Настоящее изобретение относится к каталитическому способу производства акролеина путем дегидратации глицерола или глицерина и к применению этого способа.
Под глицеролом понимается очищенный или неочищенный глицерол, предпочтительно, полученный из биомассы, в частности высокоочищенный глицерол или частично очищенный глицерол. Очищенный глицерол имеет чистоту, большую или равную 98%, и его получают путем дистилляции глицерина. Неочищенный или лишь частично очищенный глицерол может быть представлен в виде раствора в метиловом спирте, если он получен, например, путем переэтерификации триглицеридов, как это описано далее.
Под глицерином понимается, в частности, глицерин природного происхождения, полученный путем гидролиза растительных масел и/или животных жиров, или глицерин синтетического происхождения, получаемый из нефти, более или менее очищенный или рафинированный или же сырой с концентрацией от 80 до 85%. Таким образом, в дальнейшем описании речь главным образом идет о химическом превращении глицерола или глицерина, полученных из биомассы, но настоящее изобретение, разумеется, этим не ограничивается, и его объем распространяется на все разновидности глицерола или глицерина, независимо от источника получения и степени чистоты.
Исчерпаемость ископаемых источников энергии подводит промышленное производство к необходимости использования возобновляемого сырья, получаемого из биомассы, для получения горючего. В этом отношении горючим, производимым из растительного или животного масла, является биологическое дизельное топливо.
Этот продукт известен как "зеленый" по причине весьма приемлемого баланса CO2 по сравнению с ископаемыми источниками энергии. Diester® (EMVH, Esters Methyliques d'Huiles Vegetales - метиловые эфиры растительных масел) является биологическим дизельным топливом, производимым путем переэтерификации метанолом триглицеридов, содержащихся в маслянистых жидкостях, в частности в растительных маслах, таких как пальмовое, рапсовое и подсолнечное. При такой переэтерификации, осуществляемой рассматриваемыми способами, образуется также приблизительно 100 кг глицерола на тонну Diester®. Нелипидная часть используемого сырья, жмыхи, главным образом находит применение в откорме животных.
Такое биологическое дизельное топливо применяется в смеси с газойлем. Директивы ЕС 2001/77/ЕС и 2003/30/ЕС, которые вступят в силу в ближайшем будущем, предполагают включение в дизельное топливо в 2010 г. 7%, а к 2015 г. 10% Diester®. Столь значительное повышение количества производимого биологического дизельного топлива приведет к получению значительного количества глицерола, составляющего несколько сотен тысяч тонн в год.
Уже предложено около 1500 различных вариантов использования глицерола. Вот в качестве примера некоторые из них, присутствующие во многих весьма разных рецептурах:
гидратанты в фармации (в свечах и сиропах) или в косметологии в составе увлажняющих кремов, глицериновых мыл, зубных паст, растворители в пищевой промышленности, пластификаторы или смазки в химической промышленности.
Эти области применения окажутся явно недостаточными для утилизации количества глицерола, получаемого при производстве биодизеля, и рынок глицерола (мыло, фармация и т.д.) несмотря на свое расширение будет неспособен утилизировать все излишки. Таким образом, крайне важно найти новые области применения, позволяющие утилизировать весьма значительные объемы глицерола.
Имея в виду эту насущную необходимость, в последние годы было исследовано множество выходов из ситуации (см. М. Pagliaro et al., Angew. Chem. Int. Ed. (2007) 46, 4434-4440, М. Pagliaro, М. Rossi: The Future of Glycerol, RSC Publishing, Cambridge (2008)), предусматривающих, в частности, следующие шесть путей утилизации:
- превращение в 1,3-пропандиол и 1,2-пропандиол, используемые, в частности, как исходные мономеры для синтеза полиэфиров и полиуретанов,
- превращение в моноэфиры для производства смазок,
- превращение в полиглицеролы, используемые как пищевые добавки-эмульгаторы,
- превращение в акролеин (путем дегидратации) и акриловую кислоту (путем дегидратации и окисления),
- прямая утилизация в качестве кормовых добавок для животных.
Акролеин и акриловую кислоту традиционно производят путем контролируемого окисления пропилена в газовой фазе кислородом воздуха в присутствии катализаторов на основе оксидов молибдена и/или висмута. Полученный таким образом акролеин может быть либо напрямую задействован в двухэтапном процессе производства акриловой кислоты, либо использоваться как промежуточный продукт синтеза. Таким образом, производство этих двух мономеров напрямую связано с пропиленом, который, в основном, получают путем крекинга с водяным паром или каталитического крекинга нефтяных фракций.
Рынок акролеина, одного из самых простых ненасыщенных альдегидов, и акриловой кислоты огромен, поскольку эти мономеры входят в состав многочисленных продуктов массового производства.
Кроме того, акролеин, будучи благодаря своему строению весьма реакционно-способным веществом, находит многочисленные применения, в частности, как промежуточное соединение при синтезе других продуктов. Например, его используют в синтезе D,L-метионина и его гидроксиакалога - 2-гидрокси-4-метилтиобутановой кислоты (ГМТБК, ГМТБК). Эти кормовые добавки находят массовое применение, поскольку входят в состав улучшителей кормов, необходимых для роста животных (птицы, свиней, жвачных, рыбы и т.д.) В некоторых случаях он может быть полезен для повышения или даже для обеспечения существующих производственных мощностей, расширяя спектр пригодного сырья. Таким образом, представляется весьма полезным увеличить производство акролеина, сократив при этом зависимость от пропилена как от ресурса, получаемого из нефти.
Задачей настоящего изобретения является создание мощных, активных, селективных и регенерируемых катализаторов, позволяющих получить акролеин напрямую из глицерола или из глицерина, в частности, полученных из биомассы, в соответствии с реакцией:
НО-СН2-СН(ОН)-СН2-ОН→СН2=СН-СНО+2Н2O
Эта альтернатива также предоставляет собой конкурентоспособный способ синтеза акролеина, не зависящий от пропилена как нефтяного ресурса, из другого, возобновляемого сырья.
Эта возможность является особенно перспективной для синтеза метионина и его аналогов, таких как его гидроксианалог (ГМТБК), напрямую из биомассы.
Настоящее изобретение относится также к применению этой реакции для синтеза 3-метилтиопропионового альдегида, 2-гидрокси-4-метилтиобутиронитрила (ГМТБН), метионина и его аналогов, таких как 2-гидрокси-4-метилтиобутановая кислота (ГМТБК), эфиров ГМТБК, таких как изопропиловый эфир; 2-оксо-4-метилтиобутановой кислоты из акролеина.
Метионин, ГМТБК, ее эфиры и аналоги используют при откорме животных и в промышленных процессах синтеза. Акролеин обычно получают путем окисления пропилена и/или пропана. Окисление пропилена в акролеин с помощью воздуха в присутствии водяного пара является частичным и получаемый продукт-сырец на основе акролеина содержит также не вступившие в реакцию пропилен и пропан, воду и побочные продукты окисления, такие как кислоты, альдегиды и спирты.
Глицерол (называемый также глицерином), давно известен как источник акролеина (термическое превращение). Это вещество широко распространено в природе в форме эфиров (триглицеридов), в частности в составе всех масел, а также животных и растительных жиров, что делает его реактивом, доступным в большом количестве и поэтому промышленно применимым. Хорошо известно, что глицерол разлагается с образованием акролеина при нагреве до температуры выше 280°С. Эта низкоселективная реакция сопровождается образованием многочисленных побочных продуктов, в частности уксусного альдегида, гидроксиацетона, а также продуктов полного окисления СО, CO2. Следовательно, необходимо контролировать реакцию превращения глицерина в акролеин во избежание избыточного расхода этого ресурса и необходимости дополнительной энергоемкой очистки полученного акролеина. Кроме того, побочные продукты, по большей части ароматические, часто являются причиной образования слоя кокса на поверхности катализатора, что со временем приводит к порче катализатора, и часто приходится регенерировать катализатор для восстановления удовлетворительной каталитической активности.
Многие исследователи в фундаментальной и прикладной области изучали эту реакцию. Было, в частности, предложено использовать сверхкритическую воду в качестве реакционной среды. Использование сверхкритического растворителя в промышленном масштабе оказывается затруднительным при непрерывном процессе по причине особо сложного оборудования, в частности, автоклавов, которые работают при очень высоком давлении. Напротив, осуществление непрерывного или осуществляющегося с перерывами производства оказывается возможным в случае предложения производительной, селективной и стабильной каталитической системы.
В связи с растущим интересом к такой химической альтернативе в литературе описано множество исследований, связанных с использованием 2 каталитических систем на основе иммобилизованных фосфо- или кремневольфрамовых гетерополикислот, смешанных оксидов и цеолитов, применимых в непрерывных или осуществляющихся с перерывами способах производства в жидкой или газовой фазе.
Так, документы WO-A-2006087083 и WO-A-2006087084 описывают способ каталитической дегидратации глицерина с получением акролеина в газовой фазе в присутствии молекулярного кислорода и сильнокислотного катализатора, выбираемого из цеолитов, нафиона (Nation*), оксидов металлов, выбираемых из алюминия, циркония, титана, ниобия, тантала, кремния, импрегнированных кислотными группами в форме сульфатных, боратных, вольфраматных, силикатных и фосфатных групп.
Документ WO-A-2007132926 описывает способ превращения глицерола в акролеин в присутствии катализатора, выбираемого из кислых кристаллических металлосиликатов, таких как цеолиты структурного типа MFI или ВЕА, содержащих кремний и элемент, предпочтительно выбираемый из Al, Fe и Ga.
В отличие от известных способов согласно описываемому предложен способ получения акролеина из глицерола или из глицерина путем каталитической дегидратации глицерина в присутствии катализатора, который, обеспечивая превращение всего исходного глицерина, в то же время может быть очень легко регенерирован и обладает долгим сроком службы. Авторы настоящего изобретения открыли, что такими свойствами обладает катализатор на основе оксида циркония, состоящий, по меньшей мере, из:
а) смешанного оксида циркония и по меньшей мере одного металла М, где указанный металл выбран из ниобия, тантала и ванадия,
б) оксида циркония и по меньшей мере одного оксида металла М, где указанный металл выбран из ниобия, тантала и ванадия,
в) оксида кремния и смешанного оксида циркония и по меньшей мере одного металла М, где указанный металл выбран из вольфрама, церия, марганца, ниобия, титана, ванадия и кремния,
г) оксида кремния и смешанного оксида циркония и по меньшей мере одного оксида металла М, где указанный металл выбран из вольфрама, церия, марганца, ниобия, тантала, ванадия и титана,
д) оксида титана и смешанного оксида циркония и по меньшей мере одного металла М, где указанный металл выбран из вольфрама, церия, марганца, ниобия, тантала, титана, ванадия и кремния.
е) оксида титана и смешанного оксида циркония и по меньшей мере одного оксида металла М, где указанный металл выбран из вольфрама, церия, марганца, ниобия, тантала, титана, ванадия и кремния.
Таким образом, настоящее изобретение относится к способу получения акролеина из глицерола или глицерина в присутствии катализатора, как определено выше, и применения такого катализатора для превращения глицерола или глицерина в акролеин. Катализатор согласно настоящему изобретению обеспечивает контролируемое превращение глицерола или глицерина в акролеин, то есть не способствует его дальнейшему превращению в акриловую кислоту. С этой целью предпочтительный катализатор согласно настоящему изобретению не содержит вообще оксида молибдена и/или оксида меди, либо не содержит эти оксиды в значительной массовой доле по отношению к каждому из прочих оксидов, составляющих катализатор.
Поэтому настоящее изобретение относится к применению по меньшей мере одного из катализаторов а), б), в), г), д) и е), как определено выше, для превращения глицерола или глицерина в акролеин.
Катализатор могут получать различными способами (путем совместного осаждения, гидротермального синтеза и т.п.). Эффективный способ был описан в литературе (Kantcheva et al., Catalysis Communications (2008), 9(5), p.874-879 и в патентах FR 2907444 и FR 2907445).
Вышеописаный катализатор также может соответствовать предпочтительным характеристикам, приведенным ниже, рассматриваемым по отдельности или в сочетании: катализаторы от а) до е) состоят только из вышеописанных оксидов и смешанных оксидов, при этом по меньшей мере один оксид, смешанный или нет, в составе катализаторов от а) до е) иммобилизован; молярное отношение Zr/сумма других элементов, составляющих указанные катализаторы от а) до е), отличных от Zr, то есть выбранных среди Si, Ti и М, составляет от 0,5 до 200, более предпочтительно, от 1 до 100. Как указано выше, катализатор согласно настоящему изобретению выгодно отличается тем, что может легко быть регенерирован без снижения выхода реакции дегидратации и селективности в отношении акролеина.
Реакция согласно настоящему изобретению может быть осуществлена в газовой или жидкостной фазе, предпочтительно в газовой фазе. При осуществлении реакции в газовой фазе способ могут осуществлять с помощью различных технологий, а именно в неподвижном слое, в псевдоожиженном слое или в псевдоожиженном слое с циркуляцией. В двух первых вариантах, в неподвижном слое или в псевдоожиженном слое, регенерация катализатора может быть отделена от каталитической реакции. Регенерацию могут осуществлять ex situ общепринятыми методами, такими как сжигание на воздухе или в газовой смеси, содержащей молекулярный кислород. В соответствии со способом согалсно настоящему изобретению регенерацию могут осуществлять in situ, поскольку температура и давление, при которых происходит регенерация, близки к условиям реакции, осуществляемой этим способом.
В жидкой фазе реакцию могут осуществлять в обычном реакторе для реакций в жидкой фазе на твердом катализаторе, а также в реакторе с каталитической дистилляцией, принимая во внимание значительную разницу температуры кипения глицерола (290°С) и акролеина (53°С). Также есть смысл рассматривать осуществление реакции в жидкой фазе при относительно низкой температуре, которая обеспечивает непрерывную отгонку произведенного акролеина, ограничивая таким образом последующие реакции разрушения акролеина.
Экспериментальными условиями реакции в газовой фазе являются температура от 250 до 400°С и давление от 1 до 10 бар. В жидкой фазе реакцию проводят при температуре от 150 до 350°С и давлении в пределах от 3 до 70 бар.
Другое достоинство способа согласно настоящему изобретению состоит в том, что исходный глицерол или глицерин может быть представлен в чистом или частично очищенном виде либо в виде раствора, в частности водного. Предпочтительно используют водный раствор глицерола. В водном растворе концентрация глицерола предпочтительно составляет не менее 1%, лучше, если она составляет от 10 до 50 масс.% и предпочтительно от 15 до 30 масс.% в реакторе. Концентрация глицерола не должна быть слишком высокой во избежание побочных реакций, снижающих выход акролеина, таких как образование эфиров глицерола или ацетализация произведенного акролеина с непревращенным глицерином. С другой стороны, раствор глицерола не должен быть и слишком разбавленным с учетом неизбежного расхода энергии, вызываемого испарением глицерола. Во всех случаях концентрацию раствора глицерола легко можно довести до нужной путем частичной или полной утилизации воды, получаемой в ходе реакции.
Энергетическая оптимизация в рамках синтеза может состоять в утилизации теплоты на выходе реакции для испарения потока глицерола, поступающего в реактор.
Другим объектом настоящего изобретения является способ получения из акролеина 3-(метилтио)пропионового альдегида, 2-гидрокси-4-метилтиобутиронитрила (ГМТБН), метионина, 2-гидрокси-4-метилтиобутановой кислоты (ГМТБК), эфиров последней, в частности изопропилового эфира, и 2-оксо-4-метилтиобутановой (ОМТБК) кислоты, согласно которому акролеин получают способом, описанным выше. Если сравнивать с общепринятым способом получения акролеина путем контролируемого окисления пропилена, акролеин, полученный вышеуказанным способом, может содержать примеси, отличающиеся от обычных, как с точки зрения их количества, так и их природы. Таким образом, для получения акриловой кислоты, метионина или его гидроксианалога можно предусмотреть предварительную очистку акролеина способами, известными специалистам.
Акролеин, полученный согласно настоящему изобретению, напрямую или после очистки, вводят в реакцию с метилмеркаптаном для получения 3-(метилтио)пропионового альдегида (МТПА). На следующем этапе МТПА обрабатывают синильной кислотой с получением 2-гидрокси-4-(метилтио)бутиронитрила (ГМТБН). После синтеза ГМТБН различные этапы синтеза приводят к получению метионина, его гидроксианалога (ГМТБК), эфиров последнего или его оксоаналога (ОМТБК). Все этапы, начиная от синтеза акролеина, хорошо знакомы специалистам.
Далее настоящее изобретение будет более подробно описано и проиллюстрировано нижеследующими примерами и рисунками без ограничения объема формулы изобретения.
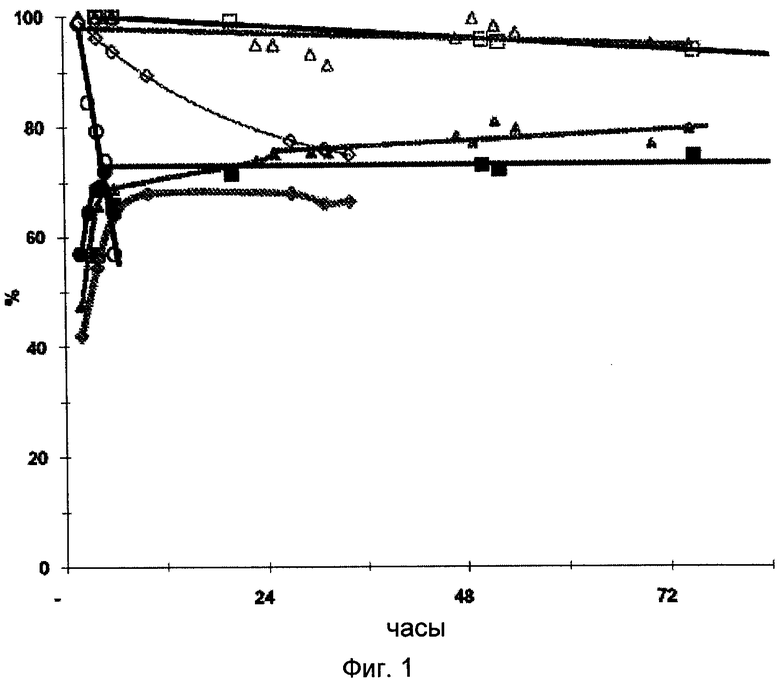
На Фигуре 1 представлена зависимость превращения глицерола и селективности по акролеину от времени для каждого из катализаторов А, В, С и D, описанных в примерах 1, 7, 8 и 9 соответственно; катализаторы А и В соответствуют настоящему изобретению, катализаторы С и D соответствуют предшествующему уровню техники. Время, указанное для каждой точки - это время окончания отбора проб, соответствующее улавливанию в течение одного часа. Условия реакции и использованные способы подсчета превращения и селективности по акролеину описаны далее.
Обозначения на этой фигуре следующие:
- превращение глицерола на катализаторе А (□), В (Δ), С (◇) или D (о)
- селективность по акролеину на катализаторе А (■), В (▲), С (♦) или D (•)
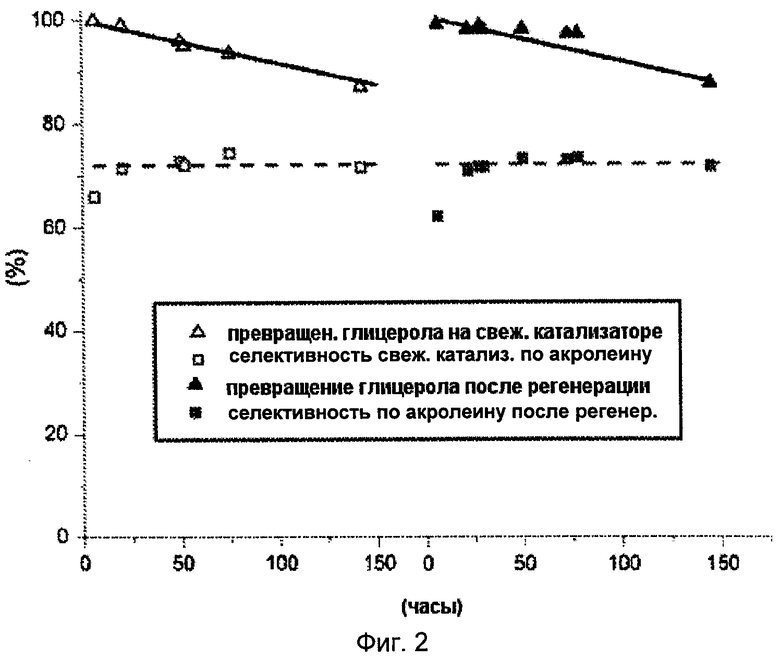
Фигура 2 иллюстрирует превращение глицерола и селективность по акролеину для катализатора А согласно настоящему изобретению до и после регенерации в потоке воздуха.
Обозначения на этой фигуре следующие:
- превращение по глицеролу на свежем катализаторе (Δ) и на регенерированном катализаторе (▲)
- селективность по акролеину на свежем катализаторе (□) и на регенерированном катализаторе (■)
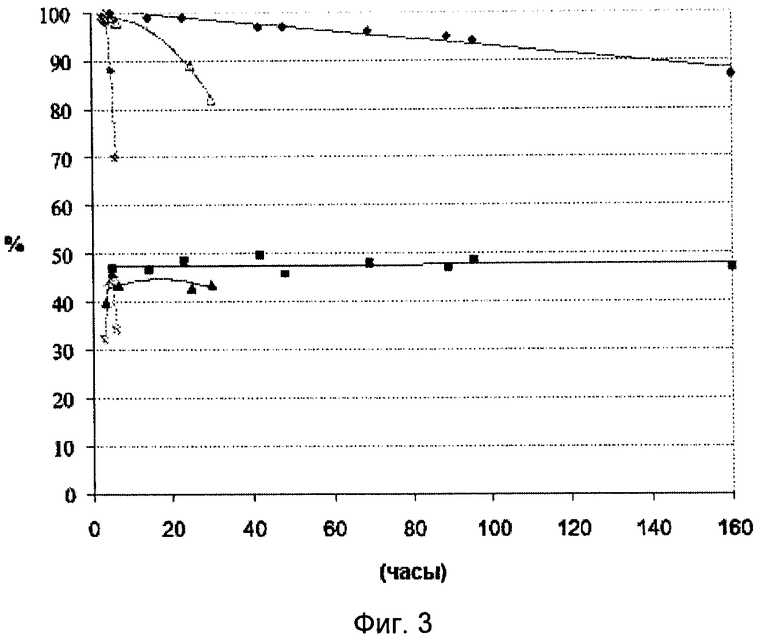
На Фигуре 3 представлено сравнение превращения глицерола и селективности этого превращения по акролеину во времени для каждого из катализаторов А', В и Г, описанных в примерах 2, 8 и 9 соответственно; катализатор А' соответствует настоящему изобретению, катализаторы В и Г соответствуют предшествующему уровню техники.
Обозначения на этой фигуре следующие:
- превращение глицерола на катализаторе A'(♦), D (•) или С (□)
- селективность по акролеину на катализаторе А'(■), D (×) или С (▲)
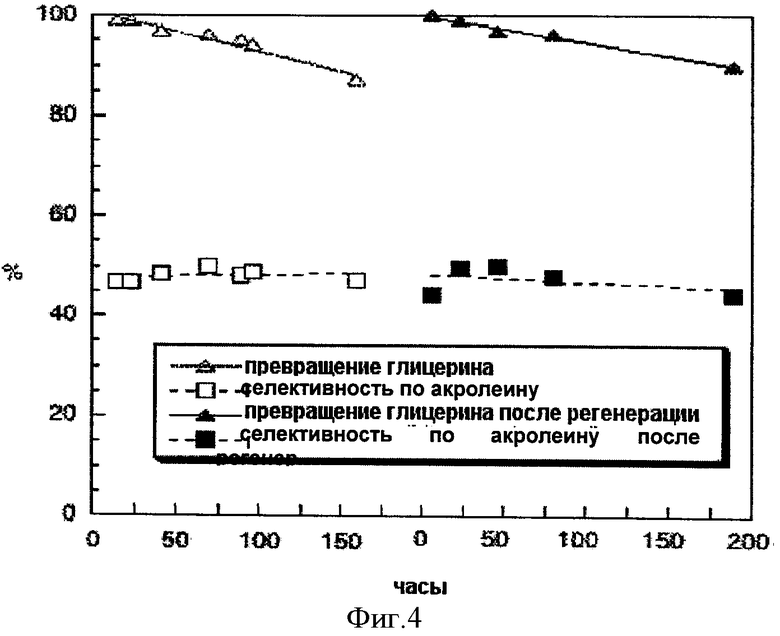
Фигура 4 иллюстрирует превращение глицерола и селективность по акролеину для катализатора А' согласно настоящему изобретению до и после регенерации в потоке воздуха.
Обозначения на этой фигуре следующие:
- превращение глицерола на свежем катализаторе (Δ) и на регенерированном катализаторе (▲)
- селективность по акролеину на свежем катализаторе (□) и на регенерированном катализаторе (•)
Время, указанное для каждой точки - это время окончания отбора проб, соответствующее улавливанию в течение одного часа. Условия реакции и использованные способы подсчета превращения и селективности по акролеину описаны далее.
Реакцию дегидратации глицерола производили на указанных катализаторах при атмосферном давлении в прямом реакторе с неподвижным слоем диаметром 18 мм. Реактор помещали в печь, которая позволяла поддерживать в катализаторе необходимую для реакции температуру, которая составляла 300°С. Объем катализатора, загруженного в реактор, составлял 4,5 мл, при этом толщина слоя составляла около 1,8 см. В реактор подавали 20%-ный (мас.) водный раствор глицерола с расходом 3,77 г/час. Водный раствор глицерола испаряли с помощью испарителя С.Е.М 15 (Controlled Evaporator Mixer) Bronkhorst* в потоке азота 75 мл/мин. Примерное молярное отношение глицерин/вода/азот составляло 2,3/46,3/51,4. Расчетное время контакта составляло порядка 1,9 с, что соответствует показателю GHSV 1930 h-1. Время контакта определяли следующим образом:
Время контакта = Объем катализатора×Pатм/(общий молярный расход×Температура×R),
где Ратм=101325 Па, Температура = 25°С, а общий молярный расход = молярный расход глицерола+молярный расход воды+молярный расход инертного газа.
По окончании реакции продукты конденсировали. Применяли две системы конденсации. В примерах 10, 11, 12, 16, 17 и 18 использовали систему с тремя ловушками, смонтированными последовательно. Первая ловушка содержала воду в определенном количестве и охлаждалась колотым льдом. Две другие ловушки содержали этанол и охлаждались в криостате при -25°С. В примерах 13, 14 и 15 использовали простую ловушку с определенной массой воды, которая охлаждалась колотым льдом. Длительность конденсации составляла 1 час, и питающий поток не прерывался при смене ловушек.
Полученные продукты анализировали хроматографически, каждый образец анализировали дважды.
Основные продукты реакции анализировали посредством газовой хроматографии в капиллярной колонке (Nukol, 30 м×0,53 мм) на хроматографе Shimadzu 2014, оборудованном детектором FID. При этом количественно определяли акролеин, уксусный альдегид, ацетон, пропионовый альдегид, гидроксипропанон, уксусную кислоту, аллиловый спирт и фенол.
Оставшийся глицерол количественно определяли посредством газовой хроматографии на хроматографе Helwett Packard, оборудованном детектором FID и капиллярной колонкой (Carbowax или ZBwax, 30 м×0,32 мм).
Превращение глицерола, селективность по акролеину и выходы различных продуктов определяли следующим образом:
Превращение глицерола (%)=100×(1 - остаточное число молей глицерола/исходное число молей глицерола)
Селективность по акролеину (%)=100×(число молей произведенного акролеина/число молей глицерола, вступившего в реакцию)
Выход Х (%) = К×100×число молей произведенного Х / исходное число молей глицерола
При этом К=1, если Х представляет собой акролеин, ацетон, гидроксипропанон, пропаналь или акриловый спирт; К=2/3, если Х представляет собой уксусный альдегид или уксусную кислоту и К=2, если Х является фенолом.
Пример 1: приготовление и характеристика катализатора А
Катализатор согласно настоящему изобретению на основе оксидов циркония и ниобия готовили из гидрата оксида циркония и оксалониобата аммония (NH4)(C2O4)2NbO.×H2O (Aldrich, 99.99%). Гидрат оксида циркония готовили путем соосаждения раствора оксонитрата циркония ZrО(NО3)2.×Н2O (Aldrich, 99%) и 28%-ного раствора аммиака при рН=8,8.
Оксалониобат аммония растворяли в пермутированной воде, подкисленной концентрированной HNО3 до рН~0,5 и нагретой до 45°С. После остывания до температуры окружающей среды добавляли гидрат оксида циркония с молярным отношением ZrO2/Nb2O5, равном 3:1, перед этим определяли уровень гидратации гидрата оксида циркония путем термогравиметрического анализа. Спустя 24 часа при перемешивании смесь фильтровали, а осадок прокаливали в потоке воздуха при 600°С. Удельная поверхность этого катализатора составляла 40 м2/г. Удельные поверхности порошков измеряли способом BET (Brunauer-Emmet-Teller) при -196°С при помощи аппарата Micromeritics ASAP 2020. Порошки предварительно подвергали десорбции при 300°С на протяжении 3 час в вакууме 5×10-5 мбар. Содержание ниобия и диоксида циркония в различных приготовленных катализаторах определяли способом ICP-OES (inductively coupled plasma optical emission spectroscopy оптическая эмиссионная спектрометрия с индуктивно связанной плазмой). Молярное соотношение Zr/Nb в катализаторе А, рассчитанное на основе проведенных анализов, составляло 9,3.
Пример 2: приготовление и характеристика катализатора А'
Катализатор согласно настоящему изобретению на основе оксидов циркония и ниобия готовили в соответствии с процедурой, описанной в литературе (Kantcheva et al., Catalysis Communications (2008), 9(5), p874-879), путем импрегнации гидрата оксида циркония.
Гидрат оксида циркония готовили путем соосаждения раствора оксонитрата циркония ZrC(NO3)2.×H2O (Aldrich, 99%) и 28%-ного раствора аммиака. Предшественник Nb(V), (NH4)(C2O4)2NbO.×H2O (Aldrich, 99.99%), добавляли при перемешивании к 35%-ному раствору перекиси водорода (Sigma Aldrich), подкисленной до рН=0,5 добавлением HNО3, концентрированной и нагретой до 50°С. Молярное отношение Н2О2/оксалат составляло 13/1. Раствор нагревали 1 час при 50°С, затем остужали до температуры окружающей среды. Далее добавляли гидрат оксида циркония до отношения ZrO2:Nb2O5=6:1, уровень гидратации гидрата оксида циркония предварительно определяли путем термогравиметрического анализа. Смесь оставляли при перемешивании на 24 часа при температуре окружающей среды, после чего жидкую фазу отгоняли при пониженном давлении при t°<70°С. Полученный остаток прокаливали в потоке воздуха при 600°С.
Удельная поверхность этого катализатора составляла 51 м2/г. Удельную поверхность порошков измеряли способом BET (Brunauer-Emmet-Teller) при -196°С при помощи аппарата Micromeritics ASAP 2020. Порошки предварительно подвергали десорбции при 300°С в течение 3 часов в вакууме 5×10-5 мбар. Содержание ниобия и циркония в разных приготовленных катализаторах определяли способом ICP-OES. Молярное отношение Zr/Nb в этом катализаторе составляло 3,3.
Пример 3: приготовление и характеристика катализатора Е
Катализатор согласно настоящему изобретению на основе оксидов циркония и ниобия готовили в соответствии с процедурой, описанной в литературе (Kantcheva et al., Catalysis Communications (2008), 9(5), стр.874-879), путем импрегнации гидрата оксида циркония раствором, содержащим смешанный оксалат аммония и ниобия.
Предшественник Nb(V), (NH4)(C2O4)2NbO.×H2O (Aldrich, 99.99%), добавляли при перемешивании к 35%-ному раствору перекиси водорода (Sigma Aldrich), подкисленной до рН=0,5 добавлением концентрированной HNО3 и нагретой до 50°С. Молярное отношение Н2О3/оксалат составляло 13/1.
Раствор нагревали 1 час при 50°С, затем остужали до температуры окружающей среды.
Далее добавляли гидрат оксида циркония, предварительно приготовленный путем совместного осаждения раствора оксонитрата циркония ZrО(NO3)2.×Н2O (Aldrich, 99%) и 28%-ного раствора аммиака в соотношении ZrO2:Nb2O5=6:1. Смесь оставляли перемешиваться на 24 часа при температуре окружающей среды, после чего жидкую фазу отгоняли при пониженном давлении при t°<70°C. Полученный остаток прокаливали в потоке воздуха при 600°С.
Удельная поверхность этого катализатора, определенная так же, как и в случае катализатора А, составляла 39 м2/г. Содержание ниобия и циркония в различных приготовленных катализаторах определяли способом ICP-OES. Молярное отношение Zr/Nb в этом катализаторе составило 3,7.
Пример 4: приготовление и характеристика катализатора F
Катализатор согласно настоящему изобретению на основе оксидов циркония, ниобия и ванадия. Предшественник ванадия готовили из NH4VO3 (Sigma, ACS Reagent 99,7%) следующим способом:
Метаванадат аммония растворяли в 9%-ном растворе перекиси водорода, содержащем щавелевую кислоту (Aldrich, 99%). Молярное отношение щавелевая кислота/NН4VО3 составляло 1,3. Через 1 час перемешивания при температуре окружающей среды раствор выпаривали при пониженном давлении, при этом получали голубой порошок. Содержание оксида ванадия в этом веществе определяли термогравиметрически.
Предшественник ниобия - смешанный оксалат ниобия-аммония (NH4)(C2O4)2NbO×H2O (Aldrich, 99.99%) и гидрат оксида циркония, полученный, как описано в примере 1, вводили в водный раствор, подкисленный концентрированной НNО3 (pН<0,5), в молярном отношении Zr/Nb/V, равном 72/22/3,2. Спустя 24 часа при перемешивани реакционную смесь фильтровали и осадок прокаливали в потоке воздуха при 600°С.
Удельная поверхность этого катализатора, определенная так же, как и в случае катализатора А, составляла 48 м2/г. Содержание ниобия, ванадия и циркония в полученном катализаторе определяли способом ICP-OES. Молярное отношение Zr/Nb/V в этом катализаторе составляло 90,4/8,4/1,2.
Пример 5: приготовление и характеризация катализатора G
Катализатор согласно настоящему изобретению на основе циркония и вольфрама с присадкой кремнезема. Приготовление этого катализатора включает три этапа. Первая стадия представляет собой синтез гидрата оксида циркония при рН=8,8. Вторая стадия состоит в стабилизации гидрата оксида циркония частицами кремнезема (Nahas et al. - Journal of Catalysis 247 (2007), p51-60). Гидроксид циркония помещали в стеклянную колбу с раствором аммиака, рН которого доводили до 11. Смесь кипятили с обратным холодильником 72 часа, затем фильтровали и промывали пермутированной водой. Последний этап состоял в реакции обмена между вольфрамовой кислотой H2WO4 (Aldrich, 99%), растворенной в перекиси водорода, и гидроксидом циркония. Вольфрамовую кислоту растворяли в 35%-ном растворе перекиси водорода при 60°С. Концентрация вольфрамовой кислоты в растворе составляла 0,04 М. Затем раствор вольфрамовой кислоты остужали при комнатной температуре и постепенно добавляли гидроксид циркония, легированный кремнеземом. Полученный осадок отделяли фильтрованием и прокаливали на воздухе при 650°С. Его удельная поверхность составляла 40 м2/г. Содержание вольфрама, кремния и циркония в катализаторе определяли способом ICP-OES. Молярное отношение W/Si/Zr в этом катализаторе составляло 4,7/1,4/93,9.
Пример 6: синтез катализатора Н
Катализатор Н готовили способом, описанным в примере 1. рН раствора азотной кислоты в случае катализатора Н подбирали так, чтобы он был несколько более кислым (pН<0,1). Полученный катализатор имеет удельную поверхность 57 м2/г и молярное отношение Zr/Nb, равное 11,8.
Пример 7: приготовление и характеристика катализатора В
Катализатор ZrTiSiW согласно настоящему изобретению был приготовлен Родиа (Rhodia) способом, описанным в патенте FR 2907445 A. Удельная поверхность этого катализатора, определенная так же, как и в случае катализатора А, составляла 105 м2/г. Массовая доля оксидов в этом катализаторе составляла 54% ZrO2, 35% ТiO2, 7,5% SiO2 3,5% WO3.
Пример 8: Приготовление и характеристика катализатора С (соответствующего предшествующему уровню техники, для сравнения)
Катализатор С представляет собою вольфрамированный диоксид циркония (89,5% ZrO2 - 10,5% WO3), синтезированный Даличи Кигенсо (Daiichi Kigenso, код поставщика: Z-1104). Удельная поверхность этого катализатора, определенная так же, как и в случае катализатора А, составляла 77 м2/г
Пример 9: приготовление и характеристика катализатора D (соответствующего предшествующему уровню техники, для сравнения)
Катализатор D представляет собой цеолит H-ZSM-5 (Zeochem, ZEOcat PZ-2/50H). Удельная поверхность этого катализатора, определенная так же, как и в случае катализатора А, составляла 406 м2/г.
Пример 10: Каталитическая дегидратация глицерола с образованием акролеина: оценка катализаторов А, В, С и D
В таблице 1 приведена производительность, достигающаяся при применении катализаторов А, В, С и D через 6 часов реакции.
Эта таблица показывает, что при равном объеме катализатора только катализаторы А и В (соответствующие настоящему изобретению) обеспечивают полное превращение глицерола. Кроме того, катализаторы согласно настоящему изобретению обладают лучшей селективностью в отношении акролеина, заметной уже через 6 часов и еще более явной через 50 часов с выходом акролеина 70% для катализатора А и 80% для катализатора B.
Таким образом, катализаторы А и В более активны и более селективны, чем катализаторы, соответствующие уровню техники.
Пример 11: Каталитическая дегидратация глицерола с образованием акролеина: изменение во времени производительности катализаторов А, В, С и D
Изменение производительности катализаторов А, В, С и D в зависимости от времени, определенное в тех же условиях, что в примере 4, представлено на Фигуре 1.
Катализаторы А и В (соответствующие настоящему изобретению) сохраняют постоянную селективность по акролеину и высокую степень превращения глицерола в течение нескольких дней, в отличие от катализаторов С и D из уровня техники, которые в значительной степени дезактивируются менее чем через 24 часа.
Таким образом, катализаторы А и В согласно настоящему изобретению более активны, более селективны в отношении акролеина, а также более долговечны, чем лучшие из катализаторов, раскрытых в предшествующем уровне техники.
Пример 12: Регенерация катализатора А
Через 143 часа инкубации реакционной смеси при 300°С с перемешиванием Катализатор А согласно настоящему изобретению регенерировали в потоке воздуха при 450°С в течение 2 час(расход воздуха 51 мл/мин). После регенерации катализатор испытывали в тех же рабочих условиях, что и до регенерации.
Полученные результаты представлены на Фигуре 2. Регенерация на воздухе при 450°С позволила катализатору А восстановить свою изначальную активность и отдачу. Таким образом, катализатор А согласно настоящему изобретению может быть регенерирован за короткое время без потери активности и селективности. Катализатор А не только активен и селективен, но может также быть легко и полностью регенерирован.
Пример 13: Каталитическая дегидратация глицерола с образованием акролеина: сравнение каталитических свойств катализаторов А', D и С
В таблице 2 приведены характеристики производительности, полученные при применении катализаторов А', С и D через 5 часов реакции при 300°С
Эта таблица показывает, что при равном объеме катализатора только катализатор А' (согласно настоящему изобретению) обеспечивает полное превращение глицерола. Кроме того, катализатор А' обладает лучшей селективностью в отношении акролеина. Таким образом, катализатор А' более активен и более селективен, чем катализаторы, соответствующие уровню техники.
Пример 14: Каталитическая дегидратация глицерола с образованием акролеина: изменение во времени производительности катализаторов А', D и С
Изменение производительности катализаторов А', D и С в зависимости от времени представлено на Фигуре 3.
Катализатор А' (соответствующий настоящему изобретению) сохраняет почти постоянную селективность по акролеину и высокую степень превращения глицерола в реакционном потоке в течение недели, в отличие от катализаторов С и D из уровня техники, которые в значительной степени дезактивируются менее чем через 24 часа.
Таким образом, катализатор А' по настоящему изобретению более активен, более селективен по акролеину, а также более долговечен, чем лучшие катализаторы, заявленные в предшествующем уровне развития техники.
Пример 15: Регенерация катализатора А'
Спустя 183 ч работы в реакционной смеси катализатор А' согласно настоящему изобретению регенерировали в потоке воздуха при 450°С в течение 1 часа (расход воздуха 51 мл/мин). После регенерации катализатор испытывали в тех же рабочих условиях, что и до регенерации.
Полученные результаты представлены на фигуре 4.
Регенерация на воздухе при 450°С позволила катализатору А' восстановить свою изначальную активность и отдачу. Таким образом, катализатор А' согласно настоящему изобретению может быть регенерирован за короткое время без потери активности и селективности. Катализатор А не только активен и селективен, но может также быть легко и полностью восстановлен.
Пример 16: Каталитическая дегидратация глицерола с образованием акролеина: оценка катализаторов Е и F (согласно настоящему изобретению)
В таблице 3 приведены характеристики катализаторов Е и F
Пример 17: Каталитическая дегидратация глицерола с образованием акролеина: оценка катализатора G (согласно настоящему изобретению)
В таблице 3 приведены характеристики катализатора.
Пример 18: Получение акролеина из неочищенного глицерола с применением катализатора Н
Производительность катализатора Н была определена с использованием неочищенного технического раствора глицерина с концентрацией 82 масс.%. Этот глицерин содержал до 15% метилового спирта. Как и в предыдущих примерах, объем катализатора в реакторе составлял 4,5 мл, расход азота - 74,5 мл/мин, а температура реакции 300°С. Расход 20%-ного (по массе) водного раствора глицерина составлял 3,77 г/час. Молярное отношение глицерол/вода/азот составляло 1,9/46,5/51,6. Полученные результаты представлены в таблице 5.
Присутствие значительного количества метилового спирта не ухудшает производительность катализатора, соответствующего настоящему изобретению.
Изобретение относится к улучшенному способу получения акролеина из глицерола. При этом дегидратацию глицерола осуществляют в присутствии катализатора на основе оксида циркония, состоящего по меньшей мере из: а) смешанного оксида циркония и по меньшей мере одного металла M, где указанный металл выбирают из ниобия, тантала и ванадия, б) оксида циркония и по меньшей мере одного оксида металла M, где указанный металл выбирают из ниобия, тантала и ванадия, в) оксида кремния и смешанного оксида циркония и по меньшей мере одного металла M, где указанный металл выбирают из вольфрама, церия, марганца, ниобия, тантала, титана, ванадия и кремния, г) оксида титана, смешанного оксида циркония и по меньшей мере одного металла M, где указанный металл выбирают из вольфрама, церия, марганца, ниобия, тантала, титана, ванадия и кремния. Способ позволяет получать акролеин путем каталитической дегидратации глицерола в присутствии катализатора, который обеспечивает превращение всего исходного глицерина и в то же время может быть легко регенерирован за короткое время без потери активности и селективности и обладает долгим сроком службы. Изобретение также относится к способу получения 3-(метилтио)пропионового альдегида из акролеина и к применению катализатора, выбранного из катализаторов а), б), в) или г) для превращения глицерола в акролеин. 3 н. и 10 з.п. ф-лы, 4 ил., 5 табл., 18 пр.
1. Способ получения акролеина из глицерола, отличающийся тем, что дегидратацию глицерола осуществляют в присутствии катализатора на основе оксида циркония, состоящего по меньшей мере из:
а) смешанного оксида циркония и по меньшей мере одного металла M, где указанный металл выбирают из ниобия, тантала и ванадия,
б) оксида циркония и по меньшей мере одного оксида металла M, где указанный металл выбирают из ниобия, тантала и ванадия,
в) оксида кремния и смешанного оксида циркония и по меньшей мере одного металла M, где указанный металл выбирают из вольфрама, церия, марганца, ниобия, тантала, титана, ванадия и кремния,
г) оксида титана, смешанного оксида циркония и по меньшей мере одного металла M, где указанный металл выбирают из вольфрама, церия, марганца, ниобия, тантала, титана, ванадия и кремния.
2. Способ по п.1, отличающийся тем, что катализатор состоит по меньшей мере из: а) смешанного оксида циркония и по меньшей мере одного металла M и б) оксида циркония и, по меньшей мере, одного оксида металла M.
3. Способ по п.1 или 2, отличающийся тем, что по меньшей мере один из оксидов в составе указанных катализаторов с а) по г) является иммобилизованным.
4. Способ по п.1, отличающийся тем, что молярное отношение Zr/сумма элементов Si, Ti и M, отличных от Zr, составляет от 0,5 до 200.
5. Способ по п.4, отличающийся тем, что указанное молярное отношение составляет от 1 до 100.
6. Способ по п.1, отличающийся тем, что глицерол находится в водном растворе с концентрацией не менее 1 масс.%.
7. Способ по п.6, отличающийся тем, что концентрация водного раствора глицерола составляет от 10 до 50 масс.%.
8. Способ по п.1, отличающийся тем, что катализатор регенерируют.
9. Способ получения 3-(метилтио)пропионового альдегида из акролеина, отличающийся тем, что акролеин получают способом по любому из пп.1-8.
10. Способ по п.1 или 9, отличающийся тем, что реакцию дегидратации осуществляют в газовой фазе.
11. Способ по п.10, отличающийся тем, что реакцию дегидратации осуществляют в реакторе с неподвижным слоем, с псевдоожиженным слоем или с циркулирующим псевдоожиженным слоем.
12. Способ по п.1 или 9, отличающийся тем, что реакцию дегидратации осуществляют в жидкой фазе.
13. Применение катализатора, состоящего по меньшей мере из одного катализатора а), б), в) или г), по любому из пп.1-5 и, возможно, 8 для превращения глицерола в акролеин.
| US20080214384 A1, 04.09.2008 | |||
| WO2006087083 A2, 24.08.2006 | |||
| JP2008266165 A, 06.11.2008 | |||
| Способ получения 2-окси-4-(метилтио)-масляной кислоты | 1984 |
|
SU1428193A3 |
| WO2008066079 A, 05.06.2008 | |||
| Способ получения акролеина из глицерина | 1936 |
|
SU52208A1 |

