ОБЛАСТЬ ТЕХНИКИ
Изобретение относится к области биомедицины, а именно к соединению тетрагидроизохинолина в качестве модулятора калиевых каналов, его получению и применению.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Калиевый канал Kv7 представляет собой тип потенциал-зависимого канала ионов калия с низким порогом активации, медленной активацией и неинактивацией. Семейство калиевых каналов Kv7 состоит из пяти членов (Kv7.1-Kv7.5) с одинаковой топологией, где функциональный канал состоит из ч етырех субъединиц, а каждая субъединица содержит шесть трансмембранных фрагментов (S1-S6). Среди них S4 представляет собой область, чувствительную к разности потенциалов, что играет важную роль в определении изменений мембранного потенциала и контроле конформационных изменений; S5-S6 являются основными компонентами области просвета канала, а также основной областью комбинирования и действия активаторов калиевых каналов. Калиевый канал Kv7.1 представляет собой ненейрональный проводящий путь, который распределяется во внешней периферической ткани, экспрессируется в сердце, опосредованно воздействуя на медленный ток Iks миокарда, и его мутация может приводить к синдрому удлиненного интервала QT. Калиевые каналы Kv7.2-Kv7.5 служат основой нейронального М-тока, широко распределены в нервной системе и обладают разнообразной физиологической активностью. Мутации генов калиевых каналов Kv7.2 и Kv7.3 могут приводить к ряду разных картин эпилепсии, таких как доброкачественные семейные неонатальные судороги (BFNC), что в полной мере демонстрирует роль M-тока в регуляции возбудимости нейронов. Калиевый канал Kv7.4 высоко экспрессируется в наружных волосковых клетках улитки и слуховом ядре мозгового ствола, и его мутация может вызывать наследственную глухоту. Калиевый канал Kv7.5 высоко экспрессируется в скелетных мышцах и головном мозге, и его мутация может вызывать ретинопатию. Особенностью многих заболеваний, таких как эпилепсия, тревожность, глухота и т.д., является высокая возбудимость мембран, а калиевые каналы Kv7, которые являются молекулярной основой М-тока, могут открываться путем определения изменений мембранного потенциала, активируя тем самым ингибирующий калиевый ток, который контролирует возбудимость мембран, благодаря чему калиевые каналы Kv7 играют важную роль при болях и психических заболеваниях, проявляющихся высокой нервной возбудимостью.
Ретигабин является препаратом для лечения эпилепсии. Регистрационное свидетельство на этот препарат было получено в Соединенном Королевстве, Германии и Дании. В исследованиях подтвердилось, что действие препарата Ретигабин связано с потенциал-зависимыми каналами ионов калия (KCNQ), и его основной механизм действия заключается в регулировании калиевых токов М-типа путем воздействия на каналы KCNQ2/3.
Ретигабин (RTG) является первым активатором калиевых каналов Kv7 для лечения эпилепсии с парциальными приступами у взрослых, который поступил на рынок в 2011 г. Помимо эпилепсии, RTG также может применяться для лечения тревожности, невралгии, нейродегенеративных заболеваний и т.д. RTG может эффективно снижать или предотвращать приступы в разных моделях эпилепсии. RTG оказывает эффективное противоэпилептическое действие как на тонические судороги при воздействии максимального электрошока (МЭШ), так и на клонические судороги, вызываемые пентилентетразолом. Кроме того, RTG также может предотвращать судороги, вызванные N-метил-D-аспартатом (NMDA), пенициллином, пикротоксином, каиновой кислотой (KA) и т.д. Модель инициации подходит для скрининга разных противоэпилептических препаратов, и RTG на этой модели проявляет более сильный эффект, чем на других моделях. Из-за сильного воздействия RTG на все калиевые каналы семейства Kv7 и на другие каналы, его низкая селективность делает его потенциально нежелательным. В широком ряде литературных источников сообщается о том, что RTG имеет высокую частоту нежелательных явлений, связанных с центральной нервной системой, что может приводить к головокружению, утомляемости, афазии, нарушениям речи, нарушениям равновесия и другим нежелательным реакциям, в том числе к камням в почках, задержке мочи и другим заболеваниям почек и мочевыделительной системы, к болезням сердца, например, к внезапной остановке сердца, неустойчивой желудочковой тахикардии, а также может вызывать обесцвечивание сетчатки, синюю или фиолетовую пигментацию на коже, ногтях и тому подобное.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Целью настоящего изобретения является предоставление соединения по формуле А, способа его получения и его использования в качестве регулятора калиевых каналов.
В первом аспекте настоящего изобретения предложено соединение по формуле А или его фармацевтически приемлемая соль,

где
R1 выбирают из замещенной или незамещенной группы, состоящей из C6-10 арильной группы и 4-7-членной гетероарильной группы, содержащей 1-3 гетероатома, выбранных из N, O или S, причем замещение означает замещение одним или несколькими заместителями, выбранными из группы, состоящей из галогена, нитрогруппы, цианогруппы, C1-6 алкильной группы, C3-6 циклоалкильной группы, C1-6 алкоксигруппы, C3-6 циклоалкоксигруппы, галогенированной C1-6 алкильной группы, галогенированной C3-6 циклоалкильной группы, галогенированной C1-6 алкоксигруппы, галогенированной C3-6 циклоалкоксигруппы, -NR8R9 и этинильной группы;
R2 и R3 независимо выбирают из группы, состоящей из водорода, дейтерия, галогена, C1-6 алкильной группы и галогенированной C1-6 алкильной группы;
либо R2 и R3 вместе с их соответствующим подсоединенным C образуют C3-10 циклоалкильную группу, при этом циклоалкильная группа может быть замещена 1-3 заместителями, выбранными из группы, состоящей из водорода, галогена, C1-6 алкильной группы и галогенированной C1-6 алкильной группы;
R4 и R5 независимо выбирают из группы, состоящей из водорода, галогена, C1-6 алкильной группы, C1-6 алкоксигруппы, галогенированной C1-6 алкильной группы и галогенированной C1-6 алкоксигруппы;
R6 выбирают из замещенной или незамещенной группы, состоящей из C1-6 алкильной группы, C3-6 циклоалкильной группы, C1-6 алкоксигруппы и C3-6 циклоалкоксигруппы, причем замещение означает замещение одним или несколькими заместителями, выбранными из группы, состоящей из галогена, нитрогруппы, цианогруппы, C1-6 алкильной группы, C1-6 алкоксигруппы, галогенированной C1-6 алкильной группы, галогенированной C1-6 алкоксигруппы, C2-6 алкенильной группы, C2-6 алкинильной группы и C3-6 циклоалкильной группы;
n выбирают из группы чисел, состоящей из 1, 2 и 3;
W представляет собой C-R7 или N;
R7 выбирают из группы, состоящей из водорода, дейтерия, галогена, цианогруппы, C1-6 алкильной группы, галогенированной C1-6 алкильной группы, C1-6 алкоксигруппы и галогенированной C1-6 алкоксигруппы;
R8 и R9 независимо выбирают из группы, состоящей из водорода, C1-6 алкильной группы и галогенированной C1-6 алкильной группы;
либо R8 и R9 вместе с их соответствующим подсоединенным N образуют 3-10-членную азациклоалкильную группу.
В другом предпочтительном варианте осуществления
R1 представляет собой замещенную или незамещенную фенильную группу, причем замещение означает замещение одним или несколькими заместителями, выбранными из группы, состоящей из галогена, цианогруппы, C1-6 алкильной группы, C3-6 циклоалкильной группы, C1-6 алкоксигруппы, C3-6 циклоалкоксигруппы, галогенированной C1-6 алкильной группы, галогенированной C3-6 циклоалкильной группы, галогенированный C1-6 алкоксигруппы, галогенированной C3-6 циклоалкоксигруппы, -NR8R9 и этинильной группы;
R2 и R3 независимо выбирают из группы, состоящей из водорода и дейтерия;
R4 и R5 представляют собой метил;
R6 выбирают из замещенной или незамещенной группы, состоящей из C1-6 алкильной группы и C3-6 циклоалкильной группы, причем замещение означает замещение одним или несколькими заместителями, выбранными из группы, состоящей из галогена, C1-6 алкильной группы и C3-6 циклоалкильной группы;
n является 1;
W представляет собой C-R7;
R7 выбирают из группы, состоящей из водорода и галогена;
R8 и R9 независимо друг от друга являются C1-6 алкильной группой.
В другом предпочтительном варианте осуществления
R1 представляет собой замещенную или незамещенную фенильную группу, причем замещение означает замещение одним или несколькими заместителями, выбранными из группы, состоящей из галогена, цианогруппы, C1-6 алкильной группы, C3-6 циклоалкильной группы, C1-6 алкоксигруппы, C3-6 циклоалкоксигруппы, галогенированной C1-6 алкильной группы, галогенированной C3-6 циклоалкильной группы, галогенированный C1-6 алкоксигруппы, галогенированной C3-6 циклоалкоксигруппы, -NR8R9 и этинильной группы;
R2 и R3 независимо выбирают из группы, состоящей из водорода и дейтерия;
R4 и R5 представляют собой метил;
R6 выбирают из замещенной или незамещенной группы, состоящей из C1-6 алкильной группы и C3-6 циклоалкильной группы, причем замещение означает замещение одним или несколькими заместителями, выбранными из группы, состоящей из галогена и C1-6 алкильной группы;
n является 1;
W представляет собой C-R7;
R7 выбирают из группы, состоящей из водорода и галогена;
R8 и R9 независимо друг от друга являются C1-6 алкильной группой.
В другом предпочтительном варианте осуществления
R1 представляет собой замещенную или незамещенную фенильную группу, причем замещение означает замещение одним или несколькими заместителями, выбранными из группы, состоящей из галогена, цианогруппы, C1-6 алкильной группы, C3-6 циклоалкильной группы, C1-6 алкоксигруппы, C3-6 циклоалкоксигруппы, галогенированной C1-6 алкильной группы, галогенированной C3-6 циклоалкильной группы, галогенированный C1-6 алкоксигруппы, галогенированной C3-6 циклоалкоксигруппы, -NR8R9 и этинильной группы;
R2 и R3 независимо выбирают из группы, состоящей из водорода и дейтерия;
R4 и R5 представляют собой метил;
R6 выбирают из группы, состоящей из  ,
, 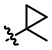 и
и 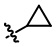 ;
;
n является 1;
W представляет собой C-R7;
R7 выбирают из группы, состоящей из водорода и галогена;
R8 и R9 независимо друг от друга являются C1-6 алкильной группой.
В другом предпочтительном варианте осуществления
R1 представляет собой замещенную или незамещенную фенильную группу, причем замещение означает замещение одним или несколькими заместителями, выбранными из группы, состоящей из галогена, цианогруппы, C1-6 алкильной группы, C1-6 алкоксигруппы, галогенированной C1-6 алкильной группы, -NR8R9 и этинильной группы;
R2 и R3 независимо выбирают из группы, состоящей из водорода и дейтерия;
R4 и R5 представляют собой метил;
R6 выбирают из группы, состоящей из , и ;
n является 1;
W представляет собой C-R7;
R7 выбирают из группы, состоящей из водорода и галогена;
R8 и R9 независимо друг от друга являются C1-6 алкильной группой.
В другом предпочтительном варианте осуществления R1 представляет собой замещенную или незамещенную фенильную группу, причем замещение означает замещение одним или несколькими заместителями, выбранными из группы, состоящей из галогена, цианогруппы, C1-6 алкильной группы, C1-6 алкоксигруппы, галогенированной C1-6 алкильной группы, -NR8R9 и этинильной группы;
R2 и R3 независимо выбирают из группы, состоящей из водорода и дейтерия;
R4 и R5 представляют собой метил;
R6 представляет собой  ;
;
n является 1;
W представляет собой C-R7;
R7 выбирают из группы, состоящей из водорода и галогена;
R8 и R9 независимо друг от друга являются C1-6 алкильной группой.
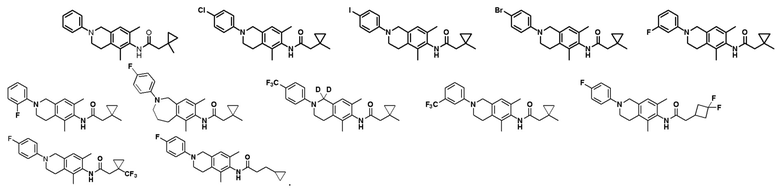
В другом предпочтительном варианте соединение выбрано из группы, состоящей из:


Во втором аспекте настоящего изобретения предлагается способ получения соединения согласно первому аспекту изобретения или его фармацевтически приемлемой соли, состоящий из следующих этапов:

где:
X выбирают из группы, состоящей из галогена, -B(OH)2 и -OTf;
R1, R2, R3, R4, R5, R6, n и W определены, как указано в первом аспекте изобретения.
В третьем аспекте настоящего изобретения предлагается фармацевтическая композиция, содержащая один или несколько фармацевтически приемлемых носителей и терапевтически эффективное количество одного или нескольких соединений в соответствии с первым аспектом настоящего изобретения или их фармацевтически приемлемых солей.
В четвертом аспекте настоящего изобретения предлагается использование соединения по первому аспекту настоящего изобретения или его фармацевтически приемлемой соли для получения лекарственного препарата для профилактики и/или лечения заболеваний, реагирующих на активность калиевых каналов.
В другом предпочтительном воплощении заболевание, реагирующее на активность калиевых каналов, представляет собой заболевание центральной нервной системы.
В другом предпочтительном варианте осуществления заболевание центральной нервной системы выбрано из группы, состоящей из эпилепсии, судорог, воспалительной боли, нейропатической боли, мигрени, депрессии, тревожного расстройства, инсульта, болезни Альцгеймера, нейродегенеративного заболевания, злоупотребления кокаином, никотиновой абстиненции, алкогольной абстиненции и шума в ушах.
Следует понимать, что в настоящем изобретении все технические признаки, конкретно описанные выше и ниже (например, в приведенных примерах), могут комбинироваться друг с другом, образуя тем самым новые или предпочтительные технические решения, которые не будут описываться здесь по отдельности.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
После продолжительных и углубленных исследований авторами настоящего изобретения было неожиданно получено соединение по формуле А, имеющее высокую активность в отношении открывания калиевых каналов, превосходную фармакокинетику (например, по показателям мозгового кровообращения и т.д.), эффективность in vivo, безопасность и новую структуру, полученную путем структурной оптимизации. На основании этого изобретателями было совершено настоящее изобретение.
ТЕРМИНЫ
В настоящем изобретении, если не указано иное, используемые термины имеют общепринятые значения, которые хорошо известны специалистам в данной области.
В настоящем изобретении термин «галоген» означает F, Cl, Br или I.
В настоящем изобретении «C1-C6 алкильная группа» означает линейную или разветвленную алкильную группу, включающую 1-6 атомов углерода, например, метильную, этильную, пропильную, изопропильную, бутильную, изобутильную, трет-бутильную, неопентильную, трет-пентильную или аналогичные группы.
В настоящем изобретении термин «C2-C6 алкенильная группа» означает алкенильную группу с прямой или разветвленной цепью, имеющую 2-6 атомов углерода, содержащую двойную связь, куда включаются, помимо прочего, винильная, пропенильная, бутенильная, изобутенильная, пентенильная, гексенильная и другие группы.
В настоящем изобретении термин «C2-C6 алкинильная группа» означает линейную или разветвленную алкинильную группу, имеющую 2-6 атомов углерода, содержащую тройную связь, куда включаются, помимо прочего, этинильная, пропинильная, бутинильная, изобутинильная, пентинильная и гексинильная и другие группы.
В настоящем изобретении термин «C3-C8 циклоалкильная группа» означает циклическую алкильную группу, имеющую 3-8 атомов углерода в кольце, куда включаются, помимо прочего, циклопропильная, циклобутильная, циклопентильная, циклогексильная, циклогептильная, циклооктильная и другие группы. Термины «C3-C6 циклоалкильная группа» и «C3-C10 циклоалкильная группа» имеют аналогичные значения.
В настоящем изобретении термин «C1-C6 алкоксигруппа» означает алкоксигруппу с прямой или разветвленной цепью, имеющую 1-6 атомов углерода, куда включаются, помимо прочего, метоксигруппа, этоксигруппа, пропоксигруппа, изопропоксигруппа и бутоксигруппа и другие группы. Предпочтительно указывается C1-C4 алкоксигруппа.
В настоящем изобретении термин «ароматическое кольцо» или «арильная группа» имеет одинаковое значение, предпочтительно указывается «C6-C10 арильная группа». Термин «C6-C10 арильная группа» означает ароматическое кольцо, имеющее 6-10 атомов углерода в кольце, которое не содержит гетероатомов, например, фенильную группу, нафтильную группу и другие группы. Термины «C6-10 арильная группа» и «C6-C10 арильная группа» имеют одинаковое значение, и другие схожие термины имеют аналогичную эквивалентность.
В настоящем изобретении термин «ароматический гетероцикл» или «гетероарильная группа» имеет одинаковое значение и означает гетероароматическую группу, содержащую один или несколько гетероатомов. Например, «C3-C10 гетероарильная группа» означает ароматический гетероцикл, содержащий от 1 до 4 гетероатомов, выбранных из группы, состоящей из кислорода, серы и азота и 3-10 атомов углерода. Неограничивающими примерами являются фурильная, тиенильная, пиридильная, пиразолильная, пирролильная, N-алкилпирролильная, пиримидинильная, пиразинильная, имидазолильная, тетразолильная и другие группы. Гетероарильное кольцо может быть конденсировано с арильным, гетероциклильным или циклоалкильным кольцом, где кольцо, присоединенное к исходной структуре, представляет собой гетероарильное кольцо. Гетероарильная группа может быть может быть замещенной или незамещенной.
В настоящем изобретении термин «галогенированный» означает замещение на галоген.
В настоящем изобретении термин «замещенный» означает один или несколько атомов водорода в отдельно взятой группе, замещенных отдельно взятыми заместителями. Отдельно взятыми заместителями являются заместители, описанные выше, или заместители, встречающиеся в соответствующих примерах. Если не указано иное, замещенная группа может иметь заместитель, выбранный из отдельно взятой группы, в любом замещаемом положении группы, при этом заместитель в каждом положении может быть одинаковым или разным. Специалистам в данной области должно быть понятно, ч то комбинации заместителей, предусмотренные настоящим изобретением, представляют собой такие комбинации, которые являются стабильными или которые можно получить путем химического синтеза. Заместители представляют собой, в качестве примера (но не ограничиваясь этим): галоген, гидроксильную группу, карбоксильную группу (-COOH), C1-C6 алкильную группу, C2-C6 алкенильную группу, C2-C6 алкинильную группу, C3-C8 циклоалкильную группу, 3-12-членную гетероциклильную группу, арильную группу, гетероарильную группу, C1-C8 альдегидную группу, C2-C10 ацильную группу, C2-C10 сложноэфирную группу, аминогруппу, C1-C6 алкоксигруппу, C1-C10 сульфонильную группу и т.п.
В настоящем изобретении термин «несколько» независимо означает 2, 3, 4, 5 или 6.
В настоящем изобретении термин 1-6 означает 1, 2, 3, 4, 5 или 6. Аналогичные значения имеют и другие схожие термины.
Соединение
Изобретение относится к соединению, представленному формулой A, или к его фармацевтически приемлемой соли,
R1, R2, R3, R4, R5, R6, n и W определены, как указано выше.
В другом предпочтительном варианте осуществления любой элемент из R1, R2, R3, R4, R5, R6, n и W в соединении представляет собой соответствующую группу в отдельно взятом соединении.
Используемый здесь термин «фармацевтически приемлемая соль» означает соль, образованную соединением по настоящему изобретению с кислотой или основанием, подходящими для использования в качестве лекарственной субстанции. Фармацевтически приемлемые соли включают неорганические соли и органические соли. Предпочтительным классом солей являются соли соединений по изобретению, образованные кислотами. Подходящими кислотами для образования солей являются, без ограничения, неорганические кислоты, например, хлористоводородная кислота, бромистоводородная кислота, фтористоводородная кислота, серная кислота, азотная кислота, фосфорная кислота; органические кислоты, например, муравьиная кислота, уксусная кислота, трифторуксусная кислота, пропионовая кислота, щавелевая кислота, малоновая кислота, янтарная кислота, фумаровая кислота, малеиновая кислота, молочная кислота, яблочная кислота, винная кислота, лимонная кислота, пикриновая кислота, бензойная кислота, метилсульфоновая кислота, этансульфокислота, п-толуолсульфокислота, бензолсульфокислота, нафталинсульфокислота; а также аминокислоты, например, пролиновая, фенилаланиновая, аспарагиновая и глутаминовая кислота.
Другим предпочтительным классом солей являются соли соединений по изобретению, образованные основаниями, например, соли щелочных металлов (например, соли натрия или калия), соли щелочноземельных металлов (например, соли магния или кальция), соли аммония (например, низкомолекулярные алканол-аммониевые соли и другие фармацевтически приемлемые аминовые соли), например, метиламиновая соль, этиламиновая соль, пропиламиновая соль, диметиламиновая соль, триметиламиновая соль, диэтиламиновая соль, триэтиламиновая соль, трет-бутиламиновая соль, этилендиаминовая соль, гидроксиэтиламиновая соль, дигидроксиэтиламиновая соль, тригидроксиэтиламиновая соль и аминовая соль, образованная из морфолина, пиперазина и лизина, соответственно.
Способ получения
Способ получения соединения формулы А по настоящему изобретению более детально описан ниже, но эти отдельно описанные способы не представляют собой каких-либо ограничений. Соединения по настоящему изобретению также могут быть с легкостью получены при необходимости путем комбинирования разных способов синтеза, описанных в описании, либо известных в данной области, и такие комбинации могут быть легко получены специалистами в области, к которой относится настоящее изобретение.
В большинстве случаев процесс получения соединений по настоящему изобретению является следующим, в котором используемые исходные материалы и реактивы являются коммерчески доступными, если не указано иное.
где:
X выбирают из группы, состоящей из галогена, -B(OH)2 и -OTf;
R1, R2, R3, R4, R5, R6, n и W определены, как указано выше.
Фармацевтическая композиция и способ введения
Фармацевтическая композиция по настоящему изобретению содержит безопасное и эффективное количество соединения по настоящему изобретению или его фармакологически приемлемой соли, а также фармакологически приемлемое вспомогательное вещество или носитель. Термин «безопасное и эффективное количество» означает, ч то количество соединения является достаточным для существенного улучшения состояния, не вызывая при этом серьезных побочных эффектов. Как правило, фармацевтическая композиция содержит 1-2000 мг соединения по настоящему изобретению на дозу, более предпочтительно 5-1000 мг соединения по настоящему изобретению на дозу. Предпочтительно «доза» является капсулой или таблеткой.
«Фармацевтически приемлемый носитель» означает один или несколько совместимых твердых, жидких или гелеобразных вспомогательных веществ, пригодных для использования в медицине, и они должны быть в должной мере ч истыми и малотоксичными. «Совместимость» означает, ч то каждый компонент композиции можно смешивать с соединениями по настоящему изобретению и друг с другом без существенного снижения эффективности соединений. Некоторые примеры фармацевтически приемлемых носителей включают целлюлозу и ее производные (например, натрия карбоксиметилцеллюлозу, натрия этилцеллюлозу, целлюлозы ацетат и т.д.), желатин, тальк, твердые смазывающие вещества (например, стеариновую кислоту, магния стеарат), кальция сульфат, растительные масла (например, соевое масло, кунжутное масло, арахисовое масло, оливковое масло и т.д.), полиолы (например, пропиленгликоль, глицерин, маннит, сорбит и т.д.), эмульгаторы (например, Твин®), смачивающие вещества (например, натрия додецилсульфат), красители, ароматизаторы, стабилизаторы, антиоксиданты, консерванты, апирогенную воду и др.
Фармацевтическая композиция представляет собой лекарственную форму для инъекций, капсулы, таблетки, пилюли, порошки или гранулы.
Способ введения соединения или фармацевтической композиции по настоящему изобретению не ограничен особенным образом, и типичными способами введения являются, без ограничения, прием внутрь, внутриопухолевое, ректальное, парентеральное (внутривенное, внутримышечное или подкожное) и местное введение.
Твердые лекарственные формы для приема внутрь включают капсулы, таблетки, пилюли, порошки и гранулы. В этих твердых лекарственных формах действующее вещество смешивают по меньшей мере с одним обычным инертным вспомогательным веществом (или носителем), например, с натрия цитратом или кальция гидрофосфатом, либо смешивают с каким-либо из следующих компонентов: (а) наполнителями или веществами, улучшающими совместимость, например, с крахмалом, лактозой, сахарозой, глюкозой, маннитолом и кремния диоксидом; (b) связующими веществами, например, с гидроксиметилцеллюлозой, альгинатом, желатином, поливинилпирролидоном, сахарозой и аравийской камедью; (c) с увлажняющими веществами, например, с глицерином; (d) с разрыхлителями, например, с агаром, кальция карбонатом, картофельным крахмалом или тапиоковым крахмалом, альгиновой кислотой, некоторыми смешанными силикатами и натрия карбонатом; (e) с веществами, замедляющими высвобождение, например, с парафином, (f) с ускорителями впитывания, например, с четвертичным соединением аммония; (g) со смачивающими веществами, например, с цетиловым спиртом и глицерина моностеаратом; (h) с адсорбирующими веществами, например, с каолином; и (i) со смазывающими веществами, например, с тальком, кальция стеаратом, магния стеаратом, твердым полиэтиленгликолем, натрия додецилсульфатом, либо их смесью. Лекарственные формы в виде капсул, таблеток и пилюль могут также содержать буферные средства.
Твердые лекарственные формы, например, таблетки, драже, капсулы, пилюли и гранулы, могут быть в оболочке, например, в кишечнорастворимой оболочке, а также содержать другие материалы, применяемые в данной области. Они могут содержать замутнители, и высвобождение действующего вещества или соединения в таких композициях может происходить с задержкой в определенной части пищеварительного тракта. Примерами компонентов для включения, которые могут использоваться, являются полимерные материалы и восковые материалы. При необходимости действующее вещество может быть также в микрокапсулированной форме с одним или несколькими из вышеупомянутых вспомогательных веществ.
Жидкие лекарственные формы для приема внутрь включают фармацевтически приемлемые эмульсии, растворы, суспензии, сиропы или настойки. Помимо действующего вещества жидкая лекарственная форма может содержать инертные разбавители, обычно применяемые в данной области, например, воду или другие растворители, солюбилизаторы и эмульгаторы, например, этанол, изопропанол, этилкарбонат, этилацетат, пропиленгликоль, 1,3-бутандиол, диметилформамид и масла, в частности, хлопковое масло, арахисовое масло, масло зародышей кукурузы, оливковое масло, касторовое масло и кунжутное масло, либо смеси этих веществ.
Кроме этих инертных разбавителей в композициях также могут быть вспомогательные вещества, например, смачивающие, эмульгирующие и суспендирующие вещества, подсластители, ароматизаторы и вкусовые добавки.
Помимо действующего вещества в суспензии также может быть суспендирующее вещество, например, этоксилированный изооктадеканол, полиоксиэтиленсорбит и дегидратированный сложный эфир сорбитана, микрокристаллическая целлюлоза, алюминия метоксид и агар, либо их смесь, и т.д.
В композициях для парентерального введения могут быть физиологически приемлемые стерильные водные или безводные растворы, дисперсии, суспензии или эмульсии и стерильные порошки, которые могут быть восстановлены в стерильных растворах или дисперсиях для инъекций. Подходящими водными и неводными носителями, разбавителями, растворителями или вспомогательными веществами являются вода, этанол, полиолы и любые подходящие их смеси.
Лекарственными формами соединений по изобретению для местного применения являются мази, порошки, пластыри, спреи и лекарственные формы для ингаляций. Действующее вещество смешивают в стерильных условиях с физиологически приемлемым вспомогательным веществом и какими-либо консервантами, буферными растворами или распыляющими веществами, которые могут потребоваться при необходимости.
Соединения по настоящему изобретению можно вводить отдельно или в комбинации с другими фармацевтически приемлемыми соединениями.
Метод лечения по настоящему изобретению можно применять отдельно или в комбинации с другими лечебными процедурами или терапевтическими средствами.
При использовании фармацевтической композиции безопасное и эффективное количество соединения по настоящему изобретению вводят млекопитающему (например, человеку), нуждающемуся в лечении, при этом дозировка при введении представляет собой фармацевтически эффективную дозу. Для людей с массой тела 60 кг суточная доза обычно составляет 1~2000 мг, предпочтительно 5~1000 мг. Конечно, в конкретных дозах также следует учитывать такие факторы, как путь введения, состояние здоровья пациента и т.д., находящиеся в компетенции квалифицированного врача.
По сравнению с предшествующим уровнем техники настоящее изобретение имеет следующие основные преимущества:
(1) соединение имеет лучшие фармакокинетические характеристики, например, лучшее соотношение концентрации в крови и мозговой ткани, период полувыведения, экспозиция, метаболическая стабильность и другие характеристики;
(2) соединение имеет более высокую активность открытия калиевых каналов, более высокую селективность ионных каналов, более высокую эффективность in vivo и более высокую безопасность;
(3) ожидается, что соединение будет использоваться для лечения и/или профилактики заболеваний и состояний, на которые влияет активность калиевых каналов.
Настоящее изобретение будет дополнительно проиллюстрировано ниже с указанием отдельных примеров. Следует понимать, что эти примеры используются только для иллюстрации изобретения и не предназначены для ограничения объема изобретения. Для экспериментальных методов в следующих примерах, в которых конкретные условия не указаны, они обычно соответствуют общепринятым, например, условиям, описанным в Sambrook et al., Molecular Cloning: Laboratory Manual (New York: Cold Spring Harbour Laboratory Press, 1989), или условиям, рекомендованным производителем. Если не указано иное, процентные значения и части рассчитываются по массе.
Если не указано иное, вся профессиональные и научные термины, используемые в тексте, имеют то же значение, которое известно специалистам в данной области. Кроме того, все способы и материалы, аналогичные или идентичные приведенным примерам, могут применяться к способам изобретения. Описанный здесь способ предпочтительного варианта осуществления и материал представлены только для демонстрационных целей.
Материалы и реактивы, используемые в следующих примерах, могут быть получены из коммерческих источников, если не указано иное.
Пример 1: Получение соединения A

Этап 1. Соединение 2
Соединение 1 (2 г, 8,05 ммоль, 1,0 экв.) растворяли в смешанном растворителе из дихлорметана (100 мл) и метанола (10 мл), затем добавляли BnMe3NBr3 (6,28 г, 16,1 ммоль, 2,0 экв.) и кальция карбонат (2,01 г, 20,1 ммоль, 2,5 экв.), и реакционный раствор перемешивали при комнатной температуре в течение 0,5 ч. Реакционный раствор фильтровали и фильтрат концентрировали с получением остатка, который очищали колоночной хроматографией на нейтральном алюминия оксиде (петролейный эфир:этилацетат = 15:1) с получением соединения 2 (2,9 г, выход 90%) в виде твердого вещества белого цвета.
ВЭЖХ-МС: [M+H]+ = 406,9.
Этап 2. Соединение 3
Соединение 2 (1 г, 2,46 ммоль, 1,0 экв.) растворяли в 1,4-диоксане (50 мл), затем добавляли метилборную кислоту (589 мг, 9,84 ммоль, 4,0 экв.), калия фосфат (2,089 г, 9,84 ммоль, 4,0 экв.) и Pd(dppf)Cl2 (0,1 г, 0,14 ммоль, 0,06 экв.), реакционный раствор нагревали до 120°C в защитной среде азота и перемешивали в течение 4 ч. После охлаждения до комнатной температуры реакционный раствор фильтровали, фильтрат разбавляли водой, затем экстрагировали этилацетатом (3×40 мл), органическую фазу промывали насыщенным раствором натрия хлорида, сушили над безводным натрия сульфатом и концентрировали с получением остатка, который очищали колоночной хроматографией на силикагеле (петролейный эфир:этилацетат = 10:1) с получением соединения 3 (504 мг, выход 74%) в виде твердого вещества белого цвета.
ВЭЖХ-МС: [M+H]+ = 277,2.
Этап 3. Соединение 5
Соединение 3 (600 мг, 2,17 ммоль, 1,0 экв.) растворяли в дихлорметане (15 мл), охлаждали до 0°C, добавляли триэтиламин (0,78 мл, 5,43 ммоль, 2,5 экв.) и соединение 4 (438 мг, 3,26 ммоль, 1,5 экв.), температуру реакционного раствора доводили до комнатной температуры и перемешивали в течение 0,5 ч. Реакционный раствор разбавляли водой, затем экстрагировали дихлорметаном (3×10 мл), органическую фазу промывали насыщенным раствором натрия хлорида, сушили над безводным натрия сульфатом и концентрировали, остаток суспендировали и очищали смешанным растворителем (петролейный эфир/этилацетат = 30 мл/3 мл) в течение получаса, затем фильтровали и полученное твердое вещество сушили с получением соединения 5 (680 мг, выход 84%) в виде твердого вещества белого цвета.
Этап 4. Соединение 6
Соединение 5 (680 мг, 1,82 ммоль, 1,0 экв.) и раствор гидрохлорида метанола (4M, 13 мл) добавляли в одногорлую колбу и перемешивали при комнатной температуре в течение 0,5 ч. Реакционный раствор концентрировали и разбавляли этилацетатом (10 мл), затем промывали водным раствором натрия карбоната (0,5 М, 20 мл) и отделенную водную фазу экстрагировали этилацетатом (3×10 мл). Объединенную органическую фазу промывали насыщенным раствором натрия хлорида, сушили над безводным натрия сульфатом и концентрировали органический слой с получением остатка, который растворяли в тетрагидрофуране, сушили над безводным натрия сульфатом и концентрировали с получением соединения 6 (370 мг, выход 74%) в виде твердого вещества белого цвета.
ВЭЖХ-МС: [M+H]+ = 275,2
Этап 5. Соединение A
Соединение 6 (150 мг, 0,547 ммоль, 1,5 экв.) растворяли в толуоле (50 мл) и затем последовательно добавляли цезия карбонат (237 мг, 0,728 ммоль, 2,0 экв.), X-Phos (35 мг, 0,0728 ммоль, 0,2 экв.), соединение 7 (81 мг, 0,364 ммоль, 1,0 экв.) и Pd2(dba)3 (34 мг, 0,0364 ммоль, 0,1 экв.), и реакционный раствор перемешивали при 90°C в течение 16 ч в защитной среде азота. Реакционный раствор охлаждали до комнатной температуры и фильтровали, фильтрат разбавляли водой, затем экстрагировали этилацетатом (3×20 мл), органическую фазу промывали насыщенным раствором натрия хлорида, сушили над безводным натрия сульфатом и концентрировали. с получением остатка, который очищали колоночной хроматографией на силикагеле (петролейный эфир:этилацетат = 2:1) с получением соединения А (6,0 мг, 3%) в виде твердого вещества белого цвета.
ВЭЖХ-МС: [M+H]+ = 369,2
1H-ЯМР (400 МГц, ДМСО) δ 9,10 (синглет, 1H), 7,08-7,02 (мультиплет, 4H), 6,90 (синглет, 1H), 4,26 (синглет, 2H), 3,49 (синглет, 2H), 2,74-2,71 (мультиплет, 2H), 2,21 (синглет, 2H), 2,11 (синглет, 3H), 2,01 (синглет, 3H), 1,06 (синглет, 9H).
Пример 2: Получение соединения B

Этап 1. Соединение B
Соединение 1 (600 мг, 2,19 ммоль, 1,0 экв.), соединение 2 (3,6 г, 27,3 ммоль, 12,4 экв.) и цезия карбонат (7,14 г, 21,9 ммоль, 10 экв.) добавляли к N-метилпирролидону (60 мл), реакционный раствор нагревали до 180°С в СВЧ-реакторе и перемешивали в течение 4 ч. Реакционный раствор охлаждали до комнатной температуры и разбавляли этилацетатом (150 мл), затем промывали насыщенным раствором натрия хлорида (80 мл × 4), сушили над безводным натрия сульфатом и концентрировали, и полученный остаток очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат = 10/1) с получением соединения В (83,9 мг, выход 10%) в виде твердого вещества белого цвета.
ВЭЖХ-МС: [M+H]+ = 387,1
1H-ЯМР (400 МГц, ДМСО-d6) δ 9,11 (синглет, 1H), 7,21-7,15 (мультиплет, 1H), 6,94-6,84 (мультиплет, 2H), 6,80-6,69 (мультиплет, 1H), 4,19 (синглет, 2H), 3,44-3,36 (мультиплет, 2H), 2,76-2,71 (мультиплет, 2H), 2,21 (синглет, 2H), 2,10 (синглет, 3H), 2,01 (синглет, 3H), 1,05 (синглет, 9H).
Пример 3: Получение соединения C
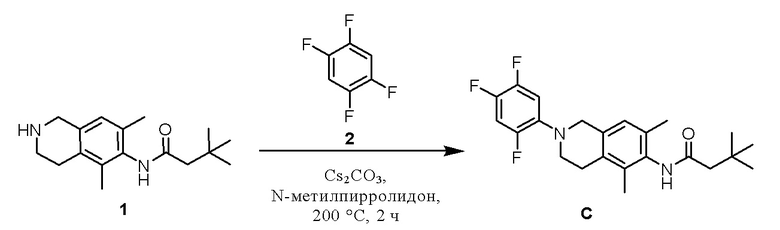
Этап 1. Соединение C
Соединение 1 (100 мг, 0,36 ммоль, 1,0 экв.) растворяли в N-метилпирролидоне (5 мл), затем добавляли цезия карбонат (1,0 г, 3,1 ммоль, 8,6 экв.) и соединение 2 (1,0 г, 6,67 ммоль, 18,5 экв.). Реакционный раствор нагревали до 200°C в СВЧ-реакторе и перемешивали в течение 2 ч, охлаждали до комнатной температуры и фильтровали, твердое вещество промывали этилацетатом (3×5 мл), и фильтрат промывали насыщенным раствором натрия хлорида (3×10 мл). Органическую фазу сушили над безводным натрия сульфатом и концентрировали с получением остатка, который очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат = 2/1) с получением соединения C (15,5 мг, выход 10%) в виде твердого вещества белого цвета.
ВЭЖХ-МС: [M+H]+ = 405,1
1H-ЯМР (400 МГц, ДМСО-d6) δ 9,13 (синглет, 1H), 7,58-7,46 (мультиплет, 1H), 7,23-7,16 (мультиплет, 1H), 6,86 (синглет, 1H), 4,15 (синглет, 2H), 3,37-3,33 (мультиплет, 2H), 2,79-2,72 (мультиплет, 2H), 2,22 (синглет, 2H), 2,11 (синглет, 3H), 2,02 (синглет, 3H), 1,07 (синглет, 9H).
Пример 4: Получение соединения D
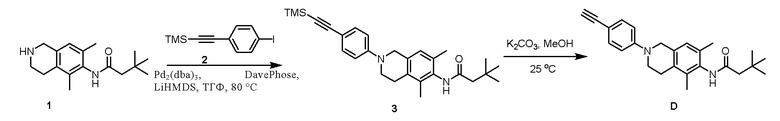
Этап 1. Соединение 3
Соединение 1 (50 мг, 0,182 ммоль, 1,0 экв.) растворяли в тетрагидрофуране (5 мл), а затем последовательно добавляли соединение 2 (164 мг, 0,547 ммоль, 3,0 экв.), Pd2(dba)3 (17 мг, 0,018 ммоль, 0,1 экв.), Dave-Phos (14 мг, 0,036 ммоль, 0,2 экв.) и LiHMDS (1 М, 1,8 мл, 1,8 ммоль, 10 экв.). Реакционный раствор перемешивали при 80°C в защитной среде азота в течение 2 ч, охлаждали до комнатной температуры, разбавляли этилацетатом (150 мл) и затем промывали насыщенным раствором натрия хлорида (80 мл×4). Полученную органическую фазу сушили над безводным натрия сульфатом и затем концентрировали. Полученный остаток очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат = 3/1) с получением соединения 3 (46 мг, выход 28%) в виде твердого вещества желтого цвета.
ВЭЖХ-МС: [M+H]+ = 447,2
Этап 2. Соединение D
Соединение 3 (20 мг, 0,045 ммоль, 1,0 экв.) и калия карбонат (62 мг, 0,447 ммоль, 10 экв.) добавляли к метанолу (3 мл) и реакционный раствор перемешивали при 25°С в течение 2 ч. Реакционный раствор концентрировали, полученный остаток растворяли в этилацетате (20 мл), затем промывали насыщенным раствором натрия хлорида (10 мл×2), полученную органическую фазу сушили над безводным натрия сульфатом и концентрировали и остаток очищали методом препаративной ВЭЖХ (0,1% муравьиная кислота/ацетонитрил/вода) с получением соединения D (4,2 мг, выход 7%) в виде твердого вещества красноватого цвета.
ВЭЖХ-МС: [M+H]+ = 375,2
1H-ЯМР (400 МГц, ДМСО-d6) δ 9,11 (синглет, 1H), 7,31 (дублет, J =8,8 Гц, 2H), 7,03-6,88 (мультиплет, 3H), 4,38 (синглет, 2H), 3,90 (синглет, 1H), 3,61 (синглет, 2H), 2,75-2,72 (мультиплет, 2H), 2,21 (синглет, 2H), 2,10 (синглет, 3H), 2,02 (синглет, 3H), 1,06 (синглет, 9H).
Пример 5: Получение соединения E

Этап 1. Соединение E
Соединение 1 (100 мг, 0,36 ммоль, 1,0 экв.) растворяли в диметилсульфоксиде (2 мл), затем последовательно добавляли соединение 2 (66 мг, 0,55 ммоль, 1,5 экв.) и калия карбонат (151 мг, 1,09 ммоль, 3,0 экв.), и реакционный раствор перемешивали при 100°С в течение 16 ч. После охлаждения до комнатной температуры к реакционному раствору добавляли воду (20 мл) и экстрагировали этилацетатом (3×20 мл). Объединенную органическую фазу промывали насыщенным раствором натрия хлорида и сушили над безводным натрия сульфатом. Остаток, полученный после концентрирования, очищали колоночной хроматографией на силикагеле (дихлорметан/метанол = 50/1) с получением соединения Е (50,3 мг, выход 37%) в виде твердого вещества белого цвета.
ВЭЖХ-МС: [M+H]+ = 376,2
1H-ЯМР (400 МГц, ДМСО-d6) δ 9,13 (синглет, 1H), 7,60-7,57 (мультиплет, 2H), 7,08-7,05 (мультиплет, 2H), 6,94 (синглет, 1H), 4,49 (синглет, 2H), 3,69 (синглет, 2H), 2,77 (триплет, J =5,6 Гц, 2H), 2,21 (синглет, 2H), 2,11 (синглет, 3H), 2,03 (синглет, 3H), 1,06 (синглет, 9H).
Пример 6: Получение соединения F

Этап 1. Соединение F
Соединение 1 (120 мг, 0,44 ммоль, 1,0 экв.) растворяли в ацетонитриле (20 мл), добавляли соединение 2 (148 мг, 1,31 ммоль, 3,0 экв.) и диизопропилэтиламин (339 мг, 2,63 ммоль, 6,0 экв.), нагревали в СВЧ-реакторе до 160°С и проводили реакцию смеси в течение 1 ч. Реакционный раствор концентрировали после охлаждения до комнатной температуры, разбавляли водой, затем экстрагировали дихлорметаном (3×100 мл), объединенную органическую фазу промывали насыщенным раствором натрия хлорида (50 мл), сушили над безводным натрия сульфатом и остаток, полученный после концентрирования, очищали колоночной хроматографией на силикагеле (дихлорметан/метанол = 10/1) с получением соединения F (40 мг, выход 26%) в виде твердого вещества желтого цвета.
ВЭЖХ-МС: [M+H]+ = 352,2
Пример 7: Получение соединения G

Этап 1. Соединение 3
Соединение 1 (600 мг, 3,0 ммоль, 1,0 экв.) и соединение 2 (780 мг, 3,9 ммоль, 1,3 экв.) растворяли в растворе аммиака в метаноле (1 М, 10,5 мл), реакционный раствор нагревали до 90°С в СВЧ-реакторе и проводили реакцию раствора в течение получаса. Реакционный раствор охлаждали до комнатной температуры, затем концентрировали, экстрагировали этилацетатом (50 мл×3) и насыщенным водным раствором натрия бикарбоната, органические фазы объединяли и сушили над безводным натрия сульфатом и остаток, полученный после концентрирования, очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат = 100/15) с получением соединения 3 (350 мг, выход 42%) в виде твердого вещества белого цвета.
Этап 2. Соединение 4
Соединение 3 (350 мг, 1,25 ммоль, 1,0 экв.) и палладиевую ч ернь (10%, 100 мг) растворяли в метаноле (30 мл) и перемешивали в течение 1 ч при комнатной температуре в атмосфере водорода под давлением 1 атм. Реакционный раствор фильтровали и фильтрат концентрировали с получением соединения 4 (300 мг, выход 96%) в виде бесцветного маслянистого вещества.
ВЭЖХ-МС: [M+H]+ = 250,1.
Этап 3. Соединение 5
Соединение 4 (300 мг, 1,2 ммоль, 1,0 экв.), бензилтриметиламмоний трибромид (936 мг, 2,4 ммоль, 2,0 экв.) и кальция карбонат (300 мг, 3,0 ммоль, 2,5 экв.) растворяли в смешанном растворителе дихлорметан/метанол (10/1,40 мл/4 мл) и перемешивали при комнатной температуре в течение получаса. Реакционный раствор фильтровали, фильтрат концентрировали и очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат = 10/1) с получением соединения 5 (160 мг, выход 33%) в виде твердого вещества белого цвета.
ВЭЖХ-МС: [M + Na]+ = 429,9.
Этап 4. Соединение 7
Соединение 5 (160 мг, 0,4 ммоль, 1,0 экв.) растворяли в 1,4-диоксане (15 мл), добавляли метилборную кислоту (96 мг, 1,6 ммоль, 4,0 экв.), Pd(dppf)Cl2 (58 мг, 0,08 ммоль, 0,2 экв.) и фосфат калия (339 мг, 1,6 ммоль, 4,0 экв.). Реакционный раствор нагревали до 120°С в защитной среде азота и проводили реакцию раствора в течение 4 ч. Реакционный раствор охлаждали до комнатной температуры и фильтровали. Остаток, полученный после концентрирования фильтрата, очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат = 65/35) с получением соединения 7 (80 мг, выход 72%) в виде твердого вещества белого цвета.
ВЭЖХ-МС: [M+H]+ = 278,1
Этап 5. Соединение 9
Соединение 7 (80 мг, 0,3 ммоль, 1,0 экв.) и триэтиламин (116 мг, 0,9 ммоль, 3,0 экв.) растворяли в дихлорметане (15 мл), медленно добавляли соединение 8 (60 мг, 0,45 ммоль, 1,5 экв.) в защитной среде азота и перемешивали при комнатной температуре в течение 1 ч. Реакционный раствор экстрагировали этилацетатом (50 мл×3) и насыщенным водным раствором натрия бикарбоната. Объединенную органическую фазу сушили над безводным натрия сульфатом и концентрировали. Остаток очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат = 65/35) с получением соединения 9 (80 мг, 71%) в виде твердого вещества светло-желтого цвета.
ВЭЖХ-МС: [M+H]+ = 376,3
Этап 6. Соединение 10
Соединение 9 (80 мг, 0,21 ммоль, 1,0 экв.) растворяли в растворе хлористого водорода в метаноле (4 М, 4 мл) и перемешивали в течение 1 ч при комнатной температуре. После концентрирования реакционного раствора получали соединение 10 (70 мг, выход 96%) в виде маслянистого вещества бледно-желтого цвета.
ВЭЖХ-МС: [M+H]+ = 276,2
Этап 7. Соединение G
Соединение 10 (30 мг, 0,11 ммоль, 1,0 экв.) и соединение 11 (38 мг, 0,17 ммоль, 1,5 экв.) растворяли в толуоле (10 мл), и затем последовательно добавляли Pd2(dba)3 (20 мг, 0,022 ммоль, 0,2 экв.), X-Phos (10 мг, 0,022 ммоль, 0,2 экв.) и цезия карбонат (90 мг, 0,275 ммоль, 2,5 экв.), реакционный раствор нагревали до 100°C в защитной среде азота и проводили реакцию раствора в течение 16 ч. Реакционный раствор охлаждали до комнатной температуры и затем концентрировали. Полученный остаток очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат = 1/1) с получением соединения G (5,3 мг, выход 13%) в виде твердого вещества белого цвета.
ВЭЖХ-МС: [M+H]+ = 370,1
1H-ЯМР (400 МГц, CDCl3) δ 7,02-6,94 (мультиплет, 4H), 6,80 (синглет, 1H), 4,35 (синглет, 2H), 3,50 (триплет, J =5,6 Гц, 2H), 2,84 (триплет, J =5,6 Гц, 2H), 2,48 (синглет, 3H), 2,33 (синглет, 2H), 2,15 (s,3H), 1,16 (синглет, 9H).
Пример 8: Получение соединения H

Этап 1. Соединение 3
Соединение 1 (5,0 г, 24,63 ммоль, 1,0 экв.) растворяли в толуоле (100 мл), добавляли соединение 2 (3,88 г, 36,945 ммоль, 1,5 экв.), реакционный раствор нагревали до 140°С и перемешивали в течение 16 ч. После охлаждения до комнатной температуры реакционный раствор концентрировали с получением соединения 3 (7 г, выход 98%) в виде маслянистого вещества коричневого цвета, которое непосредственно без очистки использовали в следующей реакции.
Этап 2. Соединение 4
Соединение 3 (3 г, 10,344 ммоль, 1,0 экв.) растворяли в метаноле (30 мл), охлаждали до 0°C и добавляли натрия борогидрид (0,27 г, 7,241 ммоль, 0,7 экв.), температуру реакционного раствора доводили до комнатной температуры и перемешивали в течение 2 ч. К реакционному раствору добавляли ледяную воду, метанол удаляли путем концентрирования и экстрагировали этилацетатом (3×30 мл). Объединенную органическую фазу промывали насыщенным раствором натрия хлорида, сушили над безводным натрия сульфатом и остаток, полученный после концентрирования, очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат = 4/1) с получением соединения 4 (2,5 г, выход 82%) в виде твердого вещества белого цвета.
ВЭЖХ-МС: [M+H]+ = 292,0
Этап 3. Соединение 5
Соединение 4 (7,45 г, 25,5 ммоль, 1,0 экв.) растворяли в дихлорметане (200 мл), последовательно добавляли 4-диметиламинопиридин (156 мг, 1,28 ммоль, 0,05 экв.) и триэтиламин (5,16 г, 51,0 ммоль, 2,0 экв.). реакционный раствор охлаждали до 0°C в защитной среде азота и добавляли п-метилбензолсульфонилхлорид (5,10 г, 26,8 ммоль, 1,05 экв.). Температуру реакционного раствора доводили до комнатной температуры и перемешивали в течение 16 ч, затем останавливали реакцию ледяной водой (200 мл) и водную фазу экстрагировали дихлорметаном (200 мл). Отделенную органическую фазу сушили над безводным натрия сульфатом и концентрировали. Полученный остаток очищали колоночной хроматографией на силикагеле (петролейный эфир:этилацетат = 10:1) с получением соединения 5 (11,0 г, выход 96%).
ВЭЖХ-МС: [M+Na]+ = 469,9
Этап 4. Соединение 6
Дихлорметан (300 мл) добавляли в реакционную колбу, содержащую алюминия трихлорид (22,0 г, 165 ммоль, 6,7 экв.), охлажденный до 0°C в защитной среде азота, после ч его добавляли соединение 5 (11,0 г, 24,65 ммоль, 1,0 экв.) в растворе метиленхлорида (100 мл). Температуру реакционного раствора доводили до комнатной температуры и перемешивали в течение 16 ч, затем останавливали реакцию ледяной водой (100 мл), значение рН раствора доводили до 10 аммиачной водой и водную фазу экстрагировали дихлорметаном (200 мл). Объединенную органическую фазу сушили над безводным натрия сульфатом и остаток, полученный после концентрирования, очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат = 4/1) с получением соединения 6 (2,6 г, выход 47%).
ВЭЖХ-МС: [M+H]+ = 225,9
Этап 5. Соединение 7
Соединение 6 (2,6 г, 11,5 ммоль, 1,0 экв.) растворяли в уксусной кислоте (40 мл) и добавляли натрия борогидрид (1,3 г, 34,5 ммоль, 3,0 экв.) после охлаждения до 0°C в защитной среде азота. Температуру реакционного раствора доводили до комнатной температуры и перемешивали в течение 2 ч, разбавляли водой (200 мл), охлаждали до 0°C, значение pH раствора доводили до 10 натрия карбонатом, после ч его экстрагировали этилацетатом (3×200 мл). Объединенную органическую фазу промывали насыщенным раствором натрия хлорида, сушили над безводным натрия сульфатом и концентрировали с получением неочищенного соединения 7 (3,0 г), которое непосредственно без очистки использовали в следующей реакции.
ВЭЖХ-МС: [M+H]+ = 230,0
Этап 6. Соединение 8
Соединение 7 (3,0 г, 11,5 ммоль, 1,0 экв.) растворяли в тетрагидрофуране (40 мл), добавляли диизопропилэтиламин (743 мг, 5,75 ммоль, 0,5 экв.) и Boc2O (3,0 г, 13,8 ммоль, 1,2 экв.). Реакционный раствор перемешивали при комнатной температуре в течение 16 ч и затем концентрировали, полученный остаток очищали колоночной хроматографией на силикагеле (петролейный эфир:этилацетат = 10:1) с получением соединения 8 (3,0 г, выход от двух этапов: 79%).
ВЭЖХ-МС: [M+Na]+ = 352,0
Этап 7. Соединение 10
Соединение 8 (3,0 г, 9,09 ммоль, 1,0 экв.) и соединение 9 (3,3 г, 18,7 ммоль, 2,0 экв.) растворяли в толуоле (100 мл), после ч его в защитной среде азота добавляли цезия карбонат (8,88 г, 27,26 ммоль, 3,0 экв.), BINAP (1,13 г, 1,82 ммоль, 0,2 экв.) и Pd2(dba)3 (832 мг, 0,09 ммоль, 0,1 экв.), реакционный раствор нагревали до 80°C и перемешивали в течение 20 ч, затем нагревали до 100°С и перемешивали в течение 2 ч. После охлаждения до комнатной температуры реакционный раствор фильтровали, твердое вещество промывали этилацетатом (200 мл), объединенный фильтрат промывали водой и насыщенным раствором натрия хлорида, затем сушили над безводным натрия сульфатом и концентрировали с получением неочищенного соединения 10 (6,0 г), которое использовали непосредственно без очистки в следующей реакции.
ВЭЖХ-МС: [M+H]+ = 431,1
Этап 8. Соединение 11
Соединение 10 (неочищенное, 6,0 г, 9,09 ммоль, 1,0 экв.) растворяли в дихлорметане (200 мл), добавляли водный раствор хлористоводородной кислоты (4 М, 100 мл) и перемешивали при комнатной температуре в течение 2 ч. Реакционный раствор отделяли, органическую фазу промывали водным раствором хлористоводородной кислоты (4 М, 3×30 мл) и рН объединенной водной фазы доводили до 8 натрия карбонатом с получением водного раствора соединения 11 (около 200 мл).
ВЭЖХ-МС: [M+H]+ = 167,1
Этап 9. Соединение 12
Boc2O (4,0 г, 18 ммоль, 2,0 экв.) в тетрагидрофуране (100 мл) добавляли к водному раствору соединения 11 (около 200 мл) и реакционный раствор перемешивали при комнатной температуре в течение 4 ч. Реакционный раствор экстрагировали этилацетатом (3×50 мл), объединенную органическую фазу промывали насыщенным раствором натрия хлорида, сушили над безводным натрия сульфатом и остаток, полученный после концентрирования, очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат = 5/1) с получением соединения 12 (3,0 г, выход от трех этапов: 90%).
ВЭЖХ-МС: [M+Na]+ = 389,1
Этап 10. Соединение 13
Соединение 12 (2,8 г, 7,65 ммоль, 1,0 экв.) растворяли в растворе хлористого водорода в метаноле (4 М, 40 мл) и смесь перемешивали при комнатной температуре в течение 2 ч. Реакционный раствор концентрировали с получением соединения 13 (2,0 г) в виде твердого вещества белого цвета.
ВЭЖХ-МС: [M+H]+ = 167,1
Этап 11. Соединение 14
Соединение 13 (2,0 г) растворяли в тетрагидрофуране (50 мл), значение pH доводили до 8 водным раствором натрия карбоната и добавляли Boc2O (1,5 г, 6,88 ммоль, 0,9 экв.). Реакционный раствор перемешивали при комнатной температуре в течение 10 мин, после ч его экстрагировали этилацетатом (3×60 мл), объединенную органическую фазу промывали насыщенным раствором натрия хлорида, сушили над безводным натрия сульфатом и остаток, полученный после концентрирования, очищали. колоночной хроматографией на силикагеле (петролейный эфир:этилацетат = 3:1) с получением соединения 14 (0,85 г, выход от двух этапов: 42%).
ВЭЖХ-МС: [M+H]+ = 267,1
Этап 12. Соединение 15
Соединение 14 (0,8 г, 3,0 ммоль, 1,0 экв.) растворяли в смешанном растворителе из дихлорметана (100 мл) и метанола (10 мл), добавляли кальция карбонат (2,1 г, 21,03 ммоль, 7,0 экв.), температуру снижали до 0°C в защитной среде азота, и к реакционному раствору порциями медленно добавляли BnMe3NBr3 (4,5 г, 11,5 ммоль, 4,0 экв.), температуру реакционного раствора доводили до комнатной температуры и перемешивали в течение получаса. Реакционный раствор фильтровали и твердое вещество промывали дихлорметаном (100 мл). Объединенный фильтрат промывали смешанным водным раствором натрия бикарбоната и натрия сульфита, органическую фазу сушили над безводным натрия сульфатом и концентрировали, а остаток очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат = 5/1) с получением соединения 15 (1,1 г, выход 80%) в виде твердого вещества белого цвета.
ВЭЖХ-МС: [M+Na]+ = 446,8
Этап 13, соединение 17
Соединение 17 (1,0 г, 2,36 ммоль, 1,0 экв.) растворяли в 1,4-диоксане (100 мл), добавляли метилборную кислоту (1,13 г, 18,86 ммоль, 4,0 экв.), калия фосфат (2,5 г, 11,79 ммоль, 5,0 экв.).) и Pd(dppf)Cl2 (289 мг, 0,35 ммоль, 0,15 экв.), реакционный раствор нагревали до 120°C в защитной среде азота и перемешивали в течение 8 ч. Реакционный раствор охлаждали до комнатной температуры и затем фильтровали. После концентрирования фильтрата его растворяли в воде и этилацетате и отделяли. Водную фазу экстрагировали этилацетатом. Объединенную органическую фазу промывали насыщенным раствором натрия хлорида, сушили над безводным натрия сульфатом и остаток, полученный после концентрирования, очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат = 6/1) с получением соединения 17 (650 мг, выход 85%) в виде твердого вещества белого цвета.
ВЭЖХ-МС: [M+H]+ = 295,1
Этап 14. Соединение 19
Соединение 17 (600 мг, 2,04 ммоль, 1,0 экв.) растворяли в дихлорметане (20 мл), охлаждали до 0°C, и добавляли триэтиламин (515 мг, 5,10 ммоль, 2,5 экв.) и соединение 18 (411 мг, 3,06 ммоль, 1,5 экв.). Температуру реакционного раствора доводили до комнатной температуры и перемешивали в течение 0,5 ч, реакционный раствор разбавляли водой и затем экстрагировали дихлорметаном (3×50 мл). Объединенную органическую фазу промывали водой, сушили над безводным натрия сульфатом, остаток, полученный после концентрирования, суспендировали со смешанным растворителем (петролейный эфир/этилацетат = 40/1) в течение получаса, и полученное после фильтрации твердое вещество высушивали с получением соединения 19 (700 мг, выход 87%) в виде твердого вещества белого цвета.
ВЭЖХ-МС: [M+Na]+ = 415,2
Этап 15. Соединение 20
Раствор соединения 19 (700 мг, 1,78 ммоль, 1,0 экв.) и хлористого водорода (4 М, 15 мл) в метаноле добавляли в одногорлую колбу и перемешивали в течение 0,5 ч при комнатной температуре. Реакционный раствор центрифугировали, добавляли этилацетат для разбавления, после ч его значение рН доводили до 8 водным раствором натрия карбоната и водную фазу экстрагировали этилацетатом. Объединенную органическую фазу промывали насыщенным раствором натрия хлорида, сушили над безводным натрия сульфатом и концентрировали с получением соединения 20 (400 мг, 77%).
ВЭЖХ-МС: [M+H]+ = 293,2
Этап 16. Соединение H
Соединение 20 (180 мг, 0,61 ммоль, 1,0 экв.), соединение 21 (1,08 г, 8,18 ммоль, 13,0 экв.) и цезия карбонат (2,0 г, 6,1 ммоль, 10 экв.) добавляли к N-метилпирролидону (10 мл), смесь нагревали до 180°C в СВЧ-реакторе в защитной среде азота и проводили реакцию смеси в течение 4 ч. Реакционный раствор охлаждали до комнатной температуры, разбавляли этилацетатом (150 мл), затем промывали насыщенным раствором натрия хлорида (80 мл×4), объединенную органическую фазу сушили над безводным натрия сульфатом и остаток, полученный после концентрирования, очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат = от 30/1 до 2/1) с получением соединения Н (4,2 мг, выход 17%) в виде твердого вещества белого цвета.
ВЭЖХ-МС: [M+Na]+ = 405,1
1H-ЯМР (400 МГц, ДМСО-d6) δ 9,30 (синглет, 1H), 7,21-7,18 (мультиплет, 1H), 7,00-6,96 (мультиплет, 1H), 6,82-6,77 (мультиплет, 1H), 4,23 (синглет, 2H), 3,44-3,36 (мультиплет, 2H), 2,82-2,73 (мультиплет, 2H), 2,24 (синглет, 2H), 2,04 (синглет, 3H), 2,01 (синглет, 3H), 1,07 (синглет, 9H).
Пример 9: Получение соединения I

Этап 1. Соединение 5
Соединение 3 (1,2 г, 10,8 ммоль, 1,5 экв.) растворяли в безводном диметилформамиде (5 мл), последовательно добавляли соединение 4 (2,0 г, 7,2 ммоль, 1,0 экв.), HATU (6,84 г, 18 ммоль, 2,5 экв.) и диизопропилэтиламин (6 мл, 36 ммоль, 5,0 экв.), реакционный раствор нагревали до 45°C и перемешивали в течение 16 ч. Реакционный раствор охлаждали до комнатной температуры, разбавляли водой и экстрагировали этилацетатом (3×10 мл). Объединенную органическую фазу промывали насыщенным раствором натрия хлорида, сушили над безводным натрия сульфатом и остаток, полученный после концентрирования, очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат = 2/1) с получением соединения 5 (2,5 г, выход 62%) в виде твердого вещества белого цвета.
ВЭЖХ-МС: [M + Na]+ = 395,2.
Этап 2. Соединение 6
Соединение 5 (2,5 г, 6,7 ммоль, 1,0 экв.) и раствор хлористого водорода в метаноле (4 М, 55 мл) последовательно добавляли в одногорлую колбу, и смесь перемешивали при комнатной температуре в течение 1 ч. Реакционный раствор концентрировали, полученный остаток растворяли в смешанном растворителе дихлорметан/метанол (10/1), затем промывали водным раствором натрия карбоната (0,5 М, 50 мл), и водную фазу экстрагировали смешанным растворителем дихлорметан/метанол (10/1,3×20 мл). Объединенную органическую фазу промывали насыщенным раствором натрия хлорида, сушили над безводным натрия сульфатом и концентрировали с получением соединения 6 (1,6 г, выход 85%) в виде твердого вещества серого цвета.
ВЭЖХ-МС: [M+H]+ = 273,2
Этап 3. Соединение I
Соединение 6 (1,4 г, 5,1 ммоль, 1,0 экв.) растворяли в толуоле (200 мл) и последовательно добавляли цезия карбонат (3,3 г, 10,2 ммоль, 2,0 экв.), соединение 7 (1,7 г, 7,7 ммоль, 1,5 экв.), X-Phos (486 мг, 1,02 ммоль, 0,2 экв.) и Pd2(dba)3 (467 мг, 0,51 ммоль, 0,1 экв.), температуру повышали до 100°C и смесь перемешивали в течение 16 ч в защитной среде азота. Реакционный раствор охлаждали до комнатной температуры, затем фильтровали, фильтрат разбавляли водой и экстрагировали этилацетатом (3×20 мл), объединенную органическую фазу промывали насыщенным раствором натрия хлорида, сушили над безводным натрия сульфатом и остаток, полученный после концентрирования, очищали колоночной хроматографией на нейтральном алюминия оксиде (петролейный эфир/этилацетат = 5/1) с получением соединения I, неочищенное соединение I далее очищали методом препаративной ВЭЖХ (0,1% муравьиная кислота/ацетонитрил/вода) с получением соединения I (377,0 мг, 20%) в виде твердого вещества белого цвета.
ВЭЖХ-МС: [M+H]+ = 367,2
1H-ЯМР (400 МГц, ДМСО-d6) δ 9,02 (синглет, 1H), 7,06-7,03 (мультиплет, 4H), 6,91 (синглет, 1H), 4,26 (синглет, 2H), 3,50 (синглет, 2H), 2,74-2,72 (мультиплет, 2H), 2,21 (синглет, 2H), 2,11 (синглет, 3H), 2,01 (синглет, 3H), 1,15 (синглет, 3H), 0,56-0,53 (мультиплет, 2H), 0,33-0,31 (мультиплет, 2H).
Пример 10: Получение соединения J

Этап 1. Соединение J
Соединение 1 (600 мг, 2,20 ммоль, 1,0 экв.), соединение 2 (3,6 г, 27,3 ммоль, 12,4 экв.) и цезия карбонат (7,19 г, 22,0 ммоль, 10 экв.) добавляли к N-метилпирролидону (60 мл), и реакционную смесь нагревали до 180°С в СВЧ-реакторе и перемешивали в течение 4 ч. После охлаждения до комнатной температуры реакционный раствор разбавляли этилацетатом (100 мл), затем промывали насыщенным раствором натрия хлорида (80 мл×2), сушили над безводным натрия сульфатом и остаток, полученный после концентрирования, очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат = 10/1) с получением соединения J (56,0 мг, 0,146 ммоль, выход 7%) в виде твердого вещества белого цвета.
ВЭЖХ-МС: [M+H]+ = 385,1
1H-ЯМР (400 МГц, ДМСО-d6) δ 9,01 (синглет, 1H), 7,19-7,12 (мультиплет, 1H), 6,92-6,82 (мультиплет, 2H), 6,75-6,67 (мультиплет, 1H), 4,16 (синглет, 2H), 3,37 (триплет, J =5,6 Гц, 2H), 2,71 (триплет, J =5,6 Гц, 2H), 2,18 (синглет, 2H), 2,07 (синглет, 3H), 1,97 (синглет, 3H), 1,11 (синглет, 3H), 0,52-0,50 (мультиплет, 2H), 0,29-0,27 (мультиплет, 2H).
Пример 11: Получение соединения K
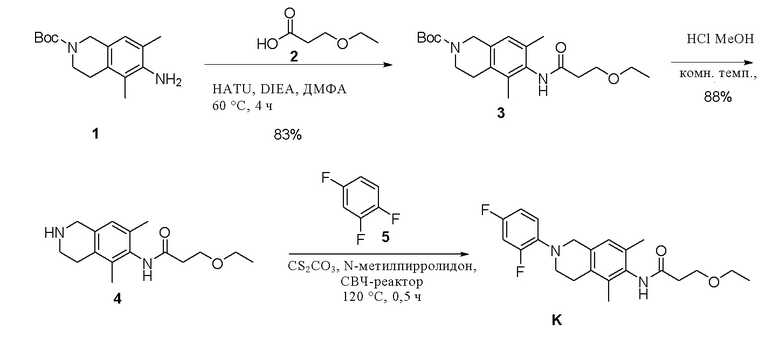
Этап 1. Соединение 3
Соединение 1 (200 мг, 0,724 ммоль, 1,0 экв.) растворяли в диметилформамиде (5 мл), добавляли соединение 2 (128 мг, 1,086 ммоль, 1,5 экв.), HATU (412 мг, 1,086 ммоль, 1,5 экв.) и диизопропилэтиламин (280 мг, 2,172 ммоль, 3,0 экв.), смесь нагревали до 60°C и перемешивали в течение 4 ч. Реакционный раствор охлаждали до комнатной температуры, разбавляли водой (20 мл) и водную фазу экстрагировали этилацетатом (3×20 мл). Объединенную органическую фазу промывали насыщенным раствором натрия хлорида, сушили над безводным натрия сульфатом и остаток, полученный после концентрирования, очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат = 1/1) с получением соединения 3 (220 мг, выход 83%) в виде твердого вещества желтого цвета.
ВЭЖХ-МС: [M+H]+ = 377,2
Этап 2. Соединение 4
Соединение 3 (280 мг, 0,745 ммоль, 1,0 экв.) растворяли в растворе хлористого водорода в метаноле (4 М, 10 мл) и смесь перемешивали в течение 1 ч при комнатной температуре. После концентрирования к остатку добавляли насыщенный водный раствор натрия карбоната для доведения значения рН до 8-9, экстрагировали этилацетатом (3×30 мл), объединенную органическую фазу промывали насыщенным раствором натрия хлорида, сушили над безводным натрия сульфатом и концентрировали с получением соединения 4 (180 мг, выход 88%) в виде твердого вещества желтого цвета.
ВЭЖХ-МС: [M+H]+ = 277,2
Этап 3. Соединение K
Соединение 4 (10 мг, 0,036 ммоль, 1,0 экв.) растворяли в N-метилпирролидоне (1 мл), добавляли цезия карбонат (117 мг, 0,36 ммоль, 10,0 экв.) и соединение 5 (58 мг, 0,446 ммоль, 12,4 экв.), реакционный раствор нагревали до 120°С в СВЧ-реакторе, проводили реакцию раствора в течение 0,5 ч, охлаждали до комнатной температуры и затем концентрировали, остаток очищали методом препаративной ВЭЖХ с получением соединения К.
ВЭЖХ-МС: [M+H]+ = 389,1
Пример 12: Получение соединения L
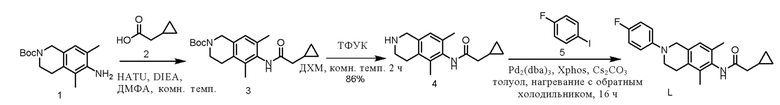
Этап 1. Соединение 3
Соединение 1 (100 мг, 0,54 ммоль, 1,0 экв.) растворяли в диметилформамиде (3 мл), последовательно добавляли соединение 2 (81 мг, 0,81 ммоль, 1,5 экв.), HATU (314 мг, 0,81 ммоль, 1,5 экв.) и диизопропилэтиламин (209 мг, 1,62 ммоль, 3,0 экв.), реакцию смеси проводили при комнатной температуре в течение 1 ч. Реакционный раствор экстрагировали этилацетатом (50 мл×3) и насыщенным раствором натрия хлорида, объединенную органическую фазу сушили над безводным натрия сульфатом и остаток, полученный после концентрирования, очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат). = 3/2) с получением соединения 3 (130 мг, выход 67%) в виде твердого вещества белого цвета.
ВЭЖХ-МС: [M + Na]+ = 381,1.
Этап 2. Соединение 4
Соединение 3 (130 мг, 0,36 ммоль, 1,0 экв.) растворяли в дихлорметане (10 мл), добавляли трифторуксусную кислоту (2 мл) и смесь перемешивали при комнатной температуре в течение 2 ч. Реакционный раствор концентрировали, экстрагировали этилацетатом и насыщенным водным раствором натрия бикарбоната, органическую фазу сушили над безводным натрия сульфатом и концентрировали с получением соединения 4 (80 мг, выход 86%) в виде маслянистого вещества светло-желтого цвета.
ВЭЖХ-МС: [M+H]+ = 259,1
Этап 3. Соединение L
Соединение 4 (80 мг, 0,31 ммоль, 1,0 экв.) и соединение 5 (103 мг, 0,47 ммоль, 1,5 экв.) растворяли в толуоле (10 мл), и добавляли Pd2(dba)3 (57 мг, 0,062 ммоль, 0,2 экв.), x-Phos (30 мг, 0,062 ммоль, 0,2 экв.) и цезия карбонат (253 мг, 0,78 ммоль, 2,5 экв.), реакционный раствор нагревали до 100°C в защитной среде азота и проводили реакцию раствора в течение 18 ч. Реакционный раствор охлаждали до комнатной температуры и затем концентрировали. Полученный остаток очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат = 1/1) с получением соединения L (8,8 мг, выход 8%) в виде твердого вещества белого цвета.
ВЭЖХ-МС: [M+H]+ = 353,1
1H-ЯМР (400 МГц, CDCl3) δ 7,23 (синглет, 1H), 7,02-6,93 (мультиплет, 4H), 6,90 (синглет, 1H), 4,27 (синглет, 2H), 3,50-3,47 (мультиплет, 2H), 2,84-2,82 (мультиплет, 2H), 2,39 (дублет, J =7,6 Гц, 2H), 2,23 (синглет, 3H), 2,13 (синглет, 3H), 1,19-1,11 (мультиплет, 1H), 0,77-0,67 (мультиплет, 2H), 0,36-0,33 (мультиплет, 2H).
Пример 13: Получение соединения M
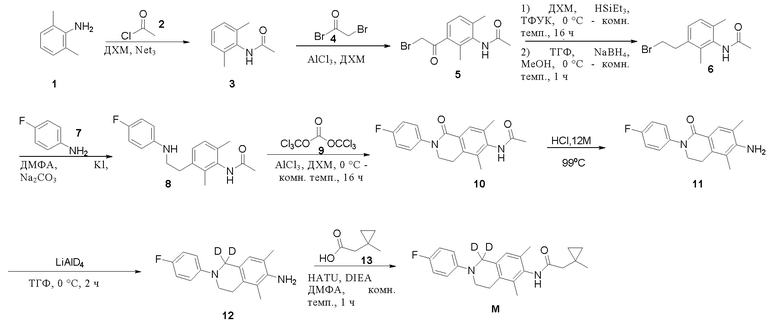

Этап 1. Соединение 3
Соединение 1 (6,0 г, 49,5 ммоль, 1,0 экв.) и диизопропилэтиламин (12,8 г, 99,0 ммоль, 2,0 экв.) добавляли к метиленхлориду (100 мл), указанный выше раствор охлаждали до 0°C в защитной среде азота, к реакционному раствору медленно добавляли соединение 2 (5,83 г, 74,3 ммоль, 1,5 экв.) и проводили реакцию раствора при комнатной температуре в течение 2 ч в защитной среде азота. Реакционный раствор концентрировали, и полученный остаток очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат = 1:1) с получением твердого вещества белого цвета 3 (6,0 г, выход 74%).
ВЭЖХ-МС: [M + H]+ =164,1.
Этап 2. Соединение 5
Алюминия трихлорид (9,6 г, 72,0 ммоль, 3,0 экв.) добавляли к дихлорметану (100 мл), реакционную смесь охлаждали до 0°C в защитной среде азота и медленно добавляли соединение 3 (4,0 г, 24,0 ммоль, 1,0 экв.) и соединение 4. (7,4 г, 36,6 ммоль, 1,5 экв.) в растворе дихлорметана (50 мл). Реакционный раствор медленно нагревали до комнатной температуры и перемешивали в течение 16 ч, разбавляли дихлорметаном (100 мл) и затем промывали ледяной водой (5×200 мл). Отделенную органическую фазу сушили над безводным натрия сульфатом и концентрировали, получая неочищенное соединение 5 (6,0 г). Неочищенное соединение 5 суспендировали со смешанным растворителем дихлорметан/петролейный эфир (20 мл/40 мл) и фильтровали с получением соединения 5 (4,0 г, выход 58%) в виде твердого вещества белого цвета.
ВЭЖХ-МС: [M+H]+ = 284,0
Этап 3. Соединение 6
Соединение 5 (2,0 г, 7,0 ммоль, 1,0 экв.) растворяли в дихлорметане (10 мл), добавляли HSiEt3 (10 мл), реакционный раствор охлаждали до 0°C в защитной среде азота, медленно добавляли трифторуксусную кислоту (10 мл), реакционный раствор медленно нагревали до комнатной температуры и перемешивали в течение 16 ч. Указанный выше реакционный раствор концентрировали, полученный остаток растворяли в тетрагидрофуране (30 мл), затем раствор охлаждали до 0°C в защитной среде азота, медленно добавляли натрия борогидрид (532 мг, 14 ммоль, 2,0 экв.), нагревали до комнатной температуры и смесь перемешивали в течение 0,5 ч. Указанный выше реакционный раствор охлаждали до 0°C в защитной среде азота, медленно по каплям добавляли метанол (10 мл) и остаток, полученный после концентрирования реакционного раствора, очищали колоночной хроматографией на силикагеле (петролейный эфир:этилацетат). = 2:1) с получением соединения 6 (2,0 г, выход 100%) в виде твердого вещества белого цвета.
ВЭЖХ-МС: [M + H] + =270,0.
Этап 4. Соединение 8
Соединение 6 (2,0 г, 7,0 ммоль, 1,0 экв.), соединение 7 (1,65 г, 14,8 ммоль, 2,1 экв.), йодид калия (1,23 г, 7,0 ммоль, 1,0 экв.) и калия карбонат (2,35 г, 22 ммоль, 3,0 экв.) добавляли к диметилформамиду (10 мл) в защитной среде азота, реакционный раствор нагревали до 90°C и перемешивали в течение 16 ч. После охлаждения до комнатной температуры реакционный раствор разбавляли этилацетатом (200 мл) и фильтровали, фильтрат промывали насыщенным раствором натрия хлорида, органическую фазу сушили над безводным натрия сульфатом и остаток, полученный после концентрирования, очищали колоночной хроматографией на силикагеле (петролейный эфир:этилацетат = 2:1) с получением соединения 8 (1,0 г, выход 45%) в виде твердого вещества белого цвета.
ВЭЖХ-МС: [M+H]+ = 301,1
Этап 5. Соединение 10
Соединение 8 (600 мг, 2,0 ммоль, 1,0 экв.) растворяли в дихлорметане (20 мл), добавляли соединение 9 (296 мг, 1,0 ммоль, 0,5 экв.) и перемешивали при комнатной температуре в течение 1 ч в защитной среде азота. Реакционный раствор охлаждали до 0°C, добавляли алюминия трихлорид (800 мг, 6,0 ммоль, 3,0 экв.), температуру медленно повышали до комнатной температуры и реакционный раствор перемешивали в течение 16 ч. Реакционный раствор разбавляли дихлорметаном (100 мл), промывали ледяной водой, органическую фазу сушили над безводным натрия сульфатом и остаток, полученный после концентрирования, очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат = 2/1) с получением соединения 10 (200 мг, выход 30%) в виде твердого вещества белого цвета.
ВЭЖХ-МС: [M+H]+ = 327,1
Этап 6. Соединение 11
Соединение 10 (190 мг, 0,6 ммоль) растворяли в концентрированной хлористоводородной кислоте (12М, 15 мл), реакционный раствор нагревали до 99°С и перемешивали в течение 64 ч в защитной среде азота. После охлаждения до комнатной температуры реакционный раствор концентрировали, и полученный остаток подвергали азеотропной обработке толуолом и этанолом для удаления воды с получением неочищенного соединения 11 (200 мг) в виде твердого вещества белого цвета.
ВЭЖХ-МС: [M+H]+ = 285,2
Этап 7. Соединение 12
Соединение 11 (200 мг, 0,6 ммоль) растворяли в тетрагидрофуране (50 мл), охлаждали до 0°C, медленно добавляли LiAlD4 (252 мг, 6,0 ммоль, 10,0 экв.), затем температуру повышали до комнатной температуры и смесь перемешивали в течение 16 ч. Реакционный раствор разбавляли тетрагидрофураном (250 мл) и охлаждали до 0°С, добавляли натрия сульфата декагидрат (10 г). Реакционный раствор перемешивали в течение 0,5 ч и остаток, полученный после фильтрации и концентрирования реакционного раствора, очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат = 2/1) с получением соединения 12 (25 мг, выход от двух этапов: 15%) в виде твердого вещества желтого цвета.
ВЭЖХ-МС: [M+H]+ = 273,1
Этап 8. Соединение M
Соединение 13 (21 мг, 0,183 ммоль, 2,0 экв.), HATU (70 мг, 0,183 ммоль, 2,0 экв.) и диизопропилэтиламин (36 мг, 0,275 ммоль, 3,0 экв.) растворяли в диметилформамиде (5 мл), и реакционный раствор перемешивали при комнатной температуре в течение 0,5 ч в защитной среде азота. К указанному выше реакционному раствору добавляли соединение 12 (25 мг, 0,092 ммоль, 1,0 экв.), реакционный раствор нагревали до 45°C и реакционный раствор перемешивали в течение 16 ч. Реакционный раствор разбавляли этилацетатом (50 мл), промывали насыщенным раствором натрия хлорида, органическую фазу сушили над безводным натрия сульфатом и остаток, полученный после концентрирования, очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат = 2/1) с получением соединения М (2,3 мг, выход 6,8%) в виде твердого вещества белого цвета.
ВЭЖХ-МС: [M+H]+ = 369,2
1H-ЯМР (400 МГц, CDCl3) δ 7,18 (синглет, 1H), 7,01-6,86 (мультиплет, 5H), 3,47 (триплет, J = 5,6 Гц, 2H), 2,81 (триплет, J = 5,6 Гц, 2H), 2,38 (синглет, 2H), 2,22 (синглет, 3H), 2,12 (синглет, 3H), 1,28 (синглет, 3H), 0,60-0,50 (мультиплет, 4H).
Пример 14: Получение соединения N
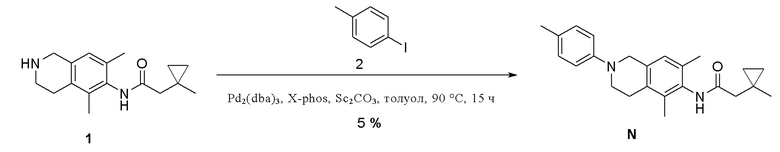
Этап 1. Соединение N
Соединение 1 (30 мг, 0,11 ммоль, 1,0 экв.) растворяли в толуоле (13 мл) и последовательно добавляли цезия карбонат (384 мг, 0,33 ммоль, 3,0 экв.), соединение 2 (37 мг, 0,17 ммоль, 1,5 экв.), x-Phos (11 мг, 0,02 ммоль, 0,2 экв.) и Pd2(dba)3 (11 мг, 0,01 ммоль, 0,1 экв.), температуру повышали до 90°C и смесь перемешивали в течение 15 ч в защитной среде азота. Реакционный раствор охлаждали до комнатной температуры, разбавляли водой, экстрагировали этилацетатом (3×20 мл), органическую фазу объединяли, промывали насыщенным раствором натрия хлорида, сушили над безводным натрия сульфатом, и полученный после концентрирования остаток очищали методом препаративной ВЭЖХ (0,1% муравьиная кислота/ацетонитрил/вода) с получением соединения N (2 мг, выход 5%) в виде твердого вещества белого цвета.
ВЭЖХ-МС: [M+H]+ = 363,2
1H-ЯМР (400 МГц, ДМСО-d6) δ 9,02 (синглет, 1H), 7,04-7,02 (мультиплет, 2H), 6,93-6,91 (мультиплет, 3H), 4,25 (синглет, 2H), 3,49 (синглет, 2H), 2,51-2,49 (мультиплет, 2H), 2,21(синглет, 2H), 2,20 (синглет, 3H), 2,11 (синглет, 3H), 2,01 (синглет, 3H), 1,15 (синглет, 3H), 0,56-0,53 (мультиплет, 2H), 0,33-0,31 (мультиплет, 2H).
Пример 15: Получение соединения O

Этап 1. Соединение O
Соединение 1 (50 мг, 0,183 ммоль, 1,0 экв.) растворяли в диметилсульфоксиде (5 мл), добавляли соединение 2 (33 мг, 0,275 ммоль, 1,5 экв.) и калия карбонат (76 мг, 0,275 ммоль, 1,5 экв.), смесь нагревали до 100°C и перемешивали в течение 16 ч. После охлаждения до комнатной температуры смесь разбавляли водой, экстрагировали этилацетатом (3×20 мл), объединенную органическую фазу промывали насыщенным раствором натрия хлорида, сушили над безводным натрия сульфатом и остаток, полученный после концентрирования, очищали методом препаративной ВЭЖХ (0,1% аммиак/ацетонитрил/вода) с получением соединения О (4,2 мг, выход 6%) в виде твердого вещества белого цвета.
ВЭЖХ-МС: [M+H]+ = 374,1
1H-ЯМР (400 МГц, ДМСО-d6) δ 9,02 (синглет, 1H), 7,56 (дублет, J =9,0 Гц, 2H), 7,04 (дублет, J =9,1 Гц, 2H), 6,92 (синглет, 1H), 4,47 (синглет, 2H), 3,66 (синглет, 2H), 2,74 (триплет, J =5,7 Гц, 2H), 2,18 (синглет, 2H), 2,09 (синглет, 3H), 2,00 (синглет, 3H), 1,12 (синглет, 3H), 0,51 (q, J =4,2 Гц, 2H), 0,29 (q, J =4,1 Гц, 2H).
Пример 16: Получение соединения P

Этап 1. Соединение P
Соединение 1 (30 мг, 0,110 ммоль, 1,0 экв.) растворяли в толуоле (5 мл), добавляли соединение 2 (45 мг, 0,165 ммоль, 1,5 экв.), калия трет-бутоксид (37 мг, 0,330 ммоль, 3,0 экв.), X-Phos (11 мг, 0,022 ммоль, 0,2 экв.) и Pd2(dba)3 (10 мг, 0,011 ммоль, 0,1 экв.), смесь нагревали до 100°C и перемешивали в течение 16 ч в защитной среде азота. После охлаждения до комнатной температуры реакционный раствор разбавляли этилацетатом (30 мл), затем промывали поочередно водой и насыщенным раствором натрия хлорида, органическую фазу сушили над безводным натрия сульфатом, а остаток, полученный после концентрирования, очищали методом препаративной ВЭЖХ (0,1% муравьиная кислота/ацетонитрил/вода) с получением соединения Р (6,3 мг, выход 13%) в виде твердого вещества белого цвета.
ВЭЖХ-МС: [M+H]+ = 417,1
1H-ЯМР (400 МГц, ДМСО-d6) δ 9,05 (синглет, 1H), 7,51 (дублет, J =8,7 Гц, 2H), 7,12 (дублет, J =8,7 Гц, 2H), 6,96 (синглет, 1H), 4,46 (синглет, 2H), 3,68 (синглет, 2H), 2,77 (синглет, 2H), 2,22 (синглет, 2H), 2,12 (синглет, 3H), 2,03 (синглет, 3H), 1,15 (синглет, 3H), 0,55 (синглет, 2H), 0,33-0,31 (мультиплет, 2H).
Пример 17: Получение соединения Q

Этап 1. Соединение Q
Соединение 1 (40 мг, 0,15 ммоль, 1,0 экв.) и соединение 2 (51,6 мг, 0,22 ммоль, 1,5 экв.) растворяли в толуоле (5 мл), добавляли Pd2(dba)3 (14 мг, 0,015 ммоль, 0,1 экв.), Xant-Phos (14 мг, 0,03 ммоль, 0,2 экв.) и цезия карбонат (143 мг, 0,45 ммоль, 3,0 экв.), реакционный раствор нагревали до 90°C и проводили реакцию раствора в течение 16 ч в защитной среде азота. После охлаждения до комнатной температуры реакционный раствор концентрировали и полученный остаток очищали с помощью препаративной ВЭЖХ (0,1% муравьиная кислота/ацетонитрил/вода) с получением соединения Q (9,7 мг, выход 18%) в виде твердого вещества белого цвета.
ВЭЖХ-МС: [M+H]+ = 379,2
1H-ЯМР (400 МГц, CDCl3) δ 7,18 (синглет, 1H), 6,98 (дублет, J =8,8 Гц, 2H), 6,92-6,83 (мультиплет, 3H), 4,23 (синглет, 2H), 3,78 (синглет, 3H), 3,44 (триплет, J =5,6 Гц, 2H), 2,83 (триплет, J =5,6 Гц, 2H), 2,40 (синглет, 2H), 2,23 (синглет, 3H), 2,13 (синглет, 3H), 1,30 (синглет, 3H), 0,61-0,54 (мультиплет, 4H).
Пример 18: Получение соединения R

Этап 1. Соединение R
Соединение 1 (30 мг, 0,11 ммоль, 1,0 экв.), соединение 2 (33 мг, 0,17 ммоль, 1,5 экв.), натрия трет-бутоксид (32 мг, 0,33 ммоль, 3,0 экв.), Dave-Phos (17 мг, 0,044 ммоль, 0,4 экв.) и Pd2(dba)3 (20 мг, 0,022 ммоль, 0,2 экв.) растворяли в 1,4-диоксане (4 мл) и трет-бутаноле (2 мл), реакционный раствор нагревали до 99°C и проводили реакцию раствора в течение 18 ч в защитной среде азота. После охлаждения до комнатной температуры реакционный раствор экстрагировали этилацетатом и насыщенным раствором натрия хлорида, органическую фазу сушили над безводным натрия сульфатом и остаток, полученный после концентрирования, очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат = 3/1) с получением соединения R (26 мг, выход 60%) в виде желтого твердого вещества.
ВЭЖХ-МС: [M+H]+ = 392,2
1H-ЯМР (400 МГц, MeOD) δ 7,15 (синглет, 1H), 7,01 (дублет, J =7,6 Гц, 2H), 6,93 (дублет, J =8,0 Гц, 2H), 4,50 (синглет, 2H), 2,93-2,90 (мультиплет, 2H), 2,85-2,83 (мультиплет, 2H), 2,81 (синглет, 3H), 2,80 (синглет, 3H), 2,23 (синглет, 2H), 2,11 (синглет, 3H), 2,02 (синглет, 3H), 1,12 (синглет, 3H), 0,53-0,50 (мультиплет, 2H), 0,33-0,31 (мультиплет, 2H).
Пример 19: Получение соединения S
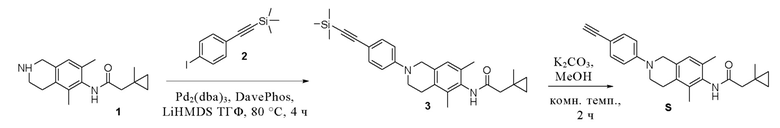
Этап 1. Соединение 3
Соединение 1 (50 мг, 0,18 ммоль, 1,0 экв.) растворяли в тетрагидрофуране (15 мл), добавляли соединение 2 (83 мг, 0,26 ммоль, 1,5 экв.), Pd2(dba)3 (17 мг, 0,018 ммоль, 0,1 экв.), Dave-Phos (14 мг, 0,036 ммоль, 0,2 экв.) и LiHMDS (1 М, 1,8 мл, 1,8 ммоль, 10 экв.), реакционный раствор нагревали до 80°C и смесь перемешивали в течение 4 ч в защитной среде азота. Реакционный раствор охлаждали до комнатной температуры и затем экстрагировали этилацетатом и насыщенным раствором натрия хлорида, после ч его органическую фазу сушили над безводным натрия сульфатом и остаток, полученный после концентрирования, очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат = 5/1) с получением соединения 3 (4 мг, выход 5%) в виде твердого вещества желтого цвета.
ВЭЖХ-МС: [M+H]+ = 445,2
Этап 2. Соединение S
Соединение 3 (4 мг, 0,009 ммоль, 1,0 экв.) и калия карбонат (4 мг, 0,027 ммоль, 3,0 экв.) добавляли к метанолу (5 мл) и смесь перемешивали при комнатной температуре в течение 2 ч. Реакционный раствор фильтровали и полученный раствор концентрировали с получением соединения S (3 мг, выход 90%) в виде твердого вещества желтого цвета.
ВЭЖХ-МС: [M+H]+ = 373,2
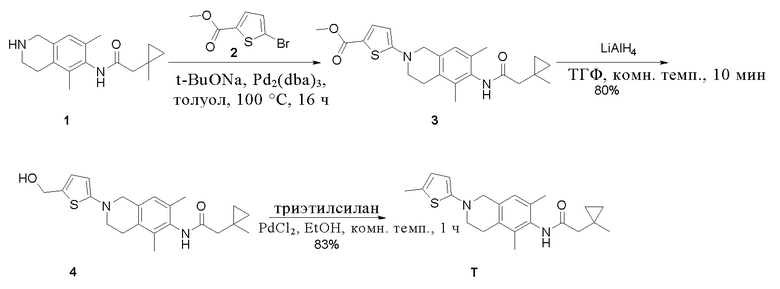
Пример 20: Получение соединения T
Этап 1. Соединение 3
Соединение 1 (80 мг, 0,293 ммоль, 1,0 экв.) растворяли в толуоле (5 мл), добавляли соединение 2 (129 мг, 0,587 ммоль, 2,0 экв.), натрия трет-бутоксид (56 мг, 0,587 ммоль, 2,0 экв.), BINAP (37 мг, 0,0587 ммоль, 0,2 экв.) и Pd2(dba)3 (27 мг, 0,0293 ммоль, 0,1 экв.), смесь нагревали до 100°C и перемешивали в течение 16 ч в защитной среде азота. После охлаждения до комнатной температуры реакционный раствор разбавляли этилацетатом (30 мл), затем поочередно промывали водой и насыщенным раствором натрия хлорида, органическую фазу сушили над безводным натрия сульфатом, и остаток, полученный после концентрирования, очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат = 2/1) с получением соединения 3 (30 мг, выход 25%) в виде твердого вещества коричневого цвета.
ВЭЖХ-МС: [M+H]+ = 413,1
Этап 2. Соединение 4
Соединение 3 (20 мг, 0,0484 ммоль, 1,0 экв.) растворяли в тетрагидрофуране (3 мл), добавляли лития тетрагидроалюминат (9 мг, 0,2424 ммоль, 5,0 экв.), смесь перемешивали при комнатной температуре в течение 10 мин в защитной среде азота, добавляли тетрагидрофуран для разбавления (5 мл), реакционный раствор охлаждали до 0°C, добавляли натрия сульфата декагидрат (0,3 г) и смесь перемешивали при 0°C в течение 15 мин. Реакционный раствор фильтровали и концентрировали с получением соединения 4 (15 мг, выход 80%) в виде твердого вещества коричневого цвета.
ВЭЖХ-МС: [M+H]+ = 385,1
Этап 2. Соединение T
Соединение 4 (10 мг, 0,026 ммоль, 1,0 экв.) растворяли в безводном этаноле (1 мл), добавляли триэтилсилан (3 мг, 0,026 ммоль, 1,0 экв.) и палладия хлорид (2 мг, 0,013 ммоль, 0,5 экв.) и перемешивали при комнатной температуре в течение 1 ч в защитной среде азота. Реакционный раствор фильтровали и концентрировали с получением соединения Т (8 мг, 83%) в виде твердого вещества коричневого цвета.
ВЭЖХ-МС: [M+H]+ = 369,1
Пример 21: Получение соединения U

Этап 1. Соединение U
Соединение 1 (120 мг, 0,4 ммоль, 1,0 экв.) растворяли в толуоле (40 мл), добавляли соединение 2 (143 мг, 0,6 ммоль, 1,5 экв.), цезия карбонат (428,1 мг, 1,31 ммоль, 3,0 экв.), X-Phos (41,7 мг, 0,08 ммоль, 0,2 экв.) и Pd2(dba)3 (40,1 мг, 0,04 ммоль, 0,1 экв.), температуру повышали до 90°C и перемешивали в течение 16 ч в защитной среде азота. После охлаждения до комнатной температуры реакционный раствор фильтровали, фильтрат разбавляли водой (30 мл) и экстрагировали этилацетатом (3×50 мл). Объединенную органическую фазу промывали насыщенным раствором натрия хлорида, сушили над безводным натрия сульфатом и остаток, полученный после концентрирования, очищали колоночной хроматографией на нейтральном алюмосиликагеле (петролейный эфир/этилацетат = 5/1) с получением соединения U (42,3 мг, выход 26%) в виде твердого вещества белого цвета.
ВЭЖХ-МС: [M+H]+ = 365,2
1H-ЯМР (400 МГц, ДМСО) δ 9,09 (синглет, 1H), 7,03 (дублет, J =8,0 Гц, 2H), 6,93-6,90 (мультиплет, 3H), 4,24 (синглет, 2H), 3,50-3,47 (мультиплет, 2H), 2,72-2,66 (мультиплет, 2H), 2,21 (синглет, 2H), 2,19 (синглет, 3H), 2,11 (синглет, 3H), 2,00 (синглет, 3H), 1,06 (синглет, 9H).
Пример 22: Получение соединения V
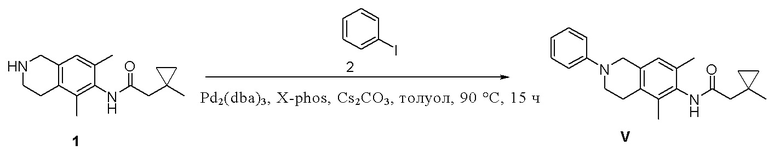
Соединение V получают по способу синтеза соединения N (выход 15%).
ВЭЖХ-МС: [M+H]+ = 349,2
1H-ЯМР (400 МГц, ДМСО-d6) δ 9,02 (синглет, 1H), 7,24-7,20 (мультиплет, 2H), 7,02-7,00 (мультиплет, 2H), 6,93 (синглет, 1H), 6,78-6,73 (мультиплет, 1H), 4,31 (синглет, 2H), 3,55 (синглет, 2H), 2,75-2,72 (мультиплет, 2H), 2,21 (синглет, 2H), 2,12 (синглет, 3H), 2,02 (синглет, 3H), 1,15 (синглет, 3H), 0,56-0,53 (мультиплет, 2H), 0,33-0,31 (мультиплет, 2H).
Пример 23: Получение соединения W
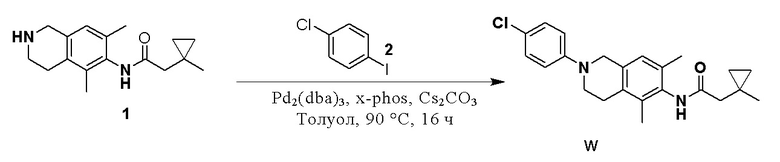
Этап 1. Соединение W
Соединение 1 (50 мг, 0,183 ммоль, 1,0 экв.) растворяли в толуоле (5 мл) в защитной среде азота, последовательно добавляли соединение 2 (44 мг, 0,183 ммоль, 1,0 экв.), Pd2(dba)3 (17 мг, 0,0183 ммоль, 0,1 экв.), X-phos (17 мг, 0,0366 ммоль, 0,2 экв.) и Cs2CO3 (119 мг, 0,366 ммоль, 2,0 экв.), реакционный раствор нагревали до 90°C и перемешивали в течение 16 ч. После охлаждения до 25°С реакционный раствор разбавляли этилацетатом (50 мл), после ч его поочередно промывали водой и насыщенным раствором натрия хлорида. Органическую фазу сушили над безводным натрия сульфатом и концентрировали. Полученный остаток очищали методом препаративной ВЭЖХ (0,1% муравьиная кислота/ацетонитрил/вода) с получением соединения W (3,2 мг, 5%) в виде твердого вещества белого цвета.
ВЭЖХ-МС: [M+H]+ = 383,1
1H-ЯМР (400 МГц, ДМСО-d6) δ 9,03 (синглет, 1H), 7,25-7,21 (мультиплет, 2H), 7,04-6,99 (мультиплет, 2H), 6,92 (синглет, 1H), 4,32 (синглет, 2H), 3,55 (синглет, 2H), 2,73-2,71 (мультиплет, 2H), 2,21 (синглет, 2H), 2,11 (синглет, 3H), 2,01 (синглет, 3H), 1,15 (синглет, 3H), 0,56-0,53 (мультиплет, 2H), 0,33-0,31 (мультиплет, 2H).
Пример 24: Получение соединения X

Этап 1. Соединение X
Соединение 1 (300 мг, 1,101 ммоль, 1,0 экв.) растворяли в толуоле (15 мл), последовательно добавляли соединение 2 (727 мг, 2,202 ммоль, 2,0 экв.), цезия карбонат (1,07 г, 3,304 ммоль, 3,0 экв.), X-PHOS (105 мг, 0,2202 ммоль, 0,2 экв.) и Pd2(dba)3 (101 мг, 0,1101 ммоль, 0,1 экв.), реакционный раствор нагревали до 90°C и перемешивали в течение 6 ч в защитной среде азота. После охлаждения до 25°С реакционный раствор разбавляли этилацетатом (30 мл), промывали поочередно водой и насыщенным раствором натрия хлорида, органическую фазу сушили над безводным натрия сульфатом, концентрировали и остаток очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат = 5/1) с получением соединения X (6,3 мг, 1%) в виде твердого вещества.
ВЭЖХ-МС: [M+H]+ = 475,0
1H-ЯМР (400 МГц, ДМСО-d6) δ 9,02 (синглет, 1H), 7,49-7,46 (мультиплет, 2H), 6,92 (синглет, 1H), 6,86-6,84 (мультиплет, 2H), 4,31 (синглет, 2H), 3,55 (синглет, 2H), 2,72 (триплет, J = 5,8 Гц, 2H), 2,21 (синглет, 2H), 2,11 (синглет, 3H), 2,01 (синглет, 3H), 1,14 (синглет, 3H), 0,55-0,53 (мультиплет, 2H), 0,33-0,30 (мультиплет, 2H).
Пример 25: Получение соединения Y

Этап 1. Соединение Y
Соединение 1 (50 мг, 0,183 ммоль, 1,0 экв.) растворяли в толуоле (5 мл) и добавляли соединение 2 (87 мг, 0,367 ммоль, 2,0 экв.), цезия карбонат (179 мг, 0,551 ммоль, 3,0 экв.), Pd2(dba)3 (17 мг, 0,018 ммоль, 0,1 экв.) и X-PHOS (17 мг, 0,036 ммоль, 0,2 экв.). Реакционный раствор нагревали до 90°С и перемешивали в течение 16 ч, охлаждали до 25°С, после ч его добавляли воду (20 мл) для разбавления и затем экстрагировали этилацетатом (3×20 мл). Объединенную органическую фазу промывали насыщенным раствором натрия хлорида и сушили над безводным натрия сульфатом. Остаток, полученный после концентрирования, очищали методом препаративной ВЭЖХ (0,1% муравьиная кислота/ацетонитрил/вода) с получением соединения Y (4,1 мг, 5%) в виде твердого вещества белого цвета.
ВЭЖХ-МС: [M+H]+ = 427,1
1H-ЯМР (400 МГц, CDCl3) δ 7,35-7,33 (мультиплет, 2H), 7,18 (синглет, 1H), 6,91 (синглет, 1H), 6,84 (дублет, J = 8,9 Гц, 2H), 4,31 (синглет, 2H), 3,53 (триплет, J = 5,6 Гц, 2H), 2,83 (триплет, J = 5,6 Гц, 2H), 2,39 (синглет, 2H), 2,24 (синглет, 3H), 2,14 (синглет, 3H), 1,29 (синглет, 3H), 0,61-0,59 (мультиплет, 2H), 0,56 - 0,54 (мультиплет, 2H).
Пример 26: Получение соединения Z
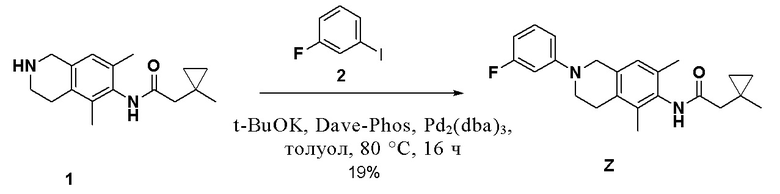
Этап 1. Соединение Z
Соединение 1 (20 мг, 0,074 ммоль, 1,0 экв.) растворяли в толуоле (5 мл), последовательно добавляли соединение 2 (24,5 мг, 0,11 ммоль, 1,5 экв.), калия трет-бутоксид (25 мг, 0,220 ммоль, 3,0 экв.), Dave-Phos (6 мг, 0,015 ммоль, 0,2 экв.) и Pd2(dba)3 (7 мг, 0,0074 ммоль, 0,1 экв.), температуру повышали до 80°C и смесь перемешивали в течение 16 ч в защитной среде азота. После охлаждения до 25°С реакционный раствор разбавляли этилацетатом (30 мл), затем поочередно промывали водой и насыщенным раствором натрия хлорида, органическую фазу сушили над безводным натрия сульфатом и остаток, полученный после концентрирования, очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат = 2/1) с получением соединения Z (5,0 мг, 19%) в виде твердого вещества белого цвета.
ВЭЖХ-МС: [M+H]+ = 367,2
1H-ЯМР (400 МГц, CDCl3) δ 7,23-7,17 (мультиплет, 2H), 6,93 (синглет, 1H), 6,72 (дублет, J = 8,4 Гц, 1H), 6,63 (дублет, J = 12,8 Гц, 1H), 6,49 (триплет, J = 8,0 Гц, 1H), 4,35 (синглет, 2H), 3,56 (триплет, J = 5,6 Гц, 2H), 2,84 (триплет, J = 5,6 Гц, 2H), 2,40 (синглет, 2H), 2,24 (синглет, 3H), 2,15 (синглет, 3H), 1,30 (синглет, 3H), 0,62-0,54 (мультиплет, 4H).
Пример 27: Получение соединения A12

Этап 1. Соединение A1
Соединение 1 (200 мг, 0,734 ммоль, 1,0 экв.) растворяли в толуоле (50 мл) и последовательно добавляли соединение 2 (1,63 г, 7,34 ммоль, 10,0 экв.), цезия карбонат (1,2 г, 3,67 ммоль, 5,0 экв.) X-Phos (140 мг, 0,294 ммоль, 0,4 экв.) и Pd2(dba)3 (134 мг, 0,147 ммоль, 0,2 экв.). Реакционный раствор нагревали до 130°С и перемешивали в течение 2 ч в защитной среде азота. Реакционный раствор охлаждали до 25°С, разбавляли этилацетатом (20 мл), затем промывали водой и насыщенным раствором натрия хлорида. Органическую фазу сушили над безводным натрия сульфатом и концентрировали. Остаток очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат = 3/1) с получением соединения A1 (24,3 мг, 9%) в виде твердого вещества белого цвета.
ВЭЖХ-МС: [M+H]+ = 367,2
1H-ЯМР (400 МГц, ДМСО-d6) δ 9,04 (синглет, 1H), 7,19-7,06 (мультиплет, 3H), 7,01-6,94 (мультиплет, 1H), 6,88 (синглет, 1H), 4,17 (синглет, 2H), 3,44-3,35 (мультиплет, 2H), 2,79-2,70 (мультиплет, 2H), 2,22 (синглет, 2H), 2,12 (синглет, 3H), 2,02 (синглет, 3H), 1,15 (синглет, 3H), 0,56-0,54 (мультиплет, 2H), 0,34-0,31 (мультиплет, 2H).
Пример 28: Получение соединения A2
Этап 1. Соединение 2
Соединение 1 (10,0 г, 82,5 ммоль, 1,0 экв.) растворяли в дихлорметане (100 мл), добавляли трифторуксусный ангидрид (26 г, 123,75 ммоль, 1,5 экв.), реакционный раствор нагревали до 50°С и перемешивали в течение 1,5 ч. После охлаждения до 25°C реакционный раствор концентрировали, остаток растворяли в этилацетате (300 мл), после ч его значение pH доводили до 8-9 насыщенным водным раствором натрия бикарбоната. Отделенную органическую фазу промывали насыщенным раствором натрия хлорида и сушили над безводным натрия сульфатом, после ч его концентрировали с получением соединения 2 (16 г, 89%) в виде твердого вещества белого цвета.
ВЭЖХ-МС: [M+H]+ = 218,1
Этап 2. Соединение 4
Алюминия трихлорид (12,9 г, 96,69 ммоль, 3,0 экв.) растворяли в дихлорметане (100 мл) и охлаждали до 0°C, добавляли смешанный раствор соединения 2 (7,0 г, 32,23 ммоль, 1,0 экв.) и соединения 3 (6,14 г, 48,34 ммоль, 1,5 экв.) в дихлорметане (20 мл), реакционный раствор нагревали до 25°C и перемешивали в течение 16 ч в защитной среде азота. Для разбавления реакционного раствора добавляли дихлорметан (1 л), после ч его реакционный раствор выливали в ледяную воду (1,0 л). Органическую фазу отделяли, и водную фазу экстрагировали этилацетатом (5×300 мл). Объединенную органическую фазу сушили над безводным натрия сульфатом и концентрировали. Остаток очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат = 10/1) с получением соединения 4 (3,2 г, 32%) в виде твердого вещества белого цвета.
ВЭЖХ-МС: [M+H]+ = 308,1
Этап 3. Соединение 5
Соединение 4 (3,0 г, 9,75 ммоль) растворяли в трифторуксусной кислоте (25 мл) и медленно по каплям добавляли триэтилсилан (25 мл). После этого реакционный раствор нагревали до 40°С и перемешивали в течение ночи. Реакционный раствор концентрировали, остаток растворяли в этилацетате (300 мл), затем последовательно промывали насыщенным водным раствором натрия бикарбоната и насыщенным раствором натрия хлорида, органическую фазу сушили над безводным натрия сульфатом, а остаток, полученный после концентрирования, очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат = 10/1) с получением соединения 5 (0,8 г, 28%).
ВЭЖХ-МС:[M+H]+ =294,1
Этап 4. Соединение 7
Соединение 5 (800 мг, 2,72 ммоль, 1,0 экв.) растворяли в N,N-диметилформамиде (10 мл), добавляли йодид калия (452 мг, 2,72 ммоль, 1,0 экв.) и соединение 6 (600 мг, 5,45 ммоль, 2,0 экв.), смесь перемешивали при 25°C в течение 0,5 ч, затем добавляли натрия карбонат (1,15 г, 10,89 ммоль, 4,0 экв.) и продолжали перемешивание в течение 16 ч. Реакционный раствор разбавляли этилацетатом (300 мл), затем поочередно промывали водой и насыщенным раствором натрия хлорида. Органическую фазу сушили над безводным натрия сульфатом и концентрировали. Остаток очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат = 10/1) с получением соединения 7 (900 мг, 90%).
ВЭЖХ-МС: [M+H]+ = 369,1
Этап 5. Соединение 8
Соединение 7 (800 мг, 1,09 ммоль, 1,0 экв.) растворяли в концентрированной хлористоводородной кислоте (12 М, 25 мл) и затем смесь перемешивали при 99°С (температура масляной бани) в течение ночи. После охлаждения реакционного раствора до комнатной температуры реакционный раствор использовали непосредственно для следующей реакции.
ВЭЖХ-МС: [M+H]+ = 273,2
Этап 6. Соединение 9
Соединение 8 из предыдущего этапа добавляли к водному раствору формальдегида (37%, 88 мг, 2,66 ммоль, 2,4 экв.) при температуре 25°C, нагревали до 60°C и смесь перемешивали в течение 1 ч. Вышеуказанный раствор концентрировали непосредственно при 55°C, затем добавляли смешанный растворитель толуол/этанол (1:1, 200 мл) и концентрировали, операцию повторяли дважды с получением неочищенного соединения 9 (400 мг) в виде твердого вещества желтого цвета.
ВЭЖХ-МС: [M+H]+ = 285,2
Этап 7. Соединение A2
Соединение 9 (неочищенное, 200 мг, 0,7 ммоль, 1 экв.) и соединение 10 (241 мг, 2,11 ммоль, 3,0 экв.) растворяли в N,N-диметилформамиде (10 мл), добавляли пиридин (1,11 г, 14,07 ммоль, 20,0 экв.) и 1-пропилфосфоновой кислоты ангидрид в растворе этилацетата (50%, 4,48 г, 7,03 ммоль, 10,0 экв.) в защитной среде азота. Реакционный раствор нагревали до 50°С в защитной среде азота и перемешивали в течение 3 ч. Реакционный раствор разбавляли этилацетатом (300 мл) и затем промывали насыщенным раствором натрия хлорида (3×200 мл). Органическую фазу сушили над безводным натрия сульфатом и концентрировали и полученный остаток очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат = от 4/1 до 2/1) с получением соединения A2 (23,2 мг, выход от трех этапов: 4,5%) в виде твердого вещества белого цвета.
ВЭЖХ-МС: [M+H]+ = 381,2
1H-ЯМР (400 МГц, ДМСО-d6) δ 8,93 (синглет, 1H), 7,09 (синглет, 1H), 6,93-6,85 (мультиплет, 2H), 6,78-6,74 (мультиплет, 2H), 4,53 (синглет, 2H), 3,75-3,56 (мультиплет, 2H), 2,92-2,90 (мультиплет, 2H), 2,18 (синглет, 2H), 2,07 (синглет, 3H), 2,05 (синглет, 3H), 1,82-1,66 (мультиплет, 2H), 1,13 (синглет, 3H), 0,59 - 0,47 (мультиплет, 2H), 0,34-0,27 (мультиплет, 2H).
Пример 29: Получение соединения A3
Способ синтеза такой же, как для соединения М, соединение A3 было получено в виде твердого вещества белого цвета.
ВЭЖХ-МС: [M+H]+ = 419,3
1H-ЯМР (400 МГц, CD3OD) δ 7,47 (дублет, J = 8,6 Гц, 2H), 7,07 (дублет, J = 8,7 Гц, 2H), 6,99 (синглет, 1H), 3,70-3,67 (мультиплет, 2H), 2,87-2,84 (мультиплет, 2H), 2,32 (синглет, 2H), 2,21 (синглет, 3H), 2,13 (синглет, 3H), 1,23 (синглет, 3H), 0,62-0,60 (мультиплет, 2H), 0,43-0,40 (мультиплет, 2H).
Пример 30: Получение соединения A4
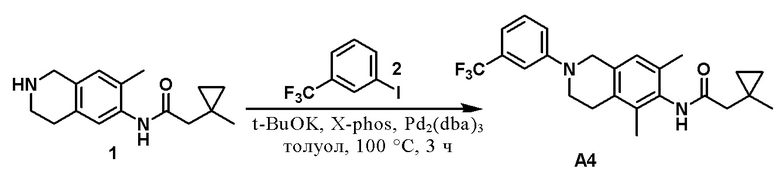
Этап 1. Соединение A4
Соединение 1 (150 мг, 0,55 ммоль, 1,0 экв.) растворяли в толуоле (3 мл), последовательно добавляли соединение 2 (224 мг, 0,826 ммоль, 1,5 экв.), калия трет-бутоксид (185 мг, 1,65 ммоль, 3,0 экв.), X-phos (52 мг, 0,11 ммоль, 0,2 экв.) и Pd2(dba)3 (50 мг, 0,05 ммоль, 0,1 экв.), реакционный раствор нагревали до 100°C в защитной среде азота и перемешивали в течение 3 ч. Реакционный раствор охлаждали до 25°C, разбавляли этилацетатом (50 мл) и промывали поочередно водой и насыщенным раствором натрия хлорида. Органическую фазу сушили над безводным натрия сульфатом и концентрировали, и остаток очищали с помощью препаративной ВЭЖХ (0,1% муравьиная кислота/ацетонитрил/вода) с получением соединения A4 (36,5 мг, 16%) в виде твердого вещества белого цвета.
ВЭЖХ-МС: [M+H]+ = 417,2
1H-ЯМР (400 МГц, ДМСО-d6) δ 9,03 (синглет, 1H), 7,42 (триплет, J =8,0 Гц, 1H), 7,30-7,27 (мультиплет, 1H), 7,23 (синглет, 1H), 7,02 (дублет, J =7,6 Гц, 1H), 6,97 (синглет, 1H), 4,41 (синглет, 2H), 3,63 (синглет, 2H), 2,77 (триплет, J =5,7 Гц, 2H), 2,21 (синглет, 2H), 2,12 (синглет, 3H), 2,03 (синглет, 3H), 1,15 (синглет, 3H), 0,56-0,53 (мультиплет, 2H), 0,33 - 0,31 (мультиплет, 2H).
Пример 31: Получение соединения A5
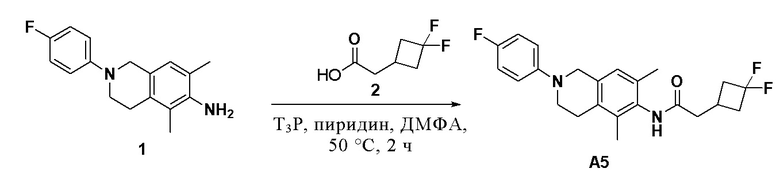
Этап 1. Соединение A5
Соединение 1 (100 мг, 0,368 ммоль, 1,0 экв.) растворяли в ДМФА (6 мл), последовательно добавляли соединение 2 (66 мг, 0,44 ммоль, 1,2 экв.), 1-пропилфосфоновой кислоты ангидрид (50%, 1,17 г, 1,84 ммоль, 5,0 экв.) и пиридин (582 мг, 7,36 ммоль, 20,0 экв.), реакционный раствор нагревали до 50°C в атмосфере азота и перемешивали в течение 2 ч. Реакционный раствор охлаждали до 25°C, после ч его разбавляли этилацетатом (50 мл). Полученный раствор поочередно промывали водой и насыщенным раствором натрия хлорида. Органическую фазу сушили над безводным натрия сульфатом и затем концентрировали, полученный остаток очищали методом препаративной ВЭЖХ (0,1% муравьиная кислота/ацетонитрил/вода) с получением соединения A5 (84,2 мг, 58%) в виде твердого вещества белого цвета.
ВЭЖХ-МС: [M+H]+ = 403,1
1H-ЯМР (400 МГц, ДМСО-d6) δ 9,25 (синглет, 1H), 7,08-7,00 (мультиплет, 4H), 6,91 (синглет, 1H), 4,26 (синглет, 2H), 3,49 (синглет, 2H), 2,77-2,72 (мультиплет, 4H), 2,68-2,67 (мультиплет, 1H), 2,46 - 2,44 (мультиплет, 1H), 2,40-2,36 (мультиплет, 2H), 2,33 -2,30 (мультиплет, 1H), 2,08 (синглет, 3H), 1,98 (синглет, 3H).
Пример 32: Получение соединения A6
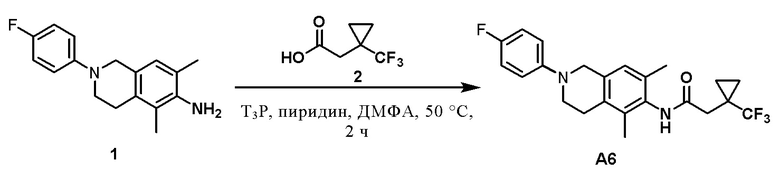
Этап 1. Соединение A6
Соединение 1 (50 мг, 0,185 ммоль, 1,0 экв.) растворяли в ДМФА (3 мл), последовательно добавляли соединение 2 (37 мг, 0,222 ммоль, 1,2 экв.), 1-пропилфосфоновой кислоты ангидрид (50%, 2,4 г, 3,700 ммоль, 20,0 экв.) и пиридин (146 мг, 1,850 ммоль, 10,0 экв.), реакционный раствор нагревали до 50°C и перемешивали в течение 2 ч. После охлаждения до 25°C реакционный раствор разбавляли водой (10 мл) и затем экстрагировали этилацетатом (20×3). Объединенную органическую фазу промывали насыщенным раствором натрия хлорида, сушили над безводным натрия сульфатом и остаток, полученный после концентрирования, очищали с помощью препаративной ВЭЖХ (0,1% муравьиная кислота/ацетонитрил/вода) с получением соединения A6 (8,9 мг, 11%) в виде твердого вещества белого цвета.
ВЭЖХ-МС: [M+H]+ = 421,2
1H-ЯМР (400 МГц, ДМСО-d6) δ 9,31 (синглет, 1H), 7,08-7,00 (мультиплет, 4H), 6,91 (синглет, 1H), 4,26 (синглет, 2H), 3,49 (синглет, 2H), 2,72 (триплет, J = 5,7 Гц, 2H), 2,67 (синглет, 2H), 2,08 (синглет, 3H), 1,99 (синглет, 3H), 1,01 (синглет, 2H), 0,98 (синглет, 2H).
Пример 33: Получение соединения A7
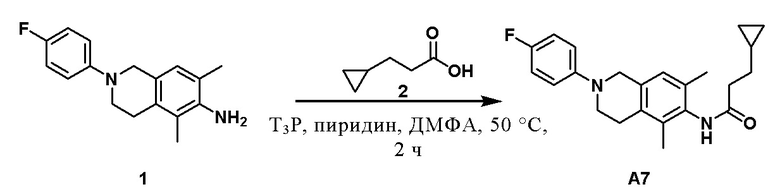
Этап 1. Соединение A7
Соединение 1 (50 мг, 0,18 ммоль, 1,0 экв.) растворяли в ДМФА (2 мл), последовательно добавляли соединение 2 (21 мг, 0,18 ммоль, 1,0 экв.), 1-пропилфосфоновой кислоты ангидрид (50%, 1,15 г, 1,8 ммоль, 10,0 экв.) и пиридин (284 мг, 1,8 ммоль, 10,0 экв.), реакционный раствор нагревали до 50°C и перемешивали в течение 2 ч. После охлаждения до 25°С реакционный раствор разбавляли водой (10 мл) и затем экстрагировали этилацетатом (3×20 мл). Объединенную органическую фазу промывали насыщенным раствором натрия хлорида, сушили над безводным натрия сульфатом и остаток, полученный после концентрирования, очищали с помощью препаративной ВЭЖХ (0,1% муравьиная кислота/ацетонитрил/вода) с получением соединения A7 (4,9 мг, 7%) в виде твердого вещества белого цвета.
ВЭЖХ-МС: [M+H]+ = 367,1
1H-ЯМР (400 МГц, ДМСО-d6) δ 9,18 (синглет, 1H), 7,09-6,98 (мультиплет, 4H), 6,90 (синглет, 1H), 4,25 (синглет, 2H), 3,49 (синглет, 2H), 2,72 (триплет, J = 5, 6 Гц, 2H), 2,39 (триплет, J = 7,6 Гц, 2H), 2,09 (синглет, 3H), 1,99 (синглет, 3H), 1,54-1,48 (мультиплет, 2H), 0,78-0,71 (мультиплет, 1H), 0,45-0,37 (мультиплет, 2H), 0,11-0,04 (мультиплет, 2H).
Пример 34: Анализ активности активатора калиевых каналов (обнаружение с помощью FDSS/μCELL)
1. Методика эксперимента:
1.1 Процедура проведения эксперимента
Подготовка клеток: клетки CHO-KCNQ2 культивировали в культуральном флаконе 175 см2, и когда плотность клеток возрастала до 60~80%, питательную среду удаляли, промывали 7 мл ФСБР (фосфатно-солевого буферного раствора) один раз, затем для гидролиза добавляли 3 мл 0,25% раствора трипсина. После завершения гидролиза добавляли 7 мл питательной среды (90% DMEM/F12 + 10% FBS + 500 мкг/мл G418) для нейтрализации и центрифугировали в течение 3 мин при 800 об/мин. Супернатант аспирировали, затем для ресуспендирования добавляли 5 мл питательной среды, после ч его проводили подсчет клеток.
Посев клеток на планшеты: с учетом результатов подсчета клеток плотность доводили до 3×104 клеток на лунку. После выдерживания при комнатной температуре в течение 30 мин планшет помещали в инкубатор с CO2 средой при 37°С и инкубировали до следующего дня в течение 16-18 ч. Плотность клеток достигала около 80%.
Инкубация с флуоресцентным красителем: среду для культивирования клеток удаляли, добавляли буфер для внесения 80 мкл/лунку и инкубировали в темноте при комнатной температуре в течение 60 мин.
Инкубация с соединением: буфер для внесения удаляли, добавляли приготовленный раствор соединения 80 мкл/лунку и инкубировали в темноте при комнатной температуре в течение 20 мин.
Сбор данных флуоресценции: регистрацию сигналов флуоресценции в реальном времени проводили с помощью прибора FDSS/μCELL при длине волны возбуждения 480 нм и длине волны испускания 540 нм с частотой регистрации 1 раз в секунду, и буфер для стимуляции 20 мкл/лунку начинали добавлять после регистрации в течение 10 с для исходного уровня и затем продолжали для регистрации до окончания 180 с.
1.2 Приготовление раствора
Буфер для внесения: 10 мл/планшет, способ приготовления был следующим:
Образец буфера для анализа: 100 мл/планшет, способ приготовления был следующим:
Буфер для стимуляции: 5 мл/планшет, способ приготовления был следующим:
Вышеупомянутый буфер был взят из коммерчески доступного набора FluxOR Potassium Ion Channel Assay.
1.3 Приготовление соединения
Готовили исходный раствор соединения в ДМСО 20 мМ, 10 мкл исходного раствора соединения 20 мМ помещали в 20 мкл раствора ДМСО, последовательно разбавляли в 3 повторностях до 8 промежуточных концентраций; затем брали промежуточные концентрации соединения и добавляли в буфер для анализа, разбавляли в 200 раз для получения окончательной концентрации для анализа, отбирали 80 мкл и добавляли в планшет для анализа.
Самая высокая концентрация для анализа составляла 100 мкМ, концентрации были следующие: 100, 33,33, 11,11, 3,70, 1,23, 0,41, 0,137, 0,045 мкМ, всего 8 концентраций. Для каждой концентрации устанавливали 3 дублирующие лунки.
Содержание ДМСО в окончательной концентрации для анализа не превышало 0,5%. Такая концентрация ДМСО не влияет на калиевый канал KCNQ2.
Анализ данных
Экспериментальные данные были проанализированы с помощью программного обеспечения Excel 2007 и GraphPad Prism 5.0, и было рассчитано соотношение 180 секунд для расчета эффекта агонизма. Эффект агонизма соединения рассчитывали по следующей формуле:

1.5 Контроль качества
Окружающая среда: температура ~ 25°C
Реактив: набор FluxOR™ Detection Kit (Invitrogen, номер по каталогу F0017)
Экспериментальные данные в отчете должны соответствовать следующим критериям: Z’ Factor > 0,5
1.6. Результаты: см. табл. 1, где меньшее значение EC50 соответствует большей активности соответствующего соединения.
Ссылки на вышеуказанные методики анализа:
Zhaobing Gao et al. Journal of Biological Chemistry. 2010, 285(36): 28322-28332.
Пример 35: Исследование фармакокинетики на мышах
1) Цели исследования: получение фармакокинетических характеристик анализируемых соединений и прохождение ч ерез гематоэнцефалический барьер на самцах мышей линии ICR
2) Содержание эксперимента
Брали шесть здоровых самцов мышей линии ICR (с массой тела в диапазоне 18-22 г), которых разделяли на 2 группы по 3 мыши в группе. После ночного голодания (только для группы с введением внутрь) исследуемое соединение вводили внутривенно в дозе 0,5 мг/кг и давали внутрь в дозе 10 мг/кг, кровь собирали ч ерез пункцию яремной вены во временных точках 0,083 (только для группы с внутривенным введением), 0,25, 0,5, 1, 2, 4, 6 (только для группы с введением внутрь), 8 и 24 ч, и в пробирку с антикоагулянтом ЭДТА-К2 собирали не менее 0,3 мл цельной крови. Плазму центрифугировали (6000 об/мин, 8 мин, 4°С) в течение получаса и замораживали при -20°С для последующего использования.
Исследование гематоэнцефалического барьера: брали 6 здоровых самцов мышей линии ICR (с массой тела в диапазоне 18-22 г), которых разделяли на 2 группы по 3 мыши в группе. После ночного голодания исследуемое соединение (10 мг/кг) давали внутрь соответственно. Кровь собирали ч ерез пункцию сердца во временных точках 2 и 4 ч, в пробирку с антикоагулянтом ЭДТА-К2 собирали не менее 0,5 мл цельной крови. Плазму центрифугировали (6000 об/мин, 8 мин, 4°С) в течение получаса и замораживали при -20°С для последующего использования. В то же время собирали мозговую ткань, которую промывали физиологическим раствором, после ч его удаляли влагу с помощью впитывающей бумаги, взвешивали и замораживали при -20°C для последующего использования.
Результаты эксперимента: согласно полученным данным о концентрации препарата в крови для расчета фармакокинетических показателей после введения использовали некомпартментную модель в программном обеспечении WinNonlin® 7.0 (Pharsight, США).
* Внутривенная (в/в) доза составляла 1 мг/кг.
Соотношение концентрации в крови и мозговой ткани является очень важным показателем для антиконвульсантов, и из таблицы 3 видно, ч то соединения по настоящему изобретению имеют отличное соотношение концентрации в крови и мозговой ткани, за счет ч его обеспечивается более высокая эффективность и высокая безопасность лекарственного препарата. При сравнении соотношения концентрации в крови и мозговой ткани соединения А, соединения I, соединения В и соединения J, можно заметить, ч то при замене трет-бутильной группы соединения А или соединения В на , соотношение концентрации в крови и мозговой ткани у мышей становится значительно выше (более ч ем в 2 раза).
Пример 36: Исследование фармакокинетики на крысах
1) Цель исследования: получение фармакокинетических характеристик анализируемых соединений на самцах крыс линии SD.
2) Содержание эксперимента
Фармакокинетическое исследование: 6 здоровых самцов крыс линии SD (со статусом SPF) были разделены на 2 группы по 3 крысы в группе. После ночного голодания (только для группы с введением внутрь) соединение А вводили внутривенно в дозе 0,3 мг/кг и давали внутрь в дозе 5 мг/кг, кровь собирали ч ерез пункцию яремной вены во временных точках 0,083 (только для группы с внутривенным введением), 0,25, 0,5, 1, 2, 4, 6 (только для группы с введением внутрь), 8 и 24 ч, и в пробирку с антикоагулянтом ЭДТА-К2 собирали не менее 0,3 мл цельной крови. Плазму центрифугировали (6000 об/мин, 8 мин, 4°С) в течение получаса и замораживали при -20°С для последующего использования.
Результаты эксперимента: согласно полученным данным о концентрации препарата в крови для расчета фармакокинетических параметров после введения использовали некомпартментную модель в программном обеспечении WinNonlin® 7.0 (Pharsight, США).
Все литературные источники, упомянутые в настоящем изобретении, включены в настоящий документ в виде ссылок, как если бы они были включены в виде отдельных ссылок. Кроме того, следует понимать, ч то после изучения вышеприведенного описания специалистами в данной области техники могут быть внесены многочисленные изменения и модификации, и такие эквиваленты также попадают в объем, определяемый прилагаемой формулой изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| СОЕДИНЕНИЕ, ОБЛАДАЮЩЕЕ НЕЙРОПРОТЕКТОРНЫМ ДЕЙСТВИЕМ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2020 |
|
RU2802457C1 |
| СОЕДИНЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ 2-АМИНОПИРИМИДИНА И ИХ ПРИМЕНЕНИЕ | 2020 |
|
RU2795914C1 |
| ПОЛИЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ, ДЕЙСТВУЮЩЕЕ КАК ИНГИБИТОР КИНАЗЫ | 2021 |
|
RU2826488C1 |
| ПРОИЗВОДНЫЕ ПИРРОЛИДИНА В КАЧЕСТВЕ АГОНИСТОВ PPAR | 2017 |
|
RU2711991C1 |
| 2,3-ДИГИДРО-1H-ПИРРОЛИЗИН-7-ФОРМАМИДНОЕ ПРОИЗВОДНОЕ И ЕГО ПРИМЕНЕНИЕ | 2019 |
|
RU2792726C2 |
| ПРОИЗВОДНЫЕ ГАЛОГЕНАЛЛИЛАМИНА И ИХ ПРИМЕНЕНИЕ | 2020 |
|
RU2793850C2 |
| СОЕДИНЕНИЯ, КОМПОЗИЦИИ И СПОСОБЫ ДЛЯ ПОВЫШЕНИЯ АКТИВНОСТИ CFTR | 2015 |
|
RU2767460C2 |
| ПРОИЗВОДНЫЕ ТРИАЗОЛА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ИНСЕКТИЦИДНОЕ И АКАРИЦИДНОЕ СРЕДСТВО | 1994 |
|
RU2131421C1 |
| СОЕДИНЕНИЯ И СПОСОБЫ ЛЕЧЕНИЯ ПАРАЗИТАРНЫХ ЗАБОЛЕВАНИЙ | 2018 |
|
RU2793122C2 |
| НОВЫЙ АГОНИСТ β-РЕЦЕПТОРА ТИРЕОИДНЫХ ГОРМОНОВ | 2021 |
|
RU2839610C1 |
Изобретение относится к новым соединениям тетрагидроизохинолина, представленным формулой (А), их получению и применению в качестве модулятора калиевых каналов. Технический результат: получены новые соединения, которые могут быть пригодны для профилактики и/или лечения заболеваний, реагирующих на активность калиевых каналов. 4 н. и 6 з.п. ф-лы, 7 табл., 192 пр.
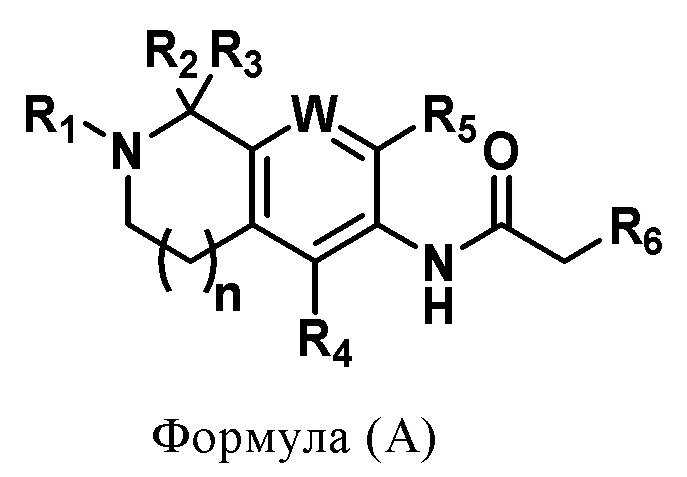
1. Соединение, представленное формулой A, или его фармацевтически приемлемая соль
 ,
,
где
R1 представляет собой замещенную или незамещенную фенильную группу, причем замещение означает замещение одним или несколькими заместителями, выбранными из группы, состоящей из галогена, цианогруппы, C1-6 алкильной группы, C1-6 алкоксигруппы, галогенированной C1-6 алкильной группы, -NR8R9 и этинильной группы;
R2 и R3 независимо выбирают из группы, состоящей из водорода, дейтерия;
R4 и R5 независимо выбирают из группы, состоящей из C1-6 алкильной группы;
R6 выбирают из замещенной или незамещенной группы, состоящей из C3-6 циклоалкильной группы, причем замещение означает замещение одним или несколькими заместителями, выбранными из группы, состоящей из галогена, C1-6 алкильной группы, C1-6 алкоксигруппы, и галогенированной C1-6 алкильной группы;
n выбирают из группы, состоящей из 1 и 2;
W представляет собой C-R7;
R7 представляет собой водород;
R8 и R9 независимо выбирают из группы, состоящей из C1-6 алкильной группы.
2. Соединение по п. 1 или его фармацевтически приемлемая соль, где:
R1 представляет собой замещенную или незамещенную фенильную группу, причем замещение означает замещение одним или несколькими заместителями, выбранными из группы, состоящей из галогена, цианогруппы, C1-6 алкильной группы, C1-6 алкоксигруппы, галогенированной C1-6 алкильной группы, -NR8R9 и этинильной группы;
R2 и R3 независимо выбирают из группы, состоящей из водорода и дейтерия;
R4 и R5 представляют собой метил;
R6 выбирают из замещенной или незамещенной группы, состоящей из C3-6 циклоалкильной группы, причем замещение означает замещение одним или несколькими заместителями, выбранными из группы, состоящей из галогена и C1-6 алкильной группы;
n является 1;
W представляет собой C-R7;
R7 представляет собой водород;
R8 и R9 независимо друг от друга являются C1-6 алкильной группой.
3. Соединение по п. 2 или его фармацевтически приемлемая соль, где:
R1 представляет собой замещенную или незамещенную фенильную группу, причем замещение означает замещение одним или несколькими заместителями, выбранными из группы, состоящей из галогена, цианогруппы, C1-6 алкильной группы, C1-6 алкоксигруппы, галогенированной C1-6 алкильной группы, -NR8R9 и этинильной группы;
R2 и R3 независимо выбирают из группы, состоящей из водорода и дейтерия;
R4 и R5 представляют собой метил;
R6 выбирают из группы, состоящей из  и
и  ;
;
n является 1;
W представляет собой C-R7;
R7 представляет собой водород;
R8 и R9 независимо друг от друга являются C1-6 алкильной группой.
4. Соединение по п. 3 или его фармацевтически приемлемая соль, где:
R1 представляет собой замещенную или незамещенную фенильную группу, причем замещение означает замещение одним или несколькими заместителями, выбранными из группы, состоящей из галогена, цианогруппы, C1-6 алкильной группы, C1-6 алкоксигруппы, галогенированной C1-6 алкильной группы, -NR8R9 и этинильной группы;
R2 и R3 независимо выбирают из группы, состоящей из водорода и дейтерия;
R4 и R5 представляют собой метил;
R6 представляет собой из ;
n является 1;
W представляет собой C-R7;
R7 представляет собой водород;
R8 и R9 независимо друг от друга являются C1-6 алкильной группой.
5. Соединение по п. 1 или его фармацевтически приемлемая соль, где соединение выбирают из группы, состоящей из:
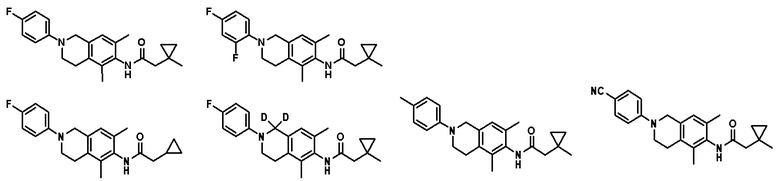
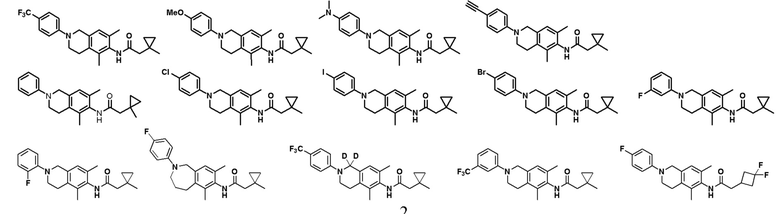
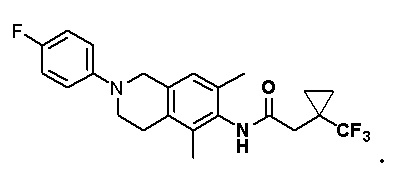
6. Способ получения соединения по п. 1 или его фармацевтически приемлемой соли, включающий следующие этапы:
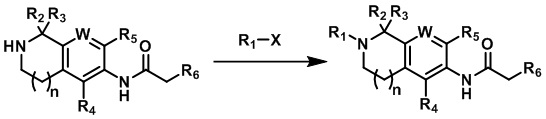 ,
,
где:
X выбирают из группы, состоящей из галогена, -B(OH)2 и -OTf;
R1, R2, R3, R4, R5, R6, n и W определены, как указано в п. 1.
7. Фармацевтическая композиция для профилактики и/или лечения заболеваний, реагирующих на активность калиевых каналов, содержащая один или несколько фармацевтически приемлемых носителей и терапевтически эффективное количество соединения по п. 1 или его фармацевтически приемлемой соли.
8. Применение соединения по п. 1 или его фармацевтически приемлемой соли для получения лекарственного препарата для профилактики и/или лечения заболеваний, реагирующих на активность калиевых каналов.
9. Применение по п. 8, где заболевание, реагирующее на активность калиевых каналов, представляет собой заболевание центральной нервной системы.
10. Применение по п. 9, где заболевание центральной нервной системы выбрано из группы, состоящей из эпилепсии, судорог, воспалительной боли, нейропатической боли, мигрени, депрессии, тревожного расстройства, инсульта, болезни Альцгеймера, нейродегенеративного заболевания, злоупотребления кокаином, никотиновой абстиненции, алкогольной абстиненции и шума в ушах.
| CN 103508960 A, 15.01.2014 | |||
| 1-ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ ТЕТРАГИДРОИЗОХИНОЛИНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2008 |
|
RU2468010C2 |
| CN 107814785 A, 30.06.2016 | |||
| Прибор для определения силы ветра | 1929 |
|
SU23649A1 |

