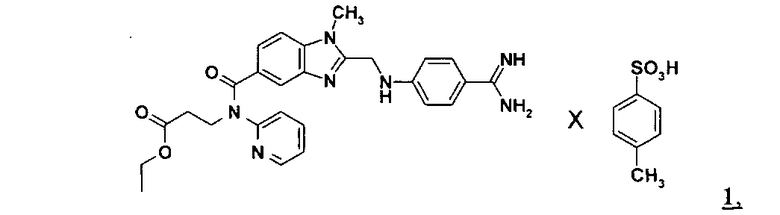
Настоящее изобретение относится к способу получения соединения формулы 1
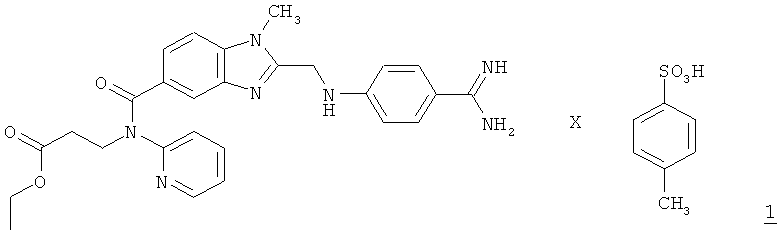 ,
,
представляющего собой ценный промежуточный продукт при синтезе фармацевтического активного вещества - этексилата дабигатрана.
Уровень техники
Этексилат дабигатрана известен из уровня техники и впервые был описан в WO 98/37075. Способы получения этексилата дабигатрана известны, кроме того, из WO 2006/000353, а также из публикации Hauel и др. (Journ. Med. Chem, 45, 2002, с.1757).
Как указано в WO 98/37075, а также в WO 2006/000353, при синтезе этексилата дабигатрана главная роль в качестве промежуточного продукта отводится соединению формулы 1, а именно: паратолуолсульфонату N-(2-пиридил)-N-(2-этоксикарбонилэтил)амида 1-метил-2-[N-[4-амидинофенил]аминометил]бензимидазол-5-илкарбоновой кислоты.
Наряду с указанными публикациями WO 98/37075 и WO 2006/000353 некоторые аспекты касательно возможных способов получения этексилата дабигатрана представлены также в WO 2007/071742 А1 и WO 2007/071743 А1.
В WO 98/37075 предлагается получать замещенные (4-бензимидазол-2-илметиламино)бензамидины взаимодействием соответствующих замещенных (4-бензоимидазол-2-илметиламино)бензонитрилов с аммиаком. Однако этот способ с технологической точки зрения связан с исключительно высокими затратами и приводит к значительному загрязнению окружающей среды кислотами, которые требуется утилизировать.
Согласно WO 2006/000353 A1, WO 2007/071742 А1 и WO 2007/071743 А1 соединение формулы 1 получают путем синтеза продукта конденсации формулы 4 в соответствии с приведенной ниже схемой 1.
Схема 1
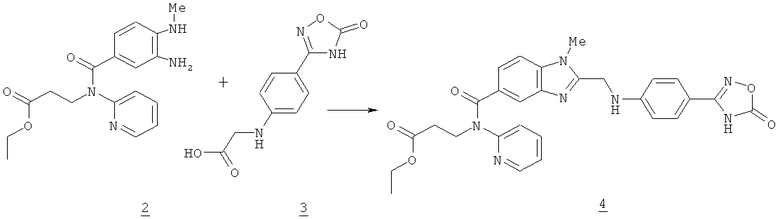
Согласно известным из уровня техники способам соединение формулы 4 сначала выделяют, а затем гидрируют.
При проведении реакции конденсации в соответствии со схемой 1 часто образуется побочный продукт формулы 5
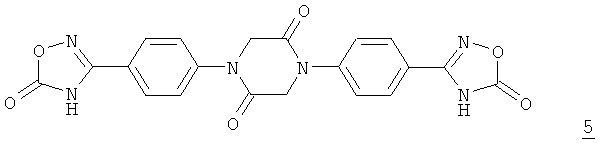 ,
,
который перед выделением продукта конденсации формулы 4 приходится удалять путем сложной и связанной с высокими затратами фильтрации в горячем состоянии.
Кроме того, последующая реакция по получению соединения формулы 1. требует замены растворителей и помимо этого требует связанных со значительными затратами времени и средств выделения и сушки промежуточного продукта формулы 4. По этим причинам возможно значительное снижение выхода конечного продукта.
В основу настоящего изобретения была положена задача разработать способ получения соединения формулы 1, который позволял бы эффективнее синтезировать такое соединение в промышленном масштабе и позволял бы избежать рассмотренных выше недостатков.
Подробное описание изобретения
Настоящее изобретение относится к способу получения соединения формулы 1
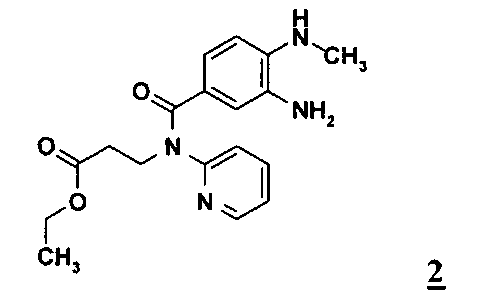
в промышленном масштабе, отличающемуся тем, что диамин формулы 2
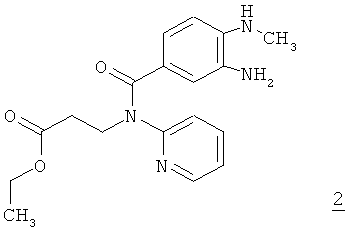
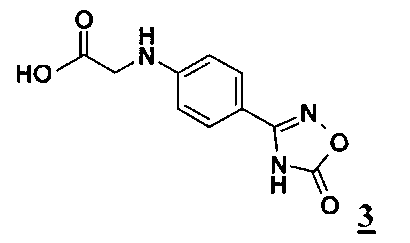
взаимодействием с оксадиазолоном формулы 3
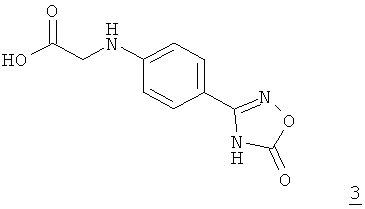
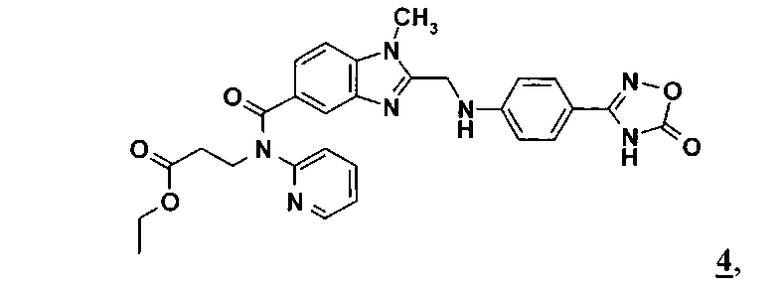
превращают в соединение формулы 4
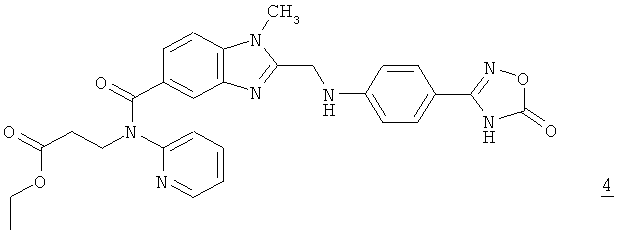 ,
,
которое без выделения превращают путем гидрирования и добавления паратолуолсульфокислоты и аммиака в амидин формулы 1.
Исходные соединения формул 2 и 3 можно получать способом, описанным в WO 2006/000353.
При проведении предлагаемой в изобретении реакции соединения формул 2 и 3 растворяют в инертном органическом растворителе и конденсируют в присутствии связывающего воду средства. В качестве инертного органического растворителя предпочтительно использовать апротонный растворитель. Его можно, например, выбирать из группы, включающей алифатические и ароматические, необязательно галогенированные углеводороды, простые эфиры, амиды и их смеси. В качестве апротонных неполярных растворителей предпочтительно использовать разветвленные или неразветвленные алифатические С5-С8алканы, С4-С10циклоалканы, алифатические С1-С6галогеналканы, ароматические С6-С10алканы либо их смеси. Особенно предпочтительно использовать алканы, такие как пентан, гексан или гептан, циклоалканы, такие как циклогексан или метилциклогексан, галогеналканы, такие как дихлорметан, ароматические алканы, такие как бензол, толуол или ксилол, либо их смеси. Для применения в качестве апротонных растворителей пригодны далее полярные простые эфиры, такие, например, как тетрагидрофуран (ТГФ), метилтетрагидрофуран, диоксан, трет-бутилметиловый эфир или диметоксиэтиловый эфир, амиды, такие, например, как диметилформамид, или лактамы, такие, например, как N-метилпирролидон.
В качестве связывающих воду средств можно использовать гигроскопические соли, неорганические или органические кислоты, соответственно их хлорангидриды, ангидриды неорганических или органических кислот, ангидриды алканфосфоновых кислот, молекулярные сита или производные мочевины. Предпочтительны при этом 1,1'-карбонилдиимидазол и алканфосфоновые ангидриды, а наиболее предпочтительны алканфосфоновые ангидриды. Среди последних особое значение согласно изобретению придается пропанфосфоновому ангидриду (ПФА, 2,4,6-триоксид 2,4,6-трипропил-[1,3,5,2,4,6]триоксатрифосфинана). Согласно изобретению при использовании алканфосфоновых ангидридов предпочтительно добавлять органическое основание, особенно предпочтительно третичный амин, наиболее предпочтительно диизопропилэтиламин.
На моль применяемого соединения формулы 2 указанный выше инертный органический растворитель предпочтительно использовать в количестве от 0,5 до 2,5 л, особенно предпочтительно от 1,0 до 2,0 л, наиболее предпочтительно от 1,3 до 1,5 л. На моль применяемого соединения формулы 2 соединение формулы 3 предпочтительно далее использовать в стехиометрических количествах. Особенно предпочтительно использовать соединение формулы 3 в небольшом избытке. На моль применяемого соединения формулы 2 соединение формулы 3 используют в количестве от 1,0 до 2,0 молей, особенно предпочтительно от 1,0 до 1,5 моля, наиболее предпочтительно от 1,1 до 1,3 моля.
Соединения формул 2 и 3 растворяют в указанном выше инертном органическом растворителе при температуре в интервале от 10 до 50°С, предпочтительно от 20 до 40°С, особенно предпочтительно от 25 до 35°С.Затем при постоянной температуре согласно особенно предпочтительному варианту осуществления изобретения добавляют третичный амин. На моль применяемого соединения формулы 2 третичный амин предпочтительно использовать в по меньшей мере стехиометрических количествах. Особенно предпочтительно, однако, использовать третичный амин в значительном избытке. В соответствии с этим на моль применяемого соединения формулы 2 третичный амин используют в количестве от 1,5 до 5,0 молей, особенно предпочтительно от 2,0 до 4,0 молей, наиболее предпочтительно от 2,3 до 2,7 моля.
По завершении добавления третичного амина затем при температуре предпочтительно в интервале от 10 до 40°С, особенно предпочтительно от 20 до 30°С, дозируют алканфосфоновый ангидрид, предпочтительно указанный выше ПФА. На моль применяемого соединения 2 алканфосфоновый ангидрид предпочтительно использовать в по меньшей мере стехиометрических количествах. Особенно предпочтительно использовать алканфосфоновый ангидрид в небольшом избытке. На моль применяемого соединения формулы 2 алканфосфоновый ангидрид предпочтительно использовать в количестве от 1,0 до 2,0 молей, особенно предпочтительно от 1,0 до 1,7 моля, наиболее предпочтительно от 1,1 до 1,4 моля. Предпочтительно используемый согласно изобретению ПФА целесообразно добавлять в разбавленном виде. В одном из предпочтительных вариантов ПФА для добавления растворяют в используемом инертном органическом растворителе. Особенно предпочтительно при этом добавлять ПФА в виде 30-60%-ного (мас.%), наиболее предпочтительно в виде 50%-ного, раствора в тетрагидрофуране или этилацетате.
По завершении добавления ПФА смесь перемешивают при постоянной температуре в течение примерно 0,25-4 ч. После этого добавляют слабую органическую кислоту в количестве, которое в пересчете на применяемое количество соединения формулы 2 предпочтительно составляет по меньшей мере 0,5 эквивалента. Под такой слабой органической кислотой в предпочтительном варианте подразумевается лимонная или уксусная кислота. Такую кислоту, однако, можно также использовать в избытке. В соответствии с этим на моль применяемого соединения формулы 2 указанную кислоту применяют в количестве от 0,5 до 4,0 эквивалента, более предпочтительно от 1,0 до 3 эквивалентов, особенно предпочтительно от 1,0 до 2,0 эквивалентов. При необходимости реакционную смесь можно разбавлять одним из вышеуказанных инертных органических растворителей, предпочтительно тем же самым растворителем. Для разбавления смеси растворитель предпочтительно добавлять в количестве, достигающем 50%, особенно предпочтительно в количестве от 10 до 30%, от уже ранее использовавшегося его количества.
После добавления кислоты и необязательного разбавления реакционной смеси затем при повышенной температуре и при необходимости при повышенном давлении проводят реакцию конденсации с получением соединения формулы 4. Температуру при этом согласно изобретению предпочтительно поддерживать выше 50°С, более предпочтительно в интервале от 60 до 100°С, особенно предпочтительно от 65 до 85°С. При использовании растворителя, который уже кипит в указанном интервале температур, давление повышают до уровня, при котором несмотря на более низкую температуру кипения растворителя реакцию можно проводить при указанной температуре. Давление, при котором проводят реакцию, предпочтительно устанавливать на уровне 1-3 бар.
Протекание реакции контролируют обычными методами, например, с помощью тонкослойной хроматографии или жидкостной хроматографии высокого разрешения (ЖХВР). По завершении реакции реакционную смесь медленно охлаждают, предпочтительно до температуры в интервале от 10 до 50°С, особенно предпочтительно до температуры примерно 20-30°С.
Далее к реакционной смеси без ее последующей переработки добавляют соответствующий катализатор гидрирования. Для применения в качестве катализаторов гидрирования обычно пригодны переходные металлы, такие, например, как никель, платина или палладий либо их соли или оксиды. Предпочтительны же никель Ренея, оксид платины и палладий на инертном носителе, прежде всего палладий на активированном угле (Pd/C). В одном из предпочтительных вариантов используют увлажненный водой 10%-ный Pd/C. На моль применяемого соединения формулы 2 такой катализатор предпочтительно использовать в количестве от 2 до 35 г, более предпочтительно примерно от 4 до 25 г, особенно предпочтительно примерно от 8 до 18 г. После добавления катализатора гидрирования добавляют воду. Добавляемое количество воды определяется преимущественно общим количеством используемого инертного органического растворителя. Воду предпочтительно добавлять в количестве, которое составляет от 50 до 100% (об./об.), особенно предпочтительно от 70 до 90% (об./об.), от всего используемого количества растворителя.
После добавления воды реакционную смесь нагревают до температуры в пределах от 30 до 70°С, особенно предпочтительно до температуры порядка 40-60°С, и при перемешивании гидрируют при давлении водорода 2-6 бар, предпочтительно 3-5 бар.
По завершении реакции катализатор отфильтровывают, фильтрат при необходимости разбавляют водой в количестве примерно от 5 до 30% от ее ранее использовавшегося количества и при температуре в пределах от 10 до 60°С, особенно предпочтительно от 20 до 40°С, смешивают с паратолуолсульфокислотой. Паратолуолсульфокислоту можно добавлять в виде твердого вещества, необязательно в виде ее моногидрата или же в водном растворе. На моль применяемого соединения формулы 2 паратолуолсульфокислоту предпочтительно использовать по меньшей мере в стехиометрических количествах. Особенно предпочтительно использовать паратолуолсульфокислоту в небольшом избытке. На моль применяемого соединения формулы 2 паратолуолсульфокислоту предпочтительно использовать в количестве от 1,0 до 2,0 молей, более предпочтительно от 1,0 до 1,7 моля, особенно предпочтительно от 1,0 до 1,4 моля, прежде всего от 1,1 до 1,3 моля.
После этого добавляют аммиак, который используют либо в газообразном виде, либо в виде водных растворов. Согласно изобретению аммиак предпочтительно использовать в виде водных растворов, особенно предпочтительно в виде водного раствора с содержанием аммиака (NH4OH) примерно 25% (мас./мас). Добавлять аммиак можно, например, при температуре в интервале от 30 до 65°С. На моль применяемого соединения формулы 2 аммиак на этой стадии предпочтительно добавлять в количестве от 2 до 20 молей, особенно предпочтительно от 6 до 16 молей, наиболее предпочтительно примерно от 9 до 13 молей.
В процессе добавления аммиака соединение формулы 1 начинает выкристаллизовываться. Реакционную смесь охлаждают до температуры в пределах от 0 до 40°С, особенно предпочтительно примерно от 15 до 25°С, после чего соединение формулы 1 отфильтровывают и промывают водой либо ацетоном. При необходимости к маточному раствору для дальнейшего выкристаллизовывания соединения формулы 1 можно повторно добавлять аммиак. В этом случае на моль применяемого соединения формулы 2 аммиак предпочтительно добавлять в количестве от 1 до 10 молей, особенно предпочтительно от 2 до 8 молей, наиболее предпочтительно от 3 до 5 молей.
При создании изобретения неожиданно было установлено, что в результате добавления аммиака соединение формулы 1 почти полностью выпадает в осадок из реакционной смеси. Благодаря этому перед известными из уровня техники способами достигается целый ряд преимуществ, некоторые из которых более подробно рассмотрены ниже. Одно из таких преимуществ состоит в значительном повышении выхода соединения формулы 1. Помимо этого при получении промежуточного соединения формулы 4 отпадает необходимость в фильтрации в горячем состоянии, которая при определенных условиях необходима для удаления загрязняющего соединения формулы 5. Кроме того, снижается расход необходимых для осуществления предлагаемого в изобретении способа растворителей и реагентов, что существенно упрощает синтез прежде всего в промышленном масштабе. Еще одно преимущество заключается в том, что в отличие от уровня техники отпадает необходимость в связанных с высокими затратами времени выделении и сушке промежуточного продукта.
Выше и ниже используются следующие сокращения:
Ниже на примерах более подробно поясняется один из возможных вариантов осуществления предлагаемого в изобретении способа синтеза. Эти примеры служат лишь для иллюстрации изобретения и не ограничивают его объем.
Пример 1
24,20 г соединения формулы 2 и 19,95 г соединения формулы 3 практически полностью растворяют в 100 мл ТГФ при температуре около 30°С. Затем при этой температуре добавляют 24,92 г ДИПЭА. После этого при КТ дозируют 57,89 г 50%-ного раствора ПФА в ТГФ и перемешивают в течение примерно 2 ч. После добавления 13,44 г лимонной кислоты и 20 мл ТГФ затем при температуре около 90°С под давлением проводят реакцию конденсации с получением невыделяемого промежуточного соединения формулы 4. По завершении реакции реакционную смесь охлаждают до КТ и смешивают с 1,21 г увлажненного водой 10%-ного Pd/C и 100 мл воды. Затем суспензию нагревают до примерно 50°С и гидрируют в атмосфере водорода (при давлении около 4 бар). Далее Pd/C отфильтровывают и промывают 25 мл воды. После добавления 25 мл воды реакционную смесь при температуре около 50°С смешивают с 20,40 г 65%-ного водного раствора ПТСК и 60 мл 25%-ного водного раствора аммиака. При этом начинается выпадение тозилата в осадок. Его охлаждают до КТ, продукт формулы 1 отфильтровывают и промывают водой. Сушку проводят при 60°С или при температуре вплоть до 95°С под вакуумом.
Выход: 41,4 г (87,2%).
Чистота: выше 99%, определяемая по площади пика на ЖХВР-хроматограмме.
Пример 2
24,20 г соединения формулы 2 и 19,95 г соединения формулы 3 практически полностью растворяют в 87 мл ТГФ при КТ. Затем при этой температуре добавляют 24,92 г ДИПЭА. После этого при КТ дозируют 57,89 г 50%-ного раствора ПФА в ТГФ, промывают 13 мл ТГФ и перемешивают в течение примерно 2 ч. После добавления 6,72 г лимонной кислоты и 20 мл ТГФ затем при температуре около 90°С под давлением проводят реакцию конденсации с получением невыделяемого промежуточного соединения формулы 4. По завершении реакции реакционную смесь охлаждают до КТ и смешивают с 1,24 г увлажненного водой 10%-ного Pd/C и 60 мл воды. Затем суспензию нагревают до примерно 50°С и гидрируют в атмосфере водорода (при давлении около 4 бар). Далее Pd/C отфильтровывают и промывают 50 мл смеси ТГФ и воды (в соотношении 7:3). После добавления 20 мл смеси ТГФ и воды (в соотношении 7:3) реакционную смесь при температуре около 50°С смешивают с 39,93 г твердой ПТСК и 60 мл 25%-ного водного раствора аммиака. При этом начинается выпадение тозилата в осадок. Его охлаждают до КТ, продукт формулы 1 отфильтровывают и промывают водой. Сушку проводят при 40°С или при температуре вплоть до 95°С под вакуумом.
Выход: 42,2 г (88,9%).
Чистота: выше 99%, определяемая по площади пика на ЖХВР-хроматограмме.
Пример 3
24,20 г соединения формулы 2 и 19,95 г соединения формулы 3 практически полностью растворяют в 87 мл ТГФ при КТ. Затем при этой температуре добавляют 24,92 г ДИПЭА. После этого при КТ дозируют 57,89 г 50%-ного раствора ПФА в EtOАс, промывают 13 мл ТГФ и перемешивают в течение примерно 2 ч. После добавления 6,72 г лимонной кислоты и 20 мл ТГФ затем при температуре около 90°С под давлением проводят реакцию конденсации с получением невыделяемого промежуточного продукта, представляющего собой оксаамидин соединения BIBR 1048. По завершении реакции реакционную смесь охлаждают до КТ и смешивают с 1,21 г увлажненного водой 10%-ного Pd/C и 75 мл воды. Затем суспензию нагревают до примерно 50°С и гидрируют в атмосфере водорода (при давлении около 4 бар). Далее Pd/C отфильтровывают и промывают 50 мл смеси ТГФ и воды (в соотношении 1:1). После добавления 25 мл ТГФ и 10 мл воды реакционную смесь при температуре около 50°С смешивают с 39,93 г твердой ПТСК и 60 мл 25%-ного водного раствора аммиака. При этом начинается выпадение тозилата в осадок. Его охлаждают до КТ, продукт формулы 1 отфильтровывают и промывают водой. Сушку проводят при 40°С или при температуре вплоть до 95°С под вакуумом.
Выход: 43,1 г (90,8%).
Чистота: выше 99%, определяемая по площади пика на ЖХВР-хроматограмме.
Пример 4
24,20 г соединения формулы 2 и 19,95 г соединения формулы 3 практически полностью растворяют в 87 мл ТГФ при КТ. Затем при этой температуре добавляют 24,92 г ДИПЭА. После этого при КТ дозируют 57,89 г 50%-ного раствора ПФА в ЕtOАс, промывают 13 мл ТГФ и перемешивают в течение примерно 2 ч. После добавления 4,20 г АсОН и 20 мл ТГФ затем при температуре около 90°С под давлением проводят реакцию конденсации с получением невыделяемого промежуточного соединения формулы 4. По завершении реакции реакционную смесь охлаждают до КТ и смешивают с 1,25 г увлажненного водой 10%-ного Pd/C и 60 мл воды. Затем суспензию нагревают до примерно 50°С и гидрируют в атмосфере водорода (при давлении около 4 бар). Далее Pd/C отфильтровывают и промывают 50 мл смеси ТГФ и воды (в соотношении 7:3). После добавления 20 мл смеси ТГФ и воды (в соотношении 7:3) реакционную смесь при температуре около 50°С смешивают с 39,93 г твердой ПТСК и 60 мл 25%-ного водного раствора аммиака. При этом начинается выпадение тозилата в осадок. Его охлаждают до КТ, продукт формулы 1 отфильтровывают и промывают водой. Сушку проводят при 45°С или при температуре вплоть до 95°С под вакуумом.
Выход: 36,4 г (76,7%).
Чистота: выше 99%, определяемая по площади пика на ЖХВР-хроматограмме.
Пример 5
24,20 г соединения формулы 2 и 19,95 г соединения формулы 3 практически полностью растворяют в 86 мл ТГФ при температуре около 30°С. Затем при этой температуре добавляют 24,92 г ДИПЭА. После этого при КТ дозируют 57,89 г 50%-ного раствора ПФА в ТГФ и перемешивают в течение примерно 2 ч. После добавления 10,50 г L-винной кислоты и 20 мл ТГФ затем при температуре около 90°С под давлением проводят реакцию конденсации с получением невыделяемого промежуточного соединения формулы 4. По завершении реакции реакционную смесь охлаждают до КТ и смешивают с 1,21 г увлажненного водой 10%-ного Pd/C и 100 мл воды. Затем суспензию нагревают до примерно 50°С и гидрируют в атмосфере водорода (при давлении около 4 бар). Далее Pd/C отфильтровывают и промывают 30 мл воды. После добавления 20 мл воды реакционную смесь при температуре около 50°С смешивают с 22,25 г 65%-ного водного раствора ПТСК и 60 мл 25%-ного водного раствора аммиака. При этом начинается выпадение тозилата в осадок. Его охлаждают до КТ, продукт формулы 1 отфильтровывают и промывают водой. Сушку проводят при 90°С или при температуре вплоть до 95°С под вакуумом.
Выход: 39,3 г (82,8%).
Чистота: выше 99%, определяемая по площади пика на ЖХВР-хроматограмме.
Пример 6
24,20 г соединения формулы 2 и 19,95 г соединения формулы 3 практически полностью растворяют в 100 мл ТГФ при температуре около 30°С. Затем при этой температуре добавляют 23,07 г ДИПЭА. После этого при 25°С дозируют 57,89 г 50%-ного раствора ПФА в ТГФ и перемешивают в течение примерно 30 мин. После добавления 20,17 г лимонной кислоты и 20 мл ТГФ затем при температуре около 75°С под давлением проводят реакцию конденсации с получением невыделяемого промежуточного соединения формулы 4. По завершении реакции реакционную смесь охлаждают до КТ и смешивают с 1,21 г увлажненного водой 10%-ного Pd/C и 100 мл воды. Затем суспензию нагревают до примерно 50°С и гидрируют в атмосфере водорода (при давлении около 4 бар). Далее Pd/C отфильтровывают и промывают 30 мл воды. После добавления 20 мл воды реакционную смесь при температуре 28-38°С смешивают с 22,25 г 65%-ного водного раствора ПТСК. Затем при температуре в интервале от 38°С до температуры перегонки (64-65°С) дозируют 60 мл 25%-ного водного раствора аммиака. При этом начинается выпадение тозилата в осадок. Его охлаждают до КТ и добавляют еще 20 мл 25%-ного водного раствора аммиака. Продукт формулы 1 отфильтровывают и промывают водой. Сушку проводят при 60°С или при температуре вплоть до 95°С под вакуумом.
Выход: 42,4 г (89,3%).
Чистота: выше 99%, определяемая по площади пика на ЖХВР-хроматограмме.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ 4-(БЕНЗИМИДАЗОЛИЛМЕТИЛАМИНО)БЕНЗАМИДИНОВ | 2005 |
|
RU2401264C2 |
| УЛУЧШЕННЫЙ СПОСОБ ПОЛУЧЕНИЯ 4-(БЕНЗИМИДАЗОЛИЛМЕТИЛАМИНО)-БЕНЗАМИДОВ И ИХ СОЛЕЙ | 2006 |
|
RU2455292C2 |
| УЛУЧШЕННЫЙ СПОСОБ ПОЛУЧЕНИЯ СОЛЕЙ 4-(БЕНЗИМИДАЗОЛИЛМЕТИЛАМИНО)-БЕНЗАМИДОВ | 2006 |
|
RU2425827C2 |
| СПОСОБ ПОЛУЧЕНИЯ ЗАМЕЩЕННОГО ИМИДАЗОПИРИДИНА | 2001 |
|
RU2275372C2 |
| СПОСОБ КРОСС-СОЧЕТАНИЯ ИНДОЛОВ | 2005 |
|
RU2430916C2 |
| СПОСОБ ПОЛУЧЕНИЯ ОЛИГОМЕРОВ | 1967 |
|
SU191798A1 |
| 2-САХАРИНИЛМЕТИЛ(АРИЛ ИЛИ АРИЛОКСИ) АЦЕТАТЫ, ИЛИ ИХ СОЛИ, ОБРАЗОВАННЫЕ ДОБАВЛЕНИЕМ КИСЛОТ К ЭТИМ СОЕДИНЕНИЯМ ОСНОВНОГО ХАРАКТЕРА, ИЛИ ИХ СОЛИ, ОБРАЗОВАННЫЕ ДОБАВЛЕНИЕМ ОСНОВАНИЙ К ЭТИМ СОЕДИНЕНИЯМ КИСЛОТНОГО ХАРАКТЕРА, И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ИНГИБИРУЮЩАЯ АКТИВНОСТЬ ПРОТЕОЛИТИЧЕСКИХ ФЕРМЕНТОВ | 1992 |
|
RU2081871C1 |
| ПРОИЗВОДНЫЕ 5-АМИНО-4-ОКСИГЕКСАНОВОЙ КИСЛОТЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1992 |
|
RU2067585C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ О-ХЛОРМЕТИЛФЕНИЛГЛИОКСИЛОВОЙ КИСЛОТЫ | 1997 |
|
RU2177472C2 |
| УСОВЕРШЕНСТВОВАННЫЙ СПОСОБ ПОЛУЧЕНИЯ 5-(2,6-ДИ-4-МОРФОЛИНИЛ-4-ПИРИМИДИНИЛ)-4-ТРИФТОРМЕТИЛПИРИДИН-2-АМИНА | 2013 |
|
RU2646760C2 |
Изобретение относится к способу получения соединения формулы 1, заключающемуся в обработке диамина формулы 2 в апротонном растворителе оксадиазолоном формулы 3, который в присутствии связывающего воду средства в смеси с третичным амином, при температуре от 10 до 100°C и давлении от 1 до 6 бар, превращают в соединение формулы 4, которое затем без выделения превращают путем гидрирования и добавления паратолуолсульфокислоты и аммиака в амидин формулы 1. Способ позволяет проводить синтез без выделения промежуточного продукта и замены растворителя, исключить применение горячей фильтрации и повысить выход продукта. 2 з.п. ф-лы, 6 пр.
1. Способ получения соединения формулы 1
отличающийся тем, что диамин формулы 2
обрабатывают в апротонном растворителе, который выбирают из группы, включающей тетрагидрофуран (ТГФ), метилтетрагидрофуран, диоксан, трет-бутилметиловый эфир, диметоксиэтиловый эфир, диметилформамид и N-метилпироллидон, оксадиазолоном формулы 3
который в присутствии связывающего воду средства, которое выбирают из группы, включающей 1,1'-карбонилдиимидазол и ангидрид пропанфосфоновой кислоты, в смеси с третичным амином, при температуре от 10 до 100°C и давлении от 1 до 6 бар, превращают в соединение формулы 4
которое затем без выделения превращают путем гидрирования и добавления паратолуолсульфокислоты и аммиака в амидин формулы 1.
2. Способ по п.1, отличающийся тем, что реакцию между соединениями формул 2 и 3 с получением промежуточного соединения формулы 4 проводят в тетрогидрофуране (ТГФ).
3. Способ по п.1 или 2, отличающийся тем, что связывающее воду средство представляет собой ангидрид пропанфосфоновой кислоты в смеси с диизопропилэтиламином.
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| ЗАМЕЩЕННЫЕ АМИДЫ ФЕНИЛЦИКЛОГЕКСАНКАРБОНОВОЙ КИСЛОТЫ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ И ЛЕКАРСТВЕННОЕ СРЕДСТВО НА ИХ ОСНОВЕ | 2000 |
|
RU2246490C9 |

