Область техники
Настоящее изобретение относится к способу и набору для детекции микроорганизмов, содержащихся в продуктах питания, биологических образцах и образцах окружающей среды, например образцах промышленных вод и водопроводной воды. Конкретно, настоящее изобретение относится к способу и набору для детекции микроорганизмов, которые обеспечивают селективную детекцию жизнеспособных клеток микроорганизмов, содержащихся в продуктах питания, биологических образцах, мазках и образцах окружающей среды, таких как промышленные воды и водопроводная вода.
Предшествующий уровень техники
Как правило, для определения общего количества жизнеспособных бактерий в продуктах питания, биологических образцах, мазках или образцах окружающей среды используют способ культуры на твердой среде. Однако в случае способа культуры на твердой среде для получения результата требуется приблизительно от двух суток до одного месяца.
В результате улучшения способов стерилизации и способов обработки продуктов питания возрастает необходимость различать жизнеспособное состояние микроорганизмов, находящихся в тестируемых образцах, от мертвого состояния микроорганизмов даже для случаев, когда клетки существуют в невероятно низком количестве. В областях санитарной проверки продуктов питания и клинического тестирования, в частности в качестве быстрого способа детекции бактерий, предпринимались попытки определить присутствие или отсутствие бактерий или количество бактерий с помощью амплификации генов, специфичных для бактерий, посредством ПЦР до такого количества, что эти гены можно наблюдать визуально. Однако если целью является бактериальная ДНК, также определяют фон из мертвых клеток, исходно содержащихся в тестируемом образце и, таким образом, положительный результат, получаемый посредством ПЦР, не обязательно означает присутствие жизнеспособных бактерий. Таким образом, текущая ситуация в областях пищевой санитарии и клинического тестирования такова, что ПЦР широко не используется, хотя она представляет собой высокочувствительный и быстрый способ.
В настоящее время предпринимаются попытки детектировать и определить количество в тестируемом образце только жизнеспособных клеток микроорганизмов, получая кДНК с использованием обратной транскриптазы с мРНК в качестве мишени и проводя ПЦР с праймерами, специфичными для различных бактерий. Однако в этом способе нет препятствий для обратной транскрипции мРНК погибших клеток, и когда в тестируемом образце содержится 104 КОЕ/мл или 104 КОЕ/г или более погибших клеток, детектируют фон погибших клеток. Таким образом, нельзя сказать, что этого способа достаточно в качестве способа определения жизнеспособного и мертвого состояний.
В частности, в качестве способа различения жизнеспособного состояния и мертвого состояния микроорганизмов, таких как бактерии, с использованием способа ПЦР, предоставлены способы, описанные в патентном документе 1 и 2. Однако в этих способах различения жизнеспособного и мертвого состояния микроорганизмов, таких как бактерии, с использованием способа ПЦР указанные ниже проблемы остаются.
В качестве способа, описанного в патентном документе 1, указаны примеры различения погибших клеток, содержащихся в кипяченых продуктах питания, подвергаемых долговременной температурной стерилизации при 100°C в течение от 10 до 30 минут, и микроорганизмов, содержащихся в продуктах питания, подвергаемых стерилизации этанолом или формальдегидом. Однако, особенно для обработки последнего типа, продуктов питания, в действительности подвергаемых пастеризационной обработке такого типа, не существует. Кроме того, не существует предполагаемого способа детекции только жизнеспособных микроорганизмов в продуктах питания, подвергаемых основным современным способам стерилизации в пищевой промышленности, низкотемпературной долговременной пастеризации (пастеризация LTLT), высокотемпературной кратковременной пастеризации (пастеризация HTST) или ультравысокотемпературной пастеризации (пастеризация UHT) и способа детекции только жизнеспособных специфических патогенных бактерий в клинических образцах у пациентов с инфекционными заболеваниями, которым вводят антибиотики. Кроме того, в случае тестируемого образца продукта питания или клинического образца, содержащего фон погибших клеток в концентрации 104 КОЕ/мл или выше, количества конечных продуктов амплификации ПЦР, получаемые от погибших клеток, превосходят предел детекции способа по патентному документу 1, и, таким образом, определить получен ли положительный ответ в тестируемом образце, получаемом посредством ПЦР, от жизнеспособных клеток или погибших клеток невозможно.
Кроме того, в качестве способа по патентному документу 2 описан способ различения жизнеспособных клеток и погибших клеток с использованием относительного снижения молярного отношения РНК/ДНК погибших клеток по сравнению с молярным отношением РНК/ДНК жизнеспособных клеток. В этом способе выделяют тотальную РНК, получают комплементарную ДНК с использованием реакции обратной транскрипции, затем проводят ПЦР для расчета ее значения Ct и с использованием отдельно полученной калибровочной кривой получают молярную концентрацию РНК. Отдельно посредством ПЦР амплифицируют область хромосомной ДНК, соответствующую области РНК с получением ее значения Ct, и рассчитывают молярную концентрацию хромосомной ДНК на основе калибровочной кривой, получая молярное отношение РНК/ДНК. То есть указанный выше способ требует проведения сложного выделения тотальной РНК и использования двух стадий реакции обратной транскрипции и ПЦР. Таким образом, этот способ по эффективности и скорости количественного анализа хуже обычной ПЦР на основе ДНК. Кроме того, РНК в жизнеспособных клетках непрерывно синтезируется, тогда как РНК, получаемая из погибших клеток, разрушается с течением времени на ранней стадии. Таким образом, в этом способе отсутствует стабильность. Кроме того, этим способом в продукте питания или клиническом образце, содержащих погибшие клетки с высокой концентрацией, можно детектировать только 1/10 жизнеспособных клеток от этой концентрации. Таким образом, этот способ в областях санитарной проверки продуктов питания и клинического тестирования, где необходима скорость, высокая чувствительность и точность, использовать трудно.
В патентном документе 3 описан способ селективной детекции жизнеспособных клеток (жизнеспособные-и-культивируемые клетки) микроорганизмов посредством различения их с погибшими клетками и поврежденными клетками (жизнеспособные-но-некультивируемые клетки (клетки VNC)). Способ, описываемый в патентном документе 3, представляет собой способ, включающий стадию обработки тестируемого образца ингибитором топоизомеразы и/или ингибитором ДНК-гиразы, стадию выделения ДНК из тестируемого образца и амплификации области-мишени выделенной ДНК посредством ПЦР и стадию анализа продукта амплификации, и в качестве примера ингибитора топоизомеразы или ингибитора ДНК-гиразы приведен моноазид этидия.
Способ, в котором используют моноазид этидия, также описан в непатентном документе 1. Этот способ представляет собой способ, включающий стадию добавления в тестируемый образец моноазида этидия и облучения образца светом, стадию выделения ДНК из образца после облучения и стадию амплификации конкретной области посредством ПЦР с использованием выделенной ДНК в качестве матрицы. Кроме того, в непатентном документе 1 описан способ полуколичественного определения количества жизнеспособных клеток посредством комбинирования культивирования микроорганизмов и ПЦР с детекцией в реальном времени.
Кроме того, в качестве способа для еще более четкого различения жизнеспособных клеток и поврежденных клеток микроорганизмов описан способ, описанный в патентном документе 4. Этот способ представляет собой способ, включающий стадию добавления сшивающего линкера, способного к образованию поперечных сшивок ДНК при облучении тестируемого образца светом с длиной волны от 350 нм до 700 нм, стадию облучения тестируемого образца, в который добавляют сшивающий линкер светом с длиной волны от 350 нм до 700 нм, стадию удаления сшивающего линкера, содержащегося в тестируемом образце, облученном светом, стадию добавления в тестируемый образец, из которого удаляют сшивающий линкер, среды и инкубации тестируемого образца, стадию повторного добавления к инкубируемому тестируемому образцу сшивающего линкера, способного к образованию поперечных сшивок ДНК при облучении светом с длиной волны от 350 нм до 700 нм, стадию облучения тестируемого образца, в который добавляют сшивающий линкер, светом с длиной волны от 350 нм до 700 нм, стадию выделения ДНК из тестируемого образца и амплификации области-мишени выделенной ДНК способом амплификации нуклеиновых кислот и стадию анализа продукта амплификации.
При этом допускается возможность, что при амплификации нуклеиновых кислот посредством ПЦР подавлять активность ингибирования ингибитора ПЦР или способствовать реакциям ПЦР может альбумин (непатентный документ 2). Кроме того, также полагают, что реакции ПЦР ингибирует кальций, но ингибирование ПЦР кальцием можно сделать приемлемым, добавляя ионы магния (непатентный документ 3).
Кроме того, описан способ проведения реакций ПЦР с использованием в качестве матричной бактериальной ДНК, где реакции ПЦР проводят без выделения ДНК из бактерий (непатентный документ 4, патентный документ 5). В патентном документе 5 описано, что в бактериях в способе фингерпринтинга ДНК у бактерий проводят ПЦР с амплификацией случайных последовательностей и в качестве компонентов состава буфера для синтеза нуклеиновой кислоты указаны фосфаты и додецилсульфаты.
Ссылки на известный уровень техники
Патентные документы
Патентный документ 1: Внутренняя выложенная публикация японского перевода заявки PCT (KOHYO) № 2003-530118.
Патентный документ 2: Международная патентная публикация WO2002/052034.
Патентный документ 3: Международная патентная публикация WO2007/094077.
Патентный документ 4: Международная патентная публикация WO2009/022558.
Патентный документ 5: Международная патентная публикация WO2004/104196.
Непатентные документы
Непатентный документ 1: Rudi, K., et al., Letters in Applied Microbiology, 2005, Vol. 40, pp.301-306.
Непатентный документ 2: Forbes, B. E., et al., Journal of Clinical Microbiology, 1996, 34 (9), pp.2125-2128.
Непатентный документ 3: Bickley, J., et al., Letter in Applied Microbiology, 1996, 22, pp.153-158.
Непатентный документ 4: Kimberly, A., et al., BioTechniques, 31, 2001, pp.598-607.
Описание изобретения
Задачи, подлежащие решению посредством изобретения
С помощью указанного выше способа с использованием ингибитора топоизомеразы и/или ингибитора ДНК-гиразы или сшивающего линкера можно селективно с высокой чувствительностью детектировать жизнеспособные клетки микроорганизмов, особенно жизнеспособные клетки бактерий Klebsiella, Citrobacter, Listeria, Salmonella и т.д. Однако желателен новый улучшенный способ, особенно способ для высокочувствительной или высокоточной детекции жизнеспособных клеток бактерий Escherichia или Salmonella.
Целью настоящего изобретения является создание нового способа для более селективной детекции жизнеспособных клеток микроорганизмов, содержащихся в продукте питания или в биологическом образце в сравнении с погибшими клетками или поврежденными клетками микроорганизма, и набора для проведения такого способа.
Средства решения задач
Авторы настоящего изобретения провели широкие исследования на способах различения жизнеспособных и погибших микроорганизмов, которые можно использовать в различных способах стерилизации и которые подходят для санитарной проверки продуктов питания с высокой чувствительностью детекции, и на способах детекции специфических патогенов у пациентов с инфекцией в госпитальной и клинической практике. В результате авторы изобретения обнаружили, что высокочувствительное различие может быть достигнуто посредством добавления в тестируемый образец средства, способного к ковалентному связыванию с ДНК или РНК при облучении светом с длиной волны от 350 нм до 700 нм; облучения тестируемого образца светом с длиной волны от 350 нм до 700 нм; добавления средства для подавления действия вещества, ингибирующего амплификацию нуклеиновых кислот, соли магния и соли органической кислоты или соли фосфорной кислоты и амплификации хромосомной ДНК микроорганизмов из клеток посредством реакции амплификации нуклеиновых кислот. Таким образом, осуществляют настоящее изобретение.
А именно, настоящее изобретение относится к способу детекции жизнеспособных клеток микроорганизмов в тестируемом образце, различая жизнеспособные клетки и погибшие клетки или поврежденные клетки, который включает стадии:
a) добавления в тестируемый образец средства, способного к ковалентному связыванию с ДНК или РНК при облучении светом с длиной волны от 350 нм до 700 нм;
b) облучения тестируемого образца, в который добавляют средство, светом с длиной волны от 350 нм до 700 нм;
c) амплификации области-мишени ДНК или РНК микроорганизма, содержащегося в тестируемом образце, способом амплификации нуклеиновых кислот в присутствии средства подавления действия вещества, ингибирующего амплификацию нуклеиновых кислот, без выделения нуклеиновых кислот из клеток; и
d) анализа продуктов амплификации.
В предпочтительном варианте осуществления настоящего изобретения амплификацию области-мишени проводят в клетках микроорганизмов.
В предпочтительном варианте осуществления указанного выше способа на указанной выше стадии c) амплификацию области-мишени проводят в присутствии одного или нескольких классов, выбранных из поверхностно-активного вещества, соли магния и соли органической кислоты или соли фосфорной кислоты.
В предпочтительном варианте осуществления указанного выше способа перед указанной выше стадией c) повторно проводят стадии a) и b).
В предпочтительном варианте осуществления указанного выше способа, перед указанной выше стадий a), проводят следующую стадию e):
e) обработка тестируемого образца ферментом с активностью разрушения клеток, отличных от клеток микроорганизма, коллоидных частиц белка, липидов или сахаридов, присутствующих в тестируемом образце.
В предпочтительном варианте осуществления указанного выше способа фермент выбран из протеазы, фермента, разрушающего липиды, и фермента, разрушающего сахариды.
В предпочтительном варианте осуществления указанного выше способа тестируемый образец представляет собой любой из продукта питания, биологического образца, питьевой воды, промышленных вод, воды из окружающей среды, сточных вод, почвы и мазка.
В предпочтительном варианте осуществления указанного выше способа микроорганизм представляет собой бактерию или вирус.
В предпочтительном варианте осуществления указанного выше способа бактерия представляет собой грамотрицательную бактерию.
В предпочтительном варианте осуществления указанного выше способа средство, способное к ковалентному связыванию с ДНК или РНК при облучении светом с длиной волны от 350 нм до 700 нм, выбрано из моноазида этидия, диазида этидия, моноазида пропидия, псоларена, 4,5',8-триметилпсоларена и 8-метоксипсоларена.
В предпочтительном варианте осуществления указанного выше способа средство для подавления действия вещества, ингибирующего амплификацию нуклеиновых кислот, состоит из одного или нескольких классов, выбранных из альбумина, декстрана, белка гена 32 T4, ацетамида, бетаина, диметилсульфоксида, формамида, глицерина, полиэтиленгликоля, соевого ингибитора трипсина, α2-макроглобулина, тетраметилхлорида аммония, лизоцима, фосфорилазы и лактатдегидрогеназы.
В предпочтительном варианте осуществления указанного выше способа соль органической кислоты выбрана из соли уксусной кислоты, соли пропионовой кислоты и соли лимонной кислоты.
В предпочтительном варианте осуществления указанного выше способа соль фосфорной кислоты представляет собой соль пирофосфорной кислоты.
В предпочтительном варианте осуществления указанного выше способа область-мишень представляет собой область-мишень, содержащую от 50 до 5000 нуклеотидов.
В предпочтительном варианте осуществления указанного выше способа область-мишень представляет собой область-мишень, соответствующую гену, выбранному из гена 5S рРНК, гена 16S рРНК, гена 23S рРНК и гена тРНК ДНК тестируемого образца.
В предпочтительном варианте осуществления указанного выше способа способ амплификации нуклеиновых кислот представляет собой ПЦР, LAMP, SDA, LCR, TMA, TRC, HC или способ с применением микропанелей.
В предпочтительном варианте осуществления указанного выше способа ПЦР проводят в виде ПЦР с детекцией в реальном времени с одновременном проведением ПЦР и анализа продукта амплификации.
В предпочтительном варианте осуществления указанного выше способа анализ продуктов амплификации проводят с использованием стандартной кривой, представляющей собой зависимость количества микроорганизмов и продуктов амплификации и полученной с использованием стандартных образцов микроорганизмов.
В качестве набора по настоящему изобретению предоставлен набор для детекции жизнеспособных клеток микроорганизмов в тестируемом образце при различении жизнеспособных клеток и погибших клеток или поврежденных клеток способом амплификации нуклеиновых кислот, содержащий следующие компоненты:
1) средство, способное к ковалентному связыванию с ДНК или РНК при облучении светом с длиной волны от 350 нм до 700 нм;
2) средство для подавления действия вещества, ингибирующего амплификацию нуклеиновых кислот; и
3) праймер или праймеры для амплификации области-мишени ДНК или РНК микроорганизма, подлежащего детекции способом амплификации нуклеиновых кислот.
В предпочтительном варианте осуществления указанного выше набора набор дополнительно содержит один или несколько классов, выбранных из поверхностно-активного вещества, соли магния и соли органической кислоты или соли фосфорной кислоты.
В предпочтительном варианте осуществления указанного выше набора набор дополнительно содержит фермент с активностью разрушения клеток, отличных от клеток микроорганизма, коллоидных частиц белка, липидов или сахаридов, присутствующих в тестируемом образце.
В предпочтительном варианте осуществления указанного выше набора способ амплификации нуклеиновых кислот представляет собой ПЦР, ПЦР-РВ, LAMP, SDA, LCR, TMA, TRC, HC или способ с использованием микропанелей.
В предпочтительном варианте осуществления указанного выше набора средство, способное к ковалентному связыванию с ДНК или РНК при облучении светом с длиной волны от 350 нм до 700 нм, выбрано из моноазида этидия, диазида этидия, моноазида пропидия, псоларена, 4,5',8-триметилпсоларена и 8-метоксипсоларена.
В предпочтительном варианте осуществления указанного выше набора средство для подавления действия вещества, ингибирующего амплификацию нуклеиновых кислот, состоит из одного или нескольких классов, выбранных из альбумина, декстрана, белка гена 32 T4, ацетамида, бетаина, диметилсульфоксида, формамида, глицерина, полиэтиленгликоля, соевого ингибитора трипсина, α2-макроглобулина, тетраметилхлорида аммония, лизоцима, фосфорилазы и лактатдегидрогеназы.
В предпочтительном варианте осуществления указанного выше набора соль органической кислоты выбрана из соли уксусной кислоты, соли пропионовой кислоты и соли лимонной кислоты.
В предпочтительном варианте осуществления указанного выше набора соль фосфорной кислоты представляет собой соль пирофосфорной кислоты.
В предпочтительном варианте осуществления указанного выше набора фермент выбран из протеазы, фермента, разрушающего липиды, и фермента, разрушающего сахариды.
Краткое описание чертежей
[Фиг. 1] Фотографии электрофореза продуктов амплификации ПЦР, полученных способом по настоящему изобретению, "жизнеспособные" означает жизнеспособные клетки, "поврежденные" означает поврежденные клетки.
[Фиг. 2] Фотографии электрофореза, демонстрирующие результаты детекции жизнеспособных клеток микроорганизмов, проводимой способом по настоящему изобретению.
[Фиг. 3] Фотографии электрофореза, демонстрирующие результаты детекции жизнеспособных клеток микроорганизмов, проводимой общепринятым способом. "Жизнеспособные" представляет собой жизнеспособные клетки, "поврежденные" представляет собой поврежденные клетки.
[Фиг. 4] Микрофотография флуоресценции и стереоскопическая микрофотография не подвергнутой нагреванию суспензии бактерий Enterobacter sakazakii в физиологическом растворе.
[Фиг. 5] Микрофотография флуоресценции и стереоскопическая микрофотография супернатанта не подвергнутой нагреванию суспензии бактерий Enterobacter sakazakii в физиологическом растворе.
[Фиг. 6] Микрофотография флуоресценции и стереоскопическая микрофотография суспензии бактерий Enterobacter sakazakii в физиологическом растворе после повторения термических циклов.
[Фиг. 7] Микрофотография флуоресценции и стереоскопическая микрофотография супернатанта суспензии бактерий Enterobacter sakazakii в физиологическом растворе после повторения термических циклов.
[Фиг. 8] Микрофотография флуоресценции и стереоскопическая микрофотография бактерии Enterobacter sakazakii, суспендированной в растворе средства для предварительной обработки без нагревания.
[Фиг. 9] Микрофотография флуоресценции и стереоскопическая микрофотография супернатанта бактерии Enterobacter sakazakii, суспендированной в растворе средства для предварительной обработки без нагревания.
[Фиг. 10] Микрофотография флуоресценции и стереоскопическая микрофотография бактерии Enterobacter sakazakii, суспендированной в растворе средства для предварительной обработки после повторения термических циклов.
[Фиг. 11] Микрофотография флуоресценции и стереоскопическая микрофотография супернатанта бактерии Enterobacter sakazakii, суспендированной в растворе средства для предварительной обработки после повторения термических циклов.
[Фиг. 12] Диаграммы, демонстрирующие результаты проточной цитометрии суспензий бактерии Enterobacter sakazakii в физиологическом растворе или их супернатантов в состоянии без нагревания или после повторения термических циклов.
[Фиг. 13] Диаграммы, демонстрирующие результаты проточной цитометрии бактерии Enterobacter sakazakii, суспендированной в растворе средства для предварительной обработки, или его супернатанта в состоянии без нагревания или после повторения термических циклов.
[Фиг. 14] Фотография электрофореза продуктов амплификации генов 16S-23S рРНК, полученных способом по настоящему изобретению с использованием бактерии Enterobacter sakazakii, обработанной различными фиксирующими растворами. Значения Ct указаны в виде среднего и SD, а SD указан в круглых скобках.
L: Лестничный маркер ДНК с шагом 100 п.н.
A: Фиксирующий раствор A
B: Фиксирующий раствор B
C: Фиксирующий раствор C
S: Отсутствие фиксации
[Фиг. 15] Фотография электрофореза продукта амплификации гена ompA, полученного способом по настоящему изобретению с использованием бактерии Enterobacter sakazakii, обработанной различными фиксирующими растворами. Значения Ct указаны в виде среднего и SD, а SD указан в круглых скобках.
A: Фиксирующий раствор A
B: Фиксирующий раствор B
L: Лестничный маркер ДНК с шагом 100 п.н.
[Фиг. 16] Фотографии электрофореза, полученные перед и после ПЦР (реакция амплификации генов 16S-23S рРНК) с использованием бактерии Enterobacter sakazakii.
Дорожки 2 и 3: супернатант реакционной смеси ПЦР.
Дорожки 5 и 6: ДНК, выделенная из осадка дважды отмытого с помощью центрифугирования после реакции ПЦР.
Дорожки 7 и 8: ДНК, непосредственно выделенная из клеток.
Дорожки 9 и 10: ДНК, выделенная из клеток, фактически используемых в тесте, непосредственно перед ПЦР.
Дорожки 13 и 14: ДНК, выделенная из клеток, отмытых после добавления продукта ПЦР.
L: Лестничный маркер ДНК с шагом 100 п.н.
B: Фиксирующий раствор B
S: Отсутствие фиксации
[Фиг. 17] Фотографии электрофореза суспензий бактерии Enterobacter sakazakii, подвергнутой тепловой обработке в присутствии физиологического раствора или средства для предварительной обработки, и их супернатантов после центрифугирования.
L: Лестничный маркер ДНК с шагом 100 п.н.
Способы осуществления изобретения
Далее в настоящем документе подробно описаны предпочтительные варианты осуществления настоящего изобретения. Однако настоящее изобретение не ограничено приведенными ниже предпочтительными вариантами осуществления, и его можно без ограничений модифицировать в объеме настоящего изобретения. Если не указано иначе, проценты в настоящем описании выражены в виде процентов по массе.
Мишень для детекции способом по настоящему изобретению включает все классы нуклеиновых кислот, а именно: одноцепочечная ДНК, двухцепочечная ДНК, одноцепочечная РНК и двухцепочечная РНК, при условии, что мишень в конечном итоге можно амплифицировать. Из них мишенью для детекции предпочтительно является ДНК, особенно предпочтительно двухцепочечная ДНК.
<1> Способ по настоящему изобретению
Способ по настоящему изобретению представляет собой способ детекции жизнеспособных клеток микроорганизма в тестируемом образце при различении жизнеспособных клеток и погибших клеток или поврежденных клеток, который включает стадии:
a) добавления средства, способного к ковалентному связыванию с ДНК или РНК при облучении светом с длиной волны от 350 нм до 700 нм в тестируемом образце;
b) облучения тестируемого образца, в который добавляют средство, светом с длиной волны от 350 нм до 700 нм;
c) амплификации области-мишени ДНК или РНК микроорганизма, содержащегося в тестируемом образце, способом амплификации нуклеиновых кислот в присутствии средства подавления действия вещества, ингибирующего амплификацию нуклеиновых кислот, без выделения нуклеиновых кислот из клеток; и
d) анализ продуктов амплификации.
В описании настоящего изобретения термин "тестируемый образец" относится к объекту, содержащему жизнеспособные клетки микроорганизма для детекции. Тестируемый образец конкретно не ограничен при условии, что присутствие микроорганизма можно детектировать с помощью амплификации конкретной области хромосомной ДНК или РНК способом амплификации нуклеиновых кислот. Его предпочтительные примеры включают продукты питания, биологические образцы, питьевую воду, промышленные воды, воду из окружающей среды, сточные воды, почву, мазки и т.д.
В частности, предпочтительные примеры продуктов питания включают: напитки, такие как безалкогольные напитки, газированные безалкогольные напитки, жидкие пищевые добавки, напитки на основе фруктовых соков и молочнокислые напитки (включая концентраты и порошки этих напитков); мороженные кондитерские изделия, такие как мороженое, фруктовое мороженое и ледяная стружка; молочные продукты, такие как переработанное молоко, молочные напитки, молочнокислые продукты и масло; энтеральное питание, жидкая пища, молоко для младенцев, спортивные напитки; функциональное питание, такое как питание для применения при определенном состоянии здоровья, и пищевые добавки, и т.д.
Примеры биологических образцов включают образцы крови, образцы мочи, образцы цереброспинальной жидкости, образцы синовиальной жидкости, образцы плеврального выпота, образцы мокроты, образцы кала, образцы слизистой полости носа, образцы слизистой гортани, образцы после промывания желудка, образцы гноя, образцы слизистой оболочки кожи, образцы слизистой полости рта, образцы слизистой оболочки органов дыхания, образцы слизистой оболочки пищеварительных органов, образцы конъюнктивы глаза, образцы плаценты, образцы репродуктивных клеток, образцы родового канала, образцы материнского молока, образцы слюны, рвоты, содержимого волдырей и т.д.
Примеры воды из окружающей среды включают водопроводную воду, грунтовую воду, речную воду, дождевую воду и т.д.
В настоящем изобретении тестируемый образец может представлять собой любой из продуктов питания, биологических образцов, питьевой воды, промышленных вод, воды из окружающей среды, сточных вод, почвы, мазков, как указано выше, и т.д. или может представлять собой любой из их разведенных или концентрированных продуктов или любой из продуктов, получаемых с помощью предварительной обработки, отличной от способа по настоящему изобретению. Примеры предварительной обработки включают тепловую обработку, фильтрование, центрифугирование и т.д.
Кроме того, в тестируемом образце можно удалять примеси, такие как клетки, отличные от микроорганизмов, коллоидные частицы белка, липиды, сахариды и т.д., или снижать их содержание посредством обработки ферментов с разрушающей их активностью или т.п. В случае, когда тестируемый образец представляет собой любой из молока, молочных продуктов и продуктов питания, получаемых из молока или молочных продуктов, примеры клеток в тестируемом образце, отличных от микроорганизмов, включают лейкоциты коровы, эпителиоциты молочной железы и т.д. При этом, в случае, когда тестируемый образец представляет собой любой из биологических образцов, таких как образцы крови, образцы мочи, образцы цереброспинальной жидкости, образцы синовиальной жидкости и образцы плеврального выпота, примеры клеток включают эритроциты, лейкоциты (такие как гранулоциты, нейтрофилы, базофилы, моноциты и лимфоциты), тромбоциты и т.д.
Фермент при условии, что он может разрушать примеси и не повреждает жизнеспособные клетки микроорганизма, подлежащего детекции, конкретно не ограничен, и его примеры включают, например, ферменты, разрушающие липиды, ферменты, разрушающие белки и ферменты, разрушающие сахариды. Из них можно использовать один фермент, или два, или более ферментов, но предпочтительно использовать и фермент, разрушающий липиды, и фермент, разрушающий белки, или все из фермента, разрушающего липиды, фермента, разрушающего белки и фермента, разрушающего сахариды.
Примеры ферментов, разрушающих липиды, включают липазу, фосфатазу и т.д., примеры ферментов, разрушающих белки, включают сериновую протеазу, цистеиновую протеазу, протеиназу K, проназу и т.д., и примеры фермента, разрушающего сахариды, включают амилазу, целлюлазу и т.д.
"Микроорганизм" представляет собой объект, подлежащий детекции способом по настоящему изобретению, и он конкретно не ограничен при условии, что его можно детектировать способом амплификации нуклеиновой кислоты, и средство, способное к ковалентному связыванию с ДНК или РНК при облучении светом с длиной волны от 350 нм до 700 нм действует на жизнеспособные клетки микроорганизма способом, отличным от способа для погибших клеток и поврежденных клеток микроорганизма. Предпочтительные примеры включают бактерии, грибы, дрожжи, вирусы и т.д. Бактерии включают грамположительные бактерии и грамотрицательные бактерии. Примеры грамположительных бактерий включают бактерии Staphylococcus, такие как Staphylococcus epidermidis, бактерии Streptococcus, такие как Streptococcus pneumoniae, бактерии Listeria, такие как Listeria monocytogenes, бактерии Bacillus, такие как Bacillus cereus и Bacillus anthracis, бактерии Mycobacterium, такие как Mycobacterium tuberculosis, Mycobacterium bovis и Mycobacterium avium, бактерии Clostridium, такие как Clostridium botulinum и Clostridium perfringens и т.д. Примеры грамотрицательных бактерий включают энтеробактерии, из которых типичными примерами являются бактерии Escherichia, такие как Escherichia coli, бактерии Enterobacter, такие как Enterobacter sakazakii, бактерии Citrobacter, такие как Citrobacter koseri, и бактерии Klebsiella, такие как Klebsiella oxytoca, и бактерии Salmonella, бактерии Vibrio, бактерии Pseudomonas, бактерии Legionella и т.д. Примеры вирусов включают вирусы с оболочкой, такие как вирус гриппа, и вирусы без оболочки, а только с нуклеокапсидом, такие как норовирусы, ротавирусы и аденовирусы.
В отношении вирусов существует известный способ определения активации и инактивации вируса в воде, в котором фотореактивному линкеру, сшивающему нуклеиновые кислоты (EMA), позволяют взаимодействовать с тестируемым образцом, а затем посредством ПЦР-РВ определяют только активированные вирусы (http://www.recwet.t.u-tokyo.ac.jp/furumailab/j/sotsuron/H21/H21sotsuron.html, Development of ethidium monoazide (EMA)-RT-PCR for selective detection of enteric viruses, 15th International Symposium on Health-Related Water Microbiology, (May 31-Jun 05, 2009, Ursulines Conference Centre, Naxos, Greece)). То есть полагают, что EMA не проникает в активированные вирусы, но проникает только в инактивированные вирусы с сильными физическими поврежденными нуклеокапсидами, и, таким образом, с применением EMA можно различать активированные вирусы (жизнеспособные) и неактивированные вирусы (погибшие). Таким образом, полагают, что настоящее изобретение можно применять не только для бактерий, нитевидных грибов и дрожжей, но также и для вирусов.
В настоящем изобретении "жизнеспособная клетка" относится к клетке в состоянии, в котором клетка может пролиферировать и демонстрирует метаболическую активность микроорганизма (жизнеспособное-и-культивируемое состояние), когда ее культивируют в основном в предпочтительных условиях культивирования, и по существу у клетки отсутствует повреждение клеточной стенки. В метаболической активности, указанной выше, можно привести примеры активности АТФ, эстеразной активности и т.д. В настоящем изобретении вирусные частицы для удобства также называют "клетками". В отношении вирусов, "жизнеспособная клетка" относится к вирусу в состоянии, когда он может инфицировать клетку млекопитающего и пролиферировать.
"Погибшая клетка" представляет собой клетку в состоянии, когда она не может пролиферировать и не проявляет метаболической активности (мертвое состояние), даже если ее культивируют в оптимальных условиях культивирования. Кроме того, она находится в состоянии, в котором хотя структура клеточной стенки сохраняется, сама клеточная стенка сильно повреждена, и средства, окрашивающие ядро, демонстрирующие слабую проницаемость, такие как йодид пропидия, могут входить или проникать через клеточную стенку. В отношении вирусов "погибшая клетка" относится к вирусу в состоянии, когда он не может инфицировать клетку млекопитающего.
"Поврежденная клетка" (поврежденная клетка или жизнеспособная-но-не культивируемая клетка) представляет собой клетку в состоянии, в котором даже когда ее культивируют в основном в предпочтительных условиях культивирования, почти не пролиферирует вследствие того, что она повреждена в результате искусственного стресса или стресса вследствие воздействия окружающей среды, и она демонстрирует метаболическую активность на меньшем уровне по сравнению с жизнеспособной клеткой, но на значительном уровне по сравнению с погибшей клеткой. В отношении вирусов "поврежденная клетка" относится к вирусу в состоянии, когда даже если он инфицирует клетку млекопитающего, он не может пролиферировать в клетке.
В этом описании, если конкретно не указано, "жизнеспособная клетка", "погибшая клетка" и "поврежденная клетка" означают соответственно жизнеспособную клетку, погибшую клетку и поврежденную клетку микроорганизма.
Привлекает внимание детекция бактерий, демонстрирующих состояние повреждения клеток вследствие умеренной тепловой обработки или введения антибиотиков, в частности в областях санитарной проверки продуктов питания и клинического тестирования, и настоящее изобретение относится к способу детекции микроорганизмов, позволяющему не только детектировать жизнеспособные клетки, но также и различать жизнеспособные клетки и погибшие клетки или поврежденные клетки.
Единицы количества клеток, как правило, представляют собой количество клеток (клетки)/мл для всех жизнеспособных клеток, поврежденных клеток и погибших клеток. В этом описании количество клеток представлено логарифмом, и "log10 клеток/мл" означает 10a клеток/мл.
Количество жизнеспособных клеток можно аппроксимировать количеством образуемых колоний (КОЕ/мл (колониеобразующие единицы/мл)), получаемых при культивировании клеток в оптимальных условиях на подходящей среде для чашек. Стандартный образец поврежденных клеток микроорганизма можно получать, например, подвергая суспензию жизнеспособных клеток тепловой обработке, например тепловой обработке в кипящей воде. Количество поврежденных клеток в таком образце можно аппроксимировать КОЕ/мл в суспензии жизнеспособных клеток перед тепловой обработкой. Хотя время тепловой обработки в кипящей воде для получения поврежденных клеток варьирует в зависимости от типа микроорганизма, поврежденные клетки бактерий, описанные в примерах, можно получать, например, тепловой обработкой в течение приблизительно 50 секунд. Кроме того, стандартный образец поврежденных клеток микроорганизма также можно получать с помощью обработки антибиотиком. В таком случае количество поврежденных клеток можно аппроксимировать на основе количества образуемых колоний (КОЕ/мл), наблюдаемых при культивировании клеток в оптимальных условиях на подходящей среде для чашек, т.е. для расчета количества поврежденных клеток, обработанных антибиотиком, суспензию жизнеспособных клеток обрабатывают антибиотиком, затем антибиотик удаляют, измеряют светопроницаемость суспензии для видимого света (длина волны: 600 нм), т.е. мутность суспензии, и измеренную мутность можно сравнивать с суспензиями жизнеспособных клеток с известной плотностью жизнеспособных клеток.
В отношении вирусов единицы количества клеток представлены бляшкообразующими единицами (бое или БОЕ).
Способ по настоящему изобретению предназначен для детекции жизнеспособных клеток микроорганизмов, а клетками микроорганизмов, отличающимися от жизнеспособных клеток, могут быть поврежденные клетки или погибшие клетки.
В настоящем изобретении, "детекция жизнеспособных клеток" включает определение наличия или отсутствия жизнеспособных клеток в тестируемом образце и определение количества жизнеспособных клеток в тестируемом образце. Количество жизнеспособных клеток не ограничено абсолютным количеством и может представлять собой относительное количество относительно количества в контрольном образце. Кроме того, "детекция жизнеспособных клеток при различении жизнеспособных клеток и погибших клеток или поврежденных клеток" означает более селективную детекцию жизнеспособных клеток в сравнении с погибшими клетками или поврежденными клетками. Кроме того, "различение жизнеспособных клеток и погибших клеток или поврежденных клеток" включает отличие жизнеспособных клеток и от погибших клеток, и от поврежденных клеток.
Ниже в настоящем документе будет описан способ по настоящему изобретению на каждой стадии. Как описано выше, способ по настоящему изобретению перед описанными ниже стадиями может включать стадию обработки тестируемого образца ферментом с активностью разрушения клеток, отличных от клеток микроорганизма, коллоидных частиц белков, липидов или сахаридов, присутствующих в тестируемом образце.
(1) Стадия a)
В тестируемый образец добавляют средство, способное к ковалентному связыванию с ДНК или РНК при облучении светом с длиной волны от 350 нм до 700 нм. То есть микроорганизмы в тестируемом образце обрабатывают средством.
Как описано ниже, средство интеркалирует в двухцепочечную ДНК или РНК и при облучении светом ковалентно связывается с ДНК или РНК со сшиванием молекул. Кроме того, полагают, что средство при облучении светом ковалентно связывается с одноцепочечной ДНК или РНК с ингибированием реакции ПЦР. Далее средство можно также просто обозначать как "сшивающий линкер".
Сшивающий линкер предпочтительно обладает отличным действием на жизнеспособные клетки микроорганизма по сравнению с поврежденными или погибшими клетками микроорганизма и с соматическими клетки, такими как лейкоциты коровы, лейкоциты или тромбоциты. Более конкретно, сшивающий линкер предпочтительно представляет собой сшивающий линкер, который может более легко проходить через клеточные стенки поврежденных или погибших клеток микроорганизма и клеточные мембраны соматических клеток, таких как лейкоциты коровы, лейкоциты или тромбоциты, по сравнению с клеточными стенками жизнеспособных клеток микроорганизма.
Примеры сшивающего линкера включают моноазид этидия, диазид этидия, псоларен, 4,5',8-триметилпсоларен, 8-метоксипсоларен, моноазид пропидия и т.д. Можно использовать один класс из этих сшивающих линкеров отдельно или можно использовать два или более класса из них в комбинации.
Можно устанавливать подходящие условия обработки сшивающим линкером. Например, условия для более легкого различения жизнеспособных клеток микроорганизма и погибших или поврежденных клеток микроорганизма можно определять с помощью: добавления в суспензии жизнеспособных клеток микроорганизма, подлежащего детекции, и погибших или поврежденных клеток микроорганизма различных концентраций сшивающего линкера; обеспечения нахождения суспензии в покое в течение различных периодов времени; разделения клеток с помощью центрифугирования и анализа клеток способом амплификации нуклеиновых кислот. Кроме того, условия для более легкого различения жизнеспособных клеток микроорганизма и различных клеток можно определять с помощью: добавления в суспензии жизнеспособных клеток микроорганизма, подлежащего детекции, и соматических клеток, таких как лейкоциты коровы или тромбоциты, различных концентраций сшивающего линкера; обеспечения нахождения суспензии в покое в течение различных периодов времени; разделения клеток и различных клеток с помощью центрифугирования и анализа клеток способом амплификации нуклеиновых кислот. В частности, примерами являются: условия обработки моноазидом этидия в конечной концентрации от 1 до 100 мкг/мл в течение периода от 5 минут до 48 часов при температуре от 4 до 10°C; условия обработки диазидом этидия в конечной концентрации от 1 до 100 мкг/мл в течение периода от 5 минут до 48 часов при температуре от 4 до 10°C; условия обработки моноазидом пропидия в конечной концентрации от 1 до 100 мкг/мл в течение периода от 5 минут до 48 часов при температуре от 4 до 10°C; условия обработки псолареном в конечной концентрации от 1×10-5 до 10 мкг/мл в течение периода от 5 минут до 48 часов при температуре от 25 до 37°C; условия обработки 4,5',8-триметилпсолареном в конечной концентрации от 1×10-5 до 10 мкг/мл в течение периода от 5 минут до 48 часов при температуре от 25 до 37°C; и условия обработки 8-метоксипсолареном в конечной концентрации от 1×10-5 до 10 мкг/мл в течение периода от 5 минут до 48 часов при температуре от 25 до 37°C.
(2) Стадия b)
Затем каждый тестируемый образец, содержащий сшивающий линкер, облучают светом с длиной волны от 350 нм до 700 нм.
Сшивающий линкер может более легко проходить через клеточные стенки погибших клеток и поврежденных клеток по сравнению с клеточными стенками жизнеспособных клеток микроорганизма. Таким образом, полагают, что сшивающий линкер по существу не проходит через клеточные стенки жизнеспособных клеток микроорганизма, но проходит через клеточные стенки поврежденных или погибших клеток микроорганизма или погибших соматических клеток при условии, что обработку проводят в пределах указанных выше периодов времени. Полагают, что в результате сшивающий линкер попадает в погибшие соматические клетки, погибшие и поврежденные клетки микроорганизма, а затем формирует водородные связи c хромосомной ДНК или РНК, а затем сшивающий линкер, облученный светом с длиной волны от 350 нм до 700 нм, сшивает молекулы ДНК или ковалентно связывается с РНК и, в результате, происходит деформация хромосомной ДНК или модификация РНК сшивающим линкером, в конечном итоге приводящая к разрушению (фрагментации/расщеплению) хромосомной ДНК или РНК, которые перестают служить в качестве матрицы в реакции амплификации нуклеиновых кислот.
Свет с длиной волны от 350 нм до 700 нм может включать по меньшей мере свет с длиной волны от 350 нм до 700 нм, и свет может представлять собой свет с одной длиной волны или свет с несколькими длинами волны. Кроме того, все составляющие света могут находиться в диапазоне от 350 нм до 700 нм или свет может включать коротковолновый свет с длиной волны короче 350 нм и/или длинноволновый свет с длиной волны более 700 нм. Пик распределения интенсивностей предпочтительно находится в диапазоне от 350 нм до 700 нм. Предпочтительно свет не включает коротковолновой составляющей на таком уровне, что хромосомная ДНК микроорганизма расщепляется только под действием облучения светом.
Когда хромосомная ДНК поврежденных или погибших клеток разрушается с большим предпочтением, чем хромосомная ДНК жизнеспособных клеток микроорганизма, происходит амплификация области-мишени хромосомной ДНК жизнеспособных клеток способом амплификации нуклеиновых кислот, тогда как область-мишень поврежденных или погибших клеток разрушается (расщепляется), что приводит к ингибированию реакций амплификации нуклеиновых кислот. В результате жизнеспособные клетки микроорганизма можно детектировать с большей селективностью, чем поврежденные или погибшие клетки.
Кроме того, когда сшивающий линкер с большим предпочтением модифицирует РНК поврежденных или погибших клеток, чем РНК жизнеспособных клеток микроорганизма, происходит амплификация области-мишени РНК жизнеспособных клеток способом амплификации нуклеиновых кислот, тогда как область-мишень РНК поврежденных или погибших клеток модифицируется, что приводит к ингибированию реакций амплификации нуклеиновых кислот. В результате жизнеспособные клетки микроорганизма можно детектировать с большей селективностью, чем поврежденные или погибшие клетки.
В предпочтительном варианте осуществления настоящего изобретения сшивающий линкер представляет собой моноазид этидия, и способ включает стадию облучения тестируемого образца, в который добавляют моноазид этидия, светом с длиной волны от 350 нм до 700 нм. Моноазид этидия (EMA) может более легко проходить через клеточные стенки поврежденных или погибших клеток по сравнению с клеточными стенками жизнеспособных клеток микроорганизма. Таким образом, полагают, что EMA по существу не проходит через клеточные стенки жизнеспособных клеток микроорганизма, но проходит через клеточные стенки поврежденных или погибших клеток микроорганизма или клеточные мембраны погибших соматических клеток.
Следует отметить, что в случае, когда лейкоциты и тромбоциты в крови являются жизнеспособными клетками, EMA может более легко проходить через клеточные стенки клеток в стерилизованной воде или гипотоническом солевом растворе.
Относительно ДНК, EMA, в частности, проникает в погибшие соматические клетки и поврежденные и погибшие клетки микроорганизма и случайным образом интеркалирует в ядерную ДНК, и при облучении светом с длиной волны от 350 нм до 700 нм интеркалированный EMA преобразуется в нитрен и ковалентно связывается с ядерной ДНК, сшивая молекулы ДНК. Затем, как полагают, EMA, который в хромосомной ДНК во многих положениях ковалентно связывается с основаниями и дезоксирибозой, вызывает большие деформации в хромосомной ДНК, что приводит к разрушению (фрагментации) хромосомной ДНК.
Кроме того, относительно двухцепочечной РНК (включая частично двухцепочечную РНК), EMA проникает в погибшие соматические клетки и поврежденные и погибшие клетки микроорганизма и случайным образом интеркалирует в РНК, а затем при облучении светом с длиной волны от 350 нм до 700 нм интеркалированный EMA преобразуется в нитрен и ковалентно связывается с РНК с сшиванием молекул РНК. Затем, как полагают, EMA, который во многих положениях ковалентно связывается с основаниями РНК, вызывает большие деформации в РНК, что приводит к разрушению (фрагментации) РНК.
Кроме того, относительно одноцепочечной ДНК или РНК, полагают, что EMA проникает в погибшие соматические клетки и поврежденные и погибшие клетки микроорганизма, а затем при облучении светом с длиной волны от 350 нм до 700 нм преобразуется в нитрен, который ковалентно связывается с ДНК или РНК.
Также можно использовать сшивающий линкер, отличный от моноазида этидия, при условии, что сшивающий линкер может более легко проходить через клеточные стенки поврежденных или погибших клеток по сравнению с клеточными стенками жизнеспособных клеток микроорганизма и может сшивать ДНК или ковалентно связывается с РНК при облучении светом с длиной волны от 350 нм до 700 нм (длинноволновой ультрафиолетовый свет или видимый свет), таким образом разрушая хромосомную ДНК или модифицируя РНК.
Можно устанавливать подходящие условия обработки EMA. Например, условия, которые обеспечивают простое различение жизнеспособных клеток микроорганизма и погибших клеток и поврежденных клеток, можно определять, добавляя в суспензии жизнеспособных клеток и поврежденных клеток или погибших клеток микроорганизма, подлежащего детекции, EMA в различных концентрациях, оставляя их на различные периоды времени, затем облучая их видимым светом, при необходимости удаляя клетки посредством центрифугирования или т.п. и проводя анализ способом амплификации нуклеиновой кислоты. Также соответствующим образом можно устанавливать предпочтительные условия облучения светом, проводя такой же эксперимент, как указано выше, с использованием различных периодов облучения. Конкретные примеры условий облучения светом включают облучение светом от 100 до 750 Вт и указанной выше длины волны в течение периода от 5 минут до 2 часов с расстояния от 10 до 50 см от тестируемого образца. Облучение светом предпочтительно проводят при низкой температуре, например с охлаждением образца на льду.
Добавление сшивающего линкера на указанной выше стадии a) и обработку посредством облучения светом на стадии b) можно повторять в течение 2 или более циклов. В таком случае концентрацию сшивающего линкера на стадии a) в первый раз предпочтительно делают более высокой по сравнению с концентрацией сшивающего линкера на стадии a) во второй раз или далее и делают меньше на стадии a) во второй раз или далее по сравнению с концентрацией сшивающего линкера на стадии a) в первый раз.
Например, если позволять EMA действовать при высокой концентрации, например, 10 мкг/мл или более, то хотя проницаемость клеточной стенки или клеточной мембраны погибших клеток становится выше, проницаемость жизнеспособных клеток также становится высокой (Microbiology and Immunology, 2007, 51, pp.763-775; Journal of Clinical Microbiology, 2008, 46, pp.2305-2313). С другой стороны, если позволять EMA действовать при низкой концентрации, например, менее 10 мкг/мл, то хотя можно избежать проникновения в жизнеспособные клетки, также снижается скорость проникновения в погибшие клетки, и, таким образом, погибшие клетки также можно детектировать посредством реакций амплификации нуклеиновых кислот. Таким образом, предпочтительно использовать высокую концентрацию сшивающего линкера на стадии a) в первый раз и делать концентрацию сшивающего линкера низкой на стадии a) во второй раз и далее.
В частности, конечная концентрация сшивающего линкера на стадии a) в первый раз составляет, например, от 10 до 100 мкг/мл в случае моноазида этидия, от 10 до 100 мкг/мл в случае диазида этидия, от 10 до 100 мкг/мл в случае моноазида пропидия, от 2×10-5 до 10 мкг/мл в случае псоларена, от 2×10-5 до 10 мкг/мл в случае 4,5',8-триметилпсоларена или от 2×10-5 до 10 мкг/мл в случае 8-метоксипсоларена. Кроме того, конечная концентрация сшивающего линкера на стадии a) во второй раз и далее составляет, например, от 1 до 10 мкг/мл в случае моноазида этидия, от 1 до 10 мкг/мл в случае диазида этидия, от 1 до 10 мкг/мл в случае моноазида пропидия, от 1×10-5 до 9 мкг/мл в случае псоларена, от 1×10-5 до 9 мкг/мл в случае 4,5',8-триметилпсоларена или от 1×10-5 до 9 мкг/мл в случае 8-метоксипсоларена.
Кроме того, время обработки на стадии a) в первый раз предпочтительно делают более коротким, чем на стадии а) во второй раз и далее. В частности, время обработки на стадии a) в первый раз составляет, например, от 5 минут до 1 часа в случае моноазида этидия, от 5 минут до 1 часа в случае диазида этидия, от 5 минут до 1 часа в случае моноазида пропидия, от 5 минут до 1 часа в случае псоларена, от 5 минут до 1 часа в случае 4,5',8-триметилпсоларена или от 5 минут до 1 часа в случае 8-метоксипсоларена. Время обработки на стадии a) во второй раз и далее составляет, например, от 6 минут до 48 часов в случае моноазида этидия, от 6 минут до 48 часов в случае диазида этидия, от 6 минут до 48 часов в случае моноазида пропидия, от 6 минут до 48 часов в случае псоларена, от 6 минут до 48 часов в случае 4,5',8-триметилпсоларена или от 6 минут до 48 часов в случае 8-метоксипсоларена.
Между стадией b) предыдущего цикла и стадией a) следующего цикла, можно добавлять стадию удаления непрореагировавшего сшивающего линкера. Кроме того, стадию удаления сшивающего линкера можно добавлять между стадией b) и стадией c), описанными ниже. Сшивающий линкер, который не реагирует на стадии a), как правило, в значительной степени инактивируется на стадии b). Таким образом, в качестве способа удаления сшивающего линкера предусмотрен способ центрифугирования тестируемого образца с разделением преципитатов, содержащих микроорганизм, и супернатанта, содержащего сшивающий линкер, и удаления супернатанта. В этом случае после удаления сшивающего линкера можно необязательно добавлять стадию отмывки микроорганизма отмывающим средством.
(3) Стадия c)
Затем, после обработки посредством облучения светом способом амплификации нуклеиновых кислот в присутствии средства подавления действия вещества, ингибирующего амплификацию нуклеиновых кислот, без выделения нуклеиновых кислот из клеток, амплифицируют область-мишень ДНК или РНК микроорганизма, содержащегося в тестируемом образце.
В частности, в раствор для реакции амплификации нуклеиновых кислот, содержащий тестируемый образец, добавляют средство для подавления действия вещества, ингибирующего амплификацию нуклеиновых кислот, и проводят реакцию амплификации нуклеиновых кислот.
Кроме того, в раствор для реакции амплификации в дополнение к средству для подавления действия вещества, ингибирующего амплификацию нуклеиновых кислот, можно добавлять поверхностно-активное вещество, соль магния или соль органической кислоты или соль фосфорной кислоты. Эти вещества можно использовать независимо или в качестве комбинации любых двух или более классов из них. Особенно предпочтительно, добавлять все эти вещества. Порядок добавления средства для подавления действия вещества, ингибирующего амплификацию нуклеиновых кислот, поверхностно-активного вещества, соли магния и соли органической кислоты или соли фосфорной кислоты не ограничен, и их можно добавлять одновременно.
Вещество, ингибирующее амплификацию нуклеиновых кислот, представляет собой вещество, ингибирующее реакцию амплификации нуклеиновых кислот или реакцию достройки нуклеиновых кислот, и примеры включают положительно заряженные ингибирующие вещества, адсорбирующиеся на матричную нуклеиновую кислоту (ДНК или РНК), отрицательно заряженные ингибирующие вещества, адсорбирующиеся на ферменты биосинтеза нуклеиновых кислот (ДНК-полимераза и т.д.) и т.д. Примеры положительно заряженных ингибирующих веществ включают ионы кальция, полиамины, гем и т.д. Примеры отрицательно заряженных ингибирующих веществ включают фенол, соединения типа фенола, гепарин, клеточная стенка грамотрицательных бактерий с наружными мембранами и т.д. Следует отметить, что такие вещества, которые ингибируют реакцию амплификации нуклеиновых кислот, в большом количестве содержатся в продуктах питания или тестируемых клинических образцах.
Примеры средства для подавления действия вещества, ингибирующего амплификацию нуклеиновых кислот, указанного выше, включают один или несколько классов, выбранных из альбумина, декстрана, белка гена 32 T4, ацетамида, бетаина, диметилсульфоксида, формамида, глицерина, полиэтиленгликоля, соевого ингибитора трипсина, α2-макроглобулина, тетраметилхлорида аммония, лизоцима, фосфорилазы и лактатдегидрогеназы. Примеры полиэтиленгликоля включают полиэтиленгликоль 400 и полиэтиленгликоль 4000. Примеры бетаина включают триметилглицин, его производные и т.д. Примеры фосфорилазы и лактатдегидрогеназы включают мышечную гликогенфосфорилазу и лактатдегидрогеназу кролика. В качестве гликогенфосфорилазы предпочтительна гликогенфосфорилаза b. Особенно предпочтительно использовать альбумин, декстран, белок гена 32 T4 или лизоцим.
В качестве попытки уменьшения ингибирующего действия вещества, ингибирующего амплификацию нуклеиновых кислот, содержащегося в тестируемом образце, в качестве которого полагают кровь, экскременты и мясо, оценивают уменьшение ингибирующего действия посредством добавления в реакционную смесь ПЦР таких веществ, как описано выше (Abu Al-Soud, W. et al, Journal of Clinical Microbiology, 38:4463-4470, 2000).
Предполагают возможность того, что альбумин, характерным примером которого является BSA (бычий сывороточный альбумин), может снижать ингибирование амплификации нуклеиновых кислот, связываясь с веществом, ингибирующим амплификацию нуклеиновых кислот, таким как гем (Abu Al-Soud et al., как указано выше). Кроме того, рассматривают две стороны возможности, то есть белок гена 32 T4 представляет собой белок, связывающий одноцепочечную ДНК, и он предварительно связывается с одноцепочечной ДНК, служащей в качестве матрицы в процессе амплификации нуклеиновых кислот так, что не происходит разрушения матрицы нуклеазой и не происходит ингибирования реакции амплификации нуклеиновых кислот, а обеспечивается поддержка реакции или он связывается с веществом, ингибирующим амплификацию нуклеиновых кислот, таким как BSA, так, что не происходит ингибирования реакции амплификации нуклеиновых кислот, а обеспечивается поддержка реакции (Abu Al-Soud et al., как указано выше). Кроме того, предполагают возможность того, что BSA, белок гена 32 T4 и ингибитор протеиназы связываются с протеиназой с уменьшением активности протеолиза, обеспечивая максимальную степень действия ферментов биосинтеза нуклеиновой кислоты. Фактически, в коровьем молоке или крови могут сохраняться протеиназы, и также опубликован пример, что, в таком случае, посредством добавления BSA или ингибитора протеиназы (например, соевого ингибитора трипсина и α2-макроглобулина) избегали разрушения ферментов биосинтеза нуклеиновой кислоты и реакция амплификации нуклеиновых кислот в значительной степени улучшалась (Abu Al-Soud et al., как указано выше). Кроме того, декстран в целом представляет собой полисахарид, который синтезируют посредством молочнокислых бактерий с использованием в качестве исходного вещества глюкозы, и также опубликовано, что комплекс подобного полисахарида и пептида, называемый муцин, прикрепляется к слизистой кишечника (Ruas-Madiedo, P., Applied and Environmental Microbiology, 74:1936-1940, 2008). Таким образом, можно предполагать, что декстран предварительно адсорбируется на отрицательно заряженные ингибирующие вещества (адсорбируется на ферменты биосинтеза нуклеиновых кислот) или положительно заряженные ингибирующие вещества (адсорбируется на нуклеиновые кислоты), а затем связывается с этими ингибирующими веществами.
Кроме того, предполагают, что лизоцим адсорбирует вещества, ингибирующие амплификацию нуклеиновых кислот, которые считают содержащимися в коровьем молоке в большом количестве (Abu Al-Soud et al., как указано выше).
Из указанного выше можно считать, что такие вещества, как указано выше, представленные альбумином, белком гена 32 T4, декстраном и лизоцимом, являются средствами, подавляющими действие веществ, ингибирующих амплификацию нуклеиновых кислот.
Примеры альбумина включают бычий сывороточный альбумин, овальбумин, молочный альбумин, сывороточный альбумин человека и т.д. Из них предпочтительным является бычий сывороточный альбумин. Альбумин может представлять собой очищенный продукт и в случаях, когда ухудшается эффект настоящего изобретения, он может содержать другие компоненты, такие как глобулин. Кроме того, альбумин также может представлять собой продукт фракционирования. Концентрация альбумина в тестируемом образце (раствор для реакции амплификации нуклеиновых кислот), как правило, составляет, например, от 0,0001 до 1% масс., предпочтительно от 0,01 до 1% масс., более предпочтительно от 0,2 до 0,6% масс.
Примеры декстрана включают декстран 40, декстран 500 и т.д. Из них предпочтительным является декстран 40. Концентрация декстрана в тестируемом образце (раствор для реакции амплификации нуклеиновых кислот), как правило, составляет, например, от 1 до 8%, предпочтительно от 1 до 6%, более предпочтительно от 1 до 4%.
Концентрация белка гена 32 T4 (например, производимого Roche A.G., также называемого gp32) в тестируемом образце (раствор для реакции амплификации нуклеиновых кислот), как правило, составляет от 0,01 до 1%, предпочтительно от 0,01 до 0,1%, более предпочтительно от 0,01 до 0,02%.
Примеры лизоцима включают лизоцим, получаемый из яичного белка. Концентрация лизоцима в тестируемом образце (раствор для реакции амплификации нуклеиновых кислот), как правило, составляет, например, от 1 до 20 мкг/мл, предпочтительно, от 6 до 15 мкг/мл, более предпочтительно, от 9 до 13 мкг/мл.
Примеры поверхностно-активного вещества включают неионные поверхностно-активные вещества, такие как поверхностно-активные вещества из ряда Triton (зарегистрированный товарный знак Union Carbide), Nonidet (Shell), Tween (зарегистрированный товарный знак ICI) и Brij (зарегистрированный товарный знак ICI), анионные поверхностно-активные вещества, такие как SDS (додецилсульфат натрия), и катионные поверхностно-активные вещества, такие как стеарилдиметилбензилхлорид аммония. Примеры поверхностно-активных веществ из ряда Triton включают Triton X-100 и т.д., примеры поверхностно-активных веществ из ряда Nonidet включают Nonidet P-40 и т.д., примеры поверхностно-активных веществ из ряда Tween включают Tween 20, Tween 40, Tween 60, Tween 80 и т.д. и примеры поверхностно-активных веществ из ряда Brij включают Brij 56 и т.д.
Тип и концентрация поверхностно-активного вещества в растворе для реакции амплификации нуклеиновых кислот конкретно не ограничены при условии, что обеспечено прохождение реагентов для ПЦР в клетки микроорганизма, и реакция амплификации нуклеиновых кислот не ингибируется в значительной степени. В частности, концентрация SDS, как правило, составляет, например, от 0,0005 до 0,01%, предпочтительно от 0,001 до 0,01%, более предпочтительно от 0,001 до 0,005%, еще более предпочтительно от 0,001 до 0,002%. Относительно других поверхностно-активных веществ, например, в случае Nonidet P-40, концентрация, как правило, составляет от 0,001 до 1,5%, предпочтительно от 0,002 до 1,2%, более предпочтительно от 0,9 до 1,1%. В случае Tween 20 концентрация, как правило, составляет от 0,001 до 1,5%, предпочтительно от 0,002 до 1,2%, более предпочтительно от 0,9 до 1,1%. В случае Brij 56 концентрация, как правило, составляет 0,1 до 1,5%, предпочтительно от 0,4 до 1,2%, более предпочтительно от 0,7 до 1,1%.
Когда в растворе фермента, используемом для реакции амплификации нуклеиновых кислот, содержится поверхностно-активное вещество, поверхностно-активное вещество может состоять только из поверхностно-активного вещества, находящегося в растворе фермента, или можно дополнительно добавлять поверхностно-активное вещество того же типа или другого типа.
Примеры соли магния включают хлорид магния, сульфат магния, карбонат магния и т.д. Концентрация соли магния в тестируемом образце (раствор для реакции амплификации нуклеиновых кислот), как правило, составляет, например, от 1 до 10 мМ, предпочтительно от 2 до 6 мМ, более предпочтительно от 2 до 5 мМ.
Примеры соли органической кислоты включают соли лимонной кислоты, винной кислоты, пропионовой кислоты, масляной кислоты и т.д. Примеры типа соли включают натриевую соль, калиевую соль и т.д. Кроме того, примеры соли фосфорной кислоты включают соли пирофосфорной кислоты и т.д. Их можно использовать независимо или в виде смеси двух или трех или более классов из них. Концентрация соли органической кислоты или соли фосфорной кислоты в тестируемом образце (раствор для реакции амплификации нуклеиновых кислот), как правило, составляет, например, от 0,1 до 20 мМ, предпочтительно от 1 до 10 мМ, более предпочтительно от 1 до 5 мМ относительно общего количества.
По настоящему изобретению выделение нуклеиновых кислот из клеток, которое проводят перед реакцией амплификации нуклеиновых кислот в общепринятых способах, не проводят. Выделение нуклеиновых кислот из клеток означает, например, получение или очистку нуклеиновых кислот из клеток, разрушенных или лизированных ферментативными или физическими способами. По настоящему изобретению такую обработку для выделения нуклеиновых кислот из клеток, например обработку с получением или очисткой нуклеиновых кислот посредством разрушения или лизиса клеток ферментативными или физическими способами, не проводят.
Область-мишень ДНК или РНК, которая присутствует в клетке, амплифицируют способом амплификации нуклеиновых кислот в присутствии средства для подавления действия вещества, ингибирующего амплификацию нуклеиновых кислот, и, если необходимо, других компонентов. В качестве матрицы для амплификации нуклеиновых кислот используют суспензию клеток микроорганизма или суспензию клеток микроорганизма, обработанную ферментом, разрушающим белки, ферментом, разрушающим липиды, ферментом, разрушающим сахариды и т.д., а выделение нуклеиновых кислот для получения матрицы не проводят. Способ амплификации нуклеиновых кислот, предпочтительно, включает стадию термической денатурации нуклеиновых кислот при высокой температуре, например от 90 до 95°C, предпочтительно от 93 до 95°C, более предпочтительно, от 94 до 95°C.
Амплификацию области-мишени, предпочтительно, проводят в клетках микроорганизма. По настоящему изобретению амплификация в клетках микроорганизма является достижимой с высокой вероятностью, как показано в примерах. То есть предполагают, что при высокотемпературной обработке в реакции амплификации нуклеиновых кислот и, в предпочтительном варианте осуществления, под действием указанных выше компонентов морфология клеток сохраняется так, что хромосомная ДНК сохраняется в клетках, но в клеточных мембранах или клеточных стенках микроорганизма формируются отверстия или поры, таким образом, в клетки поступают праймеры, ферменты, необходимые для амплификации нуклеиновых кислот и т.д., в клетках происходит реакция амплификации, а затем часть продукта амплификации остается в клетках или выходит из клеток в зависимости от длины продукта амплификации гена. Однако также нельзя исключать возможность того, что из клеток через отверстия или поры клеточных мембран или клеточных стенок выйдет экстремально малая часть хромосомной ДНК или РНК.
В любом случае приток в клетки компонентов, необходимых для амплификации нуклеиновых кислот, таких как праймеры, без существенного разрушения или лизиса клеток, задержка в клетках или выход из клеток части продуктов амплификации и выход из клеток хромосомной ДНК или РНК не включены в "выделение нуклеиновых кислот". Кроме того, хотя нельзя исключить существование механизма, отличного от механизма, описанного выше, даже в таком случае, способ соответствует определению, что "выделение нуклеиновых кислот не проводят" при условии, что обработку полученных или очищенных нуклеиновых кислот посредством разрушения или лизиса клеток ферментативными или физическими способами, не проводят.
Кроме того, даже когда хромосомная ДНК или РНК, выходящая из клеток, служит в качестве матрицы и реакция амплификации нуклеиновых кислот проходит вне клеток, если большинство продукта амплификации образуется в клетках, можно сказать, что реакцию амплификации нуклеиновых кислот "проводят в клетках микроорганизма". В частности, например, если 80% или более, предпочтительно 90% или более, более предпочтительно 99% или более продукта амплификации образуется в клетках микроорганизма, можно считать, что реакцию амплификации нуклеиновых кислот проводят в клетках микроорганизма.
Примеры способа амплификации нуклеиновых кислот включают способ ПЦР (White, T.J. et al., Trends Genet., 5, 185 (1989)), способ LAMP (изотермическая амплификация с формированием петель: Principal and application of novel gene amplification method (method LAMP), Tsugunori Notomi, Toru Nagatani, BIO INDUSTRY, Vol. 18, No. 2, 15-23, 2001), способ SDA (амплификация с замещением цепей: Edward L. Chan, et al., Arch. Pathol. Lab. Med., 124:1649-1652, 2000), способ LCR (лигазная цепная реакция: Barany, F., Proc. Natl. Acad. Sci. USA, Vol.88, p.189-193, 1991), способ TMA (опосредуемая транскрипцией амплификация: Sarrazin C. et al., J. Clin. Microbiol., vol. 39: pp.2850-2855 (2001)), способ TRC (пособ согласованных транскрипции - обратной транскрипции: Nakaguchi Y. et al., J. Clin. Microbiol., vol. 42: pp.4284-4292 (2004)), способ HC (улавливание гибридов: Nazarenko I., Kobayashi L. et al., J. Virol. Methods, vol. 154: pp.76-81, 2008), способ с использованием микропанелей (Richard P. Spence, et al., J. Clin. Microbiol., Vol. 46, No. 5, pp.1620-1627, 2008) и т.д. Хотя по настоящему изобретению особенно предпочтительно используемым способом является ПЦР, способ амплификации нуклеиновых кислот им не ограничен.
По настоящему изобретению "область-мишень" конкретно не ограничена, при условии, что выбрана область хромосомной ДНК или РНК, которую можно амплифицировать посредством ПЦР с использованием праймеров, применяемых по настоящему изобретению и которая обеспечивает детекцию микроорганизма, подлежащего детекции, и ее можно соответствующим образом выбирать в зависимости от цели. Например, когда в тестируемом образце содержатся клетки типа, отличного от клеток микроорганизма, подлежащего детекции, область-мишень предпочтительно содержит последовательность, специфичную для микроорганизма, подлежащего детекции. Кроме того, в зависимости от цели область-мишень может представлять собой область-мишень, содержащую последовательность, общую для нескольких классов микроорганизмов. Кроме того, область-мишень может состоять из одной области или двух или более областей. Если применяют набор праймеров, подходящий для области-мишени, специфичной для микроорганизма, подлежащего детекции, и набор праймеров, подходящий для хромосомной ДНК широкого спектра микроорганизмов, в качестве объекта детекции можно одновременно определять количество жизнеспособных клеток микроорганизма и количество жизнеспособных клеток широкого спектра микроорганизмов. Длина области-мишени, как правило, составляет, например, от 50 до 5000 нуклеотидов.
Праймеры для использования в амплификации нуклеиновых кислот можно выбирать на основе принципов различных способов амплификации нуклеиновых кислот, и они конкретно не ограничены при условии, что с этими праймерами можно специфично амплифицировать указанную выше область-мишень.
Предпочтительные примеры областей-мишеней включают различные конкретные гены, такие как ген 5S рРНК, ген 16S рРНК, ген 23S рРНК, ген тРНК и ген патогена. Мишенью может служить любой из этих генов или часть любого из этих генов или мишенью может служить область на протяжении двух или более генов. Например, в отношении бактерий кишечной группы и бактерий семейства Enterobacteriaceae, часть гена 16S рРНК можно амплифицировать с использованием набора праймеров, представленного SEQ ID NO: 1 и 2. Кроме того, с использованием набора праймеров, представленного SEQ ID NO: 3 и 4 можно амплифицировать область на протяжении части гена 16S рРНК, гена тРНК и части гена 23S рРНК.
В случае, когда микроорганизм, являющийся мишенью детекции, представляет собой патогенную бактерию, область-мишень может представлять собой патогенный ген. Примеры патогенного гена включают: ген листериолизина O (hlyA) бактерии Listeria; ген энтеротоксин и ген инвазии (invA) бактерии Salmonella; гены веротоксинов патогенных Escherichia coli O-157, O-26, O-111 и т.д.; ген белка наружной мембраны A (ompA) (Enterobacter sakazakii) и оперон макромолекулярного синтеза (MMS) (Enterobacter sakazakii) бактерии Enterobacter; ген белка инвазии в макрофаги (mip) бактерии Legionella; ген термоустойчивого гемолизина и ген токсина, подобного термоустойчивому гемолизину Vibrio parahaemolyticus; ген ipa (ген антигена плазмиды инвазивности) и ген invE (ген инвазии) Shigella dysenteriae и энтероинвазивной Escherichia coli; ген энтеротоксина Staphylococcus aureus; ген цереулида и ген энтеротоксина Bacillus cereus; различные гены токсинов Clostridium botulinum и т.д. Кроме того, примеры праймеров для патогенных генов включают, например, набор праймеров, представленный SEQ ID NO:5 и 6, для гена hlyA бактерии Listeria; набор праймеров, представленный SEQ ID NO: 7 и 8, для гена ompA Enterobacter sakazakii; и набор праймеров, представленный SEQ ID NO:9 и 10, для оперона MMS Enterobacter sakazakii.
Кроме того, в случае вируса гриппа с оболочкой, примеры области-мишени включают ген гемагглютинина (белка H) и ген нейраминидазы (белка N), ген РНК-полимеразы вирусов семейства Calicivirus, характерные примеры которых включают норовирусы, генетические области, кодирующие различные белки капсида и т.д. В качестве отравляющих пищевые продукты вирусов, кроме норовирусов, также представлены ротавирусы и аденовирусы, и в качестве гена-мишени, в качестве области-мишени, как в случае норовирусов, можно использовать гены РНК-полимеразы и генетические области, кодирующие их различные белки капсида.
Если используют праймеры, подходящие для двух или более классов микроорганизмов, в тестируемом образце можно детектировать жизнеспособные клетки двух или более классов микроорганизмов. Кроме того, если используют праймер(ы), специфичный для конкретной бактерии, в тестируемом образце можно детектировать жизнеспособные клетки конкретной бактерии.
Условия реакции амплификации нуклеиновых кислот конкретно не ограничены при условии, что нуклеиновую кислоту можно специфически амплифицировать на основе принципов различных способов амплификации нуклеиновых кислот (таких как ПЦР, LAMP, SDA, LCR, TMA, TRC, HC, способ с использованием микропанелей и т.д.), и можно соответствующим образом подобрать условия.
(4) Стадия d)
Затем анализируют продукты амплификации, полученные способом амплификации нуклеиновых кислот. Анализ продуктов амплификации, в зависимости от способа амплификации нуклеиновых кислот, применяемого на стадии c), проводят после стадии c) или проводят одновременно со стадией c). Например, в случае использования ПЦР с детекцией в реальном времени стадию d) можно проводить одновременно со стадией c).
Способ анализа конкретно не ограничен при условии, что этим способом можно детектировать или количественно определять продукты амплификации нуклеиновых кислот, и их примеры включают электрофорез. Следует отметить, что в случае применения в качестве способа амплификации нуклеиновых кислот способа ПЦР можно использовать способ ПЦР с детекцией в реальном времени (Nogva et al., Appl. Environ. Microbiol., vol.66, 2000, pp.4266-4271; Nogva et al., Appl. Environ. Microbiol., vol. 66, 2000, pp.4029-4036).
Электрофорез позволяет оценивать количества и размеры продуктов амплификации нуклеиновых кислот. Кроме того, ПЦР с детекцией в реальном времени позволяет быстрое количественное определение продуктов амплификации ПЦР.
В случае, когда используют ПЦР с детекцией в реальном времени, если количество циклов амплификации находится в диапазоне от 1 до 10, изменения интенсивности флуоресценции в основном находятся на уровне шума и приблизительно нуля. Таким образом, эту интенсивность рассматривают в качестве контрольных образцов, не содержащих продуктов амплификации. Рассчитывают стандартное отклонение (SD) изменений, и полученное при умножении значения SD на 10 значение определяют как пороговое значение. Количество циклов ПЦР, при котором впервые достигают значения, большего чем пороговое значение, обозначают как "значение порогового цикла (значение Ct)". Таким образом, чем больше исходное количество матричной ДНК в растворе для реакции ПЦР, тем меньше значение Ct, тогда как чем меньше количество матричной ДНК, тем больше значение Ct. Даже если количества матричной ДНК являются одинаковыми, то чем большая часть матрицы, служащей областью-мишенью при ПЦР, расщепляется, тем большим является значение Ct в реакциях ПЦР для этой области.
Кроме того, присутствие или отсутствие продукта амплификации также можно определять посредством анализа профиля температуры плавления (TM) продукта амплификации.
Все указанные выше способы также можно использовать для оптимизации различных условий для способа по настоящему изобретению.
Когда способом по настоящему изобретению детектируют жизнеспособные клетки микроорганизма, точность определения присутствия или отсутствия жизнеспособных клеток микроорганизма и количественное определение микроорганизма в анализе продуктов амплификации ПЦР можно улучшить с использованием стандартной кривой, представляющей собой зависимость количества микроорганизма и продукта амплификации, которую получают с использованием стандартных образцов микроорганизма, в которых определяют микроорганизм. Хотя можно использовать предварительно полученную стандартную кривую, предпочтительно использовать стандартную кривую, полученную посредством проведения стадий способа по настоящему изобретению со стандартными образцами в то же время, что и с тестируемым образцом. Кроме того, если предварительно определена зависимость между количеством микроорганизма и количеством ДНК или РНК, в качестве стандартного образца можно использовать ДНК или РНК, выделенные из микроорганизма.
<2> Набор по настоящему изобретению
Набор по настоящему изобретению представляет собой набор для детекции жизнеспособных клеток микроорганизма в тестируемом образце способом амплификации нуклеиновых кислот при различении жизнеспособных клеток и погибших клеток или поврежденных клеток и содержит средство, способное к ковалентному связыванию с ДНК или РНК при облучении светом с длиной волны от 350 нм до 700 нм, средство для подавления действия вещества, ингибирующего амплификацию нуклеиновых кислот, и праймер(ы) для амплификации области-мишени ДНК или РНК микроорганизма, подлежащего детекции способом амплификации нуклеиновых кислот. Набор по настоящему изобретению можно использовать для осуществления способа по настоящему изобретению.
Кроме того, набор по настоящему изобретению может дополнительно содержать любой один или несколько классов, выбранных из поверхностно-активного вещества, соли магния и соли органической кислоты или соли фосфорной кислоты.
Кроме того, набор по настоящему изобретению может дополнительно содержать фермент с активностью разрушения клеток, отличных от клеток микроорганизма, коллоидных частиц белка, липидов или сахаридов, присутствующих в тестируемом образце.
Фермент, средство, способное к ковалентному связыванию с ДНК или РНК, и средство для подавления действия вещества, ингибирующего амплификацию нуклеиновых кислот, а также поверхностно-активное вещество, соль магния и соль органической кислоты или соль фосфорной кислоты, в соответствии с требованиями, могут находиться в форме одной композиции, содержащей все из этих компонентов, или двух или более растворов или композиций, содержащих компоненты в произвольных комбинациях.
Реакция амплификации нуклеиновых кислот предпочтительно представляет собой ПЦР, LAMP, SDA, LCR, TMA, TRC, HC или способ с использованием микропанелей. Сшивающий линкер и среда в наборе являются такими, как сшивающий линкер и среда описанные в способе по настоящему изобретению.
В предпочтительном варианте осуществления набора по настоящему изобретению средство, способное к ковалентному связыванию с ДНК или РНК, предпочтительно выбрано из моноазида этидия, диазида этидия, моноазида пропидия, псоларена, 4,5',8-триметилпсоларена и 8-метоксипсоларена. Особенно предпочтительно использовать моноазид этидия.
Кроме того, примеры средства для подавления действия вещества, ингибирующего амплификацию нуклеиновых кислот, включают один или несколько классов, выбранных из альбумина, декстрана, белка гена 32 T4, ацетамида, бетаина, диметилсульфоксида, формамида, глицерина, полиэтиленгликоля, соевого ингибитора трипсина, α2-макроглобулина, тетраметилхлорида аммония, лизоцима, фосфорилазы и лактатдегидрогеназы.
Кроме того, примеры соли магния включают хлорид магния, сульфат магния, карбонат магния и т.д.
Кроме того, примеры соли органической кислоты включают соли лимонной кислоты, винной кислоты, пропионовой кислоты, масляной кислоты и т.д. Примеры типа соли включают натриевую соль, калиевую соль и т.д. Кроме того, примеры соли фосфорной кислоты включают соли пирофосфорной кислоты и т.д. Их можно использовать независимо или в виде смеси двух или трех или более классов из них.
Кроме того, фермент конкретно не ограничен при условии, что он может разрушать примеси, такие как клетки, отличные от микроорганизмов, коллоидные частицы белка, липиды и сахариды, присутствующие в тестируемом образце, и не разрушает жизнеспособные клетки микроорганизма, подлежащего детекции, и его примеры включают ферменты, разрушающие липиды, ферменты, разрушающие белки, и ферменты, разрушающие сахариды. Из них можно использовать один фермент или два или более ферментов, но предпочтительно использовать и фермент, разрушающий липиды, и фермент, разрушающий белки, или все из фермента, разрушающего липиды, фермента, разрушающего белки, и фермента, разрушающего сахариды.
Примеры ферментов, разрушающих липиды, включают липазу, фосфатазу и т.д., примеры ферментов, разрушающих белки, включают сериновую протеазу, цистеиновую протеазу, протеиназу K, проназу и т.д., а примеры фермента, разрушающего сахариды, включают амилазу, целлюлазу и т.д.
Набор по настоящему изобретению может дополнительно содержать разбавляющий раствор, реакционный раствор для реакции средства, способного к ковалентному связыванию с ДНК или РНК, ферменты и реакционные растворы для амплификации нуклеиновых кислот, инструкцию, описывающую способ по настоящему изобретению, и т.д.
Примеры
Далее в настоящем документе настоящее изобретение описано более конкретно на основании приведенных ниже примеров. Однако настоящее изобретение не ограничено этими примерами.
[Пример 1]
C использованием Enterobacter sakazakii в качестве характерной бактерии бактерий кишечной группы, тестировали условия для четкого различения жизнеспособных клеток и погибших клеток.
1. Тестовые материалы и способ культивирования
1-1) Используемые штаммы и способ культивирования
Enterobacter sakazakii ATCC51329 культивировали при 37°C в течение 16 часов с использованием бульона с сердечно-мозговым экстрактом (бульон BHI, Eiken Chemical Co., Ltd., Tokyo, Japan). Бульон с культурой в объеме 5 мл помещали в пробирку Falcon 15 мл (Becton Dickinson Labware, NJ) и подвергали центрифугированию с охлаждением при 4°C и 3000 x g в течение 10 минут, удаляли супернатант, а затем к осадку добавляли 5 мл физиологического раствора с получением исходной суспензии жизнеспособных клеток Enterobacter sakazakii (8,95±0,01 log10 клеток/мл, n=2). Кроме того, эту суспензию жизнеспособных клеток разбавляли в 10 раз физиологическим раствором с получением суспензия жизнеспособных клеток Enterobacter sakazakii (7,95±0,01 log10 клеток/мл, n=2).
Кроме того, 1 мл указанной выше исходной суспензии жизнеспособных клеток помещали в микропробирку объемом 1,5 мл (Eppendorf, Hamburg, Germany), пробирку помещали в кипящую воду на период 50 секунд и резко охлаждали, а затем подтверждали, что бактерия не формирует на стандартной среде на агаре (Eiken, Tokyo, Japan) никаких колоний, получая исходную суспензию поврежденных клеток Enterobacter sakazakii. Количество жизнеспособных клеток Enterobacter sakazakii в суспензии жизнеспособных клеток подсчитывали на стандартной среде на агаре и одновременно проводили нефелометрические измерения при длине волны 600 нм с использованием спектрофотометра U-2800A (Hitachi, Japan) с получением зависимости между количеством жизнеспособных клеток и мутностью.
Кроме того, исходную суспензию жизнеспособных клеток в 10 раз разбавляли купленным пастеризованным коровьим молоком с получением суспензии жизнеспособных клеток Enterobacter sakazakii в молоке (7,95±0,01 log10 клеток/мл, n=2).
Кроме того, исходную суспензию поврежденных клеток в 10 раз разбавляли купленным пастеризованным коровьим молоком с получением суспензии поврежденных клеток Enterobacter sakazakii в молоке (7,95±0,01 log10 клеток/мл, n=2).
1-2) Обработка моноазидом этидия (EMA) и обработка посредством облучения светом
Моноазид этидия (EMA, Sigma, St. Louis, MO) растворяли при 1000 мкг/мл с использованием стерилизованной воды и подвергали стерилизации фильтрованием с использованием 0,20-мкм фильтра (Minisart-plus, Sartorius AG, Gottingen, Germany) с получением исходного раствора, который хранили при -20°C с защитой от света.
В суспензии жизнеспособных клеток и поврежденных клеток (1 мл) Enterobacter sakazakii добавляли раствор EMA (1000 мкг/мл) в объеме 10 мкл, и суспензии оставляли при 4°C в течение 10 минут с защитой от света. Затем суспензии помещали на расстоянии 20 см от источника видимого света (100V PRF 500W Flood eye, Iwasaki Electric Co., Ltd., Tokyo, Japan) и облучали светом в течение 5 минут на льду. Каждый обработанный EMA образец подвергали центрифугированию с охлаждением при 4°C и 15000 x g в течение 10 минут, удаляли супернатант, затем к осадку добавляли 1 мл физиологического раствора для отмывки, к осадкам (к клеткам) добавляли 10 мкл стерилизованной воды для суспендирования клеток в стерилизованной воде, и 5 мкл суспензии применяли в качестве образца для амплификации ПЦР.
Суспензии жизнеспособных клеток и поврежденных клеток Enterobacter sakazakii в молоке (1 мл) подвергали обработке EMA и обработке посредством облучения светом следующими способами. Сначала каждую из суспензий жизнеспособных клеток и поврежденных клеток Enterobacter sakazakii в молоке (1 мл) подвергали центрифугированию с охлаждением при 4°C и 15000 x g в течение 10 минут, удаляли супернатант, а затем добавляли 1 мл физиологического раствора. Добавляли протеазу (полученную из бактерии Bacillus, Sigma) в объеме 3 мкл с обработкой клеток протеазой при 37°C в течение 1 часа, затем суспензию подвергали центрифугированию с охлаждением (4°C, 15000 x g, 10 минут), удаляли супернатант, добавляли 1 мл физиологического раствора, а затем добавляли 10 мкл раствора EMA (1000 мкг/мл) с защитой от света. Используемые затем способы являлись такими же, как способы, описанные для указанных выше "суспензий жизнеспособных клеток и поврежденных клеток Enterobacter sakazakii (1 мл)".
1-3) Амплификация ПЦР
Для амплификации ПЦР в 5 мкл образца добавляли средство, содержащее дигидрат трицитрата натрия (TSC, Kanto Kagaku) и гексагидрат хлорида магния (Nakarai-Tesque), а кроме того, содержащее одно или несколько, выбранных из бычьего сывороточного альбумина (BSA, Sigma), декстрана (низкомолекулярный, м.м.: 50000-70000, Nakarai-Tesque), ген белка 32 T4 (gp32, Nippon Gene), лаурилсульфат натрия (SDS, Nakarai-Tesque), Brij 56 (Sigma) и лизоцим яичного белка (Wako Pure Chemical Industries). Каждое средство, добавляемое в образец для амплификации ПЦР и имеющее каждый состав, также можно обозначать как средство для предварительной обработки. Ниже представлены составы средств для предварительной обработки.
Состав 1:
2% BSA: 5 мкл
50 мМ TSC: 1 мкл
100 мМ MgCl2: 1,5 мкл
0,05% SDS: 1 мкл
Состав 2:
2% BSA: 5 мкл
50 мМ TSC: 1 мкл
100 мМ MgCl2: 1,5 мкл
Состав 3:
20% Декстран: 2,5 мкл
50 мМ TSC: 1 мкл
100 мМ MgCl2: 1,5 мкл
Состав 4:
0,1% gp32: 5 мкл
50 мМ TSC: 1 мкл
100 мМ MgCl2: 1,5 мкл
Состав 5:
2% BSA: 5 мкл
50 мМ TSC: 1 мкл
100 мМ MgCl2: 1,5 мкл
4% Brij 56: 12,6 мкл
Состав 6:
2% BSA: 5 мкл
50 мМ TSC: 1 мкл
100 мМ MgCl2: 1,5 мкл
500 мкг/мл лизоцим яичного белка: 1,0 мкл
Состав 7:
2% BSA: 5 мкл
50 мМ TSC: 1 мкл
100 мМ MgCl2: 1,5 мкл
0,05% SDS: 1 мкл
4% Brij 56: 12,6 мкл
500 мкг/мл лизоцим яичного белка: 1,0 мкл
Состав 8:
2% BSA: 5 мкл
50 мМ TSC: 1 мкл
100 мМ MgCl2: 1,5 мкл
4% Brij 56: 12,6 мкл
500 мкг/мл лизоцим яичного белка: 1,0 мкл
Состав 9:
2% BSA: 5 мкл
Состав 10:
Только буфер для ПЦР, содержащий компоненты a)-g), указанные ниже, не содержащий компонентов составов 1-9.
Для амплификации ПЦР в качестве праймеров для ПЦР применяли праймер F: прямой праймер 16S_10F для детекции гена 16S рРНК (5'-AGTTTGATCCTGGCTC-3', SEQ ID NO:1) и праймер R: обратный праймер 16S_1500R для детекции гена 16S рРНК (5'-GGCTACCTTGTTACGA-3', SEQ ID NO:2).
Кроме того, для максимизации вариации (первичный дифференциальный пик) количества флуоресцентного вещества в зависимости от температура для проведения высокочувствительной детекции в анализе плавления продуктов амплификации после ПЦР с детекцией в реальном времени, получали буфер для ПЦР, состоящий из приведенных ниже компонентов a)-g), и амплификацию ПЦР проводили, добавляя этот буфер для ПЦР в смесь образца для амплификации ПЦР и средства для предварительной обработки.
Мишень указанных выше праймеров представляла собой длинный отрезок ДНК (1491 п.н.), содержащий от 10-го до 1500-го нуклеотида гена 16S рРНК.
a) Праймер F (10 пмоль/мкл): 4 мкл
b) Праймер R (10 пмоль/мкл): 4 мкл
c) Ex-Taq (5 Ед/мкл, Takara-Bio): 0,5 мкл (содержащая 0,5% Tween 20, 0,5% Nonidet P-40 и 50% глицерин)
d) 10 x буфер Ex-Taq (Takara-Bio): 5 мкл
e) смесь dNTP (Takara-Bio): 4 мкл
f) 10 x SYBR Green I (BMA): 8 мкл
g) Стерилизованная вода: объем, необходимый для получения общего объема 55 мкл, включая 5 мкл образца для амплификации ПЦР и средство для предварительной обработки
ПЦР с детекцией в реальном времени проводили в соответствии с приведенными ниже условиями термических циклов ПЦР с использованием устройства для ПЦР с детекцией в реальном времени (iCycler iQ, Bio-Rad, Hercules, CA).
1) 4°C в течение 3 минут (1 цикл)
2) 94°C в течение 30 секунд (1 цикл)
3) 94°C в течение 20 секунд; 55°C в течение 30 секунд; 72°C в течение 1 минуты и 30 секунд (50 циклов)
4) 95°C в течение 3 минут (1 цикл)
Затем в соответствии с протоколом анализа плавления продуктов амплификации ПЦР (температуру поднимали с интервалами 0,1°C от 60°C, каждую температуру поддерживали в течение 8 секунд, и эту процедуру повторяли 350 раз в совокупности до конечной температуры 95°C) измеряли температуру плавления продуктов амплификации ПЦР.
В качестве положительного контроля использовали суспензию жизнеспособных клеток Enterobacter sakazakii с 8 log10 клеток/мл. Кроме того, в качестве образца с отрицательным контролем для ПЦР использовали сам буфер для ПЦР, к которому ничего не добавляли.
2. Результаты
Результаты ПЦР с детекцией в реальном времени представлены в таблице 1.
Символы a)-f), используемые в таблице 1, означают то, что указано ниже. Кроме того, "лизо", используемый для указания средства для предварительной обработки означает лизоцим яичного белка.
a) Количества жизнеспособных клеток и количества поврежденных клеток Enterobacter sakazakii: 7,95±0,01 log10 клеток/мл (в физиологическом растворе и в купленном пастеризованном коровьем молоке)
b) Поврежденные клетки получали, погружая жизнеспособные клетки в кипящую воде на период 50 секунд.
c) Означает отсутствие обработки EMA.
d) Обработка EMA (10 мкг/мл, 10 минут, 4°C с защитой от света) + облучение видимым светом (5 минут)
e) Значение Ct ПЦР с детекцией в реальном времени указано как среднее ± SD (n=2)
f) н.д. означает, что амплификации гена-мишени посредством ПЦР с детекцией в реальном времени не получали.
Как видно из результатов для составов 1 и 2, приведенных в таблице 1, в этой системе, где ПЦР проводили непосредственно в бактериях, добивались явного различения жизнеспособного состояния и поврежденного состояния (различение жизнеспособных клеток и поврежденных клеток) Enterobacter sakazakii в физиологическом растворе и различения жизнеспособного состояния и поврежденного состояния Enterobacter sakazakii в коровьем молоко, и по оценке на основе значения Ct, служащей в качестве показателя скорости реакции ПЦР с детекцией в реальном времени, значимых отличий между значениями Ct Enterobacter sakazakii в физиологическом растворе и коровьем молоке (жизнеспособные клетки, не обработанные EMA) или между значениями Ct жизнеспособных клеток Enterobacter sakazakii в физиологическом растворе и коровьем молоке, подвергнутых обработке EMA в различных условиях, не наблюдали. На основе этих результатов выявлено, что не имеет никакого значения, содержится ли поверхностно-активное вещество SDS в средстве для предварительной обработки или не содержится. То же явление, как указано выше, также наблюдали при сравнении результатов, полученных с составами 7 и 8.
Как видно из сравнения результатов, полученных с составами 2, 3 и 4, во всех случаях, где в составе содержался любой из альбумина, декстрана и гена белка 32 T4, добивались явного различения жизнеспособного состояния и поврежденного состояния Enterobacter sakazakii (в физиологическом растворе и коровьем молоке) и значимых отличий между значениями Ct Enterobacter sakazakii в физиологическом растворе и коровьем молоке (жизнеспособные клетки, не обработанные EMA) или между значениями Ct жизнеспособных клеток Enterobacter sakazakii в физиологическом растворе и коровьем молоке, подвергнутых обработке EMA в различных условиях, не наблюдали. На основе этих данных выявлено, что можно использовать любой из альбумина, декстрана и гена белка 32 T4.
Как видно из сравнения результатов, полученных с составами 2 и 5, добивались явного различения жизнеспособного состояния и поврежденного состояния Enterobacter sakazakii с обоими составами (в физиологическом растворе и коровьем молоке), но предполагали, что добавление неионного поверхностно-активного вещества Brij 56 обеспечивало значимое снижение значений Ct Enterobacter sakazakii (жизнеспособных клеток, не обработанных EMA и обработанных EMA), особенно клеток в коровьем молоке, и, таким образом, улучшали чувствительность детекции жизнеспособных клеток. Кроме того, при сравнении результатов, полученных с составами 2 и 6, наблюдали, что добивались явного различения жизнеспособного состояния и поврежденного состояния с обоими составами (в физиологическом растворе и коровьем молоке), но существовала тенденция, что добавление лизоцима больше увеличивало чувствительность детекции жизнеспособных клеток в коровьем молоке (не обработанных EMA и обработанных EMA).
На основе сравнения результатов, полученных с составами 5, 6 и 8, выявлено, что добивались явного различения жизнеспособного состояния и поврежденного состояния со всеми составами (в физиологическом растворе и коровьем молоке), но совместное присутствие Brij 56 и лизоцима яичного белка однозначно улучшало чувствительность детекции жизнеспособных клеток (не обработанных EMA и обработанных EMA), как видно из результатов, полученных с составом 8. Лизоцим яичного белка непосредственно действует на пептидогликаны грамположительных бактерий с гидролизом полисахаридов (связи β-1,4 N-ацетилглюкозамина и N-ацетилмурамовая кислота), однако в случае грамотрицательных бактерий снаружи пептидогликана (со стороны, где действует лизоцим яичного белка), содержащего эти полисахариды, присутствует наружная мембрана, и, таким образом, лизоцим яичного белка не может действовать на него. Принимая в расчет этот механизм действия, можно предполагать, что лизоцим яичного белка в составе 8 не способствовал лизису (разрушению) клеток Enterobacter sakazakii, которая является грамотрицательной бактерией, но предварительно сильно действовал на клеточные стенки погибших клеток грамположительных бактерий, присутствующих в коровьем молоке (≥ 5 log10 клеток/мл) в присутствии Brij 56 с физико-химическим изменением структур поверхности клеточной стенки грамположительных бактерий, которые, как правило, считают ингибирующими ПЦР компонентами, и, таким образом, структуры поверхности клеточной стенки больше не могли функционировать в качестве ингибирующих ПЦР компонентов. Фактически, при сравнении результатов, полученных с составами 5, 6 и 8, можно видеть, что в составе 8 чувствительность детекции жизнеспособных клеток в коровьем молоке значительно улучшена, но среди жизнеспособных клеток, суспендированных в физиологическом растворе, грамположительные бактерии в виде примесей не присутствуют, и никаких данных, указывающих на то, что состав 8 особенно способствует реакциям ПЦР для жизнеспособных клеток Enterobacter sakazakii, которая является грамотрицательной бактерией, не получено.
Из указанного выше можно предполагать, что лизоцим яичного белка не принимает участия в лизисе грамотрицательных бактерий в присутствии Brij 56, но физико-химически изменяет клеточные стенки грамположительных бактерий, исходно содержащихся в тестируемом образце и рассматриваемых как загрязнители для ПЦР, делая их нефункциональными в качестве ингибирующих ПЦР компонентов, и, таким образом, в результате это значительно улучшает чувствительность детекции жизнеспособных клеток Enterobacter sakazakii, которая является грамотрицательной бактерией, как объекта. Это также согласуется с тем фактом, что при сравнении результатов, полученных с составами 2 и 8, не наблюдали никакого значимого отличия в чувствительности (значение Ct) детекции жизнеспособных клеток в физиологическом растворе, но получали крайне высокую чувствительность детекции жизнеспособных клеток в коровьем молоке (содержащем множество поврежденных клеток и погибших клеток грамположительных бактерий) с составом 8. Кроме того, полагают, что лизоцим яичного белка адсорбируется на вещество, ингибирующее амплификацию нуклеиновых кислот, с обычным улучшением реакции амплификации нуклеиновых кислот.
На основе сравнения результатов, полученных с составами 1, 2 и 9, предполагают, что если функцию вещества, подавляющего реакцию амплификации нуклеиновых кислот, можно подавлять BSA, различение жизнеспособных клеток и поврежденных клеток возможно, даже если не присутствует соль магния или соль органической кислоты. Кроме того, с составом 9 значения Ct для жизнеспособных клеток (не обработанных EMA и обработанных EMA) и поврежденных клеток (не обработанных EMA) отставали приблизительно на 3, и, таким образом, с точки зрения реакционноспособности составы 1 и 2, содержащие соль магния или соль органической кислоты, являются более предпочтительными.
И наконец, как видно из сравнения результатов, полученных с составами 2 и 10, хотя различение жизнеспособного состояния и поврежденного состояния Enterobacter sakazakii, суспендированных в физиологическом растворе только с буфером для ПЦР, являлось возможным, но чувствительность (значение Ct) для жизнеспособных клеток (не обработанных EMA и обработанных EMA) являлась крайне неудовлетворительной, кроме того, в случае допущения в качестве типичного тестируемого образца коровьего молока, жизнеспособные клетки (не обработанные EMA и обработанные EMA) и поврежденные клетки, не обработанные EMA, только с буфером ПЦР детектировать невозможно, и, таким образом, полагают, что предпочтительно, чтобы присутствовало по меньшей мере одно средство, которое может снижать влияние ингибирующего ПЦР вещества, из которых характерным примером является альбумин и соль магния и соль органической кислоты или соль фосфорной кислоты.
[Пример 2]
Проводили различение жизнеспособных клеток и поврежденных клеток бактерий кишечной группы и бактерий Enterobacteriaceae.
1. Тестовые материалы и способы тестирования
1-1) Используемые штаммы и способ культивирования
Культивировали Бактерии 16 родов бактерий кишечной группы и 1 рода Enterobacteriaceae, не принадлежащего к бактериям кишечной группы, Klebsiella oxytoca JCM1665, Citrobacter koseri JCM1658, Enterobacter sakazakii ATCC51329, Serratia fonticola JCM1242, Budvicia aquilia JCM3902, Rahnella aquatilis NBRC13544, Hafnia alvei JCM1666, Leclercia adecarboxylata JCM1667, Yokenella regensburgei JCM2403, Pantoea agglomerans JCM1236, Buttiauxella agrestis JCM1090, Kluyvera ascorbata JCM2107, Cedecea davisae JCM1685, E. coli (Escherichia coli) DH5α и Salmonella enteritidis IID604, при 37°C в течение 16 часов с использованием бульона с сердечно-мозговым экстрактом (бульон BHI, Eiken Chemical Co., Ltd., Tokyo, Japan).
Кроме того, культивировали бактерии 2 видов, принадлежащих 2 родам бактерий кишечной группы, Ewingella americana JCM4911 и Moellerella wisconsensis JCM5894, при 30°C в течение 16 часов с использованием бульона BHI.
После культивирования каждый бульон с культурой в объеме 5 мл собирали в пробирку Falcon 15 мл (Becton Dickinson Labware, NJ) и подвергали центрифугированию с охлаждением при 4°C и 3000 x g в течение 10 минут, удаляли супернатант, затем к преципитатам (осадок) добавляли 5 мл физиологического раствора и суспензию разбавляли в 10 раз физиологическим раствором с получением суспензии жизнеспособных клеток каждого штамма.
Кроме того, 1 мл указанной выше суспензии жизнеспособных клеток помещали в микропробирку объемом 1,5 мл (Eppendorf, Hamburg, Germany), и пробирку помещали в кипящую воду на период 50 секунд и резко охлаждали. Подтверждали, что каждая суспензия, помещенная в кипящую воду, не формировала никаких колоний на стандартной среде на агаре (Eiken, Tokyo, Japan) с получением суспензий поврежденных клеток бактерий кишечной группы и бактерий Enterobacteriaceae.
Суспензии жизнеспособных клеток и суспензии поврежденных клеток, полученные как описано выше, применяли в качестве тестируемых образцов для указанных ниже тестов.
Количества жизнеспособных клеток бактерий кишечной группы и бактерии Enterobacteriaceae в суспензиях жизнеспособных клеток подсчитывали на стандартной среде на агаре, и одновременно проводили нефелометрические измерения при длине волны 600 нм с использованием спектрофотометра U-2800A (Hitachi, Japan) с получением зависимости между количеством жизнеспособных клеток и мутностью.
1-2) Обработка моноазидом этидия (EMA) и обработка посредством облучения светом
Моноазид этидия (EMA, Sigma, St. Louis, MO) растворяли при 1000 мкг/мл с использованием стерилизованной воды и подвергали стерилизации фильтрованием с использованием 0,20-мкм фильтра (Minisart-plus, Sartorius AG, Gottingen, Germany) с получением исходного раствора, который хранили при -20°C с защитой от света.
В каждый тестируемый образец (суспензия жизнеспособных клеток или суспензия поврежденных клеток) в объеме 1 мл добавляли раствор EMA (1000 мкг/мл) в объеме 10 мкл, и смесь оставляли при 4°C в течение 10 минут с защитой от света.
Затем тестируемый образец помещали на расстоянии 20 см от источника видимого света (100V PRF 500W Flood eye, Iwasaki Electric Co., Ltd., Tokyo, Japan) и облучали видимым светом в течение 5 минут на льду.
Тестируемый образец, обработанный EMA и облученный видимым светом, подвергали центрифугированию с охлаждением при 4°C и 15000 x g в течение 10 минут, удаляли супернатант, затем к осадку добавляли 1 мл физиологического раствора для отмывки, и суспензию подвергали дополнительному центрифугированию с охлаждением для сбора преципитатов. Такую отмывку повторяли несколько раз, а затем к преципитатам (клетки) добавляли 10 мкл стерилизованной воды для суспендирования клеток в стерилизованной воде с получением образца для амплификации ПЦР.
1-3) Амплификация ПЦР
В каждый образец для амплификации ПЦР добавляли средство, содержащее бычий сывороточный альбумин (BSA, Sigma), дигидрат трицитрата натрия (TSC, Kanto Kagaku) и гексагидрат хлорида магния (Nakarai-Tesque) в составах 1)-3), указанных ниже, и в 5 мкл образца для амплификации ПЦР добавляли поверхностно-активное вещество 4), содержащее лаурилсульфат натрия (SDS, Nakarai-Tesque).
В приведенных ниже описаниях комбинацию, состоящую из средства, содержащего составы 1)-3) и поверхностно-активное вещество 4), также можно обозначать как средство для предварительной обработки.
1) 2% BSA: 5 мкл
2) 50 мМ TSC: 1 мкл
3) 100 мМ MgCl2: 1,5 мкл
4) 0,05% SDS: 1 мкл
Для амплификации ПЦР в качестве праймеров для ПЦР применяли праймер F: прямой праймер 16S_10F для детекции гена 16S рРНК (SEQ ID NO:1) и праймер R: обратный праймер 16S_1500R для детекции гена 16S рРНК (SEQ ID NO:2).
Кроме того, для максимизации вариации (первичный дифференциальный пик) количества флуоресцентного вещества в зависимости от температура для проведения высокочувствительной детекции в анализе плавления продуктов амплификации после ПЦР с детекцией в реальном времени получали буфер для ПЦР, состоящий из приведенных ниже компонентов a)-g), и амплификацию ПЦР проводили, добавляя этот буфер для ПЦР в смесь образца для амплификации ПЦР и средства для предварительной обработки.
a) Праймер F (10 пмоль/мкл): 4 мкл
b) Праймер R (10 пмоль/мкл): 4 мкл
c) Ex-Taq (5 Ед/мкл, Takara-Bio): 0,5 мкл (содержащая 0,005% Tween 20 и 0,005% Nonidet P-40)
d) 10 x буфер Ex-Taq (Takara-Bio): 5 мкл
e) смесь dNTP (Takara-Bio): 4 мкл
f) 10 x SYBR Green I (BMA): 8 мкл
g) Стерилизованная вода: 16 мкл
ПЦР с детекцией в реальном времени проводили в соответствии с приведенными ниже условиями термических циклов ПЦР с использованием устройства для ПЦР с детекцией в реальном времени (iCycler iQ, Bio-Rad, Hercules, CA).
1) 4°C в течение 3 минут (1 цикл)
2) 94°C в течение 30 секунд (1 цикл)
3) 94°C в течение 20 секунд; 55°C в течение 30 секунд; 72°C в течение 90 секунд (50 циклов)
4) 95°C в течение 3 минут (1 цикл)
Затем в соответствии с протоколом анализа плавления продуктов амплификации ПЦР (температуру поднимали с интервалами 0,1°C от 60°C, каждую температуру поддерживали в течение 8 секунд, и эту процедуру повторяли 350 раз в совокупности до конечной температуры 95°C) измеряли температуру плавления продуктов амплификации ПЦР.
В качестве положительного контроля для проведения амплификации ПЦР таким же способом использовали суспензию жизнеспособных клеток Enterobacter sakazakii (8 log10 клеток/мл). Кроме того, в качестве образца с отрицательным контролем для проведения амплификации ПЦР использовали сам буфер для ПЦР, к которому ничего не добавляли.
1-4) Электрофорез в геле
Получали 2% агарозный гель (2% агароза Seakem GTG, FCM BioProducts, Rockland, ME) с использованием 0,5 x TAE.
На агарозный гель наносили продукты амплификации ПЦР в объеме 10 мкл и проводили электрофорез.
Гель окрашивали раствором с 1 мкг/мл бромистого этидия и наблюдали в виде денситограммы, и получали его изображение и хранили с использованием AE-6905H Image Saver HR (Atto Co., Japan).
2. Результаты
Результаты ПЦР с детекцией в реальном времени представлены в таблице 2. Кроме того, на фиг. 1 приведены результаты электрофореза конечных продуктов амплификации ПЦР.
Значения символов, используемых на фиг. 1, являются такими, как указано ниже.
EMA+: обработка EMA (10 мкг/мл, 10 минут, 4°C с защитой от света) + облучение видимым светом (5 минут)
EMA-: отсутствие обработки EMA
PC: Положительный контроль, в котором использовали суспензию жизнеспособных клеток Enterobacter sakazakii с 8 log10 клеток/мл в объеме 5 мкл
NC: Отрицательный контроль, в котором вместо матричной ДНК использовали стерилизованную воду
M: Лестничный маркер ДНК с шагом 100 п.н.
Поврежденная клетка: Использовали поврежденные клетки, полученные погружением суспензии жизнеспособных клеток в кипящую воду на период 50 секунд.
Количества клеток в суспензиях жизнеспособных клеток каждого штамма являются такими, как указано ниже.
E. coli: Escherichia coli DH5α (7,91±0,20 log10 клеток/мл)
S. enteritidis: Salmonella enteritidis IIP 604 (8,07±0,02 log10 клеток/мл)
K. oxytoca: Klebsiella oxytoca JCM1665 (8,38±0,08 log10 клеток/мл)
C. koseri: Citrobacter koseri JCM1658 (8,02±0,06 log10 клеток/мл)
E. sakazakii: Enterobacter sakazakii ATCC51329 (7,95±0,01 log10 клеток/мл)
S. fonticola: Serratia fonticola JCM1242 (7,47±0,01 log10 клеток/мл)
B. aquilia: Budvicia aquilia JCM3902 (6,98±1,50 log10 клеток/мл)
R. aquatilis: Rahnella aquatilis NBRC13544 (7,38±0,14 log10 клеток/мл)
E. americana: Ewingella americana JCM4911 (7,47±0,43 log10 клеток/мл)
H. alvei: Hafnia alvei JCM1666 (8,04±0,22 log10 клеток/мл)
L. adecarboxylata: Leclercia adecarboxylata JCM1667 (7,46±0,20 log10 клеток/мл)
M. wisconsensis: Moellerella wisconsensis JCM5894 (7,85±0,34 log10 клеток/мл)
Y. regensburgei: Yokenella regensburgei JCM2403 (8,03±0,13 log10 клеток/мл)
P. agglomerans: Pantoea agglomerans JCM1236 (7,67±0,78 log10 клеток/мл)
B. agrestis: Buttiauxella agrestis JCM1090 (7,76±0,00 log10 клеток/мл)
K. ascorbata: Kluyvera ascorbata JCM2107 (7,80±0,02 log10 клеток/мл)
C. davisae: Cedecea davisae JCM1685 (7,56±0,10 log10 клеток/мл).
Символы a)-f), используемые в таблице 2, имеют приведенные ниже значения.
a) Количества жизнеспособных клеток бактерий кишечной группы/бактерий Enterobacteriaceae
числовые значения в колонках означают значения Ct при ПЦР с детекцией в реальном времени.
b) Поврежденные клетки получали, погружая жизнеспособные клетки в кипящую воду на период 50 секунд.
c) Означает отсутствие обработки EMA.
d) Означает конечную концентрацию EMA 10 мкг/мл.
e) Значение Ct указано как среднее ± SD (n=2).
f) н.д. означает, что ПЦР проводили дважды, но ген-мишень не амплифицируется в обеих ПЦР.
Как видно из результатов таблицы 2, значения Ct (количество циклов, соответствующих подъему кривой ПЦР с детекцией в реальном времени) необработанной EMA группы жизнеспособных клеток составляли 13-22, а значения Ct обработанной EMA группы жизнеспособных клеток составляли 16-24. Кроме того, значения Ct необработанной EMA группы поврежденных клеток составляли 15-22, и все группы обеспечивали хорошие результаты амплификация ПЦР. Однако в обработанных EMA группах поврежденных клеток всех бактерий кишечной группы и бактерий Enterobacteriaceae, амплификации гена-мишени не получали.
Кроме того, как видно из результатов электрофореза, представленного на фиг. 1, никакой полосы, указывающей на положительный результат продуктов амплификации ПЦР, нельзя было детектировать только для обработанных EMA групп поврежденных клеток всех бактерий кишечной группы и бактерий Enterobacteriaceae.
На основе приведенных выше результатов становится понятным, что при осуществлении способа по настоящему изобретению обеспечено различение жизнеспособных клеток и поврежденных клеток, а также различение жизнеспособных клеток и погибших клеток для 16 родов бактерий кишечной группы и 2 родов семейства Enterobacteriaceae. Кроме того, также сделано возможным различение жизнеспособных клеток и поврежденных клеток для видов, детекция которых, как правило, является сложной, например для бактерий Escherichia и бактерий Salmonella.
[Пример 3]
Проводили различение жизнеспособных клеток и поврежденных клеток бактерий кишечной группы и бактерий Enterobacteriaceae, инокулированных в продукт питания, т.е. коровье молоко.
1. Тестовые материалы и способы тестирования
1-1) Используемые штаммы и способ культивирования
Культивировали бактерии 1 рода семейства Enterobacteriaceae и 12 родов бактерий кишечной группы, Kluyvera ascorbata JCM2107, Cedecea davisae JCM1685, Citrobacter koseri JCM1658, Klebsiella pneumoniae NRBC3321, Serratia fonticola JCM1242, Yokenella regensburgei JCM2403, Rahnella aquatilis NBRC13544, Hafnia alvei JCM1666, Leclercia adecarboxylata JCM1667, Pantoea agglomerans JCM1236, Enterobacter sakazakii ATCC51329, E. coli DH5α и Salmonella enteritidis IID604 при 37°C в течение 16 часов с использованием бульона с сердечно-мозговым экстрактом (бульон BHI, Eiken Chemical Co., Ltd., Tokyo, Japan).
Каждый бульон с культурой после культивирования в объеме 5 мл собирали в пробирку Falcon 15 мл (Becton Dickinson Labware, NJ), и подвергали центрифугированию с охлаждением при 4°C и 3000 x g в течение 10 минут, удаляли супернатант, затем к преципитатам (осадок) добавляли 5 мл физиологического раствора и суспензию разбавляли в 10 раз физиологическим раствором с получением суспензии жизнеспособных клеток каждого штамма.
Суспензии жизнеспособных клеток, полученные как описано выше, применяли в качестве тестируемого образца для приведенных ниже тестов.
Количества жизнеспособных клеток бактерий кишечной группы и бактерий Enterobacteriaceae в суспензиях жизнеспособных клеток подсчитывали на стандартной среде на агаре и одновременно проводили нефелометрические измерения при длине волны 600 нм с использованием спектрофотометра U-2800A (Hitachi, Japan) с получением зависимости между количеством жизнеспособных клеток и мутностью.
1-2) Инокуляция суспензии бактерий в продукт питания и получение преципитатов
В 22,2 мл купленного пастеризованного коровьего молока (жизнеспособные клетки способом культивирования не детектируют) инокулировали от 9 до 25 клеток с использованием суспензий жизнеспособных клеток различных бактерий кишечной группы и бактерий Enterobacteriaceae, полученных выше.
В 22,2 мл коровьего молока инокулировали один класс бактерий кишечной группы или бактерий Enterobacteriaceae.
Кроме того, в качестве образца отрицательного контроля получали 22,2 мл коровьего молока, в которое не добавляли никакой суспензии жизнеспособных клеток (бактерии не инокулировали).
Коровье молоко, в которое инокулировали жизнеспособные клетки бактерий кишечной группы и бактерий Enterobacteriaceae, и коровье молоко, в которое не инокулировали никаких бактерий, как получено выше, подвергали центрифугированию при 37°C и 3000 x g в течение 5 минут, и липидный слой на поверхности супернатанта и водный слой, присутствующий в виде промежуточного слоя, удаляли посредством декантации со сбором преципитатов.
Собранные преципитаты (осадки) коровьего молока, в которые инокулировали жизнеспособные клетки бактерий кишечной группы или бактерий Enterobacteriaceae и в которые не инокулировали никаких бактерий, содержали погибшие клетки бактерий, исходно существующие в купленном коровьем молоке и убитые стерилизацией (грамотрицательный бактерии или грамположительный бактерии, включающие бактерии кишечной группы и т.д. (≥ 6 log10 клеток)).
Таким образом, полагали, что преципитаты, полученные из коровьего молока, в которое инокулировали жизнеспособные клетки бактерий кишечной группы или бактерий Enterobacteriaceae, содержали погибшие клетки и жизнеспособные клетки.
1-3) Ферментативная обработка
К каждому из преципитатов (тестируемый образец), полученному из коровьего молока, в которое добавляли жизнеспособные клетки бактерии кишечной группы или бактерии Enterobacteriaceae, как получено выше, добавляли 10 мл бульона с сердечно-мозговым экстрактом (BHI) предварительно подогреваемого при 37°C, и суспендировали в нем клетки. В суспензию добавляли 25 мкл разбавленного раствора фермента, полученного разбавлением раствора протеиназы K (эквивалентно 1250 Ед/мл, EC.3.4.21.64, Sigma) физиологическим раствором в 50 раз (25 Ед/мл) с проведением ферментативной обработки при 37°C в течение 3 часов.
Обработанный ферментом тестируемый образец центрифугировали при 37°C и 3000 x g в течение 5 минут, удаляли супернатант и снова собирали преципитаты.
1-4) Обработка моноазидом этидия (EMA) и обработка посредством облучения светом
Затем к преципитатам после ферментативной обработки добавляли 1 мл физиологического раствора и смесь перемешивали, в смесь добавляли 10 мкл раствора EMA (1000 мкг/мл), полученного тем же способом, как и в примере 2, и смесь оставляли при 4°C в течение 10 минут с защитой от света.
Затем проводили облучение видимым светом и отмывку тем же способом, как и в примере 2, и в преципитаты добавляли 5 мкл стерилизованной воды с получением образца для амплификации ПЦР.
1-5) Амплификация ПЦР
В 5 мкл образца для амплификации ПЦР добавляли средство для предварительной обработки, как в примере 2.
Для амплификации ПЦР в качестве праймеров для ПЦР применяли праймер F: прямой праймер 16S_1234F для детекции гена 16S рРНК (5'-CTACAATGGCGCATACAAAGAGAAG-3', SEQ ID NO:3) и праймер R: обратный праймер 23S_1703R для детекции гена 23S рРНК (5'-CCTTCTCCCGAAGTTACGGCACCAT-3', SEQ ID NO: 4).
Кроме того, для максимизации вариации (первичный дифференциальный пик) количества флуоресцентного вещества в зависимости от температура для проведения высокочувствительной детекции в анализе плавления продуктов амплификации после ПЦР с детекцией в реальном времени, получали буфер для ПЦР, содержащий приведенные ниже компоненты a)-g), и амплификацию ПЦР проводили, добавляя 41,5 мкл этого буфера для ПЦР в смесь образца для амплификации ПЦР и средства для предварительной обработки.
Праймеры для ПЦР содержали нуклеотиды положений 1234-1258 гена 16S рРНК, тРНК ген (76 п.н.) и нуклеотиды положений 1-1703 гена 23S рРНК, и их мишень представляла собой длинный отрезок ДНК (приблизительно 2450 п.н.), содержащий спейсерную область (приблизительно 364 п.н.).
a) Праймер F (10 пмоль/мкл): 4 мкл
b) Праймер R (10 пмоль/мкл): 4 мкл
c) Ex-Taq (5 Ед/мкл, Takara-Bio): 0,5 мкл (содержащая 0,5% Tween 20, 0,5% Nonidet P-40 и 50% глицерин)
d) 10 x буфер Ex-Taq (Takara-Bio): 5 мкл
e) смесь dNTP (Takara-Bio): 4 мкл
f) 10 x SYBR Green I (BMA): 8 мкл
g) Стерилизованная вода: 16 мкл
ПЦР с детекцией в реальном времени проводили в соответствии с приведенными ниже условиями термических циклов ПЦР с использованием устройства для ПЦР с детекцией в реальном времени (iCycler iQ, Bio-Rad, Hercules, CA).
1) 95°C в течение 3 минут (1 цикл)
2) 95°C в течение 30 секунд; 60°C в течение 40 секунд; 68°C в течение 3 минут (40 циклов)
3) 95°C в течение 3 минут (1 цикл)
Затем в соответствии с протоколом анализа плавления продуктов амплификации ПЦР (температуру поднимали с интервалами 0,1°C от 60°C, каждую температуру поддерживали в течение 8 секунд, и эту процедуру повторяли 350 раз в совокупности до конечной температуры 95°C) измеряли температуру плавления продуктов амплификации ПЦР.
В качестве положительного контроля использовали суспензию жизнеспособных клеток Enterobacter sakazakii с 8 log10 клеток/мл с проведением амплификации ПЦР таким же способом. Кроме того, в качестве образца с отрицательным контролем для проведения амплификации ПЦР использовали сам буфер для ПЦР, к которому не добавляли никакого тестируемого образца.
1-6) Электрофорез в геле
Получали 0,8% агарозный гель (агароза Seakem GTG, FCM BioProducts, Rockland, Me) с использованием 0,5 x TAE.
На агарозный гель для проведения электрофореза наносили продукты амплификации ПЦР в объеме от 5 до 10 мкл.
После электрофореза агарозные гель на 15 минут помещали в раствор, полученный разбавлением красителя нуклеиновых кислот в геле SYBR Gold (Invitrogen, Eugene, Oregon, USA) в 10000 раз 0,5 x TAE, и таким образом окрашивали, затем гель наблюдали в виде денситограммы, и получали его изображение и хранили с использованием AE-6905H Image Saver HR (Atto Co., Japan).
2. Результаты
Результаты ПЦР с детекцией в реальном времени приведены в таблице 3. Кроме того, на фиг. 2 приведены результаты электрофореза конечных продуктов амплификации ПЦР.
Значения символов, используемых на фиг. 2, являются такими, как указано ниже.
KP: K. pneumoniae
CK: C. koseri
EC: E. coli.
SE: S. enteritidis
KA: K. ascorbata
CD: C. davisae
SF: S. fonticola
YR: Y. regensburgei
RA: R. aquatilis
HA: H. alvei
LA: L. adecarboxylata
PA: P. agglomerans
ES: E. sakazakii
Milk: Купленное пастеризованное коровье молоко, в которое не инокулировали бактерии кишечной группы
Положительный: Положительный контроль (Enterobacter sakazakii, в качестве матрицы для ПЦР использовали 5 мкл 8 log10 КОЕ/мл)
Отрицательный: Отрицательный контроль (в качестве матрицы для ПЦР использовали 5 мкл стерилизованной воды)
L: лестничный маркер ДНК с шагом 100 п.н.
Значения символов a)-c), используемых в таблице 3, являются такими, как указано ниже.
a) Количества жизнеспособных клеток бактерий кишечной группы, измерения проводили с n =2-8.
b) Количества положительных результатов к общему количеству измерений продуктов амплификации посредством анализа плавления (профиль TM) на основе ПЦР с детекцией в реальном времени
c) Количества положительных результатов к общему количеству измерений продуктов амплификации ПЦР посредством способа электрофореза и окрашивания геля (SYBR Gold).
На основании результатов, приведенных в таблице 3, в образцах, полученных посредством инокуляции от 9 до 25 клеток различных бактерий кишечной группы (жизнеспособных клеток) в 22,2 мл пастеризованного коровьего молока, с последующим центрифугированием молока и проведением обработки протеиназой K в бульоне BHI и инкубацией (стадия пролиферации жизнеспособных бактерий кишечной группы), количество бактерий кишечной группы (жизнеспособных клеток) возрастало до количества от 90 до 2900 клеток.
Кроме того, в результате проведения анализа плавления продуктов амплификации ПЦР и электрофореза продуктов амплификации ПЦР было возможно детектировать жизнеспособные клетки всех 13 штаммов. Кроме того, продукты амплификация ПЦР, полученные из коровьего молока без инокуляции бактерий кишечной группы (жизнеспособных клеток), продемонстрировали отрицательный результат в обоих анализах плавления и электрофореза.
На основании приведенных выше результатов выявлено, что способом по настоящему изобретению можно сделать возможным детекцию жизнеспособных клеток бактерий кишечной группы и бактерий Enterobacteriaceae, инокулированных в продукт питания, такое как коровье молоко, с различением их от погибших клеток (детекция жизнеспособных клеток). Таким образом, становится возможным широко применять такое различение для продуктов питания, таких как коровье молоко, и детектировать жизнеспособные клетки с высокой чувствительностью, даже для различных клеток, для которых различение жизнеспособных клеток и погибших клеток являлось трудным (бактерии, вирусы и т.д.).
[Контрольный пример 1] Детекция жизнеспособных клеток общепринятым способом
Высокую концентрацию поврежденных клеток бактерий кишечной группы (также включавших бактерии клеток Enterobacteriaceae) детектировали способом ПЦР с EMA с мишенью 16S рРНК (длинный участок ДНК), в котором клетки подвергали обработке EMA, а затем ДНК очищали посредством выделения ДНК и использовали в качестве матрицы.
1. Тестовые материалы и способы тестирования
1-1) Используемые штаммы и способ культивирования
Способ в данном тестировании проводили в соответствии со способом патента Японии №4217797 (международная патентная публикация №WO2002/052034).
Культивировали Escherichia coli DH5α, Salmonella enteritidis IID604, Klebsiella oxytoca JCM1665 и Citrobacter koseri JCM1658 при 37°C с использованием бульона с сердечно-мозговым экстрактом (бульон BHI, Eiken, Tokyo).
Из каждого бульона с культурой, в котором клетки находились в логарифмической фазе, отбирали аликвоту предопределенного объема (10 мл) и подвергали центрифугированию с охлаждением при 4°C и 8000 x g в течение 15 минут. Затем супернатант удаляли, к осадку добавляли 10 мл физиологического раствора для повторного суспендирования клеток, проводили подобную отмывку, затем к осадку добавляли 10 мл физиологического раствора и полученную суспензию использовали как суспензию жизнеспособных клеток. Количество жизнеспособных клеток измеряли с использованием среды с агаром для чашек L.
Поврежденные клетки получали, помещая 1 мл суспензии жизнеспособных клеток в микропробирку 1,5 мл и погружая микропробирку в кипящую воду на период 50 секунд (суспензия поврежденных клеток). Поврежденные клетки, полученные посредством этой обработки, не формировали колоний на стандартной среде на агаре.
1-2) Обработка бактерий EMA и обработка бактерий посредством облучения светом
EMA (Sigma, St. Louis, MO, USA) растворяли при 1000 мкг/мл с использованием стерилизованной воды и подвергали стерилизации фильтрованием с использованием 0,20-мкм фильтра (Minisart-plus, Sartorius AG, Gottingen, Germany).
Раствор EMA (1000 мкг/мл) в объеме 10 мкл добавляли в суспензии жизнеспособных клеток и поврежденных клеток (1 мл) E. coli DH5α (7,91±0,20 log10 клеток/мл) и оставляли при 4°C в течение 10 минут с защитой от света.
Затем суспензии помещали на расстоянии 20 см от источника видимого света (100V PRF 500W Flood глаз, Iwasaki Electric Co., Ltd., Tokyo, Japan) и облучали светом в течение 5 минут на льду.
Каждый обработанный EMA образец подвергали центрифугированию с охлаждением при 4°C и 15000 x g в течение 10 минут, удаляли супернатант, а затем проводили подобную отмывку 1 мл физиологического раствора.
Также обработке EMA, подобной обработке, используемой для E. coli DH5α, подвергали суспензии жизнеспособных клеток и суспензии поврежденных клеток Salmonella enteritidis IID604 (8,47±0,02 log10 клеток/мл), Klebsiella oxytoca JCM1665 (8,38±0,08 log10 клеток/мл) и Citrobacter koseri JCM1658 (8,02±0,06 log10 клеток/мл).
1-3) Выделение ДНК из бактерий кишечной группы (включая бактерии Enterobacteriaceae)
Супернатант каждой суспензии, полученной после обработки EMA, удаляли, затем к преципитатам (клетки) добавляли 0,5 мл 10 мМ Tris-HCl (pH 8,0), к смеси добавляли 10 мкл раствора протеиназы K (соответствующих 1,250 Ед/мл, Sigma), к смеси добавляли 200 мкл 10% (масс./об.) раствора SDS и смесь инкубировали в течение ночи при 50°C.
Затем проводили выделение ДНК способом с фенолом/хлороформом и осаждением этанолом (EP).
К выделенной и очищенной ДНК добавляли стерилизованную воду в объеме 150 мкл и определяли концентрацию на основе оптической плотности при UV 260 нм (OD260). Кроме того, определяли чистоту на основе OD260/OD280.
1-4) ПЦР с детекцией в реальном времени
С использованием праймера F: прямой праймер 16S_10F для детекции гена 16S рРНК (SEQ ID NO:1) и праймера R: обратный праймер 16S_1500R для детекции гена 16S рРНК (SEQ ID NO:2), получали буфер для ПЦР с составом, представленным ниже.
a) Ex-Taq (5 Ед/мкл, Takara-Bio): 0,5 мкл
b) 10 x буфер Ex-Taq (Takara-Bio): 5 мкл
c) смесь dNTP (Takara-Bio): 4 мкл
d) Праймер F (10 пмоль/мкл): 4 мкл
e) Праймер R (10 пмоль/мкл): 4 мкл
f) SYBR Green I (2x) (BMA): 10 мкл
g) Стерилизованная вода: 22,5 мкл
В 50 мкл указанного выше буфера для ПЦР добавляли матричную ДНК в количестве, эквивалентном 150 нг, и проводили ПЦР с детекцией в реальном времени в соответствии с приведенными ниже условиями термических циклов ПЦР с использованием устройства для ПЦР с детекцией в реальном времени (iCycler iQ, Bio-Rad, Hercules, CA).
1) 4°C в течение 3 минут (1 цикл)
2) 94°C в течение 30 секунд (1 цикл)
3) 94°C в течение 20 секунд; 55°C в течение 30 секунд; 72°C в течение 90 секунд (50 циклов)
4) 95°C в течение 3 минут (1 цикл)
Затем в соответствии с протоколом анализа плавления продуктов амплификации ПЦР (температуру поднимали с интервалами 0,1°C от 60°C, каждую температуру поддерживали в течение 8 секунд, и эту процедуру повторяли 350 раз в совокупности до конечной температуры 95°C) измеряли температуру плавления продуктов амплификации ПЦР.
1-5) Электрофорез в геле
Получали 2% агарозный гель (2% Seakem GTG агароза, FCM BioProducts, Rockland, ME) с использованием 0,5 x TAE.
На агарозный гель наносили продукты амплификации ПЦР в объеме 10 мкл и проводили электрофорез.
Гель окрашивали раствором 1 мкг/мл бромистого этидия и наблюдали в виде денситограммы, получали его изображение и хранили с использованием AE-6905H Image Saver HR (Atto Co., Japan).
2. Результаты
Значения Ct (количество циклов ПЦР, при котором кривая амплификации превосходит пороговое значение), полученные при проведении ПЦР с детекцией в реальном времени, представлены в таблице 4. Кроме того, на фиг. 3 представлены результаты электрофореза.
Значения символов, используемых на фиг. 3, являются такими, как указано ниже.
Бактерия Klebsiella: K. oxytoca JCM1665 (8,38±0,08 log10 клеток/мл)
Бактерия Citrobacter: C. koseri JCM1658 (8,02±0,06 log10 клеток/мл)
Бактерия Escherichia: E. coli DH5α (7,91±0,20 log10 клеток/мл)
Бактерия Salmonella: S. enteritidis IIP604 (8,47±0,02 log10 клеток/мл)
EMA+: обработка EMA (10 мкг/мл, 10 минут, 4°C с защитой от света) + облучение видимым светом (5 минут)
EMA-: Отсутствие обработки EMA
NC: Отрицательный контроль, в котором вместо матричной ДНК использовали стерилизованную воду
M: лестничный маркер ДНК с шагом 100 п.н.
Поврежденная клетка: Поврежденные клетки, полученные погружением суспензии жизнеспособных клеток в кипящую воду на период 50 секунд.
Символы a)-f), используемые в таблице 4, имеют приведенные ниже значения.
a) Количества жизнеспособных клеток бактерии Klebsiella, бактерии Citrobacter, бактерии Escherichia и бактерии Salmonella
числовые значения в колонках представляют собой средние значения Ct, полученные в ПЦР с детекцией в реальном времени.
b) Поврежденные клетки, полученные погружением суспензии жизнеспособных клеток в кипящую воду на период 50 секунд.
c) Означает отсутствие обработки EMA.
d) Означает конечную концентрацию EMA 10 мкг/мл.
e) значение Ct указано как среднее ± SD (n=2).
f) н.д. означает, что реакция амплификации ПЦР не проходила, и получить значение Ct являлось невозможным.
В соответствии с результатами, представленными в таблице 4, значимого изменения значения Ct в ПЦР с детекцией в реальном времени при проведении обработки EMA для жизнеспособных клеток E. coli и S. enteritidis не обеспечивалось. Кроме того, в случае поврежденных клеток обработанные EMA клетки E. coli продемонстрировали значение Ct, приблизительно на 18 большее, обработанные EMA S. enteritidis клетки продемонстрировали значение Ct, приблизительно на 14 большее, по сравнению со значением Ct необработанных клеток, таким образом, наблюдали тенденцию, что амплификация ПЦР подвергалась подавлению, но ПЦР продемонстрировала положительную реакцию (значение Ct: 40±1,4 и 34±1,1).
Как видно на основании результатов различения жизнеспособных клеток и поврежденных клеток с использованием конечных продуктов амплификации ПЦР (фиг. 3) также получали полосу гена-мишени в образцах обработанных EMA поврежденных клеток E. coli DH5α и S. enteritidis IID604, и, таким образом, провести различение жизнеспособных клеток и поврежденных клеток в достаточной степени невозможно.
С другой стороны, в отношении бактерии Klebsiella и бактерии Citrobacter, когда жизнеспособные клетки подвергали обработке EMA, никаких явлений, относящихся к значимому увеличению значений Ct, не наблюдали, а когда обработке EMA подвергали поврежденные клетки, реакция амплификации ПЦР полностью ингибировалась, таким образом, значения Ct было невозможно измерить, и, таким образом, различение жизнеспособных клеток и поврежденных клеток было возможным.
[Пример 4]
Исследовали степень, до которой подвергались лизированию (лизис) клетки Enterobacter sakazakii в присутствии средства для предварительной обработки после 50 повторений термических циклов ПЦР.
1. Способы тестирования
Клетки штамма Enterobacter Sakazakii ATCC51329 (ES) в количестве 108 клеток/мл суспендировали в физиологическом растворе или растворе средства для предварительной обработки, указанном в таблице 5 (далее также обозначаемого как "DB (прямой буфер)") с получением суспензий (0,25 мл). Каждую суспензию разделяли на части по 25 мкл, и их помещали в 200-мкл пробирки для ПЦР, соответственно, подвергали стадию повторения термических циклов ПЦР с использованием цикла 95°C в течение 15 секунд, 60°C в течение 20 секунд и 72°C в течение 30 секунд (50 повторений) и снова объединяли в единое целое (всего 0,25 мл) в качестве образца амплификации ПЦР. Из указанного выше образца 0,25 мл отбирали часть 2,5 мкл и добавляли в 12,25 мкл раствора средства для предварительной обработки, указанного в таблице 5 (при условии, что объем стерилизованной воды изменяли на 2,7 мкл) и 12,75 мкл буфера для ПЦР, указанного ниже, добавляли в смесь для проведения ПЦР (соответствующей суспензии I, указанной в таблице 6). В качестве праймеров использовали, ompA_F: прямой праймер для детекции гена ompA (5'-ggatttaaccgtgaacttttcc-3', SEQ ID NO:7), и ompA_R: обратный праймер для детекции гена ompA (5'-cgccagcgatgttagaaga-3', SEQ ID NO:8).
Состав буфера для ПЦР:
a) ompA_F (10 пмоль/мкл): 2 мкл
b) ompA_R (10 пмоль/мкл): 2 мкл
c) Ex-Taq (5 Ед/мкл, Takara-Bio): 0,25 мкл (содержащая 0,5% Tween 20, 0,5% Nonidet P-40 и 50% глицерин)
d) 10 x буфер Ex-Taq (Takara-Bio): 2,5 мкл
e) смесь dNTP (Takara-Bio): 2 мкл
f) 10 x SYBR Green I (BMA): 4 мкл
Затем 247,5 мкл оставшегося образца для амплификации ПЦР (образец для амплификации ПЦР после отбора части 2,5 мкл из объема 0,25 мл) подвергали центрифугированию с охлаждением (10000 x g, 5 минут, 4°C) и 12,25 мкл раствора средства для предварительной обработки и 12,75 мкл буфера для ПЦР добавляли к 2,5 мкл супернатанта для проведения ПЦР (супернатант I). Затем к осадку, полученному указанным выше центрифугированием, добавляли 0,25 мл физиологического раствора или раствора средства для предварительной обработки, указанного в таблице 5, с получением суспензий, и к 2,5 мкл каждой суспензии добавляли 12,25 мкл раствора средства для предварительной обработки и 12,75 мкл буфера для ПЦР для проведения ПЦР (суспензия II). Остатки суспензий подвергали центрифугированию с охлаждением в тех же условиях, как указано выше, и к 2,5 мкл супернатанта, добавляли 12,25 мкл раствора средства для предварительной обработки, указанного в таблице 5, и 12,75 мкл буфера для ПЦР для проведения ПЦР (супернатант II). Затем ту же процедуру повторяли до получения суспензии IV и супернатанта IV, и с каждой из них проводили ПЦР.
ПЦР с детекцией в реальном времени проводили в соответствии с приведенными ниже условиями термических циклов ПЦР с использованием устройства для ПЦР с детекцией в реальном времени (iCycler iQ, Bio-Rad, Hercules, CA).
1) 4°C в течение 3 минут (1 цикл)
2) 95°C в течение 15 секунд; 60°C в течение 20 секунд; 72°C в течение 30 секунд (50 циклов)
3) 95°C в течение 3 минут (1 цикл)
Затем в соответствии с протоколом анализа плавления продуктов амплификации ПЦР (температуру поднимали с интервалами 0,1°C от 60°C, каждую температуру поддерживали в течение 8 секунд, и эту процедуру повторяли 350 раз в совокупности до конечной температуры 95°C) измеряли температуру плавления продуктов амплификации ПЦР.
2. Результаты
Результаты для значений Ct, определенных амплификацией ПЦР с детекцией в реальном времени, представлены в таблице 6. Обозначение "без нагревания" в таблице означает группу, в которой тепловую обработку в соответствии с термическим циклом ПЦР (50 раз повторения термического цикла, состоящего из реакций при 95°C, 60°C и 72°C) не проводили, а "нагревание с использованием термического цикла" означает группу, в которой 50 раз проводили тепловую обработку в соответствии с термическим циклом ПЦР. Количество жизнеспособных клеток Enterobacter sakazakii в суспензии IV, указанное a), составляло 107,6 клеток/мл, как определяли на стандартной среде на чашках с агаром, а количество жизнеспособных клеток Enterobacter sakazakii в супернатанте I, указанное b), составляло 105,7 клеток/мл, как определено тем же способом.
На основе определяющих характеристик этого теста результаты определения количества жизнеспособных клеток в суспензии IV и супернатанте I позволили выявить, что в супернатанте, полученном центрифугированием суспензии Enterobacter sakazakii в физиологическом растворе с охлаждением, также содержаться жизнеспособные клетки в отношении 1%. В отношении группы E. sakazakii (в физиологическом растворе) без нагревания, когда сравнивали значения Ct, полученные для суспензий I или II и супернатанта I, значения Ct для группы суспензий были приблизительно на 5 ниже, и полагают, что это в основном происходит потому, что жизнеспособные клетки в значительной степени собирались в преципитатах (осадок), а в супернатантах также собиралось только их экстремально небольшое количество. То есть такое явление означает, что жизнеспособные клетки распределены между преципитатами и супернатантом в конкретном отношении.
Результаты для подвергаемой нагреванию с использованием термического цикла группы E. sakazakii (в физиологическом растворе) означают результаты тестирования, полученные посредством подвергания суспензии жизнеспособных клеток Enterobacter sakazakii в физиологическом растворе 50 повторениям термического цикла ПЦР, а затем проведения реакции амплификации ПЦР с супернатантом и осадком, и если бы бактериальные клетки при повторении термического цикла ПЦР подвергались лизису и хромосомная ДНК в основном выходила бы из клеток, измерить значения Ct для суспензий II-IV в основном должно было становиться невозможным. Однако все значения Ct были ниже 20, и, таким образом, реакции ПЦР благополучно проводили. Таким образом, в указанных ниже примерах приняли решение определить, до какой степени клетки Enterobacter sakazakii подвергались лизису после подвергания их 50 повторениям термических циклов ПЦР в физиологическом растворе.
Кроме того, в отношении клеток Enterobacter sakazakii "без нагревания" в присутствии средства для предварительной обработки отношение клеток Enterobacter sakazakii, выходящих в супернатант при центрифугировании с охлаждением по сравнению с клетками Enterobacter sakazakii, полученными с физиологическим раствором, было уменьшено в 10 раз, т.е. отношение составляло от 0,1 до 0,2%, и эффективность выделения в виде осадка была значительно улучшена. Если клетки Enterobacter sakazakii лизируют, только оставляя их в растворе средства для предварительной обработки, значение Ct значительно возрастает, так как процесс проходит от стадии для суспензии I до стадии для суспензии IV, и если они подвергаются лизису полностью, то хромосомная ДНК после центрифугирования с охлаждением возвращается в супернатант и, таким образом, определение значения Ct должно становиться невозможным. Однако результаты, приведенные в таблице 6, не поддерживают такого предположения.
То, что следует особенно отметить для случая, когда клетки Enterobacter sakazakii подвергали 50 повторениям термических циклов ПЦР в растворе средства для предварительной обработки, включает то, что значение Ct суспензии I являлось меньшим, чем значение Ct супернатанта I более чем на 8, что значение Ct суспензии II сходным образом являлось меньшим, чем значение Ct супернатанта I более чем на 8 и что значения Ct суспензий III и IV не увеличивалось несмотря на увеличение количества отмывок осадка. Если клетки Enterobacter sakazakii при повторении термического цикла ПЦР полностью подвергаются лизису в растворе средства для предварительной обработки, и хромосомная ДНК полностью выходит во внешний раствор, значения Ct суспензии I и супернатанта I по существу должны являться теми же значениями, и так как затем количество отмывок осадка возрастает, значение Ct суспензии должно значительно возрастать или его определение должно становиться невозможным. Однако результаты, приведенные в таблице 6, не поддерживают такого предположения. С другой стороны, значение Ct суспензии I являлось меньшим, чем значение Ct супернатанта I более чем на 8, рассчитано, что отношение хромосомной ДНК, выходящей в супернатант, составляло приблизительно от 0,1 до 0,5% максимум (как для исходного значения Ct супернатанта I в супернатант может возвращаться экстремально малая часть клеток Enterobacter sakazakii, удельная плотность которых снижается вследствие повторения термического цикла), и рассчитано, что 99% или более исходного значения Ct суспензии I составляет ДНК в клетках Enterobacter sakazakii. То есть полагают, что даже после того, как клетки Enterobacter sakazakii подвергали 50 повторениям термического цикла ПЦР в присутствии средства для предварительной обработки, 99% или более из них не подвергались лизису.
Кроме того, значения Ct суспензии I и супернатанта I группы E. sakazakii (в DB) "без нагревания", приведенные в таблице 6, являются значительно меньшими, чем значения Ct, группы с "нагреванием с использованием термического цикла", соответственно. При допущении, что большинство клеток Enterobacter sakazakii подвергалось лизису при 50 повторениях термических циклов ПЦР в присутствии средства для предварительной обработки и хромосомная ДНК выходила в раствор, значения Ct суспензии I и супернатанта I группы с "нагреванием с использованием термического цикла" должны быть сравнимыми. Кроме того, в суспензии II, так как клетки Enterobacter sakazakii находились в состоянии, когда они не содержат хромосом, и, таким образом, клетки должны находиться в состоянии, когда значение Ct определить невозможно. Однако результаты, приведенные в таблице 6, не поддерживают такого предположения. Кроме того, на основании гипотезы о том, что значения Ct указанных выше суспензии I и супернатанта I в группе "без нагревания" значимо меньшие, чем значения Ct группы с "нагреванием с использованием термического цикла", обусловлены денатурацией вследствие 50 повторений термических циклов ПЦР, так как в средстве для предварительной обработки содержались белки, такие как лизоцим яичного белка и бычий сывороточный альбумин, кроме того, проводили эксперимент, приведенный в таблице 7. Ниже представлена конкретная процедура эксперимента.
Получали десять частей по 25 мкл раствора средства для предварительной обработки с составом, приведенном в таблице 5, подвергали 50 повторениям термических циклов ПЦР и комбинировали с получением 250 мкл раствора средства для предварительной обработки, подвергнутого нагреванию c использованием термического цикла. Затем в физиологический раствор, раствор средства для предварительной обработки или раствор средства для предварительной обработки, подвергнутый нагреванию с использованием термического цикла, при плотности 106-109 клеток/мл добавляли осадок жизнеспособных клеток (отмытых) штамма Enterobacter sakazakii ATCC51329, полученный из 50 мкл бульона с культурой, в которой клетки предварительно культивировали в течение ночи, и суспендировали в них (получали 50 мкл каждой суспензии). Каждую суспензию в объеме 2,5 мкл добавляли в 12,25 мкл раствора средства для предварительной обработки, указанного в таблице 5 (при условии, что объем стерилизованной воды изменяли на 2,7 мкл), и, кроме того, добавляли 12,75 мкл буфера для ПЦР для проведения ПЦР (определение проводили дважды). Результаты приведены в таблице 7.
То, что значение Ct жизнеспособных клеток Enterobacter sakazakii, суспендированных в растворе средства для предварительной обработки и подвергнутых нагреванию с использованием термического цикла, было значимо большим, чем значение Ct, полученное с физиологическим раствором или раствором средства для предварительной обработки, не подвергнутым повторению термического цикла считать нельзя, и, таким образом, подтверждали, что это не являлось причиной, по меньшей мере, возрастания значений Ct суспензии I и супернатанта I группы с "нагреванием с использованием термического цикла", приведенных в таблице 6. На основе этих результатов, можно предполагать, что хромосомная ДНК в клетках Enterobacter sakazakii, которую получали, подвергая клетки Enterobacter sakazakii 50 повторениям термических циклов ПЦР в присутствии средства для предварительной обработки, охлаждая их до 4°C, затем возвращая их до комнатной температуры и подвергая их центрифугированию с охлаждением с получением хромосомной ДНК в осадке и супернатанте, сложным образом переплетена вокруг связывающих денатурированную ДНК белков и ферментов денатурации, и сначала не может функционировать в качестве матрицы для ПЦР. Даже если значение Ct супернатанта I происходило из того, что при 50 повторениях термических циклов ПЦР в присутствии средства для предварительной обработки лизису подвергалось менее 1% клеток Enterobacter sakazakii, полагают, что при обработке термическими циклами ПЦР при 94°C по указанной выше причине в начале ПЦР ДНК была неполностью разделена на отдельные цепи.
[Пример 5]
С использованием образцов, полученных перед повторением термических циклов ПЦР и после него, на основе флуоресцентной микроскопии и стереоскопической микроскопии с использованием средства окрашивания ядер и проточной цитометрии, которая позволяет подсчитывать клетки, остающиеся после повторения термических циклов ПЦР, оценивали, подвергались ли в присутствии средства для предварительной обработки клетки Enterobacter sakazakii лизированию (лизису) при 50 повторениях термических циклов ПЦР.
A. Флуоресцентная микроскопия и стереоскопическая микроскопия
1. Экспериментальные способы
Таким же образом, как и в способе примера 4, 109 клеток/мл штамма Enterobacter Sakazakii ATCC51329 (ES) суспендировали в физиологическом растворе или растворе средства для предварительной обработки, указанном в таблице 5, с получением суспензий (0,25 мл). Каждую суспензию разделяли на части по 25 мкл и помещали 200 мкл пробирки для ПЦР, соответственно, и подвергали стадии повторения термического цикла ПЦР (95°C в течение 15 секунд, 60°C в течение 20 секунд и 72°C 30 секунд, 50 повторений), а затем части снова объединяли в одно целое (всего 0,25 мл). Каждую суспензию разделяли на две половины, и из одной из них собирали супернатанты, как есть, а другую подвергали центрифугированию с охлаждением (3000 x g, 10 минут, 4°C). К 0,125 мл каждой из указанных выше суспензий и супернатантов, которую подвергали каждому процессу, добавляли SYTO9 в отношении 1,5 мкл/мл, смесь поддерживали при 4°C в течение 15 минут с защитой от света, и 2,5 мкл смеси помещали на предметное стекло, накрывали покровным стеклом, помещали на флуоресцентный/стереоскопический микроскоп AxiosKop2 motplus (линзы: Plan-NEOFLUAR 100x/1,30 масло ∞/0,17, источник света: KublercoDIX ebq 100, отдельный, программное обеспечение: AxioVision Rel. 4.6.3, фильтр: FITC и DIC3, время выдержки: FITC 347 мсек, фиксированное; DIC3 20 мсек, выдержанное, LEJ Leistungs elektronik Jena GmbH, Germany), и проводили наблюдения для подтверждения того, происходило ли у бактериальных клеток флуоресцентное излучение зеленого света 530 нм с использованием в качестве излучения возбуждения излучения аргонового лазера 488 нм.
2. Результаты
Изображения при флуоресцентной микроскопии суспензий Enterobacter sakazakii в физиологическом растворе без нагревания или с нагреванием с использованием 50 повторений термических циклов ПЦР и супернатантов, полученных посредством их центрифугирования с охлаждением, а также суспензий Enterobacter sakazakii в растворе средства для предварительной обработки без нагревания или с нагреванием с использованием 50 повторений термических циклов ПЦР и супернатантов, полученных посредством их центрифугирования с охлаждением, приведены на фиг.4-11, соответственно. То есть эксперименты проводили так, что изображения при флуоресцентной микроскопии должны соответствовать от суспензии I до супернатанта I стадии отмывки I, указанного в таблице 6. Вместе с этими изображениями также приведены изображения при стереоскопической микроскопии и совмещенные изображения из изображений при стереоскопической микроскопии и изображений при флуоресцентной микроскопии.
Вне зависимости от использования или неиспользования стадии термических циклов бактериальные клетки Enterobacter sakazakii также выявляли в супернатанте после центрифугирования суспензии Enterobacter sakazakii в физиологическом растворе, и это также коррелировало с результатами ПЦР, приведенными в таблице 6. Как показано на фиг. 4 и 6, даже если клетки Enterobacter sakazakii подвергали 50 повторениям термических циклов ПЦР в физиологическом растворе, большинство из них сохраняли морфологию бактериальной клетки (изображение при стереоскопической микроскопии и изображение при флуоресцентной микроскопии), получали четкие изображения окрашивания SYTO9 и, таким образом, полагали, что клетки содержали хромосомную ДНК в клетках. Так как на изображении при стереоскопической микроскопии, приведенном на фиг. 6, дебрис из клеточных стенок не выявляли, предположили возможность, что небольшие различия значений Ct суспензии I и супернатанта I в подвергаемой нагреванию с использованием термического цикла группе с суспензией Enterobacter sakazakii в физиологическом растворе, приведенные в таблице 6, в основном не являлись результатом выхода ДНК в водную фазу, вызываемого лизисом клеток Enterobacter sakazakii, но их наблюдали вследствие уменьшения удельной плотности клеток Enterobacter sakazakii при 50 повторениях термических циклов ПЦР и возрастания отношения клеток Enterobacter sakazakii, возвращающихся в супернатант (это также предполагали по результатам количественного анализа посредством проточной цитометрии, приведенных на фиг. 12, объясненных позже).
Кроме того, как показано на фиг. 8 и 10, лизис самих клеток Enterobacter sakazakii после 50 повторений термических циклов ПЦР в присутствии средства для предварительной обработки, как в случае суспензии в физиологическом растворе, не наблюдали, и считали, что клетки Enterobacter sakazakii содержали хромосомную ДНК в клетках. Однако в качестве заметного отличия от случая использования физиологического раствора наблюдали явление, что клетки Enterobacter sakazakii после проведения 50 повторений термических циклов ПЦР в присутствии средства для предварительной обработки коагулировали, но лизис бактериальных клеток не наблюдали.
B. Проточная цитометрия
1. Экспериментальные способы
Ниже представлены экспериментальные способы для проточной цитометрии. Сначала таким же образом, как в способе примера 4, 109 клеток/мл штамма Enterobacter Sakazakii ATCC51329 (ES) суспендировали в физиологическом растворе или растворе средства для предварительной обработки, указанного в таблице 5 с получением суспензий (0,25 мл). Каждую суспензию разделяли на части по 25 мкл и помещали 200 мкл пробирки для ПЦР, соответственно, и подвергали стадии повторения термического цикла ПЦР (95°C в течение 15 секунд, 60°C в течение 20 секунд и 72°C 30 секунд, 50 повторений), а затем части снова объединяли в одно целое (всего 0,25 мл). Так как суспензию каждого образца и ее супернатант используют для проточной цитометрии, для каждого образца получали три части в объеме 0,25 мл каждая. В частности, первая часть состоит из самого образца, вторую часть получали, подвергая образец центрифугированию с охлаждением (3000 x g, 10 минут, 4°C), затем удаляя супернатант и добавляя к преципитатам 0,25 мл физиологического раствора с суспендированием в нем клеток, а третья часть состоит из супернатанта, полученного посредством центрифугирования образца с охлаждением, подобного указанному выше. К каждой части добавляли SYTO9 при концентрации 1,5 мкл/мл, и смесь держали при 4°C в течение 15 минут с защитой от света и использовали в качестве тестируемого образца для проточной цитометрии.
Устройство для измерений представляло собой FACS Calibur (BECTON DICKINSON), и для получения изображений бактериальных клеток посредством FSC (измерение прямого рассеяния света) и SSC (измерение бокового рассеяния света) применяли аргоновый лазер 488 нм. Если SYTO9 интеркалировал во внутриклеточную хромосомную ДНК, флуоресценцию зеленого света можно было детектировать с использованием фильтра FL1, λмакс которого при возбуждении этим лазером составляет 530 нм, и, таким образом, также проводили получение изображений посредством FL1. Хотя никакого средства окрашивания ядер на основе йодида пропидия (PI) специально не использовали, также измеряли флуоресценцию красного света с использованием фильтра FL3 для сравнения. Подробности условий измерения проточной цитометрии приведены в таблице 8.
2. Результаты
Экспериментальные результаты для суспензии клеток Enterobacter sakazakii в физиологическом растворе и ее супернатанта (без нагревания или обработанных повторением термических циклов ПЦР) приведены на фиг. 12, а экспериментальные результаты для бактерий, суспендированных в растворе средства для предварительной обработки (включая суспензию, ресуспендированную после однократной отмывки) и ее супернатанта (без нагревания или обработанных повторением термических циклов ПЦР), представлены на фиг. 13. В отношении суспензии клеток Enterobacter sakazakii в физиологическом растворе, если большинство клеток подверглось лизису при 50 повторениях термических циклов ПЦР, они разделятся на малые части и не включатся в области окна бактерий (область, окруженная многоугольником, представляет собой область, где изображены бактерии) на диаграмме FSC-SSC, и в положительной (+) по FL1 (FL1-H на рисунке) области (правая полуобласть относительно оси X) на диаграмме ничего не будет изображено, что соответственно означает, что флуоресценция зеленого света SYTO9 или ее изображение в этой области должны значительно уменьшаться. Однако результаты для образцов суспензий клеток Enterobacter sakazakii в физиологическом растворе без нагревания или подвергаемых повторению термических циклов ПЦР, представленных на фиг. 12, не поддерживают такого предположения. С другой стороны, даже на основании простого сравнения числовых значений, можно предположить, что 95% клеток сохраняли морфологию бактерий даже после термических циклов, а также содержали хромосомную ДНК. Если принимать в расчет собственное стандартное отклонение результатов двух или более повторений измерений проточной цитометрии, численная разница с высокой вероятностью находится в диапазоне ошибки измерения, и можно считать, что после 50 повторений термических циклов ПЦР по существу 100% бактериальных клеток Enterobacter sakazakii сохраняют морфологию и содержат хромосомную ДНК.
Подобным образом, на основе результатов для клеток Enterobacter sakazakii, суспендированных в растворе средства для предварительной обработки (без нагревания или с обработкой посредством повторения термических циклов ПЦР), и суспензии, полученной отмывкой и ресуспендированием клеток (без нагревания или подвергнутых термическим циклам ПЦР) Enterobacter sakazakii, представленных на фиг. 13, трудно предположить, что бактериальные клетки Enterobacter sakazakii подвергались лизису в присутствии средства для предварительной обработки. На основе сравнения результатов, приведенных на фиг. 12 и 13, легко предположить, что окрашивание SYTO9 немного ингибируется в присутствии средства для предварительной обработки. Таким образом, закономерно, что изображения Enterobacter sakazakii, суспендированных в растворе средства для предварительной обработки (без нагревания или подвергнутых термическим циклам, диспергированные клетки) при интенсивности FL1-H (SYTO9) от 101 до 103, представленные на фиг. 13, рассматривают как изображения, полученные для клеток Enterobacter sakazakii. С такими исходными условиями очевидно, что бактериальные клетки Enterobacter sakazakii на основе данных для клеток Enterobacter sakazakii, суспендированных в растворе средства для предварительной обработки без нагревания или подвергнутых термическим циклам ПЦР, лизису не подвергались, но в отношении данных для клеток Enterobacter sakazakii, суспендированных в растворе средства для предварительной обработки (полученных посредством отмывки и ресуспендирования клеток) без нагревания или подвергнутых термическим циклам ПЦР, необходимо подтверждение. В таком случае в качестве основных исходных условий, так как изображения при окрашивания Enterobacter sakazakii SYTO9 получали для клеток после отмывки и ресуспендирования в физиологическом растворе, их нужно оценивать с использованием области FL1-H (+). В этом случае, хотя общее количество изображений в области FL1-H(+) для "диспергированных клеток" и "коагулированных клеток" после термических циклов ПЦР по-видимому остается меньшим, чем общее количество изображений для клеток без нагревания, если взять в расчет, что по расчетам на основании результата, приведенного на фиг. 10, 1 изображение "коагулированных клеток Enterobacter sakazakii" следует рассматривать как с высокой вероятностью состоящее по меньшей мере из бактериальных клеток Enterobacter sakazakii в количестве от нескольких до нескольких десятков, следует предположить, что это количество является равным или большим, чем количество клеток без нагревания, и если принять в расчет данные, полученные без отмывки, в комбинации, следует полагать, что лизису не подвергнуты по существу 100% бактериальных клеток Enterobacter sakazakii.
[Пример 6]
Измерения ПЦР с детекцией в реальном времени с использованием определенного количества бактериальных клеток Enterobacter sakazakii и очищенных хромосом в том же количестве хромосомной ДНК, которое содержится в клетках
1. Экспериментальные способы
Из бульонов с культурами штаммов Enterobacter sakazakii ATCC29544 и ATCC51329, в которых клетки пролиферировали в течение ночи, получали очищенную ДНК, не содержащую примесей РНК, способом выделения ДНК, описанным в WO2007/094077, измеряли значения ее оптической плотности при 260 нм и 280 нм (OD260 и OD280, OD260 для раствора ДНК 50 мкг/м = 1,0, длина ячейки: 1 см), концентрацию ДНК рассчитывали на основе OD260, а чистоту очищенной ДНК определяли на основе OD260/OD280.
Кроме того, клетки из указанных выше бульонов с культурами, в которых клетки пролиферировали в течение ночи, отмывали, а затем серийно разбавляли стерилизованной водой с получением суспензий жизнеспособных клеток Enterobacter sakazakii с плотностями от 4×103 до 4×108 клеток/мл. Затем, в соответствии со способом из примера 4, 2,5 мкл каждой суспензии добавляли в 12,25 мкл раствора средства для предварительной обработки, указанного в таблице 5 (при условии, что объем стерилизованной воды изменяли на 2,7 мкл), и, кроме того, в смесь добавляли 12,75 мкл буфера для ПЦР для детекции гена ompA с проведением ПЦР тем же способом, как в примере 4. Каждая пробирка для ПЦР содержала от 101 до 106 клеток Enterobacter sakazakii. Так как количество хромосомной ДНК, получаемой из 1 клетки Enterobacter sakazakii, можно считать 5 фг (5×10-15 г), количество хромосомной ДНК, содержащейся в каждой пробирке для ПЦР рассчитывали с использованием этого значения, и такое количество указанной выше очищенной ДНК (2,5 мкл) помещали в каждую пробирку для ПЦР, и таким же способом последовательно добавляли раствор средства для предварительной обработки и буфер для ПЦР с проведением ПЦР.
2. Результаты
Степени очистки ДНК приведены в таблице 9, а результаты ПЦР с детекцией в реальном времени приведены в таблице 10. Как видно из результатов, приведенных в таблице 9, значения OD260/OD280 приблизительно составляли 2,0, и, таким образом, из каждого из двух штаммов Enterobacter sakazakii, соответственно, можно получать ДНК с высокой чистотой с небольшой примесью РНК. Кроме того, как видно из результатов, приведенных в таблице 10, значимого отличия значений Ct для бактериальных клеток бактерии Enterobacter sakazakii и хромосомной ДНК в количествах, соответствующих такому же количеству в клетках, не наблюдали, и, таким образом, выявлено, что если в качестве матрицы ПЦР функционирует 100% очищенной ДНК, растворенной в пробирке, то в качестве матрицы ПЦР также функционирует 100% хромосомной ДНК бактериальных клеток Enterobacter sakazakii.
В отношения случая суспендирования клеток Enterobacter sakazakii в подобном растворе средства для предварительной обработки или физиологическом растворе (или стерилизованной воде), а затем добавления к суспензии такого же буфера для ПЦР, как представленный в примере 4, с проведением ПЦР, следует предполагать, что может возникнуть двусмысленность относительно общепринятого знания, что часть клеток Enterobacter sakazakii подвергается лизису так, что хромосомная ДНК выходит во внешний раствор и служит в качестве матрицы ПЦР, обуславливая реакции ПЦР. Однако если в этом случае принять эту гипотезу, то тогда необходимо, чтобы по существу 100% клеток Enterobacter sakazakii подвергались лизису, но очевидно, что такой факт 100% лизиса отвергается экспериментальными результатами примеров 4 и 5. То есть в отношении примера 4, при сравнении значений Ct суспензий I и II и супернатанта I клеток Enterobacter sakazakii после 50 повторений термических циклов ПЦР в присутствии средства для предварительной обработки, приведенного в таблице 6, если 100% клеток подвергались лизису, значение Ct супернатанта I должно являться значительно меньшим, чем значение Ct суспензии II, и значение Ct этого супернатанта должно быть эквивалентно значению Ct суспензии I. Однако результаты, приведенные в таблице 6, не поддерживают такого предположения. Кроме того, в отношении примера 5, на основе результатов количественного определения клеток Enterobacter sakazakii перед повторением термических циклов ПЦР и после него, приведенных на фиг. 13, которые являются результатами измерений проточной цитометрии, лизис по существу 100% клеток невозможен, и также невозможен лизис даже 10% клеток. То есть гипотеза о том, что "часть бактериальных клеток подвергается лизису, и хромосомная ДНК выходит из них во внешний раствор с индукцией ПЦР" в соответствии с общим научным знанием нельзя применять по меньшей мере к ПЦР в присутствии средства для предварительной обработки (50 повторений) по настоящему изобретению.
[Пример 7]
На основании результатов примеров 4-6, выявлено, что большинство клеток Enterobacter sakazakii не подвергались лизису даже после реакции ПЦР (50 повторений) с буфером для ПЦР в присутствии средства для предварительной обработки, и клетки содержали хромосомную ДНК в клетках. С другой стороны, при анализе профиля TM продуктов амплификации ПЦР после ПЦР с детекцией в реальном времени (измерение температуры плавления) получали температурный пик, который по расчетам являлся температурным пиком продукта гена ompA, и его считали положительным для ПЦР с детекцией в реальном времени. Однако, если быть точным, присутствуют ли продукты амплификации ПЦР в бактериальных клетках, в реакционном растворе для ПЦР или в обоих компонентах, определено не было. С точки зрения здравого смысла следует предполагать, что продукты амплификации ПЦР в основном растворены в растворе для ПЦР, но даже это точно не определено, и для ПЦР в присутствии средства для предварительной обработки по настоящему изобретению это еще менее ясно. В указанном выше примере полагают, что ПЦР в присутствии средства для предварительной обработки можно проводить в бактериальных клетках, а в приведенных ниже примерах продемонстрирована возможность того, что продукты амплификации ПЦР также остаются в бактериальных клетках.
1. Экспериментальные способы
Получали пять частей по 500 мкл бульона с культурой штамма Enterobacter sakazakii ATCC51329 (9,3×108 клеток/мл), в которых клетки пролиферировали в течение ночи, и подвергали центрифугированию с охлаждением (3000 x g, 10 минут, 4°C), супернатант удаляли, а затем к осадку добавляли 500 мкл общего фиксирующего раствора A для бактерий (4% параформальдегид) или фиксирующего раствора B (метанол/уксусная кислота = 3/1), фиксирующего раствора C (Mildform 10N, 10% формалин, дезодорированный раствор нейтрального буфера, Wako Pure Chemical Industries, Osaka) или фиксирующего раствора D (Mildform 10NM, 10% формалин, дезодорированный раствор метанола в нейтральном буфере, Wako Pure Chemical Industries, Osaka), и смесь инкубировали в течение ночи при 4°C так, что хромосомная ДНК в бактериальных клетках и белки клеточной стенки сшивались с предварительным фиксированием хромосомной ДНК в клетках. В качестве контроля с использованием вместо фиксирующего раствора 500 мкл физиологического раствора получали образец, в котором фиксирование не проводили.
Затем клетки три раза отмывали 500 мкл физиологического раствора и в завершении суспендировали в 250 мкл физиологического раствора. Так как в основном полагают, что коэффициент восстановления бактерий в виде осадка после отмывки составляет 80%, а центрифугирование проводили 4 раза, расчетная плотность клеток Enterobacter sakazakii в конечном препарате составляла 7,6×108 клеток/мл. Такую суспензию в физиологическом растворе в объеме 250 мкл дополнительно разбавляли в 10 раз. Расчетная плотность клеток Enterobacter sakazakii в этом разведении составляла 7,6×107 клеток/мл. Это разведение в объеме 2,5 мкл применяли в качестве образца для амплификации ПЦР и добавляли к 12,25 мкл раствора средства для предварительной обработки, указанного в таблице 5 (при условии, что объем стерилизованной воды изменяли на 2,7 мкл), и кроме этого в смесь добавляли 12,75 мкл буфера для ПЦР для детекции грамотрицательных бактерий, указанного ниже. Для каждого образца, фиксированного каждым фиксирующим раствором, получали порции в объеме 27,5 мкл в количестве 20. В качестве праймеров использовали прямой праймер 16S_1234F для детекции гена 16S рРНК (SEQ ID NO:3) и обратный праймер 23S_1703R для детекции гена 23S рРНК (SEQ ID NO:4), указанные в примере 3.
Состав буфера для ПЦР:
a) 16S_1234F (10 пмоль/мкл): 2 мкл
b) 23S_1703R (10 пмоль/мкл): 2 мкл
c) Ex-Taq (5 Ед/мкл, Takara-Bio): 0,25 мкл (содержащая 0,5% Tween 20, 0,5% Nonidet P-40 и 50% глицерин)
d) 10 x буфер Ex-Taq (Takara-Bio): 2,5 мкл
e) смесь dNTP (Takara-Bio): 2 мкл
f) 10 x SYBR Green I (BMA): 4 мкл
ПЦР с детекцией в реальном времени проводили в соответствии с приведенными ниже условиями термических циклов ПЦР с использованием устройства для ПЦР с детекцией в реальном времени (iCycler iQ, Bio-Rad, Hercules, CA).
1) 4°C в течение 3 минут (1 цикл)
2) 95°C в течение 15 секунд; 60°C в течение 20 секунд; 72°C в течение 3 минут (30 циклов)
3) 95°C в течение 3 минут (1 цикл)
Затем в соответствии с протоколом анализа плавления продуктов амплификации ПЦР (температуру поднимали с интервалами 0,1°C от 60°C, каждую температуру поддерживали в течение 8 секунд, и эту процедуру повторяли 350 раз в совокупности до конечной температуры 95°C) измеряли температуру плавления продуктов амплификации ПЦР.
После завершения ПЦР 20 порций раствора для реакции ПЦР для каждого фиксирующего раствора объединяли в одно целое и подвергали центрифугированию с охлаждением (3000 x g, 10 минут, 4°C). Отбирали супернатант в объеме 5 мкл и использовали для электрофореза в геле с 0,8% агарозой (окрашивание SYBR Gold, для всех последующих случаев окрашивание геля проводили посредством окрашивания SYBR Gold), а оставшийся супернатант удаляли. К осадку добавляли 200 мкл физиологического раствора для суспендирования клеток. Рассчитанное количество бактериальных клеток Enterobacter sakazakii в суспензии составляло 1,5×107 клеток/мл. В суспензию добавляли SYTO9 с концентрацией 1,5 мкл/мл, и смесь оставляли при 4°C в течение 15 минут с защитой от света и использовали для проведения проточной цитометрии в тех же условиях, как в примере 5. В качестве контроля проводили ту же процедуру при условии, что проводили 0 циклов, а не 30 циклов ПЦР, с получением контрольного образца.
Кроме того, для образцов, полученных с использованием фиксирующих растворов A и B, ПЦР проводили с использованием праймера ompA_F (SEQ ID NO:7) и праймера ompA_R (SEQ ID NO:8), описанных в примере 4, вместо 16S_1234F и 23S_1703R, условиями термического цикла ПЦР их примера 4, и способом из примера 7 за исключением указанных выше условий, электрофорез проводили для 5 мкл супернатанта, полученного после ПЦР, и измерение проточной цитометрии проводили с использованием SYTO9 для суспензии осадка, полученного посредством удаления супернатанта после ПЦР, в физиологическом растворе.
2. Результаты
Результаты электрофореза супернатантов реакции после обработки каждым из указанных выше фиксирующих растворов и ПЦР (16S-23S рРНК, 2450 п.н.) приведены на фиг. 14, а значения Ct растворов соответствующих реакций, определенные в ПЦР с детекцией в реальном времени, приведены непосредственно под ними. Результаты измерения проточной цитометрии, проводимой с использованием SYTO9 для суспензий, полученных посредством удаления супернатантов образцов после ПЦР и суспендирования осадков в физиологическом растворе, приведены в таблице 11. Подобным образом на фиг. 15 представлены изображения электрофореза супернатантов после ПЦР с геном-мишенью ompA (469 п.н.). Результаты измерения проточной цитометрии, проводимой с использование SYTO9 для суспензий, полученных посредством удаления супернатантов образцов после ПЦР и суспендирования осадков в физиологическом растворе, приведены в таблице 12. Как видно из результатов, представленных на фиг. 14, количество продукта генов 16S-23S (2450 п.н.) в супернатанте смеси ПЦР, полученном с использованием фиксирующего раствора B, являлось значительно большим, чем количество продукта генов 16S-23S, полученное с использованием других фиксирующих растворов, и интенсивность полосы была эквивалентной интенсивности полосы, полученной без фиксирования ("S" на рисунке). Так как, в качестве стандартного способа фиксации бактерий, общим является использование фиксирующего раствора A, а состав фиксирующих растворов C и D также сходен с составом фиксирующего раствора A, полагают, что хромосомная ДНК в клетках Enterobacter sakazakii и белки клеточной стенки были плотно сшиты. На основе результатов, представленных на фиг. 14, уместно предположить, что фиксирующий раствор B может обладать только функциональным эквивалентом отсутствию фиксации (S), но так как фиксирующий раствор B прочно сшивает хромосомы клеток и белки клеточных мембран млекопитающих, он в некоторой степени также демонстрирует фиксирующую функцию у бактерий.
Результаты проточной цитометрии (квадрант: НП среди 4 квадрантов, т.е. сравнение количества бактериальных клеток Enterobacter sakazakii перед ПЦР и после нее с использованием в качестве показателя количества бактерий SYTO9+), приведенные в таблице 11 (а также в таблице 12), позволяет предположить, что даже когда бактериальные клетки предварительно фиксировали с использованием каждого фиксирующего раствора или без проведения фиксации (S), а именно, хромосомную ДНК бактерий фиксировали в клетках, а затем 30 раз повторяли термический цикл ПЦР в присутствии средства для предварительной обработки, большинство бактериальных клеток не подвергались лизису, а сохраняли морфологию, и хромосомы сохранялись в клетках. Кроме того, хотя изображения электрофореза подтверждали присутствие продуктов амплификации гена-мишени также в реакционной смеси супернатанта ПЦР (фиг. 14), значения Ct, определенные в ПЦР с детекцией в реальном времени, приведенные под изображениями гелей на фиг. 14, наблюдали только для образцов, полученных с использованием фиксирующего раствора B и без фиксации (S), а в ПЦР с детекцией в реальном времени с использованием других фиксирующих растворов значения Ct не наблюдали.
Можно предположить, что в случаях фиксирующих растворов A, C и D это происходит вследствие того, что степень фиксации хромосомы и клеточной стенки являлась высокой, таким образом, не достигалась подходящая термическая денатурация при ПЦР (95°C), таким образом, хромосомная ДНК не могла разделяться на одиночные цепи, и, таким образом, отжиг праймеров с хромосомой становится плохим, но если в расчет принять результаты сходного эксперимента (30 циклов) с геном-мишенью ompA (469 п.н.), представленные на фиг. 15, значения Ct, определенные в ПЦР с детекцией в реальном времени, и интенсивности полос, полученные с использованием фиксирующих растворов A и B, не демонстрируют значимого различия, и, таким образом, приведенную выше гипотезу следует отвергнуть.
То есть полагают, что небольшие количества продуктов амплификации гена-мишень в супернатантах реакционной смеси ПЦР, полученных с использованием фиксирующих растворов A, C и D, представленные на фиг. 14, получают потому, что хотя сама по себе реакция ПЦР с использованием в качестве матрицы хромосомы, находящейся в бактериальной клетке, проходила хорошо, продукты генов были в 5 раз длиннее, чем продукты генов на фиг. 15 (2450 п.н.), и в результате, выход продуктов генов, амплифицированных в клетках, во внешний раствор был снижен. Если эта гипотеза является правильной, продукт амплификации гена длиной 2450 п.н. должен также детектироваться в бактериальных клетках Enterobacter sakazakii после ПЦР. Это подтверждали в примере 8. Как указано выше, можно заключить, что даже если предварительно проводили обработку с фиксацией бактериальной хромосомы в клетке с ингибированием выхода хромосомы во внешний раствор, по существу 100% бактериальных клеток сохраняли морфологию бактерии, и хромосома сохранялась в клетке даже после 50 раз повторения термического цикла ПЦР, при условии, что ее проводили в присутствии средства для предварительной обработки, но несмотря на это реакции ПЦР проходили, и продукты амплификации ПЦР также присутствовали во внешнем растворе, и, таким образом, реакции ПЦР проходили в основном в бактериальных клетках. Кроме того, как показано в примере 8, указанном ниже, часть продуктов ПЦР в указанных выше условиях также присутствовала в клетках.
[Пример 8]
В примерах 4-7 предполагалось, что ПЦР в присутствии средства для предварительной обработки, вероятно, проходит в бактериальных клетках, т.е. вероятно проходит ПЦР in situ. ПЦР in situ (например, Gerard J. et al., American Journal of Pathology, 139: 847-854, 1991) представляет собой способ детекции и количественного определения гена, такого как ген HPV, встроенного в хромосомную ДНК клеток человека, в котором перед детекцией и количественным определением иммуноциты человека фиксируют таким фиксирующим раствором, как указан в примере 7, со сшиванием хромосомной ДНК и мембранных белков клеток человека, и клетки обрабатывают протеазой в течение короткого периода времени или клеточные мембраны иммуноцитов человека обрабатывают микроволновым излучением.
Этот способ представляет собой способ нанесения раствора для ПЦР на фиксированные иммуноциты человека с индукцией реакций амплификации ПЦР в иммуноцитах человека, при которой даже продукт ПЦР длиной приблизительно 500 п.н. не выходит из клеток. Так как продукт ПЦР не выходит из клеток, если реакции ПЦР прекращают на ранней стадии менее 5-10 циклов, он может представлять собой способ, обеспечивающий не только детекцию гена в клетках, но и расчет количества встроенного гена.
Если ПЦР в присутствии средства для предварительной обработки по настоящему изобретению представляет собой ПЦР in situ, часть продукта амплификации может также оставаться в бактериальных клетках, и в этом примере проводили эксперимент для подтверждения этого. В примере 7, в частности, хотя хромосомная ДНК Enterobacter sakazakii фиксирующим раствором B вероятно была сшита с белками клеточной стенки, наблюдали тенденцию большего выхода продукта амплификации ПЦР в присутствии средства для предварительной обработки во внешний раствор по сравнению со случаем использования других фиксирующих растворов, и явление, сходное с явлением, наблюдаемым при использовании способа по настоящему изобретению без применения какого-либо фиксирующего раствора (способ с отсутствием фиксации (S)).
Таким образом, исследователи сосредоточились на фиксирующем растворе B и отсутствии фиксации (S), указанных в примере 7, и проверяли могут ли продукты амплификации ПЦР частично оставаться в бактериальных клетках, даже если большое количество продуктов амплификации ПЦР выходит во внешний раствор.
1. Экспериментальные способы
Получали две порции по 500 мкл бульона с культурой штамма Enterobacter sakazakii ATCC51329 (4,3×108 клеток/мл), в котором клетки пролиферировали в течение ночи, и подвергали центрифугированию с охлаждением (3000 x g, 10 минут, 4°C), удаляли супернатанты, затем к каждому осадку добавляли 500 мкл фиксирующего раствора B (метанол/уксусная кислота = 3/1), смесь инкубировали в течение ночи при 4°C для сшивания хромосомной ДНК в бактериальных клетках и белков клеточной стенки, и, таким образом, ДНК предварительно фиксировали в клетках. В качестве контроля с использованием 500 мкл физиологического раствора вместо фиксирующего раствора B получали образец, в котором не проводили фиксацию. Затем использовали те же способы, что и способы в примере 7, и в заключение образец в 10 раз разбавляли с получением 250 мкл суспензии Enterobacter sakazakii в физиологическом растворе. Расчетная плотность клеток Enterobacter sakazakii в суспензии приблизительно составляла 3,5×107 клеток/мл. Эту суспензии в объеме 2,5 мкл применяли в качестве образца для амплификации ПЦР и добавляли в 12,25 мкл раствора средства для предварительной обработки, указанного в таблице 5 (при условии, что объем стерилизованной воды изменяли на 2,7 мкл), и в смесь добавляли 12,75 мкл буфера для ПЦР для детекции грамотрицательных бактерий, указанную ниже, с проведением ПЦР в указанных ниже условиях. Во время ПЦР получали порции в объеме 27,5 мкл в количестве 20 для каждого образца, полученного с использованием фиксирующего раствора, и контрольного образца. В качестве праймеров использовали прямой праймер 16S_1234F для детекции гена 16S рРНК (SEQ ID NO:3) и обратный праймер 23S_1703R для детекции гена 23S рРНК (SEQ ID NO:4), указанные в примере 3.
После завершения ПЦР 20 порций раствора для реакции ПЦР для каждого фиксирующего раствора объединяли в одно целое и подвергали центрифугированию с охлаждением (3000 x g, 10 минут, 4°C). Отбирали супернатант в объеме 5 мкл и использовали для электрофореза в геле с 0,8% агарозой, а оставшийся супернатант удаляли. Осадок дважды отмывали 500 мкл физиологического раствора, и выделяли ДНК с использованием набора QuickGene SP для выделения ДНК их такни.
Отдельно получали две порции по 500 мкл бульона с культурой, в которой клетки пролиферировали в течение ночи (4,3×108 клеток/мл) и подвергали центрифугированию с охлаждением (3000 x g, 10 минут, 4°C), супернатанты удаляли, затем к каждому осадку добавляли 500 мкл фиксирующего раствора B (метанол/уксусная кислота = 3/1) и смесь инкубировали в течение ночи при 4°C. Клетки 3 раза отмывали 500 мкл физиологического раствора, и ДНК выделяли и очищали с использованием набора QuickGene SP для выделения ДНК из ткани (Fuji Photo Film Co., Ltd.). В качестве контроля с использованием 500 мкл физиологического раствора вместо фиксирующего раствора B получали образец, в котором не проводили фиксацию. В соответствии с указанным выше описанием количество клеток в случае использования непосредственно после фиксирования только отмывки и немедленного выделения ДНК из осадка, так как образец 4 раза подвергали центрифугированию, всего составляло 4,3×108×0,5×0,41=0,9×108 клеток, и определено, что количество бактерий, используемых для ПЦР, в виде расчетной плотности клеток приблизительно составляет 3,5×107 клеток/мл ×2,5 мкл×20 = 1,8×106 клеток, и если принять в расчет, что образец затем дополнительные 3 раза подвергали центрифугированию, рассчитано, что оно составляет 0,9×106 клеток.
Так как в каждом случае количество бактерий, используемых на стадии выделения ДНК из образца перед ПЦР, было в 100 раз большим, чем количество бактерий, находящихся в образце после ПЦР, для использования их при том же количестве клеток получали 20 порций, каждая в объеме 2,5 мкл (= 1,8×106 клеток), суспензии Enterobacter sakazakii в физиологическом растворе (3,5×107 клеток/мл) (фиксирующий раствор B и контроль S), комбинировали в одно целое без проведения ПЦР, затем в совокупности 3 раза отмывали посредством центрифугирования и использовали для выделения ДНК.
Кроме того, даже если в бактериальных клетках Enterobacter sakazakii посредством указанного выше анализа подтверждали наличие длинных продуктов амплификации ПЦР длиной приблизительно 2450 п.н., так как продукты амплификации ПЦР, когда ПЦР проводили в присутствии средства для предварительной обработки, присутствовали во внешнем растворе, как показано на фиг. 14 или 15, можно неправильно понять, что продукты амплификации ПЦР во внешнем растворе могут адсорбироваться на клеточных стенках бактерий Enterobacter sakazakii, так как, если продукты амплификации ПЦР видимо присутствуют внутри клеток. Таким образом, дополнительно проводили указанный ниже эксперимент.
Получали образцы в количестве 20, добавляя в каждую пробирку для ПЦР 2,5 мкл водного раствора ДНК, содержащего 0,44 нг ДНК (440 пг, количество хромосомной ДНК, содержащееся в 8,8×104 клеток), очищенной из штамма Enterobacter sakazakii ATCC51329, добавляя раствор средства для предварительной обработки как в указанном выше случае, а затем добавляя буфер для ПЦР до общего объема 27,5 мкл, и ПЦР проводили с каждым из них в приведенных ниже условиях. Затем содержимое 20 пробирок для ПЦР объединяли в одно целое, к комбинированному реакционному раствору добавляли 50 мкл суспензии Enterobacter sakazakii в физиологическом растворе с 3,5×107 клеток/мл (обработанной фиксирующим раствором B или с отсутствием фиксации и 3 раза отмытой в соответствии со способом примера 7), смесь в достаточной степени смешивали и подвергали центрифугированию с охлаждением (3000 x g, 10 минут, 4°C) для удаления супернатанта, и осадок дважды отмывали, и использовали для выделения ДНК.
Состав буфера для ПЦР:
a) 16S_1234F (10 пмоль/мкл): 2 мкл
b) 23S_1703R (10 пмоль/мкл): 2 мкл
c) Ex-Taq (5 Ед/мкл, Takara-Bio): 0,25 мкл (содержащая 0,5% Tween 20, 0,5% Nonidet P-40 и 50% глицерин)
d) 10 x буфер Ex-Taq (Takara-Bio): 2,5 мкл
e) смесь dNTP (Takara-Bio): 2 мкл
f) 10 x SYBR Green I (BMA): 4 мкл
ПЦР с детекцией в реальном времени проводили в соответствии с приведенными ниже условиями термических циклов ПЦР с использованием устройства для ПЦР с детекцией в реальном времени (iCycler iQ, Bio-Rad, Hercules, CA).
1) 4°C в течение 3 минут (1 цикл)
2) 95°C в течение 15 секунд; 60°C в течение 20 секунд; 72°C в течение 3 минут (30 циклов)
3) 95°C в течение 3 минут (1 цикл)
Затем в соответствии с протоколом анализа плавления продуктов амплификации ПЦР (температуру поднимали с интервалами 0,1°C от 60°C, каждую температуру поддерживали в течение 8 секунд, и эту процедуру повторяли 350 раз в совокупности до конечной температуры 95°C) измеряли температуру плавления продуктов амплификации ПЦР.
2. Результаты
Результаты представлены на фиг. 16. Результаты электрофореза супернатантов реакционных смесей, полученных в ПЦР, проводимых в присутствии средства для предварительной обработки (16S-23S: 2450 п.н.) с клетками Enterobacter sakazakii, фиксированными с использованием фиксирующего раствора B, или без фиксации представлены на дорожках 2 и 3, результаты электрофореза ДНК, полученной посредством выделения ДНК из осадка, полученного после ПЦР, указанной выше, и дважды отмытого, представлены на дорожках 5 и 6, результаты электрофореза ДНК, непосредственно выделенной из фиксированных или нефиксированных клеток Enterobacter sakazakii, которые являлись тестовыми материалы в этом эксперименте, представлены на дорожках 7 и 8, результаты электрофореза ДНК, выделенной из фиксированных или нефиксированных клеток Enterobacter sakazakii перед тем, как их использовали для реакции ПЦР, представлены на дорожках 9 и 10, и результаты электрофореза ДНК, выделенной из клеток Enterobacter sakazakii дважды отмытых после добавления предварительно полученных продуктов амплификации ПЦР, представлены на дорожках 13 и 14, соответственно.
Как видно из результатов на дорожках 13 и 14, показано, что даже если продукт ПЦР-гена (2450 п.н.) адсорбирован на наружных мембранах клеточных стенок бактерий Enterobacter sakazakii и т.д. со стороны внешнего раствора, продукт ПЦР-гена можно удалить из бактерий посредством двойной отмывки фиксированных и нефиксированных клеток. Таким образом, возможность некорректной интерпретации, что продукт ПЦР-гена, который может адсорбироваться на внешних поверхностях клеточных стенок, можно рассматривать как продукт ПЦР-гена, оставшийся в клетках устранена. Кроме того, принимая в расчет, что фрагмент, по расчетам являющийся продуктом реакции ПЦР, детектировали в дорожках 5 и 6, что означает, что результаты для ДНК, выделенной из клеток, осадок которых дважды отмывали после ПЦР и что фрагмент не является продуктом ПЦР-гена, который может быть адсорбирован на внешней стороне клеточной стенки, как видно из результатов на дорожках 13 и 14, продукт ПЦР-гена на дорожках 5 и 6 с высокой вероятностью представляет собой продукт ПЦР-гена, оставшийся в клетках, а затем выделенный. Кроме того, даже если концентрации продукта ПЦР-гена во внешнем растворе и в клетках становился одинаковым, так как клетки являлись бактериальными клетками клеточные стенки которых были повреждены, и, таким образом, продукт ПЦР-гена находился в состоянии, когда он мог свободно проходить через клеточные стенки, объем внешнего раствора был приблизительно в 1010 раз большим, чем объем клеток, и количество продукта ПЦР-гена во внешнем растворе должно быть приблизительно в 1010 раз больше, чем количество продукта ПЦР-гена в клетках, то есть в клетках должно быть распределено 1/1010 количества во внешнем растворе. Однако на основе сравнения интенсивностей полос на дорожках 2, 3, 5 и 6 нельзя считать, что количество продукта ПЦР, оставшегося в клетках, составляло 1/1010 количества. То есть предполагали, что ПЦР, проходящая в способе по настоящему изобретению, возможно представляет собой ПЦР in situ. Как видно из результатов на дорожках 7 и 8, хромосомную ДНК детектировали в фиксированных и нефиксированных клетках бактерии Enterobacter sakazakii, используемых в качестве тестовых материалов, но никаких полос хромосомной ДНК на дорожках 5, 6, 9 и 10 не получали. Это отличие вероятно обусловлено различием количества клеток бактерий Enterobacter sakazakii, используемого для выделения ДНК, в частности количества клеток 0,9×106 с высокой вероятностью недостаточно для выделения ДНК, тогда как на дорожках 7 и 8 для тестовых материалов количество клеток фактически составляло до 0,9×108 клеток.
[Пример 9]
Клетки Enterobacter sakazakii подвергали обработке кипячением в физиологическом растворе или в присутствии средства для предварительной обработки и исследовали как много хромосомной ДНК Enterobacter sakazakii выходило в каждый супернатант в зависимости от продолжительности обработки.
1. Экспериментальные способы
Клетки в бульоне с культурой штамма Enterobacter sakazakii ATCC51329 (1,1×109 клеток/мл), в котором клетки пролиферировали в течение ночи, отмывали и разбавляли в 10 раз физиологическим раствором, и суспензию подвергали центрифугированию с охлаждением (3000 x g, 10 минут, 4°C) с последующим получением осадка, к осадку добавляли равный объем (500 мкл) физиологического раствора или раствора средства для предварительной обработки с составом, приведенным в таблице 5, и клетки в достаточной степени суспендировали. Затем суспензию нагревали в кипящей вода в течение периода от 0 до 5 минут и после нагревания сразу же охлаждали. Каждую суспензию непосредственно после нагревания в объеме 5 мкл и супернатант каждой суспензии, полученный посредством центрифугирования с охлаждением, в объеме 5 мкл подвергали электрофорезу на 0,8% агарозном геле, соответственно.
2. Результаты
На фиг. 17 представлены степени выхождения хромосом Enterobacter sakazakii, подвергнутых тепловой обработке в физиологическом растворе или в присутствии средства для предварительной обработки с использованием кипящей воды, в супернатант. Во-первых, в отношении результатов на дорожках 2 и 9, хотя клетки не подвергали нагреванию, уже показано присутствие незначительного количества хромосомной ДНК, но полагают, что это происходило вследствие достижения клетками в бульоне с культурой, в котором клетки пролиферировали в течение ночи, стадии покоя, таким образом, лизировали часть погибших клеток, и их хромосомная ДНК выходила во внешний раствор. Таким образом, такое незначительное количество хромосомной ДНК при оценке игнорировали. Хотя определяли, что ДНК в физиологическом растворе вследствие нагревания выходит из бактериальных клеток Enterobacter sakazakii, никаких полос хромосомных ДНК для суспензии и супернатанта в присутствии средства для предварительной обработки не детектировали даже после нагревания в течение 5 минут. Однако когда оценку проводили на основе наблюдения за лунками на дорожках 10 и 12, в лунке наблюдали полосу в случае суспензии, но никаких полос для супернатанта не наблюдали, и это демонстрирует, что хромосомная ДНК не выходит из бактериальных клеток в присутствии средства для предварительной обработки, а остается в клетках. С другой стороны, полосы также наблюдали в лунках для супернатантов физиологического раствора (дорожки 5, 6 и 7), и полагают, что они происходят из части погибших клеток Enterobacter sakazakii, попадающих в супернатант после центрифугирования, удельная плотность которых снижается вследствие кипячения. Результаты, приведенные в таблице 6 или на фиг. 12, также поддерживают это предположение. На основе приведенных выше результатов полагают, что хотя, если быть точным, условия отличались от условий повторения термического цикла ПЦР, ДНК в присутствии средства для предварительной обработки сложнее выходить из бактериальных клеток, даже когда клетки подвергают тепловой обработке.
Применимость в промышленности
В соответствии со способом по настоящему изобретению можно детектировать жизнеспособные клетки микроорганизма с высокой чувствительностью c различением их от погибших клеток или поврежденных клеток. Настоящее изобретение обеспечивает простое и быстрое различение жизнеспособных клеток, поврежденных клеток и погибших клеток микроорганизма в продуктах питания, биологических образцах, мазках и образцах окружающей среды, таких как промышленные воды, вода из окружающей среды и сточные воды, на основе способа амплификации нуклеиновых кислот. Способ и набор по настоящему изобретению можно использовать для произвольного исследования, и они также выгодны с экономической точки зрения.
По предпочтительному варианту осуществления настоящего изобретения его также можно применять для санитарной проверки различных продуктов питания, содержащих 5 log10 клеток/мл или более поврежденных клеток или погибших клеток Escherichia coli, или для диагностики бактериемии у детей, при которой в крови циркулирует Escherichia coli.
Кроме того, по предпочтительному варианту осуществления настоящего изобретения в продуктах питания, с высокой чувствительностью (1 КОЕ/2,22 мл молока) и более быстро (7 часов и 30 минут) по сравнению с регламентированным способом (закон о пищевой санитарии/постановление министерства о стандартах ингредиентов для молока и молочных продуктов), можно детектировать только жизнеспособные клетки бактерий кишечной группы, включая бактерии Enterobacteriaceae, таким образом, предполагают их использование для определения перед отгрузкой продукции предприятия после производства на различных пищевых комбинатах, представленных молочными фабриками, и полагают, что это является очень полезным для промышленности.
Кроме того, настоящее изобретение обеспечивает быстрые детекцию и количественное определение только жизнеспособных микроорганизмов при низкой концентрации, для микроорганизмов, включающих не только бактерии кишечной группы и бактерии Enterobacteriaceae, но также различные бактерии, включая патогенные бактерии, вирусы и т.д., и, таким образом, его можно применять для различных видов санитарной проверки, клинического тестирования, технологического контроля и т.д.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ДЕТЕКЦИИ МИКРООРГАНИЗМА И НАБОР ДЛЯ ДЕТЕКЦИИ МИКРООРГАНИЗМА | 2006 |
|
RU2395583C2 |
| СПОСОБ И НАБОР ДЛЯ ДЕТЕКТИРОВАНИЯ МИКРООРГАНИЗМА | 2008 |
|
RU2410440C1 |
| СПОСОБ КОЛИЧЕСТВЕННОГО АНАЛИЗА КОЛИЧЕСТВА КЛЕТОК ПРЕДСТАВЛЯЮЩЕЙ ИНТЕРЕС БАКТЕРИИ В ЖИВОМ СОСТОЯНИИ С ПРИМЕНЕНИЕМ рРНК В КАЧЕСТВЕ МИШЕНИ | 2006 |
|
RU2420595C2 |
| СРЕДСТВО ДЛЯ РАЗРУШЕНИЯ НУКЛЕИНОВОЙ КИСЛОТЫ И СПОСОБ РАЗРУШЕНИЯ НУКЛЕИНОВОЙ КИСЛОТЫ | 2007 |
|
RU2388473C2 |
| СПОСОБ СПЕЦИФИЧЕСКОГО ВЫДЕЛЕНИЯ ПОЛНОГО ДНК-СОДЕРЖИМОГО БАКТЕРИАЛЬНЫХ ВОЗБУДИТЕЛЕЙ ИНФЕКЦИИ | 2010 |
|
RU2567809C2 |
| СПОСОБ ВИДОВОЙ ДИФФЕРЕНЦИАЦИИ ЖИЗНЕСПОСОБНЫХ РОДОКОККОВ, ИММОБИЛИЗОВАННЫХ В ГЕЛЕВОМ НОСИТЕЛЕ | 2013 |
|
RU2525934C1 |
| Способ детекции чумного микроба и набор реагентов для его осуществления | 2022 |
|
RU2796820C1 |
| ОПРЕДЕЛЕНИЕ СООТНОШЕНИЯ НУКЛЕИНОВЫХ КИСЛОТ | 2018 |
|
RU2803339C2 |
| СПОСОБ ОПРЕДЕЛЕНИЯ УРОВНЯ ЭКСПРЕССИИ ХИМЕРНОГО ГЕНА Trim5a | 2015 |
|
RU2592675C1 |
| АНАЛИТИЧЕСКИЙ СПОСОБ ДЛЯ КОЛИЧЕСТВЕННОЙ ОЦЕНКИ ЖИЗНЕСПОСОБНЫХ БАКТЕРИЙ, СОДЕРЖАЩИХСЯ В КОМПОЗИЦИЯХ ДЛЯ ТЕРАПИИ ДЛЯ ВОССТАНОВЛЕНИЯ МИКРОБИОТЫ (MRT) | 2017 |
|
RU2714841C1 |




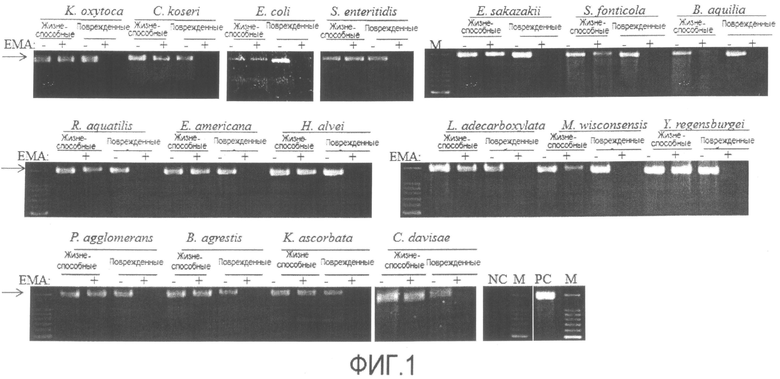

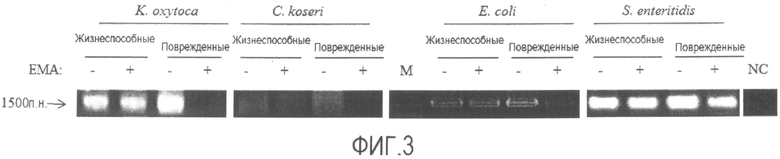
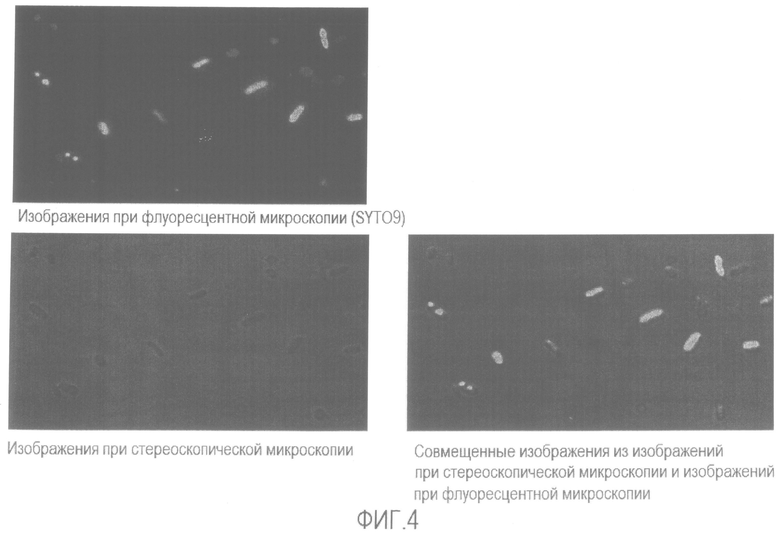
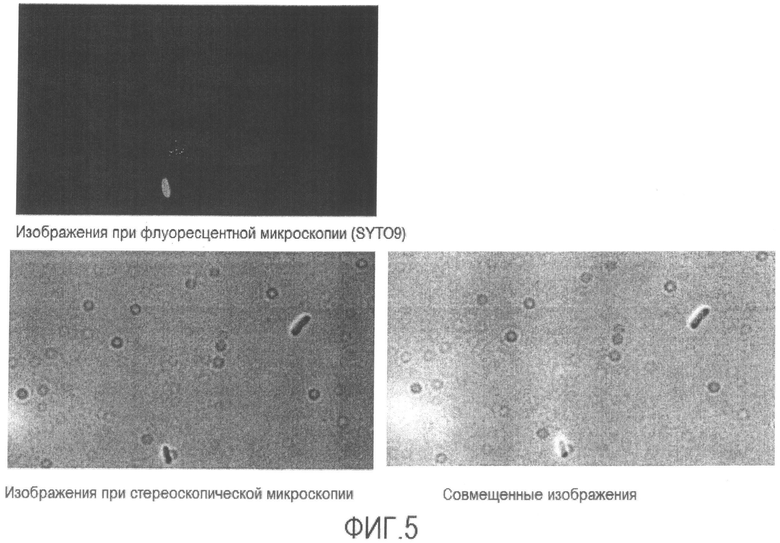
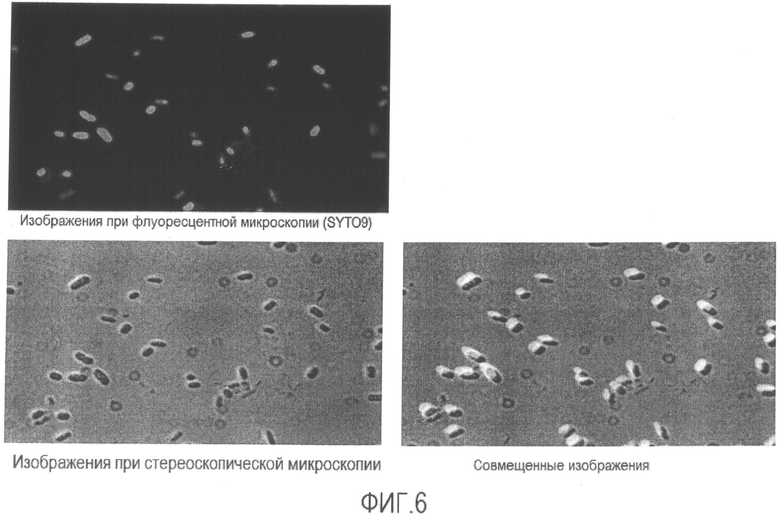
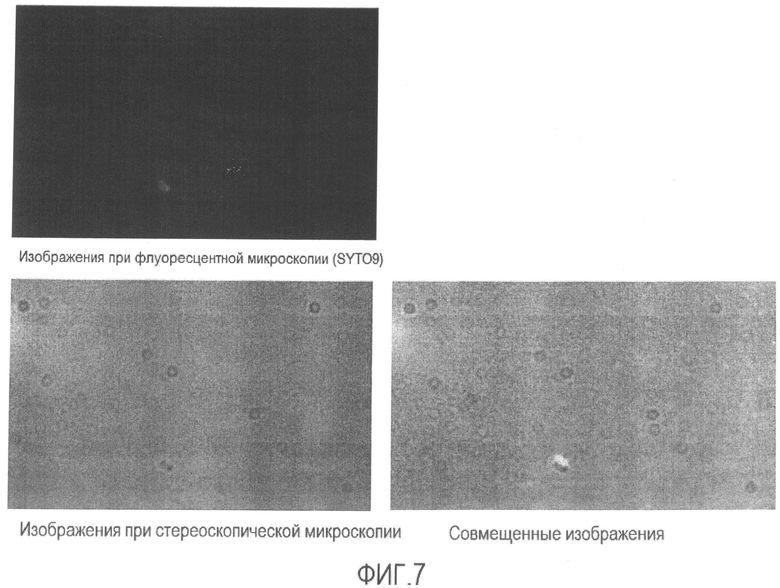
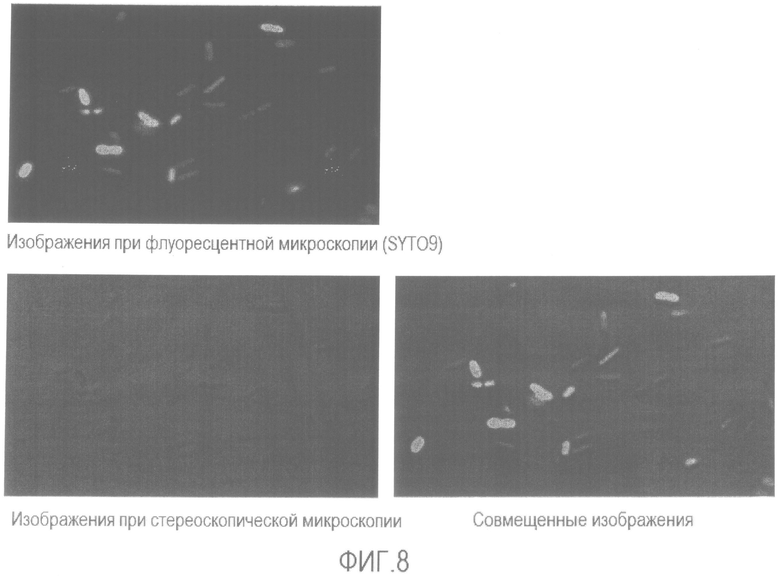
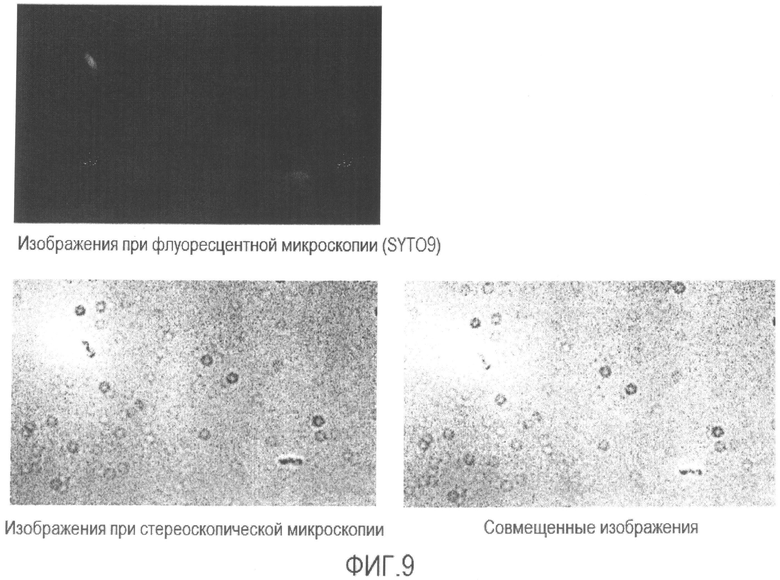
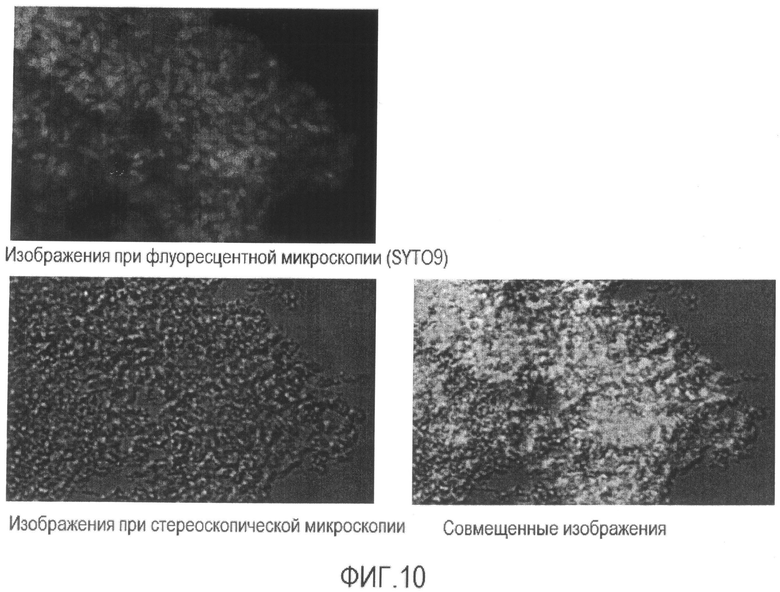
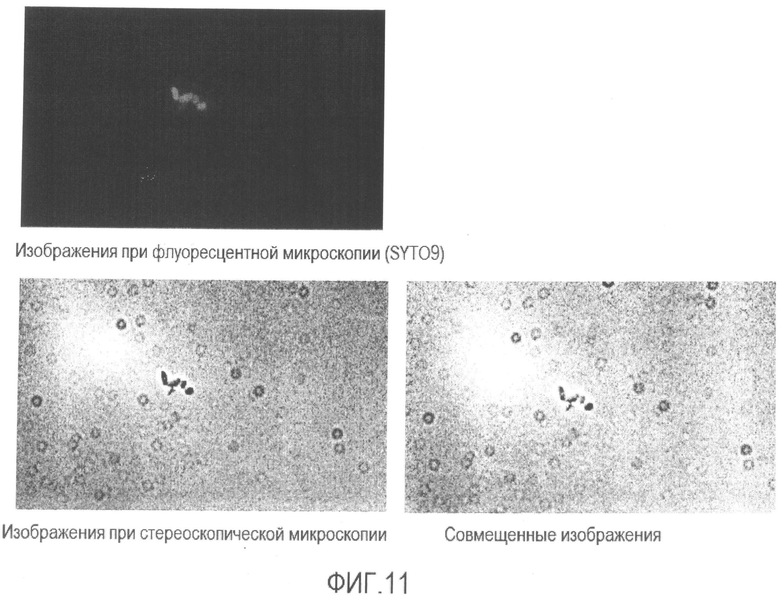
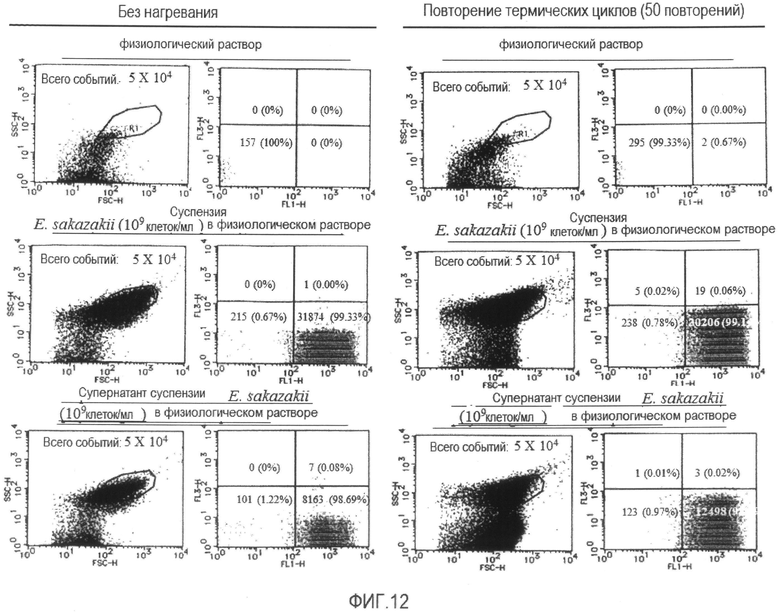
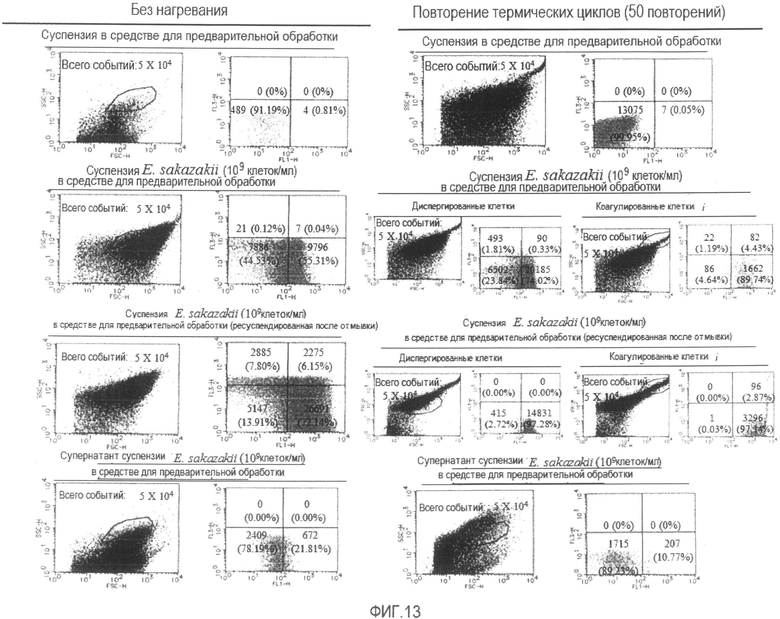
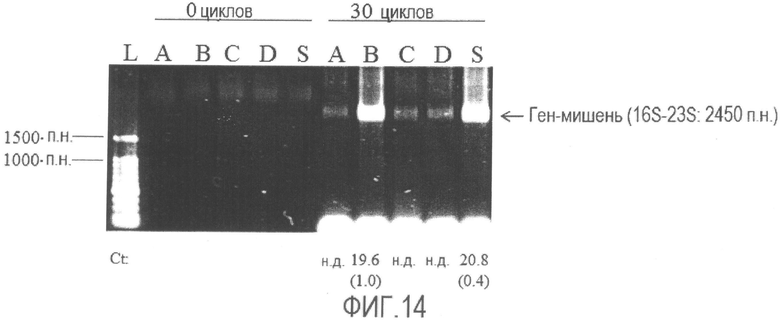
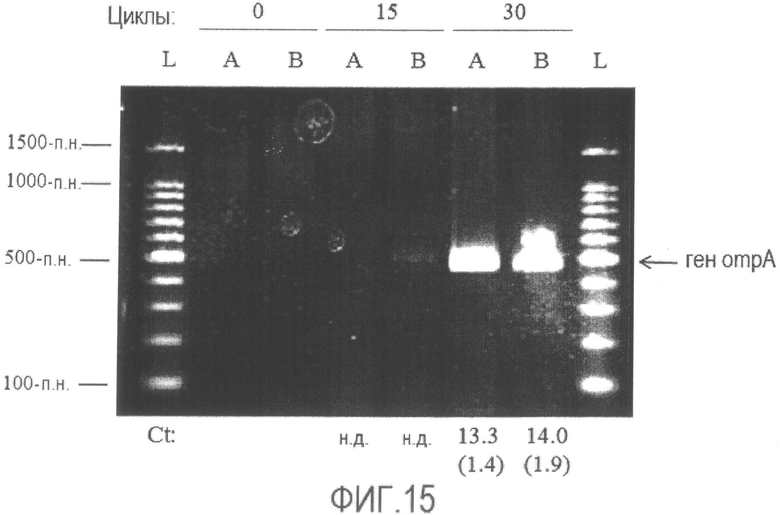
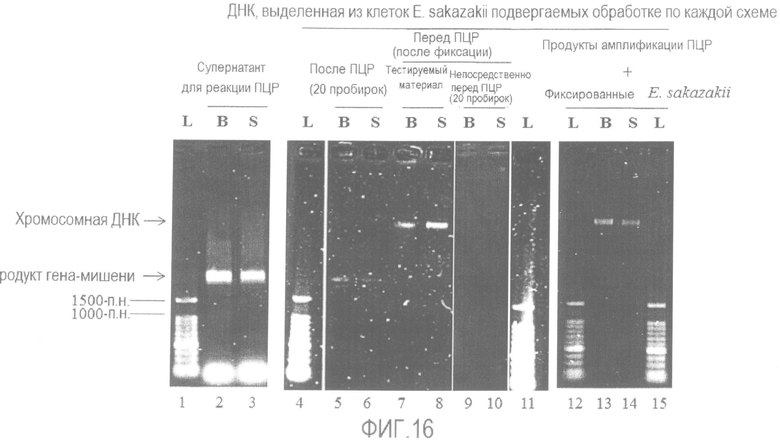
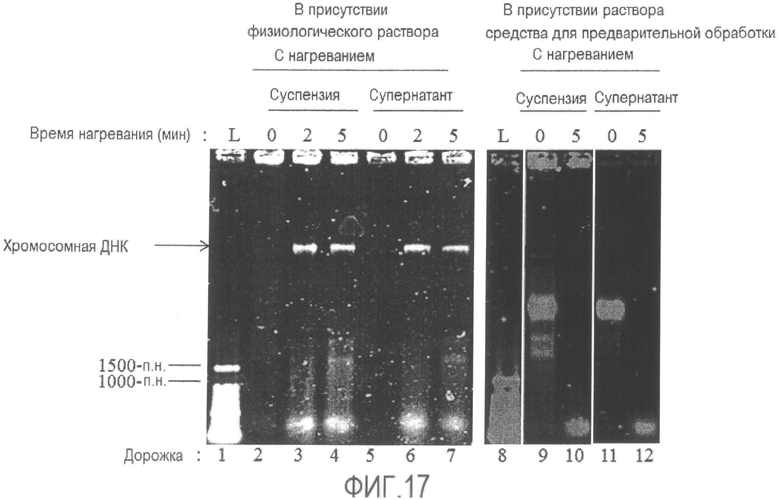
Группа изобретений относится к области микробиологии и биотехнологии. Способ детекции живых клеток микроорганизма в тестируемом образце путем отличия живых клеток от мертвых клеток или поврежденных клеток предусматривает добавление в тестируемый образец средства, способного к ковалентному связыванию с ДНК или РНК при облучении светом с длиной волны от 350 нм до 700 нм; облучение тестируемого образца; амплификацию мишеневой области ДНК или РНК микроорганизма, содержащегося в тестируемом образце, способом амплификации нуклеиновых кислот в присутствии средства подавления действия вещества, ингибирующего амплификацию нуклеиновых кислот, соли магния, соли органической кислоты или соли фосфорной кислоты, без выделения нуклеиновых кислот из клеток и анализа продукта амплификации. Способ может быть осуществлен посредством применения набора, состоящего из средства, способного к ковалентному связыванию с ДНК или РНК при облучении светом с длиной волны от 350 нм до 700 нм, средства подавления действия вещества, ингибирующего амплификацию нуклеиновых кислот и праймеров для амплификации мишеневой области. При помощи данных изобретений можно с высокой чувствительностью и точностью детектировать живые клетки микроорганизмов в различных биологических образцах. 2 н. и 25 з.п. ф-лы, 17 ил., 12 табл., 9 пр.
1. Способ детекции живых клеток микроорганизма в тестируемом образце путем отличия живых клеток от мертвых клеток или поврежденных клеток, который включает стадии:
a) добавления в тестируемый образец средства, способного к ковалентному связыванию с ДНК или РНК при облучении светом с длиной волны от 350 нм до 700 нм;
b) облучения тестируемого образца, в который добавляют средство, светом с длиной волны от 350 нм до 700 нм;
c) амплификации мишеневой области ДНК или РНК микроорганизма, содержащегося в тестируемом образце, способом амплификации нуклеиновых кислот в присутствии средства подавления действия вещества, ингибирующего амплификацию нуклеиновых кислот, соли магния, и соли органической кислоты или соли фосфорной кислоты, без выделения нуклеиновых кислот из клеток; и
d) анализа продукта амплификации.
2. Способ по п.1, где амплификацию мишеневой области проводят в клетках микроорганизма.
3. Способ по п.1 или 2, где на стадии с) амплификацию мишеневой области проводят в присутствии поверхностно-активного вещества.
4. Способ по п.1 или 2, где перед стадией с), повторно проводят стадии а) и b).
5. Способ по п.1 или 2, где перед стадией а), проводят следующую стадию е):
e) обработка тестируемого образца ферментом с активностью разрушения клеток, отличных от клеток микроорганизма, коллоидных частиц белка, липидов или сахаридов, присутствующих в тестируемом образце.
6. Способ по п.5, где фермент выбран из фермента, разрушающего белки, фермента, разрушающего липиды, и фермента, разрушающего сахариды.
7. Способ по п.1 или 2, где тестируемый образец представляет собой любое из продукта питания, биологического образца, питьевой воды, промышленных вод, воды из окружающей среды, сточных вод, почвы и мазка.
8. Способ по п.1 или 2, где микроорганизм представляет собой бактерию или вирус.
9. Способ по п.8, где бактерия представляет собой грамотрицательную бактерию.
10. Способ по п.1 или 2, где средство, способное к ковалентному связыванию с ДНК или РНК при облучении светом с длиной волны от 350 нм до 700 нм, выбрано из моноазида этидия, диазида этидия, моноазида пропидия, псоларена, 4,5',8-триметилпсоларена и 8-метоксипсоларена.
11. Способ по п.1 или 2, где средство для подавления действия вещества, ингибирующего амплификацию нуклеиновых кислот, представляет собой один или несколько классов, выбранных из альбумина, декстрана, белка гена 32 Т4, ацетамида, бетаина, диметилсульфоксида, формамида, глицерина, полиэтиленгликоля, соевого ингибитора трипсина, α2-макроглобулина, тетраметилхлорида аммония, лизоцима, фосфорилазы и лактатдегидрогеназы.
12. Способ по п.1 или 2, где соль органической кислоты выбрана из соли уксусной кислоты, соли пропионовой кислоты и соли лимонной кислоты.
13. Способ по п.1 или 2, где соль фосфорной кислоты представляет собой соль пирофосфорной кислоты.
14. Способ по п.1 или 2, где мишеневая область представляет собой мишеневую область длиной от 50 до 5000 нуклеотидов.
15. Способ по п.14, где мишеневая область представляет собой мишеневую область, соответствующую гену, выбранному из гена 5S рРНК, гена 16S рРНК, гена 23S рРНК и гена тРНК ДНК тестируемого образца.
16. Способ по п.1 или 2, где способ амплификации нуклеиновых кислот представляет собой ПЦР, ПЦР-РВ, LAMP, SDA, LCR, ТМА, TRC, НС или способ с использованием микропанелей.
17. Способ по п.16, где ПЦР проводят в виде ПЦР с детекцией в реальном времени с одновременным проведением ПЦР и анализа продуктов амплификации.
18. Способ по п.1 или 2, где анализ продуктов амплификации проводят с использованием стандартной кривой, представляющей собой зависимость количества микроорганизма и продукта амплификации, которую получают с использованием стандартных образцов микроорганизмов.
19. Набор для детекции живых клеток микроорганизма в тестируемом образце путем отличия живых клеток от мертвых клеток или поврежденных клеток способом амплификации нуклеиновых кислотпо п.1 или 2, где набор содержит следующие компоненты:
1) средство, способное к ковалентному связыванию с ДНК или РНК при облучении светом с длиной волны от 350 нм до 700 нм;
2) средство для подавления действия вещества, ингибирующего амплификацию нуклеиновых кислот, соль магния, и соль органической кислоты или соль фосфорной кислоты; и
3) праймер или праймеры для амплификации области-мишени ДНК или РНК микроорганизма, подлежащего детекции способом амплификации нуклеиновых кислот.
20. Набор по п.19, который дополнительно содержит поверхностно-активное вещество.
21. Набор по п.19 или 20, который дополнительно содержит фермент с активностью разрушения клеток, отличных от клеток микроорганизма, коллоидных частиц белка, липидов или сахаридов, присутствующих в тестируемом образце.
22. Набор по п.19 или 20, где способ амплификации нуклеиновых кислот представляет собой ПЦР, ПЦР-РВ, LAMP, SDA, LCR, ТМА, TRC, НС или способ с использованием микропанелей ДНК.
23. Набор по п.19 или 20, где средство, способное к ковалентному связыванию с ДНК или РНК при облучении светом с длиной волны от 350 нм до 700 нм, выбрано из моноазида этидия, диазида этидия, моноазида пропидия, псоларена, 4,5',8-триметилпсоларена и 8-метоксипсоларена.
24. Набор по п.19 или 20, где средство для подавления действия вещества, ингибирующего амплификацию нуклеиновых кислот, представляет собой один или несколько классов, выбранных из альбумина, декстрана, белка гена 32 Т4, ацетамида, бетаина, диметилсульфоксида, формамида, глицерина, полиэтиленгликоля, соевого ингибитора трипсина, α2-макроглобулина, тетраметилхлорида аммония, лизоцима, фосфорилазы и лактатдегидрогеназы.
25. Набор по п.19 или 20, где соль органической кислоты выбрана из соли уксусной кислоты, соли пропионовой кислоты и соли лимонной кислоты.
26. Набор по п.19 или 20, где соль фосфорной кислоты представляет собой соль пирофосфорной кислоты.
27. Набор по п.21, где фермент выбран из фермента, разрушающего белки, фермента, разрушающего липиды, и фермента, разрушающего сахариды.

