Предпосылки создания изобретения
1. Область техники, к которой относится изобретение
Настоящее изобретение относится к способу получения необязательно замещенного 4-(бензимидазол-2-илметиламино)бензамидина, заключающемуся в том, что
(а) необязательно соответствующим образом замещенный диаминобензол подвергают реакции конденсации с 2-[4-(1,2,4-оксадиазол-5-он-3-ил)фениламино]уксусной кислотой,
(б) полученный таким путем продукт гидрируют и
(в) при необходимости карбонилируют амидиновую группу.
2. Уровень техники
Замещенные (4-бензимидазол-2-илметиламино)бензамидины, прежде всего N-(2-пиридил)-N-(2-этоксикарбонилэтил)амид 1-метил-2-[N-[4-(N-н- гексилоксикарбониламидино)фенил]аминометил]бензимидазол-5-илкарбоновой кислоты, уже известны из публикации WO 98/37075 по их применению в качестве действующих веществ, обладающих ингибирующим тромбин и увеличивающим продолжительность тромбинового времени действием.
Основным показанием к применению соединения приведенной ниже химической формулы I является послеоперационная профилактика тромбозов глубоких вен.
В указанной публикации WO 98/37075 замещенные (4-бензимидазол-2-илметиламино)бензамидины предлагается получать взаимодействием соответствующих замещенных (4-бензимидазол-2-илметиламино)бензонитрилов с аммиаком. Этот способ связан со значительными производственно-техническими затратами и с образованием больших количеств кислот, которые требуется утилизировать.
В основу настоящего изобретения была положена задача разработать способ получения замещенных (4-бензимидазол-2-илметиламино)бензамидинов, который позволял бы избежать необходимости в проведении указанной выше, связанной с высокими производственно-техническими затратами стадии.
Краткое изложение сущности изобретения
При создании изобретения неожиданно было установлено, что замещенные 4-(бензимидазол-2-илметиламино)бензамидины можно получать с высоким выходом и с использованием недорогих вспомогательных веществ,
(а) подвергая необязательно соответствующим образом замещенный диаминобензол реакции конденсации с 2-[4-(1,2,4-оксадиазол-5-он-3-ил)фениламино]уксусной кислотой,
(б) гидрируя полученный таким путем продукт и
(в) при необходимости карбонилируя амидиновую группу, предпочтительно алкилгалогенформиатом в присутствии основания, прежде всего гексилхлорформиатом.
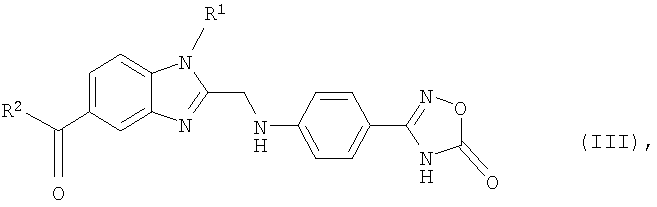
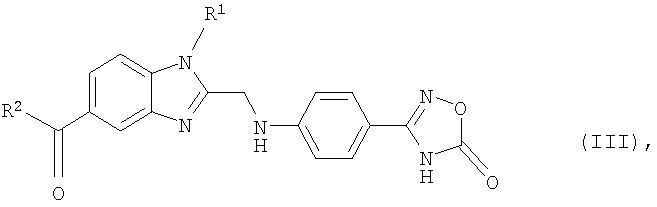
Еще одним объектом изобретения являются новые, образующиеся при осуществлении предлагаемого в изобретении способа промежуточные продукты формулы (III)
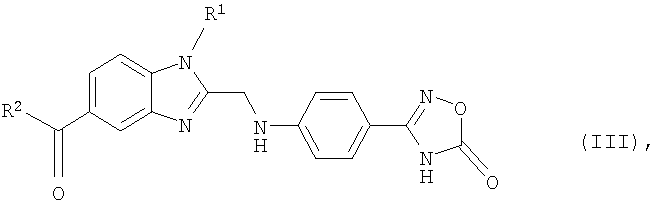
в которой R1 и R2 имеют значения, указанные ниже для соединений формулы (I), а также 2-[4-(1,2,4-оксадиазол-5-он-3-ил)фениламино]уксусная кислота и 4-(1,2,4-оксадиазол-5-он-3-ил)анилин.
Подробное описание изобретения
В предпочтительном варианте в изобретении предлагается способ получения необязательно замещенного 4-(бензимидазол-2-илметиламино)бензамидина формулы (I)
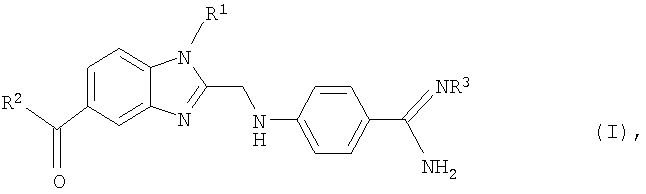
в которой R1 представляет собой C1-С6алкильную или С3-С7:циклоалкильную группу,
R2 представляет собой
1) C1-С6-алкильную группу или С3-С7циклоалкильную группу, необязательно замещенную C1-С3алкильной группой, которая в свою очередь может быть дополнительно замещена карбоксильной группой либо переводимой in vivo в карбоксигруппу группой, либо
2) R21NR22 -группу, где
R21 обозначает С1-С6алкильную группу, которая может быть замещена карбоксигруппой, C1-С6алкоксикарбонильной группой, бензилоксикарбонильной группой, C1-С3алкилсульфониламинокарбонильной группой, фенилсульфониламинокарбонильной группой, трифторметилсульфониламиногруппой, трифторметилсульфониламинокарбонильной группой или 1H-тетразолильной группой,
замещенную гидроксигруппой, фенил-С1-С3алкоксигруппой, карбокси-С1-С3алкиламиногруппой, C1-С3алкоксикарбонил-C1-С3-алкиламиногруппой, N-(С1-С3алкил)карбокси-С1-С3алкиламиногруппой или N-(С1-С3алкил)-С1-С3алкоксикарбонил-C1-С3алкиламиногруппой С2-С4алкильную группу, при этом в вышеуказанных группах расположенный непосредственно рядом с атомом азота α-атом углерода не может быть замещен, или необязательно замещенную C1-С3алкильной группой пиперидинильную группу, а
R22 обозначает атом водорода, C1-С6алкильную группу, необязательно замещенную C1-С3алкильной группой С3-С7циклоалкильную группу, С3-С6алкенильную или С3-С6алкинильную группу, при этом ненасыщенный фрагмент не может быть непосредственно соединен с атомом азота R21NR22 -группы,
необязательно замещенную атомом фтора, хлора либо брома, C1-С3алкильной группой или C1-С3алкоксигруппой фенильную группу, необязательно замещенную C1-С3алкильной группой бензильную, оксазолильную, изоксазолильную, тиазолильную, изотиазолильную, пиразолильную, пиридинильную, пиримидинильную, пиразинильную, пиридазинильную, пирролильную, тиенильную или имидазолильную группу или
R21 и R22 совместно с расположенным между ними атомом азота представляют собой необязательно замещенную карбоксигруппой или С1-С4алкоксикарбонильной группой 5-7-членную циклоалкилениминогруппу, с которой дополнительно может быть сконденсировано фенильное кольцо, и
R3 представляет собой атом водорода, С1-С9алкоксикарбонильную, циклогексилоксикарбонильную, фенил-С1-С3алкоксикарбонильную, бензоильную, n-С1-С3алкилбензоильную или пиридиноильную группу, при этом этоксильный фрагмент в положении 2 вышеуказанной С1-С9алкоксикарбонильной группы дополнительно может быть замещен
C1-С3алкилсульфонильной или 2-(С1-С3алкокси)этильной группой,
заключающийся в том, что на стадии (а) фенилдиамин формулы (II)
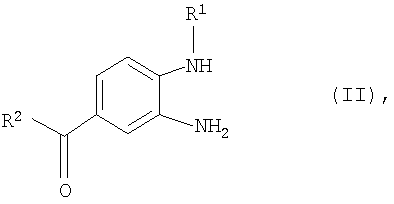
в которой R1 и R2 имеют указанные для формулы (I) значения, подвергают взаимодействию с 2-[4-(1,2,4-оксадиазол-5-он-3-ил)фениламино]уксусной кислотой, затем полученный таким путем продукт формулы (III)
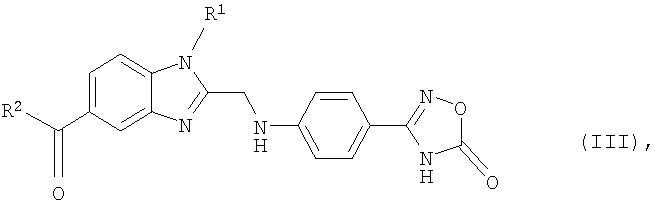
в которой R1 и R2 имеют указанные для формулы (I) значения, на стадии (б) гидрируют и после этого на стадии (в) при необходимости полученное таким путем соединение формулы (I), в которой R3 представляет собой водород, подвергают взаимодействию с соединением формулы (IV)

в которой R3 имеет указанные для формулы (I) значения, а Х обозначает приемлемую уходящую группу.
В особенно предпочтительном варианте в изобретении предлагается способ получения соединений формулы (I), в которой
R1 представляет собой C1-С3алкильную группу,
R2 представляет собой R21NR22-группу, где
R21 обозначает С1-С3алкильную группу, которая может быть замещена карбоксигруппой или C1-С3алкоксикарбонильной группой, а
R22 обозначает атом водорода, C1-С3алкильную группу, необязательно замещенную C1-С3алкильной группой пиридинильную группу, и
R3 представляет собой атом водорода или С1-С8алкоксикарбонильную группу.
В наиболее предпочтительном варианте в изобретении предлагается способ получения соединения формулы (I), в которой
R1 представляет собой метильную группу,
R2 представляет собой R21NR22-группу, где
R21 обозначает этильную группу, замещенную этоксикарбонильной группой, а
R22 обозначает пиридин-2-ильную группу, и
R3 представляет собой гексилоксикарбонильную группу.
Ниже рассмотрены предпочтительные варианты (А)-(Д) осуществления предлагаемого в изобретении способа.
(А) Реакцию конденсации на стадии (а) проводят в присутствии инертного разбавителя и связывающего воду (водопоглощающего) агента.
Соответствующим образом замещенные диаминобензолы формулы (II) известны, например, из публикации WO 98/37075 или их можно получать аналогично описанным в этой публикации. Особенно предпочтительно использовать амиды 3-амино-4-метиламинобензойной кислоты, прежде всего N-(2-пиридил)-N-(2-этоксикарбонилэтил)амид 3-амино-4-метиламинобензойной кислоты.
В качестве инертных разбавителей можно использовать апротонные неполярные растворители, такие, например, как алифатические или ароматические, необязательно галогенированные углеводороды, и апротонные полярные растворители, такие, например, как простые эфиры и/или амиды, соответственно лактамы, и/или их смеси. В качестве апротонных неполярных растворителей предпочтительно использовать разветвленные либо неразветвленные С5-С8алифатические алканы, С4-С10циклоалканы, C1-С6алифатические галогеналканы, С6-С10ароматические алканы или же их смеси. Особенно предпочтительно использовать такие алканы, как пентан, гексан или гептан, циклоалканы, такие как циклогексан или метилциклогексан, галогеналканы, такие как дихлорметан, ароматические алканы, такие как бензол, толуол или ксилол, или же их смеси. В качестве апротонных растворителей можно использовать простые полярные эфиры, такие, например, как тетрагидрофуран (ТГФ), метилтетрагидрофуран, диоксан, трет-бутилметиловый эфир или диметоксиэтиловый эфир, амиды, такие, например, как диметилформамид, или лактамы, такие, например, как N-метилпирролидон.
В качестве связывающих воду агентов можно использовать гигроскопические соли, неорганические либо органические кислоты, соответственно их хлорангидриды, ангидриды неорганических либо органических кислот, ангидриды алканфосфоновых кислот, молекулярные сита или производные мочевины. Предпочтительны 1,1'-карбонилдиимидазол и ангидриды алканфосфоновых кислот, а особенно предпочтительны ангидриды алканфосфоновых кислот.
В одном из предпочтительных вариантов 1,1'-карбонилдиимидазол суспендируют в ТГФ и нагревают. Затем добавляют 2-[4-(1,2,4-оксадиазол-5-он-3-ил)фениламино]уксусную кислоту. Далее добавляют соответствующим образом замещенный диаминобензол в ТГФ. Реакционную смесь перемешивают при температуре около 50°С, а затем после добавления уксусной кислоты концентрируют, смешивают с водой и твердое вещество отфильтровывают, промывают и сушат.
В другом, особенно предпочтительном варианте ангидриды алканфосфоновых кислот в присутствии органического основания, предпочтительно третичного амина, такого, например, как ДИПЭА (диизопропилэтиламин), добавляют к раствору 2-[4-(1,2,4-оксадиазол-5-он-3-ил)фениламино]уксусной кислоты и соответствующим образом замещенного диаминобензола в ТГФ. Реакционную смесь перемешивают предпочтительно при температуре в интервале от -10 до 50°С, и затем после добавления уксусной кислоты концентрируют. Далее смешивают с этанолом и фильтруют в горячем состоянии. Затем выпавшее в осадок из охлажденного раствора вещество отфильтровывают, промывают и сушат.
(Б) Гидрирование на стадии (б) проводят в присутствии инертного разбавителя и катализатора гидрирования.
Особенно предпочтителен вариант, в котором гидрирование проводят при температуре в интервале от 0 до 100°С, предпочтительно от 0 до 50°С, прежде всего от 10 до 30°С.
Предпочтителен далее вариант, в котором гидрирование проводят при давлении в интервале от 0,5 до 100 бар, предпочтительно от 1 до 10 бар, прежде всего при давлении от 1 до 2 бар.
В качестве инертных разбавителей можно использовать протонные растворители, такие, например, как спирты, карбоновые кислоты и/или вода, и апротонные полярные растворители, такие, например, как простые эфиры и/или амиды, соответственно лактамы, и/или их смеси. Ко всем растворителям при необходимости можно добавлять воду. В качестве протонных растворителей предпочтительно использовать разветвленные либо неразветвленные С1-С8алканолы, C1-С3карбоновые кислоты или же их смеси. Особенно предпочтительно использовать низшие спирты, такие как метанол, этанол, н-пропанол и изопропанол, карбоновые кислоты, такие как муравьиная кислота, уксусная кислота и пропионовая кислота, или же их смеси. В качестве реакционной среды особенно предпочтительно использовать этанол и/или уксусную кислоту, которые при необходимости могут содержать воду. В качестве апротонных растворителей можно использовать простые полярные эфиры, такие, например, как тетрагидрофуран и диметоксиэтиловый эфир, амиды, такие, например, как диметилформамид, или лактамы, такие, например, как N-метилпирролидон. Предпочтительно использовать растворители с пониженной воспламеняемостью (трудновоспламеняющиеся растворители).
Для использования в качестве катализаторов гидрирования пригодны, как правило, переходные металлы, такие, например, как никель, платина или палладий, или их соли либо оксиды. Предпочтительны никель Ренея, оксид платины и палладий на инертном носителе, прежде всего палладий на активированном угле (Pd/C).
Предпочтительны варианты, в которых продукт со стадии (а) и катализатор гидрирования используют при гидрировании в массовом соотношении между ними от 1:1 до 1000:1, предпочтительно от 5:1 до 100:1.
В одном из предпочтительных вариантов продукт со стадии (а) растворяют в этаноле и после добавления уксусной кислоты гидрируют при давлении водорода 2 бара при комнатной температуре в присутствии увлажненного водой 10%-ного Pd/C. Затем катализатор отфильтровывают и добавляют n-толуолсульфоновую кислоту, растворенную в 90 мл этанола либо в 90 мл воды.
Предпочтительно использовать водный раствор и-толуолсульфоновой кислоты. Тозилат полученного 4-(бензимидазол-2-илметиламино)бензамидина выпадает в осадок, который отфильтровывают и промывают несколькими порциями этанола.
В одном из особенно предпочтительных вариантов продукт со стадии (а) растворяют в смеси этанола и воды и гидрируют при давлении водорода 2 бара при комнатной температуре в присутствии увлажненного водой 10%-ного Pd/C. Затем катализатор отфильтровывают и добавляют n-толуолсульфоновую кислоту (в твердом виде либо в растворенном в 90 мл этанола или в 90 мл воды виде). Предпочтительно использовать n-толуолсульфоновую кислоту в твердом виде. Тозилат полученного 4-(бензимидазол-2-илметиламино)бензамидина выпадает в осадок, который отфильтровывают и промывают несколькими порциями этанолом.
(В) Для получения 2-[4-(1,2,4-оксадиазол-5-он-3-ил)фениламино]уксусной кислоты 4-(1,2,4-оксадиазол-5-он-3-ил)анилин подвергают взаимодействию с эфиром 2-галогенуксусной кислоты, предпочтительно с этиловым эфиром бромуксусной кислоты, в присутствии слабого основания, предпочтительно третичного амина, такого, например, как триэтиламин или карбонат щелочного металла, такого, например, как карбонат натрия, в инертном растворителе и полученный эфир 2-[4-(1,2,4-оксадиазол-5-он-3-ил)фениламино]уксусной кислоты омыляют.
В качестве инертных разбавителей можно использовать протонные растворители, такие, например, как спирты и/или вода, и апротонные полярные растворители, такие, например, как простые эфиры и/или амиды, соответственно лактамы, и/или их смеси. Ко всем растворителям при необходимости можно добавлять воду. В качестве протонных растворителей предпочтительно использовать воду или разветвленные либо неразветвленные C1-С8алканолы или же их смеси. Особенно предпочтительно использовать воду или низшие спирты, такие как метанол, этанол, н-пропанол и изопропанол, или же их смеси. В качестве реакционной среды особенно предпочтительно использовать этанол, который при необходимости может содержать воду. В равной мере возможно использование изопропанола, необязательно вместе с водой. Однако наиболее пригодным растворителем является вода. В качестве апротонных растворителей можно использовать простые полярные эфиры, такие, например, как тетрагидрофуран и диметоксиэтиловый эфир, амиды, такие, например, как диметилформамид, или лактамы, такие, например, как N-метилпирролидон.
В одном из особенно предпочтительных вариантов этиловый эфир бромуксусной кислоты добавляют в дозированных количествах к суспензии 4-(1,2,4-оксадиазол-5-он-3-ил)анилина и карбоната натрия в смеси воды и изопропанола либо предпочтительно в смеси воды и этанола и перемешивают.После охлаждения суспензию подвергают вакуум-фильтрации, несколькими порциями промывают водой и этанолом и сушат.
Омыление предпочтительно проводить в протонном растворителе с использованием гидроксида щелочного либо щелочноземельного металла, прежде всего гидроксида лития, натрия или калия.
В одном из особенно предпочтительных вариантов эфир 2-[4-(1,2,4-оксадиазол-5-он-3-ил)фениламино]уксусной кислоты суспендируют в воде, предпочтительно, однако, в смеси воды и этанола, и при комнатной температуре медленно смешивают с водным раствором NaOH. Суспензия при этом превращается в раствор, который нагревают до 45-75°С. Полученный таким путем раствор смешивают с НСl до достижения значения рН порядка 5 или предпочтительно 3. Твердое вещество выделяют и промывают холодной водой, а также холодным этанолом и МТБЭ (метил-трет-бутиловым эфиром).
(Г) Для получения 4-(1,2,4-оксадиазол-5-он-3-ил)анилина оксим 4-аминофениламида подвергают взаимодействию с диалкилкарбонатом, предпочтительно с диметилкарбонатом или диэтилкарбонатом, в присутствии основания, предпочтительно алкоголята щелочного металла, прежде всего метилата натрия, этилата натрия либо трет-бутанолята калия.
Оксим 4-аминофениламида можно получать, например, взаимодействием 4-аминобензонитрила с гидрохлоридом гидроксиламина.
В одном из особенно предпочтительных вариантов метилат натрия или предпочтительно этилат натрия при температуре 65-75°С, более предпочтительно 70-75°С, добавляют к суспензии оксима 4-аминофениламида в этаноле и затем промывают этанолом. После 15-минутного перемешивания по каплям добавляют диэтилкарбонат или предпочтительно диметилкарбонат. После протекания реакции в течении 2-4 ч смесь охлаждают и этанол отгоняют при давлении 120 мбар и температуре 40°С. Остаток растворяют в воде и после нагревания рН добавлением полуконцентрированного раствора едкого натра устанавливают на значение 10-12, затем подкислением концентрированной соляной кислотой устанавливают на значение менее 6, предпочтительно менее 4, особенно предпочтительно на значение 2-3, и медленно охлаждают. Раствор при этом превращается в суспензию, которую фильтруют и несколько раз промывают холодной водой и этанолом.
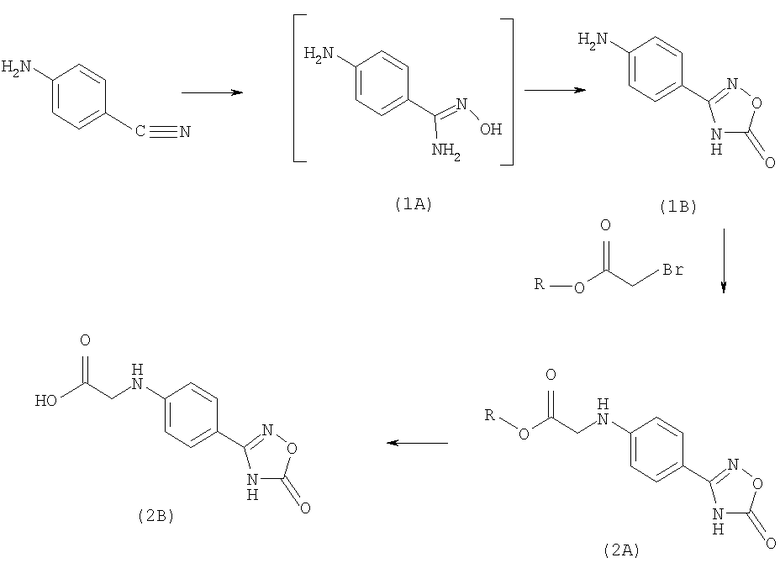
Получение необходимой для использования в качестве промежуточного продукта 2-[4-(1,2,4-оксадиазол-5-он-3-ил)фениламино]уксусной кислоты из 4-аминобензонитрила проиллюстрировано на приведенной ниже реакционной схеме.
Схема I
(Заключенные в квадратные скобки, невыделяемые промежуточные продукты в некоторых случаях могут быть разными в различных вариантах осуществления предлагаемого в изобретении способа. На схеме представлен предпочтительный вариант.)
Получение 4-(бензимидазол-2-илметиламино)бензамидина в качестве примера проиллюстрировано на приведенной ниже реакционной схеме.
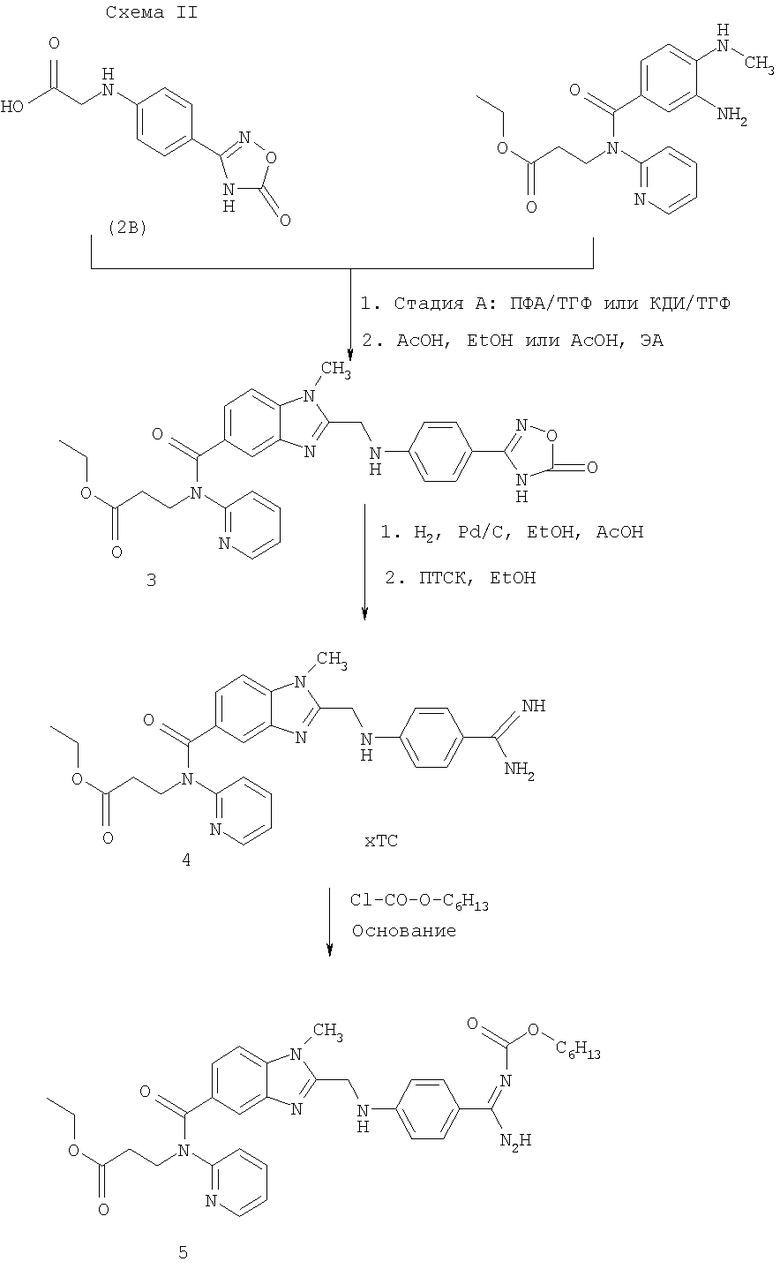
Отдельные реакционные смеси можно подвергать обычной переработке, состоящей, например, в отделении вспомогательных реакционных веществ, удалении растворителя и выделении из остатка чистого конечного продукта путем кристаллизации, перегонки, экстракции или хроматографии.
По завершении описанного выше способа полученное таким путем соединение формулы (I) можно превращать в его физиологически совместимую соль. Физиологически совместимые соли могут представлять собой соли с неорганическими либо органическими кислотами либо - при наличии в соединении карбоксигруппы - с неорганическими или органическими основаниями. В качестве кислот, пригодных для образования подобных солей, пригодны, например, метансульфоновая кислота, соляная кислота, бромистоводородная кислота, серная кислота, фосфорная кислота, фумаровая кислота, янтарная кислота, молочная кислота, лимонная кислота, винная кислота и малеиновая кислота. В качестве оснований, пригодных для образования указанных солей, пригодны среди прочих гидроксид натрия, гидроксид калия, циклогексиламин, этаноламин, диэтаноламин и триэтаноламин. Соединение формулы (5) предпочтительно переводить в его мезилат.
Ниже предлагаемый в изобретении способ более подробно поясняется на примерах. Эти примеры лишь иллюстрируют изобретение и не ограничивают его объем.
Примеры
Используемые выше и в последующем описании сокращения имеют следующие значения:
Пример 1
Получение 4-(1,2,4-оксадиазол-5-он-3-ил)анилина (1)
Вариант 1
Получение соединения (1А)
В реакционный сосуд предварительно помещают 118,6 г (1 моль) 4-аминобензонитрила и 68,9 г (0,65 моля) карбоната натрия в 500 мл этанола и 100 мл воды и нагревают до 60°С. К этой суспензии медленно по каплям добавляют 76,4 г (1,1 моля) гидрохлорида гидроксиламина, растворенного в 100 мл воды. Затем смесь оставляют перемешиваться на ночь при 60°С. Далее при охлаждении смеси до 0-5°С в осадок выпадает твердое вещество, которое отфильтровывают и несколько раз промывают в общей сложности 150 мл холодной воды и 100 мл холодного этанола. В завершение промывают 50 мл МТБЭ, получая в результате 178,4 г влажного продукта. Этот продукт сушат в вакууме при 35°С.
Выход: 135,4 г вещества светло-бежевого цвета (89,5% от теории), tпл>169,5°С (разл.), чистота: >98%, по площади пика на ЖХВР-хроматограмме.
Получение соединения (1В)
К суспензии 60,5 г продукта (1А) (0,4 моля) в 400 мл этанола при 70-75°С порциями добавляют 25,02 г (0,46 моля) метилата натрия и промывают 20 мл этанола. После 15-минутного перемешивания по каплям добавляют 47,25 г (0,4 моля) диэтилкарбоната. После протекания реакции в течение 3 ч смесь охлаждают до 40°С и этанол отгоняют при давлении 120 мбар и температуре 40°С. В результате получают темный остаток. Этот остаток растворяют при 40-45°С в 350 мл воды и после нагревания до 70°С значение рН сначала за счет медленного добавления полуконцентрированного раствора едкого натра устанавливают на 11, а затем за счет подкисления концентрированной соляной кислотой устанавливают на 5,5 и медленно охлаждают. Раствор превращается при этом в суспензию, которую фильтруют и несколько раз промывают в общей сложности 150 мл холодной воды и 50 мл этанола. Таким путем получают 88,7 г влажного вещества, которое сушат в вакууме при 35°С.
Выход: 62 г темного вещества (87,5% от теории), tпл>178°C (разл.), чистота: >98% по площади пика на ЖХВР-хроматограмме.
Вариант 2
Получение соединения (1А)
В реакционный сосуд предварительно помещают 41,3 г (0,35 моля) 4-аминобензонитрила и 36,5 г (0,53 моля) гидрохлорида гидроксиламина в 175 мл этанола и нагревают до 60°С. К этой суспензии медленно по каплям добавляют 170,1 г (0,53 моля) раствора метилата натрия (примерно 21%-ного в этаноле). Затем смесь оставляют перемешиваться на ночь при 60°С. Далее при охлаждении смеси до 0-5°С в осадок выпадает твердое вещество, которое отфильтровывают и несколько раз промывают в общей сложности 70 мл холодного этанола. В результате получают примерно 86 г влажного продукта. Этот продукт непосредственно используют в последующей реакции.
Получение соединения (1В)
К суспензии 86 г продукта (1А) в 270 мл этанола добавляют 32 г (0,35 моля) диметилкарбоната. Далее при 65-75°С добавляют 125 г (0,38 моля) раствора метилата натрия (примерно 21%-ного в этаноле) и промывают 20 мл этанола. После протекания реакции в течение 3 ч смесь охлаждают до 40°С и этанол отгоняют при давлении 120 мбар и температуре 40°С. В результате получают темный остаток. Этот остаток растворяют при 40-45°С в 280 мл воды и после нагревания до 70°С значение рН сначала за счет медленного добавления полуконцентрированного раствора едкого натра устанавливают на 11, а затем за счет подкисления концентрированной соляной кислотой устанавливают на 3-4 или предпочтительно на 2-3 и медленно охлаждают. Раствор при этом превращается в суспензию, которую фильтруют и несколько раз промывают в общей сложности 50 мл холодной воды и 20 мл этанола. Таким путем получают примерно 88 г влажного вещества, которое сушат в вакууме при температуре максимум 50°С.
Выход: 48 г вещества бежевого цвета (77,5% от теории), tпл>178°С (разд.), чистота: >98% по площади пика на ЖХВР-хроматограмме.
Пример 2
Получение 2-[4-(1,2,4-оксадиазол-5-он-3-ил)фениламино]уксусной кислоты (1)
Вариант 1
Получение соединения (2А)
При комнатной температуре к суспензии 70,86 г (0,4 моля) продукта (1В) и 26,5 г (0,25 моля) карбоната натрия в 600 мл смеси воды и изопропанола дозируют 83,5 г (0,5 моля) этилового эфира бромуксусной кислоты и смесь оставляют перемешиваться на ночь. Реакционная смесь приобретает при этом цвет от красновато-коричневого до оранжевого. Охлажденную до 0°С суспензию подвергают вакуум-фильтрации, промывают несколькими порциями 300 мл воды и 150 мл этанола (106 г влажного вещества светло-коричневого цвета) и сушат в вакууме при 35°С.
Выход: 92,44 г коричневатого вещества (87,7% от теории), tпл>186,1°С (разл.), чистота: >98% по площади пика на ЖХВР-хроматограмме.
Получение соединения (2В)
Полученный описанным выше путем эфир (2А) (86,9 г, 0,33 моля) суспендируют в 400 мл воды и при КТ к суспензии медленно по каплям добавляют 120 г 45%-ного NaOH. Суспензия превращается при этом в раствор и приобретает красноватый цвет (рН 12,5). Далее нагревают до температуры порядка 60°С и омыляют в течение 1 ч. Полученный раствор порциями смешивают с НСl (37%-ной или предпочтительно с концентрированной НСl) до достижения значения рН 5. Затем смесь охлаждают до 0°С. Твердое вещество отделяют вакуум-фильтрацией и несколькими порциями промывают в общей сложности 400 мл холодной воды, а также по 40 мл холодного этанола и МТБЭ. В результате получают 81,4 г влажного темного вещества. В завершение это вещество сушат в вакууме при 35°С.
Выход: 76,7 г требуемого вещества (98% от теории), tпл>193°С (разл.), чистота: >99% по площади пика на ЖХВР-хроматограмме.
Вариант 2
Получение соединения (2А)
При 45°С к суспензии 53,2 г (0,3 моля) продукта (1В) и 19,1 г (0,18 моля) карбоната натрия в 500 мл смеси воды и этанола (в соотношении в пределах от 90:10 до 95:5) добавляют 60,2 г (0,36 моля) этилового эфира бромуксусной кислоты и при необходимости оставляют перемешиваться на ночь. Реакционная смесь приобретает при этом цвет от красновато-коричневого до оранжевого. Охлажденную до 0°С суспензию подвергают вакуум-фильтрации, несколькими порциями промывают 100 мл этанола и сушат в вакууме при температуре максимум 50°С.
Выход: 69,5 г вещества бежево-коричневого цвета (87,7% от теории), tпл>186,1°С (разл.), чистота: >98% по площади пика на ЖХВР-хроматограмме.
Получение соединения (2В)
Полученный описанным выше путем эфир (2А) (86,9 г, 0,33 моля) суспендируют в 400 мл воды или предпочтительно в смеси этанола и воды (в соотношении 1:1) и при КТ к суспензии медленно добавляют по каплям 120 г 45%-ного NaOH. Суспензия превращается при этом в раствор и приобретает красноватый цвет (рН 12,5). Далее ее нагревают до температуры порядка 60°С и омыляют в течение 1 ч. Полученный раствор порциями смешивают с НСl (37%-ной или предпочтительно с концентрированной НСl) до достижения значения рН 3. Затем смесь охлаждают до 0°С. Твердое вещество отделяют вакуум-фильтрацией и несколькими порциями промывают в общей сложности 400 мл холодной воды, а также 40 мл холодного этанола. В результате получают 81,4 г влажного вещества. Это вещество сушат в вакууме при 35°С.
Выход: 76,7 г требуемого вещества (98% от теории), tпл>193°С (разл.), чистота: >99% по площади пика на ЖХВР-хроматограмме.
Пример 3
Получение N-(2-пиридил)-N-(2-этоксикарбонилэтил)амида 1-метил-2-[N-[4-(1,2,4-оксадиазол-5-он-3-ил)фенил]аминометил]бензимидазол-5-илкарбоновой кислоты (3)
Вариант А: использование КДИ в качестве агента сочетания
11,35 г (70 ммолей) 1,1'-карбонилдиимидазола суспендируют в 100 мл ТГФ и нагревают до 50°С. Затем к суспензии порциями добавляют 14,23 г (60,5 ммоля) продукта (2В). Далее при нагревании до 50°С в 37 мл ТГФ растворяют 17,1 г (50 ммолей) ПААМБК. Через примерно 90 мин суспензию добавляют к раствору ПААМБК и промывают 20 мл ТГФ. Реакционную смесь перемешивают в течение примерно 18 ч и затем после добавления 100 мл уксусной кислоты нагревают с обратным холодильником, отгоняя таким путем ТГФ. По истечении примерно 1 ч примешивают 400 мл воды и перемешивают. Раствор охлаждают, выпавшее в осадок твердое вещество розового цвета отфильтровывают и двумя порциями промывают 20 мл воды, после чего сушат в вакууме при температуре максимум 50°С. Выделенное вещество представляет собой диацетат соединения (3).
Выход: 24,8 г требуемого вещества (75% от теории), tпл>167°С с разл. (согласно дифференциальной сканирующей калориметрии), чистота: >95% по площади пика на ЖХВР-хроматограмме.
Вариант Б: использование ПФА в качестве агента сочетания
34,2 г (0,1 моля) ПААМБК, 27,5 г (0,12 моля) продукта (2В) и 30,3 г (0,23 моля) ДИПЭА предварительно добавляют в 170 мл ТГФ и охлаждают до температуры чуть ниже комнатной. К этой смеси добавляют 85 г (0,13 моля) ПФА (в виде примерно 50%-ного раствора в ЭА). Смесь перемешивают еще в течение 90 мин, после чего растворитель отгоняют. После этого добавляют 73,5 г уксусной кислоты и нагревают до внутренней температуры 90°С. Затем смешивают с 400 мл этанола или предпочтительно с 400 мл смеси этанола и воды (в соотношении приблизительно 85:15) и фильтруют в горячем состоянии. Раствор охлаждают, выпавшее в осадок твердое вещество отфильтровывают, двумя порциями промывают 50 мл этанола и в завершение сушат в вакууме при температуре максимум 50°С. Выделенное вещество представляет собой диацетат соединения (3).
Выход: 56 г требуемого вещества (85% от теории), tпл>167°С с разл. (согласно дифференциальной сканирующей калориметрии), чистота: >95% по площади пика на ЖХВР-хроматограмме.
Вариант В: использование пивалоилхлорида в качестве агента сочетания
96 г (0,41 моля) продукта (2В) суспендируют при 0°С в 250 мл N-МП и 550 мл ТГФ. Жидкую суспензию последовательно смешивают с 48 г (0,4 моля) пивалоилхлорида и 52 г (0,4 моля) ДИПЭА и перемешивают в течение 30 мин. Затем добавляют 125 г (0,36 моля) ПААМБК, растворенного в 800 мл уксусной кислоты, и реакционную смесь в течение 3 ч нагревают с обратным холодильником. Под низким вакуумом отгоняют ТГФ и при повышенной температуре добавляют 1600 мл воды. Твердое вещество выделяют при 5°С, промывают 550 мл воды и в течение ночи сушат в сушильном шкафу с циркуляцией воздуха при температуре максимум 50°С.
Выход: 183 г (76% от теории), чистота: >95% по площади пика на ЖХВР-хроматограмме.
Пример 4
Получение N-(2-пиридил)-N-(2-этоксикарбонилэтил)амида 1-метил-2-[N-[4-амидинофенил]аминометил]бензимидазол-5-илкарбоновой кислоты (4)
Вариант А: гидрирование соединения (3) в этаноле
37,3 г (56,4 ммоля) соединения (3) растворяют в 900 мл этанола и после добавления 10 мл уксусной кислоты гидрируют при КТ и при давлении водорода 2 бара в присутствии 4 г увлажненного водой 10%-ного Pd/C. Затем катализатор отфильтровывают и добавляют 17 г (89,4 ммоля) ПТСК, растворенной в 180 мл этанола. Выпавший в осадок тозилат соединения (4) отфильтровывают и несколькими порциями промывают 150 мл этанола. В результате получают влажное вещество, которое сушат в вакууме при 30°С.
Выход: 34,5 г требуемого вещества светло-бежевого цвета (91,3% от теории), tпл 187°С (согласно дифференциальной сканирующей калориметрии), чистота: >98% по площади пика на ЖХВР-хроматограмме.
Вариант Б: гидрирование соединения (3) в смеси этанола и воды
37,3 г (56,4 ммоля) соединения (3) растворяют в 400 мл смеси этанола и воды (в соотношении 90:10) и гидрируют при КТ и при давлении водорода 2 бара в присутствии 4 г увлажненного водой 10%-ного Pd/C. Затем катализатор отфильтровывают и добавляют 11,5 г (60,6 ммоля) ПТСК. При концентрировании в осадок выпадает тозилат соединения (4). Суспензию охлаждают, требуемое вещество отфильтровывают и несколькими порциями промывают 150 мл смеси этанола и воды. В результате получают влажное вещество, которое сушат в вакууме при 35°С.
Выход: 33,7 г вещества светло-бежевого цвета (89% от теории), tпл 187°С (согласно дифференциальной сканирующей калориметрии), чистота: >98% по площади пика на ЖХВР-хроматограмме.
Вариант В: гидрирование соединения (3) в смеси ТГФ и воды
30,0 г (45,3 ммоля) соединения (3) растворяют при КТ в 90 мл смеси ТГФ и воды (в соотношении 1:1), смешивают с 4 г увлажненного водой 10%-ного Pd/C и гидрируют при давлении 4 бара и температуре 60°С. Катализатор отфильтровывают, промывают примерно 40 мл смеси ТГФ и воды (в соотношении 1:1) и фильтрат без дальнейшей переработки используют на следующей стадии, соответственно аналогично описанной выше процедуре выделяют путем добавления 13,6 г (72 ммоля) ПТСК, растворенной в 100 мл воды, и охлаждения.
Пример 5
Получение N-(2-пиридил)-N-(2-этоксикарбонилэтил)амида 1-метил-2-[N-[4-(N-н-гексилоксикарбониламидино)фенил]аминометил]бензимидазол-5-илкарбоновой кислоты (5)
Полученное в примере 4 соединение подвергают по известной методике взаимодействию с гексилхлорформиатом в присутствии соответствующего основания.
Вариант А: ацилирование соединения (4) в смеси ацетона и воды
55 г (81,9 ммоля) соединения (4), растворенного в 437 мл ацетона и 273 мл воды, смешивают в присутствии 34 г (246 ммолей) карбоната калия при температуре порядка 15°С с 16,4 г (99,6 ммоля) гексилхлорформиата. По завершении реакции выпавший в осадок продукт отфильтровывают и промывают смесью ацетона и воды. При необходимости продукт можно повторно при нагреве растворять в примерно 270 мл ацетона и затем фильтровать. После фильтрации требуемое вещество снова кристаллизуют добавлением 220 мл воды. Выделенное вещество сушат в вакууме при 45°С.
Выход: 42-48 г (82-94% от теории).
Вариант Б: ацилирование соединения (4) в смеси ацетона и воды с разделением фаз
55 г (81,9 ммоля) соединения (4), растворенного в 437 мл ацетона и 273 мл воды, смешивают в присутствии 67 г (486 ммолей) карбоната калия при температуре порядка 15°С с 16,4 г (99,6 ммоля) гексилхлорформиата. По завершении реакции суспензию нагревают до примерно 50°С. После разделения фаз водную фазу отбрасывают, а ацетон заменяют на 440 мл этилацетата. Отделенную затем водную фазу отбрасывают, а органическую фазу несколькими порциями последовательно промывают разбавленным раствором карбоната калия и водой. Продукт кристаллизуют путем охлаждения, выделяют и промывают этилацетатом. Выделенное вещество сушат в вакууме при 45°С.
Выход: 42-48 г (82-94% от теории).
Пример 6
Получение мезилата N-(2-пиридил)-N-(2-этоксикарбонилэтил)амида 1-метил-2-[N-[4-(N-н-гексилоксикарбониламидино)фенил]аминометил]бензимидазол-5-илкарбоновой кислоты (5)
100 г (0,16 моля) соединения (5) растворяют при нагреве в 890 мл ацетона и смешивают с раствором 15 г (0,16 моля) метансульфоновой кислоты в 200 мл ацетона. Затем раствор фильтруют и после добавления 77 мл ацетона охлаждают до примерно 20°С. Выпавший в осадок продукт выделяют и промывают ацетоном. В завершение продукт сушат в вакуумном сушильном шкафу при температуре максимум 50°С.
Выход: 90-98% (103-113 г).
| название | год | авторы | номер документа |
|---|---|---|---|
| УЛУЧШЕННЫЙ СПОСОБ ПОЛУЧЕНИЯ 4-(БЕНЗИМИДАЗОЛИЛМЕТИЛАМИНО)-БЕНЗАМИДОВ И ИХ СОЛЕЙ | 2006 |
|
RU2455292C2 |
| УЛУЧШЕННЫЙ СПОСОБ ПОЛУЧЕНИЯ СОЛЕЙ 4-(БЕНЗИМИДАЗОЛИЛМЕТИЛАМИНО)-БЕНЗАМИДОВ | 2006 |
|
RU2425827C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОМЕЖУТОЧНОГО ПРОДУКТА ДЛЯ СИНТЕЗА ЭТЕКСИЛАТА ДАБИГАТРАНА | 2009 |
|
RU2524212C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛА И СОДЕРЖАЩИЕ ЭТИ СОЕДИНЕНИЯ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2000 |
|
RU2243226C2 |
| ИНГИБИТОРЫ СЕРИНПРОТЕАЗЫ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБЫ ЛЕЧЕНИЯ НА ИХ ОСНОВЕ | 2000 |
|
RU2252935C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ПРОЯВЛЯЮЩАЯ АКТИВНОСТЬ АНТАГОНИСТА АНГИОТЕНЗИНА II И СПОСОБ АНТАГОНИЗИРОВАНИЯ АНГИОТЕНЗИНА II У МЛЕКОПИТАЮЩИХ | 1992 |
|
RU2104276C1 |
| ПРОИЗВОДНОЕ АЛЬФА-АМИНОГИДРОКСАМОВОЙ КИСЛОТЫ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2000 |
|
RU2265592C2 |
| ПРОИЗВОДНЫЕ БЕНЗАМИДИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1998 |
|
RU2194044C2 |
| Способ получения 3-амино- 2-пиразолиновых производных или их солей | 1970 |
|
SU470959A3 |
| ПРОИЗВОДНЫЕ ОКСАЗОЛИДИНОНА, ФУНГИЦИДНАЯ КОМПОЗИЦИЯ И СПОСОБ БОРЬБЫ С ГРИБКОВЫМИ ЗАБОЛЕВАНИЯМИ | 1990 |
|
RU2092051C1 |
Настоящее изобретение относится к способу получения замещенного 4-(бензимидазол-2-илметиламино)бензамидина формулы (I)
или его физиологически совместимой соли,
в которой R1 представляет собой C1-С3алкильную группу, R2 представляет собой R21NR22 -группу, где R21 обозначает C1-С3алкильную группу, замещенную С1-С8алкоксикарбонильной группой, а R22 обозначает пиридинильную группу, и R3 представляет собой С1-С8алкоксикарбонильную группу, обладающего ингибирующим тромбин и увеличивающим продолжительность тромбинового времени действием. Способ заключается в том, что на стадии (а) фенилдиамин формулы (II)
в которой R1 и R2 имеют указанные для формулы (I) значения, подвергают взаимодействию с 2-[4-(1,2,4-оксадиазол-5-он-3-ил)фениламино]уксусной кислотой, затем полученный таким путем продукт формулы (III)
в которой R1 и R2 имеют указанные для формулы (I) значения, на стадии (б) гидрируют и после этого на стадии (в) при необходимости полученное таким путем соединение формулы (I), в которой R3 представляет собой водород, подвергают взаимодействию с соединением формулы (IV)
в которой R3 имеет указанные для формулы (I) значения, а Х обозначает приемлемую уходящую группу, и при необходимости превращают в физиологически совместимую соль. Кроме того, изобретение относится к новым промежуточным продуктам - соединению формулы (III), в которой R1 и R2 имеют указанные для формулы (I) значения, а также к 2-[4-(1,2,4-оксадиазол-5-он-3-ил)фениламино]уксусной кислоте, 4-(1,2,4-оксадиазол-5-он-3-ил)анилину и толуолсульфонату N-(2-пиридил)-N-(2-этоксикарбонилэтил)амида 1-метил-2-[N-[4-амидинофенил]аминометил]бензимидазол-5-илкарбоновой кислоты. 5 н. и 7 з.п. ф-лы.
1. Способ получения замещенного 4-(бензимидазол-2-илметиламино)бензамидина формулы (I)
или его физиологически совместимой соли,
в которой R1 представляет собой C1-С3алкильную группу,
R2 представляет собой R21NR22 -группу, где
R21 обозначает C1-С3алкильную группу, замещенную C1-С8алкоксикарбонильной группой, а
R22 обозначает пиридинильную группу, и
R3 представляет собой С1-С8алкоксикарбонильную группу, заключающийся в том, что на стадии (а) фенилдиамин формулы (II)
в которой R1 и R2 имеют указанные для формулы (I) значения, подвергают взаимодействию с 2-[4-(1,2,4-оксадиазол-5-он-3-ил)фениламино]уксусной кислотой, затем полученный таким путем продукт формулы (III)
в которой R1 и R2 имеют указанные для формулы (I) значения, на стадии (б) гидрируют и после этого на стадии (в) при необходимости полученное таким путем соединение формулы (I), в которой R3 представляет собой водород, подвергают взаимодействию с соединением формулы (IV)
в которой R3 имеет указанные для формулы (I) значения, а Х обозначает приемлемую уходящую группу, и при необходимости превращают в физиологически совместимую соль.
2. Способ по п.1 получения соединения формулы (I), в которой
R1 представляет собой метильную группу,
R2 представляет собой R21NR22 -группу, где
R21 обозначает этильную группу, замещенную этоксикарбонильной группой, а
R22 обозначает пиридин-2-ильную группу, и R3 представляет собой гексилоксикарбонильную группу.
3. Способ по п.1, отличающийся тем, что реакцию конденсации на стадии (а) проводят в присутствии инертного разбавителя и связывающего воду агента.
4. Способ по п.1, отличающийся тем, что гидрирование на стадии (б) проводят в присутствии инертного разбавителя и катализатора гидрирования.
5. Способ по п.1, отличающийся тем, что на стадии (а) используют 2-[4-(1,2,4-оксадиазол-5-он-3-ил)фениламино]уксусную кислоту, полученную взаимодействием 4-(1,2,4-оксадиазол-5-он-3-ил)анилина с эфиром 2-галогенуксусной кислоты в присутствии слабого основания, с последующим омылением полученного эфира 2-[4-(1,2,4-оксадиазол-5-он-3-ил)фениламино]уксусной кислоты.
6. Способ по п.5, отличающийся тем, что 4-(1,2,4-оксадиазол-5-он-3-ил)анилин получают взаимодействием оксима 4-аминофениламида с диалкилкарбонатом в присутствии основания.
7. Способ по п.1, отличающийся тем, что полученное соединение формулы (I) превращают затем в физиологически совместимую соль.
8. Способ по п.7, отличающийся тем, что физиологически совместимая соль представляет собой метансульфонат.
9. Соединение формулы (III)
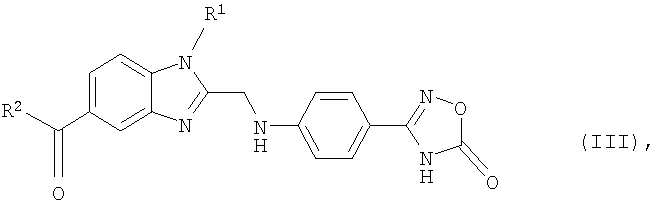
в которой R1 и R2 имеют указанные в одном из пп.1 и 2 значения.
10. 2-[4-(1,2,4-Оксадиазол-5-он-3-ил)фениламино]уксусная кислота.
11. 4-(1,2,4-Оксадиазол-5-он-3-ил)анилин.
12. Толуолсульфонат N-(2-пиридил)-N-(2-этоксикарбонилэтил)амида 1-метил-2-[N-[4-амидинофенил]аминометил]бензимидазол-5-илкарбоновой кислоты формулы
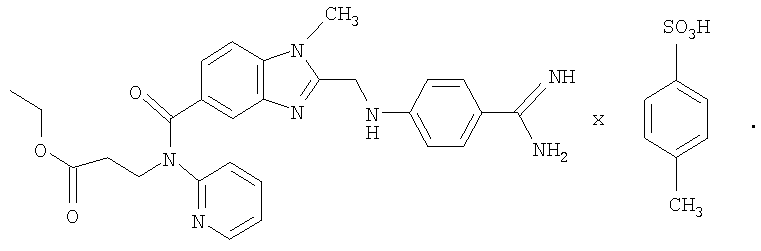
| WO 00/01704 A2, 13.01.2000 | |||
| DE 19962329 A1, 28.06.2001 | |||
| Дорожная спиртовая кухня | 1918 |
|
SU98A1 |
| RU 2001135715 A, 20.08.2003. | |||

