ПЕРЕКРЕСТНЫЕ ССЫЛКИ НА РОДСТВЕННЫЕ ЗАЯВКИ
Настоящая заявка согласно 35 U.S.C. § 119 претендует на приоритет предварительной заявки на патент США №61/014648, поданной 18 декабря 2007, которая включена в настоящую заявку посредством ссылки.
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к способу синтеза предшественников иммунологического адъюванта E6020 через β-кетоамидный интермедиат. Кроме того, настоящее изобретение относится к промежуточным соединениям в указанном способе синтеза и, для двух соединений, к их кристаллическим формам.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
Было доказано, что применение вакцин является успешным способом профилактики инфекционных заболеваний. Как правило, применение вакцин является экономически эффективным и не вызывает устойчивости к антибиотикам у целевого патогена или не влияет на нормальную микрофлору, присутствующую в организме-хозяине. Во многих случаях, например, при стимулировании противовирусного иммунитета, вакцины могут предотвратить заболевания, для которых отсутствуют реальные способы лечения или облегчения.
Вакцины действуют за счет инициирования реакции иммунной системы на тот или иной агент или антиген, как правило, инфекционный микроорганизм или его часть, которые вводятся в организм в не инфекционной или не патогенной форме. Если иммунная система однажды была «обучена» или настроена на микроорганизм, последующий контакт иммунной системы с этим микроорганизмом в форме инфекционного патогена приводит к быстрому и надежному иммунному ответу, который разрушит патоген, прежде чем он сможет размножится и инфицировать в организме-хозяине достаточное для проявления симптомов заболевания количество клеток. Агент или антиген, применяемый для обучения иммунной системы, может представлять собой целый микроорганизм с меньшей инфекционностью, известный как ослабленный микроорганизм, или, в некоторых случаях, компоненты этого микроорганизма, например, углеводы, белки или пептиды, представляющие собой различные структурные компоненты этого микроорганизма.
Во многих случаях необходимо улучшить иммунную реакцию на антигены, присутствующие в вакцине, чтобы в достаточной степени стимулировать иммунную систему с целью сделать вакцину эффективной, т.е. вызвать появление иммунитета. Многие антигены, представляющие собой белки и, в наибольшей степени, пептиды и углеводы, введенные сами по себе, не вызывают достаточного для появления иммунитета образования антител. Эти антигены необходимо представить иммунной системе таким образом, чтобы они были распознаны, как инородные объекты и вызвали иммунный ответ. С этой целью были разработаны добавки (адъюванты), которые стимулируют, усиливают и/или направляют иммунный ответ на выбранный антиген.
Лучший из известных адъювантов - полный адъювант Фрейнда - состоит из смеси микобактерий в эмульсии масло/вода. Адъювант Фрейнда действует по двум направлениям: во-первых, за счет усиления клеточно- и гуморально-опосредованного иммунитета и, во-вторых, путем блокирования быстрого распространения провоцирующего антигена («эффект депо»). Однако, вследствие часто встречающихся токсических физиологических и иммунологических реакций на этот препарат, адъювант Фрейнда не следует применять к людям.
Другой молекулой, которая, как было показано, обладает иммуностимулирующей или адъювантной активностью является эндотоксин, известный также как липополисахарид (LPS). LPS стимулирует иммунную систему путем инициирования «природной» иммунной реакции, т.е. реакции, которая способствует тому, чтобы организм распознал эндотоксин (и проникающую в организм бактерию, компонентом которой он является), причем нет необходимости в том, чтобы организм ранее подвергался его действию. В то время как LPS является слишком токсичным, чтобы реально применяться в качестве адъюванта, молекулы, структурно родственные этому эндотоксину, например, монофосфориллипид A (“MPL”), проходили тестирование в качестве адъювантов в клинических исследованиях. Было показано, что как LPS, так и MPL являются агонистами человеческого toll-подобного рецептора 4 (TLR-4). Тем не менее, в настоящее время единственным адъювантом, одобренным FDA для применения в отношении человека, является персульфат алюминия, именуемый также Alum, который используют для «депонирования» антигенов путем их осаждения. Кроме того, Alum стимулирует иммунный ответ на антигены.
E6020 является мощным агонистом рецептора TLR-4 и, следовательно, применим в качестве иммунологического адъюванта, вводимого совместно с антигенами, например, вакцинами против бактериальных и вирусных заболеваний. Например, E6020 может применяться в комбинации с любым подходящим антигеном или компонентом вакцины, например, антигенным агентом, выбранным из группы, состоящей из антигенов из патогенных и не патогенных микроорганизмов, вирусов и грибков. В качестве еще одного примера, E6020 может применяться в комбинации с белками, пептидами, антигенами и вакцинами, которые проявляют фармакологическую активность в отношении таких заболеваний и состояний, как натуральная оспа, желтая лихорадка, рак, собачья чума, холера, птичья оспа, скарлатина, дифтерия, столбняк, коклюш, грипп, бешенство, свинка, корь, заболевания ступней ног и рта, и полиомиелит, а также вирусных заболеваний, таких как герпес и родственные ему заболевания, а также гепатит и родственные ему заболевания. При применении в качестве вакцины, E6020 и антиген присутствуют в количествах, эффективных для стимулирования иммунного ответа, в случае введения в организм животного-хозяина, эмбрион или яйцеклетку, которые вакцинируют указанным составом.
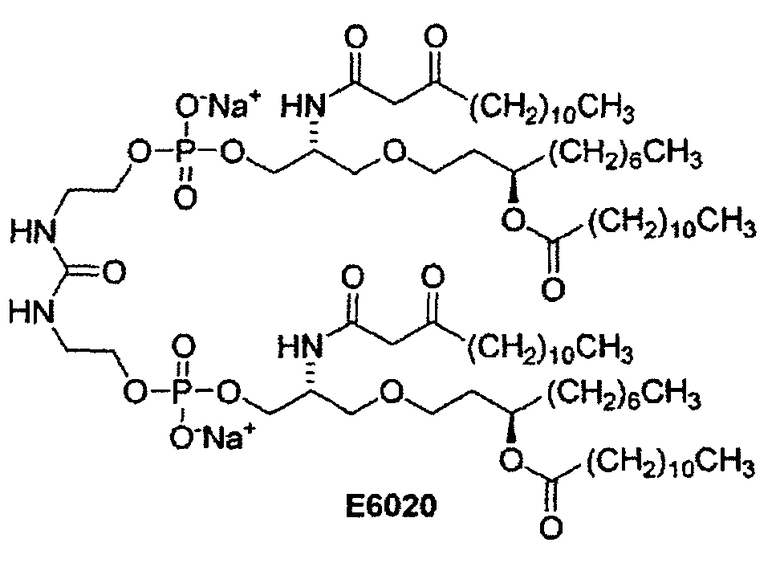
Благодаря способности стимулировать более надежный иммунный ответ по сравнению с антигеном в чистом виде, такие соединения как E6020 являются важными в иммунологическом отношении. Существует потребность в разработке способов синтеза, предназначенных для получения таких соединений, как E6020 и их синтетических предшественников, которые могут вводиться вместе с антигенами в составе вакцин. Новые способы синтеза включают новые соединения в качестве интермедиатов, и новые реакции в качестве стадий способа. В изобретении разработан улучшенный способ синтеза интермедиатов и предшественников агонистов рецептора TLR-4, например, E6020.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
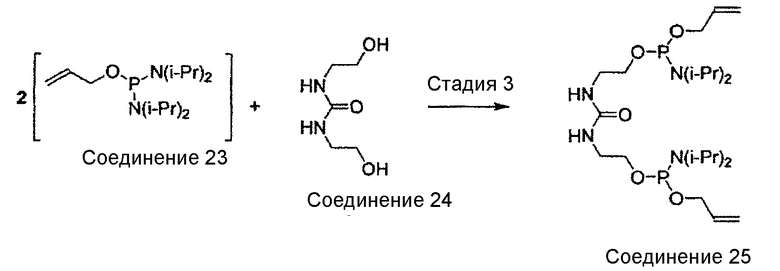
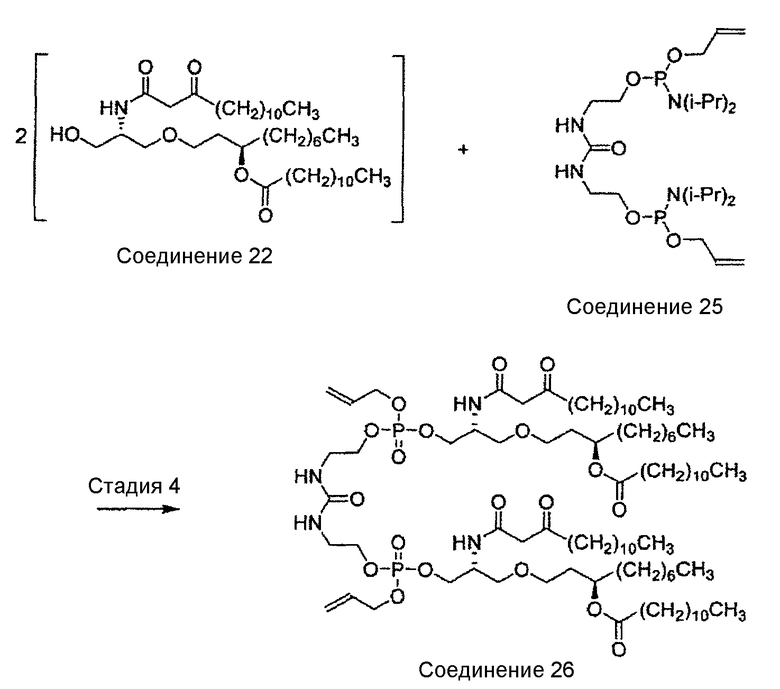
Настоящее изобретение относится к новым способам синтеза, интермедиатам и синтетическим предшественникам, ведущим к соединению 26, которое является предшественником E6020, через β-кетоамидный интермедиат, а именно, соединение 22. Способы синтеза по настоящему изобретению начинаются с соединения 14, которое служит для получения соединения 22, которое затем вводят во взаимодействие с соединением 25 для получения соединения 26 - непосредственного предшественника иммунологического адъюванта E6020. Соединение 22 и соединение 25, а также их кристаллические формы представляют собой отдельные варианты осуществления настоящего изобретения.
В другом варианте осуществления изобретение относится к соединению формулы (3):
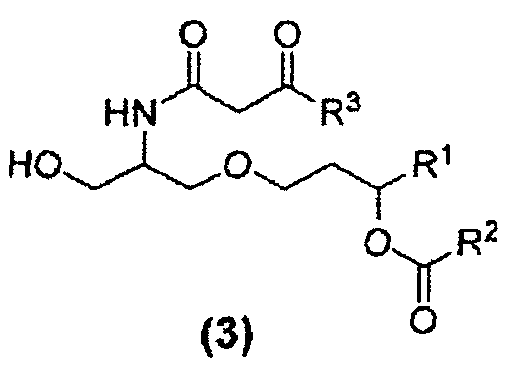
где R1,R2 и R3 в каждом случае независимо представляют собой C5-C15 алкильную группу, C5-C15 алкенильную группу или C5-C15 алкинильную группу. Кроме того изобретение относится к способу получения соединений формулы (3). β-кетоамидный интермедиат, т.е. соединение 22, принадлежит к числу соединений формулы (3).
Другим вариантом осуществления изобретения является соединение формулы (4):
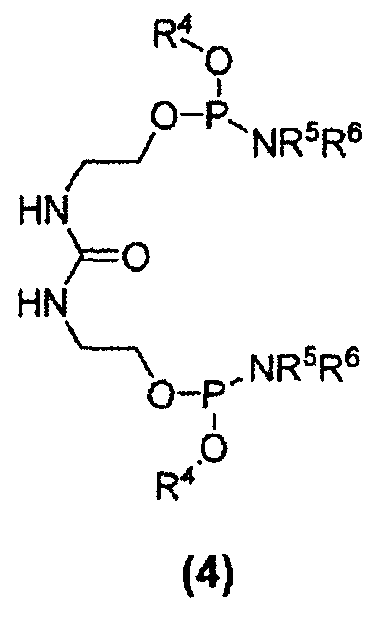
где R4 в каждом случае независимо представляет собой защитную группу, например, C1-C6 алкильную группу, C3-C5 алкенильную группу, арильную группу, бензильную группу или другую подходящую защитную группу; и каждый из заместителей R5 и R6 в каждом случае независимо представляет собой C1-C6 алкильную группу или же эти заместители, совместно с атомом азота, к которому они присоединены, образуют 5- или 6-членный гетероцикл. Кроме того, настоящее изобретение относится к способу получения соединений формулы (4). Соединение 25 является соединением формулы (4).
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
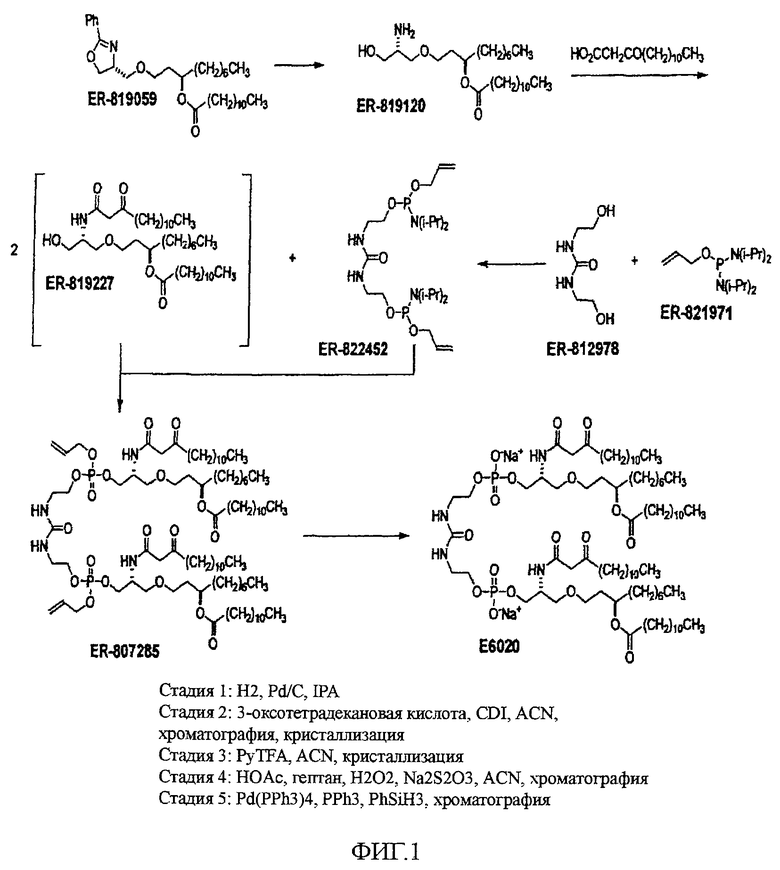
На фиг.1 в общих чертах изображен β-кетоамидный синтез соединения E6020 по настоящему изобретению.
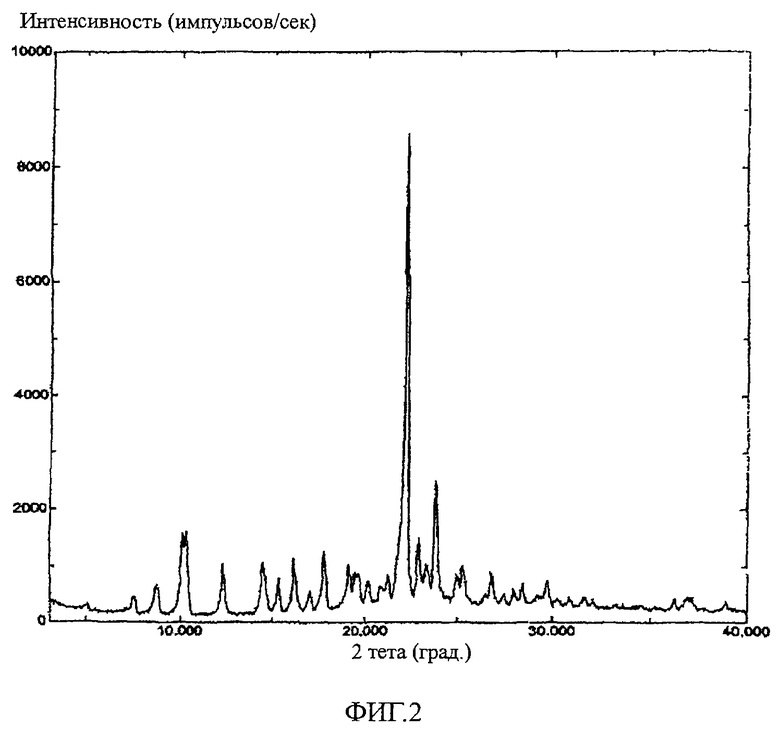
На фиг.2 показана картина дифракции рентгеновских лучей на кристаллическом порошке соединения 22.
На фиг.3 изображена DSC-термограмма кристаллического соединения 22.
На фиг.4 показана картина дифракции рентгеновских лучей на кристаллическом порошке соединения 25.
На фиг.5 показана DSC-термограмма кристаллического соединения 25.
На фиг.6 показано ORTEP-изображение кристаллического соединения 25, на котором отмечены различные атомы.
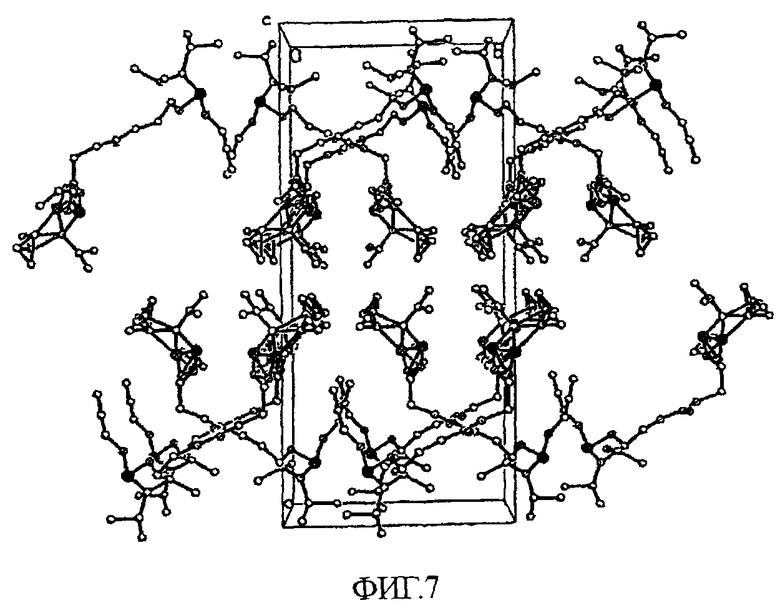
На фиг.7 показана диаграмма кристаллической упаковки кристаллического соединения 25 вдоль оси c.
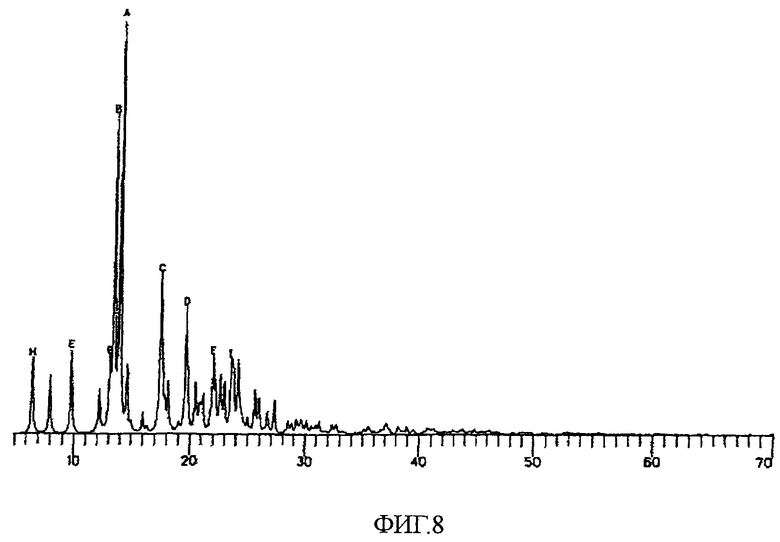
На фиг.8 показана смоделированная картина дифракции рентгеновских лучей на порошке для кристаллического соединения 25.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В заявке на патент США №11/477936 “Compounds for preparing Immunological Adjuvant”, поданной 30 июня 2006 (опубликована 12 апреля 2007 под номером US 2007-0082875 A1) описан синтез соединения E6020, и эта заявка включена в настоящую заявку посредством ссылки. Описанный синтез проходит через стадию получения уреидо димера эфира фосфорной кислоты, а именно соединения 19. На схемах 1-3 заявки US 2007-0082875 A1 показан синтез E6020 через соединение 19. Исходными веществами, показанными на схеме 1, являются ER-028694 (1,3-декандиол, коммерчески доступный от продавца Mitsui & Co. (US), New York, NY; производитель Nippon Fine Chemicals) и ER-807277 (4-оксазолметанол, 4,5-дигидро-2-фенил-,(4R)-пропандиол, коммерчески доступный от Catalytica Pharmaceuticals, Boonton, NJ). Непосредственным предшественником соединения E6020 является соединение 26.
Настоящее изобретение относится к новому способу синтеза, интермедиатам и предшественникам, ведущим к соединению 26, являющемуся предшественником соединения E6020, через β-кетоамидный интермедиат, т.е. соединение 22. Синтез по настоящему изобретению начинается с соединения 14, которое используют для получения соединения 22, которое затем вводят в реакцию с соединением 25 с получением соединения 26, т.е. синтетического предшественника иммунологического адъюванта E6020. Эти стадии синтеза по настоящему изобретению, т.е. получение β-кетоамидного интермедиата (3), с последующей конденсацией β-кетоамида (3) и дифосфорамидита мочевины (4), показаны на схеме 4 и затем подробно описаны. Соединения 22 и 25 и их кристаллические формы являются отдельными вариантами осуществления настоящего изобретения.
Получение кристаллической формы соединения, например, соединения 22 или 25 является чрезвычайно полезным при разработке лекарственных средств. Твердые формы соединения (кристаллические или аморфные) могут иметь различные физические и химические свойства, например, растворимость, стабильность или различаться возможностью воспроизведения. Эти свойства часто делают возможным оптимизацию производственных процессов, особенно, когда образуется кристаллический интермедиат. В многостадийном синтезе, например, синтезе, описанном в настоящем изобретении, получают интермедиаты, причем нежелательные побочные продукты или загрязнения могут переходить в продукт из более ранних стадий. В синтез вводят частые стадии фильтрования, разделения и/или очистки для удаления нежелательных побочных продуктов или загрязнений. Введение этих стадий не только может увеличить затраты на производство, но также способно уменьшить итоговый выход продукта синтеза. Присутствие в многостадийном синтезе кристаллического интермедиата дает возможность решить эти проблемы. Кристаллический интермедиат обеспечивает определенные преимущества - интермедиат высокой степени чистоты может уменьшить необходимость очистки на других стадиях и снизить себестоимость способа синтеза.
Схема 1
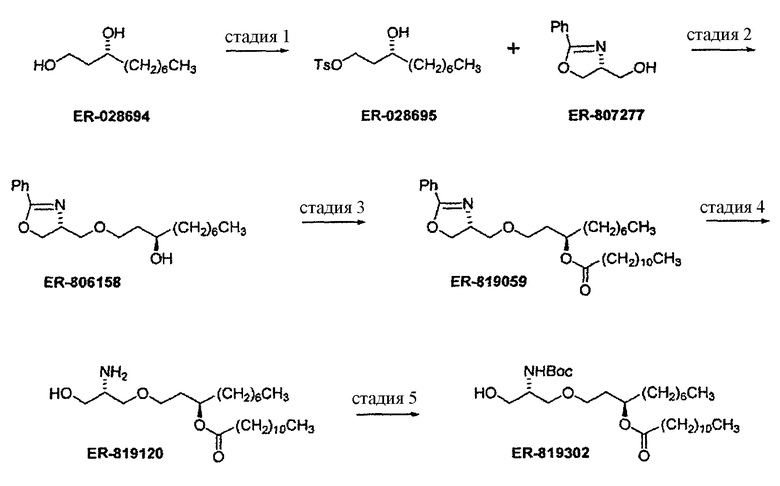
Стадия 1: TsCl, Et3N, ТГФ.
Стадия 2: NaHMDS, ТГФ, хроматография, кристаллизация.
Стадия 3: лауриновая кислота, EDC, DMAP, CH2Cl2, хроматография.
Стадия 4: H2, 10% Pd/C, хроматография.
Стадия 5: Boc2O, ТГФ, хроматография.
Схема 2
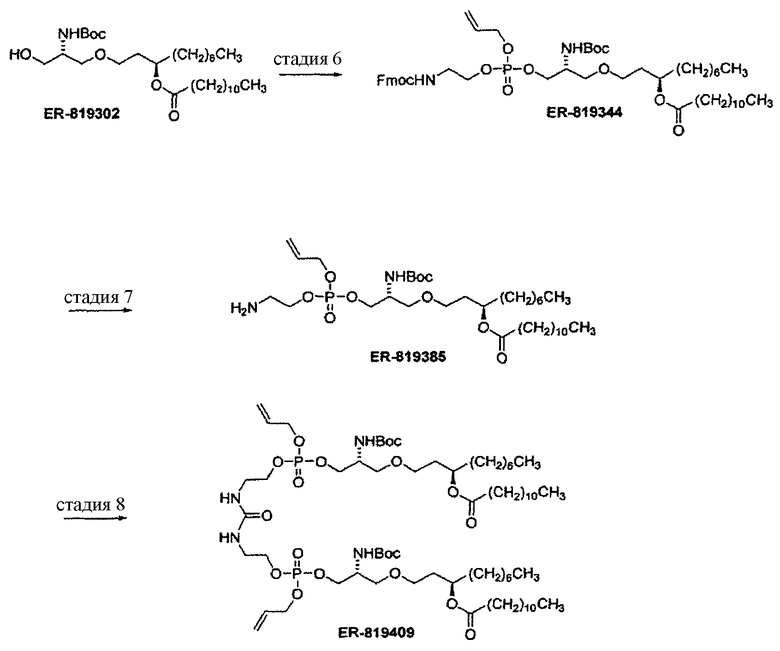
Стадия 6: a. диизопропиламин; Py·TFA, ((i-Pr)2N)2P(OAllyl), CH2Cl2.
b. HOAc, Py·TFA, FmocNH(CH2)2OH
c. H2O2, хроматография.
Стадия 7: диметиламин, ТГФ.
Стадия 8: 20% фосген в толуоле, насыщенный водный NaHCO3,
хроматография
Схема 3
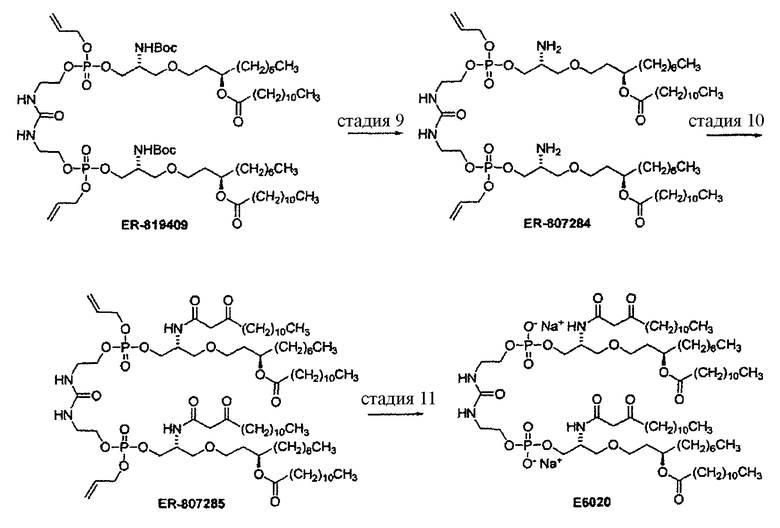
Стадия 9: TFA, CH2Cl2.
Стадия 10: 3-оксотетрадекановая кислота, EDC, ДМФА, хроматография.
Стадия 11: Pd(PPh3)4, PPh3, PhSiH3, ТГФ, хроматография.
Схема 4
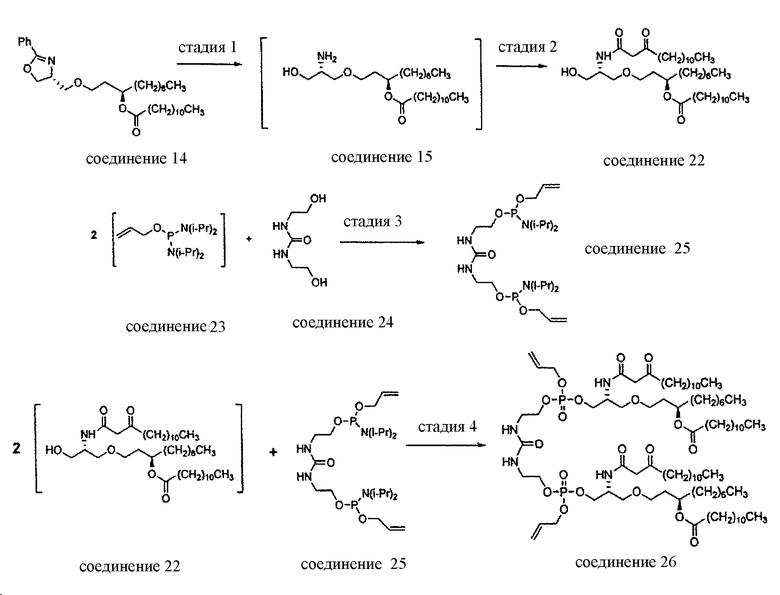
Стадия 1: H2, Pd/C, IPA.
Стадия 2: 3-оксотетрадекановая кислота, CDI, ACN, кристаллизация.
Стадия 3: Py·TFA, ACN, кристаллизация.
Стадия 4: ACN, гептан, HOAc, H2O2, Na2S2O3, хроматография.
Получение β-кетоамидоспиртового интермедиата (3)
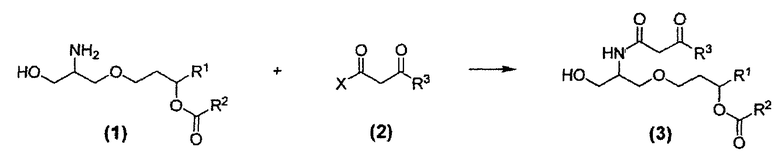
Синтез соединения 26 и аналогичных соединений включает на первом этапе взаимодействие α-гидроксиамина формулы (1) с соединением формулы (2) с получением β-кетоамида формулы (3) в подходящих условиях проведения реакции. Реакция показана на схеме 5.
Схема 5
 а
а
Получение соединения (3) согласно схеме 5 может быть осуществлено с применением различных способов. Группы R1, R2 и R3 соответствуют данным ниже определениям. Поскольку заместители R1, R2 и R3 могут меняться независимо друг от друга, соединение (3) может включать симметричные или асимметричные группы, обозначенные как R1, R2 и R3. В соединении (2) X представляет собой подходящую уходящую группу, как, например, OH, Cl, F, имидазолидил, карбонат и сложноэфирный фрагмент. Предпочтительные уходящие группы включают OH, Cl, F, имидазолидил, триметилацетокси, этилкарбонат, метилкарбонат, изобутилкарбонат или группу формулы Z:
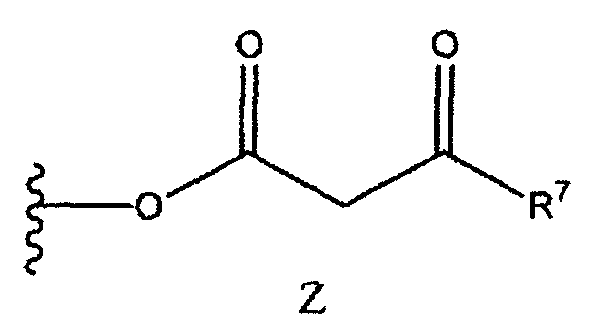
где группа R7 и группа R3 в формуле (2) являются одинаковыми, так что соединение формулы (2) представляет собой симметричный ангидрид бета-кетоэфира. Такие ангидриды могут быть получены путем конденсации двух идентичных молекул бета-кетокислоты.
В одном из способов получения, соединение (3) могло бы быть получено путем конденсации соединения (1) с соединением (2), в котором X представляет собой имидазолид. Последнее соединение получают при активации соединения (2), в котором X представляет собой OH (карбоновой кислоты) действием реагента CDI (карбонилдиимидазола) в полярном апротонном растворителе, например, ацетонитриле.
Во втором способе получении, соединение 3 могло бы также быть получено при конденсации соединения (1) с соединением (2), в котором X представляет собой OH (карбоновой кислотой) в присутствии реагента, способствующего образованию амидной связи, например, HBTU (гексафторфосфата O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония) или другого сшивающего реагента того же семейства и третичного амина (например, основания Хюнига, N,N-диизопропилэтиламина), в таком растворителе, как ДМФА или CH2Cl2. Кроме того, для активации соединения (2), в котором X представляет собой OH (карбоновой кислоты), могут применяться карбодимидные реагенты, например, EDC (гидрохлорид N-(3-диметиламинопропил)-N-этил карбодиимида).
Третий способ получения соединения (3) представляет собой конденсацию соединения (1) с соединением (2), в котором X означает F (ацилфторидом), в присутствии третичного амина (например, основания Хюнига) в таком растворителе, как CH2Cl2. Соединение (2), в котором X означает F (ацилфторид) может быть получено при активации соединения (2), в котором X означает OH (карбоновой кислоты) действием реагента TFFH (гексафторфосфата фтор-N,N,N',N'-тетраметилформамидиния) в таком растворителе, как CH2Cl2.
Соединение (3) могло бы быть получено также согласно четвертому способу синтеза, путем конденсации соединения (1) с соединением (2), в котором X представляет собой Cl (ацилхлоридом) в присутствии третичного амина (например, основания Хюнига) в таком растворителе, как CH2Cl2. Соединение (2), в котором X представляет собой Cl (ацилхлорид) может быть получено при активации соединения (2), в котором X означает OH (карбоновой кислоты) действием оксалилхлорида в таком растворителе, как CH2Cl2.
Пятый способ позволяет получить соединение (3) путем конденсации соединения (1) с соединением (2) в форме ангидрида карбоновой кислоты, в котором X представляет собой карбонат, в присутствии третичного амина (например, основания Хюнига) в таком растворителе, как CH2Cl2. Указанный ангидрид получают путем активации соединения (2), в котором X представляет собой OH (карбоновой кислоты) этилхлорформиатом в присутствии третичного амина (например, триэтиламина) в таком растворителе, как CH2Cl2.
Согласно шестому способу, соединение (3) можно получить конденсацией соединения (1) со смешанным ангидридом (2), в котором X означает сложноэфирную группу, в присутствии третичного амина (например, основания Хюнига) в таком растворителе, как CH2Cl2. Указанный смешанный ангидрид получают путем активации соединения (2), в котором X означает OH (карбоновой кислоты) пивалоилхлоридом в присутствии третичного амина (например, триэтиламина) в таком растворителе, как CH2Cl2.
Соединение (3) также можно было бы получить по седьмому способу, в котором соединение (2), где X означает OH (карбоновую кислоту), во-первых, превращают в симметричный ангидрид действием оксалилхлорида и третичного амина (например, триэтиламина) в таком растворителе, как CH2Cl2. Полученный ангидрид конденсируют с соединением (1) в присутствии третичного основания (например, триэтиламина) в таком растворителе, как CH2Cl2.
Как показано ниже в примере 1, соединение формулы (1) может быть получено гидрированием соединения формулы Y:
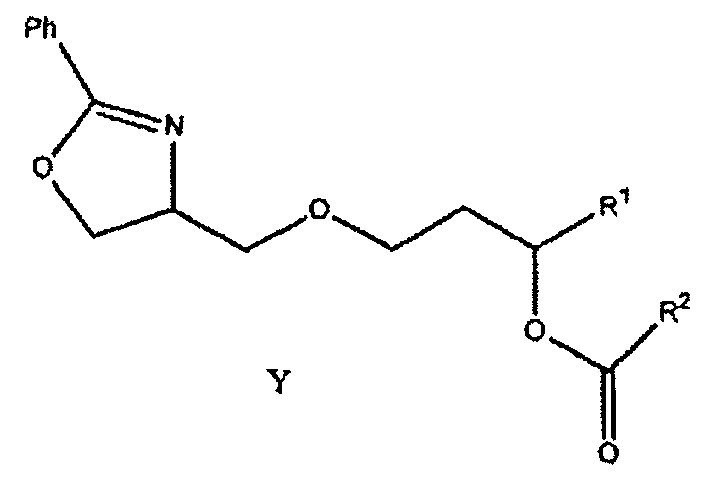
где заместители R1 и R2 соответствуют данным выше определениям. Гидрирование можно осуществлять в любом подходящем растворителе, например, этаноле, изопропаноле и других растворителях, известных в технике, но предпочтительно проводить его в изопропаноле.
В формулах (1), (2) и (3) каждый из заместителей R1, R2 и R3 независимо в каждом случае может представлять собой C5-C15 алкильную группу, C5-C15 алкенильную группу или C5-C15 алкинильную группу. Эта алкильная группа, алкенильная группа и алкинильная группа могут быть замещенными или незамещенными, линейными или разветвленными и предпочтительно имеют линейную цепь. В предпочтительном варианте осуществления R1 представляет собой C5-C12 алкильную группу, C5-C12 алкенильную группу или C5-C12 алкинильную группу и, наиболее предпочтительно, C5-C9 алкильную группу, C5-C9 алкенильную группу или C5-C9 алкинильную группу, в то время как каждый из заместителей R2 и R3 независимо в каждом случае представляет собой C7-C14 алкильную группу или C7-C14 алкенильную группу и более предпочтительно C9-C13 алкильную группу или C9-C13 алкенильную группу. Предпочтительно R1 является C7 алкильной группой, наиболее предпочтительно незамещенной н-гептильной группой, и обе группы R2 и R3 являются C11 алкильными группами, наиболее предпочтительно незамещенной н-ундецильными группами.
Алкильные, алкенильные и алкинильные группы, упомянутые в качестве конкретных вариантов осуществления различных групп R1,R2 и R3, и рассмотренные выше с точки зрения настоящего изобретения, могут быть замещенными или незамещенными. Примеры заместителей включают, не ограничиваясь этим, галогены (например, F, Cl, Br и I); C1-C6 алкоксигруппы (например, -OCH3, -OCH2CH3, -OCH(CH3)2 и т.п.); C1-C6 галогеналкильные группы (например, -CF3, -CH2CF3, -CHCl2 и т.п.); C1-C6 алкилтиогруппы; амиды; -NO2; и -CN. Если не указано иное, алкильные, алкенильные и алкинильные группы могут иметь линейные или разветвленные цепи, причем линейные цепи являются в основном предпочтительными.
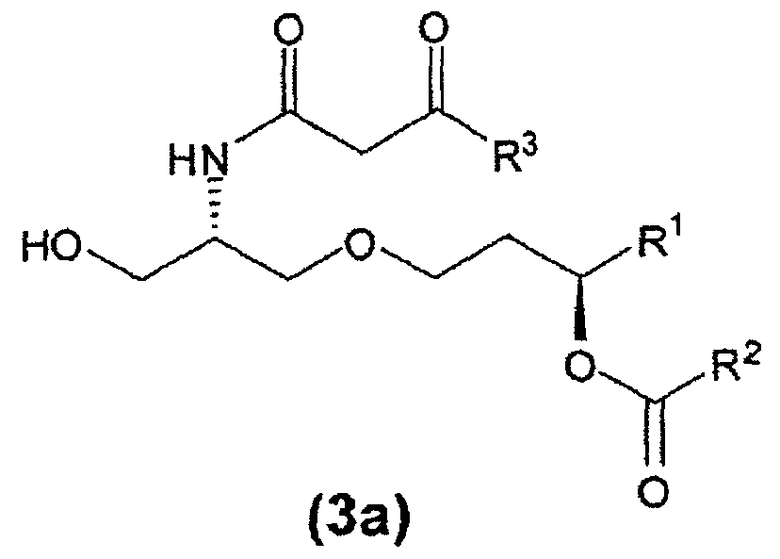
β-кетоамидоспирты формулы (3) являются новыми соединениями и они применимы, в том числе, как интермедиаты при получении таких соединений, как E6020. Соединения формулы (3) являются отдельным вариантом осуществления настоящего изобретения. β-кетоамидоспирты, имеющие стереохимию формулы (3a) являются особенно предпочтительными.
В случае синтеза соединения E6020, β-кетоамидоспирт представляет собой соединение 22. Соединение 22 и его кристаллическая форма также являются отдельными вариантами осуществления настоящего изобретения. Синтез соединения 22, исходя из соединения 14, описан ниже в примере 1, и получение характеристик твердой кристаллической формы соединения 22 описано ниже в примере 2.
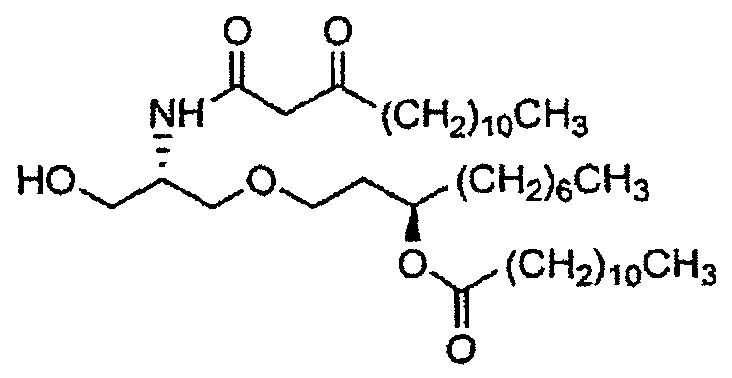
соединение 22
Получение дифосфорамидита мочевины (4)
Дифосфорамидит мочевины формулы (4) представляет собой другое новое промежуточное соединение в β-кетоамидоспиртовом способе синтеза соединения E6020. Следовательно, соединения формулы (4) являются отдельным вариантом осуществления настоящего изобретения. Дифосфорамидиты мочевины формулы (4) могут быть получены взаимодействием 1,3-бис(2-гидроксиэтил)мочевины с двумя молекулами фосфордиамидита формулы (7), как показано на схеме 6, где заместители R4, R5 и R6 соответствуют данному ниже описанию.
Схема 6
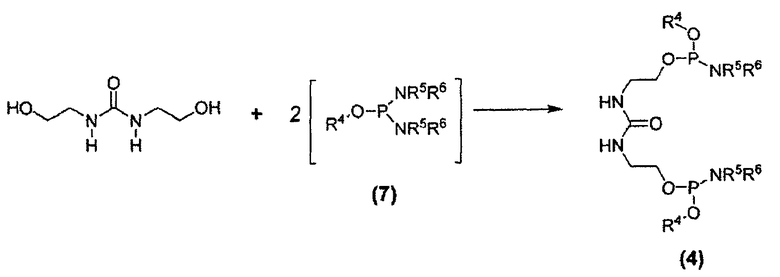
В некоторых вариантах осуществления взаимодействие 1,3-бис(2-гидроксиэтил)мочевины с фосфордиамидитом осуществляют взаимодействием бис-гидроксисоединения с подходящим активирующим агентом, например, известным специалисту в данной области техники, и затем добавляют фосфордиамидит к полученной реакционной смеси. В некоторых вариантах осуществления упомянутый активирующий агент выбран из группы, состоящей из (1H)-тетразола и трифторацетата пиридиния. В некоторых вариантах осуществления активирующий агент представляет собой трифторацетат пиридиния. В некоторых вариантах осуществления взаимодействие по схеме 6 проводят в растворителе, выбранном из группы, состоящей из ацетонитрила, дихлорметана, дихлорэтана и трет-бутилметилового эфира. В некоторых вариантах осуществления для проведения реакции по схеме 6 в качестве растворителя применяют ацетонитрил.
Конденсация β-кетоамидоспирта (3) с дифосфорамидитом мочевины (4)
Как показано выше на схеме 4, синтез соединения 26 включает реакцию конденсации β-кетоамидоспирта формулы (3) с дифосфорамидитом мочевины формулы (4) с образованием фосфита β-кетоамидомочевины формулы (5), который может быть окислен до фосфата β-кетоамидомочевины формулы (6). Это показано ниже на схеме 7.
Схема 7
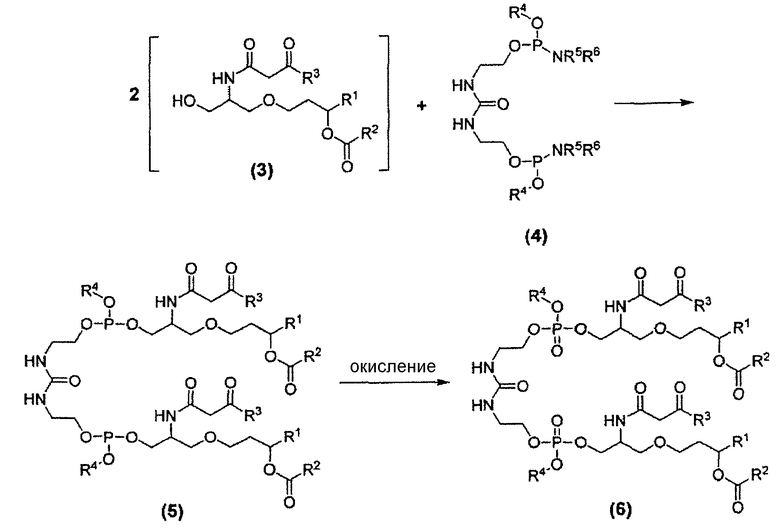
В формулах (4), (5) и (6) заместители R1, R2 и R3 соответствуют приведенному выше описанию, включая предпочтительные варианты их осуществления, и независимы друг от друга. В каждом случае заместители R1, R2 и R3 могут быть одинаковыми или различными. Другими словами, соединение формулы (6) может включать одинаковые или различные алкильные, алкенильные и/или алкинильные группы, в каждом из положений, где находятся заместители R1, R2 и R3. Каждая из групп R4 может независимо являться защитной группой, включая, но не ограничиваясь этим, такие группы, как: алкильная группа или замещенная алкильная группа, например, метил, этил, изопропил, трет-бутил и т.п., предпочтительно C1-C6 алкильная группа; алкенильная группа или замещенная алкенильная группа, например, аллил, 2-метилпропенил, бутенил и т.п., предпочтительно C3-C5 алкенильная группа; алкинильная группа, предпочтительно C3-C5 алкинильная группа; циклоалкильная группа, например, циклогексил; этильная группа, замещенная в положение 2, например, 2-цианоэтил, 2-циано-1,1-диметилэтил, 2-(триметилсилил)этил и т.п.; галогенэтильная группа, например, 2,2,2-трихлорэтил, 2,2,2-трихлор-1,1-диметилэтил, 2,2,2-трибромэтил и т.п.; бензильная или замещенная бензильная группа, например, бензил, 4-нитробензил, 4-хлорбензил и т.п.; арильная или замещенная арильная группа, например, фенил, 4-нитрофенил, 4-хлорфенил, 2-хлорфенил, 2-метилфенил, 2,6-диметилфенил, 2-бромфенил и т.п.; и силильная группа, например, триметилсилил и т.п. Особенно предпочтительными защитными группами, обозначенными символом R4, являются метил, этил, трет-бутил, аллил, 2-метилпропенил, бутенил, 2-цианоэтил (NCCH2CH2-), 2-(триметилсилил)этил ((CH3)3SiCH2CH2-) и 2,2,2-трихлорэтил (Cl3CCH2-). Как и для заместителей R1, R2 и R3, в каждом случае заместители R4 могут отличаться друг от друга, в рамках определения заместителя R4.
Заместители R5 и R6 в каждом случае независимо представляют собой C1-C6 алкильную группу, С3-С6 алкенильную группу или C3-C6 алкинильную группу, где указанные алкильная, алкенильная и алкинильная группы могут являться замещенными или незамещенными, или же совместно с атомом азота, к которому они присоединены, заместители R5 и R6 могут образовывать 5- или 6-членный гетероцикл. Этот гетероцикл может включать дополнительные гетероатомы, например, N, O и/или S; может быть насыщенным или ненасыщенным, а также может быть замещенным или незамещенным. Примеры заместителей включают, не ограничиваясь перечисленными, галогены (например F, Cl, Br и I); С1-С6 алкоксигруппы (например, -OCH3, -OCH2CH3, -OCH(CH3)2 и т.п.); С1-С6 галогеналкильные группы (например, -CF3, -CH2CF3, -CHCl2 и т.п.); С1-С6 алкилтиогруппы; группу -NO2; и группу -CN. Подходящие гетероциклические группы включают, но не ограничиваются этим, пиперидил, морфолинил, тиоморфолинил, пирролидил и т.п. Алкильные, алкенильные и алкинильные группы, обозначаемые символами R4, R5 и R6, могут быть замещенными или незамещенными, линейными или разветвленными. Примеры заместителей включают, не ограничиваясь перечисленными галогены (например F, Cl, Br и I); С1-С6 алкоксигруппы (например, -OCH3, -OCH2CH3, -OCH(CH3)2 и т.п.); С1-С6 галогеналкильные группы (например, -CF3, -CH2CF3, -CHCl2 и т.п.); С1-С6 алкилтиогруппы; группу -NO2; и группу -CN. В предпочтительном варианте осуществления заместитель R4 представляет собой C3-C5 алкенильную группу, и каждый из заместителей R5 и R6 независимо представляют собой разветвленную C1-C6 алкильную группу. Предпочтительно, R4 является аллильной группой, и R5 и R6 являются изопропильными группами.
В некоторых вариантах осуществления соединение формулы (3) вводят в реакцию с соединением формулы (4), добавляя растворитель к смеси соединения формулы (3) и соединения формулы (4), осуществляя перемешивание до полного растворения всех твердых веществ и добавляя к полученной смеси уксусную кислоту. В некоторых вариантах осуществления растворитель является безводным и может представлять собой смесь сорастворителей. В некоторых вариантах осуществления эта смесь сорастворителей включает ацетонитрил и углеводородный растворитель и в предпочтительном варианте осуществления растворитель может представлять собой, например, смесь безводного ацетонитрила и безводного гептана. В некоторых вариантах осуществления смесь сорастворителей включает гептан и ацетонитрил. В некоторых вариантах осуществления масса гептана в смеси сорастворителей в 4-6 раз превышает массу соединения формулы (3). В некоторых вариантах осуществления масса гептана в смеси сорастворителей в 4,5-5,5 раз превышает массу соединения формулы (3). В некоторых вариантах осуществления масса ацетонитрила в смеси сорастворителей в 1-2 раза превышает массу соединения формулы (3). В некоторых вариантах осуществления масса ацетонитрила в смеси сорастворителей в 1,2-1,5 раза превышает массу соединения формулы (3). В некоторых вариантах осуществления масса использованной в реакции уксусной кислоты составляет от 1 до 2 мольных эквивалентов количества соединения формулы (3). В некоторых вариантах осуществления масса использованной в реакции уксусной кислоты составляет от 1 до 2 мольных эквивалентов количества соединения формулы (3). В некоторых вариантах осуществления количество использованной в реакции уксусной кислоты составляет от 1,2 до 5 мольных эквивалентов количества соединения формулы (3). В некоторых вариантах осуществления температуру реакционной смеси во время добавления уксусной кислоты поддерживают около примерно 20-25°C.
В некоторых вариантах осуществления соединение формулы (5), полученное по способу схемы 7, окисляют до соединения формулы (6) действием окисляющего агента. В некоторых вариантах осуществления этим окисляющим агентом является пероксид водорода, окислитель Oxone®, mCPBA (мета-хлорпербензойная кислота) и т.п. В некоторых вариантах осуществления пероксид водорода применяют в виде 30% раствора (масс.) H2O2 в воде. В некоторых вариантах осуществления реакционную смесь, содержащую образовавшееся соединение формулы (5), разбавляют перед стадией окисления дополнительным количеством гептана. В некоторых вариантах осуществления масса этого дополнительно добавленного гептана составляет от 5 до 10 масс использованного соединения формулы (3). В некоторых вариантах осуществления масса этого дополнительно добавленного гептана примерно в 8 раз превышает массу использованного соединения формулы (3). В некоторых вариантах осуществления реакционную смесь, содержащую образовавшееся соединение формулы (5), охлаждают до примерно -5-10°C после добавления дополнительного гептана и перед добавлением окисляющего агента. В некоторых вариантах осуществления реакционную смесь, содержащую образовавшееся соединение формулы (5), охлаждают до примерно 0-5°C после добавления дополнительного гептана и перед добавлением окисляющего агента. В некоторых вариантах осуществления температуру реакционной смеси после добавления окисляющего агента поддерживают около примерно -5-10°C. В некоторых вариантах осуществления температуру реакционной смеси после добавления окисляющего агента поддерживают около примерно 0-2°C. В некоторых вариантах осуществления реакционную смесь после добавления окисляющего агента перемешивают при температуре примерно -5-10°C до завершения реакции. В некоторых вариантах осуществления реакционную смесь после добавления окисляющего агента перемешивают при температуре примерно -1-2°C до завершения реакции. В некоторых вариантах осуществления за ходом реакции наблюдают с помощью ВЭЖХ, чтобы определить момент ее завершения. В некоторых вариантах осуществления реакционную смесь после завершения реакции гасят пентагидратом тиосульфата натрия для разрушения избытка пероксида.
В случае синтеза соединения 26, соединение формулы (4) представляет собой соединение 25. Соединение 25 и его кристаллическая форма также являются отдельными вариантами осуществления настоящего изобретения. Синтез соединения 25, начиная с дигидроксимочевины 24, описан ниже в примере 4, и получение характеристик твердой формы кристаллического соединения 25 описано ниже в примере 5.
В примере 6 ниже описано получение синтетического предшественника E6020 - соединения 26 (варианта соединения формулы (6), где R1 представляет собой гептил, оба заместителя R2 и R3 являются н-ундецилами и R4 представляет собой аллил) путем взаимодействия соединения 22 и соединения 25.
Описанный в настоящем описании способ синтеза может быть адаптирован для получения любого из всех возможных стереоизомеров соединения 26 и, соответственно E6020, например, показанных ниже соединений (A) и (B) соответственно, имеющих следующие структуры:
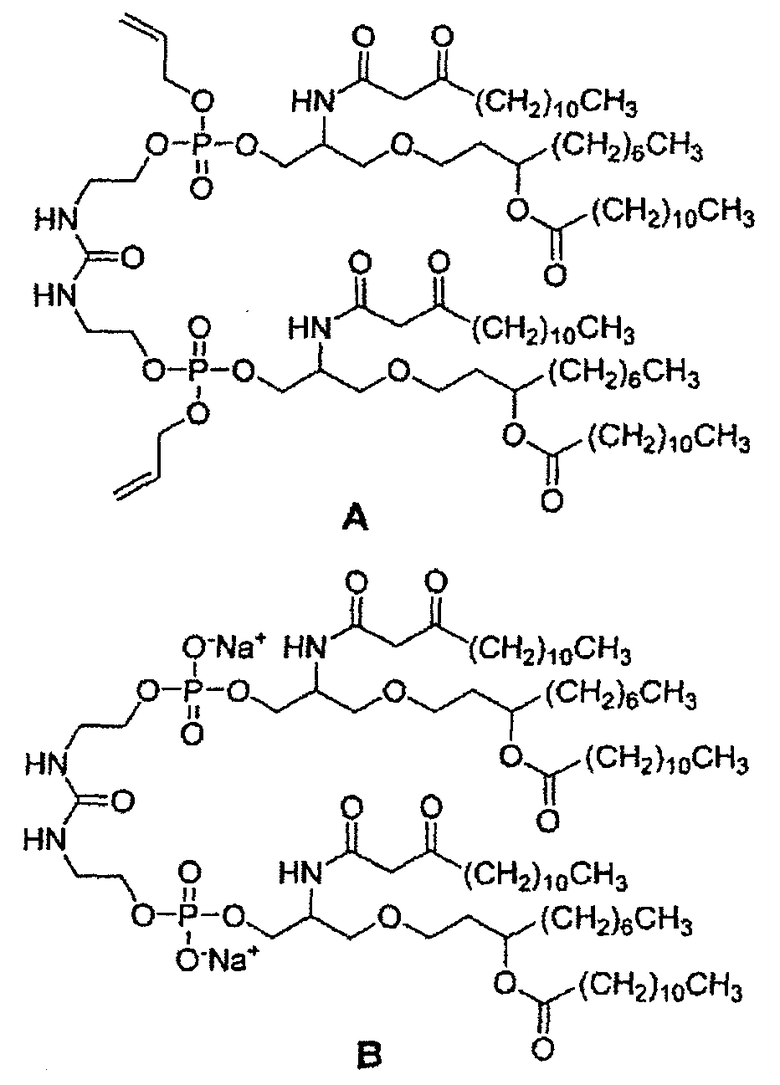
Хотя приведенные ниже примеры раскрывают получение конкретного стереоизомера, считается, что способы получения других стереоизомеров соединения 26 входят в объем настоящего изобретения. Способы синтеза по настоящему изобретению хорошо подходят для получения чистых стереоизомеров, для которых определена начальная стереохимия исходного соединения (3) и используется 2-кратный избыток соединения (3) по отношению к дифосфорамидиту мочевины (4), как показано на схеме 7 (выше). Этот способ мог бы дать возможность получения четырех чистых стереоизомеров, в т.ч. E6020 с конфигурацией (R,R,R,R) в положениях 1,6,22,27; ER-824156 (S,S,S,S); ER-804053 (R,S,S,R); и ER-824095 (S,R,R,S). Для получения других стереоизомеров (ER-826685 (R,R,S,R); ER-824887 (R,R,R,S); ER-826682 (R,R,S,S); ER-827905 (R,S,R,S); ER-826683 (R,S,S,S); ER-804097 (S,S,R,S)), можно было бы приготовить смесь исходных продуктов, в которой использовался бы 1 эквивалент исходного соединения (3) с определенной конфигурацией в положениях 2 и 7, тогда как второй эквивалент исходного соединения (3) мог бы иметь другую конфигурацию в положениях 2 и 7. В итоге реакция (по схеме 7) могла бы привести к 3 различным стереоизомерам соединения (6), которые можно разделить колоночной хроматографией и определить структуру с помощью сравнения данных 1H-ЯМР и ВЭЖХ с достоверными данными для стереоизомеров, полученных ранее по первому способу синтеза, описанному выше, и отдельному способу, описанному в патенте США №6290973. Патенты США №№6551600; 6290973 и 6521776, в которых раскрыты другие пути синтеза E6020 и родственных соединений, предоставляют полезную дополнительную информацию относительно получения некоторых реагентов и исходных соединений и включены в настоящую заявку посредством ссылки.
ПРИМЕРЫ
В приведенных ниже примерах, в случаях, когда для описания реактора (например, реакционного сосуда, колбы, стеклянного реактора и т.п.) используется термин «создана инертная атмосфера», подразумевается, что воздух в реакторе был заменен в основном свободным от влаги или сухим инертным газом (например, азотом, аргоном и т.п.). Термин «эквивалент» (сокращение: экв.) в настоящей заявке описывает стехиометрию (мольное отношение) реагента или вступающего в реакцию соединения путем сравнения с количеством предварительно выбранного исходного соединения. Термин «массовая единица» (сокращение: масс.ед.) в настоящей заявке соответствует отношению массы вещества или группы веществ к массе конкретного химического компонента реакции или методики очистки, конкретно указанного в приведенных ниже примерах. Это отношение рассчитывают как г/г или кг/кг. Термин «объемная единица» (сокращение: объемн.ед.) в настоящей заявке соответствует отношению объема данного вещества или группы веществ к массе или объему предварительно выбранного химического компонента реакции или методики очистки. Единицы, используемые в уравнении для расчета этого параметра, должны соответствовать друг другу по порядку величины. Например, соотношение рассчитывают как: мл/мл, мл/г, л/л или л/кг. В настоящей заявке используются следующие сокращения:
Пример 1: Получение соединения 22
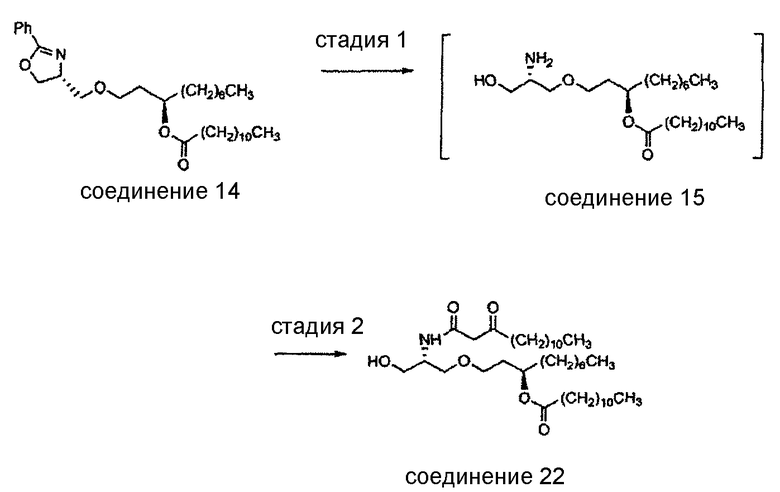
Получение соединения 15 (стадия 1)
Общая методика: как показано выше на схеме стадии 1, соединение 15 может быть получено следующим образом. В реакционный сосуд, в котором создана инертная атмосфера, добавляют соединение 14 (1 масс.ед., 1 экв.), Pd/C (предпочтительно примерно 0,1-0,2 масс.ед., 0,025-0,050 экв.) и подходящий для гидрирования растворитель (например, неводный полярный растворитель, такой как ТГФ, предпочтительно полярный протонный растворитель, более предпочтительно спирт низкой молекулярной массы; наиболее предпочтительно этанол или изопропанол) (приблизительно 6-7 объемн.ед.). Инертный газ в сосуде заменяют водородом (предпочтительно под давлением от примерно 1 атмосферы до примерно 120 фунтов/кв.дюйм) и реакционную смесь перемешивают при комнатной температуре до того, как реакция в основном завершится (примерно 3-8 дней). Затем водород в реакционном сосуде заменяют инертным газом и отделяют катализатор фильтрованием (например, с использованием целита). После этого концентрируют фильтрат с получением соединения 15 (примерно 0,8 ед.массы) в виде бледно-желтого масла. Приведенные ниже методики описывают гидрирование соединения 14 до соединения 15 в этаноле и изопропаноле.
Стадия 1(a1): Гидрирование в этаноле: В круглодонную колбу, в которой была создана инертная атмосфера, снабженную магнитной мешалкой, добавляли соединение 14 (3,00 г, 1,0 масс.ед., 1,0 экв.), 10% Pd/C (Degussa type E101 NE/W, 0,300 г, 0,10 масс.ед., 0,024 экв., Aldrich) и этанол (20,0 мл, 6,7 объемн.ед.). Инертный газ, а именно азот, в сосуде заменяли водородом (1 атмосфера) и реакционную смесь перемешивали при комнатной температуре до завершения реакции, момент которого определяли с помощью ТСХ (приблизительно 4 дня). Затем водород в реакционном сосуде заменяли азотом, катализатор удаляли фильтрованием на целите и использовали для промывания небольшое количество этанола.
Стадия 1 (a2) Соединение 15, как правило, можно использовать in situ для получения соединения 22. С целью установления характеристик, отфильтрованный раствор соединения 15 в этаноле концентрировали с получением соединения 15 (2,5 г, 0,83 масс.ед.) в виде бледно-желтого масла, вновь растворяли в минимальном количестве 10% раствора метанола в дихлорметане, вводили в колонку с силикагелем (KP-Sil, 13,0 масс.ед.), элюировали 10% раствором метанола в дихлорметане и концентрировали в вакууме с получением очищенного соединения 15 (0,1 масс.ед.).
Аналитические данные для очищенного соединения 15: 1H ЯМР (400 МГц, CDCl3) δ: 5,03-5,10 (м, 1H), 3,63 (дд, J=10,5 Гц, 1H), 3,42-3,52 (м, 3H), 3,34-3,39 (м, 2H), 3,02-3,07 (м, 1H), 2,29 (т, J=7 Гц, 3H), 1,73-1,94 (м, 6H), 1,48-1,66 (м, 4H), 1,20-1,36 (м, 26H), 0,89 (т, J=7 Hz, 6H).
МС-ESI (M+H)+ Теоретический расчет для C25H52NO4: 430,4; реальный результат: 430,4.
Стадия 1 (b1) Гидрирование в изопропаноле: В стеклянную 12л колбу, в которой была создана инертная атмосфера, загружали соединение 14 (2,0 кг, 1,0 масс.ед.) и изопропанол (8,0 кг, 4,0 масс.ед.) и смесь перемешивали до растворения соединения 14. В реактор объемом 5 галлонов, снабженный механической мешалкой, в котором была создана инертная атмосфера, загружали Pd/C (Johnson Matthey type A402028-10, 10% Pd/C, влажность 50%; 0,411 кг, 0,206 масс.ед., 0,05 экв.) и раствор соединения 14 в изопропаноле. Изопропанол (1,1 кг, 0,55 масс.ед.) использовали для смывания остатков соединения 14 в 5-галлоный реактор. В 5-галлонный реактор нагнетали азот до давления 10-15 фунтов/кв.дюйм при перемешивании со скоростью ~750 об/мин. Азот выпускали при перемешивании со скоростью ~250 об/мин. Цикл нагнетания и выпускания азота повторяли еще два раза. В реактор нагнетали водород (марки 5,0) до давления 25 фунтов/кв.дюйм при перемешивании со скоростью ~750 об/мин, и затем выпускали при перемешивании со скоростью ~250 об/мин. Затем в реактор нагнетали водород до давления 120 фунтов/кв.дюйм при перемешивании со скоростью ~750 об/мин. После этого реакционную смесь перемешивали при комнатной температуре в течение 65 ч, после чего выпускали водород. Затем в реактор нагнетали азот до давления 10-15 фунтов/кв.дюйм, после чего азот выпускали из реактора. Процесс нагнетания-выпускания азота повторяли еще два раза. Исследование реакционной смеси с помощью ВЭЖХ показало полное завершение реакции. После этого катализатор удаляли фильтрованием через два фильтра CUNO (фильтрующий картридж CUNO CTG Klean 1,0 мкм и фильтрующий картридж CUNO CTg Klean 0,2 мкм, соединенные последовательно) которые были предварительно промыты изопропанолом (3,9 кг, 1,95 масс.ед.). Реактор промывали изопропанолом (5,0 кг, 2,5 масс.ед.) и полученный в результате промывания раствор фильтровали через фильтры CUNO. Фильтраты объединяли с получением бесцветного прозрачного раствора соединения 15 в изопропаноле (15,8 кг, 7,9 масс.ед.). Примерно 15 кг (7,5 масс.ед.) (=0,95×15,8 и 7,9 соответственно) этого раствора концентрировали при пониженном давлении до массы 6,55 кг (3,28 масс.ед.) и использовали в стадии 2a без дополнительной очистки.
Получение соединения 22 (стадия 2a)
Общая методика: На стадии 2, показанной на схеме выше, получения соединения 22 может применяться следующая общая методика (все массовые единицы, эквиваленты и объемные единицы приведены относительно массы соединения 14, использованного на стадии 1): В реактор, снабженный мешалкой, в котором создана инертная атмосфера, помещают карбонилдиимидазол (CDI) (0,34 масс.ед., 1,1 экв.) и безводный ацетонитрил (7,4 масс.ед.). Начинают перемешивание и охлаждают смесь примерно до 0°C. Добавляют 3-оксо-тетрадекановую кислоту (0,514 масс.ед., 1,1 экв.) и продолжают перемешивание, поддерживая температуру примерно 0°C. За образованием имидазола следят с помощью ВЭЖХ. Когда установлено, что реакция завершилась, добавляют раствор соединения 15 в IPA (3-8 масс.ед.), полученный на стадии 1, поддерживая температуру около 0°C. Реакционную смесь перемешивают при температуре около 0°C и следят за ходом реакции при помощи ВЭЖХ. После завершения реакции добавляют ледяную уксусную кислоту (0,24 масс.ед., 2,1 экв.) поддерживая температуру реакционной смеси ≤15°C. Температуру реакционной смеси доводят примерно до 20°C и объем смеси уменьшают примерно до 5-7,5 объемн.ед. частичным выпариванием растворителя в вакууме при 20-25°C, получая прозрачный оранжевый раствор. Добавляют гептан (8,5 масс.ед.) и промывают полученную смесь раствором NH4Cl (0,15 масс.ед.) в воде (5,2 масс.ед.). Затем слой гептана промывают раствором хлорида натрия (1,0 масс.ед.) в воде (3,2 масс.ед.). Гептановый слой концентрируют в вакууме приблизительно при 25-30°C до объема примерно 2,2-2,4 объемн.ед., после чего добавляют изопропилацетат (9,0 масс.ед.) и полученный раствор концентрируют в вакууме при температуре примерно 25-30°C, получая прозрачное оранжевое масло (2,5-5,6 объемн.ед. или 2,0-5,0 масс.ед.).
Конкретный пример: В приведенной ниже методике описано получение соединения 22, исходя из 95% того количества соединения 15, которое было получено при использовании 2,0 кг соединения 14 на стадии 1 (b1). Таким образом, для этого примера 1,9 кг = 1,0 масс.ед. В 50 л стеклянный реактор, снабженный механической мешалкой, в котором была создана инертная атмосфера, загружали карбонилдиимидазол (CDI) (0,65 кг, 0,34 масс.ед. 1,1 экв.) и безводный ацетонитрил (14,1 кг, 7,4 масс.ед.). Начинали перемешивание и охлаждали реакционную смесь до 0°C. После этого при непрерывном перемешивании и температуре примерно равной 0°C добавляли 3-оксо-тетрадекановую кислоту (0,98 кг, 0,516 масс.ед., 1,1 экв.; приобретенную у DSM Pharmaceutical Products, Parsippany, NJ). За образованием имидазола наблюдали с помощью ВЭЖХ. После завершения реакции, спустя примерно 10,5 ч, в течение 1 минуты добавляли раствор соединения 15 в IPA (6,55 кг, 3,45 масс.ед.), полученный на стадии 1 (b1), и температура оставалась в районе 0°C. В качестве растворителя для промывания емкости использовали IPA (1,7 кг, 0,89 масс.ед.). Реакционную смесь перемешивали в течение ночи при 0-3°C. Данные ВЭЖХ подтвердили завершение реакции. После этого добавляли ледяную уксусную кислоту (0,46 кг, 0,24 масс.ед., 2,1 экв.), поддерживая при этом температуру реакционной смеси ≤15°C. (Добавление уксусной кислоты осуществляли примерно через 12,25 ч после добавления соединения 15). Реакционную смесь нагревали до 20°C, и ее объем уменьшали примерно до 13 л (6,8 объемн.ед.) частичным выпариванием растворителя в вакууме при 20-22°C, что приводило к получению прозрачного оранжевого раствора. Добавляли гептан (16,2 кг, 8,53 масс.ед.) и полученную смесь промывали раствором NH4Cl (0,29 кг, 0,15 масс.ед.) в воде (9,9 кг, 5,2 масс.ед.) с последующим промыванием раствором хлорида натрия (1,9 кг, 1,0 масс.ед.) в воде (6,1 кг, 3,2 масс.ед.). Гептановый слой концентрировали в вакууме при 25-30°C до объема ~7л (3,7 объемн.ед.). После этого добавляли изопропилацетат (17,0 кг, 8,95 масс.ед.), и полученный раствор концентрировали в вакууме при 25-30°C с получением прозрачного оранжевого масла ~7л (3,7 объемн.ед., чистота (ВЭЖХ): 83,8% по площади пика). Условия проведения ВЭЖХ для анализа соединения 22 (ВЭЖХ TM1 соединения 22):
B: 2,0 мл 28-30% водного раствора NH4OH в 1 л CH3CN
1,0 мл/мин
Кристаллизация соединения 22 (стадия 2b)
Общая методика: Соединение 22 можно кристаллизовать из смеси изопропилацетат/ацетонитрил. Например, соединение 22 (2,2-5,6 объемн.ед. по отношению к соединению 14) растворяют в изопропилацетате (3,5 масс.ед.) в стеклянном сосуде. Полученный раствор фильтруют и добавляют изопропилацетат до общей массы примерно 7,4 масс.ед. Затем переносят полученный раствор в стеклянный реактор подходящего размера с рубашкой, оборудованный перемешивающим устройством, и добавляют ацетонитрил (5,0 масс.ед.). В инертной атмосфере азота, полученную смесь охлаждают примерно до 5-8°C, получая кристаллы соединения 22. Температуру повышают примерно до 15°C для растворения мелких кристаллов. Поддерживают температуру около 15°C в течение примерно 2 ч, медленно охлаждают примерно до -12°C и затем предпочтительно выдерживают при -12°C в течение еще 1 ч. Соединение 22 фильтруют и твердый остаток промывают холодной смесью (~ -20°C) изопропилацетат/ацетонитрил (1:1 (объем/объем), 1-2 масс.ед.). Полученную лепешку осадка высушивают, получая соединение 22 в виде белого твердого вещества (приблизительно 0,9-1,5 масс.ед.).
Конкретный пример: В следующей методике описана кристаллизация соединения 22, в количестве, которое соответствует 1,9 кг исходного соединения 14 (т.е. 1,9 кг=1,0 масс.ед.): В стеклянном сосуде неочищенное соединение 22 в виде масла, полученного на стадии 2a (3,7 объемн.ед. относительно соединения 14) растворяли в изопропилацетате (6,7 кг, 3,5 масс.ед.). Полученный раствор фильтровали, и избыток растворителя удаляли в вакууме при 25-30°C. Затем оставшийся раствор (7,23 кг (3,8 масс.ед.)) переносили в 50 л стеклянный реактор с рубашкой, снабженный механической мешалкой и доводили массу раствора примерно до 14,0 кг (7,4 масс.ед.) добавлением изопропилацетата (6,27 кг, 3,3 масс.ед.). После этого добавляли ацетонитрил (9,6 кг, 5,1 масс.ед.). В инертной атмосфере азота начинали перемешивание и охлаждали полученную смесь до 7,3°C, в результате чего образовывались кристаллы соединения 22. Затем температуру повышали до 15,1°C для растворения мелких кристаллов. Поддерживали температуру 15°C в течение примерно 2 ч., медленно охлаждали до -11,7°C в течение ночи и затем выдерживали примерно при -12°C в течение приблизительно 1 ч. Полученную смесь фильтровали и твердый остаток промывали холодной (~-20°C) смесью изопропилацетат/ацетонитрил (1:1 (объем/объем), 2,6 кг, 1,4 масс.ед.). Полученную влажную лепешку осадка сушили в вакууме с получением соединения 22 в виде белого твердого вещества (1,83 кг, 0,96 масс.ед.; чистота: 92,5% по площади пика (ВЭЖХ ТМ1 соединения 22); выход 69% в пересчете на соединение 14).
Перекристаллизация соединения 22 (стадия 2c)
Общая методика: В стеклянном реакторе подходящего размера с рубашкой, в котором создана инертная атмосфера, снабженным мешалкой, смешивают соединение 22 (1,0 масс.ед. (количества компонентов даны относительно соединения 22)), изопропилацетат (4,4-4,9 масс.ед.) и ацетонитрил (3,9-4,5 масс.ед.). Полученную смесь перемешивают и нагревают до 20-25°C с получением прозрачного раствора. Этот прозрачный раствор охлаждают до 5-10°C, что вызывает образование кристаллов. Затем температуру повышают до 16-20°C для растворения мелких кристаллов и выдерживают примерно при 17°C в течение приблизительно 2 ч. Температуру медленно линейно понижают примерно до -3-8°C в течение приблизительно 10-11 ч. Предпочтительно температуру -3-8°C поддерживают в течение еще 2 ч. Твердый осадок фильтруют и промывают холодным (-20°C) раствором изопропилацетат/ацетонитрил (1:1 (объем/объем), 1-3 масс.ед.) и высушивают в вакууме, получая соединение 22 в виде белого порошка (примерно 0,8-0,95 масс.ед.).
Конкретный пример: В приведенной ниже методике описана перекристаллизация соединения 22: 1,79 кг соединения 22, полученного после кристаллизации на стадии 2b (чистота 92,5%=1,65 кг соединения; 1,65 кг=1,0 масс.ед.) смешивали с изопропилацетатом (8,0 кг, 4,8 масс.ед.) и ацетонитрилом (7,37 кг, 4,5 масс.ед.) в 30 л стеклянном реакторе с рубашкой, оборудованном механической мешалкой, в котором была создана инертная атмосфера. Полученную смесь перемешивали и нагревали до 24,3°C с получением прозрачного раствора. Этот прозрачный раствор охлаждали до 10,4 для образования кристаллов. Затем для растворения небольших кристаллов температуру повышали до 16,2°C и выдерживали при 16,2-17,3°C в течение примерно 2 ч. Температуру медленно понижали в течение ночи до -3,4°C. На следующий день смесь выдерживали при этой температуре еще в течение 20 мин. Твердое вещество отделяли фильтрованием, промывали холодной (~-20°C) смесью изопропилацетат/ацетонитрил (1:1 (объем/объем), 4,6 кг, 2,8 масс.ед.) и сушили в вакууме (18-25°C, 5 ч), с получением соединения 22 в виде белого твердого вещества (1,54 кг, 0.93 масс.ед.; чистота 99,4% (ВЭЖХ ТМ1 соединения 22); выход:92,8%).
Аналитические данные для соединения 22: 1H ЯМР (400 МГц, CDCl3): 7,24 (д, J=7 Гц, 1H), 5,05-5,14 (м, 1H), 4,03-4,11 (м, 1H), 3,72 (дд, J=5,5 Гц, 2H), 3,42-3,60 (м, 3H), 3,41 (с, 2H), 3,26-3,38 (м, 1H), 2,53 (т, J=7 Гц, 3H), 2,29 (т, J=7 Гц, 3H), 1,79-1,89 (м, 1H), 1,48-1,77 (м, 7H), 1,19-1,38 (м,42H), 0,88 (т, J=7 Гц, 3H). ESI-МС (M+H)+ Теоретическое значение для C39H76NO6: 654,6; реальный результат: 654,6.
Пример 2: Получение характеристик твердого кристаллического соединения 22
A. Дифракция рентгеновских лучей на порошке.
Используя стеклянную кювету, регистрировали данные на рентгеновском дифрактометре Scintag Diffractometer в стандартных условиях регистрации дифракционной картины на порошке в диапазоне углов 2-тета 3-40 градусов, используя излучение медного анода. Коррекцию на фоновый сигнал не применяли. На фиг. 2 показана картина дифракции рентгеновских лучей на порошке (PXRD) кристаллического соединения 22. На картине PXRD наблюдаются пики при углах 10,3±0,2°2Θ, 12,3±0,2°2Θ, 14,5±0,2°2Θ, 15,3±0,2°2Θ, 16,2±0,2°2Θ, 17,8±0,2°2Θ, 22,2±0,2°2Θ, 22,9±0,2°2Θ, 23,8±0,2°2Θ, 25,3±0,2°2Θ и 26,8±0,2°2Θ. Кристаллическое соединение 22 может быть охарактеризовано подмножеством пиков, показанных на фиг.2. Например, для кристаллического соединения 22 характерны следующие пики: 12,3±0,2°2Θ, 14,5±0,2°2Θ, 16,2±0,2°2Θ, 17,8±0,2°2Θ, 22,2±0,2°2Θ и 23,8±0,2°2Θ. Другие комбинации перечисленных или показанных на фиг.2 пиков также могут использоваться для идентификации соединения 22.
Условия измерений
B. Определение характеристик способом DSC
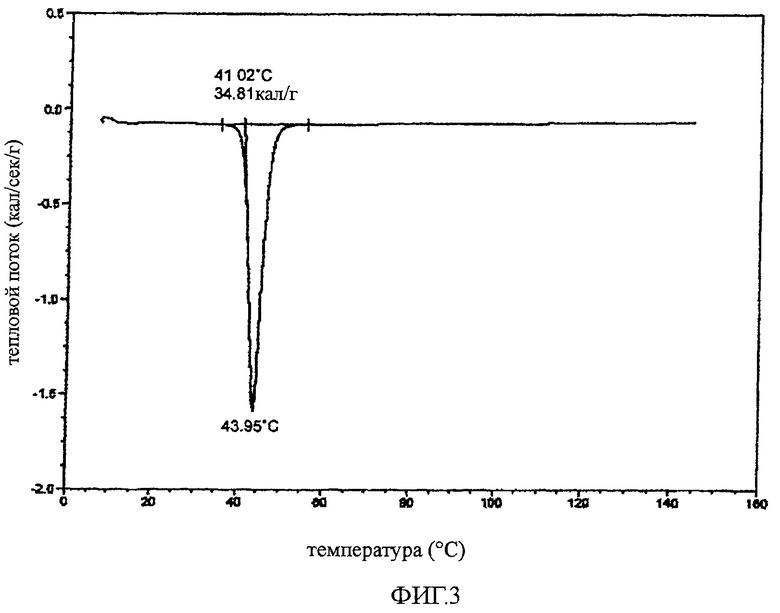
Определяли характеристики твердого кристаллического соединения 22 способом дифференциальной сканирующей калориметрии (DCS, методика с алюминиевым тиглем). DSC осуществляли с помощью калориметра 2920 DSC V2.5F, при нагревании до 150°C со скоростью 10°C/мин в алюминиевом тигле в токе азота 50 мл/мин, используя образец кристаллического соединения 22 массой 2,91 мг. На фиг.3 показана термограмма кристаллического соединения 22 с температурой плавления 41°C (температура начала плавления).
Эксперимент по определению температуры плавления проводили также с использованием прибора Electrothermal Mel.Temp Apparatus с цифровым термометром Fluke 51 II Digital Thermometer. Соединение 22 помещали в капиллярную трубку (1,5-1,8×90 мм, Kimble Product KIMAX-51, part no 34505). Наблюдаемая температура плавления составляла 41-42°C.
Пример 3: Синтез соединения 25 и получение затравок кристаллов
A. Синтез соединения 25 и получение неочищенных кристаллов (стадия 3a)
Синтез соединения 25: В стеклянный реактор с рубашкой, в котором была создана инертная атмосфера, оборудованный механической мешалкой, помещали соединение 24 (N,N'-бис(2-гидроксиэтил)мочевину; 8,00 г, 1,00 масс.ед., 1,00 экв.; приобретено у поставщика Mitsui&Co.(USA),Inc., New York,NY; производитель Yoyu Labs), трифторацетат пиридиния (0,5 г, 0,063 масс.ед.; 0,05 экв.) и ацетонитрил (59,7 г, 7,5 масс.ед.). Начинали перемешивание и добавляли аллил тетраизопропилфосфордиамидит (35,0 г, 4,4 масс.ед., 2,24 экв.; приобретен у Digital Speciality Chemicals, Inc., Dublin, NH). Затем полученную смесь перемешивали при 20-25°C в течение ночи (17 ч). ВЭЖХ-мониторинг продемонстрировал завершение реакции. Температуру понижали до 0°C, после чего появлялся осадок. Температуру медленно повышали до 16°C в течение 2 ч для растворения большей части образовавшегося твердого осадка. Затем смесь перемешивали при 16°C в течение 1,5 ч, охлаждали до -17,4°C со скоростью 5°C/ч и перемешивали в течение ночи при этой температуре с получением густой суспензии. Затем температуру повышали до 16°C в течение 2,3 ч и поддерживали около 16°C в течение 2,25 ч. Температуру понижали до 3,5°C в течение 1,2 ч и затем быстро (примерно за 5-10 мин) повышали до 10°C. Поддерживали температуру около 10°C в течение 2 ч и охлаждали в течение ночи со скоростью примерно 1,5°C/ч до конечной температуры -16°C. Образовавшееся твердое вещество отделяли фильтрованием, промывали холодным ацетонитрилом (2×6,3 г, 2×0,79 масс.ед.) и сушили в вакууме. Соединение 25 (19,7 г, 2,46 масс.ед.) получали в виде белого твердого вещества.
B. Перекристаллизация соединения 25 (стадия 3b)
В стеклянный реактор с рубашкой, в котором была создана инертная атмосфера, оборудованный механической мешалкой, помещали соединение 25, полученное на стадии 3a (19,5 г, 1,00 масс.ед.) и ацетонитрил (55,0 г, 2,8 масс.ед.). Полученную смесь перемешивали при 20-25°C в течение 15 мин. Температуру понижали до 10°C, причем при этой температуре перемешивание происходило с трудом. Температуру повышали до 20°C и поддерживали ее на этом уровне в течение 2 ч. Вновь осуществляли охлаждение до 10°C со скоростью примерно 2°C/ч. Продолжали охлаждение до 1°C со скоростью примерно 3°C/ч, после чего охлаждали до -19°C со скоростью примерно 5°C/ч. Образовавшееся твердое вещество отделяли фильтрованием, промывали холодным ацетонитрилом (32 г, 1,6 масс.ед.) и сушили в вакууме. Получали соединение 25 (16,06 г, 0,82 масс.ед.) в виде белого твердого вещества.
C. Получение затравок кристаллов соединения 25 (стадия 3c)
Следующая методика может применяться и фактически применялась для получения затравок кристаллов, которые использовались ниже на стадии 4a и стадии 4b: В стеклянный реактор с рубашкой, в котором была создана инертная атмосфера, оборудованный механической мешалкой, помещали соединение 25, полученное на стадии 3b (5,59 г, 1,00 масс.ед.) и раствор, состоящий из гептана и TBME (9:1 (объем/объем), 120 мл, 21,5 объемн.ед.). Полученную смесь перемешивали при 20-25°C и добавляли дополнительную порцию TBME (3,0 мл, 0,54 объемн.ед.) для полного растворения соединения 25. Температуру понижали до 10°C в течение примерно 1,5 ч, в результате чего происходило образование белого осадка. Температуру повышали до 16°C и поддерживали на этом уровне в течение примерно 2 ч. Реактор охлаждали со скоростью примерно 2°C/ч до 0°C и затем охлаждали со скоростью примерно 3°C/ч до -18,8°C, после чего поддерживали температуру около примерно -18,8°C. Эту схему охлаждения реализовывали в течение ночи. Образовавшийся осадок отделяли фильтрованием, промывали холодной смесью гептана и TBME (9,1 (объем/объем), 16 г, 2,9 масс.ед.) и сушили в вакууме. Получали соединение 25 (5,23 г, 0,936 масс.ед.; чистота: 94,7% (по площади пика)) в виде белого твердого вещества. Условия проведения ВЭЖХ для анализа соединения 25 (ВЭЖХ ТМ2 соединения 25):
B: CH3CN
Пример 4: Получение соединения 25
Стадия 3: Py·TFA, ACN, кристаллизация
Получение соединения 25 (стадия 4a)
Общая методика получения соединения 25: (примечание: в данной методике 1,0 масс.ед. соответствует массе соединения 24, используемого в качестве исходного соединения для получения порции соединения 25). В стеклянном реакторе подходящего размера с рубашкой, в котором создана инертная атмосфера и который снабжен мешалкой, смешивают производное мочевины соединение 24 (1,0 масс.ед., 1,0 экв.), трифторацетат пиридиния (0,07 масс.ед., 0,05 экв.) и безводный ацетонитрил (7,0 масс.ед.). Начинают перемешивание и добавляют аллил тетраизопропилфосфордиамидит (4,4 масс.ед., 2,3 экв.). Реакционную смесь перемешивают при 18-26°C в течение нескольких часов до завершения взаимодействия. Полученную смесь охлаждают приблизительно до 9-11°C, предпочтительно вносят затравки кристаллов соединения 25 (~0,005 масс.ед.; получение кристаллических затравок соединения 25 описано выше в примере 3) и поддерживают указанную температуру в течение примерно 2 ч. Затем смесь охлаждают примерно до 0 -15°C со скоростью приблизительно 1,5°C/час, затем охлаждают примерно до -20°C со скоростью приблизительно 2,5°C/час. После поддержания температуры на этом уровне в течение приблизительно 2 часов, твердое вещество отделяют фильтрованием в атмосфере аргона, промывают холодным (-20°C) безводным ацетонитрилом (2-3 масс.ед.) и сушат в атмосфере азота. Соединение 25 (примерно 2,3-2,5 масс.ед.) образуется в описанной методике в виде белого твердого вещества. До очистки соединение 25 предпочтительно хранят в атмосфере сухого азота при низкой температуре (например, -20°C).
Конкретный пример: В приведенной ниже методике описан синтез соединения 25, исходя из соединения 24 (0,50 кг, что в данной методике соответствовало 1,0 масс.ед.): В 30 л стеклянном реакторе с рубашкой, в котором создана инертная атмосфера и который снабжен механической мешалкой, смешивали производное мочевины 24 (0,50 кг, 1,0 масс.ед., 1,0 экв.), трифторацетат пиридиния (0,035 кг, 0,07 масс.ед., 0,05 экв.) и безводный ацетонитрил (3,5 кг, 7,0 масс.ед.). Начинали перемешивание и добавляли аллил тетраизопропилфосфордиамидит (2,22 кг, 4,4 масс.ед., 2,3 экв.). Реакционную смесь перемешивали при 18,4-25,6°C. За ходом реакции следили с помощью ВЭЖХ, данные которой подтвердили завершение реакции через 18 часов. Полученную смесь охлаждали до температуры 11,1°C и сохраняли температуру в пределах 9,3-11,1°C в течение примерно 50 мин. В полученную смесь вводили затравки кристаллов соединения 25 (1,9 г, 0,004 масс.ед.; получение затравок кристаллов соединения 25 описано выше в примере 3) и выдерживали в диапазоне температур 9-11°C в течение примерно 2 ч. Затем смесь охлаждали до -7,9°C со скоростью примерно 1,5°C/час. После этого скорость охлаждения повышали примерно до 2,5°C/час, и понижали температуру до -18°C. Поддерживали температуру в диапазоне от -18 до -19,2°C в течение примерно 2ч, после чего отделяли образовавшееся твердое вещество фильтрованием в атмосфере азота, промывали холодным (-20°C) безводным ацетонитрилом (1,5 кг, 3,0 масс.ед.) и сушили в токе азота. Получали соединение 25 (1,18 кг, 2,36 масс.ед.; чистота 93,6% по площади пика на хроматограмме (ВЭЖХ ТМ2 соединения 25)) в виде белого твердого вещества, которое хранили в атмосфере азота при -20°C до очистки.
Перекристаллизация соединения 25 (стадия 4b)
Общая методика перекристаллизации соединения 25: В стеклянном реакторе подходящего размера с рубашкой, в котором создана инертная атмосфера и который снабжен устройством для перемешивания, мочевину дифосфорамидит, т.е. соединение 25 (1,0 масс.ед., где 1,0 масс.ед. в данной методике соответствует массе неочищенного соединения 25, осажденного из реакционной смеси, в которой оно образовалось) растворяют в TBME (1,5-2,0 масс.ед.). Не растворившиеся частицы удаляют фильтрованием. Фильтрат переносят в стеклянный реактор с рубашкой подходящего размера, в котором создана инертная атмосфера, и который снабжен устройством для перемешивания. Добавляют гептан (13,2 масс.ед.), полученный раствор охлаждают примерно до 9-11°C и перемешивают в этом диапазоне температур в течение приблизительно 1,5-2,5 ч. Образуется белая суспензия (для облегчения кристаллизации соединения 25 может оказаться полезным введение затравок кристаллов; получение затравок кристаллов соединения 25 описано выше в примере 3). Смесь охлаждают до температуры примерно от 0 до -15°C со скоростью приблизительно 1,5°C/час, и затем охлаждают примерно до -20°C со скоростью около 2,5°C/час. Эту температуру поддерживают стабильной в течение приблизительно 2 часов, после чего твердое вещество отделяют фильтрованием, промывают холодным (~-20°C) раствором, состоящим из гептана и TBME (7:1 (объем/объем), 1,3-1,5 масс.ед.) и высушивают в токе азота. Соединение 25 получают в виде белого твердого вещества (примерно 0,5-0,6 масс.ед.). Фильтрат концентрируют в вакууме при температуре примерно 20-30°C и подвергают перекристаллизации по описанной выше методике. Таким образом также может быть получена вторая порция соединения 25 (0,2-0,3 масс.ед.).
Конкретный пример: В приведенной ниже методике описана перекристаллизация соединения 25 (в данной методике 1,18 кг соответствуют 1,0 масс.ед.). В 22 л стеклянной колбе Rotavap в атмосфере азота дифосфорамидитное производное мочевины - соединение 25, полученное на стадии 4a (1,18 кг, 1,0 масс.ед.), растворяли в TBME (1,81 кг, 1,5 масс.ед.). Нерастворимые частицы удаляли фильтрованием, используя TBME (0,60 кг, 0,50 масс.ед.) для промывания. Фильтрат переносили в 30 л стеклянный реактор с рубашкой, в котором была создана инертная атмосфера, и который был снабжен механической мешалкой. Добавляли гептан (15,54 кг, 13,2 масс.ед.) и полученный раствор охлаждали до 10,3°C. Через 5 минут начиналось осаждение соединения 25. Продолжали перемешивание при 9-11°C в течение примерно 130 мин. Затем полученную смесь охлаждали со скоростью около 1,5°C,час в течение ночи до температуры примерно -15°C. На следующий день продолжали охлаждение со скоростью примерно 1,5°C/час до температуры -17,2°C, после чего в течение примерно 2 часов поддерживали температуру в диапазоне от -17,0 до -20°C. Затем образовавшееся твердое вещество отделяли фильтрованием, промывали холодным (~-20°C) раствором, состоящим из гептана и TBME (7:1 (объем/объем), 1,56 кг, 1,3 масс.ед.) и сушили в токе азота в течение примерно 22,5 ч. Соединение 25 получали в виде белого твердого вещества (0,675 кг, 0,57 масс.ед., чистота 93,9% по площади пика на хроматограмме (ВЭЖХ ТМ2 соединения 25); выход 36,0% исходя из соединения 24). Фильтрат концентрировали в вакууме при 20-30°C и подвергали перекристаллизации по описанной методике. В итоге получали вторую порцию соединения 25 в виде белого твердого вещества (0,225 кг, 0,19 масс.ед., чистота: 92,6% по площади пика на хроматограмме (ВЭЖХ ТМ2 соединения 25); выход 11,8% исходя из соединения 24).
Аналитические данные для соединения 25: 1H ЯМР (400 МГц, CDCl3) δ: 5,90-6,01 (м, 2H), 5,30 (д, J=17 Гц, 2H), 5,16 (d, J=10 Гц, 2H), 4,79 (т, J=5 Гц, 2H), 4,06-4,26 (м, 4H), 3,66-3,79 (м, 4H), 3,54-3,66 (м, 4H), 3,33-3,47 (м, 4H), 1,18 (д, J=7 Гц, 24H). ESI-МС (M+Na)+ Теоретический расчет для C23H48N4NaO5P2: 545,3; реальный результат: 545,4.
Пример 5: Получение характеристик твердого кристаллического соединения 25
A. Дифракция рентгеновских лучей на порошке.
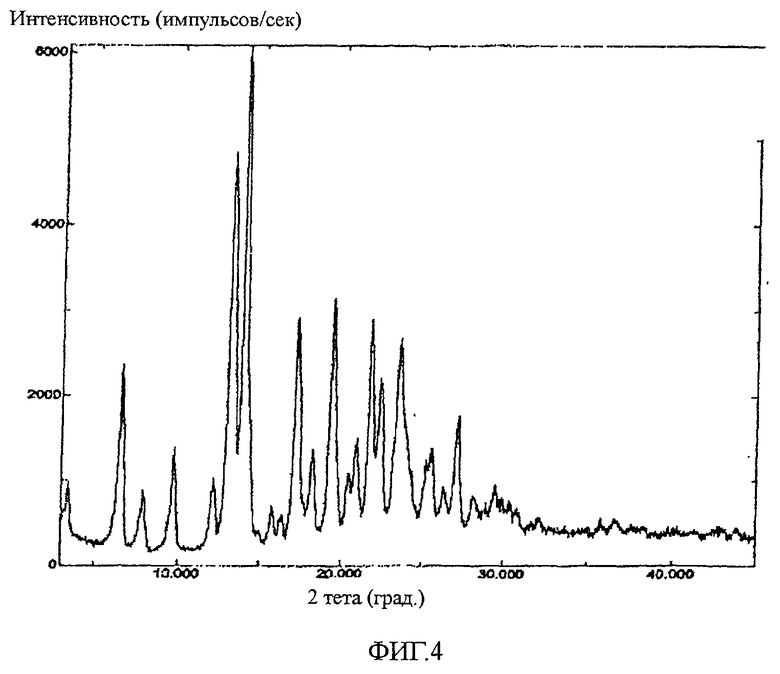
Картину дифракции рентгеновских лучей на кристаллическом порошке (PXRD) получали по той же методике, которая описана выше в примере 2A. На фиг. 4 показана картина PXRD перекристаллизованного соединения 25. На картине PXRD наблюдаются пики при углах 6,6±0,3°2Θ, 13,2±0,3°2Θ, 14,0±0,3°2Θ, 17,3±0,3°2Θ, 19,4±0,3°2Θ, 21,8±0,3°2Θ, 22,4±0,3°2Θ, 23,6±0,3°2Θ и 27,2±0,3°2Θ. Кристаллическое соединение 25 может быть охарактеризовано подмножеством пиков, показанных на фиг.4. Например, для кристаллического соединения 25 характерны следующие пики: 6,6±0,3°2Θ, 14,0±0,3°2Θ, 17,3±0,3°2Θ, 19,4±0,3°2Θ. Другие комбинации перечисленных или показанных на фиг.4 пиков также могут использоваться для идентификации соединения 25.
B. Определение характеристик способом DSC
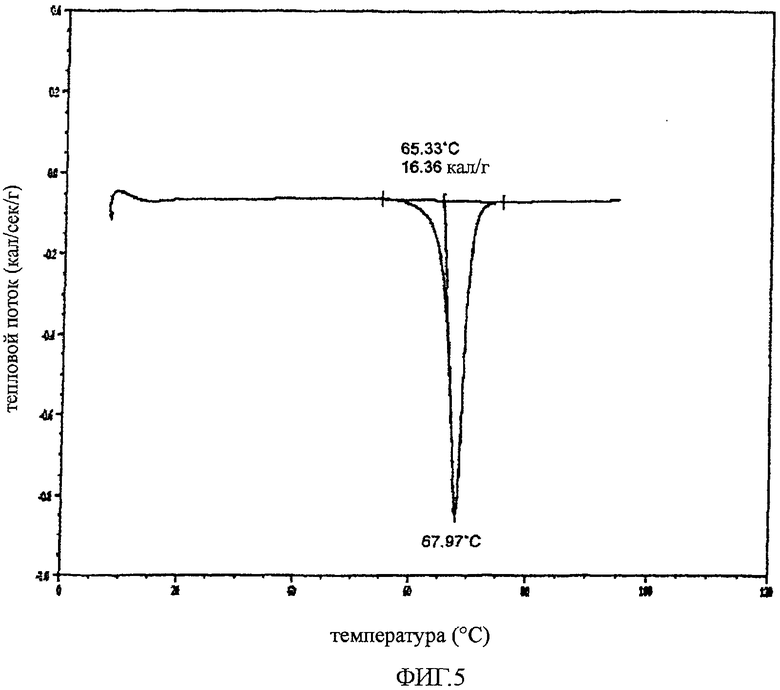
Определяли характеристики твердого кристаллического соединения 25 способом дифференциальной сканирующей калориметрии (DCS, капиллярная методика). DSC осуществляли с помощью калориметра 2920 DSC V2.5F, при нагревании до 100°C со скоростью около 10°C/мин в алюминиевом тигле в атмосфере азота, используя образец кристаллического соединения 25 массой 1,88 мг. На фиг.5 показана термограмма кристаллического соединения 25, где образец соединения 25 плавился при 65°C (температура начала плавления).
Эксперимент по определению температуры плавления проводили также с использованием прибора Electrothermal Melt Temp Apparatus с цифровым термометром Fluke 51 II Digital Thermometer. Соединение 25 (2-3 мг) помещали в капиллярную трубку (1,5-1,8×90 мм, Kimble Product KIMAX-51, part no 34505). Наблюдаемая температура плавления составляла 65-67°C.
C. Дифракция рентгеновских лучей на монокристалле
Соединение 25 в количестве 150 мг растворяли в ацетонитриле в количестве 1 мл при температуре примерно 25°C. Раствор охлаждали приблизительно до 0°C в течение примерно 425 минут и затем выдерживали в течение примерно 1000 минут. Получали монокристалл, пригодный для исследования дифракции рентгеновских лучей. Полученный кристалл был бесцветным, имел игольчатую форму и размеры 0,18×0,06×0,06 мм. Кристалл закрепляли на 0,2 мм нейлоновой петле с использованием небольшого количества паратонового масла.
Регистрировали данные с использованием дифрактометра на основе Bruker SMART CCD (прибора с зарядовой связью), оборудованного низкотемпературным устройством Oxford Cryostream, работавшим при температуре примерно 193°K. Регистрировали данные с шагом омега-сканирования 0,3° на фрейм при времени регистрации фрейма примерно 45 секунд, так, чтобы собрать данные для полусферы. В общей сложности был зарегистрирован 1271 фрейм с максимальным разрешением 0,76 Å. Первые 50 фреймов были зарегистрированы повторно в конце сеанса сбора данных для наблюдения за разрушением кристалла. Параметры кристаллической ячейки определяли с использованием программы SMART (SMART V 5.625 (NT) Software for the CCD Detector System; Bruker Analytical X-ray Systems, Madison, WI (2001)), и уточняли с использованием программы SAINT для всех зафиксированных отражений. Предварительную обработку данных осуществляли с использованием программы SAINT (SAINT V 6.22 (NT) Software for the CCD Detector System; Bruker Analytical X-ray Systems, Madison, WI (2001)), которая позволяет скорректировать Lp и разрушение. Применяли коррекцию на поглощение с использованием методики мультисканирования SADABS (смотрите SADABS, Program for absorption corrections using Siemens CCD, Blessing, R.H. Acta Cryst. A51 1995, 33-38). Структуру расшифровывали прямым способом, используя программу SHELXS-97 (смотрите Sheldrick, G.M. SHELXS-90, Program for the Solution of Crystal Structure, University of Göttingen, Germany, 1990) и уточняли методом наименьших квадратов по F2 с помощью программы SHELXL-97 (смотрите Sheldrick, G.M. SHELXL-97, Program for the Refinement of Crystal Structure, University of Göttingen, Germany, 1997), включенной в SHELXTL-PC V6.10, (SHELXTL 6.1 (PC-Version), Program library for Structure Solution and Molecular Graphics; Bruker Analytical X-ray Systems, Madison, WI (2000)). Структура кристалла продемонстрировала признаки наличия двойниковых структур, что подтверждалось исходными параметрами ячейки и формой пиков. Использование программы Cell_Now показало, что имел место двукратный поворот, причем минорный двойниковый компонент ориентирован вдоль и повернут вокруг обратной оси -0,010 1,000-0,040 и реальной оси 0,001 1,000-0,002. Данные о двойниковых структурах были включены в расчет и подвергнуты процедуре уточнения. Было найдено, что процентная величина уточнения с учетом двойниковых структур составляла менее 1,5%.
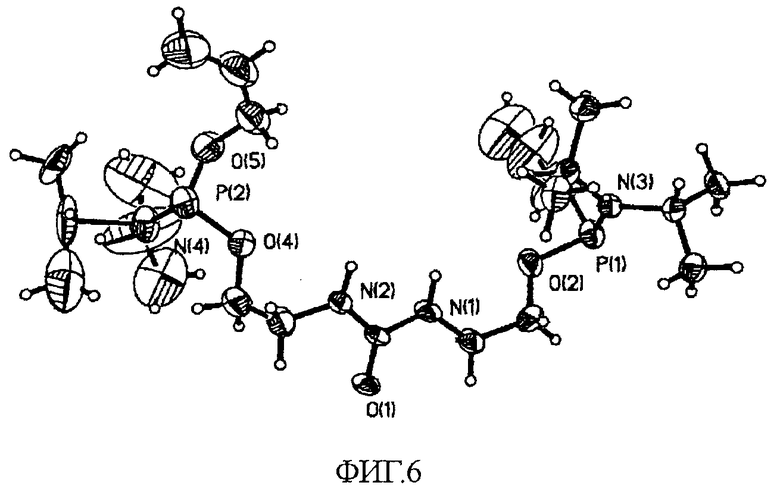
Согласно результатам расшифровки, структура кристалла соответствует пространственной группе P21/c (#14). Положения всех неводородных атомов уточняли в анизотропном приближении. Положение атомов водорода рассчитывали геометрическими способами и включали в уточнение в модели «наездника». Кристалл, использованный для дифракционных исследований, не продемонстрировал разложения за время регистрации данных. На всех рисунках приведены эллипсоиды, соответствующие 50% вероятности нахождения атома. OPTEP-изображение кристаллической структуры соединения 25, на котором отмечены различные атомы, приведено на фиг.6. На фиг.7 показана диаграмма кристаллической упаковки вдоль оси c. Сведения о параметрах кристаллической структуры и уточненные структурные данные приведены в таблице 2.
b=12,135(6)Å β=93,156(10)°
c=9,332(5)Å γ=90°
Картина XPRD для соединения 25, смоделированная на основе рентгеноструктурных данных для монокристалла, показана на фиг.8. Параметры дифракции на порошке вычисляли из данных по интенсивности для монокристалла с использованием программы XPOW (XPOW, Simulated Powder Diffraction Pattern, Version 5.101, Bruker-AXS, 1997-1998). Ключевые параметры для расчета включали длину волны, причем использовалась длина волны меди, равная 1,54Å, и параметры кристаллической ячейки, полученные в результате финальных уточнений. Ширина и интенсивность линий находятся в зависимости в соответствии с уравнением:
Ширина = интенсивность/[1+4*x*x(w=v*tan(θ))]
x=2θ/2θ0, где 2θ0 представляет собой Брэгговский угол для отражения. Переменные w и v являются параметрами формы линии. Для проведенных расчетов использовали значения 0,02 для обоих параметров w и v. В приведенной ниже таблице приведены девять пиков с наибольшей интенсивностью.
Пример 6: Получение соединения 26
Стадия 4: ACN, гептан, HOAc, H2O2, Na2S2O3, хроматография.
Получение соединения 26 (стадия 5а)
Общая методика синтеза соединения 26: (примечание: 1,0 масс.ед. в настоящей методике соответствует массе соединения 22, использованного в качестве исходного вещества для получения порции соединения 26). В стеклянный реактор подходящего размера, снабженный мешалкой, в котором создана инертная атмосфера, помещают соединение 22, например, соединение 22, полученное на стадиях 2b-2c, предпочтительно на стадии 2c, (1,0 масс.ед., 1,0 экв.) и соединение 25, например, соединение 25, полученное на любой из стадий 4a-4b, предпочтительно на стадии 4b (0,42 масс.ед., 0,52 экв.). Добавляют безводный ацетонитрил (5,0 масс.ед) и безводный гептан (1,4 масс.ед.) и начинают перемешивание. Смесь нагревают до температуры примерно 20-25°C для растворения всех твердых веществ. Медленно добавляют уксусную кислоту (0,12 масс.ед., 1,3 экв.) поддерживая температуру около примерно 20-25°C. Реакционную смесь перемешивают примерно при 20-25°C в течение нескольких часов до завершения реакции. В реактор добавляют безводный гептан и температуру понижают примерно до 0-5°C. Медленно в течение примерно 0,5 ч добавляют H2O2 30% (масс.) в воде (0,17 масс.ед., 1,1 экв.), поддерживая температуру около ≤5°C. Реакционную смесь перемешивают при температуре ≤5°C до завершения реакции. После этого реакционную смесь охлаждают примерно до 0°C и гасят оставшиеся пероксиды добавлением водного раствора пентагидрата тиосульфата натрия (1/1 (масса/масса), 0,26 масс.ед., 0,34 экв.). Перемешивание при температуре 0-4°C продолжают до тех пор, пока гептановый слой не даст отрицательный результат теста на наличие пероксидов. Температуру повышают до комнатной, после чего соли отделяют фильтрованием и промывают гептаном (2,3 масс.ед.). Полученный фильтрат помещают в новый стеклянный реактор и дают слоям разделиться в течение примерно 15 мин. Образуются три слоя, причем два нижних слоя (водный и ацетонитрильный) отделяют и выбрасывают. Верхний гептановый слой дважды промывают ацетонитрилом (1,8 масс.ед.) и концентрируют до полного удаления летучих компонентов при температуре примерно 18-25°C. Неочищенное соединение 26 (1,2-1,5 масс.ед.) получают в виде густого масла с янтарным оттенком.
Конкретный пример: в приведенной ниже методике описано получение соединения 26 В количестве, которое образуется из 0,565 кг соединения 22 с чистотой 99,4%. (Термин «1,0 масс.ед.» в данном примере относится к массе 0,56 кг, которая представляет собой действительную массу соединения 22, применяемого в качестве исходного вещества при получении соединения 26). В 30 л стеклянный реактор с рубашкой, в котором была создана инертная атмосфера и который был снабжен механической мешалкой помещали соединение 22, полученное на стадии 2c (0,56 кг (=0,565×0,994), 1,0 масс.ед., 1,0 экв.) и соединение 25, полученное на стадии 4b (0,248 кг, чистота: 93,9%, 0,42 масс.ед., 0,52 экв.). Добавляли безводный ацетонитрил (2,8 кг, 5,0 масс.ед.) и безводный гептан (0,79 кг, 1,4 масс.ед.) и начинали перемешивание. Смесь перемешивали и нагревали примерно до 20-22°C для растворения всех твердых веществ. В течение примерно 5 мин добавляли уксусную кислоту (0,068 кг, 0,12 масс.ед., 1,3 экв.), поддерживая при этом температуру около примерно 22-24°C. Реакционную смесь перемешивали примерно при 20-24°C в течение приблизительно 24 ч и подтверждали завершение реакции с помощью ВЭЖХ. Добавляли в реактор безводный гептан (4,49 кг, 8,0 масс.ед.) и понижали температуру примерно до 0-5°C. Медленно в течение примерно 26 минут добавляли 30% раствор (масс./масс.) H2O2 в воде (0,106 кг, 0,19 масс.ед., 1,1 экв.), поддерживая температуру в пределах 0-2°C. Полученную реакционную смесь перемешивали в течение примерно 3,5 ч, сохраняя температуру в диапазоне от -1 до 2°C. Завершение окисления подтверждали с помощью ВЭЖХ. Остатки пероксидов гасили при температуре около 0°C добавлением водного раствора пентагидрата тиосульфата натрия (1/1 (масс./масс.), 0,146 кг, 0,26 масс.ед., 0,34 экв.). Продолжали перемешивание в течение ночи (17,25 ч) при температуре примерно 0-2°C. Гептановый слой тестировали на присутствие пероксидов (EM Quant peroxides test, EM Science, Gibbstown, NJ), получая отрицательный результат. Температуру повышали примерно до 20°C. Соли отделяли фильтрованием и промывали их гептаном (1,26 кг, 2,25 масс.ед.). Полученный фильтрат помещали в новый стеклянный реактор и давали слоям разделиться в течение примерно 15 мин. Происходило образование трех слоев, и два нижних слоя (водный и ацетонитрильный) отделяли и выбрасывали. Верхний гептановый слой дважды промывали ацетонитрилом (1,0 кг, 1,8 масс.ед.) и концентрировали в вакууме до удаления всех летучих компонентов при температуре примерно 18-23°C. Получали неочищенное соединение 26 (0,79 кг, <1,4 масс.ед., чистота 91,1% по площади пика) в виде густого масла с янтарным оттенком, содержащего 5,6% остаточного гептана. Условия проведения ВЭЖХ для анализа соединения 26 (ВЭЖХ ТМ3 соединения 26):
Очистка соединения 26 (стадия 5b)
Общая методика очистки соединения 26: (примечание: 1,0 масс.ед. в данной методике соответствует количеству соединения 22, использованного для получения неочищенного соединения 26). Неочищенное соединение 26, например, полученное на стадии 5a (приблизительно 1,2-1,5 масс.ед.), растворяют в смеси изопропанола и этилацетата (1/99 (объем/объем), 2 масс.ед.) и полученный раствор вводят в колонку с силикагелем Biotage (примерно 10 масс.ед. SiO2), предварительно приведенную в равновесие смесью изопропанола и этилацетата (1/99 (объем/объем), примерно 55 масс.ед.). Разделение выполняют с использованием смеси изопропанол/этилацетат (3/97 (объем/объем), примерно 80 масс.ед.) и затем смеси изопропанол/этилацетат (10/90 (объем/объем), приблизительно 110 масс.ед.). Собирают предпочтительно около 35 фракций ~4 масс.ед., включая, например, фракции, содержащие ≥1% от теоретического выхода соединения 26 (≥0,0127 масс.ед. соединения 26 (оценка по данным ВЭЖХ)). Желаемые фракции объединяют и концентрируют в вакууме при температуре примерно 20-30°C с получением прозрачного масла. Это прозрачное масло растворяют в гептане (примерно 7,0 масс.ед.) и полученный раствор концентрируют до удаления всех летучих компонентов в вакууме при температуре примерно 20-30°C. Обработку гептаном повторяют еще раз и получают соединение 26 (1,0-1,15 масс.ед.; выход:75-85%) в виде бесцветного прозрачного масла.
Конкретный пример: Очистка неочищенного соединения 26 (0,79 кг). Примечание: 1,0 масс.ед. в этой методике соответствует количеству соединения 22 (0,56 кг), использованному для получения неочищенного соединения 26. Неочищенное соединение 26, полученное на стадии 5a (0,79 кг, <1,4 масс.ед.), растворяли в смеси изопропанола и этилацетата (1/99 (объем/объем), 1,18 кг, 2,1 масс.ед.) и полученный раствор вводили в колонку Biotage KP-Sil 150L (5,0 кг оксида кремния, 8,9 масс.ед.) предварительно приведенную в равновесие смесью изопропанола и этилацетата (1/99 (объем/объем), 31,5 кг, 56,3 масс.ед.). Через колонку пропускали смесь изопропанол/этилацетат (3/97 (объем/объем), 43,9 кг, 78,4 масс.ед.) и затем смесь изопропанол/этилацетат (10/90 (объем/объем), 62,3 кг, 111,3 масс.ед.). Собирали 36 фракций приблизительно по 2,3 кг (приблизительно 4,1 масс.ед.) каждая. Объединяли фракции, содержащие ≥1% от теоретического выхода соединения 26 (≥7,1 г соединения 26), т.е. в данном случае фракции 7-31, и концентрировали в вакууме при температуре 21-28°C с получением прозрачного масла. Это прозрачное масло растворяли в гептане (3,9 кг, 7,0 масс.ед.) и полученный раствор концентрировали в вакууме до удаления всех летучих компонентов при 24-30°C. Обработку гептаном повторяли еще один раз и получали соединение 26 (0,60 кг, 1,07 масс.ед.; чистота 93,4% (ВЭЖХ ТМ3 соединения 26); выход 78,8%) в виде бесцветного прозрачного масла.
Аналитические данные для соединения 26: 1H ЯМР (400 МГц, CDCl3) δ: 7,4-7,57 (м, 2H), 6,06 (т, J=5 Гц, 2H), 5,84-6,04 (м, 2H), 5,36 (д, J=17 Гц, 2H), 5,25 (д, J=10 Гц, 2H), 5,02 (м, 2H), 4,54 (м, 4H), 4,24 (м, 2H), 3,96-4,20 (м, 8H), 3,38-3,55 (м, 16H), 2,52 (т, J=7 Гц, 4H), 2,28 (т, J=1 Гц, 4M), 1,68-1,91 (м, 4H), 1,47-1,67 (м, 12H), 1,18-1,37 (м, 84H), 0,89 (т, J=7 Гц, 18H). ESI-МС (M+Na)+ Теоретический расчет для C89H168N4NaO19P2: 1682,2; реальный результат: 1682,3.
Хотя настоящее изобретение было описано с упоминанием конкретных вариантов его осуществления, специалисты в данной области должны понимать, что в изобретении могут быть произведены различные изменения и эквивалентные замены без отступления от сути изобретения и выхода за его объем. Кроме того, может быть осуществлено большое число модификаций для адаптации конкретной ситуации, материала, композиции, способа, стадии или стадий способа к сути и объему настоящего изобретения. Предполагается, что все эти модификации включены в объем формулы изобретения, приложенной к настоящей заявке.
Все патенты и публикации, упомянутые выше в тексте описания, включены в настоящую заявку посредством ссылки.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ФУРО[3,2-В]ПИРАНА, ПРИМЕНИМЫЕ В СИНТЕЗЕ АНАЛОГОВ | 2011 |
|
RU2579511C2 |
| ПОЛУЧЕНИЕ ПРОИЗВОДНЫХ ГИДРОКСИ-БЕНЗИЛБЕНЗОЛА | 2014 |
|
RU2671493C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО ИНДОЛОПИРРОЛОКАРБАЗОЛА | 2003 |
|
RU2337105C2 |
| СПОСОБЫ, ПРЕДНАЗНАЧЕННЫЕ ДЛЯ СИНТЕЗА АНАЛОГОВ ГАЛИХОНДРИНА B | 2014 |
|
RU2676486C1 |
| СПОСОБ ДЕАЦЕТИЛИРОВАНИЯ α-АМИНОАЦЕТАЛЕЙ | 2008 |
|
RU2477270C2 |
| ПРОИЗВОДНЫЕ ФЕНИЛАЛКАНАМИДА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И СЕЛЬСКОХОЗЯЙСТВЕННЫЙ ИЛИ САДОВЫЙ ФУНГИЦИД | 1996 |
|
RU2156235C2 |
| НОВЫЕ ГИДРОКСИСЛОЖНОЭФИРНЫЕ ПРОИЗВОДНЫЕ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2016 |
|
RU2734418C2 |
| ПРОИЗВОДНЫЕ АМИДА N-СУЛЬФОНИЛКАРБОНОВОЙ КИСЛОТЫ, ВКЛЮЧАЮЩИЕ N-СОДЕРЖАЩЕЕ 6-ЧЛЕННОЕ АРОМАТИЧЕСКОЕ КОЛЬЦО, ФУНГИЦИДНАЯ И ГЕРБИЦИДНАЯ КОМПОЗИЦИИ И СПОСОБЫ БОРЬБЫ С СОРНЯКАМИ И ФИТОПАТОГЕННЫМИ ГРИБКАМИ | 1993 |
|
RU2117662C1 |
| МОДУЛЯТОРЫ СЕРИН-ТРЕОНИНПРОТЕИНКИНАЗ И PARP | 2013 |
|
RU2681209C2 |
| КОНДЕНСИРОВАННЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, КОМПОЗИЦИЯ НА ИХ ОСНОВЕ, СПОСОБ БОРЬБЫ С ГРИБКАМИ | 1993 |
|
RU2098408C1 |
Изобретение относится к соединениям формулы 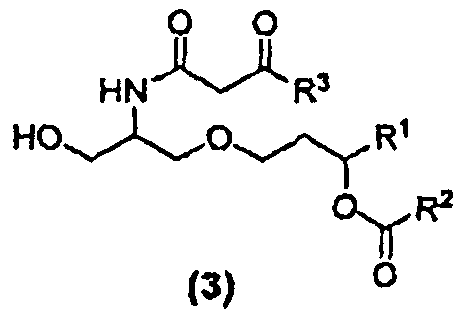 , которые могут использоваться в способе синтеза предшественников иммунологического адъюванта Е6020. В формуле (3) R1, R2, R3 представляют собой C5-C15 алкильную группу, C5-C15 алкенильную группу или C5-C15 алкинильную группу. Кроме того, изобретение относится к соединениям формулы (4), которые также могут использоваться в синтезе предшественников указанного адъюванта. В формуле
, которые могут использоваться в способе синтеза предшественников иммунологического адъюванта Е6020. В формуле (3) R1, R2, R3 представляют собой C5-C15 алкильную группу, C5-C15 алкенильную группу или C5-C15 алкинильную группу. Кроме того, изобретение относится к соединениям формулы (4), которые также могут использоваться в синтезе предшественников указанного адъюванта. В формуле 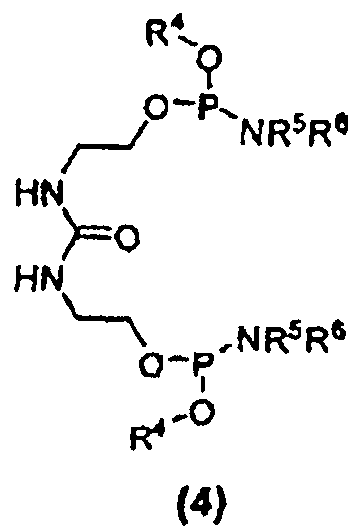 R4 представляет собой C1-С6 алкильную группу, С3-С5 алкенильную группу, С3-С5 алкинильную группу, циклоалкильную группу, этильную группу, замещенную в положении 2, галогенэтильную группу, арильную группу, бензильную группу или силильную группу; R5, R6 независимо представляют собой C1-С6 алкильную группу, С3-С6 алкенильную группу или С3-С6 алкинильную группу; или же R5 и R6 совместно с атомом азота, к которому они присоединены, образуют 5- или 6-членный гетероцикл. Изобретение относится также к способам синтеза соединений формул (3) и (4) и к кристаллическим формам их конкретных представителей. 7 н. и 27 з.п. ф-лы, 8 ил., 2 табл., 6 пр.
R4 представляет собой C1-С6 алкильную группу, С3-С5 алкенильную группу, С3-С5 алкинильную группу, циклоалкильную группу, этильную группу, замещенную в положении 2, галогенэтильную группу, арильную группу, бензильную группу или силильную группу; R5, R6 независимо представляют собой C1-С6 алкильную группу, С3-С6 алкенильную группу или С3-С6 алкинильную группу; или же R5 и R6 совместно с атомом азота, к которому они присоединены, образуют 5- или 6-членный гетероцикл. Изобретение относится также к способам синтеза соединений формул (3) и (4) и к кристаллическим формам их конкретных представителей. 7 н. и 27 з.п. ф-лы, 8 ил., 2 табл., 6 пр.
1. Соединение формулы (3)
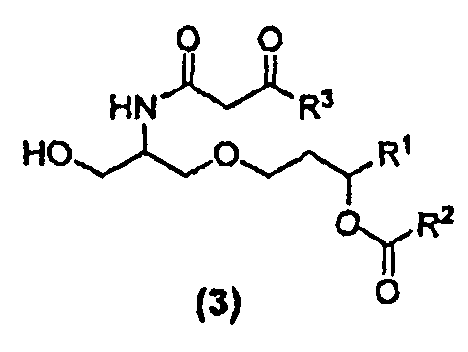
где R1 представляет собой C5-C15 алкильную группу, C5-C15 алкенильную группу или C5-C15 алкинильную группу;
R2 представляет собой C5-C15 алкильную группу, C5-C15 алкенильную группу или C5-C15 алкинильную группу; и
R3 представляет собой C5-C15 алкильную группу, C5-C15 алкенильную группу или C5-C15 алкинильную группу.
2. Соединение по п.1, где
R1 представляет собой C5-C12 алкильную группу,
R2 представляет собой С7-С14 алкильную группу и
R3 представляет собой C7-C14 алкильную группу.
3. Соединение по п.2, где
R1 представляет собой C7 алкильную группу,
R2 представляет собой С11 алкильную группу и
R3 представляет собой С11 алкильную группу.
4. Соединение по п.3, где
R1 представляет собой н-гептильную группу,
R2 представляет собой н-ундецильную группу и
R3 представляет собой н-ундецильную группу.
5. Соединение по п.1, где по меньшей мере один из заместителей R1, R2 и R3 отличается от остальных.
6. Соединение по пп.1, 2, 3, 4 или 5, имеющее стереохимию формулы (3а):
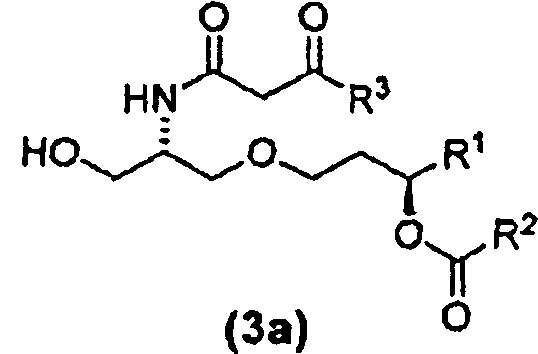
7. Соединение по п.6, которое является соединением 22
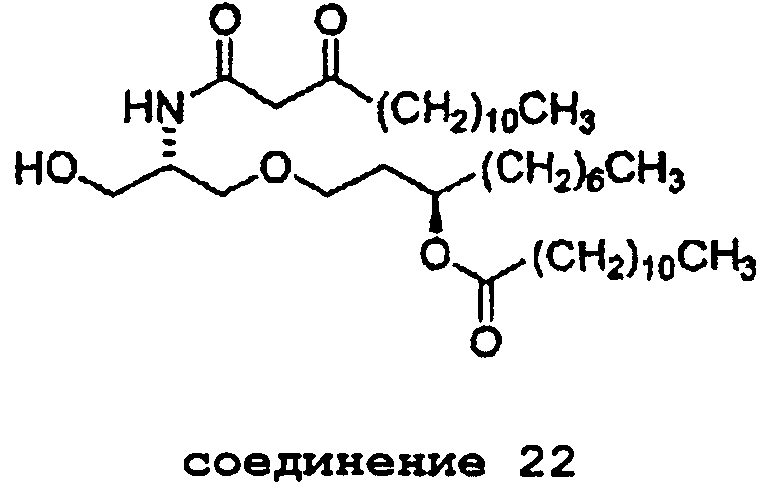
8. Способ получения соединения формулы (3) взаимодействием соединения формулы (1) с соединением формулы (2), как показано ниже
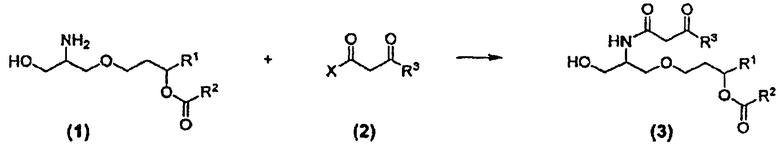
где R1 представляет собой C5-C15 алкильную группу, С5-С15 алкенильную группу или C5-C15 алкинильную группу;
R2 представляет собой C5-C15 алкильную группу, C5-C15 алкенильную группу или C5-C15 алкинильную группу;
R3 представляет собой C5-C15 алкильную группу, C5-C15 алкенильную группу или C5-C15 алкинильную группу; и
Х означает уходящую группу.
9. Способ по п.8, где
R1 представляет собой C5-C12 алкильную группу,
R2 представляет собой C7-C14 алкильную группу,
R3 представляет собой C7-C14 алкильную группу и
Х представляет собой ОН, Cl, F, имидазолидил, триметилацетокси, этилкарбонат, метилкарбонат, изобутилкарбонат или группу формулы Z:
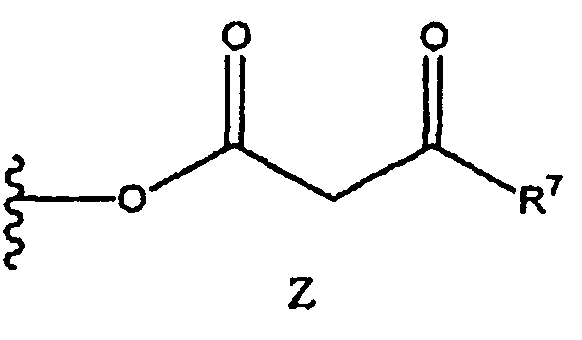
где заместитель R7 идентичен заместителю R3 в формуле (2), так что соединение формулы (2) является симметричным ангидридом бета-кетоэфира.
10. Способ по п.9, где
R1 представляет собой C7 алкильную группу;
R2 представляет собой С11 алкильную группу; и
R3 представляет собой С11 алкильную группу.
11. Способ по п.10, где
R1 представляет собой н-гептильную группу;
R2 представляет собой н-ундецильную группу; и
R3 представляет собой н-ундецильную группу.
12. Способ по п.8, где по меньшей мере один из заместителей R1, R2 и R3 отличается от остальных.
13. Способ по п.8, дополнительно включающий стадию гидрирования соединения формулы Y:
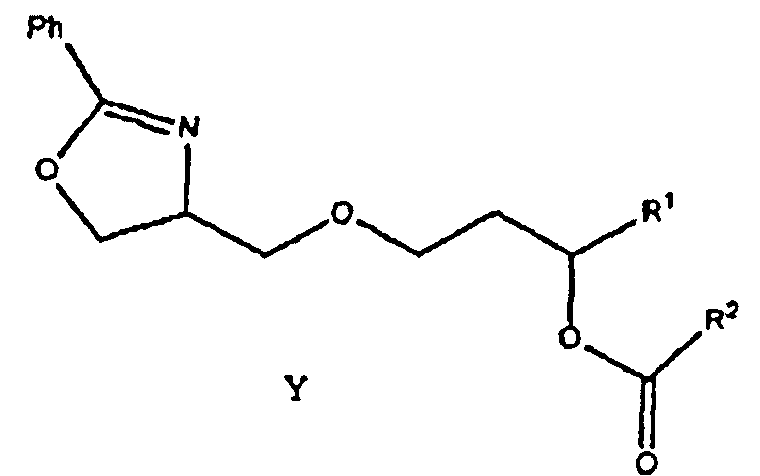
в изопропаноле для получения соединения формулы (1).
14. Соединение формулы (4):
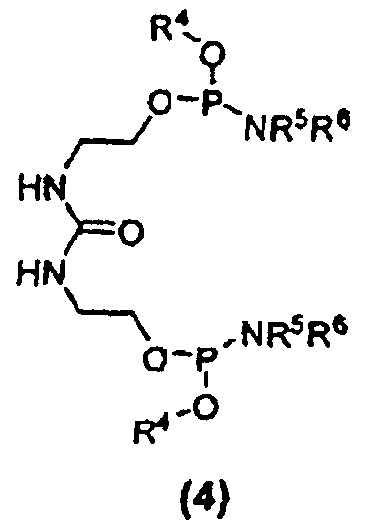
где R4 в каждом случае независимо представляет собой С1-С6 алкильную группу, С3-С5 алкенильную группу, С3-С5 алкинильную группу, циклоалкильную группу, этильную группу, замещенную в положении 2, галогенэтильную группу, арильную группу, бензильную группу или силильную группу;
R5 в каждом случае независимо представляет собой C1-С6 алкильную группу, С3-С6 алкенильную группу или С3-С6 алкинильную группу; и
R6 в каждом случае независимо представляет собой C1-С6 алкильную группу, С3-С6 алкенильную группу или С3-С6 алкинильную группу; или же R5 и R6 совместно с атомом азота, к которому они присоединены, образуют 5- или 6-членный гетероцикл.
15. Соединение по п.14, где
R4 в каждом случае независимо представляет собой С3-С5 алкенильную группу, 2-цианоэтильную группу, 2-(триметилсилил)этильную группу или 2,2,2-трихлорэтильную группу;
R5 в каждом случае независимо представляет собой С3-С6 алкильную группу; и
R6 в каждом случае независимо представляет собой С3-С6 алкильную группу.
16. Соединение формулы (4) по п.15, где
R4 в каждом случае представляет собой аллильную группу;
R5 в каждом случае означает изопропильную группу; и
R6 в каждом случае означает изопропильную группу.
17. Соединение по п.16, которое является соединением 25:
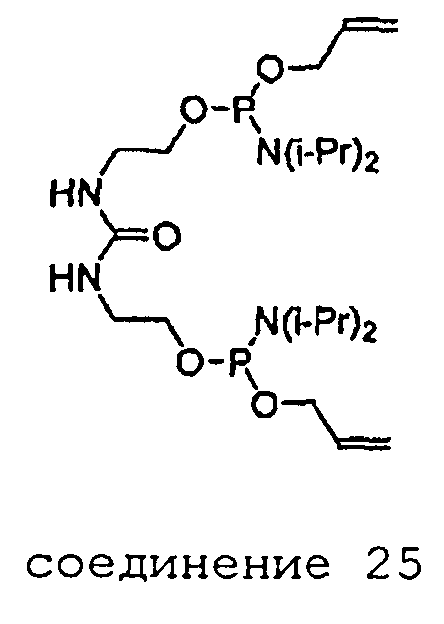 .
.
18. Способ получения соединения формулы (4) реакцией конденсации, показанной ниже:
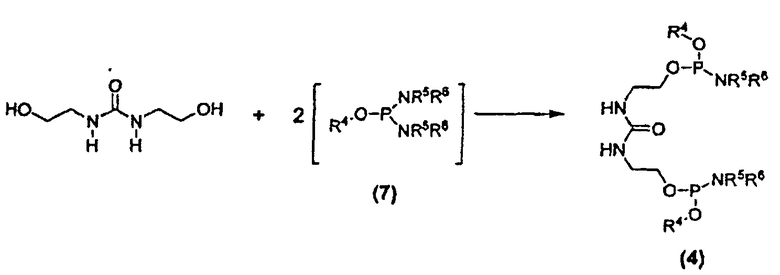
где R4 в каждом случае независимо представляет собой защитную группу;
R5 в каждом случае независимо представляет собой C1-С6 алкильную группу, С3-С6 алкенильную группу или С3-С6 алкинильную группу; и
R6 в каждом случае независимо представляет собой C1-С6 алкильную группу, С3-С6 алкенильную группу или С3-С6 алкинильную группу; или же R5 и R6 совместно с атомом азота, к которому они присоединены, образуют 5- или 6-членный гетероцикл.
19. Способ по п.18, где
R4 в каждом случае независимо представляет собой С3-С5 алкенильную группу, 2-цианоэтильную группу, 2-(триметилсилил)этильную группу или 2,2,2-трихлорэтильную группу;
R5 в каждом случае независимо представляет собой С3-С6 алкильную группу; и
R6 в каждом случае независимо представляет собой С3-С6 алкильную группу.
20. Способ по п.18, где
R4 в каждом случае представляет собой аллильную группу;
R5 в каждом случае означает изопропильную группу; и
R6 в каждом случае означает изопропильную группу.
21. Способ получения соединения формулы (6), включающий стадии:
i) взаимодействия соединения формулы (3) с соединением формулы (4) с образованием соединения формулы (5), как показано ниже:
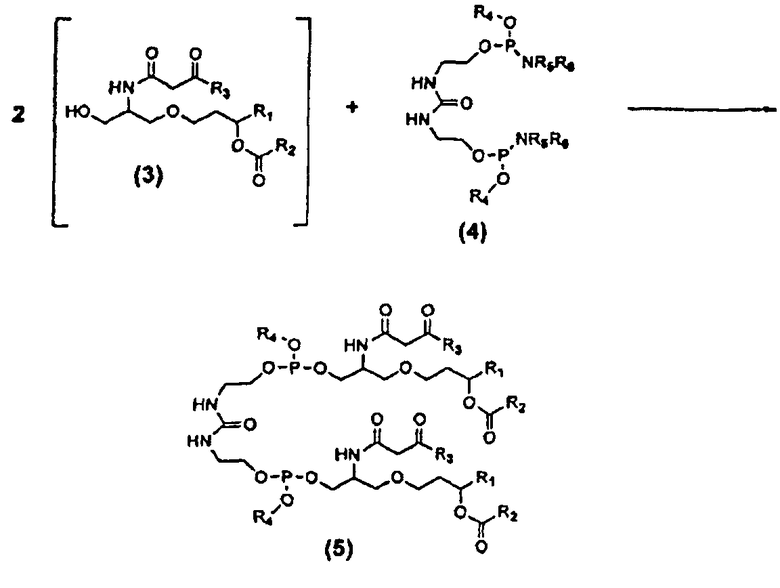 ; и
; и
ii) окисления полученного соединения формулы (5) с образованием соединения формулы (6), как показано ниже
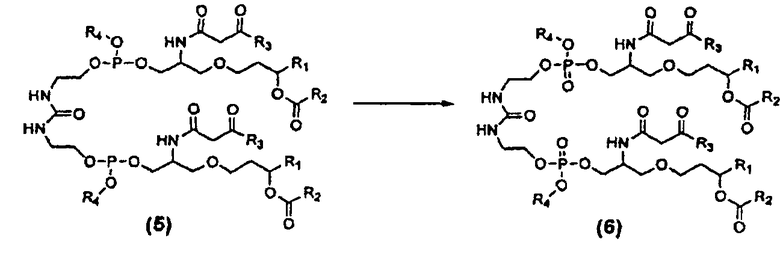 ;
;
где R1 представляет собой C5-C15 алкильную группу, С5-С15 алкенильную группу или C5-C15 алкинильную группу;
R2 представляет собой C5-C15 алкильную группу, C5-C15 алкенильную группу или C5-C15 алкинильную группу;
R3 представляет собой C5-C15 алкильную группу, C5-C15 алкенильную группу или C5-C15 алкинильную группу;
R4 в каждом случае независимо представляет собой защитную группу;
R5 в каждом случае независимо представляет собой C1-С6 алкильную группу, С3-С6 алкенильную группу или С3-С6 алкинильную группу; и
R6 в каждом случае независимо представляет собой C1-С6 алкильную группу, С3-С6 алкенильную группу или С3-С6 алкинильную группу; или же R5 и R6 совместно с атомом азота, к которому они присоединены, образуют 5- или 6-членный гетероцикл.
22. Способ по п.21, где
R1 представляет собой C5-C12 алкильную группу;
R2 представляет собой С7-С14 алкильную группу;
R3 представляет собой C7-C14 алкильную группу;
R4 в каждом случае независимо представляет собой С3-С5 алкенильную группу, 2-цианоэтильную группу, 2-(триметилсилил)этильную группу или 2,2,2-трихлорэтильную группу;
R5 в каждом случае независимо представляет собой С3-С6 алкильную группу; и
R6 в каждом случае независимо представляет собой С3-С6 алкильную группу.
23. Способ по п.21, где
R1 представляет собой С7 алкильную группу;
R2 представляет собой С11 алкильную группу;
R3 представляет собой С11 алкильную группу;
R4 означает аллильную группу;
R5 означает изопропильную группу; и
R6 означает изопропильную группу.
24. Способ по п.23, где
R1 представляет собой н-гептильную группу;
R2 представляет собой н-ундецильную группу;
R3 представляет собой н-ундецильную группу;
R4 означает аллильную группу;
R5 означает изопропильную группу; и
R6 означает изопропильную группу.
25. Способ по п.21, где по меньшей мере один из заместителей R1, R2 и R3 отличается от остальных.
26. Способ по п.21, где получение соединения формулы (5) включает стадии:
i) перемешивания смеси соединения формулы (3) и соединения формулы (4) в смеси сорастворителей гептана и ацетонитрила до полного растворения твердых веществ;
ii) медленного добавления от 1,0 до 2,0 эквивалентов уксусной кислоты при подержании температуры от 20ºС до 25°С; и
iii) перемешивания полученной смеси до завершения реакции.
27. Способ по п.26, где масса использованного гептана в 4-6 раз превышает массу введенного в реакцию соединения формулы (3).
28. Способ по п.26, где масса использованного ацетонитрила в 1-2 раза превышает массу введенного в реакцию соединения формулы (3).
29. Способ по любому из пп.21-28, где окисление образовавшегося соединения (5) с целью получения соединения формулы (6) включает стадии:
i) добавления гептана к реакционной смеси, содержащей образовавшееся соединение формулы (5);
ii) охлаждения полученной смеси до температуры от -5°С до 10°С;
iii) медленного добавления 30% (мас./мас.) раствора Н2О2 в воде с такой скоростью, чтобы температура реакционной смеси оставалась в диапазоне от -5°С до 10°С; и
iv) перемешивания полученной смеси при температуре от -5°С до 10°С до завершения реакции.
30. Способ по п.29, где
реакционную смесь на стадии ii) охлаждают до температуры от 0ºС до 5°С;
температуру реакционной смеси на стадии iii) поддерживают в пределах от 0ºС до 2°С; и
температуру реакционной смеси на стадии iv) поддерживают в пределах от -1ºС до 2°С.
31. Кристаллическое соединение 22
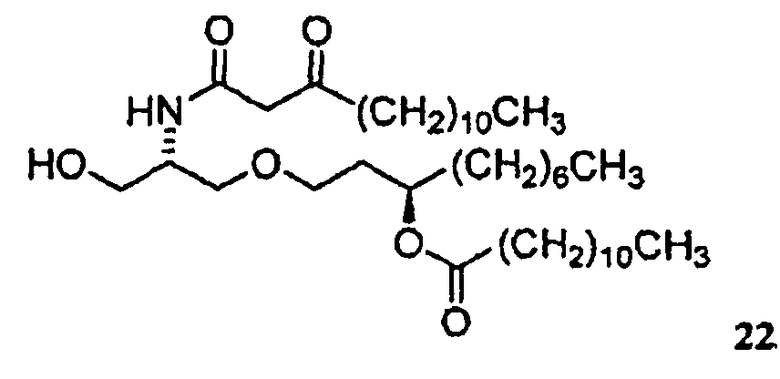 ,
,
характеризуемое картиной дифракции рентгеновских лучей на кристаллическом порошке, полученной с использованием излучения меди, имеющей пики при углах 12,3±0,2°2Θ, 14,5±0,2°2Θ, 16,2±0,2°2Θ, 17,8±0,2°2Θ, 22,2±0,2°2Θ и 23,8±0,2°2Θ.
32. Кристаллическое соединение 22 по п.31, дополнительно характеризуемое температурой плавления, составляющей примерно 41°С.
33. Кристаллическое соединение 25
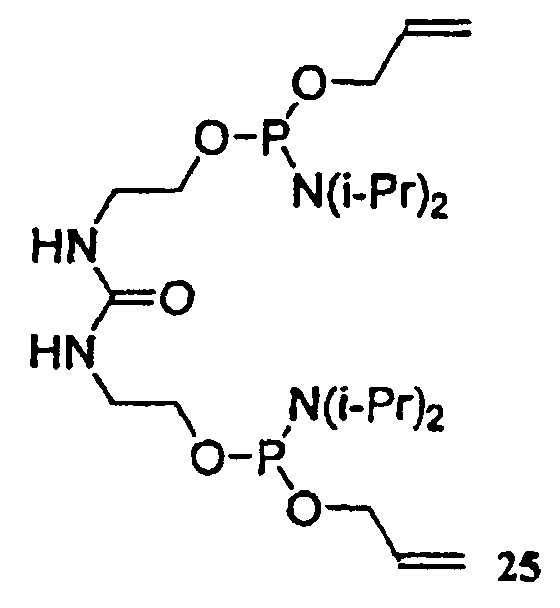 ,
,
характеризуемое картиной дифракции рентгеновских лучей на кристаллическом порошке, полученной с использованием излучения меди, имеющей пики при углах 6,6±0,3°2Θ, 14,0±0,3°2Θ, 17,3±0,3°2Θ и 19,4±0,3°2Θ.
34. Кристаллическое соединение 25 по п.33, дополнительно характеризуемое температурой плавления, составляющей примерно 65°С.
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| Способ приготовления ациалцетильных двупроизводных ароматических диаминов | 1926 |
|
SU5483A1 |

