Область техники
Настоящее изобретение относится к новым соединениям, оказывающим возбуждающее действие на рецептор активатора пролиферации пероксисом подтипа δ (PPAR δ), к способу получения указанных соединений, к лекарственным средствам, содержащим указанные соединения, и к применению указанных соединений для лечения и предотвращения сердечно-сосудистых заболеваний и т.п. Настоящее изобретение также относится к новому промежуточному соединению новых соединений и к способу получения указанного промежуточного соединения.
Уровень техники
В настоящее время наряду с высокой скоростью развития и ростом жизненных стандартов люди потребляют избыточное количество жиров и белков, что приводит к широкому распространению метаболического синдрома, характеризующегося ожирением, резистентностью к инсулину (диабет II типа), расстройством липидного обмена и гипертензией. Это представляет значительную угрозу для здоровья человека. В дополнение к генетическим характеристикам индивидуума, возрасту, половой принадлежности, физиологическим особенностям, состоянию питания, пристрастию к пище и т.д., метаболический синдром связан с дисбалансом липидного обмена, обмена энергии и углеводов in vivo. Таким образом, эффективным способом лечения метаболического синдрома является терапевтический режим, направленный на поддержание или восстановление баланса липидов, углеводов и энергии in vivo. Поскольку ядерные рецепторы (NRs) играют ключевую роль в поддержании баланса энергии, липидов и углеводов в клетке in vivo, а также в организме человека в целом, указанные рецепторы оказались в центре внимания исследователей. Лишь после активации различными физиологическими лигандами (например, насыщенными жирными кислотами, их метаболитами и различными синтетическими соединениями) ядерные рецепторы способны оказывать регулирующее воздействие на системы транскрипции чувствительных генов, тем самым влияя на физиологическую активность (Kasuga, J. et al., Bioorg. Med. Сhem. 2007, 15, 5177-5190).
Среди семейств ядерных рецепторов повышенное внимание в течение более чем десяти лет привлекают рецепторы активатора пролиферации пероксисом (PPARs), которые являются ядерными факторами транскрипции, активируемыми лигандами, и действуют как основные регуляторные факторы при метаболическом синдроме (Guan, Y. J. Am. Soc. Nephrol, 2004, 15, 2801-2815). Следовательно, PPARs играют важную роль в возникновении, развитии и контроле таких заболеваний, как резистентность к инсулину, нарушенная толерантность к глюкозе, диабет II типа, ожирение, гиперлипидемия, гипертензия, ангиокардиопатия, атеросклероз и т.д.
PPARs подразделяют на три подтипа: PPARα, PPARδ и PPARγ, которые регулируют экспрессию генов путем связывания со специфической последовательностью ДНК (Berger, J. et al., The Journal of Biological Chemistry, 1999, 274 (10), 6718-6725). PPARα главным образом экспрессируется в печени, сердце, желудочно-кишечном тракте, почках и макрофагах и после активации могут усиливать метаболизм жирных кислот, уменьшать воспалительный ответ в макрофагах и снижать содержание холестерина в липопротеинах низкой плотности; PPARγ экспрессируется в адипоцитах, плацентоме и других тканях и после активации может не только понижать уровень глюкозы в крови и увеличивать чувствительность к инсулину, но также играет ключевую роль в липидном обмене, противодействии цитокинам, противовоспалительных процессах, иммунорегуляции и регуляции кровяного давления и т.д. (Kasuga, J. et al., Bioorg. Med. Chem. 2007, 15, 5177-5190). В противоположность двум другим подтипам, физиологическая функция PPARδ до сих пор не ясна. Вместе с тем, в последних исследованиях в ходе фармакологических экспериментов на животных моделях показано, что PPARδ может усиливать разобщение катаболизма жирных кислот и энергии в жировой ткани и мышцах и может подавлять воспаление, опосредованное макрофагами. Вследствие различных функций в контроле прибавления в весе тела, повышения выносливости, увеличения чувствительности к инсулину и улучшения состояния при атеросклерозе, лиганды PPARδ могут являться эффективными лекарственными средствами для лечения гиперлипидемии, ожирения, резистентности к инсулину и атеросклероза.
В настоящее время ни один из агонистов рецепторов PPARδ не является коммерчески доступным в качестве лекарственного средства. Среди современных исследований агонистов PPARδ клиническое исследование GW501516, разработанного GlaxoSmithKline, показало, что GW501516 может повышать уровень холестерина в липопротеинах высокой плотности (ЛПВП) до 80%, понижать уровень холестерина в липопротеинах низкой плотности (ЛПНП) до 29%, снижать уровень триглицеридов (ТГ) до 56%, снижать уровень инсулина до 48% (Oliver, W.; Jr.; Shenk, J. L. et al, Natl. Acad. Sci. U.S.A. 2001, 98, 5306-5311). Таким образом, полагают, что GW501516 может стать эффективным лекарственным средством для лечения ожирения и сердечно-сосудистых заболеваний (WO01/00603A1, Bioord. Med. Chem. Lett. 2003, 13, 1517). Однако проект GW501516 в настоящее время приостановлен вследствие неблагоприятных результатов, полученных на втором этапе его клинических испытаний.
Таким образом, существует насущная необходимость в обеспечении нового соединения, которое обладает агонистическим действием на рецептор активатора пролиферации пероксисом подтипа δ (PPARδ) и намного лучшей способностью регулировать содержание липидов в крови, чем GW501516.
Сущность изобретения
Одной из задач настоящего изобретения является обеспечение соединения формулы (I) и/или фармацевтически приемлемых солей и/или сольватов указанного соединения:
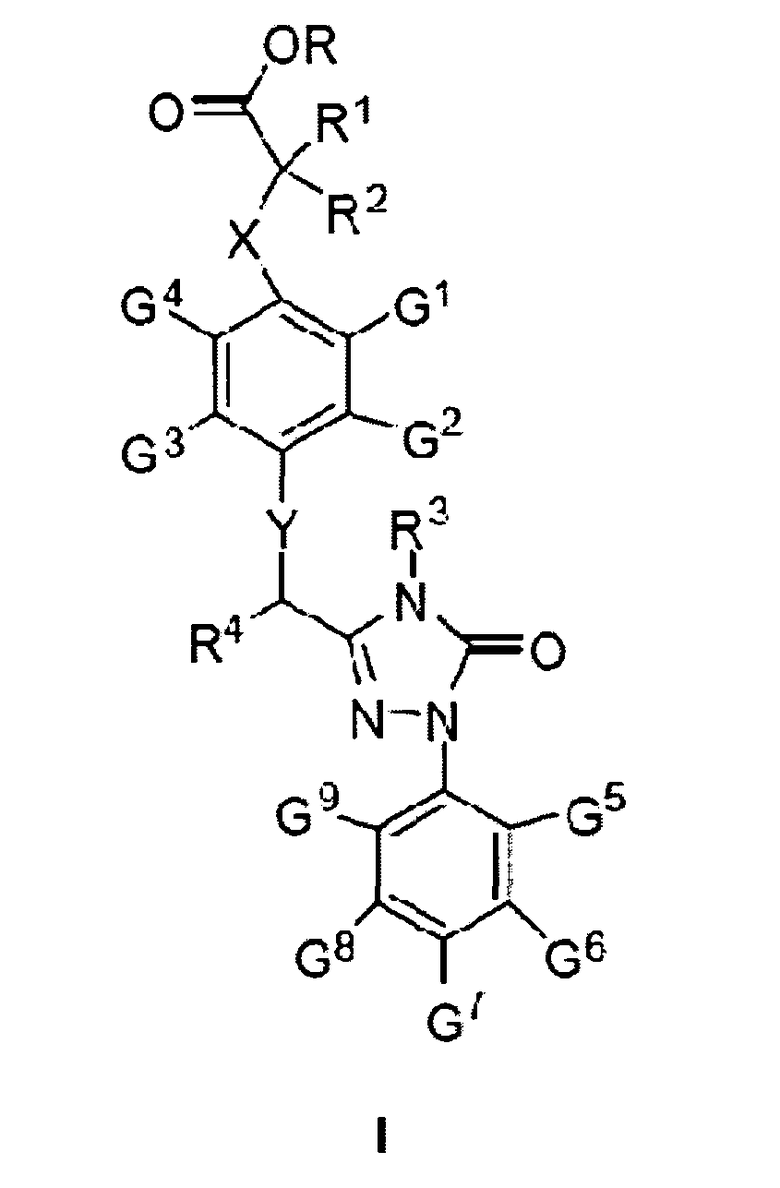
Где
1) Х представляет собой О, S, N или (СН2)n, в котором n равно целому числу от 1 до 4, Х предпочтительно представляет собой О, S или СН2;
2) Y представляет собой О, S или N, предпочтительно О или S;
3) R представляет собой Н или С1-С9 алкил, предпочтительно Н, метил или этил;
4) R1 и R2 независимо друг от друга представляют собой Н или С1-С4 алкил, и по меньшей мере один из R1 и R2 представляет собой Н; предпочтительно, R1 и R2 независимо друг от друга представляют собой метил, этил или Н;
5) R3 представляет собой Н, С1-С9 алкил, предпочтительно Н или С1-С4 алкил, например метил, этил, изопропил, и более предпочтительно - метил;
6) R4 представляет собой Н, С1-С9 алкил, С3-С7 циклоалкил, фенил или замещенный фенил, причем группа-заместитель в замещенном фениле выбрана из С1-С9 алкила, гидроксила, С1-С9 алкокси, меркапто, С1-С9 алкилтио, трифторметила, F, CI, Вr, нитро, NR5R6, COOR5, NR5COR6, или CONR5R6; R5 и R6 независимо друг от друга представляют собой Н или С1-С9 алкил, а если R4 представляет собой замещенный фенил, группа-заместитель предпочтительно представляет собой 4-метокси или 4-метил;
7) G1 и G4 по отдельности представляют собой Н, С1-С9 алкил, гидроксил, С1-С9 алкокси, меркапто, С1-С9 алкилтио, трифторметил, F, CI, Вr, нитро, NR5R6, COOR5, NR5COR6 или CONR5R6; R5 и R6 независимо друг от друга представляют собой Н или С1-С9 алкил; предпочтительно, G1 и G4 по отдельности представляют собой метил или этил;
8) G2 и G3 по отдельности представляют собой Н, С1-С9 алкил, гидроксил, С1-С9 алкокси, меркапто, С1-С9 алкилтио, трифторметил, F, CI, Вr, нитро, NR5R6, COOR5, NR5COR6 или CONR5R6; R5 и R6 независимо друг от друга представляют собой Н или С1-С9 алкил; предпочтительно, G2 и G3 по отдельности представляют собой метил, этил или Н;
9) G5, G6, G8 и G9 по отдельности представляют собой Н, С1-С9 алкил, гидроксил, С1-С9 алкокси, меркапто, С1-С9 алкилтио, трифторметил, F, CI, Вr, нитро, NR5R6, COOR5, NR5COR6 или CONR5R6; R5 и R6 независимо друг от друга представляют собой Н или С1-С9 алкил; предпочтительно, G5, G6, G8 и G9 по отдельности представляют собой Н, F, Cl, Вr, метил, этил или метокси; и
10) G7 представляет собой Н, С1-С9 алкил, гидроксил, С1-С9 алкокси, меркапто, С1-С9 алкилтио, трифторметил, F, CI, Вr, нитро, NR5R6, COOR5, NR5COR6 или CONR5R6; R5 и R6 независимо друг от друга представляют собой Н или С1-С9 алкил; предпочтительно, G7 представляет собой трифторметил, изопропил, этил, метил или Cl; более предпочтительно, G7 представляет собой трифторметил.
В случае, когда соединение согласно настоящему изобретению находится в форме эфира, указанное соединение далее в описании изобретения обозначено "соединение I (эфир)". Предпочтительное соединение I (эфир) согласно настоящему изобретению включает следующие соединения:
Этил-2-(2-метил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1Н-1,2,4-триазолил))-бензилокси)-фенокси)-ацетат (далее называемый "Е-1");
Этил-2-(2-метил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1H-1,2,4-триазолил))-бензилокси)-фенилтио)-ацетат (далее называемый "Е-2");
Этил-2-(3-метил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1Н-1,2,4-триазолил))-бензилокси)-фенилтио)-ацетат (далее называемый "Е-3");
Этил-2-(2-метил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1Н-1,2,4-триазолил))-бензилтио)-фенокси)-ацетат (далее называемый "Е-4");
Этил-2-(2-этил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1Н-1,2,4-триазолил))-бензилокси)-фенокси)-ацетат (далее называемый "Е-5");
Этил-2-(3-метил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1Н-1,2,4-триазолил))-бензилокси)-фенокси)-ацетат (далее называемый "Е-6");
Этил-2-(3-метил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1H-1,2,4-триазолил))-бензилтио)-фенокси)-ацетат (далее называемый "Е-7");
Этил-2-(2-метил-4-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1H-1,2,4-триазолил)-метилтио)-фенокси)-ацетат (далее называемый "Е-8");
Этил-2-(2-метил-4-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1Н-1,2,4-триазолил)-метокси)-фенокси)-ацетат (далее называемый "Е-9");
Этил-2-(2,5-диметил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметил-фенил)-4,5-дигидро-1H-1,2,4-триазолил))-бензилтио)-фенокси)-ацетат (далее называемый "Е-10");
Этил-2-(2,5-диметил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметил-фенил)-4,5-дигидро-1H-1,2,4-триазолил))-бензилокси)-фенилтио)-ацетат (далее называемый "Е-11");
Этил-2-(3-метил-4-(3-(4-метил-5-оксо-1-(4-трифторметил-фенил)-4,5-дигидро-1Н-1,2,4-триазолил)-метокси)-фенилтио)-ацетат (далее называемый "Е-12");
Метил-2-метил-2-(2-метил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметил-фенил)-4,5-дигидро-1H-1,2,4-триазолил))-бензилтио)-фенокси)-ацетат (далее называемый "Е-13");
Этил-2-(2-этил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметил-фенил)-4,5-дигидро-1H-1,2,4-триазолил))-бензилтио)-фенокси)-ацетат (далее называемый "Е-14");
Этил-2,2-диметил-2-(3-метил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметил-фенил)-4,5-дигидро-1H-1,2,4-триазолил))-бензилокси)-фенилтио)-ацетат (далее называемый "Е-15");
Этил-2,2-диметил-2-(2-метил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметил-фенил)-4,5-дигидро-1H-1,2,4-триазолил))-бензилтио)-фенокси)-ацетат (далее называемый "Е-16");
Этил-2-(3-метил-4-(1-(3-(4-n-бутил-5-оксо-1-(4-трифторметил-фенил)-4,5-дигидро-1H-1,2,4-триазолил))-бензилокси)-фенилтио)-ацетат (далее называемый "Е-17");
Этил-2,2-диметил-2-(2-метил-4-(1-(3-(4-n-бутил-5-оксо-1-(4-трифторметил-фенил)-4,5-дигидро-1Н-1,2,4-триазолил))-бензилтио)-фенокси)-ацетат (далее называемый "Е-18");
Этил-2-(2,5-диметил-4-(1-(3-(4-n-бутил-5-оксо-1-(4-трифторметил-фенил)-4,5-дигидро-1Н-1,2,4-триазолил))-бензилтио)-фенокси)-ацетат (далее называемый "Е-19").
Соединение I (эфир) может быть гидролизовано в щелочных условиях с получением его в форме кислоты, далее в описании изобретения называемой "соединение I (кислота)". Предпочтительно, щелочные условия формируют с помощью щелочей, являющихся гидроксидами щелочных металлов, включая гидроксид натрия, гидроксид лития, гидроксид калия и т.п., но не ограничиваясь указанными; система растворителя, используемая при гидролизе, представляет собой С1-С4 спирт (например, метанол, этанол, пропанол, бутанол и т.п.) - воду (соотношение спирт:вода=9-1:1 (об./об.)), тетрагидрофуран (ТГФ) - воду (соотношение ТГФ:вода=9-1:1 (об./об.)), или спирт-дихлорметан-воду (соотношение спирт:дихлорметан:вода=9-1:9-1:1 (об./об.)); температура реакции составляет 0-80°С; предпочтительно 20-40°С; время реакции составляет 1-12 часов, предпочтительно 2-4 часа.
Предпочтительное соединение I (кислоты) по настоящему изобретению включает следующие соединения:
2-(2-метил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1H-1,2,4-триазолил))-бензилокси)-фенокси)-уксусную кислоту (далее называемую "А-1");
2-(2-метил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметилфен ил)-4,5-дигидро-1Н-1,2,4-триазолил))-бензилокси)-фенилтио)-уксусную кислоту (далее называемую "А-2");
2-(3-метил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1H-1,2,4-триазолил))-бензилокси)-фенилтио)-уксусную кислоту (далее называемую "А-3");
2-(2-метил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1H-1,2,4-триазолил))-бензилтио)-фенокси)-уксусную кислоту (далее называемую "А-4");
2-(2-этил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1H-1,2,4-триазолил))-бензилокси)-фенокси)-уксусную кислоту (далее называемую "А-5);
2-(3-метил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1H-1,2,4-триазолил))-бензилокси)-фенокси)-уксусную кислоту (далее называемую "А-6);
2-(3-метил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1Н-1,2,4-триазолил))-бензилтио)-фенокси)-уксусную кислоту (далее называемую "А-7");
2-(2-метил-4-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1Н-1,2,4-триазолил)-метилтио)-фенокси)-уксусную кислоту (далее называемую "А-8");
2-(2-метил-4-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1Н-1,2,4-триазолил)-метокси)-фенокси)-уксусную кислоту (далее называемую "А-9");
2-(2,5-диметил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1H-1,2,4-триазолил))-бензилтио)-фенокси)-уксусную кислоту (далее называемую "А-10");
2-(2,5-диметил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1Н-1,2,4-триазолил))-бензилокси)-фенилтио)-уксусную кислоту (далее называемую "А-11");
2-(3-метил-4-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1H-1,2,4-триазолил)-метокси)-фенилтио)-уксусную кислоту (далее называемую "А-12");
2-метил-2-(2-метил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1Н-1,2,4-триазолил))-бензилтио)-фенокси)-уксусную кислоту (далее называемую "А-13");
2-(2-этил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1H-1,2,4-триазолил))-бензилтио)-фенокси)-уксусную кислоту (далее называемую "А-14");
2,2-диметил-2-(3-метил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1H-1,2,4-триазолил))-бензилокси)-фенилтио)-уксусную кислоту (далее называемую "А-15");
2,2-диметил-2-(2-метил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1H-1,2,4-триазолил))-бензилтио)-фенокси)-уксусную кислоту(далее называемую "А-16");
2-(3-метил-4-(1-(3-(4-n-бутил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1Н-1,2,4-триазолил))-бензилокси)-фенилтио)-уксусную кислоту (далее называемую "А-17");
2,2-диметил-2-(2-метил-4-(1-(3-(4-n-бутил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1H-1,2,4-триазолил))-бензилтио)-фенокси)-уксусную кислоту(далее называемую "А-18");
2-(2,5-диметил-4-(1-(3-(4-n-бутил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1H-1,2,4-триазолил))-бензилтио)-фенокси)-уксусную кислоту (далее называемую "А-19").
Наиболее предпочтительное соединение I (кислота) согласно настоящему изобретению включает следующие соединения:
2-(3-метил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1H-1,2,4-триазолил))-бензилокси)-фенилтио)-уксусную кислоту (А-3);
2-(2-метил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1H-1,2,4-триазолил))-бензилтио)-фенокси)-уксусную кислоту (А-4);
2-(2,5-диметил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1Н-1,2,4-триазолил))-бензилокси)-фенилтио)-уксусную кислоту (А-11);
2-(2-этил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1H-1,2,4-триазолил))-бензилтио)-фенокси)-уксусную кислоту (А-14);
2,2-диметил-2-(3-метил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1Н-1,2,4-триазолил))-бензилокси)-фенилтио)-уксусную кислоту (А-15);
2,2-диметил-2-(2-метил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1Н-1,2,4-триазолил))-бензилтио)-фенокси)-уксусную кислоту (А-16);
2-(3-метил-4-(1-(3-(4-n-бутил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1H-1,2,4-триазолил))-бензилокси)-фенилтио)-уксусную кислоту(А-17);
2,2-диметил-2-(2-метил-4-(1-(3-(4-n-бутил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1Н-1,2,4-триазолил))-бензилтио)-фенокси)-уксусную кислоту (А-18);
2-(2,5-диметил-4-(1-(3-(4-n-бутил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1Н-1,2,4-триазолил))-бензилтио)-фенокси)-уксусную кислоту (А-19).
Фармацевтически приемлемая соль соединения формулы I согласно настоящему изобретению относится к солям указанного соединения, образованным щелочными или щелочноземельными металлами. Предпочтительными являются соли калия, натрия и кальция.
Сольват соединения формулы 1 согласно настоящему изобретению относится к гидрату указанного соединения или органическому сольвату, предпочтительно гидрату или алкоголяту. Гидрат может содержать 1-4 молекулы воды. Алкоголят может включать алкоголят, образованный метанолом, этанолом и пропанолом.
Еще одной целью изобретения является обеспечение нового соединения, т.е. соединения III:

в котором
Z представляет собой Cl или Вr;
R3 представляет собой Н или С1-С9 алкил;
R4 представляет собой Н, С1-С9 алкил, С3-С7 циклоалкил, фенил или замещенный фенил; группа-заместитель фенила выбрана из С1-С9 алкила, гидроксила, С1-С9 алкокси, меркапто, С1-С9 алкилтио, трифторметила, F, CI, Вr, нитро, NR5R6, COOR5, NR5COR6 или CONR5R6; и
G5, G6, G7, G8, и G9 независимо друг от друга представляют собой Н, С1-С9 алкил, гидроксил, С1-С9 алкокси, меркапто, С1-С9 алкилтио, трифторметил, F, Cl, Вr, нитро, NR5R6, COOR5, NR5COR6 или CONR5R6; R5 и R6 независимо друг от друга представляют собой Н или С1-С9 алкил.
Предпочтительное соединение III согласно настоящему изобретению включает следующие соединения:
3-(1'-бромбензил)-4-метил-1-(4-трифторметил)-фенил-1Н-1,2,4-триазол-5(4Н)-он (далее называемый "III-1")
3-(1'-бромбензил)-4-n-бутил-1-(4-трифторметил)-фенил-1Н-1,2,4-триазол-5(4Н)-он (далее называемый "III-2");
3-бромметил-4-метил-1-(4-трифторметилфенил)-1,4-дигидро-1,2,4-триазол-5-он (далее называемый "III-3").
Соединение III согласно настоящему изобретению может применяться в качестве промежуточного соединения для получения соединения формулы 1 согласно настоящему изобретению.
Еще одной целью изобретения является обеспечение способа получения соединения формулы III.
Соединение формулы III согласно настоящему изобретению может быть синтезировано в ходе реакции соединения формулы VI с хлорирующим или бромирующим реагентом:
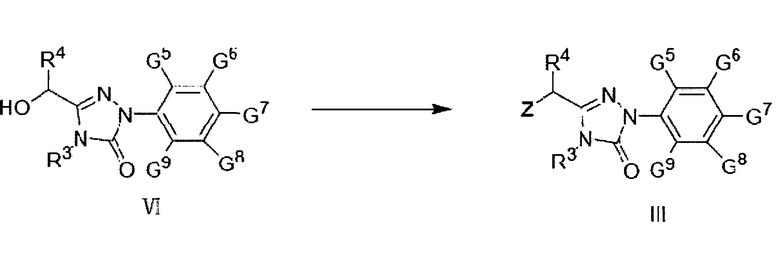
где
Z представляет собой Сl или Br; R3, R4, G5-G9 являются такими, как определено для соединения формулы III.
Хлорирующий или бромирующий реагент выбирают из:
(1) N-бромсукцинимида (NВS)/трифенилфосфина (Рh3Р);
(2) тионилхлорида (SОСl2) или тионилбромида (SOBr2);
(3) N-хлорсукцинимида (NCS)/трифенилфосфина (Рh3Р);
(4) тетрахлорметана (ССl4) или тетрабромметана (СВr4)/трифенилфосфина (Рh3Р);
(5) пентахлорида фосфора (PCl5) или пентабромида фосфора (PBr5);
(6) оксихлорида фосфора (РОСl3) или оксибромида фосфора (РОВr3); и
(7) трихлорида фосфора (РСl3) или трибромида фосфора (РВr3).
Растворитель выбирают из дихлорметана, хлороформа, тетрахлорметана и любой их смеси; температура реакции составляет 10-80°С; время реакции составляет 2-8 часов.
Соединение формулы III предпочтительно синтезируют согласно следующей схеме:

Вышеприведенная схема включает следующие этапы:
1) при использовании тетрагидрофурана (ТГФ) в качестве растворителя и гидрида натрия (NaH) в качестве щелочи бензиловый спирт 1 реагирует с бромидом 2, образуя эфир 3;
2) при использовании этанола (ЕtOН) в качестве растворителя эфир 3 аминируют амином 4 с образованием амида 5;
3) при использовании толуола (РhСН3) в качестве растворителя амид 5 реагирует с фенилгидразином 6 под действием оксихлорида фосфора (РОСl3), образуя гидразон 7;
4) при использовании тетрагидрофурана (ТГФ) в качестве растворителя гидразон 7 реагирует с карбонилдиимидазолом (CDI), образуя соединение 8;
5) при использовании этанола (ЕtOН) в качестве растворителя и палладированного угля (Pd/C) в качестве катализатора бензил отщепляют от соединения 8 путем гидрогенирования при нормальном давлении с образованием спирта 9;
6) при использовании дихлорметана (ДХМ) в качестве растворителя спирт 9 превращали в соединение III под действием хлорирующего или бромирующего реагента.
Например, если R4, G5, G6, G8, G9 представляют собой Н, R3 представляет собой метил, а G7 представляет собой трифторметил, схема синтеза выглядит следующим образом:
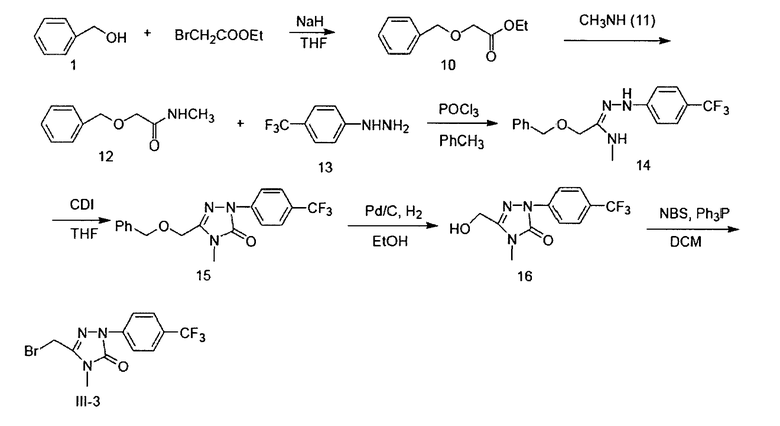
Вышеприведенная схема включает следующие этапы:
1) при использовании тетрагидрофурана (ТГФ) в качестве растворителя и гидрида натрия (NaH) в качестве щелочи, при нагревании с обратным холодильником бензиловый спирт 1 реагирует с этилбромацетатом, образуя этилбензилоксиацетат 10;
2) при использовании этанола (ЕtOН) в качестве растворителя этилбензилоксиацетат 10 аминируют метиламином 11 при комнатной температуре с образованием бензилоксиацетометиламина 12;
3) при использовании толуола (РhСН3) в качестве растворителя бензилоксиацетометиламин 12 реагирует с ρ-трифторметилфенилгидразином 13 при 80°С под действием оксихлорида фосфора (РОСl3), образуя гидразон 14;
4) при использовании тетрагидрофурана (ТГФ) в качестве растворителя гидразон 14 реагирует с карбонилдиимидазолом (CDI), образуя соединение 15;
5) при использовании этанола (ЕtOН) в качестве растворителя и палладированного угля (Pd/C) в качестве катализатора бензил отщепляют от соединения 15 при комнатной температуре путем гидрогенирования при нормальном давлении с образованием спирта 16;
6) при использовании дихлорметана (ДХМ) в качестве растворителя и N-бромсукцинимила (NBS) в качестве бромирующего реагента спирт 16 превращают в соединение III-3 под действием трифенилфосфина (Рh3Р).
Соединение формулы III также можно синтезировать в ходе реакции соединения формулы VI с хлорирующим или бромирующим агентом; при этом растворитель представляет собой хлороформ или тетрахлорметан, бромирующий агент - N-бромсукцинимид (NBS), хлорирующий агент - N-хлорсукцинимид (NCS), катализатор - дибензоилпероксид, температура реакции составляет 40-80°С, время реакции - 2-8 часов.
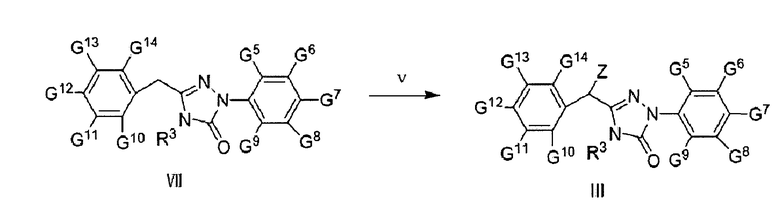
где
Z представляет собой Сl или Вr;
R3, G5-G9 являются такими, как определено для соединения формулы III;
G10-G14 независимо друг от друга или одновременно друг с другом представляют собой Н, С1-С9 алкил, гидроксил, С1-С9 алкокси, меркапто, С1-С9 алкилтио, трифторметил, F, Cl, Вr, нитро, NR5R6, COOR5, NR5COR6 или CONR5R6;
R5 и R6 независимо друг от друга представляют собой Н или С1-С9 алкил.
Соединение формулы III предпочтительно получают согласно следующей схеме:
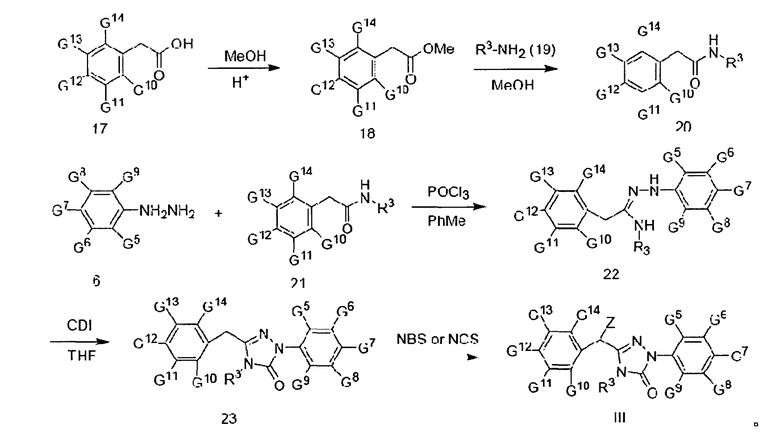
Вышеприведенная схема включает следующие этапы:
1) при использовании метанола (МеОН) в качестве растворителя и серной кислоты в качестве катализатора кислоту 17 подвергают этерификации для получения эфира 18;
2) при использовании метанола (МеОН) в качестве растворителя эфир 18 аминируют амином 19 с образованием амида 20;
3) при использовании толуола (РhСН3) в качестве растворителя амид 20 реагирует с фенилгидразином 6 под действием оксихлорида фосфора (РhСl3), образуя гидразон 22;
4) при использовании тетрагидрофурана (ТГФ) в качестве растворителя гидразон 22 реагирует с карбонилдиимидазолом (CDI), образуя соединение 23;
5) при использовании хлороформа в качестве растворителя, N-бромсукцинимида (NBS) в качестве бромирующего реагента (или N-хлорсукцинимида (NCS) в качестве хлорирующего реагента) и дибензоилпероксида в качестве катализатора соединение 23 бромируют (или хлорируют) с образованием соединения III.
Например, если R4 представляет собой фенил, G5, G6, G8, G9 представляют собой Н, R3 представляет собой метил, а G7 представляет собой трифторметил, схема синтеза имеет следующий вид:
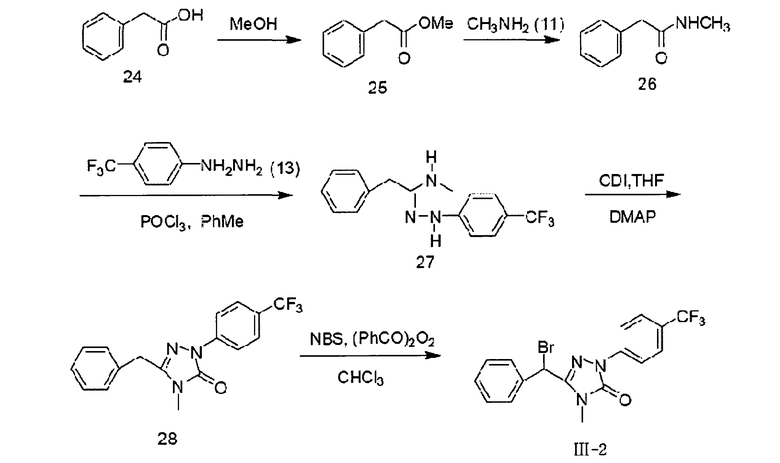
Вышеприведенная схема включает следующие этапы:
1) при использовании метанола (МеОН) в качестве растворителя и серной кислоты в качестве катализатора, при нагревании с обратным холодильником кислоту 24 подвергают этерификации с образованием эфира 25;
2) при использовании метанола (МеОН) в качестве растворителя эфир 25 аминируют амином 11 при комнатной температуре с образованием амида 26;
3) при использовании толуола (РhСН3) в качестве растворителя амид 26 реагирует с ρ-трифторметилфенилгидразином 13 под действием оксихлорида фосфора (РОСl3) при 80°С, образуя гидразон 27;
4) при использовании тетрагидрофурана (ТГФ) в качестве растворителя гидразон 27 реагирует с карбонилдиимидазолом (CDI) при комнатной температуре, образуя соединение 28;
5) при использовании хлороформа в качестве растворителя, N-бромсукцинимида (NBS) в качестве бромирующего реагента и дибензоилпероксида в качестве катализатора, при нагревании с обратным холодильником соединение 28 бромируют с образованием соединения III-1.
Еще одной целью изобретения является обеспечение способа получения соединения формулы (I).
Соединение формулы (I) согласно изобретению получают способом, изложенным ниже в Схеме 1 или Схеме 2:
Схема 1:

Где:
X, Y, R1-R4, G1-G9 являются такими, как определено для соединения формулы I, R представляет собой Н или С1-С9 алкил, предпочтительно метил или этил.
На Схеме 1 промежуточные соединения II и III подвергают взаимодействию под действием щелочи с образованием соединения I (эфира). Щелочь представляет собой органическую или неорганическую щелочь, причем неорганическая щелочь может включать карбонаты щелочных металлов, растворимые карбонаты щелочноземельных металлов, карбонат аммония и т.д. или любую их смесь; примеры неорганической щелочи включают карбонат натрия, карбонат калия, карбонат стронция, карбонат аммония и т.д., а органическая щелочь может представлять собой триэтиламин и т.п.; раствор предпочтительно выбирают из ацетонитрила, диметилформамида (ДМФ), диметилсульфоксида (ДМСО), тетрагидрофурана (ТГФ), диоксана и т.д. или любых смесей этих веществ. Предпочтительно, температура реакции составляет 0-100°С; предпочтительно - 40-80°С; время реакции составляет 1-12 часов, предпочтительно 4-8 часов.
На вышеприведенной схеме соединение формулы II может быть полученным коммерческим путем или синтезируемым согласно стандартному способу, описанному в уровне техники (например, способ, изложенный в М. L. Sznaidman, Curt D. Haffner, Patric R. et al., Bioorg. Med. Chem. Lett. 2003, 13, 1517-1521; Zhi-liang wei et al., J. Org. Chem. 2003, 68, 9116-9118; Org. Syn. Coll. Vol 1, 102, 1941; Org. Syn. Coll. Vol 2, 290, 1943; Handbook of Fine Organic Chemical Raw Materials and Intermediate. XU Ke-xun (Eds), Scientific & Technological Industry Press, 3-426-3-584).
Соединение формулы II включает, но не ограничивается следующими соединениями:
Этил-2-(2-метил-4-гидрокси)-феноксиацетат (далее называемое "II-1");
Этил-2-(3-метил-4-гидрокси)-феноксиацетат (далее называемое "II-2");
Этил-2-(2-этил-4-гидрокси)-феноксиацетат (далее называемое "II-3");
Этил-2-(3-метил-4-гидрокси)-фенилтиоацетат (далее называемое "II-4");
Этил-2-(2-метил-4-гидрокси)-фенилтиоацетат (далее называемое "II-5");
Этил-2-(2,5-диметил-4-гидрокси)-фенилтиоацетат (далее называемое "II-6").
Схема 2:

Где:
X, Y, Z, R1-R4, G1-G9 являются такими, как определено для соединения формулы I;
R представляет собой Н или С1-С9 алкил, предпочтительно метил или этил.
На Схеме 2 в ходе непрерывной реакции с карбонатом в качестве щелочи соединение III сначала реагирует с соединением IV, а затем - с соединением V, без необходимости разделения промежуточных соединений в ходе реакции. Кроме ацетонитрила, в качестве растворителя можно использовать тетрагидрофуран (ТГФ), диоксан и т.п. или любые смеси указанных веществ.
Кроме карбоната калия, в качестве карбоната можно использовать карбонат натрия, карбонат стронция, карбонат аммония и т.п. или их любые смеси. И промежуточное соединение IV, и промежуточное соединение V коммерчески доступны.
На Схеме 2 соединение I (эфир) может подвергаться гидролизу в щелочных условиях, образуя соединение I (кислоту), если R представляет собой С1-С9 алкил, указанные щелочные условия можно получить с помощью щелочи, например гидроксида щелочного металла, включая, но не ограничиваясь указанными, гидроксид натрия, гидроксид лития, гидроксид калия и т.п., или любую смесь указанных соединений; система растворителя, используемая при гидролизе, представляет собой С1-С4 спирт (например, метанол, этанол, пропанол, бутанол и т.п.) - воду (спирт: вода=9-1:1 (об./об.)), ТГФ-воду (ТГФ: вода=9-1:1 (об./об.)) или спирт-дихлорметан-воду (спирт: дихлорметан: вода=9-1:9-1:1 (об./об.)); температура реакции составляет 0-80°С, предпочтительно 20-40°С; время реакции составляет 1-12 часов, предпочтительно 2-4 часа.
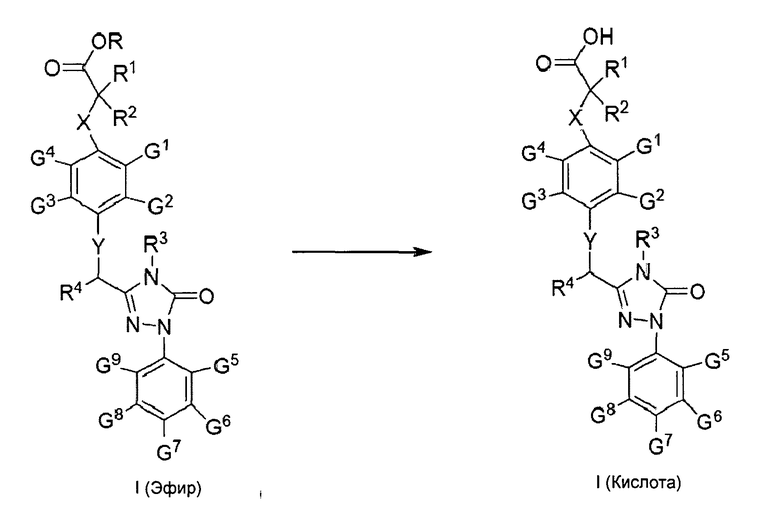
Где:
X, Y, Z, R1-R4, G1-G9 являются такими, как определено для соединения формулы I;
R представляет собой Н или С1-С9 алкил, предпочтительно метил или этил.
Еще одной целью изобретения является разработка фармацевтической композиции, содержащей вышеупомянутое соединение формулы (I) в качестве активного ингредиента.
Фармацевтическая композиция согласно настоящему изобретению, содержащая вышеупомянутое соединение формулы (I) в качестве активного ингредиента, содержит соединение формулы (I) и общепринятые вспомогательные вещества, применяемые для приготовления фармацевтических составов.
Общепринятые вспомогательные вещества, используемые для приготовления фармацевтических составов, относятся к одобренным компетентным ведомством для введения лекарственных средств и отвечают критериям для фармацевтических вспомогательных веществ. Их подразделяют на две группы на основе их различающейся функциональности: одна группа вспомогательных веществ необходима для обработки и производства фармацевтических составов и включает разбавители, связующие агенты, глиданты, суспендирующие агенты, смазочные вещества и т.д.; функция другой группы вспомогательных веществ, включающей разрыхлители, сорастворители и т.д., заключается в стимуляции переваривания и поглощения лекарственного средства in vivo. Они не обладают активностью in vivo в организме человека и не проявляют ни терапевтического, ни токсического действия.
Среди вышеупомянутых вспомогательных веществ разбавители могут быть выбраны из любой смеси любых двух или более из следующих веществ: крахмал, модифицированный крахмал, сахароза, моногидрат лактозы, безводная лактоза, глюкоза, маннит и различные микрокристаллические модификации целлюлозы.
Среди вышеупомянутых вспомогательных веществ связующие агенты могут быть выбраны из любой смеси любых двух или более из следующих веществ: гидроксипропилметилцеллюлоза, прежелатинизированный крахмал, поливидон (поливинилпирролидон), карбоксиметилцеллюлоза и их производные, метилцеллюлоза, этилцеллюлоза, крахмал, углеводы и т.п.; предпочтительно из гидроксипропилметилцеллюлозы, прежелатинизированного крахмала и поливидона.
Среди вышеупомянутых вспомогательных веществ скользящие вещества могут быть выбраны из любой смеси любых двух или более из следующих веществ: стеарат магния, порошкообразный тальк и гидрогенизированные растительные масла I типа.
Среди вышеупомянутых вспомогательных веществ суспендирующие агенты могут быть выбраны из любой смеси любых двух или более из следующих веществ: желатин, пектин, гуммиарабик, альгинат натрия, метилцеллюлоза, этилцеллюлоза, гидроксипропилцеллюлоза, карбоксиметилцеллюлоза и метилцеллюлоза.
Среди вышеупомянутых вспомогательных веществ разрыхлители могут быть выбраны из любой смеси любых двух или более из следующих веществ: крахмал, гидроксипропилцеллюлоза с низкой степенью замещения, натрий-карбоксиметилкрахмал, кальций-карбоксиметилцеллюлоза, поперечно сшитый поливидон, поперечно сшитая целлюлоза и поперечно сшитая натрий-карбоксиметилцеллюлоза.
Среди вышеупомянутых вспомогательных веществ сорастворители могут быть выбраны из любой смеси любых двух или более из следующих веществ: серия Span, серия Твин, серия полиэтиленгликоль, соевый лецитин и т.п.
Вышеупомянутая фармацевтическая композиция может быть представлена в любой форме из следующих лекарственных форм для перорального введения: 1, гладкие таблетки; 2, таблетки, покрытые пленочной оболочкой; 3, драже; 4, таблетки с энтеросолюбильным покрытием; 5, диспергируемые таблетки; 6, капсулы; 7, гранулы; 8, суспензии; и 9, растворы.
Вышеупомянутые лекарственные формы можно изготовить согласно общепринятой технологии приготовления лекарственного средства.
Еще одной целью изобретения является обеспечение применения соединения формулы (I) для приготовления лекарственного средства для лечения или предотвращения заболевания, которое можно лечить или предотвратить путем активации рецептора активатора пролиферации пероксисом подтипа δ (PPARδ). Указанное заболевание включает метаболический синдром, ожирение, дислипидемию, патологическую гликемию, резистентность к инсулину, сенильную деменцию или опухоли и т.д.
Согласно изобретению, обеспечивается применение соединения формулы (I) для приготовления лекарственного средства для лечения или предотвращения заболевания, которое можно лечить или предотвратить путем активации рецептора активатора пролиферации пероксисом подтипа δ (PPARδ) δ (PPARδ), причем указанное заболевание выбирают из одного или нескольких из следующих заболеваний: метаболический синдром, ожирение, дислипидемия, патологическая гликемия, резистентность к инсулину, сенильная деменция или опухоли и т.д.
Согласно изобретению, обеспечивается способ лечения или предотвращения заболевания, которое можно лечить или предотвратить путем активации рецептора активатора пролиферации пероксисом подтипа δ (PPARδ), причем указанный способ включает этап введения пациенту терапевтически или профилактически эффективного количества соединения формулы (I), а указанное заболевание выбирают из одного или нескольких следующих заболеваний: метаболического синдрома, ожирения, дислипидемии, патологической гликемии, резистентности к инсулину, сенильной деменции или опухолей и т.д.
После ознакомления с описанием изобретения для специалиста в области техники будут очевидны другие преимущества и варианты применения настоящего изобретения.
ОПИСАНИЕ ФИГУР
Фигура 1 представляет кривую изменений массы тела животного со временем, которая демонстрирует действие лекарственного средства на массу тела животного и потребление пищи. На оси абсцисс показано время взвешивания (взвешивание каждые 2 суток); на оси ординат показана масса тела животного. Положительный контроль представляет собой орлистат (две группы, получавшие препарат в дозах 30 мг/кг и 10 мг/кг). В настоящем изобретении животных, получавших составы лекарственного средства (А-3 и А-11), разделили на две группы по дозировке (10 мг/кг и 3,3 мг/кг).
Фигура 2 представляет кривую изменений уровня глюкозы в крови животных, которая демонстрирует результаты перорального теста на толерантность к глюкозе. На оси абсцисс показано время отбора образцов крови - 0 мин, 30 мин, 60 мин, 120 мин после перорального приема глюкозы; на оси ординат представлен соответствующий уровень глюкозы в крови (ммоль/л).
Фигура 3 представляет кривую лекарственное средство-время для А-3 (HS060098) и А-1 (HS060001) согласно настоящему изобретению. На оси абсцисс показано время (ч) отбора образцов крови; на оси ординат показаны концентрации (мкг/мл) лекарственного средства в плазме животных в разное время.
ВАРИАНТЫ РЕАЛИЗАЦИИ ИЗОБРЕТЕНИЯ
Настоящее изобретение подробно описано ниже со ссылками на конкретные примеры. Следует понимать, что в свете описания изобретения в настоящей заявке, специалисты в области техники могут вносить различные изменения и совершенствовать настоящее изобретение, не выходя за рамки настоящего изобретения, определенные прилагаемой формулой изобретения. Кроме того, следует понимать, что примеры, приведенные в настоящей заявке, предназначены только для иллюстративных целей и не должны интерпретироваться в качестве ограничения настоящего изобретения каким-либо образом.
В настоящей заявке радикал С1-С9 (алкил, алкокси, алкилтио и т.п.) включает С1-С2, С1-С3, С1-С4, С1-С5, С1-С6, С1-С7, С1-С8 и С1-С9 радикал (алкил, алкокси, алкилтио и т.п.) и т.д.
Модель для скрининга лекарственного средства
Скрининг агента, активирующего ядерный рецептор in vitro
Процедура эксперимента в рамках модели скрининга состоит в следующем.
1. Краткое описание модели скрининга, ассоциированной с ядерным рецептором
Модель скрининга для исследования агониста ядерного рецептора в живой клетке разработали с использованием методик экспериментов с репортерными генами на основе представления о том, что активированный ядерный рецептор способен активировать транскрипцию гена, расположенного справа от его сайта инициации. Сконструировали плазмиду с репортерным геном, в которой последовательность ДНК, связывающая ядерный рецептор (НРЭ), вставлена слева от гена люциферазы, так что экспрессия гена люциферазы находится под контролем ядерного рецептора. Затем в клетку одновременно ввели плазмиду с репортерным геном и ядерный рецептор; активация ядерного рецептора происходила при наличии агониста ядерного рецептора в культуральной среде. Активированный рецептор был способен индуцировать экспрессию гена люциферазы, при том что количество люциферазы можно было определять по ее светящемуся субстрату. Таким образом можно было определить интенсивность активации ядерного рецептора соединением, регистрируя интенсивность люминесценции. Для определения ошибки исследования, вызванной такими факторами, как эффективность трансфекции, количество высеянных клеток, токсичность соединения и т.д., в качестве внутреннего стандарта клетки одновременно трансфицировали плазмидой, экспрессирующей GFP, и при анализе результатов эксперимента значения люминесценции во всех проверенных лунках калибровали по значениям GFP. Результаты эксперимента выражали в относительных единицах активации, где значение 1 соответствовало контролю с растворителем, а увеличение относительной единицы означало увеличение способности к активации.
2. Процедура эксперимента
Подробную процедуру эксперимента в рамках модели скрининга можно найти в следующей публикации: "Design, synthesis and evaluation of a new class of noncyclic 1,3-dicarbonyl compounds as PPARa selective activators", Bioorg Med Chem Lett. 2004; 14(13): 3507-11. Конкретное описание эксперимента выглядит следующим образом:
Реагенты для эксперимента: исследуемые соединения (20 мМ, растворы в ДМСО, хранение при -80°С).
(1) Сутки 1: Культивирование и посев клеток
Клетки гепатокарциномы HepG2 (полученные из Американской коллекции типовых культур АТСС) культивировали на среде DMEM, обогащенной 10% эмбриональной бычьей сыворотки, инактивированной нагреванием (FBs, Invitogen, Гранд-Айленд, штат Нью-Йорк, США) в культуральных сосудах Т-75 (Greiner, Германия) в инкубаторе при 37°С, относительной влажности 100% и 5% СO2. Когда клеточная культура в культуральном сосуде достигала 80-90% конфлюэнтности, ее гидролизовали 0,25% трипсином (с ЭДТА) в течение 3 мин и высевали в 96-луночный культуральный планшет с плотностью посева 2000 клеток/100 мкл/лунку.
(2) Сутки 2: Трансфекция клеток
На следующие сутки, когда рост клеток на 96-луночных культуральных планшетах достигал 50-80% конфлюэнтности, осуществляли трансфекцию клеток. Система совместной трансфекции клеток включает трансфицирующий агент FuGene6 (Roche Molecular Biochemicals, Индианаполис, штат Индиана, США) и 60 нг ДНК (10 нг hRXR, 10 нг pCMV βGal, 10 нг плазмиды, экспрессирующей ядерный рецептор RXR/PPARδ, 30 нг плазмиды, несущей репортерный ген GFP, соответственно).
(3) Обработка лекарственным средством
Культуральную среду удаляли через 24 часа после трансфекции и заменяли 200 мкл свежей среды DMEM, содержащей исследуемое лекарственное средство (с 10% FBS, обработанной активированным углем). Конечные концентрации исследуемого лекарственного средства составляли 10, 5, 1, 0,1, 0,01, 0,001 и 0 мкМ, положительный контроль представлял собой 0,05 мкМ 2-бромстеариновой кислоты (приобретенной в Sigma, США) с ДМСО в каждой лунке (конечная концентрация составляла 0,1%).
(4) Анализ киназной активности
Через 24 ч после обработки лекарственным средством клетки лизировали в лизирующем растворе (буфер для лизирования клеточных культур, Promega), центрифугировали и собирали надосадочную жидкость. Надосадочную жидкость обрабатывали реагентами из комплекта для обнаружения флуоресценции (Promega), регистрировали флуоресценцию на флуориметре (Ascent Fluoroskan FL reader, Thermo Labsystems, Финляндия) и определяли относительную активность люциферазы. Для анализа активности β-галактозидазы, использованной в эксперименте в качестве внутреннего стандарта (калибровка по внутреннему стандарту для оценки эффективности трансфекции), 50 мкл надосадочной жидкости переносили в чистый микропланшет, обрабатывали реактивами из комплекта Promega и считывали при длине волны 405 нм на планшет-ридере (Bio-tech Instruments Inc., Винуски, штат Вермонт, США) (Sauerberg, P.; Olsen, G. S.; Jeppesen, L; Mogensen, J. P. etal., J. Med. Chem., 2007, 50, 1495-1503).
3. Анализ:
Медианная эффективная концентрация (ЕС50) образца представляет собой концентрацию, при которой образец оказывает 50% фармакологический эффект. Это значение представляет собой один из важных параметров для оценки фармакологического эффекта соединения. В настоящем исследовательском процессе ЕС50 образца рассчитывали в соответствии с активацией рецептора образцом в шести различных концентрациях.
4. Результат скрининга
Было получено соединение формулы I, способное активировать рецептор PPARδ.
Данные по активности соединения формулы I (кислоты) in vitro
Активность соединения формулы I (кислоты) in vitro измеряли согласно следующим методикам: образец соединения формулы (I) (кислоты) растворяли и разбавляли до различных концентраций, активность образца в отношении активации рецептора PPARδ тестировали при его различных концентрациях, получали соответствие концентрации и эффекта и вычисляли соответствующее значение для медианной эффективной концентрации (ЕС50). Результаты исследования показаны в Таблице 1.
Примечание: чем ниже значение EC50 (нМ), тем выше активность.
На основе данных в Таблице 1 можно видеть, что все соединения согласно настоящему изобретению обладают агонистической активностью по отношению к PPARδ.
СКРИНИНГ ФАРМАКОДИНАМИЧЕСКОЙ АКТИВНОСТИ СОЕДИНЕНИЯ IN VIVO
1. Скрининг активности в отношении контроля липидов в крови
Анализ активности части соединений формулы I (кислот) по настоящему изобретению in vivo:
Из А-1 - А-19 выбрали четыре соединения и подвергли их анализу на активность in vivo. Эффекты взаимодействия лекарственных средств наблюдали на крысах линии Спраг Доули, мышах АроЕ, Mesocricetus auratus и др. моделях с гиперлипидемией, вызванной питанием с высоким содержанием жиров. Результаты показаны в Таблице 2.
Из Таблицы 2 можно видеть, что четыре соединения, которые были выбраны и подвергнуты анализу активности in vivo, способны снижать уровень холестерина (ОХ), триглицеридов (ТГ) и липопротеинов низкой плотности (ЛПНП), а также повышать уровень липопротеинов высокой плотности (ЛПВП), что указывает на исключительное действие соединений согласно изобретению на регуляцию липидов в крови. В то же время эти четыре соединения также оказывали аналогичное фармакологическое действие моделей на основе крысы линии Спраг Доули и мыши АроЕ и оказывали лучшее действие на регуляцию липидов в крови по сравнению с GW501516.
В частности, соединение А-3 способно не только снижать уровень холестерина (ОХ) до 40%, триглицеридов (ТГ) до 65% и липопротеинов низкой плотности (ЛПНП) до 51%, но и повышать уровень липопротеинов высокой плотности (ЛПВП) до 43%.
Впоследствии эффективность медикамента А-3 подтвердили на модели гиперлипидемии на основе Масаса rhesus. Через три месяца введения из бедренной вены отбирали образцы крови для определения гематологических показателей, а также определения инсулина, аполипопротеина А-1 (ароА-1) и аполипопротеина В-100 (ароВ-100) с помощью твердофазного ИФА. Результаты показали, что по сравнению с контрольной группой моделей гиперлипидемии, в сыворотке группы животных, подвергавшихся введению А-3, содержание общего холестерина (ОХ) снизилось на 45%, содержание липопротеинов низкой плотности (ЛПНП) снизилось на 38%, а содержание липопротеинов высокой плотности (ЛПВП) увеличилось на 67%. При этом с помощью твердофазного ИФА было обнаружено, что концентрация инсулина существенно увеличилась; концентрация ароА-1 существенно увеличилась, концентрация ароВ-100 существенно снизилась, а соотношение ароА-1 и ароВ-100 достигло уровня, характерного для нормальных животных, что полностью соответствовало результатам гематологического определения ЛПНП и ЛПВП. Вышеприведенные результаты показали, что после того как животным, страдающим гиперлипидемией, вводили лекарственное средство А-3, все показатели, определенные с помощью гематологического анализа и твердофазного ИФА, существенно менялись в направлении нормы, что, в свою очередь, демонстрировало, что терапевтическое действие этого медикамента было значительно большим, чем у GW501516.
2. Скрининг фармакологической активности в отношении снижения массы тела
Методика эксперимента
В качестве экспериментальных животных были выбраны самцы мышей C57BL/6J возрастом 3 недели, 15 из них были случайно выбраны для кормления стандартным кормом, а оставшихся кормили кормом с высоким содержанием жиров, причем каждая из мышей была помечена надрезом на ухе. Массу тела и корм, полученный мышами, взвешивали каждую неделю. После кормления в течение 15 недель мыши из группы, получавшей корм с высоким содержанием жиров, весили в среднем 42 г, мыши из группы, получавшей стандартный корм, весили в среднем 28 г. В дальнейшем мышей и из модельной группы, и из группы, получавшей препараты, кормили кормом с высоким содержанием жиров. После введения соединений согласно изобретению в течение четырех недель наблюдали действие лекарственного средства на уровень липидов и глюкозы в крови, массу тела и потребление корма животными, в то время как для изучения действия лекарственного средства на толерантность к глюкозе у животных выполняли анализ толерантности к глюкозе.
(1) Действие медикаментов на уровни липидов и глюкозы в крови животных
Как показано в Таблице 3, А-3 и А-11 способны существенно снижать уровни ТГ и глюкозы в сыворотке животных-моделей, демонстрируя лучший эффект, чем орлистат.
(2) Действие лекарственного средства на массу тела и потребление пищи животными
Как показано в Таблице 4 и на Фиг,1, А-3 и А-11 способны существенно снижать массу тела животных и не оказывают существенного влияния на потребление пищи животными, что указывает на исключительное действие лекарственного средства в отношении стимуляции потери веса.
(3) Пероральный анализ на толерантность к глюкозе
Результаты показаны на Фиг.2, т.е. начальный уровень глюкозы не может быть восстановлен в течение 2 часов у животной модели, страдающей ожирением, кривая толерантности к глюкозе не соответствует норме и толерантность к глюкозе снижена, в то время как А-3 и А-11 способны существенно улучшить толерантность к глюкозе у животных, страдающих ожирением.
3. Фармакокинетическое исследование
1. Кривая "концентрация в плазме - время"
Методика эксперимента:
Отобрали 36 крыс линии Спраг Доули с массой тела в диапазоне 150-170 г при соотношении полов 1:1, распределили их на две экспериментальные группы и вводили им А-1 и А-3, соответственно; в каждой экспериментальной группе выделили 3 экспериментальные подгруппы по шесть животных в каждой при соотношении полов 1:1, получавшие концентрации лекарственного средства 50, 10 и 2 мг/кг. Все крысы в течение 12 часов содержались без пищи и с неограниченным доступом к воде. Объем дозы составил 10 мл/кг, образцы крови объемом 0,4-0,5 мл отбирали из задней глазничной вены бодрствующих животных в различные моменты времени после промывания желудка и центрифугировали при 5000 об/мин в течение 10 мин. Точно отмеряли 200 мкл образца плазмы, добавляли 200 мкл ацетонитрила, встряхивали до равномерности, оставляли постоять в течение 5 мин и центрифугировали (14000 об/мин, 5 мин). Надосадочную жидкость собирали и вновь центрифугировали (14000 об/мин, 5 мин). Полученную надосадочную жидкость собирали и подвергали ВЭЖХ. Образцы крови отбирали в моменты времени, соответствующие 0, 0,5, 1, 2, 4, 6, 8, 10, 12, 24, 36, 48 и 72 ч, площадь пика для каждого момента времени усредняли для образцов, отобранных у 6 крыс в один и тот же момент времени. Рассчитывали концентрации в плазме и составляли график "концентрация лекарственного средства - время".
Результаты показаны на Фиг.3.
Результаты показывают, что t1/2 для А-3 составило 10-12 ч, t1/2 для А-1 составило 26-28 ч, биодоступность А-3 составила 86%, а биодоступность А-1 составила 15%.
2. Распределение в тканях
18 крыс линии Спраг Доули в течение 16 ч содержали без пищи со свободным доступом к воде, распределили их на 3 группы по 6 животных/группу при соотношении полов 1:1 и вводили им HS060098 (10 мг/кг), затем через 1, 10 и 24 ч после введения животных убивали и быстро удаляли у каждого животного сердце, печень, селезенку, легкие, почки. С поверхности каждого органа немедленно смывали кровь физиологическим раствором. Ткани органа вырезали, тщательно промывали, удаляли из них жидкость, взвешивали, добавляли физиологический раствор в количестве 400 мг/мл, гомогенизировали на ледяной бане в течение 1 мин и центрифугировали при 3750 об/мин в течение 20 мин. Отбирали пипеткой ровно 400 мкл надосадочной жидкости, затем добавляли 40 мкл внутреннего стандарта (в качестве внутреннего стандарта использовали HS060001 в молярной концентрации 5 ммоль/л), разбавляли 3 мл очищенной воды, перемешивали до гомогенности и загружали в колонку (колонку SPE С18 вначале промывали 3 мл очищенной воды, а затем 3 мл 10% метанола для активации). Колонку промывали 3 мл очищенной воды при скорости 1 мл/мин и элюировали 3 мл элюирующего агента (метанол: очищенная вода=9:1). Элюат собирали и сушили в потоке азота при 45°С. Остаток растворяли в 100 мкл ацетонитрила, помещали в ВЭЖХ-инжектор и затем вводили образец.
Результаты показаны в Таблице 5. Результаты означают, что А-3 и А-1 распределяются главным образом в печени, а А-3 распределяется относительно равномерно.
ПРИМЕР
Получение промежуточного соединения II:
Пример 1: Получение соединения II-1
Получение этил-2-(2-метил-4-гидрокси)-феноксиацетата (II-1)

В одногорлую колбу объемом 500 мл последовательно добавляли при перемешивании 3-метил-4-гидроксиацетофенон (25 г, 166,5 ммоль), 250 мл ацетонитрила и карбонат калия (К2СО3, 26 г, 188,2 ммоль). Затем по каплям добавляли этилбромацетат (20 мл, 172,2 ммоль) в ацетонитриле (50 мл). Реакционную смесь перемешивали при комнатной температуре в течение 8 часов. После завершения реакции реакционную смесь разбавляли этилацетатом (200 мл), фильтровали и выпаривали, получая желтое масло.
В еще одной одногорлой колбе объемом 500 мл к перемешиваемому раствору вышеуказанного желтого масла в дихлорметане (CH2Cl2, 300 мл) добавляли 3-хлорнадбензойную кислоту (46 г, 199,9 ммоль) и каталитическое количество 4-метилбензолсульфоновой кислоты. Полученную смесь перемешивали в течение ночи при комнатной температуре. После завершения реакции реакционную смесь фильтровали и промывали полученный фильтрационный осадок 100 мл дихлорметана. Объединенный фильтрат промывали насыщенным водным раствором гипосульфита натрия (Na2S2SO4) и гидрокарбоната натрия (NаНСО3) и затем выпаривали, получая бледно-желтое масло.
Неочищенный продукт, полученный в ходе вышеописанной реакции, переносили в еще одну одногорлую колбу объемом 500 мл и добавляли 80 мл этанола и каталитическое количество 4-метилбензолсульфоновой кислоты. Раствор нагревали с обратным холодильником в течение 6 ч и затем выпаривали в вакууме, получая бледно-желтое твердое вещество. Указанное бледно-желтое твердое вещество перекристаллизовывали из петролейного эфира/этилацетата (2:1 об./об.), получая этил-2-(2-метил-4-гилрокси)-феноксиацетат (16 г, 45,8% в три стадии) в виде белого твердого вещества.
Структуру полученного соединения описывали на основе следующих данных, полученных с помощью спектроскопии ядерного магнитного резонанса:
1Н ЯМР (400 МГц, CDCl3) δ 1,34 (t, J=7,14 Гц, 3Н), 2,27 (s, 3Н), 4,30 (q, J=7,14 Гц, 2H), 4,61 (s, 2H), 6,61-6,63 (m, 1H), 6,65 (s, 1H), 6,67-6,68 (m, 1H), 7,31 (s, 1H).
Пример 2: Получение соединения II-2
Используя 3-метил-4-гидроксиацетофенон в качестве исходного материала, получали этил-2-(3-метил-4-гидрокси)-феноксиацетат (II-2) согласно методике, аналогичной методике, описанной в Примере 1, в виде белого твердого вещества с выходом 40,2%.
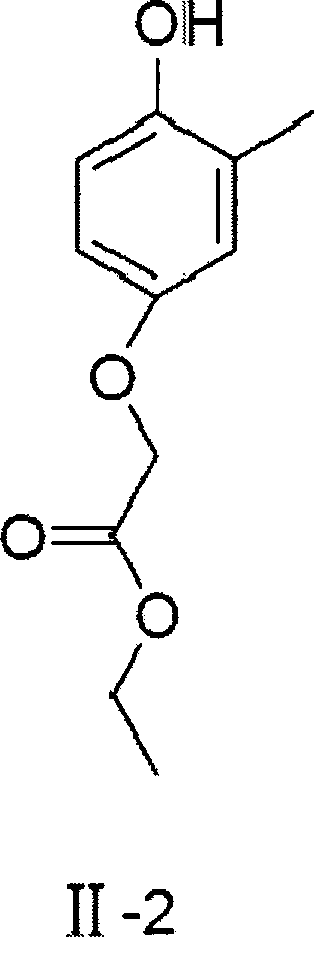
Структуру полученного соединения описывали на основе следующих данных, полученных с помощью спектроскопии ядерного магнитного резонанса:
1H ЯМР (400 МГц, ДМСО) δ 1,23 (t, J=7,12 Гц, 3Н), 2,29 (s, 3Н), 4,18 (q, J=7,14 Гц, 2H), 4,64 (s, 2H), 6,57-6,60 (m, 1H), 6,68 (s, 1H), 6,69-6,70 (m, 1H), 8,87 (s, 1H); 13С ЯМР (100 МГц, ДМСО) δ 14,5, 16,6, 60,9, 65,9, 112,2, 112,8, 115,4, 117,6, 125,2, 150,2, 150,8, 169,6.
Пример 3: Получение соединения II-3
Используя 3-этил-4-гидроксиацетофенон в качестве исходного материала, получали этил-2-(2-этил-4-гидрокси)-феноксиацетат (11-3) согласно методике, аналогичной методике, описанной в Примере 1, в виде бледно-желтого твердого вещества с выходом 74,5%.

Структуру полученного соединения описывали на основе следующих данных, полученных с помощью спектроскопии ядерного магнитного резонанса:
1H ЯМР (400 МГц, СDСl3) δ 1,23 (t J=7,53 Гц, 3Н), 1,34 (t J=7,12 Гц, 3Н), 2,69 (q, J=7,52 Гц, 2Н), 4,30 (q, J=7,14 Гц, 2Н), 4,61 (s, 2H), 5,35 (s, 1H), 6,6 (dd, J=8,65 Гц, 2,84 Гц, 1H), 6,65 (d, J=8,63 Гц, 1H), 6,72 (d, J=2,84 Гц, 1H); 13С ЯМР (100 МГц, CDCI3) δ 14,0, 14,1, 23,1, 61,3, 66,7, 112,6, 113,1, 116,5, 134,9, 149,9, 150,4, 169,8.
Пример 4: Получение соединения II-4
Получение этил-2-(3-метил-4-гидрокси)-фенилтиоацетата (II-4)

В трехгорлую колбу объемом 500 мл последовательно добавляли при перемешивании о-крезол (15 г, 138,7 ммоль), тиоцианат натрия (NaSCN, 34 г, 419,3 ммоль), бромид натрия (NaBr, 16 г, 155,5 ммоль) и метанол (200 мл). Смесь охлаждали до 0°С и по каплям добавляли раствор брома (8,6 мл, 167 ммоль) в метаноле (30 мл). Через 1 час перемешивания при 0°С смесь нагревали до комнатной температуры и перемешивали в течение следующих 4 часов при комнатной температуре. После завершения реакции к реакционной смеси добавляли 200 мл насыщенного водного раствора гидрокарбоната натрия (NaHCО3) и перемешивали в течение 10 мин. Смесь экстрагировали этилацетатом (2 Х 500 мл). Объединенный органический слой высушивали над безводным сульфатом магния, фильтровали, концентрировали при пониженном давлении, получая желтое масло. Выполняли очистку методом колоночной хроматографии на силикагеле (силикагель Н: 300-400 меш; петролейный эфир/этилацетат=8:1 об./об.), получая 3-метил-4-гидрокси-фенилтиоциановую кислоту в виде белого твердого вещества (16 г, выход: 69,8%).
Структуру полученного соединения описывали на основе следующих данных, полученных с помощью спектроскопии ядерного магнитного резонанса:
1H ЯМР (400 МГц, ДМСО) δ 2,17 (s, 3Н), 6,92(d, J=8,4 Гц, 1Н), 7,37 (dd, J=8,4 Гц, 2,48 ГЦ, 1 Н), 7,44 (d,J=2,48 ГЦ, 1Н), 10,1 (s, 1Н); 13С ЯМР (100 МГц, ДМСО) δ 16,2, 111,1, 113,1, 116,8, 127,3, 132,2, 135,4, 158,3.
В трехгорлой колбе объемом 500 мл к перемешиваемой смеси алюмотетрагидрида лития (LiAlH4, 3,5 г, 92,2 ммоль) и тетрагидрофурана (ТГФ, 50 мл) по каплям добавляли раствор 3-метил-4-гидроксифенилтиоциановой кислоты (6,13 г, 37,1 ммоль) в тетрагидрофуране (ТГФ, 30 мл) при 0°С. Через 30 мин перемешивания при 0°С смесь нагревали до комнатной температуры и затем перемешивали в течение 1 часа при комнатной температуре. Реакцию останавливали добавлением этанола (10 мл). Значение рН смеси доводили до 3-4 добавлением 6 М соляной кислоты на водоледяной бане. Затем экстрагировали водный слой этилацетатом (3 Х 100 мл). Комбинированный органический слой высушивали над безводным сульфатом магния, фильтровали и выпаривали в вакууме, получая неочищенный продукт 4-гидрокси-3-метил тиофенола в виде желтого масла.
В одногорлой колбе объемом 250 мл к раствору вышеуказанного неочищенного продукта 4-гидрокси-3-метилтиофенола в ацетонитриле (80 мл) при перемешивании добавляли этил-2-бромацетат (4,3 мл, 37 ммоль), с последующим добавлением карбоната калия (5,1 г, 36,9 ммоль). Полученную смесь перемешивали в течение 8 часов при комнатной температуре. После завершения реакции реакционную смесь разбавляли этилацетатом (80 мл), фильтровали и выпаривали, получая остаток. Остаток очищали методом колоночной хроматографии на силикагеле (силикагель Н:300-400 меш; петролейный эфир/этилацетат=8:1 об./об.), получая 6,38 г этил-2-(3-метил-4-гидрокси)-фенилтиоацетата в виде желтого масла (выход: 76%).
Структуру полученного соединения описывали на основе следующих данных, полученных с помощью спектроскопии ядерного магнитного резонанса:
1Н ЯМР (400 МГц, CDCl3) δ 1,24 (t J=7,2 Гц, 3Н), 2,18 (s, 3Н), 3,49 (s, 2H), 4,15 (q, J=7,14 Гц, 2H), 5,74 (s, 1Н), 6,63 (d, J=8,27 Гц, 1Н), 7,17 (dd, J=8,16 Гц, 2,28 ГЦ, 1Н), 7,25 (d, J=2,28 Гц, 1Н); 13С ЯМР (100 МГц, СDСl3) δ 14,1, 15,7, 38,8, 61,6, 115,6, 124,1, 125,1, 131,9,135,7, 154,5, 170,6.
Пример 5: Получение соединения II-5
Используя m-крезол в качестве исходного материала, получали этил 2-(2-метил-4-гидрокси)-фенилтиоацетат (II-5) согласно методике, аналогичной методике, описанной в Примере 4 в виде желтого масла с выходом 63%.

Структуру полученного соединения описывали на основе следующих данных, полученных с помощью спектроскопии ядерного магнитного резонанса:
1Н ЯМР (400 МГц, СDCl3) δ 1,23 (t J=7,1 Гц, 3Н), 2,39 (s, 3H), 3,44 (s, 2H), 4,14 (q, J=7,11 Гц, 2H), 6,38 (s, 1Н), 6,54 (dd, J=8,36 Гц, 2,72 Гц, 1Н), 6,64 (d, J=2,71 Гц, 1Н), 7,31 (d, J=8,37H4, 1Н).
Пример 6: Получение соединения II-6
Используя 2,5-диметилфенол в качестве исходного материала, получали этил 2-(2,5-диметил-4-гидрокси)-фенилтиоацетат (II-6) согласно методике, аналогичной методике, описанной в Примере 4 в виде белого твердого вещества с выходом 72%.

Структуру полученного соединения описывали на основе следующих данных, полученных с помощью спектроскопии ядерного магнитного резонанса:
1H ЯМР (400 МГц, CDCI3) δ 1,23 (t J=7,16 Гц, 3Н), 2,16 (s, 3H), 2,38 (s, 3H), 3,43 (s, 2H), 4,13 (q, J=7,13 Гц, 2Н), 5,19 (s, 1H), 6,58 (s, 1H), 7,23 (s, 1H); 13С ЯМР (100 МГц, CDCl3) δ 14,1, 15,1, 20,2, 37,9, 61,4, 116,9, 122,1, 123,3, 136,9, 140,0, 154,3, 170,3.
Получение промежуточного соединения III:
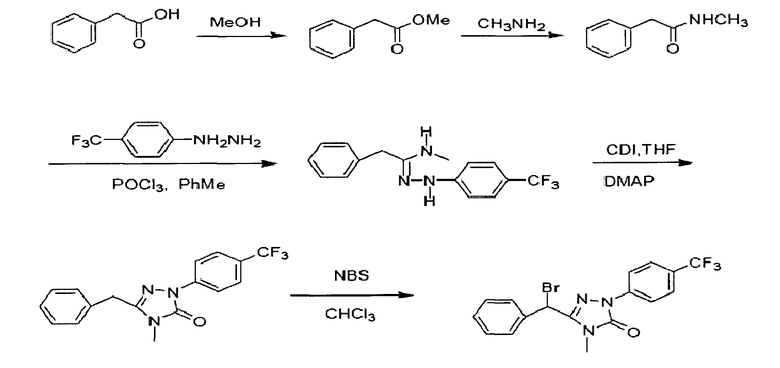
Пример 7: Получение соединения III-1
3-(1'-бромбензил)-4-метил-1-(4-трифторметил)-фенил-1H-1,2,4-триазол-5(4Н)-он (соединение 111-1) можно получить согласно следующей реакционной схеме:
Получение N-метил фенилацетамида
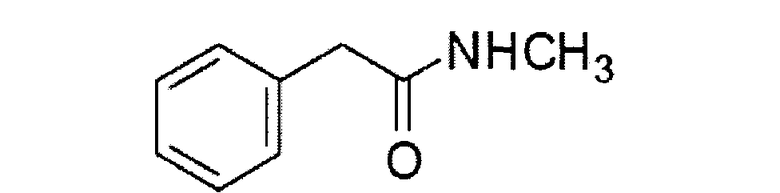
Фенилуксусную кислоту (10 г, 73,5 ммоль) и метанол (60 мл) добавляли в трехгорлую колбу объемом 250 мл с последующим добавлением 10 капель концентрированной серной кислоты. Полученный раствор нагревали с обратным холодильником в течение 2 часов. Затем раствор охлаждали до комнатной температуры, после чего добавляли 25-30% водный раствор метиламина (50 мл, приблизительно 403 ммоль) и нагревали с обратным холодильником в течение следующих 3 часов. После завершения реакции смесь охлаждали до комнатной температуры. Большую часть растворителя удаляли на ротационном вакуумном испарителе, добавляли 30 мл воды и экстрагировали смесь этилацетатом (3 Х 80 мл). Объединенный органический слой высушивали над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении, получая 10,1 г N-метилфенилацетамида (белое твердое вещество, выход: 91,8%).
Структуру полученного соединения описывали на основе следующих данных, полученных с помощью спектроскопии ядерного магнитного резонанса:
1H ЯМР (400 МГц, CDCl3) δ 2,69 (d, J=4,84 Гц, 3Н), 3,51 (s, 3H), 5,95 (wide, 1H), 7,21-7,24 (m, 3Н), 7,28-7,32 (m, 3Н); 13С ЯМР (100 МГц, CDCl3) δ 26,4, 43,6, 127,2, 128,9, 129,4, 135,1, 171,8.
Получение 3-бензил-4-метил-1-(4-трифторметил)-фенил-1Н-1,2,4-триазол-5(4Н)-она
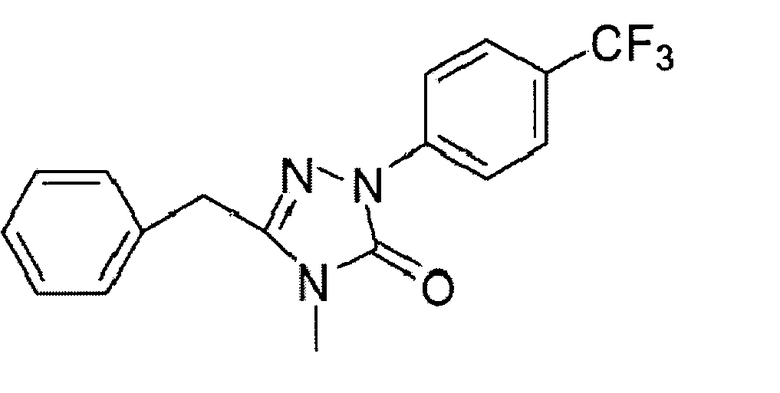
В трехгорлую колбу объемом 1000 мл к перемешиваемому раствору N-метилфенилацетамида (25 г, 167,7 ммоль) и (4-трифторметил)-фенилгидразина (30 г, 170 ммоль) в толуоле (250 мл) при 80-90°С добавляли по каплям раствор оксихлорида фосфора (POCl3, 15 мл, 180 ммоль) в толуоле (50 мл). После завершения добавления смесь перемешивали в течение 5 часов при 80-90°С, затем, после завершения реакции, охлаждали до комнатной температуры и фильтровали. Оставшийся отфильтрованный осадок промывали этилацетатом, пока он не приобретал желтовато-коричневую окраску. Фильтрат промывали насыщенным водным раствором гидрокарбоната натрия, высушивали над безводным сульфатом магния, концентрировали при пониженном давлении и повторно фильтровали. Отфильтрованный осадок промывали этилацетатом. Отфильтрованные осадки смешивали и высушивали в вакууме. В результате получали 43,5 г N-метил-2-фенил-n-(4-трифторметил)-фенилацетамида гидразона (желтовато-коричневое твердое вещество, выход: 84,4%).
В трехгорлой колбе объемом 1000 мл к перемешиваемой смеси N-метил-2-фенил-n-(4-трифторметил)-фенилацетамида гидразона (58,6 г, 190,9 ммоль) и N,N-диметиламинопиридина (ДМАП, 0,5 г, 4,0 ммоль) в тетрагидрофуране (ТГФ, 500 мл) медленно добавляли карбонилдиимидазол (CDI, 46,43 г, 286,4 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 12 часов. После завершения реакции смесь переносили в стакан объемом 2000 мл, содержащий 300 мл воды, доводили рН до 2-4 6 н. соляной кислотой при перемешивании и фильтровали. Отфильтрованный осадок промывали дистиллированной водой и высушивали в вакууме, получая 56,7 г 3-бензил-4-метил-1-(4-трифторметил)-фенил-1Н-1,2,4-триазол-5(4Н)-она (бледно-желтое твердое вещество, выход: 89,2%).
Структуру полученного соединения описывали на основе следующих данных, полученных с помощью спектроскопии ядерного магнитного резонанса:
1H ЯМР (400 МГц, CDCl3) δ 3,11 (s, 3H), 4,0 (s, 2H), 7,25-7,38 (m, 5H), 7,67 (d, J=8,68 Гц, 2H), 8,18 (d, J=8,68 Гц, 2H); 13C ЯМР (100 МГц, CDCI3) δ 27,6, 32,7, 118,0, 122,8, 125,5, 126,6, 127,7, 128,4, 129,1, 133,6, 140,7, 146,8, 152,6.
Получение 3-(1'-бромбензил)-4-метил-1-(4-трифторметил)-фенил-1H-1,2,4-триазол-5(4Н)-она (III-1)

В трехгорлую колбу объемом 500 мл при перемешивании добавляли 3-бензил-4-метил-1-(4-трифторметил)-фенил-1H-1,2,4-триазол-5(4Н)-он (15 г, 45 ммоль) и хлороформ (СНСl3, 250 мл). К перемешиваемому раствору добавляли N-бромсукцинимид (NBS, 12 г, 68 ммоль) и пероксид бензоила (0,5 г, 2,25 ммоль). Смесь осторожно нагревали с обратным холодильником в течение 12 часов. После завершения реакции смесь фильтровали. Фильтрат концентрировали при пониженном давлении, получая остаток, который очищали методом флэш-хроматографии на колонке с силикагелем (силикагель Н:300-400 меш; петролейный эфир/этилацетат=8:1 об./об.), получая 14,6 г 3-(1'-бромбензил)-4-метил-1-(4-трифторметил)-фенил-1Н-1,2,4-триазол-5(4Н)-она в виде красновато-коричневого масла (выход: 79,7%).
Структуру полученного соединения описывали на основе следующих данных, полученных с помощью спектроскопии ядерного магнитного резонанса:
1H ЯМР (400 МГц, СDCl3) δ 3,28 (s, 3H), 6,07 (s, 1H), 7,25-7,58 (m, 5H), 7,66 (d, J=8,68 Гц, 2Н), 8,13 (d, J=8,68 Гц, 2Н); 13С ЯМР (100 МГц, CDCl3) δ 28,4, 42,1, 69,6, 118,3, 126,2, 126,8, 127,3, 129,1, 129,9 133,9, 140,3, 145,7, 152,4.
Пример 8: Получение соединения III-2
Соединение 3-(1'-бромбензил)-4-n-бутил-1-(4-трифторметил)-фенил-1H-1,2,4-триазол-5(4Н)-она (соединение III-2) может быть получено в соответствии со следующей реакционной схемой

Получение N-n-бутилфенилацетамида
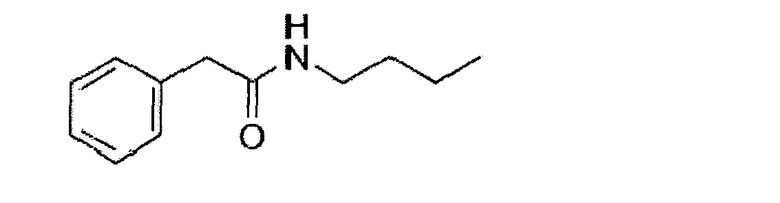
Фенилуксусную кислоту (16 г, 117,6 ммоль) и метанол (60 мл) добавляли в трехгорлую колбу объемом 250 мл с последующим добавлением 5 капель концентрированной серной кислоты. Полученный раствор нагревали с обратным холодильником в течение 6 часов. Затем смесь охлаждали до комнатной температуры с последующим добавлением n-бутиламина (12 мл, 121,4 ммоль) и нагревали с обратным холодильником в течение следующих 3 часов. После завершения реакции смесь охлаждали до комнатной температуры. Большую часть растворителя удаляли с помощью ротационного вакуумного испарителя, добавляли 30 мл воды и экстрагировали смесь этилацетатом (3 Х 80 мл). Смешанный органический слой высушивали над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении, получая 15,9 г N-n-бутилфенилацетамида (белое твердое вещество, выход: 70,4%).
Структуру полученного соединения описывали на основе следующих данных, полученных с помощью спектроскопии ядерного магнитного резонанса:
1Н ЯМР (400 МГц, СDCl3) δ 0,86 (t, J=7,28 Гц, 3Н), 1,21-1,29 (m, 2H), 1,36-1,44 (m, 2H), 3,2 (q, J=7,04 Гц, 2H), 5,62 (wide, 1H), 7,24-7,29 (m, 3Н), 7,32-7,36 (m, 2H); 13С ЯМР (100 МГц, CDCl3) δ 13,7, 19,9, 31,5, 39,4, 43,8, 127,2, 128,9, 129,4, 135,1, 170,9.
Получение 3-бензил-4-n-бутил-1-(4трифторметил)-фенил-1Н-1,2,4-триазол-5(4Н)-она
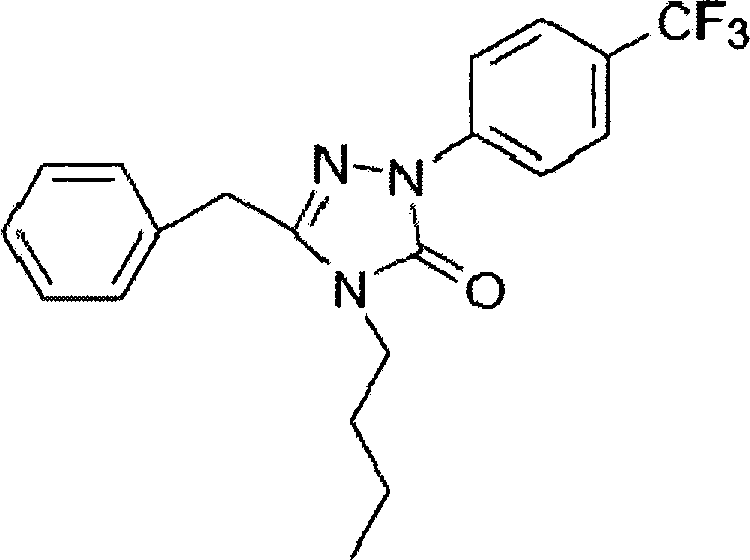
В трехгорлой колбе объемом 500 мл к перемешиваемому раствору N-n-бутилфенилацетамида (11 г, 57,5 ммоль) в толуоле (150 мл) при 80-90°С добавляли по каплям раствор оксихлорида фосфора (РОСl3, 17 мл, 75,1 ммоль) в толуоле (20 мл). После завершения добавления смесь перемешивали в течение 5 часов при 80-90°С и затем охлаждали до комнатной температуры после завершения реакции и фильтровали. Полученный отфильтрованный осадок промывали этилацетатом, пока он не приобретал желтовато-коричневую окраску. Фильтрат промывали насыщенным водным раствором гидрокарбоната натрия, высушивали над безводным сульфатом магния, фильтровали, концентрировали при пониженном давлении и повторно фильтровали. Отфильтрованный осадок повторно промывали этилацетатом. Полученные отфильтрованные осадки смешивали и высушивали под вакуумом. В конце процедуры получали 15,7 г N-n-бутил-2-фенил-n-(4-трифторметил)-фенилацетамид гидразона (желтовато-коричневое твердое вещество).
В трехгорлую колбу объемом 500 мл к перемешиваемой смеси N-n-бутил-2-фенил-n-(4-трифторметил)-фенилацетамида гидразона (15,7 г) и N,N-диметиламинопиридина (ДМАП, 0,5 г, 4,0 ммоль) в тетрогидрофуране (ТГФ, 250 мл) медленно добавляли карбонилдиимидазол (CDI, 7,43 г, 45,8 ммоль) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 12 часов. После завершения реакции смесь переливали в стакан объемом 500 мл, содержащий 100 мл воды, доводили рН до 2-4 6 н. соляной кислотой при перемешивании и отфильтровывали. Отфильтрованный осадок промывали дистиллированной водой и затем высушивали в вакууме, получая 6,28 г 3-бензил-4-n-бутил-1-(4-трифторметил)-фенил-1H-1,2,4-триазол-5(4Н)-она (бледно-желтое твердое вещество, выход в две стадии: 29,2%).
Структуру полученного соединения описывали на основе следующих данных, полученных с помощью спектроскопии ядерного магнитного резонанса и масс-спектроскопии:
1Н ЯМР (400 МГц, CDCl3) 6 0,86 (t, J=7,3 Гц, 3Н), 1,22-1,29 (m, 2H), 1,37-1,44 (m, 2H), 3,49 (q, J=7,6 Гц, 2H), 4,0 (s, 2H), 7,26-7,38 (m, 5H), 7,67 (d, J=8,70 Гц, 2H), 8,19 (d, J=8,65 Гц, 2H); 13С ЯМР (100 МГц, CDCl3) δ 13,5, 19,8, 30,6, 32,8, 41,7, 117,9, 126,1, 126,9, 127,2, 127,8, 128,6, 129,1, 134,0, 140,7, 146,6, 152,5; MS (ESI) m/z 376,29 (М+1)+.
Получение 3-(1'-бромбензил)-4-n-бутил-1-(4-трифторметил)-фенил-1H-1,2,4-триазол-5(4Н)-она (III-2)

В трехгорлую колбу объемом 250 мл добавляли 3-бензил-4-n-бутил-1-(4-трифторметил)-фенил-1H-1,2,4-триазол-5(4Н)-он (3,75 г, 10 ммоль) и хлороформ (100 мл). К перемешиваемому раствору добавляли N-бромосукцинимид (NBS, 3,56 г, 20 ммоль) и пероксид бензоила (0,4 г, 1,65 ммоль). Полученную смесь осторожно нагревали с обратным холодильником в течение 6 часов. После завершения реакции растворитель удаляли с помощью ротационного вакуумного испарителя и осадок подвергали колоночной хроматографии (силикагель 1-1:300-400 меш; петролейный эфир/этилацетат=8:1 об./об.), получая 3,9 г 3-(1'-бром-бензил)-4-n-бутил-1-(4-трифторметил)-фенил-1Н-1,2,4-триазол-5(4Н)-она (III-2) (рыжевато-бурая вязкая жидкость, выход: 85,9%).
Структуру полученного соединения описывали на основе следующих данных, полученных с помощью спектроскопии ядерного магнитного резонанса:
1H ЯМР (400 МГц, CDCl3) δ 0,90 (t, J=7,36 Гц, 3Н), 1,27-1,35 (m, 2H), 1,47-1,53 (m, 1H), 1,63-1,66 (m, 1H), 3,66-3,72 (m, 2H), 6,00 (s, 1H), 7,41-7,46 (m, 3Н), 7,61 (dd, J=7,99 Гц, 1,83 Гц, 2H) 7,66 (d, J=8,69 Гц, 2H), 8,15 (d, J=8,57 Гц, 2H); 13С ЯМР (100 МГц, СDСl3) δ 13,5, 19,9, 30,3, 41,5, 42,4, 118,2, 126,2, 126,8, 127,3, 128,6, 128,9, 129,5, 134,8, 140,4, 145,6, 152,2.
Пример 9: Получение соединения III-3
Соединение III-3 может быть получено в соответствии со следующей реакционной схемой:

Получение N-метилбензоксоацетамида

60% гидрид натрия (3,7 г, 92,5 ммоль) и тетрагидрофуран (20 мл) добавляли в трехгорлую колбу объемом 250 мл с последующим нагреванием и добавлением по каплям 20 мл раствора бензилового спирта (10 г, 96,1 ммоль) в тетрагидрофуране. После завершения добавления реакционную смесь нагревали с обратным холодильником в течение 1 часа. Затем добавляли этилбромацетат (11 мл, 94,8 ммоль) и перемешивали реакционную смесь в течение следующих 4 часов при нагревании с обратным холодильником. После охлаждения до комнатной температуры добавляли 25 мл водного раствора метиламина и проводили реакцию при нагревании с обратным холодильником в течение следующих 6 часов. После завершения реакции смесь концентрировали при пониженном давлении и подвергали колоночной хроматографии (силикагель Н: 300-400 меш; петролейный эфир/этилацетат=4:1 об./об.), получая 5,3 г N-метилбензоксоацетамида (белое твердое вещество, выход в две стадии: 32%).
Структуру полученного соединения описывали на основе следующих данных, полученных с помощью спектроскопии ядерного магнитного резонанса:
1H ЯМР (400 МГц, CDCl3) δ 2,83 (d, J=5,16 Гц, 3Н), 3,98 (s, 2H), 4,55 (S, 2h), 6,61 (s, 1H), 7,30-7,39 (m,5H).
Получение 3-бензоксометил-4-метил-1-(4-трифторметилфенил)-1,4-дигидро-1,2,4-триазол-5-она

В трехгорлую колбу объемом 250 мл к перемешиваемому раствору N-метилбензилоксиацетамида (6,4 г, 3,57 ммоль) в толуоле (40 мл) при 80-90°С по каплям добавляли оксихлорид фосфора (3,6 мл, 38,5 ммоль) в толуоле (20 мл). Полученную реакционную смесь перемешивали при 80-90°С в течение 5 часов. После завершения реакции смесь охлаждали до комнатной температуры и фильтровали. Отфильтрованный осадок промывали этилацетатом, до получения алой окраски. Фильтрат промывали насыщенным водным раствором гидрокарбоната натрия, высушивали над безводным сульфатом магния, фильтровали, концентрировали при пониженном давлении и повторно фильтровали. Отфильтрованный осадок повторно промывали этилацетатом. Полученные отфильтрованные осадки смешивали и высушивали под вакуумом. В конце процедуры получали 4,5 г алого твердого вещества.
Вышеупомянутое алое твердое вещество в количестве 4,5 г переносили в еще одну трехгорлую колбу объемом 500 мл, добавляли 200 мл тетрагидрофурана (ТГФ), перемешивали и медленно добавляли карбонилдиимидазол (CDI) (5 г, 30,8 ммоль). Реакционную смесь после добавления перемешивали при комнатной температуре в течение 12 часов. После завершения реакции смесь концентрировали при пониженном давлении и подвергали колоночной хроматографии (силикагель Н: 300-400 меш; петролейный эфир/этилацетат=4:1 об./об.), получая 3 г 3-бензоксометил-4-метил-1-(4-трифторметилфенил)-1,4-дигидро-1,2,4-триазол-5-она (светло-бурое гелеобразное вещество, выход в две стадии: 23%).
Структуру полученного соединения описывали на основе следующих данных, полученных с помощью спектроскопии ядерного магнитного резонанса:
1H ЯМР (400 МГц, CDCl3) δ 3,35 (s, 3Н), 4,50 (s, 2Н), 4,59 (s, 2H), 7,32-7,37 (m, 5H), 7,66 (d, J=8,46 Гц, 2H), 8,14 (d, J=8,46 Гц, 2H); 13С ЯМР (100 МГц, CDCl3) 5 26,9, 27,8, 62,8, 72,9, 118,1, 126,1, 126,2, 126,3, 128,3, 128,5, 136,6, 140,5, 144,7, 152,5.
Получение 3-гидроксиметил-4-метил-1 -(4-трифторметилфенил)-1,4-дигидро-1,2,4-триазол-5-она
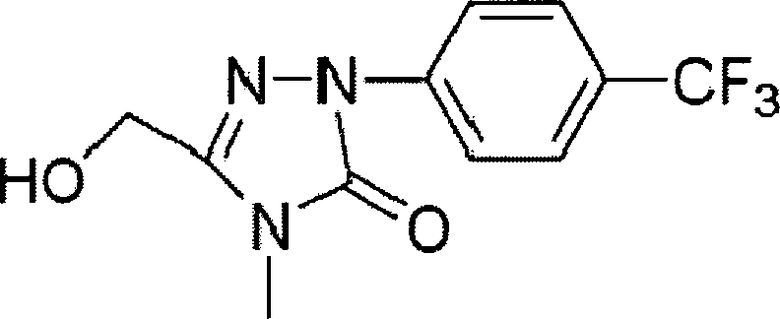
В трехгорлую колбу объемом 250 мл к перемешиваемому раствору вышеупомянутого продукта 3-бензоксометил-4-метил-1-(4-трифторметилфенил)-1,4-дигидро-1,2,4-триазол-5-она (3,0 г, 8,3 ммоль) в этаноле (60 мл) добавляли 1 г 10% палладированного угля. 1 г 10% палладированного угля в колбе замещали N2 и затем вводили газообразный водород. Реакционную смесь перемешивали при комнатной температуре в течение 24 часов. После завершения реакции смесь фильтровали и концентрировали в вакууме, получая 2,1 г 3-гидроксиметил-4-метил-1-(4-трифторметилфенил)-1,4-дигидро-1,2,4-триазол-5-она (белое твердое вещество, выход: 93%).
Структуру полученного соединения описывали на основе следующих данных, полученных с помощью спектроскопии ядерного магнитного резонанса:
1H ЯМР (400 МГц, CDCl3) δ 3,34 (s, 3Н), 4,52 (d, J=5,84 Гц, 2H), 5,77 {t, J=5,84 Гц, 1H), 7,85 (d, J=8,59 Гц, 2H), 8,17 (d, J=8,59 Гц, 2H); 13С ЯМР (100 МГц, CDCl3) δ 27,9, 55,2, 118,0, 125,2, 125,5, 126,0, 141,3, 149,0, 152,5.
Получение 3-бромметил-4-метил-1-(4-трифторметилфенил)-1,4-дигидро-1,2,4-триазол-5-она (III-3)

В трехгорлую колбу объемом 250 мл к перемешиваемому раствору вышеупомянутого продукта (2,1 г, 7,77 ммоль) в дихлорметане (100 мл) добавляли N-бромсукцинимид (NBS, 1,8 г, 10,1 ммоль) с последующим медленным добавлением трифенилфосфина (2,4 г, 9,15 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 4 часов. После завершения реакции смесь подвергали колоночной хроматографии (силикагель Н: 300-400 меш; петролейный эфир/этилацетат=5:1 об./об.), получая 2,3 г 3-бромметил-4-метил-1-(4-трифторметилфенил)-1,4-дигидро-1,2,4-триазол-5-она (бледно-желтое твердое вещество, выход: 89%).
Структуру полученного соединения описывали на основе следующих данных, полученных с помощью спектроскопии ядерного магнитного резонанса и масс-спектроскопии:
1H ЯМР (400 МГц, CDCl3) δ 3,42 (s, 3H), 4,34 (s, 2H), 7,67 (d, J=8,63 Гц, 2Н), 8,12 (d, J=8,63 Гц, 2H); 13C ЯМР (100 МГц, CDCl3) δ 19,1, 26,9, 27,9, 118,2, 126,2, 140,2, 143,5, 152,1; MS (ESI) m/z 332,27 (M-4)+.
Получение соединения (I) (эфира):
Пример 10: Получение соединения Е-1
Получение этил 2-(2-метил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1H-1,2,4-триазолил))-бензилокси)-фенокси)-ацетата (Е-1)
В одногорлую колбу объемом 150 мл к перемешиваемому раствору 3-(1-бромбензил)-4-метил-1-(4-трифторметил)-фенил-1Н-1,2,4-триазол-5(4Н)-она (111-1) (36,3 г, 88,1 ммоль) в ацетонитриле (150 мл) добавляли этил-(2-метил-4-гидрокси)-феноксиацетат (11-1) (17,5 г, 83,3 ммоль), N,N-диметиламинопиридин (ДМАП, 0,5 г, 4,09 ммоль) и карбонат калия (К2СО3, 13,8 г, 99,9 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 8 часов. После завершения реакции смесь фильтровали. Отфильтрованный осадок промывали этилацетатом (3Х50 мл), после чего выбрасывали. Фильтраты смешивали, удаляли растворитель на ротационном вакуумном испарителе и затем подвергали колоночной хроматографии (силикагель Н: 300-400 меш; петролейный эфир/этилацетат=8:1 об./об.), получая бесцветное гелеобразное вещество (которое постепенно становилось белым твердым веществом при хранении) (33,5 г, выход: 74,3%).
Структуру полученного соединения описывали на основе следующих данных, полученных с помощью спектроскопии ядерного магнитного резонанса и масс-спектроскопии:
1H ЯМР (400 МГц, СDСl3) δ 1,28 (t, J=7,11 Гц, 3Н), 2,26 (s, 3H), 3,20 (s, 3H), 4,26 {q, J=7,11 Гц, 2H), 4,61 (s, 2H), 6,28 (s, 1H), 6,65 (d, J=8,8 Гц, 1Н), 6,81 (d, J=8,75 Гц, 1Н), 6,93 (d, J=3,0 Гц, 1 Н), 7,25-7,45 (m, 3H), 7,50 (d, J=7,32, 2H), 7,67 (d, J=8,48 Гц, 2H), 8,14 (d, J=8,48 Гц, 2H); 13С ЯМР (100 МГц, CDCl3) δ 14,1, 16,7, 28,2, 61,3, 65,9, 75,1, 112,8, 113,2, 118,3, 119,3, 122,7, 125,8, 126,2, 127,1, 128,9, 129,0, 129,4, 135,1, 140,4, 146,3, 151,5,152,8, 169,1; MS (ESI)m/z 558,9 (M+NH4 +).
Пример 11: Получение соединения Е-2
Используя соединения III-1 и II-5 в качестве исходных материалов, получали этил-2-(2-метил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1Н-1,2,4-триазолил))-бензилокси)-фенилтио)-ацетат (Е-2) согласно химической методике, аналогичной методике, описанной в Примере 9. Полученное соединение представляло собой белое твердое вещество с выходом 60%.
Структуру полученного соединения описывали на основе следующих данных, полученных с помощью спектроскопии ядерного магнитного резонанса и масс-спектроскопии:
1Н ЯМР (400 МГц, CDCl3) δ 1,19 (t, J=7,12 Гц, 3Н), 2,45 (s, 3H), 3,21 (s, 3H), 3,50 (s, 2H), 4,12 (q, J=7,12 Гц, 2H), 6,39 (s, 1Н), 6,92 (dd, J=8,59 Гц, 2,82 Гц, 1Н), 7,0, (d, J=2,82 Гц, 1 Н), 7,40-7,53(m, 3Н), 7,52 (d, J=7,48 Гц, 2H), 7,69 (d, J=8,76 Гц, 2H), 8,18 (d, J=8,76 Гц, 2H); 13С ЯМР (100 МГц, CDCl3) δ 14,1, 20,9, 28,3, 37,1, 61,4, 74,6, 113,6, 117,9, 118,2, 125,4, 125,8, 126,2, 126,9, 127,3, 128,96, 129,1, 134,2, 134,7, 140,5, 142,2, 145,9, 152,6, 169,7; MS (ESI) m/z 558,01 (M+H)+.
Пример 12: Получение соединения Е-3
Используя соединения III-1 и II-4 в качестве исходных материалов, получали этил-2-(3-метил-4-(1-(3-(4-метил-5-оксо-1 -(4-трифторметилфенил)-4,5-дигидро-1 Н-1,2,4-триазолил))-бензилокси)-фенилтио)-ацетат (Е-3) согласно химической методике, аналогичной методике, описанной в Примере 9. Полученное соединение представляло собой белое твердое вещество с выходом 71%.
Структуру полученного соединения описывали на основе следующих данных, полученных с помощью спектроскопии ядерного магнитного резонанса и масс-спектроскопии:
1H ЯМР (400 МГц, CDCl3) δ 1,20 (t, J=7,10 Гц, 3Н), 2,37 (s, 3Н), 3,19 (s, 3H), 3,53 (s, 2H), 4,14 (q, J=7,10 Гц, 2H), 6,39 (s, 1H), 6,94 (d, J=8,58 Гц, 1Н), 7,24 (dd, J=8,58 Гц, 2,4 Гц, 1H), 7,34 (d, J=2,4 Гц, 1H), 7,40-7,46 (m, 3Н), 7,50 (d, J=7,52 Гц, 2H), 7,69 (d, J=8,84 Гц, 2H), 8,17 (d, J=8,84 Гц, 2H); 13С ЯМР (100 МГц, CDCl3) δ 14,1, 16,5, 28,2, 38,0, 61,4, 74,5, 113,1, 118,2, 122,7, 125,4, 125,8, 126,2, 126,3, 127,1, 127,4, 128,0, 128,96, 129,1, 130,8, 134,8, 140,4, 145,9, 152,6, 169,8; MS (ESI) m/z 557,97 (М+Н)+.
Пример 13: Получение соединения Е-4
Получение этил-2-(2-метил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1Н-1,2,4-триазолил))-бензилтио)-фенокси)-ацетата (Е-4)
В трехгорлую колбу объемом 250 мл с капельной воронкой постоянного давления к перемешиваемой смеси алюмотетрагидрида лития (LiAlH4, 1,0 г, 26,3 ммоль) и тетрагидрофурана (30 мл) при 0°С добавляли по каплям раствор 3-метил-4-гидроксифенилтиоциановой кислоты (1,7 г, 10,3 ммоль) в тетрагидрофуране (ТГФ, 30 мл). Спустя 30 мин перемешивания при 0°С и следующие 2 часа перемешивания при комнатной температуре реакцию останавливали добавлением измельченного льда в водоледяную баню. Значение рН смеси доводили до 3-4, добавляя 6 М соляную кислоту, и затем экстрагировали водную фазу этилацетатом (3 Х 100 мл). Объединенный органический слой промывали насыщенным раствором хлорида натрия (2 Х 100 мл), высушивали над безводным сульфатом магния, фильтровали и выпаривали в вакууме, получая неочищенный продукт 3-метил-4-гидроксимеркаптобензола (желтая жидкость).
В одногорлую колбу объемом 150 мл к перемешиваемому раствору 3-(1'-бромбензил)-4-метил-1-(4-трифторметил)-фенил-1Н-1,2,4-триазол-5(4Н)-она (4,01 г, 9,73 ммоль) в ацетонитриле (30 мл) добавляли свежеполученный неочищенный продукт 3-метил-4-гидроксимеркаптобензола в ацетонитриле (30 мл), N,N-диметиламинопиридин (ДМАП, 0,07 г, 0,57 ммоль) и карбонат калия (К2СО3, 1,28 г, 9,27 ммоль). После перемешивания реакционной смеси при комнатной температуре в течение 4 часов добавляли карбонат калия (К2СО3, 1,4 г, 10,1 ммоль) и этилбромацетат (1,1 мл, 9,5 ммоль). Реакционную смесь перемешивали в течение следующих 8 часов. После завершения реакции смесь фильтровали. Отфильтрованный осадок промывали этилацетатом (3 Х 30 мл), после чего выбрасывали. Фильтраты смешивали, удаляли растворитель на ротационном вакуумном испарителе и затем подвергали колоночной хроматографии (силикагель Н: 300-400 меш; петролейный эфир/этилацетат=5:1 об./об.), получая 4,4 г белого твердого вещества (выход: 81%).
Структуру полученного соединения описывали на основе следующих данных, полученных с помощью спектроскопии ядерного магнитного резонанса и масс-спектроскопии:
1H ЯМР (400 МГц, СDСl3) δ 1,26 (t, J=7,14 Гц, 3Н), 2,19 (s, 3Н), 3,15 (s, 3H), 4,23 (q, J=7,14 Гц, 2Н), 4,58 (s, 2Н), 5,17 (s, 1H), 6,54 (d, J=8,44 Гц, 1Н), 7,11 (dd, J=8,32 Гц, 2,28 Гц, 1H), 7,17 (d, J=2,28 Гц, 1H), 7,33-7,38 (m, 5H), 7,66 (d, J=8,56 Гц, 2Н), 8,10 (d, J=8,56 Гц, 2Н); 13C ЯМР (100 МГц, CDCl3) δ 14,1, 16,1, 28,0, 50,6, 61,4, 65,4, 113,6, 118,2, 122,8, 125,5, 126,2, 126,8, 127,1, 128,2, 128,5, 128,9, 133,8, 135,2, 137,8, 140,5, 146,5, 152,6, 157,1, 168,5; MS (ESI) m/z 558,03(M+H)+.
Пример 14: Получение соединения Е-5
Используя соединения III-1 и II-3 в качестве исходных материалов, получали этил-2-(2-этил-4-(1-(3-(4-метил -5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1H-1,2,4-триазолил))-бензилокси)-фенилокси)-ацетат (Е-5) согласно химической методике, аналогичной методике, описанной в Примере 9. Полученное соединение представляло собой белое твердое вещество с выходом 87,3%.
Структуру полученного соединения описывали на основе следующих данных, полученных с помощью спектроскопии ядерного магнитного резонанса и масс-спектроскопии:
1H ЯМР (400 МГц, СDCl3) δ 1,19 {t, J=7,24 Гц, 3Н), 1,27 (t, J=7,24 Гц, 3Н), 2,67 (m, 2Н), 3,20 (s, 3Н), 4,24 (q, J=7,24 Гц, 2Н), 4,57 (s, 2Н), 6,29 (s, 1H), 6,81 (dd, J=8,88 Гц, 3,08 Гц, 1H), 6,95 (d, J=3,08 Гц, 1H), 7,36-7,45 (m, 3Н), 7,52 (d, J=7,60, 2Н), 7,67 (d, J=8,76 Гц, 2Н), 8,16 (d, J=8,76 Гц, 2Н); 13С ЯМР (100 МГц, CDCl3) δ 13,9, 14,1, 23,2, 23,3, 28,3, 61,3, 66,3, 75,3, 112,5, 112,6, 112,96, 113,2, 116,5, 117,6, 118,2, 125,4, 125,9, 126,2, 127,3, 128,8, 128,96, 135,2, 135,3, 140,5, 146,4, 150,6, 151,3, 151,6, 152,8, 169,2; MS (ESI) m/z 556,29(M+H)+.
Пример 15: Получение соединения Е-6
Используя соединения III-1 и II-2 в качестве исходных материалов, получали этил-2-(3-метил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1Н-1,2,4-триазолил))-бензилокси)-фенокси)-ацетат (Е-6) согласно химической методике, аналогичной методике, описанной в Примере 9. Полученное соединение представляло собой белое твердое вещество с выходом 98%.
Структуру полученного соединения описывали на основе следующих данных, полученных с помощью спектроскопии ядерного магнитного резонанса и масс-спектроскопии:
1H ЯМР (400 МГц, CDCl3) δ 1,32 (t, J=7,02 Гц, 3Н), 2,24 (s, 3H), 3,25 (s, 3H), 4,29 (q, J=7,02 Гц, 2H), 4,59 (s, 2H), 6,35 (s, 1H), 6,56-6,75 (m, 1H), 6,88 (d, J=2,95 Гц, 1Н), 6,95 (d, J=8,94 Гц, 1H), 7,42-7,55(m, 3Н), 7,58 (d, J=7,49 Гц, 2H), 7,72 (d, J=8,69 Гц, 2H), 8,23 (d, J=8,56 Гц, 2H); 13С ЯМР (100 МГц, СDCl3) δ 14,1, 16,7, 28,2, 61,3,65,96,75,1, 112,1, 112,8, 113,9, 115,4, 117,8, 118,2, 118,5, 125,5, 125,8, 126,2, 126,9, 127,2, 128,8, 129,0, 135,3, 140,5, 146,4, 149,9, 152,7, 169,1; MS (ESI) m/z 541,9(M+H+); 558,9 (M+NH4 +).
Пример 16: Получение соединения Е-7
Получение этил-2-(3-метил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1 Н-Л,2,4-триазолил))-бензилтио)-фенокси)-ацетата (Е-7)
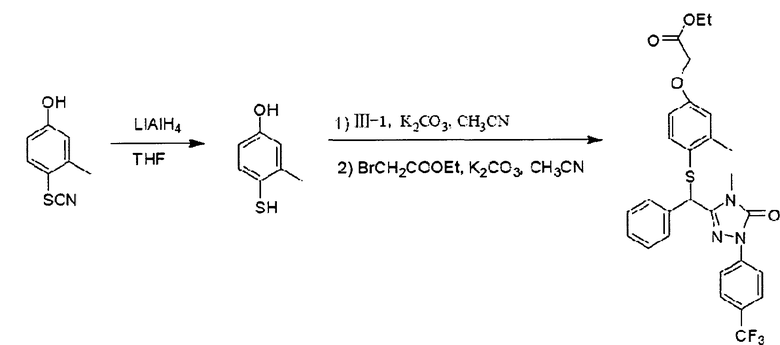
В трехгорлую колбу объемом 250 мл к перемешиваемой смеси алюмотетрагидрида лития (LiAlH4, 0,8 г, 21 ммоль) и тетрагидрофурана (30 мл) при 0°С (охлаждение холодной водой) добавляли по каплям раствор 3-метил-4-гидроксифенилтиоциановой кислоты (1,1 г, 6,6 ммоль) в тетрагидрофуране (ТГФ 30 мл). Спустя 30 мин перемешивания при 0°С и следующие 2 часа перемешивания при комнатной температуре реакцию останавливали добавлением измельченного льда в водоледяную баню. Значение рН смеси доводили до 3-4, добавляя 6 н. соляную кислоту, и затем экстрагировали водную фазу этилацетатом. Объединенный органический слой промывали насыщенным раствором хлорида натрия (2 Х 100 мл), высушивали над безводным сульфатом магния, фильтровали и выпаривали в вакууме, получая неочищенный продукт 3-метил-4-гидроксимеркаптобензола (желтая жидкость).
В еще одной одногорлой колбе объемом 250 мл к перемешиваемому раствору полученного 3-метил-4-гидроксимеркаптобензола в ацетонитриле (30 мл) добавляли 3-(1'-бромбензил)-4-метил-1-(4-трифторметил)-фенил-1Н-1,2,4-триазол-5(4Н)-он (III-1, 2,5 г, 6,06 ммоль), ацетонитрил (30 мл), N,N-диметиламинопиридин (ДМАП, 0,1 г, 0,82 ммоль) и карбонат калия (К2СО3, 0,85 г, 6,15 ммоль). После перемешивания реакционной смеси при комнатной температуре в течение 5 часов добавляли карбонат калия (К2СО3, 1,6 г, 11,6 ммоль) и этилбромацетат (1,2 мл, 10,3 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение следующих 8 часов. После завершения реакции смесь фильтровали. Отфильтрованный осадок промывали этилацетатом (3 Х 50 мл), после чего выбрасывали. Фильтраты смешивали, удаляли растворитель на ротационном вакуумном испарителе и затем подвергали колоночной хроматографии (силикагель Н: 300-400 меш; петролейный эфир/этилацетат=8:1-5:1 об./об.), получая 2,0 г белого твердого вещества (выход: 59%).
Структуру полученного соединения описывали на основе следующих данных, полученных с помощью спектроскопии ядерного магнитного резонанса и масс-спектроскопии:
1H ЯМР (400 МГц, CDCl3) δ 1,27 (t, J=7,18 Гц, 3Н), 2,31 (s, 3Н), 3,13 (s, 3H), 4,24 (q, J=7,18 Гц, 2Н), 4,55 (s, 2Н), 5,09 (s, 1H), 6,59 (dd, J=8,56 Гц, 2,88 Гц, 1Н), 6,78, (d, J=2,88 Гц, 1 Н), 7,24 (d, J=8,68 Гц, 1H), 7,30-7,34 (m, 4Н), 7,66 (d, J=8,66 Гц, 2Н), 8,10 (d, J=8,66 Гц, 2Н); 13С ЯМР (100 МГц, CDCl3) δ 14,1, 20,9, 28,1, 49,8, 61,5, 65,2, 112,6, 117,0, 118,2, 123,1, 125,5, 126,2, 126,8, 127,1, 128,0, 128,5, 128,9, 135,1, 137,9, 140,5, 144,5, 146,4, 152,5, 168,5; MS (ESI) m/z 558,05 (M+H)+.
Пример 17: Получение соединения Е-8
Получение этил 2-(2-метил-4-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1H-1,2,4-триазолил)-метилтио)-фенокси)-ацетата (Е-8)

В трехгорлую колбу объемом 250 мл к перемешиваемой смеси алюмотетрагидрида лития (LiAlH4, 1,0 г, 26,3 ммоль) и тетрагидрофурана (30 мл) при 0°С добавляли по каплям раствор 3-метил-4-гидроксифенилтиоциановой кислоты (1,38 г, 8,35 ммоль) в тетрагидрофуране (20 мл). Спустя 30 мин перемешивания при 0°С реакционной смеси позволяли нагреться до комнатной температуры и перемешивали в течение следующих 2 часов при комнатной температуре. Затем реакцию останавливали добавлением этанола (10 мл). Значение рН смеси доводили до 3-4, добавляя 6 М соляную кислоту на водоледяной бане, и затем экстрагировали водную фазу этилацетатом (3 Х 80 мл). Объединенный органический слой высушивали над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении, оставляли на воздухе в течение суток и очищали методом колоночной хроматографии (силикагель Н: 300-400 меш; петролейный эфир/этилацетат=5:1 об./об.), получая 1,6 г связанного соединения 3-метил-4-гидроксимеркаптобензола (желтая гелеподобная жидкость, выход: 68,8%).
Структуру полученного соединения описывали на основе следующих данных, полученных с помощью спектроскопии ядерного магнитного резонанса и масс-спектроскопии:
1H ЯМР (400 МГц, CDCl3) δ 2,2 (s, 3Н), 5,35 (s, 1H), 6,69 (d, J=8,28 Гц, 1Н), 7,18 (dd, J=8,23 Гц, 2,23 Гц, 1H), 7,24 (d, J=2,24 Гц, 1H); 13C ЯМР (100 МГц, CDCl3) δ 15,7, 115,5, 125,0, 128,3, 130,2, 134,0, 154,4; MS (ESI) m/z 278,33 (М+H+).
В одногорлую колбу объемом 150 мл к перемешиваемому раствору 3-метил-4-гидроксимеркаптобензола (1,6 г, 5,76 ммоль) в ацетонитриле (60 мл) добавляли этилбромацетат (1,4 мл, 60,7 ммоль) и карбонат калия (К2СО3, 1,6 г, 11,6 ммоль). После перемешивания реакционной смеси при комнатной температуре в течение 12 часов реакционную смесь разбавляли, добавляя 60 мл этилацетата, фильтровали и выпаривали, получая остаток. Этот остаток очищали методом колоночной хроматографии (силикагель Н: 300-400 меш; петролейный эфир/этил ацетат=5:1 об./об.), получая 2,0 г бледно-желтой гелеподобной жидкости (выход: 77,5%).
В трехгорлую колбу объемом 150 мл к перемешиваемому раствору вышеописанного продукта (2,0 г, 4,44 ммоль) в этаноле (15 мл) добавляли воду (15 мл) и концентрированный хлоргидрат (7 мл). Медленно добавляли порошок цинка (10 г, 153 ммоль) при перемешивании. После добавления реакционную смесь перемешивали при комнатной температуре в течение 30 мин и затем экстрагировали дихлорметаном (3 Х 60 мл). Органические фазы смешивали, высушивали над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении, получая бледно-желтую жидкость.
В одногорлую колбу объемом 150 мл к перемешиваемому раствору вышеупомянутого неочищенного продукта в 30 мл ацетонитрила добавляли 30 мл 3-(1-бромбензил)-4-метил-1-(4-трифторметил)-фенил-1Н-1,2,4-триазол-5(4Н)-она III-3 (1,3 г, 3,9 ммоль) и карбонат калия (К2СО3, 3,4 г, 24,6 ммоль). Полученную реакционную смесь перемешивали при комнатной температуре в течение 12 часов. После завершения реакции реакционный раствор разбавляли добавлением 50 мл этилацетата и фильтровали. Фильтрат концентрировали при пониженном давлении и подвергали колоночной хроматографии (силикагель Н: 300-400 меш; петролейный эфир/этилацетат=3:1 об./об.), получая 1,7 г белого твердого вещества (выход: 91%).
Структуру полученного соединения описывали на основе следующих данных, полученных с помощью спектроскопии ядерного магнитного резонанса и масс-спектроскопии:
1H ЯМР (400 МГц, СDCl3) δ 1,25 (t, J=7,12 Гц, 3Н), 2,22 (s, 3Н), 3,3 7 (s, 3H), 3,87 (s, 2Н), 4,21 (q, J=7,12 Гц, 2Н), 4,58 (s, 2H), 6,58 (d, J=8,37 Гц, 1H), 7,16 (dd, J=8,37 Гц, 2,34 Гц, 1H), 7,23 (d, J=2,34 Гц, 1H), 7,63 (d, J=8,57 Гц, 2Н), 7,98 (d, J=8,57 Гц, 2Н); 13С ЯМР (100 МГц, CDCl3) δ 14,1, 16,1, 27,8, 31,3, 61,3, 65,5, 111,6, 118,1, 122,7, 123,5, 125,4, 126,1, 128,1, 128,6, 132,4, 136,3, 140,5, 144,6, 152,4, 156,8, 168,5; MS (ESI) m/z 482,49 (М+Н)+.
Пример 18: Получение соединения Е-9
Используя соединения III-3 и II-2 в качестве исходных материалов, получали этил-2-(2-метил-4-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1H-1,2,4-триазолил)-метокси)-фенокси)-ацетат (Е-9) согласно химической методике, аналогичной методике, описанной в Примере 9. Полученное соединение представляло собой белое твердое вещество с выходом 73,5%.
Структуру полученного соединения описывали на основе следующих данных, полученных с помощью спектроскопии ядерного магнитного резонанса и масс-спектроскопии:
1H ЯМР (400 МГц, СDCl3) δ 1,32 (t, J=7,14 Гц, 3Н), 2,33 (s, 3H), 3,45 (s, 3H), 4,28 (q, J=7,14 Гц, 2H), 4,63 (s, 2H), 5,01 (s, 2H), 6,71 (d, J=8,85 Гц, 1 Н), 6,8 (dd, J=8,85 Гц, 3,08 Гц, 1Н), 6,88 (d, J=3,08 Гц, 1Н), 7,70 (d, J=8,56 Гц, 2H), 8,17 (d, J=8,55 Гц, 2H); 13С ЯМР (100 МГц, CDCl3) δ 14,1, 16,2, 61,2, 61,9, 66,4, 112,0, 112,7, 117,7, 118,2, 118,3, 126,2, 126,3, 129,3, 140,5, 143,9, 151,6, 151,8, 152,4, 169,1; MS (ESI) m/z 464,39 (M-1), 466,48 (M+H)+.
Пример 19: Получение соединения Е-10
Используя соединения III-1 и II-6 в качестве исходных материалов, получали этил-2-(2,5-диметил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1H-1,2,4-триазолил))-бензилокси)-фенилтио)-ацетат (Е-10) согласно химической методике, аналогичной методике, описанной в Примере 9. Полученное соединение представляло собой желтую гелеподобную жидкость с выходом 72,2%.
Структуру полученного соединения описывали на основе следующих данных, полученных с помощью спектроскопии ядерного магнитного резонанса и масс-спектроскопии:
1H ЯМР (400 МГц, CDCl3) δ 1,29 (t J=7,16 Гц, 3Н), 2,32 (s, 3Н), 2,37 (s, 3Н), 3,18 (s, 3Н), 3,51 (s, 2H), 4,25 (q, J=7,16 Гц, 2H), 6,39 (s, 1Н), 6,88 (s, 1Н), 7,31 (s, 1Н), 7,39-7,52 (m, 5H), 7,69 (d, J=8,67 Гц, 2H), 8,16 (d, J=8,67 Гц, 2H); 13C ЯМР (100 МГц, CDCl3) δ 16,0, 20,7, 28,2, 37,2, 74,4, 114,7, 118,2, 125,4, 125,6, 125,7, 126,3, 128,9, 129,1, 134,9, 135,7, 139,5, 140,4, 146,1, 152,7, 155,0, 168,7; MS (ESI) m/z 573,07 (M+H)+.
Пример 20: Получение соединения Е-11
Используя соединения III-3 и II-4 в качестве исходных материалов, получали этил-2-(3-метил-4-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1H-1,2,4-триазолил)-метокси)-фенилтио)-ацетат (Е-11) согласно химической методике, аналогичной методике, описанной в Примере 9. Полученное соединение представляло собой бледно-желтое твердое вещество с выходом 69,7%.
Структуру полученного соединения описывали на основе следующих данных, полученных с помощью спектроскопии ядерного магнитного резонанса и масс-спектроскопии:
1H ЯМР (400 МГц, СDCl3) δ 1,32 (t, J=7,14 Гц, 3Н), 2,19 (s, 3Н), 3,43 (s, 3H), 3,55 (s, 2H), 4,28 (q, J=7,14 Гц, 2H), 5,03 (s, 2H), 6,91 (d, J=8,77 Гц, 3Н), 7,29-7,32 (m, 2H), 7,67 (d, J=8,66 Гц, 2H), 8,12 (d, J=8,66 Гц, 2H); 13С ЯМР (100 МГц, CDCl3) δ 16,1, 28,1, 38,1, 61,4, 111,8, 118,3, 126,2, 126,3, 127,2, 127,5, 127,9, 130,8, 134,8, 140,3, 143,6, 152,5, 155,4, 169,2; MS (ESI) m/z 481,23 (M-1)-, 482,34 (M)-, 483,35 (M+H)-.
Пример 21: Получение соединения Е-12
Получение этил 2-(2,5-диметил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1Н-1,2,4-триазолил))-бензилтио)-фенокси)-ацетата(Е-12)

В трехгорлую колбу объемом 250 мл к перемешиваемой смеси алюмотетрагидрида лития (LiAlH4, 1,0 г, 26,3 ммоль) и тетрагидрофурана (40 мл) при 0°С добавляли по каплям раствор 2,5-диметил-4-гидроксифенилтиоциановой кислоты (1,2 г, 6,7 ммоль) в 20 мл тетрагидрофурана. Спустя 30 мин перемешивания при 0°С смеси позволяли нагреться до комнатной температуры и перемешивали в течение следующего 1 часа при комнатной температуре. Реакцию останавливали добавлением этанола (10 мл). Значение рН смеси доводили до 3-4, добавляя 6 М соляную кислоту на водоледяной бане, и затем экстрагировали водную фазу этилацетатом (3 Х 60 мл). Объединенный органический слой высушивали над безводным сульфатом магния, фильтровали и выпаривали в вакууме, получая неочищенный продукт 2,5-диметил-4-гидроксимеркаптобензола (желтая жидкость).
В еще одной одногорлой колбе объемом 250 мл к перемешиваемому раствору полученного неочищенного продукта 2,5-диметил-4-гидроксимеркаптобензола в ацетонитриле (60 мл) добавляли 3-(1-бромбензил)-4-метил-1-(4-трифторметил)-фенил-1H-1,2,4-триазол-5(4Н)-он (111-1) (1,6 г, 3,88 ммоль) и карбонат калия (K2СО3, 0,54 г, 3,9 ммоль). После перемешивания реакционной смеси при комнатной температуре в течение 4 часов добавляли этилбромацетат (1,8 мл, 15,5 ммоль) и карбонат калия (К2СО3, 0,54 г, 3,9 ммоль). Смесь перемешивали при комнатной температуре в течение ночи. После завершения реакции реакционный раствор разбавляли 200 мл этилацетата и фильтровали. Фильтрат выпаривали, получая остаток, который очищали методом колоночной хроматографии (силикагель Н: 300-400 меш; петролейный эфир/этилацетат=5:1 об./об.), получая 1,1 г желтой гелеподобной жидкости (выход в три стадии: 50%).
Структуру полученного соединения описывали на основе следующих данных, полученных с помощью спектроскопии ядерного магнитного резонанса и масс-спектроскопии:
1H ЯМР (400 МГц, СDCl3) δ 1,28 (t J=7,16 Гц, 3Н), 2,12 (s, 3H), 2,25 (s, 3H), 3,13 (s, 3Н), 4,25 (о, J=7,16 Гц, 2Н), 4,59 (s, 2Н), 5,08 (s, 1H), 6,51 (s, 1H), 7,13 (s, 1H), 7,27-35 (m, 5H), 7,66 (d, J=8,75 Гц, 2Н), 8,10 (d, J=8,75 Гц, 2Н); 13С ЯМР (100 МГц, CDCl3) δ 14,1, 15,5, 20,7, 28,0, 50,0, 61,3, 65,5, 113,1, 117,9, 118,2, 122,4, 125,9, 126,1, 126,2, 126,8, 128,1, 128,4, 128,8, 129,1, 135,2, 138,9, 140,5, 141,5, 146,5, 152,6, 157,0, 168,6; MS (ESI) m/z 588,31 (M+NH4 +).
Пример 22: Получение соединения Е-13
Получение этил-2-метил-2-(2-метил-4-(1-(3-(4-метил-5-оксо-1 -(4-трифторметилфенил)-4,5-дигидро-1Н-1,2,4-триазолил))-бензилтио)-фенокси)-ацетата (Е-13)

В трехгорлую колбу объемом 250 мл к перемешиваемой смеси алюмотетрагидрида лития (LiAlH4, 1,0 г, 26,4 ммоль) и тетрагидрофурана (ТГФ 30 мл) добавляли по каплям раствор 3-метил-4-гидроксифенилтиоциановой кислоты (1,38 г, 8,35 ммоль) в 20 мл тетрагидрофурана при 0°С. Спустя 30 мин перемешивания при 0°С реакционной смеси позволяли нагреться до комнатной температуры и перемешивали в течение следующих 2 часов при комнатной температуре. Затем реакцию останавливали добавлением этанола (10 мл). Значение рН смеси доводили до 3-4, добавляя 6 н. соляную кислоту на водоледяной бане, и затем экстрагировали водную фазу этилацетатом (3 Х 80 мл). Объединенный органический слой высушивали над безводным сульфатом магния, фильтровали, концентрировали при пониженном давлении и очищали методом колоночной хроматографии (силикагель Н: 300-400 меш; петролейный эфир/этилацетат=5:1 об./об.), получая 1,44 г дисульфида (желтая гелеподобная жидкость, выход: 62%).
Структуру полученного соединения описывали на основе следующих данных, полученных с помощью спектроскопии ядерного магнитного резонанса и масс-спектроскопии:
1H ЯМР (400 МГц, CDCl3) δ 2,2 (s, 3H), 5,35 (s, 1H), 6,69 (d, J=8,28 Гц, 1Н), 7,18 (dd, J=8,23 Гц, 2,23 Гц, 1H), 7,24 (d, J=2,24 Гц, 1H); 13С ЯМР (100 МГц, CDCl3) δ 15,7, 115,5, 125,0, 128,3, 130,2, 134,0, 154,4; MS (ESI) m/z 278,33 (M+H)+.
В одногорлую колбу объемом 250 мл к перемешиваемому раствору полученного неочищенного продукта (0,7 г, 2,59 ммоль) в ацетонитриле (60 мл) добавляли метил-2-бромпропионат (0,7 мл, 6,0 ммоль) и карбонат калия (К2СО3, 2 г, 14,5 ммоль). Полученную смесь перемешивали при комнатной температуре в течение ночи. После завершения реакции реакционную смесь разбавляли этилацетатом (100 мл) и фильтровали. Объединенный раствор выпаривали при пониженном давлении, получая остаток, который очищали методом хроматографии (силикагель Н: 300-400 меш; петролейный зфир/этилацетат=5:1 об./об.), получая 1,0 г желтой гелеподобной жидкости (выход: 86%).
Структуру полученного соединения описывали на основе следующих данных, полученных с помощью спектроскопии ядерного магнитного резонанса:
1H ЯМР (400 МГц, CDCl3) δ 1,63 (d, J=6,84 Гц, 1Н); 2,34 (s, 3H), 3,75 (s, 3H), 4,73 (д, J=6,84 Гц, 1Н), 6,57-6,61 (m, 1Н), 7,19-7,27 (m, 2H).
В одногорлую колбу объемом 250 мл к перемешиваемому раствору вышеописанного продукта(1,0 г, 2,22 ммоль) в этаноле (15 мл) добавляли 15 мл воды и 5 мл концентрированной соляной кислоты. Медленно добавляли порошок цинка (10 г, 153 ммоль). После добавления реакционную смесь перемешивали при комнатной температуре в течение 30 мин и затем экстрагировали дихлорметаном (3 Х 50 мл). Органические фазы объединяли, высушивали над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении, получая бледно-желтую жидкость.
В одногорлую колбу объемом 150 мл к перемешиваемому раствору вышеупомянутого неочищенного продукта в 30 мл ацетонитрила добавляли раствор 3-(1-бромбензил)-4-метил-1-(4-трифторметил)-фенил-1H-1,2,4-триазол-5(4Н)-она (111-1) (1,8 г, 4,36 ммоль) в ацетонитриле (30 мл) и карбонат калия (К2СО3, 2,5 г, 18,1 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 6 часов. После завершения реакции реакционную смесь разбавляли добавлением 100 мл этилацетата, фильтровали, концентрировали при пониженном давлении и подвергали колоночной хроматографии (силикагель Н:300-400 меш; петролейный эфир/этилацетат=5:1 об./об.), получая 1,2 г Е-13 (пару диастереоизомеров) (бледно-желтая гелеобразная жидкость, выход: 50%).
Структуру полученного соединения описывали на основе следующих данных, полученных с помощью спектроскопии ядерного магнитного резонанса:
1H ЯМР (400 МГц, CDCl3) δ 1,61 (d, J=6,84 Гц, 1Н); 2,18 (s, 3H), 3,15 (s, 3H), 3,70 (d, J=12,61 Гц, 3H); 4,70 (q, J=6,82 Гц, 1H); 5,18 (s, 1Н); 6,51 (d, J=8,46 Гц, 1Н), 7,0-7,11 (m, 1Н), 7,10-7,15 (m, 1Н), 7,34-7,37 (m, 5H), 7,67 (d, J=8,74 Гц, 2H), 8,11 (d, J=8,74 Гц, 2H); 13С ЯМР (100 МГц, CDCl3) δ 16,1, 18,5, 28,1, 50,6, 52,2, 72,7, 112,1, 118,2, 123,0, 125,5, 126,2, 126,8, 127,1, 128,2, 128,5, 128,9, 133,8, 135,2, 137,7, 140,6, 146,5, 152,6, 156,9, 172,2.
Пример 23: Получение соединения Е-14
Получение этил-2-(2-этил-4-(1 -(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1Н-1,2,4-триазолил))-бензилтио)-фенокси)-ацетата (Е-14)

В трехгорлую колбу объемом 500 мл последовательно добавляли о-этилфенол (5 г, 40,9 ммоль), тиоцианат натрия (10 г, 123,3 ммоль), бромид натрия (4,3 г, 41,79 ммоль) и метанол (100 мл). Смесь охлаждали до 0°С на ледяной бане и затем по каплям добавляли раствор брома (2,6 мл, 50,6 ммоль) в метаноле (50 мл). Спустя 1 час перемешивания при 0 °С смеси позволяли нагреться до комнатной температуры и перемешивали в течение следующих 4 часов при комнатной температуре. К реакционной смеси медленно добавляли 200 мл насыщенного водного раствора гидрокарбоната натрия и перемешивали в течение 10 мин. Смесь экстрагировали этилацетатом (2 Х 300 мл). Объединенный органический слой высушивали над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении, получая красновато-коричневую вязкую жидкость. Проводили колоночную хроматографию (силикагель Н: 300-400 меш; петролейный эфир/этилацетат=5:1 об./об.), получая 6,8 г 3-этил-4-гидрокси-фенилтиоциановой кислоты (красновато-коричневая вязкая жидкость, выход: 93%).
Структуру полученного соединения описывали на основе следующих данных, полученных с помощью спектроскопии ядерного магнитного резонанса:
1H ЯМР (400 МГц, ДМСО) δ 1,25 (t J=7,28 Гц, 3Н), 2,64 (q, J=7,53 Гц, 2Н), 5,46 (s, 1 Н), 6,80 (d, J=8,36 Гц, 1 Н), 7,28 (dd, J=8,36 Гц, 2,52 Гц, 1 Н), 7,34 (d, J=2,48,36 Гц, 1 Н).
В трехгорлую колбу объемом 150 мл к перемешиваемой смеси алюмотетрагидрида лития (LiAlH4, 1,0 г, 26,2 ммоль) и тетрагидрофурана (40 мл) добавляли по каплям раствор 3-этил-4-гидроксифенилтиоциановой кислоты (1,3 г, 7,25 ммоль) в 20 мл тетрагидрофурана при 0°С. Спустя 30 мин перемешивания при 0°С реакционной смеси позволяли нагреться до комнатной температуры и перемешивали в течение следующего 1 часа при комнатной температуре. Реакцию останавливали добавлением этанола (10 мл). Значение рН смеси доводили до 3-4, добавляя 6 н. соляную кислоту на водоледяной бане, и затем экстрагировали водную фазу этилацетатом (3 Х 60 мл). Объединенный органический слой высушивали над безводным сульфатом магния, фильтровали и выпаривали в вакууме, получая неочищенный продукт 3-этил-4-гидроксимеркаптобензола (желтая жидкость).
В еще одной одногорлой колбе объемом 150 мл к перемешиваемому раствору полученного неочищенного продукта 3-этил-4-гидроксимеркаптобензола в ацетонитриле (20 мл) добавляли карбонат калия (К2СО3, 1,0 г, 7,23 ммоль) с последующим добавлением по каплям раствора 3-(1'-бромбензил)-4-метил-1-(4-трифторметил)-фенил-1Н-1,2,4-триазол-5(4Н)-она (111-1) (1,6 г, 3,88 ммоль) в 20 мл ацетонитрила. После перемешивания смеси в течение 6 часов при комнатной температуре добавляли карбонат калия (К2СО3, 1,0 г, 7,23 ммоль) и этилбромацетат (1,6 мл, 13,8 ммоль). Смесь перемешивали в течение следующих 8 ч при той же температуре. После завершения реакции реакционную смесь разбавляли этилацетатом (50 мл) и фильтровали. Фильтрат выпаривали до остатка, который очищали методом колоночной хроматографии (силикагель Н:300-400 меш; петролейный эфир/этилацетат=5:1 об./об.), получая 1,3 г Е-14 (желтое гелеобразное вещество, выход в три стадии: 58,6%).
Структуру полученного соединения описывали на основе следующих данных, полученных с помощью спектроскопии ядерного магнитного резонанса и масс-спектроскопии:
1H ЯМР (400 МГц, ДМСО) δ 1,09 (t J=7,48 Гц, 3Н), 1,26 (t J=7,28 Гц, 3Н), 2,59 (q, J=7,52 Гц, 2Н), 3,15 (s, 3Н), 4,22 (q, J=7,20 Гц, 2Н), 4,58 (s, 2Н), 5,18 (s, 1H), 6,56 (d, J=8,20 Гц, 1H), 7,15 (dd, J=8,32 Гц, 2,28 Гц, 1H), 7,31-7,36 (m, 5H), 7,66 (d, J=8,72 Гц, 1H), 8,11 (d, J=8,52 Гц, 1H); 13С ЯМР (100 МГц, CDCl3) δ 13,7, 14,1, 23,0, 28,1, 50,6, 61,3, 65,4, 111,5, 118,2, 123,3, 126,1, 126,8, 127,1, 128,2, 128,5, 128,9, 133,8, 134,3, 135,2, 136,2, 140,5, 146,5, 152,6, 156,7, 168,5; MS (ESI) m/z 572,14 (M+H+); 589,1 (M+NH4 +).
Пример 24: Получение соединения Е-15
Получение этил-2,2-диметил-2-(3-метил-4-(1-(3-(4-метил-5-оксо-1 -(4-трифторметилфенил)-4,5-дигидро-1H-1,2,4-триазолил))-бензилокси)-фенилтио)-ацетата (Е-15)

В одногорлую колбу объемом 150 мл к перемешиваемому раствору 3-метил-4-гидроксимеркаптобензола (0,52 г, 1,87 ммоль) в ацетонитриле (30 мл) добавляли бромид III-1 (1,53 г, 3,71 ммоль) и карбонат калия (К2СО3, 2,5 г, 18,1 ммоль). Смесь перемешивали при комнатной температуре в течение 12 часов. После завершения реакции реакционную смесь разбавляли этилацетатом (100 мл) и фильтровали. Фильтрат выпаривали до остатка, который подвергали колоночной хроматографии (силикагель Н:300-400 меш; петролейный эфир/этилацетат=5:1 об./об.), получая 1,4 г бледно-желтой гелеподобной жидкости.
В трехгорлую колбу объемом 150 мл к перемешиваемому раствору вышеописанного продукта(1,4 г, 1,48 ммоль) в этаноле (15 мл) добавляли 15 мл воды и 5 мл концентрированной соляной кислоты. Медленно добавляли порошок цинка (10 г, 153 ммоль). После добавления реакционную смесь перемешивали при комнатной температуре в течение 30 мин и затем экстрагировали дихлорметаном (3 Х 50 мл). Органические фазы объединяли, высушивали над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении, получая бледно-желтую жидкость.
В одногорлую колбу объемом 150 мл к перемешиваемому раствору вышеупомянутого неочищенного продукта в ацетонитриле (30 мл) добавляли этил-2-бромизопропионат (3 мл, 20,2 ммоль) в ацетонитриле (30 мл) и карбонат калия (К2СО3, 2,0 г, 14,4 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 12 часов. После завершения реакции реакционную смесь разбавляли этилацетатом (100 мл) и фильтровали. Фильтрат выпаривали до остатка, который очищали методом колоночной хроматографии (силикагель Н:300-400 меш; петролейный эфир/этилацетат=5:1 об./об.), получая 0,58 г Е-15 (бесцветное гелеобразное вещество, выход в три стадии: 26,6%).
Структуру полученного соединения описывали на основе следующих данных, полученных с помощью спектроскопии ядерного магнитного резонанса и масс-спектроскопии:
1Н ЯМР (400 МГц, CDCl3) δ 1,19 (t, J=7,20 Гц, 3Н), 1,45 (s, 6H), 2,37 (s, 3H), 3,20 (s, 3Н), 4,10 (q, J=7,20 Гц, 2Н), 6,41 (s, 1H), 6,94 (d, J=8,52 Гц, 1Н), 7,25 (dd, J=8,52 Гц, 1,8 Гц, 1H), 7,32 (d, J=1,8 Гц, 1H), 7,39-7,52 (m, 5H), 7,69 (d, J=8,52 Гц, 2Н), 8,17 (d, J=8,52 Гц, 2Н); 13С ЯМР (100 МГц, CDCl3) δ 14,1, 16,4, 25,8, 28,2, 50,9, 61,1, 74,5, 112,4, 118,2, 124,2, 125,7, 126,2, 126,3, 127,4, 129,0, 129,1, 134,7, 136,1, 139,9, 140,4, 145,9, 152,6, 156,2, 173,9; MS (ESI) m/z 584,71 (M-H)-.
Пример 25: Получение соединения Е-16
Получение этил-2,2-диметил-2-(3-метил-4-(1-(2-(4-метил-5-оксо-1-(4-трифторметил-фенил)-4,5-дигидро-1H-1,2,4-триазолил))-бензилтио)-фенокси)-ацетата (Е-16)
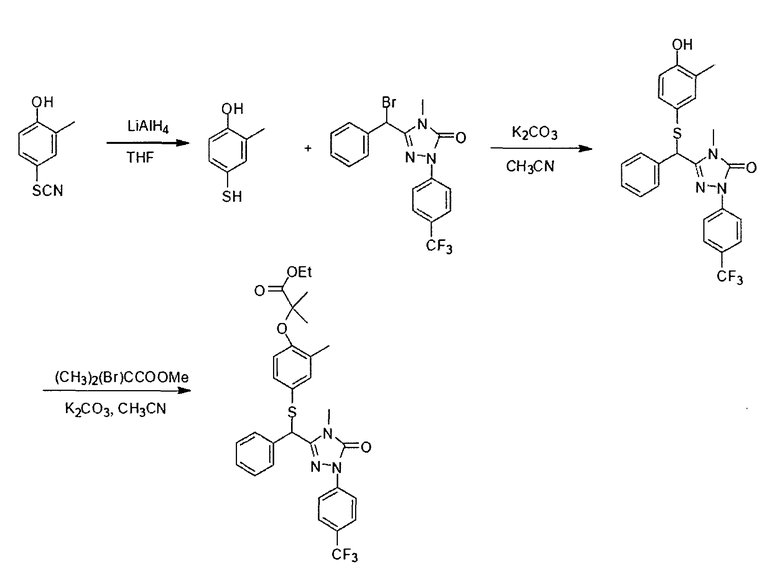
В трехгорлую колбу объемом 250 мл к перемешиваемой смеси алюмотетрагидрида лития (LiAlH4, 2,0 г, 52,7 ммоль) и тетрагидрофурана (40 мл) добавляли по каплям раствор 3-метил-4-гидроксифенилтиоциановой кислоты (2,5 г, 15,13) в тетрагидрофуране (30 мл) при 0°С. Спустя 30 мин перемешивания при 0°С реакционной смеси оставляли для нагревания до комнатной температуры и перемешивали в течение следующих 2 часов при комнатной температуре. Затем реакцию останавливали добавлением этанола (10 мл). Значение рН смеси доводили до 3-4, добавляя 6 М соляную кислоту на водоледяной бане, и затем экстрагировали водную фазу этилацетатом (3 Х 100 мл). Объединенный органический слой высушивали над безводным сульфатом магния, фильтровали и выпаривали в вакууме, получая желтую жидкость.
В одногорлую колбу объемом 250 мл к перемешиваемому раствору полученной желтой жидкости в ацетонитриле (30 мл) добавляли бромид III-1 (3,2 г, 7,76 ммоль) и карбонат калия (К2СО3, 1,15 г, 8,32 ммоль). Смесь перемешивали при комнатной температуре в течение 6 часов. После завершения реакции реакционную смесь разбавляли этилацетатом (100 мл) и фильтровали. Фильтрат выпаривали до остатка, который подвергали колоночной хроматографии (силикагель Н:300-400 меш; петролейный эфир/этилацетат=5:1 об./об.), получая 3,6 г желтой гелеподобной жидкости.
В одногорлую колбу объемом 250 мл к перемешиваемому раствору вышеописанного продукта (2,2 г, 4,67 ммоль) в ацетонитриле (60 мл) добавляли этил-2-бромизопропионат (2 мл, 13,5 ммоль) и карбонат калия (К2СО3) (1,5 г, 10,8 ммоль). Реакционную смесь нагревали с обратным холодильником в течение 36 часов. После завершения реакции реакционную смесь разбавляли этилацетатом (100 мл) и фильтровали. Фильтрат выпаривали до остатка, который очищали методом колоночной хроматографии (силикагель Н:300-400 меш; петролейный эфир/этилацетат=5:1 об./об.), получая 1,0 г Е-16 (желтое гелеподобное вещество, выход в три стадии: 44%).
Структуру полученного соединения описывали на основе следующих данных, полученных с помощью спектроскопии ядерного магнитного резонанса и масс-спектроскопии:
1H ЯМР (400 МГц, CDCl3) δ 1,20 (t, J=7,14 Гц, 3Н), 1,55 (d, J=5,76 Гц, 6Н), 2,13 (s, 3Н), 3,15 (s, 3Н), 4,18 (q, J=7,14 Гц, 2Н), 5,19 (s, 1Н), 6,49 (d, J=8,46 Гц, 1Н), 7,03 (dd, J=8,46 Гц, 2,36 Гц, 1Н), 7,16 (d, J=2,36 Гц, 1Н), 7,32-7,37 (m, 5H), 7,66 (d, J=8,74 Гц, 2Н), 8,10 (d, J=8,74 Гц, 2Н); MS (ESI) m/z 586,39(M+H+).
Пример 26: Получение соединения Е-17
Используя соединения II 1-2 и 11-4 в качестве исходных материалов, получали этил-2-(3-метил-4-(1-(3-(4-n-бутил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1 Н-1,2,4-триазолил))-бензилокси)-фенилтио)-ацетат (Е-17) согласно химической процедуре, аналогичной описанию в Примере 9. Полученное соединение представляло собой бледно-желтое твердое вещество с выходом 81,2%.
Структуру полученного соединения описывали на основе следующих данных, полученных с помощью спектроскопии ядерного магнитного резонанса и масс-спектроскопии:
1H ЯМР (400 МГц, CDCl3) δ 0,76 (t, J=7,30 Гц, 3Н), 0,92-1,02 (m, 1Н), 1,03-1,29 (m, 5H), 1,35-1,48(m, 1Н), 2,37 (s, 3Н), 3,47-3,56 (m, 3Н), 3,63-3,67 (m, 1Н), 4,11-4,16 (m, 2Н), 6,38 (s, 1Н), 6,97 (d, J=8,59 Гц, 1Н), 7,24 (dd, J=8,57 Гц, 2,25 Гц, 1Н), 7,34 (d, J=2,04 Гц, 1Н), 7,39-7,52 (m, 5H), 7,69 (d, J=8,73 Гц, 2Н), 8,19 (d, J=8,56 Гц, 2Н); 13С ЯМР (100 МГц, CDCl3) δ 13,5, 14,1, 16,5, 19,9, 30,1, 38,0, 42,4, 61,4, 74,7, 113,1, 118,2, 125,6, 126,2, 126,3, 128,0, 128,9, 129,1, 130,8, 134,9, 135,3, 140,5, 145,9, 152,6, 155,0, 169,8; MS (ESI) m/z 600,0 (M)+, 601,2 (M+1)+, 602,2 (M+2)+, 603,2 (M+3)+.
Пример 27: Получение соединения Е-18
Получение этил 2,2-диметил-2-(3-метил-4-(1-(2-(4-n-бутил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1H-1,2,4-триазолил))-бензилтио)-фенокси)-ацетата (Е-18)

В трехгорлую колбу объемом 250 мл к перемешиваемой смеси алюмотетрагидрида лития (LiAlH4, 1,2 г, 31,6 ммоль) и тетрагидрофурана (50 мл) добавляли по каплям раствор 3-метил-4-гидроксифенилтиоциановой кислоты (1,4 г, 8,47 ммоль) в тетрагидрофуране (20 мл) при 0°С. Спустя 30 мин перемешивания при 0°С смеси оставляли для нагревания до комнатной температуры и перемешивали в течение следующих 2 часов при комнатной температуре. Реакцию останавливали добавлением этанола (10 мл). Значение рН смеси доводили до 3-4, добавляя 6 н. соляную кислоту на водоледяной бане, и затем экстрагировали водную фазу этилацетатом (3 Х 100 мл). Объединенный органический слой высушивали над безводным сульфатом магния, фильтровали и выпаривали в вакууме, получая желтую жидкость
В одногорлой колбе объемом 250 мл к перемешиваемому раствору полученной желтой жидкости в ацетонитриле (60 мл) добавляли бромид III-2 (1,4 г, 3,08 ммоль) и карбонат калия (К2СО3, 0,42 г, 3,04 ммоль). После перемешивания реакционной смеси при комнатной температуре в течение 6 часов добавляли этил-2-бромизопропионат (6 мл, 40,5 ммоль) в ацетонитриле (10 мл) и карбонат калия (К2СО3, 2,4 г, 17,4 ммоль). Смесь нагревали с обратным холодильником в течение 36 часов. После завершения реакции реакционную смесь разбавляли этилацетатом (100 мл) и фильтровали. Фильтрат выпаривали до остатка, который подвергали колоночной хроматографии (силикагель Н:300-400 меш; петролейный эфир/этилацетат=10:1 об./об.), получая 1,52 г Е-18 (желтое гелеподобное вещество, выход в три стадии: 28,6%).
Структуру полученного соединения описывали на основе следующих данных, полученных с помощью спектроскопии ядерного магнитного резонанса и масс-спектроскопии:
1H ЯМР (400 МГц, CDCl3) δ 0,80 {t, J=7,30 Гц, 3Н), 1,16-1,36(m, 7H), 1,56 (d, J=2,84 Гц, 6H), 2,12 (s, 3Н), 3,48-3,57 (m, 2H), 4,17-4,23 (m, 2H), 5,10 (s, 1H), 6,48 (d, J=8,44 Гц, 1H), 7,01 (dd, J=8,46 Гц, 2,28 Гц, 1H), 7,17 (d, J=2,08 Гц, 1H), 7,30-7,35 (m, 5H), 7,67 (d, J=8,81 Гц, 2H), 8,15 (d, J=8,66 Гц, 2H); 13С ЯМР (100 МГц, CDCl3) δ 13,5, 14,0, 16,5, 19,8, 25,3, 29,7, 30,4, 41,9, 50,3, 61,5, 79,2, 116,4, 118,1, 124,5, 126,1, 128,4, 128,5, 128,8, 130,3, 133,3, 135,9, 137,8, 140,6, 146,4, 152,3, 154,9, 174,1; MS (ESI) m/z 626,1 (M-2)-, 627,1 (M-1)-, 628,1 (M)-.
Пример 28: Получение соединения Е-19
Получение этил-2-(2,5-диметил-4-(1-(3-(4-n-бутил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1Н-1,2,4-триазолил))-бензилтио)-фенокси)-ацетата(Е-19)
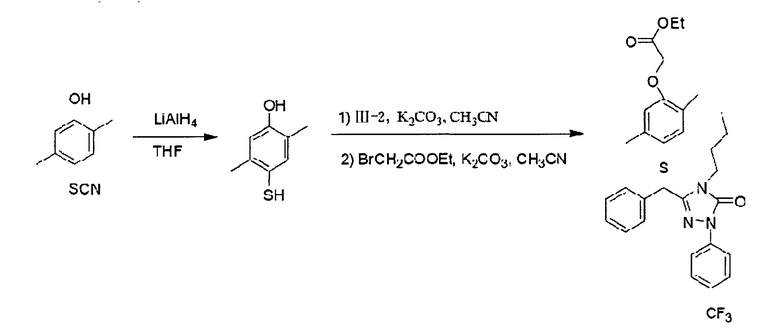
В трехгорлую колбу объемом 250 мл к перемешиваемой смеси алюмотетрагидрида лития (LiAlH4, 1,0 г, 26,3 ммоль) и тетрагидрофурана (40 мл) добавляли по каплям раствор 2,5-диметил-4-гидроксифенилтиоциановой кислоты (1,02 г, 5,69 ммоль) в тетрагидрофуране (20 мл) при 0°С. Спустя 30 мин перемешивания при 0°С реакционной смеси оставляли для нагревания до комнатной температуры и перемешивали в течение следующего 1 часа при комнатной температуре. Реакцию останавливали добавлением этанола (10 мл). Значение рН смеси доводили до 3-4, добавляя 6 М соляную кислоту на водоледяной бане, и затем экстрагировали водную фазу этилацетатом (3 Х 60 мл). Объединенный органический слой высушивали над безводным сульфатом магния, фильтровали и выпаривали в вакууме, получая неочищенный продукт 2,5-диметил-4-гидроксимеркаптобензола (желтая жидкость).
В одногорлую колбу объемом 250 мл к перемешиваемому раствору полученного неочищенного продукта 2,5-диметил-4-гидроксимеркаптобензола в ацетонитриле (60 мл) добавляли 3-(1'-бромбензил)-4-n-бутил-1-(4-трифторметил)-фенил-1H-1,2,4-триазол-5(4Н)-он III-2 (1,1 г, 2,42 ммоль) и карбонат калия (К2СО3, 0,35 г, 2,35 ммоль). После перемешивания реакционной смеси при комнатной температуре в течение 4 часов добавляли этилбромацетат (1,8 мл, 15,5 ммоль) и карбонат калия (К2СО3, 1,5 г, 10,8 ммоль), а затем реакционную смесь перемешивали при комнатной температуре в течение ночи. После завершения реакции реакционную смесь разбавляли этилацетатом (200 мл) и фильтровали. Фильтрат выпаривали в вакууме до остатка, который подвергали колоночной хроматографии (силикагель Н:300-400 меш; петролейный эфир/этилацетат=8:1 об./об.), получая 0,8 г Е-19 (желтое гелеподобное вещество, выход в три стадии: 22,9%).
Структуру полученного соединения описывали на основе следующих данных, полученных с помощью спектроскопии ядерного магнитного резонанса:
1H ЯМР (400 МГц, CDCl3) δ 0,80 (t, J=7,26 Гц, 3Н), 1,18-1,33 (m, 7H), 2,13 (s, 3H), 2,22 (s, 3Н), 3,50-3,52 (m, 2H), 4,23-4,29 (m, 2H), 4,59 (s, 2H), 5,01 (s, 1H), 6,50 (s, 1H), 7,13 (s, 1H), 7,30-7,32 (m, 5H), 7,68 (d, J=8,64 Гц, 2H), 8,16 (d, J=8,52 Гц, 2H); 13С ЯМР (100 МГц, CDCl3) δ 13,5, 14,2, 15,5, 19,8, 20,8, 29,7, 30,4, 41,8, 49,7, 61,4, 65,5, 112,7, 118,1, 122,7, 125,8, 126,2, 128,3, 128,4, 128,8, 135,9, 139,0, 140,6, 141,6, 146,5, 152,3, 156,9, 168,7.
Получение соединения (I) (кислоты):
Пример 29: Получение соединения А-1
Получение 2-(2-метил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1H-1,2,4-триазолил))-бензилокси)-фенокси)-уксусной кислоты (А-1)
К перемешиваемому раствору этил-2-(2-метил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1H-1,2,4-триазолил))-бензилокси)-феноксиацетата (28 г, 51,8 ммоль) в дихлорметане (50 мл) и этаноле (50 мл) добавляли 50 мл водного раствора гидроксида натрия (10 г, 250 ммоль). Реакционную смесь перемешивали в течение 12 часов при комнатной температуре. После завершения реакции значение рН смеси доводили до 2-3, добавляя 6 н. соляную кислоту. Полученную смесь экстрагировали дихлорметаном (3 Х 100 мл). Объединенный органический слой высушивали над безводным сульфатом магния, фильтровали и выпаривали до остатка. Остаток очищали методом колоночной хроматографии (силикагель Н:300-400 меш; петролейный эфир/этилацетат=4:1→1:1 об./об.), получая 18,2 г А-1 в виде белого твердого вещества (выход: 68,5%).
Структуру полученного соединения описывали на основе следующих данных, полученных с помощью спектроскопии ядерного магнитного резонанса и масс-спектроскопии:
1H ЯМР (400 МГц, CDCl3) δ 2,29 (s, 3Н), 3,25 (s, 3Н), 4,62 (s, 2H), 6,32 (s, 1H), 6,69 (d, J=8,8 Гц, 1 Н), 6,86 (d, J=8,75 Гц, 1 Н), 6,98 (s, 1 Н), 7,43-7,50 (m, 3Н), 7,55-7,58 (m, 2H), 7,72 (d, J=8,48 Гц, 2H), 8,19 (d, J=8,48 Гц, 2H); 13C ЯМР (100 МГц, CDCl3) δ 16,4, 18,1, 28,3, 58,4, 65,9, 75,3, 112,8, 113,2, 118,3, 119,3, 122,7, 125,8, 126,2, 127,1, 128,9, 129,0, 129,4, 135,1, 140,4, 146,3, 151,5, 152,8, 173,2; MS (ESI) m/z 514,2 (M+H+); 531 (М+NH4 +).
Пример 30: Получение соединения А-2
Используя соединение Е-2 в качестве исходного материала, получали 2-(2-метил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1H-1,2,4-триазолил))-бензилокси)-фенилтио)-уксусную кислоту (А-2) согласно способу синтеза, аналогичному способу, описанному в Примере 29. Полученное соединение представляло собой бледно-желтое твердое вещество с выходом 46%.
Структуру полученного соединения описывали на основе следующих данных, полученных с помощью спектроскопии ядерного магнитного резонанса и масс-спектроскопии:
1Н ЯМР (400 МГц, CDCl3) δ 2,42 (s, 3Н), 3,18 (s, 3Н), 3,51 (s, 2H), 6,76 (s, 1H), 6,90 (d, J=8,4 Гц, 1H), 6,98 (d, J=2,76 Гц, 1H), 7,25-7,52 (m, 5H), 7,67 (d, J=8,67 Гц, 2H), 8,14 (d, J=8,58 Гц, 2H); 13С ЯМР (100 МГц, CDCl3) δ 20,9, 28,3, 36,9, 74,6, 113,7, 118,0, 118,3, 125,8, 126,2, 126,3, 126,6, 127,1, 128,98, 129,01, 134,1, 134,6, 140,4, 142,2, 145,9, 152,6, 156,8, 174,6; MS (ESI) m/z 528 (M-H)-; 529 (M-); 530 1 (M+H)-.
Пример 31: Получение соединения А-3
Используя соединение Е-3 в качестве исходного материала, получали 2-(3-метил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1H-1,2,4-триазолил))-бензилокси)-фенилтио)-уксусную кислоту (А-3) согласно процессу синтеза, аналогичному описанию в Примере 29. Полученное соединение представляло собой белое твердое вещество с выходом 24,2%.
Структуру полученного соединения описывали на основе следующих данных, полученных с помощью спектроскопии ядерного магнитного резонанса и масс-спектроскопии:
1Н ЯМР (400 МГц, CDCl3) δ 2,41 (s, 3H), 3,24 (s, 3H), 3,61 (s, 2H), 6,43 (s, 1H), 6,99 (d, J=8,58 Гц, 1H), 7,22-7,29 (m, 1H), 7,38 (d, J=2,28 Гц, 1H), 7,38-7,56 (m, 5H), 7,73 (d, J=8,68 Гц, 2H), 8,21 (d, J=8,68 Гц, 2H); 13С ЯМР (100 МГц, CDCl3) δ 16,5, 28,2, 37,8, 74,5, 113,2, 118,2, 125,7, 126,2, 126,6, 127,1, 128,2, 128,99, 129,1, 130,8, 134,7, 140,4, 145,9, 152,7, 155,0, 174,2; MS (ESI) m/z 556,9 (M+CO)+.
Пример 32: Получение соединения А-4
Используя соединение Е-4 в качестве исходного материала, получали 2-(2-метил-4-(1-(3-(4-метил-5-оксо-1-(4трифторметилфенил)-4,5-дигидро-1H-1,2,4-триазолил))-бензилтио)-фенокси)-уксусную кислоту (А-4) согласно способу синтеза, аналогичному способу, описанному в Примере 29. Полученное соединение представляло собой белое твердое вещество с выходом 52,9%.
Структуру полученного соединения описывали на основе следующих данных, полученных с помощью спектроскопии ядерного магнитного резонанса и масс-спектроскопии:
1Н ЯМР (400 МГц, CDCl3) δ 2,18 (s, 3H), 3,13, (s, 3H), 4,63 (s, 2H), 5,19 (s, 1H), 6,57 (d, J=8,43 Гц, 1H), 7,13 (dd, J=6,07 Гц, 2,26 Гц, 1H), 7,18 (d, J=1,8 Гц, 1H), 7,32-7,36 (m, 5H), 7,66 (d, J=8,73 Гц, 2H), 8,10 (d, J=8,69 Гц, 2H); 13C ЯМР (100 МГц, CDCl3) δ 16,0, 28,1, 50,5, 64,9, 118,3, 123,6, 126,1, 126,2, 128,2, 128,4, 128,6, 128,9, 133,9, 135,0, 137,8, 140,4, 146,5, 156,7, 172,6; MS (ESI) m/z 530 (M+H)+, 531 (M+2), 532 (M+3).
Пример 33: Получение соединения А-5
Используя соединение Е-5 в качестве исходного материала, получали 2-(2-этил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1H-1,2,4-триазолил))-бензилокси)-фенокси)-уксусную кислоту (А-5) согласно способу синтеза, аналогичному способу, описанному в Примере 29. Полученное соединение представляло собой белое твердое вещество с выходом 62,8%.
Структуру полученного соединения описывали на основе следующих данных, полученных с помощью спектроскопии ядерного магнитного резонанса и масс-спектроскопии:
1H ЯМР (400 МГц, CDCl3) δ 1,19 (t, J=7,55 Гц, 3H), 2,66 (q, J=7,51 Гц, 2H), 3,19 (s, 3H), 4,62 (s, 2H), 6,29 (s, 1H), 6,66 (d, J=8,88 Гц, 1H), 6,83 (dd, J=8,87 Гц, 3,09 Гц, 1H), 6,96 (d, J=3,09 Гц, 1Н), 7,38-7,53 (m, 5Н), 7,67 (d, J=8,85 Гц, 2H), 8,15 (d, J=8,78 Гц, 2Н); 13С ЯМР (100 МГц, CDCl3) δ 13,9, 23,2, 28,3, 65,7, 75,2, 112,6, 113,0, 117,7, 118,2, 125,8, 126,2, 127,0, 127,4, 128,8, 129,01, 135,1, 135,2, 140,4, 146,3, 150,9, 151,8, 152,8, 173,3; MS (ESI) m/z 525,9 (M-1)-, 527 (M)-, 528 (M+1)-.
Пример 34: Получение соединения А-6
Используя соединение Е-6 в качестве исходного материала, получали 2-(3-метил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1H-1,2,4-триазолил))-бензилокси)-фенокси)-уксусную кислоту (А-6) согласно способу синтеза, аналогичному способу, описанному в Примере 29. Полученное соединение представляло собой бледно-желтое твердое вещество с выходом 42,4%.
Структуру полученного соединения описывали на основе следующих данных, полученных с помощью спектроскопии ядерного магнитного резонанса и масс-спектроскопии:
1Н ЯМР (400 МГц, CDCl3) δ 2,40 (s, 3H), 3,25 (s, 3H), 4,65 (s, 3H), 6,34 (s, 1Н), 6,69 (d d, J=8,86 Гц, 3,0 Гц, 1Н), 6,88 (d, J=3,0 Гц, 1Н), 6,95 (d, J=8,88 Гц, 2H), 7,43-7,58 (m, 5Н), 7,73 (d, J=8,63 Гц, 2H), 8,21 (d, J=8,58 Гц, 2H); 13С ЯМР (100 МГц, CDCl3) δ 16,8, 28,3, 65,4, 75,1, 112,2, 113,8, 118,3, 118,5, 125,7, 126,2, 126,3, 127,1, 128,9, 129,1, 135,1, 140,4, 146,3, 150,2, 152,4, 152,8, 173,1; MS (ESI) m/z 513 (M)-, 512 (M-1)-, 514 (M+1)-.
Пример 35: Получение соединения А-7
Используя соединение Е-7 в качестве исходного материала, получали 2-(3-метил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1H-1,2,4-триазолил))-бензилтио)-фенокси)-уксусную кислоту (А-7) согласно способу синтеза, аналогичному способу, описанному в Примере 29. Полученное соединение представляло собой бледно-желтое твердое вещество с выходом 42%.
Структуру полученного соединения описывали на основе следующих данных, полученных с помощью спектроскопии ядерного магнитного резонанса и масс-спектроскопии:
1H ЯМР (400 МГц, СDCl3) δ 2,33 (s, 3H), 3,11 (s, 3H), 4,65 (s, 2H), 5,11 (s, 1Н), 6,62 (dd, J=8,53 Гц, 2,88 Гц, 1 Н), 6,78 (d, J=2,88 Гц, 1 Н), 7,27-7,34 (m, 5Н), 7,66 (d, J=8,79 Гц, 2H), 8,09 (d, J=8,71 Гц, 2H); 13С ЯМР (100 МГц, CDCl3) δ 21,0, 28,1, 49,7, 64,6, 112,6, 116,9, 118,3, 123,1, 126,2, 128,1, 128,6, 129,0, 135,0, 137,9, 140,4, 144,6, 146,5, 152,6, 158,3, 172,1; MS (ESI) m/z 530 (M)+, 531 (M+1)+.
Пример 36: Получение соединения А-8
Используя соединение Е-8 в качестве исходного материала, получали 2-(2-метил-4-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1Н-1,2,4-триазолил)-метилтио)-фенокси)-уксусную кислоту (А-8) согласно способу синтеза, аналогичному способу, описанному в Примере 29. Полученное соединение представляло собой белое твердое вещество с выходом 76%.
Структуру полученного соединения описывали на основе следующих данных, полученных с помощью спектроскопии ядерного магнитного резонанса и масс-спектроскопии:
1Н ЯМР (400 МГц, ДМСО) δ 2,11 (s, 3Н), 3,3 (s, 3Н), 4,13 (s, 2H), 4,68 (s, 2H), 6,80 (d, J=8,13 Гц, 1H), 7,25-7,27 (m, 2H), 7,80 (d, J=8,85 Гц, 2H), 7,99 (d, J=8,68 Гц, 2H); 13С ЯМР (100 МГц, ДМСО) δ 16,2, 28,0, 30,4, 62,5, 65,3, 112,5, 118,1, 120,6, 123,3, 123,6, 123,9, 124,9, 125,2, 125,6, 125,9, 126,8, 127,5, 128,7, 132,1, 135,6, 141,1, 146,3, 152,3, 156,6, 170,5; MS (ESI) m/z 452,8 (M).
Пример 37: Получение соединения А-9
Используя соединение Е-9 в качестве исходного материала, получали 2-(2-метил-4-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1H-1,2,4-триазолил)-метокси)-фенокси)-уксусную кислоту (А-9) согласно способу синтеза, аналогичному способу, описанному в Примере 29. Полученное соединение представляло собой белое твердое вещество с выходом 69,2%.
Структуру полученного соединения описывали на основе следующих данных, полученных с помощью спектроскопии ядерного магнитного резонанса и масс-спектроскопии:
1H ЯМР (400 МГц, CDCl3) δ 2,28 (s, 3Н), 3,43 (s, 3Н), 4,64 (s, 2H), 4,99 (s, 2H), 6,69 (d, J=8,40 Гц, 1 Н), 6,78 (dd, J=8,82 Гц, 3,06 Гц, 1 Н), 6,85 (dd, J=2,96 Гц, 1 Н), 7,67 (d, J=8,60 Гц, 2H), 8,13 (d, J=8,55 Гц, 2H); 13С ЯМР (100 МГц, CDCl3) δ 16,4, 28,1, 61,9, 65,9, 112,1, 112,8, 118,2, 118,3, 118,4, 126,2, 126,3, 129,3, 143,9, 151,2, 152,0, 152,5, 170,4; MS (ESI) m/z 436 (M-1), 437 (M), 438 (M+1).
Пример 38: Получение соединения А-10
Используя соединение Е-10 в качестве исходного материала, получали 2-(2,5-диметил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1Н-1,2,4-триазолил))-бензилтио)-фенокси)-уксусную кислоту (А-10) согласно способу синтеза, аналогичному способу, описанному в Примере 29. Полученное соединение представляло собой белое твердое вещество с выходом 88,9%.
Структуру полученного соединения описывали на основе следующих данных, полученных с помощью спектроскопии ядерного магнитного резонанса и масс-спектроскопии:
1Н ЯМР (400 МГц, CDCl3) δ 2,09 (s, 3Н), 2,25 (s, 3Н), 3,09 (s, 3H), 4,59 (s, 2H),5,09 (s, 1Н), 6,52 (s. 1H), 7,13 (s, 1H), 7,31-7,37 (m, 5H), 7,65 (d, J=8,70 Гц, 2Н), 8,09 (d, J=8,70 Гц, 2Н); 13С ЯМР (100 МГц, CDCl3) δ 15,4, 20,7, 28,0, 49,7, 65,2, 113,2, 118,4, 122,7, 126,2, 126,7, 128,2, 128,5, 128,9, 135,1, 139,0, 140,4, 141,6, 146,7, 152,6, 156,8, 173,0; MS (ESI) m/z 542,42 (M-1)-, 543,54 (M)-, 544,43 (M+H)-.
Пример 39: Получение соединения А-11
Используя соединение Е-11 в качестве исходного материала, получали 2-(2,5-диметил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1H-1,2,4-триазолил))-бензилокси)-фенилтио)-уксусную кислоту (А-11) согласно способу синтеза, аналогичному способу, описанному в Примере 29. Полученное соединение представляло собой белое твердое вещество с выходом 70,8%.
Структуру полученного соединения описывали на основе следующих данных, полученных с помощью спектроскопии ядерного магнитного резонанса и масс-спектроскопии:
1H ЯМР (400 МГц, CDCl3) δ 2,32 (s, 3Н), 2,37 (s, 3Н), 3,18 (s, 3Н), 3,51 (s, 2Н), 6,39 (s. 1H), 6,88 (s, 1H), 7,31 (s, 1H), 7,39-7,52 (m, 5H), 7,69 (d, J=8,67 Гц, 2Н), 8,16 (d, J=8,67 Гц, 2Н); 13С ЯМР (100 МГц, CDCl3) δ 16,0, 20,7, 28,2, 37,2, 74,4, 114,7, 118,2, 125,4, 125,6, 125,7, 126,3, 128,9, 129,1, 134,9, 135,7, 139,5, 140,4, 146,1, 152,7, 155,0, 174,7; MS (ESI) m/z 541,96 (M-1)-, 543,04 (M)-, 544,07 (M+H)-.
Пример 40: Получение соединения А-12
Используя соединение Е-12 в качестве исходного материала, получали 2-(3-метил-4-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1H-1,2,4-триазолил)-метокси)-фенилтио)-уксусную кислоту (А-12) согласно способу синтеза, аналогичному способу, описанному в Примере 29. Полученное соединение представляло собой белое твердое вещество с выходом 47,9%.
Структуру полученного соединения описывали на основе следующих данных, полученных с помощью спектроскопии ядерного магнитного резонанса и масс-спектроскопии:
1Н ЯМР (400 МГц, СDCl3) δ 2,19 (s, 3Н), 3,43 (s, 3Н), 3,55 (s, 2Н), 5,03 (s, 2Н), 6,91 (d, J=8,77 Гц, 3Н), 7,29-7,32 (m, 2Н), 7,67 (d, J=8,66 Гц, 2Н), 8,12 (d, J=8,66 Гц, 2Н); 13С ЯМР (100 МГц, CDCl3) δ 16,1, 28,1, 38,1, 61,4, 111,8, 118,3, 126,2, 126,3, 127,2, 127,5, 127,9, 130,8, 134,8, 140,3, 143,6, 152,5, 155,4, 174,8; MS (ESI) m/z 452,23 (M-1)-, 453,34 (M)-, 454,35 (M+H)-.
Пример 41: Получение соединения А-13
Используя соединение Е-13 в качестве исходного материала, получали 2-метил-2-(2-метил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1H-1,2,4-триазолил))-бензилтио)-фенокси)-уксусную кислоту (А-13) согласно способу синтеза, аналогичному способу, описанному в Примере 29. Полученное соединение представляло собой белое твердое вещество с выходом 76,9%.
Структуру полученного соединения описывали на основе следующих данных, полученных с помощью спектроскопии ядерного магнитного резонанса и масс-спектроскопии:
1H ЯМР (400 МГц, CDCl3) δ 1,55 (d, J=6,56 Гц, 3Н), 2,12 (s, 3H), 3,06 (s, 3H), 4,62 (q, J=6,56 Гц, 1H), 5,17 (s, 1H), 6,56 (d, J=9,04 Гц, 1Н), 7,14-7,17 (m, 2H), 7,31-7,38 (m, 5H), 7,65 (d, J=8,68 Гц, 2H), 8,09 (d, J=8,68 Гц, 2H); MS (ESI) m/z 542,24 (M-1)-, 543,26 (M)-, 544,24 (M+H)-.
Пример 42: Получение соединения А-14
Используя соединение Е-14 в качестве исходного материала, получали 2-(2-этил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1Н-1,2,4-триазолил))-бензилтио)-фенокси)-уксусную кислоту (А-14) согласно способу синтеза, аналогичному способу, описанному в Примере 29. Полученное соединение представляло собой белое твердое вещество с выходом 63,1%.
Структуру полученного соединения описывали на основе следующих данных, полученных с помощью спектроскопии ядерного магнитного резонанса и масс-спектроскопии:
1H ЯМР (400 МГц, CDCl3) δ 1,07 (t, J=7,56 Гц, 3Н), 2,56 (q, J=7,56 Гц, 2H), 3,09 (s, 3Н), 4,59 (s, 2H), 5,18 (s, 1H), 6,56 (d, J=9,04 Гц, 1H), 7,14-7,17 (m, 2H), 7,31-7,38 (m, 5H), 7,65 (d, J=8,68 Гц, 2H), 8,09 (d, J=8,68 Гц, 2H); 13С ЯМР (100 МГц, CDCl3) δ 13,7, 22,9, 28,0, 50,4, 65,0, 111,6, 118,3, 123,6, 125,4, 126,1, 126,2, 127,3, 127,5, 128,3, 128,5, 128,9, 133,9, 134,2, 135,1, 136,3, 140,4, 146,6, 152,5, 156,4, 172,7; MS (ESI) m/z 544,14 (M+H)+, 561,93(M+NH4)+.
Пример 43: Получение соединения А-15
Используя соединение Е-15 в качестве исходного материала, получали 2,2-диметил-2-(3-метил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1Н-1,2,4-триазолил))-бензилокси)-фенилтио)-уксусную кислоту (А-15) согласно способу синтеза, аналогичному способу, описанному в Примере 29. Полученное соединение представляло собой белое твердое вещество с выходом 70,6%.
Структуру полученного соединения описывали на основе следующих данных, полученных с помощью спектроскопии ядерного магнитного резонанса и масс-спектроскопии:
1H ЯМР (400 МГц, СDCl3) δ 1,46 (s, 6Н), 2,35 (s, 3Н), 3,18 (s, 3H), 6,40 (s, 1H), 6,96 (d, J=8,56 Гц, 1H), 7,26-7,27 (m, 1H),7,35-7,50 (m, 6Н), 7,67 (d, J=8,72 Гц, 2Н), 8,15 (d, J=8,64 Гц, 2Н); 13С ЯМР (100 МГц, СDCl3) δ 16,4, 25,4, 28,2, 50,7, 74,5, 112,6, 118,2, 123,7, 125,7, 126,2, 127,4, 128,1, 128,9, 129,1, 134,7, 136,1, 139,8, 140,4, 145,9, 152,6, 156,4, 179,1; MS (ESI) m/z 556,69 (M)+, 558,04 (M+1)+.
Пример 44: Получение соединения А-16
Используя соединение Е-16 в качестве исходного материала, получали 2,2-диметил-2-(2-метил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1H-1,2,4-триазолил))-бензилтио)-фенокси)-уксусную кислоту (А-16) согласно способу синтеза, аналогичному способу, описанному в Примере 29. Полученное соединение представляло собой белое твердое вещество с выходом 52,6%.
Структуру полученного соединения описывали на основе следующих данных, полученных с помощью спектроскопии ядерного магнитного резонанса и масс-спектроскопии:
1Н ЯМР (400 МГц, СDCl3) δ 1,57 (d, J=5,25 Гц, 6Н), 2,12 (s, 3Н), 3,10 (s, 3H), 5,21 (s, 2Н), 6,63 (d, J=8,47 Гц, 1Н),7,05 (dd, J=8,47 Гц, 2,06 Гц, 1H), 7,19 (d, J=1,99 Гц, 1H), 7,25-7,37 (m, 5H), 7,65 (d, J=8,76 Гц, 2Н), 8,09 (d, J=8,62 Гц, 2Н); 13C ЯМР (100 МГц, СDCl3) δ 16,6, 25,1, 25,3, 28,1, 50,3, 79,4, 117,3, 118,3, 124,1, 126,2, 126,3, 128,2, 128,5, 128,9, 130,8, 133,2, 134,9, 137,8, 140,4, 146,6, 152,6, 154,5, 177,4; MS (ESI) m/z 558,06 (M)+
Пример 45: Получение соединения А-17
Используя соединение Е-17 в качестве исходного материала, получали 2-(3-метил-4-(1-(3-(4-n-бутил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1H-1,2,4-триазолил))-бензилокси)-фенилтио)-уксусную кислоту (А-17) согласно способу синтеза, аналогичному способу, описанному в Примере 29. Полученное соединение представляло собой белое твердое вещество с выходом 62%.
Структуру полученного соединения описывали на основе следующих данных, полученных с помощью спектроскопии ядерного магнитного резонанса и масс-спектроскопии:
1H ЯМР (400 МГц, CDCl3) δ 0,75 (t, J=7,29 Гц, 3Н), 1,03-1,05 (m, 1H), 1,11-1,18(m, 2Н),1,19-1,26 (m, 1H), 1,40-1,50 (m, 1H), 2,36 (s, 3Н), 3,47-3,53 (m, 1H), 3,56 (s, 2H), 3,62-3,69 (m, 1H), 6,38 (s, 1H), 6,97 (d, J=8,58 Гц, 1H), 7,24 (dd, J=8,47 Гц, 1,92 Гц, 1H), 7,33 (d, J=1,99 Гц, 1H), 7,39-7,51 (m, 5H), 7,68 (d, J=8,74 Гц, 2H), 8,17 (d, J=8,61 Гц, 2H); 13C ЯМР (100 МГц, СDCl3) δ 13,5, 16,6, 19,9, 37,8, 42,4, 74,7, 113,2, 118,2, 125,6, 126,2, 126,3, 128,2, 128,9, 129,1, 130,9, 134,8, 135,1, 140,4, 145,9, 152,6, 155,3, 174,4; MS (ESI) m/z 569,93 (M-2)-, 570,9 (M-1)-.
Пример 46: Получение соединения А-18
Используя соединение Е-18 в качестве исходного материала, получали 2,2-диметил-2-(2-метил-4-(1-(3-(4-n-бутил-5-оксо-1 -(4-трифторметилфенил)-4,5-дигидро-1Н-1,2,4-триазолил))-бензилтио)-фенокси)-уксусную кислоту (А-18) согласно способу синтеза, аналогичному способу, описанному в Примере 29. Полученное соединение представляло собой белое твердое вещество с выходом 61,3%.
Структуру полученного соединения описывали на основе следующих данных, полученных с помощью спектроскопии ядерного магнитного резонанса и масс-спектроскопии:
1H ЯМР (400 МГц, CDCl3) δ 0,79 (t, J=7,26 Гц, 3Н), 1,17-1,32(m, 6H), 1,59 (s, 6H), 2,13 (s, 3Н), 3,48-3,53 (m, 2H), 5,13 (s, 1H), 6,63 (d, J=8,44 Гц, 1H), 7,05 (dd, J=8,47 Гц, 2,04 Гц, 1H), 7,17 (d, J=2,08 Гц, 1H), 7,31-7,35 (m, 5H), 7,67 (d, J=8,84 Гц, 2H), 8,14 (d, J=8,60 Гц, 2H); 13C ЯМР (100 МГц, CDCl3) δ 13,5, 16,6, 19,8, 25,2, 25,3, 29,7, 30,4, 41,8, 50,1, 79,4, 117,3, 118,3, 124,4, 126,1, 128,4, 128,5, 128,8, 130,7, 133,4, 135,7, 137,9, 140,5, 146,5, 152,4, 154,5, 177,9; MS (ESI) m/z 597,96 (M-2)-, 599,0 (M-1)-, 600,0 (M)-.
Пример 47: Получение соединения А-19
Используя соединение Е-19 в качестве исходного материала, получали 2-(2,5-диметил-4-(1-(3-(4-n-бутил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1H-1,2,4-триазолил))-бензилтио)-фенокси)-уксусную кислоту (А-19) согласно способу синтеза, аналогичному способу, описанному в Примере 29. Полученное соединение представляло собой белое твердое вещество с выходом 31,4%.
Структуру полученного соединения описывали на основе следующих данных, полученных с помощью спектроскопии ядерного магнитного резонанса и масс-спектроскопии:
1H ЯМР (400 МГц, CDCl3) δ 0,79 (t, J=7,27 Гц, 3Н), 1,13-1,23 (m, 2H), 1,28-1,36 (m, 2H), 2,12 (s, 3Н), 2,25 (s, 3Н), 3,47-3,55 (m, 2H), 4,66 (s, 2H), 5,03 (s, 1H), 6,53 (s, 1H), 7,15 (s, 1H), 7,29-7,36 (m, 5H), 7,68 (d, J=8,66 Гц, 2H), 8,16 (d, J=8,56 Гц, 2H); 13С ЯМР (100 МГц, CDCl3) δ 13,5, 15,5, 19,8, 20,8, 30,4, 41,9, 49,6, 64,9, 112,9, 118,3, 123,1, 125,7, 126,2, 128,3, 128,5, 128,8, 135,8, 139,1, 140,5, 141,7, 146,5, 152,4, 156,6, 173,2; MS (ESI) m/z 584,03 (M-1)-, 584,98 (M)-.
| название | год | авторы | номер документа |
|---|---|---|---|
| Производные тетрагидротриазолопиримидина в качестве ингибиторов нейтрофильной эластазы человека | 2012 |
|
RU2622643C2 |
| БИЦИКЛИЧЕСКИЕ ЛАКТАМЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2827714C1 |
| БЕНЗОКСАЗЕПИНОВЫЕ ИНГИБИТОРЫ PI3 И СПОСОБЫ ПРИМЕНЕНИЯ | 2010 |
|
RU2654068C1 |
| БЕНЗОКСЕПИНОВЫЕ ИНГИБИТОРЫ PI3 И СПОСОБЫ ПРИМЕНЕНИЯ | 2010 |
|
RU2557658C2 |
| БЕНЗОКСАЗЕПИНОВЫЕ ИНГИБИТОРЫ PI3K И СПОСОБЫ ПРИМЕНЕНИЯ | 2010 |
|
RU2600927C2 |
| Соединения формул (I) и (A), фармацевтическая композиция, лекарственное средство, применение и способ получения соединения формулы (I) | 2018 |
|
RU2822758C2 |
| БИЦИКЛИЧЕСКИЕ КЕТОНЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2018 |
|
RU2797922C2 |
| ГЕТЕРОАРИЛЬНЫЕ СОЕДИНЕНИЯ, СОДЕРЖАЩИЕ ИХ КОМПОЗИЦИИ И СПОСОБЫ ЛЕЧЕНИЯ С ПРИМЕНЕНИЕМ ЭТИХ СОЕДИНЕНИЙ | 2007 |
|
RU2478635C2 |
| ДИФЕНИЛЬНЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ ЛЕЧЕНИЯ | 1997 |
|
RU2175319C2 |
| ЗАМЕЩЕННЫЕ СОЕДИНЕНИЯ ПИРИДИН АЗОЛОПИРИМИДИН-5-(6Н)-ОНА | 2013 |
|
RU2653054C2 |
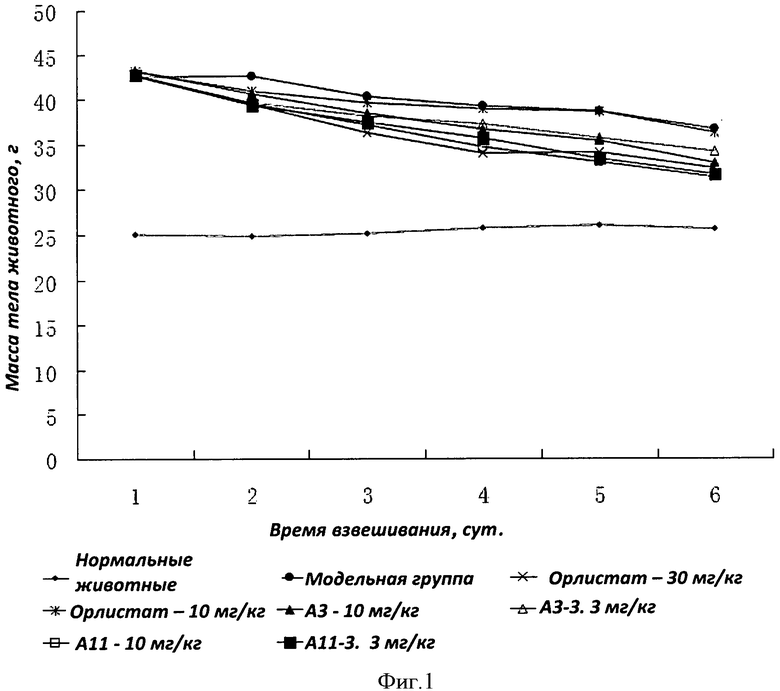
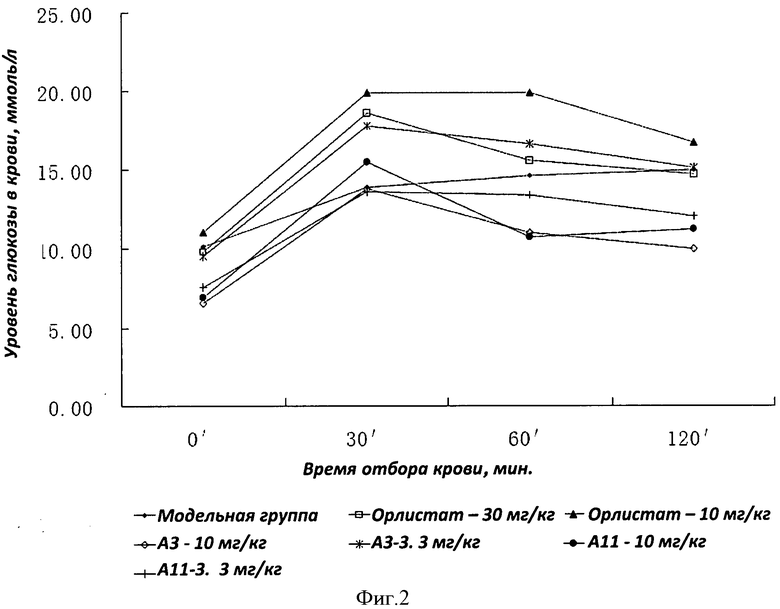

Описывается новое соединение формулы I
 ,
,
где X и Y означают О или S
R, R3, G1- G3 означают Н или С1-9алкил;
R1 и R2 - Н или С1-4алкил;
R4 - Н или фенил;
G4- G6 и G8- G9 - Н;
G7 - трифторметил, фармацевтическая композиция, его содержащая, способы получения, исходное соединение в описываемых способах и способ получения исходного соединения и применение соединения формулы I для лечения или профилактики заболевания путем активации рецептора δ(РPARδ); указанное заболевание представляет собой метаболический синдром, ожирение, дислипидемию, патологическую гликемию, резистентность к инсулину, сенильную деменцию и опухоли.
9 н. и 11 з.п. ф-лы, 47 пр., 3 ил., 5 табл.

1. Соединение формулы (I) или фармацевтически приемлемые соли указанного соединения

где
X представляет собой O или S;
Y представляет собой O или S;
R представляет собой H или C1-C9 алкил;
R1 и R2 независимо друг от друга представляют собой H или C1-C4 алкил;
R3 представляет собой Н или C1-C9 алкил;
R4 представляет собой Н или фенил;
G1, G2 и G3 независимо друг от друга представляют собой H или C1-C9 алкил;
G4, G5, G6, G8 и G9 представляют собой Н; и
G7 представляет собой трифторметил.
2. Соединение по п.1, отличающееся тем, что R представляет собой H, метил или этил.
3. Соединение по п.1, отличающееся тем, что R1 представляет собой H, метил или этил.
4. Соединение по п.1, отличающееся тем, что R2 представляет собой H, метил или этил.
5. Соединение по п.1, отличающееся тем, что R3 представляет собой H или C1-C4 алкил.
6. Соединение по п.1 или 5, отличающееся тем, что R3 представляет собой метил.
7. Соединение по п.1, отличающееся тем, что G1, G2 и G3 представляют собой H.
8. Соединение по п.1, выбранное из следующих соединений:
Этил-2-(2-метил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1Н-1,2,4-триазолил))-бензилокси)-фенокси)-ацетат;
Этил-2-(2-метил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1Н-1,2,4-триазолил))-бензилокси)-фенилтио)-ацетат;
Этил-2-(3-метил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1Н-1,2,4-триазолил))-бензилокси)-фенилтио)-ацетат;
Этил-2-(2-метил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1Н-1,2,4-триазолил))-бензилтио)-фенокси)-ацетат;
Этил-2-(2-этил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1Н-1,2,4-триазолил))-бензилокси)-фенокси)-ацетат;
Этил-2-(3-метил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1Н-1,2,4-триазолил))-бензилокси)-фенокси)-ацетат);
Этил-2-(3-метил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1Н-1,2,4-триазолил))-бензилтио)-фенокси)-ацетат;
Этил-2-(2-метил-4-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1H-1,2,4-триазолил)-метилтио)-фенокси)-ацетат;
Этил-2-(2-метил-4-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1H-1,2,4-триазолил)-метокси)-фенокси)-ацетат;
Этил-2-(2,5-диметил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1H-1,2,4-триазолил))-бензилтио)-фенокси)-ацетат;
Этил-2-(2,5-диметил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1H-1,2,4-триазолил))-бензилокси)-фенилтио)-ацетат;
Этил-2-(3-метил-4-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1H-1,2,4-триазолил)-метокси)-фенилтио)-ацетат;
Метил-2-метил-2-(2-метил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1Н-1,2,4-триазолил))-бензилтио)-фенокси)-ацетат;
Этил-2-(2-этил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1Н-1,2,4-триазолил))-бензилтио)-фенокси)-ацетат;
Этил-2,2-диметил-2-(3-метил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1H-1,2,4-триазолил))-бензилокси)-фенилтио)-ацетат;
Этил-2,2-диметил-2-(2-метил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1H-1,2,4-триазолил))-бензилтио)-фенокси)-ацетат;
Этил-2-(3-метил-4-(1-(3-(4-n-бутил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1H-1,2,4-триазолил))-бензилокси)-фенилтио)-ацетат;
Этил-2,2-диметил-2-(2-метил-4-(1-(3-(4-n-бутил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1Н-1,2,4-триазолил))-бензилтио)-фенокси)-ацетат;
Этил-2-(2,5-диметил-4-(1-(3-(4-n-бутил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1H-1,2,4-триазолил))-бензилтио)-фенокси)-ацетат;
2-(2-метил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1H-1,2,4-триазолил))-бензилокси)-фенокси)-уксусная кислота;
2-(2-метил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1Н-1,2,4-триазолил))-бензилокси)-фенилтио)-уксусная кислота;
2-(2-этил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1H-1,2,4-триазолил))-бензилокси)-фенокси)-уксусная кислота;
2-(2-метил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1H-1,2,4-триазолил))-бензилтио)-фенокси)-уксусная кислота;
2-(3-метил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1Н-1,2,4-триазолил))-бензилокси)-фенилтио)-уксусная кислота;
2-(3-метил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1H-1,2,4-триазолил))-бензилокси)-фенокси)-уксусная кислота;
2-(3-метил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1H-1,2,4-триазолил))-бензилтио)-фенокси)-уксусная кислота;
2-(2-метил-4-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1H-1,2,4-триазолил)-метилтио)-фенокси)-уксусная кислота;
2-(2-метил-4-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1H-1,2,4-триазолил)-метокси)-фенокси)-уксусная кислота;
2-(2,5-диметил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1H-1,2,4-триазолил))-бензилтио)-фенокси)-уксусная кислота;
2-(2,5-диметил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1H-1,2,4-триазолил))-бензилокси)-фенилтио)-уксусная кислота;
2-(3-метил-4-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1H-1,2,4-триазолил)-метокси)-фенилтио)-уксусная кислота;
2-метил-2-(2-метил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1H-1,2,4-триазолил))-бензилтио)-фенокси)-уксусная кислота;
2-(2-этил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1Н-1,2,4-триазолил))-бензилтио)-фенокси)-уксусная кислота;
2,2-диметил-2-(3-метил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1H-1,2,4-триазолил))-бензилокси)-фенилтио)-уксусная кислота;
2,2-диметил-2-(2-метил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1Н-1,2,4-триазолил))-бензилтио)-фенокси)-уксусная кислота;
2-(3-метил-4-(1-(3-(4-п-бутил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1Н-1,2,4-триазолил))-бензилокси)-фенилтио)-уксусная кислота;
2,2-диметил-2-(2-метил-4-(1-(3-(4-n-бутил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1Н-1,2,4-триазолил))-бензилтио)-фенокси)-уксусная кислота;
2-(2,5-диметил-4-(1-(3-(4-n-бутил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1Н-1,2,4-триазолил))-бензилтио)-фенокси)-уксусная кислота.
9. Соединение по п.1, выбранное из следующих соединений:
2-(2-метил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1H-1,2,4-триазолил))-бензилтио)-фенокси)-уксусная кислота;
2-(3-метил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1H-1,2,4-триазолил))-бензилокси-фенилтио)-уксусная кислота;
2-(2,5-диметил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1Н-1,2,4-триазолил))-бензилокси)-фенилтио)-уксусная кислота;
2-(2-этил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1Н-1,2,4-триазолил))-бензилтио)-фенокси)-уксусная кислота;
2,2-диметил-2-(3-метил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1H-1,2,4-триазолил))-бензилокси)-фенилтио)-уксусная кислота;
2,2-диметил-2-(2-метил-4-(1-(3-(4-метил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1Н-1,2,4-триазолил))-бензилтио)-фенокси)-уксусная кислота;
2-(3-метил-4-(1-(3-(4-n-бутил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1Н-1,2,4-триазолил))-бензилокси)-фенилтио)-уксусная кислота;
2,2-диметил-2-(2-метил-4-(1-(3-(4-n-бутил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1Н-1,2,4-триазолил))-бензилтио)-фенокси)-уксусная кислота;
2-(2,5-диметил-4-(1-(3-(4-n-бутил-5-оксо-1-(4-трифторметилфенил)-4,5-дигидро-1H-1,2,4-триазолил))-бензилтио)-фенокси)-уксусная кислота.
10. Фармацевтическая композиция для активации рецептора δ, активируемого пролифератором пероксисом (PPARδ), содержащая соединение по любому из пп. 1-9 и подходящие вспомогательные вещества.
11. Фармацевтическая композиция по п.10, в лекарственной форме, выбранной из группы, включающей обычные таблетки, таблетки, покрытые пленочной оболочкой, драже, таблетки с энтеросолюбильным покрытием, диспергируемые таблетки, капсулы, гранулы, растворы для перорального введения и суспензии для перорального введения.
12. Применение соединения по любому из пп. 1-9 для приготовления лекарственного средства для лечения или профилактики заболевания, которое можно лечить или предотвратить путем активации рецептора δ, активируемого пролифератором пероксисом (PPARδ).
13. Применение по п.12, отличающееся тем, что указанное заболевание выбрано из одного или более из ожирения, дислипидемии и патологической гликемии.
14. Способ получения соединения общей формулы I по п.1, где R представляет собой C1-C9 алкил, а другие радикалы имеют указанные в п.1 значения, включающий взаимодействие соединения общей формулы II, где X, Y, R, R1, R2, G1-G4 имеют указанные в п.1 значения, с соединением общей формулы III, где R3-R4 и G5-G9 имеют указанные в п.1 значения, Z представляет собой Cl или Br,

под действием неорганического основания, такого как карбонат щелочного металла, в среде растворителя, представляющего собой ацетонитрил при комнатной температуре в течение 1-12 часов.
15. Способ получения соединения общей формулы I по п.1, где R представляет собой C1-C9 алкил, а другие радикалы имеют указанные в п.1 значения, включающий в ходе непрерывного реакционного процесса последовательное взаимодействие соединения общей формулы III, где R3, R4, G5-G9 имеют указанные в п.1 значения, сначала с соединением общей формулы IV, где X, Y, G1-G4 имеют указанные в п.1 значения, а затем с соединением общей формулы V, где R, R1, R2 имеют указанные в п.1 значения, a Z представляет собой Cl или Br, в присутствии карбоната в качестве основания, выбранного из карбоната калия или карбоната натрия, в среде ацетонитрила, без необходимости разделения интермедиатов в ходе реакции

16. Способ получения соединения общей формулы I по п.1, где R представляет собой Н, а другие радикалы имеют указанные в п.1 значения, включающий осуществление щелочного гидролиза соединения общей формулы I по п.1, где R представляет собой C1-C9 алкил, в присутствии основания, представляющего собой гидроксид щелочного металла, включая гидроксид натрия, гидроксид лития, гидроксид калия, в среде растворителя, который представляет собой систему C1-C4 спирт - дихлорметан - вода с соотношением в диапазоне 9-1:9-1:1 (об./об.), причем процесс осуществляют при комнатной температуре, в течение 1-12 часов, предпочтительно в течение 2-4 часа.
17. Соединение формулы III:
 где
где
Z представляет собой Cl или Br;
R3 представляет собой Н или C1-C9 алкил;
R4 представляет собой Н или фенил;
G5, G6, G8 и G9 представляют собой Н; и
G7 представляет собой трифторметил.
18. Соединение по п.17, выбранное из следующих соединений:
3-(1'-бромбензил)-4-метил-1-(4-трифторметил)-фенил-1Н-1,2,4-триазол-5(4Н)-она;
3-(1'-бромбензил)-4-n-бутил-1-(4-трифторметил)-фенил-1H-1,2,4-триазол-5(4Н)-она; и
3-бромметил-4-метил-1-(4-трифторметилфенил)-1,4-дигидро-1,2,4-триазол-5-она.
19. Способ получения соединения формулы III по п.17, включающий осуществление реакции соединения формулы (VI)

где
Z представляет собой Cl или Br;
R3 представляет собой Н или C1-C9 алкил;
R4 представляет собой Н или фенил;
G5, G6, G8 и G9 представляют собой H;
G7 представляет собой трифторметил;
с хлорирующим или бромирующим реагентом, выбранным из группы, включающей:
N-бромсукцинимид/трифенилфосфин и N-хлорсукцинимид/трифенилфосфин;
в растворителе, представляющем собой дихлорметан, при комнатной температуре в течение 2-8 часов.
20. Способ получения соединения формулы III по п.17, включающий осуществление реакции соединения формулы (VII)

где
Z представляет собой Cl или Br;
R3 представляет собой Н или C1-C9 алкил;
G5, G6, G8 и G9 представляют собой Н;
G7 представляет собой трифторметил; и
G10-G14 представляют собой Н;
с хлорирующим или бромирующим реагентом, таким как N-бромсукцинимид или N-хлорсукцинимид, в среде растворителя, представляющего собой хлороформ, в присутствии пероксида бензоила в качестве катализатора при температуре кипения реакционной смеcи в течение 2-8 часов.
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| RU 2005114491 A 10.02.2006 | |||

