Область, к которой относится изобретение
Настоящее изобретение относится к соединениям с иммунодепрессивными свойствами и их применению в терапии.
Предпосылки к созданию изобретения
Воспалительные или иммунно-опосредованные невропатии представляют собой разнообразную группу заболеваний, включающую такие периферические невропатии как синдром Гийена-Барре (СГБ), хроническая воспалительная демиелинизирующая полирадикулоневропатия (ХВДП), мультифокальная моторная невропатия с блоком проведения (ММН) и парапротеинемическая демиелинизирующая периферическая невропатия (ПДН). Патогенез воспалительных невропатий все еще остается предметом исследования.
Ниже обсуждается ряд демиелинизирующих периферических невропатий.
Периферические невропатии включают, соответственно, синдром Гийена-Барре, являющийся острой аутоиммунной полиневропатией, затрагивающей периферическую нервную систему, обычно инициируемой острым инфекционным процессом. Существует несколько типов СГБ, причем наиболее обычной формой является острая воспалительная демиелинизирующая невропатия (ОВДН). СГБ часто протекает тяжело и обычно выражается в виде восходящего паралича, проявляющегося слабостью в ногах, распространяющейся на верхние части тела и лицо совместно с полной потерей глубоких сухожильных рефлексов. Ответ супрессорных Т-клеток понижен, что свидетельствует о клеточно-опосредованной иммунологической реакции, направленной на периферическую нервную систему.
Мультифокальная моторная невропатия является прогрессирующим мышечным расстройством, характеризуемым мышечной слабостью в руках при различиях в конкретных затронутых мышцах между одной и другой сторонами тела. Симптомы также включают атрофию мышц, мышечные судороги и непроизвольные сокращения или подергивания ножных мышц. Мультифокальная моторная невропатия считается иммунно-опосредованным расстройством.
Парапротеинемическая демиелинизирующая невропатия является главной причиной поздней демиелинизирующей невропатии, весьма схожей с ХВДП, но более хронической. Она преимущественно затрагивает людей в возрасте 60 лет и более. Пациенты могут испытывать многочисленные симптомы, с которыми необходимо бороться, и болезнь имеет тенденцию к длительности.
Хроническая воспалительная демиелинизирующая полиневропатия (ХВДП) характеризуется прогрессирующей слабостью и нарушенной сенсорной функцией ног и рук. Эти симптомы вызываются повреждениями миелиновой оболочки периферических нервов. Она часто сопровождается симптомами, включающими покалывание или онемение (начинающееся в пальцах ног и рук), слабость рук и ног, потерю глубоких сухожильных рефлексов, усталость и нарушение чувствительности. Распространенность ХВДП составляет примерно от 2 до 4 случаев на 100000. Патогенез неясен, но может включать механизмы, опосредованные как Т-, так и В-клетками.
Протекание ХВДП широко варьируется от пациента к пациенту. У некоторых может иметь место приступ ХВДП, за которым следует самопроизвольное восстановление, тогда как у других могут наблюдаться многочисленные приступы с частичным восстановлением между рецидивами. ХВДП приводит к тяжелой инвалидности у существенного числа пациентов. Современные методы лечения направлены на модуляцию иммунного ответа с целью достижения ремиссии и поддержания функционального состояния.
В международной заявке на изобретение WO 2004/103306 и заявке на изобретение США US 2005/0014728 описаны соединения, применимые при лечении заболеваний или расстройств, опосредованных лимфоцитарными взаимодействиями. Вышеупомянутые документы включены в настоящую заявку в качестве ссылки во всей их полноте и для всех целей. В особенности это относится к следующим частям US 2005/0014728: параграфы от [0006] до [0015], от [0022] до [0042], от [0102] до [0124] и от [0126] до [0149], а также таблица 1. Следует отдельно упомянуть примеры 1-5 US 2005/0014728.
Краткое описание изобретения
В одном варианте осуществления настоящего изобретения его объектом являются соединения, как они упомянуты ниже, для применения в лечении периферической невропатии, например ХВДП. В другом варианте осуществления настоящего изобретения его объектом является способ лечения пациента, страдающего периферической невропатией, например ХВДП, включающий введение пациенту эффективного количества соединения, как оно упомянуто ниже. В еще одном варианте осуществления настоящего изобретения его объектом является применение соединения, как оно упомянуто ниже, для изготовления лекарственного средства для применения в лечении периферической невропатии, например ХВДП.
Соединения, к которым относится настоящее изобретение, представляют собой соединения, описанные в заявках на изобретение WO 04/103306 и US 2005/0014728, WO 05/000833, WO 05/103309 или WO 05/113330, например соединения формулы А1 или А2
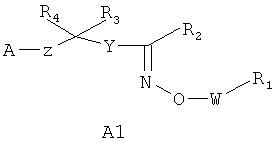
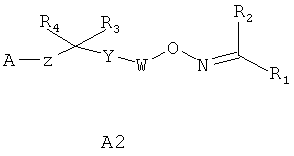 ,
,
в которых
А представляет собой COOR5, OPO(OR5)2, PO(OR5)2, SO2OR5, POR5OR5 или 1H-тетразол-5-ил, где R5 представляет собой водород или образующую сложный эфир группу, например (C1-C6)алкил;
W представляет собой связь, (C1-C3)алкилен или (C2-C3)алкенилен;
Y представляет собой (C6-C10)арил или (C2-C9)гетероарил, например (C3-C9)гетероарил, необязательно замещенный 1-3 радикалами, выбранными из галоида, OH, NO2, (C1-C6)алкила, (C1-C6)алкоксигруппы, галоидзамещенного (C1-C6)алкила и галоидзамещенной (C1-C6)алкоксигруппы;
Z выбран из
 ;
;  ;
; 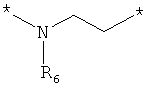 ;
; 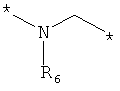 ;
; 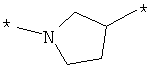 ;
; 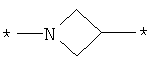 ;
;  ;
; 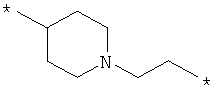 ;
;  ;
; 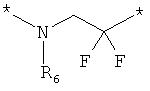 ;
;  ;
; 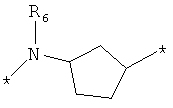 ;
;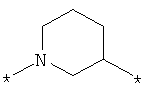 ;
; 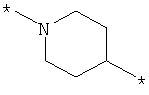 ;
; 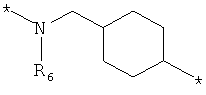 ;
; 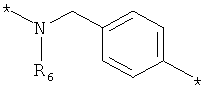 ;
; 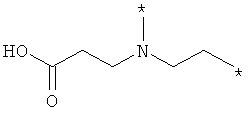 ;
; 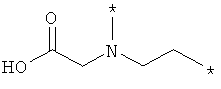 ;
; 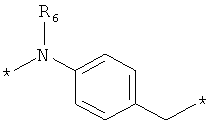 ;
; 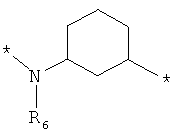 ;
; 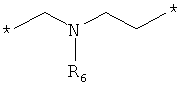 ;
; 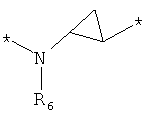 ;
; 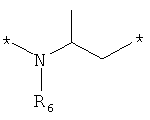 и
и 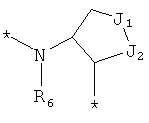 ,
,
где левая и правая звездочки Z обозначают места присоединения к С(R3)(R4)- и А в формуле А1 или А2, соответственно, R6 выбран из водорода и (C1-C6)алкила, a J1 и J2 представляют собой, независимо друг от друга, метилен или гетероатом, выбранный из серы, кислорода и NR5, где R5 выбран из водорода и (C1-C6)алкила, причем любой из алкиленов Z может быть дополнительно замещен одним-тремя радикалами, выбранными из галоида, гидроксигруппы, (C1-C6)алкила, или же R6 может быть присоединен к атому углерода Y, образуя 5-7-членный цикл;
R1 представляет собой (C6-C10)арил или (C2-C9)гетероарил, например (C3-C9)гетероарил, необязательно замещенный (C1-C6)алкилом, (C6-C10)арилом, (C6-C10)арил(C1-C4)алкилом, (C3-C9)гетероарилом, (C3-C9)гетероарил(C1-C4)алкилом, (C3-C8)циклоалкилом, (C3-C8)циклоалкил(C1-C4)алкилом, (C3-C8)гетероциклоалкилом или (C3-C8)гетероциклоалкил(C1-C4)алкилом, где любой арил, гетероарил, циклоалкил или гетероциклоалкил R1 могут быть замещены 1-5 группами, выбранными из галоида, (C1-C6)алкила, (C1-C6)алкоксигруппы и галоидзамещенных (C1-C6)алкила или (C1-C6)алкоксигруппы;
R2 представляет собой водород, (C1-C6)алкил, галоидзамещенный (C1-C6)алкил, (C2-C6)алкенил или (C2-C6)алкинил; и
каждый из R3 или R4 представляет собой, независимо от другого, водород, галоид, OH, (C1-C6)алкил, (C1-C6)алкоксигруппу или галоидзамещенные (C1-C6)алкил или (C1-C6)алкоксигруппу;
а также их N-оксидные производные или их пролекарства, или же фармакологически приемлемая соль, сольват или гидрат перечисленного.
Объектом настоящего изобретения также являются фармацевтические препараты для применения в лечении периферической невропатии, например ХВДП, включающие описываемое в настоящей заявке соединение и, необязательно, фармацевтически приемлемый разбавитель или носитель. В различных вариантах осуществления фармацевтические препараты содержат одно или несколько дополнительных терапевтических средств.
Объектом настоящего изобретения также является продукт, включающий описываемое в настоящей заявке соединение и терапевтическое средство, в качестве комбинированного препарата для одновременного, раздельного или последовательного применения в лечении периферической невропатии, например ХВДП.
Еще в одном варианте осуществления настоящего изобретения его объектом является фармацевтический препарат, включающий описываемое в настоящей заявке соединение и терапевтическое средство, каковое терапевтическое средство полезно при лечении периферической невропатии, например ХВДП.
Соединения по настоящему изобретению могут существовать в различных формах, таких как, например, кислоты в свободной форме, основания в свободной форме, сложные эфиры и другие пролекарства, соли и таутомеры, и настоящая заявка включает все варианты форм соединений.
Объем охраны патентных прав включает контрафактные и фальсифицированные продукты, содержащие соединение по настоящему изобретению или претендующие на его содержание, не зависимости от того, действительно ли они содержат подобное соединение, и вне зависимости от того, присутствует ли какое-либо из подобных соединений в терапевтически эффективном количестве.
В объем защиты патентных прав включены упаковки, содержащие описание или инструкции, указывающие на то, что упаковка содержит вещества или фармацевтический препарат по настоящему изобретению, а также продукт, который является подобным препаратом или веществами, или содержит их, или претендует на их содержание. Подобные упаковки могут быть, хотя и не обязательно, контрафактными или фальсифицированными.
Следует понимать, что свойства, объекты в целом, характеристики, вещества, химические остатки или группы, описываемые в связи с неким конкретным аспектом, вариантом осуществления или примером по настоящему изобретению, применимы и к любому другому аспекту, варианту осуществления или примеру, описываемому в контексте, если только они не являются несовместимыми с последними.
Краткое описание фигур
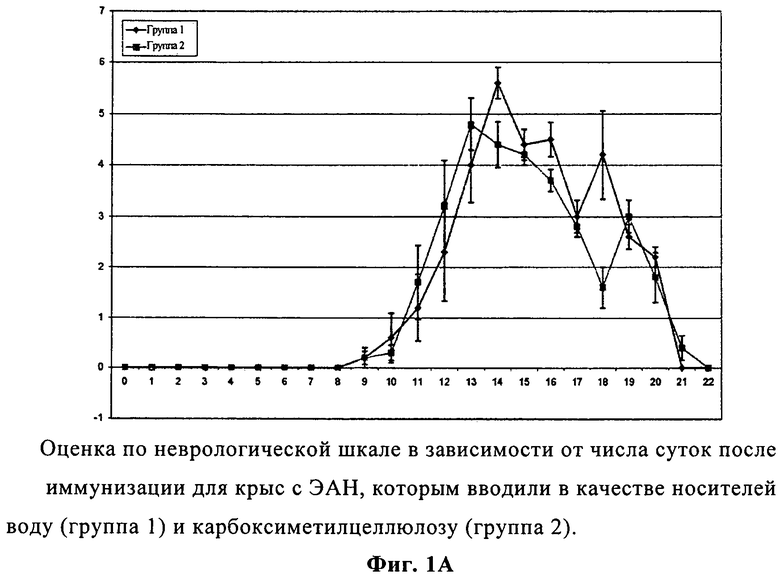
На фиг.1А представлены изменения оценки по неврологической шкале в зависимости от числа суток после иммунизации для крыс с экспериментальным аллергическим невритом, которым вводили вещества-основу воду и карбоксиметилцеллюлозу.
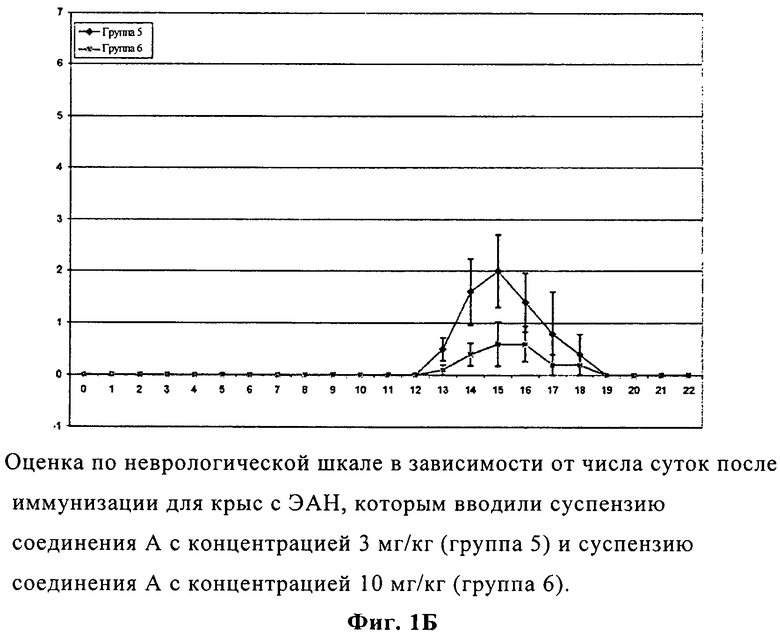
На фиг.1Б представлены изменения оценки по неврологической шкале в зависимости от числа суток после иммунизации для крыс с экспериментальным аллергическим невритом, которым вводили суспензии соединения А с концентрацией 3 и 10 мг/кг.
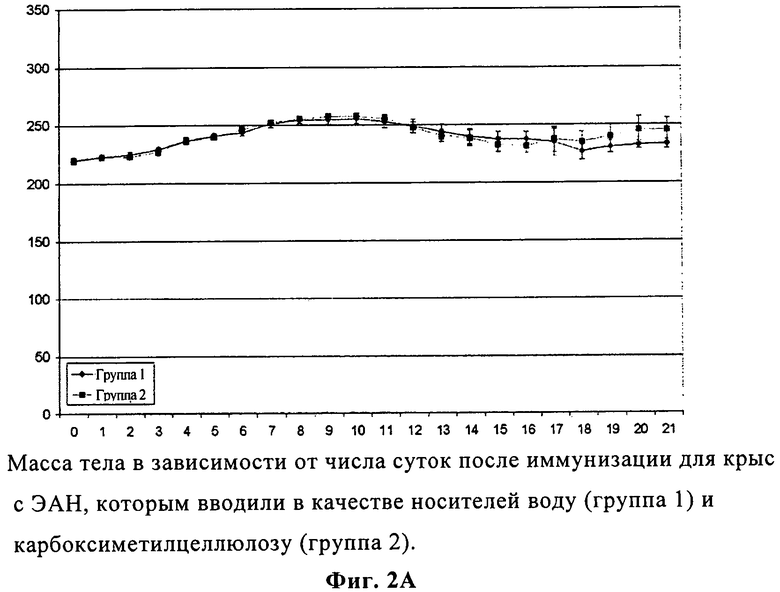
На фиг.2А представлены изменения массы тела в зависимости от числа суток после иммунизации для крыс с экспериментальным аллергическим невритом, которым вводили вещества-основу воду и карбоксиметилцеллюлозу.
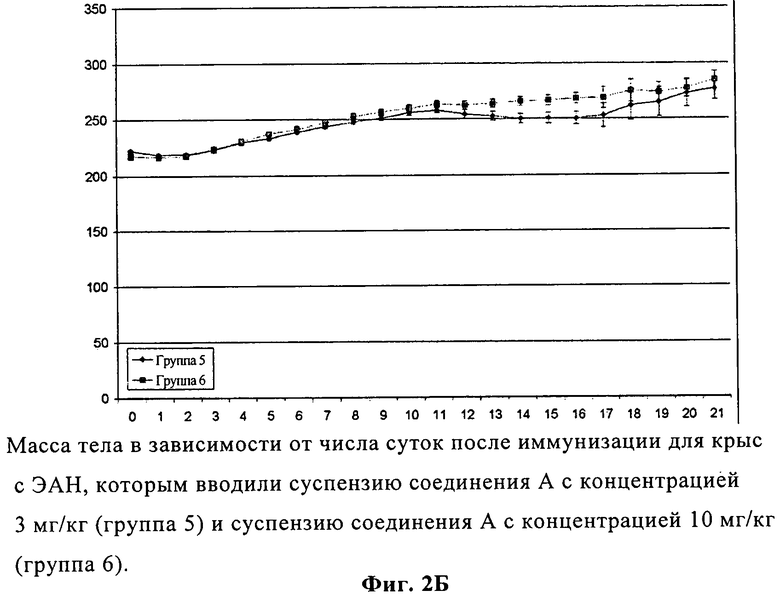
На фиг.2Б представлены изменения массы тела в зависимости от числа суток после иммунизации для крыс с экспериментальным аллергическим невритом, которым вводили суспензии соединения А с концентрацией 3 и 10 мг/кг.
Описание различных вариантов осуществления
Определения
В настоящем описании, если не дано других определений:
Алкил, алкенил, алкинил
«Алкил» в качестве группы и в качестве структурного элемента других групп, например галоидзамещенного алкила, алкоксигруппы, ацила, алкилтиогруппы, алкилсульфонильной группы и алкилсульфинильной группы, может быть как неразветвленным, так и разветвленным и, если не указано иное, может содержать от 1 до 6 атомов углерода. (C1-C6)алкильный остаток может содержать 1, 2, 3, 4, 5 или 6 атомов углерода. «Алкенил» в качестве группы и в качестве структурного элемента других групп содержит одну или несколько двойных углерод-углеродных связей и может быть как неразветвленным, так и разветвленным. Двойные связи могут находиться в цис- или транс-конфигурации. «Алкинил» в качестве группы и в качестве структурного элемента других групп и соединений содержит хотя бы одну тройную связь С≡С и может также содержать одну или несколько двойных связей С=С, а также может, если это возможно, быть как неразветвленным, так и разветвленным. Если не указано иное, алкенильный и алкинильный остатки могут содержать 2, 3, 4, 5 или 6 атомов углерода. Любая циклоалкильная группа, сама по себе или в качестве структурного элемента других групп, может содержать 3, 4, 5, 6, 7 или 8 атомов углерода, предпочтительно 3, 4, 5 или 6 атомов углерода. «Алкилен» и «алкенилен» представляют собой двухвалентные радикалы, производные от «алкильной» и «алкенильной» групп, соответственно. В настоящей заявке всякая алкильная группа из R1 необязательно является алкильной группой со вставкой, в которой имеет место замена метилена на один из остатков, выбранных из -S-, -S(O)-, -S(O)2-, -NR20- и -О- (где R20 является водородом или (C1-C6)алкилом).
Данные группы включают, например, -CH2-О-CH2-, -CH2-S(O)2-CH2-, -(CH2)2-NR20-CH2, -CH2-O-(СН2)2-. В одном из вариантов осуществления настоящего изобретения подобные алкильные группы со вставкой отсутствуют. Сюда включен класс соединений, в которых все алкильные группы не имеют вставок.
Арил
Термин «арил» означает моноциклическую или конденсированную бициклическую ароматическую систему колец, содержащую от шести до десяти атомов углерода. Например, (C6-C10)арил может представлять собой фенил, бифенил или нафтил, предпочтительно фенил. Конденсированное бициклическое кольцо может быть частично насыщенным, примером служит 1,2,3,4-тетрагидронафталин и ему подобные. Термин «арилен» означает двухвалентный радикал, производный от арильной группы. Например, арилен, как он понимается в настоящей заявке, может представлять собой фениле, бифенилен, нафтилен и им подобные.
Галоид
Термин «галоид» или «галоген» означает фтор, хлор, бром или иод, предпочтительно фтор или хлор. Галоидзамещенные алкильные группы и соединения могут быть частично галогенированы или пергалогенированы, причем в случае многократного галогенирования галоидные заместители могут быть как одинаковыми, так и различными. Предпочтительной пергалогенированной алкильной группой является, например, трифторметил или трифторметоксигруппа.
Гетероарил
Термин «гетероарил» означает арил, как он определен в настоящей заявке, с включением в кольцевую структуру хотя бы одного гетероатомного остатка, выбранного из азота, кислорода или серы, где каждый цикл включает от 5 до 6 кольцевых атомов, если не указано иное. Например, C2-гетероарил включает оксадиазол, триазол и им подобные. C9-гетероарил включает хинолин, 1,2,3,4-тетрагидрохинолин и им подобные. (C2-C9)гетероарил, как он понимается в настоящей заявке, включает тиенил, пиридинил, фуранил, изоксазолил, бензоксазолил или бензо[1,3]диоксолил, предпочтительно тиенил, фуранил или пиридинил. Термин «гетероарилен» означает гетероарил, как он определен в настоящей заявке, при том условии, что кольцевая система включает двухвалентный радикал. Конденсированная бициклическая гетероарильная кольцевая система может быть частично насыщенной, примерами являются 2,3-дигидро-1Н-изоиндол, 1,2,3,4-тетрагидрохинолин и им подобные.
Замещенный
Если не указано иное, термин «замещенный», как он употребляется в контексте в отношении остатка, означает, что один или более, особенно до 5, в особенности 1, 2 или 3, из атомов водорода в таковом остатке независимо друг от друга заменены соответствующим числом описанных заместителей. Термин «необязательно замещенный», как он употребляется в контексте, означает замещенный или незамещенный.
Следует, безусловно, понимать, что заместители находятся лишь в тех положениях, где это возможно химически, причем специалист в соответствующей области способен определить (экспериментально или теоретически) без излишних усилий, возможно ли конкретное замещение. Например, амино- или гидроксигруппы со свободными атомами водорода могут быть неустойчивы при связывании с атомами углерода ненасыщенными (т.е. олефиновыми) связями.
Фармацевтически приемлемый
Термин «фармацевтически приемлемый», как он употребляется в контексте, указывает на такие соединения, вещества, композиции и/или формы дозировки, которые, в пределах разумного медицинского назначения, подходят для применения в контакте с тканями людей или животных без излишней токсичности, раздражения, аллергической реакции или иных проблем или осложнений и сообразны с разумным соотношением между пользой и риском. Термин включает приемлемость как для человека, так и для ветеринарных целей.
Независимо
Когда два или более остатка описаны как «независимо друг от друга» выбранные из списка атомов или групп, это означает, что остатки могут быть одинаковыми или различными. Сущность каждого остатка является, таким образом, независимой от сущности одного или более других остатков.
Соединения
Объектом настоящего изобретения являются соединения, описанные в заявках на изобретение WO 04/103306 и US 2005/0014728, WO 05/000833, WO 05/103309 или WO 05/113330, например соединения формулы А1 или А2
,
в которых
А представляет собой COOR5, OPO(OR5)2, PO(OR5)2, SO2OR5, POR5OR5 или 1H-тетразол-5-ил, где R5 представляет собой водород или образующую сложный эфир группу, например (C1-C6)алкил;
W представляет собой связь, (C1-C3)алкилен или (C2-C3)алкенилен;
Y представляет собой (C6-C10)арил или (C2-C9)гетероарил, например (C3-C9)гетероарил, необязательно замещенный 1-3 радикалами, выбранными из галоида, OH, NO2, (C1-C6)алкила, (C1-C6)алкоксигруппы, галоидзамещенного (C1-C6)алкила и галоидзамещенной (C1-C6)алкоксигруппы;
Z выбран из
; ; ; ; ; ; ; ; ; ; ; ;; ; ; ; ; ; ; ; ; ; и ,
где левая и правая звездочки Z обозначают места присоединения к С(R3)(R4)- и А в формуле А1 или А2, соответственно, R6 выбран из водорода и (C1-C6)алкила, a J1 и J2 представляют собой, независимо друг от друга, метилен или гетероатом, выбранный из серы, кислорода и NR5, где R5 выбран из водорода и (C1-C6)алкила, причем любой из алкиленов Z может быть дополнительно замещен одним-тремя радикалами, выбранными из галоида, гидроксигруппы, (C1-C6)алкила, или же R6 может быть присоединен к атому углерода Y, образуя 5-7-членный цикл;
R1 представляет собой (C6-C10)арил или (C2-C9)гетероарил, например (C3-C9)гетероарил, необязательно замещенный (C1-C6)алкилом, (C6-C10)арилом, (C6-C10)арил(C1-C4)алкилом, (C3-C9)гетероарилом, (C3-C9)гетероарил(C1-C4)алкилом, (C3-C8)циклоалкилом, (C3-C8)циклоалкил(C1-C4)алкилом, (C3-C8)гетероциклоалкилом или (C3-C8)гетероциклоалкил(C1-C4)алкилом, где любой арил, гетероарил, циклоалкил или гетероциклоалкил R1 могут быть замещены 1-5 группами, выбранными из галоида, (C1-C6)алкила, (C1-C6)алкоксигруппы и галоидзамещенных (C1-C6)алкила или (C1-C6)алкоксигруппы;
R2 представляет собой водород, (C1-C6)алкил, галоидзамещенный (C1-C6)алкил, (C2-C6)алкенил или (C2-C6)алкинил; и
каждый из R3 или R4 представляет собой, независимо от другого, водород,
галоид, OH, (C1-C6)алкил, (C1-C6)алкоксигруппу или галоидзамещенные
(C1-C6)алкил или (C1-C6)алкоксигруппу;
а также их N-оксидные производные или их пролекарства, или же их фармакологически приемлемая соль, сольват или гидрат.
Ниже описаны дальнейшие варианты осуществления настоящего изобретения. Следует понимать, что характеристики, указанные в каждом из вариантов осуществления, могут быть скомбинированы с другими указанными характеристиками, что составит новые варианты осуществления.
А представляет собой COOR5, OPO(OR5)2, PO(OR5)2, SO2OR5, POR5OR5 или 1H-тетразол-5-ил, где R5 представляет собой водород или образующую сложный эфир группу, например (C1-C6))алкил. В частности, А может являться COOR5, например COOH.
W представляет собой связь, C1-, C2- или C3-алкилен или (C2-C3)алкенилен. В некоторых вариантах осуществления настоящего изобретения W является этиленом.
Y представляет собой (C6-C10)арил или (C2-C9)гетероарил, например (C3-C9)гетероарил, необязательно замещенный 1-3 радикалами, выбранными из галоида, OH, NO2, алкила, алкоксигруппы, галоидзамещенного алкила и галоидзамещенной алкоксигруппы, где алкил, как группа или как часть алкоксигруппы, являясь или не являясь галоидзамещенным, содержит 1, 2, 3, 4, 5 или 6 атомов углерода. Y, в частности, может быть фенилом или C6-гетероарилом, в каждом из случаев необязательно замещенным, как это описано выше. Таковая арильная, например фенильная, или гетероарильная группа может нести один заместитель, например один подобный алкильный заместитель. Примером алкильного заместителя является этил. Галоид, в частности, может являться фтором или хлором.
Z представляет собой остаток, как он описан выше, в частности гетероциклическую группу, как указано в международной заявке на изобретение WO 2004/103306, например азетидин, в частности азетидин, присоединенный к остальной части молекулы по положениям 1 и 3. Также следует упомянуть пирролидин и пиперидин, в каждом из случаев присоединенные к остальной части молекулы по положениям 1 и 3, а также пиперидин, дизамещенный по положениям 1 и 4 соответствующими остатками, образующими остальную часть молекулы. В некоторых соединениях данные гетероциклы являются N-замещенными (замещенными по положению 1) остатком А и замещенными по положению 3 или, в соответствующем случае, 4 группой CR3R4.
R1 представляет собой (C6-C10)арил или (C2-C9)гетероарил, например (C3-C9)гетероарил, необязательно замещенный (C1-C6)алкилом, (C6-C10)арилом, (C6-C10)арил(C1-C4)алкилом, (C3-C9)гетероарилом, (C3-C9)гетероарил(C1-C4)алкилом, (C3-C8)циклоалкилом, (C3-C8)циклоалкил(C1-C4)алкилом, (C3-C8)гетероциклоалкилом или (C3-C8)гетероциклоалкил(C1-C4)алкилом, где любой арил, гетероарил, циклоалкил или гетероциклоалкил R1 могут быть замещены 1-5 группами, выбранными из галоида, (C1-C6)алкила, (C1-C6)алкоксигруппы и галоидзамещенных (C1-C6)алкила или (C1-C6)алкоксигруппы. В частности, R1 может являться фенилом или C6-гетероарилом, необязательно замещенным, как это описано выше. В некоторых вариантах осуществления настоящего изобретения R1 несет два заместителя, выбранных из необязательно галоидзамещенного алкила, содержащего 1, 2, 3, 4, 5 или 6 атомов углерода (например, трифторметила), необязательно галоидзамещенного фенила и необязательно галоидзамещенного (C3-C8)циклоалкила (например, циклогексила). Например, R1 может нести одну необязательно галоидзамещенную алкильную группу и один необязательно галоидзамещенный циклический остаток, выбранный из фенильной и (C3-C8 (например, C6)) циклоалкильной групп. В некоторых соединениях R1 является фенилом или C6-гетероарилом, в частности фенилом, замещенным по положениям 3 и 4, как это описано выше, как, например, в случае 3-трифторметил-4-циклогексилфенила.
R2 представляет собой водород, (C1-C6)алкил, галоидзамещенный (C1-C6)алкил, (C2-C6)алкенил или (C2-C6)алкинил. Алкил, является он галоидзамещенным или нет, может содержать, соответственно, 1, 2, 3, 4, 5 или 6 атомов углерода. В частности, R2 может являться метилом.
Каждый из R3 или R4 представляет собой, независимо от другого, водород, галоид, OH, (C1-C6)алкил, (C1-C6)алкоксигруппу или галоидзамещенные (C1-C6)алкил или (C1-C6)алкоксигруппу. Алкил, является он галоидзамещенным или нет и/или является он частью алкоксигруппы или нет, может содержать, соответственно, 1, 2, 3, 4, 5 или 6 атомов углерода. В качестве примера, R3 и R4 могут независимо друг от друга являться водородом, галоидом, метилом или галоидзамещенным метилом. В частности, R3 и R4 могут оба являться водородом.
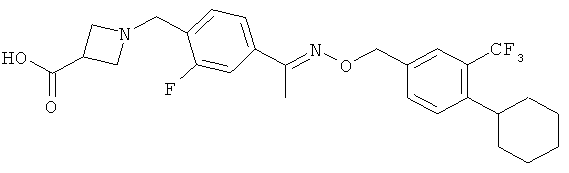
Предпочтительным соединением, полезным для целей настоящего изобретения, является 1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновая кислота:
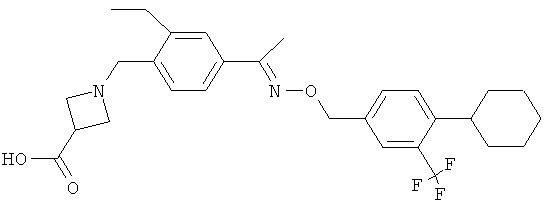 .
.
Другие соединения, полезные для целей настоящего изобретения, включают:
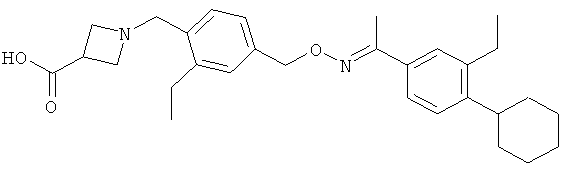 ;
;
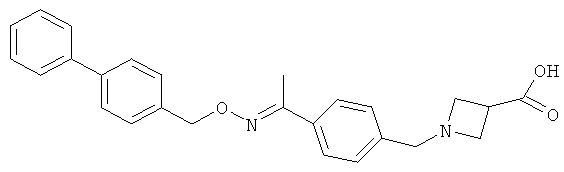 ;
;
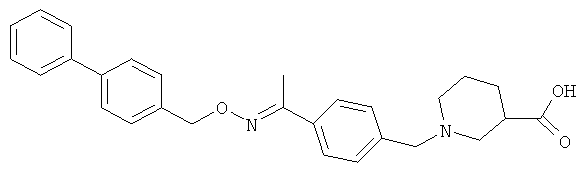 ;
;
 ;
;
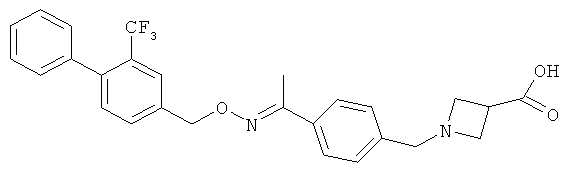 ;
;
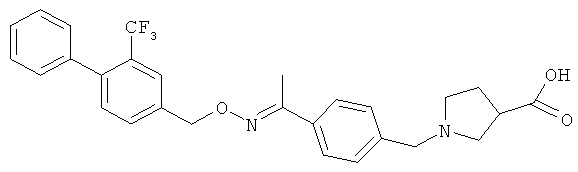 ;
;
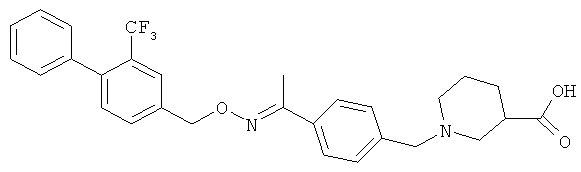 ;
;
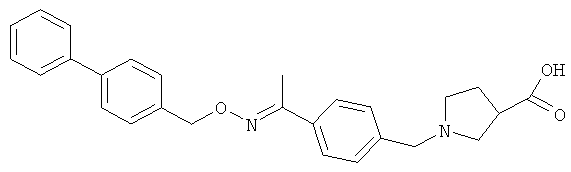 ;
;
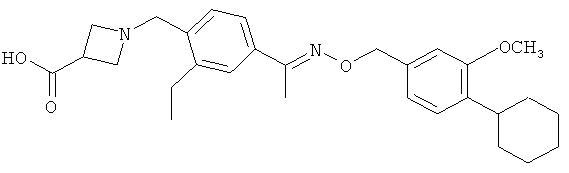 ;
;
 ;
;
 ;
;
 ;
;
 ;
;
 ;
;
 и
и
 .
.
Также следует упомянуть другие соединения из примеров и таблицы 1 международной заявки на изобретение WO 2004/103306.
Соединения по настоящему изобретению могут находиться в форме фармацевтически приемлемых солей. Фармацевтически приемлемые соли по настоящему изобретению могут быть синтезированы из исходного соединения, содержащего основный или кислотный остаток, с помощью общепринятых химических способов. Как правило, подобные соли могут быть получены посредством взаимодействия данных соединений в форме свободной кислоты или основания со стехиометрическим количеством подходящего основания или кислоты в воде или в органическом растворителе, или же в смеси двух. Как правило, предпочтительной является неводная среда, такая как диэтиловый эфир, этилацетат, этанол, изопропанол или ацетонитрил. Списки подходящих солей можно найти в справочнике Remington′s Pharmaceutical Sciences, 17-e издание, Mack Publishing Company, Easton, Pa., US, 1985, стр.1418. Соответствующее описание включено в настоящее описание в качестве ссылки во всей его полноте; см. также, "Handbook of Pharmaceutical Salts Properties Selection and Use" (Справочник по свойствам, выбору и применению фармацевтических солей) под ред. Stahl и др., Verlag Helvetica Chimica Acta и Wiley-VCH, 2002.
Настоящее изобретение включает, таким образом, фармацевтически приемлемые соли описываемых соединений, в которых исходное соединение модифицировано посредством получения его кислотных или основных солей, например, общепринятых нетоксичных солей или четвертичных аммониевых солей, образующихся, например, из неорганических или органических кислот или оснований. Примеры подобных кислотно-аддитивных солей включают ацетат, адипат, альгинат, аспартат, бензоат, бензулсульфонат, бисульфат, бутират, цитрат, камфорат, камфорсульфонат, циклопентанпропионат, диглюконат, додецилсульфат, этансульфонат, фумарат, глюкогептаноат, глицерофосфат, гемисульфат, гептаноат, гексаноат, гидрохлорид, гидробромид, гидроиодид, 2-гидроксиэтансульфонат, лактат, малеат, метансульфонат, 2-нафталинсульфонат, никотинат, оксалат, памоат, пектинат, персульфат, 3-фенилпропионат, пикрат, пивалат, пропионат, сукцинат, тартрат, тиоцианат, тозилат и ундеканоат. Соли с основаниями включают аммониевые соли, соли щелочных металлов, такие как соли натрия и калия, соли щелочноземельных металлов, такие как соли кальция и магния, соли с органическими основаниями, такие как дициклогексиламиновые соли, N-метил-D-глюкамин, а также соли с аминокислотами, такими как аргинин, лизин и т.п. Кроме того, основные азотсодержащие группы могут быть преобразованы в четвертичную форму с помощью таких веществ, как (низш.) алкилгалогениды, такие как метил-, этил-, пропил- и бутилхлориды, бромиды и иодиды, диалкилсульфаты, такие как диметил, диэтил-, дибутил- и диамилсульфаты, галогениды с длинной цепью, такие как децил-, лаурил-, миристил- и стеарилхлориды, бромиды и иодиды, арилалкилгалогениды, такие как бензил- и фенилэтилбромиды, и других.
Настоящее изобретение включает пролекарства для активных фармацевтических веществ по настоящему изобретению, в которых, например, одна или несколько функциональных групп защищены или модифицированы, но могут быть преобразованы in vivo в функциональную группу, как в случае сложных эфиров карбоновых кислот, преобразуемых in vivo в свободную кислоту, или в случае защищенных аминов, преобразуемых в свободную аминогруппу. Термин «пролекарство», как он употребляется в контексте, означает, в частности, такие соединения, которые быстро преобразуются in vivo в исходное соединение, например, путем гидролиза в крови. Подробное обсуждение приведено в Т.Higuchi и V.Stella, Pro-drugs as Novel Delivery Systems (Пролекарства как новые системы доставки) в т.14 A.C.S. Symposium Series, Bioreversible Carriers in Drug Design (Биообратимые носители в дизайне лекарств), под ред. Edward В. Roche, American Pharmaceutical Association и Pergamon Press, 1987; Design of Prodrugs (Дизайн пролекарств), под ред. Н Bundgaard, Elsevier, 1985; и Judkins и др. Synthetic Communications, 26(23), 4351-4367 (1996), каждый из документов включен в настоящее описание в качестве ссылки во всей своей полноте.
Пролекарства, таким образом, включают лекарства, содержащие функциональную группу, которая была преобразована в свое обратимо образующееся производное. Как правило, подобные пролекарства преобразуются в активное лекарственное вещество путем гидролиза. В качестве примеров можно перечислить следующие:
Пролекарства также включают соединения, преобразуемые в активное лекарственное вещество посредством окислительной или восстановительной реакции. В качестве примеров могут быть упомянуты:
Окислительная активация
- N- и О-деалкилирование
- окислительное дезаминирование
- N-окисление
- эпоксидирование Восстановительная активация
- восстановление азогруппы
- восстановление сульфоксидной группы
- восстановление дисульфидной группы
- биовосстановительное алкилирование
- восстановление нитрогруппы
Также в качестве метаболической активации пролекарств следует упомянуть нуклеотидную активацию, активацию фосфорилированием и активацию декарбоксилированием. Дополнительную информацию можно найти в "The Organic Chemistry of Drug Design and Drug Action" (Органическая химия дизайна и действия лекарств), R В Silverman (в частности, глава 8, стр.497-546), монография включена в настоящее описание в качестве ссылки во всей своей полноте.
Применение защитных групп полностью описано в справочниках ′Protective Groups in Organic Chemistry′ (Защитные группы в органической химии), под ред. J W F McOmie, Plenum Press (1973), и ′Protective Groups in Organic Synthesis′ (Защитные группы в органическом синтезе), 2-е издание, Т W Greene & Р G М Wutz, Wiley-Interscience (1991).
Таким образом, специалистам в соответствующей области будет понятно, что хотя защищенные производные соединений по настоящему изобретению могут не обладать фармакологической активностью в таковой форме, они могут быть введены, например, парэнтерально или перорально, после чего будут метаболизированы в организме, преобразуясь в соединения по настоящему изобретению, являющиеся фармакологически активными. Подобные производные являются, таким образом, примерами «пролекарств». Все пролекарства описываемых соединений включены в объем настоящего изобретения.
Некоторые упоминаемые в контексте группы (в особенности те, которые содержат гетероатомы и сопряженные связи) могут существовать в таутомерных формах, и все таковые таутомеры включены в объем настоящего изобретения. В более общем случае, многие соединения могут находиться в состоянии равновесия, например, в случае органических кислот и соответствующих их анионов. Ссылка в контексте на соединения включает, соответственно, ссылки на все их равновесные формы.
Соединения по настоящему изобретению могут также содержать один или несколько асимметричных атомов углерода и могут, таким образом, характеризоваться оптической изомерией и/или диастереоизомерией. Все диастереоизомеры могут быть разделены с помощью общепринятых методик, например хроматографии или фракционной кристаллизации. Различные стереоизомеры могут быть выделены посредством разделения рацемической или иной смеси соединений с помощью общепринятых методик, например фракционной кристаллизации или высокоэффективной жидкостной хроматографии (ВЭЖХ). Альтернативно, искомые оптические изомеры могут быть получены посредством взаимодействия соответствующих оптически активных исходных веществ в таких условиях, которые не приведут к рацемизации или эпимеризации, или же посредством, например, образования производного с гомохиральной кислотой с последующим разделением диастереомерных производных общепринятыми способами (например, ВЭЖХ, хроматография на силикагеле). Все стереоизомеры включены в объем настоящего изобретения. В тех случаях, когда описывается некий один энантиомер или диастереомер, в объем изобретения также входят прочие энантиомеры и диастереомеры, а также рацематы. В этом отношении делается особая ссылка на перечисляемые в контексте соединения.
У соединений по настоящему изобретению также могут наличествовать геометрические изомеры. Настоящее изобретение включает в свой объем различные геометрические изомеры и их смеси, являющиеся результатом различного расположения заместителей около двойной углерод-углеродной связи, и обозначает эти изомеры как Z- или Е-конфигурации, где термин «Z» относится к заместителям на одной и той же стороне относительно двойной углерод-углеродной связи, а термин «Е» относится к заместителям на противоположных сторонах относительно двойной углерод-углеродной связи.
Таким образом, настоящее изобретение включает все варианты определенных в заявке соединений, например любой таутомер или любую фармацевтически приемлемую соль, сложный эфир, кислоту или другой вариант определенных в заявке соединений и их таутомеров, равно как и тех соединений, которые способны после введения прямо или косвенно приводить к образованию соединение, как оно определено выше, или же к образованию веществ, способных существовать в равновесии с таковым соединением.
Синтез
Соединения могут быть синтезированы так, как это описано в упомянутых выше патентных спецификациях, например в заявках на изобретение WO 04/103306 и US 2005/0014728.
Введение и фармацевтические препараты
Соединения по настоящему изобретению могут, как правило, вводиться перорально, внутривенно, подкожно, трансбуккально, перректально, трансдермально, интраназально, интратрахеально, интрабронхиально, любым другим парэнтеральным путем, в виде орального или назального спрея или посредством ингаляции. Соединения могут вводиться в форме фармацевтических препаратов, содержащих пролекарство или активное соединение как в виде свободного соединения, так и, например, в виде фармацевтически приемлемой нетоксичной аддитивной соли с органической или неорганической кислотой или основанием, в фармацевтически приемлемой форме дозировки. Соединения могут вводиться в варьируемых дозах в зависимости от подвергаемых лечению пациента и расстройства, а также пути введения.
Таким образом, как правило, фармацевтические композиции по настоящему изобретению могут вводиться пациенту перорально или парэнтерально (термин «парэнтерально», как он употребляется в контексте, относится к режимам введения, включающим внутривенные, внутримышечные, внутрибрюшинные, внутригрудинные, подкожные и внутрисуставные инъекции и вливания). В случае более крупных животных, таких как люди, соединения могут вводиться отдельно в качестве альтернативы введению в виде композиций в сочетании с фармацевтически приемлемыми разбавителями, наполнителями или носителями.
Действующие уровни дозировки активных ингредиентов в фармацевтических композициях по настоящему изобретению могут варьироваться таким образом, чтобы обеспечить эффективное количество активного соединения (соединений) с точки зрения достижения желаемого терапевтического отклика для конкретных пациента, композиций и режима введения. Выбранный уровень дозировки будет зависеть от активности конкретного соединения, пути введения, тяжести подвергаемого лечению состояния и состояния и предшествующего анамнеза подвергаемого лечению пациента. Однако в соответствующей области допустимо начинать дозировку соединения с более низких уровней, чем требующиеся для достижения желаемого терапевтического эффекта, и последовательно повышать дозировку пока желаемый эффект не будет достигнут.
При лечении, профилактике, контроле, снижении интенсивности или облегчении симптома периферической невропатии подходящий уровень дозировки будет, как правило, составлять приблизительно от 0,01 до 500 мг на килограмм массы тела пациента в сутки, количество может вводиться одной или несколькими дозами. Уровень дозировки может составлять от приблизительно 0,1 до приблизительно 250 мг/кг в сутки, например от приблизительно 0,5 до приблизительно 100 мг/кг в сутки. Подходящий уровень дозировки может составлять приблизительно от 0,01 до 250 мг/кг в сутки, приблизительно от 0,05 до 100 мг/кг в сутки или приблизительно от 0,1 до 50 мг/кг в сутки. В данном диапазоне дозировка может составлять от 0,05 до 0,5, от 0,5 до 5 или от 5 до 50 мг/кг в сутки. Для перорального введения могут употребляться композиции в форме таблеток, содержащих от 1,0 до 1000 миллиграмм активного ингредиента, в частности 1,0, 5,0, 10,0, 15,0, 20,0, 25,0, 50,0, 75,0, 100,0, 150,0, 200,0, 250,0, 300,0, 400,0, 500,0, 600,0, 750,0, 800,0, 900,0 или 1000,0 миллиграмм активного ингредиента. Соединения могут вводиться в режиме от 1 до 4 раз в сутки, предпочтительно один или два раза в сутки. Режим дозировки может быть подобран таким образом, чтобы обеспечить оптимальный терапевтический отклик.
Согласно еще одному варианту осуществления настоящего изобретения его объектом является фармацевтическая композиция, содержащая соединение по настоящему изобретению в смеси с фармацевтически приемлемым адъювантом, разбавителем или носителем.
Фармацевтические композиции по настоящему изобретению для парэнтеральной инъекции содержат, соответствующим образом, фармацевтически приемлемые водные или неводные растворы, дисперсии, суспензии или эмульсии, а равно и стерильные порошки для воссоздания стерильных инъецируемых растворов или дисперсий непосредственно перед применением. Примеры подходящих водных и неводных носителей, разбавителей, растворителей или основ включают воду, этанол, полиолы (такие как глицерин, пропиленгликоль, полиэтиленгликоль и им подобные) и их смеси, растительные масла (такие как оливковое масло) и подходящие для инъекций органические сложные эфиры, такие как этилолеат. Должная текучесть может поддерживаться посредством, например, использования веществ, образующих покрытия, таких как лецитин, обеспечения требуемого размера частиц в случае дисперсий и применения поверхностно активных веществ.
Данные композиции могут также содержать адъюванты, такие как консерванты, смачивающие вещества, эмульгаторы и диспергирующие средства. Предотвращение действия микроорганизмов может быть обеспечено посредством включения различных антибактериальных и противогрибковых средств, например парабена, хлорбутанола или фенолсорбиновой кислоты. Может также оказаться желательным включение изотонических веществ, таких как, например, сахар или хлорид натрия. Пролонгированное всасывание инъецируемой фармацевтической формы может быть достигнуто посредством введения замедляющих всасывание веществ (например, моностеарата алюминия и желатина).
В некоторых случаях для пролонгации эффекта лекарственного средства оказывается желательно замедлить всасывание лекарства из подкожной или внутримышечной инъекции. Это может быть обеспечено посредством использования жидкой суспензии кристаллического или аморфного вещества с плохой водорастворимостью. Скорость всасывания лекарственного вещества зависит в этом случае от скорости его растворения, которая, в свою очередь, зависит от размера кристалла и кристаллической формы. Альтернативно, замедленное всасывание вводимой парэнтерально лекарственной формы достигается посредством растворения или суспендирования лекарственного вещества в масляной основе.
Инъецируемые депо-формы соответствующим образом изготавливают посредством образования микроинкапсулированных матриц лекарственного вещества в рассасывающихся полимерах, например полилактид-полигликолиде. В зависимости от соотношения между лекарственным веществом и полимером и от природы конкретного задействованного полимера, скорость высвобождения лекарственного вещества можно контролировать. Примеры других рассасывающихся полимеров включают полиортоэфиры и полиангидриды. Инъецируемые депо-препарты могут также быть изготовлены посредством захвата лекарственного вещества лизосомами или микроэмульсиями, совместимыми с тканями организма. Инъецируемые препараты могут быть стерилизованы посредством, например, фильтрования через удерживающий бактерии фильтр или посредством введения стерилизующих веществ в форме стерильных твердых композиций, которые могут быть растворены или диспергированы в стерильной воде или иной пригодной для инъекций стерильной среде непосредственно перед применением.
Твердые формы дозировки для перорального введения включают капсулы, таблетки, пилюли, порошки и гранулы. В подобных твердых формах дозировки активное соединение, как правило, смешано с хотя бы одним инертным фармацевтически приемлемым наполнителем или носителем, таким как цитрат натрия или дигидрат ортофосфата кальция, и/или одним или несколькими из следующих веществ: а) заполнителями или наполнителями, такими как крахмалы, лактоза, сахароза, глюкоза, манит и кремниевая кислота, б) связующими веществами, такими как карбоксиметилцеллюлоза, альгинаты, желатин, поливинилпирролидон, сахароза и камедь акации, в) увлажняющими веществами, такими как глицерин, г) веществами, способствующими распадению, такими как агар-агар, карбонат кальция, картофельный крахмал или крахмал тапиоки, альгиновая кислота, некоторые силикаты и карбонат натрия, д) замедлителями загустевания раствора, такими как парафин, е) ускорителями всасывания, такими как четвертичные аммониевые соединения, ж) смачивающими веществами, такими как цетиловый спирт и моностеарат глицерина, з) абсорбентами, такими как каолин и бентонитовая глина, и и) скользящими веществами, такими как тальк, стеарат кальция, стеарат магния, твердые полиэтиленгликоли, лаурилсульфат натрия и их смеси. В случае капсул, таблеток и пилюль форма дозировки может также включать буферные вещества. Твердые композиции аналогичного типа могут быть также задействованы в качестве заполнителей в мягких и твердых желатиновых капсулах при использовании таких наполнителей, как лактоза или молочные сахара, равно как и, например, полиэтиленгликоль с высокой молекулярной массой.
Препараты для перорального приема содержат, соответствующим образом, вспомогательное вещество, облегчающее растворение. Ограничений на сущность облегчающего растворение вспомогательного вещества не накладывается при условии его фармацевтической приемлемости. Примеры включают неионные поверхностно-активные вещества, такие как сложные эфиры сахарозы и жирных кислот, сложные эфиры глицерина и жирных кислот, сложные эфиры сорбита и жирных кислот (например, триолеат сорбита), полиэтиленгликоль, полиоксиэтилен-гидрогенизированное касторовое масло, сложные эфиры полиоксиэтиленсорбита и жирных кислот, простые алкиловые эфиры полиоксиэтилена, простые алкиловые эфиры метоксиполиоксиэтилена, простые алкилфениловые эфиры полиоксиэтилена, сложные эфиры полиэтиленгликоля и жирных кислот, полиоксиэтиленалкиламины, алкилтиоэфиры полиоксиэтилена, сополимеры полиоксиэтилена и полиоксипропилена, сложные эфиры полиоксиэтиленглицерина и жирных кислот, сложные эфиры пентаэритрита и жирных кислот, сложные моноэфиры пропиленгликоля и жирных кислот, сложные эфиры полиоксиэтиленсорбита и жирных кислот, алкилоламиды жирных кислот и алкиламиноксиды; желчную кислоту и ее соли (например, хенодезоксихолевую кислоту, холевую кислоту, дезоксихолевую кислоту, дегидрохолевую кислоту и их соли, а также их глициновые или тауриновые конъюгаты); ионные поверхностно-активные вещества, такие как лаурилсульфат натрия, детергенты на основе жирных кислот, алкилсульфонаты, алкилфосфаты, эфирофосфаты, соли жирных кислот или основных аминокислот; детергенты на основе триэтаноламина и четвертичные алкиламмониевые соли; а также амфотерные поверхностно-активные вещества, такие как бетаины и соли аминокарбоновых кислот.
Твердые формы дозировки в виде таблеток, драже, капсул, пилюль и гранул могут быть получены с покрытиями и оболочками, такими как кишечнорастворимые покрытия и другие покрытия, хорошо известные в области изготовления фармацевтических препаратов. Они могут необязательно содержать контрастные вещества и могут также иметь такой состав, что они будут высвобождать активный ингредиент (ингредиенты) исключительно или преимущественно в определенной части желудочно-кишечного тракта и/или замедленным образом. Примеры матричных составов включают полимерные вещества и воски.
Активные соединения могут также находиться, если то будет уместно, в микроинкапсулированной форме с одним или более из вышеупомянутых наполнителей.
Активное соединение может находиться в тонкоизмельченной форме, например может быть микронизировано.
Жидкие формы дозировки для перорального введения включают фармацевтически приемлемые эмульсии, растворы, суспензии, сиропы и эликсиры. Помимо активных соединений жидкие формы дозировки могут содержать инертные разбавители, обычно используемые в соответствующей области, такие как вода или другие растворители, солюбилизаторы и эмульгаторы, такие как этиловый спирт, изопропиловый спирт, этилкарбонат, этилацетат, бензиловый спирт, бензилбензоат, пропиленгликоль, 1,3-бутиленгликоль, диметилформамид, масла (в частности, хлопковое, арахисовое, масло ростков пшеницы, оливковое, касторовое и кунжутное масло), глицерин, тетрагидрофуриловый спирт, полиэтиленгликоли и сложные эфиры сорбита и жирных кислот, а также их смеси. Помимо инертных разбавителей, композиции для перорального применения могут также содержать адъюванты, такие как смачивающие вещества, эмульгаторы и суспендирующие средства, подсластители, вкусовые ароматизаторы и отдушки. Суспензии могут содержать, помимо активных соединений, суспендирующие вещества, такие как этоксилированные изостеариловые спирты, сложные эфиры полиоксиэтиленсорбита и сорбитана, микрокристаллическая целлюлоза, метагидроксид алюминия, бентонит, агар-агар и смолу трагакант, а также их смеси.
Композиции для перректального и вагинального применения предпочтительно представляют собой суппозитории, которые могут быть получены посредством смешения соединений по настоящему изобретению с подходящими не раздражающими наполнителями или носителями, такими как масло какао, полиэтиленгликоль или воск для суппозиториев, которые находятся в твердом виде при комнатной температуре, но в жидком при температуре тела и, вследствие этого, плавятся в заднем проходе или влагалище и высвобождают активное соединение.
Соединения по настоящему изобретению могут также вводиться в форме липосом. Как известно в соответствующей области, липосомы являются, как правило, производными от фосфолипидов или других липидных веществ. Липосомы формируются моно- или мультиламеллярными гидратированными жидкими кристаллами, диспергированными в водной среде. Может быть задействован любой нетоксичный, физиологически приемлемый и метаболизируемый липид, способный к образованию липосом. Настоящие композиции в форме липосом могут содержать, помимо соединения по настоящему изобретению, стабилизаторы, консерванты, наполнители и им подобные. Предпочтительными липидами являются фосфолипиды и фосфатидилхолины (лецитин) как природные, так и синтетические. Способы получения липосом известны в соответствующей области, см., например, Methods in Cell Biology (Методы в клеточной биологии) под ред. Prescott, т.XIV, Academic Press, New York, N.Y. (1976), стр.33 и последующие.
Предпочтительно соединения по настоящему изобретению могут быть активны при пероральном введении, характеризоваться быстрым проявлением активности и низкой токсичностью.
Соединения по настоящему изобретению могут иметь то преимущество, что они оказываются более эффективными, менее токсичными, более длительно действующими, располагающими более широким диапазоном активности, более действенными, производящими меньше побочных эффектов, легче всасываемыми или располагающими другими полезными фармакологическими свойствами по сравнению с соединениями, известными в соответствующей области ранее.
Способы комбинационной терапии
Соединения по настоящему изобретению могут вводиться в сочетании с одним или несколькими дополнительными терапевтическими средствами. В соответствии с этим объектом настоящего изобретения является фармацевтическая композиция, содержащая дополнительное средство. Объектом настоящего изобретения также является продукт, содержащий соединение по настоящему изобретению и средство в качестве комбинированного препарата для одновременного, раздельного или последовательного применения в терапии.
В частности, композиция или продукт по настоящему изобретению могут дополнительно содержать терапевтическое средство, применимое при лечении периферической невропатии, например демиелинизирующей периферической невропатии, в сочетании с которым будет вводиться соединение по настоящему изобретению. В качестве примеров таких средств могут быть упомянуты иммунодепрессант (например, циклоспорин А, циклоспорин G, FK.-506, АВТ-281, ASM981, рапамицин, 40-O-(2-гидрокси)этилрапамицин, кортикостероиды, циклофосфамид, азатиоприн, метотрексат, лефлуномид, мизорибин, мофетила микофенолят или 15-дезоксиспергуалин), стероид (например, преднизон или гидрокортизон), иммуноглобулин или интерферон типа 1. Соединение по настоящему изобретению и второе средство могут вводиться одновременно или последовательно. В тех случаях, когда соединение по настоящему изобретению и второе средство вводятся одновременно, из них может быть изготовлен препарат в виде единой композиции или отдельных композиций.
Применение
Соединения по настоящему изобретению могут быть применимы в терапии разнообразных периферических невропатий, в частности острых или хронических демиелинизирующих невропатий. Таким образом, соединения по настоящему изобретению могут быть применимы в терапии одного или нескольких из следующих заболеваний: синдрома Гийена-Барре (СГБ), хронической воспалительной демиелинизирующей полирадикулоневропатии (ХВДП), мультифокальной моторной невропатий с блоком проведения (ММН) и парапротеинемической демиелинизирующей периферической невропатий (ПДН). В частности, невропатия может являться ХВДП. Эффективность соединений может варьироваться от пациента к пациенту.
Термин «терапия» включает лечение, имеющее целью облегчение одного или более симптомов периферической невропатии или замедление развития подобного заболевания посредством, например, предотвращения или замедления демиелинизации, например периферической демиелинизации. Он также включает лечение, имеющее целью излечение заболевания, приведение пациента в функциональное состояние и/или поддержание пациента в функциональном состоянии, или же отдаление рецидива.
Терапевтическое применение соединения может включать профилактическое применение, имеющее целью предотвращение, контроль или снижение тяжести периферической невропатии, риском каковой характеризуется пациент, равно как и лечение, имеющее целью предотвращение, контроль или снижение тяжести наличествующего заболевания. Соединение может вводиться до появления симптомов, но может вводиться и после появления симптомов. Оно может вводиться пациенту, характеризуемому риском периферической невропатии.
Варианты лечения, в которых может быть задействовано соединение, могут, таким образом, улучшить или поддержать медицинское состояние и/или комфорт или задержать ухудшение такового у пациента, страдающего периферической невропатией, подозреваемого в ее наличии или характеризуемого риском ее появления.
Примеры
Нижеследующие примеры иллюстрируют изобретение
Пример 1 (синтез соединения А)
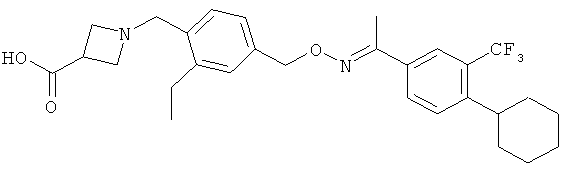
1-{4-[1-(4-Циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновая кислота (соединение А)
К суспензии MnO2 (10 экв.) в диоксане добавляют O-(4-циклогексил-3-трифторметилбензил)оксим 1-(3-этил-4-гидроксиметилфенил)этанона (1 экв.). Получаемую таким образом смесь кипятят с обратным холодильником в течение 10 минут. После фильтрования и концентрирования растворяют остаток в МеОН и обрабатывают азетидин-3-карбоновой кислотой (2 экв.) и Et3N (1,5 экв.). Получаемую после этого смесь нагревают до температуры 50°C в течение 30 минут. После охлаждения до комнатной температуры добавляют порциями HaBH3CH (3 экв.). Очистка с помощью препаративной жидкостной хроматографии с масс-спектрометрическим контролем (ЖХ-МС) приводит к 1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновой кислоте. Данные ядерного магнитного резонанса (1H ЯМР) (400 МГц, CD3OD) δ 1,24 (t, 3H), 1,30-1,60 (m, 5H), 1,74-1,92 (m, 5H), 2,28 (s, 3H), 2,79 (q, 2H), 2,92 (m, 1H), 3,68 (m, 1H), 4,32 (m, 4H), 4,51 (s, 2H) 5,22 (s, 2H), 7,38 (d, 1H), 7,50-7,68 (m, 5H). МС (ионизация электрораспылением, режим положительных ионов, ИЭР+): 517.3 (М+1)+.
Пример 2 (супрессорное влияние соединения А на экспериментальный аутоиммунный неврит (ЭАН))
Самцов крыс линии Льюис (8-10 недель, 180-200 г, Elevage-Janvier, Франция) содержали в условиях цикла 12 ч светлого времени - 12 ч темного времени и свободного доступа к пище и воде. Все манипуляции с животными производились в соответствии с методикой, одобренной местным официальным комитетом административного округа. Были предприняты все усилия для минимизации числа животных и их страданий.
Стимуляция ЭАН
Для стимуляции ЭАН крыс иммунизировали посредством подкожной инъекции в подушечки стопы обеих задних лап 100 мкл инокулята, содержавшего 100 мкг синтетического невритогенного пептида Р2 57-81 (GeneScript Corporation, Скотч Плэйнс, Нью-Джерси, США). Пептид растворяли в забуференном фосфатом солевом растворе (PBS) (2 мг/мл), а затем эмульгировали с равным объемом полного адъюванта Фрейнда (ПАФ), содержащего 2 мг/мл микобактерии туберкулеза, обеспечивая конечную концентрацию 1 мг/мл.
Тяжесть ЭАН ежедневно оценивали по следующей клинической шкале: 0 = нормальное состояние, 1 = пониженный тонус хвоста, 2 = вялый хвост, нарушенное выпрямление, 3 = отсутствие выпрямления, 4 = нарушение координации походки, 5 = слабый парез задних конечностей, 6 = умеренный парез, 7 = тяжелый парез или паралич задних конечностей, 8 = тетрапарез, 9 = агония и 10 = смерть (Zhang и др., 2009 А).
Лечение соединением А
Соединение А отдельно исследовали в двух концентрациях суспензии (3 и 10 мг/кг, суспендированной в 1% карбоксиметилцеллюлозы (КМК, бланоза, Hercules-Aqualon, Дюссельдорф, Германия)). Суспензии соединения А вводили внутрижелудочно непосредственно после стимуляции, а затем раз в сутки вплоть до 22 суток (5 крыс на группу). Контрольным крысам с ЭАН давали тот же объем 1% КМК в воде.
Иммуногистохимия
Для оценки инфильтрации воспалительных клеток и патологических изменений в периферической нервной системе (ПНС) умертвили пять крыс, которым вводили соединение А (в обеих концентрациях) и пять контрольных крыс с ЭАН, взятых на 16 сутки. Крыс подвергали глубокой анестезии эфиром и вливали им внутрисердечно 4% раствор параформальдегида в PBS с температурой 4°C. Быстро отделяли левый и правый седалищные нервы и затем выдерживали их в течение ночи в 4% растворе формальдегида при температуре 4°C. Затем седалищные нервы разрезали на два участка равной длины, заключали в парафин, делали последовательные срезы (3 мкм) и помещали на покрытые силаном предметные стекла.
После удаления парафина срезы седалищных нервов кипятили (в микроволновой печи при мощности 600 Вт) в течение 15 минут в цитратном буфере (2,1 г/л цитрата натрия, рН б). Эндогенную пероксидазу ингибировали 1% H2O2 в метаноле в течение 15 минут. Срезы инкубировали с 10% нормальной свиной сывороткой (Biochrom, Берлин, Германия) с целью блокировки неспецифического связывания иммуноглобулинов, а затем со следующими моноклональными антителами W3/13 (1:50; Serotec, Оксфорд, Великобритания) на Т-лимфоциты, 0Х22 (1:200; Serotec, Оксфорд, Великобритания) на В-клетки, ED1 на активированные макрофаги (1:100; Serotec, Оксфорд, Великобритания). Связывание антител со срезами ткани визуализировали с помощью фрагментов вторичного антитела биотинилированный IgG (иммноглобулин-гамма) F(ab)2 (кроличье противомышиное или кроличье противокозье; 1:400; DAKO, Гамбург, Германия). После этого срезы инкубировали с комплексом стрептавидин-авидин-биотин (DAKO, Гамбург, Германия), а затем обрабатывали диаминобензидиновым субстратом (ДАБ) (Fluka, Ной-Ульм, Германия). В заключение срезы контрастно окрашивали гемалюмом Майера.
Для обработки данных иммуноокрашивания рассчитывали процентное соотношение площади иммунореактивности (ИР) к площади срезов седалищных нервов. Изображения сечений седалищных нервов получали при пятидесятикратном увеличении с помощью микроскопа Nikon Coolscope (Nikon, Дюссельдорф, Германия) при фиксированных параметрах. Изображения анализировали с помощью программы MetaMorph Offline 7.1 (Molecular Devices, Торонто, Канада). Площади ИР выбирали с помощью разбивки по порогу окрашивания, причем все параметры были фиксированы для всех изображений. Площади срезов седалищных нервов выбирали вручную. Для каждой крысы с ЭАН анализировали четыре среза из корневых и средних уровней на обеих сторонах. Результаты представляли в виде средних арифметических процентных соотношений площади ИР к площади срезов седалищных нервов и стандартных отклонений (CO).
Для обнаружения миелина использовали стандартное окрашивание люксолом быстрым синим (LFB). Гистологические различия между крысами, которым вводили соединение А, и контрольными крысами с ЭАН определяли с помощью стандартного полуколичественного способа. Вкратце, анализировали четыре среза из корневого и среднего уровня на обеих сторонах для крыс с ЭАН. Все околососудистые области, присутствующие на срезах, оценивали два наблюдателя, не осведомленные о лечении, и степень патологических изменений ранжировалась полуколичественно согласно следующей шкале: 0 = нормальная околососудистая область, 1 = слабый клеточный инфильтрат рядом с сосудом, 2 = клеточная инфильтрация, а также демиелинизирование в непосредственной близости к сосуду и 3 = клеточная инфильтрация и демиелинизирование по всему срезу. Результаты представляли в виде средней оценки по гистологической шкале (Hartung и др., 1988).
Обработка данных и статистический анализ
Осуществляли проверку с помощью непарного t-теста для оценки различий между соединением А и контрольными крысами с ЭАН (программа Graph Pad Prism 4,0 для Windows). Уровни значимости для всех статистических анализов выбирались как р<0,05.
Результаты
Супрессивное лечение ЭАН соединением А
Развитие ЭАН стимулировали подкожной инъекцией невритогенного синтетического пептида Р2. В качестве супрессивного лечения вводили перорально 1% КМК в воде (контрольная группа) или соединение А немедленно после иммунизации, а затем один раз в сутки вплоть до 22 суток. Первые неврологические признаки (пониженный тонус хвоста) были обнаружены у контрольных крыс с ЭАН на 9 сутки (средний показатель по клинической шкале: 0,20±0,13). Неврологическая тяжесть ЭАН быстро росла в контрольной группе с максимальным показателем на 13 сутки (средний показатель по клинической шкале: 4,80±0,51). После этого тяжесть ЭАН медленно понижалась, и крысы полностью восстанавливались на 22 сутки (средние показатели по клинической шкале: 0±0). У крыс с ЭАН, которым вводили соединение А, существенно пониженные неврологические признаки были замечены на 14 сутки при максимальном показателе на 15 сутки и полном восстановлении крыс, отмеченном на 19 сутки. Таким образом, введение соединения А практически предотвратило развитие клинических признаков ЭАН, существенно задержало их появление, понизило неврологическую тяжесть и сократило длительность ЭАН весьма эффективным образом по сравнению с основой вода-КМК (фиг.1А и 1Б).
Дополнительным проявлением ЭАН является прогрессирующая потеря массы после появления заболевания. И у контрольных, и у получавших соединение А крыс с ЭАН до появления ЭАН (9 сутки) наблюдалась медленная и непрерывная прибавка в весе. После этого контрольные крысы с ЭАН продемонстрировали существенную потерю массы в период неврологического заболевания с 10 по 18 сутки после иммунизации с последующим набором веса в период восстановления (фиг.2А). Напротив, у крыс с ЭАН, получавших более разбавленную суспензию соединения А, отмечался пониженный уровень потери веса между 11 и 16 сутками на пике проявления заболевания, что, опять же, свидетельствовало о много менее тяжелом течении. Данный эффект был более выражен для суспензии с большей дозировкой, где наблюдалась небольшая потеря веса между 11 и 12 сутками с последующим набором веса (фиг.2Б).
Влияние введения супрессорного соединения А на гистопатологические изменения в пораженных ЭАН седалищных нервах
Инфильтрацию различных типов воспалительных клеток в седалищных нервах у контрольных и получавших соединение А крыс с ЭАН анализировали иммуногистохимически на 16 сутки (n=5). Инфильтрация Т-клеток (W3/13+), В-клеток (0Х22+) и макрофагов (ED1+) отмечалась в седалищных нервах контрольных крыс с ЭАН. Преобладающими инфильтрующимися клетками были макрофаги, чьи площади ИР занимали около 2% общей площади седалищных нервов на срезах. Эти результаты представлены в нижеприведенной таблице 1.
В седалищных нервах крыс с ЭАН соединение А существенно подавляет инфильтрацию Т-клеток, В-клеток и макрофагов.
В седалищных нервах средние оценки по гистологической шкале, измеренные с помощью окрашивания LFB, были заметно ниже у тех крыс с ЭАН, которым вводили соединении А. Эти результаты представлены в нижеприведенной таблице 2.
Данные результаты показывают, что супрессивное лечение соединением А практически предотвратило ЭАН и ингибировало паралич в результате существенного снижения инфильтрации лимфоцитов и макрофагов в периферические нервы совместно со снижением степени местной демиелинизации.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ГЛИКОПЕПТИДА ИЛИ ИХ СОЛИ, СПОСОБ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1995 |
|
RU2145609C1 |
| ТРИЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ ЯНУС-КИНАЗЫ 1, ИХ КОМПОЗИЦИИ, СПОСОБЫ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2019 |
|
RU2824349C2 |
| ПРОИЗВОДНЫЕ ТЕТРАГИДРОБЕНЗИМИДАЗОЛА В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ TNF | 2014 |
|
RU2684637C1 |
| ПРОИЗВОДНЫЕ ИМИДАЗОТРИАЗИНА В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ TNF | 2014 |
|
RU2677699C1 |
| НОВЫЕ ПРОИЗВОДНЫЕ ИМИДАЗОЛИДИНА, ИХ ПОЛУЧЕНИЕ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ АНТАГОНИСТОВ VLA-4 | 2002 |
|
RU2318815C2 |
| ПИРАЗОЛОПИРИМИДИНОВЫЕ СОЕДИНЕНИЯ-ИНГИБИТОРЫ JAK И СПОСОБЫ | 2009 |
|
RU2539568C2 |
| НОВЫЕ ПЯТИЧЛЕННЫЕ ГЕТЕРОЦИКЛЫ, ИХ ПОЛУЧЕНИЕ, ИХ ПРИМЕНЕНИЕ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ ПРЕПАРАТЫ | 1998 |
|
RU2240326C2 |
| КОНДЕНСИРОВАННЫЕ БИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЯ | 2015 |
|
RU2741387C2 |
| ТЕРАПЕВТИЧЕСКИЕ СОЕДИНЕНИЯ И СПОСОБЫ | 2018 |
|
RU2837261C2 |
| ПРОИЗВОДНЫЕ ПУРИНА В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ TNF | 2014 |
|
RU2684644C1 |
Изобретение относится к применению 1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)-этил]-2-этилбензил}азетидин-3-карбоновой кислоты или ее фармакологически приемлемой соли при лечении демиелинизирующей периферической невропатии, выбранной из хронической воспалительной демиелинизирующей полирадикулоневропатии, мультифокальной моторной невропатии с блоком проведения или парапротеинемической демиелинизирующей периферической невропатии. Технический результат - применение 1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)-этил]-2-этилбензил}азетидин-3-карбоновой кислоты для лечения демиелинизирующей периферической невропатии. 4 ил., 2 табл., 2 пр.
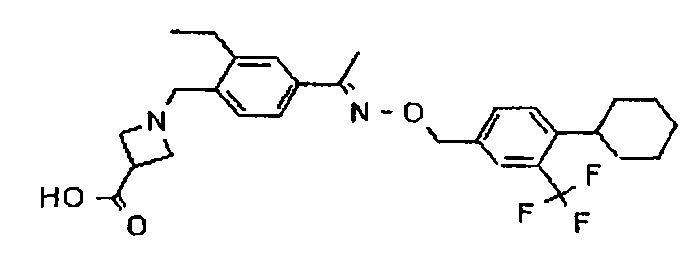
Применение 1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)-этил]-2-этилбензил}азетидин-3-карбоновой кислоты
или ее фармакологически приемлемой соли при лечении демиелинизирующей периферической невропатии, выбранной из хронической воспалительной демиелинизирующей полирадикулоневропатии, мультифокальной моторной невропатии с блоком проведения или парапротеинемической демиелинизирующей периферической невропатии.
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |
| НОВЫЕ ПРОИЗВОДНЫЕ МИНДАЛЬНОЙ КИСЛОТЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ТРОМБИНА | 2001 |
|
RU2300521C2 |

