УРОВЕНЬ ТЕХНИКИ
Рецептор минералокортикоидов (MR) представляет собой ядерный рецептор гормона, который активируется альдостероном и регулирует экспрессию многих генов, участвующих в гомеостазе электролитов и в сердечно-сосудистом заболевании. Повышенный циркулирующий альдостерон повышает кровяное давление через свое действие на натрийурез с потенциально возможными дополнительными эффектами на мозг, сердце и сосудистую сеть. Кроме того, гиперальдостеронизм связан со многими патофизиологическими процессами, приводящими к заболеваниям почек и сердечно-сосудистому заболеванию. Хотя гиперальдостеронизм обычно вызывается продуцирующими альдостерон аденомами, пациенты с резистентной гипертензией часто страдают повышенными уровнями альдостерона, которые часто называют как "Альдостероновый прорыв", в результате увеличений уровня калия в сыворотке или остаточной активности AT1R. Гиперальдостеронизм и альдостероновый прорыв обычно приводят к повышенной активности MR, и было показано, что MR антагонисты являются эффективными как гипотензивные средства, а также в лечении сердечной недостаточности и первичного гиперальдостеронизма.
Кроме того, в висцеральных тканях, таких как почка и кишечник, MR регулирует задержание натрия, экскрецию калия и водный баланс в ответ на альдостерон. Экспрессия MR в мозге также, по-видимому, играет роль в контроле нейронной возбудимости, в регуляции отрицательной обратной связи гипоталамо-гипофизарно-надпочечниковой оси и в когнитивных аспектах поведенческой работы (Castren et al., J. of Neuroendocrinology, 3, 461-66 (1993)).
Эплеренон и спиронолактон - два MR антагониста, показавшие себя эффективными в лечении сердечно-сосудистого заболевания, особенно артериальной гипертензии и сердечной недостаточности (RALES InvestigatorS(1999) The effect of spironolactone on morbidity and mortality in patients with severe heart failure, N. Engl. J. Med., 1999, 341(10):709-717; Pitt B, et al., EPHESUS investigator (2003) Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction, N. Engl. J. Med., 348(14):1309-1321; Funder JW., (2010) Eplerenone in chronic renal disease: the EVALUATE trial, Hypertens. Res., 33(6):539-40.) Moreover, multiple studies have shown that treatment with spironolactone or eplerenone significantly lower systolic blood pressure in mild-moderate, obese, systolic, PHA, and resistant hypertensive patientS(Calhoun DA, et al., (2008) Effectiveness of the selective aldosterone blocker, eplerenone, in patients with resistant hypertension, J. Am. Soc. Hypertens., 2008 Nov-Dec;2(6):462-8; Huang BS, et al., (2010) Central neuronal activation and pressor responses induced by circulating ANG II: role of the brain aldosterone-"ouabain" pathway, Am. J. Physiol. Heart. Circ. Physiol., (2):H422-30; The RALES Investigators. (1996) Effectiveness of spironolactone added to an angiotensin-converting enzyme inhibitor and a loop diuretic for severe chronic congestive heart failure, (The Randomized Aldactone Evaluation Study [RALES]), Am. J. Cardiol., 1996;78:902-907; Pitt B, et al., EPHESUS Investigators, Serum potassium and clinical outcomes in the Eplerenone Post-Acute Myocardial Infarction Heart Failure Efficacy and Survival Study (EPHESUS), Circulation, 2008 Oct 14;118(16):1643-50; Bomback AS et al., (2009), Low-dose spironolactone, added to long-term ACE inhibitor therapy, reduces blood pressure and urinary albumin excretion in obese patients with hypertensive target organ damage, Clin. Nephrol., 72(6):449-56; Williams JS, Hypertension: spironolactone and resistant hypertension, Nat. Rev. Endocrinol., 2010 May;6(5):248-50; Nishizaka MK, et al., The role of aldosterone antagonists in the management of resistant hypertension. Curr Hypertens Rep. 2005 Oct;7(5):343-7. Review; Gaddam K, et al., (2010) Rapid reversal of left ventricular hypertrophy and intracardiac volume overload in patients with resistant hypertension and hyperaldosteronism: a prospective clinical study, Hypertension, 55(5):1137-42; Zannad F, et al., (2010) Rationale and design of the Eplerenone in Mild Patients Hospitalization And SurvIval Study in Heart Failure (EMPHASIS-HF), Eur. J. Heart Fail., 12(6):617-22).
Данные, полученные в доклинических моделях, также позволяют предположить, что MR антагонисты могут быть эффективными в лечении метаболического синдрома и атеросклероза (Takai, S. et al, (2005) Eplerenone inhibits atherosclerosis in nonhuman primates. Hypertension. 46(5):1135-9; Tirosh, A. et al., GK. (2010) Mineralocorticoid receptor antagonists and the metabolic syndrome. Curr Hypertens Rep. 2010 Aug;12(4):252-7).
Кроме того, в опубликованной заявке РСТ WO2002/17895 раскрыто, что антагонисты альдостерона могут быть использованы в лечении пациентов, страдающих одной или более когнитивными дисфункциями, включая, но не ограничиваясь ими, психоз, когнитивные нарушения (такие как расстройства памяти), нарушения настроения (такие как депрессия и биполярное нарушение), тревожные нарушения и изменения личности.
Повышение уровней альдостерона, OR избыточная стимуляция минералокортикоидных рецепторов, связано с несколькими физиологическими нарушениями или патологическими болезненными состояниями, включая синдром Конна, первичный и вторичный гиперальдостеронизм, повышенная задержка натрия, увеличенная экскреция магния и калия (диурез), повышенная задержка воды, гипертензии (изолированная систолическая и комбинированная систолическая/диастолическая), аритмии, миокардиальный фиброз, инфаркт миокарда, синдром Барттера и нарушения, связанные с избыточными уровнями катехоламина. (Hadley, M.E., ENDOCRINOLOGY, 2nd Ed., pp. 366-81, (1988); и Brilla et al., Journal of Molecular and Cellular Cardiology, 25 (5), pp. 563-75 (1993). Соединения и/или фармацевтические композиции, которые действуют как MR антагонисты, должно иметь значение в лечении любого из вышеупомянутых состояний.
Несмотря на значительные терапевтические подвижки в лечении артериальной гипертензии и сердечной недостаточности, текущий стандарт лечения является подоптимальным, и существует чистая невыполненная медицинская потребность в дополнительных терапевтических/фармакологических вмешательствах. Это изобретение обращается к этим потребностям, предоставляя соединения, композиции и способы для лечения или профилактики артериальной гипертензии, сердечной недостаточности, других сердечно-сосудистых нарушений и других альдостероновых нарушений.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к соединениям, которые имеют активность антагониста рецептора минералокортикоида (MR), которые являются ценными фармацевтически активными соединениями для терапии и профилактики заболеваний, например, для лечения опосредуемых альдостероном нарушений, включая сердечно-сосудистое заболевание. Настоящее изобретение относится к соединениям Формулы I
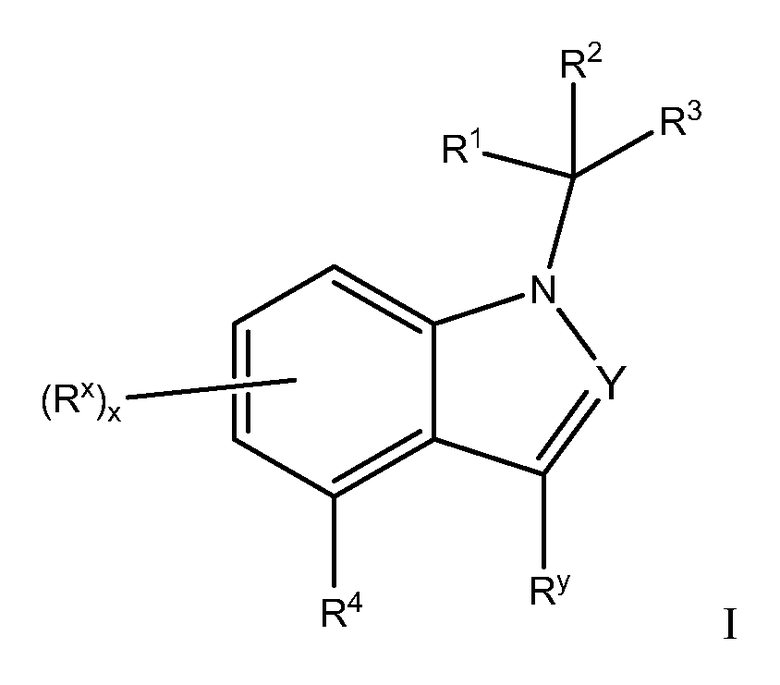
или к их фармацевтически приемлемым солям. Изобретение кроме того относится к способам лечения и профилактики вышеупомянутых заболеваний и к способам получения соединений Формулы I, и к фармацевтическим препаратам, которые включают соединения Формулы I.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Изобретение относится к соединениям Формулы I:
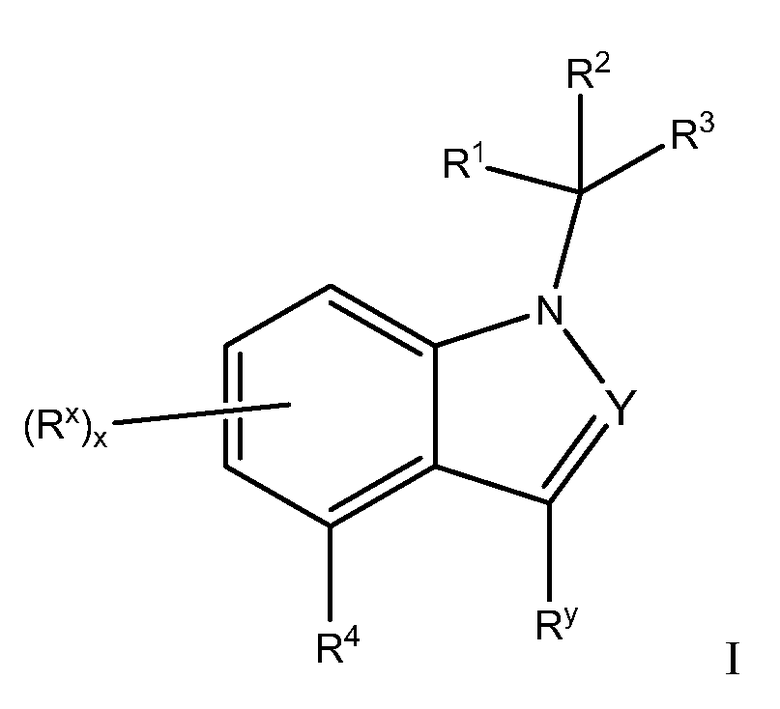
или к их фармацевтически приемлемым солям, в которой
Y обозначает N или CRy;
Каждый Ry независимо обозначает Н или C1-C6 алкил, причем указанный алкил необязательно замещен галогеном или ОН;
Каждый Rx независимо обозначает Н, галоген, OR, C1-C6 алкил, (CR2)0-1CN, C(O)OR11, C3-C10 циклоалкил, NR6COR, NR6SO2R8 или NH2, причем указанные алкил и циклоалкил необязательно замещены 1-3 заместителями, выбранными из галогена, OR и C1-C6 алкила;
Каждый R независимо обозначает Н, CF3, C1-C6 алкил или арил, причем указанные алкил и арил необязательно замещены 1-3 заместителями, выбранными из галогена, арила и C1-C6 алкила;
R1 обозначает:
1) 5-членный гетероарил или гетероциклил, причем указанный гетероарил или гетероциклил необязательно замещены одним - тремя R5,
2) C1-C6 алкил, причем указанный алкил необязательно замещен одним - тремя заместителями, представляющими собой C1-C6 алкил, OR, NR2, CF3, SR, OS(O)2R8, CN или галоген,
3) -(CRa 2)nC(O)OR11,
4) -(CRa 2)nC(O)NRR7,
5) -CN;
6) (CRa 2)0-4C(O)Rc,
7) C3-C10 циклоалкил-R5, или
8) -(CRa 2)nOC(O)Rc;
R2 обозначает:
1) Н,
2) C1-C6 алкил,
3) -(CRb 2)m-C3-C6 циклоалкил,
4) -(CRa 2)m-C(O)OR11,
5) -(CRb 2)m-C2-C6 алкенил,
6) -(CRb 2)m-C2-C6 алкинил,
7) -(CRb 2)m-арил, или
8) -(CRb 2)m-гетероарил;
Где указанные алкил, циклоалкил, алкенил, алкинил, арил и гетероарил необязательно замещены одной - тремя группами, выбранными из R12;
необязательно , R1 и R2 могут быть соединены с образованием циклического кольца, как проиллюстрировано:
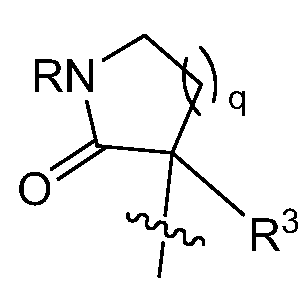 , где целое число q = 1 или 2;
, где целое число q = 1 или 2;
R3 обозначает арил, причем указанный арил необязательно замещен одним - тремя R9;
R4 обозначает
1) Н,
2) -NR6S(O)2R8,
3) C1-C6 алкил,
4) C3-C6 циклоалкил,
5) -N(O)2,
6) -(CH2)0-1-CN,
7) галоген,
8) -C(O)OR11,
9) -NH2,
10) -OR,
11) -(CRa 2)t-SO2R10,
12) -NR6C(O)R10,
13) -NR6C(O)OR10,
14) -NR62,
15) арил,
16) гетероциклил, или
17) гетероарил;
где указанные алкил, циклоалкил, арил, гетероциклил или гетероарил, необязательно, могут быть замещены одним - тремя заместителями, представляющими собой галоген, OR или C1-C6 алкил;
Каждый R5 независимо обозначает Н, OR, CN, арил, гетероарил, C(O)OR11, C(O)NRR7, C1-C6 алкил, CF3 или C3-C10 циклоалкил, где указанные алкил, циклоалкил, арил или гетероарил, необязательно, могут быть замещены одним - тремя галогенами, OR или CF3;
Каждый R6 независимо обозначает Н, C1-C6 алкил, C(O)OR11 или С(O)2R8;
Каждый R7 независимо обозначает
1) Н,
2) C1-C6 алкил, необязательно замещенный 1-3 заместителями, выбранными из галогена, OR, CN, CF3, арила и C3-C10 циклоалкила, где указанные арил и циклоалкил необязательно замещены арилом,
3) C3-C10 циклоалкил, необязательно замещенный один - тремя заместителями, представляющими собой OR, CN, CF3, арил или галоген,
4) -(CRa 2)nC(O)OR11,
5) -(CRa 2)nC(O)Rc, или
6) -(CRa 2)nC(O)NR2;
Каждый R8 независимо обозначает C1-C6 алкил, C3-C10 циклоалкил, NRR7, арил или CF3, причем указанные алкил, арил и циклоалкил необязательно замещены одним - тремя заместителями, представляющими собой галоген, CN, OR или NH2;
Каждый R9 независимо обозначает галоген, CN, CF3, OCF3, C1-C6 алкил, OR, NH2, арил или гетероарил, где указанные алкил, арил или гетероарил, необязательно, могут быть замещены одним - тремя заместителями, представляющими собой галоген, CN, OCF3, OR, C1-C6 алкил или NH2;
Каждый R10 независимо обозначает C1-C6 алкил, арил, или CF3, причем указанный, алкил необязательно замещен 1-3 заместителями, представляющими собой галоген;
Каждый R11 независимо обозначает Н, C1-C6 алкил или арил;
Каждый R12 независимо обозначает галоген, CN, CF3, OCF3, C(O)OR11, C1-C6 алкил, OR, NH2, арил или гетероарил, где указанные алкил, арил или гетероарил необязательно замещены одним - тремя заместителями, представляющими собой галоген, CN, OCF3, OR, C1-C6 алкил или NH2;
Каждый Ra независимо обозначает Н или C1-C6 алкил, причем указанный алкил необязательно замещен одним - тремя заместителями, представляющими собой галоген;
Каждый Rb независимо обозначает Н, OR, галоген или C1-C6 алкил, причем указанный алкил необязательно замещен одним - тремя заместителями, представляющими собой галоген;
Rc обозначает C1-C6 алкил или гетероциклил, причем указанные алкил и гетероциклил необязательно замещены одним - тремя заместителями, представляющими собой галоген, CN, OCF3, OR, C1-C6 алкил или NH2;
m = 0 или 1;
n = 0, 1, 2, 3 или 4;
t = 0, 1, 2 или 3; и
x = 0, 1, 2 или 3.
В одном варианте осуществления, изобретение относится к соединениям Формулы I:
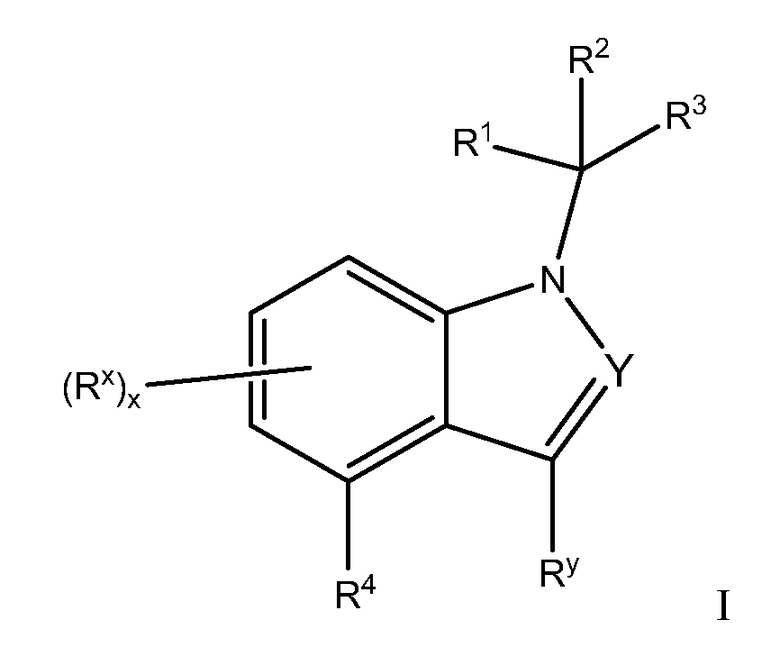
или к их фармацевтически приемлемым солям, в которой
Y обозначает N или CRy;
Каждый Ry независимо обозначает Н или C1-C6 алкил, причем указанный алкил необязательно замещен галогеном или ОН;
Каждый Rx независимо обозначает Н, галоген, OR, C1-C6 алкил, (CR2)0-1CN, OR, C(O)OR11, C3-C10 циклоалкил, NR6COR, NR6SO2R8 или NH2, причем указанные алкил и циклоалкил необязательно замещены 1-3 заместителями, выбранными из галогена, OR и C1-C6 алкила;
Каждый R независимо обозначает Н, CF3, C1-C6 алкил или арил, причем указанные алкил и арил необязательно замещены 1-3 заместителями, выбранными из галогена, арила и C1-C6 алкила;
R1 обозначает:
1) 5-членный гетероарил или гетероциклил, причем указанный гетероарил или гетероциклил необязательно замещены одним - тремя R5,
2) C1-C6 алкил, причем указанный алкил необязательно замещен одним - тремя заместителями, представляющими собой OR, CN или галоген,
3) -(CRa 2)nC(O)OR11,
4) -(CRa 2)nC(O)NRR7,
5) -(CRa 2)1-4C(O)Rc, или
6) -CN;
R2 обозначает:
1) Н,
2) C1-C6 алкил, причем указанный алкил необязательно замещен одним - тремя заместителями, представляющими собой OR, CN или галоген,
3) -(CRb 2)m-C3-C6 циклоалкил, причем указанный циклоалкил необязательно замещен 1-3 заместителями, выбранными из OR, галогена или NH2,
4) -(CRa 2)m-C(O)OR11,
5) -(CRb 2)m-C2-C6 алкенил,
6) -(CRb 2)m-C2-C6 алкинил,
7) -(CRb 2)m-арил, или
8) -(CRb 2)m-гетероарил;
необязательно, R1 и R2 могут быть соединены c образованием циклического кольца, как проиллюстрировано:
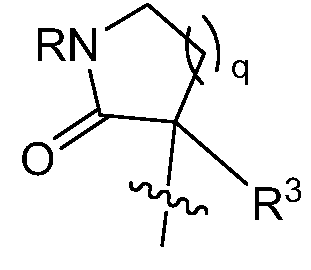 , где целое число q = 1 или 2;
, где целое число q = 1 или 2;
R3 обозначает арил, причем указанный арил необязательно замещен одним - тремя R9;
R4 обозначает
1) Н,
2) -NR6S(O)2R8,
3) C1-C6 алкил,
4) C3-C6 циклоалкил,
5) -N(O)2,
6) -(CH2)0-1-CN,
7) галоген,
8) -C(O)OR11,
9) -NH2,
10) -OR,
11) -(CRa 2)t-SO2R10,
12) -NR6C(O)R10,
13) арил,
14) гетероциклил, или
15) гетероарил;
где указанные алкил, циклоалкил, арил, гетероциклил или гетероарил, необязательно, могут быть замещены одним - тремя заместителями, представляющими собой галоген, OR или C1-C6 алкил;
Каждый R5 независимо обозначает Н, арил, гетероарил, C(O)NRR7, C1-C6 алкил, CF3 или C3-C10 циклоалкил, где указанные алкил, циклоалкил, арил или гетероарил, необязательно, могут быть замещены одним - тремя галогенами, OR или CF3;
Каждый R6 независимо обозначает Н, C1-C6 алкил или С(O)2R8;
Каждый R7 независимо обозначает
1) Н,
2) C1-C6 алкил, необязательно замещенный 1-3 заместителями, выбранными из галогена, OR, CN, CF3, арила и C3-C10 циклоалкила, где указанный арил и циклоалкил необязательно замещены арилом,
3) C3-C10 циклоалкил, необязательно замещенный одним - тремя заместителями, представляющими собой OR, CN, CF3, арил или галоген,
4) -(CRa 2)nC(O)OR11,
5) -(CRa 2)nC(O)Rc, или
6) -(CRa 2)nC(O)NR2;
Каждый R8 независимо обозначает C1-C6 алкил, C3-C10 циклоалкил, арил или CF3, причем указанные алкил, арил и циклоалкил необязательно замещены одним - тремя заместителями, представляющими собой галоген, CN, OR или NH2;
Каждый R9 независимо обозначает галоген, CN, OCF3, C1-C6 алкил, OR, NH2, арил или гетероарил, где указанные алкил, арил или гетероарил, необязательно, могут быть замещены одним - тремя заместителями, представляющими собой галоген, CN, OCF3, OR, C1-C6 алкил или NH2;
Каждый R10 независимо обозначает C1-C6 алкил, арил или CF3, причем указанный алкил необязательно замещен 1-3 заместителями, представляющими собой галоген;
Каждый R11 независимо обозначает Н, C1-C6 алкил или арил;
Каждый Ra независимо обозначает Н или C1-C6 алкил, причем указанный алкил необязательно замещен одним - тремя заместителями, представляющими собой галоген;
Каждый Rb независимо обозначает Н, галоген или C1-C6 алкил, причем указанный алкил необязательно замещен одним - тремя заместителями, представляющими собой галоген;
Rc обозначает гетероциклил;
m = 0 или 1;
n = 0, 1 или 2;
t = 0 или 1; и
x = 0, 1, 2 или 3.
В одном варианте Формулы I, Ry обозначает Н, и все другие переменные имеют значения, определенные ранее в Формуле I.
В другом варианте осуществления, настоящее изобретение относится к соединению Формулы I, как иллюстрируется Формулой II:
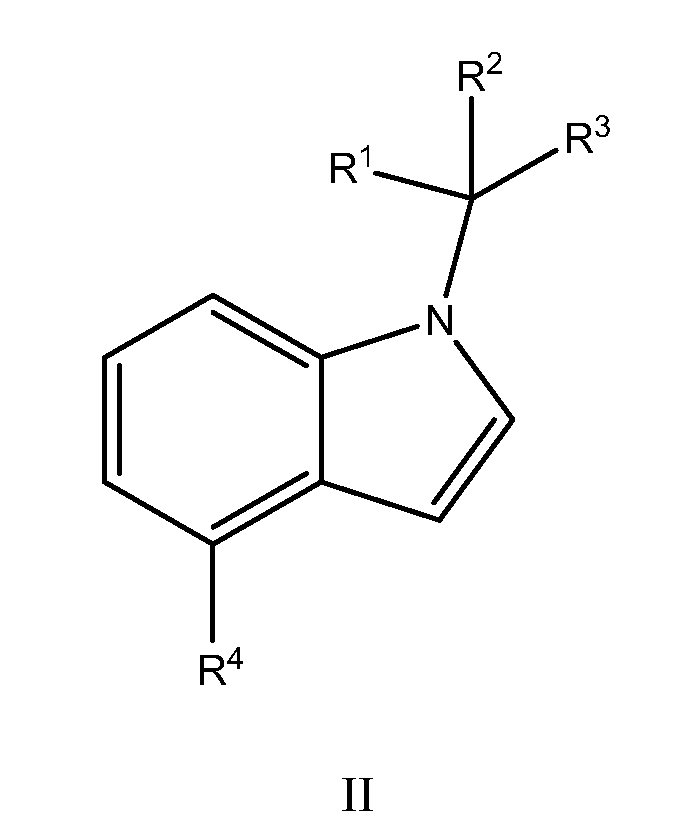
или к его фармацевтически приемлемой соли, в которой:
R2 обозначает:
1) Н,
2) C1-C6 алкил, причем указанный алкил необязательно замещен одним - тремя заместителями, представляющими собой OR, CN или галоген, или
3) -(CRb 2)m-C3-C6 циклоалкил, причем указанный циклоалкил необязательно замещен одним - тремя заместителями, представляющими собой OR, галоген или NH2;
R3 обозначает фенил, причем указанный фенил необязательно замещен одним - тремя R9;
R4 обозначает:
1) -NR6S(O)2R8,
2) -(CRa 2)t-SO2R10, или
3) -NR6C(O)R10;
Каждый R9 независимо обозначает галоген или OR;
и все другие переменные имеют значения, определенные ранее в Формуле I.
В другом варианте осуществления, настоящее изобретение относится к соединению Формулы I, как иллюстрируется Формулой III:
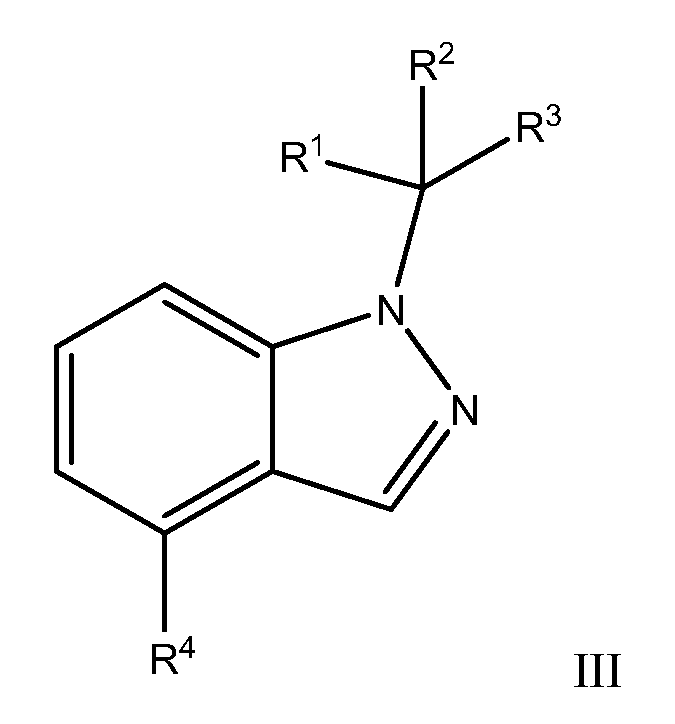
или к его фармацевтически приемлемой соли, в которой:
R2 обозначает:
1) Н,
2) C1-C6 алкил, причем указанный алкил необязательно замещен одним - тремя заместителями, представляющими собой OR, CN или галоген, или
3) -(CRb 2)m-C3-C6 циклоалкил, причем указанный циклоалкил необязательно замещен одним - тремя заместителями, представляющими собой OR, галоген или NH2;
R3 обозначает фенил, причем указанный фенил необязательно замещен одним - тремя R9;
R4 обозначает:
1) -NR6S(O)2R8,
2) -(CRa 2)t-SO2R10, или
3) -NR6C(O)R10;
Каждый R9 независимо обозначает галоген или OR;
и все другие переменные имеют значения, определенные ранее в Формуле I.
В другом варианте осуществления, настоящее изобретение относится к соединению Формулы I, как иллюстрируется Формулой IV:
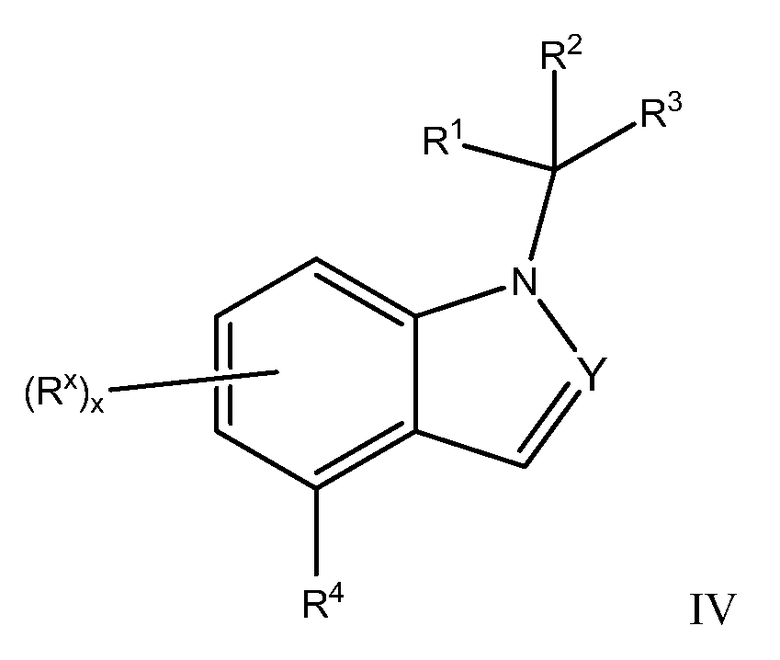
или к его фармацевтически приемлемой соли, в которой:
Y обозначает N или CRy;
Каждый Ry независимо обозначает Н или C1-C6 алкил, причем указанный алкил необязательно замещен галогеном или ОН;
Каждый Rx независимо обозначает Н, галоген, OR или C1-C6 алкил, причем указанный алкил необязательно замещен 1-3 заместителями, выбранными из галогена, OR и C1-C6 алкила;
Каждый R независимо обозначает Н, CF3, C1-C6 алкил или арил, причем указанные алкил и арил необязательно замещены 1-3 заместителями, выбранными из галогена, арила и C1-C6 алкила;
R1 обозначает:
1) 5-членный гетероарил или гетероциклил, причем указанный гетероарил или гетероциклил необязательно замещены одним - тремя R5,
2) C1-C6 алкил, причем указанный алкил необязательно замещен одним - тремя заместителями, представляющими собой C1-C6 алкил, OR, NR2, CF3, SR, OS(O)2R8, CN или галоген,
3) -(CRa 2)nC(O)OR11,
4) (CRa 2)0-4C(O)Rc,
5) C3-C10 циклоалкил-R5, или
6) -(CRa 2)nOC(O)Rc;
R2 обозначает
1) C1-C6 алкил,
2) -(CRb 2)m-C3-C6 циклоалкил,
3) -(CRa 2)m-C(O)OR11,
4) -(CRb 2)m-C2-C6 алкенил, или
5) -(CRb 2)m-C2-C6 алкинил,
Где указанные алкил, циклоалкил, алкенил и алкинил необязательно замещены одной - тремя группами, выбранными из R12;
R3 обозначает фенил, причем указанный фенил необязательно замещен одним - тремя R9;
R4 обозначает
1) -NR6S(O)2R8,
2) C1-C6 алкил,
3) галоген,
4) -C(O)OR11,
5) -NH2,
6) -OR, или
7) -(CRa 2)t-SO2R10,
где указанный алкил, необязательно, может быть замещен одним - тремя заместителями, представляющими собой галоген, OR или C1-C6 алкил;
Каждый R5 независимо обозначает Н, OR, CN, C(O)OR11, C(O)NRR7, C1-C6 алкил, CF3 или C3-C10 циклоалкил, где указанный алкил, циклоалкил, арил и гетероарил, необязательно, могут быть замещены одним - тремя галогенами, OR или CF3;
Каждый R6 независимо обозначает Н, C1-C6 алкил, C(O)OR11 или С(O)2R8;
Каждый R7 независимо обозначает
1) Н,
2) C1-C6 алкил, необязательно замещенный 1-3 заместителями, выбранными из галогена, OR, CN, CF3, арила и C3-C10 циклоалкила, где указанный арил и циклоалкил необязательно замещены арилом,
3) C3-C10 циклоалкил, необязательно замещенный одним - тремя заместителями, представляющими собой OR, CN, CF3, арил или галоген,
4) -(CRa 2)nC(O)OR11,
5) -(CRa 2)nC(O)Rc, или
6) -(CRa 2)nC(O)NR2;
Каждый R8 независимо обозначает C1-C6 алкил, C3-C10 циклоалкил, NRR7 или CF3, причем указанные алкил, арил и циклоалкил необязательно замещены одним - тремя заместителями, представляющими собой галоген, CN, OR или NH2;
Каждый R9 независимо обозначает галоген, CN, CF3, OCF3, C1-C6 алкил, OR или NH2, где указанный алкил, необязательно, может быть замещен одним - тремя заместителями, представляющими собой галоген, CN, OCF3, OR, C1-C6 алкил или NH2;
Каждый R10 независимо обозначает C1-C6 алкил, арил или CF3, причем указанный алкил необязательно замещен 1-3 заместителями, представляющими собой галоген;
Каждый R11 независимо обозначает Н или C1-C6 алкил;
Каждый R12 независимо обозначает галоген, CN, CF3, OCF3, C(O)OR11, C1-C6 алкил, OR, NH2, где указанный алкил, необязательно, может быть замещен одним - тремя заместителями, представляющими собой галоген, CN, OCF3, OR, C1-C6 алкил или NH2;
Каждый Ra независимо обозначает Н или C1-C6 алкил, причем указанный алкил необязательно замещен одним - тремя заместителями, представляющими собой галоген;
Каждый Rb независимо обозначает Н, OR, галоген или C1-C6 алкил, причем указанный алкил необязательно замещен одним - тремя заместителями, представляющими собой галоген;
Rc обозначает C1-C6 алкил или гетероциклил, причем указанные алкил и гетероциклил необязательно замещены одним - тремя заместителями, представляющими собой галоген, CN, OCF3, OR, C1-C6 алкил или NH2;
m = 0 или 1;
n = 0, 1, 2, 3 или 4;
t = 0, 1 или 2; и
x = 0 или 1.
В одном варианте осуществления, изобретение представляет собой соединение из числа следующих соединений
или его фармацевтически приемлемую соль.
В другом варианте осуществления, изобретение представляет собой соединение из числа следующих соединений:
(R)-N-{1-[(2R)-2-(4-хлорфенил)-1-гидроксибутан-2-ил]-1Н-индол-4-ил}метансульфонамид;
N-(1-(1-(4-хлорфенил)-1-(2-цианоциклопропил)пропил)-1Н-индазол-4-ил)метансульфонамид;
N-(1-(1-(4-хлорфенил)-1-(3-этил-1,2,4-оксадиазол-5-ил)пропил)-1Н-индол-4-ил)метансульфонамид;
N-(1-(2-(4-хлорфенил)-1-гидроксибутан-2-ил)-1Н-индазол-4-ил)метансульфонамид;
N-(1-(1-(4-хлорфенил)-1-(1-гидроксициклопропил)пропил)-1Н-индол-4-ил)метансульфонамид;
N-(1-(3-(4-хлорфенил)-2-гидроксипентан-3-ил)-6-фтор-1Н-индол-4-ил)метансульфонамид;
N-(1-(2-(4-хлорфенил)-1-гидроксибутан-2-ил)индолин-4-ил)метансульфонамид;
N-{1-[3-(4-хлорфенил)-2-гидроксипентан-3-ил]-1Н-индазол-4-илметансульфонамид;
N-(1-(2-амино-3-(4-хлорфенил)пентан-3-ил)-1Н-индол-4-ил)метансульфонамид;
1-(1-(3-(4-хлорфенил)-2-гидрокси-2-метилпентан-3-ил)-1Н-индазол-4-ил)-3-метил-сульфонилмочевина;
{4-[(метилсульфонил)амино]-1Н-индол-1-ил}(фенил)метилацетат;
N-(1-(3-(4-хлорфенил)-2-гидрокси-2-метилпентан-3-ил)-6-фтор-1Н-индол-4-ил)метансульфонамид;
N-(1-(3-(4-хлорфенил)-2-гидрокси-2-метилпентан-3-ил)-1Н-индол-4-ил)метансульфонамид;
N-(1-(3-(4-хлорфенил)-2-гидрокси-2-метилпентан-3-ил)-1Н-индазол-4-ил)метансульфонамид;
N-(1-(2-гидрокси-2-метил-3-(4-(трифторметил)фенил)пентан-3-ил)-1Н-индазол-4-ил)метансульфонамид;
N-(1-(2-(4-хлорфенил)-1-метоксибутан-2-ил)-1Н-индол-4-ил)метансульфонамид;
N-(1-(3-(4-хлорфенил)-2-оксопентан-3-ил)-1Н-индазол-4-ил)метансульфонамид;
N-(1-(2-гидрокси-3-(4-(трифторметил)фенил)пентан-3-ил)-1Н-индазол-4-ил)метансульфонамид;
(E)-N-(1-(3-(4-хлорфенил)-1-цианопент-1-ен-3-ил)-6-фтор-1Н-индазол-4-ил)метансульфонамид;
N-(1-(3-(4-хлорфенил)-1-цианопентан-3-ил)-1Н-индазол-4-ил)метансульфонамид;
N-(1-(1-циано-3-(4-метоксифенил)пентан-3-ил)-1Н-индол-4-ил)метансульфонамид;
N-(1-(3-(4-хлорфенил)-1-цианопентан-3-ил)-1Н-индол-4-ил)метансульфонамид;
N-(1-(1-циано-3-(4-(трифторметил)фенил)пентан-3-ил)-1Н-индазол-4-ил)метансульфонамид;
N-(1-(3-(4-хлорфенил)-1-цианопентан-3-ил)-6-фтор-1Н-индол-4-ил)метансульфонамид;
N-(1-(1-циано-3-фенилпентан-3-ил)-6-фтор-1Н-индазол-4-ил)метансульфонамид;
N-(1-(3-(4-Хлорфенил)-1-гидроксипентан-3-ил)-1Н-индол-4-ил)метансульфонамид;
N-(1-(3-(4-хлорфенил)-6-гидроксигексан-3-ил)-1Н-индол-4-ил)метансульфонамид;
N-(1-(3-(4-хлорфенил)пентан-3-ил)-1Н-индол-4-ил)метансульфонамид;
или его фармацевтически приемлемую соль.
В другом варианте осуществления, изобретение представляет собой соединение из числа следующих соединений:
(R)-N-{1-[(2R)-2-(4-хлорфенил)-1-гидроксибутан-2-ил]-1Н-индол-4-ил}метансульфонамид;
N-(1-(1-(4-хлорфенил)-1-(2-цианоциклопропил)пропил)-1Н-индазол-4-ил)метансульфонамид;
N-(1-(1-(4-хлорфенил)-1-(3-этил-1,2,4-оксадиазол-5-ил)пропил)-1Н-индол-4-ил)метансульфонамид;
N-(1-(2-(4-хлорфенил)-1-гидроксибутан-2-ил)-1Н-индазол-4-ил)метансульфонамид;
N-(1-(1-(4-хлорфенил)-1-(1-гидроксициклопропил)пропил)-1Н-индол-4-ил)метансульфонамид;
N-(1-(3-(4-хлорфенил)-2-гидроксипентан-3-ил)-6-фтор-1Н-индол-4-ил)метансульфонамид;
N-(1-(2-(4-хлорфенил)-1-гидроксибутан-2-ил)индолин-4-ил)метансульфонамид;
N-{1-[3-(4-хлорфенил)-2-гидроксипентан-3-ил]-1Н-индазол-4-илметансульфонамид;
N-(1-(2-амино-3-(4-хлорфенил)пентан-3-ил)-1Н-индол-4-ил)метансульфонамид;
1-(1-(3-(4-хлорфенил)-2-гидрокси-2-метилпентан-3-ил)-1Н-индазол-4-ил)-3-метил-сульфонилмочевина;
или его фармацевтически приемлемую соль.
В рамках изобретения, если не указано иное, "алкил" включает насыщенные алифатические углеводородные группы как с разветвленной, так и с прямой цепью, имеющие указанное число атомов углерода. Термин "циклоалкил" означает карбоциклы, не содержащие никаких гетероатомов. Примеры циклоалкила включают циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, декагидронафтил и т.п. Обычно используемые сокращения для алкильных групп используются по всему описанию, например, метил может быть представлен обычными сокращениями, включая "Ме" или
CH3 или символ, который является расширенной связью без определенной концевой группы, например, 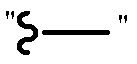 , этил может быть представлен "Et" или
CH2CH3, пропил может быть представлен “Pr” или
CH2CH2CH3, бутил может быть представлен "Bu" или
CH2CH2CH2CH3, и т.д. “C1-6 алкил” (или “C1-C6 алкил”), например, означает алкильную группу с прямой или разветвленной цепью, включая все изомеры, имеющие указанное число атомов углерода. C1-6 алкил включает все из изомеров гексила и пентила, а также н-, изо-, втор- и трет-бутил, н- и изопропил, этил и метил. “C1-4 алкил” означает н-, изо-, втор- и трет-бутил, н- и изопропил, этил и метил. Если никакое число не определено, прямая или разветвленная алкильная группа включает 1-10 атомов углерода. Фраза "C1-C6 алкил, причем указанный алкил может быть замещен одной - тремя" группами относится к алкильным группам, имеющим 0, 1, 2 или 3 заместителя, присоединенные к одному или более атомам углерода. Например, замещенная бутильная группа (C4 алкил) может иметь 1, 2 или 3 заместителя на одном, двух, трех или четырех из атомов углерода бутильной группы. Кроме того, группа "CF3", например, представляет собой метильную группу, имеющую три атома фтора, присоединенные к одному и тому же атому углерода.
, этил может быть представлен "Et" или
CH2CH3, пропил может быть представлен “Pr” или
CH2CH2CH3, бутил может быть представлен "Bu" или
CH2CH2CH2CH3, и т.д. “C1-6 алкил” (или “C1-C6 алкил”), например, означает алкильную группу с прямой или разветвленной цепью, включая все изомеры, имеющие указанное число атомов углерода. C1-6 алкил включает все из изомеров гексила и пентила, а также н-, изо-, втор- и трет-бутил, н- и изопропил, этил и метил. “C1-4 алкил” означает н-, изо-, втор- и трет-бутил, н- и изопропил, этил и метил. Если никакое число не определено, прямая или разветвленная алкильная группа включает 1-10 атомов углерода. Фраза "C1-C6 алкил, причем указанный алкил может быть замещен одной - тремя" группами относится к алкильным группам, имеющим 0, 1, 2 или 3 заместителя, присоединенные к одному или более атомам углерода. Например, замещенная бутильная группа (C4 алкил) может иметь 1, 2 или 3 заместителя на одном, двух, трех или четырех из атомов углерода бутильной группы. Кроме того, группа "CF3", например, представляет собой метильную группу, имеющую три атома фтора, присоединенные к одному и тому же атому углерода.
"Алкенил", если не указано иное, означает углеродные цепи, которые содержат по меньшей мере одну углерод-углеродную двойную связь и которые могут быть прямыми или разветвленными, или их комбинации. Примеры алкенила включают, но не ограничены ими, винил, аллил, изопропенил, пентенил, гексенил, гептенил, 1-пропенил, 2-бутенил, 2-метил-2-бутенил и т.п. Термин "циклоалкенил" означает карбоциклы, не содержащие никаких гетероатомов, имеющие по меньшей мере одну углерод-углеродную двойную связь.
Термин “алкинил” относится к прямому, разветвленному или циклическому углеводородному радикалу, содержащему от 2 до 10 атомов углерода и по меньшей мере одну углерод-углеродную тройную связь. Могут присутствовать до трех углерод-углеродных тройных связей. Таким образом, “C2-C6 алкинил” означает алкинильный радикал, имеющий от 2 до 6 атомов углерода. Алкинильные группы включают этинил, пропинил, бутинил, 3-метилбутинил и так далее. Прямая, разветвленная или циклическая часть алкинильной группы может содержать тройные связи и может быть замещена, если указана замещенная алкинил группа.
"Арил", если не указано иное, означает моно- и бициклические ароматические кольца, содержащие 6-12 атомов углерода. Примеры арила включают, но не ограничены ими, фенил, нафтил, инденил и т.п. "Арил" также включает моноциклические кольца, конденсированные с арильной группой. Примеры включают тетрагидронафтил, инданил и т.п. Предпочтительным арилом является фенил.
"Гетероарил", если не указано иное, означает моно- или бициклическое ароматическое кольцо или кольцевую систему, имеющую от 5 до 10 атомов и содержащую по меньшей мере один гетероатом, выбранный из O, S и N. Примеры включают, но не ограничены ими, пирролил, изоксазолил, изотиазолил, пиразолил, пиридил, пиридинил, оксазолил, оксадиазолил, тиадиазолил, тиазолил, имидазолил, триазолил, тетразолил, фуранил, триазинил, тиенил, пиримидил, пиримидинил, пиридазинил, пиразинил и т.п. Гетероарил также включает ароматические гетероциклические группы, конденсированные в гетероциклы, которые являются неароматическими или частично ароматическими, и ароматические гетероциклические группы, конденсированные с циклоалкильными кольцами. Дополнительные примеры гетероарилов включают, но не ограничены ими, дигидрофуранил, индазолил, тиенопиразолил, имидазопиридазинил, пиразолопиразолил, пиразолопиридинил, имидазопиридинил и имидазотиазолил. Гетероарил также включает такие группы в заряженную форму, например, пиридиний.
"Гетероциклил", если не указано иное, означает 4-, 5- или 6-членное моноциклическое насыщенное кольцо, содержащее по меньшей мере один гетероатом, выбранный из N, S и O, в котором точкой присоединения может быть углерод или азот. Примеры "гетероциклила" включают, но не ограничены ими, азетидинил, пирролидинил, пиперидинил, пиперазинил, морфолинил, имидазолидинил, 2,3-дигидрофуро(2,3-b)пиридил, бензоксазинил и т.п. Этот термин также включает частично ненасыщенные моноциклические кольца, которые не являются ароматическими, такие как 2- или 4-пиридоны, присоединенные через азот или N-замещенные-(1H,3H)-пиримидин-2,4-дионы (N-замещенные урацилы). Гетероциклил, кроме того, включает такие группы в заряженной форме, например, пиперидиний.
"Галоген", если не указано иное, включает фтор, хлор, бром и йод. В одном варианте осуществления, галоген представляет собой фтор или хлор.
Под "оксо" понимают функциональную группу "=O", которая является атомом кислорода, связанным с молекулой через двойную связь, такую как, например, (1) "C=(O)", т.е., карбонильная группа; (2) "S=(O)", то есть, сульфоксидная группа; и (3) "N=(O)", то есть, N-оксидная группа, такая как пиридил-N-оксид.
Если явно не указано иное, замещение названным заместителем возможно на любом атоме в кольце (например, арильном, гетероарильном кольце или насыщенном гетероциклическом кольце), если такое замещение в кольце химически осуществимо и приводит к стабильному соединению. "Стабильное" соединение представляет собой соединение, которое может быть получено и выделено и чья структура и свойства остаются или могут быть сохранены по существу неизменными в течение времени, достаточного для того, чтобы позволить использование соединения в целях, описанных здесь (например, терапевтическое или профилактическое введении пациенту).
Ссылка на соединения структурной формулы I включает соединения других родовых структурных формул, попадающих в рамки Формулы I, включая, но не ограничиваясь ими, Формулу II, Формулу III и/или Формулу IV.
Когда любая переменная (например, R, Ra, Rx и т.д.) встречается больше чем один раз в любом компоненте или в Формуле I, ее определение в каждом случае независимо от ее определения в каждом другом случае. Кроме того, комбинации заместителей и/или переменных допустимы, только если такие комбинации приводят к стабильным соединениям.
В рамках стандартной номенклатуры, используемой на протяжении этого раскрытия, сначала описывается концевая часть определяемой боковой цепи, сопровождаемая смежной функциональной группой к месту присоединения. Например, C1-5 алкилкарбониламино C1-6 алкильный заместитель эквивалентен следующему
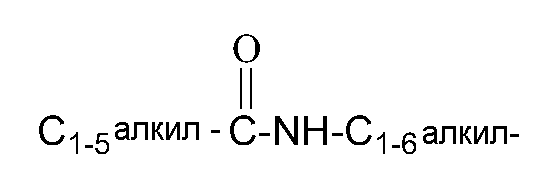
При выборе соединений согласно настоящему изобретению, специалисту будет понятно, что различные заместители, то есть, Ra, Rb, R1, R2 и т.д., должны быть выбраны в соответствии с известными принципами возможности соединения и стабильности химической структуры.
Термин "замещенный" включает множественные степени замещения названным заместителем. Если раскрыты или заявлены множественные группы заместителя, замещенное соединение может быть независимо замещено одной или более раскрытых или заявленных групп заместителя, отдельно или во множественном числе. Независимо замещенный означает, что эти (два или более) заместители могут быть одинаковыми или разными.
Если заместитель или переменная имеют множественные определения, следует понимать, что этот заместитель или переменная определяются как выбираемые из группы, состоящей из обозначенных определений.
Оптические изомеры - Диастереоизомеры - Геометрические изомеры - Таутомеры - Атропизомеры:
Соединения структурной формулы I могут содержать один или несколько центров асимметрии и могу таким образом находиться в форме рацематов и рацемических смесей, отдельных энантиомеров, диастереоизомерных смесей и индивидуальных диастереоизомеров. Настоящее изобретение включает все такие изомерные формы соединений структурной формулы I.
Соединения структурной формулы I могут быть разделены на их индивидуальные диастереоизомеры, например, фракционной кристаллизацией из подходящего растворителя, например, метанола или этилацетата, или их смеси, или хиральной хроматографией с использованием оптически активной стационарной фазы. Абсолютная стереохимия может быть определена Рентгеновской кристаллографией кристаллических продуктов или кристаллических промежуточных соединений, которые дериватизуют, в случае необходимости, с реактивом, содержащим центр асимметрии известной абсолютной конфигурации.
Альтернативно, любой стереоизомер или изомеры соединения общей структурной формулы I могут быть получены стереоспецифическим синтезом с использованием оптически чистых исходных материалов или реактивов известной абсолютной конфигурации.
Если желательно, рацемические смеси соединений могут быть разделены так, чтобы были выделены индивидуальные энантиомеры. Разделение может быть выполнено способами, известными в данной области техники, такими как сочетание рацемической смеси соединений с энантиомерно чистым соединением с образованием диастереоизомерной смеси с последующим разделением индивидуальных диастереоизомеров стандартными методами, такими как фракционная кристаллизация или хроматография. Реакция сочетания часто представляет собой формирование солей с использованием энантиомерно чистых кислоты или основания. Диастереомерные производные могут затем быть превращены в чистые энантиомеры расщеплением добавленного хирального остатка. Рацемическая смесь соединений может также быть разделена непосредственно хроматографическими способами с использованием хиральных стационарных фаз, причем эти способы являются известными в данной области техники.
Для соединений, описанных здесь, которые содержат олефиновые двойные связи, если не указано иное, они включают геометрические изомеры E и Z.
Некоторые из соединений, описанных здесь, могут существовать как таутомеры, которые имеют различные точки присоединения водорода, сопровождаемого одним или более сдвигами двойной связи. Например, кетон и его енольная форма представляют собой кето-енольные таутомеры. Индивидуальные таутомеры, а также их смеси, входят в рамки соединений согласно настоящему изобретению.
В соединениях структурной формулы I, атомы могут показывать их естественную изотопную распространенность, или один или несколько атомов могут быть искусственно обогащены специфическим изотопом, имеющем то же самое атомное число, но атомную массу или массовое число, отличное от атомной массы или массового числа, преимущественно встречающегося в природе. Настоящее изобретение включает все подходящие изотопные вариации соединений структурной формулы I. Например, различные изотопные формы водорода (H) включают протий (1H) и дейтерий (2H, также обозначенный как D). Протий является преобладающим изотопом водорода, обнаруживаемым в природе. Обогащение дейтерием может предоставить определенные терапевтические преимущества, такие как увеличение периода полужизни in vivo или ослабление требований по дозировке, или может дать соединение, пригодное в качестве стандарта для исследования биологических образцов. Изотопно обогащенные соединения в пределах структурной формулы I могут быть получены без дополнительного экспериментирования обычными методиками, известными специалисту в данной области техники, или способами, аналогичными описанным в Схемах и Примерах, приведенных здесь, с использованием подходящих изотопно обогащенных реактивов и/или промежуточных соединений.
Настоящее изобретение включает все стереоизомерные формы соединений Формулы I. Центры асимметрии, которые присутствуют в соединениях Формулы I, могут, независимо от друг друга, иметь конфигурацию S или конфигурацию R. Изобретение включает все возможные энантиомеры и диастереомеры и смеси двух или более стереоизомеров, например, смеси энантиомеров и/или диастереомеров, во всех соотношениях. Таким образом, энантиомеры являются объектом изобретения в энантиомерно чистой форме, как левовращающие и как правовращающие антиподы, в форме рацематов и в форме смесей этих двух энантиомеров во всех соотношениях. В случае изомерии цис/транс изобретение включает как форму цис, так и форму транс, а также смеси этих форм во всех соотношениях. Получение индивидуальных стереоизомеров может быть осуществлено, если желательно, разделением смеси обычными способами, например, хроматографией или кристаллизацией, при помощи стереохимически однородных исходных материалов для синтеза или стереоселективным синтезом. В случае необходимости, перед разделением стереоизомеров может быть выполнена дериватизация. Разделение смеси стереоизомеров может быть осуществлено на стадии соединений Формулы I или на стадии промежуточного соединения в ходе синтеза. Настоящее изобретение также включает все таутомерные формы соединений Формулы I.
Настоящее изобретение включает все атропизомерные формы соединений Формулы I. Атропизомеры представляют собой стереоизомеры, являющиеся следствием затрудненного вращения относительно простых связей, где пространственный барьер для вращения является достаточно высоким, чтобы позволить выделение конформеров. Атропизомеры показывают осевую хиральность. Разделение атропизомеров возможно осуществить способами хирального разделения, такими как селективная кристаллизация.
Соли:
Следует понимать, что, в рамках изобретения, ссылки на соединения структурной формулы I также включают фармацевтически приемлемые соли, а также соли, которые не являются фармацевтически приемлемыми, когда они используются как предшественники свободных соединений или их фармацевтически приемлемых солей или в других синтетических манипуляциях.
Соединения согласно настоящему изобретению могут вводиться в форме фармацевтически приемлемой соли. Термин "фармацевтически приемлемая соль" относится к солям, полученным из фармацевтически приемлемых нетоксичных оснований или кислот, включая неорганические или органические основания и неорганические или органические кислоты. Соли основных соединений, охватываемых термином "фармацевтически приемлемая соль", относятся к нетоксичным солям соединений по изобретению, которые обычно получают, вводя свободное основание в реакцию с подходящей органической или неорганической кислотой. Репрезентативные соли основных соединений согласно настоящему изобретению включают, но не ограничены ими, следующее: ацетат, аскорбат, бензолсульфонат, бензоат, бикарбонат, бисульфат, битартрат, борат, бромид, бутират, камфорат, камфорсульфонат, камсилат, карбонат, хлорид, клавуланат, цитрат, дигидрохлорид, эдетат, эдисилат, эстолат, эзилат, фумарат, глюцептат, глюконат, глутамат, гликолиларсанилат, гексилрезорцинат, гидрабамин, гидробромид, гидрохлорид, гидроксинафтоат, йодид, изотионат, лактат, лактобионат, лаурат, малат, малеат, манделат, мезилат, метилбромид, метилнитрат, метилсульфат, метансульфонат, мукат, напсилат, нитрат, N-метилглюкамин аммониевая соль, олеат, оксалат, памоат (эмбонат), пальмитат, пантотенат, фосфат/дифосфат, полигалактуронат, пропионат, салицилат, стеарат, сульфат, субацетат, сукцинат, таннат, тартрат, теоклат, тиоцианат, тозилат, триэтиодид, валерат и т.п. Кроме того, если соединения по изобретению несут кислотную группу, их подходящие фармацевтически приемлемые соли включают, но не ограничены ими, соли, полученные из неорганических оснований, включая алюминий, аммоний, кальций, медь, железо (3), железо (2), литий, магний, марганец (3), марганец (2), калий, натрий, цинк и т.п. Особенно предпочтительными являются соли аммония, кальция, магния, калия и натрия. Соли, полученные из фармацевтически приемлемых органических нетоксичных оснований, включают соли первичных, вторичных и третичных аминов, циклических аминов, дициклогексил аминов и основные ионообменные смолы, такие как аргинин, бетаин, кофеин, холин, N,N-дибензилэтилендиамин, диэтиламин, 2-диэтиламиноэтанол, 2-диметиламиноэтанол, этаноламин, этилендиамин, N-этилморфолин, N-этилпиперидин, глюкамин, глюкозамин, гистидин, гидрабамин, изопропиламин, лизин, метилглюкамин, морфолин, пиперазин, пиперидин, полиаминные смолы, прокаин, пурины, теобромин, триэтиламин, триметиламин, трипропиламин, трометамин и т.п.
Кроме того, в случае группы карбоновой кислоты (-COOH) или спиртовой группы, присутствующей в соединениях согласно настоящему изобретению, могут использоваться фармацевтически приемлемые сложные эфиры производных карбоновой кислоты, такие как метиловый, этиловый или пивалоилоксиметиловый, или ацильные производные спиртов, такие как O-ацетил, O-пивалоил, O-бензоил и O-аминоацил. Включены такие сложные эфиры и ацильные группы, которые известны в данной области техники как модифицирующие растворимость или гидролитические характеристики для использования в качестве составов замедленного высвобождения или пролекарств.
Сольваты, включая, но не ограничиваясь им, этилацетатный сольват, и в частности, гидраты соединений структурной формулы I также включены в настоящее изобретение.
Если соединения Формулы I одновременно содержат кислотные и основные группы в молекуле, изобретение также включает, в дополнение к упомянутым формам соли, внутренние соли или бетаины (цвиттерионы). Соли могут быть получены из соединений Формулы I обычными способами, которые известны специалисту в данной области техники, например, комбинацией с органической или неорганической кислотой или основанием в растворителе или диспергирующем агенте, или анионным обменом или катионным обменом из других солей. Настоящее изобретение также включает все соли соединений Формулы I, которые, вследствие низкой физиологической совместимости, не являются непосредственно подходящими для использования в фармацевтических препаратах, но которые могут использоваться, например, как промежуточные соединения для химических реакций или для получения физиологически приемлемых солей. Термины "физиологически приемлемая соль(и)" и "фармацевтически приемлемая соль(и)" имеют одно и то же значение и используются здесь взаимозаменяемо.
При необходимости, следующие варианты осуществления могут относиться к структурным формулам I, II, III и/или IV.
В одном варианте осуществления, каждый Rx независимо обозначает Н, галоген, OR, C1-C6 алкил, (CR2)0-1CN, NR6COR, NR6SO2R8 или NH2. В другом варианте осуществления, каждый Rx независимо обозначает Н, галоген, OR, C1-C6 алкил или (CR2)0-1CN. В другом варианте осуществления, каждый Rx независимо обозначает Н или галоген.
В одном варианте осуществления, каждый Ry независимо обозначает Н или C1-C6 алкил. В другом варианте осуществления, каждый Ry обозначает Н.
В одном варианте осуществления, R1 обозначает
1) 5-членный гетероарил или гетероциклил, причем указанный гетероарил или гетероциклил необязательно замещены одним - тремя R5,
2) C1-C6 алкил, причем указанный алкил необязательно замещен одним - тремя заместителями, представляющими собой C1-C6 алкил, OR, NR2, CF3, SR, OS(O)2R8, CN или галоген,
3) -(CRa 2)nC(O)OR11,
4) -(CRa 2)nC(O)NRR7,
5) -CN;
6) (CRa 2)0-4C(O)Rc,
7) C3-C10 циклоалкил-R5, или
8) -(CRa 2)nOC(O)Rc.
В другом варианте осуществления, R1 обозначает
1) 5-членный гетероарил или гетероциклил, причем указанный гетероарил или гетероциклил необязательно замещены одним - тремя R5,
2) C1-C6 алкил, причем указанный алкил необязательно замещен одним - тремя заместителями, представляющими собой C1-C6 алкил, OR, NR2, CF3, SR, OS(O)2R8, CN или галоген,
3) -(CRa 2)nC(O)OR11,
4) (CRa 2)0-4C(O)Rc,
5) C3-C10 циклоалкил-R5, или
6) -(CRa 2)nOC(O)Rc.
В другом варианте осуществления, R1 обозначает
1) 5-членный гетероарил, причем указанный гетероарил необязательно замещен одним - тремя R5,
2) C1-C6 алкил, причем указанный алкил необязательно замещен одним - тремя заместителями, представляющими собой C1-C6 алкил, OR, NR2, CF3, SR, OS(O)2R8, CN или галоген,
3) -(CRa 2)nC(O)OR11,
4) (CRa 2)0-4C(O)Rc, или
5) C3-C10 циклоалкил-R5.
В одном варианте осуществления, R2 обозначает
1) C1-C6 алкил,
2) -(CRb 2)m-C3-C6 циклоалкил,
3) -(CRa 2)m-C(O)OR11,
4) -(CRb 2)m-C2-C6 алкенил,
5) -(CRb 2)m-C2-C6 алкинил,
6) -(CRb 2)m-арил, или
7) -(CRb 2)m-гетероарил;
где указанный алкил, циклоалкил, алкенил, алкинил, арил и гетероарил необязательно замещен одной - тремя группами, выбранными из R12.
В другом варианте осуществления, R2 обозначает 1) C1-C6 алкил, 2) -(CRb 2)m-C3-C6 циклоалкил, 3) -(CRa 2)m-C(O)OR11, 4) -(CRb 2)m-C2-C6 алкенил, или 5) -(CRb 2)m-C2-C6 алкинил, где указанный алкил, циклоалкил, алкенил или алкинил необязательно замещены одной - тремя группами, выбранными из R12. В другом варианте осуществления, R2 обозначает 1) C1-C6 алкил, 2) -(CRb 2)m-C3-C6 циклоалкил, 3) -(CRb 2)m-C2-C6 алкенил или 4) -(CRb 2)m-C2-C6 алкинил, где указанный алкил, циклоалкил, алкенил или алкинил необязательно замещены одной - тремя группами, выбранными из R12. В другом варианте осуществления, R2 обозначает C1-C6 алкил.
В одном варианте осуществления, R3 обозначает фенил, необязательно замещенный одной - тремя группами R9. В другом варианте осуществления, R3 обозначает фенил, необязательно замещенный одним - тремя галогенами, OR, CF3 или C1-C6 алкильными группами.
В одном варианте осуществления, R4 обозначает
1) -NR6S(O)2R8,
2) C1-C6 алкил,
3) C3-C6 циклоалкил,
4) -N(O)2,
5) -(CH2)0-1-CN,
6) галоген,
7) -C(O)OR11,
8) -NH2,
9) -OR,
10) -(CRa 2)t-SO2R10,
11) -NR6C(O)R10,
12) -NR6C(O)OR10,
13) -NR62,
где указанный алкил и циклоалкил, необязательно, могут быть замещены одним - тремя заместителями, представляющими собой галоген, OR или C1-C6 алкил.
В другом варианте осуществления, R4 обозначает 1)-NR6S(O)2R8, 2) C1-C6 алкил, 3) C3-C6 циклоалкил, 4)-C(O)OR11, 5) -(CRa 2)t-SO2R10, 6)-NR6C(O)R10, 7)-NR6C(O)OR10, или 8) -NR6 2, где указанный алкил и циклоалкил, необязательно, могут быть замещены одним - тремя заместителями, представляющими собой галоген, OR или C1-C6 алкил. В другом варианте осуществления, R4 обозначает 1)-NR6S(O)2R8, 2) -(CRa 2)t-SO2R10, или 3) -NR6 2. В другом варианте осуществления, R4 обозначает -NR6S(O)2R8.
В одном варианте осуществления, t = 0, 1, 2 или 3. В другом варианте осуществления, t = 0, 1 или 2.
В одном варианте осуществления, x = 0, 1, 2 или 3. В другом варианте осуществления, x = 0, 1 или 2. В другом варианте осуществления, x = 0 или 1.
Настоящее изобретение также относится к способам получения соединений Формулы I, которые описаны в следующей части описания, с помощью которого соединения по изобретению могут быть получены.
Соединения Формулы I согласно изобретению являются конкурентными антагонистами минералокортикоидного рецептора (MR), и они поэтому представляют собой средства, пригодные для терапии и профилактики нарушений, связанных с увеличенными уровнями альдостерона. Способность соединений Формулы I оказывать антагонистическое действие в отношении MR может быть исследована, например, в тесте активности, описанном ниже.
Один аспект изобретения, который представляет интерес, относится к соединению в соответствии с формулой I или к его фармацевтически приемлемой соли для применения в способе лечения человека или животных путем терапии.
Другой аспект изобретения, который представляет интерес, относится к соединению в соответствии с формулой I или к его фармацевтически приемлемой соли для применения в качестве антигипертензивного средства у человека или животного.
Другой аспект изобретения, который представляет интерес, представляет собой способ лечения сердечно-сосудистого заболевания, сердечной недостаточности, гипертензии, атеросклероза, первичного гиперальдостеронизма или связанного с ним состояния у пациента-человека, включающий введение терапевтически эффективного количества соединения формулы I или его фармацевтически приемлемой соли.
Другой аспект изобретения, который представляет интерес, относится к способу лечения метаболического синдрома у млекопитающего, включающему введение терапевтически эффективного количества соединения формулы I или его фармацевтически приемлемой соли.
Другой аспект изобретения, который представляет интерес, относится к способу лечения физиологического или патологического заболевания, выбранного из синдрома Конна, первичного и вторичного гиперальдостеронизма, увеличенной задержки натрия, увеличенной экскреции магния и калия (диурез), увеличенной задержки воды, гипертензии (изолированной систолической и комбинированной систолической/диастолической), аритмий, миокардиального фиброза, инфаркта миокарда, синдрома Барттера и нарушений, связанных с избыточными уровнями катехоламина у пациента-человека, включающий введение пациенту терапевтически эффективного количества соединения формулы I или его фармацевтически приемлемой соли.
Другой аспект изобретения, который представляет интерес, представляет собой способ лечения отказа почек у пациента-человека, включающий введение терапевтически эффективного количества соединения формулы I или его фармацевтически приемлемой соли.
Другой аспект изобретения, который представляет интерес, представляет собой способ лечения гипертензии, включая, но не ограничиваясь ими, эссенциальную гипертензию, резистентную гипертензию, систолическую гипертензию, легочную артериальную гипертензию и т.п.
Дополнительно, другой аспект изобретения представляет собой способ лечения гипертензии у страдающего ожирением животного или человека.
Дополнительно, другой аспект изобретения представляет собой способ лечения гипертензии у страдающего диабетом животного или человека.
Соединения Формулы I и их фармацевтически приемлемые соли могут вводиться животным, предпочтительно млекопитающим, и в особенности человеку, как фармацевтические средства сами по себе, в смесях друг с другом или в форме фармацевтических препаратов. Термин "пациент" включает животных, предпочтительно млекопитающих, и особенно человека, которые используют активные средства по изобретению для профилактики или лечения медицинского состояния. Введение лекарственного средства пациенту включает как самостоятельное введение, так и введение пациенту другим человеком. Пациенту может требоваться лечение существующего заболевания или медицинского состояния, или может требоваться профилактическое лечение для предотвращения или уменьшения риска указанного заболевания или медицинского состояния.
Объектом настоящего изобретения поэтому также являются соединения Формулы I и их фармацевтически приемлемые соли для применения в качестве фармацевтических средств, их применение для антагонистического действия в отношении минералокортикоидных рецепторов и в особенности их применение в терапии и профилактике вышеупомянутых синдромов, а также их применение для получения лекарственных средств для этих целей.
Термины "терапевтически эффективное количество" и подобные описания, такие как "количество, эффективное для лечения", означают количество фармацевтического лекарственного средства, которое вызывает биологический или медицинский ответ ткани, системы, животного или человека, который изыскивается исследователем, ветеринаром, врачом или другим клиническим специалистом. Термины "профилактически эффективное количество" и подобные описания, такие как "количество, эффективное для профилактики", означает количество фармацевтического лекарственного средства, которое предотвращает или уменьшает риск биологического или медицинского события, которое исследователь, ветеринар, врач или другой клинический специалист желает предотвратить в ткани, системе, у животного или человека. Режим дозировки с использованием соединения согласно настоящему изобретению выбирают в соответствии с различными факторами, включая тип, вид, возраст, массу тела, сексуальное и медицинское состояние пациента; серьезность состояния, подлежащего лечению; потенциал соединения, выбранного для введения; путь введения; и почечную и печеночную функцию пациента. Рассмотрение этих факторов лежит в пределах области знаний клинического врача обычной квалификации, относящихся к определению терапевтически эффективного или профилактически эффективного количества дозировки для предотвращения, противодействия или остановки развития состояния. Следует понимать, что особая суточная доза может одновременно быть и терапевтически эффективным количеством, например, для лечения гипертензии, и профилактически эффективным количеством, например, для профилактики инфаркта миокарда.
Кроме того, объектом настоящего изобретения являются фармацевтические препараты (или фармацевтические композиции), которые включают в качестве активного компонента эффективную дозу по меньшей мере одного соединения Формулы I и/или его фармацевтически приемлемой соли и обычный фармацевтически приемлемый носитель, то есть, один или более фармацевтически приемлемых носителей и/или добавок.
Таким образом, объектом изобретения является, например, указанное соединение и его физиологически или фармацевтически приемлемые соли для применения в качестве фармацевтических средств, фармацевтических препаратов, которые включают в качестве активного компонента эффективную дозу указанного соединения и/или его физиологически (или фармацевтически) приемлемой соли и обычный фармацевтически приемлемый носитель, и применения указанного соединения и/или его физиологически (или фармацевтически) приемлемой соли в терапии или профилактике вышеупомянутых синдромов, а также их применение для получения лекарственных средств для этих целей.
Фармацевтические средства согласно изобретению могут вводиться перорально, например, в форме пилюль, таблеток, лакированных таблеток, таблеток с сахарным покрытием, гранул, твердых и мягких желатиновых капсул, водных, спиртовых или масляных растворов, сиропов, эмульсий или суспензий, или ректально, например, в форме суппозиториев. Введение может также быть осуществлено парентерально, например, подкожно, внутримышечно или внутривенно, в форме растворов для инъекции или инфузии. Другими подходящими формами введения являются, например, чрескожное или топическое введение, например, в форме мазей, тинктур, спреев или трансдермальных терапевтических систем, или введение ингаляцией в форме назальных спреев или аэрозольных смесей, или, например, микрокапсул, имплантатов или прутков. Предпочтительная форма введения зависит, например, от заболевания, которое подлежит лечению, и его серьезности.
Количество активного соединения Формулы I и/или физиологически (или фармацевтически) приемлемых солей в фармацевтических препаратах обычно составляет от 0,2 до 700 мг, предпочтительно от 1 до 500 мг, на дозу, но в зависимости от типа фармацевтического препарата оно может также быть более высоким. Фармацевтические препараты обычно включают от 0,5 до 90 весовых процентов соединений Формулы I и/или их физиологически (или фармацевтически) приемлемых солей. Получение фармацевтических препаратов может быть осуществлено таким образом, который является известным per se. С этой целью одно или более соединений Формулы I и/или их фармацевтически приемлемых солей, вместе с одним или более твердыми или жидкими фармацевтическими веществами носителя и/или добавками (или вспомогательными веществами) и, если желательно, в комбинации с другими фармацевтически активными соединениями, имеющими терапевтическое или профилактическое действие, составляют в подходящую форму для введения или лекарственную форму, которая может затем использоваться в качестве фармацевтического средства в медицине или ветеринарии.
Для получения пилюль, таблеток, таблеток с сахарным покрытием и твердых желатиновых капсул возможно использовать, например, лактозу, крахмал, например, кукурузный крахмал, или производные крахмала, тальк, стеариновую кислоту или ее соли и т.д. Носители для мягких желатиновых капсул и суппозиториев, например, жиры, воски, полутвердые и жидкие многоатомные спирты, натуральные или гидрогенизированные масла и т.д. Подходящими носителями для получения растворов, например, растворов для инъекций, или эмульсий или сиропов являются, например, вода, физиологический раствор хлорида натрия, спирты, такие как этанол, глицерин, многоатомные спирты, сахароза, инвертированный сахар, глюкоза, маннит, растительные масла и т.д. Также возможно лиофилизировать соединения Формулы I и их физиологически (или фармацевтически) приемлемые соли и использовать полученные лиофилизаты, например, для получения препаратов для инъекции или инфузии. Подходящими носителями для микрокапсул, имплантатов или штифтов являются, например, сополимеры гликолевой кислоты и молочной кислоты.
Помимо активных соединений и носителей, фармацевтические препараты могут также содержать обычные добавки, например, наполнители, разрыхлители, связующие, лубриканты, смачивающие вещества, стабилизаторы, эмульгаторы, диспергирующие агенты, консерванты, подсластители, красители, вкусовые агенты, ароматизаторы, загустители, разбавители, буферные вещества, растворители, солюбилизаторы, средства для эффекта депо, соли для изменения осмотического давления, средства для получения покрытий или антиоксиданты.
Дозировка вводимого активного соединения Формулы I и/или его фармацевтически приемлемой соли зависит от индивидуального случая и, как обычно, может быть адаптирована к индивидуальным обстоятельствам для достижения оптимального эффекта. Таким образом, она зависит от природы и серьезности нарушения, которое подвергают лечению, а также от пола, возраста, массы и индивидуальной чувствительности человека или животного, получающего лечение, от эффективности и продолжительности действия используемых соединений, от того, является ли терапия острой или хронической или профилактической, или от того, вводят ли другие активные соединения в дополнение к соединениям Формулы I. В общем, суточная доза приблизительно от 0,01 до 100 мг/кг, предпочтительно от 0,01 до 10 мг/кг, в частности, от 0,3 до 5 мг/кг (в каждом случае мг на кг массы тела) является подходящей для введения взрослому, имеющему массу тела приблизительно 75 кг, для получения желаемых результатов. Суточная доза может вводиться в единственной дозе или, в особенности когда вводят большие количества, может быть разделена на несколько, например, две, три или четыре индивидуальных дозы. В некоторых случаях, в зависимости от индивидуальной реакции, может быть необходимо отклониться вверх или вниз от данной суточной дозы.
Соединения Формулы I связываются с минералокортикоидным рецептором и противодействуют биологическим действиям альдостерона и кортизола. Из-за этого свойства, кроме использования в качестве фармацевтически активных соединений в медицине и ветеринарии, они могут также быть использованы как научный инструмент или как вспомогательное вещество для биохимических исследований, в которых требуется такой эффект на минералокортикоидный рецептор, в также в диагностических целях, например, в диагностике in vitro образцов клеток или образцов тканей. Соединения Формулы I и их соли могут кроме того использоваться, как уже упомянуты выше, как промежуточные соединения для получения других фармацевтически активных соединений.
Одно или несколько дополнительных фармакологически активных средств могут вводиться в комбинации с соединением Формулы I. Дополнительное активное средство (или средства) означает фармацевтически активное средство (или средства), отличное от соединения Формулы I. Вообще, любое подходящее дополнительное активное средство или средства, включая, но не ограничиваясь ими, антигипертензивные средства, антиатеросклеротические средства, такие как липидмодифицирующее соединение, антидиабетические средства и/или средства против ожирения, может использоваться в любой комбинации с соединением Формулы I в общем составе дозировки (комбинация лекарственных средств с фиксированной дозой) или может вводиться пациенту в одном или нескольких отдельных составах дозировки, которая позволяет осуществлять параллельное или последовательное введение активных средств (совместное введение отдельных активных средств).
Вышеупомянутые соединения находят также применение в комбинации с другими фармакологически активными соединениями. Дополнительные активные соединения, которые могут использоваться в комбинации с соединениями согласно настоящему изобретению, как при совместном введении, так и в фиксированной комбинации, включают, но не ограничены ими, ингибиторы ангиотензинпревращающего фермента (например, алацеприл, беназеприл, каптоприл, церонаприл, цилазаприл, делаприл, эналаприл, эналаприлат, фосиноприл, имидаприл, лизиноприл, мовелтиприл, периндоприл, квинаприл, рамиприл, спираприл, темокаприл или трандолаприл), антагонисты рецептора ангиотензина II (например, лосартан, валсартан, кандесартан, олмесартан, телмесартан), ингибиторы нейтральной эндопептидазы (например, тиорфан и фосфорамидон), антагонисты альдостерона, ингибиторы ренина (например, карбамидные производные ди- и трипептидов (См. патент США 5116835), аминокислоты и производные (Патенты США 5095119 и 5104869), аминокислотные цепи, связанные непептидными связями (Патент США 5114937), ди- и трипептидные производные (Патент США 5106835), пептидиламинодиолы (Патенты США 5063208 и 4845079) и пептидил-бета-аминоациламинодиол карбаматы (Патент США 5089471); также различные другие пептидные аналоги как раскрыто в следующих Патентах США 5071837; 5064965; 5063207; 5036054; 5036053; 5034512 и 4894437, и малые молекулы-ингибиторы ренина (включая диолсульфонамиды и сульфинилы (Патент США 5098924), производные N-морфолино (Патент США 5055466), N-гетероциклические спирты (Патент США 4885292) и пирролимидазолоны (патент США 5075451); также пепстатиновые производные (патент США 4980283) и фтор- и хлор-производные статон-содержащих пептидов (Патент США 5066643), эналкреин, RO 42-5892, A 65317, CP 80794, ES 1005, ES 8891, SQ 34017, алискирен (2(S),4(S),5(S),7(S)-N-(2-карбамоил-2-метилпропил)-5-амино-4-гидрокси-2,7-диизопропил-8-[4-метокси-3-(3-метоксипропокси)-фенил]-октанамид гемифумарат) SPP600, SPP630 и SPP635), антагонисты рецептора эндотелина, сосудорасширяющие средства, блокаторы кальциевых каналов (например, амлодипин, нифедипин, верапармил, дилтиазем, галлопамил, нилудипин, нимодипины, никардипин), активаторы калиевых каналов (например, никорандил, пинацидил, кромакалим, миноксидил, априлкалим, лопразолам), диуретики (например, гидрохлортиазид, хлорталидон, фуросемид), симпатолитики, бета-адреноблокаторы (например, пропранолол, атенолол, бисопролол, карведилол, метопролол или метопролол тартрат), альфа-адреноблокаторы (например, доксазоцин, празоцин или альфа-метилдофа) центральные альфа-адренергические агонисты, периферические сосудорасширяющие средства (например, гидралазин), липидпонижающие средства (например, ниацин, ингибиторы HMG Co-A редуктазы), средства, изменяющие метаболизм, включая сенсибилизаторы инсулина и родственные соединения (например, мураглитазар, глипизид, метформин, розиглитазон), или с другими лекарственными средствами, пригодными для профилактики или лечения вышеупомянутых заболеваний, включая нитропруссид и диазоксид.
Примеры других активных ингредиентов, которые могут вводиться в комбинации с соединением Формулы I, отдельно или в той же самой фармацевтической композиции, включают, но не ограничены ими:
(a) агонисты и частичные агонисты PPAR гамма, включая как глитазоны, так и не-глитазоны (например, троглитазон, пиоглитазон, энглитазон, МСС-555, розиглитазон, балаглитазон, нетоглитазон, T-131, LY-300512, LY-818 и соединения, раскрытые в WO02/08188, WO2004/020408 и WO2004/020409.
(b) бигуаниды, такие как метформин и фенетилбигуанид;
(c) ингибиторы протеинтирозинфосфатазы-1B (PTP-1B);
(d) ингибиторы дипептидил пептидазы IV (DPP-4), такие как ситаглиптин, саксаглиптин, вилдаглиптин и алоглиптин;
(e) инсулин или миметики инсулина;
(f) сульфонилмочевины, такие как толбутамид, глимепирид, глипизид и родственные материалы;
(g) ингибиторы α-глюкозидазы (такие как акарбоза);
(h) средства, которые улучшают липидный профиль пациента, такие как (i) ингибиторы HMG-CoA редуктазы (ловастатин, симвастатин, розувастатин, правастатин, флувастатин, аторвастатин, ривастатин, итавастатин, ZD-4522 и другие статины), (ii) секвестранты желчных кислот (холестирамин, колестипол и диалкиламиноалкильные производные поперечно сшитого декстрана), (iii) агонисты рецептора ниацина, никотиниловый спирт, никотиновая кислота или их соль, (iv) агонисты PPARα, такие как производные фенофиброевой кислоты (гемфиброзил, клофибрат, фенофибрат и безафибрат), (v) ингибиторы абсорбции холестерина, такие как эзетимиб, (vi) ингибиторы ацил CoA:холестерин ацилтрансферазы (ACAT), такие как авасимиб, (vii) ингибиторы CETP, такие как торцетрапиб, и (viii) фенольные антиоксиданты, такие как пробукол;
(i) двойные агонисты PPARα/γ, такие как мураглитазар, тесаглитазар, фарглитазар и JT-501;
(j) агонисты PPARδ, такие как раскрытые в WO97/28149;
(k) соединения против ожирения, такие как фенфлурамин, дексфенфлурамин, фентирамин, субитрамин, орлистат, ингибиторы нейропептида Y Y5, агонисты MC4R, антагонисты/обратные агонисты каннабиноидного рецептора 1 (СВ-1) (например, римонабант и таранабант) и агонисты β3 адренергического рецептора;
(l) ингибиторы кишечного транспортера желчных кислот;
(m) средства, предназначенные для использования при воспалительных состояниях, такие как аспирин, нестероидные противовоспалительные лекарственные средства, глюкокортикоиды, азулфидин и селективные ингибиторы циклооксигеназы-2 (Сох-2);
(n) антагонисты рецептора глюкагона;
(o) GLP-1;
(p) GIP-1;
(q) аналоги и производные GLP-1, такие как эксендины, (например, эксенатид и лируглатид), и
(r) ингибитор 11 β-гидроксистероид дегидрогензы-1 (HSD-1).
Одно или несколько дополнительных активных средств могут вводиться с соединениями, описанными здесь. Дополнительное активное средство или средства могут представлять собой липидмодифицирующие соединения или средства, имеющие другие фармацевтические активности, или средства, которые имеют как липидмодифицирующие эффекты, так и другие фармацевтические активности. Примеры дополнительных активных средств, которые могут использоваться, включают, но не ограничены ими, ингибиторы HMG-CoA редуктазы, которые включают статины в их лактонизированной или дигидрокси открытой кислотной форме и их фармацевтически приемлемые соли и сложные эфиры, включая, но не ограничиваясь ими, ловастатин (см. патент США 4342767), симвастатин (см. патент США 4444784), дигидрокси открытый кислотный симвастатин, особенно его соли аммония или кальция, правастатин, особенно его натриевая соль (см. патент США 4346227), флувастатин, особенно его натриевая соль (см. патент США 5354772), аторвастатин, особенно его соль кальция (см. патент США 5273995), питавастатин, также называемый NK-104 (см. международную публикацию РСТ WO97/23200), и росувастатин, также известный как CRESTOR®; см. патент США 5260440); ингибиторы HMG-CoA синтазы; ингибиторы сквален эпоксидазы; ингибиторы сквален синтетазы (также известные как ингибиторы сквален синтазы), ацил-коэнзим A: ингибиторы холестерин ацилтрансферазы (ACAT), включая селективные ингибиторы ACAT-1 или ACAT-2, а также двойные ингибиторы ACAT-1 и -2; ингибиторы микросомального белка переноса триглицеридов (MTP); ингибиторы эндотелиальной липазы; секвестранты желчных кислот; индукторы рецептора ЛПНП; ингибиторы агрегации тромбоцитов, например, антагонисты рецептора гликопротеида IIb/IIIa фибриногена и аспирин; агонисты гамма-рецепторов, активируемых пролифератором человеческой пероксисомы (PPAR-гамма), включая соединения, обычно называемые глитазонами, например, пиоглитазон и розиглитазон, и включая соединения, включенные в структурный класс, известный как тиазолидин дионы, а также PPAR-гамма агонисты вне структурного класса тиазолидин дионов; агонисты PPAR-альфа, такие как клофибрат, фенофибрат, включая микронизированный фенофибрат, и гемфиброзил; витамин B6 (также известный как пиридоксин) и его фармацевтически приемлемые соли, такие как соль HCl; витамин B12 (также известный как цианокобаламин); фолиевая кислота или ее фармацевтически приемлемая соль или эфир, такой как натриевая соль и соль метилглюкамина; антиоксидантные витамины, такие как витамин С и E и бета-каротин; бета-блокаторы; диуретики (например, хлорталидон, гидрохлортиазид), симпатолитики, антагонисты эндотелина; средства, которые усиливают экспрессию гена ABCA1; соединения, ингибирующие белок переноса сложного эфира холестерила (CETP), соединения, ингибирующие белок, активирующий 5-липооксигеназу (FLAP), соединения, ингибирующие 5-липооксигеназу (5-LO), лиганды фарнезоидного X рецептора (FXR), включая как антагонисты, так и агонисты; лиганды X Рецептора печени (LXR)-альфа, лиганды LXR-бета, бисфосфатные соединения, такие как алендронат натрия; ингибиторы циклооксигеназы-2, такие как рофекоксиб и целекоксиб; и соединения, которые облегчают воспаление сосудов.
Соединения Формулы I могут синтезироваться в соответствии с общими схемами, приведенными ниже, где R1, R2 и R9 имеют значения, определенные выше (если не указано иное), принимая во внимание частные примеры, которые приведены далее. Во всех схемах синтеза и примерах, аббревиатуры используются со следующими значениями, если не указано иное:
ABCA1 = аденозилтрифосфат-связывающая кассета семейства A1
Ac = ацетат, ацетил
AIBN - 2,2'-азобис(2-метилпропионитрил);
Водн. Означает водный раствор;
Ar обозначает Арил;
Bn обозначает бензил;
Вос обозначает трет-бутилкарбамоил;
br обозначает уширенный;
Bu обозначает бутил;
tBu обозначает трет-бутил;
CDI обозначает карбонилдиимидазол;
целит обозначает диатомовую землю Celite®;
CHO обозначает яичник китайского хомячка
cpm обозначает отсчеты в минуту;
°C обозначает градусы Цельсия;
δ обозначает химический сдвиг;
cPr обозначает циклопропил;
DAST обозначает трифторид диэтиламиносеры;
DBU обозначает 1,8-диазабицикло[5,4,0]ундец-7-ен;
DCM обозначает дихлорметан;
d обозначает дублет;
DEA обозначает диэтиламин;
DEAD обозначает диэтилазодикарбоксилат;
DIAD обозначает диизопропилазодикарбоксилат;
DIBAL-Н обозначает диизобутилалюминий гидрид;
DIPEA обозначает диизопропилэтиламин;
DMAP обозначает 4-диметиламинопиридин;
DME обозначает 1,2-диметоксиэтан;
DMF обозначает N,N-диметилформамид;
dppf обозначает 1,1'-бис(дифенилфосфино)ферроцен;
ДМСО обозначает диметилсульфоксид;
ЕА обозначает этилацетат;
EDC обозначает гидрохлорид N-(3-диметиламинопропил)-N'-этилкарбодиимида;
EDCI обозначает гидрохлорид 1-этил-3-(3-диметиламинопропил)карбодиимида;
EDTA обозначает этилендиамин тетрауксусная кислота;
ES-MS обозначает ион-массовую спектроскопию с электрораспылением;
Et обозначает этил;
Et2O обозначает простой диэтиловый эфир;
EtOH обозначает этанол,
EtOAc обозначает этилацетат;
FBS обозначает эмбриональная бычья сыворотка
FXR обозначает фарнезоидный X рецептор;
галоген означает галоген (предпочтительно фтор или хлор),
HATU обозначает O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфат;
HetAr или HAR обозначает гетероарил;
HMG-CoA обозначает 3-гидрокси-3-метил-глутарил коэнзим A;
HMPA обозначает триамид гексаметилфосфорной кислоты;
1НЯМР обозначает протонный ядерный магнитный резонанс;
HOAt обозначает 1-гидрокси-7-азабензотриазол;
HOBt обозначает 1-гидроксибензотриазол;
ВЭЖХ обозначает высокоэффективную жидкостную хроматографию;
Гц обозначает герц;
i обозначает Iso;
iPr обозначает изопропил;
IP обозначает точку перегиба для данной кривой титрования доза-ответ;
J обозначает константу межъядерного связывания;
кг обозначает килограмм;
LiHMDS обозначает бис(триметилсилил)амид лития;
LG обозначает уходящую группу;
LTB4 обозначает лейкотриен B4;
LXR обозначает X рецептор печени;
m обозначает мультиплет;
М обозначает моль;
Ме обозначает метил;
мкг обозначает микрограмм;
MeCN обозначает ацетонитрил;
MeOH обозначает метанол;
МГц обозначает мегагерц;
мм обозначает миллиметр;
мкл обозначает микролитр;
мМ обозначает миллимоль;
мкМ обозначает микромоль;
ммоль обозначает миллимоль;
Ms обозначает метансульфонил;
MS обозначает масс-спектр, и масс-спектр, полученный с помощью ES-MS, может быть обозначен “ES”;
MsCl обозначает метансульфонил хлорид;
m/z обозначает отношение массы к заряду;
n обозначает нормальный;
NBS обозначает N-бромсукцинимид
нм обозначает нанометр;
NMM обозначает N-метилморфолин;
NMO обозначает N-метилморфолин-N-оксид;
NMP обозначает N-метилпирролидин-2-он;
nPr обозначает н-пропил;
p обозначает пентет;
п обозначает пара-;
Pd/C обозначает палладий на углероде;
РЕ обозначает петролейный эфир;
ПЭГ обозначает полиэтиленгликоль;
Ph обозначает фенил;
Phen.H2O обозначает моногидрат 1,10-фенантролина;
Phth обозначает фталимидоил;
PPARα обозначает альфа-рецептор, активируемый пролифератором пероксисомы;
Pr обозначает пропил;
iPr обозначает изопропил;
Пре-ВЭЖХ обозначает препаративную высокоэффективную жидкостную хроматографию;
Pt/C обозначает платину на углероде;
PtO2 обозначает оксид платины (IV);
PyBOP обозначает бензотриаксол-1-ил-окси-трис-пирролидино-фосфоний гексафторфосфат;
q обозначает квартет;
rt или RT обозначает комнатную температуру;
s обозначает синглет; втор. обозначает вторичный;
(s) обозначает твердый
SEC обозначает хроматографию с исключением по размеру;
SEM обозначает 2-(триметилсилил)этоксиметил;
SFC обозначает надкритическую жидкостную хроматографию;
t обозначает триплет;
t-Bu обозначает трет-бутил;
tBuOH обозначает трет-бутанол;
трет.- обозначает третичный;
Tf обозначает трифторметансульфонил;
ТЕА обозначает триэтил амин;
TFA обозначает трифторуксусную кислоту;
TFAA обозначает трифторуксусный ангидрид;
THF обозначает тетрагидрофуран;
TLC обозначает тонкослойную хроматографию;
TMS обозначает триметилсилил;
Ts обозначает тозил;
Ед. обозначает единицы
УФ обозначает ультрафиолет;
x g обозначает изменяющуюся во времени силу тяжести
% вес./вес. - весовой процент данного твердого реагента
СХЕМЫ
Реакционные схемы A-N иллюстрируют способы, используемые в синтезе соединений Формулы I. Все аббревиатуры имеют значения, определенные выше, если не указано иное. На этих Схемах все заместители имеют значения, определенные выше в Формуле I, если не указано иное.
Реакционная схема A иллюстрирует способ синтеза соединений типа 2. В этом способе производное фенилуксусной кислоты типа 1 обрабатывают источником брома, таким как N-бромсукцинимид или подобным, в присутствии подходящего инициатора, такого как AIBN, получая α-бромфенилацетат типа 2. Реакцию проводят в инертном растворителе, таком как тетрахлорметан или бензол, при повышенных температурах от 70°C до температуры кипения растворителя.
Схема A
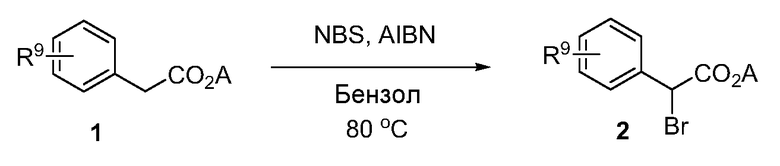
А обозначает Me, Et; jPr или tBu
Реакционная схема B иллюстрирует способ синтеза соединений типа 4. В этом способе замещенный индол или индазол типа 3 обрабатывают подходящим основанием, таким как гидрид натрия в примерах, включающих замещенные индолы, или карбонат цезия в примерах, включающих замещенные индазолы, с последующей реакцией с электрофилами, такими как α-бромфенилацетаты типа 2, получая соединения типа 4. Реакцию осуществляют в полярном апротонном растворителе, таком как DMF или ДМСО, при температурах от 0°C до комнатной температуры. Продукт представляет собой соединение типа 4, которое может быть переработано в соединения согласно настоящему изобретению (I) как описано в последующих схемах.
Схема B
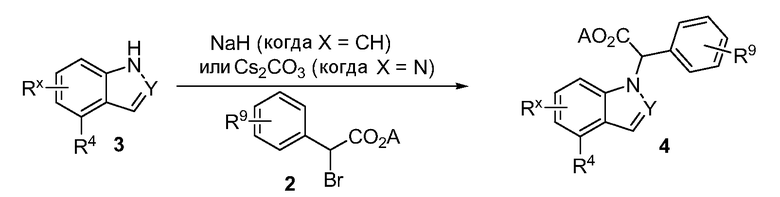
А обозначает Me, Et; jPr или tBu
Реакционная схема C иллюстрирует способ получения соединения типа 6 путем обработки соединений типа 4a, в которых R4 = NO2. В этом способе соединение 4a обрабатывают подходящим катализатором, таким как Pt/C (Pd/C может использоваться, когда соединение 4a не содержит дополнительную реактивную функциональную группу, такую как замещение галогеном), в атмосфере водорода (давление баллона) в подходящем растворителе, таком как этилацетат или подобном, получая аминоиндол типа 5. Альтернативно, нитрогруппа может быть восстановлена обработкой соединений типа 4a хлоридом олова (II) в присутствии сильной кислоты, такой как концентрированная HCl или серная кислота, в протонном растворителе, таком как этанол или подобном, при повышенных температурах от 50°C до температуры кипения растворителя. Продукты типа 5 можно обработать ди-трет-бутилбикарбонатом в основных условиях, таких как условия реакции, известные как условия Schotten-Baumen. Продукт представляет собой соединение типа 6, которое может быть переработано в соединения согласно настоящему изобретению (I) как описано в последующих схемах.
Схема C
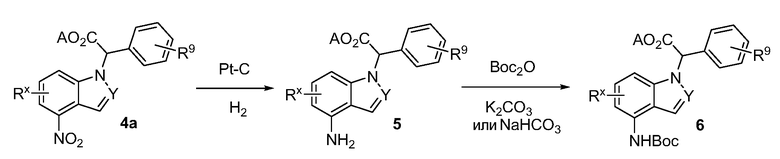
А обозначает Me, Et; jPr или tBu
Реакционная схема D иллюстрирует способ синтеза соединений типа 8. В этом способе субстраты типа 6 обрабатывают подходящим основанием, таким как гидрид натрия, с последующей реакцией соответствующего аниона с электрофилами типа 7, получая желаемый продукт. Большинство условий включает медленное добавление растворов 6 к смеси, содержащей основание, до добавления электрофила (7). Реакцию проводят в полярном апротонном растворителе, таком как DMF или подобном, при температурах от -20°C до 0°C, и продукт реакции представляет собой соединение типа 8, которое может быть переработано в соединения согласно настоящему изобретению (I) как описано в последующих схемах.
Схема D
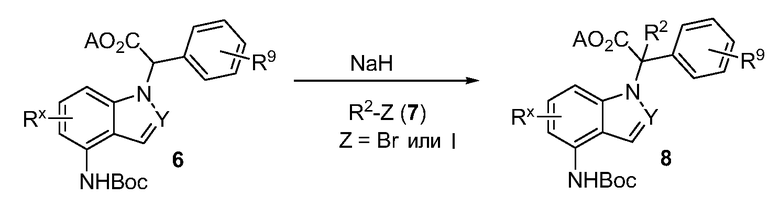
А обозначает Me, Et; jPr или tBu
Реакционная схема E иллюстрирует способ синтеза соединений типа 9. В этом способе соединения типа 8 сначала обрабатывают в сильно кислотных условиях, таких как трифторуксусная кислота или концентрированная HCl, получая аминоиндолы или аминоиндазолы типа 5a, которые затем вводят в реакцию с подходящим электрофильным источником, таким как сульфонил хлорид или ацилгалогенид или ацил ангидрид, в присутствии подходящего основания, такого как 4-метилморфолин или диизопропилэтиламин или подобного. Продукт реакции представляет собой соединение типа 9, которое может быть переработано в соединения согласно настоящему изобретению (I) как описано в последующих схемах.
Схема E
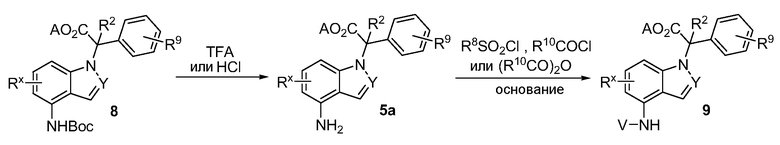
А обозначает Me, Et; jPr или tBu
V обозначает -COR10 или -SO2R8 имеет значения, определенные для R4 для настоящего изобретения (I)
Реакционная схема F иллюстрирует способ синтеза соединений структурной формулы 12 через катализируемую реакцию перекрестной связи органопереходного металла, обычно называемую реакцией Suzuki. В этом способе арильное или гетероарильное соединение типа 10 вводят в реакцию с бороновой кислотой (11) или боронатом (12) в качестве партнера сочетания в присутствии подходящего катализатора на основе палладия, такого как [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (II) или тетракистрифенилфосфинпалладий (0) или подобного, и основания, такого как водный раствор карбоната натрия или водный раствор трехосновного фосфата натрия или подобного (Pure Appl. Chem. 1991, 63, 419-422). Реакцию осуществляют в инертном органическом растворителе, таком как смесь толуол-EtOH или диоксан, при температурах выше комнатной температуры, в течение 3-24 часов. Недавние усовершенствования реакции Suzuki позволили осуществлять этот тип перегруппировки во многих случаях при комнатной температуре (например, см.: J. Am. Chem. Soc. 2000, 122, 4020-4028 и ссылки, цитируемые там). Продукт реакции представляет собой соединение типа 13, которое может быть переработано с получением других соединений согласно настоящему изобретению (I) как описано в последующих схемах.
Схема F
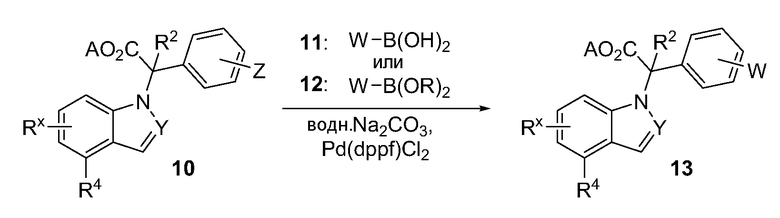
А обозначает Me, Et; jPr или tBu
Z = подходящая уходящая группа, такая как OTf, Br или I
W = арильная или гетероарильная группа, как определено для R9 в формуле I или группа, которая может быть превращена в арильную или гетероарильную группу R9 в формуле I
Реакционная схема G иллюстрирует способ синтеза соединений структурной формулы 15 согласно способам, подобным предварительно описанным в Схеме F. Соединения типа 15, которые получают при входных условиях типа 14, которые содержат дополнительные функциональные группы, могут быть переработаны с получением других соединений согласно настоящему изобретению (I).
Схема G
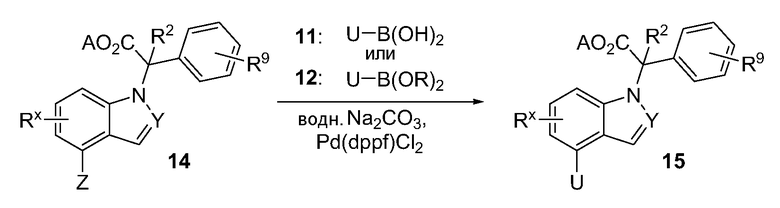
А обозначает Me, Et; jPr или tBu
Z = подходящая уходящая группа, такая как OTf, Br или I
W = арильная или гетероарильная группа, как определено для R4 в формуле I или группа, которая может быть превращена в арильную или гетероарильную группу R4 в формуле I
Реакционная схема Н иллюстрирует способ синтеза соединений структурной формулы 17. В этом способе соединения типа 16 могут быть гидролизованы до карбоновых кислот типа 17 с использованием различных способов, известных специалисту в области органического синтеза. Карбоновая кислота продукта структурной формулы 17 может использоваться в различных способах, известных в органическом синтезе, для получения соединений согласно настоящему изобретению (I).
Схема Н
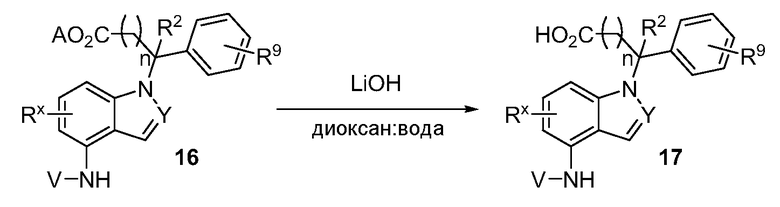
n=0, 1, 2
Y обозначает Me, Et; jPr или tBu
V обозначает -COR10 или -SO2R8, как определено для R4 в формуле (I)
Реакционная схема I иллюстрирует способ синтеза соединений структурной формулы 20. В самом общем случае, соединения типа 18 обрабатывают амином типа 19, получая амид типа 20. Реакцию сочетания амидной связи, иллюстрируемую на реакционной схеме I, проводят в подходящем инертном растворителе, таком как DMF, DCM или подобное, и она может быть выполнена с различными реактивами, подходящих для реакции сочетания амидной связи, такими как HATU, EDC или PyBOP. Условия для реакции сочетания амидной связи, показанной на реакционной схеме I, известны специалисту в области органического синтеза. Такие модификации могут включать, но не ограничены этим, использование основных реактивов, таких как триэтиламин, DIPEA или NMM, или добавление аддитива, такого как HOAt или HOBt. Альтернативно, 19 может быть обработано активированным сложным эфиром или производным хлорангидрида кислоты 18, что также дает 20. Сочетание амидной связи, показанное на реакционной схеме I, обычно проводят при температурах от 0°C до комнатной температуры, иногда при повышенных температурах, и реакцию сочетания проводят в течение от 1 до 24 часов. Продукт реакции представляет собой соединение типа 20, которое может быть переработано с получением других соединений согласно настоящему изобретению (I) как описано в последующих схемах.
Схема I
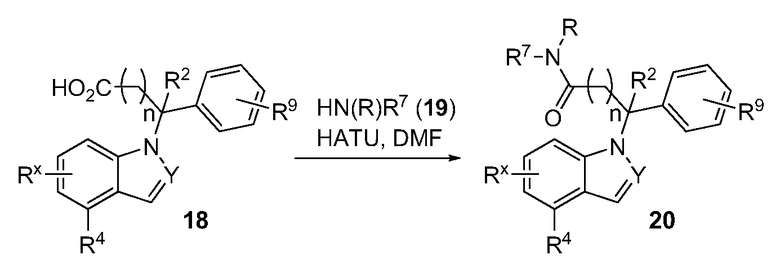
Реакционная схема J иллюстрирует способ синтеза соединений типа 22. В этом способе первичный амид типа 21 обрабатывают дегидрирующим реактивом, таким как хлорангидрид циануровой кислоты или подобным, получая нитрил типа 22. Реакцию осуществляют в инертном растворителе, таком как DCM или DMF, при температуре от 0°C до комнатной температуры. Продукт реакции представляет собой соединение типа 22, которое может быть переработано с получением других соединений согласно настоящему изобретению (I) как описано в последующих схемах.
Схема J
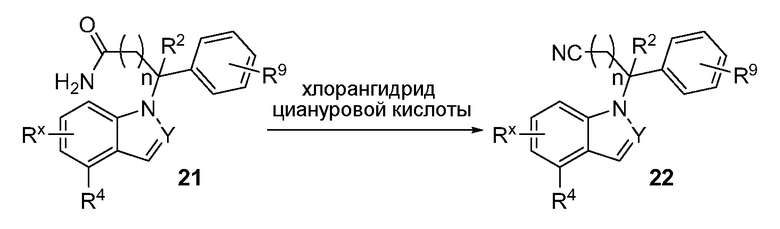
Схема K иллюстрирует в наиболее обобщенном виде, как соединения типа 23 могут быть переработаны в различные гетероциклические производные структурной формулы 24 с использованием известных способов органического синтеза. Частные примеры таких превращений показаны в разделе Примеров.
Такие превращения описаны в 1) Joule, J.A; Mills, K and Smith, G.F. Heterocyclic Chemistry, Chapman & Hall, 1995, 3rd Edn., и цитируемые там ссылки; 2) Katritzky, A.R.; Rees, C.W. (Eds), Comprehensive Heterocyclic Chemistry: The Structure, Reactions, Synthesis, and Uses of Heterocyclic Compounds, Pergamon Press, Oxford, 1984, 8v, и цитируемые там ссылки; и 3) Comprehensive Heterocyclic Chemistry II: Review of the Literature 1982-1995: The Structure, Reactions, Synthesis and Uses of Heterocyclic Compounds, Pergamon Press, New York, 2nd Edn., 1996, 11v, и цитируемые там ссылки. (Comprehensive Heterocyclic Chemistry, vol. 4-6 Pergamon Press, New York, 1984, и цитируемые там ссылки).
Схема K
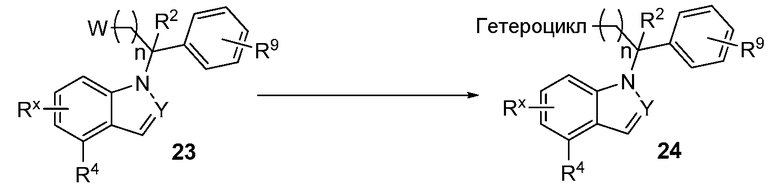
Схема L иллюстрирует способ синтеза соединений типа 26. В этом способе сложный эфир типа 25 обрабатывают сильным восстановителем, таким как литий боргидрид или литий-алюминийгидрид или подобным, в эфирном растворителе, таком как THF или простой диэтиловый эфир, при температуре от 0°C до комнатной температуры. Продукт реакции представляет собой спирт типа 26, который может быть переработан с получением других соединений согласно настоящему изобретению (I) как описано в последующих схемах.
Схема L
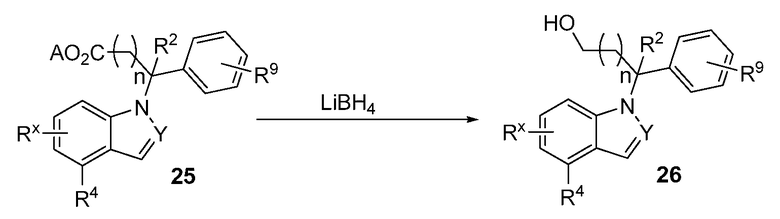
n=0, 1, 2
A = Me или Et
Схема М иллюстрирует способ синтеза соединений типа 28. В этом способе цианоэфир типа 27 обрабатывают избытком восстановителя, такого как боргидрид натрия, в присутствии неорганического ускорителя, такого как хлорид кобальта (II), с получением промежуточного амина (не показан), который подвергается внутримолекулярной циклизации на эфирной группе, что приводит к лактаму типа 28. Этот продукт может быть переработан с получением других соединений согласно настоящему изобретению (I) как описано в последующих схемах.
Схема М
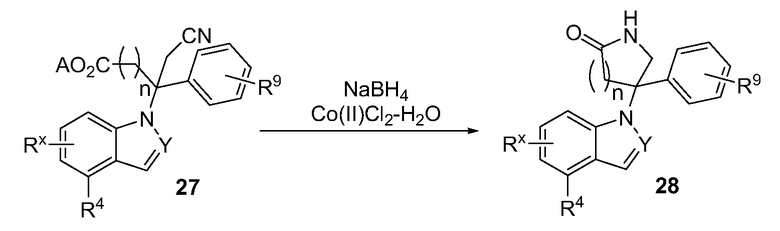
n=0, 1, 2
A = Me или Et
Схема N иллюстрирует способ разделения рацемического соединения структурной формулы 29, в котором отмеченный звездочкой углерод является центром хиральности. Вообще, конечные продукты или промежуточные соединения на пути их получения могут быть разделены с получением энантиомерно чистых соединений, таких как 30 и 31, методиками жидкостной хроматографии с хиральными стационарными фазами или другими подходящими способами, известными в органическом синтезе.
Схема N
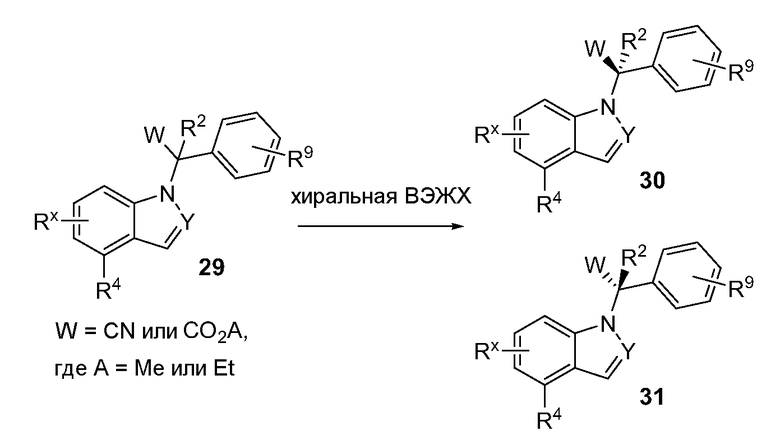
Следующие примеры приведены для того, чтобы изобретение могло бы быть более полно понято. Если не указано иное, исходные материалы являются коммерчески доступными. Они не должны рассматриваться как ограничение изобретения.
РЕПРЕЗЕНТАТИВНЫЕ ПРИМЕРЫ
Следующие примеры приведены для более полной иллюстрации настоящего изобретения, и они не должны рассматриваться как какое-либо ограничение объема изобретения. Если не указано иное:
1) Все операции осуществляли при комнатной температуре или температуре окружающей среды, то есть, при температуре в диапазоне 18-25°C;
2) Реакции обычно осуществляют, используя коммерчески доступные безводные растворители в инертной атмосфере, либо азота, либо аргона;
3) Микроволновые реакции осуществляли, используя систему Biotage Initiator™ или CEM Explorer®;
4) Выпаривание растворителя осуществляли, используя роторный испаритель при пониженном давлении (4,5-30 мм рт.ст.) с температурой ванны до 50°C;
5) Протекание реакций отслеживали с помощью тонкослойной хроматографией (TLC) и/или тандемной высокоэффективной жидкостной хроматографией (ВЭЖХ) с последующей масс-спектроскопией с электрораспылением (MS), называемой здесь LCMS, и любое время реакции приводится только для иллюстрации;
6) Структуру всех конечных соединений проверяли по меньшей мере одной из следующих методик: MS или спектрометрия протонного ядерного магнитного резонанса (1H ЯМР), и чистоту проверяли по меньшей мере одной из следующих методик: TLC или ВЭЖХ;
7) Спектры 1H ЯМР регистрировали на приборе Varian Unity или Varian Inova при 400, 500 или 600 МГц, используя обозначенный растворитель; при перечислении в линию, данные ЯМР находятся в форме значений дельта для главных диагностических протонов, приведенных в частях на миллион (ppm) относительно пиков остаточного растворителя (мультиплетность и число атомов водорода); обычные аббревиатуры, используемые для формы сигнала: c. синглет; д. дублет (видимый); т. Триплет (видимый); м. мультиплет; шир. уширенный; и т.д.;
8) Данные MS регистрировали на блоке Waters Micromass, связанным с помощью интерфейса с прибором для ВЭЖХ Hewlett-Packard (Agilent 1100) и эксплуатирующемся на программном обеспечении MassLynx/OpenLynx; ионизация с электрораспылением использовалась с детекцией положительного (ES+) или отрицательного иона (ES-); и детекцией диодной матрицы.
9) Очистку соединений препаративной ВЭЖХ с обратной фазой осуществляли на системе Gilson, используя колонку YMC-Pack Pro C18 (150×20 мм внутр.д.), элюируя со скоростью 20 мл/мин. с градиентом смеси вода/ацетонитрил (0,1% TFA) (от 5% ацетонитрила до 95% ацетонитрила) или на системе Shimadzu, используя колонку Sunfire Prep C18 OBD 5 мкМ колонка (100×30 мм внутр.д.), элюируя со скоростью 50 мл/мин. с градиентом смеси вода/ацетонитрил (0,1% TFA);
10) Очистку соединений препаративной тонкослойной хроматографией (PTLC) проводили на стеклянных планшетах 20×20 см, покрытых силикагелем, коммерчески доступных от Analtech; или E. Merck.
11) Флэш-хроматографию осуществляли на стеклянной колонке с силикагелем, используя Kieselgel 60, 0,063-0,200 мм (SiO2), или на системе картриджа Biotage SiO2, используя системы Biotage Horizon и Biotage SP-1; или картридже Teledyne Isco SiO2, используя систему CombiFlashRf;
12) Химические символы имеют их обычные значения, и следующие аббревиатуры также использовались: ч (часы), мин. (минуты), об. (объем), вес. (вес), т.к. (температура кипения), т.пл. (температура плавления), л (литр(ы)), мл (миллилитры), г (грамм(ы)), мг (миллиграммы(ы)), моль (моль), ммоль (ммоль), экв. (эквивалент(ы)), IC50 (молярная концентрация, которая приводит к 50% максимального возможного ингибирования), EC50 (молярная концентрация, которая приводит к 50% максимальной возможной эффективности), мкМ (микромоль), нМ (наномоль).
ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ
Промежуточные соединения, используемые в синтезе соединений по изобретению, могут быть получены с использованием следующих процедур. В Таблицах, связанных со следующими Схемами, были искусственно получены соединения, имеющие массовые спектральные данные.
ПРИМЕР I-1A
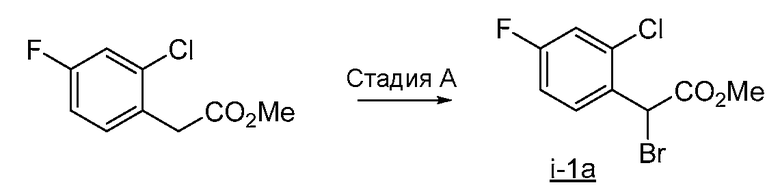
Стадия A: Получение бром(2-хлор-4-фторфенил)метилацетата (i-1a)
2-хлор-4-фторфенилметилацетат (3,15 г, 15,6 ммоль), N-бромсукцинимид (2,77 г, 15,6 ммоль) и AIBN (255 мг, 1,56 ммоль) суспендировали в бензоле (50,0 мл) и дегазировали потоком N2. Полученную смесь нагревали до 80°C в течение 12 часов. После охлаждения до комнатной температуры, смесь частично концентрировали в вакууме и разделяли между простым эфиром и водой. Слои разделяли, и органический слой промывали водой и солевым раствором. Органические фракции высушивали (MgSO4), фильтровали и концентрировали в вакууме. Полученное сырое масло очищали флэш-хроматографией на силикагеле (элюирование в градиенте; 0%-20% EtOAc/гексаны в качестве элюента), получая целевое соединение i-1a.
1H ЯМР (500 МГц, CDCl3): δ 7,82 (дд, 1H, J=6,0, 8,8 Гц), 7,17 (дд, 1H, J=2,6, 8,3 Гц), 7,08 (м, 1H), 5,88 (с, 1H), 3,84 (с, 3H).
Следуя процедурам, подобным описанным на Схеме i-1, можно получить следующие дополнительные соединения, представленные в Таблице i-1:
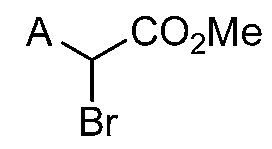
Таблица i-1. Данные 1H ЯМР и m/z (МН)+ исходного иона для соединений.
Для i-1a: бром(2,4-дихлорфенил)метилацетат: 1H ЯМР (500 МГц, CDCl3): δ 7,75 (д, 1H, J=8,6 Гц), 7,42 (с, 1H), 7,33 (д, 1H, J=8,5 Гц), 5,86 (с, 1H), 3,83 (с, 3H).
Для i-1b: бром(4-фторфенил)метилацетат: 1H ЯМР (500 МГц, CDCl3): δ 7,58 (м, 2H), 7,08 (м, 2H), 5,38 (с, 1H), 3,82 (с, 3H).
Для i-1c: бром(4-хлорфенил)метилацетат: 1H ЯМР (500 МГц, CDCl3): δ 7,51 (д, 2H, J=8,5 Гц), 7,37 (д, 2H, J=8,5 Гц), 5,35 (с, 1H), 3,82 (с, 3H).
Для i-1d: бром(4-метоксифенил)метилацетат: 1H ЯМР (500 МГц, CDCl3): δ 7,49 (д, 2H, J=9,0 Гц), 6,88 (д, 2H, J=9,0 Гц), 5,40 (с, 1H), 3,76 (с, 3H), 3,75 (с, 3H).
Для i-1e: бром(4-бромфенил)метилацетат: 1H ЯМР (500 МГц, CDCl3): δ 7,53 (д, 2H, J=8,6 Гц), 7,45 (д, 2H, J=8,5 Гц), 5,33 (с, 1H), 3,82 (с, 3H).
Для i-1f: бром(3,5-дифторфенил)метилацетат: 1H ЯМР (500 МГц, CDCl3): δ 7,13 (м, 2H), 6,84 (м, 1H), 5,30 (с, 1H), 3,84 (с, 3H).
Для i-1g: бром(3,5-дихлорфенил)метилацетат: 1H ЯМР (500 МГц, CDCl3): δ 7,46 (м, 1H), 7,20 (м, 2H), 5,26 (с, 1H), 3,84 (с, 3H).
Для i-1i: бром(3-бромфенил)метилацетат: m/z (ES) 307 (МН)+.
В Таблицах в следующих Примерах были искусственно получены соединения, имеющие массовые спектральные данные.
ПРИМЕР 1
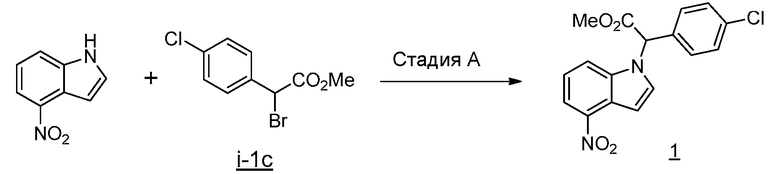
Стадия A: Получение (4-хлорфенил)(4-нитро-1Н-индол-1-ил)метилацетата (1)
Раствор 4-нитроиндола (6,89 г, 42,5 ммоль) в DMF (20 мл) добавляли по каплям к перемешиваемой суспензии гидрида натрия (1,70 г 60%-ой (вес/вес) дисперсии в минеральном масле, 42,5 ммоль) в DMF (100 мл) при 0°C. Через 10 минут добавляли медленно по каплям раствор i-1с (11,2 г, 42,5 ммоль) в DMF (20 мл), и полученную смесь перемешивали при 0°C в течение 3 часов. Реакционную смесь гасили добавлением насыщенного водного раствора хлорида аммония и экстрагировали простым эфиром. Объединенные органические фракции промывали водой и солевым раствором, высушивали (MgSO4), фильтровали и концентрировали в вакууме. Полученное сырое масло очищали флэш-хроматографией на силикагеле (элюирование в градиенте; 0%-20% EtOAc/гексаны в качестве элюента), получая целевое соединение 1. m/z (ES) 345 (МН)+; IP = C оценка значений.
Следуя процедурам, подобным описанным в Примере 1, получали следующие дополнительные соединения, представленные в Таблицах 1 и 1A:
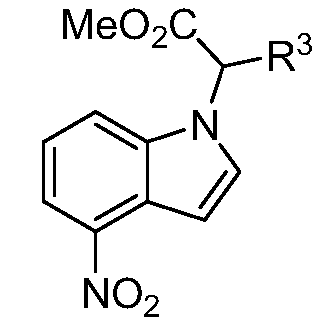
Таблица 1. Данные m/z (МН)+ исходного иона и Первичного IP Теста для соединений.
2: (2-хлор-4-фторфенил)(4-нитро-1Н-индол-1-ил)метилацетат: m/z (ES) 363 (МН)+; IP = B оценка значений.
3: (2,4-дихлорфенил)(4-нитро-1Н-индол-1-ил)метилацетат:
1H ЯМР (500 МГц, CDCl3): δ 8,20 (дд, 1H, J=4,6, 7,9 Гц), 7,72 (д, 1H, J=8,2 Гц), 7,53 (м, 1H), 7,36 (м, 4H), 7,22 (м, 1H), 6,64 (с, 1H), 3,88 (с, 3H). IP = B оценка значений.
4: (3-бромфенил)(4-нитро-1Н-индол-1-ил)метилацетат:
1H ЯМР (500 МГц, CDCl3): δ 8,21 (д, 1H, J=8,0 Гц), 7,66 (д, 1H, J=8,3 Гц), 7,60 (д, 1H, J=8,0 Гц), 7,52 (с, 1H), 7,46 (д, 1H, J=3,3 Гц), 7,35 (м, 3H), 7,27 (д, 1H, J=7,9 Гц), 6,29 (с, 1H), 3,88 (с, 3H). IP = C оценка значений.
5: (4-метоксифенил)(4-нитро-1Н-индол-1-ил)метилацетат:
1H ЯМР (500 МГц, CDCl3): δ 8,18 (д, 1H, J=8,3 Гц), 7,68 (д, 1H, J=8,1 Гц), 7,36 (д, 1H, J=3,4 Гц), 7,32 (д, 2H, J=8,4 Гц), 7,30 (м, 1H), 7,28 (д, 1H, J=3,3 Гц), 6,98 (д, 2H, J=8,7 Гц), 6,26 (с, 1H), 3,86 (с, 3H), 3,85 (с, 3H). IP = C оценка значений.
Таблица 1A. Данные m/z (МН)+ исходного иона и Первичного IP Теста для соединений
6: (2-хлор-4-фторфенил)(1H-индол-1-ил)метилацетат: m/z (ES) 318 (МН)+; IP = C оценка значений.
7: (2-хлор-4-фторфенил)(4-циано-1Н-индол-1-ил)метилацетат: m/z (ES) 343 (МН)+; IP = B оценка значений.
8: 1-[1-(2-хлор-4-фторфенил)-2-метокси-2-оксоэтил]-1Н-индол-4-этилкарбоксилат: m/z (ES) 390 (МН)+; IP = C оценка значений.
9: (2-хлор-4-фторфенил)(4-хлор-1Н-индол-1-ил)метилацетат: m/z (ES) 352 (МН)+; IP = C оценка значений.
10: (2-хлор-4-фторфенил)(4-фтор-1Н-индол-1-ил)метилацетат: m/z (ES) 336 (МН)+; IP = C оценка значений.
11: (2-хлор-4-фторфенил)(5-циано-1Н-индол-1-ил)метилацетат: m/z (ES) 343 (МН)+; IP = C оценка значений.
12: (2-хлор-4-фторфенил)(5-хлор-1Н-индол-1-ил)метилацетат: m/z (ES) 352 (МН)+; IP = C оценка значений.
13: (2-хлор-4-фторфенил)(5-фтор-1Н-индол-1-ил)метилацетат: m/z (ES) 336 (МН)+; IP = C оценка значений.
14: (2-хлор-4-фторфенил)(6-хлор-1Н-индол-1-ил)метилацетат: m/z (ES) 352 (МН)+; IP = C оценка значений.
15: (2-хлор-4-фторфенил)(7-фтор-1Н-индол-1-ил)метилацетат: m/z (ES) 336 (МН)+; IP = C оценка значений.
16: [7-(бензилокси)-1Н-индол-1-ил](2-хлор-4-фторфенил)метилацетат: m/z (ES) 424 (МН)+; IP = C оценка значений.
ПРИМЕР 2
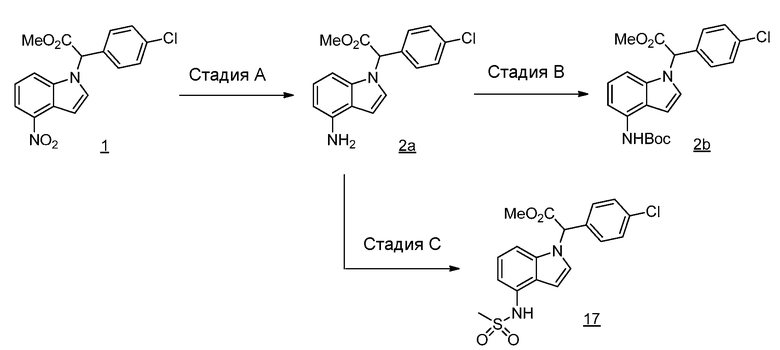 Стадия A: Получение (4-амино-1Н-индол-1-ил)(4-хлорфенил)метилацетата (2a)
Стадия A: Получение (4-амино-1Н-индол-1-ил)(4-хлорфенил)метилацетата (2a)
Раствор 1 (8,28 г, 24,0 ммоль) в этилацетате (240 мл) дегазировали, в этот момент добавляли платину на углероде (1,41 г 10%-ой (вес/вес) смеси, 0,721 ммоль), и полученную суспензию перемешивали в атмосфере водорода (баллон). Через 3 часа реакционную смесь фильтровали через короткую колонку для элюирования CELITE® с EtOAc. Объединенные органические фракции концентрировали в вакууме, получая целевое соединение 2a. m/z (ES) 315 (МН)+.
Стадия B: Получение {4-[(трет-бутоксикарбонил)амино]-1Н-индол-1-ил}(4-хлорфенил)метилацетата (2b)
Ди-трет-бутил дикарбонат (5,24 г, 24,0 ммоль) добавляли к перемешиваемому раствору 2a (7,56 г, 24,0 ммоль) в диоксане (60 мл) и насыщенном водном растворе бикарбоната натрия (15 мл). Полученную смесь перемешивали при комнатной температуре в течение ночи, после чего реакционную смесь частично концентрировали в вакууме. Полученную смесь разделяли между этилацетатом и насыщенным водным раствором хлорида аммония. Слои разделяли, и водный слой экстрагировали этилацетатом. Объединенные органические фракции высушивали (Na2SO4), фильтровали и концентрировали в вакууме. Полученное сырое масло очищали флэш-хроматографией на силикагеле (элюирование в градиенте; 0%-20% EtOAc/гексаны в качестве элюента), получая целевое соединение 2b m/z (ES) 437 (MNa)+.
Стадия C: Получение (4-хлорфенил){4-[(метилсульфонил)амино]-1Н-индол-1-ил}метилацетата (17)
Метансульфонил хлорид (159 мкл, 2,04 ммоль) добавляли к перемешиваемому раствору 2b (320 мг, 1,02 ммоль) и триэтиламина (213 мкл, 1,53 ммоль) в DCM (3 мл) при комнатной температуре. Через 1 час реакционную смесь лили в воду и экстрагировали DCM. Объединенные органические фракции промывали солевым раствором, высушивали (Na2SO4), фильтровали и концентрировали в вакууме. Сырой остаток очищали препаративной ВЭЖХ с обратной фазой на стационарной фазе YMC Pack Pro C18 (CH3CN/H2O в качестве элюента, 0,05% TFA в качестве модификатора) с последующей лиофилизацией очищенных фракций, получая целевое соединение 17. m/z (ES) 393 (МН)+ IP = C оценка значений.
Следуя процедурам, подобным описанным выше в Примере 2, получали следующее соединение:
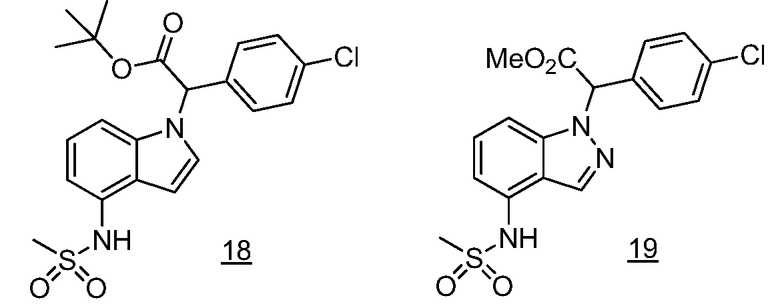
18: (4-хлорфенил){4-[(метилсульфонил)амино]-1Н-индол-1-ил}трет-бутилацетат: m/z (ES) 435 (МН)+; IP = C оценка значений.
19: (4-хлорфенил){4-[(метилсульфонил)амино]-1Н-индазол-1-ил}метилацетат: m/z (ES) 394 (МН)+; IP = C оценка значений.
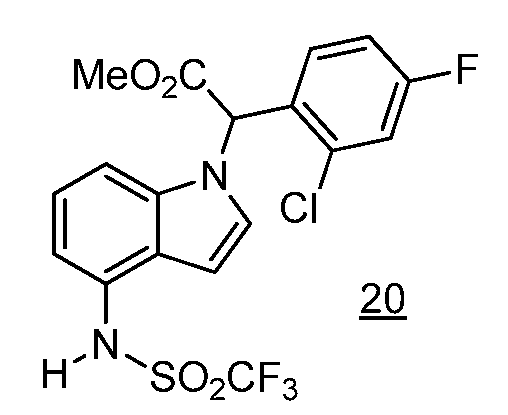
20: (2-хлор-4-фторфенил)(4-{[(трифторметил)сульфонил]амино}-1Н-индол-1-ил)метилацетат: m/z (ES) 465 (МН)+; IP = C оценка значений.
Следуя процедурам, подобным описанным в Примере 2, получали следующие дополнительные соединения, представленные в Таблице 2:
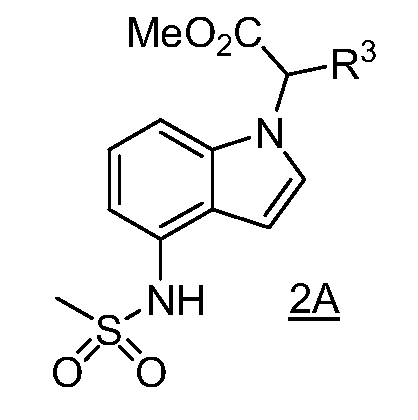
Таблица 2. Данные m/z (МН)+ исходного иона и Первичного IP Теста для соединений.
21: {4-[(метилсульфонил)амино]-1Н-индол-1-ил}(фенил)метилацетат: m/z (ES) 381 (MNa)+; IP = C оценка значений.
22: (2-хлор-4-фторфенил){4-[(метилсульфонил)амино]-1Н-индол-1-ил}метилацетат: m/z (ES) 411 (МН)+; IP = B оценка значений.
23: (2,4-дихлорфенил){4-[(метилсульфонил)амино]-1Н-индол-1-ил}метилацетат: m/z (ES) 427 (МН)+; IP = А оценка значений.
24: (4-бромфенил){4-[(метилсульфонил)амино]-1Н-индол-1-ил}метилацетат: m/z (ES) 437 (МН)+; IP = C оценка значений.
ПРИМЕР 3
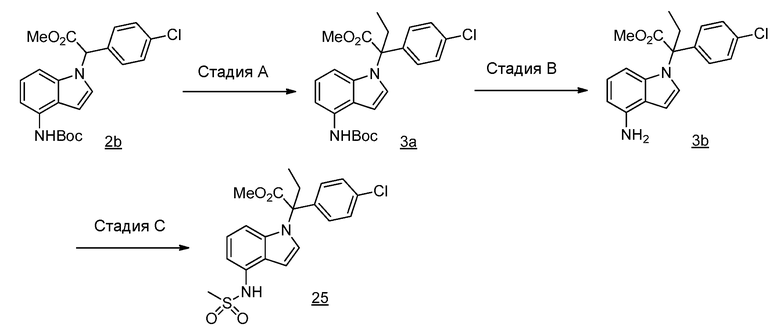
Стадия A: Получение 2-{4-[(трет-бутоксикарбонил)амино]-1Н-индол-1-ил}-2-(4-хлорфенил)метилбутаноата (3a)
Раствор 2b (10,0 г, 24,1 ммоль) в DMF (20 мл) добавляли через шприцевой насос (1,6 мл/минуты) к перемешиваемой суспензии гидрида натрия (964 мг 60%-ой (вес/вес) дисперсии в минеральном масле, 24,1 ммоль) в DMF (60 мл) при 0°C. Через 15 минут добавляли быстро по каплям йодэтан (2,34 мл, 28,9 ммоль), и полученную смесь перемешивали при 0°C в течение 2 часов. Реакционную смесь гасили добавлением насыщенного водного раствора хлорида аммония и экстрагировали простым эфиром. Объединенные органические фракции промывали водой и солевым раствором, высушивали (MgSO4), фильтровали и концентрировали в вакууме. Полученное сырое масло очищали флэш-хроматографией на силикагеле (элюирование в градиенте; 0%-20% EtOAc/гексаны в качестве элюента), получая целевое соединение 3a. m/z (ES) 465 (MNa)+.
Стадия B: Получение 2-(4-амино-1Н-индол-1-ил)-2-(4-хлорфенил)метилбутаноата (3b)
Трифторуксусную кислоту (37,5 мл, 487 ммоль) добавляли быстро по каплям к перемешиваемому раствору 3a (8,62 г, 19,5 ммоль) в DCM (100 мл) при 0°C. Через 1 час реакционную смесь гасили аккуратным добавлением насыщенного водного раствора бикарбоната натрия, затем NaHCO3 (с) и экстрагировали простым эфиром. Органические фракции промывали насыщенным водным раствором бикарбоната натрия, высушивали (MgSO4), фильтровали и концентрировали в вакууме, получая целевое соединение 3b. m/z (ES) 343 (МН)+.
Стадия C: Получение 2-(4-хлорфенил)-2-{4-[(метилсульфонил)амино]-1Н-индол-1-ил}метилбутаноата (25)
Метансульфонил хлорид (1,59 мл, 20,4 ммоль) добавляли к перемешиваемому раствору 3b (6,67 г, 19,5 ммоль) и 4-метилморфолина (2,78 мл, 25,3 ммоль) в DCM (100 мл) при 0°C. Через 30 минут добавляли дополнительную часть метансульфонил хлорида (0,80 мл, 10,2 ммоль). Через 1 час реакционную смесь разделяли между DCM и насыщенным водным раствором хлорида аммония. Слои разделяли, и водный слой экстрагировали DCM. Объединенные органические фракции промывали солевым раствором, высушивали (Na2SO4), фильтровали и концентрировали в вакууме. Сырой остаток очищали флэш-хроматографией на силикагеле (элюирование в градиенте; 0%-50% EtOAc/гексаны в качестве элюента), получая целевое соединение 25. m/z (ES) 421 (МН)+; IP = А оценка значений
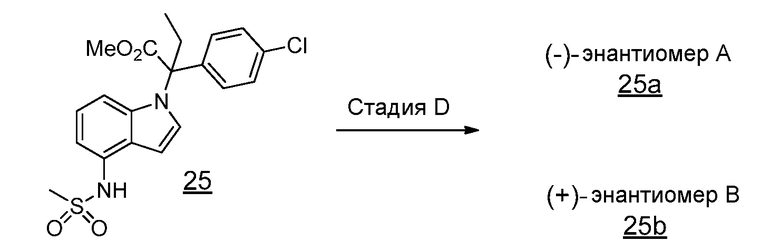
Энантиомеры 25a и 25b разделяли, используя препаративную хиральную ВЭЖХ с нормальной фазой. Раствор 25 в смеси MeOH/ацетонитрил вводили в полупрепаративную колонку для ВЭЖХ CHIRALCEL® AS-H (Chiral Technologies, Inc, Exton, Pa.) (250×30 мм) (элюируя 45% [(2:1) MeOH:ацетонитрил]/CO2 с температурой колонки 35°C при 70 мл/мин. с УФ-детекцией при 220 нм). Энантиомеры разделяли, причем более быстро элюирующий энантиомер 25a имел время удерживания 2,61 мин. и более медленно элюирующий энантиомер 25b имел время удерживания 3,13 мин., причем время удерживания получали в результате инъекции на колонку для аналитической ВЭЖХ CHIRALCEL® AS-H (4,6×250 мм, 2,1 мл/мин., 40% [(2:1) MeOH:ацетонитрил]/CO2, 35°C). Отделенные фракции концентрировали, получая энантиомеры 25a и 25b. В условиях разделения, описанных выше, более быстро элюирующий (R)-энантиомер 25a является предпочтительным для получения конечных продуктов. Для 25a: m/z (ES) 421 (МН)+; IP = А оценка значений. Для 25b: m/z (ES) 421 (МН)+; IP = B оценка значений
Следуя процедурам, подобным описанным выше в Примере 3, получали следующее соединение:
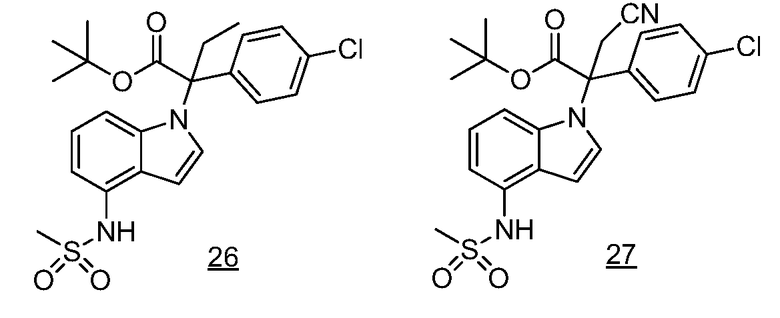
26: 2-(4-хлорфенил)-2-{4-[(метилсульфонил)амино]-1Н-индол-1-ил}трет-бутилбутаноат: m/z (ES) 463 (МН)+; IP = B оценка значений.
27: 2-(4-хлорфенил)-3-циано-2-{4-[(метилсульфонил)амино]-1Н-индол-1-ил}трет-бутилпропаноат: m/z (ES) 496 (MNa)+; IP = B оценка значений.
Следуя процедурам, подобным описанным выше в Примере 3, получали следующие дополнительные соединения:
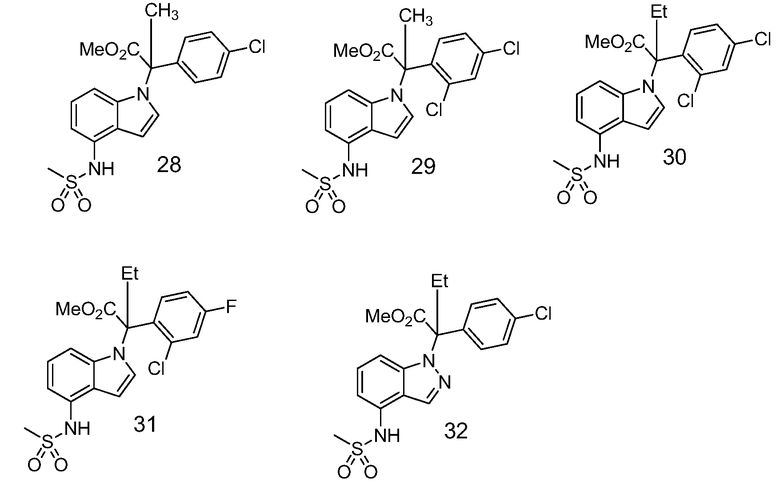
28: 2-(4-хлорфенил)-2-{4-[(метилсульфонил)амино]-1Н-индол-1-ил}метилпропаноат: m/z (ES) 407 (МН)+; IP = B оценка значений.
29: 2-(2,4-дихлорфенил)-2-{4-[(метилсульфонил)амино]-1Н-индол-1-ил}метилпропаноат: m/z (ES) 441 (МН)+; IP = C оценка значений.
30: 2-(2,4-дихлорфенил)-2-{4-[(метилсульфонил)амино]-1Н-индол-1-ил}метилбутаноат: m/z (ES) 455 (МН)+; IP = B оценка значений.
31: 2-(2-хлор-4-фторфенил)-2-{4-[(метилсульфонил)амино]-1Н-индол-1-ил}метилбутаноат: m/z (ES) 439 (МН)+; IP = B оценка значений.
32: 2-(4-хлорфенил)-2-{4-[(метилсульфонил)амино]-1Н-индазол-1-ил}метилбутаноат: m/z (ES) 422 (МН)+; IP = B оценка значений.
Были получены дополнительные соединения, как показано в Таблице 3:
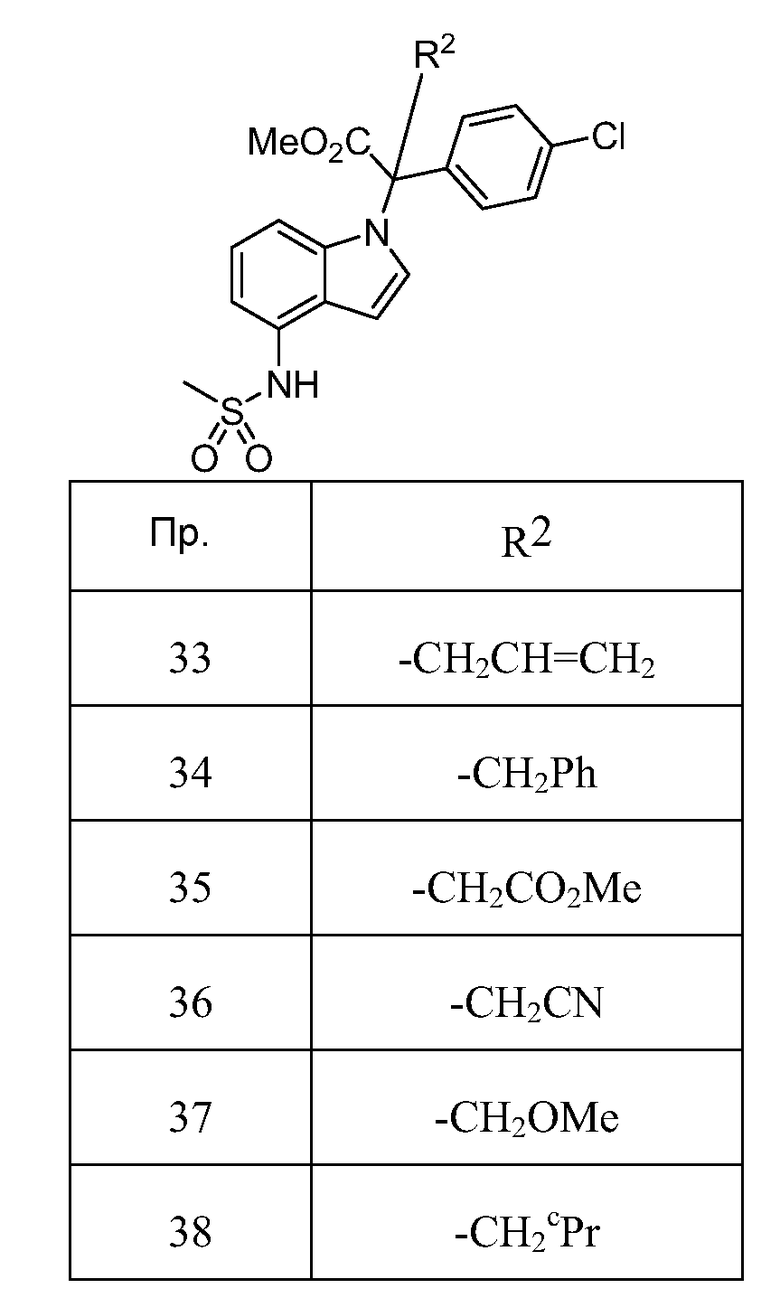
Таблица 3. Данные m/z (МН)+ исходного иона и Первичного IP Теста для соединений.
33: 2-(4-хлорфенил)-2-{4-[(метилсульфонил)амино]-1Н-индол-1-ил}метилпент-4-еноат: m/z (ES) 433 (МН)+; IP = B оценка значений.
34: 2-(4-хлорфенил)-2-{4-[(метилсульфонил)амино]-1Н-индол-1-ил}-3-метилфенилпропаноат: m/z (ES) 483 (МН)+; IP = B оценка значений.
35: 2-(4-хлорфенил)-2-{4-[(метилсульфонил)амино]-1Н-индол-1-ил}диметилбутандиоат: m/z (ES) 487 (MNa)+; IP = А оценка значений.
36: 2-(4-хлорфенил)-3-циано-2-{4-[(метилсульфонил)амино]-1Н-индол-1-ил}метилпропаноат: m/z (ES) 432 (МН)+; IP = А оценка значений.
37: 2-(4-хлорфенил)-3-метокси-2-{4-[(метилсульфонил)амино]-1Н-индол-1-ил}метилпропаноат: m/z (ES) 437 (МН)+; IP = C оценка значений.
38: 2-(4-хлорфенил)-3-циклопропил-2-{4-[(метилсульфонил)амино]-1Н-индол-1-ил}метилпропаноат: m/z (ES) 469 (MNa)+; IP = А оценка значений.
ПРИМЕР 4
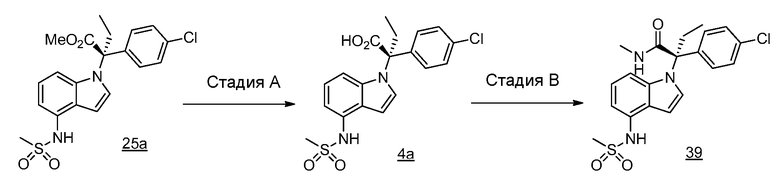
Стадия A: Получение (R)-2-(4-хлорфенил)-2-{4-[(метилсульфонил)амино]-1Н-индол-1-ил}бутановой кислоты (4a)
Гидроксид лития (484 мг, 20,2 ммоль) добавляли к перемешиваемому раствору 325a (850 мг, 2,02 ммоль) в диоксане (16 мл) и воде (4 мл), и полученную смесь нагревали до 70°C в течение 3 часов. После охлаждения до комнатной температуры, реакционную смесь вливали в 1M HCl и экстрагировали этилацетатом. Объединенные органические фракции промывали солевым раствором, высушивали (Na2SO4), фильтровали и концентрировали в вакууме, получая целевое соединение 4a. m/z (ES) 407 (МН)+.
Стадия B: Получение (R)-2-(4-хлорфенил)-N-метил-2-{4-[(метилсульфонил)амино]-1Н-индол-1-ил}бутанамида (39)
O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфат (1,34 г, 3,56 ммоль) добавляли к перемешиваемому раствору 4a (967 мг, 2,38 ммоль) и 4-метилморфолина (523 мкл, 4,75 ммоль) в DMF (12 мл). Через 5 минут добавляли метиламин (3,56 мл 2,0 М раствора в метаноле, 7,13 ммоль), и полученную смесь перемешивали при комнатной температуре. Через 3 часа реакционную смесь разделяли между этилацетатом и водой. Слои разделяли, и органические фракции промывали водой и солевым раствором, высушивали (Na2SO4), фильтровали и концентрировали в вакууме. Сырой остаток очищали флэш-хроматографией на силикагеле (элюирование в градиенте; 0%-85% EtOAc/гексаны в качестве элюента), получая целевое соединение 39. m/z (ES) 420 (МН)+; IP = B оценка значений.
Следуя процедурам, подобным описанным выше в Примере 4, получали следующие дополнительные соединения, представленные в Таблице 4:
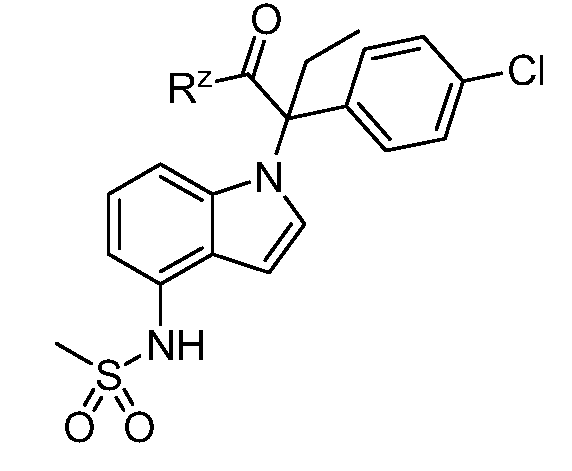

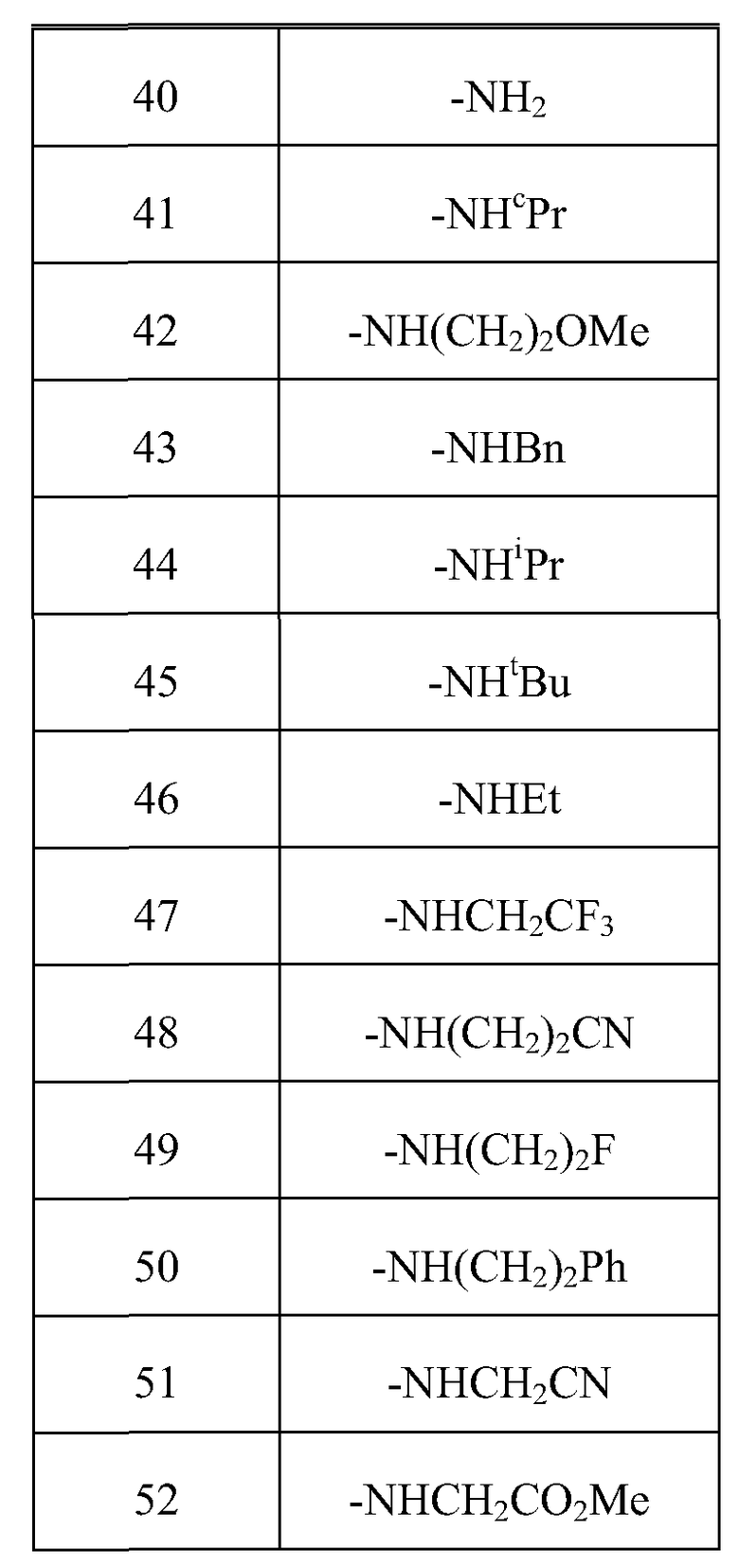
Таблица 4. Данные m/z (МН)+ исходного иона и Первичного IP Теста для соединений.
40: (R)-2-(4-хлорфенил)-2-{4-[(метилсульфонил)амино]-1Н-индол-1-ил}бутанамид: m/z (ES) 406 (МН)+; IP = B оценка значений.
41: (R)-2-(4-хлорфенил)-N-циклопропил-2-{4-[(метилсульфонил)амино]-1Н-индол-1-ил}бутанамид: m/z (ES) 446 (МН)+; IP = B оценка значений.
42: 2-(4-хлорфенил)-N-(2-метоксиэтил)-2-{4-[(метилсульфонил)амино]-1Н-индол-1-ил}бутанамид: m/z (ES) 464 (МН)+; IP = C оценка значений.
43: (R)-N-бензил-2-(4-хлорфенил)-2-{4-[(метилсульфонил)амино]-1Н-индол-1-ил}бутанамид: m/z (ES) 496 (МН)+; IP = C оценка значений.
44: (R)-2-(4-хлорфенил)-N-(1-метилэтил)-2-{4-[(метилсульфонил)амино]-1Н-индол-1-ил}бутанамид: m/z (ES) 448 (МН)+; IP = B оценка значений.
45: (R)-N-трет-бутил-2-(4-хлорфенил)-2-{4-[(метилсульфонил)амино]-1Н-индол-1-ил}бутанамид: m/z (ES) 462 (МН)+; IP = C оценка значений.
46: 2-(4-хлорфенил)-N-этил-2-{4-[(метилсульфонил)амино]-1Н-индол-1-ил}бутанамид: m/z (ES) 434 (МН)+; IP = B оценка значений.
47: (R)-2-(4-хлорфенил)-2-{4-[(метилсульфонил)амино]-1Н-индол-1-ил}-N-(2,2,2-трифторэтил)бутанамид: m/z (ES) 488 (МН)+; IP = А оценка значений.
48: 2-(4-хлорфенил)-N-(2-цианоэтил)-2-{4-[(метилсульфонил)амино]-1Н-индол-1-ил}бутанамид: m/z (ES) 459 (МН)+; IP = C оценка значений.
49: 2-(4-хлорфенил)-N-(2-фторэтил)-2-{4-[(метилсульфонил)амино]-1Н-индол-1-ил}бутанамид: m/z (ES) 474 (MNa)+; IP = B оценка значений.
50: 2-(4-хлорфенил)-2-{4-[(метилсульфонил)амино]-1Н-индол-1-ил}-N-(2-фенилэтил)бутанамид: m/z (ES) 532 (MNa)+; IP = C оценка значений.
51: 2-(4-хлорфенил)-N-(цианометил)-2-{4-[(метилсульфонил)амино]-1Н-индол-1-ил}бутанамид: m/z (ES) 467 (MNa)+; IP = А оценка значений.
52: N-[2-(4-хлорфенил)-2-{4-[(метилсульфонил)амино]-1Н-индол-1-ил}бутаноил]метилглицинат: m/z (ES) 478 (МН)+; IP = B оценка значений.
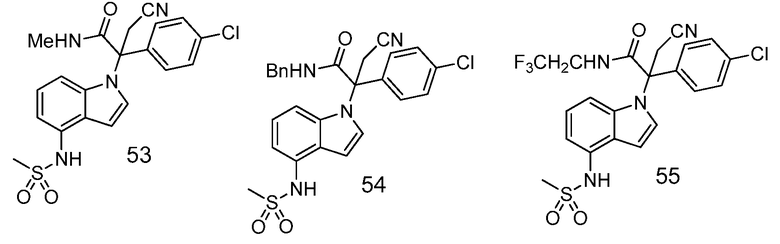
53: 2-(4-хлорфенил)-3-циано-N-метил-2-{4-[(метилсульфонил)амино]-1Н-индол-1-ил}пропанамид: m/z (ES) 431 (МН)+; IP = B оценка значений.
54: N-бензил-2-(4-хлорфенил)-3-циано-2-{4-[(метилсульфонил)амино]-1Н-индол-1-ил}пропанамид: m/z (ES) 507 (МН)+; IP = B оценка значений.
55: 2-(4-хлорфенил)-3-циано-2-{4-[(метилсульфонил)амино]-1Н-индол-1-ил}-N-(2,2,2-трифторэтил)пропанамид: m/z (ES) 499 (МН)+; IP = B оценка значений.
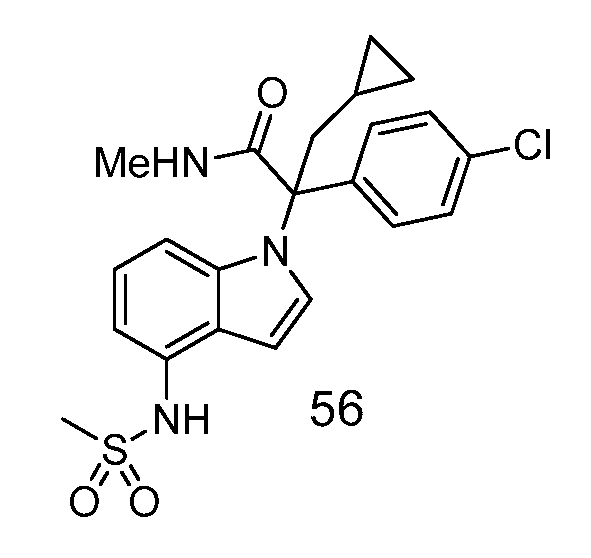
56: 2-(4-хлорфенил)-3-циклопропил-N-метил-2-{4-[(метилсульфонил)амино]-1Н-индол-1-ил}пропанамид: m/z (ES) 468 (MNa)+; IP = B оценка значений.
ПРИМЕР 5
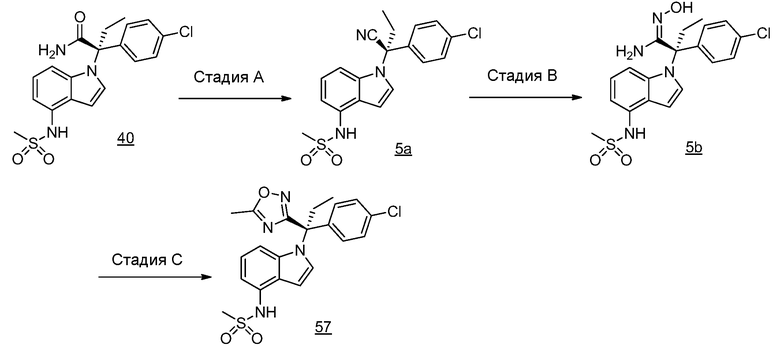
Стадия A: Получение (R)-N-{1-[1-(4-хлорфенил)-1-цианопропил]-1Н-индол-4-ил}метансульфонамида (5a)
Хлорангидрид циануровой кислоты (17,3 мг, 0,094 ммоль) добавляли к перемешиваемому раствору 40 (38,0 мг, 0,094 ммоль) в DMF (0,50 мл), и реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь лили в солевой раствор, и полученную смесь экстрагировали этилацетатом. Органические фракции высушивали (Na2SO4), фильтровали и концентрировали в вакууме. Сырой остаток очищали флэш-хроматографией на силикагеле (элюирование в градиенте; 0%-50% EtOAc/гексаны в качестве элюента), получая целевое соединение 5a. m/z (ES) 361 (М. CN) +; IP = А оценка значений.
Стадия B: Получение (R)-(1E)-2-(4-хлорфенил)-N'-гидрокси-2-{4-[(метилсульфонил)амино]-1Н-индол-1-ил}бутанимидамида (5b)
Смесь 5a (30,0 мг, 0,077 ммоль), гидроксиламина (156 мкл 50%-ого (вес/вес) водного раствора, 2,55 ммоль) и карбоната калия (1,1 мг, 7,7 ммоль) в этаноле (1,2 мл) нагревали в микроволновом реакторе при 120°C в течение 10 мин. После охлаждения до комнатной температуры, реакционную смесь концентрировали в вакууме, и полученный сырой остаток разделяли между этилацетатом и водой. Слои разделяли, и органические фракции промывали солевым раствором, высушивали (Na2SO4), фильтровали и концентрировали в вакууме, получая целевое соединение 5b. m/z (ES) 421 (МН)+.
Стадия C: Получение (R)-N-{1-[1-(4-хлорфенил)-1-(5-метил-1,2,4-оксадиазол-3-ил)пропил]-1Н-индол-4-ил}метилметансульфонамида (57)
(Бензилтриазол-1-илокси)трис(диметиламино)фосфоний гексафторфосфат (37 мг, 0,083 ммоль) добавляли к смеси 5b (35 мг, 0,083 ммоль), уксусной кислоты (4,8 мкл, 0,083 ммоль) и N,N-диизопропилэтиламина (29 мкл, 0,166 ммоль) в ацетонитриле (1,50 мл). Полученную смесь перемешивали при комнатной температуре в течение 30 сек, затем нагревали в микроволновом реакторе при 150°C в течение 20 мин. После охлаждения до комнатной температуры, реакционную смесь разбавляли водой, и смесь очищали препаративной ВЭЖХ с обратной фазой на стационарной фазе YMC Pack Pro C18 (CH3CN/H2O в качестве элюента, 0,05% TFA в качестве модификатора), с последующей лиофилизацией очищенных фракций, получая целевое соединение 57. m/z (ES) 445 (МН)+; IP = А оценка значений
Следуя процедурам, подобным описанным выше в Примере 5, получали следующие дополнительные соединения, представленные в Таблице 5:
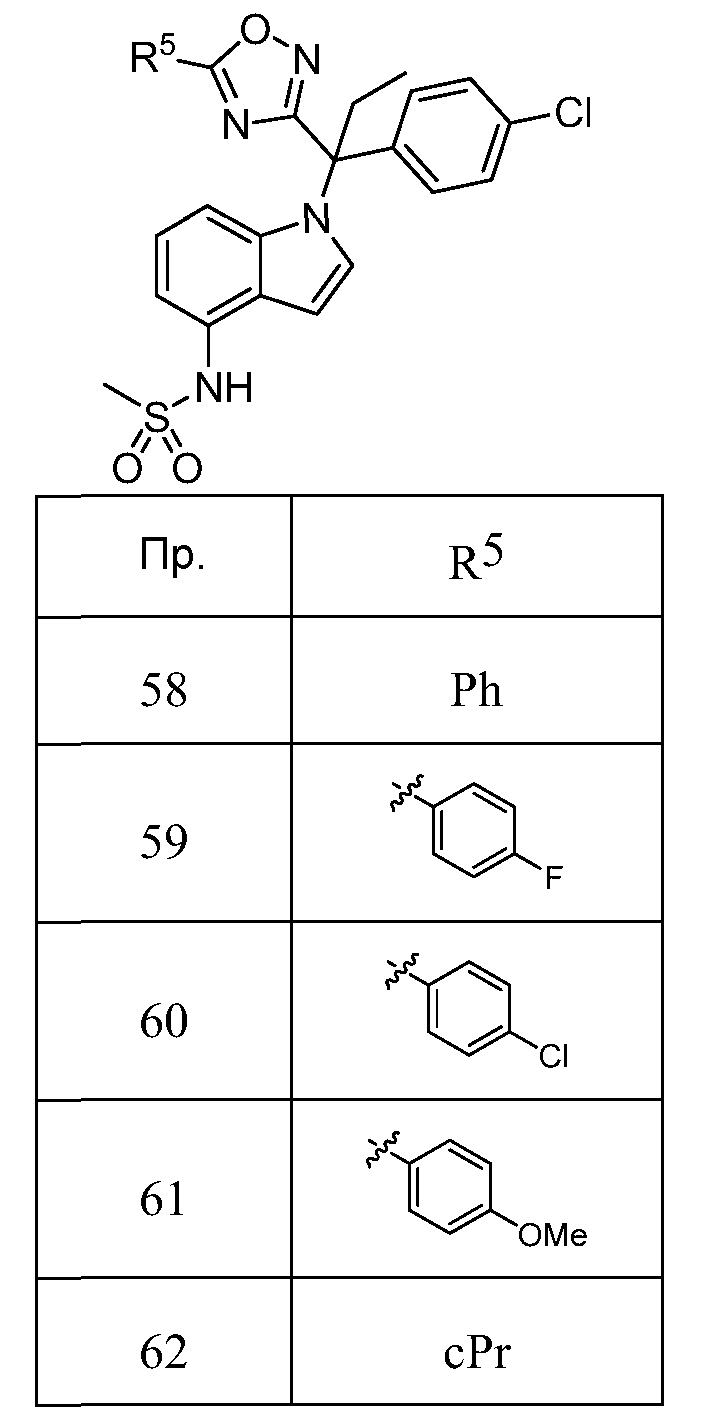
Таблица 5. Данные m/z (МН)+ исходного иона и Первичного IP Теста для соединений.
58: (R)-N-{1-[1-(4-хлорфенил)-1-(5-фенил-1,2,4-оксадиазол-3-ил)пропил]-1Н-индол-4-ил}метансульфонамид: m/z (ES) 507 (МН)+; IP = C оценка значений.
59: N-(1-{1-(4-хлорфенил)-1-[5-(4-фторфенил)-1,2,4-оксадиазол-3-ил]пропил}-1Н-индол-4-ил)метансульфонамид: m/z (ES) 525 (МН)+; IP = А оценка значений.
60: N-(1-{1-(4-хлорфенил)-1-[5-(4-хлорфенил)-1,2,4-оксадиазол-3-ил]пропил}-1Н-индол-4-ил)метансульфонамид: m/z (ES) 541 (МН)+; IP = А оценка значений.
61: (R)-N-(1-{1-(4-хлорфенил)-1-[5-(4-метоксифенил)-1,2,4-оксадиазол-3-ил]пропил}-1Н-индол-4-ил)метансульфонамид: m/z (ES) 537 (МН)+; IP = А оценка значений.
62: (R)-N-{1-[1-(4-хлорфенил)-1-(5-циклопропил-1,2,4-оксадиазол-3-ил)пропил]-1Н-индол-4-ил}метансульфонамид: m/z (ES) 471 (МН)+; IP = А оценка значений.
ПРИМЕР 6
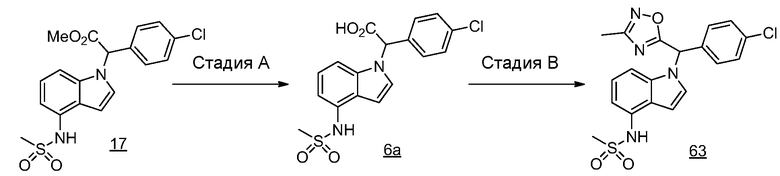
Стадия A: Получение (4-хлорфенил){4-[(метилсульфонил)амино]-1Н-индол-1-ил}уксусной кислоты (6a)
Соединение 6a получали из соединения 17 согласно процедурам, подобным предварительно описанным в Примере 4, стадия A.
Стадия B: Получение N-{1-[(4-хлорфенил)(3-фенил-1,2,4-оксадиазол-5-ил)метил]-1Н-индол-4-ил}метансульфонамида (63)
N'-гидроксибензолкарбоксимидамид (49 мг, 0,36 ммоль) добавляли к перемешиваемому раствору соединения 6a (90 мг, 0,24 ммоль), гидрохлорид N-(3-диметиламинопропил)-N'-этилкарбодиимида (91 мг, 0,48 ммоль), 1-гидроксибензотриазола (44 мг, 0,29 ммоль) и N,N-диизопропилэтиламина (50 мкл, 0,29 ммоль) в DMF (2,4 мл). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, затем реакционную смесь разделяли между этилацетатом и водой. Слои разделяли, и органический слой промывали водой и солевым раствором, высушивали над MgSO4, фильтровали и концентрировали до сырого масла, которое растворяли в толуоле (2,0 мл). Полученную смесь нагревали до 100°C в течение 1 часа, после чего реакционную смесь концентрировали в вакууме, и полученный сырой остаток очищали препаративной ВЭЖХ с обратной фазой на стационарной фазе YMC Pack Pro C18 (CH3CN/H2O в качестве элюента, 0,05% TFA в качестве модификатора), с последующей лиофилизацией очищенных фракций, получая целевое соединение 63. m/z (ES) 479 (МН)+; IP = C оценка значений.
ПРИМЕР 7
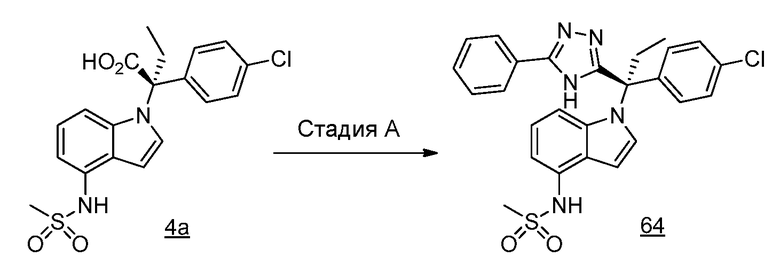
Стадия A: Получение (R)-N-{1-[1-(4-хлорфенил)-1-(3-фенил-1Н-1,2,4-триазол-5-ил)пропил]-1Н-индол-4-ил}метансульфонамида (64)
Соединение 64 синтезировали из соединения 4a согласно процедурам, подобным представленным в Примере 6, заменяя N'-гидробензолкарбоксимидамид [имино(фенил)метил]гидразиний йодидом. m/z (ES) 506 (МН)+; IP = C оценка значений.
Следуя процедурам, подобным описанным выше в Примере 7, получали следующие дополнительные соединения, представленные в Таблице 7:
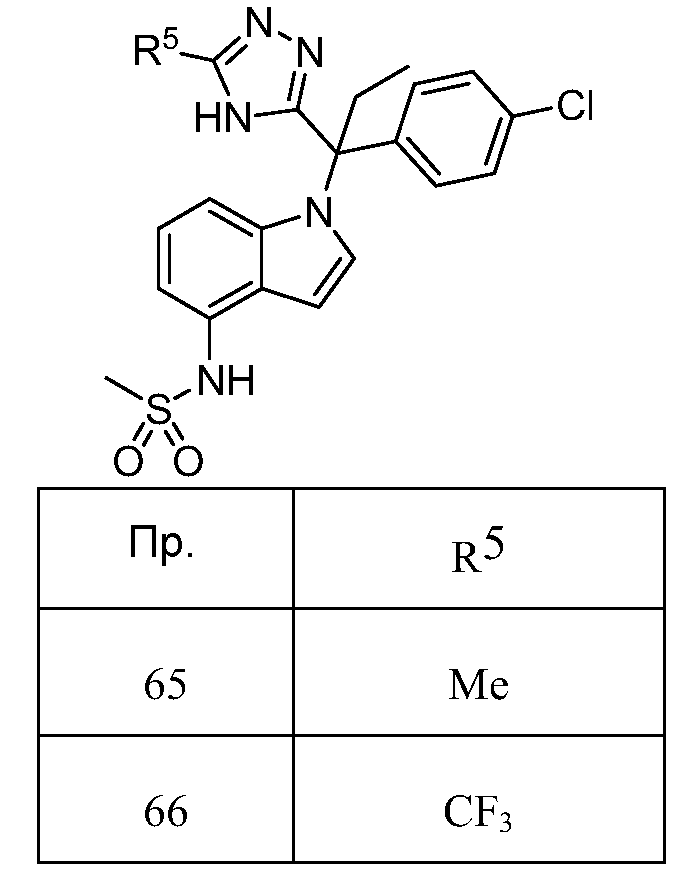
Таблица 7. Данные m/z (МН)+ исходного иона и Первичного IP Теста для соединений.
65: (R)-N-{1-[1-(4-хлорфенил)-1-(5-метил-4H-1,2,4-триазол-3-ил)пропил]-1Н-индол-4-ил}метансульфонамид: m/z (ES) 444 (МН)+; IP = B оценка значений.
66: (R)-N-(1-{1-(4-хлорфенил)-1-[5-(трифторметил)-4H-1,2,4-триазол-3-ил]пропил}-1Н-индол-4-ил)метансульфонамид: m/z (ES) 428 (М. CF3) +; IP = B оценка значений.
ПРИМЕР 8
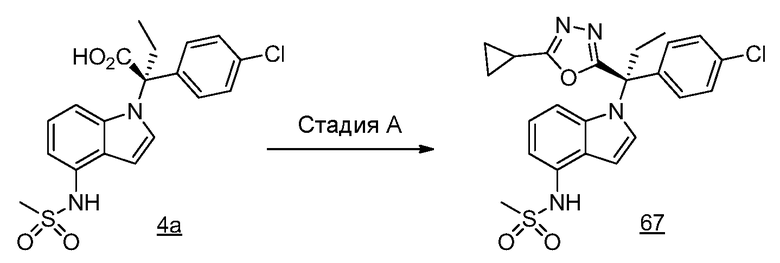
Стадия A: Получение (R)-N-{1-[1-(4-хлорфенил)-1-(5-циклопропил-1,3,4-оксадиазол-2-ил)пропил]-1Н-индол-4-ил}метансульфонамид (67)
Раствор 4a (60 мг, 0,147 ммоль), гидразида циклопропилкарбоновой кислоты (15,5 мг, 0,155 ммоль), O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфата (84 мг, 0,221 ммоль) и N,N-диизопропилэтиламина (77 мкл, 0,442 ммоль) в DMF (1 мл) перемешивали при комнатной температуре в течение 1,5 часов. Реакционную смесь разделяли между этилацетатом и водой. Слои разделяли, и органические фракции промывали водой и солевым раствором, высушивали (Na2SO4), фильтровали и концентрировали в вакууме, получая сырой остаток, который растворяли в THF (2 мл).
Добавляли (Метоксикарбонилсульфамоил)триэтиламмоний гидроксид (105 мг, 0,442 ммоль), и полученную смесь нагревали при 60°C в течение 2 часов. После охлаждения до комнатной температуры, реакционную смесь концентрировали в вакууме, и полученный сырой остаток очищали препаративной ВЭЖХ с обратной фазой на стационарной фазе YMC Pack Pro C18 (CH3CN/H2O в качестве элюента, 0,05% TFA в качестве модификатора), с последующей лиофилизацией очищенных фракций, получая целевое соединение 67. m/z (ES) 471 (МН)+; IP = B оценка значений.
Следуя процедурам, подобным описанным выше в Примере 8, получали следующие дополнительные соединения, представленные в Таблице 8:
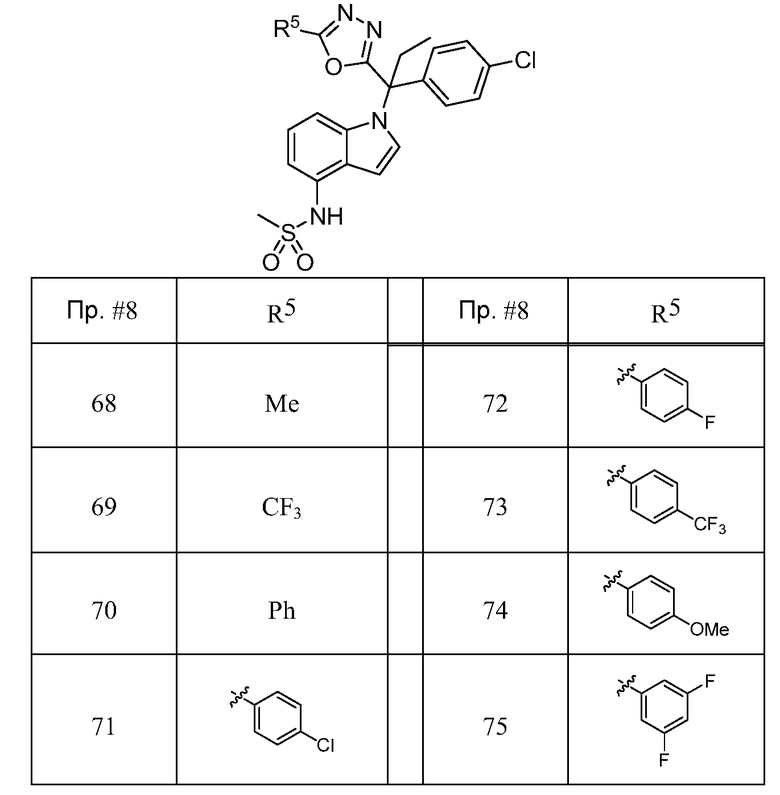
Таблица 8. Данные m/z (МН)+ исходного иона и Первичного IP Теста для соединений.
68: N-{1-[1-(4-хлорфенил)-1-(5-метил-1,3,4-оксадиазол-2-ил)пропил]-1Н-индол-4-ил}метансульфонамид: m/z (ES) 467 (MNa)+; IP = B оценка значений.
69: N-(1-{1-(4-хлорфенил)-1-[5-(трифторметил)-1,3,4-оксадиазол-2-ил]пропил}-1Н-индол-4-ил)метансульфонамид: m/z (ES) 499 (MNa)+; IP = B оценка значений.
70: N-{1-[1-(4-хлорфенил)-1-(5-фенил-1,3,4-оксадиазол-2-ил)пропил]-1Н-индол-4-ил}метансульфонамид: m/z (ES) 529 (MNa)+; IP = C оценка значений.
71: N-(1-{1-(4-хлорфенил)-1-[5-(4-хлорфенил)-1,3,4-оксадиазол-2-ил]пропил}-1Н-индол-4-ил)метансульфонамид: m/z (ES) 563 (MNa)+; IP = А оценка значений.
72: N-(1-{1-(4-хлорфенил)-1-[5-(4-фторфенил)-1,3,4-оксадиазол-2-ил]пропил}-1Н-индол-4-ил)метансульфонамид: m/z (ES) 547 (MNa)+; IP = B оценка значений.
73: N-{1-[1-(4-хлорфенил)-1-{5-[4-(трифторметил)фенил]-1,3,4-оксадиазол-2-ил}пропил]-1Н-индол-4-ил}метансульфонамид: m/z (ES) 597 (MNa)+; IP = B оценка значений.
74: N-(1-{1-(4-хлорфенил)-1-[5-(4-метоксифенил)-1,3,4-оксадиазол-2-ил]пропил}-1Н-индол-4-ил)метансульфонамид: m/z (ES) 537 (МН)+; IP = B оценка значений.
75: N-(1-{1-(4-хлорфенил)-1-[5-(3,5-дифторфенил)-1,3,4-оксадиазол-2-ил]пропил}-1Н-индол-4-ил)метансульфонамид: m/z (ES) 565 (MNa)+; IP = C оценка значений.
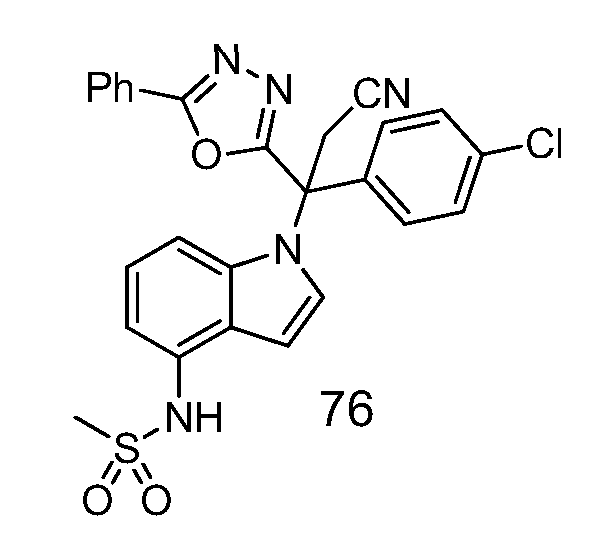
76: N-{1-[1-(4-хлорфенил)-2-циано-1-(5-фенил-1,3,4-оксадиазол-2-ил)этил]-1Н-индол-4-ил}метансульфонамид: m/z (ES) 540 (MNa)+; IP = B оценка значений.
ПРИМЕР 9
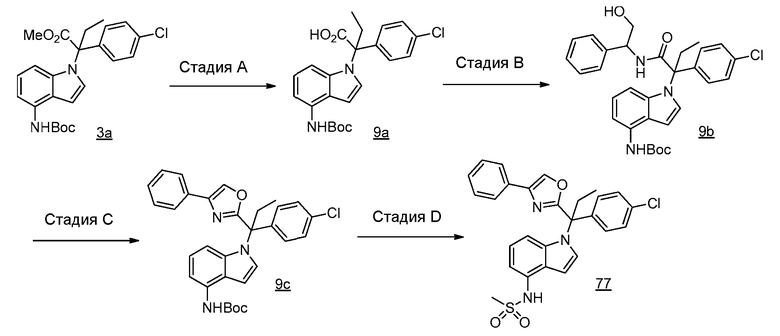
Стадия A: Получение 2-{4-[(трет-бутоксикарбонил)амино]-1Н-индол-1-ил}-2-(4-хлорфенил)бутановой кислоты (9a)
Соединение 9a получали из соединения 3a согласно процедурам, подобных описанным в Примере 4, стадия A, заменяя соединение 3c соединением 3a. m/z (ES) 429 (МН)+.
Стадия B: Получение (1-{2-(4-хлорфенил)-1-[(2-гидрокси-1-фенилэтил)амино]-1-оксобутан-2-ил}-1Н-индол-4-ил)трет-бутилкарбамата (9b)
Соединение 9b получали из соединения 9a согласно процедурам, подобным описанным в Примере 4, стадия B, заменяя соединение 4a соединением 9a. m/z (ES) 548 (МН)+.
Стадия C: Получение {1-[1-(4-хлорфенил)-1-(4-фенил-1,3-оксазол-2-ил)пропил]-1Н-индол-4-ил}трет-бутилкарбамата (9c)
Перйодинан Десса-Мартина (46 мг, 0,109 ммоль) добавляли к перемешиваемому раствору 9b (40 мг, 0,073 ммоль) в DCM (0,73 мл) при 0°C. Через 2 часа, реакционную смесь гасили добавлением насыщенного водного раствора тиосульфата натрия и экстрагировали DCM. Объединенные органические фракции промывали водой и солевым раствором, высушивали (Na2SO4), фильтровали и концентрировали в вакууме, получая сырой остаток, который растворяли в DCM (1 мл) и добавляли по каплям к перемешиваемому раствору, в котором трифенилфосфин (172 мг, 0,657 ммоль), затем триэтиламин (31 мкл, 0,220 ммоль), добавляли к йоду (28 мг, 0,110 ммоль) в DCM (1 мл) при 0°C. Полученную смесь нагревали до комнатной температуры и через 1 час гасили насыщенным водным раствором тиосульфата натрия. Смесь экстрагировали этилацетатом, и объединенные органические фракции промывали водой и солевым раствором, высушивали (Na2SO4), фильтровали и концентрировали в вакууме, получая сырой остаток, который очищали флэш-хроматографией на силикагеле (элюирование в градиенте; 0%-50% EtOAc/гексаны в качестве элюента), получая целевое соединение 9c. m/z (ES) 528 (МН)+.
Стадия D: Получение N-{1-[1-(4-хлорфенил)-1-(4-фенил-1,3-оксазол-2-ил)пропил]-1Н-индол-4-ил}метансульфонамида (77)
Соединение 77 получали из соединения 9c в две стадии согласно процедурам, описанным в Примере 3, стадия B, заменяя соединение 3a соединением 9c, продукт которых превращали в соединение 77 согласно процедурам, описанным в Примере 3, стадия C. m/z (ES) 528 (MNa)+; IP = C оценка значений.
ПРИМЕР 10
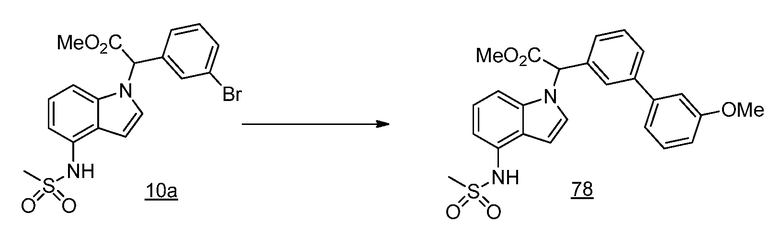
Стадия A: Получение (3-бромфенил){4-[(метилсульфонил)амино]-1Н-индол-1-ил}метилацетата (10a)
Соединение 10a получали, первоначально вводят в реакцию i-1i с 4-нитроиндолом следующими процедурами как описано в Примере 1. Продукт реакции вводили в реакцию в условиях, описанных в Примере 2, стадия A, затем Пример 2, стадия C, получая целевое соединение 10a. m/z (ES) 437 (МН)+.
Стадия B: Получение 2-(3'-метоксибифенил-3-ил)-2-{4-[(метилсульфонил)амино]-1Н-индол-1-ил}метилбутаноата (78)
[1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (II)(17 мг, 0,24 ммоль) добавляли к перемешиваемому раствору 10a (50 мг, 0,114 ммоль) и 3-метоксифенилбороновой кислоты (21 мг, 0,140 ммоль) в диоксане (0,6 мл) и насыщенном водном растворе бикарбоната натрия (0,125 мл) при комнатной температуре. Полученный раствор облучали в микроволновом реакторе при 120°C в течение 10 мин. После охлаждения до комнатной температуры, реакционную смесь фильтровали через короткую колонку для элюирования CELITE® с EtOAc. Полученное сырое масло очищали препаративной ВЭЖХ с обратной фазой на стационарной фазе YMC Pack Pro C18 (CH3CN/H2O в качестве элюента, 0,05% TFA в качестве модификатора), с последующей лиофилизацией очищенных фракций, получая целевое соединение 78. m/z (ES) 465 (МН)+; IP = C оценка значений.
Следуя процедурам, подобным описанным выше в Примере 10, получали следующие дополнительные соединения, представленные в Таблице 10:
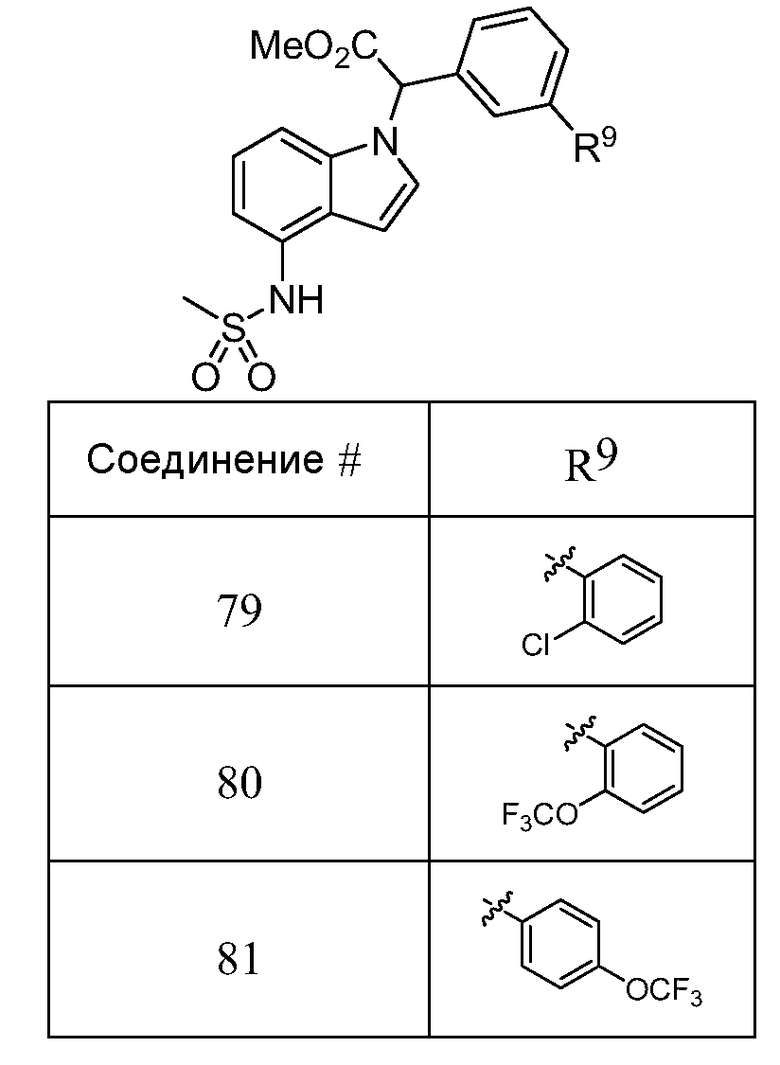
Таблица 10. Данные m/z (МН)+ исходного иона и Первичного IP Теста для соединений
79: (2'-хлорбифенил-3-ил){4-[(метилсульфонил)амино]-1Н-индол-1-ил}метилацетат: m/z (ES) 469 (МН)+; IP = C оценка значений.
80: {4-[(метилсульфонил)амино]-1Н-индол-1-ил} [2'-(трифторметокси)бифенил-3-ил]метилацетат: m/z (ES) 519 (МН)+; IP = C оценка значений.
81: {4-[(метилсульфонил)амино]-1Н-индол-1-ил}[4'-(трифторметокси)бифенил-3-ил]метилацетат: m/z (ES) 519 (МН)+; IP = C оценка значений.
ПРИМЕР 11
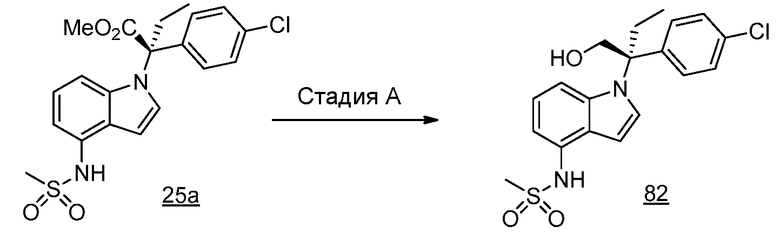
Стадия A: Получение (R)-N-{1-[(2R)-2-(4-хлорфенил)-1-гидроксибутан-2-ил]-1Н-индол-4-ил}метансульфонамида (82)
Боргидрид лития (15 мг, 0,713 ммоль) добавляли к перемешиваемому раствору 25a (200 мг, 0,475 ммоль) в THF (2,4 мл) при 0°C, и реакционной смеси давали нагреться до комнатной температуры в течение ночи. Избыток боргидрида лития гасили аккуратным добавлением 1M HCl, и полученную смесь разделяли между этилацетатом и солевым раствором. Слои разделяли, и органический слой высушивали (Na2SO4), фильтровали и концентрировали в вакууме. Полученный сырой остаток очищали флэш-хроматографией на силикагеле (элюирование в градиенте; 0%-60% EtOAc/гексаны в качестве элюента), получая целевое соединение 82. m/z (ES) 393 (МН)+; IP = А оценка значений
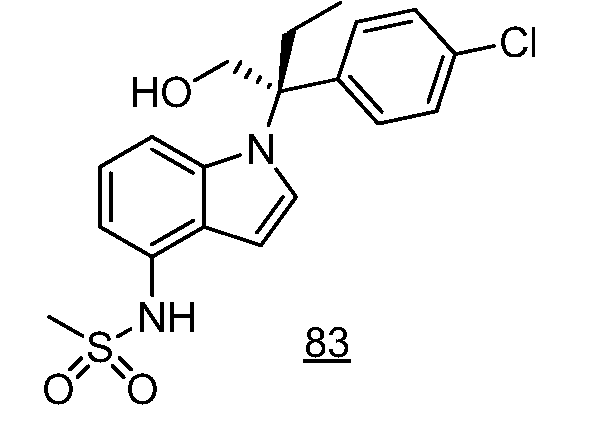
Следуя процедурам, подобным описанным выше в Примере 11, получали соединение 83, заменяя соединение 25a соединением 25b. m/z (ES) 393 (МН)+; IP = B оценка значений.
ПРИМЕР 12
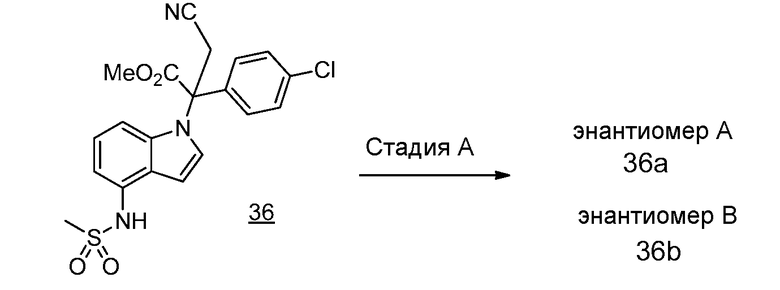
Энантиомеры 36a и 36b разделяли, используя препаративную хиральную ВЭЖХ с нормальной фазой. Раствор 36 в смеси DCM/MeOH/ацетонитрил вводили на полупрепаративную (250×30 мм) колонку для ВЭЖХ CHIRALCEL® OD-H (доступную от Chiral Technologies, Inc., Exton, Pa.) (элюируя 40% [(2:1) MeOH:ацетонитрил]/CO2 с температурой колонки 35°C со скоростью 70 мл/мин. с УФ-детекцией при 230 нм). Энантиомеры разделяли, причем более быстро элюирующий энантиомер 36a имел время удерживания 2,33 мин. и более медленно элюирующий энантиомер 36b имел время удерживания 2,93 мин., причем время удерживания получали в результате инъекции на колонку для аналитической ВЭЖХ CHIRALCEL® OD-H (4,6×250 мм, 2,1 мл/мин., 40% [(2:1) MeOH:ацетонитрил]/CO2, 35°C). Отделенные фракции концентрировали, чтобы получить энантиомеры 36a и 36b. В условиях разделения, приведенных выше, более медленно элюирующий энантиомер 36b был предпочтительным для получения конечных продуктов. Для 36a: m/z (ES) 432 (МН)+; IP = C оценка значений. Для 36b: m/z (ES) 432 (МН)+; IP = А оценка значений.
ПРИМЕР 13
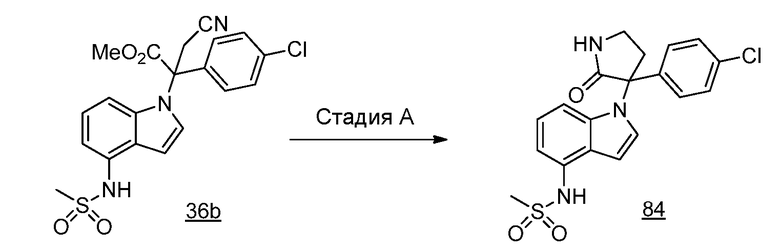
Стадия A: Получение N-{1-[3-(4-хлорфенил)-2-оксопирролидин-3-ил]-1Н-индол-4-ил}метансульфонамида (84)
Боргидрид натрия (13 мг, 0,347 ммоль) добавляли в нескольких частях к перемешиваемому раствору 36b (30 мг, 0,069 ммоль) и гексагидрата хлорида кобальта (II) (8,3 мг, 0,035 ммоль) в метаноле (0,35 мл), и реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь гасили аккуратным добавлением 1M HCl и разбавляли ацетонитрилом. Полученный раствор очищали непосредственно препаративной ВЭЖХ с обратной фазой на стационарной фазе YMC Pack Pro C18 (CH3CN/H2O в качестве элюента, 0,05% TFA в качестве модификатора) с последующей лиофилизацией очищенных фракций, получая целевое соединение 84. m/z (ES) 404 (МН)+. IP = B оценка значений
ПРИМЕР 14
N-(1-(2-(4-хлорфенил)-1-гидроксибутан-2-ил)-6-фтор-1Н-индол-4-ил)метансульфонамид (Соединение 85)
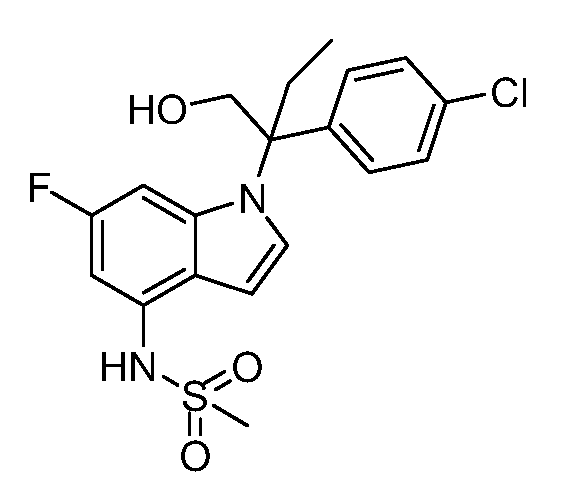
Стадия A: 5-фтор-2-метил-1,3-динитробензол
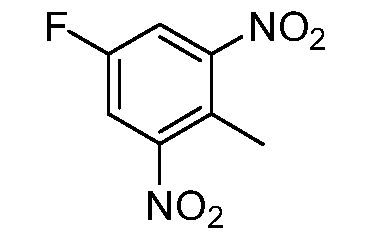
Дымящую серную кислоту (105 мл) добавляли по каплям к 4-фтор-1-метил-2-нитробензолу (30 г, 0,194 моль) при температуре от -5°C до ~ 0°C, и смесь дымящей серной кислоты (54 мл) и дымящей азотной кислоты (18 мл) добавляли по каплям при температуре от -5°C до ~ 0°C в течение 3 часов. После полного добавления, реакционную смесь перемешивали в течение 3 часов при температуре окружающей среды. TLC показала полное превращение исходного материала в продукт. Реакционную смесь лили в лед и экстрагировали дихлорметаном. Объединенные органические слои промывали водой (600 мл), насыщ. раствором бикарбоната натрия (600 мл) и солевым раствором (600 мл). Их высушивали над безводным сульфатом магния и концентрировали при пониженном давлении, получая сырой продукт, который очищали хроматографией на колонках с силикагелем (РЕ:ЕА = 20: 1), получая целевое соединение.
Стадия B: 2-(4-фтор-2,6-динитрофенил)-N,N-диметилэтенамин
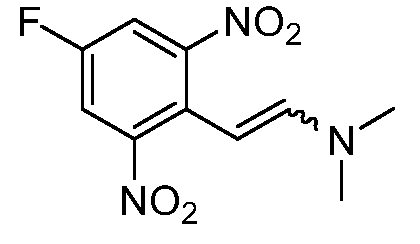
К раствору 5-фтор-2-метил-1,3-динитробензола (15 г, 0,075 моль) в N,N-диметилформамиде (300 мл) добавляли диметилформамид диметилацеталь (89 г, 0,75 моль). Ярко-красную реакционную смесь нагревали при 120°C в течение 5 часов, затем концентрировали в вакууме, получая 18 г сырого продукта, который использовали на следующей стадии без дальнейшей очистки. LC/MS m/z = 256,2 [M+H]+.
Стадия C: 6-фтор-1Н-индол-4-амин
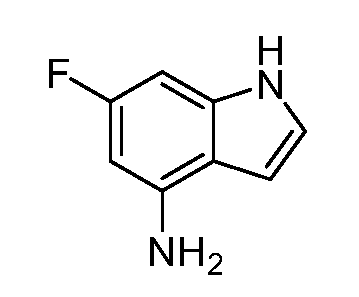
2-(4-фтор-2,6-динитрофенил)-N,N-диметилэтенамин (18 г, 0,071 моль) растворяли в этаноле (1000 мл) и загружали на 10% палладий на углероде (2,0 г). Смесь гидрировали при комнатной температуре в течение ночи. Смесь фильтровали через целит и промывали этанолом. Фильтрат объединяли и упаривали в вакууме. Остаток очищали флэш-хроматографией на силикагеле (РЕ:ЕА = 2: 1), получая целевое соединение. LC/MS m/z = 151,2 [M+H]+.
Стадия D: 1-(4-хлорбензил)-6-фтор-1Н-индол-4-амин
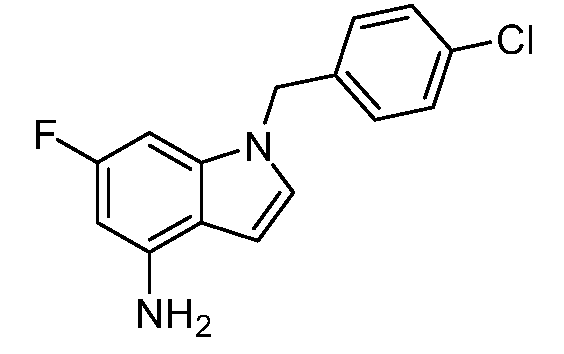
Смесь 6-фтор-1Н-индол-4-амина (5 г, 0,033 моль), 4-хлорбензил хлорида (5,55 г, 0,035 моль) и гидроксида натрия (1,32 г, 0,033 моль) нагревали при 60°C в течение 3 часов. Растворитель выпаривали, и остаток растворяли в ЕА (150 мл) и воде (70 мл). Водный слой экстрагировали ЕА и промывали солевым раствором (100 мл), высушивали над сульфатом натрия и концентрировали в вакууме. Остаток очищали флэш-хроматографией на силикагеле (РЕ:ЕА = 8:1), получая целевое соединение. LC/MS m/z = 275,1 [M+H]+.
Стадия E: 1-(4-хлорбензил)-6-фтор-1Н-индол-4-ил-трет-бутилкарбамат
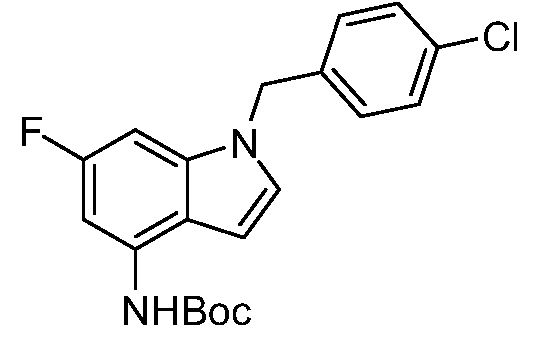
Смесь 1-(4-хлорбензил)-6-фтор-1Н-индол-4-амина (7 г, 0,026 моль) и ди-трет-бутил бикарбоната (11 г, 0,051 моль) в трет-бутиловом спирте (100 мл) нагревали при 50°C в течение 16 часов. Растворитель выпаривали, и твердое вещество фильтровали и промывали РЕ, получая целевое соединение. LC/MS m/z = 375,1 [M+H]+.
Стадия F: 2-(4-(трет-бутоксикарбониламино)-6-фтор-1Н-индол-1-ил)-2-(4-хлорфенил)уксусная кислота
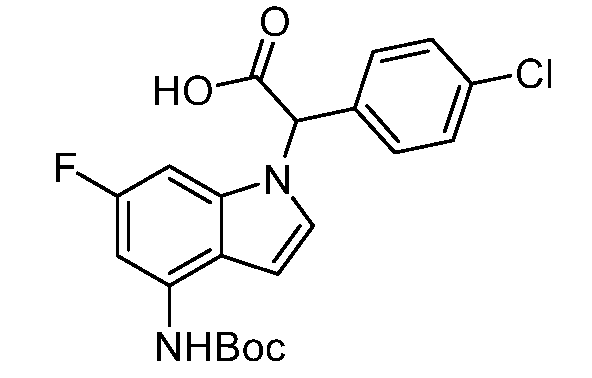
К смеси 1-(4-хлорбензил)-6-фтор-1Н-индол-4-ил-трет-бутилкарбамата (8 г, 0,021 моль) в THF (300 мл) добавляли по каплям BuLi (2,5 м., 34 мл, 0,084 моль) при -78°C и смеси перемешивали в течение еще 30 минут при -78°C. Добавляли избыток твердой углекислоты, и раствор медленно нагревали до комнатной температуры. Полученную смесь разделяли между этилацетатом и насыщенным водным раствором хлорида аммония. Водный слой экстрагировали ЕА и промывали солевым раствором (100 мл), высушивали над сульфатом натрия и концентрировали в вакууме, получая целевое соединение (8 г, выход 89%). LC/MS m/z = 363,1 [M-tBu+H]+.
Стадия G: 2-(4-амино-6-фтор-1Н-индол-1-ил)-2-(4-хлорфенил)метилацетат
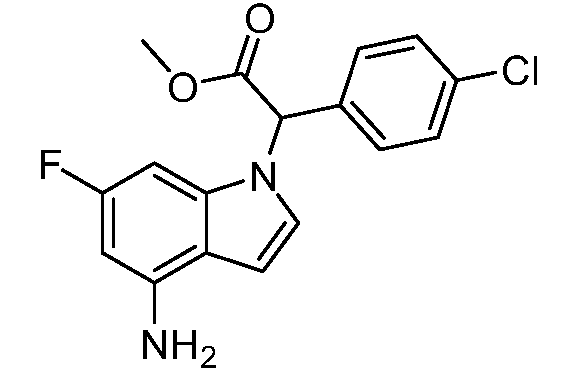
К раствору 2-(4-(трет-бутоксикарбониламино)-6-фтор-1Н-индол-1-ил)-2-(4-хлорфенил)уксусной кислоты (8 г, 0,019 моль) в метаноле (100 мл) добавляли серную кислоту (1 мл), и смесь перемешивали в течение 2 часов при 50°C. Полученную смесь разделяли между этилацетатом и насыщенным водным раствором бикарбоната натрия. Водный слой экстрагировали ЕА и промывали солевым раствором (100 мл), высушивали над сульфатом натрия и концентрировали в вакууме, получая сырое целевое соединение (5,7 г), которое использовали на следующей стадии без дальнейшей очистки. LC/MS m/z = 333,1 [M+H]+.
Стадия Н: 2-(4-(трет-бутоксикарбониламино)-6-фтор-1Н-индол-1-ил)-2-(4-хлорфенил)метилацетат
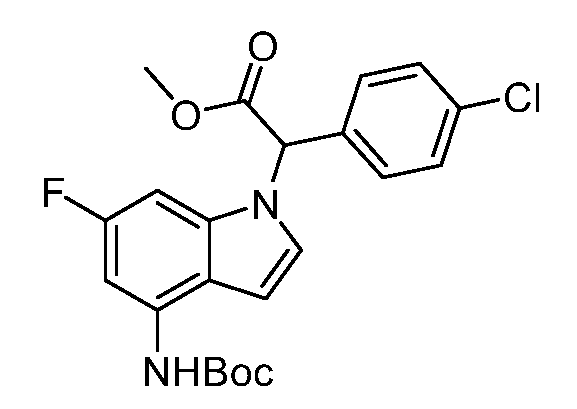
Смесь 2-(4-амино-6-фтор-1Н-индол-1-ил)-2-(4-хлорфенил)метилацетата (5,7 г, 0,017 моль) и ди-трет-бутил бикарбоната (7,4 г, 0,034 моль) в трет-бутиловом спирте (80 мл) нагревали при 50°C в течение 10 часов. Растворитель выпаривали, и остаток очищали флэш-хроматографией на силикагеле (РЕ:ЕА = 8:1), получая целевое соединение. LC/MS m/z = 377,1 [M-tBu+H]+.
Стадия I: 2-(4-(трет-бутоксикарбониламино)-6-фтор-1Н-индол-1-ил)-2-(4-хлорфенил)метилбутаноат
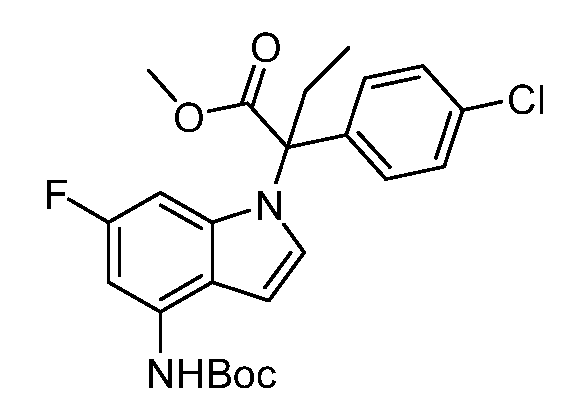
LiHMDS(20 мл, 0,020 моль) добавляли по каплям к перемешиваемому раствору 2-(4-(трет-бутоксикарбониламино)-6-фтор-1Н-индол-1-ил)-2-(4-хлорфенил)метилацетат (4,32 г, 0,010 моль) в THF (80 мл) и HMPA (20 мл) при -78°C. Полученную смесь перемешивали еще 1 час при -78°C. К вышеописанному раствору добавляли йодэтан (2,3 г, 0,015 моль). Реакционную смесь нагревали до -20°C и перемешивали в течение 20 мин. Смесь гасили насыщенным раствором хлорида аммония и экстрагировали ЕА, промывали солевым раствором и высушивали над сульфатом натрия и концентрировали в вакууме. Остаток очищали флэш-хроматографией на силикагеле (РЕ:ЕА = 8:1), получая целевое соединение. LC/MS m/z = 405,1 [M-tBu+H]+.
Стадия J: 2-(4-амино-6-фтор-1Н-индол-1-ил)-2-(4-хлорфенил)метилбутаноат
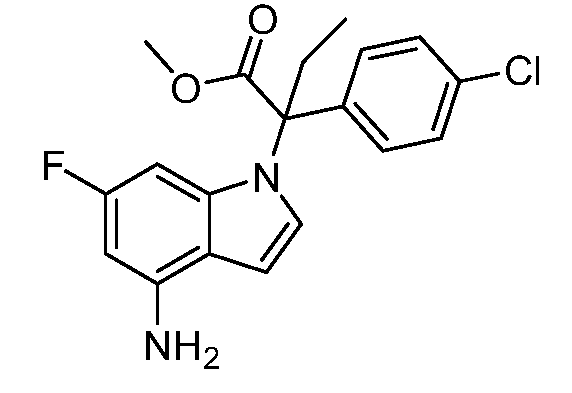
Трифторуксусную кислоту (4 мл) добавляли быстро по каплям к перемешиваемому раствору 2-(4-(трет-бутоксикарбониламино)-6-фтор-1Н-индол-1-ил)-2-(4-хлорфенил)метилбутаноата (0,92 г, 2 ммоль) в DCM (10 мл) при 0°C. Через 1 час реакционную смесь гасили аккуратным добавлением насыщенного водного раствора бикарбоната натрия с последующей экстракцией простым эфиром. Органические слои промывали насыщенным водным раствором бикарбоната натрия, высушивали над сульфатом натрия, фильтровали и концентрировали в вакууме, получая целевое соединение. LC/MS m/z = 361,1 [M+H]+.
Стадия K: 2-(4-хлорфенил)-2-(6-фтор-4-(метилсульфонамидо)-1Н-индол-1-ил)метилбутаноат
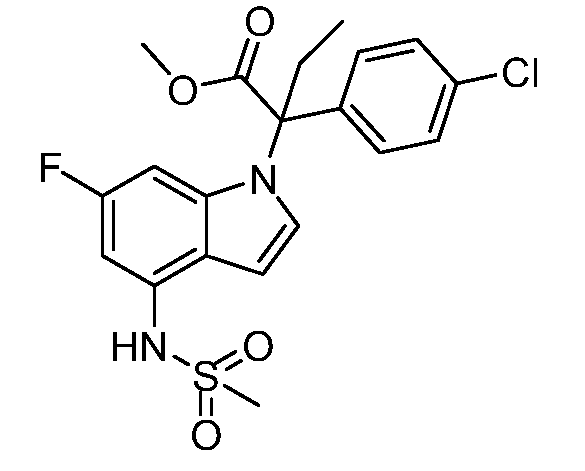
Метансульфонил хлорид (0,23 г, 2 ммоль) добавляли к перемешиваемому раствору 2-(4-амино-6-фтор-1Н-индол-1-ил)-2-(4-хлорфенил)метилбутаноата (0,6 г, 1,7 ммоль) и 4-метилморфолина (0,25 г, 2,5 ммоль) в DCM (8 мл) при 0°C. Через 30 минут добавляли дополнительную часть метансульфонил хлорида (0,1 г, 1 ммоль). Через 1 час реакционную смесь разделяли между DCM и насыщенным водным раствором хлорида аммония. Слои разделяли, и водный слой экстрагировали DCM. Объединенные органические слои промывали солевым раствором, высушивали над сульфатом натрия, фильтровали и концентрировали в вакууме. Остаток очищали флэш-хроматографией на силикагеле (РЕ:ЕА = 2:1), получая целевое соединение. LC/MS m/z = 439,1 [M+H]+.
Стадия L: N-(1-(2-(4-хлорфенил)-1-гидроксибутан-2-ил)-6-фтор-1Н-индол-4-ил)метансульфонамид (Соединение 85)
К раствору 2-(4-хлорфенил)-2-(6-фтор-4-(метилсульфонамидо)-1Н-индол-1-ил)метилбутаноата (0,5 г, 1,14 ммоль) в THF (5 мл) добавляли по каплям раствор литий-алюминийгидрида (1,4 мл, 1 М в THF) при -78°C. Реакционную смесь перемешивали в течение 0,5 часа при -78°C. Реакцию завершали под контролем TLC и LCMS, затем 3M раствор HCl добавляли аккуратно в реакционную смесь до рН = 2~3 в ванне со льдом. Органический слой отделяли, и водный слой экстрагировали ЕА. Объединенный органический слой промывали солевым раствором и высушивали над безводным сульфатом натрия. Полученную смесь фильтровали и концентрировали в вакууме. Остаток очищали флэш-хроматографией на силикагеле (РЕ:ЕА = 2:1), получая целевое соединение. LC/MS m/z = 411,1 [M+H]+.
Используя процедуру, описанную в Примере 14, но на стадии J заменяя 2-(4-(трет-бутоксикарбониламино)-6-фтор-1Н-индол-1-ил)-2-(4-хлорфенил)бутаноат 2-(4-(трет-бутоксикарбониламино)-1Н-индол-1-ил)-2-(4-хлорфенил)метилбутаноатом, 2-(4-(трет-бутоксикарбониламино)-1Н-индол-1-ил)-2-(2,4-дихлорфенил)метилбутаноатом, 2-(4-(трет-бутоксикарбониламино)-1Н-индазол-1-ил)-2-(4-хлорфенил)метилбутаноатом, 2-(4-(трет-бутоксикарбониламино)-1Н-индазол-1-ил)-2-(4-(трифторметил)фенил)метилбутаноатом, получали соединения 86-89 из представленных в Таблице 11, соответственно.
Соединение 86 получали, используя энантиомер в Примере 3 стадии D в качестве исходного материала.
Соединение 88 получали в приведенных ниже условиях хирального разделения:
Колонка: AS-H
Мобильная фаза: A: Гексан, B: EtOH (0,1% DEA), A:B = 70:30 при 1,0 мл/мин.
Температура колонки: 40,2°C
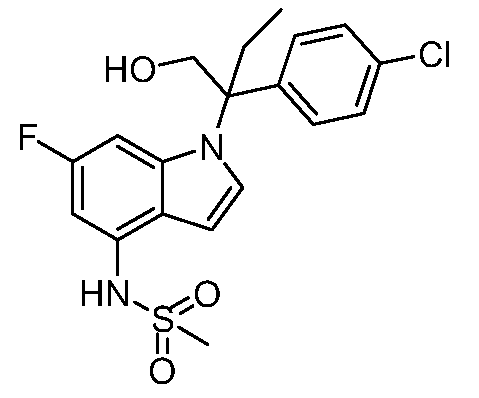
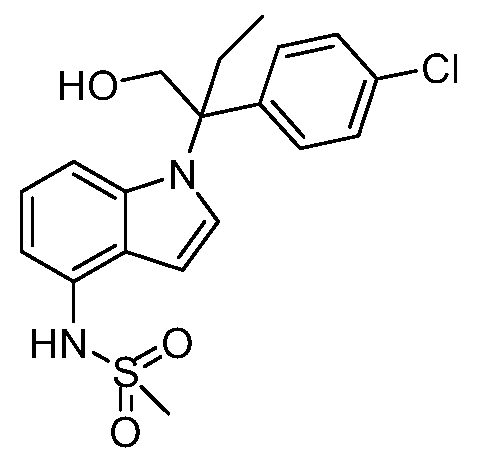
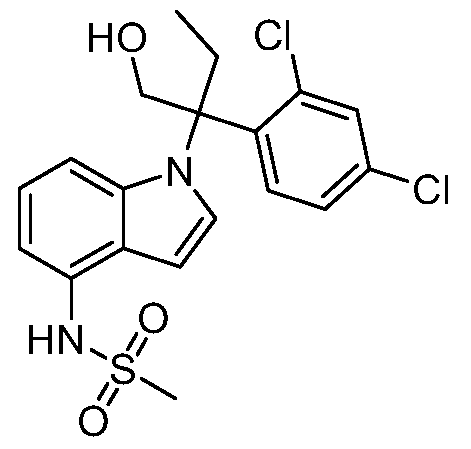
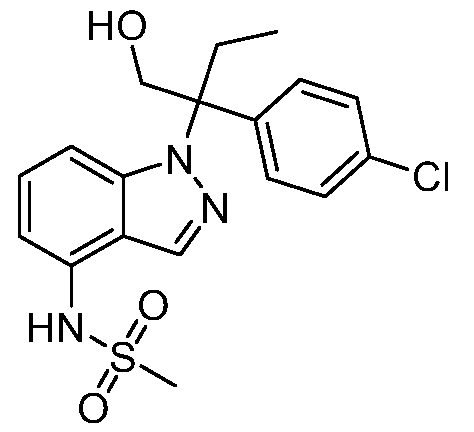
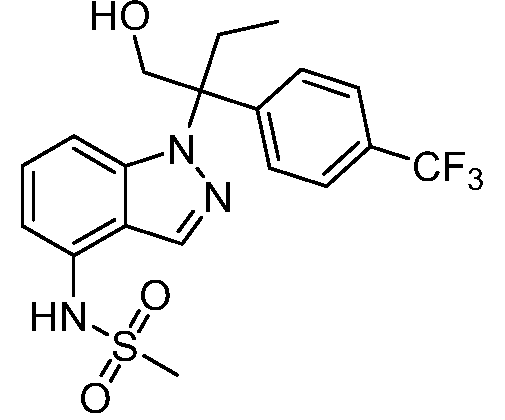
ПРИМЕР 15
N-(1-(3-(4-хлорфенил)-2-гидрокси-2-метилпентан-3-ил)-6-фтор-1Н-индол-4-ил)метансульфонамид (Соединение 90)
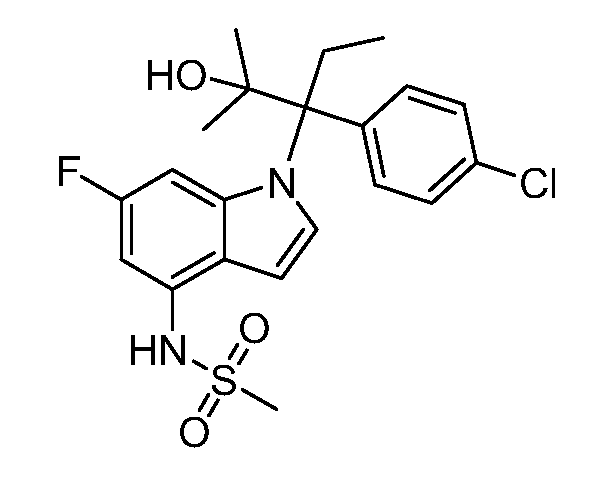
Стадия A: 1-(3-(4-хлорфенил)-2-гидрокси-2-метилпентан-3-ил)-6-фтор-1Н-индол-4-ил-трет-бутилкарбамат
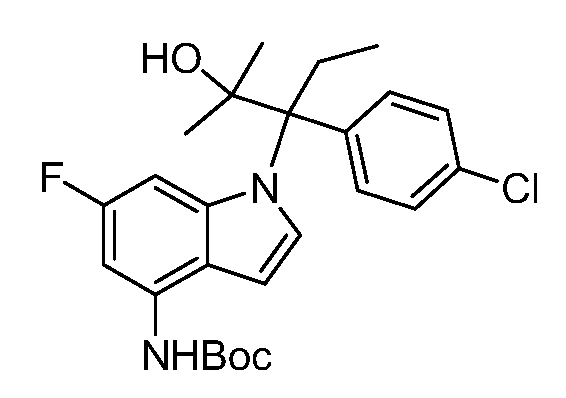
К раствору 2-(4-(трет-бутоксикарбониламино)-6-фтор-1Н-индол-1-ил)-2-(4-хлорфенил)метилбутаноата (1,0 г, 2,17 ммоль), как описано в Примере 14 Стадия I, в простом эфире (5 мл) добавляли CH3Li (7,2 мл, 21,7 ммоль, 3,0 М в простом эфире) по каплям при 0°C и перемешивали в течение 30 мин. Смесь гасили насыщенным раствором хлорида аммония, и экстрагировали этилацетатом. Органические слои промывали солевым раствором, высушивали над сульфатом натрия, фильтровали и упаривали. Остаток очищали Prep-TLC (элюент: этилацетат/петролейный эфир = 4:1), получая целевое соединение. LC/MS m/z = 461,2 [M+H]+.
Стадия B: 3-(4-амино-6-фтор-1Н-индол-1-ил)-3-(4-хлорфенил)-2-метилпентан-2-ол
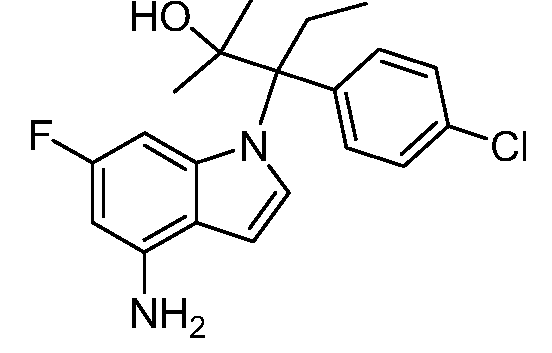
Смесь 1-(3-(4-хлорфенил)-2-гидрокси-2-метилпентан-3-ил)-6-фтор-1Н-индол-4-ил-трет-бутилкарбамата (250 мг, 0,543 ммоль) и TFA/DCM (об./об.=1/4, 10 мл) перемешивали при комнатной температуре в течение 2 часов. После удаления растворителя остаток растворяли в этилацетате (20 мл), значение рН доводили до 9~10 насыщенным раствором бикарбоната натрия, высушивали над сульфатом натрия, затем фильтровали. После удаления органического растворителя, сырое соединение получали без очистки. LC/MS m/z = 361,2 [M+H]+.
Стадия C: N-(1-(3-(4-хлорфенил)-2-гидрокси-2-метилпентан-3-ил)-6-фтор-1Н-индол-4-ил)метансульфонамид (Соединение 90)
К смеси соединения 3-(4-амино-6-фтор-1Н-индол-1-ил)-3-(4-хлорфенил)-2-метилпентан-2-ола (176 мг, 0,489 ммоль) и NMM (145 мг, 1,467 ммоль) в DCM (10,0 мл) добавляли по каплям метансульфонил хлорид (112 мг, 0,978 ммоль). Затем смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь гасили насыщенным раствором хлорида аммония и экстрагировали этилацетатом (50 мл). Органические слои промывали солевым раствором, высушивали над сухим сульфатом натрия, фильтровали и упаривали. Остаток очищали Prep-TLC (элюент: этилацетат/нефть = 4:1), получая целевое соединение. LC/MS m/z = 438,9 [M+H]+.
1H ЯМР (400 МГц, MeOD) δ 8,02 (с, 1H), 7,57-7,25 (м, 4H), 6,86 (дд, J=10,9, 2,1 Гц, 1H), 6,74 (д, J=3,6 Гц, 1H), 5,85 (д, J=11,1 Гц, 1H), 3,00 (с, 3H), 2,84-2,73 (м, 1H), 2,61-2,47 (м, 1H), 1,31 (с, 3H), 1,27 (с, 3H), 0,64 (т, J=7,3 Гц, 3H).
Используя процедуру, описанную в Примере 15, но на стадии А заменяя 2-(4-(трет-бутоксикарбониламино)-6-фтор-1Н-индол-1-ил)-2-(4-хлорфенил)бутаноат 2-(4-(трет-бутоксикарбониламино)-1Н-индол-1-ил)-2-(4-хлорфенил)метилбутаноатом, 2-(4-(трет-бутоксикарбониламино)-1Н-индазол-1-ил)-2-(4-хлорфенил)метилбутаноатом, 2-(4-(трет-бутоксикарбониламино)-1Н-индазол-1-ил)-2-(4-(трифторметил)фенил)метилбутаноатом, получали соединения 91-93 представленных в Таблице 12, соответственно. Также с использованием процедуры, описанной в Примере 15, но на стадии C заменяя метансульфонил хлорид метилсульфамоил хлоридом, диметилсульфамоил хлоридом, получали соединения 94 и 95 представленных в Таблице 12, соответственно.
Соединение 90 получали в приведенных ниже условиях хирального разделения:
Колонка: AS-H
Мобильная фаза: A: CO2, B: MeOH (0,1% DEA), A:B =70:30 при 3,0 мл/мин.
Температура колонки: 40°C
Соединение 91 получали в приведенных ниже условиях хирального разделения:
Колонка: OJ-Н
Мобильная фаза: A: N-гексан, B: EtOH (0,1% DEA), A:B =70:30 при 1,0 мл/мин.
Температура колонки: 40°C
Соединение 92 получали в приведенных ниже условиях хирального разделения:
Колонка: OJ-Н
Мобильная фаза: A: N-гексан, B: MeOH (0,1% DEA), A:B =70:30 при 3,0 мл/мин.
Температура колонки: 37,7°C
Соединение 93 получали в приведенных ниже условиях хирального разделения:
Колонка: AS-H
Мобильная фаза: A: CO2, B: MeOH, A:B = 70:30 при 3,0 мл/мин.
Температура колонки: 38,1°C
Соединения 94 и 95 получали, используя энантиомер А промежуточного соединения (пример 18 стадия D) в качестве исходного материала.
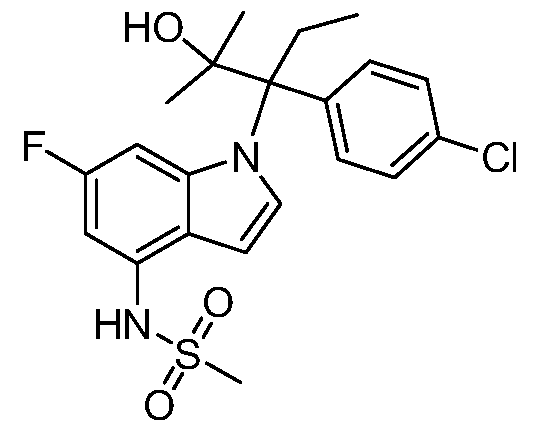
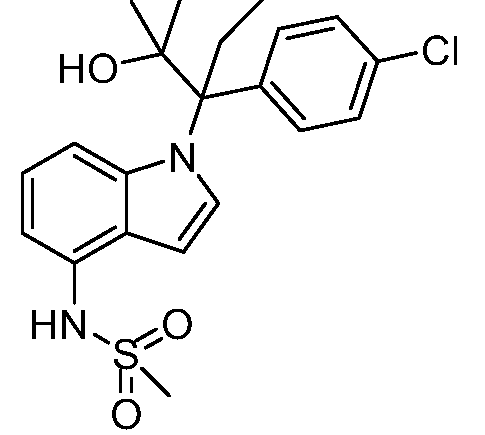
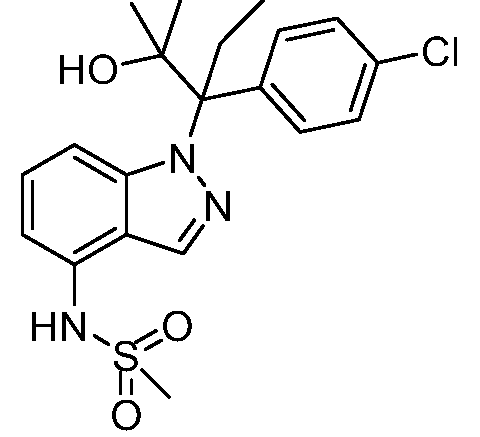
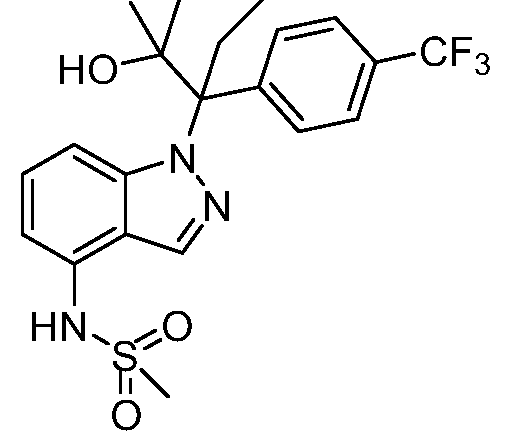
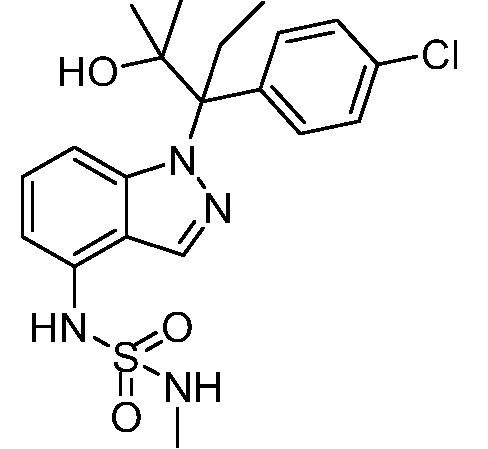
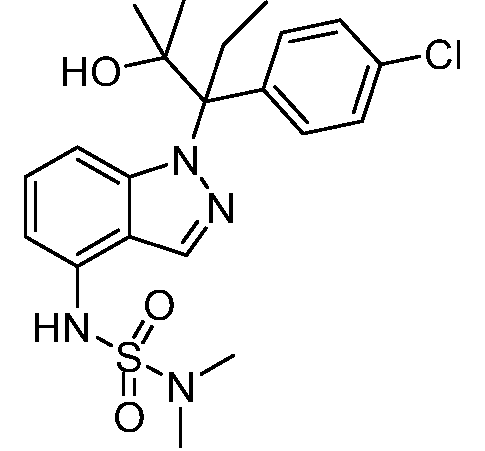
ПРИМЕР 16
N-(1-(2-(4-хлорфенил)-1-метоксибутан-2-ил)-1Н-индол-4-ил)метансульфонамид
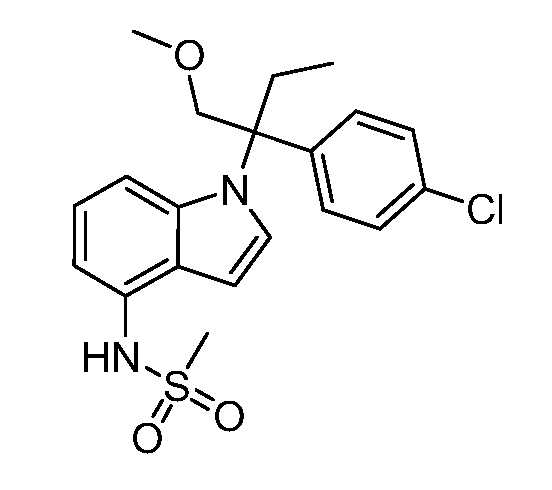
Стадия A: 2-(4-амино-1Н-индол-1-ил)-2-(4-хлорфенил)метилбутаноат
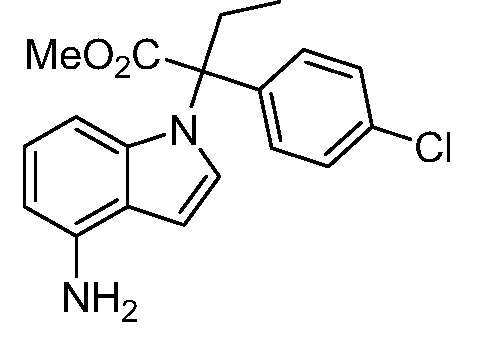
Смесь 2-(4-(трет-бутоксикарбониламино)-1Н-индол-1-ил)-2-(4-хлорфенил)метилбутаноата (500 мг), как описано в Примере 3 Стадия A, в 10 мл 4н HCl/MeOH перемешивали при комнатной температуре в течение 1 часа. Затем растворитель удаляли. Остаток растворяли в ЕА (30 мл), и значение рН доводили до 9~10 насыщенным водным раствором NaHCO3. Органические слои промывали водой (10 мл), солевым раствором, высушивали над сульфатом натрия и концентрировали, получая целевой продукт в форме твердого вещества светло-коричневого цвета. LC/MS m/z = 343,2 [M+H]+.
Стадия B: 2-(4-хлорфенил)-2-(4-(2,5-диметил-1Н-пиррол-1-ил)-1Н-индол-1-ил)метилбутаноат
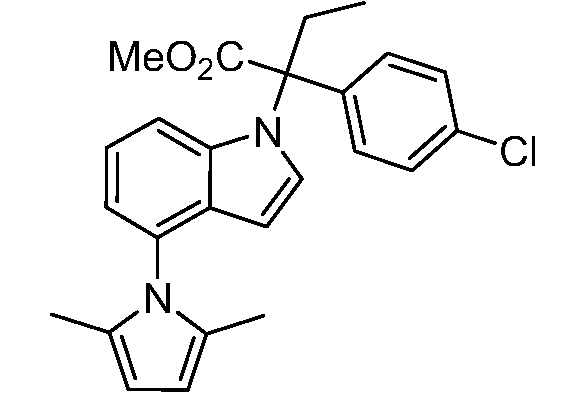
Смесь 2-(4-амино-1Н-индол-1-ил)-2-(4-хлорфенил)метилбутаноата (140 мг), гексан-2,5-диона (166 мг) и 4-метилбензолсульфоновой кислоты (3 мг) в толуоле (5 мл) перемешивали при нагревании с обратным холодильником в течение 30 мин. Концентрировали, остаток очищали хроматографией на колонке с силикагелем (5-15% этилацетата в гексанах), получая целевое соединение в форме твердого вещества желтого цвета. LC/MS m/z = 421,2 [M+H]+.
Стадия C: 2-(4-Хлорфенил)-2-(4-(2,5-диметил-1Н-пиррол-1-ил)-1Н-индол-1-ил)бутан-1-ол
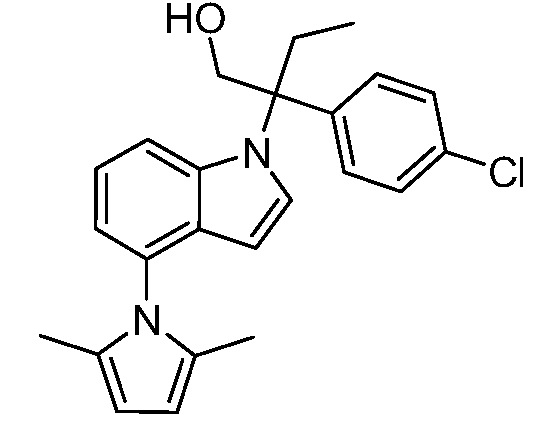
К суспензии LiBH4 (22 мг) в THF (3 мл) добавляли MeOH (32 мг, 5,43 ммоль) и раствор 2-(4-хлорфенил)-2-(4-(2,5-диметил-1Н-пиррол-1-ил)-1Н-индол-1-ил)метилбутаноата (105 мг) в THF (3 мл). Смесь перемешивали при комнатной температуре в течение 2 часов. Раствор гасили насыщенным раствором хлорида аммония и экстрагировали этилацетатом (20 мл). Органический слой промывали солевым раствором, высушивали над сульфатом натрия и упаривали. Остаток очищали хроматографией на силикагеле (25-35% этилацетата в гексанах), получая целевое соединение в форме твердого вещества светло-желтого цвета. LC/MS m/z = 393,2 [M+H]+.
Стадия D: 1-(2-(4-Хлорфенил)-1-метоксибутан-2-ил)-4-(2,5-диметил-1Н-пиррол-1-ил)-1Н-индол
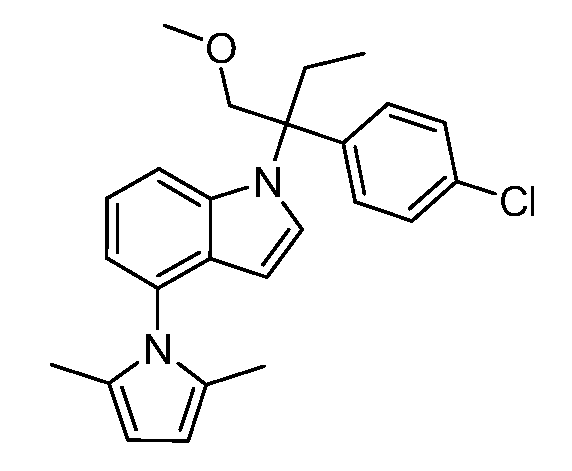
Раствор 2-(4-хлорфенил)-2-(4-(2,5-диметил-1Н-пиррол-1-ил)-1Н-индол-1-ил)бутан-1-ола (89 мг) в сухом DMF (3 мл) обрабатывали NaH (60% в масле, 18 мг) при 0°C в течение 30 минут, затем добавляли йодметан (97 мг). Смесь перемешивали при комнатной температуре в течение 2 часов. Раствор гасили насыщенным раствором хлорида аммония и экстрагировали этилацетатом (25 мл). Органические слои промывали солевым раствором, высушивали над сульфатом натрия и упаривали, получая целевое соединение в форме твердого вещества белого цвета. LC/MS m/z = 407,1 [M+H]+.
Стадия E: 1-(2-(4-Хлорфенил)-1-метоксибутан-2-ил)-1Н-индол-4-амин
К смеси 1-(2-(4-хлорфенил)-1-метоксибутан-2-ил)-4-(2,5-диметил-1Н-пиррол-1-ил)-1Н-индола (70 мг) в смеси этанол/H2O (3 мл/1 мл) добавляли NH2OH-HCl (178 мг) и триэтиламин (87 мг), и реакционную смесь нагревали с обратным холодильником в течение 18 часов в масляной бане, охлаждали до комнатной температуры, концентрировали и экстрагировали этилацетатом (30 мл). Органические слои объединяли, высушивали над безводным сульфатом натрия и концентрировали под вакуумом. Остаток очищали, используя колонку с силикагелем (20-40% этилацетата в гексане), получая целевое соединение в форме твердого вещества коричневого цвета. LC/MS m/z = 329,1 [M+H]+.
Стадия F: N-(1-(2-(4-хлорфенил)-1-метоксибутан-2-ил)-1Н-индол-4-ил)метансульфонамид
К смеси 1-(2-(4-хлорфенил)-1-метоксибутан-2-ил)-1Н-индол-4-амина (30 мг) и 4-метилморфолина (46 мг) в DCM (3 мл) по каплям добавляли метансульфонил хлорид (16 мг). Затем смесь перемешивали при комнатной температуре в течение ночи. Раствор гасили насыщенным раствором хлорида аммония и экстрагировали этилацетатом (20 мл). Органический слой промывали солевым раствором, высушивали над сульфатом натрия и упаривали. Остаток очищали хроматографией на силикагеле (20-30% этилацетата в гексане), получая целевое соединение в форме твердого вещества белого цвета. LC/MS m/z = 407,1 [M+H]+.
Используя процедуру, описанную в Примере 13, но на стадии А заменяя 2-(4-(трет-бутоксикарбониламино)-1Н-индол-1-ил)-2-(4-хлорфенил)метилбутаноат 2-(4-(трет-бутоксикарбониламино)-1Н-индазол-1-ил)-2-(4-хлорфенил)метилбутаноатом, получали соединение 97 представленных в Таблице 13.
Соединение 96 и соединение 97 были получены в приведенных ниже условиях хирального разделения:
Колонка: OZ-H
Мобильная фаза: A: CO2, B: MeOH (0,1% DEA), A:B = 70:30 при 3,0 мл/мин.
Температура колонки: 40,2°C
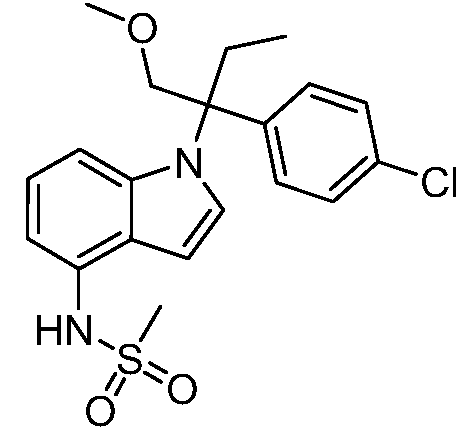
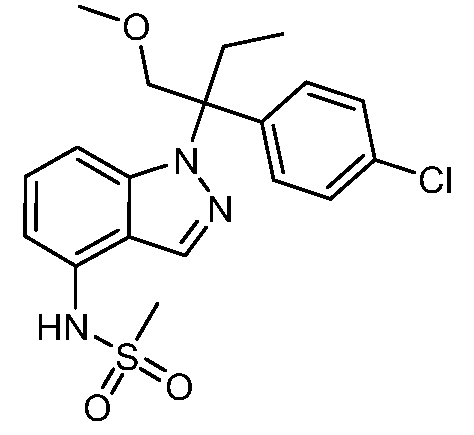
ПРИМЕР 17
N-(1-(3-(4-хлорфенил)-2-оксопентан-3-ил)-6-фтор-1Н-индол-4-ил)метансульфонамид (Соединение 98),
Стадия A: 1-(3-(4-хлорфенил)-2-оксопентан-3-ил)-6-фтор-1Н-индол-4-ил-трет-бутилкарбамат
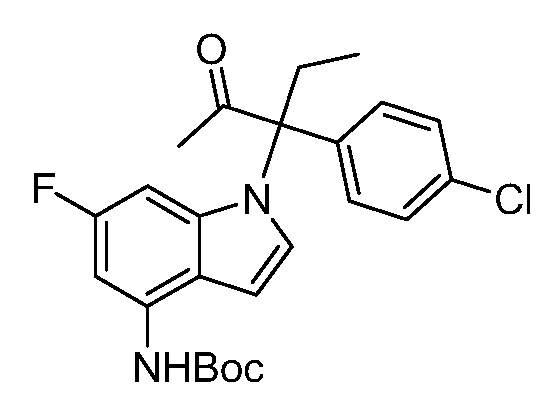
Оба продукта, 1-(3-(4-хлорфенил)-2-оксопентан-3-ил)-6-фтор-1Н-индол-4-ил-трет-бутилкарбамат и 1-(3-(4-хлорфенил)-2-гидрокси-2-метилпентан-3-ил)-6-фтор-1Н-индол-4-ил-трет-бутилкарбамат, были получены в Примере 15 на стадии A.
Стадия B: 3-(4-амино-6-фтор-1Н-индол-1-ил)-3-(4-хлорфенил)пентан-2-он
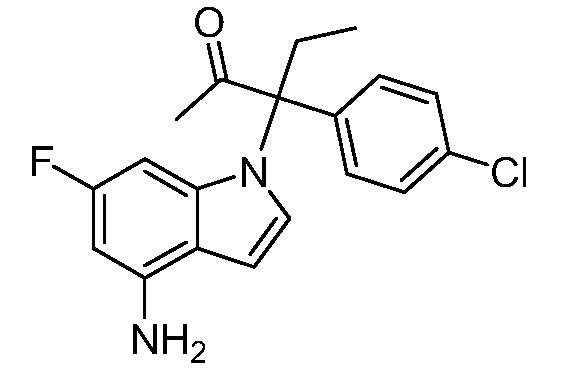
Используя ту же самую процедуру, описанную для Примера 15, стадия B, но заменяя 1-(3-(4-хлорфенил)-2-гидрокси-2-метилпентан-3-ил)-6-фтор-1Н-индол-4-ил-трет-бутилкарбамат 1-(3-(4-хлорфенил)-2-оксопентан-3-ил)-6-фтор-1Н-индол-4-ил-трет-бутилкарбаматом, получали целевое соединение. LC/MS m/z = 345,2 [M+H]+.
Стадия C: N-(1-(3-(4-хлорфенил)-2-оксопентан-3-ил)-6-фтор-1Н-индол-4-ил)метансульфонамид (Соединение 98)
Используя ту же самую процедуру, описанную для Примера 15, стадия C, но заменяя 3-(4-амино-6-фтор-1Н-индол-1-ил)-3-(4-хлорфенил)-2-метилпентан-2-ол 3-(4-амино-6-фтор-1Н-индол-1-ил)-3-(4-хлорфенил)пентан-2-оном, получали целевое соединение. LC/MS m/z = 423,2 [M+H]+.
Используя процедуру, описанную в Примере 17, но на стадии А заменяя 2-(4-(трет-бутоксикарбониламино)-6-фтор-1Н-индол-1-ил)-2-(4-хлорфенил)бутаноат 2-(4-(трет-бутоксикарбониламино)-1Н-индол-1-ил)-2-(4-хлорфенил)метилбутаноатом, 2-(4-(трет-бутоксикарбониламино)-1Н-индазол-1-ил)-2-(4-хлорфенил)метилбутаноатом, 2-(4-(трет-бутоксикарбониламино)-1Н-индол-1-ил)-2-(4-(трифторметил)фенил)метилбутаноатом, 2-(4-(трет-бутоксикарбониламино)-1Н-индазол-1-ил)-2-(4-(трифторметил)фенил)метилбутаноатом, получали соединения 99-104 представленных в Таблице 14, соответственно. Также с использованием процедуры, описанной в Примере 17, но на стадии C заменяя метансульфонил хлорид метилсульфамоил хлоридом, получали соединение 105 представленных в Таблице 14.
Соединение 100 и соединение 101 были получены в приведенных ниже условиях хирального разделения:
Колонка: OJ-Н
Мобильная фаза: A: Гексан, B: MeOH, A:B = 60:40 при 3,0 мл/мин.
Температура колонки: 40,7°C
Соединение 102 и соединение 103 были получены в приведенных ниже условиях хирального разделения:
Колонка: AD-H
Мобильная фаза: A: CO2, B: MeOH, A:B = 85:15 при 3,0 мл/мин.
Температура колонки: 41,3°C
Соединение 104 получали в приведенных ниже условиях хирального разделения:
Колонка: OJ-Н
Мобильная фаза: A: CO2, B: MeOH (0,1% DEA), A:B = 70:30 при 3,0 мл/мин.
Температура колонки: 39,9°C
Соединение 105 получали, используя энантиомер А в Примере 18 стадия D как исходный материал.
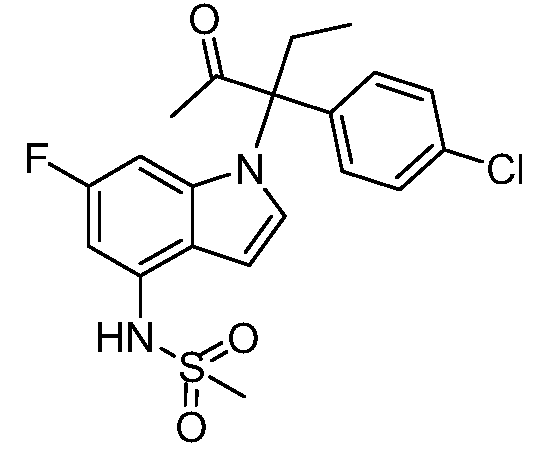
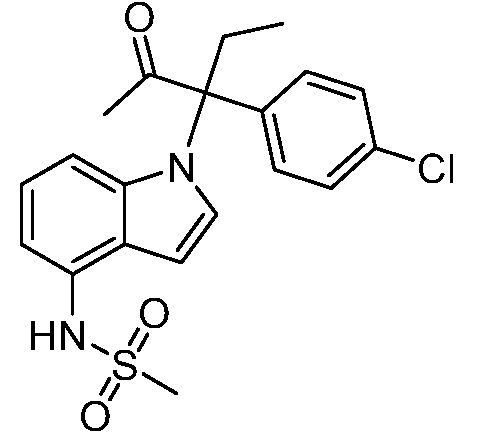
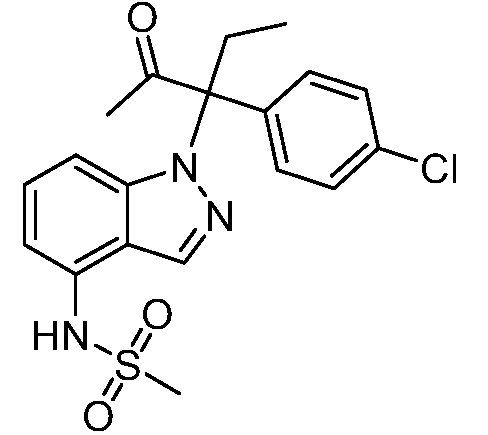
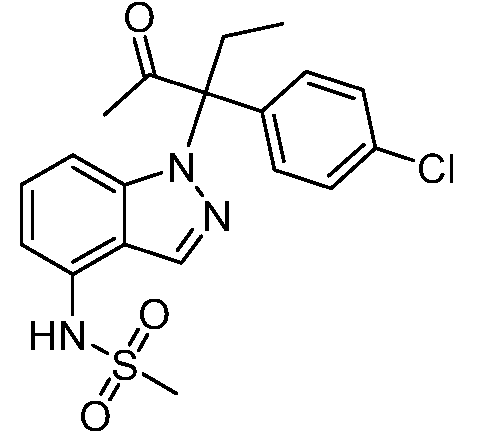
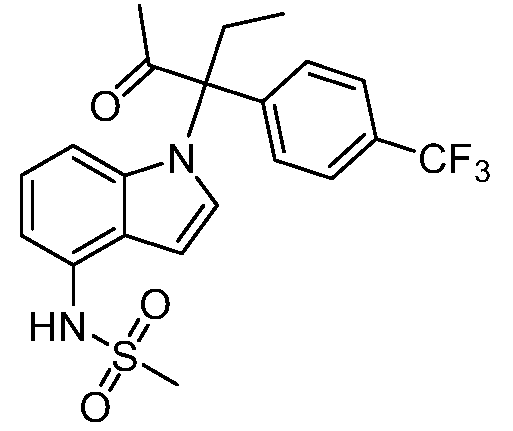
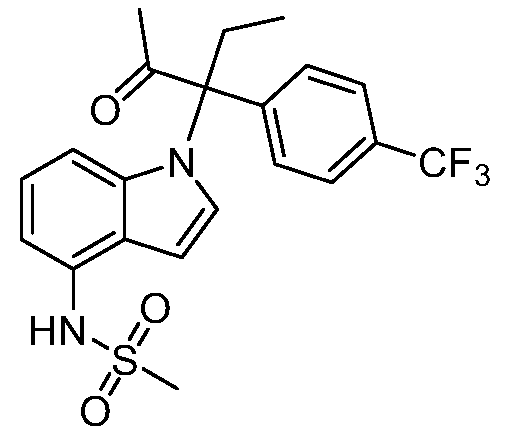
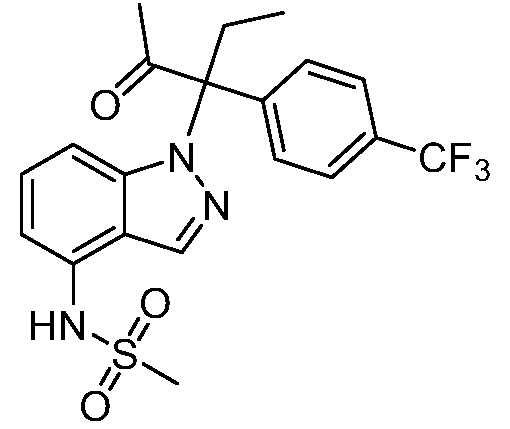
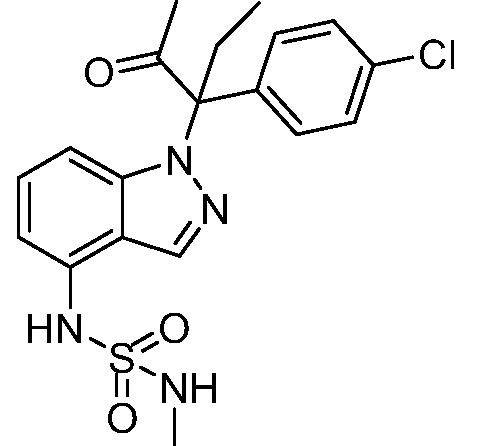
ПРИМЕР 18
N-{1-[3-(4-хлорфенил)-2-гидроксипентан-3-ил]-1Н-индазол-4-илметансульфонамид (Соединение 106),
Стадия A: смесь 2-(4-хлорфенил)-2-(4-нитро-1Н-индазол-1-ил)метилацетата и 2-(4-хлорфенил)-2-(4-нитро-2Н-индазол-2-ил)метилацетата
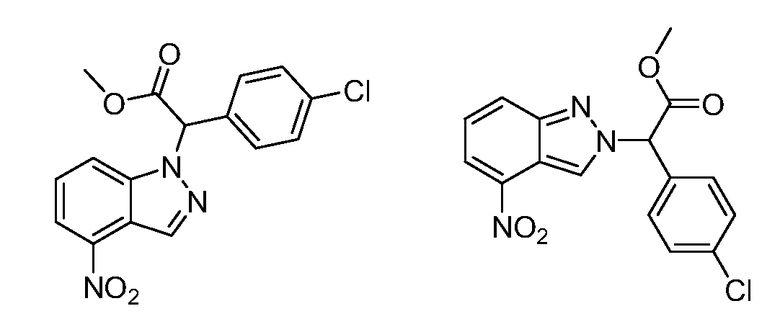
В колбу загружали K2CO3 (85 г, 613 ммоль) в CH3CN (500 мл), и затем добавляли раствор 4-нитро-1Н-индазола (50 г, 307 ммоль) в CH3CN (500 мл). После того, как раствор перемешивали в течение 30 минут, раствор 2-бром-2-(4-хлорфенил)метилацетата (100 г, 379 ммоль), как описано в Примере, 1 Стадия A, в CH3CN (250 мл) добавляли по каплям при комнатной температуре. Смесь перемешивали в течение ночи и затем фильтровали через слой целита. Фильтрат концентрировали, и остаток (черное масло) замораживали, получая твердое вещество. Добавляли простой эфир (150 мл), и смесь перемешивали в течение 15 мин. Затем ее фильтровали, и твердое вещество промывали РЕ (100 мл). Сырое целевое соединение (39 г, 37%) использовали непосредственно на следующей стадии.
Стадия B: 2-(4-амино-1Н-индазол-1-ил)-2-(4-хлорфенил)метилацетат
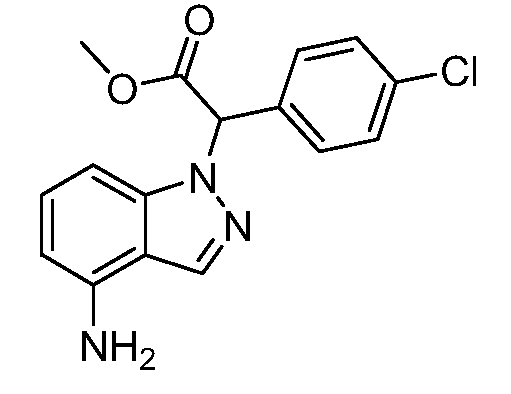
Смесь продукта со Стадии А (39 г, 113 ммоль) растворяли в этаноле (400 мл) и загружали 10% платины на углероде (4,0 г). Смесь гидрировали при комнатной температуре 2 дня. Смесь фильтровали через целит и промывали этанолом. Фильтрат объединяли и упаривали в вакууме. Сырой продукт (22 г, 62%) использовали непосредственно на следующей стадии. LC/MS m/z = 316,1 [M+H]+.
Стадия C: 2-(4-(трет-бутоксикарбониламино)-1Н-индазол-1-ил)-2-(4-хлорфенил)метилацетат
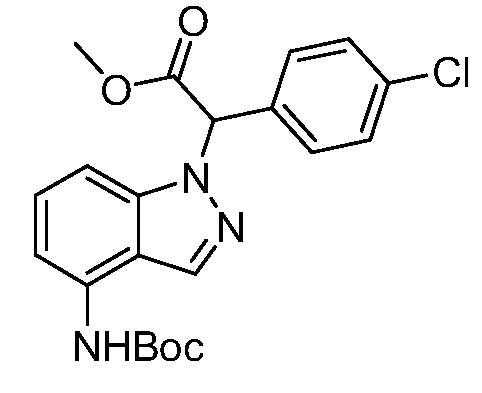
Соединение со стадии B (22 г, 70 ммоль) и (Вос)2O (30 г, 140 ммоль) растворяли в диоксане (260 мл). Затем насыщенный водный раствор Na2CO3 (50 мл) добавляли к смеси при 0°C. Смесь перемешивали при комнатной температуре в течение 2 дней. Затем растворитель удаляли при пониженном давлении, и остаток растворяли в EtOAC(500 мл), промывали H2O (100 мл), солевым раствором (100 мл). Органический слой высушивали над безводным Na2SO4, концентрировали, и остаток очищали на колонке с силикагелем (РЕ:EA = от 20:1 до 8:1), получая целевое соединение (23 г, 80%) LC/MS m/z = 416,1 [M+H]+.
Стадия D: 2-(4-(трет-бутоксикарбониламино)-1Н-индазол-1-ил)-2-(4-хлорфенил)метилбутаноат
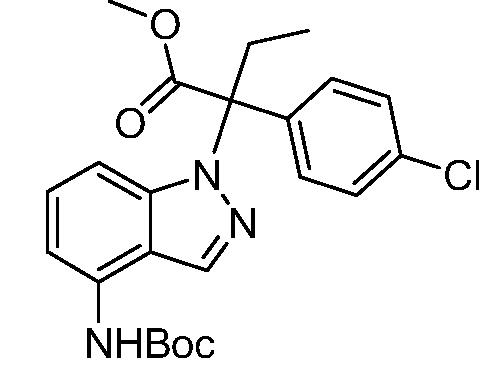
Соединение со стадии C (1 г, 2,4 ммоль) и EtI (42 мг, 2,6 ммоль) растворяли в безводном DMF (10 мл). Затем NaH (144 мг, 3,6 ммоль, 60%) добавляли к смеси медленно при 0°C. Через 30 минут реакционную смесь гасили насыщенным NH4Cl (10 мл), экстрагировали ЕА, высушивали над безводным Na2SO4, фильтровали, и фильтрат концентрировали при пониженном давлении. Остаток очищали на колонке с силикагелем (РЕ:ЕА = от 20:1 до 5:1, получая целевое соединение (0,608 г, 57%). LC/MS m/z = 444,1 [M+H]+. Эти два энантиомера разделяли хиральным разделением SFC.
Энантиомер A и энантиомер B получали в приведенных ниже условиях хирального разделения:
Колонка: AD-H
Мобильная фаза: A: CO2, B: MeOH, A:B = 80:20 при 1,0 мл/мин.
Температура колонки: 38,3°C
энантиомер A, время удерж. = 3,71 мин., энантиомер B, время удерж. = 4,73 мин.
Стадия E: 1-(2-(4-хлорфенил)-1-гидроксибутан-2-ил)-1Н-индазол-4-ил-трет-бутилкарбамат
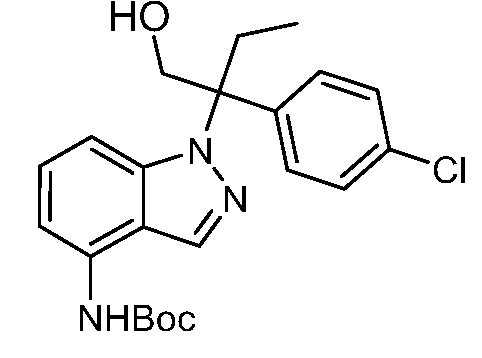
К раствору 2-[4-(трет-бутоксикарбониламино)-1Н-индазол-1-ил]-2-(4-хлорфенил)метилбутаноата (3,5 г, 7,90 ммоль) в THF (140 мл) при 0°C добавляли LiBH4 (0,7 г, 31,6 ммоль). Смесь перемешивали в течение ночи при комнатной температуре. Затем ее аккуратно гасили насыщенным NH4Cl (200 мл) и экстрагировали CH2Cl2. Объединенные органические слои промывали солевым раствором (80 мл), высушивали над безводным сульфатом натрия, фильтровали, и растворитель удаляли в вакууме, получая желаемый продукт. LC/MS m/z = 416,3 [M+H]+.
Стадия F: 1-[2-(4-хлорфенил)-1-оксобутан-2-ил]-1Н-индазол-4-ил-трет-бутилкарбамат
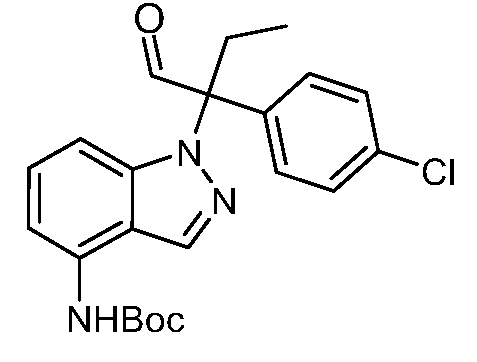
К раствору перйодинана Десса-Мартина (8,12 г, 19,15 ммоль) в DCM (90 мл) при 0°C добавляли раствор 1-[2-(4-хлорфенил)-1-гидроксибутан-2-ил]-1Н-индазол-4-ил-трет-бутилкарбамата (3,18 г, 7,67 ммоль) в DCM (20 мл). Смесь перемешивали при комнатной температуре в течение 2 часов. Затем ее фильтровали, и фильтрат концентрировали в вакууме, получая сырой продукт. Продукт очищали хроматографией на колонках (0-10% EtOAc/гексаны), получая желаемый продукт.
Стадия G: 1-[3-(4-хлорфенил)-2-гидроксипентан-3-ил]-1Н-индазол-4-ил-трет-бутилкарбамат
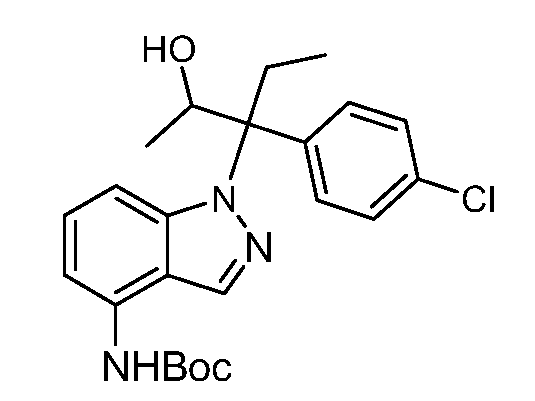
К раствору 1-[2-(4-хлорфенил)-1-оксобутан-2-ил]-1Н-индазол-4-ил-трет-бутилкарбамата (2,50 г, 6,05 ммоль) в безводном THF (50 мл) добавляли метилмагний бромид (2,5 М в простом эфире, 7,3 мл, 18,15 ммоль) при 0°C. Смесь перемешивали еще 30 минут при 0°C, затем добавляли насыщенный NH4Cl (100 мл), и смесь экстрагировали ЕА. Объединенные органические слои промывали солевым раствором (50 мл), высушивали над безводным сульфатом натрия, фильтровали, и растворитель удаляли в вакууме, получая желаемый продукт.
Стадия Н: 3-(4-амино-1Н-индазол-1-ил)-3-(4-хлорфенил)пентан-2-ол
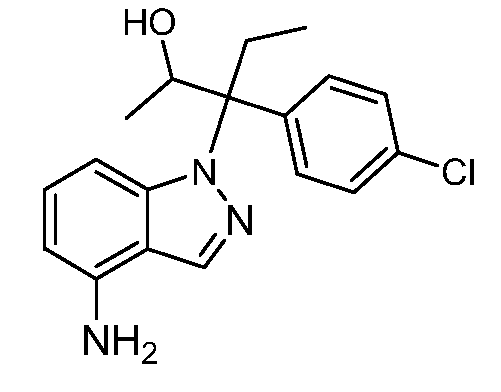
Используя ту же самую процедуру, описанную для Примера 15, стадия B, но заменяя 1-(3-(4-хлорфенил)-2-гидрокси-2-метилпентан-3-ил)-6-фтор-1Н-индол-4-ил-трет-бутилкарбамат 1-[2-(4-хлорфенил)-1-оксобутан-2-ил]-1Н-индазол-4-ил-трет-бутилкарбаматом, получали целевое соединение.
Стадия I: N-(1-(3-(4-хлорфенил)-2-гидроксипентан-3-ил)-1Н-индазол-4-ил)метансульфонамид
Используя ту же самую процедуру, описанную для Примера 15, но на стадии C заменяя 3-(4-амино-6-фтор-1Н-индол-1-ил)-3-(4-хлорфенил)-2-метилпентан-2-ол 3-(4-амино-1Н-индазол-1-ил)-3-(4-хлорфенил)пентан-2-олом, получали целевое соединение. LC/MS m/z = 408,2 [M+H]+.
Используя процедуру, описанную в Примере 18, но на стадии E заменяя 2-[4-(трет-бутоксикарбониламино)-1Н-индазол-1-ил]-2-(4-хлорфенил)метилбутаноат 2-(4-(трет-бутоксикарбониламино)-1Н-индол-1-ил)-2-(4-хлорфенил)метилбутаноатом, 2-(4-(трет-бутоксикарбониламино)-1Н-индол-1-ил)-2-(2,4-дихлорфенил)метилбутаноатом, 2-(4-(трет-бутоксикарбониламино)-6-фтор-1Н-индол-1-ил)-2-(2,4-дихлорфенил)метилбутаноатом, 2-(4-(трет-бутоксикарбониламино)-6-фтор-1Н-индазол-1-ил)-2-(4-(трифторметил)фенил)метилбутаноатом, 2-(4-(трет-бутоксикарбониламино)-1Н-индол-1-ил)-2-(4-(трифторметил)фенил)метилбутаноатом, 2-(4-(трет-бутоксикарбониламино)-1Н-индазол-1-ил)-2-(4-(трифторметил)фенил)метилбутаноатом, получали соединения 108-117 представленных в Таблице 15, соответственно. Также с использованием процедуры, описанной в Примере 18, но на стадии E заменяя метансульфонил хлорид метилсульфамоил хлоридом и диметилсульфамоил хлоридом, получали соединение 118 и соединение 119 представленных в Таблице 15, соответственно.
Соединение 106 и соединение 107, были получены в приведенных ниже условиях хирального разделения:
Колонка: IC
Мобильная фаза: A: Гексан, B: EtOH (0,1% DEA), A:B = 70:30 при 1,0 мл/мин.
Температура колонки: 40°C
Соединение 108 и соединение 109 были получены в приведенных ниже условиях хирального разделения:
Колонка: IA
Мобильная фаза: A: Гексан, B: EtOH (0,1% DEA), A:B = 70:30 при 1,0 мл/мин.
Температура колонки: 30°C
Соединение 111 получали в следующих условиях хирального разделения:
Колонка: IA
Мобильная фаза: A: Гексан, B: EtOH (0,1% DEA), A:B = 70:30 при 1,0 мл/мин.
Температура колонки: 30°C
Соединение 113 и соединение 114 были получены в приведенных ниже условиях хирального разделения:
Колонка: AD-H
Мобильная фаза: A: CO2, B: MeOH (0,1% DEA), A:B = 50:50 при 3,0 мл/мин.
Температура колонки: 38,4°C
Соединение 115, 116 и соединение 117 были получены в приведенных ниже условиях хирального разделения:
Колонка: AS-H
Мобильная фаза: A: CO2, B: MeOH (0,1% DEA), A:B = 93:7 при 3,0 мл/мин.
Температура колонки: 41,3°C
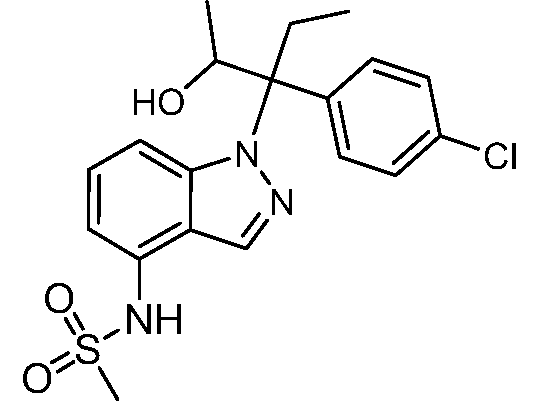
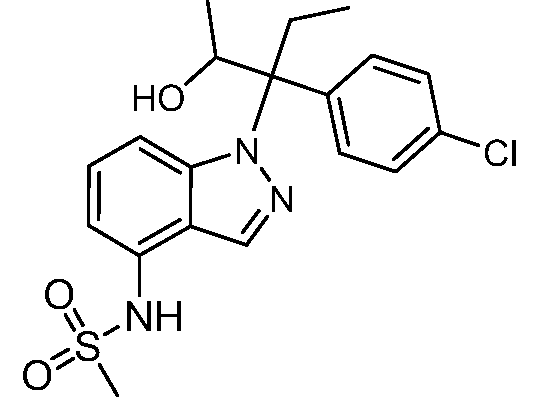
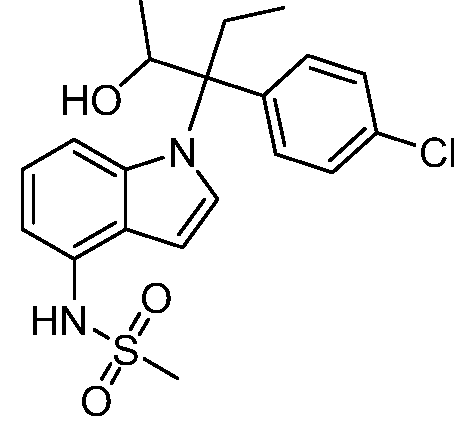
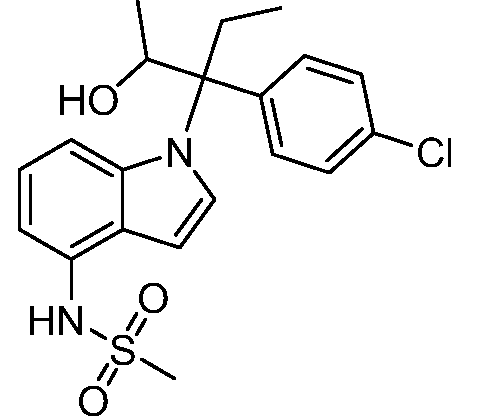
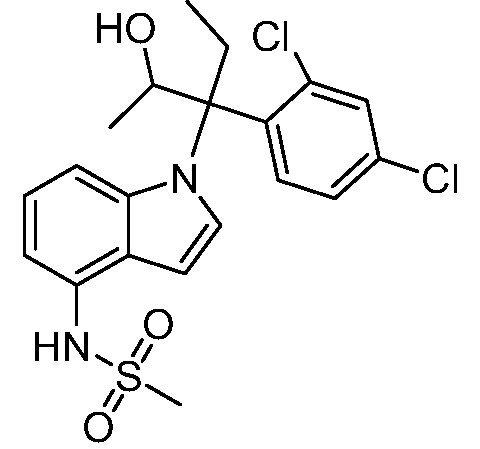
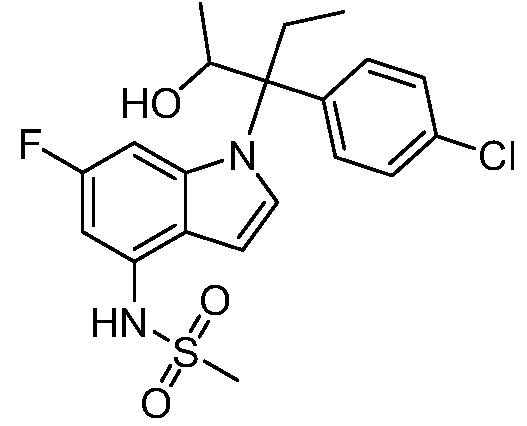
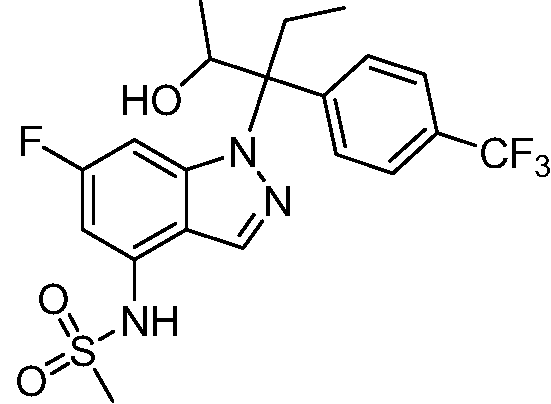
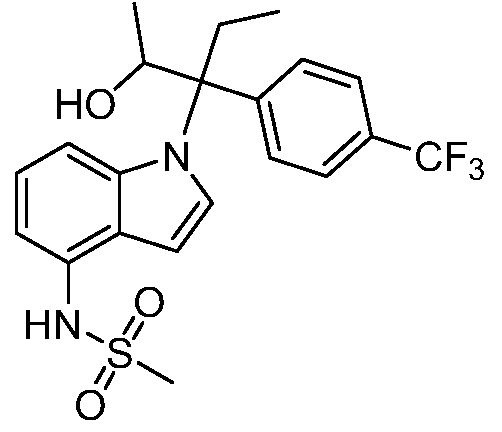
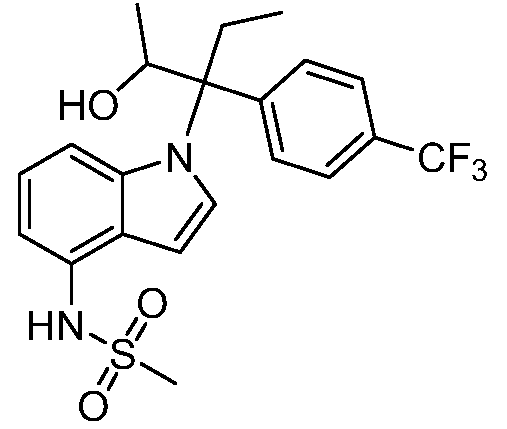
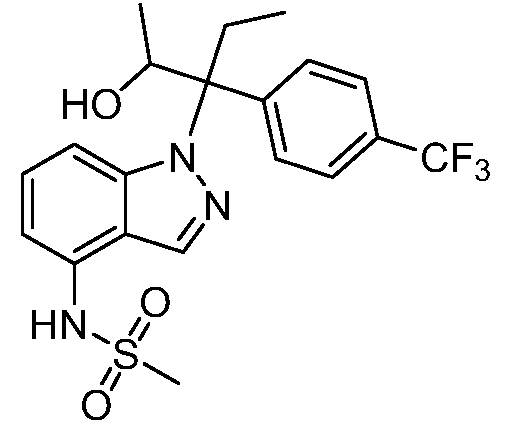
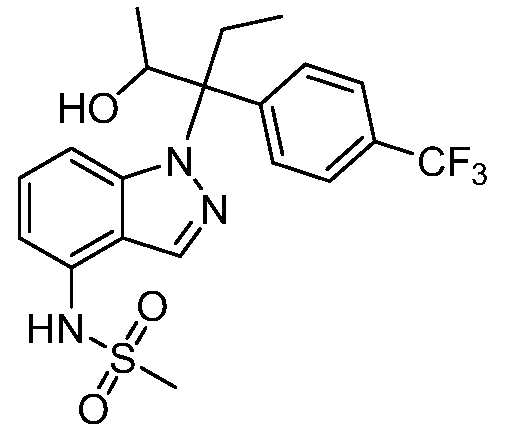
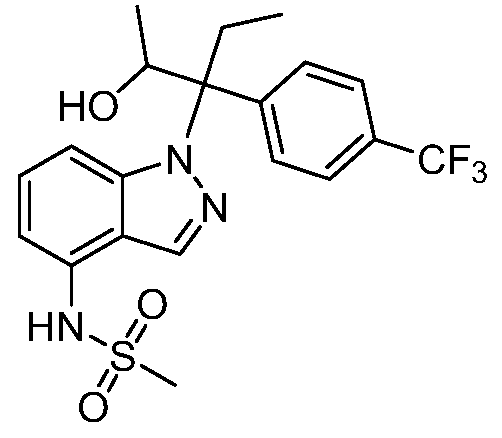
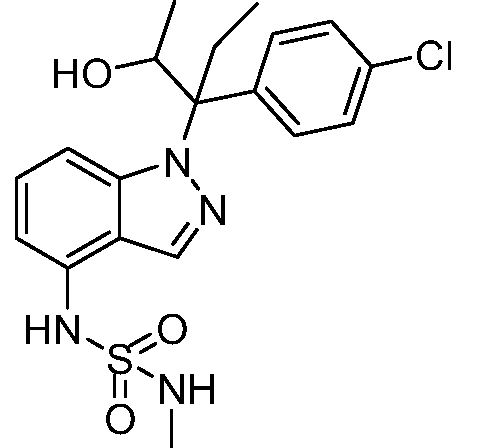
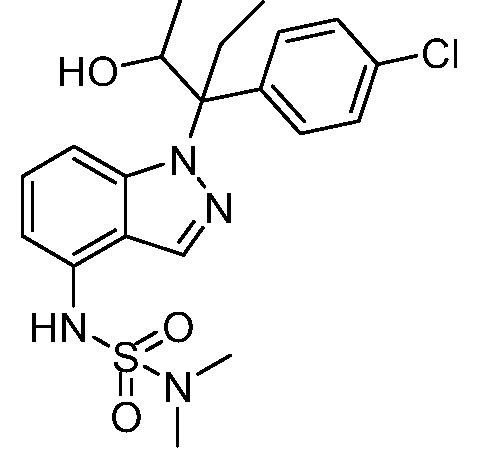
ПРИМЕР 20
N-(1-(1-(4-хлорфенил)-1-(2-цианоциклопропил)пропил)-1Н-индазол-4-ил)метансульфонамид (Соединение 120),
Стадия A: 2-(4-амино-1Н-индазол-1-ил)-2-(4-хлорфенил)метилбутаноат
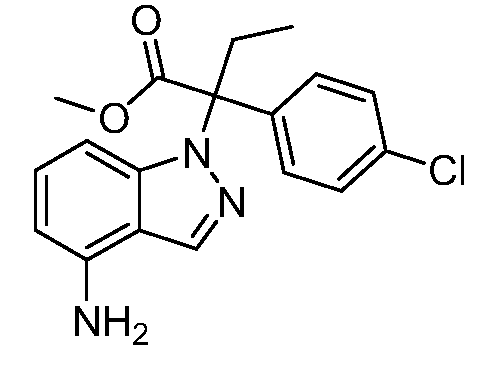
Смесь 2-(4-(трет-бутоксикарбониламино)-1Н-индазол-1-ил)-2-(4-хлорфенил)метилбутаноата (3,0 г, 6,8 ммоль), как описано в Примере 18 Стадия D, в 4н HCl/MeOH (20 мл) перемешивали при комнатной температуре в течение 2 часов. После удаления растворителя, остаток растворяли в этилацетате (80 мл), значение рН доводили до 9~10 насыщенным раствором бикарбоната натрия, высушивали над сульфатом натрия, затем фильтровали. После удаления органического растворителя, остаток использовали для следующей стадии без очистки. LC/MS m/z = 344,1 [M+H]+.
Стадия B: 2-(4-хлорфенил)-2-(4-(метилсульфонамидо)-1Н-индазол-1-ил)метилбутаноат
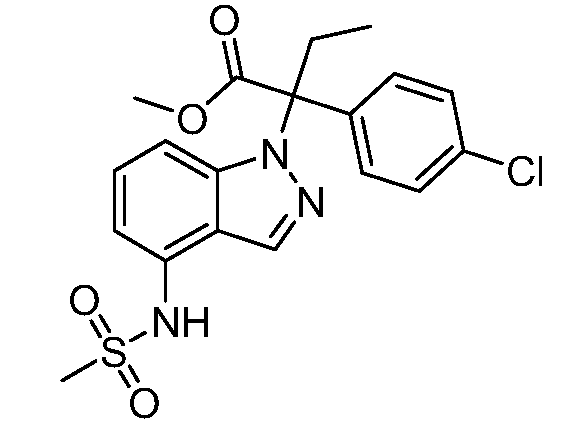
К смеси 2-(4-амино-1Н-индазол-1-ил)-2-(4-хлорфенил)метилбутаноата (1,0 г, 2,9 ммоль) и NMM (0,9 г, 8,7 ммоль) в DCM (10 мл) добавляли по каплям MsCl (0,4 г, 3,2 ммоль). Затем смесь перемешивали при комнатной температуре в течение ночи. Раствор гасили насыщенным раствором NH4Cl и экстрагировали этилацетатом (50 мл). Органический слой промывали солевым раствором, высушивали над сульфатом натрия, фильтровали и упаривали. Остаток очищали хроматографией на силикагеле (элюент: РЕ/ЕА = 4/1), получая целевое соединение в форме твердого вещества желтого цвета. LC/MS m/z = 422,1 [M+H]+.
Стадия C: 2-(4-хлорфенил)-2-(4-(N-((2-(триметилсилил)-этокси)метил)метилсульфонамидо)-1Н-индазол-1-ил)метилбутаноат
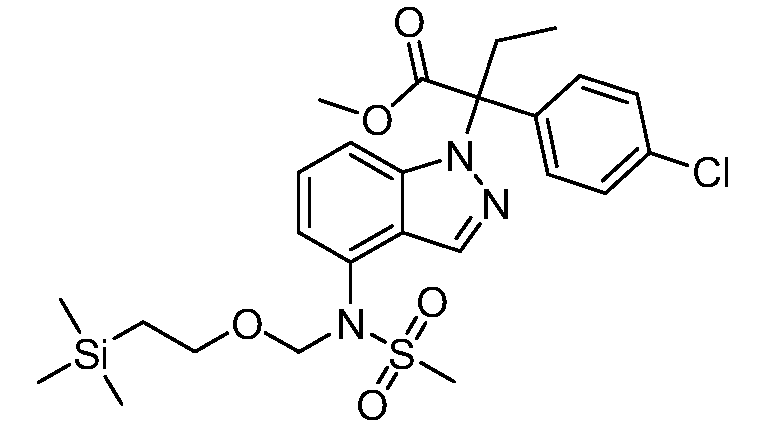
Раствор 2-(4-хлорфенил)-2-(4-(метилсульфонамидо)-1Н-индазол-1-ил)метилбутаноата (500 мг, 1,19 ммоль) в сухом THF (8 мл) обрабатывали NaH (95 мг, 2,38 ммоль, 60% в масле) при 0°C в течение 30 минут, затем по каплям добавляли (2-(хлорметокси)этил)триметилсилан (295 мг, 1,78 ммоль). Смесь перемешивали при комнатной температуре в течение 2 часов. Раствор гасили насыщенным раствором NH4Cl и экстрагировали этилацетатом (30 мл). Органический слой промывали солевым раствором, высушивали над сухим сульфатом натрия, фильтровали и упаривали. Остаток очищали хроматографией на силикагеле (РЕ/ЕА = 6/1), получая целевое соединение в форме твердого вещества светло-желтого цвета. LC/MS m/z = 551,8 [M+H]+.
Стадия D: N-(1-(2-(4-хлорфенил)-1-гидроксибутан-2-ил)-1Н-индазол-4-ил)-N-((2-(триметилсилил)этокси)метил)-метансульфонамид
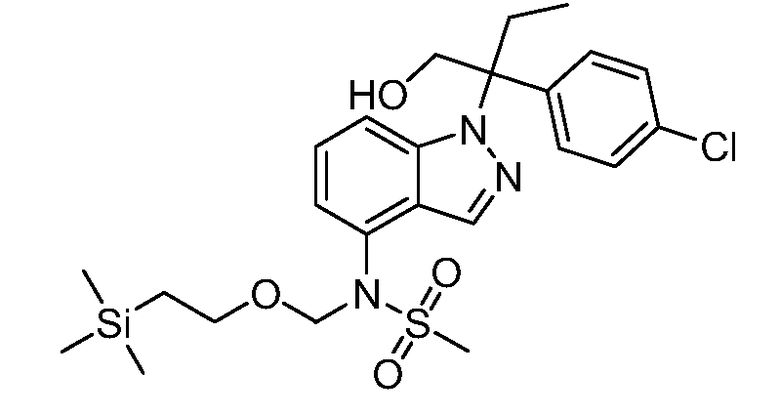
К суспензии LiBH4 (120 мг, 5,43 ммоль) в THF (5 мл) добавляли MeOH (174 мг, 5,43 ммоль) и раствор 2-(4-хлорфенил)-2-(4-(N-((2-(триметилсилил)этокси)метил)метилсульфонамидо)-1Н-индазол-1-ил)метилбутаноата (600 мг, 1,09 ммоль) в THF (8 мл). Смесь перемешивали при комнатной температуре в течение 2 часов. Раствор гасили насыщенным раствором NH4Cl и экстрагировали этилацетатом (30 мл). Органический слой промывали солевым раствором, высушивали над сульфатом натрия, фильтровали и упаривали. Остаток очищали хроматографией на силикагеле (РЕ/ЕА = 3/1), получая целевое соединение в форме твердого вещества светло-желтого цвета. LC/MS m/z = 524,2 [M+H]+.
Стадия E: N-(1-(2-(4-хлорфенил)-1-оксобутан-2-ил)-1Н-индазол-4-ил)-N-((2-(триметилсилил)этокси)метил)метансульфонамид
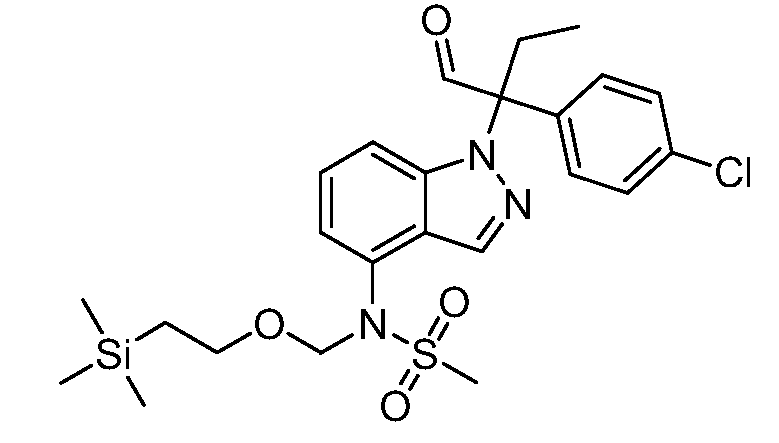
Раствор N-(1-(2-(4-хлорфенил)-1-гидроксибутан-2-ил)-1Н-индазол-4-ил)-N-((2-(триметилсилил)этокси)метил)-метансульфонамида (560 мг, 1,07 ммоль) в DCM (10 мл) добавляли частями к перйодинану Десса-Мартина (590 мг, 1,39 ммоль) при 0°C и перемешивали в течение 1,5 часов. Смесь разбавляли водой и экстрагировали этилацетатом. Органический слой промывали солевым раствором, высушивали над сульфатом натрия, фильтровали и упаривали. Остаток очищали хроматографией на силикагеле (РЕ/ЕА = 8/1), получая целевое соединение в форме твердого вещества желтого цвета. LC/MS m/z = 522,1 [M+H]+.
Стадия F: (E)-N-(1-(3-(4-хлорфенил)-1-цианопент-1-ен-3-ил)-1Н-индазол-4-ил)-N-((2-(триметилсилил)этокси)метил)-метансульфонамид
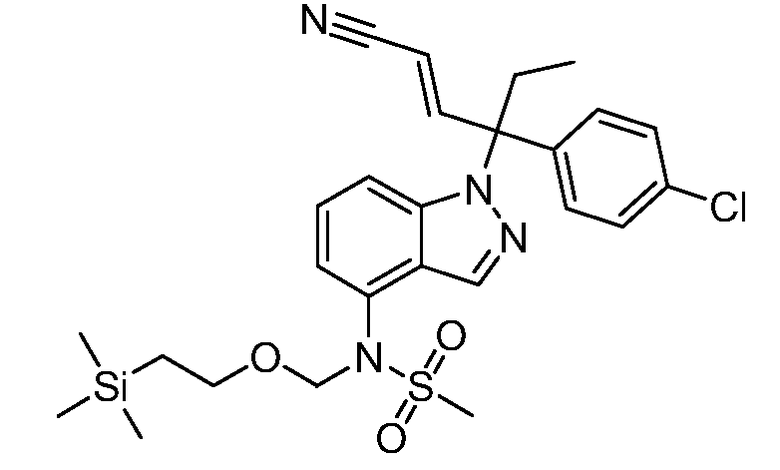
К смеси диэтилцианометилфосфоната (20 мг, 0,12 ммоль), LiCl (5 мг, 0,12 ммоль) и DBU (18 мг, 0,12 ммоль) в MeCN (2 мл) добавляли раствор N-(1-(2-(4-хлорфенил)-1-оксобутан-2-ил)-1Н-индазол-4-ил)-N-((2-(триметилсилил)этокси)метил)-метансульфонамида (50 мг, 0,10 ммоль) в MeCN (1 мл). Смесь перемешивали при комнатной температуре в течение 2 часов и упаривали. Остаток очищали хроматографией на силикагеле (РЕ/ЕА = 6/1), получая целевое соединение в форме твердого вещества белого цвета. LC/MS m/z = 545,2 [M+H]+.
Стадия G: N-(1-(1-(4-хлорфенил)-1-(2-цианоциклопропил)-пропил)-1Н-индазол-4-ил)-N-((2-(триметилсилил)этокси)метил)-метансульфонамид
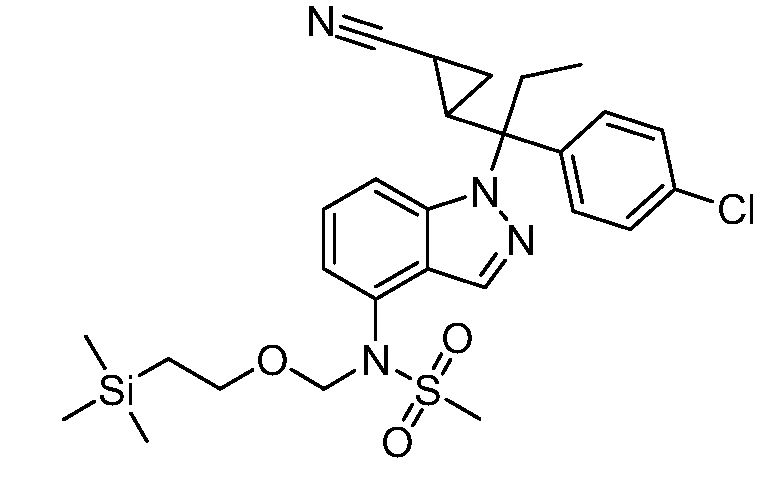
К суспензии NaH (22 мг) в безводном ДМСО (0,9 мл) добавляли триметилсульфоксоний йодид (128 мг) при 0°C, и смесь перемешивали при комнатной температуре в течение 3 часов, пока раствор не стал прозрачным. Затем добавляли раствор (E)-N-(1-(3-(4-хлорфенил)-1-цианопент-1-ен-3-ил)-1Н-индазол-4-ил)-N-((2-(триметилсилил)этокси)метил)метансульфонамида (42 мг) в безводном ДМСО (0,1 мл), и реакционную смесь перемешивали при 70°C в течение 16 часов. Раствор гасили насыщенным раствором NH4Cl, экстрагировали этилацетатом (15 мл). Органический слой промывали солевым раствором, высушивали над сухим сульфатом натрия, фильтровали и упаривали. Остаток очищали хроматографией на силикагеле (РЕ/ЕА = 4/1), получая целевое соединение в форме твердого вещества белого цвета. LC/MS m/z = 559,2 [M+H]+.
Стадия Н: N-(1-(1-(4-хлорфенил)-1-(2-цианоциклопропил)-пропил)-1Н-индазол-4-ил)метансульфонамид (Соединение 120)
Смесь N-(1-(1-(4-хлорфенил)-1-(2-цианоциклопропил)пропил)-1Н-индазол-4-ил)-N-((2-(триметилсилил)этокси)метил)метансульфонамида (14 мг) в 2н HCl/EtOH (1 мл/1 мл) перемешивали при 50°C в течение 3 часов, охлаждали до комнатной температуры, концентрировали и экстрагировали этилацетатом (10 мл). Органические слои объединяли, высушивали над безводным сульфатом натрия и концентрировали в вакууме. Остаток очищали, используя колонку с силикагелем (РЕ/ЕА = 3/1), получая целевое соединение в форме твердого вещества коричневого цвета. LC/MS m/z = 429,1 [M+H]+.
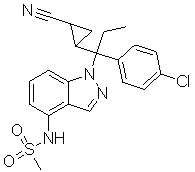
ПРИМЕР 21
N-(1-(2-(4-хлорфенил)-1-цианобутан-2-ил)-1Н-индол-4-ил)метансульфонамид
Стадия A: 1-(2-(4-хлорфенил)-1-гидроксибутан-2-ил)-1Н-индол-4-ил-трет-бутилкарбамат
К раствору 2-(4-(трет-бутоксикарбониламино)-1Н-индол-1-ил)-2-(4-хлорфенил)метилбутаноата (5 г, 11,3 ммоль), как описано в Примере 3 Стадия A, в тетрагидрофуране (50 мл) добавляли алюмогидрид лития (644 мг, 16,95 ммоль) при 0°C. Смесь постепенно нагревали до комнатной температуры и перемешивали в течение 4 часов. К реакционной смеси аккуратно добавляли воду (10 мл), затем экстрагировали этилацетатом. Объединенные органические слои промывали солевым раствором (30 мл), высушивали над безводным сульфатом натрия, фильтровали и концентрировали. Сырой продукт очищали хроматографией на силикагеле с элюированием смесью РЕ/ЕА (от 30/1 до 10/1, об./об.), получая целевое соединение в форме бесцветного аморфного твердого вещества. LC/MS m/z = 359,9 [M-tBu+H]+.
Стадия B: 1-(2-(4-хлорфенил)-1-оксобутан-2-ил)-1Н-индол-4-ил-трет-бутилкарбамат
К продукту со стадии (2 г, 4,82 ммоль) в дихлорметане (30 мл) добавляли перйодинан Десса-Мартина (3,06 г, 7,23 ммоль) при 0°C. Смесь перемешивали в течение 2 часов. Растворитель удаляли при пониженном давлении. Сырой продукт очищали хроматографией на силикагеле с элюированием смесью РЕ/ЕА (от 30/1 до 15/1, об./об.), получая целевое соединение в форме бесцветного аморфного твердого вещества. LC/MS m/z = 357,9 [M-tBu+H]+.
Стадия C: 1-(2-(4-хлорфенил)-1-цианобутан-2-ил)-1Н-индол-4-ил-трет-бутилкарбамат
К суспензии t-BuOK (683 мг, 6,10 ммоль) в THF (30 мл) добавляли раствор тозилметил изоцианида (239 мг, 1,22 ммоль) в THF (5 мл) при -78°C. Смесь перемешивали в течение 15 минут, затем обрабатывали по каплям раствором продукта со стадии B (419 мг, 1,02 ммоль) в THF (10 мл) и продолжали перемешивать в течение 1,5 часов при -78°C. К охлажденной реакционной смеси добавляли метанол (100 мл). Смесь фильтровали через короткую колонку с силикагелем и упаривали. Остаток растворяли в MeOH (40 мл) и добавляли затем t-BuOK (137 мг, 1,22 ммоль). Полученную смесь нагревали с обратным холодильником в течение 30 минут. После удаления растворителя, остаток растворяли в воде (50 мл) и экстрагировали EtOAc. Объединенные органические слои промывали солевым раствором (10 мл), высушивали над Na2SO4, фильтровали и упаривали при пониженном давлении. Сырой продукт очищали хроматографией на колонках с силикагелем с элюированием смесью РЕ/ЕА (от 20/1 до 5/1, об./об.), получая целевое соединение в форме твердого вещества белого цвета. LC/MS m/z = 446,1 [М+Na]+.
Стадия D: 3-(4-амино-1Н-индол-1-ил)-3-(4-хлорфенил)пентаннитрил
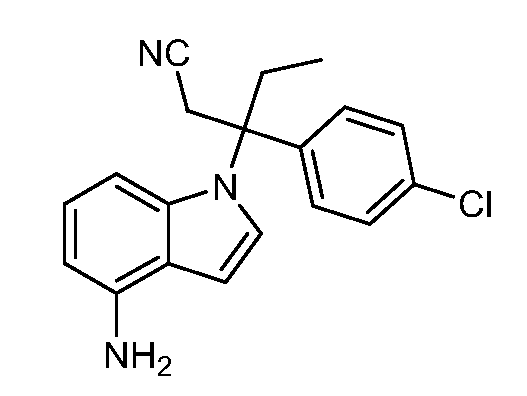
Смесь продукта со стадии C (250 мг, 0,59 ммоль) и 4н HCl/MeOH (10 мл) перемешивали при комнатной температуре в течение 2 часов. После того, как растворитель удаляли в вакууме, остаток растворяли в этилацетате (20 мл), значение рН доводили до 9~10 насыщенным раствором бикарбоната натрия, высушивали над сульфатом натрия, затем фильтровали. После удаления органического растворителя, получали целевое соединение в форме бесцветного масла. LC/MS m/z = 324,1 [M+H]+.
Стадия E: N-(1-(2-(4-хлорфенил)-1-цианобутан-2-ил)-1Н-индол-4-ил)метансульфонамид
К раствору продукта со стадии D (170 мг, 0,52 ммоль) и N-метилморфолина (105 мг, 1,04 ммоль) в дихлорметане (15 мл) добавляли метансульфонил хлорид (89 мг, 0,79 ммоль) при комнатной температуре. Смесь перемешивали в течение 2 часов, затем разбавляли водой (15 мл) и экстрагировали этилацетатом. Объединенные органические слои промывали солевым раствором (10 мл), высушивали над Na2SO4, фильтровали и концентрировали. Сырой продукт очищали PREP-TLC с элюированием смесью РЕ/ЕА (3/1, об./об.), получая целевое соединение в форме бесцветного аморфного твердого вещества. LC/MS m/z = 402,1 [M+H]+.
Используя процедуру, описанную в Примере 21, но на стадии А заменяя 2-(4-(трет-бутоксикарбониламино)-1Н-индол-1-ил)-2-(4-хлорфенил)бутаноат 2-(4-(трет-бутоксикарбониламино)-1Н-индазол-1-ил)-2-(4-хлорфенил)метилбутаноатом, 2-(4-(трет-бутоксикарбониламино)-6-фтор-1Н-индол-1-ил)-2-(4-хлорфенил)метилбутаноатом, 2-(4-(трет-бутоксикарбониламино)-1Н-индол-1-ил)-2-(4-(трифторметил)фенил)метилбутаноатом, получали соединения 123-126 представленных в Таблице 18, соответственно.
Соединение 121 и соединение 122 были получены в приведенных ниже условиях хирального разделения:
Колонка: RegisCell-OD
Мобильная фаза: A: CO2, B: MeOH, A:B = 60:40 при 1,0 мл/мин.
Температура колонки: 39,9°C
Соединение 123 получали в приведенных ниже условиях хирального разделения:
Колонка: AD-H
Мобильная фаза: A: CO2, B: MeOH (0,1% DEA), A:B = 55:45 при 3,0 мл/мин.
Температура колонки: 39,7°C
Соединение 124 и соединение 126 были получены в приведенных ниже условиях хирального разделения:
Колонка: OJ-Н
Мобильная фаза: A: N-гексан, B: EtOH, A:B = 70:30 при 1,0 мл/мин.
Температура колонки: 40°C
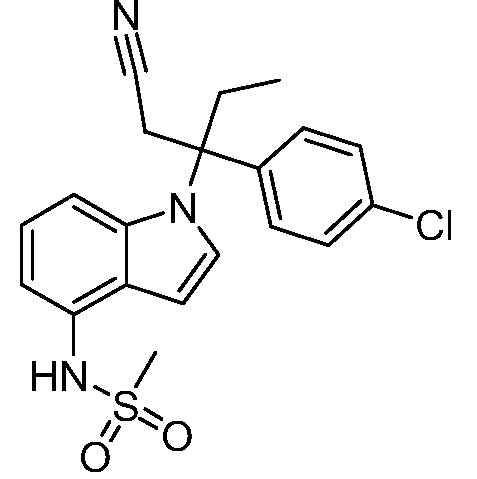
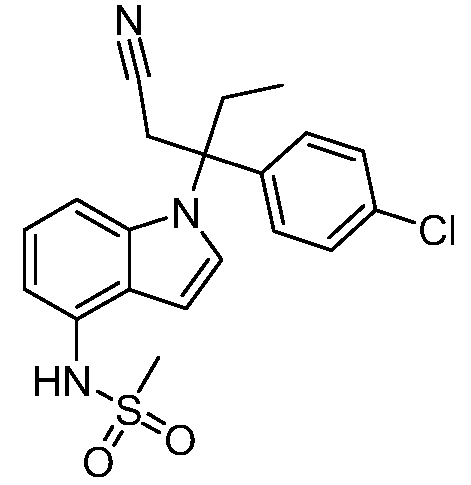
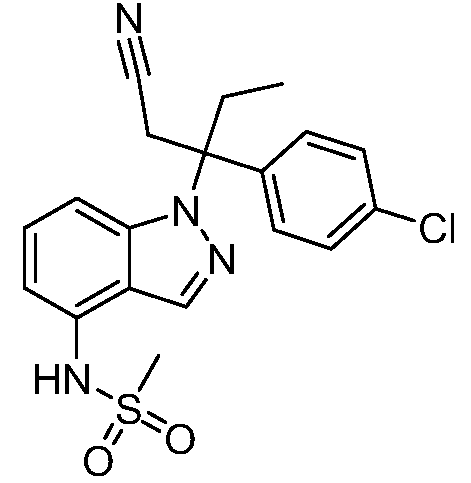
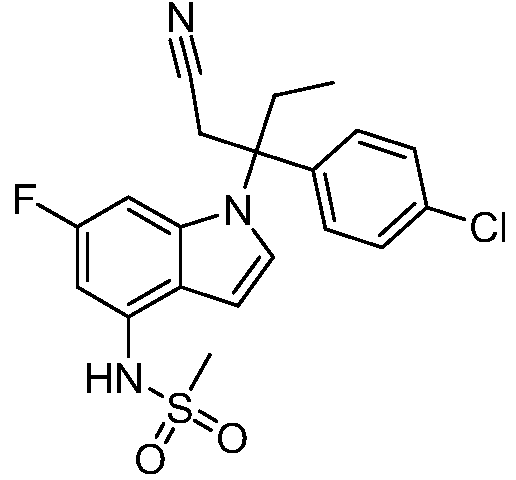
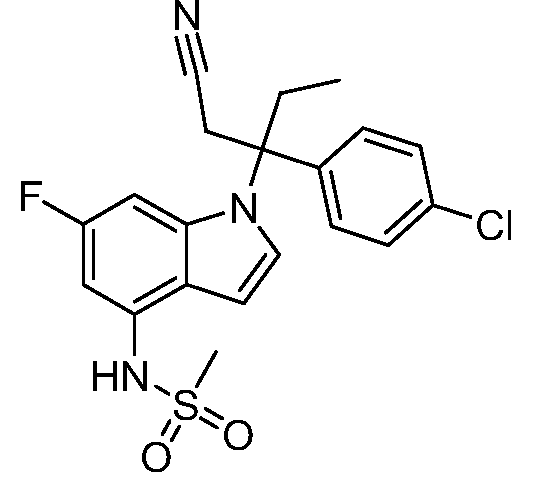
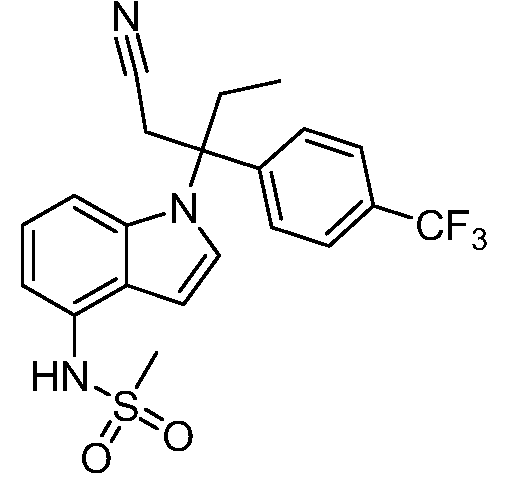
ПРИМЕР 22
(E)-N-(1-(3-(4-хлорфенил)-1-цианопент-1-ен-3-ил)-1Н-индол-4-ил)метансульфонамид (Соединение 127),
Стадия A: (E)-1-(3-(4-хлорфенил)-1-цианопент-1-ен-3-ил)-1Н-индол-4-илтрет-бутилкарбамат
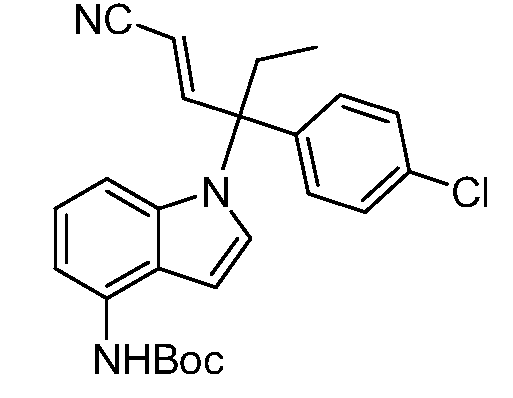
К смеси диэтил цианометилфосфоната (145 мг, 0,76 ммоль), LiCl (43 мг, 1,01 ммоль) и DBU (255 мг, 1,01 ммоль) в ацетонитриле (15 мл) добавляли раствор 1-(2-(4-хлорфенил)-1-оксобутан-2-ил)-1Н-индол-4-ил-трет-бутилкарбамата (210 мг, 1,02 ммоль), как описано в Примере 21 Стадия B, в ацетонитриле (3 мл). Смесь перемешивали при комнатной температуре в течение 2 часов, и летучую фракцию выпаривали. Остаток очищали хроматографией на силикагеле с элюированием смеьсю РЕ/ЕА (от 10/1 до 3/1, об./об.), получая целевое соединение в форме твердого вещества белого цвета. LC/MS m/z = 458,1 [М+Na]+.
Стадия B: (E)-4-(4-амино-1Н-индол-1-ил)-4-(4-хлорфенил)гекс-2-еннитрил
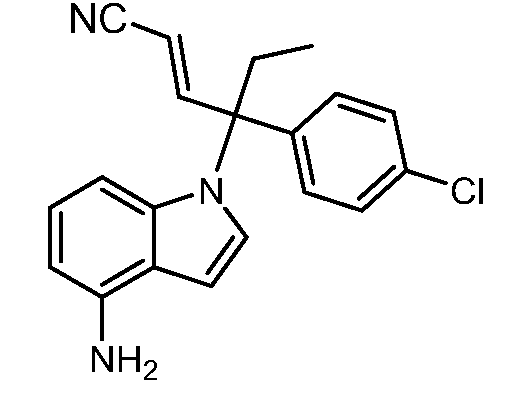
Смесь продукта со стадии A (177 мг, 0,406 ммоль) и 4н HCl/MeOH (10 мл) перемешивали при комнатной температуре в течение 2 часов. После удаления растворителя, остаток высушивали в вакууме, получая целевое соединение в форме твердого вещества белого цвета. LC/MS m/z = 336,1 [M+H]+.
Стадия C: (E)-N-(1-(3-(4-хлорфенил)-1-цианопент-1-ен-3-ил)-1Н-индол-4-ил)метансульфонамид
К смеси соединения со стадии B (123 мг, 0,367 ммоль) и NMM (74 мг, 0,734 ммоль) в DCM (15 мл) добавляли по каплям метансульфонил хлорид (62 мг, 0,550 ммоль). Затем смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь гасили насыщенным раствором хлорида аммония и экстрагировали этилацетатом (20 мл). Органический слой промывали солевым раствором, высушивали над сульфатом натрия, фильтровали и упаривали. Остаток очищали хроматографией на силикагеле с элюированием смесью РЕ/ЕА (от 10/1 до 3/1, об./об.), получая целевое соединение в форме твердого вещества белого цвета. LC/MS m/z = 436,1 [М+Na]+.
Используя процедуру, описанную в Примере 22, но на стадии А заменяя 1-(2-(4-хлорфенил)-1-оксобутан-2-ил)-1Н-индол-4-ил-трет-бутилкарбамат 1-(2-(4-метоксифенил)-1-оксобутан-2-ил)-1Н-индол-4-ил-трет-бутилкарбаматом, 1-(2-(4-хлорфенил)-1-оксобутан-2-ил)-1H-индазол-4-ил-трет-бутилкарбаматом, 1-(2-(4-хлорфенил)-1-оксобутан-2-ил)-6-фтор-1H-индазол-4-ил-трет-бутилкарбаматом, получали соединения 128-130 представленных в Таблице 19, соответственно. Используя процедуру, описанную в Примере 22, но на стадии A заменяя диэтил цианометилфосфонат диэтил 1-цианоэтилфосфонатом, получали соединение 131.
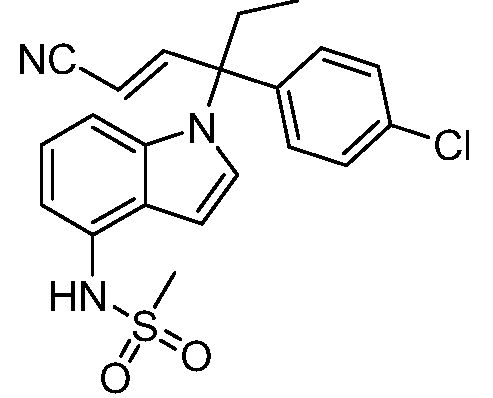
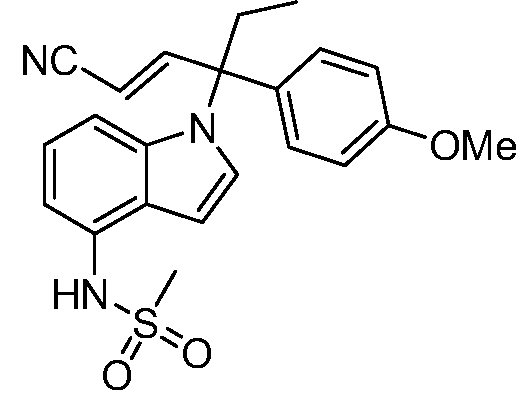
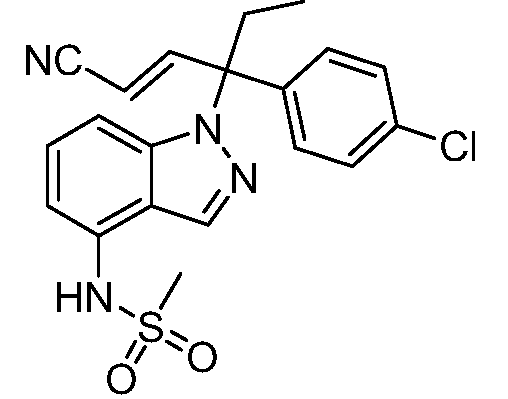
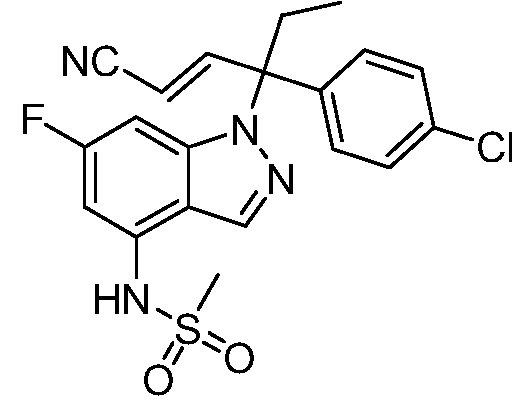
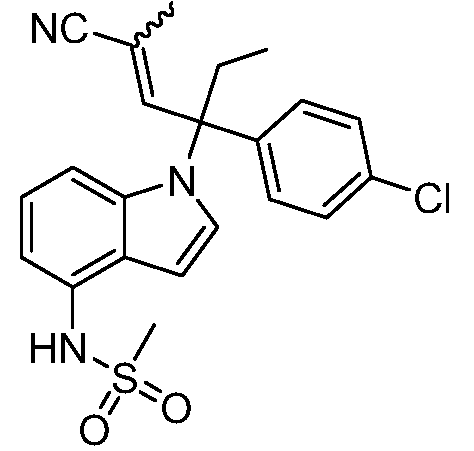
ПРИМЕР 23
N-(1-(3-(4-Хлорфенил)-1-циано-2-метилпент-1-ен-3-ил)-1Н-индол-4-ил)метансульфонамид (Соединение 132)
Стадия A: N-(1-(3-(4-хлорфенил)-2-оксопентан-3-ил)-1Н-индол-4-ил)метансульфонамид
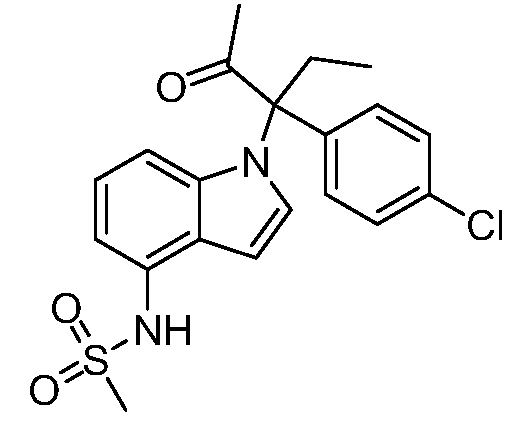
К раствору 2-(4-хлорфенил)-2-(4-(метилсульфонамидо)-1Н-индол-1-ил)метилбутаноата (1 г, 2,38 ммоль), как описано в Примере 3 Стадия C, в THF (30 мл) по каплям при 0°C добавляли CH3Li (3,2 мл, 4,76 ммоль, 1,5 М в простом эфире) и перемешивали в течение 0,5 часов. Смесь гасили насыщенным раствором хлорида аммония и экстрагировали этилацетатом. Органический слой промывали солевым раствором, высушивали над сульфатом натрия, фильтровали и упаривали. Остаток очищали хроматографией на силикагеле с элюированием смесью РЕ/ЕА (от 15/1 до 6/1, об./об.), получая целевое соединение в форме бесцветного аморфного твердого вещества. LC/MS m/z = 427,1 [М+Na]+.
Стадия B: N-(1-(3-(4-хлорфенил)-1-циано-2-гидрокси-2-метилпентан-3-ил)-1Н-индол-4-ил)метансульфонамид
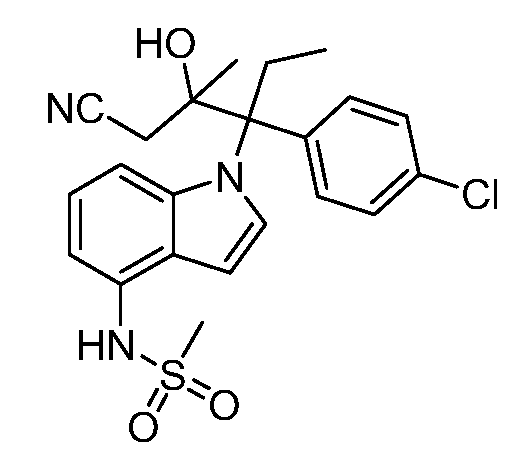
К раствору ацетонитрила (244 мг, 5,94 ммоль) в THF (20 мл) по каплям при -78°C добавляли n-BuLi (2,6 мл, 6,5 ммоль, 2,5 М в гексане). После перемешивания в течение 0,5 часов, раствор продукта со стадии А (480 мг, 1,19 ммоль) в THF (5 мл) добавляли к смеси при -78°C. Полученную смесь перемешивали при -78°C в течение 0,5 часов и затем гасили насыщенным раствором хлорида аммония, экстрагировали этилацетатом. Органический слой промывали солевым раствором, высушивали над сульфатом натрия, фильтровали и упаривали. Остаток очищали хроматографией на силикагеле с элюированием смесью РЕ/ЕА (от 15/1 до 4/1, об./об.), получая целевое соединение в форме бесцветного аморфного твердого вещества. LC/MS m/z = 468,0 [М+Na]+.
Стадия C: N-(1-(3-(4-Хлорфенил)-1-циано-2-метилпент-1-ен-3-ил)-1Н-индол-4-ил)метансульфонамид (Соединение 132)
Смесь продукта со стадии B (200 мг, 0,449 ммоль), TFAA (283 мг, 1,35 ммоль), триэтиламина (136 мг, 1,35 ммоль) и DMAP (165 мг, 1,35 ммоль) в дихлорметане (20 мл) перемешивали при комнатной температуре в течение 2 часов. Растворитель выпаривали, и остаток очищали PREP-TLC с элюированием смесью РЕ/ЕА (3/1, об./об.), получая целевое соединение в форме бесцветного аморфного твердого вещества. LC/MS m/z = 450,0 [М+Na]+.
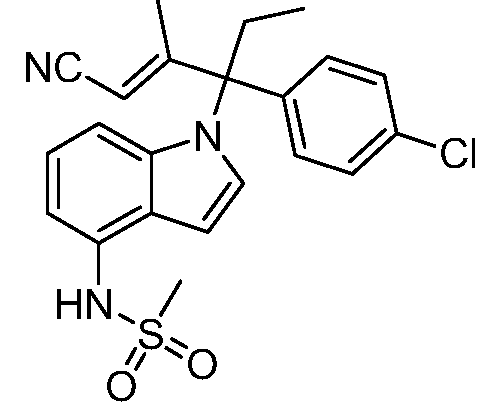
ПРИМЕР 24
(E)-4-(4-амино-1Н-индазол-1-ил)-4-(4-хлорфенил)гекс-2-еннитрил (Соединение 133)
Используя ту же самую процедуру, описанную для Примера 22, но на стадии А заменяя 1-(2-(4-хлорфенил)-1-оксобутан-2-ил)-1Н-индол-4-ил-трет-бутилкарбамат 1-(2-(4-хлорфенил)-1-оксобутан-2-ил)-1Н-индазол-4-ил-трет-бутилкарбаматом, 1-(2-(4-хлорфенил)-1-оксобутан-2-ил)-6-фтор-1Н-индазол-4-ил-трет-бутилкарбаматом, получали соединение 133 и соединение 134.
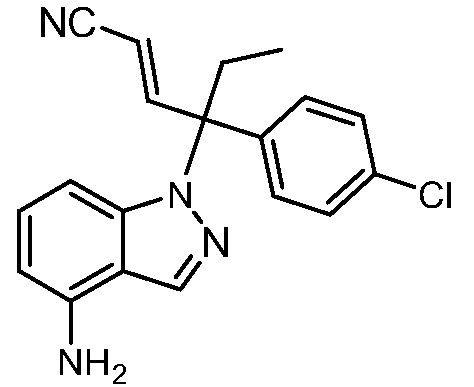
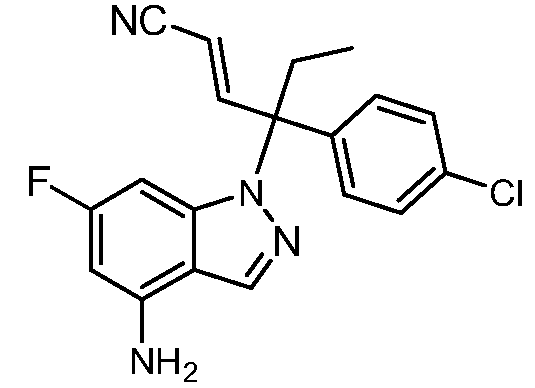
ПРИМЕР 25
(E)-1-(3-(4-хлорфенил)-1-цианопент-1-ен-3-ил)-1Н-индазол-4-ил(метилсульфонил)трет-бутилкарбамат (Соединение 135)
Стадия A: (E)-1-(3-(4-хлорфенил)-1-цианопент-1-ен-3-ил)-1Н-индазол-4-ил-трет-бутилкарбамат
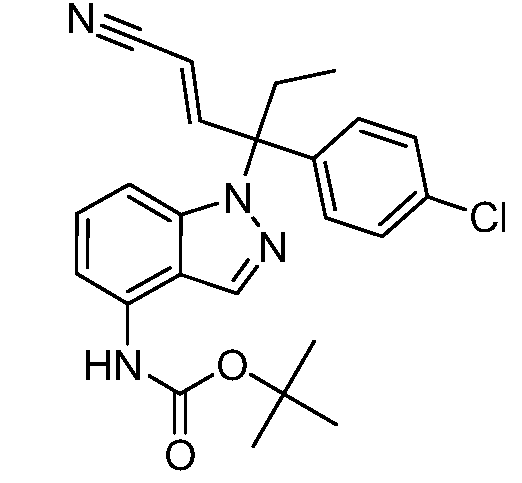
К смеси диэтил цианометилфосфоната (305 мг), LiCl (74 мг) и DBU (327 мг) в MeCN (15 мл) добавляли раствор (E)-1-(3-(4-хлорфенил)-1-цианопент-1-ен-3-ил)-1Н-индазол-4-ил-трет-бутилкарбамата (600 мг), как описано в Примере 18 Стадия F, в MeCN (5 мл). Смесь перемешивали при комнатной температуре в течение 2 часов и упаривали. Остаток очищали хроматографией на силикагеле (РЕ/ЕА = 6/1), получая целевое соединение в форме твердого вещества белого цвета. LC/MS m/z = 437,1 [M+H]+.
Стадия B: N-(1-(2-(4-хлорфенил)-1-гидроксибутан-2-ил)-1Н-индазол-4-ил)-N-((2-(триметилсилил)этокси)метил)метансульфонамид
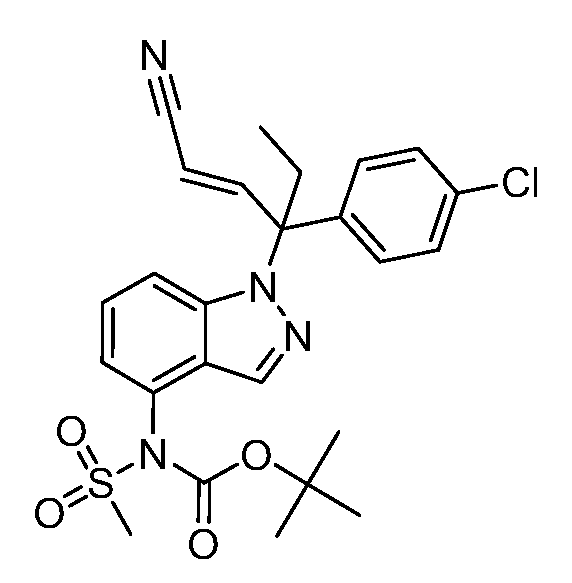
Раствор (E)-1-(3-(4-хлорфенил)-1-цианопент-1-ен-3-ил)-1Н-индазол-4-ил-трет-бутилкарбамата (50 мг, 1,19 ммоль) в безводном DMF (2 мл) обрабатывали NaH (9 мг, 60% в масле) при 0°C в течение 30 минут, затем добавляли метансульфонил хлорид (50 мг). Смесь перемешивали при комнатной температуре в течение 1 часа. Раствор гасили насыщенным раствором NH4Cl, экстрагировали этилацетатом (15 мл). Органический слой промывали водой, солевым раствором, высушивали над сульфатом натрия, фильтровали и упаривали. Остаток очищали хроматографией на силикагеле (РЕ/ЕА = 6/1), получая целевое соединение в форме твердого вещества белого цвета. LC/MS m/z = 515,2 [M+H]+.
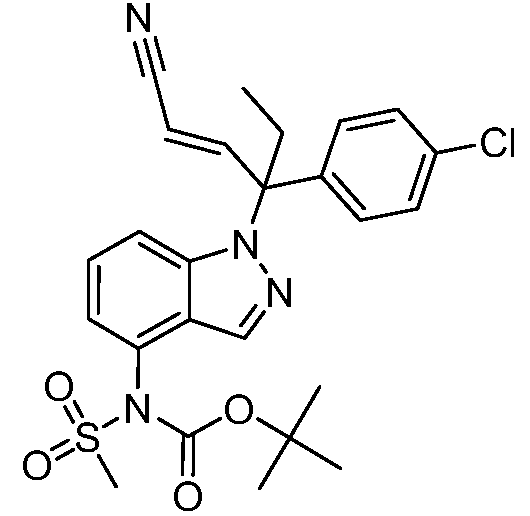
ПРИМЕР 26
(E)-N-(1-(3-(4-хлорфенил)-1-цианопент-1-ен-3-ил)-6-фтор-1Н-индазол-4-ил)-N-(метилсульфонил)метансульфонамид (Соединение 136)
Используя ту же самую процедуру, описанную для Примера 22, стадия C, но заменяя (E)-4-(4-амино-1Н-индол-1-ил)-4-(4-хлорфенил)гекс-2-еннитрил (E)-4-(4-амино-6-фтор-1Н-индазол-1-ил)-4-(4-хлорфенил)гекс-2-еннитрилом, получали соединение 136.
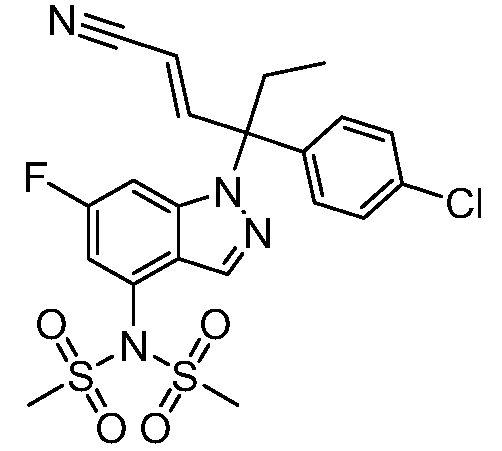
ПРИМЕР 27
N-(1-(3-(4-хлорфенил)-1-цианопентан-3-ил)-1Н-индазол-4-ил)метансульфонамид
Стадия A: (E)-4-(4-амино-1Н-индазол-1-ил)-4-(4-хлорфенил)гекс-2-еннитрил
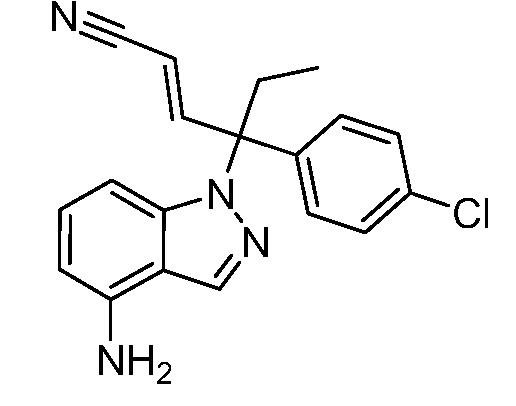
Смесь (E)-трет-бутил-1-(3-(4-хлорфенил)-1-цианопент-1-ен-3-ил)-1Н-индазол-4-илметилкарбамата (500 мг), как описано в Примере 25 Стадия A, в 15 мл 4н HCl/MeOH перемешивали при комнатной температуре в течение 1 часа. Затем растворитель удаляли. Остаток растворяли в ЕА (30 мл), и значение рН доводили до 9~10 насыщенным водным раствором NaHCO3. Органический слой промывали водой (10 мл), солевым раствором, высушивали (Na2SO4) и концентрировали. Остаток очищали на ВЭЖХ с обратной фазой (0,05% NH4HCO3 в H2O - MeCN), получая целевой продукт в форме твердого вещества светло-коричневого цвета. LC/MS m/z = 337,2 [M+H]+.
Стадия B: (E)-N-(1-(3-(4-хлорфенил)-1-цианопент-1-ен-3-ил)-1Н-индазол-4-ил)метансульфонамид
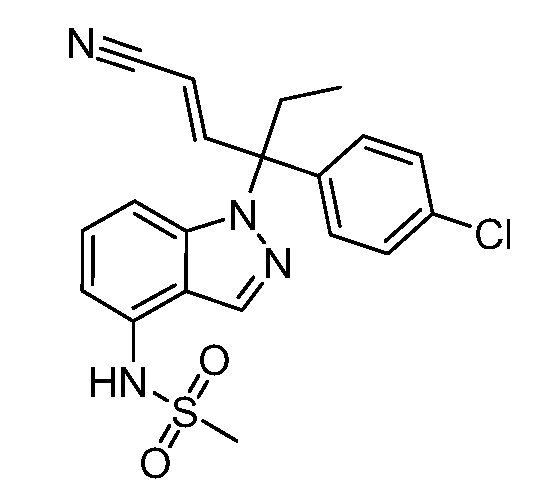
К смеси (E)-4-(4-амино-1Н-индазол-1-ил)-4-(4-хлорфенил)гекс-2-еннитрила (130 мг) и NMM (117 мг) в DCM (5 мл) добавляли MsCl (53 мг). Затем смесь перемешивали при комнатной температуре в течение 2 часов. Раствор гасили насыщенным раствором NH4Cl и экстрагировали этилацетатом (30 мл). Органический слой промывали солевым раствором, высушивали над сухим сульфатом натрия, фильтровали и упаривали. Остаток очищали хроматографией на силикагеле (элюент: РЕ/ЕА = 5/1), получая целевое соединение в форме твердого вещества светло-желтого цвета. LC/MS m/z = 415,2 [M+H]+.
Стадия C: N-(1-(3-(4-хлорфенил)-1-цианопентан-3-ил)-1Н-индазол-4-ил)метансульфонамид
Смесь (E)-N-(1-(3-(4-хлорфенил)-1-цианопент-1-ен-3-ил)-1Н-индазол-4-ил)метансульфонамида (60 мг) и 10%-ый Pd/C в ЕА (4 мл) перемешивали при комнатной температуре под атмосферой H2 в течение 16 часов. Смесь фильтровали и упаривали, получая целевое соединение в форме твердого вещества светло-желтого цвета. LC/MS m/z = 417,2 [M+H]+.
Используя процедуру, описанную в Примере 27, но на стадии B заменяя (E)-4-(4-амино-1Н-индазол-1-ил)-4-(4-хлорфенил)гекс-2-еннитрил (E)-4-(4-амино-1Н-индазол-1-ил)-4-(4-метоксифенил)гекс-2-еннитрилом, (E)-4-(4-амино-1Н-индол-1-ил)-4-(4-хлорфенил)гекс-2-еннитрилом, (E)-4-(4-амино-1Н-индазол-1-ил)-4-(4-(трифторметил)фенил)гекс-2-еннитрилом, (E)-4-(4-амино-6-фтор-1Н-индол-1-ил)-4-(4-хлорфенил)гекс-2-еннитрилом, (E)-4-(4-амино-6-фтор-1Н-индазол-1-ил)-4-фенилгекс-2-еннитрилом, (E)-4-(4-амино-6-фтор-1Н-индазол-1-ил)-4-(4-хлорфенил)гекс-2-еннитрилом, получали соединения 139-145, соответственно.
Соединение 137 и соединение 138 были получены в приведенных ниже условиях хирального разделения:
Колонка: AS-H
Мобильная фаза: A: CO2, B: MeOH, A:B = 60:40 при 3,0 мл/мин.
Температура колонки: 38,8°C
Соединение 140 получали в приведенных ниже условиях хирального разделения:
Колонка: AD-H
Мобильная фаза: A: CO2, B: MeOH, A:B = 60:40 при 3,0 мл/мин.
Температура колонки: 38,6°C
Соединение 141 получали в приведенных ниже условиях хирального разделения:
Колонка: AD-H
Мобильная фаза: A: CO2, B: MeOH (0,1% DEA), A:B = 93:7 при 2,97 мл/мин.
Температура колонки: 40,1°C
Соединение 142 и соединение 144 были получены в приведенных ниже условиях хирального разделения:
Колонка: AS-H
Мобильная фаза: A: CO2, B: MeOH, A:B = 50:50 при 3,0 мл/мин.
Температура колонки: 39,9°C
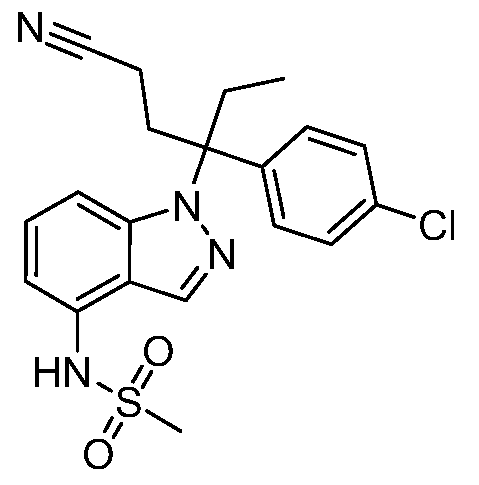
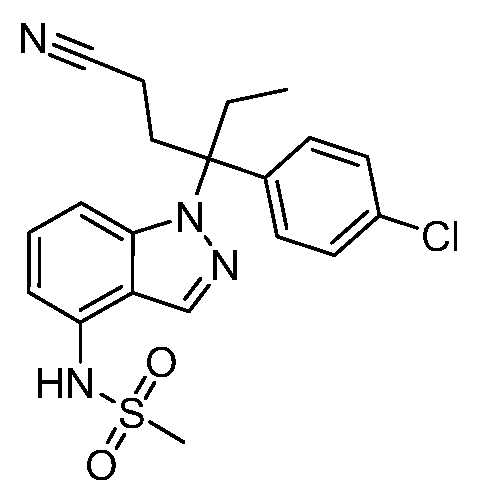
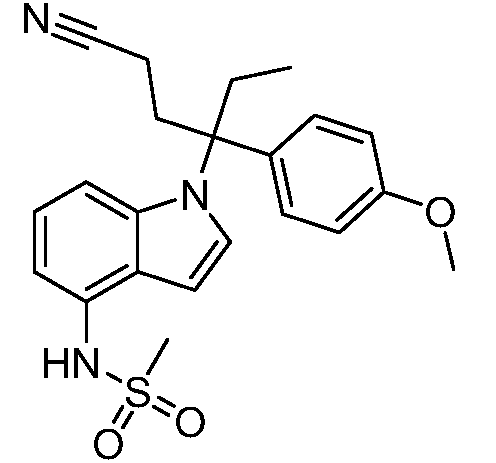
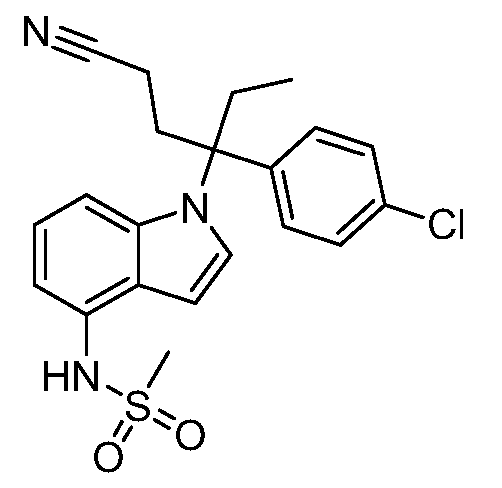
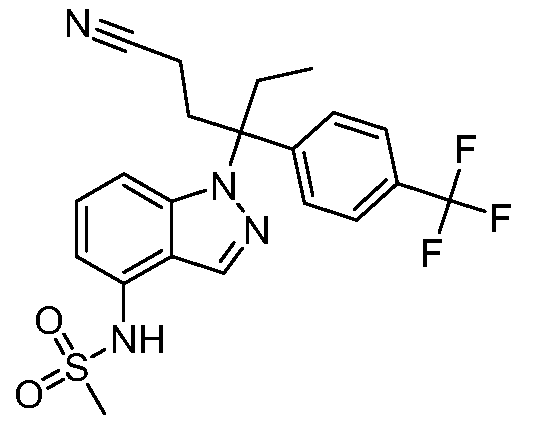
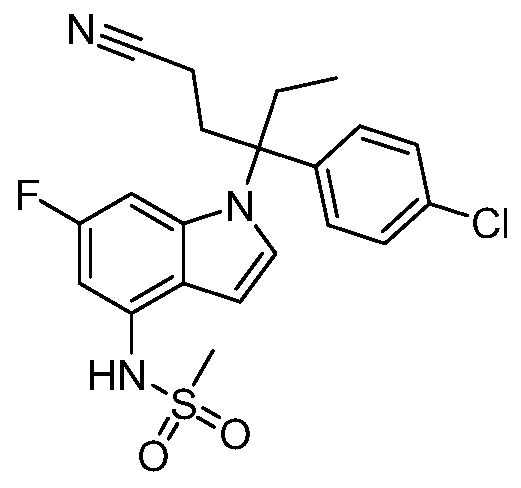
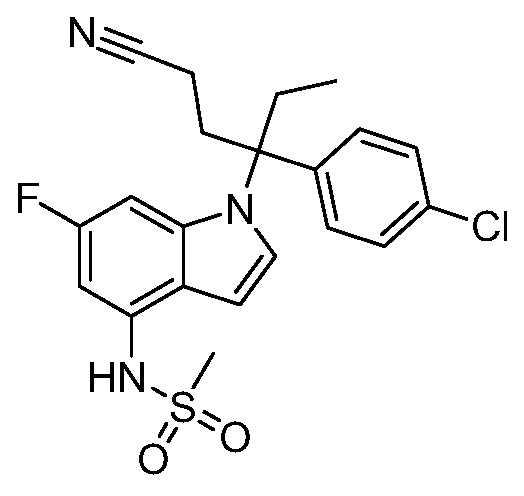
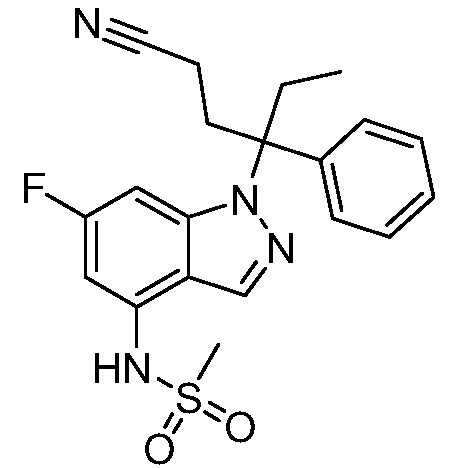
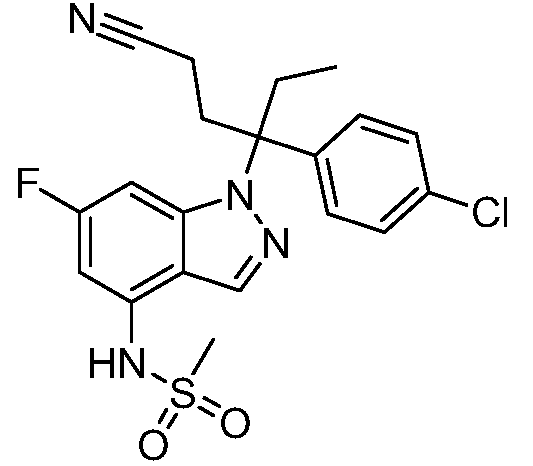
ПРИМЕР 28
N-(1-(3-(4-Хлорфенил)-1-циано-2-оксопентан-3-ил)-1Н-индол-4-ил)метансульфонамид (Соединение 146)
Стадия A: N-(1-(3-(4-Хлорфенил)-1-циано-2-оксопентан-3-ил)-1Н-индол-4-ил)метансульфонамид
К раствору ацетонитрила (180 мг, 4,4 ммоль) в тетрагидрофуране (3 мл) добавляли BuLi (1,9 мл, 4,8 ммоль, 2,5 М в гексане) при -78°C. После перемешивания в течение 30 минут, раствор 2-(4-хлорфенил)-2-(4-(метилсульфонамидо)-1Н-индол-1-ил)метилбутаноата (840 мг, 2,0 ммоль, Энантиомер A), как описано в Примере 3 стадия D, в тетрагидрофуране (3 мл) добавляли к смеси при -78°C. Полученную смесь перемешивали при -78°C еще 1 час и затем гасили водой (15 мл), экстрагировали этилацетатом. Объединенные органические фракции промывали солевым раствором (15 мл), высушивали над Na2SO4, фильтровали и концентрировали. Сырой продукт очищали хроматографией на силикагеле с элюированием смесью РЕ/ЕА (от 5/1 до 3/1, об./об.), получая целевое соединение в форме бесцветного аморфного твердого вещества (Энантиомер A). LC/MS m/z = 429,8 [M+H]+.
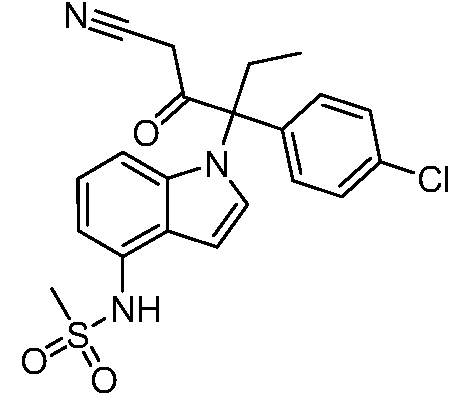
ПРИМЕР 29
N-(1-(1-(4-Хлорфенил)-1-(3-циано-4,5-дигидрофуран-2-ил)пропил)-1Н-индол-4-ил)метансульфонамид (Соединение 147)
К раствору N-(1-(3-(4-хлорфенил)-1-циано-2-оксопентан-3-ил)-1Н-индол-4-ил)метансульфонамида (50 мг, 0,116 ммоль, энантиомер A), как описано в Примере 28 стадия A, в DMF (2 мл) добавляли NaH (9 мг, 0348 ммоль, 60% в масле) при 0°C. После перемешивания в течение 30 минут, 1,2-дибромэтан (22 мг, 0,116 ммоль) добавляли к смеси при 0°C. Полученную смесь перемешивали при 0°C еще 1 час и затем гасили водой (15 мл), экстрагировали этилацетатом. Объединенные органические слои промывали солевым раствором (10 мл), высушивали над Na2SO4, фильтровали и концентрировали. Сырой продукт очищали хроматографией на силикагеле с элюированием смесью РЕ/ЕА (от 5/1 до 3/1, об./об.), получая целевое соединение в форме бесцветного аморфного твердого вещества. LC/MS m/z = 456,1 [M+H]+.
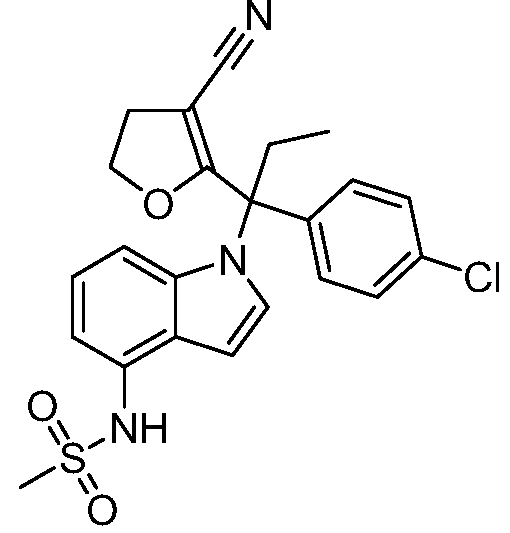
ПРИМЕР 30
N-(2-хлорэтил)-2-(4-хлорфенил)-2-(4-(метилсульфонамидо)-1Н-индол-1-ил)бутанамид (Соединение 148)
Стадия A: 2-(4-хлорфенил)-2-(4-(метилсульфонамидо)-1Н-индол-1-ил)бутановая кислота
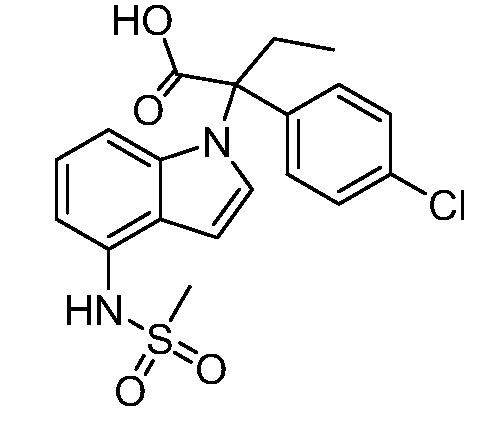
Смесь 2-(4-хлорфенил)-2-(4-(метилсульфонамидо)-1Н-индол-1-ил)метилбутаноата (840 мг, 2 ммоль), как описано в Примере 3 стадия C, и 2М LiOH (5 мл) в метаноле (5 мл) нагревали до 50°C в течение 5 часов. Реакционную смесь подкисляли 1M HCl, и летучую фракцию удаляли при пониженном давлении. Остаток растворяли в этилацетате (80 мл) и воде (20 мл). Органический слой промывали солевым раствором (10 мл) и высушивали над Na2SO4, фильтровали и концентрировали в вакууме, получая целевое соединение в форме бесцветного твердого вещества. LC/MS 407,0 (M+1).
Стадия B: N-(2-хлорэтил)-2-(4-хлорфенил)-2-(4-(метилсульфонамидо)-1Н-индол-1-ил)бутанамид
Смесь продукта Стадии А (40 мг, 0,1 ммоль), HATU (57 мг, 0,15 ммоль), N,N-диизопропилэтиламина (26 мг, 0,2 ммоль) и 2-хлорэтанамина (16 мг, 0,2 ммоль) в N,N-диметилформамиде (2 мл) перемешивали при комнатной температуре в течение ночи. Сырой продукт очищали CombiFlash (Мобильная фаза: ацетонитрил/вода (0,03% TFA)), получая целевое соединение в форме аморфного твердого вещества белого цвета. LC/MS m/z = 467,9 [M+H]+.
Следуя той же самой процедуре, как для ПРИМЕРА 30 стадия B, но с использованием 2-аминоацетонитрила вместо 2-хлорэтанамина, получали соединение 149 представленных в Таблице 27.
ПРИМЕР 31
N-(1-(1-(4-хлорфенил)-1-(3-метил-1,2,4-оксадиазол-5-ил)пропил)-1Н-индол-4-ил)метансульфонамид (Соединение 150)
Стадия A: 2-(4-хлорфенил)-N-(1-(гидроксиимино)этил)-2-(4-(метилсульфонамидо)-1Н-индол-1-ил)бутанамид
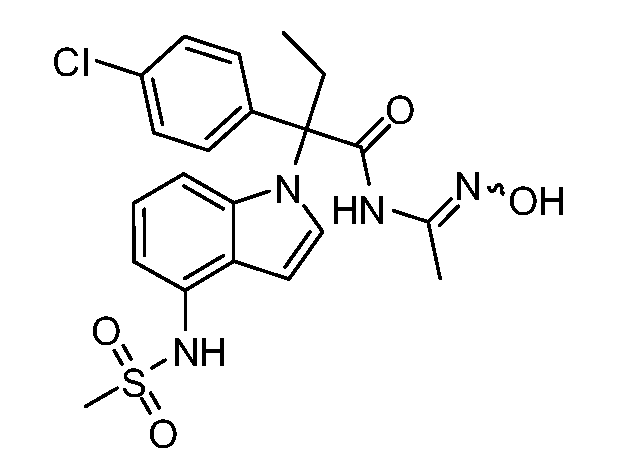
К раствору 2-(4-хлорфенил)-2-[4-(метилсульфонамидо)-1Н-индол-1-ил]бутановой кислоты (200 мг, 0,49 ммоль), как описано в Примере 30 Стадия A, в THF (5 мл) при комнатной температуре добавляли EDCI (141 мг, 0,74 ммоль), триэтиламин (75 мг, 0,74 ммоль) и HOBT (13 мг, 0,098 ммоль). Смесь перемешивали в течение 15 минут и затем добавляли N'-гидроксиацетимидамид (73 мг, 1,47 ммоль). Через 4 часа полученную смесь разбавляли насыщенным водным раствором хлорида аммония (10 мл) и экстрагировали ЕА (20 мл). Объединенные органические слои промывали солевым раствором (10 мл), высушивали над безводным сульфатом натрия, фильтровали, и растворитель удаляли в вакууме. Остаток использовали на следующей стадии без дальнейшей очистки.
Стадия B: N-(1-(1-(4-хлорфенил)-1-(3-метил-1,2,4-оксадиазол-5-ил)пропил)-1Н-индол-4-ил)метансульфонамид
Раствор 2-(4-хлорфенил)-N-(1-(гидроксиимино)этил)-2-(4-(метилсульфонамидо)-1Н-индол-1-ил)бутанамида (148 мг, 0,32 ммоль) в THF (5мкл) нагревали при 70°C в течение 2 дней. Затем растворитель удаляли в вакууме, получая сырой продукт. Продукт очищали хроматографией на колонках (50% EtOAc/гексаны), затем подвергали хиральному разделению в условиях, приведенных ниже, получая целевое соединение как единственный энантиомер.
Используя ту же самую процедуру, описанную для Примера 31, но заменяя N'-гидроксиацетимидамид на стадии А N'-гидроксипропионимидамидом, получали целевое соединение 151.
Соединение 150 получали в приведенных ниже условиях хирального разделения:
Колонка: OJ-Н
Мобильная фаза: A: N-гексан, B: EtOH (0,1% DEA), A:B =70:30 при 1,0 мл/мин.
Температура колонки: 40°C
Соединение 151 получали в приведенных ниже условиях хирального разделения:
Колонка: AS-H
Мобильная фаза: A: CO2, B: MeOH (0,1% DEA), A:B = 70:30 при 3,0 мл/мин.
Температура колонки: 39,8°C
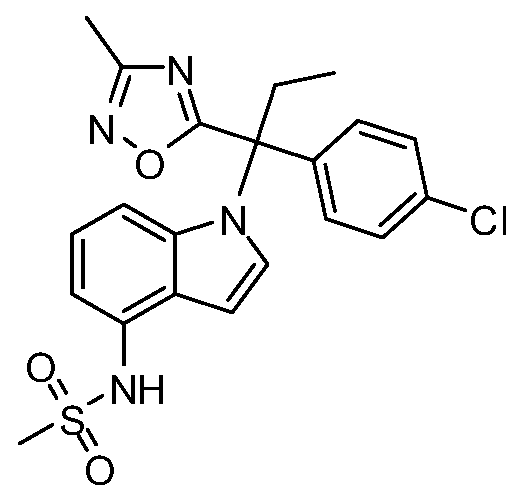
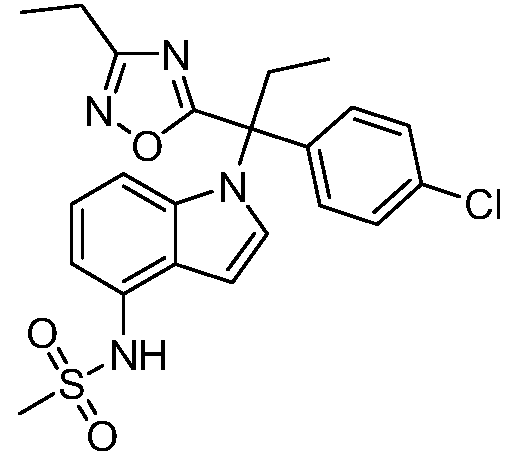
ПРИМЕР 32
N-(1-(1-(4-хлорфенил)-1-(2-этил-2Н-1,2,3-триазол-4-ил)пропил)-1Н-индол-4-ил)метансульфонамид (Соединение 152)
Стадия A: 1-(3-(4-хлорфенил)пент-1-ин-3-ил)-1Н-индол-4-ил-трет-бутилкарбамат
Смесь 1-(2-(4-хлорфенил)-1-оксобутан-2-ил)-1Н-индол-4-ил-трет-бутилкарбамата (500 мг, 1,21 ммоль), как описано в Примере 21 Стадия B, и 1-диазо-2-оксопропилдиметилфосфоната (244 мг, 1,27 ммоль) в MeOH (10 мл) перемешивали при комнатной температуре в течение 4 часов. После удаления растворителя в вакууме, остаток очищали хроматографией на колонках (0-30% EtOAc/гексаны), получая целевое соединение.
Стадия B: 1-(1-(4-хлорфенил)-1-(2H-1,2,3-триазол-4-ил)пропил)-1Н-индол-4-ил-трет-бутилкарбамат
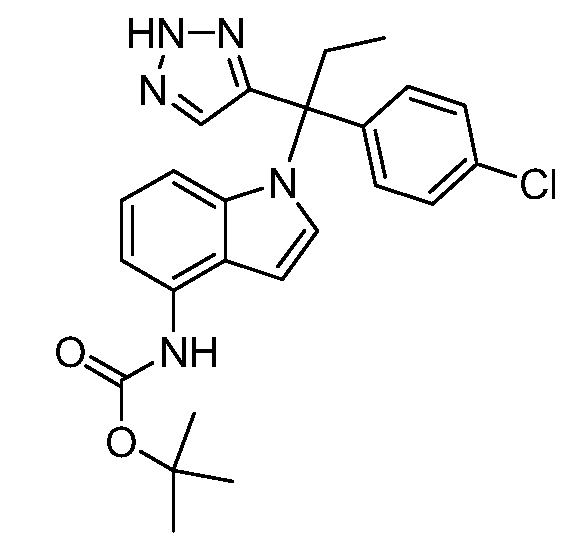
Раствор 1-(3-(4-хлорфенил)пент-1-ин-3-ил)-1Н-индол-4-ил-трет-бутилкарбамата (200 мг, 0,5 ммоль) в триметилсилил азиде (3 мл) нагревали при 100°C в микроволновом реакторе в течение 3 часов. Полученную смесь разбавляли солевым раствором (10 мл) и экстрагировали ЕА. Объединенные органические слои высушивали над безводным сульфатом натрия, фильтровали, и растворитель удаляли в вакууме, получая сырой продукт. Продукт очищали хроматографией на колонках (75% EtOAc/гексаны), получая целевой продукт.
Стадия C: 1-(1-(4-хлорфенил)-1-(2-этил-2Н-1,2,3-триазол-4-ил)пропил)-1Н-индол-4-ил-трет-бутилкарбамат
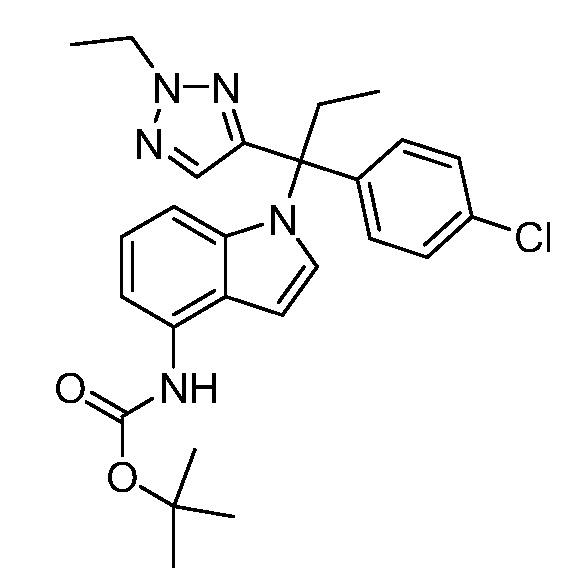
К раствору 1-(1-(4-хлорфенил)-1-(2H-1,2,3-триазол-4-ил)пропил)-1Н-индол-4-ил-трет-бутилкарбамата (150 мг, 0,33 ммоль) в DMF (5 мл) добавляли карбонат калия (91 мг, 0,66 ммоль) и йодэтан (62 мг, 0,40 ммоль). После перемешивания в течение 2 часов, смесь разбавляли насыщенным водным раствором хлорида аммония (10 мл) и экстрагировали ЕА. Объединенные органические слои промывали солевым раствором (10 мл), высушивали над безводным сульфатом натрия, фильтровали, и растворитель удаляли в вакууме, получая сырой продукт. Продукт очищали хроматографией на колонках (50% EtOAc/гексаны), получая целевой продукт.
Стадия D: 1-(1-(4-хлорфенил)-1-(2-этил-2Н-1,2,3-триазол-4-ил)пропил)-1Н-индол-4-амин
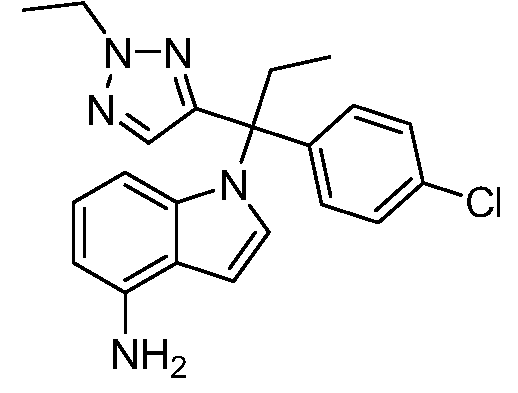
Используя ту же самую процедуру, описанную для Примера 14, Стадия G, но заменяя 2-(4-(трет-бутоксикарбониламино)-6-фтор-1Н-индол-1-ил)-2-(4-хлорфенил)уксусную кислоту 1-(1-(4-хлорфенил)-1-(2-этил-2Н-1,2,3-триазол-4-ил)пропил)-1Н-индол-4-ил-трет-бутилкарбаматом, получали целевое соединение.
Стадия E: N-(1-(1-(4-хлорфенил)-1-(2-этил-2Н-1,2,3-триазол-4-ил)пропил)-1Н-индол-4-ил)метансульфонамид (Соединение 152)
Используя ту же самую процедуру, описанную для Примера 14, стадия K, но заменяя 2-(4-амино-6-фтор-1Н-индол-1-ил)-2-(4-хлорфенил)метилбутаноат 1-(1-(4-хлорфенил)-1-(2-этил-2Н-1,2,3-триазол-4-ил)пропил)-1Н-индол-4-амином, получали целевое соединение.
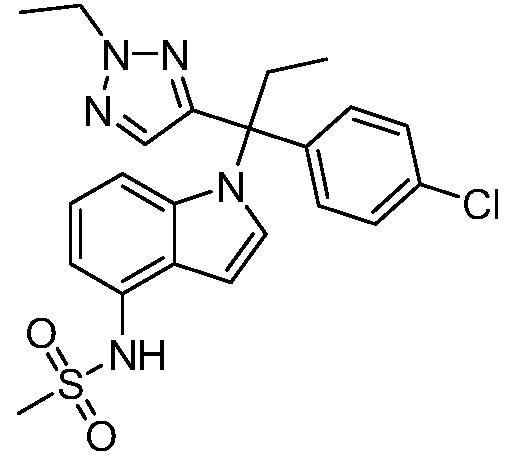
ПРИМЕР 33
2-(4-хлорфенил)-2-(4-(2-(метилсульфонил)этил)-1Н-индол-1-ил)метилбутаноат (Соединение 153)
Стадия A: 2-(4-хлорфенил)-2-(4-формил-1Н-индол-1-ил)метилацетат
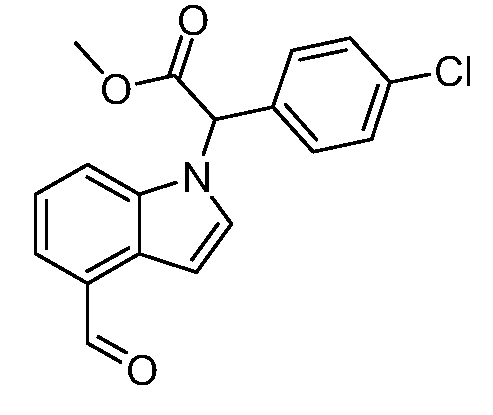
К раствору 1H-индол-4-карбальдегида (2,0 г, 13,79 ммоль) в DMF (50 мл) добавляли NaH (0,66 г, 16,55 ммоль) при 0°C. После перемешивания в течение 30 минут при 0°C, добавляли по каплям раствор 2-бром-2-(4-хлорфенил)-метилацетата (4,67 г, 17,92 ммоль) в DMF (20 мл). Реакционной смеси давали нагреться до комнатной температуры и перемешивали в течение дополнительного 1 часа. Затем ее разбавляли EtOAC (200 мл) и гасили водой. Органический слой отделяли и промывали солевым раствором (50 мл), высушивали над Na2SO4, фильтровали, и растворитель удаляли в вакууме. Остаток очищали хроматографией на колонках (0-20% EtOAc/гексаны), получая целевое соединение.
Стадия B: 2-(4-хлорфенил)-2-(4-формил-1Н-индол-1-ил)метилбутаноат
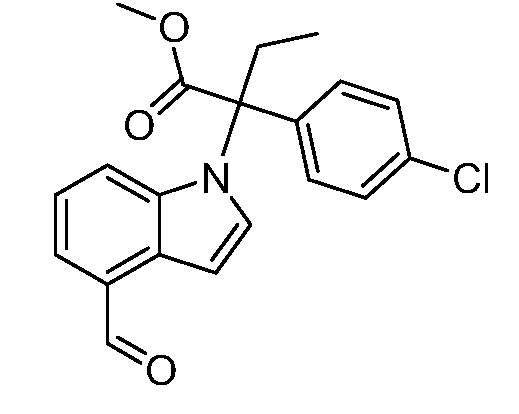
К раствору 2-(4-хлорфенил)-2-(4-формил-1Н-индол-1-ил)-метилацетата (2,34 г, 7,17 ммоль) в THF (60 мл) и HMPA (15 мл) по каплям при 0°C добавляли LiHMDS (1 М в THF, 11,0 мл, 11 ммоль). Смесь перемешивали при 0°C в течение 30 минут перед добавлением йодэтана (1,23 г, 7,88 ммоль). После перемешивания в течение еще 30 минут при 0°C, смесь разбавляли EtOAC (120 мл) и затем гасили водой (20 мл). Органический слой отделяли и промывали солевым раствором (20 мл), высушивали над Na2SO4, фильтровали, и растворитель удаляли в вакууме. Остаток очищали хроматографией на колонках (0-15% EtOAc/гексаны), получая целевое соединение.
Стадия C: (E)-2-(4-хлорфенил)-2-(4-(2-(метилсульфонил)-винил)-1Н-индол-1-ил)метилбутаноат
К раствору диэтил метилсульфонилметилфосфоната (1,05 г, 4,56 ммоль) в CH3CN (30 мл) при комнатной температуре добавляли LiCl (0,16 г, 3,80 ммоль) и DBU (2,40 г, 9,56 ммоль). Через 5 минут добавляли по каплям раствор 2-(4-хлорфенил)-2-(4-формил-1Н-индол-1-ил)метилбутаноата (1,35 г, 3,80 ммоль) в CH3CN (10 мл). Полученную смесь перемешивали в течение 4 часов при комнатной температуре. Затем ее разбавляли DCM (50 мл), и органический слой промывали водой (30 мл), высушивали над Na2SO4, фильтровали. После удаления растворителя, остаток очищали хроматографией на колонках (0-30% EtOAc/гексаны), получая целевое соединение. LC/MS m/z = 432,0 [M+H]+.
Стадия D: 2-(4-хлорфенил)-2-(4-(2-(метилсульфонил)этил)-1Н-индол-1-ил)метилбутаноат
К раствору (E)-2-(4-хлорфенил)-2-(4-(2-(метилсульфонил)-винил)-1Н-индол-1-ил)метилбутаноата (1,18 г, 2,73 ммоль) в EtOAC (50 мл) добавляли Pd/C (10% на углероде, 0,2 г). Смесь перемешивали под атмосферой H2 в течение 30 мин. Затем реакционную смесь фильтровали через слой целита, и фильтрат концентрировали в вакууме. Остаток очищали хроматографией на колонках (0-30% EtOAc/гексаны), получая целевое соединение. LC/MS m/z = 434,1 [M+H]+.
ПРИМЕР 34
N-(1-(1-(4-хлорфенил)-1-(1-гидроксициклопропил)пропил)-1Н-индол-4-ил)метансульфонамид (Соединение 154)
Стадия A: 1-(1-(4-хлорфенил)-1-(1-гидроксициклопропил)-пропил)-1Н-индол-4-ил-трет-бутилкарбамат
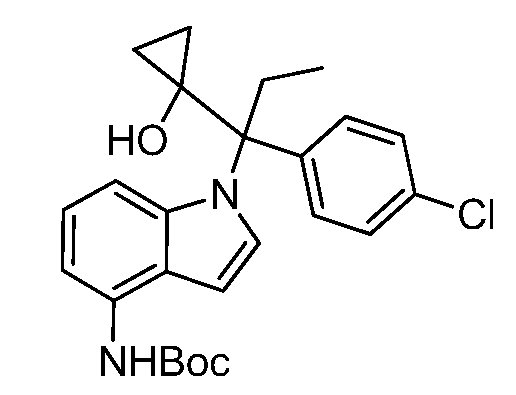
К раствору 2-{4-[(трет-бутоксикарбонил)амино]-1Н-индол-1-ил}-2-(4-хлорфенил)метилбутаноата (0,44 г, 1 ммоль), как описано в Примере 3 стадия A, в THF (20 мл) добавляли изопропилат титана (IV) (0,3 г, 1 ммоль), и раствор охлаждали до 0°C. Затем 3н этилмагний бромид в простом эфире (1,8 мл, 5 ммоль) добавляли через шприц в течение 1 часа, и реакционную смесь перемешивали в течение дополнительных 30 минут при этой температуре. Смесь нагревали при комнатной температуре и перемешивали в течение ночи. Конечную смесь разбавляли ЕА, обрабатывали насыщенным раствором бикарбоната натрия, экстрагировали ЕА и DCM, высушивали над сульфатом натрия и концентрировали. Остаток очищали флэш-хроматографией на силикагеле (элюируя от гексаны/ЕА 95:5 до ЕА), получая целевое соединение. LC/MS m/z = 441,0 [M+H]+.
Стадия B: 1-(1-(4-амино-1Н-индол-1-ил)-1-(4-хлорфенил)-пропил)циклопропанол
Соединение со стадии А (96 мг, 0,22 ммоль) обрабатывали 10 мл 4н раствора HCl/MeOH в течение ночи. Растворители удаляли при пониженном давлении, получая белый продукт. LC/MS m/z = 341,0 [M+H]+.
Стадия C: N-(1-(1-(4-хлорфенил)-1-(1-гидроксициклопропил)-пропил)-1Н-индол-4-ил)метансульфонамид
К смеси соединения со стадии B (83 мг, 0,22 ммоль) и NMM (44 мг, 0,44 ммоль) в DCM (5 мл) добавляли по каплям метилсульфонил хлорид (37 мг, 0,3 ммоль). Затем смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь гасили насыщенным раствором хлорида аммония и экстрагировали этилацетатом (20 мл). Органический слой промывали солевым раствором, высушивали над сульфатом натрия, фильтровали и упаривали. Остаток очищали Pre-ВЭЖХ, получая целевое соединение в форме твердого вещества белого цвета. LC/MS m/z = 419,1 [M+H]+.
Соединение 154 получали в приведенных ниже условиях хирального разделения:
Колонка: OD-H
Мобильная фаза: A: CO2, B: MeOH, A:B = 70:30 при 3,0 мл/мин.
Температура колонки: 40,2°C
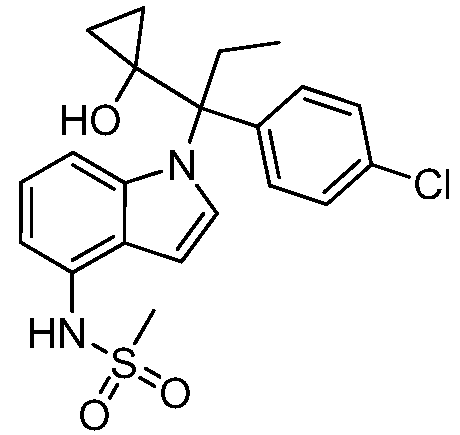
ПРИМЕР 35
N-(1-(3-(4-хлорфенил)-1-цианопент-1-ин-3-ил)-1Н-индол-4-ил)метансульфонамид
Стадия A: 4-(4-хлорфенил)-4-(4-(N-((2-(триметилсилил)-этокси)метил)метилсульфонамидо)-1Н-индол-1-ил)метилгекс-2-иноат
К раствору соединения N-(1-(3-(4-хлорфенил)пент-1-ин-3-ил)-1Н-индол-4-ил)-N-((2-(триметилсилил)этокси)метил)метансульфонамида (2,9 г, 5,6 ммоль, энантиомер A), как описано в Примере 55 Стадия A, в сухом THF (10 мл) добавляли BuLi (4,5 мл, 11,2 ммоль, 2,5 М в гексане) при -78°C. После перемешивания в течение 30 минут, метилхлоридат углерода (0,88 мл, 6,2 ммоль) добавляли к смеси при -78°C. Полученную смесь перемешивали в течение еще 2 часов и затем гасили насыщенным раствором NH4Cl (10 мл), экстрагировали этилацетатом, промывали солевым раствором (50 мл), высушивали над сульфатом натрия, затем фильтровали. После удаления органического растворителя, остаток очищали хроматографией на колонках (ЕА/РЕ = 1:10), получая целевое соединение. LC/MS m/z = 597,0 [M+Na]+.
Стадия B: 4-(4-хлорфенил)-4-(4-(N-((2-(триметилсилил)-этокси)метил)метилсульфонамидо)-1Н-индол-1-ил)гекс-2-иновая кислота
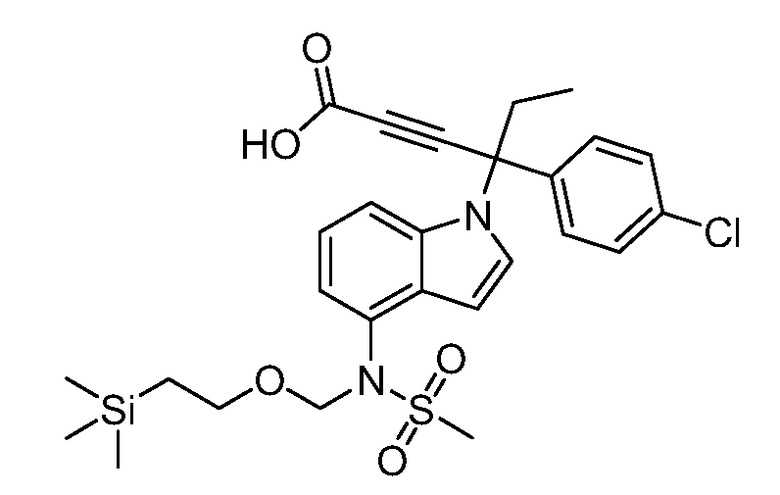
К раствору соединения со стадии А (100 мг, 0,17 ммоль, энантиомер A) в MeOH (5 мл) добавляли H2O (1 мл), LiOH (72 мг, 1,7 ммоль). Смесь перемешивали при комнатной температуре в течение ночи. После удаления органического растворителя, остаток растворяли в H2O (5 мл) и подкисляли до рН = 4 с помощью 1M HCl, затем экстрагировали этилацетатом и промывали солевым раствором (20 мл), высушивали над сухим сульфатом натрия, затем фильтровали. Растворитель выпаривали, получая целевое соединение. LC/MS m/z = 583,0 [M+Na]+.
Стадия C: 4-(4-хлорфенил)-4-(4-(N-((2-(триметилсилил)этокси) метил)метилсульфонамидо)-1Н-индол-1-ил)гекс-2-инамид
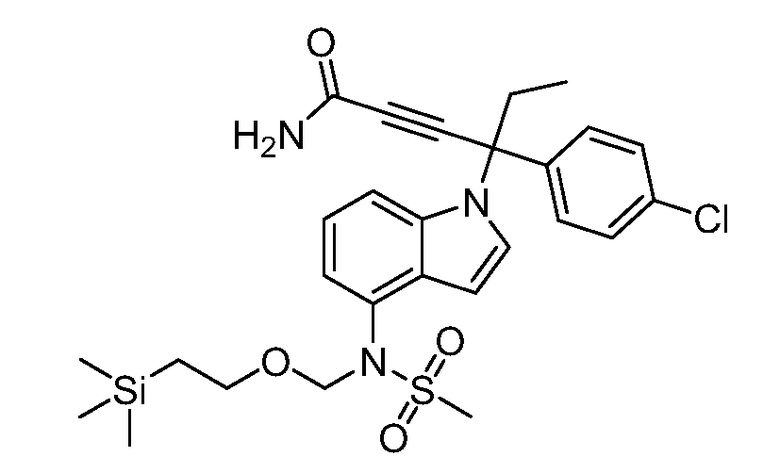
К раствору продукта со стадии B (40 мг, 0,071 ммоль, энантиомер A) в DMF (5 мл) добавляли DIPEA (18 мг, 0,142 ммоль), HATU (40 мг, 0,11 ммоль), NH4Cl (38 мг, 0,71 ммоль). Смесь перемешивали при комнатной температуре в течение ночи. После удаления органического растворителя, остаток растворяли в H2O (20 мл), экстрагировали этилацетатом и промывали солевым раствором, высушивали над сульфатом натрия, затем фильтровали. Растворитель выпаривали, и остаток очищали Prep-TLC (ЕА/РЕ = 2:1), получая целевое соединение. LC/MS m/z = 582,0 [M+Na]+.
Стадия D: N-(1-(3-(4-хлорфенил)-1-цианопент-1-ин-3-ил)-1Н-индол-4-ил)-N-((2-(триметил силил)этокси)метил)метансульфонамид
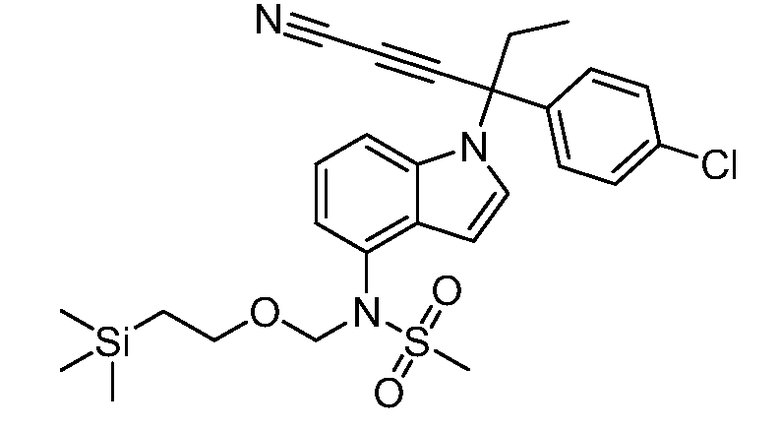
К раствору продукта со стадии C (20 мг, 0,036 ммоль, энантиомер A) в пиридине (5 мл) добавляли POCl3 (0,05 мл). Смесь перемешивали при комнатной температуре в течение 2 часов. Остаток очищали Combiflash (Мобильная фаза: MeOH/H2O (0,08% NH4HCO3), получая целевое соединение. LC/MS m/z = 564,0 [M+Na]+.
Стадия E: N-(1-(3-(4-хлорфенил)-1-цианопент-1-ин-3-ил)-1Н-индол-4-ил)метансульфонамид
К раствору продукта со стадии D (15 мг, 0,028 ммоль, энантиомер A) в EtOH (2 мл) добавляли 2н HCl (0,5 мл). Смесь перемешивали при 50°C в течение ночи. Летучую фракцию удаляли при пониженном давлении, и остаток очищали Combiflash (Мобильная фаза: MeOH/H2O (0,08% NH4HCO3), получая целевое соединение. LC/MS m/z = 412,1 [M+H]+.
1H ЯМР (400 МГц, CDCl3) δ 7,53 (д, J=3,6 Гц, 1H), 7,33 (д, J=8,8 Гц, 2H), 7,17 (д, J=8,0 Гц, 1H), 7,12 (д, J=8,4 Гц, 2H), 7,01 (т, J=8,0 Гц, 1H), 6,73 (д, J=8,4 Гц, 1H), 6,70 (д, J=3,6 Гц, 1H), 6,62 (шир., 1H), 3,04 (с, 3H), 2,80-2,76 (м, 1H), 2,52-2,48 (м, 1H), 1,05 (т, J=7,2 Гц, 3H).
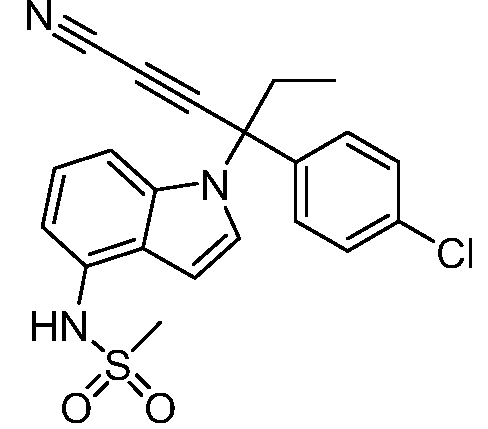
ПРИМЕР 36
2-(4-хлорфенил)-4-фтор-2-(4-(метилсульфонамидо)-1Н-индол-1-ил)метилбутаноат (Соединение 156)
Стадия A: 2-(4-(трет-бутоксикарбониламино)-1Н-индол-1-ил)-2-(4-хлорфенил)-4-фторметилбутаноат
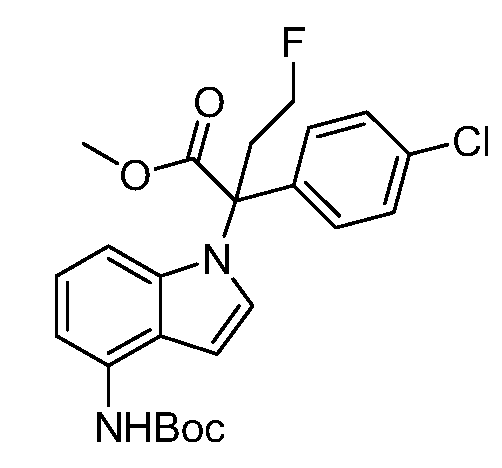
К суспензии NaH (60%-ая дисперсия в минеральном масле) (22 мг, 0,55 ммоль) в безводном DMF (3 мл) под атмосферой азота добавляли 2-(4-(трет-бутоксикарбониламино)-1Н-индол-1-ил)-2-(4-хлорфенил)метилацетат (207 мг, 0,5 ммоль), как описано в Примере 2 стадия B, в безводном DMF (2 мл) и смесь затем перемешивали при 0°C в течение 1 часа. К этой смеси добавляли 1-фтор-2-йодэтан (17,4 мг, 1 ммоль), и смесь затем перемешивали при комнатной температуре в течение 4 часов. Летучую фракцию выпаривали. Остаток очищали хроматографией на силикагеле с элюированием смесью РЕ/ЕА (от 10/1 до 3/1, об./об.), получая целевое соединение в форме твердого вещества белого цвета. LC/MS m/z = 405,1 [M+H]+.
Стадия B: 2-(4-амино-1Н-индол-1-ил)-2-(4-хлорфенил)-4-фторметилбутаноат
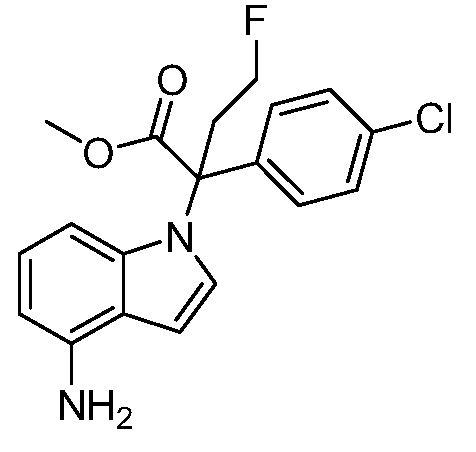
Смесь продукта со стадии А (100 мг, 0,217 ммоль) и 4н HCl/MeOH (10 мл) перемешивали при комнатной температуре в течение 2 часов. После удаления растворителя, остаток высушивали в вакууме, получая целевое соединение в форме коричневого масла. LC/MS m/z = 361,1 [M+H]+.
Стадия C: 2-(4-хлорфенил)-4-фтор-2-(4-(метилсульфонамидо)-1Н-индол-1-ил)метилбутаноат
К смеси соединения со стадии B (68 мг, 0,189 ммоль) и NMM (38 мг, 0,37 ммоль) в DCM (7 мл) добавляли по каплям метилсульфонил хлорид (32 мг, 0,283 ммоль). Затем смесь перемешивали при комнатной температуре в течение 4 часов. Реакционную смесь гасили насыщенным раствором хлорида аммония, экстрагировали этилацетатом (50 мл). Органический слой промывали солевым раствором, высушивали над сульфатом натрия, фильтровали и упаривали. Остаток очищали хроматографией на силикагеле с элюированием смесью РЕ/ЕА (от 10/1 до 3/1, об./об.), получая целевое соединение в форме твердого вещества белого цвета. LC/MS m/z = 439,0 [M+H]+.
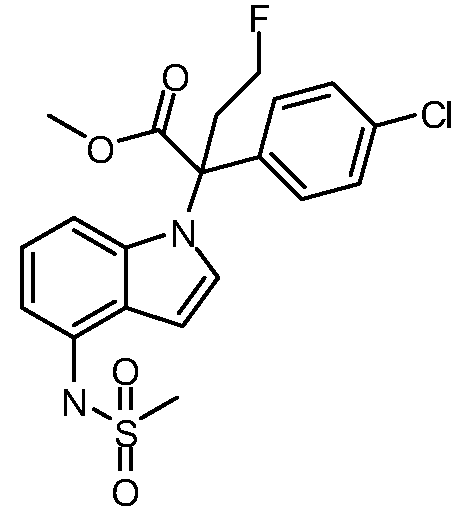
ПРИМЕР 37
5-(1-(4-хлорфенил)-1-(4-(метилсульфонамидо)-1Н-индазол-1-ил)пропил)-1,2,4-оксадиазол-3-карбоксамид
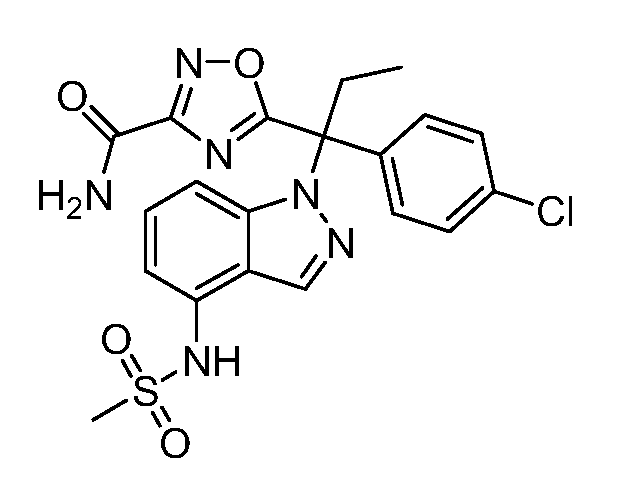
Стадия A: 2-(4-((трет-бутоксикарбонил)амино)-1Н-индазол-1-ил)-2-(4-хлорфенил)бутановая кислота
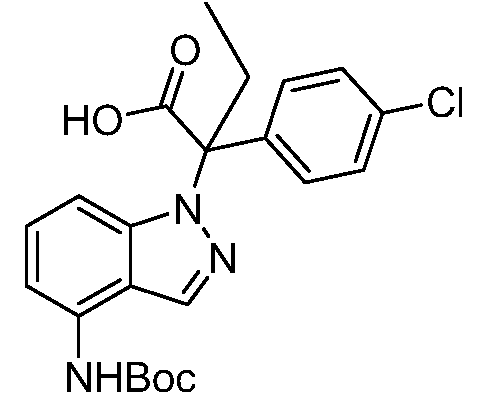
2-(4-((трет-бутоксикарбонил)амино)-1Н-индазол-1-ил)-2-(4-хлорфенил)метилбутаноат (0,5 г, 1,13 ммоль) (энантиомер А промежуточного соединения), как описано в примере 18 стадия D, растворяли в смеси 1,4-диоксана (1,0 мл) и воды (0,25 мл) с последующим добавлением LiOH (0,27 г, 11,26 ммоль). Смесь перемешивали при 70°C в течение 5 часов, затем концентрировали, чтобы удалить растворитель. Остаток растворяли в воде (20 мл) и подкисляли 1н водным раствором HCl (11,3 мл), получая белый осадок. Твердое вещество отфильтровывали, промывали водой и высушивали под вакуумом, получая целевое соединение. LC/MS m/z = 430,1 [M+H]+.
Стадия B: 2-амино-2-(((2-(4-((трет-бутоксикарбонил)амино)-1Н-индазол-1-ил)-2-(4-хлорфенил)бутаноил)окси)имино)этилацетат
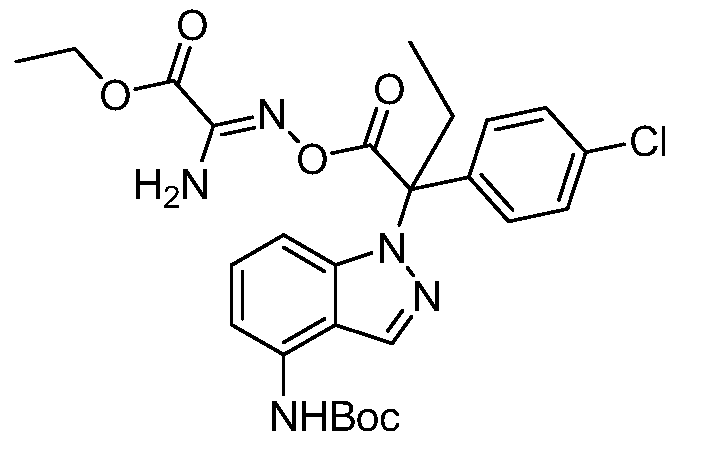
К суспензии 2-(4-((трет-бутоксикарбонил)амино)-1Н-индазол-1-ил)-2-(4-хлорфенил)бутановой кислоты со стадии А (0,16 г, 0,37 ммоль) в DCM (1,5 мл) добавляли 1,1'-карбонилдиимидазол (0,13 г, 0,78 ммоль), получая прозрачный раствор. Смесь перемешивали в течение 7,5 часов при комнатной температуре с последующим добавлением этил-2-оксиминооксамата (0,13 г, 0,93 ммоль). После перемешивания при комнатной температуре в течение ночи, полученную смесь разбавляли EtOAc, промывали насыщенным раствором NaHCO3, водой и солевым раствором. Органический слой отделяли и концентрировали. Полученный сырой продукт повторно растворяли в смеси ацетонитрила и воды и высушивали в лиофилизированном, получая целевое соединение в форме твердого вещества светло-желтого цвета. LC/MS m/z = 544,1 [M+H]+.
Стадия C: 5-(1-(4-((трет-бутоксикарбонил)амино)-1Н-индазол-1-ил)-1-(4-хлорфенил)пропил)-1,2,4-оксадиазол-3-этилкарбоксилат
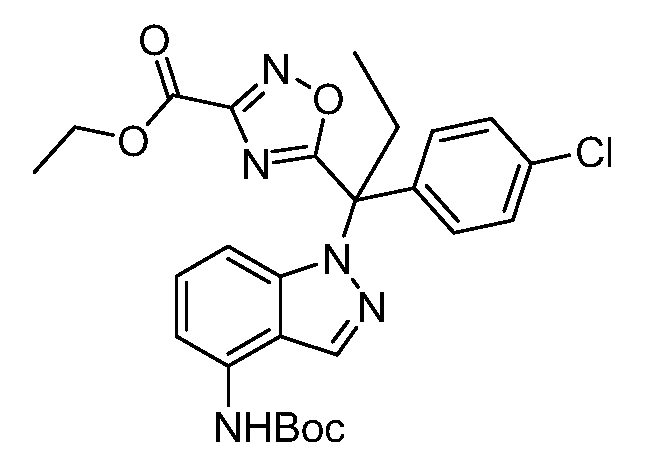
К раствору продукта со стадии B, описанной выше (0,18 г, 0,33 ммоль), в пиридине (9,0 мл) добавляли хлорангидрид фосфорной кислоты (0,25 г, 1,65 ммоль). Смесь нагревали при 70°C в течение 7 часов и перемешивали при 35°C в течение ночи, затем концентрировали досуха при пониженном давлении. Остаток растворяли в EtOAc и промывали насыщенным раствором NaHCO3, водой и солевым раствором. Органический слой отделяли и высушивали над Na2SO4. Сырой продукт очищали на колонке с силикагелем, используя градиент 0-50% этилацетата в гексанах, получая целевое соединение в форме твердого вещества светло-коричневого цвета. LC/MS m/z = 526,1 [M+H]+.
Стадия D: 5-(1-(4-амино-1Н-индазол-1-ил)-1-(4-хлорфенил)пропил)-1,2,4-оксадиазол-3-этилкарбоксилат
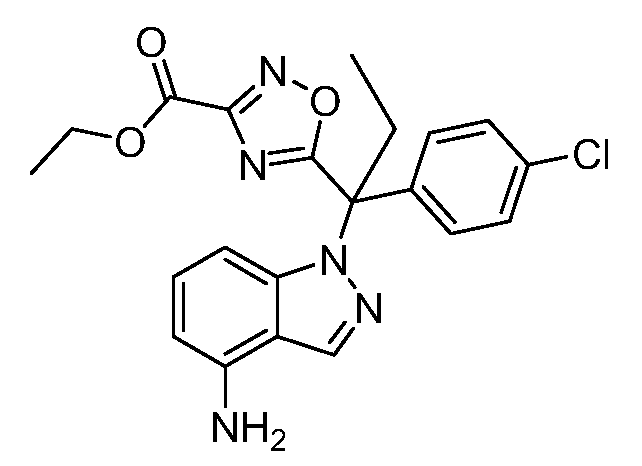
К раствору продукта со стадии C, описанной выше (0,083 г, 0,16 ммоль), и анизола (0,068 г, 0,63 ммоль) в DCM (1,0 мл) добавляли TFA (0,18 г, 1,58 ммоль). После перемешивания в течение ночи при комнатной температуре, реакционную смесь концентрировали досуха при пониженном давлении. Полученный остаток повторно растворяли в смеси ацетонитрила и воды с последующим добавлением 1н водного раствора HCl (0,16 мл). Раствор высушивали в лиофилизированном, получая сырое целевое соединение. LC/MS m/z = 426,1 [M+H]+.
Стадия E: 5-(1-(4-хлорфенил)-1-(4-(метилсульфонамидо)-1Н-индазол-1-ил)пропил)-1,2,4-оксадиазол-3-этилкарбоксилат
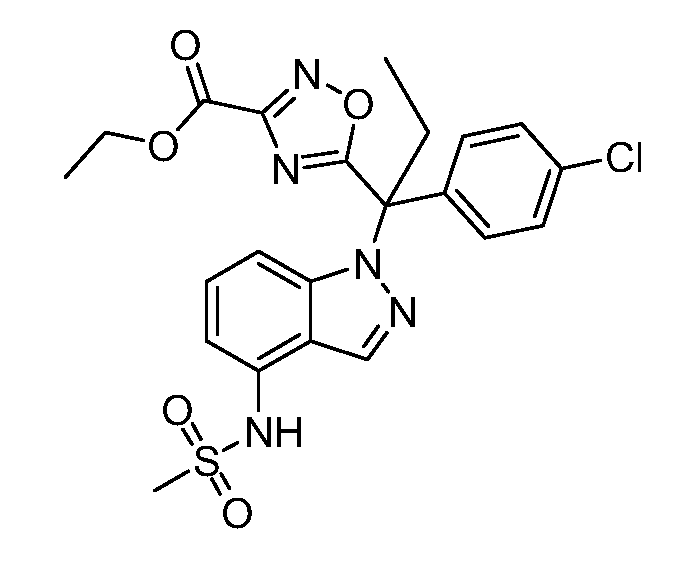
К раствору продукта со стадии D, описанной выше (0,080 г, 0,17 ммоль), и N-метилморфолина (0,035 г, 0,35 ммоль) в DCM (1,0 мл) добавляли метансульфонил хлорид (0,019 г, 0,17 ммоль) при 0°C. После перемешивания при комнатной температуре в течение 30 минут, реакционную смесь концентрировали при пониженном давлении. Полученный остаток разбавляли диоксаном/водой, подкисляли TFA и очищали непосредственно на ВЭЖХ с обратной фазой, получая целевое соединение. LC/MS m/z = 504,1 [M+H]+.
Стадия F: 5-(1-(4-хлорфенил)-1-(4-(метилсульфонамидо)-1Н-индазол-1-ил)пропил)-1,2,4-оксадиазол-3-карбоксамид
Продукт со стадии E, описанной выше (0,015 г, 0,030 ммоль), растворяли в 7н растворе аммиака в метаноле (2,6 мл). После перемешивания при комнатной температуре в течение 45 минут, полученную смесь концентрировали при пониженном давлении. Остаток растворяли в диоксане/воде, подкисляли TFA и очищали непосредственно на ВЭЖХ с обратной фазой, получая целевое соединение. LC/MS m/z = 475,0 [M+H]+.
1Ή ЯМР (500 МГц, CD3OD): δ (м.д.) 8,34 (с, 1H), 7,43 (д, J=8,8 Гц, 2H), 7,37 (д, J=8,8 Гц, 2H), 7,16-7,14 (м, 2H), 6,48-6,45 (м, 1H), 3,20-3,12 (м, 1H), 3,14-3,05 (м, 1H), 3,04 (с, 3 H), 0,89 (т, J=7,4 Гц, 3H).
ПРИМЕР 38
N-(1-(1-(4-хлорфенил)-1-(3-циано-1,2,4-оксадиазол-5-ил)пропил)-1Н-индазол-4-ил)метансульфонамид
К раствору продукта из Примера 37 стадия F, описанного выше (6,8 мг, 0,014 ммоль), в пиридине (1,0 мл) добавляли POCl3 (32,8 мг, 0,28 ммоль) при 0°C. После перемешивания при комнатной температуре в течение ночи, реакционную смесь концентрировали при пониженном давлении. Полученный остаток разбавляли диоксаном/водой, подкисляли TFA и очищали непосредственно на ВЭЖХ с обратной фазой, получая целевое соединение. LC/MS m/z = 457,1 [M+H]+.
ПРИМЕР 39
N-(1-(3-(4-хлорфенил)-1,1,1-трифторпентан-3-ил)-1Н-индазол-4-ил)метансульфонамид (Соединение 159)
Стадия A: 2-(4-(трет-бутоксикарбониламино)-1Н-индазол-1-ил)-2-(4-хлорфенил)-4,4,4-трифторметилбутаноат
В колбу загружали NaH (580 мг, 14,4 ммоль, 60% в масле) и затем раствор 2-(4-(трет-бутоксикарбониламино)-1Н-индазол-1-ил)-2-(4-хлорфенил)метилацетата (3 г, 7,22 ммоль), как описано в Примере 18 Стадия C, и 2,2,2-трифторэтил трифторметансульфонат (3,35 г, 14,4 ммоль) в THF (40 мл) при 0°C. Смесь перемешивали в течение 30 минут, гасили насыщенным раствором NH4Cl (10 мл), экстрагировали этилацетатом. Органические слои промывали солевым раствором (15 мл), высушивали над сульфатом натрия, фильтровали и упаривали. Остаток очищали хроматографией на силикагеле (РЕ/ЕА = 3:1), получая целевое соединение в форме бесцветного масла. LC/MS m/z = 498,1 [M+H]+.
Стадия B: 1-(2-(4-хлорфенил)-4,4,4-трифтор-1-гидроксибутан-2-ил)-1Н-индазол-4-ил-трет-бутилкарбамат
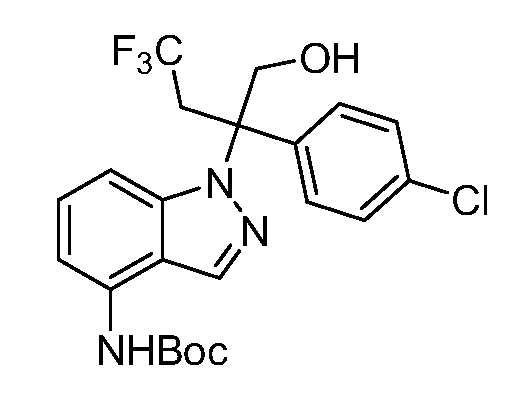
К раствору продукта Стадии А (300 мг, 0,6 ммоль) в THF (5 мл) добавляли LiAlH4 (38 мг, 0,96 ммоль) при 0°C. Смесь перемешивали при комнатной температуре в течение 1 часа и гасили H2O (5 мл), экстрагировали этилацетатом и промывали солевым раствором (10 мл), высушивали над сульфатом натрия, затем фильтровали. После того, как растворитель удаляли, остаток очищали хроматографией на силикагеле (ЕА/РЕ = 1:3), получая целевое соединение. LC/MS m/z = 470,1 [M+H]+.
Стадия C: 1-(2-(4-хлорфенил)-4,4,4-трифтор-1-оксобутан-2-ил)-1Н-индазол-4-ил-трет-бутилкарбамат
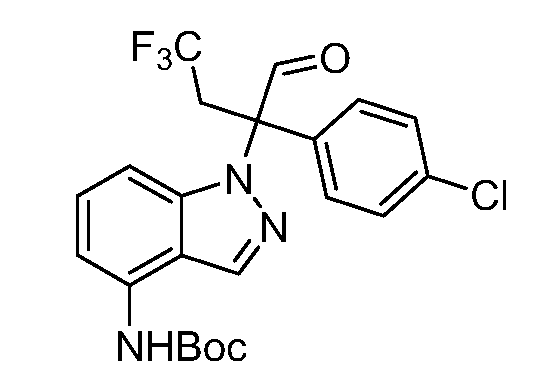
К раствору продукта Стадии B (200 мг, 0,42 ммоль) в DCM (5 мл) добавляли перйодинан Десса-Мартина (350 мг, 0,84 ммоль) частями при комнатной температуре. Смесь перемешивали 0,5 часа и затем разбавляли водой (5 мл), экстрагировали этилацетатом. Органические слои промывали солевым раствором (5 мл), высушивали над сульфатом натрия, фильтровали и упаривали. Остаток очищали хроматографией на силикагеле (ЕА/РЕ = 1:8), получая целевое соединение. LC/MS m/z = 468,2 [M+H]+.
Стадия D: 1-(3-(4-хлорфенил)-5,5,5-трифторпент-1-ин-3-ил)-1Н-индазол-4-ил-трет-бутилкарбамат
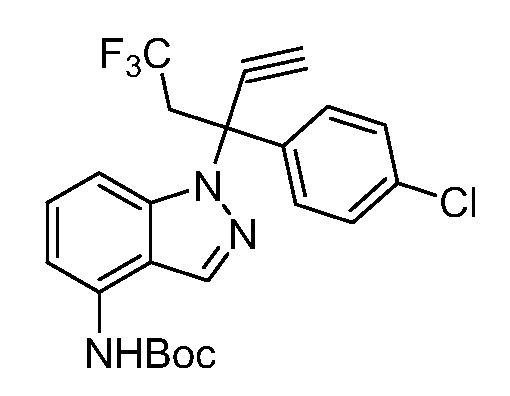
К раствору продукта Стадии C (120 мг, 0,25 ммоль) в метаноле (5 мл) добавляли K2CO3 (69 мг, 0,5 ммоль), 1-диазо-2-оксопропилэтанфосфонат (96 мг, 0,5 ммоль). Смесь перемешивали при комнатной температуре в течение 2 часов. После удаления органического растворителя, к остатку добавляли H2O (10 мл), экстрагировали этилацетатом и промывали солевым раствором (10 мл), высушивали над сульфатом натрия, затем фильтровали. Органический растворитель выпаривали, и остаток очищали хроматографией на силикагеле (ЕА/РЕ = 1:8), получая целевое соединение в форме бесцветного масла. LC/MS m/z = 464,1 [M+H]+.
Стадия E: 1-(3-(4-хлорфенил)-1,1,1-трифторпентан-3-ил)-1Н-индазол-4-ил-трет-бутилкарбамат
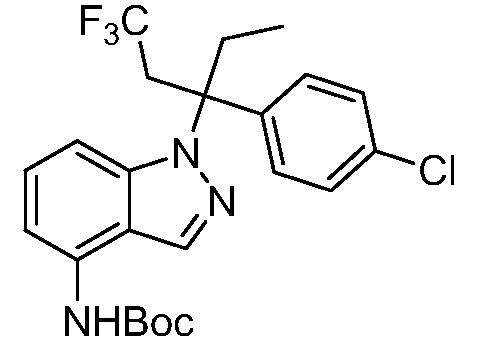
К раствору продукта Стадии D (100 мг, 0,21 ммоль) в этаноле (3 мл) добавляли PtO2 (10 мг, 0,044 ммоль) при комнатной температуре под атмосферой H2 в течение 2 часов. Твердое вещество отфильтровывали, и фильтрат концентрировали в вакууме, получая целевое соединение в форме бесцветного масла. LC/MS m/z = 468,1 [M+H]+.
Стадия F: 1-(3-(4-хлорфенил)-1,1,1-трифторпентан-3-ил)-1Н-индазол-4-амин
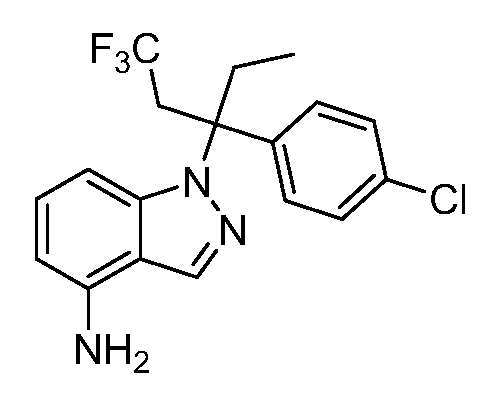
Смесь продукта Стадии E (80 мг, 0,17 ммоль) и 4н HCl/MeOH (2 мл) перемешивали при комнатной температуре в течение 2 часов. После удаления растворителя, остаток высушивали в вакууме, получая целевое соединение в форме бесцветного масла. LC/MS m/z = 368,1 [M+H]+.
Стадия G: N-(1-(3-(4-хлорфенил)-1,1,1-трифторпентан-3-ил)-1Н-индазол-4-ил)метансульфонамид
К смеси продукта Стадии F (50 мг, 0,13 ммоль) и NMM (31 мг, 0,3 ммоль) в DCM (3 мл) добавляли метансульфонил хлорид (20 мг, 0,21 ммоль). Смесь перемешивали при комнатной температуре в течение 3 часов затем гасили насыщенным раствором хлорида аммония (10 мл) и экстрагировали этилацетатом. Органические слои промывали солевым раствором (10 мл), высушивали над сульфатом натрия, фильтровали и упаривали. Остаток очищали Pre-ВЭЖХ (Мобильная фаза: ацетонитрил/вода (0,03% TFA)), получая целевое соединение. LC/MS m/z = 446,1 [M+H]+.
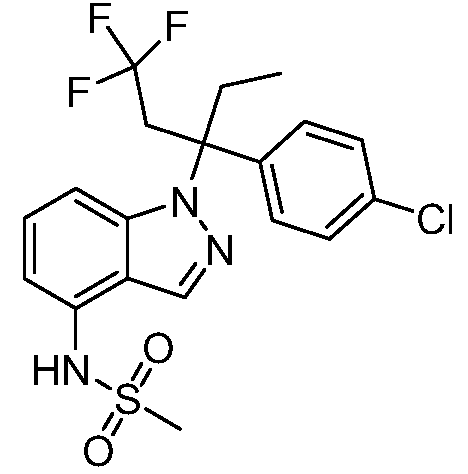
ПРИМЕР 40
N-(1-(3-(4-Хлорфенил)-1-гидроксипентан-3-ил)-1Н-индол-4-ил)метансульфонамид
Стадия A: 4-трет-бутил-1-метил-2-(4-(трет-бутоксикарбониламино)-1Н-индол-1-ил)-2-(4-хлорфенил)сукцинат
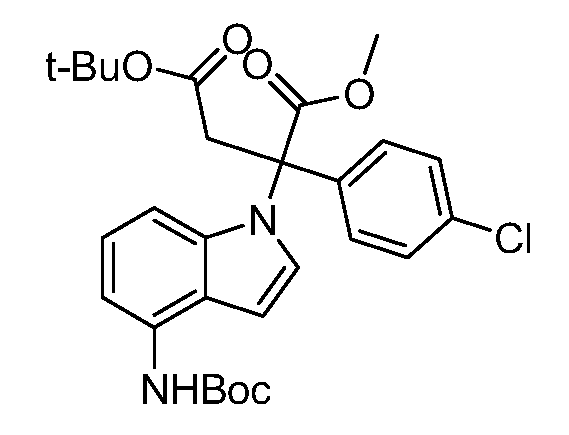
Раствор 2-(4-(трет-бутоксикарбониламино)-1Н-индазол-1-ил)-2-(4-хлорфенил)метилацетата (2 г, 4,83 ммоль), как описано в Примере 2 Стадия B, и 2-бром-трет-бутилацетат (1,4 г, 7,25 ммоль) в THF (10 мл) добавляли к NaH (290 мг, 7,25 ммоль, 60% в масле) по каплям при 0°C и перемешивали в течение 30 мин. Смесь гасили насыщенным раствором NH4Cl и экстрагировали этилацетатом. Органический слой промывали солевым раствором (20 мл), высушивали над сульфатом натрия, фильтровали и упаривали. Остаток очищали хроматографией на силикагеле (PE:EA = 3:1), получая целевое соединение. LC/MS m/z = 529,2 [M+H]+.
Стадия B: 4-трет-бутокси-2-(4-(трет-бутоксикарбониламино)-1Н-индол-1-ил)-2-(4-хлорфенил)-4-оксобутановая кислота
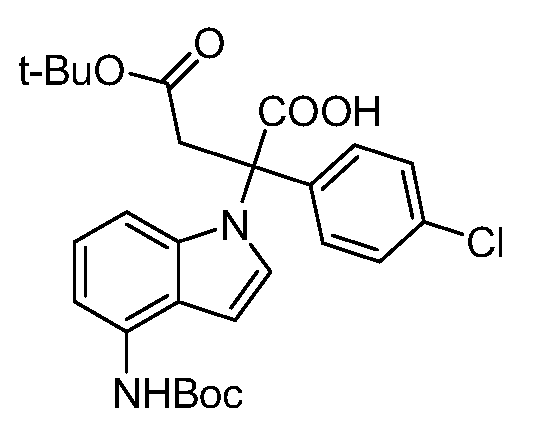
Смесь продукта стадии А (1,8 г, 3,4 ммоль) и 3н LiOH (410 мг, 17 ммоль) перемешивали при комнатной температуре в течение 3 часов. После того, как растворитель выпаривали, остаток растворяли в этилацетате (20 мл), и значение рН доводили до 6-7 с помощью 1М HCl. Органический слой промывали солевым раствором, высушивали над сульфатом натрия, фильтровали и концентрировали в вакууме, получая целевое соединение в форме бесцветного масла. LC/MS m/z = 515,2 [M+H]+.
Стадия C: 3-(4-(трет-бутоксикарбониламино)-1Н-индол-1-ил)-3-(4-хлорфенил)-4-гидрокси-трет-бутилбутаноат
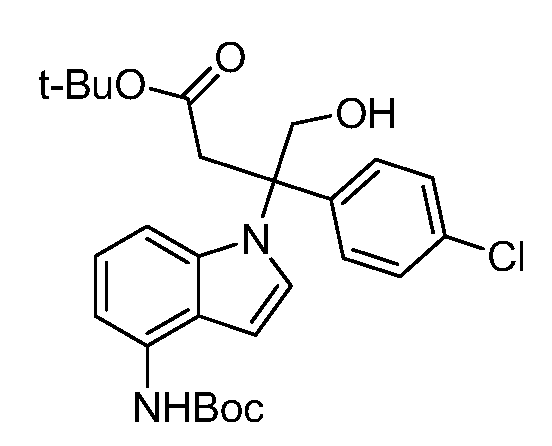
К смеси продукта стадии B (1,5 г, 2,9 ммоль) и NMM (590 мг, 5,83 ммоль) в DME (15 мл) по каплям при 0°C добавляли изобутилхлоридат углерода (793 мг, 5,83 ммоль). После перемешивания в течение 30 мин., NaBH4 добавляли к смеси при 0°C. Полученную смесь перемешивали при комнатной температуре еще 1 час и затем гасили насыщенным раствором хлорида аммония (10 мл), экстрагировали этилацетатом. Органические слои промывали солевым раствором (10 мл), высушивали над сульфатом натрия, фильтровали и упаривали. Остаток очищали хроматографией на силикагеле (PE:EA = 3:1), получая целевое соединение в форме бесцветного масла. LC/MS m/z = 501,2 [M+H]+.
Стадия D: 3-(4-(трет-бутоксикарбониламино)-1Н-индол-1-ил)-3-(4-хлорфенил)-4-оксо-трет-бутилбутаноат
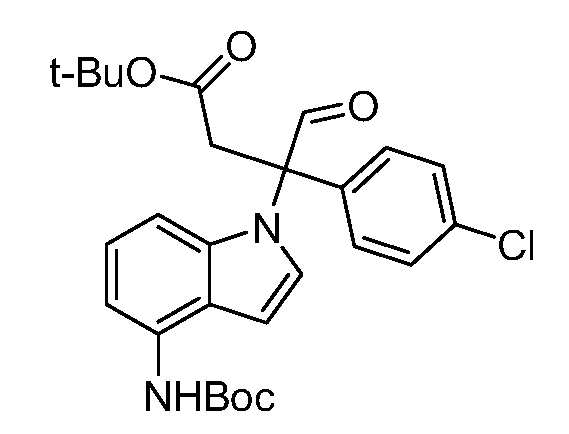
К раствору продукта Стадии C (1,2 г, 2,4 ммоль) в DCM (10 мл) добавляли перйодинан Десса-Мартина (2,03 г, 4,8 ммоль) частями при комнатной температуре. После перемешивания в течение 0,5 часов, смесь разбавляли водой (15 мл), экстрагировали этилацетатом. Органические слои промывали солевым раствором (10 мл), высушивали над сульфатом натрия, фильтровали и упаривали. Остаток очищали хроматографией на силикагеле (ЕА/РЕ = 1:8), получая целевое соединение. LC/MS m/z = 499,2 [M+H]+.
Стадия E: 3-(4-(трет-Бутоксикарбониламино)-1Н-индол-1-ил)-3-(4-хлорфенил)трет-бутилпент-4-иноат
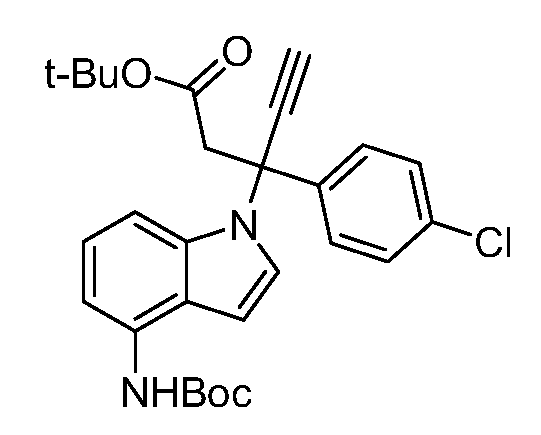
К раствору продукта Стадии D (750 мг, 1,5 ммоль) в MeOH (10 мл) добавляли K2CO3 (414 мг, 3 ммоль) и 1-диазо-2-оксопропилдиметилфосфонат (576 мг, 3 ммоль). Смесь перемешивали при комнатной температуре в течение 2 часов. После удаления органического растворителя, к остатку добавляли H2O (10 мл), экстрагировали этилацетатом и промывали солевым раствором (10 мл), высушивали над сульфатом натрия, затем фильтровали. Растворитель выпаривали в вакууме, и остаток очищали хроматографией на силикагеле (ЕА/РЕ = 1:8), получая целевое соединение в форме бесцветного твердого вещества. LC/MS m/z = 495,2 [M+H]+.
Стадия F: 3-(4-(трет-бутоксикарбониламино)-1Н-индол-1-ил)-3-(4-хлорфенил)трет-бутилпентаноат
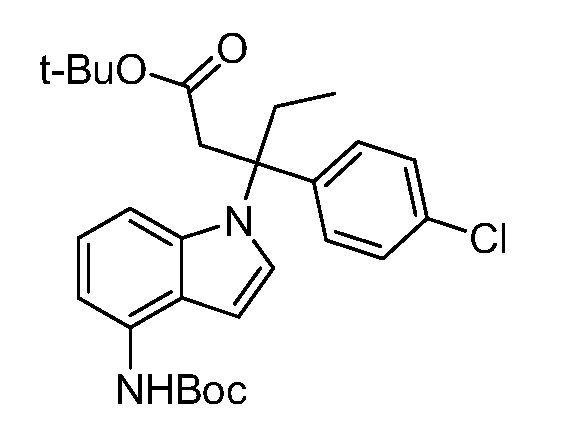
К раствору продукта Стадии E (150 мг, 0,3 ммоль) в этаноле (3 мл) добавляли PtO2 (6,8 мг, 0,03 ммоль) при комнатной температуре в атмосфере H2 в течение 2 часов. Твердое вещество отфильтровывали, и фильтрат концентрировали в вакууме, получая целевое соединение в форме бесцветного масла. LC/MS m/z = 499,2 [M+H]+.
Стадия G: 3-(4-амино-1Н-индол-1-ил)-3-(4-хлорфенил)метилпентаноат
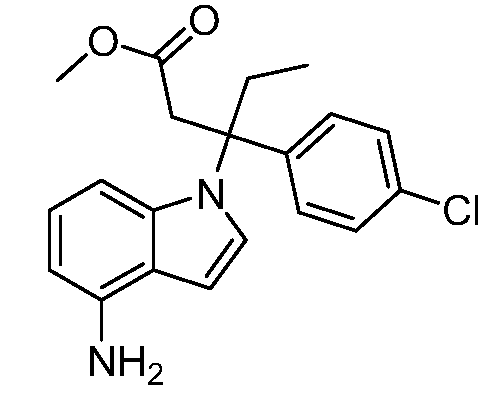
Смесь продукта Стадии F (120 мг, 0,24 ммоль) и 4н HCl/MeOH (2 мл) перемешивали при комнатной температуре в течение 2 часов. Летучую фракцию удаляли в вакууме, получая целевое соединение в форме бесцветного масла. LC/MS m/z = 357,1 [M+H]+.
Стадия Н: 3-(4-хлорфенил)-3-(4-(метилсульфонамидо)-1Н-индол-1-ил)метилпентаноат
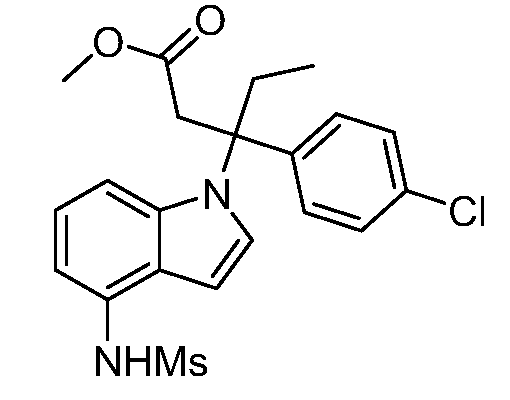
К смеси продукта Стадии G (75 мг, 0,21 ммоль) и NMM (42 мг, 0,42 ммоль) в DCM (3 мл) добавляли метансульфонил хлорид (36 мг, 0,38 ммоль). Затем смесь перемешивали при комнатной температуре в течение 3 часов, гасили насыщенным раствором хлорида аммония и экстрагировали этилацетатом. Органические слои промывали солевым раствором (10 мл), высушивали над сульфатом натрия, фильтровали и упаривали. Остаток очищали хроматографией на силикагеле (РЕ/ЕА = 3/1), получая целевое соединение в форме бесцветного масла. LC/MS m/z = 435,1 [M+H]+.
Стадия I: N-(1-(3-(4-Хлорфенил)-1-гидроксипентан-3-ил)-1Н-индол-4-ил)метансульфонамид
К раствору продукта Стадии Н (60 мг, 0,138 ммоль) в THF (5 мл) добавляли LiAlH4 (10,4 мг, 0,276 ммоль) при 0°C. Смесь перемешивали в течение 1 часа и гасили H2O (2 мл), экстрагировали этилацетатом и промывали солевым раствором (5 мл), высушивали над сульфатом натрия, затем фильтровали. После удаления органического растворителя, остаток очищали хроматографией на силикагеле (ЕА/РЕ = 1:3), получая рацемическое целевое соединение. Рацемическое соединение подвергали хиральному разделению SFC, используя колонку AS-H (50% EtOH/CO2, 4,6×250 мм, 39,3°C), получая два отделенных энантиомера. LC/MS m/z = 406,9 [M+H]+.
Соединение 160 и соединение 161 были получены в приведенных ниже условиях хирального разделения:
Колонка: AS-H
Мобильная фаза: A: CO2, B: MeOH (0,1% DEA), A:B = 55:45 при 3,0 мл/мин.
Температура колонки: 40,2°C
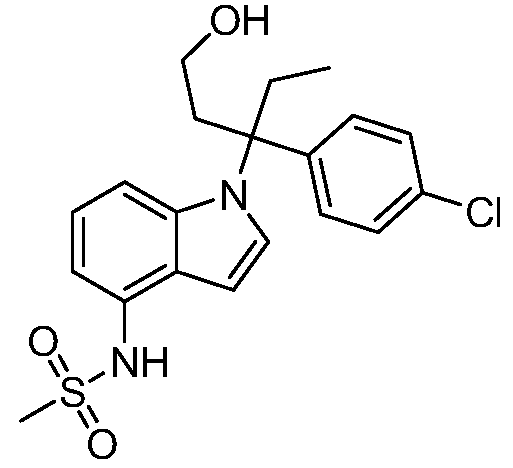
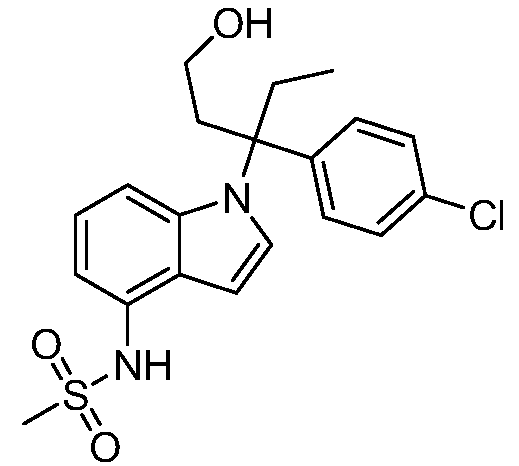
ПРИМЕР 41
N-(1-(3-(4-хлорфенил)-1-фторпентан-3-ил)-1Н-индол-4-ил)метансульфонамид
Стадия A: N-(1-(3-(4-Хлорфенил)-1-фторпентан-3-ил)-1Н-индол-4-ил)метансульфонамид
К раствору N-(1-(3-(4-хлорфенил)-1-гидроксипентан-3-ил)-1Н-индол-4-ил)метансульфонамида (20 мг, 0,05 ммоль), как описано в Примере 40 Стадия I, в DCM (5 мл) добавляли DAST (16 мг, 0,1 ммоль) при 0°C. Смесь перемешивали в течение 1 часа, затем гасили насыщенным водным раствором хлорида аммония (5 мл) и экстрагировали этилацетатом. Объединенные органические фракции промывали водой (5 мл) и солевым раствором (5 мл), высушивали над MgSO4, фильтровали и концентрировали. Полученный сырой продукт очищали Pre-ВЭЖХ (Мобильная фаза: ацетонитрил/вода (0,03% TFA)), получая рацемическое целевое соединение в форме твердого вещества белого цвета. Рацемическое соединение подвергали хиральному разделению SFC, используя IC (30% MeOH (0,1% DEA)/CO2, 4,6×250 мм, 41°C), получая два отделенных энантиомера. LC/MS m/z = 409,1 [M+H]+.
Соединение 162 и соединение 163 были получены в приведенных ниже условиях хирального разделения:
Колонка: IC
Мобильная фаза: A: CO2, B: MeOH (0,1% EDA), A:B = 70:30 при 3,0 мл/мин.
Температура колонки: 39,1°C
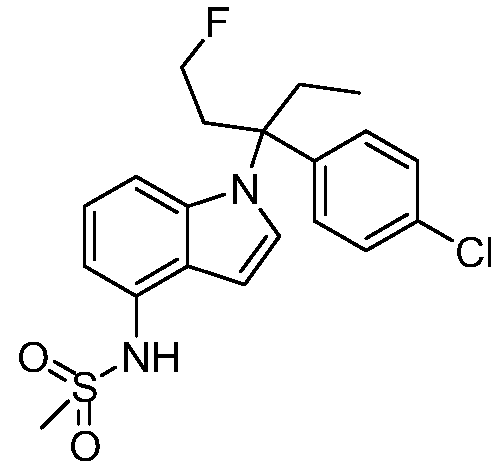
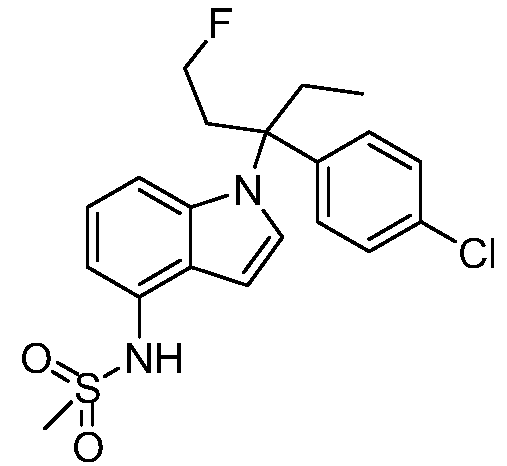
ПРИМЕР 42
3-(4-Хлорфенил)-3-(4-(метилсульфонамидо)-1Н-индол-1-ил)пентил метансульфонат (Соединение 164)
Стадия A: 3-(4-Хлорфенил)-3-(4-(метилсульфонамидо)-1Н-индол-1-ил)пентил метансульфонат
К смеси N-(1-(3-(4-хлорфенил)-1-гидроксипентан-3-ил)-1Н-индол-4-ил)метансульфонамида (20 мг, 0,05 ммоль), как описано в Примере 40 Стадия I, и NMM (10 мг, 0,1 ммоль) в DCM (3 мл) добавляли метансульфонил хлорид (7 мг, 0,06 ммоль). Затем смесь перемешивали при комнатной температуре в течение 1 часа, гасили насыщенным раствором хлорида аммония (5 мл) и экстрагировали этилацетатом. Органические слои промывали солевым раствором (5 мл), высушивали над сульфатом натрия, фильтровали и упаривали. Остаток очищали Prep-TLC(РЕ/ЕА = 4/1), получая целевое соединение в форме бесцветного масла. LC/MS m/z = 485,1 [M+H]+.
ПРИМЕР 43
N-(1-(3-(4-Хлорфенил)-1-метоксипентан-3-ил)-1Н-индол-4-ил)метансульфонамид (Соединение 165)
Стадия A: N-(1-(3-(4-Хлорфенил)-1-метоксипентан-3-ил)-1Н-индол-4-ил)метансульфонамид
К смеси 3-(4-хлорфенил)-3-(4-(метилсульфонамидо)-1Н-индол-1-ил)пентил метансульфоната (10 мг, 0,02 ммоль), как описано в Примере 40 Стадия I, в метаноле (2 мл) добавляли CH3ONa (21 мг, 0,4 ммоль) при комнатной температуре. Затем смесь нагревали до 40°C в течение 2 ч. Раствор гасили насыщенным раствором хлорида аммония (10 мл) и экстрагировали этилацетатом. Органические слои промывали солевым раствором, высушивали над сульфатом натрия, фильтровали и упаривали. Остаток очищали PREP-TLC (Мобильная фаза: ацетонитрил/вода (0,03% TFA)), получая целевое соединение в форме твердого вещества белого цвета. LC/MS m/z = 421,1 [M+H]+.
Следуя той же самой процедуре для ПРИМЕРА 43, но с использованием метантиолата натрия, получали Соединение 166 из представленного в Таблице 40.
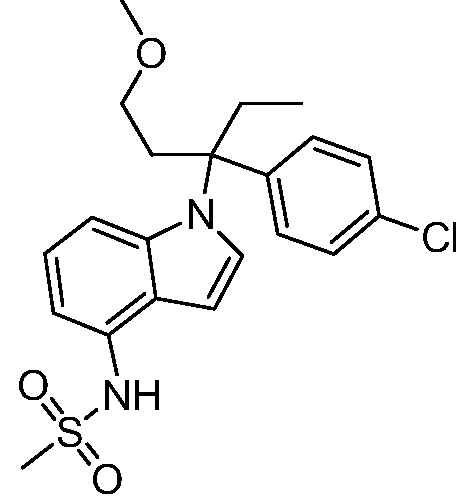
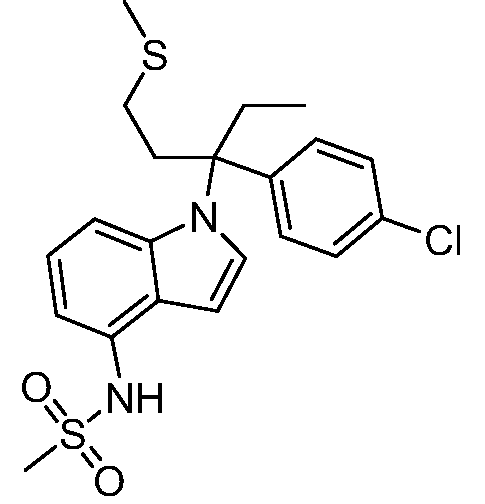
ПРИМЕР 44
4-(4-хлорфенил)-4-(4-(метилсульфонамидо)-1Н-индол-1-ил)метилгексаноат (Соединение 167)
Стадия A: 1-(3-(4-хлорфенил)пент-1-ин-3-ил)-1Н-индол-4-ил-трет-бутилкарбамат
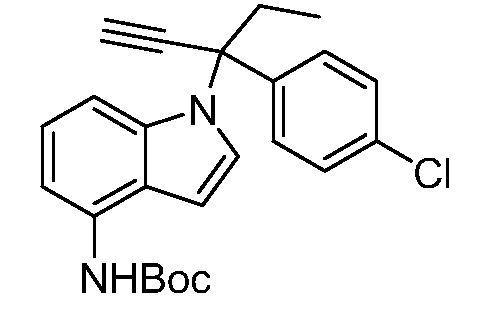
К смеси (1-(2-(4-хлорфенил)-1-оксобутан-2-ил)-1Н-индол-4-ил)трет-бутилкарбамат (412 мг, 1 ммоль), как описано в Примере 21 Стадия B, и K2CO3 (276 мг, 2 ммоль) в метаноле (10 мл) добавляли 1-диазо-2-оксопропилдиметилфосфонат (230 мг, 1,2 ммоль) при комнатной температуре. Смесь перемешивали в течение 2 часов, и твердое вещество отфильтровывали, и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали хроматографией на силикагеле с элюированием смесью РЕ/ЕА (от 20/1 до 10/1, об./об.), получая целевое соединение в форме бесцветного аморфного твердого вещества. LC/MS m/z = 352,9 [M-tBu+H]+.
Стадия B: 4-(4-(трет-бутоксикарбониламино)-1Н-индол-1-ил)-4-(4-хлорфенил)метилгекс-2-иноат
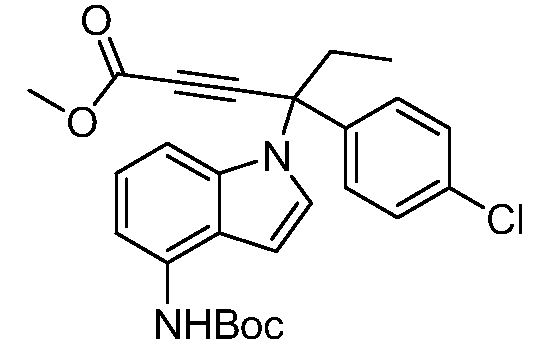
К раствору продукта Стадии А (436,5 мг, 1,07 ммоль) в тетрагидрофуране (10 мл) добавляли BuLi (0,9 мл, 2,25 ммоль, 2,5 М в гексане) при -78°C. После перемешивания при -78°C в течение 30 минут, к смеси добавляли метилхлоридат углерода (121 мг, 1,28 ммоль). Полученную смесь перемешивали еще 1 час, затем гасили водным раствором NH4Cl (10 мл) и экстрагировали этилацетатом. Объединенные органические слои промывали солевым раствором (15 мл), высушивали над Na2SO4, фильтровали и концентрировали. Сырой продукт очищали хроматографией на силикагеле с элюированием смесью РЕ/ЕА (от 20/1 до 10/1, об./об.), получая целевое соединение в форме бесцветного аморфного твердого вещества. LC/MS m/z = 410,9 [M-tBu+H]+.
Стадия C: 4-(4-(трет-бутоксикарбониламино)-1Н-индол-1-ил)-4-(4-хлорфенил)метилгексаноат

Смесь продукта Стадии B (200 мг, 0,43 ммоль), PtO2 (10 мг, 0,043 ммоль) и метанола (2 мл) дегазировали три раза и продували водородом. Смесь перемешивали при комнатной температуре в течение 2 часов в атмосфере водорода. Твердое вещество отфильтровывали, и фильтрат концентрировали в вакууме, получая целевое соединение в форме бесцветного масла. LC/MS m/z = 414,9 [M-tBu+H]+.
Стадия D: 4-(4-амино-1Н-индол-1-ил)-4-(4-хлорфенил)метилгексаноат
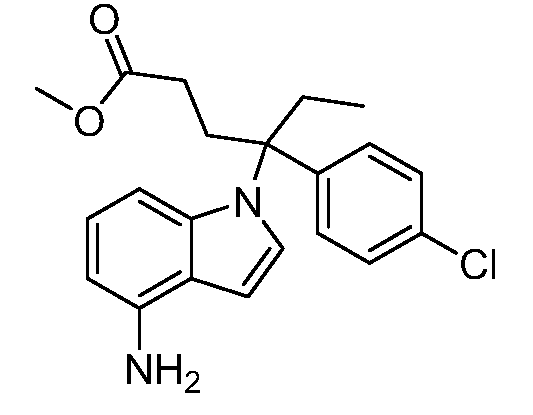
Смесь продукта Стадии C (190 мг, 0,4 ммоль) и MeOH/HCl (2 мл, 4 M) перемешивали при комнатной температуре в течение 2 часов. Летучую фракцию удаляли в вакууме, получая целевое соединение в форме бесцветного масла. LC/MS m/z = 371,1 [M+H]+.
Стадия E: 4-(4-амино-1Н-индол-1-ил)-4-(4-хлорфенил)метилгексаноат
К раствору продукта Стадии D (100 мг, 0,27 ммоль) и N-метилморфолина (41 мг, 0,4 ммоль) в дихлорметане (3 мл) добавляли метансульфонил хлорид (40 мг, 0,35 ммоль) при комнатной температуре. Смесь перемешивали в течение 2 часов, затем разбавляли водой (15 мл) и экстрагировали этилацетатом. Объединенные органические слои промывали солевым раствором (10 мл), высушивали над Na2SO4, фильтровали и концентрировали. Сырой продукт очищали PREP-TLC с элюированием смесью РЕ/ЕА (4/1, об./об.), получая целевое соединение в форме бесцветного аморфного твердого вещества. LC/MS m/z = 471,0 [М+Na]+.
Следуя той же самой процедуре для ПРИМЕРА 44, но на стадии A используя (1-(2-(4-хлорфенил)-1-оксобутан-2-ил)-1Н-индазол-4-ил)трет-бутилкарбамат, получали соединение 168 из представленного в Таблице 41.
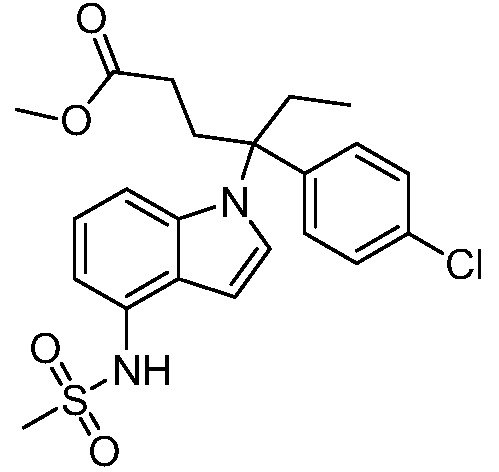
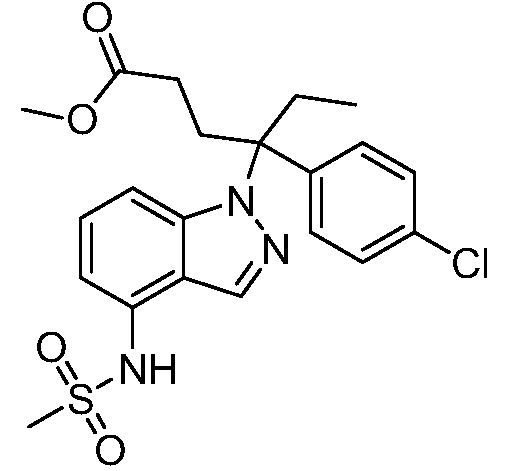
ПРИМЕР 45
N-(1-(3-(4-хлорфенил)-6-гидроксигексан-3-ил)-1Н-индол-4-ил)метансульфонамид
Стадия A: N-(1-(3-(4-хлорфенил)-6-гидроксигексан-3-ил)-1Н-индол-4-ил)метансульфонамид
К раствору 4-(4-хлорфенил)-4-(4-(метилсульфонамидо)-1Н-индол-1-ил)метилгексаноата (20 мг, 0,045 ммоль), как описано в Примере 44 Стадия E, в тетрагидрофуране (2 мл) добавляли алюмогидрид лития (2,5 мг, 0,0675 ммоль) при 0°C. Смесь постепенно нагревали до комнатной температуры и перемешивали в течение 4 часов. К реакционной смеси аккуратно добавляли воду (1 мл) и экстрагировали этилацетатом. Объединенные органические слои промывали солевым раствором (30 мл), высушивали над безводным сульфатом натрия, фильтровали и концентрировали. Сырой продукт очищали PREP-TLC с элюированием смесью РЕ/ЕА (2/1, об./об.), получая целевое соединение в форме бесцветного масла. LC/MS m/z = 421,0 [M+H]+.
Соединение 169 получали в приведенных ниже условиях хирального разделения:
Колонка: OD-H
Мобильная фаза: A: CO2, B: MeOH, A:B = 70:30 при 3,0 мл/мин.
Температура колонки: 39,9°C
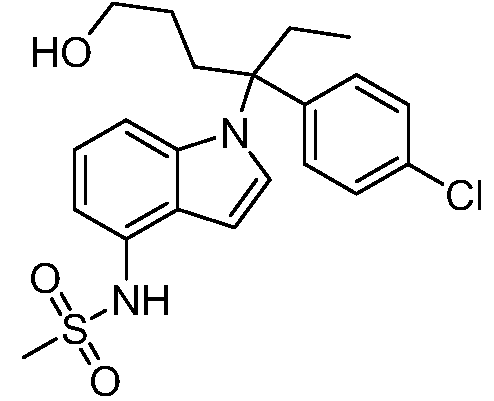
ПРИМЕР 46
N-(1-(3-(4-хлорфенил)-6-фторгексан-3-ил)-1Н-индол-4-ил)метансульфонамид (Соединение 170)
Стадия A: N-(1-(3-(4-хлорфенил)-6-фторгексан-3-ил)-1Н-индол-4-ил)метансульфонамид
К раствору N-(1-(3-(4-хлорфенил)-6-гидроксигексан-3-ил)-1Н-индол-4-ил)метансульфонамида (21 мг, 0,05 ммоль), как описано в Примере 45 Стадия A, в дихлорметане (2 мл) добавляли DAST (16 мг, 0,1 ммоль) при -78°C. Смесь постепенно нагревали до комнатной температуры и перемешивали в течение 3 часов, затем гасили метанолом (0,5 мл). Сырой продукт очищали PREP-TLC с элюированием смесью РЕ/ЕА (2/1, об./об.), чтобы получить целевое соединение в форме бесцветного масла. LC/MS m/z = 444,7 [М+Na]+.
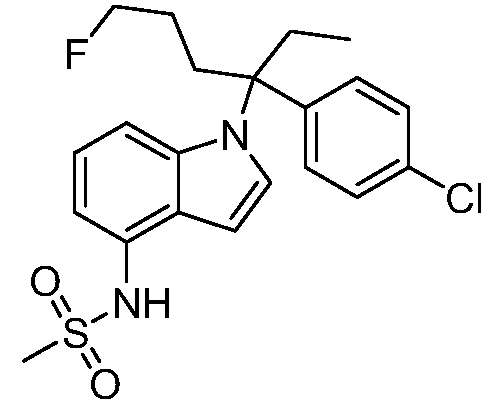
ПРИМЕР 47
(E)-4-(4-хлорфенил)-4-(4-(метилсульфонамидо)-1Н-индазол-1-ил)метилгекс-2-еноат (Соединение 171)
Стадия A: (E)-4-(4-амино-1Н-индазол-1-ил)-4-(4-хлорфенил)метилгекс-2-еноат
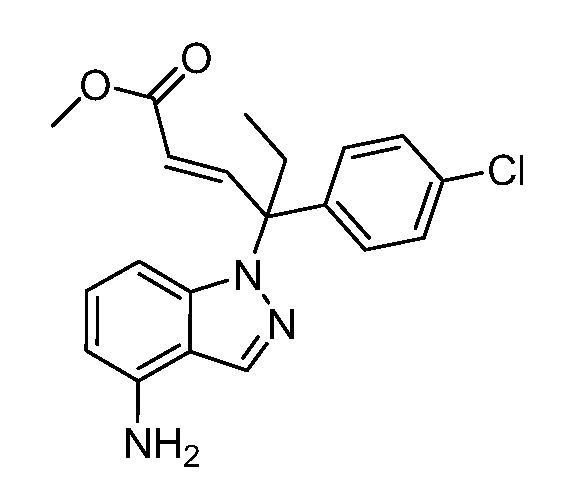
Смесь (E)-трет-бутил-1-(3-(4-хлорфенил)-1-цианопент-1-ен-3-ил)-1Н-индазол-4-илметилкарбамата (500 мг), как описано в Примере 22 Стадия A, в 15 мл 4н HCl/MeOH перемешивали при комнатной температуре в течение 1 часа. Затем растворитель удаляли. Остаток растворяли в ЕА (30 мл), и значение рН доводили до 9~10 насыщенным водным раствором NaHCO3. Органический слой промывали водой (10 мл), солевым раствором, высушивали (Na2SO4) и концентрировали. Остаток очищали на ВЭЖХ с обратной фазой (0,05% NH4HCO3 в H2O - MeCN), получая целевой продукт в форме твердого вещества светло-коричневого цвета. LC/MS m/z = 370,2 [M+H]+.
Стадия B: (E)-4-(4-хлорфенил)-4-(4-(метилсульфонамидо)-1Н-индазол-1-ил)метилгекс-2-еноат
К смеси (E)-4-(4-амино-1Н-индазол-1-ил)-4-(4-хлорфенил)метилгекс-2-еноата (120 мг) и NMM (98 мг) в DCM (5 мл) добавляли MsCl (45 мг). Затем смесь перемешивали при комнатной температуре в течение 2 часов. Раствор гасили насыщенным раствором NH4Cl и экстрагировали этилацетатом (30 мл). Органический слой промывали солевым раствором, высушивали над сухим сульфатом натрия, фильтровали и упаривали. Остаток очищали хроматографией на силикагеле (РЕ/ЕА = 5/1), получая целевое соединение в форме твердого вещества светло-желтого цвета. LC/MS m/z = 448,2 [M+H]+.
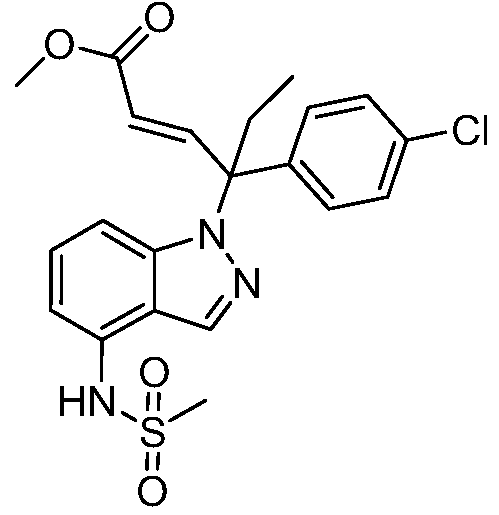
ПРИМЕР 48
N-(1-(3-(4-хлорфенил)-6-оксогептан-3-ил)-1Н-индазол-4-ил)метансульфонамид (Соединение 172)
Стадия A: N-(1-(3-(4-хлорфенил)-6-оксогептан-3-ил)-1Н-индазол-4-ил)метансульфонамид
Смесь 4-(4-хлорфенил)-4-(4-(метилсульфонамидо)-1Н-индазол-1-ил)метилгексаноата (50 мг), как описано в Примере 44 Стадия E, в 2 мл безводного THF добавляли CH3Li (1,5 М в простом эфире, 0,2 мл) по каплям при 0°C и перемешивали в течение 30 мин. Раствор гасили насыщенным раствором NH4Cl и экстрагировали этилацетатом (20 мл). Органический слой промывали солевым раствором, высушивали над сухим сульфатом натрия, фильтровали и упаривали. Остаток очищали ВЭЖХ с обратной фазой (0,05% TFA в H2O - MeCN), получая целевой продукт в форме твердого вещества белого цвета. LC/MS m/z = 434,1 [M+H]+.
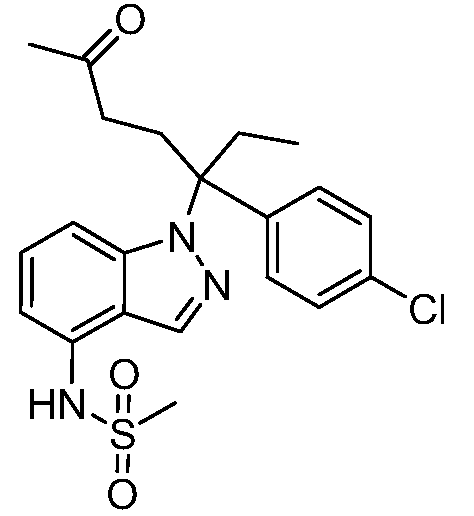
ПРИМЕР 49
N-(1-(3-(4-хлорфенил)-2-гидрокси-1-метоксипентан-3-ил)-1Н-индол-4-ил)метансульфонамид (Соединение 173)
Стадия A: (1-(1-(4-хлорфенил)-1-(оксиран-2-ил)пропил)-1Н-индол-4-ил)трет-бутилкарбамат
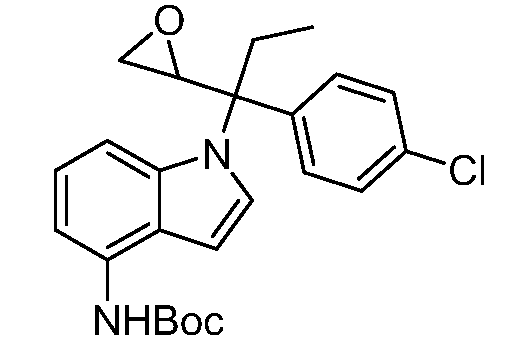
К раствору NaH (41 мг, 0,764 ммоль, 60% в масле) в ДМСО (15 мл) при 0°C добавляли триметилсульфоксоний йодид (235 мг, 1,07 ммоль), разделенный на несколько частей. Смесь перемешивали при комнатной температуре, пока смесь не стала прозрачной. К раствору 1-(2-(4-хлорфенил)-1-оксобутан-2-ил)-1Н-индол-4-ил-трет-бутилкарбамата (210 мг, 0,509 ммоль), как описано в Примере 21 Стадия B, в THF (15 мл) добавляли вышеописанный раствор по каплям при 0°C. Затем смесь перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь гасили насыщенным раствором хлорида аммония и экстрагировали этилацетатом. Органический слой промывали солевым раствором (15 мл), высушивали над сульфатом натрия, фильтровали и упаривали. Остаток очищали хроматографией на силикагеле с РЕ/ЕА (от 10/1 до 3/1, об./об.), получая целевое соединение в форме бесцветного твердого вещества. LC/MS m/z = 448,9 [М+Na]+.
Стадия B: 3-(4-амино-1Н-индол-1-ил)-3-(4-хлорфенил)-1-метоксипентан-2-ол
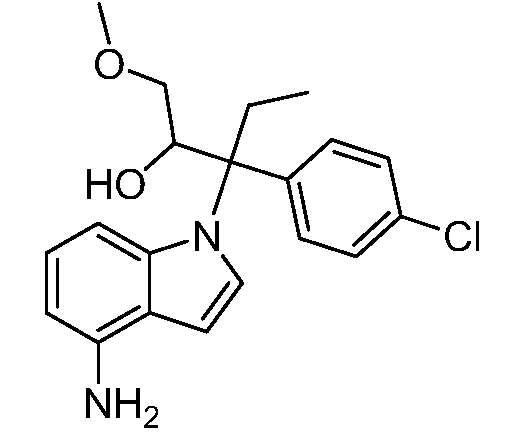
К раствору продукта со стадии А (60 мг, 0,140 ммоль) в MeOH (3 мл) добавляли MeONa (15 мг, 0,281 ммоль). Смесь перемешивали при 120°C в микроволновом реакторе в течение 0,5 часов. Раствор удаляли, и остаток очищали PREP-TLC с элюированием смесью РЕ/ЕА (1/1, об./об.), получая целевое соединение в форме бесцветного аморфного твердого вещества. LC/MS m/z = 359,1 [M+H]+.
Стадия C: N-(1-(3-(4-хлорфенил)-2-гидрокси-1-метоксипентан-3-ил)-1Н-индол-4-ил)метансульфонамид
К смеси продукта со стадии B (39 мг, 0,108 ммоль) и NMM (22 мг, 0,216 ммоль) в DCM (8 мл) добавляли по каплям метилсульфонил хлорид (18 мг, 0,163 ммоль). Затем смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь гасили насыщенным раствором хлорида аммония и экстрагировали этилацетатом (10 мл). Органический слой промывали солевым раствором, высушивали над сульфатом натрия, фильтровали и упаривали. Остаток очищали PREP-TLC с элюированием смесью РЕ/ЕА (1/1, об./об.), получая целевое соединение в форме бесцветного аморфного твердого вещества. LC/MS m/z = 437,1 [M+H]+.
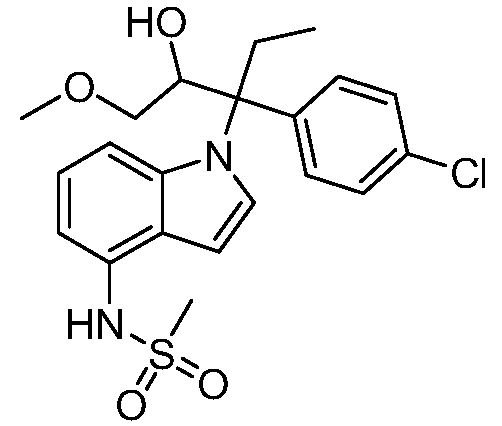
ПРИМЕР 50
N-(1-(3-(4-хлорфенил)пентан-3-ил)-1Н-индол-4-ил)метансульфонамид (Соединение 174)
Стадия A: 1-(3-(4-хлорфенил)пентан-3-ил)-1Н-индол-4-ил-трет-бутилкарбамат
Смесь 1-(3-(4-хлорфенил)пент-1-ин-3-ил)-1Н-индол-4-ил-трет-бутилкарбамат (100 мг, 0,245 ммоль), как описано в Примере 44 Стадия A, PtO2 (5,5 мг, 0,0245 ммоль) и метанола (2 мл) дегазировали три раза и продували дигидрогеном. Смесь перемешивали при комнатной температуре в течение 2 часов в атмосфере дигидрогена. Твердое вещество отфильтровывали, и фильтрат концентрировали в вакууме, получая целевое соединение в форме бесцветного масла. LC/MS m/z = 357,0 [M-tBu+H]+.
Стадия B: 1-(3-(4-хлорфенил)пентан-3-ил)-1Н-индол-4-амин
Смесь продукта Стадии А (90 мг, 0,218 ммоль) и MeOH/HCl (2 мл, 4 M) перемешивали при комнатной температуре в течение 2 часов. Летучую фракцию удаляли в вакууме, получая целевое соединение в форме бесцветного масла. LC/MS m/z = 313,1 [M+H]+.
Стадия C: N-(1-(3-(4-хлорфенил)пентан-3-ил)-1Н-индол-4-ил)метансульфонамид
К раствору продукта Стадии B (50 мг, 0,16 ммоль) и N-метилморфолина (32 мг, 0,32 ммоль) в дихлорметане (2 мл) добавляли метансульфонил хлорид (23 мг, 0,24 ммоль) при комнатной температуре. Смесь перемешивали в течение 2 часов, затем разбавляли водой (15 мл) и экстрагировали этилацетатом. Объединенные органические слои промывали солевым раствором (10 мл), высушивали над Na2SO4, фильтровали и концентрировали. Сырой продукт очищали PREP-TLC с элюированием смесью РЕ/ЕА (2/1, об./об.), получая целевое соединение в форме бесцветного твердого вещества. LC/MS m/z = 391,0 [M+H]+.
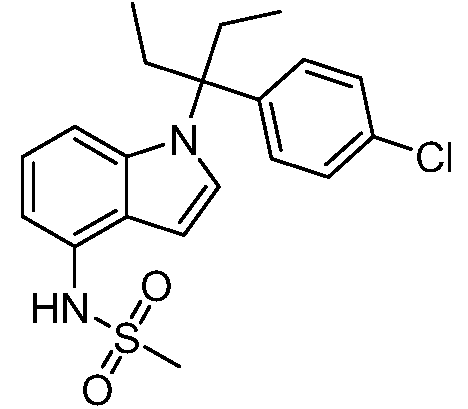
ПРИМЕР 51
N-(1-(3-(4-хлорфенил)пент-1-ен-3-ил)-1Н-индол-4-ил)метансульфонамид (Соединение 175)
Стадия A: 1-(3-(4-хлорфенил)пент-1-ен-3-ил)-1Н-индол-4-ил-трет-бутилкарбамат
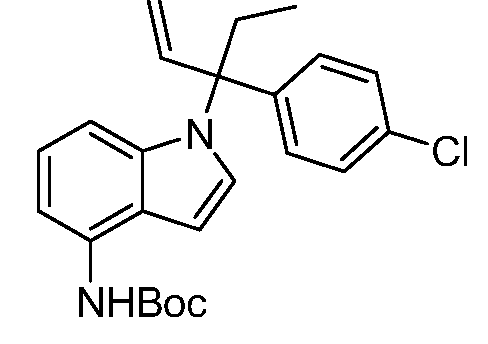
Смесь 1-(3-(4-хлорфенил)пент-1-ен-3-ил)-1Н-индол-4-ил-трет-бутилкарбамата (100 мг, 0,245 ммоль), как описано в Примере 44 Стадия A, катализатора Линдлара (10 мг) и метанола (2 мл) дегазировали три раза и продували водородом. Смесь перемешивали при комнатной температуре в течение 2 часов в атмосфере водорода. Твердое вещество отфильтровывали, и фильтрат концентрировали в вакууме, получая целевое соединение в форме бесцветного масла. LC/MS m/z = 355,0 [M-tBu+H]+.
Стадия B: 1-(3-(4-хлорфенил)пент-1-ен-3-ил)-1Н-индол-4-амин
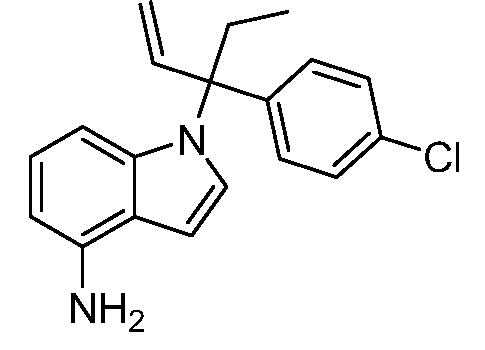
Смесь продукта Стадии А (95 мг, 0,232 ммоль) и MeOH/HCl (4 мл, 2M) перемешивали при 0°C в течение 2 часов. Летучую фракцию удаляли в вакууме, получая целевое соединение в форме бесцветного масла. LC/MS m/z = 311,1 [M+H]+.
Стадия C: N-(1-(3-(4-хлорфенил)пент-1-ен-3-ил)-1Н-индол-4-ил)метансульфонамид
К раствору продукта Стадии B (40 мг, 0,128 ммоль) и N-метилморфолина (26 мг, 0,256 ммоль) в дихлорметане (2 мл) добавляли метансульфонил хлорид (18 мг, 0,192 ммоль) при комнатной температуре. Смесь перемешивали в течение 2 часов, затем разбавляли водой (10 мл) и экстрагировали этилацетатом. Объединенные органические слои промывали солевым раствором (10 мл), высушивали над Na2SO4, фильтровали и концентрировали. Сырой продукт очищали PREP-TLC с элюированием смесью РЕ/ЕА (3/1, об./об.), получая целевое соединение в форме бесцветного твердого вещества. LC/MS m/z = 388,9 [M+H]+.
Соединение 175 получали в приведенных ниже условиях хирального разделения:
Колонка: AD-H
Мобильная фаза: A: CO2, B: MeOH, A:B = 70:30 при 3,0 мл/мин.
Температура колонки: 38,6°C
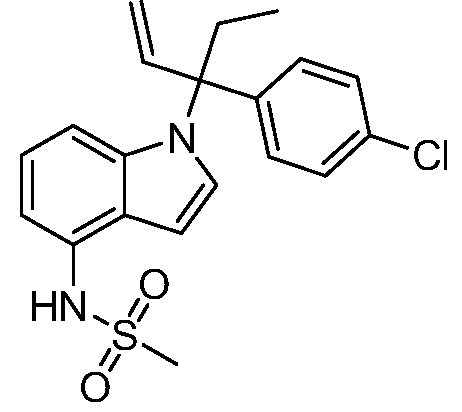
ПРИМЕР 52
N-(1-(2-амино-3-(4-хлорфенил)пентан-3-ил)-1Н-индол-4-ил)метансульфонамид (Соединение 176)
Стадия A: N-(1-(3-(4-хлорфенил)-2-(гидроксиимино)пентан-3-ил)-1Н-индол-4-ил)метансульфонамид
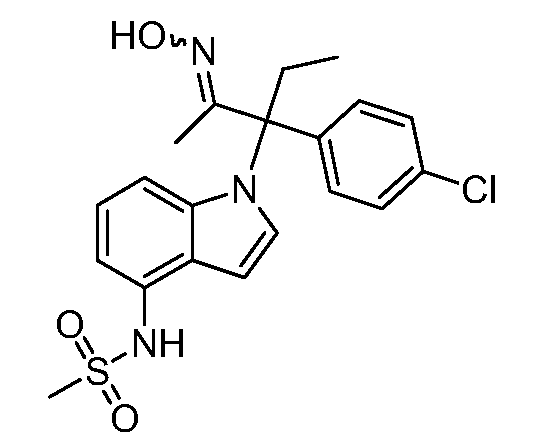
К раствору N-(1-(3-(4-хлорфенил)-2-оксопентан-3-ил)-1Н-индол-4-ил)метансульфонамида (200 мг, 0,49 ммоль), как описано в Примере 23 Стадия A, в EtOH (5 мл) добавляли гидрохлорид гидроксиламина (68 мг, 0,99 ммоль) и K2CO3 (137 мг, 0,99 ммоль). Смесь перемешивали при 130°C в микроволновом реакторе в течение 2 часов. После удаления растворителя, остаток растворяли в этилацетате (30 мл). Органический слой промывали насыщенным раствором бикарбоната натрия (10 мл) и солевым раствором (10 мл), высушивали над сульфатом натрия и затем фильтровали. После удаления органического растворителя, остаток очищали хроматографией на силикагеле с элюированием смесью РЕ/ЕА (от 10/1 до 5/1, об./об.), получая целевое соединение в форме твердого вещества белого цвета. LC/MS m/z = 441,9 [M+Na]+.
Стадия B: N-(1-(2-амино-3-(4-хлорфенил)пентан-3-ил)-1Н-индол-4-ил)метансульфонамид
Смесь продукта со стадии А (68 мг, 0,162 ммоль) и никеля Ренея (1 мг) в MeOH (10 мл) продували воздухом три раза и затем продували H2. Смесь перемешивали при температуре окружающей среды в течение ночи. Суспензию фильтровали, и фильтрат концентрировали, получая целевое соединение в форме бесцветного твердого вещества. LC/MS m/z = 406,0 [M+H]+.
Соединение 176 получали в приведенных ниже условиях хирального разделения:
Колонка: AS-H
Мобильная фаза: A: CO2, B: MeOH, A:B = 70:30 при 3,0 мл/мин.
Температура колонки: 39,8°C
Используя процедуру, описанную в Примере 39, но на стадии А заменяя N-(1-(3-(4-хлорфенил)-2-оксопентан-3-ил)-1Н-индол-4-ил)метансульфонамид N-(1-(2-(4-хлорфенил)-1-оксобутан-2-ил)-1Н-индол-4-ил)метансульфонамидом, получали соединение 177 из представленного в Таблице 49.
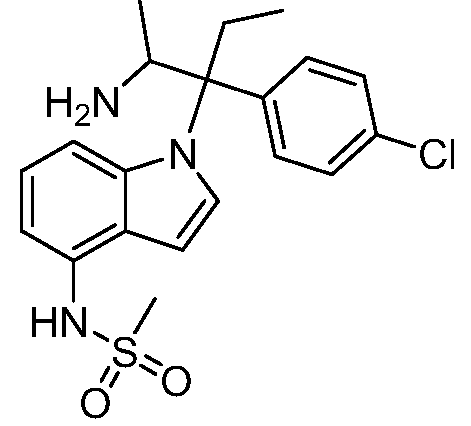
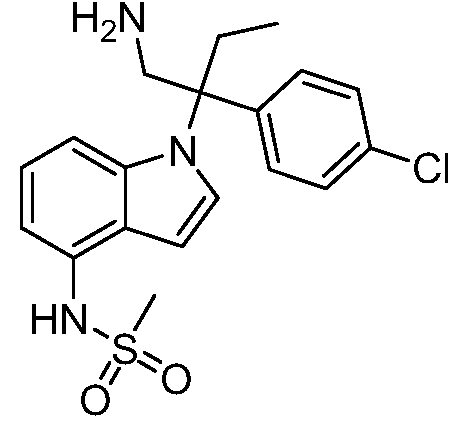
ПРИМЕР 53
N-(1-(1-(4-хлорфенил)-1-цианопропил)-1Н-индол-4-ил)этансульфонамид (Соединение 178)
Стадия A: 2-(4-хлорфенил)-2-(4-(этилсульфонамидо)-1Н-индол-1-ил)метилбутаноат
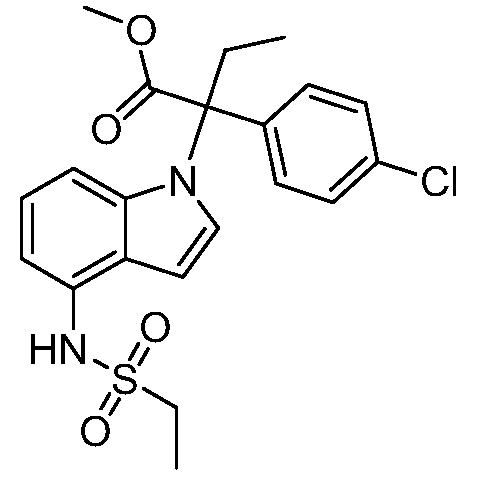
Этансульфонил хлорид (0,26 г, 2 ммоль) добавляли к перемешиваемому раствору 2-(4-амино-1Н-индол-1-ил)-2-(4-хлорфенил)метилбутаноата (0,58 г, 1,7 ммоль), как описано в Примере 3 Стадия B, и 4-метилморфолина (0,25 г, 2,5 ммоль) в DCM (8 мл) при 0°C. Через 30 минут добавляли дополнительную часть этансульфонил хлорида (0,1 г, 1 ммоль). Через 1 час, реакционную смесь разделяли между DCM и насыщенным водным раствором хлорида аммония. Слои разделяли, и водный слой экстрагировали DCM. Объединенные органические слои промывали солевым раствором, высушивали сульфатом натрия, фильтровали и концентрировали в вакууме. Остаток очищали флэш-хроматографией на силикагеле (РЕ:ЕА = 2:1), получая целевое соединение. LC/MS m/z = 435,1 [M+H]+.
Стадия B: 2-(4-хлорфенил)-2-(4-(этилсульфонамидо)-1Н-индол-1-ил)бутанамид
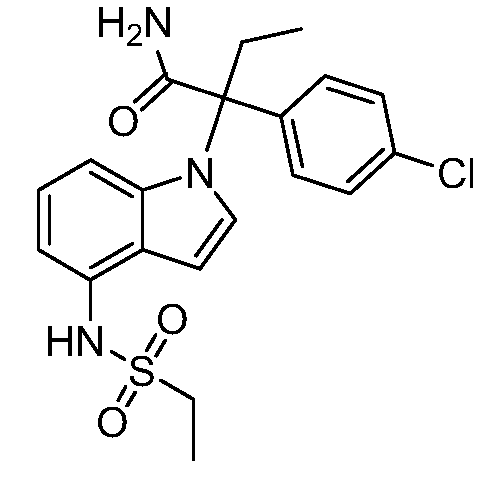
Продукт со стадии А (0,43 г, 1,0 ммоль) растворяли в 7н растворе аммиака в метаноле (25 мл). После перемешивания при комнатной температуре в течение 10 часов, полученную смесь концентрировали при пониженном давлении. Остаток растворяли в диоксане/воде, подкисляли TFA и очищали непосредственно на ВЭЖХ с обратной фазой, получая целевое соединение. LC/MS m/z = 420,1 [M+H]+. Эти два энантиомера разделяли хиральной SFC.
Энантиомер A и энантиомер B были получены в приведенных ниже условиях хирального разделения:
Колонка: IA
Мобильная фаза: A: CO2, B: MeOH (DEA 0,1%), A:B = 60:40 при 1,0 мл/мин.
Температура колонки: 39,6°C
энантиомер A, время удерж. = 2,1 минуты, энантиомер B, время удерж. = 3,54 минуты
Стадия C: N-(1-(1-(4-хлорфенил)-1-цианопропил)-1Н-индол-4-ил)этансульфонамид
Хлорангидрид циануровой кислоты (92 мг, 0,5 ммоль) добавляли к перемешиваемому раствору энантиомера продукта со стадии B (210 мг, 0,5 ммоль) в DMF (3 мл), и реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь лили в солевой раствор, и полученную смесь экстрагировали этилацетатом, высушивали над сульфатом натрия и концентрировали в вакууме. Остаток очищали флэш-хроматографией на силикагеле (РЕ:ЕА = 2:1), получая целевое соединение. LC/MS m/z = 402,0 [M+H]+.
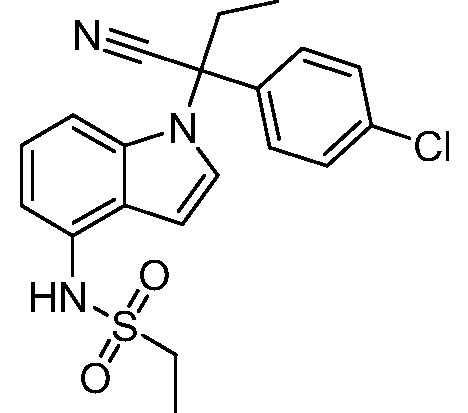
ПРИМЕР 54
N-(1-(1-(4-хлорфенил)-1-(2-этил-2Н-тетразол-5-ил)пропил)-1Н-индол-4-ил)метансульфонамид
Стадия A: N-(1-(1-(4-хлорфенил)-1-цианопропил)-1Н-индол-4-ил)метансульфонамид
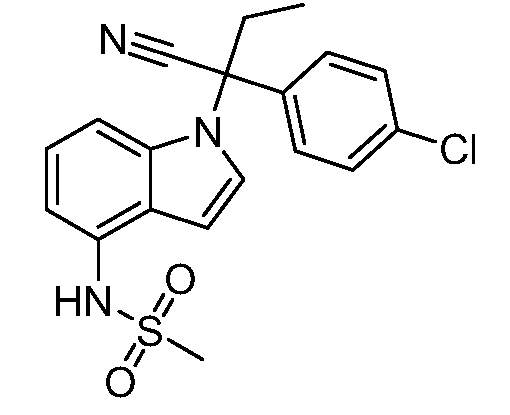
Целевое соединение получали согласно процедуре, описанной в Примере 53.
Стадия B: N-(1-(1-(4-хлорфенил)-1-(2H-тетразол-5-ил)пропил)-1Н-индол-4-ил)метансульфонамид
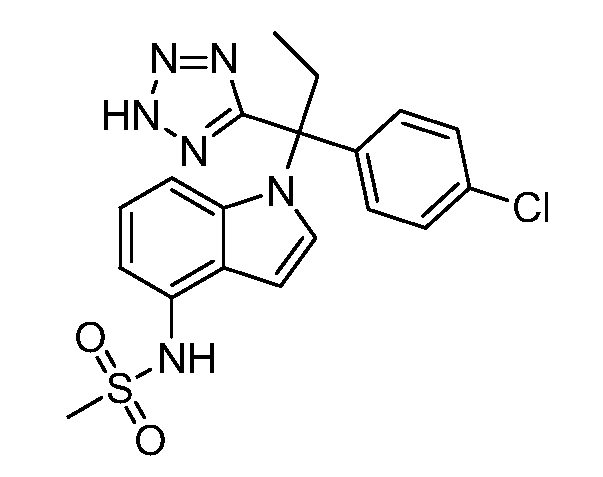
Азид натрия (117,6m г, 1,809 ммоль) добавляли к перемешиваемому раствору N-(1-(1-(4-хлорфенил)-1-цианопропил)-1Н-индол-4-ил)метансульфонамида (100 мг, 0,258 ммоль) и хлорида аммония (110,6 г, 2,067 ммоль) в DMF (2 мл) и нагревали при 130°C в течение 3 часов в микроволновом реакторе. Реакционную смесь гасили водой и экстрагировали этилацетатом (100 мл). Органический слой промывали солевым раствором, высушивали над сульфатом натрия, фильтровали и упаривали. Остаток очищали хроматографией на силикагеле с элюированием смесью РЕ/ЕА (от 5/1 до 1/1, об./об.), получая целевое соединение в форме твердого вещества желтого цвета. LC/MS m/z = 431,0 [M+H]+.
Стадия C: N-(1-(1-(4-хлорфенил)-1-(2-этил-2Н-тетразол-5-ил)пропил)-1Н-индол-4-ил)метансульфонамид
Смесь продукта со стадии B (65 мг, 0,151 ммоль), K2CO3 (41,7 мг, 0,302 ммоль) и йодэтана (30,8 мг 0,181 ммоль) в DMF (1 мл) перемешивали при 80°C в течение ночи. Летучую фракцию выпаривали. Остаток экстрагировали этилацетатом (50 мл). Органический слой промывали водой и солевым раствором, высушивали над сульфатом натрия, фильтровали и упаривали. Остаток очищали хроматографией на силикагеле с элюированием смесью РЕ/ЕА (от 5/1 до 1/1, об./об.), получая целевое соединение в форме твердого вещества желтого цвета. LC/MS m/z = 459,0 [M+H]+.
Соединение 179 получали в приведенных ниже условиях хирального разделения:
Колонка: AS-H
Мобильная фаза: A: CO2, B: MeOH, A:B = 70:30 при 3,0 мл/мин.
Температура колонки: 38,2°C
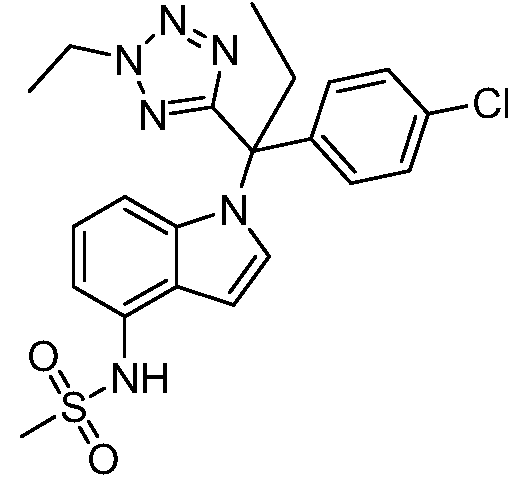
ПРИМЕР 55
N-(1-(3-(4-хлорфенил)-6,6,6-трифторгексан-3-ил)-1Н-индол-4-ил)метансульфонамид (Соединение 180)
Стадия A: N-(1-(3-(4-хлорфенил)пент-1-ин-3-ил)-1Н-индол-4-ил)-N-((2-(триметилсилил)этокси)метил)метансульфонамид
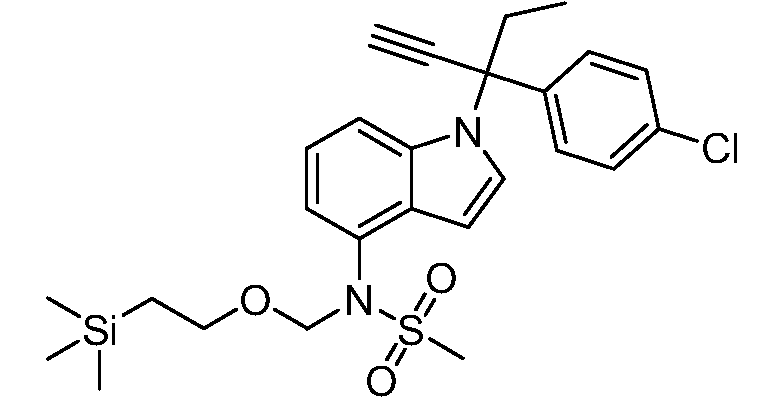
Целевое соединение получали согласно процедуре в Примере 32, Стадия А, с использованием N-(1-(2-(4-хлорфенил)-1-оксобутан-2-ил)-1Н-индазол-4-ил)-N-((2-(триметилсилил)этокси)метил)-метансульфонамида (энантиомер A), описанного в Примере 20 Стадия E, в качестве исходного материала.
Стадия B: N-(1-(3-(4-хлорфенил)-6,6,6-трифторгекс-4-ин-3-ил)-1Н-индол-4-ил)-N-((2-(триметилсилил)этокси)-метил)метансульфонамид
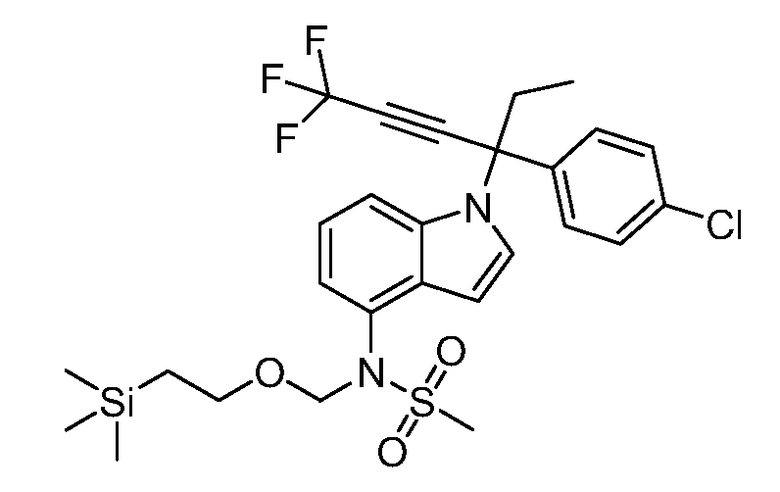
В трехгорлую круглодонную колбу загружали CuI (38 мг, 0,2 ммоль), Phen·H2O (40 мг, 0,2 ммоль), KF (58 мг, 1,0 ммоль) и стержни для перемешивания. Колбу высушивали при 130°C в вакууме в течение 3 часов. После охлаждения до комнатной температуры, колбу продували воздухом и добавляли TMSCF3 (142 мг, 1,0 ммоль), DMF (2 мл). Затем колбу нагревали до 130°C в течение 30 мин. Раствор N-(1-(3-(4-хлорфенил)пент-1-ин-3-ил)-1Н-индол-4-ил)-N-((2-(триметилсилил)этокси)метил)метансульфонамид (102 мг, 0,2 ммоль, энантиомер A) в DMF (1 мл) медленно добавляли к смеси при 130°C за 1 час. Полученную смесь перемешивали еще 1 час при 130°C. Сырой продукт очищали Combiflash (мобильная фаза: CH3CN/вода (0,08% NH4HCO3)), получая целевое соединение. LC/MS m/z = 606,7 [M+Na]+.
Стадия C: N-(1-(3-(4-хлорфенил)-6,6,6-трифторгексан-3-ил)-1Н-индол-4-ил)-N-((2-(триметилсилил)этокси)метил)-метансульфонамид
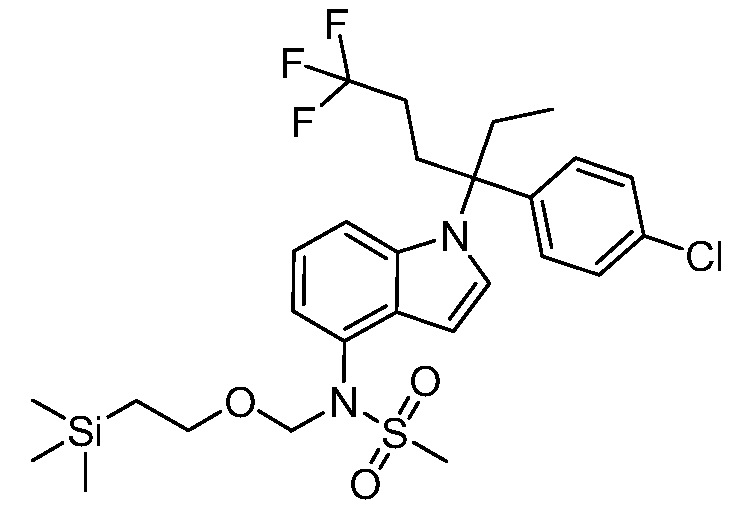
Смесь продукта со стадии B (63 мг, 0,108 ммоль, энантиомер A) и PtO2 (2,4 мг, 0,0108 ммоль) в MeOH (3 мл) очищали воздухом три раза и затем продували H2. Смесь перемешивали при комнатной температуре в течение ночи. Суспензию фильтровали, и фильтрат концентрировали, получая целевое соединение, которое использовали для следующей стадии без очистки. LC/MS m/z = 610,7 [M+Na]+.
Стадия D: N-(1-(3-(4-хлорфенил)-6,6,6-трифторгексан-3-ил)-1Н-индол-4-ил)метансульфонамид
Смесь продукта со стадии C (20 мг, 0,034 ммоль, энантиомер A), 2М HCl (1 мл) и EtOH (2 мл) нагревали до 50°C в течение 3 часов. Летучую фракцию удаляли при пониженном давлении. Остаток очищали Combiflash (мобильная фаза: MeOH/вода (0,08% NH4HCO3)), получая целевое соединение. LC/MS m/z = 458,7 [M+H]+.
1H ЯМР (400 МГц, CDCl3) δ 7,50 (д, J=3,6 Гц, 1H), 7,29 (д, J=8,4 Гц, 2H), 7,10 (д, J=7,6 Гц, 1H), 7,05 (д, J=8,8 Гц, 2H), 6,86 (т, J=8,1 Гц, 1H), 6,59-6,58 (м, 2H), 6,40 (д, J=8,4 Гц, 1H), 3,03 (с, 3H), 2,74-2,67 (м, 1H), 2,55-2,45 (м, 2H), 2,26-2,16 (м, 1H), 1,89-1,86 (м, 1H), 1,45-1,38 (м, 1H), 0,67 (т, J=7,2 Гц, 3H).
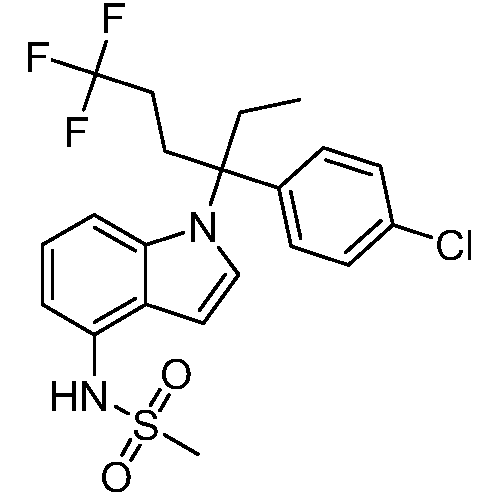
БИОЛОГИЧЕСКИЙ ТЕСТ
Активность соединений согласно настоящему изобретению в отнощении антагонизма минералокортикоидного рецептора может быть оценена с использованием следующего теста.
ИССЛЕДОВАНИЕ ПОТЕНЦИАЛА MR В ТЕСТЕ HMR NH PRO
Тест MR NH Pro человека является коммерчески доступным тестом взаимодействия Белок:Белок PathHunter™ (DiscoveRx; http://www.discoverx.com/nhrs/prod-nhrs.php), который измеряет способность соединений к антагонистическому действию в отношении связывания полноразмерного минералокортикоидного рецептора человека (MR) с пептидом-коактиватором. Клетки CHO-K1 PathHunter™, которые суперэкспрессируют человеческий MR (Cat #93-0456C2, Lot No: 09B0913) культивировали в средах для выращивания (F12K w/Глутамин и фенольный красный (Gibco 11765-047), дополненный 10% HI FBS (Gibco 16000); 0,25 мг/мл гигромицина в PBS (Invitrogen 10687-010, 50 мг/мл); 100 М.Ед./мкл и 100 мкг/мкл Pen/Strep (Gibco 15140-122); 0,6 мг/мл Geneticin).
Соединения оценивали на антагонистическую активность MR, инкубируя клетки с дозой титрования соединения в среде F12K w/Глутамин и фенольного красного (Invitrogen 11765-047), дополненной 1% углем/обработанным декстраном FBS (Hyclone #SH30068,01) и альдостероном (0,3 нМ) в течение 6 часов при 37°C. Клетки затем обрабатывали реагентом детекции DiscoveRx в течение 1 часа при комнатной температуре и считывали с использованием люминисцентного планшет-ридера Envision. Активность в % измеряли в отношении клеток, обработанных только альдостероном, и значения IC50 вычисляли, используя программное обеспечение ADA.
1. Среды для выращивания:
F12K w/Глутамин и фенольный красный (Gibco 11765-047)
10% HI FBS (Gibco 16000)
0,25 мг/мл Гигромицина в PBS (Invitrogen 10687-010, 50 мг/мл)
100 М.Ед./мкл и 100 мкг/мл Pen/Strep (Gibco 15140-122)
0,6 мг/мл Генетицина (Gibco 10131, 50 мг/мл)
2. Среды для тестирования:
F12K w/Глутамин и фенольный красный (Invitrogen 11765-047)
1%-ый уголь/обработанный декстраном FBS (Hyclone #SH30068,01)
3. 3х Реактивы для детекции PathHunter (Cat# 93-0001) (необходимо ~6 мл/планшет). Нельзя замораживать и оттаивать реактивы более чем 3 раза.
19х Буфер для клеточного теста PathHunter
5х Emerald II
1х Galacton Star
4. Контрольный агонист: Альдостерон: Sigma cat# A9477
Получали сток-раствор - 10 мкМ в ДМСО, сохраняли при -20°C
для теста, разбавляли в средах для теста до 1,8 или 6 нМ (6х конечной концентрации = от приблизительно 0,3 нМ до приблизительно 1,0 нМ)
5. Линия клеток: клетки PathHunter CHO-K1 MR Cat #93-0456C2, Lot No: 09B0913, от оперативного стока в жидком азоте.
6. Контрольный антагонист: Спиронолактон: Sigma #S-3378 и Эплеренон Sigma #107724-20-9 (10 мМ стоковую концентрацию также получали в ДМСО и сохраняли при -20°C).
Способы:
Настройки для теста и вычисления:
1. Клетки выращивали в F12+FBS+Гигромицин+pen/strep+Генетин
2. Клетки собирали с 0,05% трипсина, и суспензию клеток центрифугировали и повторно суспендировали в объеме F12+1,5% CD-FBS и подсчитывали.
3. Клетки повторно суспендировали в количестве 4×105 клеток/мл.
4. Клетки (25 мкл/лунка) добавляли в лунки 384-луночного планшета.
5. Планшет затем инкубировали при 37°C в течение ночи в увлажненном термостате с 5%-ым CO2.
6. Тестируемые соединения титровали, начиная при 4,4 мМ, титрование с 10 точками в разведении 1:3.
7. Альдостерон разбавляли в средах для теста до 1,8 нМ или 6 нМ из 10 мкМ стока (конечная концентрация составляет от приблизительно 0,3 нМ до приблизительно 1,0 нМ)
Протокол для формата планшета с 384 лунками: обработка в течение 6 часов:
1. Помещают 10К экспоненциально растущих клеток/лунка (25 мкл), повторно суспендированных в средах для теста, в каждую лунку с использованием Multidrop (Thermo Electron) (используют тестовые планшеты с белыми стенками и прозрачным дном (Costar #3570) и инкубируют в течение ночи при 37°C, 5% CO2.
2. Добавляют 0,25 мкл 120x тестируемого соединения (конечная концентрация в ДМСО должна быть <1%) к каждой лунке n=2, титрование с 10 точками, начиная с конечной концентрации 36,7 мкМ.
3. Добавляют 5 мкл 6x агониста (конечная концентрация альдостерона должна быть от приблизительно 0,3 нМ до приблизительно 1,0 нМ) ко всем лункам (используя PlateMate Plus) (ThermoFisher) (кроме лунок в столбцах 23 и 24)
4. Добавляют 5 мкл среды для теста ко всем лункам в столбце 23 и 24.
5. Инкубируют 6 часов при 37°C, 5% CO2.
6. Добавляют 15 мкл 3x реактива для детекции DiscoveRx к каждой лунке.
7. Инкубируют 1 час при комнатной температуре (планшеты хранят в темноте).
8. Считывают планшеты на люминисцентном планшет-ридере Envision (PerkinElmer) и анализируют с использованием ADA.
Способ LC/MS: (способ LC2M_Low/Med_Positive).
Условия LC: 5-98% CH3CN/H2O + v 0,1% TFA в течение 1,25 мин.; Объемная скорость потока = 1,5 мл/мин., длина волны УФ 254 нм; Колонка: Waters MS XTerra® C18 3,5 мкм 2,1×20 мм IS™
Как можно видеть в Примерах, приведенных выше, соединения согласно настоящему изобретению, которые были проверены и имели IP значение более 0 нМ, но менее 100 нМ, получали оценку значений "A". Соединения согласно настоящему изобретению, которые были проверены и имели IP значение, равное или больше чем 100 нМ, но меньшее чем 500 нМ, получали оценку значений "B". Соединения согласно настоящему изобретению, которые были проверены и имели IP значение, равное или большее чем 500 нМ, но меньшее чем 5000 нМ, получали оценку значений "C".
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ СОЕДИНЕНИЯ И КОМПОЗИЦИИ ДЛЯ ИНГИБИРОВАНИЯ FASN | 2014 |
|
RU2737434C2 |
| ПРОИЗВОДНОЕ ГЕТЕРОАРИЛ[4,3-с]ПИРИМИДИН-5-АМИНА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО МЕДИЦИНСКИЕ ПРИМЕНЕНИЯ | 2018 |
|
RU2764655C2 |
| ИНГИБИТОРЫ СЕРИН/ТРЕОНИНОВЫХ КИНАЗ | 2013 |
|
RU2650501C2 |
| ЦИТОСКЕЛЕТНО-АКТИВНЫЕ ИНГИБИРУЮЩИЕ RHO-КИНАЗУ СОЕДИНЕНИЯ, ИХ КОМПОЗИЦИИ И ПРИМЕНЕНИЕ | 2007 |
|
RU2470928C2 |
| АМИДНЫЕ ПРОИЗВОДНЫЕ И ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, СПОСОБ ИХ ПОЛУЧЕНИЯ И МЕДИЦИНСКОЕ ПРИМЕНЕНИЕ | 2015 |
|
RU2681537C2 |
| 5-ЗАМЕЩЕННЫЕ ИНДАЗОЛЫ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ | 2008 |
|
RU2487873C2 |
| СПОСОБ ЛЕЧЕНИЯ ВОСПАЛЕНИЯ ИЛИ СВЯЗАННОГО С ВОСПАЛЕНИЕМ ЗАБОЛЕВАНИЯ У СОБАК | 1996 |
|
RU2253456C2 |
| АЗЕТИДИНИЛДИАМИДЫ В КАЧЕСТВЕ ИНГИБИТОРОВ МОНОАЦИЛГЛИЦЕРИН-ЛИПАЗЫ | 2010 |
|
RU2569298C2 |
| 2,6-ЗАМЕЩЕННЫЕ-4-МОНОЗАМЕЩЕННЫЙ АМИНО-ПИРИМИДИНЫ КАК АНТАГОНИСТЫ РЕЦЕПТОРА ПРОСТАГЛАНДИНА D2 | 2005 |
|
RU2417990C2 |
| ПРОИЗВОДНЫЕ КАРБОНОВЫХ КИСЛОТ (ВАРИАНТЫ), ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ СЕЛЕКТИВНОГО ИНГИБИРОВАНИЯ СВЯЗЫВАНИЯ αβ ИНТЕГРИНА У МЛЕКОПИТАЮЩЕГО | 2000 |
|
RU2263109C2 |
Изобретение относится к соединениям формулы I и его фармацевтически приемлемым солям, которые могут быть использованы для лечения опосредуемых альдостероном заболеваний. Изобретение также относится к фармацевтическим композициям, которые включают соединения формулы I. 3 н. и 8 з.п. ф-лы, 52 табл., 55 пр.
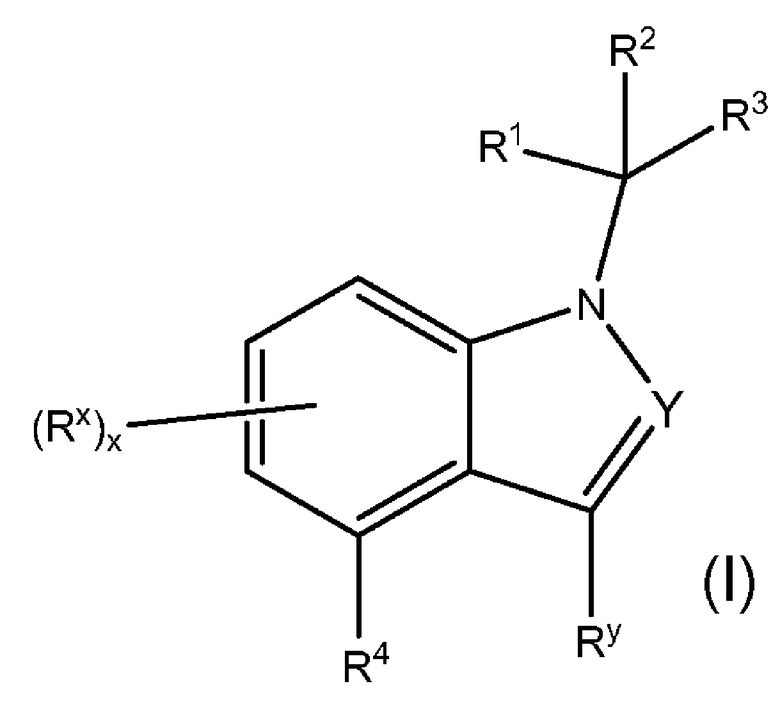
1. Соединение формулы IV:
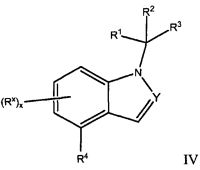
или его фармацевтически приемлемые соли, в которой
Y обозначает N или CRy;
каждый Ry независимо обозначает Н или C1-C6 алкил, причем указанный алкил необязательно замещен галогеном или ОН;
каждый Rx независимо обозначает Н, галоген, OR;
каждый R независимо обозначает Н, CF3, C1-C6 алкил, причем указанный алкил необязательно замещен 1-3 заместителями, представляющими фенил;
R1 обозначает:
1) 5-членный гетероарил, содержащий по меньшей мере один гетероатом, выбранный из О, S, N, или 5-членный гетероциклил, содержащий по меньшей мере один гетероатом, представляющий собой О, причем указанный гетероарил или гетероциклил необязательно замещен одним-тремя R5,
2) C1-C6 алкил, причем указанный алкил необязательно замещен одним-тремя заместителями, представляющими собой C1-C6 алкил, OR, NR2, CF3, SR, OS(O)2R8,
3) -(CRa 2)nC(O)OR11,
4) (CRa 2)0-4C(O)Rc,
5) C3-C10 циклоалкил-R5,
R2 обозначает
1) C1-C6 алкил,
2) -(CRb 2)m-C3-C6 циклоалкил,
3) -(CRa 2)m-C(O)OR11,
4) -(CRb 2)m-C2-C6 алкенил или
5) -(CRb 2)m-C2-C6 алкинил,
где указанные алкил, циклоалкил, алкенил и алкинил необязательно замещены одной-тремя группами, выбранными из R12;
R3 обозначает фенил, причем указанный фенил необязательно замещен одним-тремя R9;
R4 обозначает
1) -NR6S(O)2R8,
2) -NH2,
3) -(CRa 2)t-SO2R10,
каждый R5 независимо обозначает OR, CN, C(O)NRR7, C1-C6 алкил, CF3 или C3-C10 циклоалкил, где указанный алкил, циклоалкил, необязательно, могут быть замещены одним-тремя галогенами, OR или CF3;
каждый R6 независимо обозначает Н, С(О)OR11;
каждый R7 независимо обозначает
1) Н,
2) C1-C6 алкил, необязательно замещенный 1-3 заместителями, выбранными из галогена, OR, CN, CF3, фенила,
3) -(CRa 2)nC(O)OR11;
каждый R8 независимо обозначает C1-C6 алкил, NRR7 или CF3;
каждый R9 независимо обозначает галоген, CF3, OR;
каждый R10 независимо обозначает C1-C6 алкил;
каждый R11 независимо обозначает Н или C1-C6 алкил;
каждый R12 независимо обозначает галоген, CN, С(О)OR11, OR;
каждый Ra независимо обозначает Н;
каждый Rb независимо обозначает Н;
Rc обозначает C1-C6 алкил;
m=0 или 1;
n=0, 1, 2, 3 или 4;
t=0, 1 или 2; и
х=0 или 1.
2. Соединение по п. 1, где Rx независимо обозначает Н или галоген.
3. Соединение по п. 1 или 2, где Ry обозначает Н.
4. Соединение по п. 1, где R2 обозначает C1-C6 алкил.
5. Соединение по п. 1, где R3 обозначает фенил, необязательно замещен одним-тремя заместителями, представляющими собой галоген, OR, CF3.
6. Соединение по п. 1, где R4 обозначает -NR6S(O)2R8.
7. Соединение, выбранное из следующих соединений (название IUPAC):
(4-хлорфенил)(4-нитро-1Н-индол-1-ил)метилацетат;
(2-хлор-4-фторфенил)(4-нитро-1Н-индол-1-ил)метилацетат;
(2,4-дихлорфенил)(4-нитро-1Н-индол-1-ил)метилацетат;
(3-бромфенил)(4-нитро-1Н-индол-1-ил)метилацетат;
(4-метоксифенил)(4-нитро-1Н-индол-1-ил)метилацетат;
(2-хлор-4-фторфенил)(1H-индол-1-ил)метилацетат;
(2-хлор-4-фторфенил)(4-циано-1Н-индол-1-ил)метилацетат;
1-[1-(2-хлор-4-фторфенил)-2-метокси-2-оксоэтил]-1Н-индол-4-этилкарбоксилат;
(2-хлор-4-фторфенил)(4-хлор-1Н-индол-1-ил)метилацетат;
(2-хлор-4-фторфенил)(4-фтор-1Н-индол-1-ил)метилацетат;
(2-хлор-4-фторфенил)(5-циано-1Н-индол-1-ил)метилацетат;
(2-хлор-4-фторфенил)(5-хлор-1Н-индол-1-ил)метилацетат;
(2-хлор-4-фторфенил)(5-фтор-1Н-индол-1-ил)метилацетат;
(2-хлор-4-фторфенил)(6-хлор-1Н-индол-1-ил)метилацетат;
(2-хлор-4-фторфенил)(7-фтор-1Н-индол-1-ил)метилацетат;
[7-(бензилокси)-1Н-индол-1-ил](2-хлор-4-фторфенил)метилацетат;
(4-хлорфенил){4-[(метилсульфонил)амино]-1Н-индол-1-ил}метилацетат;
(4-хлорфенил){4-[(метилсульфонил)амино]-1Н-индол-1-ил}трет-бутилацетат;
(4-хлорфенил){4-[(метилсульфонил)амино]-1Н-индазол-1-ил}метилацетат;
(2-хлор-4-фторфенил)(4-{[(трифторметил)сульфонил]амино}-1Н-индол-1-ил)метилацетат;
{4-[(метилсульфонил)амино]-1Н-индол-1-ил}(фенил)метилацетат;
(2-хлор-4-фторфенил){4-[(метилсульфонил)амино]-1Н-индол-1-ил}метилацетат;
(2,4-дихлорфенил){4-[(метилсульфонил)амино]-1Н-индол-1-ил}метилацетат;
(4-бромфенил){4-[(метилсульфонил)амино]-1Н-индол-1-ил}метилацетат;
2-(4-хлорфенил)-2-{4-[(метилсульфонил)амино]-1Н-индол-1-ил}метилбутаноат;
(R)-2-(4-хлорфенил)-2-{4-[(метилсульфонил)амино]-1Н-индол-1-ил}метилбутаноат;
(S)-2-(4-хлорфенил)-2-{4-[(метилсульфонил)амино]-1Н-индол-1-ил}метилбутаноат;
2-(4-хлорфенил)-2-{4-[(метилсульфонил)амино]-1Н-индол-1-ил}трет-бутилбутаноат;
2-(4-хлорфенил)-3-циано-2-{4-[(метилсульфонил)амино]-1Н-индол-1-ил}трет-бутилпропаноат;
2-(4-хлорфенил)-2-{4-[(метилсульфонил)амино]-1Н-индол-1-ил}метилпропаноат;
2-(2,4-дихлорфенил)-2-{4-[(метилсульфонил)амино]-1Н-индол-1-ил}метилпропаноат;
2-(2,4-дихлорфенил)-2-{4-[(метилсульфонил)амино]-1Н-индол-1-ил}метилбутаноат;
2-(2-хлор-4-фторфенил)-2-{4-[(метилсульфонил)амино]-1Н-индол-1-ил}метилбутаноат;
2-(4-хлорфенил)-2-{4-[(метилсульфонил)амино]-1Н-индазол-1-ил}метилбутаноат;
2-(4-хлорфенил)-2-{4-[(метилсульфонил)амино]-1Н-индол-1-ил}метилпент-4-еноат;
2-(4-хлорфенил)-2-{4-[(метилсульфонил)амино]-1Н-индол-1-ил}-3-фенилметилпропаноат;
2-(4-хлорфенил)-2-{4-[(метилсульфонил)амино]-1Н-индол-1-ил}диметилбутандиоат;
2-(4-хлорфенил)-3-циано-2-{4-[(метилсульфонил)амино]-1Н-индол-1-ил}метилпропаноат;
2-(4-хлорфенил)-3-метокси-2-{4-[(метилсульфонил)амино]-1Н-индол-1-ил}метилпропаноат;
2-(4-хлорфенил)-3-циклопропил-2-{4-[(метилсульфонил)амино]-1H-индол-1-ил}метилпропаноат;
{R)-2-(4-хлорфенил)-N-метил-2-{4-[(метилсульфонил)амино]-1Н-индол-1-ил}бутанамид;
{R)-2-(4-хлорфенил)-2-{4-[(метилсульфонил)амино]-1Н-индол-1-ил}бутанамид;
(R)-2-(4-хлорфенил)-N-циклопропил-2-{4-[(метилсульфонил)амино]-1Н-индол-1-ил}бутанамид;
2-(4-хлорфенил)-N-(2-метоксиэтил)-2-{4-[(метилсульфонил)амино]-1Н-индол-1-ил}бутанамид;
(R)-N-бензил-2-(4-хлорфенил)-2-{4-[(метилсульфонил)амино]-1Н-индол-1-ил}бутанамид;
(R)-2-(4-хлорфенил)-N-(1-метилэтил)-2-{4-[(метилсульфонил)амино]-1Н-индол-1-ил}бутанамид;
(R)-N-трет-бутил-2-(4-хлорфенил)-2-{4-[(метилсульфонил)амино]-1Н-индол-1-ил}бутанамид;
2-(4-хлорфенил)-N-этил-2-{4-[(метилсульфонил)амино]-1Н-индол-1-ил}бутанамид;
(R)-2-(4-хлорфенил)-2-{4-[(метилсульфонил)амино]-1Н-индол-1-ил}-N-(2,2,2-трифторэтил)бутанамид;
2-(4-хлорфенил)-N-(2-цианоэтил)-2-{4-[(метилсульфонил)амино]-1H-индол-1-ил}бутанамид;
2-(4-хлорфенил)-N-(2-фторэтил)-2-{4-[(метилсульфонил)амино]-1Н-индол-1-ил}бутанамид;
2-(4-хлорфенил)-2-{4-[(метилсульфонил)амино]-1Н-индол-1-ил}-N-(2-фенилэтил)бутанамид;
(R)-2-(4-хлорфенил)-N-(цианометил)-2-{4-[(метилсульфонил)амино]-1Н-индол-1-ил}бутанамид;
N-[2-(4-хлорфенил)-2-{4-[(метилсульфонил)амино]-1Н-индол-1-ил}бутаноил]метилглицинат;
2-(4-хлорфенил)-3-циано-N-метил-2-{4-[(метилсульфонил)амино]-1Н-индол-1-ил}пропанамид;
N-бензил-2-(4-хлорфенил)-3-циано-2-{4-[(метилсульфонил)амино]-1Н-индол-1-ил}пропанамид;
2-(4-хлорфенил)-3-циано-2-{4-[(метилсульфонил)амино]-1Н-индол-1-ил}-N-(2,2,2-трифторэтил)пропанамид;
2-(4-хлорфенил)-3-циклопропил-N-метил-2-{4-[(метилсульфонил)амино]-1Н-индол-1-ил}пропанамид;
(R)-метил-N-{1-[1-(4-хлорфенил)-1-(5-метил-1,2,4-оксадиазол-3-ил)пропил]-1Н-индол-4-ил}метансульфонамид;
(R)-N-{1-[1-(4-хлорфенил)-1-(5-фенил-1,2,4-оксадиазол-3-ил)пропил]-1Н-индол-4-ил}метансульфонамид;
(R)-N-(1-{1-(4-хлорфенил)-1-[5-(4-фторфенил)-1,2,4-оксадиазол-3-ил]пропил}-1Н-индол-4-ил)метансульфонамид;
(R)-N-(1-{l-(4-хлорфенил)-1-[5-(4-хлорфенил)-1,2,4-оксадиазол-3-ил]пропил}-1Н-индол-4-ил)метансульфонамид;
(R)-N-(1-{1-(4-хлорфенил)-1-[5-(4-метоксифенил)-1,2,4-оксадиазол-3-ил]пропил}-1Н-индол-4-ил)метансульфонамид;
(R)-N-{1-[1-(4-хлорфенил)-1-(5-циклопропил-1,2,4-оксадиазол-3-ил)пропил]-1Н-индол-4-ил}метансульфонамид;
N-{1-[(4-хлорфенил)(3-фенил-1,2,4-оксадиазол-5-ил)метил]-1Н-индол-4-ил}метансульфонамид;
{R)-N-{1-[1-(4-хлорфенил)-1-(3-фенил-1Н-1,2,4-триазол-5-ил)пропил]-1Н-индол-4-ил}метансульфонамид;
(R)-N-{1-[1-(4-хлорфенил)-1-(5-метил-4Н-1,2,4-триазол-3-ил)пропил]-1Н-индол-4-ил}метансульфонамид;
(R)-N-(1-{1-(4-хлорфенил)-1-[5-(трифторметил)-4Н-1,2,4-триазол-3-ил]пропил}-1Н-индол-4-ил)метансульфонамид;
(R)-N-{1-[1-(4-хлорфенил)-1-(5-циклопропил-1,3,4-оксадиазол-2-ил)пропил]-1Н-индол-4-ил}метансульфонамид;
N-{1-[1-(4-хлорфенил)-1-(5-метил-1,3,4-оксадиазол-2-ил)пропил]-1Н-индол-4-ил}метансульфонамид;
N-(1-{1-(4-хлорфенил)-1-[5-(трифторметил)-1,3,4-оксадиазол-2-ил]пропил}-1Н-индол-4-ил)метансульфонамид;
(R)-N-{1-[1-(4-хлорфенил)-1-(5-фенил-1,3,4-оксадиазол-2-ил)пропил]-1Н-индол-4-ил}метансульфонамид;
(R)-N-(1-{1-(4-хлорфенил)-1-[5-(4-хлорфенил)-1,3,4-оксадиазол-2-ил]пропил}-1Н-индол-4-ил)метансульфонамид;
(R)-N-(1-{1-(4-хлорфенил)-1-[5-(4-фторфенил)-1,3,4-оксадиазол-2-ил]пропил}-1Н-индол-4-ил)метансульфонамид;
(R)-N-{1-[1-(4-хлорфенил)-1-{5-[4-(трифторметил)фенил]-1,3,4-оксадиазол-2-ил}пропил]-1Н-индол-4-ил}метансульфонамид;
(R)-N-(1-{1-(4-хлорфенил)-1-[5-(4-метоксифенил)-1,3,4-оксадиазол-2-ил]пропил}-1Н-индол-4-ил)метансульфонамид;
(R)-N-(1-{l-(4-хлорфенил)-1-[5-(3,5-дифторфенил)-1,3,4-оксадиазол-2-ил]пропил}-1Н-индол-4-ил)метансульфонамид;
N-{1-[1-(4-хлорфенил)-2-циано-1-(5-фенил-1,3,4-оксадиазол-2-ил)этил]-1Н-индол-4-ил}метансульфонамид;
N-{1-[1-(4-хлорфенил)-1-(4-фенил-1,3-оксазол-2-ил)пропил]-1Н-индол-4-ил}метансульфонамид;
2-(3′-метоксибифенил-3-ил)-2-{4-[(метилсульфонил)амино]-1Н-индол-1-ил}метилбутаноат;
(2′-хлорбифенил-3-ил){4-[(метилсульфонил)амино]-1Н-индол-1-ил}метилацетат;
{4-[(метилсульфонил)амино]-1Н-индол-1-ил}[2′-(трифторметокси)бифенил-3-ил]метилацетат;
{4-[(метилсульфонил)амино]-1Н-индол-1-ил}[4′-(трифторметокси)бифенил-3-ил]метилацетат;
N-{1-[(2R)-2-(4-хлорфенил)-1-гидроксибутан-2-ил]-1Н-индол-4-ил}метансульфонамид;
N-{1-[(2S)-2-(4-хлорфенил)-1-гидроксибутан-2-ил]-1Н-индол-4-ил}метансульфонамид;
2-(4-хлорфенил)-3-циано-2-{4-[(метилсульфонил)амино]-1Н-индол-1-ил}метилпропаноат;
N-{1-[3-(4-хлорфенил)-2-оксопирролидин-3-ил]-1Н-индол-4-ил}метансульфонамид;
N-(1-(2-(4-хлорфенил)-1-гидроксибутан-2-ил)-6-фтор-1Н-индол-4-ил)метансульфонамид;
N-(1-(2-(2,4-дихлорфенил)-1-гидроксибутан-2-ил)-1Н-индол-4-ил)метансульфонамид;
N-(1-(2-(4-хлорфенил)-1-гидроксибутан-2-ил)-1Н-индазол-4-ил)метансульфонамид;
N-(1-(1-гидрокси-2-(4-(трифторметил)фенил)бутан-2-ил)-1Н-индазол-4-ил)метансульфонамид;
N-(1-(3-(4-хлорфенил)-2-гидрокси-2-метилпентан-3-ил)-6-фтор-1H-индол-4-ил)метансульфонамид;
N-(1-(3-(4-хлорфенил)-2-гидрокси-2-метилпентан-3-ил)-1Н-индазол-4-ил)метансульфонамид;
N-(1-(2-гидрокси-2-метил-3-(4-(трифторметил)фенил)пентан-3-ил)-1Н-индазол-4-ил)метансульфонамид;
1-(1-(3-(4-хлорфенил)-2-гидрокси-2-метилпентан-3-ил)-1Н-индазол-4-ил)-3-метил-сульфонилмочевина;
1-(1-(3-(4-хлорфенил)-2-гидрокси-2-метилпентан-3-ил)-1Н-индазол-4-ил)-3,3-диметил-сульфонилмочевина;
N-(1-(2-(4-хлорфенил)-1-метоксибутан-2-ил)-1Н-индол-4-ил)метансульфонамид;
N-(1-(2-(4-хлорфенил)-1-метоксибутан-2-ил)-1Н-индазол-4-ил)метансульфонамид;
N-(1-(3-(4-хлорфенил)-2-оксопентан-3-ил)-6-фтор-1Н-индол-4-ил)метансульфонамид;
N-(1-(3-(4-хлорфенил)-2-оксопентан-3-ил)-1Н-индол-4-ил)метансульфонамид;
N-(1-(3-(4-хлорфенил)-2-оксопентан-3-ил)-1Н-индазол-4-ил)метансульфонамид;
N-(1-(3-(4-хлорфенил)-2-оксопентан-3-ил)-1Н-индазол-4-ил)метансульфонамид;
N-(1-(2-оксо-3-(4-(трифторметил)фенил)пентан-3-ил)-1Н-индол-4-ил)метансульфонамид;
N-(1-(2-оксо-3-(4-(трифторметил)фенил)пентан-3-ил)-1Н-индазол-4-ил)метансульфонамид;
1-(1-(3-(4-хлорфенил)-2-оксопентан-3-ил)-1Н-индазол-4-ил)-3-метил-сульфонилмочевина;
N-{1-[3-(4-хлорфенил)-2-гидроксипентан-3-ил]-1Н-индазол-4-илметансульфонамид;
N-{1-[3-(4-хлорфенил)-2-гидроксипентан-3-ил]-1Н-индазол-4-илметансульфонамид;
N-(1-(3-(4-хлорфенил)-2-гидроксипентан-3-ил)-1Н-индол-4-ил)метансульфонамид;
N-(1-(3-(4-хлорфенил)-2-гидроксипентан-3-ил)-1Н-индол-4-ил)метансульфонамид;
N-(1-(3-(2,4-дихлорфенил)-2-гидроксипентан-3-ил)-1Н-индол-4-ил)метансульфонамид;
N-(1-(3-(4-хлорфенил)-2-гидроксипентан-3-ил)-6-фтор-1Н-индол-4-ил)метансульфонамид;
N-(6-фтор-1-(2-гидрокси-3-(4-(трифторметил)фенил)пентан-3-ил)-1Н-индазол-4-ил)метансульфонамид;
N-(1-(2-гидрокси-3-(4-(трифторметил)фенил)пентан-3-ил)-1Н-индол-4-ил)метансульфонамид;
N-(1-(2-гидрокси-3-(4-(трифторметил)фенил)пентан-3-ил)-1Н-индазол-4-ил)метансульфонамид;
N-(1-(2-гидрокси-3-(4-(трифторметил)фенил)пентан-3-ил)-1Н-индазол-4-ил)метансульфонамид;
1-(1-(3-(4-хлорфенил)-2-гидроксипентан-3-ил)-1Н-индазол-4-ил)-3-метил-сульфонилмочевина;
3-(1-(3-(4-хлорфенил)-2-гидроксипентан-3-ил)-1Н-индазол-4-ил)-1,1-диметил-сульфонилмочевина;
N-(3-(3-(4-хлорфенил)-2-гидроксипентан-3-ил)-1Н-индазол-7-ил)метансульфонамид;
N-(1-(1-(4-хлорфенил)-1-(2-цианоциклопропил)пропил)-1Н-индазол-4-ил)метансульфонамид;
N-(1-(2-(4-хлорфенил)-1-цианобутан-2-ил)-1Н-индол-4-ил)метансульфонамид;
N-(1-(2-(4-хлорфенил)-1-цианобутан-2-ил)-1Н-индазол-4-ил)метансульфонамид;
N-(1-(2-(4-хлорфенил)-1-цианобутан-2-ил)-6-фтор-1Н-индол-4-ил)метансульфонамид;
N-(1-(2-(4-хлорфенил)-1-цианобутан-2-ил)-6-фтор-1Н-индол-4-ил)метансульфонамид;
N-(1-(1-циано-2-(4-(трифторметил)фенил)бутан-2-ил)-1Н-индол-4-ил)метансульфонамид;
(Е)-N-(1-(3-(4-хлорфенил)-1-цианопент-1-ен-3-ил)-1Н-индол-4-ил)метансульфонамид;
(Е)-N-(1-(1-циано-3-(4-метоксифенил)пент-1-ен-3-ил)-1Н-индол-4-ил)метансульфонамид;
(Е)-N-(1-(3-(4-хлорфенил)-1-цианопент-1-ен-3-ил)-1Н-индазол-4-ил)метансульфонамид;
(Е)-N-(1-(3-(4-хлорфенил)-1-цианопент-1-ен-3-ил)-6-фтор-1Н-индазол-4-ил)метансульфонамид;
N-(1-(3-(4-хлорфенил)-5-цианогекс-4-ен-3-ил)-1Н-индол-4-ил)метансульфонамид;
N-(1-(3-(4-Хлорфенил)-1-циано-2-метилпент-1-ен-3-ил)-1Н-индол-4-ил)метансульфонамид;
(Е)-4-(4-амино-1Н-индазол-1-ил)-4-(4-хлорфенил)гекс-2-еннитрил;
(Е)-4-(4-амино-6-фтор-1Н-индазол-1-ил)-4-(4-хлорфенил)гекс-2-еннитрил;
N-(1-(2-(4-хлорфенил)-1-гидроксибутан-2-ил)-1Н-индазол-4-ил)-N-((2-(триметилсилил)этокси)метил)метансульфонамид;
(Е)-N-(1-(3-(4-хлорфенил)-1-цианопент-1-ен-3-ил)-6-фтор-1Н-индазол-4-ил)-N-(метилсульфонил)метансульфонамид;
N-(1-(3-(4-хлорфенил)-1-цианопентан-3-ил)-1Н-индазол-4-ил)метансульфонамид;
N-(1-(3-(4-хлорфенил)-1-цианопентан-3-ил)-1Н-индазол-4-ил)метансульфонамид;
N-(1-(1-циано-3-(4-метоксифенил)пентан-3-ил)-1Н-индол-4-ил)метансульфонамид;
N-(1-(3-(4-хлорфенил)-1-цианопентан-3-ил)-1Н-индол-4-ил)метансульфонамид;
N-(1-(1-циано-3-(4-(трифторметил)фенил)пентан-3-ил)-1Н-индазол-4-ил)метансульфонамид;
N-(1-(3-(4-хлорфенил)-1-цианопентан-3-ил)-6-фтор-1Н-индол-4-ил)метансульфонамид;
N-(1-(3-(4-хлорфенил)-1-цианопентан-3-ил)-6-фтор-1Н-индол-4-ил)метансульфонамид;
N-(1-(1-циано-3-фенилпентан-3-ил)-6-фтор-1Н-индазол-4-ил)метансульфонамид;
N-(1-(3-(4-хлорфенил)-1-цианопентан-3-ил)-6-фтор-1Н-индазол-4-ил)метансульфонамид;
N-(1-(3-(4-хлорфенил)-1-циано-2-оксопентан-3-ил)-1Н-индол-4-ил)метансульфонамид;
N-(1-(1-(4-хлорфенил)-1-(3-циано-4,5-дигидрофуран-2-ил)пропил)-1Н-индол-4-ил)метансульфонамид;
N-(2-хлорэтил)-2-(4-хлорфенил)-2-(4-(метилсульфонамидо)-1Н-индол-1-ил)бутанамид;
2-(4-хлорфенил)-N-(цианометил)-2-(4-(метилсульфонамидо)-1Н-индол-1-ил)бутанамид;
N-(1-(1-(4-хлорфенил)-1-(3-этил-1,2,4-оксадиазол-5-ил)пропил)-1Н-индол-4-ил)метансульфонамид;
N-(1-(1-(4-хлорфенил)-1-(3-этил-1,2,4-оксадиазол-5-ил)пропил)-1Н-индол-4-ил)метансульфонамид;
N-(1-(1-(4-хлорфенил)-1-(2-этил-2Н-1,2,3-триазол-4-ил)пропил)-1Н-индол-4-ил)метансульфонамид;
2-(4-хлорфенил)-2-(4-(2-(метилсульфонил)этил)-1Н-индол-1-ил)метилбутаноат;
N-(1-(1-(4-хлорфенил)-1-(1-гидроксициклопропил)пропил)-1Н-индол-4-ил)метансульфонамид;
2-(4-хлорфенил)-4-фтор-2-(4-(метилсульфонамидо)-1Н-индол-1-ил)метилбутаноат;
5-(1-(4-хлорфенил)-1-(4-(метилсульфонамидо)-1Н-индазол-1-ил)пропил)-1,2,4-оксадиазол-3-карбоксамид;
N-(1-(1-(4-хлорфенил)-1-(3-циано-1,2,4-оксадиазол-5-ил)пропил)-1Н-индазол-4-ил)метансульфонамид;
N-(1-(3-(4-хлорфенил)-1,1,1-трифторпентан-3-ил)-1Н-индазол-4-ил)метансульфонамид;
N-(1-(3-(4-хлорфенил)-1-гидроксипентан-3-ил)-1Н-индол-4-ил)метансульфонамид;
N-(1-(3-(4-хлорфенил)-1-гидроксипентан-3-ил)-1Н-индол-4-ил)метансульфонамид;
N-(1-(3-(4-хлорфенил)-1-фторпентан-3-ил)-1Н-индол-4-ил)метансульфонамид;
N-(1-(3-(4-хлорфенил)-1-фторпентан-3-ил)-1Н-индол-4-ил)метансульфонамид;
3-(4-хлорфенил)-3-(4-(метилсульфонамидо)-1Н-индол-1-ил)пентил метансульфонат;
N-(1-(3-(4-Хлорфенил)-1-метоксипентан-3-ил)-1Н-индол-4-ил)метансульфонамид;
N-(1-(3-(4-хлорфенил)-1-(метилтио)пентан-3-ил)-1Н-индол-4-ил)метансульфонамид;
4-(4-амино-1Н-индол-1-ил)-4-(4-хлорфенил)метилгексаноат;
4-(4-хлорфенил)-4-(4-(метилсульфонамидо)-1Н-индазол-1-ил)метилгексаноат;
N-(1-(3-(4-хлорфенил)-6-гидроксигексан-3-ил)-1Н-индол-4-ил)метансульфонамид;
N-(1-(3-(4-хлорфенил)-6-фторгексан-3-ил)-1Н-индол-4-ил)метансульфонамид;
(Е)-метил-4-(4-хлорфенил)-4-(4-(метилсульфонамидо)-1Н-индазол-1-ил)гекс-2-еноат;
N-(1-(3-(4-хлорфенил)-6-оксогептан-3-ил)-1Н-индазол-4-ил)метансульфонамид;
N-(1-(3-(4-хлорфенил)-2-гидрокси-1-метоксипентан-3-ил)-1Н-индол-4-ил)метансульфонамид;
N-(1-(3-(4-хлорфенил)пентан-3-ил)-1Н-индол-4-ил)метансульфонамид;
N-(1-(3-(4-хлорфенил)пент-1-ен-3-ил)-1Н-индол-4-ил)метансульфонамид;
N-(1-(2-амино-3-(4-хлорфенил)пентан-3-ил)-1Н-индол-4-ил)метансульфонамид;
N-(1-(1-амино-2-(4-хлорфенил)бутан-2-ил)-1Н-индол-4-ил)метансульфонамид;
N-(1-(1-(4-хлорфенил)-1-цианопропил)-1Н-индол-4-ил)этансульфонамид;
N-(1-(1-(4-хлорфенил)-1-(2-этил-2Н-тетразол-5-ил)пропил)-1Н-индол-4-ил)метансульфонамид;
N-(1-(3-(4-хлорфенил)-6,6,6-трифторгексан-3-ил)-1Н-индол-4-ил)метансульфонамид;
N-(1-(3-(4-хлорфенил)-1-цианопент-1-ин-3-ил)-1Н-индол-4-ил)метансульфонамид;
или его фармацевтически приемлемая соль.
8. Соединение по п. 7, которое представляет собой одно из соединений:
(R)-N-{1-[(2R)-2-(4-хлорфенил)-1-гидроксибутан-2-ил]-1Н-
индол-4-ил}метансульфонамид;
N-(1-(1-(4-хлорфенил)-1-(2-цианоциклопропил)пропил)-1Н-индазол-4-ил)метансульфонамид;
N-(1-(1-(4-хлорфенил)-1-(3-этил-1,2,4-оксадиазол-5-ил)пропил)-1H-индол-4-ил)метансульфонамид;
N-(1-(2-(4-хлорфенил)-1-гидроксибутан-2-ил)-1H-индазол-4-ил)метансульфонамид;
N-(1-(1-(4-хлорфенил)-1-(1-гидроксициклопропил)пропил)-1H-индол-4-ил)метансульфонамид;
N-(1-(3-(4-хлорфенил)-2-гидроксипентан-3-ил)-6-фтор-1Н-индол-4-ил)метансульфонамид;
N-(1-(2-(4-хлорфенил)-1-гидроксибутан-2-ил)индолин-4-ил)метансульфонамид;
N-{1-[3-(4-хлорфенил)-2-гидроксипентан-3-ил]-1H-индазол-4-илметансульфонамид;
N-(1-(2-амино-3-(4-хлорфенил)пентан-3-ил)-1H-индол-4-ил)метансульфонамид;
1-(1-(3-(4-хлорфенил)-2-гидрокси-2-метилпентан-3-ил)-1Н-индазол-4-ил)-3-метил-сульфонилмочевина;
{4-[(метилсульфонил)амино]-1H-индол-1-ил}(фенил)метилацетат;
N-(1-(3-(4-хлорфенил)-2-гидрокси-2-метилпентан-3-ил)-6-фтор-1Н-индол-4-ил)метансульфонамид;
N-(1-(3-(4-хлорфенил)-2-гидрокси-2-метилпентан-3-ил)-1Н-индол-4-ил)метансульфонамид;
N-(1-(3-(4-хлорфенил)-2-гидрокси-2-метилпентан-3-ил)-1Н-индазол-4-ил)метансульфонамид;
N-(1-(2-гидрокси-2-метил-3-(4-(трифторметил)фенил)пентан-3-
ил)-1H-индазол-4-ил)метансульфонамид;
N-(1-(2-(4-хлорфенил)-1-метоксибутан-2-ил)-1H-индол-4-ил)метансульфонамид;
N-(1-(3-(4-хлорфенил)-2-оксопентан-3-ил)-1H-индазол-4-ил)метансульфонамид;
N-(1-(2-гидрокси-3-(4-(трифторметил)фенил)пентан-3-ил)-1Н-индазол-4-ил)метансульфонамид;
(E)-N-(1-(3-(4-хлорфенил)-1-цианопент-1-ен-3-ил)-6-фтор-1H-индазол-4-ил)метансульфонамид;
N-(1-(3-(4-хлорфенил)-1-цианопентан-3-ил)-1H-индазол-4-ил)метансульфонамид;
N-(1-(1-циано-3-(4-метоксифенил)пентан-3-ил)-1H-индол-4-ил)метансульфонамид;
N-(1-(3-(4-хлорфенил)-1-цианопентан-3-ил)-1H-индол-4-ил)метансульфонамид;
N-(1-(1-циано-3-(4-(трифторметил)фенил)пентан-3-ил)-1H-индазол-4-ил)метансульфонамид;
N-(1-(3-(4-хлорфенил)-1-цианопентан-3-ил)-6-фтор-1H-индол-4-ил)метансульфонамид;
N-(1-(1-циано-3-фенилпентан-3-ил)-6-фтор-1H-индазол-4-ил)метансульфонамид;
N-(1-(3-(4-Хлорфенил)-1-гидроксипентан-3-ил)-1H-индол-4-ил)метансульфонамид;
N-(1-(3-(4-хлорфенил)-6-гидроксигексан-3-ил)-1H-индол-4-ил)метансульфонамид;
N-(1-(3-(4-хлорфенил)пентан-3-ил)-1H-индол-4-ил)метансульфонамид;
или его фармацевтически приемлемая соль.
9. Соединение по п. 8, которое представляет собой одно из соединений:
(R)-N-{1-[(2R)-2-(4-хлорфенил)-1-гидроксибутан-2-ил]-1H-индол-4-ил}метансульфонамид;
N-(1-(1-(4-хлорфенил)-1-(2-цианоциклопропил)пропил)-1H-индазол-4-ил)метансульфонамид;
N-{1-(1-(4-хлорфенил)-1-(3-этил-1,2,4-оксадиазол-5-ил)пропил)-1H-индол-4-ил)метансульфонамид;
N-(1-(2-(4-хлорфенил)-1-гидроксибутан-2-ил)-1H-индазол-4-ил)метансульфонамид;
N-(1-(1-(4-хлорфенил)-1-(1-гидроксициклопропил)пропил)-1H-индол-4-ил)метансульфонамид;
N-(1-(3-(4-хлорфенил)-2-гидроксипентан-3-ил)-6-фтор-1H-индол-4-ил)метансульфонамид;
N-(1-(2-(4-хлорфенил)-1-гидроксибутан-2-ил)индолин-4-ил)метансульфонамид;
N-{1-[3-(4-хлорфенил)-2-гидроксипентан-3-ил]-1H-индазол-4-илметансульфонамид;
N-(1-(2-амино-3-(4-хлорфенил)пентан-3-ил)-1H-индол-4-ил)метансульфонамид;
1-(1-(3-(4-хлорфенил)-2-гидрокси-2-метилпентан-3-ил)-1H-индазол-4-ил)-3-метил-сульфонилмочевина;
или его фармацевтически приемлемая соль.
10. Фармацевтическая композиция, обладающая активностью антагониста рецептора минералокортикоида (MR), содержащая терапевтически эффективное количество соединения по п. 1 и
фармацевтически приемлемого носителя.
11. Соединение по п. 1 или его фармацевтически приемлемая соль для применения в терапии опосредуемых альдостероном нарушений.
| US2007072897 A1, 29.03.2007 | |||
| WO2005118539 A1, 15.12.2005 | |||
| ИНДОЛИЛПРОИЗВОДНЫЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ ПЕЧЕНОЧНОГО Х-РЕЦЕПТОРА | 2005 |
|
RU2368612C2 |
| Раздатчик-смеситель кормов | 1990 |
|
SU1732887A1 |
| Способ экстрагирования масла из семян хлопчатника | 1989 |
|
SU1730127A1 |
| WO 02060438 A1, 08.08.2002 | |||
| Приспособление для спуска затвора через заранее определенное время | 1933 |
|
SU37472A1 |
| Способ диагностики системной красной волчанки | 1978 |
|
SU775694A1 |
| WO 2010124114 A1, 28.10.2010 | |||
| Генератор прямоугольных импульсов | 1978 |
|
SU705652A1 |
| BAYINDIR SINAN ET AL.: 'An efficient synthesis of new | |||

