ПЕРЕКРЕСТНЫЕ ССЫЛКИ НА РОДСТВЕННЫЕ ЗАЯВКИ
[0001] Данная заявка испрашивает приоритет на основании предварительной заявки на патент США №61/591236, поданной 26 января 2012 г. под названием «ПЕПТИДЫ-АНТАГОНИСТЫ ПЕПТИДНЫХ ГОРМОНОВ ИЗ СЕМЕЙСТВА КАЛЬЦИТОНИНА (CGRP) И ИХ ПРИМЕНЕНИЕ» ("PEPTIDE ANTAGONISTS OF THE CALCITONIN CGRP FAMILY OF PEPTIDE HORMONES AND THEIR USE"), которая включена в настоящую заявку во всей полноте посредством ссылки.
ПЕРЕЧЕНЬ ПОСЛЕДОВАТЕЛЬНОСТЕЙ
[0002] Настоящая заявка подается вместе с перечнем последовательностей в электронной форме. Перечень последовательностей представлен в виде файла под названием CSOAR001WOSEQLIST.TXT, созданного 24 января 2012 г., размер которого составляет приблизительно 17 КБ. Информация в электронной форме перечня последовательности включена в настоящую заявку во всей полноте посредством ссылки.
УРОВЕНЬ ТЕХНИКИ
Область изобретения
[0003] Варианты реализации настоящего изобретения относятся к пептидным антагонистам пептидных гормонов из семейства кальцитонина/связанного с геном кальцитонина пептида (CT/CGRP) и их терапевтическому применению. Описание предшествующего уровня техники
[0004] Семейство пептидов CT/CGRP включает связанный с геном кальцитонина пептид (CGRP), адреномедуллин (ADM), интермедии (IM), кальцитонин (СТ) и амилин. Биологическое действие указанных пептидов опосредуется связыванием двух близкородственных G-белок-связанных рецепторов II типа - рецептора кальцитонина (CTR) и кальцитонин рецептор-подобного рецептора (CRLR) (Christopoulos, et al. 1999, Mol. Pharmacol. 56:235-242; Poyner et al. 2002 Pharmacol. Rev. 54:233-246). Хотя рецептор кальцитонина является основным медиатором действия кальцитонина, он предпочтительно связывается с амилином, когда рецептор ассоциирован с белком, модифицирующим активность рецептора (RAMP) (см., например, Tilikaratne, et al. 2000, J. Pharmacol. Exp. Ther. 294(l):61-72). Клонирование и исследования функции показали, что CGRP, ADM, IM и, в меньшей степени, амилин взаимодействуют с разными сочетаниями CRLR и трех белков, модифицирующих активность рецептора (RAMP-1, RAMP-2 и RAMP-3); (см., например, McLatchie et al. 1998, Nature 393:333-339 и Roh et al. 2004, JBC 279(8):7264-7274). Фактически для генерирования функциональных рецепторов для связанного с геном кальцитонина пептида (CGRP), адреномедуллина (ADM) и интермедина (IM) требуется совместная экспрессия кальцитонин рецептор-подобного рецептора (CRLR) и белков, модифицирующих активность рецептора (RAMP). Образование гетеродимеров между RAMP и CRLR важно для точного нацеливания на поверхность клетки и фармакологических свойств рецепторов CGRP, ADM и IM. Совместная экспрессия RAMP-1 с CRLR приводит к образованию рецептора CGRP, тогда как совместная экспрессия RAMP-2 и RAMP-3 с CRLR приводит к образованию рецепторов ADM и IM, соответственно. (Miret, et al. 2002, JBC 277(9):6881-6887). Было показано, что IM является неселективным агонистом для всех трех корецепторов RAMP/CRLR.
[0005] Физиологические функции пептидных гормонов в семействе CT/CGRP определяются специфичностью связывания с рецепторами и профилем экспрессии в тканях индивидуальных лигандов и соответствующих им рецепторов, и было показано, что они участвуют в морфогенезе сердечно-сосудистой системы, сенсорной нервной передаче, реакциях воспаления, ноцицептивном поведении и гомеостазе глюкозы (см, например, Hay, et al. 2001, Trends Pharmacol. Sci. 22:57-59; Shindo, et al. 2001, Circulation 104:1964-1971; Zhang et al. 2001, Pain 89:265-273; Salmon et al. (1999) Neuroreport 10:849-854; Salmon, et al. 2001, Nat. Neurosci. 4: 357-358; and Mulder, et al. 2000, Am. J. Physiol. 278:E684-E691).
[0006] CGRP (связанный с геном кальцитонина пептид) - хорошо изученный пептид в семействе пептидных гормонов CT/CGRP - представляет собой сенсорный нейропептид с мощным сосудорасширяющим и кардиотоническим действием, как описано в патенте США №4530838, выданном Evans, et al. CGRP присутствует как в центральной, так и в периферической нервной системе и концентрируется в тех областях организма, которые получают сенсорный вход импульсов из заднего рога с ограниченным числом, ассоциированным с вегетативным входом. В головном мозге данный пептид присутствует в ядрах чувствительных и двигательных черепно-мозговых нервов и в телах клеток в гипоталамусе, в предзрительном поле, вентромедиальном таламусе, гиппокампе и подобных областях (Poyner, D. 1992, Pharmac. Ther. 56:23-51).
[0007] Предполагается, что ингибиторы CGPR на уровне рецепторов подходят для лечения патофизиологических состояний, при которых имеет место избыточная активация рецептора CGRP. Некоторые из них включают нейрогенную вазодилатацию, нейрогенное воспаление, мигрень, кластерную головную боль и другие типы головной боли, термическое повреждение, циркуляторный шок, приливы в менопаузе и астму. В частности, было показано, что активация рецептора CGRP участвует в патогенезе головных болей при мигрени (Edvinsson L. 2001, CNS Drugs 15(10):745-53; Williamson, D.J. 2001 Microsc. Res. Tech. 53:167-178.; Grant, A.D. 2002, Brit. J Pharmacol. 135:356-362). Известно, что мигрень характеризуется сильными головными болями, которые возникают при данной патологии. Предполагается, что головная боль, ассоциированная с мигренью, возникает вследствие сильного расширения сосудов головного мозга, сопровождающего приступы мигрени. Содержащие CGRP нервные волокна иннервируют сосуды головного мозга и твердой мозговой оболочки, и считается, что CGRP продлевает расширение сосудов (Moskowitz 1992, Trends Pharmacol. Sci. 13:307-311). Кроме того, уровень CGRP в сыворотке во время мигрени повышен (Goadsby, et al. 1990, Ann. Neurol. 28:183-7), а лечение противомигренозными препаратами возвращает уровень CGRP к норме и ослабляет головную боль (Gallai, et al. 1995, Cephalalgia 15:384-90). Лица, страдающие мигренью, демонстрируют повышенный базальный уровень CGRP по сравнению с контролем (Ashina, et al., 2000, Pain 86(1-2)133-8). Внутривенное введение CGRP приводит к длительным головным болям у лиц, страдающих мигренью (Lassen, et al. 2002, Cephalalgia 22(1):54-61). Таким образом, антагонисты CGRP стали центром внимания последних исследований как способ блокирования цереброваскулярных рецепторов CGRP и, как следствие, блокирования расширения сосудов, вызывающего мигрень.
[0008] Известны как низкомолекулярные, так и пептидные антагонисты рецепторов CGRP. Они включают, например, олцегепант (BIBN4096 BS) для внутривенного введения и телкагепант (МК-0974) для перорального введения, производимые компаниями «Boehringer Ingelheim Pharmaceuticals» и «Merck & Co., Inc.», соответственно. Было показано, что оба указанных низкомолекулярных антагониста CGRP были безопасны, эффективны и хорошо переносились в ранних клинических исследованиях при лечении острой мигрени (см., например, Tepper and Stillman, 2008, Headache 48(8):1259-1268; и Durham and Vause 2010, CNS Drugs 24(7):539-548.) Однако недавно испытания II фазы по оценке применения низкомолекулярного антагониста CGRP - MK-3207 - для предотвращения мигреней, было прекращено компанией «Merck & Co., Inc.» в связи с выявлением бессимптомных аномальных результатов исследований функции печени у некоторых пациентов в продолжении фармакологического испытания I фазы («Merck Updates Status of Clinical Development Programs for Investigational CGRP Receptor Antagonist Treatments for Acute Migraine; MK-3207 Clinical Development Discontinued» Sep. 10, 2009. Merck & Co., Inc. Web. June 1, 2011).
[0009] Известны другие молекулы, конкурирующие за связывание с рецепторами CGRP, - это пептиды, содержащие последовательность CGRP, но лишенные по меньшей мере первых семи аминокислот из последовательности аминокислот CGRP, например, включая, но не ограничиваясь перечисленным, CGRP (8-37), CGRP (28-37), [Tyr°]CGRP (28-37) и CGRP (12-37). Другие антагонисты CGRP включают h-α-CGRP (9-37), h-α-CGRP (10-37), h-α-CGRP (11-37) (Mimeault, M. et al., 1992, J. Med. Chem. 35:2163-2168). Еще некоторые антагонисты CGRP включают [Ala 9]-h-α-CGRP (8-37), [Ala 10]-h-α-CGRP (8-37), [Ala 11]-h-α-CGRP (8-37) и [Ala 12]-h-α-CGRP (8-37), id. Дополнительные антагонисты CGRP включают h-α-CGRP (19-37), h-α-CGRP (23-37) и ацетил-h-α-CGRP (19-37) (Rovero, P. et al. 1992, Peptides 13:1025-1027).
[0010] Хотя было показано, что ряд пептидных антагонистов рецепторов CGRP эффективно конкурирует с CGRP in vitro, указанные антагонисты не показали аналогичного эффекта в моделях мигренеподобных патологий in vivo.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
[0011] Неожиданно было обнаружено, что некоторые конкретные аминокислоты в N-концевой части связанного с геном кальцитонина пептида, как раскрыто и описано в настоящей заявке, отвечают за агонистическую активность указанного пептида. Кроме того, замена определенных аминокислот в N-концевой части связанного с геном кальцитонина пептида может приводить к перестройке активности с агонистической на антагонистическую. Более того, было обнаружено, что дополнительные замены или модификации могут приводить к получению дополнительных желаемых свойств у антагонистов согласно настоящему изобретению.
[0012] Согласно некоторым вариантам реализации изобретения предложен антагонист на основе модифицированного связанного с геном кальцитонина пептида, причем указанный антагонист имеет структуру согласно формуле I:
где: 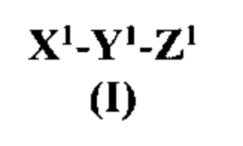
X1 представляет собой N-концевой фрагмент модифицированного связанного с геном кальцитонина пептида или другого члена семейства пептидов CT/CGRP, содержащий по меньшей мере от пяти до семи остатков аминокислот, где два остатка аминокислот указанного N-концевого фрагмента являются цистеинами (Cys), где концевой остаток является Cys и где остаток, идущий непосредственно перед концевым остатком Cys является заменой остатка треонина (Thr) на не треонин;
Y1 является центральной структурой, содержащим от 15 до более чем 24, от 15 до 24, от 15 до 22, 18-22 или 19-20 остатков, причем по меньшей мере некоторые остатки центральной структуры способны при физиологических условиях образовывать α-спираль, причем по меньшей мере одна аминокислота центральной структуры является аргинином (Arg) или лизином (Lys), и указанное центральная структура содержит α-спираль; и
Z1 является модифицированным С-концевым фрагментом модифицированного связанного с геном кальцитонина пептида или другого члена семейства пептидов CT/CGRP, содержащего от пяти до семи остатков аминокислот с С-концевым амидом, где по меньшей мере один остаток аминокислот указанного С-концевого фрагмента является фенилаланином (Phe), тирозином (Tyr), пролином (Pro) или гидроксипролином (Hyp);
или его фармацевтически приемлемая соль.
[0013] Согласно некоторым вариантам реализации изобретения предложен антагонист на основе модифицированного связанного с геном кальцитонина пептида, содержащий:
последовательность аминокислот, по меньшей мере на 80% идентичную последовательности аминокислот в соответствии с SEQ ID №: 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14 или 15, причем указанный пептид сохраняет антагонистическую активность.
[0014] Согласно некоторым вариантам реализации изобретения предложена фармацевтическая композиция, содержащая фармацевтически приемлемое вспомогательное вещество и антагонист на основе модифицированного связанного с геном кальцитонина пептида, как раскрыто и описано в настоящей заявке.
[0015] Согласно некоторым вариантам реализации изобретения предложен способ лечения состояния, ассоциированного с аномальным уровнем CGRP, включающий введение антагониста на основе модифицированного связанного с геном кальцитонина пептида, как раскрыто и описано в настоящей заявке, субъекту, способ, включающий введение указанному субъекту эффективного количества антагониста на основе модифицированного связанного с геном кальцитонина пептида, как раскрыто и описано в настоящей заявке.
[0016] Согласно некоторым вариантам реализации изобретения предложен антагонист на основе модифицированного связанного с геном кальцитонина пептида, имеющий структуру, выбираемую из следующих последовательностей пептидов, перечисленных в таблице 1.


[0017] Согласно некоторым вариантам реализации изобретения предложен способ доставки терапевтического агента в клетку. Терапевтический агент связан с антагонистом на основе модифицированного связанного с геном кальцитонина пептида, как раскрыто и описано в настоящей заявке, который селективно связывается с членом семейства рецепторов CGRP.
[0018] Согласно некоторым вариантам реализации изобретения предложен конъюгат, который содержит терапевтический агент, связанный с антагонистом на основе связанного с геном кальцитонина пептида, который раскрывается и описан в настоящей заявке, и который селективно связывается с членом семейства рецепторов CGRP. Согласно некоторым вариантам реализации изобретения предложен способ идентификации рецептора CGRP, связывающегося с лигандом, путем получения антагониста на основе модифицированного связанного с геном кальцитонина пептида, связанного с рецептором CGRP, получения тестируемого соединения или библиотеки тестируемых соединений и идентификации соединений, которые способны вызывать диссоциацию антагониста на основе связанного с геном кальцитонина пептида от рецептора CGRP. Такие соединения, идентифицированные посредством указанного способа, можно в дальнейшем подвергать скринингу на другие рецепторы CGRP и рецептор CGRP-связывающие агенты для выявления лигандов, селективно связывающих рецепторы CGRP.
ПОДРОБНОЕ ОПИСАНИЕ ПРЕДПОЧТИТЕЛЬНЫХ ВАРИАНТОВ РЕАЛИЗАЦИИ ИЗОБРЕТЕНИЯ
[0019] Согласно некоторым вариантам реализации изобретения предложен антагонист на основе модифицированного связанного с геном кальцитонина пептида, причем указанный антагонист имеет структуру согласно формуле I:
где: 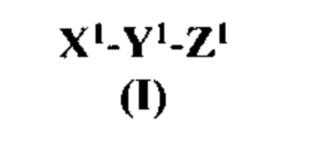
X1 представляет собой модифицированный N-концевой фрагмент связанного с геном кальцитонина пептида или другого члена семейства пептидов CT/CGRP, содержащий по меньшей мере от пяти до семи остатков аминокислот, где два остатка аминокислот указанного N-концевого фрагмента являются цистеинами (Cys), где С-концевой остаток является Cys и где остаток, идущий непосредственно перед С-концевым остатком Cys является заменой остатка треонина (Thr) на нетреонин;
Y1 является центральной структурой, причем по меньшей мере одна аминокислота центральной структуры является аргинином (Arg) или лизином (Lys), и указанная центральная структура содержит α-спираль; и
Z1 является модифицированным С-концевым фрагментом связанного с геном кальцитонина пептида или другого члена семейства пептидов CT/CGRP, содержащим от пяти до семи остатков аминокислот с С-концевым амидом, где по меньшей мере один остаток аминокислот указанного С-концевого фрагмента является фенилаланином (Phe), тирозином (Tyr), пролином (Pro) или гидроксипролином (Hyp);
или его фармацевтически приемлемая соль.
[0020] Согласно некоторым вариантам реализации изобретения X1 характеризуется тем, что остаток, который идет раньше С-концевого цистеина на четыре, пять или шесть положений аминокислот, также является цистеином, так что два вышеуказанных цистеина могут образовать дисульфидную связь. Остатки между двумя остатками Cys, участвующими в образовании дисульфидной связи, не ограничены по последовательности за исключением того, что остаток, идущий перед остатком Cys в С-концевом фрагменте не должен представлять собой Thr, как упоминалось выше, и что среди 7 С-концевых остатков фрагмента X1 может быть не более двух цистеинов. Вышеуказанная дисульфидная связь стабилизирует структуру X1, облегчая как образование альфа-спирали в Y1, описанной ниже, так и связывание X1 с трансмембранным компонентом целевого рецептора при конкурировании с CGRP.
[0021] Введение остатка, отличного от Thr в положении непосредственно между N-концом и вторым цистеином во фрагменте X1, описанном выше, приводит к утрате активирующей активности указанной молекулы при взаимодействии с рецептором CGRP или членом семейства рецепторов CT/CGRP по сравнению с молекулой дикого типа, которая содержит Thr в указанном положении, но может не влиять на связывание с рецептором. Как следствие, такие замены приводят к получению молекулы, которая может занимать рецептор, но которая выступает в роли антагониста, а не активирует путь сигнальной трансдукции, делая рецептор недоступным для связывания с агонистами сигнальной трансдукции.
[0022] Добавление остатков на N-конце к X1 может не оказывать влияния на активность антагониста согласно некоторым вариантам реализации изобретения. Согласно некоторым вариантам реализации изобретения добавление остатков на N-конце к X1, например, 864-ого остатка последовательности XTENS, содержащей Ala, Glu, Gly, Pro, Ser и Thr, может влиять на стабильность лекарственного средства (Schellenberger et al., 2009, Nature Biotechnology 27 (12): 1186-1192). Согласно некоторым вариантам реализации изобретения добавление остатков на N-конце может увеличивать время полувыведения вводимого лекарственного средства. Указанные изменения рассматриваются в настоящей заявке; специалист в данной области техники должен знать, как это можно сделать.
[0023] Согласно некоторым вариантам реализации изобретения антагонист, раскрываемый в настоящей заявке, содержит центральную структуру Y1, содержащую от 15 до 22 остатков. Согласно некоторым вариантам реализации изобретения антагонист, раскрываемый в настоящей заявке, содержит центральную структуру Y1, содержащую более 24, от 15 до 24, от 15 до 22, 18-22, или 19-20 остатков, причем по меньшей мере некоторые остатки центральной структуры способны при физиологических условиях образовывать α-спираль. Четвертый остаток от N-конца данной центральной структуры часто является положительно заряженным остатком, либо аргинином (Arg), либо лизином (Lys). Восемнадцатый остаток часто является аргинином. Длина центральной структуры ограничена не количеством остатков самим по себе, а пространственными соображениями, которые требуют, чтобы X1 и Z1 располагались так, чтобы они могли взаимодействовать с целевым рецептором на поверхности мембраны клетки, и на внеклеточном домене, соответственно, при конкурировании с CGRP.
[0024] Z1 является модифицированным С-концевым фрагментом модифицированного связанного с геном кальцитонина пептида или другого члена семейства пептидов CT/CGRP, содержащим от пяти до семи остатков аминокислот или более, с С-концевым амидом, где по меньшей мере одна аминокислота указанного С-концевого фрагмента является фенилаланином (Phe), пролином (Pro), тирозином (Tyr) или гидроксипролином (Hyp). Как и описанный выше Y1, Z1 ограничен не по последовательности, а функциональными требованиями. В случае Z1, для которого требуется, чтобы он взаимодействовал с целевым рецептором в сайте в его внеклеточном домене, как в случае связывания антагониста с рецептором CGRP, X1 располагается так, чтобы взаимодействовать с рецептором на поверхности клетки, a Z1 взаимодействует с частью RAMP рецептора.
[0025] Целый пептид может быть доставлен один или в виде его фармацевтически приемлемой соли.
[0026] Согласно некоторым вариантам реализации изобретения предложен антагонист на основе связанного с геном кальцитонина пептида, содержащий: последовательность аминокислот, идентичную по меньшей мере на 80% последовательности аминокислот в соответствии с SEQ ID №: 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14 или 15, причем указанный пептид сохраняет антагонистическую активность.
[0027] Некоторые варианты реализации изобретения включают антагонист с центральной структурой из 18-22 остатков.
[0028] Согласно некоторым вариантам реализации изобретения указанного антагониста на основе модифицированного связанного с геном кальцитонина пептида, имеющего структуру в соответствии с формулой I, N-концевой фрагмент содержит:
Х11-Х12-Х13-Х14-Х15-Х16-Х17 (SEQ ID №: 16), где:
X11 может быть выбран из группы, состоящей из аланина (Ala), цистеина (Cys), глицина (Gly), изолейцина (Ile), лейцина (Leu), метионина (Met), фенилаланина (Phe), пролина (Pro), триптофана (Trp) и валина (Val);
X12 может быть выбран из группы, состоящей из цистеина (Cys), серина (Ser и тирозина (Tyr);
X13 может быть выбран из группы, состоящей из аргинина (Arg), аспарагина (Asn), аспарагиновой кислоты (Asp), цистеина (Cys), глутаминовой кислоты (Glu), глутамина (Gln), гистидина (His), лизина (Lys), серина (Ser), треонина (Thr), тирозина (Tyr) и валина (Val);
X14 может быть выбран из группы, состоящей из аргинина (Arg), аспарагина (Asn), аспарагиновой кислоты (Asp), глутаминовой кислоты (Glu), глутамина (Gln), гистидина (His), лейцина (Leu), лизина (Lys), фенилаланина (Phe), серина (Ser), треонина (Thr), тирозина (Tyr) и валина (Val);
X15 может быть выбран из группы, состоящей из аланина (Ala), глицина (Gly), изолейцина (Ile), лейцина (Leu), метионина (Met), фенилаланина (Phe), серина (Ser), триптофана (Тур) и валина (Val);
X16 может быть выбран из группы, состоящей из аланина (Ala), глицина (Gly), изолейцина (Ile), лейцина (Leu), метионина (Met), фенилаланина (Phe), серина (Ser), триптофана (Тур) и валина (Val); и
X17 представляет собой цистеин (Cys), и он способен образовывать дисульфидный мостик с остатком цистеина в X11, X12 или X13; и
с тем дополнительным ограничением, что только два остатка X1 (то есть, X17 и только один из X11, X12 и X13) являются остатками цистеина.
[0029] Согласно некоторым вариантам реализации указанного антагониста на основе модифицированного связанного с геном кальцитонина пептида, имеющего структуру в соответствии с формулой I, X11 выбран из группы, состоящей из Ala, Cys, и Gly. Согласно некоторым вариантам указанного антагониста на основе модифицированного связанного с геном кальцитонина пептида, имеющего структуру в соответствии с формулой I, X12 выбран из группы, состоящей из Cys и Ser, с оговоркой, что только один из X11 и X12 может быть Cys. Согласно некоторым вариантам реализации указанного антагониста на основе модифицированного связанного с геном кальцитонина пептида, имеющего структуру в соответствии с формулой I, X13 выбран из группы, состоящей из Arg, Asn, Asp и Val. Согласно некоторым вариантам реализации указанного антагониста на основе модифицированного связанного с геном кальцитонина пептида, имеющего структуру в соответствии с формулой I, X14 выбран из группы, состоящей из Leu, Phe и Thr. Согласно некоторым вариантам реализации указанного антагониста на основе модифицированного связанного с геном кальцитонина пептида, имеющего структуру в соответствии с формулой I, X15 выбран из группы, состоящей из Ala, Gly и Ser. Согласно некоторым вариантам реализации указанного антагониста на основе модифицированного связанного с геном кальцитонина пептида, имеющего структуру в соответствии с формулой I, X15 выбран из группы, состоящей из Ala, Ile, Leu, Ser и Val.
[0030] Согласно некоторым вариантам реализации указанного антагониста на основе модифицированного связанного с геном кальцитонина пептида, имеющего структуру в соответствии с формулой I, Х11-Х12-Х13-Х14-Х15-Х16-Х17 выбран из группы, состоящей из NH2-Ala-Cys-Asp-Thr-Ala-Ala-Cys (SEQ ID №: 17), NH2-Ala-Cys-Asp-Thr-Ala-Ser-Cys (SEQ ID №: 18), NH2-Ala-Cys-Asp-Thr-Ala-Val-Cys (SEQ ID №: 19), NH2-Ala-Cys-Asn-Thr-Ala-Ala-Cys (SEQ ID №: 20), NH2-Ala-Cys-Val-Leu-Gly-Ala-Cys (SEQ ID №: 21), NH2-Ala-Cys-Arg-Phe-Gly-Ala-Cys (SEQ ID №: 22), NH2-Ala-Cys-Asp-Leu-Ser-Ala-Cys (SEQ ID №: 23), NH2-Ala-Cys-Asn-Leu-Ser-Ala-Cys (SEQ ID №: 24), NH2-Cys-Ser-Asn-Thr-Ala-Ala-Cys (SEQ ID №: 25), NH2-Ala-Cys-Asp-Thr-Ala-Leu-Cys (SEQ ID №: 26), NH2-Ala-Cys-Asp-Thr-Ala-Ile-Cys (SEQ ID №: 27), NH2-Ala-Cys-Asp-Thr-Ala-Leu-Cys (SEQ ID №: 28), NH2-Ala-Cys-Asp-Thr-Ala-Ile-Cys (SEQ ID №: 29), NH2-Ala-Cys-Asp-Leu-Ser-Val-Cys (SEQ ID №: 30), NH2-Ala-Cys-Asp-Leu-Ser-Val-Cys (SEQ ID №: 31), NH2-Ala-Cys-Asn-Leu-Ser-Val-Cys (SEQ ID №: 32) и NH2-Cys-Ser-Asn-Thr-Ala-Val-Cys (SEQ ID №: 33).
[0031] Согласно некоторым вариантам реализации изобретения один или более остатков присоединены на N-конце к X11, в результате чего образуется полипептид с ответвлением из остатков аминокислот на N-конце после X1. Согласно некоторым вариантам реализации изобретения данное ответвление влияет на стабильность указанного антагониста после введения.
[0032] Согласно некоторым вариантам реализации указанного антагониста на основе модифицированного связанного с геном кальцитонина пептида, имеющего структуру в соответствии с формулой I, центральная структура содержит фрагмент кальцитонина человека или лососевых рыб. Согласно некоторым вариантам реализации изобретения указанный фрагмент кальцитонина человека или лососевых рыб содержит от 18 до 21 аминокислот. Согласно некоторым вариантам реализации изобретения указанный фрагмент кальцитонина человека или лососевых рыб содержит от 18 до 20 аминокислот. Согласно некоторым вариантам реализации изобретения указанного антагониста на основе модифицированного связанного с геном кальцитонина пептида, имеющего структуру в соответствии с формулой I, Y1 содержит от 19 до 20 аминокислот. Согласно некоторым вариантам реализации указанного антагониста на основе модифицированного связанного с геном кальцитонина пептида, имеющего структуру в соответствии с формулой I, Y1 представляет собой последовательность -Val-Leu-Gly-Arg-Leu-Ser-Gln-Glu-Leu-His-Arg-Leu-Gln-Thr-Tyr-Pro-Arg-Thr-Asn- (SEQ ID №: 34) или -Val-Leu-Gly-Lys-Leu-Ser-Gln-Glu-Leu-His-Lys-Leu-Gln-Thr-Tyr-Pro-Arg-Thr-Asn- (SEQ ID №: 35). Согласно некоторым вариантам реализации указанного антагониста на основе модифицированного связанного с геном кальцитонина пептида, имеющего структуру в соответствии с формулой I, Y1 на 95% идентичен последовательности -Val-Leu-Gly-Arg-Leu-Ser-Gln-Glu-Leu-His-Arg-Leu-Gln-Thr-Tyr-Pro-Arg-Thr-Asn- (SEQ ID №: 34) или -Val-Leu-Gly-Lys-Leu-Ser-Gln-Glu-Leu-His-Lys-Leu-Gln-Thr-Tyr-Pro-Arg-Thr-Asn- (SEQ ID №: 35).
[0033] Согласно некоторым вариантам реализации указанного антагониста на основе модифицированного связанного с геном кальцитонина пептида, имеющего структуру в соответствии с формулой I, центральная структура содержит фрагмент кальцитонина любого из целого ряда видов. Согласно некоторым вариантам реализации изобретения Y1 может быть на 60%, 65%, 70%, 75%, 80%, 85%, 90% или 95% идентичен последовательности Y1 в последовательности SEQ ID №: 34 (Val-Leu-Gly-Arg-Leu-Ser-Gln-Glu-Leu-His-Arg-Leu-Gln-Thr-Tyr-Pro-Arg-Thr-Asn-). Согласно некоторым вариантам реализации указанного антагониста на основе связанного с геном кальцитонина пептида, имеющего структуру в соответствии с формулой I, Y1 может представлять собой последовательность -Val-Leu-Gly-Lys-Leu-Ser-Gln-Glu-Leu-His-Lys-Leu-Gln-Thr-Tyr-Pro-Arg-Thr-Asn- (SEQ ID №: 35) или -Val-Leu-Gly-Lys-Leu-Ser-Gln-Glu-Leu-His-Lys-Leu-Gln-Thr-Tyr-Pro-Arg-Thr-Asp- (SEQ ID №: 36), или - Val-Leu-Gly-Lys-Leu-Ser-Gln-Glu-Leu-His-Lys-Leu-Gln-Thr-Phe-Pro-Arg-Thr-Asn- (SEQ ID №: 37), или -Val-Leu-Gly-Lys-Leu-Ser-Gln-Asp-Ile-His-Lys-Leu-Gln-Thr-Tyr-Pro-Arg-Thr-Asn- (SEQ ID №: 38), или -Val-Leu-Gly-Lys-Leu-Ser-Gln-Glu-Leu-His-Lys-Met-Gln-Thr-Tyr-Pro-Arg-Thr-Asp- (SEQ ID №: 39), или - Leu-Leu-Gly-Lys-Leu-Ser-Gln-Glu-Leu-His-Arg-Leu-Gln-Thr-Tyr-Thr-Arg-Thr-Asp- (SEQ ID №: 40), или - Val-Leu-Gly-Lys-Leu-Ser-Gln-Asp-Leu-His-Lys-Leu-Gln-Thr-Phe-Pro-Arg-Thr-Asp- (SEQ ID №: 41), или -Met-Leu-Gly-Lys-Leu-Ser-Gln-Asp-Leu-His-Lys-Leu-Gln-Thr-Phe-Pro-Arg-Thr-Asp- (SEQ ID №: 42), или - Val-Leu-Gly-Lys-Leu-Ser-Gln-Asp-Ile-His-Lys-Leu-Gln-Thr-His-Pro-Arg-Thr-Asp- (SEQ ID №: 43). Согласно некоторым вариантам реализации изобретения Y1 может быть на 60% или более процентов идентичен любой последовательности Y1 из только что упомянутой последовательности.
[0034] Согласно некоторым вариантам реализации изобретения предложены полипептиды Y1, которые по меньшей мере приблизительно на 60%, в качестве альтернативы по меньшей мере приблизительно на 61%, в качестве альтернативы по меньшей мере приблизительно на 62%, в качестве альтернативы по меньшей мере приблизительно на 63%, в качестве альтернативы по меньшей мере приблизительно на 64%, в качестве альтернативы по меньшей мере приблизительно на 65%, качестве альтернативы по меньшей мере приблизительно на 66%, в качестве альтернативы по меньшей мере приблизительно на 67%, в качестве альтернативы по меньшей мере приблизительно на 68%, качестве альтернативы по меньшей мере приблизительно на 69%, в качестве альтернативы по меньшей мере приблизительно на 70%, в качестве альтернативы по меньшей мере приблизительно на 71%, в качестве альтернативы по меньшей мере приблизительно на 72%, в качестве альтернативы по меньшей мере приблизительно на 73%, в качестве альтернативы по меньшей мере приблизительно на 74%, в качестве альтернативы по меньшей мере приблизительно на 75%, качестве альтернативы по меньшей мере приблизительно на 76%, в качестве альтернативы по меньшей мере приблизительно на 77%, в качестве альтернативы по меньшей мере приблизительно на 78%, качестве альтернативы по меньшей мере приблизительно на 79%, в качестве альтернативы по меньшей мере приблизительно на 80%, в качестве альтернативы по меньшей мере приблизительно на 81%, в качестве альтернативы по меньшей мере приблизительно на 82%, в качестве альтернативы по меньшей мере приблизительно на 83%, в качестве альтернативы по меньшей мере приблизительно на 84%, в качестве альтернативы по меньшей мере приблизительно на 85%, качестве альтернативы по меньшей мере приблизительно на 86%, в качестве альтернативы по меньшей мере приблизительно на 87%, в качестве альтернативы по меньшей мере приблизительно на 88%, качестве альтернативы по меньшей мере приблизительно на 89%, в качестве альтернативы по меньшей мере приблизительно на 90%, в качестве альтернативы по меньшей мере приблизительно на 91%, в качестве альтернативы по меньшей мере приблизительно на 92%, в качестве альтернативы по меньшей мере приблизительно на 93%, в качестве альтернативы по меньшей мере приблизительно на 94%, в качестве альтернативы по меньшей мере приблизительно на 95%, качестве альтернативы по меньшей мере приблизительно на 96%, в качестве альтернативы по меньшей мере приблизительно на 97%, в качестве альтернативы по меньшей мере приблизительно на 98%, качестве альтернативы по меньшей мере приблизительно на 99% идентичны последовательности фрагмента полипептида Y1 из упомянутой выше последовательности.
[0035] Согласно некоторым вариантам реализации указанного антагониста на основе модифицированного связанного с геном кальцитонина пептида, имеющего структуру в соответствии с формулой I, Z1 содержит последовательность Z11-Z12-Z13-Z14-Z15-Z16 (SEQ ID №: 45), где:
Z11 выбран из группы, состоящей из Ala, Gly, Ile, Leu, Met, Phe, Pro, Trp и Val;
Z12 выбран из группы, состоящей из Ala, Gly, Ile, Leu, Met, Phe, Pro, Trp, и Val;
Z13 выбран из группы, состоящей из серина (Ser), и тирозина (Tyr);
Z14 выбран из группы, состоящей из Arg, Asn, Asp, Glu, Gln, His, Lys, Ser, Thr и Tyr;
Z15 выбран из группы, состоящей из Ala, Gly, Ile, Leu, Met, Phe, Pro, Trp и Val;
и
Z16 выбран из группы, состоящей из Ala, Gly, Ile, Leu, Met, Phe, Pro, Trp, и Val. Согласно некоторым вариантам реализации изобретения Z11 является Val. Согласно некоторым вариантам реализации изобретения Z12 является Gly. Согласно некоторым вариантам реализации изобретения Z13 является Ser. Согласно некоторым вариантам реализации изобретения Z14 является Lys. Согласно некоторым вариантам реализации изобретения Z15 является Ala. Согласно некоторым вариантам реализации изобретения Z16 является Phe. Согласно некоторым вариантам реализации изобретения Z11-Z12-Z13-Z14-Z15-Z16 является последовательностью -Val-Gly-Ser-Lys-Ala-Phe, такой, что С-конец указанного полипептида является карбоксильной группой (SEQ ID №: 46) или последовательностью -Val-Gly-Ser-Lys-Ala-Phe-NH2, такой, что С-конец указанного полипептида является карбоксамидной группой (SEQ ID №: 47).
[0036] Согласно некоторым вариантам реализации изобретения С-концевой остаток Z1 является фенилаланином, тирозином, пролином или гидроксипролином.
Согласно некоторым вариантам реализации изобретения С-концевой остаток Z1 является фенилаланином.
[0037] Согласно некоторым вариантам реализации изобретения Z1 содержит по меньшей мере один остаток Phe.
[0038] Согласно некоторым вариантам реализации изобретения С-конец Z1 модифицирован таким образом, что он связан амидированной карбоксильной группой (-C(=O)NH2).
[0039] Согласно некоторым вариантам реализации указанного антагониста на основе модифицированного связанного с геном кальцитонина пептида, имеющего структуру в соответствии с формулой I, X1 выбран из группы, состоящей из NH2-Ala-Cys-Asp-Thr-Ala-Ala-Cys- (SEQ ID №: 17), NH2-Ala-Cys-Asp-Thr-Ala-Ser-Cys- (SEQ ID №: 18), NH2-Ala-Cys-Asp-Thr-Ala-Val-Cys- (SEQ ID №: 19), NH2-Ala-Cys-Asn-Thr-Ala-Ala-Cys-(SEQ ID №: 20), NH2-Ala-Cys-Val-Leu-Gly-Ala-Cys-, NH2-Ala-Cys-Arg-Phe-Gly-Ala-Cys- (SEQ ID №: 21), NH2-Ala-Cys-Arg-Phe-Gly-Ala-Cys- (SEQ ID №: 22), NH2-Ala-Cys-Asp-Leu-Ser-Ala-Cys- (SEQ ID №: 23), NH2-Ala-Cys-Asn-Leu-Ser-Ala-Cys- (SEQ ID №: 24), Cys-Ser-Asn-Thr-Ala-Ala-Cys- (SEQ ID №: 25), NH2-Ala-Cys-Asp-Thr-Ala-Leu-Cys- (SEQ ID №: 26), NH2-Ala-Cys-Asp-Thr-Ala-Ile-Cys- (SEQ ID №: 27), NH2-Ala-Cys-Asp-Thr-Ala-Leu-Cys- (SEQ ID №: 28), NH2-Ala-Cys-Asp-Thr-Ala-Ile-Cys- (SEQ ID №: 29), NH2-Ala-Cys-Asp-Leu-Ser-Val-Cys- (SEQ ID №: 30), NH2-Ala-Cys-Asp-Leu-Ser-Val-Cys- (SEQ ID №: 31), NH2-Ala-Cys-Asn-Leu-Ser-Val-Cys (SEQ ID №: 32) и NH2-Cys-Ser-Asn-Thr-Ala-Val-Cys- (SEQ ID №: 33); Y1 может быть последовательностью -Val-Leu-Gly-Arg-Leu-Ser-Gln-Glu-Leu-His-Arg-Leu-Gln-Thr-Tyr-Pro-Arg-Thr-Asn- (SEQ ID №: 34) или -Val-Leu-Gly-Lys-Leu-Ser-Gln-Glu-Leu-His-Lys-Leu-Gln-Thr-Tyr-Pro-Arg-Thr-Asn- (SEQ ID №: 35); и Z1 может представлять собой последовательность -Val-Gly-Ser-Lys-Ala-Phe, содержащей карбоксильный конец (SEQ ID №: 46) или -Val-Gly-Ser-Lys-Ala-Phe-NH2 (SEQ ID №: 47).
[0040] Согласно некоторым вариантам реализации указанного антагониста на основе модифицированного связанного с геном кальцитонина пептида, имеющего структуру в соответствии с формулой I, указанный антагонист содержит от 28 до 35 остатков аминокислот, от 31 до 37 остатков аминокислот, от 31 до 33 остатков аминокислот или 32 остатка аминокислот.
[0041] Согласно некоторым вариантам реализации указанного антагониста на основе модифицированного связанного с геном кальцитонина пептида, имеющего структуру в соответствии с формулой I, указанный антагонист содержит мотив -Ala-Cys-Asp-Thr-Ala-X16-Cys- (SEQ ID №: 49), в котором X16 представляет собой остаток любой аминокислоты, кроме Thr.
[0042] Согласно некоторым вариантам реализации указанного антагониста на основе модифицированного связанного с геном кальцитонина пептида, имеющего структуру в соответствии с формулой I, указанный антагонист содержит первый пептидный фрагмент, содержащий семь или менее остатков аминокислот, причем указанный первый пептидный фрагмент содержит последовательность из модифицированного связанного с геном кальцитонина пептида. Согласно некоторым вариантам реализации указанного антагониста на основе модифицированного связанного с геном кальцитонина пептида, имеющего структуру в соответствии с формулой I, указанный антагонист содержит второй пептидный фрагмент, содержащий семь или менее остатков аминокислот, причем указанные первый и второй пептидные фрагменты не являются смежными, и каждый из них независимо содержит последовательность, которая может быть модифицирована из связанного с геном кальцитонина пептида. Согласно некоторым вариантам реализации указанного антагониста на основе модифицированного связанного с геном кальцитонина пептида, имеющего структуру в соответствии с формулой I, указанный антагонист содержит третий пептидный фрагмент, содержащий 20 или менее остатков аминокислот, причем указанный третий пептидный фрагмент содержит последовательность из кальцитонина лососевых рыб. Согласно некоторым вариантам реализации указанного антагониста на основе модифицированного связанного с геном кальцитонина пептида, имеющего структуру в соответствии с формулой I, второй и третий пептидные фрагменты являются смежными.
[0043] Согласно некоторым вариантам реализации изобретения указанный антагонист имеет структуру, выбираемую из перечня структур, включающего NH2-Ala-Cys-Asp-Thr-Ala-Ala-Cys-Val-Leu-Gly-Arg-Leu-Ser-Gln-Glu-Leu-His-Arg-Leu-Gln-Thr-Tyr-Pro-Arg-Thr-Asn-Val-Gly-Ser-Lys-Ala-Phe-NH2 (SEQ ID №: 1), NH2-Ala-Cys-Asp-Thr-Ala-Ser-Cys-Val-Leu-Gly-Arg-Leu-Ser-Gln-Glu-Leu-His-Arg-Leu-Gln-Thr-Tyr-Pro-Arg-Thr-Asn-Val-Gly-Ser-Lys-Ala-Phe-NH2 (SEQ ID №: 2), NH2-Ala-Cys-Asp-Thr-Ala-Val-Cys-Val-Leu-Gly-Arg-Leu-Ser-Gln-Glu-Leu-His-Arg-Leu-Gln-Thr-Tyr-Pro-Arg-Thr-Asn-Val-Gly-Ser-Lys-Ala-Phe-NH2 (SEQ ID №: 3), NH2-Ala-Cys-Asn-Thr-Ala-Ala-Cys-Val-Leu-Gly-Arg-Leu-Ser-Gln-Glu-Leu-His-Arg-Leu-Gln-Thr-Tyr-Pro-Arg-Thr-Asn-Val-Gly-Ser-Lys-Ala-Phe-NH2 (SEQ ID №: 4), NH2-Ala-Cys-Val-Leu-Gly-Ala-Cys-Val-Leu-Gly-Arg-Leu-Ser-Gln-Glu-Leu-His-Arg-Leu-Gln-Thr-Tyr-Pro-Arg-Thr-Asn-Val-Gly-Ser-Lys-Ala-Phe-NH2 (SEQ ID №: 5), NH2-Ala-Cys-Arg-Phe-Gly-Ala-Cys-Val-Leu-Gly-Arg-Leu-Ser-Gln-Glu-Leu-His-Arg-Leu-Gln-Thr-Tyr-Pro-Arg-Thr-Asn-Val-Gly-Ser-Lys-Ala-Phe-NH2 (SEQ ID №: 6), NH2-Ala-Cys-Asn-Leu-Ser-Ala-Cys-Val-Leu-Gly-Arg-Leu-Ser-Gln-Glu-Leu-His-Arg-Leu-Gln-Thr-Tyr-Pro-Arg-Thr-Asn-Val-Gly-Ser-Lys-Ala-Phe-NH2 (SEQ ID №: 7), NH2-Cys-Ser-Asn-Thr-Ala-Ala-Cys-Val-Leu-Gly-Arg-Leu-Ser-Gln-Glu-Leu-His-Arg-Leu-Gln-Thr-Tyr-Pro-Arg-Thr-Asn-Val-Gly-Ser-Lys-Ala-Phe-NH2 (SEQ ID №: 8), NH2-Ala-Cys-Asp-Thr-Ala-Leu-Cys-Val-Leu-Gly-Arg-Leu-Ser-Gln-Glu-Leu-His-Arg-Leu-Gln-Thr-Tyr-Pro-Arg-Thr-Asn-Val-Gly-Ser-Lys-Ala-Phe-NH2 (SEQ ID №: 9), NH2-Ala-Cys-Asp-Thr-Ala-Ile-Cys-Val-Leu-Gly-Arg-Leu-Ser-Gln-Glu-Leu-His-Arg-Leu-Gln-Thr-Tyr-Pro-Arg-Thr-Asn-Val-Gly-Ser-Lys-Ala-Phe-NH2 (SEQ ID №: 10), NH2-Ala-Cys-Asn-Leu-Ser-Val-Cys-Val-Leu-Gly-Arg-Leu-Ser-Gln-Glu-Leu-His-Arg-Leu-Gln-Thr-Tyr-Pro-Arg-Thr-Asn-Val-Gly-Ser-Lys-Ala-Phe-NH2 (SEQ ID №: 11), NH2-Cys-Ser-Asn-Thr-Ala-Val-Cys-Val-Leu-Gly-Arg-Leu-Ser-Gln-Glu-Leu-His-Arg-Leu-Gln-Thr-Tyr-Pro-Arg-Thr-Asn-Val-Gly-Ser-Lys-Ala-Phe-NH2 (SEQ ID №: 12) или NH2-Ala-Cys-Asn-Leu-Ser-Ala-Cys-Val-Leu-Gly-Arg-Leu-Ser-Gln-Glu-Leu-His-Arg-Leu-Gln-Thr-Tyr-Pro-Thr-Asn-Thr-Gly-Ser-Gly-Thr-Pro-NH2 (SEQ ID №: 13), Ala-Cys-Val-Leu-Gly-Ala-Cys-Val-Leu-Gly-Arg-Leu-Ser-Gln-Glu-Leu-His-Arg-Leu-Gln-Thr-Tyr-Pro-Val-Asp-Pro-Ser-Ser-Pro-His-Ser-Tyr-NH2 (SEQ ID №: 14) или Ala-Cys-Asp-Thr-Ala-Ala-Cys-Val-Thr-His-Arg-Leu-Ala-Gly-Leu-Leu-Ser-Arg-Ser-Gly-Gly-Val-Val-Lys-Asn-Asn-Phe-Val-Pro-Thr-Asn-Val-Gly-Ser-Lys-Ala-Phe-NH2 (SEQ ID №: 15) или является их фармацевтически приемлемой солью. Подходящий антагонист может представлять собой отдельное соединение из приведенного выше списка.
[0044] Согласно некоторым вариантам реализации изобретения указанный антагонист имеет структуру NH2-Ala-Cys-Asp-Thr-Ala-Ala-Cys-Val-Leu-Gly-Arg-Leu-Ser-Gln-Glu-Leu-His-Arg-Leu-Gln-Thr-Tyr-Pro-Arg-Thr-Asn-Val-Gly-Ser-Lys-Ala-Phe-NH2 (SEQ ID №: 1) или является ее фармацевтически приемлемой солью. Согласно некоторым вариантам реализации изобретения указанный антагонист имеет структуру NH2-Ala-Cys-Asp-Thr-Ala-Ser-Cys-Val-Leu-Gly-Arg-Leu-Ser-Gln-Glu-Leu-His-Arg-Leu-Gln-Thr-Tyr-Pro-Arg-Thr-Asn-Val-Gly-Ser-Lys-Ala-Phe-NH2 (SEQ ID №: 2) или является ее фармацевтически приемлемой солью. Согласно некоторым вариантам реализации изобретения указанный антагонист имеет структуру NH2-Ala-Cys-Asp-Thr-Ala-Val-Cys-Val-Leu-Gly-Arg-Leu-Ser-Gln-Glu-Leu-His-Arg-Leu-Gln-Thr-Tyr-Pro-Arg-Thr-Asn-Val-Gly-Ser-Lys-Ala-Phe-NH2 (SEQ ID №: 3) или является ее фармацевтически приемлемой солью. Согласно некоторым вариантам реализации изобретения указанный антагонист имеет структуру NH2-Ala-Cys-Asn-Thr-Ala-Ala-Cys-Val-Leu-Gly-Arg-Leu-Ser-Gln-Glu-Leu-His-Arg-Leu-Gln-Thr-Tyr-Pro-Arg-Thr-Asn-Val-Gly-Ser-Lys-Ala-Phe-NH2 (SEQ ID №: 4) или является ее фармацевтически приемлемой солью. Согласно некоторым вариантам реализации изобретения указанный антагонист имеет структуру NH2-Ala-Cys-Val-Leu-Gly-Ala-Cys-Val-Leu-Gly-Arg-Leu-Ser-Gln-Glu-Leu-His-Arg-Leu-Gln-Thr-Tyr-Pro-Arg-Thr-Asn-Val-Gly-Ser-Lys-Ala-Phe-NH2 (SEQ ID №: 5) или является ее фармацевтически приемлемой солью. Согласно некоторым вариантам реализации изобретения указанный антагонист имеет структуру NH2-Ala-Cys-Arg-Phe-Gly-Ala-Cys-Val-Leu-Gly-Arg-Leu-Ser-Gln-Glu-Leu-His-Arg-Leu-Gln-Thr-Tyr-Pro-Arg-Thr-Asn-Val-Gly-Ser-Lys-Ala-Phe-NH2 (SEQ ID №: 6) или является ее фармацевтически приемлемой солью. Согласно некоторым вариантам реализации изобретения указанный антагонист имеет структуру NH2-Ala-Cys-Asn-Leu-Ser-Ala-Cys-Val-Leu-Gly-Arg-Leu-Ser-Gln-Glu-Leu-His-Arg-Leu-Gln-Thr-Tyr-Pro-Arg-Thr-Asn-Val-Gly-Ser-Lys-Ala-Phe-NH2 (SEQ ID №: 7) или является ее фармацевтически приемлемой солью. Согласно некоторым вариантам реализации изобретения указанный антагонист имеет структуру NH2-Cys-Ser-Asn-Thr-Ala-Ala-Cys-Val-Leu-Gly-Arg-Leu-Ser-Gln-Glu-Leu-His-Arg-Leu-Gln-Thr-Tyr-Pro-Arg-Thr-Asn-Val-Gly-Ser-Lys-Ala-Phe-NH2 (SEQ ID №: 8) или является ее фармацевтически приемлемой солью. Согласно некоторым вариантам реализации изобретения указанный антагонист имеет структуру NH2-Ala-Cys-Asp-Thr-Ala-Leu-Cys-Val-Leu-Gly-Arg-Leu-Ser-Gln-Glu-Leu-His-Arg-Leu-Gln-Thr-Tyr-Pro-Arg-Thr-Asn-Val-Gly-Ser-Lys-Ala-Phe-NH2 (SEQ ID №: 9) или является ее фармацевтически приемлемой солью. Согласно некоторым вариантам реализации изобретения указанный антагонист имеет структуру NH2-Ala-Cys-Asp-Thr-Ala-Ile-Cys-Val-Leu-Gly-Arg-Leu-Ser-Gln-Glu-Leu-His-Arg-Leu-Gln-Thr-Tyr-Pro-Arg-Thr-Asn-Val-Gly-Ser-Lys-Ala-Phe-NH2 (SEQ ID №: 10) или является ее фармацевтически приемлемой солью. Согласно некоторым вариантам реализации изобретения указанный антагонист имеет структуру NH2-Ala-Cys-Asn-Leu-Ser-Val-Cys-Val-Leu-Gly-Arg-Leu-Ser-Gln-Glu-Leu-His-Arg-Leu-Gln-Thr-Tyr-Pro-Arg-Thr-Asn-Val-Gly-Ser-Lys-Ala-Phe-NH2 (SEQ ID №: 11) или является ее фармацевтически приемлемой солью. Согласно некоторым вариантам реализации изобретения указанный антагонист имеет структуру NH2-Cys-Ser-Asn-Thr-Ala-Val-Cys-Val-Leu-Gly-Arg-Leu-Ser-Gln-Glu-Leu-His-Arg-Leu-Gln-Thr-Tyr-Pro-Arg-Thr-Asn-Val-Gly-Ser-Lys-Ala-Phe-NH2 (SEQ ID №: 12) или является ее фармацевтически приемлемой солью. Согласно некоторым вариантам реализации изобретения указанный антагонист имеет структуру или NH2-Ala-Cys-Asn-Leu-Ser-Ala-Cys-Val-Leu-Gly-Arg-Leu-Ser-Gln-Glu-Leu-His-Arg-Leu-Gln-Thr-Tyr-Pro-Thr-Asn-Thr-Gly-Ser-Gly-Thr-Pro-NH2 (SEQ ID №: 13) или является ее фармацевтически приемлемой солью. Согласно некоторым вариантам реализации изобретения указанный антагонист имеет структуру Ala-Cys-Val-Leu-Gly-Ala-Cys-Val-Leu-Gly-Arg-Leu-Ser-Gln-Glu-Leu-His-Arg-Leu-Gln-Thr-Tyr-Pro-Val-Asp-Pro-Ser-Ser-Pro-His-Ser-Tyr-NH2 (SEQ ID №: 14) или является ее фармацевтически приемлемой солью. Согласно некоторым вариантам реализации изобретения указанный антагонист имеет структуру или Ala-Cys-Asp-Thr-Ala-Ala-Cys-Val-Thr-His-Arg-Leu-Ala-Gly-Leu-Leu-Ser-Arg-Ser-Gly-Gly-Val-Val-Lys-Asn-Asn-Phe-Val-Pro-Thr-Asn-Val-Gly-Ser-Lys-Ala-Phe-NH2 (SEQ ID №: 15) или является ее фармацевтически приемлемой солью. Антагонист согласно настоящему раскрытию также может представлять собой фармацевтическую композицию, содержащую одно из перечисленных выше соединений. Указанную фармацевтическую композицию можно применять при реализации способа лечения головной боли у субъекта, причем указанный способ включает введение субъекту эффективного количества антагониста на основе модифицированного связанного с геном кальцитонина пептида.
[0045] Согласно некоторым вариантам реализации указанного антагониста на основе модифицированного связанного с геном кальцитонина пептида, имеющего структуру в соответствии с формулой I, Y1 включает -Ala-Glu-Ala-Ala-Ala-Lys-Glu-Ala-Ala-Ala-Lys-Glu-Ala-Ala-Ala-Lys-Ala- (SEQ ID №: 50), -Ala-Lys-Ala-Ala-Ala-Glu-Lys-Ala-Ala-Ala-Glu-Lys-Ala-Ala-Ala-Glu-Ala- (SEQ ID №: 51), -Ala-Glu-Ala-Ala-Lys-Ala-Glu-Ala-Ala-Lys-Ala-Glu-Ala-Ala-Lys-Ala- (SEQ ID №: 52) или -Ala-Lys-Ala-Ala-Glu-Ala-Lys-Ala-Ala-Glu-Ala-Lys-Ala-Ala-Glu-Ala- (SEQ ID №: 53).
[0046] Согласно некоторым вариантам реализации изобретения предложен антагонист на основе модифицированного связанного с геном кальцитонина пептида, имеющий структуру пептида с последовательностью согласно Таблице 1.
[0047] Согласно некоторым вариантам реализации изобретения предложен антагонист на основе модифицированного связанного с геном кальцитонина пептида, содержащий последовательность аминокислот, которая по меньшей мере на 80% идентична последовательности аминокислот SEQ ID №: 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14 или 15, причем указанный пептид сохраняет антагонистическую активность. Согласно некоторым вариантам реализации изобретения указанная последовательность аминокислот по меньшей мере на 90% идентична последовательности аминокислот SEQ ID №: 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14 или 15, причем указанный пептид сохраняет антагонистическую активность. Согласно некоторым вариантам реализации изобретения указанная последовательность аминокислот по меньшей мере на 95% идентична последовательности аминокислот SEQ ID №: 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14 или 15, причем указанный пептид сохраняет антагонистическую активность. Согласно некоторым вариантам реализации изобретения указанная последовательность аминокислот по меньшей мере на 97%о идентична последовательности аминокислот SEQ ID №: 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14 или 15, причем указанный пептид сохраняет антагонистическую активность.
[0048] Согласно некоторым вариантам реализации изобретения предложена фармацевтическая композиция, содержащая фармацевтически приемлемое вспомогательное вещество и актуальный антагонист на основе модифицированного связанного с геном кальцитонина пептида, как раскрыто и описано в настоящей заявке.
[0049] Согласно некоторым вариантам реализации изобретения предложен способ лечения головной боли у субъекта, способ, включающий введение указанному субъекту эффективного количества актуального антагониста на основе модифицированного связанного с геном кальцитонина пептида, как раскрыто и описано в настоящей заявке. Согласно некоторым вариантам реализации изобретения указанный способ может также включать идентификацию субъекта, страдающего головной болью. Согласно некоторым вариантам реализации изобретения головная боль представляет собой мигрень.
[0050] Согласно некоторым вариантам реализации изобретения предложен способ лечения состояния, связанного с аномальным уровнем CGRP, включающий введение актуального антагониста на основе модифицированного связанного с геном кальцитонина пептида, как раскрыто и описано в настоящей заявке, субъекту, способ, включающий введение указанному субъекту эффективного количества антагониста на основе модифицированного связанного с геном кальцитонина пептида, как раскрыто и описано в настоящей заявке. Согласно некоторым вариантам реализации изобретения указанное состояние представляет собоймигрень.
[0051] Согласно некоторым вариантам реализации изобретения предложен конъюгат, который содержит терапевтический агент, связанный с актуальным антагонистом на основе модифицированного связанного с геном кальцитонина пептида, как раскрыто и описано в настоящей заявке, который селективно связывается с членом семейства рецепторов CGRP. Согласно некоторым вариантам реализации изобретения терапевтический агент может представлять собой визуализирующий агент.
[0052] Согласно некоторым вариантам реализации изобретения предложен способ идентификации связывающегося с рецептором CGRP лиганда путем получения актуального антагониста на основе модифицированного связанного с геном кальцитонина пептида, связанного с рецептором CGRP, получения тестируемого соединения или библиотеки тестируемых соединений, и идентификации соединений, которые способны диссоциировать антагонист на основе модифицированного связанного с геном кальцитонина пептида от рецептора CGRP. Такие соединения, идентифицированные при помощи указанного способа, можно в дальнейшем подвергнуть скринингу относительно других рецепторов CGRP и CGRP-рецептор-связывающими агентами, чтобы выявить селективные лиганды, связывающиеся с рецептором CGRP.
[0053] Согласно некоторым вариантам реализации изобретения в настоящей заявке описан модифицированный антагонист CGRP, который сохраняет последовательность агониста, которая содержит X1 - N-концевую область, которая связывается с рецептором CGRP на мембране клетки, и на С-конце, инициирующем и стабилизирующим спираль через дисульфидную связь - Y1, структурный мотив спирали; и Z1 - С-концевую связывающую область, но которая отличается всего на один остаток от последовательности агониста. Согласно предпочтительным вариантам реализации изобретения частью структуры, применяемой для повышения эффективности настоящего антагониста, является спираль из кальцитонина лососевых рыб.
Определения
[0054] Следующие определения представлены, чтобы проиллюстрировать и установить значения и область разных терминов, применяемых для описания вариантов реализации изобретения.
[0055] В настоящей заявке термин «модифицированный» относится к полипептиду, который сохраняет общую структуру родственного полипептида, но который отличается от указанного родственного пептида по меньшей мере одним остатком. В настоящей заявке термин «модифицированный С-конец» относится к С-концу полипептида, который имеет химическую структуру, отличную от стандартной карбоксильной группы пептида, и примером такого модифицированного С-конца является С-концевой карбоксамид.
[0056] В настоящей заявке термин «агонист» относится к биологически активному лиганду, который связывается с комплементарным ему биологически активным рецептором и активирует последний, либо вызывая биологическую реакцию в рецепторе, либо усиливая ранее существующую биологическую активность рецептора.
[0057] В настоящей заявке термин «антагонист» относится к биологически активному лиганду, который связывается с комплементарным ему биологически активным рецептором и подавляет физиологическую реакцию рецептора.
[0058] В настоящей заявке термин «фармацевтически приемлемая соль» относится к нетоксичным солям щелочных металлов, щелочноземельных металлов и солям аммония, обычно применяемым в фармацевтической промышленности, включая соли натрия, калия, лития, кальция, магния, бария, аммония и протамина цинка, которые получают при помощи способов, хорошо известных в области техники. Данный термин также включает соли присоединения нетоксичных кислот, которые, как правило, получают посредством реакции антагонистов на основе модифицированного связанного с геном кальцитонина пептида, раскрываемых в настоящей заявке, с подходящими органическими или неорганическими кислотами. Репрезентативные соли включают гидрохлорид, гидробромид, сульфат, бисульфат, оксалат, валериат, олеат, лаурат, борат, бензоат, лактат, фосфат, тосилат, цитрат, малеат, фумарат, сукцинат, тартрат, напсилат и подобные. Таким образом, данный термин относится к солям, которые сохраняют биологическую эффективность и свойства свободных оснований и которые не являются биологически или другим образом нежелательными, образуемым с неорганическими кислотами, такими как соляная кислота, бромистоводородная кислота, серная кислота, фосфорная кислота и подобные, и органическими кислотами, такими как уксусная кислота, пропионовая кислота, гликолевая кислота, пировиноградная кислота, щавелевая кислота, яблочная кислота, малоновая кислота, янтарная кислота, малеиновая кислота, фумаровая кислота, винная кислота, лимонная кислота, бензойная кислота, коричная кислота, миндальная кислота, метансульфоновая кислота, этансульфоновая кислота, р-толуолсульфоновая кислота, салициловая кислота и подобные. Описание фармацевтически приемлемых солей в качестве пролекарств можно найти в Bundgaard, Н. ed., 1985 Design of Prodrugs, Elsevier Science Publishers, Amsterdam.
[0059] В настоящей заявке термин «фармацевтически приемлемый эфир» относится к эфиру, который сохраняет при гидролизе эфирной связи биологическую эффективность и свойства карбоновой кислоты или спирта, и не является нежелательным с биологической или иной точки зрения. Описание фармацевтически приемлемых эфиров в качестве пролекарств можно найти в Bundgaard, Н. ed., 1985 Design of Prodrugs, Elsevier Science Publishers, Amsterdam. Указанные эфиры, как правило, образованы соответствующей карбоновой кислотой и спиртом. Обычно образования эфира можно достичь посредством традиционных способов синтеза. Например, см. March, 1992 Advanced Organic Chemistry, 4th Ed., John Wiley & Sons, New York, p.p. 393-396 и ссылки, цитируемые в настоящей заявке, и Mark, et al. 1980 Encyclopedia of Chemical Technology, John Wiley & Sons, New York. Спиртовой компонент эфира обычно содержит (i) алифатический спирт С2-С12, который может содержать или не содержать одну или более двойных связей, и может содержать или не содержать разветвленные углеродные скелеты или (ii) ароматические или гетероароматические спирты С7-С12.
[0060] В настоящей заявке термин «С-концевой амид» относится к амидной группе, которая замещает С-концевую гидроксильную группу, обычно присутствующую на карбоксильном конце полипептида, таким образом, что полипептид
заканчиваетсякарбоксамидом (например, C(=O)-NH2, а не С-концевая карбоксильная группа (т.е. С(=O)-ОН)). Описание фармацевтически приемлемых амидов в качестве пролекарств можно найти в Bundgaard, Н. ed., 1985 Design of Prodrugs, Elsevier Science Publishers, Amsterdam. Указанные амиды, как правило, образуются из соответствующей карбоновой кислоты и амина. Обычно образования амида можно достичь посредством традиционных способов синтеза. Например, см. March, 1992 Advanced Organic Chemistry, 4th Ed., John Wiley & Sons, New York, p. 393 и Mark, et al. 1980 Encyclopedia of Chemical Technology, John Wiley & Sons, New York.
[0061] В настоящей заявке термин «фармацевтически приемлемая основа» относится к веществу-основе, которое не препятствует эффективности биологической активности активных ингредиентов и которое не токсично для хозяина или пациента.
[0062] В настоящей заявке термин «стереоизомер» относится к структуре, обладающей такой же молекулярной массой, химическим составом и связанной последовательностью, как другая структура, но ее атомы сгруппированы в пространстве вокруг одного или более хиральных центров по-другому. То есть стереоизомеры с одной химической формулой будут содержать идентичные химические группы, расположенные в разной пространственной ориентации относительно по меньшей мере одного хирального центра. В чистом виде стереоизомеры обладают способностью к вращению плоскополяризованного света. Однако некоторые чистые стереоизомеры могут обладать таким низким оптическим вращением, которое нельзя определить современными инструментами. Антагонисты на основе модифицированного связанного с геном кальцитонина пептида, описываемые в настоящей заявке, могут содержать один или более ассиметричных атомов углерода и, следовательно, включать разные стереоизомеры. Все стереоизомеры включены в область настоящих вариантов реализации изобретения.
[0063] В настоящей заявке термин «терапевтически» или «фармацевтически эффективное количество» применительно к композициям, раскрываемым в настоящей заявке, относится к количеству композиции, достаточному для того, чтобы вызвать желаемый биологический результат. Указанным результатом может быть ослабление признаков, симптомов или причин заболевания или любое другое изменение биологической системы.
[0064] В настоящей заявке подразумевается, что термины «остаток пептида» или «пептидная структура» включают в себя пептиды, содержащие существующие в природе L-аминокислоты и соответствующие D-аминокислоты, а также производные пептидов, аналоги пептидов и пептидомиметики структур, содержащих природные L-аминокислоты. Способы конструирования аналогов, производных и миметиков пептидов известны в области техники. Например, см. Farmer, P.S. in: Drug Design E.J. Ariens, ed. Academic Press, New York, 1980, vol. 10, pp. 119-143; Ball J.B. & Alewood, P.F. 1990 J. Mol. Recognition 3:55; Morgan, B.A. & Gainor, J.A. 1989 Ann. Rep. Med. Chem. 24:243; и Freidinger, R.M. 1989 Trends Pharmacol. Sci. 10:270; Luthman, et al. 1996 A Textbook of Drug Design and Development, 14:386-406, 2nd Ed., Harwood Academic Publishers; Joachim Grante, Angew. 1994 Chem. Int. Ed. Engl. 33:1699-1720; Fauchere, J. 1986 Adv. Drug Res. 15:29; Veber and Freidinger 1985 TINS p. 392; Evans, et al. 1987 J. Med. Chem. 30:229, каждая из которых включена в настоящую заявку посредством ссылки на полную версию. Пептидомиметики, которые структурно сходны с терапевтически применимыми пептидами, можно применять для получения эквивалентного или усиленного терапевтического или профилактического эффекта, при помощи способов, известных в области техники и дополнительно описанных в следующих источниках: Spatola, A.F. 1983 in: Chemistry and Biochemistry of Amino Acids, Peptides, and Proteins, B. Weinstein, eds., Marcel Dekker, New York, p. 267; Spatola, A.F. 1983 Vega Data, Vol. 1, Issue 3, Peptide Backbone Modifications (general review); Morley, 1980 Trends. Pharm. Sci. pp. 463-468, (general review); Hudson, et al. 1979 Int. J. Pept. Prot. Res. 14:177-185 (-CH2NH-, CH2CH2-); Spatola, et al. 1986 Life Sci. 38:1243-1249 (-CH2-S); Hann, 1982 J. Chem. Soc. Perkin. Trans. I 307-314 (-CH-CH-, cis and trans); Almquist, et al. 1980 J. Med. Chem. 23:1392-1398, (-COCH2-); Jennings-White, et al. 1982 Tetrahedron Lett. 23:2533 (-COCH2-); Szelke, et al. 1982 European Appln. EP 45665 (-CH(OH)CH2-); Holladay, et al. 1983 Tetrahedron Lett. 24:4401-4404 (-C(OH)CH2-); и Hruby, 1982 Life Sci. 31:189-199 (-CH2-S-); каждая из которых включена в настоящую посредством ссылок на их полную версию.
[0065] Для создания более стабильных пептидов можно применять систематическую замену одной или более аминокислот в консенсусной последовательности на D-аминокислоты того же типа (например, D-лизин вместо L-лизина). Кроме того, связанные пептиды, содержащие консенсусную последовательность или вариации, по существу идентичные консенсусной последовательности, можно создатьпри помощи способов, известных в областитехники (Rizo, et al. 1992 Ann. Rev. Biochem. 61:387, включена в настоящую заявку посредством ссылки на ее полную версию); например, путем добавления внутренних остатков цистеина, способных к образованию внутримолекулярных дисульфидных мостиков, которые замыкают пептид в цикл, или путем применения остатков аминоизомасляной кислоты (Aib) для стабилизации спирали.
[0066] Термин «синтетические или несуществующие в природе аминокислоты» относится к аминокислотам, которые не встречаются в природе in vivo, но которые, тем не менее, могут быть включены в пептидные структуры, описываемые в настоящей заявке.
[0067] В настоящей заявке термин «производное» соединения, например, пептида или аминокислоты, относится к форме такого соединения, в которой одна или более реактивных групп в соединении были преобразованы с введением групп-заместителей. Примеры производных пептидов включают пептиды, в которых были преобразованы боковая цепь из аминокислот, пептидный остов или амино- или карбоксильный конец (например, пептидные соединения с метилированными амидными связями или гидроксилированными аминокислотами, или остатками аминокислот).
[0068] В настоящей заявке термин «аналог» соединения относится к соединению, которое сохраняет химическую структуру эталонного соединения, необходимую для функциональной активности указанного соединения, которое дополнительно содержит определенные химические структуры, отличные от эталонного соединения. Пример аналога существующего в природе пептида представляет собой пептид, который включает одну или более несуществующих в природе аминокислот или консервативных замен аминокислот, таких как, например, замена, показанная в Таблице 3 ниже. В настоящей заявке термин «миметик» соединения относится к соединению, в котором химические структуры эталонного соединения, необходимые для функциональной активности указанного соединения, были заменены другими химическими структурами, которые имитируют конформацию эталонного соединения. Примеры пептидомиметиков включают пептидные соединения, в которых пептидный остов заменен одной или более молекулой бензадиазепина (см., например, James, G.L. et al. 1993 Science 260:1937-1942, которая включена в настоящую заявку посредством ссылки на ее полную версию), пептиды, в которых все L-аминокислоты заменены на D-аминокислоты, и «ретро-инверсо»-пептиды (см. патент США №4522752, выданный Sisto, который включен в настоящую заявку посредством ссылки на его полную версию), также описаны дополнительно ниже James, G.L. et al. 1993 Science 260:1937-1942, и Goodman et al. 1981 Perspectives in Peptide Chemistry, pp. 283-294, которые включены в настоящую заявку посредством ссылки на их полную версию. Также подробное описание «ретро-инверсо» пептидов можно найти в патенте США №4522 752, выданном Sisto, который включен в настоящую заявку посредством ссылки на его полную версию. Другие производные включают С-концевые гидроксиметил-производные, О-модифицированные производные (например, С-концевой гидроксиметиловый эфир) и N-концевые модифицированные производные, включающие замещенные амиды, такие как алкиламиды и гидразиды.
[0069] Подразумевается, что в настоящей заявке термин «структура аминокислоты» (например, «структура лейцина», «структура фенилаланина» или «структура глутамина») включает аминокислоту, а также аналоги, производные и миметики аминокислоты, которые сохраняют функциональную активность указанного соединения. Например, подразумевается, что термин «структура фенилаланина» включает фенилаланин, а также пиридилаланил и гомофенилаланин. Подразумевается, что термин «структура лейцина» включает лейцин, а также замену на валин, изолейцин или другую природную или неприродную аминокислоту, содержащую алифатическую боковую цепь, такую как норлейцин.
[0070] Амино- и/или карбоксильный конец модифицированных пептидных соединений, раскрываемых в настоящей заявке, может быть стандартным амино- или карбоксильным концом, которые наблюдаются в большинстве белков. В качестве альтернативы амино- и/или карбоксильный конец пептидного соединения может быть химически измененным путем добавления или замены производной группы. Амино-производные группы, которые могут присутствовать на N-конце пептидного соединения (т.е., могут представлять собой Y1), включают ацетильную, арильную, аралкильную, ацильную, эпоксисукцинильную и холестерильную группы. Карбокси-производные группы, которые могут присутствовать на С-конце пептидного соединения (например, могут представлять собой Y2) включают спиртовые, альдегидные, эпоксисукцинатные, кислые галогеновые, карбонильные, галометановые, диазометановые группы и карбоксиамид. Предпочтительным является карбоксиамид.
[0071] В настоящей заявке термин «детектируемая метка» или «визуализирующий агент» относится к веществам, которые при ковалентном связывании с соединением, позволяют проводить детекцию указанного соединения, включая, но не ограничиваясь приведенным, детекцию у пациента in vivo, которому ввели антагонист на основе модифицированного связанного с геном кальцитонина пептида. Подходящие детектируемые метки хорошо известны в области техники и включают, в качестве примера, радиоизотопы, флуоресцентные метки (например, флуоресцеин) и подобные. Конкретная применяемая детектируемая метка не является критичной, и ее выбирают в зависимости от количества метки, которое требуется применить, а также токсичности метки при указанном количестве применяемой метки. Выбор метки в зависимости от таких факторов хорошо известен специалистам в данной области техники.
[0072] Ковалентного присоединения детектируемой метки к пептиду или пептидомиметику достигают при помощи традиционных способов, хорошо известных в области техники. Например, когда в качестве детектируемой метки применяют радиоизотоп 125I, ковалентного присоединения 125I к пептиду или к пептидомиметику можно достичь путем включения аминокислоты тирозина в пептид или пептидомиметик, а затем йодирования указанного пептида (см., например, Weaner, et al. 1994 Synthesis and Applications of Isotopically Labelled Compounds, pp. 137-140). Если в указанном пептиде или пептидомиметике тирозин не присутствует, включения тирозина на N- или С-конце пептида или пептидомиметика можно достичь при помощи хорошо известных химических реакций. Аналогично 32Р можно включить в пептид или пептидомиметик в виде фосфатной группы, например, при помощи гидроксильной группы в пептиде или пептидомиметике с применением традиционных химических реакций.
[0073] В настоящей заявке термин «терапевтический агент» означает агент, способный оказывать желаемое терапевтическое действие в отношении показания специфического заболевания, включая, но не ограничиваясь приведенным, агент, для уменьшения мигрени или боли.
[0074] В настоящей заявке термин «α-спираль» означает структурный компонент, который образует α-спиральную структуру белка или любой другой структурный аналог, который приводит к аналогичному позиционированию доменов X1 и Z1 в рецепторе.
Получение пептидов и пептидомиметиков
1. Твердофазный синтез
[0075] Антагонисты на основе модифицированного связанного с геном кальцитонина пептида, описываемые в настоящей заявке, можно получить при помощи классических способов, известных в области техники, например, путем применения стандартных твердофазных способов. Например, см. Merrifield, 1963 J. Am. Chem. Soc. 85:2149, включенную в настоящую заявку посредством ссылки на ее полную версию.
[0076] Указанные процедуры твердофазного синтеза пептидов хорошо известны в области техники и более подробно описаны в J.M. Stewart and J.D. Young, 1984 Solid Phase Peptide Syntheses 2nd Ed., Pierce Chemical Company.
2. Синтетические аминокислоты
[0077] Указанные процедуры также можно применять для синтеза пептидов, в которых аминокислоты, отличные от 20 существующих в природе, генетически кодируемых аминокислот, заменены в одном, двух или более положениях любого из антагонистов на основе модифицированного связанного с геном кальцитонина пептида, которые раскрываются в настоящей заявке. Например, нафтилаланин может быть заменен триптофаном, что облегчает синтез. Другие синтетические аминокислоты, которые можно заменять в пептидах, согласно вариантам реализации настоящего изобретения включают L-гидроксипропил, L-3,4-дигидрокси-фенилаланин, d-аминокислоты, такие как L-d-гидроксилизил и D-d-метилаланин, L-α-метилаланин, β-аминокислоты, и изохинолил. D аминоксилоты и не существующие в природе синтетические аминокислоты также можно включать в пептиды согласно вариантам реализации настоящего изобретения (см., например, Roberts, et al. 1983 Unusual Amino/Acids in Peptide Synthesis 5:341-449).
[0078] Согласно некоторым вариантам реализации изобретения существующие в природе боковые цепи 20 генетически кодируемых аминокислот или любую другую боковую цепь, раскрываемую в настоящей заявке, можно перенести на атом азота аминокислоты, вместо α-углерода, который обычно обнаруживается в пептидах.
[0079] Таблица 2: Однобуквенные обозначения для канонических аминокислот. В скобках показаны трехбуквенные сокращения.

[0080] Номенклатура и символы обозначения аминокислот и пептидов согласно Объединенной Комиссии по биохимической номенклатуре (JCBN) UPAC-IUB была опубликована в следующих документах: Biochem. J., 1984, 219, 345-373; Eur. J. Biochem., 1984, 138, 9-5 37; 1985, 152, I; 1993, 213, 2; Internal J. Pept. Prot. Res., 1984, 24, following p 84; J. Biol. Chem., 1985, 260, 14-42; Pure Appl. Chem., 1984, 56, 595-624; Amino Acids and Peptides, 1985, 16, 387-410; Biochemical Nomenclature and Related Documents, 2nd edition, Portland Press, 1992, pages 39-69.
[0081] Согласно некоторым вариантам реализации изобретения последовательность аминокислот в подходящем антагонисте на основе модифицированного связанного с геном кальцитонина пептида можно модифицировать относительно последовательности согласно SEQ ID №: 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14 или 15, так, чтобы указанная модификация снизила восприимчивость актуального антагониста на основе модифицированного связанного с геном кальцитонина пептида к ферментативному протеолизу. Согласно некоторым вариантам реализации изобретения указанная модификация может включать добавление на N-конце последовательности, содержащей все или часть из 864 остатков полипептида XTENS - полипептида, который, как было показано, повышает стабильность белка после введения его субъекту. См., например, Schellenberger, et al., 2009, Nature Biotechnology 27(12): 1186-1192, которая включена в настоящую заявку посредством ссылки на ее полную версию.
[0082] Согласно некоторым вариантам реализации изобретения актуальный модифицированный антагонист на основе связанного с геном кальцитонина пептида может включать один или более остатков D-аминокислот. Согласно некоторым вариантам реализации изобретения последовательность аминокислот в актуальном антагонисте на основе модифицированного связанного с геном кальцитонина пептида можно модифицировать относительно последовательности согласно SEQ ID №: 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14 или 15, так, чтобы указанная модификация включала замену одну или более L-аминокислот на соответствующие D-аминокислоты.
[0083] Согласно некоторым вариантам реализации изобретения последовательность аминокислот антагониста на основе модифицированного связанного с геном кальцитонина пептида может быть модифицирована относительно с последовательности согласно SEQ ID №: 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14 или 15, так, чтобы указанная модификация включала консервативные замены аминокислот.
[0084] Существующие в природе остатки можно подразделить на классы в зависимости от общих свойств боковых цепей:
гидрофобные: норлейцин (Nor), Met, Ala, Val, Leu, Ile;
нейтральные гидрофильные: Cys, Ser, Thr, Asn, Gln;
кислые: Asp, Glu;
основные: His, Lys, Arg;
остатки, которые влияют на ориентацию цепи: Gly, Pro; и
ароматические: Trp, Tyr, Phe.
[0085] Под консервативными заменами аминокислот может подразумеваться замена члена определенного класса на другой член того же класса. Консервативные замены аминокислот могут охватывать остатки несуществующих в природе аминокислот, которые, как правило, включают посредством химического синтеза пептидов, но не посредством синтеза в биологических системах. Они включают пептидомиметики и другие обратные или инвертированные формы аминокислот.
[0086] Согласно некоторым вариантам реализации изобретения консервативные замены могут включать замену одного остатка неполярной (гидрофобной) аминокислоты, такой как изолейцин, валин, лейцин, норлейцин, аланин или метионин, на другой остаток, замену одного остатка полярной (гидрофильной) аминокислоты на другой, например, замена между аргинином и лизином, между глутамином и аспарагином, между треонином и серином, замену одного остатка основной аминокислоты, такой как лизин, аргинин или гистидин, на другой, или замену одного остатка кислой аминокислоты, такой как аспарагиновая кислота или глутаминовая кислота на другой. Фраза «консервативная замена аминокислоты» также включает применение химически преобразованного остатка вместо не преобразованного остатка при условии, что такой полипептид проявляет требуемую антагонистическую активность.
[0087] В Таблице 3 приведены примеры замен остатков аминокислот, которые могут быть полезны в соответствии с вариантами реализации настоящего изобретения.

[0088] Согласно некоторым вариантам реализации изобретения основную группу аминокислот, раскрываемых в настоящей заявке, такую как гуанидин или Arg, можно заменить на основной биоизоэфир.
[0089] Термин «гидроксипролин» относится к любым и всем известным формам гидроксилирования пролина либо в форме свободной аминокислоты, либо в форме, включенной в полипептид. Он включает (2S,4R)-4-гидроксипролин, а также остатки пролина с разными стереохимическими свойствами или гидроксилированными атомами углерода.
[0090] Термин «O-карбокси»-группа относится к группе «RC(=O)O-», в которой R может быть водородом, алкилом, алкенилом, алкинилом, циклоалкилом, циклоалкенилом, циклоалкинилом, арилом, гетероарилом, гетероалициклилом, аралкилом или (гетероалициклил)алкилом, как определено в настоящей заявке. О-карбоксильная группа может быть замещенной или незамещенной.
[0091] Термин «С-карбокси»-группа относится к группе «-C(=O)OR», в которой R может быть такой же группой, которые перечислены в отношении О-карбоксигруппы. С-карбоксильная группа может быть замещенной или незамещенной.
[0092] Термин «С-амидо»-группа относится к группе «-C(=O) NRARB», в которой RA и RB могут быть или могут не быть одинаковыми, и могут быть такими же как R в определении О-карбоксигруппы. С-амидогруппа может быть замещенной или незамещенной.
[0093] Термин «N-амидо»-группа относится к группе «RC(=O)NRA-», в которой R и RA могут быть или не быть одинаковыми, и могут быть такими же, как R в определении О-карбоксигруппы. N-амидогруппа может быть замещенной или незамещенной.
[0094] В настоящей заявке термин «амид» относится к группе «-C(=O)NRARB», в которой RA и RB могут быть или не быть одинаковыми, и могут быть такими же как R в определении О-карбоксигруппы. Согласно некоторым вариантам реализации изобретения RA и RB могут представлять собой водород.
[0095] В настоящей заявке термин «амин» относится к «-NRARB», в которой RA и RB могут быть или могут не быть одинаковыми, и могут быть такими же, как R в определении О-карбоксигруппы.
[0096] В настоящей заявке термин «карбамид» относится к -NRAC(=O)NRB2, где каждый из RA и RB по отдельности могут быть такими, как R в определении О-карбоксигруппы.
[0097] Можно легко модифицировать пептиды согласно актуальным вариантам реализации изобретения посредством фосфорилирования (см., например, W. Bannwarth, et al. 1996 Biorganic and Medicinal Chemistry Letters 6:2141-2146), и других способов получения пептидных производных соединений согласно вариантам реализации настоящего изобретения, которые описаны у Hruby, et al. 1990 Biochem. J. 268:249-262. Таким образом, пептиды, как раскрыто в настоящей заявке, также служат основой для получения пептидомиметиков со сходной биологической активностью.
3. Модификации концов
[0098] Специалисту в данной области техники будет понятно, что существует целый ряд способов для конструирования пептидомиметиков с одинаковой или сходной желаемой биологической активностью, что и у соответствующего антагониста на основе модифицированного связанного с геном кальцитонина пептидом, но с более благоприятной активностью, чем эталонный пептид, в том, что касается растворимости, стабильности и восприимчивости к гидролизу и протеолизу. См., например, Morgan, et al. 1989 Ann. Rep. Med. Chem. 24:243-252. Далее описаны способы получения пептидомиметиков, модифицированных по N-концевой аминогруппе, С-концевой карбоксильной группе и/или с заменой одной или более амидных связей в пептиде на неамидные связи. Следует понимать, что две или более таких модификации можно объединять в структуре одного пептидомиметика (например, модификация в С-концевой карбоксильной группе и включение -CH2-карбаматной связи между двумя аминокислотами в пептиде).
1). N-концевые модификации
[0099] Пептиды, как правило, синтезируют в форме свободной кислоты, но как отмечалось выше, их можно легко получить в форме амида или эфира. Также можно модифицировать амино- и/или карбоксильный конец пептидного соединения с получением других полезных соединений. Модификации N-конца включают метилирование (т.е. -NHCH3 или -NH(СН3)2), ацетилирование, добавление бензилоксикарбонильной группы или блокирование N-конца любой из блокирующих групп, содержащих карбоксилатную функциональную группу, определяемую как RCOO-, где R выбран из группы, включающей нафтильную, акридинильную, стероидильную и сходные группы.
[0100] Модификации по N-концу перечислены выше и включают алкилирование, ацетилирование, добавление карбобензоильной группы, образование сукцинимидной группы и др. (см., например, Murray, et al. 1995 Burger's Medicinal Chemistry and Drug Discovery 5th ed., Vol. 1, Manfred E. Wolf, ed., John Wiley and Sons, Inc., которые включены в настоящую заявку посредством ссылки на их полную версию).
[0101] Также можно модифицировать N-конец посредством добавления по меньшей мере одного остатка к N-концу фрагмента X1. Способы оценки влияния удлинения N-конца на пептиды известны в области техники и описаны, например, в Schellenberger, et al., 2009, Nature Biotechnology 27(12): 1186-1192, которая включена в настоящую заявку посредством ссылки на ее полную версию.
2). С-концевые модификации
[0102] Модификации по карбоксильному концу включают замену свободной кислоты на карбоксиамидную группу или образование циклического лактама на карбоксильном конце для введения структурных ограничений. При получении пептидомиметиков, в которых С-концевая карбоксильная группа заменена амидом -C(O)NR3R4, в качестве твердой подложки для синтеза пептида применяют бензгидриламиновую смолу. После завершения синтеза обработка фтористым водородом для освобождения пептида с подложки напрямую приводит к образованию амида свободного пептида (т.е. С-конец представляет собой -C(O)NH2). В качестве альтернативы применение хлорметилированной смолы во время синтеза пептидов в сочетании с реакцией с аммиаком для отщепления пептида, защищенного по боковым цепям, от подложки, приводит к образованию амида свободного пептида, и реакция с алкиламином или диалкиламином приводит к образованию алкиламида или диалкиламида, защищенных по боковым цепям (т.е. С-конец представляет собой -C(O)NRR1, где R и R1 соответствуют определению, данному выше). Затем защиту боковых цепей снимают обычным способом путем обработки фтористым водородом для получения свободных амидов, алкиламидов или диалкиламидов.
[0103] Помимо перечисленных выше модификаций по N-концу модифицированные антагонисты пептидов, описанные в настоящей заявке, включая пептидомиметики, можно с пользой модифицировать или ковалентно присоединять к одному или более полимерам из целого ряда гидрофильных полимеров. Было показано, что когда пептиды преобразованы с применением гидрофильного полимера, их растворимость и время полувыведения из системы кровообращения увеличиваются, а их иммуногенность маскируется. Достаточно неожиданно, что вышеизложенные преобразования могут сопровождаться небольшим, если вообще сопровождаться, снижением их активности связывания. Согласно некоторым вариантам реализации изобретения антагонисты на основе модифицированного связанного с геном кальцитонина пептида, как раскрыто и описано в настоящей заявке, можно преобразовывать или присоединять к таким полимерам при помощи любого из способов, описанных у Zallipsky, S. 1995 Bioconjugate Chem 6:150-165; Monfardini, С, et al. 1995 Bioconjugate Chem 6:62-69; в Патентах США №4640835; 4496689; 4301144; 4670417; 4791192; 4179337 или WO 95/34326, все из которых включены в настоящую заявку посредством ссылки на их полную версию.
4. Модификации остова
[0104] Другие способы получения пептидных производных соединений описаны у Hruby, et al. 1990 Biochem. J. 268(2):249-262, которая включена в настоящую заявку посредством ссылки на ее полную версию. Таким образом, пептидные соединения также служат структурными моделями для непептидных соединений со сходной биологической активностью. Специалисты в данной области техники понимают, что существует целый ряд способов конструирования соединений с такой же или сходной желаемой биологической активностью, как у пептидного соединения - образца, но с более благоприятной активностью, чем у образца, в том, что касается растворимости, стабильности и восприимчивости к гидролизу и протеолизу. См., Morgan, et al. 1989 Ann. Rep. Med. Chem. 24:243-252, включенную в настоящую заявку посредством ссылки на ее полную версию.
5. Образование дисульфидных связей
[0105] Указанные соединения могут существовать в циклической форме с внутримолекулярной дисульфидной связью между тиольными группами цистеинов.
[0106] Другие варианты реализации изобретения включают аналоги указанных дисульфидных производных, в которых один из атомов серы был заменен группой СН2 или другой изостерой серы. Указанные аналоги можно получать путем внутримолекулярного или межмолекулярного замещения с применением способов, известных в области техники.
[0107] В качестве альтернативы амино-конец пептида можно кеппировать альфа-замещенной уксусной кислотой, причем альфа-заместителем является выходная группа, такая как α-галоген-уксусная кислота, например, α-хлоруксусная кислота, α-бромуксусная кислота или α-йодоуксусная кислота. Пептиды согласно настоящим вариантам реализации изобретения можно циклизовать или димеризовать посредством замещения выходной группы серой остатка цистеина или гомоцистеина. См., например, Andreu, et al. 1994, Meth. Mol. Bio. 35(7):91-169; Barker, et al. 1992, J. Med. Chem. 35:2040-2048; и Or, et al. 1991, J. Org. Chem. 56:3146-3149, каждая из которых включена в настоящую заявку посредством ссылки на их полную версию.
[0108] Согласно некоторым вариантам реализации изобретения антагонисты на основе модифицированного связанного с геном кальцитонина пептида, как раскрыто и описано в настоящей заявке, можно также получить посредством технологий рекомбинантных ДНК, хорошо известных в области техники.
[0109] Другие варианты реализации изобретения включают фармацевтические композиции, содержащие в качестве активного ингредиента по меньшей мере один из подходящих модифицированных пептидов или пептидомиметиков, раскрываемых в настоящей заявке, в сочетании с фармацевтически приемлемой основой или растворителем. Указанные фармацевтические композиции можно вводить любыми путями, известными специалистам в данной области техники, и которые включают без ограничений пероральный, легочный, парентеральный (внутримышечные, внутрибрюшинные, внутривенные или подкожные инъекции), ингаляционный (в форме мелкодисперсного порошка или аэрозоля), чрескожный, интраназальный или сублингвальный пути введения, причем указанные композиции можно преобразовывать в лекарственные формы, подходящие для каждого пути введения. См., например, Bernstein, et al. публикация патента РСТ № WO 93/25221, опубликованная 23 декабря 1993 г.; Pitt, et al. публикация патента РСТ № WO 94/17784, опубликованная 18 августа 1994 г.; и Pitt, et al. заявка на Европейский патент 613 683, опубликованная 7 сентября 1994 г., каждая из которых включена в настоящую заявку посредством ссылки на их полную версию. Указанные соединения также можно вводить в составе лекарственной формы с замедленным или контролируемым высвобождением, включая, но не ограничиваясь приведенным, инъекции веществ замедленного всасывания, осмотические насосы, пластыри для чрескожного введения (в том числе и на основе электротранспорта) и подобные, для пролонгированного и/или синхронизированного, импульсного введения с заранее заданной скоростью.
[0110] Фармацевтические композиции согласно вариантам реализации настоящего изобретения могут быть произведены способом, который сам по себе известен, например, посредством традиционных способов смешивания, растворения, гранулирования, образования драже, растирания в порошок, эмульгирования, инкапсулирования, захвата или таблетирования.
[0111] Таким образом, фармацевтические композиции для применения согласно вариантам реализации настоящего изобретения можно преобразовывать в лекарственные формы традиционным путем с применением одной или более физиологически приемлемых основ, содержащих носители и вспомогательные вещества, которые облегчают обработку активных соединений в препараты, которые можно применять в фармацевтических целях. Подходящая лекарственная форма зависит от избранного пути введения. Любые хорошо известные способы, основы и носители могут быть использованы как подходящие и известные в области техники; например, в Remington's Pharmaceutical Sciences, указанном выше.
[0112] Формы для инъекций могут быть приготовлены в традиционном виде, либо в виде жидких растворов, либо суспензий, твердых форм, подходящих для растворения или получения суспензии в жидкости перед инъекцией, или в виде эмульсий. Подходящие вспомогательные вещества включают, например, воду, физиологический раствор, декстрозу, маннитол, лактозу, лецитин, альбумин, глутамат натрия, гидрохлорид цистеина и подобные. Кроме того, при желании, фармацевтические композиции для инъекций могут содержать незначительные количества нетоксических вспомогательных веществ, таких как смачивающие агенты, рН-буферы и подобные. Физиологически совместимые буферы включают без ограничений раствор Ханка, раствор Рингера или физиологический раствор с буфером. При желании можно применять препараты, усиливающие всасывание (например, липосомы).
[0113] Для введения через слизистые оболочки в составе лекарственной формы можно применять проникающие вещества, соответствующие барьеру, который должно преодолеть лекарственное средство.
[0114] Фармацевтические формы для парентерального введения, например, посредством болюсных инъекций или непрерывной инфузии, включают водные растворы активных соединений в водорастворимой форме. Кроме того, суспензии активных веществ можно готовить в виде соответствующих масляных суспензий для инъекций. Подходящие липофильные растворители или основы включают жирные масла, такие как кунжутное масло, или другие органические масла, такие как соевое, грейпфрутовое или миндальное масло, или синтетические эфиры жирных кислот, такие как этилолеат или триглицериды, или липосомы. Водные суспензии для инъекций могут содержать вещества, которые увеличивают вязкость суспензии, такие как натриевая соль карбоксиметилцеллюлозы, сорбитол или декстран. Необязательно указанная суспензия может также содержать подходящие стабилизаторы или агенты, которые увеличивают растворимость актуальных соединений, которые создают возможность проникновения высококонцентрированных растворов. Лекарственные формы для инъекций могут быть представлены в виде лекарственных форм с единичной дозировкой, например, в ампулах или в контейнерах с несколькими дозами, с добавлением консерванта. Указанные композиции могут принимать такие формы, как суспензии, растворы или эмульсии на масляных или водных основах, и могут содержать такие вспомогательные вещества, как суспендирующие, стабилизирующие и/или диспергирующие агенты. В качестве альтернативы активный ингредиент может быть в форме порошка для смешивания перед применением с подходящей основой, например, стерильной апирогенной водой.
[0115] Для перорального введения актуальные соединения можно преобразовывать в лекарственную форму путем сочетания активных соединений с фармацевтически приемлемыми основами, такими, как раскрываются у D.J. Sarubbi, Oral GLP-1 Formulations, в заявке на патент США №2010/0016229 А1, опубликованной 21 января 2010 г., которая включена в настоящую заявку посредством ссылки на ее полную версию. Как обсуждалось в настоящей заявке, препараты для перорального введения могут быть представлены в форме таблеток или капсул из фармацевтически приемлемых основ, смешанных с лекарственным средством. Дополнительные подходящие агенты для доставки, описанные у Goldberg, 2009, Compositions for Delivering Parathyroid Hormone and Calcitonin, в заявке на патент США № A1, опубликованной 22 октября 2009 г. (которая включена в настоящую заявку посредством ссылки на ее полную версию), включают известные жидкие или твердые лекарственные формы.
[0116] Для введения путем ингаляции актуальные соединения для применения согласно вариантам реализации настоящего изобретения традиционно доставляют в форме аэрозоля из упаковок под давлением или распылителя, с применением подходящего газа-вытеснителя, например, дихлордифторметана, трихлорфторметана, дихлортетрафторэтана, углекислого газа или других подходящих газов. В случае аэрозоля, находящегося под давлением, единичная доза может быть определена при помощи клапана для выделения дозированного количества. Капсулы и картриджи, например, из желатина для применения в ингаляторе или инсуффляторе могут быть составлены так, чтобы они содержали смесь порошка соединения и подходящей порошковой основы, такой как лактоза или крахмал. В качестве примера препараты для введения путем ингаляции можно получить в соответствии с описанием в Quay, et al., патент США №7812120 В2, выданный 12 октября 2010 г, который включен в настоящую заявку посредством ссылки на его полную версию.
[0117] Также в настоящей заявке раскрываются разные фармацевтические композиции, хорошо известные в фармацевтике для применения, которое включает внутриглазную, интраназальную и внутриушную доставку. Подходящие проникающие вещества для указанных вариантов применения широко известны в области техники. Фармацевтические композиции для внутриглазного введения включают водные растворы активных соединений в водорастворимой форме для введения в глаз, такие как глазные капли, или в геллановой камеди (Shedden et al., 2001, Clin. Ther., 23(3):440-50), или в гидрогеле (Mayer et al., 1996, Ophthalmologica, 210(2):101-3); глазные мази; глазные суспензии, такие как микрочастицы, содержащие лекарственное вещество малые полимерные частицы, которые распределяются в жидкой среде основы (Joshi, A., J. Ocul. Pharmacol., 1994 10(1):29-45), жирорастворимые лекарственные формы (Alm et al., 1989 Prog. Clin. Biol. Res., 312:447-58) и микросферы (Mordenti, 1999, Toxicol. Sci., 52(1):101-6); и глазные вкладыши. Каждая из перечисленных выше ссылок включена в настоящую заявку посредством ссылки на их полную версию. Такие подходящие лекарственные формы наиболее часто и преимущественно составляют так, чтобы они были стерильными, изотоническими и забуференными с целью стабильности и комфорта. Фармацевтические композиции для интраназальной доставки могут также включать капли и спреи, которые часто готовят для стимуляции назальной секреции во многих аспектах, чтобы поддерживать нормальную функцию ресничек, и такие композиции включают, например и без ограничения, назальные растворы, раскрываемые у Azria, et al., в патенте США №5733569, выданном 31 марта 1998 г., который включен в настоящую заявку посредством ссылки на его полную версию. Как раскрывается в Remington's Pharmaceutical Sciences, 18th Ed., Mack Publishing Co., Easton, PA (1990), который включен в настоящую заявку посредством ссылки на его полную версию, и хорошо известно специалистам в данной области техники, подходящие лекарственные формы чаще всего и преимущественно изотоничные, слегка забуферены для поддержания рН 5,5-6,5, и чаще всего и преимущественно включают антимикробные консерванты и соответствующие лекарственные стабилизаторы. Лекарственные формы для внутриушной доставки включают суспензии и мази для местного нанесения в ухо. Часто используемые растворители для таких ушных лекарственных форм включают глицерин и воду.
[0118] Помимо лекарственных форм, описанных ранее, подходящие соединения также могут быть представлены в виде препаратов с замедленным всасыванием. Такие препараты длительного действия можно вводить путем имплантации (например, подкожно или внутримышечно) или посредством внутримышечной инъекции. Таким образом, указанные соединения можно преобразовывать в лекарственную форму с добавлением подходящих полимерных или гидрофобных материалов (например, эмульсии в соответствующем масле) или ионообменных смол, или в виде плохо растворимых производных, например, в виде плохо растворимой соли.
[0119] В зависимости от химической природы и биологической стабильности терапевтического реагента можно применять дополнительные стратегии стабилизации пептидов.
[0120] В указанные фармацевтические композиции можно включать дополнительные терапевтические или диагностические агенты. В качестве дополнения или альтернативы фармацевтические композиции можно комбинировать с другими композициями, которые содержат другие терапевтические или диагностические агенты.
[0121] Неограничивающие примеры способов введения включают, среди прочих, (а) введение пероральным путем, такое как описано в М. Goldberg, публикации патента США №2009/0264368 А1, опубликованной 22 октября 2009 г, которая включена в настоящую заявку посредством ссылки на ее полную версию, и в D. Sarubbi, публикации патента США № US 2010/0016229 A1, опубликованной 21 января 2010 г., которая включена в настоящую заявку посредством ссылки на ее полную версию; (b) введение также можно проводить не пероральным путем, например, внутриглазным, интраназальным или внутриушным, при которых введение включает введение водной суспензии, масляного препарата или подобного, или в форме капель, спрея, крема, мази или подобных; (с) введение посредством инъекций, подкожных, внутрибрюшинных, внутривенных, внутримышечных, внутрикожных, внутриорбитальных или подобных, включая доставку при помощи инфузионного насоса; (d) локальное введение, такое как инъекции непосредственно в полость черепа, например, при помощи имплантата с замедленным всасыванием; а также (е) местное введение; что найдут целесообразным специалисты в данной области техники для доставки пептида согласно вариантам реализации настоящего изобретения в живые ткани. Неограничивающий пример назального введения описан у Quay, et al., патент США №7812120 В2, выданный 12 октября 2010 г., который включен в настоящую заявку посредством ссылки на его полную версию.
[0122] Точный состав, путь введения и дозировку фармацевтических композиций согласно вариантам реализации настоящего изобретения врач может выбирать индивидуально в зависимости от состояния пациента, (см., например, Fingl et al. 1975, в "The Pharmacological Basis of Therapeutics", который включен в настоящую заявку посредством ссылки на его полную версию, в частности ссылка на Ch. 1, р. 1). Как правило, диапазон доз композиции, вводимых пациенту, может быть от 0,000001 до 100 мг/кг массы тела пациента. Доза может быть однократной или серией из двух или более доз, вводимых как курс в течение одного или более дней, в зависимости от потребности пациента. В тех случаях, когда дозы соединений для человека установлены по меньшей мере для некоторого состояния, варианты реализации настоящего изобретения основаны на применении тех же доз, или доз, которые составляют от 0,1% до 500%, более предпочтительно приблизительно от 25% до 250% от установленной для человека дозы. В случаях, когда доза для человека не установлена, как в случае вновь открытых фармацевтических соединений, подходящую дозу для человека можно вывести из значений ED50 (доза, эффективная у 50% особей) или ID50 (доза, токсичная для 50% особей), или других соответствующих значений из исследований in vitro или in vivo, которые рассматриваются как исследования токсичности и исследования эффективности на животных.
[0123] Следует отметить, что лечащий врач должен знать, как и когда прекратить, приостановить или скорректировать введение препарата в связи с токсичностью или нарушением функции органов. И, наоборот, лечащий врач должен знать, как скорректировать лечение до более высокого уровня, если клиническая реакция была неадекватной (предупредить токсичность). Величина вводимой дозы при лечении рассматриваемого расстройства может варьировать в зависимости от тяжести состояния, которое требуется лечить, и от пути введения. Тяжесть состояния, например, можно оценить, частично, при помощи стандартных способов прогностической оценки. Кроме того, доза, и, вероятно, частота введения доз могут варьировать в зависимости от возраста, массы тела и реакции индивидуального пациента. Программу, сравнимую с той, что описана выше, можно применять и в ветеринарной медицине.
[0124] Хотя точную дозировку следует определять отдельно для каждого конкретного лекарственного препарата, в большинстве случаев можно сделать некоторые обобщения относительно дозировок. Режим дозирования раз в день для взрослого пациента может включать, например, внутривенное, подкожное или внутримышечное введение дозы каждого активного ингредиента в примерном диапазоне от 0,001 мг до 100 мг, или в примерном диапазоне от 0,005 мг до 5 мг. В случаях введения фармацевтически приемлемой соли дозировки можно рассчитать для свободного основания. Согласно некоторым вариантам реализации изобретения указанную композицию вводят от 1 до 4 раз в день или в форме однократной ударной дозы, например, для облегчения боли, такой, как боль, ассоциированная с мигренью. В качестве альтернативы, композиции, описываемые в настоящей заявке, можно вводить путем непрерывной внутривенной инфузии, предпочтительно при дозе каждого активного ингредиента до 1000 мг в день. Как должны понимать специалисты в данной области техники, в определенных ситуациях может быть необходимо вводить пептиды, раскрываемые в настоящей заявке, в количествах, которые превышают или даже значительно превышают установленный выше примерный диапазон доз, чтобы эффективно и агрессивно воздействовать на конкретные агрессивные заболевания или инфекции. Согласно некоторым вариантам реализации изобретения пептиды следует вводить в течение определенного периода в форме непрерывной терапии, например, в течение недели или более, или месяцев, или лет.
[0125] Концентрацию дозы и интервал можно корректировать индивидуально, чтобы достичь уровня активного компонента в плазме, который эффективен для поддержания модулирующего действия, или минимальной эффективной концентрации (МЭК). МЭК может варьировать для каждого из соединений, но ее можно оценить на основании данных, полученных in vitro. Дозировка, необходимая для достижения МЭК, будет зависеть от индивидуальных характеристик и пути введения. Однако для определения концентраций в плазме можно применять анализ методом ВЭЖХ (высокоэффективной жидкостной хроматографии).
[0126] Интервалы между дозированием также можно определить на основании значения МЭК. Композиции следует вводить согласно режиму, при котором поддерживается уровень вещества в плазме выше МЭК в течение 10-90% времени, предпочтительно от 30 до 90% и наиболее предпочтительно 50-90%.
[0127] В случаях местного введения или селективного захвата эффективная локальная концентрация лекарственного средства может быть не связана с концентрацией его в плазме.
[0128] Количество вводимой подходящей композиции может зависеть от субъекта, которого требуется лечить, от массы субъекта, тяжести заболевания, способа введения и мнения назначающего врача.
[0129] Соединения, раскрываемые в настоящей заявке, можно оценивать по эффективности и токсичности с применением известных способов. Например, токсикологическая оценка конкретного соединения или подгруппы соединений, обладающих определенными общими химическими фрагментами, можно изучить путем определения токсичности in vitro в отношении линии клеток, например, клеток млекопитающих и предпочтительно линии клеток человека. Результаты таких исследований часто позволяют прогнозировать токсичность у животных, таких как млекопитающие, или, более конкретно, у человека. В качестве альтернативы при помощи известных способов можно определять токсичность конкретных соединений в моделях на животных, таких как мыши, крысы, кролики или обезьяны. Эффективность конкретного соединения можно определить при помощи нескольких признанных способов, таких как способы in vitro, модели на животных или клинические исследования с участием людей. Почти для каждого класса состояний существуют признанные модели in vitro, включая, но не ограничиваясь приведенным, рак, сердечно-сосудистые заболевания и разные нарушения функции иммунной системы. Аналогично, для определения эффективности химических веществ, при лечении таких состояний можно применять приемлемые модели на животных. При выборе модели для определения эффективности опытный специалист может принять решение по выбору соответствующей модели, дозы, пути введения и режима на основании уровня техники. Конечно, клинические исследования с участием людей также можно применять для определения эффективности соединения у человека.
[0130] При желании актуальные композиции можно поместить в упаковку или выдающее устройство, которое может содержать одну или более единичную дозу, содержащую активный ингредиент.
[0131] Во всем описании любое перечисление конкретного соединения должно подразумевать включение в данное понятие любой (другой) его фармацевтически приемлемой соли.
[0132] Актуальные композиции, содержащие указанные соединения, можно вводить для профилактики и/или для лечения. При терапевтическом применении композиции вводят пациенту, уже страдающему заболеванием, как было описано выше, в количестве, достаточном для излечения или по меньшей мере частичного прекращения симптомов указанного заболевания и его осложнений. Количество, адекватное для достижения данной цели, называют «терапевтически эффективной дозой». Количества, эффективные для такого применения, будут зависеть от тяжести заболевания, а также массы и общего состояния пациента.
[0133] Композиции, описываемые в настоящей заявке, также можно подвергать микроинкапсуляции, например, при помощи способа согласно Tice and Bibi (в: Treatise on Controlled Drug Delivery, ed. A. Kydonieus, Marcel Dekker, N.Y. 1992, pp. 315-339), которая включена в настоящую заявку посредством ссылки на ее полную версию.
[0134] При профилактическом применении композиции, содержащие соединения, раскрываемые в настоящей заявке, вводят пациенту, предрасположенному к определенному заболеванию или с другими факторами риска его развития. Такое количество определяют как «профилактически эффективная доза». При таком применении точное количество опять же будет зависеть от состояния здоровья пациента и его массы, и его легко может определить специалист в данной области техники.
[0135] Количества актуального антагониста, необходимые для эффективной терапии, будут зависеть от многих других факторов, включая пути введения, орган-мишень, физиологическое состояния пациента и другие вводимые лекарственные препараты. Таким образом, лечебные дозировки следует титровать так, чтобы оптимизировать безопасность и эффективность. Как правило, дозировки, применяемые in vitro, могут быть полезным руководством в выборе количеств, полезных для введения in situ указанных реагентов. Исследования на животных эффективных доз для лечения конкретных расстройств может дать дополнительные предсказывающие указания для выбора доз у человека. Разные соображения описаны, например, в: Gilman, et al. (eds.), 1990 Goodman and Gilman's: The Pharmacological Basis of Therapeutics 8th ed., Pergamon Press; и Remington's Pharmaceutical Sciences, 7th Ed., Mack Publishing Co., Easton, Pa. (1985), каждая из которых включена в настоящую заявку посредством ссылки на ее полную версию. В частности, дозировку следует корректировать, чтобы адаптировать ее для способов доставки, таких как внутримышечная инъекция, подкожная инъекция, пероральное введение или подкожное введение, введение антагониста без иглы.
[0136] Антагонисты пептиды и пептидомиметики, описываемые в настоящей заявке, эффективны при лечении состояний, опосредуемых рецептором CGRP, когда их вводят в примерном диапазоне доз от приблизительно 0,01 мг до приблизительно 50 мг/кг массы тела в день. Специфическую применяемую дозу корректируют в зависимости от конкретного состояния, которое требуется лечить, и пути введения, а также по решению лечащего врача в зависимости от таких факторов, как тяжесть состояния, возраст и общее состояние пациента, и подобные. Специалист в данной области техники может легко определить такие дозы.
[0137] Для парентерального введения пептиды можно преобразовывать, например, в раствор, суспензию, эмульсию или лиофилизированный порошок в ассоциации с фармацевтически приемлемой основой для парентерального введения. Примеры таких основ включают воду, физиологический раствор, раствор Рингера, раствор декстрозы и 5% сывороточный альбумин человека. Также можно применять липосомы и неводные основы, такие как нелетучие масла. Основа или лиофилизированный порошок могут содержать добавки, которые помогают поддерживать изотоничность (например, хлорид натрия, маннитол) и химическую стабильность (например, буферы и консерванты). Указанную лекарственную форму стерилизуют общеизвестными способами. Например, композицию для парентерального введения, подходящую для введения путем инъекций, готовят путем растворения 1,5% по массе активного ингредиента в 0,9% растворе хлорида натрия.
[0138] Фармацевтические композиции, описываемые в настоящей заявке, можно вводить как однократную дозу или многократные дозы; вводить либо как отдельные терапевтические агенты, или в сочетании с другими терапевтическими агентами; и сочетать с традиционные средствами лечения, которые можно вводить последовательно или одновременно.
[0139] Указанные соединения можно вводить в виде формы с замедленным высвобождением, например, в композиции, которая включает полимер для замедленного высвобождения. Активные соединения можно получить с основами, которые будут защищать указанное соединение от быстрого высвобождения, например, как в форме замедленного высвобождения, включая имплантаты и микроинкапсулированные системы доставки. Может быть применены биоразлагаемые, биосовместимые полимеры, такие как этиленвинилацетат, полиангидриды, полигликолевая кислоты, коллаген, полиортоэфиры, полимолочная кислота и полилактидные, полигликолевые сополимеры (PLG). Многие способы получения таких лекарственных форм общеизвестны специалистам в данной области техники.
[0140] Из соединений, описываемых в настоящей заявке, может быть составлена фармацевтическая композиция, в которой указанное соединение является единственным активным агентом. В качестве альтернативы фармацевтическая композиция может содержать дополнительные активные агенты. Кроме того, указанное соединение-пептид можно сочетать с одним или более другими агентами, которые обладают модулирующим действием на активность рецепторов CGRP. Другие области применения
[0141] Соединения, описываемые в настоящей заявке, применимы in vitro, в качество уникальных средств для понимания биологической роли рецепторов CGRP, включая оценку многих факторов, которые, как предполагают, влияют и находятся под влиянием выработки лигандов-эфринов и процесса связывания с рецепторами. Также настоящие соединения применимы при разработке других соединений, которые связываются и активируют рецепторы CGRP, поскольку настоящие соединения предоставляют важную информацию о взаимосвязи между структурой и активностью, облегчающую такую разработку.
[0142] Также указанные соединения являются конкурентными связывающими веществами в пробах, применяемых при скрининге новых антагонистов рецепторов CGRP. Согласно таким аналитическим вариантам реализации изобретения соединения, описываемые в настоящей заявке, можно применять без модификации или можно модифицировать разными путями; например, путем мечения, такого как ковалентное или нековалентное присоединение химической группы, которая прямо или косвенно обеспечивает детектируемый сигнал. В любой из указанных проб указанные материалы к тому же можно соединять с меткой либо напрямую, либо косвенно. Возможности прямого мечения включают такие группы-метки, как: радиоактивные метки, такие как 125I, ферменты (Патент США №3645090), такие как пероксидаза и щелочная фосфатаза, и флуоресцентные метки (Патент США №3940475), позволяющие отслеживать изменение интенсивности флуоресценции, сдвига длины волны или поляризации флуоресценции. Возможности непрямого мечения включают биотинилирование одного компонента, после которого проводят связывание с авидином, соединенным с одной из перечисленных выше групп-меток. Также указанные соединения могут включать разделители или линкеры в тех случаях, когда соединения требуется присоединить к твердой подложке.
[0143] Спектроскопия на основе ядерно-магнитного резонанса (ЯМР) известна в связи с тем, что она позволяет охарактеризовать макромолекулярные структуры, и она является способом исследования как статических, так и переходных свойств лиганда, связывающегося с молекулой-мишенью (Pellecchia, et al. 2002 Nature Rev Drug Disc 1:211). ЯМР-спектроскопия является полезным инструментом определения связывания лигандов с молекулами-мишенями, и имеет то преимущество, что позволяет определять и количественно оценивать взаимодействия с высокой чувствительностью, не требуя предварительных знаний о функции белка. Кроме того, ЯМР-спектроскопия может предоставить структурную информацию как о мишени, так и о лиганде, что помогает впоследствии оптимизировать импульсы от слабых связей в ориентировках с высоким сродством.
[0144] Способы определения связывания лиганда с биомолекулой-мишенью путем генерирования первого и второго ядерно-магнитно-резонансного корреляционного спектра от биомолекул-мишеней, которые были однородно помечены, описаны в патентах США №5698401 и 5804390. Первый спектр генерируют из данных, полученных для целевого вещества в отсутствии лигандов, а второй спектр - в присутствии одного или более лигандов. Сравнение двух спектров позволяет определять, какие соединения в смеси предполагаемых лигандов связывается с биомолекулой-мишенью.
[0145] Кроме того, на основании способности селективно связываться с рецепторами CGRP пептиды, описываемые в настоящей заявке, можно применять в качестве реагентов для селективного определения рецепторов CGRP в живых клетках, фиксированных клетках, в биологических жидких средах, в гомогенатах тканей, в очищенных, природных биологических материалах и т.д.
[0146] Согласно некоторым вариантам реализации изобретения предложены модифицированные пептидные антагонисты, которые по меньшей мере приблизительно на 60%, в качестве альтернативы по меньшей мере приблизительно на 61%, в качестве альтернативы по меньшей мере приблизительно на 62%, в качестве альтернативы по меньшей мере приблизительно на 63%, в качестве альтернативы по меньшей мере приблизительно на 64%, в качестве альтернативы по меньшей мере приблизительно на 65%, в качестве альтернативы по меньшей мере приблизительно на 66%, в качестве альтернативы по меньшей мере приблизительно на 67%, в качестве альтернативы по меньшей мере приблизительно на 68%, в качестве альтернативы по меньшей мере приблизительно на 69%, в качестве альтернативы по меньшей мере приблизительно на 70%, в качестве альтернативы по меньшей мере приблизительно на 70%, в качестве альтернативы по меньшей мере приблизительно на 71%, в качестве альтернативы по меньшей мере приблизительно на 72%, в качестве альтернативы по меньшей мере приблизительно на 73%, в качестве альтернативы по меньшей мере приблизительно на 74%, в качестве альтернативы по меньшей мере приблизительно на 75%, в качестве альтернативы по меньшей мере приблизительно на 76%, в качестве альтернативы по меньшей мере приблизительно на 77%, в качестве альтернативы по меньшей мере приблизительно на 78%, в качестве альтернативы по меньшей мере приблизительно на 79%, в качестве альтернативы по меньшей мере приблизительно на 80%, в качестве альтернативы по меньшей мере приблизительно на 81%, в качестве альтернативы по меньшей мере приблизительно на 82%, в качестве альтернативы по меньшей мере приблизительно на 83%, в качестве альтернативы по меньшей мере приблизительно на 84%, в качестве альтернативы по меньшей мере приблизительно на 85%, в качестве альтернативы по меньшей мере приблизительно на 86%, в качестве альтернативы по меньшей мере приблизительно на 87%, в качестве альтернативы по меньшей мере приблизительно на 88%, в качестве альтернативы по меньшей мере приблизительно на 89%, в качестве альтернативы по меньшей мере приблизительно на 90%, в качестве альтернативы по меньшей мере приблизительно на 91%, в качестве альтернативы по меньшей мере приблизительно на 92%, в качестве альтернативы по меньшей мере приблизительно на 93%, в качестве альтернативы по меньшей мере приблизительно на 94%, в качестве альтернативы по меньшей мере приблизительно на 95%, в качестве альтернативы по меньшей мере приблизительно на 96%, в качестве альтернативы по меньшей мере приблизительно на 97%, в качестве альтернативы по меньшей мере приблизительно на 98%, в качестве альтернативы по меньшей мере приблизительно на 99% идентичны по последовательности аминокислот полноразмерному пептиду, раскрываемому в настоящей заявке (например, SEQ ID №: 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14 или 15) или любому другому специально оговоренному фрагменту полноразмерного полипептида, раскрываемого в настоящей заявке.
[0147] Термин «процент (%) идентичности последовательностей аминокислот» применительно к последовательностям полипептидов, идентифицируемых в настоящей заявке, относится к проценту остатков аминокислот в кандидатной последовательности, которые идентичны остаткам аминокислот в последовательности специфического полипептида, после выравнивания последовательностей и введения разрывов, при необходимости, для достижения максимального процента идентичности последовательностей и без учета каких-либо консервативных замен как части идентичности последовательности. Выравнивания в целях определения процента идентичности последовательностей аминокислот можно достичь разными путями, которые известны в области техники, например, с применением общедоступных компьютерных программ, таких как BLAST, BLAST-2, ALIGN или Megalign (DNASTAR). Специалисты в данной области техники могут определить соответствующие параметры измерения выравнивания, включая любые алгоритмы, нужные для достижения максимального выравнивания по всей длине последовательностей, которые сравнивают. Однако в целях настоящей заявки значения % идентичности последовательностей аминокислот генерируют с применением компьютерной программы для сравнения последовательностей ALIGN-2, в которой полный исходный код программы для ALIGN-2 доступен и описан в настоящей заявке. Компьютерная программа для сравнения последовательностей ALIGN-2 была разработана компанией «Genentech, Inc.», и исходный код программы был подан в документации для пользователей в Бюро регистрации авторских прав США, Washington D.C., 20559, где он был зарегистрирован под номером свидетельства о регистрации авторских прав США № TXU510087. Программа ALIGN-2 общедоступна от компании «Genentech, Inc.», Южный Сан-Франциско, Калифорния, или может быть скомпилирована по исходному коду программы, представленному в Таблице 4 ниже. Программу ALIGN-2 следует компилировать для применения в операционной системе UNIX, предпочтительно цифровой UNIX V4.0D. Все параметры сравнения последовательностей в программе ALIGN-2 предустановленны, и их нельзя менять.
[0148] В ситуациях, когда для сравнения последовательностей аминокислот применяют программу ALIGN-2, % идентичности последовательностей аминокислот для заданной последовательности аминокислот А и заданной последовательности аминокислот В (что в качестве альтернативы можно сформулировать как заданная последовательность аминокислот А, которая обладает определенным % идентичности последовательности с последовательностью аминокислот В) рассчитывают следующим образом:
100 умножить на отношение X/Y
где X - число остатков аминокислот, засчитанных как идентичные совпадения программой выравнивания последовательностей ALIGN-2, когда программа выравнивает А и В, a Y - общее число остатков аминокислот в В. Следует понимать, что в тех случаях, когда длина последовательности А не равна длине последовательности В, % идентичности последовательностей аминокислот в А и В не будет равен % идентичности последовательностей аминокислот в В и А. В качестве примера, расчет % идентичности последовательностей аминокислот посредством данного способа, продемонстрированного в настоящей заявке, является способом расчета % идентичности последовательностей аминокислот в сравнении с назначенной последовательностью аминокислот. «Пептид сравнения» представляет собой последовательность аминокислот полипептида, относительного которого будут проводить сравнение рассматриваемого полипептида, а «X», «Y» и «Z» представляют собой разные гипотетические остатки аминокислот.
[0149] Если особо не оговаривается другое, все значения % идентичности последовательностей аминокислот, применяемые в настоящей заявке, получены, как описано в предыдущем параграфе, с применением компьютерной программы ALIGN-2. Однако значения % идентичности последовательностей аминокислот также могут быть получены, как описано ниже, с применением компьютерной программы WU-BLAST-2 (Altschul et al., 1996, Methods in Enzymology, 266:460-480). Большинство параметров поиска в программе WU-BLAST-2 установлены как значения «по умолчанию». Параметры, которые не являются значениями «по умолчанию», т.е. регулируемые параметры, устанавливают следующим образом: протяженность перекрывания = 1, доля перекрывания = 0,125, кодовый порог (Т) = 11 и матрица замен = BLOSUM62. Когда применяют WU-BLAST-2, значение % идентичности последовательностей аминокислот определяют путем деления (а) числа совпадающих остатков аминокислот между последовательностью аминокислот рассматриваемого полипептида, имеющего последовательность, полученную из указанного полипептида, и сравнения рассматриваемой последовательности аминокислот, как определено в WU-BLAST-2, на (b) общее число остатков аминокислот в рассматриваемом полипептиде. Например, при утверждении, что полипептид содержит последовательность аминокислот А, которая по меньшей мере на 80% идентична последовательности аминокислот В, последовательность аминокислот А является последовательностью аминокислот сравнения, а последовательность аминокислот В является последовательностью аминокислот в рассматриваемом полипептиде.
[0150] Также процент идентичности последовательностей аминокислот можно определить при помощи программы для сравнения последовательностей NCBI-BLAST2 (Altschul et al., 1997, Nucleic Acids Res., 25:3389-3402). Программу для сравнения последовательностей можно загрузить или другим путем получить от Национального института здоровья, Вифезда, Мериленд. В программе NCBI-BLAST2 применяются несколько параметров поиска, причем все из них установлены как значения «по умолчанию», включая, например, немаскируемые сегменты = да, нить = все, ожидаемая частотность = 10, минимальная длина низкой сложности = 15/5, многовводное е-значение = 0,01, константа для многих входов = 25, сокращение конечного выравнивания с пропусками = 25 и матрица замен = BLOSUM62.
[0151] В случаях, когда для сравнения последовательностей аминокислот применяют NCBI-BLAST2, % идентичности последовательностей аминокислот между заданной последовательностью аминокислот А и заданной последовательностью аминокислот В (что в качестве альтернативы можно сформулировать как заданная последовательность аминокислот А, которая обладает определенным % идентичности последовательности с последовательностью аминокислот В) рассчитывают следующим образом:
100 умножить на отношение X/Y
[0152] где X - число остатков аминокислот, засчитанных как идентичные совпадения программой выравнивания последовательностей NCBI-BLAST2 при выравнивании в программе А и В, a Y - общее число остатков аминокислот в В. Следует понимать, что в тех случаях, когда длина последовательности А не равна длине последовательности В, % идентичности последовательностей аминокислот в А и В не будет равен % идентичности последовательностей аминокислот в В и А.
[0153] Вариации в последовательности пептидов-антагонистов, описываемых в настоящей заявке, можно получить, например, с применением любого из способов и рекомендаций по консервативным и неконсервативным мутациям, перечисленным, например, в патенте США №5364934 (Drayna et al., выдан 15 ноября 1994 г.), который включен в настоящую заявку посредством ссылки на его полную версию. Вариации могут включать замену, делецию или вставку одного или более кодонов, кодирующих пептиды-антагонисты, которые приводят к изменению последовательности аминокислот указанных пептидов-антагонистов по сравнению с пептидами-антагонистами с эталонной последовательностью. Вариации могут соответствовать тем, которые приведены в Таблице 3, приведенной выше.
ПРИМЕРЫ
[0154] Следующие примеры приведены с целью дальнейшей иллюстрации актуальных вариантов реализации изобретения. Они не предназначены для того, чтобы ограничить область вариантов реализации изобретения.
ПРИМЕР 1
[0155] При помощи анализа потока кальция определяли дозозависимую ингибиторную реакцию пептидов-антагонистов согласно настоящему изобретению на рецептор амилина, AMY1, комплекс рецептор СТ (кальцитонина)/RAMP1 (рецептор-модифицирующий активность). В данной пробе применяли линию рекомбинантных клеток CHO-K1/AMY1/Gα15 («GenScript», Пискатауэй, Нью-Джерси, каталожный номер М00475). Активность пептидов-антагонистов определяли по два раза в пяти разных концентрациях, начиная с 1 мкМ и с последовательными пятикратными разведениями, в DMSO (диметилсульфоксид). АС187 (SEQ ID №: 55) - известный антагонист рецептора амилина (поставляемый, например, компанией «Tocris Biosciences Миннеаполис, Миннесота, каталожный номер 3419) применяли в данной пробе в качестве положительного контроля, а α-CGRP человека - известный агонист рецептора амилина (SEQ ID №:56) (поставляемый, например, компанией «Tocris Bioscience», Миннеаполис, Миннесота, каталожный номер 3012) применяли в данной пробе в качестве положительного контроля. Применяли набор для анализа FLIPR® Calcium 4 («Molecular Devices», Саннивейл, Калифорния).
Контрольные реагенты
[0156] В качестве отрицательного и положительного контроля применяли, соответственно, α-CGRP человека с последовательностью аминокислот NH2-ACDTATCVTHRLAGLLSRSGGVVKNNFVPTNVGSKAF-NH2 (SEQ ID №:56) и AC187 с последовательностью VLGKLSQELHKLQTYPRTNTGSNTY (SEQ ID №:55). Матричные растворы дополнительно разводили в буфере HBSS-HEPES, получая 5 × конечных контрольных растворов.
Другие реагенты
[0157] В Таблице 5 перечислены дополнительные материалы, применяемые при проведении проб.

Подготовка культур клеток
[0158] Клетки CHO-K1/AMY1/Gα15 засевали в лунки планшета на 384 лунки с черными стенками и прозрачным дном с плотностью 20000 клеток на 20 мкл питательной среды за 20 часов до проведения эксперимента и хранили их при 37°С/5% СО2.
Протокол анализа
[0159] По протоколу, прилагаемому к набору для анализа, в планшет для анализа добавляли раствор для загрузки красителя (2 × конечную концентрацию), 20 мкл на лунку. В планшет для анализа добавляли раствор соединения (5 х конечную концентрацию), 10 мкл на лунку. Планшет для анализа помещали в инкубатор с 37°С на 1 час, затем оставляли на 15 мин при комнатной температуре. Планшет с агонистом (при 5 × ЕС80 концентрации) помещали в Источник 2. Общее время считывания составляло 120 с. Агонист добавляли спустя 20 секунд после считывания исходного уровня, и захватывали сигнал флуоресценции еще в течение 100 секунд (21 с - 120 с).
[0160] При скрининге клетки, простимулированные буфером для анализа, выбирали в качестве фона; клетки, простимулированные агонистом (при концентрации ЕС90) выбирали в качестве отрицательного контроля. Анализ данных
[0161] Данные регистрировали при помощи Screen Works (версия 3.1) в виде файлов FMD и хранили в компьютерной сети «GenScript» для анализа в автономном режиме. Захват данных и анализ проводили при помощи программы Screen Works (версия 3.1) и экспортировали в Excel. В качестве считывания исходного уровня рассчитывали среднее значение из 20 (1 с - 20 с), и относительные единицы интенсивности флуоресценции (ΔОЕФ) рассчитывали, вычитая из среднего значения исходного уровня максимальные единицы флуоресценции (214 с - 120 с).
[0162] Значение IC50 для АС 187 было 8 мкМ. Ингибиторную активность тестируемых пептидов-антагонистов нормировали на отрицательный контроль (АС 187) и приводили как % ингибирования, рассчитанный по следующей формуле:
% ингибирования = (ΔОЕФсоединсния - ΔОЕФфона)/(ΔОЕФотрицательного контроля - ΔОЕФфона) * 100%
Затем ингибирование откладывали на графике по оси Y, а тестируемую концентрацию - по оси X. Применяемые пептиды перечислены в Таблице 1, приведенной выше. Результаты указанных экспериментов представлены в Таблице 6, а результаты для положительного контроля перечислены в Таблице 7, приведенной ниже. Указанные пептиды тестировали при указанных концентрациях, и стимуляцию проводили при 32 нМ (EC80) α-CGRP (среднее +/- СО (стандартное отклонение), n=2). Указанные результаты демонстрируют неожиданно высокую активность избранных пептидов, например, многие из них демонстрируют концентрации IC50 в нижнем наномолярном диапазоне по сравнению с концентрацией IC50 в нижнем микромолярном диапазоне положительного контроля - АС187.
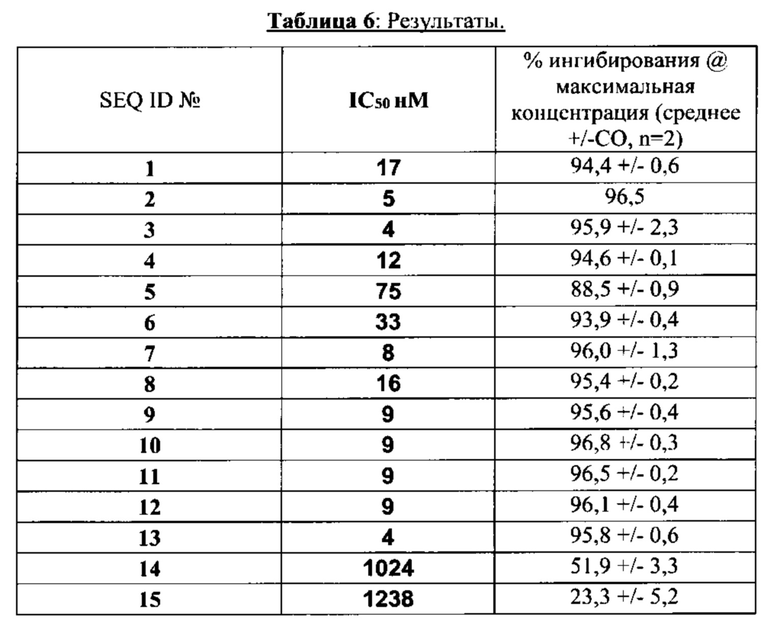

[0163] В то время как варианты реализации настоящего изобретения были описаны подробно в целях прояснения и понимания, специалисты в данной области техники должны понимать, что можно вносить разные изменения по форме и детали, не отклоняясь от настоящего объема вариантов реализации. Все рисунки, таблицы и приложения, а также патенты, заявки и публикации, на которые здесь даны ссылки, таким образом, включены в настоящую заявку посредством ссылки на их полные версии.
--->
ПЕРЕЧЕНЬ ПОСЛЕДОВАТЕЛЬНОСТЕЙ
<110> СОАРЕС, Кристофер Джозеф
<120> ПЕПТИДНЫЕ АНТАГОНИСТЫ ПЕПТИДНЫХ ГОРМОНОВ ИЗ СЕМЕЙСТВА КАЛЬЦИТОНИНА (CGRP) И ИХ ПРИМЕНЕНИЕ
<130> CSOAR.001C2
<140> 15/478053
<141> 2017-04-03
<150> 14/948,032
<151> 2015-11-20
<150> 13/821,936
<151> 2013-03-08
<150> PCT/US2013/023260
<151> 2013-01-25
<150> 61/591236
<151> 2012-01-26
<160> 63
<170> FastSEQ for Windows Version 4.0
<210> 1
<211> 32
<212> PRT
<213> Artificial Sequence
<220>
<223> Synthetic peptide
<400> 1
Ala Cys Asp Thr Ala Ala Cys Val Leu Gly Arg Leu Ser Gln Glu Leu
1 5 10 15
His Arg Leu Gln Thr Tyr Pro Arg Thr Asn Val Gly Ser Lys Ala Phe
20 25 30
<210> 2
<211> 32
<212> PRT
<213> Artificial Sequence
<220>
<223> Synthetic peptide
<400> 2
Ala Cys Asp Thr Ala Ser Cys Val Leu Gly Arg Leu Ser Gln Glu Leu
1 5 10 15
His Arg Leu Gln Thr Tyr Pro Arg Thr Asn Val Gly Ser Lys Ala Phe
20 25 30
<210> 3
<211> 32
<212> PRT
<213> Artificial Sequence
<220>
<223> Synthetic peptide
<400> 3
Ala Cys Asp Thr Ala Val Cys Val Leu Gly Arg Leu Ser Gln Glu Leu
1 5 10 15
His Arg Leu Gln Thr Tyr Pro Arg Thr Asn Val Gly Ser Lys Ala Phe
20 25 30
<210> 4
<211> 32
<212> PRT
<213> Artificial Sequence
<220>
<223> Synthetic peptide
<400> 4
Ala Cys Asn Thr Ala Ala Cys Val Leu Gly Arg Leu Ser Gln Glu Leu
1 5 10 15
His Arg Leu Gln Thr Tyr Pro Arg Thr Asn Val Gly Ser Lys Ala Phe
20 25 30
<210> 5
<211> 32
<212> PRT
<213> Artificial Sequence
<220>
<223> Synthetic peptide
<400> 5
Ala Cys Val Leu Gly Ala Cys Val Leu Gly Arg Leu Ser Gln Glu Leu
1 5 10 15
His Arg Leu Gln Thr Tyr Pro Arg Thr Asn Val Gly Ser Lys Ala Phe
20 25 30
<210> 6
<211> 32
<212> PRT
<213> Artificial Sequence
<220>
<223> Synthetic peptide
<400> 6
Ala Cys Arg Phe Gly Ala Cys Val Leu Gly Arg Leu Ser Gln Glu Leu
1 5 10 15
His Arg Leu Gln Thr Tyr Pro Arg Thr Asn Val Gly Ser Lys Ala Phe
20 25 30
<210> 7
<211> 32
<212> PRT
<213> Artificial Sequence
<220>
<223> Synthetic peptide
<400> 7
Ala Cys Asn Leu Ser Ala Cys Val Leu Gly Arg Leu Ser Gln Glu Leu
1 5 10 15
His Arg Leu Gln Thr Tyr Pro Arg Thr Asn Val Gly Ser Lys Ala Phe
20 25 30
<210> 8
<211> 32
<212> PRT
<213> Artificial Sequence
<220>
<223> Synthetic peptide
<400> 8
Cys Ser Asn Thr Ala Ala Cys Val Leu Gly Arg Leu Ser Gln Glu Leu
1 5 10 15
His Arg Leu Gln Thr Tyr Pro Arg Thr Asn Val Gly Ser Lys Ala Phe
20 25 30
<210> 9
<211> 32
<212> PRT
<213> Artificial Sequence
<220>
<223> Synthetic peptide
<400> 9
Ala Cys Asp Thr Ala Leu Cys Val Leu Gly Arg Leu Ser Gln Glu Leu
1 5 10 15
His Arg Leu Gln Thr Tyr Pro Arg Thr Asn Val Gly Ser Lys Ala Phe
20 25 30
<210> 10
<211> 32
<212> PRT
<213> Artificial Sequence
<220>
<223> Synthetic peptide
<400> 10
Ala Cys Asp Thr Ala Ile Cys Val Leu Gly Arg Leu Ser Gln Glu Leu
1 5 10 15
His Arg Leu Gln Thr Tyr Pro Arg Thr Asn Val Gly Ser Lys Ala Phe
20 25 30
<210> 11
<211> 32
<212> PRT
<213> Artificial Sequence
<220>
<223> Synthetic peptide
<400> 11
Ala Cys Asn Leu Ser Val Cys Val Leu Gly Arg Leu Ser Gln Glu Leu
1 5 10 15
His Arg Leu Gln Thr Tyr Pro Arg Thr Asn Val Gly Ser Lys Ala Phe
20 25 30
<210> 12
<211> 32
<212> PRT
<213> Artificial Sequence
<220>
<223> Synthetic peptide
<400> 12
Cys Ser Asn Thr Ala Val Cys Val Leu Gly Arg Leu Ser Gln Glu Leu
1 5 10 15
His Arg Leu Gln Thr Tyr Pro Arg Thr Asn Val Gly Ser Lys Ala Phe
20 25 30
<210> 13
<211> 31
<212> PRT
<213> Artificial Sequence
<220>
<223> Synthetic Peptide
<400> 13
Ala Cys Asn Leu Ser Ala Cys Val Leu Gly Arg Leu Ser Gln Glu Leu
1 5 10 15
His Arg Leu Gln Thr Tyr Pro Thr Asn Thr Gly Ser Gly Thr Pro
20 25 30
<210> 14
<211> 32
<212> PRT
<213> Artificial Sequence
<220>
<223> Synthetic Peptide
<400> 14
Ala Cys Val Leu Gly Ala Cys Val Leu Gly Arg Leu Ser Gln Glu Leu
1 5 10 15
His Arg Leu Gln Thr Tyr Pro Val Asp Pro Ser Ser Pro His Ser Tyr
20 25 30
<210> 15
<211> 37
<212> PRT
<213> Synthetic Peptide
<400> 15
Ala Cys Asp Thr Ala Ala Cys Val Thr His Arg Leu Ala Gly Leu Leu
1 5 10 15
Ser Arg Ser Gly Gly Val Val Lys Asn Asn Phe Val Pro Thr Asn Val
20 25 30
Gly Ser Lys Ala Phe
35
<210> 16
<211> 7
<212> PRT
<213> Artificial Sequence
<220>
<223> Synthetic peptide
<220>
<221> VARIANT
<222> (1)...(1)
<223> Xaa is selected from the group consisting of Ala,
Cys, Gly, Ile, Leu, Met, Phe, Pro, Typ, and Val
<220>
<221> VARIANT
<222> (2)...(2)
<223> Xaa is selected from the group consisting of Cys,
Ser, and Tyr
<220>
<221> VARIANT
<222> (3)...(3)
<223> Xaa is selected from the group consisting of Arg,
Asn, Asp, Cys, Glu, Gln, His, Lys, Ser, Thr, Tyr,
and Val
<220>
<221> VARIANT
<222> (4)...(4)
<223> Xaa is selected from the group consisting of Arg,
Asn, Asp, Cys, Glu, Gln, His, Leu, Lys, Phe, Ser,
Thr, Tyr, and Val
<220>
<221> VARIANT
<222> (5)...(5)
<223> Xaa is selected from the group consisting of Ala,
Gly, Ile, Leu, Met, Phe, Ser, Typ, and Val
<220>
<221> VARIANT
<222> (6)...(6)
<223> Xaa is selected from the group consisting of Ala,
Cys, Asp, Glu, Phe, Gly, His, Ile, Lys, Leu, Met,
Asn, Pro, Gln, Arg, Ser, Val, Trp, and Tyr
<400> 16
Xaa Xaa Xaa Xaa Xaa Xaa Cys
1 5
<210> 17
<211> 7
<212> PRT
<213> Artificial Sequence
<220>
<223> Synthetic peptide
<400> 17
Ala Cys Asp Thr Ala Ala Cys
1 5
<210> 18
<211> 7
<212> PRT
<213> Artificial Sequence
<220>
<223> Synthetic peptide
<400> 18
Ala Cys Asp Thr Ala Ser Cys
1 5
<210> 19
<211> 7
<212> PRT
<213> Artificial Sequence
<220>
<223> Synthetic peptide
<400> 19
Ala Cys Asp Thr Ala Val Cys
1 5
<210> 20
<211> 7
<212> PRT
<213> Artificial Sequence
<220>
<223> Synthetic peptide
<400> 20
Ala Cys Asn Thr Ala Ala Cys
1 5
<210> 21
<211> 7
<212> PRT
<213> Artificial Sequence
<220>
<223> Synthetic peptide
<400> 21
Ala Cys Val Leu Gly Ala Cys
1 5
<210> 22
<211> 7
<212> PRT
<213> Artificial Sequence
<220>
<223> Synthetic peptide
<400> 22
Ala Cys Arg Phe Gly Ala Cys
1 5
<210> 23
<211> 7
<212> PRT
<213> Artificial Sequence
<220>
<223> Synthetic peptide
<400> 23
Ala Cys Asp Leu Ser Ala Cys
1 5
<210> 24
<211> 7
<212> PRT
<213> Artificial Sequence
<220>
<223> Synthetic peptide
<400> 24
Ala Cys Asn Leu Ser Ala Cys
1 5
<210> 25
<211> 7
<212> PRT
<213> Artificial Sequence
<220>
<223> Synthetic Peptide
<400> 25
Cys Ser Asn Thr Ala Ala Cys
1 5
<210> 26
<211> 7
<212> PRT
<213> Artificial Sequence
<220>
<223> Synthetic peptide
<400> 26
Ala Cys Asp Thr Ala Leu Cys
1 5
<210> 27
<211> 7
<212> PRT
<213> Artificial Sequence
<220>
<223> Synthetic peptide
<400> 27
Ala Cys Asp Thr Ala Ile Cys
1 5
<210> 28
<211> 7
<212> PRT
<213> Artificial Sequence
<220>
<223> Synthetic peptide
<400> 28
Ala Cys Asp Thr Ala Leu Cys
1 5
<210> 29
<211> 7
<212> PRT
<213> Artificial Sequence
<220>
<223> Synthetic peptide
<400> 29
Ala Cys Asp Thr Ala Ile Cys
1 5
<210> 30
<211> 7
<212> PRT
<213> Artificial Sequence
<220>
<223> Synthetic peptide
<400> 30
Ala Cys Asp Leu Ser Val Cys
1 5
<210> 31
<211> 7
<212> PRT
<213> Artificial Sequence
<220>
<223> Synthetic peptide
<400> 31
Ala Cys Asn Leu Ser Val Cys
1 5
<210> 32
<211> 7
<212> PRT
<213> Artificial Sequence
<220>
<223> Synthetic Peptide
<400> 32
Ala Cys Asn Leu Ser Val Cys
1 5
<210> 33
<211> 7
<212> PRT
<213> Artificial Sequence
<220>
<223> Synthetic peptide
<400> 33
Cys Ser Asn Thr Ala Val Cys
1 5
<210> 34
<211> 19
<212> PRT
<213> Artificial Sequence
<220>
<223> Synthetic peptide
<400> 34
Val Leu Gly Arg Leu Ser Gln Glu Leu His Arg Leu Gln Thr Tyr Pro
1 5 10 15
Arg Thr Asn
<210> 35
<211> 19
<212> PRT
<213> Artificial Sequence
<220>
<223> Synthetic peptide
<400> 35
Val Leu Gly Lys Leu Ser Gln Glu Leu His Lys Leu Gln Thr Tyr Pro
1 5 10 15
Arg Thr Asn
<210> 36
<211> 19
<212> PRT
<213> Artificial Sequence
<220>
<223> Synthetic Peptide
<400> 36
Val Leu Gly Lys Leu Ser Gln Glu Leu His Lys Leu Gln Thr Tyr Pro
1 5 10 15
Arg Thr Asn
<210> 37
<211> 19
<212> PRT
<213> Artificial Sequence
<220>
<223> Synthetic Peptide
<400> 37
Val Leu Gly Lys Leu Ser Gln Glu Leu His Lys Leu Gln Thr Tyr Pro
1 5 10 15
Arg Thr Asp
<210> 38
<211> 19
<212> PRT
<213> Artificial Sequence
<220>
<223> Synthetic Peptide
<400> 38
Val Leu Gly Lys Leu Ser Gln Glu Leu His Lys Leu Gln Thr Phe Pro
1 5 10 15
Arg Thr Asn
<210> 39
<211> 19
<212> PRT
<213> Artificial Sequence
<220>
<223> Synthetic Peptide
<400> 39
Val Leu Gly Lys Leu Ser Gln Asp Ile His Lys Leu Gln Thr Tyr Pro
1 5 10 15
Arg Thr Asn
<210> 40
<211> 19
<212> PRT
<213> Artificial Sequence
<220>
<223> Synthetic Peptide
<400> 40
Val Leu Gly Lys Leu Ser Gln Glu Leu His Lys Met Gln Thr Tyr Pro
1 5 10 15
Arg Thr Asp
<210> 41
<211> 19
<212> PRT
<213> Artificial Sequence
<220>
<223> Synthetic Peptide
<400> 41
Leu Leu Gly Lys Leu Ser Gln Glu Leu His Arg Leu Gln Thr Tyr Thr
1 5 10 15
Arg Thr Asp
<210> 42
<211> 19
<212> PRT
<213> Artificial Sequence
<220>
<223> Synthetic Peptide
<400> 42
Val Leu Gly Lys Leu Ser Gln Asp Leu His Lys Leu Gln Thr Phe Pro
1 5 10 15
Arg Thr Asp
<210> 43
<211> 19
<212> PRT
<213> Artificial Sequence
<220>
<223> Synthetic Peptide
<400> 43
Met Leu Gly Lys Leu Ser Gln Asp Leu His Lys Leu Gln Thr Phe Pro
1 5 10 15
Arg Thr Asp
<210> 44
<211> 19
<212> PRT
<213> Artificial Sequence
<220>
<223> Synthetic Peptide
<400> 44
Val Leu Gly Lys Leu Ser Gln Asp Ile His Lys Leu Gln Thr His Pro
1 5 10 15
Arg Thr Asp
<210> 45
<211> 6
<212> PRT
<213> Artificial Sequence
<220>
<223> Synthetic peptide
<220>
<221> VARIANT
<222> (1)...(1)
<223> Xaa is selected from the group consisting of Ala,
Gly, Ile, leu, Met, Phe, Pro, Trp, and Val
<220>
<221> VARIANT
<222> (2)...(2)
<223> Xaa is selected from the group consisting of Ala,
Gly, Ile, leu, Met, Phe, Pro, Trp, and Val
<220>
<221> VARIANT
<222> (3)...(3)
<223> Xaa is selected from the group consisting of Cys,
Ser, and Tyr
<220>
<221> VARIANT
<222> (4)...(4)
<223> Xaa is selected from the group consisting of Arg,
Asn, Asp, Glu, Gln, His, Lys, Ser, Thr, and Tyr
<220>
<221> VARIANT
<222> (5)...(5)
<223> Xaa is selected from the group consisting of Ala,
Gly, Ile, leu, Met, Phe, Pro, Trp, and Val
<220>
<221> VARIANT
<222> (6)...(6)
<223> Xaa is selected from the group consisting of Ala,
Gly, Ile, leu, Met, Phe, Pro, Trp, and Val
<400> 45
Xaa Xaa Xaa Xaa Xaa Xaa
1 5
<210> 46
<211> 6
<212> PRT
<213> Artificial Sequence
<220>
<223> Synthetic peptide
<400> 46
Val Gly Ser Lys Ala Phe
1 5
<210> 47
<211> 6
<212> PRT
<213> Artificial Sequence
<220>
<223> Synthetic Peptide
<400> 47
Val Gly Ser Lys Ala Phe
1 5
<210> 48
<211> 19
<212> PRT
<213> Artificial Sequence
<220>
<223> Synthetic peptide
<400> 48
Val Leu Gly Arg Leu Ser Gln Glu Leu His Arg Leu Gln Thr Tyr Pro
1 5 10 15
Arg Thr Asn
<210> 49
<211> 19
<212> PRT
<213> Artificial Sequence
<220>
<223> Synthetic Peptide
<400> 49
Val Leu Gly Lys Leu Ser Gln Glu Leu His Lys Leu Gln Thr Tyr Pro
1 5 10 15
Arg Thr Asn
<210> 50
<211> 7
<212> PRT
<213> Artificial Sequence
<220>
<223> Synthetic peptide
<220>
<221> VARIANT
<222> (6)...(6)
<223> Xaa = any amino acid residue other than Thr
<400> 50
Ala Cys Asp Thr Ala Xaa Cys
1 5
<210> 51
<211> 17
<212> PRT
<213> Artificial Sequence
<220>
<223> Synthetic peptide
<400> 51
Ala Glu Ala Ala Ala Lys Glu Ala Ala Ala Lys Glu Ala Ala Ala Lys
1 5 10 15
Ala
<210> 52
<211> 17
<212> PRT
<213> Artificial Sequence
<220>
<223> Synthetic peptide
<400> 52
Ala Lys Ala Ala Ala Glu Lys Ala Ala Ala Glu Lys Ala Ala Ala Glu
1 5 10 15
Ala
<210> 53
<211> 16
<212> PRT
<213> Artificial Sequence
<220>
<223> Synthetic peptide
<400> 53
Ala Glu Ala Ala Lys Ala Glu Ala Ala Lys Ala Glu Ala Ala Lys Ala
1 5 10 15
<210> 54
<211> 16
<212> PRT
<213> Artificial Sequence
<220>
<223> Synthetic peptide
<400> 54
Ala Lys Ala Ala Glu Ala Lys Ala Ala Glu Ala Lys Ala Ala Glu Ala
1 5 10 15
<210> 55
<211> 25
<212> PRT
<213> Artificial Sequence
<220>
<223> Synthetic peptide
<400> 55
Val Leu Gly Lys Leu Ser Gln Glu Leu His Lys Leu Gln Thr Tyr Pro
1 5 10 15
Arg Thr Asn Thr Gly Ser Asn Thr Tyr
20 25
<210> 56
<211> 37
<212> PRT
<213> Artificial Sequence
<220>
<223> Synthetic peptide
<400> 56
Ala Cys Asp Thr Ala Thr Cys Val Thr His Arg Leu Ala Gly Leu Leu
1 5 10 15
Ser Arg Ser Gly Gly Val Val Lys Asn Asn Phe Val Pro Thr Asn Val
20 25 30
Gly Ser Lys Ala Phe
35
<210> 57
<211> 32
<212> PRT
<213> Artificial Sequence
<220>
<223> Synthetic Peptide
<400> 57
Ala Cys Asp Leu Ser Ala Cys Val Leu Gly Arg Leu Ser Gln Glu Leu
1 5 10 15
His Arg Leu Gln Thr Tyr Pro Arg Thr Asn Val Gly Ser Lys Ala Phe
20 25 30
<210> 58
<211> 32
<212> PRT
<213> Artificial Sequence
<220>
<223> Synthetic Peptide
<400> 58
Ala Cys Asp Leu Ser Val Cys Val Leu Gly Arg Leu Ser Gln Glu Leu
1 5 10 15
His Arg Leu Gln Thr Tyr Pro Arg Thr Asn Val Gly Ser Lys Ala Phe
20 25 30
<210> 59
<211> 18
<212> PRT
<213> Artificial Sequence
<220>
<223> Synthetic Peptide
<400> 59
Val Leu Gly Arg Leu Ser Gln Glu Leu His Arg Leu Gln Thr Tyr Pro
1 5 10 15
Thr Asn
<210> 60
<211> 18
<212> PRT
<213> Artificial Sequence
<220>
<223> Synthetic Peptide
<400> 60
Val Leu Gly Arg Leu Ser Gln Glu Leu His Arg Leu Gln Thr Tyr Pro
1 5 10 15
Val Asp
<210> 61
<211> 24
<212> PRT
<213> Artificial Sequence
<220>
<223> Synthetic Peptide
<400> 61
Val Thr His Arg Leu Ala Gly Leu Leu Ser Arg Ser Gly Gly Val Val
1 5 10 15
Lys Asn Asn Phe Val Pro Thr Asn
20
<210> 62
<211> 6
<212> PRT
<213> Artificial Sequence
<220>
<223> Synthetic Peptide
<400> 62
Thr Gly Ser Gly Thr Pro
1 5
<210> 63
<211> 7
<212> PRT
<213> Artificial Sequence
<220>
<223> Synthetic Peptide
<400> 63
Pro Ser Ser Pro His Ser Tyr
1 5
<---
| название | год | авторы | номер документа |
|---|---|---|---|
| ПЕПТИДНЫЕ АНТАГОНИСТЫ ПЕПТИДНЫХ ГОРМОНОВ ИЗ СЕМЕЙСТВА КАЛЬЦИТОНИНА (КАЛЬЦИТОНИН ГЕН-РОДСТВЕННЫХ ПЕПТИДОВ (CGRP)) И ИХ ПРИМЕНЕНИЕ | 2013 |
|
RU2624016C2 |
| СПОСОБЫ И КОМПОЗИЦИИ ДЛЯ ЛЕЧЕНИЯ ЖЕЛУДОЧНО-КИШЕЧНЫХ РАССТРОЙСТВ | 2014 |
|
RU2763796C2 |
| РЕКОМБИНАНТНЫЕ ВИРУСНЫЕ ЧАСТИЦЫ С МОДИФИЦИРОВАННЫМ ТРОПИЗМОМ И ПУТИ ИХ ПРИМЕНЕНИЯ ДЛЯ НАЦЕЛЕННОГО ВВЕДЕНИЯ ГЕНЕТИЧЕСКОГО МАТЕРИАЛА В КЛЕТКИ ЧЕЛОВЕКА | 2018 |
|
RU2811426C2 |
| ОТБОР ПАЦИЕНТОВ С ГОЛОВНОЙ БОЛЬЮ, ВОСПРИИМЧИВЫХ К АНТИТЕЛАМ, НАПРАВЛЕННЫМ ПРОТИВ КАЛЬЦИТОНИН ГЕН-РОДСТВЕННОГО ПЕПТИДА | 2018 |
|
RU2770066C2 |
| СПОСОБЫ И КОМПОЗИЦИИ ДЛЯ ЛЕЧЕНИЯ ЖЕЛУДОЧНО-КИШЕЧНЫХ РАССТРОЙСТВ | 2004 |
|
RU2543350C2 |
| СПОСОБЫ И КОМПОЗИЦИИ ДЛЯ ЛЕЧЕНИЯ ЖЕЛУДОЧНО-КИШЕЧНЫХ РАССТРОЙСТВ | 2004 |
|
RU2353383C2 |
| МЕТОДЫ ПРОФИЛАКТИКИ И ЛЕЧЕНИЯ СЕРДЕЧНЫХ ЗАБОЛЕВАНИЙ | 2018 |
|
RU2830232C2 |
| Т-КЛЕТКИ С КОСТИМУЛИРУЮЩИМ ХИМЕРНЫМ АНТИГЕННЫМ РЕЦЕПТОРОМ, НАЦЕЛЕННЫЕ НА IL13Rα2 | 2015 |
|
RU2749922C2 |
| АНТИТЕЛА К PAC1 И ВАРИАНТЫ ИХ ПРИМЕНЕНИЯ | 2019 |
|
RU2781553C2 |
| АНТИТЕЛА С ПРИВИТЫМ МОЗГОВЫМ НАТРИЙУРЕТИЧЕСКИМ ПЕПТИДОМ | 2019 |
|
RU2839970C2 |
Изобретение относится к области биотехнологии, конкретно к пептидным антагонистам связанного с геном кальцитонина пептида (CGRP), и может быть использовано в медицине для лечения состояния, связанного с повышенным уровнем CGRP, в том числе мигрени. Предложен пептид, имеющий структуру X1- Y1- Z1, где X1 является модифицированным N-концевым фрагментом CGRP, содержащим от 5 до 7 остатков аминокислот, где 2 аминокислоты указанного N-концевого фрагмента представляют собой остатки цистеина (Cys), при этом С-концевой остаток аминокислоты N-концевого фрагмента является Cys, остаток, расположенный непосредственно перед С-концевым остатком Cys указанного N-концевого фрагмента, является заменой остатка треонина (Thr) на не треонин, причем два остатка цистеина разделены 4, 5 или 6 аминокислотами и при этом указанные 2 остатка Cys могут образовывать дисульфидную связь; Y1 выбран из Val-Leu-Gly-Arg-Leu-Ser-Gln-Glu-Leu-His-Arg-Leu-Gln-Thr-Tyr-Pro-Thr-Asn, Val-Leu-Gly-Arg-Leu-Ser-Gln-Glu-Leu-His-Arg-Leu-Gln-Thr-Tyr-Pro-Val-Asp; a Z1 выбран из Thr-Gly-Ser-Gly-Thr-Pro-NH2, Pro-Ser-Ser-Pro-His-Ser-Tyr-NH2 или Val-Gly-Ser-Lys-Ala-Phe-NH2. Изобретение обеспечивает получение эффективного антагониста CGRP. 3 н. и 10 з.п. ф-лы, 7 табл., 1 пр.
1. Антагонист на основе связанного с геном кальцитонина пептида или его фармацевтически приемлемая соль, причем указанный антагонист имеет структуру согласно формуле I:
где: 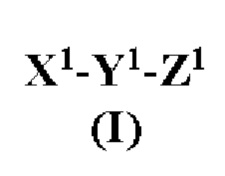
X1 является модифицированным N-концевым фрагментом связанного с геном кальцитонина пептида, содержащим от пяти до семи остатков аминокислот, где две аминокислоты указанного N-концевого фрагмента представляют собой остатки цистеина (Cys), при этом С-концевой остаток аминокислоты N-концевого фрагмента является цистеином (Cys), при этом остаток, расположенный непосредственно перед С-концевым остатком Cys указанного N-концевого фрагмента, является заменой остатка треонина (Thr) на не треонин, причем два остатка цистеина разделены четырьмя, пятью или шестью аминокислотами и при этом указанные два остатка цистеина могут образовывать дисульфидную связь;
Y1 выбран из группы, состоящей из Val-Leu-Gly-Arg-Leu-Ser-Gln-Glu-Leu-His-Arg-Leu-Gln-Thr-Tyr-Pro-Thr-Asn (SEQ ID NO: 59), Val-Leu-Gly-Arg-Leu-Ser-Gln-Glu-Leu-His-Arg-Leu-Gln-Thr-Tyr-Pro-Val-Asp (SEQ ID NO: 60); и
Z1 выбран из группы, состоящей из Thr-Gly-Ser-Gly-Thr-Pro-NH2 (SEQ ID NO: 62), Pro-Ser-Ser-Pro-His-Ser-Tyr-NH2 (SEQ ID NO: 63) и Val-Gly-Ser-Lys-Ala-Phe-NH2 (SEQ ID NO: 46).
2. Антагонист по п. 1, отличающийся тем, что X1 содержит NH2-Ala-Cys-X1-X2-X3-X4-Cys (SEQ ID NO: 64), причем X1 представляет собой Asp, Asn, Val или Arg, X2 представляет собой Thr, Leu или Phe, Х3 представляет собой Ala, Gly или Ser и X4 представляет собой Ala, Leu, Ile или Val.
3. Антагонист по п. 2, отличающийся тем, что X1 выбран из группы, состоящей из NH2-Ala-Cys-Asp-Thr-Ala-Ala-Cys- (SEQ ID NO: 17), NH2-Ala-Cys-Asp-Thr-Ala-Ser-Cys- (SEQ ID NO: 18), NH2-Ala-Cys-Asp-Thr-Ala-Val-Cys- (SEQ ID NO: 19), NH2-Ala-Cys-Asn-Thr-Ala-Ala-Cys- (SEQ ID NO: 20), NH2-Ala-Cys-Val-Leu-Gly-Ala-Cys- (SEQ ID NO: 21), NH2-Ala-Cys-Arg-Phe-Gly-Ala-Cys- (SEQ ID NO: 22), NH2-Ala-Cys-Asp-Leu-Ser-Ala-Cys- (SEQ ID NO: 23), NH2-Ala-Cys-Asn-Leu-Ser-Ala-Cys- (SEQ ID NO: 24), NH2-Cys-Ser-Asn-Thr-Ala-Ala-Cys- (SEQ Ш NO: 25), NH2-Ala-Cys-Asp-Thr-Ala-Leu-Cys- (SEQ ID NO: 26), NH2-Ala-Cys-Asp-Thr-Ala-Ile-Cys- (SEQ ID NO: 27), NH2-Ala-Cys-Asp-Thr-Ala-Leu-Cys- (SEQ ID NO: 28), NH2-Ala-Cys-Asp-Thr-Ala-Ile-Cys- (SEQ ID NO: 29), NH2-Ala-Cys-Asp-Leu-Ser-Val-Cys-(SEQ ID NO: 30), NH2-Ala-Cys-Asn-Leu-Ser-Val-Cys- (SEQ ID NO: 32) и NH2-Cys-Ser-Asn-Thr-Ala-Val-Cys- (SEQ ID NO: 33).
4. Антагонист по п. 3, отличающийся тем, что Y1 представляет собой Val-Leu-Gly-Arg-Leu-Ser-Gln-Glu-Leu-His-Arg-Leu-Gln-Thr-Tyr-Pro-Thr-Asn (SEQ ID NO: 59).
5. Антагонист по п. 4, отличающийся тем, что Z1 представляет собой Thr-Gly-Ser-Gly-Thr-Pro-NH2 (SEQ ID NO: 62).
6. Антагонист по п. 3, отличающийся тем, что Y1 представляет собой Val-Leu-Gly-Arg-Leu-Ser-Gln-Glu-Leu-His-Arg-Leu-Gln-Thr-Tyr-Pro-Val-Asp (SEQ ID NO: 60).
7. Антагонист по п. 6, отличающийся тем, что Z1 представляет собой Рrо-Ser-Ser-Pro-His-Ser-Tyr-NH2 (SEQ ID NO: 63).
8. Антагонист по любому из пп. 4-7, отличающийся тем, что X1 выбран из группы, состоящей из Ala-Cys-Asp-Thr-Ala-Ala-Cys (SEQ ID NO: 17), Ala-Cys-Val-Leu-Gly-Ala-Cys (SEQ ID NO: 21) и Ala-Cys-Asn-Leu-Ser-Ala-Cys (SEQ ID NO: 24).
9. Антагонист по п. 3, выбранный из группы, состоящей из Ala-Cys-Asn-Leu-Ser-Ala-Cys-Val-Leu-Gly-Arg-Leu-Ser-Gln-Glu-Leu-His-Arg-Leu-Gln-Thr-Tyr-Pro-Thr-Asn-Thr-Gly-Ser-Gly-Thr-Pro-NH2 (SEQ ID NO: 13), Ala-Cys-Val-Leu-Gly-Ala-Cys-Val-Leu-Gly-Arg-Leu-Ser-Gln-Glu-Leu-His-Arg-Leu-Gln-Thr-Tyr-Pro-Val-Asp-Pro-Ser-Ser-Pro-His-Ser-Tyr-NH2 (SEQ ID NO: 14) и Ala-Cys-Asp-Thr-Ala-Ala-Cys-Val-Thr-His-Arg-Leu-Ala-Gly-Leu-Leu-Ser-Arg-Ser-Gly-Gly-Val-Val-Lys-Asn-Asn-Phe-Val-Pro-Thr-Asn-Val-Gly-Ser-Lys-Ala-Phe-NH2 (SEQ ID NO: 15).
10. Способ лечения состояния, связанного с аномальным уровнем CGRP, включающий введение субъекту антагониста на основе связанного с геном кальцитонина пептида по любому из пп. 1 или 9, причем указанный способ включает введение указанному субъекту эффективного количества антагониста на основе связанного с геном кальцитонина пептида.
11. Способ по п. 10, отличающийся тем, что состояние представляет собой мигрень.
12. Способ лечения головной боли у субъекта, включающий введение указанному субъекту эффективного количества антагониста на основе связанного с геном кальцитонина пептида по любому из пп. 1 или 9.
13. Способ по п. 12, отличающийся тем, что указанная головная боль представляет собой мигрень.
| WO 2006105527 A2, 05.10.2006 | |||
| Карбюратор для двигателей внутреннею горения | 1936 |
|
SU52011A1 |
| TAYLOR C.K | |||
| ET AL., PHARMACOLOGICAL CHARACTERIZATION OF NOVEL ALPHA-CALCITONIN GENE-RELATED PEPTIDE (CGRP) RECEPTOR PEPTIDE ANTAGONISTS THAT ARE SELECTIVE FOR HUMAN CGRP RECEPTORS, JOURNAL OF PHARMACOLOGY AND EXPERIMENTAL | |||

