Настоящее изобретение относится к пролекарствам, превращающимся печенью, и более конкретно относится к лечению заболеваний с применением пролекарств, превращающихся ферментом(ами) печени в активные лекарства. Оно также относится к способу установления полезности пролекарства для лечения заболевания.
Иоддезоксиуридин (ИУдР) был синтезирован в качестве протиоопухолевого средства в 1959 г. В.Г.Прюсофф, (Biochem. Biophys. Acta, 1959, 32, 295 - 296) и был первым аналогом тимидина, использованным клинически в качестве антигерпесного средства (Archs. Ophthalmol., 1962, 68, 235 - 239). Токсичность иоддезоксиуридина при системном применении ограничила его клиническое использование. Иоддезоксиуридин был также признан в качестве потенциального клинического радиосенсибилизатора при химиотерапии рака (J. Radiation Oncology Biol. Phys, 1984, 10, 1399 - 1406). Степень радиосенсибилизации прямо зависит от количества замещенного тимидина в ДНК его аналогами (Clin. Pharmacol, Ther. , 1988, 44, 369 - 375). Некоторый успех имело внутрипеченочное вливание иоддезоксиуридина с последующим облучением при лечении опухолевых клеток печени (Cancer. Res. 1989, 49, 6437 - 6442).
При попытке разработать селективные средства против вируса герпеса на основе более широкого спектра специфичности субстрата тимидинкиназы вируса герпеса по сравнению с тимидинкиназой человека была синтезирована 5-иод-2-пиримидинон-дезоксирибоза (ИПдР), которая отличается от ИУдР оксогруппой и в положении 4 основания. Было найдено, что ИПдР имеет сильную активность против вирусов герпеса HSV-1 и HSV-2 в клеточных культурах и против HSV-2 в мышах (Antimicrob. Agents Chemother, 1989, 33, 340 - 344). Это средство не было токсичным ни для неинфецированных клеток, ни для мышей при пероральном введении в использованных дозах (Antimocrob. Agents Chemither, 1989, 33, 340 - 344). Так как ИПдР и ИУдР структурно близки, была проверена возможность превращения ИПдР в ИУдР. Ранее было показано, что ИПдР не может быть превращена в ИУдР ксантиноксидазой (Antimicrob. Agents Chemother., 1989, 33, 340 - 344).
Патент США N 4 895 937 раскрывает нуклеозид 1-(2-дезокси- β -D-рибофуранозил)-5-иод-2-пиримидинон (ИПдР) как средство против вируса герпеса, например HSV-2. Полное содержание патента США N 4 895 937 включено в рассмотрение.
Номенклатура
IUdR: иоддезоксиуридин (ИУдР)
FUdR: фтордезоксиуридин (ФУдР)
IPdR: 5-иод-2-пиримидинондезоксирибоза (ИПдР)
HSV: вирус герпеса
HPLC: высокоэффективная жидкостная хроматография (ВЭЖХ)
IU: иодурацил (ИУ)
EPdR: 5-этинил-2-пиримидинондезоксирибоза (ЭПдР)
IP: 5-иод-2-пиримидинон (ИП)
BPdR: 5-бром-2-пиримидинондезоксирибоза (БПдР)
MPdR: 5-метил-2-пиримидинондезоксирибоза (МПдР)
EtPdR: 5-этил-2-пиримидинондезоксирибоза (ЭтПдР)
BudR: 5-бромдезоксиуридин (БУдР)
HBV: вирус гепатита B
FU: 5 - фторурацил (ФУ)
FP: 5-фтор-2-пиримидинон (ФП)
ddI: дидезоксиинозин (ддИ)
ddG: дидезоксигуанин (ддГ)
DHPG: гансикловир (9-/1,3-дигидрокси-2-пропокси)метил/гуанин) ДГПГ)
ACV: (S)-N-/N-(5-амино-5-карбокси-1-оксопентил)-L-цистеинил/-D- валин (АЦВ)
D4T: 21,31-дидезокси-21,31- дидегидротимидин (Д4Т)
AZT: 31-азидо-31-дезокситимидин (АЗТ)
Цель настоящего изобретения - обеспечить препараты для применения в качестве пролекарств, которые превращаются в печени млекопитающих в биологически активное вещество, особенно биологически активное вещество, которое, как полагают, проявит свое биологическое действие в печени или которое не может быть введено перорально.
Другая цель настоящего изобретения - обеспечить препараты, содержание замещенные в положении 5 аналоги ПдР, особенно ИПдР, для использования в качестве пролекарств, превращающихся в печени млекопитающих (особенно печени человека) in situ в соответствующие биологически активные замещенные в положении 5 производные УдР.
Следующая цель настоящего изобретения - использовать такие замещенные в положении 5 аналоги ПдР для лечения связанных с печенью заболеваний, особенно в качестве радиосенсибилизатора для лечения печеночно-клеточного рака.
Следующая цель настоящего изобретения - обеспечить препараты, отличные от замещенных в положении 5 аналогов ПдР, для применения в качестве пролекарств, превращающихся в печени млекопитающих в биологически активное вещество, особенно биологически активное вещество, которое, как полагают, проявляет свое биологическое действие в печени или которое не может быть введено перорально.
Следующая цель настоящего изобретения - обеспечить такие препараты, отличные от замещенных в положении 5 аналогов ПдР, которые являются аналогами замещенного в положении 5 пиримидинона, особенно фторпимидиноном.
Следующая цель настоящего изобретения - обеспечить такие препараты, отличные от замещенных в положении 5 аналогов ПдР, которые являются пролекарствами для образования биологически активных нуклеозидов или оснований нуклеозидов, отличных от УдР и урацила, предпочтительно аналогов гуанозина, цитидина, инозина или тимина.
Следующая цель настоящего изобретения - обеспечить улучшение в лечении заболевания.
Следующая цель настоящего изобретения - обеспечить метод установления полезности пролекарства для лечения заболевания.
Следующая цель настоящего изобретения - обеспечить метод определения превращаемости пролекарства ферментом печени в биологически активное вещество, действующее на любые клетки организма.
Следующая цель настоящего изобретения - обеспечить метод лечения заболевания с помощью пролекарств.
Следующая цель настоящего изобретения - обеспечить способ синтеза химического соединения с помощью фермента альдегидоксидазы.
Вышеприведенные цели так же, как другие цели, замыслы и преимущества, удовлетворяют настоящему изобретению.
Настоящее изобретение относится к способу установления полезности пролекарства для лечения заболеваний у, например, млекопитающих, предпочтительно человека, заключающемуся в определении будет или нет нетоксичное пролекарство превращаться в клетках печени, in vitro, печеночным ферментом альдегидоксидазой, в котором если пролекарство превращается в активное лекарство или другой полезный метаболит, то это эффективное пролекарство для лечения заболевания.
Установление и использование пролекарств, эффективных против заболеваний, связанных с печенью, и опухолевых заболеваний, является предметом особого интереса.
Настоящее изобретение также относится к способу лечения заболеваний у животных, заключающемуся во введении животным, предпочтительно человеку, фармацевтически эффективного количества нетоксичного аналога нуклеозида или аналога основания нуклеозида, или его соли, или эфира либо отдельно, либо в смеси с фармацевтически приемлемым носителем, причем аналоговое пролекарство способно превращаться в клетках печени альгедидоксидазой в активное лекарство или другой полезный метаболит.
Фиг. 1 показывает хроматограмму ВЭЖХ превращения ИПдР в ИУдР гомогенатом печени. ИПдР культивируют на гомогенате печени крысы. Для контрольных реакций часть надосадочного раствора кипятят 5 минут для инактивации всех ферментов перед использованием. Условия анализа описаны ниже за исключением того, что 60 мкл 100 000 г • 60 мин надосадочного раствора (эквивалентен приблизительно 0,5 мг белка/мл) используют в реакционном объеме 1500 мкл. Аликвоты (300 мкл) отбирают в различные моменты времени (0 мин, 15 мин и 30 мин от (1) до (3) соответственно) в течение периода культивирования при 37oC. Время удерживания составляет 9,5 мин для ИПдР (A), 8,0 мин для ИУдР (B) и 6,5 мин для иодурацила (C) соответственно.
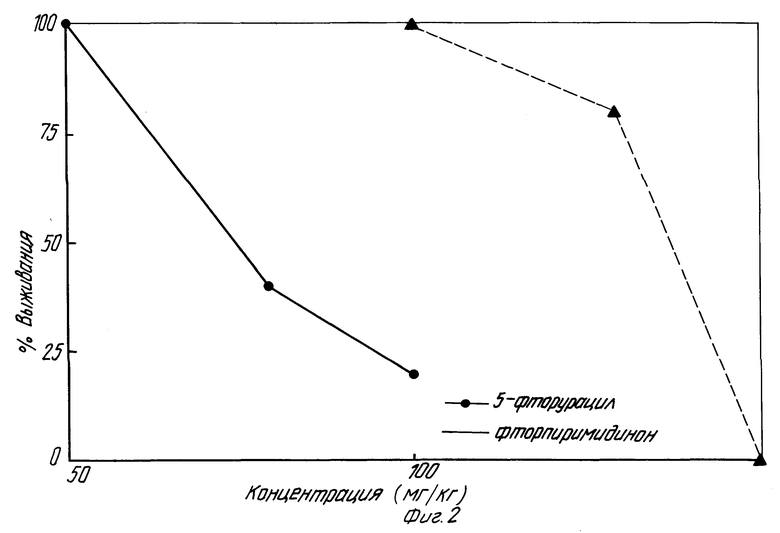
Фиг. 2 показывает коэффициент выживания мышей, которым давали перорально ежедневные дозы 100 мг/кг фторурацила и фторпиримидинона в течение 5 дней.
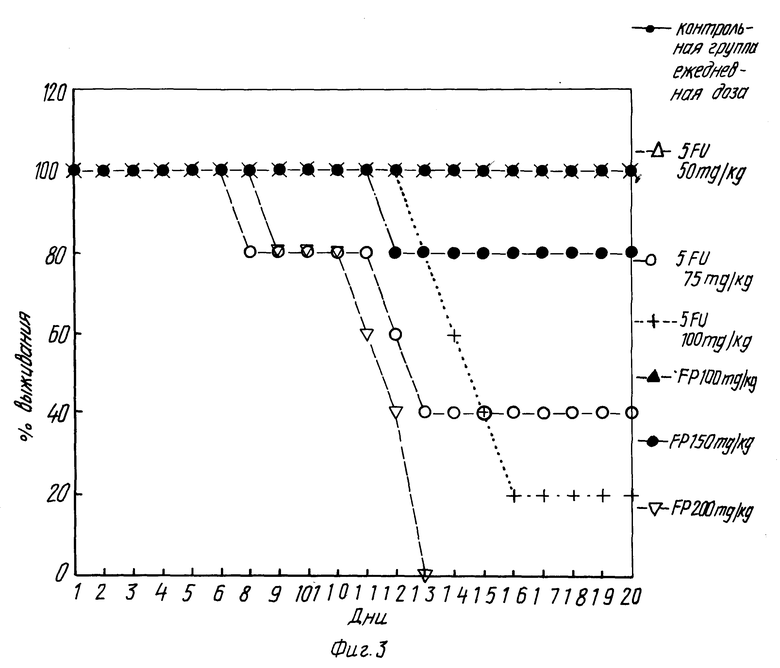
Фиг. 3 показывает коэффициент выживания мышей, которым давали перорально различные дозы фторурацила и фторпиримидинона.
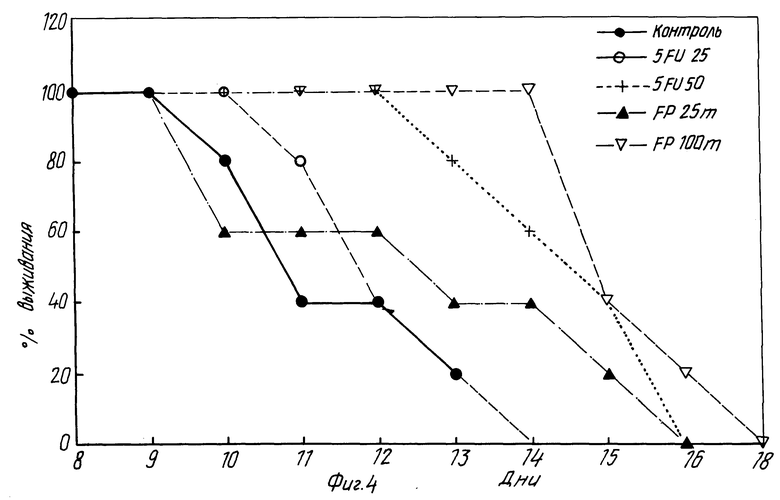
Фиг. 4 показывает коэффициент выживания мышей, сначала инъецированных клетками лейкемии, затем обработанных перорально различными концентрациями фторурацила и фторпиримидинона.
Фиг. 5 показывает изменение массы опухоли толстой кишки, обработанной различными дозами фторурацила и фторпиримидина на протяжении некоторого времени. Все цифры масс даны в сравнении с начальной контрольной цифрой.
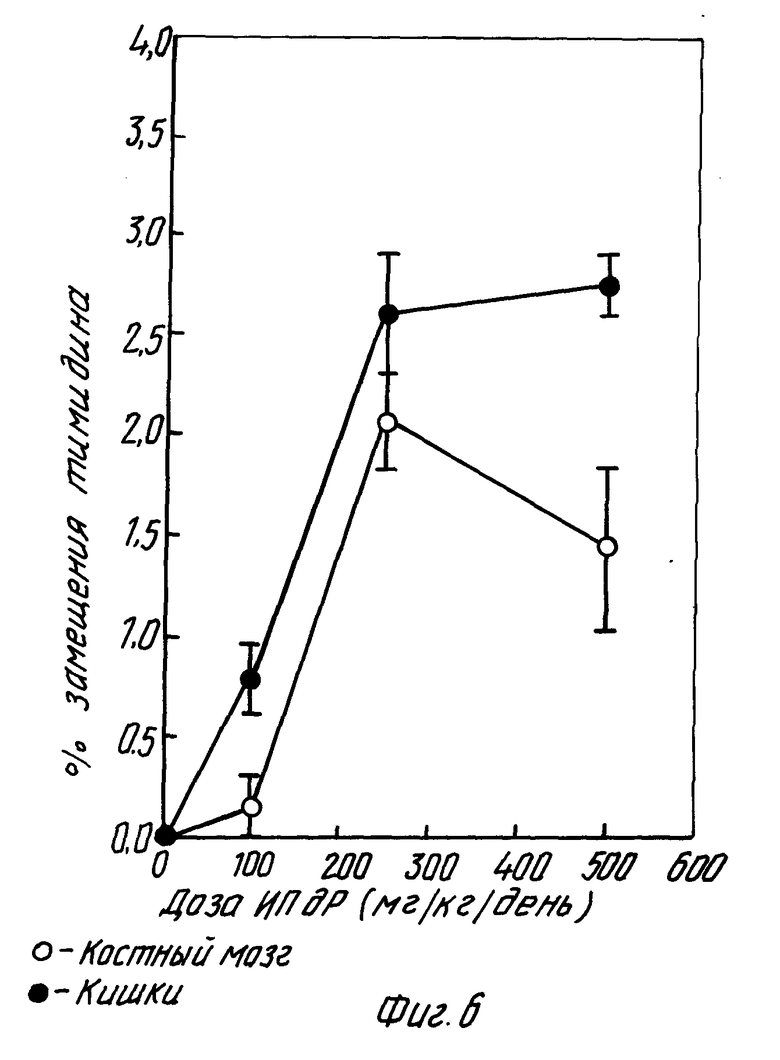
Фиг. 6 показывает относительно низкое включение ИПдР в ткани атимных безволосых мышей, такие как костный мозг и кишки, причем плато замещения тимидина составляет 250 мг/кг/день.
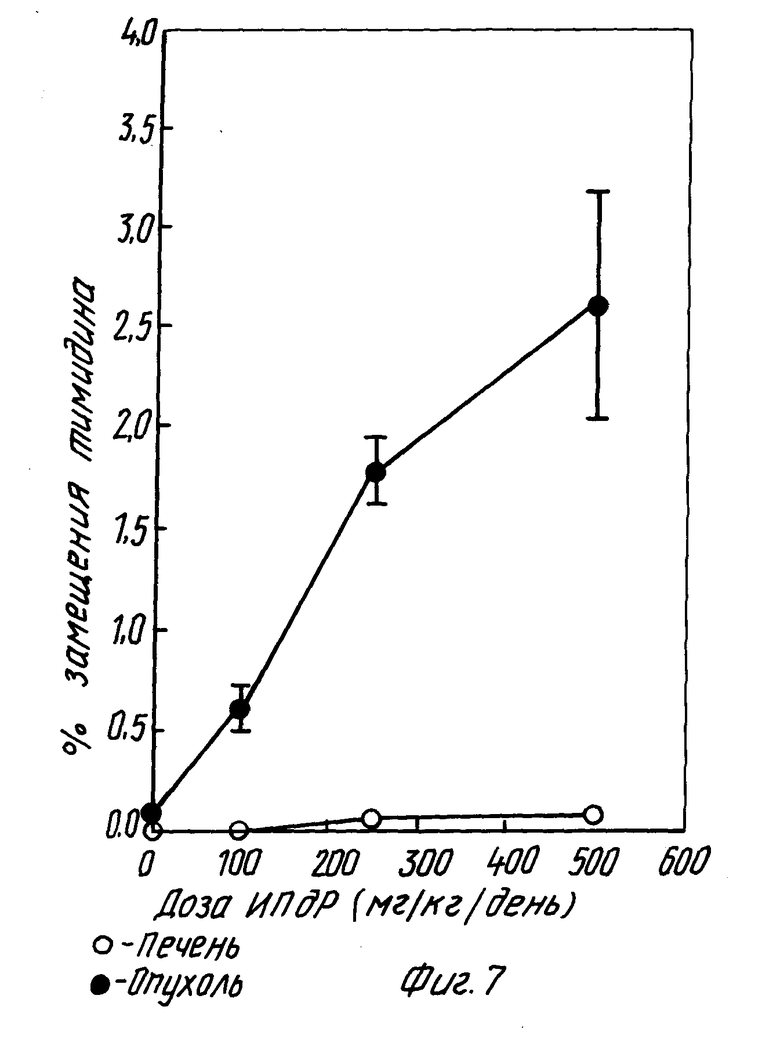
Фиг. 7 показывает сравнительное замещение ИПдР в тканях печени и опухоли атимных безволосых мышей между тканью печени и метаболической опухолью, причем включение в ткань печени ничтожно по сравнению с включением в опухоль.
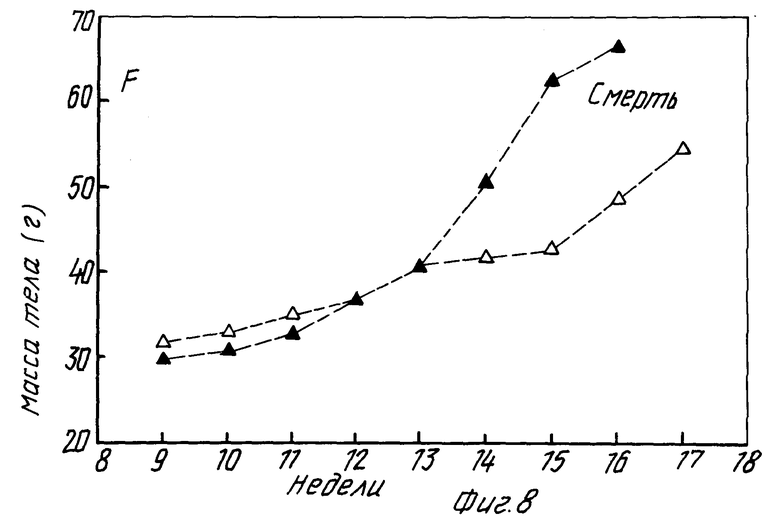
Фиг. 8 показывает результаты обработки фторпиримидиноном самок трансгенных мышей, экспресирующих антиген SV 40 большой опухоли, вызывающий различные раковые заболевания, причем обработанные фторпиримидиноном мыши жили дольше и имели меньший прирост массы тела, чем контрольные.
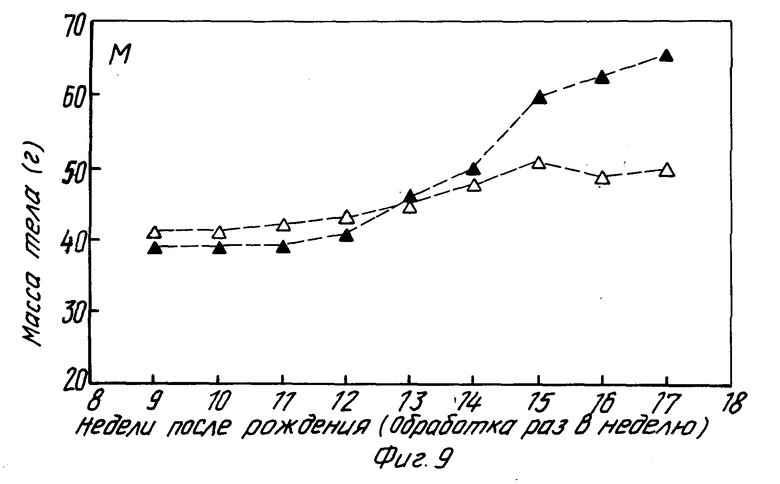
Фиг. 9 показывает результаты обработки фторпиримидиноном самцов трансгенных мышей, экспресирующих антиген SV 40 большой опухоли, вызывающий различные раковые заболевания, причем обработанные фторпиримидиноном мыши имели меньший прирост массы тела и жили дольше, чем контрольные.
Настоящее изобретение основано на открытии того, что пролекарство активируется альдегидоксидазой in vitro и in vivo и становятся активными лекарствами или метаболитами, достигая высокой селективности и терапевтического индекса. Выше описана следующая схема реакции:
Пролекарство  активное лекарство или метаболит
активное лекарство или метаболит
Одна из схем реакции вышеприведенной схемы имеет следующий вид: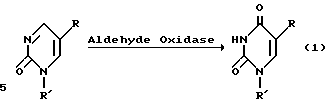
в которой R - I, F, Br, Cl, H, -CH3, -OR1, CF3, NO2, -SR1, -CH=CR2R3, -C ≡ CR2 или -N=N+-n-, R1 - C1-C5-алкил, предпочтительно алкил имеет один атом углерода, R2 и R3, независимо друг от друга, водород, C1-C5-алкил или галоген, и R' - водород, остаток сахара, такой как рибоза или дезоксирибоза, группа -CH2-O-CH2-CH2OH, -CH2-O-CH(CH2OH), замещенный или незамещенный алкил, арил, циклоалкил, циклоарил или любая другая группа, которая имеет размер, не препятствующий стерически действию альдегидоксидазы печени. Показано, что эти остатки не мешают желаемому действию альдегидоксидазы печени.
Другие примеры реакционных схем следующие: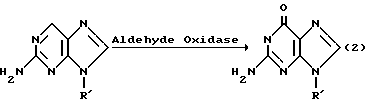
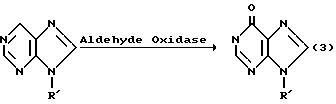

в которой R' определен выше.
Выбор пролекарства, которое может быть испытано или использовано в соответствии с настоящим изобретением, ограничен только структурой активного лекарства, полученного действием альдегидоксидазы печени. Представительные примеры полезных пролекарств включают аналоги нуклеозидов и аналоги оснований нуклеозидов. В этой ситуации трансформирующееся пролекарство превращают в продукт, который токсичен или иначе рассматривается таким, чтобы проявлять желаемое биологическое действие (такое как радиосенсибилизация), только по отношению к разрастающимся клеткам (таким, как раковые клетки) или к реплицирующемуся вирусу, но не токсичен или по меньшей мере менее токсичен, или не проявляет того же биологического действия, или проявляет меньше того же биологического действия на неразрастающиеся клетки.
Альдегидоксидаза печени широко распространена среди ферментов млекопитающих. Этот фермент катализирует окисление различных алифатических и ароматических альдегидов, а также многих неальдегидных гетероциклических соединений, таких как N1-метилникотинамид, 4-аминоантифолаты и метотрексат и их аналоги. Находкой авторов изобретения является окисление замещенных в положении 5 пиримидинонов в их урациловые или уридиновые двойники альдегидоксидазой человека или крысы, которое приводит к совершенно новой категории субстратов, на которые действует фермент. Это открытие также позволяет разработать лекарства, превращающиеся в печени, которые могут подавлять и разрушать раковые клетки, вирусы, паразиты и другие нежелательные микробные патогены.
Чтобы предложить пролекарство, полезное в настоящем изобретении, необходимо только выбрать биоактивное соединение, например соединение, проявляющее желаемый биологический эффект, которое содержит кетогруппу в структуре и которое желают образовать in situ в печени животного, особенно млекопитающего, наиболее предпочтительно человека. Любое такое биоактивное соединение с кетогруппой в структуре - потенциальный кандидат для настоящего изобретения. Когда такое биоактивное соединение выбрано, то можно провести простой анализ in vitro, чтобы определить будет ли соответствующее пролекарство окисляться в обсуждаемое биоактивное соединение альдегидоксидазой печени.
Пролекарство образуют синтезом соединения, соответствующего желаемому биоактивному соединению, но имеющему кетогруппу в восстановленной форме. Это потенциальное пролекарство затем подвергают анализу in vitro, похожему на анализ, описанный в примерах 2 и 3, чтобы определить может ли оно служить субстратом для альдегидоксидазы печени. Если пролекарство превращается в предопределенное биоактивное соединение оксидазой печени, то это пролекарство может рассматриваться как соответствующее настоящему изобретению и может быть использовано в соответствии с настоящим изобретением.
В то время как замещенные в положении 5 аналоги ПДР предпочтительны, и как было показано, превращаются в соответствующие замещенные соединения УдР in vivo, также установлено, что фрагмент дезоксирибозы не является необходимым для специфичности субстрата и что фторопиримидинон также превращается во фторурацил альдегидоксизазой in vitro так же, как in vivo. Так как большинство нуклеозидов и оснований нуклеозидов содержит кетогруппы в их формулах и многие структурно близки к структуре урацила, то ожидают, что биоактивные соединения, являющиеся аналогами нуклеозидов или оснований нуклеозидов, являются первыми кандидатами в биоактивные соединения, для которых могут быть разработаны пролекарства в соответствии с настоящим изобретением, превращающиеся альдегидоксидазой в такие соединения. Таким образом, в дополнение к аналогам урацила и уридина могут быть использованы в качестве основы образования пролекарств в соответствии с настоящим изобретением аналоги других нуклеозидов и оснований нуклеозидов, такие как аналоги цитидина, гуанозина, 6-азауридина и 6-азагуанина. Примеры биоактивных соединений этой категории включают ддГ, ДГПГ, АЦВ, ддИ, Д4Т и А3Т.
Дополнительные примеры коммерчески доступных аналогов пиримидинов и пуринов, которые могут служить биоактивными лекарствами для разработки соответствующих пролекарств в согласии с настоящим изобретением, перечислены в таблицах 1 и 2.
Таблица 1. Аналоги пиримидина:
N1-Ацетилцитидин
31-Ацетилтимидин
Аденозин-N1-оксид
Аллопуринолрибозид
4-Амино-5-аминометил-2-метилпиримидин
1-Аминобарбитуровая кислота
2-Амино-5-бром-6-метилпиримидинол
4-Амино-5-карбэтокси-2-этилмеркаптопиримидин
5-Амино-6-карбокси-2,4-дигидроксипиримидин
2-Амино-4-хлор-6-метилпиримидин
3'-Амино-3'-дезокситимидин
5'-Амино-5'-дезокситимидин
5'-Амино-2'-дезоксиуридин
5'-Амино-2',5'-дидезокси-5-иодцитидин
5'-Амино-2',5'-дидезоксиуридин
4-Амино-2,6-дигидрокси-5-нитрозопиримидин
2-Амино-4,6-дигидроксипиримидин
4-Амино-2,6-дигидроксипиримидин
5-Амино-2,4-дигидроксипиримидин
4-Амино-1,3-диметил-2,6-диокси-5-нитрозопиримидин
2-Амино-4,6-диметилпиримидин
4-Амино-2-гидрокси-5-гидроксиметилпиримидин
4-Амино-6-гидрокси-2-меркапто-5-нитрозопиримидин
4-Амино-6-гидрокси-2-меркаптопиримидин
2-Амино-4-гидрокси-6-метилпиримидин
4-Амино-2-гидрокси-5-метилпиримидин
2-Амино-4-гидроксипиримидин
4-Амино-2-гидроксипиримидин
4-Амино-6-гидрокси-2-тиопиримидин
2-Амино-4-метилпиримидин
4-Аминооротовая кислота
4-Амино-2-тиопиримидин
6-Амино-2-тиоурацил
5-Амино-2,4,6-тригидроксипиримидин
4-Аминоурацил
5-Аминоурацил
6-Аминоурацил
5-Аминоурацинамобарбитал
2',3'-Ангидротимидин
5-Азацитидин
6-Азацитидин
5-Азацитозин
6-Азацитозин
5-Аза-2'-дезоксицитидин
6-Аза-2'-дезоксиуридин
6-Аза-2-тиотимин
6-Азатимин
5-Азаурацил
6-Азаурацилрибозид
6-Азауридин
2'-Азидо-2'-дезоксицитидин
3'-Азидо-3'-дезокситимидин
2'-Азидо-2'-дезоксиуридин
Барбитуровая кислота
3'-0-Бензоилтимидин
5'-Бензоилуридин
5-Бромцитидин
5-Бромцитозин
5-Бром-2'-дезоксицитидин
5-Бром-2,3'-дидезоксиуридин
5-Бром-2,4-дигидроксипиримидин
5-Бром-2',3'-изопропилиденуридин
5-Бром-1-метилурацил
5-Броморотовая кислота
5-Бромурацил
5-Бромуридин
(E)-5-(2-Бромвинил)уридин
3-Бутилурацил
5-Карбэтоксицитозин
5-Карбэтокси-2,4-дигидроксипиримидин
5-Карбэтокси-2-этилмеркапто-4-гидроксипиримидин
5-Карэтокси-2-тиоурацил
5-Карбэтоксиурацил
5-Карбэтоксицитозин
5-Карбокси-2,4-дигидроксипиримидин
6-Карбокси-2,4-дигидроксипиримидин
5-Карбокси-2-этилмеркапто-4-гидроксипиримидин
5-Карбокси-4-гидрокси-2-тиопиримидин
Карбоксиметилурацил
6-Карбокси-5-нитро-2,4-диоксипиримидин
5-Карбокси-2-тиолаурил
5-Карбоксиурацил
5-Хлорцитозинарабинозид
5'-Хлор-5'-дезоксицитидин
2'-Хлор-2'-дезокси-4-тиоуридин
2'-Хлор-2'-дезоксиуридин
5'-Хлордезоксиуридин
2-Хлор-4,5-диаминопиримидин
6-Хлор-2,4-Диметоксипиримидин
2-Хлорпиримидин
5-Хлорурацил
4,5-Диамино-2-хлорпиримидин
4,5-Диамино-2,6-дигидроксипиримидин
2,5-Диамино-4,6-дигидроксипиримидин
4,6-Диамино-2-этилмеркаптопиримидин
4,5-Диамино-5-формиламинопиримидин
4,5-Диамино-6-гидрокси-2-меркаптопиримидин
4,6-Диамино-2-гидрокси-5-нитрозопиримидин
4,5-Диамино-6-гидроксипиримидин
2,4-Диамино-6-гидроксипиримидин
4,6-Диамино-2-гидроксипиримидин
4,6-Диамино-2-метилмеркаптопиримидин
2,4-Диамино-6-метил-5-нитропиримидин
4,5-Диамино-6-метил-2-тиопиримидин
2,4-Диамино-5-нитропиримидин
4,5-Диаминопиримидин
4,5-Диамино-2-тиопиримидин
4,5-Диамино-6-тиопиримидин
4,6-Диамино-2-тиопиримидин
5,6-Диаминоурацил
5-Диазо-2'-дезоксиуридин
5-Диазоурацил
4,6-Дихлор-5-аминопиримидин
2,4-Дихлор-6-метилпиримидин
2,4-дихлорпиримидин
4,6-Дихлорпиримидин
2',3'-Дидезоксицитидин
2',3'-Дидезоксиуридин
2,4-Диэтоксипиримидин
5,6-Дигидродезоксиуридин
5,6-Дигидро-2,4-дигидрокси-6-метилпиримидин
5,6-Дигидро-2,4-дигидроксипиримидин
Дигидро-6-метилурацил
Дигидротимидин
Дигидротимин
Дигидроурацил
Дигидроуридин
2,6-Дигидрокси-4-амино-5-нитрозопиримидин
2,6-Дигидрокси-4-аминопиримидин
2,4-Дигидрокси-6-метил-5-нитропиримидин
2,4-Дигидрокси-6-метилпиримидин
2,4-Дигидрокси-5-нитропиримидин
4,6-Дигидрокси-5-нитрозо-2-тиопиримидин
4,6-Дигидроксипиримидин
2,4-Дигидроксипиримидин-6-метилсульфон
2,4-Дигилрокси-2-тиопиримидин
1,5-Диметилцитозин
N,N-Диметил-2'-дезоксицитидин
1,3-Диметилурацил
5,6-Диоксиурацил
2,4-Дитиопиримидин
3,N'-Этеноцитидин
5-Этил-2'-дезоксиуридин
2-Этил-2'-дезоксиуридин
2-Этилмеркапто-4,6-диминопиримидин
5-фтор-2'-дезоксиуридин
Гексобарбитал
5-Гидроксиметилцитозин
5-Гидроксиметил-2'-дезоксиуридин
4-Гидрокси-6-метил-2-тиопиримидин
5-Гидроксиметилуридин
4-Гидроксипиразоло/3,4-d/пиримидин
2-Гидроксипиримидин
4-Гидроксипиримидин
4-Гидрокси-2-тиопиримидин
5-Гидроксиурацил
5-Гидроксиуридин
6-Гидроксиуридин
5-Иодцитидин
5-Иодцитозин
5-Иод-2-дезоксицитид
5-Иодоротовая кислота
5-Иодурацил
5-Иодурадин
2',3'-O-Изопропилиденцитидин
2',3'-Изопропилиденуридин-5-трифосфат
5-Меркаптоурацил
2'-O-Метилцитидин
3'-O-Метилцитидин
5-Метилцитидин
5-Метилцитозин
5-Метил-2'-дезоксицитидин
5-Метил-2-тиоцитозин
4-Метил-2-тиоурацил
2-O-Метилтимидин
3-Метилтимидин
4-O-Метилтимидин
1-Метилурацил
3-Метилурацил
6-Метилурацил
2'-O-Метилуридин
3-Метилуридин
3'-O-Метилуридин
5-Метилуридин
5-Нитробарбитуровая кислота
5-Нитро-6-метилурацил
5-Нитрооротовая кислота
5-Нитрозотиобарбитуровая кислота
5-Нитрозо-2,4,6-триаминопиримидин
5-Нитроурацил
3'-Оксаурацил
5-Пропил-2-тиоурацил
6-н-Пропил-2-тиоурацил
РибавиринТМ
5-Сульфаминоурацил
2-Сульфанилаиноиримидин
Тетрагидроуридин
2-Тио-6-азауридин
2-Тио-5-карбоксиурацил
2-Тиоцитидин
2-Тиоцитозин
4-Тио-2'-дезоксиуридин
Тиометилурацил
2-Тиопиримидин
2-Тиоурацил
5-Тиоурацил
2-Тиоурацил-5-карбоновая кислота
4-Тиоуридин
2,4,5-Триамино-6-гидроксипиримидин
4,5,6-Триамино-2-гидроксипиримидин
2,4,6-Триамино-5-нитрозопиримидин
2,4,6-Триаминопиримидин
4,5,6-Триаминопиримидин
2,4,6-Трихлорпиримидин
Трифтортимидин
2,4,5-Тригидроксипиримидин
УрамилТМ
Таблица 2. Аналоги пурина
3'-O-Ацетил-2'-дезоксиаденозин
3'-O-Ацетил-2'-дезоксицитидин
N'-Ацетил-2'-лезоксицитидин
N'-Ацетилгуанин
2-Амино-6-бензилмеркаптопурин
2-Амино-6-бензилтиопурин
2-Амино-8-бром-6-гидроксипурин
2-Амино-6-( α -карбоксиэтил)меркаптопурин
2-Амино-6-карбоксиметилмеркаптопурин
2-Амино-6-хлорпурин
2-Амино-6-хлорпуринрибозид
6-Амино-2,8-дигидроксипурин
8-Аминогуанозин
2-Амино-6-меркаптопурин
6-Амино-2-метилпурин
6-Амино-3-метилпурин
2-Аминопурин
8-Азаксантин
8-Азидоаденозин
6-Бензиламинопурин
6-Бензиламинопуринрибозин
1-Бензилинозин
8-Бромаденозин
8-Бромаденин
8-Бром-2'-дезоксигуанозин
8-Бромгуанозин
8-Бромгуанин
8-Броминозин
6-Бромпурин
6-Карбоксиметилмеркаптопурин
2-Хлораденозин
5'-Хлор-5'-дехоксиаденозин
5'-Хлор-5'-дезоксиинозин
8-Хлор-2,6-дигидроксипурин
6-Хлоргуанин
6-Хлоргуанинрибозид
6-Хлоргуанозин
6-Хлорпурин
6-Хлорпуринрибозид
8-Хлорксантин
КордицепинТМ
6-Цианопурин
2,6-Дихлорпурин
2', 3'-Дидезоксиаденозин
2', 3'-Дидезоксигуанозин
2,8-Дигидроксиаденин
2,6-Дигидрокси-1-метилпурин
2,6-Дигидроксипурин
6-Диметиламинопурин
6-Диметиламинопурин-9-рибозид
1,1-Диметилгуанидин
1,7-Диметилгуанин
1,7-Диметилгуанозин
N'-Диметилгуанозин
1,7-Диметилксантин
3,7-Диметилксантин
2,8-Дитио-6-оксипурин
2,6-Дитиопурин
1,N'-Этеноаденозин
6-Этоксипурин
9-Этиладенин
5'-(N-Этил)карбоксамидоаденозин
9-Этилгуанин
6-Этилмеркаптопурин
6-н-Гептилмеркаптопурин
6-н-Гексиламинопурин
6-Гистаминопурин
N'-(2-Гидроксиэтил)аденозин
6-(β- Гидроксиэтиламино)пурин
1-Гидрокси-изо-гуанин
2-Гидрокси-6-меркаптопурин
6-Гидрокси-2-меркаптопурин
2-Гидрокси-6-метилпурин
6-Гидрокси-1-метилпурин
2-Гидроксипурин
6-Гидроксипурин
2-Гидрокси-6-тиопурин
6-Гидрокси-2-тиопурин
5'-Иод-5'-дезоксиаденозин
6-Иодпурин
N-(Δ′- Изопентенил)аденозин
6-Изопропоксипурин
2',3'-O-Изопропилиденаденозин
2',3'-O-Изопропилиденгуанозин
2',3'-О-Изопропилиденинозин
2'',3'-O-Изопропилиден-6-Тиоинозин
2-Меркаптоинозин
2-Меркаптопурин
6-Меркаптопурин
6-Меркаптопуринарабинозид
6-Меркаптопурин-2'-дезоксирибозид
6-Меркаптопуринприбозид
2-Меркаптопиримидин
6-Метоксипурин
6-Метоксипуриприбозид
1-Метиладенин
2-Метиладенин
3-Метиладенин
1-Метиладенозин
2'-O-Метиладенозин
3'-O-Метиладенозин
6-Метиламинопурин
1-Метилгуанин
7-Метилгуанин
1-Метилгуанозин
2'-O-Метилгуанозин
3'-O-Метилгуанозин
7-Метилгуанозин
1-Метилгипоксантин
1-Метилинозин
7-Метилинозин
Метилмеркаптогуанин
6-Метилмаркаптопурин
6-Метилимеркаптопуринрибозид
6-Метилпурин
6-н-Пропоксипурин
6-н-Пропилмеркаптопурин
6-Селеногуанозин
6-Селеноинозин
6-Селенопурин
6-Тиогуанин
6-Тиогуанозин
Тиогидроксипурин
2-Тиоксантин
6-Тиоксантин
2,6,8-Трихлор-7-метилпурин
2,6,8-Трихлорпурин
1,3,9-Триметилксантин
2,6,8-Триоксипурин
Примерами пролекарств, превращающихся альдегидоксидазой печени в соответствующие уридиновые, тимидиновые, цитидиновые, гуанозиновые, 8-азагуаниновые или 6-азауридиновые аналоги, являются следующие соединения: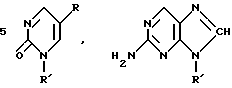
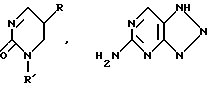
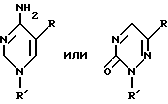
в которых R - I, F, Br, Cl, H, -CH3, -OR', -CF3, NO2, SR', -CH=CR2R3, -C≡CR2 или -N=N+ -N-, R1 - алкил с 1 - 5 атомами углерода, предпочтительно с одним атомом углерода, R2 и R3, независимо друг от друга, водород, C1-C5-алкил или галоген, и R'-водород, остаток сахара, такой как рибоза или дезоксирибоза, группа -CH2-O-CH2-CH2OH, -CH2-O-CH(CH2OH), замещенный или незамещенный алкил, арил, циклоалкил, циклоарил или любой другой желаемый остаток, размеры которого не препятствуют стерически действию альдегидоксидазы печени. Показано, что эти остатки не мешают нужному действию альдегидоксидазы печени на эти соединения. Предпочтительными группами формулы -CH= CR2R3 являются -CН= СF2 и -CH=CH2. Сахарные остатки предпочтительно имеют формулы: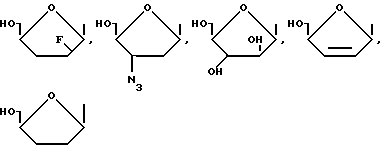
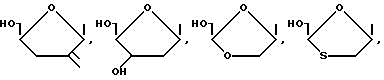
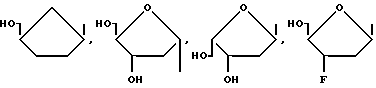
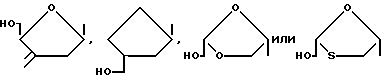
Сложные эфиры аналогов сахаров, используемых в изобретении, включают сложные эфиры, в которых H группы HOCH2 аналога замещена группой -COR4, где некарбонильный радикал R4 выбирают из водорода, нормальный или изоалкил (например, метил, этил, н-пропил, трет-бутил, н-бутил), алкоксиалкил (например, метоксиметил), аралкил (например, бензил), арилоксиалкил (например, феноксиметил), арил (например, фенил, возможно замещенный галогеном, C1-C4-алкилом или -алкоксигруппой); замещенный дигидропиридинил (например, N-метилдигидропиридинил); сульфонатные эфиры, такие как алкил- или аралкилсульфонил (например, метансульфонил); сульфатные эфиры; аминокислотные эфиры (например L-валил или L-изолейцил) и моно-, ди- или трифосфатные эфиры.
Также включенными в пределы таких эфиров, используемых в изобретении, оказываются эфиры, произведенные из полифункциональных кислот, содержащих больше одной карбоксигруппы, например дикарбоновых кислот HO2C(CH2)nCO2H, где n=1-10 (например, янтарной кислоты) или фосфорной кислоты. Способы получения таких эфиров хорошо известны.
Что касается описанных выше сложных эфиров, если не оговорено особо, то любой присутствующий алкильный остаток преимущественно содержит 1 - 16 атомов углерода, предпочтительно 1 - 4 атома углерода, и может содержать одну или больше двойных связей. Любой арильный остаток, присутствующий в таких эфирах, преимущественно содержит фенил.
В особенности эфирами могут быть C1-C8-алкиловые эфиры, незамещенные бензоиловые эфиры или бензоиловые эфиры, замещенные по меньшей мере одним галогеном (бромом, хлором, фтором или иодом), насыщенным или ненасыщенным C1-C6-алкилом, насыщенной или ненасыщенной C1-C6-алкоксигруппой, нитрогруппой или трифторметилом.
Фармацевтически приемлемые соли вышеописанных аналогов включают соли, произведенные из фармацевтически приемлемых органических кислот и оснований. Примеры подходящих кислот включают хлористоводородную, бромистоводородную, серную, азотную, перхлорную, фумаровую, малеиновую, фосфорную, гликолевую, молочную, салициловую, янтарную, п-толуолсульфоновую, винную, уксусную, лимонную, метансульфоновую, муравьиную, бензойную, малоновую, нафталин-2-сульфоновую и бензолсульфоновую кислоты.
Термин "аналог" (или "активный ингредиент") включает сам аналог так же, как его сложные эфиры или соли.
Соли, произведенные из соответствующих оснований, включают щелочнометаллические (например, натриевые), щелочноземельные (например, магниевые), аммониевые и NR
Выбор биоактивного соединения, для которого пролекарство может быть образовано в соответствии с настоящим изобретением, не ограничен только аналогами нуклеозидов или оснований нуклеозидов, фактически любое соединение, имеющее кетогруппу в структуре, является кандидатом в пролекарство, превращающееся альдегидоксидазой печени в такое соединение. Это можно установить простым анализом, обсужденным выше.
Термин "Биоактивное соединение" или "биологически активное вещество", как полагают, включает соединения, регулирующие любой аспект метаболизма животного, которому оно введено, или что соединение вторгается в организм животного, которому оно введено. Термин охватывает, без ограничений, антидепрессанты, антибиотики, лекарства, регулирующие давление крови, анальгетики противоопухолевые средства, антивирусные средства и т.д.
Для терапевтического лечения заболеваний, связанных с печенью, особенно предпочтительно использовать пролекарства настоящего изобретения вследствие того факта, что альдегидоксидаза найдена исключительно в печени. Таким образом, особое преимущество пролекарств настоящего изобретения заключается в устранении и уменьшении побочных эффектов, которые будут возникать при системном введении биоактивного соединения, хотя эффект желают иметь только в печени. Многие соединения, такие как ИУдР, полезные при лечении разрастания клеток, обладают существенной системной токсичностью, ограничивающей их клиническое использование. Соответствующее пролекарство обычно не будет иметь той же активности, что и биоактивное соединение. Таким образом, такие пролекарства обычно нетоксичны, если не превращаются метаболизмом в желаемые биоактивные соединения. Например, замещенные в положении 5 пиримидиноновые предшественники ИУдР или фторурацила не являются субстратами для тимидинкиназы и тимидинфосфорилазы человека и существенно нетоксичны. При лечении рака печени, например, ИПдР будет превращаться в печени альдегидоксидазой в ИУдР, которая затем предпочтительно будет восприниматься раковыми клетками печени до существенного распространения ИУдР по другим тканям. Таким образом, терапевтический индекс замещенных в положении 5 производных ПдР для первичного рака печени или метастазного рака печени будет много лучше, чем для их партнеров УдР.
Наряду с лечением заболеваний, связанных с печенью, существуют другие случаи, когда введение пролекарства будет предпочтительным по сравнению с введением биоактивного соединения, образующегося из такого пролекарства. Например, многие лекарства не могут быть введены перрорально по разным причинам. Например, фторурацил, известное противораковое лекарство, нельзя вводить перрорально. Вторпиримидинон является пролекарством, превращающимся альдегидроксидазой печени во фторурацил. Фторпиримидинон можно вводить пациенту перорально. Как показано в примерах ниже, фторпиримидинон показывает терапевтическую эффективность при лейкемии и раке толстой кишки.
В настоящее время ИУдР и БУдР используются в качестве радиосенсибилизаторов. Однако их эффективность ограничивается их цитотоксичностью и катаболизмом до свободных оснований с последующим дегалогенированием (Clin. Pharmacol, Ther., 1988, 44, 369 - 375). Так как ИПдР и его аналоги действительно нетоксичны и не являются субстратами для тимидинфосфорилазы, то использование ИПдР и его аналогов вместо ИУдР или БУдР позволит обойти трудности токсичности и разложения, связанные с радиационной терапией.
"Болезни, связанные с печенью" включают вирусный гепатит, например гепатит A, гепатит B, гепатит C, гепатит D; гепатому; раковые метастазы в или из печени; инфекцию вирусом цитомегалии или другими вирусами, паразитарные инфекции, например шистосомоз, клонорхоз, фасциолез, описторхоз; и инфекции трематодами и гельминтами, микробами, например грибковые или бактериальные инфекции, такие как Paracoccidioides brasiliensis, и другие болезни печени, такие как цирроз или отторжение печеночного трансплантата.
Метаболические состояния или заболевания также поддаются лечению при селективном использовании пролекарства, превращающегося в активное соединение альдегидоксидазой печени. Лекарства могут быть такими, которые нацелены на рецепторы в или на клетках печени или непеченочных клетках. Эти лекарства полностью или частично имеют активность агонистов или антагонистов. Большое разнообразие лекарств, действующих на рецепторы, можно создать введением соответствующего пролекарства субъекту или в препарат альдегидоксидазы in vitro.
Так, пролекарства лекарств, действующих на отдаленные места, такие как сердце или мозг, также можно использовать в соответствии с настоящим изобретением. Далее, если клетки печени испытывают недостаток в целевых рецепторах, то это снижает потенциал нежелательной токсичности, обусловленный высокой концентрацией в печени. Использование пролекарств, превращающихся ферментом печени в активные лекарства, имеет широкое применение.
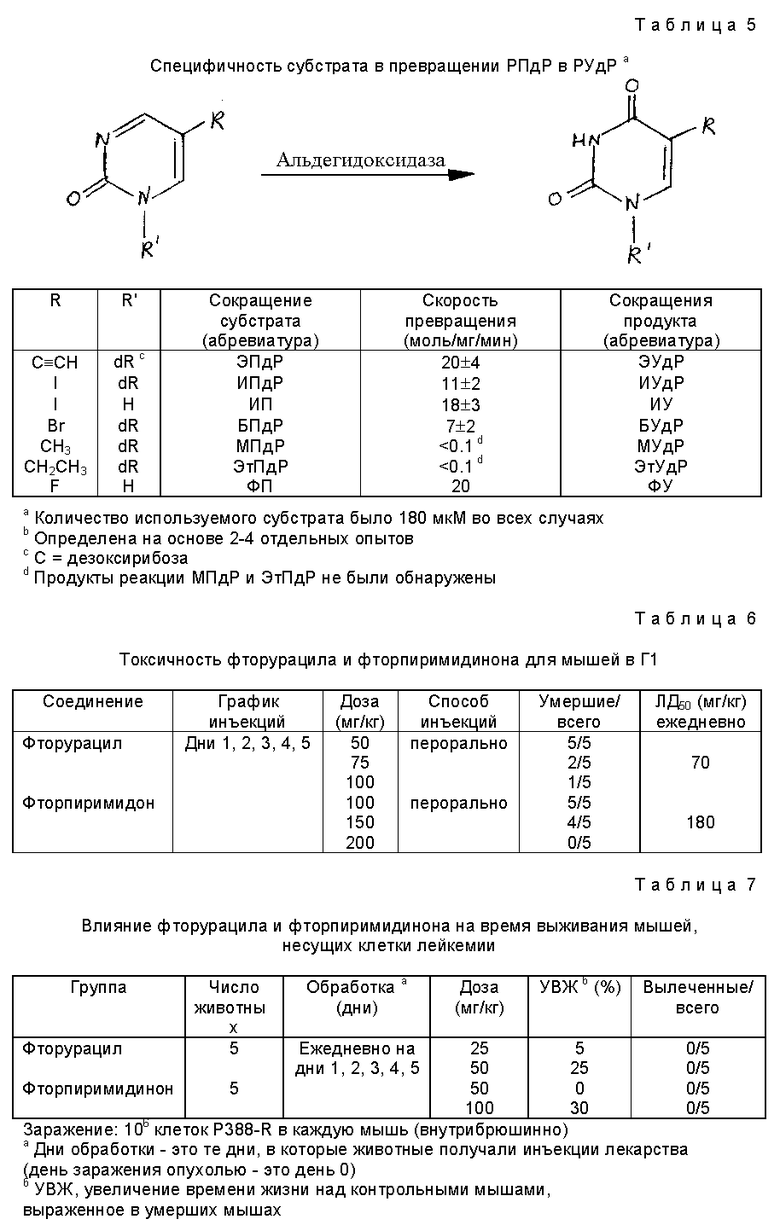
В зависимости от уровня активности фермента по отношению к пролекарству можно достичь медленной скорости образования или медленного выделения активного лекарства в организм. Соединения, приведенные в примерах, подвергаются быстрому метаболизму, но их можно модифицировать, чтобы замедлить метаболизм. Это должно привести к менее частому приему пролекарства по сравнению с лекарством со всеми вытекающими преимуществами, связанными с состоянием пациента и постоянством дозы. Если метаболизм в активное лекарство достаточно медленный, то совершенно новые классы соединений можно использовать в терапии, которые нельзя было использовать прежде из-за проблем токсичности, связанных с приемом шариков. Например, из таблицы 5 ниже можно видеть, что природа группы R пиримидинового субстрата существенно влияет на скорость превращения. Таким образом, ясно, что можно предложить пролекарство с предопределенной скоростью метаболизма.
Примером лекарства, которое можно улучшить за счет медленного превращения in vivo, является кетонсодержащее лекарство сурамин. Эффективная противораковая доза при метастазном раке простаты очень близка к дозе, приводящей к параличу, что было случайно и неудачно обнаружено во время испытаний. Прием лекарства в виде шариков, как было найдено, значительно менее эффективен. Как следствие, пациенты, нуждающиеся в таком лечении, госпитализировались, непрерывно наблюдались и подвергались вливанию для поддерживания узкого интервала эффективной и приемлемой концентрации. При использовании альдегидной или другой пролекарственной формы сурамина, превращающейся альдегидоксидазой в активный сурамин, стало возможным давать пациенту лекарство в виде шариков без необходимости в постоянной госпитализации.
Как упоминалось выше, перорально приемлемые пролекарства можно использовать вместо их активного продукта, неприемлемого перорально. Пролекарство может отличаться от активного продукта повышенной стабильностью в кислотных условиях, устойчивостью к пищеварительным ферментам, лучшей всасываемостью, меньшей раздражительной способностью или токсичностью по отношению к пищеварительной системе. Всасываясь, пролекарство превращается в активное лекарство альдегидоксидазой печени, тем самым обходя пищеварительный тракт. Преимущество перорального введения над парентеральным очевидно для всех опытных ученых.
Независимо от пути введения любое лекарство имеет ограниченный полупериод существования, обусловленный почечным клиренсом, ферментативным разложением и слишком большим или слишком малым связыванием с белками сыворотки и т. п. Применение пролекарств расширяет возможности разработки лекарств, уравновешивая такие проблемы.
Количество аналога, описанного выше, для применения в настоящем изобретении будет изменяться не только от отдельного выбранного соединения, но также от пути введения, природы излечиваемого состояния, возраста и состояния пациента и будет, в конечном итоге, определяться опытом лечащего терапевта или ветеринара. В общем, однако, подходящие дозы будут находиться в интервале 1 - 100 мг/кг массы тела в день, предпочтительно 2 - 50 мг/кг/день, наиболее предпочтительно 2 - 10 мг/кг/день.
Желаемая доза может обычно быть представлена единой дозой или раздельными дозами, вводимыми с соответствующими промежутками, например, как две, три, четыре или больше поддоз в день.
Аналог, описанный выше, обычно вводят единичной дозой, например, содержащей 0,5 - 50 мг, предпочтительно 20 - 1000 мг, наиболее предпочтительно 50 - 700 мг, активного ингредиента на единичную дозировочную форму.
В идеале, активный ингредиент должен быть введен, чтобы достичь пика концентрации в плазме активного ингредиента 1 - 75 мкМ, предпочтительно 2 - 50 мкМ, наиболее предпочтительно 3 - 30 мкМ. Этого можно достичь, например, внутривенной инъекцией 0,1 - 5% раствора пролекарства, возможно в физиологическом растворе, или введением в виде шарика, содержащего 0,1 - 50 мг/кг активного ингредиента.
Согласно оценке различные пролекарства могут быть использованы в разной дозировке. Далее, лечение заболеваний различных тканей или органов также требует разной дозировки. Эти дозировки легко определимы опытным сотрудником известными методами.
Возможно, что для применения в терапии, аналог, описанный выше, можно вводить в виде сырого химиката, но предпочтительно присутствие пролекарства в смеси с фармацевтически приемлемым носителем в форме фармацевтического препарата.
Изобретение также относится к фармацевтическим препаратам, содержащим аналог, описанный выше, вместе с одним или больше фармацевтически приемлемым носителем для него и, возможно, другими терапевтическими и/или профилактическими ингредиентами. Носитель(и) должны быть приемлемыми в том смысле, что должны быть совместимы с другими ингредиентами препарата и безвредны для реципиента.
Фармацевтические препараты включают препараты, пригодные для перорального, ректального, носового, местного (включая трансбуккальное, сублингвальное и трансдермальное), вагинального или парентерального (включая внутримышечное, подкожное и внутривенное) введения, или препараты, пригодные для введения ингаляцией или инсуффляцией. Препараты могут быть обычно представлены в дискретных дозировочных единицах и могут быть приготовлены любыми методами, хорошо известными в фармации. Все методы включают стадию смешивания активного компонента с жидкими носителями или тонко измельченными твердыми носителями или с теми и другими, затем стадию, если необходимо, формования продукта в желаемый препарат. Инкапсулирование химиката в липосомы или пузырьки также можно использовать, если показано для целей доставки или стабилизации.
Фармацевтические препараты, пригодные для перорального введения, обычно могут быть представлены в форме дискретных единиц, таких как капсулы, пакетики или таблетки, содержащие определенное количество активного компонента; таких как порошки или гранулы; как растворы; как суспензии; или как эмульсии. Активные компоненты также могут иметь форму шариков, электуария или пасты. Таблетки и капсулы для перорального введения могут содержать обычные наполнители, такие как связующие средства, смазки, дезинтегрирующие средства или смачивающие средства. Таблетки можно покрывать известными методами. Пероральные жидкие препараты могут иметь форму, например, водной или масляной суспензии, растворов, эмульсий, сиропов или элексиров, или могут быть сухими продуктами, смешиваемыми с водой или другим подходящим носителем перед использованием. Такие жидкие препараты могут содержать обычные добавки, такие как суспендирующие средства, эмульгаторы, неводные носители (включая съедобные масла) или консерванты.
Активный компонент также может быть включен в парентеральные препараты (например, инъекции, такие как инъекции ударной дозы или непрерывное вливание), имеющие единичную дозировочную форму, такую как ампулы, предварительно заполненные шприцы, малые объемы для вливания или многодозные контейнеры с добавленным консервантом. Препараты могут иметь такие формы, как суспензии, растворы или эмульсии в масляном или водном носителях, и могут содержать вспомогательные средства, такие как суспендирующие, стабилизирующие и/или диспергирующие средства. Альтернативно, активные компоненты могут иметь порошковую форму, полученную асептическим выделением стерильного твердого вещества или лиофилизацией из раствора, для смешивания с подходящим носителем, например стерильной, свободной от пирогенных веществ водой, перед использованием.
Фармацевтические препараты, пригодные для ректального введения с твердым носителем, наиболее предпочтительно представлены суппозиториями с единичной дозой. Подходящие носители включают какао-масло и другие обычно используемые материалы. Суппозитории обычно получают смешиванием активного компонента с размягченным или расплавленным носителем с получающим охлаждением и формованием.
Препараты, пригодные для вагинального введения, могут быть представлены пессариями, тампонами, кремами, гелями, пастами, пенами или разбрызгивающимися средствами, содержащими в дополнение к активному компоненту известные носители.
Для внутриносового применения активный компонент используют как жидкий разбрызгиваемый раствор или диспергируемый порошок или капли. Капли могут быть составлены из водной или неводной основы, содержащей один или больше диспергирующий, солюбилизирующий или суспендирующий агент. Жидкие разбрызгиваемые растворы обычно подают из аэрозольной упаковки.
Для введения ингаляций активный компонент обычно подают из инсуффлятора, распылителя или аэрозольной упаковки или других обычных средств подачи аэрозоля. Аэрозольные упаковки могут содержать обычный пропеллент, такой как дихлордифторметан, трихлорфторметан, дихлортетрафторэтан, диоксид углерода или другой подходящий газ. В случае аэрозоля под давлением дозировочную единицу можно определить с помощью клапана для подачи отмеренного количества.
Альтернативно, для введения ингаляцией или инсуффляцией активный компонент может иметь форму сухого порошка, например порошковой смеси, (активного) соединения и подходящей порошковой основы, такой как лактоза или крахмал. В случае порошкового препарата единичной дозировочной формой могут быть, например, капсулы или патроны, или желатиновой или пузырчатой упаковки, из которой порошок вводят с помощью ингалятора или инсуффлятора.
Если желательно, то описанные препараты можно модифицировать для замедления выделения активного компонента.
Фармацевтические препараты для использования в соответствии с изобретением могут также содержать другие активные компоненты, такие как антимикробные средства или консерванты.
Фармацевтические препараты для использования в соответствии с изобретением могут также содержать инертные компоненты, такие как осушители, вещества, обеспечивающие легкость переработки, красители, отдушки и покрытия для облегчения проглатывания.
Активный компонент можно также использовать в смеси с другими терапевтическими средствами, например другими антиинфекционными средствами. В частности, соединения формулы I можно применять вместе с известными антивирусными средствами, например аденинарабинозидом или α- интерфероном.
Изобретение, таким образом, в дополнительном аспекте обеспечивает способ использования аналогов, описанных выше, с другими терапевтически активными средствами, в частности средствами против вируса гепатита B.
Если активный компонент, генерированный из пролекарства, является противораковым средством, то другие противораковые или иммуномодулирующие средства можно применять вместе с ним, также возможна любая другая совместимая комбинация лекарств, будет ли их активности синергическими, дополнительными или отдельными.
Комбинации, рассмотренные выше, могут обычно быть представлены в форме фармацевтического препарата, и, таким образом, использование фармацевтических препаратов, содержащих комбинацию, определенную выше, вместе с фармацевтически приемлемым носителем для нее, составляет дополнительный аспект изобретения.
Индивидуальные компоненты таких комбинаций можно вводить либо последовательно, либо одновременно в раздельных или объединенных фармацевтических препаратах.
Если аналог, описанный выше, применяют в комбинации с вторым терапевтическим средством для того же заболевания, например активные против одного вируса, то доза каждого соединения может быть той же самой, либо отличной от дозы, когда аналог используют отдельно. Соответствующую дозу можно легко оценить опытному сотруднику.
Альдегидоксидаза выделяется из печени и может быть иммобилизована на твердой фазе для легкого отделения ферментного катализатора от реакционной смеси. Целевые биоактивные оптически активные соединения затем отделяют от соединений-предшественников и регенерируют. Методики очистки фермента хорошо известны и можно использовать любую подходящую. Методики иммобилизации фермента на твердой фазе, будет ли это адсорбция, улавливание, химическое связывание или удерживание за полупроницаемой мембраной, также хорошо известны.
Содержание всех ссылок, упоминаемых в этой заявке, включено в описание изобретения в качестве ссылок. Изобретение описано следующими примерами, иллюстрирующими изобретение, но не ограничивающие его.
Примеры
Пример 1. Препараты ткани
Ткань печени крысы промывают охлажденным до температуры льда 1,15% KCl и промокают досуха. Ткань затем гомогенизируют в гомогенизаторе в 1,15% растворе KCl, взятом в 3-кратном количестве в расчете на массу ткани, с образованием 25% (масса/объем) гомогената. Гомогенат затем центрифугируют при 10 000 g в течение 10 мин при 4oC. Образующий надосадочный раствор фильтруют через Мираклос (подобна сырной ткани), затем подвергают диализу в течение ночи на фоне 50 мМ буфера Трис-HCl с pH 7,5 и сохраняют при -80oC до использования. Гепатоциты крысы получают техникой перфузии, клетки экстрагируют 10 мМ фосфатным буфером с pH 7,5, содержащим 1 М KCl, и диализуют 4 часа на фоне 50 мМ буфера Трис-HCl с pH 7,5.
Пример 2. Условия анализа
В Стандартных условиях анализа реакционная смесь содержала 50 мМ Трис-HCl с pH 7,5, 1 мМ ЭДТФ, 180 мкМ ИПдР (или его аналогов) и приблизительно 0,01 мг белка надосадочного раствора гомогената ткани (полученного при 10 000 g) в конечном объеме 500 мкл и выдерживали при 37oC 10 минут, если не оговорено особо. В конце выдерживания удаляли 300 мкл реакционной смеси, смешивали с 630 мкл ацетонитрила и перемешивали. Осажденный белок удаляли центрифугированием и надосадочный раствор лиофилизировали до сухости. Образцы восстанавливали до первоначального объема аликвоты буфером подвижной фазы ВЭЖХ и анализировали на колонке PP-18 Олтех. ИПдР, ИУдР и иодурацил определяли при длине волны УФ поглощения 230 нм, ИПдР также определяли при длине волны 335 нм. Подвижной фазой была смесь 10% ацетонитрила /90% мМ ацетата аммония с pH 6,8 и скорость потока 1 мл/минут. Стандартные кривые ИПдР и ИУдР установлены на основе интегральных величин известных концентраций.
Пример 3. Превращение 5-иод-2-пиримидинон-2'-дезоксирибозы (ИПдР) в 5-иоддезоксиуридин (ИУдР)
Чтобы изучить превращение ИПдР в ИУдР ферментом печени метаболиты ИПдР анализировали после выдерживания с надосадочным раствором гомогената печени крысы, используя технику ВЭЖХ обращенных фаз. ИПдР и ИУдР можно детектировать при длине волны поглощения 230 нм, но только ИПдР можно детектировать при 335 нм. Для ограничения фосфоролитического расщепления ИУдР до иодурацила тимидинфосфорилазой (Radiation Oncology Biol. Phis., 1984, 10, 1399 - 1406) в анализе применяли буфер Трис-HCl. Как показано на фиг. 1, существует временная зависимость превращения ИПдР в ИУдР и ИУдР оказывается единственным продуктом, производимым ИПдР. Идентификация ИУдР подтверждается временами удерживания на колонке С-18 (8 мин против 9,5 мин для ИПдР) и УФ спектром так же, как спектроскопией ядерного магнитного резонанса (результаты не показаны).
Пример 4. Свойства активности "ИПдР-оксидазы"
Активность "ИПдР-оксидазы" не требует экзогенных кофакторов, она значительно меньше в экстрактах почек и селезенки, чем печени (таблица 3) и не обнаружена в экстрактах легких или кишечника крыс. Печень человека содержит подобное количество этого фермента. Экстракты гепатоцитов, которые составляют 80% количества клеток печени, имели очень близкую удельную активность в сравнении с экстрактом всей печени, что позволяет предположить, что окисление ИПдР происходит, главным образом, в этих клетках. В попытке локализовать активность фермента конверсии ИПдР использовали дифференциальное фракционное центрифугирование. Для каждой фракции был выполнен анализ активности фермента. Единственная фракция, которая показала активность фермента, возникла как надосадочный раствор при 100 000 g в течение 60 минут. Это значит, что фермент находится в растворимой фракции цитозоля. Для дальнейшей очистки применяли колоночную хроматографию на ДЭАЭ-целлюлозе, колоночную хроматографию на Сефарозе блю и центрифугирование с градиентом плотности глицерина в этом же порядке. Активность ИПдР-оксидазы возросла в 380 раз по сравнению с исходным неочищенным экстрактом печени крысы. Частично очищенный фермент катализирует синтез ИУдР со скоростью 3,8 мкмоль в мин на миллиграмм белка при 37oC в этих условиях. Средняя молекулярная масса этого фермента как у крысы, так и у человека, как определено центрифугированием с градиентом 20 - 40% глицерина, составляет приблизительно 280000. Как было найдено, ни кофактор, ни двухвалентный катион не требуется для фермента печени крысы или фермента печени человека.
Пример 5. Идентификация "ИПдР-оксидазы"
Для определения фермента, ответственного за катализ окисления ИПдР, ряд известных оксидо-редуктаз, имеющих сходную способность окислять атом углерода у соседней аминогруппы в карбонил, был изучен на способность катализировать окисления ИПдР. Ксантиноксидаза (Боэрингер Манхайм), выделенная из коровьего молока и катализирующая окисления гипоксантина в ксантин, не превращала ИПдР в ИУдР. Оксидазная система смешанной функции, полученная из микросом печени крысы (и активная) к широкому спектру субстратов, зависит от NADPH. Однако эта микросомная фракция из печени крыс не показала активности ИПдР-оксидазы, причем независимо от того добавлен ли NADPH или нет. Алкогольдегидрогеназа (Сигма) и алкогольоксидаза (Сигма) находятся в растворимой фракции экстрактов клеток печени, но конверсии ИПдР в ИУдР не обнаружено с очищенными препаратами каждого фермента в тех же условиях, в которых они превращают природные субстраты эффективно. Саркозиноксидаза (Боэрингер Манхайм), катализирующая превращение N'-метилглицина в глицин, не обладает активностью ИПдР-оксидазы. Фенилаланингидроксилаза, уроканаза, цистатионин -γ- лиаза, L-глутаматдегидрогеназа, цистатионаза и некоторые другие оксидоредуктазы были исключены на основе субстратконкурентного анализа, их различной специфичности к кофактору или по некоторым другим признакам, известным из литературы (Biochemistry, 1977, 16, 2916-2921; Enr. J, Biochem, 1976, 64, 341-350; J.Biol. Chem, 1979, 254, 843-851; J. Biol. Chem., 1970, 245, 528-537; Science, 1955, 121, 603-604, Eur. J. Biochem., 1971, 20, 269-275).
Так как альдегидоксидаза имеет одинаковую молекулярную массу, находится в цитозольной фракции клеток печени и имеет широкую специфичность к субстратам (J. Biol. Chem, 1962, 237, 922-928), то считают, что она является ферментом "ИПдР-оксидазой", ответственной за превращение ИПдР в ИУдР. Печеночная альдегидоксидаза, катализирующая окисление различных альдегидов в соответствующие кислоты, также превращает N'-метилникотинамид (Сигма) в N'-метил-2-пиридон-5-карбоксамид и N'-метил-4-пиридон-5-карбоксамид (J. Biоl., Chem., 1962, 237, 922-928; J. Biol. Chem., 1964, 239, 2022-2035; Arohs of Biochem and Biophy, 1971, 145, 27-34; Arohs of Biochem and Biophy, 1971, 145, 35-42; J. Biol. Chem., 1973, 248, 2580-2587; Biochemistry, 1982, 21, 3561-3568; J. Biol. Chem, 1981, 256, 3479-3486). Активность альдегидоксидазы, как было положено, стимулирует феррицианид калия и буфер Трис, но не MgCl2 (J. Biol, Chem, 1973, 248, 2580-2587). Этот фермент можно ингибировать 2-меркаптоэтанолом, дитиотреитолом и другими тиольными веществами (J. Biol. Chem, 1964, 239, 2022-2035). Не было ингибирования цистеином при 5 мМ, однако при 50 мМ наблюдалось сильное ингибирование активности фермента (J. Biol, Chem, 1973, 248, 2580-2587). Катионы двухвалентных металлов, такие как Cu++, Zn++ и Fe++, вызывают сильное ингибирование (J. Biol, Chem., 1962, 237, 922-928). Активность можно ингибировать ацетальдегидом, но не аллопуринолом или формальдегидом (J. Biol. Chem, 1964, 239, 2022-2035); Archs of Biochem and Biophy, 1971, 145, 35-42; J. Biol. Chem, 1981, 256, 3479-3486). Поэтому ряд соединений был проверен по их действию на окисление ИПдР в ИУдР, и было найдено, что профиль ингибирования соединений с активностью "ИПдР-оксидазы" (таблица 4) существенно идентичен характеристическим образцам альдегидоксидазы. Далее, на всех стадиях очистки альдегидоксидаза не могла быть отделена от активности "ИПдР-оксидазы".
Пример 6. Специфичность субстрата
Несколько аналогов 2-пиримидинодезоксирибозы были проверены на превращение в их дезоксиуридиновые двойники. Константа Михаэлиса К для ИПдР в реакции при pH 7,5 и pH 9,5 составляет 150 мкМ и 87 мкМ соответственно. К для 5-этинил-2-пиримидинондезоксирибозы (ЭПдР) в реакции при pH 7,5 и pH 9,5 составляет 77 мкМ и 46 мкМ соответственно. Тем не менее относительная Vмакс. для ИПдР в реакции при pH 7,5 и pH 9,5 одинакова. 5-Иод-2-пиримидинон (ИП), агликоза ИПдР был превосходным субстратом для альдегидоксидазы. Синтетические субстраты для альдегидоксидазы оказались лучшими, чем природные субстраты, N'-метилникотинамид и ацетальдегид, судя по силе ингибирования ими реакции окисления ИПдР (таблица 4). Скорость реакции фермента печени с различными аналогами ИПдР следует порядку ЭПдР, иодпиримидинон, ИПдР, 5-бром-2-пиримидинондезоксирибоза (БПдР) и 5-метил-2-пиримидинондезоксирибоза (МПдР) или 5-этил-2-пиримидинондезоксирибоза (ЭтПдР) (таблица 5). Электроотрицательные заместители в положении 5, как оказалось, увеличивают активность субстрата в этой окислительной реакции.
Пример 7. Токсичность фторурацила и фторпиримидинона
Мышам BDFI вводили ежедневно перорально дозу либо 50, 75 или 100 мг/кг фторурацила либо 100, 150 или 200 мг/кг фторпиримидинона. Коэффициенты выживаемости каждой представлены в таблице 6 и на фиг. 2 и 3. Токсичность фторпиримидинона значительно меньше при более высоких дозах введения.
Пример 8. Влияние фторурацила и фторпиримидинона на клетки лейкемии
Мышам вводили 100000 клеток лейкемии Р388-Р, чтобы вызвать лейкемию. Эти клетки устойчивы к Адриамицину. Этим лейкемическим мышам ежедневно перорально давали по 25 и 50 мг/кг фторурацила и по 50 и 100 мг/кг фторпиримидинона и измеряли время выживания. Результаты приведены в таблице 7 и на фиг. 4. Время выживания было одинаковым при использовании как фторурацила, так и фторпиримидинона.
Пример 9. Влияние фторурацила и фторпиримидинона на рак толстой кишки
Мышам вводили клетки 33 толстой кишки, часть не лечили, остальным давали ежедневно перорально по 25 и 50 мг/кг фторурацила и по 50 и 100 мг/кг фторпиримидинона и измеряли в течение времени изменение массы опухоли. Данные приведены на фиг. 5. Уменьшение размера опухоли или уменьшение скорости ее роста сравнимы для фторурацила и фторпиримидинона
Пример 10. Влияние включения ИПдР в ткани атимных безволосых мышей
Мышам вводили ежедневно перорально дозы 0, 100, 250 и 500 мг/кг. Процент замещения тимидина, как указывающий на включение ИПдР в костный мозг, ткани кишок и печени, анализировали известными методами. Как показано на фиг. 6, относительно небольшое количество тимидина замещается ИПдР в костном мозгу и кишках, причем плато замещения тимидина имеет место при уровне дозы 250 мг/кг/день. Включения ИПдР в печень не найдено. Результаты, представленные на фиг. 6, являются средним процентом замещения тимидин ± среднеквадратическая ошибка. 11 больше или равен 3 для каждой дозы. Эти результаты показывают, что нет существенного замещения тимидина на ИУдР в печени и очень малый процент замещения найден в костном мозге и кишках. Эти результаты устанавливают, что введение ИПдР в качестве пролекарства в соответствии с настоящим изобретением подходит для лечения млекопитающих, включая человека.
Пример 11. Включение ИПдР в ткани атимных безволосых мышей, имеющих метастазные опухоли
Мышам вводили ежедневно дозы 0, 100, 250 и 500 мг/кг, чтобы определить замещение тимидина в ткани печени и опухоли. Результаты, приведенные на фиг. 7, представляют средний процент замещения тимидина ± среднеквадратичная ошибка, N больше или равен 3 (контрольные опухоли N = 2). В то время как очень малый процент включения обнаружен в нормальной печени, повышенный процент замещения найден в опухоли, показывая, что использование ИПдР в качестве пролекарства для лечения опухоли даст, как можно ожидать, хорошие результаты.
Примеры 12. Лечение трансгенных мышей фторпиримидиноном
Трансгенным мышам, полученным по методу Сепулведа (Cancer Research 1989, 49, 6108-6117), вводили препарат, начиная с девяти недель после рождения, отдельно самцам и самкам, причем группа обработки получала 100 мг/кг два раза в день один раз в неделю. Фиг. 8 и 9 показывают изменение массы тела в течение 9 - 17 недель для самок и самцов. Как самки, так и самцы, подвергшиеся обработке, показывают значительное уменьшение прибавления в массе после 13 недели, коррелирующее с увеличением срока жизни. Обработанные самки переживают контрольных крыс, умирающих от осложнений рака на 16 неделе. Приведенные данные дают основание предположить, что применение фторпиримидинона в качестве средства лечения опухоли в соответствии с настоящим изобретением даст, как ожидают, хорошие результаты.
Цитированные выше ссылки включены в рассмотрение независимо от того, конкретны они или нет. Приоритетные заявки США 07/701 462, от 15 мая 1991 и 07/829 474 от 3 февраля 1992 также включены в рассмотрение.
Имея полное описание этого изобретения, опытной сотрудник может оценить, что то же самое может быть выполнено внутри широкого круга эквивалентных параметров, концентраций и условий без отступления от духа и границ изобретения и без чрезмерных экспериментов.
В то время как это изобретение описано в связи с конкретным вариантом его, следует понимать, что возможны дополнительные модификации. Эта заявка предполагает охватить любые изменения, применения или приспособления изобретений, следующих, в целом, принципам этого изобретения и включающих такие отклонения от раскрытого содержания, которые попадают в область известной и обычной практики данной области знаний и которые могут быть отнесены к существенным признакам, изложенным далее в рамках приложенных притязаний.


Изобретение относится к медицине. Способ основан на введении активного лечебного соединения в восстановленной форме (напр. 5-замещенный урацил), которое превращается в клетках печени альдегидоксидазой в биоактивное дикетосодержащее лекарство или метаболит. Показано, что пролекарства эффективны при лечении заболеваний, как и сами лекарства, причем оказывают многочисленные благоприятные воздействия и лишены многих побочных эффектов. 15 з. п. ф-лы, 9 ил., 7 табл.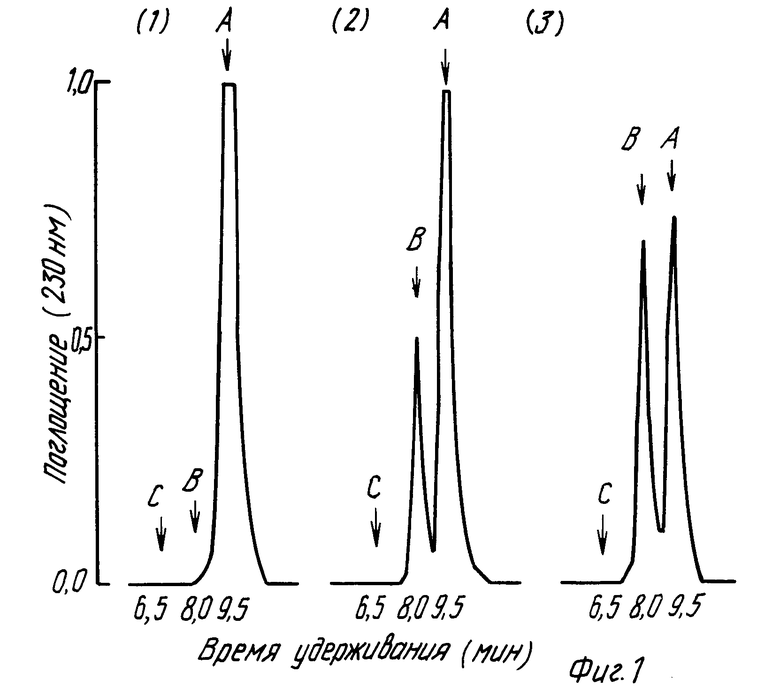
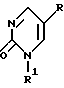
где R - J, F, Br, Cl, H, -CH3, -CF3, NO2, SR1, OR1, -CH=CR2R3,  или -N= N+= N-;
или -N= N+= N-;
R1 - C1- C5-алкил;
R2 и R3 независимо друг от друга являются водородом, C1-C5-алкилом или галогеном и R1 - водород, остаток сахара, -CH2-O-CH2-CH2OH, -CH2-O-CH(CH2OH), замещенный или незамещенный алкил, арил, циклоалкил, циклоарил или любой другой требуемый остаток, причем такой остаток, чтобы стерически не мешать действию альдегидоксидазы печени. или -N= N+= N-;
или -N= N+= N-;
R1 - C1- C5-алкил;
R2 и R3 независимо друг от друга являются водородом, C1-C5-алкилом или галогеном и R1 - водород или аналог сахара. или -N= N+= N-;
или -N= N+= N-;
R1 - C1- C5-алкил; R2 и R3 независимо друг от друга водород, C1- C5-алкил или галоген.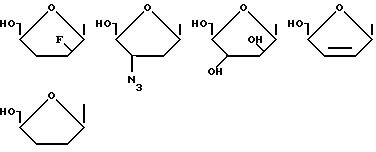
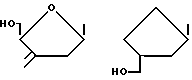
14. Способ по п.7, отличающийся тем, что R1 является водородом или остатком сахара.
| Способ получения производных 4(3Н)-оксо-5,6,7,8-тетрагидропиридо(2,3- @ )пиримидина или их таутомерных форм | 1987 |
|
SU1581222A3 |
| Способ химиотерапии рака яичников | 1981 |
|
SU969272A1 |
| US 4895937, А, 23.01.90. | |||
