Настоящее изобретение относится к биохимии, конкретнее к биологически активным пептидам, обладающим ненаркотическим типом анальгетического действия, которые могут найти применение в медицине и фармакологии в качестве обезболивающих анальгетических препаратов.
Известны различные обезболивающие препараты, которые по своей химической природе и механизму действия подразделяются на наркотические (морфин и близкие к нему структуры) и ненаркотические анальгетики (производные салициловой кислоты, пиразолона, анилина и др.). Все вышеперечисленные анальгетики обладают теми или иными недостатками, которые резко сужают возможности их применения в медицине (М.Д. Машковский. Лекарственные средства, Харьков: из-во «Торсинг», 1997, издание 13, с.144-145).
Известны пептидные анальгетики - синтетические аналоги природных энкефалинов и эндорфинов, - опиоидные пептиды (Casy A.F., Parfitt А.С., Opioid analgesics: Chemistry and receptors. New York, Plenum Press, 1986, 445-502; Lierz P., Stefan Punsmann S., 2008). Их основным недостатком является то, что обезболивающая активность сопровождается привыканием и наркотическим действием. Кроме того, наркотические анальгетики эффективны не при всех болевых синдромах (Fallon M. When morphine does not work. Support Care Cancer. 2008 Feb 15).
Также известны пептидные анальгетики, обладающие ненаркотическим типом обезболивания, не вызывающие привыкания и наркотического действия. Их обезболивающее действие развивается через неопиоидные рецепторы и нейромедиаторы. В этой группе препаратов самое широкое распространение получили синтетические и, в последнее время, рекомбинантные кальцитонины, обезболивающее действие которых реализуется через специфические кальцитониновые рецепторы и серотонинэргическую систему мозга (Yasushi Kuraishi /Neuropeptide action of calcitonin-analgesic effect/ in Magazine Kidney and Metabolic Bone Disease, V.14 No03). Наиболее часто используют синтетическую последовательность, соответствующую кальцитонину лосося, как наиболее активному из всех известных кальцитонинов. Кальцитонин лосося - полипептидный гормон, состоящий из 32 остатков аминокислот с молекулярным весом 3454,93 дальтон. Его структура представляет собой альфа-спираль (Andreotti G. et al, 2006). Первичная структура (последовательность аминокислотных остатков) кальцитонина лосося выглядит следующим образом:
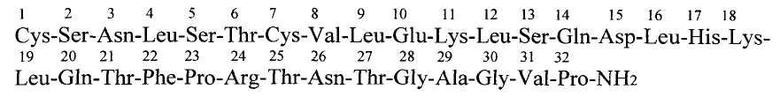
Кальцитонин лосося обладает длительным обезболивающим действием и существует сейчас в различных лекарственных формах: в виде спрея или капель для интраназального использования, для орального и внутримышечного введения, а также в виде свечей.
Однако полноразмерные кальцитонины обладают рядом принципиальных недостатков, в том числе:
1) Гормональная активность, влияние на кальциевый и фосфорный обмены. В этой связи кальцитонины не могут быть использованы во время беременности и обезболивания родов, так как возможны тератогенные эффекты и отдаленные последствия на потомство.
2) Иммунологическая активность. В силу этого при длительном использовании кальцитонина, как это имеет место при лечении и профилактике остеопороза, образуются нейтрализующие антитела, что снижает эффективность использования кальцитонина (Levy F et al., Formation of Neutralizing Antibodies During Intranasal Synthetic Salmon Calcitonin Treatment of Pagets Disease. 1988,67,3,541-545).
3) Полноразмерные кальцитонины содержат амилоидообразующую последовательность Gly2-Glnl4, общую для многих амилоидообразующих белков (Steven S.-S. Wang1, Theresa A. Good2 and Dawn L. Rymer3).
4) Стоимость синтеза полноразмерного кальцитонина и стоимость лечения этим препаратом очень велика. Поэтому кальцитонины относят к орфановым лекарствам, к которым обращаются только тогда, когда нет альтернативных путей лечения, например, при болезни Пейджета (Maresca V. Human calcitonin in the Manangment of osteoporosis: A multicenter Study. - J.Int.Med.Res.,1985, 13, 311-316).
С целью устранения отмеченных недостатков нами был выделен фрагмент кальцитонина лосося, состоящего из 16-21 аминокислот кальцитонина лосося (далее CT16-21), названный «активный центр» кальцитонина (G.P. Vlasov, V.R. Glushenkova, A.M. Kotin et al (1989) "Search of Active Centre of Calcitonin", Chemistry of Peptides and Proteins 4, 89):
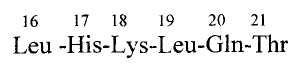
Было показано, что природный фрагмент кальцитонина лосося - пептид СТ16-21 обладает высокой анальгетической активностью в формалиновом тесте на крысах, позволяющем выявить ненаркотический тип обезболивания, и при этом не обладает иммунологической активностью, не влияет на кальциевый обмен и не содержит амилоидообразующей последовательности. Сравнение с аналогичными последовательностями (16-21) кальцитонинов человека, свиньи, быка, крысы выявило большую активность, по сравнению с последними. (А.М. Котин, Г.П. Власов и др. (1988) «Поиск «активного центра» и сравнительное изучение полноразмерного кальцитонина и последовательности 16-21 различных кальцитонинов в разных физиологических тестах». Тезисы докладов симпозиума. «Физиология пептидов». Ленинград, 106).
Задача, на решение которой направлено предлагаемое изобретение, состоит в расширении ассортимента эффективных средств, обладающих ненаркотическим типом анальгетического действия и получаемых простым синтезом.
Поставленная задача решается тем, что предложены синтетические пептиды общей формулы 1 [SEQ ID NO:1]
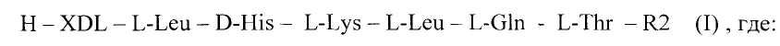
Н - водород,
XDL - отсутствие аминокислоты или L-Tyr,
R2 -ОМе или NH2,
или пептиды - ретроинверсии формулы (I), имеющие обратную последовательность аминокислот с заменой L-формы аминокислот на D-форму и D-формы аминокислот на L-форму, общей формулы 2 [SEQ ID NO:2]

Н - водород,
XDL1 - отсутствие аминокислоты или D-Tyr,
R2 - ОМе или NH2,
в качестве обезболивающих препаратов с ненаркотическим типом анальгетического действия.
Предложенные пептиды обладают обезболивающим действием, в том числе при системном или интраназальном введении.
Сущность изобретения заключается в том, что экспериментальным путем было установлено, что заявляемые пептиды, имеющие простую структуру, что облегчает их получение химическим путем, обладают высокой обезболивающей активностью, проверенной анальгетическими тестами, проведенными на животных.
Некоторые пептиды общей формулы I, II представлены в таблице 1:
Все вышеперечисленные пептиды обладали анальгетической активностью. Ниже приведены примеры, подтверждающие это.
Синтез пептидов осуществляли методами пептидной химии, твердофазным методом синтеза с использованием L и D аминокислот.
Пример 1. Синтез пептида Н-L-Leu-D-His-L-Lys-L-Leu-L-Gln-L-Thr-NH2 (Пептид №2 из Таблицы №1)
Пептид Н-L-Leu-D-His-L-Lys-L-Leu-L-Gln-L-Thr-NHb получали методом автоматического твердофазного синтеза по Fmoc-схеме на полимере Ринка (Rink Amide Resin, 0,6 ммоль амино-групп на 1 г полимера) с использованием DCC/HOBt (N,N′-дициклогексилкарбодиимид/1-гидроксибензотриазол) метода активации аминокислот. Деблокирование производили путем обработки раствором piperidine/DMF (пиперидин/N,N-диметилформамид) (1:4) в течение 7 минут. Защиту групп боковых цепей производили следующими группами: tBu (трет-бутиловый эфир) для тирозина, треонина, Trt (тритил или трифенилметил) для глутамина и для гистидина, Вое (т-бутилоксикарбонил) для лизина. Пептиды отщепляли от полимера и деблокировали смесью TFA/H2O/EDT (трифторуксусная кислота/вода/1,2-этандитиол) (90:5:5). Очистку пептидов проводили путем обратнофазовой ВЭЖХ (колонка С18), Элюент - ацетонитрил - вода (0,1М дигидрофосфата калия) в соотношении 6:4. Пептиды были охарактеризованы с помощью масс-спектрометра. Была произведена замена аминокислот в отдельных позициях пептида СТ16-21 и выяснено, как это влияет на обезболивающие свойства полученных пептидов. Анальгетическую активность вновь синтезированных петидов проверяли в «формалином тесте», позволяющем выявить ненаркотический тип обезболивания (Wheeler-Aceto H., Porrea F., A.Cowan. The rat paw formalin test: comparison of noxious agents. Pain, 40 (1990), 229-238).
Пример 2. Проверка анальгетической активности вновь синтезированных пептидов. Анальгетическую активность вновь синтезированных пептидов проверяли следующим образом. Крысам массой 180-200 г под эфирным наркозом субокципитально при помощи микродозатора в 10 мкл физиологического раствора вводили исследуемый пептид. Контрольным животным сходным образом вводили равное количество физиологического раствора. Через 20 минут в дорзальную поверхность правой задней лапы вводили 50 мкл раствора формалина в разведении 1:50. Время введения пептида и разведение формалина были отработаны нами ранее. Каждую крысу использовали только один раз. Наиболее четкие поведенческие показатели болевой реакции выражались в поджатии, вылизывании, покусывании и потряхивании лапы. При этом, после первой острой реакции на боль, продолжающейся у контрольных животных 6-7 мин, следует период покоя: крыса опускает лапу, исчезает груминг и покусывание. Затем реакция повторяется с не меньшей экспрессией - вторая фаза болевой реакции. Для получения количественных данных визуально фиксировали момент поджатия лапы (начало 1-ой фазы болевой реакции), продолжительность этой реакции, продолжительность покоя и время наступления второй фазы реакции - повторного поджатия лапы, либо ее отсутствие. Пептид вводили за 20 мин до формалина при субокципитальном способе введения пептида и за 30 мин при интраназальном.
I. Были синтезированы в соответствии с Примером 1 пептиды, с заменой природной аминокислоты L-гистидин в 17-м положении пептида СТ16-21 на D-гистидин и модификацией концевой последовательности пептида добавлением метилового эфира или гидразида, отсутствующих в природном фрагменте кальцитонина. Активность синтезированных синтетических пептидов исследовали в соответствии с методикой, описанной в Примере 2. Результаты приведены в Таблицах 2 и 3, где: СТ16-21 это L-Leu-L-His-L-Lys-L-Leu-L-Gln-L-Thr
Пептид №1 из Таблицы 1 это L-Leu-D-His-L-Lys-L-Leu-L-Gln-L-Thr-OMe
Пептид №2 из Таблицы 1 это L-Leu-D-His-L-Lys-L-Leu-L-Gln-L-Thr-NH2
ство крыс
Основной вывод, который можно сделать из данных, представленных в Таблицах 2 и 3, следующий:
1. Замена природной аминокислоты L-гистидин в 17-м положении пептида CT16-21 на D-гистидин и модификация концевой последовательности пептида добавлением метилового эфира или гидразида, отсутствующих в природном фрагменте кальцитонина, приводит не только к сохранению, но и к возрастанию обезболивающей активности пептида.
II. Были синтезированы в соответствии с Примером 1 пептиды, с заменой природной аминокислоты L-гистидин в 17-м положении пептида CTie-21 на D-гистидин, модификацией концевой последовательности пептида добавлением метилового эфира или гидразида, а также с добавлением на N-конце последовательности аминокислоты L-Tyr, отсутствующей в природном фрагменте кальцитонина. Активность синтезированных синтетических пептидов исследовали в соответствии с методикой, описанной в Примере 2. Результаты приведены в Таблице 4, где:
CT16-21 это L-Leu-L-His-L-Lys-L-Leu-L-Gln-L-Thr
Пептид №3 из Таблицы 1 это L-Tyr-L-Leu-D-His-L-Lys-L-Leu-L-Gln-L-Thr-OMe
Пептид №4 из Таблицы 1 это L-Tyr-L-Leu-D-His-L-Lys-L-Leu-L-Gln-L-Thr-NH2
Основной вывод, который можно сделать из данных, представленных в Таблице 4, следующий:
2. Синтезированные пептиды, с заменой природной аминокислоты L-гистидин в 17-м положении пептида СТ16-21 на D-гистидин, модификацией концевой последовательности пептида добавлением метилового эфира или гидразида, а также с добавлением на N-конце последовательности аминокислоты L-Tyr, отсутствующей в природном фрагменте кальцитонина, не только сохраняют обезболивающую активность, но и более эффективны в предотвращении второго пика болевой реакции по сравнению с CT16-21.
III. Были синтезированы в соответствии с Примером 1 пептиды - ретроинверсии пептидов №№1-4 из Таблицы 1, обладающие обратной последовательностью аминокислот с заменой L-форм аминокислот на D-формы и D-форм аминокислот на L-формы. Такие пептиды отличаются высокой устойчивостью к всевозможным пептидазам (Mariotti и др., European Patent EP0393786). В частности, были синтезированы последовательности:
Пептид №5 из Таблицы 1 это D-Thr-D-Gln-D-Leu-D-Lys-L-His-D-Leu-OMe
Пептид №6 из Таблицы 1 это D-Thr-D-Gln-D-Leu-D-Lys-L-His-D-Leu-NH2
Пептид №7 из Таблицы 1 это D-Thr-D-Gln-D-Leu-D-Lys-L-His-D-Leu-D-Tyr-OMe
Пептид №8 из Таблицы 1 это D-Thr-D-Gln-D-Leu-D-Lys-L-His-D-Leu-D-Tyr-NH2
Активность синтезированных синтетических пептидов сравнивали как с контролем (физиологический раствор), так и с СТ16-21 в соответствии с методикой, описанной в Примере 2. Результаты приведены в Таблице 5.
во крыс
Основной вывод, который можно сделать из данных представленных в Таблице 4, следующий:
3. Пептиды - ретроинверсии №5-8 из Таблицы №1, обладающие обратной последовательностью аминокислот с заменой L-форм аминокислот на D-формы и D-форм аминокислот на L-формы по сравнению с пептидами №1-4 из Таблицы №1, также обладают высокой анальгетической активностью.
Таким образом, техническим результатом предлагаемого изобретения является высокая анальгетическая активность предложенных пептидов, что позволяет рассматривать их в качестве основы для создания безопасных лекарственных анальгетических средств, с ненаркотическим типом анальгетического действия.



Изобретение относится к области биотехнологии, конкретно к синтетическим пептидам, обладающим ненаркотическим типом анальгетического действия, общей формулы 1
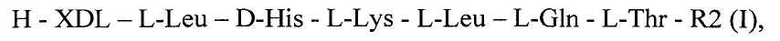
где H - водород, XDL - отсутствие аминокислоты или L-Tyr, R2 - OMe или NH2,
а также пептиды - ретроинверсии формулы (I), имеющие обратную последовательность аминокислот с заменой L-формы аминокислот на D-форму и D-формы аминокислот на L-форму, общей формулы 2
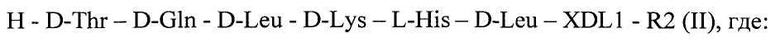
H - водород, XDL1 - отсутствие аминокислоты или D-Tyr, R2 - OMe или NH2. Заявленное изобретение позволяет получить безопасные лекарственные анальгетические средства с ненаркотическим типом анальгетического действия. 5 табл., 2 пр.
Синтетические пептиды общей формулы 1

где Н - водород,
XDL - отсутствие аминокислоты или L-Tyr,
R2 - ОМе или NH2,
или пептиды - ретроинверсии формулы (I), имеющие обратную последовательность аминокислот с заменой L-формы аминокислот на D-форму и D-формы аминокислот на L-форму, общей формулы 2

где Н - водород,
XDL1 - отсутствие аминокислоты или D-Tyr,
R2 - ОМе или NH2,
в качестве обезболивающих препаратов с ненаркотическим типом анальгетического действия.
| ANDREEV N.A | |||
| et al, Synthesis and study of the biological activity of fragments salmon calcitonin II, 1993, J | |||
| Chemistry of Natural Compounds, v.29, i.1, p.107-112 | |||
| ROGACHEVSKII I.V | |||
| et al., Synthesis and steric structure of H-Leu-His-Lys-Leu-Gln-Thr-NH2 and H-Ala-D-Ala-Lys-Leu-Ala-Thr-NH2 Peptides, Russian Journal of General Chemistry, 2005, v.75, |

