,25.
Соединение 7 можно получить с использованием кислоты. Например: к раствору соединения 7 (0,6 г, 2,0 ммоль) в 10 мл смеси 1,4-диоксан/ДМФА (9:1) при 90°C добавляют MsOH (0,14 мл, 2,1 ммоль), с получением густого белого осадка. Фильтруют и растирают в диэтиловом эфире, с получением 0,5 г (63%) мезилата 7 в виде не совсем белого твердого вещества.
1H ЯМР (300 МГц, ДМСО-d6): 1,55-1,58 (м, 2H), 1,78-1,82 (м, 2H), 2,15 (с, 3H), 3,15-3,50 (м, 4H), 3,63-3,65(м, 1H), 4,04 (с, 2H), 7,24 (д, J=8,5 Гц, 1H), 7,51-7,66 (м, 2H), 11,73 (с, 1H). Анализ. Вычислено для C15H17N5O2•1CH3SO3H•2H2O: C, 44,54; H, 5,84; N, 16,23, С, 7,43; найдено: C, 44,48; H, 5,76; N, 16,27, С, 7,60.
Следующие соединения синтезируют аналогично методикам получения соединения 7, используя соответствующие амины.
Получение 8-(4-фенилпиперазин-1-илметил)-2,9-дигидро-1,2,7,9-тетраазафенален-3-она, 8
Синтезируют по общей методике D, используя 1-фенилпиперазин. Общий выход с двух последних стадий 52%.
МС (ES+): 361; 1H ЯМР (300 МГц, ДМСО-d6): 2,65-2,68 (м, 4H), 3,19-3,22 (м, 4H) 3,39 (с, 2H); 6,78 (т, J=7,2 Гц, 1H); 6,95 (д, J=8,0 Гц, 2H), 7,19 (т, J=7,2 Гц, 2H), 7,48-7,51 (м, 1H), 7,62-7,64 (д, J=7,2 Гц, 1H), 7,75 (т, J=8,0 Гц, 1H), 11,23 (ушир.с, 1H), 11,78 (с, 1H). Анализ. Вычислено для C20H20N6O1•2,0H2O: C, 60,59; H, 6,10; N, 21,20; найдено: C, 60,48; H, 6,05; N, 21,35.
Получение 8-(4-бензилпиперидин-1-илметил)-2,9-дигидро-1,2,7,9-тетраазафенален-3-она, 9
Синтезируют по общей методике D, используя 1-бензилпиперазин. Общий выход с двух последних стадий 20%.
МС (ES-): 372; 1H ЯМР (300 МГц, ДМСО-d6): 1,22-1,50 (м, 5H), 2,45-2,55 (м, 4H), 2,85 (д, 2H), 3,28 (с, 2H), 7,14-7,19 (м, 3H), 7,25-7,30 (м, 2H), 7,50 (д, J=7,0 Гц, 1H), 7,62 (д, J=7,7 Гц, 1H), 7,75 (т, J=7,7 Гц 1H), 11,25 (ушир.с, 1H), 11,76 (с, 1H). Анализ. Вычислено для C22H23N5O1: C, 70,76; H, 6,21; N, 18,75; найдено: C, 70,36; H, 6,18; N, 18,63.
Получение 8-феноксиметил-2,9-дигидро-1,2,7,9-тетраазафенален-3-она, 10
Синтезируют по общей методике D, используя фенол. Общий выход 60% с двух последних стадий.
МС (ES+): 293; 1H ЯМР (300 МГц, ДМСО-d6): 4,90 (ушир.с, 3H), 7,00 (т, J=6,6 Гц, 1H), 7,08 (д, J=8,2 Гц, 2H), 7,34 (т, J=1,1 Гц, 2H), 7,45 (д, J=1,1 Гц, 1H), 7,65 (д, J=7,7 Гц, 1H), 7,76 (т, J=7,2 Гц, 1H), 11,20 (ушир.с, 1H), 11,80 (с, 1H). Анализ. Вычислено для C16H12N4O2•0,75H2O•0,25N2H4: C, 61,24; H, 4,66; N, 20,08; найдено: C, 61,06; H, 4,27; N, 20,13.
Получение 8-[4-(4-фторфенил)-3,6-дигидро-2H-пиридин-1-илметил]-2,9-дигидро-1,2,7,9-тетраазафенален-3-она, 11
Синтезируют по общей методике D, используя гидрохлорид 4-(4-фторфенил)-1,2,3,6-тетрагидропиридина. Общий выход с двух последних стадий 24%.
МС (ES+): 376; 1H ЯМР (400 МГц, ДМСО-d6): 2,51-2,53 (ушир.с, 2H), 2,77 (т, J=5,4 Гц, 2H), 3,24 (ушир.с, 2H), 3,46 (с, 2H), 6,16 (м, 1H), 7,16 (т, J=8,8 Гц, 2H), 7,46-7,52 (м, 3H), 7,63 (д, J=7,8 Гц, 1H), 7,44 (т, J=7,8 Гц, 1H), 11,18 (ушир.с, 1H), 11,79 (с, 1H).
Получают мезилат соединения 11.
1H ЯМР (400 МГц, ДМСО-d6): 2,34 (с, 3H), 2,84 (ушир.с, 2H), 3,66 (м, 2H), 4,11 (м, 2H), 4,36 (с, 2H), 6,21 (м, 1H), 7,25 (т, J=8,8 Гц, 2H), 7,43 (д, J=7,4 Гц, 1H), 7,56-7,59 (м, 2H), 7,72 (д, J=7,4 Гц, 1H), 7,82 (т, J=7,5 Гц, 1H), 11,25 (ушир.с, 1H), 11,76 (с, 1H). Анализ. Вычислено для C21H18FN5O1•1,0CH3SOH•0,2H2O: C, 55,62; H, 4,75; N, 14,74; S, 6,75; найдено: C, 55,65; H, 4,71; N, 14,73; S, 6,74.
Получение 8-[4-(4-хлорфенил)пиперазин-1-илметил]-2,9-дигидро-1,2,7,9-тетраазафенален-3-она, 12
Синтезируют по общей методике D, используя 1-(4-хлорфенил)пиперазин. Общий выход с двух последних стадий 23%.
Получают мезилат соединения 12.
МС (ES+): 396; 1H ЯМР (400 МГц, ДМСО-d6): 2,33 (с, 3H), 4,31 (с, 2H), 7,03 (д, J=9,3 Гц, 2H), 7,31 (д, J=9,3 Гц, 2H), 7,43 (д, J=8,5 Гц, 1H), 7,72 (д, J=8,5 Гц, 1H), 7,82 (т, J=7,9 Гц, 1H), 11,23 (ушир.с, 1H), 11,90 (с, 1H). Анализ. Вычислено для C20H19ClN6O1•1,0CH3SOH: C, 51,37; H, 4,72; N, 17,12; S, 6,53; найдено: C, 51,27; H, 4,91; N, 17,03; S, 6,48.
Получение 8-(4-фенил-3,6-дигидро-2H-пиридин-1-илметил)-2,9-дигидро-1,2,7,9-тетраазафенален-3-она, 13
Синтезируют по общей методике, используя 4-фенил-1,2,3,6-тетрагидропиридин D. Общий выход с двух последних стадий 80%.
МС (ES+): 358; 1H ЯМР (400 МГц, ДМСО-d6): 2,56 (м, 2H), 2,78 (т, J=5,5 Гц, 2H), 3,25 (д, J=2,6 Гц, 2H), 3,47 (с, 2H), 6,19 (с, 1H), 7,23-7,27 (м, 1H), 7,24 (т, J=7,6 Гц, 2H), 7,45 (д, J=7,1 Гц, 2H), 7,51 (д, J=8,9 Гц, 1H), 7,62 (д, J=7,1 Гц, 1H), 7,75 (т, J=8,0 Гц, 1H), 11,27 (ушир.с, 1H), 11,78 (с, 1H).
Получают мезилат соединения 13.
1H ЯМР (400 МГц, ДМСО-d6): 2,34 (с, 3H), 2,84-2,88 (м, 2H), 3,65-3,69 (м, 2H), 4,13 (с, 2H), 4,37 (с, 2H), 6,21-6,25 (м, 1H), 7,32-7,44 (м, 4H), 7,53 (д, J=8,6 Гц, 2H), 7,72 (д, J=7,3 Гц, 1H), 7,82 (т, J=8,1 Гц, 1H), 11,30 (ушир.с, 1H), 11,93 (с, 1H). Анализ. Вычислено для C21H19N5O•1,0CH3SOH•0,4H2O: C, 57,35; H, 5,21; N, 15,20; S, 6,96;: найдено: C, 57,30; H, 5,16; N, 15,29; S, 7,10.
Получение 8-[(3,4-дихлорбензиламино)метил]-2,9-дигидро-1,2,7,9-тетраазафенален-3-она, 14
Синтезируют по общей методике D, используя 3,4-дихлорбензиламин. Общий выход с двух последних стадий 10%.
Получают мезилат соединения 14.
МС (ES+): 375; 1H ЯМР (300 МГц, ДМСО-d6): 2,33 (с, 3H), 4,06 (с, 2H), 4,33 (с, 2H), 7,39 (д, J=8,0 Гц, 1H), 7,53-7,57 (м, 1H), 7,69-7,88 (м, 4H), 11, 31 (ушир.с, 1H), 11,91 (с, 1H).
Получение 8-{[2-(3-Фторфенил)этиламино]метил}-2,9-дигидро-1,2,7,9-тетраазафенален-3-она, 15
Синтезируют по общей методике D, используя 3-фторфенетиламин. Общий выход с двух последних стадий 12%. Получают мезилат соединения 15.
МС (ES+): 338; 1H ЯМР (300 МГц, ДМСО-d6): 2,34 (с, 3H), 3,02-3,08 (м, 2H), 3,34-3,38 (м, 2H), 4,14 (с, 2H), 7,08-7,18 (м, 3H), 7,37-7,44 (м, 2H), 7,71 (д, J=7,8 Гц, 1H), 7,82 (т, J=7,8 Гц, 1H), 11,92 11,35 (ушир.с, 1H), (с, 1H).
Получение 8-[(3-трифторметилбензиламино)метил]-2,9-дигидро-1,2,7,9-тетраазафенален-3-она, 16
Синтезируют по общей методике D, используя 3-(трифторметил)бензиламин. Общий выход с двух последних стадий 14%. Получают мезилат соединения 16.
МС (ES+): 374; 1H ЯМР (300 МГц, ДМСО-d6): 2,33 (с, 3H), 4,10 (с, 2H), 4,43 (с, 2H), 7,39 (д, J=7,6 Гц, 1H), 7,69-7,86 (м, 5H), 7,99 (с, 1H), 11,25 (ушир.с, 1H), 11,91 (с, 1H). Анализ. Вычислено для C19H18F3N5O•1,0CH3SOH•1,0H2O: C, 46,82; H, 4,14; N, 14,37; S, 6,58; найдено: C, 46,81; H, 4,17; N, 14,64; S, 6,35.
Получение 8-(1,4-диокса-8-азаспиро[4.5]дец-8-илметил)-2,9-дигидро-1,2,7,9-тетраазафенален-3-она, 17
Синтезируют по общей методике D, используя 4-пиперидонэтиленкеталь. Общий выход с двух последних стадий 10%.
МС (ES-): 370; 1H ЯМР (300 МГц, ДМСО-d6): 1,69-1,71 (м, 4H), 2,57 (ушир.с, 4H), 3,35 (с, 2H), 3,87 (с, 4H), 7,51 (д, J=7,8 Гц, 1H), 7,62 (д, J=7,7 Гц, 1H), 7,74 (т,J=7,8 Гц, 1H), 11,23 (ушир.с, 1H), 11,76 (с, 1H). Анализ. Вычислено для C17H19N5O3•0,2H2O: C, 59,19; H, 5,67; N, 20,30; найдено: C, 59,03; H, 5,60; N, 20,63.
Получение 8-{[2-(3,4-дихлорфенил)этиламино]метил}-2,9-дигидро-1,2,7,9-тетраазафенален-3-она, 18
Синтезируют по общей методике D, используя 3,4-дихлорфенетиламин. Общий выход с двух последних стадий 17%. Получают мезилат соединения 18.
МС (ES-): 387; 1H ЯМР (300 МГц, ДМСО-d6): 2,36 (с, 3H), 3,04 (т, J=8,2 Гц, 2H), 3,37 (т, J=8,1 Гц, 2H), 4,14 (с, 2H), 7,30-7,43 (м, 2H), 7,61-7,75 (м, 3H), 7,79-7,84 (м, 1H), 11,31 (ушир.с, 1H), 11,91 (с, 1H).
Получение 8-{[2-(3-трифторметилфенил)этиламино]метил}-2,9-дигидро-1,2,7,9-тетраазафенален-3-она, 19
Синтезируют по общей методике D, используя 2-(3-трифторметилфенил)этиламин. Общий выход с двух последних стадий 39%. Получают мезилат соединения 19.
МС (ES-): 387; 1H ЯМР (300 МГц, ДМСО-d6): 3,74 (с, 3H), 3,13 (т, J=8,1 Гц, 2H), 3,30 (т, J=8,2 Гц, 2H), 4,15 (с, 2H), 7,40-7,43 (м, 1H), 7,62-7,72 (м, 4H), 7,79-7,85 (м, 1H), 11,35 (ушир.с, 1H), 11,92 (с, 1H).
Получение 8-[(1-азабицикло[2.2.2]окт-3-иламино)метил]-2,9-дигидро-1,2,7,9-тетраазафенален-3-она, 20
Синтезируют по общей методике D, используя (S)-(-)-3-аминохинукледин. Общий выход с двух последних стадий 23%. Получают мезилат соединения 20.
МС (ES+): 325; 1H ЯМР (300 МГц, ДМСО-d6): 1,97-2,03 (м, 3H), 2,20-2,35 (м, 1H), 2,35-2,44 (м, 2H), 2,42 (с, 3H), 3,72-3,80 (м, 6H), 4,15-4,21 (м, 1H), 4,38 (с, 2H), 7,46 (д, J=7,6, 1H), 7,69-7,72 (м, 1H), 7,78-7,84 (м, 1H), 8,63 (ушир.с, 3H).
Получение 8-(4-этилпиперазин-1-илметил)-2,9-дигидро-1,2,7,9-тетраазафенален-3-она, 21
Синтезируют по общей методике D, используя этилпиперазин. Общий выход с двух последних стадий 35%. Получают мезилат соединения 21.
МС (ES+): 313; 1H ЯМР (300 МГц, ДМСО-d6): 1,25, (т, J=7,4 Гц, 3H), 2,41 (с, 6H), 2,51-3,87 (м, 10 H), 3,87 (с, 2H), 7,70 (д, J=8,0 Гц, 1H), 7,81 (д, J=7,9 Гц, 1H), 7,91 (т, J=8,1 Гц, 1H), 9,82 (с, 1H), 11,96 (с, 1H); 13C ЯМР (ДМСО-d6): 157,40, 155,99, 140,65, 135,96, 133,84, 126,72, 119,71, 118,65, 115,85, 56,09, 50,30, 49,05, 48,66, 8,51. Анализ. Вычислено для C16H20N6O•2,0CH3SO3H•1,2H2O: C, 40,84; H, 5,43; N, 15,79; найдено: C, 41,09; H, 5,82; N, 15,97.
Получение 8-(4-метилпиперазин-1-илметил)-2,9-дигидро-1,2,7,9-тетраазафенален-3-она, 22
Синтезируют по общей методике D, используя метилпиперазин. Общий выход с двух последних стадий 29%. Получают мезилат соединения 22.
МС (ES+): 299; 1H ЯМР (400 МГц, ДМСО-d6): 2,38 (с, 3H), 2,58-2,63 (м, 2H), 3,09-3,18 (м, 4H), 3,40-3,45 (м, 2H), 3,51 (с, 2H), 7,50 (д, J=7,8 Гц, 1H), 7,67 (д, J=7,8 Гц, 1H), 7,79 (т, J=7,8 Гц, 1H), 9,53 (ушир.с, 1H), 11,85 (с, 1H). Анализ. Вычислено для C15H18N6O•1,15CH3SO3H•1,0H2O: C, 45,44; H, 5,81; N, 19,69; S, 8,64; найдено: C, 45,18; H, 5,88; N, 19,83; S, 8,68.
Получение 8-(4-бензил-[1,4]диазепан-1-илметил)-2,9-дигидро-1,2,7,9-тетраазафенален-3-она, 23
Синтезируют по общей методике D, используя 1-бензил-[1,4]диазепан. Общий выход с двух последних стадий 24%.
Т.пл.: 140-142°C; МС (ES-): 387; 1H ЯМР (400 МГц, CDCl3): 1,88 (м, 2H), 2,77 (м, 4H), 2,89 (м, 4H), 3,62 (с, 2H), 3,69 (с, 2H), 7,20-7,42 (м, 6H), 7,45(ушир.с, 1H), 7,74 (т, J=7,8 Гц, 1H), 7,87 (д, J=7,6 Гц, 1H), 11,50 (ушир.с, 1H). Анализ. Вычислено для C22H24N6O•1,35H2O: C, 64,01; H, 6,52; N, 20,36; найдено: C, 64,18; H, 6,59; N, 20,46.
Получают соль HCl соединения 23: через раствор 23 (0,5 г) в 20 мл диоксана барботируют газообразный HCl в течение 30 мин. Раствор перемешивают при комнатной температуре в течение ночи. После фильтрования осадок промывают диоксаном, с получением 0,25 г (48%) химически чистой соли HCl соединения 23 в виде не совсем белого твердого вещества.
1H ЯМР (400 МГц, D2O): 2,08 (м, 2H), 3,36 (м, 4H), 3,56 (м, 4H), 4,04 (с, 2H), 4,24 (с, 2H), 7,02 (д, 1H), 7,20-7,35 (м, 5H); 7,36 (д, 1H), 7,45(т, 1H). Анализ. Вычислено для C22H24N6O•2,0HCl•1,15H2O: C, 54,81; H, 5,92; N, 17,43; найдено: C, 54,81; H, 5,92; N, 17,36.
Получение трет-бутилового эфира 4-(3-оксо-2,9-дигидро-3H-1,2,7,9-тетраазафенален-8-илметил)-[1,4]диазепан-1-карбоновой кислоты, 24
Синтезируют по общей методике D, используя трет-бутиловый эфир [1,4]диазепан-1-карбоновой кислоты. Общий выход с двух последних стадий 30%.
Т.пл.: 219-221°C; МС (ES-): 397; 1H ЯМР (400 МГц, CDCl3): 1,46 (с, 9H); 1,88 (м, 2H); 2,83 (м, 4H); 3,50 (м, 4H); 3,59 (с, 2H); 7,63 (м, 1H), 7,72-7,86 (м, 3H), 11,90 (ушир.с, 1H). Анализ. Вычислено для C20H26N6O3•0,5H2O: C, 58,95; H, 6,68; N, 20,62; найдено: C, 58,83; H, 6,69; N, 20,60.
Получение 8-[4-(4-фторбензил)-[1,4]диазепан-1-илметил]-2,9-дигидро-1,2,7,9-тетраазафенален-3-она, 25
Синтезируют по общей методике D, используя 1-(4-фторбензил)-[1,4]диазепан. Общий выход с двух последних стадий 35%.
Т.пл.: 163-165°C; МС (ES-): 405; 1H ЯМР (400 МГц, CDCl3): 1,87 (м, 2H), 2,72 (м, 4H), 2,88 (м, 4H), 3,63 (с, 2H), 3,65 (с, 2H), 6,99 (т, J=8,4 Гц, 2H), 7,30 (м, 3H), 7,61 (ушир.с, 1H), 7,78 (м, 1H), 7,93 (д, J=7,3 Гц, 1H), 10,82 (ушир.с, 1H). Анализ. Вычислено для C22H23N6O•1,5H2O: C, 60,96; H, 6,05; N, 19,39; найдено: C, 61,07; H, 5,97; N, 19,59.
Получают мезилат соединения 25.
1H ЯМР (400 МГц, D2O): 2,06 (м, 2H), 2,70 (с, 3H), 3,06 (м, 2H), 3,24 (м, 2H), 3,46 (м, 4H), 3,65 (с, 4H), 3,74 (с, 2H), 4,33 (с, 2H), 7,25 (м, 3H), 7,46 (м, 3H), 7,62 (т, J=8,4 Гц, 1H). Анализ. Вычислено для C22H23FN6O•1,3CH3SO3H•0,5C4H8O2•2,0H2O: C, 49,70; H, 5,97; N, 13,74; S, 6,82; найдено: C, 49,40; H, 5,97; N, 13,37; S, 6,65.
Получение 8-[1,4]диазепан-1-илметил-2,9-дигидро-1,2,7,9-тетраазафенален-3-она, 26
Синтезируют из соединения 24. К раствору соединения 24 (1,5 г, 3,7 ммоль) в 30 мл CH2Cl2 добавляют 6 мл TFA, перемешивая при комнатной температуре. Через 30 минут растворители упаривают и остаток промывают ацетонитрилом, получая 1,0 г (90%) химически чистого белого твердого вещества. Т.пл.: 147-149°C;
Т.пл.: 147-149°C; МС (ES-): 297; 1H ЯМР (400 МГц, D2O): 1,96 (м, 2H), 2,82 (т, 2H), 3,01 (т, 2H), 3,28 (т, 4H), 3,53 (с, 2H), 7,22 (д, 1H), 7,47 (д, 1H), 7,61 (т, 1H). Анализ. Вычислено для C15H18N6O•1,1CF3CO2H•1,0H2O: C, 46,76; H, 4,81; N, 19,02; найдено: C, 46,64; H, 4,98; N, 19,02.
Получение 8-[4-(2-трифторметилбензоил)-[1,4]диазепан-1-илметил]-2,9-дигидро-1,2,7,9-тетраазафенален-3-она, 27
Синтезируют из соединения 26. К раствору соединения 26 (0,2 г, 0,6 ммоль) в 5 мл CH2Cl2 добавляют 1 ммоль TEA и 0,8 ммоль 2-трифторметилбензоилхлорида. Реакционную смесь перемешивают в течение ночи при комнатной температуре. После упаривания растворителей остаток очищают полупрепаративной ВЭЖХ, получая твердое вещество (выход 15%).
Т.пл.: 140-142°C; МС (ES-): 469; 1H ЯМР (400 МГц, CDCl3): 1,92-2,10 (м, 2H), 2,91-3,10 (м, 4H), 3,36-3,44 (м, 2H), 3,64-3,74 (м, 2H), 3,93 (м, 2H), 7,38 (м, 1H), 7,57 (м, 3H), 7,79 (м, 2H), 7,93 (м, 1H). Анализ. Вычислено для C23H21F3N6O2•0,9HCl: C, 54,89; H, 4,39; N, 16,70; найдено: C, 54,93; H, 4,43; N, 16,34.
Получение 8-[4-(3-хлорбензоил)-[1,4]диазепан-1-илметил]-2,9-дигидро-1,2,7,9-тетраазафенален-3-она, 28
Синтезируют из соединения 26. К раствору соединения 26 (0,2 г, 0,6 ммоль) в 5 мл CH2Cl2 добавляют 1 ммоль TEA и 0,8 ммоль 3-хлорбензоилхлорида. Реакционную смесь перемешивают в течение ночи при комнатной температуре. После упаривания растворителей остаток очищают полупрепаративной ВЭЖХ, получая твердое вещество (выход 16%).
Т.пл.: 147-149°C; МС (ES-): 436; 1H ЯМР (400 МГц, CDCl3): 1,88-2,08 (м, 2H), 2,86-3,07 (м, 4H), 3,52-3,71 (м, 4H), 3,81-3,89 (м, 2H), 7,33-7,43 (м, 4H), 7,62 (д, 1H), 7,81 (т, 1H), 7,90 (т, 1H). Анализ. Вычислено для C22H21ClN6O2•0,7H2O: C, 54,89; H, 4,39; N, 16,70; найдено: C, 54,93; H, 4,43; N, 16,34.
Получение 8-(4-пиридин-2-илпиперазин-1-илметил)-2,9-дигидро-1,2,7,9-тетраазафенален-3-она, 30
Синтезируют по общей методике D, используя 1-пиридин-2-илпиперазин. Общий выход с двух последних стадий 20%. Получают мезилат соединения 30.
МС (ES-): 360; 1H ЯМР (400 МГц, ДМСО-d6): 2,37 (с, 6H), 3,52 (ушир.с, 4H), 3,93 (ушир.с, 4H), 4,30 (с, 2H), 6,93 (т, J=6,6 Гц, 1H), 7,25 (д, J=8,6 Гц, 1H), 7,47 (д, J=7,8 Гц, 1H), 7,73 (д, J=7,8 Гц, 1H), 7,82-7,91 (м, 2H), 8,16-8,18 (м, 1H), 11,96 (с, 1H). Анализ. Вычислено для C19H19N7O•1,9CH3SO3H•1,2H2O: C, 44,38; H, 5,17; N, 17,33; S, 10,77; найдено: C, 44,21; H, 5,19; N, 17,28; S, 10,68.
Получение 8-{[2-(2-фторфенил)этиламино]метил}-2,9-дигидро-1,2,7,9-тетраазафенален-3-она, 31
Синтезируют по общей методике D, используя 2-(2-фторфенил)этиламин. Общий выход с двух последних стадий 20%. Получают мезилат соединения 31.
МС (ES-): 336; 1H ЯМР (400 МГц, ДМСО-d6): 2,41 (с, 5H), 3,02 (т, J=7,6 Гц, 2H), 3,32 (т, J=8,3 Гц, 2H), 4,16 (с, 2H), 7,19 (т, J=8,8 Гц, 2H), 7,32-7,35 (м, 2H), 7,42 (д, J=7,8 Гц, 1H), 7,71 (д, J=7,8 Гц, 1H), 7,82 (т,J=8,1 Гц, 1H), 9,10 (ушир.с, 1H), 11,92 (с, 1H). Анализ. Вычислено для C18H16FN5O•1,75CH3SO3H•0,75H2O: C, 45,70; H, 4,76; N, 13,49; S, 10,81; найдено: C, 45,45; H, 4,69; N, 13,42; S, 11,10.
Получение 8-[4-(4-фторфенил)пиперазин-1-илметил]-2,9-дигидро-1,2,7,9-тетраазафенален-3-она, 32
Синтезируют по общей методике D, используя 4-(4-фторфенил)пиперазин. Общий выход с двух последних стадий 57%. Получают мезилат соединения 32.
МС (ES-): 377; 1H ЯМР (400 МГц, ДМСО-d6): 2,40 (с, 5H), 3,45 (ушир.с, 4H), 3,59 (ушир.с, 4H), 4,37 (с, 2H), 7,03-7,15 (м, 4H), 7,44 (д, J=7,8 Гц, 1H), 7,72 (д, J=7,8 Гц, 1H), 7,83 (т, J=7,8 Гц, 1H), 9,8 (ушир.с, 1H), 11,94 (с, 1H). Анализ. Вычислено для C20H19FN6O•1,65CH3SO3H: C, 46,85; H, 5,01; N, 15,14; S, 9,53; найдено: C, 46,74; H, 5,15; N, 15,14; S, 9,53.
Получение 8-{2-(4-Фтор-фенил)этиламино]метил}-2,9-дигидро-1,2,7,9-тетраазафенален-3-она, 33
Синтезируют по общей методике D, используя 2-(4-фторфенил)этиламин. Общий выход с двух последних стадий 19%. Получают мезилат соединения 33.
МС (ES-): 336; 1H ЯМР (400 МГц, ДМСО-d6): 2,38 (с, 6H), 3,06-3,10 (м, 2H), 3,30-3,34 (м, 2H), 4,18 (с, 2H), 7,19-7,22 (м, 2H), 7,34-7,42 (м, 3H), 7,71 (д, J=8,6 Гц, 1H), 7,82 (т, J=7,8 Гц, 1H), 9,6 (ушир.с, 1H), 11,92 (с, 1H). Анализ. Вычислено для C18H16FN5O•2,0CH3SO3H: C, 45,36; H, 4,57; N, 13,22; S, 12,11; найдено: C, 45,34; H, 4,58; N, 13,16; S, 11,88.
Получение 8-(4-ацетил-[1,4]диазепан-1-илметил)-2,9-дигидро-1,2,7,9-тетраазафенален-3-она, 34
Синтезируют по общей методике D, используя [1,4]диазепан-1-илэтанон. Общий выход с двух последних стадий 16%.
Т.пл.: 191-193°C; МС (ES-): 339; 1H ЯМР (400 МГц, CDCl3): 2,11 (с, 3H), 2,84-2,93 (м, 4H), 3,56-3,76 (м, 6H), 7,66 (м, 1H), 7,83-7,92 (м, 2H), 9,3 (ушир.с, 1H), 11,3 (ушир.с, 1H). Анализ. Вычислено для C17H20N6O2•0,6H2O: C, 58,14; H, 6,08; N, 23,93; найдено: C, 58,09; H, 6,18; N, 24,08.
Получение 8-(фенетиламинометил)-2,9-дигидро-1,2,7,9-тетраазафенален-3-она, 35
Синтезируют по общей методике D, используя фенeтиламин. Общий выход с двух последних стадий 29%. Получают мезилат соединения 35.
МС (ES-): 358; 1H ЯМР (400 МГц, ДМСО-d6): 2,32 (с, 3H), 3,00-3,04 (м, 2H), 3,31-3,36 (м, 2H), 4,15 (с, 1H), 7,27-7,42 (м, 6H), 7,71 (д, J=7,8 Гц, 1H), 7,82 (т, J=7,8 Гц, 1H), 9,70 (ушир.с, 1H), 11,92 (с, 1H). Анализ. Вычислено для C18H17N5O•1,0CH3SO3H•1,8H2O: C, 50,95; H, 5,54; N, 15,64; S, 7,16; найдено: C, 50,95; H, 5,54; N, 15,64; S, 7,16.
Получение 8-(4-фенилпиперидин-1-илметил)-2,9-дигидро-1,2,7,9-тетраазафенален-3-она, 36
Синтезируют по общей методике D, используя 4-фенилпиперидин. Общий выход с двух последних стадий 33%.
МС (ES-): 318; 1H ЯМР (400 МГц, ДМСО-d6): 1,87-1,93 (м, 4H), 2,37-2,46 (м, 2H), 2,56 (м, 1H), 3,10-3,14 (м, 2H), 3,54 (с, 2H), 7,17-7,34 (м, 5H), 7,56 (ушир.с, 1H), 7,76 (т, J=7,8 Гц, 1H), 7,93 (д, J=7,8 Гц, 1H), 11,10 (ушир.с, 1H), 11,76 (с, 1H).
Получают мезилат соединения 36.
1H ЯМР (400 МГц, D2O): 2,08 (м, 4H), 2,95 (м, 1H), 3,34 (м, 2H), 3,84 (м, 2H), 4,23 (с, 2H), 7,21-7,39 (м, 6H), 7,59 (м, 1H), 7,70 (м, 1H). Анализ. Вычислено для C21H21N5O•1,3CH3SO3H•0,5H2O: C, 54,29; H, 5,56; N, 14,19; S, 8,45; найдено: C, 54,03; H, 5,65; N, 13,98; S, 8,64.
Получение 8-(1,3-дигидроизоиндол-2-илметил)-2,9-дигидро-1,2,7,9-тетраазафенален-3-она, 37
Синтезируют по общей методике D, используя изоиндолин. Общий выход с двух последних стадий 40%.
МС (ES-): 316; 1H ЯМР (400 МГц, ДМСО-d6): 3,77 (с, 2H), 4,04 (с, 4H), 7,20-7,30 (м, 4H), 7,49 (д, J=7,8 Гц, 1H), 7,6 (д, J=7,8 Гц, 1H), 7,74 (т, J=7,8 Гц, 1H), 11,34 (ушир.с, 1H), 11,78 (с, 1H).
Получают мезилат соединения 37.
1H ЯМР (400 МГц, ДМСО-d6): 2,34 (с, 3H), 4,64 (с, 2H), 4,87 (с, 4H), 7,39-7,46 (м, 5H), 7,72 (д, J=7,8 Гц, 1H), 7,83 (т, J=8,1 Гц, 1H), 11,30 (ушир.с, 1H), 11,95 (с, 1H). Анализ. Вычислено для C18H15N5O•1,25CH3SO3H•2,0H2O: C, 48,83; H, 5,11; N, 14,79; S, 8,46; найдено: C, 48,80; H, 5,11; N, 14,97; S, 8,71.
Получение 8-(4-бензолсульфонил-[1,4]диазепан-1-илметил)-2,9-дигидро-1,2,7,9-тетраазафенален-3-она, 38
Синтезируют из соединения 26. К раствору соединения 26 (0,2 г, 0,67 ммоль) в 5 мл CH2Cl2 добавляют TEA (2 ммоль) и бензолсульфонилхлорид (1 ммоль). Смесь перемешивают при комнатной температуре в течение ночи. После упаривания растворителей остаток выливают в 10 мл H2O, затем продукт очищают препаративной ВЭЖХ, получая химически чистое белое твердое вещество (выход 5%).
Т.пл.: 265-268°C; МС (ES-): 437; 1H ЯМР (400 МГц, ДМСО-d6): 1,79 (м, 2H), 2,50 (м, 4H), 2,79 (м, 4H), 3,51 (с, 2H), 7,44 (д, 1H), 7,62-7,79 (м, 7H), 11,1 (ушир.с, 1H), 11,75 (с, 1H). Анализ. Вычислено для C21H22N6O3S•0,5H2O: C, 56,36; H, 5,18; N, 18,78; S, 7,17; найдено: C, 56,44; H, 5,12; N, 19,00; S, 7,19.
Получают мезилат соединения 38.
МС (ES+): 439; 1H ЯМР (400 МГц, D2O): 2,18 (м, 2H), 2,35 (с, 6H), 3,36 (м, 2H), 3,65 (м, 6H), 4,3 (с, 2H), 7,24 (д, 1H), 7,51-7,71 (м, 7H). Анализ. Вычислено для C21H22N6O3S•1,8CH3SO3H•1,0H2O: C, 43,50; H, 5,00; N, 13,35; S, 14,26; найдено: C, 43,61; H, 5,00; N, 13,15; S, 14,59.
Получение 8-(4-пиридин-4-илпиперазин-1-илметил)-2,9-дигидро-1,2,7,9-тетраазафенален-3-она, 39
Синтезируют по общей методике D, используя 1-(4-пиридил)пиперазин. Общий выход с двух последних стадий 10%.
МС (ES-): 360; 1H ЯМР (400 МГц, ДМСО-d6): 2,80 (т, J=5,0 Гц, 4H), 3,61 (т, J=5,0 Гц, 4H), 3,99 (с, 2H), 6,83 (д, J=7,1 Гц, 2H), 7,42-7,45 (м, 1H), 7,73-7,81 (м, 2H), 8,26 (д, J=7,1 Гц, 2H), 11,20 (ушир.с, 1H), 11,90 (с, 1H).
Получают соль HCl соединения 39.
1H ЯМР (400 МГц, D2O): 2,74-2,77 (м, 4H), 3,43 (с, 2H), 3,35-3,69 (м, 4H), 6,93 (д, J=7,1 Гц, 2H), 7,13 (д, J=8,0 Гц, 1H), 7,37 (д, J=7,8 Гц, 1H), 7,58 (т, J=7,8 Гц, 1H), 7,92 (д, J=7,1 Гц, 2H). Анализ. Вычислено для C19H19N7O•1,0HCl•2,5H2O: C, 51,53; H, 5,69; N, 22,14; Cl, 8,00; найдено: C, 51,46; H, 5,69; N, 21,90; Cl, 8,27.
Получение 8-(4-бензилпиперазин-1-илметил)-2,9-дигидро-1,2,7,9-тетраазафенален-3-она, 40
Синтезируют по общей методике D, используя 4-бензилпиперазин. Общий выход с двух последних стадий 12%.
МС (ES-): 373; 1H ЯМР (400 МГц, ДМСО-d6): 2,44 (ушир.с, 4H), 3,35 (ушир.с, 4H), 3,48 (с, 2H), 7,23-7,34 (м, 5H), 7,49 (д, J=8,8 Гц, 1H), 7,62 (д, J=7,8 Гц, 1H), 7,74 (т, J=7,8 Гц, 1H), 11,10 (ушир.с, 1H), 11,77 (с, 1H).
Получают соль HCl соединения 40.
1H ЯМР (400 МГц, D2O): 2,54-2,70 (м, 2H), 3,10-3,50 (м, 6H), 3,48 (с, 2H), 4,35 (с, 2H), 7,20 (д, J=8,1 Гц, 1H), 7,45 (т, J=7,8 Гц, 1H), 7,47-7,51 (м, 5H), 7,62 (т, J=8,1 Гц, 1H). Анализ. Вычислено для C21H22N6O•1,0HCl•2,5H2O: C, 55,32; H, 6,19; N, 18,43; найдено: C, 55,54; H, 6,08; N, 18,32.
Получение 8-(4-метил-[1,4]диазепан-1-илметил)-2,9-дигидро-1,2,7,9-тетраазафенален-3-она, 41
Синтезируют по общей методике D, используя 1-метил-[1,4]-диазепан. Общий выход с двух последних стадий 24%.
МС (ES-): 311; 1H ЯМР (400 МГц, ДМСО-d6): 1,75 (м, 2H), 2,26 (с, 3H), 2,55 (м, 4H), 2,79 (м, 4H), 3,48 (с, 2H), 7,52 (д, J=8,2 Гц, 1H), 7,64 (д, J=7,2 Гц, 1H), 7,75 (т, J=8,1 Гц, 1H), 11,55 (с, 1H). Анализ. Вычислено для C16H20N6O•0,95H2O: C, 58,33; H, 6,70; N, 25,51; найдено: C, 58,32; H, 6,65; N, 25,53.
Получение 8-[4-(1H-индол-3-ил)-пиперидин-1-илметил]-2,9-дигидро-1,2,7,9-тетраазафенален-3-она, 42
Синтезируют по общей методике D, используя 3-пиперидин-4-ил-1H-индол. Общий выход с двух последних стадий 19%.
МС (ES-): 397; 1H ЯМР (400 МГц, ДМСО-d6): 1,83-1,94 (м, 4H), 2,31 (м, 2H), 2,50 (с, 2H), 2,79-2,99 (м, 3H), 6,96-7,09 (м, 3H), 7,32 (д, J=8,1 Гц, 1H), 7,54-7,63 (м, 3H), 7,75 (т,J=7,3 Гц, 1H), 10,79 (с, 1H), 11,80 (с, 1H).
Получают мезилат соединения 42.
1H ЯМР (400 МГц, ДМСО-d6): 2,15 (м, 4H), 2,32 (с, 3H), 3,11 (м, 1H), 3,52 (м, 2H), 3,73 (м, 2H), 4,29 (с, 2H), 7,10-7,18 (м, 3H), 7,36 (д, 1H); 7,46(д, J=8,2 Гц, 1H), 7,69-7,83 (м, 3H), 10,91 (с, 1H), 11,93 (с, 1H). Анализ. Вычислено для C23H22N6O•1,0CH3SO3H•1,25H2O: C, 55,23; H, 5,62; N, 16,91; S, 6,14; найдено: C, 55,27; H, 5,53; N, 16,95; S, 6,00.
Получение 8-[(2-пиридин-4-илэтиламино)метил]-2,9-дигидро-1,2,7,9-тетраазафенален-3-она, 43
Синтезируют по общей методике D, используя 4-этиламинопиридин. Общий выход с двух последних стадий 10%. Получают соль HCl соединения 43.
МС (ES-): 319; 1H ЯМР (400 МГц, D2O): 3,28 (т, J=7,8 Гц, 2H), 3,53 (т,J=7,8 Гц, 2H), 4,09 (с, 2H), 7,02 (д, J=8,0 Гц, 1H), 7,35 (д, J=8,0 Гц, 1H), 7,52 (т, J=8,0 Гц, 1H), 7,70 (д, J=5,3 Гц, 2H), 8,52 (д, J=5,3 Гц, 2H). Анализ. Вычислено для C17H16N6O•1,3HCl•2,6H2O•0,1N2H4: C, 47,52; H, 5,38; N, 20,27; найдено: C, 47,12; H, 5,26; N, 20,67.
Получение 8-(3,4-дигидро-1H-изохинолин-2-илметил)-2,9-дигидро-1,2,7,9-тетраазафенален-3-она, 44
Синтезируют по общей методике D, используя 1,2,3,4-тетрагидроизохинолин. Общий выход с двух последних стадий 30%.
МС (ES-): 330; 1H ЯМР (400 МГц, ДМСО-d6): 2,81-2,90(м, 4H); 3,52 (с, 2H), 3,72 (с, 2H), 7,05-7,25 (м, 4H), 7,51 (д, J=7,8 Гц, 1H), 7,63 (д, J=8,0 Гц, 1H), 7,74 (т, J=8,0 Гц, 1H), 11,30 (ушир.с, 1H), 11,91 (с, 1H). Анализ. Вычислено для C19H17N5O: C, 68,87; H, 5,17; N, 21,13; найдено: C, 68,34; H, 5,19; N, 21,30.
Получают мезилат соединения 44.
МС (ES-): 330; 1H ЯМР (400 МГц, D2O): 2,80 (с, 3H), 3,31 (т, 2H), 3,85 (м, 2H), 4,47 (с, 2H), 4,68 (с, 2H), 7,23 (д, J=7,8 Гц, 1H), 7,28-7,42 (м, 4H), 7,67 (д, J=8,0 Гц, 1H); 7,80 (т, J=7,9 Гц, 1H). Анализ. Вычислено для C19H17N5O•1,12CH3SO3H•2,0H2O: C, 50,87; H, 5,41; N, 14,74; S, 7,56; найдено: C, 50,89; H, 5,47; N, 14,84; S, 7,63.
Получение 8-(5,6-диметокси-3,4-дигидро-1H-изохинолин-2-илметил)-2,9-дигидро-1,2,7,9-тетраазафенален-3-она, 45
Синтезируют по общей методике D, используя 5,6-диметокси-1,2,3,4-тетрагидроизохинолин. Общий выход с двух последних стадий 29%.
МС (ES-): 311; 1H ЯМР (400 МГц, ДМСО-d6): 2,79 (с, 4H), 3,49 (с, 2H), 3,61 (с, 2H), 3,67 (с, 3H), 3,70 (с, 3H), 6,69 (д, J=8,8 Гц, 2H), 7,48 (д, J=7,6 Гц, 1H), 7,63 (д, J=7,8 Гц, 1H), 7,74 (т, J=7,6 Гц, 1H), 11,55 (с, 1H). Анализ. Вычислено для C21H21N5O3: C, 64,44; H, 5,41; N, 17,89; найдено: C, 64,24; H, 5,43; N, 17,98.
Получают мезилат соединения 45.
МС (ES-): 330; 1H ЯМР (400 МГц, D2O): 2,82 (с, 3H), 3,21 (т, 2H), 3,65-3,85 (м, 8H), 4,48 (с, 2H), 4,60 (с, 2H), 6,75 (с, 1H), 6,83 (с, 1H), 7,38 (д, 1H), 7,71 (д, 1H), 7,82 (т, 1H). Анализ. Вычислено для C21H21N5O3•1,18CH3SO3H•1,75H2O: C, 49,70; H, 5,49; N, 13,07; S, 7,03; найдено: C, 49,77; H, 5,49; N, 13,17; S, 7,03.
Получение 8-[4-(3-Трифторметилбензолсульфонил)-[1,4]диазепан-1-илметил]-2,9-дигидро-1,2,7,9-тетраазафенален-3-она, 46
Синтезируют из соединения 26. К раствору соединения 26 (0,2 г, 0,67 ммоль) в 5 мл CH2Cl2 добавляют TEA (2 ммоль) и 3-трифторметилбензолсульфонилхлорид (1 ммоль). Смесь перемешивают при комнатной температуре в течение ночи. После упаривания растворителей остаток выливают в 10 мл H2O, затем продукт очищают препаративной ВЭЖХ, получая химически чистое белое твердое вещество (выход 15%).
МС (ES+): 507; 1H ЯМР (400 МГц, ДМСО-d6): 1,82 (м, 2H), 2,73-2,81 (м, 4H), 3,25-3,42 (м, 6H), 7,44 (д, J=7,8 Гц, 1H), 7,63 (д, J=7,2 Гц, 1H), 7,74 (т, J=7,8 Гц, 1H), 7,89 (т, J=8,2 Гц, 1H), 8,04-8,13 (м, 3H), 11,10 (ушир.с, 1H), 11,75 (с, 1H). Анализ. Вычислено для C22H21F3N6O3S•1,1H2O: C, 50,21; H, 4,44; N, 15,97; S, 6,09; найдено: C, 50,19; H, 4,54; N, 15,50; S, 5,97.
Схема 2
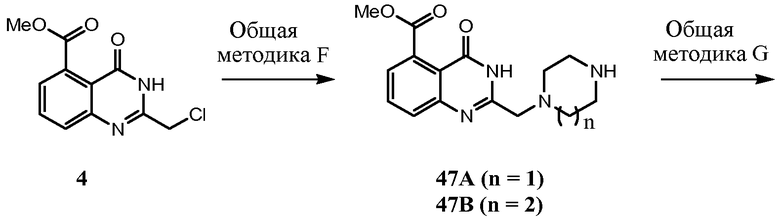
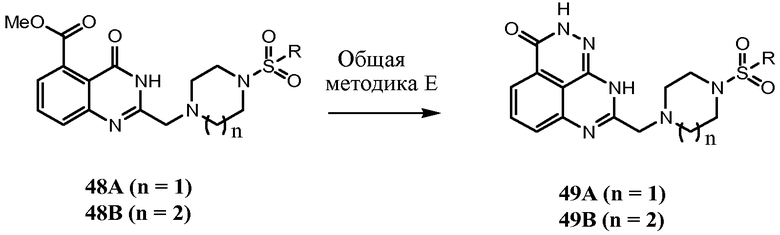
Общая методика F: Получение соединений 47A и 47B
Замена хлора в соединении 4 пиперазином или [1,4]диазепаном с использованием общего способа F дает соединение 47A или 47B. К перемешиваемому раствору соединения 4 (1 экв.) в ацетонитриле добавляют пиперазин или [1,4]диазепан (большой избыток) в атмосфере азота. Раствор оставляют перемешиваться в течение ночи и затем упаривают досуха. Неочищенное вещество очищают на слое диоксида кремния с использованием смеси 9:1 дихлорметан:метанол, с получением белого твердого вещества, метиловый эфир 4-оксо-2-пиперазин-1-илметил-3,4-дигидрохиназолин-5-карбоновой кислоты, 47A, или метиловый эфир 2-[1,4]диазепан-1-илметил-4-оксо-3,4-дигидрохиназолин-5-карбоновой кислоты, 47B.
Общая методика G: Получение соединений 48A и 48B
Взаимодействие амина 47A или 47B с разными сульфонилхлоридами дает сульфониламид 48A или 48B. К перемешиваемому раствору соединения 47A или 47B (1,0 экв.) в пиридине добавляют разные сульфонилхлориды (1,1 экв.). Реакционную смесь оставляют перемешиваться в течение ночи и затем упаривают досуха. Остаток экстрагируют дихлорметаном и промывают насыщенным раствором соли. Продукт упаривают досуха и используют без дополнительной очистки.
Общая методика E: Получение соединений 49A и 49B
2,9-дигидро-1,2,7,9-тетраазафенален-3-оновое кольцо можно получить путем конденсации соединения 48A или 48B с гидразином. К раствору соединения 6 в абсолютном этаноле при комнатной температуре добавляют избыток безводного гидразина. Раствор нагревают при кипячении с обратным холодильником в течение в течение ночи и затем охлаждают до комнатной температуры. После добавления охлажденной на льду воды выделяется белое твердое вещество. Твердое вещество собирают вакуумным фильтрованием и промывают водой и небольшим количеством метанола, с получением продуктов 6 в виде белого твердого вещества с выходом 40-90%. Пример приводится для получения соединений 49A и 49B.
Пример 2
Получение 8-[4-(4-метоксибензолсульфонил)пиперазин-1-илметил]-2,9-дигидро-1,2,7,9-тетраазафенален-3-она, 50
К перемешиваемому раствору соединения 4 (2,2 г, 8,73 ммоль, 1 экв.) в 200 мл ацетонитрила добавляют пиперазин (14 г, 0,162 моль, большой избыток) в атмосфере азота. Раствор оставляют перемешиваться в течение ночи и затем упаривают досуха. Неочищенное вещество очищают на слое диоксида кремния с использованием смеси 9:1 дихлорметан:метанол, с получением 2,0 г метилового эфира 4-оксо-2-пиперазин-1-илметил-3,4-дигидрохиназолин-5-карбоновой кислоты, 47A, в виде рыхлого белого твердого вещества.
МС (ES-): 301; 1H ЯМР (400 МГц, ДМСО-d6): 2,40-2,43 (м, 4H), 2,69-2,72 (м, 4H), 3,41 (с, 2H), 3,83 (с, 3H), 7,44 (д, J=7,2 Гц, 1H), 7,74 (д, J=8,2 Гц, 1H), 7,82 (т, J=7,8 Гц, 1H).
К перемешиваемому раствору соединения 47A (170 мг, 0,56 ммоль, 1 экв.) в 5 мл пиридина добавляют 4-метоксибензолсульфонилхлорид (130 мг, 0,62 ммоль, 1,1 экв), с получением ярко-желтого раствора. Реакционную смесь оставляют перемешиваться в течение ночи и затем упаривают досуха. Затем воскообразный остаток экстрагируют дихлорметаном и промывают насыщенным раствором соли. Неочищенное вещество растворяют в 10 мл EtOH и 5 мл моногидрата гидразина (большой избыток). Полученный раствор нагревают при кипячении с обратным холодильником в течение ночи, при этом образуется густой белый осадок, который отфильтровывают, промывают этиловым эфиром и сушат, получая не совсем белое твердое вещество. Полученное твердое вещество очищают хроматографией, с получением 112 мг химически чистого соединения 50. Получают мезилат соединения 50. Общий выход с последних трех стадий 8%.
МС (ES+): 455; 1H ЯМР (400 МГц, ДМСО-d6): 2,34 (с, 3H), 3,19 (ушир.с, 4H), 3,44 (ушир.с, 4H), 3,89 (с, 3H), 4,20 (с, 2H), 7,25 (д, J=9,0 Гц, 2H), 7,72 (д, J=7,8 Гц, 1H), 7,70-7,83 (м, 4H), 11,20 (ушир.с, 1H), 11,93 (с, 1H). Анализ. Вычислено для C21H22N6O4S•1,5CH3SO3H•3,0H2O•0,1N2H4: C, 41,20; H, 5,29; N, 13,24; S, 12,22; найдено: C, 41,07; H, 5,09; N, 13,53; S, 12,62.
Следующие соединения синтезируют аналогично методикам получения соединения 50, используя соответствующие сульфонилхлориды.
Получение 8-[4-(3-фторбензолсульфонил)пиперазин-1-илметил]-2,9-дигидро-1,2,7,9-тетраазафенален-3-она, 51
Синтезируют по общей методике G, используя 3-фтор-бензолсульфонилхлорид и соединение 47A. Получают мезилат соединения 51. Общий выход с последних трех стадий 35%.
МС (ES-): 441; 1H ЯМР (400 МГц, ДМСО-d6): 2,31 (с, 3H), 3,25 (ушир.с, 4H), 3,39 (ушир.с, 4H), 4,15 (с, 2H), 7,42 (д, J=7,8 Гц, 1H), 7,65-7,71 (м, 4H), 7,78-7,82(м, 2H), 11,78 (с, 1H). Анализ. Вычислено для C20H19N6O3S•1,25CH3SO3H•2,4H2O: C, 42,43; H, 4,87; N, 13,87; S, 11,91; найдено: C, 42,13; H, 4,79; N, 13,48; S, 11,89.
Получение 8-[4-(толуол-4-сульфонил)пиперазин-1-илметил]-2,9-дигидро-1,2,7,9-тетраазафенален-3-она, 52
Синтезируют по общей методике G, используя толуол-4-сульфонилхлорид и соединение 47A. Получают мезилат соединения 52. Общий выход с последних трех стадий 38%.
МС (ES-): 438; 1H ЯМР (400 МГц, ДМСО-d6): 2,36 (с, 3H), 2,45 (с, 3H), 3,20 (ушир.с, 4H), 3,46 (ушир.с, 4H), 4,22 (с, 2H), 7,43 (д, J=7,8 Гц, 1H), 7,54 (д, J=7,8 Гц, 2H), 7,68-7,81 (м, 4H), 11,90 (с, 1H). Анализ. Вычислено для C21H22N6O3S•1,3CH3SO3H•4,0H2O: C, 42,25; H, 5,58; N, 13,22; S, 11,61; найдено: C, 42,63; H, 5,53; N, 13,40; S, 11,90.
Получение 8-(4-бензолсульфонилпиперазин-1-илметил)-2,9-дигидро-1,2,7,9-тетраазафенален-3-она, 53
Синтезируют по общей методике G, используя бензолсульфонилхлорид и соединение 47A. Получают мезилат соединения 53. Общий выход с последних трех стадий 30%.
МС (ES-): 438; 1H ЯМР (400 МГц, ДМСО-d6): 2,70 (с, 3H), 3,36 (ушир.с, 4H), 3,51 (ушир.с, 4H), 4,14 (с, 2H), 7,11 (д, J=8,0 Гц, 1H), 7,40-7,70 (м, 7H), 11,90 (с, 1H). Анализ. Вычислено для C20H20N6O3S•1,2CH3SO3H•2,5H2O•0,08N2H4: C, 43,36; H, 5,17; N, 14,67; S, 12,01; найдено: C, 43,00; H, 5,17; N, 15,05; S, 12,40.
Получение 8-[4-(3-трифторметилбензолсульфонил)пиперазин-1-илметил]-2,9-дигидро-1,2,7,9-тетраазафенален-3-она, 54
Синтезируют по общей методике G, используя 3-трифторбензолсульфонилхлорид и соединение 47A. Получают мезилат соединения 4 Общий выход с последних трех стадий 15%.
МС (ES-): 438; 1H ЯМР (400 МГц, ДМСО-d6): 2,32 (с, 3H), 3,26-3,35 (м, 8H), 4,10 (с, 2H), 7,43 (д, J=8,0 Гц, 1H), 7,69-7,80 (м, 2H), 7,98-8,26 (м, 4H), 11,92 (с, 1H). Анализ. Вычислено для C21H19F3N6O3S•1,3CH3SO3H•2,0H2O: C, 40,99; H, 4,35; N, 12,86; S, 11,29; найдено: C, 40,71; H, 4,60; N, 12,68; S, 11,50.
Получение 8-[4-(4-хлорбензолсульфонил)пиперазин-1-илметил]-2,9-дигидро-1,2,7,9-тетраазафенален-3-она, 55
Синтезируют по общей методике G, используя 4-хлорбензолсульфонилхлорид и соединение 47A. Получают мезилат соединения 55. Общий выход с последних трех стадий 15%.
МС (ES-): 458; 1H ЯМР (400 МГц, ДМСО-d6): 2,31 (с, 3H), 3,18 (ушир.с, 4H), 3,40 (ушир.с, 4H), 3,98 (с, 2H), 7,43 (д, J=7,7 Гц, 1H), 7,68-7,84 (м, 5H), 11,90 (с, 1H). Анализ. Вычислено для C20H19ClN6O3S•1,3CH3SO3H•2,0H2O: C, 41,27; H, 4,59; N, 13,56; S, 11,90; найдено: C, 41,07; H, 4,66; N, 13,30; S, 11,89.
Получение 8-[4-(4-фторбензолсульфонил)пиперазин-1-илметил]-2,9-дигидро-1,2,7,9-тетраазафенален-3-она, 56
Синтезируют по общей методике G, используя 4-фторбензолсульфонилхлорид и соединение 47A. Получают мезилат соединения 56. Общий выход с последних трех стадий 42%.
МС (ES-): 441; 1H ЯМР (400 МГц, ДМСО-d6): 2,31 (с, 3H), 3,18 (ушир.с, 4H), 3,40 (ушир.с, 4H), 3,98 (с, 2H), 7,43 (д, J=7,8 Гц, 1H), 7,68-7,84 (м, 5H), 11,90 (с, 1H).
Получение 8-[4-(4-изопропилбензолсульфонил)пиперазин-1-илметил]-2,9-дигидро-1,2,7,9-тетраазафенален-3-она, 57
Синтезируют по общей методике G, используя 4-изопропилбензолсульфонилхлорид и соединение 47A. Получают мезилат соединения 57. Общий выход с последних трех стадий 22%.
МС (ES-): 465; 1H ЯМР (400 МГц, ДМСО-d6): 1,26 (д, J=6,8 Гц, 6H), 2,33 (с, 3H), 3,01-3,05 (м, 1H), 3,16-3,32 (м, 10H), 7,41 (д, J=8,1 Гц, 1H), 7,59 (д, J=8,6 Гц, 2H), 7,68-7,80 (м, 4H), 11,89 (с, 1H). Анализ. Вычислено для C23H26N6O3S•1,35CH3SO3H•1,75H2O•0,1N2H4: C, 46,35; H, 5,64; N, 13,76; S, 11,94; найдено: C, 46,01; H, 5,62; N, 13,80; S, 12,33.
Получение 8-[4-(4-трет-бутилбензолсульфонил)пиперазин-1-илметил]-2,9-дигидро-1,2,7,9-тетраазафенален-3-она, 58
Синтезируют по общей методике G, используя 4-трет-бутилбензолсульфонилхлорид и соединение 47A. Получают мезилат соединения 58. Общий выход с последних трех стадий 23%.
МС (ES-): 480; 1H ЯМР (400 МГц, ДМСО-d6): 1,25 (с, 9H), 2,21 (с, 3H), 3,05-3,15 (м, 8H), 3,99 (ушир.с, 2H), 7,32 (д, J=8,1 Гц, 1H), 7,59-7,72 (м, 6H), 11,81 (с, 1H). Анализ. Вычислено для C24H28N6O3S•1,5CH3SO3H•2,75H2O: C, 45,42; H, 5,90; N, 12,46; S, 11,89; найдено: C, 45,23; H, 5,76; N, 12,84; S, 12,17.
Получение 8-[4-(4-изопропилбензолсульфонил)-[1,4]диазепан-1-илметил]-2,9-дигидро-1,2,7,9-тетраазафенален-3-она, 59
Синтезируют по общей методике G, используя 4-изопропилбензолсульфонилхлорид и соединение 47B. общий выход с двух последних стадий 22%.
МС (ES-): 479; 1H ЯМР (400 МГц, ДМСО-d6): 1,23 (д, 6H), 1,79 (м, 2H), 2,40-2,55 (м, 4H), 2,71-2,90 (м, 4H), 3,00 (м, 1H), 3,48 (с, 2H), 7,48 (м, 3H), 7,73 (м, 4H), 11,80 (с, 1H). Анализ. Вычислено для C24H28N6O3S: C, 59,98; H, 5,87; N, 17,49; S, 6,67; найдено: C, 60,02; H, 5,85; N, 17,55; S, 6,52.
Получение 8-[4-(4-хлорбензолсульфонил)-[1,4]диазепан-1-илметил]-2,9-дигидро-1,2,7,9-тетраазафенален-3-она, 60
Синтезируют по общей методике G, используя 4-хлор-бензолсульфонилхлорид и соединение 47B. Общий выход с последних трех стадий 8%.
МС (ES-): 472; 1H ЯМР (400 МГц, ДМСО-d6): 1,80 (м, 2H), 2,73-2,78 (м, 4H), 3,50 (м, 4H), 3,69 (с, 2H), 7,45 (д, J=8,2 Гц, 1H), 7,71-7,83 (м, 6H), 10,95 (ушир.с, 1H), 11,76 (с, 1H).
Получают мезилат соединения 60.
1H ЯМР (400 МГц, D2O): 1,92 (м, 2H), 2,73 (с, 5H), 3,50-3,77 (м, 8H), 4,36 (с, 2H), 7,49 (д, J=12 Гц, 1H), 7,75 (т, J=8,1 Гц, 2H), 7,78-7,93 (м, 4H). Анализ. Вычислено для C21H21ClN6O3S•1,61CH3SO3H: C, 39,57; H, 4,99; N, 12,25; S, 12,20; найдено: C, 39,50; H, 5,29; N, 12,57; S, 12,47.
Получение 8-[4-(3-фторбензолсульфонил)-[1,4]диазепан-1-илметил]-2,9-дигидро-1,2,7,9-тетраазафенален-3-она, 61
Синтезируют по общей методике G, используя 3-фторбензолсульфонилхлорид и соединение 47B. Общий выход с двух последних стадий 16%.
МС (ES+): 457; 1H ЯМР (400 МГц, ДМСО-d6): 1,79 (м, 2H), 2,70-2,81 (м, 4H), 3,26-3,40 (м, 4H), 3,48 (с, 2H), 7,45 (д, J=7,3 Гц, 1H); 7,55-7,74 (м, 6H), 11,10 (ушир.с, 1H), 11,75 (с, 1H). Анализ. Вычислено для C21H21FN6O3S•1,15H2O: C, 52,85; H, 4,92; N, 17,61; S, 6,72; найдено: C, 52,88; H, 4,93; N, 17,43; S, 6,48.
Получение 8-[4-(4-метоксибензолсульфонил)-[1,4]диазепан-1-илметил]-2,9-дигидро-1,2,7,9-тетраазафенален-3-она, 62
Синтезируют по общей методике G, используя 4-метоксибензолсульфонилхлорид и соединение 47B. Общий выход с двух последних стадий 21%.
МС (ES+): 469; 1H ЯМР (400 МГц, ДМСО-d6): 1,78 (м, 2H), 2,72-2,79 (м, 4H), 3,30-3,39 (м, 4H), 3,48 (с, 2H), 3,84 (с, 3H), 7,14 (д, J=8,2 Гц, 2H), 7,48 (д, J=8,1 Гц, 1H), 7,63 (д, J=7,2 Гц, 1H); 7,09-7,22 (м, 3H), 11,10 (ушир.с, 1H), 11,80 (с, 1H). Анализ. Вычислено для C22H24N6O4S•1,0H2O: C, 54,31; H, 5,39; N, 17,27; S, 6,59; найдено: C, 54,38; H, 5,34; N, 17,28; S, 6,19.
Получение 8-[4-(4-трет-бутилбензолсульфонил)-[1,4]диазепан-1-илметил]-2,9-дигидро-1,2,7,9-тетраазафенален-3-она, 63
Синтезируют по общей методике G, используя 4-т-бутил-бензолсульфонилхлорид и соединение 47B. Общий выход с двух последних стадий 14%.
МС (ES-): 493; 1H ЯМР (400 МГц, ДМСО-d6): 1,31 (с, 9H), 1,79 (м, 2H), 2,73-2,86 (м, 4H), 3,26-3,41 (м, 4H), 3,48 (с, 2H), 7,45 (д, J=8,6 Гц, 1H), 7,62-7,76 (м, 6H), 11,20 (ушир.с, 1H), 11,80 (с, 1H). Анализ. Вычислено для C25H30N6O3S: C, 60,71; H, 6,11; N, 16,99; S, 6,48; найдено: C, 60,78; H, 6,10; N, 17,08; S, 6,36.
Получение 8-[4-(4-аминобензолсульфонил)-[1,4]диазепан-1-илметил]-2,9-дигидро-1,2,7,9-тетраазафенален-3-она, 64
Синтезируют по общей методике G, используя 4-нитробензолсульфонилхлорид и соединение 47B. Общий выход с двух последних стадий 14%.
МС (ES-): 452; 1H ЯМР (400 МГц, ДМСО-d6): 1,76 (м, 2H), 2,71-2,79 (м, 4H), 3,21-3,31 (м, 4H), 3,46 (с, 2H), 6,01 (с, 2H), 6,64 (д, J=8,6 Гц, 2H), 7,39 (д, J=8,6 Гц, 2H), 7,48 (д, J=8,0 Гц, 1H), 7,63 (д, J=7,9 Гц, 1H), 7,74 (т, J=7,8 Гц, 1H), 11,10 (ушир.с, 1H), 11,75 (с, 1H). Анализ. Вычислено для C21H23N7O3S•0,5H2O: C, 54,53; H, 5,23; N, 21,20; S, 6,93; найдено: C, 54,50; H, 5,24; N, 20,84; S, 6,74.
Получение 8-[4-(бифенил-4-сульфонил)-[1,4]диазепан-1-илметил]-2,9-дигидро-1,2,7,9-тетраазафенален-3-она, 65
Синтезируют по общей методике G, используя бифенил-4-сульфонилхлорид и соединение 47B. Общий выход с двух последних стадий 10%.
МС (ES-): 513; 1H ЯМР (400 МГц, ДМСО-d6): 1,82 (м, 2H), 2,73-2,83 (м, 4H), 3,29-3,41 (м, 4H), 3,48 (с, 2H), 7,47-7,53 (м, 4H), 7,62 (д, J=8,1 Гц, 1H), 7,68-7,78 (м, 3H), 7,82-7,93 (м, 4H), 11,00 (ушир.с, 1H), 11,75 (с, 1H). Анализ. Вычислено для C27H26N6O3S•2,3H2O: C, 58,32; H, 5,55; N, 15,11; S, 5,77; найдено: C, 58,24; H, 4,89; N, 15,10; S, 5,79.
Получение 8-[4-(4-аминобензолсульфонил)пиперазин-1-илметил]-2,9-дигидро-1,2,7,9-тетраазафенален-3-она, 66
Синтезируют по общей методике G, используя 4-нитробензолсульфонилхлорид и соединение 47A. Получают мезилат соединения 66. Общий выход с последних трех стадий 35%.
МС (ES-): 438; 1H ЯМР (400 МГц, ДМСО-d6): 2,32 (с, 3H), 3,13 (ушир.с, 4H), 3,42 (ушир.с, 4H), 4,18 (с, 2H), 6,71 (д, J=8,8 Гц, 2H), 7,40-7,43 (м, 3H), 7,70-7,80 (м, 2H), 11,20 (ушир.с, 1H), 11,92 (с, 1H). Анализ. Вычислено для C20H21N7O3S•1,3CH3SO3H•2,75H2O: C, 41,67; H, 5,20; N, 15,97; S, 12,01; найдено: C, 41,76; H, 5,25; N, 15,92; S, 12,22.
Схема 3
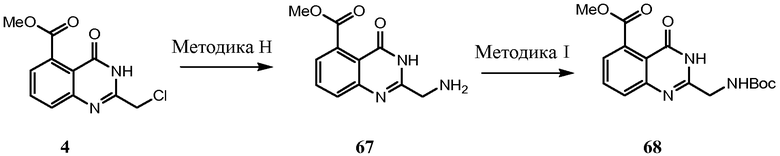
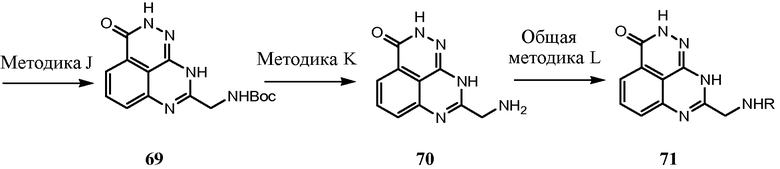
Общую методику L используют для получения соединения 71. К перемешиваемому раствору соединения 70 (1,0 экв.) в ТГФ в атмосфере азота добавляют TEA (1 мл, избыток) и либо сульфонилхлорид, либо хлорангидрид (1,2 экв.). Реакционную смесь оставляют перемешиваться в течение четырех часов, затем упаривают, экстрагируют смесью CH2Cl2/H2O, сушат и концентрируют. Неочищенное вещество очищают колоночной хроматографией, используя смесь 9:1 CH2Cl2/MeOH, с получением химически чистых продуктов 71.
Пример 3
Получение (3-оксо-2,9-дигидро-3H-1,2,7,9-тетраазафенален-8-илметил)карбаминовой кислоты трет-бутилового эфира, 69
Методику H используют для получения метилового эфира 2-аминометил-4-оксо-3,4-дигидрохиназолин-5-карбоновой кислоты, 67. К 7н. раствору NH3 (большой избыток) в MeOH объемом 25 мл при 0°C добавляют соединение 4 (1,0 г, 4,0 ммоль) и пробирку герметично закрывают. Затем смесь нагревают при 60°C в течение 4 часов. Смесь упаривают досуха, растворяют и снова упаривают в 2×50 мл CH2Cl2. Продукт используют как таковой, без дополнительной очистки.
Методику I используют для получения метилового эфира 2-(трет-бутоксикарбониламинометил)-4-оксо-3,4-дигидрохиназолин-5-карбоновой кислоты, 68. К 50 мл раствора 2 мл TEA (избыток), каталитического количества DMAP и соединения 67 (полученного по способу H) в CH2Cl2 добавляют boc-ангидрид (2,6 г, 3 экв.) при комнатной температуре. Реакционную смесь оставляют перемешиваться в течение 60 минут, при этом все твердые вещества переходят в раствор. Раствор упаривают досуха, очищают колоночной хроматографией, используя CH2Cl2 и 5% MeOH, с получением 0,5 г химически чистого соединения 68.
Методику J используют для получения соединения 69. 5 г соединения 68 растворяют в 10 мл моногидрата гидразина и 25 мл этанола. Смесь нагревают при кипячении с обратным холодильником в течение четырех часов, пока исходное вещество не перестанет обнаруживаться методом ТСХ. Реакционную смесь охлаждают, выливают в 100 мл холодной воды и экстрагируют 2×25 мл EtOAc. Органические слои сушат насыщенным раствором соли и затем сульфатом магния. Очищают колоночной хроматографией, используя смесь 9:1 CH2Cl2/MeOH, с получением 2,7 г химически чистого соединения 69.
МС (ES-): 314; 1H ЯМР (400 МГц, CDCl3): 1,45 (с, 9H), 3,90 (с, 2H), 6,15 (ушир.с, 1H), 6,94-7,30 (м, 3H), 12,38-12,43 (ушир.м, 2H). Анализ. Вычислено для C15H17N5O3•0,2H2O: C, 56,49; H, 5,50; N, 21,96; найдено: C, 56,61; H, 5,60; N, 21,85.
Получение 8-аминометил-2,9-дигидро-1,2,7,9-тетраазафенален-3-она, 70
Методику K используют для получения соединения 70. 250 мг соединения 69 растворяют в 10 мл CH2Cl2 вместе с 4 мл TFA. Реакционную смесь оставляют перемешиваться при комнатной температуре в течение ночи, получая густой белый осадок, который отфильтровывают, промывают CH2Cl2, сушат в вакууме и с количественным выходом получают химически чистое вещество, соль TFA соединения 70.
МС (ES+): 216; 1H ЯМР (400 МГц, D2O): 3,97 (с, 2H), 6,91 (д, J=8,2 Гц, 1H), 7,23 (д, J=7,8 Гц, 1H), 7,43 (т, J=8,0 Гц, 1H). Анализ. Вычислено для C10H9N5O•1,3CF3COOH•0,2H2O: C, 41,27; H, 2,86; N, 19,10; найдено: C, 41,00; H, 3,04; N, 19,25.
Получение 4-метил-N-(3-оксо-2,9-дигидро-3H-1,2,7,9-тетраазафенален-8-илметил)бензолсульфонамида, 72
Синтезируют по общей методике L, используя 4-метилбензолсульфонилхлорид и соединение 70. Выход соединения 72 составляет 20%.
МС (ES-): 368; 1H ЯМР (400 МГц, CDCl3): 2,27 (с, 3H), 3,93 (с, 2H), 7,28-7,43 (м, 3H), 7,67-7,78 (м, 4H), 11,43 (с, 1H), 11,83 (с, 1H). Анализ. Вычислено для C17H15N5O3S: C, 55,27; H, 4,09; N, 18,96; S, 8,68; найдено: C, 54,93; H, 4,09; N, 18,63; S, 8,33.
Получение N-(3-оксо-2,9-дигидро-3H-1,2,7,9-тетраазафенален-8-илметил)бензолсульфонамида, 74
Синтезируют по общей методике L, используя бензолсульфонилхлорид и соединение 70. Выход соединения 74 составляет 25%.
МС (ES-): 354; 1H ЯМР (400 МГц, CDCl3): 3,95 (д, J=5,0 Гц, 2H), 7,44 (д, J=7,8 Гц, 1H), 7,45-7,60 (м, 3H), 7,67-7,77 (м, 2H), 7,91-7,93 (м, 2H), 8,24 (т, J=8,8 Гц, 1H), 11,24 (с, 1H), 11,83 (с, 1H). Анализ. Вычислено для C16H13N5O3S•1,0H2O: C, 51,47; H, 4,05; N, 18,76; S, 8,59; найдено: C, 51,17; H, 4,20; N, 18,73; S, 8,31.
Получение N-(3-оксо-2,9-дигидро-3H-1,2,7,9-тетраазафенален-8-илметил)ацетамида, 75
Синтезируют по общей методике L, используя уксусный ангидрид и соединение 70. Выход соединения 75 составляет 22%.
МС (ES-): 256; 1H ЯМР (400 МГц, ДМСО-d6): 1,91 (с, 3H), 4,05 (с, 2H), 7,32 (д, J=8,1 Гц, 1H), 7,49-7,81 (м, 2H), 8,40 (т, J=8,3 Гц, 1H), 11,25 (с, 1H), 11,75 (с, 1H). Анализ. Вычислено для C12H11N5O2•0,5H2O: C, 54,13; H, 4,54; N, 26,30; найдено: C, 54,14; H, 4,52; N, 26,00.
Получение 4-нитро-N-(3-оксо-2,9-дигидро-3H-1,2,7,9-тетраазафенален-8-илметил)бензамида, 76
Синтезируют по общей методике L, используя 4-нитробензоилхлорид и соединение 70. Выход соединения 76 составляет 25%.
МС (ES-): 363; 1H ЯМР (400 МГц, CDCl3): 3,99 (с, 2H), 7,18-7,20 (м, 1H), 7,34-7,38 (м, 1H), 7,75-7,90 (м, 2H), 8,20-8,32 (м, 2H), 8,40-8,48 (м, 3H).
PARP-ингибирующая активность in vitro - IC 50
Традиционным способом определения IC5O соединения, ингибирующего PARP, является анализ PARP с использованием очищенного рекомбинантного человеческого PARP от Trevigan (Gaithersburg, Md.): ферментный анализ PARP проводят на льду в объеме 100 микролитров, который содержит 100 мМ Tris-HCl (pH 8,0), 1 мМ MgCl2, 28 мМ KCl, 28 мМ NaCl, 3,0 мкг/мл ДНКаза I-активированной ДНК спермы сельди (Sigma, Mo.), 30 микромолярный [3H]никотинамидадениндинуклеотид (62,5 мКи/ммоль), 15 микрограмм/мл фермента PARP и тестируемые соединения в разных концентрациях. Реакцию инициируют путем добавления фермента, затем смесь инкубируют при 25°C. После 2 минут инкубации реакцию останавливают добавлением 500 микролитров охлажденной на льду 30% (масс/об) трихлоруксусной кислоты. Образовавшийся осадок переносят на фильтр из стекловолокна (Packard Unifilter-GF/C) и промывают три раза 70% этанолом. После сушки фильтра определяют радиоактивность путем подсчета сцинтилляций. С помощью описанного анализа ингибирования обнаружено, что соединения настоящего изобретения обладают высокой ферментативной активностью с IC50 в интервале от нескольких наномолей до 20 микромолей.
С помощью описанного выше анализа PARP получают приблизительные значения IC50 для следующих соединений:
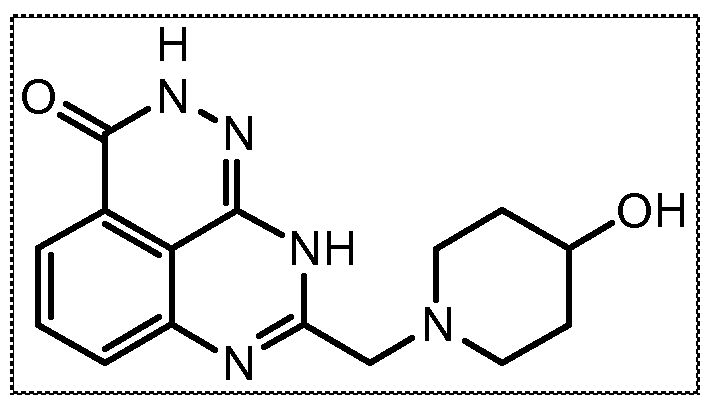
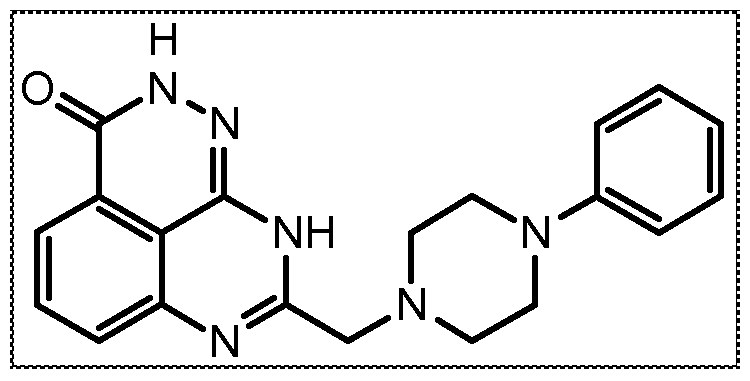
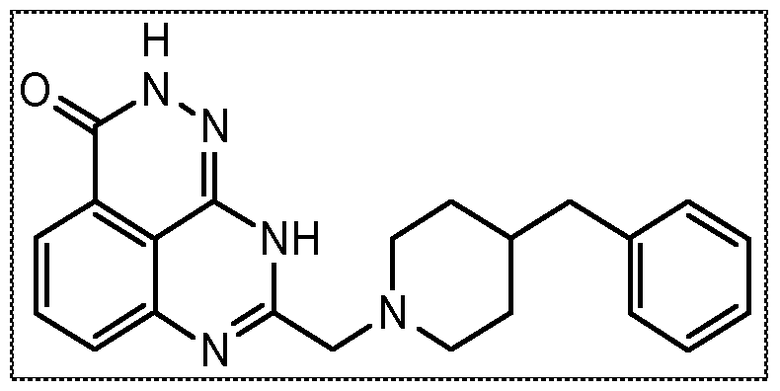
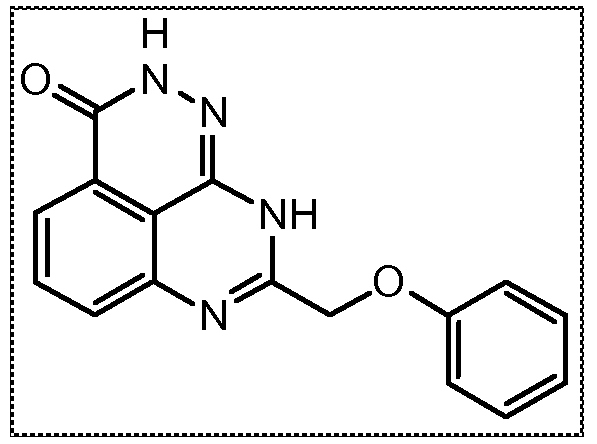
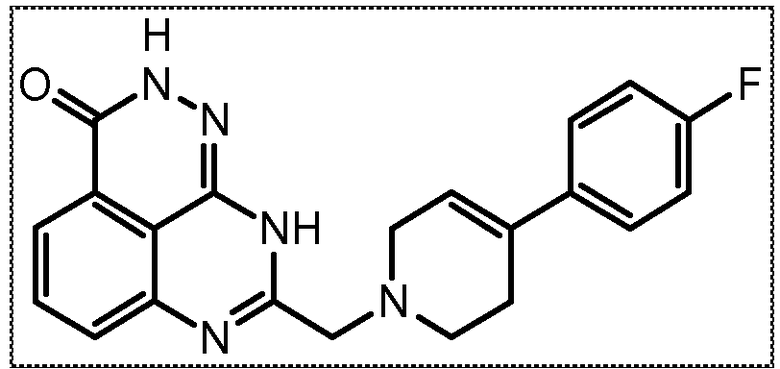
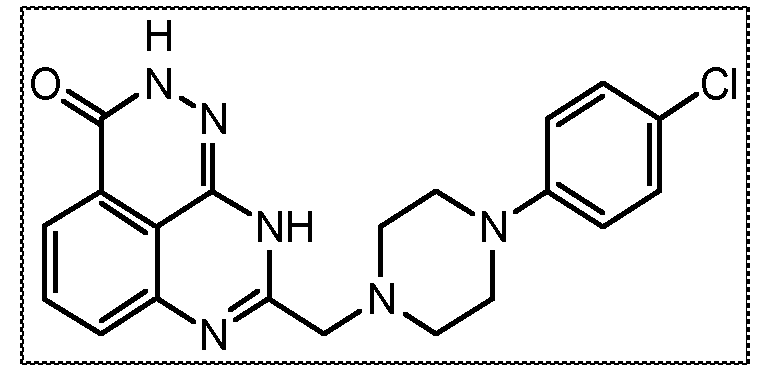
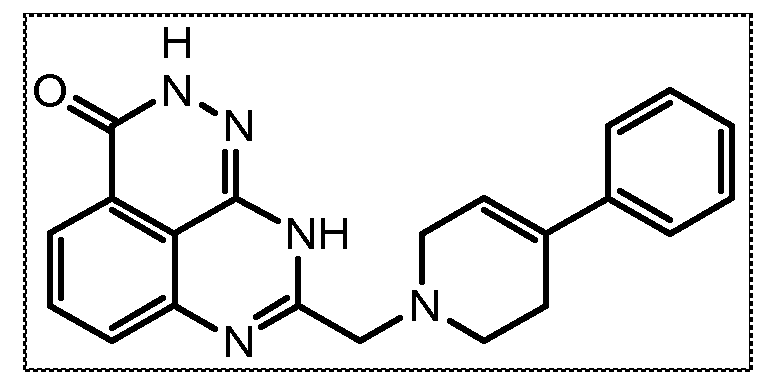
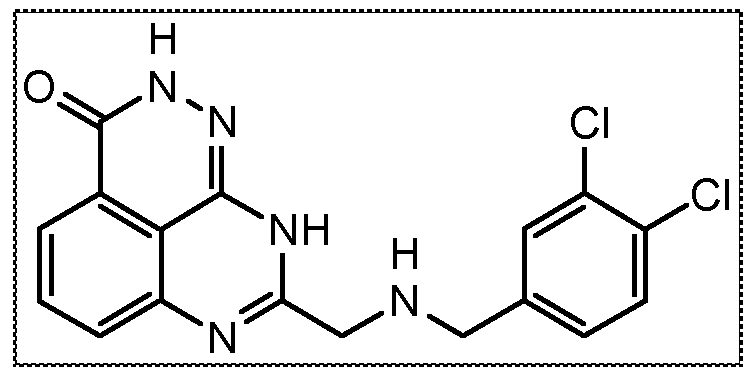
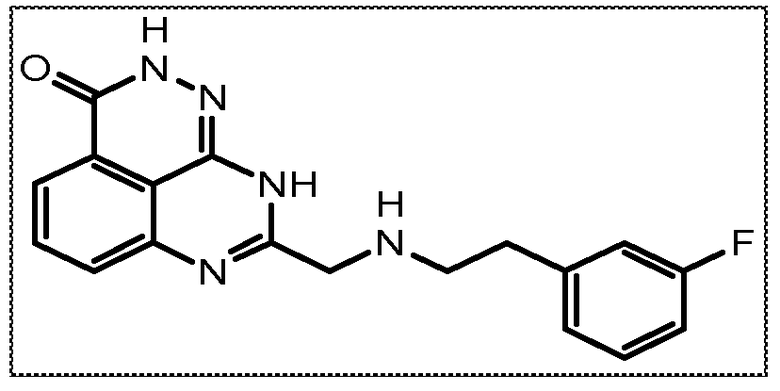
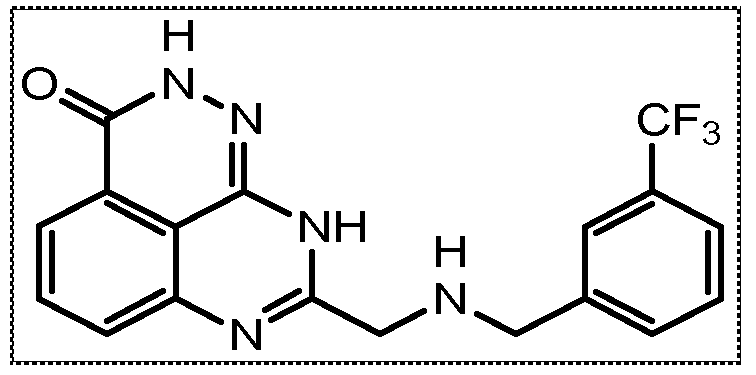
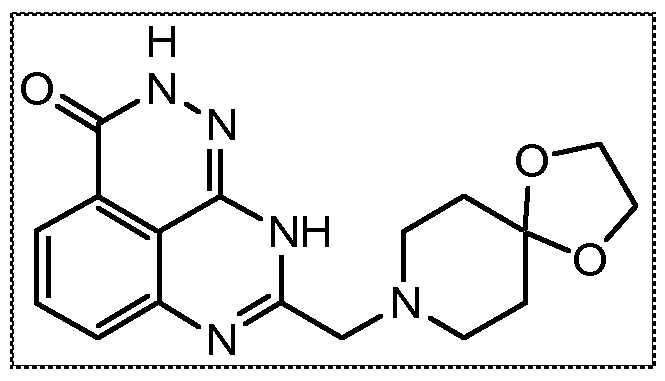
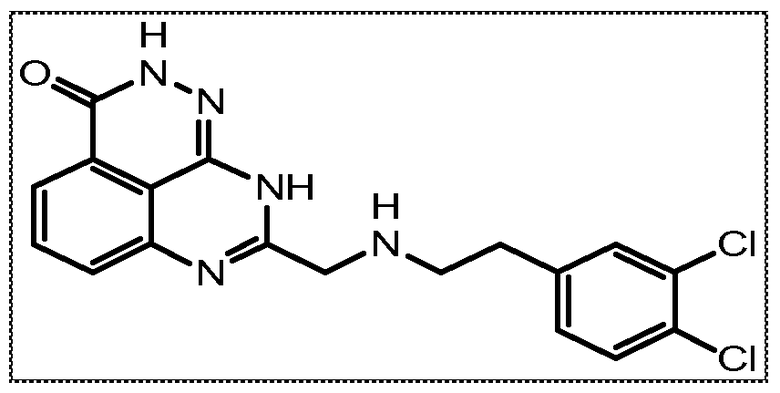
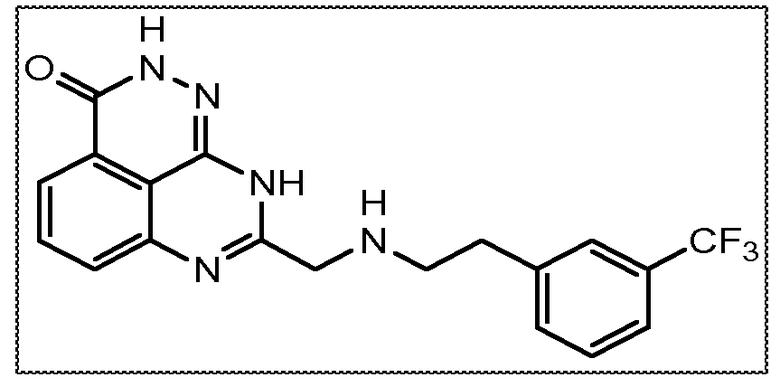
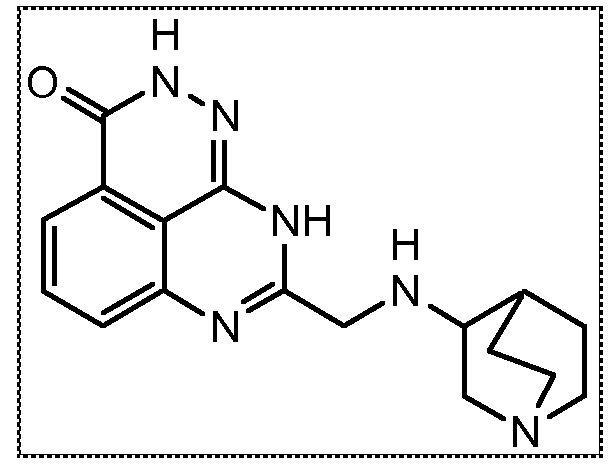

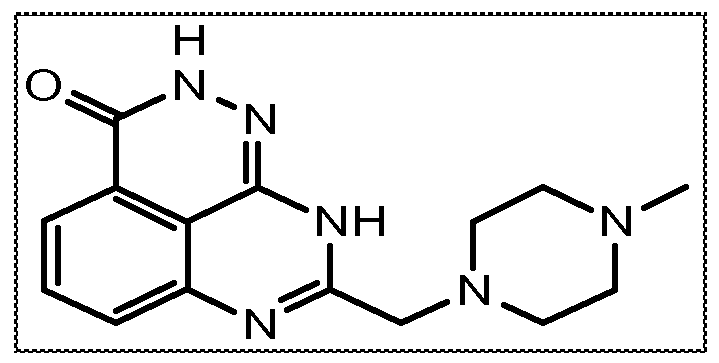
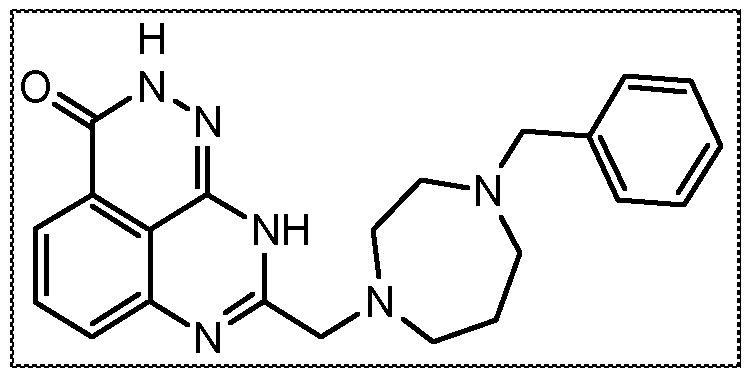
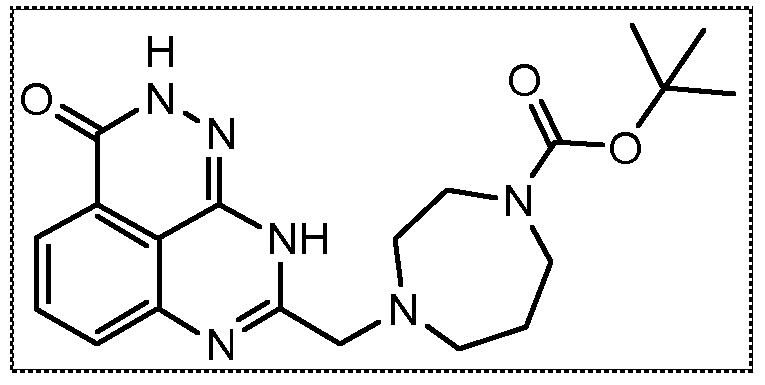
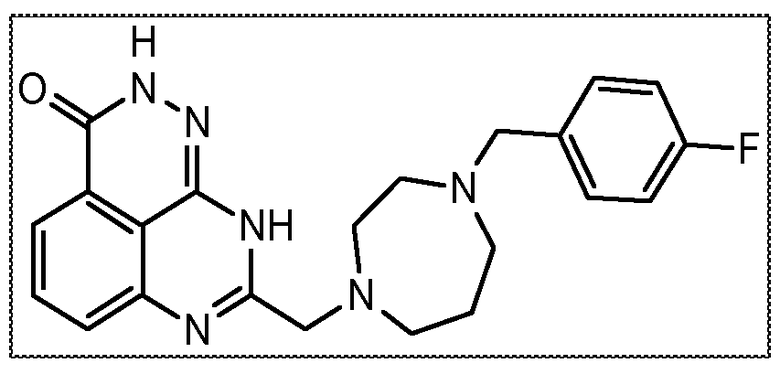
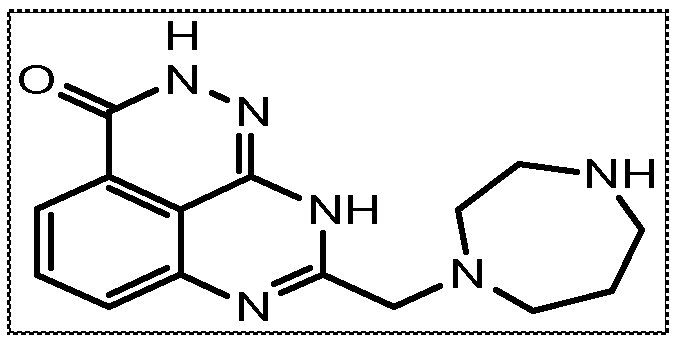
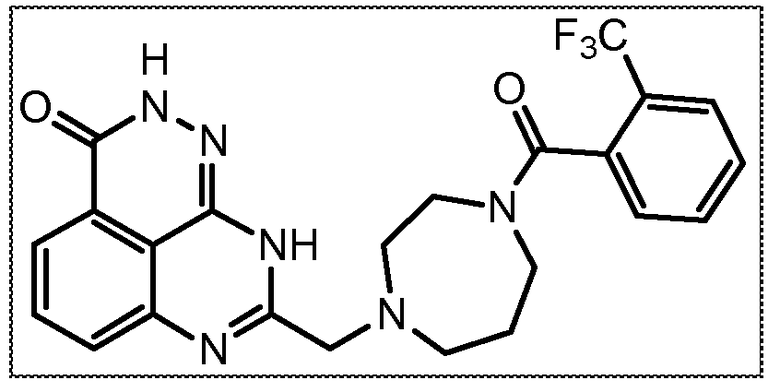
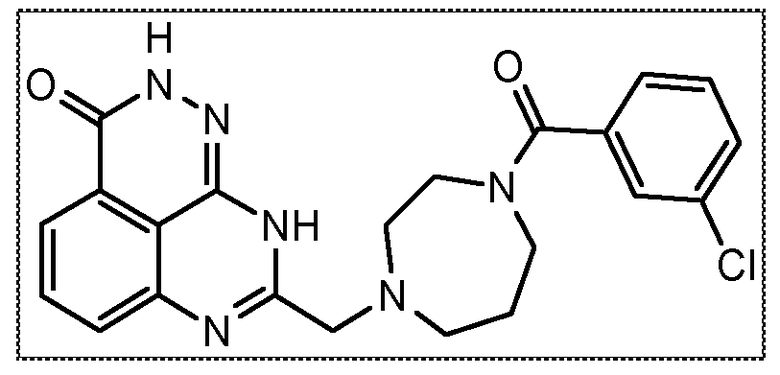
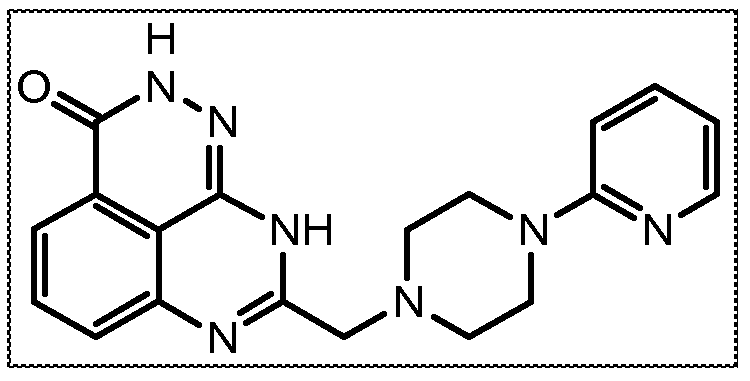
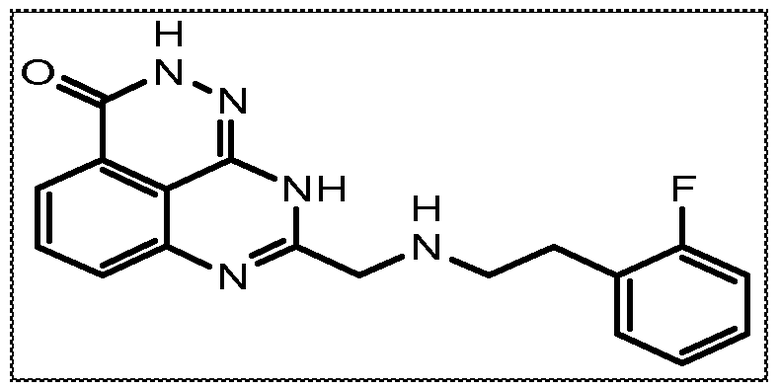
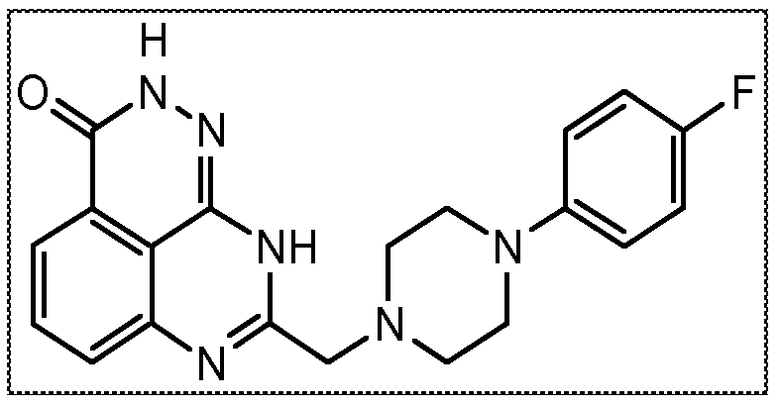
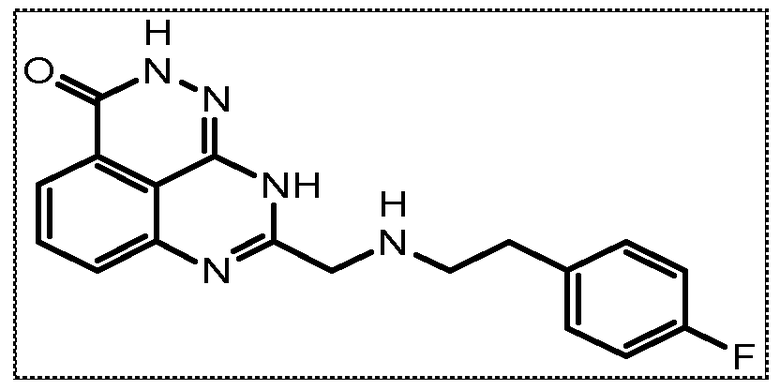

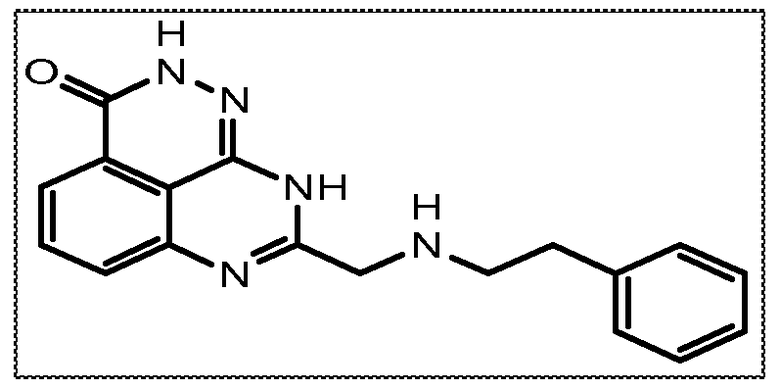
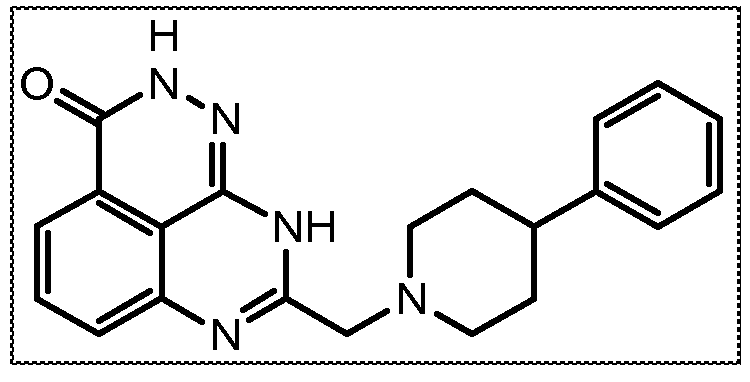
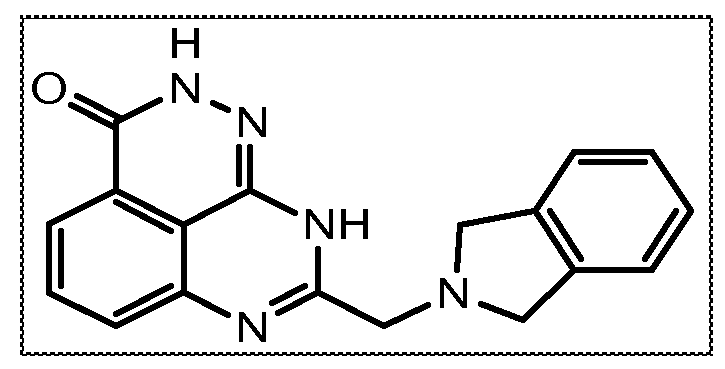
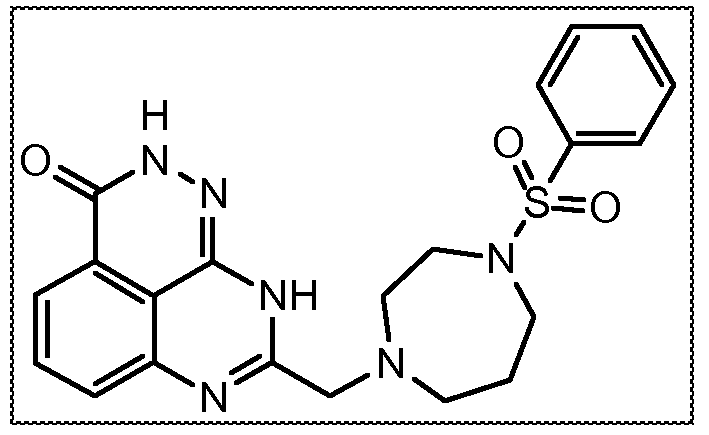
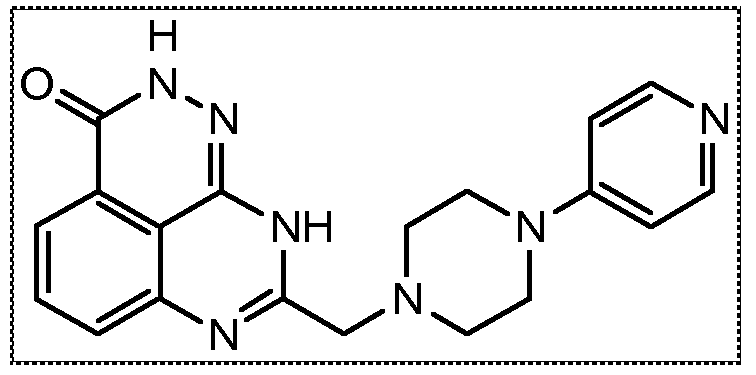

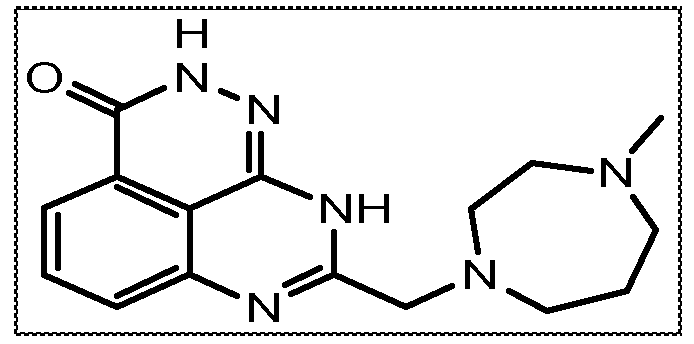
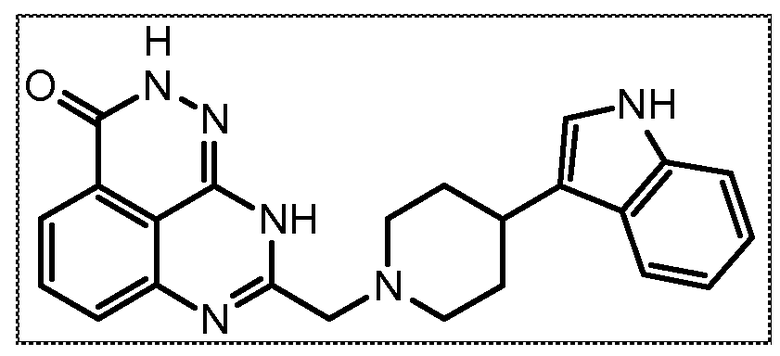
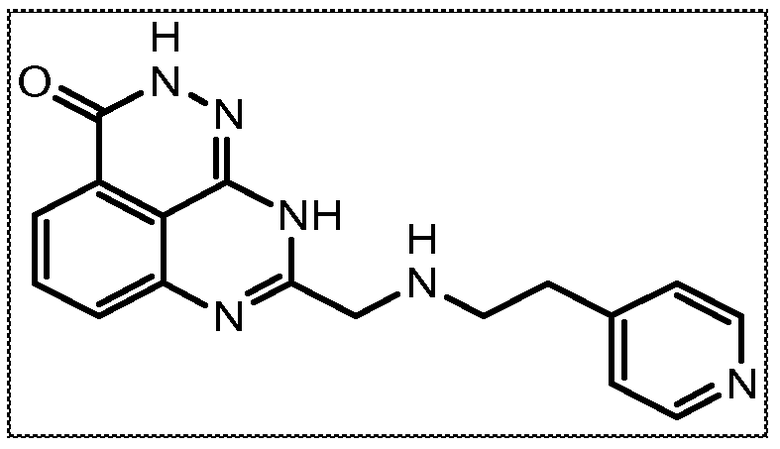
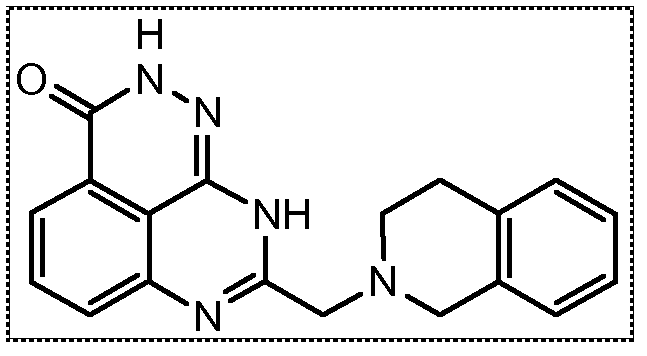
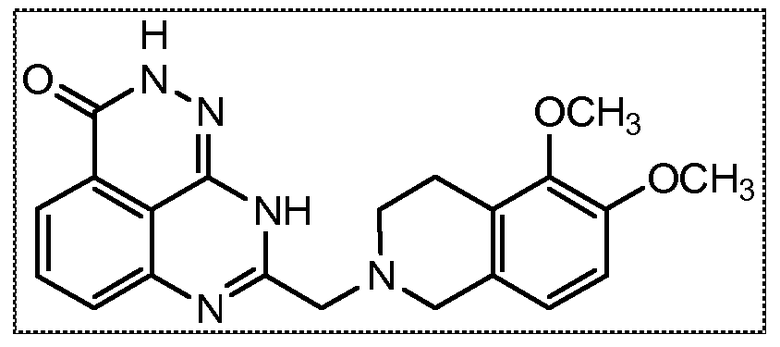
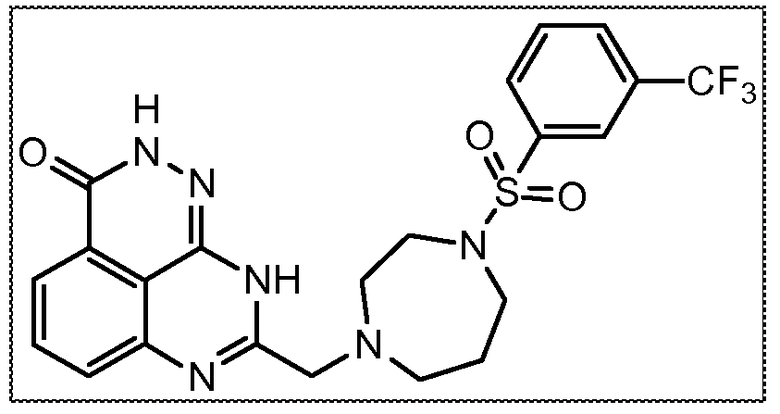
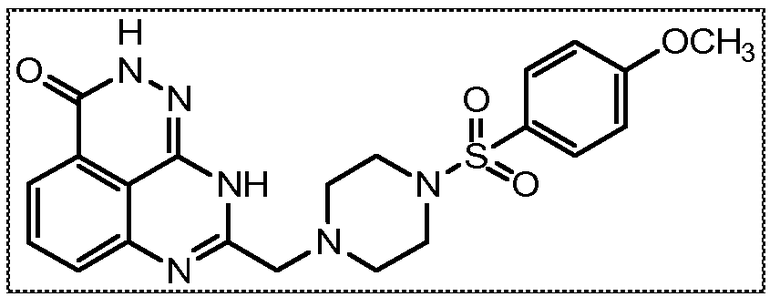
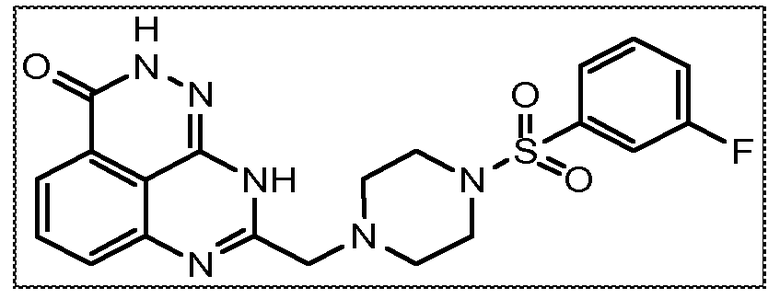
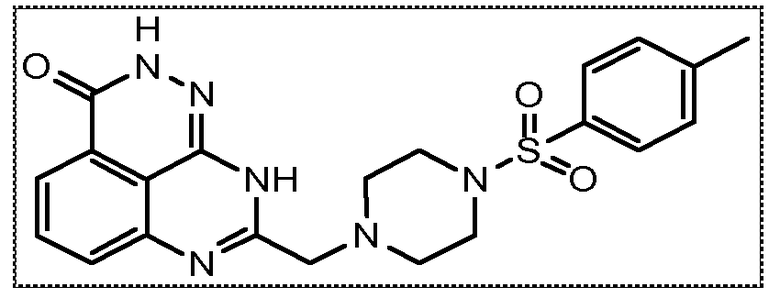
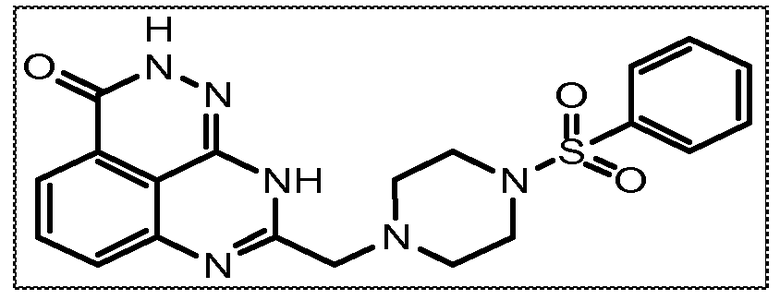
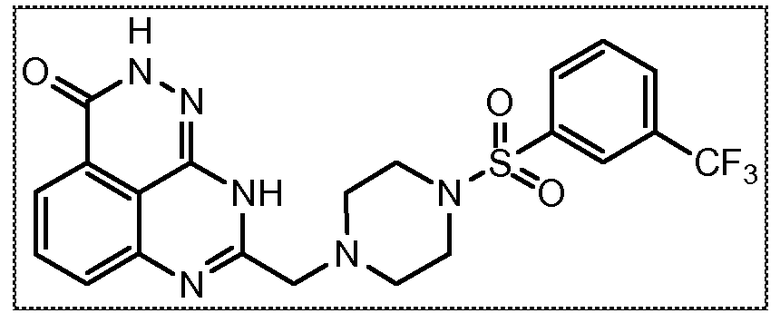

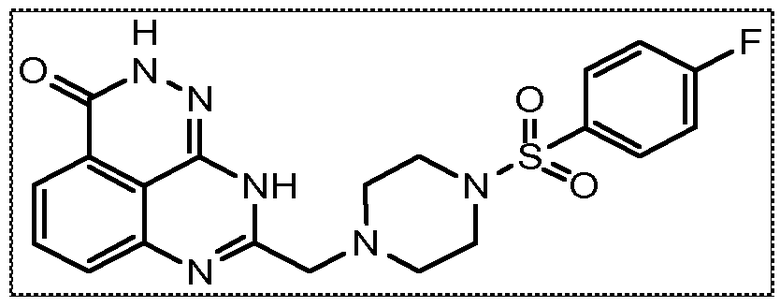
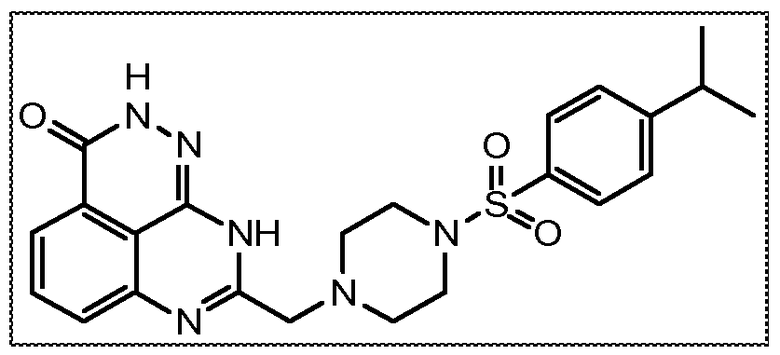
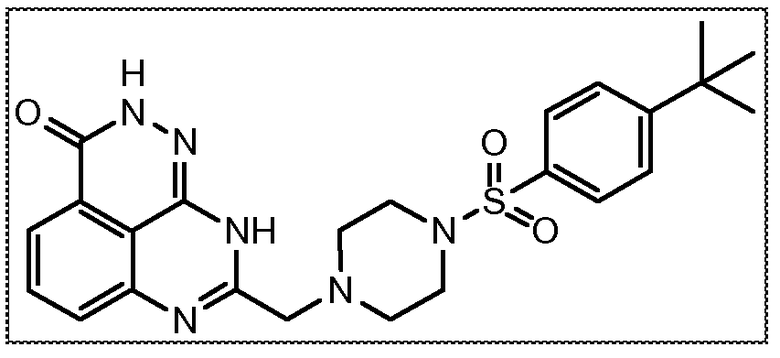
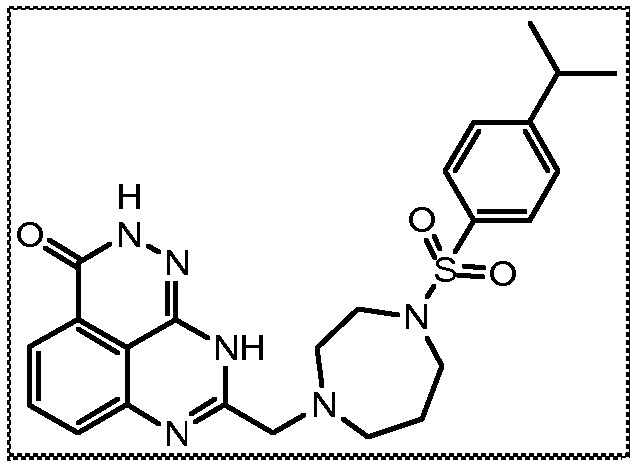
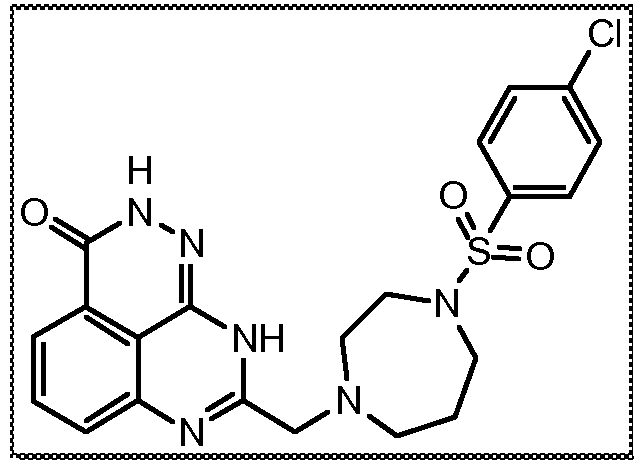
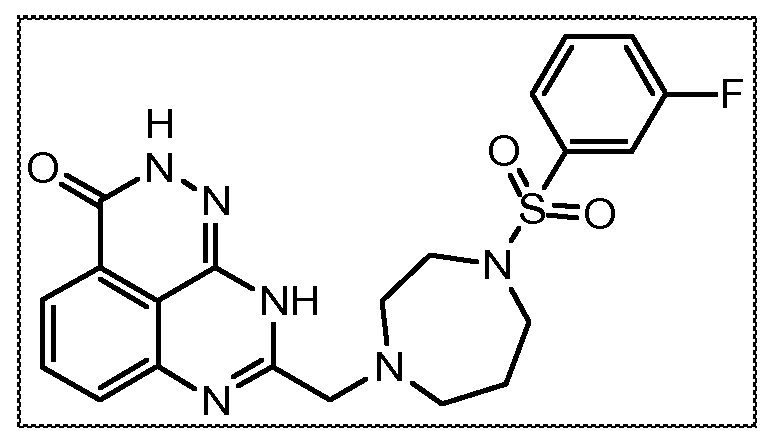
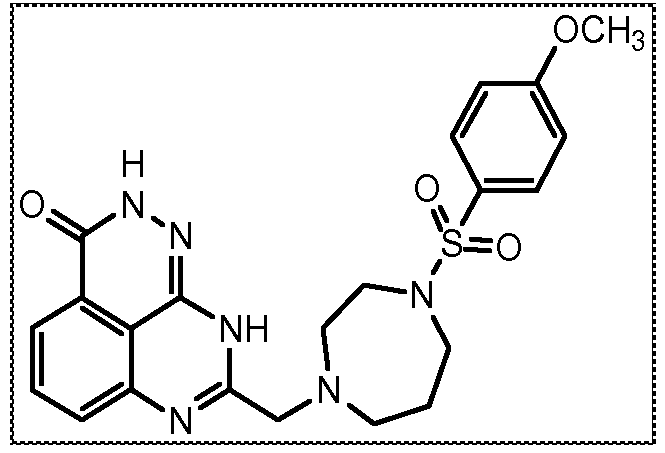
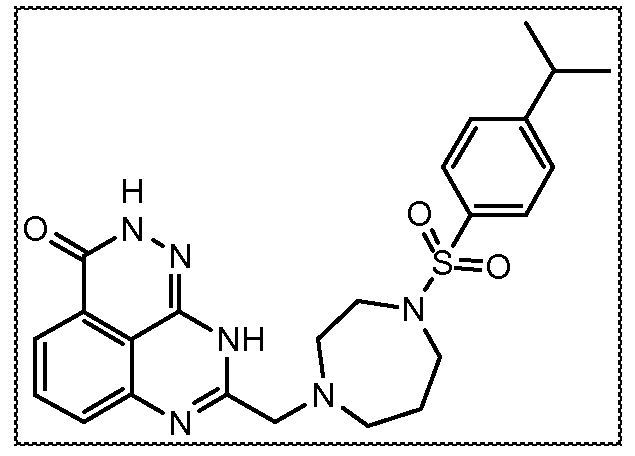
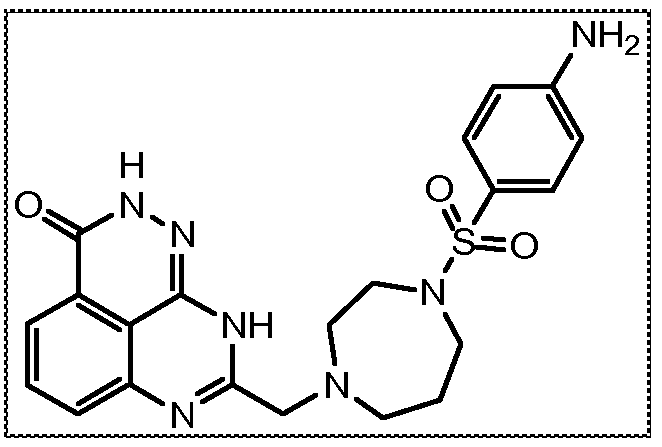
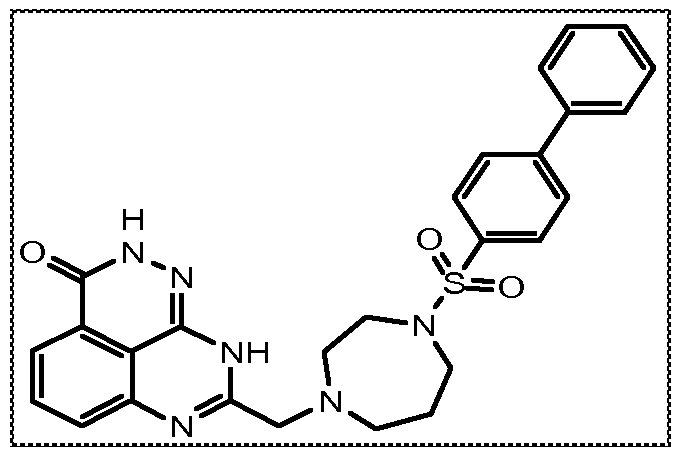
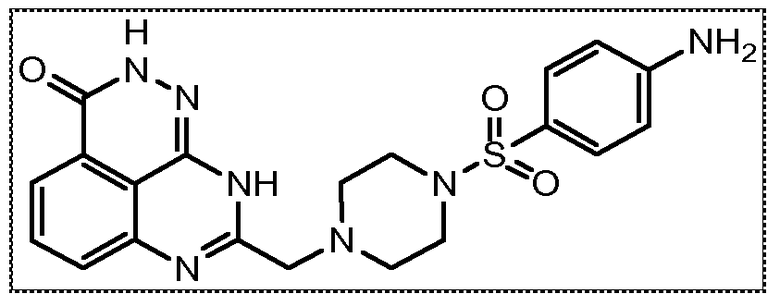
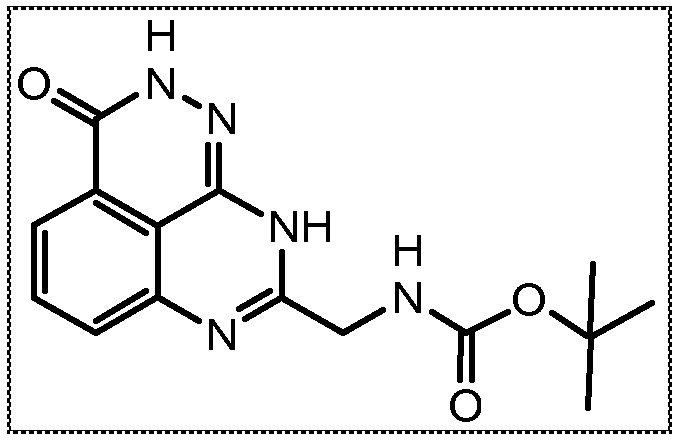
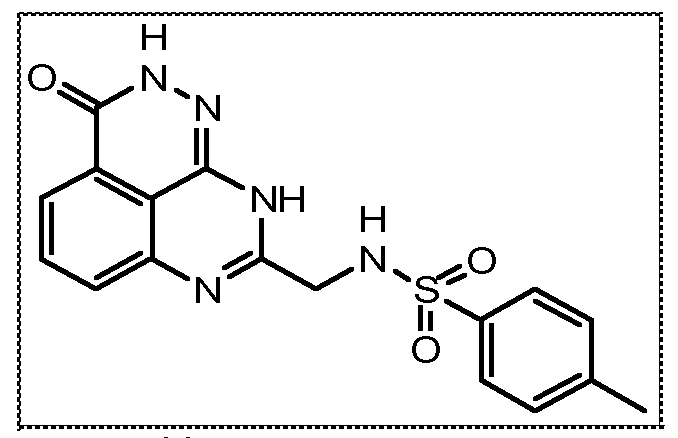
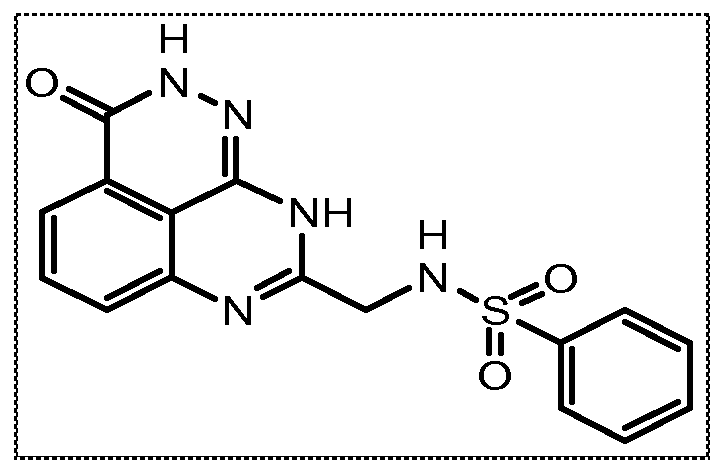
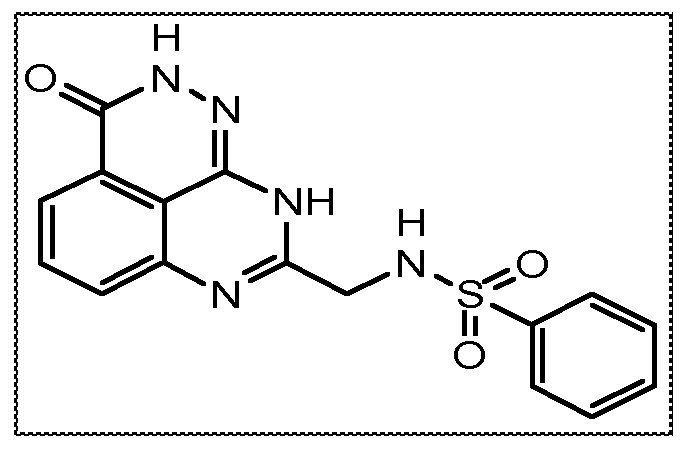

Эффективность соединения 13 in vivo
1) Мышиная внутричерепная модель меланомы B16
Клеточную линию мышиной меланомы B16, полученную из C57BL/6J (H-2b/H-2b), культивируют в среде RPMI-1640, содержащей 10% фетальной телячьей сыворотки (Invitrogen, Milan, Italy), 2 мM L-глутамин, 100 единиц/мл пенициллина и 100 мкг/мл стрептомицина (Flow Laboratories, Mc Lean, VA), при 37°C во влажной атмосфере, содержащей 5% CO2. TMZ получают от Schering-Plough Research Institute (Kenilworth, NJ). Соединение 13 растворяют в 70 мM PBS, не содержащем калия.
Чтобы провести внутричерепную трансплантацию, клетки (104 в 0,03 мл RPMI-1640) вводят интракраниально (и.к.) через центрально-среднюю зону лобной кости на глубину 2 мм с помощью стеклянного микрошприца объемом 0,1 мл и съемной инъекционной иглы 27 калибра. Клетки мышиной меланомы B16 (104) вводят и.к. самцам мышей B6D2F1 (C57BL/6×DBA/2). Перед введением опухоли животных анестезируют кетамином (100 мг/кг) и ксилазином (5 мг/кг) в 0,9% растворе NaCl (10 мл/кг/в.б.). Гистологический анализ роста опухоли в мозге проводят через 1-5 дней после введения опухоли, чтобы определить сроки лечения.
Соединение 13 вводят перорально за 15 мин до TMZ. Все контрольные мыши получают наполнитель для лекарственного средства. Лечение мышей, несущих опухоль, начинают через 48 часов после заражения, когда инфильтрация опухоли в ткань, окружающую мозг, становится гистологически очевидной. Мышей обрабатывают соединением 13 через желудочный зонд один раз в день в течение пяти дней, в дозе 10 мг/кг.
Лечение мышей, несущих опухоль, начинают на 2 день после заражения, когда инфильтрация опухоли в ткань, окружающую мозг, становится гистологически очевидной. Мышей ежедневно обрабатывают соединением 13 и TMZ в течение 5 дней и регистрируют смертность в течение 90 дней. Определяют среднее время выживания (MST) и рассчитывают процент увеличения продолжительности жизни (ILS): {[MST (дни) опытных мышей/MST (дни) контрольных мышей]-1}×100. Эффективность лечения определяют путем сравнения кривых выживания мышей из опытных и контрольных групп.
Эксперименты с использованием мышей и уход за мышами осуществляют в соответствии с национальными и международными руководствами (Инструкция европейского экономического общественного совета 86/109, OLJ318, 1 декабря 1987, и Руководство национальных институтов здравоохранения по уходу за лабораторными животными и их применению, 1985).
Кривые выживания получают по методу Каплана-Мейера продукт-лимит, и статистические различия среди разных групп (8 животных/группа) оценивают с использованием log-рангового анализа с поправкой Йейтса (программное обеспечение Primer of Biostatistics, McGraw-Hill, New York, NY). Статистическую значимость определяют на уровне p=0,05. Различия считаются статистически значимыми, если P<0,05.
Результаты показывают, что пероральное введение 10 мг/кг соединения 13 значительно увеличивает продолжительность выживания мышей, поскольку продолжительность выживания у мышей, получающих комбинацию соединение 13+TMZ, существенно выше, чем у мышей, получающих только TMZ (P<0,0001). Существенные различия в продолжительности выживания среди контрольных групп и групп, получающих TMZ, не наблюдаются (фиг.1).
2) Мышиная внутричерепная ксенотрансплантатная модель глиомы SJGBM2
Соединение 13 анализируют на внутричерепной ксенотрансплантатной модели глиомы SJGBM2 мышей (Tentori, et al. Clin. Cancer Reser. 2003, 9, 5370). Для этой цели соединение 13 вводят однократно за 15 мин до введения TMZ в дозе 10 мг/кг, п.о.
Обнаружено, что доза соединения 13 10 мг/кг является эффективной (фиг.2). Ее сочетание с TMZ увеличивает MTS с 22,5 дней (только TMZ) до 25 дней (P=0,002).
Эффективность соединения 37 in vivo
1) Мышиная внутричерепная модель меланомы B16
Эксперимент проводят по способу, описанному выше для соединения 13. Исследуют, действительно ли пероральное введение соединения 37 (5 мг/кг или 12,5 мг/кг) повышает эффективность TMZ в отношении меланомы B16, растущей в участке ЦНС. Результаты, полученные на мышах, несущих меланому B16, показывают, что продолжительность выживания в группах, получающих комбинацию соединение 37, 12,5 мг/кг+TMZ, значительно выше, чем у животных, получающих только TMZ (фиг.3).
2) Мышиная внутричерепная ксенотрансплантатная модель глиомы SJGBM2
Затем исследуют эффективность соединения 37 с использованием ортотопической модели ксенотрансплантата человеческой мультиформной глиобластомы (SJGBM2) на голых мышах. На фиг.4 показан ответ SJGBM2 на TMZ, вводимый в качестве единственного средства или в комбинации с соединением 37 (10 мг/кг или 20 мг/кг) в течение пяти дней, или в комбинации с соединением 37 (MGI25036), 10 мг/кг, в течение пяти дней, с последующим введением только соединения 37 в дозе 100 мг/кг в течение 14 дней. Результаты показывают, что пероральное введение соединения 37 (10 мг/кг или 20 мг/кг)+TMZ значительно продлевает продолжительность выживания мышей, несущих опухоль, по сравнению с контрольными животными или животными, получающими TMZ. Следует отметить, что в отношении данной модели опухоли TMZ является неэффективным. Введение 10 мг/кг соединения 37+TMZ в течение пяти дней, с последующим введением высокой дозы соединения 37 (100 мг/кг) в течение 14 дней значительно повышает выживание животных по сравнению с введением 10 мг/кг соединения 37+TMZ в течение пяти дней.
3) Усиление влияния облучения на плоскоклеточную карциному головы и шеи
Используют клеточную линию JHU012 человеческой HNSCC, предварительно охарактеризованную генетически и изначально полученную в лабораториях головы и шеи Университета Джонса Гопкинса из эксплантатов человеческих опухолей. Клеточную линию поддерживают в среде RPMI 1640, содержащей 10% фетальной бычьей сыворотки и 1% пенициллина/стрептомицина, во влажных инкубаторах при 5% CO2 и 37°C. Эксперименты проводят на самцах голых мышей BALB/c nu/nu возрастом 6 недель. Животных случайным образом распределяют на группы, получающие следующую обработку: группа 1 - контроли, группа 2 - только облучение (2 грей (гр)/день в течение 2 дней), группа 3 - только 100 мг/кг соединения 37 перорально (п.о.) qdxl7, группа 4 - 30 мг/кг соединения 37 п.о.+облучение, группа 5 - 100 мг/кг соединения 37 п.о.+облучение, причем каждая группа состоит из 8 мышей. Мышей анестезируют путем внутрибрюшинного введения 3-5 мл трибромэтанола. Опухоли приживляют с правого бока путем подкожной инъекции 1×107 клеток. Через четырнадцать дней после введения клеток опухоли экспонируют хирургическим путем и измеряют их размеры в 3 направлениях с помощью штангенциркуля. Затем опытным группам 3-5 перорально вводят соединение 37. В группах 4 и 5 животные получают соединение 37 за 15 минут до облучения (2 гр/день в течение 2 дней). На 31 день после инокуляции опухолевых клеток опухоли снова экспонируют хирургическим путем и измеряют их размеры в 3 направлениях с помощью штангенциркуля. Значительное ингибирование роста опухоли наблюдается в группе 5, получающей перорально 100 мг/кг соединения 37+облучение (объем опухоли в конце эксперимента равен 209,04 мм3) по сравнению с контрольной группой 1 (объем опухоли в конце эксперимента=585,9 мм3 p<0,01) (фиг.5). Соединение 37 в дозе 30 мг/кг в комбинации с облучением не оказывает существенного влияния на ингибирование роста опухоли по сравнению с облучением (фиг.5). Кроме того, введение только 100 мг/кг соединения 37 п.о. qdx17 не оказывает существенного влияния на ингибирование роста опухоли по сравнению с контролями, получающими наполнитель (фиг.5). Это свидетельствует о том, что комбинация повышенной дозы соединения 37 с облучением оказывает более сильный эффект, чем указанные виды обработки по отдельности.
4) Влияние соединения 37 на рост опухоли у мышей, несущих BRCA-1-дефицитные опухоли
1×106 клеток, дефицитных по BRCA-1, вводят подкожно в правый бок самок мышей nu/nu (возрастом 6-7 недель; Harlan Sprague Dawley, Indianapolis Indiana). Примерно через 10-14 дней объем опухолей достигает приблизительно 100 мм3. Мышей делят на группы так, чтобы средний размер опухолей во всех группах был примерно одинаковым с минимальными стандартными отклонениями. Введение начинают на следующий день после деления на группы, и затем объем опухолей регистрируют три раза в неделю. У опухолей измеряют два диаметра и объем, рассчитываемый по формуле (l×w)2/2. Мышей отстраняют от эксперимента, если объем опухоли достигает 1500 мм3. "Время до наступления конечного момента" или TTE (число дней, необходимое для того, чтобы объем опухоли достиг 1500 мм3 или больше) представляет собой конечный момент исследования. Соединение 37 отвешивают каждые 2-3 дня и солюбилизируют стерильной бутилированной воде (J.T. Baker, Ultrapure Bioreagent 4221-02), с получением концентрации 10 мг/мл. Соединение вводят перорально, ежедневно в течение 28 дней, начиная с 1 дня исследования. В качестве положительного контроля используют хорошо известный ингибитор PARP, который заведомо является эффективным в отношении моделей BRCA при отдельном введении (Bryant et al). Средство, используемое в качестве положительного контроля, вводят в дозе 25 мг/кг в.б. qdx5 после начала эксперимента. Соединение 37 в дозе 100 мг/кг эффективно замедляет рост опухоли на модели, дефицитной по BRCA-1, в оба анализируемых промежутка времени. Если введение соединения 37 прекращают на 28 день, опухоли начинают расти спустя примерно 10-14 дней. Соединение 37 не только значительно замедляет рост опухоли по сравнению с контролями, получающими наполнитель, но и по сравнению с положительным контролем (p<0,05) в обоих экспериментах.
Исследование проводят с целью сравнения биодоступности и уровней в плазме мозга разных млекопитающих, получающих описываемые соединения и подобное соединение известного уровня техники. Соединение известного уровня техники имеет следующую формулу:
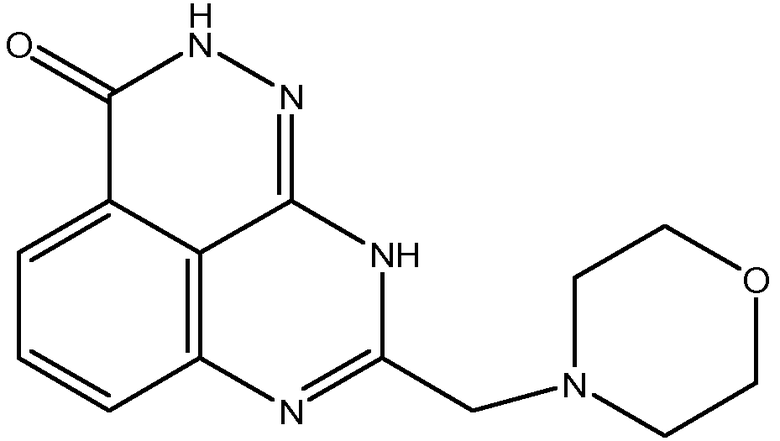
Сравнительный анализ проводят следующим образом
Ингибиторы PARP в водных растворах вводят либо путем болюсной (1 мин) внутривенной инъекции, либо перорально через зонд. В случае собак, внутривенное и пероральное введение проводят по перекрестной схеме с периодом вымывания одна неделя между способами введения. Анализируемая доза составляет 30 мг/кг для каждого соединения. В случае мышей, трех животных для каждого момента времени умерщвляют путем асфиксии в атмосфере CO2 и собирают кровь путем пункции сердца. В случае крыс и собак, серийные образцы крови берут в разные моменты времени у указанного числа животных. В случае крыс, объем полученного образца крови сразу заменяют 2× объемом 1:1 донорной крысиной крови: гепаринизированный физиологический раствор. Образцы крови переносят в гепаринизированные контейнеры, быстро перемешивают и хранят на льду до центрифугирования и получения плазмы. Плазму переносят в свежие контейнеры и хранят при -70°C до биоанализа. В некоторых случаях, после умерщвления собирают мозг или опухолевую ткань и хранят при ≤-70°C до биоанализа.
Образцы плазмы осаждают ацетонитрилом, упаривают и повторно растворяют. Образцы мозга и опухолевой ткани гомогенизируют в забуференном фосфатом физиологическом растворе, pH 7,4, осаждают ацетонитрилом, затем упаривают и повторно растворяют. Повторно растворенные образцы анализируют по сравнению с матричными калибровочными стандартами методом ЖХ-МС/МС. Результаты биоаналитического метода подтверждаются результатами анализа с использованием образцов контроля качества. Как правило, нижняя граница определяемых в плазме количеств составляет 5 нг/мл. Нижняя граница определяемых в ткани количеств зависит от степени разбавления в процессе гомогенизации, но обычно составляет от 15 до 20 нг/г.
Результаты, полученные при определении концентраций в плазме, мозге и опухоли обрабатывают путем фармакокинетического анализа без компартментализации, с помощью программного обеспечения WinNonlin, профессиональная версия 4.1. AUC рассчитывают, используя линейно-логарифмический принцип. Временные точки для фазы Lambda Z выбирают путем визуального исследования. Наклон конечных фаз рассчитывают путем невзвешенной линейной регрессии.
Селективные ингибиторы PARP тестируют по основным фармакокинетическим свойствам в плазме и ткани мышей, крыс и собак. Результаты анализа показывают, что данное семейство соединений является биодоступным после перорального введения у всех видов и способно проникать в мозг и опухолевую ткань. В таблице 1 суммированы значения пероральной биодоступности (п.о.) для соединений 8, 13, 36 и 37 и соединения сравнения у мышей и крыс, а также значения соотношения мозг/плазма (М/П) для указанных пяти соединений у мышей и крыс.
Результаты сравнительного анализа приведены в таблице II. Результаты показывают, что, хотя соединение известного уровня техники обладает хорошей биодоступностью, отношение уровней соединения известного уровня техники в мозге и плазме является очень низким. Неожиданно было обнаружено, что раскрытые соединения формулы (I) имеют хорошее отношение уровней в мозге и плазме по сравнению с соединением известного уровня техники. Полученные результаты показывают, что описываемые соединения обладают неожиданной доступностью для центральной нервной системы, что необходимо для проявления терапевтического эффекта, по сравнению с соединением известного уровня техники.
Сравнение биодоступности (п.о.) и отношение уровней в мозге и плазме (М/П) для отдельных соединений формулы (I) по сравнению с родственным соединением известного уровня техники
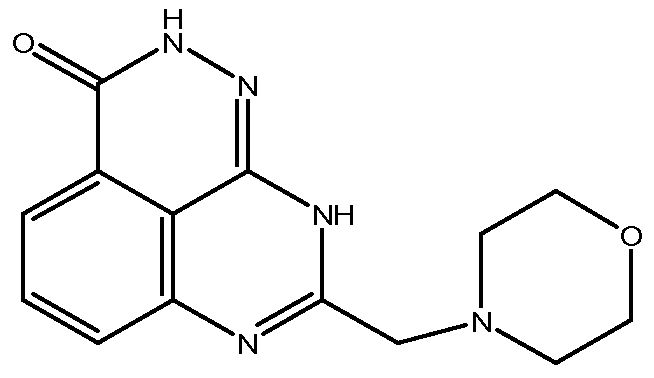
Сравнительное соединение
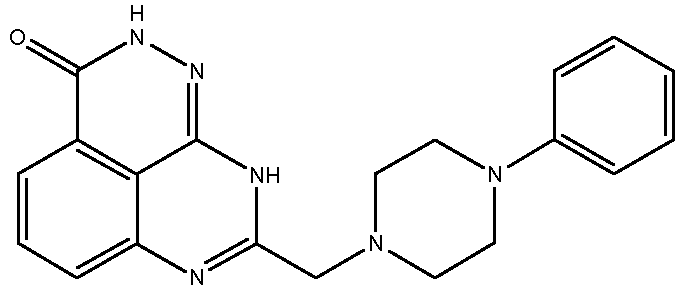
8
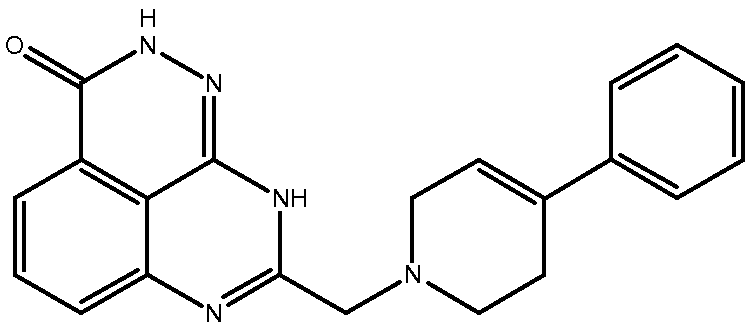
13
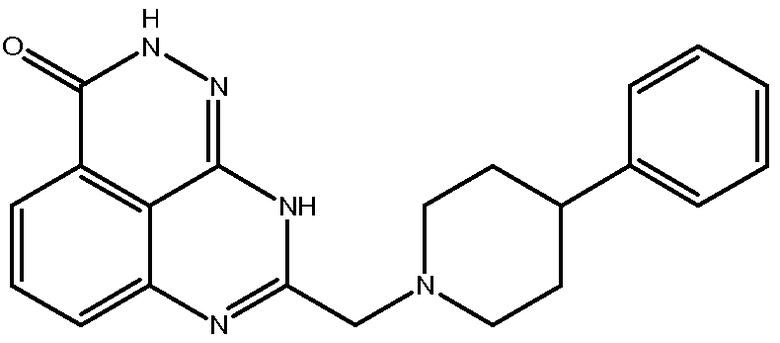
36
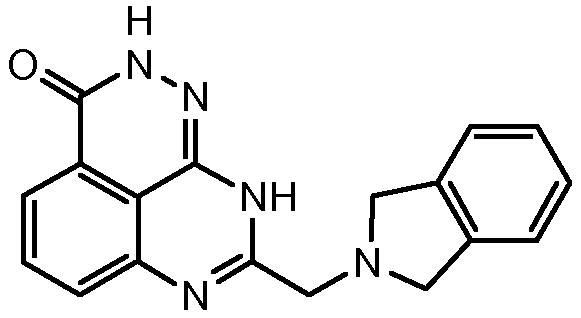
37
Очевидно, что могут существовать различные вариации раскрытого изобретения. Такие вариации не следует расценивать как отступление от сущности и объема настоящего изобретения, и предполагается, что все такие модификации входят в объем приведенной ниже формулы изобретения.
Включение посредством ссылки
Все цитируемые в настоящем описании публикации, патенты и патентные заявки, опубликованные через 18 месяцев с даты приоритета, включены в данное описание посредством ссылки, и, во всех случаях, как если бы было специально и индивидуально указано, что каждая отдельная публикация или патентная заявка включена в данное описание посредством ссылки. В случае противоречия настоящее изобретение имеет преимущественную силу.
| название | год | авторы | номер документа |
|---|---|---|---|
| ДИГИДРОПИРИДОФТАЛАЗИНОНОВЫЕ ИНГИБИТОРЫ ПОЛИ(АДФ-РИБОЗА)ПОЛИМЕРАЗЫ | 2009 |
|
RU2514937C2 |
| СОЕДИНЕННЫЕ МОСТИКОВОЙ СВЯЗЬЮ N-ЦИКЛИЧЕСКИЕ СУЛЬФОНАМИДО-ИНГИБИТОРЫ ГАММА-СЕКРЕТАЗЫ | 2006 |
|
RU2422443C2 |
| ПЕСТИЦИДНЫЕ КОМПОЗИЦИИ | 2009 |
|
RU2513723C2 |
| ПРОИЗВОДНЫЕ 2-ЗАМЕЩЕННЫХ ФЕНИЛ-5, 7-ДИГИДРОКАРБИЛ-3, 7-ДИГИДРОПИРРОЛО [2, 3-d] ПИРИМИДИН-4-ОНОВ, ИХ ПОЛУЧЕНИЕ И ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2004 |
|
RU2323220C2 |
| ПРОИЗВОДНЫЕ ЦИКЛИЧЕСКОГО ИМИНОПИРИМИДИНА В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗ | 2018 |
|
RU2801302C2 |
| НОВОЕ ТРИЦИКЛИЧЕСКОЕ ПРОИЗВОДНОЕ ИЛИ ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, СПОСОБ ИХ ПОЛУЧЕНИЯ И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2009 |
|
RU2470934C1 |
| БИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ АЛЛОСТЕРИЧЕСКИХ ИНГИБИТОРОВ SHP2 | 2018 |
|
RU2776846C2 |
| АРИЛ-ИЛИ ГЕТЕРОАРИЛЗАМЕЩЕННЫЕ БЕНЗОЛЬНЫЕ СОЕДИНЕНИЯ | 2012 |
|
RU2632193C2 |
| 6-5 КОНДЕНСИРОВАННЫЕ КОЛЬЦА КАК ИНГИБИТОРЫ С5а | 2018 |
|
RU2780338C2 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛА, ИСПОЛЬЗУЕМЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕИНКИНАЗЫ | 2001 |
|
RU2340611C2 |


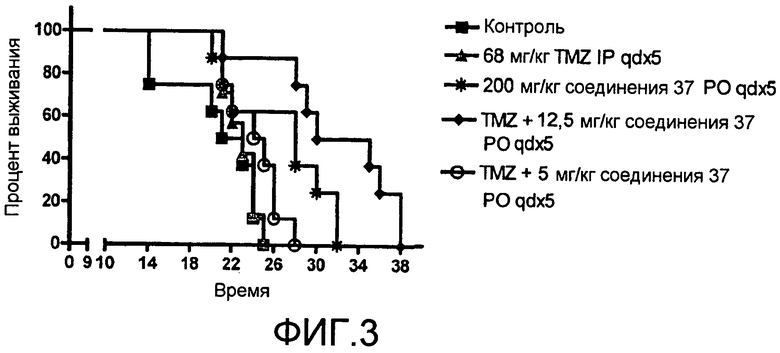
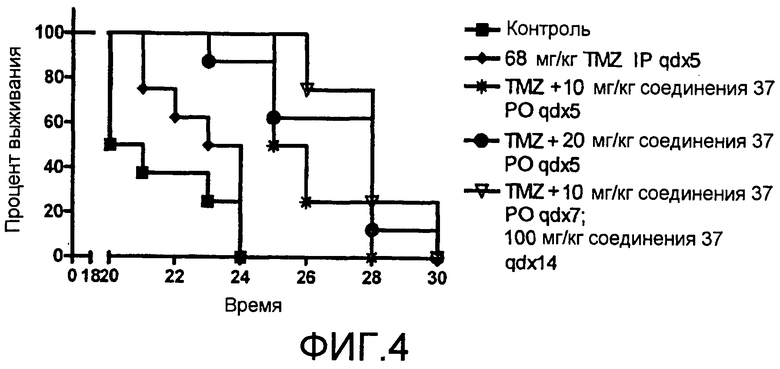

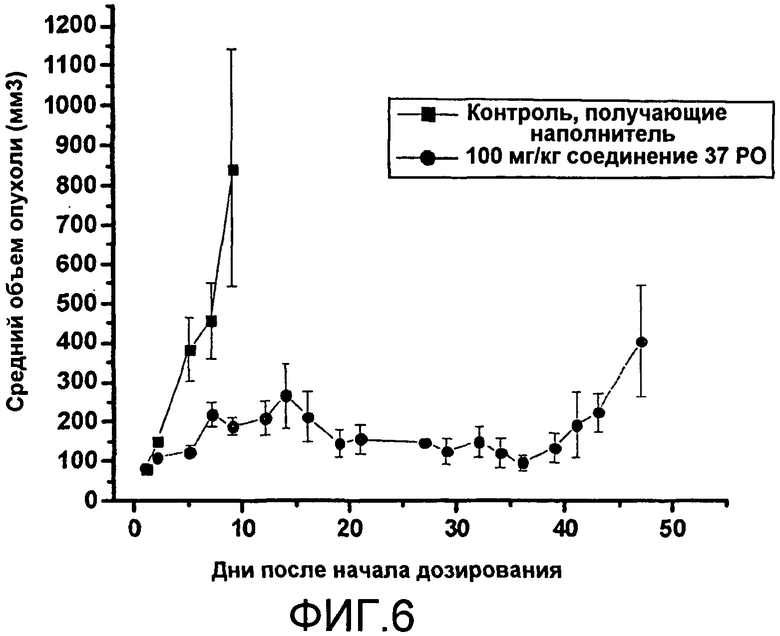
Изобретение относится к тетраазафенален-3-оновым соединениям, в частности к соединению формулы 37
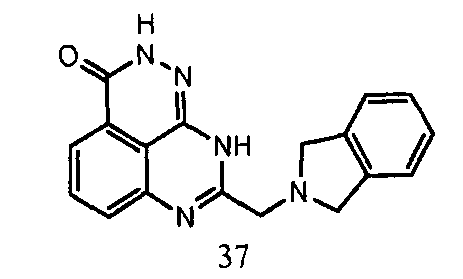
которые ингибируют поли(АДФ-рибоза)полимеразу (PARP) и могут использоваться для химиосенсибилизации в отношении противораковых химиотерапевтических средств, а также для повышения эффективности химиотерапевтических средств и при лечении раковых заболеваний, характеризующихся дефектами репарации ДНК. 2 н. и 11 з.п. ф-лы, 2 табл., 6 ил., 3 пр.
1. Соединение 37, имеющее структуру
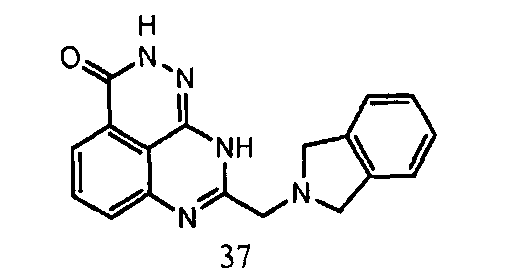
или его фармацевтически приемлемая соль, эфир, сольват или гидрат.
2. Соединение по п.1, где соединение представляет собой фармацевтически приемлемую соль соединения 37.
3. Соединение по п.2, где фармацевтически приемлемая соль содержит органическую кислоту.
4. Соединение по п.1, где соединение представляет собой соединение, имеющее структуру:
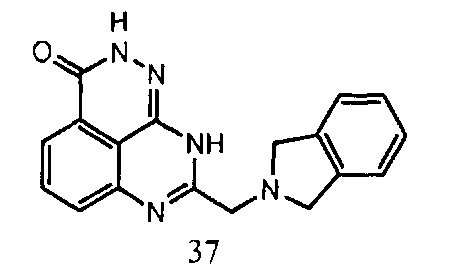
5. Фармацевтическая композиция для лечения рака, содержащая соединение по любому одному из пп.1-4 и фармацевтически приемлемый носитель.
6. Фармацевтическая композиция по п.5, содержащая, кроме того, химиотерапевтический агент, выбранный из группы, включающей темозоломид, адриамицин, камптотецин, карбоплатин, цисплатин, даунорубицин, доцетаксел, доксорубицин, интерферон-альфа, интерферон-бета, интерферон-гамма, интерлейкин 2, иринотекан, паклитаксел, топотекан, таксоид, дактиномицин, даунорубицин, 4'-дезоксидоксорубицин, блеомицин, пилкамицин, митомицин, неомицин, гентамицин, этопозид, 4-ОН- циклофосфамид, координационный комплекс платины и их смеси.
7. Фармацевтическая композиция по п.6, где указанное химиотерапевтическое средство представляет собой темозоломид или его соль.
8. Соединение по любому одному из пп.1-4, или фармацевтическая композиция по любому одному из пп.5-7 для применения в химиосенсибилизации или радиосенсибилизации раковых клеток у человека, нуждающегося в химиотерапии или радиотерапии соответственно.
9. Соединение или фармацевтическая композиция по п.8, где раковые клетки выбраны из группы, включающей клетки таких опухолей, как АКТГ-продуцирующие опухоли, острый лимфоцитарный лейкоз, острый нелимфоцитарный лейкоз, рак коры надпочечника, рак мочевого пузыря, рак мозга, рак молочной железы, рак шейки матки, хронический лимфоцитарный лейкоз, хронический миелоцитарный лейкоз, рак ободочной и прямой кишки, Т-клеточная лимфома кожи, рак эндометрия, рак пищевода, саркома Юинга, рак желчного пузыря, лейкоз ворсистых клеток, рак головы и шеи, лимфома Ходжкина, саркома Калоши, рак почки, рак печени, рак легкого (мелкоклеточный и немелкоклеточный), злокачественный перитонеальный выпот, злокачественный плевральный выпот, меланома, мезотелиома, множественная миелома, нейробластома, неходжкинская лимфома, остеосаркома, рак яичника, рак яичников (герминогенный рак), рак простаты, рак поджелудочной железы, рак полового члена, ретинобластома, рак кожи, саркома мягкой ткани, плоскоклеточная карцинома, рак желудка, рак яичка, рак щитовидной железы, трофобластические опухоли, рак матки, рак влагалища, рак наружных половых органов и опухоль Вильмса.
10. Соединение по любому одному из пп.1-4, или фармацевтическая композиция по любому одному из пп.5-7 для применения в лечении человека, страдающего от ракового заболевания, характеризующегося дефектом в пути репарации двухцепочечной ДНК посредством гомологичной рекомбинации (HR).
11. Соединение или фармацевтическая композиция по п.10, где раковые клетки имеют фенотип, выбранный из группы, включающей i) дефект BRCA-1, ii) дефект BRCA-2, iii) дефект BRCA-1 и BRC А-2 и iv) анемию Фанкони.
12. Соединение или фармацевтическая композиция по любому одному из пп.9-11, где рак выбирают из группы, включающей рак молочной железы, рак яичника или меланому.
13. Соединение по любому одному из пп.1-4 или фармацевтическая композиция по любому одному из пп.5-7 для применения при лечении рака.
| US 7268138 В2, 11.09.2007 | |||
| RU 2006100034 А, 27.08.2006 | |||
| Takayoshi Kinoshita et al | |||
| "Inhibitor-indaced structural change of the active site of human poly(ADP-ribose)polymerase", FEBS Letter, vol.556, 2004, p.p.43-46. |

