Настоящее изобретение относится к химическим соединениям, которые можно применять в лечении рака.
Противоопухолевая активность 5-фторурацила (5FU) была открыта в 1957 г. Уже прошло более пятидесяти лет с тех пор, как 5FU был впервые синтезирован, но до сих пор он продолжает широко использоваться, с одобрения FDA (Управления по контролю качества пищевых продуктов и лекарственных препаратов) в 1962 году, для лечения солидных опухолей, включая рак молочной железы, рак желудочно-кишечного тракта, злокачественное новообразование головы и шеи и рак яичника, в частности для лечения колоректального рака. Фторпиримидины 5-фторурацил (5FU) и 5-фтор-2'-дезоксиуридин (5-FdUrd) в комбинации с фолиевой кислотой используют в качестве стандартной терапии для лечения различных карцином, таких как карцинома желудка, толстой кишки и молочной железы. Кроме того, комбинация 5FU с лейковорином (leucovorin) (LV) считается стандартной химиотерапией рака толстой кишки. Лекарственные препараты 5FU обычно вводят путем внутривенной инъекции ударной дозы лекарства или путем непрерывной инфузии.
Противоопухолевая активность 5FU сравнима с противоопухолевой активностью его аналога 5-FdUrd, который отчасти действует как пролекарство 5FU. В 1970 5-FdUrd был одобрен FDA и с тех пор широко используется в клиническом лечении карцином яичника, молочной железы и желудочно-кишечного тракта. Кроме того, вследствие интенсивной деградации ферментами печени 5-FdUrd представляет собой лекарство, которое может быть использовано для химиотерапии метастазов печении путем введения через печеночную артерию, благодаря чему оно более эффективно метаболизируется в печени, чем 5FU.
Однако проблема заключается в том, что активность как 5FU, так и 5-FdUrd может быть ухудшена из-за развития устойчивости опухолевых клеток. Кроме того, было показано, что лечение рака с использованием 5FU вызывает нейротоксические и кардиотоксические побочные эффекты. Токсичность 5FU также обусловлена тем, что данное соединение не обладает опухолевой избирательностью.
Задача настоящего изобретения заключается в получении соединений, являющихся производными 5-фтор-2'-дезоксиуридина, которые обладают более высокой активностью и/или меньшей токсичностью при их использовании в лечении рака по сравнению с 5-фторацилом или 5-фтор-2'-дезоксиуридином per se.
Другая задача настоящего изобретения заключается в получении соединений, являющихся производными 5-фтор-2'-дезоксиуридина, которые характеризуются более низким уровнем устойчивости опухолевых клеток, в частности характеризуются более низким уровнем устойчивости опухолевых клеток по сравнению с 5FU или 5-FdUrd.
Согласно настоящему изобретению предложено соединение Формулы (I):
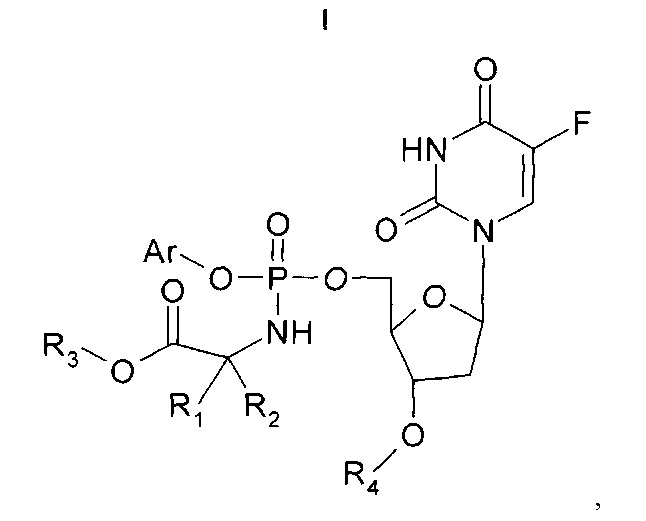
где
Ar представляет собой конденсированную бициклическую арильную группировку или моноциклическую арильную группировку, причем каждая из указанных арильных группировок является карбоциклической или гетероциклической и необязательно содержит заместители;
R3 представляет собой алкил, который необязательно содержит заместители;
R4 представляет собой атом H или алкоил; и
R1 и R2 независимо выбраны из группы, состоящей из атома H и алкила, или R1 и R2 вместе образуют алкиленовую цепь, которая вместе с атомом C, к которому присоединены R1 и R2, образует циклическую систему, или один из R1 и R2 содержит алкиленовую цепь, присоединенную к атому N, атом H, соединенный с атомом N, отсутствует и один из R1 и R2 содержит атом H или алкил, причем любая из указанных алкильных группировок или алкиленовых цепей может содержать заместители;
или фармацевтически приемлемое производное или метаболит соединения Формулы I,
при условии, что данное соединение не является соединением, содержащим, в комбинации, незамещенный фенил в качестве Ar, CH3 в качестве R3, H в качестве R4 и H в качестве одного из R1 и R2 и CH3 в качестве одного из R1 и R2.
Установлено, что соединения согласно настоящему изобретению обладают активностью, которая позволяет их использовать для профилактики или лечения рака у человека. В частности, соединения согласно настоящему изобретению обладают благоприятными свойствами, которые указывают на их способность лечить рак у пациентов, которые характеризуются более низким уровнем устойчивости опухолевых клеток. Примечательно, что соединения согласно настоящему изобретению могут обладать цитоактивностью, сравнимой с цитоактивностью 5-фторацила или более высокой, но при этом характеризуются устойчивостью, сравнимой с устойчивостью к 5-фторацилу и 5-фтор-2'-дезоксиуридину или более низкой.
ʺУстойчивостьʺ в контексте данной заявки означает низкий или сниженный уровень ответа на терапию. Устойчивость может быть врожденной или приобретенной. Врожденная устойчивость представляет собой пониженную ответную реакцию у данной особи или пациента по сравнению с другими особями или пациентами. Приобретенная устойчивость представляет собой снижение эффективности лекарства с течением времени в период проведения курса лечения у конкретного пациента не зависимо от того, наступило данное снижение эффективности в связи с терапией, включающей назначение пациенту схемы введения лекарства для лечения рака, например схемы введения лекарства, содержащего 5FU и/или 5-FdUrd, или вызвано другими причинами. Как врожденная устойчивость, так и приобретенная устойчивость могут соответствовать снижению уровня экспрессии или низкой активности белков-переносчиков, включая белки-переносчики нуклеозидов, или необходимых анаболических ферментов или повышению уровня экспрессии катаболических ферментов.
Вне связи с какой-либо теорией постулировано, что причинами устойчивости опухолевых клеток к действию 5FU и/или 5-FdUrd могут являться: а) деления активирующей киназы, например тимидинкиназы (TK), ключевого фермента, необходимого для начальной стадии фосфорилирования 5-FdUrd до 5-FdUMP, б) сверхсинтез тимидилатсинтазы (TS) и/или в) нарушение транспорта в клетки-мишени (см. более подробное обсуждение ниже).
Неожиданно было найдено, что соединения согласно настоящему изобретению могут обладать значительной цитостатической активностью в клетках с пониженным уровнем белков-переносчиков нуклеозидов, и/или в клетках, дефицитных по нуклеозидкиназе, и/или в клетках, инфицированных микоплазмой.
Благоприятное свойство соединений согласно настоящему изобретению сохранять значительную цитостатическую активность в клетках, дефицитных по нуклеозидкиназе, может давать in vivo клиническое преимущество в клеточном окружении, которое характеризуется отсутствием нуклеозидкиназ или снижением уровня нуклеозидкиназ и, соответственно, не способно эффективно активировать 5-FdUrd.
В клетках, инфицированных микоплазмой, активность нуклеозидов, таких как 5-FdUrd, значительно снижена, как считается, вследствие сверхсинтеза тимидилатсинтазы (TS). Таким образом, предложенное в настоящем изобретение применение соединений согласно настоящему изобретению в клетках, инфицированных микоплазмой, предположительно, обусловлено дополнительным благоприятным свойством соединений согласно настоящему изобретению, а именно способностью действовать в качестве TS-ингибитора, что позволяет соединениям согласно настоящему изобретению сохранять цитостатическую активность в клетках, инфицированных микоплазмой. Пролекарства, содержащие соединения согласно настоящему изобретению, вследствие их липофильной природы могут поглощаться клетками-мишенями, по меньшей мере частично, без использования переносчиков транспорта нуклеозидов, и, соответственно, могут обходить потенциальные механизмы устойчивости, обусловленные пониженным уровнем переносчиков транспорта нуклеозидов или нуклеооснований в мембране клеток-мишеней.
Кроме того, неожиданно найдено, что пролекарства, содержащие соединения согласно настоящему изобретению, являются нечувствительными к действию катаболического фермента тимидинфосфорилазы (TP), уровень экспрессии которой часто повышен в опухолевых клетках, и, соответственно, указанные пролекарства могли бы быть менее зависимы от присутствия данного катаболического фермента, чем 5-FdUrd.
Согласно наблюдениям инфицирование клеток микоплазмой может значительно уменьшить активность нуклеозидов, включая 5-FdUrd. Введение TP-ингибитора восстанавливает цитостатическую активность 5-FdUrd в клеточных культурах, инфицированных микоплазмой, что свидетельствует о негативном влиянии TP на итоговую цитостатическую активность 5-FdUrd. Это накладывает ограничение на использование нуклеозидов для лечения пациентов, инфицированных микоплазмой. В отличие от 5-FdUrd 5-FdUrd-пролекарства согласно настоящему изобретению могут сохранять высокую активность в клетках, инфицированных микоплазмой.
Соответственно, соединения согласно настоящему изобретению потенциально способны преодолеть многие из ограничений, свойственных 5-FU и 5-FdUrd.
5-Фторурацил (5FU) является одним из первых примеров противораковых лекарств. Создание 5-FU было основано на доступных биохимических данных: атом фтора и атом водорода имеют близкие размеры, однако связь углерод-фтор гораздо сильнее, чем связь углерод-водород. Тимидилатсинтаза катализирует образование тимидилата путем замещения 5-водорода дезоксиуридина монофосфата метильной группой метилентетрагидрофолата. 5FU осуществляет цитотоксическое действие, используя три разных механизма. Нуклеооснование 5FU и дезоксирибонуклеозид 5-FdUrd проникают в клетки с помощью систем облегченного переноса нуклеозидов. Одним из механизмов действия данных агентов является ингибирование фермента тимидилатсинтазы (TS). Превращение нуклеооснования 5FU в дезоксинуклеозид 5-фтор-2'-дезоксиуридин (5-FdUrd) катализируется тимидинфосфорилазой. Последующее фосфорилирование дезоксинуклеозида 5-FdURd тимидинкиназой приводит к образованию цитотоксичного нуклеотида 5-фтор-2'-дезоксиуридин-5'-монофосфата (5-FdUMP). В присутствии восстановленного фолата (5,10-метилен-тетрагидрофолата (mTHF)) данный нуклеотид (5-FdUMP) ингибирует тимидилатсинтазу (TS), так как этот фермент не способен удалить атом 5-фтора. Соответственно, первый и самый важный механизм действия 5FU и FDUR заключается в ингибировании фермента тимидилатсинтазы (TS). Тимидилатсинтаза (TS) имеет два субстрата (dUMP и mTHF), оба из которых связываются в каталитическом центре фермента для синтеза dTMP. 5-FdUMP образует ковалентный тройной комплекс с тимидилатсинтазой (TS), ингибируя активность данного фермента и вызывая истощение дезокситимидинтрифосфата, необходимого для синтеза ДНК. Альтернативно 5-FdUMP синтезируется в результате серии превращений: превращения 5FU в 5-FUMP, катализируемого OPRT (оротатфосфорибозилтрансферазой), последующего превращения в фторуридина дифосфат (FUDP) и затем в фтордезоксиуридина дифосфат (5-FdUDP) в результате действия рибонуклеотидредуктазы (RR), который в конечном итоге превращается в 5'-FdUMP. Отмечено, что после воздействия 5FU или 5-FdUrd у клеток развивается устойчивость к данным химиотерапевтическим агентам. Повышенная экспрессия тимидилатсинтазы (TS) вызывает уменьшение терапевтического эффекта TS-ингибирующих лекарств, что приводит к развитию устойчивости. Отмечено, что некоторые индивидуумы являются более устойчивыми к действию TS-направленной терапии, чем другие. Второй механизм действия заключается в том, что дезоксинуклеозид 5-фтор-2'-дезоксиуридин (5-FdUrd) может быть превращен в трифосфат 5-FdUTP, который, в свою очередь, может встраивается в ДНК, что вызывает повреждение клеток. Третий механизм действия 5FU состоит в том, что данное соединение может ингибировать также синтез РНК после его превращения в FUMP, катализируемого ферментом OPRT, и затем, в две стадии, в фторуридинтрифосфат (FUTP), который встраивается в РНК. Считается, что ингибирование синтеза РНК представляет собой другой возможный механизм действия 5FU.
Таким образом, молекулу 5FU нельзя считать оптимальным TS-ингибирующим лекарством ввиду того, что превращение 5FU в 5-FdUMP является неэффективным: для метаболической активации 5FU требуется несколько метаболических стадий. Кроме того, если клетка продуцирует избыточное количество dUMP, который конкурирует с данным лекарством за связывание с активным центром, может иметь место устойчивость.
5-FdUrd является относительно хорошим субстратом для тимидинкиназы, которая превращает 5-FdUrd сразу в 5-FdUMP. В in vitro исследованиях с использованием нескольких линий раковых клеток показано, что 5-FdURd приблизительно в 5000 раз сильнее ингибирует рост клеток, чем 5FU. Кроме того, при цитотоксических концентрациях пролекарство 5-FdURd не подвергается какому-либо значительному превращению в рибонуклеотидные метаболиты. В in vitro исследованиях показано, что значительное количество 5-FdUrd под действием тимидинфосфорилазы (фермента, характеризующегося высокой аффинностью к 5-FdUrd) деградирует с образованием своего родного основания 5FU. Быстрое фосфоролитическое расщепление 5-FdUrd до 5FU in vitro и in vivo является основным препятствием для доставки интактного 5-FdUrd к клеткам с целью повышения его цитотоксического действия. Кроме того, деградация 5-FdUrd в гомогенатах кишечника крыс и у человека, после перорального введения, дает возможность предположить, что 5-FdUrd почти не абсорбируется в виде интактного 5-FdUrd.
Другой аспект настоящего изобретения относится к применению соединения согласно настоящему изобретению в способе профилактики или лечения рака у человека. Соответственно, рак выбран из группы, включающей лейкемию, рак поджелудочной железы, предстательной железы, легкого, молочной железы и шейки матки.
В частности, предложено применение соединения согласно настоящему изобретению в способе профилактики или лечения рака у пациента, у которого развилась или может развиться устойчивость опухолевых клеток к действию 5-фторацила или 5-фтор-2'-дезоксиуридина в период профилактики или лечения рака. Например, соединение согласно настоящему изобретению можно применять в способе профилактики или лечения рака у пациента, клетки которого имеют пониженный уровень белков-переносчиков нуклеозидов, и/или клетки которого являются дефицитными по нуклеозидкиназе, и/или клетки которого инфицированы микоплазмой, в особенности, когда рак представляет собой лейкемию. Соединение согласно настоящему изобретению можно применять в способе профилактики или лечения рака у пациента, клетки которого имеют повышенный уровень тимидилатсинтазы (TS) или наряду с этим характеризуются по меньшей мере одной из перечисленных выше особенностей.
Согласно другому аспекту настоящего изобретения, предложен способ профилактики или лечения рака, включающий введение человеку, нуждающемуся в таком лечении, эффективной дозы соединения согласно настоящему изобретению. Соответственно, рак выбран из группы, включающей лейкемию, рак поджелудочной железы, предстательной железы, легкого, молочной железы и шейки матки.
В частности, настоящее изобретение включает способ лечения пациента, у которого развилась или может развиться устойчивость опухолевых клеток к действию 5-фторацила или 5-фтор-2'-дезоксиуридина в период профилактики или лечения рака. Например, способ согласно настоящему изобретению может включать лечение пациента, клетки которого имеют пониженный уровень белков-переносчиков нуклеозидов, и/или клетки которого являются дефицитными по нуклеозидкиназе, и/или клетки которого инфицированы микоплазмой, в особенности, когда рак представляет собой лейкемию. Способ согласно настоящему изобретению можно применять для лечения пациента, клетки которого имеют повышенный уровень тимидилатсинтазы (TS) или наряду с этим характеризуются по меньшей мере одной из перечисленных выше особенностей.
В контексте настоящей заявки термин ʺопухольʺ или ʺопухолевая клеткаʺ, если не указано иное, относится как к солидным опухолям и солидному раку, так как и к лейкемии.
Соединения согласно настоящему изобретению можно применять для лечения пациента, страдающего раком, либо самостоятельно de novo, либо в комбинации с другой противораковой терапией. Например, соединения согласно настоящему изобретению можно применять в схеме лечения рака в комбинации с другими противораковыми лекарствами, такими как 5-FU и/или 5-FdUrd в комбинации с лейковорином (leucovorin) (LV), или без него, и/или с другими противораковыми лекарствами. Альтернативно соединения согласно настоящему изобретению можно применять, когда у пациента уже отсутствует ответ на другие противораковые лекарства, такие как, например, 5FU и/или 5-FdUrd в комбинации с лейковорином (leucovorin) (LV) или без него, или когда у пациента уже развилась устойчивость к другим противораковым лекарствам, таким как, например, 5-FU и/или 5-FdUrd в комбинации с лейковорином (leucovorin) (LV) или без него.
Соединения согласно настоящему изобретению, у которых Ar представляет собой замещенный или незамещенный 1-нафтил, особенно подходят для применения в вышеупомянутых случаях и в способах согласно настоящему изобретению, в особенности для применения у пациента, у которого развилась или может развиться устойчивость опухолевых клеток, например, у пациента, клетки которого имеют пониженный уровень белков-переносчиков нуклеозидов, и/или клетки которого являются дефицитными по киназе, и/или клетки которого инфицированы микоплазмой, и/или у пациента, клетки которого имеют повышенный уровень тимидилатсинтазы (TS).
Другой аспект настоящего изобретения относится к фармацевтической композиции, содержащей соединение согласно настоящему изобретению в комбинации с фармацевтически приемлемым носителем, разбавителем или эксципиентом.
Согласно другому аспекту настоящего изобретения, предложен способ получения фармацевтической композиции, включающий стадию комбинирования соединения согласно настоящему изобретению с фармацевтически приемлемым эксципиентом, носителем или разбавителем.
Другой аспект настоящего изобретения относится к способу получения соединения согласно настоящему изобретению, включающему взаимодействие соединения Формулы (II)
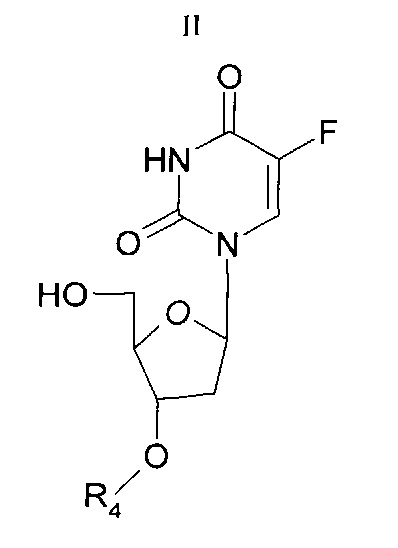
с соединением Формулы (III)
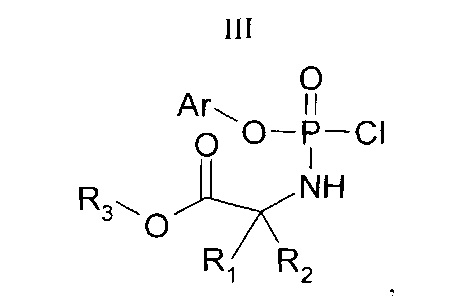
где Ar, R3, R4, R1 и R2 имеют значения, описанные выше и в п.1.
Группа Ar включает замещенную или незамещенную арильную группу, ʺарильная группаʺ и возможные заместители указанной группы являются такими, как определено в данном описании. Подходящий Ar представляет собой группировку 5-14-членных ароматических колец. Предпочтительно, когда Ar представляет собой карбоциклическое кольцо. Альтернативно одно или два кольца могут содержать 1, 2, 3 или 4 гетероатома, предпочтительно 1 гетероатом, независимо выбранные из атома О, S и N. Предпочтительно, когда Ar представляет собой конденсированную карбобициклическую арильную группировку. Более предпочтительно, когда Ar представляет собой нафтил, еще более предпочтительно 1-нафтил, то есть нафтил, связанный с атомом P через атом O, присоединенный в положении 1 нафталинового кольца. Альтернативно подходящий Ar может представлять собой фенил.
Ar может содержать один, два, три или четыре заместителя, которые могут быть одинаковыми или разными и выбраны из группы, включающей галоген, который может представлять собой -F, -Cl, -Br или -I; -NO2; -NH2; необязательно замещенный -С1-3алкил; необязательно замещенный -С1-3алкокси, предпочтительно метокси (-ОСН3); необязательно замещенный -SC1-3алкил; -CN; необязательно замещенный -COC1-3алкил и необязательно замещенный -CO2C1-3алкил; при этом указанные необязательно замещенные группы могут иметь от одного до шести заместителей, предпочтительно три заместителя, независимо выбранных из группы, включающей галоген, который может представлять собой F, Cl, Br и I, и NO2. Особенно предпочтительными заместителями у Ar являются электроноакцепторные группы, такие как атом галогена (предпочтительно атом хлора или фтора), тригалогенметил (предпочтительно трифторметил), циано- и нитро-группы.
Указанные заместители могут находиться на арильной группировке Ar в любом положении. Когда Ar представляет собой 1-нафтил, предпочтительно, чтобы единственный заместитель находился в любом из положений 2, 3, 4, 5, 6, 7 или 8. Когда Ar представляет собой фенил, предпочтительно, чтобы единственный заместитель находился в положении 2 (орто) или 4 (пара), более предпочтительно - в положении 4. Например, когда Ar представляет собой замещенный фенил, Ar может представлять собой 3,5-дихлор-фенил, пара-трифторметил-фенил, пара-циано-фенил или пара-нитро-фенил.
Подходящий R3 представляет собой первичную, вторичную или третичную C1-16 алкильную группу и может включать карбоциклические группировки; C5-7 циклическую алкильную группу или C1-6алкилC5-11арильную группу. Более предпочтительно, когда R3 представляет собой C1-10 алкильную группу или C1-3алкилC5-7арильную группу, такую как бензил (-CH2-C6H5). Циклическая алкильная группа может являться карбоциклической или может содержать в целом один, два или три кольцевых гетероатома, независимо выбранные из О, N и S. Предпочтительно, когда R3 является незамещенным. Когда R3 имеет заместители, заместители являются такими, как описано ниже.
Подходящий R4 представляет собой H или алкоил, то есть алкил-C(=O)-, где алкил представляет собой C1-C10алкил.
Когда R1 и/или R2 представляет собой алкил, каждый подходящий R1 и R2 независимо выбран из C1-C16алкила, более предпочтительно - из C1-C6алкила. Когда R1 и R2 вместе образуют алкиленовую цепь, подходящая алкиленовая цепь содержит от 1 до 6 атомов углерода (C1-C6) и может являться ненасыщенной и в целом содержать в цепи один, два или три гетероатома, независимо выбранные из О, N и S. Когда один из R1 и R2 соединен с N, подходящее общее количество кольцевых атомов, включая N и атом C, к которому присоединяются R1 и R2, составляет от 4 до 7 атомов, более предпочтительно - 5 атомов. Любая алкильная или алкиленовая цепь, содержащая R1 и/или R2, может быть замещенной и иметь один или более заместителей, описанных в данной заявке.
Когда R1 и R2 являются разными, атом C, к которому они присоединены, является хиральным. Предпочтительно, когда стереохимическая конфигурация около асимметрического центра -CR1R2 соответствует L-аминокислоте. Однако стереохимическая конфигурация около асимметрического центра -CR1R2 может соответствовать и D-аминокислоте. Альтернативно могут быть использованы смеси соединений, имеющих асимметрические центры, соответствующие L- и D-аминокислотам.
Подходящие R1 и R2 могут соответствовать группировкам, соединенным с атомом альфа-С в природной альфа-аминокислоте. Термин ʺприродная альфа-аминокислотаʺ означает аланин, аргинин, аспарагин, аспарагиновую кислоту, цистеин, цистин, глицин, глутаминовую кислоту, глутамин, гистидин, гидроксилизин, гидроксипролин, изолейцин, лейцин, лизин, метионин, фенилаланин, пролин, серин, треонин, триптофан, тирозин и валин. Один из R1 и R2 может представлять собой, соответственно, H, и один из R1 и R2 может представлять собой, соответственно, H или алкил, выбранный из перечисленных ниже группировок, или R1 и R2 вместе могут образовывать алкиленовую цепь, выбранную из перечисленных ниже группировок:
СН3-, присутствующей в аланине;
H2NC(=NH)NH[CH2]3-, присутствующей в аргинине;
NH2C(O)CH2-, присутствующей в аспарагине;
HO2CH2-, присутствующей в аспарагиновой кислоте;
HSCH2-, присутствующей в цистеине;
HO2CH(NH2)CH2SSCH2-, присутствующей в цистине;
H-, присутствующей в глицине;
HO2CH2CH2-, присутствующей в глутаминовой кислоте;
H2N(O)CCH2CH2-, присутствующей в глутамине;
C3N2HCH2-, присутствующей в гистидине;
H2NCH2CH(OH)CH2CH2-, присутствующей в гидроксилизине;
-СН2СН(ОН)СН2-, присутствующей в гидроксипролине;
СН3СН2СН(СН3)-, присутствующей в изолейцине;
(СН3)2СНСН2-, присутствующей в лейцине;
H2NCH2(CH2)3-, присутствующей в лизине;
CH3SCH2CH2-, присутствующей в метионине;
PhCH2-, присутствующей в фенилаланине;
-СН2СН2СН2-, присутствующей в пролине;
ОНСН2-, присутствующей в серине;
СН3СН(ОН)-, присутствующей в треонине;
C8NH6CH2-, присутствующей в триптофане;
НОС6Н4СН2-, присутствующей в тирозине;
(СН3)2СН-, присутствующей в валине.
Термин ʺфармацевтически приемлемое производноеʺ означает любую фармацевтически приемлемую соль, сложный эфир, соль такого сложного эфира, гидрат, сольват, или кристаллическую форму, или метаболит, или любое другое соединение, которое при введении реципиенту может быть превращено (прямо или косвенно) в соединение Формулы (I).
В контексте настоящего описания алкильная группа означает, циклический или ациклический, насыщенный или ненасыщенный (например алкенил или алкинил) гидрокарбильный радикал с разветвленной или нормальной цепью. Когда указанный гидрокарбильный радикал является циклическим, алкиленовая группа содержит предпочтительно от 3 до 12 атомов углерода (C3-C12), более предпочтительно - от 5 до 10 атомов углерода (C5-C10), более предпочтительно - от 5 до 7 атомов углерода (C5-C7). Когда указанный гидрокарбильный радикал является ациклическим, алкильная группа содержит предпочтительно от 1 до 16 атомов углерода (C1-C16), более предпочтительно - от 1 до 6 атомов углерода (C1-C6).
В контексте настоящего описания подходящая арильная группа означает ароматическую группу, содержащую от 5 до 14 кольцевых атомов. Примером Ar является фенил или нафтил. Данная ароматическая группа может представлять собой гетероароматическую группу, содержащую один, два, три или четыре гетероатома, предпочтительно один гетероатом, независимо выбранные из группы, состоящей из О, N и S. Примеры таких гетероароматических групп включают пиридил, пирролил, фуранил и тиофенил.
Алкильная и арильная группы могут быть замещенными или незамещенными. Когда указанные группы являются замещенными, обычно они содержат от одного до трех заместителей, предпочтительно - один заместитель. Заместители могут включать атомы галогенов, которые представляют собой атомы F, Cl, Br и I, и галогенметильные группы, такие как CF3 и CCl3; кислород-содержащие группы, такие как оксо, гидрокси, карбокси, карбоксиC1-16алкил, алкокси, алкоил, алкоилокси, арилокси, арилоил и арилоилокси; азотсодержащие группы, такие как амино, C1-6алкиламино, диC1-6алкиламино, циано, азид и нитро; серо-содержащие группы, такие как тиол, C1-6алкилтиол, сульфонил и сульфоксид; гетероциклические группы, которые сами могут содержать заместители; алкильные группы, такие, как определено выше, которые сами могут содержать заместители; и арильные группы, такие, как определено выше, которые сами могут являются замещенными, такие как фенил и замещенный фенил. Заместители указанных гетероциклических, алкильных и арильных групп являются такими, как определено только что выше. Заместители у R1 и/или R2 включают группировки, которые приводят к образованию соединений, у которых R1 и R2 соответствуют группировкам, соединенным с атомом альфа-C в природной альфа-аминокислоте.
В контексте настоящего описания алкокси- и арилокси-группы означают, соответственно, алкил-O- (например алкил-O-, у которого алкил представляет собой C1-C16алкил, предпочтительно C1-C6алкил) и арил-O- (например арил-O-, у которого арил представляет собой 5-14-членную ароматическую моно- или биконденсированную кольцевую группировку, необязательно содержащую 1, 2, 3 или 4 гетероатома, независимо выбранные из О, S и N, предпочтительно арил представляет собой фенил).
В контексте настоящего описания алкоильная и арилоильная группы означают, соответственно, алкил-CO- (например алкил-CO-, у которого алкил представляет собой C1-C16алкил, предпочтительно C1-C6алкил) и арил-CO- (например арил-CO-, у которого арил представляет собой 5-14-членную ароматическую моно- или биконденсированную кольцевую группировку, необязательно содержащую 1, 2, 3 или 4 гетероатома, независимо выбранные из О, S и N, предпочтительно арил представляет собой фенил).
В контексте настоящего описания алкоилокси и арилоилокси означают, соответственно, алкил-CO-O (например алкил-CO-О, у которого алкил представляет собой C1-C16алкил, предпочтительно C1-С6алкил) и арил-CO-O (например арил-CO-O, у которого арил представляет собой 5-14-членную моно- или биконденсированную ароматическую кольцевую систему, необязательно содержащую 1, 2, 3 или 4 гетероатома, независимо выбранные из О, S и N, предпочтительно арил представляет собой фенил).
В контексте настоящего описания гетероциклические группы означают группы, содержащие один или более чем один пирролил, имидазолил, пиразиолил, тиазолил, изотиазолил, оксазолил, пирролидинил, пирролинил, имидазолидинил, имидазолинил, пиразолидинил, тетрагидрофуранил, пиранил, пиронил, пиридил, пиразинил, пиридазинил, пиперидил, пиперазинил, морфолинил, тионафтил, бензофуранил, изобензофурил, индолил, оксииндолил, изоиндолил, индазолил, индолинил, 7-азаиндолил, изоиндазолил, бензопиранил, кумаринил, изокумаринил, хинолил, изохинолил, нафтридинил, циннолинил, хиназолинил, пиридопиридил, бензоксазинил, хиноксадинил, хроменил, хроманил, изохроманил и карболинил.
Согласно одному из вариантов осуществления настоящего изобретения, подходящий Ar представляет собой нафтил, особенно 1-нафтил, то есть нафтил, связанный с атомом P через атом O, присоединенный в положении 1 нафталинового кольца.
Согласно другому варианту осуществления настоящего изобретения, подходящий Ar представляет собой фенил.
Согласно одному из вариантов осуществления настоящего изобретения, Ar является замещенным. Подходящие заместители описаны в данной заявке.
Согласно одному из вариантов осуществления настоящего изобретения, Ar представляет собой незамещенный 1-нафтил.
Согласно одному из вариантов осуществления настоящего изобретения, Ar представляет собой незамещенный фенил.
Согласно одному из вариантов осуществления настоящего изобретения, R4 выбран из группы, состоящей из H и ацетила (СН3С(=O)-), особенно предпочтительно, когда R4 представляет собой Н.
Согласно одному из вариантов осуществления настоящего изобретения, R3 выбран из группы, состоящей из бензила и представителей группы, включающей C1-C10 алкилы, особенно предпочтительно, когда R3 выбран из н-пропила, н-бутила, н-пентила и н-гексила, более предпочтительно, когда R3 представляет собой н-пентил.
Согласно одному из вариантов осуществления настоящего изобретения, R1 и R2 соответствуют группировкам, соединенным с атомом альфа-С в природной альфа-аминокислоте, описанным в данной заявке. Особенно подходящая природная альфа-аминокислота представляет собой L-аланин, соответственно, один из R1 и R2 представляет собой H, один из R1 и R2 представляет собой CH3 и атом C, к которому они присоединены, является хиральным. Согласно другим вариантам осуществления изобретения, R1 и R2 соответствуют группировкам, соединенным с атомом альфа-C в неприродной альфа-аминокислоте, например оба подходящих R1 и R2 представляют собой CH3.
Конкретные отличительные признаки изобретения, описанные выше в вариантах осуществления изобретения, могут быть объединены вместе в любых без исключения комбинациях в соединениях согласно настоящему изобретению.
Особенно подходящими соединениями согласно настоящему изобретению являются соединения, у которых Ar представляет собой 1-нафтил, R3 представляет собой бензил, один из R1 и R2 представляет собой H, один из R1 и R2 представляет собой метил и атом C, к которому присоединены R1 и R2, является L-хиральным, и соединения, у которых Ar представляет собой 1-нафтил, R3 представляет собой н-пентил, один из R1 и R2 представляет собой H, один из R1 и R2 представляет собой метил и атом C, к которому присоединены R1 и R2, является L-хиральным. Для каждого из соединений наиболее предпочтительным R4 является H.
Традиционное лечение рака с использованием химиотерапии в значительной степени основано на использовании нуклеозидных аналогов. Данные молекулы созданы, чтобы имитировать природные пиримидиновые и пуриновые нуклеозиды. После поглощения клеткой эти молекулы подвергаются фосфорилированию клеточными ферментами, такими как (дезокси)цитидинкиназа (dCK), тимидинкиназа (TK) и/или нуклеозид/нуклеотид-киназы. Впоследствии данные антиметаболиты могут мешать de novo синтезу предшественников ДНК/РНК и в конечном итоге ингибируют синтез ДНК/РНК, что определяет их цитотоксическое/цитостатическое действие (Hatse et al., 1999; Galmarini et al., 2002).
Антиметаболиты на основе фторпиримидина, такие как фторурацил (5-FU), капецитабин и 5-фтор-2'-дезоксиуридин (5-FdUrd), используются главным образом в лечении карциномы толстой кишки, молочной железы и яичника (de Bruin et al., 2006; Ishikawa et al., 1998; Walko et al., 2005). Внутри клетки данные лекарства подвергаются метаболическому превращению в 5-FdUMP, который образует стабильный ингибирующий комплекс с тимидилатсинтазой (TS) и восстановленным ко-субстратом 5,10-метилентетрагидрофолатом, тем самым блокируя связывание с данным ферментом нормального субстрата dUMP (Beck et al., 1994; Tanaka et al., 2000; Longley et al., 2003). TS представляет собой фермент, отвечающий за превращение dUMP в ТМР, и поэтому является необходимым для клеточной пролиферации, это обстоятельство делает TS привлекательной мишенью при создании лекарств. Среди фторпиримидинов, перечисленных выше, только для превращения 5-FdUrd в 5-FdUMP требуется одна метаболическая стадия, а именно стадия фосфорилирования, катализируемая ТК (Longley et al., 2003). Обязательное фосфорилирование часто является стадией, лимитирующей скорость метаболизма многих противораковых лекарств (включая 5-FdUrd), и поэтому является одним из ограничивающих факторов для терапевтического использования нуклеозидных аналогов. В связи с этим для улучшения противораковой эффективности нуклеозидных аналогов были исследованы другие стратегии (Galmarini et al., 2002).
Заряд, который несут нуклеозидмонофосфаты в физиологических условиях, является причиной плохого проникновения данных молекул через клеточную мембрану, если вообще таковое имеет место (Mehellou et al., 2009). Поэтому прямое введение уже фосфорилированных молекул с целью обойти первую стадию фосфорилирования не дает какого-либо значительного терапевтического преимущества. В связи с этим для достижения более эффективной доставки лекарств изучали различные стратегии обхода стадии фосфорилирования, лимитирующей скорость метаболизма, путем использования в качестве пролекарства различных типов нуклеозид 5'-монофосфатов (Hecker & Erion, 2008). Введение липофильных фосфорамидатных нуклеотидов (ProTide) в качестве пролекарств оказалось успешным в отношении нескольких молекул, обладающих противовирусной/противораковой активностью (Harris et al., 2001; Congiatu et al., 2006; McGuigan et al., 2010). Экранирование заряда фосфатного мотива может обеспечить хорошую пассивную диффузию пролекарств через мембрану, после чего, уже внутри клетки, пролекарство быстро превращается в соответствующий нуклеозидмонофосфат в результате ферментативного расщепления (Mehellou et al., 2009).
Микоплазмы представляют собой мельчайшие самореплицирующиеся организмы на планете, которые характеризуются отсутствием клеточной стенки и очень коротким геномом (600-1200 т.п.н.). Многие из данных бактерий являются паразитирующими и присутствуют в организме человека, являясь причиной бессимптомных инфекций (Razin et al., 1998). Показано, что данные прокариоты избирательно колонизируют опухолевые ткани: Huang с соавторами (2001) сообщают, что у человека в 39,7-56% случаев раковые ткани желудка, толстой кишки, пищевода, легкого и молочной железы инфицированы микоплазмой, в то время как неопухолевые ткани инфицированы только в 20,9-30% случаев. Pehlivan с соавторами (2005) показали, что в >80% случаев образцы тканей почек пациентов, страдающих почечно-клеточной карциномой, инфицированы микоплазмой, в то время как контрольные тканевые образцы инфицированы только в 14% случаев. Chan с соавторами (1996) показали, что в 59% случаев для тканей рака яичника характерна высокая скорость инфицирования микоплазмой, в других исследованиях также наблюдали высокую скорость инфицирования микоплазмой в тканях желудка (Sasaki et al., 1995, Yang et al., 2010) и кондиломы шейки матки (Kidder et al., 1998). Вследствие небольшого количества генов у микоплазмы отсутствует путь de novo синтеза пиримидинов и пуринов, и поэтому микоплазмы экспрессируют широкий набор компенсирующих нуклеозид/нуклеотид-метаболизирующих ферментов, таких как тимидинфосфорилаза (TP), дезоксицитидиндезаминаза и так далее (Razin, 1978; Charron & Langelier, 1981; Neale et al., 1983; Tham et al., 1993). Уже в 1985 было показано, что присутствующие в зараженных клеточных культурах ферменты, кодируемые микоплазмой (например TP), вызывают уменьшение включения dTTP в лимфоцитах (Sinigaglia & Talmadge, 1985). Недавно показано, что данные ферменты, в частности кодируемая микоплазмой тимидинфосфорилаза, также препятствуют проявлению цитостатической активности некоторых химиотерапевтических лекарств, включая 5-трифтортимидин, in vitro (Bronckaers et al., 2008; Jette et al., 2008; Liekens et al., 2009). Поэтому можно предположить, что устранение микоплазмы с использованием антибиотиков или супрессия кодируемых микоплазмой ферментов в опухолевой ткани человека может повысить эффективность лечения пациентов, страдающих раком, путем использования пуриновых и пиримидиновых антиметаболитов (Liekens et al., 2009).
Настоящее изобретение представляет собой результат разработки и анализа TK-независимых фосфорамидатных пролекарств нуклеозида 5-FdUrd; в настоящем изобретении предложены соединения, которые также могут быть нечувствительны к ТР-зависимой инактивации его свободного нуклеозидного аналога. Соответственно, соединения согласно настоящему изобретению могут быть предложены в качестве пролекарств, представляющих собой нечувствительные к микоплазме нуклеозидные аналоги, которые могут повысить эффективность лечения пациентов, страдающих раком, путем использования пиримидинового антиметаболита. Из синтезированных в настоящем изобретении фосфорамидатных пролекарств нуклеозида 5-FdUrd для дополнительного более глубокого изучения выбрано соединение CPF-373 (идентифицированное далее и упоминавшееся выше в качестве конкретного подходящего соединения согласно данному изобретению, у которого R4 представляет собой Н). Данная молекула содержит нафтильную и бензилаланинильную группу для экранирования заряда 5'-фосфата на 5-FdUMP.
Ранее описаны различные механизмы устойчивости опухолевых клеток к фторпиримидинам, таким как 5FU, 5-FdUrd и трифтортимидин (TFT), включающие уменьшение активности ключевых ферментов, активирующих лекарства (например TK и оротатфосфорибозилтрансферазы), увеличение активности ферментов, инактивирующих лекарства (то есть тимидинфосфорилазы) и/или повышение уровня экспрессии ферментов-мишеней (например TS) (Agarwal et al., 1999; Murakami et al., 2000; Kosaka et al., 2004). Кроме того, описано, что высокий уровень TP, обнаруженный в некоторых типах раковых тканей, предсказывает более плохой прогноз лечения с использованием фторпиримидинов (Kamoshida et al., 2005; Ciaparrone et al., 2006; Koopman et al., 2009), хотя другие исследования не подтверждают эти данные (Ciccolini et al., 2004; Koopman et al., 2009). Настоящее изобретение представляет собой результат разработки пролекарства для 5-FdUrd, которое позволяет обойти возможные механизмы устойчивости и чувствительность к деградации катаболическими ферментами, присутствующими в микроокружении опухоли.
Соединения, описанные в вариантах осуществления настоящего изобретения, например CPF-373, представляют собой фосфорамидатные пролекарства нуклеозида 5-FdUrd и могут служить достижению целей данного изобретения. После проникновения в опухолевые клетки соединение CPF-373, например, уже внутри клетки превращается в 5-FdUMP в результате ферментативного расщепления. Анализ стабильности соединений и действия ферментов и сыворотки с использованием 31Р-ЯМР-технологии показал, что пролекарство CPF-373, например, является абсолютно стабильным в кислотных и щелочных условиях, но подвергается гидролизу в присутствии сыворотки или карбоксипептидазы Y с образованием нуклеозид 5'-фосфорамидатного производного. Несмотря на то, что TK является ключевым ферментом в активации 5-FdUrd, было найдено, что цитостатическая активация соединения CPF-373, например, совсем не зависит от TK в культурах клеток мыши (L1210) и человека (CEM). Благодаря липофильной природе молекул ProTide данные соединения могут доставлять нуклеозидмонофосфаты прямо в интактную опухолевую клетку после их превращения в соответствующее нуклеозидфосфорамидатное производное под действие таких ферментов, как карбоксиэстеразы или карбоксипептидазы (а именно карбоксипептидаза Y), устраняющего необходимость начального фосфорилирования специфическими нуклеозидкиназами, такими как TK. В связи с этим соединение CPF-373, например, может являться эффективным инструментом для лечения опухолевых клеток с модифицированной TK-активностью (которая может быть приобретенной или врожденной). Кроме того, так как экспрессия TK зависит от S-фазы репликативного цикла, можно ожидать, что соединение CPF-373, например, также может эффективно доставлять 5-FdUMP в опухолевые клетки, которые не находятся в S-фазе репликативного цикла. Анализ TS-активности показал, что соединение CPF-373, например, может ингибировать TS как в линиях клеток дикого типа, так и в линиях опухолевых клеток, дефицитных по TK, эти данные еще раз указывают на эффективную доставку 5'-монофосфат 5-FdUrd внутрь клеток и подтверждают, что метаболическая активация данного соединения в сущности не зависит от клеточной TK.
Соединения согласно настоящему изобретению, такие как CPF-373, по-видимому, не инактивируются катаболическими ферментами, участвующими в метаболизме нуклеозидов. Действительно, несмотря на то, что 5-FdUrd является чрезвычайно чувствительным к ферментативному гидролизу под действием TP, приводящему к образованию 5-FU и 2-дезоксирибоза-1-фосфату, его пролекарство, например соединение CPF-373, не является субстратом для TP прокариот (а именно Е. coli) или млекопитающих (а именно эритроцитов человека). Кроме того, уридинфосфорилаза не узнает, например, соединение CPF-373, в качестве субстрата, тогда как 5-FdUrd подвергается (плохо, но заметно) гидролизу под действием данного фермента. В нескольких исследованиях показано, что многие опухолевые клетки имеют повышенный уровень TP, что также действует как ангиогенный фактор (Koopman et al., 2009; Bronckaers et al., 2009). Кроме того, в нескольких исследованиях показано, что избирательная колонизация опухолевой ткани микоплазмой (Sasaki et al., 1995; Chan et al., 1996; Huang et al., 2001; Pehlivan et al., 2005) препятствует проявлению цитостатической активности некоторых традиционных химиотерапевтических лекарств in vitro и ответственной за данное ингибирование является TP, кодируемая микоплазмой (Bronckaers et al., 2008; Jette et al., 2008; Liekens et al., 2009). Результаты исследований, приведенные в настоящем описании, согласно которым 5-FdUrd, но, например, не соединение CPF-373, в значительной степени теряет цитостатическую активность, когда опухолевые клетки инфицированы (ТР-экспрессирующей) микоплазмой, полностью согласуется с данными наблюдениями. Поэтому введение ТР-нечувствительного противоракового пролекарства, такого как соединение CPF-373, показывает, что данное пролекарство, которое является химически стабильными при критических значения рН, может улучшить результаты химиотерапии при лечении рака. В заключение следует отметить, что ProTide, такие как соединение CPF-373, представляет собой новый интересный подход для разработки более эффективных противораковых лекарств. Например, соединение CPF-373 может иметь по меньшей мере несколько преимуществ по сравнению с исходным лекарством 5-FdUrd, а именно: цитостатическую активность, независимую от TK, и устойчивость к метаболическому расщеплению под действием TP, фермента, который часто имеет повышенный уровень экспрессии в опухолевых клетках или может дополнительно экспрессироваться микоплазмой в инфицированной опухолевой ткани.
Соединение Формулы I, или фармацевтическая композиция согласно настоящему изобретению, может быть введено человеку, нуждающемуся в таком введении, любым подходящим путем.
Лекарства для применения согласно настоящему изобретению могут быть введены пероральным или парентеральным путем, включая внутривенное, внутримышечное, интраперитонеальное, подкожное, трансдермальное введение, введение через дыхательные пути (с использованием аэрозоля), ректальное, вагинальное и местное (включая трансбуккальное и сублингвальное)введение.
Соединения согласно настоящему изобретению, предназначенные для перорального введения, обычно находятся в форме таблеток или капсул, в форме порошков или гранул или в форме водного раствора или суспензии.
Таблетки для перорального введения могут содержать активный ингредиент, смешанный с фармацевтически приемлемыми эксципиентами, такими как инертные разбавители, разрыхляющие агенты, связующие агенты, смазывающие агенты, подслащивающие агенты, корригенты, окрашивающие агенты и консерванты. Подходящие инертные разбавители включают карбонат натрия и кальция, фосфат натрия и кальция и лактозу, тогда как подходящими разрыхляющими агентами являются кукурузный крахмал и альгиновая кислота. Связующие агенты могут включать крахмал и желатин, тогда как в качестве смазывающих агентов, если такие присутствуют, обычно используют стеарат магния, стеариновую кислоту или тальк. При желании, для замедления абсорбции желудочно-кишечном тракте таблетки могут быть покрыты таким веществом, как глицерилмоностеарат или глицерилдистеарат.
Капсулы для перорального введения включают твердые желатиновые капсулы, в которых активный ингредиент смешан с твердым разбавителем, и мягкие желатиновые капсулы, в которых активный ингредиент смешан с водой или маслом, таким как арахисовое масло, вазелиновое масло или оливковое масло.
Препараты для ректального введения могут находится в форме суппозиториев с подходящей основой, содержащей, например, масло какао или салицилат.
Препараты, подходящие для вагинального введения, могут находится в форме пессариев, тампонов, кремов, гелей, паст, пен или спрея и содержат наряду с активным ингредиентом подходящие носители, известные в данной области техники.
Соединения согласно настоящему изобретению, предназначенные для внутримышечного, интраперитонеального, подкожного и внутривенного введения, обычно находятся в форме стерильных водных растворов или суспензий, забуференных до подходящих значений pH и изотоничности. Подходящие водные носители включают раствор Рингера и изотонический раствор хлорида натрия. Водные суспензии согласно настоящему изобретению могут включать суспендирующие агенты, такие как производные целлюлозы, альгинат натрия, поливинил-пирролидон и трагакантовая камедь, и увлажняющие агенты, такие как лецитин. Консерванты, подходящие для водных суспензий, включают этил- и н-пропил пара-гидроксибензоат.
Соединения согласно изобретению также могут находится в форме липосомных препаратов.
В общем случае подходящая доза находится в диапазоне от 0,1 до 300 мг на килограмм массы тела реципиента в сутки. Предпочтительно, когда более низкая доза составляет 0,5 мг на килограмм массы тела реципиента в сутки, более предпочтительно, когда более низкая доза составляет 6 мг на килограмм массы тела реципиента в сутки, еще предпочтительнее, когда более низкая доза составляет 10 мг на килограмм массы тела реципиента в сутки. Подходящая доза предпочтительно находится в диапазоне от 6 до 150 мг на килограмм массы тела в сутки, и наиболее предпочтительно - в диапазоне от 15 до 100 мг на килограмм массы тела в сутки. Предпочтительно, когда требуемую суточную дозу вводят в виде двух, трех, четырех, пяти, шести или более субдоз через подходящие интервалы времени. Данные субдозы могут быть введены в виде стандартных лекарственных форм, содержащих, например, от 10 до 1500 мг, предпочтительно от 20 до 1000 мг и наиболее предпочтительно от 50 до 700 мг активного ингредиента на стандартную лекарственную форму.
Далее варианты осуществления настоящего изобретения описаны только с помощью Примеров, со ссылкой на прилагаемые чертежи, включающие Фигуры 1-11.
На Фиг.1 приведена структурная формула 5-FdUrd и его фосфорамидатного пролекарства CPF-373.
На Фиг.2 показан результат действия тимидинфосфорилазы и уридинфосфорилазы на dThd, Urd, 5-FdUrd и CPF-373, где каждая экспериментальная точка на графике представляет собой среднее значение по меньшей мере 2 независимых экспериментов (±S.D.).
На Фиг.3 показан результат ингибирования TS соединениями 5-FdUrd и CPF-373, полученный путем измерения высвобождения трития из [5-3H]dUrd (секции А и В) и [5-3H]dCyd (секции С и D) в клеточных культурах L1210/0 и путем измерения высвобождения трития из [5-3H]dCyd (секции Е и F) в клеточных культурах L1210/TK-, где каждая экспериментальная точка на графике представляет собой среднее значение по меньшей мере 2 независимых экспериментов (±S.E.M.).
На Фиг.4 показан предложенный возможный механизм активации 5-FdUrd ProTide.
На Фиг.5 показано расщепление пролекарства CPF-373 под действием карбоксипептидазы, регистрируемое с использованием 31Р-ЯМР.
На Фиг.6 приведен 31Р-ЯМР-спектр соединения CPF-373 в сыворотке.
На Фиг.7 приведен 31Р-ЯМР-спектр соединения CPF-373 в буферном растворе (рН=1).
На Фиг.8 приведен 31Р-ЯМР-спектр соединения CPF-373 в буферном растворе (рН=8).
На Фиг.9 приведены 19F-ЯМР-спектры нуклеозида и соответствующего основания: а) 5-FdUrd в условиях фосфорилазного анализа (А); б) 5-FdUrd и основание 5FU в условиях фосфорилазного анализа в отсутствии фермента (TP) (В).
На Фиг.10 приведены 19F-ЯМР-спектры нуклеозида и основания в калий-фосфатном буфере (205 нМ): a) 5-FdUrd в условиях фосфорилазного анализа в отсутствии фермента (А); б) спектр после добавления фермент (TP) (В).
На Фиг.11 приведены спектры пролекарства CPF373 в условиях фосфорилазного анализа: а) пролекарство CPF373 в условиях фосфорилазного анализа в отсутствии фермента (TP) (А); б) пролекарство CPF373, подвергнутое действию тимидинфосфорилазы (TP) (В).
Синтез соединений
Согласно Фиг.1 и Схемам 1-3, приведенным ниже, соединения согласно настоящему изобретению (в качестве примера рассмотрено соединение CPF-373 (1)), синтезировали с использованием фосфорхлоридатной химии, соответствующие методики описаны ранее McGuigan с соавторами (1993, 1996, 1997). Например, арилфосфордихлорфосфат (2) получали в результате взаимодействия 1-нафтола (3) и оксихлорида фосфора (4) в присутствии Et3N (Схема 1) и затем подвергали взаимодействию с тозилатом бензилового эфира L-аланина (5) в присутствии Et3N с получением фосфорхлоридатного производного (6) (Схема 2). Затем нуклеозид 5-FdUrd (7) превращали в 5' ProTide, с этой целью 5-FdUrd подвергали взаимодействию с фосфорхлоридатным производным (6) в ТГФ в присутствии N-метил имидазола (NMI) с получением желаемого соединения CPF-373 (1) (Схема 3). Данное соединение получали в виде смеси двух диастереоизомеров, на что указывало присутствие двух пиков на 31Р-ЯМР-спектре.
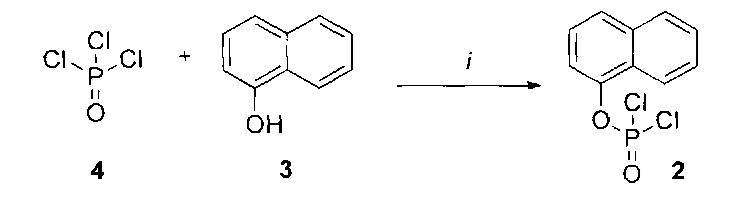
Схема 1. Реагенты и условия: (i) 1-нафтол (3), оксихлорид фосфора (4), сухой Et2O, сухой Et3N, -78°C 30 мин, затем комнатная температура 3 ч.
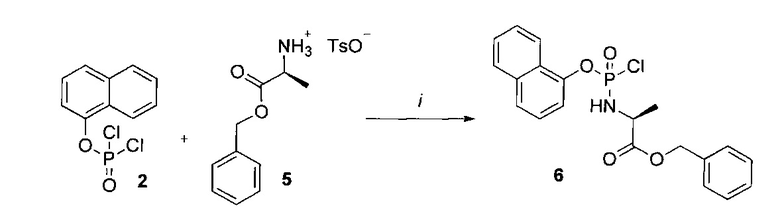
Схема 2. Реагенты и условия: (i) сухой Et3N, CH2Cl2, -78°C 1 ч, затем комнатная температура 3 ч.
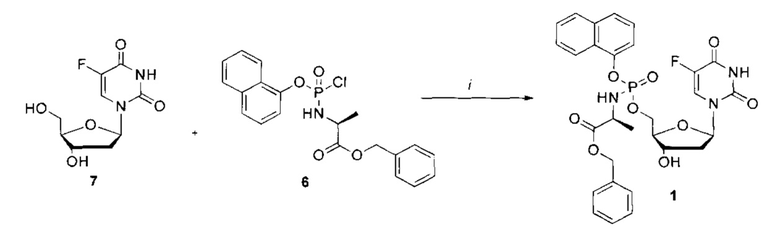
Схема 3. Реагенты и условия: (i) NMI, сухой ТГФ, 10 мин, затем фосфорхлоридат (6), комнатная температура, реакция в течение ночи.
Безводные растворители получали от Aldrich и использовали без дополнительной очистки. Все реакции проводили в атмосфере аргона. Ход реакций контролировали с помощью аналитической ТСХ (тонкослойной хроматографии) на покрытых силикагелем 60-F254 алюминиевых пластинах, используя для визуализации УФ-свет (254 нм), и/или с помощью 31Р-ЯМР-спектров. Колоночную хроматографию выполняли на силикагеле (35-70 мкм). ЯМР-спектры протонов (1H), углерода (13C), фосфора (31P) и фтора (19F) регистрировали на спектрометре Bruker Avance 500 при 25°C. Автокалибровку спектров проводили по пику дейтерированного растворителя, и все 13С-ЯМР-спектры и 31Р-ЯМР-спектры были получены в виде протон-несопряженных спектров. Аналитическую ВЭЖХ (жидкостную хроматографию высокого давления) выполняли на Varian Prostar (LC Workstation-Varian prostar 335 LC-детектор) с использованием аналитической колонки Varian Polaris С18-А (10 мкм).
Масс-спектры низкого и высокого разрешения с использованием электрораспыления (ES) получены в Бирмингемском университете. Элементный анализ CHN выполнен в MEDAC Ltd., Surrey.
Стандартная методика А: синтез дихлорфосфата (2)
К раствору 1-нафтола (1,0 экв.) в диэтиловом эфире в атмосфере аргона добавляли оксихлорид фосфора (1,0 экв.), затем добавляли по каплям безводный триэтиламин (1,0 экв.) при -78°C, и полученную реакционную смесь перемешивали в течение 1 ч. Затем реакционную смесь оставляли медленно нагреваться до комнатной температуры в течение 3 ч. Образование желаемого соединения контролировали с помощью 31P-ЯМР. Полученную смесь фильтровали и затем упаривали под вакуумом в атмосфере азота с выходом неочищенного желаемого продукта в виде бесцветного масла, который использовали на следующей стадии без дополнительной очистки.
Синтез 1-нафтил дихлорфосфата (2). Данное соединение получали в соответствии со стандартной методикой А из 1-нафтола (3,00 г, 20,81 ммоль), оксихлорида фосфора (1,94 мл, 20,81 ммоль), триэтиламина (2,9 мл, 20,81 ммоль) и безводного диэтилового эфира (70 мл). Реакционную смесь выдерживали в течение 1 ч при -78°C, затем оставляли нагреваться до комнатной температуры и перемешивали в течение 3 ч. Желаемый неочищенный продукт получали в виде масла. Полученную смесь фильтровали и затем упаривали под вакуумом, после очистки путем колоночной хроматографии с использованием для элюирования смеси гексан-EtOAc (1:1) получали бесцветное масло (4,59 г, 84%) [Rf=0,93 (гексан-EtOAc, 1:1)], 31P-ЯМР (202 МГц, CDCl3): δP 5.07; 1Н-ЯМР (500 МГц, CDCl3): δН 7.52-7.71 (m, 4Н, ArH), 7.86-7.89 (m, 1Н, ArH), 7.95-7.98 (m, 1Н, ArH), 8.16-8.19 (m, 1Н, ArH).
Стандартная методика B: синтез фосфорхлоридата (6)
К безводному триэтиламину (2,0 экв.) при -78°C добавляли по каплям в атмосфере аргона раствор арил фосфордихлоридата (1,0 экв.) и подходящей соли сложного эфира аминокислоты (1,0 экв.) в дихлорметане. Через 1 ч реакционную смесь оставляли медленно нагреваться до комнатной температуры в течение 3 ч, и образование желаемого соединения контролировали с помощью 31Р-ЯМР. Затем реакционную смесь концентрировали при пониженном давлении, остаток перерастворяли в диэтиловом эфире, фильтровали и упаривали под вакуумом в атмосфере азота с получением неочищенного бесцветного масла, которое в некоторых случаях использовали на следующей стадии без дополнительной очистки. Синтезированный арил фосфорхлоридат очищали путем колоночной хроматографии, используя для элюирования смесь гексан-EtOAc (7:3), с получением указанного в заголовке соединения в виде бесцветного масла.
Синтез 1-нафтил(бензил-L-аланинил)фосфорхлоридата (6). Данный фосфорхлоридат получали с использованием 1-нафтил дихлорфосфата (2,50 г, 9,57 ммоль), тозилатной соли бензилового эфира L-аланина (3,36 г, 9,57 ммоль), сухого триэтиламина (2,66 мл, 19,14 ммоль) и сухого дихлорметана (35,7 мл) в соответствии с общей методикой В. В результате очистки путем колоночной хроматографии с использованием для элюирования смеси гексан-EtOAc (7:3) получали указанное в заголовке соединение в виде бесцветного масла (1,82 г, 47%) [Rf=0,90 (гексан-EtOAc, 7:3)], 31Р-ЯМР (202 МГц, CDCl3, смесь диастереоизомеров): δР 7.92, 8.14 (Int.: 1.00:1.00); 1Н-ЯМР (500 МГц, CDCl3, смесь диастереоизомеров в соотношении 1:1): δН 1.42-1.45 (m, 3Н, СНСН3), 4.20-4.23 (m, 1Н, СНСН3), 4.78-4.81 (m, 1Н, NH), 5.09 (s, 2Н, OCH2Ph), 7.09-7.73 (m, 11Н, ArH), 7.97-8.12 (m, 1H, ArH).
Стандартная методика С: синтез нуклеозидфосфорамидата (1).
К NMI (5,0 экв.) при комнатной температуре в атмосфере аргона добавляли раствор подходящего нуклеозида (1,0 экв.) в сухом ТГФ (10 мл). Через 10 мин данную реакционную смесь добавляли по каплям к раствору фосфорхлоридата (3,0 экв.) в безводном ТГФ. Данную реакционную смесь перемешивали в течение ночи при комнатной температуре и упаривали под вакуумом. Полученное масло растворяли в CH2Cl2, дважды промывали H2O, затем 0,5 М раствором HCl, или альтернативно неочищенный продукт промывали диэтиловым эфиром. Затем полученный неочищенный продукт очищали путем колоночной хроматографии на силикагеле, используя для элюирования градиент CH2Cl2-МеОН, с выходом желаемого фосфорамидата.
Синтез 5-фтор-2'дезоксиуридин-5'-O-[α-нафтил(бензил-L-аланинил)] фосфата (1).
Данный фосфорамидат получали с использованием 5-фтор-2'дезоксиуридина (0,25 г, 1,01 ммоль), NMI (0,40 мл, 5,07 ммоль) и нафтил(бензил-L-аланинил) фосфорхлоридата (0,82 г, 3,04 ммоль) в соответствии с общей методикой C В результате очистки путем колоночной хроматографии с использованием для элюирования градиента CH2Cl2 (конечное соотношение растворителей: CH2Cl2:МеОН=95:5) получали указанное в заголовке соединение в виде бесцветного твердого вещества (47,0 мг, 8%) [Rf=0,19 (CH2Cl2:MeOH=95:5)], (Найдено: MNa+, 636,1520. C29H29N3O9FNaP. Вычислено: [MNa+], 636,1523); 31Р-ЯМР (202 МГц, MeOD, смесь диастереоизомеров): δР 4.24, 4.59; 19F ЯМР (470 МГц, MeOD): δF -167.36, -167.18; 1Н-ЯМР (500 МГц, MeOD): δН 1.34-1.38 (m, 3Н, СНСН3), 1.67-1.79 (m, 1Н, Н-2'), 2.08-2.17 (m, 1H, Н-2'), 4.03-4.15 (m, 2Н, СНСН3, Н-4'), 4.24-4.36 (m, 3Н, СН2ОР, Н-3'), 5.08 (d, 1Н, J=12.0 Гц, OCHHPh), 5.13 (d, 1Н, J=12.0 Гц, OCHHPh), 6.09-6.16 (m, 1H, Н-1'), 7.27-7.45 (m, 6H, ArH), 7.47-7.55 (m, 3H, ArH), 7.67-7.72 (m, 2H, ArH, H-6), 7.86-7.90 (m, 1H, ArH), 8.12-8.18 (m, 1H, ArH); 13С-ЯМР (125 МГц, MeOD): δC 20.3 (d, 3JC-P=7.6 Гц, CH3), 20.5 (d, 3JC-P=6.5 Гц, CH3), 40.8 (CH2), 40.9 (CH2), 51.8 (CH), 51.9 (CH), 67.6 (d, 2JC-P=5.3 Гц, CH2), 67.8 (d, 2JC-P=5.2 Гц, CH2), 68.0 (CH2), 68.1 (CH2), 72.0 (CH), 72.1 (CH), 86.7 (d, 3JC-P=8.1 Гц, CH), 86.8 (d, 3JC-P=8.1 Гц, CH), 86.9 (CH), 87.0 (CH), 116.2 (d, 3JC-P=3.3 Гц, CH), 116.5 (d, 3JC-P=3.5 Гц, CH), 122.6 (CH), 125.3 (CH), 125.4 (CH), 125.6 (CH), 125.7 (CH), 126.2 (CH), 126.5 (CH), 126.6 (CH), 127.6 (CH). 127.7 (CH), 127.8 (C), 127.9 (C), 128.0 (CH), 128.1 (CH), 128.9 (CH), 129.0 (CH), 129.4 (CH), 129.5 (CH), 129.6 (CH), 129.7 (CH), 136.2 (C), 137.1 (C), 137.2 (C), 141.6 (d. 1JC-F=233.8 Гц, С), 141.7 (d, 1JC-F=233.9 Гц, С), 147.8 (d, 2JC-P=7.7 Гц, C), 147.9 (d, 2JC-P=7.4 Гц, C), 150.5 (d, 4JC-F=4.0 Гц, C), 159.3 (d, 2JC-F=26.1 Гц, С), 174.6 (d, 3JC-P=5.0 Гц, С), 174.9 (d, 3JC-Р=4.3 Гц, С), m/z (ES) 636 (МН+ 100%). В результате обратнофазовой ВЭЖХ с использованием для элюирования H2O/МеОН (100/0-0/100, в течение 45 мин) получали два пика, соответствующие диастереоизомерам с tR=34,23 мин и tR=34,59 мин. Вычислено для C29H29FN3O9P: С, 56.77; Н, 4.76; N, 6.85. Найдено: С, 56.57; Н, 5.06; N,6.72.
Радиоактивные пиримидин дезоксинуклеозиды
[5-3H]dCyd (радиоактивность: 22 Ки/ммоль) и [5-3H]dUrd (радиоактивность: 15,9 Ки/ммоль) получали от Moravek Biochemicals Inc. (Brea, CA).
Стандартная методика D: синтез фосфорамидатов (NMI-методика)
К перемешиваемому раствору 5-F-dUrd (1,0 экв.) в безводном ТГФ, добавляли по каплям в атмосфере Ar подходящий фосфорхлоридат (3,0 экв.), растворенный в безводном ТГФ. К данной реакционной смеси при -78°C добавляли по каплям в течение 5 мин NMI (5,0 экв.). Через 15 мин реакционную смесь оставляли нагреваться до комнатной температуры и перемешивали в течение ночи. Растворитель удаляли под вакуумом, и остаток перерастворяли в ДХМ и три раза промывали 0,5 М раствором HCl. Органический слой сушили над MgSO4, фильтровали, упаривали досуха и очищали путем колоночной хроматографии, используя для элюирования градиент ДХМ/МеОН (99:1-97:3-95:5).
Стандартная методика E: синтез фосфорамидатов (tBuMgCl-методика)
К перемешиваемому раствору 5-FdUrd (1,0 экв.) в безводном ТГФ, добавляли по каплям в атмосфере Ar tBuMgCl (1,1 мольэкв. 1 М раствора в ТГФ), затем (через 30 мин) добавляли подходящий фосфорхлоридат (2,0 мольэкв.), растворенный в безводном ТГФ. Полученную реакционную смесь перемешивали в течение ночи при комнатной температуре. Растворитель удаляли при пониженном давлении, и остаток очищали путем колоночной хроматографии, используя для элюирования градиент ДХМ/МеОН (99:1-97:3-95:5).
5-Фтор-2'-дезоксиуридин-5'-O-[фенил(бензокси-L-аланинил)]фосфат (CPF381)
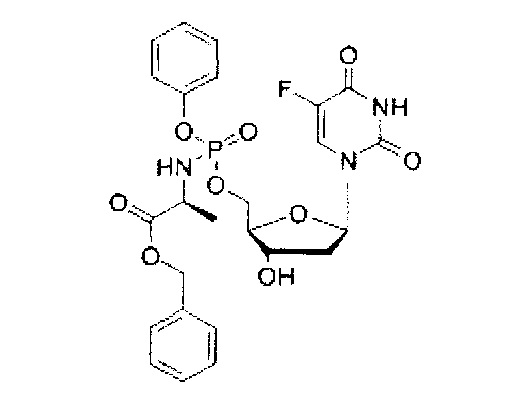
Данный фосфорамидат получали с использованием 5-фтор-2'-дезоксиуридина (0,40 г, 1,62 ммоль), хлорида трет-бутилмагния (tBuMgCl) в тетрагидрофуране (1,0 М, 2,43 мл, 2,43 ммоль) и фенил(бензокси-L-аланинил) фосфорхлоридата (1,08 г, 3,20 ммоль) в соответствии с общей методикой Е. В результате очистки путем колоночной хроматографии на силикагеле с использованием для элюирования градиента CH2Cl2 (конечное соотношение растворителей: CH2Cl2:MeOH=95:5) получали указанное в заголовке соединение в виде бесцветного твердого вещества (71,0 мг, 8%) [Rf=0,35 (CH2Cl2:MeOH=95:5)], (Найдено: MNa+, 586, 1360. C25H27N3O9NaPF. Вычислено: [MNa+], 586,1367); 31Р ЯМР (202 МГц, MeOD): ™P 3.74, 4.14; 19F ЯМР (470 МГц, MeOD): ™F -167.57, -167.46; 1Н ЯМР (500 МГц, MeOD): ™H 1.35 (d, 3Н, J=7.4 Гц, СНСН3, один диастереоизомер), 1.37 (d, 3Н, J=6.9 Гц, СНСН3, один диастереоизомер), 1.96-2.32 (m, 2Н, Н-2'), 3.95-4.08 (m, 2Н, СНСН3, Н-4'), 4.23-4.34 (m, 3Н, СН2ОР, Н-3'), 5.13 (br d, 1Н, J=12.3 Гц, OCHHPh), 5.16 (br d, 1Н, J=12.3 Гц, OCHHPh, один диастереоизомер), 5.17 (br d, 1H, J=12.2 Гц, OCHHPh, один диастереоизомер), 6.16-6.22 (m, 1H, Н-1'), 7.17-7.25 (m, 3H, ArH), 7.26-7.40 (m, 7H, ArH), 7.81-7.85 (m, 1H, H-6); 13C ЯМР (125 МГц, MeOD): ™C 20.2 (d, 3JC-P=7.5 Гц, CH3), 20.4 (d, 3JC-P=6.2 Гц, CH3), 40.6 (CH2), 40.9 (CH2), 51.6 (CH), 51.8 (CH), 67.5 (d, 2JC-P=5.3 Гц, CH2), 67.6 (d, 2JC-P=5.5 Гц, CH2), 68.0 (CH2), 71.8 (CH), 71.9 (CH), 86.6 (d, 3JC-P=8.0 Гц, CH), 86.8 (d, 3JC-P=8.3 Гц, CH), 86.9 (CH), 87.0 (CH), 121.4 (d, 3JC-P=5.1 Гц, CH), 121.5 (d, 3JC-P=5.6 Гц, CH), 125.5 (d, 5JC-P=3.2 Гц, CH), 125.8 (d, 5JC-P=3.2 Гц, CH), 126.3 (CH), 129.0 (CHx2), 129.3 (CHx2), 129.6 (CHx2), 130.8 (CHx2), 140.9 (C), 141.6 (d, 1JC-F=233.6 Гц, С), 141.7 (d, 1JC-F=233.6 Гц, С), 150.7 (d, 4JC-F=5.7 Гц, С), 152.1 (d, 2JC-F=6.5 Гц, С), 159.2 (d, 2JC-F=26.3 Гц, С), 174.6 (d, 3JC-P=4.9 Гц, С), 174.7 (d, 3JC-P=4.9 Гц, С), m/z (ES) 586 (MNa+, 100%). В результате обратнофазовой ВЭЖХ с использованием для элюирования H2O/MeOH (100/0-0/100, в течение 45 мин, 1 мл/мин, λ=275 нм) получали один пик, соответствующий смеси диастереоизомеров: tR=25,08 мин (97%).
5-Фтор-2'-дезоксиуридин-5'-O-[фенил(метокси-L-аланинил)]фосфат (CPF382) (контрольное соединение)
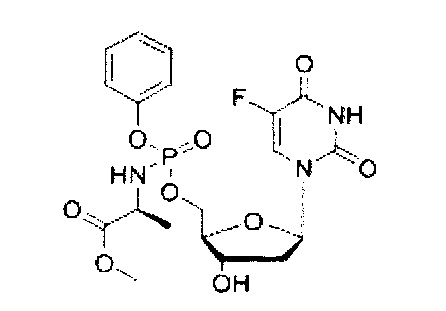
Данный фосфорамидат получали с использованием 5-фтор-2'-дезоксиуридина (0,25 г, 1,01 ммоль), N-метилимидазола (NMI) (0,40 мл, 5,07 ммоль) и фенил(метокси-L-аланинил) фосфорхлоридата (0,84 г, 3,04 ммоль) в соответствии с общей методикой D. В результате очистки путем колоночной хроматографии с использованием для элюирования градиента CH2Cl2 (конечное соотношение растворителей: CH2Cl2:МеОН=95:5) получали указанное в заголовке соединение в виде бесцветного твердого вещества (16,0 мг, 4%) [Rf=0,30 (CH2Cl2:MeOH=95:5)], (Найдено: MNa+, 510, 1045. C19H23N3O9NaPF. Вычислено: [MNa+], 510, 1054); 31Р ЯМР (202 МГц, MeOD): ™P 3.79, 4.09; 19F ЯМР (470 МГц, MeOD): ™F -167.78, -167.72; 1Н ЯМР (500 МГц, MeOD): ™H 1.34 (d, 3Н, J=7.1 Гц, СНСН3, один диастереоизомер), 1.36 (d, 3Н, J=7.1 Гц, СНСН3, один диастереоизомер), 2.02-2.16 (m, 1H, Н-2'), 2.25-2.34 (m, 1Н, Н-2'), 3.69 (s, 3Н, ОСН3, один диастереоизомер), 3.70 (s, 3Н, ОСН3, один диастереоизомер), 3.93-4.02 (m, 1Н, СНСН3), 4.08-4.13 (m, 1H, Н-4'), 4.27-4.45 (m, 3Н, СН2ОР, Н-3'), 6.20-6.29 (m, 1Н, Н-1'), 7.18-7.28 (m, 3Н, ArH), 7.35-7.40 (m, 2Н, ArH), 7.85 (d, 1Н, 3JH-F=6.4 Гц, Н-6); 13С ЯМР (125 МГц, MeOD): ™C 20.2 (d, 3JC-P=7.5 Гц, СН3), 20.5 (d, 3JC-P=6.7 Гц, СН3), 40.8 (СН2), 40.9 (СН2), 51.5 (СН3), 51.6 (СН3), 52.7 (СН), 52.8 (СН), 67.5 (d, 2JC-Р=5.5 Гц, СН2), 67.6 (d, 2JC-P=5.1 Гц, СН2), 72.0 (СН), 72.1 (СН), 86.7 (d, 3JC-P=8.2 Гц, СН), 86.8 (d, 3JC-P=8.2 Гц, СН), 86.9 (СН), 87.0 (СН), 121.2 (d, 3JC-P=4.5 Гц, СН), 121.4 (d, 3JC-P=4.7 Гц, СН), 125.6 (d, 5JC-P=2.9 Гц, СН), 125.9 (d, 5JC-P=2.9 Гц, СН), 126.2 (СН), 130.8 (СН), 130.9 (СН), 141.6 (d, 1JC-F=233.8 Гц, С), 141.7 (d, 1JC-F=233.9 Гц, С), 150.6 (d, 4JC-F=3.6 Гц, С), 152.1 (d, 2JC-P=6.8 Гц, С), 152.2 (d, 2JC-P=6.8 Гц, С), 159.4 (d, 2JC-F=26.0 Гц, С), 175.2 (d, 3JC-P=4.8 Гц, С), 175.5 (d, 3JC-P=3.7 Гц, С), m/z (ES) 510 (MNa+, 100%). В результате обратнофазовой ВЭЖХ с использованием для элюирования H2O/МеОН (100/0-0/100, в течение 45 мин, 1 мл/мин, λ=275 нм) получали два пика, соответствующие диастереоизомерам с tR=23,11 мин и tR=24,11 мин (74%:24%).
5-Фтор-2'-дезоксиуридин-5'-O-[фенил(этокси-L-аланинил)]фосфат (CPF383)
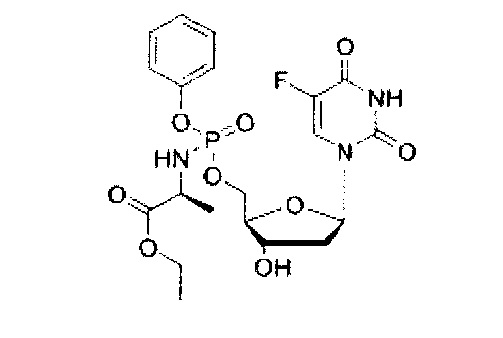
Данный фосфорамидат получали с использованием 5-фтор-2'-дезоксиуридина (0,10 г, 0,40 ммоль), N-метилимидазола (NMI) (0,16 мл, 2,03 ммоль) и фенил(этокси-L-аланинил) фосфорхлоридата (0,35 г, 1,21 ммоль) в соответствии с общей методикой D. В результате очистки путем колоночной хроматографии с использованием для элюирования градиента CH2Cl2 (конечное соотношение растворителей: CH2Cl2:МеОН=95:5) получали указанное в заголовке соединение в виде бесцветного твердого вещества (10,0 мг, 5%) [Rf=0,11 (CH2Cl2:МеОН=95:5)], (Найдено: MNa+ 524,1202. C20H25N3O9NaPF. Вычислено: [MNa+], 524,1210); 31Р ЯМР (202 МГц, MeOD): ™P 3.83, 4.11; 19F ЯМР (470 МГц, MeOD): ™F -167.67, -167.61; 1Н ЯМР (500 МГц, MeOD): ™H 1.25 (t, 3Н, J=7.1 Гц, СН2СН3, один диастереоизомер), 1.26 (t, 3Н, J=7.1 Гц, СН2СН3, один диастереоизомер), 1.34 (d, 3Н, J=7.2 Гц, СНСН3, один диастереоизомер), 1.36 (d, 3Н, J=7.2 Гц, СНСН3, один диастереоизомер), 2.02-2.15 (m, 1Н, Н-2'), 2.24-2.34 (m, 1Н, Н-2'), 3.90-4.00 (m, 1H, СНСН3,), 4.08-4.19 (m, 3Н, СН2СН3, Н-4'), 4.27-4.45 (m, 3Н, СН2ОР, Н-3'), 6.20-6.28 (m, 1Н, Н-1'), 7.18-7.28 (m, 3Н, ArH), 7.34-7.39 (m, 2Н. ArH), 7.85 (d, 1Н, 3JH-F=6.4 Гц, Н-6); 13С ЯМР (125 МГц, MeOD): ™C 14.4 (СН3), 15.4 (СН3), 20.3 (d, 3JC-P=7.6 Гц, СН3), 20.5 (d, 3JC-P=6.5 Гц, СН3), 40.8 (СН2), 40.9 (СН2), 51.6 (СН), 51.7 (СН), 62.4 (СН2), 62.5 (СН2), 67.5 (d, 2JC-P=5.4 Гц, СН2), 67.6 (d, 2JC-P=5.4 Гц, СН2), 72.0 (СН), 72.1 (СН), 86.7 (d, 3JC-P=8.1 Гц, СН), 86.8 (d, 3JC-P=8.3 Гц, СН), 86.9 (СН), 87.0 (СН), 121.3 (d, 3JC-P=4.8 Гц, СН), 121.4 (d, 3JC-P=4.6 Гц, СН), 125.6 (d, 5JC-P=4.6 Гц, СН), 125.8 (d, 5JC-P=4.8 Гц, СН), 126.3 (СН), 130.8 (СН), 130.9 (СН), 141.6 (d, 1JC-F=233.7 Гц, С), 141.8 (d, 1JC-F=233.8 Гц, С), 150.8 (br С), 152.0 (d, 2JC-P=7.1 Гц, С), 152.1 (d, 2JC-P=7.1 Гц, С), 159.6 (d, 2JC-F=26.0 Гц, С), 174.8 (d, 3JC-P=5.4 Гц, С), 175.1 (d, 3JC-P=4.4 Гц, С), m/z (ES) 524 (MNa+, 100%). В результате обратнофазовой ВЭЖХ с использованием для элюирования H2O/МеОН (100/0-0/100, в течение 45 мин, 1 мл/мин, λ=275 нм) получали два пика, соответствующие диастереоизомерам с tR=25,63 мин и tR=26.40 мин (71%:27%).
5-Фтор-2'дезоксиуридин-5'-O-[фенил(изопропокси-L-аланинил)]фосфат (CPF384)
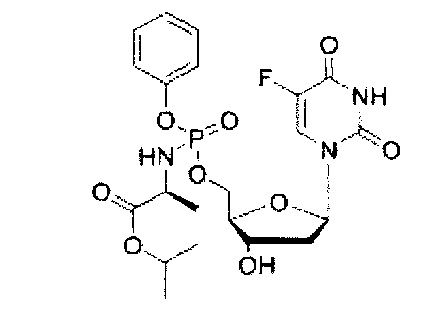
Данный фосфорамидат получали с использованием 5-фтор-2'-дезоксиуридина (0,25 г, 1,01 ммоль). N-метилимидазола (NMI) (0,40 мл, 5,07 ммоль) и фенил(изопропокси-L-аланинил) фосфорхлоридата (0,93 г, 3,04 ммоль) в соответствии с общей методикой D. В результате очистки путем колоночной хроматографии с использованием для элюирования градиента CH2Cl2 (конечное соотношение растворителей: CH2Cl2:МеОН=95:5) получали указанное в заголовке соединение в виде бесцветного твердого вещества (31,0 мг, 6%) [Rf=0,21 (CH2Cl2:МеОН=95:5)], (Найдено: MNa+, 538, 1370. C21H27N3O9NaPF. Вычислено: [MNa+], 538,1367); 31Р ЯМР (202 МГц, MeOD): ™P 3.87, 4.13; 19F ЯМР (470 МГц, MeOD): ™F -167.64, -167.56; 1Н ЯМР (500 МГц, MeOD): ™H 1.22-1.26 (m, 6Н, СН(СН3)2), 1.33 (d, 3Н, J=7.1 Гц, СНСН3, один диастереоизомер), 1.35 (d, 3Н, J=7.1 Гц, СНСН3, один диастереоизомер), 2.00-2.15 (m, 1Н, Н-2’), 2.23-2.34 (m, 1Н, Н-2’), 3.88-3.96 (m, 1Н, СНСН3), 4.08-4.14 (m, 1Н, Н-4’), 4.27-4.45 (m, 3Н, СН2ОР, Н-3’), 4.98 (гепт, 1Н, J=6.1 Гц, СН(СН3)2), 6.20-6.29 (m, 1Н, Н-1’), 7.17-7.29 (m, 3Н, Ar-H), 7.34-7.40 (m, 2Н, Ar-Н), 7.84 (d, 1Н, 3JH-F=6.4 Гц, Н-6); 13С ЯМР (125 МГц, MeOD): ™C 20.3 (d, 3JC-P=7.6 Гц, СН3), 20.5 (d, 3JC-P=6.4 Гц, СН3), 21.9 (СН3×2), 22.0 (СН3×2), 40.8 (СН2), 40.9 (СН2), 51.7 (СН), 51.8 (СН), 67.5 (d, 2JC-P=5.4 Гц, СН2), 67.6 (d, 2JC-P=5.2 Гц, СН2), 70.2 (СН), 70.3 (СН), 72.0 (СН), 72.1 (СН), 86.6 (d, 3JC-P=8.2 Гц, СН), 86.8 (d, 3JC-P=8.2 Гц, СН), 86.9 (СН), 87.0 (СН), 121.2 (d, 3JC-P=4.7 Гц, СН), 121.4 (d, 3JC-P=4.9 Гц, СН), 125.6 (d, 5JC-P=7.1 Гц, СН), 125.9 (d, 5JC-P=7.1 Гц, СН), 126.3 (СН), 130.8 (СН), 130.9 (СН), 141.8 (d, 1JC-F=234.5 Гц, С), 141.9 (d, 1JC-F=234.4 Гц, С), 150.7 (d, 4JC-F=3.7 Гц, С), 152.0 (d, 3JC-P=6.2 Гц, С), 152.1 (d, 3JC-P=6.2 Гц, С), 159.3 (d, 2JC-F=26.3 Гц, С), 159.4 (d, 2JC-F=26.0 Гц, С), 174.3 (d, 3JC-P=5.6 Гц, С), 174.6 (d, 3JC-P=4.6 Гц, С), m/z (ES) 538 (MNa+ 100%). В результате обратнофазовой ВЭЖХ с использованием для элюирования H2O/МеОН (100/0-0/100, в течение 45 мин, 1 мл/мин, λ=275 нм) получали два пика, соответствующие диастереоизомерам с tR=28,93 мин и tR 29,45 мин (44%:52%).
5-Фтор-2'дезоксиуридин-5'-O-[фенил(циклогексокси-L-аланинил)]фосфат (CPF508
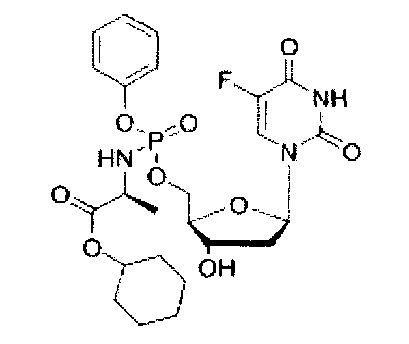
Данный фосфорамидат получали с использованием 5-фтор-2'-дезоксиуридина (0,30 г, 1,21 ммоль), N-метилимидазола (NMI) (0,48 мл, 6,09 ммоль) и фенил(циклогексокси-L-аланинил) фосфорхлоридата (1,026 г, 3,65 ммоль) в соответствии с общей методикой D. В результате очистки путем колоночной хроматографии с использованием для элюирования градиента CH2Cl2 (конечное соотношение растворителей: CH2Cl2:МеОН=95:5) получали указанное в заголовке соединение в виде бесцветного твердого вещества (6,7 мг, 3%) [Rf=0,45 (CH2Cl2:МеОН=95:5)]; (Найдено: MNa+, 565,48. C24H31N3O9NaPF. Вычислено: [MNa+], 565,49); 31Р ЯМР (202 МГц, MeOD): тмР 3.86, 4.15; 19F ЯМР (470 МГц, MeOD): ™F -167.68, -167.62; 1Н ЯМР (500 МГц, MeOD): тмН 1.26-1.40 (m, 3Н, СНСН3), 1.41-1.50 (m, 4Н, СН(СН2)5), 1.52-1.61 (m, 1Н, СН(СН2)5), 1.70-1.88 (m, 5Н, СН(СН2)5), 2.00-2.14 (m, 1Н, Н-2'), 2.23-2.34 (m, 1Н, Н-2'), 3.90-3.98 (m, 1Н, СНСН3), 4.07-4.14 (m, 1Н, Н-4'), 4.29-4.39 (m, 2Н, СН2ОР), 4.40-4.45 (m, 1Н, Н-3'), 4.72-4.78 (m, 1Н, СН(СН2)5), 6.20-6.28 (m, 1Н, Н-1'), 7.18-7.29 (m, 3Н, ArH), 7.34-7.39 (m, 2Н, ArH), 7.85 (d, 1Н, 3JH-F=6.6 Гц, Н-6); 13С ЯМР (125 МГц, MeOD): ™C 20.3 (d, 3JC-P=7.3 Гц, СН3), 20.6 (d, 3JC-P=6.5 Гц, СН3), 24.6 (СН2), 26.4 (СН2), 32.3 (СН2), 32.4 (СН2), 40.9 (СН2), 51.7 (СН), 51.9 (СН), 67.5 (d, 2JC-P=5.3 Гц, СН2), 67.7 (d, 2JC-P=5.3 Гц, СН2), 72.0 (СН), 72.1 (СН), 74.9 (СН), 86.6 (d, 3JC-P=8.5 Гц, СН), 86.8 (d, 3JC-P=8.5 Гц, СН), 86.9 (СН), 87.0 (СН). 121.3 (СН), 121.4 (СН), 121.5 (СН), 121.6 (СН), 125.6 (СН), 125.7 (СН), 125.8 (СН), 125.9 (СН), 126.3 (СН), 130.1 (СН), 141.5 (d, 1JC-F=234.0 Гц, С), 150.7 (d, 4JC-P=4.0 Гц, С), 152.0 (d, 2JC-P=7.2 Гц, С), 152.1 (d, 2JC-P=7.2 Гц, С), 159.4 (d, 2JC-F=26.3 Гц, С), 174.3 (d, 3JC-P=4.6 Гц, С), 174.5 (d, 3JC-P=4.3 Гц, С); m/z (ES) 565 (MNa+, 100%). В результате обратнофазовой ВЭЖХ с использованием для элюирования H2O/МеОН (100/0-0/100, в течение 45 мин, 1 мл/мин, λ=275 нм) получали два пика, соответствующие диастереоизомерам с tR=30,00 мин и tR=30,45 мин (33%:65%).
5-Фтор-2'дезоксиуридин-5'-O-[пара-нитро-фенил(этокси-L-аланинил)]фосфат (CPF430)
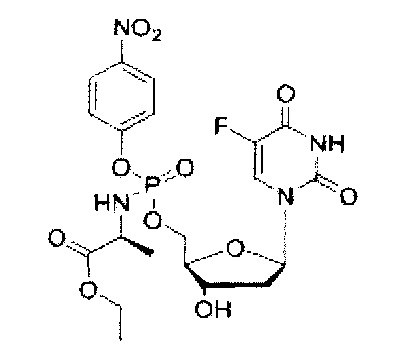
Данный фосфорамидат получали с использованием 5-фтор-2'-дезоксиуридина (0,25 г, 1,01 ммоль), N-метилимидазола (NMI) (0,40 мл, 5,07 ммоль) и пара-нитро-фенил(этокси-L-аланинил) фосфорхлоридата (1,02 г, 3,04 ммоль) в соответствии с общей методикой D. В результате очистки путем колоночной хроматографии с использованием для элюирования градиента CH2Cl2 (конечное соотношение растворителей: CH2Cl2:МеОН=95:5) получали указанное в заголовке соединение в виде бесцветного твердого вещества (77,0 мг, 14%) [Rf=0,24 (CH2Cl2:МеОН =95:5)], (Найдено: MNa+ 569,1066. C20H24N4O11NaPF. Вычислено: [MNa+], 569,1061); 31Р ЯМР (202 МГц, MeOD): ™P 3.63, 3.67; 19F ЯМР (470 МГц, MeOD): ™F -167.89, -167.82; 1Н ЯМР (500 МГц, MeOD): ™H 1.24 (t, 3Н, J=7.0 Гц, СН2СН3), 1.25 (t, 3Н, J=7.0 Гц, СН2СН3), 1.36-1.40 (m, 3Н, СНСН3), 2.16-2.25 (m, 1Н, Н-2 CH2'), 2.30-2.38 (m, 1Н, Н-2'), 3.95-4.00 (m, 1Н, СНСН3), 4.09-4.19 (m, 3Н, СН2СН3, Н-4'), 4.32-4.48 (m, 3Н, СН2ОР, Н-3'), 6.21-6.29 (m, 1Н, Н-1'), 7.46 (d, 1Н, J=8.7 Гц, ArH), 7.49 (d, 1Н, J=8.7 Гц, ArH), 7.85 (d, 1Н, 3JH-F=6.6 Гц, Н-6), 7.87 (d, 1Н, 3JH-F=6.6 Гц, Н-6), 8.29 (d, 2Н, J=8.7 Гц, ArH); 13С ЯМР (125 МГц, MeOD): тмС 14.5 (СН3), 14.6 (СН3), 20.3 (d, 3JC-P=7.5 Гц, СН3), 20.4 (d, 3JC-P=6.4 Гц, СН3), 40.8 (СН2), 51.6 (СН), 51.7 (СН), 62.5 (СН2), 67.8 (d, 2JC-P=5.5 Гц, СН2), 68.0 (d, 2JC-P=5.2 Гц, СН2), 71.8 (СН×2), 86.4 (СН), 86.5 (СН), 87.0 (d, 3JC-P=7.5 Гц, СН), 122.1 (d, 3JC-P=5.2 Гц, СН), 122.5 (d, 3JC-P=5.0 Гц, СН), 125.7 (СН), 126.0 (СН), 126.6 (СН), 141.3 (d, 1JC-F=233.6 Гц, С), 141.5 (d, 1JC-F=233.7 Гц, С), 146.2 (С), 150.6 (d, 4JC-P=4.6 Гц, С), 156.9 (d, 2JC-P=2.6 Гц, С), 157.0 (d, 2JC-P=2.6 Гц, С), 159.3 (d, 2JC-F=26.3 Гц, С), 174.6 (d, 3JC-P=4.6 Гц, С), 174.9 (d, 3JC-P=3.7 Гц, С), m/z (ES) 569 (MNa+, 100%). В результате обратнофазовой ВЭЖХ с использованием для элюирования H2O/MeOH (100/0-0/100, в течение 45 мин, 1 мл/мин, λ=275 нм) получали два пика, соответствующие диастереоизомерам с tR=31,63 мин и tR=31,89 мин (11%:85%).
5-Фтор-2'дезоксиуридин-5'-O-[1-нафтил(бензокси-L-аланинил)]фосфат(CPF373)
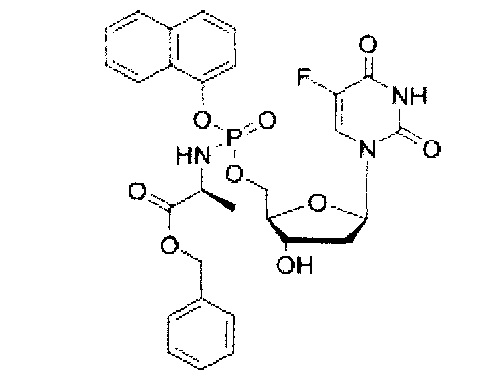
Данный фосфорамидат получали с использованием 5-фтор-2'-дезоксиуридина (0,25 г, 1,01 ммоль), N-метилимидазола (NMI) (0,40 мл, 5,07 ммоль) и 1-нафтил(бензокси-L-аланинил) фосфорхлоридата (0,82 г, 3,04 ммоль) в соответствии с общей методикой D. В результате очистки путем колоночной хроматографии с использованием для элюирования градиента CH2Cl2 (конечное соотношение растворителей: CH2Cl2:МеОН=95:5) получали указанное в заголовке соединение в виде бесцветного твердого вещества (47,0 мг, 8%) [Rf=0,19 (CH2Cl2:МеОН=95:5)], (Найдено: MNa+, 636,1520. C29H29N3O9NaPF. Вычислено: [MNa+], 636,1523); 31Р ЯМР (202 МГц, MeOD): ™P 4.24, 4.59; 19F ЯМР (470 МГц, MeOD): ™F -167.36, -167.18; 1Н ЯМР (500 МГц, MeOD): ™H 1.34-1.38 (m, 3Н, СНСН3), 1.67-1.79 (m, 1Н, Н-2'), 2.08-2.17 (m, 1Н, Н-2'), 4.03-4.15 (m, 2Н, СНСН3, Н-4'), 4.24-4.36 (m, 3Н, СН2ОР, Н-3'), 5.08 (d, 1Н, J=12.0 Гц, OCHHPh), 5.13 (d, 1Н, J=12.0 Гц, OCHHPh), 6.09-6.16 (m, 1H, Н-1'), 7.27-7.45 (m, 6H, ArH), 7.47-7.55 (m, 3H, ArH), 7.67-7.72 (m, 2H, ArH, H-6), 7.86-7.90 (m, 1H, ArH), 8.12-8.18 (m, 1H, ArH); 13С ЯМР (125 МГц, MeOD): ™C 20.3 (d, 3JC-P=7.6 Гц, CH3), 20.5 (d, 3JC-P=6.5 Гц, CH3), 40.8 (CH2), 40.9 (CH2), 51.8 (CH), 51.9 (CH), 67.6 (d, 2JC-P=5.3 Гц, CH2), 67.8 (d, 2JC-P=5.2 Гц, CH2), 68.0 (CH2), 68.1 (CH2), 72.0 (CH), 72.1 (CH), 86.7 (d, 3JC-P=8.1 Гц, CH), 86.8 (d, 3JC-P=8.1 Гц, CH), 86.9 (CH), 87.0 (CH), 116.2 (d, 3JC-P=3.3 Гц, CH), 116.5 (d, 3JC-P=3.5 Гц, CH), 122.6 (CH), 125.3 (CH), 125.4 (CH), 125.6 (CH), 125.7 (CH), 126.2 (CH), 126.5 (CH), 126.6 (CH), 127.6 (CH), 127.7 (CH), 127.8 (C), 127.9 (C), 128.0 (CH), 128.1 (CH), 128.9 (CH), 129.0 (CH), 129.4 (CH), 129.5 (CH), 129.6 (CH), 129.7 (CH), 136.2 (C), 137.1 (C), 137.2 (C), 141.6 (d, 1JC-F=233.8 Гц, С), 141.7 (d, 1JC-F=233.9 Гц, С), 147.8 (d, 2JC-P=7.7 Гц, С), 147.9 (d, 2JC-P=7.4 Гц, С), 150.5 (d, 4JC-F=4.0 Гц, С), 159.3 (d, 2JC-F=26.1 Гц, С), 174.6 (d, 3JC-P=5.0 Гц, С), 174.9 (d, 3JC-P=4.3 Гц, С), m/z (ES) 636 (MNa+, 100%). В результате обратнофазовой ВЭЖХ с использованием для элюирования H2O/MeOH (100/0-0/100, в течение 45 мин, 1 мл/мин, λ=275 нм) получали два пика, соответствующие диастереоизомерам с tR=34,23 мин и tR=34,59 мин (23%:76%).
5-Фтор-2'дезоксиуридин-5'-O-[1-нафтил(метокси-L-аланинил)]фосфат (CPF385)
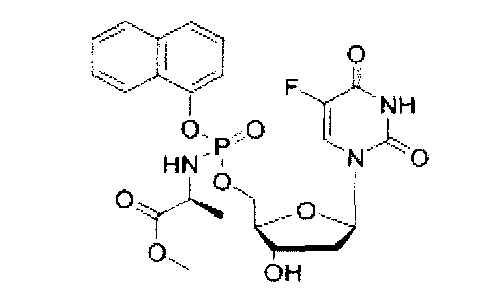
Данный фосфорамидат получали с использованием 5-фтор-2'-дезоксиуридина (0,25 г, 1,01 ммоль), N-метилимидазола (NMI) (0,40 мл, 5,07 ммоль) и 1-нафтил(метокси-L-аланинил)фосфорхлоридата (0,99 г, 3,04 ммоль) в соответствии с общей методикой D. В результате очистки путем колоночной хроматографии с использованием для элюирования градиента CH2Cl2 (конечное соотношение растворителей: CH2Cl2:МеОН=95:5) получали указанное в заголовке соединение в виде бесцветного твердого вещества (7,0 мг, 1%) [Rf=0,23 (CH2Cl2:МеОН=95:5)], (Найдено: MNa+, 560,1198. C23H25N3O9NaPF. Вычислено: [MNa+], 560,1210); 31Р ЯМР (202 МГц, MeOD): ™P 4.31, 4.56; 19F ЯМР (470 МГц, MeOD): ™F -167.51, -167.37; 1Н ЯМР (500 МГц, MeOD): ™H 1.34 (d, 3Н, J=6.7 Гц, СНСН3, один диастереоизомер), 1.36 (d, 3H, J=6.7 Гц, СНСН3, один диастереоизомер), 1.76-1.87 (m, 1Н, Н-2'), 2.12-2.22 (m, 1H, Н-2'), 3.64 (s, 3H, OCH3, один диастереоизомер), 3.65 (s, 3H, OCH3, один диастереоизомер), 4.03-4.13 (m, 2Н, СНСН3, Н-4'), 4.30-4.38 (m, 2Н, СН2ОР), 4.41 (dd, 1Н, J=2.5 Гц, J=5.8 Гц, H-3'), 6.12-6.19 (m, 1H, H-1'), 7.41-7.46 (m, 1H, ArH), 7.50-7.58 (m, 3H, ArH), 7.70-7.76 (m, 2H, Н-6, ArH), 7.87-7.91 (m, 1H, ArH), 8.15-8.20 (m, 1H, ArH); 13C ЯМР (125 МГц, MeOD): ™C 20.3 (d, JC-P=7.1 Гц, СН3), 20.4 (d, 3JC-P=6.5 Гц, CH3), 40.7 (CH2), 40.8 (CH2), 51.6 (CH3), 51.7 (CH3), 52.7 (CH), 52.8 (CH), 67.8 (d, 2JC-P=5.7 Гц, СН2), 67.5 (d, 2JC-P=5.7 Гц, СН2), 72.0 (CH), 72.1 (CH), 86.7 (d, 3JC-P=7.9 Гц, CH), 86.9 (d, 3JC-P=8.5 Гц, CH), 86.9 (CH), 87.0 (CH), 116.2 (d, 3JC-P=3.1 Гц, CH), 116.5 (d, 3JC-P=3.5 Гц,CH), 122.5 (СН), 122.6 (СН), 125.4 (СН), 125.5 (СН), 125.6 (СН), 125.7 (СН), 126.1 (СН), 126.2 (СН), 126.5 (СН), 126.6 (СН), 127.6 (СН), 127.7 (С×2), 127.8 (CH), 127.9 (СН), 128.9 (СН), 129.0 (СН), 136.3 (C), 141.6 (d, 1JC-F=233.4 Гц, С), 141.7 (d, 1JC-F=234.1 Гц, С), 147.8 (d, 2JC-P=7.9 Гц, C), 148.0 (d, 2JC-P=7.2 Гц, C), 150.6 (C), 159.4 (d, 2JC-F=27.0 Гц, С), 175.2 (d, 3JC-P=3.9 Гц, C), 175.5 (d, 3JC-P=3.9 Гц, C), m/z (ES) 560 (MNa+, 100%). В результате обратнофазовой ВЭЖХ с использованием для элюирования H2O/MeOH (100/0-0/100, в течение 45 мин, 1 мл/мин, λ=275 нм) получали два пика, соответствующие диастереоизомерам с tR=28,45 мин и tR=28,85 мин (73%:25%).
5-Фтор-2'дезоксиуридин-5'-O-[1-нафтил(этокси-L-аланинил)]фосфат (CPF386)
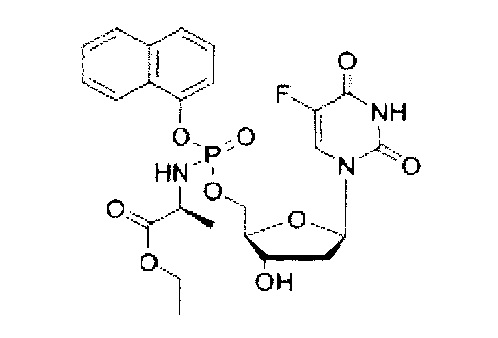
Данный фосфорамидат получали с использованием 5-фтор-2'-дезоксиуридина (0,25 г, 1,01 ммоль), N-метилимидазола (NMI) (0,40 мл, 5,07 ммоль) и 1-нафтил(этокси-L-аланинил)фосфорхлоридата (1,04 г, 3,04 ммоль) в соответствии с общей методикой D. В результате очистки путем колоночной хроматографии с использованием для элюирования градиента CH2Cl2 (конечное соотношение растворителей: CH2Cl2:MeOH=95:5) получали указанное в заголовке соединение в виде бесцветного твердого вещества (47,0 мг, 4%) [Rf=0,25 (CH2Cl2:MeOH=95:5)], (Найдено: MNa+ 574,1360. C24H27N3O9NaPF. Вычислено: [MNa+], 574,1367); 31Р ЯМР (202 МГц, MeOD): тмР 4.34, 4.55; 19F ЯМР (470 МГц, MeOD): ™F -167.31, -167.16; 1H ЯМР (500 МГц, MeOD): ™H 1.20 (t, 3H, J=7.0 Гц, CH2CH3, один диастереоизомер), 1.21 (t, 3H, J=7.0 Гц, СН2СН3, один диастереоизомер), 1.33-1.37 (m, 3H, СНСН3), 1.73-1.86 (m, 1H, Н-2'), 2.12-2.21 (m, 1H, H-2'), 4.01-4.07 (m, 1H, СНСН3), 4.08-4.13 (m, 3H, СН2СН3, H-4'), 4.31-4.43 (m, 3H, СН2ОР, Н-3'). 6.11-6.19 (m, 1H, H-1'), 7.39-7.46 (m, 1H, ArH), 7.50-7.57 (m, 3H, ArH), 7.68-7.75 (m, 2H, ArH, H-6), 7.86-7.91 (m, 1H, ArH), 8.15-8.20 (m, 1H, ArH); 13C ЯМР (125 МГц, MeOD): ™C 14.4 (CH3), 20.3 (d, 3JC-P=7.4 Гц, CH3), 20.5 (d, 3JC-P=6.2 Гц, CH3), 40.8 (CH2), 40.9 (CH2), 51.8 (CH), 51.9 (CH), 62.4 (CH2), 62.5 (CH2), 67.8 (d, 2JC-P=4.6 Гц, CH2), 67.9 (d, 3JC-P=4.6 Гц, CH2), 72.0 (CH), 72.1 (CH), 86.7 (d, 3JC-P=8.4 Гц, CH), 86.8 (d, 3JC-P=8.4 Гц, CH), 86.9 (CH), 87.0 (CH), 116.1 (d, 3JC-P=3.5 Гц, CH), 116.5 (d, 3JC-P=3.5 Гц, CH), 122.6 (CH), 125.4 (CH), 125.5 (CH), 125.7 (CH), 125.8 (CH), 126.1 (CH), 126.2 (CH), 126.5 (CH), 126.6 (CH), 127.5 (CH), 127.6 (C), 127.7 (C), 127.8 (CH), 127.9 (CH), 128.9 (CH), 129.0 (CH), 136.3 (C), 141.6 (d, 1JC-F=233.3 Гц, С), 141.7 (d, 1JC-F=233.4 Гц, C), 147.8 (d, 2JC-P=6.9 Гц, C), 148.0 (d, 2JC-P=6.9 Гц, C), 150.6 (C), 159.3 (d, 2JC-F=26.3 Гц, C), 174.8 (d, 3JC-P=4.8 Гц, C), 175.1 (d, 3JC-P=4.0 Гц, С); m/z (ES) 574 (MNa+, 100%). В результате обратнофазовой ВЭЖХ с использованием для элюирования H2O/MeOH (100/0-0/100, в течение 45 мин, 1 мл/мин, λ=275 нм) получали два пика, соответствующие диастереоизомерам с tR=30,77 мин и tR=31,20 мин (51%:48%).
5-Фтор-2'дезоксиуридин-5'-O-[1-нафтил(изопропокси-L-аланинил)]фосфат (CPF387)
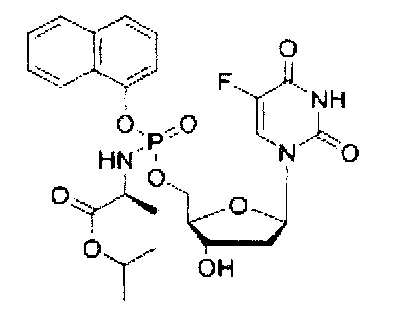
Данный фосфорамидат получали с использованием 5-фтор-2'-дезоксиуридина (0,10 г, 0,40 ммоль), хлорида трет-бутилмагния (tBuMgCl) в тетрагидрофуране (1,0 М, 0,61 мл, 0.61 ммоль) и 1-нафтил(изопропокси-L-аланинил)фосфорхлоридата (0,31 г, 0,89 ммоль) в соответствии с общей методикой E. В результате очистки путем колоночной хроматографии с использованием для элюирования градиента CH2Cl2 (конечное соотношение растворителей: CH2Cl2:MeOH=95:5) получали указанное в заголовке соединение в виде бесцветного твердого вещества (71,0 мг, 17%) [Rf=0,21 (CH2Cl2:MeOH=95:5)], (Найдено: MNa+, 588,1521. C25H29N3O9NaPF. Вычислено: [MNa+], 588,1523); 31Р ЯМР (202 МГц, MeOD): ™P 4.38, 4.58; 19F ЯМР (470 МГц, MeOD): ™F -167.43, -167.26; 1Н ЯМР (500 МГц, MeOD): ™H 1.19-1.23 (m, 6H, CH(CH3)2), 1.34-1.38 (m, 3H, СНСН3), 1.68-1.84 (m, 1H, H-2'), 2.09-2.20 (m, 1Н, Н-2'), 3.96-4.05 (m, 1H, СНСН3), 4.07-4.12 (m, 1H, H-4'), 4.29-4.38 (m, 2H, CH2OP), 4.39-4.42 (m, 1H, H-3'), 4.93-5.01 (m, 1H, CH(CH3)2), 5.10-6.18 (m, 1H, Н-1'), 7.40-7.46 (m, 1H, ArH), 7.50-7.57 (m, 3H, ArH), 7.70-7.75 (m, 2H, H-6, ArH), 7.87-7.92 (m, 1H, ArH), 8.16-8.20 (m, 1Н, ArH); 13С ЯМР (125 МГц, MeOD): ™C 20.3 (d, 3JC-Р=7.1 Гц, CH3), 20.5 (d, 3JC-Р=6.6 Гц, CH3), 21.8 (CH3), 21.9 (CH3), 22.0 (CH3), 22.1 (CH3), 40.8 (CH2), 40.9 (CH2), 51.9 (CH), 52.0 (CH), 67.8 (d, 2JC-P=4.5 Гц, CH2), 67.9 (d, 2JC-P=4.8 Гц, CH2), 70.2 (CH), 70.3 (CH), 72.0 (CH), 72.1 (CH), 86.6 (CH), 86.7 (CH), 86.9 (d, 3JC-P=8.6 Гц, CH), 87.0 (d, 3JC-P=8.6 Гц, CH), 116.2 (d, 3JC-P=2.5 Гц, CH), 116.5 (d, 3JC-P=2.7 Гц, CH), 122.6 (CH), 125.5 (CH), 125.7 (CH), 126.1 (CH), 126.2 (CH), 126.5 (CH), 127.5 (CH), 127.6 (C), 127.7 (C), 127.8 (CH), 127.9 (CH), 128.9 (CH), 129.0 (CH), 136.3 (C), 141.6 (d, 1JC-F=233.2 Гц, С), 141.7 (d, 1JC-F=233.4 Гц, C), 147.7 (d, 2JC-P=7.6 Гц, C), 147.9 (d, 2JC-P=7.7 Гц, С), 150.5 (С), 159.4 (d, 2JC-F=26.2 Гц, С), 174.4 (d, 3JC-P=5.0 Гц, C), 174.7 (d, 3JC-P=5.1 Гц, C); m/z (ES) 588 (MNa+, 100%). В результате обратнофазовой ВЭЖХ с использованием для элюирования H2O/МеОН (100/0-0/100, в течение 45 мин, 1 мл/мин, λ=275 нм) получали два пика, соответствующие диастереоизомерам с tR=32,20 мин и tR=32,80 мин (27%:69%).
5-Фтор-2'дезоксиуридин-5'-O-[1-нафтил(циклогексокси-L-аланинил)]фосфат (CPF509)
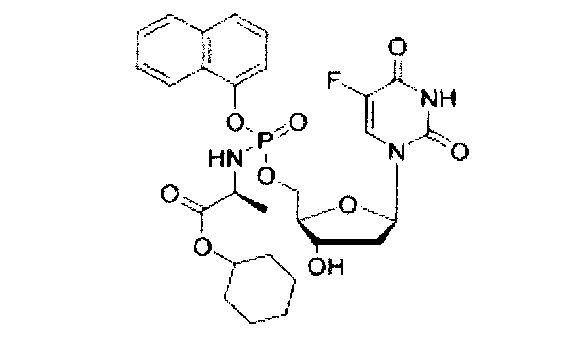
Данный фосфорамидат получали с использованием 5-фтор-2'-дезоксиуридина (0,30 г, 1,21 ммоль), N-метилимидазола (NMI) (0,48 мл, 6,09 ммоль) и фенил(циклогексокси-L-аланинил) фосфорхлоридата (1,45 г, 3,65 ммоль) в соответствии с общей методикой D. В результате очистки путем колоночной хроматографии с использованием для элюирования градиента CH2Cl2 (конечное соотношение растворителей: CH2Cl2:MeOH=95:5) получали указанное в заголовке соединение в виде бесцветного твердого вещества (6,7 мг, 3%) [Rf=0,47 (CH2Cl2:MeOH=95:5)]; (Найдено: 

5-Фтор-2'дезоксиуридин-5'-O-[фенил(бензокси-α,α-диметилглицин)]фосфат (CPF393)
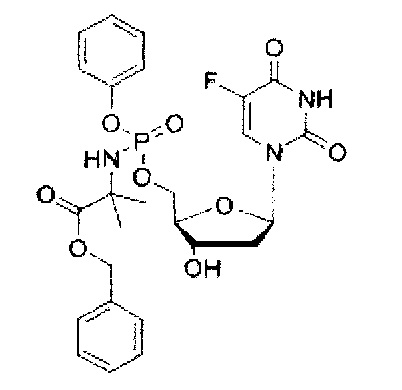
Данный фосфорамидат получали с использованием 5-фтор-2'-дезоксиуридина (0,40 г, 1,62 ммоль), хлорида трет-бутилмагния (tBuMgCl) в тетрагидрофуране (1,0 М, 2,43 мл, 2,43 ммоль) и фенил(бензокси-α,α-диметилглицин) фосфорхлоридата (1,17 г, 3,20 ммоль) в соответствии с общей методикой Е. В результате очистки путем колоночной хроматографии с использованием для элюирования градиента CH2Cl2 (конечное соотношение растворителей: CH2Cl2:MeOH=95:5) получали указанное в заголовке соединение в виде бесцветного твердого вещества (69,0 мг, 7%) [Rf=0,27 (CH2Cl2:MeOH=95:5)], (Найдено: MNa+ 600,1527. C26H29N3O9NaPF. Вычислено: [MNa+], 600,1523); 31Р ЯМР (202 МГц, MeOD): ™P 2.42, 2.47; 19F ЯМР (470 МГц, MeOD): ™F -167.80, -167.62; 1Н ЯМР (500 МГц, MeOD): ™H 1.51-1.60 (m, 6Н, C(CH3)2), 1.89-1.97 (m, 1H, Н-2', один диастереоизомер), 2.07-2.15 (m, 1H, H-2’, один диастереоизомер), 2.21 (ddd, 1Н, J=3.4 Гц, 5.9 Гц, 13.5 Гц, H-2', один диастереоизомер), 2.29 (ddd, 1H, J=3.2 Гц, 6.1 Гц, 13.5 Гц, H-2', один диастереоизомер), 4.00-4.07 (m, 1H, H-4'), 4.22-4.31 (m, 2H, СН2ОР), 4.32-4.36 (m, 1H, Н-3', один диастереоизомер), 4.37-4.41 (m, 1H, Н-3', один диастереоизомер), 5.08-5.18 (m, 2Н, OCH2Ph), 6.19-6.25 (m, 1Н, Н-1'), 7.20-7.26 (m, 3H, ArH), 7.27-7.39 (m, 7H, ArH). 7.74 (d, 3JH-F=6.4 Гц, H-6, один диастереоизомер), 7.80 (d, 3JH-F=6.4 Гц, H-6, один диастереоизомер); 13С ЯМР (125 МГц, MeOD): ™C 27.5 (СН3), 27.7 (d, 3JC-P=7.1 Гц, СН3), 27.8 (d, 3JC-P=7.1 Гц, СН3), 40.8 (СН2), 40.9 (СН2), 58.2 (С), 58.3 (С), 67.6 (d, 2JC-P=5.5 Гц, СН2), 67.7 (d, 2JC-P=5.5 Гц, СН2), 68.3 (СН2), 71.9 (СН), 72.0 (СН), 86.6 (d, 3JC-P=8.1 Гц, СН), 86.8 (d, 3JC-P=7.3 Гц, CH), 86.9 (CH), 121.4 (d, 3JC-P=4.8 Гц, СН), 121.6 (d, 3JC-P=4.5 Гц, СН), 125.6 (СН), 125.8 (СН), 125.9 (СН), 126.1 (СН), 126.2 (СН), 129.3 (СН), 129.4 (СН), 129.6 (СН), 130.7 (СН), 130.8 (СН), 137.2 (С), 137.3 (С), 141.8 (d, 1JC-F=233.7 Гц, С), 150.6 (С), 152.1 (d, 4JC-F=7.0 Гц, С), 152.1 (d, 4JC-F=7.6 Гц, С), 159.3 (d, 2JC-F=26.1 Гц, С), 159.4 (d, 2JC-F=26.1 Гц, С), 176.5 (d, 3JC-P=4.0 Гц, С), 176.6 (d, 3JC-P=3.8 Гц, С), m/z (ES) 600.1 (MNa+, 100%). В результате обратнофазовой ВЭЖХ с использованием для элюирования H2O/МеОН (100/0-0/100, в течение 35 мин, 1 мл/мин, λ=275 нм) получали один пик, соответствующий смеси диастереоизомеров с tR=17,71 (96%).
5-Фтор-2'дезоксиуридин-5'-O-[фенил(этокси-α,α-диметилглицин)]фосфат (CPF394)
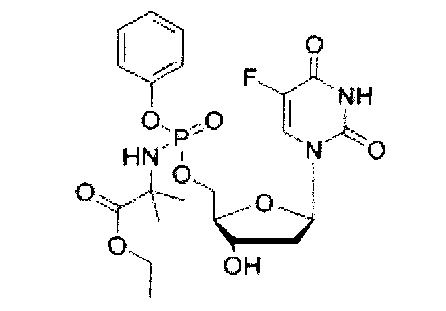
Данный фосфорамидат получали с использованием 5-фтор-2'-дезоксиуридина (0,20 г, 0,80 ммоль), N-метилимидазола (NMI) (0,31 мл, 4,0 ммоль) и фенил(этокси-α,α-диметилглицин) фосфорхлоридата (0,73 г, 2,40 ммоль) в соответствии с общей методикой D. В результате очистки путем колоночной хроматографии с использованием для элюирования градиента CH2Cl2 (конечное соотношение растворителей: CH2Cl2:MeOH=95:5) получали указанное в заголовке соединение в виде бесцветного твердого вещества (25,0 мг, 6%) [Rf=0,24 (CH2Cl2:MeOH=95:5)], (Найдено: MNa+, 538,1367. C21H27N3O9NaPF. Вычислено: [MNa+], 538,1367); 31Р ЯМР (202 МГц, MeOD): ™P 2.49, 2.52; 19F ЯМР (470 МГц, MeOD): ™F -167.62, -167.58; 1Н ЯМР (500 МГц, MeOD): ™H 1.24 (t, 3Н, J=7.1 Гц, СН2СН3, один диастереоизомер), 1.26 (t, 3Н, J=7.1 Гц, СН2СН3, один диастереоизомер), 1.44-1.54 (m, 6Н, С(СН3)2), 1.95-2.04 (m, 1Н, Н-2', один диастереоизомер), 2.13-2.21 (m, 1Н, Н-2', один диастереоизомер), 2.24 (ddd, 1Н, J=3.1 Гц, J=6.3 Гц, J=13.5 Гц, Н-2', один диастереоизомер), 2.31 (ddd, 1Н, J=3.2 Гц, J=6.1 Гц, J=13.7 Гц, Н-2', один диастереоизомер), 4.08-4.19 (m, 3H, СН2СН3, Н-4'), 4.33-4.49 (m, 3H, СН2ОР, Н-3'), 6.20-6.30 (m, 1Н, Н-1'), 7.23-7.28 (m, 3Н, ArH), 7.33-7.40 (m, 2Н, ArH), 7.80 (d, 3JH-F=6.4 Гц, Н-6, один диастереоизомер), 7.88 (d, 3JH-F=6.4 Гц, Н-6, один диастереоизомер); 13С ЯМР (125 МГц, MeOD): ™C 14.4 (CH3), 14.5 (CH3), 27.5 (d, 3JC-P=7.3 Гц, CH3), 27.7 (d, 3JC-P=7.6 Гц, CH3), 27.8 (d, 3JC-P=7.6 Гц, CH3), 40.8 (CH2), 40.9 (CH2), 58.1 (С), 62.6 (СН2), 62.7 (СН2), 67.6 (d, 2JC-P=6.7 Гц, СН2), 67.7 (d, 2JC-P=5.8 Гц, СН2), 71.9 (CH), 72.0 (СН), 86.6 (d, 3JC-P=8.1 Гц, СН), 86.8 (d, 3JC-P=7.6 Гц, СН), 86.9 (СН), 121.4 (d, 3JC-P=4.4 Гц, СН), 121.6 (d, 3JC-P=4.4 Гц, СН), 125.6 (СН), 125.8 (СН), 125.9 (СН), 126.1 (СН), 126.2 (СН), 130.7 (СН), 130.8 (СН), 130.9 (СН), 141.8 (d, 1JC-F=233.5 Гц, С), 150.6 (С), 150.7 (С), 152.2 (d, 4JC-F=7.3 Гц, С), 152.3 (d, 4JC-F=6.9 Гц, С), 159.2 (d, 2JC-F=20.3 Гц, С), 159.4 (d, 2JC-F=20.4 Гц, С), 176.6 (d, 3JC-P=4.2 Гц, С), 176.8 (d, 3JC-P=4.6 Гц, С), m/z (ES) 538.1 (MNa+, 100%). В результате обратнофазовой ВЭЖХ с использованием для элюирования H2O/МеОН (100/0-0/100, в течение 45 мин, 1 мл/мин, λ=275 нм) получали два пика, соответствующие диастереоизомерам с tR=18,76 мин и tR=20,44 мин (68%:30%).
5-Фтор-2'дезоксиуридин-5'-O-[1-нафтил(бензокси-α,α-диметилглицин)]фосфат (CPF395)
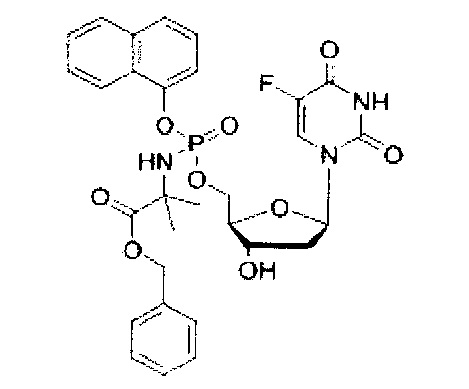
Данный фосфорамидат получали с использованием 5-фтор-2'-дезоксиуридина (0,40 г, 1,62 ммоль), N-метилимидазола (NMI) (0,64 мл, 8,0 ммоль) и 1-нафтил(бензокси-α,α-диметилглицин)фосфорхлоридата (2,00 г, 4,80 ммоль) в соответствии с общей методикой D. В результате очистки путем колоночной хроматографии с использованием для элюирования градиента CH2Cl2 (конечное соотношение растворителей: CH2Cl2:MeOH=95:5) получали указанное в заголовке соединение в виде бесцветного твердого вещества (16,4 мг, 6%) [Rf=0,15 (CH2Cl2:MeOH=95:5)], (Найдено: MNa+ 650,1678. C30H31N3O9NaPF. Вычислено: [MNa+], 650,1680); 31Р ЯМР (202 МГц, MeOD): ™P 2.87, 3.03; 19F ЯМР (470 МГц, MeOD): ™F -167.95, -167.13; 1Н ЯМР (500 МГц, MeOD): ™H 1.37-1.42 (m, 6Н, С(СН3)2), 1.61-1.69 (m, 1Н, Н-2', один диастереоизомер), 1.79-1.87 (m, 1Н, Н-2', один диастереоизомер), 2.06 (ddd, 1H, J=3.0 Гц, J=6.1 Гц, J=13.6 Гц, Н-2', один диастереоизомер), 2.15 (ddd, 1Н, J=3.2 Гц, J=5.9 Гц, J=13.7 Гц, Н-2', один диастереоизомер), 3.98-4.04 (m, 1Н, Н-4'), 4.19-4.35 (m, 3Н, СН2ОР, Н-3'), 5.09-5.13 (m, 1Н, OCHHPh), 5.18-5.19 (m, 1Н, OCHHPh), 6.05-6.15 (m, 1H, Н-1'), 7.28-7.40 (m, 7Н, ArH), 7.48-7.55 (m, 3Н, ArH), 7.62 (d, 3JH-F=6.4 Гц, Н-6, один диастереоизомер), 7.70 (d, 3JH-F=6.4 Гц, Н-6, один диастереоизомер), 7.86-7.90 (m, 1Н, ArH), 8.17-8.22 (m, 1Н, ArH); 13С ЯМР (125 МГц, MeOD): ™C 27.5 (d, 3JC-P=4.4 Гц, СН3), 27.9 (d, 3JC-P=7.3 Гц, СН3), 28.0 (d, 3JC-P=7.3 Гц, СН3), 40.7 (СН2), 40.8 (СН2), 65.2 (С), 67.8 (d, 2JC-P=6.5 Гц, СН2), 68.3 (СН2), 72.0 (СН), 72.1 (СН), 86.6 (d, 3JC-P=8.2 Гц, СН), 86.8 (d, 3JC-P=7.8 Гц, СН), 86.9 (СН), 116.3 (d, 3JC-P=3.2 Гц, СН), 116.7 (d, 3JC-P=2.9 Гц, CH), 122.8 (CH), 122.9 (CH), 125.4 (CH), 125.5 (CH), 125.6 (CH), 126.0 (CH), 126.1 (CH), 126.4 (CH), 126.5 (CH), 127.4 (CH), 127.5 (CH), 127.7 (CH), 127.8 (CH), 127.9 (C), 128.0 (CH), 128.9 (CH), 129.3 (CH), 129.4 (CH), 129.6 (CH), 136.2 (C), 137.3 (C), 141.8 (d, 1JC-F=234.4 Гц, С), 147.9 (d, 3JC-P=7.7 Гц, С), 148.0 (d, 3JC-P=8.2 Гц, С), 150.7 (d, 4JC-F=3.7 Гц, С), 159.5 (d, 2JC-F=25.8 Гц, С), 159.6 (d, 2JC-F=25.8 Гц, С), 176.5 (С), 176.6 (С), m/z (ES) 650.0 (MNa+, 100%). В результате обратнофазовой ВЭЖХ с использованием для элюирования H2O/МеОН (100/0-0/100, в течение 45 мин, 1 мл/мин, λ=275 нм) получали два пика, соответствующие диастереоизомерам с tR=20,80 мин и tR=21,00 мин (72%:24%).
5-Фтор-2'дезоксиуридин-5'-O-[1-нафтил(этокси-α,α-диметилглицин)]фосфат (CPF396)
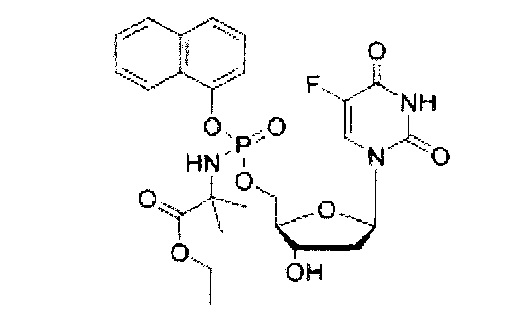
Данный фосфорамидат получали с использованием 5-фтор-2'-дезоксиуридина (0,40 г, 1,62 ммоль), хлорида трет-бутилмагния (tBuMgCl) в тетрагидрофуране (1,0 М, 2,43 мл, 2,43 ммоль) и 1-нафтил(этокси-α,α-диметилглицин)фосфорхлоридата (1,14 г, 3,20 ммоль) в соответствии с общей методикой E. В результате очистки путем колоночной хроматографии с использованием для элюирования градиента CH2Cl2 (конечное соотношение растворителей: CH2Cl2:MeOH=95:5) получали указанное в заголовке соединение в виде бесцветного твердого вещества (54,0 мг, 2%) [Rf=0,10 (CH2Cl2:MeOH=95:5)], (Найдено: MNa+, 588,1528. C25H29N3O9NaPF. Вычислено: [MNa+], 588,1523); 31Р ЯМР (202 МГц, MeOD): тмР 2.91, 3.03; 19F ЯМР (470 МГц, MeOD): ™F -167.38, -167.21; 1Н ЯМР (500 МГц, MeOD): ™H 1.24 (t, 3H, J=7.1 Гц, СН2СН3, один диастереоизомер), 1.25 (t, 3H, J=7.1 Гц, СН2СН3, один диастереоизомер), 1.50-1.55 (m, 6Н, С(СН3)2), 1.68-1.76 (m, 1Н, Н-2', один диастереоизомер), 1.87-1.94 (m, 1Н, Н-2', один диастереоизомер), 2.09 (ddd, 1Н, J=2.9 Гц, J=6.3 Гц, J=13.4 Гц, Н-2', один диастереоизомер), 2.19 (ddd, 1Н, J=3.0 Гц, J=6.3 Гц, J=13.8 Гц, Н-2', один диастереоизомер), 4.07-4.10 (m, 1Н, Н-4'), 4.16 (q, 2Н, J=7.1 Гц, СН2СН3), 4.36-4.41 (m, 3Н, СН2ОР, Н-3'), 6.10-6.18 (m, 1Н, Н-1'), 7.40-7.46 (m, 1Н, ArH), 7.50-7.59 (m, 3Н, ArH), 7.66-7.72 (m, 2Н, ArH, Н-6), 7.85-7.91 (m, 1Н, ArH), 8.18-8.24 (m, 1Н, ArH); 13С ЯМР (125 МГц, MeOD): тмС 14.4 (СН3), 27.5 (br s, СН3), 27.9 (d, 3JC-P=6.1 Гц, СН3), 28.0 (d, 3JC-P=6.1 Гц, СН3), 40.7 (СН2), 40.8 (СН2), 58.2 (С), 58.3 (С), 62.6 (СН2), 67.8 (d, 2JC-P=4.9 Гц, СН2), 67.9 (d, 2JC-P=4.5 Гц, СН2), 72.0 (СН), 72.1 (СН), 86.7 (d, 3JC-P=7.7 Гц, СН), 86.9 (d, 3JC-P=7.3 Гц, СН), 87.0 (СН), 116.3 (d, 3JC-P=3.2 Гц, СН), 116.6 (d, 3JC-P=2.9 Гц, СН), 122.8 (СН), 122.9 (СН), 125.4 (СН), 125.6 (СН), 125.7 (СН), 126.0 (СН), 126.1 (СН), 126.5 (СН), 127.4 (СН), 127.5 (СН), 127.7 (СН), 127.8 (СН), 127.9 (С), 128.0 (С), 128.9 (СН), 136.2 (С), 141.8 (d, 1JC-F=233.5 Гц, С), 148.0 (d, 2JC-P=7.3 Гц, С), 148.1 (d, 2JC-P=7.6 Гц, С), 150.5 (С). 150.6 (С), 159.3 (d, 2JC-F=26.2 Гц, С), 159.4 (d, 2JC-F=26.6 Гц, С), 176.8 (С), 176.9 (С); m/z (ES) 588,1 (MNa+, 100%). В результате обратнофазовой ВЭЖХ с использованием для элюирования H2O/МеОН (100/0-0/100, в течение 45 мин, 1 мл/мин, λ=275 нм) получали один пик. соответствующий смеси диастереоизомеров: tR=16,05 мин (96%).
5-Фтор-2'-дезоксиуридин-5'-O-[фенил(бензокси-L-пролинил)]фосфат (CPF583)
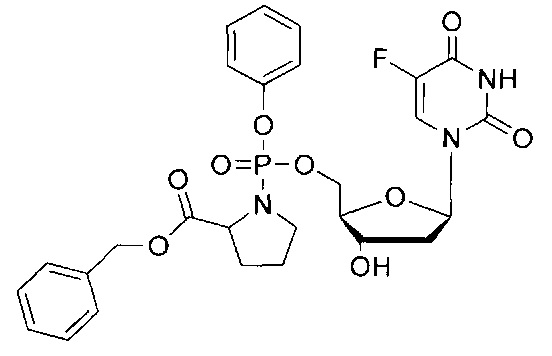
Указанное в заголовке соединение получали в соответствии со стандартной методикой D из 5-фтор-2'-дезоксиуридина (0,25 г, 1,01 ммоль), NMI (0,41 г, 5,07 ммоль, 0,40 мл) и фенил(бензокси-L-пролинил)-фосфохлоридата (0,77 г, 2,03 ммоль) в ТГФ (10 мл). В результате колоночной очистки и последующих двух очисток путем препаративной ТСХ получали желаемый продукт в виде белого твердого вещества (0,010 г, 2%).
31Р ЯМР (MeOD, 202 МГц) δ: 1.82.
19F ЯМР (MeOD, 470 МГц) δ: - 167.91.
1Н ЯМР (MeOD, 500 МГц) δ: 7.84 (d, J=7.18 Гц, 1Н, Н-основание), 7.39-7.33 (m, 7Н, H-Ar), 7.22-7.19 (m, 3Н, Н-Ar), 6.26-6.23 (m, 1Н, Н-1'), 5.22-5.13 (m, CH2Ph сложный эфир), 4.40-4.35 (m, 3Н, NCH, 2×Н-5'), 4.33-4.28 (m, 1Н, Н-3'), 4.06-4.04 (m, 1Н, Н-4'), 3.36-3.32 (m, 2Н, NCH2), 2.26-2.19 (m, 1Н, Н-2'), 2.18-2.13 (m, 1Н, CH2-L-Pro), 2.00-1.81 (m, 4Н, 3×Н, CH2-L-Pro, 1×Н, Н-2').
13С ЯМР (MeOD, 125 МГц) δ: 174.81 (С=O, сложный эфир), 159.40 (С=O, основание), 152.0 (d, 2JC-P=6.32 Гц, ОС-Ar), 150.71 (С=O, основание), 141.88 (1JC-F=232 Гц, CF, основание), 137.23 (C-Ar), 131.33, 129.70, 129.48, 129.45, 129.30, 126.45 (CH-Ar), 125.80, 125.53 (2×d, 2JC-F=29.0 Гц, CH-основание), 121.00, 120.96 (CH-Ar), 87.80 (С-1'), 86.80 (С-4'), 72.02 (С-3'), 68.16 (CH2Ph), 67.64 (d, 2JC-P=4.65 Гц, С-5'), 62.40 (d, 2JC-P=5.60 Гц, NCH), 48.03 (d, 2JC-P=4.80 Гц, NCH2), 41.07 (C-2'), 32.18, 32.11 (CH2-L-Pro), 26.29, 26.21 (CH2-L-Pro).
MC (ES+) m/e: 612 (MNa+, 100%), 590 (MH+, 1%), точно измеренная масса соответствует C27H29FN3O9P. Вычислено: 589,51.
5-Фтор-2'-дезоксиуридин-5'-O-[1-нафтил(бензокси-L-пролинил)]фосфат (CPF577)
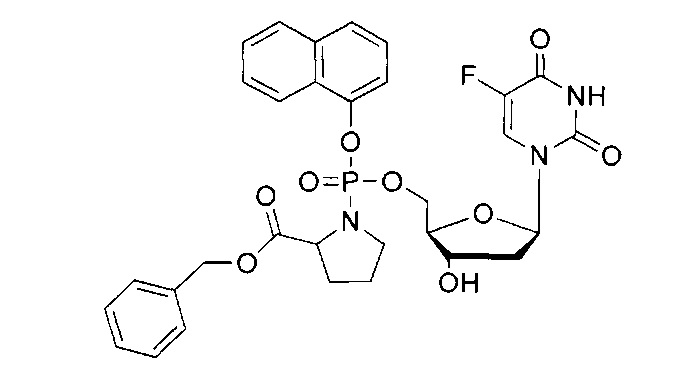
Указанное в заголовке соединение получали в соответствии со стандартной методикой D из 5-фтор-2'-дезоксиуридин (0,25 г, 1,01 ммоль), NMI (0,41 г, 5,07 ммоль, 0,40 мл) и 1-нафтил(бензокси-L-пролинил)-фосфохлоридата (0,84 г, 2,03 ммоль) в ТГФ (10 мл). В результате колоночной очистки и последующих двух очисток путем препаративной ТСХ получали желаемый продукт в виде белого твердого вещества (0,006 г, 1%).
31Р ЯМР (MeOD, 202 МГц) δ: 2.27.
19F ЯМР (MeOD, 121 МГц) δ: - 167.46.
1Н ЯМР (MeOD, 500 МГц) δ: 8.14-8.12 (m, 1H, H-Ar), 7.90-7.89 (m, 1Н, H-Ar), 7.74-7.71 (m, 2Н, 1×H-Ar, 1×H-основание), 7.56-7.42 (m, 4Н, H-Ar), 7.36-7.33 (m, 5Н, H-Ar), 6.13 (t, J=6.38 Гц, Н-1'), 5.22-5.13 (m, 2Н, CH2Ph), 4.49-4.46 (m, 1Н, NCH), 4.42-4.33 (m, 2Н, Н-5'), 4.25-4.23 (m, 1Н, Н-3'), 4.06-4.04 (m, 1Н, Н-4'), 3.36-3.34 (m, 2Н, NCH2), 2.23-2.15 (m, 1Н, СН2-L-Pro), 2.10-2.02 (m, 2Н, 1×Н, CH2-L-Pro, 1×Н, Н-2'), 1.97-1.77 (m, 2Н, СН2-L-Pro), 1.63-1.57 (m, 1Н, Н-2')
13С ЯМР (MeOD, 125 МГц) δ: 174.82 (C=O, сложный эфир), 159.52 (С=O, основание), 150.54 (С=O, основание), 147.84, 147.78 (d, 2JC-P=6.03 Гц, OC-Ar), 141.75, 139.97 (2×d, 1JC-F=232 Гц, CF, основание), 137.20, 136.34 (C-Ar), 129.76, 129.65, 129.44, 129.36, 129.27, 129.06, 128.95, 128.04, 128.75, 126.56 (CH-Ar), 125.41 (d, 2JC-F=30.0 Гц, CH-основание), 122.13 (CH-Ar), 115.76 (d, 3JC-P=3.3 Гц, CH-Ar), 87.06 (C-1'), 86.79 (С-4'), 72.23 (С-3'), 68.15 (d, 2JC-P=5.46 Гц, С-5'), 68.08 (CH2Ph), 62.53 (d, 2JC-P=5.60 Гц, NCH), 48.26 (d, 2JC-P=5.34 Гц, NCH2), 40.97 (C-2'), 32.16, 32.09 (CH2-L-Pro), 26.22, 26.15 (CH2-L-Pro).
5-Фтор-2'-дезоксиуридин-5'-O-[1-нафтил(3,3-диметил-1-бутокси-L-аланинил)]фосфат (CPF585)
Указанное в заголовке соединение получали в соответствии со стандартной методикой D из 5-фтор-2'-дезоксиуридина (0,25 г, 1,01 ммоль), NMI (0,41 г, 5,07 ммоль. 0,40 мл) и 1-нафтил-(3,3-диметил-1-бутокси-L-аланинил)-фосфохлоридата (1,21 г, 3,04 ммоль) в ТГФ (10 мл). В результате колоночной очистки и последующих двух очисток путем препаративной ТСХ получали желаемый продукт в виде белого твердого вещества (0,010 г, 2%).
31Р ЯМР (MeOD, 202 МГц) δ: 4.48, 4.33.
19F ЯМР (MeOD, 470 МГц) δ: - 167.30, - 167.47.
1Н ЯМР (MeOD, 500 МГц) δ: 8.20-8.17 (m, 1Н, H-Ar), 7.91-7.89 (m, 1Н, H-Ar), 7.77-7.72 (m, 2Н, H-Ar), 7.58-7.51 (m, 3Н, Н-основание, 2×H-Ar), 7.46-7.41 (2×t, 1Н, J=7.8 Гц, H-Ar), 6.19-6.13 (m, 1Н, Н-1'), 4.42-4.40 (m, 1Н, 1×Н-5'), 4.38-4.32 (m, 2Н, Н-3', 1×Н-5'), 4.14-4.00 (m, 4Н, Н-4', СНСН3, ОСН2СН2(СН3)3), 2.21-2.13 (m, 1Н, 1×Н-2'), 1.91-1.76 (m, 1Н, 1×Н-2'), 1.52-1.48 (m, 2Н, ОСН2СН2(СН3)3), 1.37-1.35 (m, 3Н, СНСН3), 0.92, 0.91 (2×s, 9Н, ОСН2СН2(СН3)3). 13С ЯМР (MeOD, 125 МГц) δ: 175.16, 174.84 (2×d, 3JC-P=4.75 Гц, С=O, сложный эфир), 159.56, 159.35 (С=O, сложный эфир), 150.61 (С=O, сложный эфир), 148.00, 147.86 (2×d, 2JC-P=6.25 Гц, OC-Ar), 141.78, 141.73 (2×d, 1JC-F=232 Гц, CF, основание), 136.28 (C-Ar), 128.98, 128.95, 127.92, 127.90, 127.58, 126.57, 126.20, 126.14 (CH-Ar), 125.63, 125.55 (2×d, 2JC-F=34 Гц, CH, основание), 122.65, 122.63 (CH-Ar), 116.48, 116.15 (2×d, 3JC-P=3.0 Гц, CH-Ar), 87.01, 86.94 (С-1'), 86.73, 86.68 (d, 3JC-P=7.75 Гц, С-4'), 72.18, 72.07 (С-3'), 67.87, 67.85 (2×d, 2JC-P=5.0 Гц, С-5'), 64.08, 64.05 (ОСН2СН2(СН3)3), 51.86 (d, 3JC-P=5.5 Гц, СНСН3), 42.74 (ОСН2СН2(СН3)3), 40.91, 40.83 (С-2'), 29.96 (ОСН2СН2(СН3)3), 20.50, 20.34 (2×d, 3JC-P=6.5 Гц, СНСН3).
МС (ES+) m/e: 630 (MNa+, 100%), 608 (МН+, 10%), точно измеренная масса соответствует C28H35FN3O9P. Вычислено: 607,56.
5-Фтор-2'-дезоксиуридин-5'-O-[1-нафтил-(циклобутокси-L-аланинил)]фосфат (CPF578)
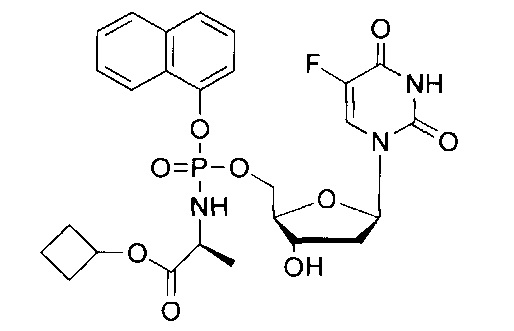
Указанное в заголовке соединение получали в соответствии со стандартной методикой D из 5-фтор-2'-дезоксиуридина (0,23 г, 0,93 ммоль), NMI (0,38 г, 4,67 ммоль, 0,37 мл) и 1-нафтил-(циклобутокси-L-аланинил)-фосфохлоридата (0,85 г, 2,33 ммоль) в ТГФ (10 мл). В результате колоночной очистки и последующей очистки путем препаративной ТСХ получали желаемый продукт в виде белого твердого вещества (0,010 г, 2%).
31Р ЯМР (MeOD, 202 МГц) δ: 4.54, 4.36.
19F ЯМР (MeOD, 470 МГц) δ: - 167.12, - 167.29.
1H ЯМР (MeOD, 500 МГц) δ: 8.18-8.17 (m, 1Н, H-Ar), 7.81-7.87 (m, 1H, H-Ar), 7.74-7.71 (m, 2H, 1×H-Ar, 1×Н-основание), 7.60-7.53 (m, 3H, H-Ar), 7.46-7.43 (2×t, J=8.0 Гц, 1H, H-Ar), 6.18-6.12 (m, 1H, Н-1'), 5.00-4.95 (m, 1H, OCH сложный эфир), 4.41-4.36 (m, 3Н, 2×H-5', H-3'), 4.11-4.00 (m, 2H, H-4', CHCH3), 2.36-2.27 (m, 2H, CH2), 2.18-1.98 (m, 3H, CH2 сложный эфир, 1×Н-2'), 1.82-1.56 (m, 3Н, CH2 сложный эфир, 1×Н-2'), 1.36-1.34 (m, 3Н, СНСН3).
13С ЯМР (MeOD, 125 МГц) δ: 175.97, 173.34 (С=O, сложный эфир), 159.88 (С=O, основание), 151.64 (С=O, основание), 146.58 (OC-Ar), 141.15 (d, 1JC-F=220 Гц, CF, основание), 136.28 (C-Ar), 128.93, 127.89, 127.54, 126.52, 126.18, 126.14 (CH-Ar), 125.53, 125.44 (2×d, 2JC-F=32.5 Гц, CH-основание), 122.63 (CH-Ar), 116.46, 116.44 (2×d, 3JC-P=2.5 Гц, CH-Ar), 86.98 (d, 3JC-P=6.25 Гц, С-4'), 86.71 (С-1'), 72.14, 72.04 (С-3'), 71.07 (OCH сложный эфир), 67.83 (d, 2JC-P=7.38 Гц, С-5'), 51.66 (d, 2JC-P=8.75 Гц, СНСН3), 40.89, 40.83 (С-2'), 31.03 (ОСНСН2), 20.43 (СНСН3), 14.23 (СН2 сложный эфир).
МС (ES+) m/e: 600 (MNa+, 100%), 578 (МН+, 10%), точно измеренная масса соответствует C26H29FN3O9P. Вычислено: 577,50.
5-Фтор-2'-дезоксиуридин-5'-O-[1-нафтил-(циклопропилметанокси-L-аланинил)]фосфат (CPF579)
Указанное в заголовке соединение получали в соответствии со стандартной методикой D из 5-фтор-2'-дезоксиуридина (0,25 г, 1,01 ммоль), NMI (0,41 г, 5,07 ммоль, 0,40 мл) и 1-нафтил-(циклопропилметанокси-L-аланинил)-фосфохлоридата (0,93 г, 2,54 ммоль) в ТГФ (10 мл). В результате колоночной очистки получали желаемый продукт в виде белого твердого вещества (0,056 г, 10%).
31Р ЯМР (MeOD, 202 МГц) δ: 4.58, 4.30.
19F ЯМР (MeOD, 470 МГц) δ: - 167.18, - 167.22.
1Н ЯМР (MeOD, 500 МГц) δ: 8.18 (d, J=7.0 Гц, 1Н, H-Ar), 7.89-7.87 (m, 1Н, H-Ar), 7.73-7.70 (m, 2Н, H-Ar), 7.58-7.53 (m, 3Н, H-Ar), 7.45-7.40 (2×t, J=8.0 Гц, 1H, H-Ar), 6.17-6.11 (m, 1Н, Н-1'), 4.43-4.41 (m, 1Н, Н-5'), 4.38-4.32 (m, 2Н, Н-5', Н-3'), 4.11-4.04 (m 2Н, Н-4', СНСН3), 3.95-3.85 (m, 2Н, ОСН2 сложный эфир), 2.19-2.11 (m, 1Н, Н-2'), 1.84-1.72 (m, 1Н, Н-2'), 1.38, 1.36 (2×d, J=5.0 Гц, 3Н, СНСН3), 1.15-1.07 (m, 1Н, OCH2CH сложный эфир), 0.59-0.50 (m, 2Н, СН2 сложный эфир), 0.30-0.24 (m, 2Н, СН2 сложный эфир).
13С ЯМР (MeOD, 125 МГц) δ: 175.25, 174.94 (2×d, 3JC-P=4.75 Гц, С=O, сложный эфир), 159.54, 159.35 (С=O, основание), 150.60, 150.56 (С=O, основание), 148.05, 147.86 (2×d, 2JC-P=7.5 Гц. OC-Ar), 141.79, 141.73 (2×d, 1JC-F=232 Гц, CF, основание), 136.29 (C-Ar), 128.94 (d, 3JC-P=4.4 Гц, CH-Ar), 127.89 (d, 4JC-P=3.7 Гц, CH-Ar), 127.56, 126.55, 126.52, 126.19, 126.16 (CH-Ar), 125.64, 125.53 (2JC-F=34 Гц, CH-основание), 122.65 (CH-Ar), 116.54, 116.24 (2×d, 4JC-P=2.6 Гц, CH-Ar), 87.04, 86.99 (С-1'), 86.90, 86.73 (2×d, 3JC-P-P=7.1 Гц, С-4'), 72.18, 72.07 (С-3'), 71.21, 71.18 (ОСН2, сложный эфир), 67.87, 67.84 (кажущийся t, 2JC-P=5.0 Гц, С-5'). 51.88 (d, 2JC-P=10.0 Гц, СНСН3), 40.91, 40.83 (С-2'), 20.60, 20.46 (2×d, 3JC-P=6.5 Гц, СНСН3), 10.69 (ОСН2СН сложный эфир), 3.70, 3.65 (2×СН2, сложный эфир).
МС (ES+) m/e: 600 (MNa+, 100%), 578 (МН+, 15%), точно измеренная масса соответствует C26H29FN3O9P. Вычислено:577,50.
HPLCb (H2O/ацетонитрил от 100/0 до 0/100 в течение 35 мин): Rt=12,91 мин.
5-Фтор-2'-дезоксиуридин-5'-O-[1-нафтил-(тетрагидропирокси-L-аланинил)]фосфат (CPF580)
Указанное в заголовке соединение получали в соответствии со стандартной методикой E из 5-фтор-2'-дезоксиуридина (0,25 г, 1,01 ммоль), tBuMgCl (1,1 мл, 1,1 ммоль) и 1-нафтил-(тетрагидропирокси-L-аланинил)-фосфохлоридата (0,80 г, 2,03 ммоль) в ТГФ (10 мл). В результате колоночной очистки и последующих двух очисток путем препаративной ТСХ получали желаемый продукт в виде белого твердого вещества (0,010 г, 1,6%).
31Р ЯМР (MeOD, 202 МГц) δ: 3.77, 3.22.
19F ЯМР (MeOD, 470 МГц) δ: - 168.27, - 168.35.
1Н ЯМР (MeOD, 500 МГц) δ: 8.60 (d, J=7.0 Гц, 2Н, H-Ar), 8.22-8.19 (m, 1Н, H-Ar), 7.92-7.91 (d, J=5.50 Гц, 1Н, H-Ar), 7.60-7.45 (m, 4Н, H-Ar, H-основание), 6.29-6.25 (m, 1Н, Н-1'), 5.25-5.17 (m, 1Н, Н-3'), 4.96-4.87 (m, 1Н, CH-сложный эфир), 4.28-4.26 (m, 1Н, Н-4'), 4.11-4.03 (m, 1Н, СНСН3), 3.88-3.66 (m, 4Н, 2×ОСН2а' сложный эфир, 2×Н-5'), 3.55-3.50 (m, 2Н, 2×ОСН2аʺ сложный эфир), 2.63-2.30 (m, 2Н, Н-2'), 1.91-1.85 (m, 2Н, 2×CH2b' сложный эфир), 1.65-1.54 (m, 2Н, CH2bʺсложный эфир), 1.39-1.35 (m, 3Н, СНСН3).
13С ЯМР (MeOD, 125 МГц) δ: 174.34 (С=O, сложный эфир), 159.24 (С=O, основание), 150.76 (С=O, основание), 148.03 (ОС-Ar), 141.97 (d, 1JC-F=238 Гц, CF, основание), 136.37 (C-Ar), 128.97, 128.56, 127.61, 127.57, 126.58, 126.23, 126.16, 126.12, 125.84 (CH-Ar), 122.70 (d, 2JC-F=24.0 Гц, CH-основание), 116.62, 116.37 (CH-Ar), 87.54 (d, 3JC-P=5.40 Гц, С-4'), 86.60, 86.57 (С-1'), 79.82, 79.47 (С-3'), 71.45 (CH-сложный эфир), 66.12, 66.08 (2×ОСН2а сложный эфир), 66.02 (С-5'), 51.83 (СНСН3), 39.97, 39.94 (С-2'), 32.65, 32.57 (2×CH2b сложный эфир), 20.45, 20.30 (СНСН3).
МС (ES+) т/е: 630 (MNa+, 100%), 608 (МН+, 10%), точно измеренная масса соответствует C27H31FN3O10P. Вычислено:607,52.
5-Фтор-2'-дезоксиуридин-5'-O-[1-нафтил-(пентокси-L-аланинил)]фосфат (CPF581)
Указанное в заголовке соединение получали в соответствии со стандартной методикой E из 5-фтор-2'-дезоксиуридина (0,25 г, 1,01 ммоль), tBuMgCl (1,1 мл, 1,1 ммоль) и 1-нафтил-(пентокси-L-аланинил)-фосфохлоридата (0,78 г, 2,03 ммоль) в ТГФ (10 мл). В результате колоночной очистки получали желаемый продукт в виде белого твердого вещества (0,047 г, 8%).
31Р ЯМР (MeOD, 202 МГц) δ: 4.48, 4.32.
19F ЯМР (MeOD, 470 МГц) δ: - 167.18, - 167.29.
1Н ЯМР (MeOD, 500 МГц) δ: 8.25-8.17 (m, 1Н, H-Ar), 8.05-7.95 (m, 2Н, H-Ar), 7.85-7.60 (m, 2Н, H-Ar, H-основание), 7.65-7.48 (m, 3Н, H-Ar), 6.30-6.18 (m, 1Н, H-1'), 4.60-4.37 (m, 3Н, 2×Н-5', Н-3'), 4.28-4.00 (m, 4Н, Н-4', СНСН3, ОСН2СН2СН2СН2СН3), 2.32-2.12 (m, 1Н, Н-2'), 1.95-1.75 (m, 1Н, Н-2'), 1.70-1.55 (m, 2Н, ОСН2СН2СН2СН2СН3), 1.50-1.28 (m, 7Н, 4×Н ОСН2СН2СН2СН2СН3, СНСН3), 0.83, 0.82 (2×d, J=7.9 Гц, 3Н, ОСН2СН2СН2СН2СН3).
13С ЯМР (MeOD, 125 МГц) δ: 175.22, 174.91 (C=O, сложный эфир), 159.5 (C=O, основание), 150.54 (C=O, основание), 147.90, 147.88 (OC-Ar), 141.75 (d,1JC-F=225 Гц, CF, основание), 136.37 (C-Ar), 128.95, 127.90, 127.56, 126.55, 126.19 (CH-Ar), 125.64, 125.53 (2×d, 2JC-F=34.0 Гц, CH-основание), 122.65 (CH-Ar), 116.51, 116.21 (CH-Ar), 87.03, 86.96 (C-1'), 86.85, 86.74 (С-4'), 72.16, 72.05 (C-3'), 67.87 (d, 2JC-P=5.0 Гц, С-5'), 66.54 (ОСН2), 51.87, 51.81 (d, 2JC-P=7.5 Гц, СНСН3), 40.87, 40.80 (С-2'), 29.35, 29.10 (СН2 сложный эфир), 23.33 (СН2 сложный эфир), 20.60, 20.43 (2×d, 3JC-P=6.5 Гц, СНСН3), 14.28 (СН3, сложный эфир).
МС (ES+) т/е: 616 (MNa+, 100%), 594 (МН+, 10%), точно измеренная масса соответствует C27H33FN3O9P. Вычислено: 593,54.
HPLCb (H2O/ацетонитрил от 100/0 до 0/100 в течение 35 мин): Rt=15,56 мин.
5-Фтор-2'-дезоксиуридин-5'-O-[1-нафтил-(циклопентокси-L-аланинил)]фосфат (CPF582)
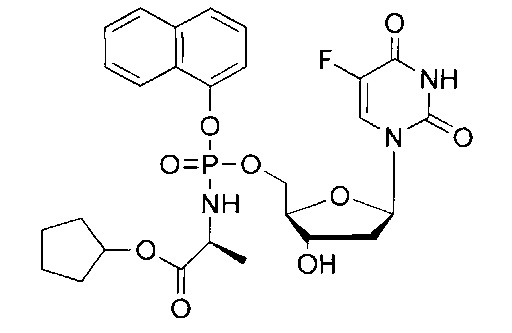
Указанное в заголовке соединение получали в соответствии со стандартной методикой E из 5-фтор-2'-дезоксиуридина (0,25 г, 1,01 ммоль), tBuMgCl (1,1 мл, 1,1 ммоль) и 1-нафтил-(циклопентокси-L-аланинил)-фосфохлоридата (0,77 г, 2,03 ммоль) в ТГФ (10 мл). В результате колоночной очистки получали желаемый продукт в виде белого твердого вещества (0,030 г, 5%).
31Р ЯМР (MeOD, 202 МГц) δ: 4.53, 4.37.
19F ЯМР (MeOD, 470 МГц) δ: - 167.07, - 167.19.
1Н ЯМР (MeOD, 500 МГц) δ: 8.18-8.16 (m, 1Н, H-Ar), 7.89-7.85 (m, 1Н, H-Ar), 7.70 (кажущийся t, J=6.50 Гц, 2Н, H-Ar), 7.57-7.50 (m, 3Н, 2×H-Ar, H-основание), 7.45-7.40 (m, 1Н, H-Ar), 6.16-6.11 (m, 1H, H-1'), 5.15-5.09 (m, 1Н,ОСН сложный эфир), 4.41-4.30 (m, 3Н, 2×Н-5', Н-3'), 4.11-4.08 (m, 1Н, Н-4'), 4.04-3.98 (m, 1Н, СНСН3), 2.19-2.10 (m, 1Н, Н-2'), 1.86-1.73 (m, 3Н, ОСНСН2 сложный эфир), 1.73-1.56 (m, 6Н, Н-2', СН2 сложный эфир), 1.35, 1.34 (2×d, J=6.57 Гц, СНСН3).
13С ЯМР (MeOD, 125 МГц) δ: 174.68, 174.64 (C=O, сложный эфир), 159.27 (C=O, основание), 150.51 (C=O, основание), 147.86 (d, 2JC-P=7.5 Гц, ОС-Ar), 141.78, 141.72 (2×d, 1JC-F=232 Гц, CF-основание), 136.30 (C-Ar), 128.95, 128.54, 127.94, 127.80, 127.60, 127.56, 127.17, 126.80, 126.54, 126.19, 126.16 (CH-Ar), 125.66, 125.53 (2×d, 2JC-F=34 Гц, СН-основание), 122.65, 122.61 (CH-Ar), 116.53, 116.22 (2×d, 4JC-P=3.75 Гц, СН-Ar), 86.99, 86.96 (С-1'), 86.70 (d, 3JC-P=7.50 Гц, С-4'), 79.64, 79.61 (ОСН сложный эфир), 72.21, 72.07 (С-3'), 67.89, 67.85 (2×d, 2JC-P=5.0 Гц, С-5'), 51.92 (d, 2JC-P=5.0 Гц, СНСН3), 40.92, 40.86 (С-2'), 33.65, 33.61, 33.52, 33.47 (2×СН2 сложный эфир), 24.68, 24.66 (СН2 сложный эфир), 20.45, 20.30 (2×d, 3JC-Р=6.25 Гц, СНСН3).
МС (ES+) m/e: 614 (MNa+, 100%), 592 (МН+, 30%), точно измеренная масса соответствует C27H31FN1O9P. Вычислено: 591,52.
HPLCb (H2O/ацетонитрил от 100/0 до 0/100 в течение 35 мин): Rt=14,03 мин.
5-Фтор-2'-дезоксиуридин-5'-O-[1-нафтил-(2-инданокси-L-аланинил)]фосфат (CPF597)
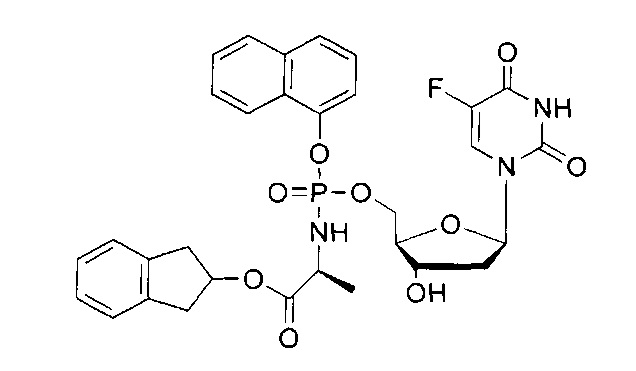
Указанное в заголовке соединение получали в соответствии со стандартной методикой E из 5-фтор-2'-дезоксиуридина (0,30 г, 1,22 ммоль), tBuMgCl (1,34 мл, 1,34 ммоль) и 1-нафтил-(2-инданокси-L-аланинил)-фосфохлоридата (1,06 г, 2,43 ммоль) в ТГФ (20 мл). В результате колоночной очистки получали желаемый продукт в виде белого твердого вещества (0,045 г, 6%).
31Р ЯМР (MeOD, 202 МГц) δ: 4.62, 4.30.
19F ЯМР (MeOD, 470 МГц) δ: - 167.14, - 167.34.
1Н ЯМР (MeOD, 500 МГц) δ: 8.15-8.12 (m, 1Н, H-Ar, Naph), 7.89-7.87 (m, 1Н, Н-Ar, Naph), 7.72-7.67 (m, 2Н, H-Ar, Naph), 7.56-7.46 (m, 3Н, 2×H-Ar, Н-основание), 7.40-7.37 (m, 1Н, H-Ar), 7.20-7.12 (m, 4Н, H-Ar, Ph), 6.14-6.08 (m, 1Н, Н-1'), 5.49-5.46 (m, 1Н, ОСН сложный эфир), 4.32-4.26 (m, 3Н, 2×Н-5', Н-3'), 4.04-3.98 (m, 1Н, Н-4', СНСН3), 3.30-3.24 (m, 2Н, 2×СН сложный эфир), 2.99-2.91 (m, 2Н, 2×СН сложный эфир), 2.14-2.07 (m, 1Н, Н-2'), 1.75-1.64 (m, 1 Н, Н-2'), 1.33-1.29 (m, 3Н, СНСН3).
13С ЯМР (MeOD, 125 МГц) δ: 175.02, 174.66 (2×d, 3JC-P=3.75 Гц, С=О, сложный эфир), 159.48 (2JC-F=25.0 Гц, С=O, основание), 150.57 (С=O, основание), 147.97, 147.80 (2×d, 2JC-P=7.5 Гц, ОС-Ar), 141.73, 141.68 (2×d, 1JC-F=232.5 Гц, CF-основание), 141.54, 141.49, 141.48, 139.10, 136.27, 136.26 (C-Ar), 129.01, 128.94, 128.91, 127.91, 127.87, 128.85, 127.80, 127.77, 127.60, 127.57, 127.50, 126.20, 126.18, 125.69 (CH-Ar), 125.50, 125.43 (2×d, 2JC-F=25 Гц, CH-основание), 122.64, 122.60, 121.85 (СН-Ar), 116.57, 116.26 (2×d, 4JC-P=2.5 Гц, СН-Ar), 86.96 (С-1'), 86.87, 86.66 (2×d, 3JC-P=7.50 Гц, С-4'), 77.85, 79. (ОСН сложный эфир), 72.21, 72.07 (С-3'), 67.77, 67.75 (2×d, 2JC-P=6.25 Гц, С-5'), 51.97, 51.82 (СНСН3), 40.91, 40.86 (С-2'), 40.44, 40.43, 40.38, 40.34 (2×СН2 сложный эфир), 20.30, 20.16 (2×d, 3JC-P=6.25 Гц, СНСН3).
5-Фтор-2'-дезоксиуридин-5'-O-[фенил-(бензокси-L-метионинил)]фосфат (CPF586)
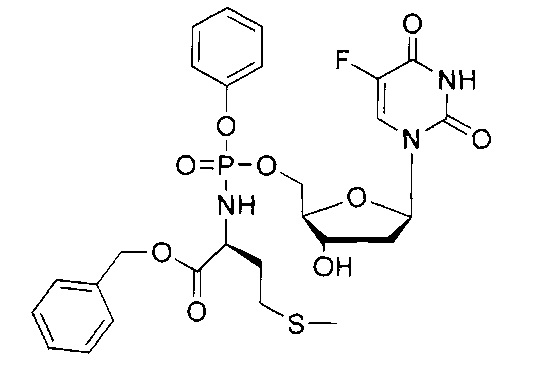
Указанное в заголовке соединение получали в соответствии со стандартной методикой D из 5-фтор-2'-дезоксиуридина (0,25 г, 1,01 ммоль), NMI (0,41 г, 5,07 ммоль, 0,40 мл) и фенил-(бензокси-L-метионинил)-фосфохлоридата (0,7 г, ммоль) в ТГФ (10 мл). В результате колоночной очистки получали желаемый продукт в виде желтоватого твердого вещества (0,014 г, 2%).
31Р ЯМР (MeOD, 202 МГц) δ: 4.34, 3.94.
19F ЯМР (MeOD, 470 МГц) δ: - 167.40, - 167.69.
1Н ЯМР (MeOD, 500 МГц) δ: 7.83-7.80 (m, 1Н, Н-Ar), 7.74-7.72 (m, 1Н, Н-Ar), 7.64-7.62 (m, 1Н, Н-Ar), 7.37-7.32 (m, 6Н, Н-Ar, Н-основание), 7.26-7.17 (m, 2Н, Н-Ar), 6.25-6.17 (m, 1Н, Н-1'), 5.18, 5.13 (АВ систем, JAB=12.0 Гц, 2Н, CH2Ph), 4.40-4.35 (m, 1Н, Н-3'), 4.32-4.22 (m, 2Н, Н-5'), 4.16-4.03 (m, 2Н, NHCH, Н-4'), 2.44, 2.36 (2×t, J=7.50 Гц, CH2S), 2.16-2.08 (m, 1Н, 1×Н-2'), 1.98-1.82 (m, 6H, 1×H-2', NHCHCH2CH2SCH3).
MC (ES+) m/e: 646 (MNa+, 100%), 624 (MH+, 10%), точно измеренная масса соответствует C27H31FN3O9PS. Вычислено: 623,56.
5-Фтор-2'-дезоксиуридин-5'-O-[1-нафтил-(бензокси-L-фенилаланинил)]фосфат (CPF587)
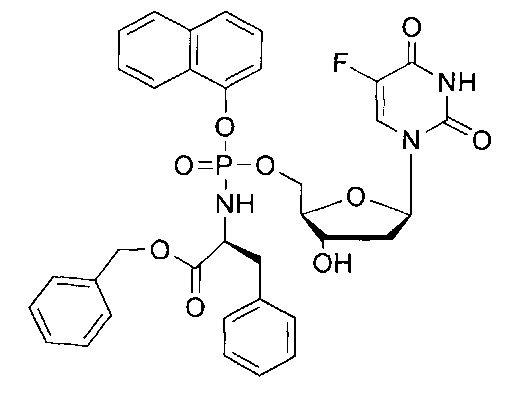
Указанное в заголовке соединение получали в соответствии со стандартной методикой D из 5-фтор-2'-дезоксиуридина (0,25 г, 1,01 ммоль), NMI (0,41 г, 5,07 ммоль, 0,40 мл) и 1-нафтил-(бензокси-L-фенилаланинил)-фосфохлоридата (1,45 г, ммоль) в ТГФ (10 мл). В результате колоночной очистки получали желаемый продукт в виде белого твердого вещества (0,007 г, 1%).
31Р ЯМР (MeOD, 202 МГц) δ: 4.27, 4.14.
19F ЯМР (MeOD, 470 МГц) δ: - 166.99, - 167.18.
1Н ЯМР (MeOD, 500 МГц) δ: 8.11-8.00 (m, 1Н, H-Ar, Ar), 7.89-7.85 (m, 1Н, H-Ar), 7.69-7.67 (m, 1Н, H-Ar), 7.60-7.49 (m, 3H, 2×H-Ar, H-основание), 7.37-7.33 (m, 2Н, H-Ar), 7.25-7.12 (m, 10Н, Н-Ar), 6.09-6.04 (m, 1Н, Н-1'), 5.11-5.01 (m, 2Н, CH2Ph), 4.29-4.18 (m, 1Н, СНСН3), 4.15-4.08 (m, 1Н, Н-3'), 4.02-3.95 (m, 2Н, Н-5'), 3.86-3.67 (m, 1Н, Н-4'), 3.14-3.10 (m, 1Н, 1×NHCHCH2Ph), 2.91-2.82 (m, 1Н, 1×NHCHCH2Ph), 2.12-2.06, 2.00-1.95 (2×m, 1Н, Н-2'), 1.68-1.62, 1.42-1.36 (2×m, 1Н, Н-2').
5-Фтор-2'-дезоксиуридин-5'-O-[1-нафтил-(2,2-диметилпропокси-L-аланинил)]фосфат (CPF588)
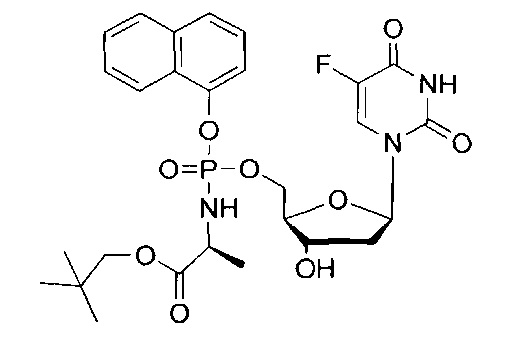
Указанное в заголовке соединение получали в соответствии со стандартной методикой D из 5-фтор-2'-дезоксиуридина (0,25 г, 1,01 ммоль), NMI (0,41 г, 5,07 ммоль, 0,40 мл) и 1-нафтил-(2,2-диметилпропокси-L-аланинил)-фосфохлоридата (0,77 г, ммоль) в ТГФ (10 мл). В результате колоночной очистки получали желаемый продукт в виде белого твердого вещества (0,006 г, 1%).
31Р ЯМР (MeOD, 202 МГц) δ: 4.56, 4.33.
19F ЯМР (MeOD, 470 МГц) δ: - 167.32, - 167.43.
1Н ЯМР (MeOD, 500 МГц) δ: 8.19-8.16 (m, 1Н, Н-Ar, Ar), 7.91-7.89 (m, 1Н, Н-Ar), 7.74-7.71 (m, 2Н, Н-Ar), 7.57-7.51 (m, 3Н, 2×Н-Ar, Н-основание), 7.46-7.41 (m, 1Н, Н-Ar), 6.17-6.10 (m, 1H, Н-1'), 4.42-4.30 (m, 3Н, Н-3', 2×Н-5'), 4.13-4.07 (m, 2Н, Н-4', СНСН3), 3.86, 3.75 (АВ систем, JAB=10.50 Гц, 2Н, СН2С(СН3)3), 2.18-2.10 (m, 1H, Н-2'), 1.81-1.70 (m, 1Н, Н-2'), 1.41-1.38 (m, 3Н, СНСН3), 0.95, 0.94 (2×s, 9Н, СН2С(СН3)3).
5-Фтор-2'-дезоксиуридин-5'-O-[1-нафтил-(бутокси-L-аланинил)]фосфат (CPF589)
Указанное в заголовке соединение получали в соответствии со стандартной методикой D из 5-фтор-2'-дезоксиуридина (0,25 г, 1,01 ммоль), NMI (0,41 г, 5,07 ммоль, 0,40 мл) и 1-нафтил-(бутокси-L-аланинил)-фосфохлоридата (0,75 г, ммоль) в ТГФ (10 мл). В результате колоночной очистки получали желаемый продукт в виде белого твердого вещества (0,006 г, 1%).
31Р ЯМР (MeOD, 202 МГц) δ: 4.52, 4.35.
19F ЯМР (MeOD, 470 МГц) δ: - 167.36, - 167.49.
1Н ЯМР (MeOD, 500 МГц) δ: 8.19-8.16 (m, 1Н, H-Ar, Naph), 7.1-7.89 (m, 1Н, Н-Ar, Naph), 7.75-7.72 (m, 2Н, H-Ar, Naph), 7.58-7.51 (m, 3Н, 2×Н-Ar, Н-основание), 7.46-7.41 (m, 1H, Н-Ar), 6.18-6.11 (m, 1H, Н-1'), 4.42-4.40 (m, 1Н, 1×Н-5'), 4.37-4.32 (m, 2Н, 1×Н-5', Н-3'), 4.12-4.01 (m, 4Н, Н-4', СНСН3, ОСН2 СН2СН2СН3), 2.20-2.12 (m, 1Н, Н-2'), 1.85-1.73 (m, 1Н, Н-2'), 1.61-1.54 (m, 2Н, ОСН2СН2СН2СН3), 1.39-1.31 (m, 5Н, ОСН2СН2СН2СН3, СНСН3), 0.93-0.89 (m, 3Н, ОСН2СН2СН2СН3).
Биологический анализ
Ниже приведены результаты экспериментов с использованием соединений, описанных в вариантах осуществления настоящего изобретения.
Клеточные культуры
Клетки мышиной лейкемии L1210/0 и Т-лимфоцитов человека СЕМ/0 получали из American Type Culture Collection (АТСС) (Rockville, MD). Клетки глиобластомы человека U87 были любезно предоставлены Dr. Е. Menue (Institut Pasteur, Paris, France). Клетки СЕМ/TK-, дефицитные по тимидинкиназе, были любезно предоставлены проф. S. Eriksson (в настоящее время работающем в Uppsala University, Uppsala, Sweden) и проф. A. Karlsson (Karolinska Institute, Stockholm, Sweden). Клетки L1210/TK-, дефицитные по тимидинкиназе, получали из клеток L1210/0 путем селекции клеток, резистентных к 5-бром-2'-dUrd (Balzarini et al., 1982). В результате инфицирования соответствующих клеточных линий микоплазмой Mycoplasma hyorhinis (АТСС) получали персистентно-инфицированные клеточные линии (названные L1210.Hyor и U87.Hyor). Все клеточные линии поддерживали в среде Игла, модифицированной по способу Дульбекко (DMEM) (Invitrogen, Carlsbad, СА), содержащей 10% фетальной телячьей сыворотки (FBS) (Biochrom AG, Berlin, Germany), 10 мМ Hepes и 1 мМ пируват натрия (Invitrogen). Клетки выращивали при 37°C во влажной камере в атмосфере 5% CO2.
Цитостатический анализ
Монослойную культуру клеток (U87 и U87.Hyor) засевали в 48-луночные планшеты для микротитрования (Nunc™, Roskilde, Denmark) в количестве 10000 клеток/на лунку. Через 24 ч добавляли равный объем свежей среды, содержащей исследуемые соединения. На пятый день клетки обрабатывали трипсином и подсчитывали на счетчике Культера (Analis, Suarlee, Belgium). Суспензию клеток (L1210/0, L1210/TK-, L1210.Hyor, CEM/0, CEM/TK-) засевали в 96-луночные планшеты для микротитрования (Nunc™) в количестве 60000 клеток/на лунку в присутствии заданного количества исследуемых соединений. Клетки выращивали и в течение 48 ч (L1210) или 72 ч (СЕМ), и затем количество клеток подсчитывали на счетчике Культера. Концентрацию, обеспечивающую 50%-е ингибирование (ИК50), определяли как концентрацию соединения, которая требуется для уменьшения количества живых клеток на 50%.
Анализ 1. Анализировали биологическую активность соединений в отношении нескольких линий опухолевых клеток (полученные данные приведены ниже в Таблице 1). Результаты представлены в виде значений цитостатической концентрации, требуемой для 50%-го ингибирования клеточной пролиферации (СС50), выраженной в μМ. Использовали следующие клеточные линии: L1210/0 (линию клеток лейкемии), FM3A/0 (линию клеток рака молочной железы), Cem/0 (линию клеток острого лимфобластной лейкемии) и HeLa (цервикальную клеточную линию).
Кроме того, в Таблице 1 для сравнения приведены данные для 5FU, 5-FdUrd и контрольных соединений CPF 382, CPF 437 и CPF 438. Структура CPF 382 приведена выше. Ниже приведены структуры CPF 437 и CPF 438:
Согласно данным, приведенным в Таблице 1, соединения, описанные в настоящем изобретении, могут обладать цитостатической активностью, сравнимой с цитостатической активностью 5-FU или более высокой, при этом данные соединения обладают значительной цитостатической активностью в клетках, дефицитных по нуклеозидкиназе. Такая цитостатическая активность в клетках, дефицитных по нуклеозидкиназе, разительно отличается от соответствующей активности 5-FdUrd.
Согласно данным, приведенным в Таблице 1, в TK--клетках активность соединений, описанных в вариантах осуществления настоящего изобретения, может быть значительно выше, чем активность контрольных соединений CPF 382, CPF 437 и CPF 438.
Анализ 2. Также анализировали активность соединений в клетках, инфицированных микоплазмой (выраженную в %), относительно их активности в неинфицированных клетках. Полученные результаты приведены ниже в Таблице 2. Данные результаты указывают на то, что соединения согласно настоящему изобретению, в отличие от 5-FdURD, могут сохранять высокую активность в клетках, инфицированных микоплазмой. Введение ингибитора тимидинфосфорилазы (TP) восстанавливает цитостатическую активность 5-FdUrd в клеточных культурах, инфицированных микоплазмой, что свидетельствует о негативном влиянии TP на итоговую цитостатическую активность 5-FdUrd. Известно, что инфицирование клеток микоплазмой значительно уменьшает цитостатическую активность нуклеозидов, включая 5-FdUrd, поэтому те из нуклеозидов, которые обладают цитостатической активность в клетках, инфицированных микоплазмой, могут быть более эффективны в лечении пациентов, инфицированных микоплазмой.
В Таблице 2 приведены значения CC50 (выраженные в μМ) 5-FdUrd и соединений, описанных в вариантах осуществления настоящего изобретения, полученные в клетках, неинфицированных и инфицированных микоплазмой, приведены также значения остаточной активности соединений, сохранившейся после инфекции микоплазмой (выраженные в %). “Остаточную активность (%)” рассчитывали как отношение значения CC50, полученного в клетках L1210, к значению СС50, полученному в клетках L1210/Hyor; остаточную активность вычисляли по формуле: CC50L1210×100/CC50L1210/Hyor.
Таблица 2
Дополнительные эксперименты (Анализы 3-8, см. ниже) проводили с использованием соединения CPF 373, описанного в одном из вариантов осуществления настоящего изобретения.
Анализ 3. Цитостатическая активность 5-FdUrd и его пролекарства CPF-373 в TK-компетентных и TK-дефицитных линиях опухолевых клеток
Цитостатическую активность 5-FdUrd и CPF-373 определяли в различных TK-экспрессирующих и TK-дефицитных линиях опухолевых клеток. Согласно данным, приведенным в Таблице 3, цитостатическая активность 5-FdUrd сильно зависит от экспрессии TK. Значение ИК50 данного нуклеозида в клетках L1210/TK- (ИК50: 3,1 μМ) в 4000 раз выше значения ИК50 в клетках дикого типа L1210/0 (ИК50: 0,0008 μМ), и значение ИК50 в клетках СЕМ/TK- (ИК50: 1,5 μМ) в 50 раз выше значения ИК50 в клетках СЕМ/0 (ИК50: 0,028 μМ). В отличие от этого цитостатическая активность CPF-373 (пролекарства нуклеозида 5-FdUrd) остается практически неизменной в клетках, дефицитных по TK, по отношению к активности в клетках дикого типа (ИК50: 0,027 и 0,011 μМ в клетках L1210/TK- и L1210/0 и 0,32 и 0,089 μМ клетках СЕМ/TK- и СЕМ/0, соответственно). Несмотря на то, что в клетках дикого типа L1210/0 и СЕМ/0 цитостатическая активность CPF-373 в 3-10 раз ниже, чем активность 5-FdUrd, в TK-дефицитных линиях опухолевых клеток цитостатическая активность CPF-373 в 5-100 раз выше, чем активность 5-FdUrd (см. Таблицу 3).
Анализ 4. Влияние инфицирования культур опухолевых клеток микоплазмой на цитостатическую активность 5-FdUrd и его пролекарства CPF-373
Культуры клеток L1210/0 инфицировали микоплазмой вида М. hyorhinis (полученные клетки получили название L1210.Hyor). Цитостатическая активность 5-FdUrd существенно снижалась, а именно в 300 раз, в отношении инфицированных микоплазмой клеток L210.Hyor, (ИК50: 0,24 μМ). Кроме того, цитостатическая активность 5-FdUrd в культурах клеток U87.Hyor была снижена в 400 раз по сравнению активностью в отношении неинфицированных клеток U87 (см. Таблицу 3). В отличие от этого соединение CPF-373 (пролекарство нуклеозида 5-FdUrd) имело сходный цитостатический потенциал как в L210/0, так и в L1210.Hyor (ИК50: 0,011 и 0,025 μМ, соответственно). Аналогичные данные были получены для данного пролекарства при исследовании его цитостатической активности в клеточных культурах U87 и U87. Hyor (ИК50: 0,035 и 0,039 μМ, соответственно). Таким образом, в то время как свободный нуклеозид 5-FdUrd значительно теряет свой цитостатический потенциал в линиях опухолевых клеток, инфицированных Mycoplasma hyorhinis, антипролиферативный потенциал его пролекарства CPF-373 не зависит от инфицирования микоплазмой.
Анализ 5. Описанные далее эксперименты проводили с целью оценки стабильности соединения CPF 373 в присутствии тимидинфосфорилазы (TP). Результаты данных экспериментов, проиллюстрированные на Фигурах 9-11, на которых приведены ЯМР-спектры, обсуждаются ниже. Настоящий анализ показал, что соединения, описанные в вариантах осуществления настоящего изобретения, не чувствительны к действию катаболического фермента TP, синтез которого часто увеличен в опухолевых клетках; поэтому активность соединений согласно настоящему изобретению в меньшей степени зависит от катаболического фермента TP, чем активность 5-FdUrd.
Фосфорилазный анализ 5-FdUrd и его ProTide, соединения CPF 373, с использованием тимидинфосфорилазы (TP), выделенной из Escherichia coli
Нуклеозид 5-FdUrd может подвергаться деградации до соответствующего основания 5FU в результате фосфорилазной реакции с использованием фосфорилазы, выделенной из Escherichia coli, а также уридинфосфорилазы, выделенной из опухоли Ehrlich ascite. Предполагается, что данная деградация (см. приведенную ниже Схему) является одной из причин, ограничивающих терапевтическую эффективность 5-FdUrd.
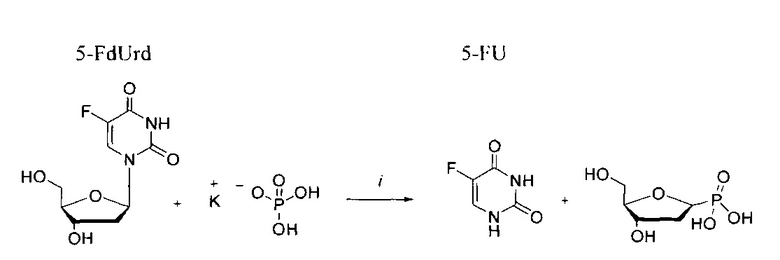
Схема
Фосфорилазный анализ выполняли путем фосфоролиза под действие тимидинфосфорилазы, выделенной из Escherichia coli, с использованием in situ 19F ЯМР. Использование ProTide, соединения CPF 373, представляло собой попытку предотвращения деградации структуры нуклеозида, позволяющую обойти действие соответствующего фермента.
В качестве донора фосфата использовали два калий-фосфатных буфера (pH=7,4, 200 нМ раствор и 300 нМ раствор, соответственно). Единицу фермента определяли как количество фермента, требуемое для гидролиза приблизительно 0,25 мг инозина (используемого в качестве стандарта) в минуту. Анализ проводили в течение 30 мин.
Фосфорилазный анализ 5-FdUrd
Сначала регистрировали 19F-ЯМР-спектры (470 МГц) 5-FdUrd и 5FU, предварительно растворенных в дейтерированном метаноле. В спектре 5-FdUrd присутствовал синглет ~ δ-167.21 м.д., а в спектре 5FU- ~δ-169.30 м.д. Фосфорилазный анализ проводили путем растворения 5-FdUrd в дейтерированном метаноле в присутствии калий-фосфатного буфера (200 нМ раствор; pH=7,4), перед добавление фермента тимидинфосфорилазы (TP) (20,7 ед.) записывали спектр холостой пробы. На 19F-ЯМР-спектре, записанном при 25°C, присутствовал синглет при ~ δ-165.17 м.д., относящийся к 5-FdUrd, и новый пик при ~ δ-169.50 м.д., относящийся к 5FU (см. Фиг.9, спектр А).
Затем для подтверждения расщепления исследуемого нуклеозида до соответствующего основания выполняли новый эксперимент, а именно растворяли эквимолярные количества нуклеозидного аналога 5-FdUrd и соответствующего основания 5FU в описанных выше условиях, но без фермента TS; результаты эксперимента приведены на Фиг.9, спектр В. На полученном спектре присутствует два синглета с теми же химическими сдвигами, которые были зарегистрированы ранее (Фиг.9, спектр А). Полученные данные подтверждают, что в спектре 5FU присутствует пик, соответствующий химическому сдвигу ~ δ-169,50 м.д., то есть имело место фосфоролитическое расщепление под действием фермента ТР. Степень превращения нуклеозида 5-FdURd в свободное основание 5FU составляла 66%.
Когда начальную концентрацию калий-фосфатного буфера увеличивали от 200 нМ до 205 нМ, субстрат 5-FdUrd полностью превращался в основание 5-FU (см. Фиг.10.)
Фосфорилазный анализ ProTide, соединения CPF 373
С целью исследования стабильности проводили фосфорилазный анализ бензил L-аланин фенильного производного CPF 373 в соответствии с методикой и условиями, описанными выше. Сравнение химических сдвигов анализируемого образца в отсутствии фермента TP (см. Фиг.11, спектр А) и в присутствии TP (см. Фиг.11, спектр В) показало, что ProTide, соединение CPF 373, является совершенно стабильным. Согласно данным 19F-ЯМР-анализа, повторно проведенного через 4 суток, ProTide, соединение CPF 373, оставалось все еще стабильным.
Данные эксперименты подтверждают, что нуклеозид 5-FdUrd в фосфорилазной реакции в присутствии тимидинфосфорилазы быстро деградирует до соответствующего основания 5FU; время полужизни этого нуклеозида составляет менее 30 мин, тогда как его пролекарство, соединение CPF 373, демонстрирует абсолютную стабильность в условиях TP-ферментативной активности в течение по меньшей мере 3 суток. Этот важный результат указывает на то, что ProTide-производные нуклеозида 5-FdUrd, описанные в вариантах осуществления настоящего изобретения, могли бы обладать лучшим терапевтическим эффектом, чем 5-FdUrd.
Анализ 6. Инкубация 5-FdUrd и CPF-373 с TP, кодируемой Е. coli, и TP и UP, кодируемыми геномом человека
Субстрат-специфичность тимидинфосфорилазы в отношении природного тимидина (dThd), уридина (Urd), 5-FdUrd и CPF-373 изучали с использованием жидкостной хроматографии высокого давления (ВЭЖХ). Реакционные смеси, содержащие исследуемое соединение (100 μМ) и рекомбинантную TP или UP (TP человека: 8,6 нг/мкл; TP Е. coli: 3,0 нг/мкл; UP человека: 4 нг/мкл; общий объем реакционного буферного раствора: 500 мкл, состав буферного раствора: 10 мМ Трис-HCl, 300 μМ NaCl, 1 мМ ЭДТА, 2 мМ KH2PO4/K2HPO4), инкубировали при комнатной температуре. В разные моменты времени (а именно через 0, 20, 40 мин) из реакционных смесей отбирали аликвоты объемом 100 мкл, которые нагревали в течение 3 мин при 95°C с целью инактивации фермента. Полученные продукты реакции разделяли на колонке для обратнофазовой хроматографии RP-8 (Merck, Darmstadt, Germany), и количество продуктов определяли с использованием ВЭЖХ-анализа (Alliance 2690, Waters, Milford, MA). Разделение dThd и тимина выполняли с использованием линейного градиента буферного раствора для разделения (50 мМ NaH2PO4 и 5 мМ гептансульфоновая кислота, pH 3,2) [(98% буферного раствора для разделения + 2% ацетонитрила) - (20% буферного раствора для разделения + 80% ацетонитрила)] (в течение 8 мин: 98% буферного раствора для разделения + 2% ацетонитрила; в течение 5 мин линейный градиент: [(98% буферного раствора для разделения + 2% ацетонитрила) - (20% буферного раствора для разделения + 80% ацетонитрила)]; в течение 10 мин: 20% буферного раствора для разделения + 80% ацетонитрила; затем колонку уравновешивали: 98% буферного раствора для разделения + 2% ацетонитрила). УФ-регистрацию выполняли при 267 нм. Разделение Urd и урацила выполняли с использованием линейного градиента буферного раствора для разделения (см. выше) [(100% буферный раствор для разделения) - (60% буферного раствора для разделения + 40% ацетонитрила)] (в течение 3 мин: 100% буферный раствор для разделения; в течение 6 мин линейный градиент: [(100% буферный раствор для разделения) - (60% буферного раствора для разделения + 40% ацетонитрила)]; в течение 6 мин: 60% буферного раствора для разделения + 40% ацетонитрила; затем колонку уравновешивали 100% буферным раствором для разделения). УФ регистрацию выполняли при 258 нм.
Фосфоролиз 5-FdUrd и CPF-373 под действием тимидинфосфорилазы и уридинфосфорилазы
5-FdUrd и его пролекарство CPF-373 инкубировали с очищенной тимидинфосфорилазой Е. coli или эритроцитов человека или с очищенной уридинфосфорилазой опухоли человека. В то время как dThd и 5-FdUrd быстро превращались под действием TP Е. coli и TP человека в соответствующие свободные основания, CPF-373 был абсолютно стабилен в присутствии данных ферментов (Фиг.2). В аналогичных экспериментальных условиях уридин превращался в урацил под действием UP человека, но был стабилен в присутствии TP Е. coli или TP человека. Когда dThd и CPF-373 инкубировали с UP, оба соединения оставались нечувствительными к действию данного фермента, тогда как 5-FdUrd подвергался незначительному гидролизу (Фиг.2, панель С).
Анализ 7. Измерение активности тимидилатсинтазы (TS)
TS-активность интактных клеток L1210/0 и L1210/TK- измеряли путем оценки высвобождения трития из [5-3H]dUMP (образованного в клетках из [5-3H]dUrd или [5-3H]dCyd) в реакции, катализируемой TS. Данная методика подробно описана Balzarini & De Clercq (1984). Вкратце, готовили клеточные культуры (культуральная среда DMEM, 500 мкл), содержащие ~3×106 клеток L1210 и подходящее количество исследуемых соединений (5-FdUrd и CPF-373). Клетки инкубировали с данными соединениями в течение 30 мин, 2 ч и 4 ч при 37°C, и затем к клеточным культурам добавляли 1 μКи [5-3H]dUrd или [5-3H]dCyd. Инкубировали в течение 30 мин, затем отбирали по 100 мкл клеточных суспензий, которые добавляли к холодной суспензии активированного угля (500 мкл, 100 мг/мл в 5% ТХУ) (VWR, Haasrode, Belgium). Через 10 мин суспензии центрифугировали при 13000 об./мин в течение 10 мин, и затем на жидкостном сцинтилляторе OptiPhase HiSafe (Perkin Elmer, Waldham, MA) подсчитывали радиоактивность супернатантов (400 мкл).
Ингибирование тимидилатсинтазы (TS) соединениями 5-FdUrd и CPF-373
Главной мишенью цитостатической активности 5-FdUrd является тимидилатсинтаза (TS). TS-активность интактных опухолевых клеток можно сразу регистрировать путем измерения высвобождения трития в интактных клеточных культурах L1210/0, которые инкубируют с [5-3Н]дезоксиуридином ([5-3H]dUrd) или [5-3Н]дезоксицитидином ([5-3H]dCyd). Действительно, в результате внутриклеточного превращения [5-3H]dUrd или [5-3H]dCyd в [5-3H]dUMP атом трития в положении С-5 пиримидинового основания высвобождается в ходе TS-катализируемого восстановительного метилирования. Поэтому оценивали способность 5-FdUrd и его пролекарства CPF-373 ингибировать высвобождение трития из [5-3H]dUrd и [5-3H]dCyd в культурах клеток L1210/0 при различных концентрациях соединений. Найдено, что 5-FdUrd является мощным ингибитором TS in situ. ИК50 данного соединения, определенная по высвобождению трития из [5-3H]dCyd и [5-3H]dUrd. приблизительно равна 0,0007-0,0009 μМ (см. Таблицу 4).
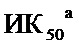
Согласно данным, полученным в экспериментах по высвобождению трития, ингибирующая активность CPF-373 (ИК50: 0,16-0,19 μМ) намного меньше (примерно в 200 раз) ингибирующей активности 5-FdUrd, особенно если предварительная инкубация с данными лекарствами продолжалась только 30 мин. Однако при увеличение времени предварительной инкубации (до 4 ч) клеток с 5-FdUrd и CPF-373 перед измерением TS-активности в интактных опухолевых клетках ингибирующая активность пролекарства в отношение TS in situ увеличивалась намного больше, чем активность 5-FdUrd (Фиг.3). Действительно, если при более продолжительной предварительной инкубации с 5-FdUrd ингибирование высвобождения 3H увеличивалось только в 2 раза, ингибирующая активность CPF-373 увеличивалось в 10 раз (Фиг.3, секции A и B, C и D).
Предварительная инкубация опухолевых клеток с 5-FdUrd и CPF-373 в течение по меньшей мере 4 ч приводит к ингибированию TS в интактных опухолевых клетках при концентрациях лекарств, которые вполне сравнимы с концентрациями данных лекарств, необходимыми для проявления их цитостатической активности, обеспечивающей 50%-е ингибирование.
Таким образом, приведенные результаты указывают на то, что для пролекарства нуклеозида 5-FdUrd требуется несколько стадий метаболических превращений до взаимодействия с TS-мишенью, приводящего к ингибированию данного фермента, и подтверждают мнение о том, что CPF-373 действует в качестве эффективного пролекарства нуклеозида 5-FdUrd, что и определяет в конечном счете его цитостатическую активность.
Активность TS в присутствии 5-FdUrd и CPF-373 также измеряли в интактных клетках L1210/TK-, используя в качестве субстрата, добавляемого в клеточную культуру, [5-3H]dCyd (так как [5-3H]dUrd не может быть использован в клетках, дефицитных по TK). Как показано в Таблице 5 и на Фиг.3 (секции E и F), концентрация 5-FdUrd, требуемая для 50%-го ингибирования TS в клетках L1210/TK-, дефицитных по TK, в 5700 раз ниже (ИК50: 1,4 μМ), чем для ингибирования TS в клетках дикого типа L1210/0 (ИК50: 0,0003 μМ). В отличие от этого ингибирующая активность CPF-373 в отношении TS остается практически неизменной в клетках L1210/TK- (ИК50: 0,053 μМ в клетках L1210/TK- versus 0,013 μМ в клетках L1210/0).
Анализ 8. Анализ стабильности
Анализ карбоксипептидазы Y (ЕС 3.4.16.1)
Стабильность пролекарства CPF-373 в условиях ферментативной активности карбоксипептидазы Y изучали с использованием in situ 31P-ЯМР (202 МГц). Данный эксперимент проводили следующим образом: соединение CPF-373 (3,0 мг) растворяли в d6-ацетоне (150 мкл), и добавляли буфер TRIZMA рН 7,6 (300 мкл). Полученный раствор помещали в пробирку для ЯМР-анализа, и при 25°C записывали 31P-ЯМР-спектр данной холостой пробы. Фермент карбоксипептидазу Y (0,2 мг) растворяли в буфере TRIZMA (150 мкл) и добавляли в пробирку ЯМР-анализа, уже содержащую раствор фосфорамидатного производного. Затем данную пробирку помещали в ЯМР-спектрометр, который настраивали на проведение 31P-ЯМР-эксперимента (64 сканирования с интервалом 4 мин в течение 14 ч при 25°C). Полученные данные обрабатывали и анализировали с использованием программы Bruker Topspin 2.1. Карбоксипептидазу Y и буфер TRIZMA получали от Sigma-Aldrich.
Сыворотка человека
Стабильность пролекарства CPF-373 в присутствии сыворотки человека изучали с использованием in situ 31P-ЯМР (202 МГц). ProTide CPF-373 (1) (5,0 мг) растворяли в ДМСО (0,05 мл) и D2O (0,15 мл). Затем полученные образцы переносили в пробирку для ЯМР-анализа, которую помещали в камеру для проведения ЯМР при 37°C (достаточное количество растворителя позволяло записать контрольный ЯМР-спектр холостой пробы). Затем к образцу в пробирке для ЯМР-анализа быстро добавляли 0,3 мл сыворотки человека. ЯМР-спектрометр программировали на запись спектров с интервалом 15 мин в течение 12,5 ч. Из-за избыточного фона и плохих профилей шиммирования (обусловленных, скорее всего, присутствием биологической среды в относительно высокой концентрации) индивидуальные спектры обрабатывали с использованием дополнительных методик. После обработки с использованием нормализованного преобразования Фурье для каждого спектра выполняли обратное преобразование (деконволюцию Лоренца-Гаусса), чтобы определить частоту и площадь спектральных пиков за вычетом фона. Полученные данные обрабатывали и анализировали с использованием программы Bruker Topspin 2.1.
Буферный раствор (pH 1)
Стабильность пролекарства CPF-373 к гидролизу при pH=1 изучали с использованием in situ 31P-ЯМР (202 МГц). ProTide CPF-373 (1) (2,6 мг) растворяли в MeODe (0,1 мл), и затем добавляли 0,5 мл буферного раствора (pH=1), полученного из равных частей 0,2 М HCl и 0,2 М KCl. Затем полученный образец переносили в пробирку для ЯМР-анализа, и 31Р-ЯМР-эксперимент проводили при 37°C, записывая спектры с интервалом 12 мин в течение 14 ч. Полученные данные обрабатывали и анализировали с использованием программы Bruker Topspin 2.1.
Буферный раствор (рН 8)
Стабильность пролекарства CPF-373 к гидролизу при pH=8 изучали с использованием in situ 31Р-ЯМР (202 МГц). ProTide CPF-373 (1) (4,9 мг) растворяли в MeODe (0,1 мл), и затем добавляли 0,5 мл буферного раствора (pH=8), полученного из 0,1 М раствора Na2HPO4, pH которого подводили 0,1 М раствором HCl. Затем полученный образец переносили в пробирку для ЯМР-анализа, и 31Р-ЯМР-эксперимент проводили при 37°C, записывая спектры с интервалом 12 мин в течение 14 ч. Полученные данные обрабатывали и анализировали с использованием программы Bruker Topspin 2.1.
Изучение стабильности
Изучение химической стабильности пролекарства CPF-373 (1) проводили с использованием in situ 31Р-ЯМР путем инкубации данного соединения с сывороткой человека и с водными буферными растворами (pH 1,0 и 8,0). В каждом эксперименте указанный ProTide растворяли в подходящем дейтерированном растворителе, и полученные образцы анализировали при 37°C, выполняя сканирование через равные интервалы времени в течение приблизительно 14 ч. Для сравнения представлены исходные спектры (нижние графики) и спектры более высокого разрешения, полученные после деконволюции (верхние графики). Согласно результатам анализа стабильности фосфорамидата CPF-373 (1) путем инкубации с сывороткой человека 73% исходного соединения подвергается гидролизу в течение 12,5 ч (см. Фиг.6).
На приведенных спектрах виден синглетный пик, соответствующий сыворотке человека (~δ 2.00), и дублет, соответствующий исходному соединению (~δ 4.50), которое через 4,25 ч подвергалось гидролизу с образованием аминоацилфосфорамидатного промежуточного соединения, которому соответствует синглетный пик (δ 7.20).
Полная стабильность пролекарства CPF-373 (1) как в кислом, так и в щелочном буфере была подтверждена путем химического гидролиза в критических экспериментальных условиях, а именно при pH 1,0 и pH 8,0 при 37°C. Спектры записывали в течение 14 ч, выполняя сканирование через равные интервалы времени (12 мин) (см. Фиг.7 и 8). В спектрах ProTide (1), полученных при pH 1,0, на протяжении всего анализа присутствовал постоянный дублет, соответствующий диастереоизомерам (δ 4.35; 4.50) (Фиг.7).
В спектрах, полученных при pH 8,0, также присутствовал постоянный пик, соответствующий пролекарству (1) (δ 4.48), и синглетный пик, соответствующий буферному раствору (δ 2.55) (Фиг.8).
Метаболизм 5-FdUrd фосфорамидатов
На Фиг.4 приведен предполагаемы механизм активации ProTide внутри клетки (после поглощения клеткой), включающий, в качестве первой стадии, ферментативную активацию, опосредованную ферментом типа карбоксипептидазы, который гидролизирует сложноэфирную связь аминоацильной группировки (стадия a), последующую спонтанную циклизацию и последующее спонтанное вытеснение арильной группы (стадия b) и стадию размыкания образовавшегося нестабильного кольца под действием воды (стадия c). Заключительная стадия включает гидролиз связи P-N, опосредованный ферментом типа фосфорамидазы (стадия d), с высвобождением нуклеозидмонофосфата в интактные клетки (Фиг 4) (McGuigan et al., 2009; Mehellou et al., 2010).
Для доказательства предложенной Схемы метаболического превращения CPF-373 (1) и доказательства отщепления сложноэфирного мотива фосфорамидатного производного нуклеозида 5-FdUrd проводили эксперимент с использованием ферментативной инкубации, имитирующей первые стадии предполагаемой активации в интактных опухолевых клетках. Соединение (1) инкубировали с карбоксипептидазой Y (также известной как катепсин А) в буфере TRIZMA, за превращением соединения (1) наблюдали с помощью 31Р-ЯМР. Спектры записывали в течение 14 ч, выполняя сканирование через равные интервалы времени (4 мин) (см. Фиг.5). Для сравнения на Фиг.5 представлены как исходные спектры (нижние графики), так и спектры более высокого разрешения, полученные после деконволюции (верхние графики).
На 31Р-ЯМР-спектрах пролекарство CPF-373 (1) представлено двумя пиками (δ 4.07 и 4.23), соответствующими двум его диастереоизомерам; считается, что данные пики принадлежат исходному соединению с характерным удвоением хирального фосфатного центра фосфорамидата. После добавления катепсина А данное соединение быстро (в течение 4 мин) подвергалось гидролизу с образованием промежуточных соединений (δ 4.95 и 5.16), которые утратили сложноэфирный мотив; однако данные промежуточные соединения не сохранялись, а, в свою очередь, быстро подвергались метаболическому превращению через потерю арильной группы (Фиг.4, стадии а-с) с образованием аминоацилфосфорамидатного промежуточного соединения, конечного продукта в данном анализе. Данное промежуточное соединение представлено на спектрах синглетным пиком (δ 6.85), обусловленным присутствием ахирального фосфатного центра. Таким образом, анализ спектров, записанных в ходе данного эксперимента, выявил быстрое метаболическое превращение исходного соединения (~δ 4.00), в том числе его полное превращение в течение 26 мин в предполагаемое промежуточное соединение, присутствие которого регистрировали на протяжении всего 14-часового анализа. Как и ожидалось, расщепление связи P-N с высвобождением нуклеозидмонофосфата не было обнаружено в эксперименте с ферментами. Данный эксперимент показывает, что первая стадия активации ProTide CPF-373 (1) может быть достаточно эффективной и поэтому может обеспечить, в конечном счете, доставку нуклеозидмонофосфатного метаболита в интактные опухолевые клетки.
Заключение
В заключение, в настоящем изобретении предложены новые фосфорамидатные нуклеотидные пролекарства нуклеозидного аналога 5-фтор-2'-дезоксиуридина (5-FdUrd), обладающего противораковой активностью, а также описан синтез данных соединений и анализ их цитостатической активности. В отличие от 5-FdUrd, который по существу теряет свою цитостатическую активность в культурах клеток мышиной лейкемии L1210, дефицитных по тимидинкиназе (TK), и в культурах лимфоцитов человека СЕМ, дефицитных по TK, соединения согласно настоящему изобретению, например CPF-373, в значительной степени сохраняют свою антипролиферативную активность в опухолевых клетках как дикого типа, так и дефицитных по TK, и, соответственно, их цитостатическая активность в основном не зависит от внутриклеточной TK-активности. Например, найдено, что CPF-373 ингибирует тимидилатсинтазу (TS) в клеточных линиях, дефицитных по TK, и в клеточных линиях дикого типа при концентрациях лекарства, которые хорошо коррелируют с его цитостатической активностью в этих клетках. CPF-373, по-видимому, является нечувствительным к инактивации катаболитическими ферментами, такими как тимидинфосфорилаза (TP) и уридинфосфорилаза (UP). Эти наблюдения согласуются с данными, согласно которым 5-FdUrd, но не CPF-373, по существу теряет свою цитостатическую активность в присутствии TP-экспрессирующей микоплазмы в культурах опухолевых клеток. Поэтому, соединения согласно настоящему изобретению, такие как CPF-373, являются новыми фосфорамидатными пролекарствами нуклеозида 5-FdUrd, которые (i) могут обходить возможные механизмы устойчивости опухолевых клеток (например уменьшение TK-активности) и (ii) не подвержены деградации катаболитическими ферментами, такими как TP, которые имеют повышенную активность в опухолевых клетках или экспрессируются в опухолевых тканях, инфицированных микоплазмой. Публикация Vande Voorde, J. et al. Biochemical Pharmacology 82 (2011) 441-452 включена в настоящее описание посредством ссылки во всей полноте.
Описанные ниже варианты осуществления настоящего изобретения раскрыты в работе McGuigan, С. et al. J. Med. Chem. 2011, 54 7247-7258 (опубликованной 05 сентября 2011), содержание которой включено в данное описание посредством ссылки во всей полноте.
В Таблице 6, приведенной ниже, представлена цитостатическая активность 5-FU, 5-FdUrd, контрольного соединения CPF382 и соединений, описанных в вариантах осуществления настоящего изобретения, в отношении линий опухолевых клеток в терминах ИК50, то есть концентрации соединения, требуемой для ингибирования пролиферации опухолевых клеток на 50%. Приведенные значения представляют собой среднее (±SD (среднеквадратическое отклонение)) по меньшей мере двух-четырех независимых экспериментов. В Таблице 6 идентифицирован фосфорамидатный мотив контрольного соединения CPF382 и соединений, описанные в вариантах осуществления настоящего изобретения, в той части, которая относится к ʺарилуʺ, который соответствует Ar в Формуле I и представляет собой либо фенил (Ph), либо 1-нафтил (Nap); ʺсложному эфируʺ, который соответствует R3 в Формуле I; и ʺААʺ, который описывает аминокислоту, у которой атом альфа-C и заместители у атома альфа-C соответствуют CR1R2 в Формуле I. В Таблице 6 раскрыты соединения согласно вариантам осуществления настоящего изобретения, не указанные ранее в Таблице 1, а также дополнительные данные для некоторых соединений, приведенных в Таблице 1.
В Таблице 7, приведенной ниже, представлена цитостатическая активность 5-FdUrd, контрольного соединения CPF382 и соединений, описанных в вариантах осуществления настоящего изобретения, измеренная в культурах клеток мышиной лейкемии L1210 дикого типа (L1210/0) и в клеточных культурах L1210, инфицированных Mycoplasma hyorhinis (L1210.Hyor), в терминах ИК50, то есть концентрации соединения, требуемой для ингибирования пролиферации опухолевых клеток на 50%. Приведенные значения представляют собой среднее (±SD) по меньшей мере двух-четырех независимых экспериментов. В Таблице 7 идентифицирован фосфорамидатный мотив контрольного соединения CPF382 и соединений, описанных в вариантах осуществления настоящего изобретения, аналогично Таблице 6, за тем исключением, что ʺNaphʺ соответствует 1-нафтилу. В Таблице 7 раскрыты соединения согласно вариантам осуществления настоящего изобретения, не указанные ранее в Таблице 2, а также дополнительные данные для некоторых соединений, приведенных в Таблице 2.
В Таблице 8, приведенной ниже, представлена цитостатическая активность 5-FdUrd и соединений, описанных в вариантах осуществления настоящего изобретения, измеренная в клеточных культурах СЕМ, содержащих переносчик hEnt1 (Cem/hEnt-1) или лишенных данного переносчика (Cem/hEnt-0), в терминах ИК50, то есть концентрации соединения, требуемой для ингибирования пролиферации опухолевых клеток на 50%. Приведенные значения представляют собой среднее (±SD) по меньшей мере двух-четырех независимых экспериментов. В Таблице 8 идентифицирован фосфорамидатный мотив соединений, описанных в вариантах осуществления настоящего изобретения, аналогично Таблице 6, за тем исключением, что ʺNaphʺ соответствует 1-нафтилу. Согласно данным Таблицы 8, цитостатическая активность соединений, описанных в вариантах осуществления настоящего изобретения, в меньшей степени зависит от присутствия переносчика hENT1, чем активность 5-FdUrd, так как антипролиферативная активность указанных соединений в отношении клеток СЕМ, дефицитных по hENT1, уменьшается только в 7-15 раз. Данные наблюдения согласуются с уменьшением цитостатической активности соединений, описанных в вариантах осуществления настоящего изобретения, в присутствии ингибиторов транспорта (а именно дипиридамола и NBMPR) только в 2-7 раз в сравнении с уменьшением антипролиферативной активности 5-FuDrd и FdUMP, определенной в аналогичных экспериментальных условиях, в 20-60 раз.
Далее приведено описание анализа соединения CPF 381.
Ферментативный фосфорилазный анализ проводили с использованием тимидинфосфорилазы (TP), выделенной из Esherichia coli, в присутствии калий-фосфатного буфера (300 нМ раствор. pH 7,4). В 19F-ЯМР-спектрах, записанных через 5 мин, 14 ч и 72 ч, не было обнаружено каких-либо доказательств фосфоролиза. В отличие от 5-FdUrd соединение CPF 381 в лучшем случае представляет собой очень плохой субстрат для тимидинфосфорилазы, если вообще является таковым.
Химический гидролиз проводили в экспериментальных условиях при pH 1 и pH 8; результаты регистрировали с использованием 31Р-ЯМР. На всем протяжении анализа (14 ч), выполняемого в кислых условиях (pH 1), регистрировали только два пика, соответствующие двум диастереоизомерам. Отсутствие новых сигналов в 31P-ЯМР-спектре указывало на то, что соединение CPF 381 является высокостабильным в кислой среде. Аналогичный результат был получен, когда соединение CPF 381 подвергали анализу в присутствии слабого основания (pH 8).
Далее приведено описание анализа соединения CPF 581.
Ферментативный анализ проводили с использованием карбоксипептидазы Y. Соединение CPF 581, карбоксипептидазу Y и буфер TRIZMA (pH 7,6) растворяли в d6-ацетоне; 31P-ЯМР-спектры (202 МГц) записывали через равные интервалы (каждые 7 мин) в течение 14 ч. Соединение CPF 581 быстро подвергалось гидролизу с образованием первого метаболита, который утратил сложноэфирную группировку (R3), при этом превращение двух диастереоизомеров происходило с приблизительно одинаковой скоростью. Дальнейшее превращение первого метаболита приводило приблизительно в течение 45 мин к образованию второго метаболита в форме аниона, утратившего Ar (то есть время полужизни первого метаболита составляло менее 5 мин). Соответственно, скорость начальной стадии активации может считаться, в общем случае, одним из условий, необходимых для хорошей биологической активности фосфорамидатов. Путем химического гидролиза соединения CPF 373 в присутствии триэтиламина и воды получали диаммонийную соль второго анионного метаболита, которую добавляли к конечному продукту данного анализа, полученному из соединения CPF 373, то есть содержащему только ферментативный второй метаболит, полученный из соединения CPF 581, в буфере TRIZMA. В 31Р-ЯМР спектре данного образца присутствовал только один пик (δР 6.85 м.д.), что является серьезным доказательством данной части метаболического пути и активации фосфорамидатных соединений согласно настоящему изобретению.
Далее приведено описание анализа соединения CPF 386.
Стабильность соединения CPF 386 в присутствии сыворотки человеке изучали с использованием in situ 31Р-ЯМР. Сначала записывали контрольные 31P-ЯМР-спектры соединения CPF 386 в ДМСО и D2O. Затем ЯМР-образец обрабатывали сывороткой человека, и сразу проводили 31P-ЯМР-анализ при 37°C. 31P-ЯМР-спектры записывали каждые 15 мин в течение 14 ч. На полученных спектрах присутствовал один пик, соответствующий сыворотке человека (~δР 2.00 м.д.) и два пика, соответствующие соединению CPF 386 (~δР 4.59 и 4.84 м.д.). Приблизительно через 6,75 ч соединение частично подвергалось гидролизу с образованием промежуточного соединения, утратившего R3 (Et), которому соответствовал один пик (δP 5.79 м.д.). Через 11,5 ч регистрировали образование второго метаболита, утратившего Ar (1-нафтил), которому соответствовал один пик (δР 7.09 м.д.). Через 13,5 ч реакционная смесь содержала 96% исходного соединения CPF 386 вместе с предполагаемым первым метаболитом (3%) и вторым метаболитом (1%).
Ссылки
Agarwal RP, Han Т, Fernandez М, Collateral resistance of a dideoxycytidine-resistant cell line to 5-fluoro-2'-deoxyuridine. Biochem Biophys Res Commun 1999; 262:657-60.
Ayusawa D, Koyama H, Iwata K, Seno T, Single-step selection of mouse FM3A cell mutants defective in thymidylate synthetase. Somatic Cell Genet 1980; 6:261-70.
Ayusawa D, Koyama H, Seno T, Resistance to methotrexate in thymidylate synthetase-deficient mutants of cultured mouse mammary tumor FM3A cells. Cancer Res 1981; 41:1497-501.
Balzarini J, De Clercq E, Torrence PF, Metres MP, Park JS, Schmidt CL, Shugar D, Barr PJ, Jones AS, Verhelst G, Walker RT, Role of thymidine kinase in the inhibitory activity of 5-substituted-2'-deoxyuridines on the growth of human and murine tumor cell lines. Biohcem Pharmacol 1982:31:1089-1095
Balzarini J and De Clercq E, Strategies for the measurement of the inhibitory effects of thymidine analogs on the activity of thymidylate synthase in intact murine leukemia L1210 cells. Biochim Biophys Acta 1984; 785:36-45.
Beck A, Etienne MC, Cheradame S, Fischel JL, Formento P, Renee N, Milano G, A role for dihydropyrimidine dehydrogenase and thymidylate synthase in tumour sensitivity to fluorouracil. Eur J Cancer 1994; 30A: 1517-22.
Bronckaers A, Balzarini J, Liekens S, The cytostatic activity of pyrimidine nucleosides is strongly modulated by Mycoplasma hyorhinis infection: Implications for cancer therapy. Biochem Pharmacol 2008; 76:188-97.
Bronckaers A, Gago F, Balzarini J, Liekens S, The dual role of thymidine phosphorylase in cancer development and chemotherapy. Med Res Rev 2009; 29:903-53.
Chan PJ, Seraj IM, Kalugdan TH, King A, Prevalence of mycoplasma conserved DNA in malignant ovarian cancer detected using sensitive PCR-ELISA. Gynecol Oncol 1996; 63:258-60.
Charron J and Langelier Y, Analysis of deoxycytidine (dC) deaminase activity in herpes simplex virus-infected or HSV TK-transformed cells: association with mycoplasma contamination but not with virus infection. J Gen Virol 1981; 57:245-50.
Ciaparrone M, Quirino M, Schinzari G, Zannoni G, Corsi DC, Vecchio FM, Cassano A, La Torre G, Barone C. Predictive role of thymidylate synthase, dihydropyrimidine dehydrogenase and thymidine phosphorylase expression in colorectal cancer patients receiving adjuvant 5-fluorouracil. Oncology 2006; 70:366-77.
Ciccolini J, Evrard A, Cuq P. Thymidine phosphorylase and fluoropyrimidines efficacy: a Jekyll and Hyde story. Curr Med Chem Anticancer Agents 2004; 4:71-81.
Congiatu C, Brancale A, Mason MD, Jiang WG, McGuigan C, Novel potential anticancer naphthyl phosphoramidates of BVdU: separation of diastereoisomers and assignment of the absolute configuration of the phosphorus center. J Med Chem 2006; 49:452-5.
de Bruin M, van Capel T, Smid K, van der Born K, Fukushima M, Hoekman K, Pinedo HM, Peters GJ, Role of platelet derived endothelial cell growth factor/thymidine phosphorylase in fluoropyrimidine sensitivity and potential role of deoxyribose-1-phosphate. Nucleosides Nucleotides Nucleic Acids 2004; 23:1485-90.
Galmarini CM, Mackey JR, Dumontet C, Nucleoside analogues and nucleobases in cancer treatment. Lancet Oncol 2002; 3:415-24.
Harris SA, McGuigan C, Andrei G, Snoeck R, De CE, Balzarini J, Synthesis and antiviral evaluation of phosphoramidate derivatives of (E)-5-(2-bromovinyl)-2'-deoxyuridine. Antivir Chem Chemother 2001; 12:293-300.
Hatse S, De CE, Balzarini J, Role of antimetabolites of purine and pyrimidine nucleotide metabolism in tumor cell differentiation. Biochem Pharmacol 1999; 58:539-55.
Hecker SJ and Erion MD, Prodrugs of phosphates and phosphonates. J Med Chem 2008; 51:2328-45.
Huang S, Li JY, Wu J, Meng L, Shou CC, Mycoplasma infections and different human carcinomas. World J Gastroenterol 2001; 7:266-9.
Ishikawa T, Utoh M, Sawada N, Nishida M, Fukase Y, Sekiguchi F, Ishitsuka H, Tumor selective delivery of 5-fluorouracil by capecitabine, a new oral fluoropyrimidine carbamate, in human cancer xenografts. Biochem Pharmacol 1998; 55:1091-7.
Jette L, Bissoon-Haqqani S, Le FB, Maroun JA, Birnboim HC, Resistance of colorectal cancer cells to 5-FUdR and 5-FU caused by Mycoplasma infection. Anticancer Res 2008; 28:2175-80.
Kamoshida S, Shiogama K, Shimomura R, Inada K, Sakurai Y, Ochiai M, Matuoka H, Maeda K, Tsutsumi Y. Immunohistochemical demonstration of fluoropyrimidine-metabolizing enzymes in various types of cancer. Oncol Rep. 2005. 14:1223-30.
Kidder M, Chan PJ, Seraj IM, Patron WC, King A. Assessment of archived paraffin-embedded cervical condyloma tissues for mycoplasma-conserved DNA using sensitive PCR-ELISA. Gynecol Oncol 1998; 71:254-257.
Kinsella AR and Smith D, Tumor resistance to antimetabolites. Gen Pharmacol 1998; 30:623-6.
Koopman M, Venderbosch S, Nagtegaal ID, van Krieken JH, Punt CJ. A review on the use of molecular markers of cytotoxic therapy for colorectal cancer, what have we learned? Eur J Cancer 2009; 45: 1935-49.
Kosaka T, Usami K, Ueshige N, Sugaya J, Nakano Y, Takashima S. Effects of thymidine phosphorylase levels in cancer, background mucosa, and regional lymph nodes on survival of advanced gastric cancer patients receiving postoperative fluoropyrimidine therapy. Oncol Rep 2004; 12:1279-86.
Liekens S, Bronckaers A, Balzarini J, Improvement of purine and pyrimidine antimetabolite-based anticancer treatment by selective suppression of mycoplasma-encoded catabolic enzymes. Lancet Oncol 2009; 10:628-35.
Longley DB, Harkin DP, Johnston PG, 5-fluorouracil: mechanisms of action and clinical strategies. Nat Rev Cancer 2003; 3:330-8.
McGuigan, C.; Pathirana, R. N.; Balzarini, J.; De Clercq, E. Intracellular delivery of bioactive AZT nucleotides by aryl phosphate derivatives of AZT. J. Med. Chem. 1993, 36, 1048.
McGuigan, C.; Cahard, D.; Hendrika, D.; Sheeka, M.; De Clercq, E.; Balzarini, J. Aryl phosphoramidate derivatives of d4T have improved anti-HIV efficacy in tissue vulture and may act by the generation of a novel intracellular metabolite. J. Med. Chem. 1996, 39, 1748.
McGuigan, C.; Tsang, H. W.; Cahardr, D. Turner, K.; Velazquez, S.; Salgado, A.; Bidois, L.; Naesens, L.; De Clercq, E.; Balzarini, J. Phosphoramidate derivatives of d4T as inhibitors of HIV: The effect of aminoacid variation. Antiviral Res. 1997, 35, 195.
McGuigan, C.; Mehellou, J.; Balzarini, J. Aryloxy phosphoramidate triesters: a technology for delivering mono-phosphorylated nucleosides and sugars into cells. Chem. Med. Chem., 2009, 4, 11, 1779.
McGuigan C, Gilles A, Madela K, Aljarah M, Holl S. Jones S, Vernachio J, Hutchins J, Ames B, Bryant KD, Gorovits E, Ganguly B, Hunley D, Hall A, Kolykhalov A, Liu Y, Muhammad J, Raja N, Walters R, Wang J, Chamberlain S, Henson G, Phosphoramidate ProTides of 2'-C-methylguanosine as highly potent inhibitors of hepatitis С virus. Study of their in vitro and in vivo properties. J Med Chem 2010; 53:4949-57.
Mehellou Y, Balzarini J, McGuigan C, Aryloxy phosphoramidate triesters: a technology for delivering monophosphorylated nucleosides and sugars into cells. ChemMedChem 2009; 4:1779-91.
Mehellou, Y.; Valente, R.; Mottram, H.; Walsby, E.; Mills, K. I.; Balzarini, J.; Mcgugan, C. Phosphoramidates of 2'-β-d-arabinouridine (AraU) as phosphate prodrugs; design, synthesis, in vitro activity and metabolism. Bioorg. Med. Chem. 2010, 18, 2439
Moertel CG, Chemotherapy for colorectal cancer. N Engl J Med 1994; 330:1136-42.
Murakami Y, Kazuno H, Emura T, Tsujimoto H, Suzuki N, Fukushima M, Different mechanisms of acquired resistance to fluorinated pyrimidines in human colorectal cancer cells. Int J Oncol 2000; 17:277-83.
Neale GA, Mitchell A, Finch LR, Enzymes of pyrimidine deoxyribonucleotide metabolism in Mycoplasma mycoides subsp. mycoides. J Bacteriol 1983; 156:1001-5.
Pehlivan M, Pehlivan S, Onay H, Koyuncuoglu M, Kirkali Z, Can mycoplasma-mediated oncogenesis be responsible for formation of conventional renal cell carcinoma? Urology 2005; 65:411-414
Peters GJ and Kohne CH, Resistance to antimetabolites. In: Fluoropyrimidines as Antifolate Drugs in Cancer Therapy. Jackman AL (ed). Humana Press Inc. 1999; pp. 101-145.
Razin S, The mycoplasmas. Microbiol Rev 1978; 42:414-70.
Razin S, Yogev D, Naot Y, Molecular biology and pathogenicity of mycoplasmas. Microbiol Mol Biol Rev 1998; 62:1094-156.
Sasaki H, Igaki H, Ishizuka T, Kogoma Y, Sugimura T, Terada M, Presence of Streptococcus DNA sequence in surgical specimens of gastric cancer. Jpn J Cancer Res 1995; 86:791-4.
Seno T, Ayusawa D, Shimizu K, Koyama H, Takeishi K, Hori T, Thymineless death and genetic events in mammalian cells. Basic Life Sci 1985; 31:241-63.
Seno T, Ayusawa D, Shimizu K, Koyama H, Takeishi K, Hori T, Thymineless death and genetic events in mammalian cells. Basic Life Sci 1985; 31:241-63.
Sinigaglia F and Talmadge KW, Inhibition of [3H]thymidine incorporation by Mycoplasma arginini-infected cells due to enzymatic cleavage of the nucleoside. Eur J Immunol 1985; 15:692-6.
Sotos GA, Grogan L, Allegra CJ, Preclinical and clinical aspects of biomodulation of 5-fluorouracil. Cancer Treat Rev 1994; 20:11-49.
Tanaka F, Fukuse T, Wada H, Fukushima M, The history, mechanism and clinical use of oral 5-fluorouracil derivative chemotherapeutic agents. Curr Pharm Biotechnol 2000; 1:137-64.
Tham TNr Ferris S, Kovacic R, Montagnier L, Blanchard A, Identification of Mycoplasma pirum genes involved in the salvage pathways for nucleosides. J Bacterid 1993; 175:5281-5.
Walko CM and Lindley C, Capecitabine: a review. Clin Ther 2005; 27:23-44.
Yang H, Qu L, Ma H, Chen L, Liu W, Liu C, Meng L, Wu J, Shou C, Mycoplasma hyorhinis infection in gastric carcinoma and its effects on the malignant phenotypes of gastric cancer cells. BMC Gastroenterol 2010; 10:132.
| название | год | авторы | номер документа |
|---|---|---|---|
| Антибактериальные средства на основе производных ципрофлоксацина | 2016 |
|
RU2636751C1 |
| Фосфорил замещенные 3-кето-андрост-4-ен-[16,17-d]пиридазины и способ их получения | 2023 |
|
RU2807870C1 |
| СПОСОБ ПОЛУЧЕНИЯ ЛИТИЕВЫХ КОМПЛЕКСНЫХ СОЛЕЙ ДЛЯ ИСПОЛЬЗОВАНИЯ В ХИМИЧЕСКИХ ИСТОЧНИКАХ ТОКА | 2000 |
|
RU2246499C2 |
| Соединения фторхинолонового ряда на основе производных пиридоксина, обладающие антибактериальными свойствами | 2019 |
|
RU2713932C1 |
| АРИЛФОСФОРАМИДАТЫ НУКЛЕОЗИДА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ПРОТИВОВИРУСНЫХ СРЕДСТВ ДЛЯ ЛЕЧЕНИЯ ВИРУСНОГО ГЕПАТИТА С | 2007 |
|
RU2464272C2 |
| ИЗОИНДОЛИНОНОВЫЕ ИНГИБИТОРЫ ВЗАИМОДЕЙСТВИЯ MDM2-P53, ОБЛАДАЮЩИЕ ПРОТИВОРАКОВОЙ АКТИВНОСТЬЮ | 2016 |
|
RU2794333C1 |
| НОВЫЕ ГЕМ-ДИФТОРИРОВАННЫЕ СОЕДИНЕНИЯ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, И ИХ ПРИМЕНЕНИЕ | 2003 |
|
RU2369612C2 |
| Способ получения @ -арилалкановых кислот | 1986 |
|
SU1574167A3 |
| ЭНАНТИОМЕРНО ЧИСТЫЕ ОСНОВНЫЕ ЭФИРЫ АРИЛ-ЦИКЛОАЛКИЛГИДРОКСИКАРБОНОВЫХ КИСЛОТ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ В ЛЕКАРСТВЕННЫХ СРЕДСТВАХ | 1997 |
|
RU2238936C2 |
| Дезоксиуридинтрифосфаты, маркированные цвитерионными индоцианиновыми красителями | 2017 |
|
RU2725884C2 |
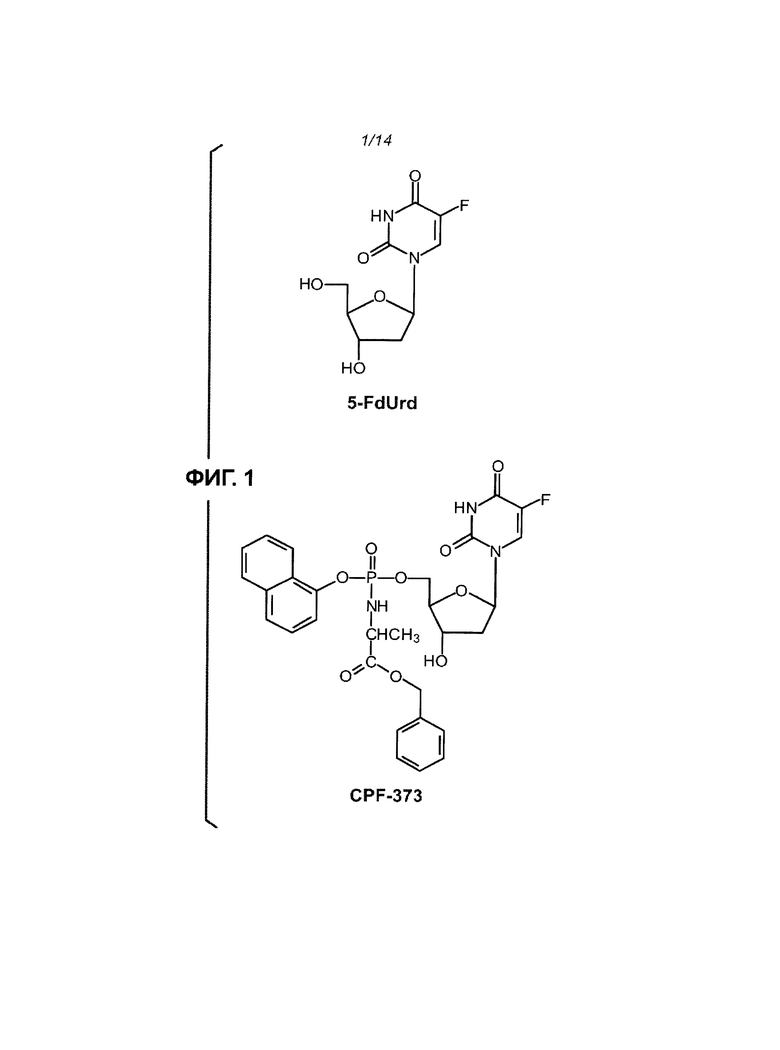
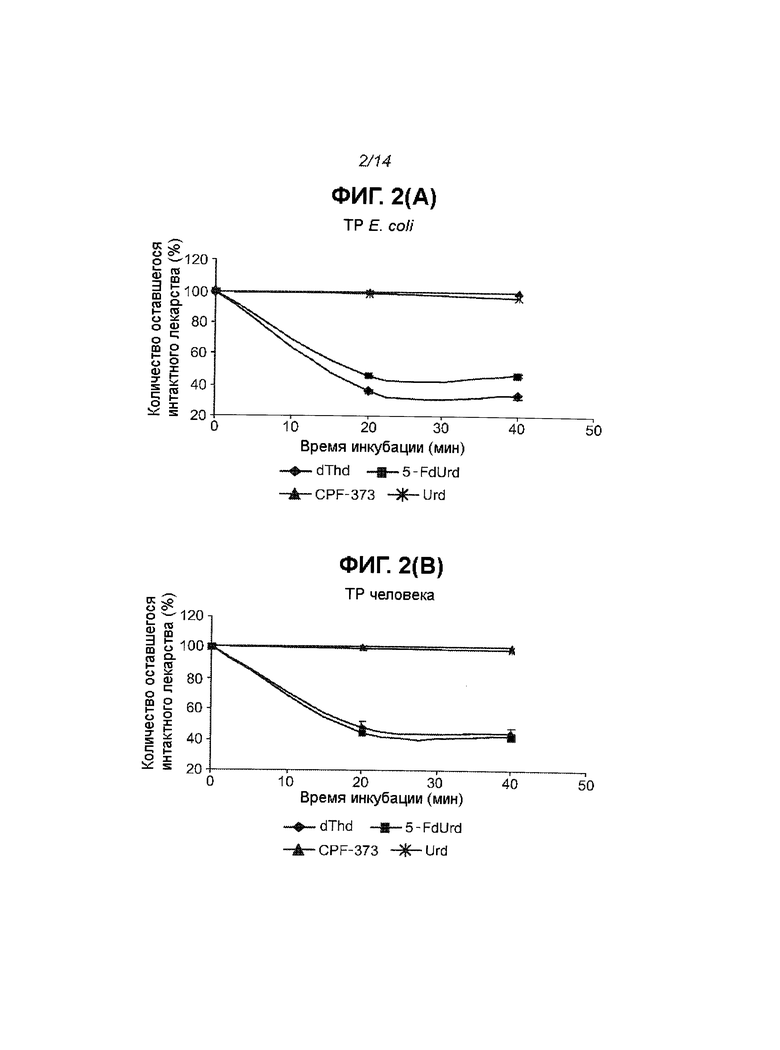
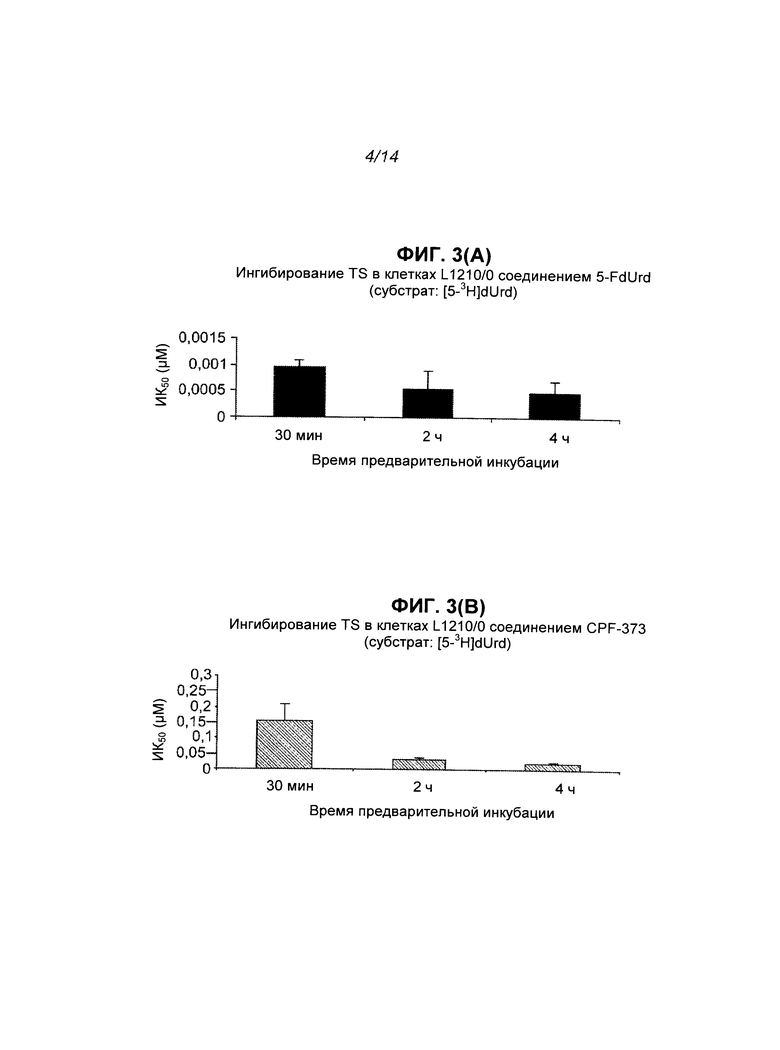
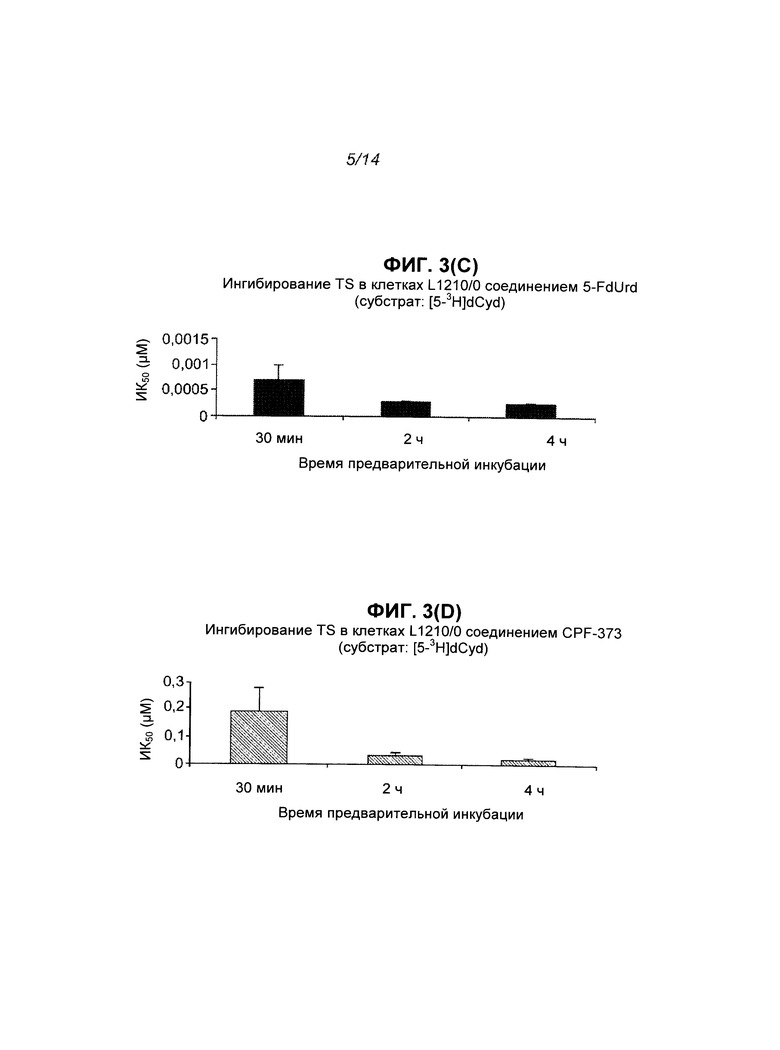
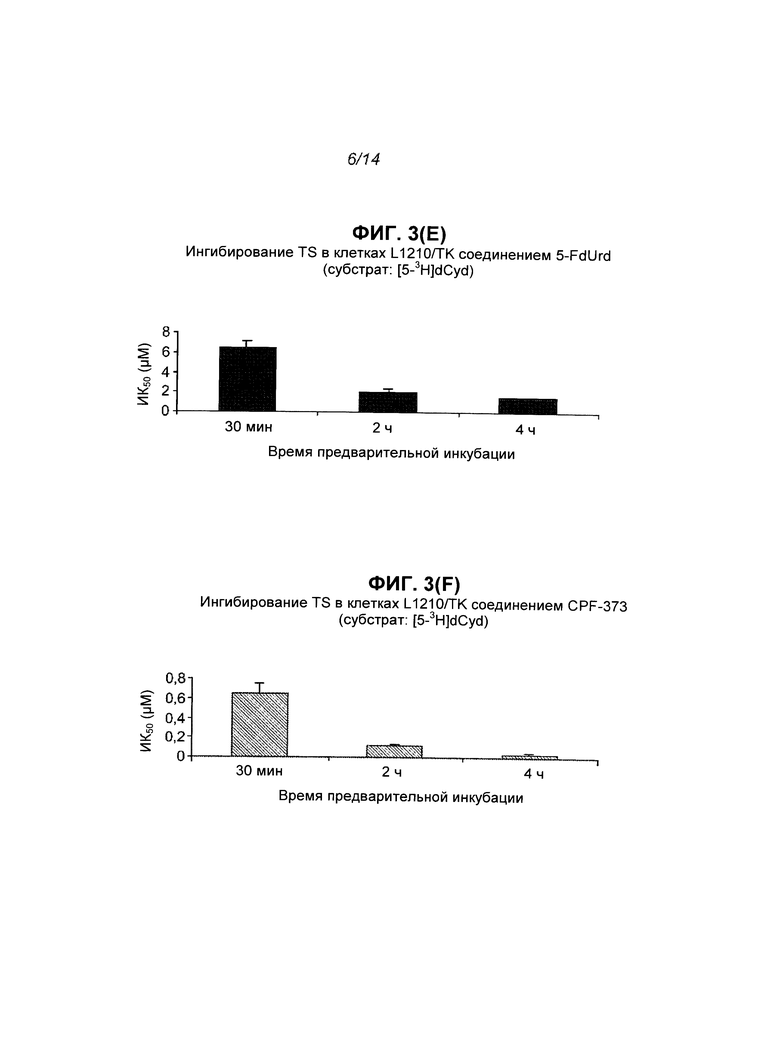
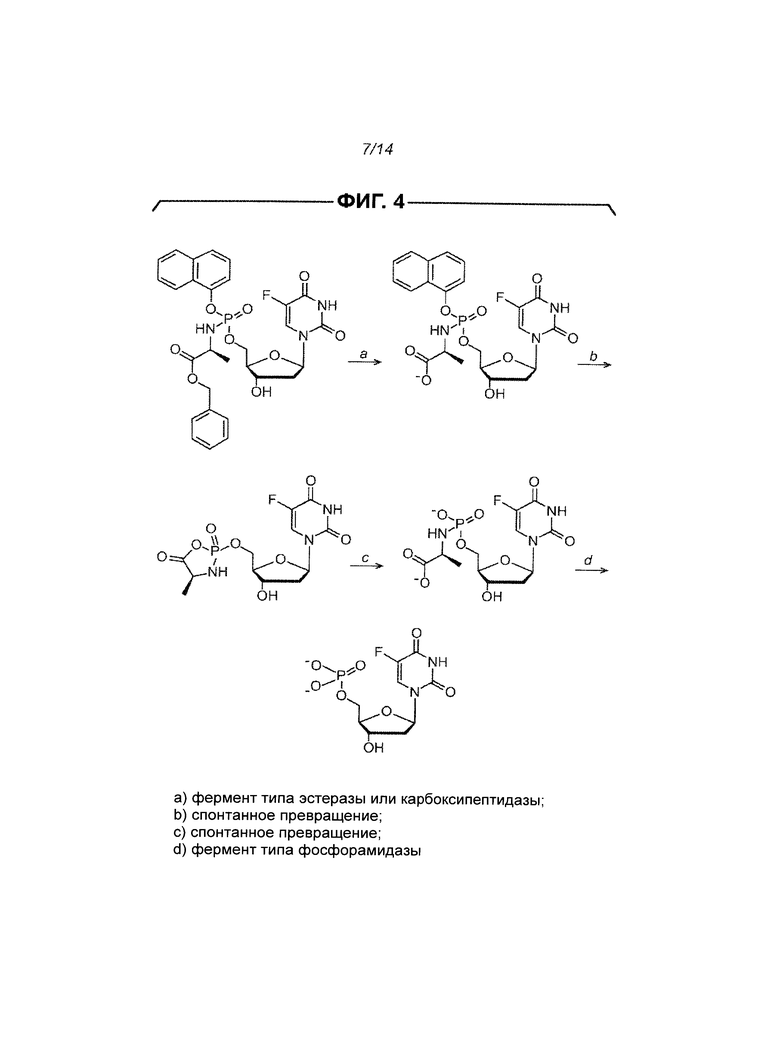
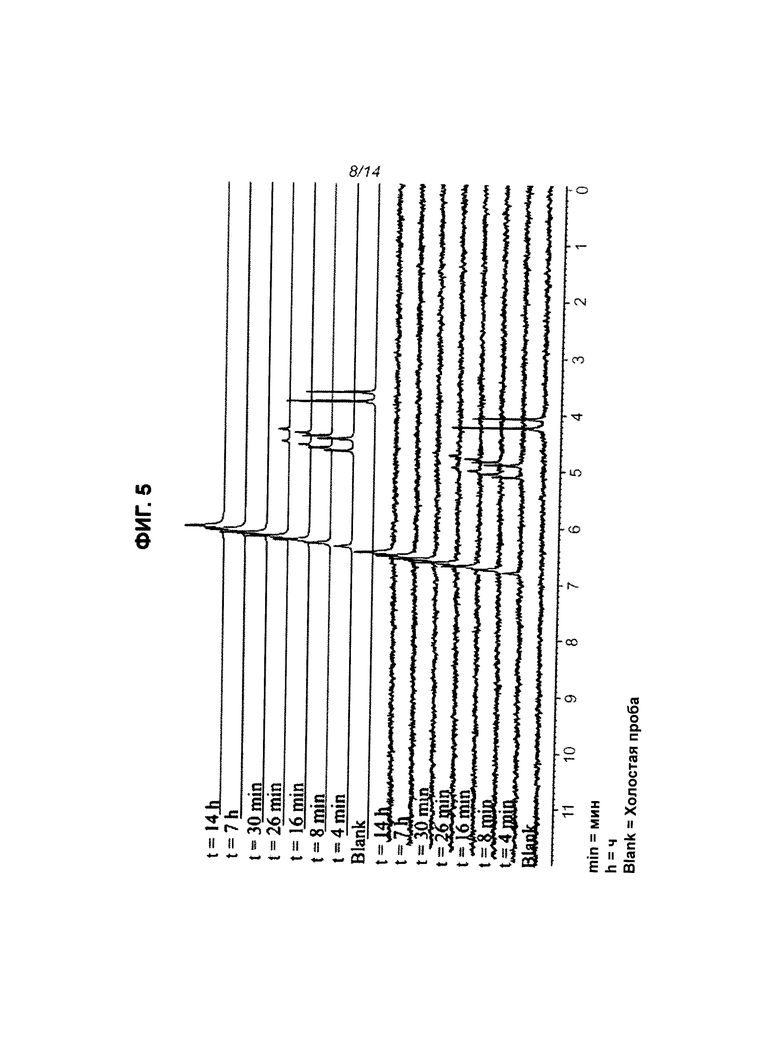
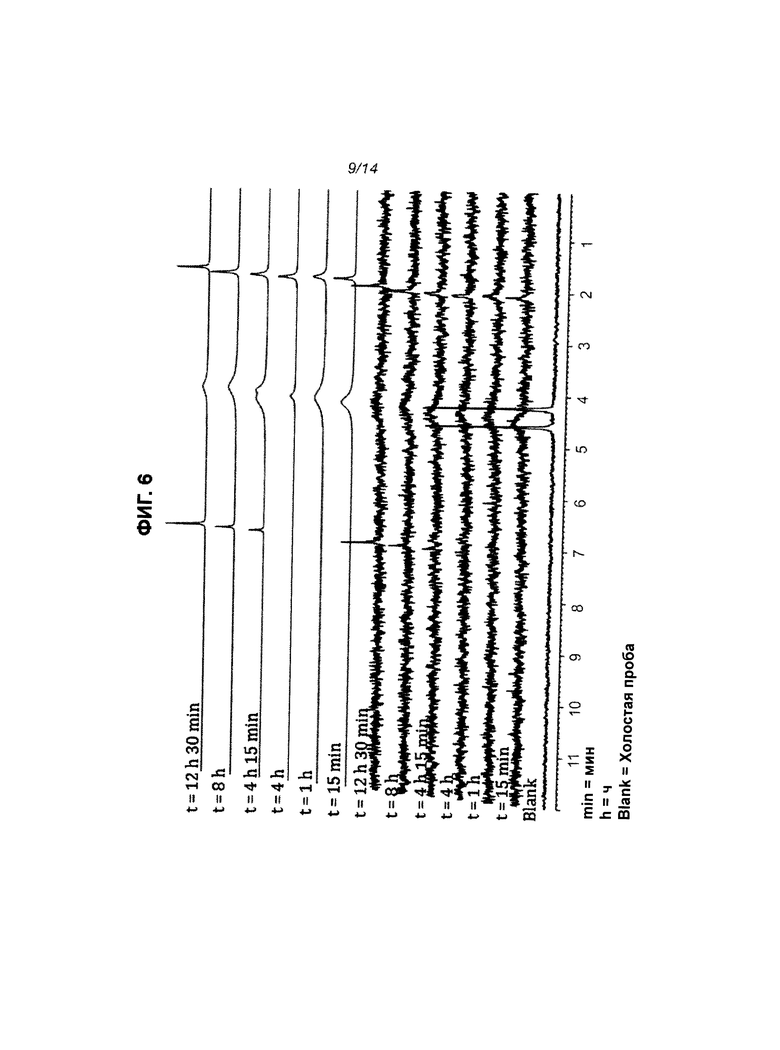
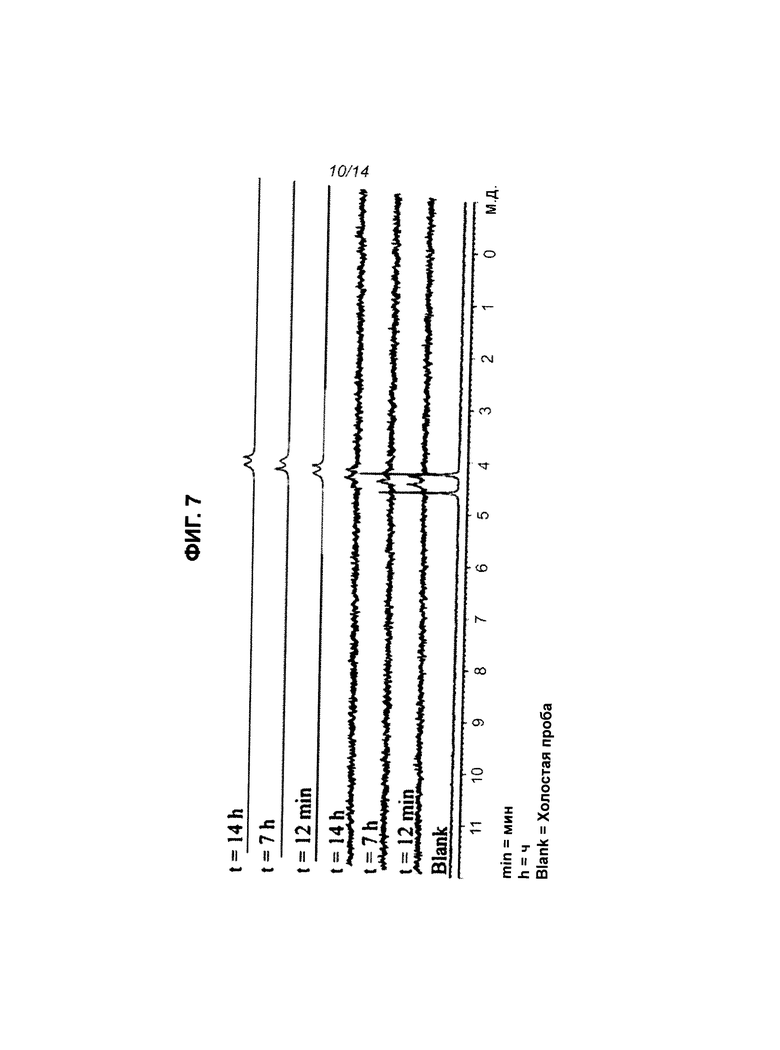
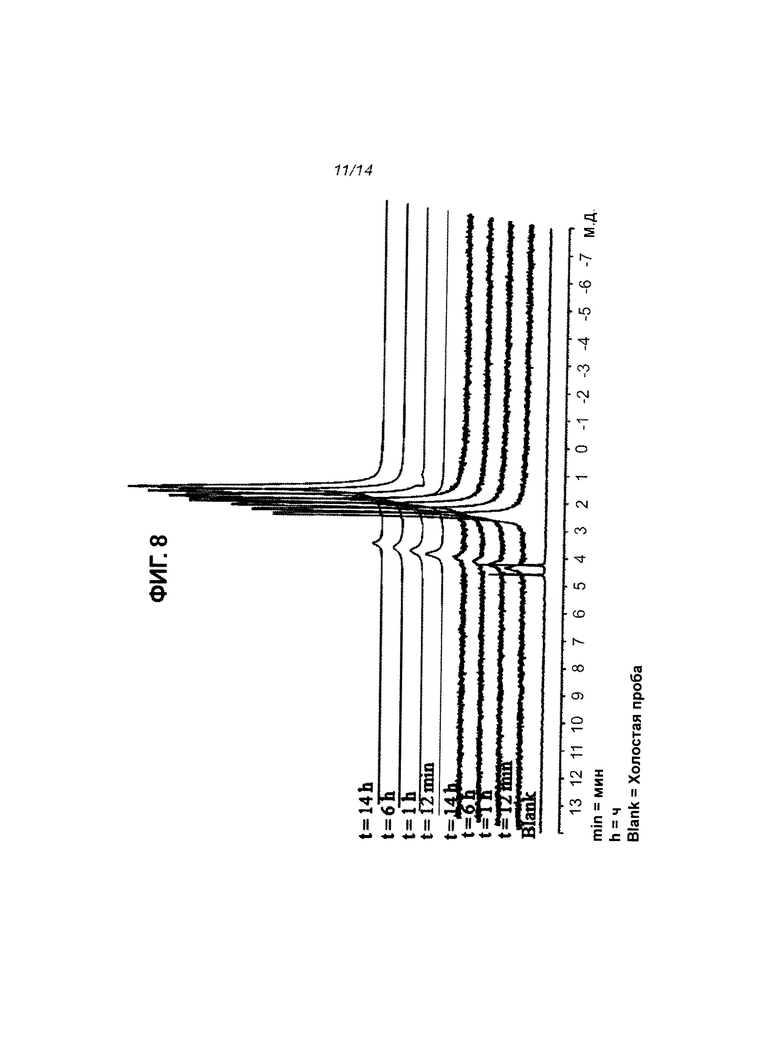
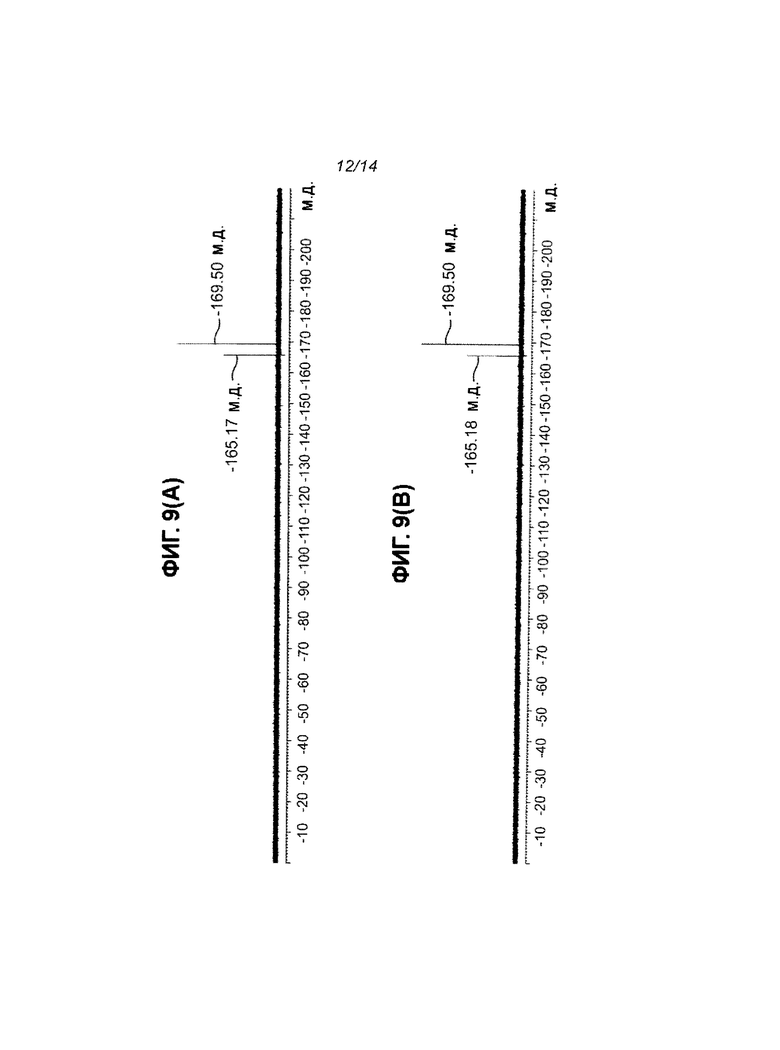
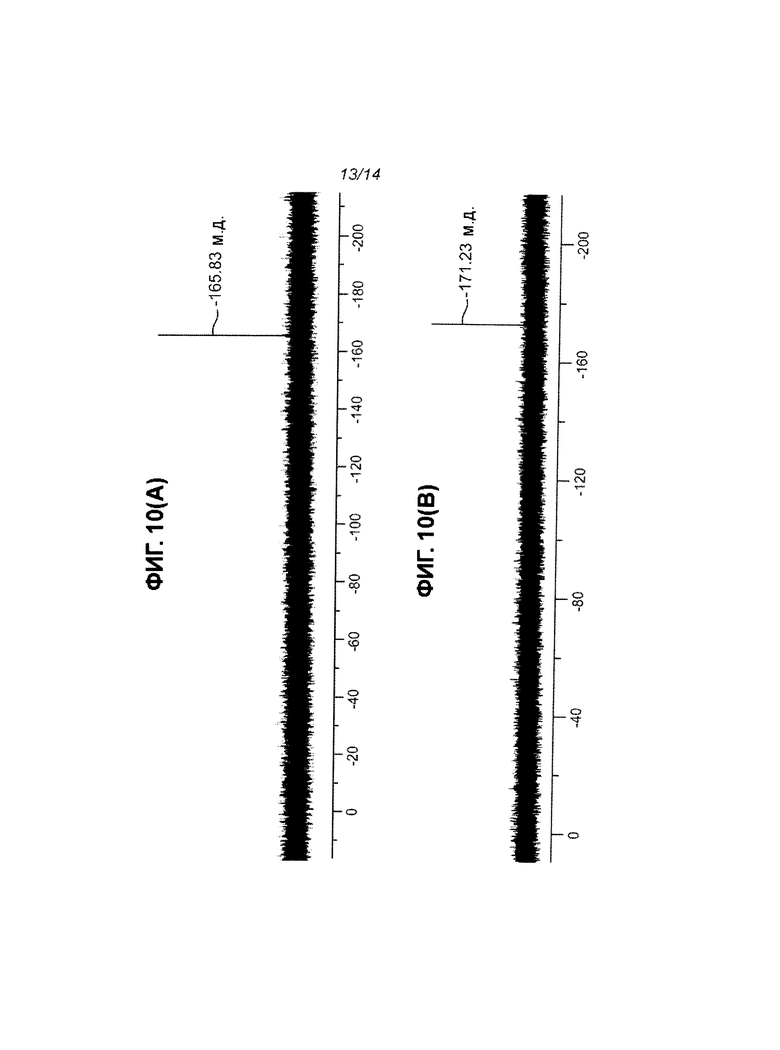
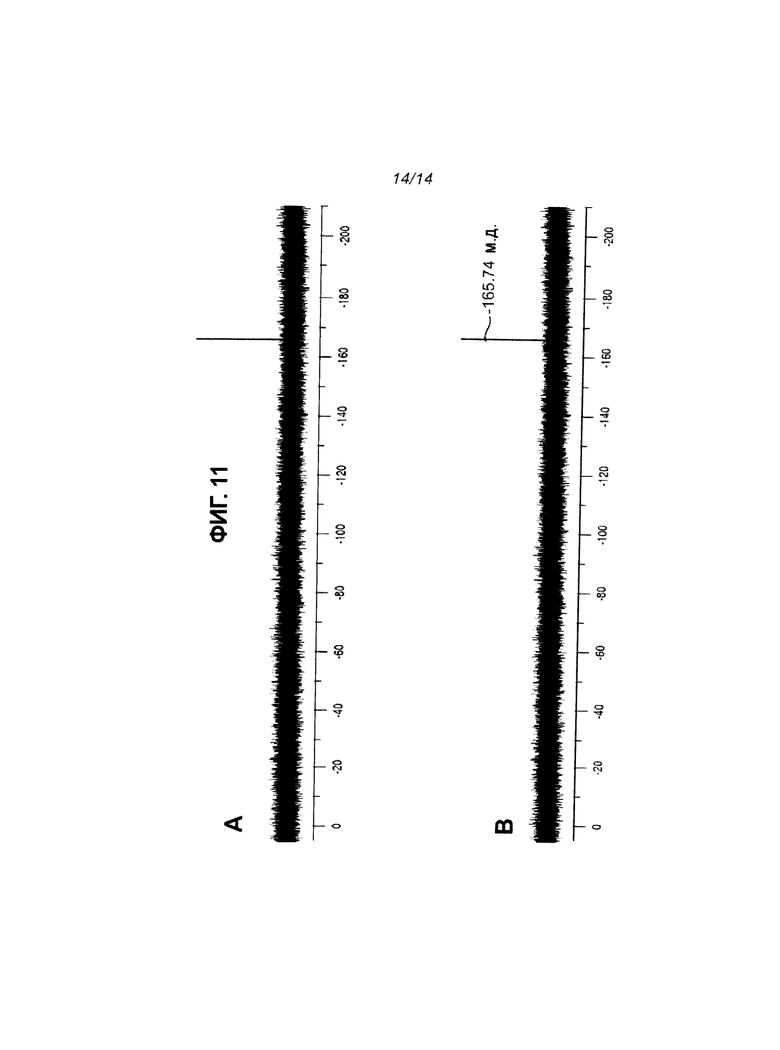
Изобретение относится к применимым в онкологии соединениям формулы (I), их фармацевтически приемлемым солям и композициям на их основе:
где Ar представляет собой фенил или нафтил и необязательно замещен -NO2; R3 выбран из бензила, метила, этила, н-пропила, н-бутила, н-пентила, н-гексила, изопропила, циклогексила, 2-инданила, 3,3-диметил-1-бутила, циклобутила, циклопропилметила, циклопентила, тетрагидропиранила и 2,2-диметилпропила; R4 представляет собой атом Н; R1 и R2 выбраны из атома H и C1-C16алкила, необязательно замещенного фенилом, или один из R1 и R2 содержит C3-алкиленовую цепь, присоединенную к атому N так, что общее количество кольцевых атомов, включая N и атом C, составляет 5 атомов, атом Н, соединенный с атомом N, отсутствует и один из R1 и R2 содержит атом Н или C1-C16алкил; или фармацевтически приемлемая соль соединения Формулы I, при условии, что данное соединение не является соединением, содержащим, в комбинации, незамещенный фенил в качестве Ar, CH3 в качестве R3, H в качестве R4, H в качестве одного из R1 и R2 и CH3 в качестве одного из R1 и R2. Предложены новые эффективные противоопухолевые средства и способ их получения. 11 н. и 56 з.п. ф-лы, 11 ил., 8 табл., 8 пр.
1. Соединение Формулы (I):
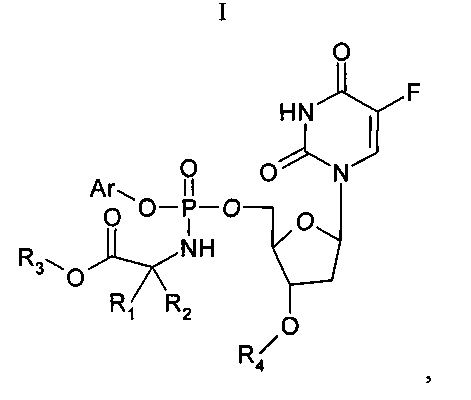
где
Ar представляет собой фенил или нафтил и необязательно замещен -NO2;
R3 выбран из группы, состоящей из бензила, метила, этила, н-пропила, н-бутила, н-пентила, н-гексила, изопропила, циклогексила, 2-инданила, 3,3-диметил-1-бутила, циклобутила, циклопропилметила, циклопентила, тетрагидропиранила и 2,2-диметилпропила;
R4 представляет собой атом Н;
R1 и R2 независимо выбраны из группы, состоящей из атома H и C1-C16алкила, необязательно замещенного фенилом, или один из R1 и R2 содержит C3-алкиленовую цепь, присоединенную к атому N так, что общее количество кольцевых атомов, включая N и атом C, составляет 5 атомов, атом Н, соединенный с атомом N, отсутствует и один из R1 и R2 содержит атом Н или C1-C16алкил;
или фармацевтически приемлемая соль соединения Формулы I,
при условии, что данное соединение не является соединением, содержащим, в комбинации, незамещенный фенил в качестве Ar, CH3 в качестве R3, H в качестве R4, H в качестве одного из R1 и R2 и CH3 в качестве одного из R1 и R2.
2. Соединение по п. 1, в котором Ar представляет собой нафтил.
3. Соединение по п. 2, в котором Ar представляет собой 1-нафтил.
4. Соединение по п. 1, в котором Ar представляет собой фенил.
5. Соединение по любому из пп. 1-4, в котором Ar замещен -NO2.
6. Соединение по любому из пп. 1-4, в котором Ar не содержит заместители.
7. Соединение по любому из пп. 1-4, в котором R3 представляет собой бензил.
8. Соединение по любому из пп. 1-4, в котором R3 представляет собой этил, н-пропил, н-бутил, н-пентил или н-гексил.
9. Соединение по п. 8, в котором R3 представляет собой н-пентил.
10. Соединение по любому из пп. 1-4, 9, в котором R1 и R2 являются разными и стереохимическая конфигурация около асимметрического центра -CR1R2 соответствует представителю, выбранному из группы, состоящей из L-аминокислоты, D-аминокислоты и смеси L- и D-аминокислот.
11. Соединение по любому из пп. 1-4, 9, в котором R1 и R2 соответствуют группировкам, соединенным с атомом альфа-С в природной альфа-аминокислоте.
12. Соединение по п. 11, в котором природная альфа-аминокислота представляет собой L-аланин.
13. Соединение по п. 1, в котором Ar представляет собой 1-нафтил, R3 представляет собой бензил, один из R1 и R2 представляет собой H, один из R1 и R2 представляет собой метил и атом C, к которому присоединены R1 и R2, является L-хиральным.
14. Соединение по п. 1, в котором Ar представляет собой 1-нафтил, R3 представляет собой н-пентил, один из R1 и R2 представляет собой H, один из R1 и R2 представляет собой метил и атом С, к которому присоединены R1 и R2, является L-хиральным.
15. Соединение, выбранное из группы, включающей
5-фтор-2'-дезоксиуридин-5'-O-[фенил(бензокси-L-аланинил)]фосфат (CPF381),
5-фтор-2'-дезоксиуридин-5'-O-[фенил(этокси-L-аланинил)] фосфат (CPF383),
5-фтор-2'дезоксиуридин-5'-O-[фенил(изопропокси-L-аланинил)] фосфат (CPF384),
5-фтор-2'дезоксиуридин-5'-O-[фенил(циклогексокси-L-аланинил)] фосфат (CPF508),
5-фтор-2'дезоксиуридин-5'-O-[пара-нитро-фенил(этокси-L-аланинил)] фосфат (CPF430),
5-фтор-2'дезоксиуридин-5'-O-[1-нафтил(бензокси-L-аланинил)] фосфат (CPF373),
5-фтор-2'дезоксиуридин-5'-O-[1-нафтил(метокси-L-аланинил)] фосфат (CPF385),
5-фтор-2'дезоксиуридин-5'-O-[1-нафтил(этокси-L-аланинил)] фосфат (CPF386),
5-фтор-2'дезоксиуридин-5'-O-[1-нафтил(изопропокси-L-аланинил)] фосфат (CPF387),
5-фтор-2'дезоксиуридин-5'-O-[1-нафтил(циклогексокси-L-аланинил)] фосфат (CPF509),
5-фтор-2'дезоксиуридин-5'-O-[фенил(бензокси-α,α-диметилглицинил)] фосфат (CPF393),
5-фтор-2'дезоксиуридин-5'-O-[фенил(этокси-α,α-диметилглицинил)] фосфат (CPF394),
5-фтор-2'дезоксиуридин-5'-O-[1-нафтил(бензокси-α,α-диметилглицинил)] фосфат (CPF395),
5-фтор-2'дезоксиуридин-5'-O-[1-нафтил(этокси-α,α-диметилглицинил)] фосфат (CPF396),
5-фтор-2'-дезоксиуридин-5'-O-[фенил(бензокси-L-пролинил)] фосфат (CPF583),
5-фтор-2'-дезоксиуридин-5'-O-[1-нафтил(бензокси-L-пролинил)] фосфат (CPF577),
5-фтор-2'-дезоксиуридин-5'-О-[1-нафтил(3,3-диметил-1-бутокси-L-аланинил)] фосфат (CPF585),
5-фтор-2'-дезоксиуридин-5'-O-[1-нафтил-(циклобутокси-L-аланинил)] фосфат (CPF578),
5-фтор-2'-дезоксиуридин-5'-O-[1-нафтил-(циклопропилметанокси-L-аланинил)] фосфат (CPF579),
5-фтор-2'-дезоксиуридин-5'-O-[1-нафтил-(тетрагидропирокси-L-аланинил)] фосфат (CPF580),
5-фтор-2'-дезоксиуридин-5'-O-[1-нафтил-(пентокси-L-аланинил)] фосфат (CPF581),
5-фтор-2'-дезоксиуридин-5'-O-[1-нафтил-(циклопентокси-L-аланинил)] фосфат (CPF582),
5-фтор-2'-дезоксиуридин-5'-O-[1-нафтил-(2-инданокси-L-аланинил)] фосфат (CPF597),
5-фтор-2'-дезоксиуридин-5'-O-[фенил-(бензокси-L-метионинил)] фосфат (CPF586),
5-фтор-2'-дезоксиуридин-5'-O-[1-нафтил-(бензокси-L-фенилаланинил)] фосфат (CPF587),
5-фтор-2'-дезоксиуридин-5'-O-[1-нафтил-(2,2-диметилпропокси-L-аланинил)] фосфат (CPF588),
5-фтор-2'-дезоксиуридин-5'-O-[1-нафтил-(бутокси-L-аланинил)] фосфат (CPF589) или его фармацевтически приемлемая соль.
16. Соединение, представляющее собой 5-фтор-2'-дезоксиуридин-5'-O-[1-нафтил(бензокси-L-аланинил)] фосфат (CPF373) или его фармацевтически приемлемую соль.
17. Соединение, представляющее собой 5-фтор-2'-дезоксиуридин-5'-O-[1-нафтил (бензокси-L-аланинил)] фосфат (CPF373).
18. Соединение по любому из пп. 1-4, 9 и 12-17 для применения в способе профилактики или лечения рака у человека.
19. Соединение по п. 18, отличающееся тем, что рак выбран из группы, включающей лейкемию, рак поджелудочной железы, предстательной железы, легкого, молочной железы и шейки матки.
20. Соединение по п. 18, отличающееся тем, что рак выбран из группы, включающей рак пищевода; рак желудочно-кишечного тракта, в том числе рак желудка, тонкого кишечника, толстой кишки и прямой кишки; рак головы и шеи и рак яичника.
21. Соединение по п. 18 для применения в способе профилактики или лечения рака у пациента, у которого развилась или может развиться устойчивость опухолевых клеток к действию 5-фторурацила или 5-фтор-2'-дезоксиуридина в период профилактики или лечения рака.
22. Соединение по п. 18 для применения в способе профилактики или лечения рака у пациента, клетки которого имеют пониженный уровень белков-переносчиков нуклеозидов.
23. Соединение по п. 18 для применения в способе профилактики или лечения рака у пациента, клетки которого являются дефицитными по нуклеозидкиназе.
24. Соединение по п. 18 для применения в способе профилактики или лечения рака у пациента, клетки которого инфицированы микоплазмой.
25. Соединение по п. 18 для применения в способе профилактики или лечения рака у пациента, клетки которого имеют повышенный уровень тимидилатсинтазы (TS).
26. Соединение по п. 18 для применения в способе профилактики или лечения рака у пациента, позволяющее обойти чувствительность к деградации нуклеозидов катаболическими ферментами.
27. Соединение по п. 26, отличающееся тем, что катаболические ферменты выбраны из группы, включающей тимидинфосфорилазу, уридинфосфорилазу и дезоксицитидиндезаминазу.
28. Соединение по п. 18 для применения в комбинации с другой противораковой терапией.
29. Способ профилактики или лечения рака, включающий введение человеку, нуждающемуся в таком лечении, эффективной дозы соединения по любому из пп. 1-17.
30. Способ по п. 29, отличающийся тем, что рак выбран из группы, включающей лейкемию, рак поджелудочной железы, предстательной железы, легкого, молочной железы и шейки матки.
31. Способ по п. 29, отличающийся тем, что рак выбран из группы, включающей рак пищевода; рак желудочно-кишечного тракта, в том числе рак желудка, тонкого кишечника, толстой кишки и прямой кишки; рак головы и шеи и рак яичника.
32. Способ по любому из пп. 29-30 для профилактики или лечения рака у пациента, у которого развилась или может развиться устойчивость опухолевых клеток к действию 5-фторурацила или 5-фтор-2'-дезоксиуридина в период профилактики или лечения рака.
33. Способ по любому из пп. 29-30, отличающийся тем, что пациент имеет клетки с пониженным уровнем белков-переносчиков нуклеозидов.
34. Способ по любому из пп. 29-30, отличающийся тем, что пациент имеет клетки, дефицитные по нуклеозидкиназе.
35. Способ по любому из пп. 29-30, отличающийся тем, что пациент имеет клетки, инфицированные микоплазмой.
36. Способ по любому из пп. 29-30, отличающийся тем, что пациент имеет клетки с повышенным уровнем тимидилатсинтазы (TS).
37. Способ по любому из пп. 29-30 и 31, позволяющий обойти чувствительность к деградации нуклеозидов катаболическими ферментами.
38. Способ по п. 37, отличающийся тем, что катаболические ферменты выбраны из группы, включающей тимидинфосфорилазу, уридинфосфорилазу и дезоксицитидиндезаминазу.
39. Способ по любому из пп. 29-30 в комбинации с другой противораковой терапией.
40. Фармацевтическая композиция для профилактики или лечения рака, содержащая соединение по любому из пп. 1-17 в комбинации с фармацевтически приемлемым носителем, разбавителем или эксципиентом.
41. Способ получения фармацевтической композиции, включающий стадию комбинирования соединения по любому из пп. 1-17 с фармацевтически приемлемым эксципиентом, носителем или разбавителем.
42. Соединение по п. 1, выбранное из группы, включающей:
5-фтор-2'-дезоксиуридин-5'-О-[фенил(этокси-L-валинил)] фосфат,
5-фтор-2'-дезоксиуридин-5'-О-[фенил(бензокси-L-лейцинил)] фосфат,
5-фтор-2'-дезоксиуридин-5'-O-[фенил(бензокси-L-изолейцинил)] фосфат,
5-фтор-2'-дезоксиуридин-5'-O-[фенил(бензокси-L-фенилаланинил)] фосфат,
5-фтор-2'-дезоксиуридин-5'-O-[фенил(пентокси-L-метионинил)] фосфат,
5-фтор-2'-дезоксиуридин-5'-O-[1-нафтил(гексокси-L-аланинил)] фосфат,
5-фтор-2'-дезоксиуридин-5'-O-[1-нафтил(циклогексокси-L-валинил)] фосфат,
5-фтор-2'-дезоксиуридин-5'-O-[1-нафтил(пентокси-L-лейцинил)] фосфат,
5-фтор-2'-дезоксиуридин-5'-O-[1-нафтил(бензокси-L-лейцинил)] фосфат,
5-фтор-2'-дезоксиуридин-5'-O-[1-нафтил(пентокси-L-изолейцинил)] фосфат,
5-фтор-2'-дезоксиуридин-5'-O-[1-нафтил(пентокси-L-фенилаланинил)] фосфат,
5-фтор-2'-дезоксиуридин-5'-O-[1-нафтил(бензокси-L-метионинил)] фосфат,
5-фтор-2'-дезоксиуридин-5'-O-[1-нафтил(пентокси-α,α-диметилглицинил)] фосфат или его фармацевтически приемлемая соль.
43. Соединение по п. 42 для применения в способе профилактики или лечения рака у человека.
44. Соединение по п. 43, отличающееся тем, что рак выбран из группы, включающей лейкемию, рак поджелудочной железы, предстательной железы, легкого, молочной железы и шейки матки.
45. Соединение по п. 43, отличающееся тем, что рак выбран из группы, включающей рак пищевода; рак желудочно-кишечного тракта, в том числе рак желудка, тонкого кишечника, толстой кишки и прямой кишки; рак головы и шеи и рак яичника.
46. Соединение по любому из пп. 43-45 для применения в способе профилактики или лечения рака у пациента, у которого развилась или может развиться устойчивость опухолевых клеток к действию 5-фторурацила или 5-фтор-2'-дезоксиуридина в период профилактики или лечения рака.
47. Соединение по любому из пп. 43-45 для применения в способе профилактики или лечения рака у пациента, клетки которого имеют пониженный уровень белков-переносчиков нуклеозидов.
48. Соединение по любому из пп. 43-45 для применения в способе профилактики или лечения рака у пациента, клетки которого являются дефицитными по нуклеозидкиназе.
49. Соединение по любому из пп. 43-45 для применения в способе профилактики или лечения рака у пациента, клетки которого инфицированы микоплазмой.
50. Соединение по любому из пп. 43-45 для применения в способе профилактики или лечения рака у пациента, клетки которого имеют повышенный уровень тимидилатсинтазы (TS).
51. Соединение по любому из пп. 43-45 для применения в способе профилактики или лечения рака у пациента, позволяющее обойти чувствительность к деградации нуклеозидов катаболическими ферментами.
52. Соединение по п. 51, отличающееся тем, что катаболические ферменты выбраны из группы, включающей тимидинфосфорилазу, уридинфосфорилазу и дезоксицитидиндезаминазу.
53. Соединение по любому из пп. 43-45, 52 для применения в комбинации с другой противораковой терапией.
54. Способ профилактики или лечения рака, включающий введение человеку, нуждающемуся в таком лечении, эффективной дозы соединения по п. 42.
55. Способ по п. 54, отличающийся тем, что рак выбран из группы, включающей лейкемию, рак поджелудочной железы, предстательной железы, легкого, молочной железы и шейки матки.
56. Способ по п. 54, отличающийся тем, что рак выбран из группы, включающей рак пищевода; рак желудочно-кишечного тракта, в том числе рак желудка, тонкого кишечника, толстой кишки и прямой кишки; рак головы и шеи и рак яичника.
57. Способ по любому из пп. 54-56 для профилактики или лечения рака у пациента, у которого развилась или может развиться устойчивость опухолевых клеток к действию 5-фторурацила или 5-фтор-2'-дезоксиуридина в период профилактики или лечения рака.
58. Способ по любому из пп. 54-56, отличающийся тем, что пациент имеет клетки с пониженным уровнем белков-переносчиков нуклеозидов.
59. Способ по любому из пп. 54-56, отличающийся тем, что пациент имеет клетки, дефицитные по нуклеозидкиназе.
60. Способ по любому из пп. 54-56, отличающийся тем, что пациент имеет клетки, инфицированные микоплазмой.
61. Способ по любому из пп. 54-56, отличающийся тем, что пациент имеет клетки с повышенным уровнем тимидилатсинтазы (TS).
62. Способ по любому из пп. 54-56, позволяющий обойти чувствительность к деградации нуклеозидов катаболическими ферментами.
63. Способ по п. 62, отличающийся тем, что катаболические ферменты выбраны из группы, включающей тимидинфосфорилазу, уридинфосфорилазу и дезоксицитидиндезаминазу.
64. Способ по любому из пп. 54-56, 63 в комбинации с другой противораковой терапией.
65. Фармацевтическая композиция для профилактики или лечения рака, содержащая соединение по п. 42 в комбинации с фармацевтически приемлемым носителем, разбавителем или эксципиентом.
66. Способ получения фармацевтической композиции, включающий стадию комбинирования соединения по п. 42 с фармацевтически приемлемым эксципиентом, носителем или разбавителем.
67. Способ получения соединения Формулы I по п. 1, включающий взаимодействие соединения Формулы (II)
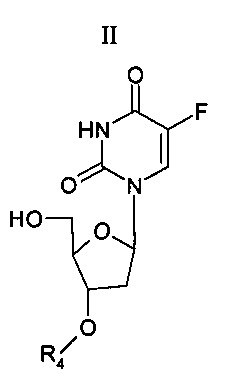
с соединением Формулы (III)
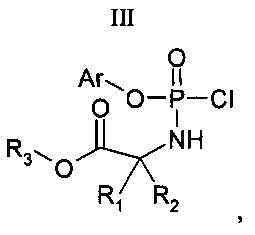
где Ar, R3, R4, R1 и R2 имеют значения, описанные в п. 1.
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, КОМПОЗИЦИЯ СТАНДАРТНОЙ ЛЕКАРСТВЕННОЙ ФОРМЫ И СПОСОБ ЛЕЧЕНИЯ ЗЛОКАЧЕСТВЕННЫХ НОВООБРАЗОВАНИЙ, ОПУХОЛЕЙ И ВИРУСНЫХ ИНФЕКЦИЙ | 1996 |
|
RU2197964C2 |
| WO 2005012327 A3, 10.02.2005 | |||
| WO 1999037753 A1, 29.07.1999 | |||
| Christopher Mcguigan et al., Journal of medicinal chemistry, 2011, vol | |||
| Видоизменение прибора для получения стереоскопических впечатлений от двух изображений различного масштаба | 1919 |
|
SU54A1 |
| Johan Vande Voorde et al., Biochemical Pharmacology, 2011, 82, 441-452. | |||

