Данное изобретение было осуществлено при поддержке правительства Соединенных Штатов по решению следующих организаций: DOE DE-FG02-84ER13183. Соединенные Штаты обладают определенными правами на это изобретение.
Здесь заявлен приоритет согласно предварительной заявке на патент, серийный номер 60/785471, зарегистрированной 24 марта 2006, которая включена в этот документ.
В настоящую эпоху уменьшения запасов нефти и политической нестабильности в странах, богатых нефтью, промышленное сообщество должно развивать пути использования мировых ресурсов обильной и возобновляемой биомассы для получения новых источников энергии и химических промежуточных продуктов [1]. Например, транспортный сектор требует топливо, которое может быть эффективно преобразовано в энергию и которое обладает высокой удельной энергией. Химическая промышленность требует функциональных молекул, таких как олефины (например, этилен, пропилен) и альдегиды (например, формальдегид), которые можно использовать для получения полимерных материалов. Особенность, которая делает полученные из биомассы углеводы особенно перспективным классом соединений для дополнения (или, в некоторых случаях, замены) нефти в упомянутых областях, состоит в том, что стехиометрические элементарные звенья этих соединений обладают атомным составом H:C:O, равным 2:1:1. Таким образом, углеводы являются идеальными кандидатами для преобразования в газовые смеси H2/CO. Эти виды газов обычно называют «синтез-газ». Синтез-газ можно преобразовать путем синтеза Фишера-Тропша (Fischer-Tropsch) на катализаторах на основе железа и кобальта [2] с получением линейных длинноцепочечных алканов для использования в качестве дизельного топлива. Синтез-газ можно также преобразовать на катализаторах на основе меди [3] с получением метанола для применения в качестве сырья для получения олефинов, формальдегида и бензина.
Хотя получение синтез-газа из биомассы в течение многих лет признавали в качестве многообещающей основы, откуда можно получить множество ценных продуктов, стандартные пути для получения синтез-газа из биомассы не являются чрезвычайно эффективными из-за того, что требуют высоких температур. Например, непосредственная каталитическая газификация биомассы требует температуру 800 K и выше [4]. Двухстадийная газификация биомассы также требует высоких температур: быстрый пиролиз биомассы (при примерно 773 K), за которым следует паровой риформинг получаемого биомасла (при примерно 1000 K) [5, 6]. Более того, газификация биомассы обычно приводит к сложному набору биопродуктов, включающему смолу (летучие органические соединения), обуглившееся вещество (твердые углеродсодержащие материалы) и легкие углеводороды, также как и соединения NOx и SOx, полученные в процессах высокотемпературного горения [1, 4-6].
Относительно недавним и быстро растущим применением биомассы является ее использование при производстве биодизельного топлива посредством переэтерификации растительных масел и животных жиров [1, 7, 8]. Реакция переэтерификации дает малоценный поток отходов глицерина, который часто содержит концентрации глицерина в воде от 50 до 80% [8]. Получающийся излишек привел к падению цен на глицерин в США от примерно 2100 долларов США за метрическую тонну в 1995 до менее 1000 долларов США за метрическую тонну в 2003 (для 97% глицерина фармакологического качества (Фармокапеи США), цены предоставлены Procter&Gamble). Настоящее производство (2006) биодизельного топлива в США и Европе составляет 108 и 2×108 литров в год, соответственно. Из-за налоговых льгот и других экономических стимулов, предоставляемых некоторыми национальными правительствами, ожидается удвоение этих величин в очень близком будущем [8, 9]. В отношении налоговых льгот для биодизельного топлива в США см. IRS Publication No. 378 и разделах 6426(c), 6427(e) и 40А Закона о внутреннем налогообложении.
Глицерин можно также получать путем ферментации Сахаров, таких как глюкоза [10]. В отличие от ферментации глюкозы с получением этанола, при которой получают этанол при концентрации только примерно 5% масс. в воде, при ферментации глюкозы с получением глицерина можно получить глицерин при концентрации примерно 25% масс. [10]. Эта более высокая концентрация глицерина в сравнении с этанолом снижает энергетические затраты, требуемые для удаления воды из кислородсодержащего углеводородного топлива. Действительно, одной из наиболее энергоемких стадий при получении этанола топливного качества из глюкозы, является стадия дистилляции [11, 12]. Другой путь получения глицерина из глюкозы и других Сахаров состоит в гидрировании глюкозы до сорбитола [13, 14], за которым следует гидрогенолиз сорбитола до полиолов, имеющих низкие молекулярные массы [15, 16].
В предыдущей работе, написанной в соавторстве с некоторыми авторами настоящего изобретения [17-19] было показано, что растворы полиолов в воде (например, этиленгликоль, глицерин, сорбитол) можно было бы преобразовать посредством риформинга в водной фазе в смесь газов H2/CO2, содержащую низкие уровни CO (например, 500 частей на миллион) на платиновых катализаторах на носителе при температуре приблизительно 500 K. Эти способы риформинга в водной фазе приводят к низким соотношениям CO:CO2 в потоке выходящего газа, так как реакция сдвига в водяном газе (СВГ) чрезвычайно благоприятна при высоких парциальных давлениях воды, образованной при этих условиях реакции (например, 2,5 МПа (25 бар)). Таким образом, условия реакции риформинга в водной фазе не являются благоприятными для получения синтез-газа, в котором требуется высокое соотношение CO:CO2. Другие исследователи изучали риформинг глицерина в паровой фазе. Czermik et al. [20] сообщали о высокой селективности выработки H2 путем парового риформинга глицерина при высоких температурах (1023 K) на промышленном катализаторе риформинга лигроина, на основе никеля. Suzuki et al. [21] также наблюдали высокую селективность выработки H2 путем парового риформинга глицерина при высоких температурах (873 K) на 3% Ru/Y2O3 катализаторе, однако они применяли высокую объемную скорость продувочного газа в экспериментах. Следовательно, остается необходимость в разработке лучшей каталитической системы для конверсии глицерина в паровой фазе при низких температурах.
Хорошо известен синтез Фишера-Тропша (Ф-Т) для получения синтетических углеводородов из синтез-газа. Его осуществляли в больших масштабах немцы во время второй мировой войны для производства жидкого топлива из угля. Основная реакция Ф-Т является следующей:

где -(CH2)- представляет основное элементарное звено углеводородных продуктов. Синтез Ф-Т является чрезвычайно экзотермическим, что приводит к тому, что теплоперенос представляет собой значительный фактор при конструировании реактора Ф-Т.
Значительное количество исследований было выполнено для максимизации способностей синтеза в реакции Ф-Т. См., например, патент US 6696501, в котором описан способ конверсии природного газа или другого ископаемого топлива в высшие углеводороды. Здесь в способе используют сочетание парового риформинга ископаемых топлив с получением синтез-газа, за которым следует синтез Ф-Т и второй паровой риформинг отходящего газа. Преобразованный отходящий газ затем подают назад в реактор Ф-Т.
См. также патент US 6976362, в котором описан способ интеграции образования синтез-газа, реакции Ф-Т и реакции сдвига в водяном газе для получения CO2, алифатических углеводородов и водорода, и затем сжигания водорода в камере сгорания газа турбины для получения электричества.
Как вкратце было отмечено выше, важным параметром для определения теоретического максимального выхода синтетических углеводородов в реакции Ф-Т является стехиометрическое число СЧ, определяемое как:

Теоретически, выход синтетических углеводородов является наибольшим, когда СЧ=2,0 и CO не реагирует дополнительно с образованием CO2 посредством реакции сдвига в водяном газе. В этом случае соотношение H2/CO будет равно СЧ, то есть 2,0, что теоретически дает наибольший выход синтетических углеводородов.
Биомасса включает в первую очередь углеводы (например, крахмал и целлюлозу). Одним из способов конверсии этих соединений в жидкое топливо является ферментация с получением жидких спиртов, таких как этанол и бутанол. Технология конверсии полученного из зерна крахмала в этанол посредством гидролиза, ферментации и дистилляции хорошо разработана, и существуют экономические преимущества в конверсии лигноцеллюлоз в этанол (например, путем разработки новых энзимов для гидролиза целлюлозы). Преимущества этанола в качестве транспортного топлива состоят в том, что он является жидкостью и имеет высокое октановое число (октановое число, согласно исследованиям, составляет 130). Однако, этанол в качестве топлива обладает несколькими значительными присущими ему недостатками по сравнению с длинноцепочечными алканами: (1) этанол обладает меньшей удельной энергией по сравнению с нефтью (например, приблизительно 2·104 БТЕ(британская тепловая единица)/литр для этанола по сравнению с 3·104 БТЕ/литр для нефти); (2) этанол полностью смешивается с водой, что приводит к значительной абсорбции воды в топливе, и (3) он обладает относительно низкой температурой кипения (73°C), что приводит к избыточному испарению при повышенных температурах. Однако наиболее важно, что способ ферментации, используемый для получения биоэтанола из углеводов, приводит к водному раствору, содержащему только примерно от 5 до 10% масс. этанола. Значительное количество энергии требуется для дистилляции этанола из воды для получения этанола топливного качества. Действительно, общий энергетический баланс производства биоэтанола не является очень благоприятным, и установлено, что количество энергии, требуемой для получения биоэтанола, приблизительно равно (или больше) энергоемкости получаемого этанола [11, 12, 36].
Длинноцепочечные алканы включают огромное большинство компонентов нефтяного транспортного топлива (разветвленные алканы в бензине, линейные алканы в дизельном топливе). Преобразование возобновляемых ресурсов биомассы в жидкие алканы является поэтому привлекательным вариантом переработки. Наиболее примечательно то, что жидкие алканы, получаемые из биомассы, (1) можно распределять, используя уже эксплуатируемую инфраструктуру для полученных из нефти продуктов; (2) можно добавлять в существующее нефтяное хранилище для дальнейшей переработки (например, смешанные топлива) и (3) можно сжигать в существующих двигателях внутреннего сгорания без изменения двигателя или топлива.
Данное изобретение представляет собой способ производства углеводородов или кислородсодержащих углеводородов, предпочтительно от C2 до C36 углеводородов и/или кислородсодержащих углеводородов. В предпочтительном воплощении изобретения жидкие алканы (то есть C5 и длиннее, линейные, разветвленные или циклические) можно получать непосредственно из соединений, полученных из биомассы, включающих полисахариды, моносахариды и полиолы (например, глицерин) посредством интегрированного способа, включающего каталитическую конверсию в газовые смеси H2/CO (синтез-газ) и синтез Фишера-Тропша (или другие реакции образования углерод-углеродных связей). Стадию синтеза Фишера-Тропша можно выполнять последовательно за стадией конверсии глицерина или, что важно, обе реакции можно соединить путем интеграции активных центров для каждой реакции внутри одного каталитического слоя. Таким образом, в одном воплощении данного изобретения, каталитическое преобразование глицерина и синтезы Фишера-Тропша соединены в реакторной системе с двумя слоями. В предпочтительном порядке действий преобразование сырьевого реагента в синтез-газ выполняют, используя катализатор, включающий Pt-Re на углеродном носителе. Затем завершают реакцию образования углерод-углеродных связей, используя катализатор, включающий Ru/TiO2.
В другом воплощении изобретения каталитическую конверсию глицерина и синтез Фишера-Тропша совмещают в одном реакторе, предпочтительно используя один слой катализатора, включающий физическую смесь Pt-Re/C и Ru/TiO2. Альтернативно, как описано в разделе подробное описание изобретения, способ можно выполнять в одном реакторе, но используя один, два или более отдельных каталитических слоев, причем каждый слой содержит один или более отдельных катализаторов. Например, способ можно выполнять, используя один каталитический слой, содержащий гомогенный катализатор двойного назначения, или способ можно выполнять, используя один каталитический слой, содержащий два или более катализаторов, физически смешанных друг с другом, или способ можно выполнять, используя два или более каталитических слоев в многослойной или ступенчатой конфигурации. Во всех этих воплощениях данного изобретения получают жидкие алканы с Sc5+ от примерно 46% до примерно 64% при содержании углерода от примерно 15% до примерно 50% в продуктах, содержащихся в органической жидкой фазе (см. Примеры). Водный жидкий продукт из интегрированного способа содержит от примерно 5% до примерно 15% масс. метанола, этанола и ацетона, которые можно отделить от воды посредством дистилляции и использовать в химической промышленности или повторно использовать для конверсии в газовые продукты. Этот интегрированный способ значительно улучшает экономику «зеленого» синтеза Фишера-Тропша путем уменьшения капитальных затрат и увеличения теплового кпд реакций. Таким образом, значительным преимуществом настоящего изобретения является то, что оно позволяет включать реакторы Фишера-Тропша меньшего масштаба в качестве компонентов биоперерабатывающего завода. Настоящее изобретение можно также использовать для переработки отходов глицерина из биодизельных установок в жидкое топливо. Более того, объединение (а) конверсии биомассы в синтез-газ и (б) синтеза Фишера-Тропша приводит к синергетическому действию этих способов. Объединяя эти две реакции в одном реакторе, избегают сильно эндотермических или экзотермических стадий, которые возникают в результате протекания реакций по отдельности.
Значительным преимуществом настоящего изобретения является то, что путем объединения реакции газификации и реакции образования углерод-углеродных связей в «одном сосуде» устраняют ингибирующий эффект парциального давления CO на скорость реакции газификации, путем потребления CO в том же реакторе (в реакции образования углерод-углеродных связей).
Таким образом, данное изобретение представляет собой способ производства углеводородов (предпочтительно), кислородсодержащих углеводородов и других органических соединений. Способ включает выполнение двух реакций, одна из которых экзотермическая, а другая эндотермическая, и использование теплоты от экзотермической реакции для поставки (по меньшей мере, частично) энергии, требующейся для проведения эндотермической реакции. Таким образом, способ включает выполнение эндотермической реакции газификации с реагирующим веществом в виде биомассы при температуре, меньшей или равной примерно 750 K, для получения синтез-газа. Способ дополнительно включает выполнение экзотермической реакции образования углерод-углеродных связей (или, вообще, любой экзотермической реакции утилизации синтез-газа, такой, как синтез метанола или синтез простого диметилового эфира) с синтез-газом, полученным в эндотермической реакции. Экзотермическую реакцию выполняют при температуре, большей или равной температуре реакции газификации, выполняемой на стадии (а). В предпочтительном воплощении в экзотермической реакции получают углеводороды (так же как и теплоту). Теплоту, выделяемую при экзотермической реакции утилизации синтез-газа, или из реакции образования углерод-углеродных связей, направляют обратно в способ (то есть, используют) для того, чтобы предоставить (по меньшей мере, частично) энергию, требуемую для проведения эндотермической реакции газификации.
Более конкретно, данное изобретение относится к способу производства углеводородов и кислородсодержащих углеводородов. Первое воплощение изобретения включает выполнение эндотермической реакции газификации с реагирующим веществом в виде биомассы (предпочтительно, полисахаридом, моносахаридом и/или полиолом) при температуре, менее или равной примерно 750 K, с получением синтез-газа. Экзотермическую реакцию утилизации синтез-газа или реакцию образования углерод-углеродных связей затем выполняют, используя синтез-газ, образованный на первой стадии, при температуре, выше или равной температуре начальной реакции газификации. В экзотермической реакции получают углеводороды или кислородсодержащие углеводороды, и теплоту. Теплоту, выделяемую при реакции утилизации синтез-газа или реакции образования углерод-углеродных связей, затем используют при проведении эндотермической газификации. Таким образом, теплоту, выделенную при экзотермической реакции, используют для проведения (по меньшей мере частично) эндотермической реакции газификации.
Предпочтительно выполнять эндотермическую реакцию газификации при температуре, менее или равной примерно 750 K (и более предпочтительно ≤ примерно 625 K, еще более предпочтительно ≤ примерно 575 K, и еще более предпочтительно ≤ 550 K). Как было замечено ранее, реакции можно выполнять в двух отдельных реакторах или в одном реакторе с одним, двумя или более отдельными каталитическими слоями и используя один, два или более катализаторов.
Другое воплощение изобретения представляет собой способ производства линейных или разветвленных углеводородов или кислородсодержащих углеводородов от C2 до C36. Способ включает выполнение эндотермической реакции газификации с потоком реагента, включающим биомассу, при температуре менее или равной примерно 750 K (и более предпочтительно примерно ≤625 K, еще более предпочтительно примерно ≤575 K, и еще более предпочтительно примерно ≤550 K) для получения синтез-газа. Экзотермическую реакцию Фишера-Тропша осуществляют так, что синтез-газ образуется при температуре выше или равной температуре реакции газификации, в которой получают углеводороды и/или кислородсодержащие углеводороды от C2 до C36 и теплоту. Теплоту, выделяемую в реакции Фишера-Тропша используют в эндотермической реакции газификации, таким образом делая способ в целом весьма энергетически эффективным.
Намного более предпочтительно, чтобы эндотермическая реакция газификации протекала при температуре, которая является оптимальной для утилизации синтез-газа или для реакции образования углерод-углеродных связей. Это условие приводит к суммарной, объединенной реакции, которая является наиболее эффективной по соответствию теплоты, выделяемой при экзотермической реакции, теплоте, требуемой для эндотермической реакции. Таким образом, когда экзотермическая реакция является реакцией Фишера-Тропша, предпочтительно, чтобы как реакция Фишера-Тропша, так и реакция газификации протекали при температуре, оптимальной для реакции Фишера-Тропша.
В предпочтительном воплощении изобретения как эндотермическая реакция газификации, так и экзотермическая реакция утилизации синтез-газа или реакция образования углерод-углеродных связей, выполняются одновременно в одном реакторе. Альтернативно, две реакции могут протекать в отдельных реакторах с тем, чтобы теплоту, выделяемую при экзотермической реакции, направлять обратно, чтобы поставить тепло в эндотермическую реакцию газификации. Реактор может быть любой известной на настоящее время или разработанной в будущем конструкции, до тех пор, пока конструкция реактора обеспечивает использование теплоты от экзотермической реакции в эндотермической реакции. В настоящем уровне техники хорошо известно конструирование реакторных систем для максимизации объединения теплоты между двумя или более реакциями, и мы не будем здесь обсуждать его подробно. См., например, ссылки (33) и (34).
Предпочтительное сырье для эндотермической реакции газификации включает биомассу или соединения, полученные из биомассы, включающие, но не ограничивающиеся этим, целлюлозные материалы, лигноцеллюлозные материалы, полисахариды, моносахариды, полиолы и им подобные. Предпочтительно сырье, включающее моносахариды и/или глицерин. Термин «биомасса», как он здесь используется, относится к органическим материалам, вырабатываемым растениями, таким как листья, корни, зерна и стебли, так же как и к отходам жизнедеятельности животных и микроорганизмов (например, навоз), без ограничений. Обычные источники биомассы включают (без ограничений): (1) сельскохозяйственные отходы, такие как стебли злаков, солома, кожура стручков, остатки сахарного тростника, жмых, скорлупа орехов и навоз крупного рогатого скота, домашней птицы и свиней; (2) древесные материалы, такие как древесина или кора, опилки, древесная стружка и отходы размольной машины; (3) городские отходы, такие как использованная бумага и изрезанные газеты, и (4) сельскохозяйственные культуры, используемые в качестве источника энергии, такие как тополя, ивы, просо, люцерна, бородатая трава, зерно, соевые бобы и подобное. Термин «реагент, полученный из биомассы» относится к любым реагентам, которые можно получить из биомассы путем любых известных в настоящее время средств или средств, которые могут быть разработаны в будущем, включающим (без ограничений) полисахариды, моносахариды, полиолы, кислородсодержащие углеводороды, сахара, крахмалы и подобное, например, этандиол, этандион, глицерин, глицеральдегид, альдотетрозы, альдопентозы, альдогексозы, кетотетрозы, кетопентозы, кетогексозы и альдитолы.
Предпочтительной экзотермической реакцией образования углерод-углеродных связей является реакция Фишера-Тропша. Реакцию газификации можно выполнить, используя один или более катализаторов, включающих металлы Группы VIIIB (Fe, Co, Ni, Ru, Rh, Pd, Os, Ir и Pt) или любой катализатор, перечисленный в разделе подробного описания изобретения. Катализатор можно нанести или не наносить на любой подходящий носитель (см. список примеров носителей в разделе подробного описания изобретения).
В предпочтительном воплощении изобретения эндотермическую реакцию газификации выполняют, используя реагирующее сырье, включающее глицерин, и экзотермическая реакция утилизации синтез-газа или реакция образования углерод-углеродных связей является реакцией Фишера-Тропша. Также можно использовать другие экзотермические реакции (такие как синтез метанола или синтез простого диметилового эфира).
Для того, чтобы более близко подобрать температуры двух реакций, предпочтительно, чтобы реакцию газификации выполняли, используя по меньшей мере один катализатор, включающий металл Группы VIIIB или сочетание металлов Группы VIIIB, и более предпочтительно, по меньшей мере еще один катализатор, включающий платину, рутений, рений (металл Группы VIIB) или их сочетание. Катализатор можно при желании нанести на носитель, что, вообще, является предпочтительным, так как такие нанесенные катализаторы становятся более стабильными во времени (см. раздел подробного описания изобретения). Можно использовать любой подходящий носитель. Предпочтительные носители включают, без ограничения, углерод, так же как и оксиды алюминия, церия, циркония и марганца и сочетания любых из них. В наиболее предпочтительном варианте изобретения используют один или более катализаторов, включающих платину, рутений, сочетание платины и рутения, или сочетание платины и рения, и катализатор наносят на носитель, выбираемый из группы, состоящей из углерода, Al2O3, CeO2, ZrO2, MgO, ZrO и их сочетания.
Таким образом, в настоящем изобретении реагирующие вещества, получаемые из биомассы, преобразуют в смесь газов H2 и CO (то есть синтез-газ); синтез-газ используют для получения топлива и химикалий посредством реакции Фишера-Тропша, синтеза метанола или других реакций образования углерод-углеродных связей. В предпочтительном варианте синтез-газ получают из глицерина над катализатором(рами), включающим платину. Как показано в приведенных здесь Примерах, настоящий способ обеспечивает выход синтез-газа с высокими процентами и высокой селективностью при температурах, которые значительно ниже по сравнению с обычной температурой газификации биомассы. В настоящем изобретении по меньшей мере часть эндотермической теплоты для стадии образования синтез-газа поставляют из экзотермической теплоты реакции образования углерод-углеродных связей. В настоящем изобретении температурные диапазоны этих каталитических процессов устанавливают так, чтобы они перекрывались или почти перекрывались, что является новой особенностью в области производства органических соединений при использовании синтез-газа в качестве сырья. Таким образом, настоящее изобретение предоставляет энергетически эффективный путь получения топлива и химических веществ из возобновляемых источников биомассы вообще и из глицерина в частности.
Получение синтез-газа из биомассы в течение многих лет рассматривалось в качестве многообещающей базы, из которой можно получить множество ценных продуктов. Однако обычное получение синтез-газа из биомассы требует очень высоких температур, которые ограничивают эффективность всего способа. Как здесь показано, каталитическое получение синтез-газа из биомассы (в частности, глицерина) можно производить так, чтобы оно происходило при температурах (примерно от 550 K до менее примерно 750 K и предпочтительно, менее примерно 620 K), которые значительно ниже, чем те, которые используют, применяя обычную непосредственную каталитическую газификацию биомассы (при 800 K) [4] или применяя обычную двухстадийную газификацию биомассы, которая включает быстрый пиролиз биомассы (при 773 K) [5, 6], за которой следует паровой риформинг получаемого био-масла (при 1000 K).
Важно, что в настоящем изобретении низкие температуры стадии газификации совпадают или почти совпадают с температурами, используемыми для синтезов Фишера-Тропша и метанола (вообще, для реакций утилизации синтез-газа). В результате по меньшей мере часть (и возможно всю) теплоту, требуемую для эндотермической стадии газификации, можно поставлять в виде теплоты, выделяемой при высоко экзотермической реакции утилизации синтез-газа или реакции образования углерод-углеродных связей. В частности, газификация глицерина (предпочтительный реагент) для получения CO и H2 протекает посредством следующей стехиометрической реакции:

Эндотермическая теплота этой реакции соответствует примерно 24% теплоты сгорания глицерина. Экзотермическая теплота, получаемая путем преобразования CO и H2 из глицерина для получения жидких алканов (например, октана) путем синтеза Фишера-Тропша соответствует примерно 28% теплоты сгорания глицерина. (Данное число включает небольшое количество теплоты, высвобождаемой при реакции сдвига в водяном газе (CO+H2O→CO2+H2), для подгонки соотношения газов H2:CO в соответствии со стехиометрией всей реакции.) Объединение стадии реакции газификации и синтеза Фишера-Тропша таким образом приводит к следующему слегка экзотермическому процессу, с теплотой, которая составляет только примерно 4% от теплоты сгорания глицерина.

Таким образом, очень значительное преимущество настоящего изобретения состоит в том, что реакция газификации работает при использовании сырья, имеющего высокую концентрацию глицерина (25% масс. и выше). Таким образом, настоящее изобретение коммерчески притягательно для создания органических соединений с дополнительными ценными характеристиками из большого (и возрастающего) количества глицерина, который получают в качестве побочного продукта при производстве биодизельного топлива. В частности, синтез-газ, получаемый из глицерина, можно использовать для получения метанола, важного реагента в процессе переэтерификации, благодаря чему уменьшают стоимость получения биодизельного топлива путем утилизации глицеринового побочного продукта.
Как показано подробно ниже, вообще предпочтительно использовать условия фазовой реакции, достигаемые путем испарения концентрированного водного раствора глицерина (например, 25-50% масс), приводящего к низкому парциальному давлению воды.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Фиг.1A, 1B, 1C и 1D представляют собой графические изображения изменений степени конверсии в газофазные продукты (Фиг.1A), частоту оборота H2 (Фиг.1B), молярное соотношение CO/CO2 (Фиг.1C), и соотношение C2 к H2, умноженное на 100 (Фиг.1D) для платиновых катализаторов, нанесенных на Al2O3 (■), CeO2/ZrO2 (▲), C (▼), ZrO2 (♦) и MgO/ZrO (o). Степень конверсии в газовую фазу вычисляют следующим образом: (число атомов углерода в потоке газофазного продукта / полное число атомов углерода, подаваемых в реактор) × 100. Реакции выполняли при 0,1 МПа (1 бар) и 623 K с подачей 0,32 см3 / мин 30% масс. водно-глицеринового раствора через 1,0 г платиновых катализаторов, нанесенных на оксид, или через 0,060 г Pt/C катализатор. ▼* показывает точку после 72 часов рабочего цикла.
Фиг.2A, 2B, 2C и 2D представляют собой графические изображения изменений степени конверсии глицерина в газофазные продукты (Фиг.2A) и молярных соотношений для риформинга в газовой фазе 30% масс. глицерина (Фиг.2B) при давлении 0,1 МПа (1 бар) (■), 50% глицерина при 0,1 МПа (1 бар) (▲) и 30% глицерина при 2 МПа (20 бар) (●) с 0,090 г Pt/C при 623 K. Изменение степени конверсии глицерина в газофазные продукты (Фиг.2C) и молярные соотношения (CO/CO2 - закрашенные значки, H2/CO - незакрашенные значки) для риформинга в газовой фазе 30% масс. глицерина (Фиг.2D) при 0,1 МПа (1 бар) с Pt:Ru/C при 548 K (треугольники: 0,435 г катализатора) и 573 K (квадраты: 0,513 г катализатора) и с Pt:Re/C при 498 K (перевернутые треугольники: 0,535 г катализатора) и 523 K (кружки: 0,535 г катализатора). Скорость подачи жидкости составляет 0,16 см3/мин для Фиг.2A и 2B и 0,08 см3/мин для Фиг.2C и 2D.
Фиг.3 представляет собой схему примерной реакционной кинетической установки, которая может быть использована для оценки настоящего изобретения. (Поток H2 используют только в течение каталитического восстановления).
Фиг.3A, 3B и 3C являются схематическими представлениями реактора и расположения каталитических слоев, которые можно использовать в настоящем изобретении. На Фиг.3A изображено расположение одного реактора с одним слоем. На Фиг.3B изображено расположение одного реактора со многими слоями, на Фиг.3C изображено расположение двух реакторов.
Фиг.4A и 4B представляют собой графические изображения изменения степени конверсии глицерина в газофазные продукты (Фиг.4A) и молярное соотношение для риформинга в газовой фазе потока 0,16 см3/мин 30% масс. глицерина (Фиг.4B) при 0,1 МПа (1 бар) и 623 K для системы с одним слоем (0,090 г Pt/C: открытые символы) и для системы с двумя слоями (0,090 г Pt/C, 1,0 г Pt/CeO2/ZrO2: закрытые символы).
Фиг.5 представляет собой графическое изображение молекулярно-массовых распределений для сухого синтез-газа (■) и совместно подаваемых воды (♦), ацетона (●), этанола (▲) и ацетола (▼). Экспериментальные условия были такими, как показаны в Табл.2. Линия АШФ нанесена с α=0,83.
Фиг.6 представляет собой графическое изображение степени конверсии в газофазные продукты (♦), молярное соотношение CO/CO2 (■) для обработки в газовой фазе 30% масс. водно-глицеринового сырья при 548 K и 0,83 МПа (8,3 бар). Степень конверсии в газовую фазу рассчитывали следующим образом (количество атомов C в потоке газофазного продукта / полное количество атомов C, подаваемых в реактор) × 100. Реакции выполняли, используя сырьевой раствор при 0,08 см3/мин через 520 мг катализатора.
Фиг.7 представляет собой графическое изображение молекулярно-массовых распределений для объединенных экспериментов по конверсии глицерина с синтезом Фишера-Тропша при использовании 1,0 г Pt-Re/C с 1,7 г Ru/TiO2 (■) и 3,0 г Ru/TiO2 (●). Линия АШФ нанесена с α=0,8.
ПОДРОБНОЕ ОПИСАНИЕ
В основе настоящего изобретения лежит регламентированная процедура реакции, в которой температура эндотермической реакции газификации приведена в соответствие с оптимальной температурой для экзотермической реакции образования углерод-углеродных связей (например, синтез Фишера-Тропша) или, в более общем смысле, реакции утилизации синтез-газа (например, синтез метанола или синтез простого диметилового эфира). Путем разумного выбора условий реакций и катализаторов для каждой реакции можно выполнять две реакции на оптимальных уровнях и при примерно одинаковой температуре (или при относительно близких температурах). Теплоту из экзотермической реакции образования углерод-углеродных связей затем используют для проведения эндотермической реакции газификации.
Таким образом, на Фиг.1 показаны результаты для рабочих характеристик различных нанесенных платиновых катализаторов для риформинга в газовой фазе при 623 K и атмосферном давлении при использовании сырьевого раствора, включающего 30% масс. глицерина в воде. Катализатор, включающий Pt, нанесенную на Al2O3, ZrO2, CeO2/ZrO2 или MgO/ZrO2, демонстрирует относительную быструю дезактивацию в течение рабочего цикла. Таким образом, хотя эти катализаторы можно использовать в настоящем изобретении, они не являются предпочтительными. Pt/C катализатор показывает стабильную конверсию глицерина в синтез-газ в течение по меньшей мере 72 часов. См. Фиг.1А. Описанный здесь катализатор с наиболее кислотным носителем, Pt/Al2O3, показывает период стабильной каталитической активности в течение первых 30 часов, за которым следует период быстрой дезактивации катализатора. Это поведение является характеристикой фронта дезактивации, который движется от входного отверстия реактора к выходному отверстию, и наступление быстрой дезактивации возникает, когда фронт достигает выхода реактора. Описанный здесь катализатор с наиболее основным носителем, MgO/ZrO2, показывает быструю дезактивацию для всего рабочего цикла. Наиболее стабильным из испытанных катализаторов на оксидном носителе, как видно, является Pt на СеО2/ZrO2. Однако, рабочая характеристика этого катализатора еще менее предпочтительна, чем у платины, нанесенной на углерод.
На Фиг.1В показана производительность по H2 на различных катализаторах в оценках частоты оборота (ЧО), где производительность нормировали к числу поверхностных атомов платины, которое определяли путем предельной адсорбции CO при 298 K. Частота оборота для получения водорода, определенная таким образом, выше (например, по меньшей мере в 10 раз), чем величины для получения водорода путем риформинга в водной фазе при 500 K с платиновым катализатором, нанесенным на оксид алюминия (см., например, 19). Производство H2 из глицерина с Pt/C катализатором соответствует приблизительно производству энергии 160 Ватт на грамм катализатора (используя более низкую теплоту сгорания H2). Так как этот катализатор производит газ с соотношением H2:CO приблизительно 1,3, содержание энергии в выходящем газе соответствует приблизительно 300 Ватт на грамм катализатора.
Различные профили дезактивации, показанные на Фиг.1А и 1В для различных испытываемых катализаторов предполагают, что носитель играет важную роль в процессе дезактивации. Соотношение H2:CO для потока продукта из Pt/C катализатора равно приблизительно 1,3 (см. Табл.1), что соответствует стехиометрии реакции 3. Напротив, соотношение H2:CO, полученное с другими катализаторами, было выше 1,5, что показывает вклад других реакций, таких как реакция сдвига в водяном газе (СВГ). Это поведение более ясно демонстрируется посредством соотношения CO:CO2, как показано на Фиг.1С. Начальное соотношение CO:CO2 для Pt/C составляет 12, в то время как оно меньше 3 для других катализаторов. Таким образом, видно, что реакции СВГ способствует присутствие оксидного носителя, как описано в других исследованиях реакции СВГ с нанесенными металлическими катализаторами (22-24). Также видно из Фиг.1С, что скорость СВГ с Pt/ZrO2 катализатором уменьшается со временем рабочего цикла.
На Фиг.1D показана производительность по C2 углеводородам (этана и этилена), нормированная к производительности по H2 для различных нанесенных платиновых катализаторов (где это соотношение умножают на 100). Только небольшие количества C2 углеводородов были образованы на Pt/C катализаторе. Напротив, катализаторы, включающие платину, нанесенную на различные оксидные носители, образуют значительные количества C2 углеводородов и соотношение (C2-ЧО):(H2-ЧО), по-видимому, возрастает со временем рабочего цикла. Это поведение позволяет предположить, что один из видов дезактивации катализатора вызывается процессами дегидратации, возникающими на оксидных каталитических носителях. Вне связи с любым специфическим лежащим в основе механизмом, это предположительно приводит к образованию ненасыщенных углеводородных молекул, которые образуют углеродсодержащие отложения на поверхности платины, тем самым уменьшая производительность по H2 и увеличивая соотношение (C2-ЧО):(H2-ЧО).
В Табл.1 показаны рабочие характеристики Pt/C катализатора при различных условиях реакции. Условия, приводящие к более низкой конверсии глицерина (то есть, к более высоким скоростям потока 30% масс. глицеринового сырья и более высоким концентрациям глицерина при постоянной скорости потока сырья), приводят к более высоким соотношениям CO:CO2. Это поведение позволяет предположить, что первичная реакция в конверсии глицерина при использовании этой каталитической системы представляет собой образование CO и H2, и выработка CO2 посредством СВГ является вторичной реакцией. Производительность по метану остается низкой для всех условий в Табл.1.

Для изучения кинетики реакций в Табл.1 использовали 0,060 г 5% масс.
Pt/C.
а Концентрация подаваемого глицерина 30% масс. 623 K и 0,1 МПа (1 бар).
б Скорость потока сырья 0,32 см3/мин, 623 K и 0,1 МПа (1 бар).
в Точка, взятая после 2 часов рабочего цикла.
г Концентрация подаваемого глицерина 30% масс. при 0,32 см3/мин и 0,1 МПа (1 бар).
д Точка, взятая после 3 часов рабочего цикла.
Результаты Табл.1 показывают, что производительность по H2 проходит через максимум по отношению к температуре реакции при постоянных условиях подачи сырья. Производительность возрастает с ростом температуры от 573 до 623 K, что соответствует энергии активации примерно 75 кДж/моль. Напротив, хотя производительность по водороду дополнительно возрастает, когда температура изначально превышает 673 K, катализатор Pt/C начинает подвергаться дезактивации в зависимости от времени рабочего цикла при этой высокой температуре. Вероятно, что процессы дегидратации являются слишком быстрыми в сравнении с реакциями образования H2 при более высоких температурах, что приводит к дезактивации катализатора.
На Фиг.2A и 2B показаны рабочие характеристики катализатора Pt/C при возрастающем давлении (от 0,1 до 2 МПа (от 1 до 20 бар)) для 30% масс. глицеринового сырья и для возрастающей концентрации глицеринового сырья (от 30 до 50% масс.) при давлении 0,1 МПа (1 бар). Катализатор показывал хорошую стабильность в течение по меньшей мере 48 часов рабочего цикла как для более высоких концентраций глицеринового сырья (50% масс), так и для более высокого давления реакции (2 МПа (20 бар)). Следует особенно отметить, что катализатор Pt/C показывает превосходную стабильность для конверсии 30% масс. глицеринового сырья для получения синтез-газа при 2 МПа (20 бар) с соотношением H2:CO (равным примерно 2), что подходит для последующего синтеза Фишера-Тропша [2] или синтеза метанола [3].
Для того, чтобы достичь эффективной передачи тепла из экзотермических стадий Фишера-Тропша или синтеза метанола (или, вообще, стадии утилизации синтез-газа) в эндотермическую стадию газификации, предпочтительно проводить стадию газификации при наименьшей возможной температуре. По этой причине исследования газификации глицерина, описанные в Примерах, выполняли при температурах 548 и 573 K, где скорость является низкой из-за загрязнения поверхности сильно адсорбированным CO. Для того чтобы достичь высокой конверсии глицерина при этих низких температурах, выбирали биметаллический катализатор, включающий Pt:Ru (с атомным соотношением Pt:Ru 1:1), наряду с биметаллическим катализатором, включающим Pt:Re (с атомным соотношением Pt:Re 1:1), так как присутствие Ru и Re уменьшает силу адсорбции CO [25]. На Фиг.2C и 2D показано, что степень конверсии глицерина, так же как и соотношения CO:CO2 и H2:CO, остаются постоянными в течение по меньшей мере 72 часов рабочего цикла при этих низких температурах с Pt:Ru/C и Pt:Re/C катализаторами. Общий баланс углерода для этих процессов находится в пределах 5%, и основное конденсируемое органическое вещество в выходящем потоке представляет собой непреобразованный глицерин (73 % мол. и 35% мол. при 548 K и 573 K, соответственно), с малыми количествами метанола (4% мол. и 15% мол. при 548 K и 573 K, соответственно) и уксусной кислоты (20% мол. и 40% мол. при 548 K и 573 K, соответственно).
Таким образом, в настоящем изобретении теплоту реакции, высвобождаемая при реакции образования углерод-углеродных связей или, в общем, из экзотермической реакции утилизации синтез-газа (и, предпочтительно, реакции Фишера-Тропша) направляют обратно для получения (по меньшей мере частично) энергии, требуемой для реакции газификации биомассы вообще (и, предпочтительно, реакции газификации глицерина). Особенным преимуществом является то, что две реакции можно осуществлять в одном реакторе, таком, как показан на Фиг.3A и Фиг.3B.
Эти результаты показывают, что газификацию биомассы вообще, и глицерина в частности, фактически можно осуществлять при температуре, заведомо находящейся в пределах диапазона температур, действующих для синтеза Фишера-Тропша и синтеза метанола [2, 3, 26], обеспечивая возможность эффективной интеграции теплоты между этими процессами. Более того, соотношение H2/CO можно отрегулировать путем добавления второго слоя катализатора, который является эффективным для СВГ, как показано в Примерах. Заметим, что производительность по H2 при 573 K с Pt:Ru/C (Фиг.2C и 2D) ниже, чем производительность с Pt/C (Табл.1) из-за более высокой конверсии глицерина и более высокого парциального давления CO, достигнутого с Pt:Ru/C катализатором.
Данное изобретение является, таким образом, низкотемпературным каталитическим способом газификации глицерина с получением газовой смеси H2:CO, которая подходит для последующего синтеза Фишера-Тропша и синтеза метанола (или, вообще, стадий экзотермической утилизации синтез-газа). Продукты реакции газификации можно использовать непосредственно в реакции образования углерод-углеродных связей, без какой-либо промежуточной обработки. (Конечно, это обязательно является случаем, когда две реакции выполняют в одном реакторе, а не в двух отдельных реакторах). Данное изобретение открывает новые возможности для объединения теплоты между стадиями утилизации синтез-газа и газификации. Эта система достигает стабильной работы в течение больших промежутков времени и предоставляет новое направление производства топлива и химических веществ из возобновляемых источников.
Для демонстрации рабочих характеристик при высокой температуре Pt-Re катализатора, нанесенного на углерод, сырьевой раствор, содержащий 30% масс. глицерина в воде, преобразовывали в синтез-газ при 548 K и 0,83 МПа (8,3 бар) с 10% масс. Pt-Re (атомное соотношение 1:1)/С. После 60 часов, в течение которых степень конверсии глицерина в газофазные продукты уменьшалась от 68% до 57%, катализатор показывал превосходную стабильность в течение дополнительных 60 часов рабочего цикла (см. Примеры и Фиг.6). Газофазный выходящий продукт включает синтез-газ с соотношением H2:CO, равным 1:6, которое можно отрегулировать посредством реакции сдвига в водяном газе так, чтобы достичь соотношения 2:1, подходящего для синтеза Фишера-Тропша [37]. Остальная часть газообразных продуктов включает CO2 (молярное соотношение CO:CO2 примерно 6) и легкие алканы (C1-C3 с углеродным соотношением CO: алканы примерно 10). При 548 K и 0,5 МПа (5 бар) распределение газофазного продукта и каталитическая стабильность были аналогичными, и степень конверсии в газофазные продукты составляла примерно 80%. Оставшийся глицерин преобразовывали в жидкие продукты, такие как метанол, этанол, н-пропанол, этиленгликоль, 1,2-пропандиол, ацетон и ацетол, которые все можно преобразовать в газовые смеси H2/CO, содержащие небольшое количество алканов [17]. Углеродный баланс находился в пределах примерно 10%. См. Примеры для описания распределения жидкофазного продукта.
Для того чтобы объединить конверсию глицерина в синтез-газ с синтезом Фишера-Тропша в реакторе с двумя слоями, требуется воздействие на катализатор Фишера-Тропша, находящийся ниже по потоку, водяным паром из водного раствора глицеринового сырья. Начальные эксперименты, использующие 30% масс. глицериновое сырье в двухслойной системе, включающей Pt-Re/C катализатор, за которым следует катализатор Фишера-Тропша на основе Co, показывают относительно низкую активность образования жидких алканов. Iglesia et al. сообщают, что небольшое количество воды может улучшить рабочую характеристику катализаторов Фишера-Тропша на основе Co [38]. Однако, наибольшее парциальное давление воды в исследовании Iglesia et al. (PH2O:PCO=3) было ниже того, которое получают при конверсии 30% масс. глицеринового сырья (PH20:PCO=8). Также исследования Iglesia et al. [38] были проведены при более высоком общем давлении (2 МПа (20 бар)). Следовательно, в настоящем изобретении преимущество составляет уменьшение отношения воды к CO. Таким образом, Pt-Re/C катализатор испытывали с 50% масс. и 80% масс. растворами глицерина от 0,1 до 1,1 МПа (от 1 до 11 бар). В Табл.2 показана степень конверсии в газофазные продукты, также как и молярные соотношения H2:CO и CO:CO2.

Степень конверсии в газофазные продукты возрастает с уменьшением концентрации глицерина в подаваемом сырье при постоянном давлении и уменьшается с увеличением давления при постоянной концентрации подаваемого сырья. Активность сдвига в водяном газе возрастает при более высоких давлениях и/или более низких концентрациях подаваемого сырья из-за увеличенного парциального давления воды, что доказано путем уменьшения соотношения CO:CO2. Эти эксперименты были выполнены при 548 K и давлениях выше точки росы для 50% масс. и 80% масс. глицерина в растворах подаваемого сырья. Однако каждое исследованное условие показывало стабильную работу для примерно 20 часов рабочего цикла и была только 6%-ная потеря активности после работы при 1,1 МПа (11 бар) с 80% масс. глицеринового сырья. Жидкая фаза содержит кислородсодержащие углеводородные продукты, похожие на те, что получаются при конверсии 30% масс. глицеринового сырья. Распределение жидкого продукта для каждого условия в Табл.2 представлено в Примерах.
Распределения продуктов для конверсии водных растворов глицерина при различных давлениях соответствуют схеме реакции, предложенной Cortright et al. [17] для риформинга полиолов в водной фазе. Схема реакции включает адсорбцию - дегидратацию глицерина, расщепление C-C связи и десорбцию CO и H2. Сдвиг в водяном газе для адсорбированного CO приводит к получению CO2 и расщеплению С-О в отличие от C-C связей, что приводит к образованию алканов и спиртов [17]. Продукты жидкой фазы можно конденсировать из синтез-газа и затем направлять обратно для дальнейшей конверсии в газообразные продукты.
Для достижения объединения энергии от эндотермической конверсии глицерина в синтез-газ и экзотермической конверсии синтез-газа в жидкие алканы, температура стадии синтеза Фишера-Тропша должна быть сравнима (или выше) с применяемой в стадии конверсии глицерина. Также давления в обеих проводимых реакциях должны быть похожими для минимизации компрессионных расходов. (То есть, реакции можно выполнять при различных давлениях, однако энергия, требуемая для сжатия газов для реакции с большим давлением, будет уменьшать энергетический кпд всего способа). Более того, когда синтез-газ из стадии конверсии глицерина поставляют непосредственно на катализатор Фишера-Тропша, данный катализатор будет подвергаться воздействию воды и кислородсодержащих углеводородных побочных продуктов. Вследствие этого были проведены серии экспериментов по синтезу Фишера-Тропша при 548 K и 0,5 МПа (5 бар) с 4 г 2,9% масс. Ru/TiO2 катализатора с 150 см3/мин сухого синтез-газа (H2:CO=2) и с совместной подачей воды или водных растворов ацетола, этанола или ацетона (наиболее обильные продукты жидкой фазы из конверсии глицерина) для моделирования условий реакторной системы с двумя слоями, которая обрабатывает 80% масс. глицериновое сырье при 0,5 МПа (5 бар). Катализатор Фишера-Тропша на основе рутения использовали из-за того, что катализатор на основе кобальта показал низкую активность в течение начальных экспериментов. В Табл.3 приведены степени конверсии CO и селективность по СН4, CO2 и C5+ углеводородам для этих экспериментов по синтезу Фишера-Тропша.

Степень конверсии CO составляет примерно 50% для синтеза Фишера-Тропша с сухим синтез-газом. Добавка воды в» подачу синтез-газа увеличивает селективность по CO2, наиболее вероятно путем увеличения скорости сдвига в водяном газе, однако степень конверсии CO и активность (определено как выход на активный центр в единицу времени) остаются такими же, как в эксперименте с сухим синтез-газом. Как степень конверсии, так и активность слегка уменьшаются с добавлением кислородсодержащих углеводородов в синтез-газ. Возможно, что адсорбированные части этих молекул подавляют реакцию Фишера-Тропша путем блокирования активных центров рутения для адсорбции CO и H2. Это уменьшение активности может быть причиной более низкой селективности по CO2 при совместной подаче кислородсодержащих углеводородов по сравнению с совместной подачей воды. Селективность по С5+ углеводородам слегка возрастает с добавлением молекул кислородсодержащего сырья по сравнению с подачей сухого синтез-газа, в то время, как селективность по метану и CO2 остается неизменной. Единственным исключением является то, что селективность по метану уменьшается более чем в два (2) раза с добавлением ацетола. Ацетол легко реагировал после добавления в реактор Фишера-Тропша. Весь подаваемый ацетол давал продукты, при том, что 30% из них превращались в ацетон, метанол и этанол в водной фазе продукта и 20% превращались в кислородсодержащие вещества в органической фазе продукта (в основном, гексаноны). Другие 10% ацетолового сырья превращались в газообразный ацетон. Следовательно, примерно 40% углерода, подаваемого в реактор в виде ацетола, поступало в рост цепи Фишера-Тропша и превращалось в жидкие углеводороды. Эти результаты показывают, что вода и кислородсодержащие углеводороды в синтез-газе, получаемые в процессе конверсии глицерина, мало влияют на селективность катализатора Фишера-Тропша. В случае ацетола, этот кислородсодержащий углеводород, по-видимому, обладает синергетическим действием при добавлении к цепочке роста углеводорода. Следовательно, описываемая здесь работа показывает объединение (1) конверсии кислородсодержащего углеводорода в синтез-газ с (2) синтезом Фишера-Тропша в двухслойной реакторной системе. Реакции будут давать требуемые продукты Фишера-Тропша без необходимости конденсировать воду и жидкие побочные продукты между двумя слоями катализатора. (И, как замечено выше, обе реакции можно выполнять в одном смешанном слое катализатора). Продукты реакции конверсии кислородсодержащего углеводорода в синтез-газ можно подавать непосредственно в реакцию Фишера-Тропша без каких-либо промежуточных стадий обработки. (Подробное описание распределения углерода для реакций Фишера-Тропша представлено в Примерах).
Активность и селективность катализаторов Фишера-Тропша могут находиться под влиянием транспортных ограничений внутри гранул катализатора [2, 38, 39]. Увеличение плотности рутениевых активных центров или радиуса гранул приводит к увеличению селективности по С5+, вызванной усиленной диффузией повторной адсорбцией а - олефинов, которая подавляет обрыв цепи [2]. Однако, эти диффузионные ограничения могут стать настолько жесткими, что они будут подавлять диффузию CO внутри гранулы, что приведет к уменьшению селективности по С5+ [2]. Iglesia et al. [2] сообщают о структурном параметре (Χ), зависящем от радиуса гранулы катализатора, распределения пор по размерам и объемной плотности поверхностных атомов рутения, который показывает степень этих диффузионных ограничений внутри катализатора (2). Для 2,9% масс. Ru/TiO2 катализатора, используемого в этих экспериментах, Χ составлял 5×1017 м-1. Это значение находится в соответствии со значением, определенным Iglesia et al. для Ru катализатора, нанесенного на TO2 (39) и лежит в промежуточном диапазоне, предполагающем, что транспортные ограничения способствуют повторной адсорбции α-олефинов, но не замедляют диффузию реагентов внутрь гранул катализатора. Действительно, катализаторы с промежуточными значениями Χ приводят к оптимальной селективности по С5+.
Более того, это явление повторной адсорбции приводит к отклонению от кинетики Андерсона-Шульца-Флори (АШФ) роста цепи. По мере того, как углеводородная цепь увеличивается в длине, диффузия через поры катализатора становится более трудной и возможность повторной адсорбции возрастает. Этот эффект увеличивает вероятность роста цепи для более длинных углеводородных цепей и приводит к кривизне на графике молекулярно-массового распределения в полулогарифмических координатах. На Фиг.5 показаны такие графики для пяти процессов Фишера-Тропша по Табл.3, и эти распределения отклоняются от кинетики АШФ, что согласуется с исследованиями Iglesia et al. [2, 38, 39]. Отношения олефинов к парафинам были относительно низкими, что соответствует длительным временам пребывания в слое (6-9 с).
Для того, чтобы показать образование жидкого топлива при объединении конверсии глицерина с синтезом Фишера-Тропша, исследовали двухслойную каталитическую систему, используя 1,0 г 10% масс. Pt-Re (1:1)/С и 1,7-3,0 г 1,0 % масс. Ru/TiO2 с 80% масс. глицеринового сырья при 548 K и 0,5 МПа (5 бар) общего давления. В Табл.4 показана селективность по C5+ углеводородам, CO, CO2 и СН4 наряду со скоростями выхода углерода в газовой, водной жидкой и органической жидкой фазах. Исходя из выработки CO из 80% масс. глицеринового сырья при 548 K и 0,5 МПа (5 бар), степень конверсии CO на слое Ru/TO2 составляла приблизительно 30% для процесса с 1,7 г, и 40% для процесса с 3,0 г, с выходами на активный центр в единицу времени 1,3 и 1,1 мин-1 соответственно. Высокая селективность по CO следует из того факта, что весь глицерин преобразовывали с Pt-Re/С катализатором, в то время, как 30-40% CO взаимодействовало на катализаторе Фишера-Тропша. Важно, что эта система дает высокую селективность по C5+ углеводородам, в сравнении с СН4 (SC5+:SCH4>1) - Значение Χ для 1,0% масс. Ru/TiO2 катализатора составляло 36×1016 м-1, что согласуется с результатами Iglesia et al. [39], и молекулярно-массовые распределения углеводородов показывали отклонение от АШФ кинетики (см. Примеры и Фиг.7 для дополнительных данных), показывая эффекты повторной адсорбции α-олефинов. Важно, что эти эксперименты показывают, что жидкие алканы можно преобразовать непосредственно из кислородсодержащих углеводородов в двухслойной реакторной системе, используя объединенные способы, при отсутствии какой-либо обработки между конверсией в синтез-газ и реакцией Фишера-Тропша. Подробное описание распределений углерода можно найти в Примерах.

Получение синтез-газа из кислородсодержащих углеводородов, таких как глицерин, в сочетании с конверсией синтез-газа для получения жидкого топлива путем синтеза Фишера-Тропша, является по сути экзотермическим способом с теплотой, которая составляет примерно 4% от наиболее низкой теплоты сгорания глицерина [37]. Представленные здесь данные показывают, что в начале обе эти стадии можно выполнять эффективно при одних и тех же условиях и в двухслойной реакторной системе, допускающей объединение конверсии кислородсодержащего углеводорода и синтеза Фишера-Тропша для использования в получении жидкого топлива из водных растворов окисленного углеводородного сырья.
Как было замечено ранее, глицерин можно преобразовывать в синтез-газ при высокой производительности и селективности при температурах, менее примерно 625 K, согласно Уравнению 3.

Этот глицерин можно получить из ферментации глюкозы, из гидролиза сорбитола или как отходы переэтерификации растительных масел и животных жиров. В настоящем изобретении эту реакцию выполняют при низкой температуре для того, чтобы дать возможность соединить эндотермическую реакцию конверсии глицерина с экзотермическим синтезом Фишера-Тропша для получения жидкого транспортного топлива согласно реакции 4:

Этот объединенный способ улучшает экономику «зеленого» синтеза Фишера-Тропша путем уменьшения стоимости, связанной с получением синтез-газа. В особенности, при использовании настоящего изобретения уменьшают капитальные затраты посредством устранения необходимости в автотермическом преобразователе, продуваемом кислородом, или в газификаторе биомассы. Также настоящее изобретение обеспечивает уменьшение размера реактора синтеза Фишера-Тропша путем получения потока неразбавленного синтез-газа и путем устранения стадий последующей очистки, требуемых при получении синтез-газа из газификации биомассы. Таким образом, настоящее изобретение позволяет включать небольшие установки для синтеза Фишера-Тропша в установку для переработки биологического сырья или в установку по переработке технологических отходов глицерина в биодизельное топливо. И, как отмечено выше, низкая температура способа конверсии делает возможным тепловое взаимодействие с реакцией синтеза Фишера-Тропша, таким образом увеличивая тепловой кпд.
Дополнительно, соединение этих реакций приводит к химическому синергизму, связанному с присутствием химических веществ от обеих реакций в одном реакторе. Например, промежуточные продукты, получаемые из конверсии глицерина (например, ацетол) могут вступать в рост углеводородной цепи на активных центрах катализатора Фишера-Тропша, и ингибирующее действие парциального давления CO на степень конверсии глицерина можно уменьшить посредством потребления CO на активных центрах Фишера-Тропша.
Для того чтобы продемонстрировать, как данное изобретение можно выполнять в одном реакторе, используя один каталитический слой, исследования конверсии глицерина выполняли, используя Pt-Re/C катализатор, который изготавливали путем пропитки начальным увлажнением сажи (Vulcan ХС-72) водным раствором H2PtCl6·6H2O (Sigma-Aldrich) и HReO4 (Strem Chemicals) для получения катализатора с содержанием 5,1% масс. Pt и 4,9 % масс. Re (атомное отношение Pt:Re 1:1). Носитель сушили на воздухе в течение 12 часов при 373 K перед пропиткой, и использовали 1,7 г раствора на грамм носителя. Катализатор сушили при 403 K в течение 12 часов на воздухе перед активацией. 1,0% масс. Ru/TiO2 катализатора синтеза Фишера-Тропша изготавливали согласно способам, описанным Iglesia et al. [40].
До измерений кинетики реакций или адсорбции газа (то есть, хемосорбции CO и O2) Pt-Re/C катализатор восстанавливали при 723 K (скорость изменения температуры 0,5 K мин-1) в течение 2 часов в потоке H2 (140 см3 (при нормальной температуре)/мин), Ru/TiO2 катализатор восстанавливали in-situ перед изучением кинетики реакций и измерениями адсорбции газа. Массовую часовую объемную скорость (МЧОС) для экспериментов по конверсии глицерина рассчитывали, используя массовый расход глицерина в реакторе и общую массу катализатора. Необратимое поглощение CO катализатором Pt-Re/C при 300 K было принято за количество каталитических активных центров (150 µмоль/г) и было измерено с использованием обычной установки для газовой адсорбции. Это число активных центров соответствует дисперсии (молярное отношение CO: общее количество металла), равной 29%. Дисперсия (CO:Ru) Ru/TiO2 катализатора была равна 0,55, как определили путем хемосорбции О2 при 195 K в статической хемосорбционной системе [41].
Установка, используемая для проведения комбинированных экспериментов по конверсии глицерина и синтезу Фишера-Тропша, показана на Фиг.3. Свежий катализатор загружали в трубчатый реактор из нержавеющей стали с внешним диаметром 12,7 мм (0,5 дюйма) и толщиной стенки 0,71 мм (0,028 дюйма). Катализаторный слой был заключен между концевой заглушкой из кварцевой ваты (Alltech) и плавлеными гранулами SiO2 (-4+16 меш; Sigma-Aldrich), которые способствовали испарению жидкого сырья. Для экспериментов, в которых объединяли конверсию глицерина с синтезом Фишера-Тропша в двухслойной системе с одним реактором, слой 1,0% масс. Ru/TiO2 смешивали с равным объемом дробленых гранул SiO2 и этот слой загружали вниз по потоку по отношению к 10% масс. Pt-Re(1:1)/C слою, смешанному с плавлеными гранулами SiO2. Для экспериментов, в которых объединяли конверсию глицерина с синтезом Фишера-Тропша в однослойной системе, катализаторный слой изготавливали путем частичного перемешивания Pt-Re/C с Ru/TiO2, то есть 0,8 г 10% масс. Pt-Re(1:1)/C смешивали с 1,7 г 1,0% масс. Ru/TiO2 и загружали под 0,2 г Pt-Re/C катализатора. Реактор нагревали печью, состоящей из тесно подогнанных алюминиевых блоков, нагреваемых извне с помощью хорошо изолированной печи (1450 Вт/115 В, Applied Test Systems series 3210). Термопары K-типа (Omega) были присоединены к внешней стороне реактора для измерения температуры реактора, которую регулировали серией температурных контроллеров типа 16А (Dwyer Instruments). Свежий катализатор восстанавливали в потоке Нг, как описано ранее. Контроллеры массового расхода (5850 Brooks Instruments) были использованы для регулирования скорости потока H2. Насос HPLC (высокоэффективной жидкостной хроматографии, ВЭЖХ) (Model 301, Alltech) использовали для введения водного сырьевого раствора в 6-ти дюймовую (15 см) иглу с наконечником «point 5-style» (Hamilton), припаянной в секцию трубки из нержавеющей стали с внешним диаметром 3,2 мм (0,125 дюйма), и эта игла была расположена выше по потоку от каталитического слоя. Выходящую жидкость конденсировали в газожидкостном сепараторе и периодически сливали для газового хроматографического (ГХ) анализа (Agilent 6890 с детектором ионизации пламени (ДИП) и колонкой HP-lnnowax или Shimadzu GC-2010 с детектором ДИП и колонкой Rtx-5) и общего органического углеродного анализа (Shimadzu ТОС - V CSH). Каждый выходящий продукт исследовали на присутствие глицерина и других жидких побочных продуктов. Трубопроводы системы ниже по потоку от печи нагревали при 373 K для того, чтобы препятствовать затвердеванию более тяжелых алканов до сепаратора.
Поток выходящего газа проходил через регулятор обратного давления (GO Regulator, Model ВР-60), который регулировал давление системы. Выходящий газ анализировали посредством газовой хроматографии: H2 при помощи газового хроматографа Carle (серия 8700), используя детектор теплопроводности (ДТП), CO и СН4, используя HP 5890 газовый хроматограф с ДТП и колонкой с отмытым молекулярным ситом 5А 80/100 (Alltech), и CO2 и легкие алканы (C2-C3), используя газовый хроматограф HP 5890 с ДТП и колонкой Porapak QS 100/120 (Alltech). Газообразные алканы (C1-С10) измеряли, используя газовый хроматограф/масс-спектрометр Varian Saturn 3 с ДИП и капиллярной колонкой GS-Q (J&W Scientific). Все сырьевые растворы изготавливали путем смешивания глицерина (99,5%, ACS реагент, Sigma Aldrich) с деионизированной водой.
Таким образом, настоящее изобретение можно выполнять в компоновке с одним реактором и одним слоем (как показано на Фиг.3A) или в компоновке с одним реактором и многими слоями (как показано на Фиг.3B), или в компоновке с двумя реакторами (как показано на Фиг.3C). На каждом из Фиг.3A, 3B и 3C реактор 14 имеет входное отверстие 10 для введения реагентов и выходное отверстие 12 для выпуска продуктов. Внутри каждого реактора присутствует один или более каталитических слоев 16.
В этих экспериментах применяли либо систему с двумя слоями катализатора, используя 1,0 г 10% масс. Pt-Re(1:1)/C, за которым следовал 1,7 г 1,0% масс. Ru/TiO2, или один слой катализатора, состоящий из частичной физической смеси двух катализаторов (как описано ранее) с подачей 80% масс. глицерина при 548 K и общем давлении от 0,5 до 1,7 МПа (от 5 до 17 бар). В Табл.5А показана селективность по С5+, СН4 и C2-С4 алканам для каждого из объединенных экспериментов, исходя исключительно из алкановых продуктов (то есть, C1-C5+). Селективность для получения C5+ алканов путем синтеза Фишера-Тропша обычно увеличивается при более высоких давлениях, и результаты для двухслойной реакторной системы следуют этой тенденции. Увеличение давления от 0,5 до 1,1 МПа (от 5 до 11 бар) приводит к увеличению селективности по C5+ углеводородам от 0,46 до 0,59, однако, дополнительное увеличение давления до 1,7 МПа (17 бар) приводит только к небольшому увеличению селективности по C5+ до 0,57. Важно, что селективность по C5+ углеводородам почти в три раза превышает селективность по CH4 при 1,1 и 1,7 МПа (11 бар и 17 бар), по сравнению с 0,5 МПа (5 бар). В Табл.5B показана общая селективность по углероду, исходя из общего количества углерода во всех продуктах.
При 0,5 МПа (5 бар) первичный продукт представлял собой CO из конверсии глицерина, и только 32% углерода пошло на алканы. Однако, увеличение давления до 1,1 и 1,7 МПа (11 бар и 17 бар) смещает распределение углерода к С1-C5+ алканам (то есть, Sалканы (селективность по алканам) возрастает до 42% и 51% при 1,1 и 1,7 МПа (11 бар и 17 бар), соответственно). Также, количество углерода в виде кислородсодержащих соединений в органическом жидком выходящем потоке (C3-С7 кетоны) увеличивается более чем в 5 раз с увеличением давления. Процентное содержание углерода в органических жидких продуктах (С5+ и органические кислородсодержащие соединения) составляло 43% при 1,7 МПа (17 бар), 35% при 1,1 МПа (11 бар) и 15% при 0,5 МПа (5 бар), при процентном содержании углерода в газообразных продуктах (CO, CO2 и C1-С10 алканы), уменьшающемся от 71% при 0,5 МПа (5 бар) до примерно 50% при 1,1 и 1,7 МПа (11 бар и 17 бар). При 0,5 МПа (5 бар) и 1,1 МПа (11 бар) 14% углерода содержится в виде кислородсодержащих веществ в водном выходящем потоке и при 1,7 МПа (17 бар) это значение слегка уменьшается до 10%. Эти водные жидкие выходящие потоки содержат от 5% масс. до 15% масс. метанола, этанола и ацетона и пригодны для дальнейшей дистилляции.
При 1,7 МПа (17 бар) количество углерода, покидающего реактор в виде CO уменьшается более чем на порядок и селективность по алканам увеличивается в сравнении с проведением реакции при 1,1 МПа (11 бар). Однако селективность по С5+ алканам слегка уменьшается. Это поведение обусловлено как увеличенной активностью сдвига в водяном газе (показанной посредством более высокой SCO2), так и увеличением скорости синтеза Фишера-Тропша при более высоких давлениях. Более высокая скорость синтеза Фишера-Тропша вызывает увеличение температуры слоя Ru/TiO2, что приводит к образованию более легких алканов (то есть, C1-С4). Поэтому распределение углерода смещается к получению более легких алканов (то есть увеличению Sалканы без соответствующего увеличения SC5+).
В Табл.6 показано процентное содержание углерода в каждой фазе продукта для объединенных экспериментов по конверсии глицерина и синтезу Фишера-Тропша в Табл.5A и 5B. Ясно, что процентное содержание выходящего углерода в жидком органическом потоке, увеличивается постепенно от 15% до 42,7% по мере того, как давление системы с двумя слоями катализатора возрастает от 0,5 МПа (5 бар) до 1,7 МПа (17 бар). Важно, что процентное содержание выходящего углерода в жидком органическом потоке возрастает до 50,7%, когда Pt-Re/C и Ru/TiO2 катализаторы частично смешаны при 1,7 МПа (17 бар). Эти результаты особенно важны, так как результаты для реакций в смешанном слое показывают, что настоящее изобретение можно использовать для получения жидкого топлива из биомассы при очень высоком выходе углерода.
Как замечено ранее, CO подавляет конверсию глицерина в синтез-газ. Наоборот, получение алканов путем гидрогенизации CO на Ru является реакцией положительного порядка по отношению к CO и высокому при высоких парциальных давлениях CO (то есть, >0,15 МПа (1,5 бар)). Более того, как установлено ранее, теплоту, получаемую из синтеза Фишера-Тропша, можно потреблять в реакции конверсии глицерина. Поэтому, когда активные центры катализа для двух реакций приходят в близкое соприкосновение, тогда Ru катализатор увеличивает активность Pt-Re катализатора при высоких давлениях путем потребления CO, который образуется на Pt-Re из глицерина. Дополнительно, реакция газификации глицерина потребляет тепло, получаемое путем гидрогенизации CO в реакции Фишера-Тропша, таким образом, сохраняя температуру в каталитическом слое при условиях, благоприятных для роста длинных цепей (то есть, при более низкой температуре в слое катализатора).
Таблицы 5A и 5B: Результаты экспериментов по объединению конверсии глицерина с синтезом Фишера-Тропша. A) Селективность по C5+, CH4 и C2-C4 в алкановых продуктах. Селективность вычисляли следующим образом: SCnHx=nFCnHx/Ftotal, где n является числом атомов углерода в алкановом продукте CnHx, FCnHx является молярной скоростью потока продукта CnHx и Ftotal является общей молярной скоростью потока углерода в алкановых продуктах. B) Общая селективность по углероду. Селективность вычисляли следующим образом: Si=Fi/Ftotal×100, где Fi является общей скоростью потока углерода в продукте i и Ftotal является общей скоростью потока углерода во всех продуктах. Реакции выполняли при 548 K, используя примерно 0,04 см3/мин подачи 80% масс. глицерина (МЧОС глицерина ≈0,86 час-1)



Данные в Табл.5A, 5B и 6 показывают, что конфигурация смешанного слоя (при использовании гомогенного катализатора или смешанного катализатора) является лучшей для выхода жидких алканов по отношению к конфигурации, включающей отделенные слои (хотя обе конфигурации включены в настоящую формулу изобретения). Смешивание слоев увеличивает селективность по алканам более чем на 10%, при этом потребляя большую часть CO, получаемого из глицерина. Высокая конверсия CO приводит к уменьшению селективности по С5+ для системы с отделенными слоями; однако, в эксперименте, использующем смешанные слои, получают алкановые продукты с более высокой S5+ (0,64) по сравнению с объединенными экспериментами. Также, количество углерода в органических жидких продуктах увеличивается от 35% для двухслойной системы при 1,1 МПа (11 бар) до 50% для смешанного слоя при 1,7 МПа (17 бар). Количество углерода в газообразных продуктах уменьшается от 50% до 43%, в то время, как количество углерода в водной фазе уменьшается в два раза (с 14% до 7%).
Селективность по С5+ , селективность по C3-С7 кетонам в органической жидкости и степень конверсии CO для объединенной конверсии глицерина с синтезом Фишера-Тропша при 1,1 МПа (11 бар) и 1,7 МПа (17 бар), являются более высокими, чем при 0,5 МПа (5 бар), несмотря на тот факт, что активность Pt-Re/C катализатора уменьшается при этих повышенных давлениях. Эти результаты показывают, что более благоприятные условия Фишера-Тропша (то есть, более высокое давление) являются более существенными в объединенном способе, чем производительность по синтез-газу. Более того, Ru/TiO2 катализатор подвергают воздействию увеличенного количества кислородсодержащих углеводородных побочных продуктов при 1,1 МПа (11 бар) и 1,7 МПа (17 бар). Однако, селективность по кислородсодержащим веществам в выходящей водной жидкости при этих давлениях такая же или меньше этой величины для выходящей водной жидкости при 0,5 МПа (5 бар). Эти результаты показывают, что кислородсодержащие углеводородные побочные продукты из глицерина реагируют со слоем Ru/TiO2, наиболее вероятно, вступая в рост цепи Фишера-Тропша. Так как распределение водных продуктов содержит широкий массив кислородсодержащих веществ, вероятно, что другие побочные продукты (например, полиолы, вторичные спирты и гидроксилкетоны) обладают похожим синергетическим влиянием на синтез Фишера-Тропша. Важно, что эти эксперименты показывают, что жидкие алканы можно получать непосредственно из глицерина, используя объединенный способ.
Конверсию глицерина и синтез Фишера-Тропша можно выполнять эффективно (и, возможно, с синергетическим эффектом) при одинаковых условиях, либо в реакторной системе с двумя слоями, состоящей из отдельных катализаторов конверсии глицерина и синтеза Фишера-Тропша, либо в системе с одним смешанным слоем, допускающей применение объединения конверсии глицерина и синтеза Фишера-Тропша для получения жидкого топлива из водно-глицериновых растворов. Оба подхода включены в настоящее изобретение. Этот «зеленый» способ представляет энергетически эффективную альтернативу получению жидкого транспортного топлива из нефти. Более того, он предоставляет возможность для улучшения экономической жизнеспособности «зеленого» синтеза Фишера-Тропша посредством снижения затрат, связанных с получением синтез-газа и путем улучшения теплового кпд процессов Фишера-Тропша.
В частности, низкая температура нашего способа конверсии глицерина допускает тепловое взаимодействие с реакцией синтеза Фишера-Тропша, таким образом увеличивая тепловой кпд. Более того, объединение этих способов приводит к химическому синергизму, связанному с присутствием в одном и том же реакторе химических веществ из обеих реакций, например, промежуточные продукты, полученные из конверсии глицерина, могут вступать в рост углеводородной цепи на активных центрах катализатора Фишера-Тропша, и ингибирующий эффект парциального давления CO на степень конверсии глицерина можно уменьшить путем потребления CO на активных центрах катализатора Фишера-Тропша.
Заметим, что предпочтительные катализаторы для использования в данном изобретении отмечены выше и в Примерах. Катализаторы, которые можно использовать в настоящем изобретении, включают, во первых, металлы, выбираемые из группы, состоящей из Ru, Co, Fe (FeC, Fe2O3, Fe3O4), Ni, Rh, Pt, Pd, Ir и их сочетания. Также катализаторы, которые можно использовать в данном изобретении, представляют собой оксиды любых перечисленных металлов и биметаллические сочетания вышеупомянутых металлов или их оксидов, так же как порошки вышеупомянутых металлов без носителя.
Когда катализаторы находятся на носителе, обычно предпочтительным носителем является углерод. Другие носители катализаторов можно также использовать в настоящем изобретении, такие, как цеолиты, полимерные носители и т.д. Носители катализаторов, которые можно использовать в настоящем изобретении, включают, без ограничения, TiO2 (предпочтительно 25%-100% анатаз), SiO2, Al2O3, MgO, ZrO2, ZrxTiyO2, ThO2, кизельгур, La2O3, MgCr2O4, TixSiyO2, TixZryO2, ZnO, Cr2O3, MnO, Nb2O5, CeO2, цеолит Y, цеолит USY, цеолит ZSM-5, цеолит MCM-41 цеолит МСМ-22, цеолит HZSM-5, цеолит Н-ВЕА, цеолит HY, Fe-замещенный цеолит LTL, деламинированный цеолит ITQ-6, деламинированный цеолит ITQ-2, молекулярные сита HMS, монтмориллонит, макропористый стирол-дивинилбензол, 4-винилпридин-дивинилбензол, антрацен, карбонизированный хинолин и их сочетания (включая смешанные оксиды, аэрогели и мезопористые формы отмеченных выше оксидов).
Промоторы, которые можно использовать в настоящем изобретении, включают, без ограничений, (с предпочтительными промоторами для каждого отдельного элемента в круглых скобках) Cu (CuO), K(K2CO3, K2O), Mn(MnO), La(La2O3), Ru, Re, Zn(ZnO), Si, Ag, Pt, Ce(CeO2), Gd2O3, ThO2, MnO, ZrO2, Pd, Ti, Co, Cr, V, Li, Na, Rb, Cs, Mo, Au, B, Cl и их сочетания.
Реакции можно выполнять в довольно большом диапазоне условий. Однако критические условия способа вообще попадают в следующие диапазоны:
Температура: ≤ примерно 750 K, с наиболее предпочтительной температурой от примерно 473 K до 625 K.
Общий диапазон давления: 0,1-3 МПа (1-30 бар).
Соотношение H2/CO в синтез-газе: 1-10.
Сырье, подаваемое совместно с синтез-газом, включает (без ограничений): воду, олефины, спирты, другие кислородсодержащие углеводородные молекулы и парафины.
ПРИМЕРЫ
Следующие примеры включены только лишь для обеспечения более полного описания изобретения, описанного и заявленного здесь. Данные Примеры не являются ограничивающими.
Пример 1 - Приготовление катализатора и определение его характеристик
Нанесенные Pt катализаторы изготавливали путем пропитки начальным увлажнением Al2O3 (Catapal B-brand от Sasol, Иоханнесбург, Южная Африка и Хьюстон, Техас), CeO2/ZrO2, MgO/ZrO2 и ZrO2, используя гексагидрат хлорплатиновой кислоты (Strem Chemicals, Ньюбэрипорт, Массачусетс) для ZrO2 и CeO2/ZrO2 и используя нитрат тетраамин платины (II) (Strem Chemicals) для Al2O3 и MgO/ZrO2. После пропитки Pt/ZrO2 и Pt/CeO2/ZrO2 катализаторы сушили на воздухе при 393 K в течение 15 часов и прокаливали при 773 K в течение 4 часов в муфельной печи. В течение прокаливания температуру увеличивали от комнатной температуры до 373 K и поддерживали в течение 1 часа, затем увеличивали с приращением 100 K до 773 K, поддерживая при каждом приращении в течение 1 часа. Нанесенные на MgO/ZrO2 и Al2O3 Pt катализаторы сушили на воздухе в течение 15 часов при 393 K и затем прокаливали в потоке O2/Не газовых смесей (20% для Pt/ MgO/ZrO2 и 10% для Pt/Al2O3, используя скорость потока 300 см3 (при нормальной температуре и давлении)/мин до 533 K (при 1,3 K в минуту) для Pt/Al2O3 и до 723 K (при 3,6 K в минуту) для Pt/MgO/ZrO2 и поддерживая эти температуры в течение 2 часов. Нанесенный на углерод платиновый катализатор и катализатор на основе Pt:Ru (атомное отношение 1:1) сплава приобретали в Е-ТЕК (подразделение PEMEAS Fuel Cell Technologies, Сомерсет, Нью Джерси). Нанесенный на углерод Pt:Re катализатор (атомное отношение 1:1) изготавливали посредством пропитки начальным увлажнением Pt/C (ЕТЕК), используя перрениевую кислоту (Strem Chemicals, Ньюбарипорт, Массачусетс). До измерений кинетики реакций или газовой адсорбции (то есть, хемосорбции CO, удельной поверхности по Брунауеру-Эмметту-Теллеру («БЭТ»)), каждый катализатор восстанавливали в потоке H2 (180 см3 (при нормальной температуре и давлении)/мин) при температуре 533 K (нанесенные на Al2O3, MgO/ZrO2 и углерод катализаторы; линейно изменяющейся на 0,5 K в минуту и поддерживаемой в течение 2 часов) или 773 K (нанесенные на CeO2/ZrO2 и ZrO2 катализаторы; скорость подъема 0,5 K в минуту и поддерживаемой в течение 1 часа). Pt:Ru/С катализатор восстанавливали в потоке H2 (140 см3 (при нормальной температуре и давлении)/мин) при температуре реакции (548 K или 573 K; скорость подъема 0,5 K в минуту), в то время, как Pt:Re/C восстанавливали при 723 K (скорость подъема 0,5 K в минуту) в течение 2 часов в потоке H2 (140 см3(при нормальной температуре и давлении)/мин). В Табл.7 показаны характеристики каждого катализатора. Необратимое поглощение CO и удельную поверхность по БЭТ измеряли на стандартной установке для газовой адсорбции, описанной в [28].
Изготовление носителей из ZrO2 и CeZrO2 детально описано в [29]. Вкратце, эти оксидные носители изготавливали путем совместного осаждения, начинающегося из водного раствора, содержащего 0,12 моля как нитрата цирконила (Sigma-Aldrich, Милуоки, Винсконсин), так и церий аммоний нитрата (Sigma-Aldrich) и используя избыток (Ме4+:ОН-=8) гидроксида аммония (28-30 % масс. NH3, Sigma-Aldrich) в качестве осаждающего агента. Осажденные вещества промывали в деионизованной (ДИ) воде и прокаливали при 773 K на воздухе в муфельной печи. Похожий способ изготовления MgO/ZrO2 описан в [30]. Носитель из MgO/ZrO2 изготавливали, используя водный раствор, содержащий 0,2 моля нитрата марганца (Sigma-Aldrich) и 0,02 моля нитрата цирконила (Sigma-Aldrich). Раствор перемешивали при комнатной температуре, добавляя при этом раствор NaOH (25% масс), до достижения pH 10 и образования соответствующего геля. Данный гель выдерживали в течение 72 часов и подвергали вакуумному фильтрованию. Образующийся осадок промывали ДИ водой до тех пор, пока концентрация натрия в фильтрате не становилась ниже 10 частей на миллион, что измеряли путем анализа с индукционно-связанной плазмой (ИСП). Образец затем сушили на воздухе при 393 K в течение 16-24 часов. Носитель MgO/ZrO2 прокаливали в потоке O2 (100 см3 (при нормальной температуре и давлении)/мин) до 873 K (3,2 K в минуту) и затем выдерживали при этой температуре в течение 3 часов.
В Табл.7 представлены свойства различных нанесенных металлических катализаторов:

Пример 2 - Измерения кинетики реакций
На Фиг.3 схематически показана установка, используемая для проведения измерений кинетики реакций. Установка, показанная на чертеже, включает трубчатый реактор, расположенный внутри печи. Плавленые гранулы из диоксида кремния и кварцевую вату используют для удерживания одного или более каталитических слоев внутри реактора. Поток, выходящий из трубчатого реактора, проходит через охлаждающую колонну, и газообразные продукты отделяют от жидких продуктов. Как газообразные, так и жидкие продукты затем удаляют из установки, показанной на Фиг.3, для дополнительного анализа. Более конкретно, свежий катализатор загружали в трубчатый реактор из нержавеющей стали с внешним диаметром 6,3 мм (0,25 дюйма). Для экспериментов при низкотемпературных условиях (то есть, 548 и 573 K с Pt:Ru/C и Pt:Re/C) использовали трубчатый реактор из нержавеющей стали с внешним диаметром 12,5 мм (0,5 дюйма). Для обоих типов трубчатых реакторов каталитический слой заключали между концевой заглушкой из кварцевой ваты (Alltech, Николасвилль, Кентукки) и плавлеными гранулами SiO2 (-4+16 меш, Sigma-Aldrich), которые способствуют испарению жидкого сырья. Порошки Pt/C, Pt:Ru/C и Pt:Re/C катализаторов смешивали с равными объемами дробленых гранул SiO2 перед загрузкой в реактор для уменьшения падения давления на слое катализатора. Термопары типа K (Omega Engineering, Стэмфорд, Коннектикут) были присоединены к внешней стороне реактора для измерения температуры реактора, которую регулировали серией температурных контроллеров типа 16А (Dwyer Instruments, Мичиган, Индиана). Свежий катализатор восстанавливали в потоке H2, как отмечено выше. Массовые расходомеры (Model 5850, Brooks Instrument, подразделение Emerson Process Management, Хатфилд, Пенсильвания) использовали для регулирования скорости потока H2. Насос ВЭЖХ (Model 301, Alltech) использовали для введения водного сырьевого раствора в блок впрыскивания жидкости выше реактора. Данный блок включает 6-ти дюймовую (15 см) иглу с наконечником «point 5-style» (Hamilton Company, Рено, Невада), припаянную в секцию 3,2 мм (0,125 дюйма) трубки из нержавеющей стали. Данная игла входит в реактор только выше плавленых гранул SiO2. Поток, выходящий из реактора, охлаждали водой в теплообменнике типа труба в трубе. Выходящую жидкость периодически сливали для газового хроматографического анализа (Aglient Model 6890 с детектором ионизации пламени (ДИП) и колонкой HP-Innowax или Shumadzu GC-20 10 с ДИП детектором и колонкой DB 5) и общего органического углеродного анализа (Shumadzu TOC-V CSH). Каждый выходящий продукт испытывали на наличие глицерина и других жидких побочных продуктов.
Поток выходящего газа проходил через регулятор обратного давления (GO Regulator, Спартанбург, Южная Каролина, Model ВР-60), с помощью которого регулировали давление системы. Выходящий газ анализировали при помощи трех различных газовых хроматографов: (1) H2 анализировали при помощи Carle GC (серия 8700), используя детектор теплопроводности (ДТП); (2) CO, СН4 и C2-углеводороды анализировали, используя НР-5890 GC с ДТП и колонкой с отмытым молекулярным ситом 5А 80/100 (Alltech); и (3) CO2 анализировали, используя Shumadzu GC-8A с ДТП и колонкой Haysep DB 100/120 (Alltech).
Все сырьевые растворы были приготовлены путем смешивания глицерина (99,5%, качество реагента Американского химического общества (ACS reagent grade), Sigma-Aldrich) с ДИ водой.
Пример 3 - Изменение соотношения H2:CO при использовании двухслойной каталитической системы
Была сформирована каталитическая система для получения синтез-газа с изменяющимися соотношениями H2:CO используя два слоя катализатора, первый из которых был слоем Pt/C для достижения 100% степени превращения глицерина с получением газовой смеси H2/CO, за которым следовал второй катализатор, который был эффективен для сдвига в водяном газе, такой, как 1,0% Pt/CeO2/ZrO2, содержащий окислительно-восстановительные активные центры для ускорения сдвига в водяном газе [31, 32]. Как показано на Фиг.4A и 4B эта система с двумя слоями катализатора достигает 100% степени конверсии глицерина, и соотношения H2:CO и CO:CO2 остаются стабильными в течение по меньшей мере 48 часов рабочего цикла. Величины 1,33 и 14 для соотношений H2:CO и CO:CO2, соответственно, для Pt/C катализатора показывают незначительный вклад реакции сдвига в водяном газе, однако, значительное увеличение соотношения H2:CO и соответствующее уменьшение соотношения CO:CO2 для системы с двумя слоями катализатора показывает, что Pt/CeO2/ZrO2 катализатор обеспечил эффективную конверсии при сдвиге в водяном газе.
Пример 4 - Объединенные реакции газификации и Фишер-Тропша в одном реакторе
Нанесенный на углерод платиново-рениевый катализатор был приготовлен так, что он содержал 5% масс. платины и атомное соотношение Pt/Re составляло 1:2,5. Этот катализатор приготавливали посредством начального смачивания водным раствором гексагидрата гексахлорплатиновой кислоты (IV) (39,85% Pt) (Alfa Aesar, дочернее предприятие, находящееся в полной собственности Johnson Matthey Company, Уорд Хилл, Массачусетс) и рениевой кислоты функционализированного перекисью водорода углерода UU 60×120 меш и сушкой при 100°C в вакууме.
9,64 г этого катализатора загружали в 12,5 мм (0,5 дюйма) реактор из нержавеющей стали и восстанавливали в потоке водорода перед реакцией. Реактор из нержавеющей стали нагревали, используя нагреватель из алюминиевых блоков для поддержания изотермических условий.
Раствор 70% масс. глицерина в воде подавали над катализатором при 260°C и 41,38 МПа избыт. (600 фунтов на кв. дюйм избыт.) при МЧОС 2,4, исходя из глицерина (2,4 грамма глицерина на грамм катализатора в час). При этих условиях реакции сырье оставалось в конденсированной форме поверх слоя катализатора.
При установленных условиях реакции превращалось 100% глицерина. 93% углерода собирали в продуктах газовой фазы. 1% углерода собирали в виде органического слоя, который анализировали посредством газового хроматографа - масс-спектрометра. Анализ этого органического слоя показывает присутствие от C9 до C20 углеводородов. См. Табл.7.
Хотя данные выходы являются низкими, этот Пример ясно показывает, что при реакции образуются углеводороды с длинной цепью. Присутствие этих углеводородов с длинной цепью показывает, что реакция Фишера-Тропша протекает внутри системы с одуим реактором.
Пример 5 - Приготовление и определение свойств катализатора
Pt-Re/C катализатор изготавливали путем пропитки методом начального смачивания сажи (Vulcan XC-72) водным раствором H2PtCl6·6H2O (Sigma-Aldrich) и HReO4 (Strem Chemicals), чтобы получить катализатор с номинальным содержанием 5,1% масс. Pt и 4,9% масс. Re (атомное соотношение 1:1). Носитель сушили в воздухе в течение 12 часов при 373 K до пропитки и 1,7 г раствора использовали на каждый грамм носителя. Катализатор сушили при 403 K в течение 12 часов на воздухе до активации. Ru/TiO2 катализаторы (1,0% масс. и 2,9% масс.) приготавливали согласно способу, использованному Iglesia et al. [40].
До измерений кинетики реакций или адсорбции газа (то есть, хемосорбции CO и O2) Pt-Re/C катализатор восстанавливали при 723 K (скорость подъема температуры 0,5 K/мин) в течение 2 часов в потоке H2 (140 см3 (при нормальных температуре и давлении)/мин). Ru/TiO2 катализаторы восстанавливали in-situ перед изучением кинетики реакций и измерениями адсорбции газа. Необратимое поглощение CO на Pt-Re/C при 300 K принимали за число каталитических активных центров (150 мкмоль/г) и измеряли, используя стандартную установку газовой адсорбции, описанную в литературе [28]. Дисперсии Ru/TiO2 катализаторов определяли путем хемосорбции О2 при 195 K в статической системе хемосорбции (41) и была рассчитаны значения Χ, равные 36x1016 м-1 и 50х1016 м-1 для 1,0% масс. Ru/TiO2 и 2,9% масс. Ru/TiO2, соответственно. В Табл.9 показаны свойства Ru/TiO2 катализаторов и эти результаты находятся в соответствии с аналогичными катализаторами, которые изучали Iglesia et al. [39]).
Пример 6 - Измерения кинетики реакций
Установка, использованная для проведения измерений кинетики реакций для Pt-Re/C, описана в [37]. Свежий катализатор загружали в трубчатый реактор из нержавеющей стали с внешним диаметром 12,5 мм (0,5 дюйма). Слой катализатора удерживали между концевой заглушкой из кварцевой ваты (Alltech) и плавлеными гранулами SiO2 (-4+16 меш, Sigma-Aldrich), которые способствовали испарению жидкого сырья. Порошок Pt-Re/C катализатора смешивали с равными объемами дробленых гранул SiO2 для того, чтобы уменьшить падение давления через слой катализатора. Для экспериментов по конверсии глицерина, объединенной с синтезом Фишера-Тропша, слой 1,0% масс. Ru/TiO2 загружали ниже по потоку от слоя Pt-Re/C. Термопары K-типа (Omega) присоединяли к внешней стороне реактора для измерения температуры реактора, которую регулировали температурным контроллером типа серии 16А (Dwyer Instruments). Свежий катализатор восстанавливали в потоке H2, как описано в предыдущем разделе. Контроллеры массового расхода (5850 Brooks Instruments) использовали для регулирования расхъода H2. ВЭЖХ насос (Model 301, Alltech) использовали для введения водного раствора сырья в 15-ти сантиметровую (6-ти дюймовую) иглу с наконечником типа «point 5 style» (Hamilton), припаянным внутрь секции 3 мм (1/8 дюйма) трубки из нержавеющей стали. Эта игла вводит растворы жидкого сырья внутрь реактора. Жидкие продукты, выходящие из реактора, охлаждали водой в теплообменнике типа «труба в трубе» и периодически сливали для газового хроматографического (ГХ) анализа (Agilent 6890 с ДИП и HP-Innowax колонкой или Scimadzu GC-2010 с детектором ионизации пламени (ДИП) и колонкой Rtx-5) и анализа общего количества органического углерода (Scimadzu TOC-V CHS). Каждый выходящий продукт исследовали на присутствие глицерина и других жидких побочных продуктов.
Поток выходящего газа проходил через регулятор обратного давления (GO Regulator, Model ВР-60), с помощью которого регулировали давление системы. Выходящий газ анализировали при помощи газовой хроматографии: H2 при помощи Carle GC (series 8700), используя детектор теплопроводности (ДТП), CO и СН4, используя HP 5890 GC с ДТП и колонкой с отмытым молекулярным ситом 5А 80/100 (Alltech), и CO2 и легкие алканы (C2-C3), используя HP 5890 GC с ДТП и колонкой Porapak QS 100/120 (Alltech). Все сырьевые растворы были изготовлены путем смешивания глицерина (99,5%, реагент АХО, Sigma-Aldrich) с деионизированной водой.
Установка, используемая для проведения экспериментов по синтезу Фишера-Тропша, аналогична установке, используемой для измерений кинетики реакций Pt-Re/C, за исключением того, что выходящие из реактора трубопроводы нагревали при 373 K. 2,9% масс. Ru/TiO2 катализатор смешивали с равным объемом дробленых гранул SiO2, чтобы способствовать рассеиванию теплоты, выделяемой в экзотермической реакции Фишера-Тропша, и загружали внутрь трубчатого реактора из нержавеющей стали с внешним диаметром 12,5 мм (0,5 дюйма). Продукты в жидкой фазе собирали в газожидкостном сепараторе и анализировали при помощи газового хроматографа (Scimadzu GC-2010 с ДИП детектором и колонкой Rtx-5). Поток выходящего газа анализировали на наличие C1-С10 углеводородов при помощи Varian GC - MS (Saturn 3) (газового хроматографа - масс-спектрометра), используя ДИП детектор и капиллярную колонку GS-Q. CO и CO2 анализировали с помощью высокоэффективного газового хроматографа ВЭГХ 5890 с ДТП и колонкой Porapak QS 100/120 (Alltech). Были использованы сверхчистые CO и H2 (Linde), и водные растворы ацетона, ацетола и этанола были введены в реактор тем же способом, как и вышеупомянутые растворы глицерина.
Пример 7 - Распределение углерода
(а) Конверсия глицерина на Pt-Re/C
На Фиг.6 показана степень конверсии в продукты газовой фазы и молярные соотношения CO/CO2 и H2/CO и в Табл.10 показано распределение выходящего углерода для конверсии 30% масс. раствора глицерина на 520 мг 10% масс. Pt-Re/C (атомное соотношение 1:1). Общая скорость входящего потока углерода (в виде глицерина) для этого эксперимента составляла 833 мкмоль/мин (скорость потока сырья 0,08 см3/мин) и общая степень конверсии глицерина составляла 91% (58% в продукты газовой фазы и 33% в продукты жидкой фазы).
В Табл.11-13 показаны распределения выходящего углерода и углеродные балансы для конверсии 50% масс. и 80% масс. растворов глицерина на 1,0 г 10% масс. Pt-Re/C (атомное соотношение 1:1) при 548 K и общем давлении от 0,1 до 1,1 МПа (1-11 бар). Скорость входящего потока жидкости составляла 0,04 см3/мин для 50% масс. раствора и 0,03 см3/мин для 80% масс. раствора. Общая степень конверсии глицерина составляла 100% для каждого условия.
(б) Синтез Фишера-Тропша на Ru/TiO2
В Табл.14 и 15 показаны распределения углерода и углеродные балансы для синтеза Фишера-Тропша 150 см3/мин смеси H2/CO с H2:CO=2 наряду с совместными подачами воды и водных растворов кислородсодержащих молекул при 548 K на 4 г 2,9% масс. Ru/TiO2. Степень конверсии совместно подаваемого ацетола в продукты составляла 100%, тогда как менее 20% этанола образовывало продукты и менее 10% ацетона образовывало продукты.
(в) Конверсия глицерина, объединенная с синтезом Фишера-Тропша
В Табл.16-18 приведено распределение углеродного продукта для конверсии 80% масс. глицеринового раствора посредством конверсии в синтез-газ с последующим синтезом Фишера-Тропша в двухслойном реакторе при 548 K и 0,5 МПа (5 бар). На Фиг.7 показано распределение молекулярной массы для этих экспериментов. Это распределение отклоняется от кинетики Андерсона-Шульца-Флори (АШФ). Скорость потока глицеринового сырья примерно 0,04 см3/мин использовали с 1,0 г 10% масс. Pt-Re(1:1)/C и 1,7-3,0 г 1,0% масс. Ru/TiO2.
Сумма веществ в таблицах распределения углерода (Табл.10-12, 14 и 16-18) может слегка отличаться от общей величины Свых в таблицах углеродного баланса (Табл.13 и 15). Это отличие происходит в результате того, что из таблиц распределения углерода опущены вещества, полученные в незначительном количестве.
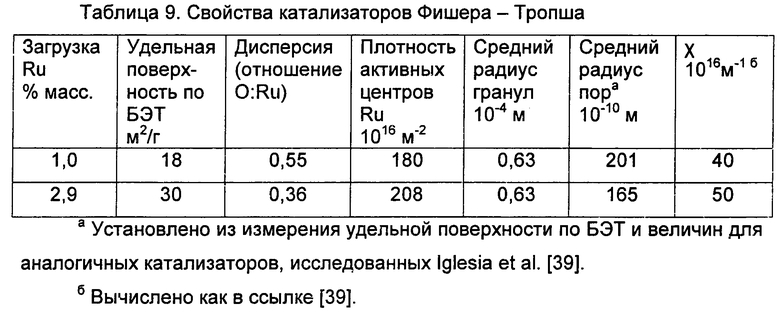







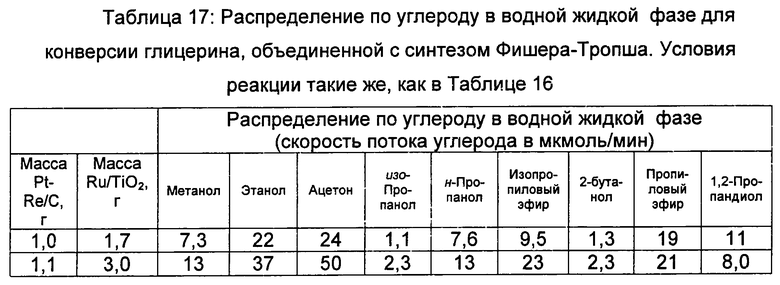

Пример 8 - Конверсия глюкозы в углеводороды
5% масс. рутениевый катализатор, нанесенный на активированный уголь, приготавливали согласно общему способу Примера 1.
38 мл водного раствора, содержащего 0,98 г нитросилнитрата рутения (III) (Alfa Aesar, 1,5% Ru), добавляли к 47,52 г активированного угля (Calgon OLC-AW, просеянный до фракции 18-40 меш). Данную смесь сушили при 100°C в вакууме. Были выполнены три дополнительных нанесения, используя 38 мл этого раствора, затем последнее нанесение, используя 14 мл этого раствора, разбавленного до 38 мл. Углеродную смесь сушили при 100°C в вакууме перед каждым нанесением.
Биметаллическую каталитическую систему, содержащую платину и рений (5% платины с молярным соотношением Pt:Re 1:2,5), нанесенные на активированный уголь (Calgon OLC-AW, просеянный до фракции 18-40 меш), изготавливали, используя метод начального смачивания, как описано в Примере 1. Активированный уголь медленно добавляли к 30% раствору перекиси водорода. После завершения добавления угля смесь оставляли на ночь. Водную фазу декантировали и уголь промывали три раза деионизированной водой, затем сушили в вакууме при 100°C. Водный раствор объемом, равным объему начального смачивания для пропитки угля, и содержащий гексагидрат гексахлорплатиновой кислоты (IV) (Alfa Aesar, 39,85% Pt) и раствор рениевой кислоты (Alfa Aesar, 76,41% HReO4), добавляли по капле во время перемешивания к углероду, функционализированному перекисью водорода. Увлажненный углерод сушили в вакууме при 100°C.
Реакторную трубку с внутренним диаметром (ID) 8,5 мм (0,334 дюйма) загружали Pt/Re и Ru катализаторами; 10,28 г Pt/Re катализатора загружали на дно реактора и 2,28 г Ru катализатора загружали в верхнюю часть реактора (используя реактор для испытаний, как показано на Фиг.3). Трубчатый реактор из нержавеющей стали помещали внутрь печи, которая содержит три зоны, нагреваемые электричеством. Температуру каждой зоны, нагреваемой электричеством, регулировали отдельным ПИД (пропорциональным интегрально-дифференциальным) регулятором, который измерял температуру поверхности секции реактора в нагреваемой зоне и регулировал электрическую мощность нагревателя, связанного с каждой нагреваемой зоной. Сырьевой раствор глюкозы непрерывно подавали в реактор, используя ВЭЖХ насос (обозначено, как «подача жидкости» на Фиг.3). Материал, выходящий из реактора, охлаждали путем пропускания через теплообменник с водяной рубашкой, и затем направляли в фазовый сепаратор для отделения жидких продуктов от газовых продуктов.
Как показано внизу Фиг.3, газы выходили из фазового сепаратора через трубопровод, который поддерживали при постоянном давлении путем подсистемы регулирования давления (не показано на Фиг.3). Количество газа, выходящего из газового сепаратора, измеряли посредством массового расходомера. Состав выходящего газа отслеживали посредством газовой хроматографии.
Уровень жидкости в фазовом сепараторе поддерживали на постоянном уровне с помощью подсистемы регулирования уровня (не показана). Водный раствор, который сливали из газового сепаратора в течение эксперимента по оценке катализатора, собирали и собранное количество измеряли гравиметрически. Были выполнены различные анализы продуктов в фазе раствора, включая pH и общую концентрацию органического углерода. Была выполнена газовая хроматография для определения концентраций непрореагировавщего субстрата и для определения специфических промежуточных и побочных продуктов.
Перед каждым экспериментом катализатор предварительно обрабатывали потоком водорода при 250°C в течение 2 часов. Температуры реакторной зоны устанавливали таким образом, что Ru/C катализатор эксплуатировали при 130°C и Pt/Re катализатор эксплуатировали при 240°C. Общее давление системы составляло 3,4 МПа избыт. (495 фунтов на кв. дюйм избыт.). 30% масс. раствор глюкозы в воде подавали в реактор с МЧОС, равной 1,2 грамма глюкозы на грамм катализатора (общие граммы катализатора) в час. В этой системе происходила полная конверсия глюкозы. В Табл.19 показаны выходы водорода, так же как и выходы углеродсодержащих продуктов. Дополнительно, в этой таблице показано образование C9-C18 компонентов, которые показывают удлинение цепи, обусловленное реакцией Фишера-Тропша.
Список литературы
В данном описании приведены ссылки на следующие работы.
[1] D.L. Klass, Biomass for Renewable Energy, Fuels and Chemicals (Academic Press, San Diego, 1998).
[2] E. Iglesia, S.C. Reyes, R.J. Madon, S.L. Soled, _4.fv. Catal 39, 221 (1993).
[3] K.A. Pokrovski, M.D. Rhodes, A.T. Bell, J. Catal. 235, 368 (2005).
[4] M. Asadullah, S.I. Ito, K. Kunimori, M. Yamada, K. Tomishige, Environ. ScL Technol. 36, 4476 (2002).
[5] F. Chornet, D. Wang, S. Czernik, D. Montane, M. Mann, "Biomass-to-hydrogen via fast pyrolysis and catalytic steam reforming," (Proceedings of the U.S. Dept. of Energy Hydrogen Program Review, 1996).
[6] R.L. Bain et al., Ind. Eng. Chem. Res. 44, 7945 (2005).
[7] J.M. Encinar, J.F. GonzMez, and A. Rodriguez-Reinares, Ind. Eng. Chem. Res. 44, 5491 (2005).
[8] Jon Van Gerpen, Fuel Proc. Technol. 86, 1097 (2005).
[9] Kahraman Bozbas, Renew. Sustain. Ener. Rev., in press.
[10] C.S. Gong, J.X. Du, N.J. Cao, G.T. Tsao, Appl. Biochem. Biotechnol. 84-86, 543 (2000).
[11] H. Shapouri, J.A. Duffield, M. Wang, The Energy Balance of Corn: An Update (report no. 814, Office of the Chief Economist, U.S. Department of Agriculture, 2002; available at http://www.usda.gov/oce/oepnu/aer-814.pdf).
[12] D. Pimentel and T.W. Patzek, Nat. Resources Res. 14, 65 (2005).
[13] S.P. Chopade et al., Published Patent Cooperation Treaty Application WO 01/66499 (PCT/US 01/00536) (published September 13, 2001; international filing date January 8, 2001).
[14] R.R. Davda, J.A. Dumesic, Chem. Comm. 1, 36 (2004).
[15] G. Gubitosa, B. Casale, Published Eur. Pat. Appl., EP 0553815 (published August 4, 1993).
[16] T.A. Werpy, J.G. Frye, Jr., A.H. Zacher, D.J. Miller, Published Patent Cooperation Treaty Application WO 03/035582 (PCT/US 02/33982) (published May 1, 2003; international filing date October 23, 2002).
[17] R.D. Cortright, R.R. Davda, J.A. Dumesic, Nature 418, 964 (2002).
[18] G.W. Huber, J.W. Shabaker, J.A. Dumesic, Science 300, 2075 (2003).
[19] J.W. Shabaker, R.R. Davda, G.W. Huber, R.D. Cortright, J.A. Dumesic, J. Catal. 215, 344 (2003).
[20] S. Czernik, R. French, C. Feik, F. Chomet, Ind. Eng. Chem. Res. 41, 4209 (2002).
[21] T. Hirai, N. Ikenaga, T. Miyake, T. Suzuki, Ener. Fuels 19,1761 (2005).
[22] A. Basinska, L. Kepinski, F. Domka, Appl. Catal, A: Gen. 183, 43 (1999).
[23] R.J. Gorte, S. Zhao, Catal. Today 104, 18 (2005).
[24] F. Esch et al., Science 309, 752 (2005).
[25] E. Christoffersen, P. Liu, A. Ruban, H.L. Skriver, J.K. Norskov, J. Catal. 199, 123 (2001).
[26] A.P. Steynberg, R.L. Espinoza, B. Jager, A.C. Vosloo, Appl. Catal, A: Gen., 186, 41 (1999).
[27] J. Greely, M. Mavrikakis, Nature Mater. 3, 810 (2004).
[28] B.F. Spiewak, J. Shen, J.A. Dumesic, J. Phys. Chem. 99, 17640 (1995).
[29] С.E. Hori et al., Appl. Catal. B: Environ. 16, 105 (1998).
[30] M.A. Aramendia et al., Colloids and Surfaces A: Physicochem. Eng. Aspects 234, 17 (2004).
[31] R.J. Gorte, S. Zhao, Catal Today 104, 18 (2005).
[32] F. Esch et al., Science 309, 752 (2005).
[33] B. Linnhoff, D.R. Vredeveld, Chem. Eng. Progress (July 1984), pp. 33-40.
[34] B. Linhoff, Chem Eng. Res. & Design 71 (A5), 503-522, (1993).
[35] C.N. Hamelinck, A.P.C. Faaij, H. den Uil and H. Boerrigter, Energy, 2004, 29, 1743-1771.
[36] T. Patzek and D. Pimentel, Critical Reviews in Plant Sciences, 2005, 24, 327-364.
[37] R.R. Soares, D.A. Simonetti and J.A. Dumesic, Angew. Chem. Int. Ed, 2006, 45, 3982-3985.
[38] E. Iglesia, Appl. Catal. A Gen., 1997, 161, 59-78.
[39] E. Iglesia, S.C. Reyes and R.J. Madon, J Catal., 1991, 129, 238-256.
[40] E. Iglesia, S.L. Soled, and R.A. Fiato, J. Catal., 1992, 137, 212-224.
[41] K.C. Taylor, J. Catal., 1975, 38, 299-306.
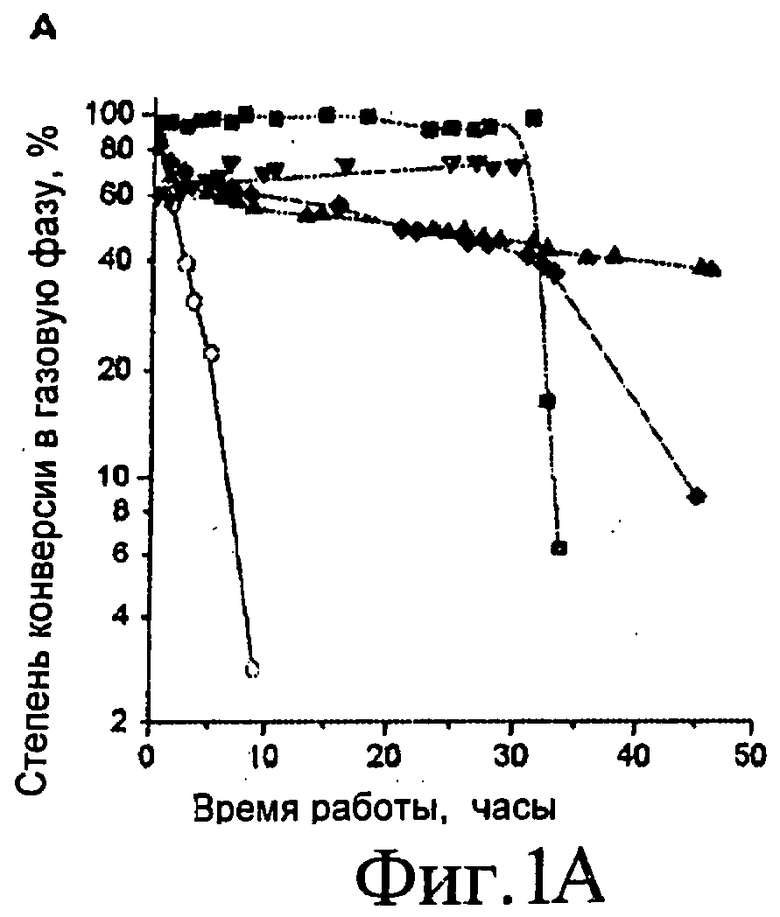


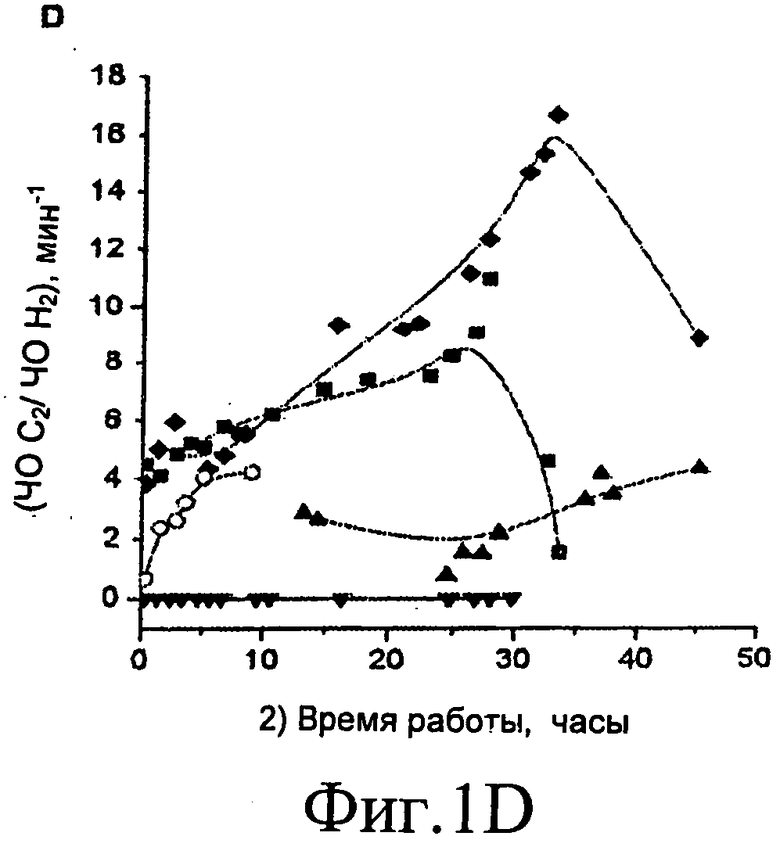

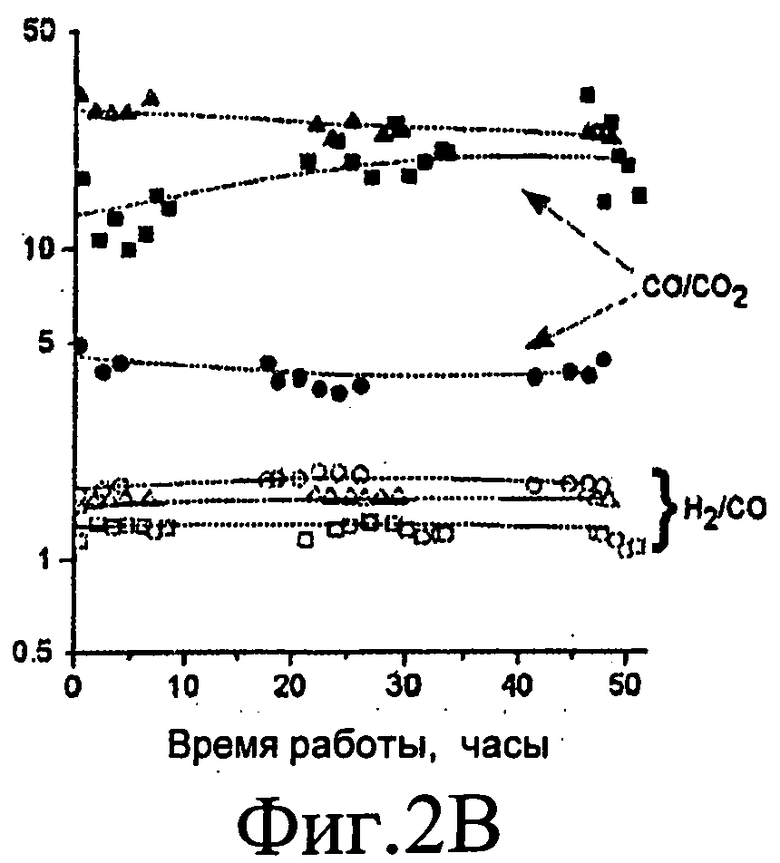

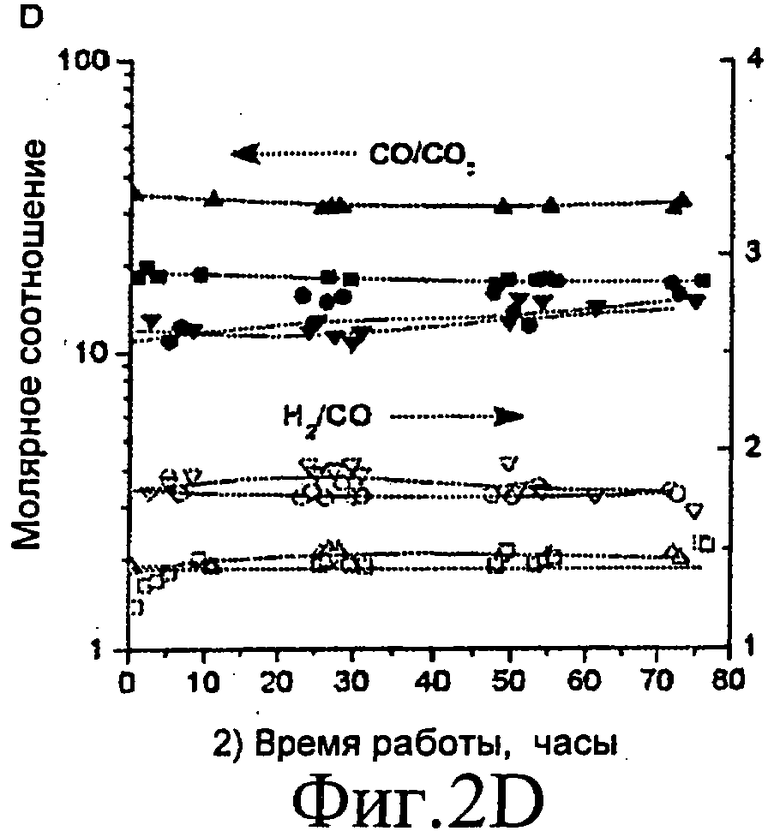

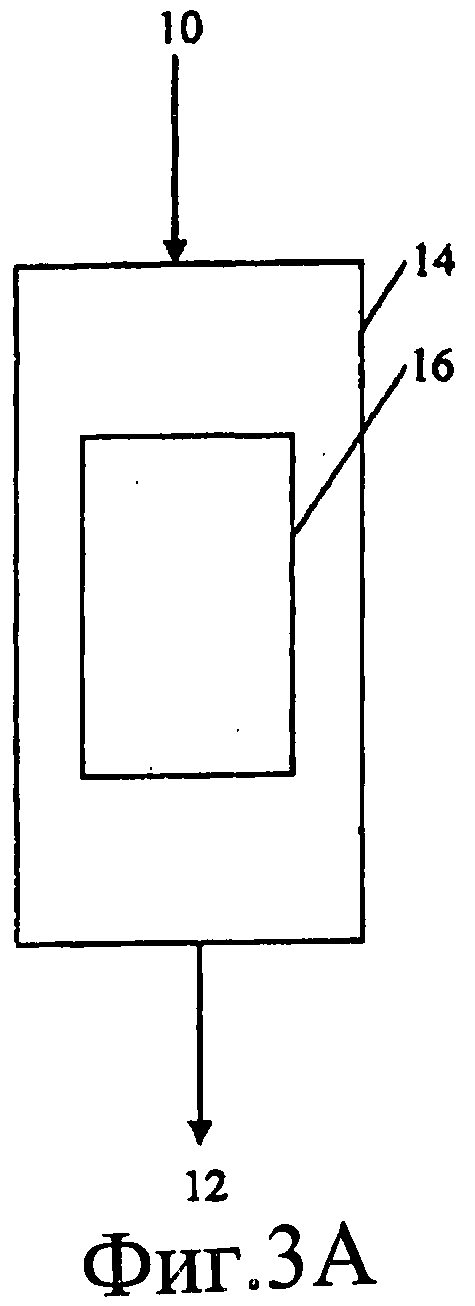

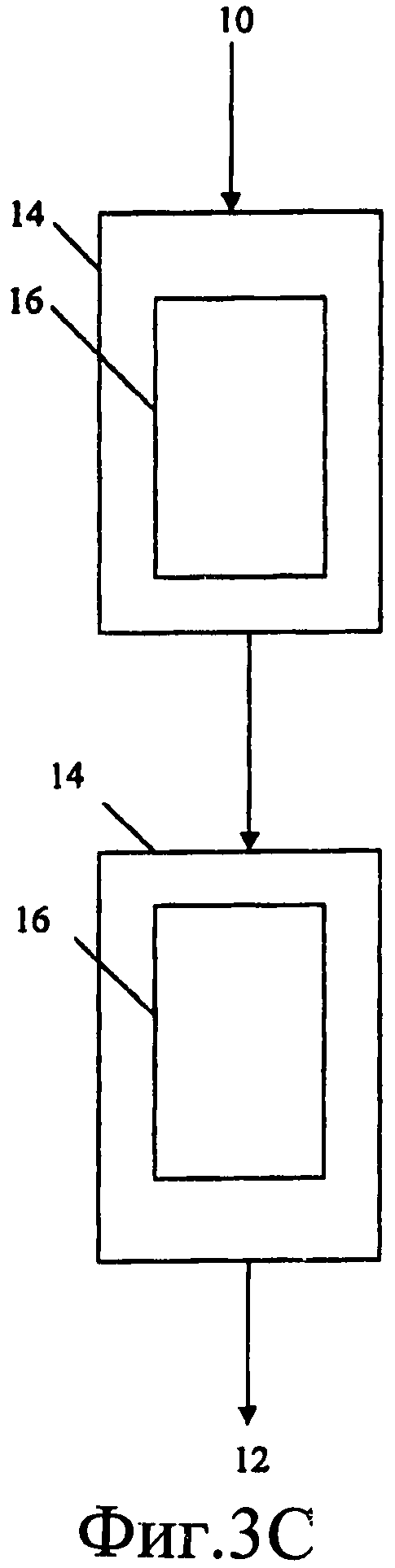
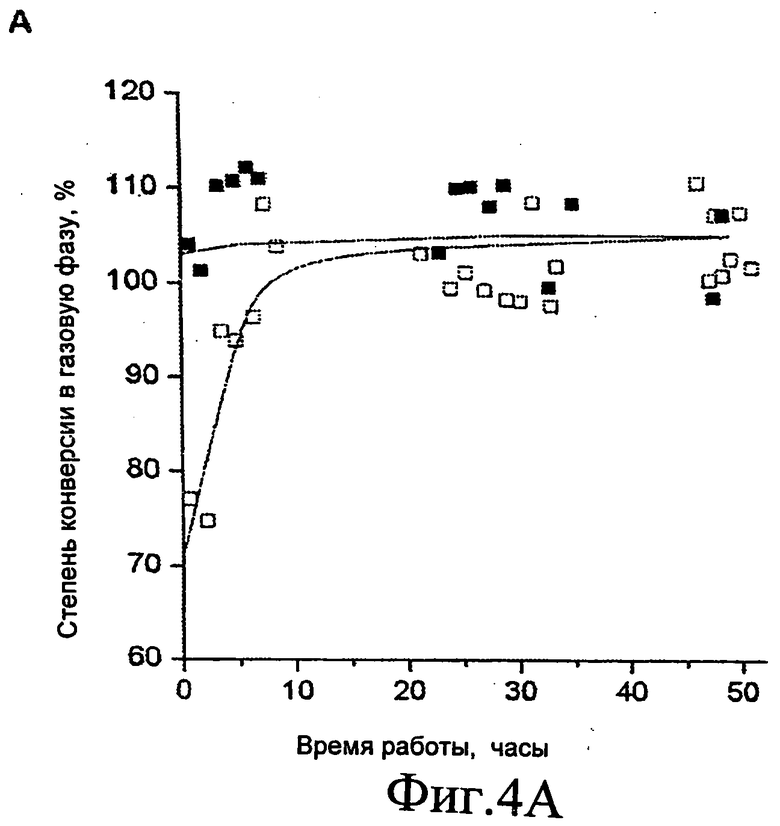

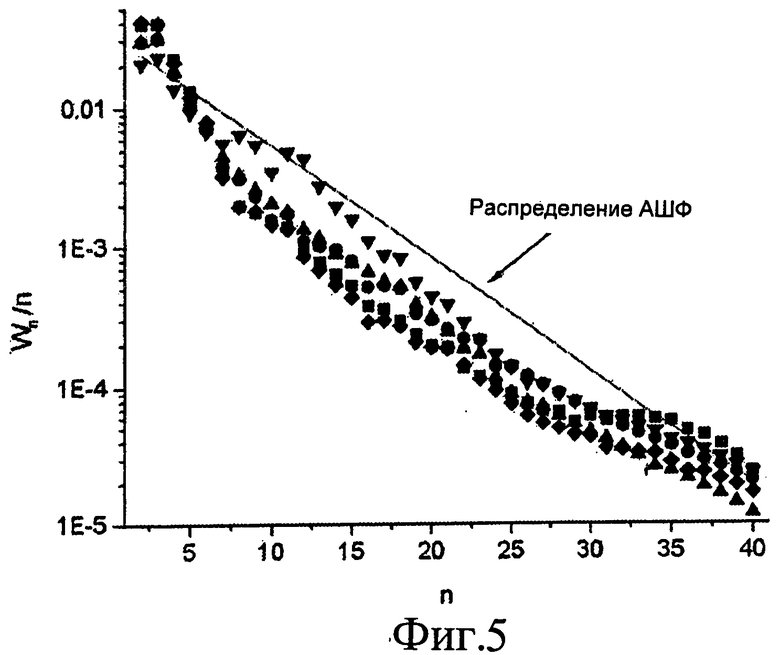


Изобретение относится к способу получения C2-C36 линейных или разветвленных углеводородов и кислородсодержащих углеводородов. Способ включает: а) проведение эндотермической реакции газификации с реагентом из биомассы при температуре менее или равной примерно 750 K, с получением синтез-газа, при этом температура является оптимальной для реакции утилизации синтез-газа или для реакции образования углерод-углеродных связей; б) проведение экзотермической реакции утилизации синтез-газа или реакции образования углерод-углеродных связей с синтез-газом стадии (а), без какой-либо промежуточной обработки синтез-газа стадии (а), при температуре выше или равной температуре реакции газификации, выполняемой на стадии (а), где реакция производит C2-C36 линейные или разветвленные углеводороды или кислородсодержащие углеводороды и теплоту, и в) использование теплоты, выделяемой при реакции утилизации синтез-газа или реакции образования углерод-углеродных связей стадии (б), в эндотермической реакции газификации стадии (а). Предложенное изобретение обеспечивает энергетически эффективный путь для получения топлива и химикатов из возобновляемых ресурсов биомассы. 16 з.п. ф-лы, 19 табл., 8 пр., 7 ил.
1. Способ получения C2-C36 линейных или разветвленных углеводородов и кислородсодержащих углеводородов, включающий:
а) проведение эндотермической реакции газификации с реагентом из биомассы при температуре менее или равной примерно 750 K с получением синтез-газа, при этом температура является оптимальной для реакции утилизации синтез-газа или для реакции образования углерод-углеродных связей;
б) проведение экзотермической реакции утилизации синтез-газа или реакции образования углерод-углеродных связей с синтез-газом стадии (а), без какой-либо промежуточной обработки синтез-газа стадии (а), при температуре выше или равной температуре реакции газификации, выполняемой на стадии (а), где реакция производит C2-C36 линейные или разветвленные углеводороды или кислородсодержащие углеводороды и теплоту, и
в) использование теплоты, выделяемой при реакции утилизации синтез-газа или реакции образования углерод-углеродных связей стадии (б), в эндотермической реакции газификации стадии (а).
2. Способ по п.1, в котором стадия (а) включает проведение эндотермической реакции газификации при температуре менее или равной примерно 625 K.
3. Способ по п.1, в котором стадия (а) включает проведение эндотермической реакции газификации при температуре менее или равной примерно 575 K.
4. Способ по п.1, в котором стадия (а) включает проведение эндотермической реакции газификации при температуре менее или равной примерно 550 K.
5. Способ по п.1, включающий выполнение стадии (а) и стадии (б) одновременно в одном реакционном сосуде.
6. Способ по п.5, включающий выполнение стадии (а) и стадии (б) с использованием только одного слоя катализатора.
7. Способ по п.1, включающий выполнение стадии (а) перед стадией (б) и выполнение стадии (а) и стадии (б) последовательно в отдельных реакционных сосудах.
8. Способ по п.1, в котором стадия (а) включает проведение эндотермической реакции газификации с реагентом, включающим одно или более соединений, выбираемых из группы, состоящей из полисахаридов, моносахаридов и полиолов.
9. Способ по п.8, в котором стадия (а) включает проведение эндотермической реакции газификации с реагентом, включающим глицерин.
10. Способ по п.1, в котором стадия (б) включает проведение реакции Фишера-Тропша.
11. Способ по п.1, в котором стадию (а) и стадию (б) выполняют, используя один или более катализаторов, включающих металл Группы VIIIB (Fe, Co, Ni, Ru, Rh, Pd, Os, Ir и Pt).
12. Способ по п.11, в котором катализатор сплавляют, соединяют или в него дополнительно включают один или более элементов, выбираемых из группы, состоящей из Cu, K, Mn, La, Re, Zn, Si, Ag, Ce, Gd, Th, Mn, Zr, Ti, Cr, V, Li, Na, Rb, Cs, Mo, Au, B и Cl, и их оксиды.
13. Способ по п.1, в котором стадию (а) и стадию (б) выполняют, используя один или более катализаторов, включающих платину, рутений, рений и их сочетания.
14. Способ по п.1, в котором стадию (а) и стадию (б) выполняют, используя один или более катализаторов, включающих метал группы VIIIB и расположенных на носителе.
15. Способ по п.14, в котором носитель выбирают из группы, состоящей из углерода и оксидов алюминия, церия, циркония и магния, и их сочетаний.
16. Способ по п.14, в котором носитель выбирают из группы, состоящей из TiO2, SiO2, Al2O3, MgO, ZrO2, ZrxTiyO2, ThO2, кизельгура, La2O3, MgCr2O4, TixSiyO2, TixZryO2, ZnO, Cr2O3, MnO, Nb2O5, CeO2, цеолита Y, цеолита USY, цеолита ZSM-5, цеолита MCM-41, цеолита MCM-22, цеолита HZSM-5, цеолита H-BEA, цеолита HY, Fe-замещенного цеолита LTL, деламинированного цеолита ITQ-6, деламинированного цеолита ITQ-2, молекулярных сит HMS, монтмориллонита, макропористого стирол-дивинилбензола, 4-винилпиридин-дивинилбензола, антрацена, карбонизированного хинолина и их сочетаний.
17. Способ по п.1, в котором стадию (а) и стадию (б) выполняют, используя по меньшей мере один катализатор, включающий платину, рутений, рений и их сочетания, и где катализатор размещают на носителе, выбираемом из группы, состоящей из углерода, Al2O3, CeO2, ZrO2, MgO, ZrO2 и их сочетаний.
| US 2003099593 A1, 29.05.2003 | |||
| US 2003220531 A1, 27.11.2003 | |||
| US 2003111410 A1, 19.06.2003 | |||
| US 4567205 A, 28.01.1986 | |||
| СПОСОБ ПРЕВРАЩЕНИЯ ПРИРОДНОГО ГАЗА В ВЫСШИЕ УГЛЕВОДОРОДЫ | 2000 |
|
RU2247701C2 |

