ОБЛАСТЬ ТЕХНИЧЕСКОГО ПРИМЕНЕНИЯ
Настоящее изобретение относится к новому моногидрату производного нафтиридина и способу его получения.
УРОВЕНЬ ТЕХНИКИ
В медицинской практике используют большое разнообразие антибиотиков и синтетических антибактериальных средств для лечения инфекционных заболеваний. Однако недавно были обнаружены резистентные бактерии, такие как метициллин-резистентные Staphylococcus aureus (MRSA), ванкомицин-резистентные Enterococcus (VRE) и пенициллин-резистентные Streptococcus pneumoniae (PRSP). Лечение пациентов, инфицированных такими резистентными микроорганизмами, представляло серьезную проблему. В дополнение, появились полирезистентные микроорганизмы, которые приобрели резистентность к многочисленным лекарственным препаратам. Инфекционные заболевания, вызываемые полирезистентными микроорганизмами, как заболевания, трудно поддающиеся лечению, представляли собой крупные общемировые проблемы.
Появление противомикробных средств, которые эффективны против этих резистентных микроорганизмов, было крайне необходимо, и, например, соединение хинолона, считающееся эффективным против MRSA, раскрыто в WO 99/07682 (ПАТЕНТНЫЙ ДОКУМЕНТ 1). Более того, соединения, раскрытые в WO 2004/002490 (ПАТЕНТНЫЙ ДОКУМЕНТ 2) и в WO 2004/002992 (ПАТЕНТНЫЙ ДОКУМЕНТ 3), известны как соединения, имеющие механизмы действия, отличные от механизмов действия существующих лекарственных средств.
ПАТЕНТНЫЙ ДОКУМЕНТ 1: Публикация международного патента, брошюра № WO 99/07682
ПАТЕНТНЫЙ ДОКУМЕНТ 2: Публикация международного патента, брошюра № WO 2004/002490
ПАТЕНТНЫЙ ДОКУМЕНТ 3: Публикация международного патента, брошюра № WO 2004/002992
РАСКРЫТИЕ ИЗОБРЕТЕНИЯ
ПРОБЛЕМЫ, КОТОРЫЕ ДОЛЖНО РЕШАТЬ ИЗОБРЕТЕНИЕ
Существует потребность в разработке лекарственного средства, обладающего высокой степенью безопасности и сильной антибактериальной активностью, направленной против грамположительных бактерий, грамотрицательных бактерий и резистентных бактерий. Кроме того, большие ожидания связывали с применимым способом получения этого лекарственного средства и генерируемыми в производстве применимыми промежуточными продуктами.
СПОСОБЫ РЕШЕНИЯ ПРОБЛЕМ
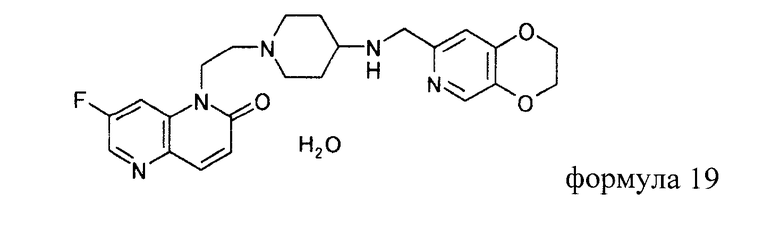
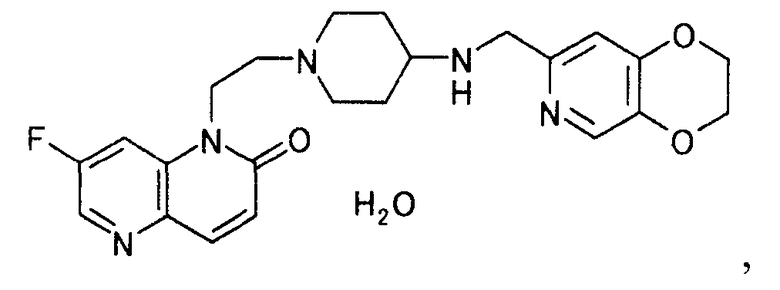
В таких условиях авторы настоящего изобретения проводили обширные исследования и обнаружили, что моногидрат 1-(2-(4-((2,3-дигидро(1,4)диоксино(2,3-c)пиридин-7-илметил)амино)пиперидин-1-ил)этил)-7-фтор-1,5-нафтиридин-2(1H)-она (1) обладает сильной антибактериальной активностью и высокой степенью безопасности; (2) не обнаруживает способности расплываться, поглощая влагу из атмосферы, или гигроскопичности; (3) легкий в обращении; (4) получается при использовании растворителя, безопасного для человеческого организма; (5) получается в условиях, которые не приносят существенного вреда окружающей среде; и (6) пригоден для массового производства.
Кроме того, авторы настоящего изобретения обнаружили, что моногидрат 1-(2-(4-((2,3-дигидро(1,4)диоксино(2,3-c)пиридин-7-илметил)амино)пиперидин-1-ил)этил)-7-фтор-1,5-нафтиридин-(1H)-она легко может быть получен посредством взаимодействия
(1) производного нафтиридина, представляемого формулой [7]
[Формула 7]
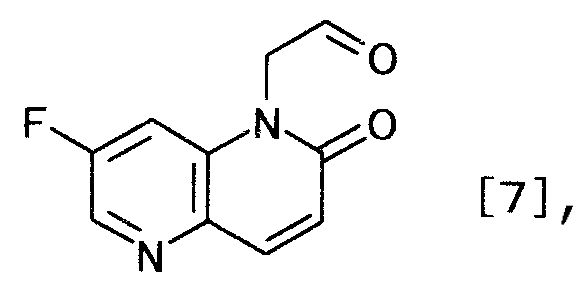
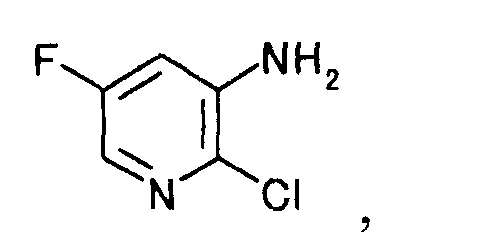
и получаемого взаимодействием производного пиридина, представляемого формулой [1]
[Формула 1]
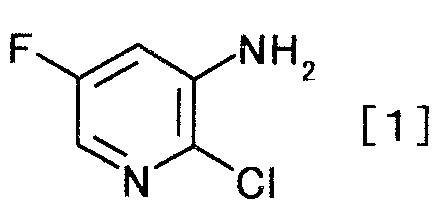 ,
,
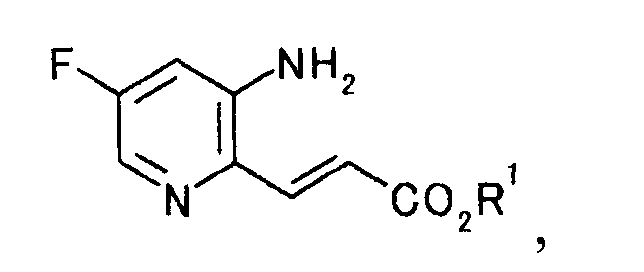
с эфиром акриловой кислоты с образованием производного акриловой кислоты, представляемого общей формулой [2]
[Формула 2]
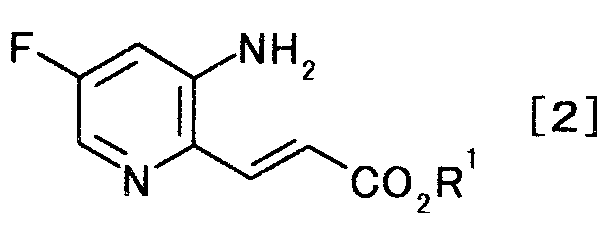 ,
,
где R1 представляет собой алкильную группу,
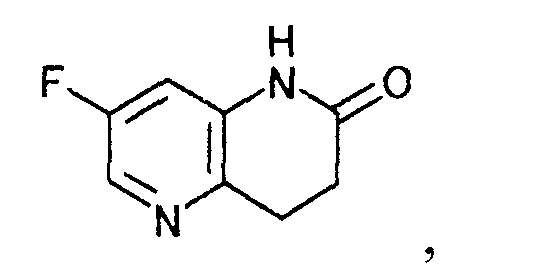
затем, восстанавливая/циклизируя полученное производное акриловой кислоты с образованием производного дигидронафтиридина, представляемого формулой [3]
[Формула 3]
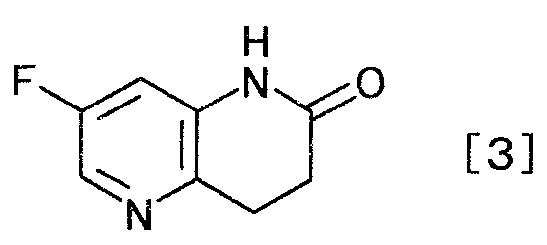 ,
,
затем взаимодействием полученного производного дигидронафтиридина с соединением, представляемым общей формулой [4]
[Формула 4]
 ,
,
где L1 представляет собой уходящую группу; Y представляет собой защищаемую карбонильную группу,
получают производное дигидронафтиридина, представляемое общей формулой [5]
[Формула 5]
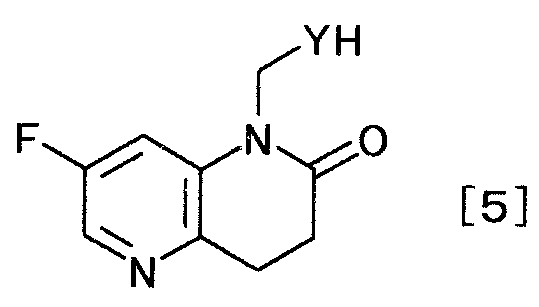 ,
,
где заместитель Y определен выше,
затем, окисляя полученное производное дигидронафтиридина с образованием производного нафтиридина, представляемого общей формулой [6]
[Формула 6]
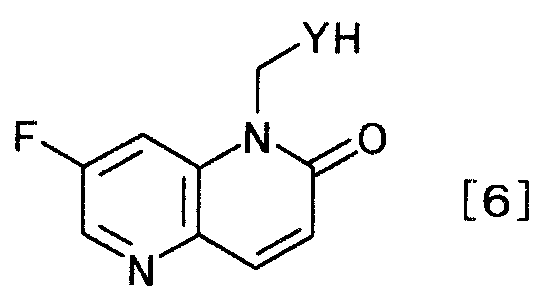 ,
,
где заместитель Y определен выше,
и затем удаляя защиту полученного производного нафтиридина с помощью
(2) производного пиперидина, представляемого общей формулой [17]
[Формула 17]
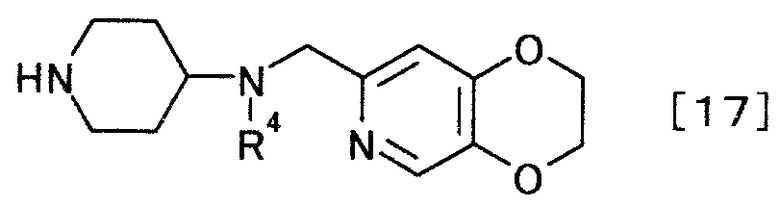 ,
,
где R4 представляет собой иминозащитную группу,
и получаемого взаимодействием производного койевой кислоты, представляемого общей формулой [8]
[Формула 8]
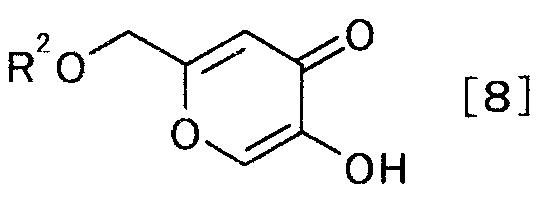 ,
,
где R2 является защитной группой гидроксила,
с соединением, представляемым общей формулой [9]
[Формула 9]
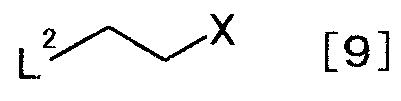 ,
,
где L2 представляет собой уходящую группу; X представляет собой уходящую группу,
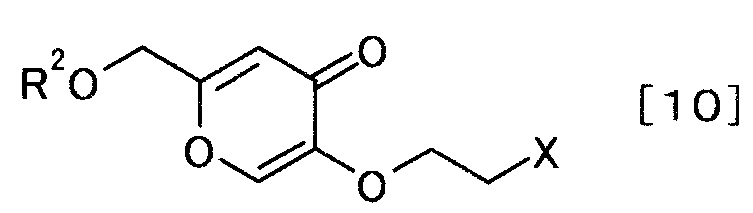
чтобы получить производное койевой кислоты, представляемое общей формулой [10]
[Формула 10]
 ,
,
где R2 и X определены выше,
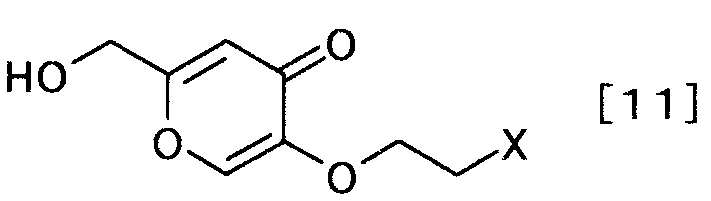
затем удаляя защиту полученного производного койевой кислоты, с образованием производного койевой кислоты, представляемого общей формулой [11]
[Формула 11]
 ,
,
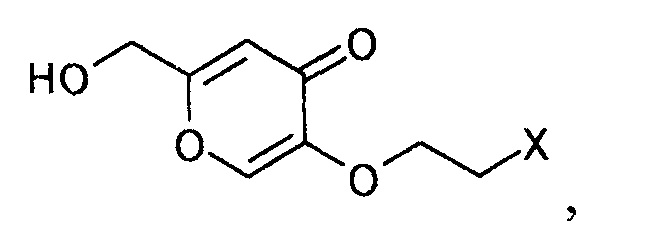
где X определен выше,
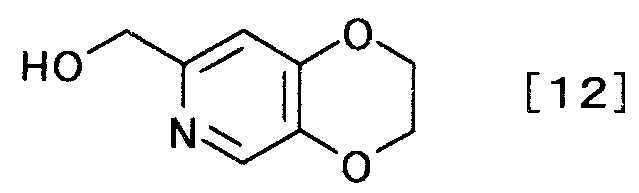
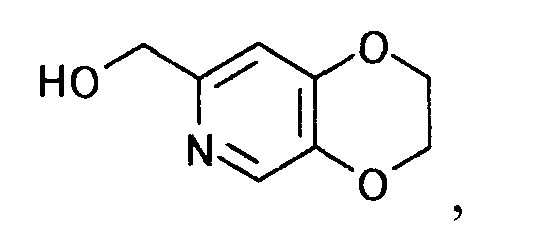
затем взаимодействием полученного производного койевой кислоты с аммиаком получают производное пиридина, представляемое формулой [12]
[Формула 12]
 ,
,
затем, окисляя полученное производное пиридина, чтобы получить производное пиридина, представляемое формулой [13] [Формула 13]
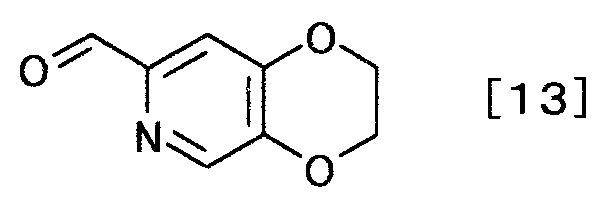 ,
,
затем взаимодействием полученного производного пиридина с производным пиперидина, представляемым общей формулой [14]
[Формула 14]
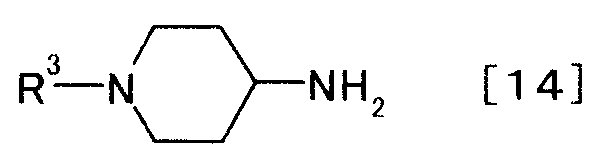 ,
,
где R3 представляет собой иминозащитную группу,
получают производное пиперидина, представляемого общей формулой [15]
[Формула 15]
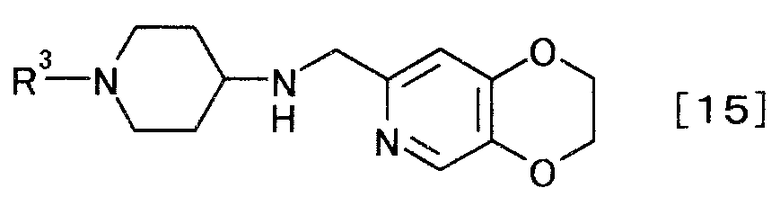 ,
,
где R3 определен выше,
затем защищают иминогруппу, чтобы получить производное пиперидина, представляемого общей формулой [16]
[Формула 16]
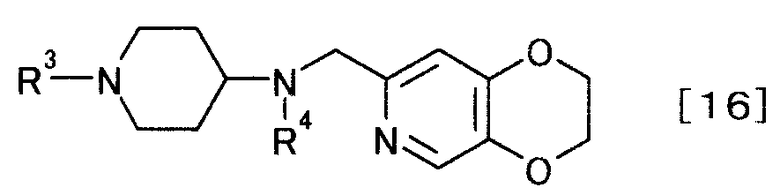 ,
,
где R4 определен выше; R3 определен выше,
и затем удаляют защиту у полученного производного пиперидина, чтобы получить (3) производное нафтиридина, представляемое общей формулой [18]
[Формула 18]
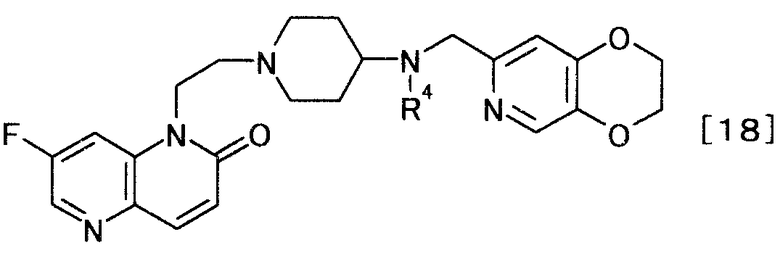 ,
,
где R4 определен выше,
и затем удаляют защиту полученного производного нафтиридина.
Авторы изобретения далее обнаружили, что производное койевой кислоты, представляемое общей формулой [19]
[Формула 19]
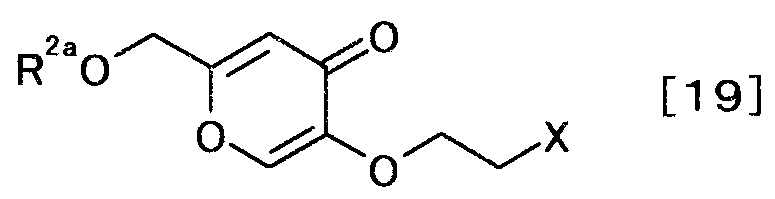 ,
,
где R2a представляет собой атом водорода или защитную группу гидроксила, X представляет собой уходящую группу, является важным промежуточным продуктом производства.
ПРЕИМУЩЕСТВА ИЗОБРЕТЕНИЯ
Моногидрат 1-(2-(4-((2,3-дигидро(1,4)диоксино(2,3-c)пиридин-7-илметил)амино)пиперидин-1-ил)этил)-7-фтор-1,5-нафтиридин-2(1H)-она по настоящему изобретению (1) обладает сильной антибактериальной активностью и высокой степенью безопасности, (2) не обнаруживает способности расплываться, поглощая влагу из атмосферы, или гигроскопичности, (3) является легким в обращении, (4) получается при использовании безопасного для человеческого организма растворителя, (5) получается в условиях, которые не приносят большого вреда окружающей среде, и (6) является пригодным для массового производства и, следовательно, применим в качестве массового фармацевтического препарата.
Способ получения по настоящему изобретению имеет такие особенности как (1) высокий выход, (2) отсутствие необходимости использования колоночной хроматографии на силикагеле, (3) следовательно, незначительное количество отходов и (4) неиспользование токсичных или неустойчивых реагентов, и, следовательно, применим для получения моногидрата 1-(2-(4-((2,3-дигидро(1,4)диоксино(2,3-c)пиридин-7-илметил)амино)пиперидин-1-ил)этил)-7-фтор-1,5-нафтиридин-2(1H)-она.
Кроме того, производное койевой кислоты, представляемое общей формулой [19]
[Формула 19]
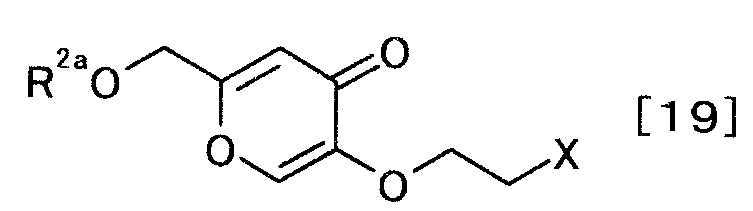 ,
,
где R2a и X определены выше, является полезным промежуточным продуктом производства.
НАИЛУЧШИЙ ПУТЬ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
В дальнейшем настоящее изобретение будет описано подробно. В настоящем описании изобретения, если не будет оговорено иначе, термин «атом галогена» обозначает, например, атом фтора, атом хлора, атом брома и атом йода. Термин «алкильная группа» обозначает, например, C1-6 алкильную группу с разветвленной или неразветвленной цепью, такую как метильная, этильная, пропильная, изопропильная, бутильная, втор-бутильная, изобутильная, трет-бутильная и пентильная группа. Термин «аралкильная группа» обозначает, например, ар-C1-6 алкильную группу, такую как бензил, дифенилметил, тритил, фенетил и нафтилметил. Термин «алкоксиалкильная группа» обозначает, например, C1-6 алкилокси C1-6 алкильную группу, такую как метоксиметил и 1-этоксиэтил. Термин «аралкилоксиалкильная группа» обозначает, например, ар-C1-6 алкилокси C1-6 алкильную группу, такую как бензилоксиметил и фенетилоксиметил. Термин «алкилсульфонильная группа» обозначает, например, C1-6 алкилсульфонильную группу, такую как метилсульфонил, трифторметилсульфонил и этилсульфонил. Термин «арилсульфонильная группа» обозначает, например, группу, такую как бензолсульфонил и толуолсульфонил. Термин «алкилсульфонилоксигруппа» обозначает, например, C1-6 алкилсульфонилоксигруппу, такую как метилсульфонилокси, трифторметилсульфонилокси и этилсульфонилокси. Термин «арилсульфонилоксигруппа» обозначает, например, такую группу, как бензолсульфонилокси и толуолсульфонилокси.
Термин «ацильная группа» обозначает, например, формильную группу, C2-6 алканоильную группу c прямой или разветвленной цепью, такую как ацетил, пропионил, бутирил, изовалерил и пивалоил, и ар-C1-6 алкилкарбонильную группу, такую как бензилкарбонил; циклическую карбонильную группу, такую как бензоил и нафтоил, и гетероциклическую карбонильную группу, такую как никотионил, теноил, пирролизинокарбонил и фуроил. Термин «алкоксикарбонильная группа» обозначает, например, C1-6 алкилоксикарбонильную группу c прямой или разветвленной цепью, такую как метоксикарбонил, этоксикарбонил, 1,1-диметилпропоксикарбонил, изопропоксикарбонил, 2-этилгексилоксикарбонил, трет-бутоксикарбонил и трет-пентилоксикарбонил. Термин «аралкилоксикарбонильная группа» обозначает, например, ар-C1-6 алкилоксикарбонильную группу, такую как бензилоксикарбонил и фенетилоксикарбонил.
Термин «кислородсодержащая гетероциклическая группа» обозначает, например, группу, такую как тетрагидропиранил и тетрагидрофуранил. Термин «серосодержащая гетероциклическая группа» обозначает, например, группу, такую как тетрагидротиопиранил. Термин «защищенная карбонильная группа» обозначает, например, группу, образованную из карбонильной группы и спирта, такую как (гидрокси)(метокси)метилен, (гидрокси)(этокси)метилен, (гидрокси)(пропокси)метилен, (гидрокси)(изопропокси)метилен, (гидрокси)(бутокси)метилен, (гидрокси)(пентилокси)метилен, (гидрокси)(гексилокси)метилен, (гидрокси)(гептилокси)метилен, (гидрокси)(октилокси)метилен, (гидрокси)(1,1-диметилпропокси)метилен, диметоксиметилен, диэтоксиметилен, дипропоксиметилен, диизопропоксиметилен, дибутоксиметилен, бис(бензилокси)метилен, 1,3-диоксолан-2-илиден и 1,3-диоксан-2-илиден, группу, образованную из карбонильной группы и тиола, такую как бис(метилтио)метилен, бис(этилтио)метилен, бис(бензилтио)метилен, 1,3-дитиолан-2-илиден и 1,3-дитиан-2-илиден, и группу, такую как оксазолин-2-илиден, имидазолидин-2-илиден и тиазолидин-2-илиден. Термин «уходящая группа» обозначает, например, атом галогена, алкилсульфонилоксигруппу и арилсульфонилоксигруппу.
Термин «гидроксилзащитная группа» охватывает все группы, которые представляют собой обычные применимые защитные группы гидроксила. Примеры включают группы, описанные в «Greene's Protective Groups in Organic Synthesis» by M. Wuts и W. Greene, 4th edition, John Wiley & Sons, INC., 2006, p.16-366. Конкретные примеры включают ацильную группу, алкоксикарбонильную группу, аралкилоксикарбонильную группу, алкильную группу, аралкильную группу, кислородсодержащую гетероциклическую группу, серосодержащую гетероциклическую группу, алкоксиалкильную группу, аралкилоксиалкильную группу, алкилсульфонильную группу и арилсульфонильную группу.
Термин «иминозащитная группа» охватывает все группы, которые используют в качестве обычной иминозащитной группы. Примеры включают группы, описанные в «Greene's Protective Groups in Organic Synthesis» by M. Wuts и W. Greene, 4th edition, John Wiley & Sons, INC., 2006, p.696-926. Конкретные примеры включают ацильную группу, алкоксикарбонильную группу, аралкилоксикарбонильную группу, аралкильную группу, алкилсульфонильную группу и арилсульфонильную группу.
Предпочтительные моногидраты 1-(2-(4-((2,3-дигидро(1,4)диоксино(2,3-c)пиридин-7-илметил)амино)пиперидин-1-ил)этил)-7-фтор-1,5-нафтиридин-2(1H)-она, используемые в настоящем изобретении, представляют собой нижеследующие соединения.
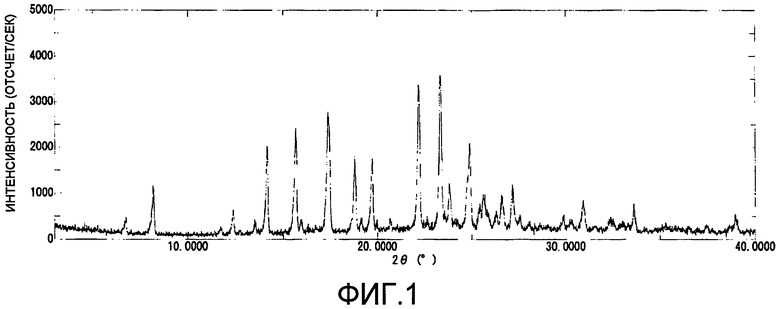
Кристаллы моногидрата 1-(2-(4-((2,3-дигидро(1,4)диоксино(2,3-c)пиридин-7-илметил)амино)пиперидин-1-ил)этил)-7-фтор-1,5-нафтиридин-2(1H)-она, имеющие углы дифракции 2θ, составляющие 6,8°; 8,2°; 14,2° и 15,7°, на порошковой рентгеновской дифрактограмме, являются предпочтительными.
В дополнение, характерные пики порошковой дифракции рентгеновских лучей могут меняться в зависимости от условий измерений. По этой причине пик порошковой дифракции рентгеновских лучей соединения по настоящему изобретению не следует строго интерпретировать.
В настоящем изобретении предпочтительные способы получения включают нижеследующие способы.
Способ получения, в котором R1 представляет собой этильную группу, пропильную группу или бутильную группу, является предпочтительным; при этом способ получения, в котором R1 является бутильной группой, является более предпочтительным.
Способ получения, в котором R2 представляет собой ацильную группу, аралкильную группу или кислородсодержащую гетероциклическую группу, является предпочтительным. При этом способ получения, в котором R2 представляет собой кислородсодержащую гетероциклическую группу, является более предпочтительным, и способ получения, в котором R2 представляет собой тетрагидропиранильную группу, является еще более предпочтительным.
Способ получения, в котором R3 является ацильной группой, алкоксикарбонильной группой или аралкильной группой, является предпочтительным. При этом способ получения, в котором R3 представляет собой аралкильную группу, является более предпочтительным, а способ получения, в котором R3 представляет собой бензильную группу, является еще более предпочтительным.
Способ получения, в котором R4 представляет собой ацильную группу, алкоксикарбонильную группу или аралкильную группу, является предпочтительным. При этом способ получения, в котором R4 представляет собой ацильную группу или алкоксикарбонильную группу, является более предпочтительным, а способ получения, в котором R4 представляет собой алкоксикарбонильную группу, является еще более предпочтительным.
Способ получения, в котором X представляет собой атом хлора, является предпочтительным.
Способ получения, в котором Y представляет собой диметоксиметиленовую группу, диэтоксиметиленовую группу, дипропоксиметиленовую группу, 1,3-диоксолан-2-илиденовую группу или 1,3-диоксан-2-илиденовую группу, является предпочтительным. При этом способ получения, в котором Y представляет собой диметоксиметиленовую группу, диэтоксиметиленовую группу или 1,3-диоксолан-2-илиденовую группу, является более предпочтительным, а способ получения, в котором Y представляет собой диметоксиметиленовую группу, является еще более предпочтительным.
Для соединения, представляемого общей формулой [19], предпочтительные соединения включают нижеследующие соединения.
Соединение, в котором R2a представляет собой атом водорода, ацильную группу, аралкильную группу или кислородсодержащую гетероциклическую группу, является предпочтительным. При этом соединение, в котором R2a представляет собой атом водорода или кислородсодержащую гетероциклическую группу, является более предпочтительным, при том, что соединение, в котором R2a представляет собой атом водорода или тетрагидропиранильную группу, является еще более предпочтительным; и соединение, в котором R2a представляет собой атом водорода, является наиболее предпочтительным.
Способ получения по настоящему изобретению описан далее.
[Способ получения 1]
 ,
,
где R1 определен выше.
(1-1)
Соединение общей формулы [2] может быть получено взаимодействием соединения формулы [1] с эфиром акриловой кислоты в присутствии катализатора, в присутствии или отсутствие основания, и в присутствии или отсутствие лиганда. Реакцию можно осуществлять, например, способом, описанным в «Chem. Rev.» by I.P.Beletskaya и A.V.Cheprakov, 2000, Vol.100, p.3009-3066; или любым способом, ему соответствующим.
(1-2)
Соединение формулы [3] может быть получено посредством восстановления/циклизации соединения общей формулы [2] в присутствии катализатора.
Реакцию восстановления можно осуществлять, например, способом, описанным в «Comprehensive Organic Transformations» by Richard C. Larock, VCH Publishers, INC., 1989, p.6-17; или любым способом, ему соответствующим.
Растворитель, используемый в реакции восстановления, может быть любым растворителем постольку, поскольку он не оказывает влияния на течение реакции. Примеры включают в себя спирты, такие как метанол, этанол, 2-пропанол и 2-метил-2-пропанол; ароматические углеводороды, такие как бензол, толуол и ксилол; простые эфиры, такие как диоксан, тетрагидрофуран, анизол, диметиловый эфир этиленгликоля, диметиловый эфир диэтиленгликоля, диэтиловый эфир диэтиленгликоля, дибутиловый эфир и монометиловый эфир этиленгликоля; сульфоксиды, такие как диметилсульфоксид; сложные эфиры, такие как этилацетат и бутилацетат; амиды, такие как N,N-диметилформамид, N,N-диметилацетамид и 1-метил-2-пирролидон; кетоны, такие как ацетон и 2-бутанон, а также воду. Эти растворители могут быть использованы в смеси. Предпочтительными примерами растворителей являются метанол и этанол.
Катализатор, используемый в реакции восстановления, включает в себя, например, палладий на угольном носителе, ацетат палладия, оксид платины, родий на угольном носителе и хлорид рутения. Предпочтительным катализатором является палладий на угольном носителе.
Восстановитель, используемый в реакции восстановления, включает в себя, например, водород; муравьиную кислоту; формиаты, такие как формиат натрия, формиат аммония и формиат триэтиламмония; и циклогексан. Предпочтительные восстановители включают водород и муравьиную кислоту.
Количество используемого катализатора может составлять массу соединения общей формулы [2], умноженную на величину, принимающую значения от 0,001 до 5, предпочтительно, массу, умноженную на величину, принимающую значения от 0,01 до 0,5.
Количество восстановителя может представлять число молей соединения общей формулы [2], умноженное на величину, принимающую значения от 1 до 100, предпочтительно, от 1 до 5.
Реакцию восстановления можно осуществлять при температуре от -30 до 150°C, предпочтительно от 0 до 100°C, в течение времени от 30 минут до 120 часов.
Растворитель, используемый в реакции циклизации, может быть любым растворителем постольку, поскольку он не оказывает влияние на течение реакции. Примеры включают в себя спирты, такие как метанол, этанол, 2-пропанол и 2-метил-2-пропанол; ароматические углеводороды, такие как бензол, толуол и ксилол; простые эфиры, такие как диоксан, тетрагидрофуран, анизол, диметиловый эфир этиленгликоля, диметиловый эфир диэтиленгликоля, диэтиловый эфир диэтиленгликоля, дибутиловый эфир и монометиловый эфир этиленгликоля; сульфоксиды, такие как диметилсульфоксид; сложные эфиры, такие как этилацетат и бутилацетат; амиды, такие как N,N-диметилформамид, N,N-диметилацетамид и 1-метил-2-пирролидон; кетоны, такие как ацетон и 2-бутанон, а также воду. Эти растворители могут быть использованы в смеси. Предпочтительные растворители включают в себя толуол и ксилол.
Реакцию циклизации можно осуществлять при температуре от 0 до 200°C, предпочтительно от 50 до 130°C, в течение времени от 30 минут до 120 часов.
(1-3)
Соединение формулы [3] может быть получено взаимодействием соединения формулы [1] с эфиром акриловой кислоты, в присутствии катализатора, в присутствии или отсутствие основания, в присутствии или отсутствие лиганда и в присутствии восстановителя. Реакция представляет собой реакцию, протекающую в одном сосуде, с получением соединения формулы [3]. Реакцию можно осуществлять, согласно способу получения (1-1) и способу получения (1-2).
[Способ получения 2]
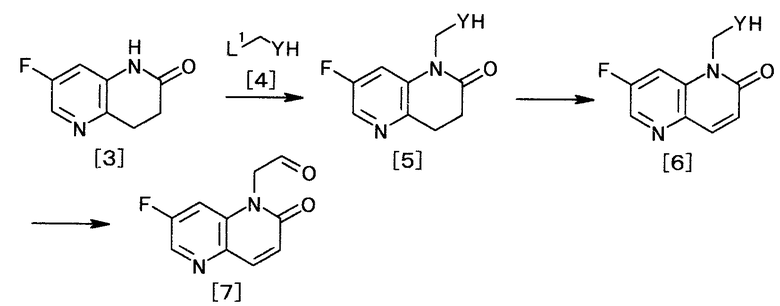 ,
,
где L1 и Y определены выше.
(2-1)
Известные соединения общей формулы [4] представляют собой, например, 2-(2-бромметил)-1,3-диоксолан, 2-бром-1,1-диэтоксиэтан и 2-бром-1,1-диметоксиэтан.
Соединение общей формулы [5] можно получить взаимодействием соединения общей формулы [4] с соединением формулы [3] в присутствии основания.
Растворитель, используемый в этой реакции, может быть любым растворителем постольку, поскольку он не оказывает влияния на течение реакции. Примеры включают в себя амиды, такие как N,N-диметилформамид, N,N-диметилацетамид и 1-метил-2-пирролидон; галогенированные углеводороды, такие как метиленхлорид, хлороформ и дихлорэтан; ароматические углеводороды, такие как бензол, толуол и ксилол; простые эфиры, такие как диоксан, тетрагидрофуран, анизол, диметиловый эфир этиленгликоля, диметиловый эфир диэтиленгликоля, диэтиловый эфир диэтиленгликоля и монометиловый эфир этиленгликоля; сульфоксиды, такие как диметилсульфоксид; сложные эфиры, такие как этилацетат, а также воду. Эти растворители могут быть использованы в смеси. Предпочтительными растворителями являются N,N-диметилацетамид и диметисульфоксид.
Основание, используемое в этой реакции, включает в себя, например, органические основания, такие как пиридин, диметиламинопиридин, триэтиламин, трет-бутилат натрия и трет-бутилат калия, а также неорганические основания, такие как гидрид натрия, гидроксид натрия, гидроксид калия, гидрокарбонат натрия, карбонат натрия, карбонат калия, фосфат калия и карбонат цезия. Предпочтительными основаниями являются карбонат калия и фосфат калия.
Количества основания и используемого соединения общей формулы [4] могут представлять число молей соединения общей формулы [3], умноженное на величину, составляющую от 1 до 50, предпочтительно от 1 до 5.
Реакцию можно осуществлять при температуре от -30 до 150°C, предпочтительно от 0 до 100°C, в течение времени от 30 минут до 48 часов.
(2-2)
Соединение общей формулы [6] можно получить окислением соединения общей формулы [5] в присутствии или отсутствие радикальных инициаторов, в присутствии или отсутствие основания. Реакцию можно осуществлять, например, способами, описанными в Chem. Rev., by Djerassi C., p.271-317, Vol.43, 1948 и «Bioorg. Med. Chem. Lett.», by Julianne A. Hunt, 2003, Vol.13, p.467-470, или любыми способами, им соответствующими.
Растворитель, используемый в этой реакции, может представлять собой любой растворитель постольку, поскольку он не оказывает влияния на течение реакции. Примеры включают амиды, такие как N,N-диметилформамид, N,N-диметилацетамид и 1-метил-2-пирролидон; галогенированные углеводороды, такие как метиленхлорид, хлороформ и дихлорэтан; ароматические углеводороды, такие как бензол и хлорбензол; простые эфиры, такие как диоксан, тетрагидрофуран, анизол, диметиловый эфир этиленгликоля, диметиловый эфир диэтиленгликоля, диэтиловый эфир диэтиленгликоля и монометиловый эфир этиленгликоля; сульфоксиды, такие как диметилсульфоксид; а также сложные эфиры, такие как этилацетат. Эти растворители можно использовать в смеси. Предпочтительным растворителем является хлорбензол.
Окислитель, используемый в этой реакции, включает в себя, например, бромид, хлор, йод, N-бромсукцинимид, N-хлорсукцинимид, N-йодсукцинимид и диоксид марганца. Предпочтительным окислителем является N-бромсукцинимид.
Радикальный инициатор, используемый в этой реакции по мере необходимости, включает в себя, например, азобисизобутиронитрил, бензоилпероксид и 2,2'-азобис(4-метокси-2,4-диметилвалеронитрил). Предпочтительным радикальным инициатором является 2,2'-азобис(4-метокси-2,4-диметилвалеронитрил).
Основание, используемое в этой реакции по мере необходимости, включает в себя, например, органические основания, такие как пиридин, диметиламинопиридин, триэтиламин, трет-бутилат натрия и трет-бутилат калия, а также неорганические основания, такие как гидрид натрия, гидроксид натрия, гидроксид калия, бикарбонат натрия, карбонат натрия, карбонат калия, карбонат бария и карбонат цезия. Предпочтительным основанием является карбонат калия.
Количество используемого окислителя представляет число молей соединения общей формулы [5], умноженное на величину, составляющую от 1 до 30, предпочтительно от 1 до 5.
Количество радикального инициатора, используемого по мере необходимости, представляет число молей соединения общей формулы [5], умноженное на величину, составляющую от 0,0001 до 0,5, предпочтительно от 0,001 до 0,1.
Реакцию можно осуществлять при температуре от -30 до 150°C, предпочтительно от 0 до 100°C, в течение времени, составляющего от 30 минут до 48 часов.
Соединение общей формулы [6] представляет собой, например, 1-(2,2-диэтоксиэтил)-7-фтор-1,5-нафтиридин-2(1H)-он. 1-(2,2-диэтоксиэтил)-7-фтор-1,5-нафтиридин-2(1H)-он можно получить взаимодействием 7-фтор-1,5-нафтиридин-2(1H)-она с 2-бром-1,1-диэтоксиэтаном.
Реакцию можно осуществлять способом, описанным в WO 2007/138974, или любым способом, ему соответствующим.
(2-3)
Соединение формулы [7] можно получить, удаляя защиту соединения общей формулы [6]. Реакцию можно осуществлять, например, способом, описанным в «Greene's Protective Groups in Organic Synthesis» by M. Wuts и W. Greene, 4th edition, John Wiley & Sons, INC., 2006, p.435-505, или любым способом, с ним согласующимся.
[Способ получения 3]
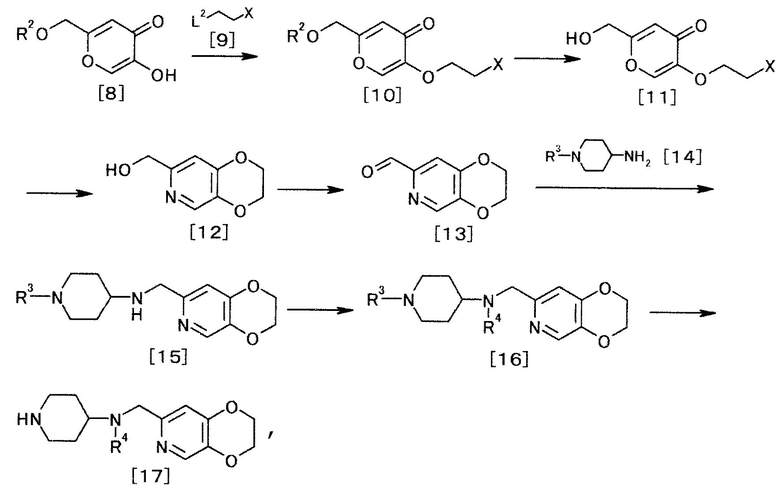
где R2, R3, R4, L2 и X определены выше.
Соединение общей формулы [8] может быть получено из койевой кислоты.
(3-1)
Известные соединения общей формулы [9] включают в себя, например, 1-бром-2-хлорэтан и 1,2-дибромэтан.
Соединение общей формулы [10] можно получать взаимодействием соединения общей формулы [9] с соединением общей формулы [8] в присутствии основания.
Реакцию можно осуществить согласно способу получения 2-1.
(3-2)
Соединение общей формулы [11] можно получать, удаляя защиту соединения общей формулы [10]. Реакцию можно осуществлять, например, способом, описанным в «Greene's Protective Groups in Organic Synthesis» by M. Wuts и W. Greene, 4th edition, John Wiley & Sons, INC., 2006, p.16-366, или любым способом, с ним согласующимся.
Кроме того, соединение общей формулы [11] может быть получено взаимодействием соединения общей формулы [9] с койевой кислотой. Реакцию можно осуществлять согласно способу получения 3-1.
(3-3)
Соединение формулы [12] может быть получено взаимодействием соединения общей формулы [11] с аммиаком.
Растворитель, используемый в этой реакции, может быть любым растворителем постольку, поскольку он не оказывает влияния на течение реакции. Примеры включают в себя спирты, такие как метанол, этанол, 2-пропанол и 2-метил-2-пропанол; ароматические углеводороды, такие как бензол, толуол и ксилол; простые эфиры, такие как диоксан, тетрагидрофуран, анизол, диметиловый эфир этиленгликоля, диметиловый эфир диэтиленгликоля, диэтиловый эфир диэтиленгликоля и монометиловый эфир этиленгликоля; сульфоксиды, такие как диметилсульфоксид; сложные эфиры, такие как этилацетат; амиды, такие как N,N-диметилформамид, N,N-диметилацетамид и 1-метил-2-пирролидон, а также воду. Эти растворители могут быть использованы в смеси. Предпочтительный растворитель включает воду.
Аммиак, используемый в этой реакции, включает в себя аммиачную воду и так далее. Количество используемого аммиака представляет число молей соединения общей формулы [11], умноженное на величину, составляющую от 1 до 100, предпочтительно от 1 до 30.
Реакцию можно осуществлять при температуре в интервале от комнатной температуры до 150°C, предпочтительно от 50 до 100°C, в течение периода времени от 30 минут до 120 часов.
(3-4)
Соединение формулы [13] можно получать окислением соединения формулы [12]. Реакцию можно осуществлять способами, описанными в «Advanced Organic Chemistry», by Jerry March, the 4th edition, John Wiley & Sons, INC., 1992, p.1167-1171; и «Comprehensive Organic Transformations» by Richard С. Larock, VCH Publishers, INC., 1989, p.604-614; или любым способом, с ними согласующимся.
Растворитель, используемый в реакции, может быть любым растворителем постольку, поскольку он не оказывает влияния на течение реакции. Примеры включают в себя галогенированные углеводороды, такие как метиленхлорид, хлороформ и дихлорэтан; простые эфиры, такие как диоксан, тетрагидрофуран, анизол, диметиловый эфир этиленгликоля, диметиловый эфир диэтиленгликоля, диэтиловый эфир диэтиленгликоля и монометиловый эфир этиленгликоля; сульфоксиды, такие как диметилсульфоксид; сложные эфиры, такие как этилацетат; амиды, такие как N,N-диметилформамид, N,N-диметилацетамид и 1-метил-2-пирролидон; кислоты, такие как уксусная кислота, а также воду. Эти растворители могут быть использованы в смеси. Предпочтительный растворитель включает тетрагидрофуран.
Окислитель, используемый в этой реакции, включает в себя, например, диметилсульфоксид, триоксид хрома, диоксид марганца и хлористый хромил. Предпочтительный окислитель включает диоксид марганца.
Количество окислителя, используемого в реакции, представляет число молей соединения общей формулы [12], умноженное на величину, составляющую от 1 до 30, предпочтительно от 1 до 5.
Реакцию можно осуществлять при температуре от -78 до 200°C, предпочтительно от 0 до 100°C, в течение периода времени от 30 минут до 48 часов.
(3-5)
Соединение общей формулы [15] может быть получено взаимодействием соединения общей формулы [14] с соединением формулы [13] в присутствии восстановителя. Реакцию можно осуществлять способами, описанными в WO 02/50061, WO 02/56882, «Advanced Organic Chemistry», by Jerry March, the 4th edition, John Wiley & Sons, INC., 1992, p.898-900; и «Comprehensive Organic Transformations» by Richard С. Larock, VCH Publishers, INC., 1989, p.421-425; или любым способом, с ними согласующимся.
Растворитель, используемый в этой реакции, может быть любым растворителем постольку, поскольку он не оказывает влияния на течение реакции. Примеры включают в себя спирты, такие как метанол, этанол, 2-пропанол и 2-метил-2-пропанол; галогенированные углеводороды, такие как метиленхлорид, хлороформ и дихлорэтан; ароматические углеводороды, такие как бензол, толуол и ксилол; простые эфиры, такие как диоксан, тетрагидрофуран, анизол, диметиловый эфир этиленгликоля, диметиловый эфир диэтиленгликоля, диэтиловый эфир диэтиленгликоля и монометиловый эфир этиленгликоля; сульфоксиды, такие как диметилсульфоксид; сложные эфиры, такие как этилацетат; амиды, такие как N,N-диметилформамид, N,N-диметилацетамид и 1-метил-2-пирролидон, а также воду. Эти растворители могут быть использованы в смеси. Предпочтительный растворитель включает в себя метанол.
Восстановитель, используемый в этой реакции, включает в себя, например, гидридные комплексы, такие как алюмогидрид лития, триацетоксиборогидрид натрия, цианоборогидрид натрия и борогидрид натрия, боран, натрий, а также амальгаму натрия. Можно также альтернативно использовать электролитическое восстановление, где в качестве катода используют медь или платину; каталитическое восстановление, в котором используют никель Ренея, оксид платины или палладиевую чернь, а также можно использовать восстановление, в котором используют состав «цинк/кислота». Предпочтительный восстановитель включает в себя борогидрид натрия. Борогидрид натрия можно использовать в твердом виде или в виде раствора.
Количества соединения общей формулы [14] и восстановителя, используемого в реакции, представляют величину, составляющую от 1 до 50, предпочтительно от 1 до 5, умноженную на число молей соединения общей формулы [13].
Реакцию можно осуществлять при температуре от -30 до 150°C, предпочтительно от 0 до 100°C, в течение периода времени от 10 минут до 120 часов.
(3-6)
Соединение общей формулы [16] может быть получено посредством защиты иминогруппы соединения общей формулы [15]. Реакцию можно осуществить, например, способом, описанным в «Greene's Protective Groups in Organic Synthesis» by M. Wuts и W. Greene, 4th edition, John Wiley & Sons, INC., 2006, p.696-926, или любым способом, с ним согласующимся.
(3-7)
Соединение общей формулы [17] может быть получено удалением защиты соединения общей формулы [16]. Реакцию можно осуществлять, например, способом, описанным в «Greene's Protective Groups in Organic Synthesis» by M. Wuts и W. Greene, 4th edition, John Wiley & Sons, INC., 2006, p.696-926; или любым способом, с ним согласующимся.
[Способ получения 4]
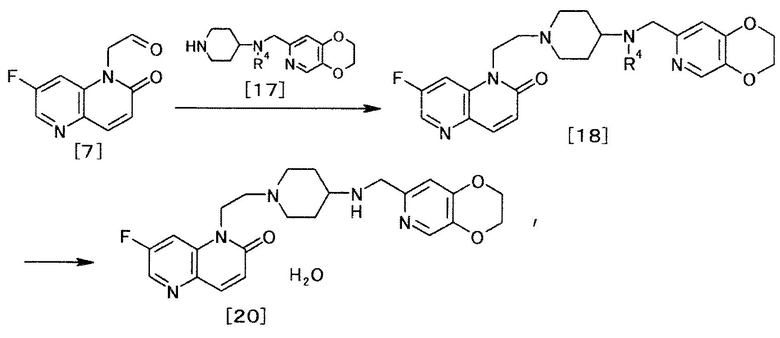
где R4 определен выше.
(4-1)
Соединение общей формулы [18] может быть получено взаимодействием соединения общей формулы [17] с соединением общей формулы [7]. Реакцию можно осуществить в соответствии со способом получения 3-5.
(4-2)
Соединение формулы [20] может быть получено удалением защиты соединения общей формулы [18], с последующей кристаллизацией в результате нейтрализации. Реакцию удаления защиты можно осуществлять, например, способом, описанным в «Greene's Protective Groups in Organic Synthesis» by M. Wuts и W. Greene, 4th edition, John Wiley & Sons, INC., 2006, p.696-926, или любым способом, с ним согласующимся.
Растворитель, используемый в этой реакции, может быть любым растворителем постольку, поскольку он не оказывает неблагоприятного воздействия на течение реакции. Примеры включают в себя смешанные растворители из воды и органических растворителей, а также воду.
Органический растворитель включает в себя, например, спирты, такие как метанол, этанол, 2-пропанол и 2-метил-2-пропанол; галогенированные углеводороды, такие как метиленхлорид, хлороформ и дихлорэтан; ароматические углеводороды, такие как бензол, толуол и ксилол; простые эфиры, такие как диоксан, тетрагидрофуран, анизол, диметиловый эфир этиленгликоля, диметиловый эфир диэтиленгликоля, диэтиловый эфир диэтиленгликоля и монометиловый эфир этиленгликоля; сульфоксиды, такие как диметилсульфоксид; сложные эфиры, такие как этилацетат; кетоны, такие как ацетон и 2-бутанон; амиды, такие как N,N-диметилформамид, N,N-диметилацетамид и 1-метил-2-пирролидон. Эти растворители могут быть использованы в смеси. Предпочтительные растворители включают смешанные растворители из воды и спиртов, а также воду; при этом вода является более предпочтительной.
Когда используют смешанные растворители из воды и органического(их) растворителя(ей), то отношение воды к органическому(им) растворителю(ям) (вода/органический растворитель) находится в диапазоне предпочтительно от 100/0 до 50/50, более предпочтительно от 100/0 до 80/10.
Реакцию можно осуществлять при температуре от -20 до 120°C, предпочтительно от 10 до 80°C, в течение периода времени от 10 минут до 120 часов.
Способ получения по настоящему изобретению обладает такими свойствами, как (1) высокий выход, (2) отсутствие необходимости в колоночной хроматографии на силикагеле (3) и, следовательно, незначительные производственные отходы, (4) неиспользование токсичных или неустойчивых реагентов, и, следовательно применим в качестве метода промышленного производства.
В способах получения 1-4 соединения формул [3], [7], [12] и [13], а также соединения общих формул [2], [5], [6], [8], [10], [11], [15], [16], [17] и [18] могут быть выделены и подвергнуты очистке, но могут быть использованы без выделения, по нижеследующей реакции.
Когда соединение по настоящему изобретению, представляемое формулой [20], используют в качестве фармацевтического продукта, обычно используемые при составлении лекарственного средства вспомогательные фармацевтические средства, такие как наполнитель, носитель и разбавитель, можно с ним смешивать по мере необходимости. Фармацевтические препараты могут быть введены орально или парентерально, согласно общепринятой методике. Их можно вводить в стандартной лекарственной форме в виде таблеток, капсул, порошков, сиропов, гранул, пилюль, суспензий, эмульсий, жидкостей/растворов, препаратов в виде крупинок, суппозиториев, глазных растворов, назальных капель, ушных капель, пластырей, мазей или препаратов для инъекций. Путь введения, дозировка и частота введения могут быть выбраны соответствующим образом, согласно возрасту, массе пациента и симптомам заболевания. Соединение в виде фармацевтического продукта обычно можно вводить орально или парентерально (например, введение путем инъекции, капельного внутривенного введения или в прямую кишку) в дозе, составляющей для взрослого пациента от 0,01 до 1000 мг/кг, от одного до нескольких раз в день.
Соединение по настоящему изобретению, представляемое формулой [20], обнаруживает значительную антибактериальную активность против грамположительных бактерий, включая резистентные бактерии, такие как полирезистентный Staphylococcus aureus, полирезистентные pneumococci и ванкомицин-резистентный Enterococcus, грамотрицательных бактерий, анаэробных или атипичных микобактерий.
Более конкретно, соединение по настоящему изобретению обнаруживает значительную антибактериальную активность против микроорганизмов, выбираемых из Staphylococcus aureus (Staphylococcus aureus Smith, Staphylococcus aureus FDA 209P, Staphylococcus aureus F-3095 (полирезистентный Staphylococcus aureus)), Staphylococcus aureus F-2161 (полирезистентный Staphylococcus aureus), Streptococcus pneumococci (Streptococcus pneumoniae IID553, Streptococcus pneumoniae D-1687 (QRSP), Streptococcus pneumoniae D-4249 (MDRSP)), Enterococcus faecalis (Enterococcus faecalis ATCC29212, Enterococcus faecalis IID682, Enterococcus faecalis D-2648 (VCM-R), Enterococcus faecalis EF-210 (VanA тип VRE), Enterococcus faecium (Enterococcus faecium NBRC 13712, Enterococcus faecium EF-211 (VanA тип VRE)), Corynebacterium diphtheriae (Corynebacterium diphtheriae ATCC 27010), Escherichia coli (Escherichia coli NIHJ), Serratia marcescens (Serratia marcescens IID5218), Haemophilus influenzae (Haemophilus influenzae ATCC 49247), Moraxella catarrhalis (Moraxella catarrhalis ATCC 25238), Pseudomonas aeruginosa (Pseudomonas aeruginosa IFO3445), Enterobacter cloacae (Enterobacter cloacae IID977), Citrobacter freundii (Citrobacter freundii NBRC 12681), Gardnerella vaginalis (Gardnerella vaginalis ATCC 14018), Neisseria gonorrhoeae (Neisseria gonorrhoeae ATCC 19424), Peptostreptococcus asaccharolyticus (Peptostreptococcus asaccharolyticus ATCC 14963), Propionibacterium acnes (Propionibacterium acnes JCM 6425), Clostridium perfringens (Clostridium perfringens ATCC 13124), Bacteroides fragilis (Bacteroides fragilis ATCC 25285), Porphyromonas gingivalis (Porphyromonas gingivalis JCM 8525), Prevotella intermedia (Prevotella intermedia JCM 7365), Fusobacterium nucleatum (Fusobacterium nucleatum JCM 8532), Legionella pneumophilia (Legionella pneumophilia ATCC 33153, Legionella pneumophilia subsp. pneumophilia ATCC 33155, Legionella pneumophilia subsp. pneumophilia ATCC 33215, Legionella pneumophilia subsp. fraseri ATCC33216) и Mycoplasma pneumoniae (Mycoplasma pneumoniae ATCC 15531).
Соединение по настоящему изобретению, представляемое формулой [20], обнаруживает достаточную степень безопасности. Степень безопасности оценивают с помощью большого набора тестов, которые выбирают из разнообразных тестов на безопасность, включающих, например, тест на цитотоксичность; тест на селективность, мишенью которого является ДНК-гираза человека и микроорганизмов; тест на селективность, мишенью которого является топоизомераза IV человека и микроорганизмов; hERG тест, исследование токсичности многократной дозы; тест на ингибирование активности цитохрома P450 (CYP); тест на метаболизм-зависимое ингибирование; in vivo тест на микроядра у мышей и in vivo USD тест печени крысы.
Соединение по настоящему изобретению, представляемое формулой [20], обладает значительной метаболической стабильностью. Метаболическую стабильность оценивают с помощью широкого набора тестов, которые выбирают из разнообразных тестов на стабильность, включая, например, тест на метаболическую стабильность микросом печени человека и тест на метаболическую стабильность S9 человека.
Применимость соединения по настоящему изобретению, представляемого формулой [20], описана далее с помощью ссылок на нижеследующие примеры тестирования.
ПРИМЕР ТЕСТИРОВАНИЯ 1
Тест на восприимчивость
В качестве соединения по настоящему изобретению было выбрано соединение примера 16.
Соединение по настоящему изобретению растворяли в диметилсульфоксиде и измеряли антибактериальную активность (MIC) методом разведения питательной среды микротитра, рекомендованным Японским обществом по химиотерапии.
В исследовании использовали бактерии Staphylococcus aureus (S.aureus Smith, FDA209P, F-3095), Enterococcus faecalis (E.faecalis D-2648) и Escherichia coli (E.coli NIHJ).
Бактериальные клетки выращивали на протяжении ночи на агаре Мюллера-Хинтона: MHA пластины при 35°C суспендировали в стерилизованном физиологическом растворе до получения эквивалентного стандарта McFarland №0,5. Для получения инокулята клеточную суспензию разводили в десять раз. Приблизительно 0,005 мл посевного материала инокулировали в катион-регулируемую питательную среду Мюллера-Хинтона (CAMHB), 100 мкл на ячейку, содержащую тестируемое вещество, и оставляли при 35°C для выращивания в течение ночи. Наиболее низкую концентрацию тестируемого вещества, при которой не наблюдали бактериального роста невооруженным глазом, определяли как MIC.
Результаты показаны в таблице 1.
Соединение по настоящему изобретению демонстрирует значительную антибактериальную активность против различных бактериальных штаммов.
ПРИМЕР ТЕСТИРОВАНИЯ 2
Тест на гигроскопичность
В качестве соединения по настоящему изобретению было выбрано соединение примера 16. В качестве сравнительного соединения было выбрано соединение сравнительного примера 1.
Соединение по настоящему изобретению и сравнительное соединение хранили в течение трех недель при комнатной температуре и относительной влажности, составляющей 97%. В результате, соединение по настоящему изобретению представляло собой порошок, в котором отсутствовали явные изменения. С другой стороны, сравнительное соединение расплылось в результате поглощения влаги из атмосферы.
Соединение по настоящему изобретению продемонстрировало высокую стабильность.
ПРИМЕР ТЕСТИРОВАНИЯ 3
Растворимость
В качестве соединения по настоящему изобретению было выбрано соединение примера 16.
Соединение по настоящему изобретению добавляли в избыточном количестве к 0,2 моль/л раствору фосфатного буфера (pH 6,5). Смесь встряхивали в термостатическом вибраторе (25°C) в течение 48 часов и центрифугировали. Надосадочную жидкость фильтровали через фильтр с размером пор 0,45 мкм, посредством чего измеряли растворимость с помощью высокоскоростной жидкостной хроматографии. В результате, растворимость в 0,2 моль/л растворе фосфатного буфера (pH 6,5) составляла 21,2 мг/мл.
ПРИМЕР
Настоящее изобретение описывают, ссылаясь на нижеследующие примеры, однако, не ограничивая его этими примерами.
Каждый сокращенный символ обозначает нижеследующее.
Boc: трет-бутоксикарбонил; Bn: бензил; Bu: бутил; Me: метил; THP: тетрагидро-2H-пиран-2-ил; ДМСО-d6: дейтерированный диметилсульфоксид
Пример 1
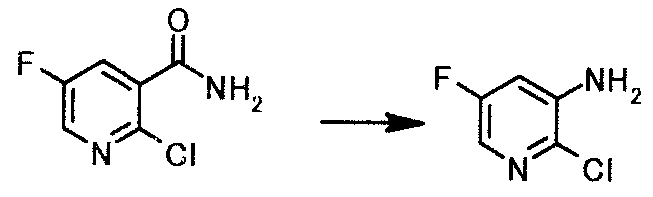
К раствору 0,11 кг гидроксида натрия в 1000 мл воды добавляли 1,4 кг 12% водного раствора гипохлорита натрия и 0,40 кг 2-хлор-5-фторникотинамида. Смесь перемешивали 2 часа 30 минут при комнатной температуре. Реакционную смесь нагревали до 45°C и перемешивали 4 часа. Реакционную смесь охлаждали до комнатной температуры и добавляли этилацетат и 6 моль/л хлористоводородной кислоты. Органический слой отделяли, и водный слой экстрагировали этилацетатом. Органический слой и экстракт объединяли, добавляли безводный сульфат магния и активированный уголь, и смесь перемешивали 30 минут при комнатной температуре. Нерастворимое вещество отфильтровывали, и растворитель выпаривали при пониженном давлении, чтобы получить 0,29 кг 2-хлор-5-фторпиридин-3-амина в виде коричневого твердого вещества.
1Н-ЯМР(CDCl3) δ величина: 4,22(2Н, с), 6,79(1Н, дд, J=9,3, 2,7 Гц), 7,67(1Н, д, J=2,7 Гц)
Пример 2
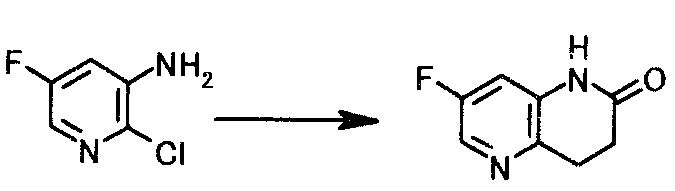
К суспензии 25,0 г 2-хлор-5-фторпиридин-3-амина, 3,8 г хлорида бис(трициклогексилфосфин)палладия(II) и 1,5 г 2-(дитрет-бутилфосфино)бифенила в 75 мл бутилакрилата добавляли 44,1 г диизопропилэтиламина; туда при комнатной температуре по каплям добавляли 15,7 г муравьиной кислоты. Смесь 3 часа кипятили с обратным холодильником. К реакционной смеси при 100°C добавляли 32,1 г диизопропилэтиламина и 11,5 г муравьиной кислоты и смесь 5 часов кипятили с обратным холодильником. Реакционную смесь охлаждали до 80°C, туда добавляли 50 мл толуола и 75 мл воды; и получающуюся в результате смесь охлаждали до комнатной температуры. Твердый продукт получали фильтрованием и промывали, используя последовательно толуол и воду, чтобы получить 18,0 г 7-фтор-3,4-дигидро-1,5-нафтиридин-2(1H)-она в виде белого твердого вещества.
1Н-ЯМР(ДМСО-d6) δ величина: 2,60(2Н, т, J=7,7 Гц), 3,00(2Н, т, J=7,7 Гц), 7,03(1Н, дд, J=9,8, 2,7 Гц), 8,07(1Н, д, J=2,7 Гц), 10,3(1Н, ш.с)
Пример 3
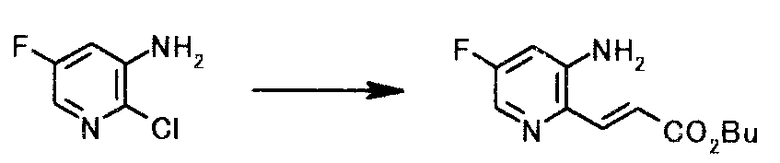
Смешанный раствор 150 мл бутилацетата и 73 мл бутилакрилата кипятили с обратным холодильником, нагревая 45 минут в атмосфере азота. Реакционную смесь охлаждали до 30°C и туда добавляли 50,0 г 2-хлор-5-фторпиридин-3-амина, 3,8 г ацетата палладия(II), 44,8 г трифенилфосфина и 36,6 г карбоната натрия. Смесь 13 часов кипятили с обратным холодильником в атмосфере азота. Реакционную смесь охлаждали до комнатной температуры, туда добавляли 150 мл воды и 700 мл бутилацетата; и смесь перемешивали 1 час. Нерастворимое вещество отфильтровывали, и осадок на фильтре промывали, используя 50 мл бутилацетата. Фильтрат и промывную жидкость объединяли. Органический слой отделяли и 800 мл растворителя выпаривали при пониженном давлении. К полученному осадку по каплям добавляли 300 мл циклогексана и 30 мл толуола; смесь охлаждали до 5°C. Твердый продукт получали фильтрованием и промывали, используя смешанный раствор толуол/циклогексан (1:2) и толуол, чтобы получить 57,8 г бутил (2E)-3-(3-амино-5-фторпиридин-2-ил)акрилата в виде желтого твердого вещества.
1Н-ЯМР(CDCl3) δ величина: 0,96(3Н, т,J=7,3 Гц), 1,38-1,48(2Н, м), 1,64-1,72(2Н, м), 4,10(2Н, ш.с), 4,21(2Н, т, J=6,6 Гц), 6,72(1Н, дд, J=9,8, 2,3 Гц), 6,86(1Н, д, J=15,1 Гц), 7,71(1Н, д, J=15,1 Гц), 7,94(1Н, д, J=2,3 Гц)
Пример 4
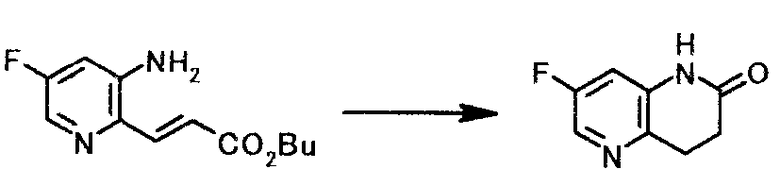
К суспензии 9,1 г бутил (2E)-3-(3-амино-5-фторпиридин-2-ил)акрилата и 0,9 г 10% палладия на углеродном носителе в 30 мл метанола добавляли 4 мл муравьиной кислоты и по каплям при охлаждении льдом добавляли 15 мл триэтиламина. Реакционную смесь 2 часа перемешивали при 60°C. Реакционную смесь охлаждали до комнатной температуры, нерастворимое вещество отфильтровывали, и осадок на фильтре промывали, используя 30 мл толуола. Фильтрат и промывную жидкость объединяли, и растворитель выпаривали при пониженном давлении. К полученному осадку добавляли 30 мл толуола. Смесь 2 часа 30 минут перемешивали при 100°C. К реакционной смеси при 45°C по каплям добавляли 30 мл воды и смесь охлаждали до 5°C. Твердый продукт получали фильтрованием и промывали последовательно водой и толуолом, чтобы получить 5,7 г 7-фтор-3,4-дигидро-1,5-нафтиридин-2(1H)-она в виде твердого белого вещества.
1Н-ЯМР(ДМСО-d6) δ величина: 2,60(2Н, т, J=7,7 Гц), 3,00(2Н, т, J=7,7 Гц), 7,03(1Н, дд, J=9,9, 2,6 Гц), 8,07(1Н, д, J=2,6 Гц), 10,3(1Н, ш.с)
Пример 5
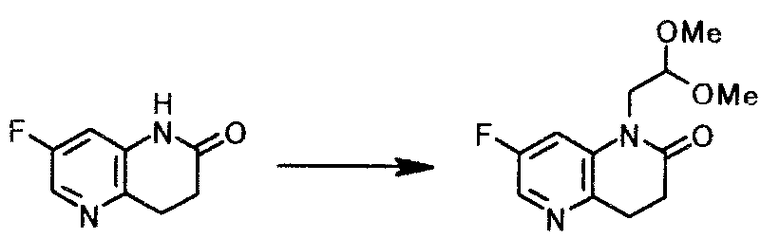
К суспензии 49,8 г фосфата калия в 90 мл диметилсульфоксида добавляли 30,0 г 7-фтор-3,4-дигидро-1,5-нафтиридин-2(1H)-она и 39,7 г 2-бром-1,1-диметоксиэтана. Смесь 3 часа перемешивали при 100°C. К смеси добавляли 7,7 г фосфата калия и 6,1 г 2-бром-1,1-диметоксиэтана и смесь 1 час перемешивали при той же температуре. Реакционную смесь охлаждали до комнатной температуры и добавляли 120 мл воды и 120 мл толуола. Смесь доводили до pH 8,5 с помощью уксусной кислоты и добавляли 3,0 г активированного угля. Нерастворимое вещество отфильтровывали, и осадок на фильтре промывали, используя 30 мл толуола и 30 мл воды. Фильтрат и промывную жидкость объединяли, органический слой отделяли, и водный слой экстрагировали 60 мл толуола. Органические слой и экстракт объединяли, чтобы выпарить растворитель при пониженном давлении. Туда добавляли 90 мл дибутилового эфира, и смесь охлаждали до -3°C. Твердый продукт получали фильтрованием и промывали, используя последовательно дибутиловый эфир и воду, чтобы получить 30,8 г 1-(2,2-диметоксиэтил)-7-фтор-3,4-дигидро-1,5-нафтиридин-2(1H)-она в виде светло-желтого твердого вещества.
1Н-ЯМР(CDCl3) δ величина: 2,73-2,83(2Н, м), 3,07-3,14(2Н, м), 3,44(6Н, с), 3,93(2Н, д, J=5,4 Гц), 4,61(1Н, т, J=5,4 Гц), 7,45(1Н, дд, J=10,5, 2,4 Гц), 8,06(1Н, д, J=2,4 Гц)
Пример 6
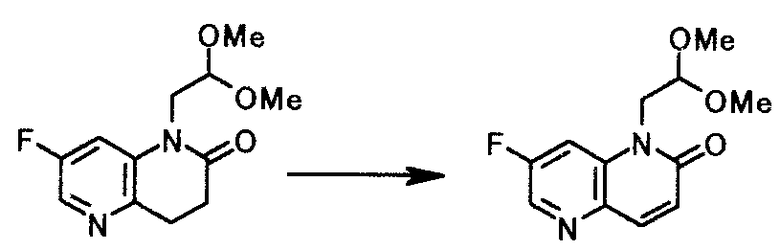
К суспензии 5,0 г 1-(2,2-диметоксиэтил)-7-фтор-3,4-дигидро-1,5-нафтиридин-2(1H)-она, 5,3 г N-бромсукцинимида и 3,0 г карбоната калия в 30 мл хлорбензола, в атмосфере азота при 50-60°C три раза ежечасно добавляли 0,12 г 2,2'-азобис(4-метокси-2,4-диметилвалеронитрил)а. После перемешивания реакционной смеси при той же температуре в течение 1 часа туда добавляли 10 мл воды. Смесь доводили до pH 12,6, используя 20% раствор гидроксида натрия. Органический слой отделяли и промывали 15 мл воды. Водный слой экстрагировали 15 мл толуола. Органический слой и экстракт объединяли, чтобы выпарить растворитель при пониженном давлении. К полученному осадку добавляли 2 мл хлорбензола и 6 мл циклогексана. Смесь перемешивали 30 минут при охлаждении льдом. Твердый продукт получали фильтрованием и промывали циклогексаном, чтобы получить 4,1 г 1-(2,2-диметоксиэтил)-7-фтор-1,5-нафтиридин-2(1H)-она в виде светло-желтого твердого вещества.
1Н-ЯМР(CDCl3) δ величина: 3,44(6Н, с), 4,30(2Н, д, J=5,3 Гц), 4,65(1Н, т, J=5,3 Гц), 6,87(1Н, д, J=9,8 Гц), 7,71(дд, 1Н, J=10,6, 2,4 Гц), 7,92(1Н, д, J=9,8 Гц), 8,41(1Н, д, J=2,4 Гц)
Пример 7
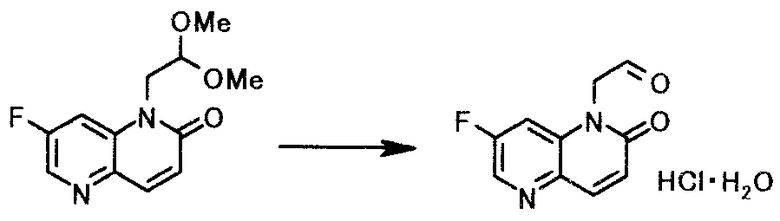
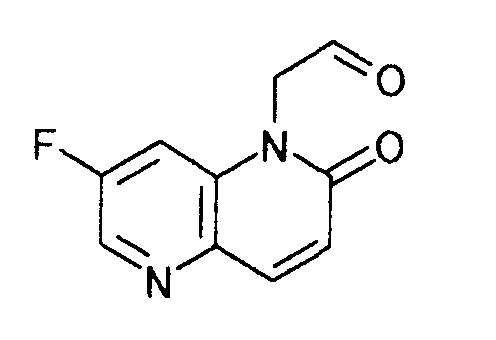
К суспензии 158 г 1-(2,2-диметоксиэтил)-7-фтор-1,5-нафтиридин-2(1H)-она в 1,26 л 2-бутанона при комнатной температуре добавляли 79 мл хлористоводородной кислоты концентрацией 12 моль/л. Смесь 3 часа кипятили с обратным холодильником. После охлаждения реакционной смеси до 10°C твердый продукт получали фильтрованием и промывали 2-бутаноном, чтобы получить 152 г моногидрата (7-фтор-2-оксо-1,5-нафтиридин-1(2H)-ил)ацетальдегида гидрохлорида в виде светло-желтого твердого вещества.
1Н-ЯМР(ДМСО-d6) δ величина: 5,27(2Н, с),6,88(1Н, д, J=9,9 Гц), 7,99-8,04(2Н, м), 8,58(1Н, д, J=2,4 Гц), 9,68(1Н, с)
Пример 8
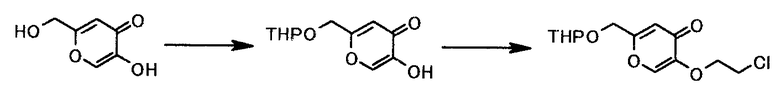
К суспензии 14,3 г койевой кислоты в 57 мл тетрагидрофурана добавляли 11 мл 3,4-дигидро-2H-пирана и 77 мг моногидрата п-толуолсульфокислоты. Смесь перемешивали 6 часов при комнатной температуре. К смеси добавляли 1 мл водного раствора гидроксида натрия концентрацией 0,5 моль/л. Растворитель выпаривали при пониженном давлении, чтобы получить 26,8 г 5-гидрокси-2-((тетрагидро-2H-пиран-2-илокси)метил)-4H-пиран-4-она в виде светло-желтого твердого вещества.
К раствору полученного 5-гидрокси-2-((тетрагидро-2H-пиран-2-илокси)метил)-4H-пиран-4-она в 45 мл N,N-диметилформамида добавляли 45 мл толуола, 20,8 мл 1-бром-2-хлорэтана и 41,6 г карбоната калия и смесь перемешивали 4 часа при 60°С. Смесь оставляли стоять при комнатной температуре в течение ночи; и растворитель выпаривали при пониженном давлении. К полученному осадку добавляли 107 мл воды и смесь 90 мл этилацетата/17 мл толуола. Органический слой отделяли, и водный слой экстрагировали, используя смесь 90 мл этилацетата/17 мл толуола. Органический слой и экстракт объединяли и растворитель выпаривали при пониженном давлении, чтобы получить 26,7 г 5-(2-хлорэтокси)-2-((тетрагидро-2H-пиран-2-илокси)метил)-4H-пиран-4-она в виде коричневого маслянистого вещества.
1Н-ЯМР(ДМСО-d6) δ величина: 1,52-1,71(4Н, м), 1,73-1,79(1Н, м), 1,80-1,88(1Н, м), 3,56(1Н, дддд, J=11,1, 4,4, 4,2, 1,4 Гц), 3,79(2Н, т, J=6,0 Гц), 3,80-3,85(1Н, м), 4,27(2Н, т, J=6,0 Гц), 4,31-4,37(1Н, м), 4,49-4,55(1Н, м), 4,73(1Н, т, J=3,4 Гц), 6,52(1Н, с), 7,75(1Н, с)
Пример 9

К раствору 314 г 5-(2-хлорэтокси)-2-((тетрагидро-2H-пиран-2-илокси)метил)-4H-пиран-4-она в 630 мл метанола добавляли 6,3 мл концентрированной хлористоводородной кислоты и смесь 6 часов перемешивали при комнатной температуре. К реакционной смеси добавляли 13 мл 28% аммиачной воды и растворитель выпаривали при пониженном давлении, чтобы получить 240 г 5-(2-хлорэтокси)-2-(гидроксиметил)-4H-пиран-4-она (неочищенный продукт) в виде коричневого маслянистого вещества.
10,6 г полученного маслянистого вещества очищали колоночной хроматографией на силикагеле [силикагель: KANTO CHEMICAL CO., INC., силикагель 60; элюат: смесь хлороформ/метанол = 95:5], чтобы получить 7,0 г 5-(2-хлорэтокси)-2-(гидроксиметил)-4H-пиран-4-она в виде светло-коричневого твердого вещества.
1Н-ЯМР(CDCl3) δ величина: 3,05(1Н,с), 3,79(2Н, т, J=5,9 Гц), 4,25(2Н, т, J=5,9 Гц), 4,50(2Н, с), 6,53(1Н, т, J=0,9 Гц), 7,75(1Н, с)
Пример 10
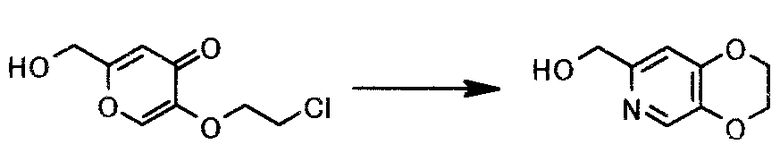
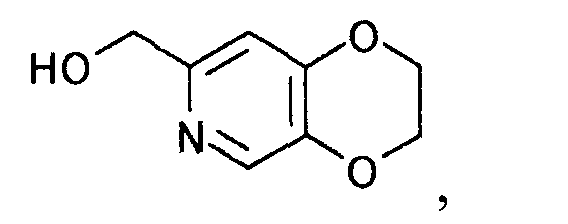
К 229 г 5-(2-хлорэтокси)-2-(гидроксиметил)-4H-пиран-4-она (неочищенный продукт) добавляли 572 мл 28% аммиачной воды. Смесь перемешивали 7 часов при 85°C и оставляли стоять при комнатной температуре на протяжении ночи. Реакционную смесь экстрагировали 4 раза 500 мл 2-пропилацетата. Органический слой сразу объединяли и растворитель выпаривали при пониженном давлении, чтобы получить 90,5 г (2,3-дигидро-(1,4)диоксино(2,3-c)пиридин-7-ил)метанола в виде коричневого маслянистого вещества.
1Н-ЯМР(CDCl3) δ величина: 4,25-4,38(4Н, м), 4,62(2Н, с), 6,76(1Н, с), 8,11(1Н, с)
Пример 11

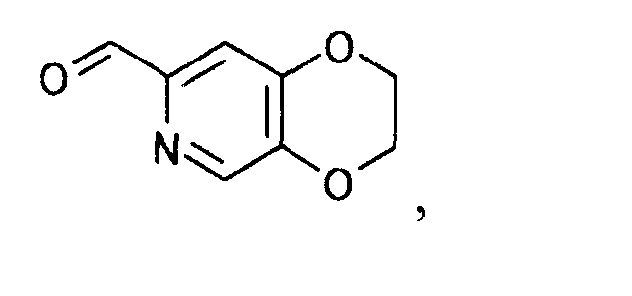
К раствору 111 г (2,3-дигидро-(1,4)диоксино(2,3-c)пиридин-7-ил)метанола в 1110 мл тетрагидрофурана добавляли 164 г диоксида марганца. Смесь перемешивали 5 часов при 70°C и перемешивали при комнатной температуре в течение ночи. Реакционную смесь подвергали фильтрованию через целит и осадок на фильтре промывали 500 мл тетрагидрофурана. Фильтрат и промывную жидкость объединяли, чтобы выпарить растворитель при пониженном давлении. Полученный осадок перекристаллизовывали из 750 мл 2-пропанола, чтобы получить 53,5 г 2,3-дигидро-(1,4)диоксино(2,3-c)пиридин-7-карбальдегида в виде светло-желтого твердого вещества.
1Н-ЯМР(CDCl3) δ величина: 4,38(4Н, с), 7,51(1Н, с), 8,31(1Н, с), 9,92(1Н, с)
Пример 12
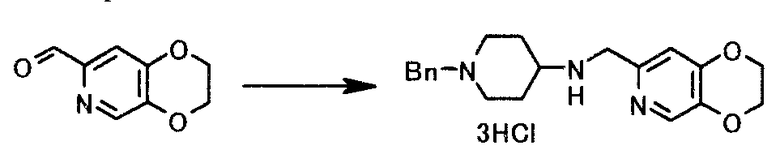
Раствор 3,0 г 2,3-дигидро-(1,4)диоксино(2,3-c)пиридин-7-карбальдегида и 3,4 г 4-амино-1-бензилпиперидина в 30 мл метанола перемешивали 50 минут при комнатной температуре и по каплям добавляли раствор 0,34 г борогидрида натрия в 30 мл раствора 0,01 моль/л гидроксида натрия/метанола, при охлаждении льдом. Смесь затем 2 часа перемешивали при охлаждении льдом, по каплям добавляли 6 мл концентрированной хлористоводородной кислоты при температуре 10°C или ниже и 1 час 30 минут перемешивали. Твердый продукт получали фильтрованием, чтобы иметь 6,8 г 1-бензил-N-(2,3-дигидро-(1,4)диоксино(2,3-c)пиридин-7-илметил)пиперидин-4-амина тригидрохлорида в виде белого твердого вещества.
1Н-ЯМР(D2O) δ величина: 1,93-2,03(2Н, м), 2,48(2Н, д, J=13,3 Гц), 3,13-3,21(2Н, м), 3,62-3,73(3Н, м), 4,37(2Н, с), 4,43-4,49(4Н, м), 4,53-4,58(2Н, м), 7,35(1Н, с), 7,49-7,57(5Н, м), 8,30(1Н, с)
Пример 13
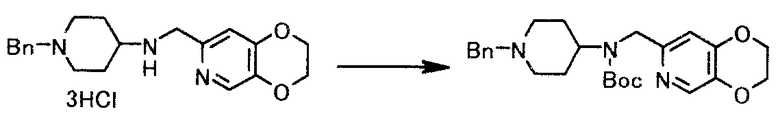
К раствору 6,8 г 1-бензил-N-(2,3-дигидро-(1,4)диоксино(2,3-c)пиридин-7-илметил)пиперидин-4-амина тригидрохлорида в смеси 20 мл воды/11 мл тетрагидрофурана при охлаждении льдом добавляли 8 мл 20% водного раствора гидроксида натрия и затем туда добавляли 3,3 г ди-трет-бутилдикарбоната. Смесь перемешивали 8 часов при комнатной температуре и туда добавляли 11 мл этилацетата. Органический слой отделяли и туда добавляли 5,1 г силикагеля (Chromatorex-NH, FUJI SILYSIA CHAMICAL LTD.). Смесь перемешивали 1 час при комнатной температуре и фильтровали, пропуская через 2,6 г силикагеля (Silica gel 60N, KANTO CHEMICAL CO., INC). Для промывания использовали 35 мл этилацетата. Фильтрат и промывную воду объединяли и растворитель выпаривали при пониженном давлении, чтобы получить 6,3 г трет-бутил(1-бензилпиперидин-4-ил)(2,3-дигидро-(1,4)диоксино(2,3-c)пиридин-7-илметил)карбамата в виде светло-желтой пены.
1Н-ЯМР(CDCl3) δ величина: 1,32-1,54(9Н, м), 1,55-1,74(4Н, м), 1,92-2,07(2Н, м), 2,87(2Н, д, J=11,5 Гц), 3,44(2Н, с), 4,07-4,18(1Н, м), 4,22-4,32(4Н, м), 4,33-4,48(2Н, м), 6,72(1Н, с), 7,20-7,24(1Н, м), 7,27-7,31(4Н, м), 8,04(1Н, с)
Пример 14

К раствору 5,9 г трет-бутил(1-бензилпиперидин-4-ил)(2,3-дигидро-(1,4)диоксино(2,3-c)пиридин-7-илметил)карбамата в 30 мл метанола добавляли 1,2 г 5% палладия на углеродном носителе и смесь перемешивали 7 часов при 60°C в атмосфере водорода. Смесь подвергали фильтрованию на целите и к фильтрату добавляли 40 мл этилацетата и 30 мл водного раствора гидроксида натрия с концентрацией 0,5 моль/л. Органический слой промывали насыщенным раствором хлорида натрия, сушили, используя сульфат натрия, и растворитель выпаривали при пониженном давлении. Полученный осадок перекристаллизовывали из смеси 5 мл этилацетата/15 мл гептана, чтобы получить 3,0 г моногидрата трет-бутил((2,3-дигидро-(1,4)диоксино(2,3-c)пиридин-7-ил)метил)(пиперидин-4-ил)карбамата в виде белого порошка.
1Н-ЯМР(CDCl3) δ величина: 1,39(9Н, с), 1,48-1,53(2Н, м), 1,63-1,68(2Н, м), 2,61-2,66(2Н, м), 3,07-3,10(2Н, м), 4,26-4,38(7Н, м), 6,75(1Н, с), 8,05(1Н, с)
Пример 15
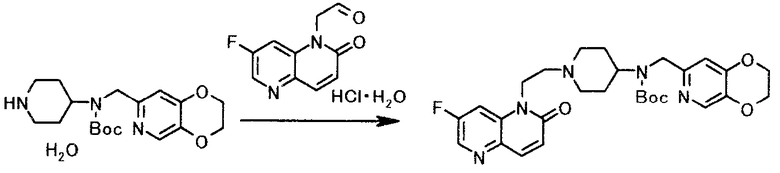
К раствору 5,0 г моногидрата трет-бутил (2,3-дигидро(1,4)диоксино(2,3-c)пиридин-7-илметил)(пиперидин-4-ил)карбамата в 40 мл N-метил-2-пирролидона добавляли 3,5 г моногидрата (7-фтор-2-оксо-1,5-нафтиридин-1(2H)-ил)ацетальдегида гидрохлорида и смесь перемешивали 1 час при комнатной температуре. К смеси при охлаждении льдом добавляли раздельно 5 порциями 4,3 г триацетоксиборогидрида натрия на протяжении периода в 80 минут и смесь 1 час 40 минут перемешивали при охлаждении льдом. После нагревания до комнатной температуры к смеси добавляли 20 мл воды и pH доводили до 11,5 с помощью 20% водного раствора гидроксида натрия. К смеси при 70-80°C добавляли 20 мл N-метил-2-пирролидона и смесь перемешивали 2 часа 30 минут при той же температуре. Реакционную смесь охлаждали до комнатной температуры и твердый продукт получали фильтрованием и промывали водой, чтобы иметь 6,5 г трет-бутил (2,3-дигидро(1,4)диоксино(2,3-c)пиридин-7-илметил)(1-(2-(7-фтор-2-оксо-1,5-нафтиридин-1(2H)-ил)этил)пиперидин-4-ил)карбамата в виде светло-коричневого твердого вещества.
1Н-ЯМР(CDCl3) δ величина: 1,30-1,80(13Н, м), 2,08-2,27(2Н, м), 2,56-2,65(2Н, м), 2,93-3,04(2Н, м), 4,02-4,19(1Н, м), 4,23-4,49(8Н, м), 6,73(1Н, с), 6,84(1Н, д, J=9,9 Гц), 7,47(1Н, дд, J=10,2, 2,3 Гц), 7,87(1Н, д, J=9,9 Гц), 8,05(1Н, с), 8,41(1Н, д, J=2,3 Гц)
Пример 16
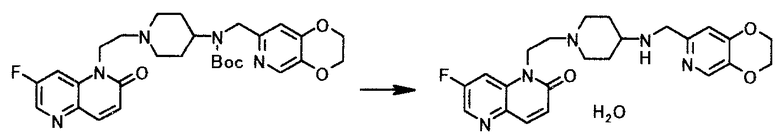
К суспензии 25,0 г трет-бутил (2,3-дигидро(1,4)диоксино(2,3-c)пиридин-7-илметил)(1-(2-(7-фтор-2-оксо-1,5-нафтиридин-1(2H)-ил)этил)пиперидин-4-ил)карбамата в 50 мл воды по каплям добавляли 18 мл концентрированной хлористоводородной кислоты при 28-39°C. Реакционную смесь перемешивали 3 часа 30 минут при 40-50°C, охлаждали до комнатной температуры, добавляли 17 мл 20% водного раствора гидроксида натрия и 25 мл воды и нагревали до 60°C. Смесь доводили до pH 3 с помощью концентрированной хлористоводородной кислоты и добавляли 25 мл воды. Нерастворимое вещество отфильтровывали при 50°C. Осадок на фильтре промывали, используя 25 мл воды. Фильтрат и промывную жидкость объединяли и нагревали до 40°C, добавляли 13,5 мл 20% водного раствора гидроксида натрия, 150 мл 2-бутанона и 25 мл воды и нагревали с обратным холодильником, чтобы растворить твердое вещество. Реакционную смесь охлаждали до 10°C и твердый продукт, полученный фильтрованием, промывали водой, чтобы иметь 19,3 г моногидрата 1-(2-(4-((2,3-дигидро(1,4)диоксино(2,3-c)пиридин-7-илметил)амино)пиперидин-1-ил)этил)-7-фтор-1,5-нафтиридин-2(1H)-она в виде светло-жделтого твердого вещества.
1Н-ЯМР(CDCl3) δ величина: 1,35-1,50(2Н, м), 1,90(2Н, д, J=12,2 Гц), 2,18(2Н, тд, J=11,5, 2,2 Гц), 2,46-2,59(1Н, м), 2,64(2Н, т, J=7,1 Гц), 2,95(2Н, д, J=12,0 Гц), 3,79(2Н, с), 4,26-4,34(6Н, м), 6,81(1Н, с), 6,85(1Н, д, J=9,8 Гц), 7,56(1Н, дд, J=10,2, 2,4 Гц), 7,88(1Н, дд, J=9,8, 0,5 Гц), 8,10(1Н, с), 8,41(1Н, д, J=2,4 Гц)
Пример 17
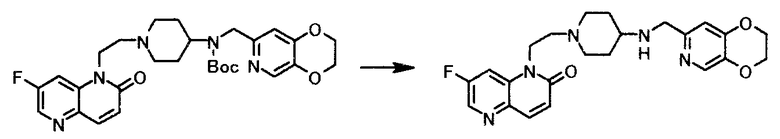
Раствор 3,03 г трет-бутил (2,3-дигидро(1,4)диоксино(2,3-c)пиридин-7-илметил)(1-(2-(7-фтор-2-оксо-1,5-нафтиридин-1(2H)-ил)этил)пиперидин-4-ил)карбамата в 45 мл трифторуксусной кислоты перемешивали 1 час 30 минут при комнатной температуре. Реакционную смесь охлаждали льдом, добавляли 30 мл воды и 30 мл этилацетата и доводили до pH 10 с помощью 2 моль/л водного раствора гидроксида натрия. Органический слой отделяли, и водный слой экстрагировали 7 раз этилацетатом. Органические слои объединяли и растворитель концентрировали при пониженном давлении до 10 мл, после чего нерастворимое вещество отфильтровывали. Растворитель выпаривали при пониженном давлении и полученный осадок очищали колоночной хроматографией на немодифицированном силикагеле [элюат; смесь хлороформ/метанол = 92:8], перекристаллизовывали из 3 мл этилацетата, чтобы получить 0,611 г 1-(2-(4-((2,3-дигидро(1,4)диоксино(2,3-c)пиридин-7-илметил)амино)пиперидин-1-ил)этил)-7-фтор-1,5-нафтиридин-2(1H)-она (дегидрат) в виде светло-желтого твердого вещества.
1Н-ЯМР(CDCl3) δ величина: 1,39-1,47(2Н, м), 1,87-1,93(2Н, м), 2,18(2Н, т, J=10,8 Гц), 2,49-2,55(1Н, м), 2,64(2Н, т, J=7,1 Гц), 2,92-2,98(2Н, м), 3,79(2Н, с), 4,26-4,29(2Н, м), 4,29-4,34(4Н, м), 6,82(1Н, с), 6,85(1Н, д, J=9,6 Гц), 7,55(1Н, д, J=9,6 Гц), 7,88(1Н, д, J=9,6 Гц), 8,10(1Н, с), 8,41(1Н, д, J=2,3 Гц)
Анал. рассчитано для (Anal. Calcd.) C23H26FN5O3: C, 62,86; H, 5,96; H, 15,94; N, 15,94; F, 4,32
Найдено: C, 62,58; H, 5,92; N, 15,80; F, 4,21
Сравнительный пример 1
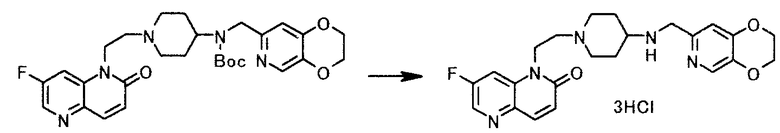
К суспензии 0,30 г трет-бутил (2,3-дигидро(1,4)диоксино(2,3-c)пиридин-7-илметил)(1-(2-(7-фтор-2-оксо-1,5-нафтиридин-1(2H)-ил)этил)пиперидин-4-ил)карбамата в 2 мл 2-пропанола добавляли 0,23 мл концентрированной хлористоводородной кислоты, и получающуюся в результате смесь перемешивали 1 час 50 минут при нагревании с обратным холодильником. Реакционную смесь охлаждали до 5°C и твердое вещество получали фильтрованием, чтобы иметь 0,28 г 1-(2-(4-((2,3-дигидро(1,4)диоксино(2,3-c)пиридин-7-илметил)амино)пиперидин-1-ил)этил)-7-фтор-1,5-нафтиридин-2(1H)-она тригидрохлорида в виде светло-желтого твердого вещества.
1Н-ЯМР(D2O) δ величина: 2,00-2,16(2Н, м), 2,52-2,61(2Н, м), 3,23-3,35(2Н, м), 3,61-3,67(2Н, м), 3,69-3,80(1Н, м), 3,98-4,07(2Н, м), 4,46-4,51(2Н, м), 4,52(2Н, с), 4,55-4,63(2Н, м), 4,71-4,96(2Н, м), 6,99(1Н, д, J=9,8 Гц), 7,44(1Н, с), 7,93-7,99(1Н, м), 8,10(1Н, д, J=9,8 Гц), 8,36(1Н, с), 8,57(1Н, д, J=2,2 Гц)
Пример получения 1
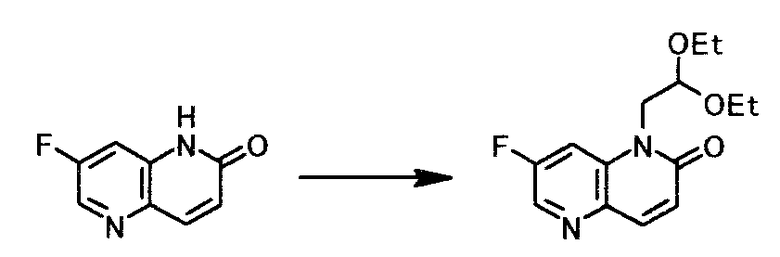
К суспензии 3,00 г 7-фтор-1,5-нафтиридин-2(1H)-она и 5,04 г фосфата калия в 12 мл диметилсульфоксида добавляли при комнатной температуре 4,68 г 2-бром-1,1-диэтоксиэтана и полученную смесь перемешивали 4,5 часа при 94°C. Реакционную смесь охлаждали и добавляли 21 мл воды и 12 мл циклопентилметилового эфира. Смесь доводили до pH 5,8 с помощью хлористоводородной кислоты с концентрацией 12 моль/л. Затем нерастворимое вещество отфильтровывали, и осадок на фильтре промывали дважды 3 мл циклопентилметилового эфира. Органические слои полученного фильтрата и промывной жидкости отделяли, и растворитель выпаривали при пониженном давлении. Полученный осадок очищали колоночной хроматографией на силикагеле, чтобы получить 3,11 г 1-(2,2-диэтоксиэтил)-7-фтор-1,5-нафтиридин-2(1H)-она в виде светло-желтого маслянистого вещества.
1Н-ЯМР(CDCl3) δ величина: 1,12(6Н, т, J=7,1 Гц), 3,47-3,55(2Н, м), 3,74-3,82(2Н, м), 4,29(2Н, д, J=5,1 Гц), 4,78(1Н, т, J=5,4 Гц), 6,86(1Н, д, J=9,8 Гц), 7,82(1Н, дд, J=10,6, 2,4 Гц), 7,92(1Н, д, J=9,8 Гц), 8,41(1Н, д, J=2,4 Гц)
Пример получения 2
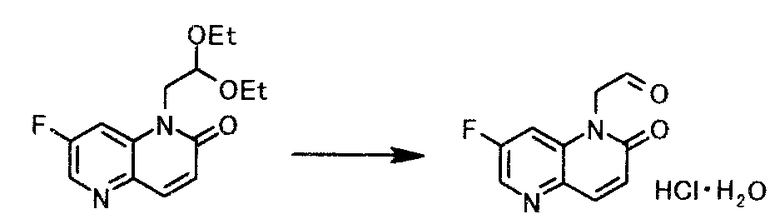
К 480 мл 2-бутанона добавляли 30 мл хлористоводородной кислоты с концентрацией 12 моль/л. Смесь нагревали до 70°C и по каплям добавляли раствор 60 г 1-(2,2-диэтоксиэтил)-7-фтор-1,5-нафтиридин-2(1H)-она в 60 мл 2-бутанона, с последующим кипячением с обратным холодильником в течение 2 часов. После охлаждения реакционной смеси до 25°C твердый продукт получали фильтрованием и промывали 2-бутаноном, чтобы в результате получить 50,3 г моногидрата гидрохлорида (7-фтор-2-оксо-1,5-нафтиридин-1(2H)-ил)ацетальдегида в виде светло-желтого твердого вещества.
КРАТКОЕ ОПИСАНИЕ РИСУНКОВ
На фиг.1 показана порошковая дифракционная рентгенограмма моногидрата 1-(2-(4-((2,3-дигидро(1,4)диоксино(2,3-c)пиридин-7-илметил)амино)пиперидин-1-ил)этил)-7-фтор-1,5-нафтиридин-2(1H)-она.
ПРИМЕНИМОСТЬ В ПРОИЗВОДСТВЕННЫХ УСЛОВИЯХ
Моногидрат 1-(2-(4-((2,3-дигидро(1,4)диоксино(2,3-c)пиридин-7-илметил)амино)пиперидин-1-ил)этил)-7-фтор-1,5-нафтиридин-2(1H)-она по настоящему изобретению (1) обладает сильной антибактериальной активностью и высокой степенью безопасности, (2) не обнаруживает расплывания, в результате поглощения влаги из воздуха, или гигроскопичности, (3) легок в обращении, (4) его получают при использовании растворителя, безопасного для человеческого организма, (5) его получают в условиях, которые являются малой нагрузкой для окружающей среды, и (6) возможно его получение в массовом производстве, тем самым он применим в качестве валового фармацевтического продукта.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВОЕ ГЕТЕРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ ИЛИ ЕГО СОЛЬ И ЕГО ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ | 2007 |
|
RU2434868C2 |
| ПРОИЗВОДНЫЕ 2-ГИДРОКСИЭТИЛ-1Н-ХИНОЛИН-2-ОНА И ИХ АЗАИЗОСТЕРИЧЕСКИЕ АНАЛОГИ С АНТИБАКТЕРИАЛЬНОЙ АКТИВНОСТЬЮ | 2010 |
|
RU2540862C2 |
| ПРОИЗВОДНЫЕ 5-АМИНО-2-(1-ГИДРОКСИЭТИЛ)ТЕТРАГИДРОПИРАНА | 2009 |
|
RU2525541C2 |
| ПРОИЗВОДНЫЕ 5-АМИНОЦИКЛИЛМЕТИЛОКСАЗОЛИДИН-2-ОНА | 2008 |
|
RU2492169C2 |
| ОКСАЗОЛИДИНИЛОВЫЕ АНТИБИОТИКИ | 2009 |
|
RU2516701C2 |
| АНТИБИОТИЧЕСКИЕ ПРОИЗВОДНЫЕ 2-ОКСО-ОКСАЗОЛИДИН-3, 5-ДИИЛА | 2012 |
|
RU2616609C2 |
| ТРИЦИКЛИЧЕСКИЕ ОКСАЗОЛИДИНОНОВЫЕ АНТИБИОТИЧЕСКИЕ СОЕДИНЕНИЯ | 2009 |
|
RU2530884C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ ПИРИДИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2004 |
|
RU2366659C2 |
| АМИНОМЕТИЛХИНОЛОНЫ, ПОЛЕЗНЫЕ ПРИ ЛЕЧЕНИИ JNK-ОПОСРЕДОВАННОГО РАССТРОЙСТВА | 2012 |
|
RU2629111C2 |
| ПРОИЗВОДНЫЕ ПИРРОЛОПИРИМИДИНА И ПИРРОЛОПИРИДИНА, ЗАМЕЩЕННЫЕ ЦИКЛИЧЕСКОЙ АМИНОГРУППОЙ КАК АНТАГОНИСТЫ CRF | 2005 |
|
RU2385321C2 |
Изобретение относится к новому соединению, а именно моногидрату 1-(2-(4-((2,3-дигидро(1,4)диоксино(2,3-с)пиридин-7-илметил)амино)пиперидин-1-ил)этил)-7-фтор-1,5-нафтиридин-2(1Н)-она, который обладает сильной антибактериальной активностью. Это соединение обладает высокой степенью безопасности и применимо в производстве лекарственных препаратов в качестве исходного лекарственного средства. Кроме того, описан способ получения моногидрата 1-(2-(4-((2,3-дигидро(1,4)диоксино(2,3-с)пиридин-7-илметил)амино)пиперидин-1-ил)этил)-7-фтор-1,5-нафтиридин-2(1Н)-она формулы 19 и способы получения промежуточных соединений.
7 н. и 1 з.п. ф-лы, 1 табл., 1 ил., 17 пр.
1. Моногидрат 1-(2-(4-((2,3-дигидро(1,4)диоксино(2,3-с)пиридин-7-илметил)амино)пиперидин-1-ил)этил)-7-фтор-1,5-нафтиридин-2(1Н)-она.
2. Способ получения моногидрата 1-(2-(4-((2,3-дигидро(1,4)диоксино(2,3-с)пиридин-7-илметил)амино)пиперидин-1-ил)этил)-7-фтор-1,5-нафтиридин-2(1Н)-она, представляемого формулой
[Формула 19]
который характеризуется взаимодействием
(1) производного нафтиридина, представляемого формулой
[Формула 7]
и получаемого по взаимодействию производного пиридина, представляемого формулой
[Формула 1]
с эфиром акриловой кислоты с образованием производного акриловой кислоты, представляемого общей формулой
[Формула 2]
где R1 представляет алкильную группу;
затем восстанавливают/циклизуют полученное производное акриловой кислоты, чтобы получить производное дигидронафтиридина, представляемое формулой
[Формула 3]
воздействуя далее на полученное производное дигидронафтиридина соединением, представляемым общей формулой
[Формула 4]
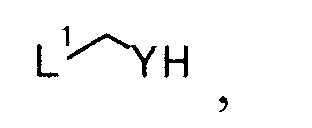
где L1 представляет уходящую группу; Y представляет защищенную карбонильную группу,
чтобы получить производное дигидронафтиридина, представляемое общей формулой
[Формула 5]
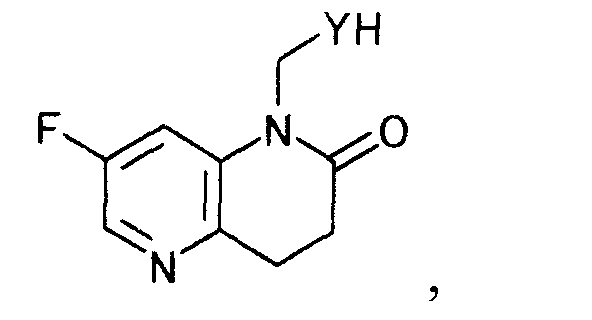
где Y определен выше,
окисляя далее полученное производное дигидронафтиридина, чтобы получить производное нафтиридина, представляемое общей формулой
[Формула 6]
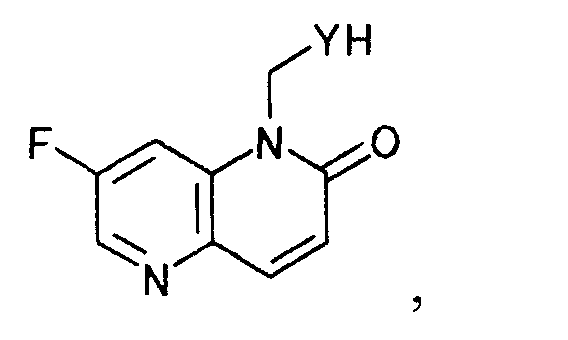
где Y определен выше; и
затем удаляя защиту полученного производного нафтиридина с помощью (2) производного пиперидина, представляемого общей формулой
[Формула 17]
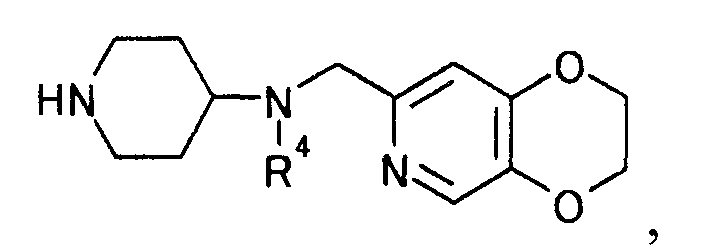
где R4 представляет иминозащитную группу,
и получаемого по взаимодействию производного койевой кислоты, представляемого общей формулой
[Формула 8]
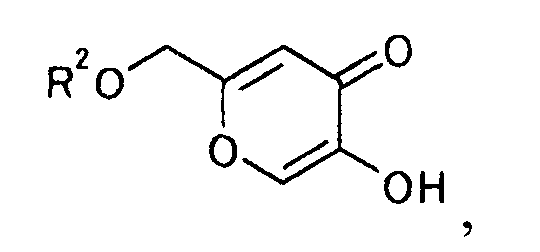
где R2 является гидроксилзащитной группой,
с соединением, представляемым общей формулой
[Формула 9]
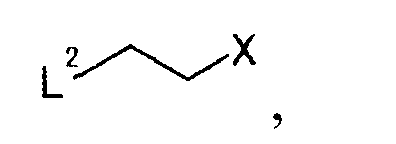
где L2 представляет уходящую группу; Х представляет уходящую группу, для получения производного койевой кислоты, представляемого общей формулой
[Формула 10]
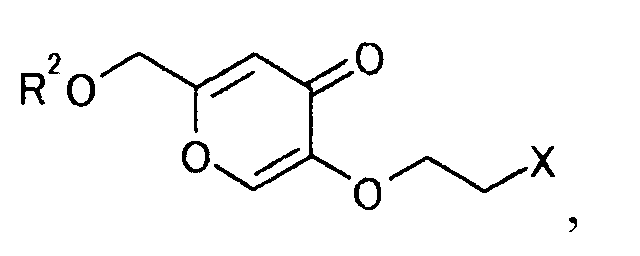
где R2 и X определены выше,
затем удаляя защиту полученного производного койевой кислоты, чтобы получить производное койевой кислоты, представляемое общей формулой
[Формула 11]
где Х определен выше,
затем воздействуя на полученное производное койевой кислоты аммиаком, чтобы получить производное пиридина, представляемого формулой
[Формула 12]
затем окисляя полученное производное пиридина с образованием производного пиридина, представляемого формулой
[Формула 13]
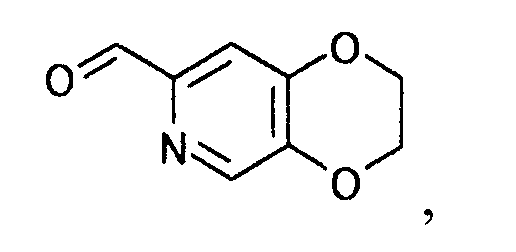
затем воздействуя на полученное производное пиридина производным пиперидина, представляемым общей формулой
[Формула 14]
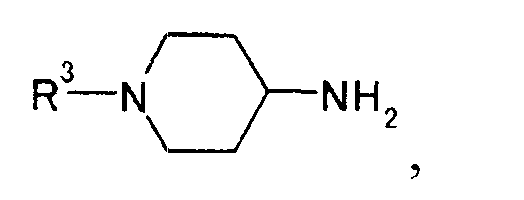
где R3 представляет иминозащитную группу,
чтобы получить производное пиперидина, представляемое общей формулой
[Формула 15]
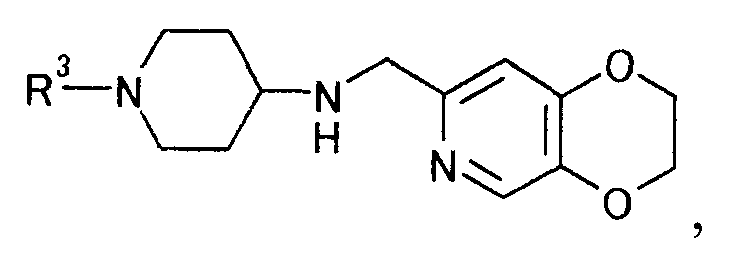
где R3 определен выше,
защищая далее иминогруппу, чтобы получить производное пиперидина, представляемое общей формулой
[Формула 16]
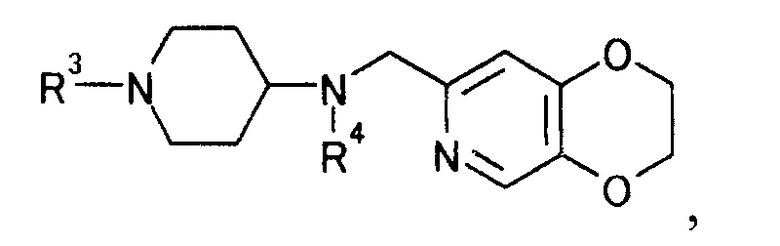
где R4 определен выше; R3 определен выше,
и затем удаляя защиту полученного производного пиперидина с образованием
(3) производного нафтиридина, представляемого общей формулой
[Формула 18]
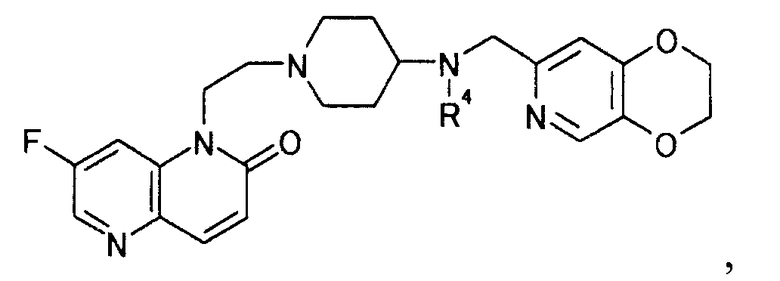
где R4 определен выше,
и затем удаляя защиту полученного производного нафтиридина.
3. Способ получения производного пиперидина, представляемого общей формулой
[Формула 29]
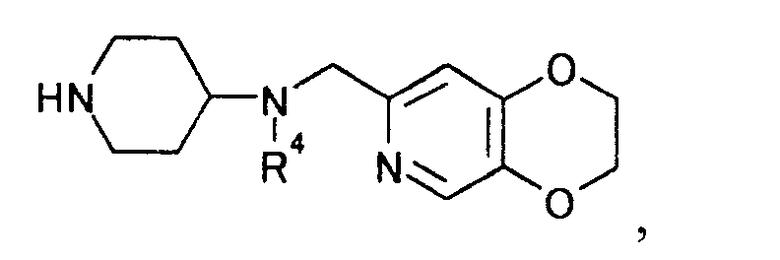
где R4 представляет иминозащитную группу,
который характеризуется
взаимодействием производного койевой кислоты, представляемого общей формулой
[Формула 20]
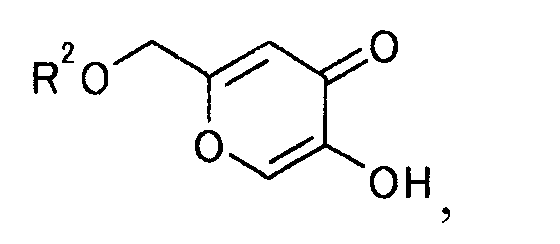
где R2 представляет гидроксилзащитную группу, с соединением, представляемым общей формулой
[Формула 21]
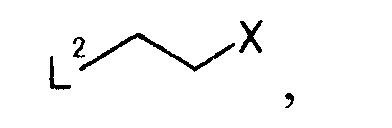
где L2 представляет уходящую группу; Х представляет уходящую группу, с образованием производного койевой кислоты, представляемого общей формулой
[Формула 22]
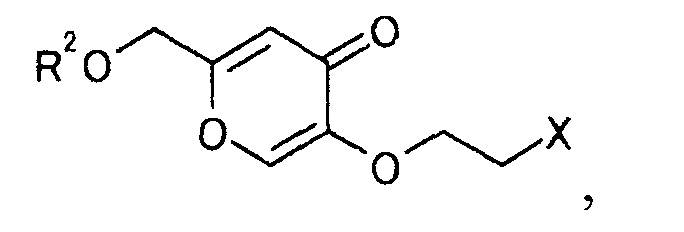
где R2 и Х определены выше,
затем удаляют защиту полученного производного койевой кислоты, чтобы получить производное койевой кислоты, представляемое общей формулой
[Формула 23]
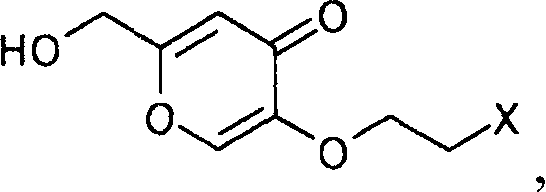
где Х определен выше,
затем воздействуя на производное койевой кислоты аммиаком, чтобы получить производное пиридина, представляемого формулой
[Формула 24]
затем окисляя полученное производное пиридина с образованием производного пиридина, представляемого формулой
[Формула 25]
затем воздействуя на полученное производное пиридина производным пиперидина, представляемым общей формулой
[Формула 26]
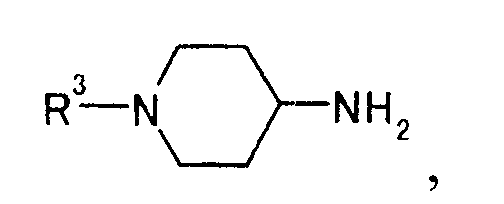
где R3 представляет иминозащитную группу,
чтобы получить производное пиперидина, представляемое общей формулой
[Формула 27]
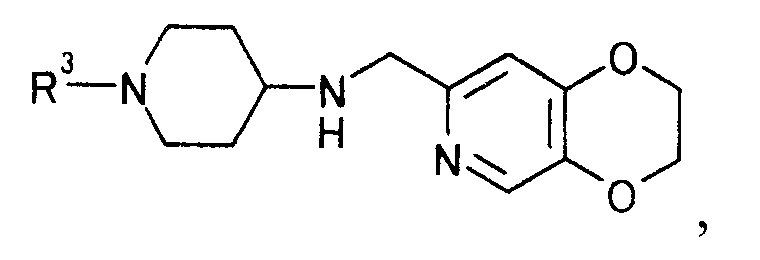
где R3 определен выше,
затем защищая иминогруппу, чтобы получить производное пиперидина, представляемое формулой
[Формула 28]
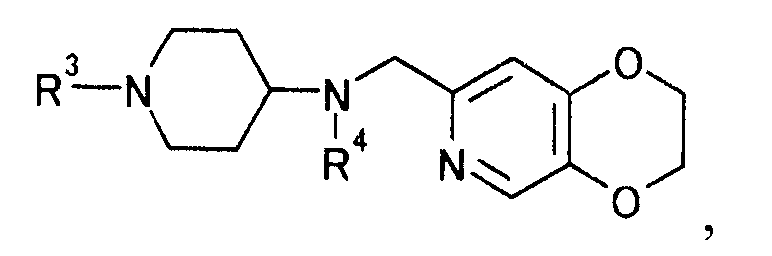
где R4 определен выше; R3 определен выше,
и затем удаляя защиту полученного производного пиперидина.
4. Способ получения по п.2 или 3, где R3 представляет собой аралкильную группу; R4 представляет ацильную группу или алкоксикарбонильную группу.
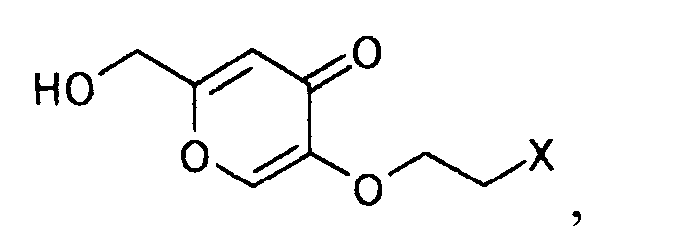
5. Способ получения производного пиридина, представляемого формулой
[Формула 34]
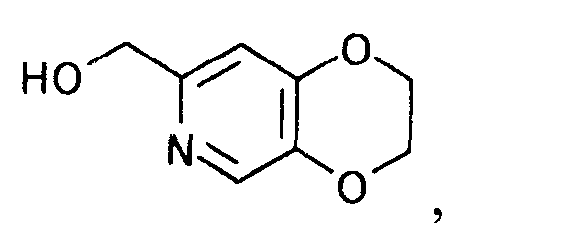
который характеризуется взаимодействием производного койевой кислоты, представляемого общей формулой
[Формула 30]
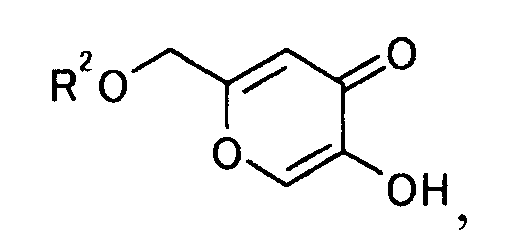
где R2 представляет гидроксилзащитную группу,
с соединением, представляемым общей формулой
[Формула 31]
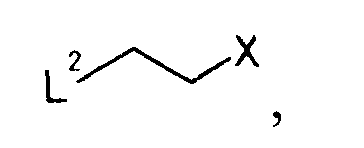
где L2 представляет уходящую группу; Х представляет уходящую группу, чтобы получить производное койевой кислоты, представляемое общей формулой
[Формула 32]
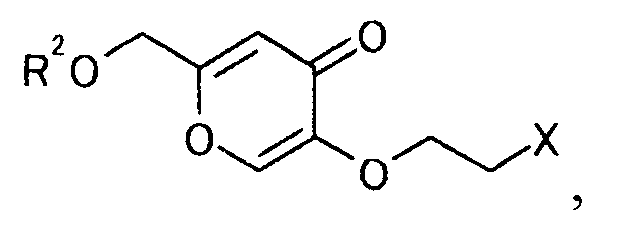
где R2 и Х определены выше,
затем удаляя защиту полученного производного койевой кислоты, чтобы получить производное койевой кислоты, представляемое общей формулой
[Формула 33]
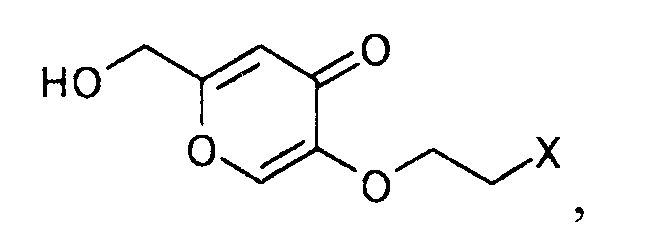
где Х определен выше,
и затем воздействуя на полученное производное койевой кислоты аммиаком.
6. Способ получения производного пиридина, представляемого формулой
[Формула 36]
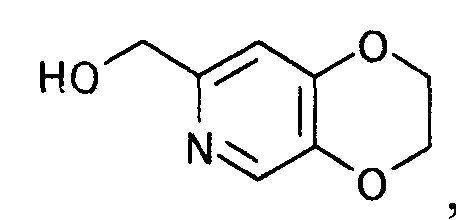
который характеризуется взаимодействием производного койевой кислоты, представляемого общей формулой
[Формула 35]
где X представляет уходящую группу,
с аммиаком.
7. Производное койевой кислоты, представляемое общей формулой
[Формула 37]
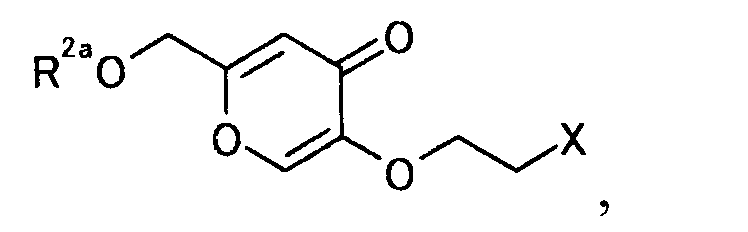
где R2a представляет собой атом водорода, тетрагидропиранильную или тетрагидрофуранильную группу; уходящая группа Х представляет собой атом галогена.
8. Антибактериальное средство, содержащее моногидрат 1-(2-(4-((2,3-дигидро(1,4)диоксино(2,3-с)пиридин-7-илметил)амино)пиперидин-1-ил)этил)-7-фтор-1,5-нафтиридин-2(1Н)-она.
| WO 2004076445 А2, 10.09.2004 | |||
| Способ получения производных 1,8-нафтиридина или их солей | 1985 |
|
SU1445558A3 |
| Способ получения производных нафтиридинов | 1977 |
|
SU637082A3 |
| Производные 6Н-2,3,7,8,9,10-гексагидро-7-оксо (1,4)-диоксино (2,3-В)карбазола | 1984 |
|
SU1203864A1 |

