Описанное здесь изобретение обеспечивает новые пептиды и их производные, которые предоставляют потенциально улучшенные терапевтические возможности для лечения опухолевых заболеваний по сравнению с доластатином-10 и -15 (патенты США 4879276 и 4816444) и соединениями, описанными в международной заявке на патент 93/23424.
Соединения данного изобретения включают новые пептиды формулы I R1R2N-CHX-CO-A-B-D-E-(G)s-K I,
где R1 означает водород, метил или этил;
R2 означает метил или этил, или
R1-N-R2 вместе образуют пирролидиновый цикл;
А означает остаток валила, изолейцила, алло-изолейцила, 2-трет-бутилглицила, 2-этилглицила, норлейцила или норвалила;
В означает остаток N-метилвалила, N-метилнорвалила, N-метил-лейцила, N-метилизолейцила, N-метил-2-трет-бутилглицила, N-метил-2-этилглицила или N-метилнорлейцила;
D означает остаток пролила, гомопролила, гидроксипролила или тиазолидин-4-карбонила;
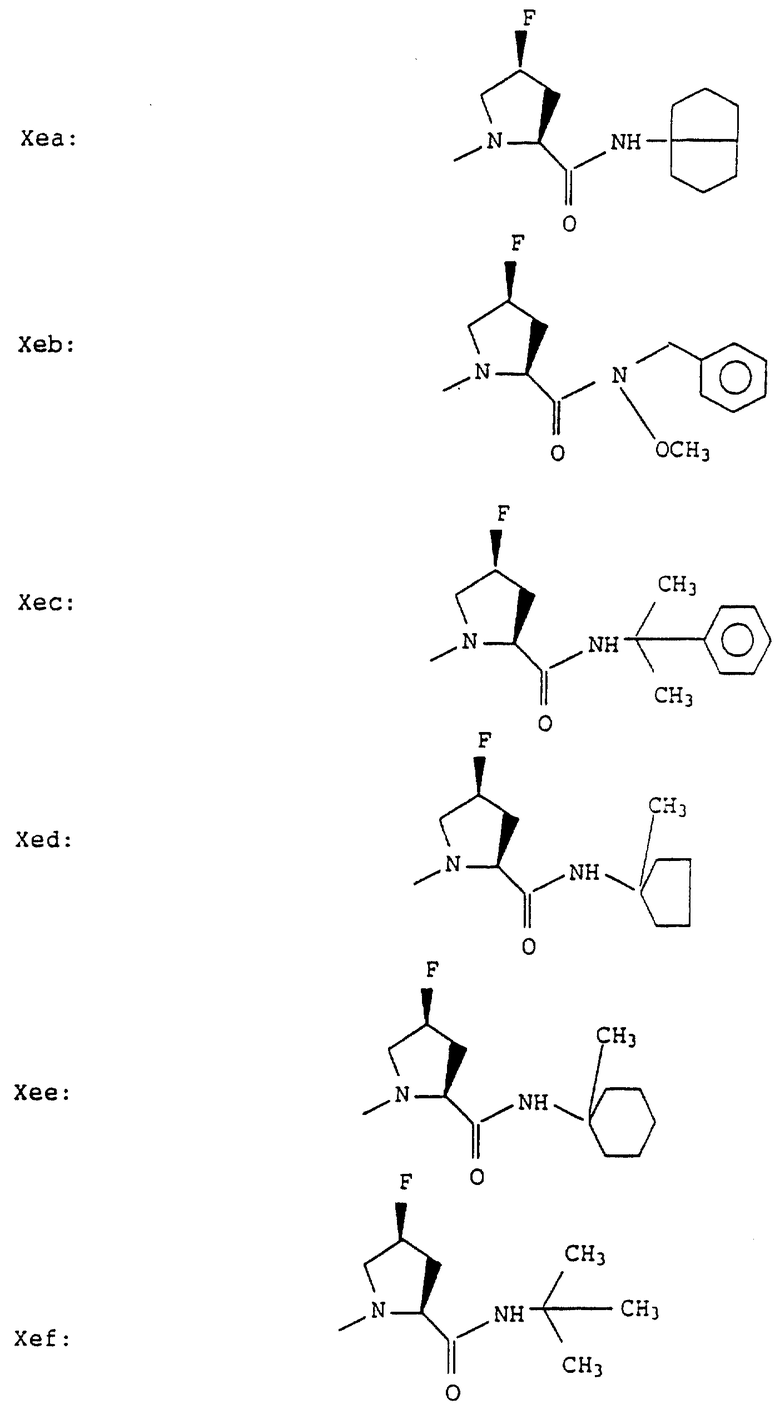
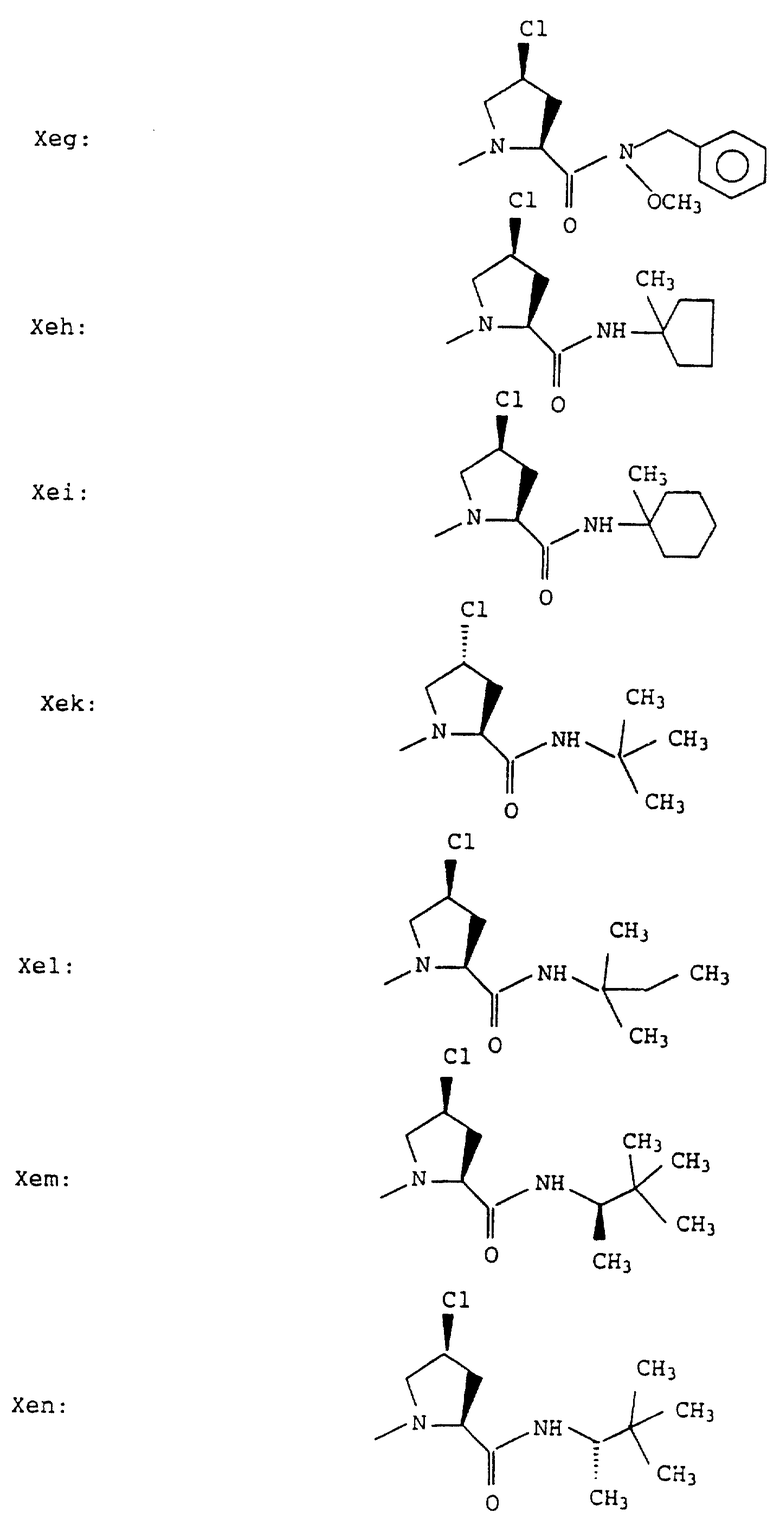
Е означает остаток пролила, гомопролила, гидроксипролила, тиазолидин-4-карбонила, транс-4-фтор-L-пролила, цис-4-фтор-L-пролила, транс-4-хлор-L-пролила или цис-4-хлор-L-пролила;
Х означает этил, пропил, бутил, изопропил, втор-бутил, трет-бутил, циклопропил или циклопентил;
G означает остаток L-2-трет-бутилглицила, D-2-трет-бутилглицила, D-валила, D-изолейцила, D-лейцила, D-норвалила, 1-аминопентил-1-карбрнила или 2,2-диметилглицила;
s означает 0 или 1;
К является алкиламиногруппой с 1-8 атомами углерода в алкиле, алкениламиногруппой с 3-8 атомами углерода в алкенильном остатке, алкиниламиногруппой с 3-8 атомами углерода в алкинильном остатке, циклоалкиламиногруппой с 6-8 атомами углерода в циклоалкиле, циклоалкилалкенаминогруппой с 1-4 атомами углерода в алкеновом остатке и с 3-8 атомами углерода в циклоалкиле, диалкиламиногруппой с 1-4 атомами углерода в одном алкиле и с 1-6 атомами углерода в другом, в которых одна метиленовая группа может быть заменена кислородом или серой, один атом водорода фенилом или циангруппой или один, два или три атома водорода могут быть заменены фтором, кроме N-метокси-N-метиламиногруппы, N-бензиламиногруппы или N-метил-N-бензиламиногруппы, или К означает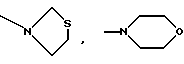
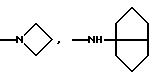
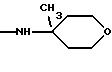
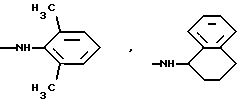
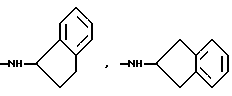
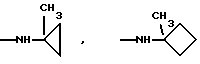
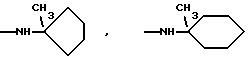
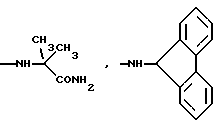
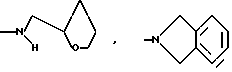
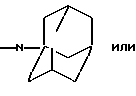
и их соли с физиологически переносимыми кислотами.
Конкретно К может быть -NНСН3, -NНСН2СН3, -NH(СH2)2СН3, -NН(СН2)3СН3, -NH(CH2)4CH3, -NH(СH2)5СН3, -NH(СH2)6СН3, -NН(СН2)7СН3, -NHCH(CH3)2, -NНСН(СН3)CH2СН3, -NНСН(СН2СН3)2, -NHCH(CH2CH2CH3)2, -NНС(СН3)3,
-NНСН(СН2СН3)СН2СН2СН3, -NHCH(CH3)CH(CH3)2, -NНСН(СН2СН3)СН(СН3)2,
-NНСН(СН3)С(СН3)3, -NH-циклогексил, -NH-циклогептил, -NH-циклооктил, -N(СН3)ОСН2СН3, -N(СН3)ОСН2СН2СН3, -N(СН3)ОСН(СН3)2, -N(СН3)O(СН2)3СН3,
-N(CH3)-СН2С6Н5, -NН(СН2)2С6Н5, -NН(СН2)3С6Н5, -NНСН(СН3)С6Н5,
-NНС(СН3)2С6Н5, -NНС(СН3)2СН2СН3, -NНС(СН3)(СН2СН3)2, -NHCH[CH(CH3)2] 2, -NHC(CH3)2CN, -NHCH(CH3)CH(OH)C6H5, -NHCH2-циклогексил, -NHCH2C(CH3)3, -NHCH2CH(CH3)2, -N(СН3)2, -N(СН2СН3)2, -N(СН2СН2СН3)2, -NHCH2CF3,
-NHCH(CH2F)2, -NHCH2CH2F, -NHCH2-СН2OСН3, -NHCH2CH2SCH3, -NHCH2CH= CH2, -NHC(CH3)2CH=CH2, -NHC(CH3)2C≡CH, -NHC(CH2CH3)2C≡CH, -NHC(CH3)2CH2CH2OH, -NН(СН2СН2O)2СН2СН3, -NНС(СН3)2СН(СН3)2,
-NHC(CH3)2CH2CH2CH3, -NHC(CH3)2CH2C6H5, -N(OCH3)CH(CH3)2, -N(ОСН3)СН2СН3, -N(ОСН3)-СН2СН2СН3, -N(ОСН3)СН2С6Н5, -N(OCH3)C6H5, -N(CH3)OC6H5, -NHCH[CH(CH3)2]2, -N(ОСH3)CН2СН2СН2СН3
или определенными циклическими системами, указанными выше.
Предпочтительны соединения формулы I, в которых заместители R1, R2, А, В, D, Е, X, G и s имеют следующие значения:
R1 - водород, метил или этил, особенно, метил;
R2 - метил или этил, особенно, метил;
А - валил, изолейцил, 2-трет-бутилглицил, 2-эгилглицил, норлейцил или норвалил, особенно, валил, изолейцил, 2-трет-бутилглицил, 2-этилглицил;
В - N-метилвалил, N-метилнорвалил, N-метилизолейцил, N-метил-2-трет-бутилглицил, N-метил-2-этилглицил или N-метилнорлейцил, особенно, N-метилвалил, N-метил-2-этилглицил, N-метилнорлейцил, N-метилизолейцил или N-метил-2-трет-бутилглицил;
D - пролил, гомопролил или тиазолидин-4-карбонил, особенно, пролил или тиазолидин-4-карбонил;
Е - пролил, гомопролил, тиазолидин-4-карбонил, транс-4-фтор-L-пролил, цис-4-фтор-L-пролил, транс-4-хлор-L-пролил или цис-4-хлор-L-пролил, особенно, пролил, транс-4-фторпролил, цис-4-фторпролил, транс-4-хлорпролил или цис-4-хлорпролил;
Х - этил, пропил, изопропил, втор-бутил, трет-бутил или циклопропил, особенно, этил, изопропил, втор-бутил или трет-бутил;
G - остаток L-2-трет-бутилглицила, D-2-трет-бутилглицила, D-валила, D-изолейцила, D-лейцила или 2,2-диметилглицила;
s - 0 или 1.
Предпочтительными значениями для К являются:
алкиламиногруппа с 1-8 атомами углерода в алкиле, циклоалкиламиногруппа с 6-8 атомами углерода в циклоалкиле, циклогексилметиленаминогруппа, диалкиламиногрупна с 1-4 и с 1-6 атомами углерода в алкилах, в которых одна метиленовая группа может быть заменена кислородом, один атом водорода фенилом или один или два атома водорода фтором, кроме N-метокси-N-метиламиногруппы, N-бензиламиногруппы или N-метил-N-бензиламиногруппы, или К означает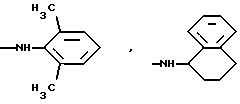
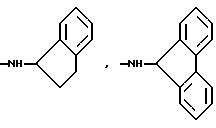
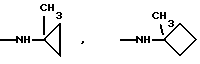
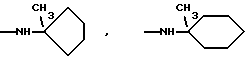
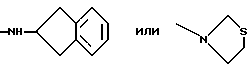
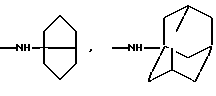
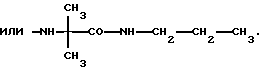
Более предпочтительно К означает
-NНСН3, -NНСН2СН3, -NH(СH2)2СН3, -NН(СН2)3СН3, -NH(CH2)4CH3, -NH(СH2)5СН3, -NH(СH2)6СН3, -NН(СН2)7СН3, -NHCH(CH3)2, -NНСН(СН3)CH2СН3,
-NНСН(СН2СН3)2, -NHCH(CH2CH2CH3)2, -NНС(СН3)3, -NНСН(СН2СН3)СН2СН2СН3, -NHCH(CH3)CH(CH3)2, -NНСН(СН2СН3)СН(СН3)2, -NНСН(СН3)С(СН3)3, -NH-циклогексил, -NH-циклогептил, -NH-циклооктил,
-N(СН3)ОСН2СН3, -N(СН3)ОСН2СН2СН3, -N(СН3)ОСН(СН3)2, -N(OСН3)CH(СН3)2,
-N(CH3)OСН2С6Н5, -NН(СН2)2С6Н5, -NН(СН2)3С6Н5, -NНСН(СН3)С6Н5,
-NНС(СН3)2С6Н5, -NНС(СН3)2СН2СН3, -NНС(СН3)(СН2СН3)2, -NHCH(CH3)CH(OH)C6H5, -NHCH2-циклогексил, -N(CH3)2, -N(СН2СН3)2, -N(СН2СН2СН3)2, -NHCH(CH2F)2,
-NHC(CH3)2CH2CH2OH, -NH(CH2CH2O)2CH2CH3, -NHC(CH3)2СH(CН3)2, NHC(CH3)CH=CH2, -NHC(CH3)2CN, 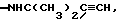 -NНС(CH3)2СONH2, -N(OCH3)C6H5, -NНСН[CH(СН3)2] 2, -N(OСН3)СН2С6H5, -N(OCH3)CH2CH3, -N(OCH3)CH2CH2CH3,
-NНС(CH3)2СONH2, -N(OCH3)C6H5, -NНСН[CH(СН3)2] 2, -N(OСН3)СН2С6H5, -N(OCH3)CH2CH3, -N(OCH3)CH2CH2CH3,
-N(OСН3)СН2СН2CH2CH3,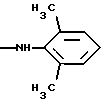
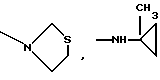
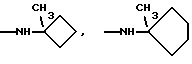
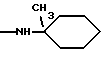
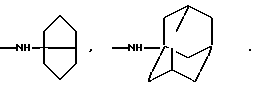
Особенно предпочтительны соединения формулы I, где
R1 и R2 означают метил,
А означает остаток валила, изолейцила или 2-трет-бутилглицила,
В означает остаток N-метилвалила, N-метилизолейцила или N-метил-2-трет-бутилглицила,
D означает остаток пролила или тиазолидин-4-карбонила, Е означает остаток пролила, цис-4-фтор-L-пролила или цис-4-хлор-L-пролила,
Х означает изипропил, втор-бутил или трет-бутил,
s означает 0 и
К означает
-NНСН(СН3)2, -NНСH(CН3)СН2СН3, -NНСН(СН2СН3)2, -NHCH(СН2СН2СН3)2, -NHC(СН3)3, -NHCH(СН2СН3)СН2СН2СН3, -NHCH(СН3)СН(СН3)2, -NНСН(СН2СН3)СН(СН3)2, -NHCH(СН3)C(СН3)3, -NH-циклогептил, -NH-циклооктил,
-N(СН3)ОСН2СН3, -N(СН3)ОСН2СН2СН3, -N(СН3)ОСН(СН3)2, -N(ОСН3)СH(СН3)2,
-N(СН3)ОСН2С6Н5, -NH(СН2)2С6Н5, -NH(СН2)3С6Н5, -NНСН(СН3)С6Н5,
-NHC(CH3)2C6H5, -NHC(CH3)2CH2CH3, -NHC(CH3)(СН2СН3)2, -NHCH(СН3)СН(ОН)C6H5, -NHCH(CH2F)2, -NНС(СН3)2СН2СН2OН, -NH(СН2СН2O)2СН2СН3, -NHC(CH3)2CH= CH2, -NНС(СН3)2СН(СН3)2, -N(ОСН3)CН2СН3, -N(ОСН3)СН2СН2СН3, -N(ОСН3)СН2СН2СН2СН3, -NHC(CH3)2CN, 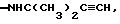 -NНСН[СН(СН3)2] 2, -NHC(CH3)2CONH2, -NНС(СН3)2СН2С6Н5, -N(ОСН3)С6Н5, -N(ОСН3)СН2С6Н5,
-NНСН[СН(СН3)2] 2, -NHC(CH3)2CONH2, -NНС(СН3)2СН2С6Н5, -N(ОСН3)С6Н5, -N(ОСН3)СН2С6Н5, 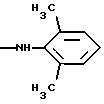
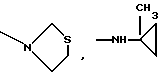
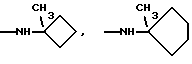
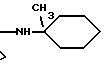
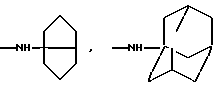
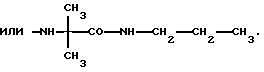
Это изобретение также предусматривает способы получения соединений формулы I, фармацевтических композиций, содержащих такие соединения, вместе с фармацевтически приемлемым носителем и способы их применения для лечения рака у млекопитающих.
Новые соединения могут быть представлены в виде солей с физиологически переносимыми кислотами, такими, как: соляная кислота, лимонная кислота, винная кислота, молочная кислота, фосфорная кислота, метансульфокислота, уксусная кислота, муравьиная кислота, малеиновая кислота, фумаровая кислота, яблочная кислота, янтарная кислота, малоновая кислота, серная кислота, L-глутаминовая кислота, L-аспарагиновая кислота, пировиноградная кислота, слизевая кислота, бензойная кислота, глюкуроновая кислота, щавелевая кислота, аскорбиновая кислота и ацетилглицин.
Новые соединения могут быть получены известными способами пептидной химии. Так, пептиды могут быть синтезированы последовательно, исходя из аминокислот, или путем связывания подходящих небольших пептидных фрагментов. При последовательном соединении, начиная с С-конца, пептидная цепь удлиняется постепенно, каждый раз на одну аминокислоту. При взаимодействии фрагментов возможно соединить вместе фрагменты различной длины, и фрагменты в свою очередь могут быть получены путем последовательного соединения из аминокислот или из них самих при взаимодействии фрагментов.
Как при последовательном соединении, так и при взаимодействии фрагментов необходимо соединять участки путем образования амидных связей. Для этого подходят ферментативные и химические способы.
Химические способы для образования амидных связей подробно описаны Мюллером в кн. "Методы органической химии", том. XV/2, стр. 1-364, издательство Thieme, Штутгарт, 1974; Стюартом, Янгом в кн. "Твердофазный пептидный синтез", стр. 31-34, 71-82, издательство Pierce Chemical Company. Рокфорд, 1984; Бодансцки, Клаузнером, Ондетти в кн. "Синтез пептидов", стр. 85-128, издательство John Wiley & Sons, Нью-Йорк, 1976; "Практика синтеза пептидов", авторы М. Бодансцки, А. Бодансцки, издательство Springer, 1994, и в других соответствующих работах по химии пептидов. Особое предпочтение отдается азидному методу, методу симметричных и смешанных ангидридов, генерированным in situ или предварительно полученным активированным сложным эфирам, применению уретан-защищенных N-карбоксиангидридов аминокислот и образованию амидной связи, используя сочетающие реагенты, в частности, дициклогексилкарбодиимид (DCC), диизопропилкарбодиимид (DIC), 1-этоксикарбонил-2-этокси- 1,2-дигидрохинолин (EEDQ), пивалоилхлорид, хлоргидрат 1-этил-3-(3-диметиламинопропил) карбодиимида (EDCI), ангидрид н-пропанфосфокислоты (РРА), N, N-бис(2-оксо-3-оксазолодинил) амидофосфорилхлорид (ВОР-С1), бром-трис- пирролидинфосфонийгексафтор -фосфат (РуВгор), дифенилфосфоразидат (DРРА), бензотриазол-1-илокси-трис(диметиламино)-фосфоний-гексафторфосфат (реагент Кастро, ВОР, РуВор), O-бензотриазолил-N, N, N', N'-тетраметилурониевые соли (HBTU), О-aзaбeнзoтpиaзoлил-N,N,N',N'-тeтpaмeтилypoниeвыe соли (HATU), диэтилфосфорилцианид (DEPCN), диоксид 2,5-дифенил-2,3-дигидро -3-оксо-4- гидрокситиофена (реагент Стеглиха, HOTDO) и 1,1'-карбонилдиимидазол (CDI). Реагенты для сочетания могут быть использованы по отдельности или в комбинации с добавками, например, N,N,-диметил-4-аминопиридином (DMAP), N-гидрокси-бензотриазолом (HOBt), N-гидроксибензотриазином (HOOBt), азабензотриазолом, N-гидроксисукцинимидом (HOSu) или 2-гидроксипиридином.
В то время, как обычно возможно обходиться без защитных групп при ферментативном пептидном синтезе, обратимая защита реакционноспособных групп, не вовлеченных в образование амидной связи, необходима для обоих реагирующих веществ в химическом синтезе. Предпочтительны три принятые методики с использованием защитных групп для химического пептидного синтеза: методики с использованием бензилоксикарбонильной(Z), трет-бутоксикарбонильной (Вос) и 9-флуоренилметилоксикарбонильной (Fmoc) защитных групп.
Определяемой в каждом случае является защитная группа при α-аминогруппе удлиняющего цепь участка. Подробный обзор защитных групп для аминокислот приводит Мюллер в кн. "Методы органической химии", том XV/1, стр. 20-906, издательство Thieme, Штутгарт, 1974. Участки, используемые для связывания в пептидную цепь, могут реагировать в растворе, в суспензии или способом, подобным описанному Мерифилдом в J.Amer.Soc. 85(1963), 2149.
Для проведения пептидного синтеза в растворе подходят все растворители, которые являются инертными в условиях реакции, в частности, вода, N,N-диметилформамид (ДМФА), диметилсульфоксид (ДМСО), ацетонитрил, дихлорметан (ДХМ), этилацетат, 1,4-диоксан, тетрагидрофуран (ТГФ), N-метил-2-пирролидон и смеси указанных растворителей.
Пептидный синтез на полимерном носителе может быть проведен во всех инертных органичских растворителях, в которых растворимы используемые аминокислотные производные. Однако, предпочтительные растворители дополнительно обладают вызывающими набухание смолы свойствами, например, ДМФА, ДХМ, N-метил-2-пирролидон, ацетонитрил и ДМСО и смеси этих растворителей. После завершения синтеза, пептид отделяют от полимерного носителя. Условия, при которых возможно отщепление от различных типов смол, описываются в литературе. Наиболее часто используемыми реакциями расщепления являются катализируемые кислотами и палладием, в частности, расщепление в жидком безводном фтористом водороде, в безводной трифторметансульфокислоте, в разбавленной или концентрированной трифторуксусной кислоте, катализируемое палладием расщепление в ТГФ или смеси ТГФ-ДХМ в присутствии слабого основания, как, например, морфолин, или расщепление в смесях уксусной кислоты/ДХМ/трифторэтанола. В зависимости от выбранных защитных групп, последние могут сохраняться или таким же образом отщепляться в условиях расщепления.
Частичное снятие защиты у пептида может также быть необходимым, когда следует провести некоторые реакции превращения.
Пептиды, диалкилированные по N-концу, могут быть получены либо при взаимодействии с соответствующими N,N-диалкиламинокислотами в растворе или на полимерном носителе, при восстановительном алкилировании связанного со смолой пептида в ДМФАЛ/% уксусной кислоте с помощью натрийцианборгидрида и соответствующих альдегидов, при гидрировании пептида в растворе в присутствии альдегида или кетона и Pd/C.
Различные не встречающиеся в природе аминокислоты, а также различные неаминокислотные участки, представленные здесь, могут быть приобретены или синтезированы из коммерчески доступных материалов, используя известные для этого способы. Например, аминокислотные блоки для построения нужных соединений с участками R1 и R2 могут быть получены согласно Е. Вунш, Губен Вейль "Методы органической химии", том. XV, 1, стр. 306 и далее, издательство Thieme, Штутгарт, 1974, и цитируемой далее литературе.
Соединения данного изобретения могут использоваться для ингибирования или, иначе, лечения солидных опухолей (например, опухолей легкого, молочной железы, толстой кишки, простаты, мочевого пузыря, прямой кишки или эндометрия) или гематологических злокачественных заболеваний (например, лейкозов, лимфом) при введении соединения млекопитающему.
Особым преимуществом новых соединений является их высокая устойчивость к ферментативному разложению и то, что их можно вводить орально.
Введение может быть осуществлено любым из способов, принятых для фармацевтических, предпочтительно онкологических, средств, включая оральные и парентеральные способы, как, например, введение подкожно, внутривенно, внутримышечно и внутрибрюшинно.
Соединения могут быть введены по отдельности или в виде фармацевтических композиций, содержащих соединение формулы I вместе с фармацевтически приемлемым носителем, подходящим желаемому способу введения. Такие фармацевтические композиции могут представлять комбинацию продуктов, то есть, могут также содержать другие терапевтически активные ингредиенты.
Доза, которую следует ввести млекопитающему, будет содержать эффективное, ингибирующее опухоль количество активного ингредиента, которое будет зависеть от общепринятых факторов, включающих биологическую активность данного используемого соединения, способы введения, возраст, здоровье и массу тела реципиента, природу и распространенность симптомов, частоту лечения, введение других лечебных препаратов и желаемый эффект. Типичная ежедневная доза будет от примерно 0,05 до 50 мг на кг массы тела при оральном введении и от примерно 0,01 до 20 мг на кг массы тела при парентеральном введении.
Новые соединения могут быть введены в виде обычных твердых или жидких фармацевтических форм для введения, например, непокрытые или покрытые тонким слоем таблетки, капсулы, порошки, гранулы, суппозитории или растворы. Эти формы готовят общепринятым способом. Активные вещества для этой цели могут быть обработаны обычными фармацевтическими, вспомогательными средствами, например, связывающими веществами для приготовления таблеток, наполнителями, консервантами, средствами для распада таблеток, регуляторами текучести, пластификаторами, смачивающими средствами, диспергаторами, эмульгаторами, растворителями, композициями для поддерживаемого высвобождения, антиоксидантами и/или диспергирующими газами (ср. Н. Sucker и др.: "Фармацевтическая технология", издательство Thieme, Штутгарт, 1978). Полученные таким образом формы для введения содержат обычно 1-90% (по массе) активного вещества.
Изобретение иллюстрируется следующими примерами. Входящие в состав белка аминокислоты сокращаются в примерах с использованием известного трехбуквенного кода. Другие примененные сокращения:
Me2Val=N,N-диметилвалин, MeVal=N-метилвалин.
А. Общие методики
I. Пептиды, заявленные в пункте 1, синтезируются либо путем классического синтеза в растворе с использованием стандартной методики с Z- и Вос-защитными группами, как описано выше, или с помощью стандартных способов твердофазного синтеза, использующего Вос- и Fmoc-защитные группы.
В случае твердофазного синтеза N, N-диалкилпента- или гексапептидные кислоты освобождаются от твердого носителя и далее соединяются с соответствующими С-концевыми аминами в растворе. N, N-Бис(2-оксо-3-оксазолидинил)амидофосфорилхлорид и бром-трис- пирролидинфосфонийгексафторфосфат использовались в качестве реагентов для присоединения аминокислоты, следующей за N-метиламинокислотами. Время реакции соответственно возрастало. Для восстановительного алкилирования по N-концу у пептида-смолы снимали защиту при N-конце, а затем подвергали реакции с 3-кратным молярным избытком альдегида или кетона в ДМФА/1% уксусной кислоте с прибавлением 3 эквивалентов натрийцианборгидрида. После завершения реакции (отрицательная проба Кайзера) смолу промывали несколько раз водой, изопропиловым спиртом, ДМФА и дихлорметаном.
При синтезе в растворе использование либо внутренних N-карбоксиангидридов Вос-защищенных аминокислот (N-карбоксиангидриды N-трет-бутоксикарбониламинокислот), N-карбоксиангидридов Z-защищенных аминокислот (N-карбоксиангидриды N-бензилоксикарбониламинокислот), либо использование пивалоилхлорида в качестве конденсирующего агента, соответственно, является наиболее выгодным для присоединения аминокислоты, следующей за N-метиламинокислотами. Восстановительное алкилирование по N-концу может, например, быть осуществлено при реакции не защищенных по N-концу пептидов или аминокислот с соответствующими альдегидами или кетонами при использовании натрийцианборгидрида или водорода в присутствии Pd/C.
II. Очистка и характеристика пептидов
Очистку проводили путем гель-хроматографии (Сефадекс G-10, G-15/10% уксусная кислота, Сефадекс LH20/метанол), хроматографии среднего давления (стационарная фаза: HD-SIL С-18, 20-45 мк, 100 ангстрем; подвижная фаза: градиент с А= 0,1% трифторуксусная кислота/метанол, Б=0,1% трифторуксусная кислота/вода) или препаративной высокоэффективной жидкостной хроматографии (стационарная фаза: Waters Delta-Pak С-18, 15 мк, 100 ангстрем; подвижная фаза: градиент с А=0,1% трифторуксусная кислота/метанол, Б=0,1% трифторуксусная кислота/вода).
Чистоту полученных в результате продуктов определяли при аналитической высокоэффективной жидкостной хроматографии (стационарная фаза: 100 2. 1 мм VYDAC С-18, 51, 300 ангстрем; подвижная фаза: градиент ацетонитрил-вода, забуферено 0,1% трифторуксусной кислотой, 40oС).
Охарактеризовывали с помощью аминокислотного анализа и масс-спектроскопии с бомбардировкой быстрыми атомами.
Б. Специфические методики
Пример 1 (последовательность 1) Me2Val-Val-MeVal-Pro-Pro-NHCH(CH3)2
a) Z-MeVal-Pro-OMe
Растворяли 66,25 г (250 ммолей) Z-MeVal-OH в 250 мл безводного дихлорметана. После прибавления 36,41 мл (262,5 ммоля) триэтиламина реакционную смесь охлаждали до -25oС и прибавляли 32,27 мл (262,5 ммоля) пивалоилхлорида. После перемешивания в течение 2,5 ч к реакционной смеси прибавляли 41,89 г (250 ммолей) Н-Рго-ОМе х НС1 в 250 мл дихлорметана, нейтрализованного 36,41 мл (262,5 ммоля) триэтиламина при 0oС. Перемешивание продолжали 2 ч при -25oС и в течение ночи при комнатной температуре. Реакционную смесь разбавляли дихлорметаном и тщательно промывали насыщенным водным раствором бикарбоната натрия (трижды), водой (один раз), 5% лимонной кислотой (трижды) и насыщенным раствором хлористого натрия. Органическую фазу сушили над сульфатом натрия и упаривали досуха. Остаток (91,24 г) перемешивали с петролейным эфиром в течение ночи и отфильтровывали. Получали 62,3 г продукта.
б) H-MeVal-Pro-OMe
Растворяли 48,9 г (130 ммолей) Z-MeVal-Рго-ОМе в 490 мл метанола. После прибавления 10,9 мл (130 ммолей) концентрированной соляной кислоты и 2,43 г 10% Pd/C реакционную смесь гидрировали. После фильтрации и упаривания досуха получали 36,43 г продукта.
в) Z-Val-MeVal-Pro-OMe
18,1 г (65 ммолей) H-MeVal-Pro-OMe, 21,6 г (78 ммолей) Z-Val-N-карбоксиангидрида и 22,8 мл (130 ммолей) диизопропилэтиламина перемешивали в 110 мл ДМФА при 40oС в течение 2 дней. После выпаривания ДМФА прибавляли дихлорметан и органическую фазу промывали насыщенным водным раствором бикарбоната натрия (трижды), один раз водой, 5% лимонной кислотой (трижды) и насыщенным раствором хлористого натрия. Органическую фазу сушили над сульфатом натрия и упаривали досуха. Продукт (29,3 г) получали в виде вязкого масла.
г) H-Val-MeVal-Рго-ОМе
Растворяли 29,3 г (61,6 ммоля) Z-Val-MeVal-Pro-OMe в 230 мл метанола. После прибавления 1,15 г 10% Pd/C реакционную смесь гидрировали. После фильтрации и упаривания досуха получали 21,96 г продукта.
д) Z-Val-Val-MeVal-Pro-OMe
Растворяли 15,29 г (61 ммоль) Z-Val-OH и 21,96 г (61 ммоль) H-Val-MeVal-Pro-OMe в 610 мл дихлорметана и охлаждали до 0oС.
После прибавления 8,16 мл (73,2 ммоля) N-метилморфолина, 2,77 г (20,3 ммоля) N-гидроксибензотриазола и 11,74 г (61 ммоля) хлоргидрата 1 -этил-3-(3-диметиламинопропил)карбодиимида реакционную смесь перемешивали в течение ночи при комнатной температуре, разбавляли дихлорметаном и тщательно промывали водным насыщенным раствором бикарбоната натрия (три раза), водой (один раз), 5% лимонной кислотой (3 раза) и насыщенным раствором хлористого натрия. Органическую фазу сушили над сульфатом натрия и упаривали досуха, получали 31,96 г продукта.
e) Z-Val-Val-MeVal-Pro-OH
Растворяли 31,96 г (57 ммолей) Z-Val-Val-MeVal-Pro-OMe в 250 мл метанола. Прибавляли 102,6 мл 1 н. раствора гидроокиси лития и смесь перемешивали в течение ночи при комнатной температуре. После прибавления 500 мл воды водную фазу промывали три раза этилацетатом, устанавливали рН 2 при 0oС и трижды экстрагировали этилацетатом. Органическую фазу сушили над сульфатом натрия и упаривали досуха, получали 30,62 г нужного продукта в виде белого твердого вещества.
ж) Z-Val-Val-MeVal-Pro-Pro-NHCH(CH3)2
Растворяли 2 г (3,35 ммоля) Z-Val-Val-MeVal-Pro-OH и 0,664 г (3,35 ммоля) Н-Рго-МНСН(СН3)2 в 34 мл безводного дихлорметана. После охлаждения до 0oС прибавляли 1,35 мл (12,1 ммоля) N-метилморфолина, 0,114 г (0,84 ммоля) N-гидроксибензотриазола и 0,645 г (3.35 ммоля) хлоргидрата 1-этил-3-(3-диметиламинопропил)-карбодиимида и реакционную смесь перемешивали в течение ночи при комнатной температуре. Прибавляли 80 мл дихлорметана и органическую фазу тщательно промывали насыщенным водным раствором бикарбоната натрия (трижды), водой (один раз), 5% лимонной кислотой (трижды) и один раз насыщенным раствором хлористого натрия. Органическую фазу сушили над сульфатом натрия и упаривали досуха, получали 1,96 г продукта, который использовали без дальнейшей очистки в следующей реакции.
з) Me2Val-Val-MeVal-Pro-Pro-NHCH(CH3)2
Растворяли 1,96 г Z-Val-Val-MeVal-Pro-Pro-NHCH(CH3)2 в 11 мл метанола, прибавляли 0,054 г 10% Pd/C в атмосфере азота и реакционную смесь гидрировали при комнатной температуре 4 ч. После прибавления 0,86 мл (11,24 ммоля) 37% водного раствора формальдегида и 0,281 г 10% Pd/C гидрирование продолжали в течение 5 ч. После фильтрации и удаления растворителя получали 2,77 г неочищенного продукта. Дальнейшую очистку осуществляли путем растворения пептида в воде, доведения рН до 2 и трехкратной экстракции водной фазы этилацетатом. Затем рН водной фазы доводили до 8-9 и четырежды экстрагировали дихлорметаном. Органическую фазу сушили над сульфатом натрия и получали 1,37 г очищенного продукта в виде белой пены. Далее вещество очищали, используя жидкостную хроматографию среднего давления (10-50% А в 10 мин; 50-90% А в 320 мин). Содержащие продукт фракции объединяли, подвергали лиофилизации, заново растворяли в воде и устанавливали рН 9 с помощью 1 н. гидрата окиси лития. После экстракции дихлорметаном органическую фазу сушили над сульфатом натрия и упаривали досуха. Лиофилизация приводила к 500 мг чистого продукта, который охарактеризовывали с помощью масс-спектрометрии с бомбардировкой быстрыми атомами ([М+Н]+=593).
Пример 2 (последовательность 1)
Me2Val-Val-MeVal-Pro-Pro-NHC(CH3)3
и) Z-Val-Val-MeVal-Pro-Pro-NHC(CH3)3
Растворяли 2 г (3,35 ммоля) Z-Val-Val-MeVal-Pro-OH и 0,692 г (3,35 ммоля) Н-Рrо-NНС(СН3)3 в 34 мл безводного дихлорметана. После охлаждения до 0oС прибавляли 1,35 мл (12,1 ммоля) N-метил-морфолина, 0,114 г (0,84 ммоля) N-гидроксибензотриазола и 0,645 г (3,35 ммоля) хлоргидрата 1-этил-3-(3-диметиламинопропил)-карбодиимида и реакционную смесь перемешивали в течение ночи при комнатной температуре. Прибавляли 80 мл дихлорметана и органическую фазу тщательно промывали три раза насыщенным водным раствором бикарбоната натрия, один раз водой, три раза 5% лимонной кислотой и один раз насыщенным раствором хлористого натрия. Органическую фазу сушили над сульфатом натрия и упаривали досуха, получали 1,8 г продукта, который использовали в следующей реакции без дальнейшей очистки.
к) Ме2Vаl-Vаl-МеVаl-Рrо-Рrо-NHС(СН3)3
Растворяли 1,8 г Z-Val-Val-MeVal-Pro-Pro-NHC(CH3)3 в 10 мл метанола, прибавляли в атмосфере азота 0,049 г 10% Pd/C и реакционную смесь гидрировали при комнатной температуре в течение 4 ч. После прибавления 0,86 мл (11,24 ммоля) 37% водного раствора формальдегида и 0,252 г 10% Pd/C гидрирование продолжали 5 ч. После фильтрации и упаривания получали 1,82 г неочищенного продукта. Далее соединение очищали, используя жидкостную хроматографию среднего давления (10-50% А в 10 мин; 50-90% А в 320 мин). Содержащие продукт фракции объединяли, подвергали лиофилизации, заново растворяли в воде и с помощью 1 н. гидрата окиси лития устанавливали рН 9. После экстракции дихлорметаном органическую фазу сушили над сульфатом натрия и упаривали досуха. Лиофилизация приводила к 547 мг чистого продукта, который был охарактеризован с помощью масс-спектрометрии с бомбардировкой быстрыми атомами ([М+Н]+= 607).
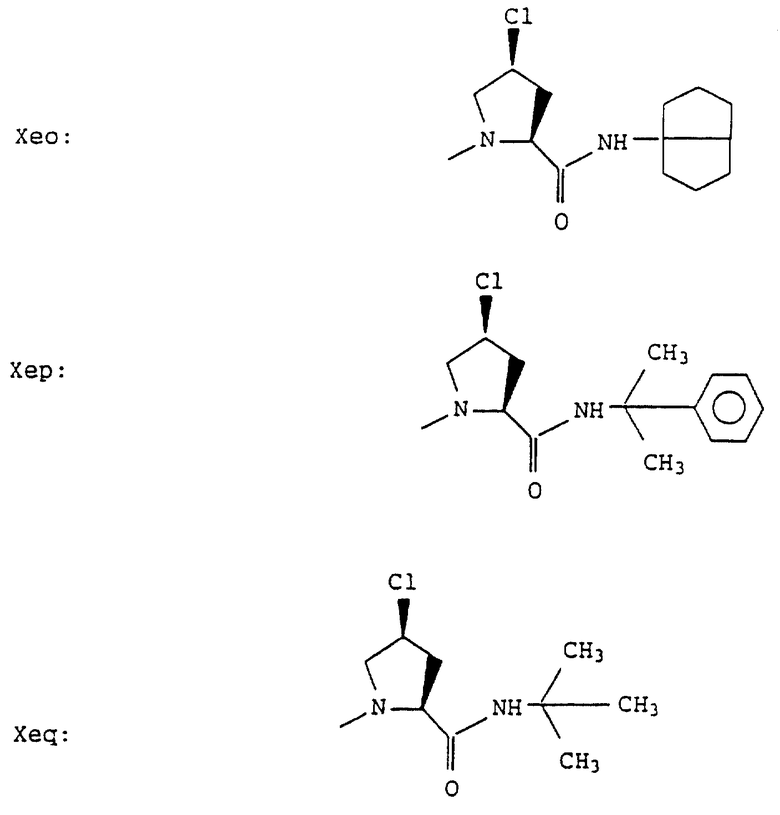
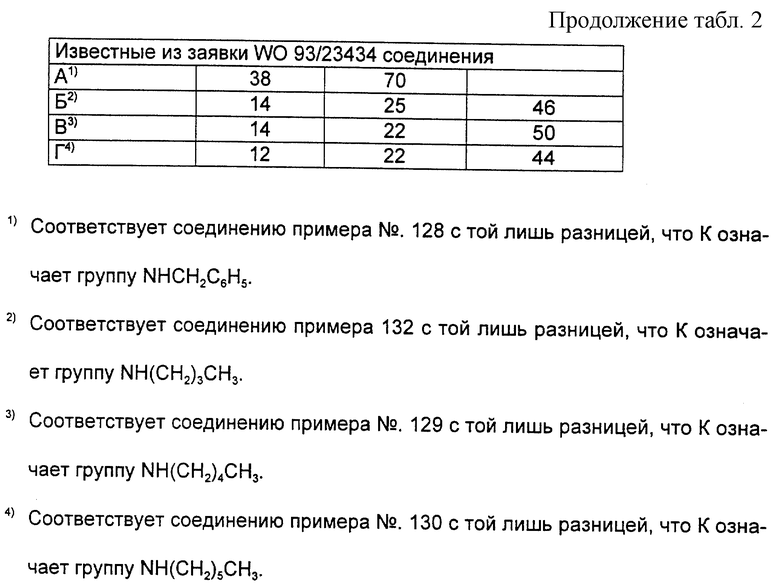
Согласно примерам 1 и 2 были получены или могут быть получены следующие соединения (см. в конце описания).
Соединения настоящего изобретения могут быть испытаны на противораковую активность с помощью общепринятых способов, включая, например, способы, описанные ниже.
A. In vitro методология
Цитотоксичность измеряли, используя стандартную методологию для сросшихся клеточных линий, например, анализ микрокультуры с использованием бромида 3-(4,5-диметилтиазол-2-ил) -2,5-дифенилтетразолия (тиазолиловый синий) (МТТ). Детали такого анализа были опубликованы (Alley M.C. и др.. Cancer Research 48: 589-601, 1988). Используются экспоненциально растущие культуры опухолевых клеток, как, например, рак толстой кишки НТ-29 или опухоль легкого LX-1 для приготовления культур для планшетов для титрования. Клетки засевают по 3000 клеток в ячейку в 96-ячеечные планшеты (в 150 мкл среды) и выращивают в течение ночи при 37oС. Соединения, подвергающиеся испытанию, прибавляются в 10-кратных разведениях, варьирующихся от 10-4 М до 10-10 М. Затем клетки инкубируют в течение 72 ч. Чтобы определить число жизнеспособных клеток в каждой ячейке, прибавляется краситель МТТ /50 мкл раствора 3-(4,5-диметилтиазол-2-ил)-2,5-дифенилтетразолия бромида в физиологическом растворе в концентрации 3 мг/мл/. Эту смесь инкубируют при 37oС в течение 5 ч, затем в каждую ячейку прибавляют 50 мкл 25% додецилсульфата натрия, рН 2. После инкубирования в течение ночи поглощение при 550 нм в каждой ячейке определяют с помощью считывающего устройства, использующегося при ферментном иммуносорбентном анализе (ELISA). Рассчитывают значения для среднего +/- стандартного отклонения данных от реплицированных ячеек, используя формулу % O/К (% обработанные жизнеспособные клетки/контроль)
Концентрация исследуемого соединения, которая дает О/К 50%-ного ингибирования роста, обозначалась как ИК50.
Б. In vivo методология
Соединения настоящего изобретения далее подвергались преклиническому испытанию на активность in vivo, что указывает на клиническую применимость. Такие испытания проводились на безволосых мышах, которым пересаживали (ксенотрансплантация) опухолевую ткань, предпочтительно человеческого происхождения, как это хорошо известно в данной области. Оценивали эффективность исследуемых соединений в качестве противоопухолевых при последующем введении мышам с ксенотрансплантатами.
Более конкретно, человеческие опухоли молочной железы (МХ-1), которые выросли у безволосых мышей с отсутствующей вилочковой железой, пересаживали новым реципиентным мышам, используя фрагменты опухоли массой около 50 мг. День трансплантации обозначали как день 0. Через 6-10 дней мышей обрабатывали исследуемыми соединениями, которые вводили в виде внутривенной инъекции или орально, в группах из 5-10 мышей на каждую дозу. Соединения вводили каждый последующий день в течение 3 недель в дозах и от 1 до 200 мг/кг массы тела.
Диаметры опухолей и массы тел измеряли дважды в неделю. Объемы опухолей рассчитывали, используя диаметры, измеренные циркулями Вернье, по формуле
(длина х ширина2)/2=объем опухоли в мм3.
Средние объемы опухолей рассчитываются для каждой подвергнутой обработке группы, и значения О/К определяются для каждой группы по отношению к необработанным контрольным опухолям.
Новые соединения обладают хорошими ингибирующими свойствами в отношении опухолей.
Соединения согласно изобретению проявляют устойчивость к расщеплению в результате воздействия энзимов, что подтверждается результатами следующего опыта, сведенными в табл.2.
Данная устойчивость намного превышает устойчивость, проявляемую соединениями, описанными в ближайшем аналоге, заявке WO 93/23434, о чем свидетельствует сравнение данных, также сведенных в нижепредставленной таблице.
Опыт
100 мкл 1,35 мкМ раствора торгового энзима пролилолигопептидазы добавляли к 1 мл 0,1 М фосфатного буфера (рН 7) и 250 мкл 810 мкМ раствора исследуемого соединения при температуре 37oС. Через 1, 2 и 4 ч отбирали пробу, которую после добавления 0,3%-ного водного раствора трифторуксусной кислоты подвергали высокоэффективной жидкостной хроматографии. При этом определяли метаболизм в %. Исследуемые соединения и результаты опыта сведены в следующей таблице.
Соединения согласно изобретению относятся к категории среднетоксичных веществ.
Перечень последовательностей см. в конце описания.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ДОЛАСТАТИНА 15 | 1998 |
|
RU2195462C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ 3-ПИРРОЛИН-2-КАРБОНОВОЙ КИСЛОТЫ | 1997 |
|
RU2199529C2 |
| ПРОИЗВОДНЫЕ ПЕПТИДА ИЛИ ИХ СОЛИ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1993 |
|
RU2116312C1 |
| ПЕНТАПЕПТИД, СПОСОБЫ ЕГО ПОЛУЧЕНИЯ И ПЕПТИДНЫЕ СОЕДИНЕНИЯ, ЯВЛЯЮЩИЕСЯ ФОРПРОДУКТАМИ ПЕНТАПЕПТИДА | 1995 |
|
RU2153504C2 |
| ПРОИЗВОДНЫЕ ДОЛАСТАТИНА-15 В КОМБИНАЦИИ С ТАКСАНАМИ | 1998 |
|
RU2218174C2 |
| ПЯТИЧЛЕННЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ АМИДИНЫ, ОБЛАДАЮЩИЕ СВОЙСТВАМИ ИНГИБИТОРОВ ТРОМБИНА, СОЕДИНЕНИЯ, СОДЕРЖАЩИЕ ПРОИЗВОДНЫЕ ПЯТИЧЛЕННОГО ГЕТЕРОЦИКЛИЧЕСКОГО АМИДИНА, В КАЧЕСТВЕ СОСТАВНОЙ ЧАСТИ ИНГИБИТОРОВ СЕРИНПРОТЕАЗЫ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ | 1997 |
|
RU2175328C2 |
| СОЕДИНЕНИЯ АГОНИСТЫ РЕЦЕПТОРА ГЛЮКАГОНОПОДОБНОГО БЕЛКА-1 (GLP-1R) | 2008 |
|
RU2432361C2 |
| ИНГИБИТОРЫ ТРОМБИНА И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДЛЯ ИХ ПОЛУЧЕНИЯ | 1995 |
|
RU2172741C2 |
| ЗАМЕЩЕННЫЕ 2-ОКСО- И 2- ТИОКСО- ДИГИДРОХИНОЛИН-3-КАРБОКСАМИДЫ В КАЧЕСТВЕ МОДУЛЯТОРОВ KCNQ2/3 | 2011 |
|
RU2581362C2 |
| КОМПОЗИЦИЯ СО СТАБИЛИЗИРОВАННЫМ СУБТИЛИЗИНОМ | 2012 |
|
RU2635355C2 |







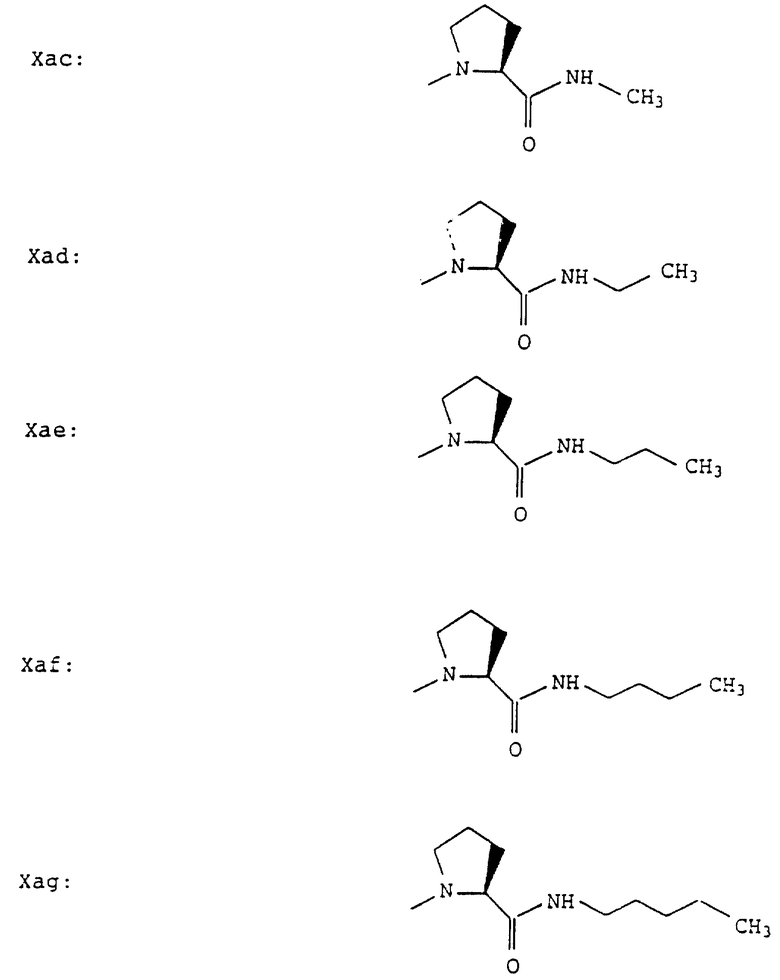
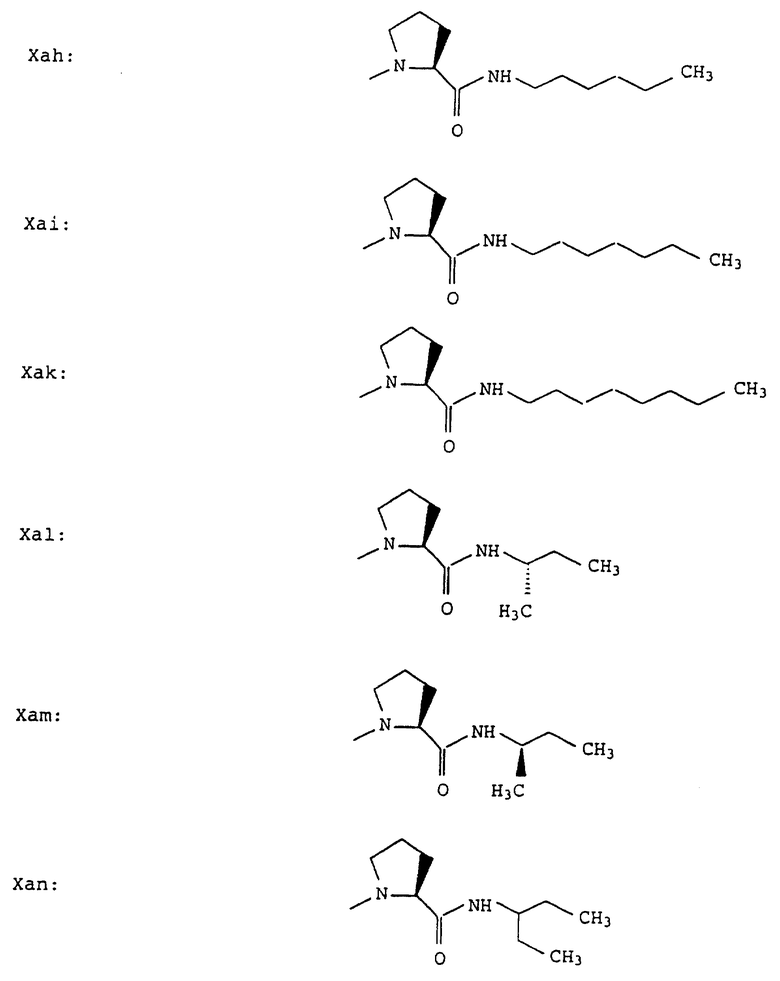
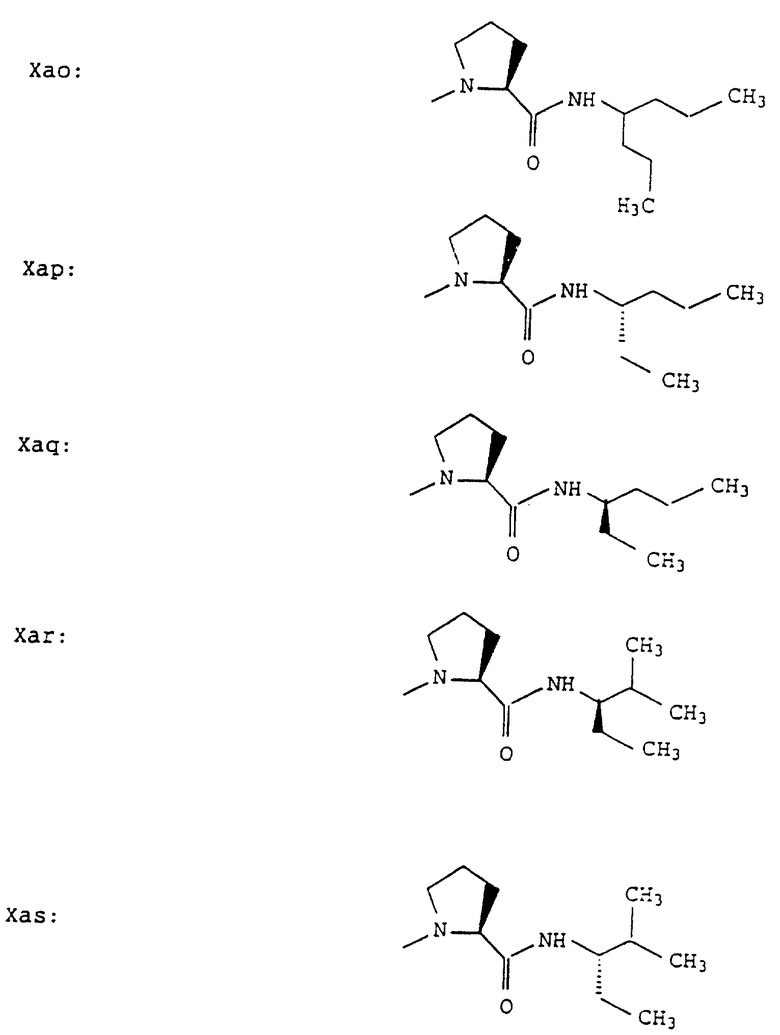
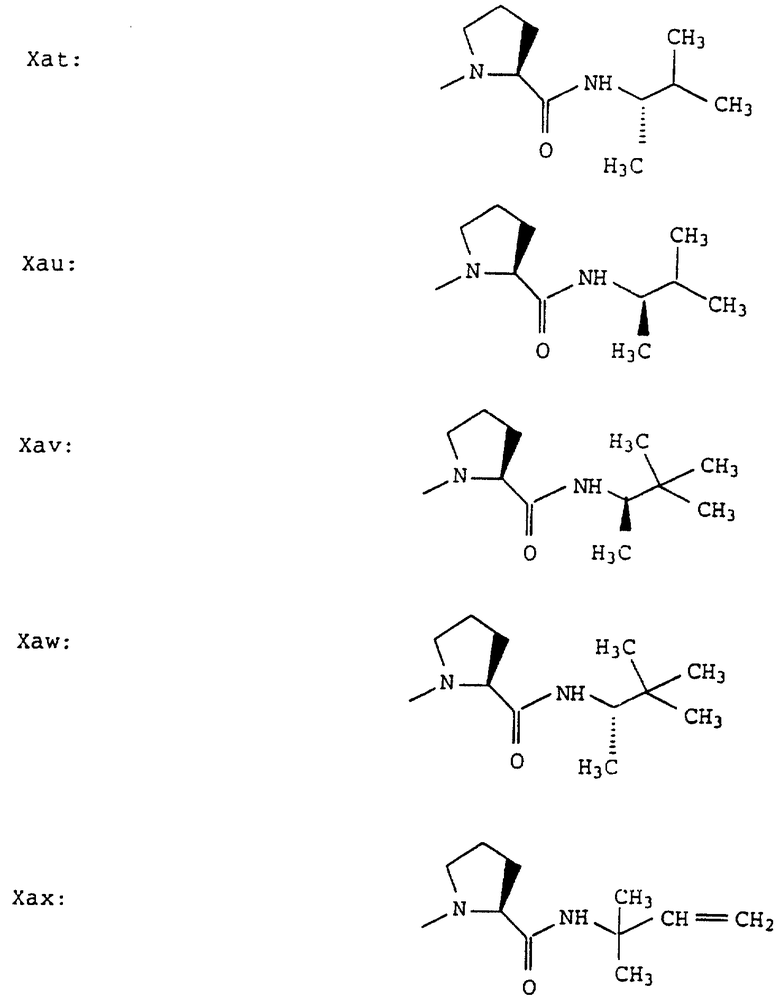
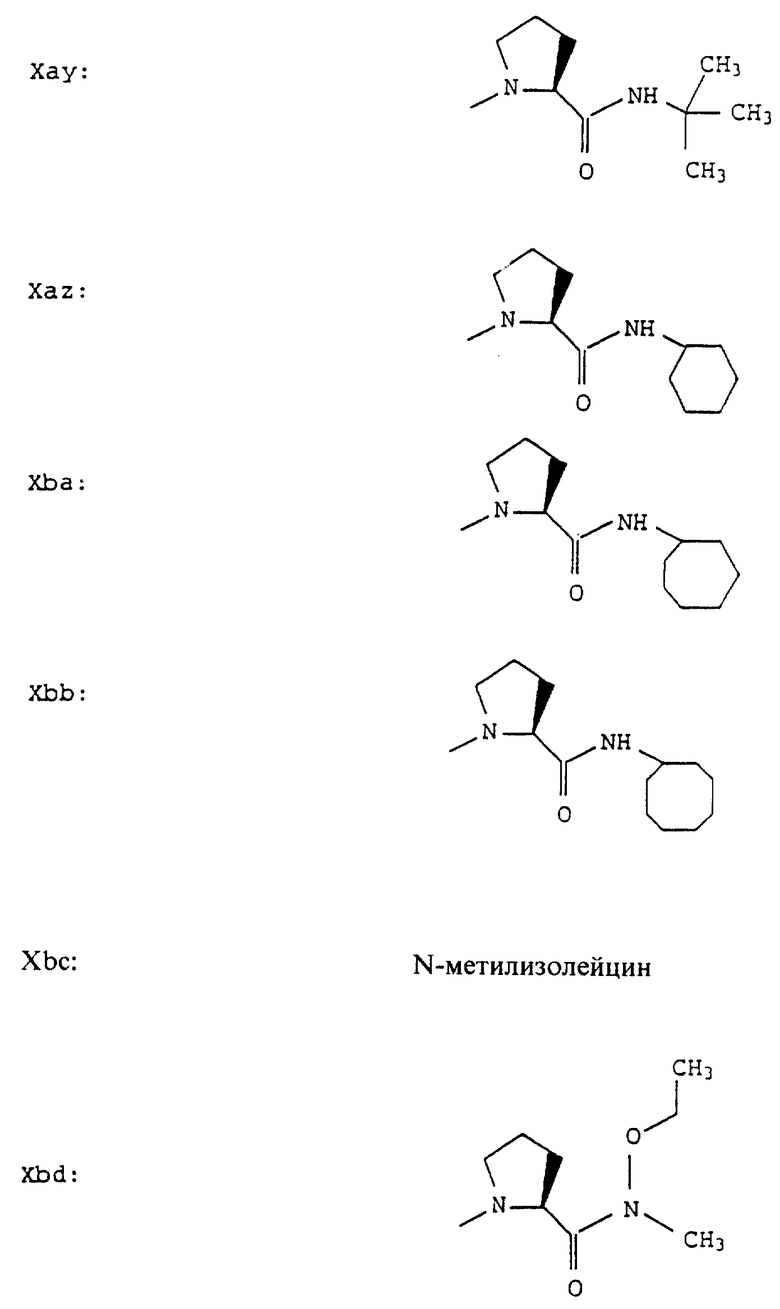

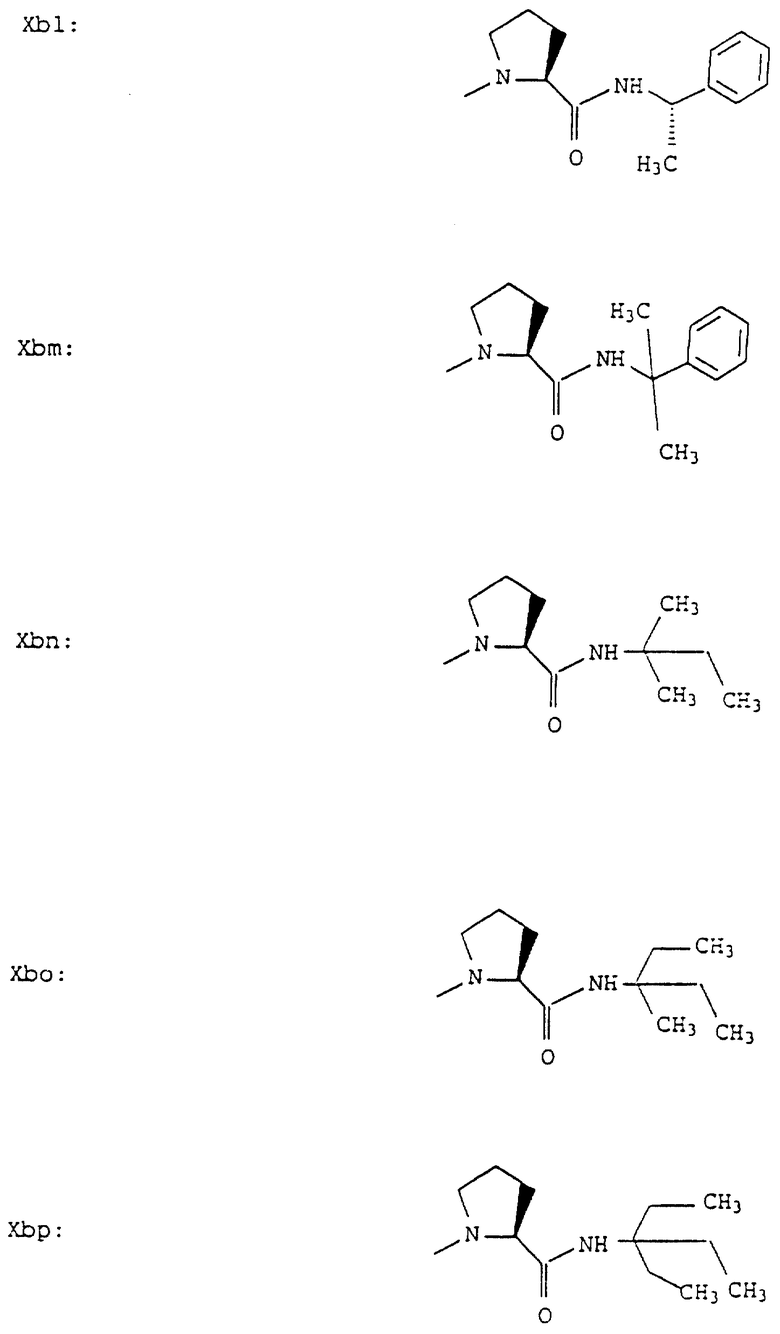
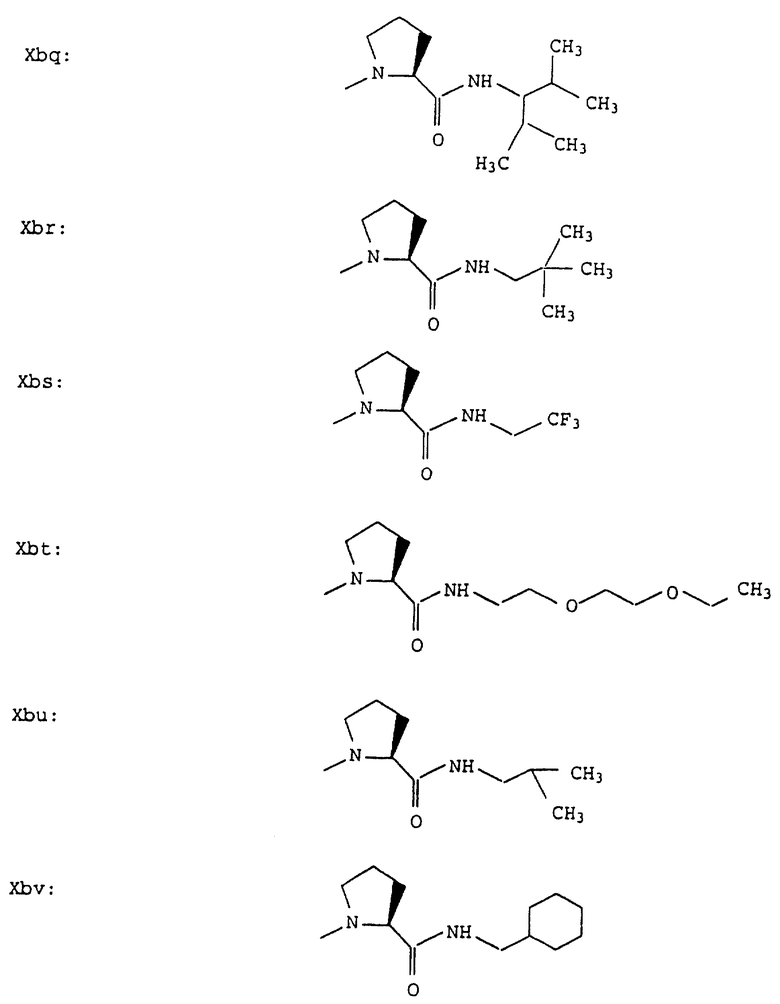
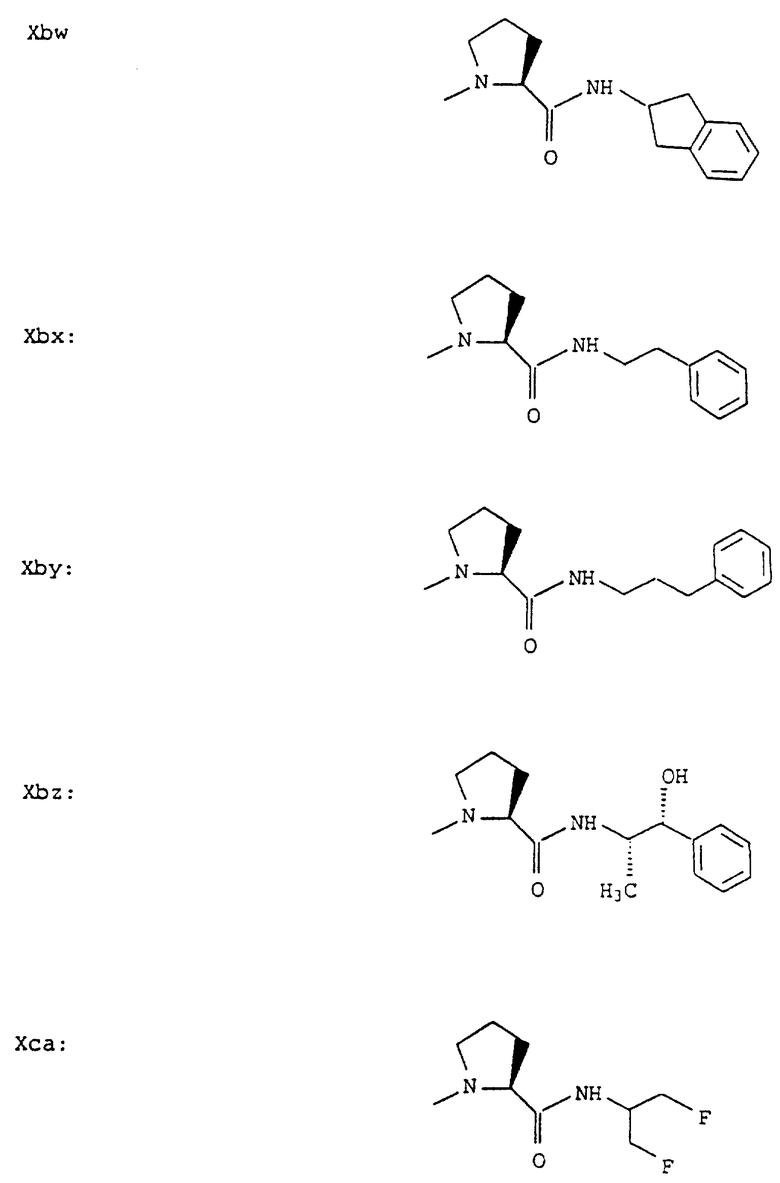
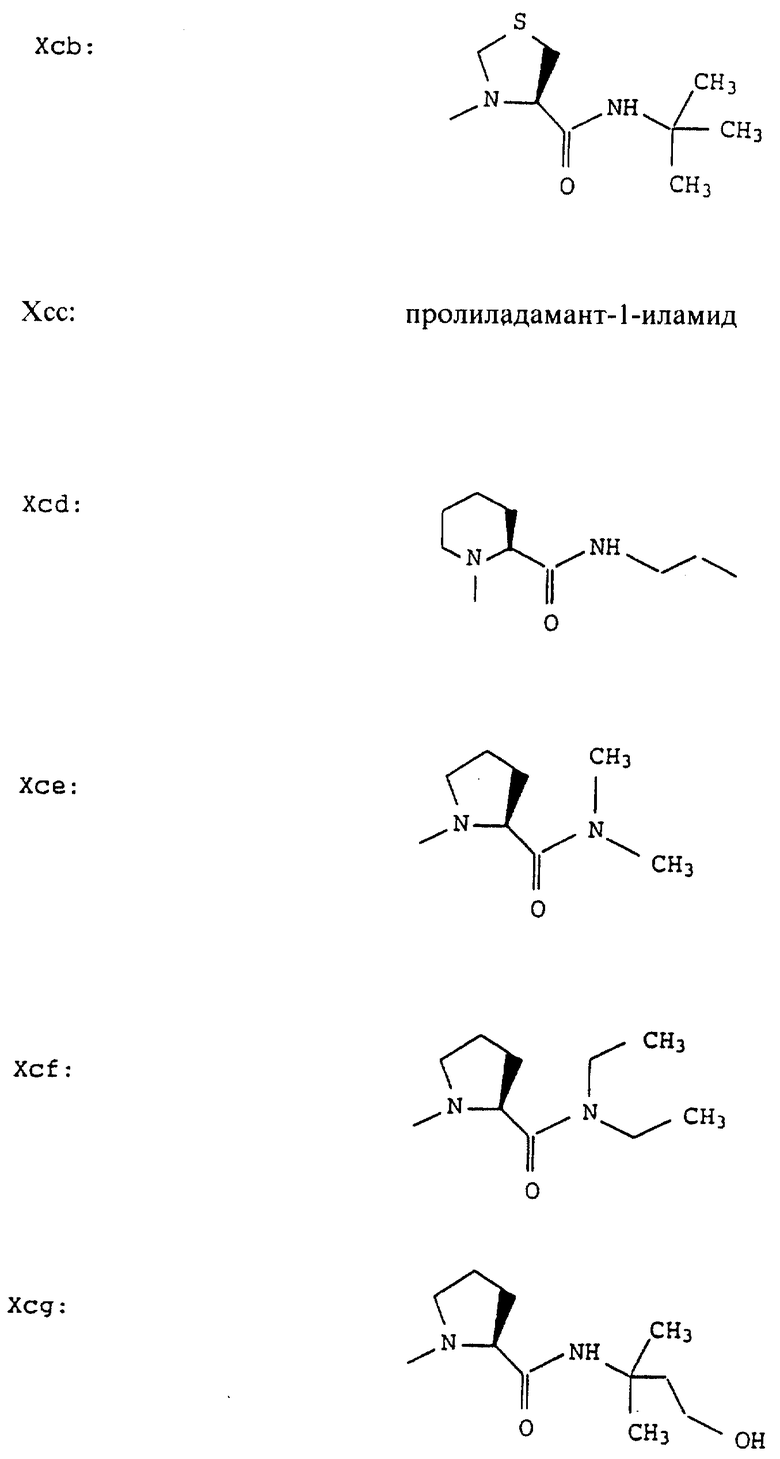
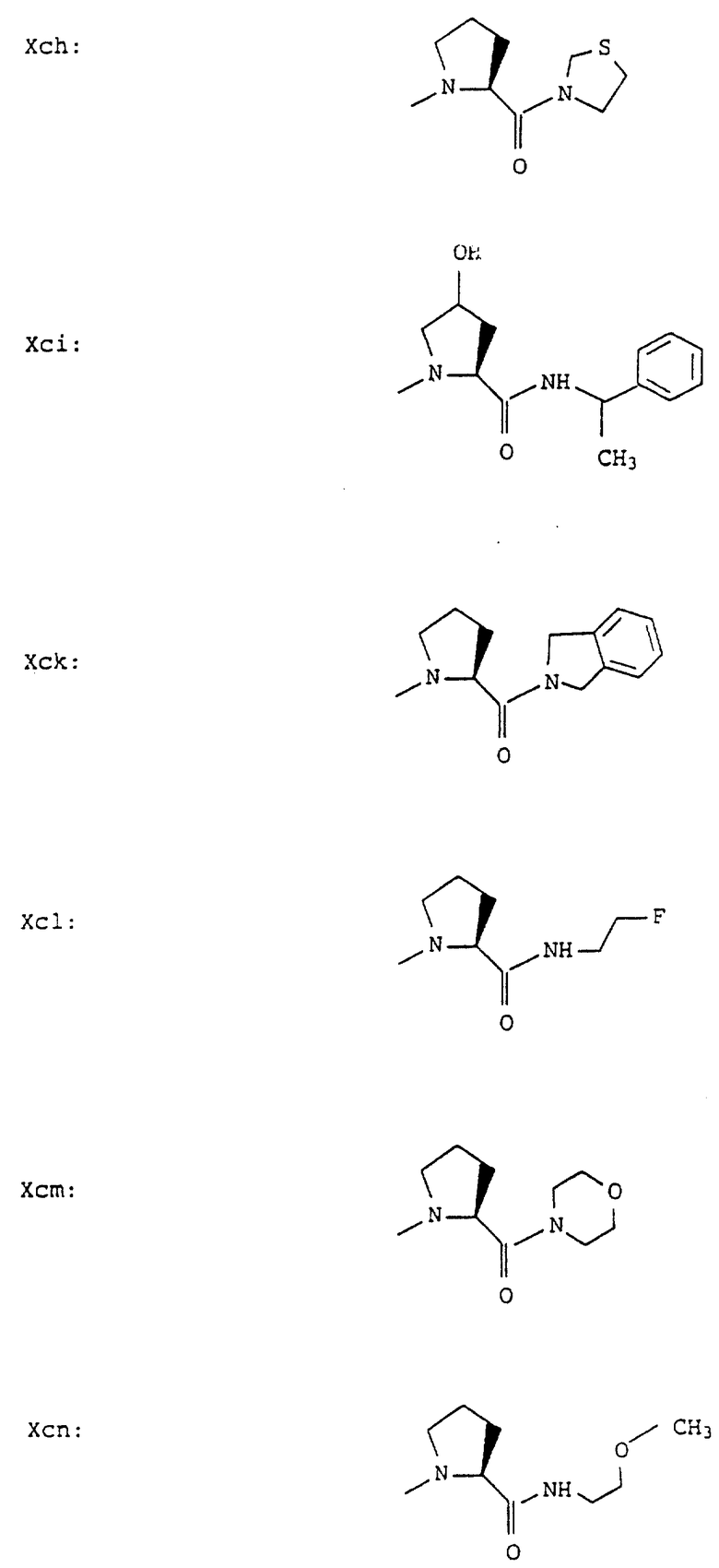
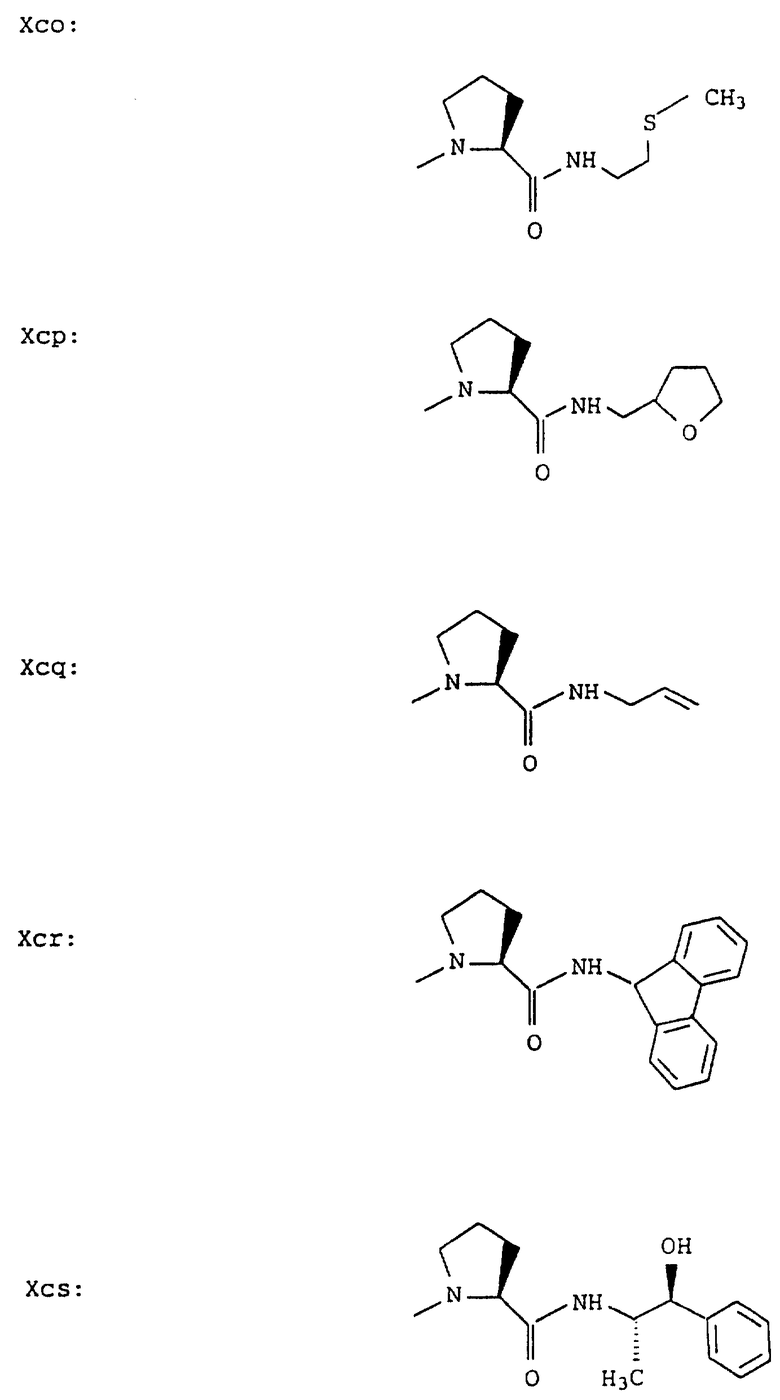
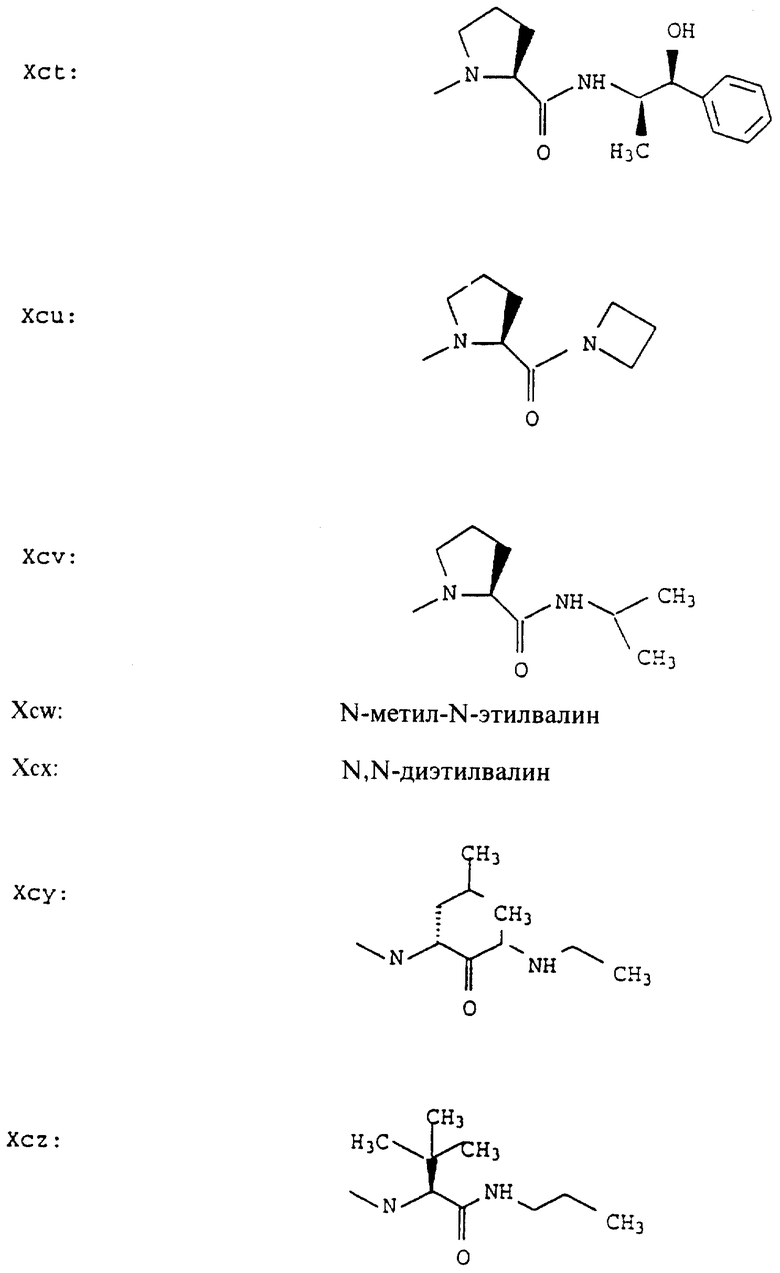
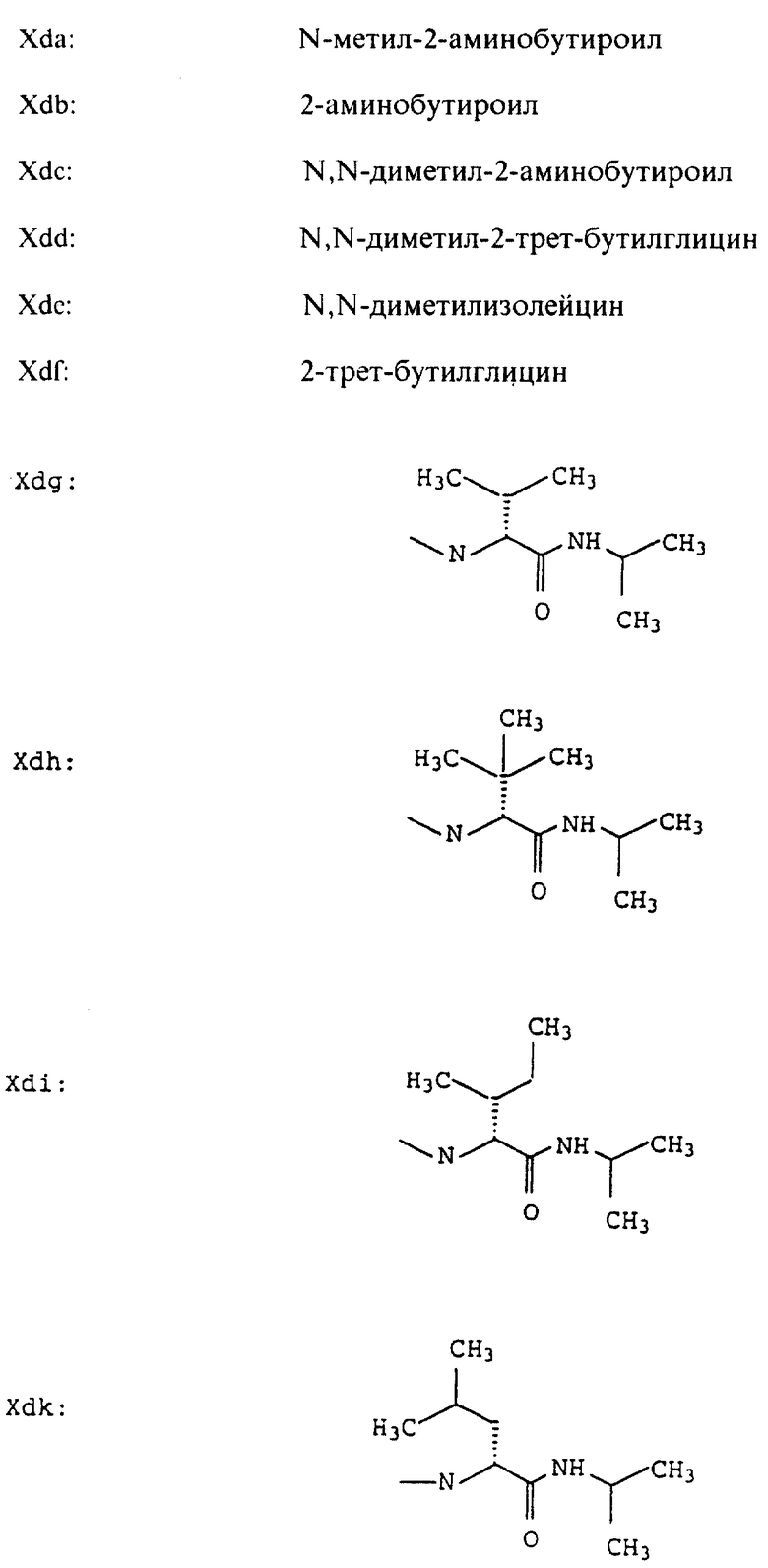
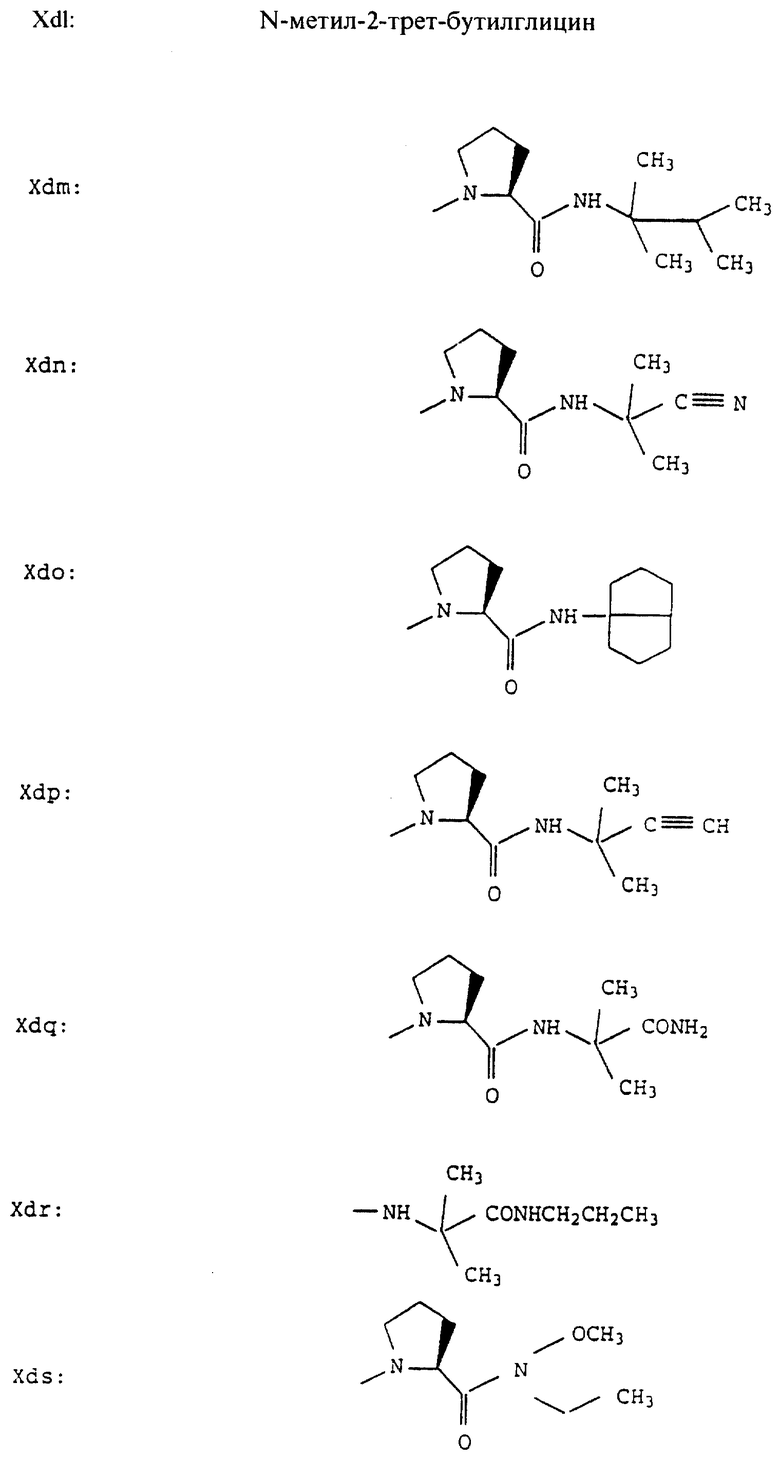





Описываются новые пептиды общей формулы (I) (CH3)2-N-CHX-CO-A-B-D-E-K, где А означает остаток валина; В означает остаток N-метилвалина; D означает остаток пролина; Е означает остаток пролина; Х означает изопропил; К означает -NНС(СН3)3, -NHCH(CH2CH2)CH(CH3)2, -NНСН(СН3)С(СН3)3, -NHCH(C2H5)2,
-NНСН(C2H5)СН(СН3)2, -NНСН(СН3)СН(СН3)2, -NНС(СН3)2C2H5, -NНС(СН3)(С2Н5)2,
-NНС(СН3)2СН(СН3)2, -NНСН(С3Н7)2, -NНСН(С2Н5)С3Н7, -NНСН(СН3)2,
-NНСН(СН3)С2Н5, -NНСН(СН3)С(СН3)3, NH-циклогексил, NH-циклогептил, -N(СН3)OСH2СН3, -N(СН3)ОС3Н7, -N(СН3)ОСН(СН3)2, -N(СН3)O(СН2)3СН3,
-N(СН3)ОСН2С6Н5, -NНС(СН3)2С6Н5, -NНС(СН3)2СН2СН3, -NНС(СН3)(СН2СН3)2,
-NНСН[СН(СН3)2] 2, -NНС(СН3)2СN, -NНСН(СН3)СН(ОН)С6Н5, -NН-С(СН3)2СН= СН2, -NHC(CH3)2C≡CH, -NHC(CH2CH3)2C≡CH, -NНС(СН3)2СН2СН2ОН, -NНС(СН3)2СН(СН3)2, -NНС(СН3)2СН2СН2СН3, -NHC(CH3)2CH2C6H5,
-N(ОСН3)СН(СН3)2, -N(ОСН3)СН2СН3, -N(ОСН3)СН2СН2СН3, -N(ОСН3)СН2С6Н5,
-N(ОСН3)С6Н5, -N(СН3)OС6Н5, -N(ОСН3)СН2СН2СН2СН3,

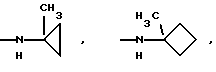

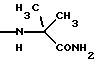
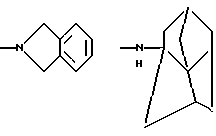
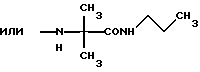
и их соли с физиологически переносимыми кислотами. Новые соединения обладают противоопухолевой активностью. В сравнении с известными аналогами, например, доластатином-10 или -15, эти соединения проявляют высокую устойчивость к разложению и могут быть использованы орально. 3 з.п. ф-лы, 2 табл.
(CH3)2-N-CHX-CO-A-B-D-E-K I,
где А означает остаток валила;
В означает остаток N-метилвалила;
D означает остаток пролила;
Е означает остаток пролила;
Х означает изопропил;
К означает -NНС(СН3)3, -NHCH(CH2CH2)CH(CH3)2, -NНСН(СН3)С(СН3)3, -NHCH(C2H5)2, -NНСН(C2H5)СН(СН3)2, -NНСН(СН3)СН(СН3)2, -NНС(СН3)2C2H5,
NНС(СН3)(С2Н5)2, -NНС(СН3)2СН(СН3)2, -NНСН(С3Н7)2, -NНСН(С2Н5)С3Н7, -NНСН(СН3)2, -NНСН(СН3)С2Н5, -NНСН(СН3)С(СН3)3, NH-циклогексил, NH-циклогептил, -N(СН3)OСH2СН3, -N(СН3)ОС3Н7, -N(СН3)ОСН(СН3)2, -N(СН3)O(СН2)3СН3,
-N(СН3)ОСН2С6Н5, -NНС(СН3)2С6Н5, -NНС(СН3)2СН2СН3, -NНС(СН3)(СН2СН3)2,
-NНСН[СН(СН3)2] 2, -NНС(СН3)2СN, -NНСН(СН3)СН(ОН)С6Н5, -NН-С(СН3)2СН= СН2, -NHC(CH3)2C≡CH, -NHC(CH2CH3)2C≡CH, -NНС(СН3)2СН2СН2ОН, -NНС(СН3)2СН(СН3)2, -NНС(СН3)2СН2СН2СН3, -NHC(CH3)2CH2C6H5,
-N(ОСН3)СН(СН3)2, -N(ОСН3)СН2СН3, -N(ОСН3)СН2СН2СН3, -N(ОСН3)СН2С6Н5,
-N(ОСН3)С6Н5, -N(СН3)OС6Н5, -N(ОСН3)СН2СН2СН2СН3,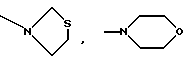
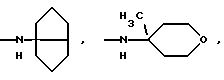
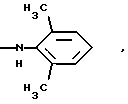

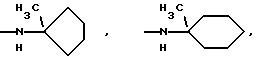
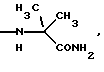
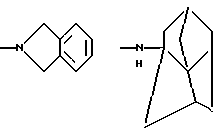

и их соли с физиологически переносимыми кислотами.
NНС(СН3)2C2H5, NНСН[СН(СН3)2] 2, NНС(СН3)2СН(СН3)2, NНСН(С3Н7)2, NНСН(C2H5)С3Н7, NH-циклогексил, NH-циклогептил, N(СН3)ОС3Н7, NНС(СН3)2C6Н5, NНС(СН3)(C2H5)2, NHC(CH3)2C≡CH, NНС(СН3)2СН2СН2OН, NHCH(CH3)2, N(ОСН3)СН2СН2С6Н5,
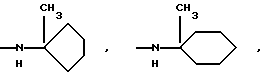
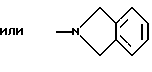
и их соли с физиологически переносимыми кислотами.
| SU 9808246 А, 23.02.1982 | |||
| DE 4415998 А, 09.11.1995 | |||
| Домовый номерной фонарь, служащий одновременно для указания названия улицы и номера дома и для освещения прилежащего участка улицы | 1917 |
|
SU93A1 |

