Область техники, к которой относится изобретение
Настоящее изобретение относится к новому способу хирального разделения 2,4-дизамещенных 1,3-оксатиоланов и их производных общей формулы (I):
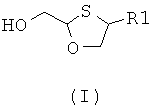
Настоящее изобретение относится к новым производным 2,4-дизамещенных 1,3-оксатиоланов.
Уровень техники
Показано, что классы соединений, известных как 2,4-дизамещенные 1,3-оксатиоланы, а именно, соединений аналогов пиримидиновых нуклеозидов, обладают сильной противовирусной активностью. В частности показано, что эти соединения действуют как сильные ингибиторы репликации ВИЧ-1 в Т-лимфоцитах в течение длительного периода времени с менее цитотоксическими побочными эффектами в сравнении с соединениями, известными в данной области техники (смотри, например, Belleau et. al. (1993) Bioorg. Med. Chem. Lett. Vol.3, No.8, 1723-1728). Показано также, что эти соединения проявляют активность против 3ТС-резистентных штаммов ВИЧ (Taylor et. al. (2000) Antiviral Chem. Chemother. Vol.11, No.4, 291-301; и Stoddart et. al. (2000) Antimicrob. Agenst Chemother. Vol.44, No.3, 783-786). Эти соединения также используют для профилактики и лечения вирусных инфекций гепатита В. Эти соединения могут быть получены способами, раскрытыми в WO 92/08717, WO 95/29176, WO 02/102796 и WO 2006/096954.
Соединения семейства 2,4-дизамещенных 1,3-оксатиолана содержат два хиральных центра. Соединения, содержащие два хиральных центра, могут существовать в виде смеси четырех стереоизомеров, в которых конфигурация при двух хиральных центрах представляет собой (R,R) или (R,S) или (S,R) или (S,S). Известно, что (R,R) и (S,S) формы являются цис-энантиомерами, поскольку они не являются зеркальными отображениями при наложении одного на другой, и известно, что (R,S) и (S,R) формы являются транс-энантиомерами по той же причине. При терапевтическом использовании для лечения людей обычно как правило необходимо выделять соединение только в одной стереоизомерной форме, также известной как хирально чистой форме. Это может быть достигнуто в том случае, когда синтез одного стереоизомера проводят из исходного материала с одним хиральным центром в энантиомерно чистой форме или из подходящего промежуточного соединения.
Например, цис-2-гидроксиметил-4-(цитозин-1'-ил)-1,3-оксатиолан может быть получен способами, описанными Mansour et al., "Anti-Human Immunodeficiency Virus and Anti-Hepatitis-B Virus Activities and Toxicities of the Enantiomers of 2'-Deoxy-3'-oxa-4'-thiacytidine and Their 5-Fluoro Analogues in vitro", J. Med. Chem., (1995), Vol.38, No. 1, 1-4, а также способами, раскрытыми в патенте US No 6,228,860, Nucleosides and Nucleotides, (1995) 14(3-5) 627-735 и Caputo et. al. в Eur. J. Org. Chem. (1999) Vol.6, 1455-1458.
Однако эти способы синтеза не всегда приводят к стереоспецифическому образованию новых хиральных центров, а вместо этого приводят к соотношению центров, известному как энантиомерный избыток (ее):
ее = [(% желаемый изомер)-(% изомер с обратным знаком)]/сумма (желаемый + изомер с обратным знаком)
Когда соединения нужны в виде единственной формы, например, если присутствуют только два (R,R) и (S,S) цис-энантиомера, единственная форма, представляющая собой либо (R,R) либо (S,S) форму, может быть получена разделением смеси двух цис-энантиомеров способом хиральной ВЭЖХ. Обзор такой технологии может быть найден в "Enantiomers, Racemates and Resolutions" by J. Jacques, A. Collet & S.H. Wilen (John Wiley & Sons, 1981).
В качестве альтернативы соединения или любое подходящее промежуточное соединение может быть подвергнуто разделению, опосредованному ферментом, содействующим протеканию энантиоселективного разложения подходящим ферментом, например, цитидиндезаминазой или селективному ферментативному разложению подходящего производного (смотри, например, Storer et. al, "The resolution and Absolute Stereochemistry of the Enantiomers of cis 1[2(Hydroxomethyl)-1,3-Oxathiolan-5-Y1)Cytosine (BCH-189): Equipotent Anti-HIV Agents", Nucleosides & Nucleotides, (1993) 12(2), 225-236).
Взаимодействие рацемической смеси соединения с оптически активной расщепляющей кислотой или основанием может быть также использовано для энантиомерного разделения соединения. Например, WO 2006/096954 раскрывает способ получения оптически активных цис-1,3-оксатиоланов. Способ состоит в (а) взаимодействии производного 1,3-оксатиолана в цис-конфигурации с хиральной кислотой, приводящем к получению двух диастереомерных солей; (b) извлечении одной из двух диастереомерных солей; и (с) обессоливании для удаления хиральной кислоты. Предпочтительные хиральные кислоты включают в себя (+)-L-винную кислоту, (1R)-(-)-10-камфорсульфоновую кислоту, (-)-2,3-дибензоилвинную кислоту или (-)-L-яблочную кислоту. Этот способ также раскрывает добавление нехиральной кислоты вместе с хиральной кислотой, приводящее к двум диастереомерным солям. Однако недостатком способа WO 2006/096954 является стадия образования солей. Эта стадия требует использования реагентов хиральных кислот и в некоторых случаях также нехиральных кислот. Стадия образования солей также требует введения дополнительной стадии обессоливания для получения желаемого оптически активного цис-продукта.
Использование дополнительных реагентов, например, хиральных кислот, и дополнительных стадий в способе, например, стадии обессоливания, необходимой в способе WO 2006/096954, нежелательно с точки зрения промышленных параметров, поскольку они повышают стоимость продукции, а также увеличивают время, необходимое для получения желаемого продукта. Кроме того, на каждой дополнительной стадии процесса имеется вероятность неэффективного извлечения конечного продукта из-за потерь, происходящих на каждой стадии процесса.
Авторы настоящего изобретения показали, что при правильном выборе групп R2, R3 и R4, оптически активное соединение общей формулы (II) или (III) может быть получено селективной перекристаллизацией. Авторы настоящего изобретения также показали, что искомый растворитель для перекристаллизации выбирают на основе групп R2, R3 и R4. Авторы настоящего изобретения избегают стадий образования солей и обессоливания, необходимые в предыдущих способах, и обеспечивают более простой, более эффективный способ получения оптически активных цис-1,3-оксатиоланов. В особенно предпочтительном аспекте настоящее изобретение предусматривает способ отделения нежелательного диастереомера, например, цис-диастереомера и повышения оптической чистоты цис-изомера перекристаллизацией. Настоящее изобретение также обеспечивает новые производные 2,4-дизамещенных 1,3-оксатиоланов.
Раскрытие изобретения
Согласно первому аспекту настоящее изобретение предоставляет способ получения соединения общей формулы (II), включающий стадии:
(а) образования 2,4-дизамещенного 1,3-оксатиолана общей формулы (II) или (III):
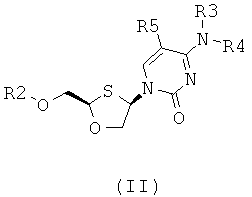 ;
; 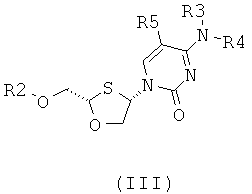
где
R2 представляет собой Н, С(O)С1-6алкил, С(O)ОС1-6алкил, С(O)С6-12арил, С(O)ОС6-12арил, С(O)С6-12арилалкил, С(O)ОС6-12арилалкил, С(O)С1-6алкиларил, C(O)OC1-6алкиларил, или силильную защитную группу общей формулы SiR7R8R9, где каждая из групп R7, R8 и R9 представляют собой независимо выбранную из С1-6алкила, арила или С1-6алкиларила;
R3 и R4 являются одинаковыми или различными и каждая индивидуально выбрана из Н или C(O)-R6;
R6 представляет собой С6-12арил или С1-6алкиларил, в котором С1-6алкиларил предпочтительно является С1-6алкилС6-12арилом; и
R5 представляет собой Н, Br, Cl, F, I или CF3; и
(b) селективной перекристаллизации соединения общей формулы (II) из растворителя, в котором растворитель представляет собой С1-6 спирт или смесь С1-6 спиртов.
В особо предпочтительном осуществлении R6 представляет собой фенил.
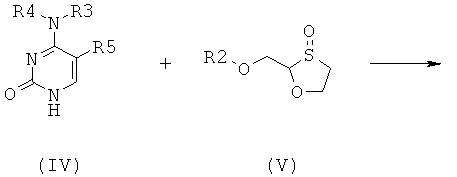
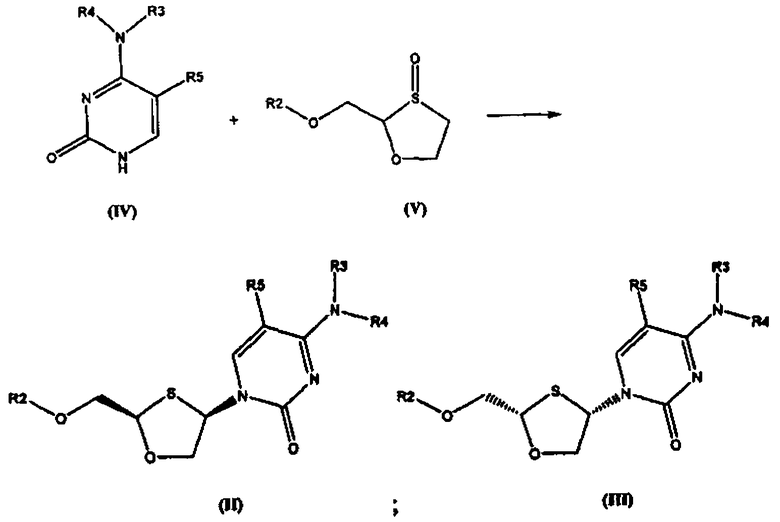
Согласно второму аспекту настоящее изобретение предоставляет способ получения соединения общей формулы (II), включающий стадии:
(а) взаимодействия основания общей формулы (IV) с 1,3-оксатиоланом общей формулы (V) с образованием соединения общей формулы (II) или (III):
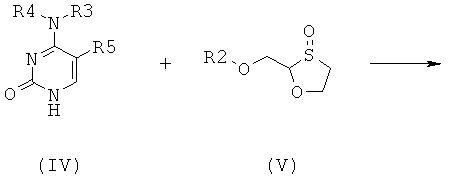
;
где
R2 представляет собой Н, С(O)С1-6алкил, С(O)ОС1-6алкил, С(O)С6-12арил, С(O)ОС6-12арил, С(O)С6-12арилалкил, С(O)ОС6-12арилалкил, С(O)С1-6алкиларил, С(O)ОС1-6алкиларил, или силильную защитную группу общей формулы SiR7R8R9, где каждая из групп R7, R8 и R9 представляют собой независимо выбранную из С1-6алкила, арила или С1-6алкиларила;
R3 и R4 являются одинаковыми или различными и каждая индивидуально выбрана из Н или C(O)-R6;
R6 представляет собой С6-12арил или С1-6алкиларил, в котором С1-6алкиларил предпочтительно является С1-6алкилС6-12арилом; и
R5 представляет собой Н, Br, Cl, F, I или CF3; и
где 1,3-оксатиолан общей формулы (V) представляет собой (R)-энантиомер, по меньшей мере, в 60% ее; и
(b) селективной перекристаллизации соединения общей формулы (II) из растворителя, в котором растворитель представляет собой С1-6 спирт или смесь С1-6 спиртов.
В особо предпочтительном осуществлении R6 является фенилом.
Согласно третьему аспекту настоящее изобретение предоставляет способ получения соединения общей формулы (VI), включающий стадии:
(а) образования 2,4-дизамещснного 1,3-оксатиолана общей формулы (II) или (III):
;
где
R2 представляет собой Н, С(O)С1-6алкил, С(O)ОС1-6алкил, С(O)С6-12арил, С(O)ОС6-12арил, С(O)С6-12арилалкил, С(O)ОС6-12арилалкил, С(O)С1-6алкиларил, С(O)ОС1-6алкиларил, или силильную защитную группу общей формулы SiR7R8R9, где R7, R8 и R9 представляют собой каждая независимо выбранную из С1-6алкила, арила или С1-6алкиларила;
R3 и R4 являются одинаковыми или различными и каждая индивидуально выбрана из Н или C(O)-R6;
R6 представляет собой С6-12арил или С1-6алкиларил, в котором С1-6алкиларил предпочтительно является С1-6алкилС6-12арилом; и
R5 представляет собой Н, Br, Cl, F, I или CF3;
(b) селективной перекристаллизации соединения общей формулы (II) из растворителя, в котором растворитель представляет собой С1-6 спирт или смесь спиртов С1-6; и
(c) снятия защитной группы в соединении общей формулы (II) для получения соединения общей формулы (VI):
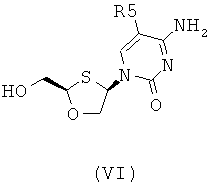
В особо предпочтительном осуществлении R6 является фенилом.
Согласно четвертому аспекту настоящее изобретение предоставляет способ получения соединения общей формулы (VI), включающий стадии:
(а) взаимодействия основания общей формулы (IV) с 1,3-оксатиоланом общей формулы (V) с образованием соединения общей формулы (II) или (III):
;
где
R2 представляет собой Н, С(O)С1-6алкил, С(O)ОС1-6алкил, С(O)С6-12арил, С(O)ОС6-12арил, С(O)С6-12арилалкил, С(O)ОС6-12арилалкил, С(O)С1-6алкиларил, С(O)ОС1-6алкиларил, или силильную защитную группу общей формулы SiR7R8R9, где R7, R8 и R9 представляют собой каждая независимо выбранную из С1-6алкила, арила или С1-6алкиларила;
R3 и R4 являются одинаковыми или различными и каждая индивидуально выбрана из Н или C(O)-R6;
R6 представляет собой С6-12арил или С1-6алкиларил, в котором С1-6алкиларил предпочтительно является С1-6алкилС6-12арилом; и
R5 представляет собой Н, Br, Cl, F, I или CF3; и
где 1,3-оксатиолан общей формулы (V) представляет собой (R)-энантиомер, по меньшей мере, в 60% ее;
(b) селективной перекристаллизации соединения общей формулы (II) из растворителя, в котором растворитель представляет собой С1-6 спирт или смесь спиртов С1-6; и
(c) снятия защитной группы в соединении общей формулы (II) для получения соединения общей формулы (VI):
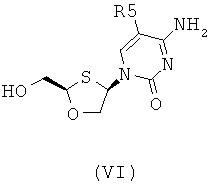
В особо предпочтительном осуществлении R6 является фенилом.
Согласно пятому аспекту настоящее изобретение предоставляет способ получения соединения общей формулы (VIII) или (IX):
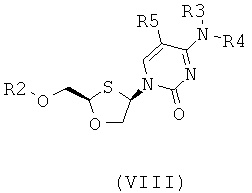 ;
; 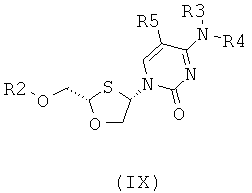
где
R2 представляет собой Н, С(O)С1-6алкил, С(O)ОС1-6алкил, С(O)С6-12арил, С(O)ОСб-12арил, С(O)С6-12арилалкил, С(O)ОС6-12арилалкил, С(O)С1-6алкиларил, С(O)ОС1-6алкиларил, или силильную защитную группу общей формулы SiR7R8R9, где R7, R8 и R9 представляют собой каждая независимо выбранную из С1-6алкила, ар ила или С1-6алкиларила;
Каждая из R3 и R4 индивидуально выбраны из Н или бензоила при условии, что когда R3 представляет собой Н, R4 не является Н и когда R4 представляет собой Н, R3 не является Н; и
R5 представляет собой Н, Br, Cl5, F, I или CF3.
Согласно шестому аспекту настоящее изобретение предоставляет способ получения соединения общей формулы (VI) или (VII) из соединения общей формулы (VIII) или (IX), соответственно, включающий стадии:
(а) снятия защитной группы в соединении общей формулы (VIII) или (IX) для получения соединения общей формулы (VI) или (VII), соответственно:
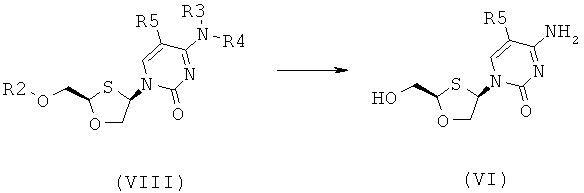
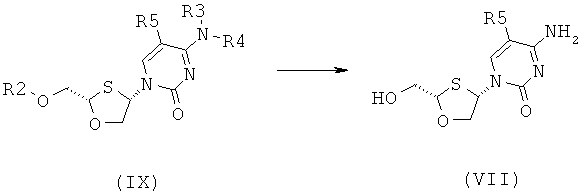
где
R2 представляет собой Н, С(O)С1-6алкил, С(O)ОС1.балкил, С(O)С6-12арил, С(O)ОС6-12арил, С(O)С6-12арилалкил, С(O)ОС6-12арилалкил, С(O)С1-6алкиларил, С(O)ОС1-6алкиларил, или силильную защитную группу общей формулы SiR7R8R9, где R7, R8 и R9 представляют собой каждая независимо выбранную из С1-6алкила, арила или С1-6алкиларила;
Каждый из R3 и R4 индивидуально выбраны из Н или бензоила при условии, что когда R3 представляет собой Н, R4 не является Н и когда R4 представляет собой Н, R3 не является Н; и
R5 представляет собой Н, Br, Cl5 F, I или CF3.
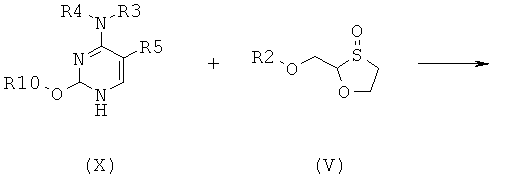
Согласно седьмому аспекту настоящее изобретение предоставляет способ получения соединения общей формулы (II), включающий стадии:
(а) взаимодействия силилированного основания общей формулы (X) с 1,3-оксатиоланом общей формулы (V) с образованием соединения общей формулы (II) или (III):
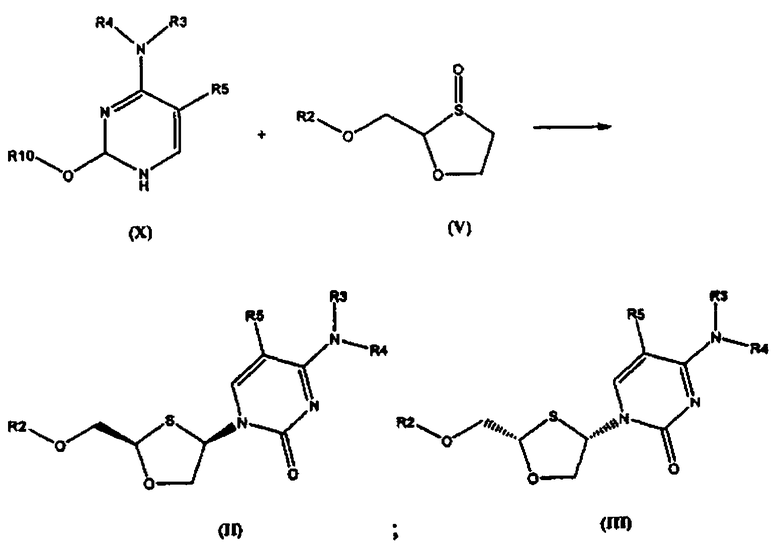
где
R2 представляет собой Н, С(O)С1-6алкил, С(O)ОС1-6алкил, С(O)С6-12арил, С(O)ОС6-12арил, С(O)С6-12арилалкил, С(O)ОС6-12арилалкил, С(O)С1-6алкиларил, С(O)ОС1-6алкиларил, или силильную защитную группу общей формулы SiR7R8R9, где R7, R8 и R9 представляют собой каждая независимо выбранную из С1-6алкила, арила или С1-6алкиларипа;
R3 и R4 являются одинаковыми или различными и каждая индивидуально выбрана из Н или C(O)-R6;
R6 представляет собой Сб-12арил или С1-6алкиларил, в котором С1-6алкиларил представляет собой предпочтительно С1-6алкилС6-12арил;
R5 представляет собой Н, Br, Cl, F, I или CF3; и
R10 представляет собой силильную защитную группу общей формулы SiR7R8R9, как определено ранее; и
где 1,3-оксатиолан общей формулы (V) представляет собой (R)-энантиомер, по меньшей мере, в 60% ее; и
(b) селективной перекристаллизации соединения общей формулы (II) из растворителя, в котором растворитель представляет собой С1-6 спирт или смесь спиртов С1-6;
В особо предпочтительном осуществлении R6 является фенилом.
Согласно восьмому аспекту настоящее изобретение предоставляет способ получения соединения общей формулы (VI), включающий стадии:
(а) взаимодействия силилированного основания общей формулы (X) с 1,3-оксатиоланом общей формулы (V) с образованием соединения общей формулы (II) или (III):
;
где
R2 представляет собой Н, С(O)С1-6алкил, С(O)ОС1-6алкил, С(O)С6-12арил, С(O)ОС6-12арил, С(O)С6-12арилалкил, С(O)ОС6-12арилалкил, С(O)С1-6алкиларил, С(O)ОС1-6алкиларил, или силильную защитную группу общей формулы SiR7R8R9, где R7, R8 и R9 представляют собой каждая независимо выбранную из С1-6алкила, арила или С1-6алкиларила;
R3 и R4 являются одинаковыми или различными и каждая индивидуально выбрана из Н или C(O)-R6;
R6 представляет собой С6-12арил или С1-6алкиларил, в котором С1-6алкиларил представляет собой предпочтительно С1-6алкилС6-12арил;
R5 представляет собой Н, Br, Cl, F, I или CF3; и
R10 представляет собой силильную защитную группу общей формулы SiR7R8R9, как определено ранее; и
где 1,3-оксатиолан общей формулы (V) представляет собой (R)-энантиомер по меньшей мере в 60% ее;
(b) селективной перекристаллизации соединения общей формулы (II) из растворителя, в котором растворитель представляет собой С1-6 спирт или смесь спиртов С1-6;
(c) снятия защитной группы в соединении общей формулы (II) для получения соединения общей формулы (VI):
В особо предпочтительном осуществлении R6 является фенилом.
Согласно девятому аспекту настоящее изобретение предоставляет способ получения соединения общей формулы (XIII) и его диастереомеров:
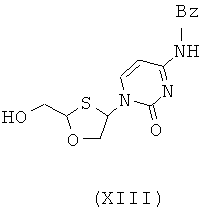
Согласно десятому аспекту настоящее изобретение предоставляет способ отделения соединения общей формулы (II) от соединения общей формулы (III), включающего стадии:
получения смеси 2,4-дизамещенных 1,3-оксатиоланов общей формулы (II) и (III):
;
где
R2 представляет собой Н, С(O)С1-6алкил, С(O)ОС1-6алкил, С(O)С6-12арил, С(O)ОС6-12арил, С(O)С6-12арилалкил, С(O)ОС6-12арилалкил, С(O)С1-6алкиларил, С(O)ОС1-6алкиларил, или силильную защитную группу общей формулы SiR7R8R9, где R7, R8 и R9 представляют собой каждая независимо выбранную из С1-6алкила, арила или С1-6алкиларила;
R3 и R4 являются одинаковыми или различными и каждый индивидуально выбран из Н или C(O)-R6;
R6 представляет собой С6-12арил или С1-6алкиларил, в котором С1-6алкиларил представляет собой предпочтительно С1-6алкилС6-12арил; и
R5 представляет собой Н, Br, Cl, F, I или CF3; и
(b) отделения соединения общей формулы (II) от соединения общей формулы (III) селективной перекристаллизацией соединения общей формулы (II) из растворителя, в котором растворитель представляет собой С1-6 спирт или смесь спиртов С1-6.
В особо предпочтительном осуществлении R6 является фенилом.
Осуществление изобретения
Настоящее изобретение будет далее описано, не ограничиваясь ссылками на предпочтительные осуществления.
Настоящее изобретение предпочтительно представляет способ получения оптически активного соединения общей формулы II или III селективной перекристаллизацией.
;
Соединения общей формулы (II) или (III) могут быть селективно перекристаллизованы из смеси двух цис-изомеров или из смеси четырех стереоизомеров, причем они представляют собой два цис-изомера (II) и (III) и два транс-изомера (XI) и (XII), как показано ниже:
цис-изомеры:
;
транс-изомеры:
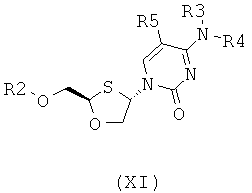 ;
; 
Согласно первому аспекту настоящее изобретение представляет способ получения соединения общей формулы II, включающий стадии:
(а) образования 2,4-дизамещенного 1,3-оксатиолана общей формулы (II) или (III):
;
где
R2 представляет собой Н, С(O)С1-6алкил, С(O)ОС1-6алкил, С(O)С6-12арил, С(O)ОС6-12арил, С(O)С6-12арилалкил, С(O)ОС6-12арилалкил, С(O)С1-6алкиларил, С(O)ОС1-6алкиларил, или силильную защитную группу общей формулы SiR7R8R9, где R7, R8 и R9 представляют собой каждая независимо выбранную из С1-6алкила, арила или С1-6алкиларила;
R3 и R4 являются одинаковыми или различными и каждая индивидуально выбрана из Н или C(O)-R6;
R6 представляет собой С6-12арил или С1-6алкиларил, в котором С1-6алкиларил предпочтительно является С1-6алкилС6-12арилом; и
R5 представляет собой Н, Br, Cl, F, I или CF3; и
(b) селективной перекристаллизации соединения общей формулы (II) из растворителя, в котором растворитель представляет собой С1-6 спирт или смесь спиртов С1-6.
В особо предпочтительном осуществлении R6 является фенилом.
Предпочтительно R2 является ацильной группой, еще более предпочтительно бензоильной группой или ацетильной группой.
Предпочтительно, каждую из R3 и R4 индивидуально выбирают из Н или бензоила, при условии, что когда R3 представляет собой Н, R4 не является Н и когда R4 представляет собой Н, R3 не является Н;
Предпочтительно, R5 представляет собой Н или F.
Предпочтительно, силильная группа общей формулы SiR7R8R9 выбрана из группы, состоящей из триметилсилила (TMS), трет-бутилдифенилсилила (TBDPS), трет-бутилдиметилсилила (TBDMS/TBS), три-изо-пропилсилила (TIPS) и триэтилсилила (TES).
Согласно второму аспекту настоящее изобретение представляет способ получения соединения общей формулы II, включающий стадии:
(а) взаимодействия основания общей формулы (IV) с 1,3-оксатиоланом общей формулы (V) с образованием соединения общей формулы (II) или (III):
где
R2 представляет собой Н, С(O)С1-6алкил, С(O)ОС1-6алкил, С(O)С6-12арил, С(O)ОС6-12арил, С(O)С6-12арилалкил, С(O)ОС6-12арилалкил, С(O)С1-6алкиларил, С(O)ОС1-6алкиларил, или силильную защитную группу общей формулы SiR7R8R9, где R7, R8 и R9 представляют собой каждая независимо выбранную из С1-6алкила, арила или С1-6алкиларила;
R3 и R4 являются одинаковыми или различными и каждая индивидуально выбрана из Н или C(O)-R6;
R6 представляет собой С6-12арил или С1-6алкиларил, в котором С1-6алкиларил предпочтительно является С1-6алкилС6-12арилом; и
R5 представляет собой Н, Br, Cl, F, 1 или CF3; и
где 1,3-оксатиолан общей формулы (V) представляет собой (R)-энантиомер, по меньшей мере, в 60% ее; и
(b) селективной перекристаллизации соединения общей формулы (II) из растворителя, в котором растворитель представляет собой С1-6 спирт или смесь спиртов С1-6.
В особо предпочтительном осуществлении R6 является фенилом.
Предпочтительно R2 является ацильной группой, еще более предпочтительно бензоильной группой или ацетильной группой.
Предпочтительно, каждую из R3 и R4 индивидуально выбирают из Н или бензоила, при условии, что когда R3 представляет собой Н, R4 не является Н и когда R4 представляет собой Н, R3 не является Н;
Предпочтительно, R5 представляет собой Н или F.
Предпочтительно, силильная группа общей формулы SiR7R8R9 выбрана из группы, состоящей из триметилсилила (TMS), трет-бутилдифенилсилила (TBDPS), трет-бутилдиметилсилила (TBDMS/TBS), три-изо-пропилсилила (TIPS) и триэтилсилила (TES).
Согласно третьему аспекту настоящее изобретение представляет способ получения соединения общей формулы (VI), включающий стадии:
(а) образования 2,4-дизамещенного 1,3-оксатиолана общей формулы (II) или (III):
;
где
R2 представляет собой Н, С(O)С1-6алкил, С(O)ОС1-6алкил, С(O)С6-12арил, С(O)ОС6-12арил, С(O)С6-12арилалкил, С(O)ОС6-12арилалкил, С(O)С1-6алкиларил, С(O)ОС1-6алкиларил, или силильную защитную группу общей формулы SiR7R8R9, где R7, R8 и R9 представляют собой каждая независимо выбранную из С1-6алкила, арила или С1-6алкиларила;
R3 и R4 являются одинаковыми или различными и каждая индивидуально выбрана из Н или C(O)-R6;
R6 представляет собой С6-12арил или С1-6алкиларил, в котором С1-6алкиларил предпочтительно является С1-6алкилС6-12арилом; и
R5 представляет собой Н, Br, Cl, F, I или CF3.
(b) селективной перекристаллизации соединения общей формулы (II) из растворителя, в котором растворитель представляет собой С1-6 спирт или смесь спиртов С1-6; и
(c) снятия защитной группы в соединении общей формулы (II) для получения соединения общей формулы (VI):
В особо предпочтительном осуществлении R6 является фенилом.
Предпочтительно R2 является ацильной группой, еще более предпочтительно бензоильной группой или ацетильной группой.
Предпочтительно, каждый из R3 и R4 индивидуально выбирают из Н или бензоила, при условии, что когда R3 представляет собой Н, R4 не является Н и когда R4 представляет собой Н, R3 не является Н; и
Предпочтительно, R5 представляет собой Н или F.
Предпочтительно, силильная группа общей формулы SiR7R8R9 выбрана из группы, состоящей из триметилсилила (TMS), трет-бутилдифенилсилила (TBDPS), трет-бутилдиметилсилила (TBDMS/TBS), три-изо-пропилсилила (TIPS) и триэтилсилила (TES).
Согласно четвертому аспекту настоящее изобретение предоставляет способ получения соединения общей формулы (VI), включающий стадии:
(а) взаимодействия основания общей формулы (IV) с 1,3-оксатиоланом общей формулы (V) с образованием соединения общей формулы (II) или (III):
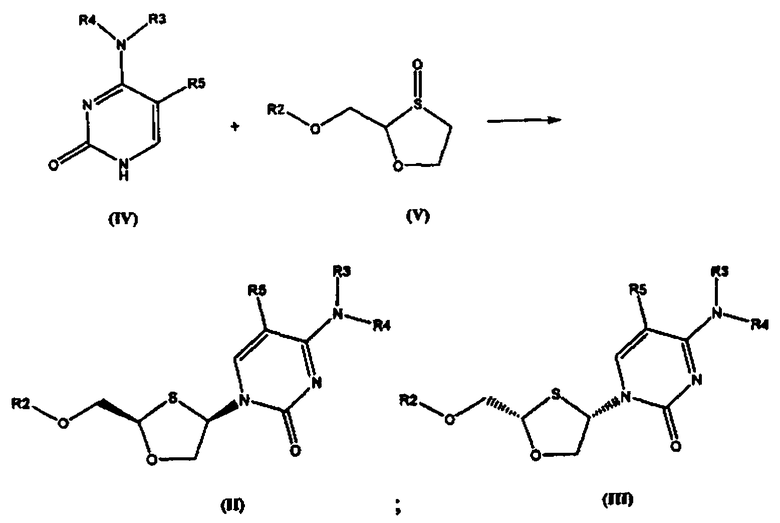
где
R2 представляет собой Н, С(O)С1-6алкил, С(O)ОС1-6алкил, С(O)С6-12арил, С(O)ОС6-12арил, С(O)С6-12арилалкил, С(O)ОС6-12арилалкил, С(O)С1-6алкиларил, С(O)ОС1-6алкиларил, или силильную защитную группу общей формулы SiR7R8R9, где R7, R8 и R9 представляют собой каждая независимо выбранную из С1-6алкила, арила или C1-6алкиларила;
R3 и R4 являются одинаковыми или различными и каждая индивидуально выбрана из Н или C(O)-R6;
R6 представляет собой С6-12арил или С1-6алкиларил, в котором С1-6алкиларил предпочтительно является С1-6алкилС6-12арилом; и
R5 представляет собой Н, Br, Cl, F, I или CF3; и
где 1,3-оксатиолан общей формулы (V) представляет собой (R)-энантиомер, по меньшей мере, в 60% ее;
(b) селективной перекристаллизации соединения общей формулы (II) из растворителя, в котором растворитель представляет собой С1-6 спирт или смесь спиртов C1-6; и
(c) снятия защитной группы в соединении общей формулы (II) для получения соединения общей формулы (VI):
В особо предпочтительном осуществлении R6 является фенилом.
Предпочтительно R2 является ацильной группой, еще более предпочтительно бензоильной группой или ацетильной группой.
Предпочтительно, каждый из R3 и R4 индивидуально выбирают из Н или бензоила, при условии, что когда R3 представляет собой Н, R4 не является Н и когда R4 представляет собой Н, R3 не является Н; и
Предпочтительно, R5 представляет собой Н или F.
Предпочтительно, силильная группа общей формулы SiR7R8R9 выбрана из группы, состоящей из триметилсилила (TMS), трет-бутилдифенилсилила (TBDPS), трет-бутилдиметилсилила (TBDMS/TBS), три-изо-пропилсилила (TIPS) и триэтилсилила (TES).
Согласно пятому аспекту настоящее изобретение представляет способ получения соединения общей формулы (VIII) или (IX):
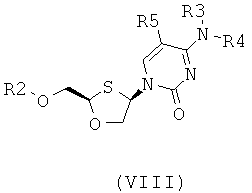 ;
; 
где
R2 представляет собой Н, С(O)С1-6алкил, С(O)ОС1-6алкил, С(O)С6-12арил, С(O)ОС6-12арил, С(O)С6-12арилалкил, С(O)ОС6-12арилалкил, С(O)С1-6алкиларил, С(O)ОС1-6алкиларип, или силильную защитную группу общей формулы SiR7R8R9, где R7, R8 и R9 представляют собой каждая независимо выбранную из C1-6алкила, арила или C1-6алкиларила;
каждый из R3 и R4 индивидуально выбирают из Н или бензоила, при условии, что когда R3 представляет собой Н, R4 не является Н, и когда R4 представляет собой Н, R3 не является Н; и
R5 представляет собой Н, Br, Cl, F, I или CF3.
Предпочтительно, R2 является ацильной группой, еще более предпочтительно бензоильной группой или ацетильной группой.
Предпочтительно, R5 представляет собой Н или F.
Согласно шестому аспекту настоящее изобретение представляет способ получения соединения общей формулы (VI) или (VII) из соединения общей формулы (VIII) или (IX, соответственно, включающий стадии:
(а) снятия защитной группы в соединении общей формулы (VIII) или (IX) для получения соединения общей формулы (VI) или (VII), соответственно:
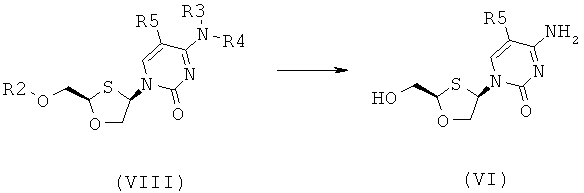
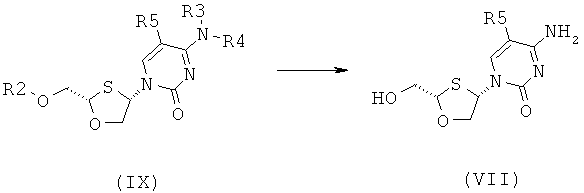
где
R2 представляет собой Н, С(O)С1-6алкил, С(O)ОС1-6алкил, С(O)С6-12арил, С(O)ОС6-12арил, С(O)С6-12арилалкил, С(O)ОС6-12арилалкил, С(O)С1-6алкиларил, С(O)ОС1-6алкиларил, или силильную защитную группу общей формулы SiR7R8R9, где R7, R8 и R9 представляют собой каждая независимо выбранную из C1-6алкила, арила или C1-6алкиларила;
каждый из R3 и R4 индивидуально выбирают из Н или бензоила, при условии, что когда R3 представляет собой Н, R4 не является Н, и когда R4 представляет собой Н, R3 не является Н; и
R5 представляет собой Н, Br, Cl, F, I или CF3.
Предпочтительно, R2 является ацильной группой, еще более предпочтительно бензоильной группой или ацетильной группой.
Предпочтительно, R5 представляет собой Н или F.
Согласно седьмому аспекту настоящее изобретение представляет способ получения соединения общей формулы (II), включающий стадии:
(а) взаимодействия силилированного основания общей формулы (X) с 1,3-оксатиоланом общей формулы (V) с образованием соединения общей формулы (II) или (III):
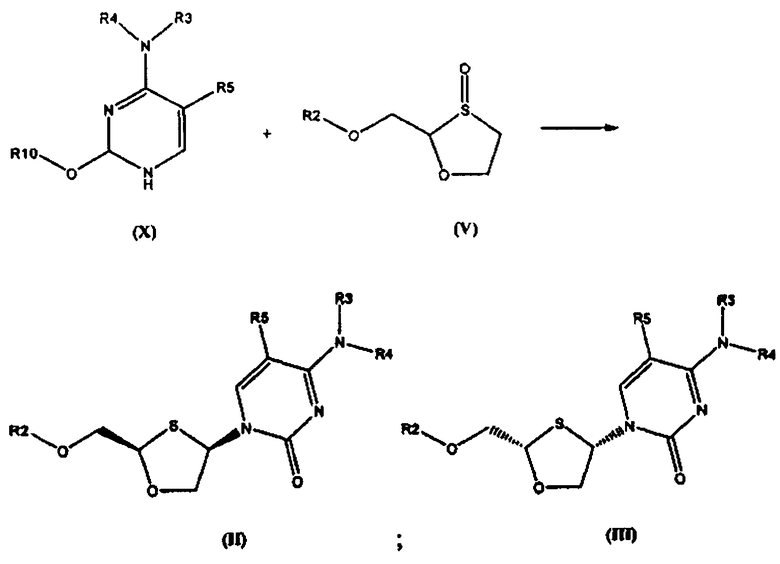
где
R2 представляет собой Н, С(O)С1-6алкил, С(O)ОС1-6алкил, С(O)С6-12арил, С(O)ОС6-12арил, С(O)С6-12арилалкил, С(O)ОС6-12арилалкил, С(O)С1-6алкиларил, С(O)ОС1-6алкиларил, или силильную защитную группу общей формулы SiR7R8R9, где R7, R8 и R9 представляют собой каждая независимо выбранную из C1-6алкила, арила или C1-6алкиларила;
R3 и R4 являются одинаковыми или различными и каждая индивидуально выбрана из Н или C(O)-R6;
R6 представляет собой С6-12арил или C1-6алкиларил, в котором С1-6алкиларил предпочтительно является С1-6алкилС6-12арилом;
R5 представляет собой Н, Br, Cl, F, I или CF3; и
R10 представляет собой силильную защитную группу общей формулы SiR7R8R9, как это было ранее определено; и
где 1,3-оксатиолан общей формулы (V) представляет собой (R)-энантиомер, по меньшей мере, в 60% ее; и
(b) селективной перекристаллизации соединения общей формулы (II) из растворителя, в котором растворитель представляет собой C1-6 спирт или смесь С1-6 спиртов.
В особо предпочтительном осуществлении R6 является фенилом.
Согласно восьмому аспекту настоящее изобретение представляет способ получения соединения общей формулы (VI), включающий стадии:
(а) взаимодействия силилированного основания общей формулы (X) с 1,3-оксатиоланом общей формулы (V) с образованием соединения общей формулы (II) или (III):
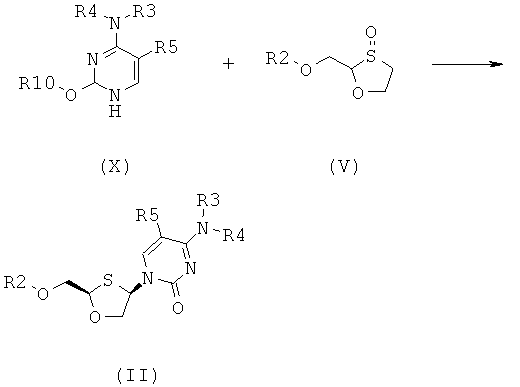 ;
;
где
R2 представляет собой Н, С(O)С1-6алкил, С(O)ОС1-6алкил, С(O)С6-12арил, С(O)ОС6-12арил, С(O)С6-12арилалкил, С(O)ОС6-12арилалкил, С(O)С1-6алкиларил, С(O)ОС1-6алкиларил, или силильную защитную группу общей формулы SiR7R8R9, где R7, R8 и R9 представляют собой каждая независимо выбранную из С1-6алкила, арила или C1-6алкиларила;
R3 и R4 являются одинаковыми или различающимися и каждая индивидуально выбрана из Н или C(O)-R6;
R6 представляет собой С6-12арил или C1-6алкиларил, в котором С1-6алкиларил предпочтительно является С1-6алкилС6-12арилом; и
R5 представляет собой Н, Br, Cl, F, I или CF3; и
R10 представляет собой силильную защитную группу общей формулы SiR7R8R9, как это было ранее определено; и
где 1,3-оксатиолан общей формулы (V) представляет собой (R)-энантиомер, по меньшей мере, в 60% ее;
(b) селективной перекристаллизации соединения общей формулы (II) из растворителя, в котором растворитель представляет собой C1-6 спирт или смесь C1-6 спиртов; и
(c) снятия защитной группы в соединении общей формулы (II) для получения соединения общей формулы (VI):
В особо предпочтительном осуществлении R6 является фенилом. Согласно девятому аспекту настоящее изобретение представляет соединение формулы (XIII) и их диастереомеры:
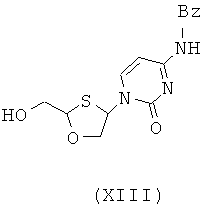
Согласно десятому аспекту настоящее изобретение представляет способ отделения соединения общей формулы (II) от соединения общей формулы (III), включающий стадии:
(а) получения смеси 2,4-дизамещенных 1,3-оксатиоланов общей формулы (II) и
;
где
R2 представляет собой Н, С(O)С1-6алкил, С(O)ОС1-6алкил, С(O)С6-12арил, С(O)ОС6-12арил, С(O)С6-12арилалкил, С(O)ОС6-12арилалкил, С(O)С1-6алкиларил, С(O)ОС1-6алкиларил, или силильную защитную группу общей формулы SiR7R8R9, где R7, R8 и R9 представляют собой каждая независимо выбранную из C1-6алкила, арила или C1-6алкиларила;
R3 и R4 являются одинаковыми или различными и каждая индивидуально выбрана из Н или C(O)-R6;
R6 представляет собой С6-12арил или C1-6алкиларил, в котором С1-6алкиларил предпочтительно является С1-6алкилС6-12арилом; и
R5 представляет собой Н, Br, Cl, F, I или CF3; и
(b) отделения соединения общей формулы (II) от соединения общей формулы (III) селективной перекристаллизацией соединения общей формулы (II) из растворителя, в котором растворитель представляет собой C1-6 спирт или смесь С1-6спиртов.
В особо предпочтительном осуществлении R6 является фенилом.
Определение
Специалистам в данной области техники будет понятно, что термин "алкил" включают в себя неразветвленные или разветвленные алкильные группы и которые могут быть необязательно замещенными. Примеры неразветвленных или разветвленных алкильных групп включают в себя, но не ограничены метильной, этильной, пропильной, бутильной, пентильной, гексильной, изо- пропильной, втор-бутильной, трет-бутильной, ди- или три-алкилированными этильной, пропильной, бутильной, пентильной или гексильной группами.
"Арил" включает в себя моно-, би- или полициклические кольцевые системы, содержащие ароматический остаток, и примеры включают в себя фенил, бифенил и нафтил. Арильная группа может быть необязательно замещенной.
Как используется здесь, термин "алкиларил" относится к моно-, би- или полициклическим (включая сопряженные и конденсированные) углеводородные кольцевые системы, предпочтительно содержащие 6-10 атомов углерода и содержащим ароматический остаток вместе с алкильной связью. Подходящие алкиларильные группы включают в себя, но не ограничены бензильной группой (т.е. -CH2 фенил).
Под термином "ацил" подразумевают группу, содержащую карбоксильный С=O остаток, не являющуюся карбоновой кислотой, сложным эфиром или амидом. Ацильные группы включают в себя, но не ограничены ацетильной группой и бензоильной группой.
"Защитные силильные группы " знакомы специалистам в данной области техники в соответствии с тем, как они представлены, например, в Greene, Т.W. and Wuts, P.G.M "Protective groups in organic synthesis" (3rd Edition) 1999 John Wiley & Sons Inc.
Типичный синтез 2,4-дизамещенных 1,3-оксатиоланов
Синтез 2,4-дизамещенных 1,3-оксатиоланов можно проводить несколькими способами, позволяющими получить смесь четырех стереоизомеров, которые являются двумя цис-изомерами и двумя транс-изомерами в соотношении, представленном в таблице 1.
Согласно настоящему изобретению соединение общей формулы (II) или (III) получают взаимодействием соединения общей формулы (IV) с 1,3-оксатиоланом общей формулы (V) для получения соединения общей формулы (II) или (III), как представлено на схеме 1:
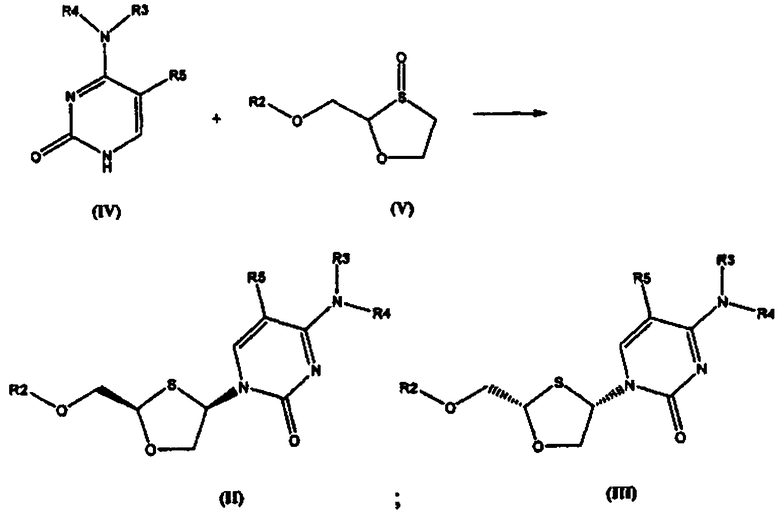
Схема 1: Синтез оптически активных соединений (II) и (III) взаимодействием основания (IV) с 1,3-оксатиоланом (V)
Соединение общей формулы (II) или (III) может также быть получено взаимодействием силилированного основания общей формулы (X) с 1,3-оксатиоланом общей формулы (V), как показано на схеме 2, где R10 представляет собой силильную защитную группу.
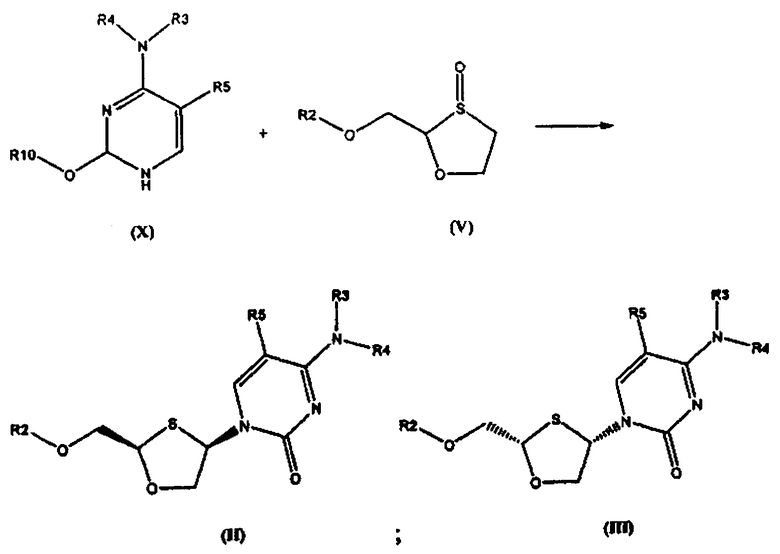
Схема 2: Синтез оптически активных соединений (II) и (III) взаимодействием силилированного основания (X) с 1,3-оксатиоланом (V)
1,3-Оксатиолан общей формулы (V) состоит, по меньшей мере, из (R)-энантиомера в 60% ее, по меньшей мере, в 70%, более предпочтительно, более чем в 85% ее, даже более предпочтительно, чем в 95% ее и еще более предпочтительно, чем в 99% ее. Опытные специалисты в данной области техники понимают, что количество соединения (II) и соединения (III), полученные по реакциям, представленным на схемах 1 и 2, зависит от % ее (R)-энантиомера соединения (V). Если % ее (R)-энантиомера соединения (V) например, больше, чем 95%, то (R,R) цис-изомер общей формулы (II) будет основным образующимся изомером. Поскольку % ее (R)-энантиомера соединения (V) уменьшается, количество образующегося соединения (II) уменьшается, а количество соединения (III) соответственно увеличивается. В процессе этой реакции также образуются транс-изомеры.
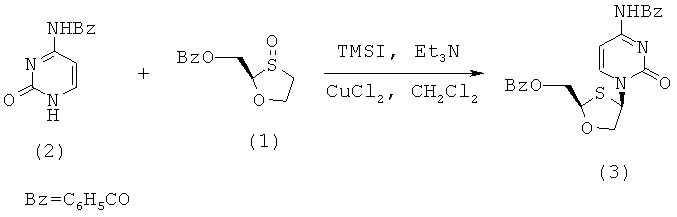
Особенно предпочтительные условия типичной реакции согласно осуществлению настоящего изобретения включают в себя присоединение N-бензоилцитозина (2) к 2-бензоилоксиметил-1,3-оксатиолан-8-оксиду (1) в присутствии триметилсилилиодида (TMSI), триэтиламина и каталитических количеств хлорида меди (II) в дихлорметане (схема 3).
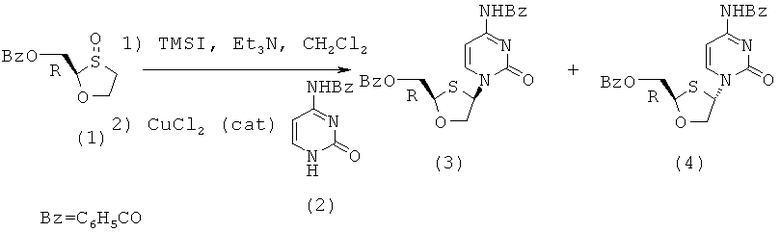
Схема 3: Взаимодействием N-бензоилцитозина с 2-бензоилоксиметил-1,3-оксатиолан-S-оксидом
Реакция происходит путем первоначальной активации (1), вступающего в перегруппировку сила-Пуммерера. Считают, что хлорид меди (II) играет некоторую роль в стимулировании β-атаки силилированного бензоилцитозина, что приводит к увеличению цис-селективности в процессе присоединения. Однако это увеличение цис-селективности невелико и количество CuCl2 может быть уменьшено без существенного воздействия на реакцию. Перегруппировку сила-Пуммерера соединения (1) проводят при -50°С и присоединение к основанию протекает при 0°С в течение ночи, перед нагреванием до комнатной температуры и гашения.
Погашенную неочищенную реакционную смесь фильтруют через целит, что снижает вероятность образования твердых веществ, которые могут осложнить процесс обработки. Отфильтрованную реакционную смесь промывают разбавленным водным аммиачным раствором и разбавленной фосфорной кислотой. Образующуюся неочищенную реакционную смесь очищают перекристаллизацией в метаноле. Перекристаллизация в метаноле приводит к получению желаемого цис-изомера с почти максимальным выходом. Перекристаллизация в метаноле также увеличивает оптическую чистоту и, используя способ захвата, оптическая чистота может быть дополнительно увеличена. Этот результат особенно полезен, когда исходный материал обладает меньшей оптической чистотой и является особенно важным результатом настоящего изобретения.
Селективная перекристаллизация
Авторы изобретения показали, что правильный выбор групп R2, R3 и R4 необходим для селективной перекристаллизации оптически активного соединения общей формулы (II) или (III) из смеси. Выбор растворителя для перекристаллизации также зависит от природы групп R2, R3 и R4.
Цитозин, защищенный ацетильной, изобутирилкарбонильной, пивалилкарбонильной, циклогексилкарбонильной, п-толуильной и бензоильной группами, присоединяют к ВОМО-сульфоксиду, как представлено на схеме 4.
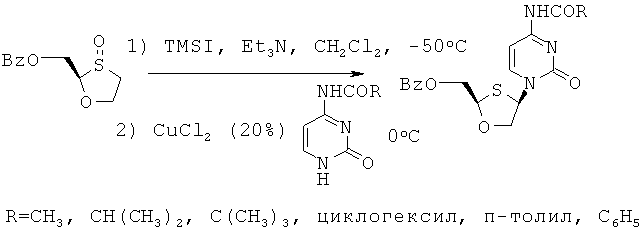
Схема 4: Присоединение защищенного цитозина к BOMO-сульфоксиду
Например, когда R2 является бензоилом, R5=H, R3=H и R4=ацетилом, перекристаллизация из смеси цис-изомеров (R,R и S,S) (соединения II и III) и транс-изомеров (R,S и S,R) (соединения XI и XII) не приводит к какому-либо разделению, как показано в таблице 2. Показано, что продукт присоединения цитозина, защищенного бензоилом, кристаллизуется эффективно с высокой цис-селективностью.
При правильном выборе групп R2, R3 и R4 могут быть выбраны такие условия, которые приводят к селективному разделению, например, соединения формулы (II) перекристаллизацией из смеси всех четырех стереоизомеров. Как показано в таблице 2, где R2 = бензоилу и R3 = H, и если R4 варьируют следующим образом, то было показано, что если R4 = бензоилу, то перекристаллизация приводит к хирально чистому соединению, в то время, если R4 = ацетилу или пивалилу или изобутирилу, то не удается селективно перекристаллизовать желаемое хирально чистое соединение.
Соответственно, в предпочтительном варианте осуществления способ согласно первому, второму, третьему, четвертому, шестому, седьмому, восьмому и десятому аспектам настоящего изобретения, R3 или R4 в соединении общей формулы II или III являются бензоилом. В предпочтительном варианте осуществления R2 в соединении общей формулы II или III является бензоилом.
Цис-селективная перекристаллизация
Одним из наиболее привлекательных преимуществ N-бензоильного направления является перекристаллизации в метаноле, которые приводят почти к максимальному выходу желаемого цис-(R,R) изомера. Предпочтительные варианты осуществления настоящего изобретения показывают, что при 5 г и 10 г масштабе реакций при использовании 0,2 эквивалентов CuCl2, реакции являются цис-селективными с селективностью в диапазоне от 2,86:1 (74%) до 2,5:1 (71%). Проведение перекристаллизации в 14-15 × объемах метанола (в сравнении с массой неочищенного вещества) дает с почти к максимальным выходом цис-изомер.
Растворители перекристаллизации
Выбранными растворителями для перекристаллизации являются С1-6 спирт или смесь спиртов C1-6. C1-6 спирт может быть спиртом с неразветвленными или разветвленными цепями. Метанол (МеОН), этанол (EtOH), пропанол и бутанол являются предпочтительными и из них наиболее предпочтительным является метанол. Как показано в таблице 3 и 4, МеОН является особенно предпочтительным для получения соединения (II) с более чем 99% ее.
Системы смешанных растворителей
Были изучены перекристаллизации с захватом в смешанных растворителях. Смеси спиртов С1-6 также являются предпочтительными растворителями для перекристаллизации, включающими в себя смеси МеОН и других спиртов C1-6, например, EtOH, пропанол, бутанол, пентанол и гексанол при различных соотношениях МеОН: спирт С2-6 приблизительно от 2:98 до 98:2, приблизительно от 5:95 до 95:5, приблизительно от 10:90 до 90:10, приблизительно от 20:80 до 80:20 или приблизительно от 30:70 до 70:30. Предпочтительным соотношением МеОН:спирт С2-6 является 90:10 и особенно предпочтительным является соотношение МеОН:спирт С2-6, равное 95:5. Предпочтительно, чтобы спирты С1-6 или их смеси содержали не более 5% воды.
При всех перекристаллизациях в раствор вносят затравки при 53°С, 14 мг затравки /г неочищенного материала, перемешивают при 1000 об./мин. с × 14 объемами растворителя. Перекристаллизации проводят с неочищенной смесью, полученной из реакционной смеси, в которой исходное соотношение ВОМО R/S равно 80/20 (фактическое соотношение R/S: 82:18). Результаты представлены в таблице 5. Однако, в отличие от результатов по перекристаллизации, представленных ниже в таблицах 10 и 11, ВЭЖХ анализ неочищенного материала дает приближенную грубую оценку соотношения R/S, равного 86/14. Однако когда соотношение R/S уменьшается, особенно ниже 90:10, для транс-изомера также появляется предрасположенность к кристаллизации (таблица 9 внизу). В этой серии экспериментов транс-изомер наблюдается постоянно в количестве приблизительно 10-12%. Поэтому наблюдаемое соотношение RR/SS корректируют на присутствие RR и SS-транс-изомеров. Независимо от этого, происходит существенное увеличение оптической активности. 100% метанол особенно предпочтителен, как показано в таблице 5. Смеси метанола, например, с 5% 1-пропанолом или 5% 1-бутанолом также особенно предпочтительны.
Увеличение оптической чистоты
Увеличение оптической чистоты является желаемым преимуществом настоящего изобретения, дающим возможность использовать исходный материал с более низкой оптической чистотой для получения перекристаллизованного продукта с более высокой оптической чистотой, что приводит к более низкой стоимости продукта.
Повторение стадии перекристаллизации и захвата
В предпочтительном варианте осуществления настоящего изобретения стадию селективной перекристаллизации (b) согласно первому, второму, третьему, четвертому, седьмому, восьмому и десятому аспектам настоящего изобретения повторяют, по меньшей мере, один раз. Повторение стадии селективной перекристаллизации может способствовать увеличению общего выхода желаемого энантиомера, так как дополнительное количество желаемого энантиомера может быть возвращено в результате повторной стадии. Однако следует иметь в виду, что стадию перекристаллизации можно также повторять для селективной перекристаллизации изомера, который не был получен на первой стадии селективной перекристаллизации. Таким образом, например, желаемый RR-энантиомер может быть селективно перекристаллизован на первой стадии селективной перекристаллизации, а SS-энантиомер может быть селективно перекристаллизован на второй стадии селективной перекристаллизации.
В особо предпочтительном варианте осуществления настоящего изобретения стадия селективной перекристаллизации (b) согласно первому, второму, третьему, четвертому, седьмому, восьмому и десятому аспектам настоящего изобретения представляет собой циклический способ захвата. Авторы настоящего изобретения неожиданно обнаружили, что выход одного предпочтительного изомера может быть увеличен путем захвата. Как показано результатами, полученными в Примере 3 Экспериментального Раздела, добавление небольшого количества RR-энантиомера облегчает перекристаллизацию R,R-энантиомера. Таким образом, такой способ внесения затравки или захвата преимущественно увеличивает выход перекристаллизации желаемого энантиомера. Результаты, полученные в Примере 3, представлены в Таблице 6.
Чтобы определить увеличение оптической чистоты при перекристаллизации исходные образцы 2-бензоилоксиметил-1,3-оксатиолан-3-оксид ВОМО (R-ВОМО (11) и S- и S-ВОМО (18)), были проанализированы при помощи хиральной ВЭЖХ.
Bz=C6H5CO
Как видно из таблицы 7, предоставленные ВОМО имеют различную чистоту.
Заметим, что образцы также часто содержат другие примеси, что является причиной различия в соотношении R/S в сравнении с анализом ВЭЖХ.
Увеличение оптической чистоты в результате двух перекристаллизаций
Результаты проведения двух перекристаллизации для получения очень чистого продукта представлены в таблице 8. Эти перекристаллизации проводят без захвата. Интересно, оказалось, что растворимость чистого продукта меньше в метаноле в сравнении с неочищенным продуктом. Это, возможно, связано с тем, что присутствующие примеси могут помочь в солюбилизации продукта. Например, неочищенный продукт может быть легко перекристаллизован уже при кипячении в метаноле (10-15 ×) с обратным холодильником. Однако с чистым продуктом, мы показали, что нелегко растворить продукт даже в ×25 объемах кипящего метанола, обычно мы используем × 40 объемов кипящего метанола (в нижеприведенном примере мы используем × 30 объемов), но все еще с высоким выходом.
«Чистый» R ВОМО (соотношение R/S равно 98,3/1,7) используют в эксперименте. Первая перекристаллизация в ×15 объемах метанола приводит к образованию продукта с соотношением RR/SS, равным 99,13/0,87. Вторая перекристаллизация (в ×30 объемах метанола) увеличивает соотношение RR/SS до 99,6/0,4 с выходом, равным 95%. Поскольку выход после второй перекристаллизации высокий, вторая перекристаллизация может также быть применимой, когда после первой перекристаллизации остается некоторое количество транс-изомера.
Таким образом, исходные ВОМО, используемые в реакции конденсации, могут иметь более низкую чистоту (т.е. соотношение R:S) поскольку проявляется способность к увеличению соотношения RR/SS в конечном продукте присоединения при использовании способа перекристаллизации настоящего изобретения.
Увеличение оптической чистоты путем захвата
Рацемическая смесь энантиомеров может быть разделена в процессе кристаллизации. Смесь энантиомеров может кристаллизоваться либо с образованием рацемического соединения, либо с образованием конгломерата. В рацемическом соединении кристаллы находятся в решетке с регулярным расположением обоих энантиомеров в равных количествах. Однако в конгломерате молекулы одного энантиомера предпочтительно притягиваются к тому же самому энантиомеру, таким образом кристаллизуясь в виде физической смеси кристаллов, принадлежащих к одному или другому энантиомеру. Если интересующий нас материал кристаллизуется в виде конгломерата, то этот способ, известный как захват (главным образом при использовании чистых кристаллов в качестве затравки для кристаллизации интересующего нас энантиомера), может быть использован для предпочтительной кристаллизации желаемого энантиомера, что увеличивает его оптическую чистоту.
На захват может влиять множество параметров:
- Температура, при которой вносят затравку: если температура слишком низкая, может начаться обычная кристаллизация, если температура слишком высокая некоторое количество добавленных кристаллов будет растворяться;
- Используемое количество кристаллов затравки;
- Чистота и размер частиц затравки;
- Скорость перемешивания в процессе кристаллизации;
- Продолжительность перемешивания;
- Объем растворителя - метанола;
- Количество неочищенного вещества также играет роль. Например, мы показали, что чистый продукт менее растворим в метаноле, чем в присутствии примесей.
Увеличение оптической чистоты при перекристаллизации, когда исходный BOMO является смесью R/S.
Смеси R/S ВОМО (при различных соотношениях в диапазоне от 100:0 R/S до 50:50) окисляют до соответствующих S-оксидов при помощи H2O2/НОАс, и затем, не кристаллизуя образующийся S-оксид, конденсируют с бензоилцитозином. Предполагают, что если сам S-оксид может кристаллизоваться путем образования конгломератов, то отделенная часть из этой негомогенной смеси может давать различные оптические отклонения по сравнению с ожидаемыми. Реакцию проводят обычным путем и неочищенное вещество перекристаллизовывают с захватом или без него.
Вначале все растворители удаляют из неочищенного материала. Затем его кипятят с обратным холодильником в 14 объемах метанола до тех пор, пока весь материал не перейдет в раствор. Далее ему дают охладиться при перемешивании, контролируя температуру. При температуре, равной приблизительно 55°С в перемешиваемую смесь добавляют затравку чистого 'R,R' изомера, доводят ее до комнатной температуры и перемешивают в течение ночи. Закристаллизованную смесь затем отфильтровывают, промывают метанолом и анализируют хиральной ВЭЖХ. В качестве контроля такой же образец кипятят с обратным холодильником в метаноле до тех пор, пока он не перейдет в раствор и затем дают охладиться без какого-либо перемешивания или внесения затравки.
В заключение, результаты таблицы 9, приведенной ниже, показывают, что при кристаллизации происходит значительное увеличение оптической чистоты, даже без захвата. Даже при соотношении R/S в исходном ВОМО, равном 80/20, соотношение RR/SS, даже без захвата увеличивается до 95/5. Однако при захвате увеличение даже выше. Такое низкое соотношение R/S в исходном ВОМО, как 90/10 дает конечную чистоту выше 99%, в то время, как соотношение R/S, равное 80/20 дает чистоту выше 98%. Эти результаты показывают, что исходный ВОМО не должен быть чрезмерно чистым для достижения высокой чистоты продукта. Однако, также интересен тот факт, что по мере того, как оптическая чистота исходного материала падает, качество цис-транс-разделения также падает. Обычно ниже соотношения R/S, равного 90/10, можно увидеть лишь присутствие следовых количеств транс-изомера (смотри строки 4 и 5, таблица 9). Однако после второй перекристаллизации следует удалять транс-изомер, как описано ранее.
Увеличение оптической чистоты при перекристаллизации продуктов взамодействия 'S' BOMO
Для настоящего конгломерата индуцируемое захватом увеличение оптической чистоты может происходить с любым из энантиомеров. Для проверки этого была проведена аналогичная реакция с сульфоксидом, полученным из ВОМО, обогащенным S-изомером (соотношение R/S равно 12/88). Неочищенную реакционную смесь, полученную конденсацией S-BOMO сульфоксида с бензоилцитозином, перекристаллизовывали с захватом или без него. Действительно, результаты, представленные в Таблице 10, показывают, что данные по перекристаллизации без захвата идентичны полученным в аналогичном эксперименте, где соотношение R/S равно 90/10.
Изучение параметров, влияющих на захват
Время
Обычно заметно влияние времени на оптическую чистоту. Оптимально и удобно оставлять смесь на ночь. Оказывается, что интервалы в 1,5 часа или 3 часа недостаточны для того, чтобы полностью провести кристаллизацию и поэтому обычно смесь оставляют на ночь (таблицы 12 и 13). Выдерживание более 1 дня, повышая чистоту, больше существенно не увеличивает чистоту.
Таблица 11 представляет результаты исследования, в котором неочищенное вещество (исходное соотношение R/S: 97/3) перекристаллизовывают в 14 объемах метанола.
Перемешивание и действие внесения затравки
Чтобы проверить влияние параметров, перемешивания и действия внесения затравки были проведены следующие эксперименты. Несмотря на то, что реакцию проводят, используя смесь ВОМО с R/S (80:20), (фактическое соотношение R/S: 82:18), для этих экспериментов само неочищенного вещество анализировали отдельно ВЭЖХ.
Для этих экспериментов вначале удаляют все растворители из неочищенного материала. Затем его кипятят в 14 объемах метанола до тех пор, пока весь материал не перейдет в раствор. Затем его охлаждают при перемешивании (при заданной скорости) или без перемешивания, контролируя температуру. При температуре, равной 55°С, в перемешиваемую смесь добавляют затравку чистого 'R,R'-цис-изомера или не добавляют затравку и перемешивают в течение ночи, давая температуре достичь комнатной. Затем отфильтровывают закристаллизованную смесь, промывают метанолом и анализируют ВЭЖХ. Результаты представлены в таблице 12. Эти результаты показывают, что перемешивание всегда лучше, чем отсутствие перемешивания. Возможно, что перемешивание может способствовать образованию затравок и распределять их по всему объему, перемешивание без добавления затравок приводит к более высокой оптической чистоте продуктов, чем перекристаллизации с добавлением затравок, но без перемешивания. Кристаллизация главным образом заканчивается за 1 день, но чистота слабо увеличивается со временем (таблица 11).
Результаты, полученные в таблице 13, возможно показывают, что при температурах выше 55°С (или близких), затравки частично растворяются. Поэтому лучше использовать температуры ниже 55°С. С другой стороны, при более низких температурах происходит обычная конкурирующая кристаллизация, особенно при высоких скоростях введения затравки. Это также может уменьшить эффективность захвата.
В еще одном предпочтительном варианте осуществления 2,4-дизамещенный 1,3-оксатиолан общей формулы (II) или общей формулы (III) полученный или образующийся на стадии (а) согласно первому, второму, третьему, четвертому, седьмому, восьмому и десятому аспектам настоящего изобретения находится в форме конгломерата. Способность этих соединений образовывать конгломераты удивительна, если принять во внимание то, что только от 5% до 10% рацематов относятся к конгломерат-образующей группе (Lorenz, Н., et. al., Journal of the University of Chemical Technology and Metallurgy (2007) 42(1): 5-16).
Предпочтительно желаемый энантиомер, который получают каким-либо из способов согласно первому, второму, третьему, четвертому, шестому, седьмому, восьмому и десятому аспектам настоящего изобретения не содержит каких-либо заметных количеств нежелаемого изомера.
Снятие защитной группы
После присоединения бензоилцитозина к ВОМО конечной стадией синтеза является удаление защитных бензоильных групп для получения дебензоилированного продукта, например, соединения (17), как показано на схеме 5.
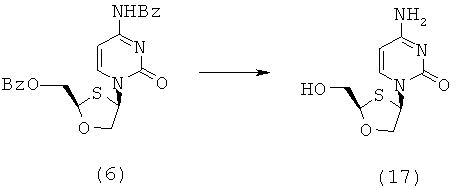
Схема 5: Реакция дебензоилирования
Подходящие способы снятия защитной группы включают: каталитический способ NaOMe/MeOH, Метанольный Аммиак и Водный Аммиак в Метаноле.
В первую очередь изучен гидролиз, опосредованный метоксидом натрия. Этот способ приводит к количественному снятию защитной группы, однако требует обработки водой для того, чтобы освободиться от солей нехроматографическим способом. Поэтому, поскольку конечные продукты, например, соединение (17), могут обладать некоторой растворимостью в воде, такая обработка уменьшает выход. Изучен также способ с метанольным аммиаком. Метанольный аммиак также чисто снимает защитную группу с молекулы после перемешивания в течение ночи и после удаления растворителя, суспендирования в ацетоне более липофильных побочных продуктов и очень чисто приводит к получению продукта. Реакция протекает одинаково успешно независимо от того, является исходный материал RR или SS-изомером (таблица 14, эксперименты 2, 3, 4). В качестве альтернативы было показано, что может быть использован также водный аммиак в метаноле. Снятие защитной группы с использованием этого способа происходит медленнее, но чисто, несмотря на то, что он включает в себя удаление воды и поэтому менее удобен в больших масштабах, чем способ с использованием метанольного аммиака.
Соединения
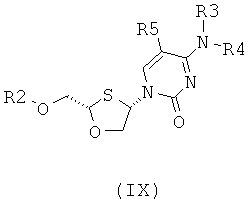
В предпочтительном варианте осуществления настоящего изобретения представлены производные 2,4-дизамещенного 1,3-оксатиолана. Соединения общей формулы VIII и IX, где R2, R3, R4 и R5 таковы, как они были ранее определены:
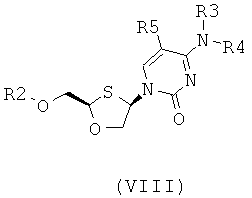 ;
; 
и предпочтительно выбраны из группы соединений, состоящих из:
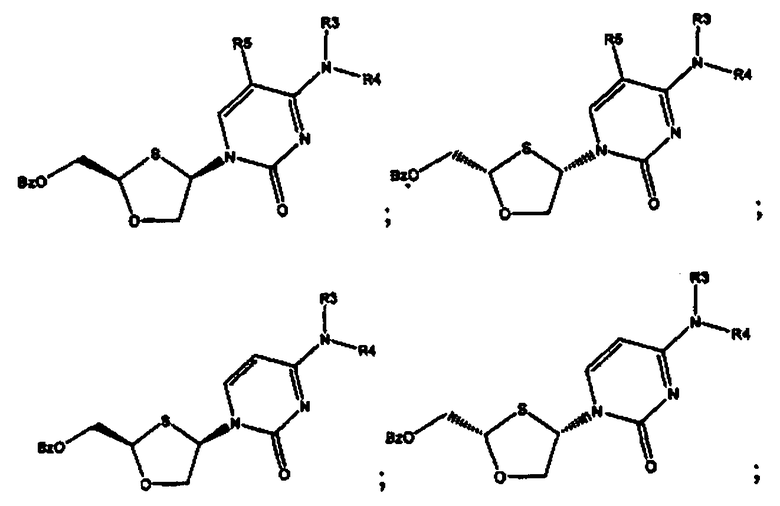
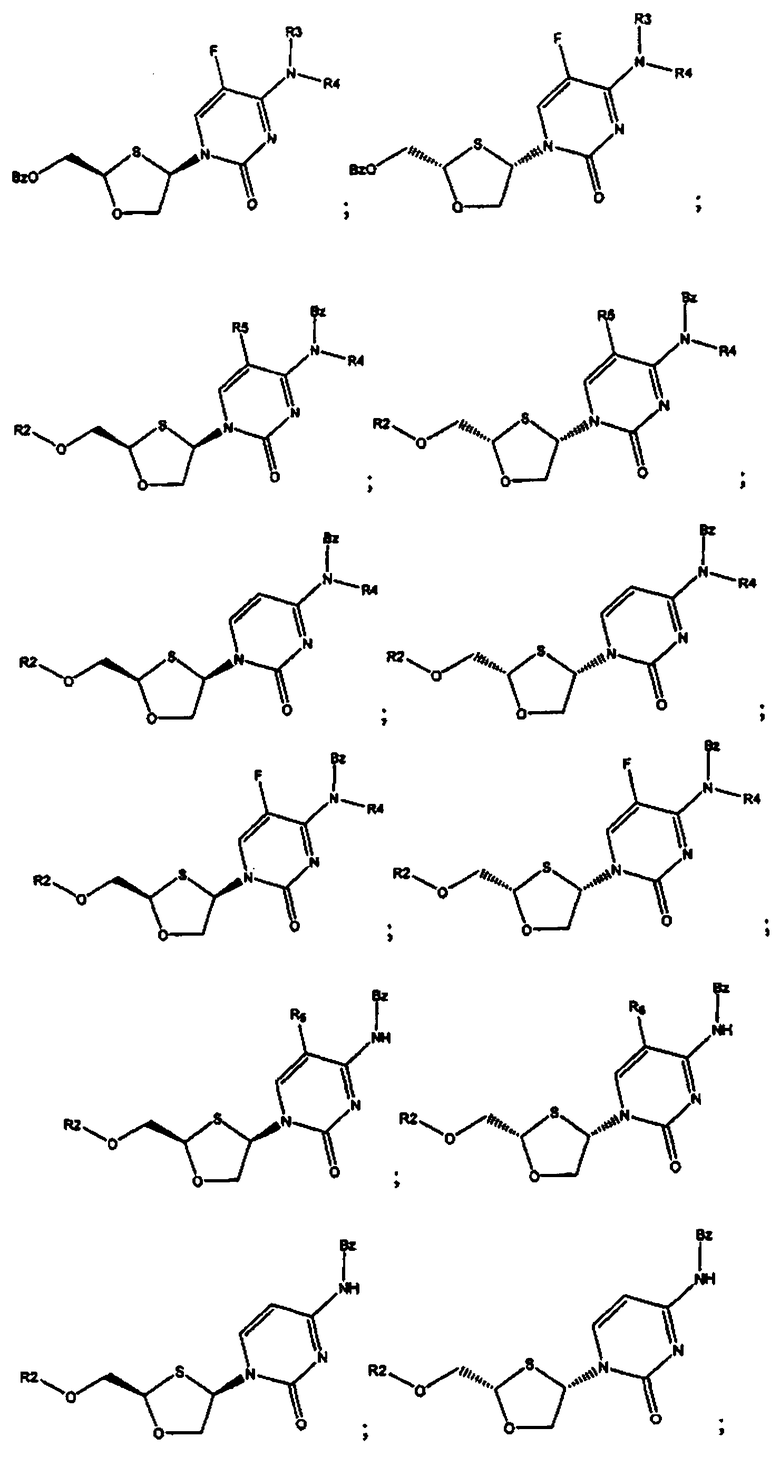
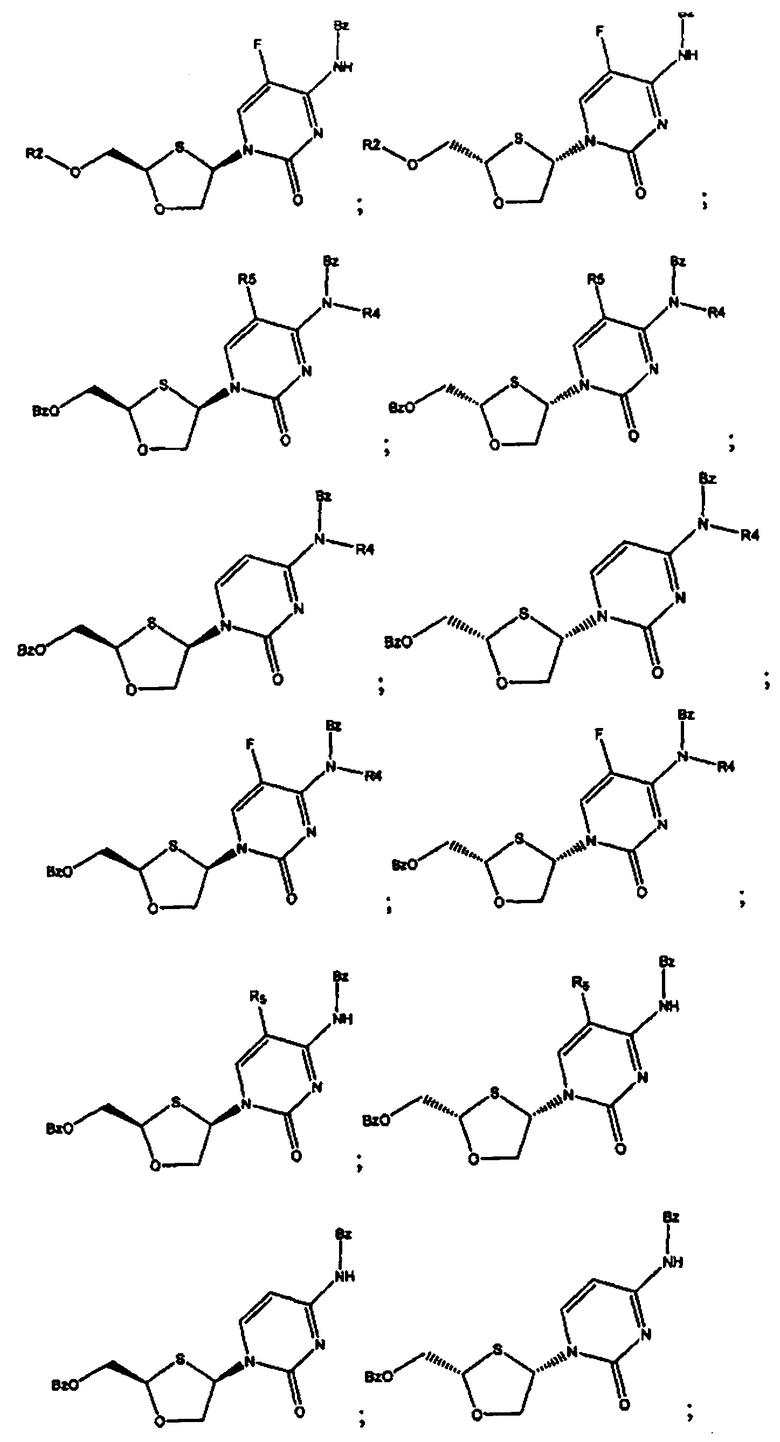

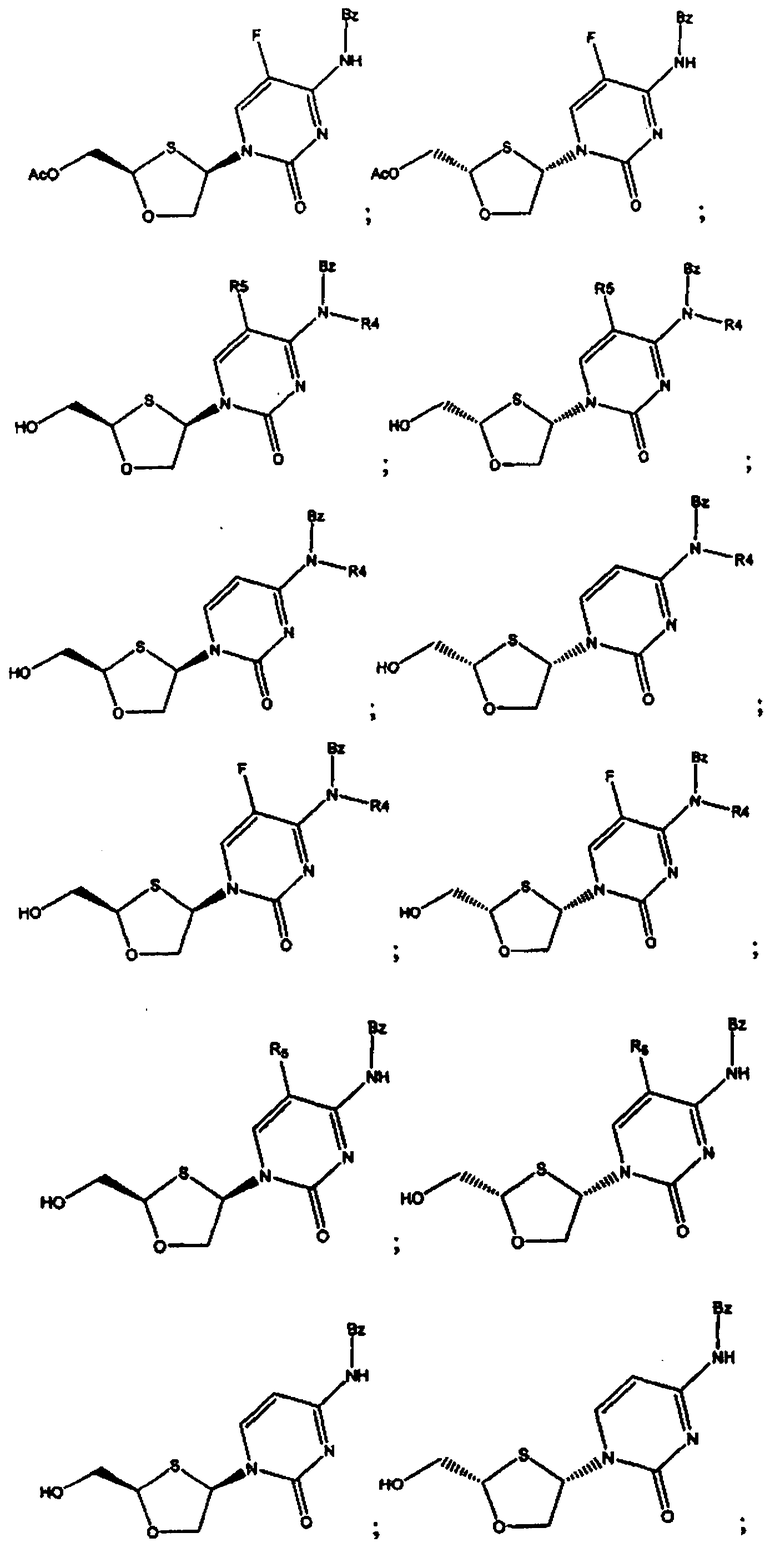
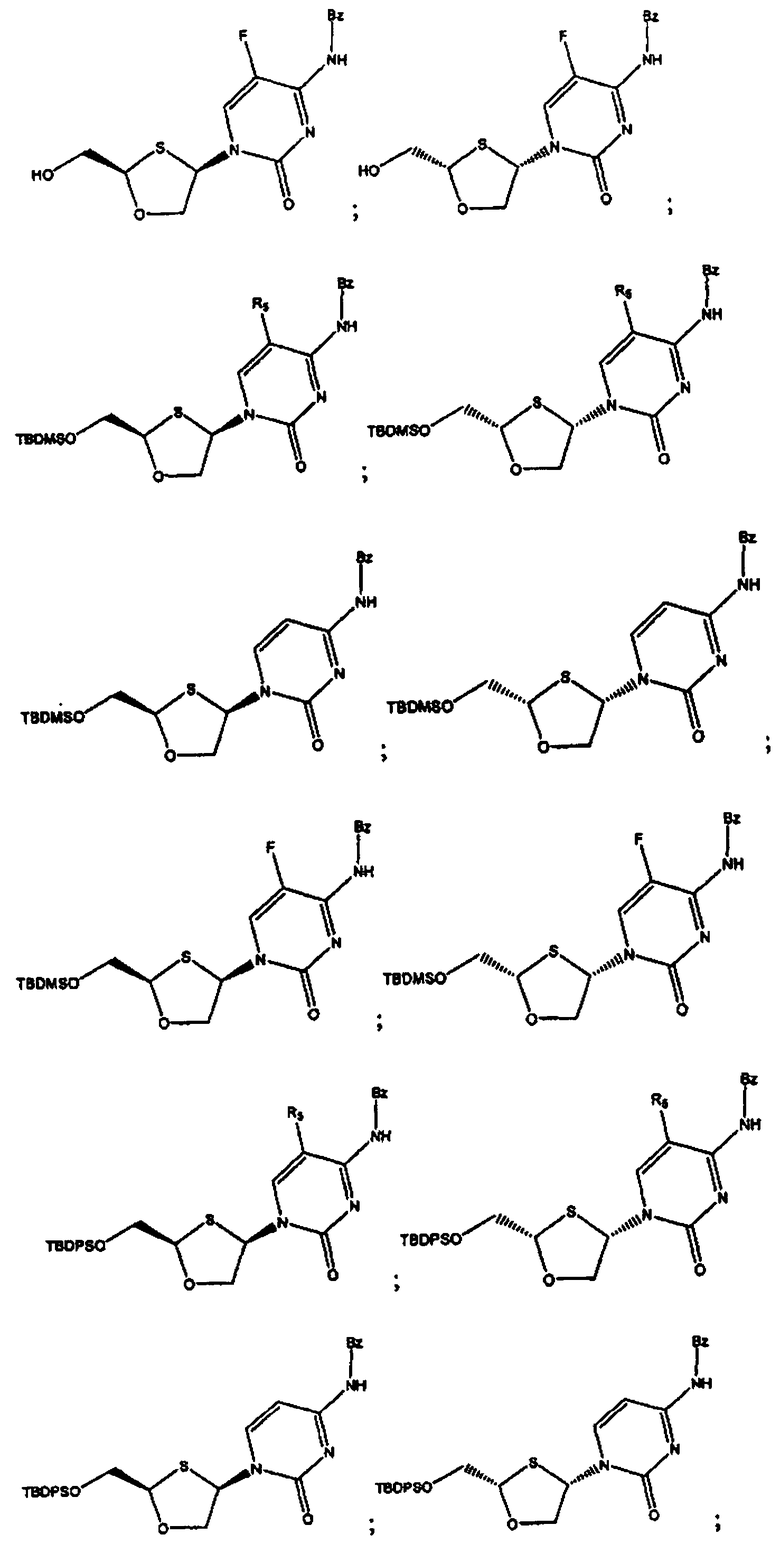
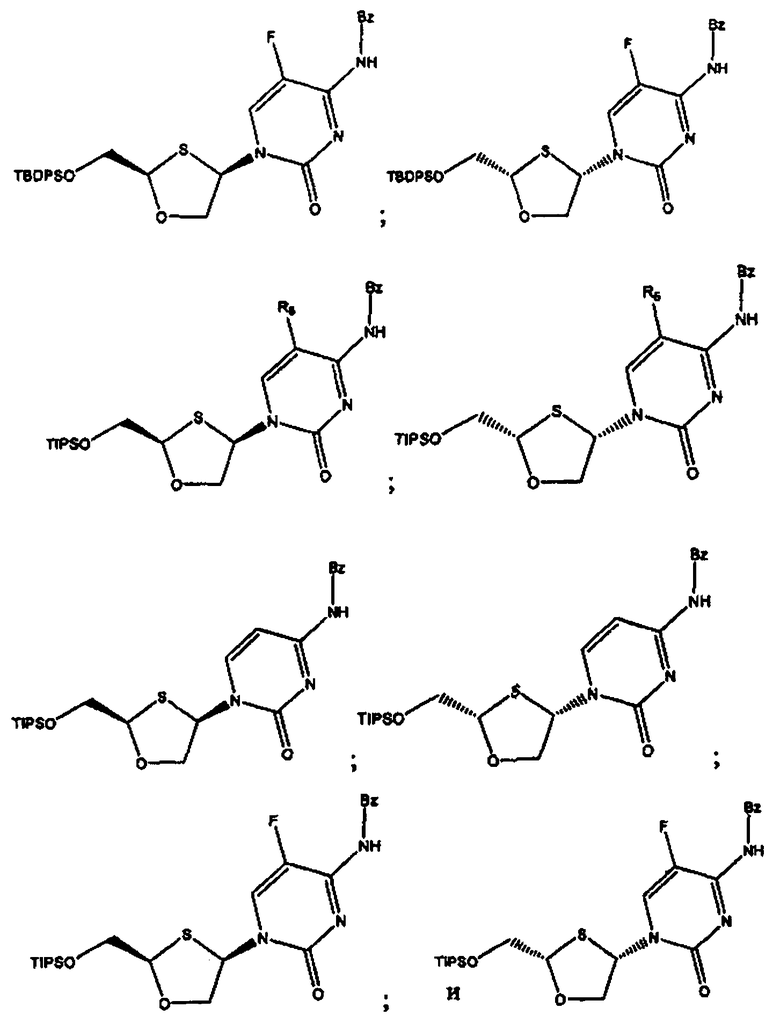
Особенно предпочтительными являются N-бензоильные производные соединений общей формулы VIII и IX, даже более предпочтительными являются N-бензоильные производные формул:
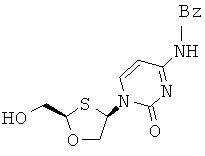 или
или 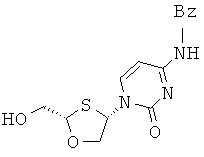
ПРИМЕРЫ
Предпочтительные варианты осуществления настоящего изобретения далее будут описаны, не ограничиваясь ссылками на следующие примеры.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Общая Часть
Все реакции проводят в атмосфере азота с использованием сухой посуды. 2-R и S-бензоилоксиметил-1,3-оксатиолан получены от Avexa. N-бензоилцитозин получен от Shanghai PI Chemicals. TMSI синтезируют сами и перегоняют после синтеза. Триэтиламин перегоняют над КОН. Растворители используют без перегонки. Для поддержания низких температур реакции используют криостат Thermo-Neslab. ЯМР спектры снимают на Varian High Field NMR spectrometer, работающем при 400 МГц. Тонкослойную хроматографию проводят на пластиковых силикагелевых пластинках (0,22 мм), предварительно покрытых Machery-Nagel. Анализы ВЭЖХ проводят в системе Waters 510 HPLC, с хиральной насадкой (Chiralpak AD), колонкой (ID) 25 см × 0,46 см, с детектированием при 254 нм. Система растворителей представляет собой 20% МеОН в ацетонитриле, подается изократически, со скоростью потока, равной 2 мл/мин.
Некоторые сокращения: Eq (эквиваленты), wrt (по отношению)
Пример 1
Синтез 2-(R)-бензоилоксиметил-1,3-оксатиолан-3-оксида
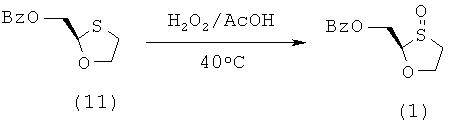
К перемешиваемой смеси 2-(R)-бензоилоксиметил-1,3-оксатиолана (118 г, 0,526 моль) и ледяной уксусной кислоты (47 г, 0,790 моль) в 500 мл колбы RB, снабженной воздушным/водяным холодильником, при 40°С, добавляют пероксид водорода (35% в воде) (65 мл, 0,736 моль) четырьмя порциями приблизительно с 10-минутным интервалом. Первые добавления являются очень экзотермическими. Смесь перемешивают при этой температуре в течение 1 часа и при комнатной температуре в течение 1 часа. Затем смесь переносят в химический стакан на 1 литр и разбавляют дихлорметаном (500 мл). При перемешивании небольшими порциями (вначале реакция проходит очень энергично) добавляют 10% раствор сульфита натрия в воде (500 мл). Органический слой отделяют и его затем перемешивают с насыщенным водным раствором карбоната натрия (500 мл) до прекращения выделения пузырьков. Органический слой отделяют, промывают насыщенным солевым раствором и сушат над сульфатом магния. Образующийся мутный бесцветный раствор фильтруют через целит для получения прозрачного раствора. Его упаривают с образованием продукта в виде бесцветного вязкого масла, которое затвердевает с образованием белого твердого вещества. Этот материал используют на следующей стадии синтеза без дальнейшей очистки. Этот материал представляет собой смесь E/Z диастереомеров в соотношении 2,4:1 (по данным ЯМР). Выход: 114 г (90%)
1Н-ЯМР (CDCl3, 400 МГц): δ=8,05 (д), 7,9 (д), 7,5 (м), 7,4 (м), 4,6-4,8 (м), 4,4 (м), 4,1 (м), 3,2 (м), 3,1 (м), 2,7 (м) м.д.. (спектр ЯМР является сложным из-за присутствия Е и Z диастереомеров)
Пример 2
Перекристаллизация 2-(R)-бензоилоксиметил-1,3-оксатиолан-S-оксида
9,6 г смеси E:Z 2-(R)-бензоилоксиметил-1,3-оксатиолан-8-оксида растворяют при кипячении с обратным холодильником в метаноле (12 мл) медленно охлаждают до комнатной температуры и оставляют на ночь. Образующиеся бесцветные игольчатые кристаллы отфильтровывают, промывают ледяным метанолом (2 мл), сушат. Выход: 3,0 г
1Н-ЯМР (CDCl3, 400 МГц): δ=7,9 (д, 2Н), 7,6 (м, 1H), 7,4 (м, 2Н), 4,6-4,8 (м, 3Н), 4,4 (м, 1Н), 3,15 (м, 1Н), 2,7 (м, 1H) м.д.
Пример 3
Синтез 2-(R)-бензоилоксиметил-4-(R)-(N-бензоилцитозин-1-ил)-1,3-оксатиолана
(а) Конденсация 2-(R)-бензоилоксиметил-1,3-оксатиолан-S-оксида с N-бензоилцитозином
2-(R)-бензоилоксиметил-1,3-оксатиолан-3-оксид (15,0 г, 0,063 моль) (оптическая чистота исходного 2-(R)-бензоилоксиметил-1,3-оксатиолана R/S: 98,3/1,7) растворяют в дихлорметане (250 мл) в трехгорлой колбе на 500 мл в атмосфере азота и охлаждают до -50°С в криостате. К смеси добавляют триэтиламин (9,6 мл, 0,069 моль, 1,1 эквивалент). Затем по каплям через капельную воронку добавляют иодотриметилсилан (18,7 мл, 0,131 моль, 2,1 эквивалента) с такой скоростью, чтобы температура внутри оставалась ниже -40°С. Образующийся светло-желтый раствор перемешивают в течение 30 минут, поддерживая температуру при -50°С. Затем к реакционной смеси вновь добавляют триэтиламин (8,7 мл, 0,062 моль, 1,0 эквивалент) с последующим повторным добавлением по каплям иодотриметилсилана (8,9 мл, 0,062 моль и 1,0 эквивалент). Затем добавляют высушенный в печи безводный хлорид меди (II) (0,84 г, 0,0062 моль) и через 5 минут добавляют N-бензоилцитозин (13,4 г, 0,062 моль). Образующуюся смесь, нагретую до 0°С, перемешивают при этой температуре в течение ночи. После ночного перемешивания реакции дают нагреться до комнатной температуры и перемешивают при комнатной температуре в течение 90 минут. Реакционную смесь гасят добавлением воды (100 мл). Ее перемешивают в течение 5 минут и фильтруют через слой целлита. Слой промывают дополнительным количеством дихлорметана (3×75 мл) и объединенные фильтраты выливают в делительную воронку. Органический слой отделяют, последовательно промывают 5% водным аммиаком (2×100 мл), 2% фосфорной кислотой (2×100 мл) и вновь 5% водным аммиаком (100 мл). Объединенные водные слои повторно экстрагируют дихлорметаном (100 мл). Затем объединенные органические слои промывают 1М тиосульфатом натрия (100 мл). Образующийся бледно-желтый раствор сушат сульфатом магния, фильтруют и упаривают с образованием светло желтого/коричневого густого масла, 24,3 г (89% выход). Эта неочищенная смесь состоит из смеси цис- и транс-продукта конденсации 75% чистоты (ЯМР) и при соотношении цис/транс-изомеров, равном 2,45:1 (ЯМР).
(b) Перекристаллизация неочищенного 2-(R)-бензоилоксиметил-1,3-оксатиолан-4-(R и S)-(N-бензоилцитозин-1-ил)-1,3-оксатиолана для получения чистого 2-(R)-бензоилоксиметил-4-(R)-(N-бензоилцитозин-1-ил)-1,3-оксатиолана
Перекристаллизация с захватом
К неочищенному (R)-бензоилоксиметил-4-(R,S)-(N-бензоилцитозин-1-ил)-1,3-океатиолану (10,3 г) в круглодонной колбе на 100 мл добавляют 14,0-кратный (по объему) метанол (144,2 мл), и смесь кипятят с обратным холодильником до тех пор, пока раствор не станет прозрачным. Затем его охлаждают при перемешивании, контролируя температуру термометром. Когда температура достигает 53°С, в раствор вносят затравку, 144 мг чистого 2-(R)-бензоилоксиметил-4-(R)-(N-бензоилцитозин-1-ил)-1,3-оксатиолан при тщательном перемешивании. После внесения затравки быстро кристаллизующуюся смесь сильно перемешивают в течение ночи. Затем образующийся кристаллический продукт отфильтровывают, и далее промывают метанолом (50 мл). После того, как весь маточный раствор и следующие за ним промывные воды пройдут через фильтр, образующийся белый кристаллический продукт вновь медленно промывают метанолом (2×100 мл) и сушат в вакууме. Анализ образующегося белого кристаллического вещества при помощи ЯМР показывает, что оно более, чем на 99% является цис-изомером. Выход выделенного вещества равен 4,4 г (44% выход после перекристаллизации в расчете на неочищенный продукт). Оптическая чистота: RR/SS: 99,3/0,7.
Перекристаллизация без захвата
К неочищенному (R)-бензоилоксиметил-4-(R)-(N-бензоилцитозин-1-ил)-1,3-оксатиолану (8,0 г) в круглодонной колбе на 100 мл добавляют приблизительно 14,0-кратное (по объему) количество метанола (112 мл) и смесь кипятят с обратным холодильником до тех пор, пока раствор не станет прозрачным. Затем его охлаждают без перемешивания в течение ночи. Затем образующийся кристаллический продукт отфильтровывают, и далее промывают метанолом (50 мл). После того, как весь маточный раствор и следующие за ним промывные воды пройдут через фильтр, образующийся белый кристаллический продукт вновь медленно промывают метанолом (2×100 мл) и сушат в вакууме. Анализ образующегося белого кристаллического вещества при помощи ЯМР показывает, что оно более, чем на 99% является цис-изомером. Выход выделенного вещества равен 3,36 г (42% выход после перекристаллизации в расчете на неочищенный продукт). Оптическая чистота: RR/SS 98,1/1,9.
Сравнение данных по перекристаллизации с захватом и без него, когда исходный 2-бензоилоксиметил-1,3-оксатиолан (ВОМО), использованный для синтеза взаимодействующего предшественника, 2-бензоилоксиметил-1,3-оксатиолан-S-оксида (2), не является энантиомерно чистым:
Сравнительные данные, представленные в следующей таблице, показывают, захват приводит к увеличению выхода RR энантиомера.
Данные анализа 2-(R)-бензоилоксиметил-4-(R)-(N-бензоилцитозин-1-ил)-1,3-оксатиолана
1Н ЯМР (CDCl3): δ 8,9 (шир с, 1H), 8,25 (д, 1Н), 8,0 (д, 2Н), 7,8 (д, 2Н), 7,6 (м, 2Н), 7,45(м, 4Н) 7,3 (плохо разрешена д, 1Н), 6,6 (д, 1Н) 5,5 (т, 1Н), 4,8 (м, 2Н), 4,5 (д, 1Н), 4,05 (дд, 1Н)
Анализ ВЭЖХ: Колонка: Chiralpak AD 0,46×25 см; Система Растворителей: ацетонитрил/метанол 80:20; Скорость потока: 2 мл/мин; Длина волны: 254 нм; Время удерживания для R,R-изомера: 11,23 мин.
Пример 4
Синтез 2-(R)-бензоилоксиметил-4-(R)-(N-бензоилцитозин-1-ил)-1,3-оксатиолана
(а) Конденсация 2-(R)-бензоилоксиметил-1,3-оксатиолана с N-бвнзоилцитозином
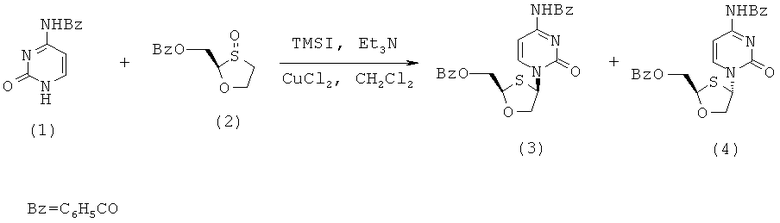
Методика:
2-(R,S)-бензоилоксиметил-1,3-оксатиолан (2) (12,0 г, 0,050 моль) растворяют в дихлорметане в трехгорлой колбе на 500 мл и охлаждают до -50°С. К раствору добавляют триэтиламин (15,3 мл, 0,110 моль) и затем иодотриметилсилан (21,4 мл, 0,150 моль) прикапыванием через капельную воронку, с такой скоростью, чтобы температур внутри находилась в диапазоне от -30°С до -50°С. Образующийся светло желтый раствор перемешивают в течение 45 минут, поддерживая температуру в диапазоне от -40°С до -50°С. Затем к реакционной смеси добавляют хлорид меди (II) (1,3 г, 0,010 моль) и еще через 5 минут добавляют N-бензоилцитозин (I) (10,1 г, 0,047 моль). Образующуюся смесь перемешивают при -50°С в течение 15 минут, а затем ей дают нагреться до 0°С в течение 1 часа. Реакционную смесь перемешивают при этой температуре в течение ночи. После ночного перемешивания реакционную смесь перемешивают при комнатной температуре в течение 1 часа, вновь охлаждают во льду, гасят добавлением воды (100 мл), а затем 5% аммиаком (100 мл). Смесь перемешивают в течение 5 минут, разбавляют дихлорметаном (50 мл) и фильтруют через слой целита. Слой промывают дополнительным количеством дихлорметана (2×50 мл) и объединенные фильтраты выливают в делительную воронку. Органический слой отделяют, промывают 2% фосфорной кислотой (2×60 мл) и вновь 2,5% аммиачным раствором (2×100 мл). Объединенные водные слои повторно экстрагируют дихлорметаном (100 мл). Объединенные органические слои сушат сульфатом магния, фильтруют и упаривают с образованием светло-желтого/коричневого густого масла, 17,8 г (86% выход). Эта неочищенная смесь состоит из цис- (3) и транс- (4) объединенного продукта конденсации 62% чистоты (ЯМР) и при соотношении цис/транс-изомеров, равном 2,86:1 (ЯМР)
(b) Перекристаллизация неочищенного 2-(R,S)-бензоилоксиметил-1,3-оксатиолан-4-(R,S)-(N-бензоилцитозин-1-ил)-1,3-оксатиолана для получения чистого 2-(R)-бензоилоксиметил-4-(R)-(N-бензоилцитозин-1-ил)-1,3-оксатиолана
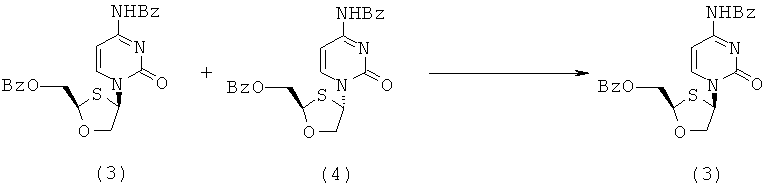
К неочищенному (R,S)-бензоилоксиметил-1,3-оксатиолан-4-(R,S)-(N-бензоилцитозин-1-ил)-1,3-оксатиолану (17,8 г) в круглодонной колбе на 500 мл добавляют 14,5-кратное количество (по объему) метанола (258 мл) и смесь кипятят с обратным холодильником до тех пор, пока раствор не станет прозрачным. Затем его постепенно охлаждают до комнатной температуры и оставляют стоять на ночь. Образующийся кристаллический продукт отфильтровывают, и далее промывают метанолом (2×100 мл) и сушат в вакууме. Анализ образующегося слегка окрашенного пушистого кристаллического твердого вещества, проведенный методом ЯМР, показывает, что оно более, чем на 99% является цис-изомером (3). Выход выделенного вещества равен 7,2 г (35% выхода цис-изомера).
1Н ЯМР (CDCl3): δ 8,5 (шир с, 1Н), 8,25 (д, 1Н), 8,0 (д, 2Н), 7,8 (д, 2Н), 7,6 (м, 2Н), 7,45 (м, 4Н) 7,3 (плохо разрешенная д, 1H), 6,6 (д, 1Н) 5,5 (т, 1Н), 4,8 (м, 2Н), 4,5 (д, 1Н), 4,05 (дд, 1H).
Анализ ВЭЖХ: Колонка: Chiralpak AD 25×0,46 см (ID); Система Растворителей: 20% МеОН в ацетонитриле 80:20; Скорость потока: 1 мл/мин; Длина волны: 254 нм; Энантиомерная Чистота: >100:1.
Пример 5
Синтез 2-(S)-бензоилоксиметил-4-(S)-(N-бензоилцитозин-1-ил)-1,3-оксатиолана
(а) Конденсация 2-(S)-бензоилоксиметил-1,3-оксатиолана с N-бензоилцитозином
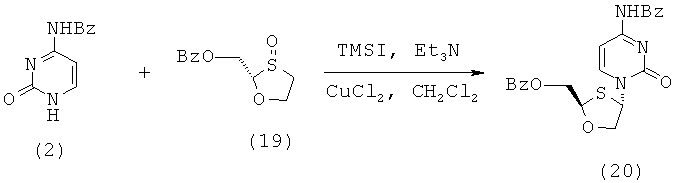
2-(R)-бензоилоксиметил-1,3-оксатиолан-8-оксид (2,0 г, 0,0083 моль) (оптическая чистота исходного 2-(S)-бензоилоксиметил-1,3-оксатиолана R/S: 12,1/87,9) растворяют в дихлорметане (40 мл) в трехгорлой колбе на 100 мл в атмосфере азота и раствор охлаждают до -50°С в криостате. К раствору добавляют триэтиламин (1,27 мл, 0,009 моль и 1,1 эквивалента). Затем по каплям добавляют иодотриметилсилан (2,5 мл, 0,017 моль, 2,1 эквивалента) через капельную воронку, с такой скоростью, чтобы температура внутри оставалась ниже -40°С. Образующийся светло-желтый раствор перемешивают в течение 30 минут, поддерживая температуру при -50°С. Затем к реакционной смеси вновь добавляют триэтиламин (1,15 мл, 0,0083 моль, 1,0 эквивалента) и далее повторно по каплям добавляют иодотриметилсилан (1,2 мл, 0,0083 моль и 1,0 эквивалента). Затем добавляют высушенный в печи безводный хлорид меди (II) (0,11 г, 0,0008 моль) и еще через 5 минут добавляют N-бензоилцитозин (1,79 г, 0,0083 моль). Образующейся смеси дают нагреться до 0°С и перемешивают при этой температуре в течение ночи. После ночного перемешивания реакционной смеси дают нагреться до комнатной температуры и перемешивают в течение 90 минут. Затем реакционную смесь гасят добавлением воды (25 мл). Смесь перемешивают в течение 5 минут и фильтруют через слой целлита. Слой промывают дополнительным количеством дихлорметана (3×25 мл) и объединенные фильтраты выливают в делительную воронку. Органический слой отделяют, последовательно промывают 5% аммиачным раствором (2×25 мл), 2% фосфорной кислотой (2×25 мл) и вновь 5% аммиачным раствором (25 мл). Объединенные водные слои повторно экстрагируют дихлорметаном (25 мл). Затем объединенные органические слои промывают 1 М тиосульфатом натрия (250 мл). Образующийся слегка желтый раствор сушат сульфатом магния, фильтруют и упаривают с образованием светло-желтого/коричневого густого масла, 2,4 г (71% выход). Эта неочищенная смесь состоит из смеси цис- и транс- продуктов конденсации 69% чистоты (ЯМР) и при соотношении цис/транс-изомеров, равном 2,4:1 (ЯМР)
(b) Перекристаллизация неочищенного 2-(S)-бензоилоксиметил-1,3-оксатгюлан-4-(R и S)-(N-бензоилцитозин-1-ил)-1,3-оксатиолана для получения чистого 2-(S)-бензоилоксиметил-4-(S)-(N-бензоилцитозин-1-ил)-1,3-оксатиолана
Перекристаллизация с захватом
К неочищенному (S)-бензоилоксиметил-1,3-оксатиолан-4-(R,S)-(N-бензоилцитозин-1-ил)-1,3-оксатиолану (2,4 г) в 100 мл круглодонной колбе добавляют приблизительно 14,0-кратное количество (по объему) метанола (34 мл) и смесь кипятят с обратным холодильником до тех пор, пока раствор не станет прозрачным. Затем его охлаждают при перемешивании, контролируя температуру термометром. Когда температура достигнет 53°С, в раствор вносят затравку, 34 мг предварительно перекристаллизованного 2-(S)-бензоилоксиметил-1,3-оксатиолан-4-(R,S)-(N-бензоилцитозин-1-ил)-1,3-оксатиолана при сильном перемешивании. После внесения затравки быстро кристаллизующуюся смесь сильно перемешивают. Затем образующийся кристаллический продукт отфильтровывают, и далее промывают метанолом (10 мл). После того, как весь маточный раствор и следующие за ним промывные воды пройдут через фильтр, образующийся белый кристаллический продукт вновь медленно промывают метанолом (2×10 мл) и сушат в вакууме. Анализ образующегося белого кристаллического вещества методом ЯМР показывает, что оно более, чем на 99% является цис-изомером. Выход выделенного вещества равен 0,96 г (40% выход после перекристаллизации в расчете на неочищенный продукт). Оптическая чистота: RR/SS 1,5/98.
Перекристаллизация без захвата
К неочищенному (S)-бензоилоксиметил-4-(R,S)-(N-бензоилцитозин-1-ил)-1,3-оксатиолану (13,95 г) в круглодонной колбе на 250 мл постепенно добавляют 14,0-кратное количество (по объему) метанола (194,6 мл) и смесь кипятят с обратным холодильником до тех пор, пока раствор не станет прозрачным. Затем его охлаждают без перемешивания в течение ночи. Затем образующийся кристаллизующийся продукт отфильтровывают с последующим промыванием метанолом (50 мл). После того, как весь маточный раствор и следующие за ним промывные воды пройдут через фильтр, образующийся белый кристаллический продукт вновь медленно промывают метанолом (50 мл) и сушат в вакууме. Анализ образующегося белого кристаллического твердого вещества, проведенный методом ЯМР, показывает, что оно более, чем на 99% является цис-изомером. Выход выделенного вещества равен 4,8 г (34% выход в результате кристаллизации в расчете на неочищенное вещество). Оптическая чистота: RR/SS: 2,2/97,8.
Пример 6
Параметры, влияющие на захват
Эксперименты проведены с неочищенным 2-(R)-бензоилоксиметил-4-(R)-(N-бензоилцитозин-1-ил)-1,3-оксатиоланом
Влияние времени
Каждый образец получают из реакционной смеси, где исходное соотношение для ВОМО равно 97 (R)/3 (S) (Соотношение R/S для неочищенного вещества не было получено для этих образцов). Неочищенное вещество перекристаллизовывают растворением при кипячении в 14 объемах метанола. Для введении в качестве затравки используют 14 мг (чистый R,R) на грамм неочищенного вещества. Введение затравки проводят при температуре в диапазоне от 55°С до 56°С. После указанного времени кристаллы отфильтровывают, промывают метанолом, сушат и анализируют ВЭЖХ. Результаты представлены в следующей таблице.
Влияние перемешивания и внесения затравки
Каждый образец получают из реакционной смеси, где исходное соотношение для ВОМО равно 82 (R)/18 (S). Неочищенное вещество перекристаллизовывают растворением при кипячении с обратным холодильником в 14 объемах метанола. Затем раствор охлаждают при перемешивании (при заданной скорости) или без перемешивания, поддерживая температуру. При температуре, равной 55°С, в перемешиваемую смесь вносят чистый 'R,R' изомер или не вносят затравку и перемешивают в течение ночи, давая возможность температуре подняться до комнатной. Затем закристаллизованную смесь отфильтровывают, промывают метанолом и анализируют ВЭЖХ. Результаты представлены в следующей таблице.

Влияние Температуры
Каждый образец получают из реакционной смеси, где исходное соотношение для ВОМО равно 82 (R)/18 (S). Неочищенное вещество перекристаллизовывают растворением при кипячении в 14 объемах метанола. Затем раствор охлаждают при перемешивании (при 1000 об/мин) или без перемешивания, поддерживая температуру. При заданной температуре, равной 55°С, в перемешиваемую смесь вносят 'R,R' чистый изомер в качестве затравки и перемешивают в течение ночи, давая возможность температуре подняться до комнатной. Затем закристаллизованную смесь отфильтровывают, промывают метанолом и анализируют ВЭЖХ. Результаты представлены в следующей таблице.
Пример 7
Перекристаллизация в системах смешанных растворителей
Все эксперименты по перекристаллизации проводят при внесении затравки при 53°С, 14 мг затравки/г неочищенного вещества, перемешивание при 1000 об/мин, × 14 объемов растворителя. Перекристаллизацию проводят с неочищенной смесью, полученной из реакционной смеси, где исходное соотношение R/S для ВОМО равно 82:18. Анализ ВЭЖХ неочищенного вещества дает приблизительное соотношение R/S, равное 86 /14. Как было рассмотрено ранее, когда соотношение R/S уменьшается, особенно становясь ниже 90:10, у транс-изомера также возникает предрасположенность к кристаллизации (таблица 11). Таким образом, во всех экспериментах транс-изомер единообразно находится в количестве приблизительно 10-12%. Поэтому в наблюдаемое соотношение RR/SS вносят поправку на присутствие RR и SS транс-изомеров. Результаты представлены в следующей таблице.
Пример 8
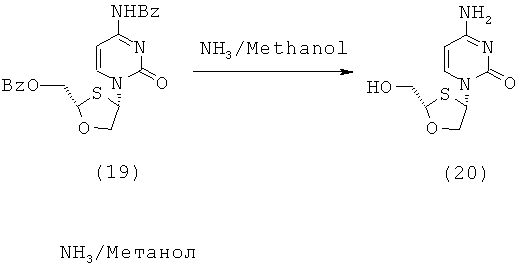
Синтез 2-(R)-гидроксиметил-4-(R)-(цитозин-1-ил)-1,3-оксатиолана

2-(R)-Бензоилоксиметил-4-(R)-(N-бензоилцитозин-1-ил)-1,3-оксатиолан (15 г, 0,028 моль) растворяют в растворе (приблизительно 2 М) метанольного аммиака (250 мл). Исходную взвесь перемешивают в течение ночи. После ночного перемешивания образующийся прозрачный раствор фильтруют через целит, упаривают досуха и суспендируют в ацетоне (100 мл). Этот образующееся не совсем белое порошкообразное вещество отфильтровывают и промывают ацетоном (2×25 мл), и сушат с образованием продукта, 6,5 г (94%).
1Н ЯМР (ДМСО): δ 7,8 (д, 1H), 7,0-7,2 (шир д, 2Н), 6,3 (д, 1Н), 5,7 (д, 1Н), 5,3 (т, 1Н) (ОН пик - не всегда разрешен), 5,1 (т, 1Н), 4,4 (д, 1Н), 3,9 (м, 1Н), 3,7 (м, 2Н), ОН пик не разрешенный.
Пример 9
Синтез 2-(R)-гидроксиметил-4-(R)-(цитозин-1-ил)-1,3-оксатиолана

Методика:
2-(R)-Бензоилоксиметил-4-(R)-(N-бензоилцитозин-1-ил)-1,3-оксатиолан (3,3 г, 0,007 моль) растворяют в смеси дихлорметане (8 мл) и метанола (10 мл) при нагревании. К раствору добавляют метоксид натрия (0,043 г, 0,0008 моль) в метаноле (2 мл) и смесь перемешивают в течение ночи. После ночного перемешивания смесь упаривают и хроматографируют на колонке из силикагеля (4×18 см) и элюируют при использовании градиента 20-50% метанола в этилацетате. Объединение и упаривание фракций приводит к получению 1,5 г (88% выход) продукта (5) в виде не совсем белого порошка.
1Н ЯМР (ДМСО): δ 7,8 (д, 1Н), 7,0-7,2 (шир д, 2Н), 6,3 (д, 1Н), 5,7 (д, 1Н), 5,1 (т, 1Н), 4,4 (д, 1Н), 3,9 (м, 1Н), 3,7 (м, 2Н), ОН пик не разрешен
Пример 10
Синтез 2-(S)-гидроксиметил-4-(S)-(цитозин-1-ил)-1,3-оксатиолана
2-(S)-Бензоилоксиметил-4-(S)-(N-бензоилцитозин-1-ил)-1,3-оксатиолан (1,0 г, 0,0022 моль) растворяют в растворе (приблизительно 2М) метанольного аммиака (20 мл). Исходную взвесь перемешивают в течение ночи. После ночного перемешивания образующийся прозрачный раствор упаривают досуха и суспендируют в ацетоне (20 мл). Это образующееся не совсем белое порошкообразное вещество отфильтровывают и промывают ацетоном (2×10 мл), и сушат с образованием продукта, 0,47 г (88%).
1Н ЯМР (ДМСО): δ 7,8 (д, 1Н), 7,0-7,2 (шир д, 2Н), 6,3 (д, 1Н), 5,7 (д, 1Н), 5,3 (т, 1Н) (ОН пик - не всегда разрешен), 5,1 (т, 1Н), 4,4 (д, 1Н), 3,9 (м, 1Н), 3,7 (м, 2Н), ОН пик не разрешен.
Пример 11
Синтез 2-(R)-бензоилоксиметил-4-(R)-(N-ацетилцитозин-1-ил)-1,3-оксатиолана
(а) Конденсация 2-(R)-бензоилоксиметил-1,3-оксатиолана с N-ацетилцитозином
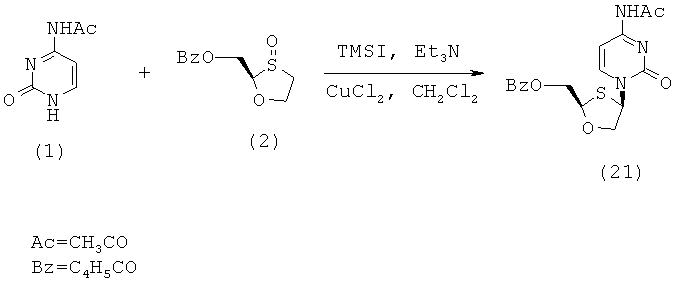
2-(R)-Бензоилоксиметил-1,3-оксатиолан (5,0 г, 0,021 моль) растворяют в дихлорметане (100 мл) в трехгорловой колбе на 500 мл в атмосфере азота и к этой смеси добавляют триэтиламин (6,08 мл, 0,044 моль). Затем раствор охлаждают до -50°С по каплям через капельную воронку добавляют иодотриметилсилан (9,22 мл, 0,065 моль) с такой скоростью, чтобы температура внутри не повышалась выше -40°С. Образующийся светло-желтый раствор перемешивают в течение 1 часа, поддерживая температуру при -50°С. Затем к реакционной смеси добавляют хлорид меди (II) (0,28 г, 0,021 моль) с последующим добавлением N-ацетилцитозин (5,0 г, 0,021 моль). Образующуюся смесь перемешивают при температуре, равной -50°С в течение 15 минут, давая смеси нагреться до 0°С и перемешивают при этой комнатной температуре в течение ночи. После ночного перемешивания реакционную смесь перемешивают при комнатной температуре в течение 1 часа, гасят добавлением воды (50 мл), после чего немедленно добавляют 5% водный аммиак (100 мл). Ее сильно перемешивают в течение 10 минут, фильтруют через слой целлита (8×3 см). Колбу и слой промывают дополнительным количеством дихлорметана (100 мл, 2×50 мл). Объединенные прозрачные фильтраты выливают в делительную воронку и отделяют органический слой. Затем органический слой промывают 2% фосфорной кислотой (100 мл). Объединенные водные слои повторно экстрагируют дихлорметаном (100 мл). Объединенные водные слои повторно экстрагируют дихлорметаном (100 мл). Объединенные органические слои сушат сульфатом магния, фильтруют и упаривают с образованием светло-желтого/коричневого вязкого масла, 6,8 г (92% выход).
Анализ методом ЯМР неочищенного вещества дает следующие результаты: Глубина протекания реакции (ЯМР определяет чистоту общего продукта конденсации): 73%; соотношение Ц/Т: 2,37:1; Количество олефинового побочного продукта (относительно общего продукта конденсации): 8%
Пример 12
Синтез 2-(R)-бензоилоксиметил-4-(R)-(N-бензоил-5-фторцитозин-1-ил)-1,3-оксатиолана
(а) Конденсация 2-(R,S)-бензоилоксиметил-1,3-оксатиолана с N-бензоил-5-фторцитозином
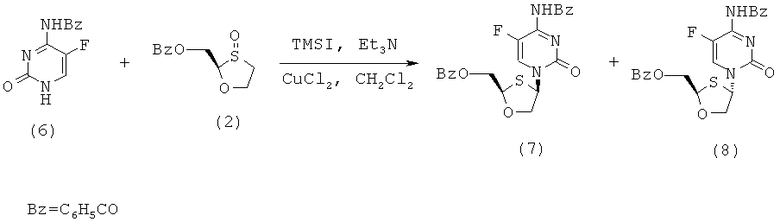
17.7 мЛ
12.2 мЛ
Методика:
Смесь 2-(R)-Бензоилоксиметил-1,3-оксатиолана и 2-(S)-Бензоилоксиметил-1,3-оксатиолана (9,6 г, 0,040 моль) растворяют в дихлорметане в трехгорлой колбе на 500 мл охлаждают до -50°С. К этой смеси добавляют триэтиламин (12,2 мл, 0,088 моль), а затем по каплям через капельную воронку иодотриметилсилан (17,7 мл, 0,124 моль) с такой скоростью, чтобы температура внутри находилась в диапазоне от -35°С до -40°С. Образующийся светло-желтый раствор перемешивают в течение 60 минут, поддерживая температуру в диапазоне от -40°С до -50°С. Затем к реакционной смеси добавляют хлорид меди (II) (1,3 г, 0,01 моль) и через 5 минут добавляют N-бензоил-5-фторцитозин (9,3 г, 0,040 моль). Образующуюся смесь перемешивают при -50°С в течение 15 минут и затем дают смеси нагреться до 0°С в течение 1 часа. Реакционную смесь перемешивают при этой температуре в течение ночи. После ночного перемешивания реакционную смесь перемешивают при комнатной температуре в течение 1 часа, охлаждают во льду, гасят добавлением насыщенного раствора бикарбоната натрия (75 мл). Ее сильно перемешивают в течение 5 минут, разбавляют дихлорметаном (50 мл) и фильтруют через слой целита. Колбу и слой промывают дополнительным количеством дихлорметана (2×50 мл) и объединенные фильтраты выливают в делительную воронку. Органический слой отделяют, промывают 5% водным аммиаком (100 мл), 2% фосфорной кислотой (2×60 мл) и вновь 5% водным аммиаком (100 мл). Объединенные водные слои повторно экстрагируют дихлорметаном (100 мл). Объединенные органические слои сушат сульфатом магния, фильтруют и упаривают с образованием светло желтого/коричневого густого масла, 14,6 г (80% выход). Эта неочищенная смесь состоит из цис (R,R и S,S) и транс (S,R и R,S) общего продукта конденсации 55% чистоты (ЯМР) с соотношением цис/транс изомеров, равном 2,65:1 (ЯМР).
(b) Перекристаллизация неочищенного 2-(R,S)-бензоилоксиметил-1,3-оксатиолан-4-(R,S)-(N-бензоил-5-фторцитозин-1-ил)-1,3-оксатиолана для получения чистого 2-(R)-бензоилоксиметил-4-(R)-(N-бензоил-5-фторцитозин-1-ил)-1,3-оксатиолана
Перекристаллизация
К неочищенному (R)-бензоилоксиметил-1,3-оксатиолан-4-(R,S)-(N-бензоил-5-фторцитозин-1-ил)-1,3-оксатиолану в трехгорлой колбе на 500 мл добавляют 10-кратное (по объему) количество метанола (146 мл) и смесь кипятят с обратным холодильником до тех пор, пока раствор не станет прозрачным. Этот раствор подвергают горячему фильтрованию и затем постепенно охлаждают до комнатной температуры и оставляют на ночь. Образующийся кристаллизующийся продукт отфильтровывают с последующим промыванием метанолом (2×100 мл) и сушат в вакууме. Анализ методом ЯМР образующегося слегка окрашенного пушистого кристаллического твердого вещества, проведенный методом ЯМР, показывает, что оно более, чем на 98% является цис-изомером. Выход выделенного вещества равен 3,5 г (19% выход расчете на цис-изомер) (7)).
1Н ЯМР (ДМСО): δ 7,95 (м, 3Н), 7,75 (м, 2Н), 7,6 (м, 2Н), 7,6-7,4 (м, 4Н), 6,22 (дд, 1Н), 5,43 (т, 1Н), 4,7 (м,2Н), 4,55 (д, 1Н), 3,95 (дд, 1Н)
Специалистам в данной области техники будет понятно, что настоящее изобретение, описанное здесь, допускает вариации и модификации, отличные от конкретно описанных. Понятно, что данное изобретение включает в себя все такие вариации и модификации, которые соответствуют сущности и объему изобретения. Настоящее изобретение также включает в себя все стадии, признаки, композиции и соединения, относящиеся к этому описанию или указанные в нем, отдельно или вместе, и любые или все сочетания любых двух или нескольких указанных стадий или признаков.
На протяжении всего описания и следующей за ней формулы изобретения, несмотря на то, что контекст требует иного толкования, слово "включать" и его производные, такие как "включает" и "включающий", будет понято как означающее включение установленного числа или стадии или группы чисел или стадий, а не исключение каких-либо чисел или стадий или группы чисел или стадий.
Ссылка в этом описании на любую предыдущую публикацию (или вытекающую из нее информацию), или вопрос, который известен, не будет рассматриваться или его не следует рассматривать как признание или разрешение или любую форму предположения, что предыдущая публикация (или вытекающая из нее информацию) или известный вопрос представляет собой часть широко известных общих знаний в той области науки, к которой относится это описание.
ССЫЛКИ
1. Belleau et. al, Bioorg. Med. Chem. Lett. (1993) Vol.3, No.8, 1723-1728.
2. Taylor et. al. Antiviral Chem. Chemother. (2000) Vol.11, No.4, 291-301.
3. Stoddart et al, Antimicrob. Agents Chemother. (2000) Vol.44, No.3, 783-786.
4. Mansour et al., J. Med. Chem., (1995) Vol.38, No. 1:1-4.
5. Nucleosides and Nucleotides (1995) 14(3-5): 627-735.
6. Caputo et al. Eur. J. Org. Chem. (1999) Vol.6: 1455-1458.
7. J. Jacques, A. Collet & S.H. Wilen "Enantiomers, Racemates and Resolutions" by (John Wiley& Sons, 1981).
8. Storer et al, Nucleosides & Nucleotides (1993) 12(2): 225-236.
9. Lorenz, H., et al. Journal of the University of Chemical Technology and Metallurgy (2007) 42(1): 5-16.
10. Greene, T.W. and Wuts, P.G.M "Protective groups in organic synthesis" (3rd Edition) 1999 John Wiley & Sons Inc.
Изобретение относится к способу получения соединения формулы (II), включающему стадии: (а) образования 2,4-дизамещенного 1,3-оксатиолана формулы (II) и (III), где заместители являются такими, как определено в формуле изобретения; (b) селективной перекристаллизации соединения общей формулы (II) из C1-6спирта или смеси C1-6спиртов. Кроме того, изобретение относится к способу получения соединения формулы (VI), включающему стадии: (а) образования 2,4-дизамещенного 1,3-оксатиолана формулы (II) и (III); (b) селективной перекристаллизации соединения общей формулы (II) из C1-6спирта или смеси C1-6спиртов; и (с) снятия защитных групп в соединении формулы (II) для получения соединения формулы (VI). Также изобретение относится к соединениям формул (VIII) и (IX), где R2 представляет собой С(O)фенил; каждая из R3 и R4 индивидуально выбрана из Н или бензоила при условии, что когда R3 представляет собой Н, R4 не является Н; и когда R4 представляет собой Н, R3 не является Н; и R5 представляет собой Н, Br, Cl, F, I или CF3. Технический результат - усовершенствованные способы получения 2,4-дизамещенных 1,3-оксатиоланов и промежуточные соединения для их получения. 6 н. и 17 з.п. ф-лы, 23 табл., 12 пр.
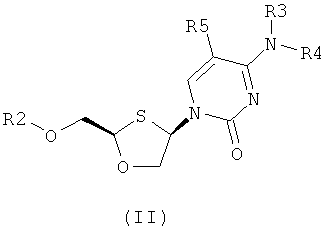 ;
; 
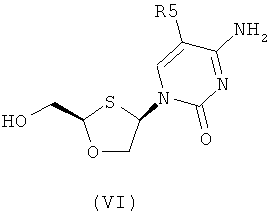 .
.
 ;
; 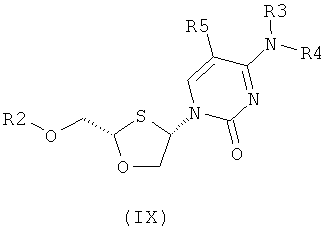
1. Способ получения соединения общей формулы (II), включающий стадии:
(а) образования 2,4-дизамещенного 1,3-оксатиолана общей формулы (II) и (III):
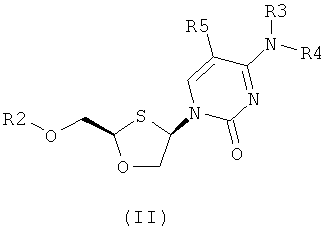 ;
; 
где
R2 представляет собой С(O)фенил;
R3 и R4 каждая индивидуально выбрана из Н или бензоила, при условии, что когда R3 представляет собой Н, R4 не является Н, и когда R4 представляет собой Н, R3 не является Н; и
R5 представляет собой Н, Br, Cl, F, I или CF3; и
(b) селективной перекристаллизации соединения общей формулы (II) из растворителя, в котором растворитель представляет собой C1-6спирт или смесь C1-6спиртов.
2. Способ получения соединения общей формулы (VI), включающий стадии:
(а) образования 2,4-дизамещенного 1,3-оксатиолана общей формулы (II) и (III):
;
где R2 представляет собой С(O)фенил;
R3 и R4 каждая индивидуально выбрана из Н или бензоила, при условии, что когда R3 представляет собой Н, R4 не является Н, и когда R4 представляет собой Н, R3 не является Н; и
R5 представляет собой Н, Br, Cl, F, I или CF3;
(b) селективной перекристаллизации соединения общей формулы (II) из растворителя, в котором растворитель представляет собой С1-6спирт или смесь С1-6спиртов; и
(c) снятия защитных групп в соединении общей формулы (II) для получения соединения общей формулы (VI):
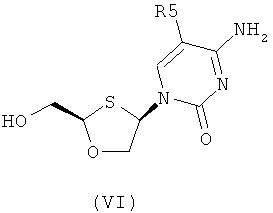 .
.
3. Способ по п.2, где (а) включает:
взаимодействие основания общей формулы (IV) с 1,3-оксатиоланом общей формулы (V) с образованием соединения общей формулы (II) и (III):
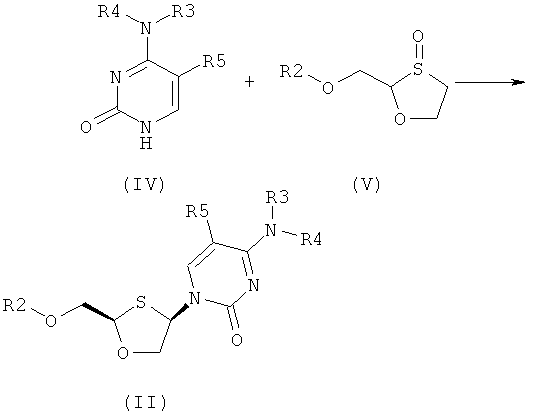 ;
; 
где 1,3-оксатиолан общей формулы (V) представляет собой (R)-энантиомер по меньшей мере в 60% ее.
4. Способ по любому из пп.1-3, где R5 представляет собой Н или F.
5. Способ получения соединения общей формулы (II), включающий стадии:
(а) взаимодействия силилированного основания общей формулы (X) с 1,3-оксатиоланом общей формулы (V) для получения соединения общей формулы (II) и (III):
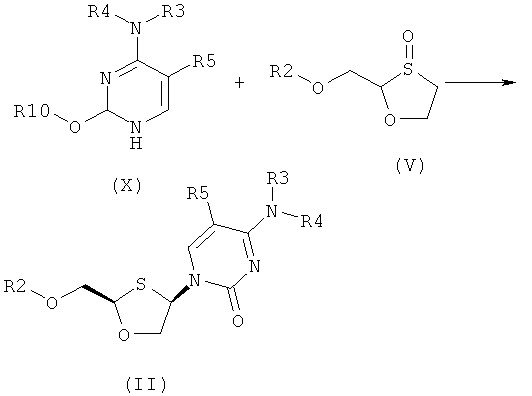 ;
; 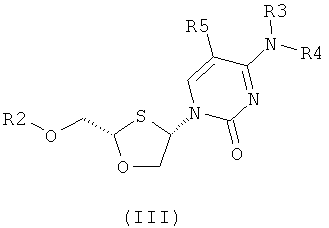
где R2 представляет собой С(O)фенил;
R3 и R4 каждая индивидуально выбрана из Н или бензоила, при условии, что когда R3 представляет собой Н, R4 не является Н, и когда R4 представляет собой Н, R3 не является Н; и
R5 представляет собой Н, Br, Cl, F, I или CF3; и
R10 представляет собой силильную защитную группу общей формулы S1R7R8R9, где R7, R8 и R9 каждый независимо выбран из С1-6алкила, арила или С1-6алкиларила; и
где 1,3-оксатиолап общей формулы (V) представляет собой (R)-энантиомер по меньшей мере в 60% ее; и
(b) селективной перекристаллизации соединения общей формулы (II) из растворителя, в котором растворитель представляет собой C1-6спирт или смесь С1-6спиртов.
6. Способ получения соединения общей формулы (VI), включающий стадии:
(а) взаимодействия силилированного основания общей формулы (X) с 1,3-оксатиоланом общей формулы (V) для получения соединения общей формулы (II) и (III):
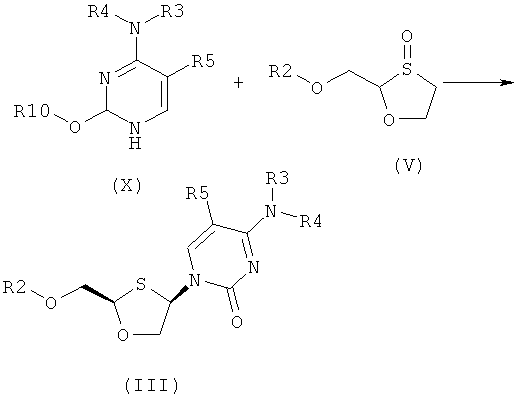 ;
; 
где R2 представляет собой С(O)фенил;
R3 и R4 каждая индивидуально выбрана из Н или бензоила, при условии, что когда R3 представляет собой Н, R4 не является Н, и когда R4 представляет собой Н, R3 не является Н; и
R5 представляет собой Н, Br, Cl, F, I или CF3; и
R10 представляет собой силильную защитную группу общей формулы S1R7R8R9, где R7, R8 и R9 каждый независимо выбран из С1-6алкила, арила или С1-6алкиларила;
где 1,3-оксатиолан общей формулы (V) представляет собой (R)-энантиомер по меньшей мере в 60% ее;
(b) селективной перекристаллизации соединения общей формулы (II) из растворителя, в котором растворитель представляет собой С1-6спирт или смесь С1-6спиртов;
(c) снятия защитных групп в соединении общей формулы (II) для получения соединения общей формулы (VI):
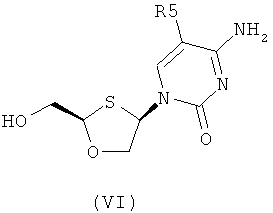 .
.
7. Способ отделения соединения общей формулы (II) от соединения общей формулы (III), включающий стадии:
(а) получения смеси 2,4-дизамещенных 1,3-оксатиоланов общей формулы (II) и (III):
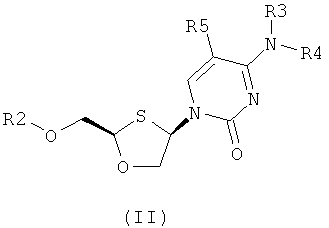 ;
; 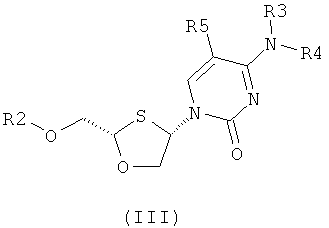
где R2 представляет собой С(O)фенил;
R3 и R4 каждая индивидуально выбрана из Н или бензоила, при условии, что когда R3 представляет собой Н, R4 не является Н, и когда R4 представляет собой Н, R3 не является Н; и
R5 представляет собой Н, Br, Cl, F, I или CF3; и
(b) отделения соединения общей формулы (II) от соединения общей формулы (III) селективной перекристаллизацией соединения общей формулы (II) из растворителя, в котором растворитель представляет собой С1-6спирт или смесь C1-6спиртов.
8. Способ по любому из пп.3, 5 или 6, где 1,3-оксатиолан общей формулы (V) имеет более 95% ее.
9. Способ по п.8, где 1,3-оксатиолан общей формулы (V) имеет более 99% ее.
10. Способ по любому из пп.1-9, где растворитель представляет собой C1-6спирт, выбранный из группы, состоящей из метанола (МеОН), этанола (EtOH), пропанола и бутанола.
11. Способ по п.10, где С1-6спиртом является метанол.
12. Способ по любому из пп.1-11, где растворитель представляет собой смесь С1-6спиртов, содержащих МеОН.
13. Способ по п.12, где С1-6спирты в смеси находятся в соотношении МеОН: С2-6спирт, равном 90:10.
14. Способ по п.13, где соотношение МеОН: С2-6спирт равно 95:5.
15. Способ по любому из пп.10-14, где растворитель содержит не более чем приблизительно 5% воды.
16. Способ по любому из пп.1-15, где стадию селективной перекристаллизации (стадию (b)) повторяют по меньшей мере один раз.
17. Способ по любому из пп.1-15, где стадия селективной перекристаллизации (стадия (b)) представляет собой способ с захватом или с циклическим захватом.
18. Способ по п.17, где способ с захватом или с циклическим захватом включает внесение затравки R,R-энантиомера.
19. Способ по п.18, где внесение затравки проводят при температуре, меньшей или равной приблизительно 55°С.
20. Способ по любому из пп.1-9, где 2,4-дизамещенный 1,3-оксатиолан общей формулы (II) или общей формулы (III), полученный или образующийся на стадии (а), находится в виде конгломерата.
21. Соединение общей формулы (VIII) или (IX):
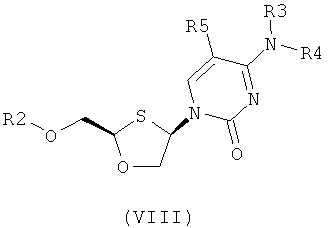 ;
; 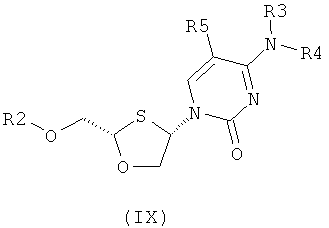
где R2 представляет собой С(O)фенил;
каждая из R3 и R4 индивидуально выбрана из Н или бензоила при условии, что когда R3 представляет собой Н, R4 не является Н; и когда R4 представляет собой Н, R3 не является Н; и R5 представляет собой Н, Br, Cl, F, I или CF3.
22. Соединение по п.21, где R5 представляет собой Н или F.
23. Соединение по п.21, где соединение выбрано из группы соединений, состоящей из:
 ;
;  ;
;
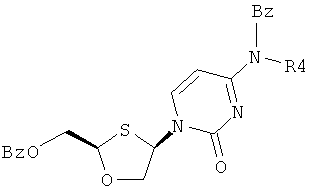 ;
; 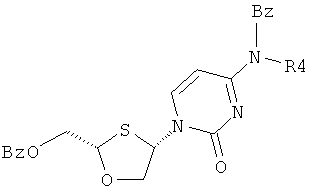 ;
;
 ;
; 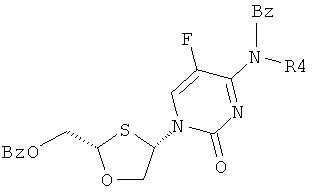 ;
;
 ;
; 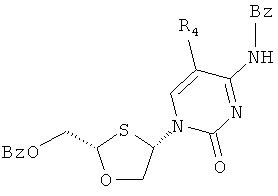 ;
;
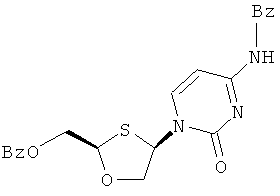 ;
; 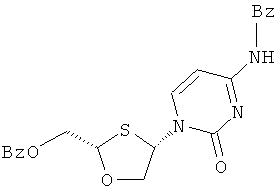 ;
;
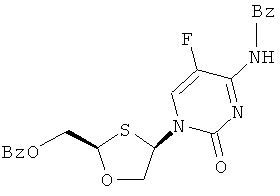 ;
;  ;
;
где R5 и R4 являются такими, как определено в п.1.
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| СПОСОБ ПОЛУЧЕНИЯ 1,3-ОКСАТИОЛАНОВОГО НУКЛЕОЗИДА, СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО 1,3-ОКСАТИОЛАНИЛ-5-ОНА | 1999 |
|
RU2244712C2 |

