Область техники
Настоящее изобретение относится к области органической химии и медицинской химии. В частности, настоящее изобретение относится к дейтерированным производным тиенопиперидина; также настоящее изобретение относится к фармацевтически приемлемым солям дейтерированных производных тиенопиперидина, к способу их получения и их применению при изготовлении лекарственного средства для лечения и предупреждения сердечно-сосудистых и цереброваскулярных заболеваний.
Предпосылки изобретения
Клопидогрел, лекарственное средство группы тиенопиридина, способно с высокой эффективностью ингибировать активность тромбоцитов и в настоящее время широко применяется в качестве антитромбоцитарного лекарственного средства при остром коронарном синдроме и при лечении пациентов, подвергающихся чрескожному коронарному вмешательству (PCI). Его структурная формула представлена ниже.
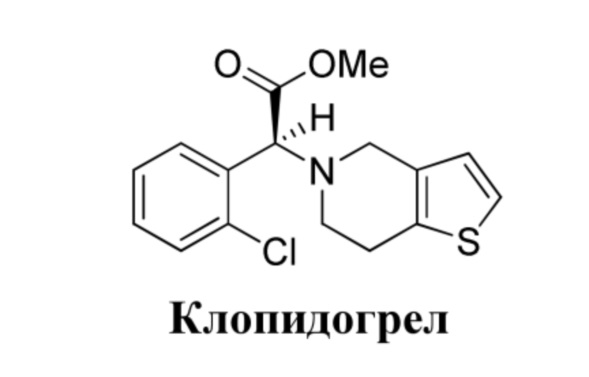
Клопидогрел представляет собой пролекарство, которое не обладает активностью, и его необходимо превратить в активный метаболит с помощью цитохрома P450 (CYP450) в печени, способ метаболизма представлен ниже:
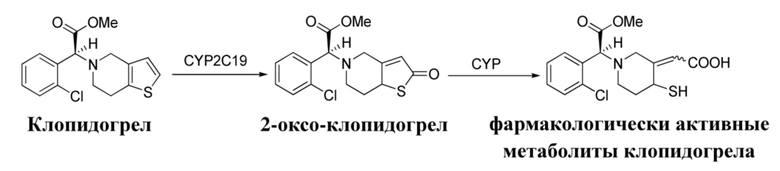
Данный метаболит связывается с рецептором P2Y12 аденозиндифосфата (ADP) на поверхности мембраны тромбоцита, и тем самым блокирует связывание ADP с рецептором тромбоцита, а также дополнительно активирует ADP-опосредованный гликопротеиновый комплекс GPIIbPIIIa, и, таким образом, ингибирует агрегацию тромбоцитов (Arterio-sclerThromb Vase Biol, 1999, 19 (8): 2002-2011). Клопидогрел способен значительно снижать частоту образования подострого тромбоза стента, снижая смертность и сердечно-сосудистые осложнения, такие как вторичный инфаркт миокарда. Тем не менее в последних исследованиях обнаружено, что у приблизительно 11%~44% (AmHeart J, 2009, 157 (2): 375-382.) пациентов наблюдалась низкая реакция или даже отсутствие реакции на клопидогрел, и данный феномен также называют «устойчивостью к клопидогрелу».
В заявке 201310428052.4 на патент Китая раскрытопроизводное тиенопиперидина со следующей структурой, которое представляет собой пролекарство на основе 2-окси-клопидогрела (метаболит клопидогрела), с целью преодолеть «устойчивость к клопидогрелу».
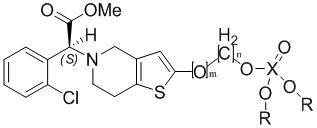
Тем не менее, данный ряд соединений все еще имеет недостатки, такие как низкая степень ингибирования агрегации тромбоцитов и высокая степень гидролиза. С целью устранения недостатков, описанных выше, разработки новых лекарственных средств, снижающих агрегацию тромбоцитов, обладающих быстрым клиническим действием, высоким терапевтическим эффектом и способностью обходить устойчивость к клопидогрелу, а также разработки соединений, которые предпочтительно составлять в состав для повышения биодоступности, уменьшения побочных эффектов и облегчения растворения, абсорбции и введения, в настоящем изобретении разработан ряд новых дейтерированных производных тиенопиперидина, способ их получения и применение на основании заявки 201310428052.4 на патент Китая. Заявка 201310428052.4 на патент Китая полностью включена в настоящее изобретение в качестве предшествующего уровня техники согласно настоящему изобретению.
Краткое описание изобретения
Технической задачей, которую необходимо решить при помощи настоящего изобретения, является устранение недостатков, описанных выше, проектирование и синтез новых оптически активных дейтерированных производных тиенопиперидина и, таким образом, разработка лекарственного средства, снижающего агрегацию тромбоцитов, с высоким терапевтическим эффектом и незначительными побочными эффектами.
Конкретно, одной из целей настоящего изобретения является получение оптически активных дейтерированных производных тиенопиперидина или их фармацевтически приемлемой соли, сольвата, полиморфа, энантиомера или рацемической смеси.
Другой целью настоящего изобретения является предоставление способа получения оптически активных дейтерированных производных тиенопиперидина или их фармацевтически приемлемой соли, сольвата, полиморфа, энантиомера, рацемической смеси или фармацевтической композиции на их основе.
Другой целью настоящего изобретения является получение фармацевтической композиции с оптически активными дейтерированными производными тиенопиперидина или их фармацевтически приемлемой солью, сольватом, полиморфом, энантиомером или рацемической смесью или фармацевтической композицией на их основе в качестве активного компонента.
Еще одной целью настоящего изобретения является предоставление применения оптически активных дейтерированных производных тиенопиперидина или их фармацевтически приемлемой соли, сольвата, полиморфа, энантиомера, рацемической смеси или фармацевтической композиции на их основе в изготовлении лекарственных средств.
Еще одной целью настоящего изобретения является предоставление способа лечения связанных заболеваний с помощью оптически активных дейтерированных производных тиенопиперидина или их фармацевтически приемлемой соли, сольвата, полиморфа, энантиомера или рацемической смеси или фармацевтической композиции на их основе или с применением фармацевтической композиции.
Для достижения вышеуказанных целей, в настоящем изобретении использовались следующие технические решения.
В настоящем изобретении предусмотрены оптически активные дейтерированные производные тиенопиперидина формулы (I) или их фармацевтически приемлемая соль, сольват, полиморф, энантиомер или рацемическая смесь:
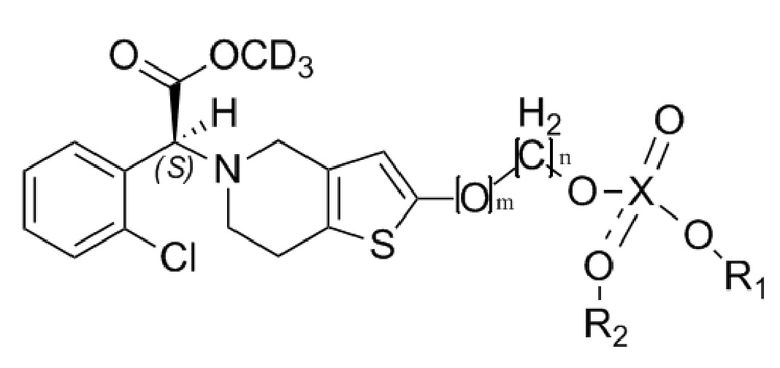
Формула (I),
где, D в CD3 представляет собой дейтерий, который представляет собой стабильный изотоп водорода, также называемый тяжелым водородом;
X представляет собой P или S; m равняется 0 или 1; n равняется 0 или 1; R1 выбран из водорода, линейного или разветвленного C1-C4алкила, замещенного или незамещенного галогеном, фенилом или замещенным фенилом; R2 не замещен или выбран из водорода, линейного или разветвленного C1-C4алкила, замещенного или незамещенного галогеном, фенилом или замещенным фенилом, при этом если R2 не замещен, то X и O образуют двойную связь.
Предпочтительно, X представляет собой P, m равняется 0, n равняется 0, R1 выбран из водорода, CH3-, CH3CH2-, изопропила, CCl3CH2- и фенила; R2 выбран из водорода, CH3-, CH3CH2-, изопропила, CCl3CH2- и фенила.
Или X представляет собой P, m равняется 1, n равняется 1, R1 выбран из водорода, CH3-, CH3CH2-, изопропила, CCl3CH2-, трет-бутила и фенила; R2 выбран из водорода, CH3-, CH3CH2-, изопропила, CCl3CH2-, трет-бутила и фенила.
Или X представляет собой S, m равняется 0, n равняется 0, R1 выбран из водорода, CH3-, CH3CH2-, изопропила, CCl3CH2-, трет-бутила и фенила; R2 не замещен, и X и O образуют двойную связь.
Дейтерированные производные тиенопиперидина по настоящему изобретению предпочтительно представляют собой следующие соединения:
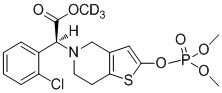
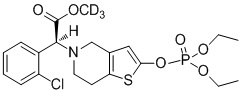
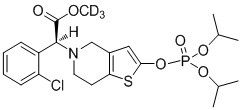
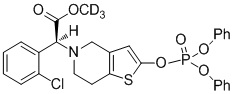
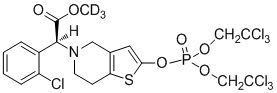
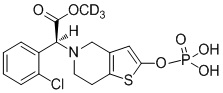
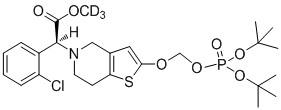
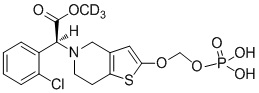
 .
.
Настоящее изобретение также включает фармацевтически приемлемую соль дейтерированных производных тиенопиперидина, при этом соль может представлять собой соль, образованную дейтерированными производными тиенопиперидина с серной кислотой, соляной кислотой, бромистоводородной кислотой, фосфорной кислотой, винной кислотой, фумаровой кислотой, малеиновой кислотой, лимонной кислотой, уксусной кислотой, муравьиной кислотой, метансульфоновой кислотой, п-толуолсульфоновой кислотой, щавелевой кислотой или янтарной кислотой.
В настоящем изобретении также предусмотрена фармацевтическая композиция, содержащая дейтерированные производные тиенопиперидина или их фармацевтически приемлемую соль, описанную в настоящем изобретении. Фармацевтическая композиция также может включать фармацевтически приемлемый носитель, если необходимо. Фармацевтически приемлемый инертный носитель может находиться в твердом состоянии или в жидком состоянии. Можно получать порошки, таблетки, диспергируемые порошки, капсулы, суппозитории и мазеподобные твердые или полутвердые фармацевтические составы, и в таком случае обычно применяют твердый носитель. Твердый носитель, который можно применять, предпочтительно представляет собой одно или более веществ, выбранных из разбавителей, ароматизаторов, солюбилизаторов, смазывающих веществ, суспендирующих средств, связующих веществ и средств, способствующих набуханию, и т. п., или может представлять собой инкапсулирующий материал. В порошкообразном составе носитель содержит 5% - 70% тонкоизмельченного активного ингредиента. Конкретные примеры подходящих твердых носителей включают карбонат магния, стеарат магния, тальк, сахарозу, лактозу, пектин, декстрин, крахмал, желатин, трагакантовую камедь, метилцеллюлозу, натрий-карбоксиметилцеллюлозу, низкокипящий воск, масло какао и т. п. Вследствие простоты введения таблетки, порошки и капсулы представляют собой твердые составы для перорального применения, которые являются наиболее предпочтительными с точки зрения абсорбции.
Жидкий состав включает раствор, суспензию и эмульсию. Например, в инъекционном составе для парентерального введения может применяться вода или смешанный раствор воды и пропиленгликоля, и его физиологические показатели, такие как изотонический коэффициент и pH, подходящие для живого организма, являются регулируемыми. Жидкий состав также можно составить в водном растворе полиэтиленгликоля. Водный раствор для перорального применения можно получать посредством растворения активного ингредиента в воде с последующим добавлением подходящих красителей, ароматизаторов, стабилизаторов и загустителей. Тонкоизмельченный активный ингредиент можно диспергировать в вязком веществе, таком как натуральный или синтетический каучук, метилцеллюлоза, натрий-карбоксиметилцеллюлоза и другие известные суспендирующие средства, с получением водной суспензии для перорального применения.
Для простоты введения и равномерности дозирования, особенно предпочтительным является, если вышеупомянутый фармацевтический состав составлен в виде единичной лекарственной формы. Единичная лекарственная форма состава относится к физически отделяемой единице, подходящей в качестве однократной дозы, при этом каждая единица содержит предварительно установленное количество активного ингредиента, рассчитанное с целью получения необходимого терапевтического эффекта. Данная единичная лекарственная форма может представлять собой упакованную форму, например, таблетки, капсулы, порошки, упакованные в ампулу или флакон, или мази, гели или кремы, упакованные в тюбик или бутыль.
Хотя количество активного ингредиента в единичной лекарственной форме может изменяться, обычно его регулируют до значения, находящегося в диапазоне 1-1000 мг в зависимости от эффективности выбранного активного ингредиента.
Специалист в данной области техники сможет определить предпочтительную дозу, подходящую для конкретного случая, в соответствии с традиционным способом. Как правило, первичная лечебная доза ниже оптимальной дозы активного ингредиента, и затем вводимую дозу постепенно увеличивают до достижения оптимального терапевтического эффекта. Для удобства общую дневную дозу можно разделять на несколько порций и принимать за несколько раз.
Применение дейтерированных производных тиенопиперидина по настоящему изобретению или их фармацевтически приемлемой соли заключается в изготовлении лекарственных средств для лечения и предупреждения сердечно-сосудистых и цереброваскулярных заболеваний, таких как сердечная недостаточность, инсульт и нестабильная стенокардия, особенно применение заключается в изготовлении лекарственных средств, снижающих агрегацию тромбоцитов.
В другом аспекте в настоящем изобретении также предусмотрен способ получения дейтерированных производных тиенопиперидина по настоящему изобретению или их фармацевтически приемлемой соли, сольвата, полиморфа, энантиомера или рацемической смеси, при этом способ получения включает следующие стадии реакции:

где заместители описаны ранее.
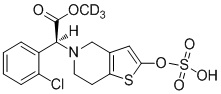
В соответствии с подробными вариантами осуществления настоящего изобретения соединение TSD-9 по настоящему изобретению можно получать следующим образом:
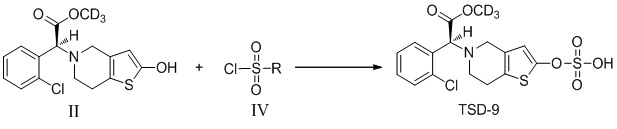
где R представляет собой хлор или гидроксил.
Краткое описание графических материалов
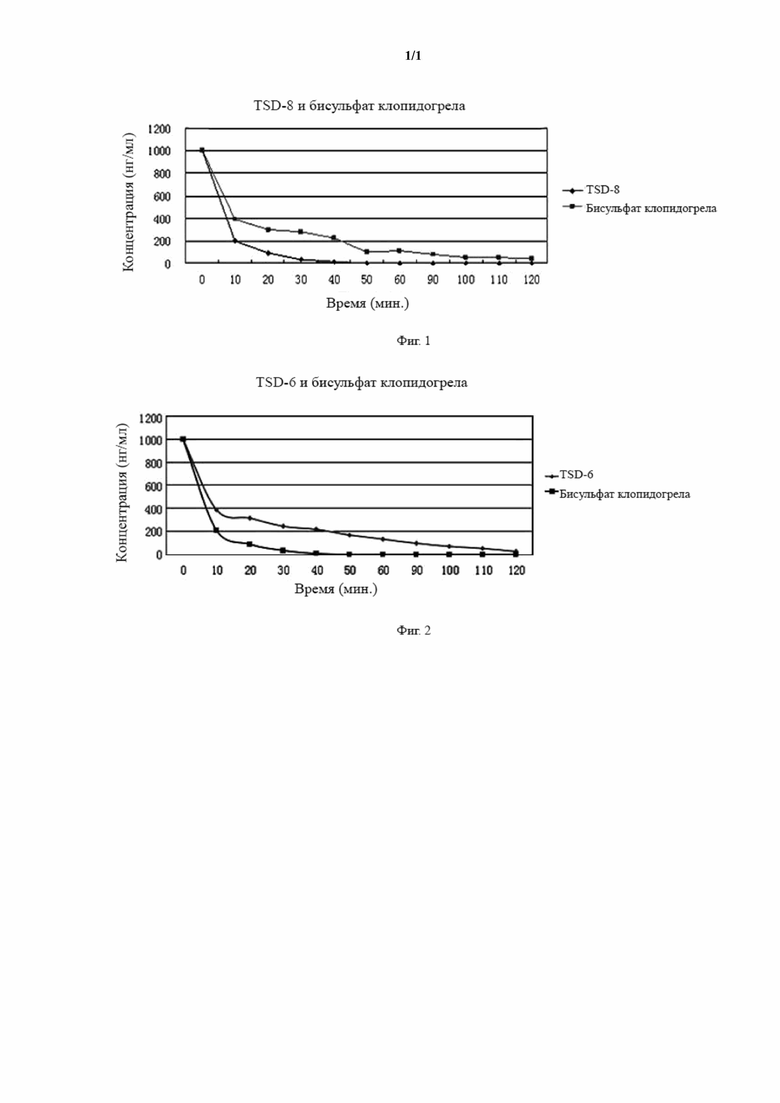
Фигура 1 представляет собой график степени гидролиза эстеразой TSD-8 и клопидогрела.
Фигура 2 представляет собой график степени гидролиза эстеразой TSD-6 и клопидогрела.
Подробное описание изобретения
Варианты осуществления приведены с целью дополнительной иллюстрации настоящего изобретения, но не с целью его ограничения.
Вариант осуществления 1
Метил-d3-(R)-o-хлорманделат

Растворяли 9,4 г (R)-o-хлорминдальной кислоты в 36 мл дейтерированного метанола, в который добавляли 1 мл раствора HCl/диоксан (4 М), нагревали с обратным холодильником в течение 5 часов и растворитель удаляли посредством выпаривания при пониженном давлении после охлаждения. Остаток растворяли с использованием метиленхлорида и полученный раствор промывали последовательно 5% водным раствором карбоната калия и водой, и раствор метиленхлорида высушивали с помощью безводного сульфата натрия. После удаления осушителя посредством фильтрации раствор выпаривали до сухого состояния с получением 9,2 г бесцветного прозрачного маслянистого продукта, представляющего собой метил-d3 (R)-o-хлорманделат, с выходом 89,7%.
Вариант осуществления 2
Метил-d3-(R)-2-(2-хлорфенил)-2-(4-нитрофенилсульфонилокси)-ацетат (II-1)
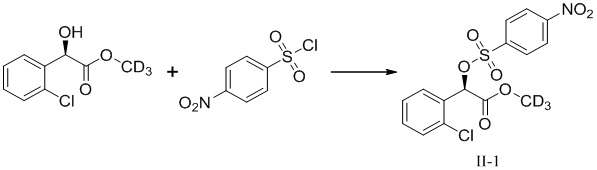
Растворяли 10,2 г метил-d3 (R)-o-хлорманделата в 50 мл безводного метиленхлорида, в который добавляли 65,6 г триэтиламина и каталитическое количество DMAP, перемешивали и охлаждали до 0°C. Добавляли 50 мл безводного раствора, содержащего 12,2 г п-нитробензолсульфонилхлорида в метиленхлориде по каплям при той же температуре с осуществлением реакции в течение 4 часов при постоянной температуре. Добавляли 100 мл воды в реакционный раствор, перемешивали, обеспечивали отстаивание с разделением жидкости. Водную фазу трижды экстрагировали с помощью 150 мл метиленхлорида и высушивали с помощью безводного сульфата натрия после объединения органических фаз. Высушивали метиленхлорид при пониженном давлении после удаления осушителя посредством фильтрации с получением 20,9 г маслянистого неочищенного продукта темно-красного цвета. Полученное вещество перекристаллизовывали с метанолом с получением 15,8 г твердого продукта (II-1), с выходом 81,3%.
Вариант осуществления 3
Метил-d3(2S)-2-(2-хлорфенил)-2-(2-оксо-7, 7a-дигидротиено-[3,2-c]пиридин-5(2H,4H,6H)-ил)ацетат (V-1)
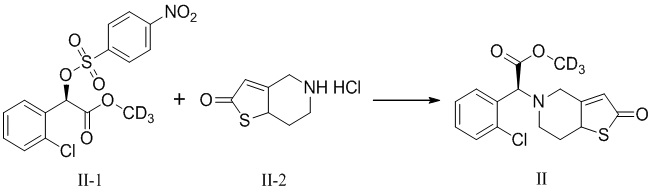
Объединяли 58,1 г (0,15 моль) метил-d3(R)-2-(2-хлорфенил)-2-(4-нитрофенилсульфонилокси)-ацетата (II-1), 32,3 г (0,17 моль) 5,6,7,7a-тетрагидротиено[3.2-c]пиридин-2(4H)-гидрохлорида (Ⅳ-1) и 37,8 г (0,38 моль) бикарбоната калия в 500 мл ацетонитрила с обеспечением реакции при перемешивании при комнатной температуре в течение 26 часов с применением системы защитной атмосферы азота. Нерастворимые вещества отфильтровывали из реакционного раствора после отстаивания с получением темно-красного маточного раствора. Растворитель высушивали при пониженном давлении, подвергали флэш-хроматографии (петролейный эфир:этилацетат = 4:1) с получением 35,4 г маслянистого продукта с выходом 70%.
Вариант осуществления 4
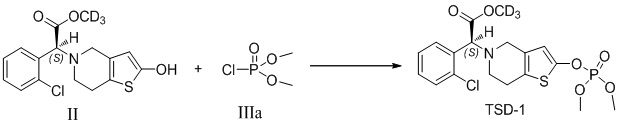
Растворяли промежуточное соединение II (200 мг, 0,6 ммоль), которое представляет собой дейтерированный 2-оксо-клопидогрел, в 5 мл безводного тетрагидрофурана, охлаждали до -20°C, в него добавляли диизопропиламид лития (2,0 М, 0,5 мл, 1 ммоль), и перемешивали в течение 20 минут. Добавляли соединение IIIa (104 мг, 0,72 ммоль) в реакционный раствор с осуществлением реакции в течение 12 часов при самонагревании процесса. Реакционную смесь гасили 4% соляной кислотой, в которую добавляли 50 мл этилацетата, и органический слой промывали соответственно бикарбонатом натрия и солевым раствором, высушивали с помощью безводного сульфата натрия, фильтровали и концентрировали. Очистку проводили с помощью колоночной хроматографии на силикагеле (PE: EA = 4:1) с получением соединения TSD-1 (245 мг, выход: 92%).
1H ЯМР (400 МГц, CDCl3): δ 7,67-7,65 (m, 1H), 7,42-7,40 (m, 1H), 7,31-7,26 (m, 2H), 6,25 (d, 1H), 4,91 (s, 1H), 3,87 (s, 3H), 3,64-3,60 (m, 1H), 3,51-3,48 (m, 1H), 2,89-2,87 (m, 2H), 2,75-2,73 (m, 2H), MS: масса/заряд 449 [M+1]+.
Вариант осуществления 5

Растворяли промежуточное соединение II (500 мг, 1,5 ммоль), которое представляет собой дейтерированный 2-оксо-клопидогрел, в 10 мл безводного тетрагидрофурана и охлаждали до -20°C, в него добавляли диизопропиламид лития (2,0 М, 1,25 мл, 2,5 ммоль), перемешивали в течение 30 минут. Добавляли соединение IIIb (311 мг, 1,8 ммоль) в реакционный раствор с осуществлением реакции в течение 12 часов при самонагревании процесса. Реакционную смесь гасили 4% соляной кислотой, в которую добавляли 100 мл этилацетата, и органический слой промывали соответственно бикарбонатом натрия и солевым раствором, высушивали с помощью безводного сульфата натрия, фильтровали и концентрировали. Очистку проводили с помощью колоночной хроматографии на силикагеле (PE: EA = 4:1) с получением соединения TSD-2 (660 мг, выход: 93%).
1H ЯМР (400 МГц, CDCl3): δ 7,69-7,66 (m, 1H), 7,43-7,41 (m, 1H), 7,33-7,28 (m, 2H), 6,27 (d, 1H), 4,91 (s, 1H), 4,27-4,18 (m, 4H), 3,65-3,61 (m, 1H), 3,52-3,49 (m, 1H), 2,90-2,87 (m, 2H), 2,76-2,74 (m, 2H), 1,39-1,36 (dt, 6H). MS: масса/заряд 477 [M+1]+.
Вариант осуществления 6
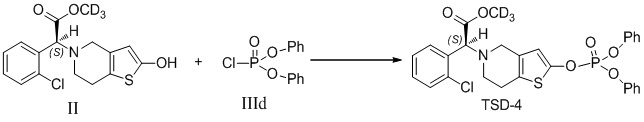
Растворяли промежуточное соединение II (100 мг, 0,3 ммоль), которое представляет собой дейтерированный 2-оксо-клопидогрел, в 5 мл безводного тетрагидрофурана и охлаждали до -20°C, в него добавляли диизопропиламид лития (2,0 М, 0,25 мл, 0,5 ммоль), перемешивали в течение 20 минут. Добавляли соединение IIId (97 мг, 0,36 ммоль) в реакционный раствор с осуществлением реакции в течение 12 часов при самонагревании процесса. Реакционную смесь гасили 4% соляной кислотой, в которую добавляли 50 мл этилацетата, и органический слой промывали соответственно бикарбонатом натрия и солевым раствором, высушивали с помощью безводного сульфата натрия, фильтровали и концентрировали. Очистку проводили с помощью колоночной хроматографии на силикагеле (PE: EA = 2:1) с получением соединения TSD-4 (162 мг, выход: 95%).
1H ЯМР (400 МГц, CDCl3): δ 7,71-7,68 (m, 1H), 7,47-7,42 (m, 5H), 7,35-7,24 (m, 10H), 6,28 (d, 1H), 4,92 (s, 1H), 2,89-2,87 (m, 2H), 2,75-2,73 (m, 2H), MS: масса/заряд 573 [M+1]+.
Вариант осуществления 7
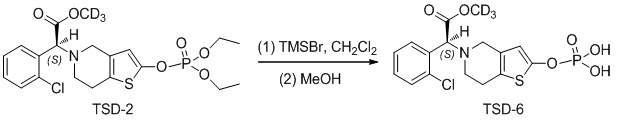
Растворяли TSD-2 (500 мг, 1,04 ммоль) в 10 мл сухого метиленхлорида, в который добавляли TMSBr (1,7 мл, 13 ммоль) с осуществлением реакции при комнатной температуре в течение 12 ч., реакцию останавливали и растворитель откачивали при пониженном давлении, затем добавляли 10 мл метанола и перемешивали в течение 1 ч. Реакционный раствор непосредственно концентрировали, очистку проводили с помощью колоночной хроматографии на силикагеле (н-бутанол:муравьиная кислота:вода = 5:5:1) с получением соединения TSD-6 (390 мг, выход: 90%).
1H ЯМР (400 МГц, DMSO): δ 7,60 (d, 1H), 7,53 (d, 1H), 7,41-7,40 (m, 2H), 6,24 (s, 1H), 4,91 (s, 1H), 3,56 (s, 2H), 2,85 (brs, 2H), 2,66 (brs, 2H), MS: масса/заряд 421 [M+1]+.
Вариант осуществления 8

Растворяли промежуточное соединение II (500 мг, 1,5 ммоль), которое представляет собой дейтерированный 2-оксо-клопидогрел, в 5 мл безводного тетрагидрофурана и охлаждали до -20°C, в него добавляли диизопропиламид лития (2,0 М, 1,25 мл, 2,5 ммоль) и перемешивали в течение 20 минут. Добавляли соединение IIIe (466 мг, 1,8 ммоль) в реакционный раствор с осуществлением реакции в течение 12 часов при самонагревании процесса. Реакционную смесь гасили 4% соляной кислотой, в которую добавляли 100 мл этилацетата, и органический слой промывали соответственно бикарбонатом натрия и солевым раствором, высушивали с помощью безводного сульфата натрия, фильтровали и концентрировали. Очистку проводили с помощью колоночной хроматографии на силикагеле (PE: EA = 2:1) с получением соединения TSD-7 (269 мг, выход: 32%).
1H ЯМР (400 МГц, CDCl3): δ 7,69-7,65 (m, 1H), 7,42-7,40 (m, 1H), 7,31-7,24 (m, 2H), 6,17 (s, 1H), 5,46 (s, 1H), 5,43 (s, 1H), 4,91 (s, 1H), 3,64-3,60 (m, 1H), 3,50-3,47 (m, 1H), 2,91-2,88 (m, 2H), 2,75-2,72 (m, 2H), 1,50 (s, 18H). MS: масса/заряд 560 [M+1]+.
Вариант осуществления 9
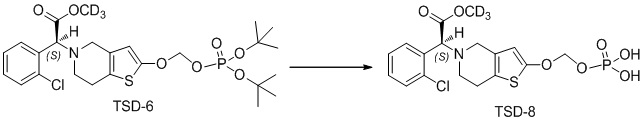
Растворяли TSD-6 (500 мг, 0,89 ммоль) в 10 мл метиленхлорида, в который добавляли трифторуксусную кислоту (2 мл), перемешивали при комнатной температуре в течение 1 ч. и концентрировали при пониженном давлении. Очистку проводили с помощью колоночной хроматографии на силикагеле (н-бутанол:муравьиная кислота:вода = 5:5:1) с получением соединения TSD-8 (140 мг, выход: 35%).
1H ЯМР (400 МГц, DMSO): δ 7,62-7,60 (m, 1H), 7,54-7,41 (m, 3H), 6,18 (s, 1H), 5,84 (s, 1H), 5,37-5,32 (d, 2H), 4,26-3,98 (m, 2H), 3,74-3,66 (m, 2H), 3,15-3,00 (m, 2H), MS: масса/заряд 451 [M+1]+.
Вариант осуществления 10

Растворяли промежуточное соединение II (150 мг, 0,45 ммоль), которое представляет собой дейтерированный 2-оксо-клопидогрел, в 5 мл безводного тетрагидрофурана и охлаждали до -20°C, в него добавляли диизопропиламид лития (2,0 M, 0,4 мл, 0,8 ммоль) и перемешивали в течение 20 минут. Добавляли соединение IIIc (108 мг, 0,54 ммоль) в реакционный раствор с осуществлением реакции в течение 12 часов при самонагревании процесса. Реакционную смесь гасили 4% соляной кислотой, в которую добавляли 50 мл этилацетата, и органический слой промывали соответственно бикарбонатом натрия и солевым раствором, высушивали с помощью безводного сульфата натрия, фильтровали и концентрировали. Очистку проводили с помощью колоночной хроматографии на силикагеле (PE: EA = 2:1) с получением соединения TSD-3 (192 мг, выход: 85%).
1H ЯМР (400 МГц, CDCl3): δ 7,68-7,67 (m, 1H), 7,41-7,39 (m, 1H), 7,34-7,28 (m, 2H), 6,28 (d, 1H), 4,92 (s, 1H), 4,74 (m, 2H), 4,26-4,17 (m, 4H), 3,64-3,61 (m, 1H), 3,53-3,49 (m, 1H),1,28 (d, 12H). MS: масса/заряд 505 [M+1]+.
Вариант осуществления 11
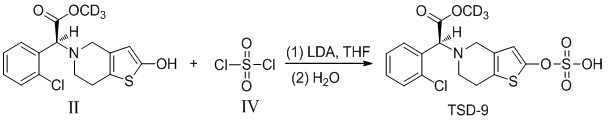
Растворяли промежуточное соединение II (500 мг, 1,5 ммоль), которое представляет собой дейтерированный 2-оксо-клопидогрел, в 5 мл безводного тетрагидрофурана и охлаждали до -20°C, в него добавляли диизопропиламид лития (2,0 М, 1,25 мл, 2,5 ммоль) и перемешивали в течение 20 минут. Добавляли соединение IV в реакционный раствор с осуществлением реакции в течение 12 часов при самонагревании процесса. Реакционную смесь гасили 4% соляной кислотой, в которую добавляли 100 мл этилацетата, органический слой промывали соответственно бикарбонатом натрия и солевым раствором, высушивали с помощью безводного сульфата натрия, фильтровали и концентрировали. Очистку проводили с помощью колоночной хроматографии на силикагеле (PE: EA = 2:1) с получением соединения TSD-9 (269 мг, выход: 32%).
1H ЯМР (400 МГц, CDCl3): δ 7,69-7,65 (m, 1H), 7,42-7,40 (m, 1H), 7,31-7,24 (m, 2H), 6,17 (s, 1H), 5,46 (s, 1H), 5,43 (s, 1H), 4,91 (s, 1H), 3,64-3,60 (m, 1H), 3,50-3,47 (m, 1H), 2,91-2,88 (m, 2H), 2,75-2,72 (m, 2H), 1,50 (s, 18H). MS: масса/заряд 563 [M+1]+.
Вариант осуществления 12. Изучение эффективности лекарственных средств на основе соединений по настоящему изобретению
Экспериментальный способ
При добавлении небольшой дозы ADP (концентрация менее чем 0,9 мкмоль/л) в суспензию тромбоцитов могло привести к быстрой агрегации тромбоцитов с последующей быстрой дезагрегацией; при добавлении умеренной дозы ADP (приблизительно 1,0 мкмоль/л) вторая необратимая фаза агрегации наступала вскоре после окончания первой фазы агрегации и дезагрегации. Максимальную степень агрегации во время необратимой фазы агрегации использовали для измерения эффекта тестовых образцов на функционирование свертываемости крови. В данном эксперименте применялся полуавтоматический анализатор агрегации тромбоцитов модели NJ4 от Precil с целью обнаружения ингибирующего эффекта тестовых образцов, предоставленных Tasly Group, в отношении агрегацию тромбоцитов.
Материалы для эксперимента
Животные: самцы крыс линии Вистар весом 230-250 г, приобретенные у Vital River Laboratory Animal Technology Co., Ltd.
Реагент: ADP, Sigma Corporation.
Тестовые образцы: 16 тестовых образцов были предоставлены Tasly Group; упомянутых в заявке 201310428052.4 на патент Китая на способ получения TSC-1~4 и TSC-6~9.
Доза введения: тестовые образцы суспендировали с помощью 0,25% CMC, вводили в количестве 3 мг/кг веса, объем введения: 2 мл.
Процедура эксперимента
Через 2 часа после введения крыс анестезировали пентобарбиталом натрия, кровь отбирали из брюшной аорты, добавляли антикоагулянт цитрат натрия в соотношении 1:9 и центрифугировали с получением тромбоцитарно-обогащенной плазмы и тромбоцитарно-обедненной плазмы с примерным соотношением тромбоцитарно-обедненной плазмы к тромбоцитарно-обогащенной плазме =3: 1.
Результаты эксперимента
Таблица 1. Эффекты соединений по настоящему изобретению в отношении максимальной степени агрегации ADP-индуцированной агрегации тромбоцитов
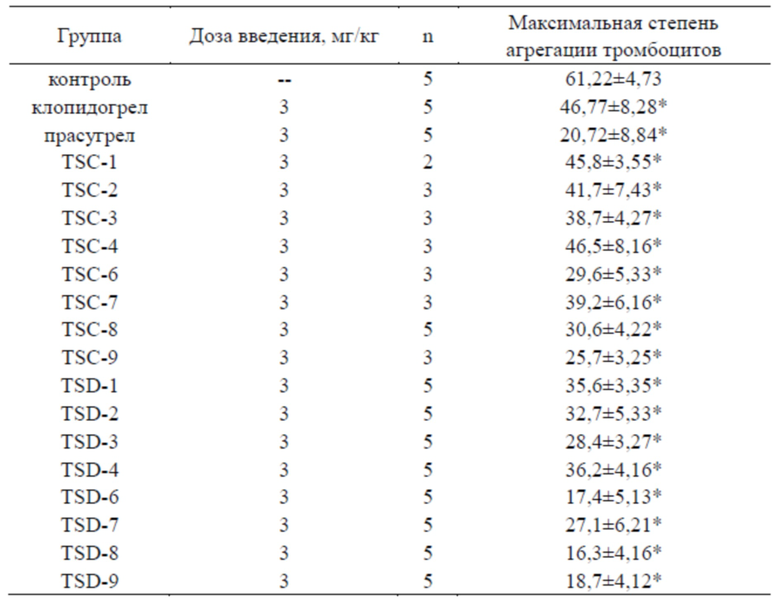
*P<0,001 по сравнению с нормальной группой.
В эксперименте с ADP-индуцированной агрегацией тромбоцитов каждый из тестовых образцов оказывал значительный эффект ингибирования в отношении агрегации тромбоцитов крыс и был способен обратить вторую фазу агрегации тромбоцитов, приводя к дезагрегации. Более того, эффекты ингибирования в отношении агрегации тромбоцитов у ряда дейтерированных производных тиенопиперидина по настоящему изобретению (TSD-1~4, 6~9) были гораздо сильнее, чем таковые для ряда недейтерированных производных тиенопиперидина (TSC-1~TSC-4, TSC-6~TSC-9).
Вариант осуществления 13
Сравнительный тест степени гидролиза эстеразой соединения формулы TSD-8 и клопидогрела
Степени гидролиза соединения формулы TSD-8 и бисульфата клопидогрела в цельной крови крыс определяли с применением способа инкубации in vitro.
Отбирали 3 мл свежей цельной крови крыс и помещали в стеклянную пробирку. Добавляли 30 мкг/мл TSD-8 и бисульфата клопидогрела (получали в физиологическом растворе) в 3 параллельных тестах для каждой группы. Пробирки встряхивали при постоянной температуре, составляющей 37℃, 100 мкл смеси отбирали в установленные моменты времени, составляющие 10 мин., 20 мин., 30 мин., 40 мин., 50 мин., 60 мин., 70 мин., 80 мин., 90 мин., 100 мин., 110 мин. и 120 мин., добавляли 900 мкл метанола с мгновенной остановкой реакции, затем последовательно добавляли 100 мкл метанола/воды (1:1, об./об.) и 100 мкл внутреннего стандарта (диазепам, 100 нг/мл). Смесь центрифугировали при 13000 об./мин. при низкой температуре в течение 10 мин., надосадочную жидкость переносили в другую пробирку для центрифугирования и 20 мкл надосадочной жидкости отбирали для введения образца.
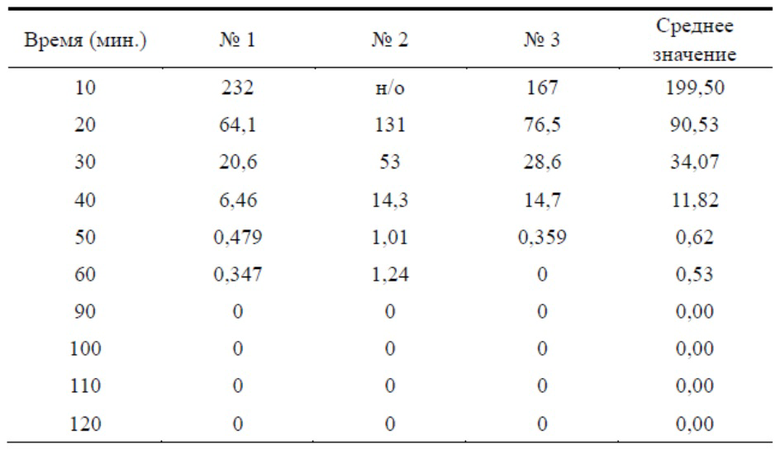
Таблица 2. Сравнительный тест степени гидролиза эстеразой TSD-8 и бисульфата клопидогрела
Результаты теста для образцов TSD-8

Результаты теста для образцов бисульфата клопидогрела
н/о означает не определено, ниже аналогично.
На фигуре 1 можно видеть, что концентрация TSD-8 в каждый момент времени была выше концентрации клопидогрела, следовательно, степень гидролиза TSD-8 в цельной крови крыс была ниже таковой клопидогрела, и примерно через 50 мин. концентрация бисульфата клопидогрела в цельной крови уже была ниже минимального обнаруживаемого значения, в то время как таковую соединения формулы TSD-8 все еще можно было обнаружить.
Вариант осуществления 14
Сравнительный тест степени гидролиза эстеразой соединения формулы TSD-6 и клопидогрела
Значения степени гидролиза соединения формулы TSD-6 и бисульфата клопидогрела в цельной крови крыс определяли с применением способа инкубации in vitro.
Отбирали 3 мл свежей цельной крови крыс и помещали в стеклянную пробирку. Добавляли 30 мкг/мл соединения формулы TSD-6 и бисульфата клопидогрела (получали в физиологическом растворе) в 3 параллельных тестах для каждой группы. Пробирки встряхивали при постоянной температуре, составляющей 37°C, 100 мкл смеси отбирали в установленные моменты времени, составляющие 10 мин., 20 мин., 30 мин., 40 мин., 50 мин., 60 мин., 70 мин., 80 мин., 90 мин., 100 мин., 110 мин. и 120 мин., в нее добавляли 900 мкл метанола с мгновенной остановкой реакции, затем последовательно добавляли 100 мкл метанола/воды (1:1, об./об.) и 100 мкл внутреннего стандарта (диазепам, 100 нг/мл). Смесь центрифугировали при 13000 об./мин. при низкой температуре в течение 10 мин., надосадочную жидкость переносили в другую пробирку для центрифугирования и 20 мкл надосадочной жидкости отбирали для введения образца.
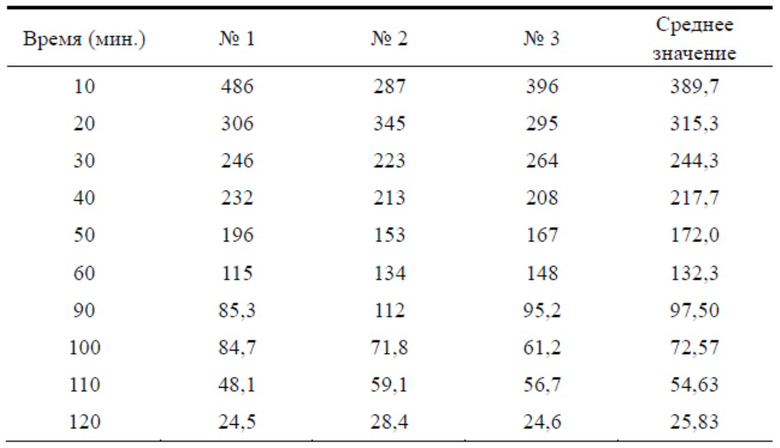
Таблица 3. Сравнительный тест степени гидролиза эстеразой TSD-6 и бисульфата клопидогрела
Результаты теста для образцов TSD-6
Результаты теста для образцов бисульфата клопидогрела

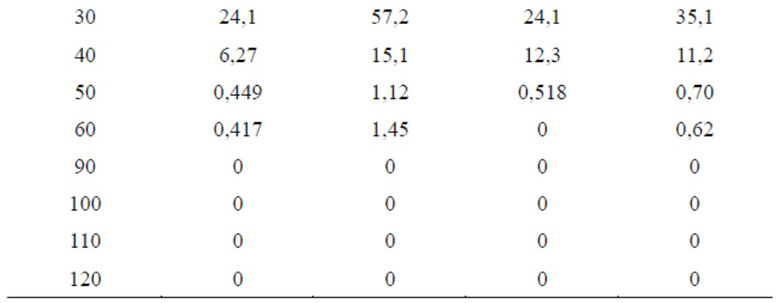
На фигуре 2 можно видеть, что концентрация TSD-6 в каждый момент времени была выше концентрации клопидогрела, степень гидролиза TSD-6 в цельной крови крыс была значительно ниже таковой клопидогрела, и примерно через 50 мин. концентрация бисульфата клопидогрела в цельной крови уже была ниже минимального обнаруживаемого значения, в то время как таковую соединения формулы TSD-6 все еще можно было обнаружить.
Вариант осуществления 15
Сравнение фармакокинетики соединений формулы TSD-6, TSC-6, I-1 и клопидогрела при метаболизировании in vivo до 2-оксо-клопидогрела у крыс; структуры соединений представлены на следующей фигуре:

Тестовые животные: 24 самца крыс линии Спрег-Доули, возрастом 6-7 недель, весом 240~290 г, приобретенные у Shanghai Slac Laboratory Animal Co., Ltd., с сертификатом на животных № 2015000514648. Перед тестом животных следует кормить в течение по меньшей мере 3 дней для адаптации к окружающей среде. Животных из группы внутривенной инъекции (IV) не ограничивали в пище; животных из группы перорального приема (PO) не кормили в течение ночи перед введением и кормили через 4 часа после введения; животным позволяли пить воду на протяжении всего теста.
Тестируемые лекарственные средства: бисульфат клопидогрела (клопидогрел), TSC-6, I-1 и TSD-6, предоставлены Tasly.
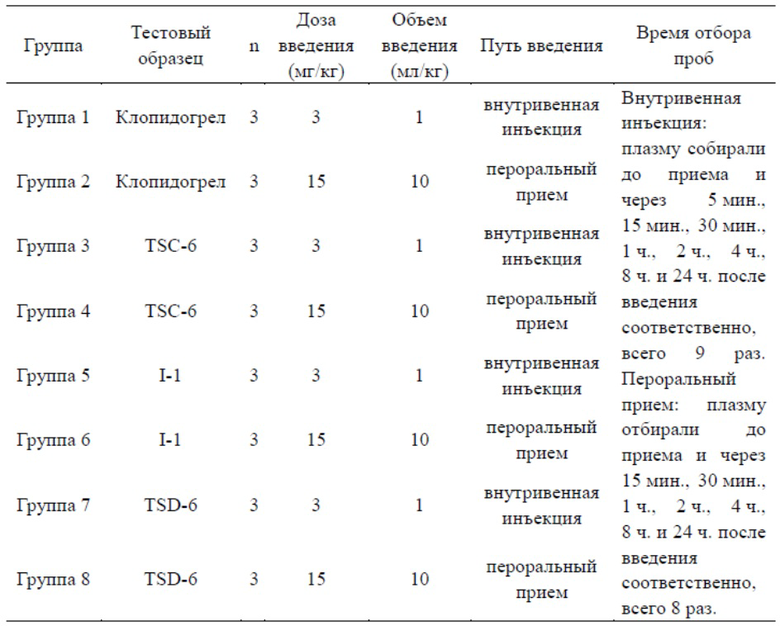
Распределение животных на группы и время отбора проб: 24 крысы линии Спрег-Доули разделили на 8 групп, по 3 в каждую группу, животным из группы внутривенной инъекции вводили 3 мг/кг тестируемых лекарственных средств в венозную дугу стопы, и животным из группы перорального приема вводили 15 мг/кг тестируемых лекарственных средств посредством кормления. См. таблицу 4 со схемой приема.
Таблица 4. Схема приема животными и отбора крови
Сбор и хранение проб: В заранее заданные моменты времени соответствующих животных обездвиживали, отбирали приблизительно 80 мкл крови из хвостовой вены, в пробу крови добавляли антикоагулянт гепарин натрия и помещали на натуральный лед. Немедленно отбирали 60 мкл пробы крови и добавляли в 600 мкл раствора внутреннего стандарта (40 нг/мл раствор диклофенака и ацетонитрила с 0,1% муравьиной кислоты) и смесь перемешивали на вортексе в течение 0,5 мин. и центрифугировали при 12000 об./мин. при 4°C в течение 5 мин. с получением надосадочной жидкости. Пробу надосадочной жидкости сначала помещали на сухой лед для хранения в замороженном состоянии и затем переносили в холодильную камеру при -70°C для длительного хранения до анализа.
Результаты теста
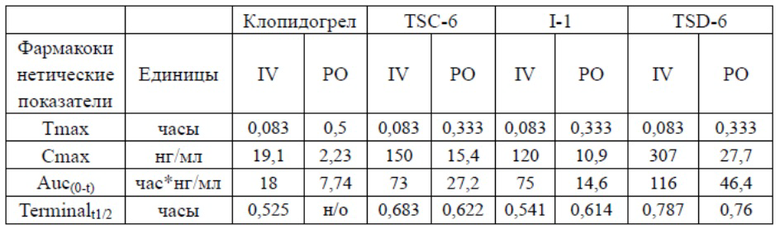
Из сравнительного исследования фармакокинетики главного метаболита 2-оксо-клопидогрела видно, что концентрация главного промежуточного метаболита разработанного дейтерированного соединения TSD-6 была значительно выше таковой недейтерированного соединения TSC-6 и аналогичного соединения I-1 подобного строения как в случае перорального введения, так и введения посредством внутривенной инъекции. Фармакокинетические данные показали, что метаболиты TSD-6 обладают лучшими характеристиками, что будет предпочтительным для усиления эффекта лекарственного средства и устранения недостатков соединений из предшествующего уровня техники.
Вариант осуществления 16
Сравнительное исследование эффектов лекарственных средств на основе соединений формулы TSC-6, I-1, TSD-6 и клопидогрела в модели кровотечения хвоста крысы
Животные и кормление. Вид, линия: крысы линии Спрег-Доули
Поставщик: Slac
Вес: 250-350 г
Пол: самцы
Количество животных: 30
Условия кормления: кормление осуществляли в асептическом отделении для животных, температура - 20,5-22,5°C, влажность - 50-65%, освещенность 150-250 лк, продолжительность дня и ночи по 12 часов (день 6:00-18:00).
Тестовые образцы, контрольное лекарственное средство и способ получения: соединения TSC-6, I-1, TSD-6 и клопидогрел растворяли в 5 мл 0,25% CMC, обрабатывали с помощью ультразвука при 37°C в течение 20 мин. и перемешивали до состояния суспензии с помощью мешалки.
Установление и обоснование дозы: доза тестовых соединений TSC-6, I-1, TSD-6 составляла 1 мг/кг. Вводимая доза соединения с подтвержденным действием, клопидогрела, составляла 10 мг/кг, определенная на основании сведений из литературы и результатов лабораторных тестов.
Путь введения: пероральный прием.
Экспериментальный способ
• Обеспечивали адаптацию животных к окружающей среде в течение 1 недели после прибытия и не кормили в течение 16 часов до эксперимента.
• Длительность кровотечения хвоста начинали измерять через 2 ч. после перорального введения тестируемых соединений и через 4 ч. после перорального введения клопидогрела.
• Крыс анестезировали пентобарбиталом натрия (50 мг/кг, внутрибрюшинно) за 10 мин. до измерения длительности кровотечения хвоста, когда крыса была полностью анестезирована и наступало время исследования, хвост отрезали ножницами на расстоянии 1,3 мм от кончика хвоста и вертикально погружали в физиологический раствор при 37°C. Отсчет времени не начинали до появления потока крови.
• Отсчет времени останавливали, если кровотечение прерывалось на более чем 20 секунд. Максимальное время исследования потока крови устанавливали на 40 минут. В случае превышения 40 минут отсчет времени останавливали и записывали время как 40 минут.
Результаты теста
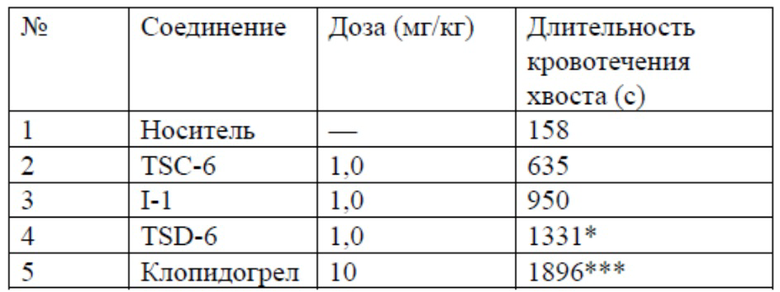
*P<0,05, ***P<0,001 по сравнению с носителем
Антикоагулянтные эффекты соединений оценивали посредством сравнения длительности кровотечения хвоста в модели кровотечения хвоста крысы между TSC-6, I-1, TSD-6 и клопидогрелом. Можно видеть, что антикоагулянтный эффект дейтерированного соединения TSD-6, разработанного авторами настоящего изобретения, был значительно выше такового для недейтерированного соединения TSC-6 и его аналога I-1 подобного строения, что показывает уникальную антикоагулянтную активность TSD-6.
Вариант осуществления 17
Сравнительное исследование эффектов лекарственных средств на основе TSC-6, I-1, TSD-6 и клопидогрела в модели тромба в артериовенозной петле у крыс
Животные и кормление. Вид, линия: крысы линии Спрег-Доули
Поставщик: Slac
Вес: 250-350 г
Пол: самцы
Количество животных: 30
Условия кормления: кормление осуществляли в асептическом отделении для животных, температура - 20,5-22,5°C, влажность - 50-65%, освещенность 150-250 лк, продолжительность дня и ночи по 12 часов (день 6:00-18:00).
Тестовые образцы, контрольное лекарственное средство и способ получения: соединения TSC-6, I-1, TSD-6 и клопидогрел растворяли в 5 мл 0,25% CMC, обрабатывали с помощью ультразвука при 37°C в течение 20 мин. и перемешивали до состояния суспензии с помощью мешалки.
Установление и обоснование дозы: Доза тестовых соединений TSC-6, I-1 и TSD-6 составляла 1 мг/кг. Вводимая доза соединения с подтвержденным действием, клопидогрела, составляла 10 мг/кг, определенная на основании сведений из литературы и результатов лабораторных тестов.
Путь введения: пероральный прием.
Оборудование и материалы
• Ватная палочка, сухой тампон, спиртовой ватный шарик, йодный тампон.
• Хирургические ножницы, глазной пинцет, кровоостанавливающий зажим, микрохирургические ножницы, микрохирургический зажим, артериальный зажим.
• 3-0 хирургическая нить, плотная полиэтиленовая трубка (внутренний диаметр/внешний диаметр = 1,14 мм/1,63 мм, длина 8 см), плотная полиэтиленовая трубка (внутренний диаметр/внешний диаметр = 0,72 мм/1,22 мм, длина 6 см).
• Операционный стол, нить для связывания, секундомер, точные электронные весы, бумага для взвешивания.
• Шприц, физиологический раствор.
Экспериментальный способ
• После прибытия обеспечивали адаптацию животных к окружающей среде в течение 1 недели и не кормили в течение 16 часов перед тестом.
• Циркуляцию крови по артериовенозной петле начинали через 2 ч. после перорального введения тестируемых соединений и через 4 ч. после перорального введения клопидогрела.
• Крыс анестезировали пентобарбиталом натрия (50 мг/кг, внутрибрюшинно) за 15 мин. до начала циркуляции крови по артериовенозной петле.
• Разделяли левую наружную яремную вену и правую сонную артерию и вводили плотные полиэтиленовые трубки соответственно.
• Две полиэтиленовые трубки соединяли с другой плотной полиэтиленовой трубкой с длиной 8 см с образованием циркуляционного канала. Плотная полиэтиленовая трубка содержала хирургическую нить (3-0) длиной 6 см.
• Через 15 мин. после открытия циркуляционного канала поток крови блокировали, нить вытаскивали и взвешивали после впитывания крови. Вес тромба получали после вычитания веса самой нити.
Результаты теста
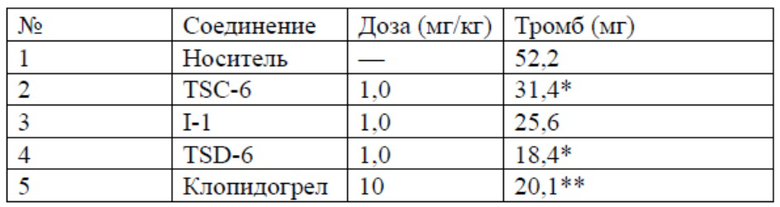
*P<0,01, **P<0,01 по сравнению с носителем
Антикоагулянтные эффекты соединений оценивали посредством сравнения значений разницы в весе тромбов, образованных в модели тромба в артериовенозной петле у крыс для TSC-6, I-1, TSD-6 и клопидогрела. Можно видеть, что вес тромба, образованного для дейтерированного соединения TSD-6, разработанного авторами настоящего изобретения, был гораздо меньше такового для недейтерированного соединения TSC-6 и его аналога I-1 подобного строения, что свидетельствует об уникальной антикоагулянтной активности TSD-6.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНОЕ СЛОЖНОГО ЭФИРА ТИЕНОПИРИДИНА, СОДЕРЖАЩЕЕ ЦИАНОГРУППУ, СПОСОБ ЕГО ПОЛУЧЕНИЯ, ЕГО ПРИМЕНЕНИЕ И КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ | 2011 |
|
RU2526624C2 |
| 2-ПИПЕРИДИНО-5-(ТИЕНИЛ-2)- И 2-ПИПЕРИДИНО-5-(ТИЕНИЛ-3)-6Н-1,3,4-ТИАДИАЗИНЫ ГИДРОБРОМИДЫ, ОБЛАДАЮЩИЕ АНТИАГРЕГАНТНЫМ ДЕЙСТВИЕМ | 2010 |
|
RU2445310C2 |
| ПРОИЗВОДНОЕ СУЛЬФОНИЛБЕНЗАМИДА И ЕГО КОНЬЮГАТЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2021 |
|
RU2839468C1 |
| ПРОИЗВОДНОЕ ЗАМЕЩЕННОГО ЦИННАМАМИДА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2012 |
|
RU2587668C2 |
| 4-ГИДРОКСИТИОБЕНЗАМИДНЫЕ ПРОИЗВОДНЫЕ ЛЕКАРСТВЕННЫХ ВЕЩЕСТВ | 2007 |
|
RU2465271C2 |
| АНТИТРОМБОТИЧЕСКИЕ ДВОЙНЫЕ ИНГИБИТОРЫ, ВКЛЮЧАЮЩИЕ БИОТИНОВУЮ МЕТКУ | 2006 |
|
RU2434876C2 |
| ПУТИ ПРИМЕНЕНИЯ ИНГИБИТОРОВ ФОСФОДИЭСТЕРАЗЫ | 2020 |
|
RU2795503C2 |
| СОЕДИНЕНИЕ, ИМЕЮЩЕЕ ДЕЙСТВИЕ ИНГИБИРОВАНИЯ АГРЕГАЦИИ ТРОМБОЦИТОВ, И ЕГО СОЛЬ, И СОДЕРЖАЩАЯ ТАКИЕ СОЕДИНЕНИЯ КОМПОЗИЦИЯ ДЛЯ ПРЕДУПРЕЖДЕНИЯ ИЛИ ЛЕЧЕНИЯ ТРОМБОТИЧЕСКИХ ЗАБОЛЕВАНИЙ | 2016 |
|
RU2739915C2 |
| ИНГИБИТОРЫ ПРОТЕИНКИНАЗ, ИХ СПОСОБ ПОЛУЧЕНИЯ И МЕДИЦИНСКОЕ ПРИМЕНЕНИЕ | 2016 |
|
RU2749437C2 |
| СПОСОБЫ ЛЕЧЕНИЯ ИЛИ ПРОФИЛАКТИКИ ТРОМБООБРАЗОВАНИЯ ИЛИ ЭМБОЛИИ | 2011 |
|
RU2611662C2 |
Изобретение относится к дейтерированным производным тиенопиперидина со структурой формулы (I) или их фармацевтически приемлемым солям
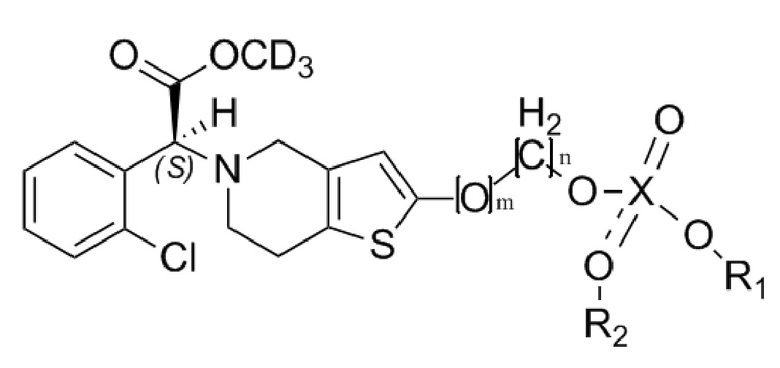
Формула (I),
где X представляет собой P или S; m равняется 0 или 1; n равняется 0 или 1; R1 выбран из водорода, линейного или разветвленного C1-C4алкила, замещенного или незамещенного галогеном или фенилом; R2 не замещен или выбран из водорода, линейного или разветвленного C1-C4алкила, замещенного или незамещенного галогеном или фенилом, при этом если R2 не замещен, то X и O образуют двойную связь, а также к способу их получения и применению для лечения и предупреждения сердечно-сосудистых и цереброваскулярных заболеваний. 4 н. и 5 з.п. ф-лы, 2 ил., 4 табл., 12 пр.
1. Дейтерированные производные тиенопиперидина со структурой формулы (I) или их фармацевтически приемлемая соль,

Формула (I),
где X представляет собой P или S; m равняется 0 или 1; n равняется 0 или 1; R1 выбран из водорода, линейного или разветвленного C1-C4алкила, замещенного или незамещенного галогеном или фенилом; R2 не замещен или выбран из водорода, линейного или разветвленного C1-C4алкила, замещенного или незамещенного галогеном или фенилом, при этом если R2 не замещен, то X и O образуют двойную связь.
2. Дейтерированные производные тиенопиперидина или их фармацевтически приемлемая соль по п. 1, где X представляет собой P, m равняется 0, n равняется 0, R1 выбран из водорода, CH3-, CH3CH2-, изопропила, CCl3CH2- и фенила; R2 выбран из водорода, CH3-, CH3CH2-, изопропила, CCl3CH2- и фенила.
3. Дейтерированные производные тиенопиперидина или их фармацевтически приемлемая соль по п. 1, где X представляет собой P, m равняется 1, n равняется 1, R1 выбран из водорода, CH3-, CH3CH2-, изопропила, CCl3CH2-, трет-бутила и фенила; R2 выбран из водорода, CH3-, CH3CH2-, изопропила, CCl3CH2-, трет-бутила и фенила.
4. Дейтерированные производные тиенопиперидина или их фармацевтически приемлемая соль по п. 1, где X представляет собой S, m равняется 0, n равняется 0, R1 выбран из водорода, CH3-, CH3CH2-, изопропила, CCl3CH2-, трет-бутила и фенила, при этом R2 не замещен, и X и O образуют двойную связь.
5. Дейтерированные производные тиенопиперидина или их фармацевтически приемлемая соль по п. 1, где они выбраны из следующих соединений:
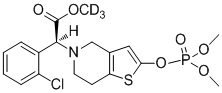
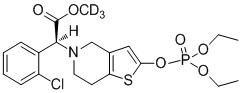
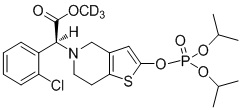
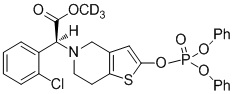
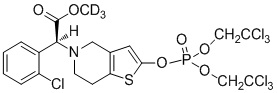

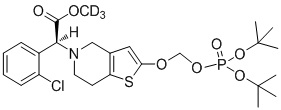
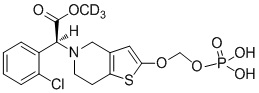
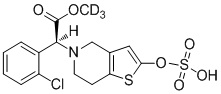 .
.
6. Дейтерированные фосфатные производные тиенопиперидина или их фармацевтически приемлемая соль по п. 1, где соль может представлять собой соль, образованную дейтерированным фосфатным производным тиенопиперидина с серной кислотой, соляной кислотой, бромистоводородной кислотой, фосфорной кислотой, винной кислотой, фумаровой кислотой, малеиновой кислотой, лимонной кислотой, уксусной кислотой, муравьиной кислотой, метансульфоновой кислотой, п-толуолсульфоновой кислотой, щавелевой кислотой или янтарной кислотой.
7. Фармацевтическая композиция для лечения и предупреждения сердечно-сосудистых и цереброваскулярных заболеваний, отличающаяся тем, что фармацевтическая композиция содержит эффективное количество дейтерированных фосфатных производных тиенопиперидина или их фармацевтически приемлемой соли по п. 1, а также содержит фармацевтически приемлемый носитель.
8. Применение дейтерированных фосфатных производных тиенопиперидина или их фармацевтически приемлемой соли по п. 1 при изготовлении лекарственных средств для лечения и предупреждения сердечно-сосудистых и цереброваскулярных заболеваний, таких как сердечная недостаточность, инсульт и нестабильная стенокардия.
9. Применение дейтерированного фосфатного производного тиенопиперидина или его фармацевтически приемлемой соли по п. 1 при изготовлении лекарственных средств, снижающих агрегацию тромбоцитов.
| CN 104447867 A 25.03.2015 | |||
| CN 103554132 A 05.02.2014 | |||
| RU 2011105422 A 20.08.2012 | |||
| НОВЫЕ СОЕДИНЕНИЯ ДЕЙСТВУЮЩИХ НАЧАЛ, СОДЕРЖАЩИХ КЛОПИДОГРЕЛ, И АНТИТРОМБИЧЕСКОЕ СРЕДСТВО | 1997 |
|
RU2184547C2 |

