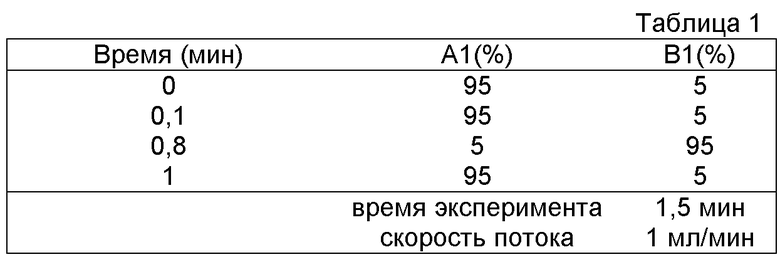
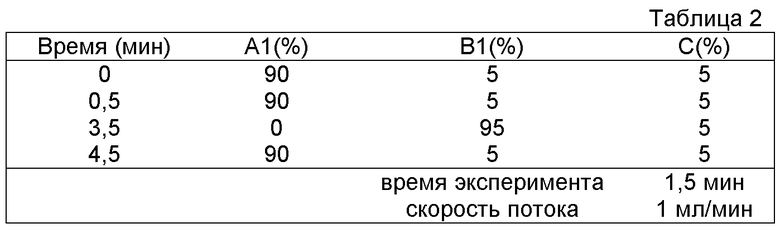
Область изобретения
Группа изобретений относится к новым оксииндольным производным формулы (I) или их фармацевтически приемлемым солям, способам их получения, фармацевтическим композициям, содержащим такие соединения, и их применению для лечения различных расстройств, которые опосредованы через мотилиновый рецептор (GPR38).
Предпосылки изобретения
GPR38 представляет собой 7-трансмембранный связанный с G-белком рецептор с высоким сродством в отношении пептида мотилина [Feighner et al., Science 1999, 284, 2184], что позволяет предположить, что эндогенный мотилин проявляет всю или бóльшую часть своей активности через этот рецептор.
Мотилин представляет собой состоящий из 22 аминокислот пептид, обнаруженный в больших количествах в эндокрино-подобных клетках желудочно-кишечного (GI) тракта, и особенно в области двенадцатиперстной кишки-тощей кишки. Известно, что при голодании этот пептид связан с началом Фазы III активности мигрирующего комплекса в желудке [Boivin et al., Dig. Dis. Sci. 1992, 37, 1562], что говорит о роли в механизмах прокинетической активности. Мотилин также высвобождается из кишечника в процессе питания, мнимого кормления, растяжения желудка или при пероральном или внутривенном введении питательных веществ [Christofides et al., Gut 1979, 20, 102; Bormans et al., Scand. J. Gastroenterol. 1987, 22, 781], что говорит о дополнительных ролях этого пептида в модуляции характера перистальтики в процессе питания.
Давно известно, что у животных или человека мотилин усиливает GI перистальтику и способствует опорожнению желудка и прохождению через кишечник в анальном направлении как в условиях голодания, так и насыщения. Считают, что эта активность проявляется преимущественно за счет способствования, по меньшей мере, холинергической возбудительной функции кишечника [Van Assche et al., Eur. J. Pharmacol. 1997, 337, 267], возможно также с вовлечением активации блуждающего нерва [Mathis & Malbert, Am. J. Physiol. 1998, 274, G80]. Кроме того, более высокие концентрации мотилина непосредственно вызывают небольшое сокращение мышц [Van Assche et al., Eur. J. Pharmacol. 1997, 337, 267].
Было показано, что антибиотик эритромицин имитирует GI активность мотилина в дополнение к его ранее описанным антибиотическим свойствам [см. Peeters, Problems of the Gastrointestinal Tract, Anaesthesia Ed., Herbert MK et al. Springer-Verlag, Berlin, Heidelberg 1999, pp 39-51]. Недавно было показано, что эритромицин активирует GPR38 рецептор, что подтверждает его способность имитировать функцию мотилина [Carreras et al., Analyt. Biochem. 2002, 300, 146]. Кроме того, доступность этого непептидного агониста мотилинового рецептора позволила осуществить, по меньшей мере, некоторые клинические исследования для определения клинического потенциала агонистов мотилинового рецептора. Эти исследования убедительно продемонстрировали способность усиливать опорожнение желудка в различных условиях, связанных с гастропарезом, таких как функциональная диспепсия и диабетический гастропарез. Кроме того, было показано, что у человека эритромицин повышает давление в нижнем эзофагеальном сфинктере, что, вместе с усилением опорожнения желудка, говорит о роли в лечении гастроэзофагеального рефлюкса (GERD). Наконец, эритромицин используется для промотирования пропульсивной активности кишечника, находя клиническое применение в лечении псевдообструкции и состояний с нарушенной перистальтикой толстой кишки [Peeters, Problems of the Gastrointestinal Tract, Anaesthesia Ed., Herbert MK et al. Springer-Verlag, Berlin, Heidelberg 1999, pp 39-51].
Следовательно, ожидают, что агонисты на GPR38 рецепторе будут имитировать активность мотилина или других веществ, действующих на этом рецепторе, таких как эритромицин, и найдут клиническое применение в лечении расстройств GI, связанных с гипокинезией, в частности функциональных кишечных расстройств, таких как GERD, функциональная диспепсия (FD) и синдром раздраженной толстой кишки (IBS). Соединения также будут полезными для лечения других GI состояний, причина которых известна и при которых GI перистальтика понижена. Такие состояния включают запор, вызванный различными заболеваниями, такими как связанные с невропатией, и/или введением других лекарственных средств, псевдообструкция кишечника, паралитическая непроходимость кишечника после хирургической операции или некоторых других манипуляций, желудочный стаз или гипокинезия, вызванная различными заболеваниями, такими как диабет, и/или введением других лекарственных средств, или возникающая у пациентов с энтеральным кормлением. Интересно, что способность мотилина или эритромицина активировать блуждающий нерв, связь этого нерва с изменениями пищевого поведения [например, Furness et al., Auton. Neurosci. 2001, 92, 28] и хромосомальная локализация GPR38 [based on Ensembl: 13q21.1 (58.46-59.46 Mb)] в маркерах (D13S257-13q14.11 до D13S258 на 13q21.33) локуса, связанного с ожирением [Feitosa et al., Am. J. Hum. Genet. 2002, 70, 72], также говорит о том, что агонисты, активные на GPR38 рецепторе, помимо промотирования GI перистальтики, будут способствовать пищевому поведению, по меньшей мере, у тех пациентов, у которых имеет место некоторое подавление аппетита или кахексия. Такая активность указывает на то, что агонисты на этом рецепторе найдут клиническое применение в лечении симптомов, связанных, например, с лечением рака или вызванных развитием рака.
Помимо способности агонистов мотилинового рецептора промотировать GI перистальтику, связь полиморфизма гена мотилина с болезнью Крона [Annese et al., Dig. Dis. ScL 1998, 43, 715-710] и изменения плотности мотилинового рецептора при колите [Depoortere et al., Neurogastroenterol. Motil. 2001, 13, 55] предполагают возможность применения агонистов мотилинового рецептора для лечения воспалительных состояний кишечника в целом.
Наконец, GPR38 также обнаружен в областях вне GI тракта. Эти области включают гипофиз, жировую ткань, мочевой пузырь и некоторые области головного мозга. Это предполагает клиническое применение для промотирования функции гипофиза, такой как высвобождение веществ, способствующих секреции гормона роста, присутствие в жировой ткани также говорит о роли в контроле массы тела, а присутствие в мочевом пузыре говорит о роли агонистов на этом рецепторе в лечении недержания мочи. Присутствие GPR38 в головном мозге подтверждает применение при расстройствах GI и нарушениях питания, указанных выше, но, кроме того, говорит о том, что рецептор вовлечен в больший спектр функций блуждающего нерва-гипоталамуса.
WO 9410185, EP 838469, WO 9823629, DE 19805822 и US 6165985 заявляют производные эритромицина, нацеленные на GPR38, для применения при расстройствах, связанных с GI перистальтикой. WO 9921846, WO 0185694, WO 0168620, WO 0168621 и WO 0168622 раскрывают ряд малых молекул-антагонистов GPR38 рецептора. JP 07138284 и EP 807639 раскрывают пептидные агонисты. JP 09249620, WO 02092592, WO 05027637, US 2005065156 и Li et al., (2004, Journal of Medicinal Chemistry, 47(7) p1704-1708) раскрывают ряд малых молекул-агонистов. WO 05012331 и WO 05012332 раскрывают макроциклические соединения, которые являются агонистами или антагонистами мотилиновых или грелиновых рецепторов млекопитающих. WO 06127252 раскрывает производные эритромицина.
Краткое описание изобретения
Техническая задача
Существует необходимость в обеспечении агонистов мотилина, которые могут быть хорошим лекарственным средством. Они должны хорошо абсорбироваться из GI тракта, быть метаболически стабильными и обладать привлекательными фармакокинетическими свойствами. Они не должны быть токсичными. Кроме того, идеальный кандидат для использования в качестве лекарственного средства должен существовать в физической форме, которая является стабильной, негигроскопичной и легко формулируемой в композицию. В частности, желательно, чтобы соединения могли сильно связываться с мотилиновым рецептором и демонстрировать функциональную активность в качестве агонистов. Настоящее изобретение обеспечивает новые соединения, которые обладают отличной агонистической активностью в отношении мотилина.
Решение задачи
WO 08/000729, WO 07/007018 и WO 07/012479 раскрывают ряд малых молекул-агонистов.
Соединения по настоящему изобретению структурно отличаются от известных соединений, раскрытых в указанных документах предшествующего уровня техники, присутствием оксииндола.
Кроме того, WO 96/13265 формально раскрывает оксииндольные соединения. Однако соединения представляют собой вещества, способствующие секреции гормона роста, которые отличаются от агонистов мотилина. Кроме того, все раскрытые соединения по настоящему изобретению, как считают, вводят D-аминокислоту в молекулы, тогда как соединения по настоящему изобретению отличаются тем, что они вводят L- (альфа-, бета- или гамма-) аминокислоту в молекулы.
WO 96/13265 раскрывает соединения, которые не содержат L-аминокислотную группу и не обладают активностью агонистов мотилина.
Был обнаружен структурно новый класс соединений с аминокислотной группой L-типа в молекулах, который обеспечивает получение агонистов мотилинового рецептора (GPR38).
Настоящее изобретение поэтому обеспечивает соединения формулы (I) и их соли:
[Химическая формула 1]
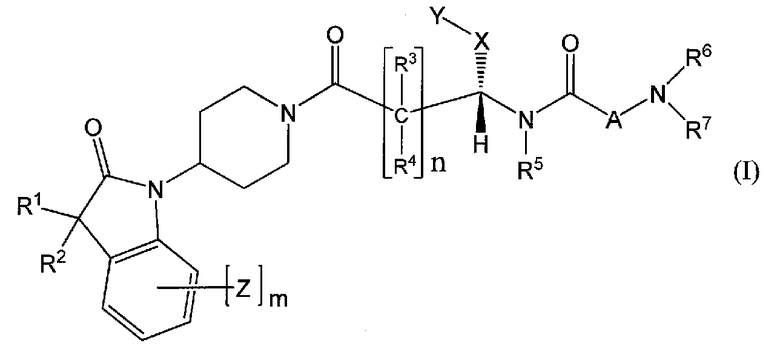
где
R1 и R2 независимо выбраны из группы, включающей водород, C1-C4 алкил и C3-C7 циклоалкил; или альтернативно, R1 и R2, вместе с атомами, с которыми они связаны, образуют 3-6-членное кольцо, которое может содержать кислород; указанное кольцо необязательно замещено 1-4 заместителями, независимо выбранными из группы, включающей галоген, гидрокси, C1-C4 алкил и C1-C4 алкокси;
R3 и R4 независимо представляют собой водород или C1-C4 алкил;
R5 представляет собой водород, C1-C4 алкил, или C1-C4 алкокси C1-C4 алкил; из них, водород или C1-C4 алкил является предпочтительным для R5;
R6 и R7 независимо выбраны из водорода, C1-C4 алкила, гидрокси C1-C4 алкила, C1-C4 алкокси C1-C4 алкила, амино C1-C4 алкила, C1-C4 алкиламино C1-C4 алкила, ди(C1-C4 алкил)амино C1-C4 алкила, насыщенного гетероциклила и насыщенного гетероциклил C1-C4 алкила; указанный насыщенный гетероциклил и алкил могут содержать независимо от 1 до 4 C1-C4 алкильных групп; или альтернативно, R6 и R7 вместе с атом азота, с которым они связаны, образуют 4-6-членное кольцо, которое может содержать азот или кислород, где указанное 4-6-членное кольцо необязательно замещено 1-4 заместителями, независимо выбранными из группы, включающей гидрокси, C1-C4 алкил, C1-C4 алкокси, C3-C7 циклоалкил, амино, C1-C4 алкиламино и ди(C1-C4 алкил)амино;
A представляет собой
[Химическая формула 2]
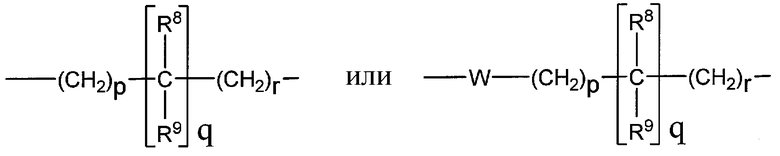
где p, q и r независимо имеют значение 0, 1, 2 или 3;
R8 и R9 независимо представляет собой водород, C1-C6 алкил или C3-C7 циклоалкил; указанный алкил и циклоалкил необязательно замещены гидрокси, C1-C4 алкилом, амино, C1-C4 алкиламино и ди(C1-C4 алкил)амино; или R8 и R9 могут быть объединены друг с другом с образованием C3-C7-членного кольца, которое может содержать кислород; или R8 и R9 независимо могут быть объединены с одной или обеими R8 и R9 группами с образованием алкиленовых мостиков между концевым азотом и алкильной частью R8 или R9 групп, где мостик содержит от 1 до 5 атомов углерода и может содержать азот или кислород; указанный мостик необязательно замещен 1-4 заместителями, независимо выбранными из группы, включающей галоген, гидрокси, C1-C4 алкил, галоген C1-C4 алкил и C1-C4 алкокси;
W представляет собой N-R10, указанный R10 представляет собой водород или C1-C4 алкил;
X представляет собой C0-C4 алкилен или C0-C4 алкилен-K-C0-C4 алкилен, где K представляет собой -O-, -NH-, NR9-, -S-, -SO-, -SO2-, -CO-, -OCO-, -C(O)O-, -CR11=CR12-,
[Химическая формула 3]
-С≡С-,
-NR11CO-, или -CONR11-; указанный алкилен необязательно замещен C1-C4 алкилом, гидрокси C1-C4 алкилом, амино C1-C4 алкилом, C1-C4 алкиламино C1-C4 алкилом, ди(C1-C4)алкиламино C1-C4 алкилом или C1-C4 алкокси C1-C4 алкилом; из них, -O-, -NH- или NR9- является предпочтительным для K; из них, указанный алкилен, необязательно замещенный C1-C4 алкилом, гидрокси C1-C4 алкилом или C1-C4 алкокси C1-C4 алкилом, является предпочтительным;
R11 представляет собой водород или C1-C4 алкил;
Y представляет собой водород, галоген или 5-10-членное кольцо; указанное кольцо необязательно замещено гидрокси, галогеном, галоген C1-C4 алкилом, C1-C4 алкилом, гидрокси C1-C4 алкилом, амино C1-C4 алкилом, C1-C4 алкокси или C1-C4 алкокси C1-C4 алкилом; из них, водород или 5-10-членное кольцо является предпочтительным для Y; из них, указанное кольцо, необязательно замещенное гидрокси, галогеном, галоген C1-C4 алкилом, C1-C4 алкилом, гидрокси C1-C4 алкилом, C1-C4 алкокси или C1-C4 алкокси C1-C4 алкилом, является предпочтительным;
Z представляет собой галоген, C1-C4 алкил, C1-C4 алкокси или гидрокси;
m имеет значение 0, 1, 2, 3 или 4; из них, 0, 1 или 2 является предпочтительным для m;
n имеет значение 0, 1 или 2; из них, 0 или 1 является предпочтительным для n.
Также настоящее изобретение обеспечивает применение соединения формулы (I) или его фармацевтически приемлемой соли, каждого как оно описано в настоящей заявке, для получения лекарственного средства для лечения состояния, опосредованного активностью мотилинового рецептора, в частности мотилиновой агонистической активностью.
Предпочтительно, настоящее изобретение также обеспечивает применение соединения формулы (I) или его фармацевтически приемлемой соли, каждого как он описан в настоящей заявке, для получения лекарственного средства для лечения заболеваний, выбранных из связанных с мотилином заболеваний.
Также настоящее изобретение обеспечивает фармацевтическую композицию, включающую соединение формулы (I) или его фармацевтически приемлемую соль, каждого как оно описано в настоящей заявке, вместе с фармацевтически приемлемым носителем для указанного соединения.
Также, настоящее изобретение обеспечивает фармацевтическую композицию, включающую соединение формулы (I) или его фармацевтически приемлемую соль, каждого как оно описано в настоящей заявке, вместе с фармацевтически приемлемым носителем для указанного соединения, и другое фармакологически активное вещество.
Кроме того, настоящее изобретение обеспечивает способ лечения состояния, опосредованного активностью мотилинового рецептора, у субъекта-млекопитающего, который включает введение млекопитающему, нуждающемуся в таком лечении, терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, каждого как он описан в настоящей заявке.
Примеры состояний, опосредованных активностью мотилинового рецептора, включают, но не ограничиваются этим, связанные с мотилином заболевания. Соединения по настоящему изобретению демонстрируют агонистическую активность в отношении мотилинового рецептора. Соединения по настоящему изобретению могут демонстрировать меньшую токсичность, хорошую абсорбцию, распределение, хорошую растворимость, меньшее сродство связывания с белком, отличным от мотилинового рецептора, меньшее взаимодействие лекарственное средство-лекарственное средство и хорошую метаболическую стабильность.
Преимущества настоящего изобретения
Фактически, в настоящем изобретении подтверждено, что, когда аминокислотную группу L-типа замещают D-аминокислотой, агонистическая активность в отношении мотилина существенно снижается. Как показано в таблице в экспериментальной части, на функциональньную активность в отношении мотилинового рецептора влияет абсолютная конфигурация аминокислотной линкерной группы, которая отличается не менее чем в 100-10000 раз между соединением с L-аминокислотной группой и соответствующими соединениями с D-аминокислотной группой.
Описание вариантов воплощения
Как он используется в настоящей заявке, термин "алкил", в качестве группы или части группы, например, алкокси или гидроксиалкил, относится к линейной или разветвленной алкильной группе во всех изомерных формах. Термин "C1-C4 алкил" относится к алкильной группе, определенной выше, содержащей, по меньшей мере, 1 и максимально 4 атомов углерода. Примеры таких алкильных групп включают метил, этил, пропил, изопропил, н-бутил, изобутил, втор-бутил или трет-бутил. Примеры таких алкоксигрупп включают метокси, этокси, пропокси, изопропокси, бутокси, изобутокси, втор-бутокси и трет-бутокси.
Термин "циклоалкил", как используется в настоящей заявке, предпочтительно означает циклоалкильную группу, содержащую 3-7 атомов углерода, и примеры включают циклопропил, циклобутил, циклопентил и циклогексил, циклогептил.
Термин "галоген" относится к фтору (F), хлору (Cl), брому (Br) или иоду (I), и термин "гало" относится к галогену: фтору (-F), хлору (-Cl), брому (-Br) и иоду (-I).
Термин "гетероциклил" означает 5- или 6-членное кольцо, которое включает один или несколько гетероатомов, выбранных из азота, кислорода и серы. Примеры таких гетероциклильных групп включают пирролидинил, пиперидинил, пиперазинил и морфолинил.
В соединениях формулы (I) присутствует хиральный атом углерода, присоединенный к X, и поэтому соединения формулы (I) в плоскостной структуре существуют в виде стереоизомеров. Настоящее изобретение характеризуется тем, что содержит один оптический изомер стереоизомерных форм вокруг атома углерода, связанного с X, в соединениях формулы (I), что показано пунктирной линией C-X связи. Предполагаемые стереоизомерные формы можно разделить или отделить друг от друга традиционными способами, или любой определенный изомер можно получить путем традиционного стереоселективного или асимметрического синтеза.
В некоторых соединениях формулы (I) могут присутствовать несколько хиральных атомов углерода, отличных от атома углерода, связанного с X. В таких случаях, соединения формулы (I) существуют в виде стереоизомеров. Настоящее изобретение охватывает все оптические изомеры, такие как стереоизомерные формы соединений формулы (I), включая энантиомеры, диастереоизомеры и их смеси, такие как рацематы. Разные стереоизомерные формы можно разделить или отделить друг от друга традиционными способами, или любой определенный изомер можно получить путем традиционного стереоселективного или асимметрического синтеза.
Некоторые соединения в настоящей заявке могут существовать в различных таутомерных формах, и должно быть понятно, что настоящее изобретение охватывает все такие таутомерные формы.
Подходящими соединениями по настоящему изобретению являются следующие:
(R)-N-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)пиперидин-3-карбоксамид;
(S)-N-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)пиперидин-2-карбоксамид;
(S)-2-амино-N-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)-3,3-диметилбутанамид;
(S)-2-(1-аминоциклобутил)-N-(1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)ацетамид;
(S)-1-(аминометил)-N-(1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)циклопропанкарбоксамид;
(S)-N-(1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)азетидин-3-карбоксамид;
(S)-N-(1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)пиперидин-4-карбоксамид;
(S)-2-(1-аминоциклопентил)-N-(1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)ацетамид;
(S)-N-(1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)-3-метил-3-(метиламино)бутанамид;
(S)-3-(циклопентиламино)-N-(1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)пропанамид;
(S)-N-(1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)-3-((2-метоксиэтил)(метил)амино)пропанамид;
(S)-N-(1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)-2-(пирролидин-1-ил)ацетамид;
(R)-N-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)-1-метилпиперидин-3-карбоксамид;
(1S,3R)-N-((S)-3-(4-хлорфенил)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксопропан-2-ил)-3-(метиламино)циклопентанкарбоксамид;
(1S,3R)-N-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-3-(2-(трифторметил)фенил)пропан-2-ил)-3-(метиламино)циклопентанкарбоксамид;
(1S,3R)-N-((S)-3-циклогексил-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксопропан-2-ил)-3-(метиламино)циклопентанкарбоксамид;
(1S,3R)-N-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-3-(нафталин-1-ил)-1-оксопропан-2-ил)-3-(метиламино)циклопентанкарбоксамид;
(1S,3R)-N-((S)-3-(2-хлорфенил)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксопропан-2-ил)-3-(метиламино)циклопентанкарбоксамид;
(1S,3R)-N-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-3-o-толилпропан-2-ил)-3-(метиламино)циклопентанкарбоксамид;
(1S,3R)-N-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-3-(4-фторфенил)-1-оксопропан-2-ил)-3-(метиламино)циклопентанкарбоксамид;
(1S,3R)-N-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-3-(3-фторфенил)-1-оксопропан-2-ил)-3-(метиламино)циклопентанкарбоксамид;
(1S,3R)-N-((S)-3-(3-хлорфенил)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксопропан-2-ил)-3-(метиламино)циклопентанкарбоксамид;
(1S,3R)-N-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-3-фенилпропан-2-ил)-3-(метиламино)циклопентанкарбоксамид;
(1S,3R)-N-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-3-(3-метоксифенил)-1-оксопропан-2-ил)-3-(метиламино)циклопентанкарбоксамид;
(1S,3R)-N-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-3-(2-фторфенил)-1-оксопропан-2-ил)-3-(метиламино)циклопентанкарбоксамид;
(1S,3R)-N-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-4-метил-1-оксопентан-2-ил)-3-(метиламино)циклопентанкарбоксамид;
(1S,3R)-N-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-3-(1H-индол-3-ил)-1-оксопропан-2-ил)-3-(метиламино)циклопентанкарбоксамид;
(1S,3R)-N-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-3-(3-(трифторметил)фенил)пропан-2-ил)-3-(метиламино)циклопентанкарбоксамид;
(1S,3R)-N-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-3-(4-(трифторметил)фенил)пропан-2-ил)-3-(метиламино)циклопентанкарбоксамид;
(1S,3R)-N-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-3-(4-метоксифенил)-1-оксопропан-2-ил)-3-(метиламино)циклопентанкарбоксамид;
(1S,3R)-N-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-3-феноксипропан-2-ил)-3-(метиламино)циклопентанкарбоксамид;
(1S,3R)-N-((S)-3-(2-хлорфенокси)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксопропан-2-ил)-3-(метиламино)циклопентанкарбоксамид;
(1S,3R)-N-((S)-4-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-4-оксо-1-фенилбутан-2-ил)-3-(метиламино)циклопентанкарбоксамид;
(R)-N-((S)-1-(4-(5-фтор-3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)пиперидин-3-карбоксамид;
(1S,3R)-N-((S)-1-(4-(5-фтор-3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)-3-(метиламино)циклопентанкарбоксамид;
(S)-N-(1-(4-(5-фтор-3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)-3-метил-3-(метиламино)бутанамид;
(S)-N-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)-3-(метиламино)пирролидин-1-карбоксамид;
1-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)-3-((S)-пирролидин-2-илметил)мочевина;
1-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)-3-((R)-пиперидин-3-ил)мочевина;
3-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)-1-метил-1-((S)-пирролидин-2-илметил)мочевина;
1-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)-3-((R)-пирролидин-2-илметил)мочевина;
1-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)-3-((R)-пирролидин-3-ил)мочевина;
1-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)-3-((R)-пирролидин-3-илметил)мочевина;
(R)-N-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)-3-метилпиперазин-1-карбоксамид;
(S)-1-(1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)-3-(4-этилпиперидин-4-ил)мочевина;
(S)-1-(1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)-3-(4-метилпиперидин-4-ил)мочевина;
1-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)-3-((S)-пирролидин-3-ил)мочевина;
1-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)-3-((S)-пиперидин-2-илметил)мочевина;
1-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)-3-((R)-пиперидин-3-илметил)мочевина;
(S)-1-(2-амино-2-метилпропил)-3-(1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)мочевина;
(S)-3-(циклопропил(метил)амино)-N-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)пирролидин-1-карбоксамид;
(S)-1-(1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)-3-(2-(пирролидин-1-ил)этил)мочевина;
(S)-1-(1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)-3-(2-(диметиламино)этил)мочевина;
(S)-N-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)-3-(диметиламино)пирролидин-1-карбоксамид;
1-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)-3-(((S)-1-метилпирролидин-2-ил)метил)мочевина;
(S)-1-(1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)-3-(1,4-диметилпиперидин-4-ил)мочевина;
(S)-N-((S)-1-(4-(5-фтор-3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)-3-(метиламино)пирролидин-1-карбоксамид;
1-((S)-1-(4-(5-фтор-3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)-3-((S)-пирролидин-2-илметил)мочевина;
(S)-1-(1-(4-(5-фтор-3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)-3-(4-метилпиперидин-4-ил)мочевина;
(S)-N-(1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-3-(2-(трифторметил)фенил)пропан-2-ил)пиперидин-4-карбоксамид;
(S)-N-(3-(2-хлорфенил)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксопропан-2-ил)пиперидин-4-карбоксамид;
(S)-N-(3-(4-хлорфенил)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксопропан-2-ил)пиперидин-4-карбоксамид;
(R)-N-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-3-фенилпропан-2-ил)пиперидин-3-карбоксамид;
(R)-N-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-3-(2-фторфенил)-1-оксопропан-2-ил)пиперидин-3-карбоксамид;
(R)-N-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-3-(3-фторфенил)-1-оксопропан-2-ил)пиперидин-3-карбоксамид;
(R)-N-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-3-(2-(трифторметил)фенил)пропан-2-ил)пиперидин-3-карбоксамид;
(R)-N-((S)-3-(2-хлорфенил)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксопропан-2-ил)пиперидин-3-карбоксамид;
(R)-N-((S)-3-(4-хлорфенил)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксопропан-2-ил)пиперидин-3-карбоксамид;
(S)-N-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-3-(3-фторфенил)-1-оксопропан-2-ил)-3-(метиламино)пирролидин-1-карбоксамид;
(S)-N-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-3-(2-(трифторметил)фенил)пропан-2-ил)-3-(метиламино)пирролидин-1-карбоксамид;
(S)-N-((S)-3-(2-хлорфенил)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксопропан-2-ил)-3-(метиламино)пирролидин-1-карбоксамид;
(S)-N-((S)-3-(4-хлорфенил)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксопропан-2-ил)-3-(метиламино)пирролидин-1-карбоксамид;
и их соли.
В объем "соединений по настоящему изобретению" включены все соли, сольваты, гидраты, комплексы, полиморфы, пролекарства, радиоактивно меченные производные, стереоизомеры и оптические изомеры соединений формулы (I).
Соединения формулы (I) могут образовывать кислотно-аддитивные соли. Должно быть понятно, что для использования в медицине соли соединений формулы (I) должны быть фармацевтически приемлемыми. Подходящие фармацевтически приемлемые соли должны быть очевидны для специалистов в данной области, и включают соли, описанные в J. Pharm. ScL, 1977, 66, 1-19, такие как кислотно-аддитивные соли, образованные с неорганическими кислотами, например с хлористоводородной, бромистоводородной, серной, азотной или фосфорной кислотой; и с органическими кислотами, например янтарной, малеиновой, муравьиной, уксусной, трифторуксусной, пропионовой, фумаровой, лимонной, винной, бензойной, п-толуолсульфоновой, метансульфоновой или нафталинсульфоновой кислотой. Некоторые соединения формулы (I) могут образовывать кислотно-аддитивные соли с одним или несколькими эквивалентами кислоты. Объем настоящего изобретения включает все возможные стехиометрические и нестехиометрические формы. Кроме того, некоторые соединения, содержащие кислотную функцию, такую как карбокси, можно выделить в форме их неорганической соли, в которой противоион может быть выбран из натрия, калия, лития, кальция, магния и т.п., а также из органических оснований.
Соединения формулы (I) и их соли можно получить в кристаллической или некристаллической форме, и, в случае кристаллической формы, они необязательно могут быть гидратированы или сольватированы. В объем настоящего изобретения включены стехиометрические гидраты или сольваты, а также соединения, содержащие различные количества воды и/или растворителя.
Соли и сольваты, содержащие не являющиеся фармацевтически приемлемыми противоионы или ассоциированные растворители, включены в объем настоящего изобретения, например, для использования в качестве промежуточных соединений в получении других соединений формулы (I) и их фармацевтически приемлемых солей.
Кроме того, соединения формулы (I) можно вводить в виде пролекарств. Как это используется в настоящей заявке, "пролекарство" соединения формулы (I) представляет собой функциональное производное соединения, которое, при введении пациенту, в конечном счете высвобождает соединение формулы (I) in vivo. Введение соединения формулы (I) в виде пролекарства может позволить квалифицированному специалисту осуществить одно или несколько из следующих: (a) модифицировать начало действия соединения in vivo; (b) модифицировать продолжительность действия соединения in vivo; (c) модифицировать транспортирование или дистрибуцию соединения in vivo; (d) модифицировать растворимость соединения in vivo; и (e) преодолеть побочный эффект или другое затруднение, связанное с соединением. Типичные функциональные производные, используемые для получения пролекарств, включают модификации соединения, которые химически или ферментативно расщепляются in vivo. Такие модификации, которые включают получение фосфатов, амидов, сложных эфиров, тиоэфиров, карбонатов и карбаматов, хорошо известны специалистам в данной области.
Настоящее изобретение также включает изотопно-меченные соединения, которые идентичны соединениям, описанным в настоящей заявке, но отличаются тем, что один или несколько атомов замещены атомом, имеющим атомную массу или массовое число, отличное от атомной массы или массового числа, обычно существующих в природе. Примеры изотопов, которые могут быть включены в соединения по настоящему изобретению, включают изотопы водорода, углерода, азота, кислорода, фосфора, фтора, иода и хлора, такие как 3H, 11C, 14C, 18F, 123I и 125I. Соединения по настоящему изобретению, которые содержат указанные выше изотопы и/или другие изотопы других атомов, включены в объем настоящего изобретения. Изотопно-меченные соединения по настоящему изобретению, например, соединения, в которые включены радиоактивные изотопы, такие как 3H, 14C, являются полезными в анализах распределения лекарственного средства и/или тканевого субстрата. Изотопы трития, т.е. 3H, и углерода-14, т.е. 14C, являются особенно предпочтительными из-за простоты их получения и возможности детекции. 11C и 18F изотопы являются особенно полезными в PET (позитрон-эмиссионная томография), а 125I изотопы являются особенно полезными в SPECT (однофотонная эмиссионная компьютерная томография), все они являются полезными для методов визуального исследования головного мозга. Кроме того, замещение более тяжелыми изотопами, такими как дейтерий, т.е. 2H, может обеспечить некоторые терапевтические преимущества в результате большей метаболической стабильности, например больший период полураспада in vivo или потребность в меньших дозах, следовательно, могут быть предпочтительными в некоторых обстоятельствах. Изотопно-меченные соединения по настоящему изобретению, как правило, можно получить путем осуществления процедур, раскрытых на Схемах и/или в Примерах ниже, с последующим замещением реагента, который не является изотопно-меченным, изотопно-меченным реагентом.
Активность и эффективность соединений по настоящему изобретению в отношении GPR38 можно определить при помощи анализа репортера, осуществляемого на человеческом клонированном рецепторе, как описано в настоящей заявке. Соединения формулы (I) продемонстрировали агонистическую активность на GPR38 рецепторе при использовании функционального анализа, описанного в настоящей заявке.
Поэтому соединения формулы (I) и их фармацевтически приемлемые соли являются полезными для лечения состояний или расстройств, которые опосредованы через GPR38 рецептор. В частности, соединения формулы (I) и их фармацевтически приемлемые соли являются полезными для лечения некоторых расстройств GI, таких как гастроэзофагеальный рефлюкс, функциональная диспепсия, синдром раздраженной толстой кишки, запор, псевдообструкция кишечника, паралитическая непроходимость кишечника после хирургической операции или других манипуляций, рвота, желудочный стаз или гипокинезия, вызванная различными заболеваниями, такими как диабет, и/или введением других лекарственных средств, или возникающая у пациентов с энтеральным кормлением, болезнь Крона, колит, кахексию, связанную с прогрессирующими заболеваниями, такими как рак, и/или их лечением, и других расстройств, таких как недержание (далее указаны как "Расстройства по настоящему изобретению").
Должно быть понятно, что "лечение", как это используется в настоящей заявке, включает профилактику, а также облегчение выявленных симптомов.
Таким образом, настоящее изобретение также обеспечивает соединения формулы (I) и их фармацевтически приемлемые соли для использования в качестве терапевтических средств, в частности, для лечения состояний или расстройств, опосредованных через GPR38 рецептор. В частности, настоящее изобретение обеспечивает соединения формулы (I) и их фармацевтически приемлемые соли для использования в качестве терапевтических средств для лечения "Расстройств по настоящему изобретению".
Настоящее изобретение, кроме того, обеспечивает способ лечения состояний или расстройств у млекопитающих, включая человека, которые могут быть опосредованы через GPR38 рецептор, который включает введение страдающему таким расстройством субъекту терапевтически надежного и эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли.
В другом аспекте, настоящее изобретение обеспечивает применение соединений формулы (I) и их фармацевтически приемлемых солей для получения лекарственного средства для применения в лечении состояний или расстройств, опосредованных через GPR38 рецептор.
Для применения соединений формулы (I) и их фармацевтически приемлемых солей в терапии, их обычно следует сформулировать в фармацевтическую композицию в соответствии со стандартной фармацевтической практикой. Настоящее изобретение также обеспечивает фармацевтическую композицию, которая включает соединение формулы (I) или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель или эксципиент.
В следующем аспекте, настоящее изобретение обеспечивает способ получения фармацевтической композиции, который включает смешивание соединения формулы (I) или его фармацевтически приемлемой соли и фармацевтически приемлемого носителя или эксципиента.
Фармацевтическая композиция по настоящему изобретению, которую можно получить путем смешивания, подходящим при температуре окружающей среды и атмосферном давлении, обычно адаптирована для перорального, парентерального или ректального введения и, как таковая, может быть в форме таблеток, капсул, пероральных жидких препаратов, порошков, гранул, лепешек, реструктурируемых порошков, растворов или суспензий для инъекций или инфузий или суппозиториев. Перорально вводимые композиции, как правило, являются предпочтительными. Таблетки и капсулы для перорального введения могут быть в стандартной лекарственной форме и могут содержать традиционные эксципиенты, такие как связующие (например, предварительно желатинизированный кукурузный крахмал, поливинилпирролидон или гидроксипропил метилцеллюлоза); наполнители (например, лактоза, микрокристаллическая целлюлоза или гидрофосфат кальция); смазывающие вещества для таблетирования (например, стеарат магния, тальк или диоксид кремния); разрыхлители (например, картофельный крахмал или натрий крахмалгликолят); и приемлемые смачивающие вещества (например, лаурилсульфат натрия). На таблетки может быть нанесено покрытие в соответствии со способами, хорошо известными в обычной фармацевтической практике.
Пероральные жидкие препараты могут быть, например, в форме водных или масляных суспензий, растворов, эмульсий, сиропов или эликсиров или могут быть в форме сухого продукта для реструктурирования водой или другим подходящим носителем перед использованием. Такие жидкие препараты могут содержать традиционные добавки, такие как суспендирующие вещества (например, сироп сорбита, производные целлюлозы или гидрированные пищевые жиры), эмульгаторы (например, лецитин или аравийская камедь), неводные носители (которые могут включать пищевые масла, например миндальное масло, масляные сложные эфиры, этиловый спирт или фракционированные растительные масла), консерванты (например, метил или пропил-п-гидроксибензоаты или сорбиновая кислота) и, если это желательно, традиционные отдушки или красители, буферные соли и подсластители, которые являются подходящими. Препараты для перорального введения могут быть подходящим образом сформулированы для получения контролируемого высвобождения активного соединения или его фармацевтически приемлемой соли.
Для парентерального введения жидкие стандартные лекарственные формы получают с использованием соединения формулы (I) или его фармацевтически приемлемой соли и стерильного носителя. Композиции для инъекций могут быть представлены в стандартной лекарственной форме, например в ампулах или в содержащей несколько доз упаковке, с использованием соединения формулы (I) или его фармацевтически приемлемой соли и стерильном носителе, необязательно с добавлением консерванта. Композиции могут принимать такие формы, как суспензии, растворы или эмульсии в масляных или водных носителях и могут содержать используемые для формулирования вещества, такие как суспендирующие, стабилизирующие и/или диспергирующие вещества. Альтернативно, активный ингредиент может быть в форме порошка для реструктурирования при помощи подходящего носителя, например стерильной апирогенной воды, перед использованием. Соединение, в зависимости от используемого носителя и концентрации, может быть либо суспендировано, либо растворено в носителе. При получении растворов соединение может быть растворено для получения препарата для инъекций и стерилизовано путем фильтрования с последующим заполнением им подходящих флаконов или ампул и запаивания. Преимущество дает использование адъювантов, таких как местный анестетик, консерванты и буферные вещества, которые растворяют в носителе. Для повышения стабильности композицию можно заморозить после заполнения флаконов и удалить воду в условиях вакуума. Парентеральные суспензии получают, по существу, таким же способом, за исключением того, что соединение суспендируют в носителе вместо его растворения и стерилизацию нельзя осуществить путем фильтрования. Соединение можно стерилизовать путем воздействия на него этиленоксида перед суспендированием в стерильном носителе. Преимущество дает использование поверхностно-активного вещества или смачивающего вещества, которые включают в композицию для облегчения равномерного распределения соединения.
Лосьоны могут быть сформулированы с водной или масляной основой и, как правило, также содержат один или несколько эмульгаторов, стабилизаторов, диспергирующих веществ, суспендирующих веществ, загустителей или красителей. Капли могут быть сформулированы с использованием водной или неводной основы, также включающей одно или несколько диспергирующих веществ, стабилизаторов, солюбилизирующих веществ или суспендирующих веществ. Они также могут содержать консервант.
Соединения формулы (I) или их фармацевтически приемлемые соли также могут быть сформулированы в композиции для ректального введения, такие как суппозитории или удерживаемые клизмы, например, содержащие традиционные основы для суппозиториев, такие как масло какао или другие глицериды.
Соединения формулы (I) или фармацевтически приемлемые соли также могут быть сформулированы в виде депо препаратов. Такие долгодействующие композиции можно вводить путем имплантации (например подкожно или внутримышечно) или путем внутримышечной инъекции. Таким образом, например, соединения формулы (I) или фармацевтически приемлемые соли могут быть сформулированы с использованием подходящих полимерных или гидрофобных веществ (например, в виде эмульсии в приемлемом масле) или ионообменных смол или в виде умеренно растворимых производных, например, в форме умеренно растворимой соли.
Для интраназального введения соединения формулы (I) или их фармацевтически приемлемые соли могут быть сформулированы в виде растворов для введения через подходящее устройство для введения отмеренной или стандартной дозы или, альтернативно, в виде порошкообразной смеси с подходящим носителем для введения с использованием подходящего устройства доставки. Таким образом, соединения формулы (I) или их фармацевтически приемлемые соли могут быть сформулированы для перорального, буккального, парентерального, местного (включая глазное и назальное), депо или ректального введения или в форме, подходящей для введения путем ингаляции или инсуффляции (либо через рот, либо через нос). Соединения формулы (I) и их фармацевтически приемлемые соли могут быть сформулированы для местного введения в форме мазей, кремов, гелей, лосьонов, пессариев, аэрозолей или капель (например, капель для введения в глаз, ухо или нос). Мази и кремы могут быть сформулированы, например, с использованием водной или масляной основы с добавлением подходящего загустителя и/или желирующего вещества. Мази для введения в глаз могут быть получены в стерильных условиях с использованием стерилизованных компонентов.
Композиция может содержать от 0,1% до 99% масс., предпочтительно от 10 до 60% масс. активного вещества, в зависимости от способа введения. Доза соединения, используемая для лечения указанных выше расстройств, варьирует обычным образом в зависимости от тяжести расстройств, массы тела субъекта, страдающего расстройством, и других подобных факторов. Однако, в качестве общих указаний, подходящие стандартные дозы могут составлять от 0,05 до 1000 мг, более подходяще от 1,0 до 500 мг или от 1,0 до 200 мг, и такие стандартные дозы можно вводить раз в день или более одного раза в день, например два или три раза в день.
Соединения формулы (I) и их фармацевтически приемлемые соли можно использовать в комбинированных препаратах. Например, соединения формулы (I) и их фармацевтически приемлемые соли можно использовать в сочетании с одним или несколькими соединениями с активностью, направленной на уменьшение желудочной кислоты; с одним или несколькими соединениями с активностью, направленной на уменьшение гастро-эзофагеального рефлюкса; с одним или несколькими соединениями с активностью, направленной на уменьшение гастро-эзофагеального раздражения или воспаления, в частности, когда их используют для облегчения эрозивного или неэрозивного эзофагита; с одним или несколькими соединениями с анальгетической активностью; и/или с одним или несколькими соединениями со смешанной активностью, действующих на сократительную способность и боль.
Примеры соединений с активностью, направленной на снижение желудочной кислоты, включают антагонистов H2 рецептора, антагонистов кислотного насоса и ингибиторов протонного насоса. Примеры соединений с активностью, направленной на уменьшение гастроэзофагеального рефлюкса, включают агонистов на GABA-B. Примеры соединений с анальгетической активностью включают соединения, активные на нейрокининовых рецепторах (NK1, 2, 3), TRPV1 и натриевых каналах. Примеры соединений со смешанной активностью, действующих на сократительную способность и боль, включают антагонистов CRF2, антагонистов 5-HT3 или октреотид или другие молекулы, активные на sst2 рецепторах.
Все публикации, включая, но не ограничиваясь этим, патенты и патентные заявки, на которые имеются ссылки в настоящем описании, включены в настоящую заявку посредством ссылки, как если бы каждая отдельная публикация была специально и индивидуально указана как включенная в настоящую заявку посредством ссылки во всей ее полноте. Следующие далее Описания и Примеры иллюстрируют получение соединений по настоящему изобретению.
Общий синтез
В тексте настоящей заявки использованы следующие аббревиатуры, которые имеют следующие значения:
BOC: трет-Бутилоксикарбонил
BOP: Гексафторфосфат 1H-бензотриазол-1-илокситрис(диметиламино)фосфония
CBZ: Бензилоксикарбонил
DCC: Дициклогексилкарбодиимид
DMF: N,N-диметилформамид
EDC: Гидрохлорид 1-этил-3-(3-диметиламинопропил)карбодиимида
FMOC: 9-Флуоренилметоксикарбонил
HOBT: 1-Гидроксибензотриазол
HBTU: Гексафторфосфат O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония
ВЭЖХ: Высокоэффективная жидкостная хроматография
МГц: мегаГерц
ЯМР: Ядерный магнитный резонанс
TFA: Трифторуксусная кислота
ТГФ: Тетрагидрофуран
ТСХ: Тонкослойная хроматография
Соединения по настоящему изобретению все содержат, по меньшей мере, один асимметричный центр, указанный звездочкой в следующей структурной Формуле I:
[Химическая формула 4]
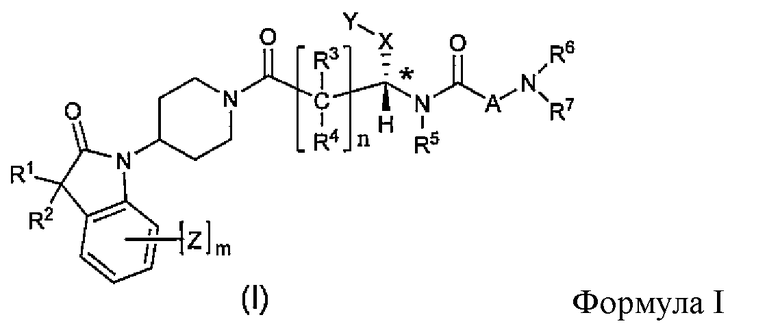
Настоящее изобретение характеризуется тем, что содержит один оптический изомер из стереоизомерных форм вокруг помеченного звездочкой углерода. Дополнительные асимметричные центры могут присутствовать в молекуле в зависимости от природы различных заместителей в молекуле. Каждый такой асимметричный центр даст оптические изомеры в количестве, равном или больше двух, и предполагается, что все такие оптические изомеры, в их разделенном виде в виде их чистых оптических изомеров охватываются настоящим изобретением.
Соединения, которые обладают активностью, большей чем около 103, в качестве агониста мотилинового рецептора, и поэтому являются предпочтительными, представляют собой такие соединения, в которых заместитель -X-Y находится ниже, а атом водорода находится выше плоскости структуры, представленной в Формуле I.
Эта конфигурация соответствует той, которая присутствуе в L-аминокислоте, включающей альфа-аминокислоту, бета-аминокислоту или гамма-аминокислоту, в случае, когда n=1,2 или 3, формулы I, соответственно. В большинстве случаев, это также обозначается как S-конфигурация, хотя при отнесении стереохимии к R- или S- обозначение меняется в зависимости от значения -X-Y. Их абсолютную стереохимию можно определить при помощи рентгеновской кристаллографии кристаллических продуктов или кристаллических промежуточных соединений, которые подвергают дериватизации, если это необходимо, содержащих асимметричный центр известной конфигурации.
Получение соединений формулы I по настоящему изобретению можно осуществить последовательным или конвергентным синтетическим путем.
Способы синтеза, подробно иллюстрирующие получение соединений формулы I последовательным путем, представлены на следующих схемах реакций.
Термин "стандартные реакционные условия пептидного связывания" неоднократно используется в настоящем описании и означает связывание карбоновой кислоты с амином с использованием кислотного активирующего агента, такого как EDC, DCC, HBTU и BOP, в инертном растворителе, таком как DMF и дихлорметан, в присутствии катализатора, такого как HOBT, и/или в присутствии основания, такого как диизопропилэтиламин или триэтиламин.
Термин "PG1", как используется далее в настоящей заявке, означает амино-защитную группу, которую выбирают из типичных амино-защитных групп, описанных в Protective Groups in Organic Synthesis edited by T. W. Greene et al. (John Wiley & Sons, 1999). Типичные амино-защитные группы включают бензил, CBZ, FMOC и BOC. Из этих групп, бензил, CBZ и BOC являются предпочтительными.
BOC и бензил широко используются в синтезе по настоящему изобретению, и условия их удаления известны специалистам в данной области. Удаление защитных групп BOC осуществляют в растворителе, таком как метиленхлорид или метанол, с использованием сильной кислоты, такой как трифторуксусная кислота (TFA) или хлористоводородная кислота (HCl). Удаление бензильной и CBZ групп можно осуществить различными способами, например каталитическим гидрированием с использованием водорода в присутствии палладиевого катализатора в протонном растворителе, таком как этанол. В случаях, когда каталитическому гидрированию препятствует присутствие другой потенциально реакционноспособной функциональной группы, удаление CBZ групп также можно осуществить путем обработки раствором бромистого водорода в уксусной кислоте или путем обработки смесью TFA и диметилсульфида.
Защищенные аминокислотные производные IV во многих случаях являются коммерчески доступными, когда амино-защитная группа представляет собой PG1. Защищенные аминокислотные производные IV, не являющиеся коммерчески доступными, можно получить способами, известными из литературы (Williams, R. M. Synthesis of Optically Active Amino Acids, Pergamon Press: Oxford, 1989).
Многие из оксииндолпиперидинов формулы II можно получить следующими способами, описанными на Схеме 13, 14 и 15. Навыки, необходимые для осуществления реакции и очистки полученных продуктов реакции, известны специалистам в данной области. Процедуры очистки включают кристаллизацию, хроматографию с нормальной фазой и/или обращенной фазой.
[Химическая формула 5]
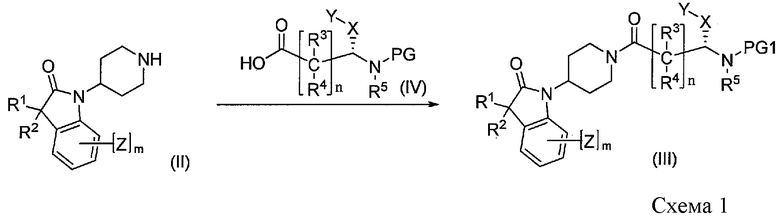
Промежуточные соединения формулы III можно синтезировать, как показано на Схеме 1. Связывание амина формулы II, получение которого описано ниже, если он не является коммерчески доступным, с защищенными аминокислотами формулы IV, где PG1 представляет собой подходящую защитную группу, удобным образом осуществляют в стандартных условиях пептидного связывания.
[Химическая формула 6]
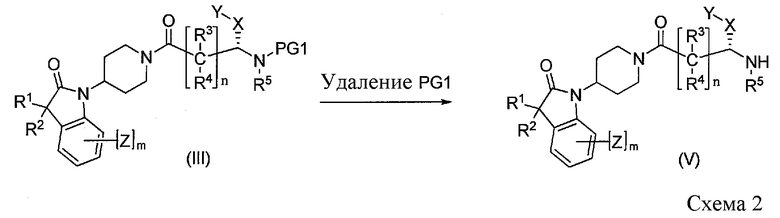
Преобразование соединения III в промежуточное соединение V можно осуществить, как проиллюстрировано на Схеме 2, путем удаления амино-защитной группы, PG1 (CBZ, BOC и т.п.).
[Химическая формула 7]
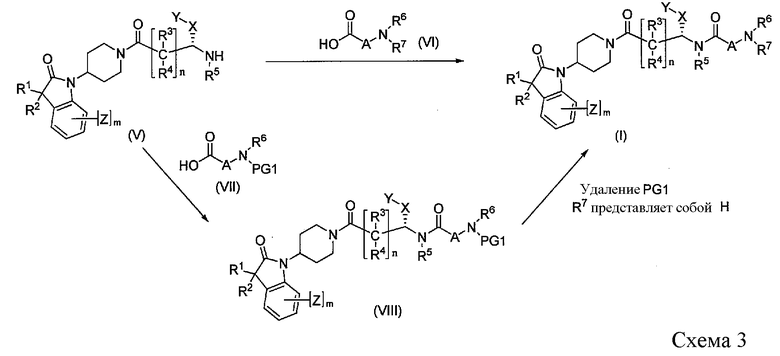
Промежуточные соединения формулы IV, где A связан с карбонилом посредством атома углерода -(CH2)pCR8R9q(CH2)r-, как показано на Схеме 3, можно подвергнуть сочетанию с промежуточными соединениями формулы V в стандартных реакционных условиях пептидного связывания. Аминокислоты VI, как аминокислоты VII, являются коммерчески доступными, или их можно синтезировать в соответствии с процедурами, известными в данной области техники. Одна из процедур описана в рабочем примере 21. Также, когда R6 или R7 представляет собой водород, используют защищенные аминокислоты VII в реакции сочетания, где PG1 представляет собой амино-защитную группу, определенную выше. Удаление PG1 в соединении VIII с получением соединения I, где R7 = атом водорода (H), можно осуществить в условиях, известных в данной области техники.
[Химическая формула 8]
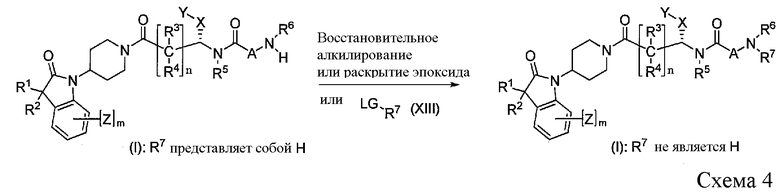
Соединения формулы I, где R6 и/или R7 представляет собой H, можно далее преобразовать в новые соединения I, которые замещены по аминогруппе, как показано на Схеме 4. Восстановительное алкилирование соединения I при помощи альдегида осуществляют в условиях, известных в данной области техники, например, путем восстановления соответствующего имина или соли иминия, либо выделенного, либо образованного in situ, при помощи водорода в присутствии платинового, палладиевого или никелевого катализаторов или с использованием химических восстановителей, таких как триацетоксиборогидрид натрия или цианоборогидрид натрия, в протонном растворителе, таком как метанол, этанол или 2-пропанол, в присутствии каталитического количества кислоты. Альтернативно, аналогичное преобразование можно осуществить через реакцию раскрытия эпоксида в присутствии подходящего основания в инертном растворителе. Альтернативно, аналогичное преобразование можно осуществить путем алкилирования с использованием реагента R7-LG в присутствии подходящего основания в инертном растворителе. При этом LG представляет собой "удаляемую группу", как это используется в настоящей заявке, и означает группу, способную к замещению нуклеофильными группами, такими как амины, металл-амиды, и примеры таких удаляемых групп включают атомы галогенов, алкилсульфонильную группу и арилсульфонильную группу, нитробензолкарбонильную группу, пергалогенбензолкарбонильную группу. Из них, атом иода, метансульфонильная группа, трифторметансульфонильная группа и 4-метилфенилсульфонильная группа являются предпочтительными.
Термин "основание" также не предполагает конкретного ограничения природы используемых оснований, и любое основание, обычно используемое в реакциях такого типа, можно в равной степени использовать в настоящем изобретении. Примеры таких оснований включают: гидроксиды щелочных металлов, такие как гидроксид лития, гидроксид натрия, гидроксид калия и гидроксид бария; гидриды щелочных металлов, такие как гидрид лития, гидрид натрия и гидрид калия; алкоксиды щелочных металлов, такие как метоксид натрия, этоксид натрия и трет-бутоксид калия; карбонаты щелочных металлов, такие как карбонат лития, карбонат натрия, карбонат калия и карбонатцезия; гидрокарбонаты щелочных металлов, такие как гидрокарбонат лития, гидрокарбонат натрия и гидрокарбонат калия; амины, такие как N-метилморфолин, триэтиламин, трипропиламин, трибутиламин, диизопропилэтиламин, N-метилпиперидин, пиридин, 4-пирролидинопиридин, пиколин, 2,6-ди(трет-бутил)-4-метилпиридин, хинолин, N,N-диметиланилин, N,N-диэтиланилин, 1,5-диазабицикло[4,3,0]нон-5-ен (DBN), 1,4-диазабицикло[2,2,2]октан (DABCO), 1,8-диазабицикло[5,4,0]ундец-7-ен (DBU), лутидин и колидин; амиды щелочных металлов, такие как амид лития, амид натрия, амид калия, диизопропиламид лития, диизопропиламид калия, диизопропиламид натрия, бис(триметилсилил)амид лития и бис(триметилсилил)амид калия. Из них, триэтиламин, диизопропилэтиламин, DBU, DBN, DABCO, пиридин, лутидин, колидин, карбонат натрия, гидрокарбонат натрия, гидроксид натрия, карбонат калия, гидрокарбонат калия, гидроксид калия, гидроксид бария и карбонат цезия являются предпочтительными.
Реакции обычно и предпочтительно осуществляют в присутствии инертного растворителя. Нет никакого конкретного ограничения, связанного с природой используемого растворителя, при условии, что он не оказывает неблагоприятного действия на реакцию или используемые в реакции реагенты, и что он может растворять реагенты, по меньшей мере, в некоторой степени. Примеры подходящих растворителей включают, но не ограничиваются этим: галогенированные углеводороды, такие как дихлорметан, хлороформ, тетрахлорид углерода и дихлорэтан; простые эфиры, такие как диэтиловый эфир, диизопропиловый эфир, ТГФ и диоксан; ароматические углеводороды, такие как бензол, толуол и нитробензол; амиды, такие как DMF, N,N-диметилацетамид и триамид гексаметилфосфорной кислоты; амины, такие как N-метилморфолин, триэтиламин, трипропиламин, трибутиламин, диизопропилэтиламин, N-метилпиперидин, пиридин, 4-пирролидинопиридин, N,N-диметиланилин и N,N-диэтиланилин; спирты, такие как метанол, этанол, пропанол, изопропанол и бутанол; нитрилы, такие как ацетонитрил и бензонитрил; сульфоксиды, такие как диметилсульфоксид (DMSO) и сульфолан; кетоны, такие как ацетон и диэтилкетон. Из этих растворителей, включая, но не ограничиваясь этим, DMF, DMSO, ТГФ, диэтиловый эфир, диизопропиловый эфир, диметоксиэтан, ацетонитрил, дихлорметан, дихлорэтан и хлороформ являются предпочтительными.
[Химическая формула 9]
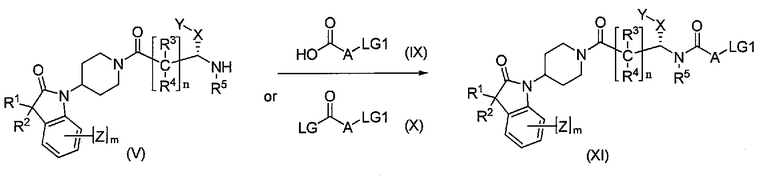
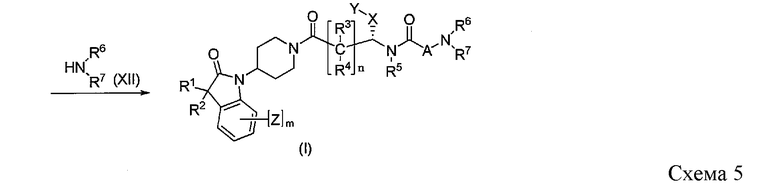
Соединения формулы I можно получить путем нуклеофильного присоединения промежуточного соединения формулы XI с использованием амина IX, как показано на Схеме 5. Промежуточное соединение формулы XI можно получить путем реакции сочетания с амином формулы V и кислотой формулы IX, которую осуществляют в стандартных реакционных условиях пептидного связывания, где LG1 представляет собой атом галогена, такой как хлор, бром и иод. Альтернативно, промежуточное соединение формулы XI можно получить путем реакции сочетания с использованием амина формулы V и эквивалента активированной кислоты формулы X, которую осуществляют в присутствии подходящих оснований в инертном растворителе.
Соединение формулы XI преобразуют в целевое соединение формулы I в присутствии амина XII и подходящего основания в инертном растворителе.
[Химическая формула 10]
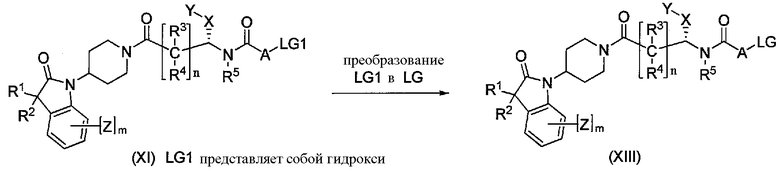
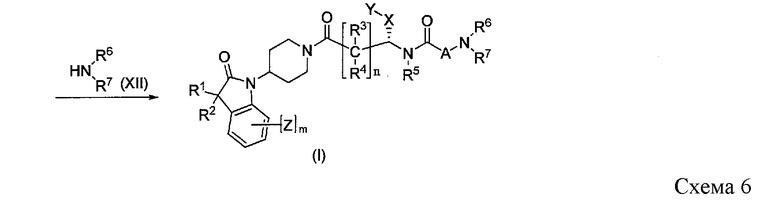
Когда LG1 представляет собой гидроксигруппу, желаемое соединение формулы I можно получить через промежуточное соединение формулы XIII, где LG имеет значение, определенное выше, как показано на Схеме 6. Преобразование гидроксигруппы соединения формулы XI в промежуточное соединение формулы XIII осуществляют в условиях, известных в данной области техники; например, путем преобразования гидроксигруппы в LG с использованием трифенилфосфина и тетрагалогенметана или N-галоген-сукцинимида в инертном растворителе. Альтернативно, электрофильное промежуточное соединение формулы XIII можно получить путем сульфонилирования. Преобразование в алкил- или арилсульфониловый эфир формулы XIII можно осуществить с использованием соответствующего сульфонилхлорида в присутствии подходящего основания в инертном растворителе.
Альтернативно, соединения формулы I можно получить, как показано на Схеме 6, путем взаимодействия соединения XIII с реагентом XII, где A-LG представляет собой -(CH2)p-C{(R8)(R9)}q-(CH2)r-1-CHO.
[Химическая формула 11]
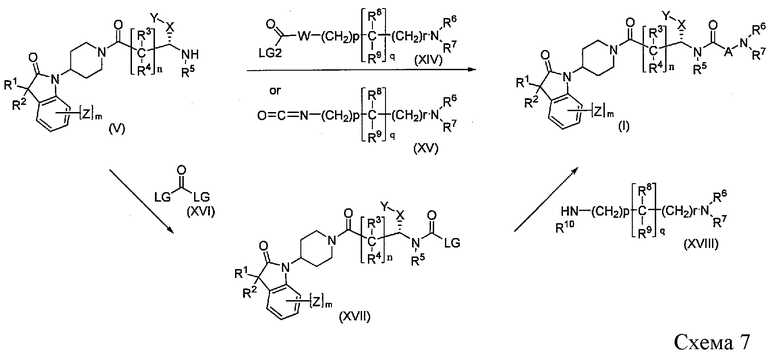
Соединения формулы I, где A представляет собой W-(CH2)p-C{(R8)(R9)}q-(CH2)r- и W представляет собой N-R10 или NH, можно получить, как показано на Схеме 7, путем взаимодействия соединения V с реагентом XIV, где LG2 представляет собой подходящую удаляемую группу, такую как Cl, Br, I, имидазол или o-нитробензол. Альтернативно, соединение V можно подвергнуть взаимодействию с изоцианатом формулы XV в инертном растворителе, таком как 1,2-дихлорэтан, что приводит к соединению формулы I, где W представляет собой NH. Соединение формулы I можно получить через промежуточное соединение формулы XVII. Промежуточное соединение XVII можно получить путем реакции сочетания соединения формулы V с реагентом формулы XVI, где LG имеет значение, определенное выше, в присутствии подходящего основания в инертном растворителе. Промежуточное соединение формулы XVII может быть выделено или образовано in situ. Преобразование из промежуточного соединения формулы XVII в желаемое соединение формулы I осуществляют при помощи реакции нуклеофильного замещения соединения формулы XVIII в присутствии подходящего основания в инертном растворителе.
[Химическая формула 12]
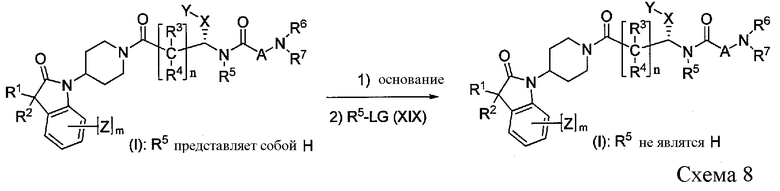
Соединение формулы I, где R5 является отличным от водорода, можно далее преобразовать в новые соединения I, которые замещены по амидной NH группе, как показано на Схеме 8. Алкилирование соединения формулы I осуществляют при помощи реакции нуклеофильного замещения реагента формулы XIX, где LG имеет значение, определенное выше, с соединением формулы I, обработанным подходящим основанием.
[Химическая формула 13]
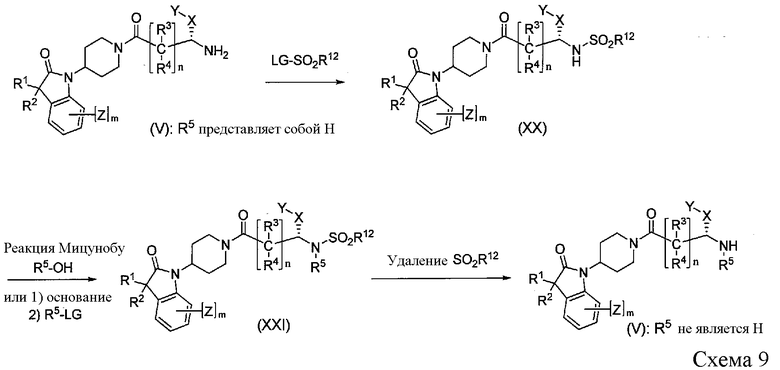
Соединение формулы V, где R5 является отличным от водорода, можно получить через сульфонамиды формулы XX, как показано на Схеме 9. Получение сульфонамидного соединения XX осуществляют путем взаимодействия соединения формулы V и алкилсульфонил- или арилсульфонилгалогенида, где R12 выбирают из типичных сульфонильных защитных групп, описаннных в Protective Groups in Organic Synthesis edited by T. W. Greene et al. (John Wiley & Sons, 1999). Примеры подходящих защитных групп включают, но не ограничиваются этим: метансульфонильную, трифторметансульфонильную, бензолсульфонильную, 2- или 4-нитробензолсульфонильную и 2,4-динитробензолсульфонильную группы. Из этих групп, 2- или 4-нитробензолсульфонил и 2,4-динитробензолсульфонил являются предпочтительными. Удаление сульфонильной группы из соединения формулы XXI осуществляют в условиях, представляющих собой известные методы, указанные в настоящей заявке.
Соединение формулы XXI можно получить в условиях реакции Мицунобу, определенных ниже, из сульфонамида формулы XX и R5-OH. Альтернативно, соединение формулы XXI также можно получить из соединения формулы XX с использованием процедуры, аналогичной показанной на Схеме 8.
Реакцию Мицунобу осуществляют в присутствии реагента (реагентов). Соответственно, нет никаких конкретных ограничений, касающихся природы используемых реагентов, и любой реагент, обычно используемый в реакциях такого типа, может быть равным образом использован в данной реакции. Примеры таких реагентов включают, но не ограничиваются этим:
(a) сочетание (a1) диалкилазодикарбоксилата, такого как диэтилазодикарбоксилат (DEAD), диметилазодикарбоксилат (DMAD) и диизопропилазодикарбоксилат (DIAD), и (a2) триалкилфосфина, такого как трибутилфосфин (TBP) или триарилфосфин, такой как трифенилфосфин (TPP);
(b) сочетание (b1) тетраалкилдиазокарбоксамида, такого как N,N,N',N'-тетраизопропилазодикарбоксамид (TIPA) и N,N,N',N'-тетраметилазодикарбоксамид (TMAD), и (b2) триалкилфосфина, такого как трибутилфосфин (TBP) или триарилфосфин, такой как трифенилфосфин (TPP);
(c) фосфоран, такой как цианометилентрибутилфосфоран (CMBP), цианометилентриметилфосфоран и диметил(трибутилфосфоранилиден)малонат (DMTP).
Реакцию обычно и предпочтительно осуществляют в присутствии растворителя. Нет никакого конкретного ограничения, касающегося природы используемого растворителя, при условии, что он не оказывает неблагоприятного действия на реакцию или используемые в реакции реагенты, и что он может растворять реагенты, по меньшей мере, до некоторой степени. Примеры подходящих растворителей включают, но не ограничиваются этим: алифатические углеводороды, такие как гексан, гептан и петролейный эфир; галогенированные углеводороды, такие как дихлорметан, хлороформ, тетрахлорид углерода и дихлорэтан; простые эфиры, такие как диэтиловый эфир, диизопропиловый эфир, ТГФ, диметоксиэтан и диоксан; ароматические углеводороды, такие как бензол, толуол, ксилол, хлорбензол, дихлорбензол и нитробензол; амиды, такие как формамид, DMF, N,N-диметилацетамид и триамид гексаметилфосфорной кислоты; амины, такие как N-метилморфолин, триэтиламин, трипропиламин, трибутиламин, диизопропилэтиламин, пиридин, 4-пирролидинопиридин, N,N-диметиланилин и N,N-диэтиланилин; нитрилы, такие как ацетонитрил и бензонитрил; сульфоксиды, такие как диметилсульфоксид и сульфолан. Из этих растворителей, толуол, бензол, ксилол, хлорбензол, дихлорбензол, ТГФ, диэтиловый эфир, диизопропиловый эфир, диметоксиэтан, ацетонитрил, дихлорметан, дихлорэтан и хлороформ являются предпочтительными.
Соединение формулы V получают путем удаления сульфонильной защитной группы из промежуточного соединения формулы XXI, как показано на Схеме 9, в соответствии с процедурами, аналогичными описанным в Protective Groups in Organic Synthesis edited by T. W. Greene et al. (John Wiley & Sons, 1999). Типичные условия для удаления защитных групп включают использование, но не ограничиваются этим:
(a) Сильной кислоты, такой как бромистый водород (HBr) (Haskell, B., E., et. al., J. Org. Chem, 41, 159 (1976)) и т.п.
(b) Сильного нуклеофила, такого как избыточное количество пропиламина, тиобензола, тиоуксусной кислоты (Fukuyama, T., et. al., Tetrahedron Lett., 36, 6373 (1995), Fukuyama, T., et. al., Tetrahedron Lett., 38, 5831 (1997)) и т.п.
(c) Восстановителя, такого как растворяющийся металл, такой как натрий в трет-бутаноле (Merlin, P., et. al. Tetrahedron Lett., 29, 1691 (1988) или литий в жидком аммиаке (Heathcock, C. H., et. al., J. Am. Chem. Soc., 108, 5022 (1986), гидридные реагенты, такие как литийалюминийгидрид (Bell, K. E., et. al. Tetrahedron Lett., 36, 8681 (1995)) и т.п.
Соединения общей формулы I по настоящему изобретению также можно получить конвергентным способом, как описано в схемах реакций 10, 11 и 12. Карбоновокислотно-защищенные аминокислотные производные XXI, во многих случаях, являются коммерчески доступными. На Схеме реакций 10, PG2 представляет собой карбокси-защитную группу. Термин "карбокси-защитная группа", как он используется в настоящей заявке, означает защитную группу, которая может быть удалена химическим путем, таким как гидрогенолиз, гидролиз, электролиз или фотолиз, и такие карбокси-защитные группы описаны в Protective Groups in Organic Synthesis edited by T. W. Greene et al. (John Wiley & Sons, 1999). Типичные карбокси-защитные группы включают, но не ограничиваются этим: метил, этил, трет-бутил, метоксиметил, 2,2,2-трихлорэтил, бензил, дифенилметил, триметилсилил, трет-бутилдиметилсилил и аллил. Из этих групп, трет-бутил или метил являются предпочтительными.
[Химическая формула 14]
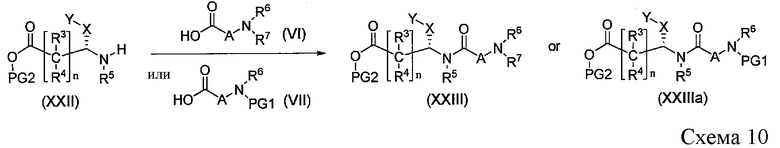
Промежуточные соединения формулы XXIII или XXIIIa можно получить, как показано на Схеме 10, путем сочетания сложных эфиров аминокислот XXII с аминокислотами формулы VI или VII. Когда присутствует мочевинная связь в соединении XXIII или XXIIIa, она может быть введена, как проиллюстрировано на Схеме 7.
[Химическая формула 15]
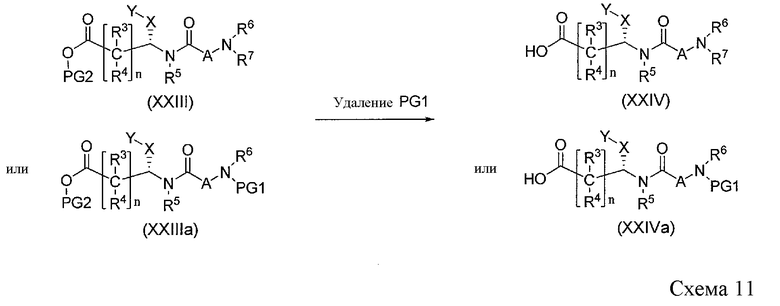
Преобразование сложного эфира XXIII или XXIIIa в промежуточные кислотные соединения XXIV или XXIVa можно осуществить различными способами, известными в данной области техники, как показано на Схеме 11. Например, метиловый и этиловый эфиры могут быть гидролизованы с использованием гидроксида лития или гидроксида натрия или гидроксида калия в протонном растворителе, таком как водный раствор спирта, такого как метанол, этанол и 2-пропанол. Кроме того, удаление бензильной группы можно осуществить различными восстановительными способами, включая гидрирование в присутствии палладиевого катализатора в протонном растворителе, таком как метанол. Расщепление аллилового эфира можно осуществить с использованием катализатора на основе тетракис-трифенилфосфин палладия в присутствии 2-этилгексановой кислоты в различных растворителях, включая этилацетат и дихлорметан (см. J. Org. Chem., 42, 587 (1982)).
[Химическая формула 16]
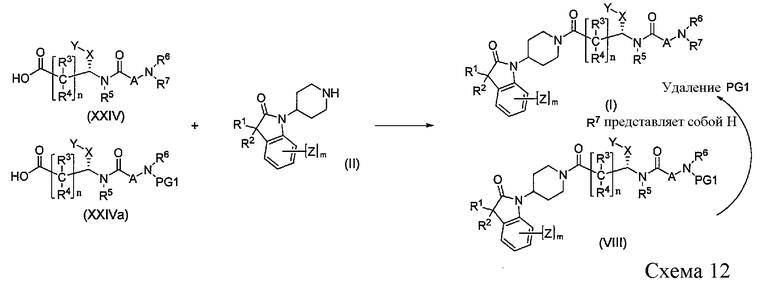
Кислоты XXIV или XXIVa затем можно преобразовать в соединение I или соединение VIII, как показано на Схеме 12. Связывание пиперидинов формулы II с кислотами формулы XXIV или XXIVa, где PG1 представляет собой подходящую защитную группу, определенную выше, удобным образом осуществляют в стандартных реакционных условиях пептидного связывания. Преобразование соединения VIII в соединение I осуществляют путем удаления защитной группы PG1. Когда R6 и/или R7 представляет собой водород, замещенные алкильные группы необязательно могут быть присоединены к атому азота, как показано на Схеме 4.
[Химическая формула 17]
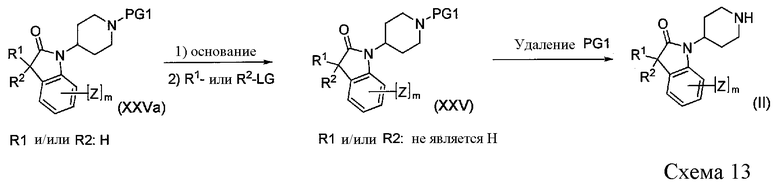
Когда R1 и/или R2 представляет собой/представляют собой водород, пиперидины формулы II можно получить из промежуточных соединений XXVa путем алкилирования, с последующим удалением защитной группы PG1, как показано на Схеме 13. При этом удаляемая группа LG и амино-защитная группа PG1 имеют значение, определенное выше.
Процедура протокола алкилирования подробно описана Dounay et al. (J. Am Chem. Soc., 125, 6261-6271 (2003)).
[Химическая формула 18]
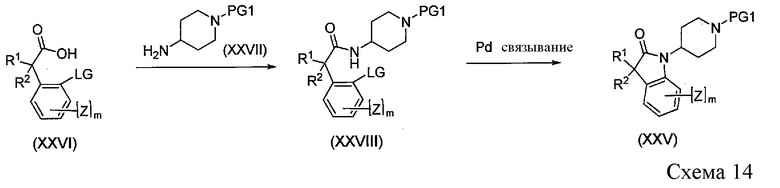
Соединения формулы XXV можно получить путем межмолекулярного опосредованного палладием связывания, как показано на Схеме 14. Промежуточные соединения формулы XXVIII получают при помощи стандартной реакции пептидного связывания кислот формулы XXVI и пиперидинов формулы XXVII. Катализируемую палладием реакцию сочетания осуществляют с использованием известной процедуры (Hoogenband et al. Tetrahedron Lett., 45 (2004) 8535).
[Химическая формула 19]
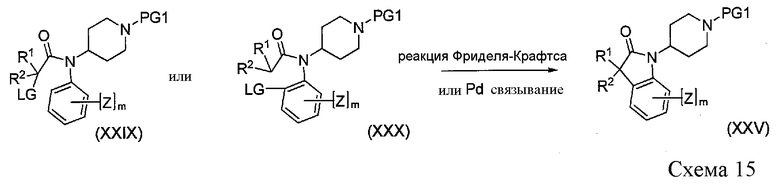
Соединения формулы XXV можно получить с использованием реакции Фриделя-Крафтса или путем межмолекулярного опосредованного палладием связывания, как показано на Схеме 15. Протоколы реакции Фриделя-Крафтса и опосредованного палладием связывания соединения формулы XXIX и XXX осуществляют с использованием известных процедур (Zaveri, N. et al. J. Med. Chem., 47, (2004), 2973-2976., Buchwald, S., J. Am. Chem. Soc., 125, (2003), 12084-12085., Lee, S. et al. J. Org. Chem. 66 (2001), 3402-3415). Получение промежуточных соединений формулы XXIX и XXX осуществляют в соответствии с протоколами, указанными в литературных источниках, на которые ссылаются выше.
Примеры
Настоящее изобретение иллюстрируется представленными ниже неограничивающими примерами, в которых, если не указано иное: все реагенты являются коммерчески доступными, все процедуры осуществляли при комнатной температуре или при температуре окружающей среды, т.е. в пределах около 18-25°C; выпаривание растворителя осуществляли с использованием роторного испарителя при пониженном давлении и при температуре бани до около 60°C; реакции отслеживали методом тонкослойной хроматографии (ТСХ), и время реакции указано только в целях иллюстрации; структуру и чистоту всех выделенных соединений подтверждали с использованием, по меньшей мере, одного из следующих методов: ТСХ (предварительно покрытые силикагелем Merck 60 F254 пластины для ТСХ, или предварительно покрытые Merck NH2 F254 пластины для высокоэффективной ТСХ), масс-спектрометрия или ядерный магнитный резонанс (ЯМР). Выходы указаны только в целях иллюстрации. Колоночную флэш-хроматографию осуществляли с использованием силикагеля 60 от компании Merck (230-400 меш ASTM) или Fuji Silysia Chromatorex (зарегистрированная торговая марка) DU3050 (Амино типа, 30-50 мкм) или диоксида кремния Biotage (32-63 мм, KP-Sil) или амино-связанного диоксида кремния Biotage (35-75 мм, KP-NH). Очистку соединений методом ВЭЖХ осуществляли с использованием следующего оборудования и условий ("способ A"); Оборудование: система Waters MS-trigger AutoPurificationTM; Колонка: Waters XTerra C18, 19Ч50 мм, размер частиц 5 мм, Способ A: Метанол или ацетонитрил/0,05% (об./об.) водный раствор муравьиной кислоты, Способ B: Метанол или ацетонитрил/0,01% (об./об.) водный раствор аммиака. Очистку методом ВЭЖХ ("Способ B") осуществляли с использованием следующего оборудования и условий: Оборудование: Система UV-trigger preparative HPLC, Waters (Колонка: XTerra MS C18, 5 мкм, 19Ч50 мм или 30Ч50 мм), Детектор: УФ 254 нм, Условия: ацетонитрил:0,05% водный раствор муравьиной кислоты или ацетонитрил:0,01% водный раствор аммиака; 20 мл/мин (19Ч50 мм) или 40 мл/мин (30Ч50 мм) при комнатной температуре. Данные масс-спектрометрии низкого разрешения (ESI) получали с использованием следующего оборудования и условий: Оборудование: система ВЭЖХ Waters Alliance с использованием ZQ или ZMD масс-спектрометра и УФ детектора. Данные ЯМР получали при 270 МГц (спектрометр JEOL JNM-LA 270) или 300 МГц (JEOL JNM-LA300) с использованием дейтерированного хлороформа (99,8% D) или диметилсульфоксида (99,9% D) в качестве растворителя, если не указано иное, относительно тетраметилсилана (TMS) в качестве внутреннего стандарта, в миллионных долях (м.д.); использовали следующие традиционные аббревиатуры: с=синглет, д=дублет, т=триплет, кв.=квартет, м=мультиплет, ушир.=уширенный и т.п. Химические символы имеют их обычные значения; мкл (микролитр(микролитров)), мкг (микрограмм), M (моль(молей) на литр), л (литр(литров)), мл (миллилитр(миллилитров)), г (грамм), мг (миллиграмм), моль (моль (молей)), ммоль (миллимоль(миллимолей)).
Условия для определения времени удерживания ВЭЖХ:
Способ A:
Оборудование: Waters Acquity Ultra Performance LC с использованием TUV Детектора и ZQ масс-спектрометра
Колонка: Waters ACQUITY C18, 2,1Ч50 мм, размер частиц 1,7 мкм
Температура колонки: 60°C
Растворители:
A1: 10 мМ водного раствора ацетата аммония
B1: ацетонитрил
Способ B:
Оборудование: Waters Acquity Ultra Performance LC с использованием TUV Детектора и ZQ масс-спектрометра
Колонка: Waters SunFire C18 2,1Ч50 мм, размер частиц 3,5 мкм
Температура колонки: 40°C
Растворители:
A: вода
B: ацетонитрил
C: 1% (об/об) водного раствора муравьиной кислоты
Способ C:
Оборудование: Waters Acquity Ultra Performance LC с использованием TUV Детектора и ZQ масс-спектрометра
Колонка: Waters ACQUITY C18, 2,1Ч100 мм, размер частиц 1,7 мкм
Температура колонки: 60°C
Растворители:
A1: 10 мМ водного раствора ацетата аммония
B1: ацетонитрил
ПРИМЕР 1
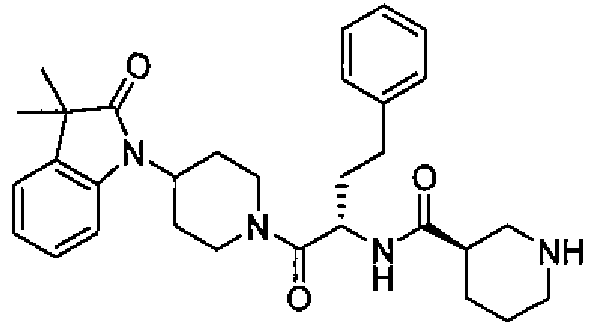
(R)-N-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)пиперидин-3-карбоксамид
[Химическая формула 20]
Стадия 1.
1-(1-бензилпиперидин-4-ил)-3,3-диметилиндолин-2-он
К перемешиваемому раствору N,N-диизопропилэтиламина (28,9 мл, 205 ммоль) и безводного тетрагидрофурана (446 мл), охлажденному до -78°C, добавляли по каплям н-бутиллитий (82,3 мл, 205 ммоль) в атмосфере азота. Реакционную смесь перемешивали при -78°C в течение 10 минут, затем добавляли по каплям 1-(1-бензилпиперидин-4-ил)индолин-2-он (21,0 г, 68,0 ммоль, Tetrahedron Letters, 2004, 50, 8535-8537) в тетрагидрофуране (258 мл). Реакционную смесь перемешивали еще в течение 30 минут и добавляли иодметан (12,8 мл, 205 ммоль). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение ночи. Реакционную смесь охлаждали до 0°C и добавляли водный раствор хлорида аммония для гашения реакции. Водный слой экстрагировали этилацетатом (Ч3). Органические слои объединяли, сушили над сульфатом натрия и концентрировали. Остаток очищали колоночной хроматографией на силикагеле с использованием 10% этилацетата в гексане с получением указанного в заголовке соединения (19,0 г, 83%).
1Н-ЯМР (CDCl3) δ: 7,31-7,25 (4H, м), 7,21-7,10 (4H, м), 7,10-6,94 (1H, м), 4,27-4,21 (1H, м), 3,49 (2H, с), 2,97-2,94 (2H, м), 2,44-2,36 (2H, м), 2,11-2,05 (2H, м), 1,62-1,58 (2H, м), 1,28 (6H, с).
Стадия 2.
Гидрохлорид 3,3-диметил-1-(пиперидин-4-ил)индолин-2-она
1-(1-Бензилпиперидин-4-ил)-3,3-диметилиндолин-2-он (17,0 г, 50,8 ммоль, Стадия 1) и палладий на углероде (19,7 г) добавляли к этанолу (442 мл). Реакционную смесь нагревали до 60°C в атмосфере водорода в течение 10 часов. Реакционную смесь фильтровали и фильтрат концентрировали до около 50 мл. Добавляли диэтиловый эфир (200 мл) и полученную смесь доводили до pH 3 при помощи насыщенного раствора хлористого водорода в метаноле. Белые твердые частицы фильтровали и сушили в вакууме с получением указанного в заголовке соединения (7,5 г, 53%).
1Н-ЯМР (CDCl3) δ: 7,33-7,29 (2H, м), 7,23-7,21 (1H, м), 7,13-7,09 (1H, м), 4,46-4,40 (1H, м), 3,59-3,56 (2H, м), 3,26-3,18 (2H, м), 2,83-2,72 (2H, м), 2,01-1,98 (2H, м), 1,34 (6H, с).
Стадия 3.
(S)-трет-бутил 1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-илкарбамат
К смеси гидрохлорида 3,3-диметил-1-(пиперидин-4-ил)индолин-2-она (2,00 г, 7,12 ммоль, ПРИМЕР 1, Стадия 2) и (S)-N-трет-бутоксикарбонил-гомофенилаланина (2,09 г, 7,48 ммоль) в дихлорметане (30 мл) при 0°C добавляли триэтиламин (3,50 мл, 24,9 ммоль), гидрохлорид 1-этил-3-(3-диметиламинопропил)-карбодиимида (1,64 г, 8,55 ммоль) и 1-гидроксибензотриазол гидрат (1,16 г, 8,55 ммоль). Затем смесь перемешивали в течение 24 часов. Реакцию гасили водой и летучие вещества удаляли в вакууме. Остаток разбавляли этилацетатом (100 мл). Органический слой промывали при помощи 0,5 моль/л хлористоводородной кислоты (40 мл Ч3), 0,5 моль/л водного раствора гидроксида натрия (40 мл Ч2) и насыщенного солевого раствора (30 мл), сушили над сульфатом магния и концентрировали с получением указанного в заголовке соединения (2,79 г, 77%) в виде твердого вещества.
1Н-ЯМР (CDCl3) δ: 7,25-7,08 (7H, м), 7,00-6,95 (2H, м), 5,45-5,35 (1H, м), 4,75-4,25 (3H, м), 3,80-3,60 (1H, м), 3,09-2,90 (1H, м), 2,70-2,55 (3H, м), 2,35-2,15 (2H, м), 2,00-1,60 (4H, м), 1,43-1,41 (9H, псевдо д), 1,29-1,22 (6H, псевдо д).
Стадия 4.
гидрохлорид (S)-1-(1-(2-амино-4-фенилбутаноил)пиперидин-4-ил)-3,3-диметилиндолин-2-она
Смесь (S)-трет-бутил 1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-илкарбамата (2,79 г, 5,52 ммоль, ПРИМЕР 1, Стадия 3) и 1 моль/л гидрохлорида в диэтиловом эфире (25 мл) перемешивали при комнатной температуре. Через 4 часа к реакционной суспензии добавляли смесь хлортриметилсилана (2 мл) и метанола (4 мл). Полученная смесь превращалась в прозрачный раствор, и раствор перемешивали в течение 3 часов. Смесь концентрировали в вакууме и остаток разбавляли диэтиловым эфиром и снова концентрировали для полного удаления метанола. Полученное твердое вещество промывали диэтиловым эфиром с получением указанного в заголовке соединения (2,05 г, 84%).
1Н-ЯМР (CDCl3) δ: 8,38-8,25 (3H, м), 7,45-6,74 (9H, м), 4,68-4,56 (2H, м), 4,45-4,10 (1H, м), 3,63-3,44 (1H, м), 3,05-2,75 (3H, м), 2,57-2,05 (5H, м), 1,65-1,44 (2H, м), 1,26-1,23 (6H, псевдо д).
MS (ESI) m/z: 406 (M+H)+.
Стадия 5.
(R)-N-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)пиперидин-3-карбоксамид
К раствору гидрохлорида (S)-1-(1-(2-амино-4-фенилбутаноил)пиперидин-4-ил)-3,3-диметилиндолин-2-она (45 мг, 0,10 ммоль, ПРИМЕР 1, Стадия 4) добавляли (R)-N-Boc-пиперидин-3-карбоновую кислоту (31 мг, 0,14 ммоль) и гексафторфосфат 2-(1H-бензотриазол-1-ил)-1,1,3,3-тетраметилурония (53 мг, 0,14 ммоль) и смесь перемешивали при 60°C в течение 6 часов, затем при комнатной температуре в течение 13 часов. Летучие вещества удаляли в вакууме.
К остатку, растворенному в 1,2-дихлорэтане (1 мл), добавляли 2,2,2-трифторуксусную кислоту (1 мл) и смесь перемешивали при комнатной температуру в течение 2 часов. Затем смесь концентрировали в вакууме. Остаток разбавляли метанолом и наносили на сильный катионообменный картридж (BondElute (зарегистрированная торговая марка) SCX, 1 г/6 мл, Varian Inc.) и твердофазную матрицу промывали метанолом (6 мл). Неочищенную смесь элюировали в трубке-сборнике при помощи 1 моль/л аммиака в метаноле (6 мл) и концентрировали в вакууме. Остаток очищали методом препаративной ЖХ-МС с получением указанного в заголовке соединения (22,7 мг, 44%).
MS (ESI) m/z: 517 (M+H) +.
ВЭЖХ время удерживания: 0,72 мин (Способ A).
ПРИМЕР 1: Альтернативный способ
Стадия 1.
(R)-трет-бутил 3-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-илкарбомоил)пиперидин-1-карбоксилат
К смеси гидрохлорида (S)-1-(1-(2-амино-4-фенилбутаноил)пиперидин-4-ил)-3,3-диметилиндолин-2-она (500 мг, 1,13 ммоль, ПРИМЕР 1, Стадия 4), (R)-N-Boc-пиперидин-3-карбоновой кислоты (389 мг, 1,70 ммоль) и триэтиламина (556 мкл, 3,96 ммоль) в диметилформамиде (8 мл) добавляли гидрохлорид 1-этил-3-(3-диметиламино)пропилкарбодиимида (347 мг, 1,81 ммоль) и 1-гидроксибензотриазол гидрат (277 мг, 1,81 ммоль). После перемешивания в течение 17 часов смесь разбавляли этилацетатом и водой. Органический слой промывали насыщенным водным раствором гидрокарбоната натрия и насыщенным солевым раствором, сушили над сульфатом натрия и концентрировали в вакууме. Остаток очищали колоночной хроматографией на силикагеле (гексан/этилацетат) и колоночной хроматографией на аминовом геле (гексан/этилацетат) с получением указанного в заголовке соединения (615 мг, 88%) в виде аморфного твердого вещества.
1Н-ЯМР (CDCl3) δ: 7,33-7,08 (м, 7H), 7,01-6,85 (м, 2H), 6,68-6,58 (м, 1H), 5,03-4,93 (м, 1H), 4,83-4,70 (м, 1H), 4,52-4,30 (м, 1H), 4,23-4,06 (м, 2H), 4,04-3,88 (м, 1H), 3,76 (м, 1H), 3,11-2,56 (м, 5H), 2,43-2,18 (м, 3H), 2,09-1,57 (м, 8H), 1,48 (с, 9H), 1,38-1,30 (м, 6H).
MS (ESI) m/z: 617 (М+H)+.
Стадия 2.
(R)-N-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)пиперидин-3-карбоксамид
Смесь (R)-трет-бутил 3-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-илкарбамоил)пиперидин-1-карбоксилата (615 мг, 0,997 ммоль, ПРИМЕР 1, Альтернативный Способ, Стадия 1) и трифторуксусной кислоты (2 мл) перемешивали в течение 20 минут и концентрировали. Остаток разбавляли дихлорметаном и подщелачивали насыщенным водным раствором гидрокарбоната натрия. Органический слой сушили над сульфатом натрия и концентрировали в вакууме. Неочищенный продукт очищали колоночной хроматографией на аминовом геле (дихлорметан/метанол, градиент) с получением указанного в заголовке соединения (430 мг, 83%) в виде аморфного твердого вещества.
1Н-ЯМР (CDCl3) δ: 8,16-7,84 (м, 1H), 7,55-6,77 (м, 9H), 5,16-4,97 (м, 1H), 4,88-4,71 (м, 1H), 4,58-4,26 (м, 1H), 3,96-3,77 (м, 1H), 3,26-2,54 (м, 9H), 2,48-2,19 (м, 3H), 2,18-1,18 (м, 14H).
MS (ESI) m/z: 517 (М+H)+.
ПРИМЕР 2-16
[Химическая формула 21]
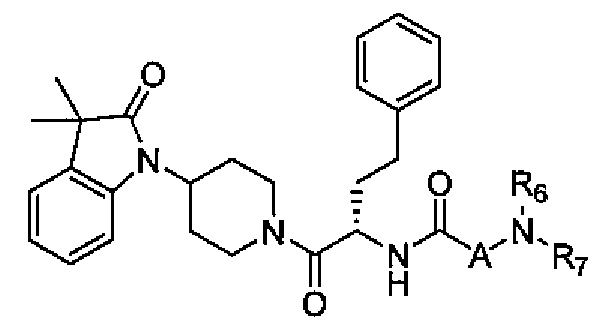
Следующие примеры, ПРИМЕР 2-16, получали в соответствии с процедурой, аналогичной описанной на Стадии 5 Примера 1, с использованием подходящего предшественника вместо (R)-1-(трет-бутоксикарбонил)пиперидин-3-карбоновой кислоты.
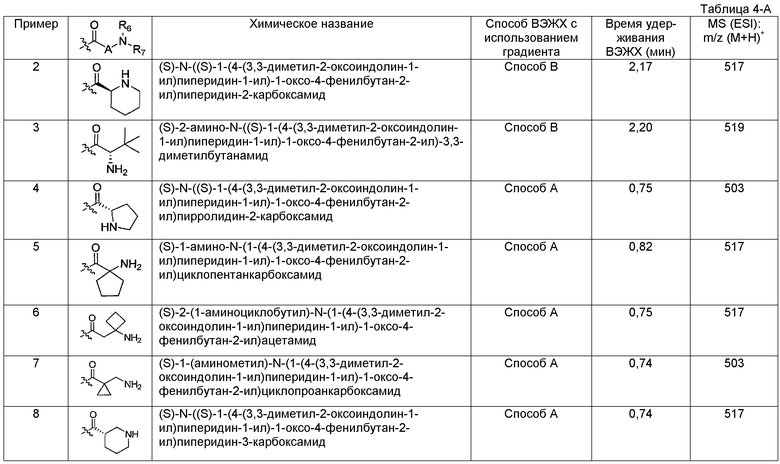
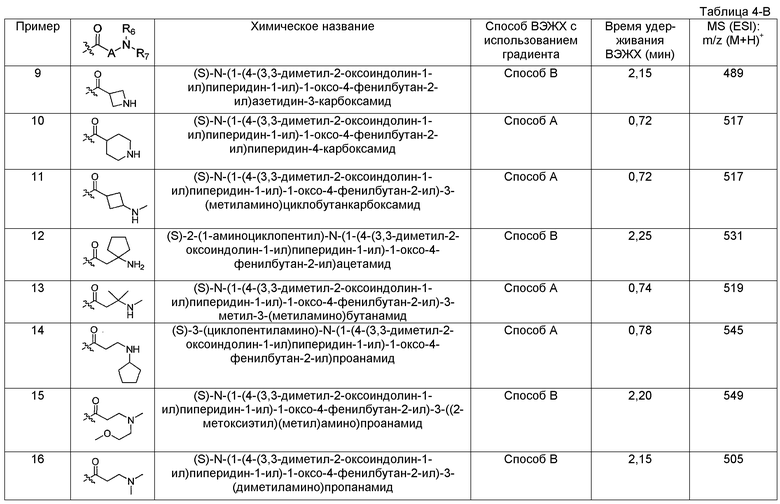
ПРИМЕР 17
(S)-N-(1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)-2-(пирролидин-1-ил)ацетамид
[Химическая формула 22]
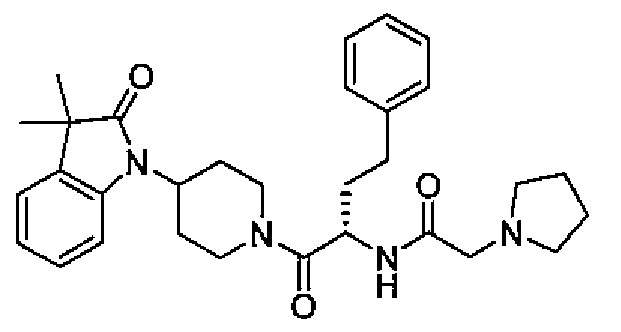
Стадия 1.
(S)-2-хлор-N-(1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)ацетамид
К раствору хлорацетилхлорида (120 мкл, 1,49 ммоль) в дихлорметане (5 мл) медленно добавляли смесь гидрохлорида (S)-1-(1-(2-амино-4-фенилбутаноил)пиперидин-4-ил)-3,3-диметилиндолин-2-она (600 мг, 1,36 ммоль, ПРИМЕР 1, Стадия 4) и триэтиламина (420 мкл, 2,99 ммоль) в дихлорметане (4 мл) при 0°C. Через 5 минут реакцию гасили водой и смесь разбавляли этилацетатом для разделения. Органический слой промывали водой (Ч2) и насыщенным солевым раствором, сушили над сульфатом натрия и концентрировали с получением указанного в заголовке соединения. Продукт разбавляли при помощи 6,0 мл тетрагидрофурана с получением 0,226 моль/л тетрагидрофуранового раствора, что предполагало 100% преобразование. Концентрированный раствор использовали на следующей стадии без дополнительной очистки.
MS (ESI) m/z: 482 M(35Cl)+, 484 M(37Cl)+.
Стадия 2.
(S)-N-(1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)-2-(пирролидин-1-ил)ацетамид
К раствору пирролидина (16,9 мкл, 0,204 ммоль) добавляли (S)-2-хлор-N-(1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)ацетамид (0,226 моль/л раствор в тетрагидрофуране, 300 мкл, 0,0679 ммоль, ПРИМЕР 17, Стадия 1) при комнатной температуре. После перемешивания при 60°C в течение 2 часов смесь концентрировали. Остаток разбавляли метанолом (4 мл). Остаток разбавляли метанолом и наносили на сильный катионо-обменный картридж (BondElute(зарегистрированная торговая марка) SCX, 1 г/6 мл, Varian Inc.) и твердофазную матрицу промывали метанолом (6 мл). Неочищенную смесь элюировали в трубке-сборнике при помощи 1 моль/л аммиака в метаноле (6 мл) и концентрировали в вакууме. Остаток очищали методом препаративной ЖХ-МС с получением указанного в заголовке соединения (12,2 мг, 35 %).
MS (ESI) m/z: 517 (M+H)+.
ВЭЖХ время удерживания: 2,19 мин. (Способ B).
ПРИМЕР 18-19
[Химическая формула 23]
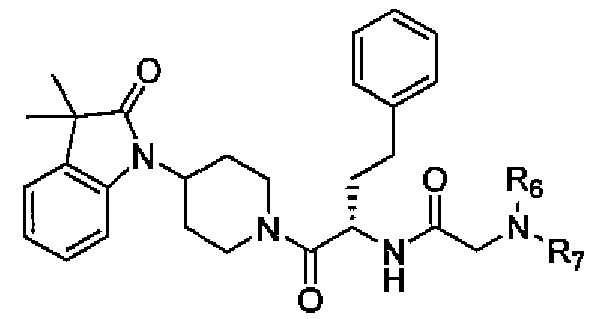
Следующие примеры, ПРИМЕР 18-19, получали в соответствии с процедурой, аналогичной описанной на Стадиях 1-2 Примера 17, с использованием подходящего предшественника вместо пирролидина.

ПРИМЕР 20
(R)-N-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)-1-метилпиперидин-3-карбоксамид
[Химическая формула 24]
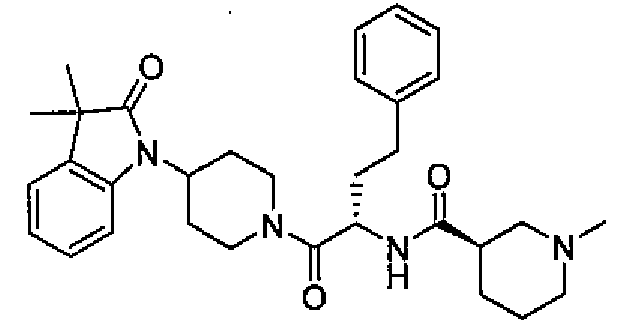
Стадия 1
(R)-N-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)-1-метилпиперидин-3-карбоксамид
Смесь гидрохлорида (S)-1-(1-(2-амино-4-фенилбутаноил)пиперидин-4-ил)-3,3-диметилиндолин-2-она (30 мг, 0,068 ммоль), (R)-N-Boc-пиперидин-3-карбоновой кислоты (31 мг, 0,14 ммоль), триэтиламина (29 мкл, 0,20 ммоль) и гексафторфосфата 2-(1H-бензотриазол-1-ил)-1,1,3,3-тетраметилурония (52 мг, 0,14 ммоль) в диметилформамиде (1 мл) перемешивали в течение 1,5 часа при комнатной температуре. Реакционную смесь разбавляли водой (2 мл) и этилацетатом (6 мл). Органический слой промывали водным раствором бикарбоната натрия (2 мл Ч2), фильтровали через колонку с сульфатом магния и концентрировали. К остатку добавляли трифторуксусную кислоту (1 мл) и перемешивали в течение 10 минут.
После удаления трифторуксусной кислоты остаток разбавляли метанолом (4 мл) и раствор фильтровали через сильный катионо-обменный картридж (BondElute(зарегистрированная торговая марка) SCX, 1 г/6 мл, Varian Inc.). Колонку промывали при помощи 1 моль/л аммиака в метаноле (2 мл Ч3) и фильтрат концентрировали. Остаток растворяли в дихлорметане (1,5 мл) и метаноле (0,15 мл). К раствору добавляли параформальдегид (6,1 мг, 0,20 ммоль) и триацетоксиборогидрид натрия (43 мг, 0,20 ммоль) при комнатной температуре. Через 1 час к смеси добавляли дополнительное количество триацетоксиборогидрида натрия (200 мг) и смесь перемешивали. Через 10 минут смесь концентрировали и остаток подщелачивали водным раствором гидрокарбоната натрия и экстрагировали этилацетатом (3 мл Ч2). Органический слой фильтровали через колонку с сульфатом магния, и фильтрат концентрировали в вакууме. Неочищенный продукт очищали методом препаративной ЖХ-МС с получением указанного в заголовке соединения (17,4 мг, 48 %).
MS (ESI) m/z: 531 (M+H)+.
ВЭЖХ время удерживания: 0,75 мин. (Способ A).
ПРИМЕР 21
(1S,3R)-N-((S)-3-(4-хлорфенил)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксопропан-2-ил)-3-(метиламино)циклопентанкарбоксамид
[Химическая формула 25]
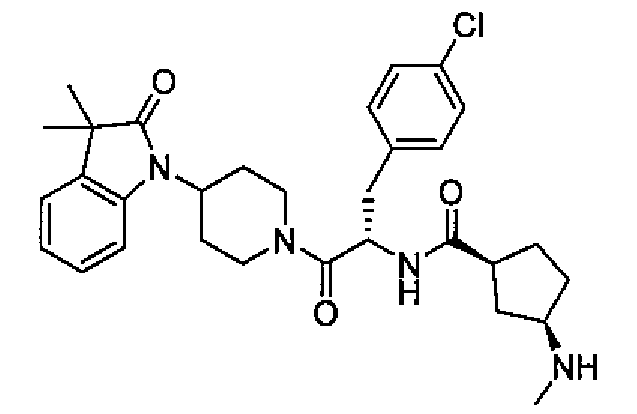
Стадия 1.
(1S,3R)-3-(трет-бутоксикарбонил(метил)амино)-циклопентанкарбоновая кислота
К перемешиваемой суспензии гидрида натрия (89 мг, 2,24 ммоль) в тетрагидрофуране (10 мл) и диметилформамиде (10 мл), охлажденной на ледяной бане, добавляли (1S,3R)-3-(трет-бутоксикарбониламино)циклопентанкарбоновую кислоту (427 мг, 1,86 ммоль, WO2006/011035) и иодметан (128 мкл, 2,05 ммоль). Смеси давали нагреться до комнатной температуры и перемешивали в течение 20 часов. Добавляли гидрид натрия (178 мг, 4,474 ммоль) и иодметан (256 мкл, 4,102 ммоль) и реакционную смесь перемешивали в течение 20 часов при комнатной температуре. Реакционную смесь подкисляли до pH 3,5 при помощи 1 моль/л хлористоводородной кислоты и экстрагировали этилацетатом. Органический слой сушили над сульфатом натрия и концентрировали при пониженном давлении. Неочищенное вещество очищали колоночной хроматографией (50% этилацетата в гексане, содержащем 1,5% уксусной кислоты) с получением желаемого соединения в виде бесцветного твердого вещества (138,0 мг, 31%).
MS (ESI) m/z: 242 (M-H)-.
Стадия 2.
(S)-1-(1-(2-амино-3-(4-хлорфенил)пропаноил)пиперидин-4-ил)-3,3-диметилиндолин-2-он
К раствору гидрохлорида 3,3-диметил-1-(пиперидин-4-ил)индолин-2-она (45 мг, 0,10 ммоль, ПРИМЕР 1, Стадия 2) в растворе 3,75% триэтиламина/N,N-диметилацетамид (1,5 мл) добавляли (S)-2-(трет-бутоксикарбониламино)-3-(4-хлорфенил)пропановую кислоту (32,9 мг, 0,11 ммоль) и гексафторфосфат 2-(1H-бензотриазол-1-ил)-1,1,3,3-тетраметилурония (38 мг, 0,15 ммоль) и смесь перемешивали при 60°C в течение 6 часов и при комнатной температуре в течение 13 часов. Летучие вещества удаляли в вакууме.
К остатку, растворенному в 1,2-дихлорэтане (1 мл), добавляли 2,2,2-трифторуксусную кислоту (1 мл) и смесь перемешивали при комнатной температуре в течение 2 часов. Затем смесь концентрировали в вакууме. Остаток разбавляли метанолом и наносили на сильный катионо-обменный картридж (BondElute (зарегистрированная торговая марка) SCX, 1 г/6 мл, Varian Inc.) и твердофазную матрицу промывали метанолом (6 мл). Неочищенную смесь элюировали в трубке-сборнике при помощи 1 моль/л аммиака в метаноле (6 мл) и концентрировали в вакууме. Остатки использовали на следующей стадии без дополнительной очистки.
Стадия 3.
(1S,3R)-N-((S)-3-(4-хлорфенил)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксопропан-2-ил)-3-(метиламино)циклопентанкарбоксамид
К раствору (S)-1-(1-(2-амино-3-(4-хлорфенил)пропаноил)пиперидин-4-ил)-3,3-диметилиндолин-2-она (ПРИМЕР 21, Стадия 2) добавляли (1S,3R)-3-(трет- бутоксикарбонил(метил)амино)циклопентанкарбоновую кислоту (26,7 мг, 0,11 ммоль, ПРИМЕР 21, Стадия 1) и гексафторфосфат 2-(1H-бензотриазол-1-ил)-1,1,3,3-тетраметилурония (53 мг, 0,14 ммоль) и смесь перемешивали при 60°C в течение 6 часов, затем при комнатной температуре в течение 13 часов. Летучие вещества удаляли в вакууме.
К остатку, растворенному в 1,2-дихлорэтане (1 мл), добавляли 2,2,2-трифторуксусную кислоту (1 мл) и смесь перемешивали при комнатной температуре в течение 2 часов. Затем смесь концентрировали в вакууме. Остаток разбавляли метанолом и наносили на сильный катионо-обменный картридж (BondElute(зарегистрированная торговая марка) SCX, 1 г/6 мл, Varian Inc.) и твердофазную матрицу промывали метанолом (6 мл). Неочищенную смесь элюировали в трубке-сборнике при помощи 1 моль/л аммиака в метаноле (6 мл) и концентрировали в вакууме. Остаток очищали методом препаративной ЖХ-МС с получением указанного в заголовке соединения (16,2 мг, 29%).
MS (ESI) m/z: 551 (M+H) +.
ВЭЖХ время удерживания: 0,74 мин (Способ A).
ПРИМЕР 22-43
[Химическая формула 26]
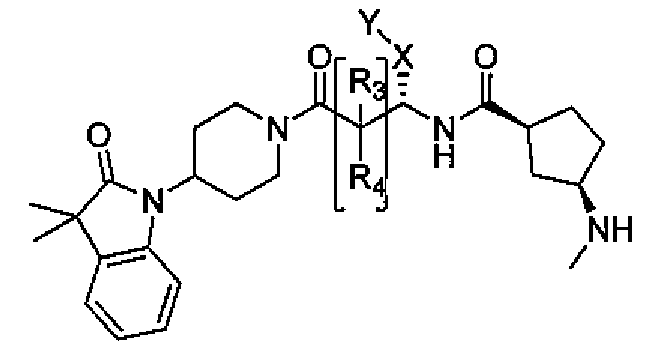
Следующие примеры, ПРИМЕР 22-43, получали в соответствии с процедурой, описанной на Стадиях 2-3 Примера 21, с использованием подходящего аминокислотного предшественника вместо (S)-2-(трет-бутоксикарбониламино)-3-(4-хлорфенил)пропановой кислоты.
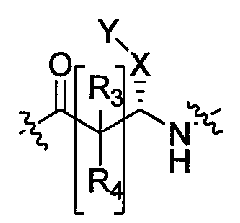
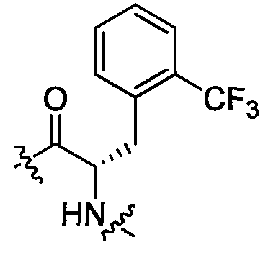
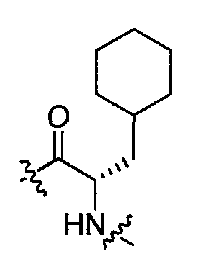
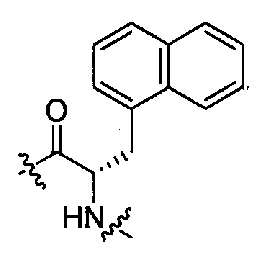
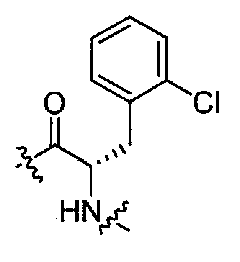
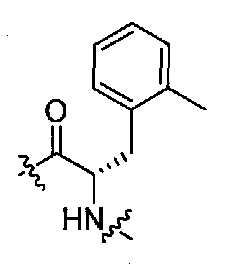
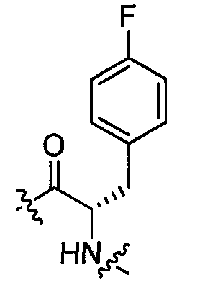
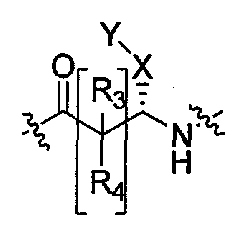
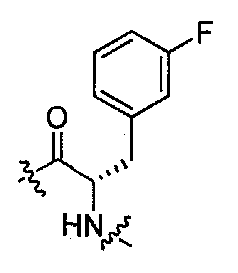
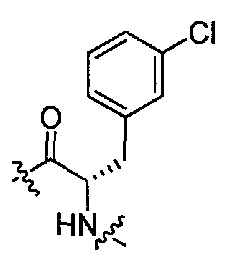
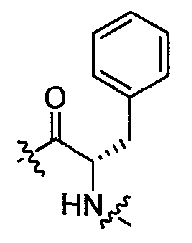
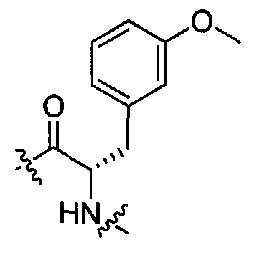
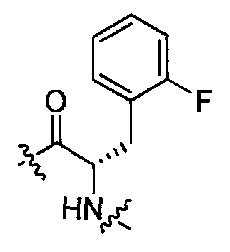
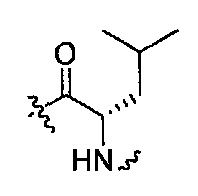
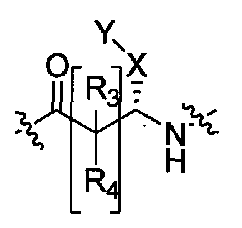
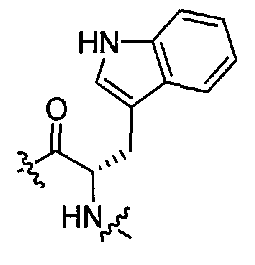
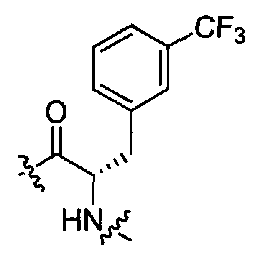
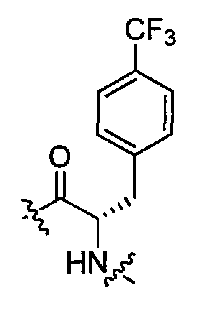
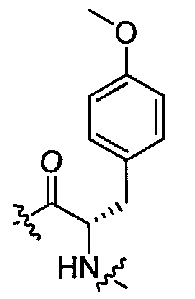
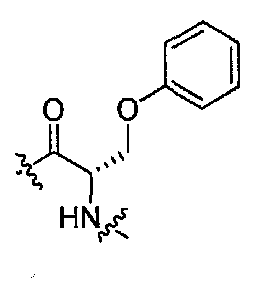
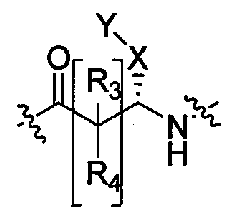
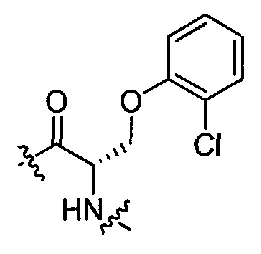
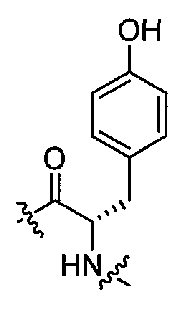
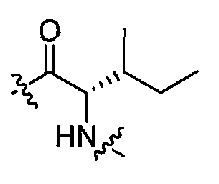
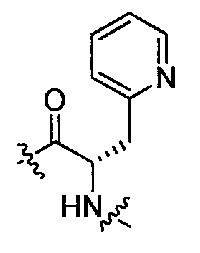
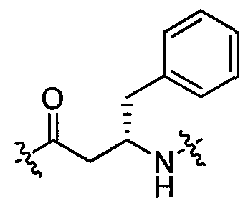
ПРИМЕР 44
(R)-N-((S)-1-(4-(5-фтор-3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)пиперидин-3-карбоксамид
[Химическая формула 27]
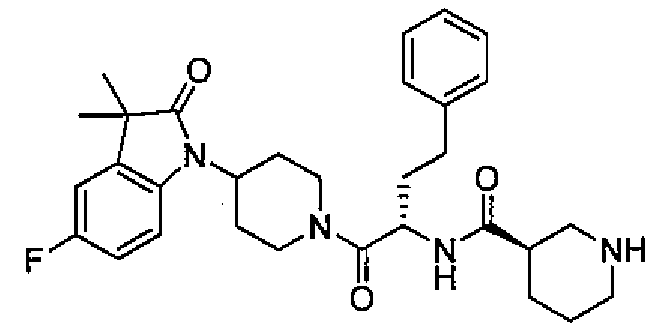
Стадия 1.
2-(2-бром-5-фторфенил)-2-метилпропановая кислота
Смесь метил 2-(2-бром-5-фторфенил)-2-метилпропаноата (0,529 г, 3,12 ммоль, J. Am. Chem. Soc., 2008,130, 15157-15166), гидроксида калия (3,2 г, 57 ммоль), воды (10 мл) и этанола (30 мл) подвергали кипячению с обратным холодильником в течение 20 часов. Смесь охлаждали до комнатной температуры и концентрировали до около 10 мл. Полученную смесь доводили до pH 4 при помощи концентрированной хлористоводородной кислоты и экстрагировали дихлорметаном (200 мл). Органическую фазу промывали водой и насыщенным солевым раствором, сушили над сульфатом магния и концентрировали в вакууме с получением указанного в заголовке соединения (0,510 г, 100%).
1Н ЯМР (CDCl3) δ: 7,55-7,51 (1Н, м), 7,18-7,13 (1H, м), 6,90-6,84 (1H, м), 1,67 (6H, с).
Стадия 2.
N-(1-бензилпиперидин-4-ил)-2-(2-бром-5-фторфенил)-2-метилпропанамид
К раствору 2-(2-бром-5-фторфенил)-2-метилпропановой кислоты (510 мг, 1,95 ммоль, ПРИМЕР 44, Стадия 1) в тетрагидрофуране (20 мл) добавляли трифосген (197 мг, 0,66 ммоль) и триэтиламин (0,288 мл, 2,05 ммоль). Смесь перемешивали при комнатной температуре в течение 30 минут и фильтровали. Фильтрат добавляли по каплям к раствору 1-бензил-4-аминопиперидина (372 мг, 1,95 ммоль) и триэтиламина (0,288 мл, 2,05 ммоль) в тетрагидрофуране (20 мл). Смесь перемешивали при комнатной температуре в течение ночи.
Осажденные вещества отфильтровывали и фильтрат концентрировали. Остаток разбавляли этилацетатом. Органический слой промывали 0,1 моль/л водного раствора гидроксида натрия, водой и насыщенным солевым раствором и сушили над сульфатом натрия и концентрировали. Остаток очищали колоночной хроматографией на силикагеле (20-100% этилацетат в гексане) с получением указанного в заголовке соединения (275 мг, 33%).
1Н ЯМР (CDCl3) δ: 7,56-7,52 (1H, м), 7,32-7,19 (6H, м), 6,92-6,86 (1H, м), 4,94-4,91 (1H, м), 3,81-3,78 (1H, м), 3,45 (2H, с), 2,75-2,71 (2H, м), 2,12-2,04 (2H, м), 1,91-1,86 (2H, м), 1,61 (6H, с), 1,38-1,23 (2H, м).
MS (ESI) m/z: 433 (М+H)+.
Стадия 3.
1-(1-бензилпиперидин-4-ил)-5-фтор-3,3-диметилиндолин-2-он
Смесь N-(1-бензилпиперидин-4-ил)-2-(2-бром-5-фторфенил)-2- метилпропанамида (275 мг, 0,635 ммоль, ПРИМЕР 44, Стадия 2), ацетата палладия (14 мг, 0,063 ммоль), фенилбороновой кислоты (15 мг, 0,13 ммоль), дициклогексил(2',4',6'-триизопропилбифенил-2-ил)фосфина (30 мг, 0,063 ммоль), карбоната калия (220 мг, 1,6 ммоль) и трет-бутилового спирта (15 мл) подвергали кипячению с обратным холодильником в атмосфере азота в течение 14 часов. Смесь охлаждали до комнатной температуры, фильтровали и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (8-66% этилацетата в гексане) с получением указанного в заголовке соединения (171 мг, 76%).
1Н ЯМР (CDCl3) δ: 7,35-7,26 (5H, м), 7,11-7,07 (1H, м), 6,94-6,89 (2H, м), 4,35-4,25 (1H, м), 3,56 (2H, с), 3,05-2,98 (2H, м), 2,50-2,34 (2H, м), 2,19-2,09 (2H, м), 1,70-1,63 (2H, м), 1,32 (6H, с).
MS (ESI) м/z: 353 (М+H)+.
TLC Rf: 0,25 (этилацетат/гексан 1:2).
Стадия 4.
5-фтор-3,3-диметил-1-(пиперидин-4-ил)индолин-2-он
Смесь 1-(1-бензилпиперидин-4-ил)-5-фтор-3,3-диметилиндолин-2-она (171 мг, 0,485 ммоль, ПРИМЕР 44, Стадия 3), гидроксида палладия (20% масс. на углероде) (85 мг), муравьиной кислоты (0,372 мл, 9,7 ммоль) и этанола (20 мл) перемешивали при 50°C в течение 1 часа.
Смесь охлаждали до комнатной температуры и фильтровали через Целит (зарегистрированная торговая марка). Фильтрат концентрировали при пониженном давлении. К остатку добавляли 2 моль/л водного раствора гидроксида натрия (1 мл) и воду (15 мл). Водную смесь экстрагировали два раза этилацетатом. Объединенные органические слои промывали насыщенным солевым раствором, сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением указанного в заголовке соединения (124 мг, 86%) в виде твердого вещества.
1Н ЯМР (CDCl3) δ: 7,12-7,07 (1H, м), 6,98-6,88 (2H, м), 4,40-4,30 (1H, м), 3,28-3,23 (2H, м), 2,80-2,72 (2H, м), 2,35-2,29 (2H, м), 1,73-1,65 (4H, м), 1,35 (6H, с).
MS (ESI) м/z: 263 (М+H)+.
Стадия 5.
(S)-трет-бутил 1-(4-(5-фтор-3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-илкарбамат
Указанное в заголовке соединение получали в соответствии с процедурой, описанной на Стадии 3 Примера 1, с использованием 5-фтор-3,3-диметил-1-(пиперидин-4-ил)индолин-2-она (ПРИМЕР 44, Стадия 4) вместо гидрохлорида 3,3-диметил-1-(пиперидин-4-ил)индолин-2-она.
1Н ЯМР (CDCl3) δ: 7,32-7,19 (5H, м), 6,95-6,73 (3H, м), 5,50-5,37 (1H, м), 4,80-4,32 (3Н, м), 3,78-3,65 (1H, м), 3,05-2,90 (1H, м), 2,73-2,63 (3Н, м), 2,28-2,17 (2H, м), 1,96-1,67 (4H, м), 1,49-1,46 (9H, псевдо д), 1,34-1,33 (6H, псевдо д).
MS (ESI) м/z: 524 (М+H)+.
Стадия 6.
(S)-1-(1-(2-амино-4-фенилбутаноил)пиперидин-4-ил)-5-фтор-3,3-диметилиндолин-2-он
Указанное в заголовке соединение получали в соответствии с процедурой, описанной на Стадии 4 Примера 1, с использованием 1-(4-(5-фтор-3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-илкарбамата (ПРИМЕР 44, Стадия 5) вместо 1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-илкарбамата.
MS (ESI) m/z: 424 (M+H)+.
Стадия 7.
(R)-N-((S)-1-(4-(5-фтор-3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)пиперидин-3-карбоксамид
Указанное в заголовке соединение получали в соответствии с процедурой, описанной на Стадии 5 Примера 1, с использованием 1-(4-(5-фтор-3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-илкарбамата (ПРИМЕР 44, Стадия 6) вместо 1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-илкарбамата.
MS (ESI) m/z: 535 (M+H)+.
ВЭЖХ время удерживания: 0,73 мин (Способ A).
ПРИМЕР 45-46
[Химическая формула 28]
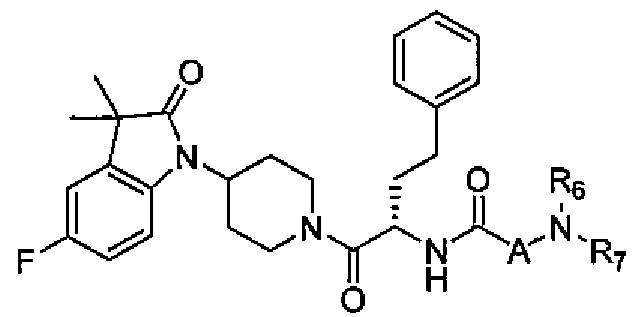
Следующие примеры, ПРИМЕР 45-46, получали в соответствии с процедурой, аналогичной описанной на Стадии 7 Примера 44, с использованием подходящего предшественника вместо (R)-1-(трет-бутоксикарбонил)пиперидин-3-карбоновой кислоты.

ПРИМЕР 47
(S)-N-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)-3-(метиламино)пирролидин-1-карбоксамид
[Химическая формула 29]
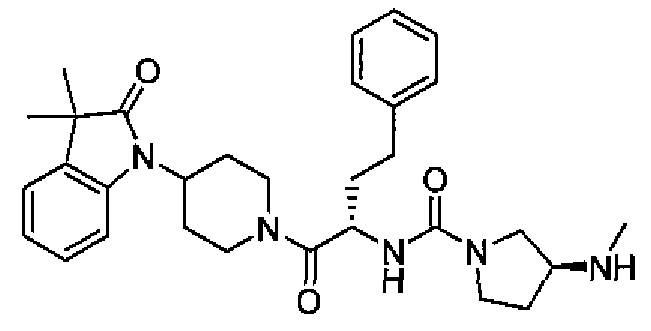
(S)-N-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)-3-(диметиламино)пирролидин-1-карбоксамид
К раствору 4-нитрофенилхлороформиата (15 мг, 0,075 ммоль) в дихлорметане (0,5 мл) добавляли смесь гидрохлорида (S)-1-(1-(2-амино-4-фенилбутаноил)пиперидин-4-ил)-3,3-диметилиндолин-2-она (30 мг, 0,068 ммоль) и триэтиламина (20 мкл, 0,143 ммоль) в дихлорметане (1 мл) при комнатной температуре. После перемешивания в течение 5 минут к смеси добавляли (S)-трет-бутил метил(пирролидин-3-ил)карбамат (27 мг, 0,136 ммоль, Bioorg. Med. Chem. Lett., 2006, 16, 4922-4930) и триэтиламин (19 мкл, 0,136 ммоль) в дихлорметане (1 мл). Полученную смесь перемешивали при комнатной температуре в течение 20 минут, затем концентрировали в вакууме.
К остатку добавляли трифторуксусную кислоту (1 мл) и полученный раствор перемешивали в течение 20 минут. Затем смесь концентрировали в вакууме. Остаток разбавляли метанолом и наносили на сильный катионо-обменный картридж (BondElute(зарегистрированная торговая марка) SCX, 1 г/6 мл, Varian Inc.) и твердофазную матрицу промывали метанолом (6 мл). Неочищенную смесь элюировали в трубке-сборнике при помощи 1 моль/л аммиака в метаноле (6 мл) и концентрировали в вакууме. Остаток очищали при помощи препаративной ЖХ-МС с получением указанного в заголовке соединения (13,6 мг, 38%).
MS (ESI) m/z: 532 (M+H)+.
ВЭЖХ время удерживания: 0,72 мин. (Способ A).
ПРИМЕР 47: АЛЬТЕРНАТИВНЫЙ СПОСОБ
Стадия 1.
трет-бутил (S)-1-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-илкарбомоил)пирролидин-3-ил(метил)карбамат
К смеси 4-нитрофенилхлороформиата (227 мг, 1,13 ммоль) в дихлорметане (3 мл) добавляли смесь (S)-1-(1-(2-амино-4- фенилбутаноил)пиперидин-4-ил)-3,3-диметилиндолин-2-она (500 мг, 1,13 ммоль) и триэтиламина (0,32 мл, 2,26 ммоль) в дихлорметане (6 мл) и реакционную смесь перемешивали в течение 30 минут при комнатной температуре.
Затем к смеси добавляли смесь (S)-трет-бутил метил(пирролидин-3-ил)карбамата (239 мг, 1,13 ммоль, Bioorg. Med. Chem. Lett., 2006, 16, 4922-4930) и триэтиламина (0,32 мл, 2,26 ммоль) в дихлорметане (6 мл) и полученный раствор перемешивали в течение 20 минут при комнатной температуре. Смесь разбавляли насыщенным водным раствором гидрокарбоната натрия и экстрагировали этилацетатом. Органический слой сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаточное масло очищали колоночной хроматографией на силикагеле с получением указанного в заголовке соединения (475 мг, 67%) в виде бесцветного масла.
1Н ЯМР (CDCl3) δ: 7,34-7,12 (7H, м), 7,09-6,83 (2H, м), 5,29-5,20 (1H, м), 4,98-4,68 (2H, м), 4,59-4,27 (1H, м), 3,85-3,71 (1H, м), 3,68-3,50 (2H, м), 3,39-3,17 (2H, м), 3,12-2,92 (1H, м), 2,87-2,58 (7H, м), 2,46-2,18 (2H, м), 2,18-1,60 (6H, м), 1,48 (9H, с), 1,35 (6H, ушир.с).
MS (ESI) м/z: 632 (М+H)+, 630 (М-H)-.
Стадия 2.
(S)-N-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)-3-(метиламино)пирролидин-1-карбоксамид
Смесь трет-бутил (S)-1-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-илкарбамоил)пирролидин-3-ил(метил)карбамата (267 мг, 0,42 ммоль, ПРИМЕР 47: Альтернативный Способ, Стадия 1) в дихлорметан-трифторуксусной кислоте (1:1, 6 мл) перемешивали при комнатной температуре в течение 1 часа. Смесь концентрировали, разбавляли дихлорметаном и промывали насыщенным водным раствором гидрокарбоната натрия. Органический слой сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаточное масло очищали методом препаративной ВЭЖХ (32-96% метанола в 0,01% водном растворе аммиака) и колоночной хроматографией на аминогеле с получением указанного в заголовке соединения (209 мг, 93%) в виде бесцветного масла.
1Н ЯМР (CDCl3) δ: 7,34-7,11 (7H, м), 7,10-6,82 (2H, м), 5,29-5,18 (1H, м), 5,00-4,88 (1H, м), 4,80-4,72 (1H, м), 4,59-4,27 (1H, м), 3,85-3,74 (1H, м), 3,66-3,45 (2H, м), 3,45-3,10 (3H, м), 3,10-2,94 (1H, м), 2,83-2,57 (3H, м), 2,46 (3H, ушир.с), 2,45-1,55 (9H, м), 1,34 (6H, ушир.с).
MS (ESI) m/z: 532 (М+H)+, 530 (М-H)-.
ПРИМЕР 48-66
Следующие примеры, 48-66, получали в соответствии с процедурой, аналогичной описанной в Примере 47, с использованием подходящих предшественников, указанных в таблице, вместо (S)-трет-бутил метил(пирролидин-3-ил)карбамата.
[Химическая формула 30]

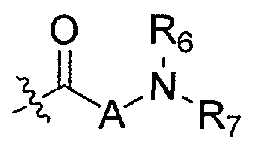
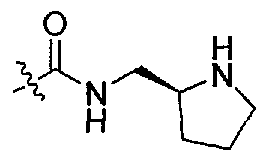
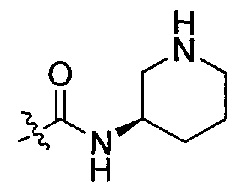
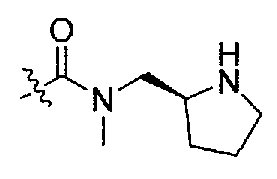
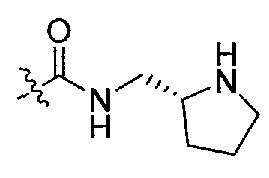
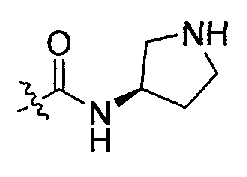
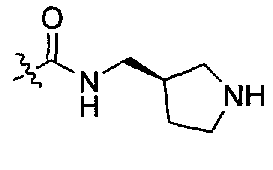
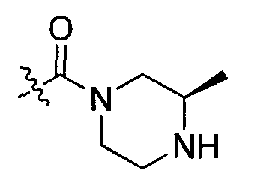
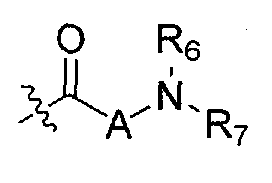
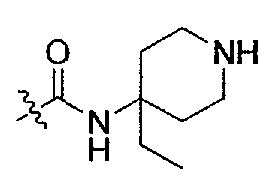
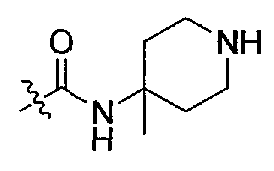
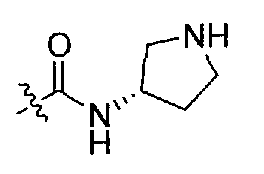
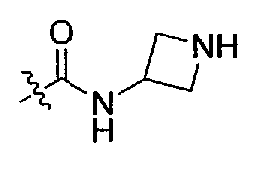
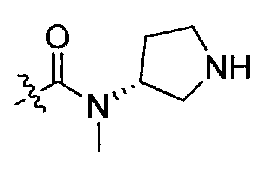
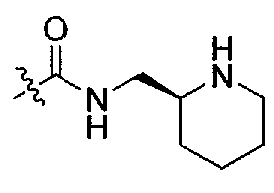
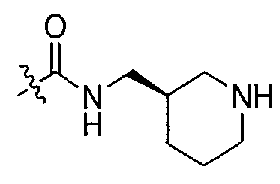
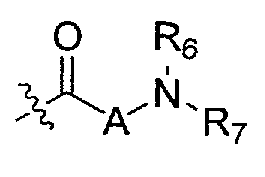
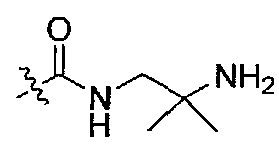
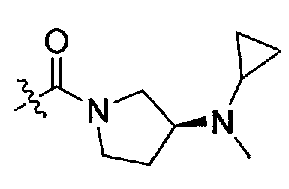
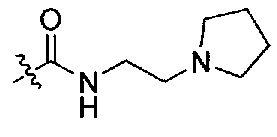
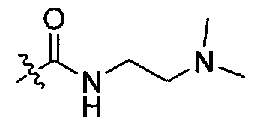
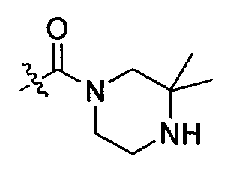
ПРИМЕР 67
(S)-N-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)-3-(диметиламино)пирролидин-1-карбоксамид
[Химическая формула 31]
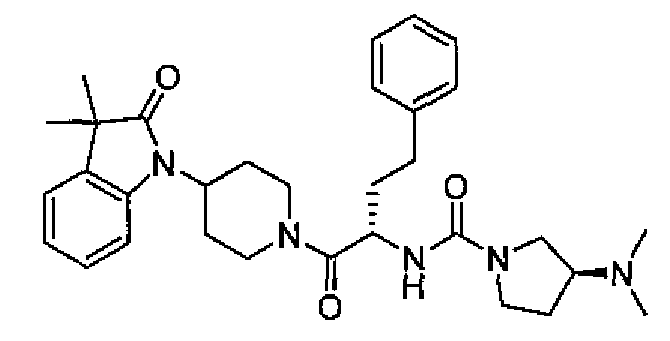
Стадия 1.
(S)-N-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)-3-(диметиламино)пирролидин-1-карбоксамид
К раствору 4-нитрофенилхлороформиата (15 мг, 0,075 ммоль) в дихлорметане (0,5 мл) добавляли смесь гидрохлорида (S)-1-(1-(2-амино-4-фенилбутаноил)пиперидин-4-ил)-3,3-диметилиндолин-2-она (30 мг, 0,068 ммоль) и триэтиламина (20 мкл, 0,143 ммоль) в дихлорметане (1 мл) при комнатной температуре. После перемешивания в течение 5 минут к смеси добавляли (S)-трет-бутил метил(пирролидин-3-ил)карбамат (27 мг, 0,136 ммоль, Bioorg. Med. Chem. Lett., 2006, 16, 4922-4930) и триэтиламин (19 мкл, 0,136 ммоль) в дихлорметане (1 мл). Полученную смесь перемешивали при комнатной температуре в течение 20 минут. Смесь концентрировали. К остатку добавляли трифторуксусную кислоту (1 мл) и полученный раствор перемешивали в течение 20 минут. Затем смесь концентрировали в вакууме. Остаток разбавляли метанолом и наносили на сильный катионо-обменный картридж (BondElute(зарегистрированная торговая марка) SCX, 1 г/6 мл, Varian Inc.) и твердофазную матрицу промывали метанолом (6 мл). Неочищенную смесь элюировали в трубке-сборнике при помощи 1 моль/л аммиака в метаноле (6 мл) и концентрировали в вакууме.
Остаток растворяли в метаноле (2 мл). К раствору добавляли параформальдегид (20 мг, 0,68 ммоль) и триацетоксиборогидрид натрия (72 мг, 0,34 ммоль) при комнатной температуре. Через 2 часа к смеси добавляли 2 капли уксусной кислоты, с последующим добавлением триацетоксиборгидрида (72 мг, 0,34 ммоль). Через 1 час смесь концентрировали и остаток подщелачивали при помощи водного раствора бикарбоната натрия и экстрагировали этилацетатом (2 мл Ч3). Объединенные органические слои фильтровали через колонку с сульфатом магния и фильтрат концентрировали в вакууме. Неочищенный продукт очищали методом препаративной ЖХ-МС с получением указанного в заголовке соединения (10,4 мг, 28 %).
MS (ESI) m/z: 546 (M+H)+.
ВЭЖХ время удерживания: 0,75 мин. (Способ A).
ПРИМЕР 68-69
[Химическая формула 32]
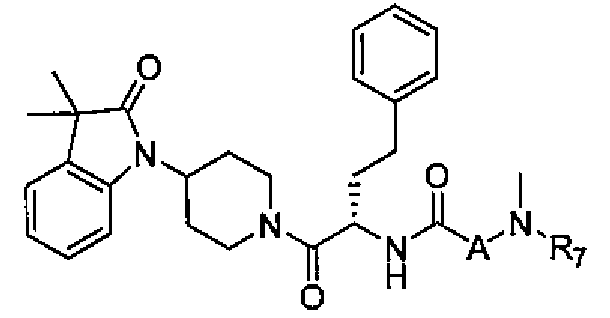
Следующие примеры, ПРИМЕР 68-69, получали в соответствии с процедурой, аналогичной описанной в Примере 67, с использованием подходящего предшественника вместо (S)-трет-бутил метил(пирролидин-3-ил)карбамата.

ПРИМЕР 70
(S)-N-((S)-1-(4-(5-фтор-3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)-3-(метиламино)пирролидин-1-карбоксамид
[Химическая формула 33]
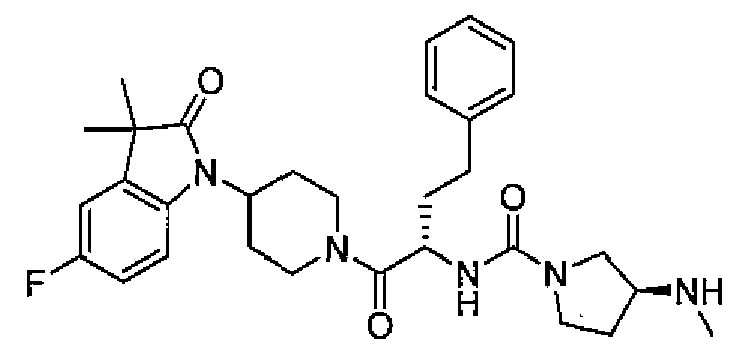
Указанное в заголовке соединение получали в соответствии с процедурой, описанной на Стадии 1 Примера 47, с использованием (S)-1-(1-(2-амино-4-фенилбутаноил)пиперидин-4-ил)-5-фтор-3,3-диметилиндолин-2-она (ПРИМЕР 44, Стадия 6) вместо гидрохлорида (S)-1-(1-(2-амино-4-фенилбутаноил)пиперидин-4-ил)-3,3-диметилиндолин-2-она.
MS (ESI) m/z: 550 (M+H) +.
ВЭЖХ время удерживания: 0,73 мин (Способ A).
ПРИМЕР 71-72
[Химическая формула 34]
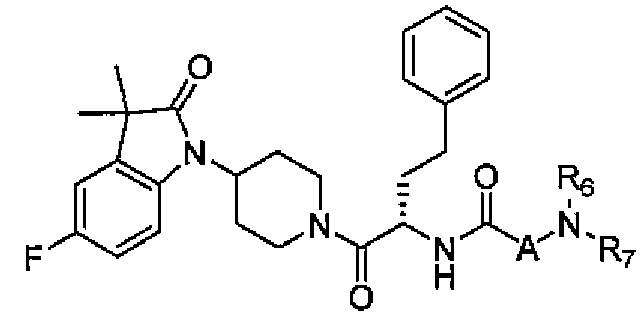
Следующие примеры, ПРИМЕР 71-72, получали в соответствии с процедурой, аналогичной описанной на Стадии 1 Примера 67, с использованием (S)-1-(1-(2-амино-4-фенилбутаноил)пиперидин-4-ил)-5-фтор-3,3-диметилиндолин-2-она (ПРИМЕР 44, Стадия 6) и подходящего аминового предшественника вместо гидрохлорида (S)-1-(1-(2-амино-4-фенилбутаноил)пиперидин-4-ил)-3,3-диметилиндолин-2-она и (S)-трет-бутил метил(пирролидин-3-ил)карбамата, соответственно.

ПРИМЕР 73
(R)-N-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)-N-метилпиперидин-3-карбоксамид
[Химическая формула 35]
Стадия 1.
(S)-N-(1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)-2-нитробензолсульфонамид
К раствору гидрохлорида (S)-1-(1-(2-амино-4-фенилбутаноил)пиперидин-4-ил)-3,3-диметилиндолин-2-она (1,00 г, 2,26 ммоль, ПРИМЕР 1, Стадия 4) в дихлорметане (10 мл) добавляли триэтиламин (954 мкл, 6,79 ммоль) и 2-нитробензолсульфонилхлорид (501 мг, 2,26 ммоль) при 0°C. После перемешивания в течение 20 минут при комнатной температуре к смеси добавляли воду и этилацетат. Органический слой промывали водным раствором гидрокарбоната натрия, 1 моль/л хлористоводородной кислоты и насыщенным солевым раствором, сушили над сульфатом натрия и концентрировали. Неочищенный продукт очищали колоночной хроматографией на силикагеле (гексан/этилацетат, градиент) с получением указанного в заголовке соединения (1,26 г, 94%) в виде аморфного твердого вещества.
1H ЯМР (CDCl3) δ: 8,20-7,62 (м, 4H), 7,39-7,10 (м, 7H), 6,84-6,78 (м, 1H), 6,66-6,48 (м, 1H), 4,56-3,97 (м, 3H), 3,50-3,30 (м, 1H), 3,00-2,73 (м, 3H), 2,56-1,80 (м, 8H), 1,46-1,31 (м, 6H).
Стадия 2.
(S)-N-(1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)-N-метил-2-нитробензолсульфонамид
К смеси (S)-N-(1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)-2-нитробензолсульфонамида (1,26 г, 2,13 ммоль, ПРИМЕР 73, Стадия 1), метанола (519 мкл, 12,8 ммоль), трифенилфосфина (1,12 г, 4,27 ммоль) в тетрагидрофуране (10 мл) добавляли диэтилазодикарбоксилат (2,2 моль/л раствор в толуоле, 3,88 мл, 8,53 ммоль) при комнатной температуре. Через 3 часа смесь концентрировали и остаток очищали колоночной хроматографией на силикагеле (гексан/этилацетат, градиент) с получением смеси указанного в заголовке соединения и трифенилфосфиноксида (3,30 г).
MS (ESI) m/z: 605 (M+H)+.
Стадия 3.
(S)-3,3-диметил-1-(1-(2-(метиламино)-4-фенилбутаноил)пиперидин-4-ил)индолин-2-он
К раствору (S)-N-(1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)-N-метил-2-нитробензолсульфонамида (3,30 г, с трифенилфосфиноксидом в виде примеси, ПРИМЕР 73, Стадия 2) в диметилформамиде (10 мл) добавляли меркаптоуксусную кислоту (491 мг, 5,33 ммоль) и гидроксид лития моногидрат (447 мг, 10,7 ммоль) при комнатной температуре. После перемешивания в течение 16 часов смесь разбавляли этилацетатом и насыщенным водным раствором гидрокарбоната натрия. Органический слой промывали насыщенным водным раствором гидрокарбоната натрия и насыщенным солевым раствором, сушили над сульфатом натрия и концентрировали в вакууме. Остаток разбавляли дихлорметаном (10 мл) и к раствору добавляли хлористый водород в метаноле. Затем смесь концентрировали. Остаток разбавляли метанолом и смешивали с сильным катионо-обменным гелем (BondeSil(зарегистрированная торговая марка) SCX, 40 г, Varian Inc.). Гель фильтровали и промывали раствором 1 моль/л аммиака в метаноле и раствор концентрировали с получением указанного в заголовке соединения (636 мг, 71%).
MS (ESI) m/z: 420 (M+H)+.
1H ЯМР (CDCl3) δ: 7,33-7,12 (м, 7H), 7,09-7,00 (м, 1H), 6,93-6,85 (м, 1H), 4,94-4,79 (м, 1H), 4,49-4,31 (м, 1H), 3,68-3,51 (м, 1H), 3,43-3,27 (м, 1H), 3,07-2,55 (м, 4H), 2,55-2,12 (м, 5H), 1,94-1,51 (м, 5H), 1,40-1,32 (м, 6H).
Стадия 4.
(R)-N-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)-N-метилпиперидин-3-карбоксамид
К раствору гидрохлорида (S)-1-(1-(2-амино-4-фенилбутаноил)пиперидин-4-ил)-3,3-диметилиндолин-2-она (44 мг, 0,1 ммоль, ПРИМЕР 73, Стадия 3) в 3,75% триэтиламина/N,N-диметилацетамид (1,5 мл) добавляли (S)-1-(трет-бутоксикарбонил)пиперидин-2-карбоновую кислоту (25,2 мг, 0,110 ммоль) и гексафторфосфат 2-(1H-бензотриазол-1-ил)-1,1,3,3-тетраметилурония (57 мг, 0,150 ммоль) и смесь перемешивали при 60°C в течение 6 часов. Затем смесь охлаждали до комнатной температуры и перемешивали еще в течение 13 часов. Летучие вещества удаляли в вакууме.
Остаток растворяли в 1,2-дихлорэтане (1 мл) и добавляли трифторуксусную кислоту (1 мл). Смесь перемешивали при комнатной температуре в течение 2 часов. Затем смесь концентрировали в вакууме. Остаток разбавляли метанолом и наносили на сильный катионо-обменный картридж (BondElute(зарегистрированная торговая марка) SCX, 1 г/6 мл, Varian Inc.) и твердофазную матрицу промывали метанолом (6 мл). Неочищенную смесь элюировали в трубке-сборнике при помощи 1 моль/л аммиака в метаноле (6 мл) и концентрировали в вакууме. Остаток очищали методом препаративной ЖХ-МС с получением указанного в заголовке соединения (4,3 мг, 8 %).
MS (ESI) m/z: 531 (M+H)+.
ВЭЖХ время удерживания: 0,74 мин (Способ A).
ПРИМЕР 74
1-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)-1-метил-3-((R)-пиперидин-3-ил)мочевина
[Химическая формула 36]
Стадия 1.
1-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)-1-метил-3-((R)-пиперидин-3-ил)мочевина
К раствору 4-нитрофенилхлороформиата (14 мг, 0,072 ммоль) в дихлорметане (1 мл) добавляли смесь (R)-трет-бутил 3-аминопиперидин-1-карбоксилата (14 мг, 0,072 ммоль) и триэтиламина (10 мкл, 0,072 ммоль) в дихлорметане (1 мл) при комнатной температуре. После перемешивания в течение 5 минут этот раствор добавляли к раствору (S)-3,3-диметил-1-(1-(2-(метиламино)-4-фенилбутаноил)пиперидин-4-ил)индолин-2-она (20 мг, 0,048 ммоль, ПРИМЕР 73, Стадия 3) и триэтиламина (13 мкл, 0,095 ммоль) в дихлорметане (1 мл). Полученную смесь перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь концентрировали. К остатку добавляли трифторуксусную кислоту (1 мл) и смесь перемешивали в течение 15 минут. Затем смесь концентрировали в вакууме. Остаток разбавляли метанолом и наносили на сильный катионо-обменный картридж (BondElute(зарегистрированная торговая марка) SCX, 1 г/6 мл, Varian Inc.) и твердофазную матрицу промывали метанолом (6 мл). Неочищенную смесь элюировали в трубке-сборнике при помощи 1 моль/л аммиака в метаноле (6 мл) и концентрировали в вакууме. Остаток очищали методом препаративной ЖХ-МС с получением указанного в заголовке соединения (2,3 мг, 9 %).
MS (ESI) m/z: 546 (M+H)+.
ВЭЖХ время удерживания: 0,73 мин (Способ A).
ПРИМЕР 75-81
[Химическая формула 37]
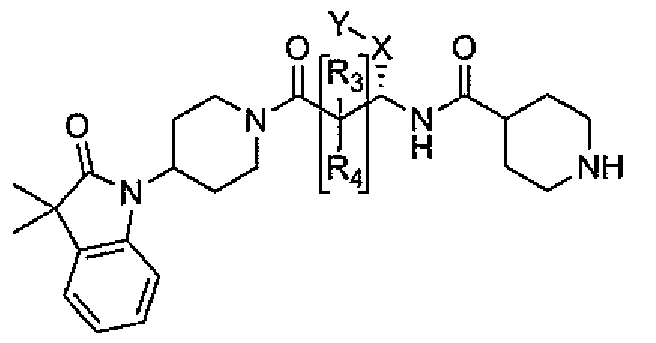
Следующие примеры, ПРИМЕР 75-81, получали в соответствии с процедурой, описанной на Стадиях 2-3 Примера 21, с использованием подходящего аминокислотного предшественника и 1-(трет-бутоксикарбонил)пиперидин-4-карбоновой кислоты вместо (S)-2-(трет-бутоксикарбониламино)-3-(4-хлорфенил)пропановой кислоты и (1S,3R)-3-(трет-бутоксикарбонил(метил)амино)-циклопентанкарбоновой кислоты, соответственно.
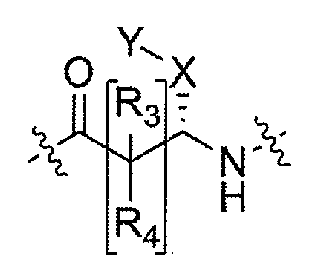
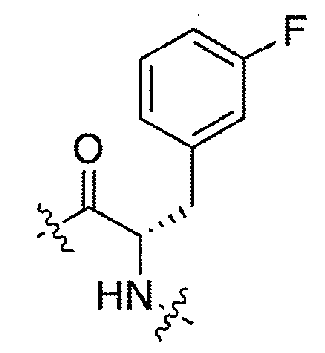
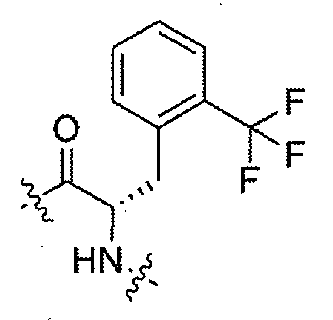
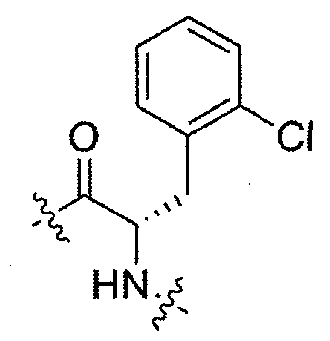
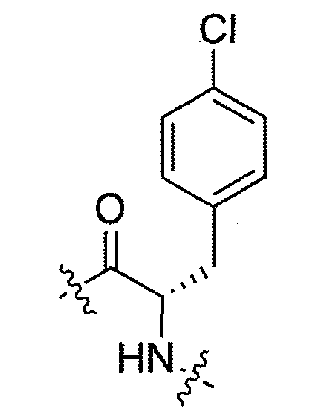

ПРИМЕР 82-88
[Химическая формула 38]
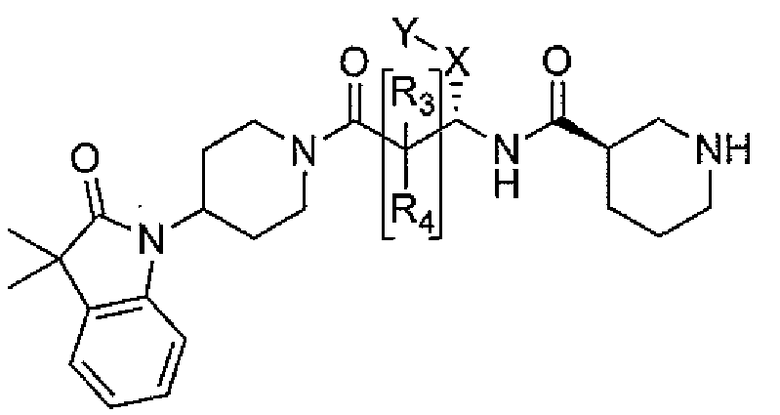
Следующие примеры, ПРИМЕР 82-88, получали в соответствии с процедурой, описанной на Стадиях 2-3 Примера 21, с использованием подходящего аминокислотного предшественника и (R)-1-(трет-бутоксикарбонил)пиперидин-3-карбоновой кислоты вместо (S)-2-(трет-бутоксикарбониламино)-3-(4-хлорфенил)пропановой кислоты и (1S,3R)-3-(трет-бутоксикарбонил(метил)амино)циклопентанкарбоновой кислоты, соответственно.
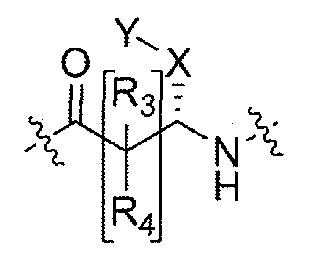
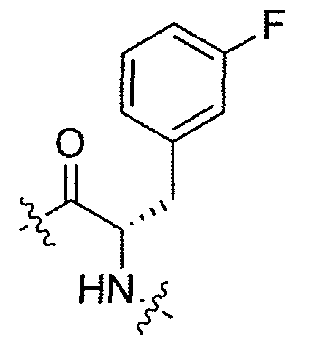
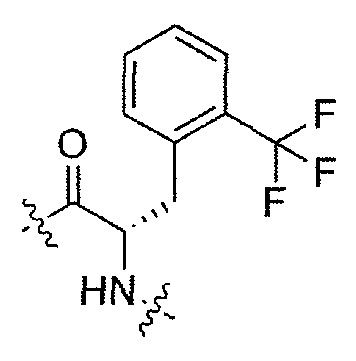
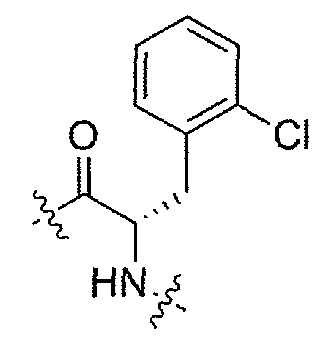
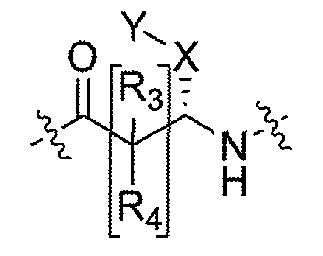
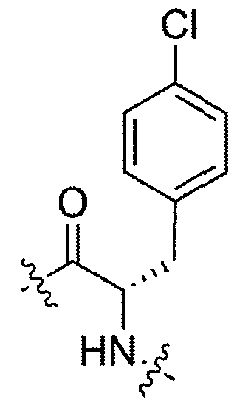
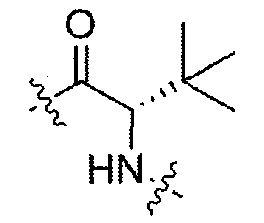
ПРИМЕР 89
(S)-N-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-3-фенилпропан-2-ил)-3-(метиламино)пирролидин-1-карбоксамид
[Химическая формула 39]
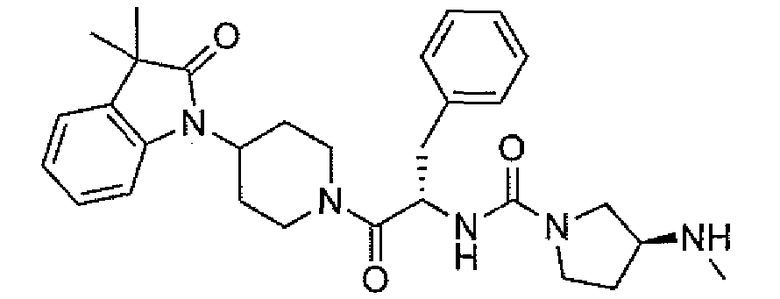
Стадия 1.
(S)-1-(1-(2-амино-3-фенилпропаноил)пиперидин-4-ил)-3,3-диметилиндолин-2-он
Указанное в заголовке соединение получали в соответствии с процедурой, описанной на Стадии 2 Примера 21, с использованием (S)-2-(трет-бутоксикарбониламино)-3-фенилпропановой кислоты вместо (S)-2-(трет-бутоксикарбониламино)-3-(4-хлорфенил)пропановой кислоты.
MS (ESI) m/z: 391 (M+H)+.
ВЭЖХ время удерживания: 1,52 мин (Способ C).
Стадия 2.
(S)-N-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-3-фенилпропан-2-ил)-3-(метиламино)пирролидин-1-карбоксамид
Указанное в заголовке соединение получали в соответствии с процедурой, описанной в Примере 47, с использованием (S)-1-(1-(2-амино-3-фенилпропаноил)пиперидин-4-ил)-3,3-диметилиндолин-2-она вместо гидрохлорида (S)-1-(1-(2-амино-4-фенилбутаноил)пиперидин-4-ил)-3,3-диметилиндолин-2-она.
MS (ESI) m/z: 516 (M+H)+.
ВЭЖХ время удерживания: 1,43 мин (Способ C).
ПРИМЕР 90-94
[Химическая формула 40]
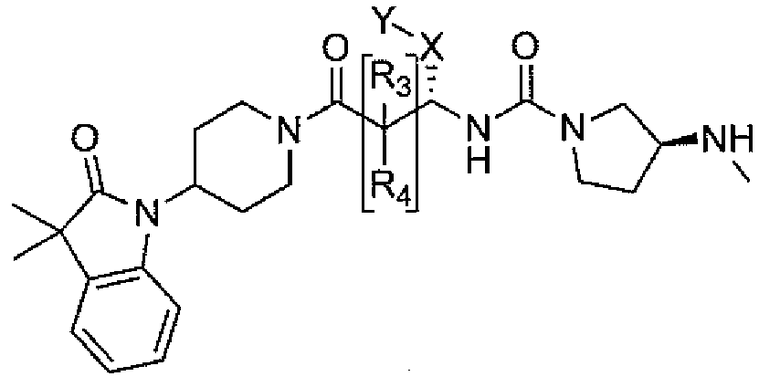
Следующие примеры, ПРИМЕР 90-94, получали в соответствии с процедурой, описанной в Примере 89, с использованием подходящего аминокислотного предшественника вместо (S)-2-(трет-бутоксикарбониламино)-3-фенилпропановой кислоты.
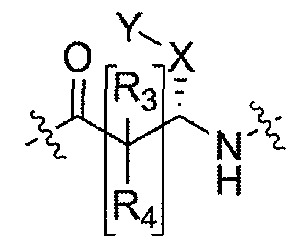
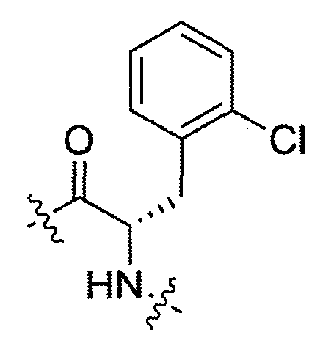
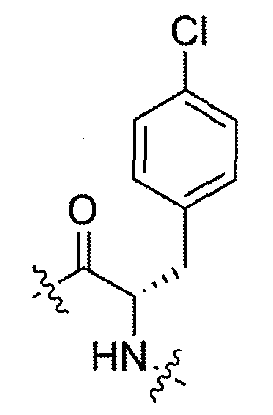
ПРИМЕР 95-97
[Химическая формула 41]
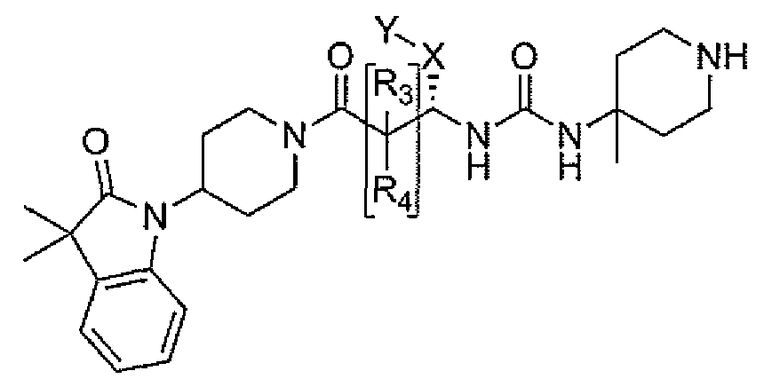
Следующие примеры, ПРИМЕР 95-97, получали в соответствии с процедурой, описанной в Примере 89, с использованием подходящего аминокислотного предшественника и трет-бутил 4-амино-4-метилпиперидин-1-карбоксилата вместо (S)-2-(трет-бутоксикарбониламино)-3-фенилпропановой кислоты и (S)-трет-бутил метил(пирролидин-3-ил)карбамата, соответственно.
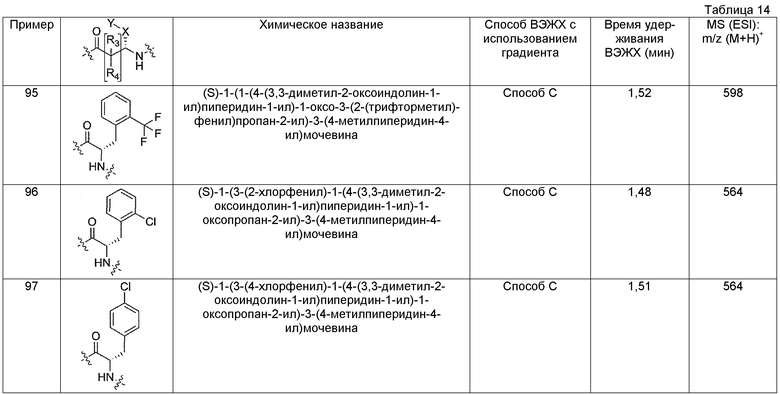
ПРИМЕР 98
(S)-N-((R)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)-3-(метиламино)пирролидин-1-карбоксамид
[Химическая формула 42]
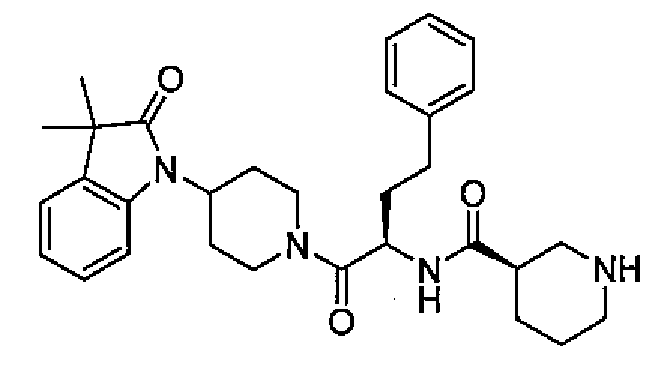
Указанное в заголовке соединение получали в соответствии с процедурой, аналогичной описанной на Стадиях 2-3 Примера 21, с использованием (R)-2-(трет-бутоксикарбониламино)-4-фенилбутановой кислоты и (R)-1-(трет-бутоксикарбонил)пиперидин-3-карбоновой кислоты вместо (S)-2-(трет-бутоксикарбониламино)-3-(4-хлорфенил)пропановой кислоты и (1S,3R)-3-(трет-бутоксикарбонил(метил)амино)циклопентанкарбоновой кислоты, соответственно.
MS (ESI) m/z: 517 (M+H)+.
ВЭЖХ время удерживания: 1,50 мин (Способ C).
ПРИМЕР 99
(S)-N-((R)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)-3-(метиламино)пирролидин-1-карбоксамид
[Химическая формула 43]
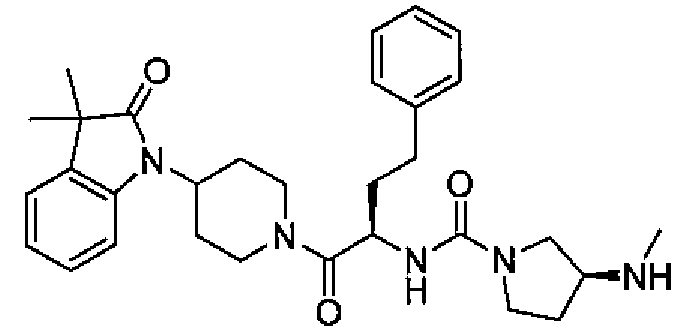
Стадия 1.
(R)-1-(1-(2-амино-4-фенилбутаноил)пиперидин-4-ил)-3,3-диметилиндолин-2-он
Указанное в заголовке соединение получали в соответствии с процедурой, аналогичной описанной на Стадиях 3-4 Примера 1, с использованием (R)-N-трет-бутоксикарбонил-гомофенилаланина вместо (S)-N-трет-бутоксикарбонил-гомофенилаланина.
MS (ESI) m/z:406 (M+H)+.
Стадия 2.
(S)-N-((R)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)-3-(метиламино)пирролидин-1-карбоксамид
Указанное в заголовке соединение получали в соответствии с процедурой, аналогичной описанной на Стадии 1 Примера 47, с использованием (R)-1-(1-(2-амино-4-фенилбутаноил)пиперидин-4-ил)-3,3-диметилиндолин-2-она (ПРИМЕР 76, Стадия 1) вместо (S)-1-(1-(2-амино-4-фенилбутаноил)пиперидин-4-ил)-3,3-диметилиндолин-2-она.
MS (ESI) m/z: 532 (M+H)+.
ВЭЖХ время удерживания: 1,49 мин (Способ C).
Анализ функциональной активности в отношении GPR38 (мотилинового рецептора)
Использовали клеточную линию CHO-K1, стабильно трансфицированную NFAT-бета-лактамазным репортером и геном человеческого мотилинового рецептора. Клетки культивировали в культуральной среде (модифицированная Дульбекко среда Игла, содержащая 10% фетальную бычью сыворотку, 1 мМ пирувата натрия, 0,1 мМ MEM заменимых аминокислот, 25 мМ HEPES буфера, 50 единиц/мл пенициллина, 50 мкг/мл стрептомицина, 100 мкг/мл зеоцина и 500 мкг/мл G418). Клетки собирали с использованием 0,05% трипсин EDTA и центрифугировали. Клеточный осадок после центрифугирования ресуспендировали в культуральной среде и высевали в черный с прозрачным дном 384-луночный планшет при плотности 8000 клеток на лунку, затем инкубировали при 37°C в атмосфере 5% CO2 в течение ночи (около 20 часов). В день анализа культуральную среду аспирировали и добавляли 20 мкл среды для анализа (модфицированная Дульбекко среда Игла, содержащая 100 единиц/мл пенициллина и 100 мкг/мл стрептомицина) в каждую лунку, затем планшеты с клетками помещали в инкубатор при 37°C и 5% CO2 на 1,5 часов. Соединения получали в 100% DMSO и разбавляли до Ч5 конечной концентрации буфером для анализа (сбалансированный солевой раствор Хэнка, 20 мМ HEPES и 0,1% BSA), затем распределяли в 384-луночный планшет (5 мкл/лунка). После инкубации при 37°C и 5% CO2 в течение 4,5 часов в каждую лунку добавляли нагрузочный раствор CCF4-AM (Invitrogen Corp.). После инкубации в течение 2 часов при комнатной температуре измеряли интенсивность флуоресценции с использованием планшет-ридера FDSS (Hamamatsu Photonics K. K.) с возбуждением 400 нм, эмиссией 465 нм/540 нм. Агонистическую активность (EC50) рассчитывали из сигмоидальной кривой доза-ответ с использованием программы GraphPad Prism (GraphPad Software, Inc.).
Примеры 1-97 по настоящему изобретению имеют EC50=<100 нМ в функциональном анализе, описанном выше.
Примеры 1-3, 6-7, 9-10, 12-15, 17, 20-39, 43-57, 60-65, 67-72, 78-80, 82-87 и 91-94 по настоящему изобретению имеют EC50=<10 нМ в функциональном анализе, описанном выше.
Как показано в таблице ниже, функциональная активность в отношении GPR38 (мотилинового рецептора) изменялась в зависимости от абсолютной конфигурации аминокислотной линкерной группы в 100-10000 раз.
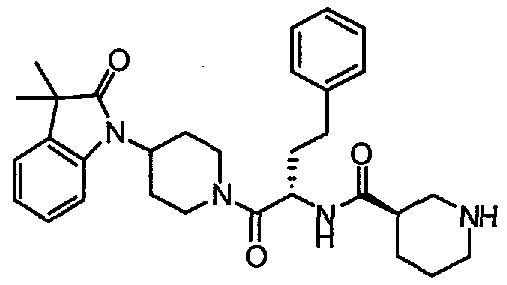
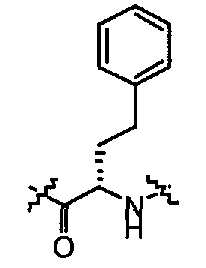
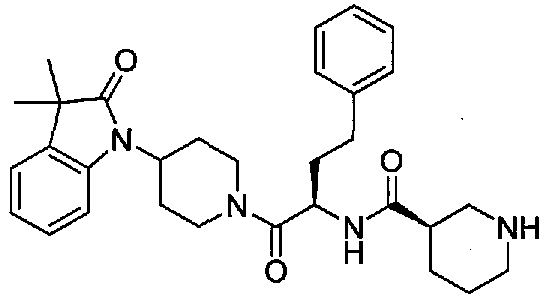
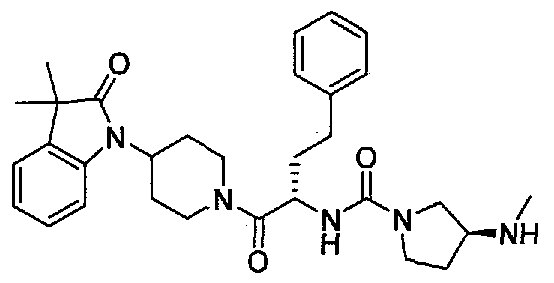
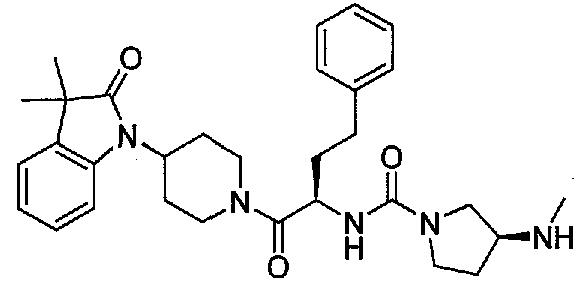
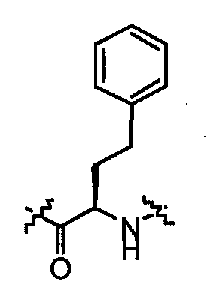
PAMPA (параллельный анализ проникновения через искусственную мембрану)
Эксперименты осуществляли в 96-луночных акцепторном и донорном планшетах. Такая 96-луночная система описана в J. Med. Chem., 1998, 41, 1007. В качестве вещества искусственной мембраны использовали 4% фосфатидилхолина и 1% стеариновой кислоты в додекане. Акцепторный планшет (96-луночный гидрофобный фильтровальный планшет (MAIPN45, Millipore)) получали путем добавления 5 pL вещества искусственной мембраны на фильтр сверху и планшет заполняли 250 pL 2-(N-морфолино)этансульфоновой кислоты (MES), забуференной сбалансированным солевым раствором Хэнка (HBSS) (pH 6,5). Донорный планшет (Transport Receiver plate (MATRNPS50, Millipore)) заполняли 35 с использованием 300 pL MES, забуференного HBSS (pH 6,5), содержащим 10 pM испытываемого соединения. Акцепторный планшет помещали на донорный планшет с образованием "сэндвичевой" конструкции и инкубировали при 30°C в течение 2,5 часов. По окончании периода инкубации акцепторный, донорный и исходный донорный раствор (контроль) анализировали при помощи ЖХ-МС/МС.
Данные представляли в виде значения эффективной проницаемости (Pe) в см Ч10-6/сек и значения удерживания на мембране.
Примеры 1, 3-4, 6-7, 13, 17, 20, 22-23, 29, 35-36, 39, 44, 47-48, 50, 54, 64-66, 68-70, 73-74, 85 и 92-94 по настоящему изобретению имеют Pe≥2,0 см Ч10-6/сек в описанном выше анализе проницаемости.
Все публикации, включая, но не ограничиваясь этим, изданные патенты, патентные заявки и журнальные статьи, на которые имеются ссылки в настоящей заявке, включены в настоящую заявку посредством ссылки, каждая во всей ее полноте. Хотя настоящее изобретение описано выше со ссылкой на раскрываемые варианты воплощения, для специалистов в данной области будет очевидным, что конкретные подробно описанные эксперименты являются лишь иллюстрацией настоящего изобретения. Должно быть понятно, что возможно осуществление различных модификаций без отступления от сути настоящего изобретения. Соответственно, настоящее изобретение ограничивается только представленной далее формулой изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| МОДУЛЯТОРЫ ПРОТЕОЛИЗА И СООТВЕТСТВУЮЩИЕ СПОСОБЫ ПРИМЕНЕНИЯ | 2019 |
|
RU2805511C2 |
| ИНГИБИТОРЫ УРИДИН-5-ДИФОСФАТ (UDP) ГЛИКОЗИЛТРАНСФЕРАЗЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2019 |
|
RU2810928C2 |
| ПОЛИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И СПОСОБЫ ЦЕЛЕНАПРАВЛЕННОЙ ДЕГРАДАЦИИ ПОЛИПЕПТИДОВ БЫСТРО УСКОРЕННОЙ ФИБРОСАРКОМЫ | 2019 |
|
RU2830173C2 |
| АНИЛИДЫ АМИНОКИСЛОТ В КАЧЕСТВЕ НИЗКОМОЛЕКУЛЯРНЫХ МОДУЛЯТОРОВ IL-17 | 2019 |
|
RU2815505C2 |
| КОНЪЮГАТ ЛИГАНДА С ЦИТОТОКСИЧЕСКИМ ЛЕКАРСТВЕННЫМ СРЕДСТВОМ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ | 2016 |
|
RU2708461C2 |
| СУЛЬФОНАМИДСОДЕРЖАЩИЕ СВЯЗЫВАЮЩИЕ СИСТЕМЫ ДЛЯ ЛЕКАРСТВЕННЫХ КОНЪЮГАТОВ | 2014 |
|
RU2729194C2 |
| ПРОИЗВОДНЫЕ ИМИДАЗОПИРАЗИНА ИЛИ ИМИДАЗОДИАЗЕПИНА, АКТИВНЫЕ В ОТНОШЕНИИ РЕЦЕПТОРА СВ2 | 2008 |
|
RU2540074C2 |
| ТРИТЕРПЕНОВЫЕ ПРОИЗВОДНЫЕ ЛУПЕОЛЬНОГО ТИПА КАК ПРОТИВОВИРУСНЫЕ ПРЕПАРАТЫ | 2010 |
|
RU2561604C2 |
| ПУРИНОВЫЕ ИНГИБИТОРЫ ФОСФАТИДИЛИНОЗИТОЛ-3-КИНАЗЫ ДЕЛЬТА ЧЕЛОВЕКА | 2013 |
|
RU2661896C2 |
| НЕСТЕРОИДНЫЕ МОДУЛЯТОРЫ ГЛЮКОКОРТИОДИДНЫХ РЕЦЕПТОРОВ ДЛЯ МЕСТНОЙ ДОСТАВКИ ЛЕКАРСТВ | 2016 |
|
RU2731618C2 |
Группа изобретений относится к новым оксииндольным производным формулы (I) или их фармацевтически приемлемым солям, фармацевтическим композициям на их основе и их применению для лечения различных расстройств, которые опосредованы через мотилиновый рецептор (GPR38). В общей формуле I R1 означает водород, C1-С4алкил или C3-С7циклоалкил; R2 означает C1-С4алкил или C3-С7циклоалкил; или R1 и R2 вместе с атомами, с которыми они связаны, образуют 3-6-членное кольцо, которое может содержать кислород; R3 и R4 означают водород или C1-С4алкил; R5 означает водород или C1-С4алкил; R6 и R7 означают водород, C1-С4алкил или C1-С4алкокси C1-С4алкил; или R6 и R7 вместе с атом азота, с которым они связаны, образуют 4-6-членное кольцо, которое может содержать азот или кислород, при этом 4-6-членное кольцо необязательно замещено 1-4 заместителями, выбранными из группы, состоящей из C1-С4алкила, амино, C1-С4алкиламино и ди(C1-С4алкил)амино; A означает
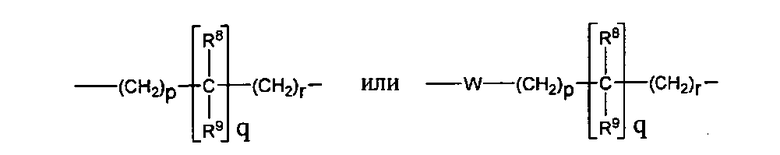
где p, q и r независимо имеют значение 0, 1 или 2; R8 и R9 означают водород или C1-С6алкил; где алкил необязательно замещен гидрокси, C1-С4алкилом, амино, C1-С4алкиламино и ди(C1-С4алкил) амино; или R8 и R9 могут быть объединены друг с другом с образованием C3-C7-членного кольца; или R8 и R9 независимо могут быть объединены с одной иди обеими R8 и R9 группами с образованием алкиленовых мостиков между концевым азотом и алкильной частью R8 или R9 групп, при этом мостик содержит от 1 до 5 атомов углерода; указанный мостик необязательно замещен 1-4 C1-C4 алкильными группами; W означает N-R10, указанный R10 означает водород или C1-С4алкил; X означает C0-С4алкилен или C0-С4алкилен-K-C0-С4алкилен, где K означает -O- и где алкилен необязательно замещен C1-С4алкилом; Y означает водород или 5-10-членное кольцо; указанное кольцо необязательно замещено гидрокси, галогеном, галоген C1-С4алкилом, C1-С4алкилом или C1-С4алкокси, причем когда X означает C0, то Y не означает водород; Z означает галоген или C1-С4алкил; m имеет значение 0, 1, 2, 3 или 4; n имеет значение 0, 1 или 2. 5 н. и 5 з.п. ф-лы, 15 табл., 99 пр.
1. Соединение следующей формулы (I):
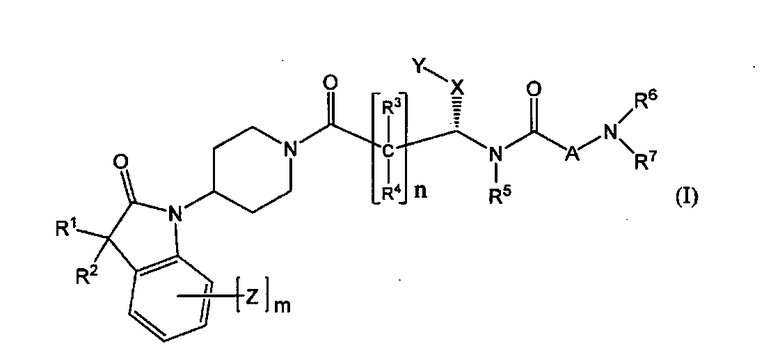
где
R1 независимо выбраны из группы, включающей водород, C1-С4алкил и C3-С7циклоалкил;
R2 независимо выбраны из группы, включающей C1-С4алкил и C3-С7циклоалкил; или, альтернативно, R1 и R2, вместе с атомами, с которыми они связаны, образуют 3-6-членное кольцо, которое может содержать кислород;
R3 и R4 независимо представляют собой водород или C1-С4алкил;
R5 представляет собой водород или C1-С4алкил;
R6 и R7 независимо выбраны из водорода, C1-С4алкила, и C1-С4алкокси C1-С4алкила; или альтернативно, R6 и R7 вместе с атом азота, с которым они связаны, образуют 4-6-членное кольцо, которое может содержать азот или кислород, при этом 4-6-членное кольцо необязательно замещено 1-4 заместителями, независимо выбранными из группы, состоящей из C1-С4алкила, амино, C1-С4алкиламино и ди(C1-С4алкил)амино;
A представляет собой
где p, q и r независимо имеют значение 0, 1 или 2;
R8 и R9 независимо представляют собой водород или C1-С6алкил; где указанный алкил необязательно замещен гидрокси, C1-С4алкилом, амино, C1-С4алкиламино и ди(C1-С4алкил) амино; или R8 и R9 могут быть объединены друг с другом с образованием C3-C7-членного кольца; или R8 и R9 независимо могут быть объединены с одной или обеими R8 и R9 группами с образованием алкиленовых мостиков между концевым азотом и алкильной частью R8 или R9 групп, при этом мостик содержит от 1 до 5 атомов углерода; указанный мостик необязательно замещен 1-4 C1-C4 алкильными группами;
W представляет собой N-R10, указанный R10 представляет собой водород или C1-С4алкил;
X представляет собой C0-С4алкилен или C0-С4алкилен-K-C0-С4алкилен, где K представляет собой -O-, где указанный алкилен необязательно замещен C1-С4алкилом;
Y представляет собой водород или 5-10-членное кольцо; указанное кольцо необязательно замещено гидрокси, галогеном, галоген C1-С4алкилом, C1-С4алкилом или C1-С4алкокси, причем когда X означает C0, то Y не означает водород;
Z представляет собой галоген или C1-С4алкил;
m имеет значение 0, 1, 2, 3 или 4;
n имеет значение 0, 1 или 2;
или его фармацевтически приемлемая соль.
2. Соединение или его фармацевтически приемлемая соль по п.1, где:
R5 представляет собой водород или C1-С4алкил;
X представляет собой C1-С4алкилен или C1-С4алкилен-K-C0-С4алкилен, где K представляет собой -O-; указанный алкилен необязательно замещен C1-С4алкилом;
m имеет значение 0, 1 или 2; и
n имеет значение 0 или 1.
3. Соединение формулы (I), которое выбрано из следующих:
(R)-N-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)пиперидин-3-карбоксамид;
(S)-N-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)пиперидин-2-карбоксамид;
(S)-2-амино-N-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)-3,3-диметилбутанамид;
(S)-2-(1-аминоциклобутил)-N-(1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)ацетамид;
(S)-1-(аминометил)-N-(1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)циклопропанкарбоксамид;
(S)-N-(1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)азетидин-3-карбоксамид;
(S)-N-(1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)пиперидин-4-карбоксамид;
(S)-2-(1-аминоциклопентил)-N-(1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)ацетамид;
(S)-N-(1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)-3-метил-3-(метиламино)бутанамид;
(S)-3-(циклопентиламино)-N-(1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)пропанамид;
(S)-N-(1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)-3-((2-метоксиэтил)(метил)амино)пропанамид;
(S)-N-(1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)-2-(пирролидин-1-ил)ацетамид;
(R)-N-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)-1-метилпиперидин-3-карбоксамид;
(1S,3R)-N-((S)-3-(4-хлорфенил)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксопропан-2-ил)-3-(метиламино)циклопентанкарбоксамид;
(1S,3R)-N-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-3-(2-(трифторметил)фенил)пропан-2-ил)-3-(метиламино)циклопентанкарбоксамид;
(1S,3R)-N-((S)-3-циклогексил-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксопропан-2-ил)-3-(метиламино)циклопентанкарбоксамид;
(1S,3R)-N-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-3-(нафталин-1-ил)-1-оксопропан-2-ил)-3-(метиламино)циклопентанкарбоксамид;
(1S,3R)-N-((S)-3-(2-хлорфенил)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксопропан-2-ил)-3-(метиламино)циклопентанкарбоксамид;
(1S,3R)-N-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-3-о-толилпропан-2-ил)-3-(метиламино)циклопентанкарбоксамид;
(1S,3R)-N-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-3-(4-фторфенил)-1-оксопропан-2-ил)-3-(метиламино)циклопентанкарбоксамид;
(1S,3R)-N-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-3-(3-фторфенил)-1-оксопропан-2-ил)-3-(метиламино)циклопентанкарбоксамид;
(1S,3R)-N-((S)-3-(3-хлорфенил)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксопропан-2-ил)-3-(метиламино)циклопентанкарбоксамид;
(1S,3R)-N-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-3-фенилпропан-2-ил)-3-(метиламино)циклопентанкарбоксамид;
(1S,3R)-N-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-3-(3-метоксифенил)-1-оксопропан-2-ил)-3-(метиламино)циклопентанкарбоксамид;
(1S,3R)-N-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-3-(2-фторфенил)-1-оксопропан-2-ил)-3-(метиламино)циклопентанкарбоксамид;
(1S,3R)-N-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-4-метил-1-оксопентан-2-ил)-3-(метиламино)циклопентанкарбоксамид;
(1S,3R)-N-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-3-(1Н-индол-3-ил)-1-оксопропан-2-ил)-3-(метиламино)циклопентанкарбоксамид;
(1S,3R)-N-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-3-(3-(трифторметил)фенил)пропан-2-ил)-3-(метиламино)циклопентанкарбоксамид;
(1S,3R)-N-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-3-(4-(трифторметил)фенил)пропан-2-ил)-3-(метиламино)циклопентанкарбоксамид;
(1S,3R)-N-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-3-(4-метоксифенил)-1-оксопропан-2-ил)-3-(метиламино)циклопентанкарбоксамид;
(1S,3R)-N-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-3-феноксипропан-2-ил)-3-(метиламино)циклопентанкарбоксамид;
(1S,3R)-N-((S)-3-(2-хлорфенокси)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксопропан-2-ил)-3-(метиламино)циклопентанкарбоксамид;
(1S,3R)-N-((S)-4-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-4-оксо-1-фенилбутан-2-ил)-3-(метиламино)циклопентанкарбоксамид;
(R)-N-((S)-1-(4-(5-фтор-3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)пиперидин-3-карбоксамид;
(1S,3R)-N-((S)-1-(4-(5-фтор-3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)-3-(метиламино)циклопентанкарбоксамид;
(S)-N-(1-(4-(5-фтор-3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)-3-метил-3-(метиламино)бутанамид;
(S)-N-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)-3-(метиламино)пирролидин-1-карбоксамид;
1-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)-3-((S)-пирролидин-2-илметил)мочевина;
1-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)-3-((R)-пиперидин-3-ил)мочевина;
3-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)-1-метил-1-((S)-пирролидин-2-илметил)мочевина;
1-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)-3-((R)-пирролидин-2-илметил)мочевина;
1-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)-3-((R)-пирролидин-3-ил)мочевина;
1-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)-3-((R)-пирролидин-3-илметил)мочевина;
(R)-N-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)-3-метилпиперазин-1-карбоксамид;
(S)-1-(1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)-3-(4-этилпиперидин-4-ил)мочевина;
(S)-1-(1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)-3-(4-метилпиперидин-4-ил)мочевина;
1-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)-3-((S)-пирролидин-3-ил)мочевина;
1-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)-3-((S)-пиперидин-2-илметил)мочевина;
1-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)-3-((R)-пиперидин-3-илметил)мочевина;
(S)-1-(2-амино-2-метилпропил)-3-(1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)мочевина;
(S)-3-(циклопропил(метил)амино)-N-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)пирролидин-1-карбоксамид;
(S)-1-(1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)-3-(2-(пирролидин-1-ил)этил)мочевина;
(S)-1-(1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)-3-(2-(диметиламино)этил)мочевина;
(S)-N-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)-3-(диметиламино)пирролидин-1-карбоксамид;
1-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)-3-(((S)-1-метилпирролидин-2-ил)метил)мочевина;
(S)-1-(1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)-3-(1,4-диметилпиперидин-4-ил)мочевина;
(S)-N-((S)-1-(4-(5-фтор-3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)-3-(метиламино)пирролидин-1-карбоксамид;
1-((S)-1-(4-(5-фтор-3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)-3-((S)-пирролидин-2-илметил)мочевина;
(S)-1-(1-(4-(5-фтор-3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-4-фенилбутан-2-ил)-3-(4-метилпиперидин-4-ил)мочевина;
(S)-N-(1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-3-(2-(трифторметил)фенил)пропан-2-ил)пиперидин-4-карбоксамид;
(S)-N-(3-(2-хлорфенил)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксопропан-2-ил)пиперидин-4-карбоксамид;
(S)-N-(3-(4-хлорфенил)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксопропан-2-ил)пиперидин-4-карбоксамид;
(R)-N-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-3-фенилпропан-2-ил)пиперидин-3-карбоксамид;
(R)-N-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-3-(2-фторфенил)-1-оксопропан-2-ил)пиперидин-3-карбоксамид;
(R)-N-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-3-(3-фторфенил)-1-оксопропан-2-ил)пиперидин-3-карбоксамид;
(R)-N-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-3-(2-(трифторметил)фенил)пропан-2-ил)пиперидин-3-карбоксамид;
(R)-N-((S)-3-(2-хлорфенил)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксопропан-2-ил)пиперидин-3-карбоксамид;
(R)-N-((S)-3-(4-хлорфенил)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксопропан-2-ил)пиперидин-3-карбоксамид;
(S)-N-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-3-(3-фторфенил)-1-оксопропан-2-ил)-3-(метиламино)пирролидин-1-карбоксамид;
(S)-N-((S)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксо-3-(2-(трифторметил)фенил)пропан-2-ил)-3-(метиламино)пирролидин-1-карбоксамид;
(S)-N-((S)-3-(2-хлорфенил)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксопропан-2-ил)-3-(метиламино)пирролидин-1-карбоксамид; и
(S)-N-((S)-3-(4-хлорфенил)-1-(4-(3,3-диметил-2-оксоиндолин-1-ил)пиперидин-1-ил)-1-оксопропан-2-ил)-3-(метиламино)пирролидин-1-карбоксамид;
или его фармацевтически приемлемая соль.
4. Фармацевтическая композиция, обладающая агонистической активностью в отношении мотилинового рецептора, включающая соединение или его фармацевтически приемлемую соль по любому из пп.1-3 и фармацевтически приемлемый носитель.
5. Фармацевтическая композиция по п.4, дополнительно включающая другое фармакологически активное вещество, выбранное из группы, состоящей из:
(1) соединений с активностью, направленной на уменьшение желудочной кислоты, выбранных из антагонистов H2 рецептора, антагонистов кислотного насоса и ингибиторов протонного насоса;
(2) GABA-B агонистов;
(3) соединений с анальгетической активностью, выбранных из агонистов нейрокининовых рецепторов (NK1, NK2, NK3), агонистов TRPV1 и агонистов натриевых каналов; и
(4) соединений со смешанной активностью, действующих на сократительную способность и боль, выбранных из антагонистов CRF2, антагонистов 5-HT3, и октреотида или других агонистов sst2 рецепторов.
6. Способ лечения состояния, опосредованого активностью агониста мотилинового рецептора, у субъекта-млекопитающего, включая человека, который включает введение млекопитающему, нуждающемуся в таком лечении, терапевтически эффективного количества соединения или его фармацевтически приемлемой соли по любому из пп.1-3.
7. Способ по п.6, где указанное состояние или расстройство представляют собой гастроэзофагеальный рефлюкс, функциональную диспепсию, синдром раздраженной толстой кишки, запор, псевдообструкцию кишечника, паралитическую непроходимость кишечника после хирургической операции или других манипуляций, рвоту, желудочный стаз или гипокинезию, вызванную различными заболеваниями, такими как диабет и/или введением других лекарственных средств, или имеющую место у пациентов с энтеральным кормлением, болезнь Крона, колит, кахексию связанную с прогрессирующими заболеваниями, такими как рак, и/или их лечением, кахексию, связанную с аппетитом/метаболизмом, и другие расстройства, такие как недержание.
8. Соединение формулы (I) или его фармацевтически приемлемая соль по любому из пп.1-3, для применения для лечения состояния, опосредованного агонистической активностью относительно мотилинового рецептора.
9. Применение соединения формулы (I) или его фармацевтически приемлемой соли по любому из пп.1-3 для получения лекарственного средства для лечения состояния, опосредованного агонистической активностью относительно мотилинового рецептора.
10. Применение по п.9, где указанное состояние или расстройство представляют собой гастроэзофагеальный рефлюкс, функциональную диспепсию, синдром раздраженной толстой кишки, запор, псевдообструкцию кишечника, паралитическую непроходимость кишечника после хирургической операции или других манипуляций, рвоту, желудочный стаз или гипокинезию, вызванную различными заболеваниями, такими как диабет, и/или введением других лекарственных средств, или возникающую у пациентов с энтеральным кормлением, болезнь Крона, колит, кахексию, связанную с прогрессирующими заболеваниями, такими как рак, и/или их лечением, кахексию, связанную с аппетитом/метаболизмом, и другие расстройства, такие как недержание.
| WO 1996013265 A1, 09.05.1996 | |||
| WO 2008000729 A1, 03.01.2008 | |||
| WO 2006090279 A1, 31.08.2006 | |||
| 1,4-ЗАМЕЩЕННОЕ ПРОИЗВОДНОЕ ЦИКЛИЧЕСКОГО АМИНА, СПОСОБЫ ЕГО ПОЛУЧЕНИЯ, ЛЕКАРСТВЕННОЕ СРЕДСТВО, ОБЛАДАЮЩЕЕ АНТАГОНИСТИЧЕСКИМ К СЕРОТОНИНУ ДЕЙСТВИЕМ, СПОСОБ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЯ, ПО ОТНОШЕНИЮ К КОТОРОМУ ЭФФЕКТИВЕН АНТАГОНИЗМ К СЕРОТОНИНУ | 1998 |
|
RU2203275C2 |

