Изобретение относится к области аналитической химии, в частности к инверсионному вольтамперометрическому способу определения антибиотика (АБ) левомицетина (хлорамфеникола), который представляет собой
D (-)-трео-2-Дихлорацетиламино-1-(4-нитрофенил)пропан-1,3-диол:
Левомицетин относится к антибактериальным веществам и обладает высокой клинической эффективностью в лечении ряда тяжелых инфекций. Его применяют в ветеринарии и животноводстве в качестве лечебно-профилактических средств. АБ добавляются, как правило, в корм на уровне 50-200 г на 1 т. Левомицетин способен переходить в мясо, молоко животных, яйца птиц, другие продукты и оказывать токсическое действие на организм человека при концентрации более 25 мкг/см3 [Позняковский В. М. Гигиенические основы питания и экспертизы продовольственных товаров. - Новосибирск: Изд-во Новосиб. ун-та, 1996. - 432 с.], что делает потенциально опасным его бесконтрольное применение.
Медико-биологические требования и санитарные нормы качества продовольственного сырья и пищевых продуктов указывают на недопустимость в продуктах питания остаточных количеств левомицетина. Максимальный уровень остатков АБ составляет 0,01 ЕД/г, что соответствует 0,01 мг/кг или 10,0 мкг/кг (предельно допустимая концентрация, ПДК) [Гигиенические требования к качеству и безопасности продовольственного сырья и пищевых продуктов - Санитарные правила и нормы СанПиН 2.3.2.560-96.]. Это в свою очередь предъявляет повышенные требования к контролю за качеством пищевого сырья и совершенствованию методов определения АБ.
Наиболее широкое применение как в научных исследованиях, так и в практических учреждениях нашли микробиологические методы [Методические указания по определению остаточных количеств антибиотиков в продуктах животноводства. Мин. здравоохранения СССР. - Москва, 1985 г.], позволяющие определять минимальные концентрации антибиотиков в исследуемом материале. Они основаны на непосредственном биологическом действии АБ на чувствительные штаммы микроорганизмов и являются сравнительно простыми, однако, отличаются недостаточной специфичностью и воспроизводимостью. Методики с их использованием чрезвычайно длительны (время анализа составляет более 36 часов), трудоемки и зачастую применяются лишь для качественного определения АБ.
Практически отсутствуют инструментальные методы определения левомицетина в пищевых продуктах. Известен способ [Меламед Д.Б., Кирпичная В.К., Ляпков Б. Г. Способ анализа левомицетина в пищевых продуктах. А.С. SU 1702302 А1, G 01 N 30/90, А 23 L 3/00] высокоэффективной жидкостной хроматографии (ВЭЖХ) в системе растворителей ацетонитрил-вода-дециламин. Для увеличения чувствительности используют ультрафиолетовое детектирование при λ = 278 нм. Количественное определение левомицетина проводят сравнением площадей пиков левомицетина - стандарта и левомицетина в образце. Несмотря на высокую чувствительность метода ВЭЖХ (10 мкг/кг), длительность анализа с учетом времени пробоподготовки, а также высокая стоимость приборов, существенно ограничивают его использование в контроле пищевых продуктов.
Имеется множество вариантов оптических методов определения левомицетина в лекарственных формах [Таблетки левомицетина. ФС 42-3679-98. - с.8.; Раствор левомицетина. ФС 42-3425-97. - с.6.; Тираспольская С.Г., Степанюк С.Н., Филипьева К. Н. Фотометрическое определение левомицетина в лекарственных формах. //Фармация,-1980.-Т.29, 6, с.48-49].
Согласно литературным данным, левомицетин поглощает в ультрафиолетовой области от 205 до 400 нм. Максимум светопоглощения находится при 278-280 нм.
Значительное место занимают колориметрические методы, основанные на различных специфических цветных реакциях [Тираспольская С.Г., Степанюк С.Н., Филипьева К. Н. Фотометрическое определение левомицетина в лекарственных формах. //Фармация. -1980.-т.29, 6, с.48-49]. Чувствительность этих методов невысока и составляет 0,1 мг/см3 и более.
Разработан метод иммуноферментного анализа хлорамфеникола в сыворотке крови человека [Колосова А. Ю., Самсонова Ж.В., Блинцов А.Н., Егоров А.М. Твердофазный иммуноферментный анализ хлорамфеникола в сыворотке крови человека. //Вопросы медиц. Химии. - 1998. - т.44., 2, с.194 - 202], основанный на измерении оптической плотности продукта ферментативной реакции в растворе антибиотика в диапазоне концентраций 10÷1000 нг/см3. В работе использовано большое количество малодоступных биохимических реагентов. Методика требует больших трудозатрат.
Много внимания в литературе уделено разработке полярографического метода определения левомицетина в фармацевтических препаратах (таблетках, капсулах, глазных каплях, инъекционных растворах и технических порошках) [Мискиджьян С. П. , Кравченюк Л.П. Полярография лекарственных препаратов. - Киев: Изд-во "Вища школа", 1976. - 230 с.]. Полярографическая деполяризация левомицетина основана на восстановлении нитрогрупп и остатка дихлорацетила на ртутном капающем электроде (р.к.э.). Левомицетин образовывал две волны, первая из которых имела потенциал полуволны E1/2=-0,35 В и соответствовала четырехэлектронному процессу необратимого восстановления нитрогруппы до гидроксиламина. E1/2=-1,10 В. Для подавления максимумов использовали тимол или желатин. Наиболее воспроизводимые результаты количественного определения левомицетина на р. к. э. в чистом препарате получены с использованием желатина [Пассет Б. В. , Антипов М.А. Практикум по техническому анализу и контролю производства химико-фармацевтических препаратов и антибиотиков. - М. : "Медицина", 1981. - 272 с.] (прототип). Сущность методики состоит в растворении 0,1 г препарата в 100 мл 95% спирта. Из полученного раствора отбирали 1 см3, добавляли ацетатного буферного фонового раствора до объема 10 см3 и 1 каплю 1% раствора желатина. После пропускания водорода снимали полярограмму, начиная с 0,0 В. Потенциал полуволны регистрировали при E1/2=-0,34 В. Аналогично готовили раствор стандартного образца левомицетина и в тех же условиях снимали его полярограмму. Минимально определяемая концентрация составляет 1000 мг/кг. В данных условиях экспрессное количественное определение левомицетина на уровне требований, определяемых нормативными документами (0,01 мг/кг), невозможно.
Одним из ограничивающих факторов применения полярографии (с точки зрения техники безопасности) является также использование больших количеств токсичной ртути в качестве электродов в электролитической ячейке. Поэтому встает задача замены р.к.э. на более безопасные. Таким образом, несмотря на возможность количественного определения левомицетина, ни один из перечисленных выше полярографических методов не применяется в анализе пищевых продуктов.
Одним из наиболее перспективных методов определения АБ, на наш взгляд, является вольтамперометрический (ВА) и в первую очередь такие его высокочувствительные варианты, как дифференциальная вольтамперометрия (ДВА). Преимущество ВА благодаря высокой чувствительности состоит в возможности работы с малыми по массе и объемам пробами в мутных и окрашенных средах. Пробоподготовка может быть существенно упрощена из-за необязательной дополнительной очистки определяемого вещества перед собственно электрохимическим анализом. Поэтому часто допустимы более простые методики предварительного выделения соединений из сложной многокомпонентной системы. Методы вольтамперометрии также ранее не применялись для КХА левомицетина в пищевых продуктах. Это, по-видимому, объясняется отставанием электроаналитической практики пищевых продуктов и недостатком аппаратурного оснащения лабораторий этой отрасли.
Задачей заявляемого изобретения является повышение чувствительности, экспрессности и селективности определения левомицетина в пищевых продуктах методом дифференциальной вольтамперометрии.
Поставленная задача достигается тем, что левомицетин переводят из пробы в раствор, проводят кислотный гидролиз и осаждают белок из гидролизата с последующим вольтамперометрическим определением антибиотика. Новым в способе является то, что проводят кислотный гидролиз 0,10-0,12 моль/дм3 НСl при температуре 25-30oС в течение 10-15 минут, затем проводят осаждение водорастворимого белка 2,5-3,0 г сульфатом аммония и 3,0-5,0 см3 этанолом, отделение осадка центрифугированием в течение 10-15 минут при скорости вращения 6000 об/мин с последующим адсорбционным вольтамперометрическим определением левомицетина в безбелковом гидролизате путем регистрации катодных пиков антибиотика на индикаторном ртутно-пленочном (РПЭ) или стеклоуглеродном (СУ) электродах в дифференциальном режиме съемки вольтамперограмм при соответствующих потенциалах -(0,67±0,05) В и -(0,60±0,03) В относительно насыщенного хлорид серебряного электрода (нас. х.с.э.) на фонах 0,1 моль/дм3 аммония лимоннокислого двузамещенного С6Н14O7N2 (рН 4,7-5,1) или 0,1 моль/дм3 (NH4)2SO4 с добавлением НCl до рН 5,1 при скорости развертки потенциала 10-25 мВ/с.
В прототипе описано использование в качестве фонов ацетатного буферного раствора (рН 4,4). Определение левомицетина в этих условиях па уровне 10 мкг/кг затруднено из-за плохой воспроизводимости и искажения формы аналитического сигнала, связанных с большим остаточным током, и регистрации дополнительного пика при потенциале Еп=-(0,48±0,05) В. Предполагаемый в заявляемом изобретении фон 0,1 моль/дм3 C6H14O7N2 (рН 4,7-5,1) позволяет определять левомицетин на уровне 2,8-30,0 мкг/кг с хорошей воспроизводимостью. Относительное стандартное отклонение для указанного диапазона концентраций изменяется от 0,20 до 0,08. Абсолютной новизной являются экспериментально подобранные фоны: смесь (NH4)2SO4 с НСl (рН 5,1); 0,1 моль/дм3 C6H14O7N2, установленный pHl раствора 4,7-5,1, от чего зависит количественное определение левомицетина. Фоновые электролиты (NH4)SO4 и C6H14O7N2 одновременно являются хорошими осадителями белковых примесей в пищевых продуктах. Для количественного определения левомицетина они ранее не применялись. Особое значение при вольтамперометрическом анализе левомицетина имеет рН среды. Оптимальным показателем является экспериментально установленное значение рН 4,7-5,1, которое соответствует изоэлектрическому состоянию молекул белков молочных и мясных продуктов. При значении рН 4,7-5,1 растворимость белковых примесей снижена и молекулы белков не перемещаются под воздействием внешнего электрического поля, что способствует стабилизации остаточных белковых примесей и получению хорошо воспроизводимых пиков восстановления левомицетина, особенно при использовании СУ индикаторного электрода. При значении рН 4,7-5,1 регистрируется один четко выраженный пик с потенциалом -(0,67±0,05) и -(0,60±0,03) В соответственно на РПЭ и СУ электродах. При 5,1<рН<4,7 белки остаточных примесей заряжены и перемещаются под воздействием внешнего электрического поля. Это способствует их адсорбции на электродах, что ухудшает воспроизводимость, снижает чувствительность определения и экспрессность.
Установленное значение рН 4,7-5,1 зависит от природы фоновых электролитов и позволяет анализировать растворы на уровне 2,8 мкг/кг (нижняя граница определяемых содержаний, Сн). Предел обнаружения Сmin.p, рассчитанный по 3σ критерию равен 1,5 мкг/кг. Зависимость аналитического сигнала от концентрации левомицетина в таких растворах прямо пропорциональна диапазону определяемых содержаний от 2,8 до 30,2 мкг/кг.
Другим отличительным признаком являются установленные условия электрохимического накопления: Еэ=-(0,45-0,47) В. При потенциалах -0,45< Еэ<-0,47 уменьшалась величина тока восстановления органического вещества, кроме того, при Еэ<-0,47 В возникал большой остаточный ток, связанный с выделением водорода в кислых растворах, и уменьшалась высота пика. Без предварительного электролиза невозможна регистрация четкого пика левомицетипа на уровне 2,8 мкг/кг.
Важным для определения левомицетина является выбор скорости развертки поляризующего напряжения. Оптимальной является скорость 10-25 мВ/с. Увеличение скорости более 25 мВ/с увеличивает чувствительность, но при этом растет остаточный ток и уменьшается разрешающая способность способа. Использование скорости более 25 мВ/с при анализе пищевых продуктов не позволяет полностью удалить определяемое вещество с поверхности электрода. Это ухудшает воспроизводимость и требует дополнительной очистки поверхности электрода, особенно СУ. При скорости менее 10 мВ/с снижается величина катодного тока и понижается чувствительность определения.
Время предварительного электролиза (τэ) для диапазона концентраций левомицетина 2,8-30,0 мкг/кг составляет 30-60 с, при этом достигается максимальное значение величины тока восстановления. При 60c<τэ<30c увеличивается ошибка определения (относительное стандартное отклонение, Sr>0,17 при определении левомицетина на уровне 10 мкг/кг).
Еще одним отличительным признаком является использование РПЭ или СУ электродов в качестве индикаторных. Ртутно-пленочный представляет собой пленку ртути толщиной 20-50 мкм, нанесенную на серебряную подложку, площадь поверхности составляет 15,0 мм2, рабочая поверхность стеклоуглеродного электрода составляет 25-30 мм2 (в прототипе применяли ртутный капающий электрод, р.к. э.). Существенным преимуществом РПЭ является возможность получения более узких и высоких пиков, служащих аналитической характеристикой определяемого вещества, за счет значительного возрастания отношения активной поверхности к объему ртути и уменьшения времени выхода определяемого вещества из тонкой пленки (в два - три раза по сравнению с р.к.э.). Использование СУ электрода обусловлено высокой химической и электрохимической устойчивостью графита, широкой областью рабочих потенциалов как в водных, так и в неводных средах, а также простотой механического обновления поверхности. Величина потенциала восстановления левомицетина определяется строением, структурой и степенью адсорбируемости на гексагонах графита. По легкости восстановления РПЭ и СУ электроды можно расположить:
РПЭ
-(0,67±0,05) В
СУ
-(0,60±0,03) В
Структурное подобие материала электрода и специфическая адсорбция, по-видимому, благоприятствуют более обратимому восстановлению левомицетина на СУ электроде, чем на РПЭ. Нижняя граница определяемых содержаний левомицетина при использовании РПЭ и СУ электродов на 2-3 порядка ниже, чем на р.к. э. , и составляет 2,8 и 3,4 мкг/кг соответственно. Пики, полученные с использованием таких электродов, характеризуются хорошей воспроизводимостью, кроме того, РПЭ является менее токсичным, чем р.к.э., а СУ - не токсичным. Оба электрода удобны в практической работе. Для определения левомицетина РПЭ и СУ ранее не применялись.
Использование дифференциального режима записи вольтамперограмм позволяет фиксировать четкие узкие пики, что повышает разрешающую способность способа и облегчает автоматизацию электродного процесса. Для определения левомицетина дифференциальный режим ранее не применялся.
В предлагаемом способе установлены условия подготовки пробы молочных продуктов, яиц, мяса и субпродуктов убойных животных и птиц. Составные компоненты растворимых белков пищевых продуктов адсорбируются на углеродных и ртутных электродах и участвуют в редокс превращениях. На поверхности электрода может образовываться слой белка толщиной 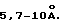 Возможны также конформационные изменения белковых макромолекул на поверхности [Тарасович М.Р., Радюшкина К. А. , Богдановская В.А. Электрохимия порфиринов. - М.: Наука, 1991. - 312 с.]. Поэтому присутствие белков затрудняет электродные процессы, снижает чувствительность, воспроизводимость и точность анализа. Для отделения белка и его распада на более простые составные части использовали кислотный гидролиз 0,10-0,12 моль/дм3 НС1 при температуре 25-30oС в течение 10-15 минут при перемешивании или встряхивании (при анализе мясных продуктов). (В прототипе гидролиз не применяли). Концентрация кислоты, время гидролиза (10-15 минут) и температура (25-30oС) подобраны экспериментально. Использование НСl с молярной концентрацией с <0,10 моль/дм3 ухудшает условия гидролиза, а при с > 0,15 моль/дм3 ухудшаются условия электрохимического определения из-за большого значения остаточного тока, связанного с выделением водорода на индикаторных электродах. Оптимальное время гидролиза (τг): 10-15 минут. При τг>15 мин снижается экспресспость анализа, а при τг<10 мин ухудшаются условия проведения гидролиза. Оптимальная температура гидролиза (tг): 25-30oС. При tг< 25oC усиливаются процессы соосаждения антибиотика с нерастворимыми белками, а при tг>30oC снижается устойчивость левомицетина в растворе, что приводит к уменьшению высоты пика и снижению точности определения.
Возможны также конформационные изменения белковых макромолекул на поверхности [Тарасович М.Р., Радюшкина К. А. , Богдановская В.А. Электрохимия порфиринов. - М.: Наука, 1991. - 312 с.]. Поэтому присутствие белков затрудняет электродные процессы, снижает чувствительность, воспроизводимость и точность анализа. Для отделения белка и его распада на более простые составные части использовали кислотный гидролиз 0,10-0,12 моль/дм3 НС1 при температуре 25-30oС в течение 10-15 минут при перемешивании или встряхивании (при анализе мясных продуктов). (В прототипе гидролиз не применяли). Концентрация кислоты, время гидролиза (10-15 минут) и температура (25-30oС) подобраны экспериментально. Использование НСl с молярной концентрацией с <0,10 моль/дм3 ухудшает условия гидролиза, а при с > 0,15 моль/дм3 ухудшаются условия электрохимического определения из-за большого значения остаточного тока, связанного с выделением водорода на индикаторных электродах. Оптимальное время гидролиза (τг): 10-15 минут. При τг>15 мин снижается экспресспость анализа, а при τг<10 мин ухудшаются условия проведения гидролиза. Оптимальная температура гидролиза (tг): 25-30oС. При tг< 25oC усиливаются процессы соосаждения антибиотика с нерастворимыми белками, а при tг>30oC снижается устойчивость левомицетина в растворе, что приводит к уменьшению высоты пика и снижению точности определения.
Отличительным признаком являются также установленные условия осаждения остаточных количеств растворимого белка из гидролизата (в прототипе осаждение не применяли). В предлагаемом способе в качестве осадителя выбрана соль аммония сернокислого (NH4)2SO4 в количестве 2,5-3,0 г с добавлением 3,0-5,0 см3 этанола. Использование больших количеств соли (более 3,0 г) и спирта (более 5,0 см3) повышает растворимость белковых примесей и ухудшает условия проведения собственно электрохимической реакции на индикаторном электроде. При добавлении соли менее 2,5 г и объема спирта менее 3,0 см3 ухудшаются условия осаждения. Осадок белка может вновь частично раствориться при стоянии пробы или после разведения пробы водой или раствором фонового электролита. Соль и спирт добавляют в гидролизат, рН которого лежит в диапазоне 4,7-5,1, что соответствует изоэлектрической точке остаточных количеств растворимых белковых примесей. При этих значениях рН растворимость белков снижена. При 5,1<рH<4,7 растворимость остаточных белковых примесей увеличивается, что ухудшает проведение электродного процесса.
После отделения осадка центрифугированием в течение 10-15 минут при скорости вращения 6000 об/мин проводили фильтрование гидролизата через бумажный фильтр с последующим дифференциально-вольтамперометрическим определением левомицетина в аликвоте гидролизата. Время центрифугирования и скорость вращения установлены экспериментально. Использование времени центрифугирования более 15 минут снижает экспрессность. Использование скорости вращения более 6000 об/мин приводит к частичной потере центрифугата, а менее 6000 об/мин (и времени менее 15 минут) недостаточно для полного отделения осадка.
Вольтамперограммы левомицетина регистрируют при максимальных значениях потенциала -(0,67±0,05) В и -(0,60±0,03) В соответственно на ртутно-пленочном и стеклоуглеродном электродах.
При определении левомицетина в лекарственных препаратах (глазные капли, таблетки, порошки) не требовалось дополнительного отделения сопутствующих веществ. После перевода пробы в раствор проводили дифференциально-вольтамперометрическое определение препарата в установленных оптимальных условиях проведения электродного процесса.
Метод вольтамперометрии для определения левомицетина в пищевых продуктах и лекарственных препаратах ранее не применялся.
Массовую долю антибиотика в пробе определяют методом добавок аттестованных смесей по общепринятой методике.
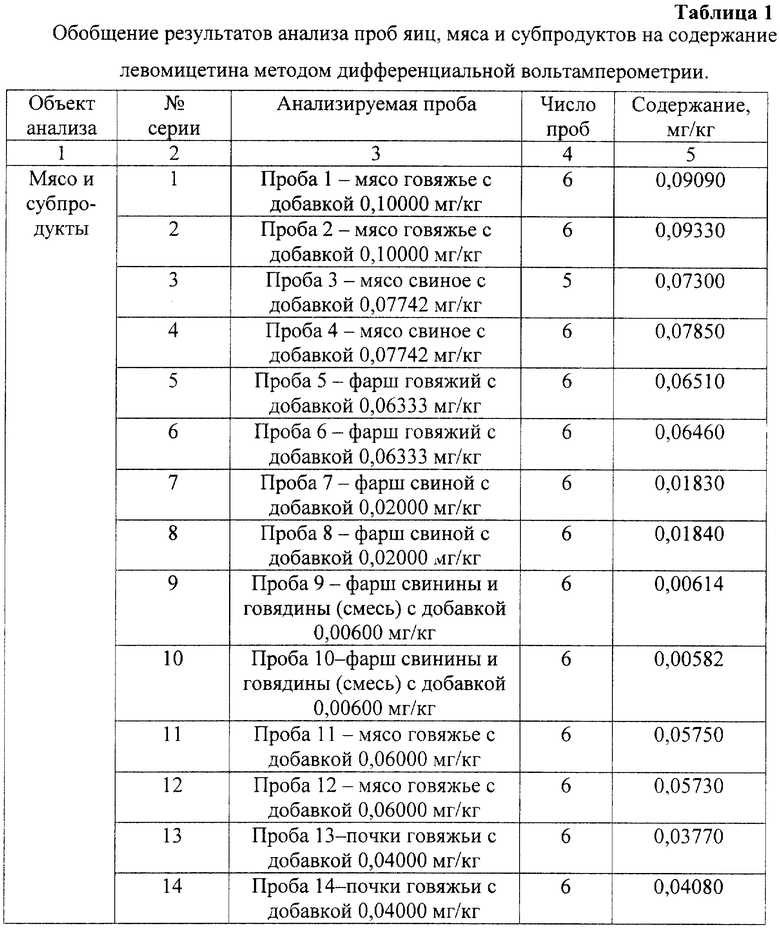
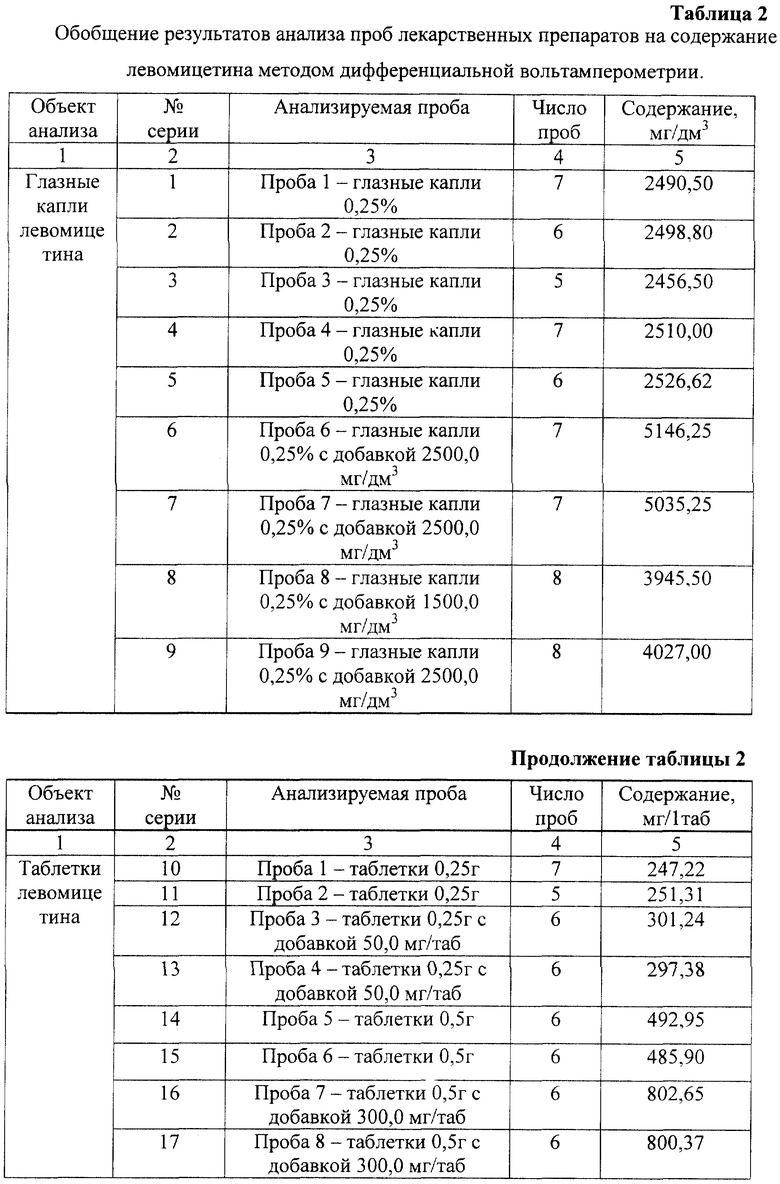
Установленные условия анализа в предлагаемом способе впервые позволили экспрессно (за 40-60 минут) количественно определять левомицетин в пищевых продуктах (таблица 1) и лекарственных препаратах (таблица 2) без предварительного отделения водорастворимых витаминов групп В, РР, аскорбиновой, фолиевой, никотиновой, лимонной, мочевой кислот, мочевины, цистина, цистеина, анионов и ионов тяжелых металлов.
Пример 1. Определение левомицетина (хлорамфеникола) в молоке и молочных продуктах.
В коническую колбу вместимостью 100,0 см3 вносят 25,0 см2 молока (или молочного продукта, взвешенного с точностью до 0,01 г) и 25,0 см2 0,1 моль/дм3 HСl. Смесь энергично встряхивают в течение 10 минут. Затем к смеси добавляют 2,5-3,0 г (NH4)2SO4 и 3,0-5,0 см3 этанола. Снова энергично встряхивают содержимое колбы в течение 5 минут. Полученную смесь переносят в центрифужные пробирки и центрифугируют 10 минут при скорости 6000 об/мин. Центрифугат отфильтровывают через бумажный фильтр в коническую колбу вместимостью 100,0 см3, добавляют С6Н14О7N2 до рH 4-5 и перемешивают до полного растворения. Аликвоту фильтрата объемом 10,0 см3 помещают в электролизер и проводят вольтамперометрические измерения при условиях: потенциал электролиза Еэ=-0,45 В, время электролиза τэ = 30 c, скорость развертки потенциала w= 10 мВ/с. Катодный пик левомицетина регистрируют с использованием ртутно-пленочного электрода в диапазоне потенциалов -(0,67±0,05) В или стеклоуглеродного индикаторного электрода в диапазоне потенциалов -(0,60±0,03) В при чувствительности прибора 1•10-9 - 5•10-10 А/мм в дифференциальном режиме съемки вольтамперограмм. Содержание антибиотика оценивают методом добавок аттестованных смесей. Время анализа одной пробы не превышает 60 минут.
Пример 2. Определение левомицетина (хлорамфеникола) в мясе и субпродуктах убойных животных.
В коническую колбу вместимостью 100,0 см3 вносят 5,0-10,0 г мелко измельченного мяса, взвешенного с точностью до 0,01 г, добавляют 20,0 см2 0,1 моль/дм3 НСl и 5,0 см3 этилового спирта. Смесь встряхивают в течение 10-15 минут, переносят в центрифужные пробирки и центрифугируют 15 минут при скорости вращения 6000 об/мин. Центрифугат отфильтровывают через бумажный фильтр в коническую колбу вместимостью 100,0 см3 и добавляют 3,0 г (NH4)2SO4. Содержимое колбы встряхивают в течение 10-15 минут. Полученную смесь переносят в центрифужные пробирки и вновь центрифугируют 15 минут при скорости вращения 6000 об/мин. Центрифугат отфильтровывают через бумажный фильтр в коническую колбу вместимостью 50,0 см3. Для анализа берут аликвоту полученного фильтрата объемом 1,0-5,0 см3, вносят в кварцевый стакан вместимостью 15,0-25,0 см3 с помощью пипетки и доводят бидистиллированной водой до объема 10,0 см3. Затем проводят вольтамперометрические измерения при условиях: потенциал электролиза Еэ= -0,45 В, время электролиза τэ = 30 c, скорость развертки потенциала w=20 мВ/с. Катодный пик левомицетина регистрируют в диапазоне потенциалов -(0,67±0,05) В или -(0,60±0,03) В соответственно на ртутно-пленочном или стеклоуглеродном электродах при чувствительности прибора 1•10-9 - 5•10-10 А/мм в дифференциальном режиме съемки вольтамперограмм. Содержание антибиотика оценивают методом добавок аттестованных смесей. Время анализа одной пробы не превышает 60-80 минут.
Пример 3. Определение левомицетина (хлорамфеникола) в яйце птицы.
В коническую колбу вместимостью 100,0 см3 вносят 5,0-10,0 г предварительно взбитого до однородной массы яйца, взвешенного с точностью до 0,01 г, добавляют 5,0 см3 этанола и 20,0 см3 0,1 моль/дм3 НСl. Смесь осторожно перемешивают покачиванием в течение 10-15 минут, переносят в центрифужные пробирки и центрифугируют в течение 15 минут при скорости вращения 6000 об/мин. Центрифугат осторожно сливают в колбу вместимостью 50,0 см3, добавляют 3,0 г (NH4)2SO4 маленькими порциями до полного растворения соли, пока не образуется яичный студень. Полученный студень энергично встряхивают до образования однородного жидкого раствора, переносят в центрифужные пробирки и центрифугируют в течение 15 минут при скорости вращения 6000 об/мин. Центрифугат отфильтровывают через бумажный фильтр в коническую колбу вместимостью 50,0 см3. Для анализа берут аликвоту полученного фильтрата пробы объемом 1,0-5,0 см3, вносят в кварцевый стакан вместимостью 15,0-25,0 см3 с помощью пипетки и доводят бидистиллированной водой до объема 10,0 см3. Затем проводят вольтамперометрические измерения при условиях: потенциал электролиза Еэ=-0,45 В, время электролиза τэ = 30 c, скорость развертки потенциала w=20 мВ/с. Катодный пик левомицетина регистрируют в диапазоне потенциалов -(0,67±0,05) В на ртутно-пленочном электроде при чувствительности прибора 1•10-9 - 5•10-10 А/мм в дифференциальном режиме съемки вольтамперограмм. Содержание антибиотика оценивают методом добавок аттестованных смесей. Время анализа одной пробы не превышает 70 минут.
Пример 4. Определение левомицетина (хлорамфеникола) в глазных каплях и таблетках.
Пробу глазных капель объемом 0,2 см3 вносят в кварцевый стаканчик вместимостью 15,0÷25,0 см3, разводят бидистиллированной водой до 10 см3. Для анализа берут аликвоту полученного раствора объемом 0,2 см3, вносят в другой полярографически чистый кварцевый стаканчик вместимостью 15,0÷25,0 см3 и доводят фоновым раствором электролита 0,1 моль/дм3 (NH4)2SO4 до 10,0 см3 и проводят вольтамперометрические измерения при условиях: потенциал электролиза Еэ=-0,45 В, время электролиза τэ = 30 c, скорость развертки потенциала w= 25 мВ/с. Катодный пик левомицетина регистрируют в диапазоне потенциалов -(0,67±0,05) В на ртутно-пленочном электроде при чувствительности прибора (1-5)•10-8 А/мм в дифференциальном режиме съемки вольтамперограмм. Содержание антибиотика оценивают методом добавок аттестованных смесей. Время анализа одной пробы не превышает 10 минут.
При анализе таблеток левомицетина измельчают в ступке 5 таблеток, берут навеску пробы 0,09 г, взвешенную с точностью до 0,001 г, вносят в мерную колбу вместимостью 100,0 см3, растворяют навеску пробы в 5.0 см3 этанола и доводят бидистиллированной водой до метки. 0,2 см3 полученного раствора вносят в кварцевый стаканчик вместимостью 15,0-25,0 см3 и добавляют бидистиллированную воду до 6,0 см3. Для анализа берут аликвоту объемом 0,3 см3, доводят фоновым раствором электролита 0,1 моль/дм3 (NH4)2SO4 до 10,0 см3 и далее вольтамперометрические измерения проводят согласно описанному выше примеру 4.
По предлагаемому способу проведена метрологическая аттестация способа количественного химического анализа проб пищевых продуктов и лекарственных препаратов на содержание массовых концентраций левомицетина (хлорамфеникола) методом дифференциальной вольтамперометрии.
Метрологическая аттестация проведена с целью установления приписанных характеристик погрешности результатов анализа, а также для назначения нормативов контроля точности. Метрологические исследования и аттестация данного способа проведены аккредитованной метрологической службой научно-исследовательской лаборатории Томского политехнического университета согласно требованиям ГОСТ Р 8.563-96 "Методики выполнения измерений" и МИ 2336-95 "Характеристики погрешности результатов количественного химического анализа. Алгоритмы оценивания".
Характеристика случайной составляющей погрешности (показатель воспроизводимости) оценена по результатам анализов различных объектов. Из каждого объекта проводили анализ 14 проб по две параллельных. При этом варьировали условия проведения анализов (время, приборы, температура, операторы и т.д.), чтобы учесть все возможные влияющие факторы. Характеристика случайной составляющей погрешности в диапазоне определяемых массовых концентраций левомицетина 3,0-30,0 мкг/кг находится в пределах от 7 до 14 отн.%.
Оценку характеристики систематической составляющей погрешности (показатель правильности) проводили по МИ 2336-95 с использованием образцов для контроля и методом добавок определенного вещества в пробу. Образцы для контроля по первому алгоритму готовили из реальных рабочих проб анализируемых объектов, в которых антибиотик отсутствовал, с добавкой в них определяемого вещества. Добавки левомицетина в пробы делались из чистых реактивов, полученных по фармакопейным статьям Минздрава России до стадии пробоподготовки.
Характеристику общей погрешности оценивали по значениям характеристик случайной и систематической составляющей.
Таким образом, сравнение характеристик КХА левомицетина в пищевых продуктах и фармпрепаратах по предлагаемому способу существенно улучшило метрологические характеристики анализа. Значительно повысилась экспрессность. Время проведения анализа фармпрепаратов сократилось в 3 раза, предложенный способ позволяет экспрессно за 1-1,5 часа определять левомицетин в пищевых продуктах. Условия, используемые в прототипе, не позволяют анализировать пищевые продукты.
Предложенный способ прост, не требует больших трудозатрат, большого количества реактивов и может быть применен в любой химической лаборатории, особенно в настоящее время, когда налажен выпуск современных компьютеризированных анализаторов типа СТА, ТА. Предложенный способ может быть использован для определения антибиотика в биосистемах (кровь, моча и др.) в фармакокинетических исследованиях, в токсикологическом и техническом анализе лекарственных средств, в кормах для животных, а также может служить основой для создания диагностической системы метаболизма антибиотика.

Изобретение относится к аналитической химии и может быть использовано для определения антибиотика в биосистемах (кровь, моча и др.) в фармакокинетических исследованиях, в токсикологическом и техническом анализе лекарственных средств, в кормах для животных, а также может служить основой для создания диагностической системы метаболизма антибиотика. Техническим результатом изобретения является повышение экспрессности, чувствительности и селективности определения левомицетина в пищевых продуктах. Сущность изобретения: левомицетин переводят из пробы в раствор, проводят кислотный гидролиз и осаждают белок из гидролизата с последующим вольтамперометрическим определением левомицетина в безбелковом гидролизате путем регистрации катодных пиков антибиотика на индикаторном ртутно-пленочном или стеклоуглеродном электродах в дифференциальном режиме съемки вольтамперограмм при соответствующих потенциалах -(0,67 ± 0,05)В и -(0,60 ± 0,03)В относительно насыщенного хлорид серебряного электрода на фонах 0,1 моль/дм3 С6Н14О7N2 (рН 4,7 - 5,1) или 0,1 моль/дм3 (NH4)2SO4 с добавлением HCl до рН 5,1 при скорости развертки потенциала 10 - 25 мВ/с. Концентрацию левомицетина определяют по высоте пика методом добавок аттестованных смесей. 2 табл.
Способ количественного определения левомицетина методом дифференциальной вольтамперометрии, заключающийся в том, что левомицетин переводят из пробы в раствор, проводят кислотный гидролиз и осаждают белок из гидролизата с последующим вольтамперометрическим определением, отличающийся тем, что вольтамперометрическое определение левомицетина осуществляют путем регистрации катодных пиков антибиотика на индикаторном ртутно-пленочном или стеклоуглеродном электродах в дифференциальном режиме съемки вольтамперограмм при соответствующих потенциалах -(0,67±0,05) В и -(0,60±0,03) В относительно насыщенного хлорид серебряного электрода на фонах 0,1 моль/дм3 аммония лимоннокислого двузамещенного (рН 4,7-5,1) или 0,1 моль/дм3 (NH4)2SO4 с добавлением HCl до рН 5,1 при скорости развертки потенциала 10-25 мВ/с и концентрацию левомицетина определяют по высоте пика методом добавок аттестованных смесей.
| ПАССЕТ Б.В., АНТИПОВ М.А | |||
| Практикум по техническому анализу и контролю производства химико-фармацевтических препаратов и антибиотиков | |||
| - М.: Медицина, 1981, с | |||
| Паровоз с приспособлением для автоматического регулирования подвода и распределения топлива в его топке | 1919 |
|
SU272A1 |
| МИСКИДЖЬЯН С.П., КРАВЧЕНОК Л.П | |||
| Полярография лекарственных препаратов | |||
| - Киев: Вища школа, 1976, с | |||
| Канальная печь-сушильня | 1920 |
|
SU230A1 |
| Способ анализа левомицетина в пищевых продуктах | 1989 |
|
SU1702302A1 |
| ВОЛЬТАМПЕРОМЕТРИЧЕСКИЙ СПОСОБ ОПРЕДЕЛЕНИЯ 5-ФТОРУРАЦИЛА | 1996 |
|
RU2106623C1 |
| ДИАГНОСТИЧЕСКОЕ УСТРОЙСТВО И СПОСОБ ДЛЯ АМПЕРОМЕТРИЧЕСКОГО ОПРЕДЕЛЕНИЯ ТОКА | 1994 |
|
RU2136204C1 |
| Способ определения хлоракона и левомицетина | 1979 |
|
SU853537A1 |
