Настоящее изобретение относится к соединениям, пригодным в качестве лекарственных препаратов, к способам получения указанных соединений, композициям, содержащим указанные соединения, и их применению для лечения и предотвращения аллергических заболеваний, таких как астма, аллергический ринит и атопический дерматит, и других воспалительных заболеваний, опосредуемых простагландином D2 (ПГD2) или другими агонистами, действующими на рецептор CRTH2, который присутствует в клетках, включая эозинофилы, базофилы и Th2-лимфоциты.
ПГD2 принадлежит к эйкозаноидам - классу химических медиаторов, синтезируемых клетками в ответ на локальное повреждение тканей, обычные раздражители или гормональные раздражители, или посредством путей активации клеток. Эйкозаноиды связываются со специфическими рецепторами на поверхности клеток в широком спектре тканей всего организма и опосредуют различные действия в указанных тканях. Известно, что ПГD2 производится тучными клетками, макрофагами и лимфоцитами Th2 и обнаруживается в высоких концентрациях в дыхательных путях пациентов, страдающих астмой, сенсибилизированных антигеном (Murray et al, (1986), N.Engl. J.Med. 315: 800-804). Инстилляция ПГD2 в дыхательные пути может провоцировать множество характерных признаков астматического ответа, включая сужение бронхов (Hardy et al, (1984) N.Engl. J.Med. 311: 209-213; Sampson et al, (1997) Thorax 52: 513-518) и накопление эозинофилов (Emery et al, (1989) J.Appl. Physiol. 67: 959-962).
Способность экзогенно применяемого ПГD2 вызывать воспалительные ответы подтверждена использованием трансгенных мышей со сверхэкспрессией человеческой ПГD2-синтазы, у которых наблюдают воспаление легких с повышенным количеством эозинофилов и продукцией цитокина Th2 в ответ на антиген (Fujitani et al, (2002) J.Immunol. 168: 443-449).
Первым из открытых рецепторов, специфических для ПГD2, был рецептор DP, связанный с повышением внутриклеточных уровней цАМФ. Однако полагают, что ПГD2 опосредует большую часть своей воспалительной активности через взаимодействие со связанным с G-белком рецептором, называемым CRTH2 (молекула, гомологичная хемоаттрактантному рецептору, экспрессированная на клетках Th2), который экспрессируют Th2 лимфоциты, эозинофилы и базофилы (Hirai et al, (2001) J.Exp.Med. 193: 255-261, и ЕР0851030 и ЕР-А-1211513 и Bauer et al, EP-A-1170594). Кажется очевидным, что действие ПГD2 на активацию Th2 лимфоцитов и эозинофилов опосредуется через CRTH2, поскольку селективные агонисты CRTH2 13,14-дигидро-15-кето-ПГD2 (ДК-ПГD2) и 15R-метил-ПГD2 могут вызывать указанный ответ, и действие ПГD2 блокируется антителом к CRTH2 (Hirai et аl, 2001; Monneret et al, (2003) J.Pharmacol. Exp.Ther. 304: 349-355). Напротив, селективный агонист рецептора DP BW245C не вызывает миграцию Th2 лимфоцитов или эозинофилов (Hirai et аl, 2001; Gervais et al, (2001) J.Allergy Clin. Immunol. 108: 982-988). На основании этого факта оказание антагонистического воздействия на взаимодействие ПГD2 и рецептора CRTH2 является привлекательным подходом для лечения воспалительной составляющей Th2-зависимых аллергических заболеваний, таких как астма, аллергический ринит и атопический дерматит.
Согласно EP-A-1170594 способ, который раскрыт в указанном документе, можно применять для идентификации соединений, применимых для лечения аллергической астмы, атопического дерматита, аллергического ринита, аутоиммунного, реперфузионного повреждения и ряда воспалительных состояний, все из которых опосредуются действием ПГD2 или других агонистов рецептора CRTH2.
Соединения, связывающиеся с CRTH2, раскрыты в WO-A-03066046 и WO-А-03066047. Указанные соединения не являются новыми, но описаны впервые, наряду с похожими соединениями, в GB 1356834, GB 1407658 и GB 1460348, где было показано, что они обладают противовоспалительным, противоаллергическим и жаропонижающим действием. Согласно WO-A-03066046 и WO-A-03066047 описанные в данных документах соединения представляют собой модуляторы активности рецептора CRTH2 и поэтому пригодны для применения при лечении или предотвращении обструктивных заболеваний дыхательных путей, таких как астма, хроническое обструктивное заболевание легких (ХОЗЛ) и ряда других заболеваний, включая различные состояния костей и суставов, кожи и глаз, ЖКТ, центральной и периферической нервной системы, и других тканей, а также отторжения аллотрансплантата. Все указанные соединения являются производными индола с заместителем - уксусной кислотой в положении 3 индольного кольца.
PL 65781 и JP 43-24418 также относятся к производным индола и уксусной кислоты в положении 3, которые по своей структуре напоминают индометацин, и подобно индометацину, как считают, обладают противовоспалительным и жаропонижающим действием. Таким образом, хотя это не учитывали во время публикации указанных документов, описанные в них соединения являются ингибиторами циклооксигеназы (ЦОГ), действие, т.е. обладают активностью, которая существенно отличается от активности соединений согласно настоящему изобретению. Действительно, ингибиторы ЦОГ противопоказаны при лечении многих заболеваний и состояний, например астмы и воспалительного заболевания кишечника, при которых соединения согласно настоящему изобретению применимы, хотя иногда их можно использовать для лечения артритических состояний.
Также из уровня техники известны соединения индол-1-уксусной кислоты, хотя они не описаны как антагонисты CRTH2. Например, WO-A-950268, WO-A-0032180, WO-A-0151849 и WO-A-0164205 все относятся к соединениям, являющимся производными индола-1 -уксусной кислоты, но как указано, описанные соединения являются ингибиторами альдозоредуктазы, используемыми при лечении сахарного диабета (WO-A-9950268, WO-A-0032180 и WO-A-0164205) или гипоурикемическими средствами (WO-A-0151849). Ни в одном из указанных документов не имеется указания, что описанные соединения могут быть полезны для лечения заболеваний и состояний, опосредуемых ПГD2 или другими агонистами рецептора CRTH2.
US 4,363,912 относится к производным индол-1-уксусной кислоты для применения в качестве ингибиторов тромбоксансинтазы, пригодных для лечения таких состояний, как тромбоз, ишемическая болезнь сердца и инсульт.
WO-A-9603376 относится к соединениям - ингибиторам сФЛА2, причем все указанные соединении имеют амидные или гидразидные заместители вместо производных карбоновой кислоты, которые присутствуют в соединениях согласно настоящему изобретению.
JP 2001247570 относится к способу получения 3-бензотиазолилметилиндолуксусной кислоты, которая является ингибитором альдозоредуктазы.
US 4,859,692 относится к соединениям, известным в качестве антагонистов лейкотриена и пригодным для лечения таких состояний, как астма, сенная лихорадка и аллергический ринит, а также некоторых воспалительных состояний, таких как бронхит, атопическая и эктопическая экзема. Некоторые из соединений согласно упомянутому документу представляют собой индол-1-уксусные кислоты, но те же авторы, в J.Med. Chem., 6(33), 1781-1790 (1990), отмечают, что указанные соединения с уксуснокислой группой при индольном атоме азота не обладают значительной пептидолейкотриеновой активностью. С этой точки зрения особенно удивительно то, что соединения согласно настоящему изобретению, все из которых содержат уксуснокислую группу при индольном атоме азота, пригодны для лечения таких состояний, как астма, сенная лихорадка и аллергический ринит.
US 4,273,782 относится к производным индол-1-уксусной кислоты, которые полезны для лечения таких состояний, как тромбоз, ишемическая болезнь сердца, инсульт, преходящее нарушение мозгового кровообращения, мигрень и сосудистые осложнения диабета. В указанном документе не упомянуты состояния, опосредуемые действием ПГD2 или других агонистов рецептора CRTH2.
US 3,557,142 относится к 3-замещенным индол-1-карбоновым кислотам и сложным эфирам, которые полезны для лечения воспалительных состояний.
WO-A-03/097598 относится к соединениям, являющимся антагонистами рецептора CRTH2. В них отсутствует ароматический заместитель в положении 3 индольного кольца.
Cross et al, J.Med. Chem. 29, 342-346 (1986) относится к способу получения производных индол-1-уксусной кислоты из соответствующих сложных эфиров. Соединения, к которым относится указанный документ, считают ингибиторами тромбоксансинтазы.
ЕР-А-0539117 относится к производным индол-1-уксусной кислоты, являющимся антагонистами лейкотриена.
US 2003/0153751 относится к производным индол-1-уксусной кислоты, являющимся ингибиторами секреторной фосфолипазы А2 (сФЛА2). Однако все приведенные соединения содержат объемные заместители в положениях 2 и 5 индольной системы и этим значительно отличаются от соединений согласно настоящему изобретению.
US 2004/011648 раскрывает производные индол-1-уксусной кислоты, являющиеся ингибиторами ИАП-1. Не предполагается, что указанные соединения могут оказывать антагонистическое действие по отношению к CRTH2.
WO 2004/058164 относится к соединениям, считающимся модуляторами астмы и аллергического воспаления. Единственные соединения, для которых показана соответствующая активность, полностью отличаются по структуре от производных индол-1-уксусной кислоты согласно настоящему изобретению.
Соединения, связывающиеся с рецептором CRTH2, раскрыты в WO-A-03/097042 и WO-A-03/097598. Указанные соединения представляют собой индолуксусные кислоты, но в WO-A-03/097042 индольная система слита в положениях 2-3 с 5-7-членным карбоциклическим кольцом. В WO-A-03/09 7598 в положении 3 индольного кольца находится пирролидиновая группа.
WO-A-03/101981, WO-A-03/101961 и WO-A-2004/007451 все относятся к производным индол-1-уксусной кислоты, считающимся антагонистами CRTH2, но отличающимся по структуре от соединений общей формулы (I), поскольку в них отсутствует группа-спейсер или имеется группа -S- или -SO2- в положении 3 индольного кольца вместо группы СН2 у соединений согласно настоящему изобретению, описанных ниже.
WO-A-2005/019171 также описывает производные индол-1-уксусной кислоты, считающиеся антагонистами CRTH2, которые считаются подходящими для лечения различных респираторных заболеваний. Все указанные соединения имеют заместитель, присоединенный по положению 3 индольного кольца через кислородный спейсер.
WO-A-2005/094816 также описывает производные индол-1-уксусной кислоты, которые в этом случае имеют алифатический заместитель в положении 3 индольного кольца. Описанные соединения считают антагонистами CRTH2.
WO-A-2006/034419 относится к антагонистам CRTH2 - индольным соединениям, имеющим гетероциклический или гетероароматический заместитель, непосредственно присоединенный в положении 3 индольной кольцевой системы.
В более ранней заявке, WO-A-2005/044260 авторов настоящего изобретения, описаны соединения, являющиеся антагонистами в отношении действия ПГD2 на рецепторы CRTH2. Указанные соединения представляют собой производные индол-1-уксусной кислоты, которые в положении 3 имеют группу CR8R9 в качестве заместителя, в которой R9 представляет собой водород или алкил, а R8 представляет собой ароматический остаток, который может иметь один или более заместитель. Соединения, описанные в указанном документе, являются мощными антагонистами ПГD2 для рецептора CRTH2 in vitro. Однако авторы настоящего изобретения обнаружили, что при тестировании in vivo фармакокинетические профили некоторых соединений не являются оптимальными, и их действие при исследовании на изменение формы эозинофилов цельной крови, результаты которого показательны для подобной активности соединений in vivo, часто заметно меньше, чем можно было предполагать на основании результатов по связыванию in vitro.
Однако неожиданно авторы настоящего изобретения обнаружили, что с помощью изменений в группе R8 соединений, описанных в WO-A-2005/044260, можно добиться улучшения показателей в тесте на изменение формы эозинофилов цельной крови in vitro, в ингибировании индуцированной ДК-ПГD2 эозинофилии крови in vivo и в фармакокинетическом профиле при пероральном введении субъекту.
Настоящее изобретение, таким образом, относится к новым соединениям, связывающимся с CRTH2, которые, благодаря этому, применимы для лечения заболеваний и состояний, опосредуемых действием ПГD2 на рецептор CRTH2.
Согласно настоящему изобретению предложены соединения общей формулы (I)
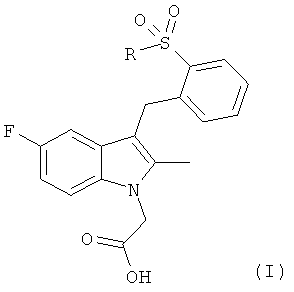 ,
,
в которой R представляет собой фенил, который может иметь один или более атом галогена в качестве заместителя;
или фармацевтически приемлемая соль, гидрат, сольват, комплекс или пролекарство указанных соединений.
Соединения общей формулы (I) представляют собой антагонисты рецептора CRTH2 и полезны для лечения состояний, опосредуемых ПГD2 или другими агонистами, связывающимися с CRTH2. Указанные состояния включают аллергические заболевания, астматические состояния и воспалительные заболевания, примерами которых являются следующие состояния:
астма, включая аллергическую астму, бронхиальную астму, обострения астмы и родственных аллергических заболеваний, вызванные вирусной инфекцией, в частности обострения, вызванные риновирусом и респираторно-синцитиальным вирусом, эндогенной, экзогенной, вызванной физической нагрузкой, медикаментозной и вызванной пылью астмы, лечение кашля, включая хронический кашель, связанный с воспалительными и секреторными состояниями дыхательных путей, и ятрогенный кашель, острый и хронический ринит, включая медикаментозный ринит, вазомоторный ринит, круглогодичный аллергический ринит, сезонный аллергический ринит, полипоз носа, острые вирусные инфекции, включая простуду, инфекцию, вызванную респираторно-синцитиальным вирусом, вирусом гриппа, коронавирусом и аденовирусом, атопический дерматит, контактная гиперчувствительность (включая контактный дерматит), экзематозный дерматит, фитодерматит, фотодерматит, себорейный дерматит, герпетиформный дерматит, красный плоский лишай, склерозирующий и атрофический лишай, гангренозная пиодермия, кожный саркоидоз, дискоидная красная волчанка, пемфигус, пемфигоид, буллезный эпидермолиз, ангионевротический отек, васкулиты, токсические эритемы, кожные эозинофилии, гнездная плешивость, облысение по мужскому типу, синдром Свита, синдром Вебера-Крисчена, полиморфная эритема, целлюлит, панникулит, кожные лимфомы, немеланомный рак кожи и другие диспластические поражения; блефарит, конъюнктивит, особенно аллергический конъюнктивит, передний и задний увеит, хориоидит, аутоиммунные, дегенеративные или воспалительные нарушения, поражающие сетчатку, офтальмит; бронхит, включая инфекционный и эозинофильный бронхит, эмфизема, бронхоэктаз, «легкое фермера», гиперчувствительный пневмонит, идиопатические интерстициальные пневмонии, осложнения после пересадки легкого, сосудистые и тромботические нарушения в сосудистой системе легких, легочная гипертензия, пищевые аллергии, гингивит, глоссит, периодонтит, эзофагит, включая рефлюкс, эозинофильный гастроэнтерит, проктит, анальный зуд, глютеновая болезнь, аллергии, связанные с пищей, воспалительное заболевание кишечника, язвенный колит и болезнь Крона, мастоцитоз, а также другие опосредуемые CRTH2 заболевания, например аутоиммунные заболевания, такие как гипер-lgE-синдром, тиреоидит Хашимото, болезнь Грейвса, болезнь Аддисона, сахарный диабет, идиопатическая тромбоцитопеническая пурпура, эозинофильный фасциит (paschiitis), антифосфолипидный синдром и системная красная волчанка, СПИД, проказа, синдром Сезари, паранеопластичекий синдром, смешанные и недифференцированные заболевания соединительных тканей, воспалительные миопатии, включая дерматомиозит и полимиозит, ревматическая полимиалгия, ювенильный артрит, ревматическая лихорадка, васкулиты, включая гигантоклеточный артериит, артериит Такаясу, синдром Черджа-Строссса, узелковый полиартериит, микроскопический полиартериит, темпоральный артериит, миастения гравис, острая и хроническая боль, нейропатические болевые синдромы, нейродегенерация, осложнения на центральную и периферическую нервную систему злокачественных, инфекционных или аутоиммунных процессов, поясничная боль, семейная средиземноморская лихорадка, синдром Макла-Уэльса, семейная ирландская лихорадка, болезнь Кикучи, псориаз, акне, множественный склероз, отторжение аллотрансплантата, реперфузионное повреждение, хроническое обструктивное заболевание легких, а также ревматоидный артрит, болезнь Стилла, анкилозирующий спондилит, реактивный артрит, недифференцированная спондилоартропатия, псориатический артрит, септический артрит и другие инфекционной природы артропатии и болезни костей и остеоартрит; острые и хронические вызванные кристаллическими отложениями синовиты, включая уратную подагру, болезнь отложения кристаллов пирофосфата кальция, связанный с (гидроксил)апатитом кальция сухожильный синдром и синовиальное воспаление, болезнь Бехчета, первичный и вторичный синдром Шегрена, системный склероз и ограниченная склеродермия; гепатит, цирроз печени, холецистит, панкреатит, нефрит, нефритический синдром, цистит и язва Ханнера (Hunner's ulcer) мочевого пузыря, острый и хронический уретрит, простатит, эпидидимит, оофорит, сальпингит, вульвовагинит, болезнь Пейрони, эректильная дисфункция, болезнь Альцгеймера и другие дементные заболевания; перикардит, миокардит, воспалительные и аутоиммунные кардиомиопатии, включая миокардиальный саркоидоз, ишемические реперфузионные повреждения, эндокардит, вальвулит, аортит, флебит, тромбоз, лечение распространенных видов рака и фиброзных состояний, таких как идиопатический легочный фиброз, включая криптогенный фиброзирующий альвеолит, келоиды, избыточное фиброзное рубцевание/спайки после операции, печеночный фиброз, включая печеночный фиброз, связанный с гепатитами В и С, маточный фиброз, саркоидоз, включая нейросаркоидоз, склеродермия, почечный фиброз, являющийся результатом диабета, фиброз, связанный с ревматоидным артритом, атеросклероз, включая церебральный атеросклероз, васкулит, миокардиальный фиброз, являющийся результатом инфаркта миокарда, кистозный фиброз, рестеноз, системный склероз, болезнь Дюпюитрена, фиброз, осложняющий противоопухолевое лечение и хронические инфекции, включая туберкулез, аспергиллез и другие грибковые инфекции, фиброз ЦНС после инсульта или способствование излечению без фиброзного рубцевания.
Описанные соединения особенно полезны для лечения или предотвращения аллергической астмы, круглогодичного аллергического ринита, сезонного аллергического ринита, атопического дерматита, контактной гиперчувствительности (включая контактный дерматит), конъюнктивита, особенно аллергического конъюнктивита, эозинофильного бронхита, пищевых аллергий, эозинофильного гастроэнтерита, воспалительного заболевания кишечника, язвенного колита и болезни Крона, мастоцитоза, боли, нейродегенеративных заболеваний, а также других заболеваний, опосредуемых ПГD2, например аутоиммунных заболеваний, таких как гипер-lgE-синдром и системная красная волчанка, псориаз, акне, множественный склероз, отторжение аллотрансплантата, реперфузионное повреждение, хроническое обструктивное заболевание легких, а также ревматоидный артрит, псориатический артрит и остеоартрит.
Улучшенные характеристики при исследовании на изменение формы эозинофилов цельной крови и фармакокинетический профиль соединений общей формулы (I) являются особенно неожиданными, поскольку некоторые из соединений, описанных в WO-A-2005/044260, близкие по структуре к соединениям общей формулы (I), не обладают указанными благоприятными свойствами. В частности, соединение из Примера 17 в WO-A-2005/044260 похоже на соединения согласно настоящему изобретению, и можно предположить, что оно обладает сходными свойствами. Однако замещение метилсульфонильной группы в положении 4 бензольного кольца в Примере 17 WO-A-2005/044260 на группу SO2R в положении 2 бензольного кольца в соединениях формулы (I) оказывает значительное влияние на фармакокинетику и активность соединений, поскольку, когда Соединение 17 из WO-A-2005/044260 вводят перорально, его фармакокинетический профиль in vivo оказывается менее подходящим, чем тот же профиль для соединений общей формулы (I).
Кроме того, для многих соединений, описанных в WO-A-2005/044260, обнаружено, что их активность в отношении изменения формы эозинофилов цельной крови in vitro часто меньше, чем можно было бы ожидать на основании их активности, измеренной в экспериментах по радиолигандному связыванию с рецептором CRTH2 in vitro.
Кроме того, улучшенная активность является весьма специфичной для группы соединений общей формулы (I), поскольку соединения, еще более тесно связанные с соединениями, описанными в WO-A-2005/044260, не обладают указанными благоприятными свойствами. Например, аналоги общей формулы (I), в которых группа SO2R находится в положениях 3 или 4 бензольного кольца, менее активны в тестах in vitro на изменение формы эозинофилов цельной крови.
В настоящем описании «C1-C6 алкил» относится к прямой или разветвленной насыщенной углеводородной цепи, содержащей от одного до шести атомов углерода и, возможно, замещенной одним или более галогеновыми заместителями или одной или более С3-С7 циклоалкильными группами. Примеры включают метил, этил, н-пропил, изо-пропил, m-бутил, н-гексил, трифторметил, 2-хлорэтил, метиленциклопропил, метиленциклобутил и метиленциклопентил.
«C1-C4 алкил» и «C1-C18 алкил» имеют сходные значения за исключением того, что они содержат от одного до четырех и от одного до восьми атомов углерода, соответственно.
Термин С3-С7 циклоалкил относится к насыщенному 3-7-членному карбоциклическому кольцу. Примеры указанных групп включают циклопропил, циклобутил, циклопентил и циклогексил.
В настоящем описании термин «галоген» относится к фтору, хлору, брому или йоду.
Термины «ароматический остаток» и «арил» в контексте настоящего описания относятся к ароматической кольцевой системе, содержащей от 5 до 14 атомов углерода в кольце и включающей до трех колец. Примерами ароматических остатков являются бензол и нафталин. Арильные группы могут быть замещенными одним или более заместителями, выбираемыми среди галогена, C1-C6 алкила, C1-C6 алкоксила, 5-7-членного гетероциклического кольца или SO2R9, где R9 такой, как определено выше.
Соответствующие фармацевтически и ветеринарно приемлемые соли соединений общих формул (I) и (II) включают соли, образованные присоединением основания, такие как соли натрия, калия, кальция, алюминия, цинка, магния и других металлов, а также соли холина, диэтаноламина, этаноламина, этилдиамина и другие широко известные соли, образованные присоединением основания.
Когда это уместно, фармацевтически или ветеринарно приемлемые соли могут также включать соли органических кислот, особенно карбоновых кислот, включая, но не ограничиваясь этим, ацетат, трифторацетат, лактат, глюконат, цитрат, тартрат, малеат, малат, пантотенат, адипинат, альгинат, аспартат, бензоат, бутират, диглюконат, циклопентаноат, глюкогептаноат, глицерофосфат, оксалат, гептаноат, гексаноат, фумарат, никотинат, памоат, пектинат, 3-фенилпропионат, пикрат, пивалат, пропионат, тартрат, лактобионат, пивалат, камфорат, ундеканоат и сукцинат, органических сульфоновых кислот, таких как метансульфонат, этансульфонат, 2-гидроксиэтансульфонат, камфорсульфонат, 2-нафталинсульфонат, бензолсульфонат, п-хлорбензолсульфонат и п-толуолсульфонат; и неорганических кислот, такие как гидрохлорид, гидробромид, гидроиодид, сульфат, бисульфат, гемисульфат, тиоцианат, персульфат, соли фосфорных и сульфоновых кислот.
Соли, не являющиеся фармацевтически или ветеринарно приемлемыми, могут все же быть полезны в качестве промежуточного продукта.
Пролекарства представляют собой любые ковалентно модифицированные соединения, которые высвобождают активный родительский лекарственный препарат, соответствующие общей формуле (I), in vivo. Примеры пролекарств включают алкильные сложные эфиры соединений общей формулы (I), например сложные эфиры общей формулы (II), описанные ниже.
Если в соединении согласно настоящему изобретению присутствует хиральный центр или другой вид центра изомерии, все формы указанного изомера или изомеров, включая энантиомеры и диастереомеры, рассматривают как входящие в объем настоящего описания. Соединения согласно настоящему изобретению, содержащие хиральный центр, можно применять в виде рацемической смеси, энантиомерно обогащенной смеси, или рацемическую смесь можно разделить с использованием широко известных методик, и можно использовать индивидуальные энантиомеры по отдельности.
В соединениях общей формулы (I) предпочтительно, чтобы фенильная группа R была незамещенной или имела один галогеновый заместитель, обычно фтор или хлор, который обычно находится в положении 4 фенильной группы R.
К наиболее предпочтительным соединениям относятся следующие:
2-{5-фтор-2-метил-3-[2-(фенилсульфонил)бензил]-1Н-индол-1-ил}уксусная кислота;
2-{3-[2-(4-хлорфенилсульфонил)бензил]-5-фтор-2-метил-1Н-индол-1-ил}уксусная кислота;
2-{5-фтор-3-[2-(4-фторфенилсульфонил)бензил]-2-метил-1Н-индол-1-ил}уксусная кислота;
или C1-С6 алкильные, арильные, (СН2)mOC(=O)С1-С6алкильные, (СН2)mN(R11)2, CH((CH2)mO(C=O)R12)2 сложные эфиры любого из вышеперечисленных соединений; в которых
m равно 1 или 2;
R11 представляет собой водород или метил;
R12 представляет собой C1-C18 алкил.
Согласно другому аспекту настоящего изобретения предложено соединение общей формулы (II):
 ,
,
где R такой, как определено для общей формулы (I); и
R1 представляет собой C1-C6 алкил, арил, (СН2)mOC(=O)С1-С6алкил, (СН2)mN(R11)2, CH((CH2)mO(C=O)R12)2;
m равно 1 или 2;
R11 представляет собой водород или метил;
R12 представляет собой C1-C18 алкил.
Соединения общей формулы (II) являются новыми и их можно применять в качестве пролекарств для соединений общей формулы (I). Когда соединение общей формулы (II) действует как пролекарство, оно впоследствии превращается в указанный лекарственный препарат под действием эстеразы в крови или в ткани пациента.
Примеры особенно подходящих групп R1 для случая, когда соединение общей формулы (II) применяют в качестве пролекарства, включают:
метил, этил, пропил, фенил, CH2OC(=O)tBu, CH2CH2N(Me)2 CH2CH2NH2 или CH(CH2O(C=O)R12)2, где R12 такой, как определено выше.
В дополнение к их использованию в качестве пролекарств, соединения формулы (II), в которых R1 представляет собой C1-C6 алкил, можно применять в способе получения соединения общей формулы (I), причем указанный процесс включает осуществление взаимодействия соединения общей формулы (II) с основанием, таким как гидроксид натрия или гидроксид лития. Реакцию можно осуществлять в водном растворителе или в органическом растворителе, или в смеси указанных двух растворителей. Типичным растворителем, применяемым в указанной реакции, является смесь тетрагидрофурана и воды.
Соединения общей формулы (II) можно получить из соединений общей формулы (III):
 ,
,
в которой R1 такой, как определено для общей формулы (II); путем осуществления взаимодействия с соединением общей формулы (IV):
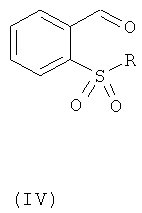 ,
,
где R такой, как определено для общей формулы (I);
в условиях кислотного восстановительного алкилирования.
Соединения общей формулы (III) легко доступны или могут быть получены способами, широко известными специалистам в данной области.
Альдегиды общей формулы (IV) можно получить снятием защиты с ацеталей общей формулы (V)
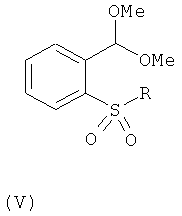 ,
,
где R такой, как определено для общей формулы (I). Снятие защиты можно осуществить путем проведения взаимодействия с водным раствором кислоты, например серной кислоты, с последующей нейтрализацией основанием, обычно твердым карбонатом калия. Описанную реакцию можно проводить при температуре от 0 до 40°С, обычно при комнатной температуре.
Соединения общей формулы (V) можно получить окислением соединения общей формулы (VI)
 ,
,
где R такой, как определено для общей формулы (I). Окисление сульфидной группы можно осуществить при помощи избыточного количества окислителя, такого как хлорпероксибензойная кислота. Реакционную смесь можно вначале охладить, например, от -5 до 5°С, а затем оставить нагреваться, обычно до комнатной температуры.
Ацеталь общей формулы (VI) можно получить путем защиты альдегида общей формулы (VII)
 ,
,
где R такой, как определено для общей формулы (I). Защиту можно установить путем осуществления взаимодействия с трифторметилортоформиатом и п-толуолсульфоновой кислотой в сухих условиях и инертной атмосфере, а затем с метилатом натрия в метаноле.
Соединения общей формулы (VII) являются коммерчески доступными. Как вариант, их можно получить путем осуществления взаимодействия соединения общей формулы (VIII):
 ,
,
где R такой, как определено для общей формулы (I), с 2-фторбензальдегидом. Реакцию можно проводить в слабощелочных условиях в полярном растворителе, таком как ДМСО, в инертной атмосфере при температуре от 80 до 110°С.
Соединения общей формулы (VIII) являются коммерчески доступными или могут быть получены способами, широко известными специалистам в данной области.
Другим путем получения соединения общей формулы (VII), как определено выше, является формилирование соединения общей формулы (X):
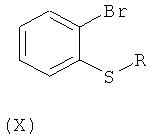 ,
,
где R такой, как определено для общей формулы (I);
при помощи н-бутиллития и диметилформамида (ДМФА) в органическом растворителе, таком как тетрагидрофуран.
Обычно указанную реакцию проводят в инертной атмосфере, например атмосфере азота, при охлаждении до -78°С во время взаимодействия с н-бутиллитием, и давая возможность нагреться до комнатной температуры после добавления ДМФА.
Соединения общей формулы (X) можно получить посредством осуществления взаимодействия 2-бромтиофенола с соединением общей формулы (XI):

в которой R такой, как определено для общей формулы (I), а Х представляет собой уходящую группу, в частности галогеновую группу, такую как хлор или бром.
Указанную реакцию можно проводить в присутствии основания, например карбоната цезия, при температуре 20-50°С, обычно 40°С.
Другим путем получения соединения общей формулы (IV) является осуществление взаимодействия натриевых солей общей формулы (IX):
 ,
,
где R такой, как определено для общей формулы (I), с 2-фторбензальдегидом. Реакцию можно проводить в таком растворителе, как диметилсульфоксид, при повышенной температуре, обычно от 80 до 110°С. Полное прохождение этой реакции может занять несколько дней.
Натриевые соли общей формулы (IX) являются коммерчески доступными.
Соединения общей формулы (IV) также можно получить непосредственно из соединений общей формулы (VII) без необходимости в стадиях постановки и снятия защиты. Согласно указанной методике охлажденный окислитель, такой как мета-хлорпероксибензойная кислота, можно добавлять к соединению общей формулы (VII), обычно при охлаждении примерно от -5 до 5°С. Реакционной смеси можно дать возможность нагреться до 15-30°С, обычно до комнатной температуры, а затем провести реакцию с метабисульфитом натрия.
Как отмечено выше, некоторые соединения общей формулы (VII) имеются в продаже.
Соединения общей формулы (I) представляют собой антагонисты рецептора CRTH2, а соединения общей формулы (II) представляют собой пролекарства для соединений общей формулы (I). Соединения общих формул (I) и (II), таким образом, можно применять в способе лечения заболеваний и состояний, опосредуемых ПГD2 или другими агонистами рецептора CRTH2, причем указанный способ включает введение пациенту, нуждающемуся в указанном лечении, подходящего количества соединения общей формулы (I) или (II).
Согласно третьему аспекту настоящего изобретения предложено соединение общей формулы (I) или (II) для применения в медицине, в частности для лечения или предотвращения заболеваний и состояний, опосредуемых ПГD2 или другими агонистами рецептора CRTH2.
Кроме того, предложено применение соединения общей формулы (I) или (II) для производства средства для лечения или предотвращения заболеваний и состояний, опосредуемых агонистами рецептора CRTH2, в частности ПГD2.
Как отмечено выше, указанные заболевания и состояния включают аллергические заболевания, астматические состояния и воспалительные заболевания, примерами которых являются: астма, включая аллергическую астму, бронхиальную астму, эндогенную, экзогенную, вызванную физической нагрузкой, медикаментозную и вызванную пылью астму, лечение кашля, включая хронический кашель, связанный с воспалительными и секреторными состояниями дыхательных путей, и ятрогенный кашель, острый и хронический ринит, включая медикаментозный ринит, вазомоторный ринит, круглогодичный аллергический ринит, сезонный аллергический ринит, полипоз носа, острые вирусные инфекции, включая простуду, инфекцию, вызванную респираторно-синцитиальным вирусом, вирусом гриппа, коронавирусом и аденовирусом, атопический дерматит, контактная гиперчувствительность (включая контактный дерматит), экзематозный дерматит, фитодерматит, фотодерматит, себорейный дерматит, герпетиформный дерматит, красный плоский лишай, склерозирующий и атрофический лишай, гангренозная пиодермия, кожный саркоидоз, дискоидная красная волчанка, пемфигус, пемфигоид, буллезный эпидермолиз, ангионевротический отек, васкулиты, токсические эритемы, кожные эозинофилии, гнездная плешивость, облысение по мужскому типу, синдром Свита, синдром Вебера-Крисчена, полиморфная эритема, целлюлит, панникулит, кожные лимфомы, немеланомный рак кожи и другие диспластические поражения; блефарит, конъюнктивит, особенно аллергический конъюнктивит, передний и задний увеит, хориоидит, аутоиммунные, дегенеративные или воспалительные нарушения, поражающие сетчатку, офтальмит; бронхит, включая инфекционный и эозинофильный бронхит, эмфизема, бронхоэктаз, «легкое фермера», гиперчувствительный пневмонит, идиопатические интерстициальные пневмонии, осложнения после пересадки легкого, сосудистые и тромботические нарушения в сосудистой системе легких, легочная гипертензия, пищевые аллергии, гингивит, глоссит, периодонтит, эзофагит, включая рефлюкс, эозинофильный гастроэнтерит, проктит, анальный зуд, глютеновая болезнь, аллергии, связанные с пищей, воспалительное заболевание кишечника, язвенный колит и болезнь Крона, мастоцитоз, а также другие опосредуемые CRTH2 заболевания, например аутоиммунные заболевания, такие как гипер-lgE-синдром, тиреоидит Хашимото, болезнь Грейвса, болезнь Аддисона, сахарный диабет, идиопатическая тромбоцитопеническая пурпура, эозинофильный фасциит (paschiitis), антифосфолипидный синдром и системная красная волчанка, СПИД, проказа, синдром Сезари, паранеопластичекий синдром, смешанные и недифференцированные заболевания соединительных тканей, воспалительные миопатии, включая дерматомиозит и полимиозит, ревматическая полимиалгия, ювенильный артрит, ревматическая лихорадка, васкулиты, включая гигантоклеточный артериит, артериит Такаясу, синдром Черджа-Строссса, узелковый полиартериит, микроскопический полиартериит, темпоральный артериит, миастения гравис, острая и хроническая боль, нейропатические болевые синдромы, нейродегенерация, осложнения на центральную и периферическую нервную систему злокачественных, инфекционных или аутоиммунных процессов, поясничная боль, семейная ирландская лихорадка, болезнь Кикучи, псориаз, акне, множественный склероз, отторжение аллотрансплантата, реперфузионное повреждение, хроническое обструктивное заболевание легких, а также ревматоидный артрит, болезнь Стилла, анкилозирующий спондилит, реактивный артрит, недифференцированная спондилоартропатия, псориатический артрит, септический артрит и другие инфекционной природы артропатии и болезни костей и остеоартрит; острые и хронические вызванные кристаллическими отложениями синовиты, включая уратную подагру, болезнь отложения кристаллов пирофосфата кальция, связанный с гидроксилапатитом кальция сухожильный синдром и синовиальное воспаление, болезнь Бехчета, первичный и вторичный синдром Шегрена, системный склероз и ограниченная склеродермия; гепатит, цирроз печени, холецистит, панкреатит, нефрит, нефритический синдром, цистит, язва Ханнера мочевого пузыря, острый и хронический уретрит, простатит, эпидидимит, оофорит, сальпингит, вульвовагинит, болезнь Пейрони, эректильная дисфункция, болезнь Альцгеймера и другие дементные заболевания; перикардит, миокардит, воспалительные и аутоиммунные кардиомиопатии, включая миокардиальный саркоидоз, ишемические реперфузионные повреждения, эндокардит, вальвулит, аортит, флебит, тромбоз, лечение распространенных видов рака и фиброзных состояний, таких как идиопатический легочный фиброз, включая криптогенный фиброзирующий альвеолит, келоиды, избыточное фиброзное рубцевание/спайки после операции, печеночный фиброз, включая печеночный фиброз, связанный с гепатитами В и С, маточный фиброз, саркоидоз, включая нейросаркоидоз, склеродермия, почечный фиброз, являющийся результатом диабета, фиброз, связанный с ревматоидным артритом, атеросклероз, включая церебральный атеросклероз, васкулит, миокардиальный фиброз, являющийся результатом инфаркта миокарда, кистозный фиброз, рестеноз, системный склероз, болезнь Дюпюитрена, фиброз, осложняющий противоопухолевое лечение и хронические инфекции, включая туберкулез, аспергиллез и другие грибковые инфекции, фиброз ЦНС после инсульта или способствование излечению без фиброзного рубцевания.
Соединения общей формулы (I) или (II) должны входить в рецептуры, разработанные соответствующим образом в зависимости от заболеваний или состояний, для лечения которых они необходимы.
Таким образом, согласно еще одному аспекту настоящего изобретения предложена фармацевтическая композиция, содержащая соединение общей формулы (I) или (II) совместно с фармацевтическим наполнителем или носителем. Также могут присутствовать другие активные вещества, которые можно считать подходящими или целесообразными для применения в отношении заболевания или состояния, подлежащего лечению или предотвращению.
Носитель, или, если присутствует более одного носителя, каждый из них, должен быть приемлемым с точки зрения совместимости с другими компонентами состава и не вредным для реципиента.
Указанные составы включают составы, подходящие для перорального, ректального, назального, бронхиального (ингаляции), местного (включая глазные капли, трансбуккальное и сублингвальное), вагинального или парентерального (включая подкожное, внутримышечное, внутривенное и внутрикожное) введения, и могут быть приготовлены любыми способами, широко известными в области фармацевтики.
Путь введения может зависеть от состояния, подлежащего лечению, но предпочтительные рецептуры композиций составлены для перорального, назального, бронхиального или местного введения.
Композицию можно приготовить, объединяя определенное выше активное средство с носителем. В общем случае, составы готовят путем однородного и тесного соединения активного средства с жидкими носителями или тонкоизмельченными твердыми носителями, или обоими этими видами носителей, и, при необходимости, придания продукту формы. Настоящее изобретение распространяется на способы производства фармацевтической композиции, включающие соединение или смешивание соединения общей формулы (I) или (II) с фармацевтически или ветеринарно приемлемым носителем или наполнителем.
Составы для перорального введения согласно настоящему изобретению могут быть представлены в виде: отдельных единиц, таких как капсулы, саше или таблетки, каждая из которых содержит заранее заданное количество активного средства; порошка или гранул; раствора или суспензии активного средства в водной жидкости или неводной жидкости; или жидкой эмульсии типа масло-в-воде или жидкой эмульсии типа вода-в-масле; или в виде болюса, и т.д.
В случае составов для перорального введения (например, таблеток или капсул) термин «приемлемый носитель» включает такие наполнители, как обычные формообразующие средства, например связующие, например, сироп, гуммиарабик, желатин, сорбит, трагакант, поливинилпирролидон (Повидон), метилцеллюлоза, этилцеллюлоза, натрий-карбоксиметилцеллюлоза, гидроксипропилметилцеллюлоза, сахароза и крахмал; наполнители и носители, например, кукурузный крахмал, желатин, лактоза, сахароза, микрокристаллическая целлюлоза, каолин, маннит, дикальцийфосфат, хлорид натрия и альгиновая кислота; и смазочные вещества, такие как стеарат магния, стеарат натрия, и другие стеараты металлов, глицерилстеарат, стеариновая кислота, силиконовая жидкость, тальк, воски, масла и коллоидный диоксид кремния. Также можно использовать ароматизаторы, такие как мята, масло гаультерии, вишневый ароматизатор и тому подобное. Может быть желательным добавление красящего средства для того, чтобы сделать дозировочную форму легко заметной. На таблетки также можно наносить покрытия способами, широко известными в данной области.
Таблетки можно изготовить путем прессования или отливки, возможно, с одним или более вспомогательными компонентами. Прессованные таблетки можно получить путем прессования в подходящей машине активного средства в легкосыпучей форме, такой как порошок или гранулы, возможно, смешанного со связующим, смазочным веществом, инертным разбавителем, консервантом, поверхностно-активным или диспергирующим средством. Формованные таблетки можно изготовить путем формования в подходящей машине смеси порошкообразных компонентов, увлажненных инертным жидким разбавителем. На таблетки, при желании, можно нанести покрытие или желобок, их можно изготовить таким образом, чтобы обеспечить замедленное или контролируемое высвобождение активного средства.
Другие составы, подходящие для перорального введения, включают леденцы, содержащие активное средство в ароматизированной основе, обычно сахарозе и гуммиарабике или трагаканте; пастилки, содержащие активное средство в инертной основе, такой как желатин и глицерин, или сахароза и гуммиарабик; и жидкости для полоскания рта, содержащие активное средство в подходящем жидком носителе.
Для местного нанесения на кожу соединения общей формулы (I) или (II) могут быть изготовлены в виде крема, мази, геля, раствора, суспензии и т.д. Составы кремов или мазей, которые можно применять в качестве лекарственных препаратов, представляют собой обычные составы, широко известные в данной области, например, описанные в стандартных пособиях по фармацевтике, таких как Британская Фармакопея.
Соединения общей формулы (I) или (II) можно применять для лечения дыхательных путей путем назального, бронхиального или трансбуккального введения, например, аэрозолей или спреев, которые могут распылять фармакологически активный компонент в виде порошка или в виде капель раствора или суспензии. Фармацевтические композиции с возможностью распыления порошка обычно содержат, кроме активного компонента, жидкий пропеллент с температурой кипения ниже комнатной температуры, и, при желании, вспомогательные вещества, такие как жидкие или твердые неионогенные или анионные поверхностно-активные вещества и/или разбавители. Фармацевтические композиции, в которых фармацевтически активный компонент находится в растворе, содержат, помимо него, подходящий пропеллент, и, кроме того, при необходимости, дополнительный растворитель и/или стабилизатор. Вместо пропеллента также можно использовать сжатый воздух, который возможно получать по необходимости с помощью подходящего устройства для сжатия и расширения.
Парентеральные композиции в общем случае должны быть стерильными.
Обычно доза соединения может составлять примерно от 0,01 до 100 мг/кг; так чтобы обеспечить концентрацию лекарственного средства в плазме крови на уровне концентрации, эффективной для ингибирования воздействия ПГD2 на рецептор CRTH2. Точное количество соединения общей формулы (I) или (II), которое является терапевтически эффективным, и наилучший путь, которым вводят указанное соединение, легко может определить средний специалист в данной области путем сравнения уровня средства в крови с концентрацией, необходимой для достижения терапевтического эффекта.
Соединения общей формулы (I) или (II) можно применять в сочетании с одним или более активным агентом, применяемым для лечения перечисленных выше заболеваний и состояний, хотя указанные активные агенты не обязательно являются ингибиторами ПГD2 на рецепторе CRTH2.
Таким образом, вышеописанные фармацевтические композиции могут дополнительно содержать один или более из указанных активных агентов.
Также предложено применение соединения общей формулы (I) или (II) для получения средства для лечения заболеваний и состояний, опосредуемых агонистами рецептора CRTH2, особенно ПГD2, причем указанное средство содержит дополнительный активный агент, применимый для лечения этих же заболеваний и состояний.
Указанные дополнительные активные агенты могут представлять собой другие антагонисты рецептора CRTH2 или могут обладать полностью отличным образом действия. Они включают существующие средства лечения аллергических и других воспалительных заболеваний, включая:
Суплатаст тозилат и похожие соединения;
Агонисты адренорецепторов от β1 до β4, такие как метапротеренол, изопротеренол, изопреналин, альбутерол, сальбутамол, формотерол, салметерол, тербуталин, орципреналин, битолтерола мезилат и пирбутерол или метилксантанины, такие как теофиллин и аминофиллин, стабилизаторы тучных клеток, такие как кромогликат натрия или антагонисты мускариновых рецепторов (М1, М2 или М4);
Антигистаминные, например антагонисты гистаминового рецептора H1, такие как лоратадин, цетиризин, дезлоратадин, фексофенадин, астемизол, азеластин и хлорфенирамин или антагонисты гистаминовых рецепторов H2 или Н4;
Агонисты адренорецепторов α1 и α2, такие как пропилгекседрин, фенилэфрин, фенилпропаноламин, псевдоэфедрин, нафазолина гидрохлорид, оксиметазолина гидрохлорид, тетрагидрозолина гидрохлорид, ксилометазолина гидрохлорид и этилнорэпинефрина гидрохлорид;
Миметики инсулиноподобного фактора роста (ИФР-1);
Ингибиторы матриксной металлопротеиназы (ММП), например ингибиторы стромелизинов, коллагеназ, желатиназ и аггреканазы, особенно коллагеназы-1, коллагеназы-2, коллагеназы-3, стромелизина-1, стромелизина-2, стромелизина-3 и ММП-12;
Модуляторы функций хемокиновых рецепторов, например, CCR1, CCR2, CCR2A, CCR2B, CCR3, CCR4, CCR5, CCR6, CCR7, CCR8, CCR9, CCR10 и CCR11 (для семейства С-С) или CXCR1, CXCR2, CXCR3, CXCR4 и CXCR5 (для семейства С-Х-С) и CX3CR1 для семейства С-Х3-С;
Противовирусные средства, такие как Вирасепт, AZT, ацикловир и фамцикловир, и антисептические соединения, такие как Valant;
Сердечнососудистые средства, например блокаторы кальциевых каналов, гиполипидемические средства, такие как статины, фибраты, бета-блокаторы, ингибиторы АПФ, антагонисты рецептора ангиотензина-2 и ингибиторы агрегации тромбоцитов;
Средства, действующие на ЦНС, например антидепрессанты, такие как сертралин, лекарственные препараты для лечения болезни Паркинсона, такие как депренил, L-допа, Реквип, Мирапекс, ингибиторы МАО-Б, такие как селегин и расагилин, ингибиторы КОМТ, такие как Тасмар, ингибиторы А-2, ингибиторы обратного захвата дофамина, антагонисты НМДА, антагонисты никотина, агонисты дофамина, ингибиторы нейрональной синтазы оксида азота и лекарственные препараты для лечения болезни Альцгеймера, такие как донепезил, такрин, ингибиторы ЦОГ-2, пропентофиллин или метрифонат;
Ингибиторы триптазы;
Антагонисты фактора активации тромбоцитов (ФАТ);
Ингибиторы интерлейкин-превращающего фермента (ICE);
Ингибиторы ИМФДГ;
Ингибиторы молекул адгезии, включая антагонисты VLA-4;
Катепсины;
Ингибиторы МАП-киназы;
Ингибиторы глюкозо-6-фосфатдегидрогеназы;
Антагонисты рецепторов кинина-B1 и B2,
Средства против подагры, такие как колхицин;
Ингибиторы ксантиноксидазы, такие как аллопуринол;
Урикозурические средства, такие как пробенецид, сульфинилпиразон и бензбромарон;
Средства, усиливающие секрецию гормона роста;
Трансформирующий фактор роста бета (ТФРβ);
Тромбоцитарный фактор роста (ТрФР);
Фактор роста фибробластов, например основной фактор роста фибробластов;
Гранулоцитарно-макрофагальный колониестимулирующий фактор (ГМ-КСФ);
Капсаицин;
Антагонисты тахикининовых рецепторов NK1 и NK3, такие как NKP-608C, талнетант и D-4418;
Ингибиторы эластазы, такие как UT-77 и ZD-0892;
Ингибиторы индуцибельной синтазы оксида азота (иСОА);
Средства против остеопороза, такие как ралоксифен, дролоксифен, лазофоксифен или фосамакс;
Антихолинергические средства, такие как ипратропий бромид, тиотропий бромид, окситропий бромид, пирензепин и телензепин;
Антагонисты лейкотриенов (антагонисты ЛТВ4, ЛТD4 и ЛТE4), такие как фенотиазин-3-оны, такие как L-651,392, амидиновые соединения, такие как CGS-25019 с, бензоксазоламины, такие как онтазоласт, бензолкарбоксимидамиды, такие как BIIL 284/260 и такие соединения, как зафирлукаст, аблукаст, монтелукаст, пранлукаст, верлукаст, RG-12525, Ro-245913, иралукаст и BAY×7195;
Ингибиторы биосинтеза лейкотриенов, такие как ингибиторы 5-липоксигеназы, ингибиторы 5-липоксигеназ-активирующего протеина (ЛОАП), такие как зилеутон, АВТ-761, фенлеутон, тепоксалин, Abbott-79175, N-(5-замещенные)-тиофен-2-алкилсульфонамиды, 2,6-ди-трет-бутилфенолгидразоны, метокситетрагидропираны, такие кaк ZD2138, SB-210661, пиридинил-замещенные-2-цианонафталиновые соединения, такие как L-739010, 2-цианохинолиновые соединения, такие как L-746,530, индольные и хинолиновые соединения, такие как МK-591, МK-886 и BAY x 1005;
Ингибиторы фосфодиэстеразы, включая ингибиторы ФДЭ4, такие как ингибиторы ФДЭ4D;
Лекарственные средства на основе антител к IgE, такие как омализумаб;
Противоинфекционные средства, такие как фусидовая кислота (особенно для лечения атопического дерматита);
Противогрибковые средства, такие как клотримазол (особенно для лечения атопического дерматита);
Иммунодепрессанты, такие как такролимус и особенно пимекролимус в случае воспалительного заболевания кожи, и, как вариант, FK-506, рапамицин, циклоспорин, азатиоприн или метотрексат;
Антипролиферативные/противоопухолевые лекарственные средства, такие как алкилирующие агенты, например цисплатин, карбоплатин, циклофосфамид, азотистый иприт, мелфалан, хлорамбуцил, бусульфан и нитрозомочевины, антиметаболиты, например, антифолаты, такие как фторпиримидины, такие как 5-фторурацил и тегафур, ралтитрексед, метотрексат, цитозина арабинозид, гидроксимочевина, гемцитабин и паклитаксел;
Противоопухолевые антибиотики, такие как антрациклины, такие как адриамицин, блеомицин, доксорубицин, дауномицин, эпирубицин, идарубицин, митомицин-С, дактиномицин и метрамицин;
Антимитотические средства, такие как алкалоиды барвинка, включая винкристин, винбластин, виндесин и винорелбин, и таксоиды, такие как таксол и таксотер, и ингибиторы топоизомеразы, такие как эпиподофиллотоксины, подобные этопозиду и тенипозиду, амсакрин, топотекан и камптотецин;
Цитостатические средства, такие как антиэстрогены, такие как тамоксифен, торемифен, ралоксифен, дролоксифен и иодоксифен, негативные регуляторы рецепторов эстрогена, такие как фулвестрант, антиандрогены, такие как бикалутамид, флутамид, нилутамид и ципротерона ацетат, антагонисты или агонисты ГНРГ (гонадотропин-высвобождающего гормона), такие как гозерелин, лейпрорелин и бусерелин, прогестероны, такие как мегестрола ацетат, ингибиторы ароматазы, такие как анастрозол, летрозол, боразол и эксеместан, и ингибиторы 5α-редуктазы, такие как финастерид;
Средства, ингибирующие инвазию раковых клеток, например ингибиторы металлопротеиназы, такие как маримастат, и ингибиторы функции рецептора урокиназного активатора плазминогена;
Ингибиторы функции фактора роста, например антитела к фактору роста, антитела к рецептору фактора роста, например антитело к erbb2 - трастузумаб и антитело к еrbb1 - цетуксимаб, ингибиторы фарнезилтрансферазы, ингибиторы тирозинкиназы и ингибиторы серин- или треонинкиназы, например ингибиторы семейства эпидермального фактора роста, такие как ингибиторы тирозинкиназы семейства ЭФР, такие как N-(3-хлор-4-фторфенил)-7-метокси-6-(3-морфолинопропокси)хиназолин-4-амин (гефитиниб), N-(3-этинилфенил)-6,7-бис(2-метоксиэтокси)хиназолин-4-амин (эрлотиниб) и 6-акриламидо-N-(3-хлор-4-фторфенил)-7-(3-морфолинопропокси)хиназолин-4-амин (Cl 1033), или ингибиторы семейств фактора роста тромбоцитов и фактора роста гепатоцитов;
Антиангиогенные средства, особенно ингибирующие действие фактора роста сосудистого эндотелия, например антитело к фактору роста клеток сосудистого эндотелия бевацизумаб, а также соединения, действующие по другим механизмам, например линомид, ингибиторы функции интегрина-avb3 и ангиостатин;
Средства, повреждающие сосуды, такие как Комбретастатин А4;
Антисмысловые лекарственные средства, такие как нацеленные на вышеперечисленные мишени, например ISIS 2503, анти-Ras антисмысловые;
Средства генной терапии, включая средства для замещения аберрантных генов, таких как аберрантный р53, или аберрантный BRCA1 или BRCA2, GDEPT (лечение пролекарствами, активируемыми генуправляемыми ферментами), ферменты цитозиндезаминаза, тимидинкиназа или бактериальная нитроредуктаза, или средства для увеличения толерантности пациента к химиотерапии или радиотерапии, такие как мультирезистентная генотерапия;
Иммунотерапевтические средства, включая способы терапии in vivo, и ex vivo, направленные на увеличение иммуногенности опухолевых клеток пациента, такие как трансфицирование цитокинами, такими как ИЛ2, ИЛ4 или ГМ-КСФ, подход к снижению инертности Т-клеток, подходы, использующие трансфицированные иммунные клетки, такие как цитокин-трансфицированные дентритные клетки, или подходы, использующие цитокин-трансфицированные опухолевые линии клеток, или антитела к идиотипам;
Кортикостероиды, такие как преднизон, преднизолон, флунисолид, триамицинолона ацетонид, беклометазона дипропионат, будесонид, флутиказона пропионат и мометазона фуроат, и гиалуроновые кислоты, такие как гиалган и синвиск, и антагонисты рецептора Р2Х7;
Лекарственные средства, способствующие ответу цитокинов Th1, такие как интерфероны, ФНО или ГМ-КСФ.
Антагонисты CRTH2 можно также сочетать с:
Другими антагонистами ПГD2, действующими на другие рецепторы, такими как антагонисты рецепторов DP;
Ингибиторами фосфодиэстеразы типа 4, такими как циломиласт;
Лекарственными средствами, модулирующими производство цитокинов, такими как: ингибиторы фермента, конвертирующего ФНОα (ТАСЕ), моноклональные антитела к ФНО, молекулы иммуноглобулина к рецептору ФНО, ингибиторы других изоформ ФНО, неселективные ингибиторы ЦОГ-1/ЦОГ-2, такие как пироксикам, диклофенак, пропионовые кислоты, такие как напроксен, флубипрофен, фенопрофен, кетопрофен и ибупрофен, фенаматы, такие как мефенаминовая кислота, индометацин, сулиндак и апазон, пиразолоны, такие как фенилбутазон, салицилаты, такие как аспирин; ингибиторы ЦОГ-2, такие как мелоксикам, целекоксиб, рофекоксиб, вальдекоксиб и эторикоксиб, низкими дозами метотрексата, лефлуномида, циклесонида, гидроксихлорохина, d-пеницилламина, ауранофин или препараты золота для перорального или парентерального введения;
Лекарственными средствами, модулирующими активность Th2 цитокинов ИЛ-4 и ИЛ-5, такими как блокирующие моноклональные антитела и растворимые рецепторы;
Агонистами РАПП-γ, такими какросиглитазон; или с
Антителами к РСВ, такими как Синагис (паливизумаб), и средствами, которые можно применять для лечения риновирусной инфекции в будущем, например, интерферон-бета и другие интерфероны.
Согласно еще одному из аспектов настоящего изобретения предложен продукт, который содержит соединение общей формулы (I) или (II) и одно или более из вышеперечисленных агентов в виде комбинированного препарата для одновременного, раздельного или последовательного применения при лечении заболевания или состояния, опосредуемого действием ПГD2 на рецептор CRTH2.
Далее, настоящее изобретение будет описано более детально со ссылками на следующие не ограничительные примеры и рисунки, в которых:
ФИГУРА 1 представляет собой график зависимости от времени концентрации Соединения 1 в крови крыс, получавших Соединение 1 перорально в дозировке 3 мг/кг.
ФИГУРА 2 представляет собой график зависимости от времени концентрации Соединения 2 в крови крыс, получавших Соединение 2 перорально в дозировке 3 мг/кг.
ФИГУРА 3 представляет собой график зависимости от времени концентрации Соединения 3 в крови крыс, получавших Соединение 3 перорально в дозировке 3 мг/кг.
ФИГУРА 4 представляет собой график зависимости от времени концентрации Соединения Сравнения А в крови крыс, получавших Соединение А перорально в дозировке 3 мг/кг.
ФИГУРА 5 представляет собой график зависимости от времени концентрации Соединения Сравнения Б в крови крыс, получавших Соединение В перорально в дозировке 3 мг/кг.
На ФИГУРЕ 6 показано воздействие доз 0,0001, 0,001, 0,01 и 0,1 мг/кг Соединения 1 на индуцированную ДК-ПГD2 эозинофилию у крыс.
На ФИГУРЕ 7 показано воздействие доз 0,001, 0,01 0,1 и 1,0 мг/кг Соединения 2 на индуцированную ДК-ПГD2 эозинофилию у крыс.
На ФИГУРЕ 8 показано воздействие доз 0,001, 0,01 0,1 и 1,0 мг/кг Соединения 3 на индуцированную ДК-ПГD2 эозинофилию у крыс.
На ФИГУРЕ 9 показано воздействие доз 0,01, 0,1 и 1,0 мг/кг Соединения А на индуцированную ДК-ПГD2 эозинофилию у крыс.
На ФИГУРЕ 10 показано воздействие доз 0,001, 0,01 0,1 и 1,0 мг/кг Соединения Б на индуцированную ДК-ПГD2 эозинофилию у крыс.
ПОЛУЧЕНИЕ СОЕДИНЕНИЙ ОБЩЕЙ ФОРМУЛЫ I
Соединения в Примерах 1-3 получены в соответствии со следующими реакционными схемами.
Схема 1
Схема 2
Схема 3

ПРИМЕР 1 - Получение 2-(5-фтор-2-метил-3-(2-(фенилсульфонил)бензил)-1Н-индол-1-ил)уксусной кислоты (Соединение 1).
Из коммерчески доступной натриевой соли бензолсульфоновой кислоты и 2-фторбензальдегида.
а) Операция И. (SNAr) для получения 2-(фенилсульфонил)бензальдегида
К раствору 2-фторбензальдегида (5,00 мл, 47,6 ммоль) в диметилсульфоксиде (45 мл) добавляют натриевую соль бензолсульфоновой кислоты (8,60 г, 52,4 ммоль) и нагревают полученную смесь до 100°С. При нагревании соль сульфоновой кислоты растворяется. Полученный раствор выдерживают нагретым до 100°С в течение 3 дней. Охлаждают реакционную смесь до комнатной температуры и добавляют воду (50 мл). Полученную смесь экстрагируют этилацетатом, объединенные органические экстракты промывают насыщенным раствором соли, высушивают над MgSO4 и концентрируют в вакууме. Черновой материал очищают высокоскоростной хроматографией на силикагеле, элюируя от 25% этилацетат:петролейный эфир (40-60°С) до 33% этилацетат:петролейный эфир (40-60°С), чтобы получить (4,12 г, 16,7 ммоль, 35%).
δн (300 МГц, d6-ДМСО) 10,68 (1Н, с, СНО), 8,26-8,17 (1Н, м, Ar), 8,08-7,99 (2Н, м, Ar), 7,99-7,89 (3Н, м, Ar) и 7,80-7,62 (3Н, м, Ar).
б) Операция Д. (Восстановительное алкилирование) для получения этилового эфира 2-(5-фтор-2-метил-3-(2-(фенилсульфонил)бензил)-1Н-индол-1-ил)уксусной кислоты.
К раствору этилового эфира 2(5-фтор-2-метил-1Н-индол-1-ил)уксусной кислоты (1,29 г, 1,49 ммоль), 2-(фенилсульфонил)бензальдегида (1,50 г, 6,10 ммоль) и триэтилсилана (4,30 мл, 27,0 ммоль) в дихлорметане (40 мл) добавляют трифторуксусную кислоту (1,25 мл, 16,5 ммоль) по каплям в атмосфере N2 при 0°С в течение 30 минут. Реакционную смесь нагревают до комнатной температуры и перемешивают в течение 2 часов. Добавляют насыщенный водный раствор гидрокарбоната натрия и экстрагируют полученный продукт дихлорметаном. Объединенные органические экстракты промывают насыщенным раствором соли, высушивают над MgSO4 и концентрируют в вакууме, получая коричневое масло, которое растирают с петролейным эфиром (40-60°С) для получения белого твердого вещества (1,34 г, 2,88 ммоль, 52%).
δн (300 МГц, CDCl3) 8,36-8,30 (1Н, м, Ar), 8,00-7,93 (2Н, м, Ar), 7,68-7,52 (3Н, м, Ar), 7,45-7,33 (2Н, м, Ar), 7,05 (1 Н, дд, J 8,6 и 4,3 Гц, Ar), 6,96-6,90 (1Н, м, Ar), 6,82 (1Н, тд, J 9,1 и 2,7 Гц, Ar), 6,24 (1Н, дд, J 9,5 и 2,4 Гц, Ar), 4,76 (2Н, с, NCH2), 4,22 (2Н, с, ArCH2Ar), 4,21 (2Н, кв, J 7,1 Гц, CH2CH3), 2,14 (3Н, с, СН3) и 1,27 (3H, T, J 7,1 Гц, СН2СН3).
в) Операция Е (Омыление) для получения 2-(5-фтор-2-метил-3-(2-(фенилсульфонил)бензил)-1Н-индол-1-ил)уксусной кислоты
К раствору сложноэфирного продукта, полученного на стадии (б) (1,33 г, 2,86 ммоль) в тетрагидрофуране (15 мл), добавляют при перемешивании водный раствор КОН (500 мг, 8,57 ммоль) в воде (15 мл). Спустя 2 часа удаляют тетрагидрофуран при пониженном давлении и щелочной водный слой промывают этилацетатом. Оставшийся водный слой подкисляют HCl (2 N) и экстрагируют этилацетатом. Объединенные органические экстракты промывают насыщенным раствором соли, высушивают над MgSO4 и концентрируют в вакууме для получения коричневого твердого вещества, которое растирают со смесью диэтилового эфира и петролейного эфира (40-60°С), чтобы получить белое твердое вещество (1,14 г, 2,61 ммоль, 91%).
δн (300 МГц, d6-ДМСО) 13,00 (1Н, уш. с, CO2H), 8,26-8,20 (1Н, м, Ar), 7,99-7,93 (2Н, м, Ar), 7,80-7,62 (3Н, м, Ar), 7,55-7,48 (2Н, м, Ar), 7,34 (1Н, дд, J 8,6 и 4,3 Гц, Ar), 6,93-6,87 (1Н, м, Ar), 6,81 (1Н, тд, J 9,1 и 2,7 Гц, Ar), 6,18 (1Н, дд, J 9,7 и 2,4 Гц, Ar), 4,95 (2Н, с, NCH2), 4,14 (2Н, с, ArCH2Ar) и 2,06 (3Н, с, СН3), Тр=4,62 мин (95%), m/z (M+H)+ 438,3.
ПРИМЕР 2 - Получение 2-(3-(2-(4-хлорфенилсульфонил)бензил)-5-фтор-2-метил-1Н-индол-1-ил)уксусной кислоты (Соединение 2).
Из продажного 2-(4-хлорфенилтио)бензальдегида.
а) Операция К. (Прямое окисление) для получения 2-(4-хлорфенилсульфонил)бензальдегида.
К раствору 2-(4-хлорфенилтио)бензальдегида (2,00 г, 8,00 ммоль) в дихлорметане (20 мл) при 0°С добавляют мета-хлорпероксибензойную кислоту (77% макс., 5,40 г, 24,17 ммоль) порциями в течение 15 минут, затем нагревают до комнатной температуры и перемешивают в течение 2 часов. Осторожно добавляют водный раствор метабисульфита натрия до тех пор, пока не закончится выделение пузырьков газа. Полученный раствор экстрагируют дихлорметаном, объединенные органические экстракты промывают NaOH (1 N), затем насыщенным раствором соли, высушивают над MgSO4 и концентрируют в вакууме, получая белое твердое вещество (1,05 г, 3,74 ммоль, 46%).
δн (300 МГц, d6-ДМСО) 10,69 (1Н, с, СНО), 8,25-8,18 (1Н, м, Ar), 8,07-8,00 (2Н, м, Ar), 8,00-7,90 (3Н, м, Ar) и 7,81-7,64 (3Н, м, Ar).
б) Операция Д. (Восстановительное алкилирование) для получения этилового эфира 2-(3-(2-(4-хлорфенилсульфонил)бензил)-5-фтор-2-метил-1Н-индол-1-ил)уксусной кислоты.
δн (300 МГц, CDCl3) 8,33-8,26 (1Н, м, Ar), 7,89-7,82 (2Н, м, Ar), 7,53-7,46 (2Н, м, Ar), 7,44-7,73 (2Н, м, Ar), 7,06 (1Н, дд, J 8,6 и 4,3 Гц, Ar), 7,02-6,96 (1Н, м, Ar), 6,84 (1Н, тд, J 9,1 и 2,7 Гц, Ar), 6,33 (1Н, дд, J 9,5 и 2,4 Гц, Ar), 4,76 (2Н, с, NCH2), 4,23 (2Н, с, ArCH2Ar), 4,22 (2Н, кв, J 7,2 Гц, CH2CH3), 2,16 (3Н, с, СН3) и 1,27 (3Н, т, J 7,2 Гц, СН2СН3).
в) Операция Е. (Омыление) для получения 2-(3-(2-(4-хлорфенилсульфонил)бензил)-5-фтор-2-метил-1Н-индол-1-ил)уксусной кислоты.
δн (300 МГц, d6-ДМСО) 13,01 (1Н, уш, с, CO2H), 8,27-8,20 (1Н, м, Ar), 7,97-7,90 (2Н, м, Ar), 7,74-7,67 (2Н, м, Ar), 7,57-7,51 (2Н, м, Ar), 7,34 (1Н, дд, J 8,7 и 4,3 Гц, Ar), 7,00-6,93 (1Н, м, Ar), 6,81 (1Н, тд, J 9,4 и 2,6 Гц, Ar), 6,16 (1Н, дд, J 9,8 и 2,6 Гц, Ar), 4,95 (2Н, с, NCH2), 4,17 (2Н, с, ArCH2Ar) и 2,12 (3Н, с, СН3), Тр=4,04 мин (96%), m/z (M+H)+ 472,0.
ПРИМЕР 3 - Получение 2-(5-фтор-3-(2-(4-фторфенилсульфонил)бензил)-2-метил-1Н-индол-1-ил)уксусной кислоты (Соединение 3).
а) Операция A. (SNAr) для получения 2-(4-фторфенилтио)бензальдегида
К суспензии 4-фторфенилтиола (0,86 мл, 8,06 ммоль) и К2СО3 (2,50 г, 18,12 ммоль) в ДМСО (5 мл) добавляют 2-фторбензальдегид (1,00 г, 8,06 ммоль) в атмосфере N2 и нагревают полученную смесь при 100°С в течение 3 часов. Реакционную смесь охлаждают до комнатной температуры и добавляют воду (20 мл). Полученную смесь экстрагируют этилацетатом, объединенные органические экстракты промывают насыщенным раствором соли, высушивают над MgSO4 и концентрируют в вакууме, чтобы получить желтое твердое вещество (1,20 г, 5,17 ммоль, 64%).
δн (300 МГц, d6-ДМСО) 10,23 (1Н, с, СНО), 7,98 (1Н, дд, J 7,3 и 1,7 Гц, Ar), 7,63-7,49 (3Н, м, Ar), 7,45-7,32 (3Н, м, Ar) и 6,88 (1Н, д, J 7,7 Гц, Ar).
б) Операция Б. (Защита альдегида) для получения (2-(диметоксиметил)фенил)(4-фторфенил)сульфида
К раствору альдегидного продукта стадии (а) (1,20 г, 5,17 ммоль) и триметилортоформиата (0,62 мл, 0,58 ммоль) в безводном метаноле (80 мл) добавляют п-толуолсульфоновую кислоту (0,10 г, 0,58 ммоль) в атмосфере N2 и перемешивают полученную смесь при комнатной температуре в течение 72 часов. Добавляют раствор метилата натрия в метаноле (0,12 мл, 25% об., 0,58 ммоль) и удаляют растворитель в вакууме, получая бесцветное масло (1,50 г). Дальнейшую очистку не проводят.
δн (300 МГц, CDCl3) 7,64 (1Н, дд, J 7,3 и 2,0 Гц, Ar), 7,38-7,30 (2Н, м, Ar), 7,29-7,19 (2Н, м, Ar), 7,15-7,10 (1Н, м, Ar), 7,07-6,99 (2Н, м, Ar), 5,72 (1Н, с, СН(СН3)2) и 3,37 (6Н, с, СН(СН3)2).
в) Операция В. (Окисление) для получения 1-(диметоксиметил)-2-(4-фторфенилсульфонил)бензола
К раствору сульфидного продукта стадии (б) (1,50 г) в дихлорметане (40 мл) добавляют 3-хлорпероксибензойную кислоту (4,60 г, 20,59 ммоль) порциями в течение 30 минут при 0°С. Реакционную смесь нагревают до комнатной температуры и перемешивают в течение 2 часов. Добавляют водный раствор метабисульфита натрия (50 мл) и экстрагируют полученный продукт дихлорметаном. Объединенные органические экстракты промывают NaOH (50 мл, 1 N), а затем насыщенным раствором соли, высушивают над MgSO4 и концентрируют в вакууме для получения желтого масла (1,40 г, 4,52 ммоль, 87% за 2 стадии).
δн (300 МГц, CDCl3) 8,11 (1Н, дд, J 8,1 и 1,5 Гц, Ar), 7,85 (2Н, дд, J 8,9 и 5,0 Гц Ar), 7,76 (1Н, дд, J 7,9 и 1,5 Гц, Ar), 7,58 (1Н, ддд, J 7,8, 7,6 и 1,4 Гц, Ar), 7,47 (1Н, ддд, J 7,8, 7,6 и 1,4 Гц, Ar), 7,14-7,05 (2Н, м, Ar), 6,12 (1Н, с, СН(СН3)2) и 3,12 (6Н, с, СН(СН3)2).
г) Операция Г. (Снятие защиты с ацеталя) для получения 2-(4-фторфенилсульфонил)бензальдегида
К раствору 1-(диметоксиметил)-2-(4-фторфенилсульфонил)бензола (1,40 г, 4,52 ммоль) в тетрагидрофуране (20 мл) добавляют водный раствор серной кислоты (20 мл, 2% раствор) и перемешивают при комнатной температуре в течение 12 часов. Добавляют твердый К2СО3 до тех пор, пока не прекратится выделение пузырьков газа и раствор не станет щелочным. Полученный раствор экстрагируют этилацетатом, объединенные органические экстракты промывают насыщенным раствором соли, высушивают над MgSO4 и концентрируют в вакууме, получая желтое твердое вещество (0,90 г, 340 ммоль, 75%).
δн (300 МГц, CDCl3) 10,85 (1Н, с, СНО), 8,21-8,16 (1Н, м, Ar), 8,08-8,02 (1Н, м, Ar), 7,97-7,90 (2Н, м, Ar), 7,80-7,74 (2Н, м, Ar) и 7,27-7,19 (2Н, м, Ar).
д) Операция Д. (Восстановительное алкилирование) для получения 2-(5-фтор-3-(2-(4-фторфенилсульфонил)бензил)-2-метил-1Н-индол-1-ил)ацетата.
δн (300 МГц, CDCl3) 8,32-8,26 (1Н, м, Ar), 8,00-7,90 (2Н, м, Ar), 7,43-7,37 (2Н, м, Ar), 7,26-7,17 (2Н, м, Ar), 7,06 (1Н, дд, J 8,8 и 4,3 Гц, Ar), 7,00-6,94 (1Н, м, Ar), 6,84 (1Н, тд, J 9,1 и 2,6 Гц, Ar), 6,32 (1Н, дд, J 9,5 и 2,6 Гц, Ar), 4,77 (2Н, с, NCH2), 4,24 (2Н, с, ArCH2Ar), 4,22 (2Н, кв, J 7,2 Гц, CH2CH3), 2,16 (3Н, с, СН3) и 1,27 (3H, T, J 7,2 Гц, СН2СН3).
е) Операция Е. (Омыление) для получения 2-(5-фтор-3-(2-(4-фторфенилсульфонил)бензил)-2-метил-1Н-индол-1-ил)уксусной кислоты.
δн (300 МГц, d6-ДМСО) 13,00 (1Н, уш, с CO2H), 8,27-8,20 (1Н, м, Ar), 8,08-8,00 (2Н, м, Ar), 7,60-7,45 (4Н, м, Ar), 7,35 (1Н, дд, J 8,7 и 4,3 Гц, Ar), 6,99-6,93 (1Н, м, Ar), 6,83 (1Н, тд, J 9,0 и 2,3 Гц, Ar), 6,17 (1Н, дд, J 9,8 и 2,6 Гц, Ar), 4,98 (2Н, с, NCH2), 4,18 (2Н, с, ArCH2Ar) и 2,13 (3Н, с, СН3), Тр=4,60 мин (95%), m/z (M+H)+ 456,3.
ПРИМЕР 4 - Тест на изменение формы эозинофилов цельной крови человека.
Исследовали действие Соединений 1-3 на индуцируемое ПГD2 изменение формы эозинофилов и сравнивали с Соединениями Сравнения А-Ж.
Соединение Сравнения А представляет собой (5-фтор-2-метил-3-хинолин-2-илметил-индол-1-ил)-уксусную кислоту.
Соединение Сравнения Б представляет собой [5-фтор-3-(4-метансульфонил-бензил)-2-метил-индол-1-ил]-уксусную кислоту.
Соединение Сравнения В представляет собой 2-{5-фтор-2-метил-3-[4-(фенилсульфонил)бензил]-1Н-индол-1-ил}уксусную кислоту (региоизомер по положению 4 Соединения 1).
Соединение Сравнения Г представляет собой 2-{3-[4-(4-хлорфенилсульфонил)бензил]-5-фтор-2-метил-1Н-индол-1-ил}уксусную кислоту (региоизомер по положению 4 Соединения 2).
Соединение Сравнения Д представляет собой 2-{5-фтор-3-[4-(4-фторфенилсульфонил)бензил]-2-метил-1Н-индол-1-ил}уксусную кислоту (региоизомер по положению 4 Соединения 3).
Соединение Сравнения Е представляет собой 2-{5-фтор-2-метил-3-[3-(фенилсульфонил)бензил]-1Н-индол-1-ил}уксусную кислоту (региоизомер по положению 3 Соединения 1).
Соединение Сравнения Ж представляет собой 2-{3-[3-(4-хлорфенилсульфонил)бензил]-5-фтор-2-метил-1Н-индол-1-ил}уксусную кислоту (региоизомер по положению 3 Соединения 2).
СПОСОБЫ
Тест на изменение формы в цельной крови.
Соединения (1 µл, конечная концентрация × 200) добавляли непосредственно к 200 µл цельной крови, хорошо перемешивали и инкубировали в течение 15 минут, 37°С, 5% CO2. Спустя указанное время фиксировали форму клеток путем добавления 300 µл буферного раствора Cytofix™ (BD Biosciences), 15 мин на льду. К фиксированным клеткам добавляли 10 мл лизисного буферного раствора RBC, инкубировали в течение 5 мин при комнатной температуре и центрифугировали, 300 g в течение 5 мин. Надклеточную жидкость (содержащую лизированные эритроциты) удаляли и повторяли стадию лизирования. Лейкоциты ресуспендировали в 250 µл среды RPMI/10% ЭТС и анализировали изменение формы при помощи FACS (флуоресцентно-активированный клеточный сортинг). Эозинофилы выделяли на основании их аутофлуоресценции и для каждого образца подсчитывали 2000 эозинофилов. Данные анализировали, повторяя трижды.
РЕЗУЛЬТАТЫ
Результаты, полученные в тесте на изменение формы эозинофилов, приведены в Таблице 1.
Все соединения связывались с CRTH2 с Ki не более 0,012 µM. Из Таблицы 1 можно видеть, что все Соединения 1-3 обладают превосходными значениями IC50 в указанном тесте. Соединение Сравнения Б имеет в указанном тесте активность, сравнимую с активностью Соединений 1-3, тогда как активность Соединение Сравнения А гораздо ниже. Однако Соединения Сравнения В-Ж, которые являются пара- и мета-региоизомерами Соединений 1-3, обладают в указанном тесте сравнительно слабой активностью (порядка от 10 до 1000 раз ниже, чем у Соединений 1-3).
ПРИМЕР 5 - Изучение фармакокинетики соединений общей формулы (I) при пероральном введении крысам
МЕТОДИКИ ЭКСПЕРИМЕНТА
а) Взвешивание крыс
Крыс взвешивали в день введения дозы.
б) Приготовление дозы
Исследуемое вещество готовили в виде 0,3 мг/мл суспензии в 1% карбоксиметилцеллюлозе (КМЦ).
в) Схема дозирования
Три группы по 3 крысы получали следующие дозы:
группы
г) Введение дозы
Дозы вводили в виде единичной пероральной дозы при помощи желудочного зонда, постоянным объемом дозы 10 мл/кг.
д) Отбор образцов крови
В каждый контрольный момент времени образцы крови (приблизительно 0,3 мл) отбирали через катетер, вставленный в боковую вену хвоста перед началом эксперимента. Окончательный образец крови (приблизительно 2 мл) у каждого животного отбирали при помощи пункции сердца под анестезией изофлураном, после чего животных умерщвляли путем обескровливания. Образцы крови отбирали в индивидуальные гепаринизированные контейнеры. Образцы крови отбирали в следующие моменты времени после введения дозы:
15, 30, 60, 120, 240,360,480,720 минут и 24 часа.
После отбора образцы крови центрифугировали (приблизительно 10000 g, 2 мин, при 4°С) и хранили плазму в виде одной аликвоты приблизительно при -20°С до осуществления анализа концентрации лекарства при помощи ЖХ-МС/МС.
е) Биоанализ образцов
Образцы плазмы анализировали на концентрации исследуемого вещества на приборе BioDynamics при помощи способа ЖХ-МС/МС, разработанного для BioDynamics.
РЕЗУЛЬТАТЫ
Фармакокинетический профиль соединений важен, поскольку он показывает, сколько соединения остается в организме, в течение какого времени, и степень воздействия на субъекта при пероральном введении соединения.
Результаты для концентраций в крови Соединений 1, 2, 3, А и Б приведены в Таблицах 2, 3, 4, 5 и 6 соответственно.
Соединение, которое предполагают вводить перорально, в идеале требует приема только один или два раза в сутки, поскольку это снижает нагрузку на пациента, и таким образом, увеличивается приверженность пациента к лечению. Поэтому гораздо предпочтительнее, чтобы спустя 12 часов концентрация оставшегося в крови лекарственного средства была, по меньшей мере, не ниже величины IC50 для данного соединения и предпочтительно была значительно выше нее. Еще более предпочтительным является такой фармакокинетический профиль, в котором концентрация лекарственного средства, оставшегося в крови спустя 24 часа, по меньшей мере, не ниже величины IC50 для указанного соединения, а предпочтительно значительно выше нее.
Результаты, приведенные в Таблице 2, показывают, что через 24 часа среднее количество Соединения 1, оставшееся в крови, составляло 47,4 нг/мл. Величина IC50 для Соединения 1 в тесте на изменение формы эозинофилов цельной крови равна 5 нМ (2,2 нг/мл) и, таким образом, имеется прибл. 21-кратное превышение IC50, полученной в тесте на изменение формы эозинофилов, через 24 часа. Полученные результаты, таким образом, показывают, что Соединение 1 особенно подходит для перорального введения пациенту.
Результаты, приведенные в Таблице 3, показывают, что через 12 часов среднее количество Соединения 2, оставшееся в крови, составляло 100,3 нг/мл. Результаты через 24 часа были менее согласующимися, и среднее для этого момента времени не определяли. Величина IC50 для Соединения 2 в тесте на изменение формы эозинофилов цельной крови составляла 2 нМ (0,9 нг/мл) и, таким образом, имеется прибл. 111-кратное превышение IC50, полученной в тесте на изменение формы эозинофилов цельной крови, через 12 часов. Полученные результаты, таким образом, показывают, что Соединение 2 подходит для перорального введения пациентам, хотя его профиль не столь благоприятный, как у Соединения 1.
Результаты, приведенные в Таблице 4, показывают, что через 24 часа среднее количество Соединения 3, оставшееся в крови, составляло 16,1 нг/мл. Величина IC50 для Соединения 3, полученная в тесте на изменение формы эозинофилов цельной крови, составляла 6 нМ (2,7 нг/мл), и таким образом, имеется прибл. 6-кратное превышение IC50, полученной в тесте на изменение формы эозинофилов цельной крови, через 24 часа. Таким образом, полученные результаты показывают, что Соединение 3 особенно подходит для перорального введения пациенту.
Результаты, приведенные в Таблице 5, показывают, что через 24 часа среднее количество Соединения (А), оставшееся в крови, составляло 43,7 нг/мл. Величина IC50 для Соединения А в тесте на изменение формы эозинофилов цельной крови составляла 103 нМ (35,8 нг/мл), и таким образом, имеется прибл. 1,2-кратное превышение IC50, полученной в тесте на изменение формы эозинофилов цельной крови, через 24 часа.
Результаты, приведенные в Таблице 6, показывают, что через 12 часов количество Соединения Б, оставшееся в крови, составляло 27,3 нг/мл. Величина IC50 для Соединения Б в тесте на изменение формы эозинофилов цельной крови составляла 8 нМ (3,0 нг/мл), и, таким образом, имеется прибл. 9,1-кратное превышение IC50, полученной в тесте на изменение формы эозинофилов цельной крови, через 12 часов.
Фигуры 1-5 представляют собой графики зависимости концентрации в крови от времени для крыс, получивших перорально дозу 3 мг/кг Соединений 1, 2, 3, А и Б соответственно. Таким образом, они являются графическими представлениями данных, приведенных в Таблицах 2-6.
Величины Cmax (максимальная концентрация в крови), Tmax (время, в которое наблюдается Cmax), ППКinf (площадь под кривой до бесконечности) и Т1/2 (период полувыведения), полученные из Фигур 1-5, приведены в Таблицах 7-11 для Соединений 1, 2, 3, А и Б соответственно.
Величины Cmax и ППКinf отражают воздействие на субъекта пероральной дозы соединения. Поэтому предпочтительно, чтобы числовые значения как для Cmax, так и для ППК были как можно больше, поскольку при этом воздействие соединения максимально.
Из Таблиц 7-11 можно видеть, что Соединения 1, 2 и 3 имеют средние величины Сmах, равные 4163, 2954 и 4076 соответственно. Напротив, средние величины для соединений А и Б составляют только 1245,9 и 381,0.
Величины ППKinf для Соединений 1, 2 и 3 составляют 19238, 11666 и 21059, тогда как для Соединений А и Б-8157,8 и 2197,6.
Таким образом, числовые значения как Cmax, так и ППKinf значительно выше для соединений согласно настоящему изобретению, чем для Соединений сравнения А и Б. Из полученных результатов очевидно, что субъекты, перорально получающие соединения А и Б, подвергаются гораздо меньшему воздействию препарата, чем субъекты, перорально получающие соединения 1, 2 и 3.
Результаты, полученные в Примерах 4 и 5, объединены в Таблице 12, в которой Превышение IC50 через 12 ч определяют как среднее количество соединения, оставшееся в крови через 12 часов, в нг/мл, деленное на IC50 для указанного соединения, в нг/мл. Подобным образом, Превышение IC50 через 24 ч определяют как среднее количество соединения, оставшееся в крови через 24 часа, в нг/мл, деленное на IC50 для указанного соединения, в нг/мл.
В Таблице 12 показано, что Соединение Сравнения А имеет более высокую IC50, чем Соединения 1, 2 и 3, и более низкое значение Превышения IC50 через 24 часа, чем Соединения 1 и 3. Воздействие лекарственного средства на субъекта после перорального введения, как указывают Cmax и ППКinf, значительно ниже для Соединения А, чем для любого из Соединений 1-3.
Соединение Б имеет величину IC50, сравнимую с величинами для соединений 1, 2 и 3. Однако Превышение IC50 через 12 и 24 часа является значительно менее благоприятным, чем для любого из Соединений 1-3, и воздействие на субъекта после перорального введения, как указывают Cmax и ППКinf, значительно ниже для Соединения Б, чем для любого из Соединений 1-3.
Каждое из Соединений Сравнения А и Б обладает некоторыми свойствами, сравнимыми с теми же свойствами Соединений 1, 2 и 3. Однако взятые в целом, их свойства менее желательные, чем свойства соединений согласно настоящему изобретению.
ПРИМЕР 6 - Оценка соединений общей формулы (I) в качестве ингибиторов индуцированной 13,14-дигидро-15-кетопростагландином D2 (ДК-ПГD2) эозинофилии крови у крыс
МЕТОДИКА
Исследуемое соединение растворяли в ДМСО и разбавляли водой для получения конечного объема дозы 2 мл/кг.
Самки крыс (175-250 г; UH colony) получали пероральную дозу исследуемого соединения (или среды).
Через 30 мин после введения дозы всех животных анестезировали изофлураном.
После введения анестезии животные получали внутрисердечную инъекцию 10 µг ДК-ПГD2 в 0,3 мл гепаринизированного (10 Ед/мл) солевого раствора. Контрольные животные получали инъекцию 0,3 мл гепаринизированного солевого раствора.
Через 60 минут после внутрисердечной инъекции животные получали инъекцию избыточной дозы пентобарибитона натрия и у них брали образцы крови (в гепарин) путем пункции сердца в то время, когда крысы находились под анестезией, но еще не умерли.
Аликвоту крови (100 µл) добавляли к раствору Тюрка и определяли общее количество лейкоцитов при помощи гематоцитометра.
Другую аликвоту крови (500 µл) смешивали с равным объемом 4% Декстрана (мол. вес 500000) и давали осесть эритроцитам. Из полученной богатой лейкоцитами фракции готовили препараты клеток осаждением на стекла (Cytospin).
Препараты клеток фиксировали метанолом (5 мин) и окрашивали красителями Май-Грюнвальда (5 мин) и Гимза (15 мин). Наконец, препараты клеток промывали фосфатным буферным раствором (рН 6,8) и высушивали на воздухе.
Указанные препараты клеток применяли для подсчета лейкоцитарных формул.
Число эозинофилов в крови определяли на основании общего числа лейкоцитов и процентного содержания эозинофилов (лейкоцитарная формула).
СХЕМА ЭКСПЕРИМЕНТА
В каждом эксперименте использовали до 12 животных. Группы включали:
Необработанный контроль
Индуцированная ДК-ПГD2 эозинофилия, животные, получавшие среду (положительный контроль)
Индуцированная ДК-ПГD2 эозинофилия, Соединение 1; дозы 0,0001, 0,001, 0,01 и 0,1 µг/кг перорально
Индуцированная ДК-ПГD2 эозинофилия, Соединение 2; дозы 0,001, 0,01, 0,1 и 1,0 µг/кг перорально
Индуцированная ДК-ПГD2 эозинофилия, Соединение 3; дозы 0,001, 0,01, 0,1 и 1,0 µг/кг перорально
Индуцированная ДК-ПГD2 эозинофилия, Соединение А; дозы 0,01, 0,1 и 1,0 мг/кг перорально
Индуцированная ДК-ПГD2 эозинофилия, Соединение Б; дозы 0,001, 0,01, 0,1 и 1,0 мг/кг перорально
Поскольку практически невозможно получить достаточное количество данных на основании одного эксперименте, данные получены из ряда повторяющихся экспериментов. Данные из указанных повторяющихся экспериментов объединяли, чтобы обеспечить, по меньшей мере, 5 животных в каждой группе, и обрабатывали кривую доза - отклик при помощи критерия достоверности (Манна-Уитни) для определения, вызывает ли ДК-ПГD2 значительное увеличение содержания эозинофилов в крови (GraphPad, Prism). Различие между группами считали значительным, если Р<0,05. Величины ED50 вычисляли на основании кривой доза - ответ.
Результаты описанных экспериментов приведены ниже на Фигурах 6-10 и в Таблице 13.
Из Таблицы 13 и Фигур 6-10 видно, что Соединения 1-3 в несколько тысяч раз мощнее, чем Соединения Сравнения А и Б при ингибировании индуцированной ДК-ПГD2 эозинофилии крови у крыс.
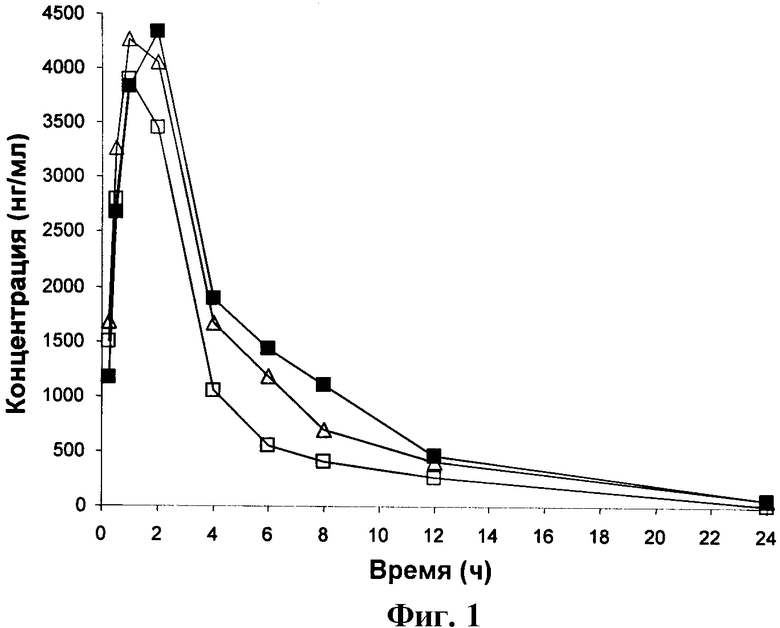

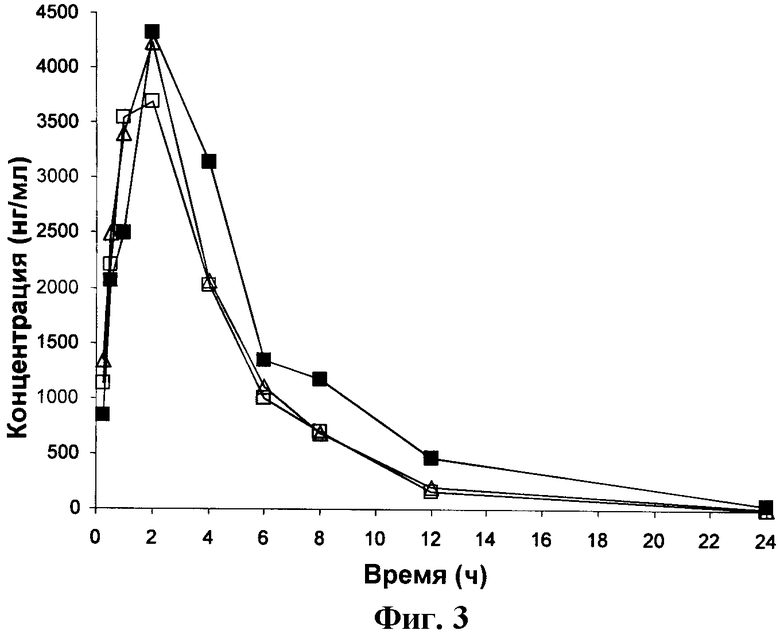
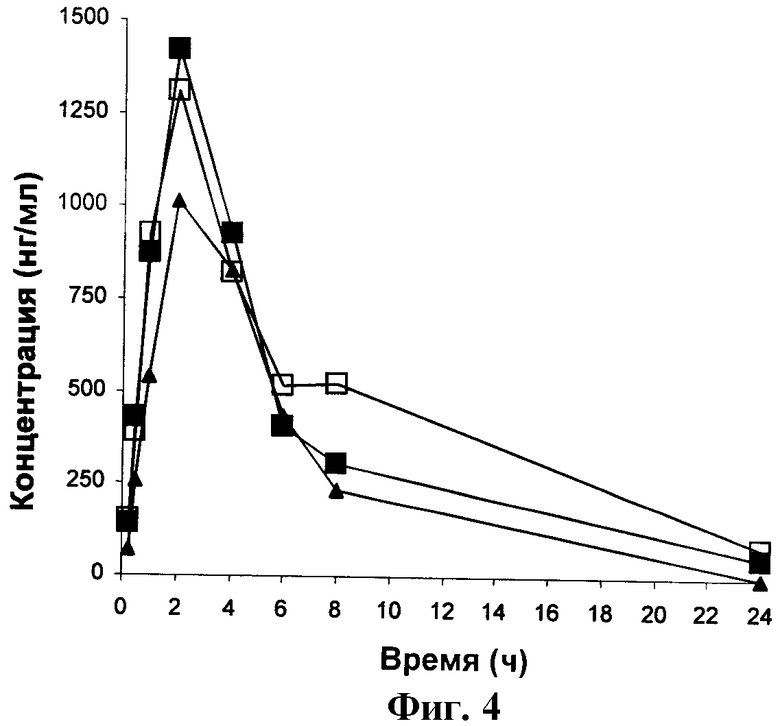
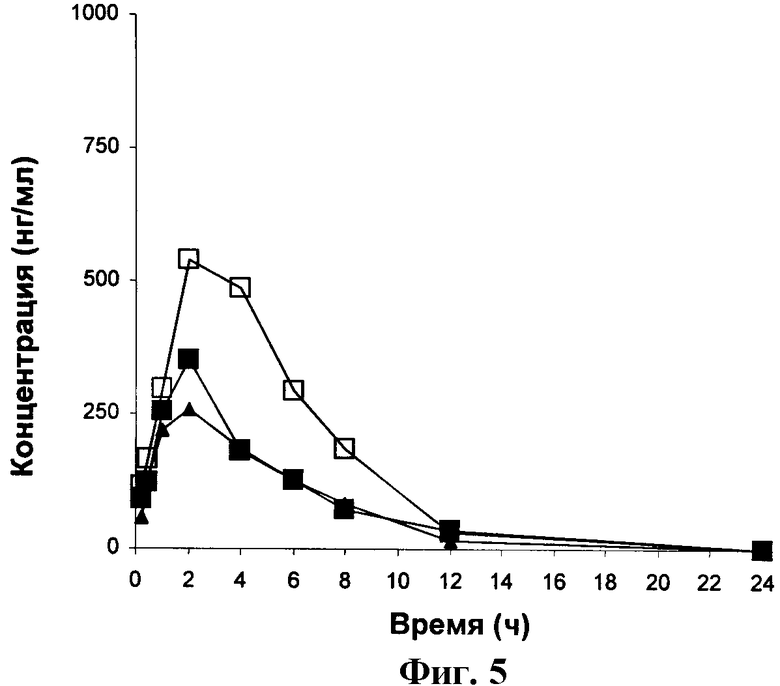

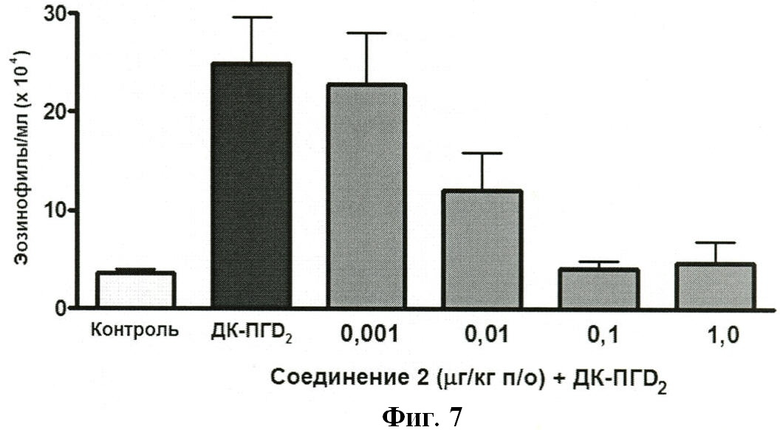
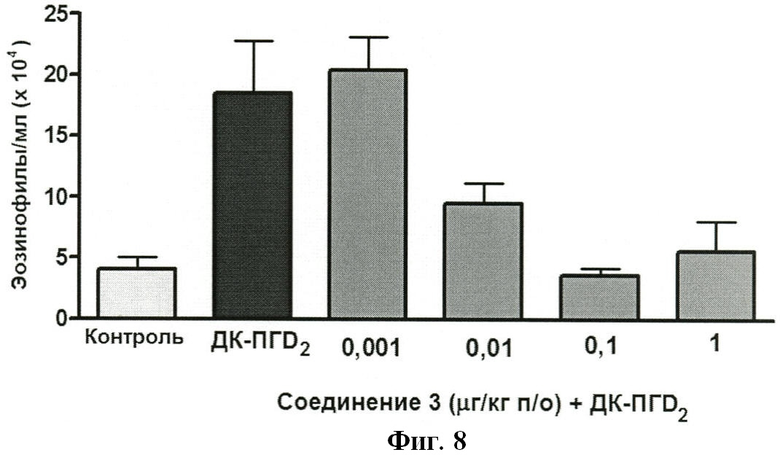
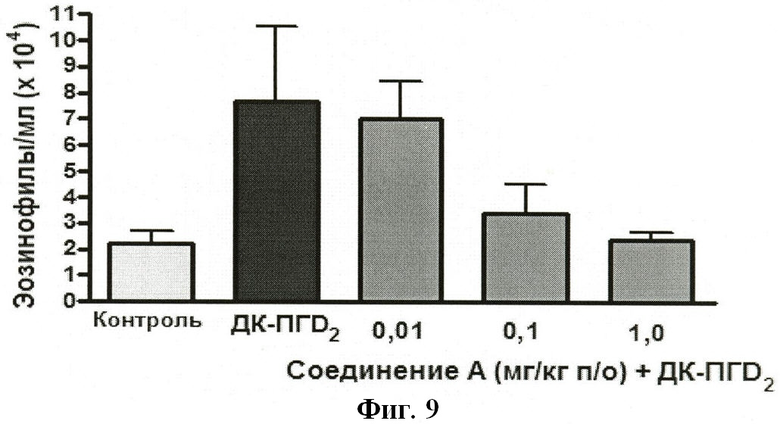
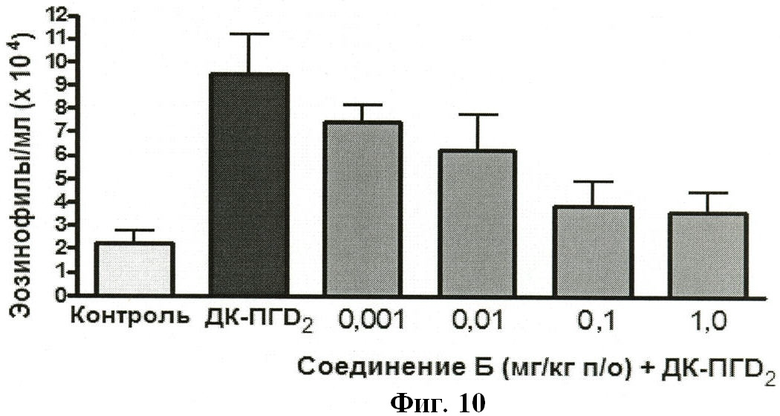
Изобретение относится к новым соединениям общей формулы (I)
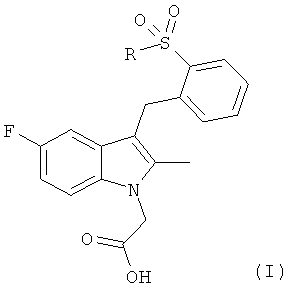
где R означает фенил, который может иметь один или более заместитель - галоген; или его фармацевтически приемлемым солям. Соединения могут быть использованы при лечении заболеваний, опосредуемых CRTH2, таких как астма, аллергический ринит и др. 6 н. и 10 з.п. ф-лы, 10 ил., 13 табл., 5 пр.
1. Соединение общей формулы (I)
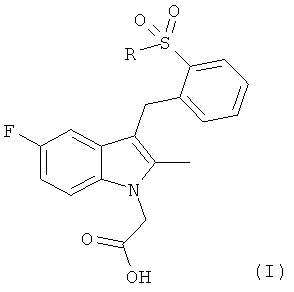 ,
,
где R представляет собой фенил, который может иметь один или более заместитель - галоген;
или его фармацевтически приемлемая соль.
2. Соединение по п.1, отличающееся тем, что фенильная группа R является незамещенной или имеет один заместитель - галоген.
3. Соединение по п.2, отличающееся тем, что указанный галоген представляет собой фтор или хлор.
4. Соединение по п.3, отличающееся тем, что указанный атом фтора или хлора находится в положении 4 фенильной группы R.
5. Соединение по п.1, которое представляет собой:
2-{5-фтор-2-метил-3-[2-(фенилсульфонил)бензил]-1Н-индол-1-ил}уксусную кислоту;
2-{3-[2-(4-хлорфенилсульфонил)бензил]-5-фтор-2-метил-1Н-индол-1-ил} уксусную кислоту;
или
2-{5-фтор-3-[2-(4-фторфенилсульфонил)бензил]-2-метил-1Н-индол-1-ил} уксусную кислоту.
6. Соединение общей формулы (II):
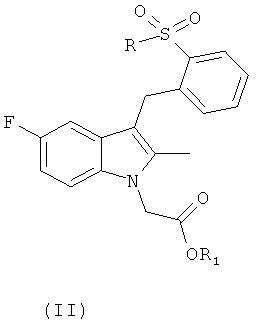 ,
,
где R такой, как определено для общей формулы (I); и R1 представляет собой C1-С6 алкил.
7. Соединение по п.6, отличающееся тем, что фенильная группа R является незамещенной или имеет один заместитель - галоген.
8. Соединение по п.7, отличающееся тем, что указанный галоген представляет собой фтор или хлор.
9. Соединение по п.8, отличающееся тем, что указанный атом фтора или хлора находится в положении 4 фенильной группы R.
10. Соединение по п.6, которое представляет собой C1-С6 алкильный сложный эфир любого из нижеперечисленных соединений:
2-{5-фтор-2-метил-3-[2-(фенилсульфонил)бензил]-1Н-индол-1-ил}уксусная кислота;
2-{3-[2-(4-хлорфенилсульфонил)бензил]-5-фтор-2-метил-1Н-индол-1-ил}уксусная кислота;
2-{5-фтор-3-[2-(4-фторфенилсульфонил)бензил]-2-метил-1Н-индол-1-ил}уксусная кислота.
11. Способ получения соединения общей формулы (I) по любому из пп.1-5, который включает осуществление взаимодействия соединения общей формулы (II) по п.6, в котором R1 представляет собой C1-С6алкил, с основанием.
12. Соединение по любому из пп.1-5 для применения при лечении заболеваний, опосредуемых CRTH2, таких как астма, аллергическая астма, обострение астмы, хронический кашель, связанный с воспалительными и секреторными состояниями дыхательных путей, круглогодичный аллергический ринит, сезонный аллергический ринит, полипоз носа, атопического дерматит, контактную гиперчувствительность, контактный дерматит, крапивницу, конъюнктивит, аллергический конъюнктивит, эозинофильный бронхит, эзофагит, эозинофильный гастроэнтерит, аллергия, связанная с пищей, воспалительное заболевание кишечника, язвенный колит, болезнь Крона, мастоцитоз, гипер-lqE-синдром, синдром Черджа-Строссса, множественный склероз, хроническое обструктивное заболевание легких, фиброзные состояния.
13. Применение соединения по любому из пп.1-5 при производстве средства для лечения или предотвращения состояния, выбранного из:
астмы, аллергической астмы, обострения астмы, хронического кашеля, связанного с воспалительными и секреторными состояниями дыхательных путей, круглогодичного аллергического ринита, сезонного аллергического ринита; полипоза носа, атопического дерматита, контактной гиперчувствительности, контактного дерматита, крапивницы, конъюнктивита, аллергического конъюнктивита, эозинофильного бронхита, эзофагита, эозинофильного гастроэнтерита, аллергии, связанной с пищей, воспалительного заболевания кишечника, язвенного колита, болезни Крона, мастоцитоза, гипер-lqЕ-синдрома, синдрома Черджа-Строссса, множественного склероза, хронического обструктивного заболевания легких, фиброзных состояний.
14. Фармацевтическая композиция для лечения заболеваний, опосредуемых CRTH2, таких как астма, аллергическая астма, обострение астмы, хронический кашель, связанный с воспалительными и секреторными состояниями дыхательных путей, круглогодичный аллергический ринит, сезонный аллергический ринит, полипоз носа, атопического дерматит, контактную гиперчувствительность, контактный дерматит, крапивницу, конъюнктивит, аллергический конъюнктивит, эозинофильный бронхит, эзофагит, эозинофильный гастроэнтерит, аллергия, связанная с пищей, воспалительное заболевание кишечника, язвенный колит, болезнь Крона, мастоцитоз, гипер-IqE-синдром, синдром Черджа-Строссса, множественный склероз, хроническое обструктивное заболевание легких, фиброзные состояния, при этом указанная композиция содержит соединения по любому из пп.1-5 совместно с фармацевтическим наполнителем или носителем.
15. Фармацевтическая композиция по п.14, в форме для перорального, ректального, назального, бронхиального (ингаляция), местного (включая глазные капли, трансбуккальное и сублингвальное), вагинального или парентерального (включая подкожное, внутримышечное, внутривенное и внутрикожное) введения.
16. Способ получения фармацевтической композиции, включающий соединение или смешивание соединения по любому из пп.1-5 с фармацевтически или ветеринарно приемлемым носителем или наполнителем.
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| US 5641800 A, 24.06.1997 | |||
| EP 1505061 A1, 09.02.2005 | |||
| RU 2005137403 A, 27.05.2006. | |||

