Объект настоящего изобретения - новое производное урацила, которое является ингибитором уридинфосфрилазы. Данное изобретение также относится к способу получения названного соединения и к фармацевтической композиции на основе этого соединения, являющейся антиметаболитом урацила.
Уридинфосфорилаза (UPh, УФаза) - ключевой фермент метаболизма пиримидина, катализирующий обратимое фосфорилирование уридина с образованием урацила и рибозо-1-фосфата [1, 2]. Фермент идентифицирован в прокариотах, дрожжах и высших организмах [3]. Физиологическая активность уридинфосфорилазы присутствует у всех живых организмов от бактерий до человека, хотя структура гена и пространственная структура белковой молекулы УФазы варьируют в широком диапазоне [4, 5, 6, 7]. Белок экспрессируется в различных клетках и тканях человека и животных. Уридинфосфорилаза играет решающую роль в так называемом быстром клиренсе, то есть выведении из организма избытков плазменного уридина. [8] Была показана индукция уридинфосфорилазной активности для клеток карциномы толстой кишки [9, 10, 11], меланомы [3], аденокарциномы молочной железы [12], гепатомы и асцитной карциномы Эрлиха [13]. Авторы [9] обнаружили увеличение уридинфосфорилазной активности в 9-ти из 12-ти исследованных образцов карциномы толстой кишки. В трех образцах активность фермента не изменялась или несколько снижалась. Katsumata и др. [10] обнаружили положительную корреляцию между уридинфосфорилазной активностью и стадией процесса при раке толстой кишки.
Ингибирование уридинфосфорилазы приводит к гибели некоторых простейших - возбудителей заболеваний человека и животных (например, Giardia lamblia, Schistosoma mansoni) [14, 15, 16], т.к. у простейших уридинфосфорилаза участвует в метаболизме уридина и тимидина [16], и большая часть пиримидиновых оснований синтезируется путем ресинтеза.
М. Iigo и соавторы [17] показали, что ингибиторы уридинфосфорилазы способствуют увеличению активности противоопухолевого фармацевтического препарата 5-фторурацила (5ФУ) при химиотерапии индуцированного рака молочной железы у мышей линии BDF1 и трансплантатов плоскоклеточного рака толстой кишки человека у безтимусных мышей. В опытах на мышах линии CD8F1 установлено, что введение BAU (240 мг/кг) позволило уменьшить дозу 5ФУ в 1.5 раза при той же терапевтической эффективности [18]. Тем самым показана возможность изменения активности 5ФУ специфическими ингибиторами уридинфосфорилазы с целью снижения токсичности основного препарата. Данный эффект связан с уменьшением катаболизма 5ФУ под действием ингибитора уридинфосфорилазы.
Противоопухолевый препарат 5-фторурацил (5ФУ) является антиметаболитом урацила из-за конкуренции за тимидилатсинтетазу. В результате действия 5ФУ нарушается синтез нуклеиновых кислот и достигается остановка пролиферации и/или гибель опухолевых клеток. 5ФУ - важный компонент современной химиотерапии - успешно применяется при раке толстой кишки, молочной железы, органов пищеварения, мочеполовой системы, а также при опухолях кожи. Для проявления цитостатического действия 5ФУ должен быть превращен в один из активных метаболитов: 5-фторуридин-5'-трифосфат (FUTP), 5-фтор-2'-дезоксиуридин-5'-монофосфат (FdUMP) или 5-фтордезоксиуридин-5'-трифосфат (FdUTP) [19].
В настоящее время используемыми в клинической практике либо проходящими стадии доклинических или клинических испытаний ингибиторами уридинфосфорилаз являются также соединения - производные пиримидинов [20, 21, 22, 23]. В основном используются соединения, синтезированные на базе ациклоуридина либо, ангидроуридинов (2.2'- или 2.3'-). Данные ингибиторы были синтезированы без использования методов компьютерного рационального конструирования молекул биологически активных соединений и не образуют связей с анионом фосфата в фосфат-связывающем центре. Этилендиаминовая группа описываемого в патенте соединения образует водородные и ионные связи с анионом фосфата, тем самым улучшается аффинность соединения к уридинфосфорилазе. Следует отметить также низкую стоимость и простоту синтеза соединения, так как схема одностадийная, а все предшественники коммерчески доступны.
Настоящее изобретение относится к новому классу сильных ингибиторов уридинфосфорилаз.
Таким образом, объектом изобретения является соединение формулы
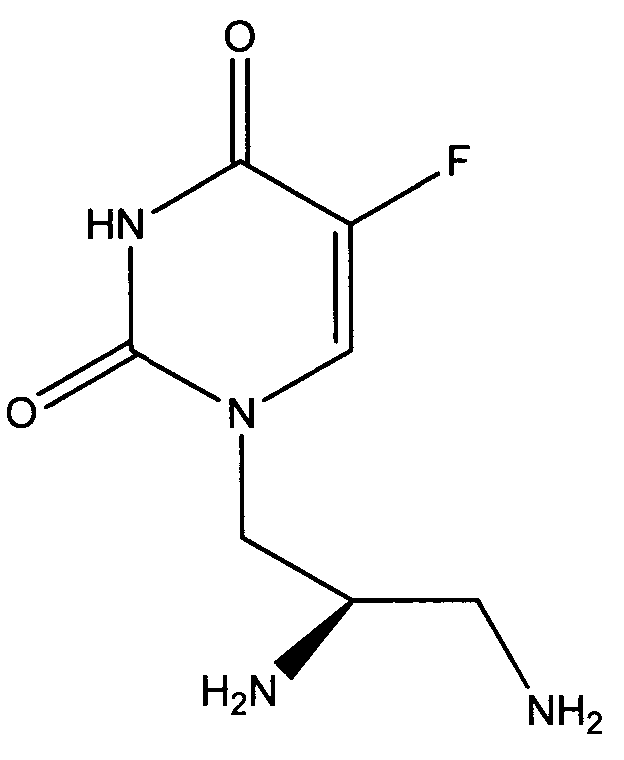
в R-энантиомерной форме, а также фармацевтически приемлемая соль соединения, например дигидрохлорид.
Способ лабораторного получения фармацевтически приемлемой соли (R)-1-(2,3-Диаминопропил)-5-фторпиримидин-2,4-диона
(R)-1-(2,3-Диаминопропил)-5-фторпиримидин-2,4-дион дигидрохлорид 3 может быть получен в соответствии со следующей схемой:
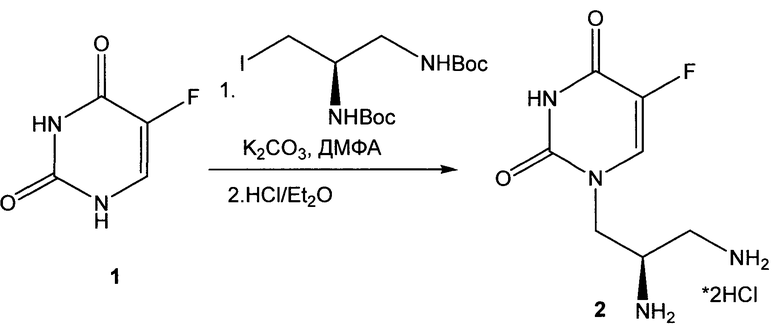
Пример 1
К раствору 1.3 г (10 ммоль) 5-фторурацила 1 в 20.0 мл ДМСО прибавляют 2.0 г (5 ммоль) (R)-трет-бутил 3-иодопропан-1,2-диилдикарбамат (полученного из (D)-2,3-диаминопропионовой кислоты по методу [Wu W., Li R., Malladi S.S., Warshakoon H.J., Kimbrell M.R., Amolins M.W., Ukani R., Datta A., David S.A. J. Med. Chem., 2010, vol.53 (8), p.3198]) и 2.8 г (0.02 моль) безводного карбоната калия и перемешивают смесь в токе аргона 2 ч при 40°C. Реакционную массу выливают в воду и при перемешивании осторожно нейтрализуют 10% раствором соляной кислоты, экстрагируют продукты этилацетатом, экстракт трижды промывают водой, сушат и упаривают в вакууме. Остаток очищают хроматографически ("Silica Gel Merck 60", хлороформ-метанол, 10:0→10:1) и кристаллизуют из 1,4-диоксана.
Выход ди(Вос)-производного 0.8 г (39%).
Т. пл.>206-8°C. Спектр ЯМР 1Н (ДМСО-d6, 400 МГц), δ, м.д.: 10.99 (1Н, уш с, NH); 7.77 (1Н, д, J=5.7 Гц, Н-6); 6.86 (1Н, т, J=6.1 Гц, NH); 6.43 (1Н, д, J=8.8 Гц, NH); 3.92-3.92 (2H, м, NCH2); 3.67-3.60 (1Н, м, СН); 3.07-2.97 (2H, м, CH2NH); 1.37 (9Н, с, t-Bu); 1.27 (9Н, с, t-Bu). Найдено: m/z (ESI), 403.1979 [M+H]+. C7H12FN4O2. Вычислено: 403.1987.
К раствору 0.7 г (1.7 ммоль) ди(Вос)-производного растворяют в теплой смеси 30.0 мл метанол-диоксан (1:1), прибавляют 30.0 мл раствора (2н) HCl в Et2O и перемешивают смесь 4 ч. Выпавший осадок отфильтровывают, промывают Et2O и сушат в вакууме.
Выход 0.4 г (84%), бесцветных кристаллов дигидрохлорида 2. Т. пл.>260°C (разл.). ВЭЖХ (LW=267 нм, элюент H2O-MeCN-HCO2NH4 (98-2-0.2)) tR=4.80 мин, чистота 98.8%. Спектр ЯМР 1Н (CD3OD, 400 МГц), δ, м.д.: 7.72 (1Н, д, J=5.13 Гц, Н-6); 4.36-4.34 (2H, м, NCH2); 4.99-4.93 (1Н, м, СН); 3.37-3.24 (2H, м, CH2NH3). Спектр ЯМР 13С (CD3OD, 100 МГц), δ, м.д.: 160.7 (C=O, J=26 Гц); 152.6 (C=O); 141.1 (CF, J=230 Гц); 126.5 (СН, J=33 Гц); 153.7 (C=O); 50.3 (NCH2); 41.5 (CH); 40.0 (CH2NH3). Найдено: m/z (ESI), 203.0921 [М+Н]+. C7H12FN4O2. Вычислено: 203.0939.
Для жидкостной хроматографии использовали системы Waters, включая линейный дегазатор, кватернарный носос Waters 600, аппликатор образцов на плашку Gilson 233 и ультрафиолетовый детектор Waters 996 PDA.
Для масс-спектрометрии использовался унифицированный квадрупольный масс-спектрометр (Micromass, Platform model), оснащенный источником электрораспыления, с разрешением 0,8 Да с 50% долиной. Калибровку осуществляли ежемесячно в области масс 80-1000 Да, используя калибровочную смесь йодида натрия и йодида рубидия в растворе смеси изопропанола/воды (1/1 об./об.).
Агрегатное состояние и цвет при стандартных условиях: бесцветные кристаллы.
Тпл.>200°C.
Применения
Соединения по настоящему изобретению могут иметь широкое терапевтическое применение. Они могут эффективно использоваться при лечении ряда онкологических заболеваний таких, как рак толстой и прямой кишки, рак молочной железы, пищевода, желудка, поджелудочной железы, первичный рак печени, рак яичников, рак шейки матки, мочевого пузыря, злокачественные опухоли головы и шеи, рак предстательной железы, рак надпочечников, рак вульвы, рак полового члена, карциноид, рак кожи (для мази).
Объектом данного изобретения также являются продукты указанной выше формулы, обладающее свойствами ингибитора уридинфосфорилаз в качестве активного компонента при изготовлении лекарственного средства, а также соли добавления фармацевтически приемлемых неорганических или органических кислот указанных продуктов формулы, а также фармацевтические композиции, содержащие в качестве активного ингредиента лекарственное средство, указанное выше, в сочетании с фармацевтически приемлемым носителем. Фармацевтические композиции, содержащие соединение по данному изобретению, должны находиться в жидкой форме в виде стерильного раствора для внутрисосудистого и внутриполостного введения. Подходящим жидким носителем может быть вода для инъекций, в качестве стабилизаторов могут выступать слабые растворы соляной кислоты и хлорида натрия.
Значения всех специальных и научных терминов, использованных в настоящем документе, хорошо понятны специалисту в данной области. Более того, все патенты (или патентные заявки), а также другие библиографические ссылки приведены в качестве ссылки.
Моделирование связывания соединения с УФазой из Salmonella typhimurium (StUPh) и Homo sapiens (hUPP1).
Для исследования механизма ингибирования уридинфосфорилаз был применен метод молекулярного докинга. Поиск оптимальных решений встраивания лиганда в сайт связывания белков-мишеней StUPh и hUPP1 проводился в соответствии с протоколом, отработанном на структуре комплекса StUPh с 5-FUra (ID PDB: 3NSR), решенного методом РСА. Молекулярный докинг проведен программой AutoDock 4.2 в варианте генетического алгоритма [24].
По результатам рентгеноструктурного анализа опорной структуры StUPh область урацилсвязывающего сайта может быть описана прямоугольным параллепипедом с длиной сторон 15.1 Ǻ × 16.4 Ǻ × 13.6 Ǻ. На основании этих данных область связывания энзимами лигандов была выбрана в форме куба с длиной ребра 22.5 Ǻ при ее 60-кратной дискретности.
Группа решений докинга объединялась в кластер, если значение r.m.s.d. координат атомов выявленных конформеров не превышало 1.5 Ǻ. После проведения молекулярного докинга проводилась оптимизация геометрии комплекса в программе Gromacs (силовое поле Gromos 96, модель воды SPC 216).
Предполагаемый механизм ингибирования уридинфосфорилаз
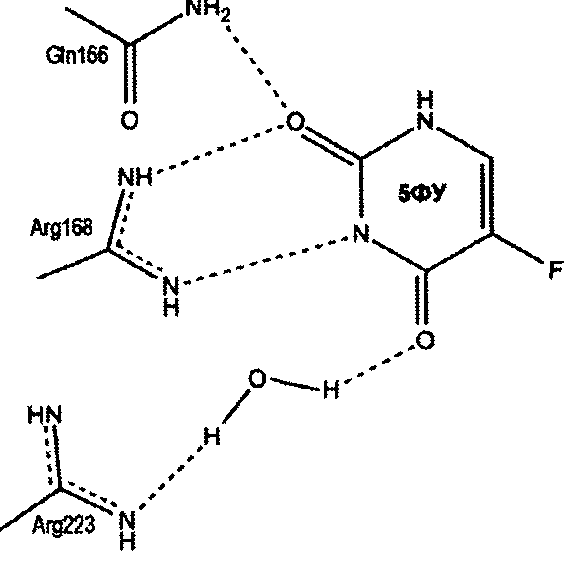
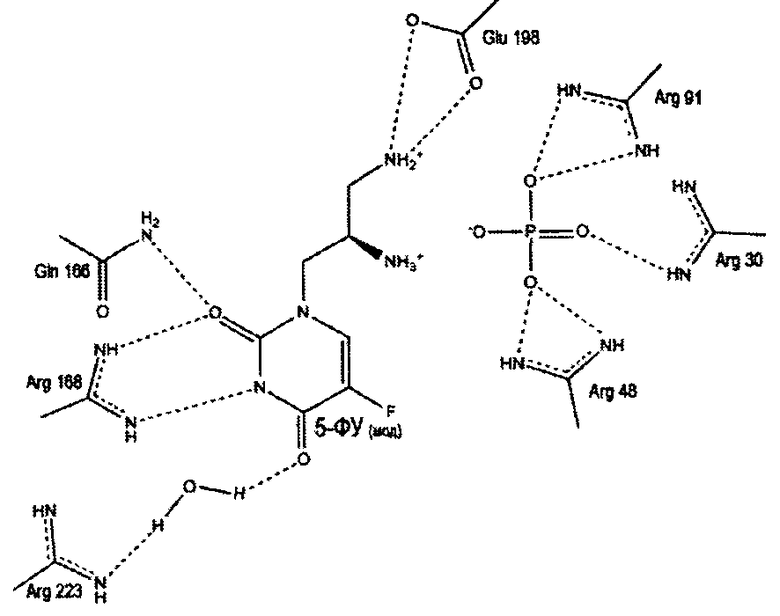
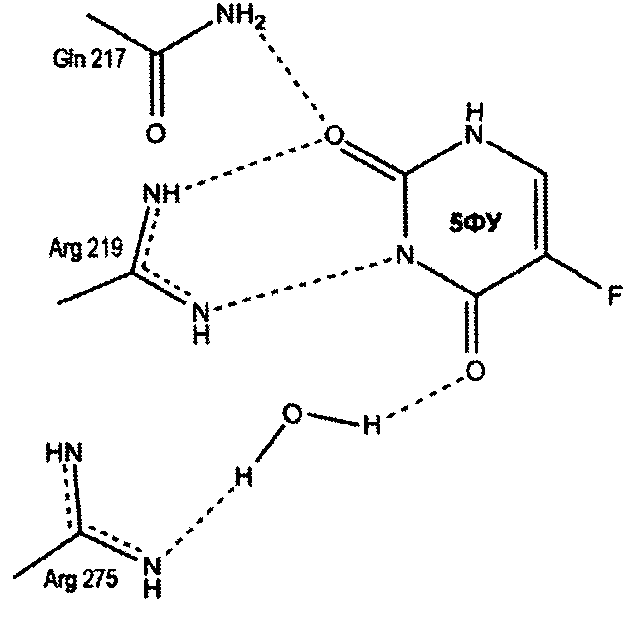
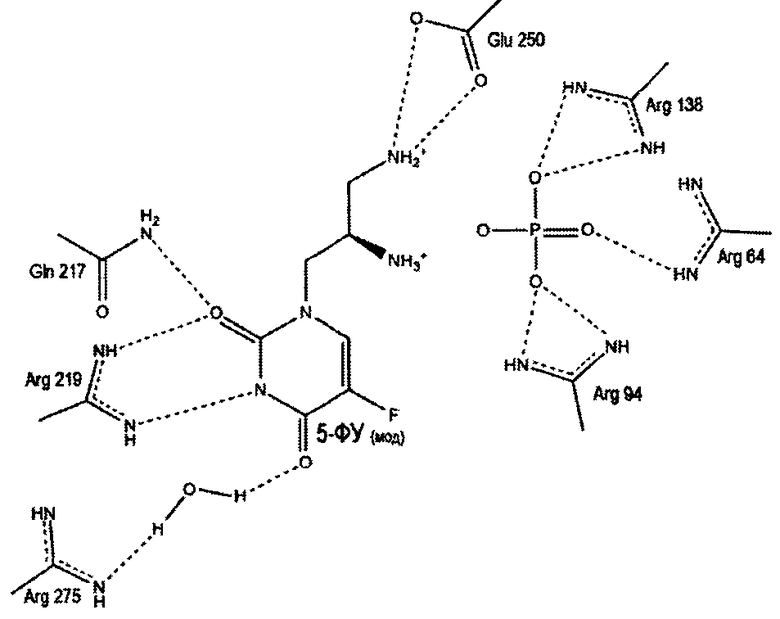
Пропилдиаминовая группа патентуемого соединения координируется фосфат-анионом, посредством ионных и водородных связей. Кроме того, дополнительная водородная связь образуется между атомом азота пропилдиаминовой группы и карбоксильным атомом кислорода боковой цепи глютаминовой кислоты (198 - StUPh, 250 - hUPP1). Тем самым достигается высокая аффинность патентуемого соединения к активному центру уридинфосфорилазы.
Источники информации
1. Leer J.C., Hammer-Jespersen К., Schwartz M. // Eur J Biochem. 1977. V.75. I.1. P.217-24.
2. Vita A., Amici A., Cacciamani T. et al. // Int J Biochem. 1986. V.18. I.5. P.431-5.
3. Leyva A., Kraal I., Lankelma J. et al. // Anticancer Res. 1983. V.3. I.4. P.227-31.
4. Liu M., Cao D., Russell R. et al. // Cancer Res. 1998. V.58. I.23. P.5418-24.
5. Takehara M., Ling F., Izawa S. et al. // Biosci Biotechnol Biochem. 1995. V.59. I.10. P.1987-90.
6. Walton L., Richards C.A., Elwell L.P. // Nucleic Acids Res. 1989. V.17. I.16. P.6741.
7. Watanabe S., Hino A., Wada K. et al. // J Biol Chem. 1995. V.270. I.20. P.12191-6.
8. Moyer J.D., Henderson J.F. // Biochem Pharmacol. 1985. V.34. I.1. P.101-5.
9. Finan P.J., Koklitis P.A., Chisholm E.M. et al. // Br J Cancer. 1984. V.50. I.5. P.711-5.
10. Katsumata K., Tomioka H., Sumi T. et al. // Cancer Chemother Pharmacol. 2003. V.51. I.2. P.155-60.
11. Luccioni С., Beaumatin J., Bardot V. et al. // Int J Cancer. 1994. V.58. I.4. P.517-22.
12. Kanzaki A., Takebayashi Y., Bando H. et al. // Int J Cancer. 2002. V.97. I.5. P.631-5.
13. Ishitsuka H., Miwa M., Takemoto K. et al. // Gann. 1980. V.71. I.1. P.112-23.
14. el Kouni M.H., Naguib F.N., Niedzwicki J.G. et al. // J Biol Chem. 1988. V.263. I.13. P.6081-6.
15. Jimenez B.M., Kranz P., Lee C.S. et al. // Biochem Pharmacol. 1989. V.38. I.21. P.3785-9.
16. Lee C.S., Jimenez B.M., O'Sullivan W.J. // Mol Biochem Parasitol. 1988. V.30. I.3. P.271-7.
17. Iigo M., Nishikata K., Nakajima Y. et al. // Biochem Pharmacol. 1990. V.39. I.7. P.1247-53.
18. Martin D.S., Stolfi R.L., Sawyer R.C. // Cancer Chemother Pharmacol. 1989. V.24. I.1. P.9-14.
19. Iigo M., Nishikata K., Hoshi A. // Jpn J Cancer Res. 1992. V.83. I.4. P.392-6.
20. Niedzwicki J.G., Chu S.H., el Kouni M.H. et al. // Biochem Pharmacol. 1982. V.31. I.10. P.1857-61.
21. Drabikowska A.K., Lissowska L., Draminski M. et al. // Z Naturforsch C. 1987. V.42. I.3. P.288-96.
22. Drabikowska A.K., Lissowska L., Veres Z.et al. // Biochem Pharmacol. 1987. V.36. I.23. P.4125-8.
23. Pizzorno G., Yee L., Burtness B.A. et al. // Clin Cancer Res. 1998. V.4. I.5. P.1165-75.
24. Morris G.M., Huey R., Lindstrom W. et al. // J Comput Chem. 2009. V.30. I.16. P.2785-91.
| название | год | авторы | номер документа |
|---|---|---|---|
| ФОСФОРОАМИДАТНЫЕ ПРОИЗВОДНЫЕ 5-ФТОР-2'-ДЕЗОКСИУРИДИНА ДЛЯ ПРИМЕНЕНИЯ ДЛЯ ЛЕЧЕНИЯ РАКА | 2012 |
|
RU2614406C2 |
| СПОСОБ СНИЖЕНИЯ ЖЕЛУДОЧНО-КИШЕЧНОЙ ТОКСИЧНОСТИ, ВОЗНИКАЮЩЕЙ В РЕЗУЛЬТАТЕ ПРИМЕНЕНИЯ ТЕГАФУРА | 2005 |
|
RU2348409C2 |
| ВВЕДЕНИЕ МНОЖЕСТВЕННЫХ БОЛЮСОВ [6R]-MTHF В ХИМИОТЕРАПИИ НА ОСНОВЕ 5-ФТОРУРАЦИЛА | 2018 |
|
RU2762596C2 |
| Ингаляционная форма на основе 3N-амино-6-метил-1Н-пиримидин-2,4-диона для лечения аденокарциномы легкого | 2024 |
|
RU2841172C1 |
| Мультифункциональные конъюгаты такрина и его аналогов с производными 1,2,4-тиадиазола, способ их синтеза и применение для лечения нейродегенеративных заболеваний | 2017 |
|
RU2675794C1 |
| [6R]-MTHF - ЭФФЕКТИВНАЯ ФОЛАТНАЯ АЛЬТЕРНАТИВА В ХИМИОТЕРАПИИ НА ОСНОВЕ 5-ФТОРУРАЦИЛА | 2018 |
|
RU2763934C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ НА ОСНОВЕ ГЕНОТИПА ИЛИ ФЕНОТИПА | 2013 |
|
RU2683260C2 |
| ТРИАЗОЛЫ, ИСПОЛЬЗУЕМЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕИНКИНАЗ | 2005 |
|
RU2393155C2 |
| ПРИМЕНЕНИЕ 3-О-СУЛЬФАМОИЛОКСИ-7β-МЕТИЛ-D-ГОМО-6-ОКСАЭСТРА-1,3,5(10),8(9)-ТЕТРАЕН-17а-ОНА ДЛЯ ЛЕЧЕНИЯ РАКА МОЛОЧНОЙ ЖЕЛЕЗЫ, ВКЛЮЧАЯ ТРИЖДЫ НЕГАТИВНУЮ ФОРМУ | 2018 |
|
RU2678845C1 |
| СИНТЕЗ ЛАКТОНОВ РЕЗОРЦИЛОВОЙ КИСЛОТЫ, ИСПОЛЬЗУЕМЫХ В КАЧЕСТВЕ ТЕРАПЕВТИЧЕСКИХ АГЕНТОВ | 2009 |
|
RU2534527C2 |
Изобретение относится к новому производному урацила, соответствующему нижеуказанной структурной формуле, и его фармацевтически приемлемой соли. Соединение обладает свойствами ингибиторов уридинфосфорилазы и может быть использовано в качестве активного компонента для получения лекарственного средства для лечения рака, такого как антиметаболит ингибитора уридинфосфорилазы клеток опухоли. Соединение имеет структурную формулу, соответствующую R-энантиомерной форме :
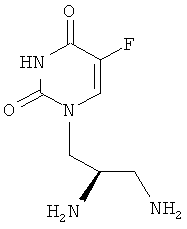 .
.
Данное изобретение также относится к способу получения названного соединения и к фармацевтической композиции на основе этого соединения. Способ получения заключается во взаимодействии 5-фторурацила с R-трет-бутил-3-иодопропан-1,2-диилдикарбаматом. 3 н. и 1 з.п. ф-лы, 1 табл., 1 пр.
1. Соединение формулы
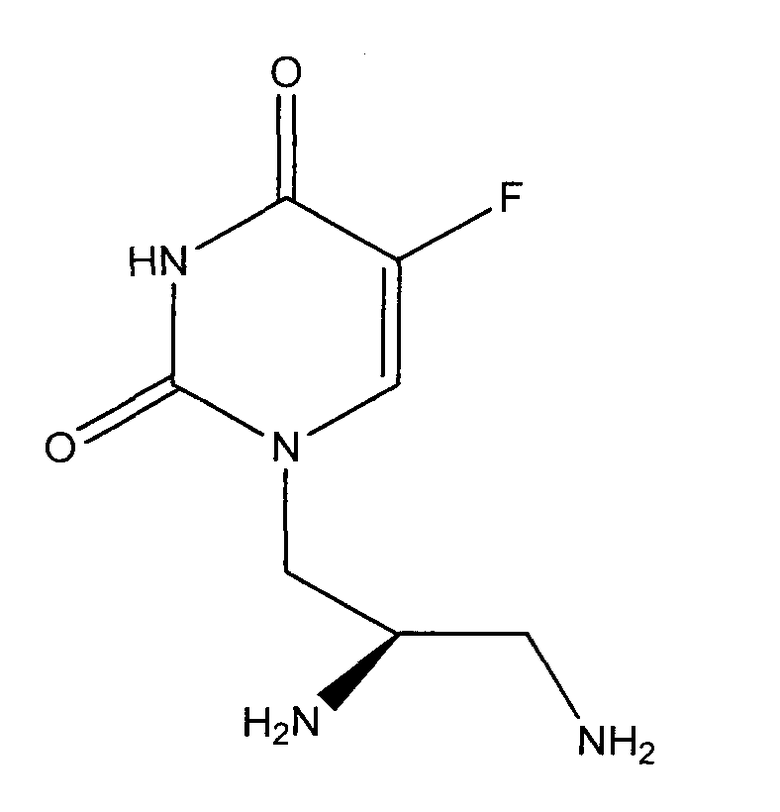
в R-энантиомерной форме, а также фармацевтически приемлемая соль соединения.
2. Соединение по п.1. обладающее свойствами ингибитора уридинфосфорилаз в качестве активного компонента при изготовлении лекарственного средства,пригодного для лечения рака, антиметаболита-ингибитора уридинфсфорилаз клеток опухоли.
3. Фармацевтическая композиция, обладающая свойствами ингибитора уридинфосфорилазы, пригодная для лечения рака, содержащая соединение по п.1 вместе с фармацевтически приемлемым разбавителем или носителем.
4. Способ получения соединения формулы (I),как определено в п.1, включающий взаимодействие 5-фторурацила с R-трет-бутил-3-иодопропан-1,2-диилдикарбаматом.
| СПОСОБ ПОЛУЧЕНИЯ ПРОТИВООПУХОЛЕВОГО ЭФФЕКТА У МЛЕКОПИТАЮЩИХ | 1992 |
|
RU2126255C1 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ ПРОТИВООПУХОЛЕВЫМ ДЕЙСТВИЕМ СО СНИЖЕННЫМИ ПОБОЧНЫМИ ЭФФЕКТАМИ, СОДЕРЖАЩАЯ ПРОТИВООПУХОЛЕВЫЙ АГЕНТ И ПРОИЗВОДНОЕ ГИДРОКСАМОВОЙ КИСЛОТЫ | 1998 |
|
RU2214238C2 |

