Область техники
Настоящее изобретение касается одной группы производных азаиндола-индола совсем новой структуры. Молекулярная структура этих производных характеризуется тем, что они связаны азаиндолами с бимолекулярными индолами на различных позициях, формируются большие π-сопряженные гетероциклические системы. Эти производные посредством различных механизмов препятствуют расстройствам роста и пролиферации клеток. Настоящее изобретение также включает в себя способы изготовления производных, в том числе фармацевтические композиции описанных производных и их использование.
Предшествующий уровень техники
Настоящий патент на изобретение представляет собой патент, следующий за следующими патентами:
1) «Специфические соединения индола, способ их изготовления и использование в лечении и профилактике рака и других заболеваний", CN 02138518, 1А, опубликованная 04.05.2005);
2) «Эмульсия для использования в нерастворимых препаратах и способ ее изготовления», CN 200410052816, 5А, приоритет 14.07.2004);
3) «Способ изготовления производных N(1)-гидрокарбил-3' оксима-индирубина (I) и их медицинское использование», CN 200510094482.2 А, опубликованная 01.08.2007;
4) «Дисперсионный агент для изготовления нерастворимых препаратов», CN 200610038112.1 А, приоритет 17.01.2006.
Мономерные соединения растений лидируют среди лекарств в борьбе с раком, как например, Камптотецин, выделенный из Камптотеки остроконечной, и Паклитаксел, полученный из Головчатотисса, являющиеся двумя хорошо известными лекарствами.
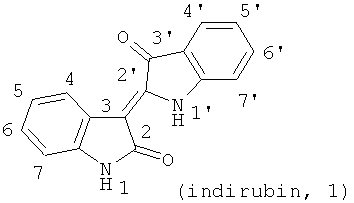
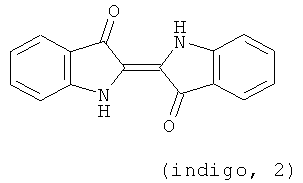
Современные фармацевтические исследования традиционных растительных лекарств показали, что содержащиеся в Индиго натуралис производные дииндола - индирубины (1, индирубин, 2',3-дииндол, пурпурный цвет) особенно эффективны для лечения хронической миелоидной лейкемии (ХМЛ) они характеризуются быстрым действием, применением небольшого количества, меньшим побочным эффектом, низкой ценой и т.д. Впоследствии провели структурные обработки и широкие научные исследования биологической активности соединений дииндола, содержащего в Индиго натуралис [в т.ч. индиго (2), 2,2'-дииндол, синий цвет; изоиндиго (3), 3,3'-дииндол, коричневый цвет], при этом было замечено, что Н-1-метил-изоиндиго имеют лучший лечебный эффект, чем индирубины, и низкую токсичность.

Дальнейшие исследования показали, что фармакодинамический механизм индирубина и его производные препятствуют распространению опухоли путем ингибирования циклинзависимой киназы, т.н. циклин-киназы (ЦЗКы).
Семейство циклинзависимых киназ (ЦЗК) является типичной киназой серин/треонина, и сигнальными молекулами роста клеток, действующими, в основном, на разные фазы клеточного цикла (процесс клеточного цикла разделен на четыре фазы G1, S, G2 и М), что позволяет клеткам расти и пролиферировать (репликации ДНК и сегрегации хромосом), покой (клетки оторвались от цикла разделения, переходит в фазу покоя роста и разделения, известный как G0 этап) или входят в апоптоз. ЦЗК также имеют функции регулирования нервов и тимуса. ЦЗК отличаются от других киназ, они играют роль катализатора и регулятора только при условии связи с соответствующими циклинами и образования сложного комплекса димера. В клетках человека, по крайней мере, существует девять членов семейства ЦЗК (ЦЗК 1~9) и 11 циклинов (А~J). Различные ЦЗК сочетаются с различными циклинами или их подрадикалами, так: ЦЗК1 связывается с циклинами А и В1-В3; ЦЗК2 связывается с циклинами A, D1-D3 и Е; ЦЗК4, ЦЗК5 и ЦЗК6 связываются с циклинами D1-D3; ЦЗК5 также связывается, главным образом, с pk35; ЦЗК7 связывается с циклином Н; ЦЗК6 связывается с циклином К, ассоциированным с D.
По результатам медико-биологических исследований Нобелевской премии в 2001 году - "Отношения клеточной пролиферации и рака", почти все опухолевые клетки имеют различные аномалии клеточного циклина киназы[1-2], раковые клетки продолжительно вступают в фазы S, G2 и М, и неограниченно пролиферируются. Например, более 85% больных раком молочной железы с ненормальным циклином Е / ЦЗК4 / 6[3]. С помощью сдерживания циклинзависимых киназ можно эффективно блокировать пролиферацию клеток (но не убивать клетки), тем самым или содействовать клеточной дифференцировке и созреванию, или способствовать апоптозу; что позволяет достичь эффекта лечения различных опухолей. Есть основания полагать, что клеточный циклин киназы, как ингибитор, представляет собой новый тип противораковых лекарств широкого спектра действия. Кроме того, эти препараты подавляют размножение клеток, но не убивают клетки, они характеризуются высокой селективностью, хорошей эффективностью и низкой токсичностью.
Исследования показали, что циклинзависимые киназы в качестве ингибитора могут эффективно сдерживать рак молочной железы, рак толстой кишки, рак простаты, рак мозга и другие раковые заболевания. Более существенное значение заключается в том, что они препятствуют пролиферации клеток, эти соединения также имеют хорошие воздействия ингибирования клеток гормональнозависимого рака простаты (РС-3, ДУ-145), клеток ряда устойчивых для гормонов и химиотерапии поздних метастатических раков предстательной железы. Таким образом, поиск ингибитора циклинзависимой киназы уже стал новой рациональной стратегией для исследования и разработки новых противораковых лекарств[4-6].
До настоящего времени около 10 видов маломолекулярных по химической структуре ингибиторов и/или контрольных агентов ЦЗК обратили на себя внимание и исследовались, это, в основном, ингибиторы ЦЗК, направленные на область АТР- [7]. Использованные в клиническом лечении индирубин и N-1-метил-изоиндиго являются одним из таких видов[8-9]. Кроме того, еще два соединения вступили в период клинических исследований: UCN-01 и флавопиридол, разработанный Национальным институтом рака (НИР) США[10].

Подводя итог, соединения дииндола типа индирубина являются важным ингибитором ЦЗК, имеют малотоксичных побочных эффектов, но этого типа соединения плохо растворяются в воде и в жире, что мешает их применению в клиническом лечении. В последние годы многие зарубежные научно-исследовательские институты и фармацевтические компании провели широкие структурные изменения соединений такого типа, однако, эффективность соединений дииндола типа индирубина против опухолей по-прежнему далека от удовлетворительной.
Короче говоря, в этой области срочно необходимо разработать новые ингибиторы, имеющие отличные способности ингибирования активности циклинзависимых киназ (ЦЗК).
Раскрытие изобретения
Цель настоящего изобретения является создание производных азаиндола-индола в качестве ингибитора ЦЗК; указанные соединения должны иметь высокую ингибирующую активность, хорошо растворяться в воде и другие преимущества.
Другой целью настоящего изобретения является создание способов изготовления указанных соединений, их фармацевтических композиций и их использование.
Согласно первому изобретению, предлагаются производные азаиндола-индола и их фармацевтически приемлемые соли, по формуле,
 ,
,
где Y представляет группа азаиндола по формуле (Y1) или (Y2);


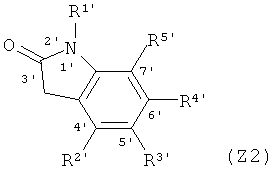
Z представляет группа индола по формуле (Z1) или (Z2);

"=" означает двойную связь, находящуюся в 3-позиции группы азаиндола Y и между 2'- и 3'-позициями группы индола-Z;
в вышеуказанных формулах Y1, Y2, Z1 и Z2, R1 и R1' независимо представляют собой Н или следующие группы, которые могут быть незамещенными или замещенными с 1 до 3 заместителями: С1~С6 алкил, арил, аралкил, ацил, ароил, гликозил или биосил, защищенный ацилом, гликозилом или биосилом; при этом описанные заместители выбраны из: галоген, гидроксил, С1-С3 алкил, нитро или амино;
R2, R3, R4, R2', R3', R4' и R5' независимо представляют собой Н, галоген, гидроксил, сульфгидрил или следующие группы, которые могут быть незамещенными или замещенными с 1 до 3 заместителями: C1~С4 алкил, нитро, амино, амидо, амиде, C1~С4 алкокси, метилтио, фенил, фенокси, арил, аралкил, трифторметил, ацил, ароил, сульфоновую группу, сульфамоил, изоцианат или алкил-изоцианат, при этом описанные заместители выбраны из: галоген, гидроксил, С1-С3 алкил, нитро или амино;
R означает кислород, серу, селен или группу NR6, или NOR6, в которой R6 представляет собой Н, или следующие группы, которые могут быть незамещенными или замещенными с 1 до 3 заместителями: С1~С6-алкил прямой связи или разветвленной связи, арил, аралкил, С3~С6 алициклическая группа, ацил, ароил, сульфонил или фосфорил; при этом описанные заместители выбраны из: галоген, гидроксил, С1-С3 алкил, нитро или амино.
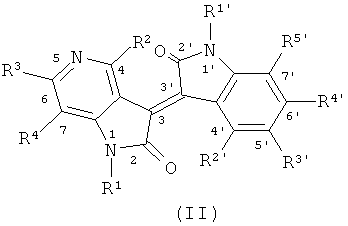
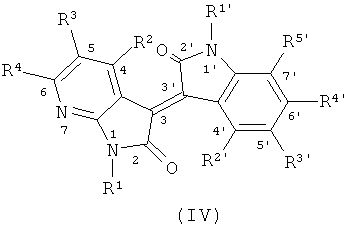
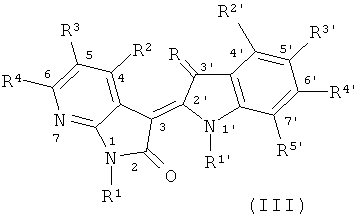
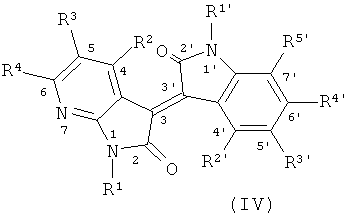
В другом оптимальном примере, описанные соединения показаны в формулах (I), (II), (III) или (IV), в которых формула (I) представляет собой производные 5-азаиндирубина, формула (II) - производные 5-азаизоиндиго, формула (III) - производные 7-азаиндирубина, и формула (IV) - производные 7-азаизоиндиго;


где R, R1, R2, R3, R4, R1', R2', R3', R4' и R5' обозначают, как указано выше.
В другом оптимальном примере, R1 и R1' независимо представляют собой Н, C1~С6 алкил, арил, аралкил, ацил, ароил, гликозил, защищенный алкилом или гликозилом;
R2, R3, R4, R2', R3', R4' и R5' независимо представляют собой Н, галоген, гидроксил, сульфгидрил, С1~С4 алкил, амино, амидо, амиде, C1~С4 алкокси, метилтио, фенил, фенокси, арил, аралкил, трифторметил, ацил, ароил, сульфоновую группу или изоцианат;
вышеупомянутые гликозилы - это арабиноза, ксилоза, рибоза, манноза или глюкоза;
R означает кислород, серу, селен или группу NR6, или NOR6, в которой R6 представляет собой Н, С1~С6-алкил прямой связи или разветвленной связи, арил, аралкил, С3~С6 алициклическая группа, ацил, ароил, сульфонил или фосфорил.
В другом оптимальном примере описанные соединения выбираются из следующих групп: производные 5-азаиндирубина; (таблица 1: соединения 1-59), производные 5-азаизоиндиго (таблица 2: соединения 60-89), производные 7-азаиндирубина (таблица 3: соединения 92-150) и производные 7-азаизоиндиго (таблица 4: соединения 151-180).
В другом оптимальном примере вышеуказанные фармацевтически приемлемые соли включают соли, образованные неорганическими кислотами или органическими кислотами, при этом неорганические кислоты включают следующие: соляная кислота, бромистоводородная кислота, фосфорная кислота, азотная кислота и серная кислота; органические кислоты включают следующие: муравьиная кислота, уксусная кислота, пропионовая кислота, янтарная кислота, нафталиндисульфокислота (1, 5), азиатская кислота, щавелевая кислота, винная кислота, молочная кислота, салициловая кислота, бензойная кислота, бутилкарбоновая кислота, диэтилуксусная кислота, малоновая кислота, янтарная кислота, фумаровая кислота, пимелиновая кислота, гександиоловая кислота, малеиновая кислота, яблочная кислота, аминосульфоновая кислота, фенилпропионовая кислота, гликоновая кислота, аскорбиновая кислота, никотиновая кислота, изоникотиновая кислота, этансульфоновая кислота, паратолуолсульфоновая кислота, лимонная кислота и аминокислота.
Во-вторых, в настоящем изобретении предоставлена Фармацевтическая композиция, содержащая: (а) соединения или их фармацевтически приемлемые соли, по п.1 формулы; (б) фармацевтически приемлемые носители.
В другом оптимальном примере формы выполнения композиции следующие: раствор для инъекций в малом, среднем и большом объеме, порошок для инъекций, эмульсия для инъекций, таблетки, пилюли, капсулы, мази, кремы, пластыри, линимент, порошок, аэрозоли, имплантаты, капли, суппозитории, мази; различные виды нано-лекарственных средств; или липосомы.
В другом оптимальном примере описанные фармацевтические композиции могут быть использованы отдельно в качестве мономерного средства или в комбинации с другими препаратами (например, совместное использование в хирургии, использование в сочетании с одним или несколькими препаратами европейской медицины или травяными лекарствами китайской медицины, или в сочетании с радиоактивным лечением и генной терапией, или совместное использование в сочетании с био-регуляторами).
В-третьих, в настоящем изобретении представлен способ изготовления фармацевтической композиции, предусматривающий следующие шаги: (а) смешивание соединений или их фармацевтически приемлемых солей по п.1 и (б) фармацевтически приемлемых носителей, тем самым формирование фармацевтической композиции.
В-четвертых, в настоящем изобретении предоставлено использование соединений или их фармацевтически приемлемых солей для лечения следующих заболеваний: заболеваний, вызванных отклонением от нормы циклинзависимой киназы, расстройством роста и пролиферации клеток, или резистентностью к инсулину.
В другом оптимальном примере описанные заболевания включают в себя злокачественные раки, псориаз, вирусные заболевания кожи, ВИЧ, дегенерацию и расстройство, и другие заболевания нервной системы, а также диабет 2 типа.
В-пятых, в настоящем изобретении предоставлена композиция, которая содержит соединения или их фармацевтически приемлемые соли по формуле IG в качестве ингибитора циклинзависимой киназы.
В другом оптимальном примере описанные композиции представляют собой композицию фармацевтических продуктов (содержащую носители, приемлемые для фармацевтических продуктов), композицию пищевых продуктов (содержащую носители, приемлемые для пищевых продуктов) и косметическую композицию (содержащую носители, приемлемые для косметики).
В-шестых, в настоящем изобретении предоставлены способы внутреннего и наружного ингибирования циклинзависимой киназы млекопитающих животных, или методы лечения заболеваний, вызванных чрезмерно высокой активностью циклинзависимой киназы, включая меры введения объектам, требующим лечения, соединения по формуле IG, или фармацевтически приемлемые соли, или фармацевтические композиции, содержащие соединения по формуле IG или фармацевтически приемлемые соли.
В другом оптимальном примере описанные заболевания, вызванные слишком высокой активностью циклинзависимой киназы, подразумевают: злокачественные опухоли, псориаз, вирусные заболевания кожи, ВИЧ, нейродегенерацию, расстройство и другие заболевания нервной системы.
Краткое описание чертежей
На Фиг.1 показана общая структурная формула производных 5- и 7-азаиндирубина и производных 5- и 7-азаизоиндиго.
На Фиг.2 показана степень ингибирования производной 5- и 7-азаиндирубина и производной 5- и 7-азаизоиндиго в отношении роста андроген-независимых раковых клеток DU145 простаты человека. С помощью различных концентраций соединений №№107, 108, 112, 115, обрабатывают раковые клетки DU145 логарифмического периода роста простаты человека на 72 часа, измеряют темпы роста клеток методом МТТ.
На Фиг.3 показано ингибирование производной 5- и 7-азаиндирубина и 5- и 7-азаизоиндиго ЦЗК андроген-независимых раковых клеток DU145 простаты человека; с помощью соединений №№124, 126 различной концентрации обрабатывают клетки рака DU145 логарифмического периода роста простаты человека на 24 часа, выводят общий объем белка клеток, измеряют методом "Вестерн-блот" фосфорно-ЦЗК2Thr160, р27 и циклин-D1, беря β-Актин в качестве внутреннего стандарта.
Лучший вариант осуществления изобретения
Изобретателем после широких и углубленных исследований впервые создана одна группа производных азаиндола-индола в качестве ингибиторов ЦЗК; эти соединения образованы азаиндолами в связи с индольными парамолекулами в различных позициях, в них сформированы большие π-сопряженные гетероциклические системы. Испытания показали, что эта группа производных азаиндола-индола может посредством различных механизмов осуществлять биологическую активность, препятствовать клеточному росту и пролиферации, подавить циклинзависимые киназы, индуктировать эндогенные ингибиторы циклин-киназы (ИЦК), а также восстанавливать преобразование сигналов инсулина и другие функции, тем самым лечить различные заболевания, вызванные расстройством роста клеток, в том числе рак, псориаз, вирусные заболевания кожи, ВИЧ и нейродегенерации, расстройство нервной системы, также диабет 2 типа, вызванный резистентностью к инсулину.
Соединения настоящего изобретения
Используемые в настоящей статье термины «соединения настоящего изобретения» или «производные азаиндола-индола» могут использоваться взаимозаменяемо, они обозначают соединения или фармацевтически приемлемые соли, показанные в общей формуле IG.
Конкретно, в настоящем изобретении осуществлено существенное структурное преобразование соединения индирубина и изоиндиго, повышены растворимость и биодоступность, усилен эффект в медикаментозной терапии, снижены дозы препарата, уменьшены негативные реакции медикамента. По сравнению с материнскими ядрами существующих соединений индирубина и изоиндиго, в материнских ядрах соединения настоящего изобретения сформированы большие π-сопряженные гетероциклические системы, тем самым, улучшена растворимость соединений настоящего изобретения в воде.
Одна группа оптимального соединения приведена в общей формуле (I), общей формуле (II), общей формуле (III) и общей формуле (IV), среди них в общей формуле (I) - производные 5-азаиндирубина, в общей формуле (II) - производные 5-азаизоиндиго, в общей формуле (III) - производные 7-азаиндирубина, в общей формуле (IV) - производные 7-азаизоиндиго.
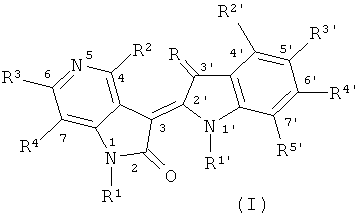
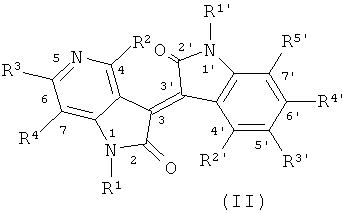
R1 и R1' независимо представляют собой Н или следующие группы, которые могут быть незамещенными или замещенными с 1 до 3 заместителями: С1~С6 алкил, арил, аралкил, ацил, ароил, глюкозил или биосил, защищенный ацилом, гликозилом или биосилом; при этом описанные заместители выбраны из: галоген, гидроксил, С1-С3 алкил, нитро или амино;
R2, R3, R4, R2', R3', R4' и R5' независимо представляют собой Н, галоген, гидроксил, сульфгидрил или следующие группы, которые могут быть незамещенными или замещенными с 1 до 3 заместителями: C1~С4 алкил, нитро, амино, амидо, амиде, C1~С4 алкокси, метилтио, фенил, фенокси, арил, аралкил, трифторметил, ацил, ароил, сульфоновую группу, сульфамоил, изоцианат или алкил-изоцианат, при этом описанные заместители выбраны из: галоген, гидроксил, С1-С3 алкил, нитро или амино;
R означает кислород, серу, селен или группу NR6 или NOR6, в которой R6 представляет собой Н, или следующие группы, которые могут быть незамещенными или замещенными с 1 до 3 заместителями: C1~С6-алкил прямой связи или разветвленной связи, арил, аралкил, С3~С6 алициклическая группа, ацил, ароил, сульфонил или фосфорил; при этом описанные заместители выбраны из: галоген, гидроксил, C1-С3 алкил, нитро или амино.
В вышеупомянутых общих формулах (I), (II), (III) и (IV) имеются более хорошие соединения, в частности: R1 и R1' независимо представляют собой Н, C1~С6 алкил, арил, аралкил, ацил, ароил, гликозил, защищенный алкилом или гликозилом;
R2, R3, R4, R2', R3', R4' и R5' независимо представляют собой Н, галоген, гидроксил, сульфгидрил, C1~С4 алкил, амино, амидо, амиде, C1~С4 алкокси, метилтио, фенил, фенокси, арил, аралкил, трифторметил, ацил, ароил, сульфоновую группу или изоцианат;
вышеупомянутые гликозилы - это арабиноза, ксилоза, рибоза, манноза или глюкоза;
R означает кислород, серу, селен или группу NR6, или NOR6, в которой R6 представляет собой Н, С1~С6-алкил прямой связи или разветвленной связи, арил, аралкил, С3~С6 алициклическая группа, ацил, ароил, сульфонил или фосфорил.
Более предпочтительными видами соединения являются производные азаиндола-индола, подготовленные в примерах реализации, а именно в следующей таблице:
Фармацевтически приемлемые соли
В настоящее изобретение также включены соединения настоящего изобретения и их фармацевтически приемлемые соли, образованные неорганическими или органическими кислотами. Неорганические кислоты включают следующие: соляная кислота, бромистоводородная кислота, фосфорная кислота, азотная кислота и серная кислота. Органические кислоты включают следующие: муравьиная кислота, уксусная кислота, пропионовая кислота, янтарная кислота, нафталиндисульфокислота (1, 5), азиатская кислота, щавелевая кислота, винная кислота, молочная кислота, салициловая кислота, бензойная кислота, бутилкарбоновая кислота, диэтилуксусная кислота, малоновая кислота, янтарная кислота, фумаровая кислота, пимелиновая кислота, гександиоловая кислота, малеиновая кислота, яблочная кислота, аминосульфоновая кислота, фенилпропионовая кислота, гликоновая кислота, аскорбиновая кислота, никотиновая кислота, изоникотиновая кислота, этансульфоновая кислота, паратолуолсульфоновая кислота, лимонная кислота и аминокислота.
В настоящем изобретении улучшены физические и химические свойства описанных солей, увеличена проницаемость клеток, способность доступа в клетки, тем самым повышена эффективность лекарства.
Активность соединений настоящего изобретения
Соединения настоящего изобретения и их соли представляют собой ингибиторы циклинзависимой киназы, и они могут индуктировать эндогенные ингибиторы циклинзависимой киназы, тем самым сдерживают рост и пролиферацию клеток, способствуют апоптозу опухолевых клеток. Соединения настоящего изобретения и их соли путем восстановления преобразования сигналов инсулина повышают чувствительность периферических тканей в использовании инсулина, уменьшают действие резистентности к инсулину. Таким образом, указанные в настоящем изобретении соединения и их соли могут быть использованы в подготовке лекарств для лечения заболеваний, отклонением от нормы циклинзависимой киназы, расстройством роста и пролиферации клеток, или резистентностью к инсулину. В эти заболевания включают рак, псориаз, вирусные кожные заболевания, ВИЧ, а также нейродегенерацию, расстройство нервной системы и сахарный диабет 2 типа.
Для облегчения понимания настоящего изобретения изобретателем предоставлено описание механизма действия соединений настоящего изобретения. Следует понимать, однако, что сфера защиты настоящего изобретения не ограничена описанным механизмом.
В настоящем изобретении, например азаиндирубин, по сути, является продуктом связывания молекулы азаиндола с молекулой индола в 3,2' позиции; например азаизоиндиго, он является продуктом сцепления молекулы азаиндола с молекулой индола в 3,3'-позиции. Замена атомом азота одного из атомов углерода бензольного кольца позволяет получить четыре следующих типа изомеров:
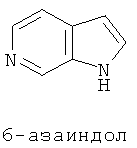
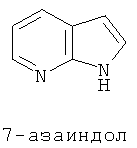
Пурин (конкретный пример ниже) представляет собой один ингибитор ЦЗК из 10 известных типов химической структуры, а также ингибитор ЦЗК[7] в первых исследованиях. С точки зрения структуры, азаиндолы и пурин имеют много сходных черт, они имеют 6-членные ароматические гетероциклы и 5-членные гетероциклы.
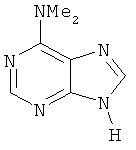

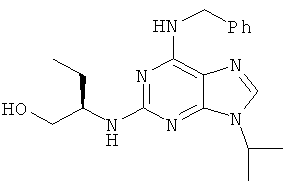
Результаты испытаний настоящего изобретения показали, что соединения настоящего изобретения имеют аналогичную активность ингибитора ЦЗК.
Инсулиннезависимый сахарный диабет (диабет 2 типа) является одним из главных заболеваний в мире, опасных для здоровья человека и приводящих к его гибели, его механизм патогенеза является резистентностью к инсулину. Исследования показали, что производные индирубина оказывают влияние на активность компонентов PI3K (фосфоинозитидного-3-киназа) на сигнальном пути инсулина путем активации (протеинкиназа) и ингибирования (мишень для рапамицина у млекопитающих), восстанавливают преобразование сигналов инсулина, тем самым повышают чувствительность периферических тканей к инсулину, снижают эффект резистентности к инсулину.
Таким образом, производные настоящего изобретения имеют эффект против опухолей, псориаза, вирусных кожных заболеваний, ВИЧ, нейродегенерации, расстройств нервной системы, а также сахарного диабета 2 типа.
Композиции и способы использования
В настоящем изобретении также предоставлены фармацевтические композиции, содержащие соединения настоящего изобретения. Описанные композиции могут быть использованы для ингибирования активности ЦЗК, индуктирования ИЦК, восстановления преобразования сигнала инсулина. Композиция настоящего изобретения может быть фармацевтической комбинацией (содержащей фармацевтически приемлемые носители), комбинацией здравоохранительных продуктов (содержащих фармацевтически приемлемые носители), комбинацией пищевых продуктов (содержащие приемлемые для продуктов питания носители) и косметической комбинацией (содержащей приемлемые для косметики носители).
Предпочтительно, если композиция этого изобретения является фармацевтической композицией, содержащей соединения настоящего изобретения (или их фармацевтически приемлемые соли), а также ряд фармацевтически приемлемых носителей или наполнителей.
Формы выполнения фармацевтической композиции настоящего изобретения не ограничены, они могут быть в любой клинически приемлемой форме. Формы выполнения композиции следующие: раствор для инъекций в малом, среднем и большом объеме, порошок для инъекций, эмульсия для инъекций, таблетки, пилюли, капсулы, мази, кремы, пластыри, линимент, порошок, аэрозоли, имплантаты, капли, суппозитории, мази; различные виды нанолекарственных средств; из соответствующих липосом, в основном, изготовлены вышеупомянутые инъекции. Как правило, различные лекарственные формы должны отвечать методам внесения лекарства.
Предпочтительно, если лекарственные композиции этого изобретения изготовлены в форме инъекции, жидких средств, твердых средств. Такие твердые лекарственные комбинации могут быть изготовлены с помощью обычных способов. Лекарственные комбинации, например, раствор для инъекций, жидкие препараты, твердые препараты должны производиться при условиях стерилизации или соответствующей очистки.
В другом оптимальном примере, в настоящем изобретении предоставлены инъекции соединения (или его приемлемых фармацевтически солей) настоящего изобретения, т.е. эмульсии, суб-микро-эмульсии, нано-эмульсии, подготовленные с использованием поверхностно-активных веществ и/или растворителя и/или масляных компонентов и/или других вспомогательных материалов.
В другом оптимальном примере, в настоящем изобретении предоставлены твердый дисперсионный агент соединения (или его приемлемых фармацевтически солей) настоящего изобретения, т.е. препараты высокой концентрации диспергированы в водорастворимых, неводорастворимых, кишечнорастворимых инертных носителях, сформирована дисперсионная система в виде твердого тела, и путем традиционных способов изготовления: капсулы, таблетки, пилюльки, мази, суппозитории, а также средства для инъекций и т.д. В результате этого соединения не только сохраняют высокую степень дисперсии, но и стабильно хранятся.
Способы введения лекарств
При использовании лекарственной комбинации вводят безопасное и эффективное количество соединения настоящего изобретения в млекопитающих; так называемое, безопасное и эффективное количество, как правило, составляет, по меньшей мере, около 1 мг/сут, и в большинстве случаев не более 10 мг/кг веса тела. А лучшая доза составляет около 1 мг/сут или около 3 мг/кг веса тела. Конечно, конкретная доза определяется также в зависимости от способов введения лекарств и состояния здоровья пациента и других факторов, эти вопросы должны находиться в рамках квалификации опытных врачей.
Соединения (или их фармацевтически приемлемые соли) настоящего изобретения могут быть использованы отдельно в качестве мономерного средства или в комбинации с другими препаратами. В выбранное комбинированное использование входят: совместное использование с хирургической операцией, использование в сочетании с одним или несколькими препаратами европейской медицины, в сочетании с травяными лекарствами китайской медицины, в сочетании с радиоактивным лечением и генной терапией, в сочетании с био-регуляторами.
Способы введения фармацевтических композиций настоящего изобретения не ограничены особыми условиями, в том числе: пероральное применение, инъекция, внутриопухолевое введение, имплантатное введение, внутриполостное введение, анальное введение, кожнопроницаемое введение, внутреннее и внешнее прикладывание.
В оптимальные инъекции входят: внутривенная инъекция, внутримышечная инъекция, подкожная инъекция, внутриполостная инъекция.
Способы изготовления
Соединение настоящего изобретения, описанного в общих формулах IG, может быть подготовлено в соответствии с процессом по следующей формуле, а также принимая во внимание известные синтетические методы в области данной технологии. В целом, в следующем процессе подготовки, все реакции должны проводиться при температуре -10° до температуры рециркуляции, обычно при комнатной температуре (около 25°) до температуры рециркуляции. Лучше, если температура реакции при температуре 5-100°, еще лучше при температурах 20-80°. Время реакции, как правило, без особых ограничений, обычно составляет 1 минута - 24 часов, лучше 1-20 часов. Растворители, как правило, должны быть полярные, такие как вода, мастер-файл препарата, спирты (например, метанол, этанол, изопропанол и т.д.). В синтезе химических соединений могут быть использованы физико-химические методы такие, как 1H ЯМР спектроскопия (1H-NMR), масс-спектрометрия (MS) и элементный анализ для структурной идентификации.
1. Промежуточные и целевые соединения 5-азаиндирубина (общая формула I)
(1) Синтез промежуточного соединения: 1-гидрокарбил-5-азаиндол-2,3-диона (А):
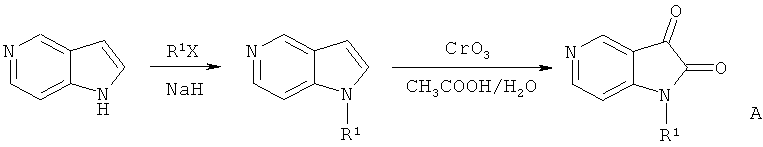
Где R1=СН3, С2Н5, n-C3H7, n-C4H9, Ph-CH2, гликозил, защищенный ацилом, и т.д..
Берут 5-азаиндол с гидрокарбилом в N-1 позиции, а затем в результате реакции окисления с CrO3 и СН3СООН получают продукт (А): 1-гидрокарбил-5-азаиндол-2,3-диона[11].
(2) Синтез промежуточного соединения: производных 1-ацетил-3-гидроксииндола (В)
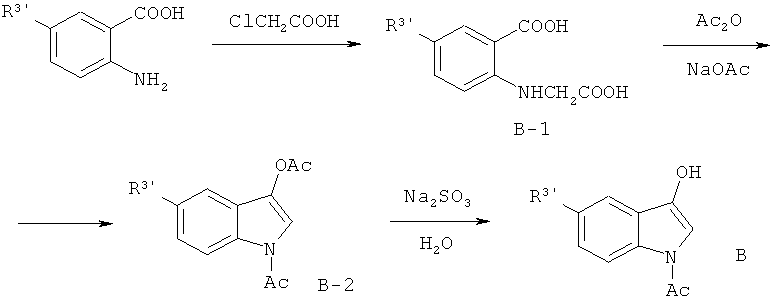
Где R3'=Н, Cl, Br, F, СН3, ОСН3, SCH3, Ph и т.д.
Продукт (В) получают следующим образом: берут замещенные производные 2-аминобензойной кислоты с хлоруксусной кислотой в качестве заместителя, производят ацилирование и замыкание кольца в присутствии ангидрида уксусной кислоты и ацетата натрия, и восстанавливают.
(3) Синтез целевого соединения: производных 5-азаиндирубина (I)
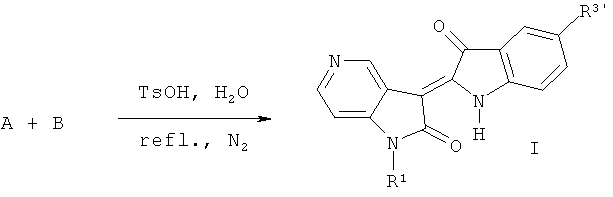
Где R1=СН3, С2Н5, n-C3H7, n-C4H9, Ph-CH2, гликозил, защищенный ацилом и т.д.; R3'=Н, Cl, Br, F, СН3, ОСН3, SCH3 и Ph и т.д.
1-гидрокарбил-5-азаиндол-2,3-диона и 1-ацетил-3-гидроксииндол или 5-галогенсодержащие-1-ацетил-3-гидроксииндол нагревают в кислых условиях, орошая соответственно N2, получают производные 1-гидрокарбил-5-азаиндирубина (I).
(4) Синтез целевого соединения: производных 3'-оксимидо-5-азаиндирубина
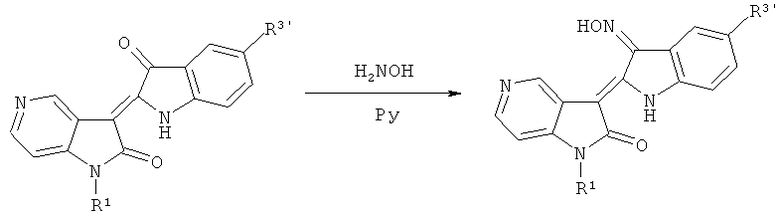
Где R1=СН3, С2Н5, n-C3H7, n-C4H9, Ph-CH2, гликозил, защищенный ацилом и т.д.; R3'=Н, Cl, Br, F, СН3, ОСН3, SCH3, Ph и т.д.
(5) Синтез целевого соединения: 5-азаиндирубин-3'-оксим эфира и т.д.
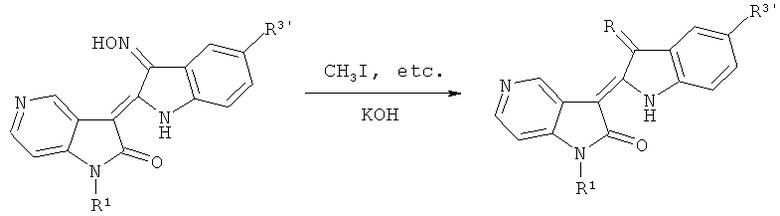
Где R=CH3ON, EtON, R1=СН3, С2Н5, n-C3H7, n-C4H9, Ph-CH2 и т.д., R3'=Н, Cl, Br и F, и так далее.
2. Целевое соединение 5-азаизоиндиго (общая формула II)
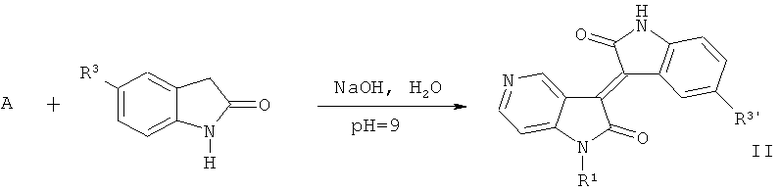
Где R1=СН3, С2Н5, n-C3H7, n-C4H9, Ph-CH2, гликозил, защищенный ацилом и т.д., R3'=Н, Cl, Br, F, ОН и ОСН3 и т.д.
Производные 1-гидрокарбил-5'-замещенные 5-азаизоиндиго (II) получают путем реакции 1-гидрокарбил-5-азаиндол-2,3-диона (А) с 5-замещенные 2-гидроксииндол в щелочных условиях.
3. Промежуточные и целевые соединения 7-азаиндирубина (общая формула III)
(1) Синтез промежуточного соединения: 1-гидрокарбил-7-Азаиндол-2,3-диона (С):
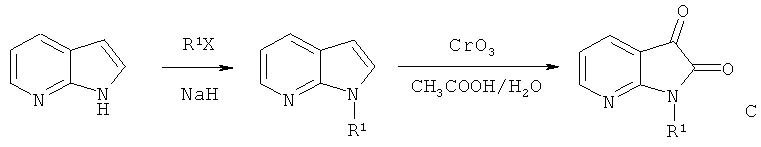
Где R1=СН3, С2Н5, n-C3H7, n-C4H9, Ph-CH2 гликозил, защищенный ацилом, и т.д.
Берут 7-азаиндол с алкильной группой в N-1 позиции, а затем в результате реакции окисления с CrO3 и СН3СООН[11] получают продукт (С): 1-гидрокарбил-7-азаиндол-2,3-диона (III).
(2) Синтез целевого соединения: 7-азаиндирубина (III)
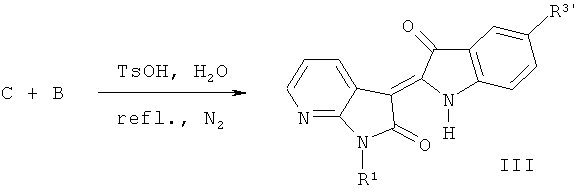
Где R1=СН3, С2Н5, n-C3H7, n-C4H9, Ph-CH2, гликозил, защищенный ацилом, и т.д.; R3'=Н, Cl, Br, F, СН3 и ОСН3, SCH3 и Ph и т.д.
1-гидрокарбил-7-азаиндол-2,3-диона и 1-ацетил-3-гидроксииндол или 5-галогенсодержащие-1-ацетил-3-гидроксииндол нагревают в кислых условиях, орошая соответственно N2, получают производные 1-гидрокарбил-7-азаиндирубина (III).
(3) Синтез целевого соединения: 3'-оксим-7-азаиндирубина.
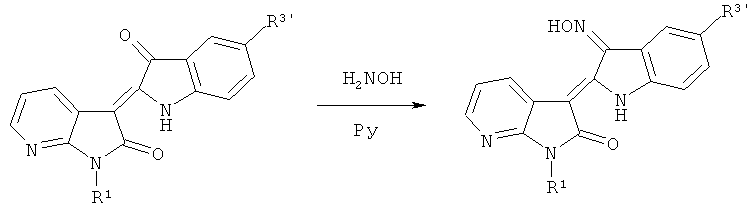
Где R1=СН3, С2Н5, n-C3H7, n-C4H9, Ph-CH2; гликозил, защищенный ацилом, и т.д.; R3'=Н, Cl, Br, F, СН3 и ОСН3, SCH3 и Ph и т.д.
(4) Синтез целевого соединения: 7-азаиндирубин-3'-оксим эфира и т.д.
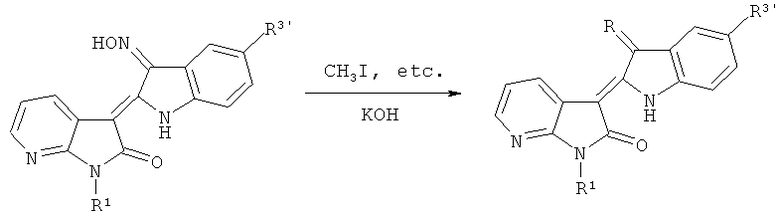
Где R=CH3ON, EtON, R1=СН3, С2Н5, n-C3H7, n-C4H9, Ph-CH2 и т.д., R3'=H, Cl, Br и т.д.
4. Целевое соединение: 7-азаизоиндиго (общая формула IV)
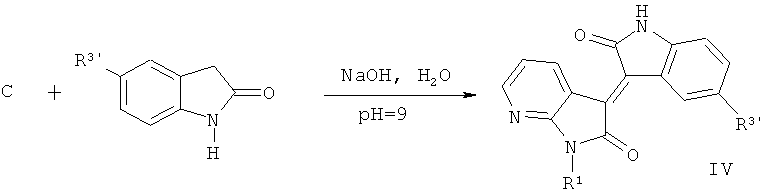
Где R1=СН3, С2Н5, n-C3H7, n-C4H9, Ph-CH2, гликозил, защищенный ацилом, и т.д.; R3'=H, Cl, Br, F, ОН и ОСН3 и т.д.
Производные 1-гидрокарбил-5'-замещенные 7-азаизоиндиго (IV) получают путем реакции 1-гидрокарбил-7-азаиндол-2,3-диона (С) с 5-замещенные 2-гидроксииндол в щелочных условиях.
Основными преимуществами этого изобретения являются:
1) Настоящее изобретение полностью изменило атомную структуру молекулы материнского ядра индирубина и изоиндиго, таким образом, сформировав класс соединений с совсем новой структурой, и улучшило электрические свойства первоначальной молекулы. Учитывая, что пиридин растворим в воде, и бензол почти не растворим в воде, настоящее изобретение улучшило растворимость предложенного соединения в воде, тем самым повысило биодоступность.
2) Соединения настоящего изобретения относится к ингибиторам циклинзависимой киназы, они индуктируют эндогенные ингибиторы циклин-киназы, тем самым препятствуют росту и пролиферации клеток, содействия апоптозу опухолевых клеток.
3) Соединения настоящего изобретения за счет восстановления преобразования сигналов инсулина делают периферийные ткани чувствительными к инсулину и уменьшают роль резистентности к инсулину.
4) Соединения настоящего изобретения обладают улучшенными физико-химическими свойствами и увеличенной проницаемостью клеток, они легко могут войти внутрь клеток, тем самым, повышая эффективность лекарства.
Конкретные примеры реализации
В дальнейшем изобретение поясняется подробным описанием примеров его осуществления. Эти примеры лишь поясняют изобретение, они не ограничивают его сущность приведенными конкретными вариантами. В экспериментальных способах, применяемых в нижеследующих примерах, все процедуры выполняются в стандартных условиях либо согласно указаниям производителя, а все части, проценты и доли указаны по массе, если прямо не указано иное.
Пример 1. Подготовка соединений
Температура плавления производных 5- или 7-азаиндирубина и 5- или 7-изоиндиго, полученных в этом примере реализации, была измерена прибором измерения температуры плавления Mel-TEMP без калибровки. Массовый спектр (МС) был определен с использованием масс-спектрометра HP1100LC/MSD.
Тонкослойные хроматографические пластины (TLC) были изготовлены из кварцевого геля GF254 (Qingdao Haiyang Chemical Co., Ltd) и 0,8% раствора CMC-Na в дистиллированной воде; составляющие перемешивали, активировали в течение 1 часа при температуре 100-110°, затем хранили в сушилке и доводили при ультрафиолетовом свете (длина волны 254 nm и 365 nm). Хроматографические колонки были упакованы гелем кварца (200-300 меш или 100-200 меш) (Qingdao Haiyang Chemical Co., Ltd) с использованием сухого метода. Водородный спектр 1H-ЯМР (1H-NMR) определяли с помощью ядерного магниторезонансного аппарата типа Bruck AV-300, внутренний стандарт TMS. Элементный анализ проводился с использованием аппаратуры Elementar ario EL III.
Реактивы были коммерчески доступными химически чистыми и аналитически чистыми продуктами сорта. Если иначе не обозначено, реактивы использовались непосредственно без дополнительных обработок.
Пример 1-1. Подготовка промежуточных соединений
(1) 1-метил-5-азаиндол-2,3-дион
1-метил-5 азаиндол (2.0 г, 15 ммол) растворяют в 70 мл АсОН, заранее суспензируют 3.2 г CrO3 в 20 мл воды, наливают в вышесказанный раствор уксусную кислоту, проводят реакцию в течение 0,5 часа при комнатной температуре, смесь разбавляют водой, экстрагируют 3 раза хлороформом; органические вещества растворяются в хлороформе, органические фазы интегрируются и промываются, после обезвоживания состав обогащается. Получают оранжевые промежуточные соединения 1-метил-5-азаиндол-2,3-диона (1.5 г, выход 62%; т.п. 140-142°С).
2) 2-(N-карбоксиметиламин)-5-хлоробензойная кислота
2-амино-5-хлорбензойная кислота (2.0 г, 11.6 ммоль) растворяли в 15 мл раствора 2 моль/л Na2CO3; хлоруксусную кислоту (0.69 г, 7.3 ммоль) растворяли в 7.5 мл раствора 2 моль/л Na2CO3, последней раствор медленно каплями добавляли в предыдущий раствор. Затем смесь перемешивали при 80°С в течение 20 часов, охлаждали до комнатной температуры, добавляли 50 мл эфира и 8 мл 2 моль/л соляной кислоты. Отделяли органические фазы и сушили с MgSO4. После концентрации получали светло-коричневое твердое тело. После очистки колоночной хроматографией на силикагеле (этилацетат/метанол, по объему, 1/1) получили белое твердое тело (2-(N-карбоксиметиламино)-5-хлорбензойная кислота) (1.58 г, выход 59%; т.п.: 182-183°С).
(3) 1-ацетил-5-хлор-3-ацетоксииндол
2-(N-карбоксиметиламино)-5-хлорбензойная кислота (1.20 г, 5.2 ммоль) и безводный ацетат натрия (0.6 г, 7.3 ммоль) растворяли в 8 мл ангидрида уксусной кислоты. После перемешивания в течение 5 часов при 60°С реакционную смесь охлаждали до комнатной температуры, и ацетат натрия отфильтровывали. После концентрации в концентрированный фильтрат добавляли 100 мл этилового ацетата для растворения, добавляли еще 100 мл воды и 20 мл насыщенного бикарбоната натрия, разделяли органические слои. Органические вещества в водном слое экстрагировали этилацетатом (50 мл × 2). Комбинированные органические фазы промывали насыщенным бикарбонатом натрия (100 мл × 2), высушивали и выпаривали, чтобы получить белое тело (1-ацетил-5-хлор-3-ацетоксииндол) (0,84 г, выход: 64%).
(4) 1-ацетил-5-хлор-3-карбоксииндол
1-ацетил-5-хлор-3-ацетоксииндол (1.0 г, 3.97 ммоль) смешивали в 20 мл воды с сульфитом натрия (1.0 г, 7.94 ммоль), смесь нагревали при 80°С в течение 3 часов, охлаждали до комнатной температуры, экстрагировали этилацетатом (100 мл × 2). Комбинированные органические фазы высушивали. После концентрации получили белое иглообразное твердое тело (1-ацетил-5-хлор-3-карбоксииндол) (0.84 г, выход 66%; т.п.: 186-188°С).
(5) 1-метил-7-азаиндол 2,3-дион
1-метил-7-азаиндол (2 г, 15 ммоль) растворяли в 70 мл уксусной кислоты, суспензировали 3.2 г CrO3 в 20 мл воды. Реакционную смесь перемешивали в течение 0,5 ч при комнатной температуре и разбавляли водой. Смесь экстрагировали с трихлорметаном три раза. Комбинированные органические фазы промывали водой, высушивали и выпаривали. Был получено оранжевое промежуточное соединение (1-метил-7-azaindole-2,3-дион) (1,73 г, выход: 71,3%; т.п.: 162-163°С).
(6) 2-(N-карбоксиметиламино)-5-бромобензойная кислота
2-амино-5-бромобензойная кислота (2 г, 9 ммоль) растворяли в 15 мл раствора 2 моль/л Na2CO3; хлоруксусную кислоту (0.69 г, 7.3 ммоль) растворяли в 7.5 мл раствора 2 моль/л Na2CO3.
Затем, после перемешивания в течение 20 часов при температуре 80°С, реакционную смесь охлаждали до комнатной температуры. 50 мл эфира и 8 мл 2 моль соляной кислоты добавляли в смесь. Органическую фазу отделяли и сушили с MgSO4. После испарения было получено светло-коричневое твердое тело.
После очистки колоночной хроматографией на силикагеле (этилацетат/метанол, объем на объем, 1/1) получили белое твердое тело (2-(N-карбоксиметиламин)-5-бромбензойная кислота) (1,55 г, выход: 60%,; т.п.: 178-180°С).
(7) 1-ацетил-5-бром-3-ацетоксииндол
2-(N-карбоксиметиламино)-5-бромобензойная кислота (0.84 г, 3.4 ммоль) и безводный ацетат натрия (0.6 г, 7.3 ммоль) растворяли в 8 мл ангидрида уксусной кислоты. После перемешивания в течение 5 часов при 60°С реакционную смесь охлаждали до комнатной температуры, и ацетат натрия отфильтровывали. После концентрации в концентрированный фильтрат добавляли 100 мл этилового ацетата для растворения, добавляли еще 100 мл воды и 20 мл насыщенного бикарбоната натрия, разделяли органические слои. Органические вещества в водном слое экстрагировали этилацетатом (50 мл × 2). Комбинированные органические фазы промывали насыщенным бикарбонатом натрия (100 мл × 2), высушивали и выпаривали, чтобы получить белое тело (1-ацетил-5-бромо-3-ацетоксииндол) (1,3 г, выход: 25,4%).
(8) 1-ацетил-5-бром-3-карбоксииндол
1-ацетил-5-бром-3-ацетоксииндол (1.0 г, 3.38 ммоль) и сульфит натрия (1.0 г, 7.94 ммоль) смешивали в 20 мл воды. После нагревания при 80°С в течение 3 часов реакционную смесь охлаждали до комнатной температуры, экстрагировали этилацетатом (50 мл × 2). Комбинированные органические фазы высушивали. После концентрации получили белое иглообразное твердое тело (1-ацетил-5-бром-3-карбоксииндол) (0.7 г, выход 82%; т.п.: 180-182°С).
Пример 1-2. Синтез целевых соединений
(1) 1-метил-5-азаиндирубин (2)
К 1-метил-5-азаиндол-2,3-дион (0.2 г, 1.23 ммоль) добавляли 1-ацетил-3-гидроксииндол (0.21 г, 1.2 ммоль), 20 мл воды и 0.02 г паратолуолсульфоновой кислоты. Реакционную смесь перемешивали и кипятили в атмосфере азота в течение 1 часа, чтобы получить пурпурно-красный раствор. После остывания смесь экстрагировали трихлорметаном, промывали водой и выпаривали. Получили фиолетовое твердое тело. После очистки колоночной хроматографией на силикагеле (трихлорметан/петролейный эфир, по объему, 3/1) его рекристаллизировали этилацетатом, получили красные иглообразные кристаллы 1-метил-5-азаиндирубин (2) (0.15 г, выход 44%; т.п.: 114-116°С).
ESI-MS: 278.1 [М+Н]+, C16H11N3O2 (277.2);
1Н NMR (AV-300, CDCl3, ppm) δ: 3.48 (s, 3Н, -СН3), 7.08 (m, 1H, 5'-Н), 7.09 (m, 1Н, 6'-Н), 7.16 (dd, 1Н, J=7.6 Hz, 4'-Н), 7.76 (d, J=7.6 Hz, 1H, 7'-Н), 8.10 (s, 1H, 4-Н), 8.26 (dd, J=5.5 Hz; 1H. 6-H), 9.10 (dd, J=5.5 Hz, 1H, 7-H), 10.4 (bs, 1H, N-H);
Аналогично для: C16H11N3O2: С, 69.31 H, 3.97 N, 15.16;
Получено: С, 69.15 Н, 4.09 N, 15.29.
(2) 1-бензил-5'-хлор-5-азаиндирубин (19)
Использовали тот же метод, как (1), 1-бензил-5-азаиндол-2,3-дион и 1-ацетил-5-хлор-3-гидроксииндол в количестве, как в (1), и 0.02 г паратолуол-сульфоновой кислоты растворяли в 20 мл воды. Реакционную смесь перемешивали и кипятили в атмосфере азота в течение 1 часа, чтобы получить пурпурно-красный раствор. После охлаждения смесь экстрагировали трихлорметаном, промывали водой и выпаривали. Получали пурпурное твердое тело. После очистки колоночной хроматографией на силикагеле (трихлорметан/петролейный эфир, по объему, 3/1) его рекристаллизировали этилацетатом, получили красные иглообразные кристаллы 1-бензил-5'-хлор-5-азаиндирубин (19) (0.18 г, выход 39%; т.п.: 110-112°С).
ESI-MS: 389 [М+Н]+, C22H14ClN3O2 (387.9);
1Н NMR (AV-300, CDCl3, ppm) δ: 5.21 (s, 2Н, N-CH2), 10.44 (s, 1H, N-H), 6.91~9.02 (m, 11H, Ar-Hs);
Аналогично для C22H14ClN3O2: С, 68.13 H, 3.64 N, 10.83;
Получено: С, 68.42 Н, 3.59 N,10.89.
(3) 1-бутил-5-азаиндирубин-3'-оксим (40)
Растворяли 1-бутил-5-азаиндирубин (0.4 г, 1.25 ммоль, получили методом (1)) в 12 мл метанола, добавив 6 мл безводного пиридина и 0.15 г гидроксиламина гидрохлорида (2.2 ммоль).
Реакционную смесь перемешивали и кипятили в течение 1 часа, охлаждали и концентрировали, чтобы удалить большую часть растворителя. Остаток сбрасывали в 100 мл измельченного льда, интенсивно перемешивая, затем фильтровали, в результате получили оранжевое твердое тело. После очистки колоночной хроматографией на силикагеле (трихлорметан/петролейный эфир, по объему, 3/1) получили оранжевый кристаллический порошок 1-бутил-5'-5-азаиндирубин-3'-оксим (40) (0.32 г, выход 90%; т.п.: 250-252°С).
ESI-MS: 335.1 [М+Н]+, C19H18N4O2 (334.3);
1Н NMR (AV-300, D6-DMSO, ppm) δ: 0.91 (t, 3Н, -СН3), 1.3l (m, 2Н, -СН2), 2.28 (m, 2Н, -СН2), 3.28 (m, 2Н, N-CH2), 7.10~8.81 (m, 7Н, Ar-Hs), 11.71 (s, 1H, N-H), 13.70 (s, 1H, N-OH);
Аналогично для C19H18N4O2: C, 68.25 H, 5.43 N,16.76;
Получено: С, 68.33 H, 5.56 N, 16.66.
(4) 1-бутил-5-азаиндирубин-3'-монооксим О-метила (53)
1-бутил-5-азаиндирубин-3'-оксим (1.5 г, 4.5 ммоль) добавляли к 50 мл 5% гидроксида калия в безводном этаноле, растворенного при теплой температуре, и фильтровали. В фильтрат добавляли каплями 5 мл СН31 при постоянном перемешивании. При реакции выделялось тепло, был получен темно-красный осадок. После перемешивания в течение 0,5 часа осадок фильтровали и промывали водой до получения нейтрального раствора, сушили и получили темно-красный сырой продукт, который рекристаллизировали ацетоном, получили темно-красные кристаллы 1-бутил-5-азаиндирубин-3'-монооксим О-метила (53) (1.20 г, выход 77%; т.п.: 209-211°С).
ESI-MS: 349.1 [М+Н]+, C20H20N4O (348.2);
1H-NMR (AV-300, D6-DMSO, ppm) δ: 0.98 (t, 3Н, -СН3), 1.46 (m, 2Н, -СН2), 2.08 (m, 2Н, -СН2), 3.86 (m, 2Н, N-CH2), 4.16 (s, 3Н, O-СН3), 7.10~9.19 (m, 7Н, Ar-Hs), 10.86 (bs, 1H, N-H);
Аналогично для C20H20N4O2: С, 68.95 Н, 5.79 N, 16.08
Получено: С, 68.81 Н, 5.62 N, 15.85.
(5) 1-изопропил-5-азаизоиндиго (73)
1-Изопропил-5 азаиндол-2,3-дион (0.4 г, 2.1 ммоль) и 2-гидроксииндол (0.28 г, 2.1 ммоль) добавляли в 10 мл этанола, корректировали рН до 9 с помощью 1 моль/л NaOH, проводили реакцию при 70°С в течение двух часов и получили коричневое тело. После охлаждения фильтровали, промывали водой и этанолом, проводили сушку в вакууме, в результате получили красно-коричневое твердое тело 1-изопропил-5-азаизоиндиго (73) (0.44 г, выход 66%; т.п.: 128~130°С).
ESI-MS: 306.1 [М+Н]+, 304.2 [М-Н]-, C18H15N3O2 (305.3);
1Н NMR (AV-300, D6-DMSO, ppm) δ: 1.52 (d, 6Н, -СН(СН3)2), 4.81 (m, 1Н, -СН(СН3)2), 6.86-9.32 (m, 7Н, Ar-Hs), 10.96 (bs, 1H, N-H);
Аналогично для C18H15N3O2: С, 70.81 Н, 4.95 N, 13.76
Получено: С, 70.62 Н, 5.10 N, 13.58.
(6) 1-метил-7-азаиндирубин (93)
К 1-метил-7-азаиндол 2,3-дион (0.2 г, 1.23 ммоль) добавляли 1-ацетил-3-гидроксииндол (0.21 г, 1.2 mmol), 20 мл воды и 0.02 г паратолуолсульфоновой кислоты. Реакционную смесь рециркулировали с азотом при перемешивании в течение 1 часа, получили пурпурно-красный раствор. После охлаждения раствор экстрагировали хлороформом, промывали водой, концентрировали, получили пурпурное тело. После очистки колоночной хроматографией на силикагеле (трихлорметан/петролейный эфир, по объему, 3/1) рекристаллизировали этилацетатом, получили красные иглообразные кристаллы 1-метил-7-азаиндирубин (93) (0.14 г, выход 41,1%; т.п.: 116~118°с).
ESI-MS: 278.1 [М+Н]+, C16H11N3O2 (277.2);
1Н NMR (AV-300, CDCl3, ppm) δ: 3.59 (s, 3Н, -СН3), 7.08 (m, 1H, 5'-Н), 7.09 (m, 1Н, 6'-Н), 7.16 (dd, 1H, J=7.6 Hz, 4-Н), 7.58 (m, 1H, 5-Н), 7.76 (d, J=7.6 Hz, 1Н, 7'-Н), 8.21 (dd, J=5.5 Hz, 1H, 4-H), 9.13 (dd, J=5.5 Hz, 1H, 6-H), 10.4 (bs, 1H, N-H);
Аналогично для C16H11N3O2: С, 69.31 H, 3.97 N, 15.16;
Получено: С, 69.05 Н, 4.18 N, 15.34.
(7) 1-бензил-5'-бром-7-азаиндирубин (109)
Тем же методом как в (6), к 1-бензил-7-азаиндол 2,3-дион и 1-ацетил-5-бром-3-гидроксииндол в количестве, как в (6), добавляли 20 мл воды и 0.02 г паратолуолсульфоновой кислоты. Реакционную смесь рециркулировали с азотом при перемешивании в течение 1 часа, получили пурпурно-красный раствор. После охлаждения раствор экстрагировали хлороформом, промывали водой, концентрировали, получили пурпурное тело. После очистки колоночной хроматографией на силикагеле (трихлорметан/петролейный эфир, по объему, 3/1) рекристаллизировали этилацетатом, получили красные иглообразные кристаллы 1-бензил-5'-бром-7-азаиндирубин (109) (0.14 г, выход 27%; т.п.: 112~114°С).
ESI-MS: 433 [М+Н]+, C22H14BrN3O2 (432.2);
1Н NMR (AV-300, CDCl3, ppm) δ: 5.24 (s, 2Н, N-CH2), 10.44 (s, 1H, N-H), 6.91~9.0 (m, 11Н, Ar-Hs);
Аналогично для C22H14BrN3O2: С, 61.13 H, 3.26 N, 9.72;
Получено: С, 60.72 Н, 3.57 N, 9.38.
(8) 1-бутил-7-азаиндирубин-3'-оксим (131)
1-бутил-7-азаиндирубин (0.4 г, 1.25 ммоль, получен методом (6)) растворяли в 12 мл метанола, добавив 6 мл безводного пиридина и 0.15 г гидроксиламина гидрохлорида (2.2 ммоль). Реакционную смесь нагревали и рециркулировали в течение 1 часа, охлаждали, концентрировали для удаления большей части растворителя, остатки сбрасывали в 100 мл измельченного льда, интенсивно перемешивая, фильтровали, в результате получали оранжевое тело. После очистки колоночной хроматографией на силикагеле (трихлорметан/петролейный эфир, по объему, 3/1) рекристаллизировали этилацетатом, получили оранжевое твердое тело 1-бутил-7-азаиндирубин-3'-оксим (131) (0.31 г, выход 87,7%; т.п.: 254~256°С).
ESI-MS: 335.1 [М+Н]+, C19H18N4O2 (334.3);
1H NMR (AV-300, D6-DMSO, ppm) δ: 0.92 (t, 3Н, -СН3), 1.31 (m, 2Н, -СН2), 2.28 (m, 2Н, -СН2), 3.32 (m, 2Н, N-CH2), 7.03~8.81 (m, 7Н, Ar-Hs), 11.7 (s, 1H, N-H), 13.7 (s, 1H, N-OH);
Аналогично для C19H18N4O2: С, 68.25 Н, 5.43 N, 16.76;
Получено: С, 68.09 Н, 5.60 N, 16.58.
(9) 1-бутил-7-азаиндирубин-3'-монооксим О-метила эфира (144)
1-бутил-7-азаиндирубин-3'-оксим (1.5 г, 4.5 ммоль) добавляли к 50 мл 5% гидроксида калия в безводном этаноле, растворенного при теплой температуре, и фильтровали. В фильтрат добавляли каплями 5 мл СН31 при постоянном перемешивании. При реакции выделялось тепло, был получен темно-красный осадок. После перемешивания в течение 0,5 часа осадок фильтровали и промывали водой до получения нейтрального раствора, сушили и получили темно-красный сырой продукт, который рекристаллизировали ацетоном, получили темно-красные кристаллы 1-бутил-7-азаиндирубин-3'-монооксим О-метила (144) (1.26 г, выход 80,5%; т.п.: 212-214°С).
ESI-MS: 349.1 [М+Н]+, C20H20N4O (348.2);
1H-NMR (AV-300, D6-DMSO, ppm) δ: 0.98 (t, 3H, -CH3), 1.46 (m, 2Н, -СН2), 2.08 (m, 2Н, -СН2), 3.88 (m, 2Н, N-CH2), 4.16 (s, 3Н, O-CH3), 7.06~9.19 (m, 7Н, Ar-Hs), 10.86 (bs, 1Н, N-H);
Аналогично для C20H20N4O2: С, 68.95 Н, 5.79 N, 16.08
Получено: С, 68.79 Н, 5.59 N, 15.88.
(10) 1-изопропил-7-азаизоиндиго (164)
1-изопропил-7 азаиндол-2,3-дион (0.4 г, 2.1 ммоль) и 2-гидроксииндол (0.28 г, 2.1 ммоль) добавляли в 10 мл этанола, корректировали рН до 9 с помощью 1 моль/л NaOH, проводили реакцию при 70°С в течение двух часов и получили коричневое тело. После охлаждения фильтровали, промывали водой и этанолом, проводили сушку в вакууме, в результате получили красно-коричневое твердое тело 1-изопропил-7-азаизоиндиго (164) (0.42 г, выход 63,2%; т.п.: 132~134°С).
ESI-MS: 306.1 [М+Н]+, 304.2 [М-Н]-, C18H15N3O2 (305.3);
1Н NMR (AV-300, D6-DMSO, ppm) δ: 1.51 (d, 6Н, -СН(СН3)2), 4.78 (m, 1H, -СН(СН3)2), 6.86-9.3 (m, 7Н, Ar-Hs), 10.99 (bs, 1H, N-H);
Аналогично для C18H15N3O2: С, 70.81 Н, 4.95 N, 13.76
Получено: С, 70.53 Н, 5.04 N, 13.52.
В соответствии с упомянутым выше методом подготовили 59 соединений 5-азаиндирубина (1), кроме того, синтезировали производные 5-азаиндирубина типа 2, 19, 40 и 53. Их структуры представлены в таблице 1, все структуры этих новых соединений подтверждены ИК-спектрометрией, ультрафиолетовой спектрометрией (УФ/VIS), масс-спектрометрией (ESI-MS), ЯМР (1H-ЯМР) и элементным анализом.

В формуле 1, R2~R4, R1', R2', R4' и R5' соответственно представляет собой Н, остальное см. таблицу 1:
В соответствии с методом подготовки 1-Изопропил-5-азаизоиндиго (73) всего синтезировано 30 соединений 5-азаизоиндиго (73), их структура представлена в таблице 2. Структуры всех этих новых соединений подтверждены ИК-спектрометрией, ультрафиолетовой спектрометрией (УФ/VIS), масс-спектрометрией (ESI-MS), ЯМР (1H-ЯМР) и элементным анализом.
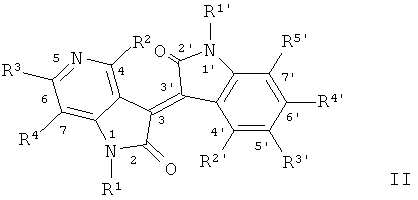
В формуле II, R2~R4, R2', R4' и R5', соответственно составляют Н, остальные см.Таблицу 2:
В соответствии с указанными выше методами подготовки производных 7-азаиндирубина 93, 109, 131 и 144 всего синтезировано 59 соединений типа 7-азаиндирубина (III), их структуры представлены в таблице 3. Структуры всех этих новых соединений подтверждены ИК-спектрометрией, ультрафиолетовой спектрометрией (УФ/VIS), масс-спектрометрией (ESI-MS), ЯМР (1H-ЯМР) и элементным анализом.
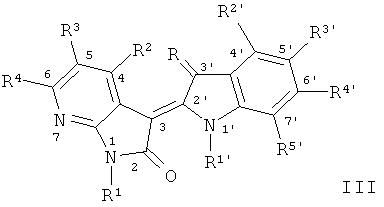
В формуле III, R2, R4, R1', R2', R4' и R5' соответственно составляет Н, остальные см. Таблицу 3:
Структура соединений 7-азаиндирубина (III)
В соответствии с указанными выше методами подготовки 1-изопропил-7-азаизоидиго (164) всего синтезировано 30 соединений 7-азаизоиндиго (IV), их структуры представлены в таблице 4. Структуры всех этих новых соединений подтверждены по ИК-спектрометрией, ультрафиолетовой спектрометрией (УФ/VIS), масс-спектрометрией (ESI-MS), ЯМР (1Н-ЯМР) и элементным анализом.
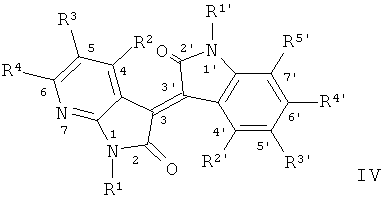
В формуле IV R2, R4, R2', R4' и R5' соответственно составляют Н, остальные см.в таблице 4.
Пример 2. Тест антиопухолевой активности (1)
1. Материалы и приборы
(i) Клеточные линии: андроген-независимые раковые клетки простаты DU145, приобретенные из США, American Type Culture Collection.
(ii) реагенты RPM1 Medium 1640 (США компания GIBCOBRL), сыворотка теленка (Ханчжоуская био-инженерная компании Сыцзицин), МТТ (компания Sigma), HEPES (ООО Шанхайская биотехнологическая компания Личжу-дунфэн), L-глютамина (импортировано из Японии), диметилсульфоксид (ДМСО, аналитический реактив).
Тестируемые образцы: выбраны 20 из новых соединений 5-азаиндирубин и 5-азаизоиндиго (собственного производства), 38 из новых соединений 7-азаиндирубин и 7-азаизоиндиго (собственного производства).
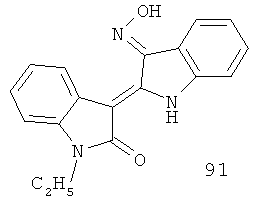
Контрольные вещества: 1-этил-индирубин (90), 1-этил-индирубин-3'-оксим (91), (собственного производства, структуры идентифицированы, обработаны ретинойной кислотой).
(iii) Подготовка реагентов
A. Среда клеточной культуры: 10.4 г из 1640 средних порошков, 2.1 г бикарбоната натрия, 0.3 г глутамина, 5.95 г HEPES, 100000 единиц пенициллина и 100000 единиц стрептомицина были добавлены к 1000 мл двойной дистиллированной воды. Смесь стерилизовалась фильтрацией с использованием микропористого мембранного фильтра, после расфасовки хранили при температуре -20°С. До использования к среде была добавлена инактивированная сыворотка теленка.
B. Сыворотка теленка: была инактивирована на водяной бане при 56°С в течение 30 минут, после упаковки хранилась при -20°С.
C. МТТ: растворено в 5 мг/мл PBS, хранили в защищенном от света месте при 4°С, действительно в течение двух недель.
D. PBS: 8.00 г хлорида натрия, 0.20 г хлорида калия, 3.4 г двенадцатигидратного динатриевого фосфата водорода, 0.20 г дикалиевого фосфата водорода растворяли на водяной бане при 37°С в двойной дистиллированной воде, фасовали в 1000 мл тару, после фасовки ранили при 4°С.
Е. Изготовлены растворы в диметилсульфоксиде 58 тестируемых образцов, вещества (90 и 91) и обработанные ретинойной кислотой и хранили при -20°С.
(iv) Основные приборы:
CO2 инкубатор (типа GB16, немецкой компании Heraeus): Стол для очистки (SW-CJ-1F, ООО Сучжоуская воздушной технологии Аньтай); горизонтальная центрифуга (типа LXJ-II, Шанхайский Медико-инструментальный завод №3); иммуноферментный анализатор (BIO RAD Модель 550, Соединенные Штаты Америки); инвертированный биологический микроскоп (XSZ-D2, Чунцинская оптико-инструментльная фабрика); устройство для быстрого перемешивания (типа SK-1, ООО Чанчжоуская электроприборная компания Гохуа); термостатный электрический бак (типа DK-8D, Шанхайский завод медицинского термостатного оборудования); проточный цитометр (FACSCalibur, американской компании B-D); платформенный вибратор (типа 752-А, Шанхайская фабрика медицинских аналитических инструментов); электронные весы (типа BS110S, немецкой фирмы Sartorius).
2. Методы.
(i) Культивирование клеток
DU145 клетки привиты в среде RPM11640 с 10% эмбриональной сывороткой телят, поставляют в инкубаторе при 5% CO2, 37°С, передаются через каждые 2-3 дней, во время эксперимента берут клетки в логарифмической фазе.
(ii) Экспериментальные группы
В экспериментах берут клетки в логарифмической фазе, создают клеточную суспензию, после равномерного смешивания определяют жизнеспособность клеток трипановым синим окрашиванием, количество жизнеспособных клеток должно быть более 98%. Клеточная суспензия разделена поровну на несколько групп: 1: пустые контрольные группы (клеточная суспензия); 2: экспериментальные группы (клеточная суспензия + лекарство).
(iii) Методом МТТ определяют значения IC50 (половина количества ингибирования)
Подготавливают резервный раствор с помощью диметилсульфоксида концентрацией 20 ммоль (эксперимент проводится в течение 4 часов). Эксперименты проводятся в стерильных условиях, в рабочем растворе на основе среды RPMI1640 с 10% эмбриональной сывороткой телят, подготавливают рабочий раствор тестированного лекарства концентрацией 80 мкМоль, концентрация лекарства постепенно увеличивается в 2 раза (1,25-20 мкМоль).
Отбирали клетки DU145 в логарифмической фазе, центрифугировали, подсчитывали и подготовили суспензию плотностью 2,5×104/мл из среды RPMI1460, содержащей 10%-ную сыворотку телят. Суспензией делали прививку в пластину с 96 отверстиями, в каждом отверстии 5000 клеток/200 мл, культивировали в течение 24 часов при 37°С, в 5% среде CO2. Затем по вышеуказанной концентрации прививали в 6 групп (в том числе в контрольную группу), в каждой группе создавали 8 донных отверстий. После выведения в течение 72 часов, жизнеспособность ячеек была измерена МТТ колориметрией. Значение поглощения (А) было измерено с длиной волны обнаружения в 540 нм, длина волны ссылки в 630 нм. Рассчитают в соответствии со следующей формулой степень ингибирования:
Коэффициент ингибирования (I) был вычислен следующим уравнением, где Т - величина спектральной поглощательной способности экспериментальных групп, а С - величина спектральной поглощательной способности чистой группы контроля:
I=(1-Т/С)×100%.
В соответствии с кривой концентрации ингибирования рассчитано уравнение регрессии, чтобы получить 50%-ную и 90%-ную концентрацию ингибирования (IC50 и IC90 µМ). Результаты представлены в таблице 5 и таблице 6 и Фиг.2.

3. Обсуждение
(i) Из результатов испытания МТТ легко увидеть, что подавляющее большинство производных 5-или 7-азаиндирубина и 5- или 7-азаизоиндиготина имеет сильную антиопухолевую активность, ингибирующие эффекты против роста опухолевых клеток намного сильнее, чем при обработке ретинойной кислотой в качестве индуцированного дифференцированного агента клеток. Еще более важным является то, что подавляющее большинство производных 5- или 7-азаиндирубина и 5- или 7-азаизоиндиготина имеют хорошие ингибирующие эффекты для андроген-независимых раковых клеток простаты DU-145, с которыми в нынешней клинической практике до сих пор бороться было нечем;
(ii) многие из производных 5- или 7-азаиндирубина и 5 - или 7-азаизоиндиго имеют значение IC50, близкое или менее IC50 контрольных веществ (90 и 91), а композиция 91 - это известный ингибитор CDKs[13]. Структуры соединений 38, 129 очень похожи на 91, разница между ними заключается только в типах атомов на 5 или 7 позициях. Также предложено, что новые синтезированные композиции настоящего изобретения могут иметь аналогичный механизм ингибирования роста опухолевых клеток;
(iii) 3'-оксимированные и 5'-галоидированные или 7-азаиндирубинированные производные демонстрируют более значительное ингибирующее влияние на рост опухолевых клеток, особенно соединения 25, 30, 116, 121 и 124, а соединения 30, 121 и 124 имеют более хорошую безопасность.
Пример 3. Тест антиопухолевой активности (2)
1. Опухолевые клетки: раковые клетки печени человека 7701 QGY, HepG-2, раковые клетки легких человека А549, клетки хронического миелолейкоза человека К562, клетки лейкоза человека СЕМ, клетки меланомы мыши KIll.
2. Используя методы, описанные в примере 2, была определена биологическая активность части новых синтезированных производных 5- или 7-азаиндирубина и 5- или 7-азаизоиндиго (20 соединений) на активность ингибирования роста различных опухолевых клеток. Результаты тестирования при 50% ингибирующей концентрации (IC50, мкм) представлены в таблице 7.
Результаты вышеуказанных тестирований на антиопухолевую активность свидетельствуют о том, что производные 5- или 7-азаиндирубина и 5- или 7-азаизоиндиго имеют многообразные биоактивности ингибирования роста опухолевых клеток, тем самым открыло новое направление по исследованию антиопухолевых соединений индирубина, обеспечена материальная база для разработки новых антиопухолевых препаратов.
Пример 4. Ингибирующие эффекты CDKs
Реактивы: Источники тестируемых соединений те же, что и в Примере 2. Если иначе не обозначено, другие химические реактивы были куплены в Американской компании Sigma. Полиакриламидный гель для электрофореза белков (SDS-PAGE), SDS, электрофорезный буфер, буферный раствор для электропередачи белков, нитроцеллюлозные мембраны и др. приобретены в американской компании American Bio-Rad Life Science Company. Набор для теста Вестерн-блот (Western bolt) и пленки приобрели в американской компании American GE Company. Phospho-CDK2Thr160 antibody, p27 антитела ингибитора эндогенных циклинкиназ, антитела CyclinD1 и β-Actin закуплены соответственно в компаниях American Cell Signaling Inc., DAKO and Santa Cruz Biochemical Technology Company.
Опухолевые клетки и их культивирование: то же, что и в примере 2.
Методом Вестерн-блот тестировали Phospho-CDK2, р27, и циклин D1:
Проводили обработку андроген-независимых раковых клеток простаты человека DU145, в соответствии с концентрацией, как показано на Фиг.3, с помощью производных 5- или 7-азаиндирубина и 5- или 7-азаизоиндиго №124 и №126 в течение 24 часов. Собирали клетки, промывали, в соответствии с методами, описанными в литературах[14], экстрагировали белки, брали необходимое количество. Брали 50 мг белка, проводили SDS-электрофорез в полиакриламидном геле. После электрофореза белки передавали в нитроцеллюлозные мембраны, используя специфическое антитело Phospho-CDK2, антитело р27 ингибитора эндогенных циклинкиназ и CyclinDl антитело, проводили тест Вестерн-блот, антитело β-Actin выбирали в качестве внутреннего стандарта, на ECI пленке записывали результаты тестирования.
Результаты и обсуждение.
По данным тестирования соединения индирудина имеют функции ингибирования CDKs раковых клеток. Вышеуказанные примеры 2 и 3 показали, что производные 5- или 7-азаиндирубина и 5- или 7-азаизоиндиго настоящего изобретения имеют сильную функцию ингибирования клеточного роста. Чтобы глубже разъяснить то, что эти соединения путем регулирования активности циклинкиназы выполняют функции ингибирования роста клеток, применяли метод Вестерн-блот, используя специфическое антитело фосфорилированого белка Cdc2 и другие антитела важных белков р27 и cyclin D1p27, регулирующих клеточный цикл; наблюдали влияние активиторов представительных соединений №124 и №126 настоящего изобретения на выражение белка р27 и cyclin D1.
Как показано на Фиг.3, после обработки с соединениями №124 и №126 в течение 24 часов активности CDK2 (фосфорилирование) в андроген-независимых раковых клетках простаты человека DU145 значительно сократились, В то же время выражение белка cyclin D1 также значительно снизилось. Напротив, в тех же экспериментальных условиях, влияние эндогенных ингибиторов циклин-киназы на выражение р27 было значительно увеличено. В результате изменений этих сигнальных белков рост клеток ингибируется. Из них производные 5- или 7-азаиндирубина и 5- или 7-азаизоиндиго индуктируют выражение р27, может быть, путем активации пути рецептора AhR[15].
Пример 5. Исследования твердых диспергентов
Пример 5-1.
Процесс технологии: полиэтиленгликоль 400 при 50°С расплавляли, добавив соединения 110 в примерах 1-2, перемешивали до равномерности, при перемешивании в то же время проводили холодную вулканизацию со льдом, сушили в эксикаторе в течение 24 часов, затем в соответствии с обычными методами изготавливали пилюли или капсулы.
Пример 5-2.
Процесс технологии: брали соединение 18 в примерах 1-2, растворяли умеренным щелочным раствором этанола, добавив полиэтиленгликоль 6000, плавили при 50°С; перемешивая до равномерности, добавляли вспомогательные материалы (лактоза/микрокристаллическая целлюлоза = 10/1), перемешивая до состояния полусухого порошка, размещали в 60°С духовке в течение 24 часов, в соответствии с обычными методами изготавливали таблетки или капсулы.
Пример 5-3.
Процесс технологии: брали соединение 18 в примерах 1-2, вливали в умеренный щелочной этанольный раствор, перемешивая до полного растворения, добавив поливинилпирролидон К-25, перемешивали до растворения, добавляли вспомогательные материалы (лактоза/микрокристаллическая целлюлоза = 10/1), перемешивая до равномерности, помещали в 60°С духовке в течение 24 часов, в соответствии с обычными методами изготовляли таблетки или капсулы.
Пример 5-4.
Процесс технологии: Брали соединение 31 в примерах 1-2, растворяли в хлороформе подходящего количества, добавляли полиоксиэтилен (35) касторовое масло, перемешивая для растворения, добавляли вспомогательные материалы (лактоза/микрокристаллическая целлюлоза = 1/4), перемешивая до равномерности. Смесь нагревали в 80°С водяной бане, чтобы улетучился трихлорметан, сушили в 60°С духовке в течение 24 часов, в соответствии с обычными методами изготавливали таблетки или капсулы.
Пример 5-5.
Процесс технологии: брали соединение 116 в примерах 1-2, растворяли в соответствующем щелочном растворе этанола, добавляли полоксамер 188, перемешивали до растворения, еще добавляли вспомогательные материалы (микрокристаллическая целлюлоза), перемешивали до равномерности, размещали в 60°С сушилке в течение 24 часов, в соответствии с обычными методами изготавливали таблетки или капсулы.
Пример 5-6.
Процесс технологии: брали соединение 18 в примерах 1-2, растворяли в умеренном щелочном растворе этанола, добавляли полоксамер 188 и полиэтиленгликоль 6000, перемешивали до растворения, добавляли вспомогательные материалы (лактоза/микрокристаллическая целлюлоза = 10/1), перемешивая до равномерности, помещали в 60°С духовке в течение 24 часов, в соответствии с обычными методами изготавливали таблетки или капсулы.
Пример 5-7.
Процесс технологии: соединение 119 в примерах 1-2 распускали в умеренном щелочном этаноле, добавив полоксамер 188, полиэтиленгликоль 6000 и поливинилпирролидон К-25, перемешивая до растворения, добавляли вспомогательные материалы, помещали в 60°С духовку в течение 24 часов, затем в соответствии с обычными методами изготавливали таблетки или капсулы.
Пример 5-8.
Процесс: Брали соединение 18 в примерах 1-2, помещали в умеренное количество щелочного этанольного раствора, добавляли полоксамер 188, полиэтиленгликоль 6000 и поливинилпирролидон К-25, перемешивая до растворения, добавляли вспомогательные материалы (лактоза/микрокристаллическая целлюлоза = 10/1), перемешивая до равномерности, и размещали в 60°С духовку в течение 24 часов, в соответствии с обычными методами изготавливали таблетки или капсулы.
Пример 5-9.
Процесс технологии: соединение 29 в примерах 1-2 растворяли в полиоксиэтилен (40) гидрогенизированное касторовое масло/раствор этанола, добавив полиэтиленгликоль 4000, нагревали до 50°С, перемешивая до растворения, выделяли растворители, отверждали ледяными мешками, помещали в 60°С духовку на 24 часа, в соответствии с обычными методами изготавливали гранулы или капсулы.
Пример 5-10.
Процесс технологии: брали соединение 121 в примерах 1-2, растворяли его полиоксиэтилен (40) гидрогенизированное касторовое масло/раствор этанола, добавляли поливинилпирролидон К-25, перемешивая до растворения, добавляли вспомогательные материалы (лактоза/микрокристаллическая целлюлоза = 7/3), перемешивая до равномерности, замораживали и сушили при температуре -50°С в течение 24 часов, в соответствии с обычными методами изготавливали таблетки или гранулы.
Пример 5-11.
Процесс технологии: растворяли соответствующим объемом этанольного раствора полиоксиэтилен (40) касторовое масло и додецилсульфат натрия, после полного растворения добавляли соединения 18 в примерах 1-2, перемешивая до полного растворения, добавляли поливинилпирролидон К-25, смешивая до растворения, добавляли вспомогательные материалы (лактоза/ микрокристаллическая целлюлоза = 10/1), перемешивая до равномерности, и помещали в 60°С духовку в течение 24 часов, в соответствии с обычными методами изготавливали пилюльки.
Пример 5-12.
Процесс технологии: Брали соединение 18 в примерах 1-2, растворяли его полиоксиэтилен (40) гидрогенизированное касторовое масло/раствор этанола, добавляли поливинилпирролидона К17, перемешивая до растворения, добавляли вспомогательные материалы (лактоза/микрокристаллическая целлюлоза = 10/1), перемешивая до равномерности, помещали в 60°С духовку в течение 24 часов, в соответствии с обычными методами изготавливали таблетки или капсулы.
Пример 5-13.
Процесс технологии: брали соединение 34 в примерах 1-2, растворяли его в сукцинат полиэтиленгликоля витамина Е/раствор этанола, добавив поливинилпирролидон К-90, перемешивая до растворения, добавляли вспомогательные материалы (лактоза/микрокристаллическая целлюлоза = 5:5), перемешивая до равномерности, и помещали в 60°С духовку в течение 24 часов, в соответствии с обычными методами изготавливали таблетки или капсулы.
Пример 6. Исследования на растворимость части твердых дисперсионных препаратов.
Приборы: RCZ-5А Интеллектуальный детектор процента растворимости, производства Tianjin University Precision Instruments Factory; UV1900 Ультрафиолетовый спектрофотометр, производства Shanghai Yayan Electronic Science And Technology Co., Ltd.
Метод испытания на растворимость: тестировали в соответствии с методом испытания на растворимость (третий способ) «Chinese Pharmacopeia)), 2005. Согласно этому методу, тестировали при скорости вращения 100 об /мин, температуре 37±0,5°С, в 100 мл водного раствора 1% додецилсульфата натрия ультразвуковой дегазацией. Пробы отбирали через 45 минут, сразу фильтровали через 0.8 Мкм пленку, фильтрат разбавляли. Светопоглощение измеряли с помощью UV1900, рассчитывали процент растворимости.
Результаты и обсуждение:
В вышеупомянутых примерах был исследован процент растворимости части твердых дисперсионных препаратов. Результаты представлены в таблице 8.
Из вышеприведенной таблицы видно, что твердые дисперсионные препараты, которые сделаны от соединений настоящего изобретения, преодолели недостатки данных соединений, заключающиеся в плохой гидрофильности и сложности для создания соответствующих препаратов. В результате значительного повышения растворимости соединений могут быть получены полезные препараты. Ввиду использования твердых дисперсионных препаратов с совместными носителями, под взаимодействием свыше одного носителя, соединения настоящего изобретения имеют более оптимальную растворимость, чем с одним носителем, растворимость соответствующих твердых дисперсионных препаратов будет значительно лучше.
Пример 7. Исследования на инъекциях
Пример 7-1.
Процесс технологии: все следующие процессы выполняются в стерильном помещении.
Масляная фаза: брали соединение 18 в примерах 1-2, вводили в диметил сульфоксид, нагревали при низкой температуре для растворения, добавив триглицериды средней цепочки. Смесь нагревали при низкой температуре, перемешивали, смешивали равномерно.
Водная фаза: глицерин, полоксамер 188, добавив воды для инъекций, нагревали при низкой температуре при интенсивном перемешивании, медленно вливали в масляную фазу и продолжали перемешивание 3 минуты. Испаряли в роторном вакуумном испарителе при низкой температуре для удаления органических растворителей.
Полученную эмульсию стерилизовали, фильтровали, в ламинарном состоянии вливали в лиофилизированный стерильный флакон, замораживали, сушили, закупоривали, закрывали крышками из алюминия для уплотнения.
Пример 7-2.
Процесс технологии: все следующие процессы выполняются в стерильном помещении.
Масляная фаза: брали соединение 25 в примерах 1-2, нагревали при низкой температуре, растворяли в соответствующем количестве диметила сульфоксида, добавив моноглицериды олеиновой кислоты, нагревали до растворения, а затем присоединяли к фосфолипидам, нагревали при низкой температуре, смешивали равномерно.
Водная фаза: глицерин, полоксамер 188, добавив воды для инъекций, нагревали при интенсивном перемешивании, постепенно добавляли в масляную фазу и продолжали перемешивание 3 минуты. Испаряли в роторном вакуумном испарителе при низкой температуре для удаления органических растворителей.
Полученную эмульсию стерилизовали, фильтровали, в ламинарном состоянии вливали в лиофилизированный стерильный флакон, замораживали, сушили, закупоривали, закрывали крышками из алюминия для уплотнения.
Пример 7-3.
Процесс технологии: все следующие процессы выполняются в стерильном помещении.
Масляная фаза: брали соединение 34 в примерах 1-2, нагревали и растворяли при низкой температуре в смеси тетрагидрофурана и метиленхлорида.
Водная фаза: водный раствор альбумина человека вливали в масляную фазу, проводили ультразвуковую обработку в течение 1 минуты, испаряли в вакуумном вращающемся испарителе при низкой температуре для удаления органических растворителей.
Полученную эмульсию стерилизовали, фильтровали, в ламинарном состоянии вливали в лиофилизированный стерильный флакон, замораживали, сушили, закупоривали, закрывали крышками из алюминия для уплотнения.
Пример 7-4.
Процесс технологии: все следующие процессы выполняются в стерильном помещении.
Масляная фаза: брали соединение 122 в примерах 1-2, растворяли в щелочном растворе этанола, добавив лецитин и полиэтиленгликоль 400, перемешивали до растворения.
Водная фаза: гидроксипропил-β-циклодекстрин растворяли в растворе 70% этанола, вливали в масляную фазу, добавив соответствующее количество воды для инъекций, размешивали до равномерности, испаряли в роторном вакуумном испарителе при низкой температуре для удаления органических растворителей.
Полученную эмульсию стерилизовали, фильтровали, в ламинарном состоянии вливали в лиофилизированный стерильный флакон, замораживали, сушили, закупоривали, закрывали крышками из алюминия для уплотнения.
Пример 7-5.
Процесс технологии: все следующие процессы выполняются в стерильном помещении.
Брали соединение 110 в примерах 1-2, растворяли в щелочном растворе этанола, добавив полоксамер 188, нагревали, размешивая до растворения, охлаждали, добавляли 0,9% раствор натрия хлорида, смешанный с соответствующим количеством этанола, добавляли лецитин, смешивая равномерно, добавляли гидроксипропил-β-циклодекстрин, смешивая равномерно, проводили ультразвуковую обработку в течение 1 минуты, испаряли в роторном вакуумном испарителе при низкой температуре для удаления органических растворителей.
Полученную эмульсию стерилизовали, фильтровали, в ламинарном состоянии вливали в лиофилизированный стерильный флакон, замораживали, сушили, закупоривали, закрывали крышками из алюминия для уплотнения.
Пример 7-6.
Процесс технологии: все следующие процессы выполняются в стерильном помещении.
Брали соединение 30 в примерах 1-2, растворяли в щелочном растворе этанола, добавив соевый лецитин и полоксамер 188 до растворения, а затем добавляли гидроксипропил-β-циклодекстрин, воду для инъекций и растворяли подходящим количеством этанола; испаряли в роторном вакуумном испарителе при низкой температуре для удаления органических растворителей.
Полученную эмульсию стерилизовали, фильтровали, в ламинарном состоянии вливали в лиофилизированный стерильный флакон, замораживали, сушили, закупоривали, закрывали крышками из алюминия для уплотнения.
Пример 7-7.
Процесс технологии: все следующие процессы выполняются в стерильном помещении.
Масляная фаза: брали соединение 18 в примерах 1-2, растворяли в щелочном растворе этанола, добавив лецитин и полиэтиленгликоль монолаурат, перемешивая до растворения.
Водная фаза: гидроксипропил-β-циклодекстрин добавляли в 70% раствор этанола, вливали в масляную фазу, перешивая; испаряли в роторном вакуумном испарителе при низкой температуре для удаления органических растворителей.
Полученную эмульсию стерилизовали, фильтровали, в ламинарном состоянии вливали в лиофилизированный стерильный флакон, замораживали, сушили, закупоривали, закрывали крышками из алюминия для уплотнения.
Пример 7-8.
Процесс технологии: все следующие процессы выполняются в стерильном помещении.
Масляная фаза: брали соединение 19 в примерах 1-2, растворяли в щелочном этаноле, добавляя лецитин и полиоксиэтилен (40) касторовое масло, перемешивая до растворения.
Водная фаза: гидроксипропил-β-циклодекстрин растворяли в 70% растворе этанола, вливали в масляную фазу, добавив подходящее количество воды для инъекций, перешивали равномерно, испаряли в роторном вакуумном испарителе при низкой температуре для удаления органических растворителей.
Полученную эмульсию стерилизовали, фильтровали, в ламинарном состоянии вливали в лиофилизированный стерильный флакон, замораживали, сушили, закупоривали, закрывали крышками из алюминия для уплотнения.
Пример 7-9.
Процесс технологии: все следующие процессы выполняются в стерильном помещении.
Масляная фаза: брали соединение 18 в примерах 1-2, растворяли в смешанном растворе тетрагидрофурана и трихлорэтилена при ультразвуковой обработке.
Водная фаза: альбумин человека растворяли в соответствующем количестве раствора 0.1 моль/л соляной кислоты, при интенсивном перемешивании вливали в масляную фазу, продолжали интенсивное перемешивание в течение 5 минут до достижения равномерности. Смесь была гомогенизирована (600 бар) и охлаждалась водой. Операция была повторена в течение шести раз. Испаряли в роторном вакуумном испарителе при низкой температуре для удаления органических растворителей.
Полученную эмульсию стерилизовали, фильтровали, в ламинарном состоянии вливали в лиофилизированный стерильный флакон, замораживали, сушили, закупоривали, закрывали крышками из алюминия для уплотнения.
Пример 7-10.
Процесс технологии: все следующие процессы выполняются в стерильном помещении.
Масляная фаза: брали соединение 120 в примерах 1-2, после растворения этанолом добавляли сукцинат полиэтиленгликоля витамина Е, среднецепочный триглицерид и полоксамер 188, нагревали до растворения.
Водная фаза: 8 мл 2,25% раствора глицерина наливали в масляную фазу, перемешивали, добавив раствор лецитина, подвергали ультразвуковой обработке в течение 10 минут, испаряли в роторном вакуумном испарителе при низкой температуре для удаления органических растворителей.
Полученную эмульсию стерилизовали, фильтровали, в ламинарном состоянии вливали в лиофилизированный стерильный флакон, замораживали, сушили, закупоривали, закрывали крышками из алюминия для уплотнения.
Пример 8. Испытание на растворимость
Соединения 129 и 91 очень похожи по структуре, разницу между ними только в типе атомов в 7-позиции (см. формулы ниже), использовали их в качестве растворителя для проверки изменения растворимости 7-азаиндирубина по сравнению с индирубином.
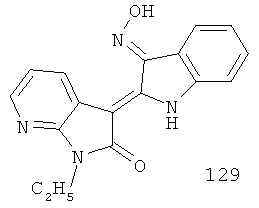
Метод испытания: при комнатной температуре (20°С) брали, соответственно, соединения 129 и 91 по 5 мг, добавляли к 2 мл соответствующего растворителя, после перемешивания растворяли, результаты представлены в таблице 9.
По результатам, представленным в таблице выше, можно увидеть, что у производных 7-азаиндирубина (129) водорастворимость значительно возросла, а растворимость в жире уменьшилась. В общем, водорастворимость и жирорастворимость индирубина не хорошие, но у обработанного индирубина жирорастворимость увеличилась, а водорастворимость снизилась, соединение 91 именно такое же. Это изменение растворимости отражает необходимость исследования производных 7-азаиндирубина. Исследования состояния поглощения лекарственных средств в организме и выбора лекарственных форм будут приносить благоприятные стороны.
Упомянутые в этом изобретении все ссылки на литературу цитируются в этой заявке в качестве пособий, как и все статьи были отдельно цитированы в качестве справок. Кроме того, следует понимать, что после чтения вышеизложенного содержания в настоящем изобретении технический персонал в этой области может сделать различного рода изменения или модификации, эти эквивалентные формы также попадают в сферу ограничения формулы, приложенной к настоящей заявке.
Ссылки
[1] Lundberg AS, Weinberg RA., Control of the cell cycle and apoptosis, Eur J Cancer, 1999, 35: 531-539.
[2] Sherr CJ. Canaer cell cycles, Science, 1996, 274: 1672-1677.
[3] Keyomarsi K., Pardee AB., Redundant cyclin overexpression and amplification in breast cancer cells, Proc Natl Acad Sci USA, 1993, 90: 1112-1116.
[4] Gray N, Detivand L, Meijer L et al., ATP-site directed inhibitors of cyclin-dependent kinases, Curr Med Chem, 1999, 6: 859-875.
[5] Malumbres M et al., Targeting cell cycle kinases for cancer therapy, Curr Med Chem, 2007: 14(9): 969-85.
[6] Rudolph J., Inhibiting transient protein-protein interactions: lessons from the Cdc25 protein tyrosine phosphatases, Nat Rev Cancer, 2007, 7(3): 202-11.
[7] Huwe A., Mazitschek R., Giannis A., Small molecules as inhibitors of cyclin-dependent kinases, Angew Chem Int Ed, 2003, 42: 2100-2138.
[8] Цзи Сюцзюань, У Кэмэй, Хуан Лян и т.д., Индиго Натуралис, см.: под редакцией Института медикоментов при Китайской академии медицинских наук. Современные исследования китайской травяной медицины (том 1), первое издание, Пекин: Совместное издательство Пекинского медицинского университета и Китайского медицинского университета «Сехэ», 1995: 227-257.
[9] Nam S., Buettner R, Turkson J. et al., Indirubin derivatives inhibit Stat3 signaling and induce apoptosis in human cancer cells, PNAS, 2005, 102(17): 5998-6003.
[10] Roy KK., Sausville EA. Early development of cyclin dependent kinase modulators, Curr Pharm Design, 2001, 7(16): 1669-1687.
[11] Doklady, Akad Nauk SSSR, 1958, 118: 302-305.
[12] Christoph M.S., Pascale H., John M. et al., Synthesis and evaluation of analogues of 10H-indolo[3,2-b]-quinoline as G-quadruplex stabilising ligands and potential inhibitors of the enzyme telomerase, Org. Biomol. Chem., 2004, 2: 981-988.
[13] Яо Цичжэн, Ван Лунгуй, Ван Чжаохой и др. Метод подготовки и медицинские применения производных N (1)-алкил-3'-оксим индирубин(I), китайский изобретательный патент CN 1329376C, 2007-8-1.
[14] Wang LG, Ossowski L., Ferrari AC, Androgen receptor level controlled by a suppressor complex lost in an androgen-independent prostate cancer cell line, Oncogene, 2004, 23: 5175-5184.
[15] Marie K., Marc В., Maryse L. et al., Independent actions on cyclin-dependent kinases and aryl hydrocarbon receptor mediate the antiproliferative effects of indirubins, Oncogene, 2004, 23: 4400-4412.
| название | год | авторы | номер документа |
|---|---|---|---|
| ФАРМАЦЕВТИЧЕСКИ АКТИВНЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ Axl | 2011 |
|
RU2573834C2 |
| СПОСОБ ПОЛУЧЕНИЯ НЕФТЕПОЛИМЕРНЫХ СМОЛ | 2008 |
|
RU2375380C1 |
| АРОМАТИЧЕСКИЕ СОЕДИНЕНИЯ КАК ПРОТИВОВОСПАЛИТЕЛЬНЫЕ, ИММУНОМОДУЛИРУЮЩИЕ И АНТИПРОЛИФЕРАТИВНЫЕ СРЕДСТВА | 2003 |
|
RU2386625C2 |
| ЗАМЕЩЕННЫЕ 4(5)-ИМИДАЗОЛЫ ИЛИ ИХ НЕТОКСИЧНЫЕ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ С КИСЛОТАМИ | 1991 |
|
RU2036193C1 |
| ПРОТИВООПУХОЛЕВЫЕ ПЕПТИДЫ | 1996 |
|
RU2182911C2 |
| ПРОИЗВОДНЫЕ ПИРИДИНА И ГЕРБИЦИДНОЕ СРЕДСТВО НА ИХ ОСНОВЕ | 2002 |
|
RU2304141C2 |
| 3-КАРБАЛКОКСИАМИНО-5-( ω -АМИНОАЦИЛ)-5Н-ДИБЕНЗ [B, F]АЗЕПИНЫ, ОБЛАДАЮЩИЕ АНТИАРИТМИЧЕСКИМ ДЕЙСТВИЕМ | 1989 |
|
RU2092480C1 |
| ПРОИЗВОДНЫЕ ПИРИДИНОНА В КАЧЕСТВЕ ИНГИБИТОРОВ ТРАНСГЛУТАМИНАЗЫ ТКАНЕЙ | 2013 |
|
RU2652987C2 |
| 1,3-ДИЗАМЕЩЕННЫЕ 4-МЕТИЛ-1Н-ПИРРОЛ-2-КАРБОКСАМИДЫ И ИХ ПРИМЕНЕНИЕ ДЛЯ ИЗГОТОВЛЕНИЯ ЛЕКАРСТВЕННЫХ СРЕДСТВ | 2007 |
|
RU2463294C2 |
| СПОСОБ ПОЛУЧЕНИЯ НЕФТЕПОЛИМЕРНЫХ СМОЛ | 2002 |
|
RU2218358C1 |


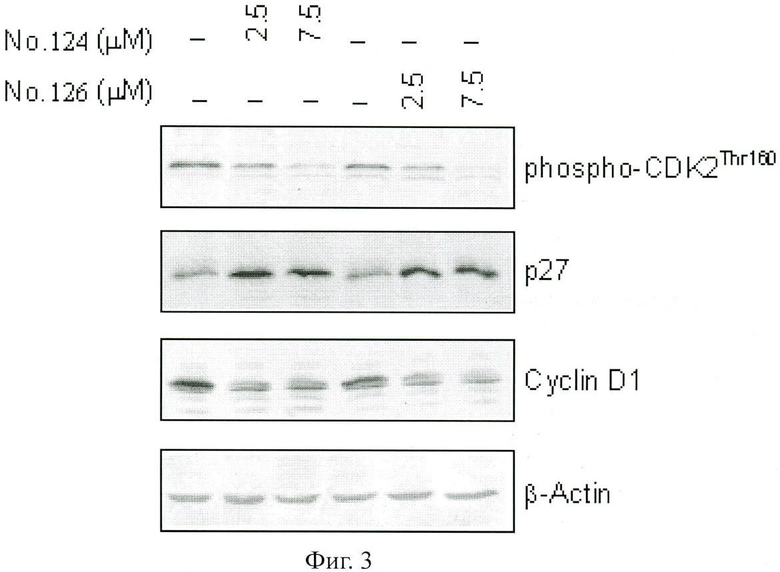
Изобретение относится к производным азаиндола-индола и их фармацевтически приемлемым солям общей формулы
где Y означает группу азаиндола формулы (Y1), (Y2);
Z означает группу индола формулы (Z1) или (Z2);
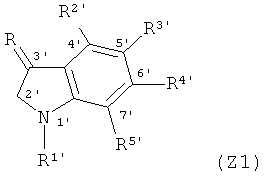
где значения «=», R, R1, R1', R2, R3, R4, R2', R3', R4', R5' приведены в пункте 1 формулы. Соединения ингибируют циклинзависимую киназу, что позволяет использовать их в фармацевтической композиции. 2 н. и 4 з.п. ф-лы, 3 ил., 9 табл., 8 пр.
1. Производные азаиндола-индола и их фармацевтически приемлемые соли по формуле

где Y означает группу азаиндола формулы (Y1), (Y2)
Z означает группу индола формулы (Z1) или (Z2)
"=" означает двойную связь, находящуюся в 3-позиции группы азаиндола Y и между 2'- и 3'-позициями группы индола-Z;
в вышеуказанных формулах Y1, Y2, Z1 и Z2, R1 и R1 независимо означают Н или следующие группы, которые могут быть незамещенными или замещенными с 1 до 3 заместителями: С1-С6 алкил, арил, аралкил, ацил, ароил, глюкозил или биосил, защищенный ацилом, глюкозилом или биосилом; при этом описанные заместители выбраны из группы: галоген, гидроксил, C1-С3 алкил, нитро или амино;
R2, R3, R4, R2', R3', R4', R5' независимо означают Н, галоген, гидроксил, сульфгидрил или следующие группы, которые могут быть незамещенными или замещенными с 1 до 3 заместителями: C1-C4 алкил, нитро, амино, амидо, амиде, C1-C4 алкокси, метилтио, фенил, фенокси, арил, аралкил, трифторметил, ацил, ароил, сульфоновую группу, сульфамоил, изоцианат или алкил-изоцианат, при этом описанные заместители выбраны из группы: галоген, гидроксил, C1-С3 алкил, нитро или амино;
R означает кислород, серу, селен или группу NR6 или NOR6, в которой R6 означает Н, или следующие группы, которые могут быть незамещенными или замещенными с 1 до 3 заместителями: С1-С6-алкил прямой связи или разветвленной связью, арил, аралкил, С3-С6 алициклическая группа, ацил, ароил, сульфонил или фосфорил; при этом описанные заместители выбраны из группы: галоген, гидроксил, С1-С3 алкил, нитро или амино.
2. Соединения или их фармацевтически приемлемые соли по п.1, как они представлены в формулах (I), (II), (III) или (IV), в которых формула (I) представляет собой производые 5-азаиндирубина, формула (II) - производные 5-азаизоиндиго, формула (III) - производные 7-азаиндирубина, и формула (IV) - производные 7-азаизоиндиго
где R, R1, R2, R3, R4, R1', R2', R3', R4' и R5' означают, как указано выше.
3. Соединения или их фармацевтически приемлемые соли по п.1, в которых R1 и R1' независимо означают Н, C1-C6 алкил, арил, аралкил, ацил, ароил, гликозил, защищенный алкилом или глюкозилом;
R2, R3, R4, R2', R3', R4' и R5' независимо означают Н, галоген, гидроксил, сульфгидрил, C1-C4 алкил, амино, амидо, амиде, C1-C4 алкокси, метилтио, фенил, фенокси, арил, аралкил, трифторметил, ацил, ароил, сульфоновую группу или изоцианат;
вышеупомянутые гликозилы - это арабиноза, ксилоза, рибоза, манноза или глюкоза;
R означает кислород, серу, селен, или группу NR6 или NOR6, в которой R6 представляет собой Н, С1-С6-алкил прямой связи или разветвленной связи, арил, аралкил, С3-С6 алициклическая группа, ацил, ароил, сульфонил или фосфорил.
4. Соединения или их фармацевтически приемлемые соли по п.1, в которых указанные соединения выбраны из следующих: производные 5-азаиндирубин (I) (таблица 1), производные 5-азаизоиндиго (II) (таблица 2), производные 7-азаиндирубин (III) (таблица 3) и производные 7-азаизоиндиго (IV) (таблица 4)

где R2-R4, R1', R2', R4' и R5' означают Н, остальные радикалы представлены в таблице 1:
Структура соединений 5-азаиндирубина (I)
Таблица 1
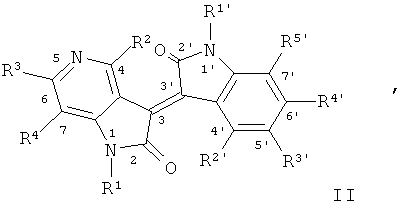
где R2-R4, R2', R4' и R5' означают Н, остальные радикалы представлены в таблице 2:
Структура соединений 5-азаизоиндиго (II)
Таблица 2
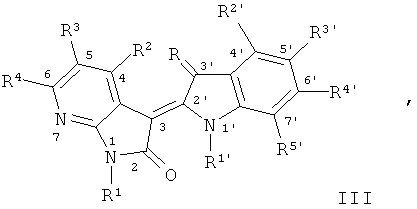
где R2, R4, R1', R2', R4' и R5' означают Н, остальные радикалы представлены в таблице 3:
Структура соединений 7-азаиндирубина (III)
Таблица 3
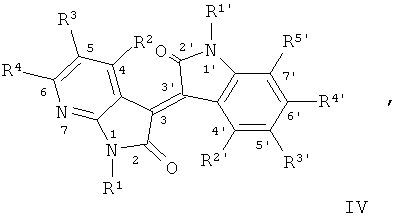
где R2, R4, R2, R4 и R5 означают Н, остальные радикалы представлены в таблице 4:
Структуры соединений 7-азаиндиго (IV)
Таблица 4
5. Соединения или их фармацевтически приемлемые соли по п.1, в которых указанные фармацевтически приемлемые соли включают соли, образованные неорганическими кислотами или органическими кислотами, при этом неорганические кислоты включают следующие: соляная кислота, бромистоводородная кислота, фосфорная кислота, азотная кислота и серная кислота; органические кислоты включают следующие: муравьиная кислота, уксусная кислота, пропионовая кислота, янтарная кислота, нафталиндисульфокислота (1, 5), азиатская кислота, щавелевая кислота, винная кислота, молочная кислота, салициловая кислота, бензойная кислота, бутилкарбоновая кислота, диэтилуксусная кислота, малоновая кислота, янтарная кислота, фумаровая кислота, пимелиновая кислота, гександиоловая кислота, малеиновая кислота, яблочная кислота, аминосульфоновая кислота, фенилпропионовая кислота, глюконовая кислота, аскорбиновая кислота, никотиновая кислота, изоникотиновая кислота, этансульфоновая кислота, паратолуолсульфоновая кислота, лимонная кислота и аминокислота.
6. Фармацевтическая композиция, ингибирующая циклинзависимую киназу, содержащая соединения или их фармацевтически приемлемые соли по п.1 и фармацевтически приемлемые носители.
| Компенсирующая муфта | 1986 |
|
SU1355789A1 |
| CN 1424312 А, 18.06.2003 | |||
| US 6566341 В, 20.05.2003 | |||
| Bioorganic & Medicinal Chemistry, 2006 v.14, n.1, p.237-246 | |||
| RU 2004101408 A, 20.04.2005. | |||

