Настоящее изобретение относится к способу получения особого производного индолинона и его фармацевтически приемлемой соли, а именно, 3-Z-[1-(4-(N-((4-метилпиперазин-1-ил)-метилкарбонил)-N-метиламино)-анилино)-1-фенилметилен]-6-метоксикарбонил-2-индолинона и его моноэтансульфоната, к новым стадиям способа получения и к новым промежуточным продуктам для этого способа.
Производное индолинона, 3-Z-[1-(4-(N-((4-метилпиперазин-1-ил)-метилкарбонил)-N-метиламино)-анилино)-1-фенилметилен]-6-метоксикарбонил-2-индолинон, и его моноэтансульфонат являются известными из следующих заявок: WO 01/027081, WO 04/013099, WO 04/017948, WO 04/096224 и WO 06/067165. В этих заявках раскрыто соединение, способ его получения, особая соль этого соединения и использование этого соединения или его соли в фармацевтической композиции, предназначенной для лечения онкологических или неонкологических заболеваний путем подавления пролиферации клеток-мишеней, по отдельности или в комбинации с другими терапевтическими средствами. Механизм действия, по которому происходит пролиферация клеток-мишеней, по существу представляет собой механизм ингибирования различных рецепторов тирозинкиназы и, в частности, ингибирование рецептора сосудистого эндотелиального фактора роста (РСЭФР).
Хотя в указанных выше заявках уже описан способ получения указанного выше производного индолинона и его моноэтансульфоната, объектом настоящего изобретения является новый и улучшенный способ получения этого соединения. Таким образом, по сравнению со способами, уже описанными в предшествующем уровне техники, способ, предлагаемый в настоящем изобретении, наряду с другими обладает указанными ниже примечательными преимуществами.
Первым преимуществом является более высокий общий выход, который можно обеспечить с помощью нового и улучшенного способа. Этот более высокий общий выход означает повышение общей эффективности способа. Это также влечет за собой экономическое преимущество.
Вторым преимуществом является то, что новый и улучшенный способ, предлагаемый в настоящем изобретении, является менее вредным для окружающей среды, чем способы, известные из предыдущего уровня техники. Это преимущество основано на том факте, что стадии способа проводят при более высоких концентрациях.
Третьим преимуществом, которое можно отметить, является применимость нового и улучшенного способа, предлагаемого в настоящем изобретении, для крупномасштабного производства. Эта применимость характеризуется наличием надежных стадий реакций, т.е. стадий реакций, менее чувствительных к количествам исходных веществ.
Эти преимущества обеспечивают требуемую высокую чистоту активного фармацевтического ингредиента.
Способ, предлагаемый в настоящем изобретении, является сходящимся способом и включает несколько вариантов, представленных на приведенной ниже общей схеме синтеза, и используется приведенная ниже номенклатура.
Общая схема синтеза
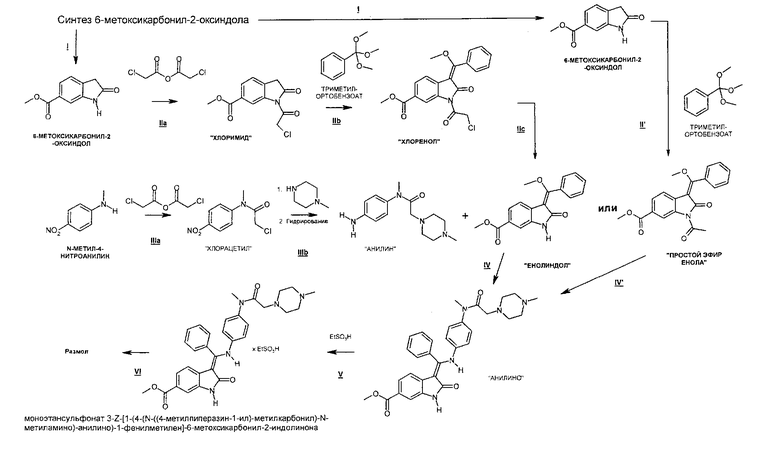
Таким образом, способ включает приведенные ниже стадии реакций.
I. Синтез 6-метоксикарбонил-2-оксиндола
6-Метоксикарбонил-2-оксиндол можно синтезировать способами, описанными на приведенных ниже схемах синтеза А или В. Эти способы описаны в предшествующем уровне техники.
Схема синтеза А
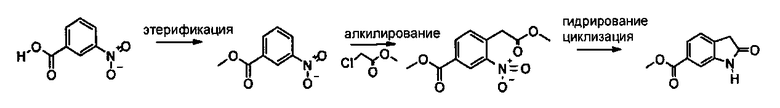
Таким образом, 6-метоксикарбонил-2-оксиндол можно получить по трехстадийной методике, включающей этерификацию 3-нитробензойной кислоты с последующим электрофильным замещением с использованием метилового эфира хлоруксусной кислоты, что приводит к образованию метилового эфира 4-метоксикарбонилметил-3-нитробензойной кислоты, и конечную последовательность реакций гидрирования-внутримолекулярного амидирования.
Схема синтеза В
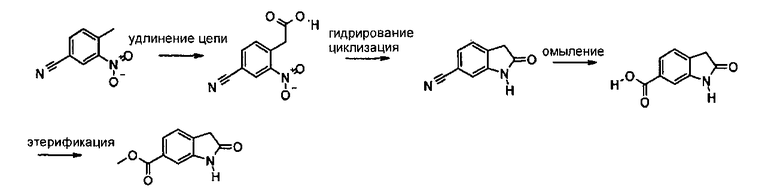
6-Метоксикарбонил-2-оксиндол также можно получить по описанной выше четырехстадийной методике. Сначала проводят удлинение цепи 4-метил-3-нитробензонитрила и восстановительную циклизацию полученной (4-циано-2-нитрофенил)уксусной кислоты с образованием оксиндольного ядра, синтез заканчивают омылением нитрильной группы и последующей этерификацией карбоксильной функциональной группы.
Альтернативно, 6-метоксикарбонил-2-оксиндол также можно синтезировать способом синтеза 2-оксиндолов, описанным в патенте US 6469181.
Альтернативно, 6-метоксикарбонил-2-оксиндол можно синтезировать способом, описанным на приведенной ниже схеме синтеза С.
Схема синтеза C
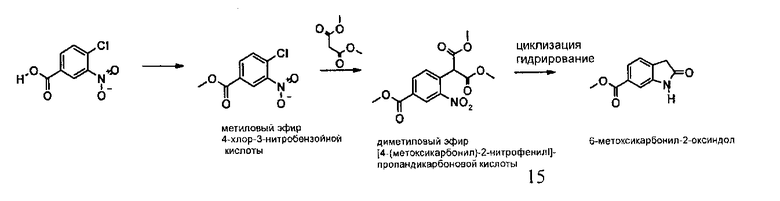
Стадии реакций схемы синтеза C, которые также являются объектом настоящего изобретения, дополнительно описаны в приведенном ниже экспериментальном примере 1, который не следует рассматривать в качестве ограничивающего настоящее изобретение.
IIа. Реакция 6-метоксикарбонил-2-оксиндола с ангидридом хлоруксусной кислоты с образованием "хлоримида" (метил-1-(хлорацетил)-2-оксоиндолин-6-карбоксилата)
Реакцию 6-метоксикарбонил-2-оксиндола с ангидридом хлоруксусной кислоты или другим соответствующим образом активированным производным хлоруксусной кислоты, например, хлорацетилхлоридом, предпочтительно проводят в высококипящем апротонном растворителе, например, толуоле, ксилоле или бутилацетате, при температуре в диапазоне от примерно 80 до примерно 130°C.
Кристаллизацию инициируют путем добавления неполярного растворителя, например, циклогексана или метилциклогексана, при температуре в диапазоне от примерно 80 до примерно 100°C, и заканчивают при температуре в диапазоне от примерно -5°C до комнатной температуры. Твердое вещество собирают, промывают, предпочтительно - полярными растворителями, такими как спирты, наиболее предпочтительно - метанолом, и сушат, и получают "хлоримид".
Алкилирующие реагенты, такие как хлорацетилхлорид или ангидрид хлоруксусной кислоты, можно приобрести у различных фирм-поставщиков. Фирмой-поставщиком больших количеств ангидрида хлоруксусной кислоты является, например, SF-Chem (Switzerland).
Описанная выше стадия реакции IIа и продукт этой реакции, т.е. "хлоримид" (метил-1-(хлорацетил)-2-оксоиндолин-6-карбоксилат), также являются объектом настоящего изобретения,
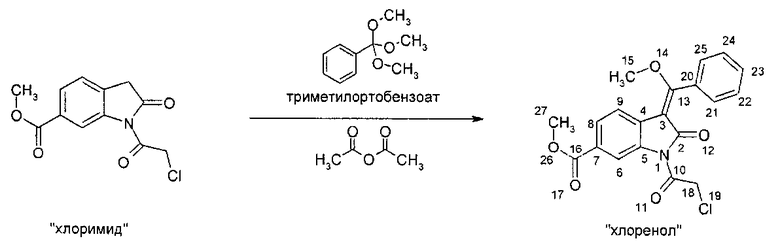
IIb. Реакция "хлоримида" с триметилортобензоатом с образованием "хлоренола" (метил- 1-(хлорацетил)-3-[метокси(фенил)метилен]-2-оксоиндолин-6-карбоксилата)
Реакцию метил-1-(хлорацетил)-2-оксоиндолин-6-карбоксилата с триметилортобензоатом проводят в высококипящем апротонном растворителе, таком как бутилацетат, N,N-диметилформамид, ксилол, или, предпочтительно - толуол, при температуре в диапазоне от примерно 100 до примерно 140°C. Реакции содействуют связывающие метанол реагенты, такие как уксусный ангидрид. В ходе реакции летучие вещества можно отгонять с заменой удаленных веществ на используемый в реакции растворитель или без такой замены. Кристаллизацию заканчивают при температуре в диапазоне от температуры окружающей среды примерно до -10°C. Твердое вещество собирают и промывают, предпочтительно - растворителями, такими как толуол, ксилол и/или этил ацетат. После сушки получают "хлоренол".
Уксусный ангидрид можно приобрести у различных фирм-поставщиков. Триметилортобензоат можно приобрести у фирмы AMI Drugs & Speciality Chemicals India Inc.
Описанная выше стадия реакции IIb и продукт этой реакции, т.е. "хлоренол" (метил-1-(хлорацетил)-3-[метокси(фенил)метилен]-2-оксоиндолин-6-карбоксилат), также являются объектом настоящего изобретения.
IIс. Реакция "хлоренола" с основаниями с образованием "енолидола" (метил-3-[метокси(фенил)метилен]-2-оксоиндолин-6-карбоксилата)
Катализируемое основанием дехлорацетилирование метил-1-(хлорацетил)-3-[метокси(фенил)метилен]-2-оксоиндолин-6-карбоксилата проводят в протонных растворителях, таких как спирты, например, изопропанол или, предпочтительно - метанол, при температуре в диапазоне примерно от 70°C до температуры окружающей среды. В качестве катализатора можно использовать неорганические основания, такие как гидроксиды щелочных металлов, или органические основания, такие как метоксид натрия. Кристаллизацию заканчивают при температуре в диапазоне от температуры окружающей среды примерно до -10°C. Твердое вещество собирают и промывают, предпочтительно - спиртами, наиболее предпочтительно - метанолом. После сушки получают "енолидол".
Описанная выше стадия реакции IIс и продукт этой реакции, т.е. "енолидол" (метил-3-[метокси(фенил)метилен]-2-оксоиндолин-6-карбоксилат), также являются объектом настоящего изобретения.
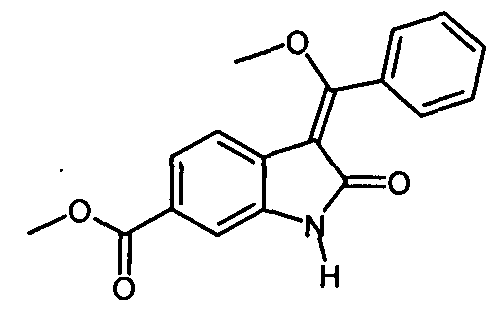
Альтернативно, 6-метоксикарбонил-2-оксиндол можно непосредственно ввести в реакцию с триметилортобензоатом в присутствии уксусного ангидрида и получить "простой эфир енола" (метил-3-[метокси(фенил)метилен]-2-оксоиндолин-6-карбоксилат). Этот альтернативный вариант осуществления описан на общей схеме синтеза как стадия II′, и его можно провести так, как описано выше для стадии реакции IIb.
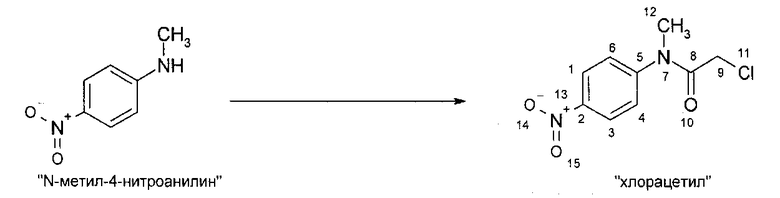
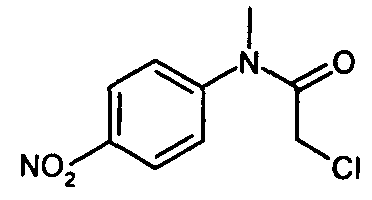
IIIa. Реакция "N-метил-4-нитроанилина с ангидридом хлоруксусной кислоты с образованием "хлорацетила" (N-(4-нитроанилино)-М-метил-2-хлорацетамида)
Хлорацетилирование N-метил-4-нитроанилина проводят в апротонных растворителях, таких как толуол или сложные эфиры, предпочтительно - в этилацетате, при температурах, равных не ниже 60°C. В качестве алкилирующего реагента можно использовать производные хлоруксусной кислоты, предпочтительно - хлорангидрид хлоруксусной кислоты, или, наиболее предпочтительно - ангидрид хлоруксусной кислоты. Кристаллизацию инициируют путем добавления неполярных растворителей, предпочтительно - циклогексана или метилциклогексана, при температуре в диапазоне от примерно 60 до примерно 80°C и заканчивают при температуре в диапазоне от температуры окружающей среды примерно до -10°C. Твердое вещество собирают и промывают, предпочтительно - неполярными растворителями, такими как метилциклогексан. После сушки получают "хлорацетил".
Алкилирующие реагенты, такие как хлорацетилхлорид или ангидрид хлоруксусной кислоты, можно приобрести у различных фирм-поставщиков. Фирмой-поставщиком больших количеств ангидрида хлоруксусной кислоты является, например, SF-Chem (Switzerland). Фирмой-поставщиком исходного вещества, N-метил-4-нитроанилина, является, например, RRJ Dyes & Intermediates Ltd (India).
Описанная выше стадия реакции Ilia также является объектом настоящего изобретения.
IIIb. Реакция "хлорацетила" с 1-метилпиперазином с образованием "нитроанилина" (N-нитрофенил)-N-метил-2-метилпиперазин-1 -ил)ацетамида) и последующее гидрирование с образованием "анилина" (N-(4-aминoфeнил)-N-мeтил-2-(4-мeтилпипepaзин-1-ил)ацетамида).
Начальную реакцию N-(4-нитpoaнилинo)-N-мeтил-2-xлopaцeтaмидa с 1-метилпиперазином проводят в апротонных растворителях, таких как сложные эфиры (например, бутилацетат) и кетоны (например, метилизобутилкетон), или в ароматических растворителях, предпочтительно - в толуоле, при температуре в диапазоне от примерно 30 до примерно 60°C. Затем органические соли удаляют путем экстракции водой или разбавленными водными растворами неорганических солей, например рассолом. Полученную реакционную смесь разбавляют спиртом, предпочтительно - изопропанолом, и гидрируют при температуре в диапазоне от примерно 20 до примерно 90°C, при давлении водорода в диапазоне примерно от 1 до 10 бар с использованием гетерогенных катализаторов гидрирования, таких как палладий на древесном угле. После удаления катализатора большую часть растворителей отгоняют при пониженном давлении и при температуре в диапазоне от примерно 40 до примерно 80°C. Остаток растворяют в толуоле или смеси толуола и сложного эфира, предпочтительно - этилацетата, при температуре в диапазоне от примерно 70 до примерно 90°C, и затем кристаллизуют путем снижения температуры до значений в диапазоне от примерно 10 до примерно -10°C. Кристаллы отделяют и промывают неполярным растворителем, предпочтительно - толуолом, и сушат, и получают "анилин".
Исходное вещество, 1-метилпиперазин, для начальной реакции замещения можно приобрести у различных фирм-поставщиков, например, у фирмы Enzal Chemicals (India) Pvt., Ltd.
Описанная выше стадия реакции IIIb также является объектом настоящего изобретения.
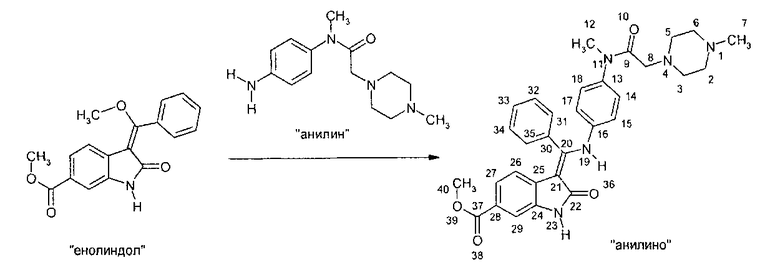
IV Реакция "анилина" с "енолидолом" с образованием "анилино" (3-Z-[1-(4-(М-((4-метилпиперазин-1-ил)-метилкарбонил)-"М-метиламино)-анилино)-1-фенилметилен]-6-метоксикарбонил-2-индолинона).
Эта реакция является стереоспецифичной по отношению к Z- и Е-изомерам. По этой реакции получают изомерную форму Z.
Реакцию метил-3-[метокси(фенил)метилен]-2-оксоиндолин-6-карбоксилата и N-(4-аминофенил)-N-метил-2-(4-метилпиперазин-1-ил)ацетамида проводят в протонных растворителях, таких как спирты, например, в этаноле, или, предпочтительно - в метаноле, или в ароматических растворителях, предпочтительно - в толуоле, или в смеси этих растворителей с высокополярными растворителями, такими как N,N-диметилацетамид, или, предпочтительно - N,N-диметилформамид, при температуре не ниже 50°C при кипячении с обратным холодильником. После завершения превращения кристаллизацию проводят при температуре не ниже температуры окружающей среды. Твердое вещество собирают и последовательно промывают протонным растворителем, таким как этанол, или, предпочтительно - метанол, или ароматическими растворителями, такими как толуол. После сушки выделяют "анилино" в виде желтых кристаллов.
Описанная выше стадия реакции IV также является объектом настоящего изобретения.
Альтернативно, "анилино" (3-Z-[1-(4-(1N-((4-метилпиперазин-1-ил)-метилкарбонил)-N-метиламино)-анилино)-1-фенилметилен]-6-метоксикарбонил-2-индолинон) можно получить по стадии реакции IV', в которой "анилин" вводят в реакцию с "простым эфиром енола" в присутствии MeONa-MeOH. Эта стадия реакции IV' также является объектом настоящего изобретения.
V Реакция "анилино" (3-Z-[1-(4-(N-((4-метилпиперазин-1-ил)-метилкарбонил)-N-метиламино)-анилино)-1-фенилметилен]-6-метоксикарбонил-2-индолинона) с ЕtSO3H с образованием моноэтансульфоната этого соединения.
Образование соли 3-Z-[1-(4-(N-((4-метилпиперазин-1-ил)-метилкарбонил)-N-метиламино)-анилино)-1-фенилметилен]-6-метоксикарбонил-2-индолинона проводят в высокополярных спиртах, например, в этаноле или, предпочтительно - в метаноле, с использованием или без использования воды в качестве сорастворителя, при температуре в диапазоне от примерно 40°C до примерно 70°C, путем добавления чистой этансульфоновой кислоты или ее водного раствора. Осаждение инициируют путем внесения затравки в полученный раствор при температуре в диапазоне от примерно 40°C до примерно 60°C и последующего добавления менее полярного спирта, такого как изопропанол. Кристаллизацию завершают при температуре не выше комнатной. Твердое вещество выделяют, промывают спиртом, таким как метанол или, предпочтительно - изопропанол, и сушат, и получают моноэтансульфонат соединения в виде желтых кристаллов.
Описанная выше стадия реакции V также является объектом настоящего изобретения.
VI. Размол моноэтансульфоната 3-Z-[1-(4-(N-((4-метилпиперазин-1-ил)-метилкарбонил)-N-метиламино)-анилино)-1-фенилметилен]-6-метоксикарбонил-2-индолинона.
Для хранения и последующей обработки моноэтансульфонат соединения, предлагаемого в настоящем изобретении, можно размолоть, например, в ударной мельнице или мельнице с классификатором. Эта стадия размола также является объектом настоящего изобретения.
Таким образом, первым объектом настоящего изобретения является приведенный ниже способ D получения соединения формулы 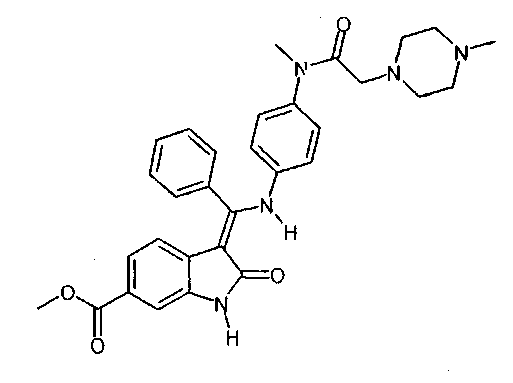 ,
,
включающий стадию реакции соединения формулы 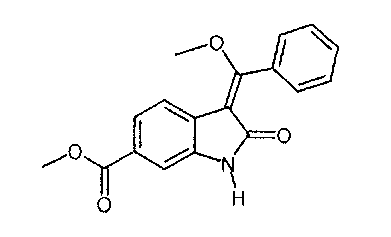
с соединением формулы 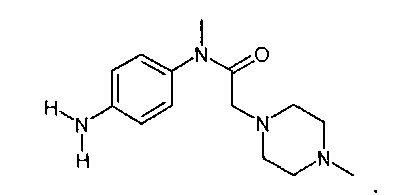
Другим объектом настоящего изобретения является соединение формулы 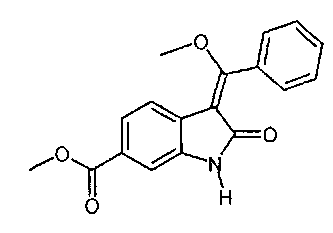 .
.
Другим объектом настоящего изобретения является приведенный ниже способ D1, основанный на описанном выше способе D, и в котором соединение формулы 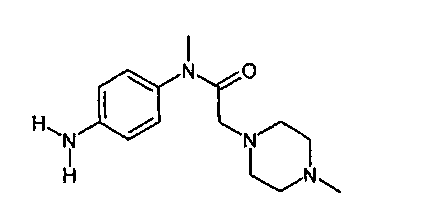
получают по реакции соединения формулы 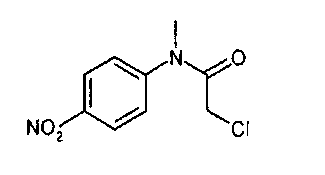
с соединением формулы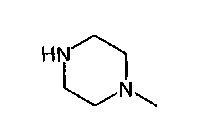
с последующим гидрированием нитрогруппы с образованием аминогруппы.
Другим объектом настоящего изобретения является приведенный ниже способ D2, основанный на описанном выше способе D1, и в котором соединение формулы 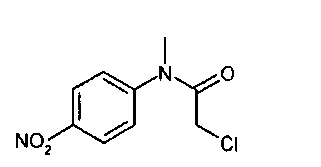
получают по реакции соединения формулы 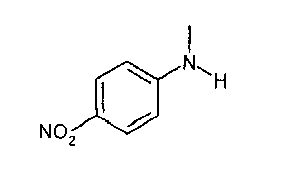
с соединением формулы 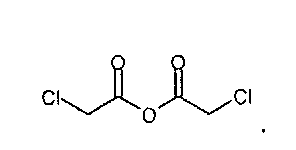
Другим объектом настоящего изобретения является приведенный ниже способ E1, основанный на описанных выше способах D, D1 или D2, и в котором соединение формулы 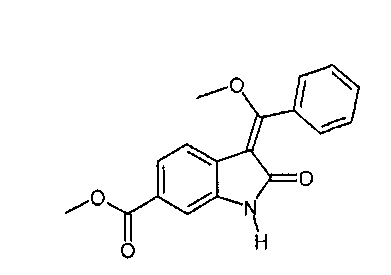
получают путем катализируемого основанием дехлорацетилирования соединения формулы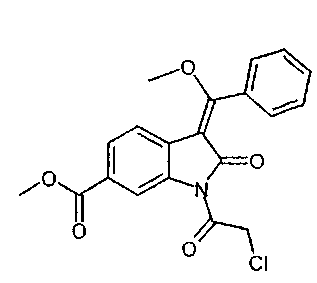 .
.
Другим объектом настоящего изобретения является соединение формулы 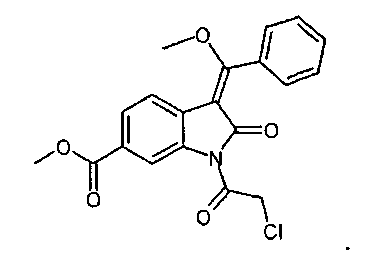
Другим объектом настоящего изобретения является приведенный ниже способ E2, основанный на описанном выше способе E1, и в котором соединение формулы
получают по реакции соединения формулы 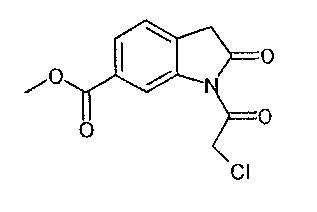
с соединением формулы 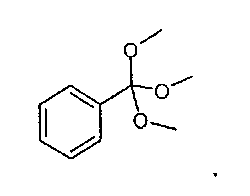
Другим объектом настоящего изобретения является соединение формулы 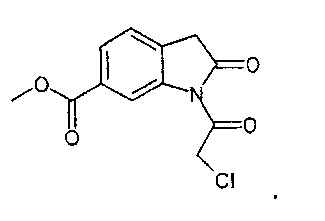
Другим объектом настоящего изобретения является приведенный ниже способ Е3, основанный на описанном выше способе Е2, и в котором соединение формулы 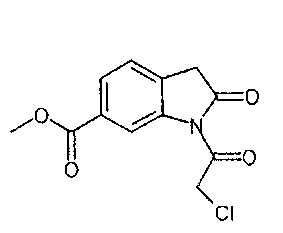
получают по реакции соединения формулы 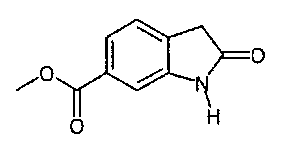
с соединением формулы 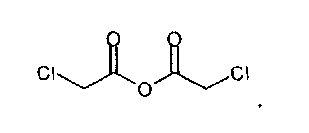
Другим объектом настоящего изобретения является приведенный ниже способ F, основанный на описанном выше способе Е3, и в котором соединение формулы 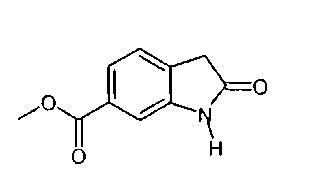 получают с помощью приведенной ниже последовательности реакций:
получают с помощью приведенной ниже последовательности реакций:
(i) этерификация соединения формулы 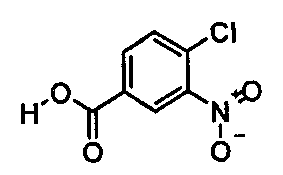
с образованием соединения формулы 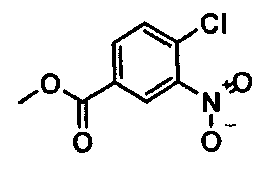 ;
;
(ii) реакция продукта реакции (i) с диметиловым эфиром малоновой кислоты с образованием соединения формулы 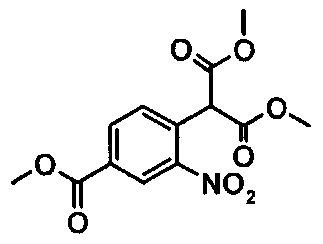 ;
;
(iii) проведение циклизации продукта реакции (ii) по реакции гидрирования.
Другим объектом настоящего изобретения является приведенный ниже способ G, основанный на описанных выше способах D, D1, D2, E1, E2, E3 или F, и в котором соединение формулы 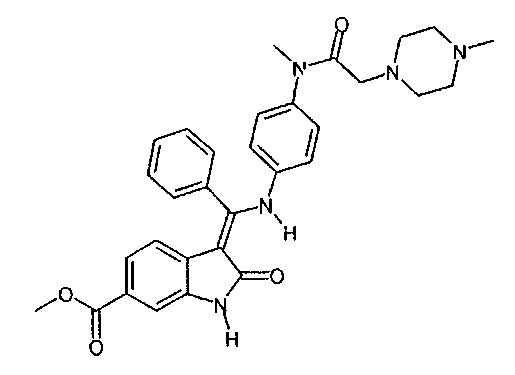
вводят в реакцию с EtSO3H, чтобы получить моноэтансульфонат этого соединения.
Другим объектом настоящего изобретения является приведенный ниже способ H, основанный на описанном выше способе G, который включает стадию размола моноэтансульфоната этого соединения.
Настоящее изобретение более подробно описано с помощью приведенных ниже примеров, которые иллюстрируют дополнительные варианты осуществления, и их не следует рассматривать в качестве ограничивающих настоящее изобретение.
Пример 1: Синтез 6-метоксикарбонил-2-оксиндола способом, описанным на схеме синтеза С.
Синтез метилового эфира 4-хлор-3-нитробензойной кислоты 20 кг 4-Хлор-3-нитробензойной кислоты (99,22 моля) суспендируют в 76 л метанола. В течение 15 мин добавляют 5,9 кг тионилхлорида (49,62 моля) и смесь кипятят с обратным холодильником в течение примерно 3 ч. После охлаждения примерно до 5°C продукт выделяют центрифугированием и сушат при 45°C
Выход: 19,0 кг (88,8% от теоретического).
Чистота (ВЭЖХ (высокоэффективная жидкостная хроматография)): 99,8%.
Синтез диметилового эфира [4-(метоксикарбонил)-2-нитрофенил]-пропандикарбоновой кислоты
12,87 кг Диметилового эфира малоновой кислоты (97,41 моля) добавляют к горячему (75°C) раствору 10,73 кг трет-амилата натрия (97,41 моля) в 35 л 1-метил-2-пирролидинона (NMP). При 75°C добавляют раствор 10 кг метилового эфира 4-хлор-3-нитробензойной кислоты (46,38 моля) в 25 л 1-метил-2-пирролидинона. После перемешивания при температуре, равной примерно 75°C, в течение 1,5 ч и охлаждения до 20°С смесь подкисляют до рН 1 с помощью 100 л разбавленной хлористоводородной кислоты. После перемешивания при температуре, равной примерно 5°C, в течение 1,5 ч продукт выделяют центрифугированием и сушат при 40°C.
Выход: 13,78 кг (95,4% от теоретического).
Чистота (ВЭЖХ): 99,9%.
Альтернативно, диметиловый эфир [4-(метоксикарбонил)-2-нитрофенил]-пропандикарбоновой кислоты можно синтезировать следующим образом:
33,1 кг Диметилового эфира малоновой кислоты (250,6 моля) и 27,0 кг метилового эфира 4-хлор-3-нитробензойной кислоты (125,3 моля) при 20°C последовательно добавляют к раствору 45,1 кг метилата натрия (250,6 моля) в 172 кг 1-метил-2-пирролидинона (NMP). После перемешивания при температуре, равной примерно 45°C, в течение 1,5 ч и охлаждения до 30°С смесь подкисляют с помощью 249 л разбавленной хлористоводородной кислоты. При такой же температуре в смесь вносят затравку, затем ее охлаждают до 0°C и перемешивают в течение еще 1 ч. Полученные кристаллы выделяют центрифугированием, промывают и сушат при 40°C.
Выход: 37,5 кг (86% от теоретического).
Чистота (ВЭЖХ): 99,7%.
Синтез 6-метоксикарбонил-2-оксиндола
Раствор 13 кг диметилового эфира [4-(метоксикарбонил)-2-нитрофенил]-пропандикарбоновой кислоты (41,77 моля) в 88 л уксусной кислоты гидрируют в присутствии 1,3 кг 10% Pd/C при 45°C и давлении, равном 40-50 фунт-сила/дюйм. После остановки гидрирования реакционную смесь нагревают при 115°C в течение 2 ч. Катализатор отфильтровывают и при температуре, равной примерно 50°C, добавляют 180 л воды. Продукт выделяют после охлаждения до 5°C, центрифугирования и сушки при 50°C.
Выход: 6,96 кг (87,2% от теоретического).
Чистота (ВЭЖХ): 99,8%.
Пример 2: Синтез "хлоримида" (метил-1-(хлорацетил)-2-оксоиндолин-6-карбоксилата) 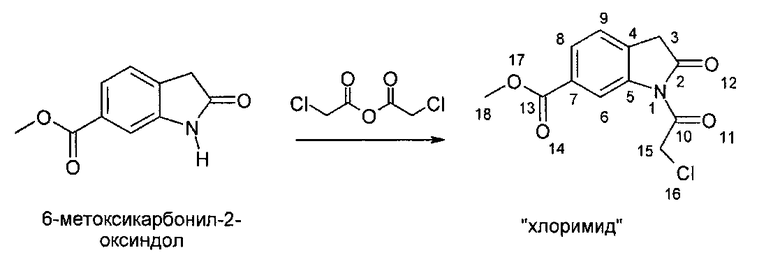
Методика 1
6-Метоксикарбонил-2-оксиндол (400 г; 2,071 моля) при комнатной температуре суспендируют в толуоле (1200 мл). К этой суспензии добавляют ангидрид хлоруксусной кислоты (540 г; 3,095 моля). Смесь кипятят с обратным холодильником в течение 3 ч, затем охлаждают до 80°C и в течение 30 мин добавляют метилциклогексан (600 мл). Затем полученную суспензию в течение 60 мин охлаждают до комнатной температуры. Маточный раствор отделяют и твердое вещество промывают охлажденным льдом метанолом (400 мл). Кристаллы сушат и получают 515,5 г (93,5%) "хлоримида" в виде белого твердого вещества. 1Н-ЯМР (ядерный магнитный резонанс) (500 МГц, ДМСО-d6) δ: 8,66 (s, 1Н, 6-Н); 7,86 (d, J=8,3 Гц, 1Н, 8-Н); 7,52 (d, J=8,3 Гц, 1Н, 9-Н); 4,98 (s, 2Н, 15-Н2); 3,95 (s, 3Н, 18-Н3); 3,88 (s, 2Н, 3-Н2). 13С-ЯМР (126 МГц, ДМСО-d6)δ: 174,7 (С-2); 36,0 (С-3); 131,0 (С-4); 140,8 (С-5); 115,7 (С-6); 128,9 (С-7); 126,1 (С-8); 124,6 (С-9); 166,6 (С-10); 165,8 (С-13); 46,1 (С-15); 52,3 (С-18). МС (масс-спектроскопия): m/z 268 (М+Н)+ Анализ: Рассчитано для C12H10ClNO4: С, 53,85; Н, 3,77; Сl, 13,25; N, 5,23. Найдено: С, 52,18; Н, 3,64; Сl, 12,89; N,5,00.
Методика 2
6-Метоксикарбонил-2-оксиндол (10 г; 0,052 моля) при комнатной температуре суспендируют в н-бутилацетате (25 мл). К этой суспензии в течение 3 мин добавляют раствор ангидрида хлоруксусной кислоты (12,8 г; 0,037 моля) в н-бутилацетате (25 мл). Смесь кипятят с обратным холодильником в течение 2 ч, затем охлаждают до 85°C и добавляют метилциклогексан (20 мл). Затем полученную суспензию охлаждают до комнатной температуры и перемешивают в течение 2 ч. Маточный раствор отделяют и твердое вещество при температуре окружающей среды промывают метанолом (400 мл). Кристаллы сушат и получают 12,7 г (91,5%) "хлоримида" в виде желтоватого твердого вещества.
Пример 3: Синтез "хлоренола" (метил-1-(хлорацетил)-3-[метокси(фенил)метилен]-2-оксоиндолин-6-карбоксилата)
Методика 1
Метил-1-(хлорацетил)-2-оксоиндолин-6-карбоксилат (12,0 г; 0,045 моля) при температуре окружающей среды суспендируют в толуоле (60 мл). К этой суспензии добавляют уксусный ангидрид (16,2 г; 0,157 моля). Смесь нагревают до температуры, равной не ниже 104°C, и в течение 60 мин добавляют триметилортобензоат (20,0 г; 0,108 моля). В ходе добавления и последующего перемешивания при такой же температуре в течение 3 ч содержащиеся в реакционной смеси летучие вещества отгоняют. Концентрацию реакционной смеси поддерживают постоянной путем замены отогнанных веществ толуолом (40 мл). Смесь охлаждают до 5°C, перемешивают в течение 1 ч и фильтруют. Твердое вещество последовательно промывают толуолом (14 мл) и смесью толуола (8 мл) и этилацетата (8 мл). После сушки выделяют 16,3 г (91,7%) "хлоренола" в виде желтоватых кристаллов. 1Н-ЯМР (500 МГц, ДМСО-d6) δ: 8,73 (d, J=1,5 Гц, 1Н, 6-Н); 8,09 (d, J=8,0 Гц, 1Н, 9-Н); 7.90 (dd, J=8,1; 1,5 Гц, 1Н, 8-Н); 7,61-7,48 (m, 5Н, 21-Н, 22-Н, 23-Н, 24-Н, 25-Н); 4,85 (s, 2Н, 18-Н3); 3,89 (s, 3Н, 27-Н3); 3,78 (s, 3Н, 15-Н3). 13С-ЯМР (126 МГц, ДМСО-d6)δ:165,9 (С-2+С16); 103,9 (С-3); 127,4; 128,6; 130,0; 135,4 (С-4+С-5+С-7+С-20); 115,1 (С-6); 126,1 (С-8); 122,5 (С-9); 166,7 (С-10); 173,4 (С-13); 58,4 (С-15); 46,4 (С-18); 128,6 (С-21+С-22+С-24+С-25); 130,5 (С-23); 52,2 (C-27). MC: m/z 386 (М+Н)+. Анализ: Рассчитано для C20H16ClNO5: С, 62,27; Н, 4,18; Cl, 9,19; N, 3,63. Найдено: С, 62,21; Н, 4,03; Cl, 8,99; N, 3,52.
Методика 2
Метил-1-(хлорацетил)-2-оксоиндолин-6-карбоксилат при температуре окружающей среды (12,0 г; 0,045 моля) суспендируют в ксилоле (60 мл). К этой суспензии добавляют уксусный ангидрид (16,2 г; 0,157 моля). Смесь кипятят с обратным холодильником, в течение 40 мин добавляют триметилортобензоат (20,0 г; 0,108 моля) и нагревание продолжают в течение 4 ч. Смесь охлаждают до 0°C и маточный раствор отделяют. Твердое вещество последовательно промывают ксилолом (14 мл) и смесью ксилола (8 мл) и этилацетата (8 мл). После сушки выделяют 14,3 г (81,0%) "хлоренола" в виде желтых кристаллов.
Методика 3
Метил-1-(хлорацетил)-2-оксоиндолин-6-карбоксилат (12,0 г; 0,045 моля) при температуре окружающей среды суспендируют в толуоле (60 мл). К этой суспензии добавляют уксусный ангидрид (16,2 г; 0,157 моля). Смесь кипятят с обратным холодильником, в течение 40 мин добавляют триметилортобензоат (20,0 г; 0,108 моля) и нагревание продолжают в течение 3 ч. Смесь охлаждают до 0°C и маточный раствор отделяют. Твердое вещество последовательно промывают толуолом (14 мл) и смесью толуола (8 мл) и этилацетата (8 мл). После сушки выделяют 15,3 г (87,3%) "хлоренола" в виде бежевых кристаллов.
Пример 4: Синтез "енолидола" (метил-3-[метокси(фенил)метилен]-2-оксоиндолин-6-карбоксилата) 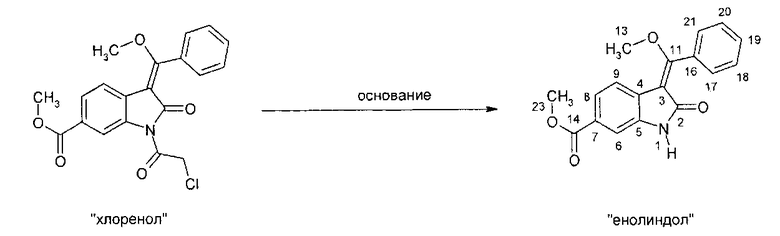
Методика 1
Раствор гидроксида калия (0,41 г, 0,006 моля) в метаноле (4 мл) при 63°C добавляют к суспензии метил-1-(хлорацетил)-3-[метокси(фенил)метилен]-2-оксоиндолин-6-карбоксилата (8,0 г; 0,020 моля) в метаноле (32 мл). Затем смесь перемешивают в течение 30 мин, охлаждают до 0°C и перемешивание продолжают в течение 2 ч. После фильтрования твердое вещество промывают метанолом (24 мл), сушат и получают 6,0 г (94,6%) "енолидола" в виде желтых кристаллов. 1H-ЯМР (500 МГц, CDCl3) 8: 8,08 (s, 1H, 1-Н); 7,88 (d, J=7,8 Гц, 1Н, 9-Н); 7,75 (m, 1H, 8-H); 7,52-7,56 (m, 3H, 18-Н, 19-Н, 20-Н); 7,40-7,45 (m, 3Н, 6-Н, 17-Н, 21-Н); 3,92 (s, 3Н, 23-Н3); 3,74 (s, 3H, 13-Н3). 13С-ЯМР (126 МГц, CDCl) 8: 168.8 (C-2); 107,4 (C-3); 130,8 (C-4); 138,2 (C-5); 109,4 (C-6); 128,2 и 128,3 (С-7, С-16); 123,5 (С-8); 123,1 (С-9); 170,1 (С-11); 57.6 (С-13); 167,2 (С-14); 128,7 и 128,9 (С-17, С-18, С-20, С-21); 130,5 (С-19); 52,1 (С-23). МС (m/z): 310 (M+Н)+ Анализ: Рассчитано для C18H15NO4: С, 69,89; Н, 4.89; N, 4,53. Найдено: С, 69,34; Н, 4,92; N, 4,56.
Методика 2
Суспензию метил-1-(хлорацетил)-3-[метокси(фенил)метилен]-2-оксоиндолин-6-карбоксилата (7,0 г; 0,018 моля) в метаноле (28 мл) кипятят с обратным холодильником. К этой суспензии в течение 3 мин добавляют раствор метоксида натрия в метаноле (0,24 г, 30% (мас./мас.), 0,001 моля). Затем смесь перемешивают в течение 30 мин, охлаждают до 5°C и перемешивание продолжают в течение 2 ч. После фильтрования твердое вещество промывают метанолом (9 мл), сушат и получают 5,4 г (89,7%) "енолидола" в виде желтых кристаллов.
Методика 3
Суспензию метил-1-(хлорацетил)-3-[метокси(фенил)метилен]-2-оксоиндолин-6-карбоксилата (8,0 г; 0,021 моля) в метаноле (32 мл) кипятят с обратным холодильником. К этой суспензии по каплям добавляют раствор метоксида натрия в метаноле (0,74 г, 30% (мас./мас.), 0,004 моля), дополнительно разбавленный метанолом (4 мл). Затем смесь перемешивают в течение 90 мин, охлаждают до 0°C и перемешивание продолжают в течение 2 ч. После фильтрования твердое вещество промывают метанолом (24 мл) и сушат и получают 5,9 г (91,2%) "енолидола" в виде желтых кристаллов.
Пример 5: Синтез "хлорацетила" (N-(4-нитроанилино)-N-метил-2-хлор-ацетамида)
Методика 1
Суспензию N-метил-4-нитроанилина (140 г; 0,920 моля) в этилацетате (400 мл) нагревают до 70°C. К этой суспензии в течение 90 мин добавляют хлорацетилхлорид (114 г; 1,009 моля). Затем полученный раствор кипятят с обратным холодильником в течение 1 ч, охлаждают до 60°C и добавляют метилциклогексан (245 мл). Затем суспензию охлаждают до 0°C и перемешивают в течение 1 ч. Реакционную смесь фильтруют, промывают метилциклогексаном (285 мл) и осадившееся вещество сушат и получают 210,4 г (92,7%) "хлорацетила" в виде белых кристаллов. 1Н-ЯМР (500 МГц, ДМСО-d6) δ: 8,29 (d, J=8,5 Гц, 2Н, 1-Н+3-Н); 7,69 (d, J=8,5 Гц, 2Н, 4-Н+6-Н); 4,35 (s, 2Н, 9-Н2); 3,33 (s, 3 Н, 12-Н3). 13C-ЯМР (126 МГц, ДМСО-d6) 5: 124,6 (C-l+С-3); 145,6 (С-2); 127,4 (С-4+С-6); 148,6 (С-5); 165,6 (С-8); 42,7 (С-9); 37,2 (С-12). МС (m/z): 229 (М+H)+. Анализ: Рассчитано для C9H9ClN2O3: С, 47,28; Н, 3,97; N, 12,25. Найдено: С, 47,26; Н, 3,99; Сl, 15,73; N, 12,29.
Методика 2
Суспензию N-метил-4-нитроанилина (20,0 г; 0,131 моля) в этилацетате (20 мл) нагревают до 60°C. К этой суспензии в течение 15 мин добавляют раствор ангидрида хлоруксусной кислоты (26,0 г; 0,151 моля) в этилацетате (60 мл). Затем полученный раствор кипятят с обратным холодильником в течение 1 ч, охлаждают до 75°C и добавляют метилциклогексан (80 мл). После внесения затравки при 60°C суспензию охлаждают до 0°C и перемешивают в течение 1 ч. Реакционную смесь фильтруют, промывают метилциклогексаном (40 мл) и осадившееся вещество сушат и получают 25,9 г (83,3%) "хлорацетила" в виде серых кристаллов.
Пример 6: Синтез "нитроанилина" (N-(4-нитрофенил)-N-метил-2-(4-метилпиперазин-1-ил)ацетамида) и "анилинп" (N(4-аминофенил)-N-метил-2-(4-метилпиперазин-1-ил)ацетамида) 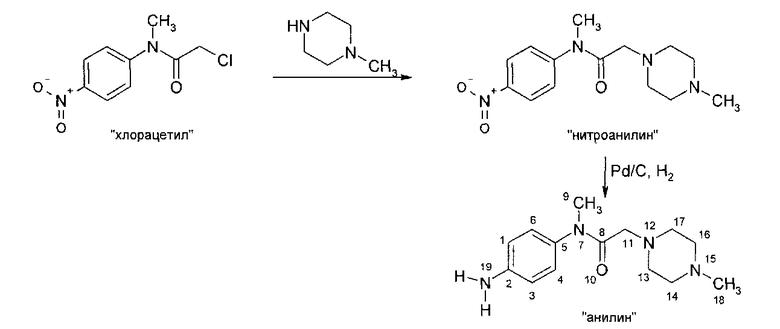
Методика 1
Суспензию N-(4-нитроанилино)-N-метил-2-хлорацетамида (20,0 г; 0,087 моля) в толуоле (110 мл) нагревают до 40°C. В течение 30 мин по каплям добавляют 1-метилпиперазин (21,9 г; 0,216 моля). После промывки капельной воронки толуолом (5 мл) реакционную смесь перемешивают при 55°C в течение 2 ч, охлаждают до температуры окружающей среды и промывают водой (15 мл). Органический слой разбавляют изопропанолом (100 мл) и добавляют Pd/C (10%; 1,0 г). После последующего гидрирования (Н2, 4 бар) при 20°C катализатор удаляют. Примерно 4/5 объема полученного раствора выпаривают при 50°C. Полученный остаток растворяют в этилацетате (20 мл) и толуоле (147 мл), нагретых до 80°C, затем охлаждают до 55°C и вносят затравку. Затем реакционную смесь охлаждают до 0°C и перемешивают при такой же температуре в течение 3 ч. После фильтрования твердое вещество промывают охлажденным льдом толуолом (40 мл), сушат и получают 20,2 г (88,0%) "анилина" в виде белых кристаллов. 1Н-ЯМР (500 МГц, ДМСО-d6) δ: 6,90 (d, J -8,5 Гц, 2Н, 4-Н+6-Н); 6,65 (d, J=8,5 Гц, 2Н, 1-Н+3-Н); 5,22 (2Н, 19-Н2); 3,04 (s, 3 Н, 9-Н3); 2,79 (s, 2 Н, 11-H2); 2,32 (m, 4 Н, 13-Н2+17-Н2); 2,23 (m, 4 Н, 14-H2+16-H2);2,10(s,3H, 18-Н2). 13C-ЯMP(126 MГц, ДMCO-d6)δ: 114,0(С-1+С-3); 148,0 (С-2); 127,6 (С-4+С-6); 131,5 (С-5); 168,9 (С-8); 36,9 (С-9); 58,5 (С-11); 52,4 (С-13+С-17); 54,6 (С-14+С-16); 45,7 (С-18). МС (m/z): 263 (M+Н)+/ Анализ: Рассчитано для C14H22N4O: С, 64,09; Н, 8,45; N, 21,36. Найдено: С, 64,05; Н, 8,43; N,21,39.
Методика 2
Суспензию N-(4-нитроанилино)-N-метил-2-хлорацетамида (14,5 г; 0,063 моля) в этилацетате (65 мл) нагревают до 40°C. В течение 30 мин по каплям добавляют 1-метилпиперазин (15,8 г; 0,156 моля). После промывки капельной воронки этилацетатом (7 мл) реакционную смесь перемешивают при 50°C в течение 90 мин, охлаждают до температуры окружающей среды и промывают водой (7 мл). Органический слой разбавляют изопропанолом (75 мл) и сушат над сульфатом натрия. После отделения твердого вещества добавляют Pd/C (10%; 2,0 г) и раствор гидрируют (Н2, 5 бар) при температуре окружающей среды без охлаждения. Затем катализатор удаляют фильтрованием и растворитель выпаривают при 60°C. Полученный остаток растворяют в этилацетате (250 мл) и перекристаллизовывают. После фильтрования и сушки выделяют 10,4 г (60,4%) "анилина" в виде белых кристаллов.
Пример 7: Синтез "анилино" (3-Z-[1-(4-(N-((4-метилпиперазин-1-ил)-метилкарбонил)-N-метиламино)-анилино)-1-фенилметилен]-6-метоксикарбонил- 2-индолинона)
Методика 1
Суспензию метил-3-[метокси(фенил)метилен]-2-оксоиндолин-6-карбоксилата (10,0 г; 0,032 моля) и N-(4-аминофенил)-М-метил-2-(4-метилпиперазин-1-ил)ацетамида (8,6 г; 0,032 моля) в смеси метанола (72 мл) и N, N-диметилформамида (18 мл) кипятят с обратным холодильником. После кипячения с обратным холодильником в течение 7 ч суспензию охлаждают до 0°C и перемешивание продолжают в течение еще 2 ч. Твердое вещество отфильтровывают, промывают метанолом (40 мл), сушат и получают 15,4 г (88,1%) "анилино" в виде желтых кристаллов. 1Н-ЯМР (500 МГц, ДМСО-d6) δ: 11,00 (s, 1H, 23-H); 12,23 (s, 19-H); 7,61 (t; J=7,1 Гц, 1Н, 33-Н); 7,57 (t, J=7,5 Гц. 2Н, 32-Н+34-Н); 7,50 (d, J=7,7 Гц, 2Н, 31-Н+35-Н); 7,43 (d, J=1,6 Гц, 1Н, 29-Н); 7,20 (dd, J=8,3; 1,6 Гц, 1Н, 27-Н); 7,13 (d, J=8,3 Гц, 2Н, 14-Н+18-Н); 6,89 (d, 8,3 Гц, 2Н, 15-Н+17-Н); 5,84 (d, J=8,3 Гц, 1Н, 26-Н); 3,77 (s, 3 Н, 40-Н3); 3,06 (m, 3 Н, 12-Н3); 2,70 (m, 2 Н, 8-Н2); 2,19 (m, 8 Н, 2-Н2, 3-Н2, 5-H2, 6-Н2); 2,11 (s, 3 Н, 7-Н3). 13С-ЯМР (126 МГц, ДМСО-d6) 5: 54,5 (С-2+С-6); 52,2 (С-3+С-5); 45,6 (С-7); 59,1 (С-8); 168,5 (С-9); 36,6 (С-12); 140,1 (С-13); 127,6 (С-14+С-18); 123,8 (С-17+С-15); 137,0 (С-16); 158,3 (С-20); 97,5 (С-21); 170,1 (С-22); 136,2 (С-24); 128,9 (С-25); 117,2 (С-26); 121,4 (С-27); 124,0(С-28); 109,4 (С-29); 131,9 (С-30); 128,4 (С-31+С-35); 129,4 (С-32+С-34); 130,4 (С-33); 166,3 (С-37); 51,7 (С-40). МС (m/z): 540 (M+H)+ Анализ: Рассчитано для C31H33N5O4: С, 69,00; Н, 6,16; N, 12,98. Найдено: С, 68,05; Н, 6,21; N, 12,81.
Методика 2
Суспензию метил-3-[метокси(фенил)метилен]-2-оксоиндолин-6-карбоксилата (20,0 г; 0,064 моля) и N-(4-аминофенил)-N-метил-2-(4-метилпиперазин-1-ил)ацетамида (17,1 г; 0,065 моля) в метаноле (180 мл) кипятят с обратным холодильником в течение 7,5 ч. Полученную суспензию в течение 1 ч охлаждают до 10°C и перемешивание продолжают в течение 1 ч. После фильтрования твердое вещество промывают охлажденным льдом метанолом (80 мл), сушат и получают 31,0 г (89,0%) "анилино" в виде желтых кристаллов.
Пример 8: Синтез моноэтансульфоната 3-Z-[1-(4-(N-((4-метилпиперазин-1-ил)-метилкарбонил)-N-метиламино)-анилино)-1-фенилметилен]-6-метоксикарбонил-2-индолинона 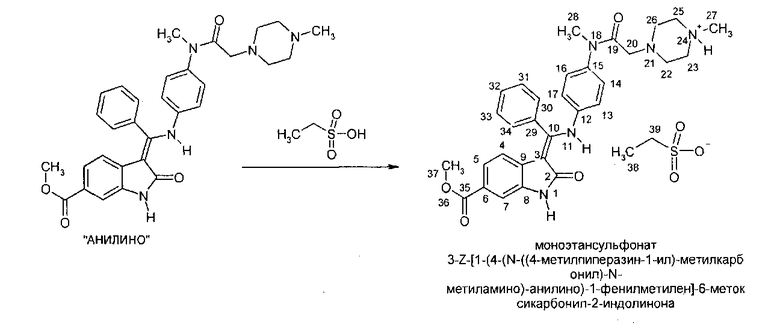
Суспензию 3-Z-[1-(4-(N-((4-метилпиперазин-1-ил)-метилкарбонил)-N-метиламино)-анилино)-1-фенилметилен]-6-метоксикарбонил-2-индолинона (30,0 г; 0,055 моля) в метаноле (200 мл) и воде (2,4 мл) нагревают до 60°C. К реакционной смеси добавляют водный раствор этансульфоновой кислоты (70% (мас./мас.); 8,75 г; 0,056 моля). Полученный раствор охлаждают до 50°C, вносят затравку и затем разбавляют изопропанолом (200 мл). Затем смесь охлаждают до 0°C и перемешивают при такой же температуре в течение 2 ч. Осадившееся вещество выделяют, промывают изопропанолом (120 мл), сушат и получают 35,1 г (97,3%) моноэтансульфоната искомого соединения в виде желтых кристаллов. 1Н-ЯМР (400 МГц, ДМСО-d6) δ: 12,26 (s, 11-Н); 10,79 (s, 1H, 1-Н);9,44 (s, 1H, 24-H); 7,64 (m, 1H, 32-H); 7.59 (m, 2H, 31-H+33-Н); 7,52 (m, 2H, 30-H+34-Н); 7,45 (d, J=1,6 Гц. 1H, 7-H); 7,20 (dd, J=8,2; 1,6 Гц, 1H, 5-H);
7,16 (m. 2H, 14-H+16-H); 6,90 (m, 2H. 13-H+17-H); 5,85 (d, J=8.2 Гц, 1H, 4-H); 3,78 (s, 3H, 37-Н3); 3,45-2,80 (широкий m, 4H, 23-Н3+25-H2); 3.08 (s, 3H. 28-Н3); 2,88 (s, 2H, 20-Н3); 2,85-2,30 (широкий m, 4H, 22-Н3+26-Н3); 2.75 (s, 3Н, 27-Н3); 2,44 (q, J=7,4 Гц, 2H, 39-Hz); 1,09 (t, J-7,4 Гц, 3Н, 38-Н3). 13С-ЯМР (126 МГц, ДМСО-d6) δ: 9,8 (С-38); 36,6 (С-28); 42,3 (С-27); 45,1 (С-39); 51,7 (С-37); 48,9 (С-22+С-26); 52,6 (С-23+С-25); 57,5 (С-20); 97,7 (С-3); 109,5 (С-7);117,3 (С-4); 121,4 (С-5); 123,8 (С-13+С-17); 124,1 (С-6); 127,7 (С-14+С-16); 128,4 (С-30+С-34); 128,8 (С-9); 129,5 (С-31+С-33); 130,5 (С-32); 132,0 (С-29); 168,5 (С-9); 136,3 (С-8); 137,3 (С-12); 139,5 (С-15); 158,1 (С-10); 166,3 (С-35); 168,0 (С-19); 170,1 (С-2). МС (m/z): 540 (М(основание)+Н)+ Анализ: Рассчитано для C33H39N5O7S: С, 60,17; Н, 6,12; N, 10,63; S, 4,87. Найдено: С, 60,40; Н, 6,15; N, 10,70; S, 4,84.
| название | год | авторы | номер документа |
|---|---|---|---|
| КОМБИНАЦИИ, ПРЕДНАЗНАЧЕННЫЕ ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ, ВКЛЮЧАЮЩИХ ПРОЛИФЕРАЦИЮ КЛЕТОК | 2005 |
|
RU2407532C9 |
| Производные индолинона, обладающих свойствами лигандов мелатонинового рецептора и их применение | 2017 |
|
RU2712036C2 |
| 1-ФЕНИЛ-БЕНЗИМИДАЗОЛЬНЫЕ СОЕДИНЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЛЕЧЕНИЯ РАССТРОЙСТВА ИЛИ ЗАБОЛЕВАНИЯ, ЧУВСТВИТЕЛЬНОГО К МОДУЛЯЦИИ ГАМК-РЕЦЕПТОРНОГО КОМПЛЕКСА ЦЕНТРАЛЬНОЙ НЕРВНОЙ СИСТЕМЫ | 1997 |
|
RU2194699C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ5,П-ДИГИДРО-6Н-ПИРИДО-[2,3-Ь]-[1,4]-БЕНЗО-ДИАЗЕПИН-6-ОНА | 1972 |
|
SU331554A1 |
| КОНДЕНСИРОВАННОЕ ПРОИЗВОДНОЕ ТИОФЕНА, ПРОИЗВОДНОЕ БЕНЗОТИОФЕНА, ПРОИЗВОДНОЕ ТИЕНОБЕНЗОКСАЗЕПИНОНА, ФАРМАЦЕВТИЧЕСКОЕ СРЕДСТВО И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1998 |
|
RU2197491C2 |
| Способ получения хлорацетанилидов | 1974 |
|
SU596162A3 |
| Способ получения производных 2-оксоиндол-1-карбоксамида | 1985 |
|
SU1445556A3 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ АМИНЫ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1992 |
|
RU2108325C1 |
| АМИДНОЕ ПРОИЗВОДНОЕ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ТЕРАПЕВТИЧЕСКИЕ СРЕДСТВА НА ЕГО ОСНОВЕ | 2003 |
|
RU2315043C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛА И СОДЕРЖАЩИЕ ЭТИ СОЕДИНЕНИЯ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2000 |
|
RU2243226C2 |
Изобретение относится к способу получения 3-Z-[1-(4-(N-((4-метилпиперазин-1-ил)-метилкарбонил)-N-метиламино)-анилино)-1-фенилметилен]-6-метоксикарбонил-2-индолинона формулы
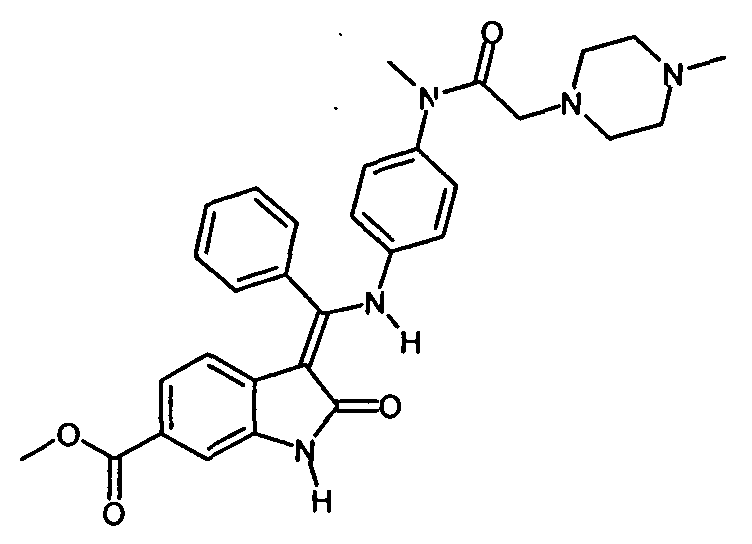
включающий стадию реакции соединения формулы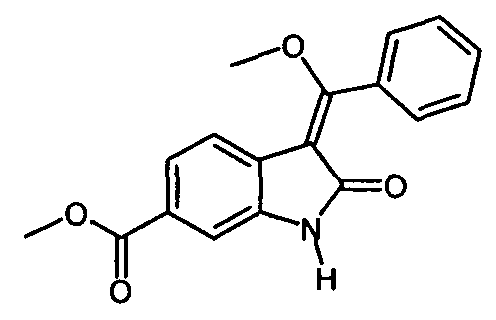 , которое получают путем катализируемого основанием дехлорацетилирования соединения формулы
, которое получают путем катализируемого основанием дехлорацетилирования соединения формулы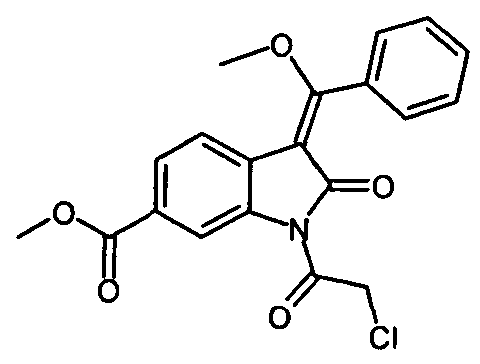 , с соединением формулы
, с соединением формулы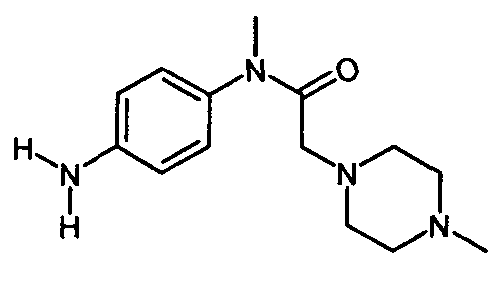 .Способ позволяет повысить выход 3-Z-[1-(4-(N-((4-метилпиперазин-1-ил)-метилкарбонил)-N-метиламино)-анилино)-1-фенилметилен]-6-метоксикарбонил-2-индолинона. 7 з.п. ф-лы, 8 пр.
.Способ позволяет повысить выход 3-Z-[1-(4-(N-((4-метилпиперазин-1-ил)-метилкарбонил)-N-метиламино)-анилино)-1-фенилметилен]-6-метоксикарбонил-2-индолинона. 7 з.п. ф-лы, 8 пр.
1. Способ получения соединения формулы
,
включающий стадию реакции соединения формулы
с соединением формулы
характеризующийся тем, что соединение формулы
получают путем катализируемого основанием дехлорацетилирования соединения формулы
.
2. Способ по п.1, в котором соединение формулы
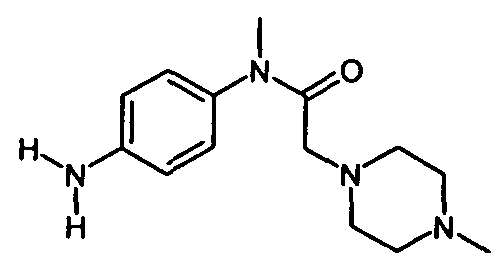
получают по реакции соединения формулы
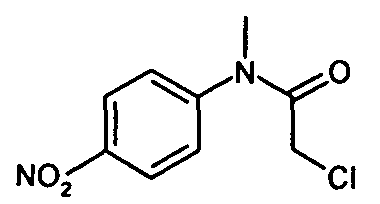
с соединением формулы
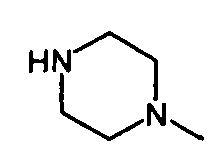
с последующим гидрированием нитрогруппы с образованием аминогруппы.
3. Способ по п.2, в котором соединение формулы
получают по реакции соединения формулы
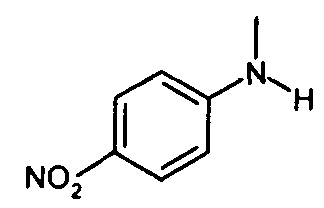
с соединением формулы
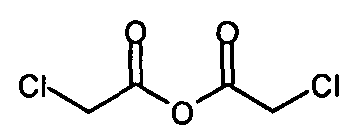 .
.
4. Способ по любому из пп.1-3, в котором соединение формулы
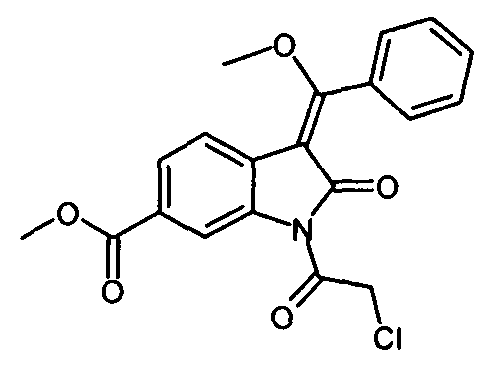
получают по реакции соединения формулы
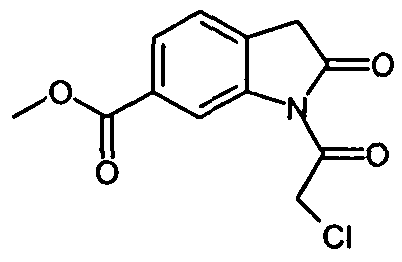
с соединением формулы
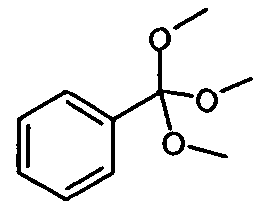 .
.
5. Способ по п.4, в котором соединение формулы
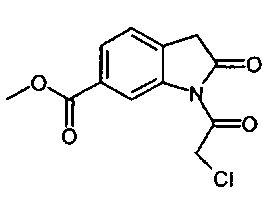
получают по реакции соединения формулы
с соединением формулы
.
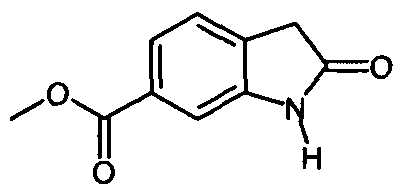
6. Способ по п.5, в котором соединение формулы
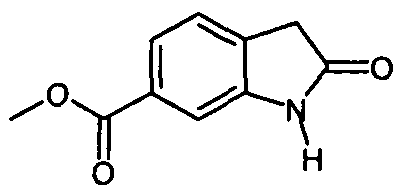
получают с помощью приведенных ниже стадий:
(i) этерификация соединения формулы
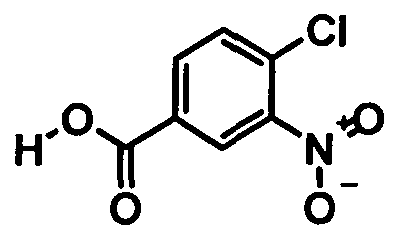
с образованием соединения формулы
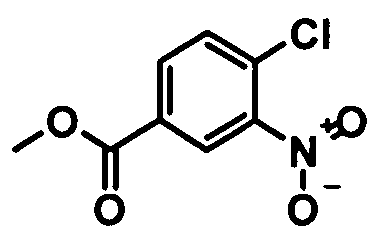 ;
;
(ii) реакция продукта реакции (i) с диметиловым эфиром малоновой кислоты с образованием соединения формулы
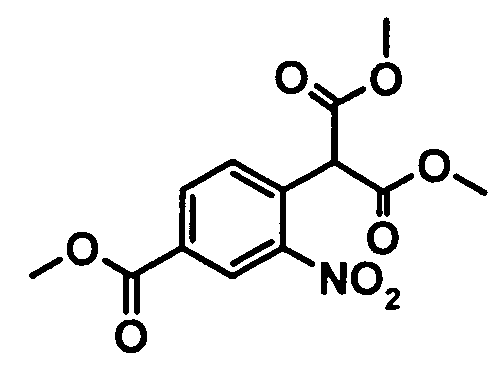 ;
;
(iii) проведение циклизации продукта реакции (ii) по реакции гидрирования.
7. Способ по п.1, в котором соединение формулы
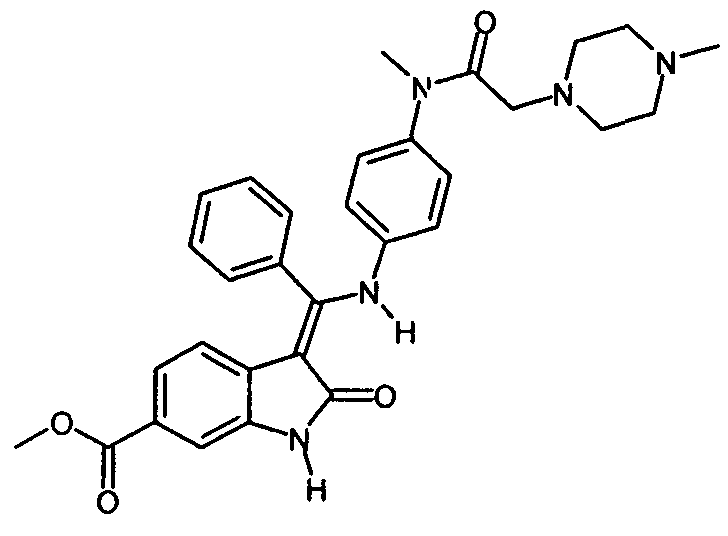
вводят в реакцию с EtSO3H чтобы получить моноэтансульфонатную соль этого соединения.
8. Способ по п.7, который дополнительно включает стадию размола моноэтансульфонатной соли соединения.
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| WO 2001027081 A1, 19.04.2001 | |||
| WO 2000018734 A1, 06.04.2000 | |||
| WO 2004026829 A3, 01.04.2004 | |||
| Прибор для нагревания | 1926 |
|
SU6080A1 |

