ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к способу получения соли необязательно замещенного 4-(бензимидазол-2-илметиламино)-бензамидина, в котором
(a) соответствующий необязательно замещенный диаминобензол конденсируют с 2-[4-(1,2,4-оксадиазол-5-он-3-ил)-фениламино]-уксусной кислотой и
(b) i) полученный таким образом продукт гидрируют,
ii) амидиновую группу необязательно карбонилируют без предварительного выделения промежуточного продукта гидрирования.
УРОВЕНЬ ТЕХНИКИ
Замещенные (4-бензимидазол-2-илметиламино)-бензамидины, в особенности N-(2-пиридил)-N-(2-этоксикарбонилэтил)-амид 1-метил-2-[N-[4-(N-н-гексилоксикарбониламидино)фенил]-аминометил]-бензимидазол-5-илкарбоновой кислоты уже известны из заявки WO 98/37075 как активные вещества, обладающие способностью ингибировать тромбин и увеличивать тромбиновое время.
Основными типами показаний для применения соединения химической формулы I являются послеоперационное предупреждение тромбоза глубоких вен и предупреждение удара (предупреждение удара, обусловленного фибрилляцией предсердий, аббревиатура ПУФП).
В WO 98/37075 предложено получать замещенные (4-бензимидазол-2-илметиламино)-бензамидины по реакции соответствующих замещенных (4-бензимидазол-2-илметиламино)-бензонитрилов с аммиаком. Эта технология является весьма сложной с производственной точки зрения и приводит к введению большого количества кислот, которые затем необходимо удалять.
В основу настоящего изобретения была положена задача разработки альтернативного способа получения замещенных (4-бензимидазол-2-илметиламино)-бензамидинов, с помощью которой можно исключить эту технологически сложную стадию.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Согласно изобретению неожиданно было установлено, что соли замещенных 4-(бензимидазол-2-илметиламино)-бензамидинов можно получить с высокими выходами и с использованием недорогих вспомогательных веществ, если
(a) соответствующий необязательно замещенный диаминобензол конденсируют с 2-[4-(1,2,4-оксадиазол-5-он-3-ил)-фениламино]-уксусной кислотой и
(b) i) полученный таким образом продукт гидрируют,
ii) амидиновую группу необязательно карбонилируют без предварительного выделения промежуточного продукта гидрирования и
iii) без предварительного выделения промежуточного продукта карбонилирования отделяют искомую соль.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Предпочтительным является способ получения соли необязательно замещенного 4-(бензимидазол-2-илметиламино)-бензамидина формулы (I) с неорганической или органической кислотой

в которой
R1 обозначает C1-С6-алкильную или С3-С7-циклоалкильную группу,
R2 (i) обозначает C1-С6-алкильную, С3-С7-циклоалкильную группу, необязательно замещенную C1-С3-алкильной группой, где C1-С3-алкильная группа может быть дополнительно замещена карбоксигруппой или группой, которая in vivo может быть превращена в карбоксигруппу,
или
(ii) обозначает группу R21NR22, в которой
R21 обозначает C1-С6-алкильную группу, которая может быть замещена карбоксигруппой, С1-С6-алкоксикарбонилом, бензилоксикарбонилом, С1-С3-алкилсульфониламинокарбонилом, фенилсульфониламинокарбонилом, трифторсульфониламиногруппой, трифторсульфониламинокарбонилом или 1Н-тетразолильной группой,
С2-С4-алкильную группу, замещенную гидроксигруппой, фенил-С1-С3-алкоксигруппой, карбокси-С1-С3-алкиламиногруппой, С1-С3-алкоксикарбонил-С1-С3-алкиламиногруппой, N-(С1-С3-алкил)-карбокси-С1-С3-алкиламиногруппой или N-(С1-С3-алкил)-С1-С3-алкоксикарбонил-С1-С3-алкиламиногруппой, где в указанных выше группах атома углерода, находящийся в α-положении по отношению к соседнему атому азота, не может быть замещенным, или пиперидинильную группу, необязательно замещенную С1-С3-алкильной группой, и
R22 обозначает атом водорода, C1-С6-алкильную группу, С3-С7-циклоалкильную группу, необязательно замещенную С1-С3-алкильной группой, или С3-С6-алкенильную или С3-С6-алкинильную группу, где ненасыщенный фрагмент может не быть непосредственно связан с атомом азота группы R21NR22, фенильную группу, необязательно замещенную атомом фтора, хлора или брома, С1-С3-алкильной или C1-С3-алкоксигруппой, или бензильную, оксазолильную, изоксазолильную, тиазолильную, изотиазолильную, пиразолильную, пиридинильную, пиримидинильную, пиразинильную, пиридазинильную, пирролильную, тиенильную или имидазолильную группу, необязательно замещенную C1-С3-алкильной группой, или
R21 и R22 вместе с расположенным между ними атомом азота обозначают 5- - 7-членную циклоалкилениминогруппу, необязательно замещенную карбоксигруппой или С1-С4-алкоксикарбонильной группой, с которым дополнительно может быть сконденсировано фенильное кольцо, и
R3 обозначает атом водорода, С1-С9-алкоксикарбонильную, циклогексилоксикарбонильную, фенил-С1-С3-алкоксикарбонильную, бензоильную, п-С1-С3-алкилбензоильную или пиридиноильную группу, где этоксигруппа в положении 2 указанной выше С1-С9-алкоксикарбонильной группы может быть дополнительно замещена С1-С3-алкилсульфонильной или 2-(С1-С3-алкокси)-этильной группой,
где на стадии (а) фенилдиамин формулы (II)

в которой R1 и R2 обладают значениями, указанными для формулы (I), вводят в реакцию с 2-[4-(1,2,4-оксадиазол-5-он-3-ил)-фениламино]-уксусной кислотой, полученный продукт формулы (III)

в которой R1 и R2 обладают значениями, указанными для формулы (I), гидрируют на стадии (b) i), затем, без какого-либо предварительного выделения продукта гидрирования, полученное таким образом соединение формулы (I), в которой R3 обозначает водород, на стадии (b) ii) необязательно вводят в реакцию с соединением формулы (IV)
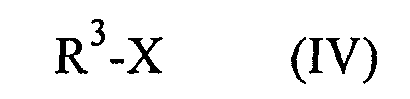
в которой R3 обладает значением, указанным для формулы (I), и
Х обозначает подходящую отщепляющуюся группу,
и затем без предварительного выделения продукта карбонилирования, на стадии (b) iii) полученное таким образом соединение формулы (I), в которой R1, R2 и R3 являются такими, как определено выше в настоящем изобретении, превращают в искомую соль, предпочтительно в фармацевтически приемлемую соль.
Особенно предпочтительными являются предлагаемые в настоящем изобретении способы получения солей соединений формулы (I), в которой
R1 обозначает С1-С3-алкильную группу,
R2 обозначает группу R21NR22, в которой
R21 обозначает C1-С3-алкильную группу, которая может быть замещена карбоксигруппой, С1-С3-алкоксикарбонилом,
R22 обозначает атом водорода, С1-С3-алкильную группу, пиридинильную группу, необязательно замещенную С1-С3-алкильной группой, и
R3 обозначает атом водорода или C1-C8-алкоксикарбонильную группу.
Наиболее предпочтительными являются предлагаемые в настоящем изобретении способы получения солей соединения формулы (I), в которой
R1 обозначает метильную группу,
R2 обозначает группу R21NR22, в которой
R21 обозначает этильную группу, которая замещена этоксикарбонильной группой,
R22 обозначает пиридин-2-ильную группу и
R3 обозначает н-гексилкарбонильную группу.
Предпочтительными солями являются метансульфонат, хлорид, малеат, тартрат, салицилат, цитрат и малонат соединения формулы (I). Особенно предпочтительной солью является метансульфонат.
Следующие варианты осуществления (А)-(F) предлагаемых в настоящем изобретении способов являются предпочтительными.
(А) Конденсацию на стадии (а) проводят в присутствии инертного разбавителя и связывающего воду агента.
Соответствующим образом замещенные диаминобензолы формулы (II) известны, например, из заявки WO 98/37075, например, из примера 25 (стадии а и b) или их можно получить аналогично описанным в этой заявке. Для гидрирования содержащего нитрогруппу предшественника с целью получения диаминобензола формулы (II) использующимся растворителем может быть, например, толуол, изопропанол, триэтиламин, этанол, бутилацетат, этилацетат, метанол или смеси этих растворителей. Гидрирование предпочтительно проводят при давлении водорода, равном от 1 до 20 бар, но возможны и более высокие давления. Концентрация ароматического азотсодержащего соединения (эдукта) предпочтительно составляет от 10 до 40 мас.%, более предпочтительно, если он содержится в концентрации, равной от 20 до 30 мас.%. Использующимся катализатором может быть, например, 5-10% палладий на древесном угле, а предпочтительно в качестве катализатора используют 2-20 мас.% влажного палладия на древесном угле в пересчете на ароматическое азотсодержащее соединение, что соответствует примерно 0,05-1 мас.% палладия в пересчете на ароматическое азотсодержащее соединение. Особенно предпочтительно использовать амиды 3-амино-4-метиламинобензойной кислоты, предпочтительно - N-(2-пиридил)-N-(2-этоксикарбонилэтил)-амид 3-амино-4-метиламинобензойной кислоты.
Использующимися инертными разбавителями могут быть апротонные неполярные растворители, такие как, например, алифатические или ароматические необязательно галогенированный углеводороды, или апротонные полярные растворители, такие как, например, простые эфиры и/или амиды или лактамы и/или их смеси. Использующимися апротонными неполярными растворителями предпочтительно являются разветвленные или неразветвленные С5-С8-алифатические алканы, С4-С10-циклоалканы, C1-С6-алифатические галогеналканы, С6-С10-ароматические алканы или их смеси. Особенно предпочтительно использовать алканы, такие как пентан, гексан или гептан, циклоалканы, такие как циклогексан или метилциклогексан, галогеналканы, такие как дихлорметан, ароматические алканы, такие как бензол, толуол или ксилол или их смеси. Подходящими апротонными растворителями являются полярные простые эфиры, такие как, например, тетрагидрофуран (ТГФ), метилтетрагидрофуран, диоксан, трет-бутилметиловый эфир или диметоксиэтиловый эфир, или амиды, такие как, например, диметилформамид, или лактамы, такие как, например, N-метилпирролидон.
Связывающими воду агентами, которые можно использовать, являются гигроскопические соли, неорганические или органические кислоты или их хлорангидриды, ангидриды алканфосфоновых кислот, молекулярные сита и производные мочевины. 1,1'-Карбонилдиимидазолы и ангидриды алканфосфоновых кислот являются предпочтительными, а ангидриды алканфосфоновых кислот являются особенно предпочтительными.
В предпочтительном варианте осуществления 1,1'-карбонилдиимидазол (КДИ) суспендируют в ТГФ и нагревают. Прибавляют 2-[4-(1,2,4-оксадиазол-5-он-3-ил)-фениламино]-уксусную кислоту. К ТГФ прибавляют соответствующим образом замещенный диаминобензол. Реакционную смесь перемешивают примерно при 50°С и затем, после прибавления уксусной кислоты, выпаривают и перемешивают с водой и твердое вещество отфильтровывают, промывают и сушат.
Во втором особенно предпочтительном варианте осуществления ангидриды алканфосфоновых кислот прибавляют в присутствии органического основания, предпочтительно трет-амина, такого как, например, ДИПЭА, к раствору 2-[4-(1,2,4-оксадиазол-5-он-3-ил)-фениламино]-уксусной кислоты и соответствующим образом замещенного диаминобензола в ТГФ. Реакционную смесь перемешивают предпочтительно при температуре от -10 до 50°С и затем, после прибавления уксусной кислоты, выпаривают. Ее объединяют со смесью этанол/вода и необязательно фильтрующим средством, например кизельгуром (например, Clarcel®), и фильтруют в горячем виде. Затем вещество, осадившееся из охлажденного раствора, отфильтровывают, промывают и сушат.
(В) Стадию гидрирования (b) i) проводят в присутствии инертного разбавителя и катализатора гидрирования.
Особенно предпочтительным является способ, в котором гидрирование проводят при температуре в диапазоне от 0 до 100°С, предпочтительно от 0 до 70°С, особенно предпочтительно от 25 до 60°С.
Также предпочтительным является способ, в котором гидрирование проводят при давлении, равном более чем от 0,5 до 100 бар, предпочтительно при давлении, равном от 1 до 10 бар, особенно предпочтительно примерно при 1-5 бар.
Инертными разбавителями могут быть протонные растворители, такие как, например, спирты, карбоновые кислоты и/или вода, или апротонные полярные растворители, такие как, например, простые эфиры и/или амиды или лактамы и/или их смеси. Ко всем растворителям необязательно можно прибавить воду. Использующимися протонными растворителями предпочтительно являются разветвленные или неразветвленные C1-C8-алканолы, С1-С3-карбоновые кислоты или их смеси. Особенно предпочтительно использовать низшие спирты, такие как метанол, этанол, н-пропанол и изопропанол, карбоновые кислоты, такие как муравьиная кислота, уксусная кислота и пропионовая кислота или их смеси. В качестве реакционной среды особенно предпочтительно использовать этанол и/или уксусную кислоту, которые необязательно могут содержать воду. Подходящие апротонные растворители включают полярные простые эфиры, такие как, например, тетрагидрофуран, диоксан или диметоксиэтиловый эфир, или амиды, такие как, например, диметилформамид, или лактамы, такие как, например, N-метилпирролидон. Особенно предпочтительными являются ТГФ и/или уксусная кислота, которые необязательно могут содержать воду в любом соотношении. Предпочтительно использовать растворители, обладающие небольшой склонностью к воспламенению. При гидрировании апротонные растворители предпочтительнее протонных растворителей.
Подходящими катализаторами гидрирования обычно являются переходные металлы, такие как, например, никель, платина или палладий, или их соли или оксиды. Предпочтительными являются никель Ренея, оксид платины и палладий на инертном носителе, предпочтительно палладий на активированном древесном угле (Pd/C).
Предпочтительными являются способы, в которых продукт, полученный на стадии (а), во время гидрирования используют в массовом отношении к количеству катализатора гидрирования, составляющем от 1:1 до 1000:1, предпочтительно от 5:1 до 100:1.
В предпочтительном варианте осуществления стадии (b) i) продукт, полученный на стадии (а), растворяют в смеси ТГФ/вода (7:3 в объемном отношении) и при давлении водорода, равном 4 бар, гидрируют с помощью увлажненного водой 10% Pd/C примерно при 40°С. Катализатор отфильтровывают, фильтр промывают смесью ТГФ/вода (7:3) и фильтрат осветляют активированным древесным углем. Древесный уголь отфильтровывают и фильтр промывают смесью ТГФ и воды. Полученный таким образом фильтрат без обработки вводят в реакцию на стадии b) ii).
(С) Необязательное последующее карбонилирование на стадии (b) ii) для получения из соединения формулы (I), в которой R3 обозначает водород, соединения формулы (I) в которой R3 не обозначает водород, без промежуточного выделения продукта гидрирования, проводят путем прямой реакции соединения формулы (I), полученного на стадии (b) i), в которой R3 обозначает водород, с карбонилирующим реагентом R3-X, где R3 обладает указанными выше значениями, кроме водорода, и Х обозначает отщепляющуюся группу. Х предпочтительно может обозначать галоген, такой как, например, хлор или бром, или п-толуолсульфонильную, метансульфонильную или трифторметанметансульфонильную группу. Для получения соединения формулы (I), в которой R3 обозначает н-гексил, особенно предпочтительным является н-гексилхлорформиат. Реакцию предпочтительно проводят при температуре от 10 до 50°С, более предпочтительно при температуре от 10 до 20°С, в присутствии основания. Использующимся основанием предпочтительно может являться карбонат щелочного металла, такой как, например, карбонат калия или карбонат натрия, гидрокарбонат щелочного металла, такой как, например, гидрокарбонат натрия или гидрокарбонат калия, или третичный амин, такой как, например, триэтиламин. Предпочтительно использовать карбонат калия. Реакцию можно, например, проводить в смесях воды и ацетона, воды и диоксана или воды и ТГФ, предпочтительной является смесь вода/ТГФ.
После завершения реакции прозрачную двухфазную смесь можно получить путем нагревания суспензии, например, примерно до 50°С, так чтобы можно было легко отделить водную фазу, которая содержит много неорганических веществ. Затем можно заменить растворитель. Подходящие растворители включают, например, кетоны или сложные эфиры, такие как МИБК, бутилацетат, этилацетат, пропилацетат, изопропилацетат или изобутилацетат. Предпочтительно используют МИБК или бутилацетат, особенно предпочтительным является бутилацетат.
В предпочтительном варианте осуществления стадии (b)ii) продукт, полученный на стадии (b)i) (фильтрат после гидрирования), при температуре окружающей среды объединяют с водным раствором карбоната калия. Затем при температуре от 10-20°С прибавляют карбонилирующий реагент, например, н-гексилхлорформиат. Суспензию нагревают при 50°С и при этом образуется прозрачная двухфазная смесь. В зависимости от результатов проверки степени превращения примерно при 50°С необязательно прибавляют дополнительное количество карбонилирующего реагента, пока реакция эдукта не завершится. Затем ТГФ отгоняют и заменяют на бутилацетат. Органическую фазу несколько раз промывают водой при нагревании до 50-70°С для удаления полярных примесей. Затем остаточную воду удаляют с помощью азеотропной перегонки.
(D) Затем до осаждения соли на стадии (b)iii) можно заменить растворитель. Для этого использующийся ранее растворитель, такой как бутилацетат, отгоняют и заменяют на растворитель, необходимый для осаждения соли. Растворители, подходящие для проведения частичной стадии (b)iii), включают, например, кетоны, такие как, например, ацетон или МИБК, простые эфиры, такие как, например, ТГФ, сложные эфиры, такие как, например, этилацетат, изопропилацетат или изобутилацетат, или спирты, такие как, например, метанол, этанол или изопропанол. Предпочтительно используют ацетон и/или этанол, особенно предпочтительно смесь этих двух растворителей. Затем, путем прибавления соответствующей кислоты, например метансульфоновой кислоты для получения метансульфоната, предпочтительно 1 экв. кислоты, искомую соль можно осадить и сразу же отделить.
В предпочтительном варианте осуществления стадии (b) iii) после замены растворителя на смесь ацетона и этанола, при температуре, равной от примерно 30-36°С, раствор соответствующей кислоты, например метансульфоновой кислоты, в ацетоне медленно прибавляют к продукту, полученному на стадии (b) ii) (раствор для карбонилирования), в присутствии затравочных кристаллов. Суспензию перемешивают и осадившийся продукт отделяют фильтрованием, промывают ацетоном и сушат при подходящих условиях.
(Е) Для получения 2-[4-(1,2,4-оксадиазол-5-он-3-ил)-фениламино]-уксусной кислоты 2-[4-(1,2,4-оксадиазол-5-он-3-ил)-анилин вводят в реакцию с эфиром 2-галогенуксусной кислоты, предпочтительно этилбромацетатом, в присутствии слабого основания, предпочтительно третичного амина, такого как, например, триэтиламин, или карбоната щелочного металла, такого как, например, карбонат натрия, в инертном растворителе и полученный эфир 2-[4-(1,2,4-оксадиазол-5-он-3-ил)-фениламино]-уксусной кислоты омыляют.
Использующимися инертными разбавителями могут быть протонные растворители, такие как, например, спирты, и/или вода или апротонные полярные растворители, такие как, например, простые эфиры, и/или амиды или лактамы и/или их смеси. Ко всем растворителям необязательно можно прибавить воду. Использующимися протонными растворителями предпочтительно являются вода или разветвленные или неразветвленные C1-C8-алканолы или их смеси. Особенно предпочтительно использовать воду или низшие спирты, такие как метанол, этанол, н-пропанол и изопропанол или их смеси. В качестве реакционной среды еще более предпочтительно использовать этанол, и он необязательно может содержать воду. Также можно использовать изопропанол, необязательно вместе с водой. Однако наиболее подходящим растворителем является вода. Подходящими апротонными растворителями являются полярные простые эфиры, такие как, например, тетрагидрофуран или диметоксиэтиловый эфир, или амиды, такие как, например, диметилформамид, или лактамы, такие как, например, N-метилпирролидон.
В особенно предпочтительном варианте осуществления этилбромацетат добавляют в суспензию 2-[4-(1,2,4-оксадиазол-5-он-3-ил)-анилина и карбоната натрия в смеси вода/изопропанол или предпочтительно в смеси вода/этанол и перемешивают при 35-45°С. Охлажденную суспензию фильтруют с отсасыванием, промывают несколькими порциями воды и этанола и сушат.
Омыление предпочтительно проводят в протонном растворителе гидроксидом щелочного металла или щелочноземельного металла, предпочтительно гидроксидом лития, натрия или калия.
В особенно предпочтительном варианте осуществления эфир 2-[4-(1,2,4-оксадиазол-5-он-3-ил)-фениламино]-уксусной кислоты суспендируют в воде или предпочтительно в смеси вода/этанол и медленно объединяют с водным раствором NaOH при температуре окружающей среды. Суспензия превращается в раствор и ее нагревают при температуре от 45 до 75°С. К полученному таким образом раствору прибавляют HCl до установления рН, равного примерно 5, или предпочтительно рН, равного 3. Твердое вещество отделяют и промывают холодной водой и холодным этанолом и МТБЭ.
(F) Для получения 2-[4-(1,2,4-оксадиазол-5-он-3-ил)-анилина 4-аминофениламидоксим вводят в реакцию с диалкилкарбонатом, предпочтительно диметилкарбонатом или диэтилкарбонатом, в присутствии основания, предпочтительно алкоксида щелочного металла, предпочтительно метоксида натрия, этоксида натрия или трет-бутоксида калия.
4-Аминофениламидоксим можно получить например, по реакции 4-аминобензонитрила с гидроксиламингидрохлоридом.
В особенно предпочтительном варианте осуществления метоксид натрия или предпочтительно этоксид натрия при 65-75°С, предпочтительно при 70-75°С, прибавляют к суспензии 4-аминофениламидоксима в этаноле и промывают этанолом. После 15 мин перемешивания по каплям прибавляют диэтилкарбонат или предпочтительно диметилкарбонат. После проведения реакции в течение 2-4 ч смесь охлаждают и этанол отгоняют при 120 мбар и 40°С. Остаток растворяют в воде и после нагревания значение рН доводят до 10-12 с помощью разбавленного вдвое концентрированного раствора гидроксид натрия, затем до рН<6, предпочтительно до рН<4, особенно предпочтительно до рН 2-3, путем подкисления концентрированной хлористоводородной кислотой и медленно охлаждают. Раствор превращается в суспензию, которую фильтруют и несколько раз промывают холодной водой и этанолом.
Получение 2-[4-(1,2,4-оксадиазол-5-он-3-ил)-фениламино]-уксусной кислоты, необходимой для использования в качестве промежуточного продукта, из 4-аминобензонитрила, иллюстрируется приведенной ниже схемой реакций.
Схема I
(Невыделенные промежуточные вещества, заключенные в квадратные скобки, в разных вариантах способа необязательно могут быть разными. Представлен предпочтительный вариант осуществления.)

Получение 4-(бензимидазол-2-илметиламино)-бензамидина в качестве примера представлено на приведенной ниже схеме реакций.
Схема II

Обработку для отдельных реакций можно проводить обычным образом, например путем отделения вспомогательных веществ реакции, удаления растворителей и выделения чистого конечного продукта из остатка с помощью кристаллизации, перегонки, экстракции или хроматографии.
На последней стадии описанного выше способа полученное таким образом соединение формулы (I) превращают в физиологически приемлемую соль. Физиологически приемлемые соли могут быть солями с неорганическими или органическими кислотами или, если соединение содержит карбоксигруппу, с неорганическими или органическими основаниями. Примеры кислот, использующихся для этой цели, включают метансульфоновую кислоту, хлористоводородную кислоту, бромистоводородную кислоту, серную кислоту, фосфорную кислоту, фумаровую кислоту, янтарную кислоту, молочную кислоту, лимонную кислоту, винную кислоту и малеиновую кислоту. Примеры оснований, которые можно использовать, включают гидроксид натрия, гидроксид калия, циклогексиламин, этаноламин, диэтаноламин и триэтаноламин. Соединение формулы (6) предпочтительно превращают в его мезилат.
Способ, предлагаемый в настоящем изобретении, проиллюстрирован приведенными ниже примерами. Специалист в данной области техники должен понимать, что эти примеры предназначены только для иллюстрации и их не следует рассматривать в качестве ограничивающих.
Примеры
Выше и ниже в настоящем изобретении используются следующие аббревиатуры:
Пример 1
Получение 2-[4-(1,2,4-оксадиазол-5-он-3-ил)-анилина (1 В)
(1А)
В сосуд для проведения реакций помещают 41,3 г (0,35 моля) 4-аминобензонитрила и 36,5 г (0,53 моля) гидроксиламингидрохлорида в 175 мл этанола и смесь нагревают при 60°С. К этой суспензии по каплям медленно прибавляют 170,1 г (0,53 моля) раствора этоксида натрия (~21% в этаноле). Затем смесь перемешивают в течение ночи при 60°С. Во время охлаждения до 0-5°С вещество осаждается, его отфильтровывают и несколько раз промывают всего с помощью 70 мл холодного этанола. Получают примерно 86 г влажного продукта. Затем его используют без обработки.
(1В)
32 г (0,35 моля) диметилкарбоната прибавляют к суспензии 86 г (1А) в 270 мл этанола. При 65-75°С прибавляют 125 г (0,38 моля) раствора этоксида натрия (~21% в этаноле) и промывают с помощью 20 мл этанола. После проведения реакции в течение 3 ч смесь охлаждают до 40°С и этанол отгоняют при 120 мбар и 40°С. Получают темный остаток. Его растворяют при 40-45°С в 280 мл воды и после нагревания до 70°С значение рН сначала доводят до 11 путем медленного прибавления разбавленного вдвое концентрированного раствора гидроксида натрия, затем до рН 3-4 или предпочтительно до рН 2-3 путем подкисления концентрированной хлористоводородной кислотой и медленно охлаждают. Раствор переходит в суспензию, которую фильтруют и несколько раз промывают всего с помощью 50 мл холодной воды и 20 мл этанола. Получают примерно 88 г влажного вещества, которое сушат максимально при 50°С в вакууме. Выход 48 г бежевого вещества (77,5% от теоретического значения); температура плавления от 178°С (с разложением); чистота >98% по площади пика в ВЭЖХ (высокоэффективная жидкостная хроматография)
Пример 2
Получение 2-[4-(1,2,4-оксадиазол-5-он-3-ил)-фениламино]-уксусной кислоты (2В)
(2А)
При 45°С 60,2 г (0,36 моля) этилбромацетата добавляют в суспензию 53,2 г (0,3 моля) (1 В) и 19,1 г (0,18 моля) карбоната натрия в 500 мл смеси вода/этанол (от 90:10 до 95:5) и необязательно перемешивают в течение ночи. Реакционная смесь обладает окраской от красновато-коричневой до оранжевой. Суспензию охлаждают до 0°С, фильтруют с отсасыванием, промывают несколькими порциями по 100 мл этанола и сушат максимально при 50°С в вакууме.
Выход 69,5 г коричневато-бежевого вещества (87,7% от теоретического значения); температура плавления от 186,1°С (с разложением); чистота >98% по площади пика в ВЭЖХ.
(2В)
Полученный таким образом сложный эфир (2А) (86,9 г; 0,33 моля) суспендируют в 400 мл воды или предпочтительно в смеси этанол/вода (1:1) и при КТ по каплям медленно прибавляют 120 г 45% NaOH. Суспензия превращается в раствор и становится красноватой (рН 12,5). Ее нагревают при ~60°С и омыляют в течение 1 ч. К полученному раствору порциями прибавляют HCl (37% или предпочтительно концентрированную HCl) до рН 3. Его охлаждают до 0°С. Твердое вещество отфильтровывают с отсасыванием и промывают несколькими порциями холодной воды, всего с помощью 400 мл, а также с помощью 40 мл холодного этанола. Получают 81,4 г влажного вещества. Его сушат при 35°С в вакууме.
Выход 76,7 г вещества (98% от теоретического значения); температура плавления от 193°С (с разложением); чистота>99% по площади пика в ВЭЖХ
Пример 3
Получение N-(2-пиридил)-N-(2-этоксикарбонилэтил)-амида 3-амино-4-метиламинобензойной кислоты (АМАБА) (3)
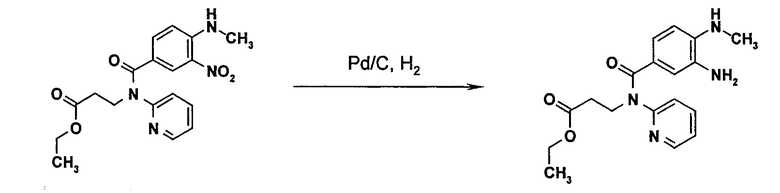
Вариант A: Pd/C 5%
150 г (0,4 моля) N-(2-пиридил)-N-(2-этоксикарбонилэтил)-амида 4-метиламино-3-нитробензойной кислоты, 12 г 5% палладия на древесном угле в качестве катализатора и 627 мл этилацетата помещают в автоклав для гидрирования. Смесь гидрируют в атмосфере водорода при давлении 3-4 бар при 35-55°С, пока потребление водорода не станет постоянным (1-2 ч). После охлаждения до 20°С после гидрирования раствор отфильтровывают от катализатора и выпаривают в вакууме с помощью роторного испарителя. Остаток растворяют в 650 мл изопропанола, отгоняют до половины исходного объема и охлаждают до 5-10°С. Через 4 ч полученную суспензию фильтруют и выделенный таким образом осадок промывают порциями всего с помощью 100 мл изопропанола. Полученное твердое вещество сушат в вакуумном сушильном шкафу при 50°С.
Выход 114,2 г (скорректированный - 83% от теоретического значения).
Вариант В: Pd/C 10%
25 г (0,07 моля) N-(2-пиридил)-N-(2-этоксикарбонилэтил)-амида 4-метиламино-3-нитробензойной кислоты, 2,5 г 10% палладия на древесном угле в качестве катализатора и 83 мл этилацетата помещают в автоклав для гидрирования. Смесь гидрируют в атмосфере водорода при давлении 3-4 бар при 50°С, пока потребление водорода не станет постоянным (4-5 ч). После охлаждения до 20°С после гидрирования раствор отфильтровывают от катализатора и выпаривают в вакууме с помощью роторного испарителя. Остаток растворяют в теплом состоянии в небольшом количестве этилацетата и объединяют с 68 мл толуола. После охлаждения до 5°С смесь выдерживают в течение 1 ч при перемешивании, затем осадок отфильтровывают и промывают толуолом. Полученный продукт сушат при 40°С в вакуумном сушильном шкафу. Выход 20,9 г (скорректированный - 91% от теоретического значения).
Пример 4
Получение N-(2-пиридил)-N-(2-этоксикарбонилэтил)-амида 1-метил-2-[N-[4-(1,2,4-оксадиазол-5-он-3-ил)фенил]-аминометил]-бензимидазол-5-илкарбоновой кислоты (4)
Вариант А: КДИ в качестве реагента сочетания
11,35 г (70 ммоля) 1,1'-карбонилдиимидазод суспендируют в 100 мл ТГФ и нагревают до 50°С. Порциями прибавляют 14,23 г (60,5 ммоля) (2 В). 17,1 г (50 ммоля) АМРВА 3 растворяют в 37 мл ТГФ при нагревании до 50°С. Примерно через 90 мин суспензию добавляют в раствор АМРВА и промывают с помощью 20 мл ТГФ. Реакционную смесь перемешивают в течение примерно 18 ч и затем, после прибавления 100 мл уксусной кислоты, кипятят с обратным холодильником, так что ТГФ отгоняется. Примерно через 1 ч смесь объединяют с 400 мл воды и перемешивают. Раствор охлаждают, осадившееся розовое твердое вещество отфильтровывают, промывают 2 порциями по 20 мл воды и сушат максимально при 50°С в вакууме. Выход 24,8 г вещества (75% от теоретического значения); температура плавления от 167°С с разложением (ДСК (дифференциальная сканирующая калориметрия)); чистота >95% по площади пика в ВЭЖХ.
Вариант В: АПС в качестве реагента сочетания
34,2 г (0,1 моля) АМАБА 3, 27,5 г (0,12 моля) (2В) и 30,3 г (0,23 моля) ДИПЭА помещают в 170 мл ТГФ и охлаждают до температуры, немного меньшей температуры окружающей среды. Затем прибавляют 85 г (0,13 моля) АПС (в виде ~50% раствора в этилацетате). Смесь перемешивают в течение еще 90 мин и затем растворитель отгоняют. В заключение прибавляют 73,5 г уксусной кислоты и смесь нагревают до внутренней температуры, равной 90°С. Затем прибавляют 400 мл этанола или предпочтительно смеси 400 мл этанол/вода (примерно 85:15) и кизельгур в качестве вспомогательного фильтрующего средства (например, Clarcel®) и смесь фильтруют в горячем виде. Раствор охлаждают, осадившееся твердое вещество отфильтровывают и промывают 2 порциями по 50 мл этанола и сушат максимально при 50°С в вакууме.
Выход 56 г вещества (85% от теоретического значения); температура плавления от 167°С с разложением (ДСК); чистота >95% по площади пика в ВЭЖХ.
Вариант С: пивалоилхлорид в качестве реагента сочетания
96 г (0,41 моля) (2 В) суспендируют в 250 мл NMP и 550 мл ТГФ при 0°С. Разбавленную суспензию последовательно объединяют с 48 г (0,4 моля) пивалоилхлорида и 52 г (0,4 моля) ДИПЭА и перемешивают в течение 30 мин. Затем прибавляют 125 г (0,36 моля) АМАБА 3, растворенного в 800 мл уксусной кислоты, и реакционную смесь кипятят с обратным холодильником в течение 3 ч. ТГФ отгоняют в слабом вакууме и при нагревании прибавляют 1600 мл воды.
Твердое вещество отделяют при 5°С, промывают с помощью 550 мл воды и сушат в течение ночи в сушильном шкафу с циркуляцией воздуха максимально при 50°С.
Выход 183 г (76%); чистота >95% по площади пика в ВЭЖХ.
Пример 5
Получение N-(2-пиридил)-N-(2-этоксикарбонилэтил)-амида 1-метил-2-N-[4-(N-н-гексилоксикарбониламидино)фенил]-аминометил]-бензимидазол-5-илкарбоновой кислоты (6) из N-(2-пиридил)-N-(2-этоксикарбонилэтил)-амида 1-метил-2-[N-[4-(l,2,4-оксадиазол-5-он-3-ил)фенил]-аминометил]-бензимидазол-5-илкарбоновой кислоты (4)
60 г (91 ммоля) N-(2-пиридил)-N-(2-этоксикарбонилэтил)-амида 1-метил-2-[N-[4-(1,2,4-оксадиазол-5-он-3-ил)фенил]-аминометил]-бензимидазол-5-илкарбоновой кислоты (4) гидрируют с помощью 3,0 г 10% палладия на древесном угле (увлажненного водой) в 126 мл ТГФ и 54 мл воды при 40°С и избыточном давлении водорода, равном 4 бар, в течение 25 мин. После гидрирования раствор фильтруют и фильтр промывают с помощью 75 г смеси ТГФ/вода (7:3). Фильтрат последовательно объединяют с 56 мл ТГФ, 260 мл воды и порциями с 75,2 г (544 ммоля) карбоната калия при температуре окружающей среды. Затем в течение 40 мин прибавляют 14,2 г (86 ммоля) н-гексилхлорформиата. После проверки степени превращения прибавляют еще 1,2 г (7,3 ммоля) н-гексилхлорформиата, чтобы в реакцию вступило все исходное вещество. Суспензию нагревают примерно при 45°С. Образуется прозрачная двухфазная смесь. Водную фазу отбрасывают и ТГФ в основном отгоняют. К суспензии прибавляют 150 мл ацетона, ее нагревают при 50°С, фильтруют и получают прозрачный фильтрат. Фильтр промывают с помощью 100 мл ацетона. Фильтрат охлаждают до температуры окружающей среды и продукт осаждают путем медленного прибавления 100 мл воды. Влажный продукт промывают с помощью 150 мл смеси ацетон/вода (1:1) и 150 мл воды и сушат в вакууме.
Выход 56,9 г (94%);
ВЭЖХ-чистота >98,8%.
Пример 6
Получение N-(2-пиридил)-N-(2-этоксикарбонилэтил)-амидмезилата 1 -метил-2-[N-[4-(N-н-гексилоксикарбониламидино)фенил]-аминометил]-бензимидазол-5-илкарбоновой кислоты (7)
100 г (0,16 моля) соединения (6) при нагревании растворяют в 890 мл ацетона и объединяют с раствором 15 г (0,16 моля) метансульфоновой кислоты в 200 мл ацетона. Раствор фильтруют и после прибавления 77 мл ацетона охлаждают примерно до 20°С. Осадившийся продукт отделяют и промывают ацетоном. Затем смесь сушат максимально при 50°С в вакуумном сушильном шкафу.
Выход 90-98% (103-113 г).
Пример 7
Получение N-(2-пиридил)-N-(2-этоксикарбонилэтил)-амидмезилата 1 -метил-2-[N-[4-(N-н-гексилоксикарбониламидино)фенил]-аминометил]-бензимидазол-5-илкарбоновой кислоты (7) из N-(2-пиридил)-N-(2-этоксикарбонилэтил)-амида 1-метил-2-[N-[4-(1,2,4-оксадиазол-5-он-3-ил)фенил]-аминометил]-бензимидазол-5-илкарбоновой кислоты (4)
60 г (91 ммоля) соединения (4) (необязательно содержащего ацетат) гидрируют с помощью 3,0 г 10% палладия на древесном угле (увлажненного водой) в 126 мл ТГФ и 54 мл воды при 40°С и избыточном давлении водорода, равном 4 бар, в течение 30 мин. После гидрирования раствор фильтруют, фильтр промывают с помощью 51 г смеси ТГФ/вода (7:3) и к фильтрату прибавляют активированный древесный уголь. Активированный древесный уголь отфильтровывают и фильтр промывают с помощью 102 мл ТГФ и 80 мл воды. Фильтрат при температуре окружающей среды объединяют с раствором 75,2 г (544 ммоля) карбоната калия в 80 мл воды и при 10-20°С в течение 1 ч прибавляют 14,6 г (88,9 ммоля) н-гексилхлорформиата. Суспензию нагревают примерно при 50°С. Образуется прозрачная двухфазная смесь, к которой после проверки степени превращения прибавляют еще 0,452 г (2,7 ммоля) н-гексилхлорформиата, чтобы в реакцию вступило все исходное вещество. После отделения водной фазы 180 мл ТГФ отгоняют и заменяют на 350 мл бутилацетата. Органическую фазу дважды экстрагируют с помощью 30 мл воды при 50-70°С, 210 мл бутилацетата отгоняют и заменяют на 300 мл ацетона и 60 мл этанола. Раствор охлаждают до 30-36°С, смешивают с затравочными кристаллами (7) (полученными, например, с помощью реакции, проведенной ранее в соответствии с примером 5, или по методике, описанной в примере 3 в WO 03/074056) и по каплям прибавляют полученный ранее раствор 7,84 г (82 ммоля) метансульфоновой кислоты в 50 мл ацетона. Суспензию перемешивают, продукт отделяют фильтрованием и промывают ацетоном. Выделенное вещество сушат при 45°С в вакууме.
Выход 56,2 г (86%);
чистота >99% по площади пика в ВЭЖХ.
Другие соединения формулы (I) и их соли можно получить по аналогии с описанными выше примерами.
| название | год | авторы | номер документа |
|---|---|---|---|
| УЛУЧШЕННЫЙ СПОСОБ ПОЛУЧЕНИЯ 4-(БЕНЗИМИДАЗОЛИЛМЕТИЛАМИНО)-БЕНЗАМИДОВ И ИХ СОЛЕЙ | 2006 |
|
RU2455292C2 |
| СПОСОБ ПОЛУЧЕНИЯ 4-(БЕНЗИМИДАЗОЛИЛМЕТИЛАМИНО)БЕНЗАМИДИНОВ | 2005 |
|
RU2401264C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ПРОЯВЛЯЮЩАЯ АКТИВНОСТЬ АНТАГОНИСТА АНГИОТЕНЗИНА II И СПОСОБ АНТАГОНИЗИРОВАНИЯ АНГИОТЕНЗИНА II У МЛЕКОПИТАЮЩИХ | 1992 |
|
RU2104276C1 |
| НОВЫЕ ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛА И СОДЕРЖАЩИЕ ЭТИ СОЕДИНЕНИЯ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2000 |
|
RU2243226C2 |
| ИНГИБИТОРЫ КАНАЛОВ С ТРАНЗИТОРНЫМ ПОТЕНЦИАЛОМ НА ОСНОВЕ ОКСАДИАЗОЛОВ | 2019 |
|
RU2818244C2 |
| ПУРИНОВЫЕ ПРОИЗВОДНЫЕ КАК ИНГИБИТОРЫ ТИРОЗИНПРОТЕИНАЗЫ SYK | 2000 |
|
RU2248977C2 |
| 2,3-ЗАМЕЩЕННЫЕ ПИРАЗИНСУЛЬФОНАМИДЫ В КАЧЕСТВЕ ИНГИБИТОРОВ CRTH2 | 2006 |
|
RU2453540C2 |
| ГЕТЕРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ АНТАГОНИЗИРОВАНИЯ АНГИОТЕНЗИНА II | 1992 |
|
RU2168510C2 |
| Способ получения 3-амино- 2-пиразолиновых производных или их солей | 1970 |
|
SU470959A3 |
| ПРОИЗВОДНЫЕ АЛЬФА-(N-СУЛЬФОНАМИДО)АЦЕТАМИДА КАК ИНГИБИТОРЫ БЕТА-АМИЛОИДА | 2002 |
|
RU2300518C2 |
Изобретение описывает способ получения необязательно замещенного 4-бензимидазол-2-илметиламино)-бензамидина формулы (I) с органическими или неорганическими кислотами 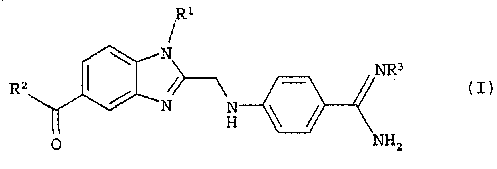
в которой R1, R2 и R3 такие, как представлено в п.1 формулы изобретения, характеризующийся тем, что (а) фенилдиамин формулы (II) 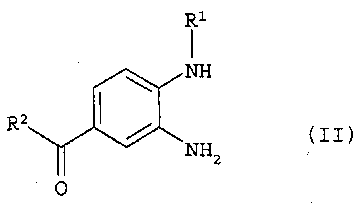
в которой R1 и R2 обладают значениями, указанными для формулы (I), конденсируют с 2-[4-(1,2,4-оксадиазол-5-он-3-ил)-фениламино]-уксусной кислотой и (b) i) полученный продукт формулы (III) 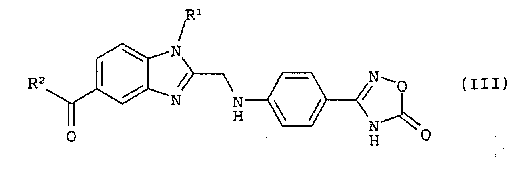
в которой R1 и R2 обладают значениями, указанными для формулы (I), гидрируют, ii) амидиновую группу необязательно карбонилируют без предварительного выделения промежуточного продукта гидрирования и iii) полученное таким образом соединение формулы (I), в которой R3 обозначает водород, без предварительного выделения промежуточного продукта карбонилирования, необязательно вводят в реакцию с соединением формулы (IV)  , в которой R3 обладает значением, указанным для формулы (I), и X обозначает подходящую отщепляющуюся группу, и отделяют искомую соль. Технический результат: описан новый способ получения замещенного 4-бензимидазол-2-илметиламино)-бензамидина, который позволяет получать искомое соединение с высоким выходом и с использованием недорогих вспомогательных веществ. 9 з.п. ф-лы.
, в которой R3 обладает значением, указанным для формулы (I), и X обозначает подходящую отщепляющуюся группу, и отделяют искомую соль. Технический результат: описан новый способ получения замещенного 4-бензимидазол-2-илметиламино)-бензамидина, который позволяет получать искомое соединение с высоким выходом и с использованием недорогих вспомогательных веществ. 9 з.п. ф-лы.
1. Способ получения соли необязательно замещенного 4-бензимидазол-2-илметиламино)-бензамидина формулы (I) с органическими или неорганическими кислотами

в которой R1 обозначает C1-С6-алкильную или С3-С7-циклоалкильную группу,
R2 обозначает
(i) С1-С6-алкильную, С3-С7-циклоалкильную группу, необязательно замещенную C1-С3-алкильной группой, где С1-С3-алкильная группа может быть дополнительно замещена карбоксигруппой или группой, которая in vivo может быть превращена в карбоксигруппу, или
(ii) группу R21NR22, в которой
R21 обозначает C1-С6-алкильную группу, которая может быть замещена карбоксигруппой, С1-С6-алкоксикарбонилом, бензилоксикарбонилом, С1-С3-алкилсульфониламинокарбонилом, фенилсульфониламинокарбонилом, трифторсульфониламиногруппой, трифторсульфониламинокарбонилом или 1H-тетразолильной группой,
С2-С4-алкильную группу, замещенную гидроксигруппой, фенил-С1-С3-алкоксигруппой, карбокси-С1-С3-алкиламиногруппой, С1-С3-алкоксикарбонил-С1-С3-алкиламиногруппой, N-(C1-С3-алкил)-карбокси-С1-С3-алкиламиногруппой или N-(C1-С3-алкил)-С1-С3-алкоксикарбонил-С1-C3-алкиламиногруппой, где в указанных выше группах атом углерода, находящийся в α-положении по отношению к соседнему атому азота, не может быть замещенным, или пиперидинильную группу, необязательно замещенную С1-С3-алкильной группой, и
R22 обозначает атом водорода, C1-С6-алкильную группу, С3-С7-циклоалкильную группу, необязательно замещенную С1-С3-алкильной группой, или С3-С6-алкенильную или С3-С6-алкинильную группу, где ненасыщенный фрагмент может не быть непосредственно связан с атомом азота группы R21NR22, фенильную группу, необязательно замещенную атомом фтора, хлора или брома, С1-С3-алкильной или С1-С3-алкоксигруппой, или бензильную, оксазолильную, изоксазолильную, тиазолильную, изотиазолильную, пиразолильную, пиридинильную, пиримидинильную, пиразинильную, пиридазинильную, пирролильную, тиенильную или имидазолильную группу, необязательно замещенную С1-С3-алкильной группой, или
R21 и R22 вместе с расположенным между ними атомом азота обозначают 5-7-членную циклоалкилениминогруппу, необязательно замещенную карбоксигруппой или С1-С4алкоксикарбонильной группой, с которым дополнительно может быть сконденсировано фенильное кольцо, и
R3 обозначает атом водорода, С1-С9-алкоксикарбонильную, циклогекси-локсикарбонильную, фенил-С1-С3-алкоксикарбонильную, бензоильную, п-С1-С3-алкилбензоильную или пиридиноильную группу, где этоксигруппа в положении 2 указанной выше С1-С9-алкоксикарбонильной группы может быть дополнительно замещена С1-С3-алкилсульфонильной или 2-(С1-С3-алкокси)этильной группой, характеризующийся тем, что
(а) фенилдиамин формулы (II)
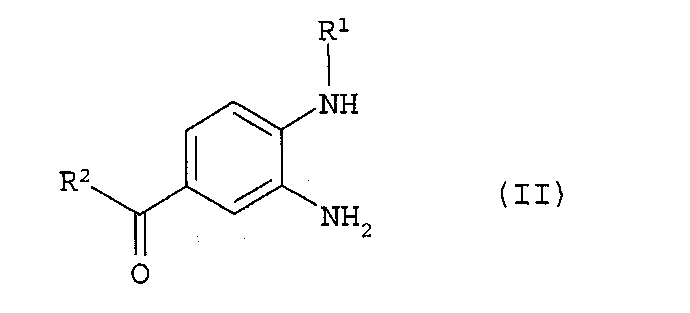
в которой R1 и R2 обладают значениями, указанными для формулы (I), конденсируют с 2-[4-(1,2,4-оксадиазол-5-он-3-ил)-фениламино]уксусной кислотой и
(b) i) полученный продукт формулы (III)
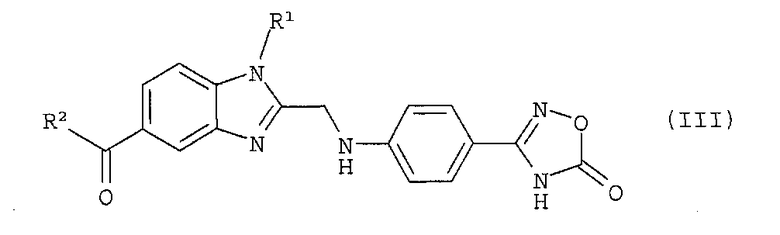
в которой R1 и R2 обладают значениями, указанными для формулы (I), гидрируют,
ii) амидиновую группу необязательно карбонилируют без предварительного выделения промежуточного продукта гидрирования и
iii) полученное таким образом соединение формулы (I), в которой R3 обозначает водород, без предварительного выделения промежуточного продукта карбонилирования, необязательно вводят в реакцию с соединением формулы (IV)
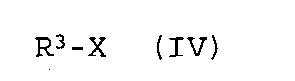
в которой R3 обладает значением, указанным для формулы (I), и X обозначает подходящую отщепляющуюся группу, и отделяют искомую соль.
2. Способ по п.1 получения соли соединения формулы (I), в которой
R1 обозначает С1-С3-алкильную группу,
R2 обозначает группу R21NR22, в которой
R21 обозначает С1-С3-алкильную группу, которая может быть замещена карбоксигруппой, С1-С3-алкоксикарбонилом, и
R22 обозначает атом водорода, С1-С3-алкильную группу, пиридинильную группу, необязательно замещенную C1-С3-алкильной группой, и
R3 обозначает атом водорода или С1-С8-алкоксикарбонильную группу.
3. Способ по п.2 получения соли соединения формулы (I), в которой
R1 обозначает метильную группу,
R2 обозначает группу R21NR22, где
R21 обозначает этильную группу, которая замещена этоксикарбонильной группой, и
R22 обозначает пиридин-2-ильную группу, и
R3 обозначает н-гексилкарбонильную группу.
4. Способ по п.1, отличающийся тем, что полученное таким образом соединение формулы (I) затем превращают в физиологически приемлемую соль.
5. Способ по п.4, отличающийся тем, что физиологически приемлемой солью является метансульфонат, гидрохлорид, малеат, тартрат, салицилат, цитрат или малонат.
6. Способ по п.5, отличающийся тем, что физиологически приемлемой солью является метансульфонат.
7. Способ по п.1, отличающийся тем, что конденсацию на стадии (а) проводят в присутствии инертного разбавителя и связывающего воду агента.
8. Способ по п.1, отличающийся тем, что стадию гидрирования (b)i) проводят в присутствии инертного разбавителя и катализатора гидрирования.
9. Способ по п.1, отличающийся тем, что для получения 2-[4-(1,2,4-оксадиазол-5-он-3-ил)-фениламино]уксусной кислоты 2-[4-(1,2,4-оксадиазол-5-он-3-ил)-анилин вводят в реакцию с эфиром 2-галогенуксусной кислоты в присутствии слабого основания и полученный эфир 2-[4-(1,2,4-оксадиазол-5-он-3-ил)-фениламино]уксусной кислоты омыляют.
10. Способ по п.1, отличающийся тем, что для получения 2-[4-(1,2,4-оксадиазол-5-он-3-ил)-анилина 4-аминофениламидоксим вводят в реакцию с диалкилкарбонатом в присутствии основания.
| Дорожная спиртовая кухня | 1918 |
|
SU98A1 |
| ПРОИЗВОДНЫЕ БЕНЗАМИДИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1998 |
|
RU2194044C2 |

