Настоящая заявка является международной патентной заявкой, по которой испрашивается приоритет на основании предварительной заявки США № 61/077426, поданной 1 июля 2008, содержание которой приведено здесь посредством ссылки.
Изобретение относится к бициклическим гетероциклам формул I и II, обладающим противораковой активностью, и, более конкретно, обладающим ингибирующей активностью в отношении киназы MEK. Изобретение относится к композициям и способам, применяемым для подавления патологического клеточного роста, лечения гиперпролиферативных расстройств или лечения воспалительных заболеваний у млекопитающего. Изобретение также относится к способам применения соединений для in vitro, in situ и in vivo диагностики или лечения клеток млекопитающих, или связанных с ними патологических состояний.
В стремлении понять, каким образом Ras передает внутриклеточные сигналы роста, MAP (митоген-активируемый белок) киназный (MAPK) путь выступает в качестве ключевого направления между мембраносвязанным Ras и ядром. Путь MAPK охватывает каскад событий фосфорилирования, включающих три основные киназы, а именно: Raf, MEK (киназа киназы MAP) и ERK (киназа MAP). Активный GTP-связанный Ras приводит к активации и косвенному фосфорилированию киназы Raf. Затем Raf фосфорилирует MEK1 и 2 по двум остаткам серина (S218 и S222 для MEK1 и S222 и S226 для MEK2) (Ahn et al., Methods in Enzymology 2001, 332, 417-431). Активный MEK далее фосфорилирует только его известные субстраты, киназы MAP, ERK1 и 2. Фосфорилирование ERK посредством MEK происходит на Y204 и T202 для ERK1 и Y185 и T183 для ERK2 (Ahn et al., Methods in Enzymology 2001, 332, 417-431). Фосфорилированный ERK димеризуется и затем транслоцируется в ядро, где он накапливается (Khokhlatchev et al., Cell 1998, 93, 605-615). В ядре ERK вовлечен в несколько важных клеточных функций, включая, но этим не ограничиваясь, ядерный транспорт, трансдукцию сигнала, репарацию ДНК, сборку нуклеосом и транслокацию, и процессинг и трансляцию мРНК (Ahn et al., Molecular Cell 2000, 6, 1343-1354). В целом, обработка клеток факторами роста приводит к активации ERK1 и 2, что приводит к пролиферации и, в некоторых случаях, дифференциации (Lewis et al., Adv. Cancer Res. 1998, 74, 49-139).
Существовали убедительные доказательства того, что генетические мутации и/или сверхэкспрессия протеинкиназ, вовлеченных в путь киназы MAP, приводят к неконтролируемой пролиферации клеток и, в конечном счете, формированию опухоли, при пролиферативных заболеваниях. Например, некоторые злокачественные опухоли содержат мутации, которые приводят к длительной активации этого пути вследствие непрерывного продуцирования факторов роста. Другие мутации могут привести к дефектам в дезактивации активированного GTP-связанного комплекса Ras, что снова приводит его к активации пути киназы MAP. Мутированные онкогенные формы Ras обнаружены в 50% случаев злокачественных опухолей толстой кишки и >90% случаев злокачественных опухолей поджелудочной железы, а также во многих других видах злокачественных образований (Kohl et al., Science 1993, 260, 1834-1837). В последнее время мутации bRaf были обнаружены в более чем в 60% случаев заболевания злокачественной меланомой (Davies, H. Et al., Nature 2002, 417, 949-954). Эти мутации bRaf приводят к конститутивной активации MAP-киназного каскада. Исследования первичных образцов опухоли и клеточных линий также показали конститутивную активацию или сверхактивацию MAP-киназного пути в злокачественных опухолях поджелудочной железы, толстого кишечника, легких, яичников и почек (Hoshino, R. Et al., Oncogene 1999, 18, 813-822).
MEK выступает в качестве привлекательной терапевтической мишени в MAP-киназном каскадном пути. MEK, на выходе Ras и Raf, является высоко специфичным для фосфорилирования MAP-киназы; фактически, единственными известными основаниями для MEK фосфорилирования являются MAP-киназы, ERK1 и 2. Ингибирование MEK, как было показано в нескольких исследованиях, обладает потенциальным терапевтическим эффектом. Например, низкомолекулярные MEK-ингибиторы подавляют рост опухоли человека, что было показано на модели лишенной волосяного покрова мыши с ксенотрансплантантом человеческой опухоли, (Sebolt-Leopold et al., Nature-Medicine 1999, 5 (7), 810-816); Trachet et al., AACR Apr. 6-10, 2002, Poster #5426; Tecle, H. IBC 2.sup.nd International Conference of Protein Kinases, Sep. 9-10, 2002), блокируют статическую аллодинию у животных (WO 01/05390, опублик. 25 января 2001) и подавляют рост лейкозных клеток при остром миелолейкозе (Milella et al., J Clin Invest 2001, 108 (6), 851-859).
Некоторые низкомолекулярные MEK-ингибиторы также были рассмотрены, например, в WO02/06213, WO 03/077855 и WO03/077914. Все еще существует потребность в новых ингибиторах MEK в качестве эффективной и безопасной терапии для лечения ряда пролиферативных болезненных состояний, таких как состояния, связанные с гиперактивностью MEK, а также заболеваний, модулированных MEK каскадом.
В целом, изобретение относится к бициклическим гетероциклам формулы I и II (и/или сольватам, гидратам и/или их солям), обладающим противоопухолевой и/или противовоспалительной активностью, и, более конкретно, ингибирующей активностью в отношении MEK киназны. Некоторые гиперпролиферативные и воспалительные расстройства характеризуются модулированием функции киназы MEK, например мутацией или сверхэкспрессией белков. Соответственно, соединения по изобретению и их композиции используются при лечении гиперпролиферативных расстройств, таких как рак и/или воспалительные заболевания, такие как ревматоидный артрит.
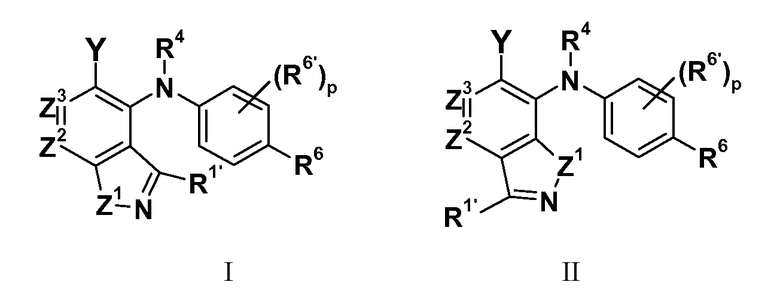
где:
Z1 представляет собой NR1, S или O;
R1 представляет собой H, C1-C3 алкил, CF3, CHF2 или циклопропил;
R1' представляет собой H, C1-C3 алкил, циклопропил, галоген, CF3, CHF2, CN, NRARA или ORB;
каждый RA независимо представляет собой H или C1-C3 алкил;
RB представляет собой H или C1-C3 алкил, необязательно замещенный одним или несколькими галогенами;
Z2 представляет собой CR2 или N;
Z3 представляет собой CR3 или N; при условии, что Z2 и Z3 одновременно не являются оба N;
R2 и R3 независимо выбраны из H, галогена, CN, CF3, -OCF3, -NO2, -(CR14R15)nC(=Y')R11, -(CR14R15)nC(=Y')OR11, -(CR14R15)nC(=Y')NR11R12, -(CR14R15)nNR11R12, -(CR14R15)nOR11, -(CR14R15)nSR11, -(CR14R15)nNR12C(=Y')R11, -(CR14R15)nNR12C(=Y')OR11, -(CR14R15)nNR13C(=Y')NR11R12, -(CR14R15)nNR12SO2R11, -(CR14R15)nOC(=Y')R11, -(CR14R15)nOC(=Y')OR11, -(CR14R15)nOC(=Y')NR11R12, -(CR14R15)nOS(O)2(OR11), -(CR14R15)nOP(=Y')(OR11)(OR12), -(CR14R15)nOP(OR11)(OR12), -(CR14R15)nS(O)R11, -(CR14R15)nS(O)2R11, -(CR14R15)nS(O)2NR11R12, -(CR14R15)nS(O)(OR11), -(CR14R15)nS(O)2(OR11), -(CR14R15)nSC(=Y')R11, -(CR14R15)nSC(=Y')OR11, -(CR14R15)nSC(=Y')NR11R12, C1-C12 алкила, C2-C8 алкенила, C2-C8 алкинила, карбоциклила, гетероциклила, арила и гетероарила;
R4 представляет собой H, C1-C6 алкил или C3-C4 карбоциклил;
Y представляет собой W-C(O)- или W';
W представляет собой
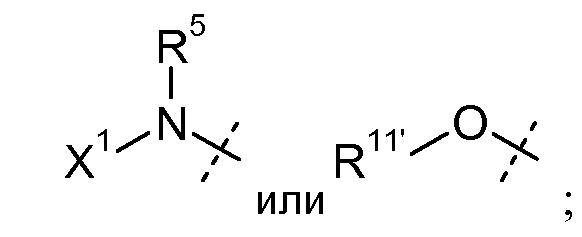
R5 представляет собой H или C1-C12 алкил;
X1 выбран из R11' и -OR11'; когда X1 представляет собой R11', X1, необязательно взятый вместе с R5 и атомом азота, к которому они присоединены, образуют 4-7-членное насыщенное или ненасыщенное кольцо, содержащее 0-2 дополнительных гетероатома, выбранных из O, S и N, где указанное кольцо необязательно замещено одним или несколькими группами, выбранными из галогена, CN, CF3, -OCF3, -NO2, оксо, -(CR19R20)nC(=Y')R16, -(CR19R20)nC(=Y')OR16, -(CR19R20)nC(=Y')NR16R17, -(CR19R20)nNR16R17, -(CR19R20)nOR16, -(CR19R20)n-SR16, -(CR19R20)nNR16C(=Y')R17, -(CR19R20)nNR16C(=Y')OR17, -(CR19R20)nNR18C(=Y')NR16R17, -(CR19R20)nNR17SO2R16, -(CR19R20)nOC(=Y')R16, -(CR19R20)nOC(=Y')OR16, -(CR19R20)nOC(=Y')NR16R17, -(CR19R20)nOS(O)2(OR16), -(CR19R20)nOP(=Y')(OR16)(OR17), -(CR19R20)nOP(OR16)(OR17), -(CR19R20)nS(O)R16, -(CR19R20)nS(O)2R16, -(CR19R20)nS(O)2NR16R17, -(CR19R20)nS(O)(OR16), -(CR19R20)nS(O)2(OR16), -(CR19R20)nSC(=Y')R16, -(CR19R20)nSC(=Y')OR16, -(CR19R20)nSC(=Y')NR16R17 и R21;
каждый R11' независимо представляет собой H, C1-C12 алкил, C2-C8 алкенил, C2-C8 алкинил, карбоциклил, гетероциклил, арил или гетероарил;
R11, R12 и R13 независимо представляют собой H, C1-C12 алкил, C2-C8 алкенил, C2-C8 алкинил, карбоциклил, гетероциклил, арил или гетероарил,
или R11 и R12 вместе с атомом азота, к которому они присоединены, образуют 3-8-членное насыщенное, ненасыщенное или ароматическое кольцо, имеющее 0-2 гетероатома, выбранные из O, S и N, где указанное кольцо необязательно замещено одним или несколькими группами, выбранными из галогена, CN, CF3, -OCF3, -NO2, C1-C6 алкила, -OH, -SH, -O(C1-C6 алкил), -S(C1-C6 алкил), -NH2, -NH(C1-C6 алкил), -N(C1-C6 алкил)2, -SO2(C1-C6 алкил), -CO2H, -CO2(C1-C6 алкил), -C(O)NH2, -C(O)NH(C1-C6 алкил), -C(O)N(C1-C6 алкил)2, -N(C1-C6 алкил)C(O)(C1-C6 алкил), -NHC(O)(C1-C6 алкил), -NHSO2(C1-C6 алкил), -N(C1-C6 алкил)SO2(C1-C6 алкил), -SO2NH2, -SO2NH(C1-C6 алкил), -SO2N(C1-C6 алкил)2, -OC(O)NH2, -OC(O)NH(C1-C6 алкил), -OC(O)N(C1-C6 алкил)2, -OC(O)O(C1-C6 алкил), -NHC(O)NH(C1-C6 алкил), -NHC(O)N(C1-C6 алкил)2, -N(C1-C6 алкил)C(O)NH(C1-C6 алкил), -N(C1-C6 алкил)C(O)N(C1-C6 алкил)2, -NHC(O)NH(C1-C6 алкил), -NHC(O)N(C1-C6алкил)2, -NHC(O)O(C1-C6 алкил) и -N(C1-C6 алкил)C(O)O(C1-C6 алкил);
R14 и R15 независимо выбраны из H, C1-C12 алкила, арила, карбоциклила, гетероциклила и гетероарила;
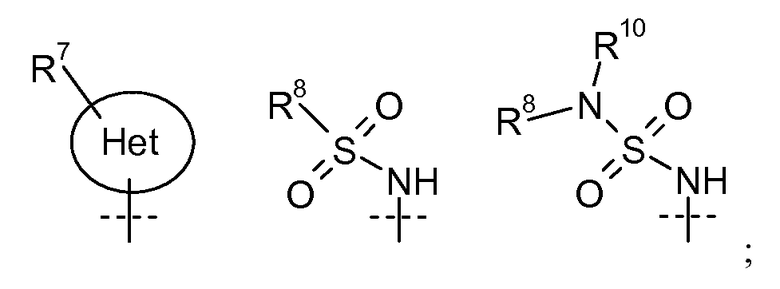
W' представляет собой
где  представляет собой
представляет собой
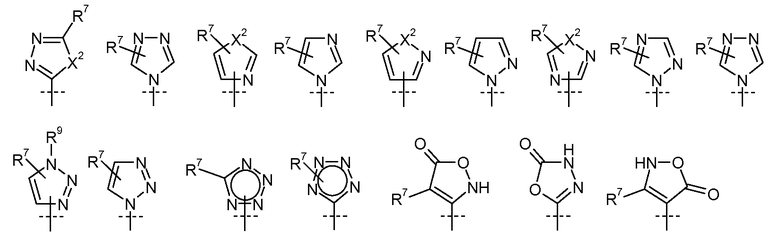
X2 представляет собой O, S или NR9;
R7 выбран из H, галогена, CN, CF3, -OCF3, -NO2, -(CR14R15)nC(=Y')R11, -(CR14R15)nC(=Y')OR11, -(CR14R15)nC(=Y')NR11R12, -(CR14R15)nNR11R12, -(CR14R15)nOR11, -(CR14R15)nSR11, -(CR14R15)nNR12C(=Y')R11, -(CR14R15)nNR12C(=Y')OR11, -(CR14R15)nNR13C(=Y')NR11R12, -(CR14R15)nNR12SO2R11, -(CR14R15)nOC(=Y')R11, -(CR14R15)nOC(=Y')OR11, -(CR14R15)nOC(=Y')NR11R12, -(CR14R15)nOS(O)2(OR11), -(CR14R15)nOP(=Y')(OR11)(OR12), -(CR14R15)nOP(OR11)(OR12), -(CR14R15)nS(O)R11, -(CR14R15)nS(O)2R11, -(CR14R15)nS(O)2NR11R12, -(CR14R15)nS(O)(OR11), -(CR14R15)nS(O)2(OR11), -(CR14R15)nSC(=Y')R11, -(CR14R15)nSC(=Y')OR11, -(CR14R15)nSC(=Y')NR11R12, C1-C12 алкил, C2-C8 алкенил, C2-C8 алкинил, карбоциклил, гетероциклил, арил и гетероарил;
R8 выбран из C1-C12 алкила, арила, карбоциклила, гетероциклила и гетероарила;
R9 выбран из H, -(CR14R15)nC(=Y')R11, -(CR14R15)nC(=Y')OR11, -(CR14R15)nC(=Y')NR11R12, -(CR14R15)qNR11R12, -(CR14R15)qOR11, -(CR14R15)qSR11, -(CR14R15)qNR12C(=Y')R11, -(CR14R15)qNR12C(=Y')OR11, -(CR14R15)qNR13C(=Y')NR11R12, -(CR14R15)qNR12SO2R11, -(CR14R15)qOC(=Y')R11, -(CR14R15)qOC(=Y')OR11, -(CR14R15)qOC(=Y')NR11R12, -(CR14R15)qOS(O)2(OR11), -(CR14R15)qOP(=Y')(OR11)(OR12), -(CR14R15)qOP(OR11)(OR12), -(CR14R15)nS(O)R11, -(CR14R15)nS(O)2R11, -(CR14R15)nS(O)2NR11R12, C1-C12 алкила, C2-C8 алкенила, C2-C8 алкинила, карбоциклила, гетероциклила, арила и гетероарила;
R10 представляет собой H, C1-C6 алкил или C3-C4 карбоциклил;
R6 представляет собой H, галоген, C1-C6 алкил, C2-C8 алкенил, C2-C8 алкинил, карбоциклил, гетероарил, гетероциклил, -OCF3, -NO2, -Si(C1-C6 алкил), -(CR19R20)nNR16R17, -(CR19R20)nOR16 или -(CR19R20)nSR16;
каждый R6' независимо представляет собой H, галоген, C1-C6 алкил, C2-C8 алкенил, C2-C8 алкинил, карбоциклил, гетероциклил, арил, гетероарил, CF3, -OCF3, -NO2, -Si(C1-C6 алкил), -(CR19R20)nNR16R17, -(CR19R20)nOR16 или -(CR19R20)nSR16; при условии, что R6 и R6' не являются оба одновременно H;
p равно 0, 1, 2 или 3;
n равно 0, 1, 2 или 3;
q равно 2 или 3;
где каждый указанный алкил, алкенил, алкинил, карбоциклил, гетероциклил, арил и гетероарил в R2, R3, R4, R5, R6, R6', R7, R8, R9, R10, R11, R11', R12, R13, R14 и R15 независимо необязательно замещены одним или несколькими группами, независимо выбранными из галогена, CN, CF3, -OCF3, -NO2, оксо, -Si(C1-C6 алкил), -(CR19R20)nC(=Y')R16, -(CR19R20)nC(=Y')OR16, -(CR19R20)nC(=Y')NR16R17, -(CR19R20)nNR16R17, -(CR19R20)nOR16, -(CR19R20)nSR16, -(CR19R20)nNR16C(=Y')R17, -(CR19R20)nNR16C(=Y')OR17, -(CR19R20)nNR18C(=Y')NR16R17, -(CR19R20)nNR17SO2R16, -(CR19R20)nOC(=Y')R16, -(CR19R20)nOC(=Y')OR16, -(CR19R20)nOC(=Y')NR16R17, -(CR19R20)nOS(O)2(OR16), -(CR19R20)nOP(=Y')(OR16)(OR17), -(CR19R20)nOP(OR16)(OR17), -(CR19R20)nS(O)R16, -(CR19R20)nS(O)2R16, -(CR19R20)nS(O)2NR16R17, -(CR19R20)nS(O)(OR16), -(CR19R20)nS(O)2(OR16), -(CR19R20)nSC(=Y')R16, -(CR19R20)nSC(=Y')OR16, -(CR19R20)nSC(=Y')NR16R17 и R21;
каждый R16, R17 и R18 независимо представляют собой H, C1-C12 алкил, C2-C8 алкенил, C2-C8 алкинил, карбоциклил, гетероциклил, арил или гетероарил, где указанные алкил, алкенил, алкинил, карбоциклил, гетероциклил, арил, или гетероарил необязательно замещены одной или несколькими группами, выбранными из галогена, CN, -OCF3, CF3, -NO2, C1-C6 алкила, -OH, -SH, -O(C1-C6 алкил), -S(C1-C6 алкил), -NH2, -NH(C1-C6 алкил), -N(C1-C6 алкил)2, -SO2(C1-C6 алкил), -CO2H, -CO2(C1-C6 алкил), -C(O)NH2, -C(O)NH(C1-C6 алкил), -C(O)N(C1-C6 алкил)2, -N(C1-C6 алкил)C(O)(C1-C6 алкил), -NHC(O)(C1-C6 алкил), -NHSO2(C1-C6 алкил), -N(C1-C6 алкил)SO2(C1-C6 алкил), -SO2NH2, -SO2NH(C1-C6 алкил), -SO2N(C1-C6 алкил)2, -OC(O)NH2, -OC(O)NH(C1-C6 алкил), -OC(O)N(C1-C6 алкил)2, -OC(O)O(C1-C6 алкил), -NHC(O)NH(C1-C6 алкил), -NHC(O)N(C1-C6 алкил)2, -N(C1-C6 алкил)C(O)NH(C1-C6 алкил), -N(C1-C6 алкил)C(O)N(C1-C6 алкил)2, -NHC(O)NH(C1-C6 алкил), -NHC(O)N(C1-C6 алкил)2, -NHC(O)O(C1-C6 алкил) и -N(C1-C6 алкил)C(O)O(C1-C6 алкил);
или R16 и R17 вместе с атомом азота, к которому они присоединены, образуют 3-8-членное насыщенное, ненасыщенное или ароматическое кольцо, имеющее 0-2 гетероатома, выбранные из O, S и N, где указанное кольцо необязательно замещено одной или несколькими группами, выбранными из галогена, CN, -OCF3, CF3, -NO2, C1-C6 алкила, -OH, -SH, -O(C1-C6 алкил), -S(C1-C6 алкил), -NH2, -NH(C1-C6 алкил), -N(C1-C6 алкил)2, -SO2(C1-C6 алкил), -CO2H, -CO2(C1-C6 алкил), -C(O)NH2, -C(O)NH(C1-C6 алкил), -C(O)N(C1-C6 алкил)2, -N(C1-C6 алкил)C(O)(C1-C6 алкил), -NHC(O)(C1-C6 алкил), -NHSO2(C1-C6 алкил), -N(C1-C6 алкил)SO2(C1-C6 алкил), -SO2NH2, -SO2NH(C1-C6 алкил), -SO2N(C1-C6 алкил)2, -OC(O)NH2, -OC(O)NH(C1-C6 алкил), -OC(O)N(C1-C6 алкил)2, -OC(O)O(C1-C6 алкил), -NHC(O)NH(C1-C6 алкил), -NHC(O)N(C1-C6 алкил)2, -N(C1-C6 алкил)C(O)NH(C1-C6 алкил), -N(C1-C6 алкил)C(O)N(C1-C6 алкил)2, -NHC(O)NH(C1-C6 алкил), -NHC(O)N(C1-C6 алкил)2, -NHC(O)O(C1-C6 алкил) и -N(C1-C6 алкил)C(O)O(C1-C6 алкил);
R19 и R20 независимо выбраны из H, C1-C12 алкила, -(CH2)n-арила, -(CH2)n-карбоциклила, -(CH2)n-гетероциклила и -(CH2)n-гетероарила;
R21 представляет собой C1-C12 алкил, C2-C8 алкенил, C2-C8 алкинил, карбоциклил, гетероциклил, арил или гетероарил, где каждый член в R21 необязательно замещен одной или несколькими группами, выбранными из галогена, оксо, CN, -OCF3, CF3, -NO2, C1-C6 алкила, -OH, -SH, -O(C1-C6 алкил), -S(C1-C6 алкил), -NH2, -NH(C1-C6 алкил), -N(C1-C6 алкил)2, -SO2(C1-C6 алкил), -CO2H, -CO2(C1-C6 алкил), -C(O)NH2, -C(O)NH(C1-C6 алкил), -C(O)N(C1-C6 алкил)2, -N(C1-C6 алкил)C(O)(C1-C6 алкил), -NHC(O)(C1-C6 алкил), -NHSO2(C1-C6 алкил), -N(C1-C6 алкил)SO2(C1-C6 алкил), -SO2NH2, -SO2NH(C1-C6 алкил), -SO2N(C1-C6 алкил)2, -OC(O)NH2, -OC(O)NH(C1-C6 алкил), -OC(O)N(C1-C6 алкил)2, -OC(O)O(C1-C6 алкил), -NHC(O)NH(C1-C6 алкил), -NHC(O)N(C1-C6 алкил)2, -N(C1-C6 алкил)C(O)NH(C1-C6 алкил), -N(C1-C6 алкил)C(O)N(C1-C6 алкил)2, -NHC(O)NH(C1-C6 алкил), -NHC(O)N(C1-C6 алкил)2, -NHC(O)O(C1-C6 алкил) и -N(C1-C6 алкил)C(O)O(C1-C6 алкил);
каждый Y' независимо представляет собой O, NR22 или S; и
R22 представляет собой H или C1-C12 алкил;
при условии, что в формуле (I), (i) когда Z1 представляет собой NR1 и Z2 представляет собой N, тогда Y не представляет собой CO2NH2; и (ii) когда Z1 представляет собой NR1, Z2 представляет собой N, R1' представляет собой H, Z3 представляет собой CR3, где R3 представляет собой H, CH3, CF3, CHF2 или CH2F,
тогда Y не представляет собой CO2Et или
Настоящее изобретение включает композицию (например, фармацевтическую композицию), содержащую соединение формулы I или II (и/или сольваты, гидраты и/или их соли) и носитель (фармацевтически приемлемый носитель). Настоящее изобретение также включает композицию (например, фармацевтическую композицию), содержащую соединение формулы I или II (и/или сольваты, гидраты и/или их соли) и носитель (фармацевтически приемлемый носитель) и, кроме того, содержащую второй химиотерапевтический и/или второй противовоспалительный агент. Настоящие композиции используются для ингибирования аномального роста клеток или при лечении гиперпролиферативного нарушения у млекопитающего (например, человека). Настоящие композиции также могут использоваться для лечения воспалительных заболеваний у млекопитающего (например, человека).
Настоящее изобретение включает способ ингибирования аномального роста клеток или лечения гиперпролиферативного нарушения у млекопитающего (например, человека), включающий введение указанному млекопитающему терапевтически эффективного количества соединения формулы I или II (и/или сольваты и их соли) или композиции на его основе, самостоятельно или в сочетании со вторым химиотерапевтическим агентом.
Настоящее изобретение включает способ лечения воспалительного заболевания у млекопитающего (например, человека), включающий введение указанному млекопитающему терапевтически эффективного количества соединения формулы I или II (и/или сольваты и их соли) или композиции на его основе, самостоятельно или в сочетании со вторым противовоспалительным агентом.
Настоящее изобретение включает способ применения настоящих соединений для in vitro, in situ и in vivo диагностики или лечения человеческих клеток, организмов или связанных патологических состояний.
Далее дано подробное описание некоторых вариантов осуществления изобретения, примеры которых иллюстрированы структурами и формулами. Хотя изобретение описано далее в сочетании с перечисленными вариантами осуществления, следует учесть, что они не предназначены для ограничения изобретения этими вариантами осуществления. Напротив, изобретение предусматривает включение всех альтернатив, модификаций и эквивалентов, которые могут входить в объем настоящего изобретения, как определено в пунктах формулы изобретения. Специалисту в данной области понятны многие способы и продукты, подобные или эквивалентные тем, которые описаны в данном документе, и которые могли бы быть использованы при практическом осуществлении настоящего изобретения. Настоящее изобретение не ограничивается каким-либо образом описанными способами и продуктами. В случае, когда один или несколько из включенных литературных источников, патентов и подобных материалов отличаются от или противоречат данной заявке, включая, но не ограничиваясь указными терминами, применение терминов, описанных методов или тому подобное, определяется данной заявкой.
Термин «алкил», как здесь используется, относится к насыщенному линейному или разветвленному моновалентному углеводородному радикалу, состоящему из от одного до двенадцати атомов углерода. Примеры алкильной группы включают, но этим не ограничиваются, метил (Me, -CH3), этил (Et, -CH2CH3), 1-пропил (н-Pr, н-пропил, -CH2CH2CH3), 2-пропил (изо-Pr, изо-пропил, -CH(CH3)2), 1-бутил (н-Bu, н-бутил, -CH2CH2CH2CH3), 2-метил-1-пропил (изо-Bu, изо-бутил, -CH2CH(CH3)2), 2-бутил (втор-Bu, втор-бутил, -CH(CH3)CH2CH3), 2-метил-2-пропил (трет-Bu, трет-бутил, -C(CH3)3), 1-пентил (н-пентил, -CH2CH2CH2CH2CH3), 2-пентил (-CH(CH3)CH2CH2CH3), 3-пентил (-CH(CH2CH3)2), 2-метил-2-бутил (-C(CH3)2CH2CH3), 3-метил-2-бутил (-CH(CH3)CH(CH3)2), 3-метил-1-бутил (-CH2CH2CH(CH3)2), 2-метил-1-бутил (-CH2CH(CH3)CH2CH3), 1-гексил (-CH2CH2CH2CH2CH2CH3), 2-гексил (-CH(CH3)CH2CH2CH2CH3), 3-гексил (-CH(CH2CH3)(CH2CH2CH3)), 2-метил-2-пентил (-C(CH3)2CH2CH2CH3), 3-метил-2-пентил (-CH(CH3)CH(CH3)CH2CH3), 4-метил-2-пентил (-CH(CH3)CH2CH(CH3)2), 3-метил-3-пентил (-C(CH3)(CH2CH3)2), 2-метил-3-пентил (-CH(CH2CH3)CH(CH3)2), 2,3-диметил-2-бутил (-C(CH3)2CH(CH3)2), 3,3-диметил-2-бутил (-CH(CH3)C(CH3)3, 1-гептил, 1-октил и тому подобное.
Термин «алкенил» относится к линейному или разветвленному моновалентному углеводородному радикалу, состоящему из от одного до двенадцати атомов углерода по меньшей мере с одним участком ненасыщенности, то есть, углерод-углерод, sp2 двойной связью, где алкенильный радикал включает радикалы, имеющие «цис» и «транс» ориентации, или, альтернативно, «E» и «Z» ориентации. Примеры включают, но этим не ограничиваются, этиленил или винил (-CH=CH2), аллил (-CH2CH=CH2) и тому подобное.
Термин «алкинил» относится к линейному или разветвленному моновалентному углеводородному радикалу, состоящему из от одного до двенадцати атомов углерода по меньшей мере с одним участком ненасыщенности, то есть, углерод-углерод, sp тройной связью. Примеры включают, но этим не ограничиваются, этинил (-C≡CH), пропинил (пропаргил, -CH2C≡CH) и тому подобное.
Термины «карбоцикл», «карбоциклил», «карбоциклическое кольцо» и «циклоалкил» относятся к моновалентному неароматическому, насыщенному или частично ненасыщенному кольцу, имеющему от 3 до 12 атомов углерода в виде моноциклического кольца или от 7 до 12 атомов углерода в виде бициклического кольца. Бициклические карбоциклы, имеющие от 7 до 12 атомов, могут быть представлены, например, в виде бицикло [4,5], [5,5], [5,6] или [6,6] системы, и бициклические карбоциклы, имеющие 9 или 10 кольцевых атомов, могут быть представлены в виде бицикло [5,6] или [6,6] системы, или в виде мостиковой системы, такой как бицикло[2.2.1]гептан, бицикло[2.2.2]октан и бицикло[3.2.2]нонан. Примеры моноциклических карбоциклов включают, но этим не ограничиваются, циклопропил, циклобутил, циклопентил, 1-циклопент-1-енил, 1-циклопент-2-енил, l-циклопент-3-енил, циклогексил, 1-циклогекс-1-енил, 1-циклогекс-2-енил, 1-циклогекс-3-енил, циклогексадиенил, циклогептил, циклооктил, циклононил, циклодецил, циклоундецил, циклододецил и тому подобное.
«Арил» подразумевает моновалентный ароматический углеводородный радикал из 6-18 атомов углерода, образуемый в результате удаления одного атома водорода от одного атома углерода первоначальной ароматической кольцевой системы. Некоторые арильные группы представлены в структурах примеров как «Ar». Арил включает бициклические радикалы, содержащие ароматическое кольцо, конденсированное с насыщенным, частично ненасыщенным кольцом или ароматическим карбоциклическим или гетероциклическим кольцом. Типичные арильные группы включают, но этим не ограничиваются, радикалы, образованные из бензола (фенил), замещенных бензолов, нафталина, антрацена, инденила, инданила, 1,2-дигидронафталина, 1,2,3,4-тетрагидронафтила и тому подобное.
Термины «гетероцикл», «гетероциклил» и «гетероциклическое кольцо» используются здесь взаимозаменяемо и относятся к насыщенному или частично ненасыщенному (то есть, имеющему один или несколько двойных и/или тройных связей в кольце) карбоциклическому радикалу, состоящему из от 3 до 18 кольцевых атомов, в котором меньшей мере один кольцевой атом представляет собой гетероатом, выбранный из азота, кислорода и серы, остальные кольцевые атомы представляют собой C, где одно или несколько кольцевых атомов необязательно независимо замещены одним или несколькими заместителями, описанными далее. Гетероцикл может представлять собой моноцикл, имеющий от 3 до 7 членов кольца (от 2 до 6 атомов углерода и от 1 до 4 гетероатомов, выбранных из N, O и S), или бицикл, имеющий от 7 до 10 членов кольца (от 4 до 9 атомов углерода и от 1 до 6 гетероатомов, выбранных из N, O и S), например: бицикло [4,5], [5,5], [5,6] или [6,6] системы. Гетероциклы описаны в работе Paquette, Leo A.; “Principles of Modern Heterocyclic Chemistry” (W. A. Benjamin, New York, 1968), в частности, главы 1, 3, 4, 6, 7, и 9; “The Chemistry of Heterocyclic Compounds, A series of Monographs” (John Wiley & Sons, New York, 1950 to present), в частности, тома 13, 14, 16, 19, и 28; и J. Am. Chem. Soc. (1960) 82:5566. Термин «гетероциклил» также включает радикалы, где гетероциклические радикалы конденсированы с насыщенным, частично ненасыщенным кольцом или ароматическим карбоциклическим или гетероциклическим кольцом. Примеры гетероциклических колец включают, но этим не ограничиваются, пирролидинил, тетрагидрофуранил, дигидрофуранил, тетрагидротиенил, тетрагидропиранил, дигидропиранил, тетрагидротиопиранил, пиперидинил, морфолинил, тиоморфолинил, тиоксанил, пиперазинил, гомопиперазинил, азетидинил, оксетанил, тиетанил, гомопиперидинил, оксепанил, тиепанил, оксазепинил, диазепинил, тиазепинил, 2-пирролинил, 3-пирролинил, индолинил, 2H-пиранил, 4H-пиранил, диоксанил, 1,3-диоксоланил, пиразолинил, дитианил, дитиоланил, дигидропиранил, дигидротиенил, дигидрофуранил, пиразолидинилимидазолинил, имидазолидинил, 3-азабицикло[3.1.0]гексанил, 3-азабицикло[4.1.0]гептанил и азабицикло[2.2.2]гексанил. Спиро группы также включены в объем данного определения. Примерами гетероциклической группы, где кольцевые атомы замещены оксо (=O) группами, являются пиримидинонил и 1,1-диоксо-тиоморфолинил.
Термин «гетероарил» относится к моновалентному ароматическому радикалу с 5- или 6-членными кольцами и включает конденсированные кольцевые системы (по меньшей мере одна из которых является ароматической), состоящие из 5-18 атомов, содержащие один или несколько гетероатомов, независимо выбранных из азота, кислорода и серы. Примерами гетероарильных групп являются пиридинил (включая, например, 2-гидроксипиридинил), имидазолил, имидазопиридинил, пиримидинил (включая, например, 4-гидроксипиримидинил), пиразолил, триазолил, пиразинил, тетразолил, фурил, тиенил, изоксазолил, тиазолил, оксазолил, изотиазолил, пирролил, хинолинил, изохинолинил, индолил, бензимидазолил, бензофуранил, циннолинил, индазолил, индолизинил, фталазинил, пиридазинил, триазинил, изоиндолил, птеридинил, пуринил, оксадиазолил, триазолил, тиадиазолил, фуразанил, бензофуразанил, бензотиофенил, бензотиазолил, бензоксазолил, хиназолинил, хиноксалинил, нафтиридинил и фуропиридинил.
Гетероцикл или гетероарильные группы могут быть присоединены по углероду (углерод-связанные) или по азоту (азот-связанные), что также возможно. В качестве примера и без ограничения, присоединенные по углероду гетероциклы или гетероарилы присоединены в положении 2, 3, 4, 5 или 6 пиридина, в положении 3, 4, 5 или 6 пиридазина, в положении 2, 4, 5 или 6 пиримидина, в положении 2, 3, 5 или 6 пиразина, в положении 2, 3, 4 или 5 фурана, тетрагидрофурана, тиофена, пиррола или тетрагидропиррола, в положении 2, 4 или 5 оксазола, имидазола или тиазола, в положении 3, 4 или 5 изоксазола, пиразола или изотиазола, в положении 2 или 3 азиридина, в положении 2, 3 или 4 азетидина, в положении 2, 3, 4, 5, 6, 7 или 8 хинолина или в положении 1, 3, 4, 5, 6, 7 или 8 изохинолина.
В качестве примера и без ограничения, присоединенные по азоту гетероциклы или гетероарилы присоединены в положении 1 азиридина, азетидина, пиррола, пирролидина, 2-пирролина, 3-пирролина, имидазола, имидазолидина, 2-имидазолина, 3-имидазолина, пиразола, пиразолина, 2-пиразолина, 3-пиразолина, пиперидина, пиперазина, индола, индолина, 1H-индазола, в положении 2 изоиндола или изоиндолина, в положении 4 морфолина, и положение 9 карбазола или β-карболина.
Термин «галоген» относится к F, Cl, Br или I. Гетероатомы, представленные в гетероариле или гетероциклиле, включают оксиленные формы, такие как N+→O-, S(O) и S(O)2.
Термины "лечить" и "лечение" относятся как к терапевтическсому лечению, так и к профилактическим или превентивным мерам, цель которых заключается в том, чтобы предотвратить или сдержать (уменьшить) нежелаемое физиологическое изменение или расстройство, такое как развитие или распространение рака. В целях настоящего изобретения выгодные или желаемые клинические результаты включают, но ими не ограничиваются, облегчение симптомов, ограничение распространения заболевания, стабилизирование (то есть не ухудшение) состояния заболевания, задержку или замедление развития заболевания, улучшение или ослабление болезненного состояния, и ремиссию (частичную или полную), выявленную или невыявленную. "Лечение" может также означать продление выживания по сравнению с ожидаемым выживанием в случае неполучения лечения. Нуждающиеся в лечении уже включают тех, кто находится в состоянии заболевания или имеет расстройство, а также тех, кто склонен к состоянию заболевания или расстройству, или тех, кто нуждается в профилактике состояния или расстройства.
Фраза "терапевтически эффективное количество" подразумевает количество соединения по настоящему изобретению, которое (i) лечит или предотвращает конкретное заболевание, состояние или расстройство, (ii) ослабляет, улучшает или устраняет один или несколько симптомов конкретного заболевания, состояния или расстройства, или (iii) предотвращает или задерживает проявление одного или нескольких симптомов конкретного заболевания, состояния или расстройства, описанных в данной документе. В случае рака, терапевтически эффективное количество лекарственного средства может уменьшать количество клеток рака; уменьшать размер опухоли; подавлять (то есть, сдерживать до некоторой степени и предпочтительно останавливать) инфильтрацию клеток рака в периферические органы; подавлять (то есть сдерживать до некоторой степени и предпочтительно останавливать) метастазирование опухоли; подавлять до некоторой степени рост опухоли; и/или облегчать до некоторой степени один или несколько симптомов, связаных с раком. Препарат может до некоторой степени препятствовать росту и/или ликвидировать существующие раковые клетки, препарат может быть цитостатическим и/или цитотоксическим средством. Для терапии рака, эффективность может быть оценена, например, путем определения времени развития заболевания (TTP) и/или определения скорости реакции (RR).
В настоящей заявке термины "патологический рост клеток" и "гиперпролиферативное расстройство" используются взаимозаменяемо. Используемая здесь фраза "патологический рост клеток", если не указано иное, относится к росту клеток, который не зависит от нормальных регуляторных механизмов (например, потеря контактного ингибирования). Это включает, например, патологический рост: (1) опухолевых клеток (опухоли), которые пролифелируют путем экспрессии мутированной тирозинкиназы или сверхэкспрессии рецепторной тирозинкиназы; (2) доброкачественных и злокачественных клеток других пролиферативных заболеваний, в которых происходит аберрантная активация тирозинкиназы; (3) любых опухолей, которые пролифелируют с помощью рецепторных тирозинкиназ; (4) любых опухолей, которые пролифелируют путем аберрантной серин/треонин киназной активации; и (5) доброкачественных и злокачественных клеток других пролиферативных заболеваний, в которых происходит аберрантная активация серин/треонин киназы.
Термины "рак" и "раковый" относятся к или описывают физиологическое состояние у млекопитающих, которое обычно характеризуется нерегулируемым ростом клеток. Термин "опухоль" включает один или несколько видов раковых клеток. Примеры рака включают, но этим не ограничиваются, карциному, лимфому, бластому, саркому и лейкемию или лимфоидные злокачественные заболевания. Более конкретные примеры таких видов рака включают плоскоклеточный рак (например, эпителиальный плоскоклеточный рак), рак легкого, включая мелкоклеточный рак легкого, не-мелкоклеточный рак легкого ("NSCLC"), аденокарциному легкого и плоскоклеточную карциному легкого, рак брюшной полости, гепатоцеллюлярный рак, рак желудка, включая гастроинтеральный рак, рак поджелудочной железы, глиобластому, цервикальный рак, рак яичников, рак печени, рак мочевого пузыря, гепатому, рак груди, рак толстой кишки, рак прямой кишки, рак ободочной и прямой кишки, эндометриальный рак или рак матки, карциному слюнных желез, рак почки или ренальный рак, рак простаты, рак влагалища, рак щитовидной железы, гепатокарциному, рак анального канала, пениальную карциному, острую лейкемию, а также рак головы/мозга и рак шеи.
"Химиотерапевтический агент" представляет собой соединение, используемое при лечении рака. Примеры химиотерапевтических агентов включает Эрлотиниб (TARCEVA®, Genentech/OSI Pharm.), Бортезомиб (VELCADE®, Millennium Pharm.), Фулвестрант (FASLODEX®, AstraZeneca), Сутент (SU11248, Pfizer), Летрозол (FEMARA®, Novartis), Иматиниб мезилат (GLEEVEC®, Novartis), PTK787/ZK 222584 (Novartis), Оксалиплатин (Eloxatin®, Sanofi), 5-FU (5-фторурацил), Лейковорин, Рапамицин (Sirolimus, RAPAMUNE®, Wyeth), Лапатиниб (TYKERB®, GSK572016, Glaxo Smith Kline), Лонафарниб (SCH 66336), Сорафениб (BAY43-9006, Bayer Labs), и Гефитиниб (IRESSA®, AstraZeneca), AG1478, AG1571 (SU 5271; Sugen), алкилирующие агенты, такие как тиотепа и CYTOXAN® циклосфосфамид; алкил сульфонаты, такие как бусульфан, импросульфан и пипосульфан; азиридины, такие как бензодопа, карбохон, метуредопа и уредопа; этиленимины и метиламеламины, включая альтретамин, триэтиленмеламин, триэтиленфосфорамид, триэтилентиофосфорамид и триметиломеламин; ацетогенины (особенно буллатацин и буллатацинон); камптотецин (включая ситетический аналог топотекан); бриостатин; каллистатин; CC-1065 (включая их ситетические аналоги адоцелезин, карцелезин и бицелезин); криптофицины (в частности, криптофицин 1 и криптофицин 8); доластатин; дуокармицин (включая ситетические аналоги, KW-2189 и CB1-TM1); элеутеробин; панкратистатин; сакродистиин; спонгистатин; мустины, такие как хлорамбуцил, хлорнафазин, хлорофосфамид, эстрамустин, ифосфамид, мехлоретамин, мехлоретамин оксид гидрохлорид, мелфалан, новембихин, фенстерин, преднимустин, трофосфамид, урамустин; нитромочевины, такие как кармустин, хлорозотоцин, фотемустин, ломустин, нимустин и ранимнустин; антибиотики, такие как энедииновые антибиотики (например, каликеамицин, особенно каликеамицин гамма lI и каликемицин омега Il (Angew Chem. Intl. Ed. Engl. (1994) 33: 183-186); динемецин, включая динемецин A; бисфосфонаты, такие как клодронат; эсперамицин; а также неокарциностатин хромофор и близкие хромопротеин энедиин антибиотические хромофоры), аклациномизины, актиномицин, аутрамицин, азасерин, блеомицины, катитомицин, карабицин, карминомицин, карцинофилин, хромомицинис, дактиномицин, даунорубицин, деторубицин, 6-диазо-5-оксо-L-норлейцин, АДРИАМИЦИН® (доксорубицин), морфолино-доксорубицин, цианоморфолино-доксорубицин, 2-пирролино-доксорубицин и деоксидоксорубицин), эпирубицин, эзорубицин, идарубицин, марцелломицин, митомицины, такие как митомицин C, микофеноловая кислота, ногаламицин, оливомицины, пепломицин, порфиромицин, пуромицин, хеламицин, родорубицин, стрептонигрин, стрептозоцин, туберцидин, убенимекс, циностатин, зорубицин; анти-метаболиты, такие как метотрексат и 5-фторурацил (5-FU); аналоги фолиевой кислоты, такие как деноптерин, метотрексат, птероптерин, триметрексат; пуриновые аналоги, такие как флударабин, 6-меркаптопурин, тиамиприн, тиогуанин; пиримидиновые аналоги, такие как анцитабин, азацитидин, 6-азауридин, кармофур, цитарабин, дидеоксиуридин, доксифлуридин, эноцитабин, флоксиридин; андрогены, такие как калустерон, дромостанолон пропионат, эпитиостанол, мепитиостан, тестолактон; анти-андреналины, такие как аминоглютетимид, митотан, трилостан; стимулятор фолевой кислоты, такой как фролиновая кислота; ацеглатон; альдофосфамид гликозид; аминолевулиновая кислота; энилурацил; амсакрин; бестрабуцил; бисантрен; эдатраксат; дефофамин; демеколцин; диазихон; эльфорнитин; эллиптиниум ацетат; эпотилон; этоглуцид; нитрат галлия; гидроксимочевина; лентинан; лонидаинин; маитансиноиды, такие как маитансин и ансамитоцины; митогуазон; митоксантрон; мопиданмол; нитраерин; пентостатин; фенамет; пирарубицин; лозоксантрон; подофиллиновая кислота; 2-этилгидразид; прокарбазин; PSK®полисахаридный комплекс (JHS Natural Products, Eugene, OR); разоксан; ризоксин; сизофиран; спирогерманий; тенуазоновая кислота; триазихон; 2,2',2"-трихлортриэтиламин; трихотецены (особенно T-2 токсин, верракурин A, роридин A и ангуидин); уретан; виндезин; дакарбазин; манномустин; митобронитол; митолактол; пипоброман; гацитозин; арабинозид ("Ara-C"); циклофосфамид; тиотепа; таксоиды, например, TAXOL® (паклитаксел; Bristol-Myers Squibb Oncology, Princeton, N.J.), ABRAXANE™ (не содержащий Cremophor), альбумин-инжиниринговые на основе наночастиц препараты паклитаксела (American Pharmaceutical Partners, Schaumberg, Illinois), и TAXOTERE® (доксетаксел; Rhône-Poulenc Rorer, Antony, France); хлоранбуцил; GEMZAR® (гемцитабин); 6-тиогуанин; меркаптопурин; метотрексат; аналоги платины, такие как цисплатин и карбоплатин; винбластин; этопозид (VP-16); ифосфамид; митоксантрон; винкристин; NAVELBINE® (винорелбин); новантрон; тенипозид; эдатрексат; дауномицин; аминоптерин; капецитабин (XELODA®); ибандронат; CPT-11; ингибитор топоизомеразы RFS 2000; дифторметилорнитин (ДМФО); ретиноиды, такие как ретиноевая кислота; и фармацевтически приемлемый соли, кислоты и производные любого из указанных выше.
В определение "химиотерапевтический агент" также включены: (i) анти-гормональные агенты, которые регулируют или ингибируют гормональное действие на опухоли, такие как анти-эстрогены и модуляторы селективных эстрогенных рецепторов (SERMs), включая, например, тамоксифен (включая NOLVADEX®; тамоксифен цитрат), ралоксифен, дролоксифен, 4-гидрокситамоксифен, триоксифен, кеоксифен, LY 117018, онапристон и FARESTON® (торемифин цитрат); (ii) ингибиторы ароматазы, которые ингибируют ферментную ароматазу, которая регулирует продукцию эстрогена в надпочечных железах, такие как, например, 4(5)-имидазолы, аминоглютетимид, MEGASE® (мегестрол ацетат), AROMASIN® (экземестан; Pfizer), форместанин, фадрозол, RTVISOR® (ворозол), FEMARA® (летрозол; Novartis) и ARIMIDEX® (анастрозол; AstraZeneca); (iii) анти-андрогены, такие как флутамид, нилутамид, бикалутамид, лейпролид и гозерелин; а также троксацитабин (аналог 1,3-диоксолан нуклеозид цитозин); (iv) ингибиторы протеин киназы; (v) ингибиторы липид киназы; (vi) антисмысловые олигонуклеотиды, в частности, такие, которые ингибируют экспрессию генов в сигнальном пути, задействованном в аберрантной пролиферации клеток, такие как, например, PKC-альфа, RaIf и H-Ras; (vii) рибозимы, такие как VEGF ингибиторы экспрессии (например, ANGIOZYME®) и HER2 ингибиторы экспрессии; (viii) вакцины, такие как вакцины генной терапии, например, ALLOVECTIN®, LEUVECTIN® и VAXID®; PROLEUKIN® rIL-2; ингибитор топоизомеразы 1, такие как LURTOTECAN®; ABARELIX® rmRH; (ix) анти-ангиогенные агенты, такие как бевацизумаб (AVASTIN®, Genentech); и (x) фармацевтически приемлемые соли, кислоты и производные любых из указанных выше. Другие анти-ангиогенные агенты включают MMP-2 (матрикс-металлопротеиназа 2) ингибиторы, MMP-9 (матрикс-металлопротеиназа 9) ингибиторы, COX-II (циклооксигеназа II) ингибиторы и ингибиторы VEGF рецепторов тирозинкиназы. Примеры таких используемых ингибиторов матрикс металлопротеиназы, которые могут быть использованы в сочетании с настоящим соединением/композицией описаны в WO 96/33172, WO 96/27583, EP 818442, EP 1004578, WO 98/07697, WO 98/03516, WO 98/34918, WO 98/34915, WO 98/33768, WO 98/30566, EP 606046, EP 931788, WO 90/05719, WO 99/52910, WO 99/52889, WO 99/29667, WO 99/07675, EP 945864, патент США 5863949, патент США 5861510 и EP 780386, все из них включены в настоящий документ в своем полном объеме в виде ссылки. Примеры ингибиторов VEGF рецепторов тирозинкиназы включают 4-(4-бром-2-фторанилино)-6-метокси-7-(1-метилпиперидин-4-илметокси)хиназолин (ZD6474; Пример 2 в WO 01/32651), 4-(4-фтор-2-метилиндол-5-илокси)-6-метокси-7-(3-пирролидин-1-илпропокси)-хиназолин (AZD2171; Пример 240 в WO 00/47212), ваталаниб (PTK787; WO 98/35985) и SU11248 (сунитиниб; WO 01/60814) и соединения, такие как описаны в публикациях PCT WO 97/22596, WO 97/30035, WO 97/32856 и WO 98/13354).
Другие примеры химиотерапевтических агентов, которые могут быть использованы в сочетании с настоящими соединениями, включают ингибиторы PI3K (фосфоинозитид-3 киназа), такие как те, что описаны в работе Yaguchi et al (2006) Jour. of the Nat. Cancer Inst. 98(8):545-556; US 7173029; US 7037915; US 6608056; US 6608053; US 6838457; US 6770641; US 6653320; US 6403588; WO 2006/046031; WO 2006/046035; WO 2006/046040; WO 2007/042806; WO 2007/042810; WO 2004/017950; US 2004/092561; WO 2004/007491; WO 2004/006916; WO 2003/037886; US 2003/149074; WO 2003/035618; WO 2003/034997; US 2003/158212; EP 1417976; US 2004/053946; JP 2001247477; JP 08175990; JP 08176070; US 6703414; и WO 97/15658, все из которых включены в данный документ во всей полноте в виде ссылки. Конкретные примеры таких PI3K ингибиторов включают SF-1126 (PI3K ингибитор, Semafore Pharmaceuticals), BEZ-235 (PI3K ингибитор, Novartis), XL-147 (PI3K ингибитор, Exelixis, Inc.) и GDC-0941 (PI3K ингибитор, PIramed и Genenetch).
Термин "воспалительные заболевания", используемый в настоящей заявке, включает, но этим не ограничивается, ревматоидный артрит, атеросклероз, хроническую головную боль, воспалительные заболевания кишечника (включая, но этим не ограничиваясь, болезнь Крона и неспецифический язвенный колит), хронические обструктивные пульмональные заболевания легких, фиброзные заболевания печени и почек, болезнь Крона, волчанку, кожные заболевания, такие как псориаз, экзема и склеродермия, остеоартрит, рассеянный склероз, астму, заболевания и расстройства, связанные с осложнениями при диабете, фиброзные органические нарушения в органах, таких как легкие, печень, почки, и воспалительные осложнения сердечно-сосудистой системы, такие как острый коронарный синдром.
"Противовоспалительный агент" представляет собой соединение, используемое при лечении воспаления. Примеры противовоспалительных агентов включают инъекционные белковые терапевтические средства, такие как Enbrel®, Remicade®, Humira® и Kineret®. Другие примеры противовоспалительных агентов включают нестероидные противовоспалительные агенты (NSAIDs), такие как ибупрофен или аспирин (которые уменьшают нарастание и снижают боль); заболевания-модифицирующие антиревматоидные лекарственные средства (DMARDs), такие как метотрексат; 5-аминосалицилаты (сульфазалазин и не содержащие сульфа агенты); кортикостероиды; иммуномодуляторы, такие как 6-меркаптопурин ("6-MP"), азатиоприн ("AZA"), циклоспорины и модификаторы биологического ответа, такие как Remicade.RTM. (инфликсимаб) и Enbrel.RTM. (этанерцепт); факторы роста фибропластов; фактор роста тромбоцитарный; ферментные блокаторы, такие как Arava.RTM. (лефлуномид); и/или хондропротективный агент, такой как гиалуроновая кислота, глюкозамин и хондроитин.
Используемый в настоящей заявке термин "пролекарство" относится к предшественнику или производному соединения по изобретению, способный ферментативно или гидролитически активироваться или преобразовываться в более активную исходную форму. Смотри, например, Wilman, "Prodrugs in Cancer Chemotherapy" Biochemical Society Transactions, 14, pp. 375-382, 615th Meeting Belfast (1986) и Stella et al., "Prodrugs: A Chemical Approach to Targeted Drug Delivery," Directed Drug Delivery, Borchardt et al., (ed.), pp. 247-267, Humana Press (1985). Пролекарства по настоящему изобретению включают, но ими не ограничиваются, сложноэфирные пролекарства, фосфатсодержащие пролекарства, тиофосфатсодержащие пролекарства, сульфатсодержащие пролекарства, пептидсодержащие пролекарства, D-амино-кислотно-модифицированные пролекарства, гликолизированные пролекарства, β-лактамсодержащие пролекарства, необязательно замещенные феноксиацетамидсодержащие пролекарства, необязательно замещенные фенилацетамидсодержащие пролекарства, 5-фторцитозин и другое 5-фторуридин пролекарства, которые могут быть преобразованы в более активное свободное цитотоксическое лекарство. Примерами цитотоксических лекарственных препаратов, которые могут быть преобразованы в пролекарственные формы для использования в данном изобретении включают, но не ограничиваются, соединения по изобретению и химиотерапевтические агенты, такие как описано выше.
«Метаболит» представляет собой продукт, который образуется в организме путем метаболизма конкретного соединения или его соли. Метаболиты соединения могут быть идентифицированы с применением обычных методов, известных в области техники и их активность можно определить с помощью таких испытаний, как описано в настоящем описании. Такие продукты могут быть получены в результате, например, окисления, гидроксилирования, восстановления, гидролиза, амидирования, дезамидирования, этерификации, деэтерификации, ферментативного расщепления и тому подобное, вводимого соединения. Соответственно, изобретение охватывает метаболиты соединений по изобретению, включая соединения, полученные способом, заключающимся в контактировании соединения по настоящему изобретению с млекопитающим в течение периода времени, достаточного для получения его метаболического продукта.
«Липосома» представляет собой небольшую везикулу, состоящую из различных типов липидов, фосфолипидов и/или поверхностно-активного вещества, которая используется для доставки препарата (такого, как ингибиторы MEK, описанные в данном документе, и, необязательно, химиотерапевтического средства) млекопитающему. Компоненты липосомы обычно расположены бислоем, подобно расположению липидов в биологических мембранах.
Используемый в данном документе термин «упаковочный вкладыш» относится к инструкциям, обычно вкладываемым в промышленно доступные упаковки терапевтических продуктов, которые содержат информацию о показаниях, применении, дозе, введении, противопоказаниях и/или предупреждениях, касающихся применения таких терапевтических продуктов.
Термин "хиральный" относится к молекулам, зеркальные отражения которых не-совпадают при наложении, тогда как термин "ахиральный" относится к молекулам, зеркальные отражения которых совпадают при наложении.
Термин «стереоизомер» относится к соединениям, котоые имеют одинаковое химическое строение и связи, но различную ориентацию атомов в пространстве, что не позволяет им быть взаимопревращаемыми при вращении вокруг одинарных связей.
«Диастереомер» относится к стереоизомеру с двумя или более центрами хиральности, и молекулы которого не являются зеркальным отражением одной для другой. Диастереомеры обладают разичными физическими свойствами, например точкой плавления, точками кипения, спектральными свойствами и реакционной способностью. Смеси диастереомеров могут быть разделены с помощью аналитических методов высокого разделения, таких как кристаллизация, электрофорез и хроматография.
«Энантиомеры» относятся к двум стереоизомерным соединениям, зеркальные отражения которых не могут быть наложены друг на друга.
Стереохимические определения и условные обозначения, используемые в данном документе, обычно соответствуют S. P. Parker, Ed., McGraw-Hill Dictionary of Chemical Terms (1984) McGraw-Hill Book Company, New York; и Eliel, E. And Wilen, S., «Stereochemistry of Organic Compounds», John Wiley & Sons, Inc., New York, 1994. Соединения по изобретению могут содержать асимметрические или хиральные центры, и поэтому существуют в виде различных стереоизомерных форм. Предполагается, что все стереоизомерные формы соединений по изобретению, включая, но этим не ограничиваясь, диастереомеры, энантиомеры и атропоизомеры, а также их смеси, такие как рацемические смеси, образуют часть настоящего изобретения. Многие органические соединения существуют в оптически активных формах, то есть, они обладают способностью к вращению плоскости плоскополяризованного света. В описании оптически активного соединения, префиксы D и L, или R и S, используются для обозначения абсолютной конфигурации молекулы относительно ее хирального центра(ов). Префиксы d и l или (+) и (-) используются для обозначения знака вращения плоскополяризованного света соединением, где (-) или l означает, что соединение является левовращающим. Соединение с префиксом (+) или d является правовращающим. Для данной химической структуры эти стереоизомеры являются идентичными, за исключением того, что они являются зеркальным отражением один другого. Конкретный стереоизомер может также быть указан как энантиомер, и смесь таких изомеров часто называют энантиомерной смесью. Смесь 50:50 энантиомеров обозначает рацемическую смесь или рацемат, которые могут образовываться, когда стереоизбирательность или стереоспецифичность в реакции или в процессе отсутствует. Термины «рацемическая смесь» и «рацемат» относятся к эквимолярной смеси двух энантиомерных образцов, не имеющих оптическую активность.
Термин «таутомер» или «таутомерная форма» относится к структурным изомерам различного энергетического уровня, которые могут взаимопревращаться при низком энергетическом барьере. Например, протонные таутомеры (также известные как прототропные таутомеры) включают взаимопревращения путем миграции протона, такие как кето-енол и имин-енамин изомеризации. Валентные таутомеры включают взаимопревращение путем перемещения некоторых электронов связи.
Фраза «фармацевтически приемлемая соль», как используется в данном документе, относится к фармацевтически приемлемой органической или неорганической соли соединения по изобретению. Примеры солей включают, но этим не ограничиваются, сульфат, цитрат, ацетат, оксалат, хлорид, бромид, йодид, нитрат, бисульфат, фосфат, кислота фосфат, изоникотинат, лактат, салицилат, кислый цитрат, тартрат, олеат, таннат, пантотенат, битартрат, аскорбат, сукцинат, малеат, гентисинат, фумарат, глюконат, глюкуронат, сахарат, формиат, бензоат, глютамат, метансульфонат «мезилат», этансульфонат, бензолсульфонат, п-толуолсульфонат, памоат (то есть, 1,1'-метилен-бис-(2-гидрокси-3-нафтоат)), соли щелочных металлов (например, натрия и калия), соли щелочноземельных металлов (например, магния) и соли аммония. Фармацевтически приемлемая соль может охватывать включение другой молекулы, такой как ион ацетата, ион сукцината или другого противоиона. Противоион может быть органической или неорганической группой, которая стабилизирует заряд первоначального соединения. Кроме того, фармацевтически приемлемая соль может иметь более одного заряженного атома в своей структуре. Например, когда многозарядные атомы являются частью фармацевтически приемлемой соли, она может иметь несколько противоионов. Таким образом, фармацевтически приемлемая соль может иметь один или несколько заряженных атомов и/или один или несколько противоионов.
Если соединение по изобретению представляет собой основание, желаемая фармацевтически приемлемая соль может быть получена любым подходящим способом, известным в данной области, например, обработкой свободного основания неорганической кислотой, такой как хлористоводородная кислота, бромистоводородная кислота, серная кислота, азотная кислота, метансульфоновая кислота, фосфорная кислота и тому подобное, или органической кислотой, такой как уксусная кислота, малеиновая кислота, янтарная кислота, миндальная кислота, фумаровая кислота, малоновая кислота, пировиноградная кислота, щавелевая кислота, гликолевая кислота, салициловая кислота, пиранозидиловая кислота, такая как глюкуроновая кислота или галактуроновая кислота, альфагидрокси кислота, такая как лимонная кислота или винно-каменная кислота, амино кислота, такая как аспарагиновая кислота или глутаминовая кислота, ароматическая кислота, такая как бензойной кислота или коричная кислота, сульфоновые кислоты, такие как п-толуолсульфоновая кислота или этансульфоновая кислота или тому подобное.
Если соединение по изобретению представляет собой кислоту, желаемая фармацевтически приемлемая соль может быть получена любым подходящим способом, например, обработкой свободной кислоты неорганическим или органическим основанием, таким как амин (первичный, вторичный или третичный), гидроксид щелочного металла или гидроксид щелочноземельного металла или тому подобное. Иллюстративные примеры подходящих солей включают, но этим не ограничиваются, органические соли производных аминокислот, таких как глицин и аргинин, аммиака, первичных, вторичных и третичных аминов и циклических аминов, таких как пиперидин, морфолин и пиперазин, и неорганические соли, производные натрия, кальция, калия, магния, марганца, железа, меди, цинка, алюминия и лития.
Фраза «фармацевтически приемлемый» указывает, что вещество или композиция должны быть химически и/или токсикологически совместимы с другими ингредиентами, составляющими препарат, и/или с млекопитающим, которого подвергают лечению этим препаратом.
«Сольват» относится к ассоциации или комплексу одного или нескольких молекул растворителя и соединения по изобретению. Примеры растворителей, которые образуют сольваты, включают, но этим не ограничиваются, воду, изопропанол, этанол, метанол, ДМСО, этилацетат, уксусную кислоту и этаноламин. Термин «гидрат» относится к комплексу, где молекулой растворителя является вода.
Термин «защитная группа» относится к заместителю, который обычно используется для блокирования или защиты конкретной функциональной группы в процессе реакции, протекающей по другим функциональным группам соединения. Например, «амино-защитная группа» представляет собой заместитель, присоединенный к амино группе, который блокирует или защищает функциональную амино группу соединения. Подходящие амино-защитные группы включают ацетил, трифторацетил, трет-бутоксикарбонил (BOC), бензилоксикарбонил (CBZ) и 9-флуоренилметиленоксикарбонил (Fmoc). Подобным образом, «гидрокси-защитная группа» относится к заместителю гидроксильной группы, который блокирует или защищает функциональную гидрокси группу. Подходящие защитные группы включают ацетил и триалкилсилил. «Карбокси-защитная группа» относится к заместителю карбоксильной группы, который блокирует или защищает функциональную карбоксильную группу. Общеизвестные карбокси-защитные группы включают фенилсульфонилэтил, цианоэтил, 2-(триметилсилил)этил, 2-(триметилсилил)этоксиметил, 2-(п-толуолсульфонил)этил, 2-(п-нитрофенилсульфенил)этил, 2-(дифенилфосфино)-этил, нитроэтил и тому подобное. Общее описание защитных группы и их применение смотри в обзоре T. W. Greene, Protective Groups in Organic Synthesis, John Wiley & Sons, New York, 1991.
Термины «соединение по данному изобретению», «соединения по настоящему изобретению» и «соединения формулы I или II», если не указано иного, включают соединения формулы I или II и их стереоизомеры, геометрические изомеры, таутомеры, сольваты, метаболиты, соли (например, фармацевтически приемлемые соли) и пролекарства. Если не указано иного, структуры, представленные в настоящем документе, подразумевают также включение соединений, которые отличаются только наличием одного или нескольких изотопно обогащенных атомов. Например, соединения формулы I или II, где один или несколько атомов водорода заменены дейтерием или тритием, или один или несколько атомов углерода заменены 13C- или 14C-обогащенным углеродом, входят в объем настоящего изобретения.
Настоящее изобретение относится к бициклическим гетероциклам формулы I и II, как описано выше, используемым в качестве ингибиторов киназы, в частности, используемым в качестве ингибиторов MEK киназы.
В соответствии с осуществлением настоящего изобретения, когда R3 представляет собой -(CR14R15)nC(=O)R11, -(CR14R15)nNR11R12, -(CR14R15)nOR11, -(CR14R15)nSR11, -(CR14R15)nS(O)R11 или -(CR14R15)nS(O)2R11; n равно 0; и Z1 представляет собой O, тогда указанный R11 или R12 не представляют собой арил; когда Z1 представляет собой O, тогда R не представляет собой CH2-арил; и все другие переменные имеют значения, указанные в формуле I.
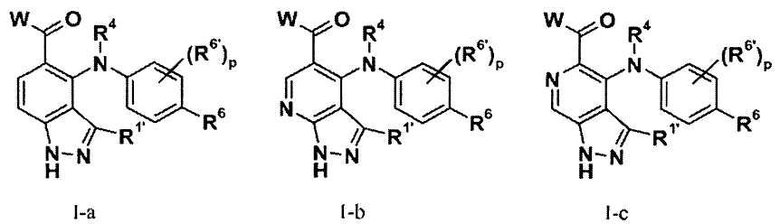
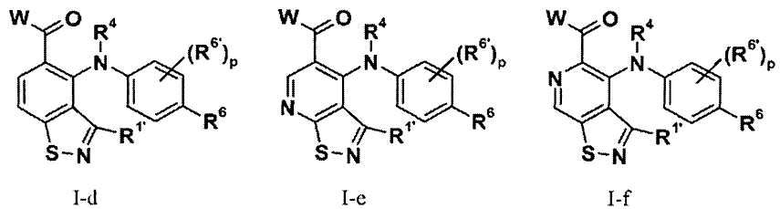
В соответствии с осуществлением настоящего изобретения, соединения имеют формулы I-a (то есть, Z1 представляет собой NH, и Z2 и Z3 представляют собой CH), I-b (то есть, Z1 представляет собой NH, Z2 представляет собой N и Z3 представляет собой CH), I-c (то есть, Z1 представляет собой NH, Z2 представляет собой CH и Z3 представляет собой N), I-d (то есть, Z1 представляет собой S, Z2 и Z3 представляют собой CH), I-e (то есть, Z1 представляет собой S, Z2 представляет собой N и Z3 представляет собой CH), I-f (то есть, Z1 is S, Z2 представляет собой CH и Z3 представляет собой N), II-a (то есть, Z1 представляет собой NH, и Z2 и Z3 представляют собой CH), II-b (то есть, Z1 представляет собой NH, Z2 представляет собой N и Z3 представляет собой CH), II-c (то есть, Z1 представляет собой NH, Z2 представляет собой CH и Z3 представляет собой N), II-d (то есть, Z1 представляет собой S, Z2 и Z3 представляют собой CH), II-e (то есть, Z1 is S, Z2 представляет собой N и Z3 представляет собой CH), или II-f (то есть, Z1 представляет собой S, Z2 представляет собой CH и Z3 представляет собой N); и все другие переменные имеют значения, указанные в формуле I или II.
В соответствии с осуществлением настоящего изобретения, Z2 представляет собой CR2 и R2 представляет собой H, галоген, CF3, или C1-C3 алкил; и все другие переменные имеют значения, указанные в формуле I или II, или как указано в каком-либо из вариантов осуществлений, описанных выше.
В соответствии с другим осуществлением настоящего изобретения, Z2 представляет собой CR2 и R2 представляет собой H, метил, CF3, F или Cl; и все другие переменные имеют значения, указанные в формуле I или II, или как указано в каком-либо из вариантов осуществлений, описанных выше.
В соответствии с другим осуществлением настоящего изобретения, Z2 представляет собой CR2 и R2 представляет собой H, F или Cl; и все другие переменные имеют значения, указанные в формуле I или II, или как указано в каком-либо из вариантов осуществлений, описанных выше.
В соответствии с другим осуществлением настоящего изобретения, Z2 представляет собой N; и все другие переменные имеют значения, указанные в формуле I или II, или как указано в каком-либо из вариантов осуществлений, описанных выше.
В соответствии с осуществлением настоящего изобретения, Z3 представляет собой CR3 и R3 представляет собой H, галоген, CF3, O-C1-C3 алкил) или C1-C3 алкил; и все другие переменные имеют значения, указанные в формуле I или II, или как указано в каком-либо из вариантов осуществлений, описанных выше.
В соответствии с другим осуществлением настоящего изобретения, Z3 представляет собой CR3 и R3 представляет собой H, метил, CF3, F, OMe, или Cl; и все другие переменные имеют значения, указанные в формуле I или II, или как указано в каком-либо из вариантов осуществлений, описанных выше.
В соответствии с другим осуществлением настоящего изобретения, Z3 представляет собой CR3 и R3 представляет собой H, F, OMe или Cl; и все другие переменные имеют значения, указанные в формуле I или II, или как указано в каком-либо из вариантов осуществлений, описанных выше.
В соответствии с другим осуществлением настоящего изобретения, Z3 представляет собой N; и все другие переменные имеют значения, указанные в формуле I или II, или как указано в каком-либо из вариантов осуществлений, описанных выше.
В соответствии с осуществлением настоящего изобретения, R1 представляет собой H, и все другие переменные имеют значения, указанные в формуле I или II, или как указано в каком-либо из вариантов осуществлений, описанных выше.
В соответствии с другим осуществлением настоящего изобретения, Z1 представляет собой NR1; и все другие переменные имеют значения, указанные в формуле I или II, или как указано в каком-либо из вариантов осуществлений, описанных выше. В другом варианте осуществления, R1 представляет собой H, и все другие переменные имеют значения, указанные в формуле I или II, или как указано в любом из вышеуказанных вариантов осуществлений.
В соответствии с другим осуществлением настоящего изобретения, Z1 представляет собой S; и все другие переменные имеют значения, указанные в формуле I или II, или как указано в каком-либо из вариантов осуществлений, описанных выше.
В соответствии с осуществлением настоящего изобретения, R4 представляет собой H или C1-C6 алкил; и все другие переменные имеют значения, указанные в формуле I или II, или как указано в любом из вышеуказанных вариантов осуществлений.
В соответствии с другим осуществлением настоящего изобретения, R4 представляет собой H или метил; и все другие переменные имеют значения, указанные в формуле I или II, или как указано в любом из вышеуказанных вариантов осуществлений. В соответствии с другим осуществлением настоящего изобретения, R4 представляет собой H; и все другие переменные имеют значения, указанные в формуле I или II, или как указано в любом из вышеуказанных вариантов осуществлений.
В соответствии с осуществлением настоящего изобретения, R5 представляет собой H или C1-C6 алкил; и все другие переменные имеют значения, указанные в формуле I или II, или как указано в любом из вышеуказанных вариантов осуществлений.
В соответствии с другим осуществлением настоящего изобретения, R5 представляет собой H или метил; и все другие переменные имеют значения, указанные в формуле I или II, или как указано в любом из вышеуказанных вариантов осуществлений.
В соответствии с другим осуществлением настоящего изобретения, R5 представляет собой H; и все другие переменные имеют значения, указанные в формуле I или II, или как указано в любом из вышеуказанных вариантов осуществлений.
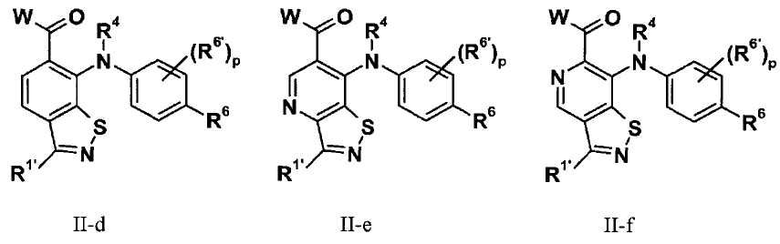
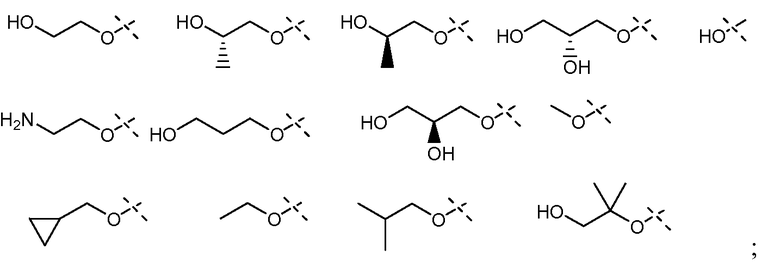
В соответствии с осуществлением настоящего изобретения, X1 представляет собой OR11', где R11' представляет собой H или C1-C12 алкил (например, C1-C6 алкил), замещенный одной или несколькими группами, независимо выбранными из галогена, CN, CF3, -OCF3, -NO2, оксо, -(CR19R20)nC(=Y')R16, -(CR19R20)nC(=Y')OR16, -(CR19R20)nC(=Y')NR16R17, -(CR19R20)nNR16R17, -(CR19R20)nOR16, -(CR19R20)nSR16, -(CR19R20)nNR16C(=Y')R17, -(CR19R20)nNR16C(=Y')OR17, -(CR19R20)nNR18C(=Y')NR16R17, -(CR19R20)nNR17SO2R16, -(CR19R20)nOC(=Y')R16, -(CR19R20)nOC(=Y')OR16, -(CR19R20)nOC(=Y')NR16R17, -(CR19R20)nOS(O)2(OR16), -(CR19R20)nOP(=Y')(OR16)(OR17), -(CR19R20)nOP(OR16)(OR17), -(CR19R20)nS(O)R16, -(CR19R20)nS(O)2R16, -(CR19R20)nS(O)2NR16R17, -(CR19R20)nS(O)(OR16), -(CR19R20)nS(O)2(OR16), -(CR19R20)nSC(=Y')R16, -(CR19R20)nSC(=Y')OR16, -(CR19R20)nSC(=Y')NR16R17 и R21; и все другие переменные имеют значения, указанные в формуле I или II, или как указано в любом из вышеуказанных вариантов осуществлений.
В соответствии с другим осуществлением настоящего изобретения, X1 представляет собой OR11', где R11' представляет собой гетероциклил (например, 4-6-членный гетероциклил), необязательно замещенный одним или несколькими группами, независимо выбранными из галогена, CN, CF3, -OCF3, -NO2, оксо, -(CR19R20)nC(=Y')R16, -(CR19R20)nC(=Y')OR16, -(CR19R20)nC(=Y')NR16R17, -(CR19R20)nNR16R17, -(CR19R20)nOR16, -(CR19R20)nSR16, -(CR19R20)nNR16C(=Y')R17, -(CR19R20)nNR16C(=Y')OR17, -(CR19R20)nNR18C(=Y')NR16R17, -(CR19R20)nNR17SO2R16, -(CR19R20)nOC(=Y')R16, -(CR19R20)nOC(=Y')OR16, -(CR19R20)nOC(=Y')NR16R17, -(CR19R20)nOS(O)2(OR16), -(CR19R20)nOP(=Y')(OR16)(OR17), -(CR19R20)nOP(OR16)(OR17), -(CR19R20)nS(O)R16, -(CR19R20)nS(O)2R16, -(CR19R20)nS(O)2NR16R17, -(CR19R20)nS(O)(OR16), -(CR19R20)n S(O)2(OR16), -(CR19R20)nSC(=Y')R16, -(CR19R20)nSC(=Y')OR16, -(CR19R20)n SC(=Y')NR16R17 и R21; и все другие переменные имеют значения, указанные в формуле I или II, или как указано в любом из вышеуказанных вариантов осуществлений.
В соответствии с другим осуществлением настоящего изобретения, X1 представляет собой OR11', где R11' представляет собой 4-6-членный гетероциклил, содержащий 1 кольцевой атом азота, где указанный гетероциклил необязательно замещен одним или несколькими группами, независимо выбранными из галогена, CN, CF3, -OCF3, -NO2, оксо, -(CR19R20)nC(=Y')R16, -(CR19R20)nC(=Y')OR16, -(CR19R20)nC(=Y')NR16R17, -(CR19R20)nNR16R17, -(CR19R20)nOR16, -(CR19R20)nSR16, -(CR19R20)nNR16C(=Y')R17, -(CR19R20)nNR16C(=Y')OR17, -(CR19R20)nNR18C(=Y')NR16R17, -(CR19R20)nNR17SO2R16, -(CR19R20)nOC(=Y')R16, -(CR19R20)nOC(=Y')OR16, -(CR19R20)nOC(=Y')NR16R17, -(CR19R20)nOS(O)2(OR16), -(CR19R20)nOP(=Y')(OR16)(OR17), -(CR19R20)nOP(OR16)(OR17), -(CR19R20)nS(O)R16, -(CR19R20)nS(O)2R16, -(CR19R20)nS(O)2NR16R17, -(CR19R20)nS(O)(OR16), -(CR19R20)nS(O)2(OR16), -(CR19R20)nSC(=Y')R16, -(CR19R20)nSC(=Y')OR16, -(CR19R20)nSC(=Y')NR16R17 и R21; и все другие переменные имеют значения, указанные в формуле I или II, или как указано в любом из вышеуказанных вариантов осуществлений.
В соответствии с другим осуществлением настоящего изобретения, X1 представляет собой:
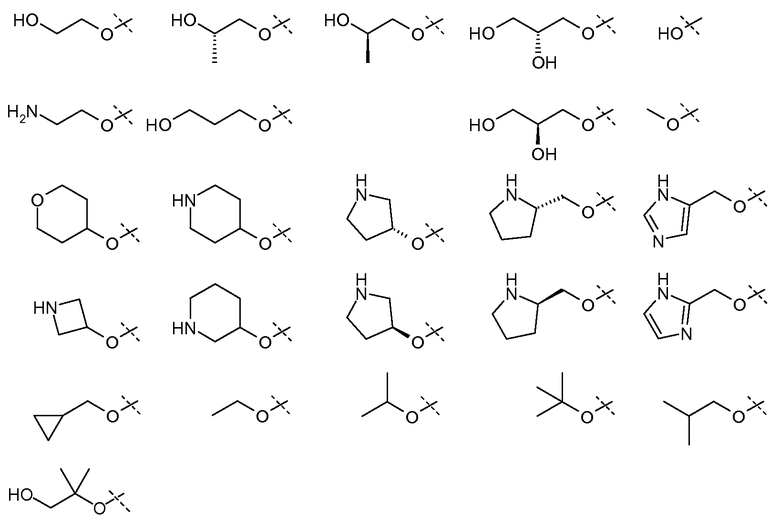
и все другие переменные имеют значения, указанные в формуле I или II, или как указано в любом из вышеуказанных вариантов осуществлений.
В соответствии с другим осуществлением настоящего изобретения, X1 представляет собой
и все другие переменные имеют значения, указанные в формуле I или II, или как указано в любом из вышеуказанных вариантов осуществлений.
В соответствии с осуществлением настоящего изобретения, W представляет собой 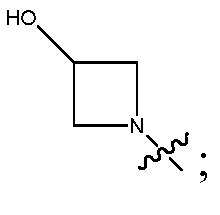 и все другие переменные имеют значения, указанные в формуле I или II, или как указано в любом из вышеуказанных вариантов осуществлений.
и все другие переменные имеют значения, указанные в формуле I или II, или как указано в любом из вышеуказанных вариантов осуществлений.
В соответствии с осуществлением настоящего изобретения, W представляет собой -OR11', где R11' представляет собой H или C1-C12 алкил; и все другие переменные имеют значения, указанные в формуле I или II, или как указано в любом из вышеуказанных вариантов осуществлений.
В соответствии с другим осуществлением настоящего изобретения, W представляет собой -OR11', где R11' представляет собой H; и все другие переменные имеют значения, указанные в формуле I или II, или как указано в любом из вышеуказанных вариантов осуществлений.
В соответствии с другим осуществлением настоящего изобретения, W представляет собой -OR11', где R11' представляет собой C1-C6 алкил; и все другие переменные имеют значения, указанные в формуле I или II, или как указано в любом из вышеуказанных вариантов осуществлений.
В соответствии с осуществлением настоящего изобретения, W представляет собой -NHSO2R8; и все другие переменные имеют значения, указанные в формуле I или II, или как указано в любом из вышеуказанных вариантов осуществлений. В соответствии с осуществлением настоящего изобретения, R8 представляет собой циклопропил; и все другие переменные имеют значения, указанные в формуле I или II, или как указано в любом из вышеуказанных вариантов осуществлений.
В соответствии с осуществлением настоящего изобретения, R6 представляет собой галоген, C2-C8 алкинил, карбоциклил, или -SR16; и все другие переменные имеют значения, указанные в формуле I или II, или как указано в любом из вышеуказанных вариантов осуществлений.
В соответствии с другим осуществлением настоящего изобретения, R6 представляет собой галоген, C2-C3 алкинил, C3-карбоциклил, или -SR16, где R16 представляет собой C1-C2 алкил; и все другие переменные имеют значения, указанные в формуле I или II, или как указано в любом из вышеуказанных вариантов осуществлений.
В соответствии с другим осуществлением настоящего изобретения, R6 представляет собой Br, I, SMe, C3-карбоциклил, или C2 алкинил; и все другие переменные имеют значения, указанные в формуле I или II, или как указано в любом из вышеуказанных вариантов осуществлений.
В соответствии с осуществлением настоящего изобретения, R6' представляет собой H, галоген, или C1-C3 алкил; и все другие переменные имеют значения, указанные в формуле I или II, или как указано в любом из вышеуказанных вариантов осуществлений.
В соответствии с осуществлением настоящего изобретения, R6' представляет собой H, F, Cl или метил; и все другие переменные имеют значения, указанные в формуле I или II, или как указано в любом из вышеуказанных вариантов осуществлений.
В соответствии с другим осуществлением настоящего изобретения, R6' представляет собой F или Cl; и все другие переменные имеют значения, указанные в формуле I или II, или как указано в любом из вышеуказанных вариантов осуществлений.
В соответствии с осуществлением настоящего изобретения, p равно 1 или 2; и все другие переменные имеют значения, указанные в формуле I или II, или как указано в любом из вышеуказанных вариантов осуществлений.
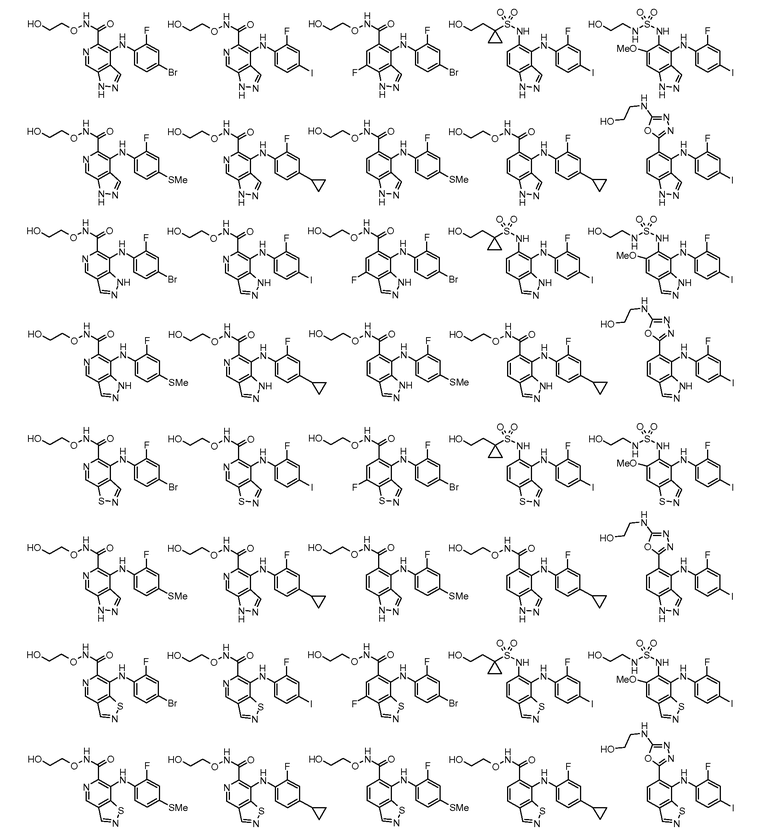
Другой вариант осуществления настоящего изобретения включает соединения, описанные в примерах 5-29 и соединения показанные ниже:
Соединения формулы I и II получали в соответствии со способами, описанными далее в схемах и примерах, или способами, известными в данной области. Например, соединения формулы I могут быть получены в соответствии со схемой 1.
Схема 1
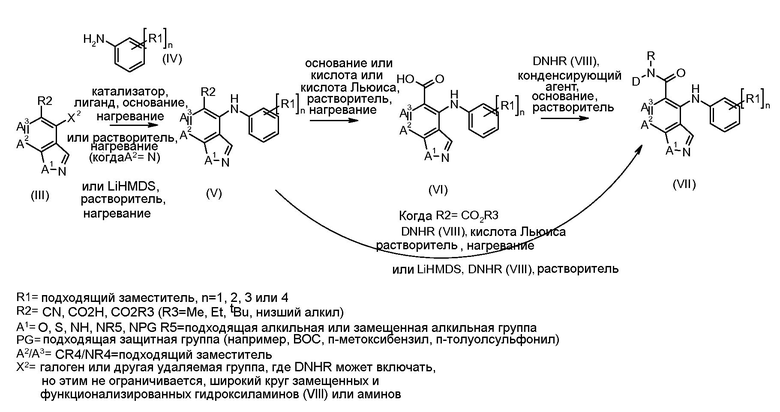
Соединения формулы (VII) могут быть получены из промежуточных соединений формулы (III) (полученных в соответствии со схемами 2, 3 и 5-8 далее). Соединения формулы (V) могут быть получены, исходя из соединений формулы (III) путем взаимодействия с анилином формулы (IV) (включая подходящие заместители R1), в присутствии катализатора, такого как трис(дибензилиденацетон)дипалладий(0) или палладий(II) ацетат, основания, такого как фосфат калия или карбонат цезия, лиганда, такого как Xantphos или 2-дициклогексилфосфино-2',6'- (диизопропокси)бифенил, в подходящем растворителе, таком как толуол или DME, при температуре от комнатной температуры до температуры кипения растворителя, или под действием микроволнового излучения при температуре от 70ºC до 150ºC. Альтернативно, соединения формулы (V) могут быть получены исходя из соединений формулы (III) путем взаимодействия анилина формулы (IV) в присутствии сильного основания, такого как литий бис(триметилсилил)амид, в растворителе, таком как ТГФ, при температуре от -78ºC до комнатной температуры. Альтернативно, и предпочтительно, когда A2 представляет собой N, анилин и соединение формулы (III) могут быть подвергнуты взаимодействию в растворителе, таком как диоксан или ДМФ, в присутствии основания, такого как карбонат калия, при температуре от 50ºC до температуры кипения.
Соединения формулы (VI) могут быть получены исходя из соединений формулы (V), где R2 представляет собой CO2R3 и R3 представляет собой Me, этил, другой алкил, путем взаимодействия с основанием, таким как гидроксид натрия, в растворителе, таком как этанол или метанол, при температуре от комнатной температуры до температуры кипения. Когда R3 представляет собой CO2 tBu, соединения формулы (VI) могут быть получены исходя из соединений формулы (V) путем обработки кислотой, такой как ТФУ, чистой, или в присутствии растворителя, такого как DCM, при температуре от 0ºC до температуры кипения.
Альтернативно, когда R3 представляет собой Me, может быть осуществлено омыление в не-основных условиях, путем обработки кислотой Льюиса, такой как бис(три-н-бутилолово)оксид, в растворителе, таком как толуол, при температуре от комнатной до температуры кипения.
Соединения формулы (VI) могут быть подвергнуты взаимодействию с функционализированным гидроксиламином формулы (VIII) (коммерчески доступный или полученный в соответствии со схемой 11) или с амином и подходящим конденсирующим агентом, таким как O-(7-аза-бензо-триазол-1-ил)-N,N,N',N'-тетра-метилуроний гексафтор-фосфат, N-(3-диметиламинопропил)-N'-этилкарбодиимид гидрохлорид или NN'-дициклогексилкарбодиимид, в присутствии N-гидрокси-1,2,3-бензотриазола, в присутствии подходящего основания, такого как диизопропилэтиламин или триэтиламин, в инертном растворителе, таком как тетрагидрофуран, N,N-диметилформамид или дихлорметан, при температуре около комнатной температуры, с получением соединений формулы (VII). Альтернативно, соединения формулы (VII) могут быть получены напрямую исходя из соединений формулы (V) путем взаимодействия с амином или гидроксиламином DΝHR (VIII) в присутствии сильного основания, такого как бис(триметилсилил)амид лития, в растворителе, таком как ТГФ, при температуре от -20ºC до комнатной температуры. Альтернативно, соединения формулы (VII) могут быть получены напрямую, исходя из соединений формулы (V) путем взаимодействия с амином или гидроксиламином DΝHR (VIII) в присутствии кислоты Льюиса, такой как триметил алюминия, в растворителе, таком как DCM, при температуре от комнатной температуры до температуры кипения.
В соединениях формулы (VII), где A1 представляет собой Ν, защитная группы (ΝPG) может быть добавлена и удалена на любой стадии синтеза, как потребуется.
Соединения формулы (III), где A1 представляет собой NH, NR5 и NPG могут быть получены в соответствии со схемой 2.
Схема 2
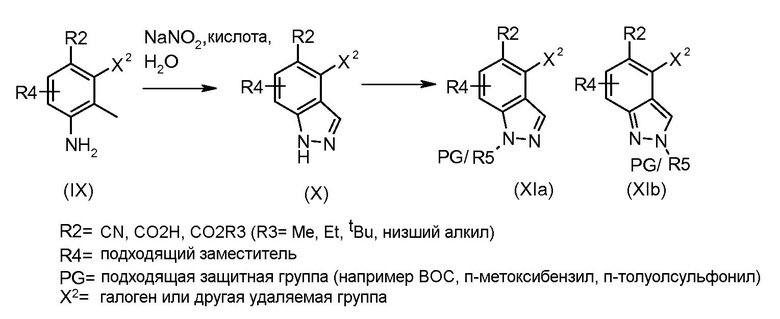
Соединения формулы (IX) могут быть получены, используя способы, описанные в литературе. Соединения формулы (X) могут быть получены исходя из соединений формулы (IX) путем взаимодействия с диазотирующим агентом, таким как нитрит натрия, в присутствии кислоты, такой как уксусная кислота или тетрафторборная кислота, и растворителя, такого как вода, при температуре от -20ºC до 50ºC. Соединения формулы (X) могут быть защищены подходящей защитной группой для получения соединений формулы (XIa) и (XIb) путем взаимодействия с подходящим сульфонилхлоридом, таким как п-толуолсульфонилхлорид, или алкилхлоридом, таким как 2-(триметилсилил)этоксиметил хлорид, в присутствии основания, такого как триэтиламин или гидрид натрия, в растворителе, таком как ТГФ, или DCM, при температуре от 0ºC до комнатной температуры. Альтернативно, соединения формулы (X) могут быть защищены карбаматой защитной группой, такой как трет-бутил карбамат, путем взаимодействия соединения формулы (X) с ди-трет-бутил дикарбонатом в присутствии основания третичного амина, такого как триэтиламин, в растворителе, таком как DCM, при температуре около комнатной. Индазолы, полученные этими способами, могут быть выделены в виде смеси изомеров (XIa) и (XIb), как показано.
Альтернативно, соединения формулы (III), где A1 представляет собой NH, NR5 или NPG, могут быть получены в соответствии со схемой 3.
Схема 3
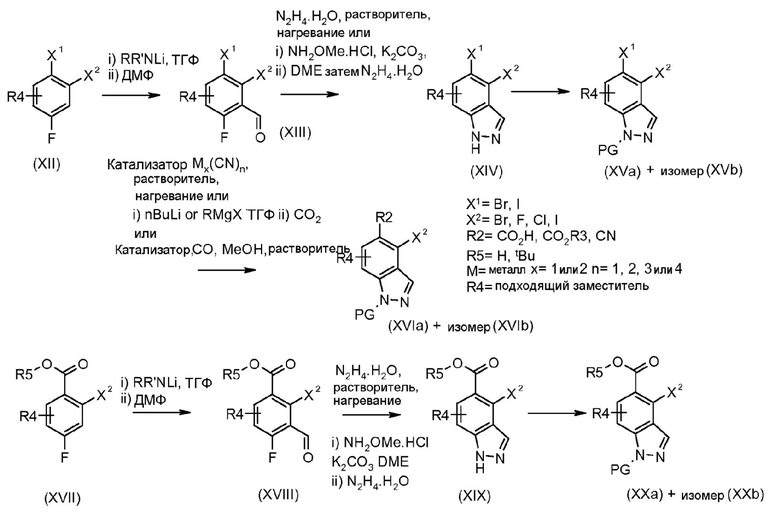
Соединения формул (XII) и (XVII) могут быть коммерчески доступными или получены, используя способы, описанные в литературе. Соединения формул (XIII) и (XVIII) могут быть получены, исходя из соединений формул (XII) и (XVII), соответственно, путем взаимодействия со стерически затрудненным сильным основанием, таким как диизопропиламид лития, в растворителе, таком как ТГФ, при температуре от -80ºC до 0ºC, затем путем гашения формилирующим реагентом, таким как ДМФ или 1-формилпиперидин. Соединения формул (XIII) и (XVIII) могут быть преобразованы в соединения формул (XIV) и (XIX) путем обработки гидразин гидратом, чистым, или в растворителе, таком как этанол или DME, при температуре от комнатной температуры до 150ºC. Альтернативно, соединения формул (XIV) и (XIX) могут быть получены, исходя из соединений формул (XIII) и (XVIII) путем преобразования в промежуточный оксим путем взаимодействия с гидроксиламином, таким как O-метилгидроксиламин, в растворителе, таком как DME, в присутствии основания, такого как карбонат калия, при температуре от комнатной до температуры кипения. Промежуточные оксимы могут быть преобразованы в индазолы формул (XIV) и (XIX) без выделения путем обработки гидразин гидратом, чистым, или в присутствии растворителя, такого как DME. Соединения формул (XIV) и (XIX) могут быть преобразованы в соединения формул (XVa/XVb) и (XXa/XXb), используя способы, описанные для преобразования соединений формулы (X) в соединения формул (XIa) и (XIb). Соединения формул (XVa/XVb), где X1 представляет собой I, Br, могут быть преобразованы в соединения формулы (XVIa/XVIb), где R2 представляет собой CO2R3 путем большого числа различных способов. Наиболее предпочтительно, соединения формул (XVIa/XVIb) могут быть получены исходя из соединений формул (XVa/XVb) путем обмена металл-галоген при обработке сильным металлоорганическим основанием, таким как н-бутилитий или реактив Гриньяра, такой как изопропил магний йодид, в растворителе, таком как ТГФ, при температуре от -80ºC до 0ºC. Промежуточные производные ариллития или арилмагния могут быть преобразованы в соединения формул (XVIa/XVIb) с помощью гашения электрофилом, таким как CO2 или метилхлорформиат. Альтернативно, соединения формул (XVIa/XVIb) могут быть получены, исходя из соединений формул (XVa/XVb) путем катализирумого переходным металлом карбонилирования, используя катализатор, такой как ацетат палладия(II), основание, такое как DIPEA, со-катализатор, такой как DMAP, в растворителе, таком как метанол, и источник монооксида углерода, такой как Mo(CO)6, при температуре от 80ºC до температуры кипения, но, предпочтительно, используя микроволновое облучение, при температуре от 150ºC до 200ºC при давлении 1-10 бар. Соединения формул (XVa/XVb), где X1 представляет собой I или Br, могут быть преобразованы в соединения формулы (XVI), где R2 представляет собой CN, путем взаимодействия с цианидом металла, таким как цианид цинка, в присутствии катализатора, такого как тетракис(трифенилфосфин) палладий(0), в растворителе, таком как ДМФ, при температуре от 50ºC до температуры кипения или используя микроволновое нагревание при температуре от 120ºC до 200ºC.
Соединения формулы (V), где A1 представляет собой NH, NR5, или NPG, могут быть также получены в соответствии со схемой 4.
Схема 4
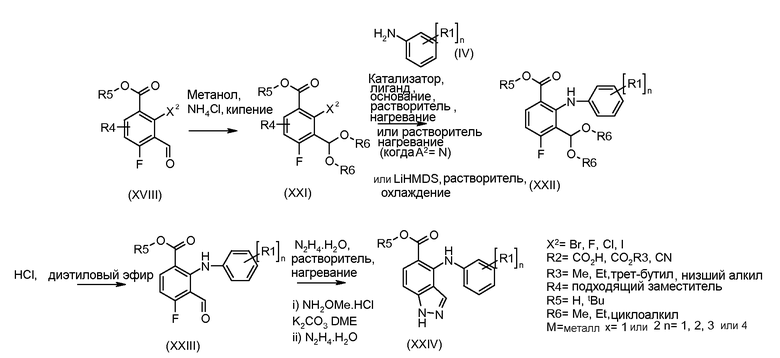
Соединения формулы (XVIII) могут быть коммерчески доступными или получены, используя способы, описанные в литературе. Соединения формулы (XVIII) могут быть преобразованы в соединения формулы (XXI) путем взаимодействия со спиртом, таким как метанол (R6= Me), в присутствии кислоты, такой как аммоний хлорид, при температуре около температуры кипения. Соединения формулы (XXI) могут быть преобразованы в соединения формулы (XXII), используя способы, описанные для преобразования соединений формулы (III) в соединения формулы (V) по схеме 1. Соединения формулы (XXII) могут быть преобразованы в соединения формулы (XXIII) путем взаимодействия с кислотой, такой как хлористоводородная кислота, в растворителе, таком как эфир, при температуре около комнатной. Соединения формулы (XXIII) могут быть преобразованы в соединения формулы (XXIV), используя способы, описанные для преобразования соединений формулы (XVIII) в соединения формулы (XIX) по схеме 3.
Соединения формулы (III), где A1 представляет собой S и R2 представляет собой CO2R3, могут быть получены в соответствии со схемой 5.
Схема 5
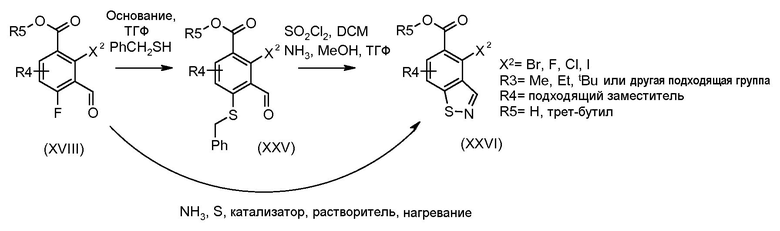
Соединения формулы (XVIII), полученные в соответствии со схемой 3, могут быть преобразованы в соединения формулы (XXVI) путем двухстадийного способа. Соединения формулы (XVIII) могут быть подвергнуты взаимодействию с бензолметантиолом в присутствии основания, такого как трет-бутоксид калия, в растворителе, таком как ТГФ, при температуре от 0ºC до температуры кипения. Промежуточные тиоэфиры формулы (XXV) могут быть преобразованы в соединения формулы (XXVI) путем обработки сульфурилхлоридом в растворителе, таком как дихлорметан, с последующим взаимодействием с аммиаком в растворителе, таком как смесь этанол/ТГФ. Альтернативно, соединения формулы (XXVI) могут быть получены, исходя из соединений формулы (XVIII) прямой обработкой элементарной серой, аммиаком или гидроксидом аммония в растворителе, такие как ДМФ или 2-метоксиэтанол, в присутствии катализатора, такого как метиламин, при температуре от 100ºC до температуры кипения или при более высокой температуре, чем температура кипения (от 150 до 200ºC), с использованием реакционного автоклава под давлением от 1-20 бар.
Соединения формулы (XXIX), где A2 представляет собой N, могут быть получены в соответствии со схемой 6.
Схема 6
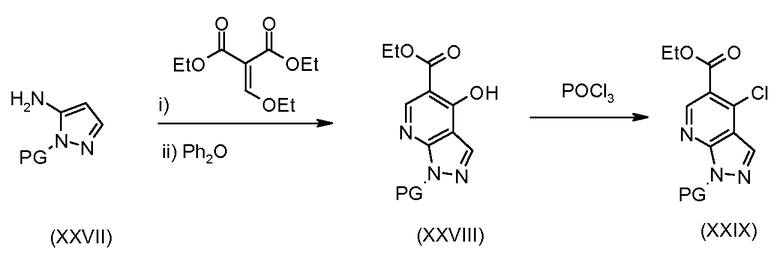
Защищенные аминопиразолы формулы (XXVII) могут быть получены, используя способы, описанные в литературе. Соединения формулы (XXVII) могут быть подвергнуты взаимодействию с эфиром 2-алкоксиметиленмалоновой кислоты, таким как диэтиловый эфир 2-этоксиметиленмалоновой кислоты, в присутствии высококипящего растворителя, такого как дифениловый эфир, при температуре от 150ºC до 300ºC с получением соединения формулы (XXVIII). Соединения формулы (XXVIII) могут быть преобразованы в соединения формулы (XXIX) путем обработки галогенирующим агентом, таким как оксихлорид фосфора, чистый, или в присутствии растворителя, такого как толуол, в присутствии или в отсутствие основания, такого как триэтиламин, при температуре от 50ºC до температуры кипения.
Соединения формулы XXXIVa и XXXIVb могут быть получены в соответствии со схемой 7.
Схема 7
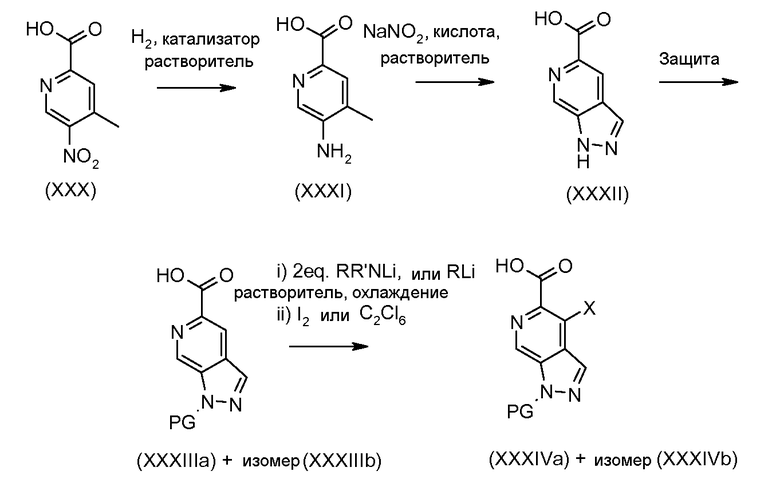
Соединения формулы (XXX) могут быть коммерчески доступными или получены, используя способы, описанные в литературе. Соединения формулы (XXX) могут быть преобразованы в соединения формулы (XXXI) путем восстановления нитрогруппы, используя катализатор, такой как никель Ренея, при давлении водорода (1-5 бар) в растворителе, таком как ТГФ, при комнатной температуре. Соединения формулы (XXXII) могут быть получены, исходя из соединений формулы (XXXI) путем обработки диазотирующим агентом, таким как нитрит натрия, в присутствии кислоты, такой как уксусная кислота или тетрафторборная кислота, и растворителя, такого как вода, при температуре от -20ºC до 50ºC. Соединения формулы (XXXII) могут быть преобразованы в соединения формулы (XXXIII), где PG представляет собой SEM (SEM=2-(триметилсилил)этоксиметил) путем обработки SEM-Cl в присутствии основания, такого как гидрид натрия, в растворителе, таком как ТГФ, в области комнатной температуры. Соединения формул (XXXIIIa/XXXIIIb) могут быть преобразованы в соединения формул (XXXIVa/XXXIVb) путем орто-литирования сильным основанием, таким как литий тетраметилпиперидин, в растворителе, таком как ТГФ, при температуре от -100ºC до -60ºC, с последующим гашением галогенирующим агентом, таким как йод или гексахлорэтан, при температуре от -100ºC до 0ºC.
Промежуточные соединения формул XXXVIIa/XXXVIIb могут быть получены в соответствии со схемой 8.
Схема 8
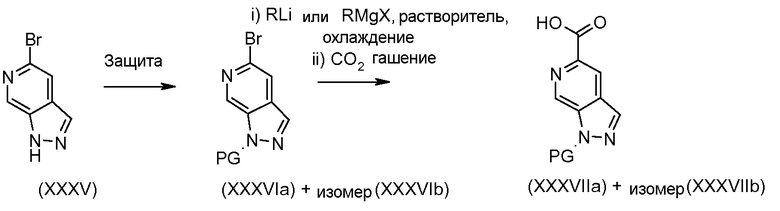
Индазолы формулы (XXXV) могут быть коммерчески доступными или получены в соответствии со способами, описанными в литературе. Соединения формул (XXXVIa/XXXVIb) могут быть получены, исходя из соединений формулы (XXXV), используя способы, описанные для преобразования соединений формулы (X) в соединения формул (XIa/XIb), как показано на схеме 2. Соединения формул (XXXVIa/XXXVIb) могут быть преобразованы в кислоты формул (XXXVIIa/XXXVIIb) путем обмена литий-галоген, используя сильное металлоорганическое основание, такое как н-бутиллитий, в растворителе, таком как ТГФ, при температуре от -100ºC до -60ºC, с последуюшим гашением электрофилом, таким как диоксид углерода при температуре от -78ºC до 0ºC. Альтернативно, соединения формул (XXXVIa/XXXVIb) могут быть преобразованы в соединения формул (XXXVIIa/XXXVIIb) путем образования промежуточного сложного эфира, полученного путем взаимодействия гетероарилгалогенида с монооксидом углерода (при давлении 1-15 бар) в присутствии катализатора, такого как ацетат палладия или 1,1'-бис(дифенилфосфино)ферроцен, и лиганда, такого как трифенилфосфин, и основания, такого как ацетат натрия, в присутствии спирта, такого как метанол, в растворителе, таком как ДМФ или метанол, при температуре от 80ºC до 200ºC.
Соединения формулы II могут быть получены в соответствии со схемой 9.
Схема 9
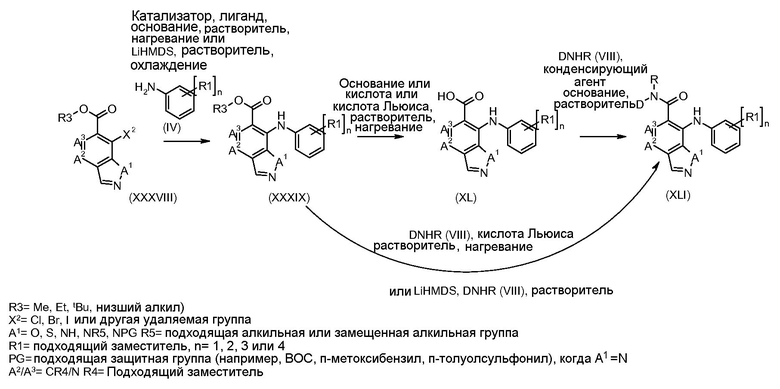
Соединения формул (XXXIX) могут быть получены из промежуточных соединений формул (XXXVIII) (полученных в соответствии со схемами 9 и 10 далее). Соединения формул (XXXIX) могут быть получены исходя из соединений формул (XXXVIII), используя способы, описанные для преобразования соединений формулы (III) в соединения формулы (V) по схеме 1. Соединения формулы (XLI) могут быть получены, исходя из соединений формулы (XXXIX) и (XL), используя способы, описанные для преобразования соединений формул (V) и (VI) в соединения формул (VII), как показано на схеме 1.
Соединения формулы (XLI), где A1 представляет собой NH защитную группу (NPG), могут быть добавлены и удалены на любой стадии синтеза, как потребуется.
Промежуточные соединения формул (XLVI) и (XLVII) могут быть получены в соответствии со схемой 10 далее.
Схема 10
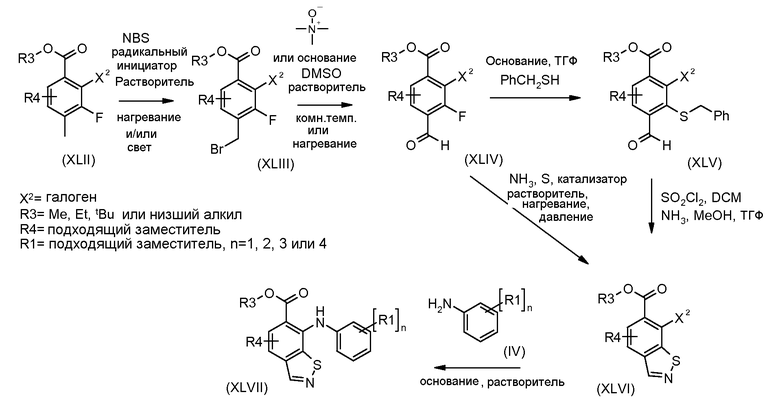
Соединения формулы (XLII) могут быть подвергнуты взаимодействию с бромирующим агентом, таким как NBS, в присутствии радикального инициатора, такого как AIBN, в растворителе, таком как тетрахлорид углерода, при температуре кипения, с активацией или без активации светом, с получением соединения формулы (XLIII). Соединения формулы (XLIII) могут быть преобразованы в соединения формулы (XLIV) путем обработки N-оксидом триметиламина, в присутствии ДМСО, в растворителе, таком как DCM, при температуре от комнатной до температуры кипения. Альтернативно, соединения формулы (XLIV) могут быть получены путем обработки соединений формулы (XLIII) основанием, таким как гидрокарбонат натрия, в ДМСО, при температуре около 100ºC. Соединения формулы (XLIV) могут быть подвергнуты взаимодействию с бензолметантиолом в присутствии основания, такого как трет-бутоксид калия, в растворителе, таком как ТГФ, при температуре от -78ºC до -30ºC с получением соединений формулы (XLV). Промежуточные тиоэфиры формулы (XLV) могут быть преобразованы в соединения формулы (XLVI) путем обработки сульфурилхлоридом в растворителе, таком как дихлорметан, с последующим взаимодействием с аммиаком в смеси растворителей, такой как метанол/ТГФ. Альтернативно, соединения формулы (XLVI) могут быть получены исходя из соединений формулы (XLIV) напрямую путем обработки элементарной серой, аммиаком или гидроксидом аммония, в растворителе, таком как ДМФ или 2-метоксиэтанол, в присутствии катализатора, такого как метиламин, при температуре от 100ºC до температуры кипения или при более высокой температуре, чем температура кипения (150-200ºC) с использованием реакционного автоклава при давлении 1-20 бар. Соединения формулы (XLVI) могут быть преобразованы в соединения формулы (XLVII), используя способы, описанные для преобразования соединений формулы (III) в соединения формулы (V) по схеме 1.
Соединения формулы (I) W'=NHSO2R8 или NHSO2NR8R10 могут быть получены в соответствии со схемой 11.
Схема 11
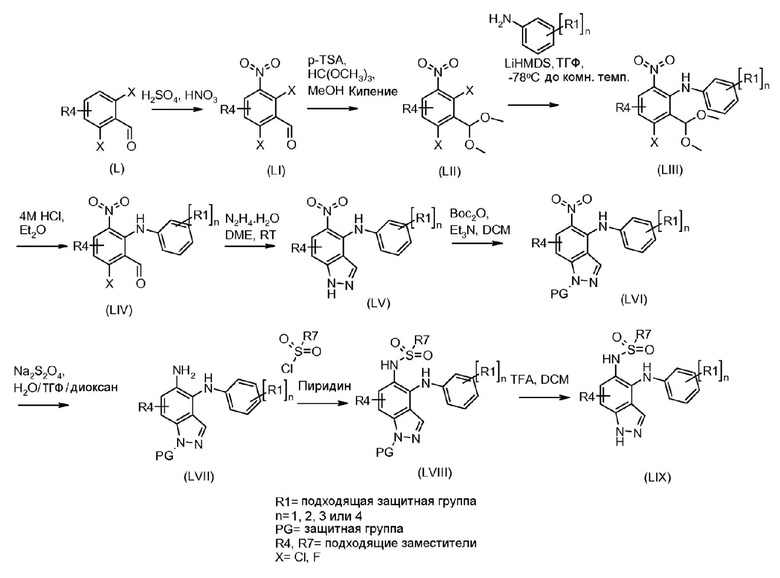
Соединения формулы (L) могут быть коммерчески доступными или получены, используя способы, описанные в литературе. Соединения формулы (L) могут быть подвергнуты нитрованию с получением соединений формулы (LI) путем обработки смесью серной и азотной кислот при температуре <5°C. Соединения формулы (LI) могут быть преобразованы в соединения формулы (LII) путем взаимодействия с ортоформиатом, таким как триметил ортоформиат, в присутствии кислоты, такой как п-толуолсульфоновая кислота, в растворителе, таком как метанол, при температуре около температуры кипения. Соединения формулы (LII) могут быть преобразованы в соединения формулы (LIII), используя способы, описанные для преобразования соединений формулы (III) в соединения формулы (V) по схеме 1. Соединения формулы (LIV) могут быть преобразованы в соединения формулы (LV), используя способы, описанные для преобразования соединений формулы (XVIII) в соединения формулы (XIX) по схеме 3. Соединения формулы (LV) могут быть преобразованы в соединения формулы (LVI), используя способы, описанные для преобразования соединения формулы (X) в соединения формул (XIa и XIb) по схеме 2. Нитросоединения формулы (LVI) могут быть восстановлены до анилинов формулы (LVII), используя восстанавливающий агент, такой как дитионит натрия, в смеси растворителей, такой как ТГФ/вода/диоксан, при температуре в области комнатной. Сульфонамиды формулы (LVIII) могут быть получены из анилинов формулы (LVII) путем взаимодействия с сульфонилхлоридом в присутствии растворителя, такого как пиридин. Соединения формулы (LIX) могут быть получены исходя из соединений формулы (LVIII) в условиях, подходящих для удаления используемой защитной группы. Например, когда PG=Boc, соединения формулы (LVIII) могут быть обработаны сильной кислотой, такой как трифторуксусная кислота, в растворителе, таком как дихлорметан, при температуре около комнатной с получением соединения формулы (LIX).
Гидроксиламины формулы (VIIIa) и (VIIIb) могут быть получены, используя способы, описанные в литературе или синтетическим путем, представленным на схеме 12.
Схема 12
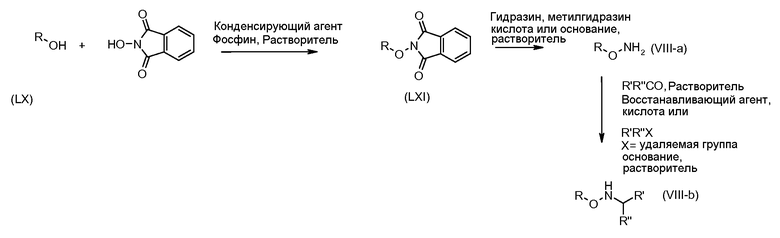
Первичные или вторичные спирты общей формулы (LX) могут быть получены, используя способы, описанные в литературе. Спирты могут быть подвергнуты взаимодействию с N-гидроксифталимидом, используя фосфин и конденсирующий реагент, такой как диэтил азодикарбоксилат, с получением соединений общей формулы (LXI). Соединения общей формулы (LXI) могут быть подвергнуты удалению защитных групп, используя гидразин, метилгидразин, кислоту, такую как хлористоводородная кислота, или основание, такое как водный аммиак, с получением гидроксиламинов общей формулы (VIII-a).
Соединения формулы (VIII-a) могут быть далее модифицированы путем восстановительного аминирования с альдегидами или кетонами, используя восстанавливающий агент, такой как триацетоксиборгидрид натрия, цианоборгидрид натрия или боран-пиридин, в растворителе, таком как дихлорэтан, при температуре от температуры окружающей среды до температуры кипения с получением гидроксиламинов общей формулы (VIII-b). Кроме того, соединения формулы (XII-a) могут быть далее модифицированы путем алкилирования алкилгалогенидом в присутствии основания, такого как триэтиламин, в растворителе, таком как дихлорметан, с получением гидроксиламинов общей формулы (VIII-b).
Альтернативно, гидроксиламины формулы (VIII-a) могут быть получены в соответствии со схемой 13.
Схема 13
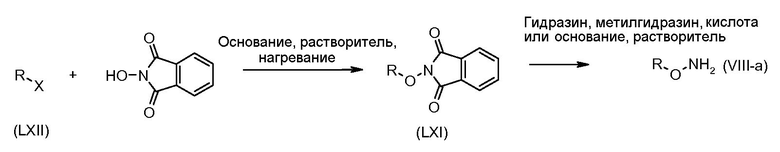
Алкилгалогениды формулы (LXII) могут быть подвергнуты взаимодействию с N-гидроксифталимидом в присутствии основания, такого как карбонат калия, в растворителе, таком как диметил сульфоксид, при температуре от 10ºC до 50ºC. Соединения формулы (LXI) могут быть преобразованы в соединения формулы (VIII-a), используя способы, описанные для преобразования соединений формулы (LXI) в соединения формулы (VIII-a) по схеме 12.
Альтернативно, соединения формулы (VIII-a) могут быть получены в соответствии со схемой 14.
Схема 14
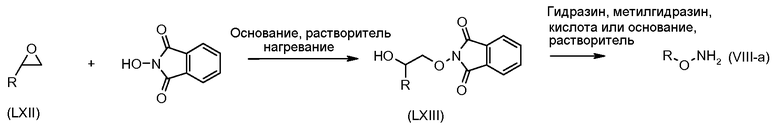
Соединения формулы (LXII) могут быть подвергнуты взаимодействию N-гидроксифталимида в присутствии каталитического количества основания, такого как DIPEA, и co-катализатора, такого как тетра-бутиламмоний бромид, в растворителе, таком как толуол, при температуре от 50ºC до температуры кипения с получением соединений формулы (LXIII). Соединения формулы (LXIII) могут быть преобразованы в соединения формулы (VIII-a), используя способы, описанные для преобразования соединений формулы (LXI) в соединения формулы (VIII-a) по схеме 12.
Анилины общей формулы (LXV), используемые в реакциях конденсации и поперечного связывания, описанных выше, могут быть получены, используя способы, описанные в литературе или в соответствии со схемой 15.
Схема 15
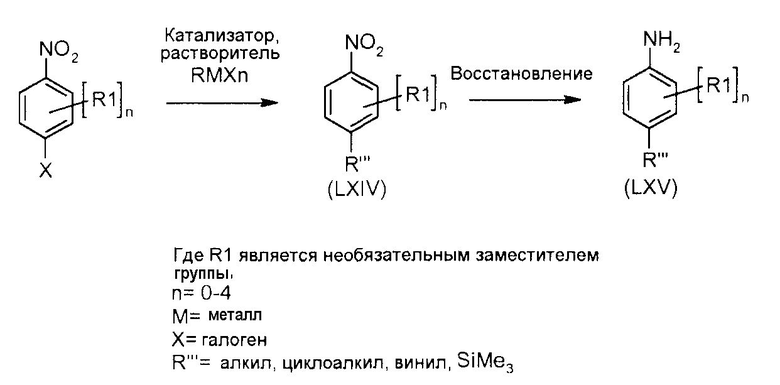
Замещенный 1-хлор-4-нитробензол может быть подвергнут взаимодействию с металл R'"MXn, таким как циклопропил борная кислота или гексаметилдисилазан, в растворителе, таком как ксилол, используя катализатор, такой как тетракис(трифенилфосфин)палладий, при температуре от комнатной до температуры кипения с получением соединений формулы (LXIV). Нитрогруппа может быть восстановлена, используя способы, описанные в литературе, такие как реакция в атмосфере водорода, при давлении от 1 до 5 атмосфер, в присутствии катализатора, такого как палладий на углероде, и в растворителе, таком как этанол или этилацетат, при комнатной температуре с получением соединения формулы (LXV).
Альтернативно, анилины формулы (LXVII) могут быть получены в соответствии со схемой 16.
Схема 16
4-Бром или йод анилины формулы (LXVI) могут быть подвергнуты взаимодействию по меньшей мере с 2 эквивалентами сильного металлоогранического основания, такого как н-бутиллитий, в растворителе, таком как ТГФ, при температуре от -100ºC до -20ºC, с последующим гашением промежуточных производных ариллития с электрофилом, таким как триметилсилил хлорид, с получением соединения формулы (LXVII).
Следует учесть, что, когда имеются подходящие функциональные группы, соединения формулы (I) или любые промежуточные соединения, используемые при их получении, могут быть затем преобразованы одним или несколькими стандартными синтетическими способами, используя реакции замещения, окисления, восстановления или отщепления. Конкретные примеры введения заместителей включают обычные способы алкилирования, арилирования, гетероарилирования, ацилирования, сульфонилирования, галогенирования, нитрования, формилирования и связывания.
Например, арил бромидные или хлоридные группы могут быть преобразованы в арилйодиды, используя реакцию Финкельштейна, используя источник йодида, такой как йодид натрия, катализатор, такой как йодид меди, и лиганд, такой как транс-N,N'-диметил-1,2-циклогексан диамин, в растворителе, таком как 1,4-диоксан, и нагревание реакционной смеси при температуре кипения. Арилтриалкилсиланы могут быть преобразованы в арилйодиды путем обработки силана источником йодида, таким как монохлорид йода, в растворителе, таком как дихлорметан, с использованием или без использования кислоты Льюиса, такой как тетрафторборат серебра, при температуре от -40ºC до температуры кипения.
В следующем примере первичные амино (-NH2) группы могут быть алкилированы с помощью способа восстановительного алкилирования, испольуя альдегид или кетон и боргидрид, например, триацетоксиборгидрид натрия или цианоборгидрид натрия, в растворителе, таком как галогенированный углеводород, например, 1,2-дихлорэтан, или спирт, такой как этанол, как требуется, в присутствии кислоты, такой как уксусная кислота, при температуре окружающей среды. Вторичные амино (-NH-) группы могут быть подобным образом алкилированы, используя альдегид.
В следующем примере, первичные амино или вторичные амино группы могут быть преобразованы в амидные группы (-NHCOR' или -NRCOR') путем ацилирования. Ацилирование можно осуществить путем взаимодействия с подходящим хлорангидридом кислоты в присутствии основания, такого как триэтиламин, в подходящем растворителе, таком как дихлорметан, или путем взаимодействия с подходящей карбоновой кислотой в присутствии подходящего конденсирующего агента, такого как HATU (O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфат), в подходящем растворителе, таком как дихлорметан. Подобным образом, амино группы могут быть преобразованы в сульфонамидные группы (-NHSO2R' или -NR”SO2R') путем взаимодействия с подходящим сульфонилхлоридом в присутствии подходящего основания, такого как триэтиламин, в подходящем растворителе, таком как дихлорметан. Первичные или вторичные амино группы могут быть преобразованы в мочевино группы (-NHCONR'R" или -NRCONR'R") путем взаимодействия с подходящим изоцианатом в присутствии подходящего основания, такого как триэтиламин, в подходящем растворителе, таком как дихлорметан.
Амин (-NH2) может быть получен путем восстановления нитро (-NO2) группы, например, путем каталитического гидрирования, используя, например, водород, в присутствии металлического катализатора, например, палладия на носителе, таком как углерод в растворителе, таком как этилацетат или спирт, например, метанол. Альтернативно, преобразование может быть осуществлено путем химического восстановления, используя, например, металл, например, олово или железо, в присутствии кислоты, такой как хлористоводородная кислота.
В следующем примере, амино (-CH2NH2) группы могут быть получены путем восстановления нитрилов (-CN), например, путем каталитического гидрирования, используя, например, водород, в присутствии металлического катализатора, например, палладия на носителе, таком как углерод или никель Ренея, в растворителе, таком как эфир, например, циклический эфир, такой как тетрагидрофуран, при температуре от -78ºC до температуры кипения растворителя.
В следующем примере, амино (-NH2) группы могут быть получены из карбоксильной группы (-CO2H) путем преобразования в соответствующий ацилазид (-CON3), перегруппировкой Курциуса и гидролизом полученного изоцианата (-N=C=O).
Альдегид группы (-CHO) может быть преобразован в амино группу (-CH2NR'R")) путем восстановительного аминирования, используя амин и боргидрид, например, триацетоксиборгидрид натрия или цианоборгидрид натрия, в растворителе, таком как галогенированный углеводород, например дихлорметан, или спирт, такой как этанол, как требуется, в присутствии кислоты, такой как уксусная кислота, при температуре окружающей среды.
В следующем примере альдегидная группа может быть преобразована в алкенильную группу (-CH=CHR') путем использования реакции Виттига или Вадсворт-Эммонса, используя подходящий фосфоран или фосфонат в обычных условиях, известных специалисту в данной области.
Альдегидные группы могут быть получены путем восстановления сложноэфирных групп (таких как -CO2Et) или нитрилы (-CN), используя гидрид диизобутилалюминия в подходящем растворителе, таком как толуол. Альтернативно, альдегидные группы могут быть получены путем окисления спиртовых групп, используя любой подходящий окислительный агент, известный специалисту в данной области.
Сложноэфирные группы (-CO2R') могут быть преобразованы в соответствующие карбоксильные группы (-CO2H) путем катализируемого кислотой или основанием гидролиза, в зависимости от природы R. Если R представляет собой трет-бутил, катализируемый кислотой или основанием гидролиз может быть осуществлен, например, путем обработки органической кислотой, такой как трифторуксусная кислота, в водном растворителе, или путем обработки неорганической кислотой, такой как хлористоводородная кислота в водном растворителе.
Карбоксильные группы (-CO2H) могут быть преобразованы в амиды (CONHR' или -CONR'R") путем взаимодействия с подходящим амином в присутствии подходящего конденсирующего агента, такого как HATU, в подходящем растворителе, таком как дихлорметан.
В следующем примере, могут быть получены гомологи карбоновых кислот, содержащие в цепи дополнительный атом углерода (то есть -CO2H в -CH2CO2H) путем преобразования в соответствующий хлорангидрид кислоты (-COCl), затем осуществляя синтез Арндта-Эйстерта.
В следующем примере, -OH группы могут быть образованы из соответствующего сложного эфира (например -CO2R') или альдегида (-CHO) путем восстановления, используя, например, комплекс гидрида металла, такой как литий алюминий гидрид, в диэтиловом эфире или тетрагидрофуране, или боргидрид натрия в растворителе, таком как метанол. Альтернативно, спирт может быть получен путем восстановления соответствующей кислоты (-CO2H), используя, например, литий алюминий гидрид в растворителе, таком как тетрагидрофуран, или путем использования борана в растворителе, таком как тетрагидрофуран.
Спиртовые группы могут быть преобразованы в удаляемые группы, такие как атомы галогена или сульфонилокси группы, такие как алкилсульфонилокси, например, трифторметилсульфонилокси или арилсульфонилокси, например п-толуолсульфонилокси группу, используя условия, известные специалисту в данной области. Например, спирты могут быть подвергнуты взаимодействию тионил хлорида в галогенированный углеводород (например, дихлорметан) с получением соответствующего хлорида. Основание (например, триэтиламин) может также быть использовано в реакции.
В другом примере, спиртовые, фенольные или амидные группы могут быть алкилированы путем связывания фенола или амида со спиртом в растворителе, таком как тетрагидрофуран, в присутствии фосфина, например, трифенилфосфина, и активатора, такого как диэтил-, диизопропил- или диметилазодикарбоксилат. Альтернативно, алкилирование может быть осуществлено путем депротонирования, используя подходящее основание, например, гидрид натрия, с последующим добавлением алкилирующего агента, такого как алкилгалогенид.
Ароматические галогенсодержащие заместители в соединениях могут быть подвергнуты обмену галоген-металл путем обработки основанием, например, литиевым основанием, таким как н-бутил или трет-бутил литий, необязательно при низкой температуре, например, в области -78°C, в растворителе, таком как тетрагидрофуран, и с последующим гашением электрофилом для введения желаемого заместителя. Так, например, формильная группа может быть введена, используя N,N-диметилформамид в качестве электрофила. Ароматические галогенсодержащие заместители могут альтернативно быть подвергнуты катализирумой металлом (например, палладий или меди) реакции, для введения, например, кислотного, сложноэфирного, циано, амидного, арильного, гетероарильного, алкенильного, алкинильного, тио- или амино заместителей. Подходящие способы, которые могут быть использованы, включают те, которые описаны Heck, Suzuki, Stille, Buchwald или Hartwig.
Ароматические галогенсодержащие заместители могут также быть подвергнуты нуклеофильной замене с последующим взаимодействием с подходящим нуклеофилом, таким как амин или спирт. Преимущественно, такую реакцию можно осуществить при повышенной температуре при микроволновом облучении.
Соединения по настоящему изобретению исследовали на предмет их способности ингибировать MEK активность и активацию (первичные исследования) и их биологическое действие на рост клеток (вторичные исследования), как описано далее. Соединения по настоящему изобретению, имеющие IC50 меньше чем 5 мкМ (более предпочтительно, меньше чем 0,1 мкМ, наиболее предпочтительно меньше чем 0,01 мкМ) в исследовании MEK активности соединения по примеру 1, IC50 меньше чем 5 мкМ (более предпочтительно, меньше чем 1 мкМ, еще более предпочтительно, меньше чем 0,1 мкМ, наиболее предпочтительно, меньше чем 0,01 мкМ) в исследовании MEK активации примера 2, EC50 меньше чем 10 мкМ (более предпочтительно, меньше чем 1 мкМ, еще более предпочтительно меньше, чем 0,5 мкМ, наиболее предпочтительно, меньше чем 0,1 мкМ) в исследовании клеточной пролиферации примера 3, и/или EC50 меньше чем 10 мкМ (более предпочтительно, меньше чем 1 мкМ, еще более предпочтительно, меньше чем 0,5 мкМ, наиболее предпочтительно, меньше чем 0,1 мкМ) в исследовании ERK фосфорилирования примера 4, используют в качестве MEK ингибиторов.
Настоящее изобретение включает композицию (например, фармацевтическую композицию), содержащую соединение формулы I или II (и/или его сольват и/или их соль) и носитель (фармацевтически приемлемый носитель). Настоящее изобретение также включает композицию (например, фармацевтическую композицию), содержащую соединение формулы I или II (и/или сольваты и/или их соли) и носитель (фармацевтически приемлемый носитель), дополнительно содержащий вспомогательный химиотерапевтический и/или вспомогательный противовоспалительный агент, например, такой, как описан в настоящем документе. Настоящие композиции применяют для ингибирования патологического роста клеток или лечения гиперпролиферативных расстройств у млекопитающего (например, человека). Настоящие композиции также применяют для лечения воспалительных заболевания у млекопитающего (например, человека).
Настоящие соединения и композиции также применяют для лечения аутоиммунного заболевания, связанного с поражением костной ткани, пролиферативных расстройств, инфекционных заболеваний, вирусных заболеваний, фиброзных заболеваний или нейродегенеративных заболеваний у млекопитающего (например, человека). Примеры таких заболеваний/расстройств включают, но ими не ограничиваются, диабет и диабетические осложнения, диабетическую ретинопатию, ретинопатию недоношенных, старческую дегенерацию желтого пятна, гемангиому, идиопатический фиброзирующий альвеолит, ринит и атопический дерматит, заболевание почек и почечную недостаточность, поликистозное заболевание, застойную сердечную недостаточность, нейрофиброматоз, отторжение трансплантированного органа, кахексию, инсульт, септический шок, сердечную недостаточность, отторжение трансплантированного органа, болезнь Альцгеймера, хроническую или нейропатическую боль и вирусные инфекции, такие как ВИЧ, вирус гепатита B (HBV), человеческий вирус папилломы (HPV), цитомегаловирус (CMV) и вирус Эпштейна-Барра (EBV). Хроническая боль, в контексте настоящего изобретения, включает, но этим не ограничивается, идиопатическую боль и боль, связанную с хроническим алкоголизмом, витаминную недостаточность, уремию, гипотириоз, воспаление, артрит и послеоперационную боль. Нейропатическая боль связана с различными состояниями, которые включают, но этим не ограничиваются, воспаление, послеоперационную боль, фантомную боль, боль при ожоге, подагру, тригеминальную невралгию, острую герпетическую и постгерпетическую боль, каузалгию, диабетическую нейропатию, разрыв сплетения, неврома, васкулит, вирусную инфекцию, повреждение раздавливанием, повреждение сжатием, повреждение ткани, ампутация конечности, боль при артрите и повреждение нерва между периферической нервной системой и центральной нервной системой.
Настоящие соединения и композиции также применяют для лечения панкреатита или заболевания почек (включая пролиферативный гломерулонефрит и вызванное диабетом заболевание почек) у млекопитающих (например, человека).
Настоящие соединения и композиции также применяют для профилактики имплантация бластоцитов у млекопитающих (например, человека).
Настоящее изобретение включает способ ингибирования патологического роста клеток или лечение гиперпролиферативного расстройства у млекопитающих (например, человека), включающий введение указанному млекопитающему терапевтически эффективного количества соединения формулы I или II (и/или его сольвата и/или соли) или их композиции. Также настоящее изобретение включает способ лечения воспалительного заболевания у млекопитающих (например, человека), включающий введение указанному млекопитающему терапевтически эффективного количества соединения формулы I или II (и/или его сольвата и/или соли) или его композиции.
Настоящее изобретение включает способ ингибирования патологического роста клеток или лечение гиперпролиферативного расстройства у млекопитающего (например, человека), включающий введение указанному млекопитающему терапевтически эффективного количества соединения формулы I или II (и/или его сольвата и/или соли) или его композиции, в сочетании с дополнительным химиотерапевтическим агентом, например, таким, как описан в настоящем документе. Настоящее изобретение также включает способ лечения воспалительного заболевания у млекопитающих (например, человека), включающий введение указанному млекопитающему терапевтически эффективного количества соединения формулы I или II (и/или его сольвата и/или соли) или его композиции, в сочетании с дополнительным противовоспалительным агентом, например, таким, как описан в настоящем документе.
Настоящее изобретение включает способ лечения аутоиммунного заболевания, расстройства поражающего костную ткань, пролиферативных расстройств, инфекционного заболевания, вирусного заболевания, фиброзного заболевания или нейродегенеративного заболевания у млекопитающих (например, человека), включающий введение указанному млекопитающему терапевтически эффективного количества соединения формулы I или II (и/или его сольвата и соли) или его композиции, и необязательно дополнительно содержащей вспомогательный терапевтический агент. Примеры таких заболеваний/расстройств включают, но этим не ограничиваются, диабет и диабетические осложнения, диабетическую ретинопатию, ретинопатию недоношенных, старческую дегенерацию желтого пятна, гемангиому, идиопатический фиброзирующий альвеолит, ринит и атопический дерматит, заболевание почек и почечную недостаточность, поликистозное заболевание почек, застойную сердечную недостаточность, нейрофиброматоз, отторжение трансплантированного органа, кахексию, инсульт, септический шок, сердечную недостаточность, отторжение трансплантированного органа, болезнь Альцгеймера, хроническую или нейропатическую боль и вирусные инфекции, такие как HIV, вирус гепатита B (HBV), человеческий вирус папилломы (HPV), цитомегаловирус (CMV) и вирус Эпштейна-Барра (EBV)
Настоящее изобретение включает способ лечения панкреатита или заболевания почек (включая пролиферативный гломерулонефрит и вызванное диабетом заболевание почек) у млекопитающих (например, человека), включающий введение указанному млекопитающему терапевтически эффективного количества соединения формулы I или II (и/или его сольвата и соли) или его композиции, и необязательно дополнительно содержащей вспомогательный терапевтический агент.
Настоящее изобретение включает способ профилактики имплантации бластоцитов у млекопитающих (например, человека), включающий введение указанному млекопитающему терапевтически эффективного количества соединения формулы I или II (и/или его сольвата и соли) или его композиции, и необязательно дополнительно содержащей вспомогательный терапевтический агент.
Настоящее изобретение включает способ применения настоящих соединений для in vitro, in situ и in vivo диагностики или лечения клеток млекопитающих, организмов или сопутствующих патологических состояний.
Также полагают, что соединения по настоящему изобретению могут способствовать усилению восприимчивости патологических клеток к воздействию облучения в целях уничтожения и/или подавления роста таких клеток. Соответственно, настоящее изобретение дополнительно относится к способу сенсибилизации патологических клеток млекопитающих (например, человека) к лечению с помощью облучения, который включает введение указанному млекопитающему соединения формулы I или II (и/или его сольвата и соли) или его композиции, в количестве, которое является эффективным для сенсибилизации патологических клеток к лечению с помощью облучения.
Введение соединений по настоящему изобретению (далее "активное соединение(я)") может быть осуществлено любым способом, который обеспечивает доставку соединений к месту приложения действия. Эти способы включают пероральные способы введения, интрадуоденальные способы введения, парентеральную инъекцию (включая внутривенную, подкожную, внутримышечную, внутрисосудистую или инфузию), местный способ введения, ингаляцию и ректальное введение.
Количество активного соединения для введения будет зависеть от субъекта лечения, тяжести расстройства или состояния, скорости введения, распределения соединения и усмотрения назначающего лечение врача. Однако эффективная доза находится в пределах от около 0,001 до около 100 мг на кг массы тела в день, предпочтительно, от около 1 до около 35 мг/кг/день, в одноразовой или дробной дозах. Для человека весом 70 кг, доза может составлять от около 0,05 до 7 г/день, предпочтительно, от около 0,05 до около 2,5 г/день. В некоторых случаях, уровни дозы ниже нижней границы в вышеуказанном пределе, могут быть более чем достаточными, тогда как в других случаях могут применяться еще большие дозы, не вызывая вредного побочного эффекта, при условии, что такие большие дозы сначала разделены на несколько небольших доз для введения в течение дня.
Активное соединение может быть использовано в качестве самостоятельной терапии или в сочетании с одним или несколькими химиотерапевтическими или противовоспалительными агентами, например, такими, как описаны в настоящем документе. Такое совместное лечение может быть достигнуто посредством одновременного, последовательного или раздельного дозирования отдельных компонентов лечения.
Фармацевтическая композиция может, например, быть в виде подходящей для перорального введения таблетки, капсулы, пилюли, порошка, композиций замедленного высвобождения, раствора, суспензии, для парентенральной инъекции в виде стерильного раствора, суспензии или эмульсии, для местного введения в виде мази или крема, или для ректального введения в виде свечи. Фармацевтическая композиция может быть в формах одноразового введения, подходящих для разового введения определенных доз. Фармацевтическая композиция будет включать обычный фармацевтический носитель или наполнитель и соединение по изобретению в качестве активного ингредиента. Кроме того, она может включать другие медицинские или фармацевтические агенты, носители, адъюванты и так далее.
Типичные формы для парентерального введения включают растворы или суспензии активных соединений в стерильных водных растворах, например, водные растворы пропиленгликоля или декстрозы. Если желательно, такие лекарственные формы могут быть соответствующим образом буферизированы.
Подходящие фармацевтические носители включают инертные разбавители или наполнители, воду и различные органические растворители. Фармацевтические композиции могут, если желательно, содержать дополнительные ингредиенты, такие как ароматизаторы, связующие вещества, наполнители и тому подобное. Таким образом, для перорального введения, таблетки, содержащие различные наполнители, такие как лимонная кислота, могут быть использованы совместно с различными разрыхляющими агентами, такими как крахмал, альгиновая кислота и некоторые сложные силикаты, и с конденсирующими агентами, такими как сахароза, желатин и камедь. Кроме того, для целей таблетирования часто используют смазывающие агенты, такие как стеарат магния, лаурилсульфат натрия и тальк. Также могут быть использованы твердые композиции аналогичного типа в мягких и твердых наполняемых желатиновых капсулах. Предпочтительные вещества, соответственно, включают лактозу или молочный сахар и полиэтиленгликоли с высоким молекулярным весом. В случае, когда требуются водные суспензии или эликсиры для перорального введения, активное соединение в них может сочетаться с различными подсластителями или ароматизаторами, красящими веществами или красителями и, если желательно, эмульгирующими агентами или суспендирующими агентами, вместе с разбавителями, такими как вода, этанол, пропиленгликоль, глицерин или их сочетания.
Способы получения различных фармацевтических композиций с определенным количеством активного соединения хорошо известны, или будут очевидны, для специалиста в данной области. Например, смотри Remington's Pharmaceutical Sciences, Mack Publishing Company, Ester, Pa., 15.sup.th Edition (1975).
ПРИМЕРЫ
Сокращения
AIBN Азобисизобутиронитрил
BOC трет-бутокси карбонил
nBuLi н-бутиллитий
CDCl3 Дейтерированный хлороформ
CD3OD Дейтерированный метанол
DCM Дихлорметан
DIAD Диизопропил азо-дикарбоксилат
DIPEA Диизопропилэтиламин
DMAP N,N'-диметил-4-аминопиридин
DME Диметиловый эфир этиленгликоля
ДМФ Диметилформамид
ДМСО Диметилсульфоксид
Dppf 1,1'-Бис(дифенилфосфино)ферроцен
EDCI l-Этил-3-(3'-диметиламинопропил)карбодиимид гидрохлорид
Et3N Триэтиламин
Et2O Диэтиловый эфир
HATU O-(7-Азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфат
HCl хлористоводородная кислота
Hyflo® диатомовая земля
HM-N Isolute ® абсорбент диатомовой земли
HOBt 1-Гидроксибензотриазол
ВЭЖХ высокоэффективная жидкостная хроматография
IMS промышленный метиловый спирт
K3PO4 трехосновный фосфат калия
LHMDS бис(триметилсилил)амид лития
MeOH Метанол
NaHCO3 Гидрокарбонат натрия
NH4Cl Хлорид аммония
NaOH Гидроксид натрия
Na2SO4 Сульфат натрия
Pd(dppf)Cl2 [1,1'-Бис(дифенилфосфино)ферроцен]-дихлорпалладий(II)
Pd(PPh3)4 Тетракис(трифенилфосфин)палладий(0)
Pd2dba3 Трис-(дибензилиденацетон)дипалладий(0)
Si-PPC укомплектованный картридж для флеш хроматографии на силикагеле: Isolute® SPE,
Biotage SNAP ® или ISCO Redisep®
SCX-2 Isolute ® сорбент на основе силикагеля с химически связанной функциональной группой пропилсульфоновой кислоты.
TBME трет-бутил-метиловый эфир
TMEDA Тетраметилэтилендиамин
ТГФ Тетрагидрофуран
ТФУ Трифторуксусная кислота
TMSCl Триметилсилилхлорид
Xantphos 4,5-Бис(дифенилфосфино)-9,9-диметилксантен
Общие условия проведения экспериментов
1H ЯМР спектр записывали при температуре окружающей среды, используя спектрометр Varian Unity Inova (400 МГц) с датчиком тройного резонанса 5 мм. Химические сдвиги выражали в миллионных долях относительно тетраметилсилана. Использовали следующие сокращения: br=уширенный сигнал, s=синглет, d=дублет, dd=двойной дублет, t=триплет, q=квартет, m=мультиплет.
Выполняли эксперименты с применением высокоэффективной жидкостной хроматографии - масс-спектрометрии (LCMS) для определения времени удерживания (RT) и ассоциированных масс ионов, используя один из следующих способов.
Способ A: Эксперименты выполняли на квадрупольном масс-спектрометре Waters Micromass ZQ, соединенным с системой Hewlett Packard HP1100 LC с детектором на диодной матрице. Эта система использует колонку Higgins Clipeus 5 мкм C18 100×3,0 мм и скорость потока 1 мл/мин. Начальной системой растворителя была смесь из 95% воды, содержащей 0,1%-ную муравьиную кислоту (растворитель A) и 5% ацетонитрила, содержащего 0,1%-ную муравьиную кислоту (растворитель B), в течение первой минуты, затем градиент до 5% растворителя A и 95% растворитель B в течение следующих 14 минут. Конечная система растворителя оставалась неизменной в течение еще 5 минут.
Способ B: Эксперименты выполняли на квадрупольном масс-спектрометре Waters Platform LC, соединенным с системой Hewlett Packard HP1100 LC с детектором на диодной матрице и 100 позиционный автосэмплер, используя колонку Phenomenex Luna C 18(2) 30×4,6 мм и скорость потока 2 мл/мин. Системой растворителя была смесь 95% воды, содержащей 0,1%-ную муравьиную кислоту (растворитель A) и 5% ацетонитрила, содержащего 0,1%-ную муравьиную кислоту (растворитель B) в течение первых 0,50 минут, затем градиент до 5% растворителя A и 95% растворителя B в течение следующих 4 минут. Конечная система растворителя оставалась неизменной в течение еще 0,50 минут.
Способ C: Эксперименты выполняли на квадрупольном масс-спектрометре PE Sciex API 150 EX, соединенным с системой Shimadzu LC-10AD LC с детектором на диодной матрице и 225 позиционных автосемплером, используя колонку Kromasil C18 50×4,6 мм и скорость потока 3 мл/мин. Системой растворителя был градиент, начиная со 100% воды с 0,05% ТФУ (растворитель A) и 0% ацетонитрила с 0,0375% ТФУ (растворитель B), повышая до 10% растворителя A и 90% растворителя B в течение 4 минут. Конечная система растворителя оставалась неизменной в течение еще 0,50 минут.
Эксперименты с применением микроволнового облучения выполняли, используя систему Personal Chemistry Emrys Iniatiator™ или Optimizer™, которая использует одномодовый резонатор и регулятор нестационарного поля, оба из них обеспечивают воспроизводимость и контроль. Может быть достигнута температура от 40 до 250ºC, и может быть достигнуто давление до 20 бар.
ПРИМЕР 1 MEK исследование (исследование MEK активности)
Конститутивно активированный мутантный MEK1 человека, экспрессированный в клетки насекомого, использовали в качестве источника ферментной активности при конечной концентрации в киназном исследовании, составляющей 15 нМ.
Исследование выполняли в течение 30 минут в присутствии 50 мкМ ATP, используя рекомбинантный GST-ERK1, полученный в E. Coli в качестве субстрата. Фосфорилирование субстрата устанавливали и количественно определяли, используя реагенты HTRF, поставляемые компанией Cisbio. Они состоят из анти-GST антитела, конъюгированного с аллофикоцианином (XL665) и анти-фосфо (Thr202/Tyr204) ERK антитела, конъюгированного с криптатом европия. Их использовали с конечными концентрациями 4 мкг/мл и 0,84 мкг/мл соответственно. Анти-фосфо антитело распознает ERK1 двойственно фосфорилированный на Thr202 и Tyr204. Когда оба антитела связаны с ERK1 (то есть когда субстрат фосфорилирован), передача энергии от криптата к аллофикоцианину происходит после возбуждения при 340 нм, приводя к излучаемой флюоресценции, то есть пропорционально количеству полученного фосфорилированного субстрата. Флюоресценцию регистрировали, используя мультилуночный флуориметр.
Соединения разбавляли в ДМСО перед добавлением в буфер для исследования, и конечная концентрация ДМСО в исследовании составляет 1%.
Значение IC50 определяли как концентрацию, при которой данное соединение достигает 50% ингибирования в контрольном образце. Значения IC50 подсчитывали, используя пакет программного обеспечения XLfit (версия 2.0.5).
Указанные в заголовке соединения примеров 5-9, 12-13, 16, 20-23, и 25-27 показали значение IC50 меньше чем 0,1 мкМ в исследовании, описанном в примере 1. Указанные в заголовке соединения примеров 10-11, 15, 17 и 24 показали значение IC50 между 0,1 и 0,6 мкМ в исследовании, описанном в примере 1.
ПРИМЕР 2 bRaf исследование (исследование MEK активации)
Конститутивно активированный мутант bRaf, экспрессированный в клетки насекомого, использовали в качестве источника ферментной активности.
Исследование выполняли в течение 30 минут в присутствии 200 мкМ ATP, используя рекомбинантный GST-MEK1, полученный в E. Coli в качестве субстрата. Фосфорилирование субстрата регистрировали и измеряли количественно, используя HTRF, и реагенты, поставляемые компанией Cisbio. Они состоят из анти-GST антитела, конъюгированного с аллофикоцианином (XL665) и анти-фосфо (Ser217/Ser221) MEK антитела, конъюгированного с криптатом европия. Анти-фосфо антитело распознает MEK двойственно фосфорилированный на Ser217 и Ser221 или однократно фосфорилированный на Ser217. Когда оба антитела связаны с MEK (то есть, когда субстрат фосфорилирован), передача энергии от криптата к аллофикоцианину происходит после возбуждения при 340 нм, приводя к излучаемой флюоресценции, то есть пропорционально количеству полученного фосфорилированного субстрата. Флюоресценцию регистрировали, используя мультилуночный флуориметр.
Соединения разбавляли в ДМСО перед добавлением к аналитическому буферу, и конечная концентрация ДМСО в исследовании составляла 1%.
Значение IC50 определяли как концентрацию, при которой данное соединение достигает 50% ингибирования в контрольном образце. Значения IC50 подсчитывают, используя пакет программного обеспечения XLfit (версия 2.0.5).
ПРИМЕР 3 Исследование клеточной пролиферации
Соединения тестировали в исследовании клеточной пролиферации, используя следующие клеточные линии:
HCT116 клетки колоноректальной карциномы человека (ATCC)
A375 клетки злокачественной меланомы человека (ATCC)
Обе клеточные линии содержали в среде DMEM/F12 (1:1) (Gibco) с добавлением 10% FCS при 37ºC в 5% CO2 влажной камеры.
Клетки высевали в 96-луночный планшеты с плотностью 2000 клеток на лунку и через 24 часа их экспонировали в другие концентрации соединения в 0,83% ДМСО. Клетки выращивали в течение еще 72 часов, и в каждую лунку добавляли одинаковый объем реагента CellTiter-Glo (Promega).
Это лизирует клетки и генерирует люминесцентный сигнал, пропорциональный количеству освобожденного ATP (и, соответственно, пропорциональный количеству клеток в лунке), который может быть обнаружен, используя мультилуночный люминометр.
Значение EC50 определяли, как концентрацию, при которой данное соединение достигает 50% ингибирования в контрольном образце. Значения EC50 подсчитывали, используя пакет программного обеспечения XLfit (версия 2.0.5).
В этом исследовании, указанные в заголовке соединения примера 5-7, 9-14, 16-17, 20-22 и 24-27 показали значение EC50 меньше чем 0,5 мкМ в клеточной линии HCT116. Указанное в заголовке соединение примера 8 показало EC50 меньше чем 0,6 мкМ в клеточной линии HCT116. Указанные в заголовке соединения примеров 5-12, 14, 16-17 и 20-27 показали значение EC50 меньше чем 0,1 мкМ в клеточной линии A375.
ПРИМЕР 4 Исследование на основе фосфо-ERK клеток
Соединения тестировали в клеточном анализе фосфо-ERK ELISA, используя следующие клеточные линии:
Клетки HCT116 колоноректальной карциномы человека (ATCC)
Клетки A375 злокачественной меланомы человека (ATCC)
Обе клеточные линии выдерживали в среде DMEM/F12 (1:1) (Gibco), с добавлением 10% FCS при 37ºC во влажной камере с 5% CO2.
Клетки высевали в 96-луночный планшеты с плотностью 2000 клеток на лунку и через 24 часа их экспонировали в другие концентрации соединения в 0,83% ДМСО. Клетки выращивали в течение еще 2 часов или 24 часов, фиксировали формальдегидом (2% конечный) и пермеабилизировали метанолом. После блокирования TBST-3% BSA, фиксированные клетки инкубировали с первичным антителом (анти-фосфо ERK от кролика) в течение ночи при 4ºC. Клетки инкубировали с пропидиум иодидом (флуоресцентный краситель ДНК) и определение клеточного p-ERK осуществляли, используя вторичное антикроличье антитело, конъюгированное с флуоресцентным красителем Alexa Fluor 488 (молекулярные зонды). Флуоресценцию анализировали, используя Acumen Explorer (TTP Labtech), лазерный сканирующий микропланшетный цитометр, и сигнал Alexa Fluor 488 нормализовали относительно сигнала PI (пропорционально количеству клеток).
Значение EC50 определяли как концентрацию, при которой данное соединение достигает половинного значения сигнала между базовой линией и максимальным ответом. Значения EC50 подсчитывали, используя пакет программного обеспечения XLfit (версия 2.0.5).
В этом исследовании, указанные в заголовке соединения примеров 5-14, 16-17, 20-22 и 24-27 показали значение EC50 меньше чем 0,1 мкМ в клеточной линии HCT116. Указанные в заголовке соединения примеров 6-12, 14, 17 и 20-27 показали значение EC50 меньше чем 0,1 мкМ в клеточной линии A375.
СИНТЕЗ ГИДРОКСИЛАМИНОВ
(S)-1-Аминоокси-пропан-2-ол гидрохлорид
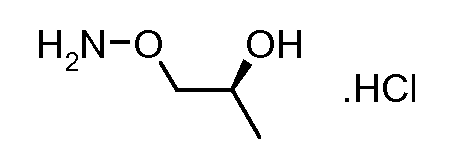
Стадия 1: Этиловый эфир (S)-2-(трет-бутил-диметил-силанилокси)-пропионовой кислоты,
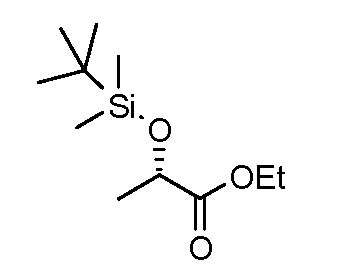
К раствору (S)-(-)-этиллактата (37,4 г, 0,32 моль) в DCM (200 мл) добавляли имидазол (25,75 г, 0,38 моль) и TBSCl (50 г, 0,33 моль). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь разбавляли водой и экстрагировали DCM (2×50 мл), объединенные органические экстракты промывали насыщенным солевым раствором, сушили (MgSO4) и концентрировали в вакууме с получением указанного в заголовке вещества в виде бесцветного масла (100%). 1H ЯМР (CDCl3, 400 МГц) 4,21 (1H, кв, J=6,73 Гц), 4,14-4,01 (2H, м), 1,29 (3H, д, J=6,79 Гц), 1,18 (3H, т, J=7,09 Гц), 0,80 (9H, с), -0,01 (6H, с).
Стадия 2: (S)-2-(трет-бутил-диметил-силанилокси)-пропан-1-ол
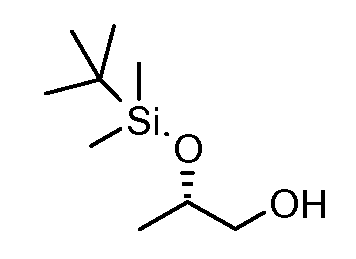
2M раствор LiBH4 (2,77 г, 0,13 моль) в ТГФ (60 мл) добавляли по каплям к раствору этилового эфира (S)-2-(трет-бутил-диметил-силанилокси)-пропионовой кислоты (22,75 г, 0,10 моль) и метанола (5,15 мл, 0,13 моль) в диэтиловом эфире (500 мл) при температуре 0ºC. Реакционную смесь перемешивали при температуре 0ºC в течение 1 часа и затем при комнатной температуре в течение 1,5 часов, далее охлаждали до температуры 0ºC и осторожно гасили водой. Реакционную смесь фильтровали и фильтрат экстрагировали диэтиловым эфиром (2×50 мл), объединенные органические экстракты промывали насыщенным солевым раствором, сушили (MgSO4) и концентрировали в вакууме с получением указанного в заголовке вещества в виде бесцветного масла (18,2 г, 97%). 1H ЯМР (CDCl3, 400 МГц) 3,86-3,77 (1H, м), 3,45-3,37 (1H, м), 3,31-3,22 (1H, м), 1,96-1,85 (1H, м), 1,03 (3H, д, J=6,23 Гц), 0,81 (9H, с), 0,00 (6H, с).
Стадия 3: 2-[(S)-2-(трет-бутил-диметил-силанилокси)-пропокси]изоиндол-1,3-дион
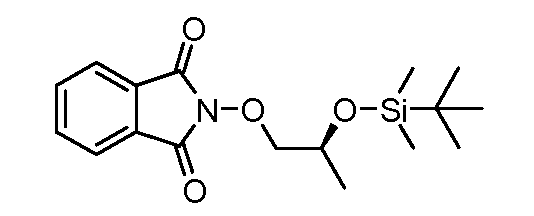
К суспензии (S)-2-(трет-бутил-диметил-силанилокси)-пропан-1-ола (53,0 г,0,28 моль), N-гидроксифталимида (47,0 г, 0,29 моль) и трифенилфосфина (77,9 г, 0,30 моль) в ТГФ (200 мл) при температуре 0ºC добавляли по каплям DIAD (56,8 мл, 0,29 моль). В процессе добавления реагенты растворялись, и раствор опять приобретал красный цвет, постепенно бледнеющий до бледно-желтого цвета по завершении добавления. Реакционную смесь перемешивали и давали нагреться до комнатной температуры в течение ночи. Реакционную смесь концентрировали в вакууме и полученный остаток вновь растворяли в диэтиловом эфире. Суспензию фильтровали, и фильтрат концентрировали в вакууме с получением сырого указанного в заголовке соединения в виде масла бледно-желтого цвета. Продукт использовали на следующей стадии без дополнительной очистки.
Стадия 4: O-[(S)-2-(трет-бутил-диметил-силанилокси)-пропил]гидроксиламин
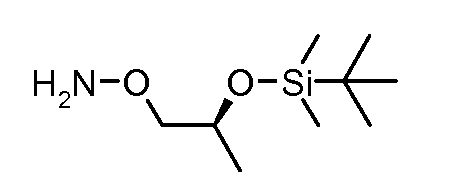
В холодный раствор сырого 2-[(S)-2-(трет-бутил-диметил-силанилокси)-пропокси]-изоиндол-1,3-диона (0,28 моль) в DCM (200 мл) при температуре 0ºC добавляли метил гидразин (14,74 мл, 0,28 моль). Реакционную смесь перемешивали при температуре 0ºC в течение 30 минут, затем фильтровали и фильтрат концентрировали в вакууме. Полученный остаток подвергали перегонке (т. кип. 108-112°C при давлении 2-10 мбар) с получением указанного в заголовке соединения в виде бесцветного масла. (42,63 г, 74% из этиллактат). 1H ЯМР (CDCl3, 400 МГц) 5,36 (2H, с), 3,95 (1H, м), 3,55-3,41 (2H, м), 1,04 (3H, д, J=6,28 Гц), 0,81 (9H, с), 0,02 (6H, м).
Стадия 5: (S)-1-аминоокси-пропан-2-ол гидрохлорид
O-[(S)-2-(трет-бутил-диметил-силанилокси)-пропил]-гидроксиламин (6,56 г, 31,9 ммоль) растворяли в IMS (20 мл) и добавляли 12н HCl (2,79 мл, 33,5 ммоль), реакционную смесь перемешивали при комнатной температуре в течение 1,5 часов. Реакционную смесь концентрировали в вакууме, и полученный остаток кристаллизовали из смеси IPA/диэтиловый эфир (1:1) с получением указанного в заголовке соединения в виде красивых белых иголок (3,2 г, 79%). 1H ЯМР (ДМСО-d6, 400 МГц) 10,93 (3H, с), 3,94-3,80 (2H, м), 2,51-2,48 (1H, м), 1,07-1,02 (3H, д, J= 6,01 Гц).
Гидрохлорид (S)-1-аминооксипропан-2-ола. Альтернативный способ
Стадия 1: 2-((S)-2-гидрокси-пропокси)-изоиндол-1,3-дион
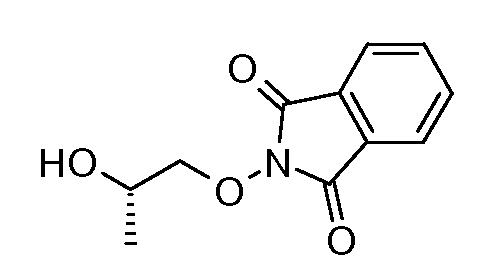
К суспензии N-гидроксифталимида (250 г, 1,53 моль), толуола (450 мл), тетрабутиламмоний бромида (24,7 г, 76,6 ммоль) и (S)-(-)-пропилен оксида (214,7 мл, 3,07 моль) добавляли DIPEA (13,3 мл, 76,6 ммоль). Реакционную смесь нагревали в атмосфере азота при температуре кипения в течение 3 часов. Реакционную смесь концентрировали в вакууме с получением твердого продукта желтого цвета. Твердый продукт растворяли в горячем этилацетате и помещали на слой флеш силикагеля (600 г) и продукт элюировали диэтиловым эфиром (4 л). Эфирный раствор концентрировали в вакууме с получением твердого продукта бледно-желтого цвета. Твердый продукт растворяли в этилацетате (150 мл) при температуре 75°C и добавляли циклогексан (300 мл). Раствор оставляли охлаждаться до комнатной температуры при перемешивании, что вызывало кристаллизацию из раствора твердого продукта белого цвета. Кристаллы собирали путем фильтрации, промывали смесью этилацетат/гексан (1:2). Твердый продукт перекристаллизовывали, используя условия, описанные ранее, с получением продукта в виде твердого кристаллического вещества белого цвета (233 г, 69%). 1H ЯМР (CDCl3, 400 МГц) 7,89-7,83 (2H, м), 7,81-7,75 (2H, м), 4,22 (1H, дд, J=11,5, 2,3 Гц), 4,11-4,02 (1H, м), 3,93 (1H, дд, J=11,3, 9,2 Гц), 3,79 (1H, д, J=3,2 Гц), 1,18 (3H, д, J=6,4 Гц).
Стадия 2: Гидрохлорид (S)-1-аминооксипропан-2-ола
Раствор 2-((S)-2-гидроксипропокси)-изоиндол-1,3-диона (20 г, 90,4 ммоль) и водной соляной кислоты (150 мл, 6н, 0,9 моль) перемешивали при комнатной температуре в течение 16 часов с получением суспензии белого цвета. Реакционную смесь фильтровали, и фильтрат концентрировали в вакууме с получением твердого продукта белого цвета. Полученный остаток кристаллизовали из горячей смеси IPA/циклогексан (10 мл/20 мл), что давало продукт, гидрохлорид (S)-1-аминооксипропан-2-ола, в виде твердого кристаллического вещества белого цвета (7,78 г, 67%). 1H ЯМР (ДМСО-d6, 400 МГц) 10,82 (2H, ушир.c), 3,92-3,78 (3H, м), 1,06 (3H, д, J=6,2 Гц).
Гидрохлорид 2-аминоокси-2-метил-пропан-1-ола
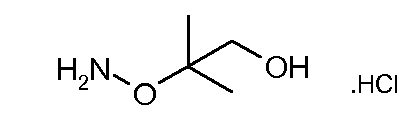
Стадия 1: этиловый эфир 2-(N-Boc-аминоокси)изомасляной кислоты
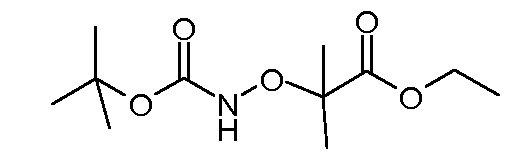
К раствору N-Boc-гидроксиламина (5,2 г, 39,05 ммоль) в этаноле (100 мл) добавляли гидроксид калия (2,63 г, 46,86 ммоль), смесь перемешивали при комнатной температуре до образования раствора. Добавляли этиловый эфир 2-бромизобутановой кислоты (6,87 мл, 46,9 ммоль) и реакционную смесь нагревали при температуре кипения в течение 18 часов. Реакционную смесь охлаждали до комнатной температуры, затем фильтровали, и фильтрат концентрировали в вакууме. Полученный маслянистый остаток распределяли между водой (75 мл) и диэтиловым эфиром, и водную фракцию экстрагировали диэтиловым эфиром (2×100 мл). Объединенные органические экстракты сушили (Na2SO4), фильтровали и концентрировали в вакууме с получением указанного в заголовке соединения в виде прозрачного масла (9,5 г, 99%). LCMS (способ C): RT=2,55 мин, [M+H]+=248. 1H ЯМР (CDCl3, 400 МГц) 4,20 (кв, 2H), 1,50 (с, 6H), 1,49 (с, 9H), 1,30 (т, 3H).
Стадия 2: 2-(N-Boc-аминоокси)-2-метилпропан-1-ол
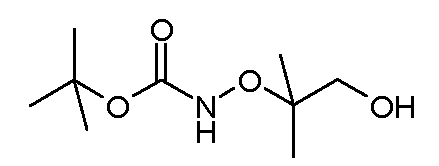
К раствору этилового эфира 2-(N-Boc-аминоокси)изобутановой кислоты (2,35 г, 9,5 ммоль) в безводном этиловом эфире (100 мл) при температуре 0ºC в атмосфере азота добавляли 1,0 M литий тетрагидроалюмината в тетрагидрофуране (17,1 мл, 17 ммоль), и реакционную смесь перемешивали при температуре 0ºC в течение 5 часов. Реакционную смесь гасили с помощью воды (25 мл) и давали нагреться до комнатной температуры. Суспензию фильтровали, остаток промывали диэтиловым эфиром и слои разделяли. Органический экстракт сушили (Na2SO4), фильтровали и концентрировали в вакууме с получением указанного в заголовке соединения в виде твердого вещества белого цвета (1,94 г, 99%). 1H ЯМР (CDCl3, 400 МГц) 3,40 (с, 2H), 1,50 (с, 9H), 1,20 (с, 6H).
Стадия 3: гидрохлорид 2-аминоокси-2-метил-пропан-1-ола
К раствору 2-(N-Boc-аминоокси)-2-метилпропан-1-ола (1,94 г, 9,45 ммоль) в безводном дихлорметане (10 мл) добавляли 4 M HCl в диоксане (47,3 мл, 200 ммоль) и смесь перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь концентрировали в вакууме, и остаток растирали в эфире (3×30 мл) с получением указанного в заголовке соединения в виде твердого продукта белого цвета (1,10 г, 82 %). 1H ЯМР (ДМСО-d6, 400 МГц) 3,58 (с, 2H), 3,48 (с, 2H), 1,34 (с, 6H).
СИНТЕЗ АНИЛИНОВ
2-Фтор-4-триметилсиланил-фениламин
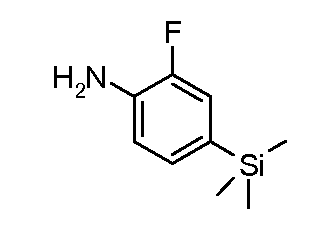
Способ A. Стадия 1: (3-фтор-4-нитрофенил)-триметилсилан
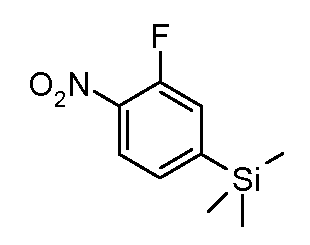
4-хлор-2-фтор-1-нитробензол (97,2 г, 0,55 моль) растворяли в ксилоле (208 мл) и добавляли гексаметилдисилан (306 г, 2,78 моль). В смесь в течение 20 минут барботировали аргон, затем добавляли Pd(PPh3)4 (16,2 г, 14 ммоль) и смесь нагревали в непрерывном токе аргона при температуре 150ºC в течение 1 часа. Затем присоединяли баллон, наполненный аргоном, и смесь нагревали при температуре 150ºC в течение еще 60 часов. После охлаждения смесь разбавляли Et2O и фильтровали через слой силикагеля в 4 см. Лепешку на фильтре промывали дополнительным количеством Et2O и объединенные органические остатки концентрировали в вакууме. Очистка полученного остатка с помощью флеш хроматографии (SiO2, элюент 98:1:1 пентан:CH2Cl2:Et20) давала 76,7 г указанного в заголовке соединения в виде масла оранжевого цвета, а также смешанные фракции. Смешанные фракции объединяли и концентрировали, затем перегоняли (110ºC, 6 мбар) с получением дополнительных 7,2 г указанного в заголовке соединения (всего 83,9 г, 71%). 1H ЯМР (ДМСО-d6) 0,30 (9H, с), 7,56 (1H, д, J=8,02 Гц), 7,67 (1H, дд, J=11,49, 1,14 Гц), 8,10 (1H, т, J=7,66 Гц).
Способ A. Стадия 2: 2-фтор-4-триметилсиланил-фениламин
Взвесь 10% масс. палладия на углероде (4,0 г) в IMS (25 мл) добавляли к раствору (3-фтор-4-нитрофенил)триметилсилана (62,0 г, 0,29 моль) в IMS (250 мл), и реакционную смесь продували азотом пять раз, затем водородом три раза. Реакционную смесь затем перемешивали под давлением водорода в 3 бар при комнатной температуре в течение 4 часов. Реакционную смесь затем опять продували азотом, затем фильтровали через слой целлита Celite®, промывая этилацетатом. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения в виде масла светло-коричневого цвета (53,0 г, количественный). 1H ЯМР (CDCl3, 400 МГц) 7,16-7,09 (1H, м), 7,10 (1H, д, J=7,75 Гц), 6,81 (1H, т, J=8,16 Гц), 3,78 (2H, с), 0,26 (9H, с).
Способ B. 2-фтор-4-триметилсиланил-фениламин
К раствору 4-бром-2-фтор-фениламина (114 г, 0,6 моль) в безводном ТГФ (750 мл) в инертной атмосфере при температуре -78°C по каплям добавляли 1,6M раствор нBuLi в гексане (1500 мл, 2,4 моль), поддерживая внутреннюю температуру ниже -60ºC. Реакционную смесь обрабатывали по каплям TMSCl (256 мл, 2,0 моль), поддерживая внутреннюю температуру ниже -60ºC. Реакционной смеси давали нагреться до 0ºC в течение 1 часа и выливали в охлажденную льдом 2M HCl (приблизительно 1 л). Смесь энергично перемешивали в течение 10 мин, затем органический слой отделяли, промывали водой, затем насыщенным раствором карбоната калия, сушили (Na2SO4), фильтровали и концентрировали с получением указанного в заголовке соединения в виде масла светло коричневого цвета (89 г, 81%).
4-Циклопропил-2-фтор-фениламин
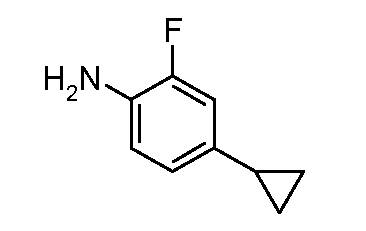
Стадия 1: 3-фтор-4-нитрофениловый эфир трифтор-метансульфоновой кислоты
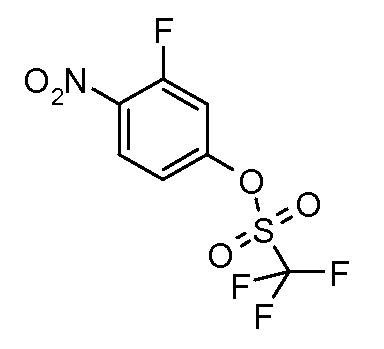
К раствору 3-фтор-4-нитрофенола (12,5 г, 80 ммоль) и ангидрида трифторметансульфоновой кислоты (26,8 мл, 160 ммоль) в DCM (300 мл) при температуре 0ºC по каплям добавляли триэтиламин (44,6 мл, 320 ммоль). Реакционную смесь перемешивали при температуре 0ºC в течение 2 часов, затем давали нагреться до комнатной температуры и перемешивали в течение 18 часов. Реакционную смесь гасили путем добавления воды, и смесь экстрагировали DCM. Органический слой отделяли, промывали водой и затем сушили (MgSO4), фильтровали и концентрировали в вакууме. Полученный остаток подвергали флеш хроматографии (Si-PPC, градиент от 0-40% этилацетата в циклогексане) с получением указанного в заголовке соединения в виде масла желтого цвета (12,8 г, 56%-ный выход). 1H ЯМР (ДМСО-d6, 400 МГц) 8,39 (1H, т, J=8,83 Гц), 8,12 (1H, дд, J=11,09, 2,65 Гц), 7,67 (1H, ддд, J=9,20, 2,62, 1,52 Гц).
Стадия 2: 4-циклопропил-2-фтор-1-нитробензол
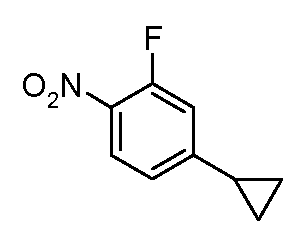
Перемешиваемую суспензию 3-фтор-4-нитрофенилового эфира трифторметансульфоновой кислоты (5,6 г, 19 ммоль), циклопропилбороновой кислоты (2,09 г, 23,3 ммоль), Pd(dppf)Cl2 (1,24 г, 1,5 ммоль) и 2M водного карбоната цезия (30 мл, 60 ммоль) в толуоле (20 мл) дегазировали, после чего нагревали при температуре 90ºC в атмосфере аргона в течение 2,5 часов. Реакционную смесь оставляли охлаждаться до комнатной температуры, затем фильтровали через слой целита Celite®, промывали этилацетатом. Фильтрат промывали (вода, насыщенный солевой раствор) и затем сушили (MgSO4), фильтровали и концентрировали в вакууме. Полученный остаток подвергали флеш хроматографии (Si-PPC, градиент 0-30% этилацетата в пентане) с получением указанного в заголовке соединения в виде твердого продукта желтого цвета (2,79 г, 81%). 1H ЯМР (ДМСО-d6, 400 МГц) 8,03 (1H, т, J=8,39 Гц), 7,28 (1H, дд, J=13,19, 1,91 Гц), 7,16 (1H, дд, J=8,61, 1,90 Гц), 2,14-2,05 (1H, м), 1,21-1,05 (2H, м), 0,92-0,82 (2H, м).
Стадия 3: 4-циклопропил-2-фторфениламин
Взвесь палладия на углероде (200 мг, 10% масс.) в IMS добавляли в дегазированный раствор 4-циклопропил-2-фтор-1-нитробензола (1,45 г, 8 ммоль) в IMS (50 мл), воздух откачивали и сосуд наполняли азотом, затем вновь вакуумировали и наполняли водородом. Реакционную смесь перемешивали под давлением водорода в 1 атмосферу при комнатной температуре в течение 24 часов, затем фильтровали через слой целита Celite®, потом промывали этилацетатом. Фильтрат концентрировали в вакууме с получением указанного в заголовке соединения в виде остатка бледно-лилового цвета (1,19 г, 98%). 1H ЯМР (CDCl3, 400 МГц) 6,72-6,63 (3H, м), 3,56 (2H, с), 1,83-1,75 (1H, м), 0,93-0,82 (2H, м), 0,59-0,54 (2H, м).
СИНТЕЗ ПРОМЕЖУТОЧНЫХ ГЕТЕРОЦИКЛИЧЕСКИХ ЯДЕР
Метиловый эфир 4-хлор-1-(толуол-4-сульфонил)-1H-индазол-5-карбоновой кислоты и метиловый эфир 4-хлор-2-(толуол-4-сульфонил)-1H-индазол-5-карбоновой кислоты
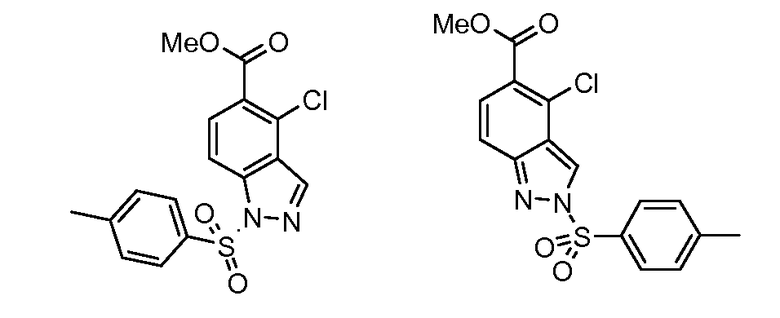
Стадия 1: метиловый эфир 4-амино-2-хлор-3-метил-бензойной кислоты
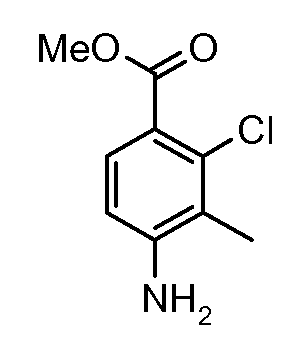
К суспензии 4-амино-2-хлор-3-метил-бензойной кислоты (0,64 г, 3,45 ммоль) в толуоле (10 мл) и метаноле (10 мл) при температуре 0ºC добавляли по каплям триметилсилилдиазометан (3,45 мл, 2 M в гексане, 6,90 ммоль). Реакционную смесь перемешивали при температуре 0ºC в течение 30 мин, в процессе чего реагенты растворялись. Реакционную смесь гасили путем добавления уксусной кислоты (1 мл), затем промывали насыщенным водным раствором бикарбоната натрия (10 мл) и водную фракцию дважды экстрагировали этилацетатом (2×10 мл). Объединенные органические экстракты промывали насыщенным солевым раствором (20 мл), сушили (MgSO4) и концентрировали в вакууме с получением указанного в заголовке соединения в виде твердого вещества бежевого цвета (0,66 г, 96%). 1H ЯМР (CDCl3, 400 МГц) 2,26 (3H, с), 3,86 (3H, с), 6,55 (1H, д, J=8,8 Гц), 7,60 (1H, д, J=8,8 Гц).
Стадия 2: метиловый эфир 4-хлор-1H-индазол-5-карбоновой кислоты
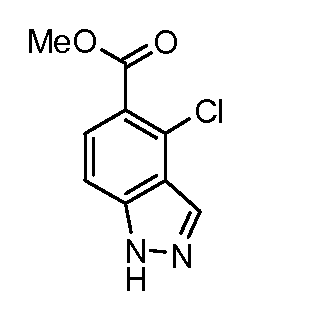
К раствору метилового эфира 4-амино-2-хлор-3-метил-бензойной кислоты (5,29 г, 26,5 ммоль) в уксусной кислоте (100 мл) добавляли изоамилнитрит (3,9 мл, 29,2 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 30 минут, затем нагревали при температуре кипения в течение 3 часов. Реакционную смесь концентрировали в вакууме, и полученный остаток подвергали флеш хроматографии (SiO2, градиент 0-100% этилацетата в циклогексане) с получением указанного в заголовке соединения в виде твердого продукта не совсем белого цвета (2,26 г, 40%). 1H ЯМР (CDCl3, 400 МГц) 3,97 (3 H, с), 7,42 (1H, д, J=8,8 Гц), 7,95 (1H, д, J=8,8 Гц), 8,29 (1H, с).
Стадия 3: метиловый эфир 4-хлор-1-(толуол-4-сульфонил)-1H-индазол-5-карбоновой кислоты и метиловый эфир 4-хлор-2-(толуол-4-сульфонил)-1H-индазол-5-карбоновой кислоты
К раствору метилового эфира 4-хлор-1H-индазол-5-карбоновой кислоты (1 г, 4,74 ммоль) в ТГФ (20 мл) добавляли п-толуолсульфонил хлорид (1,0 г, 5,2 ммоль), триэтиламин (0,8 мл, 5,7 ммоль) и DMAP (каталитическое количество). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов, в процессе чего образовывался осадок белого цвета. Реакционную смесь разбавляли водой и экстрагировали этилацетатом (3×20 мл). Объединенные органические фракции промывали насыщенным солевым раствором (20 мл), сушили (MgSO4) и концентрировали в вакууме. Полученный остаток подвергали флеш хроматографии (SiO2, градиент 0-80% этилацетата в циклогексане) с получением указанного в заголовке соединения в виде твердого продукта не совсем белого цвета (1,51 г, 82%). 1H ЯМР показывал, что продукт являлся смесью 1:1 изомеров. LCMS (Способ B): RT=4,02 мин, [M-H]-=363.
Ди-трет-бутиловый эфир 4-бром-индазол-1,5-дикарбоновой кислоты
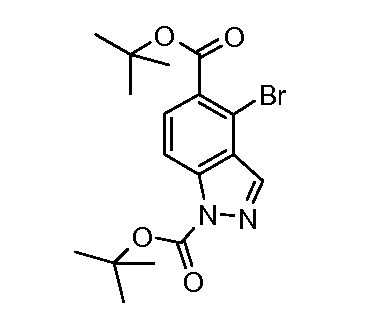
Стадия 1: трет-бутиловый эфир 2-бром-4-фтор-бензойной кислоты
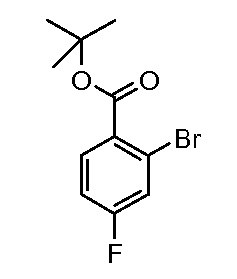
К суспензии 2-бром-4-фтор-бензойной кислоты (28,5 г, 0,13 моль) в дихлорметане (500 мл) при комнатной температуре добавляли оксалил хлорид (11,35 мл, 0,26 ммоль), затем ДМФ (0,05 мл, каталитическое количество, (ВНИМАНИЕ! Энергичное выделение газа) и реакционную смесь перемешивали в течение 3 часов. Реакционную смесь концентрировали в вакууме, и остаток растворяли в DCM (500 мл), далее обрабатывали раствором трет-бутанола (28,5 г, 0,26 моль) и пиридином (20,5 г, 0,26 моль). Полученную смесь перемешивали при комнатной температуре в течение 3 дней, затем разбавляли DCM и промывали (1M водный гидроксид натрия, вода, 0,1M водная HCl, вода) сушили (Na2SO4) фильтровали и концентрировали в вакууме с получением масла желтого цвета. Сырое масло подвергали флеш хроматографии (Si-PPC, градиент от 0 до 20% этилацетата в циклогексане) с получением указанного в заголовке соединения в виде бесцветного масла (15,2 г, 42%). 1H ЯМР (CDCl3, 400 МГц) 7,76-7,73 (1H, м), 7,37 (1H, дд, J=8,36, 2,53 Гц), 7,05 (1H, ддд, J=8,70, 7,73, 2,53 Гц), 1,60 (9H, с).
Стадия 2: трет-бутиловый эфир 2-бром-4-фтор-3-формил-бензойной кислоты
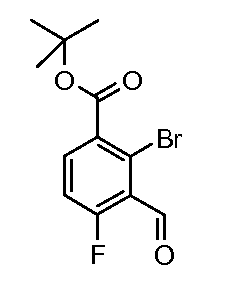
К раствору трет-бутилового эфира 2-бром-4-фтор-бензойной кислоты (10,0 г, 36 ммоль) в ТГФ (100 мл) при температуре -78ºC в атмосфере азота добавляли по каплям диизопропиламид лития (1,8M раствор, 20 мл, 36 ммоль). Реакционную смесь перемешивали в течение 1,25 часа, затем добавляли ДМФ (10 мл), затем перемешивали в течение еще 10 минут, далее гасили уксусной кислотой (3 мл). Продукты распределяли между этилацетатом и водой, органический слой отделяли, промывали насыщенным солевым раствором, сушили (Na2SO4), фильтровали и концентрировали в вакууме с получением масла желтого цвета, которое крислаллизовалось при стоянии. Сырой продукт растирали в циклогексане с получением указанного в заголовке соединения в виде твердого продукта желтого цвета (6,8 г, 62%). 1H ЯМР (CDCl3, 400 МГц) 10,38 (1H, с), 7,80-7,73 (1H, м), 7,17 (1H, т, J=9,09 Гц), 1,62 (9H, с).
Стадия 3: трет-бутиловый эфир 4-бром-1H-индазол-5-карбоновой кислоты
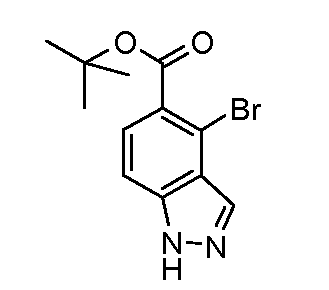
Бифазный раствор трет-бутилового эфира 2-бром-4-фтор-3-формил-бензойной кислоты (4,25 г, 14 ммоль), DME (25 мл) и гидразин гидрата (15 мл) нагревали при температуре 90ºC в течение 1 часа. После охлаждения продукты распределяли между этилацетатом и водой, водный слой экстрагировали этилацетатом и объединенные органические экстракты сушили (Na2SO4), фильтровали и концентрировали в вакууме с получением указанного в заголовке соединения в виде твердого продукта рыжеватого цвета (4,1 г, 100%). LCMS (способ B) RT=3,63 мин, [M + CH3CN + H]+=338/340, [M-H]-=295/297.
Стадия 4: ди-трет-бутиловый эфир 4-бром-индазол-1,5-дикарбоновой кислоты
К раствору трет-бутилового эфира 4-бром-1H-индазол-5-карбоновой кислоты (3,0 г, 10 ммоль) и триэтиламина (1,53 мл, 11 ммоль) в DCM (30 мл) добавляли ди-трет-бутил-дикарбонат (2,4 г, 11 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 4 часов. Реакционную смесь разбавляли DCM, промывали (насыщенный водный NaHCO3, вода), сушили (Na2SO4), фильтровали и концентрировали в вакууме с получением масла желтого/оранжевого цвета, которое кристаллизовалось при стоянии. Сырой продукт растирали в пентане с получением указанного в заголовке соединения в виде твердого вещества беловатого/желтого цвета (1,8 г, 45%). 1H ЯМР (CDCl3, 400 МГц) 8,29 (1H, с), 8,18-8,10 (1H, м), 7,92 (1H, д, J=8,74 Гц), 1,70 (9H, с), 1,63 (9H, с).
Трет-бутиловый эфир 4-бром-бензо[d]изотиазол-5-карбоновой кислоты
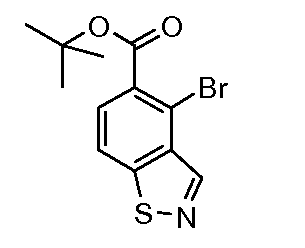
Стадия 1: трет-бутиловый эфир 4-бензилсульфанил-2-бром-3-формил-бензойной кислоты
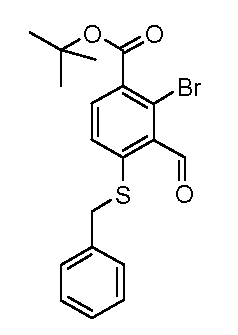
К раствору трет-бутоксида калия (318 мг, 2,84 ммоль) в ТГФ (20 мл) в атмосфере азота добавляли бензолметантиол (332 мкл, 2,84 ммоль) и смесь перемешивали при комнатной температуре в течение 10 минут. Добавляли трет-бутиловый эфир 2-бром-4-фтор-3-формил-бензойной кислоты (860 мг, 2,84 ммоль) и перемешивание продолжали в течение 30 минут, затем гасили насыщенным водным раствором NH4Cl. Продукты распределяли между этилацетатом и водой, органический слой отделяли, промывали насыщенным солевым раствором, сушили (Na2SO4), фильтровали и концентрировали в вакууме с получением указанного в заголовке соединения в виде твердого продукта желтого цвета (1,15 г, 100%). 1H ЯМР (CDCl3) 10,57 (1H, с), 7,61 (1H, д, J=8,44 Гц), 7,40-7,25 (6H, м), 4,17 (2H, с), 1,61 (9H, с).
Стадия 2: трет-бутиловый эфир 4-бром-бензо[d]изотиазол-5-карбоновой кислоты
В холодный (0ºC) раствор трет-бутилового эфира 4-бензилсульфанил-2-бром-3-формил-бензойной кислоты (1,15 г, 2,82 ммоль) в DCM (15 мл) добавляли сульфурил хлорид (453 мкл, 5,64 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 18 часов. Реакционную смесь концентрировали при пониженном давлении, и остаток подвергали азеотропной отгонке с ТГФ (×2) и затем растворяли в ТГФ (7 мл). Полученный раствор охлаждали до температуры 0-5ºC и добавляли по каплям раствор аммиака в метаноле (2M, 15 мл). Реакционную смесь перемешивали холодной в течение 30 минут, затем при комнатной температуре в течение 18 часов. Реакционную смесь концентрировали при пониженном давлении, и остаток частично растворяли в диэтиловом эфире. Эфирный раствор концентрировали в вакууме, и остаток подвергали флеш хроматографии (Si-PPC, градиент 0-10% диэтиловый эфир в пентане) с получением указанного в заголовке соединения в виде бесцветного масла (523 мг, 59%). 1H ЯМР (CDCl3) 9,13 (1H, д, J=0,88 Гц), 7,89 (1H, дд, J=8,36, 0,97 Гц), 7,83 (1H, д, J=8,37 Гц), 1,61 (9H, с).
7-Фтор-бензо[d]изотиазол-6-карбоновая кислота
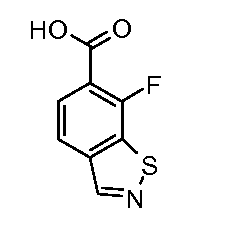
Стадия 1: трет-бутиловый эфир 2,3-дифтор-4-метил-бензойной кислоты
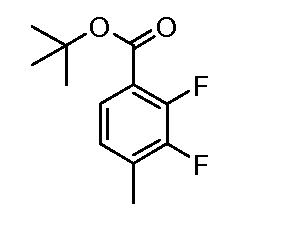
Смесь 2,3-дифтор-4-метил-бензойной кислоты (20,0 г, 116 ммоль), ди-трет-бутил дикарбоната (25,0 г, 116 ммоль) и DMAP (2,0 г, 16,4 ммоль) в трет-бутаноле (500 мл) перемешивали при температуре 45°C в течение 5 часов, после чего концентрировали в вакууме. Полученный остаток растирали в Et2O и фильтровали. Фильтрат концентрировали в вакууме с получением остатка, который распределяли между этилацетатом и 1M водным раствором соляной кислоты. Органический слой отделяли и промывали насыщенным водным раствором гидрокарбоната натрия, затем насыщенным солевым раствором, сушили (Na2SO4), фильтровали и упаривали в вакууме с получением указанного в заголовке соединения в виде бесцветного масла (17,3 г, 65%). 1H ЯМР (CDCl3, 400 МГц) 7,51 (1H, ддд, J=8,3, 6,6, 1,9 Гц), 6,95 (1H, м), 2,33 (3 H, д, J=2,3 Гц), 1,59 (9H, с).
Стадия 2: трет-бутиловый эфир 4-бромметил-2,3-дифтор-бензойной кислоты
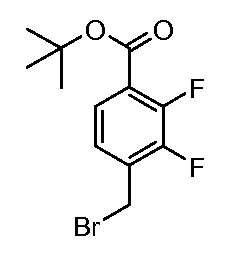
Раствор трет-бутилового эфира 2,3-дифтор-4-метил-бензойной кислоты (17,3 г, 75,9 ммоль) и N-бромсукцинимида (13,5 г, 75,9 ммоль) в тетрахлориде углерода (250 мл) дегазировали в течение 10 минут. Добавляли AIBN (1,2 г, 7,32 ммоль) и реакционную смесь перемешивали при температуре кипения в течение 18 часов, после чего охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали в вакууме с получением остатка, который подвергали флеш хроматографии (Si-PPC, градиент от 0% до 10%, TBME в циклогексане), что давало указанное в заголовке соединение в виде бесцветного масла (16,7 г, загрязненные 25% исходного продукта). 1H ЯМР (CDCl3, 400 МГц) 7,61 (1H, ддд, J=8,3, 6,4, 2,1 Гц), 7,18 (1H, ддд, J=8,1, 6,4, 1,8 Гц), 4,49 (2H, д, J=1,3 Гц), 1,59 (9H, с).
Стадия 3: трет-бутиловый эфир 2,3-дифтор-4-формил-бензойной кислоты
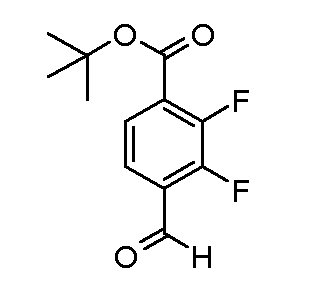
К раствору трет-бутилового эфира 4-бромметил-2,3-дифтор-бензойной кислоты (12,9 г, 42,1 ммоль) в ДМСО (80 мл) и DCM (40 мл) при температуре 0ºC добавляли N-оксид триметиламина (3,4 г, 45,3 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 18 часов, после чего концентрировали в вакууме. Полученный остаток распределяли между ледяной водой и этилацетатом. Органический слой отделяли и дважды промывали насыщенным солевым раствором, сушили (Na2SO4), фильтровали и упаривали в вакууме. Получали остаток, который подвергали флеш хроматографии (Si-PPC, градиент от 0% до 10%, TBME в циклогексане), что давало указанное в заголовке соединение в виде твердого вещества белого цвета (4,34 г, 43%). 1H ЯМР (CDCl3, 400 МГц) 10,38 (1H, д, J=0,8 Гц), 7,71 (1H, дддд, J=7,4, 5,6, 1,7, 0,8 Гц), 7,63 (1H, ддд, J=7,4, 5,6, 1,5 Гц), 1,61 (9H, с).
Стадия 4: трет-бутиловый эфир 3-бензилсульфанил-2-фтор-4-формил-бензойной кислоты
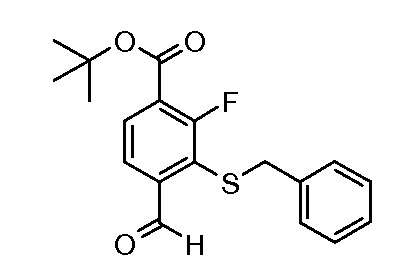
К раствору трет-бутоксида калия (2,0 г, 17,9 ммоль) в безводном ТГФ (80 мл) добавляли бензил меркаптан (2,1 мл, 17,9 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 5 минут, после чего охлаждали до температуры -30ºC. В течение 15 минут по каплям добавляли раствор трет-бутилового эфира 2,3-дифтор-4-формил-бензойной кислоты (4,34 г, 17,9 ммоль) в безводном ТГФ (20 мл) и полученную смесь перемешивали при температуре -30ºC в течение 30 минут, после чего гасили путем добавления воды и экстрагировали этилацетатом. Органический слой отделяли, промывали водой, затем насыщенным солевым раствором, сушили (Na2SO4), фильтровали и упаривали в вакууме с получением указанного в заголовке соединения в виде масла желтого цвета (6,2 г, 100%). 1H ЯМР (CDCl3, 400 МГц) 10,20 (1H, д, J=0,6 Гц), 7,84 (1H, ддд, J=8,0, 6,8, 0,7 Гц), 7,59 (1H, дд, J=8,0, 0,9 Гц), 7,19 (3 H, м), 7,05 (2H, м), 4,07 (2H, с), 1,63 (9H, с).
Стадия 5: трет-бутиловый эфир 7-фтор-бензо[d]изотиазол-6-карбоновой кислоты
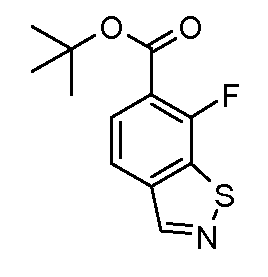
К раствору трет-бутилового эфира 3-бензилсульфанил-2-фтор-4-формил-бензойной кислоты (6,20 г, 17,9 ммоль) в DCM (100 мл) добавляли сульфурилхлорид (2,9 мл, 35,8 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа, после чего концентрировали в вакууме. Полученный остаток дважды подвергали азеотропной отгонке с толуолом и затем обрабатывали ТГФ (50 мл). Полученный раствор охлаждали до температуры 0ºC и добавляли 2M раствор аммиака в метаноле (100 мл). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа и затем концентрировали в вакууме. Полученный остаток распределяли между этилацетатом и насыщенным водным раствором гидрокарбоната натрия. Органический слой отделяли и промывали водой, затем насыщенным солевым раствором, сушили (Na2SO4), фильтровали и упаривали в вакууме. Остаток подвергали флеш хроматографии (Si- PPC, градиент от 0% до 10%, Et2O в пентане), что давало указанное в заголовке соединение в виде твердого продукта желтого цвета (2,96 г, 65%). 1H ЯМР (CDCl3, 400 МГц) 8,93 (1H, дд, J=4,1, 0,5 Гц), 7,91 (1H, ддд, J=8,3, 5,8, 0,5 Гц), 7,84 (1H, д, J=8,3 Гц), 1,64 (9H, с).
Стадия 6: 7-фтор-бензо|d]изотиазол-6-карбоновая кислота
К раствору трет-бутилового эфира 7-фтор-бензо[d]изотиазол-6-карбоновой кислоты (1,0 г, 4,0 ммоль) в DCM (10 мл) добавляли воду (0,2 мл) и ТФУ (10 мл). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа и концентрировали в вакууме. Остаток подвергали азеотропной отгонке с толуолом с получением указанного в заголовке соединения в виде твердого продукта желтого цвета (788 мг, 100%). LCMS (способ B): RT=2,86 мин, [M-H]-=196.
СИНТЕЗ ФЕНИЛАМИНОКИСЛОТ
4-(2-Фтор-4-йод-фениламино)-1-(толуол-4-сульфонил)-1H-индазол-5-карбоновая кислота
Стадия 1: метиловый эфир 4-(2-фтор-4-триметилсиланилфениламино)-1-(толуол-4-сульфонил)-1H-индазол-5-карбоновой кислоты
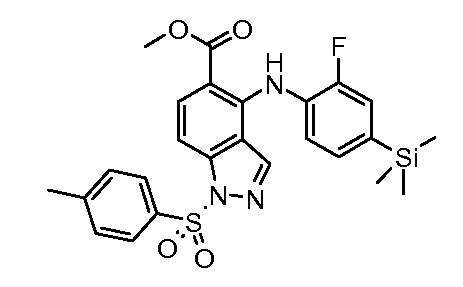
Метиловый эфир 4-хлор-1-(толуол-4-сульфонил)-1H-индазол-5-карбоновой кислоты и метиловый эфир 2-[1-хлор-1-[3-метил-1-(толуол-4-сульфонил)-1H-пиразол-4-ил]-мет-(Z)-илиден]-бут-3-еновой кислоты (1,1 г, 3,0 ммоль), 2-фтор-4-триметилсиланилфениламин (0,61 г, 3,3 ммоль), 2-дициклогексилфосфино-2'-6'-диизопропил бифенил (0,28 г, 0,60 ммоль), фосфат калия (0,70 г, 3,3 ммоль) и трис(дибензилиденацетон)дипалладий (O) (87 мг, 0,15 ммоль) суспендировали в толуоле (5 мл) и полученную смесь дегазировали. Реакционную смесь нагревали при температуре 105ºC в течение 3 часов. Реакционную смесь гасили путем добавления хлористоводородной кислоты (1M, 5 мл) и полученную смесь экстрагировали этилацетатом (3×20 мл). Объединенные органические экстракты промывали насыщенным солевым раствором (20 мл), сушили (MgSO4) и концентрировали в вакууме. Полученный остаток подвергали флеш хроматографии (Si-SPE, градиент 0-100% DCM в циклогексане) с получением указанного в заголовке соединения в виде твердого продукта бледно-коричневого цвета (0,78 г, 50%). Данные 1H ЯМР показали, что продукт является индивидуальным изомером. LCMS (Способ B): RT=5,22 мин, [M+H]+=512.
Стадия 2: метиловый эфир 4-(2-фтор-4-йодфениламино)-1-(толуол-4-сульфонил)-1H-индазол-5-карбоновой кислоты
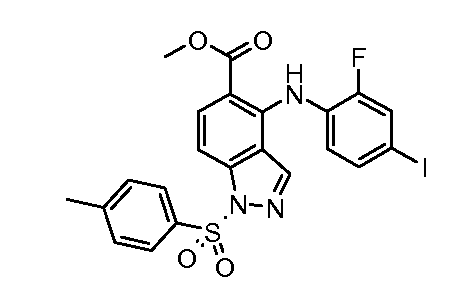
К раствору метилового эфира 4-(2-фтор-4-триметилсиланилфениламино)-1-(толуол-4-сульфонил)-1H-индазол-5-карбоновой кислоты (0,56 г, 1,1 ммоль) в DCM (3 мл) при температуре 0ºC добавляли раствор монохлорида йода в DCM (2,2 мл, 1M, 2,2 ммоль). Реакционную смесь перемешивали при температуре 0ºC в течение 30 минут. Реакционную смесь гасили путем добавления воды (10 мл), затем разбавляли насыщенным водным раствором тиосульфата натрия (10 мл). Полученную смесь экстрагировали этилацетатом (3×20 мл) и объединенные органические фракции промывали насыщенным солевым раствором (20 мл), сушили (MgSO4), затем концентрировали в вакууме. Полученный остаток подвергали флеш хроматографии (SiO2, градиент 0-100% DCM в циклогексане) с получением указанного в заголовке соединения в виде твердого вещества светло-коричневого цвета (0,51 г, 83%). LCMS (Способ B): RT=4,87 мин, [M+H]+=566.
Стадия 3: 4-(2-фтор-4-йод-фениламино)-1-(толуол-4-сульфонил)-1H-индазол-5-карбоновая кислота
К раствору метилового эфира 4-(2-фтор-4-йодфениламино)-1-(толуол-4-сульфонил)-1H-индазол-5-карбоновой кислоты (0,51 г, 0,91 ммоль) в толуоле (5 мл) добавляли бис(трибутилолово)оксид (0,92 мл, 1,8 ммоль) и реакционную смесь нагревали при температуре кипения в течение 48 часов. Реакционную смесь концентрировали в вакууме и полученный остаток подвергали флеш хроматографии (Si-PPC, градиент 0-10% метанол в DCM) с получением указанного в заголовке соединения в виде твердого вещества светло-коричневого цвета (0,46 г, 92%). LCMS (Способ B): RT=4,27 мин, [M+H]+=552.
4-(2-Фтор-4-йодфениламино)-1-(4-метоксибензил)-1H-пиразоло[3,4-b]пиридин-5-карбоновая кислота
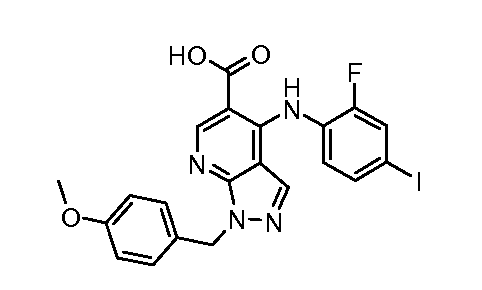
Стадия 1: этиловый эфир 4-(2-фтор-4-йодфениламино)-1-(4-метоксибензил)1H-пиразоло[3,4-b]пиридин-5-карбоновой кислоты
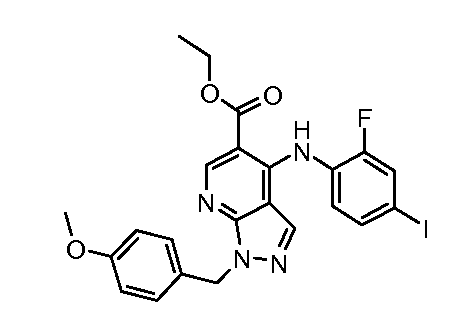
Этиловый эфир 4-хлор-1-(4-метокси-бензил)-1H-пиразоло[3,4-b]пиридин-5-карбоновой кислоты (1,8 г, 5,2 ммоль) и 2-фтор-4-йоданилин (1,5 г, 6,3 ммоль) растворяли в диоксане (20 мл) и полученную смесь перемешивали при температуре кипения в течение 16 часов. Реакционную смесь разбавляли водой (50 мл) и водный слой экстрагировали дихлорметаном (3×20 мл). Объединенные органические фракции фильтровали через гидрофобную фритту и концентрировали в вакууме. Полученный остаток подвергали флеш хроматографии (Si-PPC, градиент 0-30% этилацетата в циклогексане) с получением указанного в заголовке соединения в виде твердого вещества белого цвета (1,56 г, 56 %). LCMS (Способ B): RT=4,78 мин, [M+H]+=547.
Стадия 2: 4-(2-фтор-4-йодфениламино)-1-(4-метоксибензил)-1H-пиразоло[3,4-blпиридин-5-карбоновая кислота
К суспензии этилового эфира 4-(2-фтор-4-йодфениламино)-1-(4-метоксибензил)-1H-пиразоло[3,4-b]пиридин-5-карбоновой кислоты (380 мг, 0,71 ммоль) в IMS (10 мл) добавляли водный раствор гидроксида натрия (1,78 мл, 1M, 1,78 ммоль). Реакционную смесь нагревали при температуре 65°C в течение 2 часов, в процессе чего все твердые продукты растворялись. Легко летучие растворители удаляли в вакууме и полученный раствор подкисляли до pH ~3 осторожным добавлением водной соляной кислоты (1M), что вызывало образование осадка. Осадок собирали путем фильтрации и сушили в вакууме при температуре 45°C с получением указанного в заголовке соединения в виде твердого вещества коричневого цвета (360 мг, 100%). LCMS (Способ B): RT=3,84 мин, [M+H]+=519.
4-(4-Бром-2-фтор-фениламино)-1H-индазол-5-карбоновая кислота
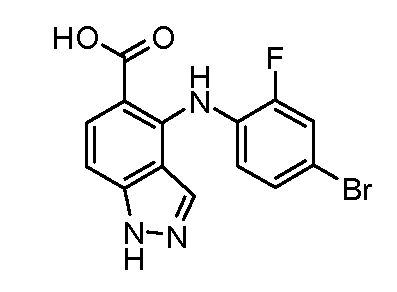
Стадия 1: ди-трет-бутиловый эфир 4-(2-фтор-4-триметилсиланил-фениламино)-индазол-1,5-дикарбоновой кислоты
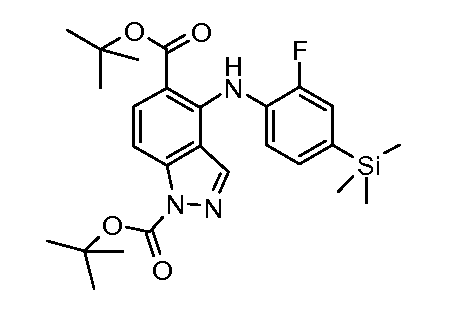
Раствор 2-фтор-4-триметилсиланил-фениламина (1,2 г, 6,5 ммоль) в толуоле (20 мл) добавляли к смеси ди-трет-бутилового эфира 4-бром-индазол-1,5-дикарбоновой кислоты (2,0 г, 5,0 ммоль), Pd2dba3 (114 мг, 2,5 мол%), Xantphos (144 мг, 5 моль%) и трехосновного фосфата калия (1,49 г, 7 ммоль) в атмосфере азота. Смесь вакуумировали и наполняли азотом и затем реакционную смесь нагревали при температуре 90ºC в течение 18 часов. Охлажденную реакционную смесь разбавляли этилацетатом, фильтровали через Celite®, и фильтрат концентрировали в вакууме. Полученный остаток подвергали флеш хроматографии (Si-PPC, сухой помещали на HM-N, градиент 0-10% этилацетата в циклогексане) с получением указанного в заголовке соединения в виде смолы оранжевого цвета (1,9 г, 76%). 1H ЯМР (CDCl3, 400 МГц) 10,03 (1H, с), 8,05 (1H, д, J=9,03 Гц), 7,57 (1H, дд, J=9,00, 0,83 Гц), 7,28-7,18 (4H, м), 1,68 (9H, с), 1,63 (9H, с), 0,28 (9H, с).
Стадия 2: ди-трет-бутиловый эфир 4-(4-бром-2-фтор-фениламино)-индазол-1,5-дикарбоновой кислоты
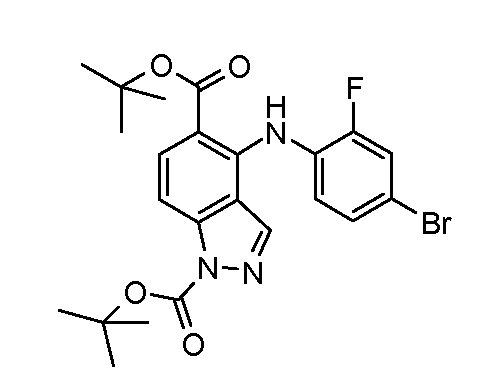
К раствору ди-трет-бутилового эфира 4-(2-фтор-4-триметилсиланил-фениламино)-индазол-1,5-дикарбоновой кислоты (850 мг, 1,7 ммоль) в DCM (10 мл) при температуре -15ºC добавляли по каплям N-бромсукцинимид (303 мг, 1,7 ммоль) в виде раствора в DCM (3 мл). Реакционную смесь перемешивали при температуре -15ºC в течение 1,25 часа, затем добавляли DCM и раствор промывали (насыщенный водный NaHCO3, затем вода), сушили (Na2SO4), фильтровали и концентрировали в вакууме. Полученный остаток подвергали флеш хроматографии (Si-PPC, градиент 0-10% этилацетата в циклогексане) с получением указанного в заголовке соединения в виде смолы оранжевого цвета (790 мг, 92%). LCMS (способ B) RT=5,41 мин, [M + CH3CN + Na]+=569/571.
Стадия 3: 4-(4-бром-2-фтор-фениламино)-1H-индазол-5-карбоновая кислота
К раствору ди-трет-бутилового эфира 4-(4-бром-2-фторфениламино)-индазол-1,5-дикарбоновой кислоты (420 мг, 0,83 ммоль) в DCM (5 мл) добавляли ТФУ (1,2 мл, 16,2 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов, после чего концентрировали в вакууме. Полученный остаток растворяли в этилацетате (10 мл), промывали насыщенным водным раствором бикарбоната натрия (10 мл) и водную фракцию экстрагировали этилацетатом (2×10 мл). Объединенные органические экстракты промывали насыщенным солевым раствором (20 мл), сушили с помощью MgSO4 и концентрировали в вакууме. Полученный остаток подвергали флеш хроматографии (Si-PPC, градиент 0-100% этилацетата в циклогексане) с получением указанного в заголовке соединения в виде твердого продукта не совсем белого цвета (232 мг, 80%). LCMS (Способ B): RT=3,22 мин, [M+H]+=350/352.
4-(2-Фтор-4-йодфениламино)-1H-индазол-5-карбоновая кислота
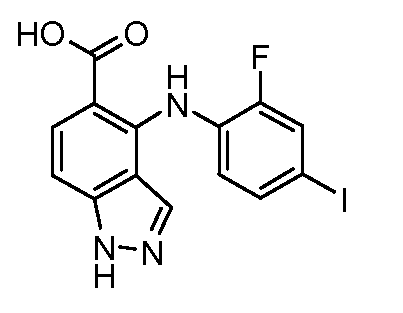
Стадия 1: ди-трет-бутиловый эфир 4-(2-фтор-4-йодфениламино)-индазол-1,5-дикарбоновой кислоты
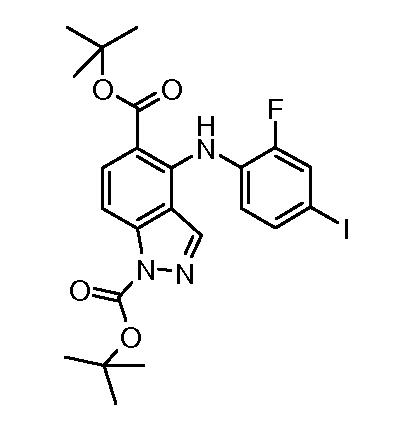
К раствору ди-трет-бутилового эфира 4-(2-фтор-4-триметилсиланилфениламино)-индазол-1,5-дикарбоновой кислоты (1,07 г, 2,14 ммоль) в DCM (10 мл) при температуре 0ºC добавляли монохлорид йода в виде раствор в DCM (4,2 мл, 1н, 4,2 ммоль). Реакционную смесь перемешивали при температуре 0ºC в течение 20 минут, затем разбавляли насыщенным водным раствора тиосульфата натрия (10 мл) и экстрагировали DCM (2×10 мл). Объединенные органические экстракты промывали насыщенным солевым раствором (20 мл), сушили (MgSO4) и концентрировали в вакууме. Полученный остаток подвергали флеш хроматографии (Si-PPC, градиент 0-100% DCM в циклогексане) с получением указанного в заголовке соединения в виде твердого вещества бледно-коричневого цвета (611 мг, 52%). LCMS (Способ B): RT=5,48 мин, [M+H]+=554.
Стадия 2: 4-(2-фтор-4-йодфениламино)-1H-индазол-5-карбоновая кислота
К раствору ди-трет-бутилового эфира 4-(2-фтор-4-йодфениламино)-индазол-1,5-дикарбоновой кислоты (611 мг, 1,10 ммоль) в DCM (5 мл) добавляли ТФУ (1,2 мл, 16,2 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов, после чего концентрировали в вакууме. Полученный остаток растворяли в этилацетате (10 мл), промывали насыщенным водным раствором бикарбоната натрия (10 мл) и водную фракцию экстрагировали этилацетатом (2×10 мл). Объединенные органические фракции промывали насыщенным солевым раствором (20 мл), сушили (MgSO4) и концентрировали в вакууме. Полученный остаток подвергали флеш хроматографии (Si-PPC, градиент 0-100% этилацетата в циклогексане) с получением указанного в заголовке соединения в виде твердого продукта не совсем белого цвета (367 мг, 84%). LCMS (Способ B): RT=3,23 мин, [M+H]+=398.
4-(2-Фтор-4-метилсульфанил-фениламино)-1H-индазол-5-карбоновая кислота
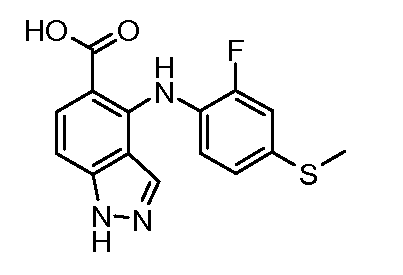
Стадия 1: 2,4-дифтор-3-формил-бензойная кислота
Раствор TMEDA (10,5 мл, 70 ммоль) в ТГФ (50 мл) охлаждали до температуры <-80ºC и обрабатывали втор-бутиллитием (50 мл, 70 ммоль). Добавляли по каплям раствор 2,4-дифторбензойной кислоты (5,0 г, 31,6 ммоль) в ТГФ, поддерживая температуру ниже -80ºC. Реакционной смеси давали нагреться до температуры -75ºC и обрабатывали ДМФ (15 мл), смеси затем давали нагреться до температуры 0ºC, затем гасили реакционную смесь водой. Водный слой отделяли, подкисляли до pH ~2 (концентрированная HCl) и дважды экстрагировали диэтиловым эфиром. Объединенные органические экстракты сушили (Na2SO4), фильтровали и концентрировали в вакууме с получением масла желтого цвета. Сырой продукт кристаллизовали из смеси циклогексан/диэтиловый эфир с получением указанного в заголовке соединения в виде твердого продукта желтого цвета (670 мг, 11%). 1H ЯМР (CDCl3, 400 МГц) 10,40 (1H, с), 8,31 (1H, ддд, J=8,96, 8,02, 6,08 Гц), 7,12 (1H, тд, J=9,09, 1,42 Гц).
Стадия 2: 3-диметоксиметил-2,4-дифтор-бензойная кислота
Смесь 2,4-дифтор-3-формил-бензойной кислоты (670 мг, 3,6 ммоль) и хлорида аммония (1,15 г, 21,6 ммоль) в метаноле (20 мл) нагревали при температуре кипения в течение 18 часов. Реакционную смесь концентрировали в вакууме, остаток частично растворяли в этилацетате и затем фильтровали. Фильтрат концентрировали в вакууме с получением указанного в заголовке соединения в виде твердого продукта желтого цвета (747 мг, 89%-ный выход). 1H ЯМР (ДМСО-d6, 400 МГц) 7,95 (1H, тд, J=8,55, 6,32 Гц), 5,62 (1H, с), 3,38 (6H, с).
Стадия 3: 4-фтор-2-(2-фтор-4-метилсульфанил-фениламино)-3-формил-бензойная кислота
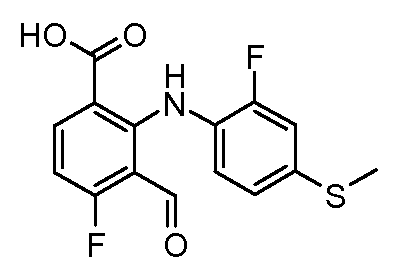
В холодный (-78ºC) раствор 2-фтор-4-метилсульфанил-фенил амина (1,55 г, 9,9 ммоль) в ТГФ (15 мл) добавляли по каплям LHMDS (9,9 мл, 1,0 M раствор в гексане 9,9 ммоль) таким образом, чтобы поддерживать температуру ниже -65ºC. После перемешивания в течение 30 минут добавляли по каплям раствор 3-диметоксиметил-2,4-дифтор-бензойной кислоты (700 мг, 3,0 ммоль) в ТГФ (15 мл), полученную смесь перемешивали при охлаждении в течение 3 часов, затем давали нагреться до комнатной температуры и перемешивали в течение 18 часов. Реакционную смесь гасили путем добавления водного раствора хлорида аммония и продукты распределяли между этилацетатом и водой. Водный слой отделяли и подкисляли до pH 1 (концентрированная HCl) и затем дважды экстрагировали этилацетатом. Объединенные органические экстракты сушили (Na2SO4), фильтровали и концентрировали в вакууме. Полученный остаток растирали диэтиловым эфиром с получением указанного в заголовке соединения в виде твердого продукта желтого цвета (105 мг, 10 %-ный выход). 1H ЯМР (CDCl3, 400 МГц) 10,26-10,23 (2H, м), 8,04 (1H, дд, J=8,76, 6,31 Гц), 7,03-6,96 (2H, м), 6,91 (1H, д, J=1,96 Гц), 6,65 (1H, дд, J=10,02, 8,77 Гц), 2,42 (3H, с).
Стадия 4: 4-(2-фтор-4-метилсульфанил-фениламино)-1H-индазол-5-карбоновая кислота
К суспензии 4-фтор-2-(2-фтор-4-метилсульфанил-фениламино)-3-формил-бензойной кислоты (105 мг, 0,32 ммоль) в DME (5 мл) добавляли гидразин гидрат (5 мл) и реакционную смесь нагревали при температуре 90ºC в течение 18 часов. Легколетучий растворитель удаляли в вакууме и остаток подкисляли концентрированной HCl при охлаждении. Полученный осадок собирали путем фильтрации и промывали водой с получением указанного в заголовке соединения в виде твердого вещества рыжеватого цвета (85 мг, 82%). LCMS (Способ B) RT 3,12 [M+H]+ 318.
4-(2-Фтор-4-йод-фениламино)-бензо[d]изотиазол-5-карбоновая кислота
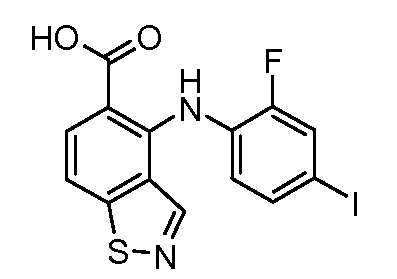
Стадия 1: трет-бутиловый эфир 4-(2-фтор-4-триметилсиланил-фениламино)бензо[d]изотиазол-5-карбоновой кислоты
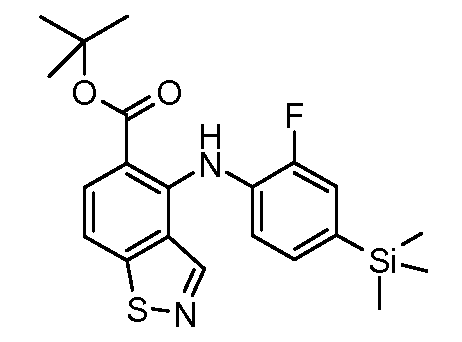
Раствор 2-фтор-4-триметилсиланил-фениламина (340 мг, 1,43 ммоль), трет-бутилового эфира 4-бром-бензо[d]изотиазол-5-карбоновой кислоты (449 мг, 1,43 ммоль), Pd2dba3 (65 мг, 0,07 ммоль), Xantphos (83 мг, 0,14 ммоль) и трехосновного фосфата калия (455 мг, 2,14 ммоль) в толуоле (5 мл) дегазировали и затем реакционную смесь нагревали при температуре 90ºC в течение 18 часов. Охлажденную реакционную смесь разбавляли этилацетатом, фильтровали через Celite®, и фильтрат концентрировали в вакууме. Полученный остаток подвергали флеш хроматографии (Si-PPC, градиент 2,5-5% диэтилового эфира в пентане) с получением указанного в заголовке соединения в виде темного масла (465 мг, 78%). LCMS (Способ B): RT=5,76 мин, [M+H]+=417.
Стадия 2: трет-бутиловый эфир 4-(2-фтор-4-йод-фениламино)-бензо[d]изотиазол-5-карбоновой кислоты
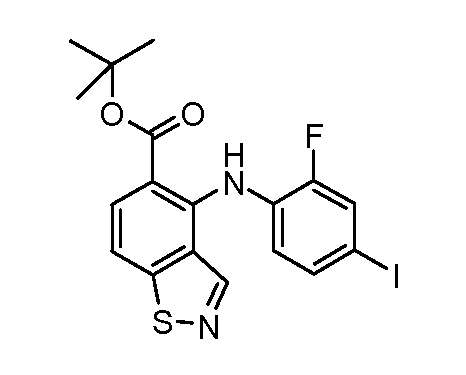
К раствору трет-бутилового эфира 4-(2-фтор-4-триметилсиланил-фениламино)-бензо[d]изотиазол-5-карбоновой кислоты (416 мг, 1,0 ммоль) в DCM (10 мл) при температуре -78°C добавляли монохлорид йода в виде раствора в DCM (2,0 мл, 1M, 2,0 ммоль). Реакционную смесь перемешивали при температуре -78°C в течение 1 часа, затем разбавляли насыщенным водным раствором тиосульфата натрия и экстрагировали DCM. Объединенные органические экстракты промывали насыщенным солевым раствором, сушили (Na2SO4) и концентрировали в вакууме с получением указанного в заголовке соединения в виде пены желтого цвета (459 мг, 98%). LCMS (Способ B): RT=5,32 мин, [M+H]+=471.
Стадия 3: 4-(2-фтор-4-йод-фениламино)-бензо[d]изотиазол-5-карбоновая кислота
К раствору трет-бутилового эфира 4-(2-фтор-4-йод-фениламино)-бензо[d]изотиазол-5-карбоновой кислоты (445 мг, 0,95 ммоль) в DCM (20 мл) добавляли воду (0,1 мл), смесь охлаждали до температуры 0ºC и добавляли ТФУ (20 мл). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, затем концентрировали в вакууме и остаток подвергали азеотропной отгонке с толуолом (×2) с получением указанного в заголовке соединения в виде твердого вещества бежевого цвета (392 мг, 100%). LCMS (Способ B): RT=3,97 мин, [M+H]+=415.
7-(2-Фтор-4-йод-фениламино)-бензо[d]изотиазол-6-карбоновая кислота
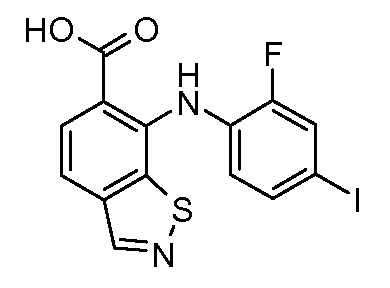
Стадия 1: трет-бутиловый эфир 7-(2-фтор-4-йод-фениламино)-бензо[d]изотиазол-6-карбоновой кислоты
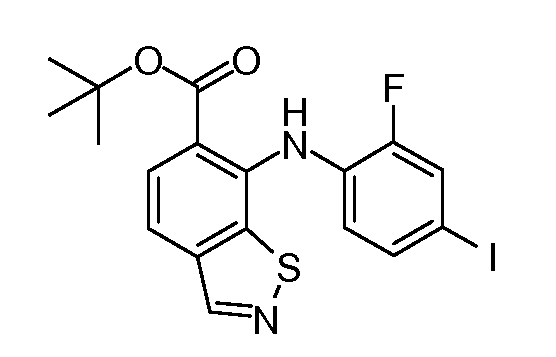
К раствору трет-бутилового эфира 7-фтор-бензо[d]изотиазол-6-карбоновой кислоты (1,26 г, 5,0 ммоль) и 2-фтор-4-йод-фениламина (1,18 г, 5,0 ммоль) в безводном ТГФ (25 мл) при температуре -78°C добавляли 1,0M раствор LHMDS в гексане (10,0 мл, 10,0 ммоль) в атмосфере азота. Реакционной смеси давали нагреться до комнатной температуры и затем перемешивали в течение 30 минут, после чего гасили путем добавления насыщенного водного раствора хлорида аммония и экстрагировали этилацетатом. Органический слой отделяли и промывали водой, затем насыщенным солевым раствором, сушили (Na2SO4), фильтровали и упаривали в вакууме. Остаток подвергали флеш хроматографии (Si-PPC, градиент от 0% до 10%, TBME в циклогексане) с получением указанного в заголовке соединения в виде твердого продукта желтого цвета (717 мг, 30%). LCMS (способ B): RT=5,14 мин, [M+H]+=471.
Стадия 2: 7-(2-фтор-4-йод-фениламино)-бензо[d]изотиазол-6-карбоновая кислота
К раствору трет-бутилового эфира 7-(2-фтор-4-йод-фениламино)-бензо[d]изотиазол-6-карбоновой кислоты (710 мг, 1,51 ммоль) в DCM (5 мл) добавляли воду (0,15 мл) и ТФУ (5 мл). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа, после чего концентрировали в вакууме. Полученный остаток подвергали азеотропной отгонке с толуолом с получением указанного в заголовке соединения в виде твердого продукта желтого цвета (571 мг, 91%). LCMS (способ B): RT=3,95 мин, [M+H]+=415.
7-(4-Бром-2-фтор-фениламино)-бензо[d]изотиазол-6-карбоновой кислоты
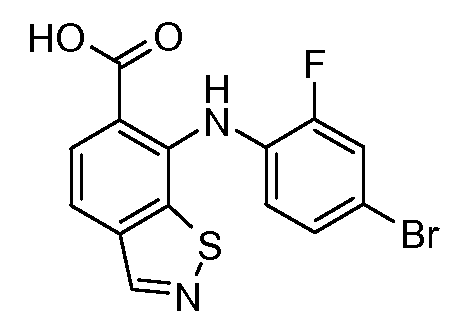
К раствору 4-бром-2-фтор-фениламина (1,05 г, 5,55 ммоль) в безводном ТГФ (20 мл) при температуре -78°C добавляли 1,0 M раствор LHMDS в гексане (8,3 мл, 8,3 ммоль) в атмосфере азота. Реакционную смесь перемешивали в течение 10 минут и добавляли по каплям суспензию 7-фтор-бензо[d]изотиазол-6-карбоновой кислоты (547 мг, 2,78 ммоль) в безводном ТГФ (20 мл). Полученную смесь перемешивали при температуре -78°C в течение 30 мин, затем температуре давали подняться до комнатной и перемешивали в течение 18 часов, после чего гасили добавлением 1M водной HCl (прибл. 50 мл) и экстрагировали этилацетатом. Органический слой отделяли и промывали водой, затем насыщенным солевым раствором, сушили (Na2SO4), фильтровали и упаривали в вакууме. Полученный остаток подвергали флеш хроматографии (Si-PPC, градиент 0%-10% MeOH в DCM) с получением указанного в заголовке соединения в виде твердого вещества коричневого цвета (584 мг, 58%). LCMS (способ B): RT=3,76 мин, [M-H]+=365/367.
7-(4-Циклопропил-2-фтор-фениламино)-бензо[d]изотиазол-6-карбоновая кислота
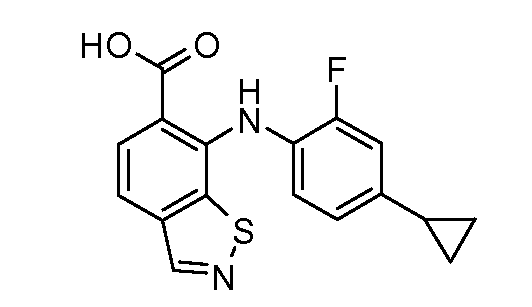
К раствору 4-циклопропил-2-фтор-фениламина (445 мг, 2,94 ммоль) в безводном ТГФ (10 мл) при температуре -78°C добавляли 1,0 M раствор LHMDS в гексане (4,4 мл, 4,4 ммоль) в атмосфере азота. Реакционную смесь перемешивали в течение 10 минут и добавляли по каплям суспензию 7-фтор-бензо[d]изотиазол-6-карбоновой кислоты (290 мг, 1,47 ммоль) в безводном ТГФ (10 мл). Полученную смесь перемешивали при температуре -78°C в течение 30 минут, затем температуре давали подняться до комнатной и перемешивали в течение 18 часов, после чего гасили добавлением 1M водной HCl и экстрагировали этилацетатом. Органический слой отделяли и промывали водой, затем насыщенным солевым раствором, сушили (Na2SO4), фильтровали и упаривали в вакууме. Полученный остаток растирали в диэтиловом эфире с получением указанного в заголовке соединения в виде твердого вещества коричневого цвета (155 мг, 32%). LCMS (способ B): RT=3,79 мин, [M+H]+=329.
7-(2-Фтор-4-метилсульфанил-фениламино)-бензо[d]изотиазол-6-карбоновая кислота
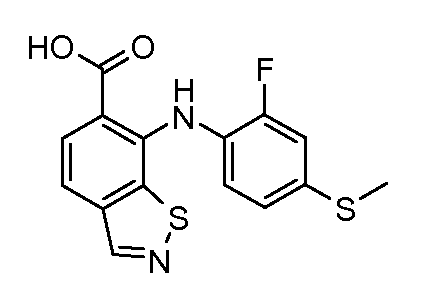
К раствору 2-фтор-4-метилсульфанил-фениламина (628 мг, 4,0 ммоль) в безводном ТГФ (15 мл) при температуре -78°C добавляли 1,0M раствор LHMDS в гексане (6,0 мл, 6,0 ммоль) в атмосфере азота. Реакционную смесь перемешивали в течение 5 минут и добавляли по каплям суспензию 7-фтор-бензо[d]изотиазол-6-карбоновой кислоты (394 мг, 2,0 ммоль) в безводном ТГФ (15 мл). Полученную смесь перемешивали при температуре -78°C в течение 30 минут, затем температуре давали подняться до комнатной и перемешивали в течение 18 часов, после чего гасили добавлением 1M водной HCl (прибл. 50 мл) и экстрагировали этилацетатом. Органический слой отделяли и промывали водой, затем насыщенным солевым раствором, сушили (Na2SO4), фильтровали и упаривали в вакууме. Полученный остаток подвергали флеш хроматографии (Si-PPC, градиент от 0% до 10%, MeOH в DCM) с получением указанного в заголовке соединения в виде твердого вещества коричневого цвета (200 мг, 30%). LCMS (способ B): RT=3,67 мин, [M+H]+=335
СИНТЕЗ 5-АМИНО-4-АНИЛИНО ИНДАЗОЛ ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ
Трет-бутиловый эфир 5-амино-4-(2-фтор-4-йод-фениламино)-индазол-1-карбоновой кислоты
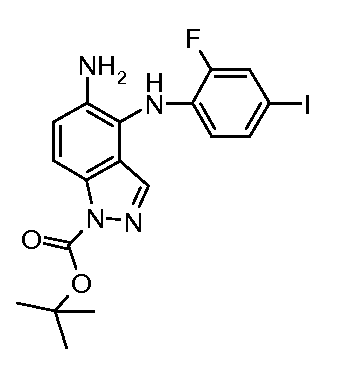
Стадия 1: 2,6-дифтор-3-нитро-бензальдегид

К перемешиваемой 69%-ной азотной кислоте (6,48 мл, 93,59 ммоль), охлажденной до температуры 5°C, добавляли по каплям концентрированную серную кислоту (4,16 мл, 78,11 ммоль), поддерживая внутреннюю температуру ниже 8°C. После завершения добавления перемешивание продолжали в течение 15 минут. Эту смесь добавляли по каплям к раствору 2,6-дифторбензальдегида (10 г, 70,37 ммоль) в концентрированной серной кислоте (50 мл) поддерживая внутреннюю температуру ниже 10ºC. После завершения добавления смесь перемешивали при температуре 5ºC в течение 15 минут, затем давали нагреться до комнатной температуры в течение 90 минут. Реакционную смесь выливали на смесь лед-вода (400 мл) и образовавшийся твердый продукт перемешивали в течение 1 часа. Твердый продукт собирали путем фильтрации, промывали водой и сушили в вакууме с получением указанного в заголовке соединения (9,27 г, 70%). 1H ЯМР (ДМСО-d6, 300 МГц): 10,21 (1H, т, J=1,02 Гц), 8,52 (1H, ддд, J=9,38, 8,60, 5,51 Гц), 7,51 (1H, тд, J=9,53, 1,76 Гц).
Стадия 2: 2-диметоксиметил-1,3-дифтор-4-нитробензол
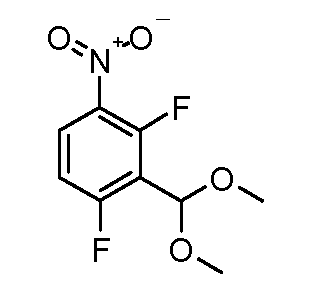
Раствор 2,6-дифтор-3-нитро-бензальдегида (9,0 г, 48,1 ммоль), моногидрата п-толуолсульфоновой кислоты (183 мг, 0,962 ммоль), триметилортоформиата (3,5 мл, 32,4 ммоль) и метанола (200 мл) нагревали при температуре кипения в течение 4 часов. Реакционную смесь оставляли охлаждаться до комнатной температуры и затем концентрировали в вакууме. Полученный остаток растворяли в DCM (250 мл) и промывали смесью воды (200 мл) и насыщенного раствора NaHCO3 (50 мл). Органический слой сушили (Na2SO4) и концентрировали в вакууме с получением указанного в заголовке соединения (10,83 г, 97%). 1H ЯМР (ДМСО-d6, 300 МГц): 8,28 (1H, ддд, J=9,34, 8,48, 5,51 Гц), 7,39 (1H, тд, J=9,34, 1,81 Гц), 5,66 (1H, с), 3,40 (6H, с).
Стадия 3: (2-диметоксиметил-3-фтор-6-нитрофенил)-(2-фтор-4-йод-фенил)-амин
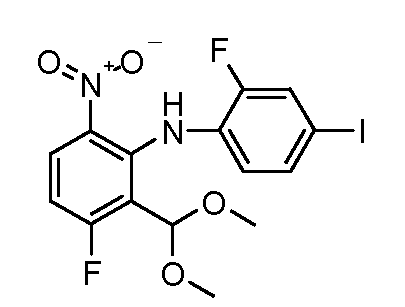
К раствору 2-фтор-4-йод-анилина (2,24 г, 9,43 ммоль) в безводном ТГФ (30 мл) в атмосфере азота добавляли 1M LHMDS в гексане (18 мл, 1M, 18,0 ммоль), поддерживая внутреннюю температуру ниже -65°C. После завершения добавления реакционную смесь перемешивали в течение 30 минут при температуре -70ºC. В реакционную смесь добавляли раствор 2-диметоксиметил-1,3-дифтор-4-нитробензола (2,0 г, 8,58 ммоль) в безводном ТГФ (20 мл), поддерживая внутреннюю температуру ниже -68ºC. После завершения добавления реакционную смесь перемешивали в течение 30 минут при температуре -70ºC, затем давали нагреться до комнатной температуры и перемешивали в течение 22 часов. Реакционную смесь гасили с помощью насыщенного водного раствора NH4Cl и экстрагировали этилацетатом (200 мл). Органический слой промывали насыщенным солевым раствором, сушили (Na2SO4), концентрировали в вакууме до минимального объема и растирали в циклогексане с получением указанного в заголовке соединения (2,18 г, 56%). LCMS (Способ B): RT=4,26 мин, [M-H]-=449.
Стадия 4: 6-фтор-2-(2-фтор-4-йод-фениламино)-3-нитро-бензальдегид
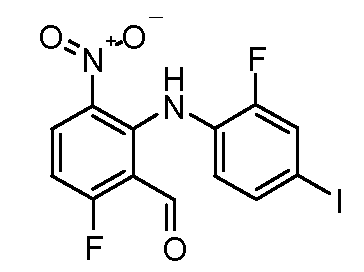
К перемешиваемому раствору (2-диметоксиметил-3-фтор-6-нитрофенил)-(2-фтор-4-йод-фенил)-амина (1,0 г, 2,22 ммоль) в ТГФ (10 мл) при комнатной температуре добавляли раствор 4M HCl. Спустя 2 часа ТГФ удаляли в вакууме и остаток разбавляли водой (20 мл), что приводило к образованию осадка. Твердый осадок собирали путем фильтрации, промывали водой и сушили в вакууме с получением указанного в заголовке соединения (864 мг, 96%). LCMS (Способ B): RT=4,06 мин, [M-H]-=403.
Стадия 5: (2-фтор-4-йод-фенил)-(5-нитро-1H-индазол-4-ил)-амин
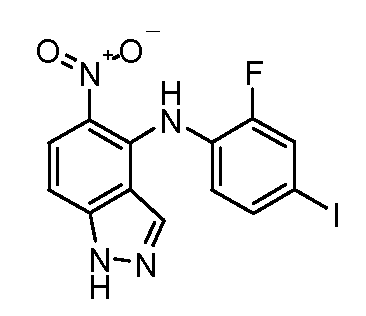
К перемешиваемому раствору 6-фтор-2-(2-фтор-4-йод-фениламино)-3-нитро-бензальдегида (864 мг, 2,14 ммоль) в DME (15 мл) при комнатной температуре медленно добавляли гидразин гидрат (10 мл) и реакционную смесь перемешивали в течение 18 часов. Органический растворитель удаляли в вакууме и смесь разбавляли водой (50 мл). Образовавшийся твердый продукт собирали путем фильтрации, промывали водой и сушили в вакууме с получением указанного в заголовке соединения (839 мг, 98%). LCMS (Способ B): RT=3,61 мин, [M+H]+=399.
Стадия 6: трет-бутиловый эфир 4-(2-фтор-4-йод-фениламино)-5-нитро-индазол-1-карбоновой кислоты
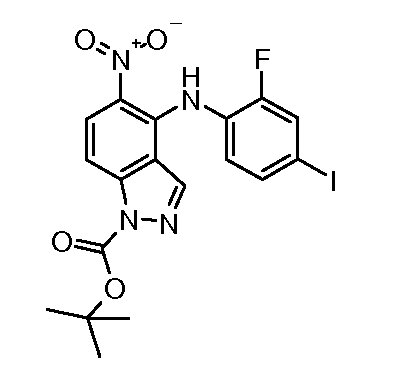
К суспензии (2-фтор-4-йод-фенил)-(5-нитро-1H-индазол-4-ил)-амина (839 мг, 2,11 ммоль) и триэтиламина (0,323 мл, 2,32 ммоль) в дихлорметане добавляли ди-трет-бутил дикарбонат (552 мг, 2,53 ммоль) и ДМФ (1 мл), полученный раствор перемешивали при комнатной температуре в течение 2 часов. Добавляли еще ди-трет-бутил дикарбонат (275 мг, 1,26 ммоль) и DMAP (25 мг, 10 моль%) и смесь перемешивали в течение 10 минут. Реакционную смесь разбавляли этилацетатом (150 мл) и промывали насыщенным водным NaHCO3 (100 мл), затем водой (100 мл) и затем насыщенным солевым раствором (100 мл), далее сушили (Na2SO4) и концентрировали в вакууме. Полученный остаток подвергали флеш хроматографии (Si-PPC градиент 0-35% этилацетат в циклогексане) с получением указанного в заголовке соединения (739 мг, 70%). LCMS (Способ B): RT=4,57 мин, [M-Boc+H]+=399.
Стадия 7: трет-бутиловый эфир 5-амино-4-(2-фтор-4-йод-фениламино)-индазол-1-карбоновой кислоты
Суспензию дитионита натрия (978 мг, 4,21 ммоль) и трет-бутилового эфира 4-(2-фтор-4-йод-фениламино)-5-нитро-индазол-1-карбоновой кислоты (700 мг, 1,4 ммоль) в воде (35 мл) обрабатывали смесью 1:1 ТГФ:диоксан (34 мл), гомогенную реакционную смесь перемешивали при комнатной температуре в течение 4 часов. Добавляли еще дитионит натрия (978 мг, 4,21 ммоль) и перемешивание продолжали в течение 20 часов. Реакционную смесь подщелачивали путем добавления насыщенного водного раствора гидрокарбоната натрия и затем экстрагировали этилацетатом (150 мл). Органический экстракт промывали водой, сушили (Na2SO4), фильтровали и концентрировали в вакууме с получением указанного в заголовке соединения в виде твердого продукта желтого цвета (758 мг, колич.). LCMS (Способ B) RT=4,09 мин, [M+H]+=469.
Трет-бутиловый эфир 5-амино-4-(2-фтор-4-йод-фениламино)-6-метокси-индазол-1-карбоновой кислоты
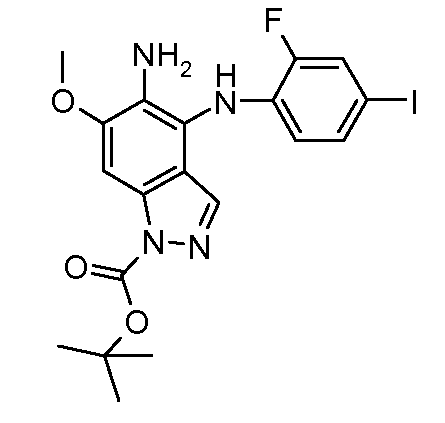
Стадия 1: 2,6-дифтор-4-метокси-3-нитро-бензальдегид
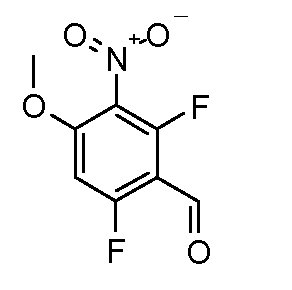
К раствору холодной (-5ºC) азотной кислоты (2,7 мл) по каплям добавляли концентрированную серную кислоту (1,75 мл, 32 ммоль), поддерживая температуру ниже 5ºC. Раствор добавляли к охлажденному раствору 2,6-дифтор-4-метокси бензальдегида (5,0 г, 29 ммоль) в серной кислоте (20 мл) в течение 15 минут, поддерживая температуру ниже 5ºC. После перемешивания при температуре 0ºC в течение 2 часов оранжевый раствор выливали на лед, образовавшийся осадок белого цвета собирали путем фильтрации с получением указанного в заголовке соединения в виде твердого вещества белого цвета (6,33 г, 100%). 1H ЯМР (CDCl3) 10,20 (1H, т, J=1,17 Гц), 6,69 (1H, дд, J=11,68, 1,93 Гц), 4,03 (3H, с).
Стадия 2: 2-диметоксиметил-1,3-дифтор-5-метокси-4-нитробензол
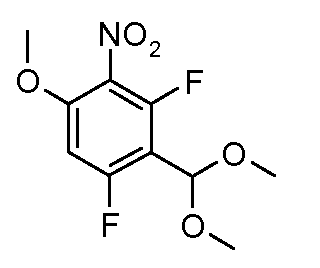
Смесь 2,6-дифтор-4-метокси-3-нитро-бензальдегида (6,3 г, 29 ммоль) в метаноле (30 мл) с п-толуолсульфоновой кислотой (110 мг, 0,58 ммоль) нагревали при температуре кипения в течение 18 часов. Реакционную смесь концентрировали в вакууме и остаток распределяли между насыщенным водным раствором гидрокарбоната натрия и этилацетатом. Водный слой экстрагировали этилацетатом и объединенные органические экстракты сушили (Na2SO4), фильтровали и концентрировали в вакууме с получением указанного в заголовке соединения в виде твердого продукта бледно-желтого цвета (6,52 г, 85%). 1H ЯМР (CDCl3) 6,59 (1H, дд, J=11,48, 2,08 Гц), 5,52 (1H, с), 3,93 (3H, с), 3,45 (6H, с).
Стадия 3: (2-диметоксиметил-3-фтор-5-метокси-6-нитрофенил)-(2-фтор-4-йод-фенил)-амин
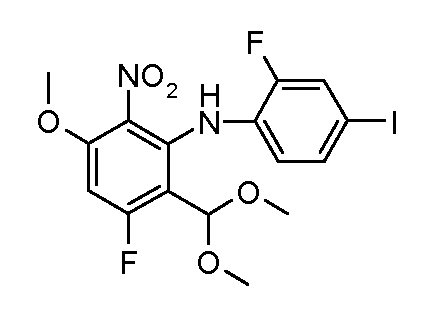
В холодный (-780C) раствор 2-фтор-4-йод-фениламина (2,97 г, 12,5 ммоль) в ТГФ (20 мл) добавляли по каплям LHMDS (24 мл, 1,0 M раствор в гексане 24 ммоль), поддерживая температуру ниже -65ºC. После перемешивания в течение 30 минут добавляли по каплям раствор 2-диметоксиметил-1,3-дифтор-5-метокси-4-нитробензола (3,0 г, 11,4 ммоль) в ТГФ (20 мл), полученную смесь перемешивали холодной (-78ºC) в течение 1 часа, затем давали нагреться до комнатной температуры и перемешивали в течение 18 часов. Реакционную смесь гасили путем добавления водного раствора хлорида аммония и дважды экстрагировали диэтиловым эфиром. Объединенные органические экстракты промывали водой, сушили (Na2SO4), фильтровали и концентрировали в вакууме с получением твердого продукта оранжевого цвета. Сырой продукт оранжевого цвета растирали в диэтиловом эфире с получением указанного в заголовке соединения в виде твердого продукта желтого цвета (4,2 г, 77%). 1H ЯМР (CDCl3) 7,36 (1H, дд, J=10,09, 1,93 Гц), 7,28-7,17 (2H, м), 6,59 (1H, т, J=8,62 Гц), 6,44 (1H, д, J=11,47 Гц), 5,49 (1H, с), 3,88 (3H, с), 3,39 (6H, с).
Стадия 4: 6-фтор-2-(2-фтор-4-йод-фениламино)-4-метокси-3-нитро-бензальдегид
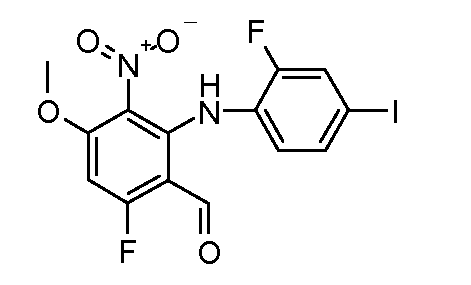
Смесь (2-диметоксиметил-3-фтор-5-метокси-6-нитрофенил)-(2-фтор-4-йод-фенил)-амина (4,2 г, 8,7 ммоль) в диэтиловом эфире (70 мл) и 4M HCl (50 мл) перемешивали при комнатной температуре в течение 8 часов. Смесь экстрагировали этилацетатом, органический экстракт промывали водой, сушили (Na2SO4), фильтровали и концентрировали в вакууме с получением указанного в заголовке соединения в виде твердого продукта желтого цвета (3,1 г, 82%). LCMS (Способ B) RT=4,00, нет молекулярного иона.
Стадия 5: (2-фтор-4-йод-фенил)-(6-метокси-5-нитро-1H-индазол-4-ил)-амин
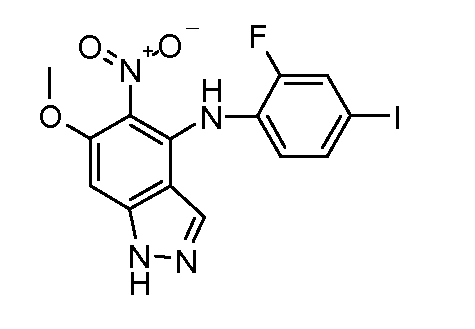
Бифазную смесь 6-фтор-2-(2-фтор-4-йод-фениламино)-4-метокси-3-нитро-бензальдегида (1,5 г, 3,46 ммоль) в гидразингидрате (10 мл) и DME (10 мл) перемешивали при комнатной температуре в течение 4 часов, затем нагревали при температуре 50ºC в течение 3 часов. Реакционную смесь концентрировали в вакууме, остаток обрабатывали водой и полученный твердый осадок собирали путем фильтрации с получением твердого продукта красного/оранжевого цвета. Твердый продукт перекристаллизовывали из IMS с получением указанного в заголовке соединения в виде твердого продукта желтого цвета (956 мг, 64%). LCMS (Способ B) RT=3,62 мин, [M+H]+=429 [M-H]-=427.
Стадия 6: трет-бутиловый эфир 4-(2-фтор-4-йод-фениламино)-6-метокси-5-нитро-индазол-1-карбоновой кислоты
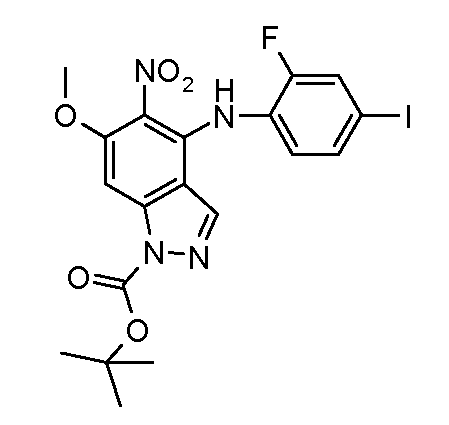
Суспензию (2-фтор-4-йод-фенил)-(6-метокси-5-нитро-1H-индазол-4-ил)-амина (900 мг, 2,1 ммоль) в DCM (10 мл) обрабатывали ди-трет-бутилдикарбонатом (550 мг, 2,5 ммоль) и триэтиламином (0,321 мл, 4,6 ммоль) и ДМФ (2 мл). Реакционную смесь перемешивали в течение 5 часов при комнатной температуре, после чего концентрировали в вакууме и остаток распределяли между этилацетатом и насыщенным водным раствором гидрокарбоната натрия. Органический слой отделяли, промывали водой, сушили (Na2SO4), фильтровали и концентрировали в вакууме. Полученный остаток подвергали флеш хроматографии (Si-PPC градиент 0-15% этилацетата в циклогексане) с получением указанного в заголовке соединения в виде твердого продукта желтого цвета (534 мг, 48%). 1H ЯМР (CDCl3) 7,84 (1H, с), 7,52 (1H, дд, J=9,63, 1,90 Гц), 7,47 (1H, с), 7,43-7,38 (1H, м), 7,29 (1H, д, J=0,78 Гц), 6,91 (1H, т, J=8,40 Гц), 4,02 (3H, с), 1,70 (9H, с).
Стадия 7: трет-бутиловый эфир 5-амино-4-(2-фтор-4-йод-фениламино)-6-метокси-индазол-1-карбоновой кислоты
Суспензию дитионита натрия (524 мг, 3,48 ммоль) и трет-бутилового эфира 4-(2-фтор-4-йод-фениламино)-6-метокси-5-нитро-индазол-1-карбоновой кислоты (434 мг, 0,87 ммоль) в воде (10 мл) обрабатывали смесью 1:1 ТГФ:диоксан (10 мл). Гомогенную реакционную смесь перемешивали при комнатной температуре в течение 18 часов. Реакционную смесь подщелачивали путем добавления насыщенного водного раствора гидрокарбоната натрия и затем дважды экстрагировали этилацетатом. Объединенные органические экстракты промывали водой, сушили (Na2SO4), фильтровали и концентрировали в вакууме с получением указанного в заголовке соединения в виде твердого продукта желтого цвета (300 мг, 73 %). LCMS (Способ B) RT 4,16 мин, [M+H]+=499 [M-H]-=497.
ПРИМЕР 5: (2-гидроксиэтокси)-амид 4-(2-фтор-4-йод-фениламино)-1H-индазол-5-карбоновой кислоты
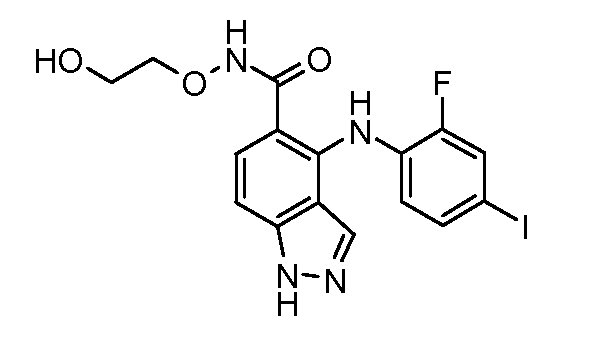
Способ A. Стадия 1: (2-винилокси-этокси)-амид 4-(2-фтор-4-йодфениламино)-1-(толуол-4-сульфонил)-1H-индазол-5-карбоновой кислоты
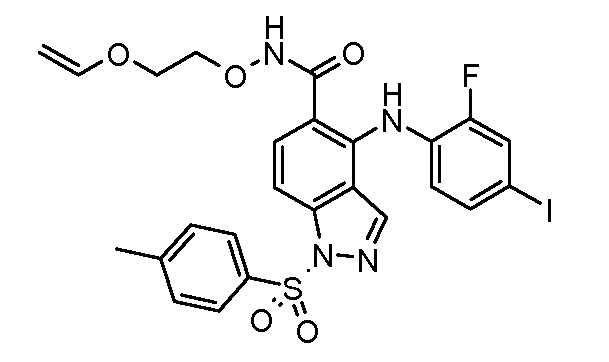
К раствору 4-(2-фтор-4-йод-фениламино)-1-(толуол-4-сульфонил)-1H-индазол-5-карбоновой кислоты (0,58 г, 1,05 ммоль) в ДМФ (10 мл) добавляли EDCI (0,22 г, 1,1 ммоль), затем HOBt (0,16 г, 1,1 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 10 минут. Добавляли O-(2-винилокси-этил)-гидроксиламин (0,12 г, 1,1 ммоль) и DIPEA (0,2 мл, 1,1 ммоль) и реакционную смесь перемешивали в течение 16 часов, после чего концентрировали в вакууме. Полученный остаток растворяли в этилацетате (10 мл), промывали насыщенным водным раствором бикарбоната натрия (10 мл), далее водную фракцию дважды экстрагировали этилацетатом (2×10 мл). Объединенные органические экстракты промывали насыщенным солевым раствором (20 мл), сушили (MgSO4) и концентрировали в вакууме. Полученный остаток подвергали флеш хроматографии (Si-PPC, градиент 0-100% этилацетата в циклогексане) с получением указанного в заголовке соединения в виде твердого вещества бледно-коричневого цвета (439 мг, 66%). LCMS (Способ B): RT=4,40 мин, [M+H]+=637.
Способ A, Стадия 2: (2-гидроксиэтокси)-амид 4-(2-фтор-4-йод-фениламино)-1H-индазол-5-карбоновой кислоты
К раствору (2-винилокси-этокси)-амида 4-(2-фтор-4-йодфениламино)-1-(толуол-4-сульфонил)-1H-индазол-5-карбоновой кислоты (200 мг, 0,31 ммоль) в метаноле (3 мл) добавляли соляную кислоту (1 мл, 1н) и реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь концентрировали в вакууме и полученный остаток растворяли в ТФУ (2 мл). Реакционную смесь нагревали при температуре 65ºC в течение 3 часов, затем при температуре 50ºC в течение 16 часов, после чего концентрировали в вакууме. Полученный остаток растворяли в этилацетате (10 мл), промывали насыщенным водным раствором бикарбоната натрия (10 мл) и водную фракцию дважды экстрагировали этилацетатом (2×10 мл). Объединенные органические фракции промывали насыщенным солевым раствором (20 мл), сушили (MgSO4) и концентрировали в вакууме. Полученный остаток подвергали обращеннофазовой препаративной ВЭЖХ (градиент 10-95% ацетонитрил/вода+0,1%-ная муравьиная кислота, Phenominex gemini PhC6, 5 микрон, 250×20 мм). Полученный продукт растворяли в этилацетате (5 мл) и промывали насыщенным водным раствором бикарбоната натрия (10 мл). Водную фракцию экстрагировали дважды этилацетатом (2×10 мл) и объединенные органические фракции промывали насыщенным солевым раствором (20 мл), сушили с помощью MgSO4 и концентрировали в вакууме с получением указанного в заголовке соединения в виде твердого вещества белого цвета (14 мг, 10%). LCMS (Способ A): RT=8,31 мин, [M+H]+=457. 1H ЯМР (ДМСО-d6, 400 МГц): 13,19 (1H, с), 11,60 (1H, с), 9,93 (1H, с), 7,66 (1H, дд, J=10,31, 1,93 Гц), 7,46 (1H, д, J=8,70 Гц), 7,42 (1H, д, J=8,56 Гц), 7,23 (1H, с), 7,01 (1H, д, J=8,77 Гц), 6,91 (1H, т, J=8,64 Гц), 4,68 (1H, с), 3,85 (2H, т, J=4,92 Гц), 3,60-3,52 (2H, м).
Способ B, Стадия 1: (2-винилоксиэтокси)-амид 4-(2-фтор-4-йодфениламино)-1H-индазол-5-карбоновой кислоты
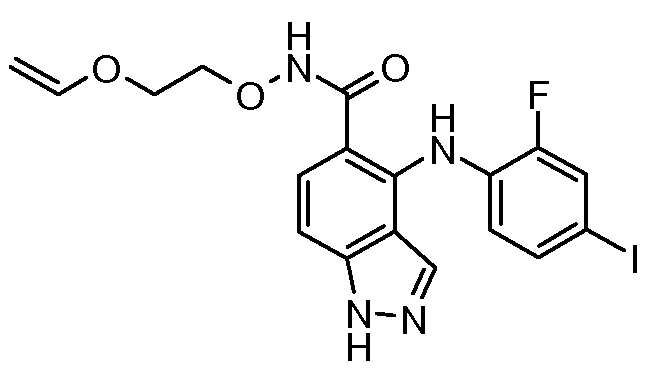
К раствору 4-(2-фтор-4-йодфениламино)-1H-индазол-5-карбоновой кислоты (2,14 г, 5,39 ммоль) и O-(2-винилокси-этил)-гидроксиламина (668 мг, 6,50 ммоль) в ДМФ (50 мл) добавляли EDCI (1,14 г, 5,93 ммоль), HOBt (0,80 г, 5,93 ммоль) и DIPEA (1 мл, 5,93 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, после чего концентрировали в вакууме. Полученный остаток растворяли в этилацетате (30 мл), промывали насыщенным водным раствором гидрокарбоната натрия (300 мл) и водную фракцию экстрагировали этилацетатом (2×20 мл). Объединенные органические экстракты промывали насыщенным солевым раствором (30 мл), сушили (MgSO4) и концентрировали в вакууме. Полученный остаток подвергали флеш хроматографии (SiO2, градиент 0-100% этилацетат в циклогексане) с получением указанного в заголовке соединения в виде твердого продукта бледно-желтого цвета (1,85 г, 71%). LCMS (Способ B): RT=3,52 мин, [M-H]-=481.
Способ B, Стадия 2: (2-гидроксиэтокси)-амид 4-(2-фтор-4-йодфениламино)-1H-индазол-5-карбоновой кислоты
К раствору (2-винилоксиэтокси)-амида 4-(2-фтор-4-йодфениламино)-1H-индазол-5-карбоновой кислоты (1,85 г, 3,84 ммоль) в метаноле (40 мл) добавляли соляную кислоту (3 мл, 1н, 3 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа, в процессе чего образовывался не совсем белый твердый осадок. Реакционную смесь концентрировали в вакууме и остаток растирали в горячей смеси метанол/вода (10 мл, 1:1). Продукт собирали путем фильтрации и сушили в вакууме с получением указанного в заголовке соединения в виде твердого продукта не совсем белого цвета (1,26 г, 72%). LCMS (Способ A): RT=8,28 мин, [M+H]+=457. 1H ЯМР (ДМСО-d6): 13,20 (1H, с), 11,61 (1H, с), 9,93 (1H, с), 7,66 (1H, дд, J=10,32, 1,95 Гц), 7,46 (1H, д, J=8,81 Гц), 7,42 (1H, дд, J=8,49, 1,84 Гц), 7,24 (1H, с), 7,01 (1H, д, J=8,78 Гц), 6,91 (1H, т, J=8,65 Гц), 4,68 (1H, с), 3,85 (2H, дд, J=5,41, 4,48 Гц), 3,56 (2H, т, J=4,85 Гц).
ПРИМЕР 6: (2-гидроксиэтокси)-амид 4-(2-фтор-4-йодфениламино)-1H-пиразоло[3,4-b]пиридин-5-карбоновой кислоты
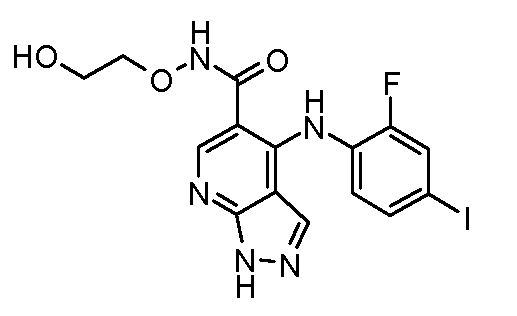
Стадия 1: (2-винилоксиэтокси)-амид 4-(2-фтор-4-йодфениламино)-1-(4-метоксибензил)-1H-пиразоло[3,4-b]пиридин-5-карбоновой кислоты
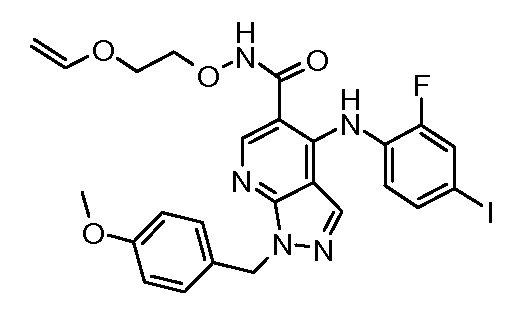
4-(2-фтор-4-йодфениламино)-1-(4-метоксибензил)-1H-пиразоло[3,4-b]пиридин-5-карбоновой кислоты (360 мг, 0,71 ммоль), O-(2-винилоксиэтил)-гидроксиламин (79 мг, 0,77 ммоль), HOBt (103 мг, 0,77 ммоль), EDCI (147 мг, 0,77 ммоль) и DIPEA (130 мкл, 0,77 ммоль) растворяли в ДМФ (10 мл). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов, затем концентрировали в вакууме. Полученный остаток растворяли в этилацетате (10 мл), промывали насыщенным водным раствором бикарбоната натрия (10 мл) и водную фракцию экстрагировали дважды этилацетатом (2×10 мл). Объединенные органические фракции промывали насыщенным солевым раствором (20 мл), сушили (MgSO4) и концентрировали в вакууме. Полученный остаток подвергали флеш хроматографии (Si-SPE, градиент 0-100% этилацетат в циклогексане) с получением указанного в заголовке соединения в виде твердого продукта желтого цвета (346 мг, 81%). LCMS (Способ B): RT=4,08 мин, [M+H]+=604.
Стадия 2: (2-гидроксиэтокси)-амид 4-(2-фтор-4-йодфениламино)-1H-пиразоло[3,4-b]пиридин 5-карбоновой кислоты
(2-Винилоксиэтокси)-амид 4-(2-фтор-4-йодфениламино)-1-(4-метоксибензил)-1H-пиразоло[3,4-b]пиридин-5-карбоновой кислоты (346 мг, 0,57 ммоль) растворяли в ТФУ (5 мл) и реакционную смесь нагревали при температуре 65°C в течение 3 часов. Реакционную смесь концентрировали в вакууме и остаток растворяли в этилацетате (10 мл), промывали насыщенным водным раствором бикарбоната натрия (10 мл) и водную фракцию экстрагировали дважды этилацетатом (2×10 мл). Объединенные органические экстракты промывали насыщенным солевым раствором (20 мл), сушили (MgSO4) и концентрировали в вакууме. Полученный остаток подвергали флеш хроматографии (Si-SPE, градиент 0-10% метанол в DCM) с получением указанного в заголовке соединения в виде твердого продукта желтого цвета (75 мг, 30%). LCMS (Способ A): RT=6,67 мин, [M+H]+=458. 1H ЯМР (ДМСО-d6, 400 МГц) 13,51 (1H, с), 11,75 (1H, с), 10,38 (1H, с), 8,45 (1H, с), 7,80 (1H, дд, J=9,67, 1,86 Гц), 7,64-7,60 (1H, м), 7,30-7,21 (1H, м), 6,74 (1H, с), 4,71 (1H, с), 3,90 (2H, т, J=4,95 Гц), 3,61-3,56 (3H, м).
ПРИМЕР 7: ((S)-2-гидроксипропокси)-амид 4-(2-фтор-4-йодфениламино)-1H-пиразоло[3,4-b]пиридин-5-карбоновой кислоты
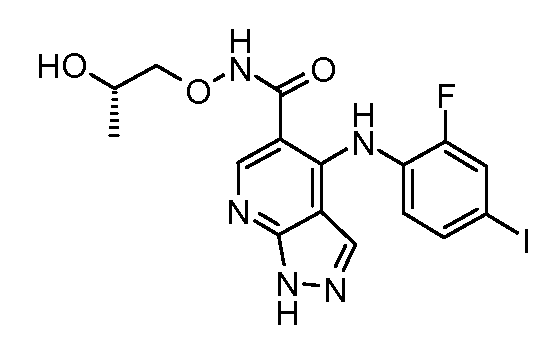
Стадия 1: ((S)-2-гидроксипропокси)-амид 4-(2-фтор-4-йодфениламино)-1-(4-метоксибензил)-1H-пиразоло[3,4-b]пиридин-5-карбоновой кислоты
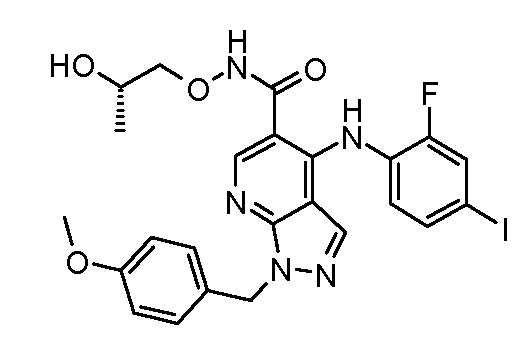
К раствору 4-(2-фтор-4-йодфениламино)-1-(4-метоксибензил)-1H-пиразоло[3,4-b]пиридин-5-карбоновой кислоты (728 мг, 1,4 ммоль), HOBt (228 мг, 1,7 ммоль) и EDCI (323 мг, 1,7 ммоль) в ДМФ (15 мл) добавляли раствор гидрохлорид (S)-1-аминооксипропан-2-ола (200 мг, 1,5 ммоль) и DIPEA (596 мкл, 3,5 ммоль) в ДМФ (5 мл). Реакционную смесь перемешивали при комнатной температуре в течение 3 часов, затем разбавляли водой (30 мл) и водный слой экстрагировали дихлорметаном (3×20 мл). Объединенные органические экстракты фильтровали через гидрофобную фритту и концентрировали в вакууме. Полученный остаток подвергали флеш хроматографии (Si-SPE, градиент 0-100% этилацетат в циклогексане) с получением указанного в заголовке соединения в виде твердого продукта кремового цвета (500 мг, 60%). LCMS (Способ B): RT=3,23 мин, [M+H]+=592.
Стадия 2: ((S)-2-гидроксипропокси)-амид 4-(2-фтор-4-йодфениламино)-1H-пиразоло[3,4-b]пиридин-5-карбоновой кислоты
((S)-2-гидроксипропокси)-амид 4-(2-фтор-4-йодфениламино)-1-(4-метоксибензил)-1H-пиразоло[3,4-b]пиридин-5-карбоновой кислоты (500 мг, 0,85 ммоль) растворяли в ТФУ (5 мл) и полученную смесь нагревали при температуре 65ºC в течение 3 часов, после чего концентрировали в вакууме. Полученный остаток растворяли в дихлорметане (10 мл) и метаноле (2 мл), затем энергично перемешивали насыщенным водным раствором бикарбоната натрия (20 мл) в течение 2 часов. Водный слой экстрагировали дихлорметаном (2×10 мл) и объединенные органические фракции фильтровали через гидрофобную фритту, затем концентрировали в вакууме. Полученный остаток очищали с помощью обратимо-фазовой ВЭЖХ (градиент 10-95% метанол/вода+0,1%-ная муравьиная кислота, Phenominex gemini PhC6, 5 микрон, 250×20 мм) с получением указанного в заголовке соединения в виде твердого вещества белого цвета (106 мг, 27%). LCMS (Способ A): RT=7,19 мин, [M+H]+=472. 1H ЯМР (ДМСО-d6, 400 МГц) 13,51 (1H, с), 10,37 (1H, с), 8,45 (1H, с), 7,80 (1H, дд, J=9,68, 1,89 Гц), 7,63-7,60 (1H, м), 7,25 (1H, т, J=8,42 Гц), 6,74 (1H, с), 3,88-3,81 (1H, м), 3,73-3,69 (2H, м), 1,05 (3H, д, J=6,34 Гц).
ПРИМЕР 8: (2-гидрокси-1,1-диметилэтокси)-амид 4-(2-фтор-4-йодфениламино)-1H-пиразоло[3,4-b]пиридин-5-карбоновой кислоты
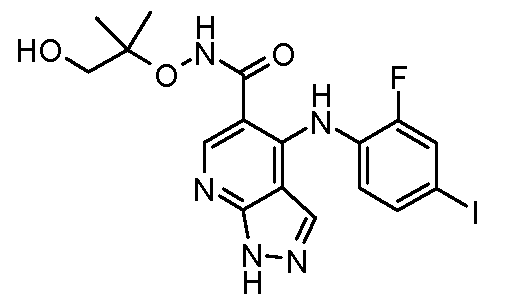
Стадия 1: (2-гидрокси-1,1-диметилэтокси)-амид 4-(2-фтор-4-йодфениламино)-1-(4-метоксибензил)-1H-пиразоло[3,4-b|пиридин-5-карбоновой кислоты
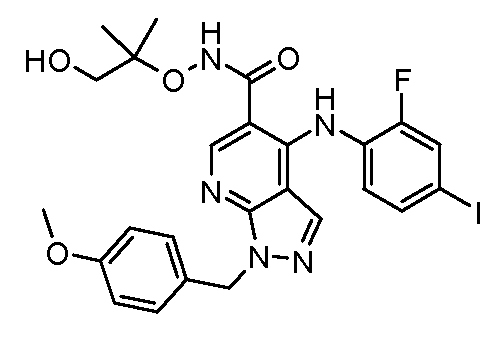
К раствору 4-(2-фтор-4-йодфениламино)-1-(4-метоксибензил)-1H-пиразоло[3,4-b]пиридин-5-карбоновой кислоты (737 мг, 1,4 ммоль), HOBt (231 мг, 1,7 ммоль) и EDCI (327 мг, 1,7 ммоль) в ДМФ (15 мл) добавляли раствор гидрохлорида 2-аминоокси-2-метил-пропан-1-ола (280 мг, 2,0 ммоль) и DIPEA (603 мкл, 3,6 ммоль) в ДМФ (5 мл). Реакционную смесь перемешивали при комнатной температуре в течение 3 дней, затем разбавляли водой (30 мл) и водный слой экстрагировали дихлорметаном (3×20 мл). Объединенные органические фракции фильтровали через гидрофобную фритту и концентрировали в вакууме. Полученный остаток подвергали флеш хроматографии (Si-SPE, градиент 0-100% этилацетат в циклогексане) с получением указанного в заголовке соединения в виде твердого продукта желтого цвета (420 мг, 49%). LCMS (Способ B): RT=3,47 мин, [M+H]+=606.
Стадия 2: (2-гидрокси-1,1-диметилэтокси)амид 4-(2-фтор-4-йодфениламино)-1H-пиразоло[3,4-b]пиридин-5-карбоновой кислоты
(2-Гидрокси-1,1-диметилэтокси)-амид 4-(2-фтор-4-йодфениламино)-1-(4-метоксибензил)-1H-пиразоло[3,4-b]пиридин-5-карбоновой кислоты (420 мг, 0,69 ммоль) растворяли в ТФУ (5 мл) и реакционную смесь нагревали при температуре 65ºC в течение 3 часов. Реакционную смесь концентрировали в вакууме и полученный остаток растворяли в дихлорметане (10 мл) и метаноле (2 мл), после чего промывали насыщенным водным раствором бикарбоната натрия (3×10 мл). Водный слой экстрагировали дихлорметаном (2×10 мл) и объединенные органические экстракты фильтровали через гидрофобную фритту, затем концентрировали в вакууме. Полученный остаток очищали с помощью обратимо фазовой ВЭЖХ (градиент 10-95% метанол/вода+0,1%-ная муравьиная кислота, Phenominex gemini PhC6, 5 микрон, 250×20 мм) с получением указанного в заголовке соединения в виде твердого вещества белого цвета (21 мг, 11%). LCMS (Способ A): RT=7,71 мин, [M+H]+=486. 1H ЯМР (ДМСО-d6, 400 МГц) 13,53 (1H, с), 11,20 (1H, с), 10,15 (1H, с), 8,49 (1H, с), 7,80 (1H, д, J=9,68 Гц), 7,63 (1H, д, J=8,38 Гц), 7,25 (1H, т, J=8,38 Гц), 6,70 (1H, с), 4,65 (1H, с), 1,17 (6H, с).
ПРИМЕР 9: ((R)-2,3-дигидрокси-пропокси)-амид 4-(2-фтор-4-йодфениламино)-1H-индазол-5-карбоновой кислоты
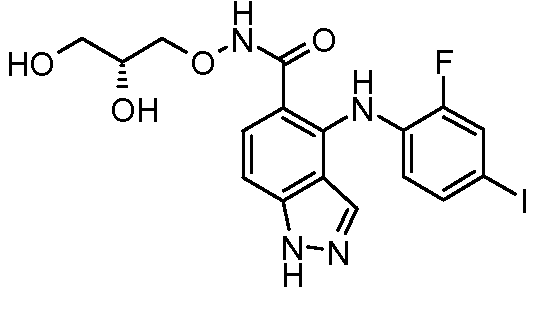
Стадия 1: ((R)-2,2-диметил-[1,3]-диоксолан-4-илметокси)амид 4-(2-фтор-4-йодфениламино)-1H-индазол-5-карбоновой кислоты
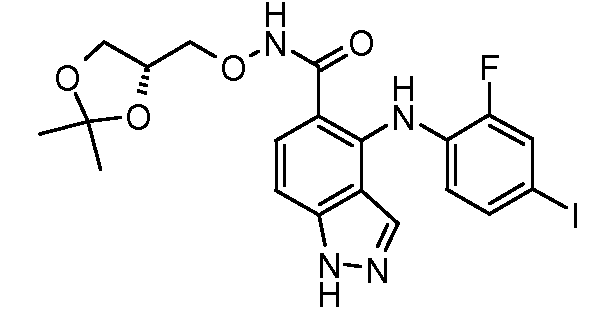
К раствору 4-(2-фтор-4-йодфениламино)-1H-индазол-5-карбоновой кислоты (150 мг, 0,38 ммоль) и O-((R)-2,2-диметил-[l,3]диоксолан-4-илметил)-гидроксиламина (83 мг, 0,57 ммоль) в ДМФ (4 мл) добавляли EDCI (80 мг, 0,42 ммоль), HOBt (56 мг, 0,42 ммоль) и DIPEA (70 мкл, 0,42 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 3,5 часов, после чего концентрировали в вакууме. Полученный остаток растворяли в этилацетате (10 мл), промывали насыщенным водным раствором бикарбоната натрия (10 мл) и водную фракцию экстрагировали дважды этилацетатом (2×10 мл). Объединенные органические экстракты промывали насыщенным солевым раствором (20 мл), сушили (MgSO4) и концентрировали в вакууме. Полученный остаток подвергали флеш хроматографии (Si-SPE, градиент 0-10% метанола в DCM) с получением указанного в заголовке соединения в виде твердого продукта бледно-желтого цвета (135 мг, 68%). LCMS (Способ B): RT=3,45 мин, [M+H]+=527.
Стадия 2: ((R)-2,3-дигидрокси-пропокси)-амид 4-(2-фтор-4-йодфениламино)-1H-индазол-5-карбоновой кислоты
К раствору ((R)-2,2-диметил-[l,3]диоксолан-4-илметокси)-амида 4-(2-фтор-4-йодфениламино)-1H-индазол-5-карбоновой кислоты (135 мг, 0,26 ммоль) в метаноле (4 мл) добавляли хлористоводородную кислоту в диоксане (2 мл, 4н, 8 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа, после чего концентрировали в вакууме. Полученный остаток подвергали флеш хроматографии (Si-SPE, градиент 0-10% метанола в DCM) с получением указанного в заголовке соединения в виде твердого продукта не совсем белого цвета (94 мг, 75%). LCMS (Способ A): RT=7,67 мин, [M+H]+=487. 1H ЯМР (ДМСО-d6, 400 МГц) 13,20 (1H, с), 11,67 (1H, с), 9,94 (1H, с), 7,66 (1H, дд, J=10,30, 1,92 Гц), 7,47 (1H, д, J=8,79 Гц), 7,45-7,41 (1H, м), 7,22 (1H, с), 7,01 (1H, д, J=8,78 Гц), 6,92 (1H, т, J=8,64 Гц), 3,92-3,85 (1H, м), 3,76-3,58 (2H, м), 3,41-3,31 (2H, м).
ПРИМЕР 10: (2-гидрокси-этокси)-амид 4-(4-бром-2-фтор-фениламино)-1H-индазол-5-карбоновой кислоты
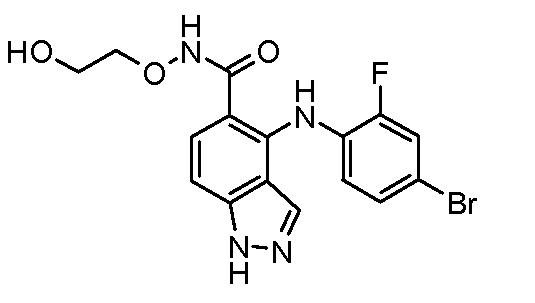
Стадия 1: (2-винилокси-этокси)-амид 4-(4-бром-2-фторфениламино)-1H-индазол-5-карбоновой кислоты
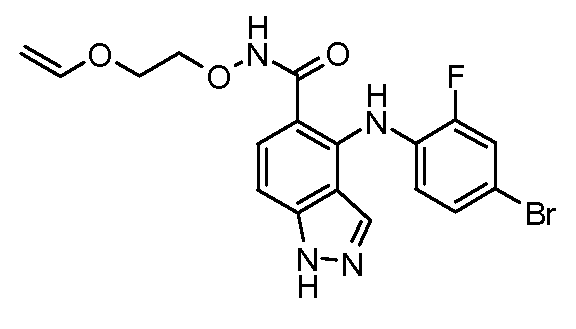
К раствору 4-(4-бром-2-фторфениламино)-1H-индазол-5-карбоновой кислоты (115 мг, 0,33 ммоль) и 0-(2-винилокси-этил)-гидроксиламина (51 мг, 0,49 ммоль) в ДМФ (3 мл) добавляли EDCI (69 мг, 0,36 ммоль), HOBt (49 мг, 0,36 ммоль) и DIPEA (61 мкл, 0,36 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 4 часов, после чего концентрировали в вакууме. Полученный остаток растворяли в этилацетате (10 мл), промывали насыщенным водным раствором бикарбоната натрия (10 мл) и водную фракцию экстрагировали дважды этилацетатом (2×10 мл). Объединенные органические экстракты промывали насыщенным солевым раствором (20 мл), сушили (MgSO4) и концентрировали в вакууме. Полученный остаток подвергали флеш хроматографии (Si-SPE, градиент 0-10% метанола в DCM) с получением указанного в заголовке соединения в виде твердого продукта бледно-желтого цвета (96 мг, 67%). LCMS (Способ B): Rτ=3,43 мин, [M-H]-=433/435.
Стадия 2: (2-гидрокси-этокси)-амид 4-(4-бром-2-фтор-фениламино)-1H-индазол-5-карбоновой кислоты
К раствору (2-винилокси-этокси)-амида 4-(4-бром-2-фторфениламино)-1H-индазол-5-карбоновой кислоты (96 мг, 0,22 ммоль) в метаноле (5 мл) добавляли хлористоводородную кислоту (1 мл, 1н, 1 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 30 минут, после чего концентрировали в вакууме. Полученный остаток растворяли в метаноле (2 мл) и добавляли несколько капель воды, что вызывало образование осадка. Продукт собирали путем фильтрации и сушили в вакууме с получением указанного в заголовке соединения твердого продукта не совсем белого цвета (50 мг, 55%). LCMS (Способ A): RT=7,99 мин, [M+H]+=409/411. 1H ЯМР (ДМСО-d6, 400 МГц) 13,20 (1H, с), 11,61 (1H, с), 9,95 (1H, с), 7,58 (1H, дд, J=10,48, 2,24 Гц), 7,47 (1H, д, J=8,77 Гц), 7,29 (1H, ддд, J=8,60, 2,21, 1,05 Гц), 7,23 (1H, с), 7,08 (1H, т, J=8,81 Гц), 7,01 (1H, д, J=8,81 Гц), 4,68 (1H, с), 3,86 (2H, т, J=4,95 Гц), 3,56 (3H, с).
ПРИМЕР 11: ((S)-2-гидрокси-пропокси)-амид 4-(4-бром-2-фтор-фениламино)-1H-индазол-5-карбоновой кислоты
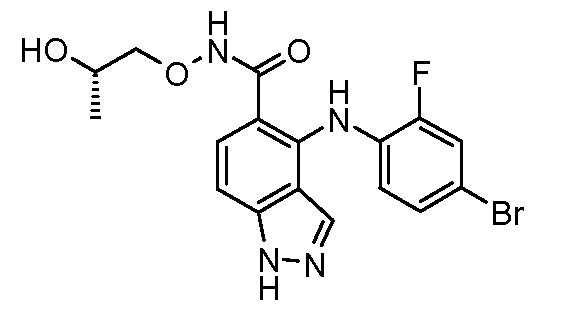
К раствору 4-(4-бром-2-фторфениламино)-1H-индазол-5-карбоновой кислоты (115 мг, 0,33 ммоль) и гидрохлорида (S)-1-аминоокси-пропан-2-ола (63 мг, 0,49 ммоль) в ДМФ (3 мл) добавляли EDCI (69 мг, 0,36 ммоль), HOBt (49 мг, 0,36 ммоль) и DIPEA (150 мкл, 0,85 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов, после чего концентрировали в вакууме. Полученный остаток растворяли в этилацетате (10 мл), промывали насыщенным водным раствором бикарбоната натрия (10 мл) и водную фракцию экстрагировали дважды этилацетатом (2×10 мл). Объединенные органические фракции промывали насыщенным солевым раствором (20 мл), сушили (MgSO4) и концентрировали в вакууме. Полученный остаток подвергали препаративной с обращенной фазой ВЭЖХ (10-90% ацетонитрил/вода 0,1%-ная муравьиная кислота, Phenominex gemini PhC6, 5 микрон, 250×20 мм). Полученный продукт растворяли в этилацетате (5 мл) и промывали насыщенным водным раствором бикарбоната натрия (10 мл). Водную фракцию экстрагировали дважды этилацетатом (2×10 мл) и объединенные органические слои промывали насыщенным солевым раствором (20 мл), сушили (MgSO4) и концентрировали в вакууме с получением указанного в заголовке соединения в виде твердого вещества белого цвета (61 мг, 44%). LCMS (Способ A): RT=8,45 мин, [M+H]+=423/425. 1H ЯМР (ДМСО-d6, 400 МГц) 13,21 (1H, с), 11,63 (1H, с), 9,90 (1H, с), 7,58 (1H, дд, J=10,47, 2,24 Гц), 7,46 (1H, д, J=8,75 Гц), 7,29 (1H, ддд, J=8,59, 2,19, 1,06 Гц), 7,23 (1H, с), 7,08 (1H, т, J=8,80 Гц), 7,02 (1H, д, J=8,78 Гц), 4,77 (1H, д, J= 4,15 Гц), 3,84-3,78 (1H, м), 3,70-3,61 (2H, м), 1,03 (3H, д, J=6,33 Гц).
ПРИМЕР 12: (2-гидрокси-этокси)-амид 4-(2-фтор-4-йод-фениламино)-бензо[d]изотиазол-5-карбоновой кислоты
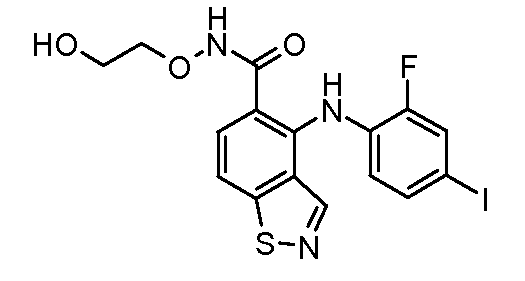
Стадия 1: (2-винилокси-этокси)-амид 4-(2-фтор-4-йод-фениламино)-бензо[d]изотиазол-5-карбоновой кислоты
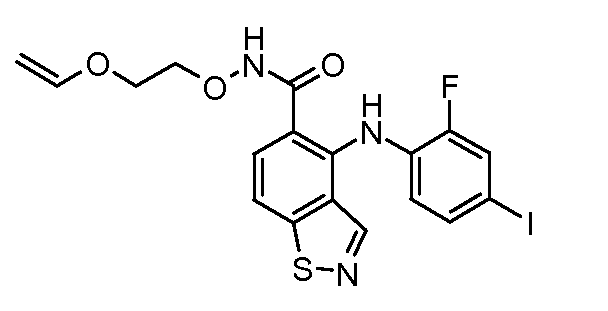
К раствору 4-(2-фтор-4-йод-фениламино)-бензо[d]изотиазол-5-карбоновой кислоты (240 мг, 0,58 ммоль) в ТГФ (6 мл) добавляли DIPEA (396 мкл, 2,34 ммоль), O-(2-винилокси-этил)-гидроксиламин (119 мг, 1,15 ммоль), HOBt (156 мг, 1,15 ммоль) и EDCI (221 мг, 1,15 ммоль), затем реакционную смесь перемешивали при комнатной температуре в течение 18 часов. Реакционную смесь концентрировали в вакууме и полученный остаток распределяли между этилацетатом и водой. Органический экстракт промывали насыщенным водным раствором бикарбоната натрия, затем водой, сушили (Na2SO4) и концентрировали в вакууме. Полученный остаток подвергали флеш хроматографии (Si-SPE, градиент 0-10% метанола в DCM) с получением указанного в заголовке соединения (258 мг, 89%). LCMS (Способ B): RT=3,83 мин, [M+H]+=500.
Стадия 2: (2-гидрокси-этокси)-амид 4-(2-фтор-4-йод-фениламино)-бензо[d]изотиазол-5-карбоновой кислоты
К суспензии (2-винилокси-этокси)-амида 4-(2-фтор-4-йод-фениламино)-бензо[d]изотиазол-5-карбоновой кислоты (258 мг, 0,52 ммоль) в метаноле (10 мл) добавляли соляную кислоту (1,0 мл, 1M раствор, 1,0 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь концентрировали в вакууме и остаток распределяли между этилацетатом и насыщенным водным NaHCO3. Органический слой отделяли и промывали водой, затем насыщенным солевым раствором, сушили (Na2SO4), фильтровали и концентрировали в вакууме. Полученный остаток подвергали флеш хроматографии (Si-SPE, градиент 0-100% этилацетата в циклогексане) с получением указанного в заголовке соединения в виде твердого вещества белого цвета (140 мг, 57%). LCMS (Способ A): RT=9,72 мин, [M+H]+=474. 1H ЯМР (CD3OD, 400 МГц) 8,57 (1H, с), 7,71 (1H, д, J=8,53 Гц), 7,69-7,61 (1H, м), 7,51 (1H, дд, J=10,43, 1,94 Гц), 7,33 (1H, д, J=8,54 Гц), 6,67 (1H, т, J=8,63 Гц), 3,92 (2H, т, J=4,60 Гц), 3,70 (2H, т, J=4,60 Гц).
Пример 13: (2-гидрокси-этокси)-амид 4-(2-фтор-4-метилсульфанил-фениламино)-1H-индазол-5-карбоновой кислоты
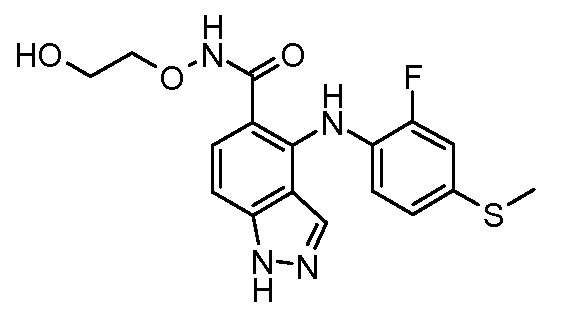
Стадия 1: (2-винилокси-этокси)-амид 4-(2-фтор-4-метилсульфанил-фениламино)-1H-индазол-5-карбоновой кислоты
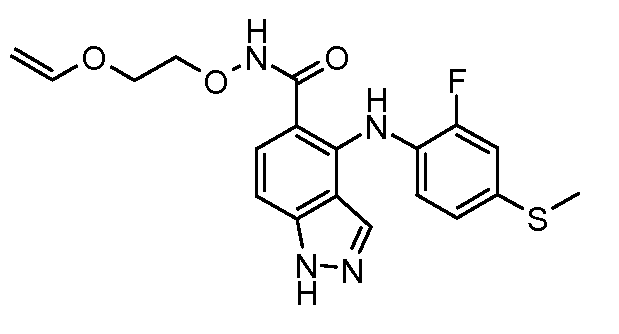
К раствору 4-(2-фтор-4-метилсульфанил)-1H-индазол-5-карбоновой кислоты (85 мг, 0,268 ммоль) и O-(2-винилоксиэтил)-гидроксиламина (33 мг, 0,32 ммоль) в ДМФ (10 мл) добавляли EDCI (66 мг, 0,32 ммоль), HOBt (47 мг, 0,32 ммоль) и DIPEA (68 мкл, 0,40 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 18 часов. Реакционную смесь разбавляли этилацетатом и промывали насыщенным водным раствором гидрокарбоната натрия, затем водой, сушили (Na2SO4), фильтровали и концентрировали в вакууме. Полученный твердый продукт подвергали флеш хроматографии (Si-PPC, градиент 0-75% этилацетата в DCM) с получением указанного в заголовке соединения в виде твердого продукта рыжевато-коричневого цвета (45 мг, 42%). LCMS (Способ A): RT=3,40 мин, [M+H]+=403.
Стадия 2: (2-гидрокси-этокси)-амид 4-(2-фтор-4-метилсульфанил-фениламино)-1H-индазол-5-карбоновой кислоты
Раствор (2-винилокси-этокси)-амида 4-(2-фтор-4-метилсульфанил-фениламино)-1H-индазол-5-карбоновой кислоты (45 мг, 0,112 ммоль) в метаноле (5 мл) обрабатывали соляной кислотой (1M, 0,225 мл, 0,22 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь затем концентрировали в вакууме и остаток растворяли в метаноле и к этому раствору добавляли воду, что вызывало образование осадка, который отфильтровывали. Фильтрат экстрагировали дважды этилацетатом, объединенные экстракты сушили (Na2SO4), фильтровали и концентрировали в вакууме. Сырой продукт объединяли с ранее выпавшим твердым осадком и подвергали флеш хроматографии (Si-PPC, градиент 0-10% метанола в DCM) с получением твердого продукта. Твердый продукт растирали в диэтиловом эфире с получением твердого продукта бледного рыжевато-коричневого цвета (23 мг, 55%). LCMS (Способ A) RT 7,79 [M+H]+ 377. 1H ЯМР (MeOD, 400 МГц): 7,52-7,44 (1H, м), 7,21-7,09 (3H, м), 7,06 (1H, дд, J=8,41, 2,08 Гц), 6,93 (1H, д, J=8,88 Гц), 4,54 (1H, с), 4,02-3,98 (2H, м), 3,75 (2H, дд, J=5,28, 4,05 Гц), 2,48 (3H, с).
ПРИМЕР 14: этокси-амид 4-(2-фтор-4-йод-фениламино)-1H-индазол-5-карбоновой кислоты
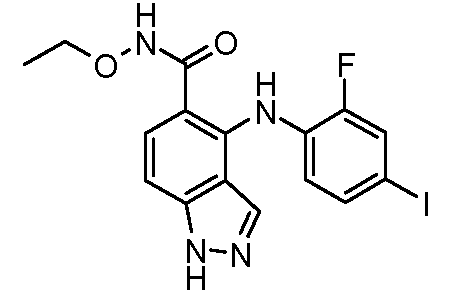
К раствору 4-(2-фтор-4-йодфениламино)-1H-индазол-5-карбоновой кислоты (100 мг, 0,252 ммоль) в ДМФ (3 мл) добавляли гидрохлорид O-этилгидроксиламина (37 мг, 0,378 ммоль), HOBt (37 мг, 0,277 ммоль) и EDCI (53 мг, 0,277 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа, затем добавляли DIPEA (91 мкл, 0,529 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 18 часов. В реакционную смесь добавляли дополнительные количества, как в начале реакции, O-этил гидроксиламин гидрохлорид, HOBt, EDCI и DIPEA и перемешивание продолжали в течение 4 часов. Реакционную смесь разбавляли этилацетатом и промывали насыщенным водным раствором гидрокарбоната натрия, твердый продукт, выпавший в смеси, отфильтровывали, промывали водой с получением указанного в заголовке соединения (41 мг, 37%). LCMS (Способ A): RT=9,65 мин, [M+H]+=441. 1H ЯМР (ДМСО-d6, 400 МГц): 13,19 (1H, с), 11,51 (1H, с), 9,97 (1H, с), 7,66 (1H, дд, J=10,35, 1,95 Гц), 7,46 (1H, д, J=8,78 Гц), 7,42 (1H, дд, J=8,47, 1,84 Гц), 7,25 (1H, с), 7,02 (1H, д, J=8,76 Гц), 6,91 (1H, т, J=8,66 Гц), 3,86 (2H, кв, J=7,04 Гц), 1,15 (3H, т, J=7,03 Гц).
ПРИМЕР 15: (тетрагидро-пиран-4-илокси)-амид 4-(2-фтор-4-йод-фениламино)-1H-индазол-5-карбоновой кислоты
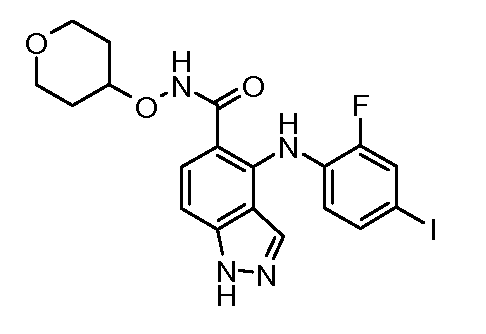
К раствору 4-(2-фтор-4-йодфениламино)-1H-индазол-5-карбоновой кислоты (100 мг, 0,252 ммоль) в ДМФ (3 мл) добавляли O-(тетрагидро-пиран-4-ил)-гидроксиламин (44 мг, 0,378 ммоль), HOBt (37 мг, 0,277 ммоль), EDCI (53 мг, 0,277 ммоль) и DIPEA (92 мкл, 0,529 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 70 часов. Реакционную смесь разбавляли этилацетатом и промывали насыщенным водным раствором гидрокарбоната натрия, затем водой, сушили (Na2SO4), фильтровали и концентрировали в вакууме. Полученный остаток подвергали флеш хроматографии (Si-PPC, градиент 30-100% этилацетата в циклогексане). Полученный твердый продукт растирали в метаноле, твердый продукт отфильтровывали, промывали метанолом с получением указанного в заголовке соединения (33 мг, 26%). LCMS (Способ A): RT=9,27 мин, [M+H]+=497. 1H ЯМР (ДМСО-d6, 400 МГц): 13,19 (1H, с), 11,44 (1H, с), 9,87 (1H, с), 7,65 (1H, дд, J=10,36, 1,95 Гц), 7,48 (1H, д, J=8,77 Гц), 7,44-7,38 (1H, м), 7,28 (1H, с), 7,03 (1H, д, J=8,75 Гц), 6,89 (1H, т, J=8,66 Гц), 4,02-3,93 (1H, м), 3,84-3,76 (2H, м), 3,36-3,28 (2H, м), 1,88-1,81 (2H, м), 1,51 (2H, дддд, J=12,96, 9,42, 8,75, 4,11 Гц).
ПРИМЕР 16: циклопропилметокси-амид 4-(2-фтор-4-йод-фениламино)-1H-индазол-5-карбоновой кислоты
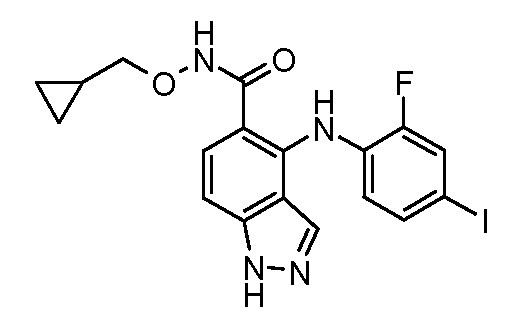
К раствору 4-(2-фтор-4-йодфениламино)-1H-индазол-5-карбоновой кислоты (70 мг, 0,176 ммоль) и O-циклопропилметилгидроксиламин (23 мг, 0,21 ммоль) в ДМФ (3 мл) добавляли EDCI (40 мг, 0,21 ммоль), HOBt (28 мг, 0,21 ммоль) и DIPEA (70 мкл, 0,42 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 18 часов. Реакционную смесь разбавляли этилацетатом и промывали насыщенным водным раствором гидрокарбоната натрия, затем водой, сушили (Na2SO4), фильтровали и концентрировали в вакууме. Полученный твердый продукт растирали в растворе горячей смеси метанол/вода/NaHCO3, твердый продукт отфильтровывали, промывали водой с получением указанного в заголовке соединения в виде твердого продукта бледно-розового цвета (24 мг, 29%). LCMS (Способ A): RT=10,42 мин, [M+H]+=467. 1H ЯМР (ДМСО-d6, 400 МГц) 13,17 (1H, с), 7,64 (1H, дд, J=10,37, 1,95 Гц), 7,48 (1H, д, J=8,76 Гц), 7,42-7,37 (1H, м), 7,26 (1H, д, J=0,95 Гц), 7,01 (1H, дд, J=8,76, 0,99 Гц), 6,87 (1H, т, J=8,66 Гц), 3,62 (2H, д, J=7,13 Гц), 1,08-1,00 (1H, м), 0,50-0,44 (2H, м), 0,23-0,17 (2H, м).
ПРИМЕР 17: метоксиамид 4-(2-фтор-4-йод-фениламино)-1H-индазол-5-карбоновой кислоты
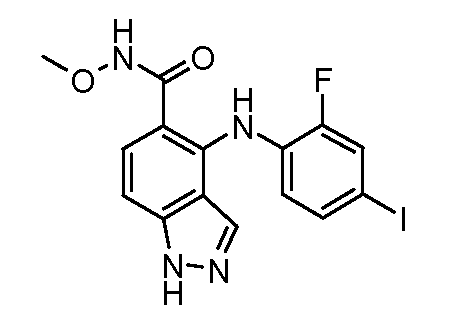
К раствору 4-(2-фтор-4-йодфениламино)-1H-индазол-5-карбоновой кислоты (70 мг, 0,176 ммоль) и O-метил-гидроксиламина (19 мг, 0,21 ммоль) в ДМФ (2 мл) добавляли EDCI (40 мг, 0,21 ммоль), HOBt (28 мг, 0,21 ммоль) и DIPEA (70 мкл, 0,42 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 18 часов. Реакционную смесь разбавляли этилацетатом и промывали насыщенным водным раствором гидрокарбоната натрия, затем водой, сушили (Na2SO4), фильтровали и концентрировали в вакууме. Полученный твердый продукт растирали в растворе горячей смеси метанол/вода/NaHCO3, твердый продукт отфильтровывали, промывали водой с получением указанного в заголовке соединения в виде твердого продукта бледно-розового цвета (33 мг, 44%). LCMS (Способ A): RT=9,09 мин, [M+H]+=427. 1H ЯМР (ДМСО-d6, 400 МГц) 13,20 (1H, с), 11,62 (1H, с), 9,99 (1H, с), 7,66 (1H, дд, J=10,33, 1,94 Гц), 7,46-7,40 (2H, м), 7,23 (1H, с), 7,01 (1H, д, J=8,78 Гц), 6,92 (1H, т, J=8,66 Гц), 3,64 (3 H, с).
ПРИМЕР 18: метокси-метил-амид 4-(2-фтор-4-йод-фениламино)-1H-индазол-5-карбоновой кислоты
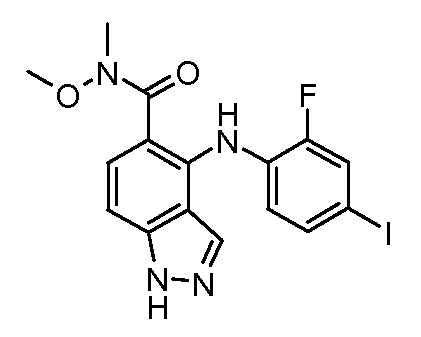
К раствору 4-(2-фтор-4-йодфениламино)-1H-индазол-5-карбоновой кислоты (70 мг, 0,176 ммоль) и N-O-диметил-гидроксиламина (21 мг, 0,21 ммоль) в ДМФ (3 мл) добавляли EDCI (40 мг, 0,21 ммоль), HOBt (28 мг, 0,21 ммоль) и DIPEA (70 мкл, 0,42 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 18 часов. Реакционную смесь разбавляли этилацетатом и промывали насыщенным водным раствором гидрокарбоната натрия, затем водой, сушили (Na2SO4), фильтровали и концентрировали в вакууме затем подвергали азеотропной отгонке с диэтиловым эфиром с получением пены бледно-рыжевато-коричневого цвета (35 мг, 45%). LCMS (Способ A): RT=9,63 мин, [M+H]+=441. 1H ЯМР (ДМСО-d6, 400 МГц) 13,16 (1H, с), 8,25 (1H, с), 7,59-7,51 (2H, м), 7,34-7,28 (2H, м), 7,14 (1H, д, J=8,55 Гц), 6,71 (1H, т, J=8,73 Гц), 3,38 (3H, с), 3,10 (3H, с).
ПРИМЕР 19: [4-(2-фтор-4-йод-фениламино)-1H-индазол-5-ил]-(3-гидрокси-азетидин-1-ил)-метанон
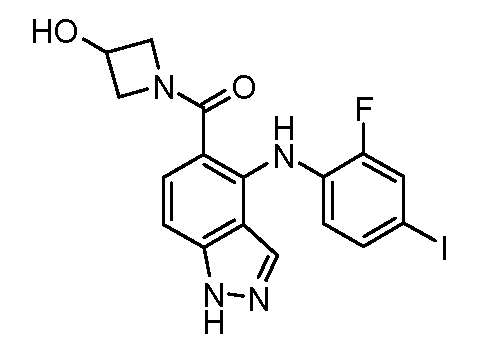
К раствору 4-(2-фтор-4-йодфениламино)-1H-индазол-5-карбоновой кислоты (70 мг, 0,176 ммоль) и гидрохлорида 3-гидроксиазетидина (23 мг, 0,21 ммоль) в ДМФ (1 мл) добавляли EDCI (40 мг, 0,21 ммоль), HOBt (28 мг, 0,21 ммоль) и DIPEA (70 мкл, 0,42 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 18 часов. Реакционную смесь разбавляли этилацетатом и промывали насыщенным водным раствором гидрокарбоната натрия, затем водой, сушили (Na2SO4), фильтровали и концентрировали в вакууме. Полученный остаток растирали в циклогексане с получением указанного в заголовке соединения в виде твердого продукта бледного рыжеватого цвета (39 мг, 49%). LCMS (Способ A): RT=8,23 мин, [M+H]+=453. 1H ЯМР (ДМСО-d6, 400 МГц) 13,17 (1H, с), 9,55 (1H, с), 7,63 (1H, дд, J=10,47, 1,94 Гц), 7,41-7,37 (1H, м), 7,37 (1H, с), 7,32 (1H, д, J=8,69 Гц), 7,05-7,00 (1H, м), 6,92-6,82 (1H, м), 5,67 (1H, д, J=6,13 Гц), 4,44-4,37 (1H, м), 4,24 (2H, ушир.с), 3,83 (2H, ушир.с).
ПРИМЕР 20: ((S)-2-гидрокси-пропокси)-амид 4-(2-фтор-4-йод-фениламино)-бензо[d]изотиазол-5-карбоновой кислоты
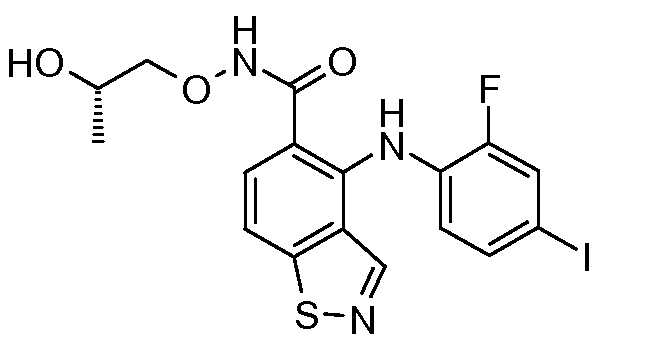
К раствору 4-(2-фтор-4-йод-фениламино)-бензо[d]изотиазол-5-карбоновой кислоты (150 мг, 0,362 ммоль), диизопропилэтиламина (0,25 мл, 1,45 ммоль), HOBt (98 мг, 0,724 ммоль) и (S)-1-аминоокси-пропан-2-ола (92 мг, 0,724 ммоль в ДМФ (2 мл) добавляли EDCI (139 мг, 0,724 ммоль). Реакционную смесь перемешивали в течение 16 часов при комнатной температуре, разбавляли этилацетатом, и промывали водой, насыщенным водным раствором гидрокарбоната натрия, затем насыщенным солевым раствором, после чего сушили (Na2SO4), фильтровали и концентрировали в вакууме. Полученный остаток подвергали флеш хроматографии (Si-PPC, градиент 30%-80% EtOAc в циклогексане), что давало указанное в заголовке соединение в виде твердого продукта (45 мг, 26%). LCMS (способ A): RT=10,26 мин, [M+H]+=488. 1H ЯМР (ДМСО-d6, 400 МГц) 11,72 (1H, с), 9,22 (1H, с), 8,69 (1H, с), 7,86-7,81 (1H, м), 7,64-7,55 (2H, м), 7,30 (1H, дд, J=8,47, 1,83 Гц), 6,63 (1H, т, J=8,72 Гц), 4,73 (1H, с), 3,76-3,68 (1H, м), 3,53 (2H, д, J=5,78 Гц), 0,99 (3H, д, J=6,33 Гц).
ПРИМЕР 21: ((R)-2,3-дигидрокси-пропокси)-амид 4-(2-фтор-4-йод-фениламино)-бензо[d]изотиазол-5-карбоновой кислоты
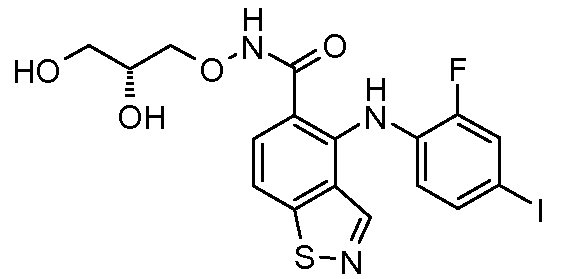
Стадия 1: ((R)-2,2-диметил-[1,3]-диоксолан-4-илметокси)-амид 4-(2-фтор-4-йод-фениламино)-бензо[d]изотиазол-5-карбоновой кислоты
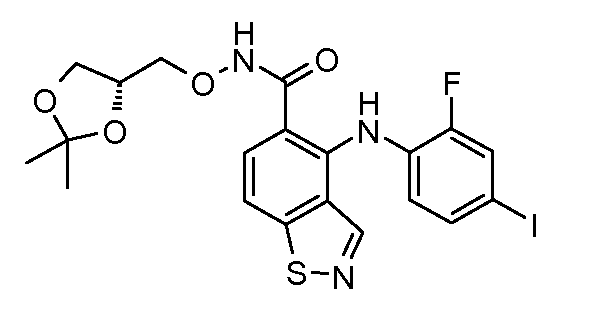
К раствору 4-(2-фтор-4-йод-фениламино)-бензо[d]изотиазол-5-карбоновой кислоты (250 мг, 0,604 ммоль), диизопропилэтиламина (0,42 мл, 2,42 ммоль), HOBt (163 мг, 0,1,21 ммоль) и O-((R)-2,2-диметил-[1,3]диоксолан-4-илметил)-гидроксиламина (178 мг, 1,21 ммоль) в ДМФ (2 мл) добавляли EDCI (232 мг, 1,21 ммоль). Реакционную смесь перемешивали в течение 16 часов при комнатной температуре, разбавляли этилацетатом и промывали водой, насыщенным водным раствором гидрокарбоната натрия, затем насыщенным солевым раствором, после чего сушили (Na2SO4), фильтровали и концентрировали в вакууме. Полученный остаток подвергали флеш хроматографии (Si-PPC, градиент 0%-50% EtOAc в циклогексане), что давало указанное в заголовке соединение в виде твердого продукта (142 мг, 43%). LCMS (способ B): RT=3,92 мин, [M+H]+=544.
Стадия 2: ((R)-2,3-дигидрокси-пропокси)-амид 4-(2-фтор-4-йод-фениламино)-бензо[d]изотиазол-5-карбоновой кислоты
К раствору ((R)-2,2-диметил-[l,3]диоксолан-4-илметокси)-амида 4-(2-фтор-4-йод-фениламино)-бензо[d]изотиазол-5-карбоновой кислоты (142 мг, 0,261 ммоль) в MeOH (2 мл) добавляли 4M раствор HCl в диоксане (2 мл). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, после чего концентрировали в вакууме. Полученный остаток обрабатывали этилацетатом, промывали насыщенным водным раствором гидрокарбоната натрия, воду, затем насыщенным солевым раствором, сушили (Na2SO4), фильтровали и концентрировали в вакууме. Полученный остаток растирали в смеси этилацетат/циклогексан, что давало указанное в заголовке соединение в виде твердого продукта (73 мг, 56%). LCMS (способ A): RT=8,97 мин, [M+H]+=504. 1H ЯМР (ДМСО-d6, 400 МГц) 11,78 (1H, с), 9,28 (1H, с), 8,64 (1H, с), 7,84 (1H, дд, J=8,48, 0,92 Гц), 7,64-7,57 (2H, м), 7,31 (1H, дд, J=8,47, 1,83 Гц), 6,65 (1H, т, J=8,71 Гц), 4,80 (1H, с), 4,54 (1H, с), 3,80 (1H, т, J=6,46 Гц), 3,68-3,61 (2H, м), 3,32 (2H, с).
ПРИМЕР 22: ((R)-2,3-дигидрокси-пропокси)-амид 7-(2-фтор-4-метилсульфанил-фениламино)-бензо[d]изотиазол-6-карбоновой кислоты
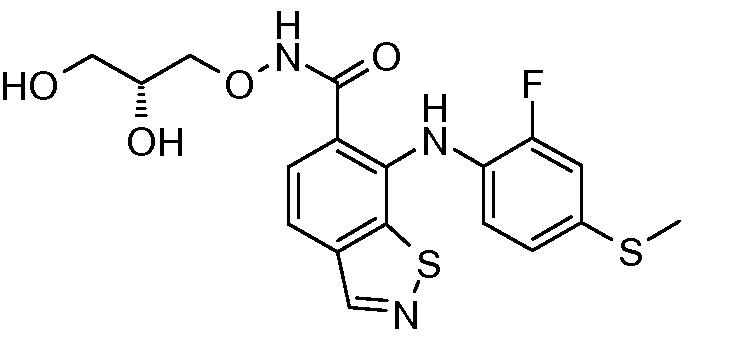
Стадия 1: ((R)-2,2-диметил-[1,3]диоксолан-4-илметокси)-амид 7-(2-фтор-4-метилсульфанил-фениламино)-бензо[d]изотиазол-6-карбоновой кислоты
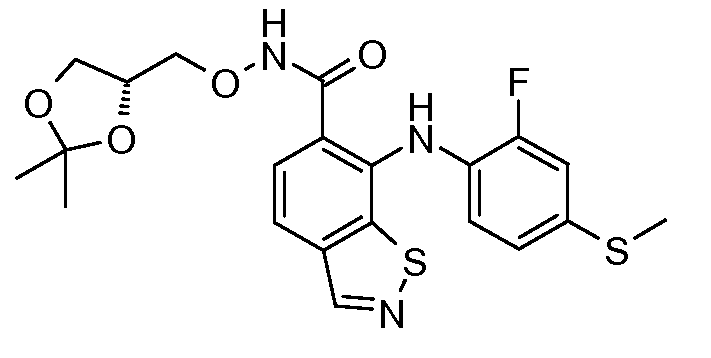
К раствору 7-(2-фтор-4-метилсульфанил-фениламино)-бензо[d]изотиазол-6-карбоновой кислоты (200 мг, 0,60 ммоль) и диизопропилэтиламина (0,31 мл, 1,80 ммоль) в ДМФ (2 мл) добавляли O-((R)-2,2-диметил-[l,3]диоксолан-4-илметил)-гидроксиламин (176 мг, 1,20 ммоль), EDCI (230 мг, 1,20 ммоль) и HOBt (162 мг, 1,20 ммоль). Реакционную смесь перемешивали в течение 18 часов при комнатной температуре, и затем разбавляли этилацетатом и промывали водой, насыщенным водным раствором гидрокарбоната натрия и насыщенным солевым раствором, после чего сушили (Na2SO4), фильтровали и концентрировали в вакууме. Полученный остаток подвергали флеш хроматографии (Si-PPC, градиент 50%-100%, Et2O в пентане), что давало указанное в заголовке соединение в виде масла желтого цвета (171 мг, 61%). LCMS (способ B): RT=3,85 мин, [M+H]+=464.
Стадия 2: ((R)-2,3-дигидрокси-пропокси)-амид 7-(2-фтор-4-метилсульфанил-фениламино)-бензо[d]изотиазол-6-карбоновой кислоты
К раствору ((R)-2,2-диметил-[l,3]диоксолан-4-илметокси)-амида 7-(2-фтор-4-метилсульфанил-фениламино)-бензо[d]изотиазол-6-карбоновой кислоты (170 мг, 0,37 ммоль) в MeOH (2 мл) добавляли 1,0M водный раствор соляной кислоты (0,80 мл). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа, после чего концентрировали в вакууме. Остаток обрабатывали этилацетатом, промывали насыщенным водным раствором гидрокарбоната натрия, затем насыщенным солевым раствором, сушили (Na2SO4), фильтровали и упаривали в вакууме. Полученный остаток подвергали флеш хроматографии (Si-PPC, градиент 0%-100% MeOH в DCM), что давало указанное в заголовке соединение в виде твердого продукта желтого цвета (42 мг, 26%). LCMS (Способ A): RT=8,80 мин, [M+H]+=478. 1H ЯМР (CD3OD) 8,81 (1H, с), 7,63-7,56 (2H, м), 7,12-7,04 (3H, м), 4,10-4,04 (1H, м), 3,98-3,86 (2H, м), 3,64-3,55 (2H, м), 2,50 (3H, с).
ПРИМЕР 23: ((R)-2,3-дигидрокси-пропокси)-амид 7-(4-циклопропил-2-фтор-фениламино)-бензо[d]изотиазол-6-карбоновой кислоты
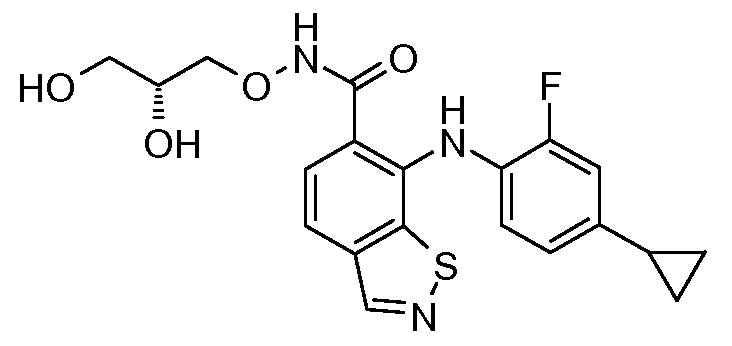
Стадия 1: ((R)-2,3-дигидрокси-пропокси)-амид 7-(4-циклопропил-2-фтор-фениламино)-бензо[d]изотиазол-6-карбоновой кислоты
К раствору 7-(4-циклопропил-2-фтор-фениламино)-бензо[d]изотиазол-6-карбоновой кислоты (155 мг, 0,47 ммоль) и диизопропилэтиламина (0,10 мл, 0,61 ммоль) в ДМФ (5 мл) добавляли O-((R)-2,2-диметил-[l,3]диоксолан-4-илметил)-гидроксиламин (97 мг, 0,66 ммоль), EDCI (117 мг, 0,61 ммоль) и HOBt (83 мг, 0,61 ммоль). Реакционную смесь перемешивали в течение 2 часов при комнатной температуре, разбавляли этилацетатом, промывали водой, насыщенным водным раствором гидрокарбоната натрия, затем насыщенным солевым раствором, после чего сушили (Na2SO4), фильтровали и концентрировали в вакууме. Полученный остаток обрабатывали MeOH (10 мл) и добавляли 4,0 M раствор хлористоводородной кислоты в диоксане (1,0 мл). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа, после чего разбавляли этилацетатом, промывали насыщенным водным раствором гидрокарбоната натрия, затем насыщенным солевым раствором, сушили (Na2SO4), фильтровали и упаривали в вакууме. Полученный остаток подвергали обращенно фазовой ВЭЖХ (Gemini 5 микрон C18 250×21,20 мм колонка, 0,1%-ная муравьиная кислота, градиент ацетонитрил/вода, 15-95%, время линейного нарастания 20 минут), что давало указанное в заголовке соединение в виде твердого продукта желтого цвета (115 мг, 59%). LCMS (способ A): RT=9,02 мин, [M+H]+=418. 1H ЯМР (ДМСО-d6, 400 МГц) 9,98 (1H, с), 8,97 (1H, с), 7,72 (1H, с), 7,61 (1H, д, J=8,35 Гц), 7,09-6,90 (3H, м), 3,95 (1H, дд, J=9,94, 3,81 Гц), 3,79 (2H, д, J=16,92 Гц), 3,40 (2H, д, J=5,46 Гц), 2,03-1,94 (1H, м), 1,03-0,97 (2H, м), 0,77-0,71 (2H, м).
ПРИМЕР 24: ((R)-2,3-дигидрокси-пропокси)-амид 7-(4-бром-2-фтор-фениламино)-бензо[d]изотиазол-6-карбоновой кислоты
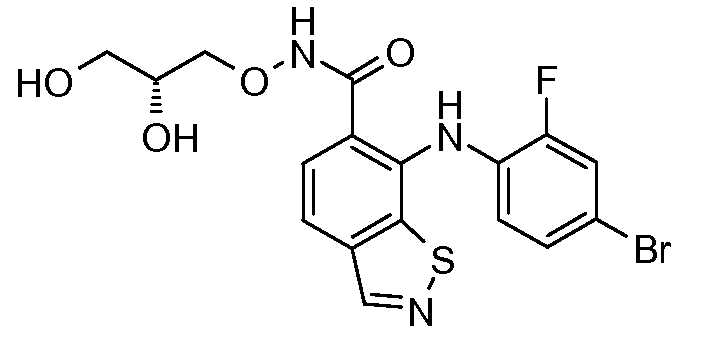
Стадия 1: ((R)-2,2-диметил-[1,3]диоксолан-4-илметокси)-амид 7-(4-бром-2-фтор-фениламино)-бензо[d]изотиазол-6-карбоновой кислоты
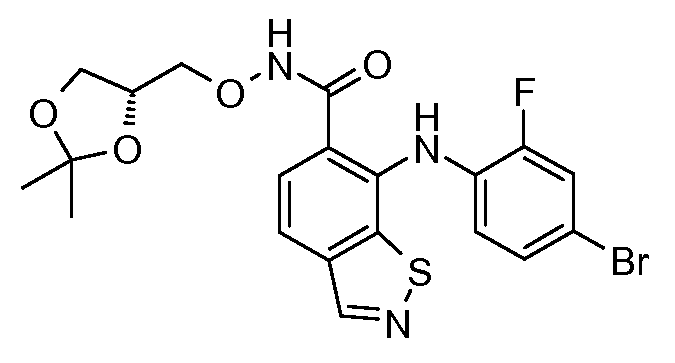
К раствору 7-(4-бром-2-фторо-фениламино)-бензо[d]изотиазол-6-карбоновой кислоты (584 мг, 1,59 ммоль) и диизопропилэтиламина (0,82 мл, 4,77 ммоль) в ДМФ (6 мл) добавляли O-((R)-2,2-диметил-[l,3]диоксолан-4-илметил)-гидроксиламин (468 мг, 3,18 ммоль), EDCI (611 мг, 3,18 ммоль) и HOBt (430 мг, 3,18 ммоль). Реакционную смесь перемешивали в течение 18 часов при комнатной температуре, разбавляли этилацетатом и промывали водой, насыщенным водным раствором гидрокарбоната натрия, затем насыщенным солевым раствором, после чего сушили (Na2SO4), фильтровали и концентрировали в вакууме. Полученный остаток подвергали флеш хроматографии (Si-PPC, градиент 50%-100%, TBME в циклогексане), что давало указанное в заголовке соединение в виде масла желтого цвета (370 мг, 47%). LCMS (способ B): RT=3,96 мин, [M+H]+=496/498.
Стадия 2: ((R)-2,3-дигидрокси-пропокси)-амид 7-(4-бром-2-фтор-фениламино)-бензо[d]изотиазол-6-карбоновой кислоты
К раствору ((R)-2,2-диметил-[l,3]диоксолан-4-илметокси)-амида 7-(4-бром-2-фтор-фениламино)-бензо[d]изотиазол-6-карбоновой кислоты (370 мг, 0,75 ммоль) в MeOH (4 мл) добавляли 1,0M водный раствор соляной кислоты (1,50 мл). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, после чего концентрировали в вакууме. Полученный остаток обрабатывали этилацетатом, промывали насыщенным водным раствором гидрокарбоната натрия, затем насыщенным солевым раствором, сушили (Na2SO4), фильтровали и упаривали в вакууме. Полученный остаток подвергали флеш хроматографии (Si-PPC, градиент 0% to 100%, MeOH в DCM), что давало указанное в заголовке соединение в виде твердого продукта желтого цвета (242 мг, 71%). LCMS (способ A): RT=8,80 мин, [M+H]+=456/458. 1H ЯМР (CD3OD, 400 МГц) 8,87 (1H, с), 7,76-7,70 (1H, м), 7,61 (1H, д, J=8,36 Гц), 7,42-7,36 (1H, м), 7,30-7,25 (1H, м), 6,95 (1H, т, J=8,66 Гц), 4,06 (1H, дд, J=10,08, 3,55 Гц), 3,96-3,83 (2H, м), 3,64-3,54 (2H, м).
ПРИМЕР 25: (2-гидрокси-этокси)-амид 7-(4-бром-2-фтор-фениламино)-бензо[d]изотиазол-6-карбоновой кислоты
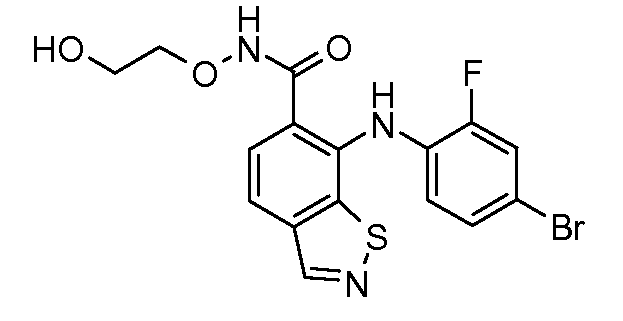
Стадия 1: (2-винилоксиэтокси)-амид 7-(4-бром-2-фтор-фениламино)-бензо[d]изотиазол-6-карбоновой кислоты
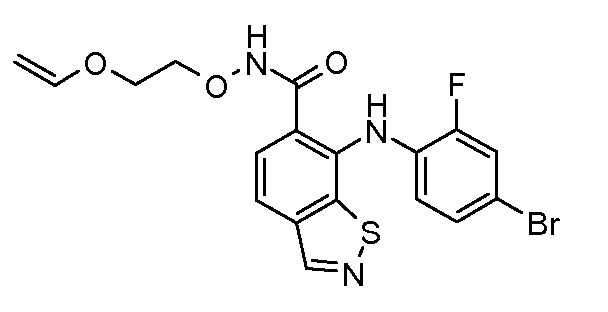
К раствору 7-(4-бром-2-фтор-фениламино)-бензо[d]изотиазол-6-карбоновой кислоты (210 мг, 0,57 ммоль) и диизопропилэтиламина (0,29 мл, 1,71 ммоль) в ДМФ (2 мл) добавляли O-(2-винилокси-этил)-гидроксиламин (117 мг, 1,14 ммоль), EDCI (220 мг, 1,14 ммоль) и HOBt (154 мг, 1,14 ммоль). Реакционную смесь перемешивали в течение 18 часов при комнатной температуре, разбавляли этилацетатом и промывали водой, насыщенным водным раствором гидрокарбоната натрия, затем насыщенным солевым раствором, после чего сушили (Na2SO4), фильтровали и концентрировали в вакууме. Полученный остаток подвергали флеш хроматографии (Si-PPC, градиент 0%-100%, TBME в циклогексане), что давало указанное в заголовке соединение в виде масла желтого цвета (95 мг, 37%). LCMS (способ B): RT=3,90 мин, [M+H]+=452/454.
Стадия 2: (2-гидрокси-этокси)-амид 7-(4-бром-2-фтор-фениламино)-бензо[d]изотиазол-6-карбоновой кислоты
К раствору (2-винилокси-этокси)-амида 7-(4-бром-2-фтор-фениламино)-бензо[d]изотиазол-6-карбоновой кислоты (95 мг, 0,21 ммоль) в MeOH (4 мл) добавляли 1,0M водный раствор соляной кислоты (0,42 мл). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа, после чего концентрировали в вакууме. Полученный остаток обрабатывали этилацетатом, промывали насыщенным водным раствором гидрокарбоната натрия, затем насыщенным солевым раствором, сушили (Na2SO4), фильтровали и упаривали в вакууме. Полученный остаток подвергали флеш хроматографии (Si-PPC, градиент 0%-100% этилацетата в DCM), что давало указанное в заголовке соединение в виде твердого продукта желтого цвета (62 мг, 70%). LCMS (способ A): RT=9,51 мин, [M+H]+=426/428. 1H ЯМР (CDCl3, 400 МГц) 9,37 (1H, с), 8,85 (1H, с), 8,76 (1H, с), 7,51 (1H, д, J=8,40 Гц), 7,43 (1H, д, J=8,40 Гц), 7,34-7,29 (1H, м), 7,28 (1H, д, J=8,75 Гц), 7,02 (1H, т, J=8,46 Гц), 4,10 (2H, т, J=4,09 Гц), 3,91 (1H, с), 3,80 (2H, с).
ПРИМЕР 26; (2-гидрокси-этокси)-амид 7-(2-фтор-4-йод-фениламино)-бензо[d]изотиазол-6-карбоновой кислоты
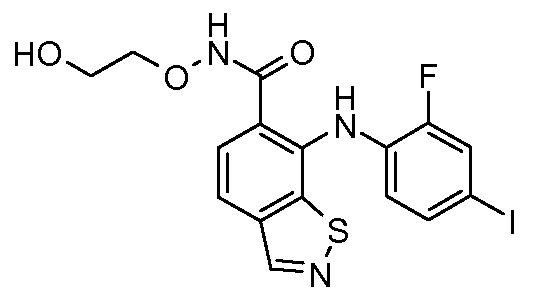
Стадия 1: (2-винилокси-этокси)-амид 7-(2-фтор-4-йод-фениламино)-бензо[d]изотиазол-6-карбоновой кислоты
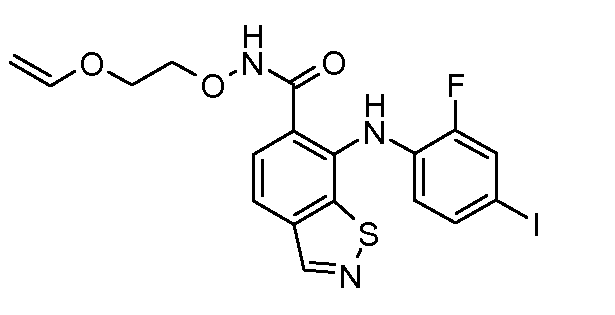
К раствору 7-(2-фтор-4-йод-фениламино)-бензо[d]изотиазол-6-карбоновой кислоты (328 мг, 0,79 ммоль) и диизопропилэтиламина (0,41 мл, 2,37 ммоль) в ДМФ (2 мл) добавляли O-(2-винилокси-этил)-гидроксиламин (163 мг, 1,58 ммоль), EDCI (303 мг, 1,58 ммоль) и HOBt (213 мг, 1,58 ммоль). Реакционную смесь перемешивали в течение 18 часов при комнатной температуре, разбавляли этилацетатом и промывали водой, затем насыщенным водным раствором гидрокарбоната натрия, затем насыщенным солевым раствором, после чего сушили (Na2SO4), фильтровали и концентрировали в вакууме. Полученный остаток подвергали флеш хроматографии (Si-PPC, градиент 0%-100%, TBME в циклогексане), что давало указанное в заголовке соединение в виде пены желтого цвета (194 мг, 49%). LCMS (способ B): RT=3,99 мин, [M+H]+=500.
Стадия 2: (2-гидрокси-этокси)-амид 7-(2-фтор-4-йод-фениламино)-бензо[d]изотиазол-6-карбоновой кислоты
К раствору (2-винилокси-этокси)-амида 7-(2-фтор-4-йод-фениламино)-бензо[d]изотиазол-6-карбоновой кислоты (187 мг, 0,37 ммоль) в MeOH (4 мл) добавляли 1,0 M водный раствор соляной кислоты (0,75 мл). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа, после чего концентрировали в вакууме. Полученный остаток обрабатывали этилацетатом, промывали насыщенным водным раствором гидрокарбоната натрия, затем насыщенным солевым раствором, сушили (Na2SO4), фильтровали и упаривали в вакууме. Полученный остаток подвергали флеш хроматографии (Si-PPC, градиент от 0% до 100%, этилацетат в DCM), что давало указанное в заголовке соединение в виде твердого продукта желтого цвета (128 мг, 72%). LCMS (способ A): RT=9,81 мин, [M+H]+=474. 1H ЯМР (CDCl3, 400 МГц) 9,33 (1H, с), 8,86 (1H, с), 8,77 (1H, с), 7,55-7,40 (4H, м), 6,84 (1H, т, J=8,30 Гц), 4,10 (2H, т, J=4,16 Гц), 3,91 (1H, т, J=6,45 Гц), 3,80 (2H, т, J=4,55 Гц).
ПРИМЕР 27; ((R)-2,3-дигидрокси-пропокси)амид 7-(2-фтор-4-йод-фениламино)-бензо[d]изотиазол-6-карбоновой кислоты
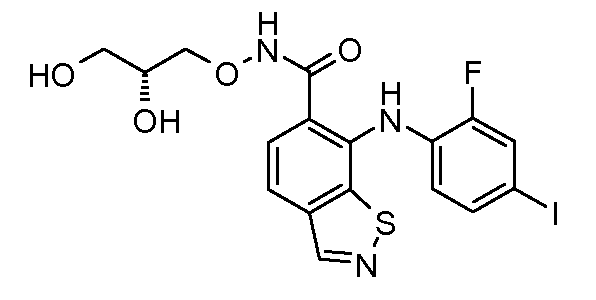
Стадия 1: ((R)-2,2-диметил-[1,3]диоксолан-4-илметокси)-амид 7-(2-фтор-4-йод-фениламино)-бензо[d]изотиазол-6-карбоновой кислоты
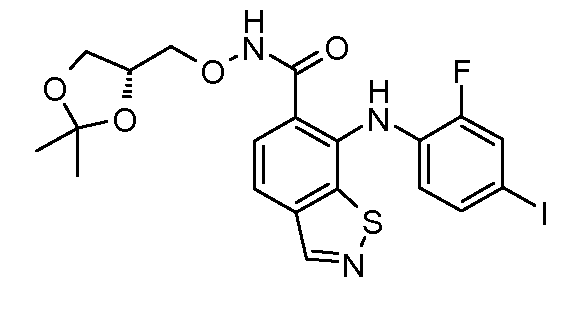
К раствору 7-(2-фтор-4-йод-фениламино)-бензо[d]изотиазол-6-карбоновой кислоты (377 мг, 0,91 ммоль) и диизопропилэтиламина (0,47 мл, 2,73 ммоль) в ДМФ (4 мл) добавляли O-((R)-2,2-диметил-[l,3]диоксолан-4-илметил)-гидроксиламин (268 мг, 1,82 ммоль), EDCI (349 мг, 1,82 ммоль) и HOBt (246 мг, 1,82 ммоль). Реакционную смесь перемешивали в течение 18 часов при комнатной температуре, разбавляли этилацетатом и промывали водой, насыщенным водным раствором гидрокарбоната натрия, затем насыщенным солевым раствором, после чего сушили (Na2SO4), фильтровали и концентрировали в вакууме. Полученный остаток подвергали флеш хроматографии (Si-PPC, градиент от 0% до 100%, TBME в циклогексане), что давало указанное в заголовке соединение в виде твердого продукта желтого цвета (266 мг, 54%). LCMS (способ B): RT=4,04 мин, [M+H]+=544.
Стадия 2: ((R)-2,3-дигидрокси-пропокси)амид 7-(2-фтор-4-йод-фениламино)-бензо[d]изотиазол-6-карбоновой кислоты
К раствору ((R)-2,2-диметил-[1,3]диоксолан-4-илметокси)-амида 7-(2-фтор-4-йод-фениламино)-бензо[d]изотиазол-6-карбоновой кислоты (263 мг, 0,48 ммоль) в MeOH (10 мл) добавляли 1,0 M водный раствор соляной кислоты (0,97 мл). Реакционную смесь перемешивали при комнатной температуре в течение 18 часов, после чего концентрировали в вакууме. Полученный остаток обрабатывали этилацетатом, промывали насыщенным водным раствором гидрокарбоната натрия, затем насыщенным солевым раствором, сушили (Na2SO4), фильтровали и упаривали в вакууме. Полученный остаток подвергали флеш хроматографии (Si-PPC, градиент от 0% до 100%, этилацетат в DCM), что давало указанное в заголовке соединение в виде твердого продукта желтого цвета (127 мг, 53%). LCMS (способ A): RT=9,05 мин, [M+H]+=504. 1H ЯМР (CD3OD, 400 МГц) 8,88 (1H, с), 7,74 (1H, д, J=8,36 Гц), 7,61 (1H, д, J=8,35 Гц), 7,53 (1H, дд, J=10,01, 1,93 Гц), 7,44 (1H, ддд, J=8,36, 1,93, 1,06 Гц), 6,76 (1H, т, J=8,53 Гц), 4,05 (1H, дд, J=10,03, 3,50 Гц), 3,96-3,83 (2H, м), 3,63-3,52 (2H, м).
ПРИМЕР 28: [4-(2-фтор-4-йод-фениламино)-1H-индазол-5-ил]-амид циклопропансульфоновой кислоты
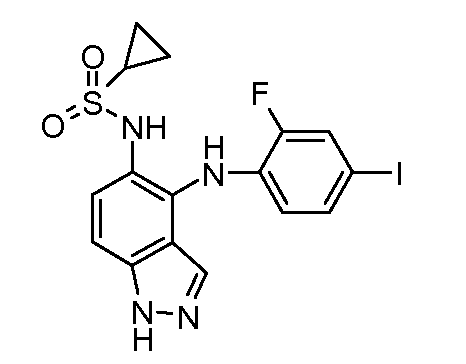
Стадия 1: трет-бутиловый эфир 5-циклопропансульфониламино-4-(2-фтор-4-йод-фениламино)-индазол-1-карбоновой кислоты
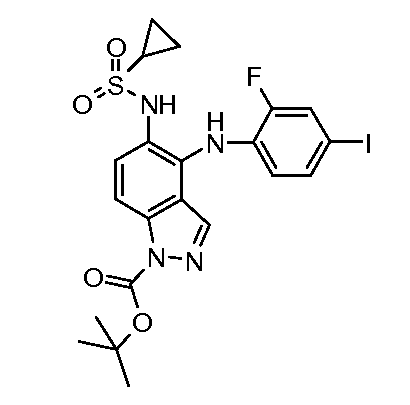
К раствору трет-бутилового эфира 5-амино-4-(2-фтор-4-йод-фениламино)-индазол-1-карбоновой кислоты (200 мг, 0,43 ммоль) в пиридине (2 мл) добавляли циклопропилсульфонил хлорид (0,218 мл, 2,14 ммоль) и смесь перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь концентрировали в вакууме и остаток распределяли между этилацетатом (100 мл) и водой (100 мл). Органический слой отделяли, промывали насыщенным солевым раствором, затем сушили (Na2SO4), фильтровали и концентрировали в вакууме. Полученный остаток подвергали флеш хроматографии (Si-PPC, градиент 10-35% этилацетата в циклогексане) с получением указанного в заголовке соединения (206 мг, 84%). LCMS (Способ B): RT=4,17 мин, [MH]+= 573.
Стадия 2: [4-(2-фтор-4-йод-фениламино)-1H-индазол-5-ил]-амид циклопропансульфоновой кислоты
К раствору трет-бутилового эфира 5-циклопропансульфониламино-4-(2-фтор-4-йод-фениламино)-индазол-1-карбоновой кислоты (206 мг, 0,36 ммоль) в DCM (8 мл) добавляли ТФУ (1 мл) и реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Добавляли дополнительное количество ТФУ (1 мл) и перемешивание продолжали еще в течение 1 часа, затем реакционную смесь концентрировали в вакууме и остаток подвергали азеотропной отгонке с DCM, метанолом и затем опять DCM. Полученный остаток растирали в диэтиловом эфире, твердый продукт отфильтровывали и сушили в вакууме при температуре 40ºC с получением указанного в заголовке соединения в виде твердого продукта желтого цвета (83 мг, 49%). LCMS (Способ A): RT=10,03 [M+H]+=437, 1H ЯМР (ДМСО-d6, 400 МГц): 13,14 (1H, с), 9,07 (1H, с), 7,80 (1H, с), 7,57 (1H, дд, J=10,84, 1,96 Гц), 7,39 (1H, с), 7,31 (1H, д, J=8,74 Гц), 7,25-7,20 (2H, м), 6,45 (1H, т, J=8,84 Гц), 2,40-2,32 (1H, м), 0,75-0,62 (4H, м), -0,05 (1H, т, J=3,33 Гц).
ПРИМЕР 29; [4-(2-фтор-4-йод-фениламино)-6-метокси-1H-индазол-5-ил]-амид циклопропансульфоновой кислоты
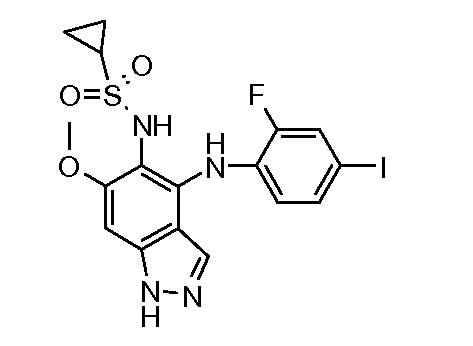
Стадия 1: трет-бутиловый эфир 5-циклопропансульфониламино-4-(2-фтор-4-йод-фениламино)-6-метокси-индазол-1-карбоновой кислоты
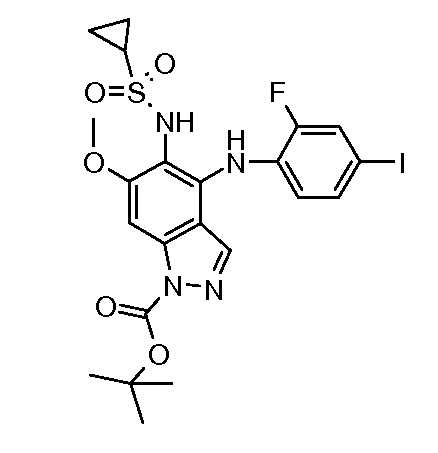
К раствору трет-бутилового эфира 5-амино-4-(2-фтор-4-йод-фениламино)-6-метокси-индазол-1-карбоновой кислоты (200 мг, 0,401 ммоль) в пиридине (2 мл) добавляли циклопропилсульфонил хлорид (281 мг, 2,0 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 18 часов. Реакционную смесь обрабатывали водой и дважды экстрагировали этилацетатом. Объединенные органические экстракты промывали водой, сушили (Na2SO4), фильтровали и концентрировали в вакууме с получением маслянистого остатка. Остаток подвергали флеш хроматографии (Si-PPC, градиент от 0 до 25% этилацетата в циклогексане) с получением указанного в заголовке соединения в виде пены не совсем белого цвета (217 мг, 89%). LCMS (Способ B) RT=4,16 мин, [M+H]+=603.
Стадия 2: [4-(2-фтор-4-йод-фениламино)-6-метокси-1H-индазол-5-ил]-амид циклопропансульфоновой кислоты
Раствор трет-бутилового эфира 5-циклопропансульфониламино-4-(2-фтор-4-йод-фениламино)-6-метокси-индазол-1-карбоновой кислоты (217 мг, 0,36 ммоль) в DCM (5 мл) обрабатывали ТФУ (2 мл) и реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь концентрировали в вакууме и остаток растирали в диэтиловои эфире с получением указанного в заголовке соединения в виде твердого продукта не совсем белого цвета (102 мг, 52 %). LCMS (способ A): RT=10,17 [M+H]+=503. 1H ЯМР (ДМСО-d6, 400 МГц): 12,92 (1H, с), 8,72 (1H, с), 7,60-7,52 (2H, м), 7,34-7,24 (2H, м), 6,71 (1H, с), 6,53 (1H, т, J=8,80 Гц), 3,84 (3H, с), 2,53-2,46 (1H, м), 0,84-0,73 (2H, м), 0,72-0,66 (2H, м).
| название | год | авторы | номер документа |
|---|---|---|---|
| 8-АНИЛИНОИМИДАЗОПИРИДИНЫ И СПОСОБЫ ИХ ИСПОЛЬЗОВАНИЯ | 2008 |
|
RU2498985C2 |
| СОЕДИНЕНИЯ АЗАБЕНЗОФУРАНИЛА И СПОСОБ ИХ ПРИМЕНЕНИЯ | 2007 |
|
RU2448111C2 |
| ИЗОИНДОЛОНЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2009 |
|
RU2495028C2 |
| 5-АНИЛИНОИМИДАЗОПИРИДИНЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2008 |
|
RU2441004C1 |
| АЗАБЕНЗОТИОФЕНИЛЬНЫЕ СОЕДИНЕНИЯ И СПОСОБЫ ПРИМЕНЕНИЯ | 2007 |
|
RU2444524C2 |
| АЗАИНДОЛИЗИНЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2008 |
|
RU2483069C2 |
| ЗАМЕЩЕННЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СУЛЬФОНАМИДНЫЕ СОЕДИНЕНИЯ, ПОЛЕЗНЫЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ TRPA 1 | 2014 |
|
RU2675792C2 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛОПИРИДИНА В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ 1β-ДРЕНЕРГИЧЕСКОГО РЕЦЕПТОРА | 2006 |
|
RU2415855C2 |
| ДИАЗАКАРБАЗОЛЫ И СПОСОБЫ ПРИМЕНЕНИЯ | 2009 |
|
RU2515972C2 |
| АНТАГОНИСТЫ РЕЦЕПТОРА МИНЕРАЛОКОРТИКОИДОВ | 2012 |
|
RU2598842C2 |
Изобретение относится к бициклическим гетероциклам формул I и II, в которых радикалы и символы имеют значения, приведенные в формуле изобретения. Данные соединения обладают ингибирующей активностью в отношении киназы МЕК. Изобретение также относится к фармацевтической композиции для лечения гиперпролиферативного расстройства или воспалительного заболевания, к способу ингибирования аномального роста клеток или лечения гиперпролиферативных нарушений и к способу лечения воспалительных заболеваний у млекопитающего. Кроме того, изобретение относится к применению фармацевтической композиции в приготовлении лекарственного средства для лечения вышеуказанных заболеваний у млекопитающего. 6 н. и 13 з.п. ф-лы, 29 пр.
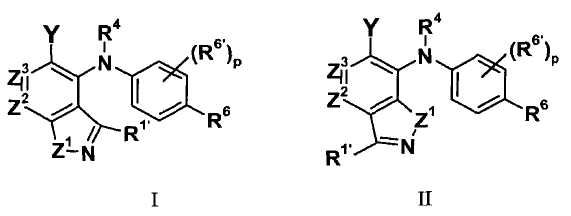
1. Соединение формулы I и II:
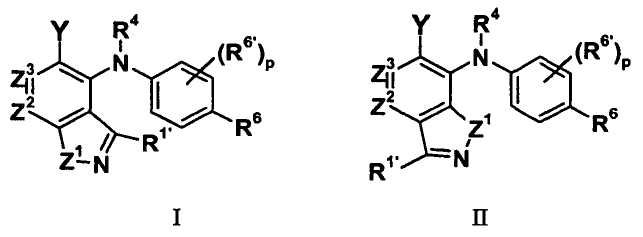
или его фармацевтически приемлемая соль, где:
Z1 представляет собой NR1 или S;
R1 представляет собой H;
R1′ представляет собой H;
Z2 представляет собой CR2 или N;
Z3 представляет собой CR3;
R2 и R3 представляют собой Н;
R4 представляет собой Н или C1-C6алкил;
Y представляет собой W-C(O)-;
W представляет собой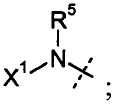
R5 представляет собой H или C1-C12алкил;
X1 выбран из R11′ и-OR11′;
R11 представляет собой Н, C1-C12алкил или тетрагидропиран-4-ил;
или R11′ и R5 вместе с азотом, к которому они присоединены, представляют собой азетидин, замещенный гидроксильной группой;
R6 представляет собой галоген, C3-C5циклоалкил или -(CR19R20)nSR16;
R6′ представляет собой галоген;
p равно 1;
n равно 0, 1, 2 или 3;
указанный алкил R11′ необязательно замещен -(CR19R20)nOR16 или R21;
R16 представляет собой H или C1-C12алкил;
R19 и R20 представляют собой Н;
R21 представляет собой C3-C5-циклоалкил.
2. Соединение по п.1, где Z2 представляет собой N и Z3 представляет собой CR3.
3. Соединение по п.1, где Z2 представляет собой CR2 и Z3 представляет собой CR3.
4. Соединение по п.2 или 3, где Z1 представляет собой NR1.
5. Соединение по п.2 или 3, где Z1 представляет собой S.
6. Соединение по п.4, где X1 выбран из:

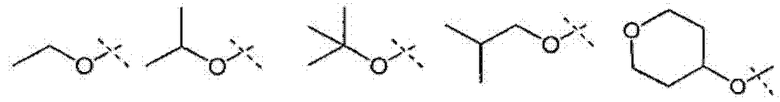
7. Соединение по п.6, где R6 выбран из галогена, -SR16, C3-C4карбоциклила.
8. Соединение по п.7, где R6 представляет собой I, Br, -SMe, C3карбоциклил.
9. Соединение по п.8, где R6′ представляет собой F или Cl.
10. Соединение по п.7, где R4 представляет собой H или метил.
11. Соединение по п.10, где R4 представляет собой H.
12. Соединение по п.10, где R5 представляет собой H или метил.
13. Соединение по п.12, где R5 представляет собой H.
14. Соединение по п.1, выбранное из группы, состоящей из
(2-гидроксиэтокси)-амида 4-(2-фтор-4-иод-фениламино)-1H-индазол-5-карбоновой кислоты;
(2-гидроксиэтокси)-амида 4-(2-фтор-4-иодфениламино)-1H-пиразоло[3,4-b]пиридин-5-карбоновой кислоты;
((S)-2-гидроксипропокси)-амида 4-(2-фтор-4-иодфениламино)-1H-пиразоло[3,4-b]пиридин-5-карбоновой кислоты;
(2-гидрокси-1,1-диметилэтокси)-амида 4-(2-фтор-4-иодфениламино)-1H-пиразоло[3,4-b]пиридин-5-карбоновой кислоты;
((R)-2,3-дигидрокси-пропокси)-амида 4-(2-фтор-4-иодфениламино)-1H-индазол-5-карбоновой кислоты;
(2-гидроксиэтокси)-амида 4-(4-бром-2-фтор-фениламино)-1H-индазол-5-карбоновой кислоты;
((S)-2-гидроксипропокси)-амида 4-(4-бром-2-фтор-фениламино)-1H-индазол-5-карбоновой кислоты;
(2-гидроксиэтокси)-амида4-(2-фтор-4-иод-фениламино)-бензо[d]изотиазол-5-карбоновой кислоты;
(2-гидроксиэтокси)-амида 4-(2-фтор-4-метилсульфанилфениламино)-1H-индазол-5-карбоновой кислоты;
этоксиамида 4-(2-фтор-4-иод-фениламино)-1H-индазол-5-карбоновой кислоты;
(тетрагидропиран-4-илокси)-амида 4-(2-фтор-4-иод-фениламино)-1H-индазол-5-карбоновой кислоты;
циклопропилметоксиамида 4-(2-фтор-4-иод-фениламино)-1H-индазол-5-карбоновой кислоты;
метоксиамида 4-(2-фтор-4-иод-фениламино)-1H-индазол-5-карбоновой кислоты;
[4-(2-фтор-4-иод-фениламино)-1H-индазол-5-ил]-(3-гидрокси-азетидин-1-ил)-метанона;
4-(2-фтор-4-иод-фениламино)-бензо[d]изотиазол-5-карбоновой кислоты ((S)-2-гидроксипропокси)-амида;
((R)-2,3-дигидроксипропокси)амида 4-(2-фтор-4-иод-фениламино)-бензо[d]изотиазол-5-карбоновой кислоты;
((R)-2,3-дигидроксипропокси)амида 7-(2-фтор-4-метилсульфанилфениламино)-бензо[d]изотиазол-6-карбоновой кислоты;
((R)-2,3-дигидрокси-пропокси)амида 7-(4-циклопропил-2-фтор-фениламино)-бензо[d]изотиазол-6-карбоновой кислоты;
((R)-2,3-дигидроксипропокси)амида 7-(4-бром-2-фтор-фениламино)-бензо[d]изотиазол-6-карбоновой кислоты;
(2-гидроксиэтокси)амида 7-(4-бром-2-фтор-фениламино)-бензо[d]изотиазол-6-карбоновой кислоты;
(2-гидроксиэтокси)амида 7-(2-фтор-4-иодфениламино)бензо[d]изотиазол-6-карбоновой кислоты; и
((R)-2,3-дигидроксипропокси)амида 7-(2-фтор-4-иод-фениламино)-бензо[d]изотиазол-6-карбоновой кислоты; или
его фармацевтически приемлемой соли.
15. Фармацевтическая композиция для лечения гиперпролиферативного расстройства или воспалительного заболевания у млекопитающего, содержащая терапевтически эффективное количество соединения по любому из пп.1-14 и фармацевтически приемлемый носитель.
16. Способ ингибирования аномального роста клеток или лечения гиперпролиферативных нарушений у млекопитающего, включающий введение указанному млекопитающему терапевтически эффективного количества фармацевтической композиции по п.15.
17. Способ лечения воспалительного заболевания у млекопитающего, включающий введение указанному млекопитающему терапевтически эффективного количества фармацевтической композиции по п.15.
18. Применение фармацевтической композиции по п.15 в приготовлении лекарственного средства для ингибирования аномального роста клеток или лечения гиперпролиферативного нарушения у млекопитающего.
19. Применение фармацевтической композиции по п.15 в приготовлении лекарственного средства для лечения воспалительного заболевания у млекопитающего.
| RU 2006109599 A, 10.10.2007 | |||
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| Топчак-трактор для канатной вспашки | 1923 |
|
SU2002A1 |

