Настоящее изобретение относится к новым N-сульфамоилпиперидинамидам и их физиологически приемлемым солям присоединения с кислотами, к включающим их фармацевтическим композициям, к способам их получения и к их применению для лечения ожирения и родственных патологических состояний.
В WO 03/088908 описаны N-сульфамоилпиперидинамиды, обладающие особым набором заместителей в пиперидиновом кольце. Соединения, указанные в WO 03/088908, предположительно применимы для лечения аритмии, связанных с IKur патологических состояний, желудочно-кишечных нарушений, диабета, нарушений познавательной способности и родственных патологических состояний.
US 2004/0167185 описаны некоторые N-сульфамоилпиперидинамиды, применимые в области лечения или предупреждения рака.
Способ получения соединений, пригодных для лечения и/или профилактики ожирения путем ингибирования липогенеза посредством ингибирования карбоангидраз у млекопитающих и людей, описан в документе WO 02/07821.
В основу настоящего изобретения была положена задача получения новых лекарственных средств, предназначенных для лечения и/или профилактики ожирения и сопутствующих ему и/или вторичных заболеваний или патологических состояний, которые являются весьма эффективными и могут быть получены простым образом.
Согласно изобретению неожиданно было установлено, что некоторые новые N-сульфамоилпиперидинамиды и их физиологически приемлемые соли присоединения с кислотами применимы для лечения и/или профилактики ожирения и сопутствующих ему и/или вторичных заболеваний или патологических состояний.
Настоящее изобретение относится к соединениям общей формулы I
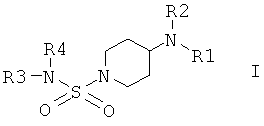
в которой R1 выбран из группы, включающей Н; алкил; циклоалкил; алкиленалкоксигруппу; алкиленциклоалкил; арил, незамещенный или замещенный одним или большим количеством следующих групп: алкил, алкоксигруппа, галоген, CF3, CN; алкиленарил; алкиленариленалкил; алкиленариленгалоген; алкиленариленоксиалкил; алкиленариленалкиламиногруппа; гетероарил; алкиленгетероарил, незамещенный или замещенный с помощью алкила, алкоксигруппы, галогена, CN, CF3;
в которой R2 выбран из группы, включающей циклоалкил; арил, незамещенный или замещенный с помощью алкила, алкоксигруппы, галогена, CN, CF3; алкиленарил, незамещенный или замещенный, но не замещенный фураном или фуранилом; алкиленалкоксигруппу; алкиленциклоалкил; гетероарил; СО-алкил; СО-циклоалкил; СО-арил, замещенный с помощью алкила, алкоксигруппы, галогена, CF3, CN; СО-алкиленарил, незамещенный или замещенный с помощью алкила, алкоксигруппы, галогена, CF3, CN; СО-гетероарил, незамещенный или замещенный с помощью алкила, алкоксигруппы, галогена, CF3, CN; СО-O-алкил; СО-O-циклоалкил; СО-O-арил, замещенный с помощью алкила, алкоксигруппы, галогена, CN, CF3; СО-O-алкиленарил, незамещенный или замещенный с помощью алкила, алкоксигруппы, галогена, CN, CF3; СО-O-гетероарил; CO-NH-алкил; CO-NH-циклоалкил; CO-NH-арил, замещенный с помощью алкила, алкоксигруппы, галогена, CN, CF3; CO-NH-алкиленарил, незамещенный или замещенный с помощью алкила, алкоксигруппы, галогена, CN, CF3; CO-NH-гетероарил; SO2-NH2; SO2-алкил; SO2-арил, незамещенный или замещенный с помощью алкила, алкоксигруппы, галогена, CF3, CN;
или в которой R1 и R2 совместно образуют 5- или 6-членное кольцо, которое необязательно может содержать от 1 до 2 дополнительных гетероатома, независимо выбранных из группы, включающей азот, кислород и/или серу; и которое необязательно может содержать 1 или 2 двойные связи; и которое также может быть замещено с помощью алкила, галогеналкила, арила, незамещенного или замещенного с помощью алкила, алкоксигруппы, гидроксигруппы, галогена, CN, CF3 и/или гетероарила; и которое также может содержать карбонильную группу; и которое также может быть сконденсировано с арилом;
в которой R3 и R4 независимо выбраны из группы, включающей Н, алкил, циклоалкил, циклоалкил, содержащий 1 или большее количество гетероатомов, выбранных из группы, включающей азот и/или кислород; циклоалкил, содержащий 1 или большее количество гетероатомов, выбранных из группы, включающей азот и/или кислород, замещенный с помощью алкила, алкоксигруппы, галогена, CF3, CN; арил; арил, замещенный с помощью алкила, алкоксигруппы, галогена, CF3, CN; гетероарил, незамещенный или замещенный с помощью алкила, алкоксигруппы, галогена, CF3, CN; алкиленарил; или в которой R3 и R4 совместно образуют 5- или 6-членное кольцо, которое необязательно может содержать от 1 до 2 гетероатомов, независимо выбранных из группы, включающей атомы азота и/или кислорода, и который также может быть замещен с помощью арила или арила, замещенного с помощью алкила, алкоксигруппы, галогена, CF3 и CN;
и их физиологически приемлемым солям присоединения с кислотами.
Соединения общей формулы I применимы для лечения и/или профилактики глаукомы, эпилепсии, биполярных нарушений, мигрени, невропатической боли, ожирения, диабета типа II, метаболического синдрома, алкогольной зависимости и/или рака и сопутствующих им и/или вторичных заболеваний или патологических состояний у млекопитающих и людей.
Более предпочтительно, если в соединениях общей формулы I R1 выбран из группы, включающей Н; алкил; циклоалкил; алкиленалкоксигруппу; алкиленциклоалкил; арил; алкиленарил; гетероарил; алкиленгетероарил, незамещенный или замещенный галогеном; R2 выбран из группы, включающей циклоалкил; арил, замещенный с помощью алкила, алкоксигруппы, галогена, CN, CF3; алкиленарил, незамещенный или замещенный, но не замещенный фураном или фуранилом; алкиленалкоксигруппу; алкиленциклоалкил; СО-алкил; СО-циклоалкил; СО-алкиленарил; СО-гетероарил; СО-O-алкил; СО-O-циклоалкил; СО-O-арил, замещенный с помощью алкила, алкоксигруппы, галогена, CN, CF3; СО-O-алкиленарил, незамещенный или замещенный с помощью алкила, алкоксигруппы, галогена, CN, CF3; СО-O-гетероарил; CO-NH-арил, замещенный с помощью алкила, алкоксигруппы, галогена, CN, CF3; СО-NH-алкиленарил, незамещенный или замещенный с помощью алкила, алкоксигруппы, галогена, CN, CF3; CO-NH-гетероарил; SO2-NH2, или R1 и R2 совместно образуют 5- или 6-членное кольцо, которое необязательно может содержать от 1 до 2 гетероатомов, независимо выбранных из группы, включающей азот, кислород и/или серу; и которое необязательно может содержать 1 или 2 двойные связи; и которое также может быть замещено с помощью алкила, галогеналкила, арила, незамещенного или замещенного с помощью алкила, алкоксигруппы, гидроксигруппы, галогена, CN, CF3, и/или гетероарила; и которое также может содержать карбонильную группу; и которое также может быть сконденсировано с арилом; R3 и R4 независимо выбраны из группы, включающей Н, алкил, циклоалкил; или R3 и R4 совместно образуют 5-или 6-членное кольцо, которое необязательно может содержать от 1 до 2 гетероатомов, независимо выбранных из группы, включающей атомы азота и/или кислорода, и которое также может быть замещено арилом.
Еще более предпочтительно, если в соединениях общей формулы I R1 выбран из группы, включающей Н, алкил; циклоалкил; алкиленалкоксигруппу; алкиленциклоалкил; арил; алкиленарил; гетероарил; алкиленгетероарил, замещенный галогеном; R2 выбран из группы, включающей алкиленалкоксигруппу; алкиленциклоалкил; СО-алкил; СО-циклоалкил; СО-алкиленарил; СО-гетероарил; CO-NH-алкиленарил; CO-NH-арил, замещенный с помощью алкила, алкоксигруппы, галогена, CN, CF3; CO-NH-алкиленарил, замещенный с помощью алкила, алкоксигруппы, галогена, CN, CF3; CO-NH-гетероарил; SO2-NH2, или R1 и R2 совместно образуют 5- или 6-членное кольцо, которое необязательно может содержать от 1 до 2 гетероатомов, независимо выбранных из группы, включающей азот, кислород и/или серу; и которое необязательно может содержать 1 или 2 двойные связи; и которое также может быть замещено с помощью алкила, галогеналкила, арила, незамещенного или замещенного с помощью алкила, алкоксигруппы, гидроксигруппы, галогена, CN, CF3, и/или гетероарила; и которое также может содержать карбонильную группу; и которое также может быть сконденсировано с арилом; R3 и R4 независимо выбраны из группы, включающей Н, алкил, циклоалкил; или R3 и R4 совместно образуют кольцо, выбранное из группы, включающей пирролидинил, пиперидинил-п-фенил, пиперазинил-п-фенил и морфолиновую группу.
В особенно предпочтительном варианте осуществления настоящего изобретения R1 обозначает только Н, если R2 не содержит группу СО.
В другом особенно предпочтительном варианте осуществления настоящего изобретения R3 и R4 оба обозначают Н.
Если в соединениях формулы I или в других соединениях, описанных в объеме настоящего изобретения, заместители представляют собой или содержат алкил, циклоалкил, алкилен, алкоксигруппу, то все они могут обладать линейной или разветвленной цепью и содержать от 1 до 8, предпочтительно от 1 до 6 и более предпочтительно от 1 до 4 атомов углерода. Подходящими являются метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, циклопропил, циклобутил, циклопентил, циклогексил, метилен, этилен, пропилен, изопропилен, бутилены, изобутилен, трет-бутилен, метоксигруппа, этоксигруппа, пропоксигруппа, изопропоксигруппа, бутоксигруппа, изобутоксигруппа и трет-бутоксигруппа.
Если заместителями в соединениях формулы I являются галогены, то подходящими являются фтор, хлор, бром или йод. Хлор и бром являются предпочтительными.
Если заместителями в соединениях формулы I являются арилы, то они означают моноциклические ароматические кольцевые системы, содержащие количество атомов водорода, зависящее от типа замещения. Однако в это определение входят конденсированные и спирановые арильные системы. Подходящими арильными заместителями являются фенил, 1H-инден, 9Н-флуорен, нафталин, антрацен и фенантрен.
Если заместителями в соединениях формулы I являются гетероарилы, то они означают арильные кольцевые системы, в которых один или большее количество атомов углерода ароматической кольцевой системы заменены на гетероатом, выбранный из группы, включающей кислород и/или азот и/или серу. Подходящими гетероарилами являются пиррол, фуран, тиофен, индолизин, индол, изоиндол, кумарон, тионафтен, пиразол, имидазол, оксазол, изооксазол, тиазол, изотиазол, триазол, тетразол, тиадиазол, пиридин, пиран, тиопиран, хинолин, изохинолин, пиридазин, пиримидин, пиразин и триазин.
Физиологически совместимые соли присоединения с кислотами соединений общей формулы I представляют собой их обычные соли с неорганическими кислотами, например серной кислотой, фосфорной кислотой или с галогенводородными кислотами, предпочтительно хлористоводородной кислотой, или с органическими кислотами, например низш. алифатическими монокарбоновыми, дикарбоновыми или трикарбоновыми кислотами, такими как малеиновая кислота, фумаровая кислота, молочная кислота, винная кислота, лимонная кислота, или с сульфоновыми кислотами, например низш. алкансульфоновыми кислотами, такими как метансульфоновая кислота или трифторметансульфоновая кислота, или с бензолсульфоновыми кислотами, необязательно замещенными по бензольному кольцу галогеном или низш. алкилом, такими как п-толуолсульфоновая кислота. Гидрохлориды соединений общей формулы I являются предпочтительными.
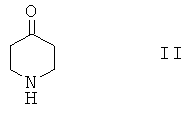
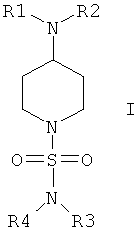
Соединения формулы I, в которой R3 и R4 оба не обозначают Н, и их физиологически приемлемые соли присоединения с кислотами можно получить по реакции соединения формулы II
с сульфамоилхлоридом формулы III
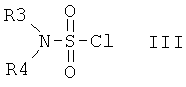
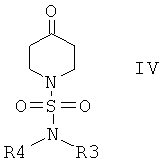
и получить соединение формулы IV
Соединения формулы IV затем вводят в реакцию с амином H2NR1 и получают соединения формулы V
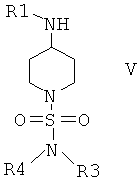
Соединения формулы V дополнительно вводят в реакцию с R2X, в котором Х выбран из группы, включающей Cl, Br и I, и получают соединения формулы I
Альтернативно, соединения формулы I, в которой R3 и R4 оба не обозначают Н, можно получить по реакции соединений формулы IV с амином HNR1R2 и получить соединения формулы I
Соединения формулы I, в которой R3 и R4 оба обозначают И, можно получить по реакции соединения формулы II
с сульфамоилхлоридом, который содержит защитную группу PG, предпочтительно трет-бутоксикарбонил или бензил, формулы VIa или с реагентом формулы VIb
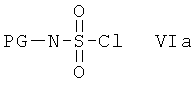
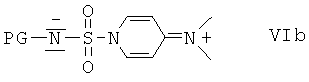
и получить соединение формулы VII
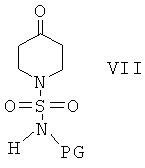
Соединения формулы VII затем вводят в реакцию с амином H2NR1 и получают соединения формулы VIII
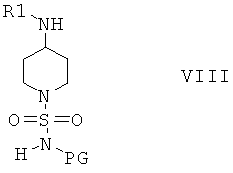
Соединения формулы VIII дополнительно вводят в реакцию с R2X, в котором Х выбран из группы, включающей Cl, Br и I, и получают соединения формулы IX
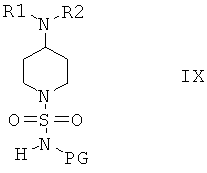
и затем отщепляют защитную группу PG от полученных промежуточных продуктов при подходящих условиях, что приводит к получению соединений формулы I, в которой R3 и R4 оба обозначают Н. Если защитной группой является трет-бутоксикарбонил, то после этого удаление PG можно провести в кислой среде, предпочтительно в присутствии хлористого водорода. Если защитной группой является бензил, то удаление PG можно провести путем гидрирования, предпочтительно в присутствии водорода и катализатора, такого как Pd.
Альтернативно, соединения формулы I, в которой R3 и R4 оба обозначают Н, можно получить по реакции соединения формулы VII с амином HNR1R2 и получить соединения формулы IX
и затем отщепить защитную группу PG от полученных промежуточных продуктов при подходящих условиях, что приводит к получению соединений формулы I, в которой R3 и R4 оба обозначают Н. Если защитной группой является трет-бутоксикарбонил, то после этого удаление PG можно провести в кислой среде, предпочтительно в присутствии хлористого водорода. Если защитной группой является бензил, то удаление PG можно провести путем гидрирования, предпочтительно в присутствии водорода и катализатора, такого как Pd.
Соединения формулы I, в которой R2 содержит метиленовую разделительную группу СН2, можно получить по реакции соединения формулы Х
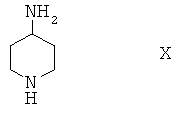
с реагентом, содержащим защитную группу PG, и получить соединение формулы XI
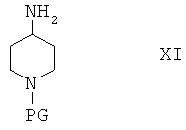
Соединения формулы XI затем вводят в реакцию с альдегидом R2'-CHO и получают соединения формулы XII
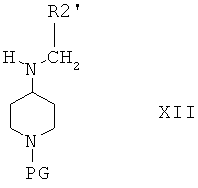
в которой R2' выбран из группы, включающей алкил; циклоалкил; алкиленарил, незамещенный или замещенный, но не замещенный фураном или фуранилом; алкиленалкоксигруппу; и алкиленциклоалкил.
Затем защитную группу PG соединений формулы XII отщепляют при подходящих условиях и после этого не содержащее защитной группы соединение вводят в реакцию с сульфамоилхлоридом ClSO2-NH2 и получают соединения формулы I. Если защитной группой является трет-бутоксикарбонил, то удаление PG можно провести в кислой среде, предпочтительно в присутствии хлористого водорода. Если защитной группой является бензил, то удаление PG можно провести путем гидрирования, предпочтительно в присутствии водорода и катализатора, такого как Pd.
Альтернативно, соединения формулы I, в которой R2 содержит метиленовую разделительную группу CH2, можно получить путем отщепления защитной группы PG от соединений формулы XII в подходящих условиях и после этого не содержащее защитной группы соединение вводят в реакцию с сульфамоилхлоридом, который содержит защитную группу PG, предпочтительно трет-бутоксикарбонил или бензил, формулы VIa или с реагентом формулы VIb
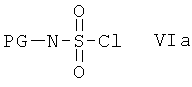
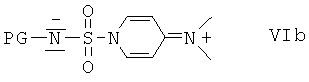
и получают соединение формулы XIII
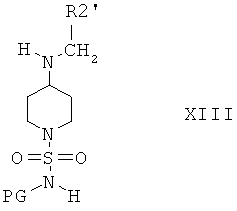
и затем отщепляют защитную группу PG от полученного промежуточного продукта при подходящих условиях, что приводит к получению соединений формулы I, в которой R3 и R4 оба обозначают Н. Если защитной группой является трет-бутоксикарбонил, то после этого удаление PG можно провести в кислой среде, предпочтительно в присутствии хлористого водорода. Если защитной группой является бензил, то удаление PG можно провести путем гидрирования, предпочтительно в присутствии водорода и катализатора, такого как Pd.
В другом альтернативном варианте осуществления соединения формулы I, в которой R2 содержит метиленовую разделительную группу CH2, можно получить по реакции соединения формулы Х
с реагентом, содержащим защитную группу PG, и получить соединение формулы XI
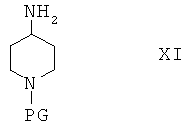
Соединения формулы XI затем вводят в реакцию с кетоном R2'-COR1' и получают соединения формулы XIV
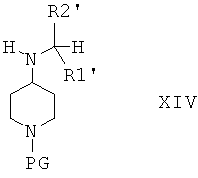
в которой R1' выбран из группы, включающей алкил; алкиленалкоксигруппу; алкиленциклоалкил; алкиленарил; алкиленариленалкил; алкиленариленгалоген; алкиленариленоксиалкил, алкиленариленалкиламиногруппу; и алкиленгетероарил, незамещенный или замещенный с помощью алкила, алкоксигруппы, галогена, CN, CF3;
в которой R2' выбран из группы, включающей алкил; циклоалкил; алкиленарил, незамещенный или замещенный, но не замещенный фураном или фуранилом; алкиленалкоксигруппу; и алкиленциклоалкил.
Защитную группу PG соединений формулы XIV затем отщепляют при подходящих условиях и после этого не содержащее защитной группы соединение вводят в реакцию с сульфамоилхлоридом, который содержит защитную группу PG, предпочтительно трет-бутоксикарбонил или бензил, формулы VIa или с реагентом формулы VIb
и получают соединение формулы XV
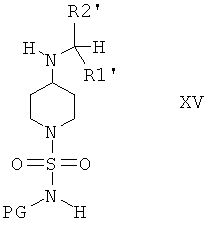
и затем отщепляют защитную группу PG от полученного промежуточного продукта при подходящих условиях, что приводит к получению соединений формулы I, в которой R3 и R4 оба обозначают Н. Если защитной группой является трет-бутоксикарбонил, то после этого удаление PG можно провести в кислой среде, предпочтительно в присутствии хлористого водорода. Если защитной группой является бензил, то удаление PG можно провести путем гидрирования, предпочтительно в присутствии водорода и катализатора, такого как Pd.
Соединения формулы I также можно получить по реакции соединения формулы II
с реагентом, содержащим защитную группу PG, и получить соединение формулы XVII
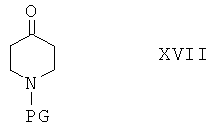
Соединения формулы XVII затем вводят в реакцию с амином NHR1R2 и получают соединения формулы XVIII
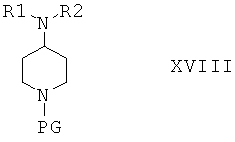
Защитную группу PG соединений формулы XVIII затем отщепляют при подходящих условиях и после этого не содержащее защитной группы соединение вводят в реакцию с сульфамидом и получают соединения формулы I, или с соединением формулы III и получают соединения формулы I, или с соединением формулы VIa или VIb и получают соединение формулы XIX
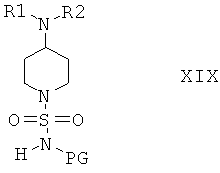
и затем отщепляют защитную группу PG от полученного промежуточного продукта при подходящих условиях, что приводит к получению соединений формулы I. Если защитной группой является трет-бутоксикарбонил, то после этого удаление PG можно провести в кислой среде, предпочтительно в присутствии хлористого водорода. Если защитной группой является бензил, то удаление PG можно провести путем гидрирования, предпочтительно в присутствии водорода и катализатора, такого как Pd.
В другом варианте осуществления соединения формулы I получают по реакции соединения формулы VXIIIa, в которой R1 обозначает Н
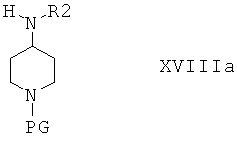
с соединением формулы XX
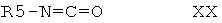
в которой R5 выбран из группы, включающей алкил; циклоалкил; арил, замещенный с помощью алкила, алкоксигруппы, галогена, CN, CF3; алкиленарил, незамещенный или замещенный с помощью алкила, алкоксигруппы, галогена, CN, CF3; и гетероарил; и получают соединение формулы XXI
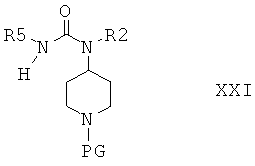
Защитную группу PG соединений формулы XXI затем отщепляют при подходящих условиях и после этого не содержащее защитной группы соединение вводят в реакцию с сульфамидом и получают соединения формулы I, или с соединением формулы III и получают соединения формулы I, или с соединением формулы VIa или VIb и получают соединение формулы XXII
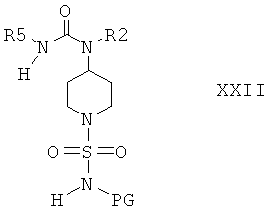
и затем отщепляют защитную группу PG от полученного промежуточного продукта при подходящих условиях, что приводит к получению соединений формулы I. Если защитной группой является трет-бутоксикарбонил, то после этого удаление PG можно провести в кислой среде, предпочтительно в присутствии хлористого водорода. Если защитной группой является бензил, то удаление PG можно провести путем гидрирования, предпочтительно в присутствии водорода и катализатора, такого как Pd.
Соединения формулы I альтернативно также можно получить по реакции соединения формулы VXIIIa, в которой R1 обозначает Н
с соединением формулы XXIII

в которой R6 выбран из группы, включающей алкил; арил, незамещенный или замещенный с помощью алкила, алкоксигруппы, галогена, CF3, CN; и получить соединение формулы XXIV
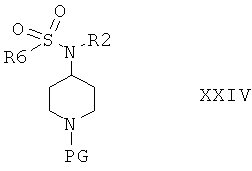
Защитную группу PG соединений формулы XXIV затем отщепляют при подходящих условиях и после этого не содержащее защитной группы соединение вводят в реакцию с сульфамидом и получают соединения формулы I, или с соединением формулы III и получают соединения формулы I, или с соединением формулы VIa или VIb и получают соединение формулы XXV
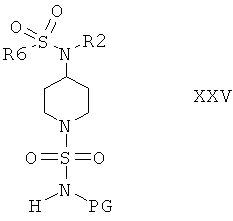
и затем отщепляют защитную группу PG от полученного промежуточного продукта при подходящих условиях и получают соединения формулы I. Если защитной группой является трет-бутоксикарбонил, то после этого удаление PG можно провести в кислой среде, предпочтительно в присутствии хлористого водорода. Если защитной группой является бензил, то удаление PG можно провести путем гидрирования, предпочтительно в присутствии водорода и катализатора, такого как Pd.
В другом варианте осуществления настоящего изобретения соединения формулы I можно получить по реакции соединения формулы XI
с соединением формулы XXIII и получить соединения формулы XXVI
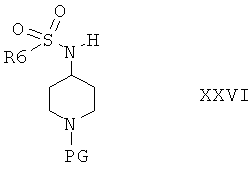
Защитную группу PG соединений формулы XXVI затем отщепляют при подходящих условиях, после этого не содержащее защитной группы соединение вводят в реакцию с сульфамидом и получают соединения формулы I, или с соединением формулы III и получают соединения формулы I, или с соединением формулы VIa или VIb и получают соединение формулы XXVII
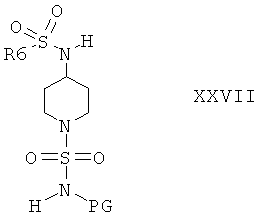
и затем отщепляют защитную группу PG от полученного промежуточного продукта при подходящих условиях и получают соединения формулы I. Если защитной группой является трет-бутоксикарбонил, то после этого удаление PG можно провести в кислой среде, предпочтительно в присутствии хлористого водорода. Если защитной группой является бензил, то удаление PG можно провести путем гидрирования, предпочтительно в присутствии водорода и катализатора, такого как Pd.
В другом варианте осуществления настоящего изобретения соединения формулы I можно получить по реакции соединения формулы VIII
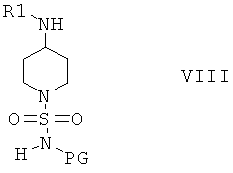
с соединениями формулы XX
и получить соединения формулы XXVIII
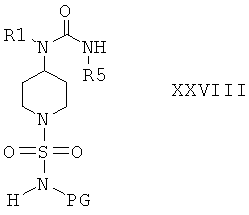
Защитную группу PG соединений формулы XXVIII затем отщепляют при подходящих условиях и получают соединения формулы I. Если защитной группой является трет-бутоксикарбонил, то после этого удаление PG можно провести в кислой среде, предпочтительно в присутствии хлористого водорода. Если защитной группой является бензил, то удаление PG можно провести путем гидрирования, предпочтительно в присутствии водорода и катализатора, такого как Pd.
В другом варианте осуществления настоящего изобретения соединения формулы I можно получить по реакции соединения формулы XI
с соединениями формулы XXIX

и получить соединения формулы XXX
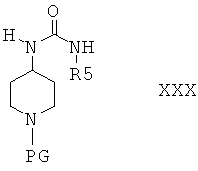
Защитную группу PG соединений формулы XXX затем отщепляют при подходящих условиях и получают соединения формулы XXXI
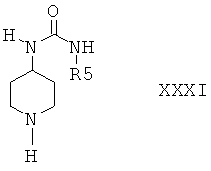
Затем соединение формулы XXXI вводят в реакцию с сульфамоилхлоридом формулы III и получают соединения формулы I или соединение формулы XXXI затем вводят в реакцию с соединением формулы VIa или VIb и получают соединение формулы XXXII
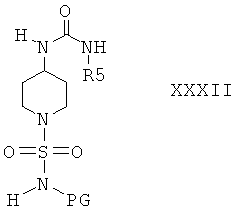
Защитную группу PG соединений формулы XXXII затем отщепляют при подходящих условиях и получают соединения формулы I. Если защитной группой является трет-бутоксикарбонил, то после этого удаление PG можно провести в кислой среде, предпочтительно в присутствии хлористого водорода. Если защитной группой является бензил, то удаление PG можно провести путем гидрирования, предпочтительно в присутствии водорода и катализатора, такого как Pd.
При необходимости полученные свободные основания соединений формулы I независимо от типа замещения с помощью R3 и R4 можно превратить в их физиологически приемлемые соли присоединения с кислотами или полученные соли присоединения с кислотами соединений формулы I независимо от типа замещения с помощью R3 и R4 можно превратить в свободные основания формулы I.
В еще одном варианте осуществления настоящее изобретение также относится к способу лечения или предупреждения глаукомы, эпилепсии, биполярных нарушений, мигрени, невропатической боли, ожирения, диабета типа II, метаболического синдрома, алкогольной зависимости и/или рака и сопутствующих им и/или вторичных заболеваний или патологических состояний у млекопитающих и людей, включающему введение нуждающемуся в нем субъекту терапевтически эффективного количества соединения общей формулы I или его физиологически совместимых солей присоединения с кислотами.
Ожирение в контексте настоящего изобретения включает любое увеличение количества жира в организме, которое приводит к увеличению массы тела, включая в качестве предпочтительной альтернативы, но не ограничиваясь только им медицинское определение ожирения. Таким образом, настоящее изобретение также относится к не обусловленному лечением уменьшению массы тела, такому как косметическое уменьшение массы тела, и включает улучшение внешнего вида тела в целом. Кроме того, термин ожирение также включает вызванное применением лекарственных средств ожирение и/или юношеское ожирение.
Заболевания, сопутствующие ожирению, и сопутствующие им и/или вторичные заболевания или патологические состояния у млекопитающих и людей в контексте настоящего изобретения, в частности, включают метаболический синдром и/или синдром Х и сердечно-сосудистые заболевания.
Термин "метаболический синдром" при использовании в настоящей заявке включает комплекс клинических проявлений, которые - наряду с центральным ожирением - в основном включает гипертензию, в частности артериальную гипертензию; резистентность к инсулину, в частности сахарный диабет типа II; непереносимость глюкозы; дислипопротеинемию, в частности, в виде гипертриглицеридемии, сопровождающейся дислипопротеинемией, возникающей при пониженном содержании ЛВП-холестерина (ЛВП - липопротеины высокой плотности), а также гиперурикемию, которая может привести к подагре. По информации, полученной от Американской ассоциации кардиологов, метаболический синдром тесно связан с резистентностью к инсулину. Некоторые люди генетические предрасположены к резистентности к инсулину. Приобретенные факторы, такие как увеличение количества жира в организме и отсутствие физической активности, у этих людей могут вызвать резистентность к инсулину и метаболический синдром. Большинство людей, у которых наблюдается резистентность к инсулину, страдают от центрального ожирения. На молекулярном уровне биологические механизмы, связывающие резистентность к инсулину и факторы риска метаболического синдрома, полностью не изучены и, видимо, являются комплексными. Одной группой людей, характеризующейся риском развития метаболического синдрома, является те страдающие диабетом, у которых наблюдается недостаточное воздействие инсулина и не может поддерживаться необходимая концентрация глюкозы в крови. Второй группой являются люди, в основном обладающие высоким артериальным давлением, которые не страдают диабетом и резистентностью к инсулину, но у которых происходит компенсация путем выработки большого количества инсулина. Это патологическое состояние известно, как гиперинсулинемия. Третьей группой являются перенесшие инфаркт миокарда, у которых, в отличие от страдающих гипертензией, наблюдается гиперинсулинемия без аномального содержания глюкозы. Метаболический синдром стал все более распространенным в США, где по оценкам им страдают примерно 20-25 взрослых. Надежно установленные критерии диагностики метаболического синдрома отсутствуют.
Критерии, предложенные в Третьем отчете Группы экспертов по диагностике, оценке и лечению высокого содержания холестерина в крови взрослых Национальной образовательной программы по профилактике атеросклероза (НОПА) (Группа экспертов по лечению взрослых (ГЛВ) III), являются самыми последними и широко применяются. В соответствии с критериями ГЛВ III метаболический синдром идентифицируется по наличию трех или большего количества следующих компонентов:
- центральное ожирение, характеризующееся обхватом талии для мужчин - более 40 дюймов; для женщин - более 35 дюймов;
- содержание триглицеридов в крови натощак, большее или равное 150 мг/дл;
- содержание ЛВП-холестерина в крови для мужчин - менее 40 мг/дл; для женщин - менее 50 мг/дл;
- артериальное давление, большее или равное 130/85 мм рт.ст.;
- содержание глюкозы натощак, большее или равное 110 мг/дл.
Термин "синдром X" тесно связан с термином "метаболический синдром" и обычно предполагается, что они означают одно и то же заболевание или патологическое состояние. Однако по информации, полученной от Американской ассоциации кардиологов Американской ассоциации кардиологов, термин "синдром X" дополнительно означает патологическое состояние сердца, при котором наблюдаются боль в груди и изменения электрокардиограммы, которые свидетельствуют об ишемической болезни сердца, но ангиография не обнаруживает ишемической болезни сердца. У пациентов, у которых наблюдается сердечный синдром X, иногда наблюдаются и липидные аномалии.
Термин "сердечно-сосудистые заболевания" в связи с ожирением обычно означает ишемическую болезнь сердца, которая может привести к сердечной недостаточности, цереброваскулярным заболеваниям, которая может, например, сопровождаться повышенным риском удара и облитерирующим артериитом.
Вследствие своих особых характеристик соединения общей формулы I или их физиологически совместимые соли присоединения с кислотами предположительно также могут быть применимы для лечения диабетических патологических состояний или заболеваний, которые не связаны с ожирением. Такие диабетические патологические состояния или заболевания включают, например, сахарный диабет типа II, диабетическую невропатию, диабетическую ретинопатию, диабетическую нефропатию, диабетическую микроангиопатию и диабетическую макроангиопатию.
Другими сопутствующими ожирению и/или вторичными заболеваниями могут быть заболевания желчного пузыря, такие как образование камней желчного пузыря, синдром апноэ во сне, ортопедические осложнения, такие как остеоартрит, и психосоциальные нарушения.
Соединения общей формулы I предположительно также могут быть применимы в качестве противосудорожных средств, предназначенных для профилактики или лечения эпилепсии у млекопитающих и людей.
Соединения общей формулы I, предлагаемые в настоящем изобретении, являются ингибиторами карбоангидраз млекопитающих, в частности изоферментов карбоангидраз человека подтипов II и/или V (hCA II и/или hCA V).
Методики фармакологических исследований
Номера примеров, приведенные в методиках фармакологических исследований, относятся к описанным ниже примерам получения.
1. Ингибирование изофермента карбоангидразы человека II (hCA II) in vitro Исследуемые соединения общей формулы I помещали в 96-луночные планшеты для микротитрования и с помощью автоматического устройства для пипетирования (CyBiWell®) разводили бидистиллированной водой. Из планшетов с разными разведениями аликвоты по 20 мкл с помощью устройства для пипетирования (Tecan Genesis®) переносили в 96-луночные черные планшеты для анализа. На второй стадии прибавляли 148 мкл калийфосфатного буфера (20 мМ, рН 7,4) и на третьей стадии 20 мкл раствора фермента (1 мкМ изофермента карбоангидразы человека II, полученного из эритроцитов (Sigma-Aldrich), растворенного в калийфосфатном буфере) инкубировали в течение 60 мин при комнатной температуре и в конце периода предварительной инкубации регистрировали флуоресцентный сигнал (FLU-1) (считывающее устройство для исследования флуоресценции Tecan Ultra®; длина волны возбуждения 280 нм; длина волны испускания 465 нм). После завершения предварительной инкубации прибавляли 20 мкл водного раствора дансиламида (1 мМ дансиламид (Sigma-Aldrich), растворенного в хлористоводородной кислоте), флуоресцентный сигнал регистрировали каждые 10 мин в течение 60 мин при 37°С. Для расчета использовали данные по флуоресценции для конца 60-минутного периода (FLU-2). Полный объем смеси для анализа составлял 208 мкл. Конечная концентрация карбоангидразы II составляла 10-7 М/л, дансиламида - 2,25×10-6 и соединений - от 10-8 М/л до 10-5 М/л. Конечная концентрация ДМСО (диметилсульфоксид), как растворителя соединения, составляла 0,1 мМ. В каждом планшете для микротитрования находились лунки с холостыми пробами, не содержащие соединения и фермента, контрольные лунки, не содержащие соединения и этоксзоламида (конечная концентрация 5×10-8 М/л). Все данные относятся к однократным исследованиям. Данные представлены в виде выраженного в % ингибирования, рассчитанного по формуле
ингибирование, %=100((1-(FLU-2соединение-FLU-2холостая проба-FLU-1соединение+FLUхолостая проба)/(FLU-2контроль-FLU-2холостая проба-FLU-1контроль-FLU-1холостая проба))
Данные по ингибированию в % для каждого соединения и соответствующие конечные концентрации использовали для расчета IC50 с помощью программного обеспечения Prism 4. Характеристики воздействия при разных концентрациях рассчитывали путем использования алгоритма Prism для нелинейного регрессионного анализа (аппроксимация кривой): сигмоидальная зависимость реакции от дозы с переменным наклоном и ограничениями: максимальное значение - 100 и минимальное - 0.
В этой модели исследования исследуемые соединения общей формулы I, приведенные в представленной ниже таблице 1, характеризуются указанными ниже значениями IC50:
2. Исследование непосредственного потребления корма мышами in vivo
Исследования проводили на самцах или самках мышей линии С57В1/6 (n=8-12 в группе). Мышей держали в обращенном цикле 12/12 ч освещение/затемнение (освещение включали в 22:00). Они в неограниченном количестве получали корм (высококалорийный кормовой рацион) и воду. Потребление корма и воды определяли ежедневно. Исследуемое соединение общей формулы I суспендировали в 1% водном растворе метилцеллюлозы и 2% (об./об.) Poloxamer 188 (Lutrol F68®) и вводили перорально через зонд в дозе 100 мг/кг/сутки. Половину дозы вводили в 7.00-9.00 ч; оставшуюся половину дозы вводили в период 15.00-15.30 ч.
В описанной выше модели исследования исследуемые соединения приводили к уменьшению 24-часового потребления корма животными, выраженному в процентах от потребления корма контрольной группой, приведенному в представленной ниже таблице 2.
Настоящее изобретение также относится к фармацевтической композиции или лекарственному средству, включающему фармакологически эффективное количество соединения общей формулы I или его физиологически совместимых солей присоединения с кислотами и дополнительно включающему фармацевтически приемлемые вспомогательные вещества и/или носители.
Подходящие фармацевтически приемлемые вспомогательные вещества и/или носители хорошо известны в данной области техники и включают фармацевтические марки крахмала, маннита, лактозы, стеарата магния, натриевой соли сахарина, талька, целлюлозы, глюкозы, сахарозы (или другого сахара), карбоната магния, желатина, масла, спирта, моющих средств, эмульгаторов или воды (предпочтительно стерильные). Композиция может являться смешанным препаратом или может являться комбинированным препаратом, предназначенным для одновременного, раздельного или последовательного применения (включая введение). Соединения, предлагаемые в настоящем изобретении, или их физиологически совместимые соли присоединения с кислотами при использовании в случае указанных выше показаний можно вводить по любому обычному пути, например перорально (в том числе ингаляционно), парентерально, в слизистую оболочку (например, трансбуккально, сублингвально, назально), ректально или чрескожно и композиции соответствующим образом изменяют. Для перорального введения соединения можно приготовить в виде жидкостей или твердых веществ, например растворов, сиропов, суспензий или эмульсий, таблеток, капсул и лепешек. Жидкий препарат обычно представляет собой суспензию или раствор соединения или физиологически приемлемой соли в подходящем водном или неводном жидком носителе (носителях), например в воде, этаноле, глицерине, полиэтиленгликоле или в масле.
Препарат также может содержать суспендирующий агент, консервант, ароматизатор или окрашивающий агент. Композицию в форме таблетки можно приготовить с использованием любого подходящего фармацевтического носителя (носителей), обычно применяющегося для приготовления твердых препаратов. Примеры таких носителей включают стеарат магния, крахмал, лактозу, сахарозу и микрокристаллическую целлюлозу. Композицию в форме капсулы можно приготовить с использованием стандартных методик капсулирования. Например, порошки, гранулы или пеллеты, содержащие активный ингредиент, можно приготовить с использованием стандартных носителей и затем поместить в капсулу из твердого желатина; альтернативно, дисперсию или суспензию можно приготовить с использованием любого подходящего фармацевтического носителя (носителей), например водных камедей, целлюлоз, силикатов или масел, и затем дисперсию или суспензию поместить в капсулу из мягкого желатина. Композиции для перорального введения можно приготовить так, чтобы обеспечить защиту активного ингредиента от разложения, когда она проходит через пищеварительный тракт, например, путем нанесения наружного покрытия на таблетку или капсулу. Типичные композиции для парентерального введения содержат раствор или суспензию соединения или физиологически совместимых солей присоединения с кислотами в стерильном водном или неводном носителе или применимом для парентерального введения масле, например, полиэтиленгликоле, поливинилпирролидоне, лецитине, арахисовом масле или кунжутном масле. Альтернативно, раствор можно лиофилизировать и затем непосредственно перед введением восстановить с помощью подходящего растворителя. Композиции для назального или перорального введения обычно можно приготовить в виде аэрозолей, капель, гелей и порошков.
Аэрозольные препараты обычно представляют собой раствор или мелкодисперсную суспензию активного вещества в физиологически приемлемом водном или неводном растворителе и обычно содержатся в виде одной дозы или множества доз в стерильной форме в герметичном контейнере, который может представлять собой емкость или повторно заполняемую емкость, предназначенную для использования в распыляющем устройстве. Альтернативно, герметичный контейнер может представлять собой единое дозирующее устройство, такое как одноразовый назальный ингалятор или аэрозольное дозирующее устройство, снабженное дозирующим клапаном, которое выбрасывают после израсходования содержимого контейнера. Если дозированная форма представляет собой аэрозольное дозирующее устройство, то она содержит фармацевтически приемлемый пропеллент. Аэрозольные дозированные формы также могут представлять собой распыляющие устройства с насосами. Композиции, пригодные для трансбуккального или сублингвального введения, включают таблетки, лепешки и пастилки, в которых активный ингредиент объединен с носителями, такими как сахар или камедь акации, трагакантовая камедь или желатин, или глицерин. Композиции для ректального или вагинального введения обычно представляют собой суппозитории (содержащие обычную основу суппозитория, такую как масло какао), пессарии, вагинальные таблетки, вспененные вещества или композиции для спринцевания. Композиции, пригодные для чрескожного введения, включают мази, гели, пластыри и композиции для инъекции, включая порошкообразные композиции. Обычно композиция находится в форме разовой дозы, такой как таблетка, капсула или ампула. Фармацевтические композиции, предлагаемые в настоящем изобретении, применимы для лечения и/или профилактики и/или предупреждения глаукомы, эпилепсии, биполярных нарушений, мигрени, невропатической боли, ожирения, диабета типа II, метаболического синдрома, алкогольной зависимости и/или рака и сопутствующих им и/или вторичных заболеваний или патологических состояний; другого обусловленного лечением уменьшения массы тела и не обусловленного лечением уменьшения массы; и/или диабетических патологических состояний или заболеваний.
Соединения, предлагаемые в настоящем изобретении, и их физиологически совместимые соли присоединения с кислотами обычно вводят в виде фармацевтических композиций, которые являются важными и новыми вариантами осуществления настоящего изобретения вследствие наличия соединений, раскрытых в настоящем изобретении. Варианты осуществления настоящего изобретения относятся к фармацевтической упаковке или набору, содержащему один или большее количество емкостей, заполненных одним или большим количеством ингредиентов фармацевтической композиции, предлагаемой в настоящем изобретении. Совместно с такой емкостью (емкостями) могут находиться различные печатные материалы, такие как инструкции по применению или примечания в форме, установленной правительственным агентством, регулирующим изготовление, применение или продажу фармацевтических продуктов, и в этом примечании сообщается об утверждении агентством изготовления, применения или продажи для использования в медицине или ветеринарии.
В еще одном варианте осуществления настоящее изобретение относится к способу приготовления фармацевтической композиции, описанной выше в настоящем изобретении. Приготовление можно проводить по стандартным технологиям, хорошо известным в данной области техники, и они включают объединение соединения, предлагаемого в настоящем изобретении, и фармацевтически приемлемых вспомогательных веществ и/или носителей. Композиция может находиться в любой форме, включая таблетку, жидкость, капсулу и порошок, или в форме пищевого продукта, например специализированного пищевого продукта. В последнем случае сам пищевой продукт может выступать в качестве фармацевтически приемлемого носителя.
Соединение или композицию предпочтительно вводить нуждающемуся в нем пациенту и в количестве, достаточном для предупреждения и/или устранения симптомов патологического состояния, нарушения или заболевания. Во всех вариантах осуществления настоящего изобретения, особенно лечебных, введение соединения или композиции характеризуется режимом дозирования, который в конечном счете определяет лечащий врач, который принимает во внимание такие факторы, как применяющееся соединение, вид животного, возраст, массу тела, тяжесть симптомов, путь введения, побочные эффекты и/или другие противопоказания. Конкретные диапазоны доз можно установить с помощью стандартных схем клинических исследований, при которых полностью регистрируется прогресс для пациента и его возвращение к норме. При таких исследованиях можно использовать режим повышения дозы, в котором для человека в качестве начальной дозы используют дозу, составляющую несколько процентов от максимальной переносимой дозы для животного. Физиологически приемлемые соединения, предлагаемые в настоящем изобретении, обычно вводят в суточном дозировочном режиме (для взрослого пациента), например, пероральную дозу, равную от 1 до 2000 мг, предпочтительно от 30 до 1000 мг, например, от 10 до 250 мг, или внутривенную, подкожную или внутримышечную дозу, равную от 0,1 до 100 мг, предпочтительно от 0,1 до 50 мг, например от 1 до 25 мг соединения общей формулы I или его физиологически приемлемой соли в пересчете на свободное основание вводят от 1 до 4 раз в сутки. Соединение, применяющееся в контексте настоящего изобретения, также можно вводить детям или подросткам и в этих случаях врач должен особенно тщательно подбирать индивидуальный режим дозирования и обычно используются меньшие дозы, чем вводимые взрослым.
Соединения предпочтительно вводить в режиме непрерывного лечения, например, в течение не менее недели, а обычно в течение более длительного времени, составляющего от нескольких недель до нескольких месяцев. Настоящее изобретение также относится к косметическому (не являющемуся лечебным) способу, предназначенному для поддержания имеющейся массы тела или для косметического уменьшения массы тела, способ включает введение соединения, соответствующего другим вариантам осуществления настоящего изобретения, предпочтительно в комбинации с фармацевтически приемлемым носителем или разбавителем.
Соединение или композицию предпочтительно вводить нуждающемуся в нем или испытывающему в нем недостаток субъекту и в количестве, достаточном для поддержания имеющейся массы тела или для косметического уменьшения массы тела.
В еще одном варианте осуществления соединения общей формулы I и их физиологически совместимые соли присоединения с кислотами с успехом можно вводить в комбинации с одним или большим количеством активных средств (в виде комбинированной фармацевтической композиции), выбранных из группы, включающей противодиабетические средства; средства против ожирения или средства, регулирующие аппетит; активные сердечно-сосудистые средства, в частности гипотензивные средства; диуретики; активные средства, изменяющие содержание липидов, в частности средства, снижающие содержание липидов; и активные ингредиенты, предназначенные для лечения и/или предупреждения осложнений, вызванных диабетом или связанных с диабетом.
Подходящие противодиабетические средства включают, например, инсулины, амилин, производные GLP-1 и GLP-2, такие как, например, описанные в WO 98/08871 и активные при пероральном введении гипогликемически активные ингредиенты. Активные при пероральном введении гипогликемически активные ингредиенты предпочтительно включают сульфонилмочевины, например толбутамид, глибенкламид, глимепирид, глипизид, глихидон, глизоксепид, глибомурил или гликлазид; бигуаниды, например метформин; меглитиниды, например репаглинид; бета-3 адрегергические агонисты; оксадиазолидиндионы; ингибиторы глюкозидазы, например ингибиторы альфа-глюкозидазы, такие как миглитол или акарбоза; антагонисты глюкагонового рецептора, агонисты GLP-1, активаторы калиевых каналов, такие как диазоксид или описанные в WO 97/26265 или WO 99/03861; антагонисты/обратные агонисты СВ-1 (каннабиноидного рецептора 1); сенсибилизаторы воздействия инсулина, такие как тиазолидиндионы, например троглитазон, циглитазон, пиоглитазон, розиглитазон или соединения, описанные в WO 97/41097, в частности 5-[[4-[(3,4-дигидро-3-метил-4-оксо-2-хиназолинилметокси]фенил]метил]-2,4-тиазолидиндион; активаторы киназы инсулинового рецептора; ингибиторы ферментов печени, участвующих в стимулировании глюконеогенеза и/или гликогенолиза, например ингибиторы гликогенфосфорилазы; и модуляторы поглощения глюкозы и выведения глюкозы.
Подходящие средства против ожирения или средства, регулирующие аппетит, включают одно или большее количество из следующих: ингибитор переноса 5-НТ (серотонин), ингибитор переноса NE (норэпинефрин), антагонист/обратный агонист СВ-1 (каннабиноидного рецептора 1), антитело к грелину, антагонист грелина, антагонист/обратный агонист Н3 (гистамина Н3), антагонист MCH1R (меламинконцентрирующего гормона 1R), антагонист/обратный агонист MCH2R (меламинконцентрирующего гормона 2R), антагонист NPY1 (нейропептид Y Y1), агонист NPY2 (нейропептид Y Y2), антагонист NPY5 (нейропептид Y Y5), лептин, производное лептина, опиоидный антагонист, антагонист орексина, агонист BRS3 (бомбезинового рецептора подтипа 3), агонист ССК-А (холецистокинин-А), CNTF (цилиарный нейротропный фактор), производное CNTF, агонист GHS (рецептор, усиливающий секрецию гормона роста), агонист SHT2c (серотониновый рецептор 2с), агонист Мс3r (меланокортиновый рецептор 3), агонист Мс4r (меланокортиновый рецептор 4), ингибитор повторного всасывания моноамина, ингибитор повторного всасывания серотонина, агонист GLP-1 (глюкагоноподобный пептид 1), топираман, фитофатм - соединение 57, ингибитор АСС2 (ацетил-СоАкарбоксилаза 2), бета-3 адрегергический агонист, ингибитор DGAT1 (диацилглицеринацилтрансфераза 1), ингибитор DGAT2 (диацилглицеринацилтрансфераза 2), ингибитор FAS (синтаза жирной кислоты), ингибитор PDE (фосфодиэстераза), агонист тиреоидного гормона В, активатор UCP-1 (разобщающий белок 1), 2 или 3, ацилэстроген, глюкокортикоидный (glucocorticoid) антагонист, ингибитор 11 HSD-1 (11-бетагидроксистероиддегидрогеназа типа 1), ингибитор SCD-1 (стеароил-СоА-десатураза 1), ингибитор дипептидилпептидазы IV (DP-IV), ингибитор липазы, ингибитор переноса жирных кислот, ингибитор переноса дикарбоксилата, ингибитор переноса глюкозы, ингибитор переноса фосфата и их фармацевтически приемлемые соли и сложные эфиры.
Подходящие средства, регулирующие аппетит (средства подавления аппетита) включают сибутрамин и моно- и бисметилированные активные метаболиты сибутрамина; фенфлурамин или дексфенфлурамин; мазиндол, диэтилпропион или фентермин; лептин или модифицированный лептин; дексамфетамин и амфетамин.
Подходящие ингибиторы липазы включают орлистат, панклицины, ингибиторы липазы, выделенные из микроорганизмов, такие как липстатин (из Streptomyces toxytricini), эбелактон В (из Streptomyces aburaviensis), синтетические производные этих соединений; производные 2-окси-4Н-3,1-бензоксазин-4-она, такие как Alizyme's ATL-962 или структурно родственные соединения; производные 2-амино-4Н-3,1-бензоксазин-4-она или экстракты растений, для которых известно, что они обладают способностью ингибировать липазу, например экстракты Alpinia officinarum или соединения, выделенные из таких экстрактов, такие как 3-метиловый эфир галангина (из A. officinarum).
Подходящие CB1-каннабиноидные антагонисты включают римонабант, SLV319, SR147778 и СР-945598.
Подходящие активные сердечно-сосудистые средства включают антагонисты ангиотензинового рецептора II, например абитесартан, бензиллосартан, кардесартан, элисартан, эмбусартан, енолтазосартан, эпросартан, фонсартан, фора, глициллосартан, ирбесартан, изотеолин, лосартан, милфасартан, олмесартан, опомисартан, пратосартан, риписартан, саприсартан, саралазин, сармезин, тазосартан, телмисартан, валсартан, золасартан; Kissei KRH-94, Lusofarmaco LR-B/057, Lusofarmaco LR-B/081, Lusofarmaco LR B/087, Searle SC-52458, Sankyo CS-866, Takeda TAK-536, Uriach UR-7247, A-81282, A-81988, BIBR-363, BIBS39, BIBS-222, BMS-180560, BMS-184698, CGP-38560A, CGP-48369, CGP-49870, CGP-63170, CI-996, CV-11194, DA-2079, DE-3489, DMP-811, DuP-167, DuP-532, GA-0056, E-4177, EMD-66397, EMD-73495, EXP-063, EXP-929, EXP-3174, EXP-6155, EXP-6803, EXP-7711, EXP-9270, FK-739, HN-65021, HR-720, ICI-D6888, ICI-D7155, ICI-D8731, KRI-1177, KT3-671, KW-3433, L-158809, L-158978, L-159282, L-159689, L-159874, L-161177, L-162154, L-162234, L-162441, L-163007, L-163017, LY-235656, LY-285434, LY-301875, LY-302289, LY-315995, ME-3221, PD-123177, PD-123319, PD-150304, RG-13647, RWJ-38970, RWJ-46458, S-8307, S-8308, SL-91.0102, U-96849, U-97018, UP-269-6, UP-275-22, WAY-126227, WK-1492.2K, WK-1360, X-6803, XH-148, XR-510, YM-358, YM-31472, ZD-6888, ZD-7155 и ZD-8731 или их любые физиологически совместимые соли, сольваты, пролекарства или сложные эфиры; даглутрил; неселективные антагонисты альфа-адренорецептора, например толазолин или феноксибензамин; селективные антагонисты альфа-адренорецептора, например доксазозин, празозин, теразозин или урапидил; антагонисты бета-адренорецептора, например, ацебутолол, алпренолол, атенолол, бетаксолол, бисопролол, бупранолол, каразолол, картеолол, целипролол, мепиндолол, метипранолол, надолол, окспренолол, пенбутолол, пиндолол, пропранолол, соталол и тимолол; смешанные антагонисты альфа- и бета-адренорецепторов, например карведилол или лабетолол; ганглиоблокаторы, например резерпин или гванетидин; агонисты альфа-2-адренорецептора (включая агонисты альфа-адренорецептора центрального действия), например клонидин, гуанфацин, гуанабенз, метилдопа и моксонидин; ингибиторы ренина, например алискирен; ингибиторы АСЕ, например беназеприл, каптоприл, цилазаприл, эналаприл, фозиноприл, имидаприл, лизиноприл, моэксиприл, хинаприл, периндоприл, рамиприл, спираприл или трандолаприл; антагонисты эндотелинового рецептора смешанного или селективного действия, например атрасентан, босентан, клазосентан, дарусентан, ситакссентан, тезосентан, BMS-193884 или J-104132; сосудорасширяющие средства прямого действия, например диазоксид, дигидралазин, гидралазин или миноксидил; смешанные ингибиторы ACE/NEP, например омпатрилат; ингибиторы ЕСЕ, например FR-901533; PD-069185; CGS-26303; CGS-34043; CGS-35066; CGS-30084; CGS-35066; SM-19712; Ro0677447; селективные ингибиторы NEP; антагонисты вазопрессина, антагонисты альдостеронового рецептора, например эплеренон или спиронолактон; ангиотензиновая вакцина; и антагонисты уротензинового рецептора II.
Подходящие диуретики включают тиазидные диуретики, например алтиазид, беметизид, бендрофлуметиазид, бензилгидрохлортиазид, бензтиазид, бутиазид, хлортиазид, циклотиазид, гидрохлортиазид, гидрофлуметиазид, метиклотиазид, парафлутизид, политиазид, теклотиазид, трихлорметиазид; диуретики - аналоги тиазида, например хлораминофенамид, хлорталидон, клофенамид, клопамид, клорексолон, фенхизон, индапамид, мефрузид, метолазон, хинетазон, трипамид, ксипамид; петлевые диуретики, например азосемид, буметанид, фуросемид, пиретанид, торсемид; калийсберегающие диуретики, например амилорид, канреноат калия, спиронолактон, триамтрен или любые физиологически совместимые таутомеры, соли, сольваты, пролекарства или сложные эфиры любого из указанных выше диуретиков.
Подходящие активные средства, которые изменяют содержание липидов, включают соединения, которые изменяют метаболизм липидов, такие как антигиперлипидемически активные ингредиенты и антилипидемически активные ингредиенты, такие как ингибиторы HMGCoA редуктазы, например аторвастатин, беривастатин, церивастатин, крилвастатин, флувастатин, гленвастатин, ловастатин, мевастатин, питавастатин, правастатин, росувастатин, симвастатин или их любые физиологически совместимые соли, сольваты, пролекарства или сложные эфиры; ингибиторы переноса холестерина и/или всасывания холестерина; ингибиторы повторного всасывания желчных кислот или ингибиторы переноса триглицерида микросомного белка (МТР); соединения, которые уменьшают потребление пищи, агонисты PPAR (активированного рецептора пролифератора пероксисом) и RXR и активные средства, которые воздействуют на АТР-зависимые калиевые каналы бета-клеток; фибриновые кислоты, например безафибрат, ципрофибрат, клофибрат, фенофибрат или гемфиброзил; холестирамин, колестипол, пробукол, эзетимиб и декстротироксин; ингибитор HMGCoA синтазы, ингибитор всасывания холестерина, ингибитор ацил-кофермент-А-холестеринацилтрансферазы (АСАТ), ингибитор белка - переносчика эфиров холестерина (СЕТР), ингибитор скваленсинтетазы, антиоксидант, агонист PPARa, модулятор рецептора FXR, агонист рецептора LXR, ингибитор синтеза липопротеинов, ингибитор ренин-ангиотензиновой системы, ингибитор переноса триглицеридов микросом, ингибитор повторного всасывания желчных кислот, агонист PEAR8, ингибитор синтеза триглицеридов, модулятор транскрипции, ингибитор скваленэпоксидазы, индуктор рецептора липопротеинов низкой плотности, ингибитор агрегации тромбоцитов, ингибитор 5-LO или FLAP, частичный агонист PPAR 8 и агонист ниацина или ниацинового рецептора и его фармацевтически приемлемые соли и сложные эфиры.
Другие активные средства, которые могут быть подходящими для применения в комбинации с соединением общей формулы I, предлагаемым в настоящем изобретении, можно выбрать из группы, включающей агонисты CART, антагонисты Н3, агонисты TNF, агонисты CRF, антагонисты CRF ВР, агонисты урокортина, агонисты бета-3, агонисты MSH (меланоцитостимулирующий гормон), ингибиторы повторного всасывания серотонина, смешанные ингибиторы повторного всасывания серотонина и норадреналина, модуляторы 5НТ, ингибиторы МАО, антагонисты галанина, гормоны роста, соединения, высвобождающие гормоны роста, агонисты TRH, модуляторы разобщающих белков 2 или 3, агонисты лелптина, агонисты допамина (бромокриптин, допрексин), модуляторы RXR, hCNTF и агонисты TR-бета.
Предпочтительные комбинированные фармацевтические композиции, предлагаемые в настоящем изобретении, включают комбинации по меньшей мере одного соединения общей формулы I и по меньшей мере одного бигуанида; по меньшей мере одного соединения общей формулы I и по меньшей мере одной фибриновой кислоты; по меньшей мере одного соединения общей формулы I и по меньшей мере одного ингибитора HMGCoA редуктазы; и по меньшей мере одного соединения общей формулы I и по меньшей мере одного сенсибилизатора воздействия инсулина.
Предпочтительными соединениями общей формулы I для комбинирования с одним или большим количеством указанных активных средств являются амид 4-фенилпиперазин-1-сульфоновой кислоты; амид 4-(2-хлорфенил)-пиперазин-1-сульфоновой кислоты; амид 4-(2-метоксифенил)-пиперазин-1-сульфоновой кислоты; амид 4-пиридин-4-илпиперазин-1-сульфоновой кислоты; амид 4-пиримидин-2-илпиперазин-1-сульфоновой кислоты; амид 4-(4-фторфенил)-пиперазин-1-сульфоновой кислоты; амид 4-(4-хлор-3-трифторметилфенил)-пиперазин-1-сульфоновой кислоты и/или амид 4-(3-хлор-5-трифторметилпиридин-2-ил)-пиперазин-1-сульфоновой кислоты.
Метформин является предпочтительным бигуанидом для комбинации по меньшей мере с одним соединением общей формулы I.
Предпочтительными фибриновыми кислотами для комбинации по меньшей мере с одним соединением общей формулы I являются безафибрат, ципрофибрат, клофибрат, фенофибрат и/или гемфиброзил. Фенофибрат является наиболее предпочтительным.
Предпочтительными ингибиторами HMGCoA редуктазы для комбинации по меньшей мере с одним соединением общей формулы I являются аторвастатин, беривастатин, церивастатин, крилвастатин, флувастатин, гленвастатин, ловастатин, мевастатин, питавастатин, правастатин, росувастатин и/или симвастатин или их любые физиологически совместимые соли, сольваты, пролекарства или сложные эфиры. Наиболее предпочтительными являются симвастатин, ловастатин и/или правастатин.
Предпочтительными сенсибилизаторами воздействия инсулина для комбинации по меньшей мере с одним соединением общей формулы I являются тиазолидиндионы, предпочтительно троглитазон, циглитазон, пиоглитазон и/или розиглитазон. Розиглитазон и пиоглитазон являются наиболее предпочтительными.
Более предпочтительными комбинациями, предлагаемыми в настоящем изобретении, являются комбинации амида 4-фенилпиперазин-1-сульфоновой кислоты с метформином; амида 4-фенилпиперазин-1-сульфоновой кислоты с фенофибратом; амида 4-фенилпиперазин-1-сульфоновой кислоты с симвастатином и амида 4-фенилпиперазин-1-сульфоновой кислоты с розиглитазоном.
В одном варианте осуществления комбинированных фармацевтических композиций, описанных выше и предлагаемых в настоящем изобретении, соединения общей формулы I можно применять и вводить вместе с разными активными средствами, например в одной комбинированной разовой дозированной форме, такой как одна таблетка или капсула, т.е. в виде физической комбинации. В такой комбинированной разовой дозированной форме соединение общей формулы I и разные активные средства можно отделить друг от друга, например, с использованием разных слоев в указанной таблетке, например, путем использования инертных промежуточных слоев, известных в данной области техники; или с использованием разных отделений в указанной капсуле. Соответствующие активные средства или их фармацевтически приемлемые соли также можно применять в форме из гидратов или с включением других растворителей, использовавшихся при кристаллизации. Разовая дозированная форма может представлять собой фиксированную комбинацию. Разовая дозированная форма, предпочтительно фиксированная комбинация соединения общей формулы I и одного или большего количества активных средств, является предпочтительной альтернативой этого варианта осуществления.
В другом варианте осуществления соединения общей формулы I и разные активные средства можно применять и вводить в виде двух или большего количества отдельных разовых дозированных форм, например в виде двух или большего количества таблеток или капсул и таблетки или капсулы физически отделены друг от друга. Две или большее количество отдельных разовых дозированных форм можно вводить одновременно или последовательно (по отдельности), например последовательно одну после другой в любом порядке. Таким образом, соединения общей формулы I и разные активные средства можно вводить в любом порядке в одно и то же время или в разные моменты времени в течение суток, оптимальный дозированный режим обычно определяется предписанием врача.
Приведенные ниже примеры предназначены для разъяснения настоящего изобретения без ограничения его объема.
Пример 1
Аналоги мочевины (R1=Н, R2=CO-NH-C6H4F; boc=трет-бутоксикарбонил)
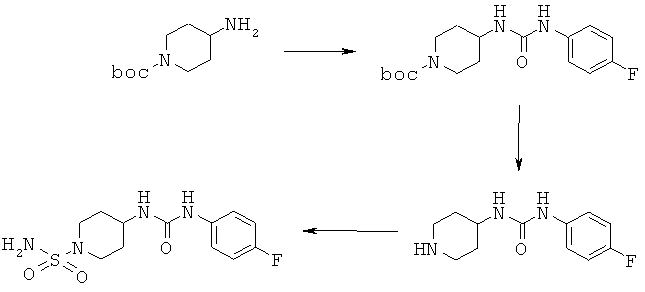
К охлаждаемому льдом раствору 0,7 г 1-фтор-4-изоцианатобензола в 25 мл дихлорметана в атмосфере азота по каплям прибавляли раствор 1,0 г трет-бутил-4-аминопиперидин-1-карбоксилата в 25 мл дихлорметана. Эту смесь перемешивали в течение 10 мин при 0°С и затем в течение 16 ч при комнатной температуре. Реакционную смесь разбавляли водой и затем промывали насыщенным водным раствором NaCl. После отделения органический слой сушили над сульфатом натрия и выпаривали при пониженном давлении и получали 1,6 г трет-бутил-4-{[[(4-фторфенил)-амино]-карбонил]амино}-пиперидин-1-карбоксилата.
1H ЯМР (δ част./млн, 400 МГц): 7,37 [2Н], 7,04 [2Н], 8,34 [1Н, NH], 6,13 [1Н, NH], 1,40 [9Н].
1,6 г трет-бутил-4-{[[(4-фторфенил)амино]карбонил]амино}пиперидин-1-карбоксилата растворяли в 50 мл дихлорметана. Этот раствор охлаждали до 0°С и затем прибавляли 2,3 мл трифторуксусной кислоты. После перемешивания в течение 40 ч при комнатной температуре реакционную смесь выпаривали при пониженном давлении и дважды обрабатывали толуолом. После сушки в вакууме получали 2,4 г 1-(4-фторфенил)-3-пиперидин-4-илмочевины в виде трифторацетата.
1Н ЯМР (δ част./млн, 400 МГц): 7,39 [2Н], 7,05 [2Н], 8,51 [1Н, NH], 6,56 [1H, NH].
2,4 г трифторацетата 1-(4-фторфенил)-3-пиперидин-4-илмочевины, 7 мл триэтиламина и 1,17 г сульфамида растворяли в 70 мл диоксана и кипятили с обратным холодильником (130°С) в течение 5 ч. После удаления растворителя при пониженном давлении неочищенный продукт очищали с помощью флэш-хроматографии с использованием 9:1 смеси дихлорметан/метанол в качестве элюента. Получали 1,0 г 4-{[[(4-фторфенил)амино]-карбонил]амино}пиперидин-1-сульфонамида, температура плавления 219,6-221,2°С.
Пример 2
Аналоги мочевины (R1=СН2С6Н5, R2=СО-NH-С6Н5; boc=трет-бутоксикарбонил)
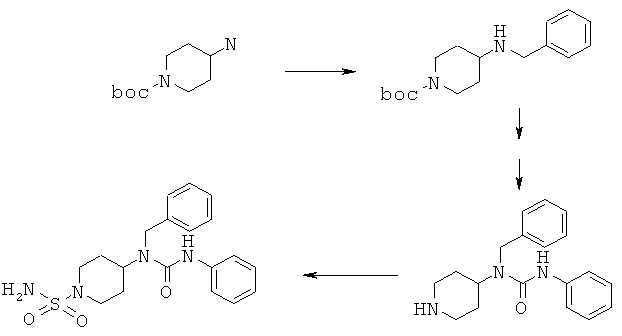
2.1 2,6 г ацетата натрия, 5,0 г трет-бутил-4-аминопиперидин-1-карбоксилата, 2,0 мл уксусной кислоты и 2,1 мл бензальдегида объединяли в 200 мл ТГФ (тетрагидрофуран) и перемешивали в течение 4 ч при комнатной температуре. После прибавления 8,8 г трисацетоксиборогидрида натрия смесь перемешивали в течение 20 ч. Затем растворитель удаляли при пониженном давлении и остаток растворяли в смеси метил-трет-бутилового эфира и воды. Водный слой подщелачивали с помощью NaOH и дважды экстрагировали метил-трет-бутиловым эфиром. Объединенные органические слои несколько раз промывали с помощью 30 мл 0,1 н. HCl и 5 раз с помощью 50 мл 0,1 н. HCl. Затем водные слои объединяли и подщелачивали с помощью NaOH, а затем 2 раза экстрагировали метил-трет-бутиловым эфиром. Органические слои промывали водой и насыщенным водным раствором NaCl, сушили над сульфатом натрия и затем выпаривали в вакууме. Получали 4,9 г трет-бутил 4-(бензиламино)пиперидин-1-карбоксилата в виде маслообразного продукта.
1H ЯМР (δ част./млн, 400 МГц): 3,82 [s, 2Н], 4,01 [2Н], 2,81 [2Н], 2,67 [1Н], 1,86 [2H], 1,30 [2H], 1,45 [s,9H].
К раствору 0,187 мл фенилизоцианата в 25 мл дихлорметана при охлаждении льдом по каплям прибавляли раствор 0,5 г трет-бутил 4-(бензиламино)пиперидин-1-карбоксилата в 20 мл дихлорметана. После 5 ч перемешивания при комнатной температуре реакционную смесь 3 раза промывали водой и насыщенным водным раствором NaCl. Органический слой отделяли, сушили над сульфатом натрия и выпаривали в вакууме. Неочищенный продукт кристаллизовали из смеси этилацетат/н-гексан в холодильнике и получали 0,55 г трет-бутил-4-[(анилинокарбонил)(бензил)амино]пиперидин-1-карбоксилата.
1Н ЯМР (δ част./млн, 400 МГц): 4,61 [1Н], 4,46 [s, 2H], 4,21 [2H], 2,83 [2H], 2,67 [1Н], 1,83 [2H], 1,56 [2H], 1,44 [s, 9H].
2.3 При охлаждении льдом 0,94 мл трифторуксусной кислоты прибавляли к раствору 0,5 г трет-бутил-4-[(анилинокарбонил)(бензил)амино]пиперидин-1-карбоксилата в 50 мл дихлорметана. После перемешивания в течение 16 ч при комнатной температуре реакционную смесь выпаривали при пониженном давлении и остаток растворяли в смеси метил-трет-бутилового эфира и воды. Водный слой подщелачивали путем прибавления раствора NaOH и 3 раза экстрагировали метил-трет-бутиловым эфиром. Объединенные слои, содержащие метил-трет-бутиловый эфир, промывали насыщенным водным раствором NaCl, сушили над сульфатом натрия и затем выпаривали в вакууме, и получали 0,23 г 1-бензил-3-фенил-1-пиперидин-4-илмочевины.
1Н ЯМР (δ част./млн, 400 МГц): 4,52 [1Н], 4,52 [s, 2H], 3,13 [2H], 2,75 [2H], 1,84 [2H], 1,59 [2H].
2.4 0,2 г 1-бензил-3-фенил-1-пиперидин-4-илмочевины и 0,075 г сульфамида растворяли в 50 мл диоксана и кипятили с обратным холодильником в течение 5 ч. Затем реакционную смесь концентрировали в вакууме и остаток растворяли в воде. После перемешивания в течение 3 ч остаток отделяли фильтрованием и перемешивали с метил-трет-бутиловым эфиром. Фильтрование и сушка в вакууме давали 0,23 г неочищенного продукта, который очищали с помощью флэш-хроматографии с использованием смеси дихлорметан/метанол (19:1) в качестве элюента. Выделяли 50 мг чистого 4-[(анилинокарбонил)-(бензил)амино] пиперидин-1-сульфонамида (температура плавления 188-189°С).
Пример 3
Замещенные амиды (R1=СН3; R2=С6Н11;4 boc=трет-бутоксикарбонил)
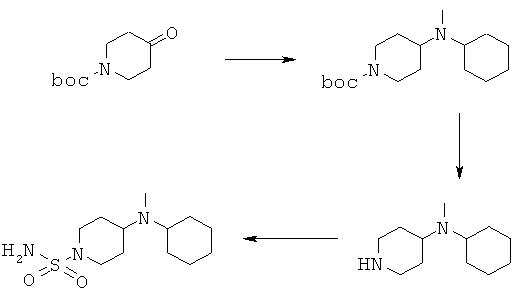
3.1 2,0 г трет-бутил 4-оксопиперидин-1-карбоксилата, 1,23 г ацетата натрия, 0,98 мл уксусной кислоты и 1,56 мл N-метилциклогексиламина растворяли в 100 мл ТГФ и перемешивали в течение 1 ч при комнатной температуре. Затем прибавляли 4,25 г трисацетоксиборогидрида натрия и реакционную смесь перемешивали в течение 18 ч при комнатной температуре. Реакционную смесь концентрировали при пониженном давлении и остаток растворяли в смеси воды и метил-трет-бутилового эфира. Водный слой подщелачивали и дважды экстрагировали метил-трет-бутиловым эфиром. В заключение органический слой 2 раза промывали с помощью 0,1 н. HCl, водные слои объединяли и подщелачивали (рН 10) путем прибавления раствора NaOH. После экстракции (2 раза) метил-трет-бутиловым эфиром органический слой сушили над сульфатом натрия и выпаривали в вакууме. Выделяли 1,3 г маслообразного трет-бутил 4-[циклогексил(метил)амино]пиперидин-1-карбоксилата и использовали без дополнительной очистки.
1H ЯМР (δ част./млн, 400 МГц): 4,12 [2Н], 2,69 [2Н], 2,63 [1Н], 2,50 [1Н], 2,23 [s, 3H], 1,45 [s, 9H].
3.2 1,25 г трет-бутил 4-[циклогексил(метил)амино] пиперидин-1-карбоксилата растворяли в 100 мл дихлорметана, обрабатывали с помощью 3,0 мл трифторуксусной кислоты и выдерживали в течение 2 дней при комнатной температуре. Затем реакционную смесь концентрировали при пониженном давлении и остаток растворяли в смеси воды и метил-трет-бутилового эфира. Водный слой насыщали хлоридом натрия и 3 раза экстрагировали метил-трет-бутиловым эфиром. Органический слой сушили над сульфатом натрия и растворитель удаляли в вакууме, и получали 0,8 г N-циклогексил-N-метилпиперидин-4-амина, который использовали без дополнительной очистки.
1Н ЯМР (δ част./млн, 400 МГц): 3,12 [2Н], 2,60 [1Н], 2,59 [2Н], 2,53 [1Н], 2,26 [s, 3Н].
3.3 0,8 г N-циклогексил-N-метилпиперидин-4-амина и 0,47 г сульфамида растворяли в 70 мл диоксана и кипятили с обратным холодильником в течение 3 ч. Затем реакционную смесь концентрировали при пониженном давлении и остаток растворяли в смеси воды и метил-трет-бутилового эфира. Органический слой промывали водой и насыщенным водным раствором NaCl, сушили над сульфатом натрия и затем выпаривали в вакууме и получали 0,7 г 4-[циклогексил(метил)амино]пиперидин-1-сульфонамида.
1Н ЯМР (δ част./млн, 400 МГц): 6,66 [s, 2Н], 3,44 [2Н], 2,52 [2Н], 2,48 [1Н], 2,48 [1H], 2,15 [s, 3H].
Соль с хлористоводородной кислотой получали путем обработки амина с помощью HCl в изопропаноле и с последующим выпариванием растворителя. Температура плавления найдена равной 240-242°С.
1Н ЯМР (δ част./млн, 400 МГц): 6,86 [2Н], 3,34 [2Н], 3,32 [s, 3Н].
Пример 4
Замещенные амиды (R1=Н; R2=С6Н11; boc=трет-бутоксикарбонил)
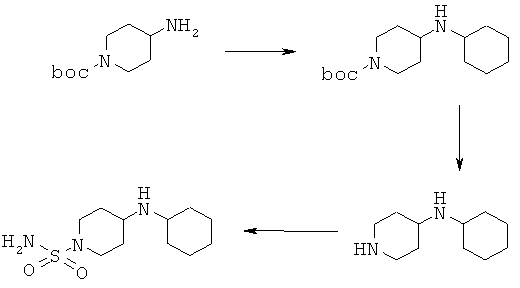
1,23 г ацетата натрия, 2,4 г трет-бутил-4-аминопиперидин-1-карбоксилата, 1,0 мл уксусной кислоты и 1,0 г циклогексанона объединяли в 150 мл ТТФ и перемешивали в течение 3 ч при комнатной температуре. Затем прибавляли 4,25 г трисацетоксиборогидрида натрия и реакционную смесь перемешивали в течение 16 ч при комнатной температуре. После концентр ирования реакционной смеси при пониженном давлении остаток растворяли в смеси метил-трет-бутилового эфира и воды, которую подщелачивали карбонатом натрия до рН 9. Органический слой 4 раза промывали с помощью 0,1 н. HCl. Затем водный слой подщелачивали с помощью NaOH и дважды экстрагировали метил-трет-бутиловым эфиром. Объединенные органические слои промывали водой и насыщенным водным раствором NaCl, сушили над сульфатом натрия и затем выпаривали при пониженном давлении, и получали 2,4 г маслообразного трет-бутил 4-(циклогексиламино)пиперидин-1 -карбоксилата.
1ЯМР (δ част./млн, 400 МГц): 4,03 [2Н], 2,78 [2Н], 2,74 [1Н], 2,56 [1Н], 1,83 [4Н], 1,73 [2Н], 1,45 [9Н].
4.2 2,3 г трет-бутил 4-(циклогексиламино)пиперидин-1-карбоксилата растворяли в 100 мл дихлорметана и при охлаждении льдом обрабатывали с помощью 6,3 мл трифторуксусной кислоты. После перемешивания в течение 16 ч при комнатной температуре реакционную смесь концентрировали при пониженном давлении. Остаток растворяли в смеси воды и метил-трет-бутилового эфира. Водный слой подщелачивали гидроксидом натрия и 5 раз экстрагировали метил-трет-бутиловым эфиром. Затем органический слой промывали водой и насыщенным водным раствором NaCl, сушили над сульфатом натрия и растворитель удаляли в вакууме. Выделяли 1,15 г маслообразного N-циклогексилпиперидин-4-амина и его использовали без дополнительной очистки.
1H ЯМР (δ част./млн, 400 МГц): 3,08 [2Н], 2,68 [1Н], 2,60 [2Н], 2,58 [1Н], 1,85 [4Н], 1,72 [2Н].
4.3 1,1 г N-циклогексилпиперидин-4-амина и 0,7 г сульфамида растворяли в 100 мл диоксана и кипятили с обратным холодильником в течение 5 ч. Затем растворитель удаляли при пониженном давлении и полученный остаток растворяли в смеси воды и дихлорметана. Органический слой промывали водой и насыщенным водным раствором NaCl, сушили над сульфатом натрия и затем выпаривали в вакууме и получали 0,8 г 4-(циклогексиламино)пиперидин-1-сульфонамида.
1Н ЯМР (δ част./млн, 400 МГц): 6,83 [2Н], 3,36 [2Н], 2,59 [1Н], 2,57 [2Н], 2,48 [1Н], 1,79 [4Н].
Соль с хлористоводородной кислотой получали путем обработки амина с помощью HCl в изопропаноле и с последующим выпариванием растворителя. Температура плавления найдена равной более 240°С.
1Н ЯМР (δ част./млн, 400 МГц): 6,83 [2Н], 3,53 [2Н], 3,25 [1Н], 3,08 [1Н], 2,56 [2Н], 2,10 [2Н].
Пример 5
Сульфонамиды (R1=СН2С6Н5; R2=SO2С6Н4СН3; boc=трет-бутоксикарбонил)
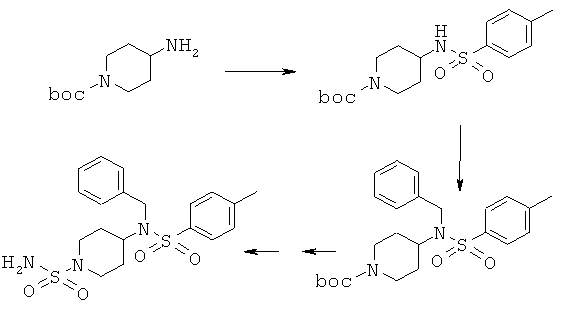
5.1 1,5 г трет-бутил-4-аминопиперидин-1-карбоксилата и 1,13 мл триэтиламина растворяли в 20 мл дихлорметана. К этой смеси в атмосфере азота при охлаждении льдом прибавляли раствор 1,57 г п-толуолсульфонилхлорида в 15 мл дихлорметана. Затем реакционную смесь перемешивали в течение 2 дней при комнатной температуре. После промывки водой, водным раствором гидрокарбоната натрия, водой и насыщенным водным раствором NaCl органический слой сушили над сульфатом натрия и выпаривали в вакууме и после сушки в вакууме получали 2,6 г трет-бутил 4-{[(4-метилфенил)сульфонил]амино}пиперидин-1-карбоксилата.
1H ЯМР (δ част./млн, 400 МГц): 7,70 [2Н], 7,67 [d, 1Н], 7,39 [2H], 3,70 [2Н], 3,13 [1Н], 2,75 [1Н], 2,39 [s, 3H], 1,36 [s, 9H].
5.2 К раствору 1,3 г трет-бутил 4-{[(4-метилфенил)сульфонил]амино}пиперидин-1-карбоксилата в 40 мл ДМФ (диметилформамид) при охлаждении льдом и в атмосфере азота прибавляли 0,62 г трет-бутилата калия. Через 30 мин по каплям прибавляли 0,69 г бензилбромида в 5 мл ДМФ. Эту смесь перемешивали при комнатной температуре в течение 20 ч. Для проведения обработки растворитель удаляли при пониженном давлении и остаток растворяли в метил-трет-бутиловом эфире. Этот раствор промывали водой и насыщенным водным раствором NaCl, сушили над сульфатом натрия и в заключение концентрировали в вакууме. Неочищенный продукт очищали с помощью флэш-хроматографии с использованием смеси н-гексана и этилацетата (9:1) в качестве элюента. После выделения 0,69 г исходного вещества выделяли 0,51 г трет-бутил 4-{бензил[(4-метилфенил)сульфонил]амино}пиперидин-1-карбоксилата.
1Н ЯМР (δ част./млн, 400 МГц): 7,76 [2Н], 7,41 [2Н], 7,38 [2Н], 7,33 [2Н], 7,25 [1Н], 4,39 [s, 2Н], 3,86 [1Н], 3,80 [2Н], 2,63 [2Н], 2,41 [s, 3H], 1,31 [s, 9H].
5.3 0,5 г трет-бутил-4-{бензил[(4-метилфенил)сульфонил]амино}пиперидин-1-карбоксилата и 0,86 мл трифторуксусной кислоты растворяли в 15 мл дихлорметана и перемешивали в течение 60 ч при комнатной температуре. Смесь разбавляли дихлорметаном и перемешивали с 0,1 н. раствором гидроксида натрия. Затем органический слой отделяли, промывали насыщенным водным раствором NaCl, сушили над сульфатом натрия и выпаривали в вакууме, и получали 0,36 г N-бензил-4-метил-N-пиперидин-4-илбензолсульфонамида.
1Н ЯМР (δ част./млн, 400 МГц): 7,74 [2Н], 7,40 [2Н], 7,39 [2Н], 7,34 [2Н], 7,25 [1Н], 4,40 [s, 2Н], 3,69 [1Н], 2,77 [2Н], 2,40 [s, 3H], 2,31 [2Н].
5.4 0,36 г N-бензил-4-метил-N-пиперидин-4-илбензолсульфонамида и 0,12 г сульфамида в течение 8 ч кипятили с обратным холодильником в 5 мл диоксана. Реакционную смесь концентрировали в вакууме и остаток растворяли в метил-трет-бутиловом эфире, затем промывали водой, раствором карбоната натрия, водой и насыщенным водным раствором NaCl. После сушки над сульфатом натрия органический слой выпаривали в вакууме и получали 0,4 г 4-{бензил[(4-метилфенил)сульфонил]амино}пиперидин-1-сульфонамида.
1Н ЯМР (δ част./млн, 400 МГц): 7,78 [2Н], 7,41 [2Н], 7,40 [2Н], 7,35 [2Н], 7,27 [1Н], 6,67 [s, 2Н], 4,42 [s, 2Н], 3,70 [1Н], 3,35 [2Н], 2,43 [2Н], 2,41 [s, 3H].
Пример 6
Сульфонамиды (R1 и R2 образуют фенилзамещенную пиперазиновую систему; boc=трет-бутоксикарбонил)
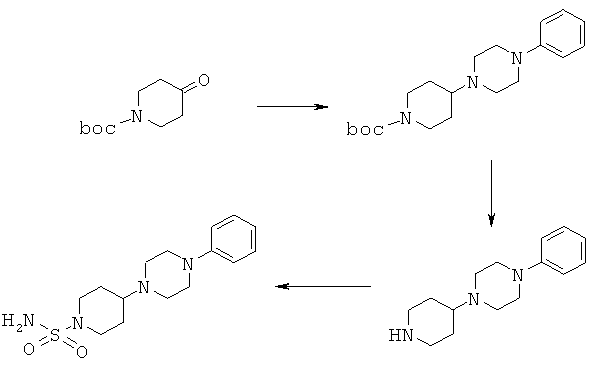
6.1 2,0 г трет-бутил 4-оксопиперидин-1-карбоксилата, 1,23 г ацетата натрия, 0,98 мл уксусной кислоты и 1,83 мл 1-фенилпиперазина объединяли в 150 мл ТГФ и перемешивали в течение 2 ч при комнатной температуре. Затем прибавляли 4,25 г трисацетоксиборогидрида натрия и перемешивали в течение еще 16 ч. Для проведения обработки реакционную смесь концентрировали в вакууме и остаток растворяли в метил-трет-бутиловом эфире и воде. Эту смесь подщелачивали путем прибавления раствора карбоната натрия (рН=10). Затем органический слой 6 раз промывали с помощью 0,1 н. HCl и водный слой (объединенные фракции 3, 4 и 5) подщелачивали путем прибавления разбавленного раствора гидроксида натрия. После экстракции метил-трет-бутиловым эфиром органический слой промывали водой и насыщенным водным раствором NaCl, сушили над сульфатом натрия и затем концентрировали в вакууме. Выделяли 2,0 г трет-бутил 4-(4-фенилпиперазин-1-ил)пиперидин-1-карбоксилата.
1H ЯМР (δ част./млн, 400 МГц): 7,26 [2Н], 6,93 [2Н], 6,85 [1Н], 4,15 [2Н], 3,20 [2Н], 2,73 [2Н], 2,72 [2Н], 2,42 [1Н], 1,46 [s, 9H].
6.2 1,9 г трет-бутил 4-(4-фенилпиперазин-1-ил)пиперидин-1-карбоксилата и 4,25 мл трифторуксусной кислоты растворяли в 100 мл дихлорметана и перемешивали в течение 24 ч. Реакционную смесь концентрировали в вакууме и полученный остаток растворяли в метил-трет-бутиловом эфире и воде. После подщелачивания раствором гидроксида натрия водный слой экстрагировали метил-трет-бутиловым эфиром. Затем осадок, содержащийся в водном слое, отделяли фильтрованием, промывали водой и сушили в вакууме при 50°С и получали 0,67 г 1-фенил-4-пиперидин-4-илпиперазина.
1H ЯМР (δ част./млн, 400 МГц): 7,25 [2Н], 6,93 [2Н], 6,85 [1Н], 3,21 [2Н], 3,16 [2Н], 2,73 [2Н], 2,61 [2Н], 2,38 [1Н].
6.3 0,65 г 1-фенил-4-пиперидин-4-илпиперазина и 0,30 г сульфамида растворяли в 50 мл диоксана и кипятили с обратным холодильником в течение 3 ч. Реакционную смесь концентрировали при пониженном давлении. При растворении остатка в воде начиналось осаждение, которое завершалось после дополнительного перемешивания в течение 1 ч. После фильтрования осадок перемешивали в присутствии дихлорметана в течение 1 ч, повторно фильтровали и сушили в вакууме при 50°С. Выделяли 0,68 г 4-(4-фенилпиперазин-1-ил)пиперидин-1-сульфонамида.
1H ЯМР (δ част./млн, 400 МГц): 7,20 [2Н], 6,92 [2Н], 6,76 [1Н], 6,69 [s, 2Н], 3,50 [2Н], 3,11 [2Н], 2,64 [2Н], 2,54 [2Н], 2,31 [1Н].
Это соединение превращали в соответствующую соль с HCl путем обработки раствором HCl/изопропанол. Перекристаллизация давала 0,52 г продукта, температура плавления 222-227°С.
Пример 7
Альтернативное введение сульфонамидной группы
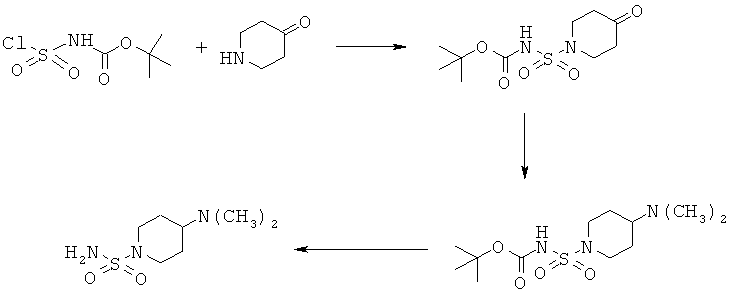
7.1 К охлаждаемому льдом раствору 4,36 мл хлорсульфонилизоцианата в 50 мл дихлорметана в течение 30 мин по каплям прибавляли раствор 4,8 мл трет-бутанола в 50 мл дихлорметана. После дополнительного перемешивания в течение 30 мин при охлаждении эту смесь по каплям прибавляли к раствору 7,68 г соли с HCl 4-пиперидонгидрата и 14,6 мл триэтиламина в 100 мл дихлорметана. После перемешивания в течение 45 мин при охлаждении реакционной смеси давали нагреться до комнатной температуры и перемешивали в течение 16 ч. Реакционную смесь разбавляли дихлорметаном, 3 раза промывали с помощью 0,1 н. HCl (порциями по 60 мл) и насыщенным водным раствором NaCl. После сушки над сульфатом натрия органический слой концентрировали в вакууме и получали 7,2 г неочищенного продукта. Очистка с помощью флэш-хроматографии с использованием смеси дихлорметан/метанол (9:1) в качестве элюента давала 6,0 г трет-бутил [(4-оксопиперидин-1-ил)сульфонил]карбамата.
1Н ЯМР (δ част./млн, 400 МГц, ДМСО-d6): 3,56 [t, 4H], 2,44 [t, 4H], 1,42 [s, 9H].
Следующие стадии реакции проводили так же, как и в любом из приведенных выше примеров 1-6.
Пример 8
Альтернативное введение сульфонамидной группы
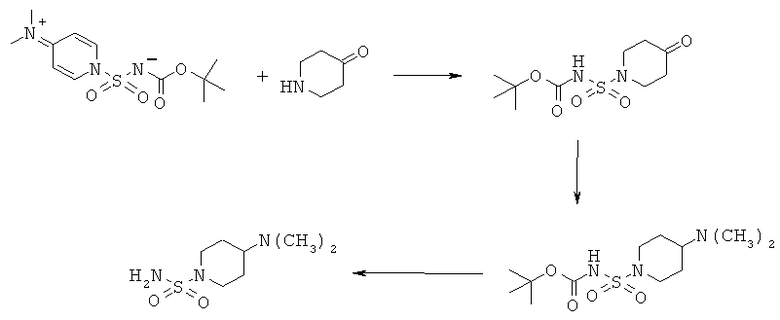
8.1 К раствору 2,6 мл трет-бутанола в 20 мл дихлорметана при охлаждении льдом в течение 15 мин по каплям прибавляли 2,4 мл хлорсульфонилизоцианата. После перемешивания в течение 15 мин прибавляли 6,9 г 4-диметиламинопиридина. Охлаждение прекращали и реакционную смесь перемешивали в течение 1 ч при комнатной температуре и в это время образовывался белый осадок. Смесь разбавляли с помощью 130 мл дихлорметана и 4 раза промывали водой и в заключение насыщенным водным раствором NaCl. После сушки над сульфатом натрия органический слой концентрировали в вакууме и получали 6,4 г кристаллического реагента (трет-бутоксикарбонил){[4-(диметилиминио)пиридин-1(4H)-ил]сульфонил}азанида.
1H ЯМР (δ част./млн, 400 МГц, CDCl3): 8,46 [d, 2H], 6,98 [d, 2H], 3,23 [s, 6H], 1,26 [s, 9H].
8.2 0,5 г (трет-бутоксикарбонил){[4-(диметилиминио)пиридин-1(4H)-ил]сульфонил}азанида, 0,26 г соли с HCl 4-пиперидонгидрата и 0,205 г 4-диметиламинопиридина растворяли в 50 мл диоксана и нагревали при 50°С в течение 4 ч. Реакционную смесь концентрировали в вакууме и остаток растворяли в дихлорметане. После двукратной промывки разбавленным раствором гидросульфата калия органический слой промывали насыщенным водным раствором NaCl и сушили над сульфатом натрия. Растворитель удаляли при пониженном давлении и получали 0,23 трет-бутил[(4-оксопиперидин-1-ил)сульфонил]карбамата. Еще 0,07 г продукта получали, экстрагируя объединенные водные слои дихлорметаном.
1Н ЯМР (δ част./млн, 400 МГц, CDCl3): 3,74 [t, 4Н], 2,58 [t, 4Н], 1,49 [s, 9H].
Следующие стадии реакции проводили так же, как и в любом из приведенных выше примеров 1-6.
Пример 9
Замещенные сульфонамиды
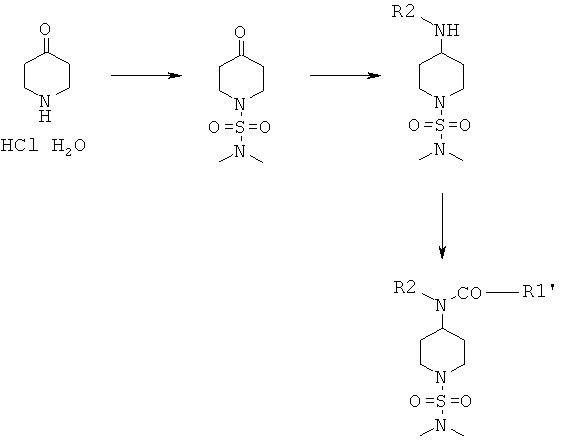
9.1 10 г (65 ммоля) пиперидонгидрохлорида и 7,7 мл (1,1 экв., 71,6 ммоля) диметилсульфамоилхлорида растворяли в смеси ацетон/вода (1:1, 400 мл). Прибавляли 20 мл (2,2 экв., 143 ммоля) триэтиламина и реакционную смесь перемешивали при комнатной температуре в течение 4 дней. Затем растворитель выпаривали и остаток растворяли в этилацетате. Затем органический слой промывали водой, сушили над сульфатом магния, растворитель удаляли в вакууме и получали искомый сульфамид в виде белого твердого вещества (12,55 г, выход 95%).
1Н ЯМР (δ част./млн, 400 МГц, CDCl3): 2,49 (t, 4H, J=6,24 Гц), 2,79 (s, 6H), 3,53 (t, 4Н, J=6,12 Гц).
9.2 0,35 г (1,7 ммоля) сульфамида, полученного на стадии 9.1, растворяли в 10 мл дихлорэтана, а затем прибавляли 1,05 экв. (1,78 ммоля) пентиламина и 1,5 экв. (2,55 ммоля) трисацетоксиборогидрида натрия. Реакционную смесь перемешивали при комнатной температуре в течение ночи и затем промывали с помощью 1 М NaOH. Водный слой экстрагировали эфиром. Объединенные органические слои промывали насыщенным водным раствором NaCl, затем сушили над сульфатом магния и растворитель удаляли в вакууме. Неочищенный продукт вводили во взаимодействие с 0,5 г толуолсульфоновой кислоты, нанесенной на полимерную подложку. Смолу промывали с помощью МеОН, затем с помощью 2 М NH3 в МеОН и извлекали чистый амин.
1Н ЯМР (δ част./млн, 400 МГц, CDCl3): 0,69 (t, 3Н, J=7 Гц), 1,20 (m, 9H), 1,72 (m, 2H), 2,38 (m, 1H), 2,42 (t, 2H, J=7 Гц), 2,60 (s, 6H), 2,65 (td, 2H, J=2, 13 Гц), 3,43 (2H, td, J=3, 13 Гц).
13С ЯМР (δ част./млн, 400 МГц, CDCl3): 14,0, 22,6, 29,6, 30,1, 32,3, 38,2, 45,2, 46,9, 54,3.
9.3 0,036 ммоля амина, полученного на стадии 9.2, растворяли в 1 мл дихлорметана и смешивали с 1,5 экв. (0,054 ммоля) хлорангидрида уксусной кислоты и 0,054 ммоля (1,5 экв., 15 мг) триэтиламина, нанесенного на полимерную подложку. Реакционную смесь встряхивали в течение 3 дней при комнатной температуре, а затем к реакционной смеси прибавляли 0,054 ммоля (1,5 экв., 26 мг) аминометилированного полистирола. Затем встряхивали в течение еще 1 дня. Смолу отфильтровывали, промывали дихлорэтаном, растворитель выпаривали в вакууме и получали искомые сульфонамиды.
Пример 10
Замещенные сульфонамиды (boc=трет-бутоксикарбонил)
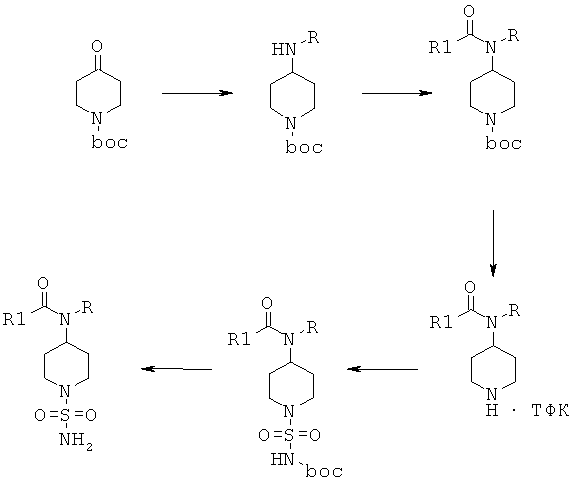
10.1 0,35 г (17,57 ммоля) трет-бутил-4-оксопиперидин-1-карбоксилата растворяли в 100 мл дихлорэтана, а затем прибавляли 1,05 экв. (18,44 ммоля) амина и 1,5 экв. (26,36 ммоля) трисацетоксиборогидрида натрия. Реакционную смесь перемешивали при комнатной температуре в течение ночи. Затем реакционную смесь промывали с помощью 1 М NaOH и водный слой экстрагировали эфиром. Объединенный органический слой промывали насыщенным водным раствором NaCl, затем сушили над сульфатом магния, растворитель удаляли в вакууме и получали искомый чистый аминозамещенный пиперидин.
10.2 18 ммоля аминозамещенного пиперидина, полученного на стадии 10.1, растворяли в 120 мл дихлорметана и смешивали с 19,8 ммоля (1,1 экв.) хлорангидрида кислоты и 27 ммоля (3,76 мл, 1,5 экв.) триэтиламина. Реакционную смесь перемешивали в течение 3 дней при комнатной температуре. Затем реакцию останавливали насыщенным водным раствором NaCl и водный слой экстрагировали дихлорметаном. Объединенный органический слой сушили над сульфатом магния и растворитель выпаривали в вакууме. Неочищенное соединение очищали с помощью флэш-хроматографии, элюируя смесью 40-50% этилацетат/гептан, и получали искомый карбонилзамещенный пиперидин.
10.3 12,21 ммоля карбонилзамещенного пиперидина, полученного на стадии 10.2, растворяли в 20% трифторуксусной кислоте и 45 мл дихлорметана. Реакционную смесь перемешивали в течение 1 ч. Растворитель выпаривали в вакууме и получали искомый карбонилзамещенный пиперидин в виде трифторацетата с количественным выходом. Соединение больше не очищали и использовали в неочищенном виде на следующей стадии.
10.4 0,84 ммоля карбонилзамещенного пиперидина в виде трифторацетата, полученного на стадии 10.3, растворяли в 20 мл дихлорэтана и к смеси прибавляли 9,51 ммоля (3 г) связанного с полимером тетраалкиламмонийкарбоната. Реакционную смесь перемешивали в течение 1 дня, затем смолу отфильтровывали и промывали дихлорметаном. Растворитель удаляли и получали карбонилзамещенный пиперидин в виде свободного основания.
1,68 ммоля (2 экв.) трет-бутанола при 0°С медленно прибавляли к раствору 1,68 ммоля (2 экв., 0,145 мл) хлорсульфонилизоцианата в 10 мл дихлорметана. Реакционную смесь нагревали до комнатной температуры в течение 1 ч, а затем охлаждали до 0°С. К реакционной смеси прибавляли 0,47 мл (3,36 ммоля; 4 экв.) триэтиламина, а затем свободное основание амина, полученное выше на стадии 10.4, в 5 мл дихлорметана. Реакционную смесь нагревали до комнатной температуры и перемешивали в течение ночи. Реакционную смесь промывали с помощью 1 н. HCl, органический слой сушили над сульфатом магния и растворитель выпаривали в вакууме. Неочищенное соединение очищали с помощью флэш-хроматографии, элюируя смесью 50% этилацетат/гептан, и получали содержащий защитную группу boc сульфонамид в виде белого твердого вещества (0,185 г, выход 55%).
10.5 0,12 ммоля содержащего защитную группу boc сульфамида, полученного на стадии 10.4, растворяли в 20% ТФК (трифторуксусная кислота) в 1 мл дихлорметана и реакционную смесь перемешивали в течение 2 ч. Затем растворитель выпаривали в вакууме и получали белое твердое вещество, искомый конечный продукт в виде трифторацетата с количественным выходом (42 мг).
Пример 11
Гетероарилзамещенные сульфонамиды (boc=трет-бутоксикарбонил)
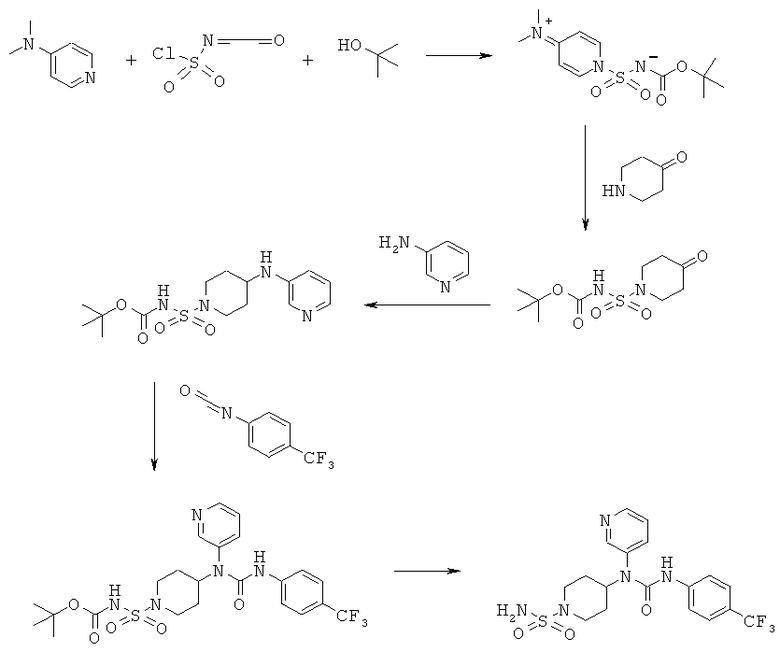
11.1 К раствору 26 мл трет-бутанол в 200 мл дихлорметана при охлаждении льдом (5°С) в течение 90 мин по каплям прибавляли 24 мл хлорсульфонилизоцианата. После перемешивания в течение 60 мин порциями прибавляли 69 г 4-диметиламинопиридина. Охлаждение прекращали и реакционную смесь перемешивали в течение 1 ч при комнатной температуре, и в это время образовывался белый осадок. Смесь разбавляли с помощью 200 мл дихлорметана и промывали с помощью 500 мл воды. Затем прибавляли 1,1 л дихлорметана, полученный раствор 4 раза промывали водой (порциями по 0,5 л) и в заключение рассолом. После сушки над сульфатом натрия органический слой концентрировали в вакууме и получали 76,5 г кристаллического реагента (трет-бутоксикарбонил){ [4-(диметилиминио)пиридин-1(4H)-ил]сульфонил}азанида, обладающего температурой плавления, равной 180-181°С.
1Н-ЯМР (δ част./млн, 400 МГц, CDCl3): 8,46 [d, 2], 6,98 [d, 2], 3,23 [s, 6], 1,26 [s, 9].
11.2 50 г (трет-бутоксикарбонил){[4-(диметилиминио)пиридин-1(4Н)-ил]сульфонил}азанида, 30,6 г соли с HCl 4-пиперидонгидрата и 24,3 г 4-диметиламинопиридина растворяли в 1,5 л диоксана и нагревали при 55°С в течение 16 ч. Реакционную смесь концентрировали в вакууме и остаток растворяли в дихлорметане. После трехкратной промывки разбавленным раствором гидросульфата калия органический слой промывали рассолом и сушили над сульфатом натрия. Растворитель удаляли при пониженном давлении и получали 32,8 г трет-бутил[(4-оксопиперидин-1-ил)сульфонил]карбамата, обладающего температурой плавления, равной 107-109°С.
1Н ЯМР (δ част./млн, 400 МГц, CDCl3): 3,74 [t, 4], 2,58 [t, 4], 1,49 [s, 9].
11.3 14 г трет-бутил-[(4-оксопиперидин-1-ил)сульфонил]карбамата растворяли в 600 мл ТГФ и прибавляли 5,7 г 3-аминопиридина и 6,2 г ацетата натрия. К этой смеси прибавляли 4,9 мл уксусной кислоты и 7,4 мл тетраизопропилортотитаната и затем реакционную смесь перемешивали в течение 5 ч при комнатной температуре. Затем порциями прибавляли 21,3 г трисацетоксиборогидрида натрия и смесь перемешивали в течение 1 6 ч при комнатной температуре. Реакционную смесь концентрировали в вакууме и остаток растворяли в смеси дихлорметана и воды. Водный слой и часть твердого вещества отделяли и перемешивали с дихлорметаном. Для лучшего разделения объединенные органические слои центрифугировали (4000 об/мин). Органический слой промывали рассолом, сушили над сульфатом натрия и после выпаривания при пониженном давлении получали 20 г светло-зеленого масла. Кристаллизация из МТБЭ (метил-трет-бутиловый эфир) (100 мл) давала 8,34 г чистого трет-бутил{[4-(пиридин-3-иламино)пиперидин-1-ил]сульфонил}карбамата, обладающего температурой плавления, равной 170°С (с разложением).
1Н ЯМР (δ част./млн, 400 МГц, ДМСО-d6): 10,92 [s, br, 1], 8,00 [d, 1], 7,76 [dd, 1], 7,11 [dd, 1], 7,02 [dd, 1], 3,62 [m, 2], 3,42 [m, 1], 2,99 [m, 2], 1,97 [m, 2], 1,44 [s, 9], 1,38 [m, 2].
11.4 28,6 г трет-бутил{[4-(пиридин-3-иламино)пиперидин-1-ил]сульфонил}карбамата почти полностью растворяли в 1,5 л дихлорметана и затем прибавляли 15 г 4-(трифторметил)фенилизоцианата. Эту реакционную смесь перемешивали в течение 24 ч при 40°С. Реакционную смесь фильтровали (выделяли 0,9 г исходного вещества) и концентрировали в вакууме. Остаток перемешивали в смеси 500 мл МТБЭ и 500 мл воды в течение 1 ч, и в это время начиналось осаждение продукта. Твердое вещество отделяли фильтрованием, промывали с помощью МТБЭ и водой и затем сушили в вакууме при 60°С. Выделяли 41,4 г трет-бутил{[4-(пиридин-3-ил{[4-(трифторметил)фенил]карбамоил}амино)пиперидин-1-ил]сульфонил}карбамата, обладающего температурой плавления, равной 145°С (вспенивание).
1Н ЯМР (δ част./млн, 400 МГц, ДМСО-d6): 10,88 [s, br, 1], 8,61 [d, 1], 8,45 [dd, 1], 8,11 [s, 1], 7,70 [dd, 1], 7,61 [d, 2], 7,55 [d, 2], 7,50 [dd, 1], 4,43 [m, 2], 3,67 [m, 1], 2,95 [m, 2], 1,92 [m, 2], 1,36 [s, 9], 1,22 [m, 2].
11.5 К раствору 1,3 г трет-бутил{[4-(пиридин-3-ил{[4-(трифторметил)фенил]карбамоил}амино)пиперидин-1-ил]сульфонил}карбамата в 100 мл дихлорметана прибавляли 1,8 мл трифторуксусной кислоты. Эту смесь перемешивали в течение 30 ч при комнатной температуре. Затем реакционную смесь концентрировали в вакууме и полученный маслообразный остаток растворяли в МТБЭ, и в это время продукт начинал кристаллизоваться. Через 2 ч твердое вещество отделяли фильтрованием и затем сушили при пониженном давлении. Полученный остаток перемешивали со смесью МТБЭ и воды. После удаления части нерастворенного продукта (0,13 г) органический слой отделяли, промывали водой и раствором гидрокарбоната натрия и осаждался продукт. Выход объединенного кристаллического вещества, 4-(пиридин-3-ил{[4-(трифторметил)фенил]карбамоил}амино)пиперидин-1-сульфонамида, обладающего температурой плавления, равной 188-191°С, составлял 0,57 г.
1Н ЯМР (δ част./млн, 400 МГц, ДМСО-d6): 8,62 [d, 1], 8,47 [dd, 1], 8,14 [s, 1], 7,71 [dd, 1], 7,62 [d, 2], 7,55 [d, 2], 7,52 [dd, 1], 4,36 [m, 2], 3,50 [m, 1], 2,62 [m, 2], 1,93 [m, 2], 1,26 [m, 2].
Указанные ниже соединения синтезировали с превосходными выходами в соответствии с любым из приведенных выше примеров 1-8.
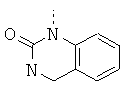
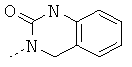
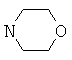
Пример 1
Капсулы, содержащие соединение 1 из примера 6:
Активное вещество, кукурузный крахмал и лактозу с использованием ЭА превращают в однородную пастообразную смесь. Пасту измельчают, полученные гранулы помещают в подходящий лоток и сушат при 45°С для удаления растворителя. Высушенные гранулы пропускают через измельчающее устройство и в смесителе смешивают со следующими вспомогательными веществами:
и затем по 400 мг помещают в капсулы (капсулы размера 0).
| название | год | авторы | номер документа |
|---|---|---|---|
| ПИПЕРИДИНИЛ-ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ ИЗОХИНОЛОНА КАК ИНГИБИТОРЫ Rho-КИНАЗЫ | 2006 |
|
RU2414467C2 |
| МОЧЕВИНА И СУЛЬФАМИДНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ TAFIA | 2007 |
|
RU2459619C2 |
| МОДУЛЯТОРЫ ПРОТЕОЛИЗА И СООТВЕТСТВУЮЩИЕ СПОСОБЫ ПРИМЕНЕНИЯ | 2019 |
|
RU2805511C2 |
| МАКРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ МОЧЕВИНЫ И СУЛЬФАМИДА В КАЧЕСТВЕ ИНГИБИТОРОВ TAFIa | 2009 |
|
RU2502736C2 |
| ПРОИЗВОДНЫЕ ТЕТРАГИДРОНАФТАЛИНА И ТЕТРАГИДРОИЗОХИНОЛИНА В КАЧЕСТВЕ РАЗРУШИТЕЛЕЙ ЭСТРОГЕНОВОГО РЕЦЕПТОРА | 2017 |
|
RU2797244C2 |
| ЗАМЕЩЕННЫЕ ИЗОХИНОЛИНОВЫЕ И ИЗОХИНОЛИНОНОВЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ Rho-КИНАЗЫ | 2007 |
|
RU2455302C2 |
| ЗАМЕЩЕННЫЕ АМИДОМ КАРБОНОВОЙ КИСЛОТЫ ПРОИЗВОДНЫЕ ФЕНИЛМОЧЕВИНЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ЛЕКАРСТВЕННЫХ СРЕДСТВ | 2002 |
|
RU2291858C2 |
| ПРОИЗВОДНЫЕ ГЕКСАГИДРОДИАЗЕПИНОНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ИХ СОДЕРЖАЩАЯ, И ПРИМЕНЕНИЕ ДЛЯ ПОЛУЧЕНИЯ ЛЕКАРСТВЕННОГО СРЕДСТВА ДЛЯ ЛЕЧЕНИЯ ИНСУЛИННЕЗАВИСИМОГО ДИАБЕТА | 2003 |
|
RU2301803C2 |
| БИ- И ПОЛИЦИКЛИЧЕСКИЕ ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ ИЗОХИНОЛИНА И ИЗОХИНОЛИНОНА, ПОЛЕЗНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ RHO-КИНАЗЫ | 2009 |
|
RU2532481C2 |
| ТАРТРАТНЫЕ ПРОИЗВОДНЫЕ ДЛЯ ПРИМЕНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ ФАКТОРА СВЕРТЫВАНИЯ КРОВИ IXA | 2007 |
|
RU2446160C2 |
Изобретение относится к соединениям формулы
в которой R1 выбран из группы, включающей Н; алкил; алкиленарил и пиридин; R2 выбран из группы, включающей циклоалкил; арил; CO-NH-циклоалкил; CO-NH-арил, незамещенный или замещенный с помощью галогена, CF3; SO2-арил, незамещенный или замещенный с помощью алкила, или в которой R1 и R2 совместно образуют 5- или 6-членное кольцо, которое необязательно может содержать 1 дополнительный гетероатом азота, и которое также может быть замещено с помощью арила; и которое также может содержать карбонильную группу; и которое также может быть сконденсировано с арилом; R3 и R4 обозначают Н; и к их физиологически приемлемым солям присоединения с кислотами.
Изобретение также относится к фармацевтической композиции, к способу получения соединения формулы I, к применению соединения формулы I, а также к способу лечения или предупреждения ожирения, диабета типа II, метаболического синдрома и сопутствующих им и/или вторичных заболеваний или патологических состояний у млекопитающих.
Технический результат - получение новых биологически активных соединений, ингибирующих изофермент карбоангидразы человека II (hCA II). 5 н. и 6 з.п. ф-лы, 3 табл.
1. Соединение формулы I
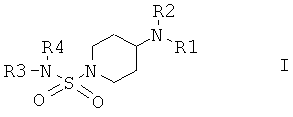
в которой R1 выбран из группы, включающей: Н; алкил; алкиленарил и пиридин; R2 выбран из группы, включающей: циклоалкил; арил; CO-NH-циклоалкил; CO-NH-арил, незамещенный или замещенный с помощью галогена, CF3; SO2-арил, незамещенный или замещенный с помощью алкила; или в которой R1 и R2 совместно образуют 5- или 6-членное кольцо, которое необязательно может содержать 1 дополнительный гетероатом азота, и которое также может быть замещено с помощью арила; и которое также может содержать карбонильную группу; и которое также может быть сконденсировано с арилом;
R3 и R4 обозначают Н;
и его физиологически приемлемые соли присоединения с кислотами.
2. Соединение по п.1, в котором R1 выбран из группы, включающей Н и пиридин; R2 выбран из группы, включающей CO-NH-арил, замещенный с помощью галогена, CF3; R3 и R4 обозначают Н.
3. Соединение по п.1, в котором R1 обозначает алкиленарил; R2 обозначает незамещенный CO-NH-арил; R3 и R4 обозначают Н.
4. Фармацевтическая композиция, ингибирующая изофермент карбоангидразы человека II (hCA II), включающая (a) фармакологически эффективное количество соединения формулы I
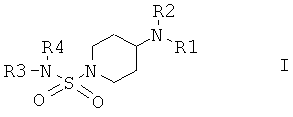
в которой R1 выбран из группы, включающей: Н; алкил; алкиленарил и пиридин; R2 выбран из группы, включающей: циклоалкил; арил; CO-NH-циклоалкил; CO-NH-арил, незамещенный или замещенный с помощью галогена, CF3; SO2-арил, незамещенный или замещенный с помощью алкила; или в которой R1 и R2 совместно образуют 5- или 6-членное кольцо, которое необязательно может содержать 1 дополнительный гетероатом азота, и которое также может быть замещено с помощью арила; и которое также может содержать карбонильную группу; и которое также может быть сконденсировано с арилом;
R3 и R4 обозначают Н; или
их физиологически приемлемые соли присоединения с кислотами; и
(b) обычные фармацевтически приемлемые вспомогательные вещества и/или носители.
5. Композиция по п.4, в которой R1 выбран из группы, включающей Н и пиридин; R2 выбран из группы, включающей CO-NH-арил, замещенный с помощью галогена, CF3; R3 и R4 обозначают Н.
6. Композиция по п.4, в которой R1 обозначает алкиленарил; R2 обозначает незамещенный CO-NH-арил; R3 и R4 обозначают Н.
7. Способ получения соединения формулы I
в которой R1 выбран из группы, включающей: Н; алкил; алкиленарил и пиридин; R2 выбран из группы, включающей: циклоалкил; арил; CO-NH-циклоалкил; CO-NH-арил, незамещенный или замещенный с помощью галогена, CF3; SO2-арил, незамещенный или замещенный с помощью алкила; или в которой R1 и R2 совместно образуют 5- или 6-членное кольцо, которое необязательно может содержать 1 дополнительный гетероатом азота, и которое также может быть замещено с помощью арила; и которое также может содержать карбонильную группу; и которое также может быть сконденсировано с арилом;
R3 и R4 обозначают Н;
и его физиологически приемлемых солей присоединения с кислотами, характеризующийся тем, что
соединения формулы I, в которой R3 и R4 оба обозначают Н, получают по реакции соединения формулы VII
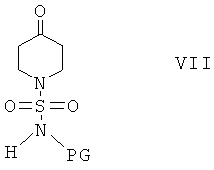
с амином HNR1R2 с получением соединения формулы IX
и затем отщепляют защитную группу PG от полученных промежуточных продуктов, что приводит к получению соединений формулы I, в которой R3 и R4 оба обозначают Н;
и при необходимости превращают полученные свободные основания формулы I в их физиологически приемлемые соли присоединения с кислотами или превращают соли присоединения с кислотами соединений формулы I в свободные основания формулы I.
8. Способ по п.7, в котором R1 выбран из группы, включающей Н и пиридин; R2 обозначает CO-NH-арил, замещенный с помощью галогена, CF3; R3 и R4 обозначают Н.
9. Способ по п.7, в котором R1 обозначает алкиленарил; гетероарил; алкиленгетероарил, замещенный галогеном; в котором R2 обозначает незамещенный CO-NH-арил; R3 и R4 обозначают Н.
10. Применение соединения формулы I
в которой R1 выбран из группы, включающей: Н; алкил; алкиленарил и пиридин; R2 выбран из группы, включающей: циклоалкил; арил; CO-NH-циклоалкил; CO-NH-арил, незамещенный или замещенный с помощью галогена, CF3; SO2-арил, незамещенный или замещенный с помощью алкила; или в которой R1 и R2 совместно образуют 5- или 6-членное кольцо, которое необязательно может содержать 1 дополнительный гетероатом азота, и которое также может быть замещено с помощью арила; и которое также может содержать карбонильную группу; и которое также может быть сконденсировано с арилом; R3 и R4 обозначают Н;
и его физиологически приемлемых солей присоединения с кислотами для приготовления лекарственного средства, предназначенного для профилактики, и/или лечения, и/или предупреждения ожирения, диабета типа II, метаболического синдрома, и сопутствующих им и/или вторичных заболеваний, или патологических состояний у млекопитающих.
11. Способ лечения или предупреждения ожирения, диабета типа II, метаболического синдрома и сопутствующих им и/или вторичных заболеваний, или патологических состояний у млекопитающих, включающий введение нуждающемуся в нем субъекту терапевтически эффективного количества соединения формулы I
в которой R1 выбран из группы, включающей: Н; алкил; алкиленарил и пиридин; R2 выбран из группы, включающей: циклоалкил; арил; CO-NH-циклоалкил; CO-NH-арил, незамещенный или замещенный с помощью галогена, CF3; SO2-арил, незамещенный или замещенный с помощью алкила; или в которой R1 и R2 совместно образуют 5- или 6-членное кольцо, которое необязательно может содержать 1 дополнительный гетероатом азота, и которое также может быть замещено с помощью арила; и которое также может содержать карбонильную группу; и которое также может быть сконденсировано с арилом;
R3 и R4 обозначают Н;
и его физиологически приемлемых солей присоединения с кислотами.
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| RU 2002128733 А, 10.03.2004. | |||

