ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
По этой не предварительной заявке испрашивается приоритет в соответствии с 35 USC §119(e) предварительной заявки США № 61/080944, поданной 15 июля 2008, которая включена посредством ссылки в полном объеме.
ОБЛАСТЬ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к конъюгатам терапевтически используемых антрациклинов с такими носителями, как поликлональные и моноклональные антитела, белки или пептиды природного или синтетического происхождения; к способам их получения, к содержащей их фармацевтической композиции и к их применению при лечении определенных опухолей у млекопитающих. Изобретение также относится к новым производным антрациклина и к их получению.
ПРЕДПОСЫЛКИ К СОЗДАНИЮ ИЗОБРЕТЕНИЯ
Антрациклины представляют собой антибиотические соединения, которые ингибируют цитотоксическую активность. Исследования показали, что антрациклины могут действовать для уничтожения клеток посредством различных механизмов, включая: 1) интеркаляцию молекул лекарственного средства в ДНК клетки, ингибируя, таким образом, ДНК-зависимый синтез нуклеиновой кислоты; 2) продуцирование лекарственным средством свободных радикалов, которые затем взаимодействуют с макромолекулами клетки, чтобы вызвать разрушение клетки, или 3) взаимодействие молекул лекарственного средства с клеточной мембраной [смотри, например, C. Peterson et al., «Transport And Storage Of Anthracycline In Experimental Systems And Human Leukemia» в Anthracycline Antibiotics In Cancer Therapy; N.R. Bachur, “Free Radical Damage” id. на стр. 97-102]. Благодаря своему цитотоксическому потенциалу антрациклины были использованы в лечении ряда злокачественных опухолей, таких как лейкоз, рак молочной железы, рак легкого, аденокарцинома яичников и саркомы [см., например, P.H- Wiernik, в Anthracycline: Current Status And New Developments p. 11]. Обычно используемые антрациклины включают доксорубицин, эпирубицин, идарубицин и дауномицин.
В последние годы было синтезировано много новых высокоцитотоксичных антрациклинов. Например, неморубицин, производное антрациклина, несущее морфолино-замещенное кольцо, связанное с положением C-3' фрагмента сахара, продемонстрировал многообещающую противоопухолевую активность на экспериментальных опухолях мыши (см. J.W. Lown, Bioactive Molecules, 55-101, (1988) vol. 6:55-101) и в настоящее время является объектом клинической фазы испытаний для лечения гепатоцеллюлярного рака [см.: C. Sessa, O. Valota, C. Geroni, Cardiovascular Toxicology (2007) 7(2):75-79]. Хотя эти соединения могут быть использованы в лечении новообразований и других патологических состояний, при которых желательно элиминировать некоторую популяцию клеток, их терапевтическая эффективность зачастую ограничивается дозозависимой токсичностью, связанной с их применением.
Были предприняты попытки улучшить терапевтическую эффективность этих соединений, путем связывания антрациклина с антителами или с различными носителями. Например, антрациклин, конъюгированный с антителами, описан, например, в EP 0328147, Bristol Myers, в WO 9202255, Farmitalia Carlo Erba или в US 5776458, Pharmacia & Upjohn.
Другие представляющие интерес трициклические морфолино-производные антрациклина, характеризующиеся высокой активностью, были описаны и заявлены в международной патентной заявке WO 98/02446 (1997), M. Caruso et al. В числе этих производных особенно активным соединением является PNU-159682, описанное Quintieri, L., Geroni, C. et al. в Clinical Cancer Research (2005) 11(4):1608-1617. Соединение PNU-159682 имеет формулу (IIA), как определено далее в настоящей заявке, и следующие химические названия:
5,12-нафтацендион, 7,8,9,10-тетрагидро-6,8,11-тригидрокси-8-(гидроксиацетил)-1-метокси-10-[[(1S,3R,4aS,9S,9aR,10aS)-октагидро-9-метокси-1-метил-1H-пирано[4',3':4,5]оксазоло[2,3-c][1,4]оксазин-3-ил]окси]-, (8S,10S)-(9CI);
5,12-нафтацендион, 7,8,9,10-тетрагидро-6,8,11-тригидрокси-8-(гидроксиацетил)-1-метокси-10-[(октагидро-9-метокси-1-метил-1H-пирано[4',3':4,5]оксазоло[2,3-c][1,4]оксазин-3-ил)окси]-, [1S-[1α,3β(8R*,10R*),4aβ,9α,9aα,10aβ]] или (8S,10S)-6,8,11-тригидрокси-8-(гидроксиацетил)-1-метокси-10-{[(1S,3R,4aS,9S,9aR,10aS)-9-метокси-1-метилоктагидро-1H-пирано[4',3':4,5][1,3]оксазоло[2,3-c][1,4]оксазин-3-ил]окси}-7,8,9,10-тетрагидротетрацен-5,12-дион.
Хорошо известна терапия антителами для направленного лечения пациентов, страдающих злокачественным заболеванием, иммунологическими и ангиогенными нарушениями (Carter, P. (2006) Nature Reviews Immunology 6:343-357). Применение конъюгатов антител с лекарственными средствами (ADC), то есть иммуноконъюгатов, для местной доставки цитотоксических или цитостатических средств, то есть лекарственных средств для уничтожения или ингибирования опухолевых клеток при лечении злокачественной опухоли, направленной доставки фрагмента лекарственного средства к опухолям и внутриклеточное накопление в них, тогда как системное введение этих неконъюгированных лекарственных средств может приводить к неприемлемым уровням токсичности для нормальных клеток, а также требуемых для выведения опухолевых клеток (Xie et al. (2006) Expert. Opin. Biol. Ther. 6(3):281-291; Kovtun et al. (2006) Cancer Res. 66(6):3214-3121; Law et al. (2006) Cancer Res. 66(4):2328-2337; Wu et al. (2005) Nature Biotech. 23(9):1137-1145; Lambert J. (2005) Current Opin. in Pharmacol. 5:543-549; Hamann P. (2005) Expert Opin. Ther. Patents 15(9):1087-1103; Payne, G. (2003) Cancer Cell 3:207-212; Trail et al. (2003) Cancer Immunol. Immunother. 52:328-337; Syrigos and Epenetos (1999) Anticancer Research 19:605-614). Таким образом, желательными являются максимальная эффективность с минимальной токсичностью. Попытки создания и очищения ADC сосредоточены на селективности моноклональных антител (mAb), а также механизме действия лекарственного средства, связывания с лекарственным средством, соотношении лекарственного средства/антитела (нагрузки) и свойствах высвобождения лекарственного средства (McDonagh (2006) Protein Eng. Design & Sel.; Doronina et al (2006) Bioconj. Chem. 17:114-124; Erickson et al (2006) Cancer Res. 66(8):1-8; Sanderson et al. (2005) Clin. Cancer Res. 11:843-852; Jeffrey et al. (2005) J. Med. Chem. 48:1344-1358; Hamblett et al. (2004) Clin. Cancer Res. 10:7063-7070). Лекарственные фрагменты могут осуществлять свои цитотоксические и цитостатические действия посредством механизмов, включающих связывание тубулина, связывание ДНК или ингибирование топоизомеразы. Некоторые цитотоксические лекарственные средства имеют тенденцию становиться неактивными или менее активными при конъюгировании с крупными антителами или белковыми лигандами рецепторов.
Считают, что аналог антрациклина, доксорубицин (АДРИАМИЦИН®) взаимодействует с ДНК путем интеркаляции и ингибирования развития образования фермента топоизомеразы II, которая раскручивает ДНК для транскрипции. Доксорубицин стабилизирует топоизомеразный комплекс II после разрушения цепи ДНК для репликации, предотвращая повторное уплотнение двойной спирали ДНК и тем самым останавливая процесс репликации. Доксорубицин и даунорубицин (ДАУНОМИЦИН) являются прототипом природных цитотоксических химиотерапевтических продуктов антрациклина (Sessa et al. (2007) Cardiovasc. Toxicol. 7:75-79). Были получены и изучены иммуноконъюгаты и пролекарства даунорубицина и доксорубицина (Kratz et al. (2006) Current Med. Chem. 13:477-523; Jeffrey et al. (2006) Bioorganic & Med. Chem. Letters 16:358-362; Torgov et al. (2005) Bioconj. Chem. 16:717-721; Nagy et al. (2000) Proc. Natl. Acad. Sci. 97:829-834; Dubowchik et al. (2002) Bioorg. & Med. Chem. Letters 12:1529-1532; King et al. (2002) J. Med. Chem. 45:4336-4343; US 6630579). Конъюгат антитело-лекарственное средство BR96-доксорубицин специфически взаимодействует с опухоль-связанным антигеном Lewis-Y и был оценен на I и II фазе испытаний (Saleh et al. (2000) J. Clin. Oncology 18:2282-2292; Ajani et al. (2000) Cancer Jour. 6:78-81; Tolcher et al. (1999) J. Clin. Oncology 17:478-484).
Морфолиновые аналоги доксорубицина и даунорубицина, образованные путем циклизации на гликозидной аминогруппе, являются более эффективными (Acton et al. (1984) J. Med. Chem. 638-645; US 4464529; US 4672057; US 5304687). Неморубицин является полусинтетическим аналогом доксорубицина с 2-метоксиморфолино группой на гликозидной аминогруппе доксорубицина и находится на клиническом испытании (Grandi et al. (1990) Cancer Treat. Rew. 17:133; Ripamonti et al. (1992) Brit. J. Cancer 65:703;), включая II/III фазу испытаний для печеночноклеточной карциномы (Sun et al. (2003) Proceedings of the American Society for Clinical Oncology 22, Abs1448; Quintieri (2003) Proceedings of the American Association of Cancer Research, 44:1st Ed, Abs 4649; Pacciarini et al. (2006) Jour. Clin. Oncology 24:14116).
Неморубицин назван как (8S,10S)-6,8,11-тригидрокси-10-((2R,4S,5S,6S)-5-гидрокси-4-((S)-2-метоксиморфолино)-6-метилтетрагидро-2H-пиран-2-илокси)-8-(2-гидроксиацетил)-1-метокси-7,8,9,10-тетрагидротетрацен-5,12-дион, с регистрационным номером CAS 108852-90-0 и имеет структуру:
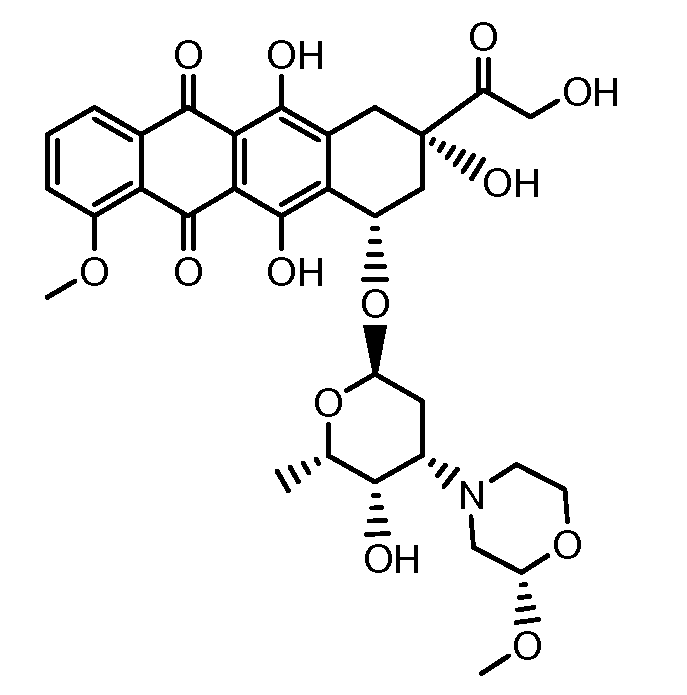
Были охарактеризованы некоторые метаболиты неморубицина (MMDX) из микросом печени, включая PNU-159682, (Quintieri et al. (2005) Clinical Cancer Research, 11(4):1608-1617; Beulz-Riche et al. (2001) Fundamental & Clinical Pharmacology, 15(6):373-378; EP 0889898; WO 2004/082689; WO 2004/082579). PNU-159682 был заметно более цитотоксичным, чем неморубицин и доксорубицин in vitro, и был эффективен в моделях опухолей in vivo. PNU-159682 (формула (IIA) называют 3'-дезамино-3'',4'-ангидро-[2''(S)-метокси-3''(R)-окси-4''-морфолинил]доксорубицин и имеет структуру:
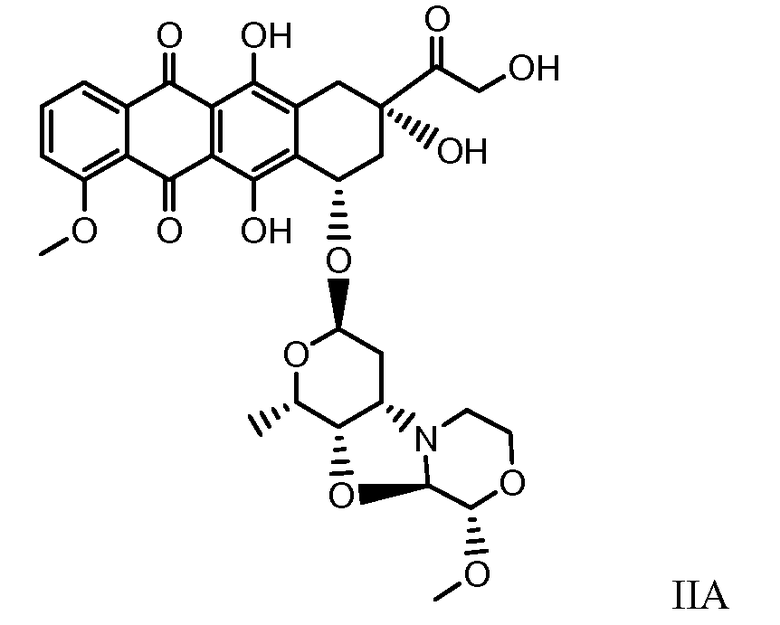
Были описаны некоторые конъюгаты PNU-159682 антитело-лекарственное средство («NEMORUBICIN METABOLTE AND ANALOG ANTIBODY-DRUG CONJUGATES AND METHODS», PCT/US2009/031199, подана 16 января 2009).
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Один из аспектов настоящего изобретения относится к новым конъюгатам производного антрациклина с носителями, такими как моноклональные или поликлональные антитела, вступающие в реакцию с выбранной популяцией клеток, белками, пептидами или другим носителям синтетического происхождения, вступающим в реакцию с рецепторными тканями.
Другим аспектом является способ получения таких конъюгатов, а также применимых промежуточных соединений.
Конъюгаты по настоящему изобретению характеризуются формулой (I)
[Ant-L-Z-]m-T, (I)
где
Ant представляет собой остаток производного антрациклина,
L представляет собой линкер,
Z представляет собой спейсер,
m представляет собой целое число от 1 до 30 и
T представляет собой носитель, такой как белок, пептид, моноклональное или поликлональное антитело или его химически модифицированное производное, подходящие для присоединения к фрагменту или фрагментам [Ant-L-Z-], или полимерному носителю;
отличающееся тем, что остаток производного антрациклина, который представляет Ant, может высвобождаться с получением производного антрациклина формулы (II):
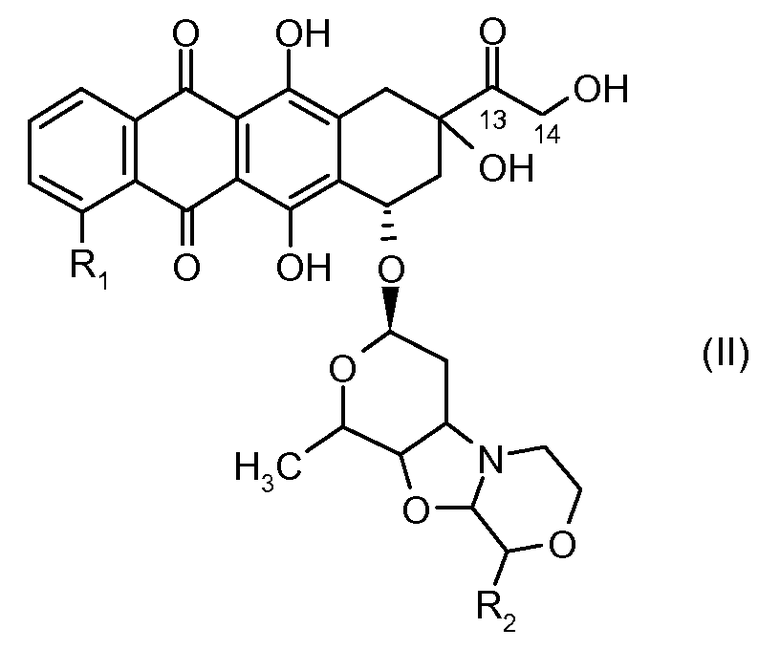
где R1 представляет собой атом водорода, гидрокси или метокси группу и R2 представляет собой C1-C5 алкокси группу, или их фармацевтически приемлемую соль.
Остаток производного антрациклина соединен с носителем через линкерный спейсер [L-Z-], и связь между производным антрациклина и линкерным плечом может расщепляться в физиологических условиях, таким образом, что высвобождается производное антрациклина формулы (II), определенное выше, которое является биологически активным соединением.
Например, конъюгаты, в которых связь между производным антрациклина и линкером является чувствительной к кислым условиям или к условиям восстановления, могут высвобождать производное антрациклина в условиях, которые обычно встречаются в клетке, например, лизосомных везикулах.
Предпочтительный способ по настоящему изобретению предназначен для лечения определенных типов злокачественных опухолей, в том числе, но не только: карциномы, например, мочевого пузыря, груди, толстой кишки, почек, печени, легких, включая мелкоклеточный рак легких, пищевода, желчного пузыря, яичников, поджелудочной железы, желудка, шейки матки, щитовидной железы и кожи, включая скваможноклеточную карциному; гематопоэтических опухолей лимфоидной линии дифференцировки, включая лейкоз, острый лимфоцитарный лейкоз, острый лимфобластный лейкоз, В-клеточную лимфому, Т-клеточную лимфому, лимфому Ходжкина, неходжкинскую лимфому, волосатоклеточную лимфому и лимфому Беркитта; гематопоэтических опухолей миелоидной линии дифференцировки, включая острый и хронический миелолейкозы, миелодиспластический синдром и промиелоцитарный лейкоз; опухолей мезенхимального происхождения, включая фибросаркому и рабдомиосаркому; опухолей центральной и периферической нервной системы, включая астроцитому, нейробластому, глиому и шванному; других опухолей, включая меланому, семиному, тератокарциному, остеосаркому, пигментную ксеродерму, кератоксантому, фолликулярный рак щитовидной железы и саркому Капоши.
Другим предпочтительным способом по настоящему изобретению является лечение специфических нарушений пролиферации, например, таких как доброкачественная гиперплазия предстательной железы, семейный аденоматоз толстой кишки, нейрофиброматоз, псориаз, пролиферация гладкомышечных клеток сосудов, связанная с атеросклерозом, фиброз легких, артрит, гломерулонефрит и стеноз после гломерулонефрита и пост-хирургический стеноз и рестеноз.
Конъюгаты производных антрациклина формулы (I) могут быть получены через способ, состоящий из стандартных синтетических трансформаций; такой способ и промежуточные соединения, используемые в таком способе, также представлены настоящим изобретением.
Настоящее изобретение также относится к фармацевтической композиции, содержащей конъюгат производного антрациклина формулы (I) или его фармацевтически приемлемую соль и фармацевтически приемлемый эксципиент или разбавитель.
Один из аспектов настоящего изобретения представляет собой производное антрациклина формулы (IIc)
где Ant представляет собой производное антрациклина, выбранное из структур:

где волнистая линия указывает на присоединение к линкеру L; Z представляет собой необязательный спейсер; m равно 0 или 1; X представляет собой реакционно-способную функциональную группу; и n равно целому числу от 1 до 6.
Один из аспектов настоящего изобретения представляет собой конъюгат антитела с лекарственным средством (ADC), соединение, содержащее антитело, ковалентно присоединенное посредством линкера L и необязательного спейсера Z к одному или нескольким фрагментам лекарственного средства производного антрациклина D, соединение имеет формулу (Ic)
или его фармацевтически приемлемая соль, где p равно целому числу от 1 до 8.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
На Фигуре 1 в графической форме показана стабильность соединения 2, представленного в таблице 1, при pH 5,2, где по оси (Y) отложено процентное содержание соединения формулы (IIA), как определено далее, и по оси (X) время в часах. Этот график демонстрирует чувствительность к кислоте ацетальной связи по настоящему изобретению, показанную повышенным высвобождением свободного соединения формулы (IIA) из конъюгата при кислом pH.
На Фигуре 2 показан приводимый в качестве примера способ получения соединения Формулы Ia путем взаимодействия производного антрациклина формулы II с простым виниловым эфиром соединения (X) или (IX) с получением ацетального соединения (XI), гидролиза до карбоксильного соединения (XII) и активации с получением сложного эфира N-гидроксисукцинимида (NHS) (XIII), готового для конъюгации с соединением-носителем.
На Фигуре 3a показан приводимый в качестве примера способ получения соединения Формулы Ia путем взаимодействия активированного сложного эфира N-гидроксисукцинимида (NHS) (XIII) с амино-тиольным реагеном (XXI) с получением XXII, которое может вступать в реакцию с промежуточным соединением (VI) пиридил-дисульфидным носителем (Т) с получением дисульфид-связанного конъюгата производного атрациклина (Ia), или вступать в реакцию с промежуточным соединением малеимидным носителем (T) (XXII) с получением малеимид-связанного конъюгата производного антрациклина (Ia).
На Фигуре 3b показан приводимый в качестве примера способ получения соединения Формулы Ia, путем взаимодействия активированного сложного эфира N-гидроксисукцинимида (NHS) (XIII) с амино-сложноэфирным реагентом (XVIII), затем сложноэфирного гидролиза с получением производного карбоксиантрациклина (XIX), которое может быть активировано как сложный эфир NHS и соединения с промежуточным соединением амино-носителем T, например антитела, с получением амид-связанного конъюгата производного антрациклина (Ia).
На Фигуре 3c показаны приводимые в качестве примера способы получения соединения Формулы Ia, путем взаимодействия активированного сложного эфира N-гидроксисукцинимида (NHS) (XIII) с амино или тиольной группой носителя T (XIV), например антителом, с получением конъюгата производного антрациклина, связанного с амидом или тиоамидом (Ia).
На Фигуре 4 показан приводимый в качестве примера процесс взаимодействия производного антрациклина (II) с производным ацилгидразида (XXV) с получением гидразона (XXVI), с последующим конъюгированием с реагентом носителем T с получением конъюгата производного антрациклина (Ib).
На Фигуре 5a показаны приводимые в качестве примера процессы: (2a) взаимодействия гидразона производного антрациклина (XXVI) с соединением тиол- или амино-носителем T (XIV) с получением конъюгата производного антрациклина (Ib); (2b) взаимодействия гидразона производного антрациклина (XXVI) с реагентом (XXVII), с последующим снятием защиты с получением (XXVIII) и соединения с соединением карбоксил-носителем T (XVII) с получением конъюгата производного антрациклина (Ib); и (2d) конденсирования незащищенного (XXVIII) с соединением альдегид-носителем T (XX) с получением конъюгата производного антрациклина (Ib).
На Фигуре 5b показан приводимый в качестве примера процесс: (2c) взаимодействия гидразона производного антрациклина (XXVI) с реагентом (XXIX), с последующим снятием защиты и соединением с амино-носителем T с получением конъюгата производного антрациклина (Ib).
На Фигуре 5c показаны приводимые в качестве примера процессы: (2e) взаимодействия гидразона производного антрациклина (XXVI) с реагентом (XXXI) с получением (XXXII), затем (2e") соединения с пиридилдисульфидным соединением-носителем (VI) с получением конъюгата производного антрациклина (Ib), и (2e') соединения (XXXII) с малеимидным соединением-носителем (V) с получением конъюгата производного антрациклина (Ib).
На Фигуре 6 показан приводимый в качестве примера процесс взаимодействия производного антрациклина (II) с ацилгидразидом, пиридилдисульфидом (XXXIII) с образованием пиридилдисульфидгидразона (XXXIV), с последующим конъюгированием с реагентом носителем T с получением конъюгата производного антрациклина (Ib).
На Фигуре 7a показаны приводимые в качестве примера процессы: (3a) взаимодействия производного антрациклина (XXXIV) с тиол-носителем (XIV) с получением конъюгата производного антрациклина (Ib); (3b) взаимодействия производного антрациклина (XXXIV) с тиольным соединением (XXXV) с получением амин дисульфидного соединения (XXXVI), которое соединено с реагентом карбоксил-носитель T с получением конъюгата производного антрациклина (Ib); и (3d) конденсирования незащищенного (XXXVI) с соединением альдегид-носитель T (XX) с получением конъюгата производного антрациклина (Ib).
На Фигуре 7b показан приводимый в качестве примера процесс (3c) взаимодействия производного антрациклина (XXXIV) с тиоловым сложным эфиром (XXXVII), с последующим снятием защиты до карбоксил-дисульфидного соединения (XXXVIII), и соединения с амино-носителем T (XIV) с получением конъюгата производного антрациклина (Ib).
На Фигуре 7c показаны приводимые в качестве примера процессы: (3e) взаимодействия производного антрациклина (XXXIV) тиольного реагента (XXXIX) с образованием дисульфид-тиола (XL), (3e') соединения дисульфид-тиола (XL) с соединением пиридилдисульфидным носителем (VI) с получением конъюгата производного антрациклина (Ib); и соединения дисульфидтиола (XL) с малеимидным носителем (V) с получением конъюгата производного антрациклина (Ib).
На Фигуре 7d показан путь синтеза промежуточного соединения лекарственного средства-линкера N-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]-L-валил-N 5-карбамоил-N-[4-({[(4-{[(2S,4S)-2,5,12-тригидрокси-7-метокси-4-{[(1S,3R,4aS,9S,9aR,10aS)-9-метокси-1-метилоктагидро-1H-пирано[4',3':4,5][1,3]оксазоло[2,3-c][1,4]оксазин-3-ил]окси}-6,11-диоксо-1,2,3,4,6,11-гексагидротетрацен-2-ил]карбонил}пиперазин-1-ил)карбонил]окси}метил)фенил]-L-орнитинамида 55
На Фигуре 8 показан график зависимости 3-дневной выживаемости клеток SK-BR-3 in vitro от нМ концентраций: непрерывного воздействия свободного лекарственного средства PNU-159682, 1-часового инкубирования с PNU-159682, NHS-кеталь-Ant 50, малеимид-кеталь-Ant 51, малеимид-гидразон-Ant 52, и тиопиридин-гидразон-Ant 53.
На Фигуре 9 показан график зависимости 3-дневной выживаемости клеток BT-474 in vitro от нМ концентраций: непрерывного воздействия свободного лекарственного средства PNU-159682, 1-часового инкубирования с PNU-159682, NHS-кеталь-Ant 50, малеимид-кеталь-Ant 51, малеимид-гидразон-Ant 52, и тиопиридин-гидразон-Ant 53.
На Фигуре 10 показан график зависимости 3-дневной выживаемости клеток BT-474 in vitro от нМ концентраций: непрерывного воздействия свободного лекарственного средства PNU-159682, 1-часового инкубирования с PNU-159682, NHS-кеталь-Ant 50, малеимид-кеталь-Ant 51, малеимид-гидразон-Ant 52, и тиопиридин-гидразон-Ant 53.
На Фигуре 11 показан график зависимости 3-дневной выживаемости клеток Her2, резистентных к доксорубицину (DoxRes) in vitro от нМ концентраций: непрерывного воздействия свободного лекарственного средства PNU-159682, 1-часового инкубирования с PNU-159682, NHS-кеталь-Ant 50, малеимид-кеталь-Ant 51, малеимид-гидразон-Ant 52 и тиопиридин-гидразон-Ant 53. Клеточная линия DoxRes Her2 также известна как «AdrRes Her2».
На Фигуре 12 показан график зависимости 3-дневной выживаемости клеток SK-BR-3 in vitro от концентрации: трастузумаба, трастузумаб-MCC-DM1 101, тио-трастузумаб (HC A114C)-малеимид-кеталь-Ant 102, тио-трастузумаб (HC A114C)-малеимид-гидразон-Ant 103 (нумерация тяжелой цепи антитела по схеме нумерации Кабата).
На Фигуре 13 показан график зависимости 3-дневной выживаемости клеток ВТ-474 in vitro от концентраций: трастузумаба, трастузумаб-MCC-DM1 101, тио-трастузумаб (HC A114C)-малеимид-кеталь-Ant 102, тио-трастузумаб (HC A114C)-малеимид-гидразон-Ant 103.
На Фигуре 14 показан график зависимости 3-дневной выживаемости клеток MCF-7 in vitro от концентраций: трастузумаба, трастузумаб-MCC-DM1 101, тио-трастузумаб (HC A114C)-малеимид-кеталь-Ant 102, тио-трастузумаб (HC A114C)-малеимид-гидразон-Ant 103.
На Фигуре 15 показан график зависимости 3-дневной выживаемости клеток Her2, резистентных к доксорубицину (DoxRes), in vitro от концентраций: трастузумаба, трастузумаб-MCC-DM1 101, тио-трастузумаб (HC A114C)-малеимид-кеталь-Ant 102, тио-трастузумаб (HC A114C)-малеимид-гидразон-Ant 103.
На Фигуре 16 показан график зависимости 3-дневной выживаемости клеток SK-BR-3 in vitro от концентраций: анти-CD22 NHS кеталь-Ant 110, трастузумаба, трастузумаб-MCC-DM1 101, тио-трастузумаб (HC A114C)-NHS-кеталь-Ant 105.
На Фигуре 17 показан график зависимости 3-дневной выживаемости клеток ВТ-474 in vitro от концентраций: анти-CD22 NHS кеталь-Ant 110, трастузумаба, трастузумаб-MCC-DM1 101, тио-трастузумаб (HC A114C)-NHS-кеталь-Ant 105.
На Фигуре 18 показан график зависимости 3-дневной выживаемости клеток MCF-7 in vitro от концентраций: анти-CD22 NHS кеталь-Ant 110, трастузумаба, трастузумаб-MCC-DM1 101, тио-трастузумаб (HC A114C)-NHS-кеталь-Ant 105.
На Фигуре 19 показан график зависимости 3-дневной выживаемости клеток Her2, резистентных к доксорубицину (DoxRes), in vitro от концентраций: анти-CD22 NHS кеталь-Ant 110, трастузумаба, трастузумаб-MCC-DM1 101, тио-трастузумаб (HC A114C)-NHS-кеталь-Ant 105.
На Фигуре 20 показан график зависимости 3-дневной выживаемости клеток SK-BR-3 in vitro от концентраций (мкг/мл): тио-трастузумаб (HC A114C)-малеимид-кеталь-Ant 102, тио-трастузумаб (HC A114C)-малеимид-гидразон-Ant 103, тио-трастузумаб (HC A114C)-тиопиридин-гидразон-Ant 104, тио-трастузумаб (HC A114C)-NHS-кеталь-Ant 105, трастузумаб-MCC-DM1 101, тио-трастузумаб (HC A114C)-MC-vc-PAB-MMAE 106.
На Фигуре 21 показан график зависимости 3-дневной выживаемости клеток SK-BR-3 in vitro от концентраций: трастузумаб, тио-анти-CD22 (HC A114C)-малеимид-кеталь-Ant 107, тио-анти-CD22 (HC A114C)-малеимид-гидразон-Ant 108, тио-анти-CD22 (HC A114C)-тиопиридин-гидразон-Ant 109, анти-CD22-NHS-кеталь-Ant 110, свободное лекарственное средство PNU-159682.
На Фигуре 22 показан график зависимости 3-дневной выживаемости клеток ВТ-474 in vitro от концентраций: тио-трастузумаб (HC A114C)-малеимид-кеталь-Ant 102, тио-трастузумаб (HC A114C)-малеимид-гидразон-Ant 103, тио-трастузумаб (HC A114C)-тиопиридин-гидразон-Ant 104, тио-трастузумаб (HC A114C)-NHS-кеталь-Ant 105, трастузумаб-MCC-DM1 101, тио-трастузумаб (HC A114C)-MC-vc-PAB-MMAE 106.
На Фигуре 23 показан график зависимости 3-дневной выживаемости клеток ВТ-474 in vitro от концентраций: трастузумаба, тио-анти-CD22 (HC A114C)-малеимид-кеталь-Ant 107, тио-анти-CD22 (HC A114C)-малеимид-гидразон-Ant 108, тио-анти-CD22 (HC A114C)-тиопиридин-гидразон-Ant 109, анти-CD22-NHS-кеталь-Ant 110, свободного лекарственного средства PNU-159682.
На Фигуре 24 показан график зависимости 3-дневной выживаемости клеток MCF-7 in vitro от концентраций: тио-трастузумаб (HC A114C)-малеимид-кеталь-Ant 102, тио-трастузумаб (HC A114C)-малеимид-гидразон-Ant 103, тио-трастузумаб (HC A114C)-тиопиридин-гидразон-Ant 104, тио-трастузумаб (HC A114C)-NHS-кеталь-Ant 105, трастузумаб-MCC-DM1 101, тио-трастузумаб (HC A114C)-MC-vc-PAB-MMAE 106.
На Фигуре 25 показан график зависимости 3-дневной выживаемости клеток MCF-7 in vitro от концентраций: трастузумаба, тио-анти-CD22 (HC A114C)-малеимид-кеталь-Ant 107, тио-анти-CD22 (HC A114C)-малеимид-гидразон-Ant 108, тио-анти-CD22 (HC A114C)-тиопиридин-гидразон-Ant 109, анти-CD22-NHS-кеталь-Ant 110, свободного лекарственного средства PNU-159682.
На Фигуре 26 показан график зависимости 3-дневной выживаемости клеток Her2, резистентных к доксорубицину (DoxRes), in vitro от концентраций: тио-трастузумаб (HC A114C)-малеимид-кеталь-Ant 102, тио-трастузумаб (HC A114C)-малеимид-гидразон-Ant 103, тио-трастузумаб (HC A114C)-тиопиридин-гидразон-Ant 104, тио-трастузумаб (HC A114C)-NHS-кеталь-Ant 105, трастузумаб-MCC-DM1 101, тио-трастузумаб (HC A114C)-MC-vc-PAB-MMAE 106.
На Фигуре 27 показан график зависимости 3-дневной выживаемости клеток Her2, резистентных к доксорубицину (DoxRes), in vitro от концентраций: трастузумаба, тио-анти-CD22 (HC A114C)-малеимид-кеталь-Ant 107, тио-анти-CD22 (HC A114C)-малеимид-гидразон-Ant 108, тио-анти-CD22 (HC A114C)-тиопиридин гидразон-Ant 109, анти-CD22-NHS-кеталь-Ant 110, свободного лекарственного средства PNU-159682.
На Фигуре 28 показан график зависимости 3-дневной выживаемости клеток Her2, резистентных к доксорубицину (DoxRes), in vitro от концентраций: тио-трастузумаб (HC A114C)-малеимид-кеталь-Ant 102, тио-трастузумаб (HC A114C)-малеимид-гидразон-Ant 103, тио-трастузумаб (HC A114C)-тиопиридин-гидразон-Ant 104, тио-трастузумаб (HC A114C)-NHS-кеталь-Ant 105, трастузумаб-MCC-DM1 101, тио-трастузумаб (HC A114C)-MC-vc-PAB-MMAE 106.
На Фигуре 29 показан график зависимости 3-дневной выживаемости клеток Her2, резистентных к доксорубицину (DoxRes), in vitro от концентраций: трастузумаба, тио-анти-CD22 (HC A114C)-малеимид-кеталь-Ant 107, тио-анти-CD22 (HC A114C)-малеимид-гидразон-Ant 108, тио-анти-CD22 (HC A114C)-тиопиридин гидразон-Ant 109, анти-CD22-NHS-кеталь-Ant 110, свободного лекарственного средства PNU-159682, все плюс верапамил.
На Фигуре 30 показан график изменения среднего объема опухоли с течением времени в ксенотрансплантатах опухолей лимфомы Беркитта Bjab-luc, инокулированных мышам CB17 SCID, после однократного внутривенного (в/в) введения в 0 день: (1) носителя, (2) тио-анти-CD22 (HC A114C)-MC-vc-PAB-MMAE 111 1 мг/кг, (3) тио-трастузумаб (HC A114C)-малеимид-кеталь-Ant 102 1 мг/кг, (4) тио-трастузумаб (HC A114C)-малеимид-кеталь-Ant 102 5 мг/кг, (5) тио-анти-CD22 (HC A114C)-малеимид-кеталь-Ant 107 1 мг/кг, (6) тио-анти-CD22 (HC A114C)-малеимид-кеталь-Ant 107 5 мг/кг, (7) тио-трастузумаб (HC A114C)-малеимид-гидразон-Ant 103 1 мг/кг, (8) тио-анти-CD22 (HC A114C)-малеимид-гидразон-Ant 108 1 мг/кг, (9) свободного лекарственного средства PNU-159682 8,77 мкг/кг (воздействие 26 мкг/м2), соответствующей дозе лекарственного средства 1 мг/кг конъюгата антитело-лекарственное средство.
На Фигуре 31 показан график изменения среднего объема опухоли с течением времени в MMTV-HER2 Fo5 аллотрансплантированных опухолях млекопитающих, инокулированных мышам CRL nu/nu после однократного в/в введения в 0 день: (1) носителя, (2) трастузумаб-MCC-DM1 101 5 /мг/кг, (3) трастузумаб-MCC-DM1 101 10 мг/кг, (4) тио-трастузумаб (HC A114C)-малеимид-кеталь-Ant 102 5 мг/кг, (5) тио-трастузумаб (HC A114C)-малеимид-кеталь-Ant 102 10 мг/кг, (6) тио-анти-CD22 (HC A114C)-малеимид-кеталь-Ant 107 5 мг/кг, (7) тио-анти-CD22 (HC A114C)-малеимид-кеталь-Ant 107 10 мг/кг, (8) трастузумаб-MCC-DM1 101 5 мг/кг + тио-трастузумаб (HC A114C)-малеимид-кеталь-Ant 102 5 мг/кг.
На Фигуре 32 показан график изменения среднего объема опухоли с течением времени в ксенотрансплантированных опухолях LnCap-Ner, инокулированных самцам мышей SCID-beige, после однократного в/в введения в 0 день: (1) носителя, (2) тио-анти-steap1 (HC A114C)-MC-vc-PAB-MMAE 112 1 мг/кг, (3) тио-анти-steap1 (HC A114C)-MC-vc-PAB-MMAE 112 3 мг/кг, (4) тио-анти-steap1 (HC A114C)-малеимид-кеталь-Ant 113 1 мг/кг, (5) тио-анти-steap1 (HC A114C)-малеимид-кеталь-Ant 113 3 мг/кг, (6) тио-анти-steap1 (HC A114C)-малеимид-кеталь-Ant 113 6 мг/кг, (7) тио-анти-CD22 (HC A114C)-малеимид-кеталь-Ant 107 1 мг/кг, (8) тио-анти-CD22 (HC A114C)-малеимид-кеталь-Ant 107 3 мг/кг, (9) тио-анти-CD22 (HC A114C)-малеимид-кеталь-Ant 107 6 мг/кг.
ПОДРОБНОЕ ОПИСАНИЕ ВАРИАНТОВ ОСУЩЕСТВЛЕНИЯ, ПРИВОДИМЫХ В КАЧЕСТВЕ ПРИМЕРА
Будет дана подробная ссылка на некоторые варианты осуществления изобретения, примеры которых проиллюстрированы в сопровождающих структурах и формулах. Хотя настоящее изобретение будет описано в отношении приведенных вариантов осуществления, будет понятно, что они не предназначены для ограничения настоящего изобретения этими вариантами осуществления. Напротив, настоящее изобретение предназначено для охвата всех альтернатив, модификаций и эквивалентов, которые могут быть включены в объем настоящего изобретения, определенного формулой изобретения. Специалисту в данной области будут понятны многочисленные способы и материалы, аналогичные или эквивалентные тем, которые описаны в настоящем документе, которые могли быть использованы при практическом осуществлении настоящего изобретения. Настоящее изобретение никаким образом не ограничивается описанными способами и материалами. Если не определено иное, технические и научные термины, используемые в настоящей заявке, имеют то же значение, которое является общепринятым у специалистов в области, к которой относится настоящее изобретение, и согласуются с: Singleton et al., (1994) Dictionary of Microbiology и Molecular Biology, 2nd Ed., J. Wiley & Sons, New York, NY; и Janeway, C., Travers, P., Walport, M., Shlomchik (2001) Immuno Biology, 5th Ed., Garland Publishing, New York.
ОПРЕДЕЛЕНИЯ
Если не указано иное, следующие термины и выражения, используемые в настоящем документе, имеют следующие значения.
При использовании торговых наименований, заявители независимо включают состав продукта торгового наименования, лекарственное средство дженерик и активный фармацевтический ингредиент(ы) продукта торгового наименования.
«Производное антрациклина» представляет собой метаболит неморубицина, или аналогичное соединение, включая, но не только, PNU-159682.
«Конъюгат производного антрациклина» представляет собой соединение, состоящее из производного антрациклина, ковалентно присоединенного через линкер к группе носителю, содержащей антитела, белки или пептиды. Конъюгат производного антрациклина включает соединения конъюгата антитела с лекарственным средством (ADC).
Термин «боковая цепь аминокислоты» включает группы, находящиеся в: (i) природных аминокислотах, таких как аланин, аргинин, аспарагин, аспарагиновая кислота, цистеин, глутамин, глутаминовая кислота, глицин, гистидин, изолейцин, лейцин, лизин, метионин, фенилаланин, пролин, серин, треонин, триптофан, тирозин и валин; (ii) второстепенных аминокислотах, таких как орнитин и цитруллин; (iii) неприродных аминокислотах, бета-аминокислотах, синтетических аналогах и производных природных аминокислотах; и (iv) все энантиомеры, диастереомеры, изомерно обогащенные, изотопно меченные (например, 2H, 3H, 14C, 15N), защищенные формы и их рацемические смеси.
Термин «антитело» в контексте настоящего изобретения используется в самом широком смысле и, в частности, включает в себя моноклональные антитела, поликлональные антитела, димеры, мультимеры, мультиспецифичные антитела (например, биспецифические антитела), и фрагменты антител, при условии, что они проявляют желаемую биологическую активность (Miller et al. (2003) Jour. of Immunology 170:4854-4861). Антитела могут представлять собой антитела мыши, человека, могут быть гуманизированными, химерными или получены из других видов. Антитело представляет собой белок, продуцируемый иммунной системой, который способен распознавать и связываться со специфическим антигеном (Janeway, C., Travers, P., Walport, M., Shlomchik (2001) Immuno Biology, 5th Ed., Garland Publishing, New York). Антиген-мишень в основном имеет многочисленные сайты связывания, также называемые эпитопами, распознаваемые CDR многочисленных антител. Каждое антитело, которое специфически связывается с другим эпитопом, имеет другую структуру. Таким образом, один антиген может иметь более одного соответствующего ему антитела. Антитело включает полноразмерную молекулу антитела или иммунологически активную часть полноразмерной молекулы иммуноглобулина, то есть, молекулы, которая содержит антиген-связывающий сайт, который иммуноспецифически связывается антигеном мишени, представляющей интерес, или его частью, такие мишени включают, но не только, злокачественную клетку или клетки, которые продуцируют аутоиммунные антитела, связанные с аутоиммунным заболеванием. Иммуноглобулин может быть любого типа (например, IgG, IgE, IgM, IgD и IgA), класса (например, IgG1, IgG2, IgG3, IgG4, IgA1 и IgA2) или подкласса молекулы иммуноглобулина. Иммуноглобулины могут быть получены из любых видов, включая человека, мышь или кролика.
«Фрагменты антитела» содержат часть полноразмерного антитела, в основном его антиген-связывающую или вариабельную область. Примеры фрагментов антитела включают фрагменты Fab, Fab', F(ab')2 и Fv; диатела; линейные антитела; фрагменты, продуцированные экспрессионной библиотекой Fab, анти-идиотипные (анти-Id) антитела, CDR (область, определяющую комплементарность) и эпитоп-связывающие фрагменты любого из указанного выше, которые иммуноспецифически связываются с антигенами злокачественной клетки, вирусными антигенами или микробными антигенами, молекулами одноцепочечных антител; и мультиспецифичные антитела, образованные из фрагментов антител.
Термин «моноклональное антитело», используемый в настоящем документе, относится к антителу, полученному из популяции по существу гомогенных антител, то есть индивидуальные антитела, составляющие эту популяцию, являются идентичными, за исключением возможных природных мутаций, которые могут присутствовать в незначительных количествах. Моноклональные антитела являются высокоспецифичными, направленными против одного антигенного сайта. Более того, в отличие от препаратов поликлональных антител, которые включают различные антитела, направленные против различных детерминант (эпитопов), каждое моноклональное антитело направлено против одной детерминанты на антигене. Помимо их специфичности, моноклональные антитела являются предпочтительными тем, что они могут быть синтезированы без примесей других антител. Определение «моноклональное» указывает на характер антитела, полученного по существу из гомогенной популяции антител, и не интерпретируется как требующее получения антитела каким-либо конкретным способом. Например, моноклональные антитела, используемые в соответствии с настоящим изобретением, могут быть получены гибридомным способом, впервые описанным Kohler et al. (1975) Nature 256:495, или могут быть получены способами рекомбинантных ДНК (смотри, US 4816567). Моноклональные антитела также могут быть выделены из фаговых библиотек антител, используя методики, описанные у Clackson et al. (1991) Nature, 352:624-628; Marks et al. (1991) J. Mol. Biol., 222:581-597.
Моноклональные антитела в настоящем изобретении специфически включают «химерные» антитела, в которых часть тяжелой и/или легкой цепи идентична или гомологична соответствующим последовательностям в антителах, полученных из конкретных видов или принадлежащих конкретному классу антител или подклассу антител, тогда как остаток цепи (цепей) идентичен или гомологичен соответствующим последовательностям в антителах, полученных из других видов или принадлежащим другому классу или подклассу антител, а также фрагменты таких антител, при условии, что они демонстрируют желаемую биологическую активность (US 4816567; и Morrison et al. (1984) Proc. Natl. Acad. Sci. USA, 81:6851-6855). Химерные антитела включают «приматизированные» антитела, содержащие вариабельный домен антиген-связывающих последовательностей, полученных от примата, не являющегося человеком (например, обезьяны Старого Света или Ape), и последовательностей константной области человека.
«Интактное антитело» в настоящей заявке представляет собой антитело, содержащее домены VL и VH, а также константный домен легкой цепи (CL) и константные домены тяжелой цепи, CH1, CH2 и CH3. Константные домены могут представлять собой нативную последовательность константных доменов (например, нативную последовательность константных доменов человека) или вариант их аминокислотной последовательности. Интактное антитело может иметь одну или несколько «эффекторных функций», которые относятся к их биологическим активностям, свойственным области Fc (нативной последовательности области Fc или варианту аминокислотной последовательности области Fc) антитела. Примеры эффекторных функций антитела включают связывание C1q; комплемент-зависимую цитотоксичность; Fc-рецепторное связывание; антитело-зависимую клеточно-опосредованную цитотоксичность (ADCC); фагоцитоз и отрицательную регуляцию рецепторов клеточной поверхности, таких как В-клеточный рецептор и BCR.
В зависимости от аминокислотной последовательности константного домена их тяжелых цепей, интактные антитела могут быть отнесены к различным «классам». Существует пять основных классов интактных антител: IgA, IgD, IgE, IgG, и IgM, и некоторые из них дополнительно могут быть разделены на «подклассы» (изотипы), например, IgG1, IgG2, IgG3, IgG4, IgA, и IgA2. Константные домены тяжелой цепи, которые соответствуют различным классам антител, называются α, δ, ε, γ, и μ, соответственно. Структуры субъединиц и трехмерные конфигурации различных классов иммуноглобулинов хорошо известны.
«ErbB рецептор» представляет собой рецепторный белок тирозинкиназу, который принадлежит семейству рецепторов ErbB, которые являются важными медиаторами роста клеток, дифференцировки и выживания. Семейство рецепторов ErbB включает четыре различных представителя, включая рецептор эпидермального фактора роста (EGFR, ErbB1, HER1), HER2 (ErbB2 или p185neu), HER3 (ErbB3) и HER4 (ErbB4 или tyro2). Рецептор ErbB в основном будет содержать внеклеточный домен, который может связываться с ErbB лигандом; липофильный трансмембранный домен; консервативный внутриклеточный домен тирозинкиназы и карбокси-концевой сигнальный домен, содержащий несколько остатков тирозина, которые могут быть фосфорилированы. Рецептор ErbB может быть рецептором ErbB «нативной последовательности» или «вариантом его аминокислотной последовательности». Рецептор ErbB может представлять собой нативную последовательность ErbB рецептора человека. Соответственно, «представителем семейства рецепторов ErbB» является EGFR (ErbB1), ErbB2, ErbB3, ErbB4 или любой другой ErbB рецептор, известный в настоящее время или идентифицированный в будущем. Скрининг идентичности последовательностей привел к идентификации двух других представителей семейства рецепторов ErbB; ErbB3 (US 5183884; US 5480968; Kraus et al. (1989) PNAS (USA) 86:9193-9197) и ErbB4 (EP 599274; Plowman et al. (1993) Proc. Natl. Acad. Sci. USA, 90:1746-1750; и Plowman et al. (1993) Nature 366:473-475). Оба этих рецептора демонстрируют повышенную экспрессию по меньшей мере на нескольких клеточных линиях рака груди. Были охарактеризованы антитела против ErbB2 (US 5677171; US 5821337; US 6054297; US 6165464; US 6407213; US 6719971; US 6800738; Fendly et al. (1990) Cancer Research 50:1550-1558; Kotts et al. (1990) In Vitro 26(3):59A; Sarup et al. (1991) Growth Regulation 1:72-82; Shepard et al. J. (1991) Clin. Immunol. 11(3):117-127; Kumar et al. (1991) Mol. Cell. Biol. 11(2):979-986; Lewis et al. (1993) Cancer Immunol. Immunother. 37:255-263; Pietras et al. (1994) Oncogene 9:1829-1838; Vitetta et al. (1994) Cancer Research 54:5301-5309; Sliwkowski et al. (1994) J. Biol. Chem. 269(20):14661-14665; Scott et al. (1991) J. Biol. Chem. 266:14300-5; D'souza et al. Proc. Natl. Acad. Sci. (1994) 91:7202-7206; Lewis et al. (1996) Cancer Research 56:1457-1465; и Schaefer et al. (1997) Oncogene 15:1385-1394.
«Гуманизированные» формы антител, не являющихся антителами человека (например, грызунов), представляют собой химерные антитела, которые содержат минимальную последовательность, полученную из иммуноглобулина, не являющегося иммуноглобулином человека. Для наибольшей части, гуманизированные антитела представляют собой иммуноглобулины человека (реципиентное антитело), в которых остатки из гипервариабельной области реципиента заменены остатками из гипервариабельной области видов, не относящихся к человеку (донорское антитело), таких как мышь, крыса, кролик или нечеловекообразный примат, обладающее желаемой специфичностью, аффинностью и емкостью. В некоторых случаях остатки каркасной области (FR) иммуноглобулина человека замещаются соответствующими остатками, не относящимися к человеку. Более того, гуманизированные антитела могут содержать остатки, которые не находятся в реципиентном антителе или в донорском антителе. Эти модификации делают для выполнения дополнительной очистки антитела. В основном, гуманизированное антитело будет содержать по существу все, по меньшей мере из одного, и обычно двух, вариабельных доменов, в которых все или по существу все гипервариабельные петли соответствуют таковым иммуноглобулина, не являющегося иммуноглобулином человека, и все, или по существу все из FR представляют собой последовательность иммуноглобулина человека. Гуманизированное антитело необязательно также будет содержать по меньшей мере часть константной области иммуноглобулина (Fc), обычно иммуноглобулина человека (Jones et al. (1986) Nature, 321:522-525; Riechmann et al. (1988) Nature 332:323-329; и Presta, (1992) Curr. Op. Struct. Biol., 2:593-596). Гуманизированные антитела против ErbB2 включают huMAb4D5-1, huMAb4D5-2, huMAb4D5-3, huMAb4D5-4, huMAb4D5-5, huMAb4D5-6, huMAb4D5-7 и huMAb4D5-8 (HERCEPTIN®, трастузумаб), описанные в таблице 3 US 5821337, специально включенной в настоящую заявку в качестве ссылки; гуманизированные 520C9 (WO 93/21319) и гуманизированные 2C4 антитела.
Термины «терапия» и «лечение» относятся как к терапевтическому лечению, так и к профилактическим или превентивным мерам, при которых целью является предупреждение или замедление (уменьшение) нежелательного физиологического изменения или нарушения, например, развития или распространения злокачественного заболевания. В целях данного изобретения, благоприятные или желательные клинические результаты включают, но этим не ограничиваются, облегчение симптомов, уменьшение степени заболевания, стабилизированное (то есть, без ухудшения) состояние заболевания, задержку или замедление прогрессирования заболевания, улучшение состояния или временное облегчение патологического состояния и ремиссию (частичную или полную), определяемую или неопределяемую. «Лечение» также означает продление выживаемости по сравнению с ожидаемой выживаемостью, без получения лечения. Нуждающиеся в лечении включают тех, у которых уже имеется это состояние или нарушение, а также тех, кто подвержен такому состоянию или нарушению, или тех, у которых это состояние или нарушение подлежит профилактике.
«Нарушение» представляет собой любое состояние, которое улучшилось бы в результате лечения в соответствии с настоящим изобретением. Это включает хронические и острые нарушения или заболевания, в том числе те патологические состояния, которые предрасполагают млекопитающее к рассматриваемому нарушению. Неограничивающие примеры нарушений, подвергаемых лечению, в настоящей заявке включают доброкачественные и злокачественные опухоли; лейкоз и лимфоидные злокачественные заболевания, в частности, рак груди, яичников, желудка, эндометрия, слюнных желез, легких, почек, толстой кишки, щитовидной железы, поджелудочной железы, предстательной железы или мочевого пузыря; нейрональные, глиальные, астроцитарные, гипоталамические и другие железистые, макрофагальные, эпителиальные, стромальные и бластоцельные нарушения; и воспалительные, ангиогенные и иммунологические нарушения. Приводимым в качестве примера нарушением, подвергаемым лечению в соответствии с настоящим изобретением, является солидная злокачественная опухоль.
Термин «терапевтически эффективное количество» относится к количеству лекарственного средства, эффективному для лечения заболевания или нарушения у млекопитающего. В случае злокачественного заболевания, терапевтически эффективное количество лекарственного средства может: (i) уменьшать число злокачественных клеток; (ii) уменьшать размер опухоли; (iii) ингибировать, задерживать, замедлять до некоторой степени и предположительно останавливать инфильтрацию злокачественных клеток в периферические органы; (iv) ингибировать (то есть, замедлять до некоторой степени и предпочтительно останавливать) метастазирование опухоли; (v) ингибировать рост опухоли; и/или (vi) облегчать до некоторой степени один или несколько симптомов, связанных со злокачественным заболеванием. До некоторой степени лекарственное средство может предотвращать рост и/или уничтожать существующие злокачественные клетки, это может быть цитостатическим и/или цитотоксическим. В моделях на животных, эффективность может оцениваться физическими измерениями опухоли во время курсового введения ADC, и путем определения частичной или полной ремиссии опухоли. Для лечения злокачественного заболевания, эффективность, например, может быть измерена путем оценки времени до прогрессирования заболевания (TTP) и/или определения скорости ответа (RR).
Термин «биодоступность» относится к системной доступности (то есть, уровни в крови/плазме) заданного количества лекарственного средства, введенного пациенту. Биодоступность является абсолютным термином, который указывает измерение как времени (скорости), так и количества (меры) лекарственного средства, которое достигает общего кровотока из введенной лекарственной формы.
Термины «злокачественное заболевание» и “злокачественный” относятся к или описывают физиологическое состояние у млекопитающих, которые обычно характеризуются нерегулируемым ростом клеток. «Опухоль» включает одну или несколько злокачественных клеток. Примеры злокачественных заболеваний включают, но этим не ограничиваются, карциному, лимфому, бластому, саркому и лейкоз или лимфоидные злокачественные заболевания. Более конкретные примеры таких злокачественных заболеваний включают сквамозно-клеточный рак (например, эпителиальный сквамозно-клеточный рак), рак легких, включая мелко-клеточный рак легких, немелкоклеточный рак легких («NSCLC»), аденокарциному легких и сквамозную карциному легких, рак брюшины, печеночноклеточный рак, рак желудка или пищеварительного тракта, включая гастроинтестинальный рак, гастроинтестинальную стромальную опухоль (GIST), рак поджелудочной железы, глиобластому, рак шейки матри, рак яичников, рак печени, рак мочевого пузыря, гепатому, рак груди, рак толстой кишки, рак прямой кишки, рак толстой и прямой кишки, карциному эндометрия или матки, карциному слюнных желез, рак почек, рак предстательной железы, рак вульвы, рак щитовидной железы, карциному печени, карциному ануса, карциному пениса, а также рак тканей головы и шеи.
«Злокачественная опухоль, экспрессирующая ErbB» представляет собой опухоль, содержащую клетки, у которых имеется белок ErbB, находящийся на их клеточной поверхности. «Злокачественная опухоль, экспрессирующая ErbB2», представляет собой опухоль, которая продуцирует достаточные уровни ErbB2 на поверхности клетки, так, что антитело против ErbB2 может с ним связываться и оказывать терапевтический эффект в отношении этой злокачественной опухоли.
Злокачественная опухоль, которая «сверхэкспрессирует» рецептор, например рецептор ErbB, представляет собой опухоль, которая имеет значительно более высокие уровни рецептора, например ErbB2, на клеточной поверхности, по сравнению с незлокачественной клеткой того же типа ткани. Такая сверхэкспрессия может быть вызвана амплификацией гена или повышенной транскрипцией или трансляцией. Сверхэкспрессия рецепторов может быть определена в диагностическом или прогностическом исследовании путем оценки повышенных уровней рецепторного белка, находящегося на поверхности клетки (например, посредством иммуногистохимического анализа; IHC). Альтернативно, или дополнительно, можно измерить уровни нуклеиновой кислоты, кодирующей рецептор, в клетке, например, с помощью флуоресценции в гибридизации in situ (FISH; см. WO 98/45479), саузерн блоттинге или методиках полимеразной цепной реакции (ПЦР), например, количественной ПЦР в режиме реального времени (RT-PCR). Сверхэкспрессию рецепторного лиганда можно определить диагностически путем оценки уровней этого лиганда (или нуклеиновой кислоты, кодирующей его) у пациента, например, при биопсии опухоли, или с помощью различных диагностических исследований, таких как IHC, FISH, саузерн блоттинг, ПЦР или in vivo анализов, описанных выше. Также можно исследовать сверхэкспрессию рецепторов, путем измерения «слущивающегося» антигена (например, внеклеточного домена ErbB) в биологической жидкости, например, в сыворотке (см., например, US 4933294; WO 91/05264; US 5401638; и Sias et al. (1990) J. Immunol. Methods 132: 73-80). Помимо приведенных выше анализов, специалисту, практикующему в данной области, доступны различные другие исследования in vivo. Например, можно подвергать клетки в организме пациента воздействию антитела, которое необязательно мечено детектируемой меткой, например, радиоактивным изотопом, и можно оценивать связывание этого антитела с клетками в организме пациента, например, путем наружного сканирования радиоактивности или анализируя материал биопсии, взятый у пациента до воздействия на него антитела.
Термин «цитотоксическое средство», используемый в настоящем документе, относится к веществу, которое ингибирует или предотвращает функционирование клеток и/или вызывает разрушение клеток. Термин включает радиоактивные изотопы (например, 211At, 131I, 125I, 90Y, 186Re, 188Re, 153Sm, 212Bi, 32P, 60C, и радиоактивные изотопы Lu), химиотерапевтические средства и токсины, такие как мелкомолекулярные токсины или ферментативно активные токсины бактериального, грибного, растительного или животного происхождения, включая их синтетические аналоги и производные.
«Химиотерапевтическое средство» представляет собой химическое соединение, применимое для лечения злокачественного заболевания, независимо от механизма действия. Классы химиотерапевтических средств включают, но этим не ограничиваются: алкилирующие агенты, антиметаболиты, растительные алкалоиды, представляющие собой веретенный яд, цитотоксические/противоопухолевые антибиотики, ингибиторы топоизомеразы, антитела, фотосенсибилизаторы и ингибиторы киназ. Химиотерапевтические средства включают соединения, используемые в «направленной терапии» и традиционной химиотерапии. Примеры химиотерапевтических средств включают: эрлотиниб (TARCEVA®, Genentech/OSI Pharm.), доцетаксел (TAXOTERE®, Sanofi-Aventis), 5-FU (фторурацил, 5-фторурацил, CAS No. 51-21-8), гемцитабин (GEMZAR®, Lilly), PD-0325901 (CAS No. 391210-10-9, Pfizer), цисплатин (цис-диамин, дихлорплатина (II), CAS No. 15663-27-1), карбоплатин (CAS No. 41575-94-4), паклитаксел (TAXOL®, Bristol-Myers Squibb Oncology, Princeton, N.J.), трастузумаб (HERCEPTIN®, Genentech), темозоломид (4-метил-5-оксо-2,3,4,6,8-пентазабицикло [4.3.0] нона-2,7,9-триен-9-карбоксамид, CAS No. 85622-93-1, TEMODAR®, TEMODAL®, Schering Plough), тамоксифен ((Z)-2-[4-(1,2-дифенилбут-1-енил)фенокси]-N,N-диметилэтанамин, NOLVADEX®, ISTUBAL®, VALODEX®), и доксорубицин (АДРИАМИЦИН®), Akti-1/2, HPPD, и рапамицин.
Большинство примеров химиотерапевтических средств включает: оксалиплатин (ELOXATIN®, Sanofi), бортезомиб (VELCADE®, Millennium Pharm.), сутент (SUNITINIB®, SU11248, Pfizer), летрозол (FEMARA®, Novartis), иматиниб мезилат (GLEEVEC®, Novartis), XL-518 (ингибитор Mek, Exelixis, WO 2007/044515), ARRY-886 (ингибитор Mek, AZD6244, Array BioPharma, Astra Zeneca), SF-1126 (ингибитор PI3K, Semafore Pharmaceuticals), BEZ-235 (ингибитор PI3K, Novartis), XL-147 (ингибитор PI3K, Exelixis), PTK787/ZK 222584 (Novartis), фулвестрант (FASLODEX®, AstraZeneca), лейковорин (фолиновая кислота), рапамицин (сиролимус, RAPAMUNE®, Wyeth), лапатиниб (TYKERB®, GSK572016, Glaxo Smith Kline), лонафарниб (SARASAR™, SCH 66336, Schering Plough), сорафениб (NEXAVAR®, BAY43-9006, Bayer Labs), гефитиниб (IRESSA®, AstraZeneca), иринотекан (CAMPTOSAR®, CPT-11, Pfizer), типифарниб (ZARNESTRA™, Johnson & Johnson), ABRAXANE™ (Кремофор- фри), альбуминовые наночастицы паклитаксела (American Pharmaceutical Partners, Schaumberg, Il), вандетаниб (rINN, ZD6474, ZACTIMA®, AstraZeneca), хлоранбуцил, AG1478, AG1571 (SU 5271; Sugen), темсиролимус (TORISEL®, Wyeth), пазопаниб (GlaxoSmithKline), канфосфамид (TELCYTA®, Telik), тиотепа и циклофосфамид (CYTOXAN®, NEOSAR®); алкилсульфонаты, такие как бусульфан, импросульфан и пипосульфан; азиридины, такие как бензодопа, карбоквон, метуредопа и уредопа; этиленимины и метилмеламины, включая алтретамин, триэтиленмеламин, триэтиленфосфорамид, триэтилентиофосфорамид и триметилмеламин; ацетогенины (главным образом буллатацин и буллатацинон); камптотецин (включая синтетический аналог топотекан); бриостатин; каллистатин; CC-1065 (включая его синтетические аналоги адозелезин, карзелезин, и бизелезин); криптофицины (главным образом криптофицин 1 и криптофицин 8); доластатин; дуокармицин (включая синтетические аналоги KW-2189 и CB1-TM1); элеутеробин; панкратистатин; саркодиктин; спонгистатин; нитроиприты, такие как хлорамбуцил, хлорнафазин, хлорфосфамид, эстрамустин, ифосфамид, мехлорэтамин, мехлоэтамин оксид гидрохлорид, мелфалан, новембихин, фенестерин, преднимустин, трофосфамид, урамустин; нитрозомочевины, такие как кармустин, хлорозотоцин, фотемустин, ломустин, нимустин и ранимустин; антибиотики, такие как энедииновые антибиотики (например, калихеамицин, калихеамицин гамма1I, калихеамицин омега I1 (Angew Chem. Intl. Ed. Engl. (1994) 33:183-186); динемицин, динемицин A; бисфосфонаты, такие как клодронат; эсперамицин; так же как и неокарциностатиновый хромофор и родственные хромофоры - хромопротеиновые энедииновые антибиотики), аклациномицины, актиномицин, аутрамицин, азасерин, блеомицины, кактиномицин, карабицин, карминомицин, карзинофилин, хромомицины, дактиномицин, даунорубицин, деторубицин, 6-диазо-5-оксо-L-норлейцин, морфолино-доксорубицин, цианоморфолино-доксорубицин, 2-пирролино-доксорубицин и дезоксидоксорубицин, эпирубицин, эзорубицин, идарубицин, марцелломицин, митомицины, такие как митомицин C, микофеноловая кислота, ногаламицин, оливомицины, пепломицин, порфиромицин, пуромицин, квеламицин, родорубицин, стрептонигрин, стрептозоцин, туберцидин, убенимекс, зиностатин, зорубицин; антиметаболиты, такие как метотрексат и 5-фторурацил (5-FU); аналоги фолиевой кислоты, такие как деноптерин, метотрексат, птероптерин, триметрексат; аналоги пурина, такие как флударабин, 6-меркаптопурин, тиамиприн, тиогуанин; аналоги пиримидина, такие как анцитабин, азацитидин, 6-азауридин, кармофур, цитарабин, дидезоксиуридин, доксифлуридин, эноцитабин, флоксуридин; андрогены, такие как калустерон, дромостанолон пропионат, эпитиостанол, мепитиостан, тестолактон; антиадреналиновые препараты, такие как аминоглутетимид, митотан, трилостан; компенсатор фолиевой кислоты, такой как фролиновая кислота; ацеглатон; альдофосфамид гликозид; аминолевулиновая кислота; энилурацил; амсакрин; бестрабуцил; бисантрен; эдатраксат; дефофамин; демеколцин; диазиквон; элфорнитин; эллиптиниум ацетат; эпотилон; этоглюцид; нитрат галлия; гидроксимочевина; лентинан; лонидаинин; майтансиноиды, такие как майтасин и ансамитоцины; митогуазон; митоксантрон; мопиданмол; нитракрин; пентостатин; фенамет; пирарубицин; лосоксантрон; подофиллиновая кислота; 2-этилгидразид; прокарбазин; PSK® полисахаридный комплекс (JHS Natural Products, Eugene, OR); разоксан; ризоксин; сизофиран; спирогерманий; тенуазоновая кислота; триазиквон; 2,2',2"-трихлортриэтиламин; трихотецены (особенно T-2 токсин, верракурин A, роридин A и ангуидин); уретан; виндезин; дакарбазин; манномустин; митобронитол; митолактол; пипоброман; гацитозин; арабинозид («Ara-C»); циклофосфамид; тиотепа; 6-тиогуанин; меркаптопурин; метотрексат; аналоги платины, такие как цисплатин и карбоплатин; винбластин; этопозид (VP-16); ифосфамид; митоксантрон; винкристин; винорелбин (NAVELBINE®); новантрон; тенипозид; эдатрексат; дауномицин; аминоптерин; капецитабин (XELODA®, Roche); ибандронат; CPT-11; ингибитор топоизомеразы RFS 2000; дифторметилорнитин (DMFO); ретиноиды, такие как ретиноевая кислота; и фармацевтически приемлемые соли, кислоты и производные любого из указанных выше соединений.
Также включены в определение «химиотерапевтического средства»: (i) противогормональные средства, которые действуют для регуляции или ингибирования действия гормонов на опухоли, такие как анти-эстрогены и селективные модуляторы рецепторов эстрогенов (SERM), в том числе, например, тамоксифен (включая NOLVADEX®; тамоксифена цитрат), ралоксифен, дролоксифен, 4-гидрокситамоксифен, триоксифен, кеоксифен, LY117018, онапристон, и FARESTON® (торемифина цитрат); (ii) ингибиторы ароматазы, которые ингибируют фермент ароматазу, которая регулирует продукцию эстрогенов в надпочечниках, такие как, например, 4(5)-имидазолы, аминоглутетимид, MEGASE® (мегестрола ацетат), AROMASIN® (экземестан; Pfizer), форместан, фадрозол, RIVISOR® (ворозол), FEMARA® (летрозол; Novartis), и ARIMIDEX® (анастрозол; AstraZeneca); (iii) анти-андрогены, такие как флутамид, нилутамид, бикалутамид, лейпролид, и гозерелин; а также троксацитабин (1,3-диоксолан аналог нуклеозида цитозин); (iv) ингибиторы протеинкиназы, такие как MEK ингибиторы (WO 2007/044515); (v) ингибиторы липидкиназа; (vi) антисмысловые олигонуклеотиды, в частности те, которые ингибирут экспрессию генов в сигнальных путях, вовлеченных в аберрантную клеточную пролиферацию, например, PKC-альфа, Raf и H-Ras, такие как облимерсен (GENASENSE®, Genta Inc.); (vii) рибозимы, такие как ингибиторы экспрессии VEGF (например, ANGIOZYME®) и ингибиторы экспрессии HER2; (viii) вакцины, такие как вакцины для генной терапии, например, ALLOVECTIN®, LEUVECTIN®, и VAXID®; PROLEUKIN® rIL-2; ингибиторы топоизомеразы 1, такие как LURTOTECAN®; ABARELIX® rmRH; (ix) анти-ангиогенные средства, такие как бевацизумаб (AVASTIN®, Genentech); и фармацевтически приемлемые соли, кислоты и производные любого из указанного выше.
Также включены в определение «химиотерапевтического средства» терапевтические антитела, такие как алемтузумаб (Campath), бевацизумаб (AVASTIN®, Genentech); цетуксимаб (ERBITUX®, Imclone); панитумумаб (VECTIBIX®, Amgen), ритуксимаб (RITUXAN®, Genentech/Biogen Idec), пертузумаб (OMNITARG™, 2C4, Genentech), трастузумаб (HERCEPTIN®, Genentech), тозитумомаб (Bexxar, Corixia), и конъюгаты антитела с лекарственным средством, гемузумаб озогамицин (MYLOTARG®, Wyeth).
Гуманизированные моноклональные антитела с терапевтической эффективностью в качестве химиотерапевтических средств в сочетании с ингибиторами PI3K по изобретению включают: алемтузумаб, аполизумаб, азелизумам, атлизумаб, бапинейзумаб, бевацизумаб, биватузумаб мертанзин, кантузумаб мертанзин, цеделизумаб, цертолизумаб пегол, цидфузитузумаб, цидтузумаб, даслизумаб, экулизумаб, эфализумаб, эпратузумаб, эрлизумаб, фелвизумаб, фонтолизумаб, гемтузумаб озогамицин, инотузумаб озогамицин, ипиламумаб, лабетузумаб, линтузумаб, матузумаб, меполизумаб, мотавизумаб, мотовизумаб, натализумаб, нимотузумаб, ноловизумаб, нумавизумаб, окрелизумаб, омализумаб, паливизумаб, пасколизумаб, пекфузитузумаб, пектузумаб, пертузумаб, пекселизумаб, раливизумаб, ранибизумаб, ресливизумаб, реслизумаб, ресивизумаб, ровелизумаб, руплизумаб, сибротузумаб, сиплизумаб, сонтузумаб, такатузумаб тетраксетан, тадоцизумаб, тализумаб тефибазумаб, тоцилизумаб, торализумаб, трастузумаб, тукотузумаб, целмолейкин, тукуситузумаб, умавизумаб, уртоксазумаб и визилизумаб.
Термин «лист-вкладыш» используется в отношении к инструкциям, обычно включенным в коммерческие упаковки терапевтических продуктов, которые содержат информацию о показаниях, применении, дозе, введении, противопоказаниях и/или предупреждениях, касающихся применению таких терапевтических продуктов.
«Алкил» представляет собой C1-C8 углеводород, содержащий номальные, вторичные, третичные или циклические атомы углерода. Примеры алкильных радикалов включают, но этим не ограничиваются: метил (Me, -CH3), этил (Et, -CH2CH3), 1-пропил (n-Pr, н-пропил, -CH2CH2CH3), 2-пропил (i-Pr, и-пропил, -CH(CH3)2), 1-бутил (n-Bu, н-бутил, -CH2CH2CH2CH3), 2-метил-1-пропил (i-Bu, и-бутил, -CH2CH(CH3)2), 2-бутил (s-Bu, втор-бутил, -CH(CH3)CH2CH3), 2-метил-2-пропил (t-Bu, трет-бутил, -C(CH3)3), 1-пентил (н-пентил, -CH2CH2CH2CH2CH3), 2-пентил (-CH(CH3)CH2CH2CH3), 3-пентил (-CH(CH2CH3)2), 2-метил-2-бутил (-C(CH3)2CH2CH3), 3-метил-2-бутил (-CH(CH3)CH(CH3)2), 3-метил-1-бутил (-CH2CH2CH(CH3)2), 2-метил-1-бутил (-CH2CH(CH3)CH2CH3), 1-гексил (-CH2CH2CH2CH2CH2CH3), 2-гексил (-CH(CH3)CH2CH2CH2CH3), 3-гексил (-CH(CH2CH3)(CH2CH2CH3)), 2-метил-2-пентил (-C(CH3)2CH2CH2CH3), 3-метил-2-пентил (-CH(CH3)CH(CH3)CH2CH3), 4-метил-2-пентил (-CH(CH3)CH2CH(CH3)2), 3-метил-3-пентил (-C(CH3)(CH2CH3)2), 2-метил-3-пентил (-CH(CH2CH3)CH(CH3)2), 2,3-диметил-2-бутил (-C(CH3)2CH(CH3)2), 3,3-диметил-2-бутил (-CH(CH3)C(CH3)3.
Термин «алкилен», используемый в настоящем документе, относится к насыщенному линейному или с разветвленной цепью двухвалентному углеводородному радикалу из одного-двенадцати атомов углерода (C1-C12), где этот алкилен радикал необязательно может быть замещен независимо одним или несколькими заместителями, описанными далее. В другом варианте осуществления, алкиленовый радикал содержит от одного до восьми атомов углерода (C1-C8), или от одного до шести атомов углерода (C1-C6). Примеры алкиленовой группы включают, но этим не ограничиваются, метилен (-CH2-), этилен (-CH2CH2-), пропилен (-CH2CH2CH2-) и тому подобное.
Термин «алкенил» относится к линейному или разветвленному моновалентному углеводородному радикалу из двух-восьми атомов углерода (C2-C8) по меньшей мере с одним участком ненасыщенности, то есть, углерод-углерод, sp2 двойной связью, где алкенильный радикал необязательно может быть замещен независимо одним или несколькими заместителями, описанными в настоящем документе, и включает радикалы, имеющие «цис» и «транс» ориентации, или альтернативно, «E» и «Z» ориентации. Примеры включают, но этим не ограничиваются, этиленил или винил (-CH=CH2), аллил (-CH2CH=CH2) и тому подобное.
Термин «алкенилен» относится к линейному или разветвленному двухвалентному углеводородному радикалу от двух до восьми атомов углерода (C2-C8) по меньшей мере с одним участком ненасыщенности, то есть, углерод-углеродной, sp2 двойной связью, где этот алкенильный радикал необязательно может быть замещен, и включает радикалы, имеющие «цис» и «транс» ориентации, или альтернативно, «E» и «Z» ориентации. Примеры включают, но этим не ограничиваются, этиленилен или винилен (-CH=CH-), аллил (-CH2CH=CH-) и тому подобное.
Термин «алкинил» относится к линейному или разветвленному моновалентному углеводородному радикалу от двух до восьми атомов углерода (C2-C8) по меньшей мере с одним участком ненасыщенности, то есть, углерод-углеродной, sp тройной связью, где радикал алкинил необязательно может быть замещен независимо одним или несколькими заместителями, описанными в настоящем документе. Примеры включают, но этим не ограничиваются, этинил (-C≡CH), пропинил (пропаргил, -CH2C≡CH) и тому подобное.
Термин «алкинилен» относится к линейному или разветвленному двухвалентному углеводородному радикалу из двух-восьми атомов углерода (C2-C8) по меньшей мере с одним участком ненасыщенности, то есть, углерод-углеродной, sp тройной связью, где радикал алкинил может быть необязательно. Примеры включают, но этим не ограничиваются, этинилен (-C≡C-), пропинилен (пропаргилен, -CH2C≡C-) и тому подобное.
Термины «карбоцикл», «карбоциклил», «карбоциклическое кольцо» и «циклоалкил» относятся к моновалентному не-ароматическому, насыщенному или частично ненасыщенному кольцу, имеющему 3-12 атомов углерода (C3-C12) в виде моноциклического кольца или 7-12 атомов углерода в виде бициклического кольца. Бициклические карбоциклы, имеющие 7-12 атомов, могут быть организованы, например, в виде бицикло [4,5], [5,5], [5,6] или [6,6] системы, а бициклические карбоциклы, имеющие 9 или 10 кольцевых атомов, могут быть организованы в виде бицикло [5,6] или [6,6] системы, или в виде системы с мостиковой связью, такие как бицикло[2.2.1]гептан, бицикло[2.2.2]октан и бицикло[3.2.2]нонан. Примеры моноциклических карбоциклов включают, но этим не ограничиваются, циклопропил, циклобутил, циклопентил, 1-циклопент-1-енил, 1-циклопент-2-енил, 1-циклопент-3-енил, циклогексил, 1-циклогекс-1-енил, 1-циклогекс-2-енил, 1-циклогекс-3-енил, циклогексадиенил, циклогептил, циклооктил, циклононил, циклодецил, циклоундецил, циклододецил и тому подобное.
«Арил» подразумевает моновалентный ароматический углеводородный радикал из 6-20 атомов углерода (C6-C20), полученный удалением одного атома водорода от единичного атома углерода исходной ароматической кольцевой системы. Некоторые арильные группы представлены в структурах, приводимых в качестве примера, как «Ar». Арил включает бициклическое радикалы, содержащие ароматическое кольцо, конденсированное с насыщенным, частично ненасыщенным кольцом, или ароматическим карбоциклическим кольцом. Типичные арильные группы включают, но этим не ограничиваются, радикалы, полученные из бензола (фенил), замещенных бензолов, нафталина, антрацена, бифенила, инденила, инданила, 1,2-дигидронафталина, 1,2,3,4-тетрагидронафтила и тому подобного. Арильные группы необязательно замещены независимо одним или несколькими заместителями, описанными в настоящем документе.
«Арилен» подразумевает двухвалентный ароматический углеводородный радикал из 6-20 атомов углерода (C6-C20), полученный путем удаления двух атомов водорода от двух атомов углерода исходной ароматической кольцевой системы. Некоторые ариленовые группы представлены в структурах, приводимых в качестве примера, как «Ar». Арилен включает бициклические радикалы, содержащие ароматическое кольцо, конденсированное с насыщенным, частично ненасыщенным кольцом, или ароматическим карбоциклическим кольцом. Типичные ариленовые группы включают, но этим не ограничиваются, радикалы, полученные из бензола (фенилен), замещенных бензолов, нафталина, антрацена, бифенилена, инденилена, инданилена, 1,2-дигидронафталина, 1,2,3,4-тетрагидронафтила и тому подобного. Ариленовые группы необязательно являются замещенными.
Термины «гетероцикл», «гетероциклил» и «гетероциклическое кольцо» используются в настоящей заявке взаимозаменяемо и относятся к насыщенному или частично ненасыщенному (то есть, имеющему одну или несколько двойных и/или тройных связей в кольце) карбоциклическому радикалу от 3 до 20 кольцевых атомов, в котором по меньшей мере один кольцевой атом представляет собой гетероатом, выбранный из азота, кислорода, фосфора и серы, оставшиеся кольцевые атомы, представляющие собой C, где один или несколько кольцевых атомов необязательно замещены независимо одним или несколькими заместителями, описаны далее. Гетероцикл может представлять собой моноцикл, имеющий от 3 до 7 атомов в кольце (от 2 до 6 атомов углерода и от 1 до 4 гетероатомов, выбранных из N, O, P и S), или бицикл, имеющий от 7 до 10 атомов в кольце (от 4 до 9 атомов углерода и от 1 до 6 гетероатомов, выбранных из N, O, P и S), например: бицикло [4,5], [5,5], [5,6] или [6,6] системы. Гетероциклы описаны у Paquette, Leo A.; "Principles of Modern Heterocyclic Chemistry" (W.A. Benjamin, New York, 1968), в частности главы 1, 3, 4, 6, 7, и 9; "The Chemistry of Heterocyclic compounds, A series of Monographs" (John Wiley & Sons, New York, 1950 до настоящего времени), в частности тома 13, 14, 16, 19, и 28; и J. Am. Chem. Soc. (1960) 82:5566. «Гетероциклил» также включает радикалы, где гетероциклические радикалы конденсированы с насыщенным, частично ненасыщенным кольцом, или ароматическим карбоциклическим или гетероциклическим кольцом. Примеры гетероциклических колец включают, но этим не ограничиваются, пирролидинил, тетрагидрофуранил, дигидрофуранил, тетрагидротиенил, тетрагидропиранил, дигидропиранил, тетрагидротиопиранил, пиперидино, морфолино, тиоморфолино, тиоксанил, пиперазинил, гомопиперазинил, азетидинил, оксетанил, тиетанил, гомопиперидинил, оксепанил, тиепанил, оксазепинил, диазепинил, тиазепинил, 2-пирролинил, 3-пирролинил, индолинил, 2H-пиранил, 4H-пиранил, диоксанил, 1,3-диоксоланил, пиразолинил, дитианил, дитиоланил, дигидропиранил, дигидротиенил, дигидрофуранил, пиразолидинилимидазолинил, имидазолидинил, 3-азабицикло[3.1.0]гексанил, 3-азабицикло[4.1.0]гептанил, азабицикло[2.2.2]гексанил, 3H-индолилхинолизинил и N-пиридилмочевину. Спиро группы также включены в объем настоящего определения. Примеры гетероциклической группы, где 2 кольцевых атома углерода замещены оксо (=O) группами, представляют собой пиримидинонил и 1,1-диоксо-тиоморфолинил. Гетероциклические группы в настоящей заявке необязательно замещены независимо одним или несколькими заместителями, описанными в настоящем документе.
Термин «гетероарил» относится к моновалентному ароматическому радикалу 5-, 6-, или 7-замещенных колец и включает конденсированные кольцевые системы (по меньшей мере одна из которых является ароматической) из 5-20 атомов, содержащие один или несколько гетероатомов, независимо выбранных из азота, кислорода и серы. Примерами гетероарильных групп являются пиридинил (включая, например, 2-гидроксипиридинил), имидазолил, имидазопиридинил, пиримидинил (включая, например, 4-гидроксипиримидинил), пиразолил, триазолил, пиразинил, тетразолил, фурил, тиенил, изоксазолил, тиазолил, оксадиазолил, оксазолил, изотиазолил, пирролил, хинолинил, изохинолинил, тетрагидроизохинолинил, индолил, бензимидазолил, бензофуранил, циннолинил, индазолил, индолизинил, фталазинил, пиридазинил, триазинил, изоиндолил, птеридинил, пуринил, оксадиазолил, триазолил, тиадиазолил, тиадиазолил, фуразанил, бензофуразанил, бензотиофенил, бензотиазолил, бензоксазолил, хиназолинил, хиноксалинил, нафтиридинил, и фуропиридинил. Гетероарильные группы необязательно замещены независимо одним или несколькими заместителями, описанными в настоящем документе.
Гетероцикл или гетероарильные группы могут быть связаны углеродной (углерод-связанные), или азотной (азот-связанные) связью, где такое возможно. В качестве примера, а не для ограничения, углерод-связанные гетероциклы или гетероарилы связаны в положении 2, 3, 4, 5 или 6 пиридина, положении 3, 4, 5 или 6 пиридазина, положении 2, 4, 5 или 6 пиримидина, положении 2, 3, 5 или 6 пиразина, положении 2, 3, 4 или 5 фурана, тетрагидрофурана, тиофурана, тиофена, пиррола или тетрагидропиррола, положении 2, 4 или 5 оксазола, имидазола или тиазола, положении 3, 4 или 5 изоксазола, пиразола, или изотиазола, положении 2 или 3 азиридина, положении 2, 3 или 4 азетидина, положении 2, 3, 4, 5, 6, 7 или 8 хинолина или положении 1, 3, 4, 5, 6, 7 или 8 изохинолина.
Для примера, а не для ограничения, азот-связанные гетероциклы или гетероарилы связаны в положении 1 азиридина, азетидина, пиррола, пирролидина, 2-пирролина, 3-пирролина, имидазола, имидазолидина, 2-имидазолина, 3-имидазолина, пиразола, пиразолина, 2-пиразолина, 3-пиразолина, пиперидина, пиперазина, индола, индолина, 1H-индазола, положении 2 изоиндола, или изоиндолина, положении 4 морфолина и положении 9 карбазола, или β-карболина.
«Линкер» или «связь» означает двухвалентную химическую группу, содержащую ковалентную связь или цепь атомов, которые ковалентно присоединяют антитело к лекарственной части. В различных вариантах осуществления формулы I, линкер обозначается как L.
Термин «хиральный» относится к молекулам, которые имеют свойство несовпадения при наложении зеркального изображения партнера, тогда как термин «ахиральный» относится к молекулам, которые совпадают при наложении на зеркальное изображение их партнера.
Термин «стереоизомеры» относится к соединениям, которые имеют идентичное химическое строение, но отличаются в отношении расположения атомов или групп в пространстве.
«Диастереомер» относится к стереоизомеру с двумя или несколькими центрами хиральности, и эти молекулы не являются зеркальными изображениями друг друга. Диастереомеры имеют различные физические свойства, например температуру плавления, температуру кипения, спектральные свойства и реакционную способность. Смеси диастереомеров можно разделить в условиях аналитических способов высокого разрешения, таких как электрофорез и хроматография.
«Энантиомеры» относятся к двум стереоизомерным соединениям, которые не совпадают при наложении зеркальных изображений друг на друга.
Стереохимические определения и условные обозначения, используемые в настоящей заявке, в основном следуют из S. P. Parker, Ed., McGraw-Hill Dictionary of Chemical Terms (1984) McGraw-Hill Book Company, New York; и Eliel, E. и Wilen, S., Stereochemistry of Organic Compounds (1994) John Wiley & Sons, Inc., New York. Многие органические соединения существуют в оптически активных формах, то есть, они обладают способностью вращать плоскость плоскополяризованного света. При описании оптически активного соединения, приставки D и L, или R и S, используют для обозначения абсолютной конфигурации молекулы вокруг ее хирального центра (центров). Приставки d и l или (+) и (-) используют для обозначения направления вращения плоскополяризованного света этим соединением, при этом (-) или l означает, что соединение является левовращающим. Соединение, имеющее впереди знак (+) или d, является правовращающим. Для заданной химической структуры эти стереоизомеры являются идентичными, за исключением того, что они являются зеркальными изображениями друг друга. Конкретный стереоизомер также может называться энантиомером, и смесь таких изомеров зачастую называют энантиомерной смесью. Смесь энантиомеров 50:50 называют рацемической смесью или рацематом, которая может встречаться в тех случаях, где отсутствовала стереоселективность или стереоспецифичность в химической реакции или процессе. Термины «рацемическая смесь» и «рацемат» относятся к эквимолярной смеси двух энантиомерных видов, свободных от оптической активности.
Фраза «фармацевтически приемлемая соль», используемая в настоящем документе, относится к фармацевтически приемлемой органической или неорганической соли ADC. Приводимые в качестве примера соли включают, но этим не ограничиваются, сульфат, цитрат, ацетат, оксалат, хлорид, бромид, йодид, нитрат, бисульфат, фосфат, кислый фосфат, изоникотинат, лактат, салицилат, кислый цитрат, тартрат, олеат, таннат, пантотенат, битартрат, аскорбат, сукцинат, малеат, гентизинат, фумарат, глюконат, глюкуронат, сахарат, формиат, бензоат, глутамат, метансульфонат, этансульфонат, бензолсульфонат, p-толуолсульфонат, и памоат (то есть, 1,1'-метилен-бис-(2-гидрокси-3-нафтоат)). Фармацевтически приемлемая соль может содержать включение другой молекулы, такой как ацетатный ион, сукцинатный ион или другой противоион. Противоионом может быть любая органическая или неорганическая группа, которая стабилизирует заряд на исходном соединении. Более того, фармацевтически приемлемая соль может иметь более одного заряженного атома в своей структуре. Случаи, где многочисленные заряженные атомы являются частью фармацевтически приемлемой соли, могут иметь многочисленные противоионы. Следовательно, фармацевтически приемлемая соль может иметь один или несколько заряженных атомов и/или один или несколько противоионов.
«Фармацевтически приемлемый сольват» относится к ассоциации одной или нескольких молекул растворителя и ADC. Примеры растворителей, которые образуют фармацевтически приемлемые сольваты, включают, но этим не ограничиваются, воду, изопропанол, этанол, метанол, ДМСО, этилацетат, уксусную кислоту и этаноламин.
ПРОИЗВОДНЫЕ АНТРАЦИКЛИНА И КОНЪЮГАТЫ ПРОИЗВОДНЫХ АНТРАЦИКЛИНА
Как указано ранее, в первом аспекте настоящее изобретение относится к конъюгатам производного антрациклина формулы (I):
где Ant, L, Z, m и T определены выше, или его фармацевтически приемлемая соль.
Во втором аспекте представлены производные антрациклина формулы (I')
где Ant, L и Z определены выше, и Q представляет собой атом водорода, C1-C6 алкил, C3-C6 циклоалкил, фенильную или бензильную группу, или их фармацевтически приемлемая соль.
Предпочтительно, остаток производного антрациклина, который представляет Ant, может высвобождаться с получением производного антрациклина формулы (IIA):

В другом предпочтительном аспекте, настоящее изобретение относится к конъюгату производного антрациклина или его фармацевтически приемлемой соли формулы (Ia):
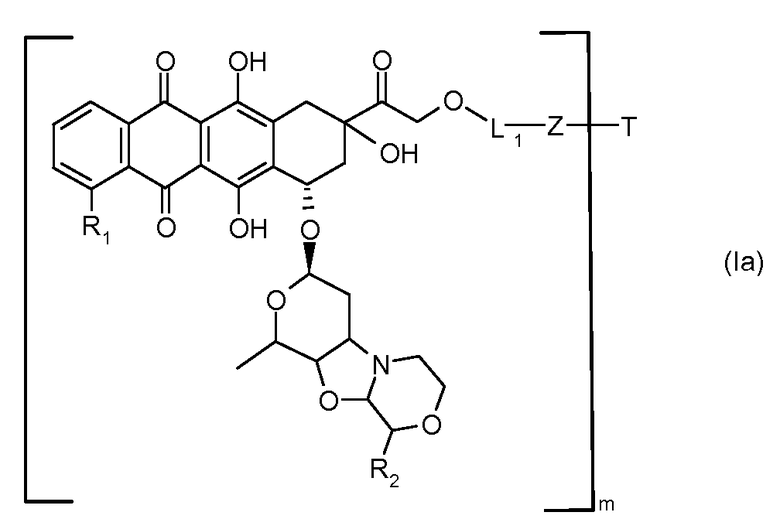
где
R1, R2, Z, m и T имеют значения, указанные выше, и L1 представляет собой линкер формулы (III) или (IV):
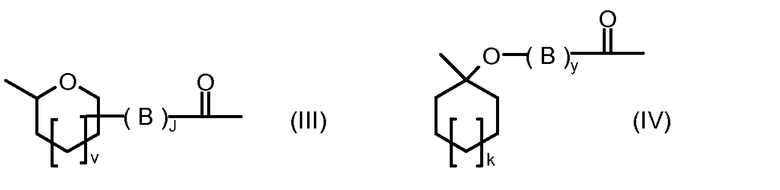
где B представляет собой C1-C6 алкиленовую группу, необязательно гетеро прерываемую, и v, j, k и y независимо равны 0 или 1.
Очевидно, что в этом случае остаток производного антрациклина, который представляет Ant, привязан к линкеру L1 через ацетальную связь, в которую вовлечен первичный спирт в положении C-14 антрациклинового скелета.
Как указано выше, конъюгат производного антрациклина или его фармацевтически приемлемой соли формулы (I) высвобождает желаемое свободное производное антрациклина, как показано далее для предпочтительных конъюгатов формулы (Ia)
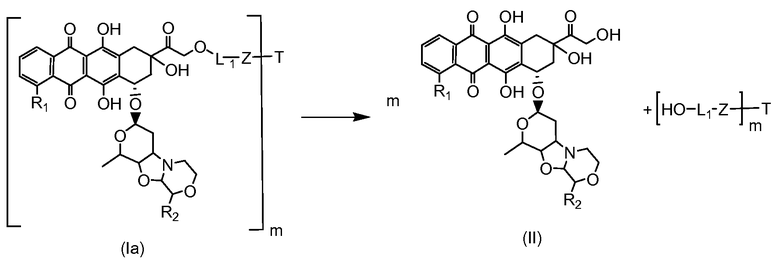
где R1, R2, L1, Z, m и T определены выше.
Предпочтительно, Z представляет собой спейсерную группу, наподобие
a) -NH-,
b) -S-,
c) аминоалкилена, тиоалкилена, аминоциклоалкилена или тиоциклоалкилена, несущих дополнительную тиольную или амино группу или карбоксильный остаток,
d) пептидного остатка, способного связывать L1 линкер с T носителем, путем образования новых связей, как например; амидные связи, дисульфидные связи.
T предпочтительно выбран из поликлонального антитела или его фрагмента, содержащего антиген-связывающий сайт, способный связываться с опухоле-связанным антигеном; моноклонального антитела или его фрагмента, содержащего антиген-связывающий сайт, способный связываться с антигеном, предпочтительно или селективно экспрессированным на популяции опухолевых клеток; пептид или белок, необязательно способный предпочтительно или селективно связываться с опухолевой клеткой; или его химически модифицированное производное, подходящее для присоединения к группе [Ant-L1-Z-] или группам, или полимерному носителю.
Особенно предпочтительными соединениями формулы (Ia) являются те, в которых спейсерная группа, которую представляет Z, представляет собой:
i) -NH-, то есть [-Z-]m-T получен из носителя формулы T-[NH2]m, где m определено выше;
ii) -S-, то есть [-Z-]m-T получен из носителя формулы T-[SH]m, где m определено выше;
iii) -NH-D-NH-CO-, где -D- представляет собой C1-C6 алкилен, C3-C6 циклоалкилен или -D-NH- представляет собой пептидный остаток, состоящий из 1-4 аминокислот, имеющих по меньшей мере одну свободную аминогруппу, то есть [-Z-]m-T получен из носителя формулы T-[COOH]m, где m определено выше;
iv) -NH-D-CO-NH-, где -D- имеет значения, указанные выше, или -D-CO- представляет собой пептидный остаток, состоящий из 1-4 аминокислот, имеющих по меньшей мере одну свободную карбоксильную группу, то есть [-Z-]m-T получен из носителя формулы T-[NH2]m, где m определено выше;
v) -NH-D-N=CH-, где -D- имеет значения, указанные выше, и -D-N- имеет значения, указанные выше для -D-NH, то есть [-Z-]m-T получен из носителя формулы T-[CHO]m, где m определено выше;
vi) -NH-D-S-CH-, где -D- имеет значения, указанные выше, или -D-S- представляет собой пептидный остаток, состоящий из 1-4 аминокислот, имеющих по меньшей мере одну свободную тиольную группу, то есть [-Z-]m-T получен из носителя, производного формулы (V);

где m определено выше;
vii) -NH-D-S-S-, где -D- и -D-S- имеют значения, указанные выше, то есть [-Z-]m-T получен из носителя, производного формулы (VI):
где m определено выше.
В дополнительном предпочтительном аспекте, настоящее изобретение относится к конъюгату производного антрациклина или его фармацевтически приемлемой соли (Ib):
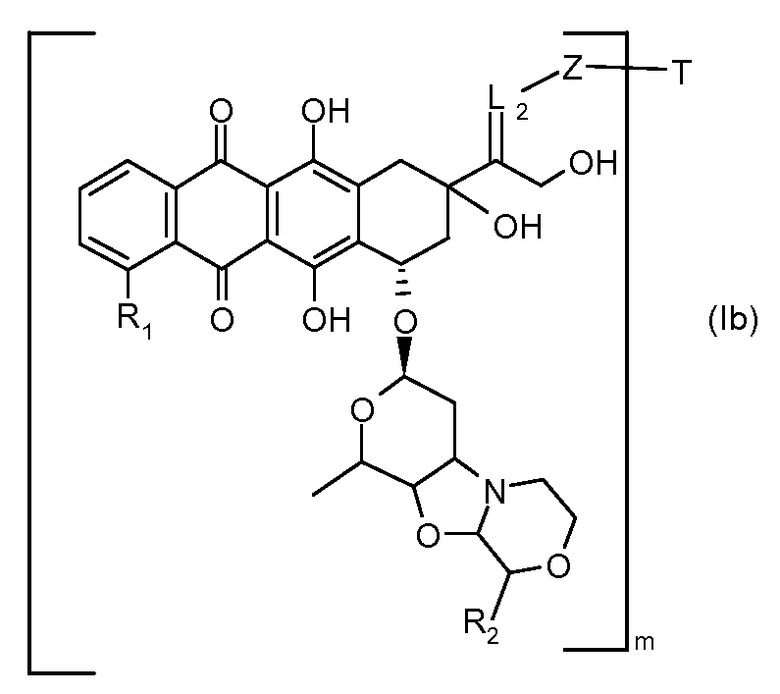
где R1 и R2, Z и T имеют значения, указанные выше, и L2 представляет собой линкер формулы (VII) или (VIII):
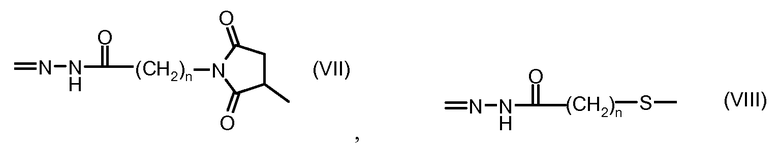
где n равно целому числу от 1 до 9.
В этом случае, высвобождение желаемого свободного производного антрациклина схематически может быть проиллюстрировано следующим образом, из предпочтительных конъюгатов формулы (Ib):
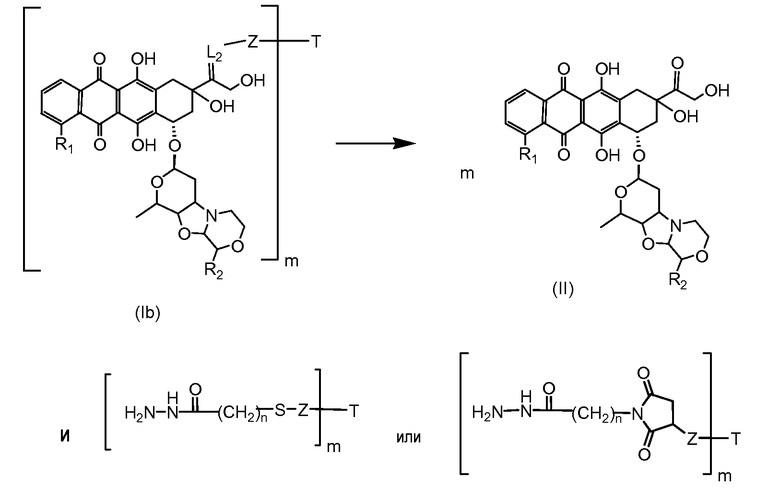
Особенно предпочтительными соединениями формулы (Ib) являются те, в которых:
a) L2 представляет собой линкер формулы (VII), как определено выше, а Z представляет собой спейсерную группу, которая:
viii) имеет значения, указанные выше в пункте i);
ix) имеет значения, указанные выше в пункте ii);
x) имеет значения, указанные выше в пункте iii);
xi) имеет значения, указанные выше в пункте iv);
xii) имеет значения, указанные выше в пункте v);
xiii) имеет значения, указанные выше в пункте vi);
xiv) имеет значения, указанные выше в пункте vii);
xv) -S-D-NH-CO-, где - D-NH-CO- имеет значения, указанные выше в пункте iii);
xvi) -S-D-CO-NH-, где - D-CO-NH имеет значения, указанные выше в пункте iv);
xvii) -S-D-N=C-, где D и D-N- имеют значения, указанные выше в пункте v);
xviii) S-D-S-CH-, где D и -D-S- имеют значения, указанные выше в пункте vi);
xix) -S-D-S-S-, где D и -D-S- имеют значения, указанные выше в пункте vii).
Другими особенно предпочтительными соединениями формулы (Ib) являются те, в которых:
b) L2 представляет собой линкер формулы (VIII), как определено выше, а Z представляет собой спейсерную группу, которая:
xx) имеет значения, указанные выше в пункте ii)
xxi) имеет значения, указанные выше в пункте xv);
xxii) имеет значения, указанные выше в пункте xvi);
xxiii) имеет значения, указанные выше в пункте xvii);
xxiv) имеет значения, указанные выше в пункте xviii);
xxv) имеет значения, указанные выше в пункте xix).
Другим особенно предпочтительным объектом по настоящему изобретению являются соединения формул (I'a) и (I'b):
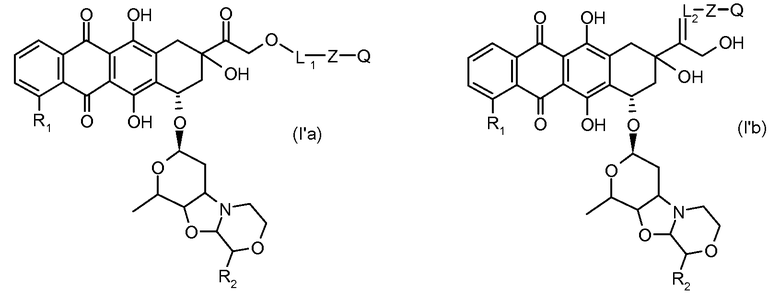
где L1, L2, R1 и R2 имеют значения, указанные выше, Z представляет собой -NH- или пептидный остаток, состоящий из 1-3 аминокислот, и Q представляет собой атом водорода, C1-C6 алкил, C3-C6 циклоалкил, фенильную или бензильную группу.
Также в этом случае, соединения формулы (I'a) и (I'b) могут высвобождаться в соответствующих условиях, давая соединение формулы (II), как определено выше.
Другим особенно предпочтительным классом соединений является соединение формулы (I) или (I'), где L представляет собой:
- группу формулы (IIIa) или (IVa),

где v и k имеют значения, указанные выше;
или группу формулы (VII) или (VIII), как определено выше, отличающуюся тем, что n равно целому числу от 2 до 5.
ЛИНКЕРЫ И СПЕЙСЕРЫ
Линкерные L и спейсерные Z звенья присоединяют носитель, например антитело, к лекарственной части D - производному антрациклина посредством ковалентной связи (связей). Линкер представляет собой бифункциональную или многофункциональную группу, которая может быть использована для соединения одной или нескольких лекарственных групп (D) и антительной единицы (Ab) с образованием конъюгатов антитела с лекарственным средством (ADC) формулы Ic. Линкер (L) может быть стабильным снаружи клетки, то есть внеклеточно, или может расщепляться под действием ферментативной активности, гидролиза или других условий метаболизма. Конъюгаты антитела с лекарственным средством (ADC) можно удобно получать, используя линкер, обладающий реактивной функциональностью для связывания с лекарственной группой и с антителом. Тиол цистеина, или амин, например N-концевой или боковой цепи аминокислоты, такие как лизин, антитела (Ab) могут образовывать связь с функциональной группой линкерного или спейсерного реагента, лекарственной группой (D) или реагеном лекарственное средство-линкер (D-L).
Многие положения на производных соединениях антрациклина могут быть использованы в качестве положения связи, в зависимости от типа связи. Например, сложноэфирные, амидные, тиоамидные, тиокарбаматные, или карбаматные связи могут быть образованы из гидроксильной группы гидроксиметилкетона в положении C14; кетальные и гидразонные связи могут быть образованы из C13 карбонильной группы на лекарственной группе; амидные, карбаматные и мочевинные связи могут быть образованы из аминогруппы на лекарственной группе D; и различные алкильные, эфирные, тиоэфирные, дисульфидные и ацильные связи могут быть образованы из фенильных и арильных колец на лекарственной группе путем реакций алкилирования и ацилирования по типу Фриделя-Крафта.
Линкеры и спейсеры предпочтительно являются стабильными внеклеточно. До транспортировки или доставки в клетку, конъюгат антитело-лекарственное средство (ADC) предпочтительно является стабильным и остается интактным, то есть антитело остается связанным с лекарственной группой. Линкеры являются стабильными вне клетки-мишени и могут расщепляться с некоторой эффективной скоростью внутри клетки. Эффективный линкер будет: (i) поддерживать специфические связывающие свойства антитела; (ii) позволять осуществлять внутриклеточную доставку конъюгата или лекарственной части; (iii) оставаться стабильным и интактным, то есть нерасщепленным, пока конъюгат не будет доставлен или транспортирован к участку-мишени; и (iv) поддерживать цитотоксическое действие, действие по уничтожению клеток или цитостатическое действие лекарственной части производного антрациклина. Стабильность ADC может быть измерена с помощью стандартных аналитических методик, таких как масс-спектроскопия, ВЭЖХ, и методика разделения/анализа LC/MS.
Ковалентное присоединение антитела и лекарственной группы требует наличия линкера и необязательного спейсера, чтобы иметь две реакционно-способные функциональные группы, то есть бивалентность в смысле реакционной способности. Бивалентные линкерные реагенты, которые используют для присоединения двух или более функциональных или биологически активных фрагментов, таких как пептиды, нуклеиновые кислоты, лекарственные средства, токсины, антитела, гаптены и репортерные группы являются известными, и были описаны способы получения их результирующих конъюгатов (Hermanson, G.T. (1996) Bioconjugate Techniques; Academic Press: New York, p 234-242).
В другом варианте осуществления, линкер или спейсер могут быть замещены группами, которые модулируют растворимость или реакционноспособность. Например, сульфонатный заместитель может повышать водную растворимость реагента и облегчать реакцию соединения линкерного реагента с антителом или лекарственной частью, или облегчать реакцию соединения Ab-L с D, или D-L с Ab, в зависимости от пути синтеза, используемого для получения ADC.
Нуклеофильные группы на антителах включают, но этим не ограничиваются: (i) N-концевые аминные группы, (ii) аминные группы боковых цепей, например, лизина, (iii) тиольные группы боковых цепей, например, цистеина, и (iv) гидроксильные или аминогруппы сахара, где антитело является гликозилированным. Аминные, тиольные и гидроксильные группы являются нуклеофильными и способными взаимодействовать с образованием ковалентных связей с электрофильными группами на линкерных частях и линкерных реагентах, в том числе: (i) активные сложные эфиры, такие как сложные эфиры NHS, сложные эфиры HOBt, галоформиаты и кислые галогениды; (ii) алкил и бензил галогениды, такие как галоацетамиды; (iii) альдегиды, кетоны, карбоксильные и малеимидные группы. Некоторые антитела имеют восстанавливаемые межцепочечные дисульфиды, то есть цистеиновые мостики. Антитела могут быть получены реакционно-способными для конъюгирования с линкерными реагентами путем обработки восстанавливающим агентом, таким как DTT (дитиотреитол). Каждый цистеиновый мостик, таким образом, будет образовывать, теоретически, два реакционно-способных тиольных нуклеофила. Дополнительные нуклеофильные группы могут быть введены в антитела путем взаимодействия лизинов с 2-иминотиоланом (реагент Трота), получая в результате превращение амина в тиол. Реакционно-способные тиольные группы могут быть введены в антитело (или его фрагмент) путем ведения одного, двух, трех, четырех или более остатков цистеина (например, получая мутантные антитела, содержащие один или несколько ненативных остатков аминокислоты цистеина). В US 2007/0092940 представлены сведения о сконструированных антителах путем введения реакционно-способной аминокислоты цистеина.
В некоторых вариантах осуществления, линкер имеет реакционно-способную нуклеофильную группу, которая является реакционно-способной в отношении электрофильной группы, находящейся на антителе. Подходящие электрофильные группы на антителе включают, но этим не ограничиваются, альдегидные и кетонные карбонильные группы. Гетероатом нуклеофильной группы линкера может взаимодействовать с электрофильной группой на антителе и образует ковалентную связь с антительной единицей. Подходящие нуклеофильные группы на линкере включают, но этим не ограничиваются, гидразид, оксим, амино, гидроксил, гидразин, тиосемикарбазон, гидразинкарбоксилат и арилгидразид. Электрофильная группа на антителе предоставляет удобное место для присоединения к линкеру.
Нуклеофильные группы на лекарственной части включают, но этим не ограничиваются: амин, тиол, гидроксил, гидразид, оксим, гидразин, тиосемикарбазон, гидразинкарбоксилат, и арилгидразидные группы, способные вступать в реакцию с образованием ковалентных связей с электрофильными группами на линкерных частях и линкерных реагентах, в том числе: (i) активные сложные эфиры, такие как сложные эфиры NHS, сложные эфиры HOBt, галоформиаты и кислые галогениды; (ii) алкил и бензил галогениды, такие как галоацетамиды; (iii) альдегиды, кетоны, карбоксильные и малеимидные группы.
Спейсеры (Z) могут быть пептидными, содержащими одно или несколько аминокислотных звеньев. Пептидные линкерные реагенты могут быть получены путем твердофазных или жидкофазных способов синтеза (E. Schröoder и K. Lübke, The Peptides, volume 1, pp 76-136 (1965) Academic Press), которые хорошо известны в области пептидной химии, включая химию t-BOC (Geiser et al. «Automation of solid-phase peptide synthesis» в Macromolecular Sequencing and Synthesis, Alan R. Liss, Inc., 1988, pp. 199-218) и химию Fmoc/HBTU (Fields, G. и Noble, R. (1990) «Solid phase peptide synthesis utilizing 9-fluoroenylmethoxycarbonyl amino acids», Int. J. Peptide Protein Res. 35:161-214), на автоматическом синтезаторе, например, пептидном синтезаторе «Rainin Symphony Peptide Synthesizer» (Protein Technologies, Inc., Tucson, AZ), или Model 433 (Applied Biosystems, Foster City, CA).
Приводимые в качестве примера аминокислотные линкеры включают дипептид, трипептид, тетрапептид или пентапептид. Приводимые в качестве примера дипептиды включают: валин-цитруллин (vc или val-cit), аланин-фенилаланин (af или ala-phe). Приводимые в качестве примера трипептиды включают: глицин-валин-цитруллин (gly-val-cit) и глицин-глицин-глицин (gly-gly-gly). Аминокислотные остатки, которые содержат аминокислотный линкерный компонент, включают те, которые встречаются в природе, а также второстепенные аминокислоты и неприродные аналоги аминокислот, такие как цитруллин. Компоненты аминокислотного линкера могут быть созданы и оптимизированы по селективности для ферментативного расщепления под действием конкретных ферментов, например, опухоле-связанной протеазы, катепсина B, C и D, или протеазы плазмина.
Боковые цепи аминокислот включают те, которые встречаются в природе, а также второстепенные аминокислоты и неприродные аналоги аминокислот, такие как цитруллин. Боковые цепи аминокислот включают водород, метил, изопропил, изобутил, втор-бутил, бензил, п-гидроксибензил, -CH2OH, -CH(OH)CH3, -CH2CH2SCH3, -CH2CONH2, -CH2COOH, -CH2CH2CONH2, -CH2CH2COOH, -(CH2)3NHC(=NH)NH2, -(CH2)3NH2, -(CH2)3NHCOCH3, -(CH2)3NHCHO, -(CH2)4NHC(=NH)NH2, -(CH2)4NH2, -(CH2)4NHCOCH3, -(CH2)4NHCHO, -(CH2)3NHCONH2, -(CH2)4NHCONH2, -CH2CH2CH(OH)CH2NH2, 2-пиридилметил-, 3-пиридилметил-, 4-пиридилметил-, фенил, циклогексил, а также следующие структуры:
В тех случаях, когда аминокислотные боковые цепи содержат иное, чем водород (глицин), атом углерода, к которому присоединена боковая цепь аминокислоты, является хиральным. Каждый атом углерода, к которому присоединена боковая цепь аминокислоты, независимо находится в (S) или (R) конфигурации, или рацемической смеси. Лекарственное средство-линкерные реагенты, следовательно, могут быть энантиомерно чистыми, рацемическими или диастереомерными.
В приводимых в качестве примера вариантах осуществления, боковые цепи аминокислот выбраны из природных и неприродных аминокислот, в том числе аланина, 2-амино-2-циклогексилуксусной кислоты, 2-амино-2-фенилуксусной кислоты, аргинина, аспарагина, аспарагиновой кислоты, цистеина, глутамина, глутаминовой кислоты, глицина, гистидина, изолейцина, лейцина, лизина, метионина, норлейцина, фенилаланина, пролина, серина, треонина, триптофана, тирозина, валина, γ-аминомасляной кислоты, α,α-диметил γ-аминомасляной кислоты, β,β-диметил γ-аминомасляной кислоты, орнитина, и цитруллина (Cit).
ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ЛЕКАРСТВЕННОЕ СРЕДСТВО-ЛИНКЕР
Настоящее изобретение включает промежуточные соединения лекарственное средство-линкер, используемые для конъюгирования с белками, пептидами и антителами. Такие промежуточные соединения лекарственное средство-линкер включают производное антрациклина формулы (IIc):
где Ant выбран из структур:
где волнистая линия указывает на присоединение к L;
L представляет собой линкер, выбранный из -N(R)-, -N(R)m(C1-C12 алкилен)-, -N(R)m(C2-C8 алкенилен)-, -N(R)m(C2-C8 алкинилен)-, -N(R)m(CH2CH2O)n-, и структуры:
где волнистые линии указывают на присоединения к Ant и Z; и
Z представляет собой необязательный спейсер, выбранный из -CH2C(O)-, -CH2C(O)NR(C1-C12 алкилен)-, и структур:
X представляет собой реакционно-способную функциональную группу, выбранную из малеимида, тиола, амино, бромида, п-толуолсульфоната, йодида, гидроксила, карбоксила, пиридилдисульфида и N-гидроксисукцинимида;
R представляет собой H, C1-C12 алкил, или C6-C20 арил;
R1 и R2 независимо выбраны из боковых цепей аминокислот;
Z1 выбран из -(C1-C12 алкилен)-, -(C2-C8 алкенилен)-, -(C2-C8 алкинилен)-, и -(CH2CH2O)n-,
m равно 0 или 1; и n равно 1-6.
Производные антрациклина формулы IIc включают структуры:
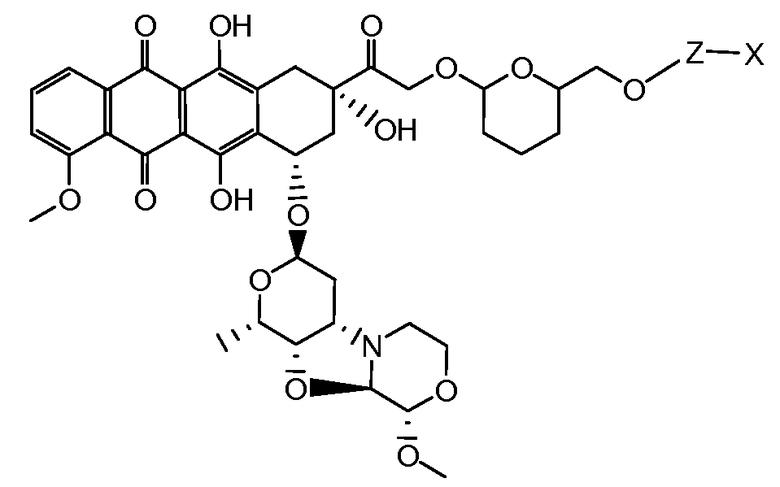
где Z-X выбран из:

Приводимые в качестве примера варианты осуществления промежуточных соединений лекарственное средство-линкер формулы IIc, содержащие часть антрациклинового лекарственного средства и звено линкер-спейсер, включают соединения 50-53:
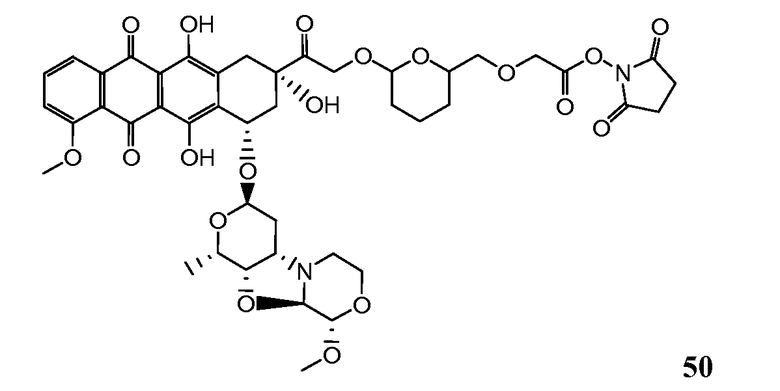
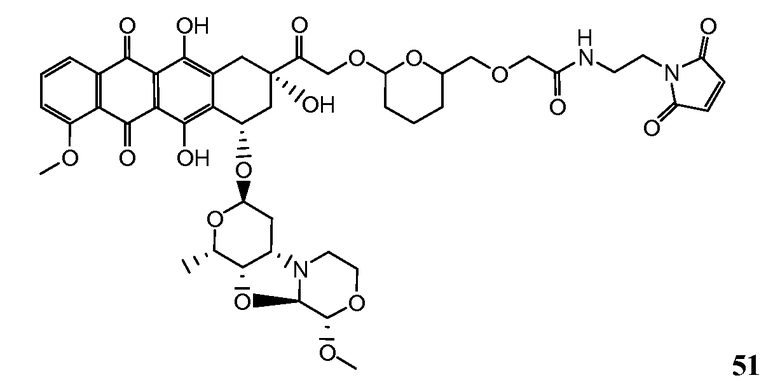
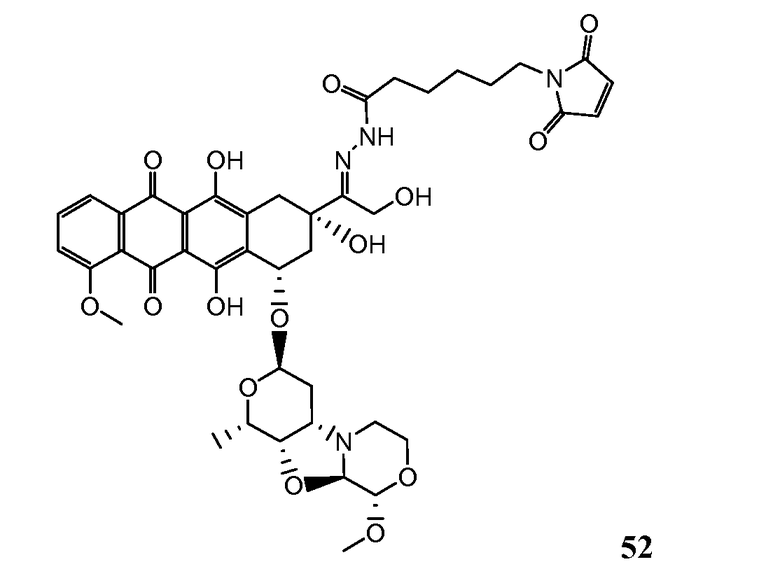
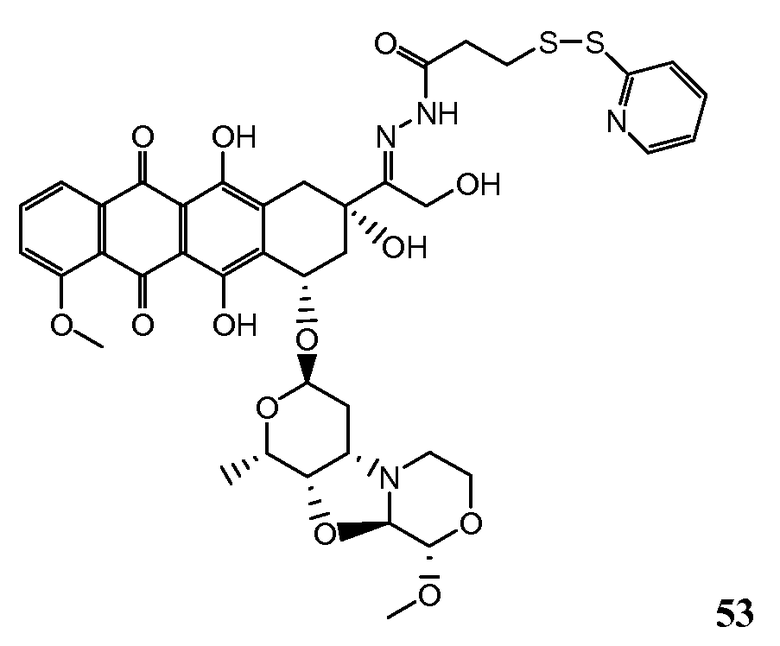
Производные антрациклинов формулы IIc включают структуру:
где Z представляет собой C1-C12 алкилен.
Приводимые в качестве примера варианты осуществления промежуточных соединений лекарственное средство-линкер формулы IIc, содержащие часть антрациклинового лекарственного средства и звено линкер-спейсер, включают соединение 54 (Пример 3a):
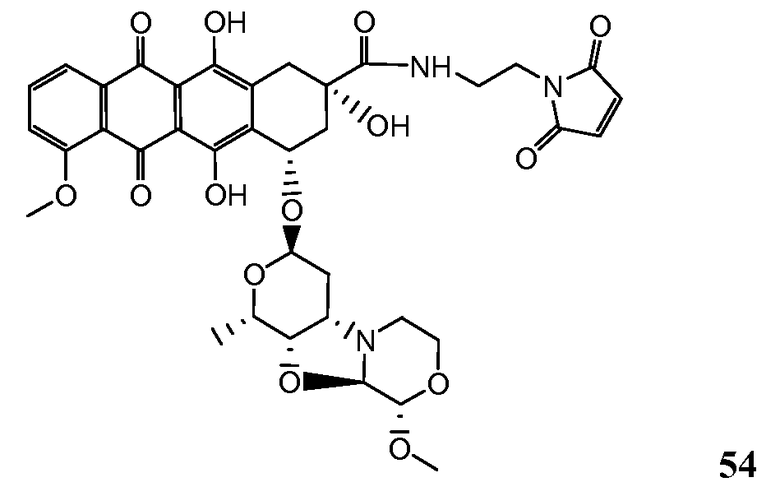
Производные антрациклинов формулы IIc включают структуры:
и где Х представляет собой малеимид:
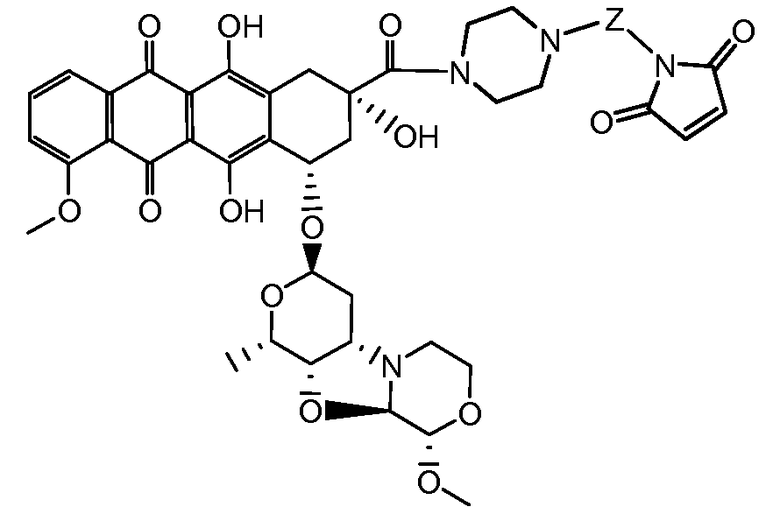
Приводимые в качестве примера варианты осуществления промежуточных соединений лекарственное средство-линкер формулы IIc, содержащие часть антрациклинового лекарственного средства и звено линкер-спейсер, включают соединение 55 (Пример 3b). Путь синтеза промежуточного соединения 55 лекарственное средство-линкер из PNU-159682 C-14 карбоксильного производного 56 и промежуточного соединения 60 линкер-спейсер, показан на Фигуре 7d.
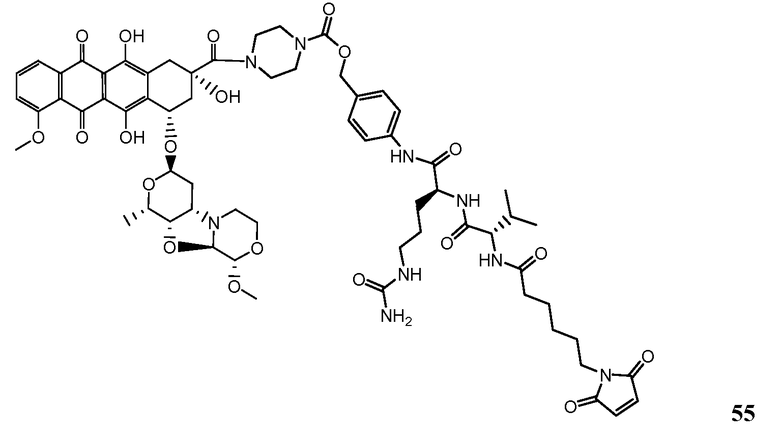
Производные антрациклинов формулы IIc включают структуры:
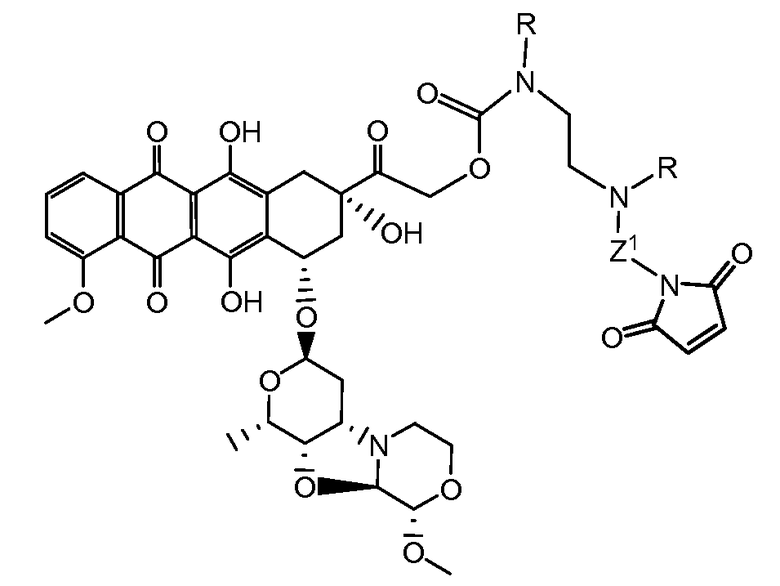
и где Z1 представляет собой -(C1-C12 алкилен)-.
Приводимые в качестве примера варианты осуществления промежуточных соединений лекарственное средство-линкер формулы IIc, содержащие часть антрациклинового лекарственного средства и звено линкер-спейсер, включают соединение:
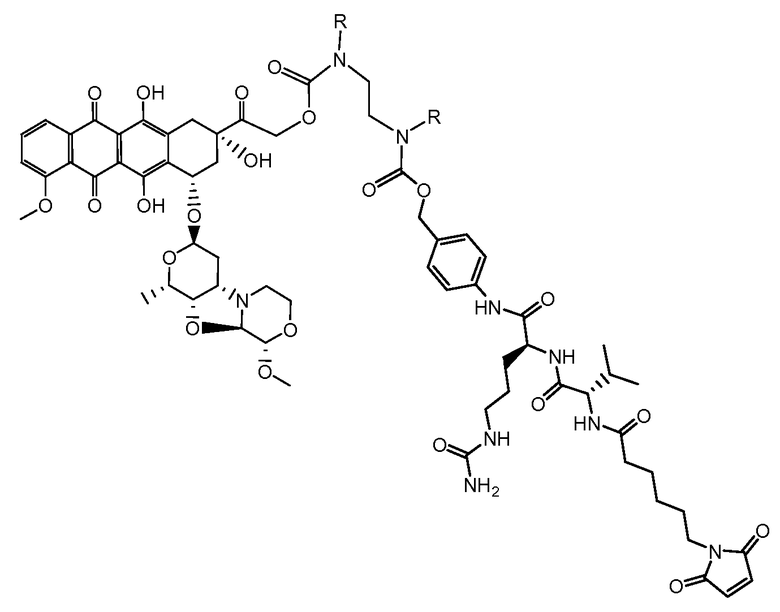
ЛИНКЕРНЫЕ И СПЕЙСЕРНЫЕ РЕАГЕНТЫ
Бета-глюкуронидные линкеры между антителом и лекарственной частью являются субстратами для расщепления под действием бета-глюкуронидазы (Jeffrey et al. (2006) Bioconjugate Chem. 17:831-840; WO 2007/011968). Ацетальная связь бета-глюкуронида высвобождает фенольный гидроксил на арильном кольце, потенциируя «саморазрушение» и 1,6-элиминацию бензилоксикарбонильной группы.
Приводимый в качестве примера валин-цитруллин (val-cit или vc) дипептидный линкерный реагент, имеющий малеимидный удлинитель и пара-аминобензилкарбамоильный (PAB) саморазрушаемый спейсер, имеет структуру:

где Q представляет собой C1-C8 алкил, -O-(C1-C8 алкил), -галоген, -NO2 или -CN; и m равно целому числу в интервале от 0 до 4.
Приводимый в качестве примера phe-lys(Mtr) дипептидный линкерный реагент, имеющий малеимидную удлиняющую единицу и п-аминобензильную саморазрушаемую спейсерную единицу, может быть получен в соответствии с Dubowchik, et al. (1997) Tetrahedron Letters, 38:5257-60, и имеет структуру:
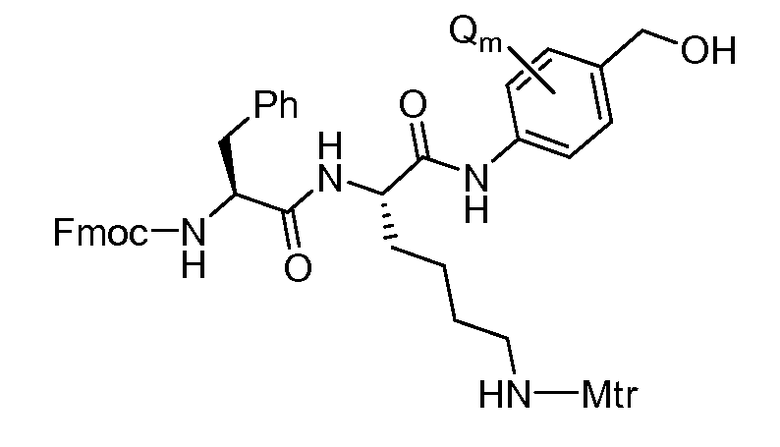
где Mtr представляет собой моно-4-метокситритил, Q представляет собой C1-C8 алкил, -O-(C1-C8 алкил), -галоген, -NO2 или -CN; и m равно целому числу в интервале от 0-4.
«Саморазрушаемый линкер», PABC или PAB (пара-аминобензилоксикарбонил), присоединяет лекарственную группу к антителу в конъюгате (Carl et al. (1981) J. Med. Chem. 24:479-480; Chakravarty et al. (1983) J. Med. Chem. 26:638-644; US 6214345; US20030130189; US20030096743; US6759509; US20040052793; US6218519; US6835807; US6268488; US20040018194; WO98/13059; US20040052793; US6677435; US5621002; US20040121940; WO2004/032828). Другие примеры саморазрушаемых спейсеров помимо PAB включают, но этим не ограничиваются: (i) ароматические соединения, которые электронно сходны с PAB группой, такие как производные 2-аминоимидазол-5-метанола (Hay et al. (1999) Bioorg. Med. Chem. Lett. 9:2237), тиазолы US 2005/0256030), многочисленные удлиненные звенья PAB (de Groot et al. (2001) J. Org. Chem. 66:8815-8830; и орто или пара-аминобензилацетали; и (ii) гомологичные стирил PAB аналоги (US 7223837). Могут быть использованы спейсеры, которые подвергаются циклизации при гидролизе амидной связи, такие как замещенные и незамещенные амиды 4-аминомасляной кислоты (Rodrigues et al. (1995) Chemistry Biology 2:223), соответственно замещенные бицикло[2.2.1] и бицикло[2.2.2] кольцевые системы (Storm et al. (1972) J. Amer. Chem. Soc. 94:5815) и амиды 2-аминофенилпропионовой кислоты (Amsberry, et al. (1990) J. Org. Chem. 55:5867). Элиминация амин-содержащих лекарственных средств, которые замещены на глицине (Kingsbury et al. (1984) J. Med. Chem. 27:1447), также являются примерами саморазрушаемых спейсеров, используемых в ADC.
Линкерные реагенты, используемые для конъюгатов антитела с лекарственным средством в соответствии с настоящим изобретением, включают, но этим не ограничиваются: BMPEO, BMPS, EMCS, GMBS, HBVS, LC-SMCC, MBS, MPBH, SBAP, SIA, SIAB, SMCC, SMPB, SMPH, сульфо-EMCS, сульфо-GMBS, сульфо-KMUS, сульфо-MBS, сульфо-SIAB, сульфо-SMCC, и сульфо-SMPB, и SVSB (сукцинимидил-(4-винилсульфон)бензоат), и в том числе бис-малеимидные реагенты: DTME, BMB, BMDB, BMH, BMOE, 1,8-бис-малеимидодиэтиленгликоль (BM(PEO)2), и 1,11-бис-малеимидотриэтиленгликоль (BM(PEO)3), которые являются коммерчески доступными от Pierce Biotechnology, Inc., ThermoScientific, Rockford, IL, и других поставщиков реагентов. Бис-малеимидные реагенты дают возможность присоединять свободную тиольную группу остатка цистеина антитела к тиол-содержащей лекарственной части, метке или линкерному промежуточному соединению, последовательным или одновременным образом. Другие функциональные группы помимо малеимида, которые являются реакционно-способными в отношении тиольной группы антитела, части производного антрациклинового лекарственного средства, или линкерного промежуточного соединения, включают йодацетамид, бромацетамид, винилпиридин, дисульфид, пиридилдисульфид, изоцианат и изотиоцианат.

Другие линкерные реагенты представляют собой: N-сукцинимидил-4-(2-пиридилтио)пентаноат (SPP), N-сукцинимидил-3-(2-пиридилдитио)пропионат (SPDP, Carlsson et al. (1978) Biochem. J. 173:723-737), сукцинимидил-4-(N-малеимидометил) циклогексан-1-карбоксилат (SMCC), иминотиолан (IT), бифункциональные производные имидо сложных эфиров (например, диметиладипимидат HCl), активных сложных эфиров (например, дисукцинимидилсуберат), альдегиды (например, глутаральдегид), бис-азидо соединения (например, бис(п-азидобензоил)гександиамин), производные бис-диазония (например, бис-(п-диазонийбензоил)-этилендиамин), диизоцианаты (например, толуол 2,6-диизоцианат), и бис-активные соединения фтора (например, 1,5-дифтор-2,4-динитробензол). Подходящие линкерные реагенты также могут быть получены через другие коммерческие источники, такие как Molecular Biosciences Inc.(Boulder, CO), или синтезированы в соответствии со способами, описанными у Toki et al. (2002) J. Org. Chem. 67:1866-1872; US 6214345; WO 02/088172; US 2003130189; US2003096743; WO 03/026577; WO 03/043583; и WO 04/032828.
Линкер может представлять собой линкер дендритного типа для ковалентного присоединения более одной лекарственной части через разветвленную многофункциональную линкерную часть к антителу (US 2006/116422; US 2005/271615; de Groot et al. (2003) Angew. Chem. Int. Ed. 42:4490-4494; Amir et al. (2003) Angew. Chem. Int. Ed. 42:4494-4499; Shamis et al. (2004) J. Am. Chem. Soc. 126:1726-1731; Sun et al. (2002) Bioorganic & Medicinal Chemistry Letters 12:2213-2215; Sun et al. (2003) Bioorganic & Medicinal Chemistry 11:1761-1768; King et al. (2002) Tetrahedron Letters 43:1987-1990). Дендритные линкеры могут увеличивать молярное отношение лекарственного средства к антителу, то есть нагрузку, которая относится к эффективности ADC. Таким образом, в тех случаях, когда антитело несет только одну реакционно-способную тиольную группу цистеина, множество лекарственных частей может быть присоединено через дендритный разветвленный линкер.
НОСИТЕЛЬ
Несущую часть конъюгата производного антрациклина получают из поликлональных и моноклональных антител, белков или пептидов природного или синтетического происхождения. Соединения-носители T-NH2, T-COOH, T-CHO, T-SH, (V) или (VI) подходят для конъюгирования с соединениями производного антрациклина. Несущие части могут быть получены из поликлональных антител, индуцированных против опухоле-связанных антигенов; или из моноклональных антител, связывающихся с антигенами, предпочтительно или селективно экспрессированных на популяциях опухолевых клеток; или из природных или рекомбинантных пептидов или белков, или факторов роста, предпочтительно или селективно связывающихся с опухолевыми клетками; или из природных или синтетических полимерных носителей, таких как полилизин, полиглутаминовая кислота, полиаспарагиновая кислота и их аналогов и производных, или таких как декстран или других полимерных углеводных аналогов и их производных; или из синтетических сополимеров, например, полученных из N-(2-гидроксипропил)метакриламида (HPMA) смотри: J. Kopecek, Macromolecules. H. Benoit & P. Rempp, Ed.: 505-520 (1982) Pergamon Press. Oxford, England; или из поли(аминокислота)сополимеров, например, поли(GluNa, Ala, Tyr), которые используют в качестве направляемых носителей лекарственных средств для легочной ткани R. Duncan et al., Journal of Bioactive and Compatible Polymers, Vol 4, July 1989. Несущая часть также может быть получена из частей вышеупомянутых пептидов или белков, полученных с помощью технологий рекомбинантных ДНК.
АНТИТЕЛА
Характерными примерами упомянутых выше антител и соответствующих возможных терапевтических применений являются: антитело против Т-клеток T101 (Royston, I. et al., J. Immunol. 1980, 125:725); антитело против CD5 OKT1 (Ortho) ATCC CRL 8000 хронический лимфоцитарный лейкоз); антитело против CD20 IgG1 ибритумомаб, (Theuer, C. P. et al. Biotechnology Annual Review 2004 неходжкинская лимфома); анти-CD33 антитело huCD33, (Drug of the future 2000 25(7):686 острый миелоидный лейкоз); антитело OKT9 против рецептора трансферрина (Ortho) ATCC CRL 8021 опухоли яичника и другие опухоли; антитело против меланомы MAb 9.2.27 (Bumol, T. F. et al., Proc. Natl. Acad. Sci. USA 1982, 79:1245 меланомы); антитело против маркеров карциномы, например: анти-CEA 1116 NS-3d ATCC CRL 8019), анти-альфа-фетопротеин OM 3-1.1 ATCC HB 134 также гепатомы), 791T/36 (Embleton, M. J. et al., Br. J. Cancer 1981, 43, 582 также остеогенная саркома), B 72.3 (US 4522918 карциномы толстой и прямой кишки и другие опухоли), антитело против карциномы яичников OVB 3 ATCC HB 9147, антитело против рака молочной железы HMGF антиген (Aboud-Pirak, E. et al., Cancer Res. 1988, 48:3188), против карциномы мочевого пузыря 1G3.10 (Yu, D. S. et al., Eur. J. Urol. 1987, 13:198), анти-CanAg антитело huC242 антитело (Olcher, Anthony W. et al., Journal of Clinical Oncology 2003, 21(2):211-222 кишечника, поджелудочной железы, желудка), антитело против предстательной железы MLN591 (Henry, Michael D.et al., Cancer Research 2004 прогрессирующий гормонорезистентный рак предстательной железы).
Характерными примерами упомянутых выше факторов роста и белков природного и рекомбинантного происхождения являются FGF, EGF, PDGF, TGF-ALPHA, ALPHA-MS, интерлейкины, интерфероны, TNF, меланотропин (MSH), Mcm2 и т.д. Носитель T-CHO предпочтительно получают из поликлональных или моноклональных антител, имеющих углеводную часть, предпочтительно расположенную в области Fc, селективно окисленную до альдегидных групп с помощью химических или ферментативных способов, как описано в US 4671958.
Антительная единица (Ab) формулы IIc включает любую единицу, тип или класс антитела, которое связывается или реактивно ассоциируется или образует комплексы с рецептором, антигеном или другим рецепторным фрагментом, связанным с заданной популяцией клеток-мишеней. Антитело может представлять собой любой белок или белково-подобную молекулу, которая связывается, образует комплекс или взаимодействует с частью клеточной популяции, которую необходимо терапевтически или иным образом биологически модифицировать. В одном аспекте, антительная единица действует для доставки лекарственной части производного антрациклина к конкретной популяции клеток-мишеней, с которой взаимодействует антительная единица. Такие антитела включают, но этим не ограничиваются, белки с большой молекулярной массой, такие как полноразмерные антитела и фрагменты антител. Антитела формулы I позволяют достичь высоких концентраций молекул активных метаболитов в злокачественных клетках. Внутриклеточное нацеливание может достигаться с помощью способов и соединений, которые дают возможность для накопления или удерживания биологически активных агентов внутри клеток. Такое эффективное нацеливание может применяться к целому ряду фармацевтических композиций и способов.
В одном варианте осуществления, ADC специфически связывается с рецептором, кодируемым геном ErbB, такие как EGFR, HER2, HER3 и HER4. ADC может специфически связываться с внеклеточным доменом рецептора HER2. ADC может ингибировать рост опухолевой клетки, которая сверхэкспрессирует рецептор HER2.
В другом варианте осуществления, антитело (Ab) формулы IIc представляет собой гуманизированное антитело, такое как huMAb4D5-1, huMAb4D5-2, huMAb4D5-3, huMAb4D5-4, huMAb4D5-5, huMAb4D5-6, huMAb4D5-7 или huMAb4D5-8 (трастузумаб).
Антитела по изобретению включает сконструированные антитела с цистеиновыми заменами, где одна или несколько аминокислот любой формы дикого типа или родительского антитела замещены аминокислотой цистеином. Сконструированная аминокислота цистеин представляет собой свободную цистеиновую кислоту и не является частью внутрицепочечного или межцепочечного дисульфидного звена. Любая форма, тип или вариант антитела может быть сконструировано таким образом, то есть мутировано. Например, фрагмент Fab родительского антитела может быть сконструирован для образования сконструированного Fab с цистеиновыми заменами, называемого здесь «ТиоFab». Подобным образом, родительское моноклональное антитело может быть сконструировано для образования «ТиоMab». Следует отметить, что односайтовая мутация дает на выходе один сконструированный цистеиновый остаток ТиоFab, тогда как односайтовая мутация дает на выходе два сконструированных цистеиновых остатка в ТиоMab, благодаря димерной природе антитела IgG. Антитела с цистеиновыми заменами по изобретению включают моноклональные антитела, гуманизированные антитела или химерные моноклональные антитела, антиген-связывающие фрагменты антител, слитые полипептиды и аналоги, которые предпочтительно связываются с полипептидами, связанными с клетками.
Антитела с цистеиновыми заменами были созданы в виде фрагментов Fab антитела (тиоFab) и экспрессированы в виде полноразмерных IgG моноклональных (тиоMab) антител (US 7521541, содержание которого включено в качестве ссылки). Антитела ТиоFab и ТиоMab были конъюгированы посредством линкеров при вновь введенных тиолах цистеина с тиол-реактивными линкерными реагентами и реагентами лекарственное средство-линкер для получения конъюгатов антитела с лекарственным средством (Тио ADC), включая анти-HER2 (US 7521541), анти-CD22 (US 2008/0050310), и анти-steap1 (WO 2008/052187), а также других антител с цистеиновыми заменами и конъюгатов антитела с лекарственным средством.
Антитела, содержащие конъюгат антитела с лекарственным средством формулы IIc, предпочтительно сохраняют антигенсвязывающую способность их природных аналогов дикого типа. Таким образом, антитела по изобретению способны связываться, предпочтительно специфически, с антигенами. Такие антигены включают, например, опухоль-связанные антигены (TAA), рецепторные белки клеточной поверхности и другие молекулы клеточной поверхности, регуляторные факторы выживаемости клетки, регуляторные факторы клеточной пролиферации, молекулы, связанные с (например, известные или предполагаемые, способствующие функциональной активности) развитием ткани или дифференцировкой, лимфокинами, цитокинами, молекулами, вовлеченными в регуляцию клеточного цикла, молекулами, вовлеченными в образование и развитие сосудов, и молекулами, связанными с (например, известными или предполагаемыми, способствующими функциональной активности) ангиогенезом. Опухоль-связанный антиген может представлять собой фактор кластера дифференцировки (то есть, белок CD). Антиген, с которым антитело по изобретению способно связываться, может быть представителем субпопуляции одной из упомянутых выше категорий, где другая субпопуляция (субпопуляции) указанной категории содержит другие молекулы/антигены, которые имеют другие характеристики (относительно антигена, представляющего интерес).
В одном варианте осуществления антитело конъюгатов антитела с лекарственным средством (ADC) формулы IIc специфически связывается с рецептором, кодируемым геном ErbB. Антитело может специфически связываться с рецептором ErbB, выбранным из EGFR, HER2, HER3 и HER4. ADC может специфически связываться с внеклеточным доменом (ECD) рецептора HER2 и ингибировать рост опухолевых клеток, которые сверхэкспрессируют рецептор HER2. Антитело ADC может представлять собой моноклональное антитело, например моноклональное антитело мыши, химерное антитело, или гуманизированное антитело. Гуманизированное антитело может представлять собой huMAb4D5-1, huMAb4D5-2, huMAb4D5-3, huMAb4D5-4, huMAb4D5-5, huMAb4D5-6, huMAb4D5-7 или huMAb4D5-8 (трастузумаб). Антитело может представлять собой фрагмент антитела, например, фрагмент Fab.
Антитела в формуле IIc конъюгатов антитела с лекарственным средством (ADC) и которые могут быть использованы при лечении злокачественного заболевания, включают, но этим не ограничиваются, антитела против рецепторов клеточной поверхности и связанных с опухолью антигенов (TAA). Такие опухоль-связанные антигены известны из уровня техники и могут быть получены для использования в получении антител, с использованием способов и информации, хорошо известных из уровня техники. В попытках обнаружить эффективные клеточные мишени для диагностики и лечения злокачественных заболеваний, исследователи стремились идентифицировать трансмембранные или иным образом связанные с опухолью полипептиды, которые специфически экспрессированы на поверхности одного или нескольких конкретных типов злокачественных клеток по сравнению с одной или несколькими конкретными типами нормальных незлокачественных клеток. Зачастую, такие связанные с опухолью полипептиды более обильно экспрессированы на поверхности злокачественных клеток по сравнению с поверхностью незлокачественных клеток. Идентификация таких антигенных полипептидов клеточной поверхности, связанных с опухолью, обусловило способность к специфическому нацеливанию на злокачественные клетки для разрушения посредством способов лечения, основанных на антителах.
Примеры TAA включают, но этим не ограничиваются, антигены, связанные с опухолями (1)-(36), перечисленные далее. Для удобства, информация, касающаяся этих антигенов, все которые известны из уровня техники, перечислены далее и включают названия, альтернативные названия, регистрационные номера Genbank и основную ссылку (ссылки), с последующими условными обозначениями для идентификации последовательностей нуклеиновой кислоты и белка Национального центра биотехнологической информации (NCBI). Последовательности нуклеиновой кислоты и белка, соответствующие TAA (1)-(36), доступны в базах данных общего пользования, таких как GenBank. Антигены, связанные с опухолью, на которые нацелены антитела, включают все варианты аминокислотных последовательностей и изоформы, по меньшей мере примерно на 70%, 80%, 85%, 90%, или 95% идентичные последовательностям, идентифицированным в процитированных ссылках, или которые демонстрируют по существу такие же биологические свойства или характеристики, что и TAA, имеющий последовательность, найденную в цитированных ссылках. Например, TAA, имеющий вариант последовательности, в основном способен специфически связываться с антителом, которое специфически связывается с TAA с соответствующей приведенной последовательностью. Последовательности и описания в ссылке, специально процитированной в настоящей заявке, специально включены путем ссылки.
АНТИГЕНЫ, СВЯЗАННЫЕ С ОПУХОЛЬЮ (1)-(36):
(1) BMPR1B (рецептор костного морфогенетического белка-тип IB, Genbank регистрационный номер NM_001203); ten Dijke,P., et al. Science 264 (5155):101-104 (1994), Oncogene 14 (11):1377-1382 (1997)); WO2004063362 (Пункт 2); WO2003042661 (Пункт 12); US2003134790-A1 (страница 38-39); WO2002102235 (Пункт 13; страница 296); WO2003055443 (страница 91-92); WO200299122 (Пример 2; страница 528-530); WO2003029421 (Пункт 6); WO2003024392 (Пункт 2; Фиг. 112); WO200298358 (Пункт 1; страница 183); WO200254940 (страница 100-101); WO200259377(страница 349-350); WO200230268 (Пункт 27; страница 376); WO200148204 (Пример; Фиг 4); NP_001194 рецептор костного морфогенетического белка, тип IB /pid=NP_001194.1; Перекрестные ссылки: MIM:603248; NP_001194.1; AY065994;
(2) E16 (LAT1, SLC7A5, Genbank регистрационный номер NM_003486); Biochem. Biophys. Res. Commun. 255 (2), 283-288 (1999), Nature 395 (6699):288-291 (1998), Gaugitsch, H.W., et al. (1992) J. Biol. Chem. 267 (16):11267-11273); WO2004048938 (Пример 2); WO2004032842 (Пример IV); WO2003042661 (Пункт 12); WO2003016475 (Пункт 1); WO200278524 (Пример 2); WO200299074 (Пункт 19; страница 127-129); WO200286443 (Пункт 27; страницы 222, 393); WO2003003906 (Пункт 10; страница 293); WO200264798 (Пункт 33; страница 93-95); WO200014228 (Пункт 5; страница 133-136); US2003224454 (Фиг. 3); WO2003025138 (Пункт 12; страница 150); US 20050107595; US 20050106644; NP_003477 растворенный носитель семейство 7 (катионный переносчик аминокислот, система y+ системы), компонент 5 /pid=NP_003477.3 - Homo sapiens; Перекрестные ссылки: MIM:600182; NP_003477.3; NM_015923; NM_003486_1;
(3) STEAP1 (шестой трансмембранный эпителиальный антиген предстательной железы, Genbank регистрационный номер NM_012449); Cancer Res. 61 (15), 5857-5860 (2001), Hubert, R.S., et al. (1999) Proc. Natl. Acad. Sci. U.S.A. 96 (25):14523-14528); WO2004065577 (Пункт 6); WO2004027049 (Фиг 1L); EP1394274 (Пример 11); WO2004016225 (Пункт 2); WO2003042661 (Пункт 12); US2003157089 (Пример 5); US2003185830 (Пример 5); US2003064397 (Фиг 2); WO200289747 (Пример 5; страница 618-619); WO2003022995 (Пример 9; Фиг 13A, Пример 53; страница 173, Пример 2; Фиг. 2A); NP_036581 шестой трансмембранный эпителиальный антиген предстательной железы; Перекрестные ссылки: MIM:604415; NP_036581.1; NM_012449_1;
(4) 0772P (CA125, MUC16, Genbank регистрационный номер AF361486); J. Biol. Chem. 276 (29):27371-27375 (2001)); WO2004045553 (Пункт 14); WO200292836 (Пункт 6; Фиг 12); WO200283866 (Пункт 15; страница 116-121); US2003124140 (Пример 16); US2003091580 (Пункт 6); WO200206317 (Пункт 6; страница 400-408); Перекрестные ссылки: GI:34501467; AAK74120.3; AF361486_1;
(5) MPF (MPF, MSLN, SMR, мегакариоцитарный потенцирующий фактор, мезотелин, Genbank регистрационный номер NM_005823); Yamaguchi, N., et al. Biol. Chem. 269 (2), 805-808 (1994), Proc. Natl. Acad. Sci. U.S.A. 96 (20):11531-11536 (1999), Proc. Natl. Acad. Sci. U.S.A. 93 (1):136-140 (1996), J. Biol. Chem. 270 (37):21984-21990 (1995)); WO2003101283 (Пункт 14); (WO2002102235 (Пункт 13; страница 287-288); WO2002101075 (Пункт 4; страница 308-309); WO200271928 (страница 320-321); WO9410312 (страница 52-57); Перекрестные ссылки: MIM:601051; NP_005814.2; NM_005823_1;
(6) Napi3b (NAPI-3B, NPTIIb, SLC34A2, растворенный носитель семейство 34 (натрия фосфат), компонент 2, тип II натрий-зависимый переносчик фосфата 3b, Genbank регистрационный номер NM_006424); J. Biol. Chem. 277 (22):19665-19672 (2002), Genomics 62 (2):281-284 (1999), Feild, J.A., et al. (1999) Biochem. Biophys. Res. Commun. 258 (3):578-582); WO2004022778 (Пункт 2); EP1394274 (Пример 11); WO2002102235 (Пункт 13; страница 326); EP875569 (Пункт 1; страница 17-19); WO200157188 (Пункт 20; страница 329); WO2004032842 (Пример IV); WO200175177 (Пункт 24; страница 139-140); Перекрестные ссылки: MIM:604217; NP_006415.1; NM_006424_1;
(7) Sema 5b (FLJ10372, KIAA1445, Мм.42015, SEMA5B, SEMAG, Семафорин 5b Hlog, sema домен, семь тромбоспондиновых повторов (тип 1 и подобные типу 1), трансмембранный домен (TM) и короткий цитоплазматический домен, (семафорин) 5B, Genbank регистрационный номер AB040878); Nagase T., et al. (2000) DNA Res. 7 (2):143-150); WO2004000997 (Пункт 1); WO2003003984 (Пункт 1); WO200206339 (Пункт 1; страница 50); WO200188133 (Пункт 1; страница 41-43, 48-58); WO2003054152 (Пункт 20); WO2003101400 (Пункт 11); Accession: Q9P283; EMBL; AB040878; BAA95969.1. Genew; HGNC:10737;
(8) PSCA hlg (2700050C12Rik, C530008O16Rik, RIKEN кДНК 2700050C12, RIKEN кДНК 2700050C12 ген, Genbank регистрационный номер AY358628); Ross et al. (2002) Cancer Res. 62:2546-2553; US2003129192 (Пункт 2); US2004044180 (Пункт 12); US2004044179 (Пункт 11); US2003096961 (Пункт 11); US2003232056 (Пример 5); WO2003105758 (Пункт 12); US2003206918 (Пример 5); EP1347046 (Пункт 1); WO2003025148 (Пункт 20); Перекрестные ссылки: GI:37182378; AAQ88991.1; AY358628_1;
(9) ETBR (рецептор эндотелина тип B, Genbank регистрационный номер AY275463); Nakamuta M., et al. Biochem. Biophys. Res. Commun. 177, 34-39, 1991; Ogawa Y., et al. Biochem. Biophys. Res. Commun. 178, 248-255, 1991; Arai H., et al. Jpn. Circ. J. 56, 1303-1307, 1992; Arai H., et al. J. Biol. Chem. 268, 3463-3470, 1993; Sakamoto A., Yanagisawa M., et al. Biochem. Biophys. Res. Commun. 178, 656-663, 1991; Elshourbagy N.A., et al. J. Biol. Chem. 268, 3873-3879, 1993; Haendler B., et al. J. Cardiovasc. Pharmacol. 20, s1-S4, 1992; Tsutsumi M., et al. Gene 228, 43-49, 1999; Strausberg R.L., et al. Proc. Natl. Acad. Sci. U.S.A. 99, 16899-16903, 2002; Bourgeois C., et al. J. Clin. Endocrinol. Metab. 82, 3116-3123, 1997; Okamoto Y., et al. Biol. Chem. 272, 21589-21596, 1997; Verheij J.B., et al. Am. J. Med. Genet. 108, 223-225, 2002; Hofstra R.M.W., et al. Eur. J. Hum. Genet. 5, 180-185, 1997; Puffenberger E. G. et al. Cell 79, 1257-1266, 1994; Attie T., et al., Hum. Mol. Genet. 4, 2407-2409, 1995; Auricchio A., et al. Hum. Mol. Genet. 5:351-354, 1996; Amiel J., et al. Hum. Mol. Genet. 5, 355-357, 1996; Hofstra R.M.W., et al. Nat. Genet. 12, 445-447, 1996; Svensson P.J., et al. Hum. Genet. 103, 145-148, 1998; Fuchs S., et al. Mol. Med. 7, 115-124, 2001; Pingault V., et al. (2002) Hum. Genet. 111, 198-206; WO2004045516 (Пункт 1); WO2004048938 (Пример 2); WO2004040000 (Пункт 151); WO2003087768 (Пункт 1); WO2003016475 (Пункт 1); WO2003016475 (Пункт 1); WO200261087 (Фиг 1); WO2003016494 (Фиг 6); WO2003025138 (Пункт 12; страница 144); WO200198351 (Пункт 1; страница 124-125); EP522868 (Пункт 8; Фиг 2); WO200177172 (Пункт 1; страница 297-299); US2003109676; US6518404 (Фиг 3); US5773223 (Пункт 1a; кол 31-34); WO2004001004;
(10) MSG783 (RNF124, гипотетический белок FLJ20315, Genbank регистрационный номер NM_017763); WO2003104275 (Пункт 1); WO2004046342 (Пример 2); WO2003042661 (Пункт 12); WO2003083074 (Пункт 14; страница 61); WO2003018621 (Пункт 1); WO2003024392 (Пункт 2; Фиг 93); WO200166689 (Пример 6); Перекрестные ссылки: LocusID:54894; NP_060233.2; NM_017763_1;
(11) STEAP2 (HGNC_8639, IPCA-1, PCANAP1, STAMP1, STEAP2, STMP, ген, связанный с раком предстательной железы, 1, белок, связанный с раком предстательной железы, 1, шестой трансмембранный эпителиальный антиген предстательной железы 2, шестой трансмембранный белок предстательной железы, Genbank регистрационный номер AF455138); Lab. Invest. 82 (11):1573-1582 (2002)); WO2003087306; US2003064397 (Пункт 1; Фиг 1); WO200272596 (Пункт 13; страница 54-55); WO200172962 (Пункт 1; Фиг 4B); WO2003104270 (Пункт 11); WO2003104270 (Пункт 16); US2004005598 (Пункт 22); WO2003042661 (Пункт 12); US2003060612 (Пункт 12; Фиг 10); WO200226822 (Пункт 23; Фиг 2); WO200216429 (Пункт 12; Фиг 10); Перекрестные ссылки: GI:22655488; AAN04080.1; AF455138_1;
(12) TrpM4 (BR22450, FLJ20041, TRPM4, TRPM4B, транзиторный рецептор катионного канала потенциала, подсемейство M, компонент 4, Genbank регистрационный номер NM_017636); Xu, X.Z., et al. Proc. Natl. Acad. Sci. U.S.A. 98 (19):10692-10697 (2001), Cell 109 (3):397-407 (2002), J. Biol. Chem. 278 (33):30813-30820 (2003)); US2003143557 (Пункт 4); WO200040614 (Пункт 14; страница 100-103); WO200210382 (Пункт 1; Фиг 9A); WO2003042661 (Пункт 12); WO200230268 (Пункт 27; страница 391); US2003219806 (Пункт 4); WO200162794 (Пункт 14; Фиг 1A-D); Перекрестные ссылки: MIM:606936; NP_060106.2; NM_017636_1;
(13) CRIPTO (CR, CR1, CRGF, CRIPTO, TDGF1, тератокарциномный фактор роста, Genbank регистрационный номер NP_003203 или NM_003212); Ciccodicola, A., et al. EMBO J. 8 (7):1987-1991 (1989), Am. J. Hum. Genet. 49 (3):555-565 (1991)); US2003224411 (Пункт 1); WO2003083041 (Пример 1); WO2003034984 (Пункт 12); WO200288170 (Пункт 2; страница 52-53); WO2003024392 (Пункт 2; Фиг 58); WO200216413 (Пункт 1; страница 94-95, 105); WO200222808 (Пункт 2; Фиг 1); US5854399 (Пример 2; Col 17-18); US5792616 (Фиг 2); Перекрестные ссылки: MIM:187395; NP_003203.1; NM_003212_1;
(14) CD21 (CR2 (рецептор комплемента 2) или C3DR (C3d/рецептор вируса Эпштейна-Барр) или Hs.73792 Genbank регистрационный номер M26004); Fujisaku et al. (1989) J. Biol. Chem. 264 (4):2118-2125); Weis J.J., et al. J. Exp. Med. 167, 1047-1066, 1988; Moore M., et al. Proc. Natl. Acad. Sci. U.S.A. 84, 9194-9198, 1987; Barel M., et al. Mol. Immunol. 35, 1025-1031, 1998; Weis J.J., et al. Proc. Natl. Acad. Sci. U.S.A. 83, 5639-5643, 1986; Sinha S.K., et al. (1993) J. Immunol. 150, 5311-5320; WO2004045520 (Пример 4); US2004005538 (Пример 1); WO2003062401 (Пункт 9); WO2004045520 (Пример 4); WO9102536 (Фиг 9.1-9.9); WO2004020595 (Пункт 1); Accession: P20023; Q13866; Q14212; EMBL; M26004; AAA35786.1;
(15) CD79b (CD79B, CD79β, IGb (иммуноглобулин-связанный бета), B29, Genbank регистрационный номер NM_000626 или 11038674); Proc. Natl. Acad. Sci. U.S.A. (2003) 100 (7):4126-4131, Blood (2002) 100 (9):3068-3076, Muller et al. (1992) Eur. J. Immunol. 22 (6):1621-1625); WO2004016225 (Пункт 2, Фиг 140); WO2003087768, US2004101874 (Пункт 1, страница 102); WO2003062401 (Пункт 9); WO200278524 (Пример 2); US2002150573 (Пункт 5, страница 15); US5644033; WO2003048202 (Пункт 1, страницы 306 и 309); WO 99/558658, US6534482 (Пункт 13, Фиг 17A/B); WO200055351 (Пункт 11, страницы 1145-1146); Перекрестные ссылки: MIM:147245; NP_000617.1; NM_000626_1;
(16) FcRH2 (IFGP4, IRTA4, SPAP1A (SH2 домен, содержащий якорный белок фосфатазы 1a), SPAP1B, SPAP1C, Genbank регистрационный номер NM_030764, AY358130); Genome Res. 13 (10):2265-2270 (2003), Immunogenetics 54 (2):87-95 (2002), Blood 99 (8):2662-2669 (2002), Proc. Natl. Acad. Sci. U.S.A. 98 (17):9772-9777 (2001), Xu, M.J., et al. (2001) Biochem. Biophys. Res. Commun. 280 (3):768-775; WO2004016225 (Пункт 2); WO2003077836; WO200138490 (Пункт 5; Фиг 18D-1-18D-2); WO2003097803 (Пункт 12); WO2003089624 (Пункт 25); Перекрестные ссылки: MIM:606509; NP_110391.2; NM_030764_1;
(17) HER2 (ErbB2, Genbank регистрационный номер M11730); Coussens L., et al. Science (1985) 230(4730):1132-1139); Yamamoto T., et al. Nature 319, 230-234, 1986; Semba K., et al. Proc. Natl. Acad. Sci. U.S.A. 82, 6497-6501, 1985; Swiercz J.M., et al. J. Cell Biol. 165, 869-880, 2004; Kuhns J.J., et al. J. Biol. Chem. 274, 36422-36427, 1999; Cho H.-S., et al. Nature 421, 756-760, 2003; Ehsani A., et al. (1993) Genomics 15, 426-429; WO2004048938 (Пример 2); WO2004027049 (Фиг 1I); WO2004009622; WO2003081210; WO2003089904 (Пункт 9); WO2003016475 (Пункт 1); US2003118592; WO2003008537 (Пункт 1); WO2003055439 (Пункт 29; Фиг 1A-B); WO2003025228 (Пункт 37; Фиг 5C); WO200222636 (Пример 13; страница 95-107); WO200212341 (Пункт 68; Фиг 7); WO200213847 (страница 71-74); WO200214503 (страница 114-117); WO200153463 (Пункт 2; страница 41-46); WO200141787 (страница 15); WO200044899 (Пункт 52; Фиг 7); WO200020579 (Пункт 3; Фиг 2); US5869445 (Пункт 3; Col 31-38); WO9630514 (Пункт 2; страница 56-61); EP1439393 (Пункт 7); WO2004043361 (Пункт 7); WO2004022709; WO200100244 (Пример 3; Фиг 4); Доступ: P04626; EMBL; M11767; AAA35808.1. EMBL; M11761; AAA35808.1;
(18) NCA (CEACAM6, Genbank регистрационный номер M18728); Barnett T., et al. Genomics 3, 59-66, 1988; Tawaragi Y., et al. Biochem. Biophys. Res. Commun. 150, 89-96, 1988; Strausberg R.L., et al. Proc. Natl. Acad. Sci. U.S.A. 99:16899-16903, 2002; WO2004063709; EP1439393 (Пункт 7); WO2004044178 (Пример 4); WO2004031238; WO2003042661 (Пункт 12); WO200278524 (Пример 2); WO200286443 (Пункт 27; страница 427); WO200260317 (Пункт 2); Доступ: P40199; Q14920; EMBL; M29541; AAA59915.1. EMBL; M18728;
(19) MDP (DPEP1, Genbank регистрационный номер BC017023); Proc. Natl. Acad. Sci. U.S.A. 99 (26):16899-16903 (2002)); WO2003016475 (Пункт 1); WO200264798 (Пункт 33; страница 85-87); JP05003790 (Фиг 6-8); WO9946284 (Фиг 9); Перекрестные ссылки: MIM:179780; AAH17023.1; BC017023_1;
(20) IL20Rα (IL20Ra, ZCYTOR7, Genbank регистрационный номер AF184971); Clark H.F., et al. Genome Res. 13, 2265-2270, 2003; Mungall A.J., et al. Nature 425, 805-811, 2003; Blumberg H., et al. Cell 104, 9-19, 2001; Dumoutier L., et al. J. Immunol. 167, 3545-3549, 2001; Parrish-Novak J., et al. J. Biol. Chem. 277, 47517-47523, 2002; Pletnev S., et al. (2003) Biochemistry 42:12617-12624; Sheikh F., et al. (2004) J. Immunol. 172, 2006-2010; EP1394274 (Пример 11); US2004005320 (Пример 5); WO2003029262 (страница 74-75); WO2003002717 (Пункт 2; страница 63); WO200222153 (страница 45-47); US2002042366 (страница 20-21); WO200146261 (страница 57-59); WO200146232 (страница 63-65); WO9837193 (Пункт 1; страница 55-59); Доступ: Q9UHF4; Q6UWA9; Q96SH8; EMBL; AF184971; AAF01320.1;
(21) Бревикан (BCAN, BEHAB, Genbank регистрационный номер AF229053); Gary S.C., et al. Gene 256, 139-147, 2000; Clark H.F., et al. Genome Res. 13, 2265-2270, 2003; Strausberg R.L., et al. Proc. Natl. Acad. Sci. U.S.A. 99, 16899-16903, 2002; US2003186372 (Пункт 11); US2003186373 (Пункт 11); US2003119131 (Пункт 1; Фиг 52); US2003119122 (Пункт 1; Фиг 52); US2003119126 (Пункт 1); US2003119121 (Пункт 1; Фиг 52); US2003119129 (Пункт 1); US2003119130 (Пункт 1); US2003119128 (Пункт 1; Фиг 52); US2003119125 (Пункт 1); WO2003016475 (Пункт 1); WO200202634 (Пункт 1);
(22) EphB2R (DRT, ERK, Hek5, EPHT3, Tyro5, Genbank регистрационный номер NM_004442); Chan, J. and Watt, V.M., Oncogene 6 (6), 1057-1061 (1991) Oncogene 10 (5):897-905 (1995), Annu. Rev. Neurosci. 21:309-345 (1998), Int. Rev. Cytol. 196:177-244 (2000)); WO2003042661 (Пункт 12); WO200053216 (Пункт 1; страница 41); WO2004065576 (Пункт 1); WO2004020583 (Пункт 9); WO2003004529 (страница 128-132); WO200053216 (Пункт 1; страница 42); Перекрестные ссылки: MIM:600997; NP_004433.2; NM_004442_1;
(23) ASLG659 (B7h, Genbank регистрационный номер AX092328); US20040101899 (Пункт 2); WO2003104399 (Пункт 11); WO2004000221 (Фиг 3); US2003165504 (Пункт 1); US2003124140 (Пример 2); US2003065143 (Фиг 60); WO2002102235 (Пункт 13; страница 299); US2003091580 (Пример 2); WO200210187 (Пункт 6; Фиг 10); WO200194641 (Пункт 12; Фиг 7b); WO200202624 (Пункт 13; Фиг 1A-1B); US2002034749 (Пункт 54; страница 45-46); WO200206317 (Пример 2; страница 320-321, пункт 34; страница 321-322); WO200271928 (страница 468-469); WO200202587 (Пример 1; Фиг 1); WO200140269 (Пример 3; страницы 190-192); WO200036107 (Пример 2; страница 205-207); WO2004053079 (Пункт 12); WO2003004989 (Пункт 1); WO200271928 (страница 233-234, 452-453); WO 0116318;
(24) PSCA (предшественник антигена стволовой клетки предстательной железы, Genbank регистрационный номер AJ297436); Reiter R.E., et al. Proc. Natl. Acad. Sci. U.S.A. 95, 1735-1740, 1998; Gu Z., et al. Oncogene 19, 1288-1296, 2000; Biochem. Biophys. Res. Commun. (2000) 275(3):783-788; WO2004022709; EP1394274 (Пример 11); US2004018553 (Пункт 17); WO2003008537 (Пункт 1); WO200281646 (Пункт 1; страница 164); WO2003003906 (Пункт 10; страница 288); WO200140309 (Пример 1; Фиг 17); US2001055751 (Пример 1; Фиг 1b); WO200032752 (Пункт 18; Фиг 1); WO9851805 (Пункт 17; страница 97); WO9851824 (Пункт 10; страница 94); WO9840403 (Пункт 2; Фиг 1B); Accession: O43653; EMBL; AF043498; AAC39607.1;
(25) GEDA (Genbank регистрационный номер AY260763); AAP14954 липома HMGIC белок, подобный слитым клеткам /pid=AAP14954.1 Homo sapiens (человек); WO2003054152 (Пункт 20); WO2003000842 (Пункт 1); WO2003023013 (Пример 3, пункт 20); US2003194704 (Пункт 45); Перекрестные ссылки: GI:30102449; AAP14954.1; AY260763_1;
(26) BAFF-R (рецептор фактора, активирующего В-клетки, BLyS рецептор 3, BR3, Genbank регистрационный номер AF116456); BAFF рецептор /pid=NP_443177.1- Homo sapiens; Thompson, J.S., et al. Science 293 (5537), 2108-2111 (2001); WO2004058309; WO2004011611; WO2003045422 (Пример; страница 32-33); WO2003014294 (Пункт 35; Фиг 6B); WO2003035846 (Пункт 70; страница 615-616); WO200294852 (Col 136-137); WO200238766 (Пункт 3; страница 133); WO200224909 (Пример 3; Фиг 3); Перекрестные ссылки: MIM:606269; NP_443177.1; NM_052945_1; AF132600;
(27) CD22 (B-клеточный рецептор CD22-B изоформа, BL-CAM, Lyb-8, Lyb8, SIGLEC-2, FLJ22814, Genbank регистрационный номер AK026467); Wilson et al. (1991) J. Exp. Med. 173:137-146; WO2003072036 (Пункт 1; Фиг 1); Перекрестные ссылки: MIM:107266; NP_001762.1; NM_001771_1;
(28) CD79a (CD79A, CD79α, иммуноглобулин-связанный альфа, специфичный белок В-клеток, который ковалентно взаимодействует с Ig бета (CD79B) и образует комплекс на поверхности с молекулами Ig M, трансдуцирует сигнал, вовлеченный в дифференцировку В-клеток); 226 aa), pI: 4,84, ММ: 25028 TM: 2 [P] ген хромосома: 19q13.2, Genbank регистрационный номер NP_001774.10); WO2003088808, US20030228319; WO2003062401 (Пункт 9); US2002150573 (Пункт 4, страницы 13-14); WO9958658 (Пункт 13, Фиг 16); WO9207574 (Фиг 1); US5644033; Ha et al. (1992) J. Immunol. 148(5):1526-1531; Mueller et al. (1992) Eur. J. Biochem. 22:1621-1625; Hashimoto et al. (1994) Immunogenetics 40(4):287-295; Preud'homme et al. (1992) Clin. Exp. Immunol. 90(1):141-146; Yu et al. (1992) J. Immunol. 148(2) 633-637; Sakaguchi et al. (1988) EMBO J. 7(11):3457-3464;
(29) CXCR5 (рецептор 1 лимфомы Беркитта, рецептор, сопряженный с G белком, то есть активированный под действием хемокина CXCL13, действует при миграции лимфоцитов и гуморальной защите, играет роль в инфицировании HIV-2 и возможно развитии СПИДа, лимфомы, миеломы и лейкоза); 372 aa), pI: 8,54 ММ: 41959 TM: 7 [P] ген хромосома: 11q23.3, Genbank регистрационный номер NP_001707.1); WO2004040000; WO2004015426; US2003105292 (Пример 2); US6555339 (Пример 2); WO200261087 (Фиг 1); WO200157188 (Пункт 20, страница 269); WO200172830 (страницы 12-13); WO200022129 (Пример 1, страницы 152-153, Пример 2, страницы 254-256); WO9928468 (Пункт 1, страница 38); US5440021 (Пример 2, кол 49-52); WO9428931 (страницы 56-58); WO9217497 (Пункт 7, Фиг 5); Dobner et al. (1992) Eur. J. Immunol. 22:2795-2799; Barella et al. (1995) Biochem. J. 309:773-779;
(30) HLA-DOB (бета субъединица молекулы MHC II класса (Ia антиген), который связывается с пептидами и представляет их CD4+ T-лимфоцитам); 273 aa, pI: 6,56 ММ: 30820 TM: 1 [P] ген хромосома: 6p21.3, Genbank регистрационный номер NP_002111.1); Tonnelle et al. (1985) EMBO J. 4(11):2839-2847; Jonsson et al. (1989) Immunogenetics 29(6):411-413; Beck et al. (1992) J. Mol. Biol. 228:433-441; Strausberg et al. (2002) Proc. Natl. Acad. Sci USA 99:16899-16903; Servenius et al. (1987) J. Biol. Chem. 262:8759-8766; Beck et al. (1996) J. Mol. Biol. 255:1-13; Naruse et al. (2002) Tissue Antigens 59:512-519; WO9958658 (Пункт 13, Фиг 15); US6153408 (Кол 35-38); US5976551 (col 168-170); US6011146 (col 145-146); Kasahara et al. (1989) Immunogenetics 30(1):66-68; Larhammar et al. (1985) J. Biol. Chem. 260(26):14111-14119;
(31) P2X5 (пуринергический рецептор P2X лиганд-зависимый ионный канал 5, ионный канал, управляемй внеклеточным АТФ, может быть вовлечен в синаптическую трансмиссию и нейрогенез, недостаточность может вносить вклад в патофизиологию идиопатической нестабильности детрузора); 422 aa), pI: 7,63, ММ: 47206 TM: 1 [P] ген хромосома: 17p13.3, Genbank регистрационный номер NP_002552.2); Le et al. (1997) FEBS Lett. 418(1-2):195-199; WO2004047749; WO2003072035 (Пункт 10); Touchman et al. (2000) Genome Res. 10:165-173; WO200222660 (Пункт 20); WO2003093444 (Пункт 1); WO2003087768 (Пункт 1); WO2003029277 (страница 82);
(32) CD72 (Антиген дифференцировки B-клеток CD72, Lyb-2); 359 aa), pI: 8,66, ММ: 40225 TM: 1 [P] ген хромосома: 9p13.3, Genbank регистрационный номер NP_001773.1); WO2004042346 (Пункт 65); WO2003026493 (страницы 51-52, 57-58); WO200075655 (страницы 105-106); Von Hoegen et al. (1990) J. Immunol. 144(12):4870-4877; Strausberg et al. (2002) Proc. Natl. Acad. Sci USA 99:16899-16903;
(33) LY64 (лимфоцитарный антиген 64 (RP105), тип I мембранный белок семейства, богатого лейциновыми повторами (LRR), регулирует B-клеточную активацию и апоптоз, утрата функции связана с повышенной патологической активностью у пациентов с системной красной волчанкой); 661 aa), pI: 6,20, ММ: 74147 TM: 1 [P] ген хромосома: 5q12, Genbank регистрационный номер NP_005573.1); US2002193567; WO9707198 (Пункт 11, страницы 39-42); Miura et al. (1996) Genomics 38(3):299-304; Miura et al. (1998) Blood 92:2815-2822; WO2003083047; WO9744452 (Пункт 8, страницы 57-61); WO200012130 (страницы 24-26);
(34) FcRH1 (белок 1, подобный Fc рецептору, a предполагаемый рецептор для Fc домена иммуноглобулина, который содержит C2 тип Ig-подобных и ITAM доменов, может играть роль в дифференцировке В-лимфоцитов); 429 aa), pI: 5,28, ММ: 46925 TM: 1 [P] ген хромосома: 1q21-1q22, Genbank регистрационный номер NP_443170.1); WO2003077836; WO200138490 (Пункт 6, Фиг 18E-1-18-E-2); Davis et al. (2001) Proc. Natl. Acad. Sci USA 98(17):9772-9777; WO2003089624 (Пункт 8); EP1347046 (Пункт 1); WO2003089624 (Пункт 7);
(35) IRTA2 (FcRH5, рецептор суперсемейства иммуноглобулинов, связанный с транслокацией 2, предполагаемый иммунорецептор с возможными ролями в развитии В-клеток и лимфомагенезе; дезрегуляция этого гена под действием транслокации встречается при некоторых В-клеточных злокачественных заболеваниях); 977 aa), pI: 6,88 ММ: 106468 TM: 1 [P] ген хромосома: 1q21, (Genbank регистрационный номер Человек: AF343662, AF343663, AF343664, AF343665, AF369794, AF397453, AK090423, AK090475, AL834187, AY358085; Мышь: AK089756, AY158090, AY506558; NP_112571.1); WO2003024392 (Пункт 2, Фиг 97); Nakayama et al. (2000) Biochem. Biophys. Res. Commun. 277(1):124-127; WO2003077836; WO200138490 (Пункт 3, Фиг 18B-1-18B-2);
(36) TENB2 (TMEFF2, томорегулин, TPEF, HPP1, TR, предполагаемый трансмембранный протеогликан, относящийся к семейству факторов роста EGF/герегулин и фоллистатину); 374 aa, Доступ в NCBI: AAD55776, AAF91397, AAG49451, NCBI RefSeq: NP_057276; NCBI Gene: 23671; OMIM: 605734; SwissProt Q9UIK5; (Genbank регистрационный номер AF179274; AY358907, CAF85723, CQ782436); WO2004074320; JP2004113151; WO2003042661; WO2003009814; EP1295944 (страницы 69-70); WO200230268 (страница 329); WO200190304; US2004249130; US2004022727; WO2004063355; US2004197325; US2003232350; US2004005563; US2003124579; US 6410506; US 6642006l; Horie et al. (2000) Genomics 67:146-152; Uchida et al. (1999) Biochem. Biophys. Res. Commun. 266:593-602; Liang et al. (2000) Cancer Res. 60:4907-12; Glynne-Jones et al. (2001) Int J Cancer. Oct 15;94(2):178-84.
Дополнительные предпочтительные соединения представляют собой соединения формулы (Ia), (Ib), (I'a) и (I'b) представленные как соединения 1-14 в таблице 1 далее:
[Ant-L-Z-]m -T (I) Ant-L-Z-Q (I')
ние
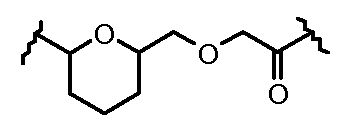
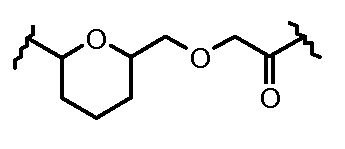

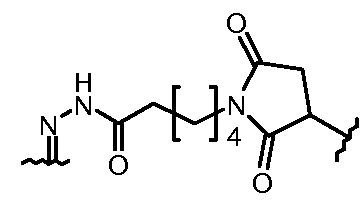
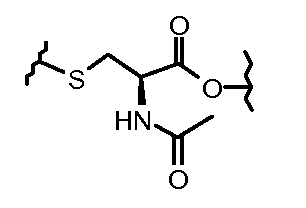
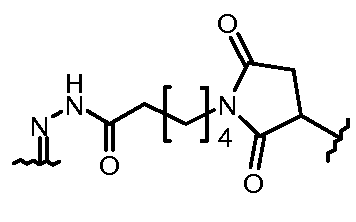
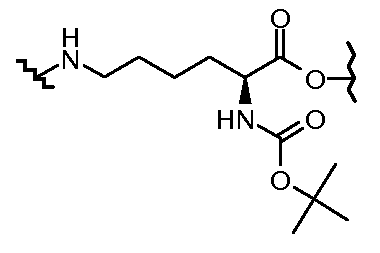
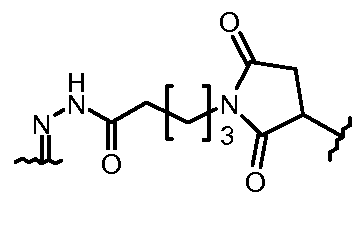
где остаток [Ant] представлен соединением формулы (IIa) или (IIb) далее, то есть [Ant] представляет собой остаток антрациклина формулы IIA, как определено выше
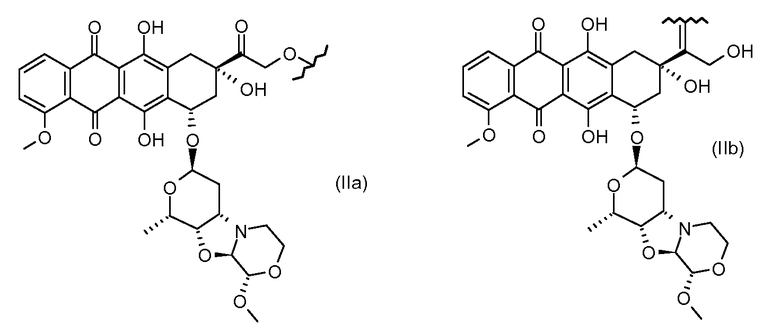
Если хиральный центр или другая форма изомерного центра находится в соединении по настоящему изобретению, все формы такого изомера или изомеров, включая энантиомеры и диастереомеры, охватываются в настоящей заявке. Соединения, содержащие хиральный центр, могут быть использованы в качестве рацемической смеси, энантиомерно обогащенной смеси, или рацемическая смесь может быть разделена, с использованием хорошо известных методик и индивидуальный энантиомер может быть использован отдельно. В случаях, в которых соединения имеют ненасыщенные углерод-углеродные двойные связи, как цис (Z), так и транс (E) изомеры входят в объем данного изобретения.
В случаях, где соединения могут существовать в таутомерных формах, таких как кето-енольные таутомеры, каждая таутомерная форма рассматривается включенной в настоящее изобретение, находящиеся в равновесии или преобладающие в одной форме.
Фармацевтически приемлемые соли производного антрациклина формулы (I') или конъюгата производных антрациклинов формулы (I) включают кислотно-аддитивные соли с неорганическими или органическими кислотами, например, азотной, соляной, бромистоводородной, серной, хлорной, фосфорной, уксусной, трифторуксусной, пропионовой, гликолевой, молочной, щавелевой, малоновой, яблочной, фумаровой, винной, лимонной, бензойной, коричной, миндальной, метансульфоновой, изетионовой и салициловой кислотой. Предпочтительно, кислотно-аддитивная соль соединений по изобретению представляет собой гидрохлорид или мезилат.
Фармацевтически приемлемые соли производного антрациклина формулы (I') или конъюгата производных антрациклинов формулы (I) также включают соли с неорганическими или органическими основаниями, например, щелочных или щелочноземельных металлов, в особенности, натрия, калия, аммония кальция или магния гидроксиды, карбонаты или бикарбонаты, ациклических или циклических аминов, предпочтительно, метиламином, этиламином, диэтиламином, триэтиламином, пиперидином и подобными.
Как используется в настоящем документе, если не указано иное, термин C1-C6 алкил означает группу, такую как, например, метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-пентил, неопентил, н-гексил, изогексил и тому подобное.
Термин C3-C6 циклоалкильная группа означает, например, циклопропил, циклобутил, циклопентил, циклогексил, циклопентенил, циклогексенил и тому подобное.
Термин C1-C6 алкиленовая группа означает двухвалентный остаток, такой как, например, метилен, этилен, н-пропилен, изопропилен, н-бутилен, изобутилен, втор-бутилен, трет-бутилен, н-пентилен, неопентилен, н-гексилен, изогексилен и тому подобное.
Термин C3-C6 циклоалкиленовая группа означает двухвалентный остаток, такой как, например, циклопропилен, циклобутилен, циклопентилен, циклогексилен, циклопентенилен, циклогексилен и тому подобное.
Специалисту в данной области ясно, что любая группа или заместители, определенные в настоящей заявке, могут быть интерпретированы по названиям групп, из которых они происходят.
В качестве примера, если особым образом не отмечено иное, в C1-C5 алкокси группе, алкильная часть включает, например, метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-пентил, неопентил и тому подобное. Приводимыми в качестве примера C1-C5 алкокси группы являются метокси (-OCH3), этокси (-OCH2CH3), пропилокси, изопропилокси, н-бутилокси, изобутилокси, втор-бутилокси, трет-бутилокси, н-пентилокси, неопентилокси и тому подобное.
«Пептидный остаток, состоящий из 1-4 амино» означает пептид, содержащий последовательность от одного до четырех природных или синтетических аминокислот.
Настоящее изобретение также относится к способам получения соединения формулы (I), как определено выше.
Соединение формулы (Ia), определенное выше, и его фармацевтически приемлемые соли могут быть получены, как изображено на Фигуре 2.
Способы получения конъюгатов производных антрациклина формулы (Ia)
Соответственно, первый способ по настоящему изобретению получения соединения формулы (Ia), как определено выше, и их фармацевтически приемлемые соли включает следующие стадии:
Стадия 1 взаимодействие соединения формулы (II), как определено выше, с соединением формулы (IX) или (X):
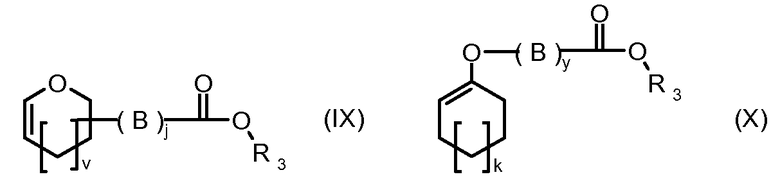
где v, j, k, y, и B имеют значения, указанные выше, и R3 представляет собой C1-C3 алкильную группу;
Стадия 2 гидролиз полученного в результате сложноэфирного промежуточного соединения (XI):
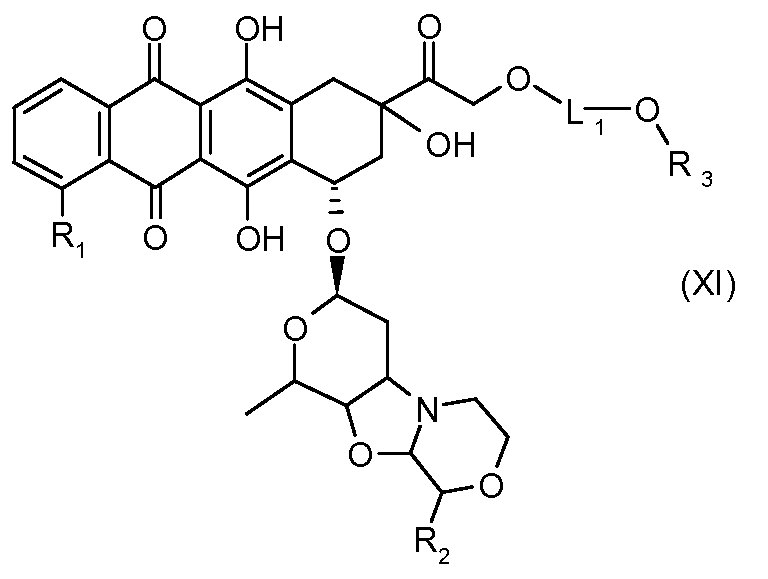
где R1, R2, R3 имеют значения, указанные выше, и L1 представляет собой группу формулы (III) или (IV), как определено выше;
Стадия 3 активация полученной в результате кислоты формулы (XII):
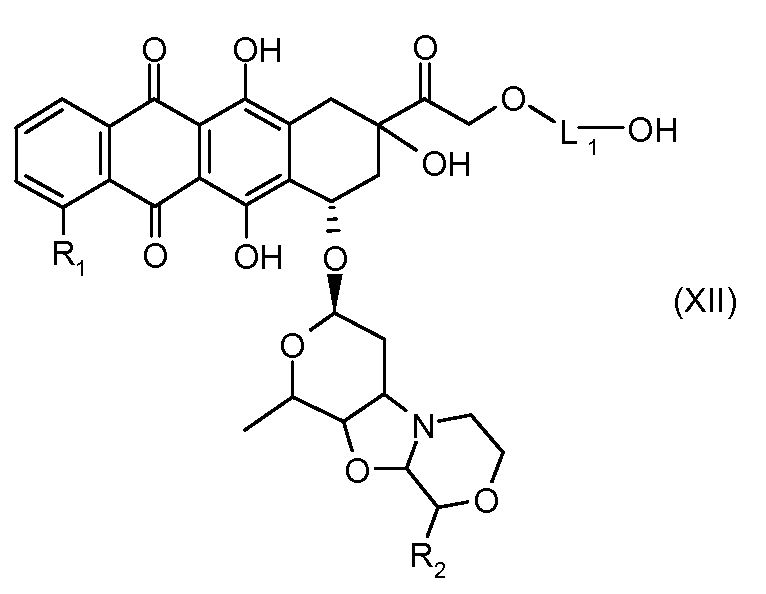
где R1, R2 и L1 имеют значения, указанные выше, и
Стадия 4 соединение полученного в результате активированного соединения формулы (XIII):
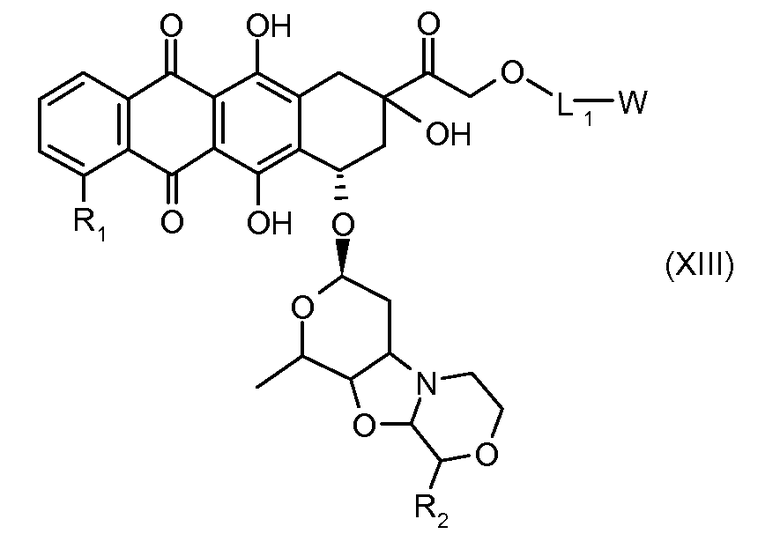
где R1, R2 и L1 имеют значения, указанные выше, и W представляет собой активирующую группу кислой группы, например, N-оксисукцинимидо, N-оксисульфосукцинимидо или 2,4-динитрофенокси или 2,3,4,5,6-пентафторфенокси или трет-бутокси карбонилокси с желаемым носителем, с получением соединения формулы (Ia), и необязательно превращение полученного в результате соединения в фармацевтически приемлемую кислоту.
Соединения формул (XI), (XII) и (XIII), определенные выше, также являются объектами настоящего изобретения.
Реакцию стадии 1 проводят в органическом растворителе, например диметоксиэтане или предпочтительно N,N-диметилформамиде (ДМФ) и в присутствии п-толуолсульфоновой кислоты при температуре в интервале от 0°C до 80°C и в течение периода времени в интервале от 1 часа до 24 часов.
Реакцию стадии 2 проводили в основных гидролитических условиях, предпочтительно, с сильным основанием, наподобие NaOH, при температуре от 0°C до комнатной температуры в течение периода времени в интервале от 1 до 48 часов.
Реакцию стадии 3 проводили в соответствии с хорошо известными способами, например, N-оксисукцинимидо производное может быть получено путем взаимодействия кислоты (XII) с N-гидроксисукцинимидом или его водорастворимой 3-замещенной сульфонат натриевой солью в присутствии N,N'-дициклогексилкарбодиимида в растворителе, таком как дихлорметан или N,N-диметилформамид при температуре от 0°C до 50°C в течение периода времени от 1 до 24 часов.
Реакцию стадии 4 можно проводить в соответствии с одним из способов, кратко изложенных на Фигурах 3a, 3b, 3c, в зависимости от желаемого получаемого соединения формулы (Ia), как определено выше:
В частности, конечная конденсация для получения соединения формулы (Ia), как определено выше, предусматривает взаимодействие соединения формулы (XIII), как определено выше, с:
1a) соединение формулы T-[X]m (XIV), где Х представляет собой -NH2 или -SH и m имеет значения, указанные выше, с получением соединения формулы (Ia), как определено выше в пункте i) или ii), соответственно.
Конденсирование проводят в условиях, способных создавать ковалентные связи амидного типа или сложного тиоэфирного типа и совместимых со структурой носителя. Предпочтительные условия охватывают применение буферных водных растворов при pH 7-9,5, температуры от 4°C до 37°C, в течение времени от нескольких часов до нескольких дней.
Например, условиями для конденсирования между соединениями формулы (XIII) и антителами T-NH2 являются: водный 0,1 M натрия фосфат и водный 0,1 M натрия хлорид при pH 8, содержащий моноклональное антитело в концентрации 1 мг/мл, обработанное 30-кратным молярным избытком 10% масс./об. раствора 6 в N,N-диметилформамиде, в течение 24 часов при 20 (градусах) C. Конъюгат очищают с помощью гель-фильтрации на колонке SEPHADEX G-25 (Pharmacia Fine Chemical, Piscataway, N.J.), элюируя с PBS (фосфат-буферным солевым раствором).
Другое конденсирование для получения соединения формулы (Ia), как определено выше, предусматривает взаимодействие соединения формулы (XIII), определенного выше, с:
1b) соединением формулы (XV) NH2-D-NH-P, где -D- или -D-NH- имеют значения, указанные выше, и P представляет собой атом водорода или предпочтительно защитную группу;
1b') снятие защиты NH функциональной группы, при необходимости, полученного в результате соединения формулы (XVI):
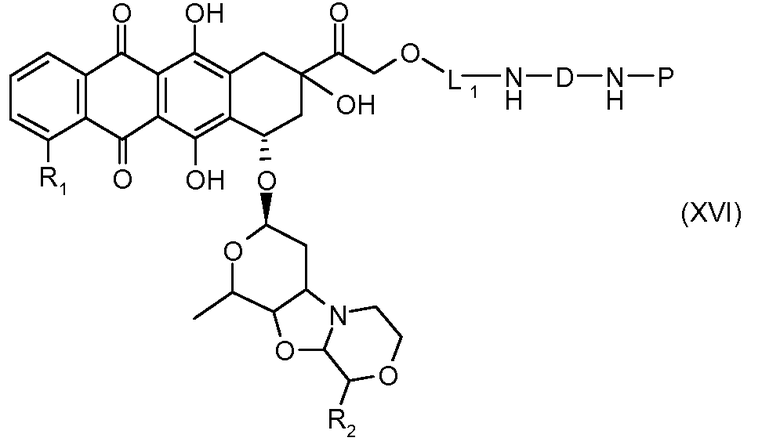
где R1, R2, L1 и D определены выше, и Р представляет собой защитную группу, а затем
1”b) соединение полученного в результате соединения формулы (XVI):
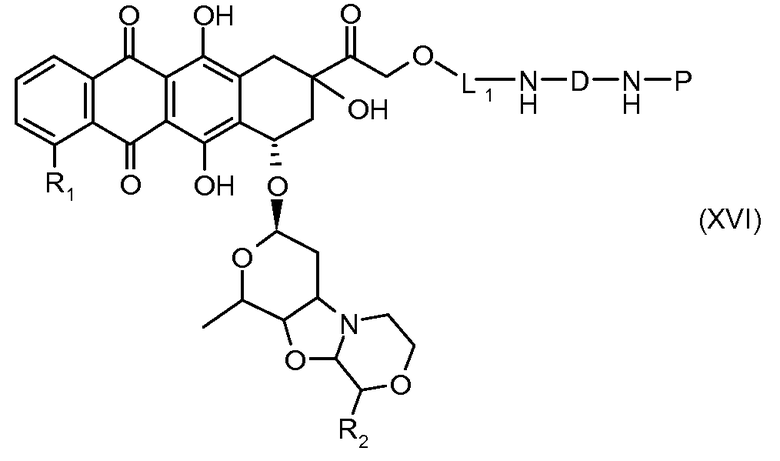
где R1, R2, L1 и D определены выше и P представляет собой атом водорода, с остатком носителя формулы T-[COOH]m (XVII), где T и m имеют значения, указанные выше, с получением соединения формулы (Ia), как определено в пункте (iii) выше, где [T-Z]- представляет собой -NH-D-NHCO-T, R1, R2, L1 и T имеют значения, указанные выше.
Реакцию стадии 1b проводят в условиях, способных создавать ковалентные связи амидного типа, хорошо известных в литературе и совместимых со структурой спейсера. Предпочтительные условия охватывают применение забуференных водных растворов с pH 7-9,5, или органических растворителей, таких как, например N,N-диметилформамид, дихлорметан, тетрагидрофуран или этилацетат, температуры в интервале от 4°C до 50°C и в течение времени от нескольких часов до нескольких дней.
Необязательное снятие защиты на стадии стадия 1'b проводят, используя хорошо известный способ, изложенный в литературе [см., например, Green T.W., Wuts P.G.M в Protective Groups in Organic Chemistry]. Реакцию присоединения на стадии 1”b проводят в органическом растворителе, предпочтительно N,N-диметилформамиде в присутствии конденсирующего агента, такого как например 1-этил-3-(3-диметиламинопропил)карбодиимид гидрохлорид 1,3-ди-трет-бутилкарбодиимид, N-(3-диметиламинопропил)-н'-этилкарбодиимид, 1-(3-диметиламинопропил)-3-этилкарбодиимид гидрохлорид, или предпочтительно, N,N'-дициклогексилкарбодиимид. Реакцию проводят при температуре в интервале от 5°C до 50°C и в течение периода времени в интервале от 1 часа до 24 часов.
В альтернативном варианте, соединение формулы (XVII) может быть активировано с использованием подходящей активирующей кислоты группы W, как описано выше, на стадии 3 выше, а затем соединено с незащищенным амином, используя те же условия, которые приведены выше.
Другое конденсирование для получения соединения формулы (Ia), как определено выше, предусматривает взаимодействие соединения формулы (XIII), как определено выше, с:
1c) соединением формулы (XVIII) NH2-D-COO-P1, где D или D-CO- имеют значения, указанные выше, и P1 представляет собой подходящую защитную кислотную группу, например сложный алкилэфир, которую удаляют после реакции соединения с получением кислого соединения формулы (XIX):
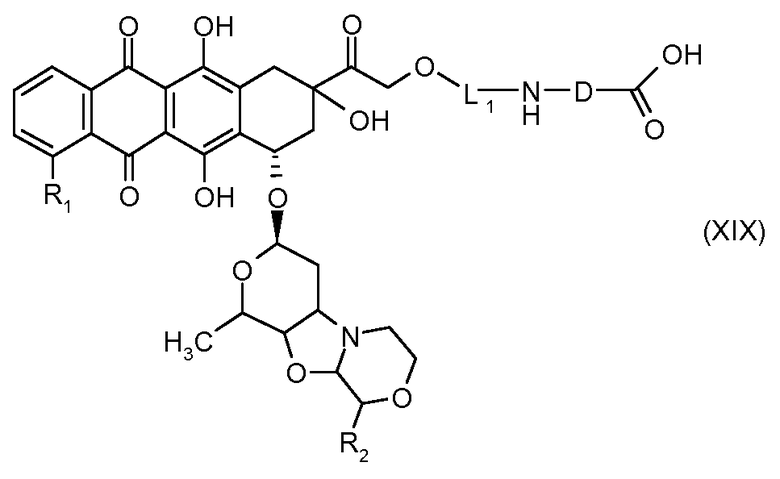
где R1, R2, L1 и D описаны выше.
Полученное в результате соединение формулы (XIX) может быть использовано как таковое, или предпочтительно активировано посредством подходящей активирующей кислотной группы, как описано выше на стадии 3, а затем соединено с группой носителя формулы (XIV) T-[X]m, где X представляет собой NH2 и m и T имеют значения, указанные выше, для получения соединения формулы (Ia), определенного в пункте (iv), где [T-Z]- представляет собой -NH-D-CONH-T, R1, R2, L1 и T имеют значения, указанные выше.
Предпочтительные условия реакции для присоединения соединения формулы (XIII) к соединению формулы (XVIII), определенного выше, такие же, как изложено на стадии 1b выше. Удаление кислой защитной группы проводят, используя хорошо проверенные способы [см., например, Green T.W., Wuts P.G.M в Protective Groups in Organic Chemistry], например, когда функциональная группа кислоты защищена в виде производного этилового эфира, снятие защиты может осуществляться в основных гидролитических условиях, предпочтительно с использованием NaOH и при температуре в интервале от 0°C до комнатной температуры и в течение времени в интервале от 1 часа до 48 часов. Реакцию соединения формулы (XIX) с соединением формулы (XIV) проводят в органическом растворителе, предпочтительно, N,N-диметилформамиде в присутствии конденсирующего агента, такого как, например, 1-этил-3-(3-диметиламинопропил)карбодиимид гидрохлорид 1,3-ди-трет-бутилкарбодиимид, N-(3-диметиламинопропил)-н'-этилкарбодиимид, 1-(3-диметиламинопропил)-3-этилкарбодиимид гидрохлорид, или предпочтительно, N,N'-дициклогексилкарбодиимид. Реакцию проводят при температуре в интервале от 5°C до 50°C и в течение интервала времени от 1 часа до 24 часов или альтернативным путем синтеза, активируя кислоту (XIX) подходящей активирующей группой, как изложено на стадии 3 этого способа, а затем соединяя активированную кислоту с соединением формулы (XIV) в условиях синтеза, изложенных выше на стадии 1.
Другое конденсирование для получения соединения формулы (Ia), определенного выше, предусматривает взаимодействие соединения формулы (XIII), как определено выше, с:
1d) соединением формулы (XV), как описано выше, а затем соединение полученного в результате промежуточного соединения формулы (XVI), определенного выше, где P представляет собой атом водорода, с группой носителя формулы T-[CHO]m (XX), где m и T имеют значения, указанные выше, для получения соединения формулы (Ia), описанного в пункте (v) выше, где [T-Z]- группа представляет собой -NH-D-N=CH-T.
Конъюгирование незащищенного амино производного формулы (XVI) с носителем формулы (XX) можно проводить в условиях, способных создавать ковалентные связи гидразонного типа и совместимых со структурой носителя. Предпочтительные условия охватывают применение забуференных водных растворов при pH 4-7,5, спиртов или их смесей, при температуре от 4°C до 37°C, в течение времени от нескольких часов до нескольких дней. Условиями для соединения между соединениями незащищенного производного формулы (XVI) и антителами T-CHO являются: водный 0,1 M ацетат натрия и водный 0,1 M хлорид натрия при pH 6, содержащий моноклональное антитело в концентрации 1 мг/мл, обрабатанное 30-кратным молярным избытком 5% масс./об. раствора 8 в том же буфере, в течение 24 часов при 20°C. Этот конъюгат очищают с помощью гель-фильтрации, как указано выше.
Другое конденсирование для получения соединения формулы (Ia), определенного выше, предусматривает взаимодействие соединения формулы (XIII), как определено выше, с:
1e) соединением формулы (XXI) NH2-D-SH, где D имеет значения, указанные выше, в тех же условиях реакции, изложенных на стадии 1 этого способа, а затем присоединение полученного в результате соединения формулы (XXII):
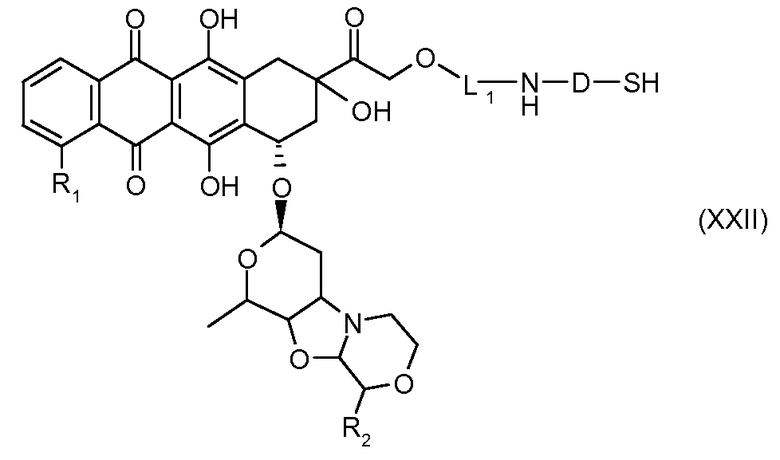
где R1, R2, L1 и D определены далее, либо с:
1e') группой носителя формулы (V):
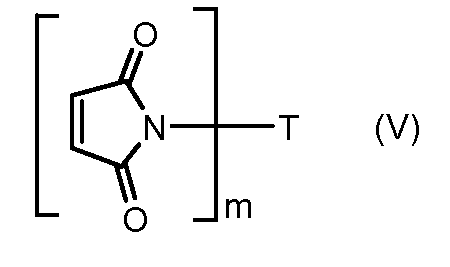
где T и m имеют значения, указанные выше, с получением после присоединения по Михаэлю соединения формулы (Ia), описанного в пункте (vi), где [T-Z]- представляет собой группу формулы (XXIII):
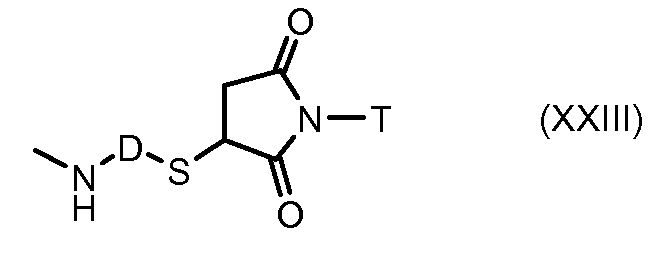
где D и T имеют значения, указанные выше; или
1e'') с группой носителя формулы (VI):
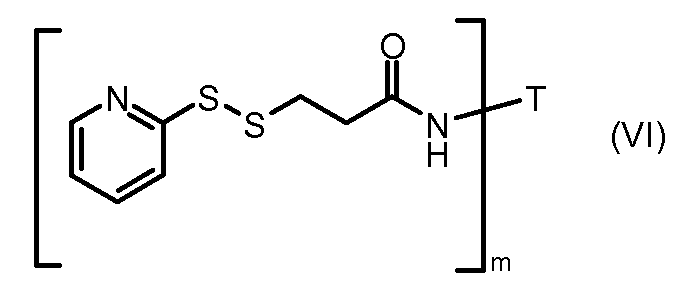
где T и m имеют значения, указанные выше, с получением после замещения пиридин-2-тиольной группы, соединения формулы (Ia), определенного в пункте (vii) выше, где [T-Z]- представляет собой группу формулы (XXIV)
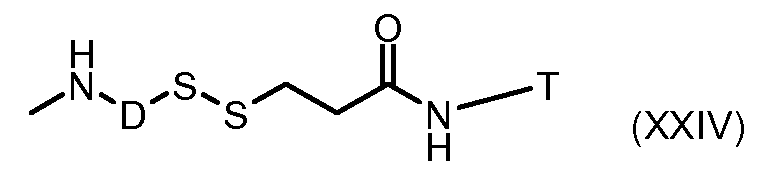
где D и T имеют значения, указанные выше.
Реакцию соединения формулы (XXII), определенного выше, с соединением формулы (V), определенного выше, можно проводить в забуференных водных растворах при pH 7-9,5, спиртах или их смесях, при температуре от 4°C до комнатной температуры и в течение периода времени от 1 до 6 часов [см., например Willner D. et al., Bioconjugate Chem. (1993) 4:521-527]. Присоединение соединения (XXII) к соединению (VI) проводят предпочтительно в смеси метанола и фосфатно-буферного раствора, при pH 7,2 с 1-1,5 эквивалентами соединения формулы (XXII), определенного выше, для каждой реакционно-способной группы соединения (VI), определенного выше. Реакционную смесь инкубируют, предпочтительно, при температуре от 4°C до комнатной температуры [см., например, EP328147].
Соединение формулы (Ib), определенное выше, и его фармацевтически приемлемые соли могут быть получены, как изображено на Фигуре 4.
Способ получения конъюгатов производного антрациклина формулы (Ib)
Соответственно, настоящее изобретение относится к способу получения соединения формулы (Ib), определенного выше, и их фармацевтически приемлемых солей, указанный способ включает следующие стадии:
Стадия 1 взаимодействие соединения формулы (II), определенного выше, с ацилгидразидным производным формулы (XXV):
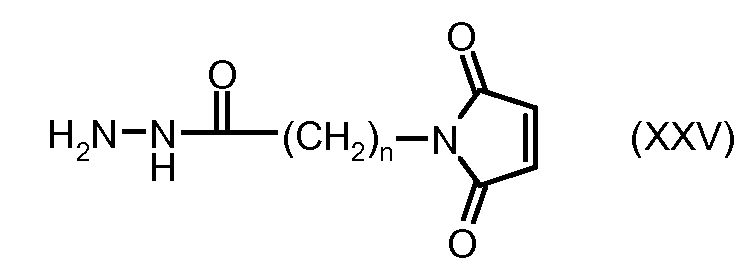
в условиях, способных создавать ковалентные связи ацилгидразонного типа и совместимых со структурой носителя; и
Стадия 2 превращение полученного в результате соединения формулы (XXVI):
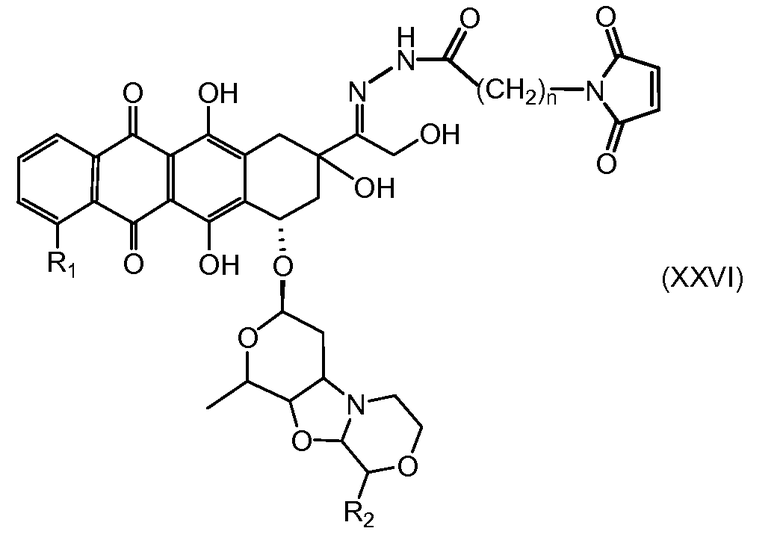
в конечное соединение формулы (Ib), определенное выше, подходящим способом.
Соединение формулы (XXVI), определенное выше, также является объектом настоящего изобретения.
Предпочтительные условия стадии 1, описанной выше, охватывают применение забуференных водных растворов при pH от 4 до 7,5 или предпочтительно органического растворителя, такого как, например, этанол, тетрагидрофуран, или более предпочтительно метанола, при температуре от 4°C до 50°C, в течение периода времени от 1 часа до нескольких дней.
Конечное превращение можно проводить в соответствии с одним из способов, кратко изложенных на Фигурах 5a, 5b, 5c:
В частности, конечное конденсирование для получения соединения формулы (Ib), определенного выше, включает взаимодействие соединения формулы (XXVI), определенного выше, с:
2a) носителем соединением формулы T-[X]m (XIV), где X представляет собой NH2 или SH и m имеет значения, указанные выше, с получением соединения формулы (Ib), определенного в пунктах (viii) и (ix) выше, где L2 представляет собой линкер формулы (VII), как определено выше, R1, R2 имеют значения, указанные выше, и [T-Z]- представляет собой T-NH- или T-S-, где T имеет значения, указанные выше. Реакцию конъюгирования проводят, используя те же условия, изложенные выше на стадии 1e.
Другое конденсирование для получения соединения формулы (Ib), как определено выше, предусматривает взаимодействие соединения формулы (XXVI), определенного выше, с:
2b) соединением формулы (XXVII) H-R4-D-NH-P, где D имеет значения, указанные выше, R4 представляет собой -NH- или -S- и P представляет собой атом водорода или, предпочтительно, амино защитную группу, которую удаляют после реакции присоединения, а затем
2'b) соединение полученного в результате ацилгидразонного производного формулы (XXVIII):
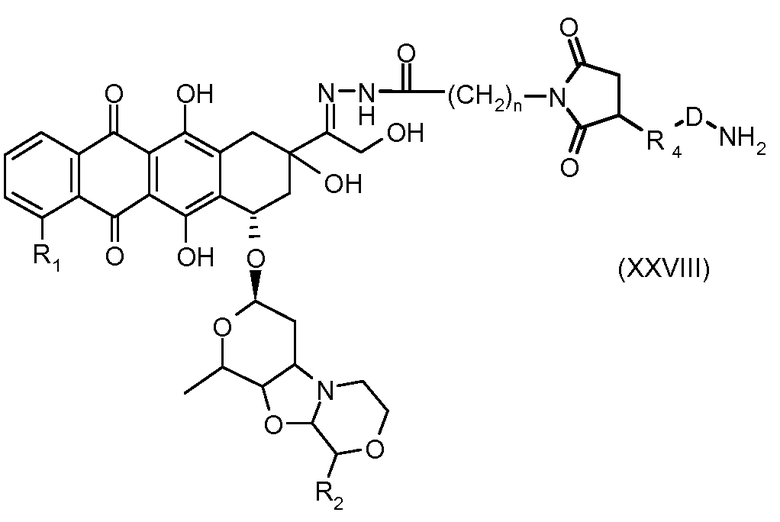
где R1, R2, R4 и D имеют значения, указанные выше, с носителем производным формулы (XVII) T-[COOH]m, где T и m имеют значения, указанные выше, с получением соединения формулы (Ib), определенного в пункте (x) и (xv), где группа [T-Z]- представляет собой -NH-D-NHCO-T или -S-D-NHCO-T и R1, R2, L2, T имеют значения, указанные выше. Реакцию конъюгирования проводят, используя те же условия, изложенные в пункте 1b выше.
Другое конденсирование для получения соединения формулы (Ib), определенного выше, включает взаимодействие соединения формулы (XXVI), как определено выше, с:
2c) соединением формулы (XXIX) H-R4-D-CO-P1, где R4, D, D-CO- и P1 имеют значения, указанные выше, и удаление защитной группы, если имеется; и
2'c) соединение полученного в результате ацилгидразонного производного формулы (XXX):
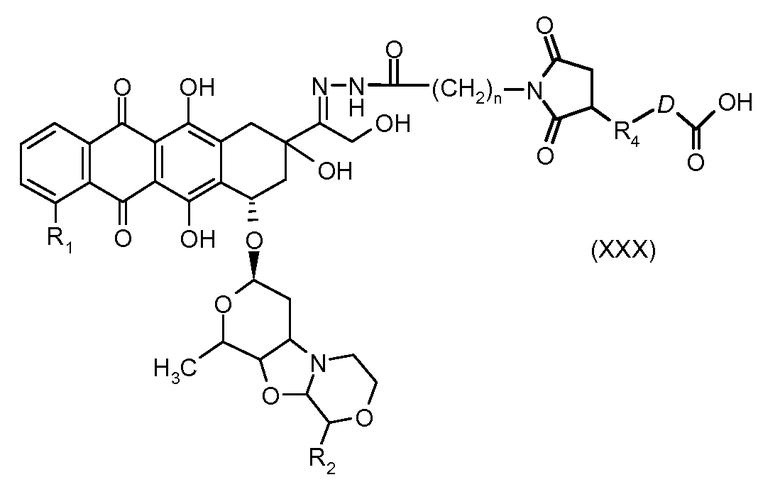
где R1, R2, R4 и D имеют значения, указанные выше, предпочтительно после активации с подходящей активирующей кислотной группой W, где W определено выше, с носителем формулы (XIV) T-[X]m, где X представляет собой NH2, и T и m имеют значения, указанные выше, с получением соединения формулы (Ib), определенного в пунктах (xi) и (xvi) выше, где группа [T-Z]- представляет собой -NH-D-CONH-T или -S-D-CONH-T и L2, R1, R2, T имеют значения, указанные выше. Реакцию конъюгирования проводят, используя те же условия, изложенные в пункте (1c) этого способа.
Другое конденсирование для получения соединения формулы (Ib), определенного выше, включает:
2d) присоединение соединения формулы (XXVIII), полученного, как описано выше, к носителю формулы (XX) T-[CHO]m, с получением соединения формулы (Ib), определенного в пунктах (xii) и (xvii) выше, где группа [T-Z]- представляет собой -NH-D-N=C-T или -S-D-N=C-T и L2, R1, R2, T имеют значения, указанные выше, используя те же условия реакции, изложенные в пункте 1d выше.
Другое конденсирование для получения соединения формулы (Ib), определенного выше, предусматривает взаимодействие соединения формулы (XXVI), как определено выше, с:
2e) соединением формулы (XXXI) H-R4-D-S-P2, где R4 и D имеют значения, указанные выше, и P2 представляет собой атом водорода или, предпочтительно, тиол-защитную группу, затем соединение полученного в результате соединения формулы (XXXII):
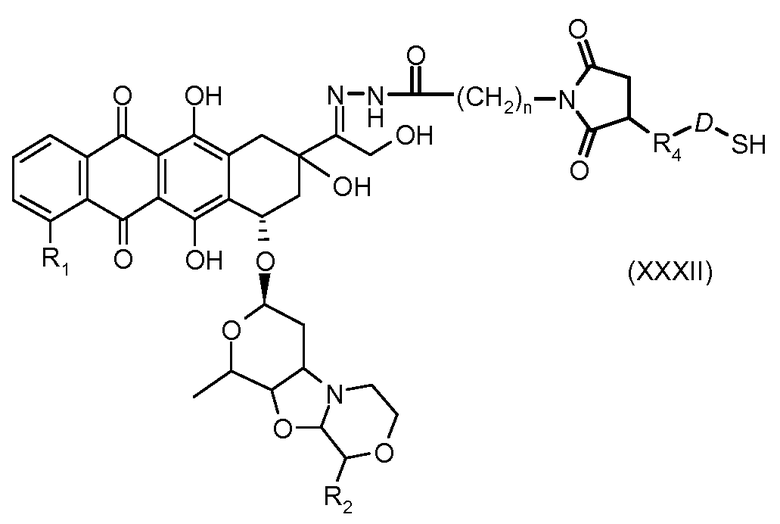
где n, R1, R2, R4 и D имеют значения, указанные выше, после удаления тиол-защитной группы, если имеется, либо с:
2e') носителем производным формулы (V), определенным выше, с получением соединения формулы (Ib), определенного в пунктах (xiii) и (xviii) выше, где L2, R1, R2, D имеют значения, указанные выше, и [T-Z]- представляет собой группу формулы (XXIII), как определено выше, или (XXIIIa) либо,
2e'') носителем производным формулы (VI), как определено выше, с получением соединения формулы (Ib), определенного в пунктах (xiv) и (xix) выше, где L2, R1, R2, D имеют значения, указанные выше и [T-Z]- представляет собой группу формулы (XXIV), как определено выше, или (XXIVa).
Реакцию конъюгирования проводят, используя те же условия, изложенные в пункте (1e) этого способа, тогда как удаление выбранной тиол-защитной группы можно проводить в условиях, изложенных в литературе [см., например Green T.W., Wuts P.G.M в Protective Groups in Organic Chemistry].
Соединение формулы (Ib), где L2 представляет собой спейсер формулы (VIII), и фармацевтически приемлемые соли могут быть получены способом, изображенным на Фигуре 6, где G представляет собой атом углерода или атом азота, предпочтительно, атом азот, R5 представляет собой галоген или атом водорода, предпочтительно, атом водорода, и n, R1 и R2 имеют значения, указанные выше.
Соответственно, настоящее изобретение относится к способу получения соединения формулы (Ib), определенного выше, где L2 представляет собой спейсер формулы (VIII), и его фармацевтически приемлемых солей, указанный способ включает следующие стадии:
Стадия 1 взаимодействие соединения формулы (II), определенного выше, с ацилгидразидным производным формулы (XXXIII):
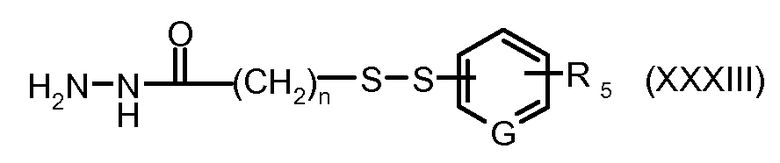
где G и R5 определены выше, используя те же условия, изложенные на стадии 1, как описано выше в способе получения соединения формулы (Ib), и
Стадия 2 превращение полученного в результате ацилгидразонного производного формулы (XXXIV):
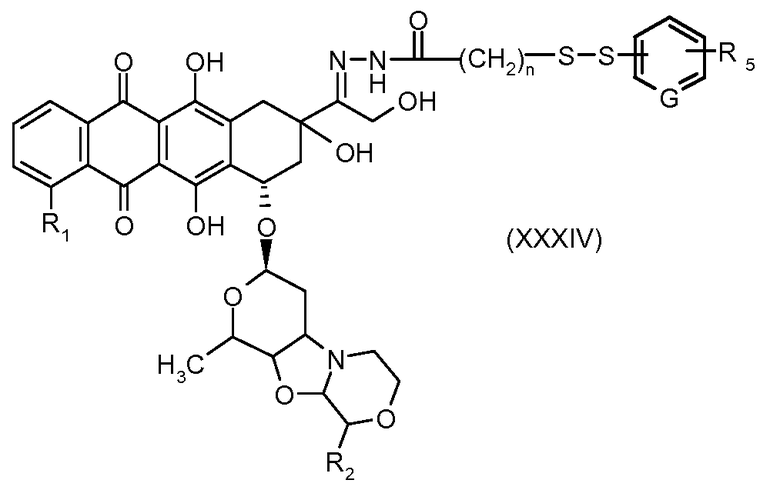
где n, R1, R2, и G имеют значения, указанные выше, в конечное соединение формулы (Ib) с помощью подходящего способа.
Соединение формулы (XXXIV), определенное выше, также является объектом настоящего изобретения.
Конечное превращение можно проводить в соответствии с одним из способов, кратко изложенных на Фигурах 7a, 7b, 7c, где n, m, T, D, P, P1, P2 и R4 имеют значения, указанные выше. В частности, конечное конденсирование для получения соединения формулы (Ib), определенного выше, включает взаимодействие соединения формулы (XXXIV), определенного выше, с:
(3a) носителем производного формулы (XIV) T-[X]m, где Х представляет собой -SH и m имеет значения, указанные выше, с получением соединения формулы (Ib), как определено в пункте (xx) выше, где L2 представляет собой линкер формулы (VIII), как определено выше, R1 и R2 имеют значения, указанные выше, и [T-Z] представляет собой -S-T, где T имеет значения, указанные выше. Условия реакции для соединения носителя с соединением формулы (XXXIV) такие же, как описано в пункте (1e”') выше.
Другое конденсирование для получения соединения формулы (Ib), как определено выше, предусматривает взаимодействие соединения формулы (XXXIV), как определено выше, с:
(3b) соединением формулы (XXXV) SH-D-NH-P, где D и P имеют значения, указанные выше, в тех же условиях, описанных в пункте 1e''выше, и
(3'b) соединение полученного в результате ацилгидразонного производного формулы (XXXVI), после удаления амино-защитной группы, если имеется:
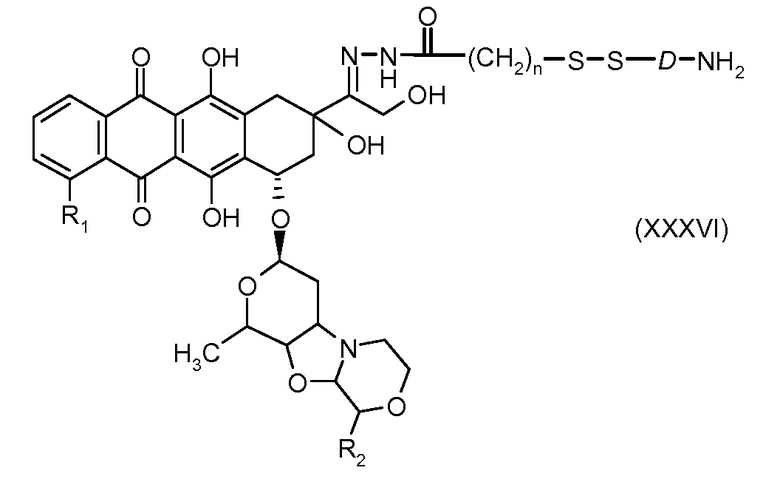
где n, R1, R2 и D имеют значения, указанные выше, с носителем производным формулы (XVII) T-[COOH]m, где T и m имеют значения, указанные выше, с получением соединения формулы (Ib) как определено в пункте (xxi) выше, где L2 имеет значения, указанные выше, и группа [T-Z]- представляет собой -S-D-NHCO-T. Реакцию проводят, используя те же условия, которые были использованы для получения конечных соединений в пункте (1b) выше.
Другое конденсирование для получения соединения формулы (Ib), как определено выше, предусматривает взаимодействие соединения формулы (XXXIV), как определено выше, с:
(3c) соединением формулы (XXXVII) HS-D-CO-OP1, где D и P1 имеют значения, указанные выше, в тех же условиях реакции, изложенных в пункте 1e'', и после удаления амино-защитной группы, если имеется;
(3'c) соединение полученного в результате ацилгидразонного производного формулы (XXXVIII), предпочтительно активированного путем взаимодействия подходящей активирующей кислотной группы W, где W представляет собой группу, определенную выше:
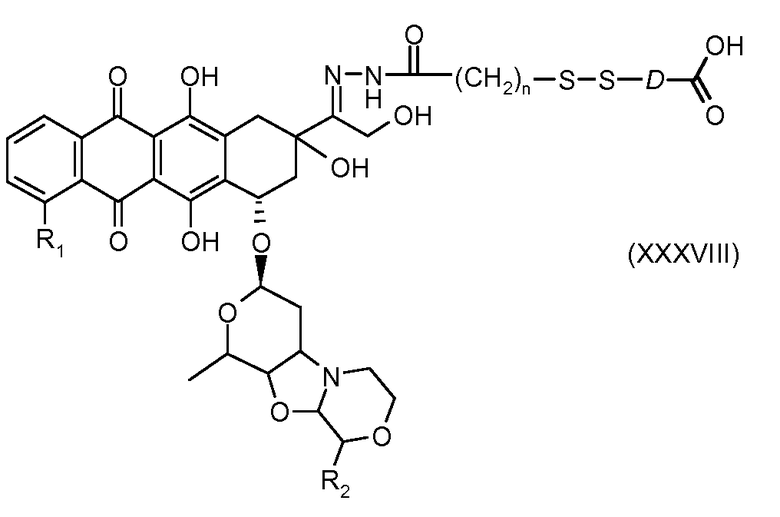
где n, R1, R2 и D имеют значения, указанные выше, с носителем формулы (XIV) T-[X]m, где X представляет собой NH2, и T и m имеют значения, указанные выше, с получением соединения формулы (Ib), как определено в пункте (xxii) выше, где группа [T-Z]- представляет собой -S-D-CONH-T и L2, R1, R2, T определены, используя те же условия, изложенные в пункте 1c выше.
Другое конденсирование для получения соединения формулы (Ib), как определено выше, предусматривает:
(3d) присоединение соединения формулы (XXXVI), полученного как описано выше, с носителем производным формулы (XX) T-[CHO]m, где T и m имеют значения, указанные выше, с получением соединения формулы (Ib), как определено в пункте (xxiii) выше, где группа [T-Z]- представляет собой -S-D-N=C-T и D, L2, R1, R2, T имеют значения, указанные выше. Условия реакции такие же, как изложено в пункте 1d выше.
Другое конденсирование для получения соединения формулы (Ib), как определено выше, предусматривает взаимодействие соединения формулы (XXXIV), как определено выше, с:
(3e) соединением формулы (XXXIX) HS-D-S-P2, где D и P2 имеют значения, указанные выше, и P2 представляет собой предпочтительно тиол-защитную группу, в тех же условиях, изложенных в пункте 1e'' выше, и после удаления защитной группы, если имеется, используя условия, известные в литературе, и соединяя полученное в результате ацилгидразонное производное формулы (XL):
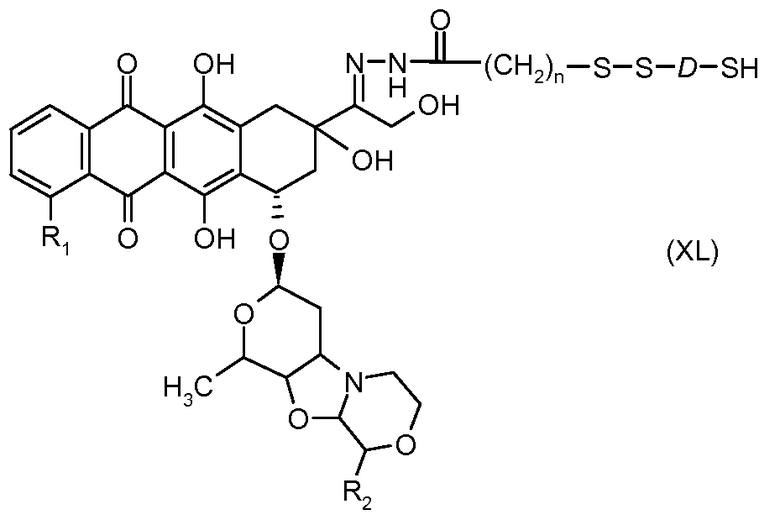
где n, R1, R2 и D имеют значения, указанные выше, либо с:
(3'e) носителем формулы (V), как определено выше, с получением соединения формулы (Ib), как определено в пункте (xiv) выше, где L2, R1, R2, D имеют значения, указанные выше, и [T-Z]- представляет собой группу формулы (XXIIIa), как определено выше, либо
(3''e) носителем формулы (VI), как определено выше, с получением соединения формулы (Ib), как определено в пункте (xxv) выше, где L2, R1, R2 и D имеют значения, указанные выше и [T-Z]- представляет собой группу формулы (XXIVa), как определено выше.
Реакцию между соединением (XL), определенным выше, с соединением формулы (VII) или (VIII), как определено выше, проводят в тех же условиях, изложенных в пункте 1e' и 1e”, соответственно.
Соединения по настоящему изобретению формулы (I'), как определено выше, где L представляет собой L1, и их фармацевтически приемлемые соли могут быть получены с помощью способа, изображенного далее на схемах 7-10, где все символы имеют те же значения, что указаны выше.
Схема 7
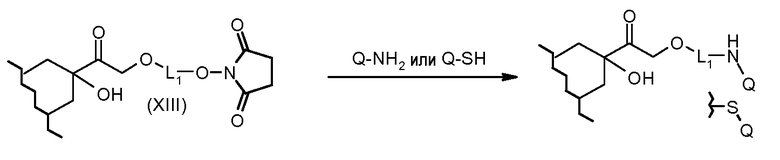
Путем взаимодействия соединения формулы (XIII) с соединением формулы Q-NH2 или Q-SH, где Q имеет значения, указанные выше, получали соединения формулы (I'), где L представляет собой L1 и R1, R2, Q и L1 имеют значения, указанные выше. Условия реакции соединения представляют собой те же, что описаны выше в пункте 1a.
Схема 8
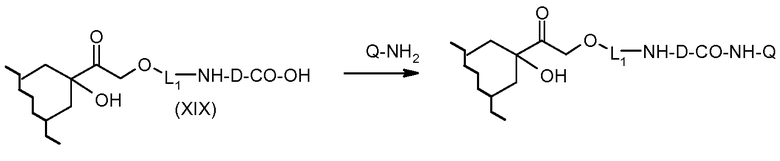
Путем взаимодействия соединения формулы (XIX) с соединением формулы Q-NH2, как определено выше, получали соединения формулы (I'), где L представляет собой L1 и R1, R2, Q и D имеют значения, указанные выше. Условия реакции присоединения такие же, как описано выше в пункте 1c.
Схема 9
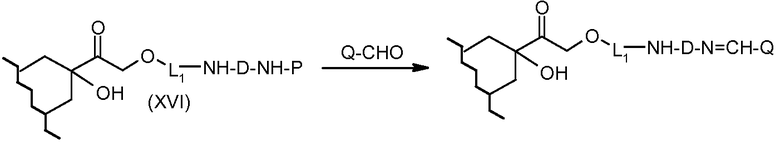
Путем взаимодействия соединения формулы (XVI), где P представляет собой атом водорода с соединением формулы Q-CHO, где Q имеет значения, указанные выше, получали соединения формулы (I'), где L представляет собой L1 и R1, R2, Q, L1 и D имеют значения, указанные выше. Условия реакции присоединения такие же, как описано выше в пункте 1d.
Схема 10
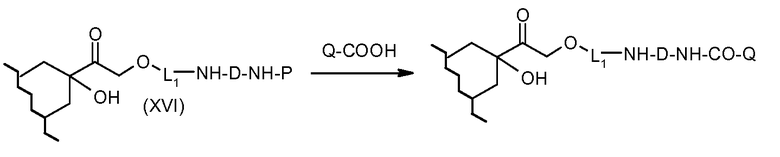
Путем взаимодействия соединения формулы (XVI), где P представляет собой атом водорода и L1 и D имеют значения, указанные выше, с соединением формулы Q-COOH, где Q имеет значения, указанные выше, получали соединения формулы (I'), где L представляет собой L1 и L1, R1, R2, Q и D имеют значения, указанные выше.
Условия реакции присоединения являются такими же, как описано выше в пункте 1b. Соединения по настоящему изобретению формулы (I') имеют значения, указанные выше, где L представляет собой L2, и их фармацевтически приемлемые соли могут быть получены способом, изображенным далее на Схемах 11-15, где все символы имеют те же значения, что указаны выше.
Схема 11
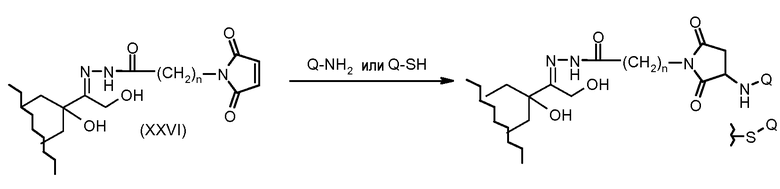
Путем взаимодействия соединения формулы (XXVI) с соединением формулы Q-NH2 или Q-SH имеет значения, указанные выше, получали соединения формулы (I'), где L представляет собой L2, и L2 представляет собой формулу (VII), как определено выше, и R1, R2, Q имеют значения, указанные выше.
Схема 12
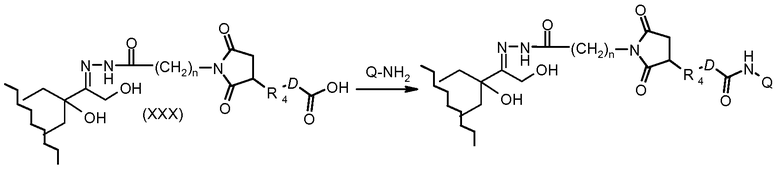
Путем взаимодействия кислотного соединения формулы (XXX) с соединением формулы Q-NH2, как определено выше, получали соединения формулы (I'), где L представляет собой L2, и L2 представляет собой линкер формулы (VII), как определено выше, и R1, R2, R4, D, Q имеют значения, указанные выше.
Схема 13
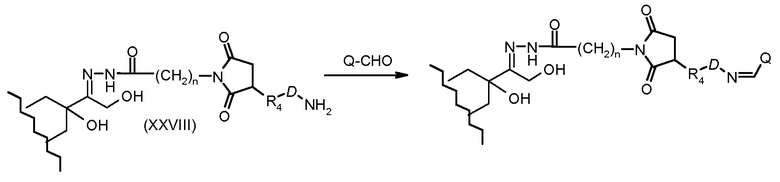
Путем взаимодействия кислотного соединения формулы (XXVIII) с соединением формулы Q-CHO, как определено выше, получали соединения формулы (I'), где L2 представляет собой линкер формулы (VII), как указано выше, и R1, R2, R4, D, Q имеют значения, указанные выше.
Схема 14
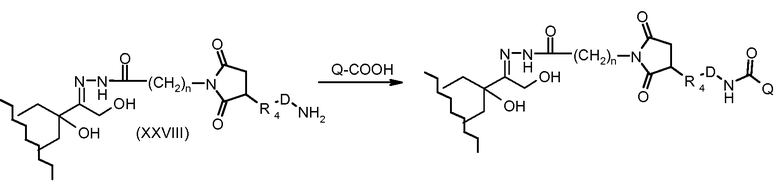
Путем взаимодействия кислотного соединения формулы (XXVIII) с соединением формулы Q-COOH, как определено выше, получали соединения формулы (I'), где L представляет собой L2, L2 представляет собой линкер формулы (VII), как определено выше, и R1, R2, R4, D, Q имеют значения, указанные выше. Условия реакции присоединения, описанные выше, представляют собой те же, что описаны в пункте 1e'.
Схема 15
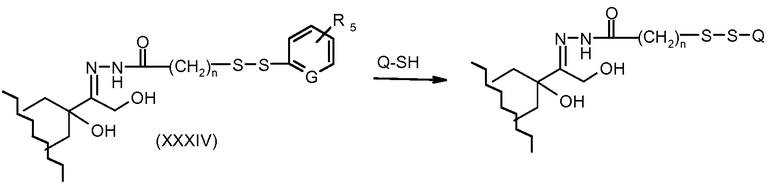
Путем взаимодействия соединения формулы (XXXIV) с соединением формулы Q-SH, как определено выше, получали соединения формулы (I'), где L представляет собой L2, L2 представляет собой линкер формулы (VIII), как определено выше, и R1, R2, Q имеют значения, указанные выше.
Схема 16
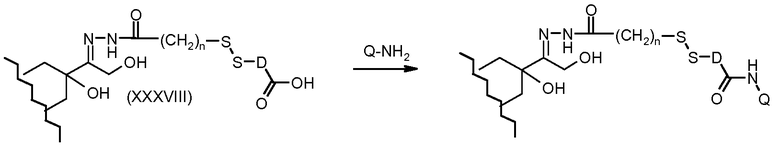
Путем взаимодействия соединения формулы (XXXVIII) с соединением формулы Q-NH2, как определено выше, получали соединения формулы (I'), где L представляет собой L2, L2 представляет собой линкер формулы (VIII) и R1, R2, Q и D имеют значения, указанные выше.
Схема 17
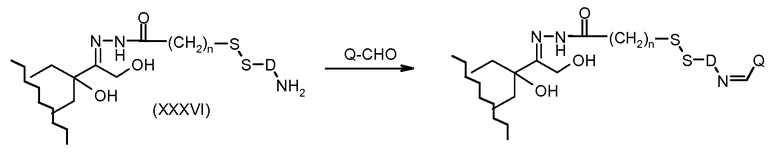
Путем взаимодействия соединения формулы (XXXVI) с соединением формулы Q-CHO, как определено выше, получали соединения формулы (I'), где L представляет собой L2, L2 представляет собой линкер формулы (VIII) и R1, R2, Q и D имеют значения, указанные выше.
Схема 18

Путем взаимодействия соединения формулы (XXXVI) с соединением формулы Q-COOH, как определено выше, получали соединения формулы (I'), где L представляет собой L2, L2 представляет собой линкер формулы (VIII) и R1, R2, Q и D имеют значения, указанные выше. Условия реакции присоединения, описанные выше, являются такими же, как описано в пункте 1e'' выше.
Исходные соединения и реагенты являются коммерчески доступными или могут быть получены в соответствии с известным способом, описанным в литературе. Например, соединения формулы (II) описаны в WO 98/02446, соединения формулы (IX) и (X) описаны в WO 9202255.
КОНЪЮГАТЫ АНТИТЕЛО-ЛЕКАРСТВЕННОЕ СРЕДСТВО
Соединения конъюгата производного антрациклина по изобретению включают соединения, обладающие противоопухолевой активностью. В одном варианте осуществления, соединения конъюгата производного антрациклина включают антитело, конъюгированное, то есть ковалентно присоединенное посредством линкера, к лекарственной части производного антрациклина, где лекарственное средство, когда оно не конъюгировано с антителом, оказывает цитотоксическое или цитостатическое действие. Биологическая активность лекарственной части, таким образом модулируется путем конъюгирования с антителом. Конъюгаты антитела с лекарственным средством (ADC) по изобретению могут селективно доставлять эффективную дозу цитотоксического средства к опухолевой ткани, тем самым с большей селективностью, то есть может достигаться более низкая эффективная доза.
В одном варианте осуществления биодоступность ADC, или внутриклеточного метаболита ADC, улучшена у млекопитающего по сравнению с соответствующим PNU-159682, соединением производным антрациклина в отдельности. Также, биодоступность ADC, или внутриклеточного метаболита ADC улучшается у млекопитающего по сравнению с соответствующим антителом в отдельности (антитело ADC, без лекарственной части или линкера).
В одном варианте осуществления, лекарственная часть производное антрациклина ADC не отщепляется от антитела до тех пор, пока конъюгат антитело-лекарственное средство не свяжется с рецептором клеточной поверхности или не войдет в клетку посредством рецептора клеточной поверхности, специфичным для этого антитела конъюгата антитело-лекарственное средство. Лекарственная часть может отщепляться от антитела после входа конъюгата антитело-лекарственное средство в клетку. Лекарственная часть производное антрациклина может внутриклеточно отщепляться у млекопитающего от антитела, или внутриклеточного метаболита, под действием ферментов, гидролиза, окисления или другого механизма.
Конъюгаты антитела с лекарственным средством по изобретению также могут быть получены путем модификации антитела для введения электрофильных групп, которые могут взаимодействовать с нуклеофильными заместителями на линкерном реагенте или лекарственном средстве. Сахара гликозилированных антител могут быть окислены, например с использованием периодатных окисляющих реагентов, с образованием альдегидных или кетонных групп, которые могут взаимодействовать с аминной группой линкерных реагентов или лекарственных частей. Полученные в результате группы имины - основания Шиффа могут образовывать стабильную связь, или могут быть восстановлены, например с использованием боргидридных реагентов с образованием стабильных аминных связей. В одном варианте осуществления, реакция углеводной части гликозилированного антитела либо с галактозоксидазой, либо мета-периодатом натрия может давать карбонильные (альдегидные и кетонные) группы в белке, которые могут взаимодействовать с соответствующими группами на лекарственном средстве (Hermanson, G.T. (1996) Bioconjugate Techniques; Academic Press: New York, p 234-242). В другом варианте осуществления, белки, содержащие N-концевые остатки серина или треонина, могут взаимодействовать с мета-периодатом натрия, приводя в результате к получению альдегида вместо первой аминокислоты (Geoghegan & Stroh, (1992) Bioconjugate Chem. 3:138-146; US 5362852). Такой альдегид может вступать в реакцию с лекарственной частью или нуклеофилом линкера.
В одном варианте осуществления, соединение конъюгата антитела с лекарственным средством (ADC) содержит антитело, ковалентно присоединенное посредством линкера L и необязательного спейсера Z к одному или нескольким лекарственным частям производного антрациклина D, указанное соединение имеет формулу (Ic)
или его фармацевтически приемлемой соли, где:
Ab представляет собой антитело;
D представляет собой производное антрациклина, выбранное из структур:
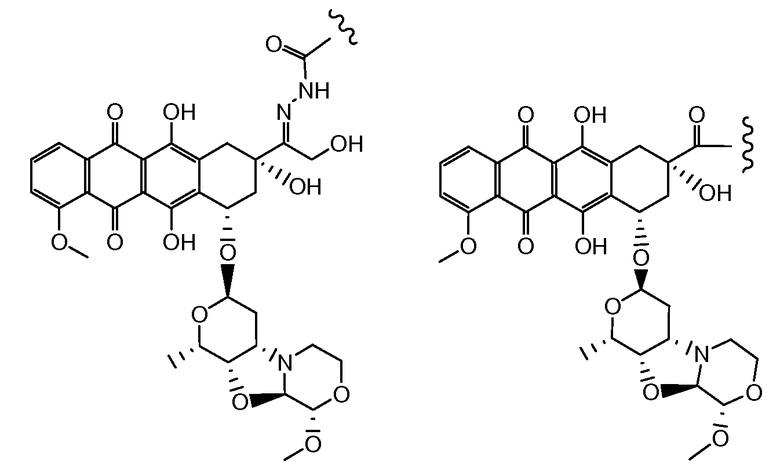
где волнистая линия указывает на присоединение к L;
L представляет собой линкер, выбранный из -N(R)-, -N(R)m(C1-C12 алкилен)-, -N(R)m(C2-C8 алкенилен)-, -N(R)m(C2-C8 алкинилен)-, -N(R)m(CH2CH2O)n-, и структур:

где волнистые линии указывают на присоединения к D и Z; и
Z представляет собой необязательный спейсер, выбранный из -CH2C(O)-, -CH2C(O)NR(C1-C12 алкилен)-, и структур:
R представляет собой H, C1-C12 алкил, или C6-C20 арил;
R1 и R2 независимо выбраны из боковых цепей аминокислот;
Z1 выбран из -(C1-C12 алкилен)-, -(C2-C8 алкенилен)-, -(C2-C8 алкинилен)- и -(CH2CH2O)n-,
m равно 0 или 1;
n равно от 1 до 6; и
p равно целому числу от 1 до 8.
Соединения формулы I включают все смеси различно нагруженных и присоединенных конъюгатов антитела с лекарственным средством, где 1, 2, 3, 4, 5, 6, 7, и 8 лекарственные части ковалентно присоединены к антителу.
Приводимые в качестве примера варианты осуществления конъюгатов антитела с лекарственным средством включают:
 валин-цитруллин (val-cit или vc)
валин-цитруллин (val-cit или vc)
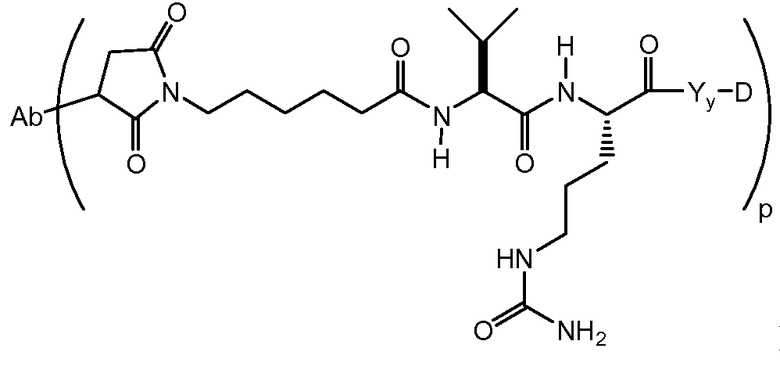 MC-val-cit
MC-val-cit
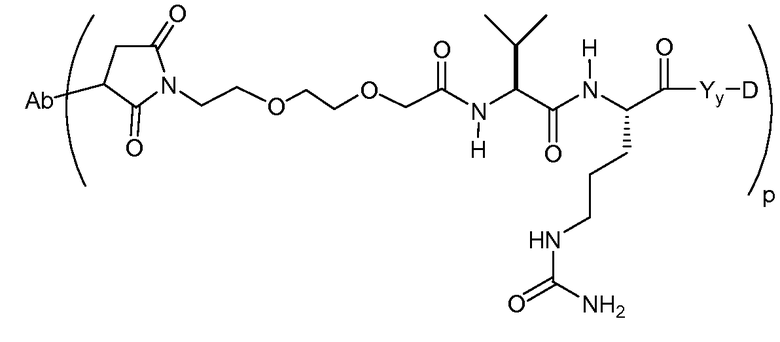 MPEG-val-cit
MPEG-val-cit
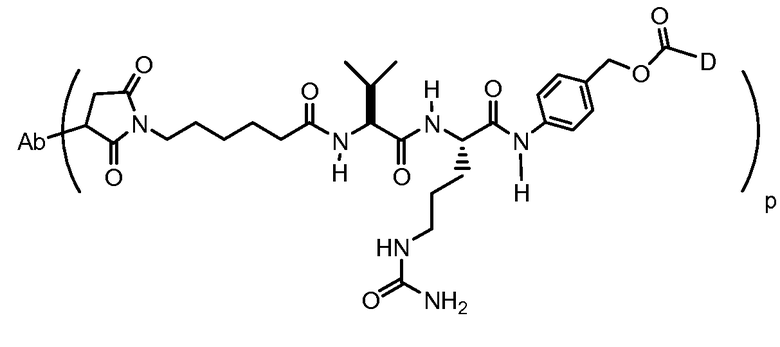 MC-val-cit-PAB
MC-val-cit-PAB
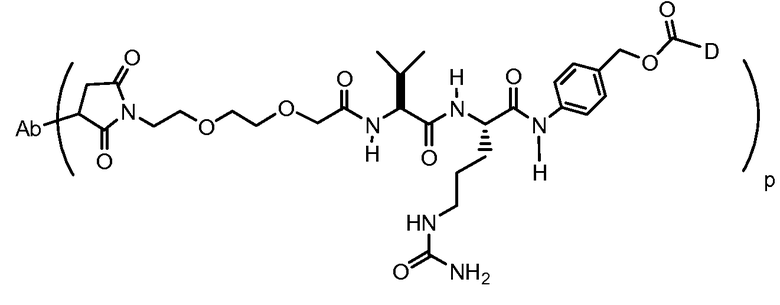 MPEG-val-cit-PAB
MPEG-val-cit-PAB
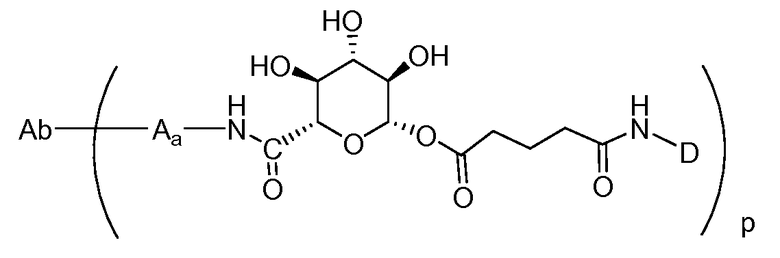
где Aa представляет собой двухвалентное звено, такое как MC (малеимидокапроил), MP (малеимидопропаноил) или MPEG (2-(2-(2-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)этокси)этокси)ацетил), способное соединять антитело (Ab) с аминокислотным звеном, таким как валин-цитруллин; и Yy представляет собой двухвалентное звено, такое как PAB (пара-аминобензилоксикарбонил), которое связывает аминокислотное звено с лекарственной частью (D) в тех случаях, когда имеется аминокислотное звено. В других вариантах осуществления, Aa связывает Yy непосредственно с лекарственной частью, в тех случаях, когда аминокислотное звено отсутствует. В других вариантах осуществления, звено Yy соединяет непосредственно лекарственную группу с антительной единицей, в тех случаях, когда отсутствуют и аминокислотное звено, и звено Aa.
Приводимые в качестве примера конъюгаты антитело-дисульфидный линкер-лекарственное средство представлены структурами:
Дисульфидный линкер SPP может быть сконструирован с линкерным реагентом N-сукцинимидил 4-(2-пиридилтио) пентаноатом.
Конъюгаты антитела с лекарственным средством формулы (I) и соединения формулы (I') включают все энантиомеры, диастереомеры, изомерно обогащенные рацемические смеси, изотопно меченные и изотопно обогащенные формы (например, 2H, 3H, 14C, 15N), и их защищенные формы.
Не ограничиваясь каким-либо конкретным механизмом действия, конъюгаты антитела с лекарственным средством формулы (I) и соединение формулы (I') по настоящему изобретению могут быть подходящими терапевтическими средствами, поскольку они содержат ацетальную связь или гидразонную связь, которая высвобождает исходное лекарственное средство (II) при гидролизе, катализируемом ионом гидрония или ферментативном расщеплении «in vivo». Хорошо известно, что в злокачественных опухолях существует высокая скорость гликолиза по сравнению с нормальной тканью, вызывающая повышение продукции лактата, и, следовательно, снижение рН в опухоли, см.: H. M. Rauen et al., Z. Naturforsch, Teil B, 23 (1968) 1461. Настоящее изобретение обеспечивает двухуровневую специфичность действия соединений, первая состоящая в предпочтительной локализации конъюгата в опухолевой ткани посредством распознавания антигена, и вторая, состоящая в предпочтительном высвобождении лекарственного средства в активной форме в опухолевой ткани посредством предпочтительного кислотного расщепления. Не ограничивая объем или применимость композиций или способов по изобретению, чувствительные к кислоте ацетальные линкеры, описанные в настоящей заявке, могут расщепляться in vivo в локализованных или системных кислых условиях, таким образом, отделяя направляющее антитело от лекарственной группы.
Конъюгаты, полученные в соответствии с описанными способами, охарактеризованы различными физико-химическими способами. Сохранение первоначальной молекулярной массы и отсутствие образования агрегатов может быть определено с помощью методик хроматографической гель-фильтрации (Yu, D. S. et al., J. Urol. 140, 415, 1988) с одновременным и независимым обнаружением антрациклина и антитела при различных длинах волн, и с помощью способов гель-электрофореза. Суммарное распределение заряда полученных соединений может быть определено с помощью способов ионобменной хроматографии. Концентрацию антрациклина можно определить с помощью спектрофотометрического титрования по стандартной калибровочной кривой, полученной из исходного антрациклина. Концентрацию белка можно определить с помощью колориметрических анализов, таких как анализ с бицинхониновой кислотой (Smith, P. K. et al., Anal. Biochem. 150, 76, 1985) или анализ с красителем Брэдфорда (Bradford, M. M., (1976) Anal. Biochem. 72:248). Сохранение антигенсвязывающей активности антител после методик конъюгирования можно определить с помощью способа ELISA (Yu, D. S. et al., J. Urol. 140, 415, 1988) и с помощью цитофлуориметрических способов (Gallego, J. et al., Int. J. Cancer 33, 737, 1984). Чувствительность к кислоте конъюгата может оцениваться хроматографическими способами после инкубирования соединений в подходящих буферных растворах.
ЛЕКАРСТВЕННАЯ НАГРУЗКА
Лекарственная нагрузка представлена p в молекуле конъюгата антитела с лекарственным средством формулы I, средним числом лекарственных средств производных антрациклина на антитело. Лекарственная нагрузка может находиться в интервале от 1 до 8 лекарственных средств (D) на антитело (Ab), то есть, где 1, 2, 3, 4, 5, 6, 7 и 8 лекарственных групп ковалентно присоединены к антителу. Композиции ADC формулы I включают множество антител, конъюгированных с рядом лекарственных средств, от 1 до 8. Среднее число лекарственных средств на антитело в препаратах ADC в результате реакций конъюгирования может быть охарактеризовано обычными способами, такими как масс-спектроскопия, анализ ELISA, электрофорез и ВЭЖХ. Также может быть определено количественное распределение ADC по величине p. С помощью ELISA можно определить среднюю величину p в конкретном препарате ADC (Hamblett et al. (2004) Clin. Cancer Res. 10:7063-7070; Sanderson et al. (2005) Clin. Cancer Res. 11:843-852). Однако величины распределения p (лекарственного средства) не различимы посредством связывания антитела с антигеном и ограничением определения ELISA. Также, анализ ELISA для обнаружения конъюгатов антитела с лекарственным средством не определяет, где лекарственные группы присоединяются к антителу, например, фрагментам тяжелой цепи или легкой цепи, или конкретным аминокислотным остаткам. В некоторых случаях, разделение, очистка и характеристика гомогенных ADC, где p равно конкретному значению из ADC с другими нагрузками лекарственного средства, могут быть достигнуты такими способами, как ВЭЖХ с обращенной фазой или электрофорез.
Для некоторых конъюгатов антитела с лекарственным средством, p может быть ограничено числом сайтов присоединения на антителе. Например, антитело может иметь только одну или несколько тиольных групп цистеина, или может иметь только одну или несколько достаточно реакционно-способных тиольных групп, через которые может быть присоединен линкер. Более высокая нагрузка лекарственного средства, например p >5, может вызывать агрегацию, нерастворимость, токсичность или недостаточную клеточную проницаемость некоторых конъюгатов антитела с лекарственным средством.
Обычно, во время реакции конъюгирования с антителом конъюгируют меньше теоретического максимума лекарственных групп. Антитело может содержать, например, много остатков лизина, которые не взаимодействуют с промежуточным соединением лекарственного средства и линкера (D-L) или линкерным реагентом. Только наиболее реакционно-способные группы лизина могут взаимодействовать с амин-реактивным линкерным реагентом. Также, только наиболее реакционно-способные тиольные группы цистеина могут взаимодействовать с тиол-реактивным линкерным реагеном. В основном, антитела не содержат много, если таковые имеются, свободных и реакционно-способных тиольных групп цистеина, которые могут быть соединены с лекарственной частью. Большинство тиолов остатков цистеина в антителах соединений существуют в виде дисульфидных мостиков и должны быть восстановлены восстанавливающим агентом, таким как дитиотреитол (DTT) или TCEP, в частичных или полностью редуцирующих условиях. Кроме того, антитело должно подвергаться денатурирующим условиям для обнаружения реакционно-способных нуклеофильных групп, таких как лизин или цистеин. Нагрузка (соотношение лекарственное средство/антитело) ADC может контролироваться несколькими различными способами, включая: (i) ограничение молярного избытка промежуточного соединения лекарственное средство-линкер (D-L) или линкерного реагента относительно антитела, (ii) ограничение времени реакции конъюгирования или температуры, и (iii) частичные или ограничивающие восстановительные условия для модификации тиола цистеина.
Цистеины могут быть сконструированы в реакционно-способных сайтах в антителе и которые не образуют внутрицепочечных или межмолекулярных дисульфидных связей (US 7521541). Сконструированные тиола цистеина могут взаимодействовать с линкерными реагентами или лекарственным средством-линкерными реагентами по настоящему изобретению, которые обладают тиол-реактивными, электрофильными группами, такими как малеимид или альфа-галоамиды с образованием ADC с антителами с цистеиновыми заменами и лекарственными группами производного антрациклина. Таким образом, локализация лекарственной части может быть разработана, контролироваться и быть известной. Лекарственная нагрузка может контролироваться, поскольку сконструированные тиольные группы цистеина обычно взаимодействуют с тиол-реактивными линкерными реагентами или лекарственным средством-линкерными реагентами с высоким выходом. Конструирование IgG антитела для введения аминокислоты цистеина путем замещения в единичном сайте на тяжелой или легкой цепи дает два новых цистеина на симметричном антителе. Может достигаться лекарственная нагрузка, близкая к 2, и близкая гомогенность продукта конъюгирования ADC.
В тех случаях, когда более одной нуклеофильной или электрофильной группы антитела взаимодействует с промежуточным соединением лекарственное средство-линкер, или линкерным реагентом, затем с реагентом лекарственной группы, затем полученный в результате продукт представляет собой смесь соединений ADC с распределением лекарственных групп, присоединенных к антителу, например 1, 2, 3, и т.д. Способы жидкостной хроматографии, такие как полимерная с обращенной фазой (PLRP) и гидрофобного взаимодействия (HIC) могут разделять соединения в смеси по величине лекарственной нагрузки. Препараты ADC с единичной величиной лекарственной нагрузки (p) могут быть выделены, однако, эти ADC с единичной величиной лекарственной нагрузки могут оставаться гетерогенными смесями, поскольку лекарственные группы могут быть присоединены через линкер, в различных сайтах на антителе.
Таким образом, композиции по изобретению конъюгат антитела с лекарственным средством, где антитело имеет одну или несколько лекарственных групп производного антациклина, и где лекарственные группы могут быть присоединены к антителу на различных аминокислотных остатках.
ПОЛУЧЕНИЕ КОНЪЮГАТОВ АНТИТЕЛА С ЛЕКАРСТВЕННЫМ СРЕДСТВОМ
ADC формулы I может быть получен несколькими путями, используя реакции органической химии, условия и реагенты, известные специалистам в данной области, в том числе: (1) реакцию нуклеофильной группы или электрофильной группы антитела с бивалентным линкерным реагентом с образованием промежуточного соединения антитело-линкер Ab-L, посредством ковалентной связи, с последующим взаимодействием с активированным реагентом лекарственной группы; и (2) реакция нуклеофильной группы или электрофильной группы реагента лекарственной группы с линкерным реагентом, с образованием реагента лекарственное средство-линкер D-L, через ковалентную связь, с последующим взаимодействием с нуклеофильной группой или электрофильной группой антитела. Способы конъюгирования (1) и (2) могут быть использованы с целым рядом антител, лекарственных групп и линкеров для получения конъюгатов антитела с лекарственным средством формулы I.
Нуклеофильные группы на антителах включают, но этим не ограничиваются: (i) N-концевые аминные группы, (ii) аминные группы боковых цепей, например, лизина, (iii) тиольные группы боковых цепей, например, цистеина, и (iv) гидроксил сахара или аминогруппы, где антитело является гликозилированным. Аминные, тиольные и гидроксильные группы являются нуклеофильными и способны вступать в реакцию с образованием ковалентных связей с электрофильными группами на линкерных звеньях и линкерных реагентах, в том числе: (i) активными сложными эфирами, такими как сложные эфиры NHS, сложные эфиры HOBt, галоформиаты и кислые галогениды; (ii) алкил и бензил галогениды, такие как галоацетамиды; (iii) альдегиды, кетоны, карбоксильные и малеимидные группы. Некоторые антитела имеют восстанавливаемые межцепочечные дисульфиды, то есть цистеиновые мостики. Антитела могут быть сделаны реакционно-способными для конъюгирования с линкерными реагентами путем обработки восстанавливающим агентом, таким как DTT (реактив Клеланда, дитиотреитол) или TCEP (трис(2-карбоксиэтил)фосфин гидрохлорид; Getz et al. (1999) Anal. Biochem. Vol 273:73-80; Soltec Ventures, Beverly, MA). Каждый цистеиновый дисульфидный мостик таким образом будет образовывать, теоретически, два реакционно-способных тиольных нуклеофила. Дополнительные нуклеофильные группы могут быть введены в антитела посредством реакции лизинов с 2-иминотиоланом (реактивом Трота), получая в результате превращение амина в тиол.
Конъюгаты антитела с лекарственным средством также могут быть получены путем модификации антитела для введения электрофильных групп, которые могут вступать в реакцию с нуклеофильными заместителями на линкерном реагенте или лекарственном средстве. Сахара гликозилированных антител могут быть окислены, например периодатными окисляющими реагентами с образованием альдегидных или кетонных групп, которые могут вступать в реакцию с аминной группой линкерных реагентов или лекарственных частей. Полученные в результате иминные группы основания Шиффа могут образовывать стабильную связь, или могут быть восстановлены, например под действием боргидридных реагентов с образованием стабильных аминных связей. В одном варианте осуществления, реакция углеводной части гликозилированного антитела либо с галактозоксидазой, либо мета-периодатом натрия может давать на выходе карбонильные (альдегидные и кетонные) группы в белке, которые могут вступать в реакцию с соответствующими группами на лекарственном средстве (Hermanson, G.T. (1996) Bioconjugate Techniques; Academic Press: New York, p 234-242). В другом варианте осуществления, белки, содержащие N-концевые остатки серина или треонина, могут вступать в реакцию с мета-периодатом натрия, приводя в результате к образованию альдегида вместо первой аминокислоты (Geoghegan & Stroh, (1992) Bioconjugate Chem. 3:138-146; US 5362852). Такой альдегид может вступать в реакцию с лекарственной группой или нуклеофилом линкера.
Аналогично, нуклеофильные группы на лекарственной составляющей включают, но этим не ограничиваются: аминные, тиольные, гидроксильные, гидразидные, оксимные, гидразинные, тиосемикарбазонные, гидразинкарбоксилатные и арилгидразидные группы, способные вступать в реакцию с образованием ковалентных связей с электрофильными группами на линкерных составляющих и линкерных реагентах, в том числе: (i) активными сложными эфирами, такими как сложные эфиры NHS, сложные эфиры HOBt, галоформиаты и кислые галогениды; (ii) алкил и бензил галогенидами, такими как галоацетамиды; (iii) альдегидами, кетонами, карбоксильными и малеимидными группами. Реакционно-способные нуклеофильные группы могут быть введены в соединения производного антрациклина путем стандартных взаимных превращений функциональных групп. Например, гидроксильные группы могут быть преобразованы в тиольные группы посредством реакций типа реакции Мицунобу, с образованием соединений тиол-модифицированных лекарственных средств.
Конъюгаты антитела с лекарственным средством в таблице 2 получали в соответствии со способами, описанными в Примерах, и тестировали на эффективность с помощью исследования клеточной пролиферации in vitro и ингибирования роста ксенотрансплантата in vivo.
Конъюгаты антитела с лекарственным средством
СКРИНИНГ КОНЪЮГАТОВ АНТИТЕЛА С ЛЕКАРСТВЕННЫМ СРЕДСТВОМ (ADC), НАПРАВЛЕННЫХ ПРОТИВ СВЯЗАННЫХ С ОПУХОЛЬЮ АНТИГЕНОВ И РЕЦЕПТОРОВ КЛЕТОЧНОЙ ПОВЕРХНОСТИ
Способы исследования для выявления злокачественных клеток включают воздействие на клетки соединения конъюгата антитела с лекарственным средством и определение степени связывания конъюгата антитело-лекарственное средство с этими клетками. Соединения ADC Формулы I, которые идентифицируют в моделях на животных и клеточных исследованиях, дополнительно могут быть протестированы у высших приматов с опухолями и в клинических испытаниях у людей.
Трансгенные животные и клеточные линии особенно применимы при скрининге конъюгатов антитела с лекарственным средством (ADC), которые эффективны в качестве профилактического или терапевтического лечения заболеваний или нарушений, в патологический процесс которых вовлечена сверхэкспрессия антигенов, связанных с опухолью, и рецепторов клеточной поверхности, например HER2 (US 6632979). Скрининг на применимые ADC может включать введение предполагаемых ADC в диапазоне доз трансгенному животному и исследование в различные моменты времени действия ADC на рассматриваемые заболевания или нарушения. Альтернативно, или дополнительно, лекарственное средство можно вводить до или одновременно с воздействием индуктора заболеваний, в случае необходимости. Предполагаемые ADC могут подвергаться скринингу партиями и по-отдельности, или параллельно в среднем или высокопроизводительном формате скрининга. Скорость, при которой может проводиться скрининг ADC на применимость для профилактического или терапевтического лечения заболеваний или нарушений, ограничена только скоростью синтеза или методикой скрининга, включая обнаружение/измерение/анализ данных.
Один из вариантов осуществления представляет собой способ скрининга, предусматривающий (a) трансплантацию клеток из стабильной клеточной линии рака груди животному, не являющемуся человеком, (b) введение потенциального лекарственного средства ADC животному, не являющемуся человеком, и (c) определение способности потенциального лекарственного средства ингибировать образование опухолей из трансплантированной клеточной линии. Настоящее изобретение также относится к способу скрининга потенциальных ADC для лечения заболевания или нарушения, характеризующегося сверхэкспрессией рецепторного белка, предусматривающему (a) контактирование клеток из стабильной клеточной линии рака груди с потенциальным лекарственным средством, и (b) оценку способности потенциального ADC ингибировать рост стабильной клеточной линии.
Один из вариантов осуществления представляет собой способ скрининга, предусматривающий (a) контактирование клетки из стабильной клеточной линии рака груди с потенциальным лекарственным средством ADC и (b) оценку способности потенциального средства ADC индуцировать гибель клеток, индуцировать апоптоз, блокировать связывание герегулина, блокировать стимулируемое лигандом фосфорилирование тирозина или блокировать лигандную активацию HER2. В другом варианте осуществления оценивают способность потенциального ADC.
Другой вариант осуществления представляет собой способ скрининга, предусматривающий (a) введение потенциального лекарственного средства ADC трансгенному млекопитающему, не являющемуся человеком, у которого сверхэкспрессируется, например, в клетках молочной железы, нативный белок человека, например HER2 или его фрагмент, где такое трансгенное млекопитающее имеет стабильно интегрированную в его геном последовательность нуклеиновой кислоты, кодирующей нативный белок человека или его фрагмент, обладающий биологической активностью нативного белка человека, оперативно связанную с регуляторными последовательностями транскрипции, направляющими его экспрессию, и развивается опухоль. Потенциальные ADC подвергают скринингу путем введения трансгенному животному в диапазоне доз и оценки физиологического ответа животного на соединение в динамике по времени. Введение может быть пероральным или посредством подходящей инъекции, в зависимости от химической природы оцениваемого соединения. В некоторых случаях, может быть целесообразно вводить соединение в сочетании с кофакторами, которые бы усиливали эффективность соединения. Если клеточные линии, полученные от рассматриваемых трансгенных животных, используют для скрининга соединений, применимых при лечении различных нарушений, связанных с сверхэкспрессией некоторых антигенных белков, связанных с опухолью, или рецепторов клеточной поверхности, например сверхэкспрессии HER2. Для идентификации соединений ADC, ингибирующих рост, которые специфически направлены на HER2, можно проводить скрининг на ADC, который ингибирует рост злокачественных клеток, сверхэкспрессирующих HER2, полученных от трансгенных животных (US 5677171).
АНАЛИЗ ПРОЛИФЕРАЦИИ КЛЕТОК IN VITRO
В основном, цитотоксическую или цитостатическую активность конъюгата антитела с лекарственным средством (ADC) измеряют путем: воздействия на клетки млекопитающих, имеющих связанные с опухолью антигены или рецепторные белки, антитела ADC в клеточной культуральной среде; культивирования клеток в течение периода времени примерно от 6 часов примерно до 5 дней и измерения жизнеспособности клеток. Клеточные исследования in vitro могут быть использованы для измерения жизнеспособности, то есть пролиферации (IC50), цитотоксичности (EC50) и индукции апоптоза (активация каспазы) ADC. Анализ жизнеспособности клеток CellTiter-Glo® Luminescent Cell Viability Assay представляет собой коммерчески доступный (Promega Corp., Madison, WI) гомогенный метод исследования, основанный на рекомбинантной экспрессии люциферазы Coleoptera (US 5583024; US 5674713; US 5700670). Этот анализ клеточной пролиферации определяет количество жизнеспособных клеток в культуре, исходя из количества присутствующего АТФ, индикатора метаболически активных клеток (Crouch et al. (1993) J. Immunol. Meth. 160:81-88; US 6602677). Анализ CellTiter-Glo® Assay проводят в 96-луночном формате, делая его пригодным для автоматического выскопроизводительного скрининга (HTS) (Cree et al. (1995) AntiCabcer Drugs 6:398-404). Методика гомогенного анализа включает добавление одного реагента (CellTiter-Glo® Reagent) непосредственно к клеткам, культивируемым в среде с добавлением сыворотки. Стадии промывания клеток, удаления среды и многочисленного пипетирования не требуются. Эти системы обнаруживают минимальное количество 15 клеток/лунку в 384-луночном формате через 10 минут после добавления реагента и перемешивания.
Оценку сохранения цитотоксичности конъюгатов по сравнению с исходным лекарственным средством можно проводить с помощью теста, основанного на количественном определении АТФ. Злокачественные клетки яичника человека A2780 и молочной железы человека MCF7 (1250 клетки/лунку) высевали в белые 384-луночные планшеты в полной среде (RPMI1640 или EMEM плюс 10% эмбриональная сыворотка теленка) и обрабатывали соединениями, растворенными в 0,1% ДМСО, через 24 ч после высевания. Клетки инкубировали при 37°C и 5% CO2 и через 72 часа планшеты обрабатывали, используя анализ CellTiter-Glo (Promega), следуя инструкциям производителя.
CellTiter-Glo представляет собой гомогенный способ, основанный на количественном определении присутствующего АТФ, индикатора метаболически активных клеток. Количественное определение АТФ проводят, используя системы, основанные на люциферазе и D-люциферине, получая в результате генерацию света. Люминесцентный сигнал является пропорциональным количеству клеток, находящихся в культуре. Вкратце, 25 мкл/лунку растворов реагентов добавляли к каждой лунке и после 5 минут встряхивания микропланшеты считывали с помощью люминометра. Люминесцентный сигнал является пропорциональным количеству клеток, находящихся в культуре.
Антипролиферативные эффекты конъюгатов антитело-лекарственное средство формулы Ic (Таблица 6) измеряли с помощью анализа CellTiter-Glo® (Пример 9) против линий опухолевых клеток, экспрессирующих HER2, на Фигурах 8-29 в 3 дневных исследованиях непрерывного воздействия.
На Фигуре 8 показан график зависимости 3-дневной выживаемости клеток SK-BR-3 in vitro от концентраций: непрерывного воздействия свободного лекарственного средства PNU-159682, 1-часового инкубирования с PNU-159682, линкер лекарственное средство: NHS-кеталь-Ant 50, линкер лекарственное средство: малеимид-кеталь-Ant 51, линкер лекарственное средство: малеимид-гидразон-Ant 52, и линкер лекарственное средство: тиопиридин-гидразон-Ant 53. Уровень экспрессии HER2 клеток SK-BR-3 составляет 3+. Пролиферация клеток SK-BR-3 наиболее эффективно ингибировалась непрерывным воздействием PNU-159682. Короткое (1 час) воздействие также эффективно ингибировало рост клеток SK-BR-3. Гидразон-связанные Ant 52 и 53 были менее эффективным, чем свободный Ant, тогда как кеталь-связанные Ant 50 и 51 промежуточные соединения линкера и лекарственного средства показали минимальную антипролиферативную активность.
На Фигуре 9 показан график зависимости 3-дневной выживаемости клеток ВТ-474 in vitro от концентраций: свободного лекарственного средства: PNU-159682 непрерывное воздействие, свободного лекарственного средства: PNU-159682 1-часовое инкубирование, линкера-лекарственного средства: NHS-кеталь-Ant 50, линкера-лекарственного средства: малеимид-кеталь-Ant 51, линкера-лекарственного средства: малеимид-гидразон-Ant 52 и линкера-лекарственного средства: тиопиридин-гидразон-Ant 53. Уровень экспрессии HER2 клеток BT-474 составляет 3+. Пролиферация клеток BT-474 наиболее эффективно ингибировалась непрерывным воздействием PNU-159682. Короткое (1 час) воздействие также эффективно ингибировало рост клеток BT-474. Гидразон-связанные Ant 52 и 53 показали минимальную антипролиферативную активность, тогда как кеталь-связанные Ant 50 и 51 были неактивными.
На Фигуре 10 показан график зависимости 3-дневной выживаемости MCF7 in vitro от концентраций: свободного лекарственного средства: PNU-159682 непрерывное воздействие, свободного лекарственного средства: PNU-159682 1-часовое инкубирование, линкера-лекарственного средства: NHS-кеталь-Ant 50, линкера-лекарственного средства: малеимид-кеталь-Ant 51, линкера-лекарственного средства: малеимид-гидразон-Ant 52 и линкера-лекарственного средства: тиопиридин-гидразон-Ant 53. Пролиферация клеток MCF7 наиболее эффективно ингибировалась непрерывным воздействием o PNU-159682. Короткое (1 час) воздействие также эффективно ингибировало рост клеток MCF7. Гидразон-связанные Ant 52 и 53 показали минимальную антипролиферативную активность, тогда как кеталь-связанные Ant 50 и 51 были неактивными.
На Фигуре 11 показан график зависимости 3-дневной выживаемости клеток Her2, резистентных к доксорубицину (DoxRes), in vitro от концентраций: свободного лекарственного средства: PNU-159682 непрерывное воздействие, свободного лекарственного средства: PNU-159682 1 часовое инкубирование, линкера-лекарственного средства: NHS-кеталь-Ant 50, линкера-лекарственного средства: малеимид-кеталь-Ant 51, линкера-лекарственного средства: малеимид-гидразон-Ant 52 и линкера-лекарственного средства: тиопиридин-гидразон-Ant 53. Уровень экспрессии HER2 клеток DoxRes HER2 составляет 3+, с высоким PgP/MDR1. Пролиферация клеток DoxRes/HER2 наиболее эффективно ингибировалась непрерывным воздействием PNU-159682. Короткое (1 час) воздействие также эффективно ингибировало рост клеток AdrRes/HER2. Гидразон-связанные Ant 52 и 53 были менее эффективными, чем свободный Ant, тогда как кеталь-связанные Ant 50 и 51 показали минимальную антипролиферативную активность. Клеточная линия DoxRes Her2 также известна как «AdrRes Her2».
На Фигуре 12 показан график зависимости 3-дневной выживаемости клеток SK-BR-3 in vitro от концентраций: трастузумаба, трастузумаб-MCC-DM1 101, тио-трастузумаб (HC A114C)-малеимид-кеталь-Ant 102 и тио-трастузумаб (HC A114C)-малеимид-гидразон-Ant 103. Тио-Tr-HC-малеимид-гидразон-Ant 103 показал наиболее сильную антипролиферативную активность в отношении клеток SK-BR-3. Тио-Tr-HC-малеимид-кеталь-Ant 102 и Tr-MCC-DM1 101 были в равной мере эффективными в значениях IC50, но обработка клеток SK-BR-3 с использованием 102 в результате приводила к большему уничтожению клеток чем 101. Все протестированные конъюгаты были более эффективными, чем трастузумаб.
На Фигуре 13 показан график зависимости 3-дневной выживаемости клеток ВТ-474 in vitro от концентраций: трастузумаба, трастузумаб-MCC-DM1 101, тио-трастузумаб (HC A114C)-малеимид-кеталь-Ant 102 и тио-трастузумаб (HC A114C)-малеимид-гидразон-Ant 103. Тио-Tr-HC-малеимид-гидразон-Ant 103 показал наиболее сильную антипролиферативную активность в отношении клеток BT-474. Тио-Tr-HC-малеимид-кеталь-Ant 102 и Tr-MCC-DM1 101 были в равной мере эффективными в ингибировании роста клеток BT-474. Все протестированные конъюгаты были более эффективными, чем трастузумаб.
На Фигуре 14 показан график зависимости 3-дневной выживаемости клеток MCF-7 in vitro от концентраций: трастузумаба, трастузумаб-MCC-DM1 101, тио-трастузумаб (HC A114C)-малеимид-кеталь-Ant 102 и тио-трастузумаб (HC A114C)-малеимид-гидразон-Ant 103. Уровень экспрессии HER2 клеток MCF7 является нормальным. Тио-Tr-HC-малеимид-гидразон-Ant 103 показал сильную антипролиферативную активность в отношении клеточной линии Her2-норма MCF7. Трастузумаб, Tr-MCC-DM1 101 и тио-Tr-HC-малеимид-кеталь-Ant 102 не были активными в отношении клеток MCF7.
На Фигуре 15 показан график зависимости 3-дневной выживаемости клеток HER2, резистентных к доксорубицину (DoxRes), in vitro от концентраций: трастузумаба, трастузумаб-MCC-DM1 101, тио-трастузумаб (HC A114C)-малеимид-кеталь-Ant 102 и тио-трастузумаб (HC A114C)-малеимид-гидразон-Ant 103. Тио-Tr-HC-малеимид-гидразон-Ant 103 показал сильную антипролиферативную активность в отношении клеток DoxRes/HER2, которые экспрессируют высокие уровни как HER2, так и переносчика MDR1/PgP со множественной резистентностью к лекарственным средствам. Тио-Tr-HC-малеимид-кеталь-Ant 103 был активным только в двух наиболее высоких тестируемых концентрациях (3,3 и 10 мкг/мл), тогда как Tr-MCC-DM1 101 и трастузумаб показали минимальную активность в отношении этой клеточной линии.
На Фигуре 16 показан график зависимости 3-дневной выживаемости клеток SK-BR-3 in vitro от концентраций: анти-CD22 NHS кеталь-Ant 110, трастузумаба, трастузумаб-MCC-DM1 101 и тио-трастузумаб (HC A114C)-NHS-кеталь-Ant 105. Тио-Tr-HC-NHS-кеталь-Ant 105 и Tr-MCC-DM1 101 показали эквивалентную эффективность в отношении пролиферации клеток SK-BR-3 в значениях IC50; обработка с использованием 105 в результате приводит к большему уничтожению клеток. Ненаправленный контроль анти-CD22-NHS-кеталь-Ant 110 также показал сильную антипролиферативную активность в отношении клеток SK-BR-3. Все протестированные конъюгаты были более эффективными, чем трастузумаб.
На Фигуре 17 показан график зависимости 3-дневной выживаемости клеток ВТ-474 in vitro от концентраций: анти-CD22 NHS кеталь-Ant 110, трастузумаба, трастузумаб-MCC-DM1 101 и тио-трастузумаб (HC A114C)-NHS-кеталь-Ant 105. Тио-Tr-HC-NHS кеталь-Ant 105 показал наиболее сильную антипролиферативную активность в отношении клеток BT-474. Обработка с использованием Tr-MCC-DM1 также в результате приводила к ингибированию роста клеток BT-474. Ненаправленный контроль анти-CD22-NHS-кеталь-Ant 110 показал сильные антипролиферативные эффекты в отношении клеток BT-474. Все протестированные конъюгаты были более эффективными, чем трастузумаб.
На Фигуре 18 показан график зависимости 3-дневной выживаемости клеток MCF-7 in vitro от концентраций: анти-CD22 NHS кеталь-Ant 110, трастузумаба, трастузумаб-MCC-DM1 101 и тио-трастузумаб (HC A114C)-NHS-кеталь-Ant 105. Тио-Tr-HC-NHS-кеталь-Ant 105 и анти-CD22-NHS-кеталь-Ant 110 показали эквивалентную активность в отношении клеток MCF7 с низкой экспрессией HER2. Трастузумаб и Tr-MCC-DM1 101 не были активными в отношении MCF7.
На Фигуре 19 показан график зависимости 3-дневной выживаемости клеток Her2, резистентных к доксорубицину (DoxRes), in vitro от концентраций: анти-CD22 NHS кеталь-Ant 110, трастузумаба, трастузумаб-MCC-DM1 101 и тио-трастузумаб (HC A114C)-NHS-кеталь-Ant 105. Тио-Tr-HC-NHS-кеталь-Ant 105 и анти-CD22-NHS-кеталь-Ant 110 показали эквивалентную активность в отношении клеток DoxRes/HER2. Tr-MCC-DM1 101 показал невысокую активность, а трастузумаб не оказывал действия на DoxRes/HER2.
На Фигуре 20 показан график зависимости 3-дневной выживаемости клеток SK-BR-3 in vitro от концентраций: тио-трастузумаб (HC A114C)-малеимид-кеталь-Ant 102, тио-трастузумаб (HC A114C)-малеимид-гидразон-Ant 103, тио-трастузумаб (HC A114C)-тиопиридин-гидразон-Ant 104, тио-трастузумаб (HC A114C)-NHS-кеталь-Ant 105, трастузумаб-MCC-DM1 101 и тио-трастузумаб (HC A114C)-MC-vc-PAB-MMAE 106.
На Фигуре 21 показан график зависимости 3-дневной выживаемости клеток SK-BR-3 in vitro от концентраций: трастузумаба, тио-анти-CD22 (HC A114C)-малеимид-кеталь-Ant 107, тио-анти-CD22 (HC A114C)-малеимид-гидразон-Ant 108, тио-анти-CD22 (HC A114C)-тиопиридин-гидразон-Ant 109, анти-CD22-NHS-кеталь-Ant 110, и свободного лекарственного средства PNU-159682. Как сообщалось ранее, трастузумаб незначительно ингибирует рост клеток SK-BR-3 посредством цитостатического механизма. Ненаправленные контрольные ADC тио-анти-CD22-HC-кеталь-Ant 107 и анти-CD22-NHS-кеталь-Ant 110 показали антипролиферативную активность только при наиболее высоких тестируемых дозах. Ненаправленные контрольные ADC тио-анти-CD22-HC-малеимид-гидразон-Ant 108 и тио-анти-CD22-H-тиопиридин-гидразон-Ant 109 показали эквивалентную эффективнсть в отношении ингибирования роста клеток SK-BR-3 как HER2-направленные ADC (На Фигуре 20), что указывает на лабильность гидразон-связанных ADC. Свободное лекарственное средство PNU-159682, вводимое в мкМ концентрациях, вызывало цитотоксичность во всех тестируемых дозах.
На Фигуре 22 показан график зависимости 3-дневной выживаемости клеток ВТ-474 in vitro от концентраций: тио-трастузумаб (HC A114C)-малеимид-кеталь-Ant 102, тио-трастузумаб (HC A114C)-малеимид-гидразон-Ant 103, тио-трастузумаб (HC A114C)-тиопиридин-гидразон-Ant 104, тио-трастузумаб (HC A114C)-NHS-кеталь-Ant 105, трастузумаб-MCC-DM1 101 и тио-трастузумаб (HC A114C)-MC-vc-PAB-MMAE 106. Все протестированные ADC показали аналогичную активность в отношении пролиферации клеток BT-474. Тио-Tr-HC-vc-MMAE 106 имел самую низкую IC50 (0,01 мкг/мл), хотя обработка с использованием тио-Tr-HC-малеимид-гидразон-Ant 103 и тио-Tr-HC-тиопиридин-гидразон-Ant 104 в результате приводила к наибольшей степени суммарного ингибирования роста.
На Фигуре 23 показан график зависимости 3-дневной выживаемости клеток ВТ-474 in vitro от концентраций: трастузумаба, тио-анти-CD22 (HC A114C)-малеимид-кеталь-Ant 107, тио-анти-CD22 (HC A114C)-малеимид-гидразон-Ant 108, тио-анти-CD22 (HC A114C)-тиопиридин-гидразон-Ant 109, анти-CD22-NHS-кеталь-Ant 110, и свободного лекарственного средства PNU-159682. Как сообщалось ранее, трастузумаб незначительно ингибирует рост клеток BT-474 посредством цитостатического механизма. Ненаправленные контрольные ADC тио-анти-CD22-HC-кеталь-Ant 107 и анти-CD22-NHS-кеталь-Ant 110 не оказывали антипролиферативного действия на клетки BT-474. Ненаправленные контрольные ADC тио-анти-CD22-HC-малеимид-гидразон-Ant 108 и тио-анти-CD22-H-тиопиридин-гидразон-Ant 109 показали эквивалентную эффективность в отношении ингибирования роста клеток BT-474 как HER2-направленные ADC (Фигура 22), указывая на лабильность гидразон-связанных ADC. Свободное лекарственное средство PNU-159682, вводимое в мкМ концентрациях, вызывало цитотоксичность во всех тестируемых дозах.
На Фигуре 24 показан график зависимости 3-дневной выживаемости клеток MCF-7 in vitro от концентраций: тио-трастузумаб (HC A114C)-малеимид-кеталь-Ant 102, тио-трастузумаб (HC A114C)-малеимид-гидразон-Ant 103, тио-трастузумаб (HC A114C)-тиопиридин-гидразон-Ant 104, тио-трастузумаб (HC A114C)-NHS-кеталь-Ant 105, трастузумаб-MCC-DM1 101 и тио-трастузумаб (HC A114C)-MC-vc-PAB-MMAE 106. Только тио-Tr-HC-малеимид-гидразон-Ant 103 и тио-Tr-HC-тиопиридин-Ant 104 оказывали антипролиферативные действия на клетки MCF7 с низким HER2, указывая на лабильность гидразон-связанных ADC. Все другие протестированные ADC (тио-Tr-HC-vc-MMAE 106, Tr-MCC-DM1 101, тио-Tr-HC-малеимид-кеталь-Ant 102 и тио-Tr-NHS-кеталь-Ant 105) не оказывали действия на рост клеток MCF7.
На Фигуре 25 показан график зависимости 3-дневной выживаемости клеток MCF-7 in vitro от концентраций: трастузумаба, тио-анти-CD22 (HC A114C)-малеимид-кеталь-Ant 107, тио-анти-CD22 (HC A114C)-малеимид-гидразон-Ant 108, тио-анти-CD22 (HC A114C)-тиопиридин-гидразон-Ant 109, анти-CD22-NHS-кеталь-Ant 110, и свободного лекарственного средства PNU-159682. Согласуясь с предыдущими сообщениями, трастузумаб был полностью неактивным в отношении клеток MCF7 с низкой экспрессией HER2. Ненаправленные контрольные ADC тио-анти-CD22-HC-малеимид-кеталь-Ant 107 и тио-анти-CD22-NHS-кеталь-Ant 110 также не ингибировали рост клеток MCF7, указывая на недостаточное высвобождение лекарственного средства из стабильных кетальных линкеров. Гидразон-связанные ненаправленные контрольные ADC 108 и 109 показали антипролиферативные действия на клетки MCF7, что указывает на высвобождение лекарственного средства из лабильных гидразон-связанных ADC. Свободное лекарственное средство PNU-159682, вводимое в мкМ дозах, вызывало цитотоксичность во всех тестируемых концентрациях.
На Фигуре 26 показан график зависимости 3-дневной выживаемости in vitro доксорубицин-резистентных (DoxRes)/HER2 от концентраций: тио-трастузумаб (HC A114C)-малеимид-кеталь-Ant 102, тио-трастузумаб (HC A114C)-малеимид-гидразон-Ant 103, тио-трастузумаб (HC A114C)-тиопиридин-гидразон-Ant 104, тио-трастузумаб (HC A114C)-NHS-кеталь-Ant 105, трастузумаб-MCC-DM1 101 и тио-трастузумаб (HC A114C)-MC-vc-PAB-MMAE 106. Хотя 106 не оказывал действия, а Tr-MCC-DM1 101 оказывал минимальное действие на рост клеток DoxRes/HER2, кеталь-связанные ADC 102 и 105 были активными только в наиболее высоких тестируемых концентрациях, тогда как гидразон-связанные ADC 103 и 104 эффективно ингибировали рост клеток DoxRes/HER2.
На Фигуре 27 показан график зависимости 3-дневной выживаемости in vitro доксорубицин-резистентных (DoxRes)/HER2 клеток от концентраций: трастузумаба, тио-анти-CD22 (HC A114C)-малеимид-кеталь-Ant 107, тио-анти-CD22 (HC A114C)-малеимид-гидразон-Ant 108, тио-анти-CD22 (HC A114C)-тиопиридин-гидразон-Ant 109, анти-CD22-NHS-кеталь-Ant 110, и свободного лекарственного средства PNU-159682. Трастузумаб отдельно не оказывал действия на рост клеток DoxRes/HER2. Кеталь-связанные не направленные контрольные ADC 107 и 110 ингибировали рост клеток DoxRes/HER2 только при наиболее высоких тестируемых концентрациях, тогда как гидразон-связанные ненаправленные контрольные ADC 108 и 109 показали сильную антипролиферативную активность в отношении клеток DoxRes/HER2. Активности всех четырех ненаправленных анти-CD22-Ant контрольных ADC были аналогичны HER2-направленным тио-Ant ADC (Фигура 26). Свободное лекарственное средство PNU-159682, вводимое в мкМ дозах, ингибировало рост во всех тестируемых концентрациях.
На Фигуре 28 показан график зависимости 3-дневной выживаемости доксорубицин-резистентных (DoxRes)/HER2 клеток in vitro от концентраций: тио-трастузумаб (HC A114C)-малеимид-кеталь-Ant 102, тио-трастузумаб (HC A114C)-малеимид-гидразон-Ant 103, тио-трастузумаб (HC A114C)-тиопиридин-гидразон-Ant 104, тио-трастузумаб (HC A114C)-NHS-кеталь-Ant 105, трастузумаб-MCC-DM1 101 и тио-трастузумаб (HC A114C)-MC-vc-PAB-MMAE 106, все введены в присутствии верапамила (10 мкг/м). Верапамил ингибирует отток различных лекарственных средств, известных в качестве субстратов транспортера MDR1/P-гликопротеина со множественной устойчивостью к лекарственным средствам (PgP), который высоко экспрессируется на клетках DoxRes/HER2. Добавление верапамила делало клетки DoxRes/HER2 чувствительными к цитотоксическим эффектам Tr-MCC-DM1 101, что указывает на то, что DM1 является субстратом для MDR1/PgP. Верапамил не оказывал действия на ответ на другие тестируемые ADC (102-106), указывая на то, что лекарственные действия этих ADC не ингибируются под действием NDR1/PgP.
На Фигуре 29 показан график зависимости 3-дневной выживаемости доксорубицин-резистентных клеток (DoxRes)/HER2 in vitro от концентраций: трастузумаба, тио-анти-CD22 (HC A114C)-малеимид-кеталь-Ant 107, тио-анти-CD22 (HC A114C)-малеимид-гидразон-Ant 108, тио-анти-CD22 (HC A114C)-тиопиридин-гидразон-Ant 109, анти-CD22-NHS-кеталь-Ant 110, и свободного лекарственного средства PNU-159682, все вводили в присутствии верапамила. Все протестированные ADC (107-110) были в равной мере активны в присутствии верапамила (сравните с Фигурой 27), указывая на то, что Ant лекарственное средство не является субстратом для MDR1/PgP.
В Таблице 3 представлены значения IC50 (мкг/мл) для ингибирования пролиферации клеток SK-BR-3, BT-474, MCF7, доксорубицин-резистентных (DoxRes) клеток и доксорубицин-резистентных (DoxRes) клеток с верапамилом тестируемых соединений Фигур 20-29. Сравнение SK-BR-3 и BT-474 с MCF7, малеимид-кеталь-Ant и NHS-кеталь-Ant ADC показывает направленное уничтожение, тогда как гидразон-связанный-Ant ADC показывает ненаправленное, неселективное уничтожение клеток.
in vitro ингибирование пролиферации клеток SK-BR-3, BT-474, MCF7, доксорубицин-резистентных (DoxRes) клеток и доксорубицин-резистентных (DoxRes) клеток с верапамилом
SK-BR-3
Фигуры 20, 21
BT-474
Фигуры 22, 23
MCF7
Фигуры 24, 25
Adr-res HER2
Фигуры 26, 27
Фигуры 28, 29
Антипролиферативные действия конъюгатов антитело-лекарственное средство формулы Ic (Таблица 6) измеряли с использованием анализа CellTiter-Glo® (Пример 9) в отношении CD22 позитивных клеточных линий B-лимфомы: BJAB, GRANTA, DoHH2 и SuDHL4 в 3-дневных исследованиях непрерывного воздействия. Клетки Jurkat (CD22 негативные) обрабатывали конъюгатом антитело-лекарственное средство в качестве отрицательного контроля.
В Таблице 4 представлены значения IC50 (мкг/мл) для ингибирования клеточных линий BJAB, GRANTA, DoHH2 SuDHL4 и Jurkat под действием соединений тестируемых конъюгатов антитела с лекарственным средством в 3-дневных исследованиях непрерывного воздействия. Конъюгаты анти-CD22 антитела с лекарственным средством 107-110 показали высокоэффективные действия по уничтожению клеток. Значительное уничтожение клеток наблюдалось с конъюгатами отрицательного контроля анти-HER2 антитела 102-105. Значительное уничтожение клеток наблюдалось с конъюгатами 107-110 анти-CD22 антитела с лекарственным средством на CD22 негативные клетки Jurkat.
in vitro ингибирование клеточных линий BJAB, GRANTA, DoHH2 SuDHL4 и Jurkat
BJAB
GRANTA
DoHH2
SuDHL4
JURKAT
IN VIVO СЫВОРОТОЧНЫЙ КЛИРЕНС И СТАБИЛЬНОСТЬ У МЫШЕЙ
Сывороточный клиренс и стабильность ADC можно исследовать у “голых” “наивные” (без опухолей, полученных путем экзогенной трансплантации) мышей в соответствии с методиками из примера 10. Различие количества суммарных антител и ADC указывает на расщепление линкера и отделение антитела от его лекарственной группы.
Стабильность конъюгатов исследовали, используя ВЭЖХ анализ. Конъюгат инкубировали в аммоний ацетатных буферах при pH 4 и 5,2 при 37°C. Каждый раствор затем периодически брали и наносили на ВЭЖХ колонку, используя способ, описанный выше. Количество вещества, высвобожденного из конъюгата, определяли и выражали как процент высвобожденного вещества.
ЭФФЕКТИВНОСТЬ IN VIVO
Терапевтическое действие соединений конъюгата антитела с лекарственным средством (ADC) и улучшение их терапевтической эффективности по сравнению с исходным лекарственным средством оценивали в моделях на животных трансплантированных опухолей человека. Мышам, несущим ксенотрансплантаты опухолей человека, вводили подходящие дозы конъюгатов антитела с лекарственным средством, свободного лекарственного средства PNU-159682, и “голого” антитела, в определенных дозах, и рост опухоли регистрировали и сравнивали в различных испытуемых группах. На Фигурах 30-32 показана эффективность конъюгата антитела с лекарственным средством формулы Ic путем ингибирования ксенотрансплантированной опухоли у мышей.
Эффективность конъюгатов антитела с лекарственным средством по изобретению может быть измерена in vivo путем имплантации аллотрансплантов или ксенотрансплантатов злокачественных клеток или первичных опухолей у грызунов и воздействия на опухоли ADC в соответствии с методиками примера 12. Ожидаются переменные результаты в зависимости от клеточной линии, специфичности связывания антитела ADC с рецепторами, находящимися на злокачественных клетках, режима дозирования и других факторов. Например, эффективность in vivo анти-HER2 ADC может быть измерена с помощью мышиной модели с трансгенным эксплантом, высоко экспрессирующим HER2. Аллотрансплантат может быть репродуцирован из Fo5 mmtv трансгенной мыши, которая не реагирует или слабо реагирует на лечение HERCEPTIN®. Испытуемых подвергали воздействию однократно ADC и наблюдали на протяжении 3-6 недель для измерения времени удвоения опухоли, log уничтожения клеток и уменьшения размера опухоли. Впоследствии дополнительно можно проводить эксперименты по дозозависимому эффекту и эксперименты с многократными дозами.
На Фигуре 30 показан график изменения среднего объема опухоли с течением времени в лимфомах Беркитта Bjab-luc ксенотрансплантированных опухолях, инокулированных мышам CB17 SCID после однократного введения дозы в 0 день: (1) носителя, (2) тио-анти-CD22 (HC A114C)-MC-vc-PAB-MMAE 111 1 мг/кг, (3) тио-трастузумаб (HC A114C)-малеимид-кеталь-Ant 102 1 мг/кг, (4) тио-трастузумаб (HC A114C)-малеимид-кеталь-Ant 102 5 мг/кг, (5) тио-анти-CD22 (HC A114C)-малеимид-кеталь-Ant 107 1 мг/кг, (6) тио-анти-CD22 (HC A114C)-малеимид-кеталь-Ant 107 5 мг/кг, (7) тио-трастузумаб (HC A114C)-малеимид-гидразон-Ant 103 1 мг/кг, (8) тио-анти-CD22 (HC A114C)-малеимид-гидразон-Ant 108 1 мг/кг, (9) свободного лекарственного средства PNU-159682 8,77 мкг/кг. Все анти-CD22 конъюгаты (107, 108 и 111) показали специфичное к мишени ингибирование опухолевого роста и ингибирующая активность кеталь-связанного анти-CD22 ADC 107 была дозозависимой. Свободное лекарственное средство PNU-159682 и ненаправленные контрольные ADC (102 и 103) в эквивалентной дозе не оказывали действия на опухолевый рост.
В Таблице 5 показан уровень воздействия лекарственного средства, средняя лекарственная нагрузка, случаи возникновения опухоли и реакции для каждой испытуемой группы тестируемого соединения Фигуры 30- в 48- дневном исследовании эффективности in vivo на ксенотрансплантированной опухоли Bjab-luc. Bjab-luc (EBV-отрицательная лимфома Беркитта, клетки Bjab, экспрессирующие люциферазу) опухоли экспрессируют рецепторный белок CD22 (Polson et al. (2009) Cancer Res. 69(6):2358-2364; US 2008/0050310; US 2005/0276812). CD22 экспрессируется только в B-клеточном компартменте и на поверхности большинства клеток NHL (D'Arena et al. (2000) Am J Hematol. 64:275-81; Olejniczak et al. (2006) Immunol Invest. 35:93-114). Эффективность в ингибировании ксентрансплантированной опухоли Bjab-luc у мышей в качестве модельной системы может прогнозировать клинический ответ в лечении пациентов с гематопоэтическими злокачественными заболеваниями, такими как неходжкинская лимфома. Все из анти-CD22 ADC (107, 108, 111) были эффективны в отношении ингибирования опухоли по сравнению с носителем и контрольным ADC (102 и 103), который не связывался с клетками Bjab-luc. Отсутствие активности с контрольным ADC указывает на то, что эта активность, наблюдаемая с направленными ADC, является специфичной, например, не вследствие системного высвобождения свободного лекарственного средства. В некоторых случаях опухоли были не только ингибированы, а также частично и полностью регрессировали под действием анти-CD22 конъюгатов (107, 108, 111). Эти данные указывают на то, что анти-CD22 поверхностный антиген является потенциально эффективной мишенью для антрациклин-производных ADC по изобретению.
Исследование эффективности in vivo в отношении Bjab-luc ксенотрансплантированной опухоли (Фигура 30)
1 мг/кг
1 мг/кг,
5 мг/кг
1 мг/кг
5 мг/кг
1 мг/кг
1 мг/кг
8,77 мкг/кг
На Фигуре 31 показан график изменения среднего объема опухоли с течением времени в аллотрансплантированных опухолях молочной железы MMTV-HER2 Fo5, инокулированным мышам CRL nu/nu после однократного введения в 0 день: (1) носителя, (2) трастузумаб-MCC-DM1 101 5 мг/кг, (3) трастузумаб-MCC-DM1 101 10 мг/кг, (4) тио-трастузумаб (HC A114C)-малеимид-кеталь-Ant 102 5 мг/кг, (5) тио-трастузумаб (HC A114C)-малеимид-кеталь-Ant 102 10 мг/кг, (6) тио-анти-CD22 (HC A114C)-малеимид-кеталь-Ant 107 5 мг/кг, (7) тио-анти-CD22 (HC A114C)-малеимид-кеталь-Ant 107 10 мг/кг, (8) трастузумаб-MCC-DM1 101 5 мг/кг + тио-трастузумаб (HC A114C)-малеимид-кеталь-Ant 102 5 мг/кг. Все анти-Her2 конъюгаты (101 и 102) показали специфичное к мишени ингибирование опухолевого роста и ингибирующая активность была дозозависимой. Ненаправленный контрольный ADC 107 в эквивалентных дозах не оказывал действия на опухолевый рост.
В Таблице 6 показано ингибирование роста опухоли на 7 день, уровень лекарственного воздействия, средняя лекарственная нагрузка, частота возникновения опухоли и ответы для каждого тестируемого соединения испытуемой группы Фигуры 31- в 38- дневном исследовании эффективности in vivo MMTV-HER2 Fo5 аллотрансплантированных опухолях молочной железы (Phillips et al. (2008) Cancer Res. 68(22):9280-9290; US 2005/0276812). Аллотрансплантированная опухоль молочной железы MMTV-HER2 Fo5 является не чувствительной к трастузумабу, клеточная линия опухоли молочной железы, сверхэкспрессирующая HER2. Направленный анти-HER2 ADC (101, 102, и сочетание 101 и 102) были эффективными при ингибировании опухоли по сравнению с носителем и контрольным ADC (107), который не связывался с клетками MMTV-HER2 Fo5. Отсутствие активности с контрольным ADC указывает на то, что активность, наблюдаемая с направленным ADC, является специфичной, например, не вследствие системного высвобождения свободного лекарственного средства. Опухоли не только ингибируются, а также частично и полностью регрессированы под действием анти-HER2 конъюгатов (101, 102, и сочетание 101 и 102). Сочетание трастузумаб-MCC-DM1 101 и тио-трастузумаб (HC A114C)-малеимид-кеталь-Ant 102 не показало дополнительного ответа, выше чем одного средства 102. Эти данные указывают на то, что анти-HER2 поверхностный антиген является потенциально эффективной мишенью для антрациклин-производного ADC по изобретению.
in vivo исследование эффективности в аллотрансплантированной опухоли молочной железы MMTV-HER2 Fo5 (Фигура 31)
5 мг/кг
10 мг/кг
5 мг/кг
10 мг/кг
5 мг/кг
10 мг/кг
На Фигуре 32 показан график изменения среднего объема опухоли с течением времени в ксенотрансплантированных опухолях LnCap-Ner, инокулированных самцам мышей SCID мышь с врожденным отсутствием естественных клеток-киллеров после однократного введения в 0: (1) носителя, (2) тио-анти-steap1 (HC A114C)-MC-vc-PAB-MMAE 112 1 мг/кг, (3) тио-анти-steap1 (HC A114C)-MC-vc-PAB-MMAE 112 3 мг/кг, (4) тио-анти-steap1 (HC A114C)-малеимид-кеталь-Ant 113 1 мг/кг, (5) тио-анти-steap1 (HC A114C)-малеимид-кеталь-Ant 113 3 мг/кг, (6) тио-анти-steap1 (HC A114C)-малеимид-кеталь-Ant 113 6 мг/кг, (7) тио-анти-CD22 (HC A114C)-малеимид-кеталь-Ant 107 1 мг/кг, (8) тио-анти-CD22 (HC A114C)-малеимид-кеталь-Ant 107 3 мг/кг, (9) тио-анти-CD22 (HC A114C)-малеимид-кеталь-Ant 107 6 мг/кг. Все анти-steap1 конъюгаты (112 и 113) показали специфичное к мишени ингибирование роста опухоли и ингибирующая активность кеталь-связанного анти-steap1 ADC 113 была дозозависимой. Ненаправленный контрольный ADC 107 в эквивалентных дозах не оказывали действия на рост опухоли.
В Таблице 7 показан уровень лекарственного воздействия, средняя лекарственная нагрузка, частота возникновения опухоли и ответы для каждого тестируемого соединения испытуемой группы Фигуры 32- в 49-дневном исследовании эффективности in vivo в ксенотрансплантированной опухоли LnCap-Ner. Steap1 (шестой трансмембранный эпителиальный антиген предстательной железы) представляет собой простата-специфический антиген клеточной поверхности, высоко экспрессированный в опухолях предстательной железы человека (Hubert et al. (1999) PNAS 96(25):14523-14528). Клеточная линия LnCap высоко экспрессирует steap1. Исследование ингибирования ксенотрансплантированной опухоли LnCap-Ner (Jin et al. (2004) Cancer Res. 64:5489-5495) у мышей может быть эффективным прогностическим фактором для лечения злокачественных опухолей предстательной железы у пациентов. Направленные анти-steap1 ADC (112 и 113) были эффективными в ингибировании опухоли по сравнению с носителем и контрольным ADC (107), который не связывался с клетками LnCap. Отсутствие активности с контрольным ADC указывает на то, что активность, наблюдаемая с направленным ADC, является специфичной, например, не вследствие системного высвобождения свободного лекарственного средства. Опухоли не только были ингибированы, а также частично регрессировали под действием анти-HER2 конъюгатов (112 и 113). Эти данные указывают на то, что анти-steap1 поверхностный антиген является потенциально эффективной мишенью для антрациклин-производного ADC по изобретению.
In vivo исследование эффективности на ксенотрансплантированной опухоли LnCap-Ner (Фигура 32)
1 мг/кг
3 мг/кг
1 /мг/кг
3 мг/кг
6 мг/кг
1 мг/кг
3 мг/кг
6 мг/кг
ТОКСИЧНОСТЬ У ГРЫЗУНОВ
Конъюгаты антитела с лекарственным средством и ADC-минус контроль, “Носитель”, можно оценивать в модели острой токсичности у крыс (Brown et al. (2002) Cancer Chemother. Pharmacol. 50:333-340) и в соответствии с Примером 11. Токсичность ADC изучают путем воздействия на самок крыс Sprague-Dawley ADC и последующего наблюдения и анализа воздействия на различные органы. Исходя из общих наблюдений (масса тела), параметров клинической патологии (химический состав сыворотки и гематология) и гистологии, можно наблюдать токсичность ADC, охарактеризовать и измерить.
Многодневное исследование острой токсичности у пубертантных самок крыс можно проводить с использованием одной или нескольких доз потенциального ADC, контрольного ADC, свободного соединения производного антрациклина (PNU-159682) и контрольного носителя (день 0). Массу тела измеряют периодически. Клинический биохимический анализ, анализ сывороточных ферментов и гематологический анализ также проводят периодически; завершая полной некропсией с гистопатологической оценкой. Сигналы токсичности включали клиническое наблюдение потери массы тела, принимая во внимание, что потеря массы тела или изменение массы тела, относящаяся к животным, которым вводили только носитель, у животных после введения ADC, является приблизительным и общим показателем системной или локализованной токсичности. Гепатотоксичность может быть измерена по: (i) повышенным печеночным ферментам, таким как AST (аспартатаминотрансфераза), ALT (аланинаминотрасфераза), GGT (g-глутамилтрансфераза); (ii) повышенному числу митотических и апоптотических фигур и (iii) некрозу гепатоцитов. Гематолимфоидную токсичность наблюдают по истощению лейкоцитов, первичных гранулоцитов (нейтрофилов) и/или тромбоцитов и вовлечению лимфоидных органов, то есть атрофии или апоптотической активности. Токсичность также отмечается по повреждениям желудочно-кишечного тракта, например, повышенному числу митотических и апоптотических фигур и дегенеративному энтероколиту.
ВВЕДЕНИЕ ФАРМАЦЕВТИЧЕСКИХ СОСТАВОВ КОНЪЮГАТА АНТИТЕЛА С ЛЕКАРСТВЕННЫМ СРЕДСТВОМ
Терапевтические конъюгаты антитела с лекарственным средством (ADC) можно вводить любым путем, соответствующим состоянию, подвергаемому лечению. ADC обычно будет вводиться парентерально, то есть инфузия, подкожная, внутримышечная, внутривенная, внутрикожная, подоболочечная, болюсная, инъекция внутрь опухоли или эпидуральная (Shire et al. (2004) J. Pharm. Sciences 93(6):1390-1402). Фармацевтические составы терапевтических конъюгатов антитела с лекарственным средством (ADC) обычно получают для парентерального введения с фармацевтически приемлемым парентеральным носителем и в форме единичной инъекционной дозы. Конъюгат антитела с лекарственным средством (ADC), имеющий желаемую степень чистоты, необязательно смешивают с фармацевтически приемлемыми разбавителями, носителями, эксципиентами или стабилизаторами, в форме лиофилизированного состава или водного раствора (Remington's Pharmaceutical Sciences (1980) 16th edition, Osol, A. Ed.).
ADC может быть составлен в фармацевтические композиции с фармацевтически приемлемым носителем или разбавителем. Может быть использован любой подходящий носитель или разбавитель. Подходящие носители и разбавители включают физиологический раствор и декстрозные растворы Рингера.
Приемлемые парентеральные носители, разбавители, носители, эксципиенты и стабилизаторы являются нетоксичными для реципиентов в используемых дозах и концентрациях и включают: (i) буферы, такие как фосфатный, цитратный, двухосновный кальций-фосфатный, стеарат магния и другие органические кислоты; (ii) антиоксиданты, в том числе аскорбиновую кислоту и метионин; (iii) консерванты (такие как октадецилдиметилбензил аммония хлорид; гексаметония хлорид; бензалкония хлорид, бензетония хлорид; фенол, бутил или бензиловый спирт; (iv) алкилпарабены, такие как метил или пропилпарабен; катехол; резорцинол; циклогексанол; 3-пентанол; и м-крезол); (v) полипептиды с низкой молекулярной массой (примерно менее 10 остатков); белки, такие как сывороточный альбумин, желатин или иммуноглобулины; (vi) гидрофильные полимеры, такие как поливинилпирролидон; (vii) амино кислоты, такие как глицин, глутамин, аспарагин, гистидин, аргинин или лизин; (viii) моносахариды, дисахариды и другие углеводы, в том числе, глюкозу, лактозу, сахарозу, маннитол, трегалозу, гликолят крахмал натрия, сорбитол, маннозу, карбоксиметилцеллюлозу, или декстрины; (ix) хелатообразующие агенты, такие как EDTA; (x) соль-образующие противоионы, например, натрия; комплексы металлов (например, комплексы Zn-белок); (xi) неионные поверхностно-активные вещества, такие как TWEEN™, PLURONICS™ или полиэтиленгликоль (ПЭГ); (xii) вещества, способствующие скольжению, или гранулирующие вещества, такие как стеарат магния, карбоксиметилцеллюлоза, тальк, диоксид кремния и гидрогенизированное растительное масло; (xiii) дезинтеграт, такой как кросповидон, гликолят крахмал натрия или кукурузный крахмал; (xiv) загустители, такие как желатин и полиэтиленгликоль; (xv) кишечно-растворимые покрытия, такие как триэтилцитрат; и/или (xvi) модификаторы вкуса или структуры, противопенные вещества, пигменты и поглотители влаги. Например, лиофилизированные составы анти-ErbB2 антитела описаны не в WO 97/04801, специально включены в настоящую заявку путем ссылки. Приводимый в качестве примера состав ADC содержит примерно 100 мг/мл трегалозы (2-(гидроксиметил)-6-[3,4,5-тригидрокси-6-(гидроксиметил)тетрагидропиран-2-ил]окси-тетрагидропиран-3,4,5-триол; C12H22O11; CAS номер 99-20-7) и примерно 0,1% TWEEN™ 20 (полисорбат 20; додекановая кислота 2-[2-[3,4-бис(2-гидроксиэтокси)тетрагидрофуран-2-ил]-2-(2-гидроксиэтокси)этокси]этиловый эфир; C26H50O10; CAS номер 9005-64-5) приблизительно при pH 6.
Фармацевтические композиции терапевтического конъюгата антитела с лекарственным средством (ADC) могут содержать определенные количества не участвующей в реакции лекарственной группы (D), промежуточного соединения антитело-линкер (Ab-L), и/или промежуточного соединения лекарственное средство-линкер (D-L), в результате неполной очистки и отделения избытка реагентов, примесей и побочных продуктов, в процессе получения ADC; или гидролиза время/температура или разрушения при хранении нерасфасованного ADC или составленной композиции ADC. Например, состав ADC может содержать определяемое количество свободного лекарственного средства. Альтернативно, или дополнительно, композиция может содержать определяемое количество промежуточного соединения лекарственное средство-линкер. Альтернативно, или дополнительно, композиция может содержать определяемое количество антитела. Активные фармацевтические ингредиенты также могут быть заключены в микрокапсулы, полученные, например, с помощью методик коацервации или полимеризации на границе раздела фаз, например, гидроксиметилцеллюлозные или желатиновые микрокапсулы и поли-(метилметакрилат)микрокапсулы, соответственно, в коллоидных системах доставки лекарственных средств (например, липосомы, альбуминовые микросферы, микроэмульсии, наночастицы и нанокапсулы) или в микроэмульсиях. Такие методики описаны в Remington's Pharmaceutical Sciences 16th edition, Osol, A. Ed. (1980).
Могут быть получены препараты замедленного высвобождения. Подходящие примеры препаратов замедленного высвобождения включают полупроницаемые матрицы твердых гидрофобных полимеров, содержащие ADC, эти матрицы находятся в форме профилированных изделий, например, пленок или микрокапсул. Примеры матриц замедленного высвобождения включают сложные полиэфиры, гидрогели (например, поли(2-гидроксиэтилметакрилат), или поли(виниловый спирт)), полилактиды (US 3773919), сополимеры L-глутаминовой кислоты и гамма-этил-L-глутамата, неразлагаемый этиленвинилацетат, разлагаемые сополимеры молочной кислоты и гликолевой кислоты, такие как LUPRON DEPOT™ (инъекционные микросферы, состоящие из сополимера молочной кислоты и гликолевой кислоты и лейпролидацетата), и поли-D-(-)-3-гидроксимасляная кислота.
Составы могут быть удобно представлены в единичной лекарственной форме и могут быть получены любым из способов, хорошо известных в области фармации. Методики и составы в основном находятся в Remington's Pharmaceutical Sciences (Mack Publishing Co., Easton, PA). Такие способы включают стадию объединения активного ингредиента с носителем, который составляет один или несколько добавочных ингредиентов. В основном составы получают в стерильных условиях и путем однородного и равномерного объединения ADC с жидкими носителями или мелкоизмельченными твердыми носителями или и тем, и другим, а затем, при необходимости, придают форму продукту.
Водные суспензии содержат активные вещества (ADC) в смеси с эксципиентами, подходящими для производства водных суспензий. Такие эксципиенты включают суспендирующей агент, такой как карбоксиметилцеллюлоза натрия, кроскармелоза, повидон, метилцеллюлоза, гидроксипропилметилцеллюлоза, альгинат натрия, поливинилпирролидон, трагакантовая камедь и акациевая камедь, и диспергирующие и увлажняющие агенты, такие как природный фосфатид (например, лецитин), продукт конденсации алкиленоксида с жирной кислотой (например, полиоксиэтиленстеаратом), продукт конденсации этиленоксида с длинноцепочечным алифатическим спиртом (например, гептадекаэтиленоксицетанол), продукт конденсации этиленоксида с частичным сложным эфиром, полученным из жирной кислоты и гекситолангидрида (например, полиоксиэтиленсорбитанмоноолеат). Водная суспензия также может содержать один или несколько консервантов, таких как этил или н-пропил п-гидроксибензоат, один или несколько красителей, один или несколько веществ, корригирующих вкус и запах, и один или несколько подсластителей, таких как сахароза или сахарин.
Фармацевтические композиции ADC могут быть в форме стерильного инъекционного препарата, такого как стерильная инъекционная водная или масляная суспензия. Эта суспензия может быть получена в соответствии со способом, известным из уровня техники, используя подходящие диспергирующие или увлажняющие агенты и суспендирующие агенты, которые были упомянуты выше. Стерильный инъекционный препарат также может представлять собой стерильный инъекционный раствор или суспензию в нетоксичном парентерально приемлемом разбавителе или растворителе, таком как раствор в 1,3-бутандиоле или получены в виде лиофилизированного порошка. Среди приемлемых носителей и растворителей, которые могут быть использованы, находятся вода, раствор Рингера и изотоничный раствор хлорида натрия. Кроме того, стерильные жирные масла обычно могут быть использованы в качестве растворителя или суспендирующей среды. Для этой цели любое легкое жирное масло может быть использовано, в том числе, синтетические моно- или диглицериды. Кроме того, жирные кислоты, такие как олеиновая кислота, таким же образом могут быть использованы в получении инъекционных препаратов.
Количество активного ингредиента, которое может быть объединено с веществом носителем для получения единичной лекарственной формы, будет варьировать в зависимости от организма, подвергаемого лечению, и конкретного способа введения. Например, водный раствор, предназначенный для внутривенной инфузии, может содержать примерно от 3 до 500 мкг активного ингредиента на миллилитр раствора для того, чтобы можно было осуществить инфузию подходящего объема со скоростью примерно 30 мл/час. Подкожное (болюсное) введение может осуществляться примерно с 1,5 мл или меньшим суммарным объемом и концентрацией примерно 100 мг ADC на мл. Для ADC, для которого требуется быстрое и постоянное введение, может быть использован подкожный путь введения, например, с помощью предварительно заполненного шприца или технологии автоматического инжекторного устройства.
В качестве общего предложения, начальное фармацевтически эффективное количество ADC, вводимого на дозу, будет находиться в интервале примерно 0,01-100 мг/кг, а именно примерно от 0,1 до 20 мг/кг массы тела пациента в день, с характерным начальным диапазоном используемого соединения от 0,3 до 15 мг/кг/день. Например, людям пациентам первоначально можно вводить дозу примерно 1,5 мг ADC на кг массы тела пациента. Эта доза может быть увеличена до максимально переносимой дозы (MTD). Схема введения может быть примерно каждые 3 недели, но в соответствии с диагностированным состоянием или ответом, частота введения может быть более или менее частой. Дозу дополнительно можно устанавливать во время курса лечения на уровне MTD или ниже, которая может быть безопасно введена во многочисленных циклах, например, около 4 или более.
Составы, подходящие для парентерального введения, включают водные и неводные стерильные инъекционные растворы, которые могут содержать антиоксиданты, буферы, бактериостатики и растворы, которые придают составу изотоничность с кровью конкретного реципиента; и водные и неводные стерильные суспензии, которые могут включать суспендирующие агенты и загустители.
Хотя пероральное введение белковых терапевтических средств в основном не одобряется вследствие плохой биодоступности вследствие ограниченной абсорбции, гидролиза или денатурации в кишечнике, составы ADC, подходящие для перорального введения, могут быть получены в виде дискретных единиц, таких как капсулы, крахмальные капсулы или таблетки, содержащие заранее определенное количество ADC.
Составы могут быть упакованы в контейнеры единичной дозы или многократной дозы, например, герметично упакованные ампулы и флаконы, и могут храниться в сублимированном (лиофилизированном) состоянии, требующем только добавления стерильного жидкого носителя, например, воды, для введения непосредственно перед применением. Приготовленные для немедленного введения инъекционные растворы и суспензии получают из стерильных порошков, гранул и таблеток ранее описанного типа. Приводимые в качестве примера единичные дозированные составы содержат суточную дозу или единичную суточную субдозу, или их соответствующую фракцию, активного ингредиента.
СПОСОБЫ ЛЕЧЕНИЯ С ИСПОЛЬЗОВАНИЕМ КОНЪЮГАТА АНТИТЕЛО-ЛЕКАРСТВЕННОЕ СРЕДСТВО
Конъюгаты антитела с лекарственным средством по изобретению используют в качестве противоопухолевых средств. Млекопитающего, например, человека или животного, следовательно можно лечить способом, включающим введение фармацевтически эффективного количества конъюгата формулы 1, как определено ранее в настоящей заявке. Состояние человека или животного можно улучшить или облегчить этим путем.
ADC Формулы I может быть использован для лечения различных заболеваний или нарушений у пациента, таких как злокачественное заболевание и аутоиммунные состояния, в том числе те, которые характеризуются сверхэкспрессией связанного с опухолью антигена. Приводимые в качестве примера состояния или нарушения включают доброкачественные или злокачественные опухоли; лейкоз и лимфоидные злокачественные заболевания; другие нарушения, такие как нейрональные, глиальные, астроцитарные, гипоталамические, железистые, макрофагальные, эпителиальные, стромальные, бластоцельные, воспалительные, ангиогенные и иммунологические нарушения. Типы злокачественных опухолей, чувствительных к лечению ADC, включают те, которые характеризуются сверхэкспрессией некоторых связанных с опухолью антигенов или рецепторов клеточной поверхности, например HER2.
Один способ предназначен для лечения злокачественного заболевания у млекопитающего, где злокачественное заболевание характеризуется сверхэкспрессией рецептора ErbB. Необязательно млекопитающее не реагирует или плохо реагирует на лечение “голым” антителом против ErbB. Способ предусматривает введение млекопитающему терапевтически эффективного количества соединения конъюгата антитела с лекарственным средством. Рост опухолевых клеток, которые сверхэкспрессируют рецептор фактора роста, такие как рецептор HER2 или рецептор EGF, может быть ингибирован путем введения пациенту ADC Формулы I, который специфически связывается с указанным рецептором фактора роста и химиотерапевтическим средством, где указанный конъюгат антитела с лекарственным средством и указанное химиотерапевтическое средство каждое вводят в количествах, эффективных для ингибирования роста опухолевых клеток у пациента.
Человека пациента, подверженного нарушению или у которого диагностировано нарушение, характеризующееся сверхэкспрессией рецептора ErbB2, можно лечить путем введения сочетания ADC Формулы I и химиотерапевтического средства. Такая избыточная активация может быть обусловлена сверхэкспрессией или повышенной продукцией рецептора ErbB или лиганда ErbB. В одном варианте осуществления, диагностическое или прогностическое исследование будет проведено для определения, характеризуется ли злокачественное заболевание пациента избыточной активацией рецептора ErbB. Например, может быть определена амплификация гена ErbB и/или сверхэкспрессия рецептора ErbB в злокачественной опухоли. Различные исследования для определения такой амплификации/сверхэкспрессии доступны из уровня техники и включают IHC, FISH и исследования «слущивающегося» антигена.
Примеры злокачественного заболевания, подвергаемого лечению, в настоящей заявке включают, но этим не ограничиваются, карциному, лимфому, бластому, саркому и лейкоз или лимфоидные злокачественные заболевания. Более конкретные примеры таких злокачественных заболеваний включают сквамозноклеточную карциному (например, эпителиальный сквамозноклеточный рак), рак легких, в том числе мелкоклеточный рак легких, немелкоклеточный рак легких, аденокарциному легких и сквамозную карциному легких, рак брюшины, печеночно клеточный рак, рак желудка или пищеварительного тракта, в том числе гастроинтестинальный рак, гастроинтестинальную стромальную опухоль (GIST), рак поджелудочной железы, глиобластому, рак шейки матки, рак яичников, рак печени, рак мочевого пузыря, гепатому, рак груди, рак толстой кишки, рак прямой кишки, рак толстой и прямой кишки, карциному эндометрия или матки, карциному слюнных желез, рак почек, рак предстательной железы, рак вульвы, рак щитовидной железы, карциному печени, карциному ануса, карциному пениса, а также рак тканей головы и шеи.
Для профилактики или лечения заболевания, подходящая доза ADC будет зависеть от типа заболевания, подвергаемого лечению, как определено выше, тяжести и течения заболевания, вводят ли молекулу с профилактической или терапевтической целью, предыдущего лечения, истории болезни пациента и реакции на антитело, и мнения лечащего врача. Состав ADC подходящим образом вводят пациенту, в одно время или на протяжении серии обработок. В зависимости от типа и тяжести заболевания, примерно от 1 мкг/кг до 15 мг/кг (например, 0,1-20 мг/кг) ADC является начальной возможной дозой для введения пациенту, например, посредством одного или нескольких отдельных введений, или путем непрерывной инфузии. Характерный режим дозирования может находиться в интервале примерно от 1 мкг/кг до 100 мг/кг или более, в зависимости от факторов, упомянутых выше. Приводимая в качестве примера доза ADC, вводимая пациенту, находится в интервале примерно от 0,1 примерно до 10 мг/кг массы тела пациента. Для повторных введений на протяжении нескольких дней или дольше, в зависимости от состояния, лечение задерживают до появления желаемой суппрессии симптомов заболевания. Приводимый в качестве примера режим дозирования включает введение начальной насыщающей дозы примерно 4 мг/кг, затем еженедельной поддерживающей дозы примерно 2 мг/кг ADC. Могут быть использованы другие режимы дозирования.
КОМБИНИРОВАННАЯ ТЕРАПИЯ
Конъюгат антитела с лекарственным средством (ADC) можно объединить в фармацевтический комбинированный состав или режим дозирования в виде комбинированной терапии со вторым соединением, обладающим противоопухолевыми свойствами. Второе соединение фармацевтического комбинированного составы или режима дозирования предпочтительно обладает комплементарными активностями в отношении ADC этого сочетания, так, что они не влияют неблагоприятным образом друг на друга.
Второе соединение может представлять собой химиотерапевтическое средство, цитотоксическое средство, цитокин, средство, ингибирующее рост, противогормональное средство, ингибитор ароматазы, ингибитор протеинкиназы, ингибитор липидкиназы, антиандрогены, антисмысловые олигонуклеотиды, рибозим, вакцину для генной терапии, антиангиогенное средство и/или кардиопротектор. Такие молекулы соответственно присутствуют в сочетании в количествах, которые являются эффективными для намеченной цели. Фармацевтическая композиция, содержащая ADC, также может иметь терапевтически эффективное количество химиотерапевтического средства, например, ингибитора образования тубулина, ингибитора топоизомеразы или ДНК связующего.
Альтернативно, или дополнительно, второе соединение может представлять собой антитело, которое связывается или блокирует лигандную активацию связанного с опухолью антигена или рецептора. Второе антитело может быть конъюгировано с цитотоксическим или химиотерапевтическим средством, например, макроциклическим депсипептидом, ауристатином, калихеамицином или 1,8 бис-нафталимидной группой. Например, это может быть желательным для дальнейшего предоставления антител, которые связываются с EGFR, ErbB2, ErbB3, ErbB4, или сосудистым эндотелиальным фактором (VEGF) в одном составе или режиме дозирования.
Комбинированная терапия может применяться в виде одновременного или последовательного режима. При последовательном введении, это сочетание можно вводить за два или более применений. Комбинированное введение включает совместное введение, с использованием отдельных составов, или одного фармацевтического состава, и последовательное введение в любом порядке, где существует период времени, когда оба (или все) активные агенты одновременно осуществляют свои биологические активности.
В одном варианте осуществления, лечение с использованием ADC по настоящему изобретению включает комбинированное введение противоопухолевого средства, идентифицированного в настоящей заявке, и одного или нескольких химиотерапевтических средств или ингибиторов роста. Получение и схемы введения для таких химиотерапевтических средств могут быть использованы в соответствии с инструкциями производителя или как определено эмпирически опытным практикующим специалистом. Получение и схемы введения для такой химиотерапии также описаны в Chemotherapy Service Ed., M.C. Perry, Williams & Wilkins, Baltimore, Md. (1992).
ADC могут быть объединены с противогормональным соединением; например, антиэстрогеном, таким как тамоксифен; антипрогестероном, таким как онапристон (EP 616812); или антиандрогеном, таким как флутамид, в дозах, известных для таких молекул. Где злокачественное заболевание, подвергаемое лечению, представляет собой гормонально независимый рак, пациент предварительно мог подвергаться антигормональной терапии и после того, как злокачественная опухоль становится гормононезависимой, анти-ErbB2 антитело (и необязательно другие средства, описанные в настоящем документе) могут быть введены пациенту. Также может быть полезным совместно вводить кардиопротектор (для профилактики или уменьшения нарушения функции миокарда, связанного с этим лечением) или один или несколько цитокинов пациенту. Дополнительно к указанным выше терапевтическим режимам, пациент может подвергаться хирургическому удалению злокачественных клеток и/или лучевой терапии.
Подходящими дозами для любого из указанных выше совместно вводимых средств являются дозы, используемые в настоящее время, и могут быть снижены благодаря комбинированному действию (синергии) только что идентифицированного средства и других химиотерапевтических средств или способов лечения.
Комбинированная терапия может обеспечивать «синергию» и оказывать «синергетический эффект», то есть эффект, достигаемый в тех случаях, когда активные ингредиенты, используемые вместе, является больше, чем сумма эффектов, которые получены в результате использования соединений по-отдельности. Синергетический эффект может достигаться, когда активные ингредиенты: (1) совместно получены и введены или доставлены одновременно в объединенной композиции единичной дозы; (2) доставлены путем чередования или параллельно в виде отдельных составов; или (3) каким-то другим образом. При доставке в попеременном лечении, синергетический эффект может достигаться, когда соединения вводят или доставляют последовательно, например, посредством различных инъекций в отдельных шприцах. В основном, во время попеременного лечения, эффективную дозу каждого активного ингредиента вводят последовательно, то есть периодически, тогда как в комбинированной терапии эффективные дозы двух или более активных ингредиентов вводят вместе.
МЕТАБОЛИТЫ КОНЪЮГАТОВ АНТИТЕЛО-ЛЕКАРСТВЕННОЕ СРЕДСТВО
Также в объем данного изобретения входят in vivo продукты метаболизма соединений ADC, описанные в настоящем документе, в тех случаях, когда такие продукты являются новыми и неочевидны по сравнению с предшествующим уровнем техники. Такие продукты могут быть результатом, например, окисления, восстановления, гидролиза, амидирования, этерификации, ферментативного расщепления и тому подобного, введенного соединения. Соответственно, настоящее изобретение включает новые и неочевидные соединения, полученные способом, включающим контактирование соединения данного изобретения с организмом млекопитающего в течение периода времени, достаточного для получения его метаболического продукта.
Продукты метаболизма могут быть идентифицированы путем получения радиоактивно меченного (например, 14C или 3H) ADC, введения его парентерально в определяемой дозе (например, примерно большей чем 0,5 мг/кг) животному, такому как крыса, мышь, морская свинка, обезьяна, или человеку, давая достаточно времени для осуществления метаболизма (обычно примерно от 30 секунд до 30 часов), и выделение продуктов его превращения из мочи, крови или других биологических образцов. Эти продукты легко выделять, поскольку они мечены (другие выделяют путем применения антител, способных связываться с эпитопами, уцелевшими в этом метаболите). Структуры метаболита определяют обычным способом, например, с помощью анализов MS, LC/MS или ЯМР. В основном, анализ метаболитов проводят тем же путем, что и обычные исследования метаболизма лекарственных средств, хорошо известные специалистам в данной области. Продукты превращения, при условии, что они иным путем не находятся in vivo, используют в диагностических анализах терапевтического дозирования соединений ADC.
Метаболиты включают продукты in vivo расщепления ADC, где происходит расщепление любой связи, которая связывает лекарственную группу с антителом. Метаболическое расщепление, следовательно, может приводить в результате к получению “голого” антитела или фрагмента антитела. Метаболит антитела может быть связан с частью линкера или со всем линкером. Метаболическое расщепление также может приводить в результате к получению лекарственной группы или ее части. Метаболит лекарственной группы может быть связан с частью или со всем линкером.
ИЗДЕЛИЯ
В другом варианте осуществления представлено изделие, или «набор», содержащий ADC и вещества, используемые для лечения нарушений, описанных выше. Изделие содержит контейнер и этикетку или лист-вкладыш на контейнере или соединенный с контейнером. Подходящие контейнеры включают, например, бутыли, флаконы, шприцы или блистерную упаковку. Контейнеры могут быть получены из целого ряда материалов, таких как стекло или пластик. Контейнер содержит композицию конъюгата антитела с лекарственным средством (ADC), которая эффективна для лечения патологического состояния и может иметь стерильное входное отверстие (например, контейнер может представлять собой мешок для внутривенного раствора или флакон, имеющий пробку, прокалываемую иглой для подкожных инъекций). По меньшей мере одним активным средством в композиции является ADC. Этикетка или лист-вкладыш указывает, что композиция используется для лечения выбранного патологического состояния, такого как злокачественное заболевание.
В одном варианте осуществления, изделие дополнительно может содержать второй (или третий) контейнер, содержащий фармацевтически приемлемый буфер, такой как бактериостатическая вода для инъекций (BWFI), фосфат-буферный солевой раствор, раствор Рингера и раствор декстрозы, и лист-вкладыш, указывающий, что первое и второе соединения могут быть использованы для лечения злокачественного заболевания. Изделие дополнительно может содержать другие вещества, желательные с коммерческой и потребительской точки зрения, в том числе, другие буферы, разбавители, фильтры, иглы и шприцы.
ПРИМЕРЫ
Соединения настоящего изобретения, полученные в соответствии со следующими примерами, были охарактеризованы с помощью аналитических данных ВЭЖХ/МС; ВЭЖХ/МС данные получали в соответствии с одним из способов 1, 2.
Аналитический способ 1 ВЭЖХ/МС
Систему Waters 2795 Alliance HT HPLC, снабженную детектором 2996 Waters PDA и Micromass mod. ZQ масс-спектрометром с одной квадрупольной линзой, оборудованным источником электрораспылительной ионизации (ESI). Контроль за прибором, получение и накопление данных и обработка данных обеспечивались программами Empower и MassLynx 4.0. ВЭЖХ проводили при 30°C со скоростью потока 1,0 мл/мин, используя колонку C18, 3 микрон Phenomenex (4,6×50 мм). Подвижная фаза A представляла собой буфер ацетат аммония 5 мМ pH 5,2 с ацетонитрилом (95:5), а подвижная фаза B представляла собой H2O/ацетонитрил (5:95); градиент был от 10 до 90% B, через 8 минут затем быстро линейно увеличивался до 100% B через 1,0 минуту. Вводимый объем составил 10 мкл. Масс-спектрометр работал в режиме определения положительных и отрицательных ионов, напряжение капилляра устанавливали на 3,5 кВ (ES+) и 28 В (ES-); температура источника составляла 120°C; конус был 14 В (ES+) и 2,8 KB (ES-); полное сканирование, диапазон масс от 100 до 1000 m/z.
Аналитический способ 2 ВЭЖХ/MC
Система Waters 2795 HPLC была оборудована детектором 996 Waters PDA и Micromass mod. ZQ масс-спектрометром с одной квадрупольной линзой, оборудованным источником электрораспылительной ионизации (ESI). Контроль за прибором, получение и накопление данных и обработка данных обеспечивались программами Empower и MassLynx 4.0. ВЭЖХ проводили при 30°C со скоростью потока 1,0 мл/мин, используя колонку RP18 Waters X Terra (4,6 мкМ×50 мм). Подвижная фаза A представляла собой буфер гидроксид аммония 0,05% pH 10 с ацетонитрилом (95:5), а подвижная фаза B представляла собой H2O/ацетонитрил (5:95); градиент был от 10 до 90% B через 8 минут, затем удерживали 100% B в течение 2 минут. Вводимый объем составил 10 мкл. Масс-спектрометр работал в режиме определения положительных и отрицательных ионов, напряжение капилляра устанавливали на 2,5 кB; температура источника составляла 120°C; конус был 10 В; полное сканирование, диапазон масс от 100 до 1000 m/z.
Пример 1 N-метил-2-[(1-{2-оксо-2-[(2S,4S)-2,5,12-тригидрокси-7-метокси-4-{[(1S,3R,4aS,9S,9aR,10aS)-9-метокси-1-метилоктагидро-1H-пирано[4',3':4,5][1,3]оксазоло[2,3-c][1,4]оксазин-3-ил]окси}-6,11-диоксо-1,2,3,4,6,11-гексагидротетрацен-2-ил]этокси}циклогексил)окси]ацетамид (Соединение 2)
Стадия 1 Синтез промежуточного этил [(1-{2-оксо-2-[(2S,4S)-2,5,12-тригидрокси-7-метокси-4-{[(1S,3R,4aS,9S,9aR,10aS)-9-метокси-1-метилоктагидро-1H-пирано[4',3':4,5][1,3]оксазоло[2,3-c][1,4]оксазин-3-ил]окси}-6,11-диоксо-1,2,3,4,6,11-гексагидротетрацен-2-ил]этокси}циклогексил)окси]ацетата 45
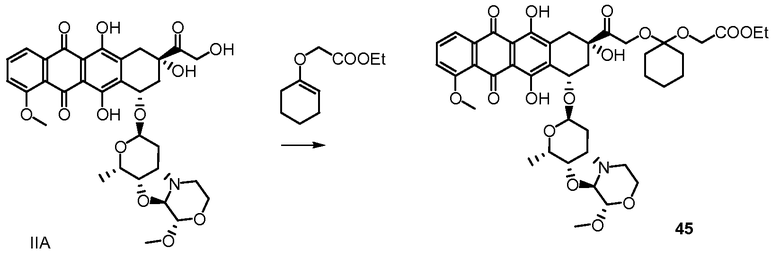
К раствору (8S,10S)-6,8,11-тригидрокси-8-(гидроксиацетил)-1-метокси-10-{[(1S,3R,4aS,9S,9aR,10aS)-9-метокси-1-метилоктагидро-1H-пирано[4',3':4,5][1,3]оксазоло[2,3-c][1,4]оксазин-3-ил]окси}-7,8,9,10-тетрагидротетрацен-5,12-диона (50 мг, 0,078 ммоль) [PNU-159682, соединение IIA, полученного, как описано в WO 9802446] в 2 мл сухого диметилформамида, который держали в атмосфере аргона, добавляли этиловый эфир (циклогекс-1-енилокси)-уксусной кислоты (0,5 мл, [полученного, как описано в J. Org. Chem. (1978) 43:1244-1245] и моногидрат п-толуолсульфоновой кислоты (5 мг, 0,026 ммоль). Реакционную смесь перемешивали в течение ночи при комнатной температуре, добавляли насыщенный раствор бикарбоната натрия (20 мл) и продукт экстрагировали дихлорметаном (2×20 мл). Объединенные органические фазы сушили над безводным сульфатом натрия, фильтровали, растворитель удаляли под вакуумом и остаток частично очищали с помощью флэш-хроматографии (DCM/MeOH 97,5:2,5) с получением 30 мг (37%) сложноэфирного производного, которое использовали как есть на стадии 2. MS (ESI): 826 [M+H]+. Время удерживания = 7,48 мин. способ 2
Стадия 2 Синтез промежуточного соединения [(1-{2-оксо-2-[(2S,4S)-2,5,12-тригидрокси-7-метокси-4-{[(1S,3R,4aS,9S,9aR,10aS)-9-метокси-1-метилоктагидро-1H-пирано[4',3':4,5][1,3]оксазоло[2,3-c][1,4]оксазин-3-ил]окси}-6,11-диоксо-1,2,3,4,6,11-гексагидротетрацен-2-ил]этокси}циклогексил)окси]уксусная кислота 46
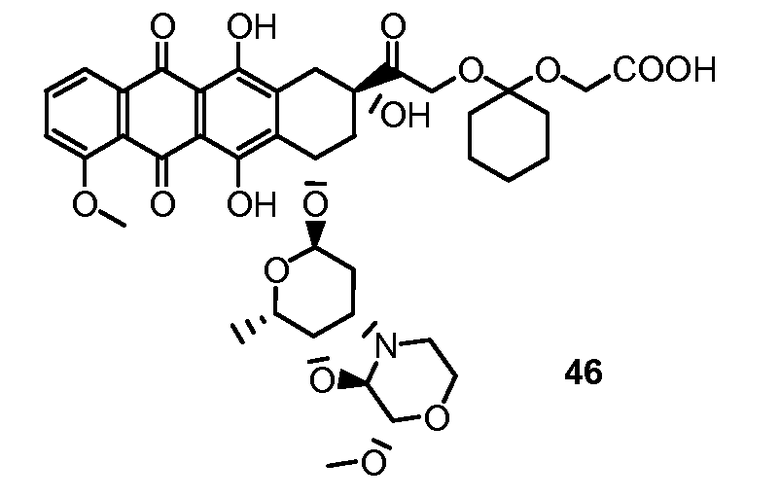
К 30 мг 45, добавляли 5 мл в 01 н. NaOH. Суспензию охлаждали при 5°C и перемешивали в атмосфере аргона в течение 3 часов. Водный раствор доводили до pH ≅8 с использованием 10% водного раствора уксусной кислоты и экстрагировали дихлорметаном (2×10 мл). Объединенные органические фазы сушили над безводным сульфатом натрия, фильтровали, растворитель удаляли под вакуумом и остаток очищали с помощью флэш-хроматографии (DCM/MeOH 90:10) с получением 5 мг (y = 8% 2 стадии) кислого промежуточного соединения 46 в виде твердого вещества красного цвета. MS (ESI): 798 [M+H]+. Время удерживания = 3,94 мин. Способ 2
Стадия 3 Синтез промежуточного соединения 1-({[(1-{2-оксо-2-[(2S,4S)-2,5,12-тригидрокси-7-метокси-4-{[(1S,3R,4aS,9S,9aR,10aS)-9-метокси-1-метилоктагидро-1H-пирано[4',3':4,5][1,3]оксазоло[2,3-c][1,4]оксазин-3-ил]окси}-6,11-диоксо-1,2,3,4,6,11-гексагидротетрацен-2-ил]этокси}циклогексил)окси]ацетил}окси)пирролидин-2,5-диона 47
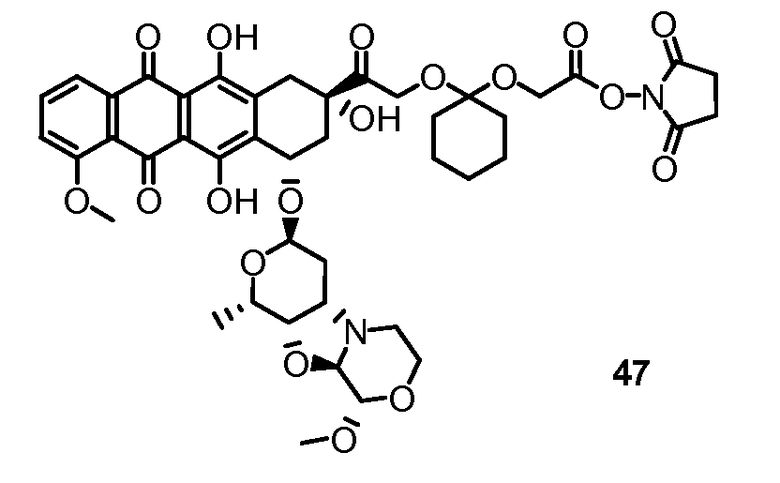
К раствору кислого промежуточного соединения 46 (4 мг, 0,005 ммоль) в сухом дихлорметане (2 мл), удерживаемого при +5°C, добавляли N-гидроксисукцинимид (2 мг, 0,017 ммоль) и N,N'-дициклогексилкарбодиимид (2 мг, 0,01 ммоль). Раствор перемешивали при комнатной температуре в течение 6 ч, растворитель упаривали под вакуумом и остаток обрабатывали этиловым эфиром (5 мл). Суспензию перемешивали в течение 30 минут, твердый продукт удаляли путем фильтрации и органический раствор концентрировали в вакууме. Очистка сырого продукта с помощью флэш-хроматографии (DCM/Ацетон 80:20) давала на выходе 1,5 мг (y=33%) 47 в виде твердого вещества красного цвета. MS (ESI): 895 [M+H]+. Время удерживания = 3,22 мин. Способ 1.
Стадия 4 Синтез указанного в заголовке соединения 2
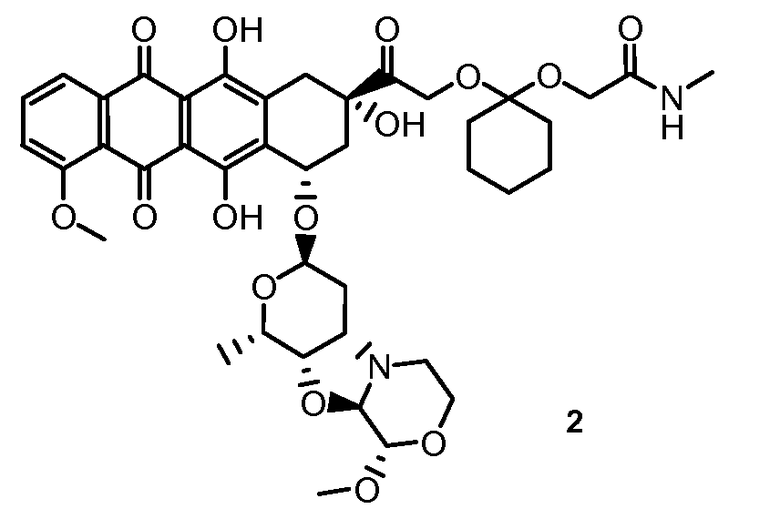
К раствору 47, полученному на стадии 3 (1 мг, 0,001 ммоль) в сухом тетрагидрофуране (2 мл), добавляли 1 M метиламина в ТГФ (3 мкл, 0,003 ммоль). Раствор перемешивали при комнатной температуре 30 минут, растворитель упаривали под вакуумом и остаток очищали с помощью флэш-хроматографии (дихлорметан/метанол 90:10) с выходом 0,9 мг (y=99%) 2 в виде твердого вещества красного цвета. MS (ESI): 811 [M+H]+. Время удерживания = 6,10 мин. Способ 2. Время удерживания = 5,99 мин. Способ 1.
С помощью аналогичного способа и используя подходящие исходные вещества, получали следующие соединения:
2-[(1-{2-оксо-2-[(2S,4S)-2,5,12-тригидрокси-7-метокси-4-{[(1S,3R,4aS,9S,9aR,10aS)-9-метокси-1-метилоктагидро-1H-пирано[4',3':4,5][1,3]оксазоло[2,3-c][1,4]оксазин-3-ил]окси}-6,11-диоксо-1,2,3,4,6,11-гексагидротетрацен-2-ил]этокси}циклогексил)окси]ацетамид (Соединение 1) MS (ESI): 887 [M+H]+. Время удерживания = 5,86 мин. Способ 2
N-бензил-2-[(1-{2-оксо-2-[(2S,4S)-2,5,12-тригидрокси-7-метокси-4-{[(1S,3R,4aS,9S,9aR,10aS)-9-метокси-1-метилоктагидро-1H-пирано[4',3':4,5][1,3]оксазоло[2,3-c][1,4]оксазин-3-ил]окси}-6,11-диоксо-1,2,3,4,6,11-гексагидротетрацен-2-ил]этокси}циклогексил)окси]ацетамид (Соединение 3) MS (ESI): 797 [M+H]+. Время удерживания = 7,16 мин. Способ 2
N 2-(трет-бутоксикарбонил)-N 6-{[(1-{2-оксо-2-[(2S,4S)-2,5,12-тригидрокси-7-метокси-4-{[(1S,3R,4aS,9S,9aR,10aS)-9-метокси-1-метилоктагидро-1H-пирано[4',3':4,5][1,3]оксазоло[2,3-c][1,4]оксазин-3-ил]окси}-6,11-диоксо-1,2,3,4,6,11-гексагидротетрацен-2-ил]этокси}циклогексил)окси]ацетил}-L-лизин (Соединение 4) MS (ESI): 1026 [M+H]+. Время удерживания = 5,26 мин. Способ 1. Время удерживания = 4,64 мин. Способ 2
Пример 2 N-метил-2-[(6-{2-оксо-2-[(2S,4S)-2,5,12-тригидрокси-7-метокси-4-{[(1S,3R,4aS,9S,9aR,10aS)-9-метокси-L-метилоктагидро-1H-пирано[4',3':4,5][1,3]оксазоло[2,3-c][1,4]оксазин-3-ил]окси}-6,11-диоксо-1,2,3,4,6,11-гексагидротетрацен-2-ил]этокси}тетрагидро-2H-пиран-2-ил)метокси]ацетамид (Соединение 7)
Стадия 1 Синтез промежуточного соединения: этил (3,4-дигидро-2H-пиран-2-илметокси)ацетата
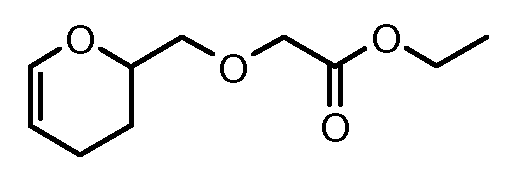
В сухой круглодонной колбе в атмосфере аргона, 60% гидрид натрия (240 мг, 6,0 ммоль) промывали три раза безводным н-пентаном. Раствор 2-гидроксиметил-3,4-дигидро-2H-пирана (570,8 мг, 5 ммоль) в тетрагидрофуране (10 мл) охлаждали при температуре 0°C, а затем добавляли в NaH. Реакционную смесь перемешивали при температуре 0°C, пока не закончится выделение водорода. Раствор этилбромацетата (1253 мг, 7,5 ммоль) в тетрагидрофуране (6 мл) добавляли в реакционную смесь и перемешивание продолжали при комнатной температуре до исчезновения исходного спирта (TLC анализ). После охлаждения, добавляли H2O, и растворитель упаривали под пониженным давлением. Остаток очищали с помощью колоночной флэш-хроматографии (AcOEt:гексан=1:12) на силикагеле (230-400 меш), получая 626 мг (выход 57%) этил(3,4-дигидро-2H-пиран-2-илметокси)ацетата в виде бесцветного масла; 1H ЯМР (401 МГц, ДМСО-d 6) δ м.д. 1,18-1,23 (м, 3 H) 3,55-3,59 (м, 2 H) 3,93 (м, J=10,08, 5,11, 5,11, 2,38 Гц, 1 H) 4,13 (кв, J=7,07 Гц, 2 H) 4,14 (с, 2 H) 4,67 (дддд, J=6,14, 4,83, 2,56, 1,34 Гц, 1 H) 6,36 (дт, J=6,13, 1,75 Гц, 1 H).
Стадия 2 Синтез промежуточного соединения: этил [(6-{2-оксо-2-[(2S,4S)-2,5,12-тригидрокси-7-метокси-4-{[(1S,3R,4aS,9S,9aR,10aS)-9-метокси-1-метилоктагидро-1H-пирано[4',3':4,5][1,3]оксазоло[2,3-c][1,4]оксазин-3-ил]окси}-6,11-диоксо-1,2,3,4,6,11-гексагидротетрацен-2-ил]этокси}тетрагидро-2H-пиран-2-ил)метокси]ацетата 48
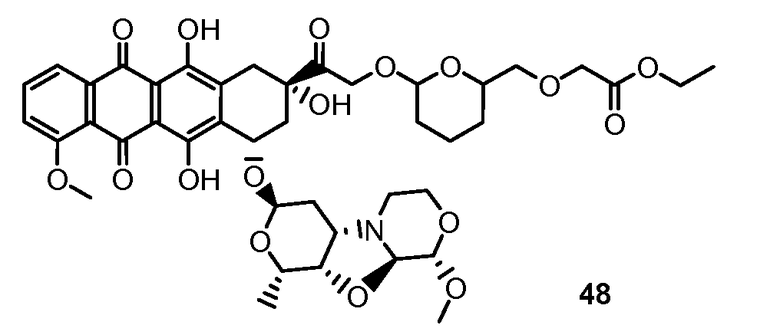
К раствору (8S,10S)-6,8,11-тригидрокси-8-(гидроксиацетил)-1-метокси-10-{[(1S,3R,4aS,9S,9aR,10aS)-9-метокси-1-метилоктагидро-1H-пирано[4',3':4,5][1,3]оксазоло[2,3-c][1,4]оксазин-3-ил]окси}-7,8,9,10-тетрагидротетрацен-5,12-диона (50 мг, 0,078 ммоль) [PNU-159682, формула IIA, полученного как описано в WO 9802446] в 4 мл сухого дихлорметана в атмосфере аргона, добавляли этил (3,4-дигидро-2H-пиран-2-илметокси)ацетат со стадии 1 (118,5 мг, 0,592 ммоль) и безводную п-толуолсульфоновую кислоту (22,3 мг, 0,12 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 4 часов, пока исходное вещество не будет определяемо (TLC анализ, MeOH:CH2Cl2 = 0,3:9,7). 10% водный раствор бикарбоната натрия затем добавляли в реакционную смесь и водную фазу экстрагировали дихлорметаном (4×20 мл). Объединенные органические фазы сушили над безводным сульфатом натрия, фильтровали и упаривали под вакуумом. Остаток очищали с помощью колоночной флэш-хроматографии (MeOH:CH2Cl2=0,3:9,7) на силикагеле (230-400 меш), получая 41 мг (красный воск, выход 62%) 48, в виде смеси четырех диастереоизомеров. MS (ESI): 842 [M+H]+. Время удерживания = 7,47, 7,83 мин (способ 2).
Стадия 3 Синтез промежуточного соединения: [(6-{2-оксо-2-[(2S,4S)-2,5,12-тригидрокси-7-метокси-4-{[(1S,3R,4aS,9S,9aR,10aS)-9-метокси-1-метилоктагидро-1H-пирано[4',3':4,5][1,3]оксазоло[2,3-c][1,4]оксазин-3-ил]окси}-6,11-диоксо-1,2,3,4,6,11-гексагидротетрацен-2-ил]этокси}тетрагидро-2H-пиран-2-ил)метокси]уксусной кислоты 49
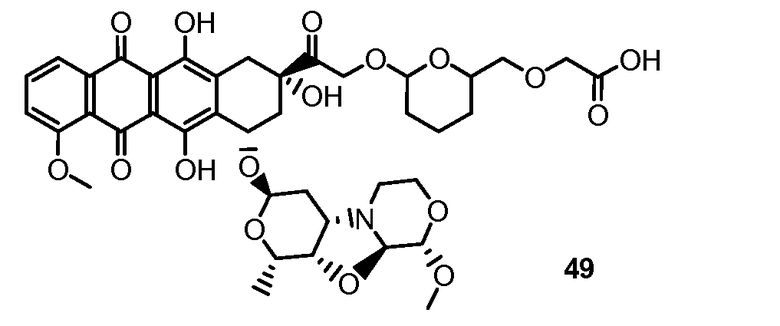
Этиловый эфир промежуточного соединения 48, полученного на стадии 2 (40 мг, 0,0475 ммоль), охлажденного при температуре 0°C, обрабатывали водным 0,1 н. гидроксидом натрия (1,5 мл) в атмосфере аргона. Реакционную смесь перемешивали при температуре 0°C в течение 2 часов. Течение реакции сопровождалось ВЭЖХ-МС с обращенной фазой. После этого реакционную смесь доводили до pH ≅8 с использованием 10% водного раствора уксусной кислоты и экстрагировали н-бутанолом, насыщенным водой (8×10 мл). Объединенные органические фазы сушили над безводным сульфатом натрия, фильтровали, и растворитель удаляли под вакуумом. Остаток очищали с помощью колоночной флэш-хроматографии (MeOH:CH2Cl2=1:9) на силикагеле (230-400 меш), получая 4,5 мг (твердого вещества красного цвета, выход 12%) 49 в виде диастереоизомерной смеси. MS (ESI): 814 [M+H]+. Время удерживания =2,99, 3,50, 3,66 (способ 2). Время удерживания = 4,24 (способ 1).
Стадия 4 Синтез промежуточного соединения: 1-({[(6-{2-оксо-2-[(2S,4S)-2,5,12-тригидрокси-7-метокси-4-{[(1S,3R,4aS,9S,9aR,10aS)-9-метокси-1-метилоктагидро-1H-пирано[4',3':4,5][1,3]оксазоло[2,3-c][1,4]оксазин-3-ил]окси}-6,11-диоксо-1,2,3,4,6,11-гексагидротетрацен-2-ил]этокси}тетрагидро-2H-пиран-2-ил)метокси]ацетил}окси)-пирролидин-2,5-диона 50
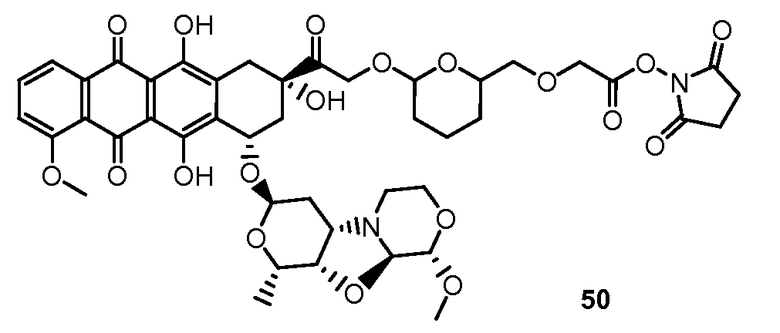
К раствору кислого промежуточного соединения 49, полученного на стадии 3 этого способа (2 мг, 0,0024 ммоль) в сухом дихлорметане (1 мл), охлажденного при температуре 0°C, добавляли N-гидроксисукцинимид (1 мг, 0,00792 ммоль) и N,N'-дициклогексилкарбодиимид (1 мг, 0,004556 ммоль). Реакционную смесь перемешивали при температуре 0°C в течение 2,5 ч, до исчезновения исходного вещества (ВЭЖХ-МС анализ). Растворитель упаривали под вакуумом, и остаток обрабатывали этиловым эфиром (2×4 мл). Суспензию перемешивали в течение 10 минут, твердый продукт удаляли путем фильтрации и органический раствор концентрировали в вакууме, получая 2 мг 50 (твердое вещество красного цвета) в виде смеси диастереоизомеров. MS (ESI): 911 [M+H]+. Время удерживания = 6,12, 6,30, 6,42 мин (способ 1).
Стадия 5 Указанное в заголовке соединение 7
К раствору 50, полученному со стадии 4 этого способа (1 мг, 0,0011 ммоль) в сухом дихлорметане (100 мкл), добавляли 2,0 M метиламин в ТГФ (65 мкл, 0,0033 ммоль). Раствор перемешивали при комнатной температуре 4 часа до тех пор, пока не обнаруживалось никакого исходного вещества (ВЭЖХ-МС анализ), и растворитель упаривали под вакуумом. Остаток очищали с помощью колоночной флэш-хроматографии (MeOH: CH2Cl2=0,3:9,7) на силикагеле (230-400 меш), получая 0,46 мг (твердое вещество красного цвета, выход 51%) 7 в виде смеси диастереоизомеров. MS (ESI): 827 [M+H]+. Время удерживания = 5,34, 5,54, 5,66 мин (способ 1).
Аналогичными способами и используя подходящие исходные вещества, получали следующие соединения:
2-[(6-{2-оксо-2-[(2S,4S)-2,5,12-тригидрокси-7-метокси-4-{[(1S,3R,4aS,9S,9aR,10aS)-9-метокси-1-метилоктагидро-1H-пирано[4',3':4,5][1,3]оксазоло[2,3-c][1,4]оксазин-3-ил]окси}-6,11-диоксо-1,2,3,4,6,11-гексагидротетрацен-2-ил]этокси}тетрагидро-2H-пиран-2-ил)метокси]ацетамид (Соединение 6) MS (ESI): 813 [M+H]+. Время удерживания=5,42 мин (способ 2).
N-бензил-2-[(6-{2-оксо-2-[(2S,4S)-2,5,12-тригидрокси-7-метокси-4-{[(1S,3R,4aS,9S,9aR,10aS)-9-метокси-1-метилоктагидро-1H-пирано[4',3':4,5][1,3]оксазоло[2,3-c][1,4]оксазин-3-ил]окси}-6,11-диоксо-1,2,3,4,6,11-гексагидротетрацен-2-ил]этокси}тетрагидро-2H-пиран-2-ил)метокси]ацетамид (Соединение 8) MS (ESI): 903 [M+H]+. Время удерживания=6,92 мин (способ 1). Время удерживания=6,94 мин (способ 2).
Пример 3 N-ацетил-3-({3-[(2E)-2-{2-гидрокси-1-[(2S,4S)-2,5,12-тригидрокси-7-метокси-4-{[(1S,3R,4aS,9S,9aR,10aS)-9-метокси-1-метилоктагидро-1H-пирано[4',3':4,5][1,3]оксазоло[2,3-c][1,4]оксазин-3-ил]окси}-6,11-диоксо-1,2,3,4,6,11-гексагидротетрацен-2-ил]этилиден}гидразинил]-3-оксопропил}дисульфанил)-L-аланина (Соединение 12)
Стадия 1 N'-{(1E)-2-гидрокси-1-[(2S,4S)-2,5,12-тригидрокси-7-метокси-4-{[(1S,3R,4aS,9S,9aR,10aS)-9-метокси-1-метилоктагидро-1H-пирано[4',3':4,5][1,3]оксазоло[2,3-c][1,4]оксазин-3-ил]окси}-6,11-диоксо-1,2,3,4,6,11-гексагидротетрацен-2-ил]этилиден}-3-(пиридин-2-илдисульфанил)пропангидразид 53
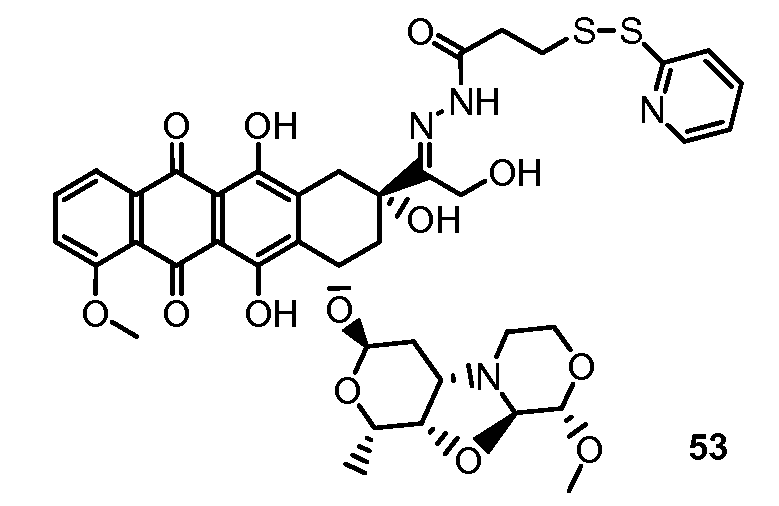
Раствор 3-(2-пиридилдитио)пропионовой кислоты гидразид HCl (41,5 мг, 0,156 ммоль) в безводном метаноле (5 мл) добавляли к (8S,10S)-6,8,11-тригидрокси-8-(гидроксиацетил)-1-метокси-10-{[(1S,3R,4aS,9S,9aR,10aS)-9-метокси-1-метилоктагидро-1H-пирано[4',3':4,5][1,3]оксазоло[2,3-c][1,4]оксазин-3-ил]окси}-7,8,9,10-тетрагидротетрацен-5,12-диону [PNU-159682, соединение IIA, полученное, как описано в WO 9802446] (50 мг, 0,078 ммоль). Раствор перемешивали в темноте при комнатной температуре в течение 20 часов. Ход реакции сопровождался ВЭЖХ-МС с обращенной фазой. После этого периода растворитель упаривали, и остаток очищали с помощью колоночной флэш-хроматографии (MeOH:CH2Cl2=0,2:9,8) на силикагеле (230-400 меш), получая 18 мг (выход 27%) 53. MS (ESI): 853 [M+H]+. Время удерживания = 5,52 мин (способ 2).
Аналогичными способами и используя подходящие исходные вещества, получали следующие соединения: 6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)-N'-{(1E)-2-гидрокси-1-[(2S,4S)-2,5,12-тригидрокси-7-метокси-4-{[(1S,3R,4aS,9S,9aR,10aS)-9-метокси-1-метилоктагидро-1H-пирано[4',3':4,5][1,3]оксазоло[2,3-c][1,4]оксазин-3-ил]окси}-6,11-диоксо-1,2,3,4,6,11-гексагидротетрацен-2-ил]этилиден}гексангидразид 52. MS (ESI): 849 [M+H]+. Время удерживания = 5,5 мин (способ 2).
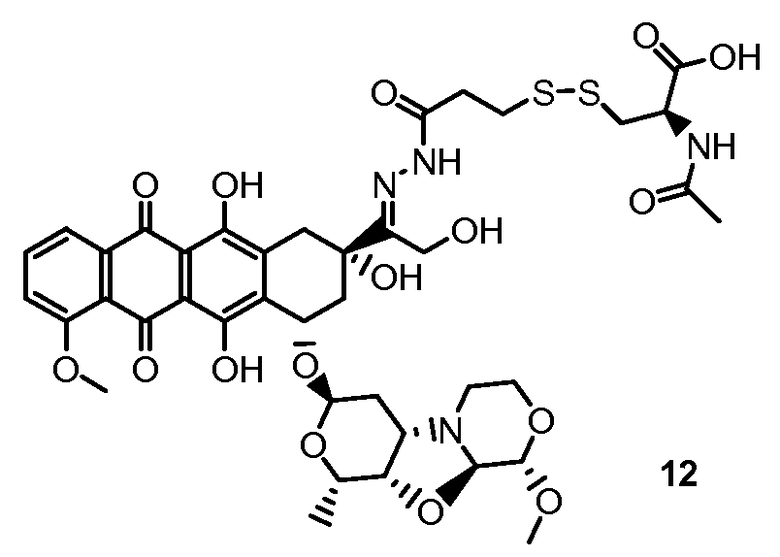
Стадия 2 К раствору 53, полученному со стадии 1 (8,5 мг, 0,01 ммоль), добавляли N-ацетилцистеин (0,32 мг, 0,02 ммоль). Раствор перемешивали при комнатной температуре в течение 24 часов, растворитель упаривали, и остаток очищали с помощью колоночной флэш-хроматографии (MeOH:CH2Cl2=2:8) на силикагеле (230-400 меш), получая 7,2 мг (выход 80%) 12 в виде твердого вещества красного цвета. MS (ESI): 905 [M+H]+. Время удерживания =3,62 мин (способ 2).
Аналогичными способами, и используя подходящие исходные вещества, получали следующие соединения:
N-ацетил-S-(1-{6-[(2E)-2-{2-гидрокси-1-[(2S,4S)-2,5,12-тригидрокси-7-метокси-4-{[(1S,3R,4aS,9S,9aR,10aS)-9-метокси-1-метилоктагидро-1H-пирано[4',3':4,5][1,3]оксазоло[2,3-c][1,4]оксазин-3-ил]окси}-6,11-диоксо-1,2,3,4,6,11-гексагидротетрацен-2-ил]этилиден}гидразинил]-6-оксогексил}-2,5-диоксопирролидин-3-ил)-L-цистеин (Соединение 9). MS (ESI): 1012 [M+H]+.
N 2-(трет-бутоксикарбонил)-N 6-(1-{6-[(2E)-2-{2-гидрокси-1-[(2S,4S)-2,5,12-тригидрокси-7-метокси-4-{[(1S,3R,4aS,9S,9aR,10aS)-9-метокси-1-метилоктагидро-1H-пирано[4',3':4,5][1,3]оксазоло[2,3-c][1,4]оксазин-3-ил]окси}-6,11-диоксо-1,2,3,4,6,11-гексагидротетрацен-2-ил]этилиден}гидразинил]-6-оксогексил}-2,5-диоксопирролидин-3-ил)-L-лизин (Соединение 10, таблица 1) MS (ESI): 1095 [M+H]+.
Пример 3a Получение (2S,4S)-N-[2-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)этил]-2,5,12-тригидрокси-7-метокси-4-{[(1S,3R,4aS,9S,9aR,10aS)-9-метокси-1-метилоктагидро-1H-пирано[4',3':4,5][1,3]оксазоло[2,3-c][1,4]оксазин-3-ил]окси}-6,11-диоксо-1,2,3,4,6,11-гексагидротетрацен-2-карбоксамида 54
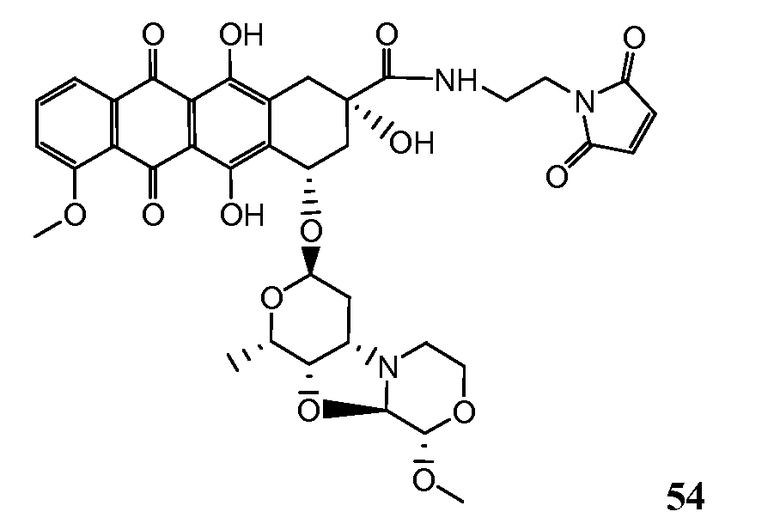
К раствору PNU-159682 (15,3 мг, 0,02038 ммоль), полученного, как описано в WO 98/02446, в 3 мл метанола и 2 мл H2O, добавляли раствор NaIO4 (5,1 мг, 0,0238 ммоль) в 1 мл H2O. Реакционную смесь перемешивали при комнатной температуре в течение 3 часов, до тех пор, пока не обнаруживалось никакого исходного вещества (TLC и ВЭЖХ анализ). Растворители удаляли под пониженным давлением и сырое твердое вещество красного цвета (2S,4S)-2,5,12-тригидрокси-7-метокси-4-{[(1S,3R,4aS,9S,9aR,10aS)-9-метокси-1-метилоктагидро-1H-пирано[4',3':4,5][1,3]оксазоло[2,3-c][1,4]оксазин-3-ил]окси}-6,11-диоксо-1,2,3,4,6,11-гексагидротетрацен-2-карбоновую кислоту 56 использовали без дополнительной очистки на следующей стадии. MS (ESI): 628 [M+H]+. Время удерживания = 2,1-3,2 мин (способ 1, Пример 3b).
К раствору сырого промежуточного соединения 56 (4,4 мг) в безводном дихлорметане (1,5 мл) в атмосфере аргона добавляли безводный триэтиламин (2,2 мг, 0,0204 ммоль), TBTU (4,4 мг, 0,01388 ммоль) и коммерчески доступную соль N-(2-аминоэтил)малеимидтрифторацетат (3,6 мг, 0,00694 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 30 мин, до исчезновения исходного вещества (ВЭЖХ-МС анализ). Растворитель упаривали под вакуумом, и остаток затем очищали с помощью колоночной флэш-хроматографии (EtOH:CH2Cl2 = 0,2:9,8) на силикагеле (230-400 меш), получая 1,1 мг (твердое вещество красного цвета, выход рассчитан на PNU-159682=21%) 54. MS (ESI): 750 [M+H]+. Время удерживания = 5,18 мин (способ 1, Пример 3b).
Пример 3b Получение N-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]-L-валил-N 5-карбамоил-N-[4-({[(4-{[(2S,4S)-2,5,12-тригидрокси-7-метокси-4-{[(1S,3R,4aS,9S,9aR,10aS)-9-метокси-1-метилоктагидро-1H-пирано[4',3':4,5][1,3]оксазоло[2,3-c][1,4]оксазин-3-ил]окси}-6,11-диоксо-1,2,3,4,6,11-гексагидротетрацен-2-ил]карбонил}пиперазин-1-ил)карбонил]окси}метил)фенил]-L-орнитинамида 55
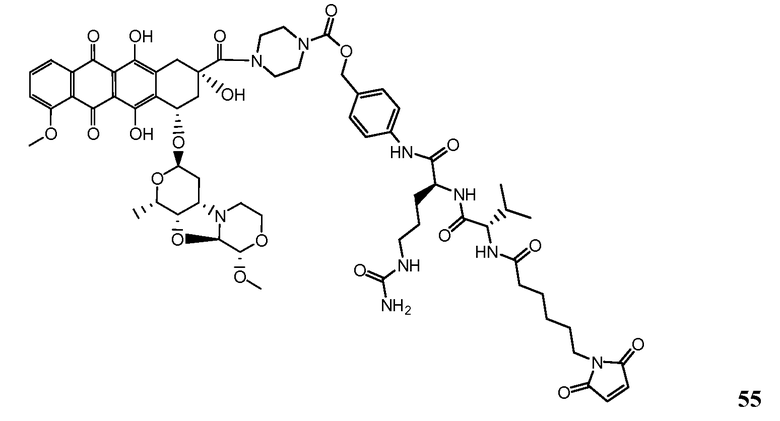
Стадия 1 4-((S)-2-((S)-2-(6-(2,5-Диоксо-2,5-дигидро-1H-пиррол-1-ил)гексанамидо)-3-метилбутанамидо)-5-уреидопентанамидо)бензил 4-нитрофенилкарбонат 58 (30 мг, 0,041 ммоль) взаимодействовал с трет-бутилпиперазин-1-карбоксилатом (5,3 мг, 0,0287 ммоль) в безводном ДМСО в атмосфере аргона при комнатной температуре (Фигура 7d). Реакционную смесь перемешивали в течение 1 ч, до исчезновения исходного вещества (ВЭЖХ-МС анализ). Диэтиловый эфир (80 мл) затем добавляли в реакционную смесь и полученный таким образом преципитат собирали путем фильтрации с получением N-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]-L-валил-N-{4-[({[4-(трет-бутоксикарбонил)пиперазин-1-ил]карбонил}окси)метил]фенил}-N 5-карбамоил-L-орнитинамида 59 в виде твердого продукта желтого цвета, 22,0 мг) выделяли и использовали без дополнительной очистки на следующей стадии. MS (ESI): 785 [M+H]+. Время удерживания = 4,87 мин (способ 1).
Стадия 2 Промежуточное соединение 59 (22,0 мг) обрабатывали трифторуксусной кислотой (327 мг, 2,87 ммоль) в безводном дихлорметане (0,12 мл). Реакционную смесь перемешивали при комнатной температуре в течение 15 минут, до исчезновения исходного вещества (ВЭЖХ-МС анализ). После этого, реакционную смесь обрабатывали диэтиловым эфиром (20 мл) и остаток, полученный таким образом, промывали диэтиловым эфиром (2×10 мл): продукт N-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]-L-валил-N 5-карбамоил-N-(4-{[(пиперазин-1-илкарбонил)окси]метил}фенил)-L-орнитинамид 60 (белый воск, 20,0 мг) таким образом выделяли и использовали без дополнительной очистки на следующей стадии. MS (ESI): 685 [M+H]+. Время удерживания = 2,97 мин (способ 1).
Стадия 3 К промежуточному соединению 60 (13,2 мг) добавляли раствор сырой (2S,4S)-2,5,12-тригидрокси-7-метокси-4-{[(1S,3R,4aS,9S,9aR,10aS)-9-метокси-1-метилоктагидро-1H-пирано[4',3':4,5][1,3]оксазоло[2,3-c][1,4]оксазин-3-ил]окси}-6,11-диоксо-1,2,3,4,6,11-гексагидротетрацен-2-карбоновой кислоты 56 (9,3 мг) в безводном дихлорметане (2,6 мл), TBTU (5,3 мг, 0,0165 ммоль), и безводный триэтиламин (2,8 мг, 0,0275 мг). Реакционную смесь перемешивали при комнатной температуре в атмосфере аргона в течение 15 минут, до исчезновения исходного вещества (ВЭЖХ-МС анализ). Растворитель затем упаривали под вакуумом и сырой продукт очищали с помощью колоночной флэш-хроматографии (EtOH:AcOEt=1,5:8,5) на силикагеле (230-400 меш), получая 4,8 мг (твердое вещество красного цвета, выход рассчитан на PNU-159682=34%) 55. MS (ESI): 1295 [M+H]+. Время удерживания = 5,51 мин (способ 1).
Соединения были охарактеризованы с помощью аналитических данных ВЭЖХ/МС; ВЭЖХ/МС данные собирали по одному из следующих способов 1 или 2.
Аналитический способ 1 ВЭЖХ/МС: Оборудование для ВЭЖХ состояло из системы Waters 2795 Alliance HT, снабженной детектором 2996 Waters PDA и Micromass mod. ZQ масс-спектрометром с одной квадрупольной линзой, оборудованным источником электрораспылительной ионизации (ESI). Контроль за прибором, получение и накопление данных и обработка данных обеспечивались программами Empower и MassLynx 4.0. ВЭЖХ проводили при 30°C со скоростью потока 1,0 мл/мин, используя колонку Waters X Terra MS C18-3,5 мкМ (4,6×50 мм). Подвижная фаза A представляла собой буфер ацетат аммония 5 мМ pH 5,2 с ацетонитрилом (95:5), а подвижная фаза B представляла собой H2O/ацетонитрил (5:95); градиент был от 10 до 90% B через 8 минут, затем быстро линейно увеличивался до 100% B через 1,0 минуту. Масс-спектрометр работал в режиме определения положительных и отрицательных ионов, напряжение капилляра устанавливали на 3,5 кВ (ES+) и 28 В (ES-); температура источника составляла 120°C; конус был 14 В (ES+) и 2,8 кB (ES-); полное сканирование, диапазон масс был установлен от 100 до 1000 m/z.
Аналитический способ 2 ВЭЖХ/MC: Оборудование для ВЭЖХ состояло из системы Waters 2795 HPLC, снабженной детектором 996 Waters PDA и Micromass mod. ZQ масс-спектрометром с одной квадрупольной линзой, оборудованным источником электрораспылительной ионизации (ESI). Контроль за прибором, получение и накопление данных и обработка данных обеспечивались программами Empower и MassLynx 4.0. ВЭЖХ проводили при 30°C со скоростью потока 1,0 мл/мин, используя колонку RP18 Waters X Terra (4,6 мкМ×50 мм). Подвижная фаза A представляла собой буфер гидроксид аммония 0,05% pH 10 с ацетонитрилом (95:5), а подвижная фаза B представляла собой H2O/ацетонитрил (5:95); градиент был от 10 до 90% B через 8 минут, затем удерживали 100% B в течение 2 минут. Масс-спектрометр работал в режиме определения положительных и отрицательных ионов, напряжение капилляра устанавливали на 2,5 кB; температура источника составляла 120°C; конус был 10 В; полное сканирование, диапазон масс был установлен от 100 до 1000 m/z.
Пример 3c Получение (8S,10S)-6,8,11-тригидрокси-1-метокси-10-{[(1S,3R,4aS,9S,9aR,10aS)-9-метокси-1-метилоктагидро-1H-пирано[4',3':4,5][1,3]оксазоло[2,3-c][1,4]оксазин-3-ил]окси}-8-(пиперазин-1-илкарбонил)-7,8,9,10-тетрагидротетрацен-5,12-диона 57
К раствору 56 (9 мг) в безводном дихлорметане (5 мл) в атмосфере аргона добавляли безводный триэтиламин (1,6 мг, 0,0158 ммоль), пиперазин (3,6 мг, 0,0424 ммоль), HOBt (2,1 мг, 0,0158 ммоль) и EDC (3,0 мг, 0,0158 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Растворитель упаривали под вакуумом, и остаток затем очищали с помощью колоночной флэш-хроматографии (DCM/MeOH/AcOH/H2O 45/4/1/0,5) на силикагеле (230-400 меш). Полученный продукт растворяли в DCM и промывали насыщенным NaHCO3 (x2) и водой (x2). Органический растворитель упаривали под вакуумом, с получением 5,0 мг 57. MS (ESI): 696 [M+H]+. Время удерживания=4,01 мин (способ 1, Пример 3b).
Пример 3d Получение N-[2-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)этил]-2-[(6-{2-оксо-2-[(2S,4S)-2,5,12-тригидрокси-7-метокси-4-{[(1S,3R,4aS,9S,9aR,10aS)-9-метокси-1-метилоктагидро-1H-пирано[4',3':4,5][1,3]оксазоло[2,3-c][1,4]оксазин-3-ил]окси}-6-метилиден-11-оксо-1,2,3,4,6,11-гексагидротетрацен-2-ил]этокси}тетрагидро-2H-пиран-2-ил)метокси]ацетамида 51
К раствору сырого промежуточного соединения 50 (103,8 мг) в безводном дихлорметане (29,7 мл) в атмосфере аргона добавляли коммерчески доступную соль N-(2-аминоэтил)малеимид трифторацетат (57,9 мг, 0,228 ммоль) и безводный триэтиламин (23,1 мг, 0,228 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч, до исчезновения исходного вещества (ВЭЖХ-МС анализ). Растворитель упаривали под вакуумом и остаток промывали смесью Et2O/н-гексан (1 мл/20 мл). Сырой продукт затем очищали с помощью колоночной флэш-хроматографии (EtOH:CH2Cl2=0,2:9,8) на силикагеле (230-400 меш), получая 12,2 мг (твердое вещество красного цвета, выход рассчитан по PNU-159682=20%) 51 в виде диастереоизомерной смеси. MS (ESI): 936 [M+H]+. Время удерживания=5,86, в соответствии со следующим способом:
Система Waters 2795 Alliance HT HPLC c детектором 2996 Waters PDA и Micromass mod. ZQ масс-спектрометром с одной квадрупольной линзой, с источником электрораспылительной ионизации (ESI). Контроль за прибором, получение и накопление данных и обработка данных осуществляли с помощью программ Empower и MassLynx 4.0. ВЭЖХ проводили при 30°C со скоростью потока 1,0 мл/мин, используя колонку Waters X Terra MS C18-3,5 мкМ (4,6×50 мм). Подвижная фаза A представляла собой буфер ацетат аммония 5 мМ pH 5,2 с ацетонитрилом (95:5), а подвижная фаза B представляла собой H2O/ацетонитрил (5:95); градиент был от 10 до 90% B через 8 минут, затем быстро линейно увеличивался до 100% B через 1,0 минуту. Масс-спектрометр работал в режиме определения положительных и отрицательных ионов, напряжение капилляра устанавливали на 3,5 кВ (ES+) и 28 В (ES-); температура источника составляла 120°C; конус был 14 В (ES+) и 2,8 кB (ES-); полное сканирование, диапазон масс от 100 до 1000 m/z.
Пример 4 Получение MCM2 конъюгата соединения 5, как указано в таблице 1.
Белок MCM2 (10-294) [Ishimi et al. (2001) Jour. Biol Chem. 276(46):42744-42752] (1,5 мг, 0,045 мкмоль) растворяли в 0,5 мл фосфатно-буферного солевого раствора (pH 7,2), значение pH доводили до 8,5 путем добавления 55 мкл 1 M NaHCO3 (pH 8,5) и 0,5 мг 1-({[(1-{2-оксо-2-[(2S,4S)-2,5,12-тригидрокси-7-метокси-4-{[(1S,3R,4aS,9S,9aR,10aS)-9-метокси-1-метилоктагидро-1H-пирано[4',3':4,5][1,3]оксазоло[2,3-c][1,4]оксазин-3-ил]окси}-6,11-диоксо-1,2,3,4,6,11-гексагидротетрацен-2-ил]этокси}циклогексил)окси]ацетил}окси)пирролидин-2,5-диона (0,55 мкмоль) [получено, как описано в примере 1 стадия 3] добавляли из 10 мг/мл раствора ацетонитрила. Реакционную смесь инкубировали в течение 1 часа при комнатной температуре, затем реакционную смесь обессоливали на колонке NAP-10, кондиционированной в фосфатно-буферном солевом растворе, и фракции, содержащие белок, собирали и объединяли.
Вступивший в реакцию белок анализировали с помощью SDS PAGE по сравнению с непрореагировавшим MCM2 и различным количеством 1-({[(1-{2-оксо-2-[(2S,4S)-2,5,12-тригидрокси-7-метокси-4-{[(1S,3R,4aS,9S,9aR,10aS)-9-метокси-1-метилоктагидро-1H-пирано[4',3':4,5][1,3]оксазоло[2,3-c][1,4]оксазин-3-ил]окси}-6,11-диоксо-1,2,3,4,6,11-гексагидротетрацен-2-ил]этокси}циклогексил)окси]ацетил}окси)пирролидин-2,5-диона, как описано выше.
С помощью аналогичных способов, и используя подходящие исходные вещества, получали следующие соединения (таблица 1): MCM2 конъюгат соединения 11; MCM2 конъюгат соединения 13; MCM2 конъюгат соединения 14.
Пример 5 Стабильность конъюгата: общая методика
К 0,5 мг конъюгата добавляли буферный раствор ацетата аммония 5 мМ pH 5,2 (200 мл). Раствор нагревали при 37°C и периодически брали образец и анализировали с помощью ВЭЖХ (способ 2). Результаты выражены в % высвобожденного вещества (8S,10S)-6,8,11-тригидрокси-8-(гидроксиацетил)-1-метокси-10-{[(1S,3R,4aS,9S,9aR,10aS)-9-метокси-1-метилоктагидро-1H-пирано[4',3':4,5][1,3]оксазоло[2,3-c][1,4]оксазин-3-ил]окси}-7,8,9,10-тетрагидротетрацен-5,12-диона (Соединение IIA) из конъюгата.
Аналогичными способами, используя буферный раствор ацетата аммония 5 мМ pH 4,5, также была определена стабильность при pH 4,5.
Аналогичными способами, стабильность была выполнена на соединении 12 (Таблица 1), показывающая после четырех часов инкубирования 90% высвобождение в pH 5,2 буферном растворе и 100% высвобождение в pH 4,2 буферном растворе, (8S,10S)-6,8,11-тригидрокси-8-(гидроксиацетил)-1-метокси-10-{[(1S,3R,4aS,9S,9aR,10aS)-9-метокси-1-метилоктагидро-1H-пирано[4',3':4,5][1,3]оксазоло[2,3-c][1,4]оксазин-3-ил]окси}-7,8,9,10-тетрагидротетрацен-5,12-диона (Соединение IIA) из конъюгата.
Пример 6 Получение конструированных антител с цистеиновыми заменами для конъюгирования путем восстановления и повторного окисления
Аминокислоты легкой цепи и тяжелой цепи нумеруются в соответствии с Kabat (Kabat et al., Sequences of proteins of immunological interest, (1991) 5th Ed., US Dept of Health and Human Service, National Institutes of Health, Bethesda, MD). Используются однобуквенные обозначения аминокислот.
Полноразмерные моноклональные антитела с цистеиновыми заменами (ТиоMabs), экспрессированные в клетках CHO, несут цистеиновые аддукты (цистеины) или глутатионилированы по сконструированным цистеиновым заменам, благодаря условиям культивирования клеток. Для высвобождения реакционно-способных тиольных групп сконструированных цистеиновых замен, тиоMab растворяют в 500 мМ бората натрия и 500 мМ хлорида натрия примерно при pH 8,0 и восстанавливали примерно 50-100-кратным избытком 1 мМ TCEP (трис(2-карбоксиэтил)фосфин гидрохлорид; Getz et al. (1999) Anal. Biochem. 273:73-80; Soltec Ventures, Beverly, MA) примерно в течение 1-2 часов при 37°C. Альтернативно, DTT может быть использован в качестве восстановителя. Образование межцепочечных дисульфидных связей отслеживали либо с помощью нередуцирующего SDS-PAGE, либо с помощью денатурирующей колоночной хроматографии с обращенной фазой HPLC PLRP. Восстановленное антитело с цистеиновыми заменами разбавляют и нагружают на колонку HiTrap S колонка в 10 мМ ацетате натрия, pH 5, и элюируют с использование PBS, содержащего 0,3 M хлорид натрия. Элюированное восстановленное антитело с цистеиновыми заменами (ТиоMab) обрабатывают 2 мМ дегидроаскорбиновой кислотой (dhAA) при pH 7 в течение 3 часов, или 2 мМ водным сульфатом меди (CuSO4) при комнатной температуре в течение ночи. Также может быть эффективным окисление окружающим воздухом. Обмен буфера проводят путем элюции на смоле Sephadex G25, и элюируют PBS с 1 мМ DTPA. Величину Тиол/Ab проверяют, путем определения концентрации восстановленного антитела по поглощению при 280 нм раствора, а концентрацию тиола путем взаимодействия с DTNB (Aldrich, Milwaukee, WI) и определения поглощения при 412 нм.
Жидкостная хроматография/масс-спектрометрический анализ проводят на квадрупольном масс-спектрометре TSQ Quantum Triple с расширенным диапазоном масс (Thermo Electron, San Jose California). Образцы хроматографировали на PRLP-S, 1000 A, микроколонке (50 мм × 2,1 мм, Polymer Laboratories, Shropshire, UK) нагревали до 75°C. Использовали линейный градиент от 30-40% B (растворитель A: 0,05% TFA в воде, растворитель B: 0,04% TFA в ацетонитриле) и растворитель для элюирования непосредственно ионизировали, используя источник электрораспыления. Данные собирали с помощью системы Xcalibur и деконволюцию проводили, используя ProMass (Novatia, LLC, New Jersey). Перед проведением анализа LC/MS, антитела или конъюгаты антитела с лекарственным средством (50 мкг) обрабатывают PNGазой F (2 единиц/мл; PROzyme, San Leandro, CA) в течение 2 часов при 37°C для удаления N-связанных углеводов.
Образцы для хроматографии гидрофобных взаимодействий (HIC) вводили в колонку Бутил HIC NPR (2,5 мкм, 4,6 мм × 3,5 см) (Tosoh Bioscience) и элюировали с линейным градиентом от 0 до 70% B при 0,8 мл/мин (A: 1,5 M аммония сульфат в 50 мМ фосфате калия, pH 7, B: 50 мМм фосфат калия pH 7, 20% изопропанол). ВЭЖХ системы серии Agilent 1100 оборудованы мультиволновым детектором и программное обеспечение Chemstation использовали для разделения и количественной оценки видов антител с различными соотношениями лекарственных средств на антитело.
Пример 7 Конъюгирование антител и промежуточных соединений лекарственного средства производного антрациклина с линкером
В основном, антитела и промежуточные соединения лекарственного средства производного антрациклина с линкером конъюгируют в соответствии со способами US 7521541; US 7498298; US 2005/0276812; US 2008/0311134; и "NEMORUBICIN METABOLITE AND ANALOG ANTIBODY-DRUG CONJUGATES AND METHODS", PCT/US2009/031199, подана 16 января 2009, каждый из которых включен путем ссылки.
Квантизационный анализ конъюгатов путем флуоресцентной детекции в геле, проводили, используя сканер ProXpress на основе CCD (PerkinElmer). Возбуждение прибора и эмиссионные фильтры устанавливали на 480/30 нм и 590/35 нм, соответственно. Квантизационный анализ проводили, используя программное обеспечение Profinder, предоставляемое с прибором, используя различное количество исходных веществ, нагруженных на гель в качестве эталонов. Общий нагруженный белок затем оценивали путем окрашивания белка Кумасси Голубым.
Пример 8 Конъюгирование антител с цистеиновыми заменами и малеимидных промежуточных соединений лекарственного средства и линкера
После восстановления и повторного окисления из примера 6, сконструированное антитело с цистеиновыми заменами растворяют в PBS буфере (фосфатно-буферном солевом растворе) и охлаждают на льду. Примерно 1,5 молярных эквивалентов относительно сконструированных цистеинов на антитело производного антрациклина с тиол-реактивной функциональной группой, такого как: пиридин-дисульфид, например, промежуточное соединение лекарственного средства с линкером 53 или бром-ацетамидо и малеимид, например, промежуточные соединения лекарственного средства с линкером 51 и 52, растворяют в ДМСО, разбавляют в ацетонитриле и воде и добавляют к охлажденному восстановленному повторно окисленному антителу в PBS. Примерно через один час избыток малеимида добавляют для остановки реакции и закрытия любых непрореагировавших тиольных групп антитела. Реакционную смесь концентрируют путем центрифужной ультрафильтрации и конъюгат сконструированного антитела с цистеиновыми заменами и лекарственного средства очищают и обессоливают путем элюции через смолу G25 в PBS, фильтруют через 0,2 мкм фильтры в стерильных условиях, и замораживают для хранения.
Способами, указанными выше, конъюгаты сконструированного антитела с цистеиновыми заменами и лекарственного средства получали: 102, 103, 104, 105, 106, 107, 108, 109, 111, 112 и 113 (Таблица 2). Каждый из конъюгатов антитела с лекарственным средством, содержащий сконструированные антитела с цистеиновыми заменами, 102, 103, 104, 105, 106, 107, 108, 109, 111, 112, и 113 являлся мутантным с цистеиновыми заменами в тяжелой цепи (HC) A114C (Kabat) (US 7521541). Для антитела трастузумаб, мутантный тип A114C по схеме нумерации Kabat является таким же, как мутантный тип A118C по EU схеме нумерации и мутантный тип A121C по схеме нумерации последовательностей.
Конъюгаты сконструированного антитела с цистеиновыми заменами и лекарственного средства с малеимидкапроилом (MC), валин-цитруллином (vc), п-аминобензилоксикарбамоилом (PAB) и лекарственным средством ауристатином (MMAE) группами лекарственное средство-линкер (106, 111 и 112) получали способом из примера 3 заявки US 2008/0311134 и Примеров 27 и 29 US 7498298, которые включены путем ссылки, с промежуточным соединением лекарственного средства и линкера MC-vc-PAB-MMAE:
Пример 9 Исследование пролиферации клеток in vitro
Опухолевые клеточные линии рака молочной железы BT-474, SKBR-3 и MCF7 получали из Американской коллекции типовых культур.
Эффективность in vitro конъюгатов антитела с лекарственным средством измеряли с помощью исследования клеточной пролиферации (Фигуры 8-29). Люминесцентный анализ жизнеспособности клеток CellTiter-Glo® является коммерчески доступным (Promega Corp., Madison, WI), гомогенный способ исследования основан на рекомбинантной экспрессии люциферазы Coleoptera (US 5583024; US 5674713; US 5700670). В этом исследовании определяют количество жизнеспособных клеток в культуре на основании количественного определения присутствующего АТФ, показателя метаболически активных клеток (Crouch et al. (1993) J. Immunol. Meth. 160:81-88; US 6602677). Анализ CellTiter-Glo® проводили в 96-луночном формате, делая его пригодным для автоматического высокопроизводительного скрининга (HTS) (Cree et al. (1995) AntiCancer Drugs 6:398-404). Методика гомогенного анализа включает добавление одного реагента (CellTiter-Glo® Reagent) непосредственно к клеткам, культивируемым в сывороточной среде.
Гомогенный формат «добавить-перемешать-измерить» в результате приводит к лизису клеток и генерированию люминесцентного сигнала, пропорционального количеству присутствующего АТФ. Субстрат, люциферин жука, окислительно декарбоксилируется под действием люциферазы светлячка с сопутствующим превращением АТФ в АМФ и генерированием фотонов. Жизнеспособные клетки отражают в относительных единицах люминесценции (RLU). Данные могут регистрироваться люминометром или визуализирующим устройством полупроводниковой камерой на приборах с зарядовой связью. Выход люминесценции представлен в виде RLU, измеренных в динамике по времени. Альтернативно, фотоны из люминесценции могут быть подсчитаны в сцинтилляционном счетчике в присутствии сцинтиллятора. Световые единицы затем могут быть представлены как CPS - импульсы в минуту.
Эффективность ADC измеряли с помощью анализа жизнеспособности клеток, используя следующий протокол, основанный на люминесцентном анализе жизнеспособности клеток CellTiter Glo, Promega Corp. Technical Bulletin TB288 и Mendoza et al. (2002) Cancer Res. 62:5485-5488:
1. Аликвоту 50 мкл клеточной культуры, содержащей примерно 1000 или более клеток, в том числе: клетки, экспрессирующие HER2, и клетки, экспрессирующие CD22, в среде, помещали в каждую лунку 96-луночного планшета с прозрачным дном и непрозрачными стенками.
2. ADC (50 мкл) добавляли в экспериментальные лунки в трех повторах до конечной концентрации 10 мкг/мл с контрольными лунками «без ADC», содержащими только среду, и инкубировали в течение 3 или более дней.
3. Планшеты уравновешивали до комнатной температуры приблизительно в течение 30 минут.
4. Добавляли реагент CellTiter-Glo (100 мкл).
5. Содержимое перемешивали в течение 2 минут на орбитальном встряхивателе для индукции лизиса клеток.
6. Планшет инкубировали при комнатной температуре в течение 10 минут для стабилизации люминесцентного сигнала.
7. Люминесценцию регистрировали и отмечали на графиках как RLU = относительные единицы люминесценции.
Пример 10 Фармакокинетические параметры - сывороточный клиренс и стабильность
Распределение конъюгатов антитело-лекарственное средство in vivo анализируют, путем измерения сывороточных концентраций антитела и лекарственного конъюгата после однократного внутривенного болюсного введения дозы крысам Sprague-Dawley. Концентрации конъюгатов антитело-лекарственное средство, содержащих по меньшей мере одно цитотоксическое лекарственное средство, измеряют с помощью ELISA, в котором используют белок внеклеточного домена (ECD) для захвата и против антрациклина и конъюгированное с пероксидазой хрена (HRP) антитело против Fc мыши для обнаружения. Суммарные концентрации антитела в сыворотке измеряли с помощью ELISA, в котором используют ECD для захвата и HRP против Fc человека, измеряя антитело, как с конъюгированным производным антрациклина, так и без него. Данные сывороточная концентрация-время от каждого животного анализируют, используя двухкамерную модель с внутривенным болюсным введением, элиминацию первого порядка и макро константы скорости (Model 8, WinNonlin Pro v.5.0.1, Pharsight Corporation, Mountain View, CA).
Пример 11 Токсичность у животных
12-дневное исследование острой токсичности у пубертантных самок крыс (100-125 г) проводят путем однократного введения конъюгата антитела с лекарственным средством в концентрации примерно 1-10 мг/кг и контрольного носителя в 1 день. Введение тестируемого вещества обычно проводят в виде внутривенного болюса. Массу тела измеряют ежедневно. Время от времени проводят анализы клинической биохимии, сывороточных ферментов и гематологический анализ. Признаки токсичности включали клиническое наблюдение потери массы тела.
Пример 12 Ингибирование роста опухоли, мышиная модель эффективности in vivo
Все исследования на животных проводили в соответствии с руководствами NIH по содержанию и использованию лабораторных животных и были одобрены Комитетом по биоэтике при Genentech.
Исследования эффективности проводили, используя мышей SCID (Charles River Laboratories). Модели эффективности для исследований, показанных на примерах на Фигурах 30-32, использовали, как описано (Polson et al. (2009) Cancer Res. 69(6):2358-2364; Phillips et al. (2008) Cancer Res. 68(22):9280-9290; US 2008/0050310; US 2005/0276812), оценивая объем опухоли после однократной внутривенной дозы. Модели трансплантатов были разработаны с использованием опухолей, иссеченных у мышей, несущих внутрибрюшинную опухоль, затем серийно пересевали мышам реципиентам. Например, клетки для имплантации промывали и суспендировали в HBSS (Hyclone) и инокулировали подкожно в боковые стороны самкам мышей CB17 ICR с тяжелым комбинированным иммунодефицитом (возраст 7-16 недель из Charles Rivers Laboratories), в объеме 0,2 мл/мышь. Для тестирования эффективности конъюгатов антитела с лекарственным средством in vivo, приблизительно несколько миллионов клеток на мышь с SCID инокулировали однократно и оставляли расти примерно в течение 10-60 дней после введения. Когда объемы опухоли достигали 150-200 мм3 (обычно с 14 дня по 21 день после инокуляции), мышей разделяли на испытуемые группы по 9-10 мышей на группу, и объем опухоли определяли у каждой мыши. Когда средний размер опухоли достигал желаемого объема, мышей делили на группы по 8-10 мышей с одинаковым размером опухоли и вводили внутривенно через хвостовую вену образцы: ADC, антитела или носитель. Дозы ADC измеряли либо как массу конъюгированного цитотоксического лекарственного средства с низкой молекулярной массой на площадь поверхности тела мыши, либо как постоянную массу ADC на массу тела мыши (например, 5 мг ADC/кг массы тела мыши). В основном, лекарственные нагрузки на антитела в любом заданном эксперименте были сходными или нормализованы, так, чтобы эти два измерения могли рассматриваться эквивалентными. Объем опухоли измеряли, используя штангенциркуль в соответствии с формулой: V (мм3)=0,5A x B2, где A и B представляют собой продольные и поперечные диаметры, соответственно. Мышей подвергали эвтаназии до достижения объема опухоли 3000 мм3 или когда опухоли демонстрировали признаки приближающегося изъязвления. Данные, полученные от каждой экспериментальной группы, выражали как среднее ± SE.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПОЛУЧЕННЫЕ С ПОМОЩЬЮ ГЕННОЙ ИНЖЕНЕРИИ АНТИТЕЛА С ЦИСТЕИНОВЫМИ ЗАМЕНАМИ И ИХ КОНЪЮГАТЫ | 2011 |
|
RU2626537C2 |
| КОНЪЮГАТЫ АНТИТЕЛ С ЛЕКАРСТВЕННЫМИ СРЕДСТВАМИ, ХАРАКТЕРИЗУЮЩИЕСЯ ВЫСОКОЙ IN VIVO ПЕРЕНОСИМОСТЬЮ | 2018 |
|
RU2764031C2 |
| ПОЛУЧЕННЫЕ С ПОМОЩЬЮ ГЕННОЙ ИНЖЕНЕРИИ АНТИТЕЛА С ЦИСТЕИНОВЫМИ ЗАМЕНАМИ И ИХ КОНЪЮГАТЫ | 2011 |
|
RU2755066C2 |
| ПЕПТИДОМИМЕТИЧЕСКИЕ СОЕДИНЕНИЯ И ИХ КОНЪЮГАТЫ АНТИТЕЛ С ЛЕКАРСТВЕННЫМИ СРЕДСТВАМИ | 2014 |
|
RU2689388C1 |
| ПИРРОЛОБЕНЗОДИАЗЕПИНОВЫЕ КОНЪЮГАТЫ АНТИТЕЛО-ЛЕКАРСТВЕННОЕ СРЕДСТВО И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2736725C1 |
| АНТИТЕЛА И ИММУНОКОНЪЮГАТЫ И ИХ ПРИМЕНЕНИЯ | 2007 |
|
RU2436796C9 |
| КОНЪЮГАТЫ АНТРАЦИКЛИН-АНТИТЕЛО | 2004 |
|
RU2349343C2 |
| АНТИ-CD79b АНТИТЕЛА И ИММУНОКОНЪЮГАТЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2009 |
|
RU2553566C2 |
| АНТИ-CD79b АНТИТЕЛА И ИММУНОКОНЪЮГАТЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2008 |
|
RU2511410C2 |
| МУЛЬТИСПЕЦИФИЧЕСКИЕ АНТИТЕЛА, АНАЛОГИ АНТИТЕЛ, КОМПОЗИЦИИ И СПОСОБЫ | 2010 |
|
RU2580038C2 |
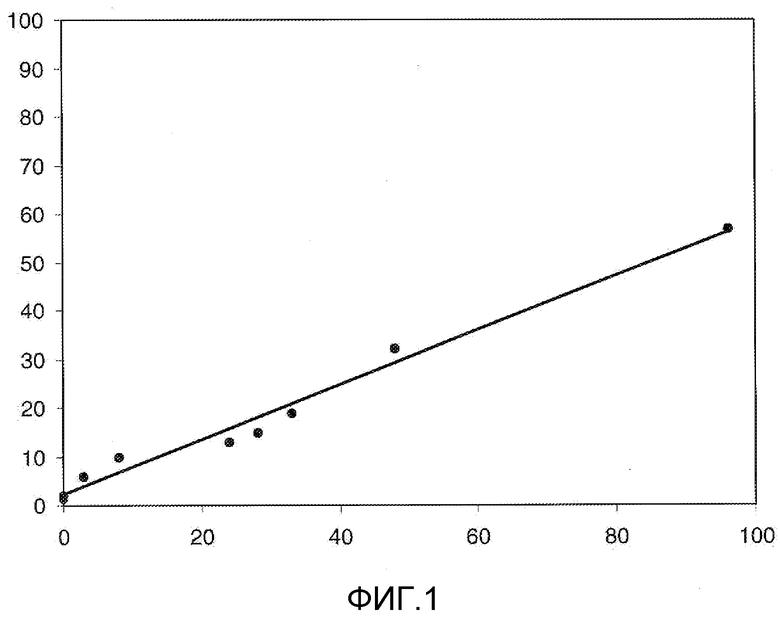
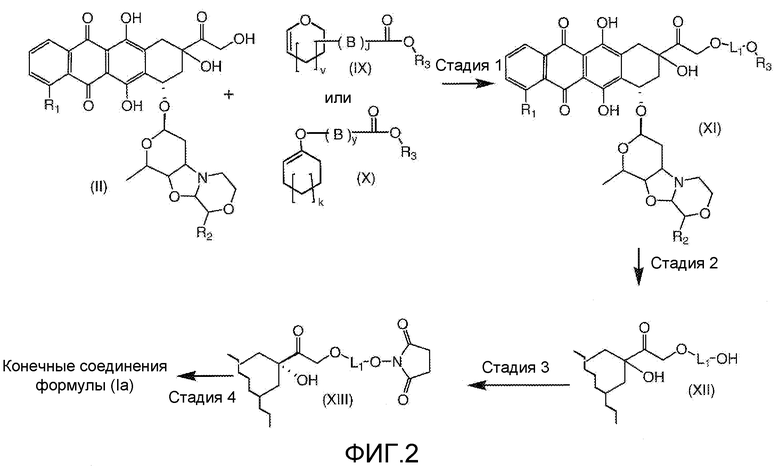
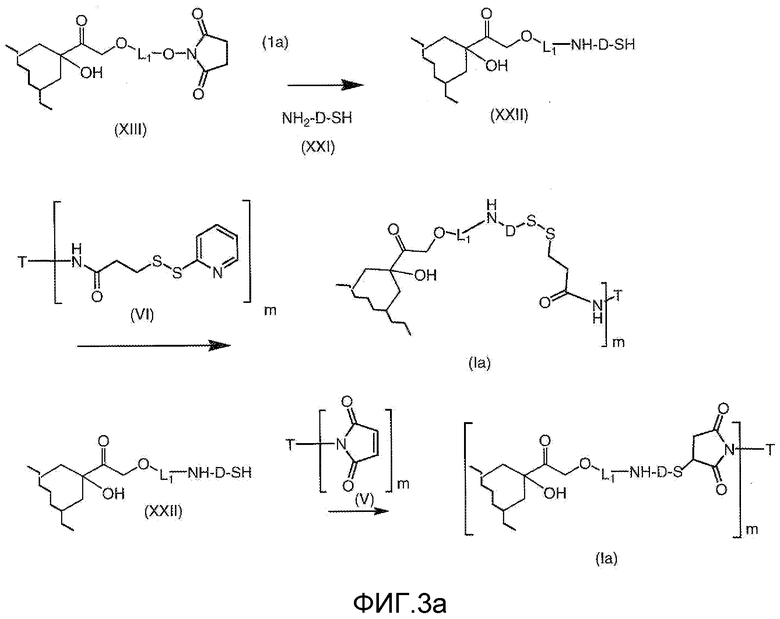
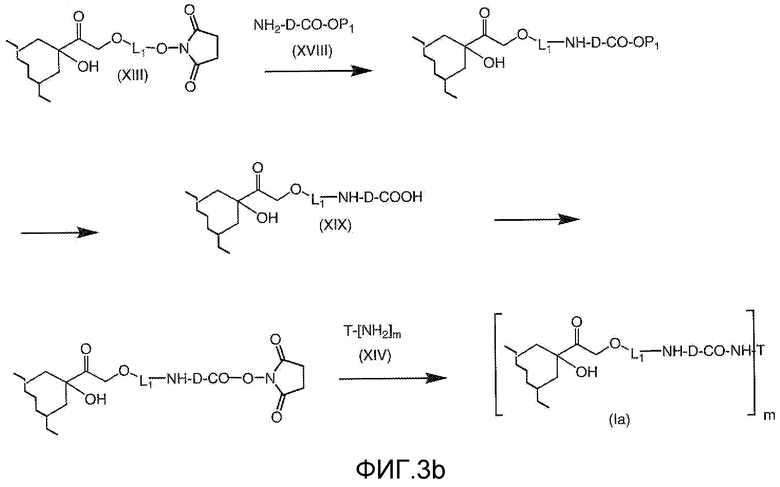
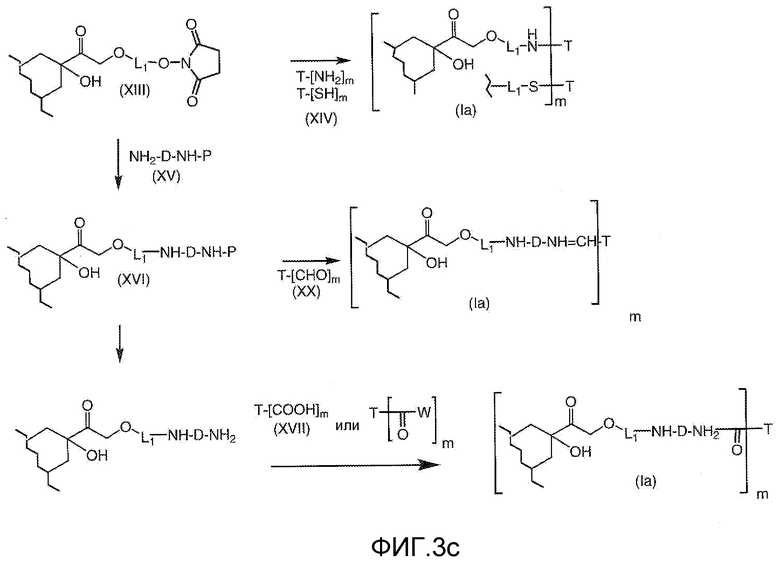
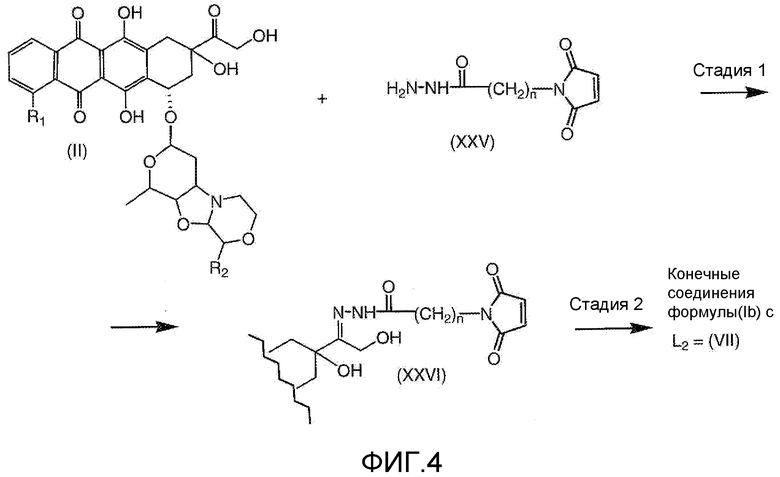
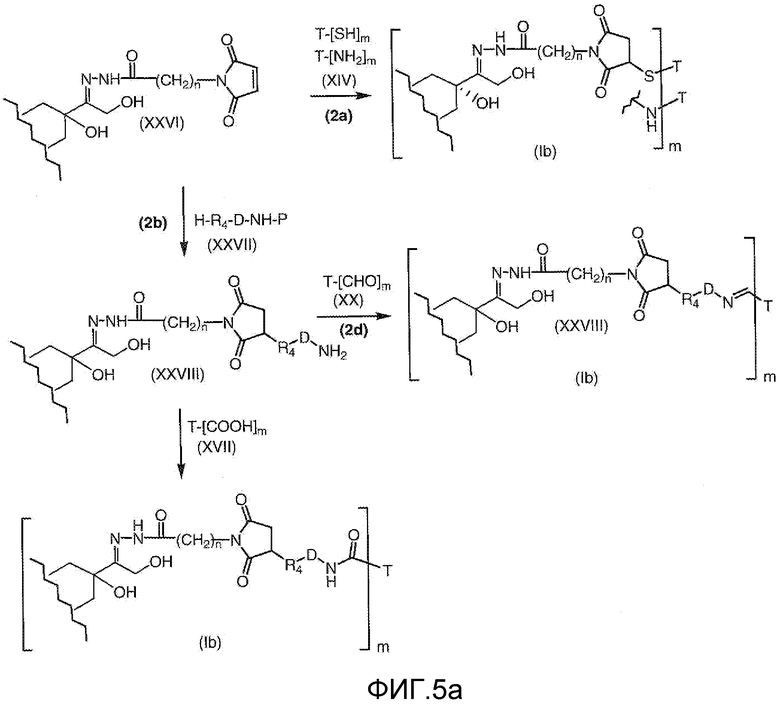
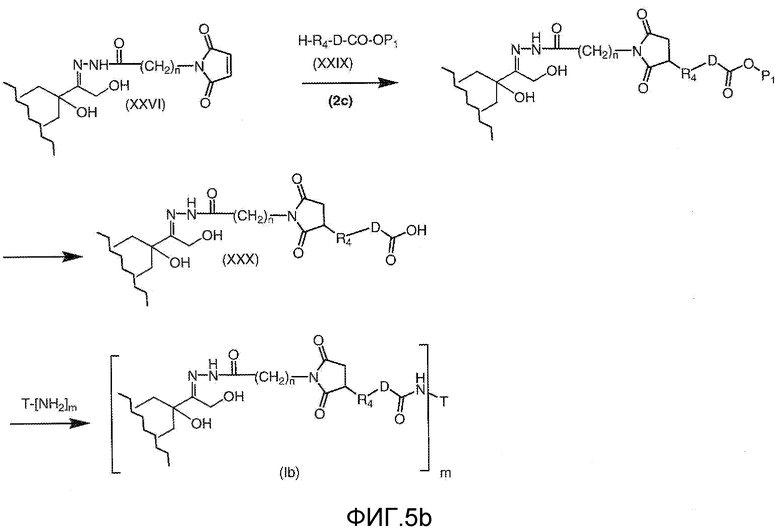
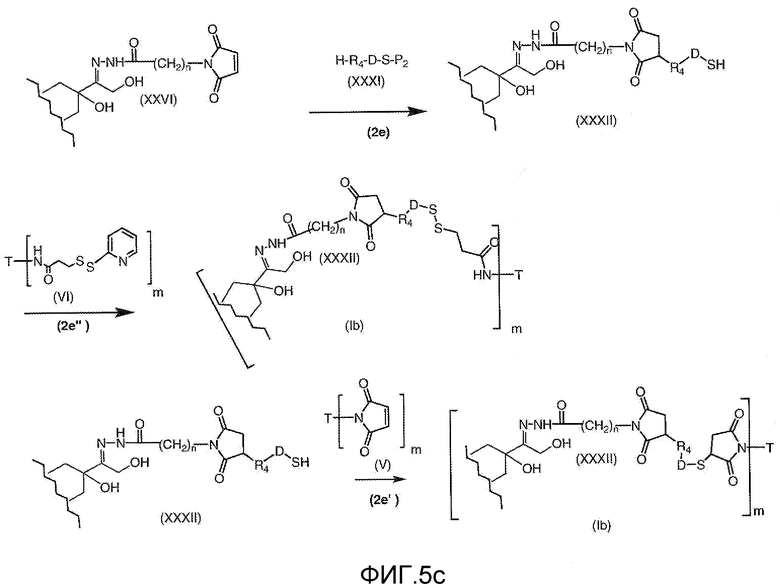
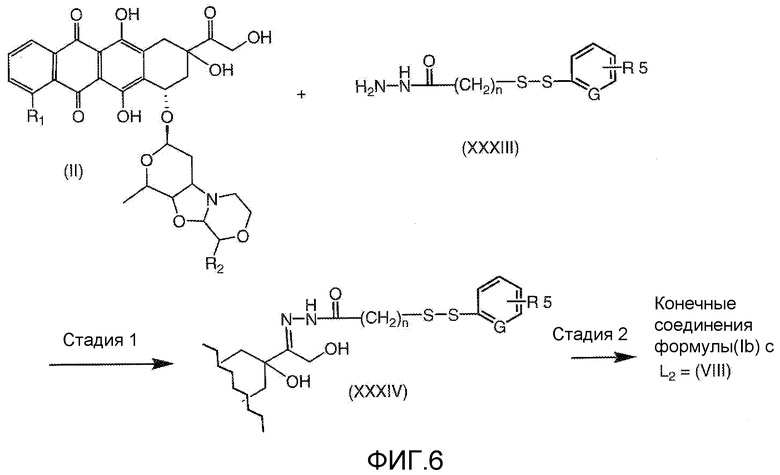
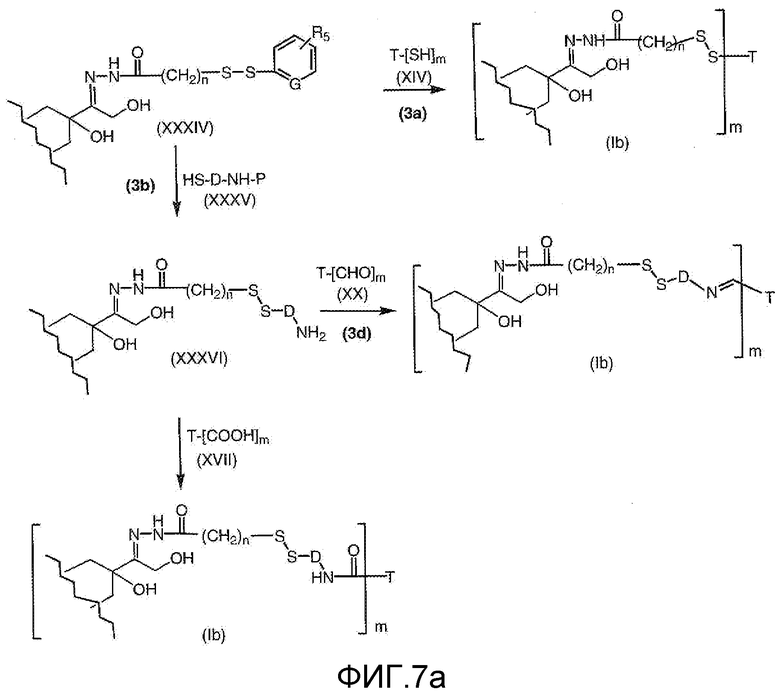
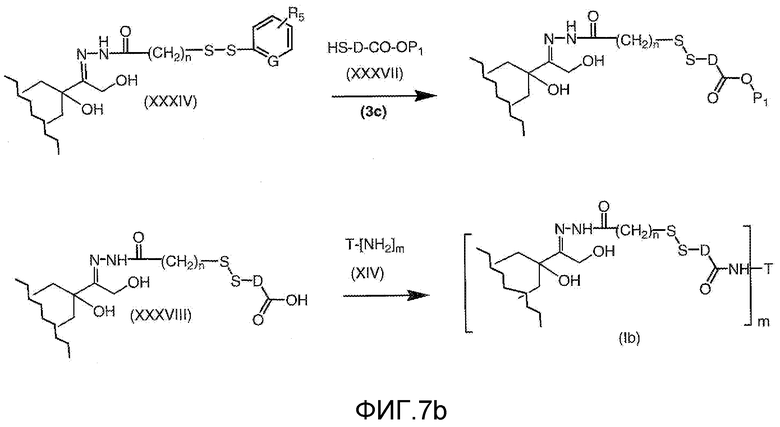
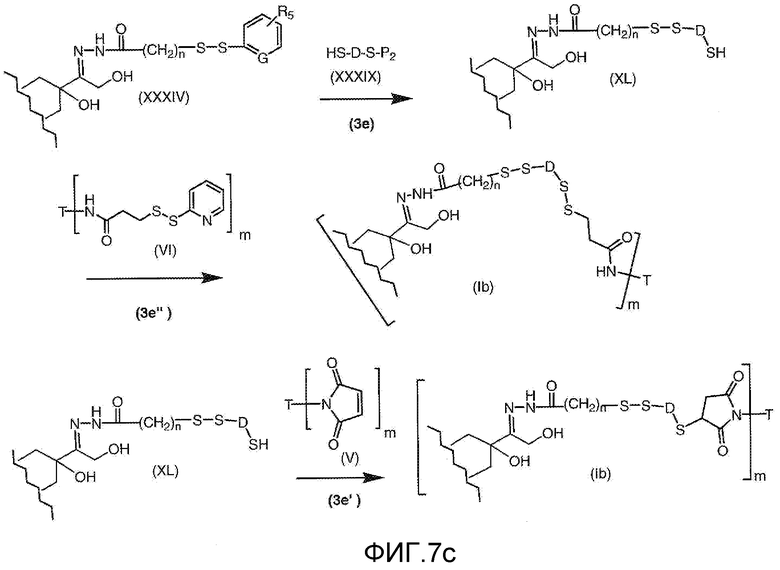
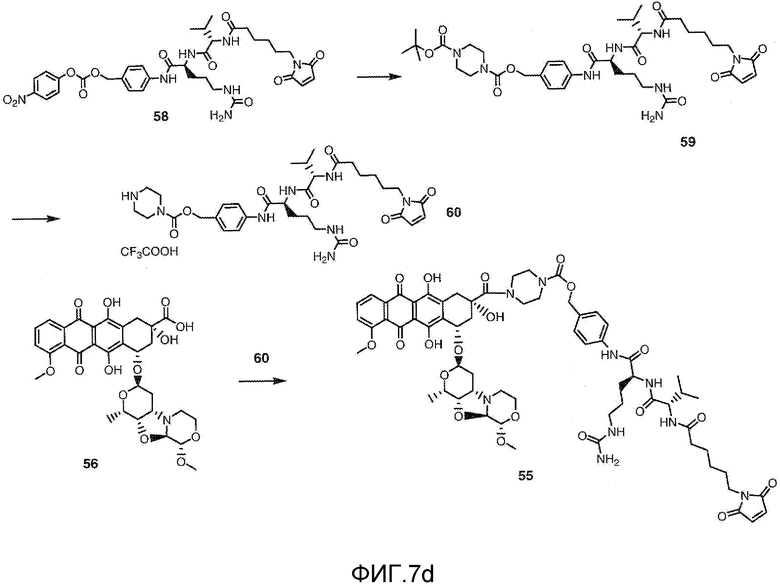
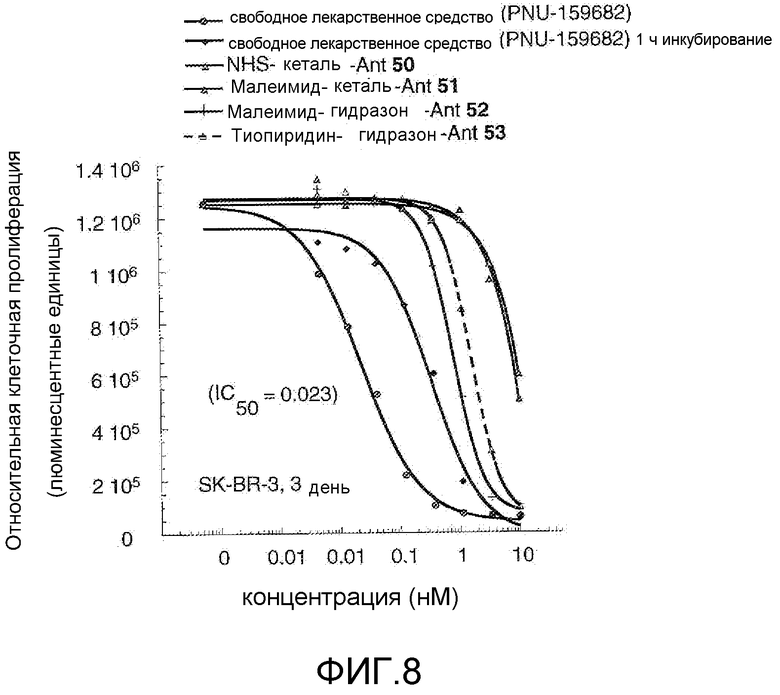
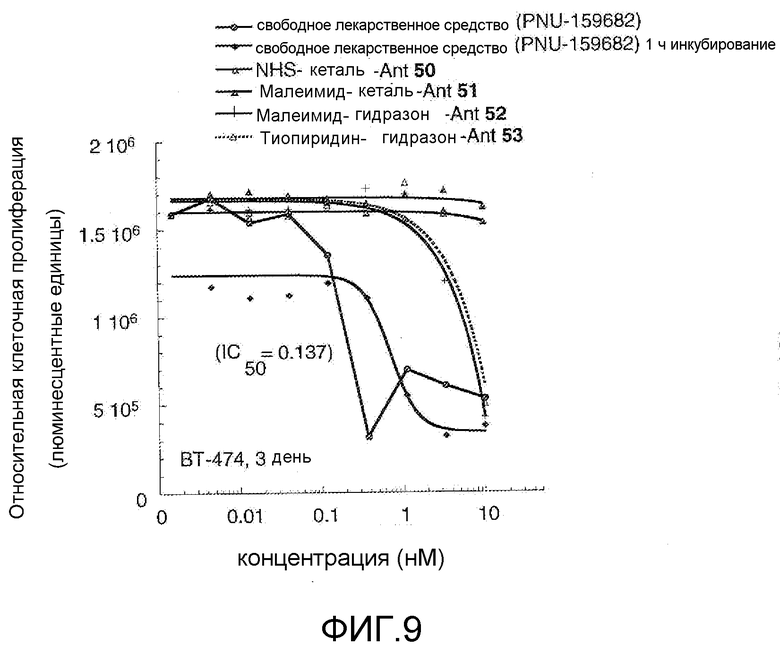
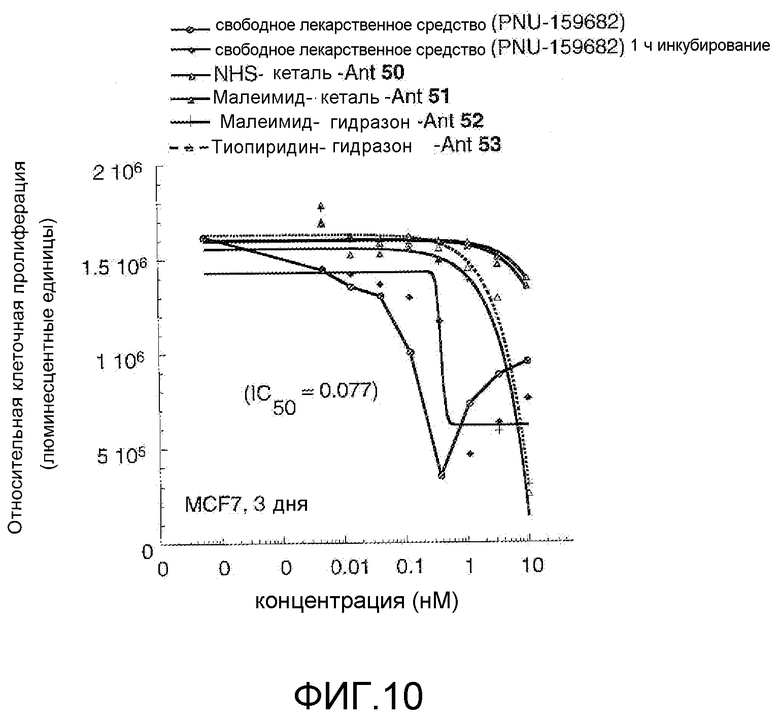
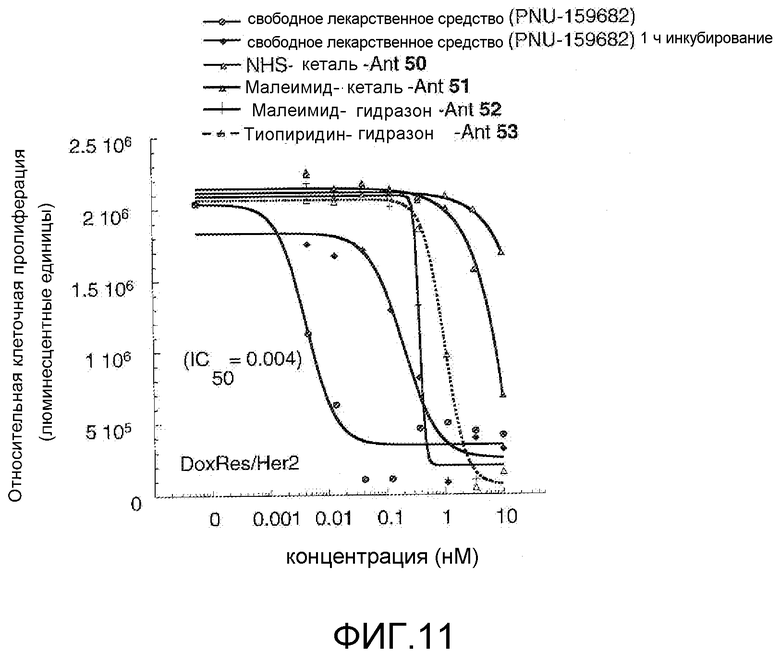
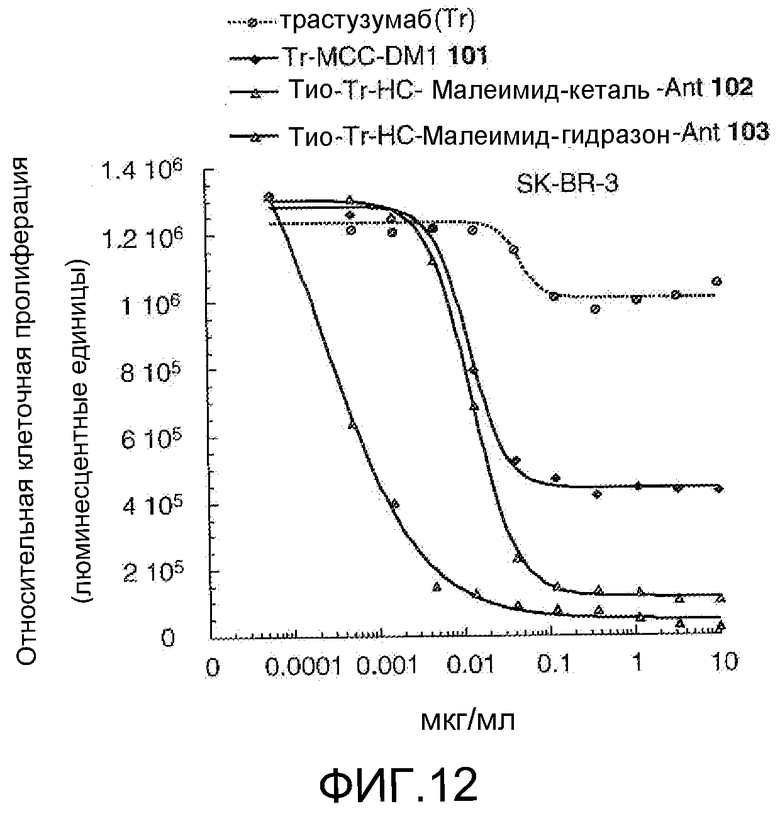
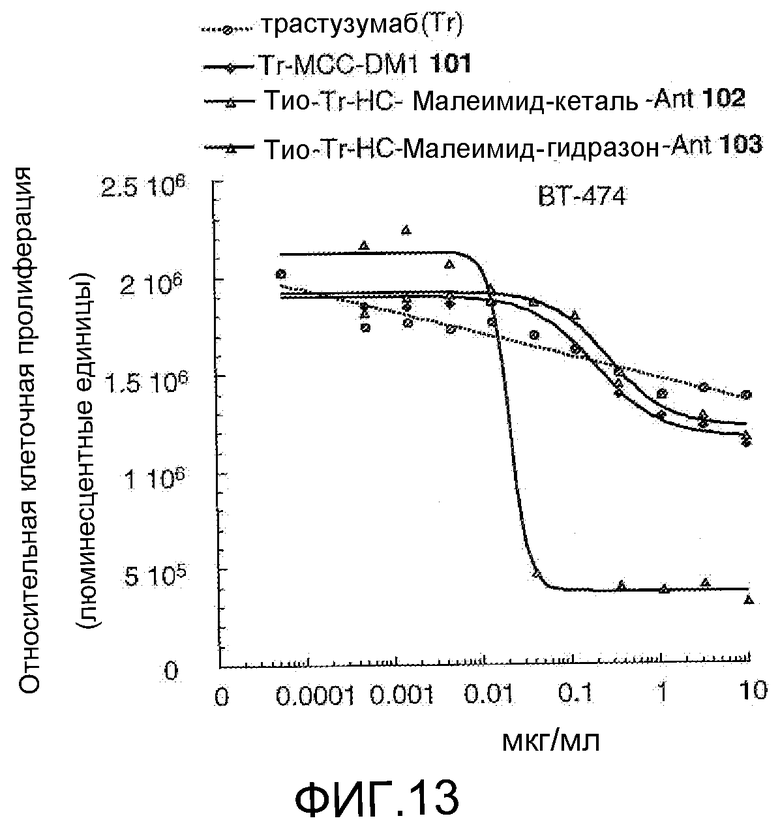

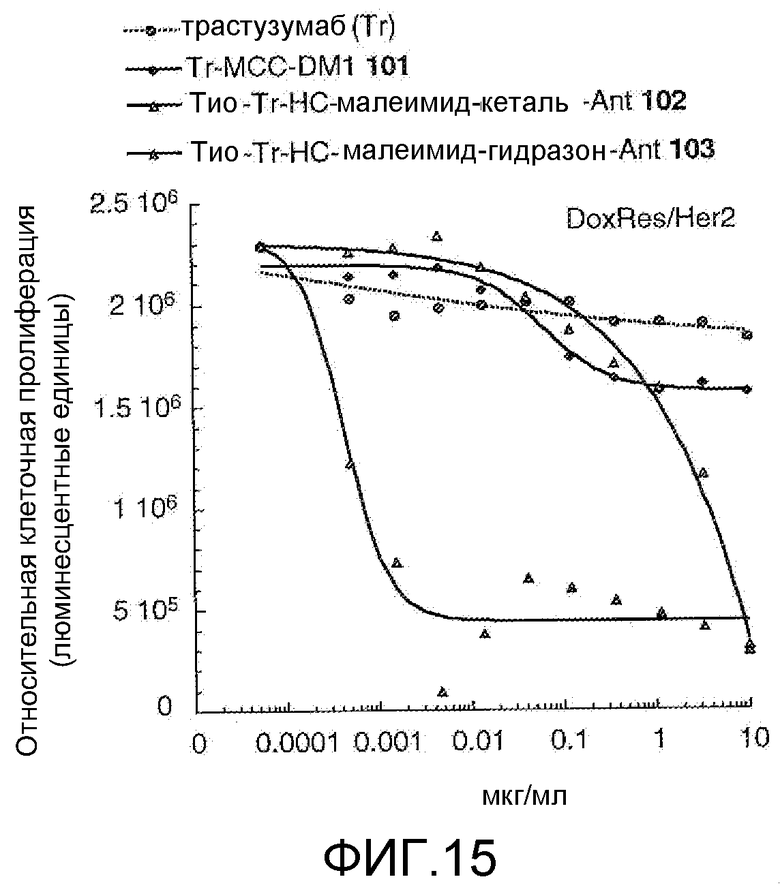
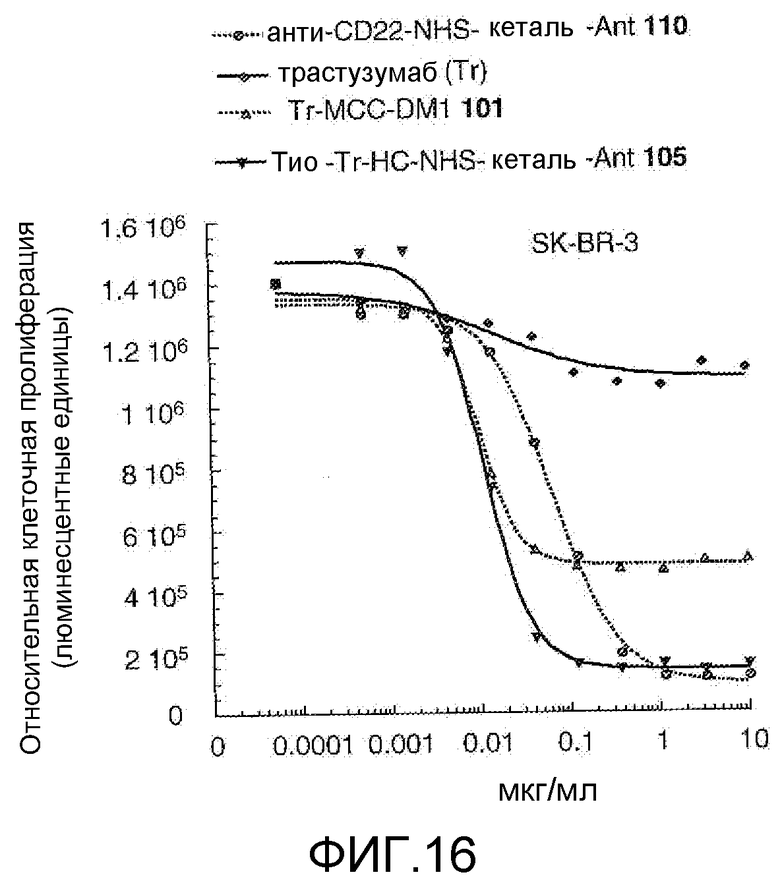
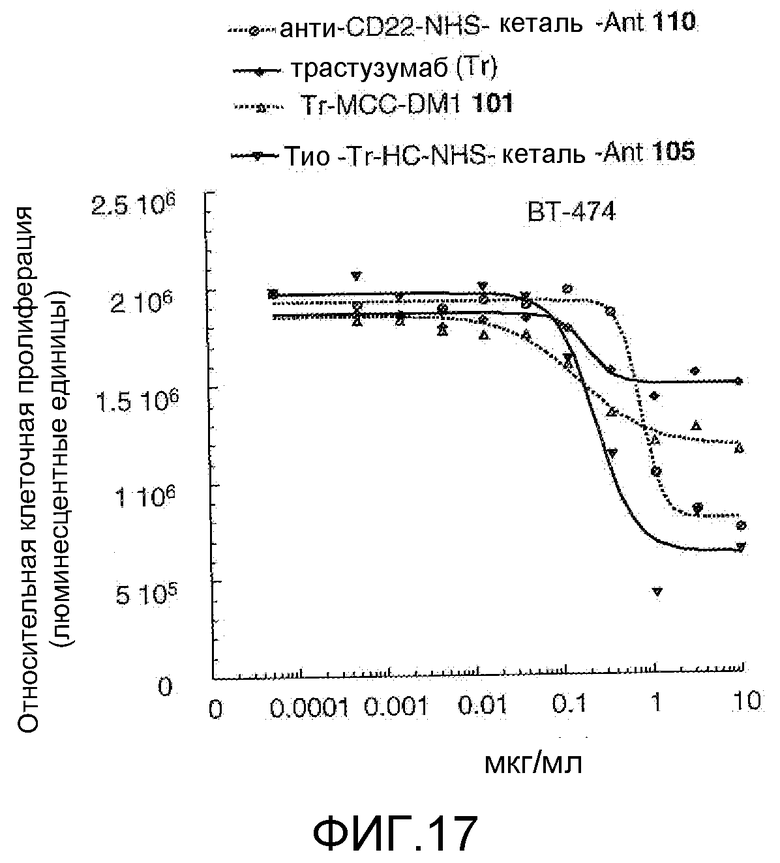
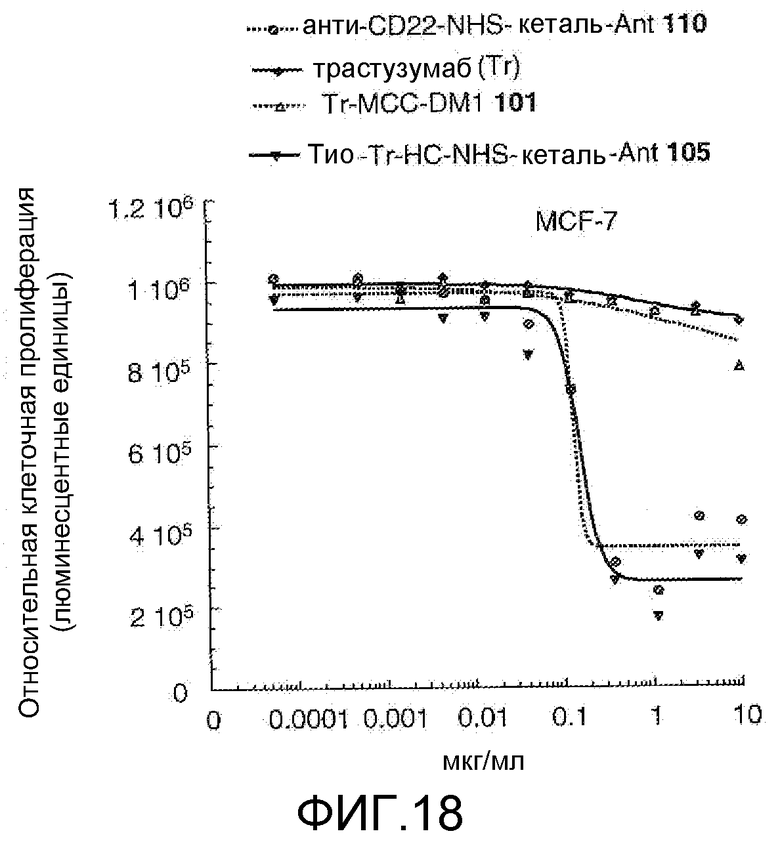
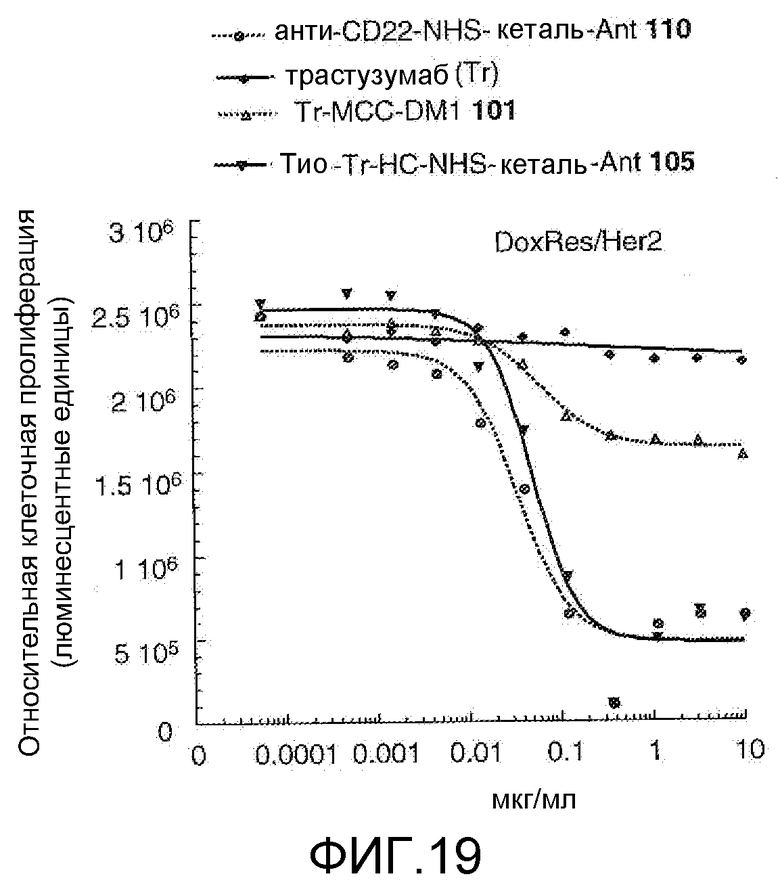

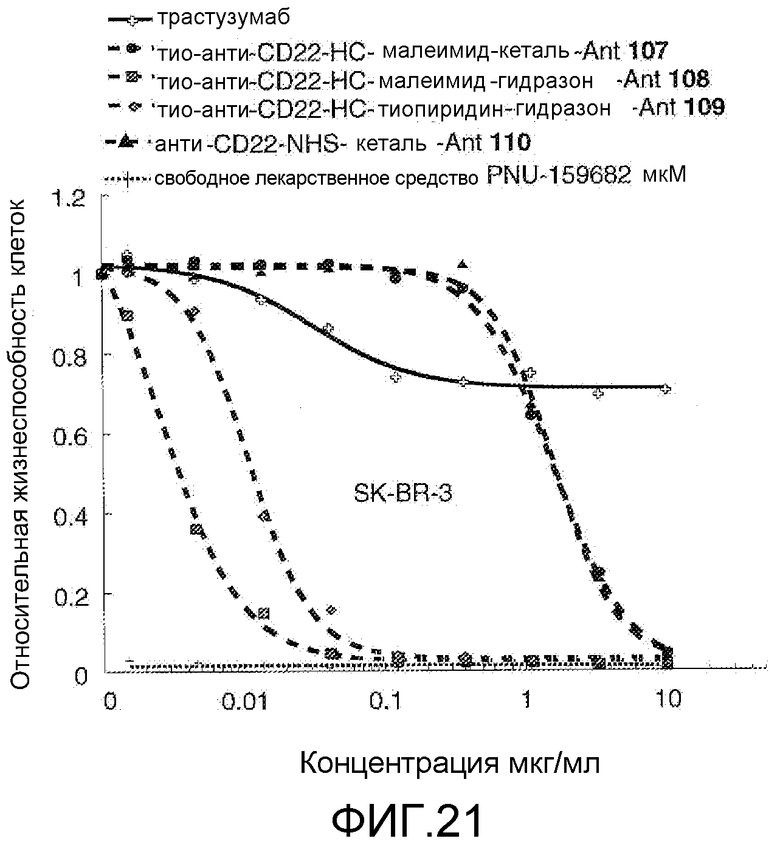
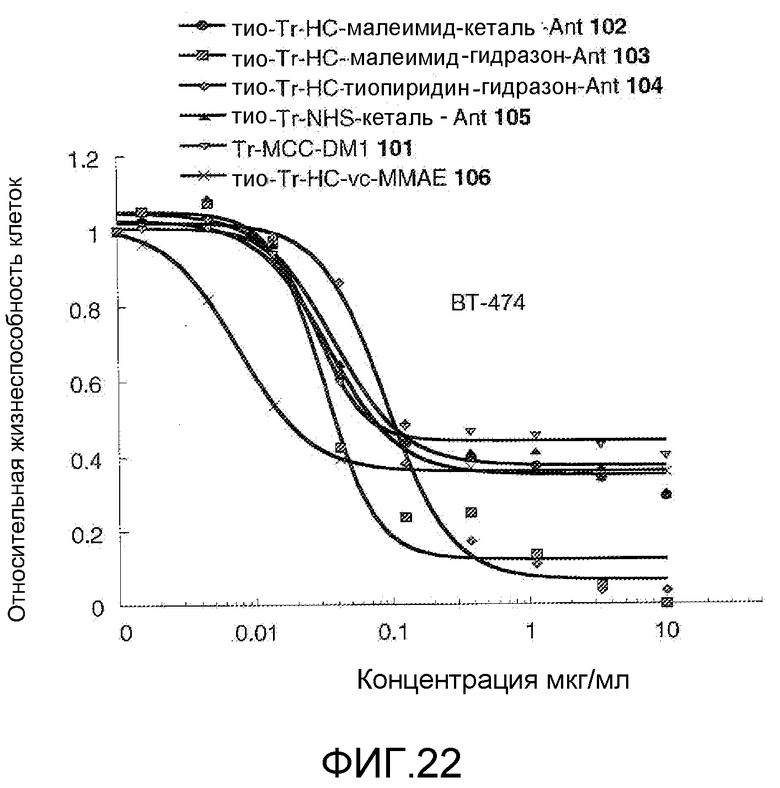
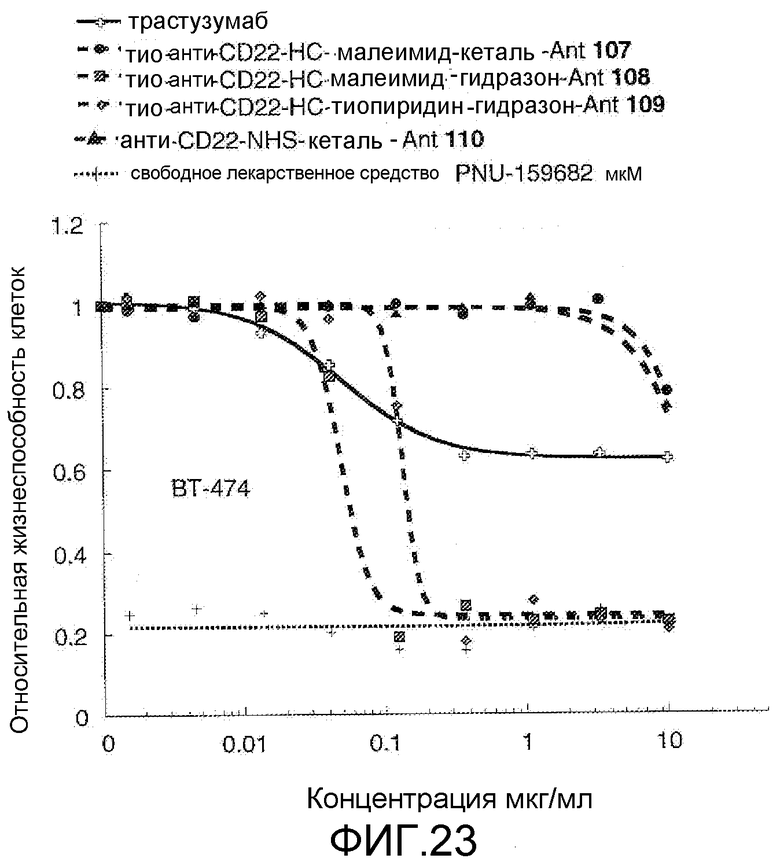
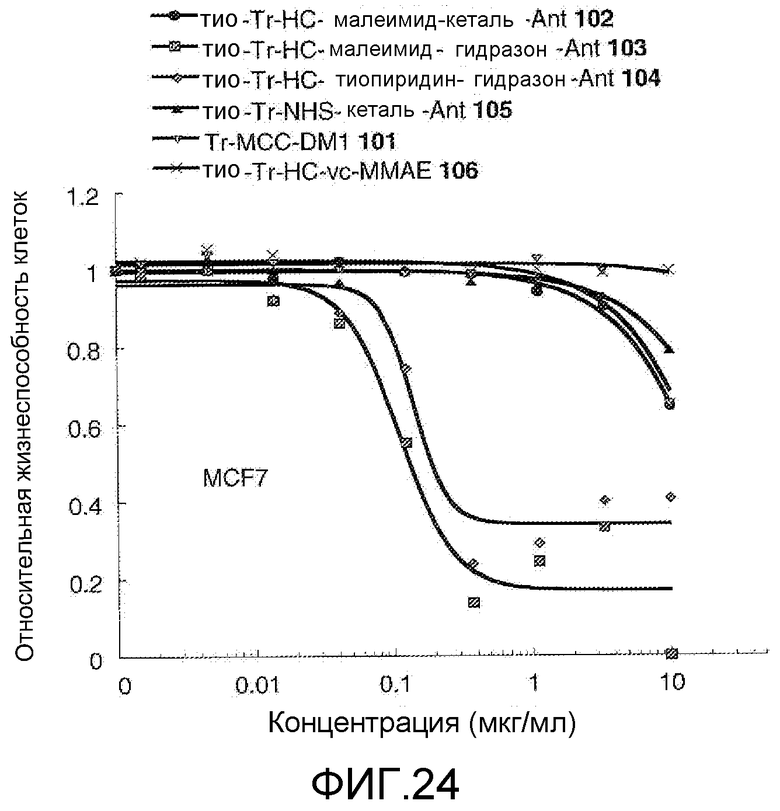
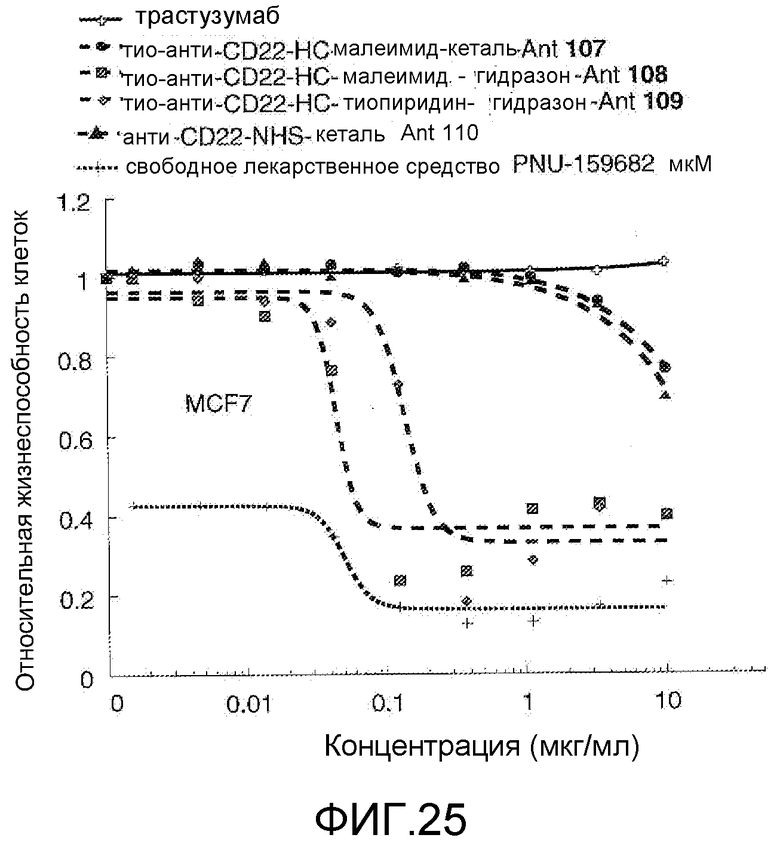
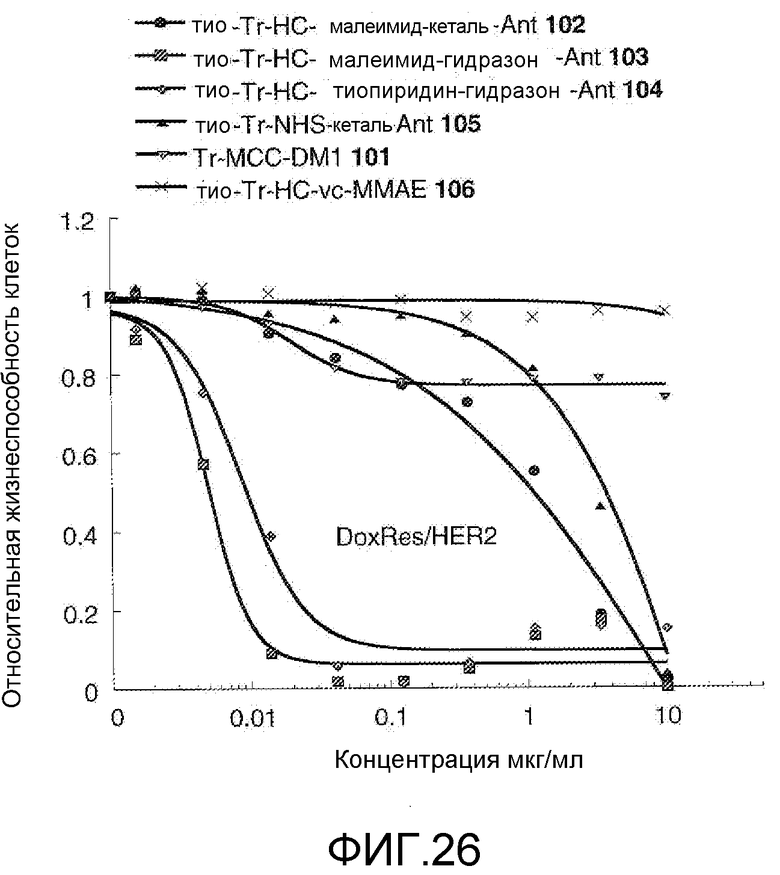
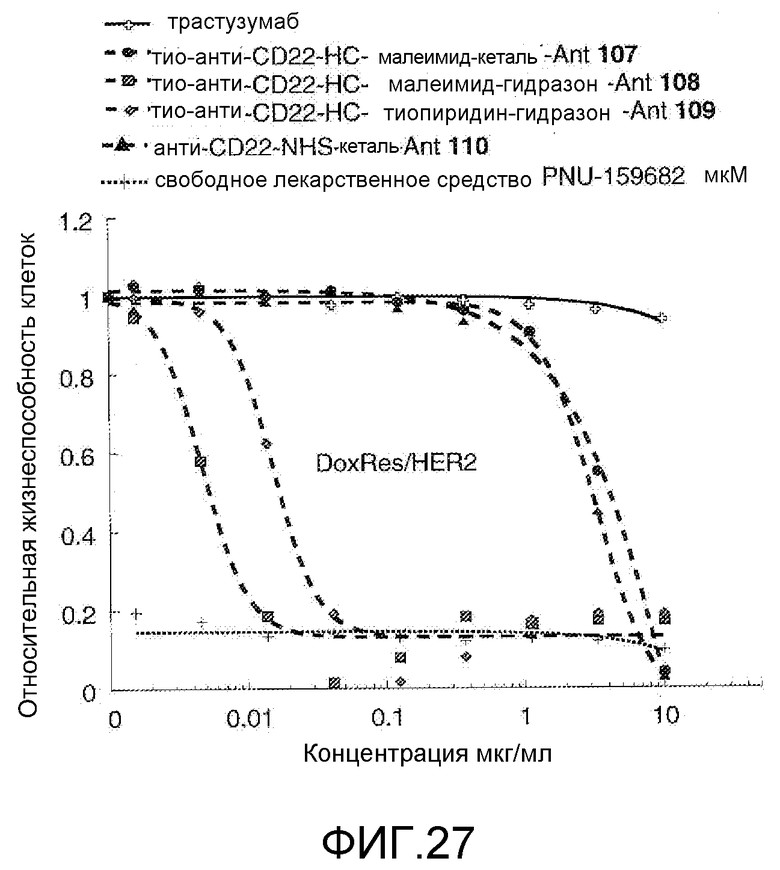
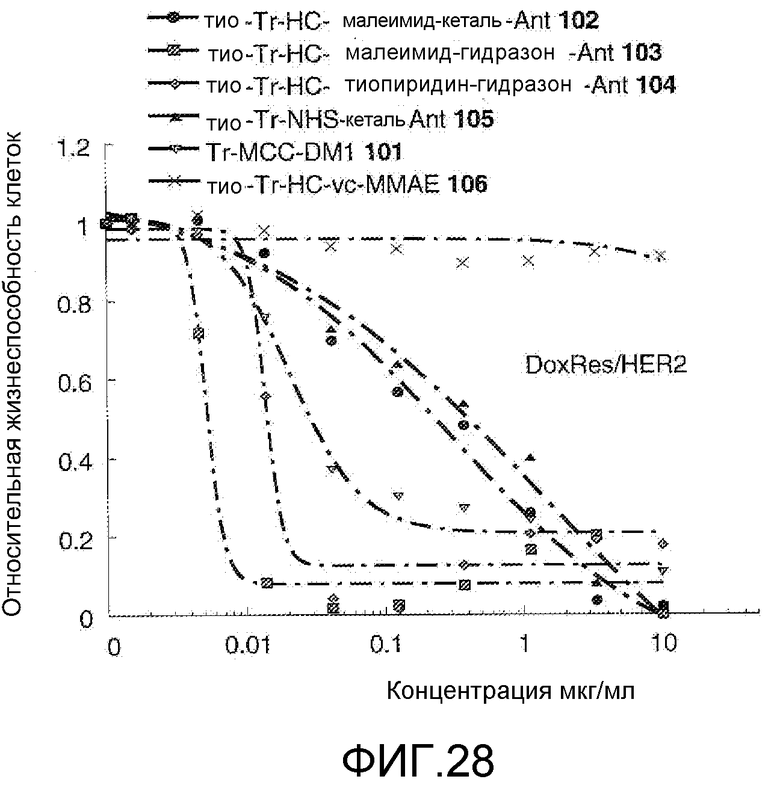
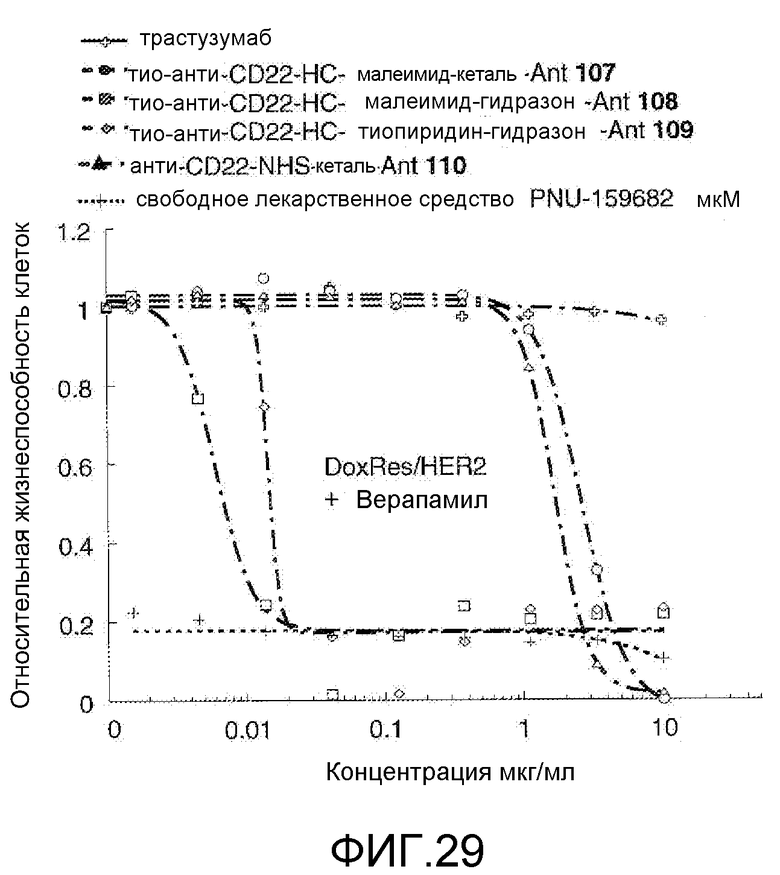
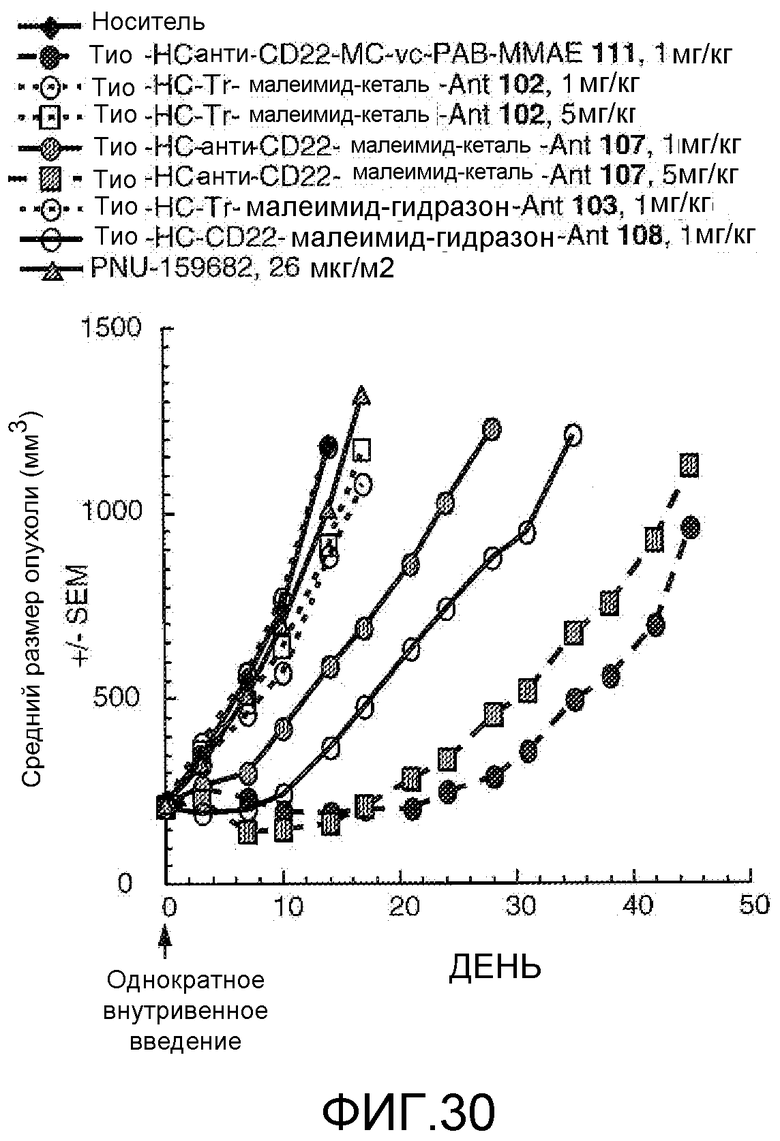
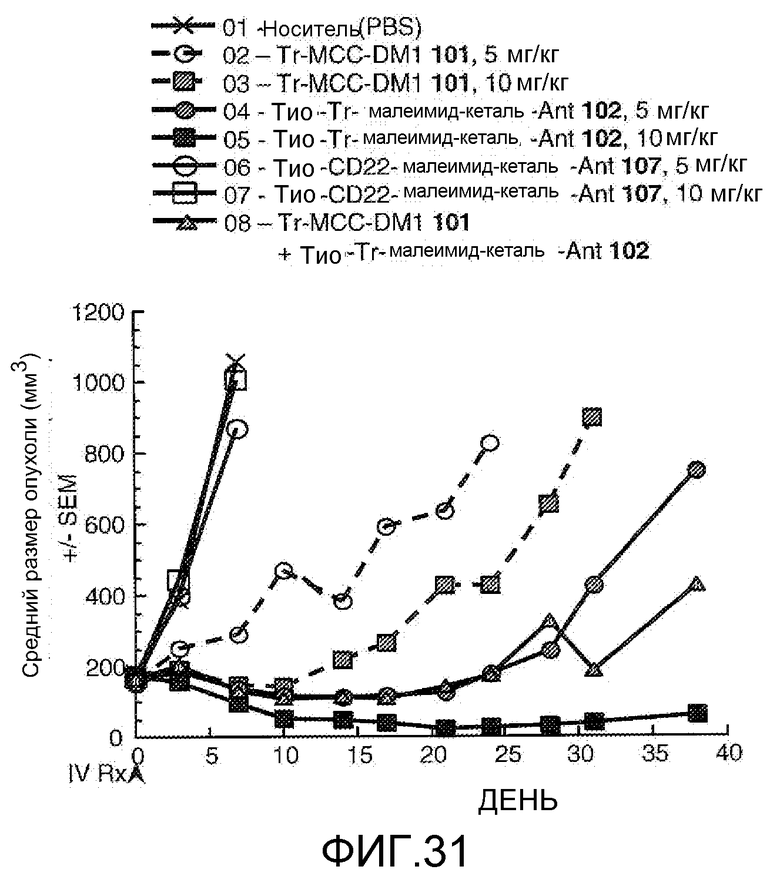
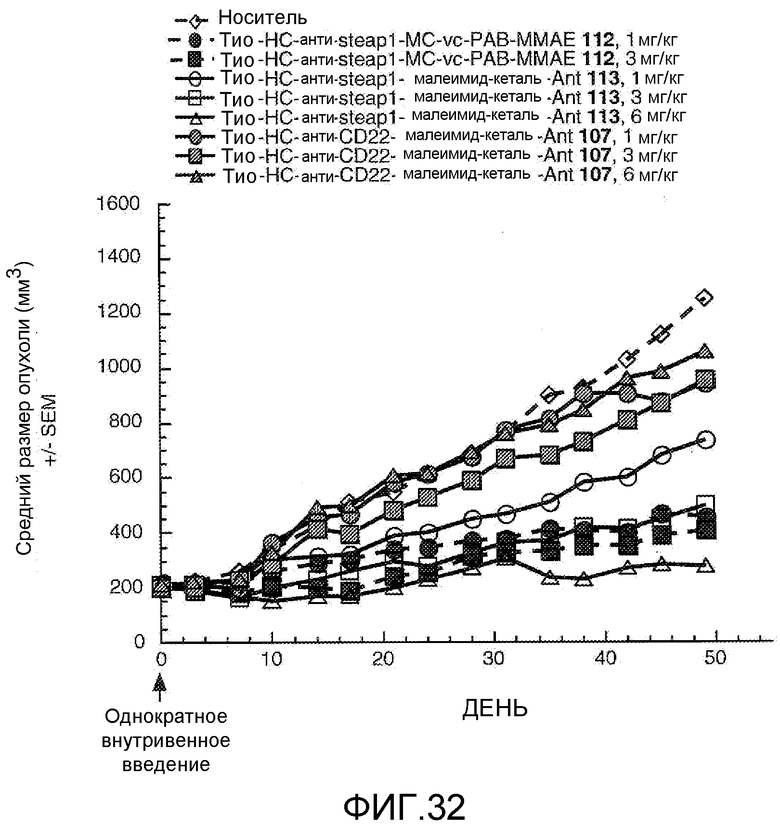
Группа изобретений относится к медицине и касается конъюгатов антрациклинов с носителями, такими как антитела; способам их получения; фармацевтической композиции, содержащей их; и их применению при лечении опухолей млекопитающих. Группа изобретений обеспечивает мощный и селективный эффект уничтожения раковых клеток. 8 н. и 28 з.п. ф-лы, 12 пр., 32 ил., 7 табл.
1. Конъюгат производного антрациклина, выбранный из группы, состоящей из
2-[(1-{2-оксо-2-[(2S,4S)-2,5,12-тригидрокси-7-метокси-4-{[(1S,3R,4аS,9S,9aR,10aS)-9-метокси-1-метилоктагидро-1H-пирано[4',3': 4,5][1,3]оксазоло[2,3-c][1,4]оксазин-3-ил]окси}-6,11-диоксо-1,2,3,4,6,11-гексагидротетрацен-2-ил]этокси}циклогексил)окси]ацетамида,
N-метил-2-[(1-{2-оксо-2-[(2S,4S)-2,5,12-тригидрокси-7-метокси-4-{[(1S,3R,4aS,9S,9aR,10aS)-9-метокси-1-метилоктагидро-1H-пирано[4',3':4,5][1,3]оксазоло[2,3-c][1,4]оксазин-3-ил]окси}-6,11-диоксо-1,2,3,4,6,11-гексагидротетрацен-2-ил]этокси}циклогексил)окси]ацетамида,
N-бензил-2-[(1-{2-оксо-2-[(2S,4S)-2,5,12-тригидрокси-7-метокси-4-{[(1S,3R,4аS,9S,9aR,10aS)-9-метокси-1-метилоктагидро-1H-пирано[4',3':4,5] [1,3]оксазоло[2,3-c][1,4]оксазин-3-ил]окси}-6,11-диоксо-1,2,3,4,6,11-гексагидротетрацен-2-ил]этокси}циклогексил)окси]ацетамида,
N2-(трет-бутоксикарбонил)-N6-{[(1-{2-оксо-2-[(2S,4S)-2,5,12-тригидрокси-7-метокси-4-{[(1S,3R,4aS,9S,9aR,10aS)-9-метокси-1-метилоктагидро-1H-пирано[4',3':4,5][1,3]оксазоло[2,3-с][1,4]оксазин-3-ил]окси}-6,11-диоксо-1,2,3,4,6,11-гексагидротетрацен-2-ил]этокси}циклогексил)окси]ацетил}-L-лизина,
N-метил-2-[(6-{2-оксо-2-[(2S,4S)-2,5,12-тригидрокси-7-метокси-4-{[(1S,3R,4аS,9S,9aR,10aS)-9-метокси-1-метилоктагидро-1H-пирано[4',3':4,5][1,3]оксазоло[2,3-c][1,4]оксазин-3-ил]окси}-6,11-диоксо-1,2,3,4,6,11-гексагидротетрацен-2-ил]этокси}тетрагидро-2H-пиран-2-ил)метокси]ацетамида,
2-[(6-{2-оксо-2-[(2S,4S)-2,5,12-тригидрокси-7-метокси-4-{[(1S,3R,4aS,9S,9aR,10aS)-9-метокси-1-метилоктагидро-1H-пирано[4',3':4,5][1,3]оксазоло[2,3-c][1,4]оксазин-3-ил]окси}-6,11-диоксо-1,2,3,4,6,11-гексагидротетрацен-2-ил]этокси}тетрагидро-2H-пиран-2-ил)метокси]ацетамида,
N-бензил-2-[(6-{2-оксо-2-[(2S,4S)-2,5,12-тригидрокси-7-метокси-4-{[(1S,3R,4aS,9S,9aR,10aS)-9-метокси-1-метилоктагидро-1Н-пирано[4',3':4,5][1,3]оксазоло[2,3-c][1,4]оксазин-3-ил]окси}-6,11-диоксо-1,2,3,4,6,11-гексагидротетрацен-2-ил]этокси}тетрагидро-2H-пиран-2-ил)метокси]ацетамида,
N-ацетил-5-(1-{6-[(2Е)-2-{2-гидрокси-1-[(2S,4S)-2,5,12-тригидрокси-7-метокси-4-{[(1S,3R,4aS,9S,9aR,10aS)-9-метокси-1-метилоктагидро-1H-пирано[4',3':4,5][1,3]оксазоло[2,3-с][1,4]оксазин-3-ил]окси}-6,11-диоксо-1,2,3,4,6,11-гексагидротетрацен-2-ил]этилиден}гидразинил]-6-оксогексил}-2,5-диоксопирролидин-3-ил)-L-цистеина,
N2-(трет-бутоксикарбонил)-N6-(1-{6-[(2Е)-2-{2-гидрокси-1-[(2S,4S)-2,5,12-тригидрокси-7-метокси-4-{[(1S,3R,4aS,9S,9aR,10aS)-9-метокси-1-метилоктагидро-1H-пирано[4',3':4,5][1,3]оксазоло[2,3-с][1,4]оксазин-3-ил]окси}-6,11-диоксо-1,2,3,4,6,11-гексагидротетрацен-2-ил]этилиден}гидразинил]-6-оксогексил}-2,5-диоксопирролидин-3-ил)-L-лизина и
N-ацетил-3-({3-[(2Е)-2-{2-гидрокси-1-[(2S,4S)-2,5,12-тригидрокси-7-метокси-4-{[(1S,3R,4aS,9S,9aR,10aS)-9-метокси-1-метилоктагидро-1H-пирано[4',3':4,5][1,3]оксазоло[2,3-с][1,4]оксазин-3-ил]окси}-6,11-диоксо-1,2,3,4,6,11-гексагидротетрацен-2-ил]этилиден}гидразинил]-3-оксопропил}дисульфанил)-L-аланина.
2. Производное антрациклина формулы (IIc)
Ant-L-(Z)m-X (IIC)
где Ant выбран из структур:
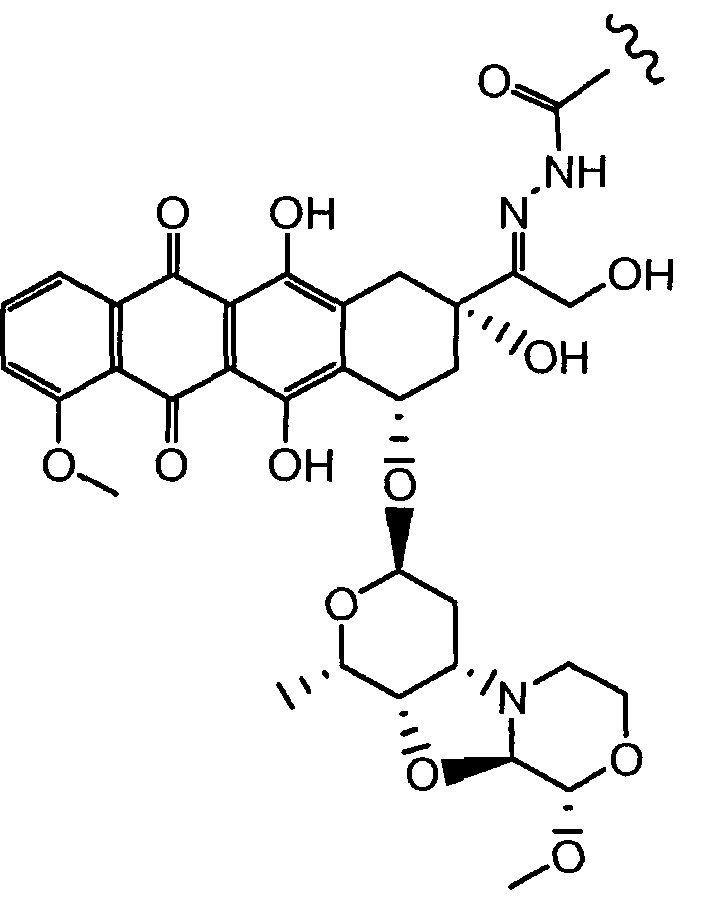
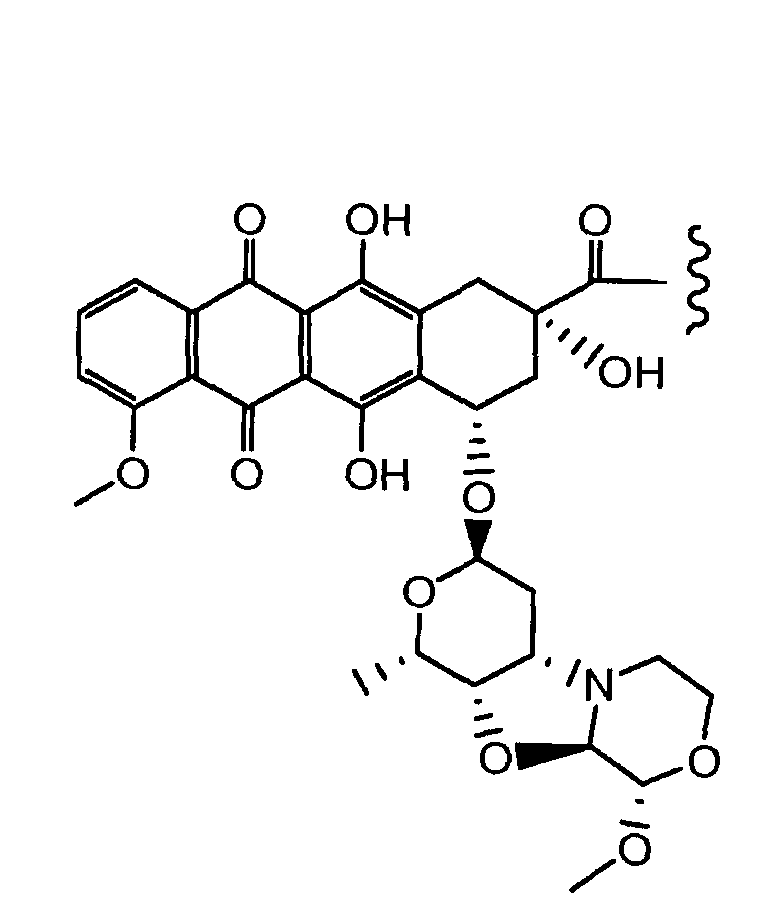
где волнистая линия указывает на присоединение к L;
L представляет собой линкер, выбранный из -CH2O-, -N(R)-, -N(R)m(C1-С12алкилен)-, -N(R)m (C2-C8алкенилен)-, -N(R)m (C2-C8алкинилен)-, -N(R)m (CH2CH2O)n-, и структур:

где волнистые линии указывают на присоединения к Ant и Z; и
Z представляет собой необязательный спейсер, выбранный из -CH2C(О)-, -(N(R))m (C1-С12алкилен)-, -СН2С(О)NR (C1-С12алкилен)-, и структур:
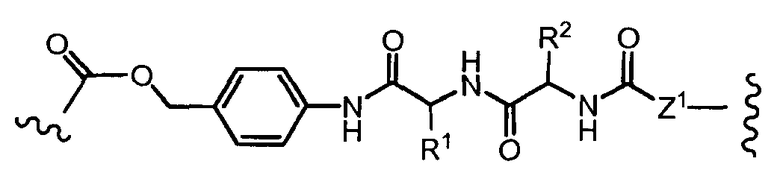

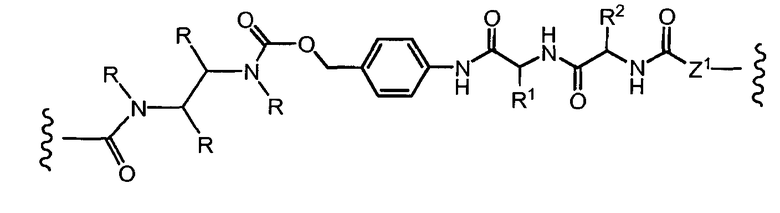
Х представляет собой реакционно-способную функциональную группу, выбранную из малеимида, тиола, амино, бромида, п-толуолсульфоната, йодида, гидроксила, карбоксила, пиридилдисульфида и N-гидроксисукцинимида;
R представляет собой Н, C1-C12алкил или C6-C20арил;
R1 и R2 независимо выбраны из боковых цепей аминокислот;
Z1 выбран из -(C1-С12алкилен)-, -(C2-C8алкенилен)-, -(C2-C8алкинилен)- и - (CH2CH2O)n-,
m равно 0 или 1; и n равно 1-6.
3. Производное антрациклина по п.2, выбранное из структур:
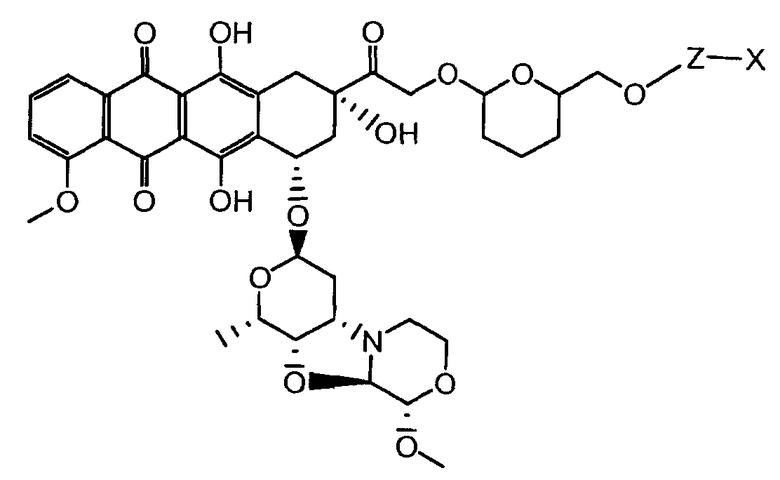
где Z-X выбран из:

4. Производное антрациклина по п.3, имеющее структуру
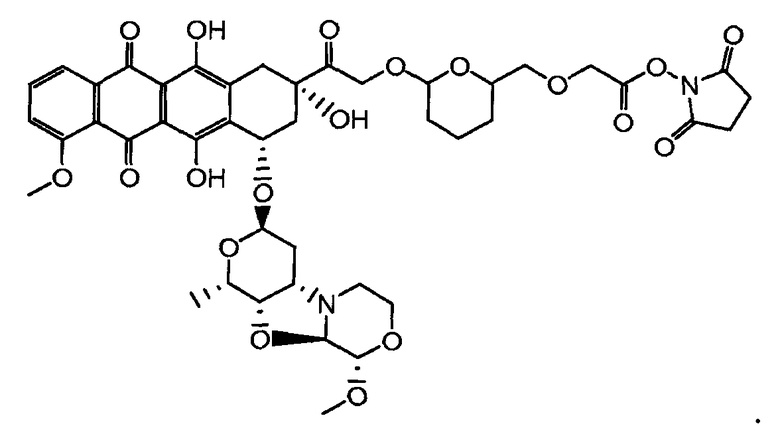
5. Производное антрациклина по п.3, имеющее структуру 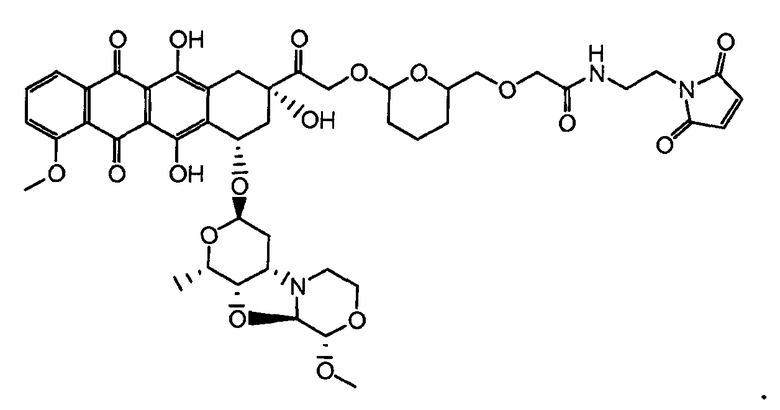
6. Производное антрациклина по п.2, имеющее структуру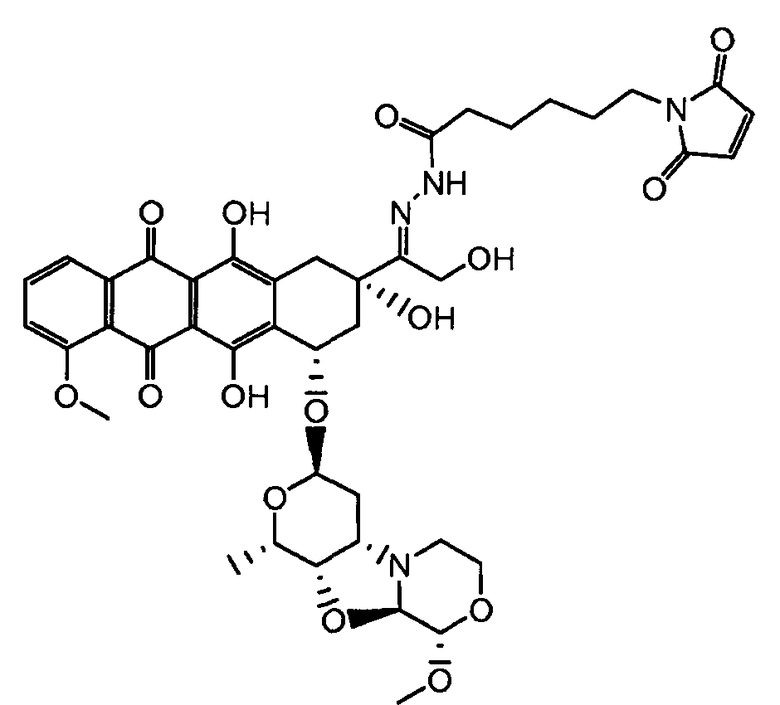
7. Производное антрациклина по п.2, имеющее структуру
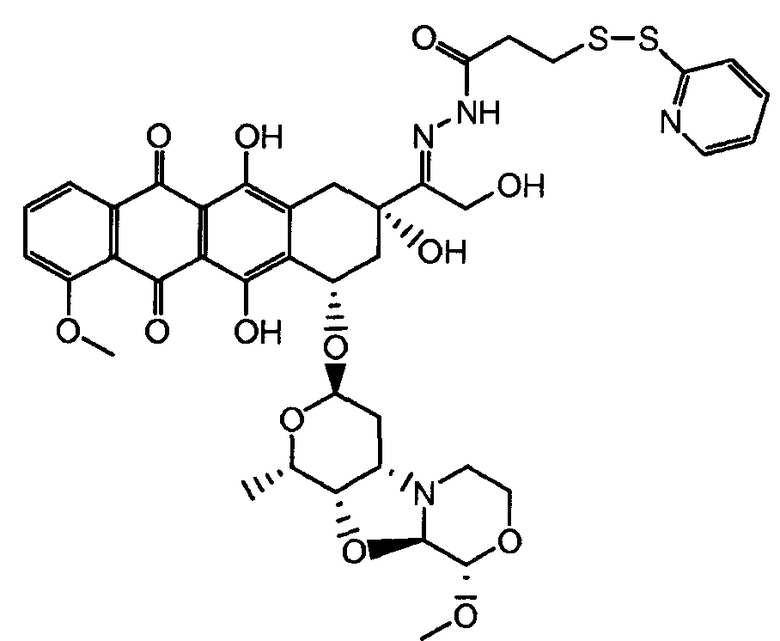
8. Производное антрациклина по п.2, имеющее структуру 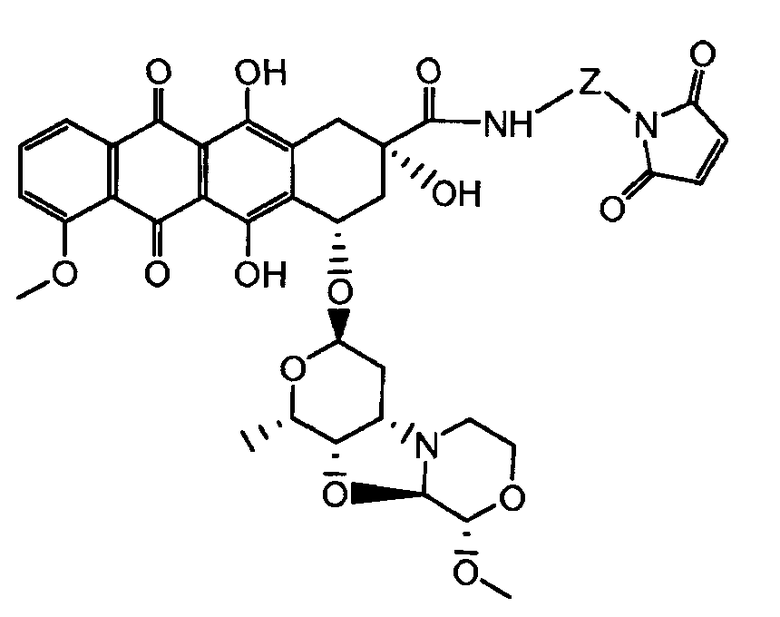
где Z представляет собой C1-С12алкилен.
9. Производное антрациклина по п.8, имеющее структуру
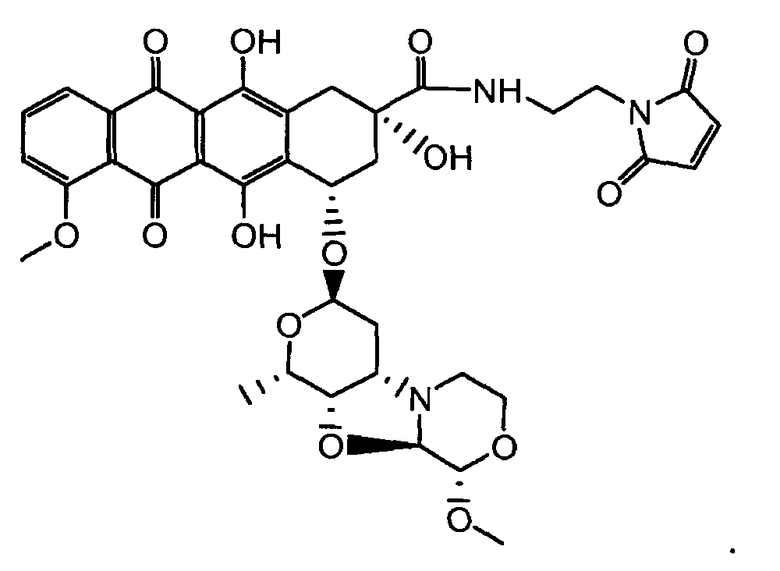
10. Производное антрациклина по п.2, имеющее структуру
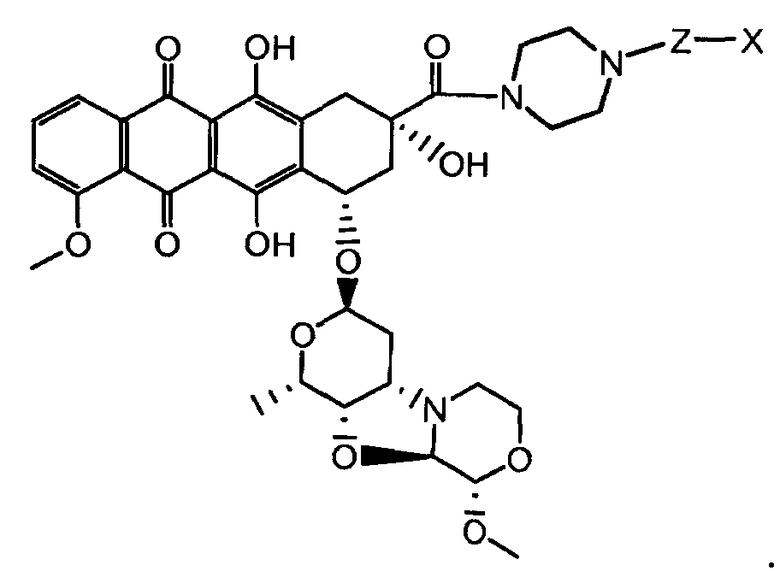
11. Производное антрациклина по п.10, имеющее структуру
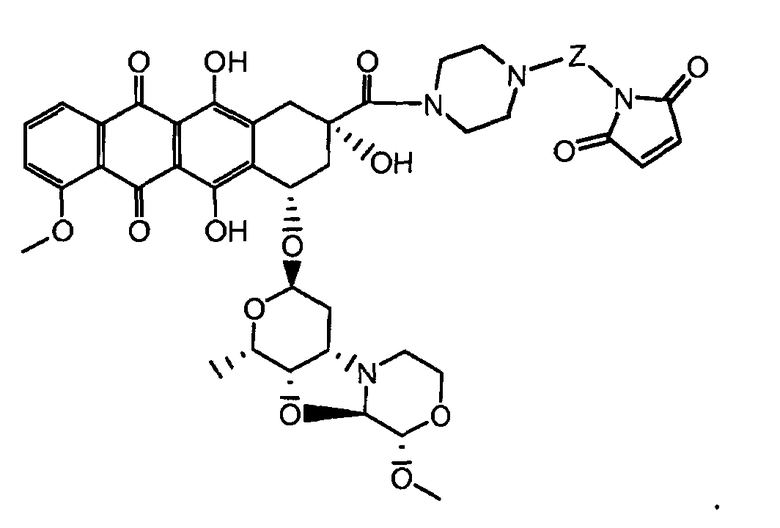
12. Производное антрациклина по п.10, имеющее структуру
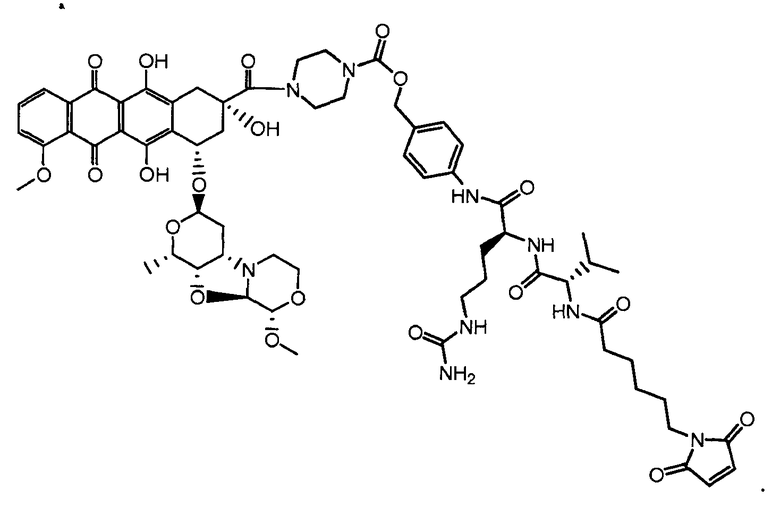
13. Производное антрациклина по п.10, выбранное из структур:

14. Производное антрациклина по п.13, где Z1 представляет собой -(C1-С12алкилен)-.
15. Производное антрациклина по п.13, имеющее структуру:
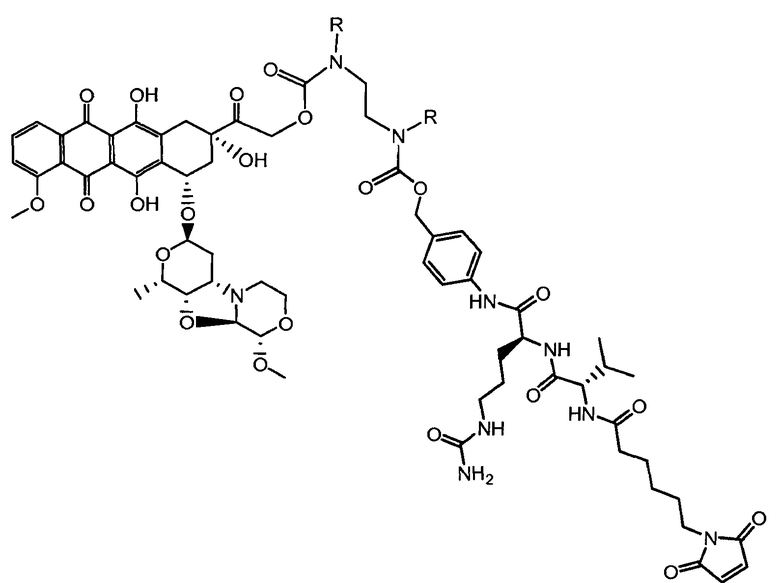
16. Производное антрациклина по п.2, где R1 и R2 независимо выбраны из водорода, метила, изопропила, изобутила, втор-бутила, бензила, п-гидроксибензила, -CH2OH, -CH(OH)CH3, -CH2CH2SCH3, CH2CONH2, -CH2COOH, -CH2CH2CONH2, -CH2CH2COOH, - (CH2)3NHC(=NH)NH2, -(CH2)3NH2, - (CH2)3NHCOCH3, - (CH2)3NHCHO, - (CH2)4NHC(=NH)NH2, (CH2)4NH2, - (CH2)4NHCOCH3, - (CH2)4NHCHO, - (CH2)3NHCONH2, (CH2)4NHCONH2, -CH2CH2CH(OH)CH2NH2, 2-пиридилметил-, 3-пиридилметил-, 4-пиридилметил-, фенила, циклогексила, и следующих структур:
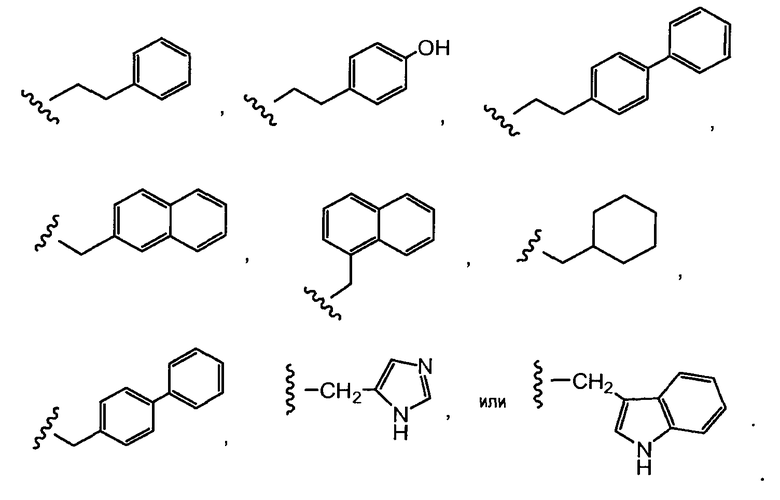
17. Конъюгат антитела с лекарственным средством, содержащее антитело, ковалентно присоединенное посредством линкера L и необязательного спейсера Z к одной или нескольким лекарственным группам производного антрациклина D, имеет формулу
(D-L-(Z)m)p-Ab,
или его фармацевтически приемлемая соль, где:
Ab представляет собой антитело;
D представляет собой производное антрациклина, выбранное из структур:

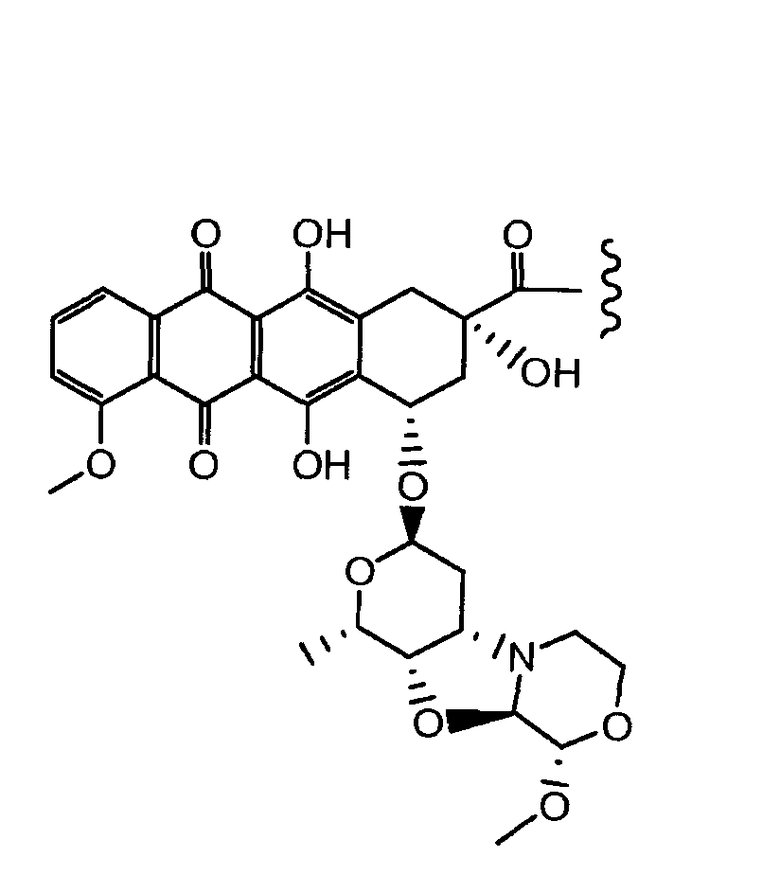
где волнистая линия указывает на присоединение к L;
L представляет собой линкер, выбранный из -CH2O-, -N(R)-, -N(R)m(C1-С12алкилен)-, -N(R)m(C2-C8алкенилен)-, -N(R)m(C2-С8алкинилен)-, -N(R)m(CH2CH2O)n-, и структур:

где волнистые линии указывают на присоединения к D и Z; и
Z представляет собой необязательный спейсер, выбранный из
-CH2C(O)-, -(N(R))m(C1-С12алкилен)-X1-, -CH2C(O)NR (C1-С12алкилен)-X1-, и структур:
R представляет собой Н, C1-C12алкил или C6-C20арил;
R1 и R2 независимо выбраны из боковых цепей аминокислот;
Z1 выбран из -X1-, -S-, -CH2C(O)-, - (CH2CH2O)nCH2C(O)-, (CH2CH2O)nCH2-X1-, -(C1-С12алкилен)-X1- и -(C1-С12алкилен)-;
X1 представляет собой
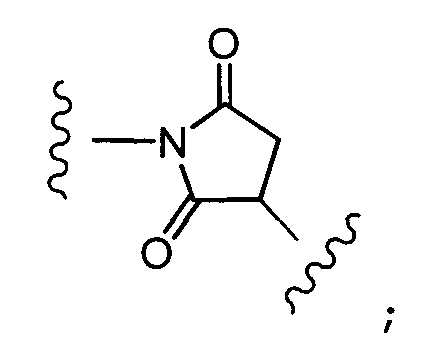
m равно 0 или 1;
n равно 1-6; и
p равно целому числу от 1 до 8.
18. Конъюгат антитела с лекарственным средством по п.17, где R1 и R2 независимо выбраны из водорода, метила, изопропила, изобутила, втор-бутила, бензила, п-гидроксибензила, -CH2OH, CH(OH)CH3, -CH2CH2SCH3, -CH2CONH2, -CH2COOH, -CH2CH2CONH2, CH2CH2COOH, -(CH2)3NHC(=NH)NH2, -(CH2)3NH2, -(CH2)3NHCOCH3, (CH2)3NHCHO, -(CH2)4NHC(=NH)NH2, -(CH2)4NH2- (CH2)4NHCOCH3, (CH2)4NHCHO, -(CH2)3NHCONH2, -(CH2)4NHCONH2, -CH2CH2CH(OH)CH2NH2, 2-пиридилметил-, 3-пиридилметил-, 4-пиридилметил-, фенила, циклогексила, и следующих структур:
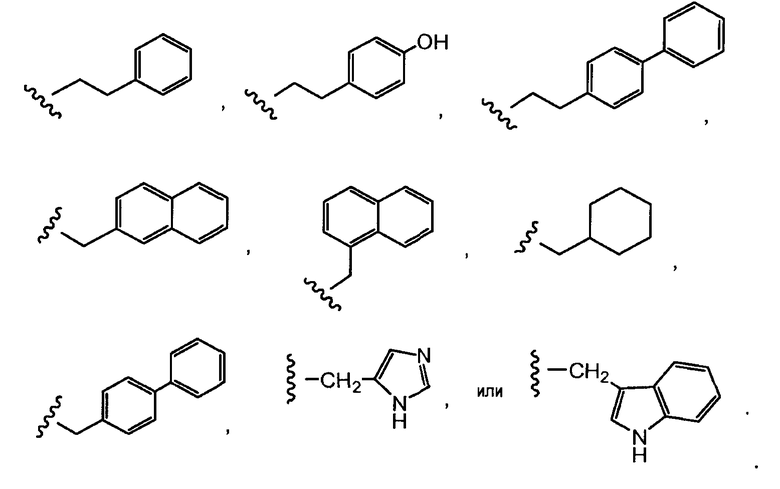
19. Конъюгат антитела с лекарственным средством по п.17, где спейсер Z содержит пара-аминобензилоксикарбонильную (РАВ) группу.
20. Конъюгат антитела с лекарственным средством по п.17, где линкер-спейсерная единица L-Z содержит малеимидокапроильную (МС) группу.
21. Конъюгат антитела с лекарственным средством по п.17, где Z содержит группу валин-цитруллин (vc).
22. Конъюгат антитела с лекарственным средством по п.17, где Ab представляет собой антитело, которое связывается с одним или несколькими антигенами, связанными с опухолью, или рецепторами клеточной поверхности, выбранными из (1)-(36):
(1) BMPR1B (рецептора костного морфогенного белка типа IB);
(2) Е16 (LAT1, SLC7A5);
(3) STEAP1 (шестого трансмембранного эпителиального антигена предстательной железы);
(4) 0772Р (CA125, MUC16);
(5) MPF (MPF, MSLN, SMR, мегакариоцитарный потенцирующий фактор, мезотелин);
(6) Napi3b (NAPI-3B, NPTIIb, SLC34A2, растворенный носитель семейства 34 (натрия фосфат), компонент 2, тип II натрий-зависимый переносчик фосфата 3b);
(7) Sema 5b (FLJ10372, KIAA1445, Мм.42015, SEMA5B, SEMAG, Семафорин 5b Hlog, sema домен, семь тромбоспондиновых повторов (тип 1 и подобные типу 1), трансмембранный домен (ТМ) и короткий цитоплазматический домен, (семафорин) 5B);
(8) PSCA hlg (2700050C12Rik, C530008016Rik, RIKEN кДНК 2700050C12, RIKEN кДНК 2700050С12 ген);
(9) ETBR (рецептор эндотелина тип В);
(10) MSG783 (RNF124, гипотетический белок FLJ20315);
(11) STEAP2 (HGNC_8639, IPCA-1, PCANAP1, STAMP1, STEAP2, STMP, ген, связанный с раком предстательной железы, 1, белок, связанный с раком предстательной железы, 1, шестой трансмембранный эпителиальный антиген предстательной железы 2, шестой трансмембранный белок предстательной железы);
(12) TrpM4 (BR22450, FLJ20041, TRPM4, TRPM4B, транзиторный рецептор катионного канала потенциала, подсемейство М, компонент 4);
(13) CRIPTO (CR, CR1, CRGF, CRIPTO, TDGF1, тератокарциномный фактор роста);
(14) CD21 (CR2 (рецептор комплемента 2) или C3DR (C3d/рецептор вируса Эпштейна-Барр) или Hs 73792);
(15) CD79b (CD79B, CD79β, IGb (иммуноглобулин-связанный бета), B29);
(16) FcRH2 (IFGP4, IRTA4, SPAP1A (SH2 домен, содержащий якорный белок фосфатазы la), SPAP1B, SPAP1C);
(17) HER2;
(18) NCA;
(19) MDP;
(20) IL20Rα;
(21) Бревикан;
(22) EphB2R;
(23) ASLG659;
(24) PSCA;
(25) GEDA;
(26) BAFF-R (рецептор фактора, активирующего В-клетки, BLyS рецептор 3, BR3);
(27) CD22 (В-клеточный рецептор CD22-B изоформа);
(28) CD79a (CD79A, CD79α, иммуноглобулин-связанный альфа);
(29) CXCR5 (рецептор 1 лимфомы Беркитта);
(30) HLA-DOB (бета субъединица молекулы МНС II класса (1а антиген));
(31) Р2Х5 (пуринергический рецептор Р2Х лиганд-зависимый ионный канал 5);
(32) CD72 (Антиген дифференцировки В-клеток CD72, Lyb-2);
(33) LY64 (лимфоцитарный антиген 64 (RP105), тип I мембранный белок семейства, богатого лейциновыми повторами (LRR));
(34) FcRH1 (белок 1, подобный Fc рецептору);
(35) IRTA2 (рецептор суперсемейства иммуноглобулинов, связанный с транслокацией 2); и
(36) TENB2 (предполагаемый трансмембранный протеогликан).
23. Конъюгат антитела с лекарственным средством по п.17, где Аb представляет собой антитело с цистеиновыми заменами.
24. Конъюгат антитела с лекарственным средством по п.17, где Аb представляет собой антитело, которое связывается с рецептором ErbB.
25. Конъюгат антитела с лекарственным средством по п.24, где Ab представляет собой трастузумаб.
26. Конъюгат антитела с лекарственным средством по п.17, где p равно 1, 2, 3 или 4.
27. Конъюгат антитела с лекарственным средством по п.17, содержащий смесь конъюгата антитела с лекарственным средством, где средняя лекарственная нагрузка на антитело в смеси конъюгата антитела с лекарственным средством составляет примерно от 2 примерно до 5.
28. Конъюгат антитела с лекарственным средством по п.26, где средняя лекарственная нагрузка на антитело в смеси конъюгата антитела с лекарственным средством составляет примерно от 3 примерно до 4.
29. Фармацевтическая композиция для лечения рака, содержащая конъюгат антитела с лекарственным средством по п.17 и фармацевтически приемлемый разбавитель, носитель или эксципиент.
30. Фармацевтическая композиция по п.29, дополнительно содержащая терапевтически эффективное количество химиотерапевтического агента.
31. Способ лечения рака, предусматривающий введение пациенту фармацевтической композиции по п.29.
32. Способ по п.31, где пациенту вводят химиотерапевтическое средство, в сочетании с конъюгатом антитела с лекарственным средством.
33. Применение конъюгата антитела с лекарственным средством по п.17 для производства лекарственного средства для лечения рака у млекопитающего.
34. Применение по п.33, где млекопитающим является человек.
35. Изделие, содержащее
конъюгат антитела с лекарственным средством по п.17;
контейнер; и
лист-вкладыш или этикетку, указывающие, что конъюгат может быть использован для лечения злокачественного заболевания.
36. Способ получения конъюгата антитела с лекарственным средством, предусматривающий взаимодействие производного антрациклина и антитела Аb с образованием конъюгата антитела с лекарственным средством п.17, где производное антрациклина имеет формулу (11с):
Ant-L-(Z)m-X (IIc),
где Ant выбран из структур:

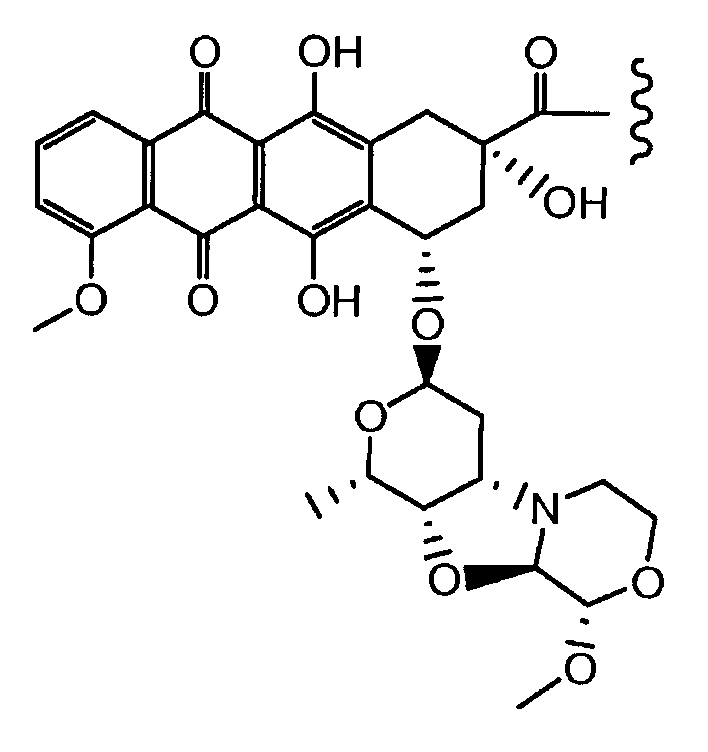
где волнистая линия указывает на присоединение к L;
L представляет собой линкер, выбранный из -СН2O-, -N(R)-,
-N(R)m(C1-С12алкилен)-, -N(R)m(C2-C8алкенилен)-, -N(R)m(C2-C8алкинилен)-, -N(R)m(CH2CH2O)n-, и структур:
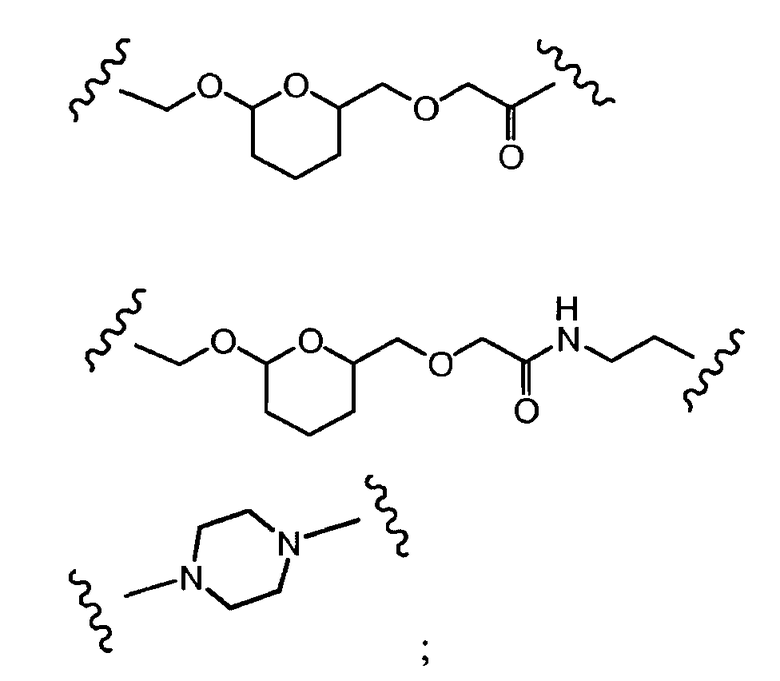
где волнистые линии указывают на присоединения к Ant и Z; и
Z представляет собой необязательный спейсер, выбранный из -CH2C(O)-, -(N(R))m(C1-С12алкилен)-, -CH2C(O)NR(C1-С12алкилен)-, и структур:

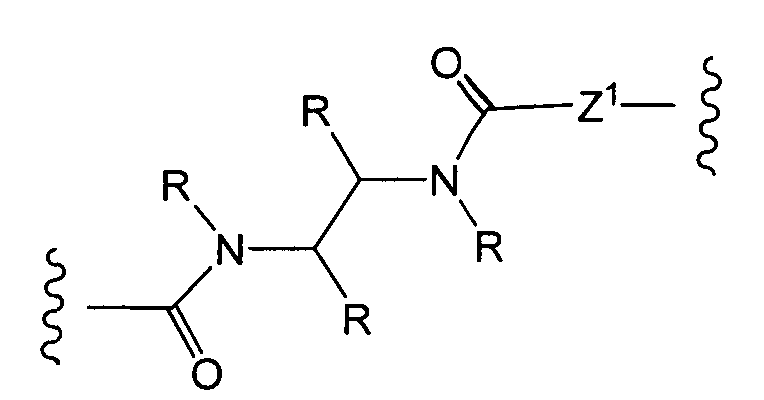
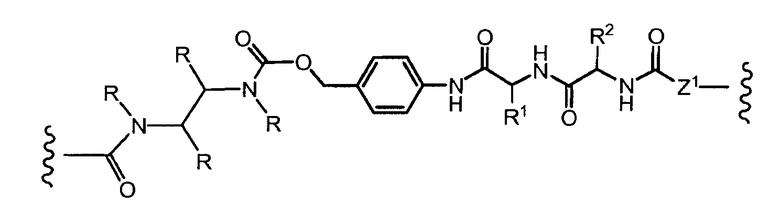
X представляет собой реакционно-способную функциональную группу, выбранную из малеимида, тиола, амино, бромида, п-толуолсульфоната, йодида, гидроксила, карбоксила, пиридилдисульфида и N-гидроксисукцинимида;
R представляет собой H, C1-C12алкил, или C6-C20арил;
R1 и R2 независимо выбраны из боковых цепей аминокислот;
Z1 выбран из -(C1-С12алкилен)-, -(C2-C8алкенилен)-, -(C2-C8алкинилен)- и -(CH2CH2O)n-,
m равно 0 или 1; и n равно 1-6.
| Кипятильник для воды | 1921 |
|
SU5A1 |
| QUINTIERI L., et al., Formation and antitumor activity of PNU-159682, a major metabolite of nemorubicin in human liver microsomes | |||
| Clin Cancer Res | |||
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |

