В настоящем изобретении заявляется о приоритете заявки с порядковым номером 61/076900, поданной 30 июня 2008 года, заявки с порядковым номером 61/076908, поданной 30 июня 2008 года, и заявки с порядковым номером 61/076915, поданной 30 июня 2008 года.
ОБЛАСТЬ ПРИМЕНЕНИЯ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к способам дифференцирования плюрипотентных стволовых клеток. В частности, настоящее изобретение относится к способам и препаратам для дифференцирования плюрипотентных стволовых клеток в клетки, экспрессирующие маркеры, характерные для линии сформированной эндодермы, включая культивирование плюрипотентных стволовых клеток в среде, содержащей достаточное количество GDF-8 для стимуляции дифференцирования плюрипотентных стволовых клеток в клетки, экспрессирующие маркеры, характерные для линии сформированной эндодермы.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Последние достижения в области заместительной клеточной терапии для лечения сахарного диабета 1 типа и нехватка островков Лангерганса для трансплантации заставили обратить внимание на разработку источников инсулин-продуцирующих клеток, или β-клеток, подходящих для трансплантации. Одним из подходов является формирование функциональных β-клеток из плюрипотентных стволовых клеток, таких как, например, эмбриональные стволовые клетки.
При эмбриональном развитии позвоночных плюрипотентные клетки дают начало группе клеток, формирующих три зародышевых листка (эктодерму, мезодерму и эндодерму) в ходе процесса, именуемого гаструляцией. Такие ткани, как, например, щитовидная железа, тимус, поджелудочная железа, кишечник и печень, будут развиваться из эндодермы через промежуточную стадию. Промежуточной стадией данного процесса является образование сформированной эндодермы. Клетки сформированной эндодермы экспрессируют ряд маркеров, таких как HNF-3beta, GATA4, MIXL1, CXCR4 и SOX17.
Формирование поджелудочной железы происходит при дифференцировании сформированной эндодермы в панкреатическую эндодерму. Клетки панкреатической эндодермы экспрессируют ген панкреатическо-дуоденального гомеобокса, PDX1. В отсутствие PDX1 развитие поджелудочной железы не идет дальше формирования вентрального и дорсального зачатков. Таким образом, экспрессия PDX1 характеризует критическую стадию органогенеза поджелудочной железы. Зрелая поджелудочная железа содержит, помимо других типов клеток, экзокринную ткань и эндокринную ткань. Экзокринная и эндокринная ткани образуются при дифференцировании панкреатической эндодермы.
По имеющимся данным, клетки, обладающие свойствами островковых клеток, были получены из эмбриональных клеток мыши. Например, в публикации Lumelsky et al. (Science 292:1389, 2001) сообщается о дифференцировании эмбриональных стволовых клеток мыши в инсулин-секретирующие структуры, сходные с островками поджелудочной железы. В публикации Soria et al. (Diabetes 49:157, 2000) сообщается, что инсулин-секретирующие клетки, полученные из эмбриональных стволовых клеток мыши, нормализовали гликемию у мышей с диабетом, вызванным стрептозотоцином.
В одном примере, в публикации Hori et al. (PNAS 99: 16105, 2002), отмечается, что обработка эмбриональных стволовых клеток мыши ингибиторами фосфоинозитид 3-киназы (LY294002) приводила к получению клеток, сходных с β-клетками.
В другом примере, в публикации Blyszczuk et al. (PNAS 100:998, 2003), сообщается о получении инсулин-продуцирующих клеток из эмбриональных стволовых клеток мыши с конститутивной экспрессией Pax4.
В публикации Micallef et al. сообщается, что ретиноевая кислота может регулировать способность эмбриональных стволовых клеток формировать Pdx1-положительную панкреатическую эндодерму. Ретиноевая кислота с наибольшей эффективностью индуцирует экспрессию Pdx1 при добавлении в культуру на 4 день дифференцирования эмбриональных стволовых клеток в течение периода, соответствующего концу гаструляции эмбриона (Diabetes 54:301, 2005).
В публикации Miyazaki et al. сообщается о линии эмбриональных стволовых клеток мыши со сверхэкспрессией Pdx1. Эти результаты показывают, что экспрессия экзогенного Pdx1, очевидно, повышает экспрессию генов инсулина, соматостатина, глюкокиназы, нейрогенина 3, p48, Pax6 и HNF6 в образующихся дифференцированных клетках (Diabetes 53: 1030, 2004).
В публикации Skoudy et al. сообщается, что активин А (входящий в суперсемейство TGF-β) повышает экспрессию экзокринных панкреатических генов (p48 и амилаза) и эндокринных генов (Pdx1, инсулин и глюкагон) в эмбриональных стволовых клетках мыши.
Максимальный эффект наблюдался при использовании 1 нМ активина A. Также авторы наблюдали, что на уровень экспрессии мРНК инсулина и Pdx1 не влияла ретиноевая кислота; однако обработка раствором 3 нМ FGF7 приводила к повышению уровня транскрипта Pdx1 (Biochem. J. 379: 749, 2004).
В работе Shiraki et al. изучались эффекты факторов роста, специфически ускоряющих дифференцирование эмбриональных стволовых клеток в Pdx1-положительные клетки. Авторы наблюдали, что TGFβ2 приводил к воспроизводимому увеличению доли Pdx1-положительных клеток (Genes Cells. 2005 June; 10(6): 503-16).
В публикации Gordon et al. продемонстрирована индукция образования brachyury-[положительных]/HNF-3beta-[положительных] эндодермальных клеток из эмбриональных стволовых клеток мыши в отсутствии сыворотки и в присутствии активина в сочетании с ингибитором сигнального пути Wnt (США № 2006/0003446A 1).
В публикации Gordon et al. (PNAS, Vol 103, page 16806, 2006) говорится: «Для образования передней первичной полоски одновременно требовались сигнальные пути Wnt и TGF-beta/nodal/активин».
Однако модель развития эмбриональных стволовых клеток на мышах может не имитировать в точности программу развития у высших млекопитающих, например у человека.
В работе Thomson et al. эмбриональные стволовые клетки выделяли из человеческих бластоцист (Science 282:114, 1998). Параллельно Gearhart и соавторы получили клеточные линии эмбриональных зародышевых клеток человека (hEG) из ткани половых желез эмбриона (Shamblott et al., Proc. Natl. Acad. Sci. USA 95:13726, 1998). В отличие от эмбриональных стволовых клеток мыши, воспрепятствовать дифференцированию которых можно путем простого культивирования с фактором торможения лейкемии (LIF), эмбриональные стволовые клетки человека должны культивироваться в очень специфических условиях (Патенты США №№ 6200806; WO 99/20741; WO 01/51616).
В публикации D'Amour et al. описывается производство обогащенных культур сформированной эндодермы, производной от эмбриональных стволовых клеток человека, в присутствии высокой концентрации активина и низкой концентрации сыворотки (D'Amour K A et al. 2005). Трансплантация этих клеток под почечную капсулу мышей привела к их дифференцированию в более зрелые клетки, обладающие характерными особенностями некоторых эндодермальных органов. Клетки сформированной эндодермы, производные от эмбриональных стволовых клеток человека, могут подвергаться дальнейшему дифференцированию в Pdx1-положительные клетки после добавления FGF-10 (США № 2005/0266554A1).
В публикации D'Amour et al. (Nature Biotechnology - 24, 1392-1401 (2006)) говорится: «Мы разработали процесс дифференцирования, преобразующий эмбриональные клетки человека (hES) в эндокринные клетки, способные синтезировать гормоны поджелудочной железы: инсулин, глюкагон, соматостатин, панкреатический полипептид и грелин. Данный процесс имитирует органогенез поджелудочной железы in vivo, проводя клетки через стадии, напоминающие образование сформированной эндодермы, эндодермы кишечной трубки, панкреатической эндодермы и превращение предшественников эндокринных клеток в клетки, экспрессирующие эндокринные гормоны».
В другом примере, в публикации Fisk et al., сообщается о системе для производства островковых клеток поджелудочной железы из эмбриональных стволовых клеток человека (США № 2006/0040387A1). В данном случае процесс дифференцирования был разделен на три стадии. Сначала эмбриональные стволовые клетки человека были дифференцированы до эндодермы с помощью сочетания н-бутирата и активина А. Далее клетки культивировались с антагонистами TGF-β, такими как Noggin, в сочетании с EGF или бетацеллюлином с получением Pdx1-положительных клеток. Окончательное дифференцирование запускалось никотинамидом.
В одном примере, в публикации Benvenistry et al., сообщается: «Мы делаем вывод, что сверхэкспрессия PDX1 увеличивала экспрессию панкреатических обогащенных генов, а для индукции экспрессии инсулина могут требоваться дополнительные сигналы, присутствующие только in vivo» (Benvenistry et al, Stem Cells 2006; 24:1923-1930).
Активин А является членом семейства TGF-beta и демонстрирует широкий спектр биологических эффектов, включая регуляцию клеточной пролиферации и дифференцирования, а также стимуляцию выживания нейронов. Выделение и очистка активина А часто сложна и часто может иметь низкий выход продукта. Например, авторы S.A. Pangas и T.K. Woodruff сообщают: «Ингибин и активин являются белковыми гормонами с различающимися физиологическими функциями, включая регуляцию секреции гипофизарного ФСГ. Подобно другим членам семейства генов трансформирующего фактора роста-β, они подвергаются процессингу из более крупных молекул-предшественников, а также объединяются в функциональные димеры. При выделении ингибина и активина из природных источников можно получать лишь ограниченное количество биологически активного белка» (J. Endocrinol. 172 (2002) 199-210).
В другом примере, в публикации K.Y. Arai et al, сообщается: «Активины представляют собой многофункциональные факторы роста, принадлежащие к суперсемейству трансформирующего фактора роста-β. Выделение активинов из природных источников включает в себя множество этапов, а получаемое количество белка ограничено. Хотя в последних работах используются рекомбинантные препараты, очистка рекомбинантных активинов, тем не менее, требует множества этапов» (Protein Expression and Purification 49 (2006) 78-82).
Следовательно, по-прежнему сохраняется значительная потребность в альтернативах активина А, используемых при дифференцировании плюрипотентных стволовых клеток.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В одном из вариантов осуществления настоящее изобретение предлагает способ дифференцирования плюрипотентных стволовых клеток в клетки, экспрессирующие маркеры, характерные для линии сформированной эндодермы, включая культивирование плюрипотентных стволовых клеток в среде, содержащей достаточное количество GDF-8 для стимуляции дифференцирования плюрипотентных стволовых клеток в клетки, экспрессирующие маркеры, характерные для линии сформированной эндодермы.
В одном из вариантов осуществления среда, содержащая достаточное количество GDF-8, также содержит по меньшей мере одно другое соединение. В одном из вариантов осуществления такое по меньшей мере одно другое соединение представляет собой анилин-пиридинотриазин. В альтернативном варианте осуществления такое по меньшей мере одно другое соединение представляет собой циклический анилин-пиридинотриазин.
КРАТКОЕ ОПИСАНИЕ РИСУНКОВ
На фиг. 1 показано дифференцирование эмбриональных стволовых клеток человека H1 в клетки, экспрессирующие маркеры, характерные для линии сформированной эндодермы. Дифференцирование определяли путем измерения числа клеток (часть A) и интенсивности сигнала SOX17 (часть B) с использованием анализатора IN Cell Analyzer 1000 (GE Healthcare). Эмбриональные стволовые клетки человека обрабатывали в течение четырех дней средой, содержащей 20 нг/мл Wnt3a и активин A в обозначенных концентрациях (черные столбцы), или средой, не содержащей Wnt3a, но содержащей активин A в обозначенных концентрациях (белые столбцы).
На фиг. 2 показана зависимость доза-эффект для активина A и GDF8, используемых для дифференцирования эмбриональных стволовых клеток человека линии H1 в клетки, экспрессирующие маркеры, характерные для линии сформированной эндодермы. Клетки обрабатывали в течение трех дней активином A или GDF8 в указанных концентрациях в сочетании с 20 нг/мл Wnt3a, добавляемого в первый день анализа. Дифференцирование определяли путем измерения интенсивности сигнала SOX17 с использованием флуоресцентного антительного зонда и одновременного многопараметрического анализа на анализаторе IN Cell Analyzer (GE Healthcare).
На фиг. 3 показаны уровни экспрессии CXCR4 в клетках после первого этапа дифференцирования, проводимого в соответствии со способами, описанными в Примере 12. Клетки H1 обрабатывали 100 нг/мл активина А или 200 нг/мл GDF-8 в течение трех дней в сочетании с 20 нг/мл Wnt3a, добавляемого в первый день, или 2,5 мкМ Соединения 34, или 2,5 мкМ Соединения 56, добавляемого в течение всех трех дней. Экспрессию CXCR4 измеряли с помощью флуоресцентного антительного зонда и проточной цитометрии, получая в результате показанную процентную долю положительных клеток.
На фиг. 4 показаны уровни экспрессии SOX17 в клетках после трех дней дифференцирования в сформированную эндодерму, проводимого в соответствии со способами, описанными в Примере 12. Клетки H1 обрабатывали в течение трех дней 100 нг/мл активина А или 200 нг/мл GDF-8 в сочетании с 20 нг/мл Wnt3a, добавляемого в первый день, или 2,5 мкМ Соединения 34, или 2,5 мкМ Соединения 56, добавляемого в течение всех трех дней. Дифференцирование определяли путем измерения интенсивности сигнала SOX17 (черные столбцы) и итогового числа клеток (белые столбцы) с использованием флуоресцентного антительного зонда и одновременного многопараметрического анализа на анализаторе IN Cell Analyzer (GE Healthcare).
На фиг. 5 показаны уровни экспрессии PDX1 и белка CDX2 в клетках после третьего этапа дифференцирования, проводимого в соответствии со способами, описанными в Примере 12. Клетки H1 обрабатывали в течение трех дней 100 нг/мл активина А или 200 нг/мл GDF-8 в сочетании с 20 нг/мл Wnt3a, добавляемого в первый день, или 2,5 мкМ Соединения 34, или 2,5 мкМ Соединения 56, добавляемого в течение всех трех дней, с последующим проведением клеток через второй и третий этапы дифференцирования. Для каждой из экспериментальных групп показаны уровни экспрессии белков и число клеток, измеренные с помощью флуоресцентных антительных зондов и одновременного многопараметрического анализа. С целью сравнения значения нормированы по отношению к обработке активином A/Wnt3a.
На фиг. 6 показаны уровни экспрессии белка PDX1 (белые столбцы) и число клеток (черные столбцы) после четвертого этапа дифференцирования, проводимого в соответствии со способами, описанными в Примере 12. Клетки H1 обрабатывали в течение трех дней 100 нг/мл активина А или 200 нг/мл GDF-8 в сочетании с 20 нг/мл Wnt3a, добавляемого в первый день, или 2,5 мкМ Соединения 34, или 2,5 мкМ Соединения 56, добавляемого в течение всех трех дней, с последующим проведением клеток через второй, третий и четвертый этапы дифференцирования. Для каждой из экспериментальных групп показаны уровни экспрессии белков и число клеток, измеренные с помощью флуоресцентных антительных зондов и одновременного многопараметрического анализа. С целью сравнения значения нормированы по отношению к обработке активином A/Wnt3a.
На фиг. 7 показаны уровни экспрессии белков инсулина и глюкагона и число клеток, дифференцированных в соответствии со способами, описанными в Примере 12. Клетки H1 обрабатывали в течение трех дней 100 нг/мл активина А или 200 нг/мл GDF-8 в сочетании с 20 нг/мл Wnt3a, добавляемого в первый день, или 2,5 мкМ Соединения 34, или 2,5 мкМ Соединения 56, добавляемого в течение всех трех дней, с последующим проведением клеток через второй, третий, четвертый и пятый этапы дифференцирования. Для каждой из экспериментальных групп показаны уровни экспрессии белков и число клеток, измеренные с помощью флуоресцентных антительных зондов и одновременного многопараметрического анализа. С целью сравнения значения нормированы по отношению к обработке активином A/Wnt3a.
На фиг. 8 показаны уровни экспрессии белка SOX17 в эмбриональных стволовых клетках человека и число этих клеток после дифференцирования в сформированную эндодерму, проводимого в соответствии со способами, описанными в Примере 13. Клетки H1 обрабатывали в течение четырех дней 100 нг/мл активина А или 100 нг/мл фактора роста GDF в сочетании с 20 нг/мл Wnt3a, добавляемого в первый день, или 2,5 мкМ Соединения 34, или 2,5 мкМ Соединения 56, добавляемого в течение первых двух дней анализа. Для каждой из экспериментальных групп показаны уровни экспрессии белка SOX17 (черные столбцы) и число клеток (белые столбцы), измеренные с помощью флуоресцентных антительных зондов и одновременного многопараметрического анализа. С целью сравнения значения нормированы по отношению к обработке активином A/Wnt3a. В части 8A показана серия контрольных сред для дифференцирования в отсутствие любых факторов роста (NONE), с обработкой активином A/Wnt3a (AA/Wnt3a) или с добавлением реагентов по отдельности. В части 8B показано дифференцирование с добавлением GDF-3, как отдельно, так и в разнообразных сочетаниях с Wnt3a, Соединением 34 или Соединением 56. В части 8С показано дифференцирование с добавлением GDF-5, как отдельно, так и в разнообразных сочетаниях с Wnt3a, Соединением 34 или Соединением 56. В части 8D показано дифференцирование с добавлением GDF-8, как отдельно, так и в разнообразных сочетаниях с Wnt3a, Соединением 34 или Соединением 56. В части 8E показано дифференцирование с добавлением GDF-10, как отдельно, так и в разнообразных сочетаниях с Wnt3a, Соединением 34 или Соединением 56. В части 8F показано дифференцирование с добавлением GDF-11, как отдельно, так и в разнообразных сочетаниях с Wnt3a, Соединением 34 или Соединением 56. В части 8G показано дифференцирование с добавлением GDF-15, как отдельно, так и в разнообразных сочетаниях с Wnt3a, Соединением 34 или Соединением 56.
На фиг. 9 показаны уровни экспрессии белка SOX17 в эмбриональных стволовых клетках человека после дифференцирования в сформированную эндодерму, проводимого в соответствии со способами, описанными в Примере 14. Клетки H1 обрабатывали в течение трех дней 100 нг/мл активина А или различными факторами роста в указанных концентрациях в сочетании с 20 нг/мл Wnt3a или 2,5 мкМ Соединения 34, добавляемого в первый день. Для каждой из экспериментальных групп показаны уровни экспрессии белка SOX17 (черные столбцы) и число клеток (белые столбцы), измеренные с помощью флуоресцентных антительных зондов и одновременного многопараметрического анализа. С целью сравнения значения нормированы по отношению к обработке активином A/Wnt3a. В части 9А показана серия контрольных сред для дифференцирования только с Wnt3a в отсутствие любых факторов роста (None) или с обработкой активином A/Wnt3a (AA/Wnt3a). В части 9B показано дифференцирование с GDF-8 (PeproTech) в указанных концентрациях в сочетании с 20 нг/мл Wnt3a. В части 9C показано дифференцирование с GDF-8 (Shenendoah) в указанных концентрациях в сочетании с 20 нг/мл Wnt3a. В части 9D показано дифференцирование с TGFβ1 в указанных концентрациях в разнообразных сочетаниях с Wnt3a или Соединением 34. В части 9E показано дифференцирование с BMP2 в указанных концентрациях в разнообразных сочетаниях с Wnt3a или Соединением 34. В части 9F показано дифференцирование с BMP3 в указанных концентрациях в разнообразных сочетаниях с Wnt3a или Соединением 34. В части 9G показано дифференцирование с BMP4 в указанных концентрациях в разнообразных сочетаниях с Wnt3a или Соединением 34.
На фиг. 10 показаны уровни экспрессии белка SOX17 в эмбриональных стволовых клетках человека после дифференцирования в сформированную эндодерму, проводимого в соответствии со способами, описанными в Примере 15. Клетки H1 обрабатывали в течение трех дней, используя различное время воздействия, 100 нг/мл активина А или 100 нг/мл GDF-8 в сочетании с 20 нг/мл Wnt3a. Определенная с помощью флуоресцентных антительных зондов и одновременного многопараметрического анализа экспрессия белка SOX17 представлена в виде суммарных значений интенсивности для каждой из экспериментальных групп: для контрольных сред дифференцирования без добавления факторов роста (без обработки), только с Wnt3a, только с активином A или GDF-8, или с обработкой активином A/Wnt3a или GDF-8/Wnt3a, причем, как показано, Wnt3a добавлялся только в первый день анализа или в течение всех трех дней анализа.
На фиг. 11 показаны уровни экспрессии белка SOX17 в эмбриональных стволовых клетках человека после дифференцирования в сформированную эндодерму, проводимого в соответствии со способами, описанными в Примере 15. Клетки H1 обрабатывали в течение трех дней, используя разное время воздействия, 100 нг/мл активина А в сочетании с тестовым соединением (Соединение 181 (часть A), Соединение 180 (часть B), Соединение 19 (часть C), Соединение 202 (часть D), Соединение 40 (часть E), Соединение 34 (часть F) или ингибитор GSK3 BIO (часть G)) в указанных концентрациях, причем тестовое соединение добавлялось только в первый день анализа. Определенная с помощью флуоресцентных антительных зондов и одновременного многопараметрического анализа экспрессия белка SOX17 представлена в виде суммарных значений интенсивности.
На фиг. 12 показаны уровни экспрессии белка SOX17 в эмбриональных стволовых клетках человека после дифференцирования в сформированную эндодерму, проводимого в соответствии со способами, описанными в Примере 15. Клетки H1 обрабатывали в течение трех дней, используя различное время воздействия, 100 нг/мл активина А в сочетании с тестовым соединением (Соединение 181 (часть A), Соединение 180 (часть B), Соединение 19 (часть C), Соединение 202 (часть D), Соединение 40 (часть E), Соединение 34 (часть F) или ингибитор GSK3 BIO (часть G)) в указанных концентрациях, причем тестовое соединение добавлялось в течение всех трех дней анализа. Определенная с помощью флуоресцентных антительных зондов и одновременного многопараметрического анализа экспрессия белка SOX17 представлена в виде суммарных значений интенсивности.
На фиг. 13 показаны уровни экспрессии белка SOX17 в эмбриональных стволовых клетках человека после дифференцирования в сформированную эндодерму, проводимого в соответствии со способами, описанными в Примере 15. Клетки H1 обрабатывали в течение трех дней, используя различное время воздействия, 100 нг/мл GDF-8 в сочетании с тестовым соединением (Соединение 181 (часть A), Соединение 180 (часть B), Соединение 19 (часть C), Соединение 202 (часть D), Соединение 40 (часть E), Соединение 34 (часть F) или ингибитор GSK3 BIO (часть G)) в указанных концентрациях, причем тестовое соединение добавлялось только в первый день анализа. Определенная с помощью флуоресцентных антительных зондов и одновременного многопараметрического анализа экспрессия белка SOX17 представлена в виде суммарных значений интенсивности.
На фиг. 14 показаны уровни экспрессии белка SOX17 в эмбриональных стволовых клетках человека после дифференцирования в сформированную эндодерму, проводимого в соответствии со способами, описанными в Примере 15. Клетки H1 обрабатывали в течение трех дней, используя различное время воздействия, 100 нг/мл GDF-8 в сочетании с тестовым соединением (Соединение 181 (часть A), Соединение 180 (часть B), Соединение 19 (часть C), Соединение 202 (часть D), Соединение 40 (часть E), Соединение 34 (часть F) или ингибитор GSK3 BIO (часть G)) в указанных концентрациях, причем тестовое соединение добавлялось в течение всех трех дней анализа. Определенная с помощью флуоресцентных антительных зондов и одновременного многопараметрического анализа экспрессия белка SOX17 представлена в виде суммарных значений интенсивности.
На фиг. 15 показан выход по числу клеток после дифференцирования эмбриональных стволовых клеток человека в сформированную эндодерму, проводимого в соответствии со способами, описанными в Примере 15. Клетки H1 обрабатывали в течение трех дней, используя различное время воздействия, 100 нг/мл активина А или 100 нг/мл GDF-8 в сочетании с 20 нг/мл Wnt3a. Определенное с помощью флуоресцентного ядерного зонда и одновременного многопараметрического анализа число клеток показано для каждой из экспериментальных групп: для контрольных сред дифференцирования без добавления факторов роста (без обработки), только с Wnt3a, только с активином A или GDF-8, или с обработкой активином A/Wnt3a или GDF-8/Wnt3a, причем, как показано, Wnt3a добавлялся только в первый день анализа или в течение всех трех дней анализа.
На фиг. 16 показан выход по числу клеток после дифференцирования эмбриональных стволовых клеток человека в сформированную эндодерму, проводимого в соответствии со способами, описанными в Примере 15. Клетки H1 обрабатывали в течение трех дней, используя различное время воздействия, 100 нг/мл активина А в сочетании с тестовым соединением (Соединение 181 (часть A), Соединение 180 (часть B), Соединение 19 (часть C), Соединение 202 (часть D), Соединение 40 (часть E), Соединение 34 (часть F) или ингибитор GSK3 BIO (часть G)) в указанных концентрациях, причем тестовое соединение добавлялось только в первый день анализа. Показаны значения выхода по числу клеток, определенные с помощью флуоресцентного ядерного зонда и одновременного многопараметрического анализа.
На фиг. 17 показан выход по числу клеток после дифференцирования эмбриональных стволовых клеток человека в сформированную эндодерму, проводимого в соответствии со способами, описанными в Примере 15. Клетки H1 обрабатывали в течение трех дней, используя различное время воздействия, 100 нг/мл активина А в сочетании с тестовым соединением (Соединение 181 (часть A), Соединение 180 (часть B), Соединение 19 (часть C), Соединение 202 (часть D), Соединение 40 (часть E), Соединение 34 (часть F) или ингибитор GSK3 BIO (часть G)) в указанных концентрациях, причем тестовое соединение добавлялось в течение всех трех дней анализа. Показаны значения выхода по числу клеток, определенные с помощью флуоресцентного ядерного зонда и одновременного многопараметрического анализа.
На фиг. 18 показан выход по числу клеток после дифференцирования эмбриональных стволовых клеток человека в сформированную эндодерму, проводимого в соответствии со способами, описанными в Примере 15. Клетки H1 обрабатывали в течение трех дней, используя различное время воздействия, 100 нг/мл GDF-8 в сочетании с тестовым соединением (Соединение 181 (часть A), Соединение 180 (часть B), Соединение 19 (часть C), Соединение 202 (часть D), Соединение 40 (часть E), Соединение 34 (часть F) или ингибитор GSK3 BIO (часть G)) в указанных концентрациях, причем тестовое соединение добавлялось только в первый день анализа. Показаны значения выхода по числу клеток, определенные с помощью флуоресцентного ядерного зонда и одновременного многопараметрического анализа.
На фиг. 19 показан выход по числу клеток после дифференцирования эмбриональных стволовых клеток человека в сформированную эндодерму, проводимого в соответствии со способами, описанными в Примере 15. Клетки H1 обрабатывали в течение трех дней, используя различное время воздействия, 100 нг/мл GDF-8 в сочетании с тестовым соединением (Соединение 181 (часть A), Соединение 180 (часть B), Соединение 19 (часть C), Соединение 202 (часть D), Соединение 40 (часть E), Соединение 34 (часть F) или ингибитор GSK3 BIO (часть G)) в указанных концентрациях, причем тестовое соединение добавлялось в течение всех трех дней анализа. Показаны значения выхода по числу клеток, определенные с помощью флуоресцентного ядерного зонда и одновременного многопараметрического анализа.
На фиг. 20 показаны уровни экспрессии различных белковых маркеров в клетках в ходе множества этапов дифференцирования, проводимого в соответствии со способами, описанными в Примере 16. Клетки H1 обрабатывали в течение трех дней 100 нг/мл активина А или 100 нг/мл GDF-8 в сочетании с 20 нг/мл Wnt3a, добавляемого в первый день, или в сочетании с 2,5 мкМ различных соединений (Соединение 19, Соединение 202, Соединение 40 или ингибитор GSK3 BIO), которые добавлялись только в первый день. На фиг. 20 в части A показано содержание маркера сформированной эндодермы, CXCR4, в клетках после первого этапа дифференцирования, измеренное методом FACS. Экспрессию CXCR4 измеряли с помощью антительного флуоресцентного зонда и проточной цитометрии, получая в результате показанную процентную долю положительных клеток. На фиг. 20 в части B показаны полученные с помощью одновременного многопараметрического анализа нормированные данные по экспрессии белка SOX17 (черные столбцы) и числу полученных клеток (белые столбцы) после первого этапа дифференцирования для различных видов обработки. На фиг. 20 в части C показано полученное с помощью одновременного многопараметрического анализа относительное число клеток, полученных из культур, которые подвергались обработке до 5 этапа дифференцирования. На фиг. 20 в части D показаны полученные с помощью одновременного многопараметрического анализа данные по экспрессии белка глюкагона в клетках из культур, которые подвергались обработке до 5 этапа дифференцирования. На фиг. 20 в части E показаны полученные с помощью одновременного многопараметрического анализа данные по экспрессии белка инсулина в клетках из культур, которые подвергались обработке до 5 этапа дифференцирования. На фиг. 20 в части F показано отношение экспрессии глюкагон/инсулин в клетках из культур, которые подвергались обработке до 5 этапа дифференцирования. Для сравнения значения экспрессии в частях B, C, D, E и F нормированы по отношению к контрольной обработке активином А и Wnt3a в ходе 1 этапа.
На фиг. 21 показаны уровни экспрессии различных белковых маркеров и маркеров ОТ-ПЦР в клетках в ходе множества этапов дифференцирования, проводимого в соответствии со способами, описанными в Примере 17. Клетки H1 обрабатывали в течение трех дней 100 нг/мл активина А или 100 нг/мл GDF-8 в сочетании с 20 нг/мл Wnt3a, добавляемого в первый день, или в сочетании с различными соединениями в указанных концентрациях (Соединение 181, Соединение 180, Соединение 19, Соединение 202, Соединение 40, Соединение 56 или ингибитор GSK3 BIO), которые добавлялись только в первый день. Показано измеренное методом FACS содержание маркера сформированной эндодермы, CXCR4, в клетках после первого этапа дифференцирования, при котором обработка включала сочетание активина A (часть A) или GDF-8 (часть B) с Wnt3a или различными соединениями. Экспрессию CXCR4 измеряли с помощью флуоресцентного антительного зонда и проточной цитометрии, получая в результате показанную процентную долю положительных клеток. В следующих частях фиг. 21 представлены нормированные данные ОТ-ПЦР по различным маркерам дифференцирования при соответствующей обработке активином A или GDF-8 на первом этапе дифференцирования: маркеры, присутствующие в конце первого этапа дифференцирования при обработке активином A (часть C) или GDF-8 (часть D); маркеры, присутствующие в конце третьего этапа дифференцирования при обработке активином A (часть E) или GDF-8 (часть F); маркеры, присутствующие в конце четвертого этапа дифференцирования при обработке активином A (часть G) или GDF-8 (часть H); маркеры, присутствующие в конце пятого этапа дифференцирования при обработке активином A (часть I) или GDF-8 (часть J). После завершения пятого этапа дифференцирования был проведен одновременный многопараметрический анализ для измерения числа полученных клеток после обработки их на первом этапе дифференцирования активином А (часть K) или GDF-8 (часть M). Одновременный многопараметрический анализ также использовался для измерения интенсивности сигнала глюкагона и инсулина в конце пятого этапа дифференцирования в полученных клеточных популяциях, которые обрабатывались на первом этапе дифференцирования активином A (часть L) или GDF-8 (часть N).
На фиг. 22 показаны уровни экспрессии различных белковых маркеров и маркеров ОТ-ПЦР в клетках, обработанных в соответствии со способами, описанными в Примере 18. Клетки H1 обрабатывали в течение трех дней 100 нг/мл активина А или 100 нг/мл GDF-8 в сочетании с 20 нг/мл Wnt3a, добавляемого в первый день, или в сочетании с 2,5 мкМ Соединения 40, или 2,5 мкМ Соединения 202, добавляемого только в первый день. На фиг. 22 в части A показано содержание маркера сформированной эндодермы, CXCR4, в клетках после первого этапа дифференцирования, измеренное методом FACS. Экспрессию CXCR4 измеряли с помощью флуоресцентного антительного зонда и проточной цитометрии, получая в результате показанную процентную долю положительных клеток. На фиг. 22 в части B представлены нормированные данные ОТ-ПЦР по различным маркерам дифференцирования в клетках, полученных после четвертого этапа дифференцирования, при соответствующей обработке сочетаниями активин A/Wnt3a или GDF-8/Соединение 40, или GDF-8/Соединение 202 на первом этапе дифференцирования.
На фиг. 23 показаны уровни C-пептида у мышей с синдромом тяжелого комбинированного иммунодефицита, обусловленного врожденным отсутствием клеток-киллеров (SCID-beige), которые получали клетки в конце четвертого этапа протокола дифференцирования, описанного в Примере 18.
На фиг. 24 в части A показаны уровни экспрессии CXCR4, измеренные методом FACS в клетках в конце первого этапа протокола дифференцирования, описанного в Примере 19. В части B показаны уровни экспрессии различных генов, измеренные методом ОТ-ПЦР в клетках в конце четвертого этапа протокола дифференцирования, описанного в Примере 19. Показаны две различные повторности эксперимента (Rep-1 и Rep-2), в которых протоколы обработки были идентичными. В части C показаны уровни C-пептида у мышей SCID-beige, получавших клетки в конце четвертого этапа протокола дифференцирования, обработанные GDF-8 и Wnt3a в ходе первого этапа дифференцирования in vitro. В части D показаны уровни C-пептида у мышей SCID-beige, получавших клетки в конце четвертого этапа протокола дифференцирования, обработанные GDF-8 и Соединением 28 в ходе первого этапа дифференцирования in vitro.
На фиг. 25 показано число клеток (часть A) и экспрессия CXCR4 (часть B) для клеток, выращенных на гранулах микроносителя и обработанных в соответствии со способами, составляющими предмет настоящего изобретения, описанными в Примере 22. Клетки выращивались на гранулах Cytodex3 без обработки (недифференцированные) или с обработкой сочетанием 100 нг/мл активина A и 20 нг/мл Wnt3a (AA/Wnt3a), или с различными типами обработки GDF-8: 50 нг/мл GDF-8 с 2,5 мкМ Соединения 34 (Cmp 34+8); или 50 нг/мл GDF-8 с 2,5 мкМ Соединения 34 и 50 нг/мл PDGF (Cmp 34+8+D); или 50 нг/мл GDF-8 с 2,5 мкМ Соединения 34 и 50 нг/мл PDGF и 50 нг/мл VEGF (Cmp 34+8+D+V); или 50 нг/мл GDF-8 с 2,5 мкМ Соединения 34 и 50 нг/мл PDGF и 50 нг/мл VEGF и 20 нг/мл мусцимола (Cmp 34+8+D+V+M).
На фиг. 26 показана пролиферация клеток после обработки соединениями, составляющими предмет настоящего изобретения, как описано в Примере 23. В частях с B по I показаны данные для обработки с применением соединения в сочетании с GDF-8, где показания оптической плотности в тесте MTS измерялись через 1, 2 и 3 дня после начала анализа дифференцирования.
На фиг. 27 показана экспрессия различных белков и генов в клетках, выращенных на гранулах микроносителя и обработанных в соответствии со способами, составляющими предмет настоящего изобретения. В части A показана процентная доля положительной экспрессии CXCR4, CD99 и CD9, измеренная методом FACS в клетках в конце первого этапа протокола дифференцирования, описанного в Примере 24. В части B показано число клеток, полученных в результате указанной обработки и дифференцированных до третьего этапа протокола дифференцирования. В части C показаны значения ddCT для различных генных маркеров, экспрессированных в клетках, прошедших указанную обработку и дифференцированных до третьего этапа протокола.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Для ясности описания, а не в ограничение изобретения, приведенное ниже подробное описание изобретения разделено на следующие подразделы, описывающие или иллюстрирующие определенные особенности, варианты осуществления или области применения настоящего изобретения.
Определения
Стволовые клетки представляют собой недифференцированные клетки, определяемые по их способности на уровне единичной клетки как самообновляться, так и дифференцироваться с образованием клеток-потомков, таких как самообновляющиеся клетки-предшественники, необновляющиеся клетки-предшественники и окончательно дифференцированные клетки. Стволовые клетки также характеризуются способностью дифференцироваться in vitro в функциональные клетки различных клеточных линий дифференцирования из нескольких зародышевых листков (эндодермы, мезодермы и эктодермы), а также после трансплантации давать начало тканям, происходящим от нескольких зародышевых листков, и вносить существенный вклад в формирование большинства, если не всех, тканей после инъекции в бластоцисты.
По потенциалу развития стволовые клетки классифицируются следующим образом: (1) тотипотентные, т.е. способные давать начало всем эмбриональным и внеэмбриональным типам клеток; (2) плюрипотентные, т.е. способные давать начало всем эмбриональным типам клеток; (3) мультипотентные, т.е. способные давать начало группе клеточных линий дифференцирования в пределах конкретной ткани, органа или физиологической системы (например, гематопоэтические стволовые клетки (HSC) могут давать таких потомков, как HSC (самообновление), олигопотентные предшественники, ограниченные клетками крови, и все типы клеток и клеточных элементов (таких как тромбоциты), являющиеся нормальными компонентами крови); (4) олигопотентные, т.е. способные давать начало более ограниченному набору клеточных линий дифференцирования, чем мультипотентные стволовые клетки; и (5) унипотентные, т.е. способные давать начало единственной клеточной линии дифференцирования (например, сперматогенные стволовые клетки).
Дифференцирование представляет собой процесс, при помощи которого неспециализированная («некоммитированная») или менее специализированная клетка приобретает свойства специализированной клетки, например нервной или мышечной клетки. Дифференцированная клетка или клетка с индуцированным дифференцированием представляет собой клетку, занявшую более специализированное («коммитированное») положение в линии дифференцирования клетки. Термин «коммитированная» применительно к процессу дифференцирования обозначает клетку, дошедшую в ходе процесса дифференцирования до стадии, от которой в нормальных условиях она продолжит дифференцироваться до определенного типа клеток или набора типов клеток и не сможет в нормальных условиях дифференцироваться в иной тип клеток или вернуться обратно к менее дифференцированному типу. Дедифференцированием называется процесс, в ходе которого клетка возвращается к менее специализированному (или коммитированному) положению в линии дифференцирования. Используемый в настоящей заявке термин «линия дифференцирования клетки» определяет наследственность клетки, то есть определяет, из какой клетки произошла данная клетка и каким клеткам она может дать начало. В линии дифференцирования клетка помещается в наследственную схему развития и дифференцирования. Маркером, специфичным для линии дифференцирования, называется характерная особенность, специфически ассоциированная с фенотипом клеток конкретной линии дифференцирования, которая может использоваться для оценки дифференцирования некоммитированных клеток в клетки данной линии дифференцирования.
Используемый в настоящей заявке термин «β-клеточная линия дифференцирования» относится к клеткам, положительным по экспрессии гена транскрипционного фактора PDX-1 и по меньшей мере одного из следующих транскрипционных факторов: NGN3, NKX2.2, NKX6.1, NEUROD, ISL1, HNF-3 beta, MAFA, PAX4 или PAX6. Клетки, экспрессирующие маркеры, характерные для β-клеточной линии дифференцирования, включают β-клетки.
Используемые в настоящей заявке термины «клетки, экспрессирующие маркеры, характерные для линии сформированной эндодермы» или «клетки стадии 1», или «стадия 1» относятся к клеткам, экспрессирующим по меньшей мере один из следующих маркеров: SOX17, GATA4, HNF-3 beta, GSC, CER1, Nodal, FGF8, Brachyury, гомеобоксный белок Mix-like, FGF4, CD48, эомезодермин (EOMES), DKK4, FGF17, GATA6, CXCR4, C-Kit, CD99 или OTX2. Клетки, экспрессирующие маркеры, характерные для линии сформированной эндодермы, включают клетки-предшественники первичной полоски, клетки первичной полоски, клетки мезэндодермы и клетки сформированной эндодермы.
Используемый в настоящей заявке термин «клетки, экспрессирующие маркеры, характерные для линии дифференцирования в клетки панкреатической эндодермы» относится к клеткам, экспрессирующим по меньшей мере один из следующих маркеров: PDX1, HNF-1 beta, PTF1 alpha, HNF6 или HB9. Клетки, экспрессирующие маркеры, характерные для линии дифференцирования в клетки панкреатической эндодермы, включают клетки панкреатической эндодермы, клетки первичной кишечной трубки и клетки задней части передней кишки.
Используемый в настоящей заявке термины «клетки, экспрессирующие маркеры, характерные для линии дифференцирования в панкреатические эндокринные клетки» или «клетки стадии 5», или «стадия 5» относятся к клеткам, экспрессирующим по меньшей мере один из следующих маркеров: NGN3, NEUROD, ISL1, PDX1, NKX6.1, PAX4 или PTF-1 alpha. Клетки, экспрессирующие маркеры, характерные для линии дифференцирования в панкреатические эндокринные клетки, включают панкреатические эндокринные клетки, панкреатические экспрессирующие гормоны клетки и панкреатические секретирующие гормоны клетки, а также клетки β-клеточной линии дифференцирования.
Используемый в настоящей заявке термин «сформированная эндодерма» относится к клеткам, обладающим характерными особенностями клеток, происходящих в ходе гаструляции от эпибласта, и формирующим желудочно-кишечный тракт и его производные. Клетки сформированной эндодермы экспрессируют следующие маркеры: HNF-3 beta, GATA4, SOX-17, церберус, OTX2, гузекоид, C-Kit, CD99 или MIXL1.
Используемый в настоящей заявке термин «внеэмбриональная эндодерма» относится к популяции клеток, экспрессирующих по меньшей мере один из следующих маркеров: SOX7, AFP или SPARC.
Используемый в настоящей заявке термин «маркеры» означает молекулы нуклеиновых кислот или полипептидов с дифференциальной экспрессией в интересующих клетках. В данном контексте под дифференциальной экспрессией подразумевается повышение уровня экспрессии для положительного маркера и понижение уровня экспрессии для отрицательного маркера. Поддающийся обнаружению уровень маркерной нуклеиновой кислоты или полипептида в интересующих клетках оказывается значительно выше или ниже по сравнению с другими клетками, что позволяет идентифицировать интересующую клетку и отличить ее от других клеток с помощью любого из множества известных в данной области способов.
Используемый в настоящей заявке термин «клетка мезэндодермы» относится к клеткам, экспрессирующим по меньшей мере один из следующих маркеров: CD48, эомезодермин (EOMES), SOX17, DKK4, HNF-3 beta, GSC, FGF17 или GATA-6.
Используемый в настоящей заявке термин «панкреатическая эндокринная клетка» или «панкреатическая экспрессирующая гормоны клетка» относится к клеткам, способным к экспрессии по меньшей мере одного из следующих гормонов: инсулин, глюкагон, соматостатин и панкреатический полипептид.
Используемые в настоящей заявке термины «клетка панкреатической эндодермы» или «клетки стадии 4», или «стадия 4» относятся к клеткам, способным к экспрессии по меньшей мере одного из следующих маркеров: NGN3, NEUROD, ISL1, PDX1, PAX4 или NKX2.2.
Используемый в настоящей заявке термин «панкреатическая продуцирующая гормоны клетка» относится к клеткам, способным производить по меньшей мере один из следующих гормонов: инсулин, глюкагон, соматостатин и панкреатический полипептид.
Используемый в настоящей заявке термин «панкреатическая секретирующая гормоны клетка» относится к клеткам, способным к секреции по меньшей мере одного из следующих гормонов: инсулин, глюкагон, соматостатин и панкреатический полипептид.
Используемые в настоящей заявке термины «клетка задней части передней кишки» или «клетки стадии 3», или «стадия 3» относятся к клеткам, способным к секреции по меньшей мере одного из следующих маркеров: PDX1, HNF1, PTF-1 alpha, HNF6, HB-9 или PROX-1.
Используемый в настоящей заявке термин «клетка-предшественник клетки первичной полоски» относится к клеткам, экспрессирующим по меньшей мере один из следующих маркеров: Nodal и FGF8.
Используемые в настоящей заявке термины «клетка первичной кишечной трубки» или «клетки стадии 2», или «стадия 2» относятся к клеткам, способным к секреции по меньшей мере одного из следующих маркеров: HNF1, HNF-4 alpha.
Используемый в настоящей заявке термин «клетка первичной полоски» относится к клеткам, экспрессирующим по меньшей мере один из следующих маркеров: Brachyury, гомеобоксный белок Mix-like или FGF4.
Выделение, размножение и культивирование плюрипотентных стволовых клеток
Характеристика плюрипотентных стволовых клеток
Плюрипотентность плюрипотентных стволовых клеток может быть подтверждена, например, путем инъекции клеток мышам с тяжелым комбинированным иммунодефицитом (SCID), фиксирования образующихся тератом с помощью 4% параформальдегида и последующего их гистологического исследования для получения доказательств наличия клеточных типов, происходящих от трех зародышевых листков. В качестве альтернативы плюрипотентность можно определить по созданию эмбриоидных телец и анализа их на предмет присутствия маркеров, ассоциирующихся с тремя зародышевыми листками.
Выращенные линии плюрипотентных стволовых клеток могут быть кариотипированы с применением стандартного способа окрашивания с использованием красителя Гимза (G-banding) и сравнения с опубликованными кариотипами соответствующих видов приматов. Желательно получить клетки, имеющие «нормальный кариотип», т.е. эуплоидные клетки, в которых все человеческие хромосомы присутствуют и не имеют видимых изменений.
Источники плюрипотентных стволовых клеток
К типам плюрипотентных стволовых клеток, которые можно использовать, относятся устойчивые линии плюрипотентных клеток, получаемые из формируемой после вынашивания плода ткани, в том числе из преэмбриональной ткани (такой как бластоциста), эмбриональной ткани или ткани плода, взятой в любой момент в ходе вынашивания, как правило, но не обязательно, до срока приблизительно 10-12 недель беременности. Неограничивающими настоящее изобретение примерами являются устойчивые линии эмбриональных стволовых клеток человека или эмбриональных зародышевых клеток человека, например линии эмбриональных стволовых клеток человека H1, H7 и H9 (WiCell). Также возможно использование описываемых в настоящей заявке составов в ходе первоначального установления или стабилизации таких клеток, в этом случае исходными клетками являются первичные плюрипотентные клетки, взятые напрямую из тканей-источников. Также соответствуют целям настоящего изобретения клетки, взятые из популяции плюрипотентных стволовых клеток, уже культивированных в отсутствие питающих клеток. Также соответствуют целям настоящего изобретения клетки мутантных линий эмбриональных стволовых клеток человека, таких как, например, BG01v (BresaGen, Атенс, Джорджия, США).
В одном из вариантов осуществления эмбриональные стволовые клетки человека готовят как описано в следующих публикациях Thomson et al. (патент США № 5843780; Science 282:1145, 1998; Curr. Top. Dev. Biol. 38:133 ff., 1998; Proc. Natl. Acad. Sci. U.S.A. 92:7844, 1995).
В одном из вариантов осуществления эмбриональные стволовые клетки человека готовят как описано в публикации Takahashi et al. (Cell 131: 1-12, 2007).
Культивирование плюрипотентных стволовых клеток
В одном из вариантов осуществления плюрипотентные стволовые клетки, как правило, культивируют на слое питающих клеток, которые поддерживают плюрипотентные клетки в различных отношениях. В качестве альтернативы плюрипотентные стволовые клетки культивируют в культуральной системе, существенно свободной от питающих клеток, но, тем не менее, способной поддерживать пролиферацию плюрипотентных стволовых клеток без существенного дифференцирования. Рост плюрипотентных стволовых клеток в свободной от питающих клеток культуральной системе без дифференцирования поддерживается путем использования среды, кондиционированной посредством предварительного культивирования клеток иного типа. В качестве альтернативы рост плюрипотентных стволовых клеток в свободной от питающих клеток культуральной системе без дифференцирования поддерживается путем использования среды с химически определенным составом.
Плюрипотентные стволовые клетки могут быть высеяны на соответствующий культуральный субстрат. В одном из вариантов осуществления соответствующим культуральным субстратом является компонент внеклеточного матрикса, такой как, например, полученный из базальной мембраны, или тот, который может участвовать в лиганд-рецепторном взаимодействии с участием молекулы адгезивного слоя. В одном из вариантов осуществления соответствующим культуральным субстратом является MATRIGEL® (Becton Dickenson). MATRIGEL® представляет собой растворимый препарат из клеток опухоли Engelbreth-Holm-Swarm, который при комнатной температуре превращается в гель, образуя восстановленную базальную мембрану.
В качестве альтернативы можно использовать другие компоненты внеклеточного матрикса и смеси компонентов. В зависимости от типа пролиферирующих клеток, это может быть ламинин, фибронектин, протеогликан, энтактин, гепарансульфат и т.п., по отдельности или в различных сочетаниях.
Плюрипотентные стволовые клетки могут высеиваться на субстрат с соответствующим распределением по поверхности и в присутствии среды, поддерживающей выживание, размножение и сохранение требуемых характеристик клеток. Все эти характеристики улучшаются при тщательном подходе к распределению клеток при посеве и могут быть определены специалистом в данной области.
Соответствующая целям настоящего изобретения культуральная среда может быть приготовлена из следующих компонентов, таких как, например, модифицированная по способу Дульбекко среда Игла (DMEM), Gibco № 11965-092; модифицированная по способу Дульбекко нокаут-среда Игла (KO DMEM), Gibco № 10829-018; основная среда Хэма F12/50% DMEM; 200 мМ L-глутамина, Gibco № 15039-027; раствор заменимых аминокислот, Gibco 11140-050; β-меркаптоэтанол, Sigma № M7522; человеческий рекомбинантный основной фактор роста фибробластов (bFGF), Gibco № 13256-029.
Образование панкреатических продуцирующих гормоны клеток из плюрипотентных стволовых клеток
В одном из вариантов осуществления настоящего изобретения предлагается способ получения панкреатических продуцирующих гормоны клеток из плюрипотентных стволовых клеток, включающий:
а. культивирование плюрипотентных стволовых клеток;
b. дифференцирование плюрипотентных стволовых клеток в клетки, экспрессирующие маркеры, характерные для линии сформированной эндодермы;
c. дифференцирование клеток, экспрессирующих маркеры, характерные для линии сформированной эндодермы, в клетки, экспрессирующие маркеры, характерные для линии дифференцирования в клетки панкреатической эндодермы; и
d. дифференцирование клеток, экспрессирующих маркеры, характерные для линии дифференцирования в клетки панкреатической эндодермы, в клетки, экспрессирующие маркеры, характерные для линии дифференцирования в панкреатические эндокринные клетки.
В одном из аспектов настоящего изобретения панкреатическая эндокринная клетка представляет собой панкреатическую продуцирующую гормоны клетку. В другом аспекте настоящего изобретения упомянутая панкреатическая эндокринная клетка представляет собой клетку, экспрессирующую маркеры, характерные для β-клеточной линии дифференцирования. Клетка, экспрессирующая маркеры, характерные для β-клеточной линии дифференцирования, экспрессирует PDX1 и по меньшей мере один из следующих факторов транскрипции: NGN3, NKX2.2, NKX6.1, NEUROD, ISL1, HNF-3 beta, MAFA, PAX4 или Pax6. В одном из аспектов настоящего изобретения клетка, экспрессирующая маркеры, характерные для β-клеточной линии дифференцирования представляет собой β-клетку.
Плюрипотентные стволовые клетки, соответствующие целям настоящего изобретения, включают, например, эмбриональные стволовые клетки человека линии H9 (код NIH: WA09), эмбриональные стволовые клетки человека линии H1 (код NIH: WA01), эмбриональные стволовые клетки человека линии H7 (код NIH: WA07) и эмбриональные стволовые клетки человека линии SA002 (Cellartis, Швеция). Также соответствуют целям настоящего изобретения клетки, которые экспрессируют по меньшей мере один из следующих маркеров плюрипотентности: ABCG2, крипто, CD9, FOXD3, коннексин43, коннексин45, OCT4, SOX2, Nanog, hTERT, UTF-1, ZFP42, SSEA-3, SSEA-4, Tra1-60 или Tra1-81.
Плюрипотентные стволовые клетки могут культивироваться на слое питающих клеток. В качестве альтернативы плюрипотентные стволовые клетки могут культивироваться на внеклеточном матриксе. Внеклеточный матрикс может представлять собой растворимую базальную мембрану, полученную экстракцией из клеток саркомы мыши (поставляется компанией BD Biosciences под торговым наименованием MATRIGELTM). В качестве альтернативы внеклеточный матрикс может представлять собой MATRIGELTM с пониженным содержанием фактора роста. В качестве альтернативы внеклеточный матрикс может представлять собой фибронектин. В альтернативном варианте осуществления плюрипотентные стволовые клетки культивируются и дифференцируются на культуральном субстрате, на который нанесен слой человеческой сыворотки.
Внеклеточный матрикс может быть растворен перед нанесением слоя на культуральный субстрат. Примеры соответствующих способов растворения внеклеточного матрикса и нанесения слоя на культуральный субстрат можно найти в публикациях H.K. Kleinman et al., Biochemistry 25:312 (1986) и M.A. Hadley et al., J.Cell.Biol. 101:1511 (1985).
В одном из вариантов осуществления внеклеточный матрикс представляет собой MATRIGELTM. В одном из вариантов осуществления на культуральный субстрат наносится слой MATRIGELTM в концентрации 1:10. В альтернативном варианте осуществления на культуральный субстрат наносится слой MATRIGELTM в концентрации 1:15. В альтернативном варианте осуществления на культуральный субстрат наносится слой MATRIGELTM в концентрации 1:30. В альтернативном варианте осуществления на культуральный субстрат наносится слой MATRIGELTM в концентрации 1:60.
В одном из вариантов осуществления внеклеточный матрикс представляет собой MATRIGELTM с пониженным содержанием фактора роста. В одном из вариантов осуществления на культуральный субстрат наносится слой MATRIGELTM с пониженным содержанием фактора роста в концентрации 1:10. В альтернативном варианте осуществления на культуральный субстрат наносится слой MATRIGELTM с пониженным содержанием фактора роста в концентрации 1:15. В альтернативном варианте осуществления на культуральный субстрат наносится слой MATRIGELTM с пониженным содержанием фактора роста в концентрации 1:30. В альтернативном варианте осуществления на культуральный субстрат наносится слой MATRIGELTM с пониженным содержанием фактора роста в концентрации 1:60.
Маркеры, характерные для линии сформированной эндодермы, выбраны из группы, включающей следующие маркеры: SOX17, GATA4, HNF-3 beta, GSC, CER1, Nodal, FGF8, Brachyury, гомеобоксный белок Mix-like, FGF4 CD48, эомезодермин (EOMES), DKK4, FGF17, GATA6, CXCR4, C-Kit, CD99 и OTX2. Соответствующей целям настоящего изобретения является клетка, которая экспрессирует по меньшей мере один из маркеров, характерных для линии сформированной эндодермы. В одном из аспектов настоящего изобретения клетка, экспрессирующая маркеры, характерные для линии сформированной эндодермы, представляет собой клетку-предшественник первичной полоски. В другом аспекте настоящего изобретения клетка, экспрессирующая маркеры, характерные для линии сформированной эндодермы, представляет собой мезэндодермальную клетку. В другом аспекте настоящего изобретения клетка, экспрессирующая маркеры, характерные для линии сформированной эндодермы, представляет собой клетку сформированной эндодермы.
Маркеры, характерные для линии панкреатической эндодермы, выбраны из группы, включающей следующие маркеры: PDX1, HNF-1 beta, PTF1 alpha, HNF6, HB9 и PROX1. Соответствующей целям настоящего изобретения является клетка, которая экспрессирует по меньшей мере один из маркеров, характерных для линии панкреатической эндодермы. В одном из аспектов настоящего изобретения клетка, экспрессирующая маркеры, характерные для линии панкреатической эндодермы, представляет собой клетку панкреатической эндодермы.
Маркеры, характерные для линии панкреатических эндокринных клеток, выбраны из группы, включающей следующие маркеры: NGN3, NEUROD, ISL1, PDX1, NKX6.1, PAX4 и PTF-1 alpha. В одном из вариантов осуществления панкреатическая эндокринная клетка способна экспрессировать по меньшей мере один из следующих гормонов: инсулин, глюкагон, соматостатин и панкреатический полипептид. Соответствующей целям настоящего изобретения является клетка, экспрессирующая по меньшей мере один из маркеров, характерных для линии панкреатических эндокринных клеток. В одном из аспектов настоящего изобретения клетка, экспрессирующая маркеры, характерные для линии панкреатических эндокринных клеток, представляет собой панкреатическую эндокринную клетку. Панкреатическая эндокринная клетка может представлять собой панкреатическую экспрессирующую гормоны клетку. В качестве альтернативы панкреатическая эндокринная клетка может представлять собой панкреатическую секретирующую гормоны клетку.
Образование клеток, экспрессирующих маркеры, характерные для линии сформированной эндодермы
В одном из аспектов настоящего изобретения плюрипотентные стволовые клетки могут быть дифференцированы в клетки, экспрессирующие маркеры, характерные для линии сформированной эндодермы, путем культивирования плюрипотентных стволовых клеток в среде, содержащей достаточное количество GDF-8 для стимуляции дифференцирования плюрипотентных стволовых клеток в клетки, экспрессирующие маркеры, характерные для линии сформированной эндодермы.
Плюрипотентные стволовые клетки могут культивироваться в среде, содержащей достаточное количество GDF-8, в течение от приблизительно одного до приблизительно семи дней. В качестве альтернативы плюрипотентные стволовые клетки могут культивироваться в среде, содержащей достаточное количество GDF-8, в течение от приблизительно одного до приблизительно шести дней. В качестве альтернативы плюрипотентные стволовые клетки могут культивироваться в среде, содержащей достаточное количество GDF-8, в течение от приблизительно одного до приблизительно пяти дней. В качестве альтернативы плюрипотентные стволовые клетки могут культивироваться в среде, содержащей достаточное количество GDF-8, в течение от приблизительно одного до приблизительно четырех дней. В качестве альтернативы плюрипотентные стволовые клетки могут культивироваться в среде, содержащей достаточное количество GDF-8, в течение от приблизительно одного до приблизительно трех дней. В качестве альтернативы плюрипотентные стволовые клетки могут культивироваться в среде, содержащей достаточное количество GDF-8, в течение от приблизительно одного до приблизительно двух дней. В качестве альтернативы плюрипотентные стволовые клетки могут культивироваться в среде, содержащей достаточное количество GDF-8, в течение приблизительно одного дня.
В одном из вариантов осуществления GDF-8 используется в концентрации от приблизительно 5 нг/мл до приблизительно 500 нг/мл. В альтернативном варианте осуществления GDF-8 используется в концентрации от приблизительно 5 нг/мл до приблизительно 50 нг/мл. В альтернативном варианте осуществления GDF-8 используется в концентрации от приблизительно 5 нг/мл до приблизительно 25 нг/мл. В альтернативном варианте осуществления GDF-8 используется в концентрации приблизительно 25 нг/мл.
В одном из вариантов осуществления среда, содержащая достаточное количество GDF-8, дополнительно содержит по меньшей мере один другой фактор. В одном из вариантов осуществления указанный по меньшей мере один другой фактор выбран из группы, включающей следующие факторы: EGF, FGF4, PDGF-A, PDGF-B, PDGF-C, PDGF-D, VEGF, мусцимол, PD98059, LY294002, U0124, U0126 и натрия бутират.
В одном из вариантов осуществления EGF используется в концентрации от приблизительно 5 нг/мл до приблизительно 500 нг/мл. В альтернативном варианте осуществления EGF используется в концентрации от приблизительно 5 нг/мл до приблизительно 50 нг/мл. В альтернативном варианте осуществления EGF используется в концентрации приблизительно 50 нг/мл.
В одном из вариантов осуществления FGF4 используется в концентрации от приблизительно 5 нг/мл до приблизительно 500 нг/мл. В альтернативном варианте осуществления FGF4 используется в концентрации от приблизительно 5 нг/мл до приблизительно 50 нг/мл. В альтернативном варианте осуществления FGF4 используется в концентрации приблизительно 50 нг/мл.
В одном из вариантов осуществления PDGF-A используется в концентрации от приблизительно 5 нг/мл до приблизительно 500 нг/мл. В альтернативном варианте осуществления PDGF-A используется в концентрации от приблизительно 5 нг/мл до приблизительно 50 нг/мл. В альтернативном варианте осуществления PDGF-A используется в концентрации приблизительно 50 нг/мл.
В одном из вариантов осуществления PDGF-B используется в концентрации от приблизительно 5 нг/мл до приблизительно 500 нг/мл. В альтернативном варианте осуществления PDGF-B используется в концентрации от приблизительно 5 нг/мл до приблизительно 50 нг/мл. В альтернативном варианте осуществления PDGF-B используется в концентрации приблизительно 50 нг/мл.
В одном из вариантов осуществления PDGF-C используется в концентрации от приблизительно 5 нг/мл до приблизительно 500 нг/мл. В альтернативном варианте осуществления PDGF-C используется в концентрации от приблизительно 5 нг/мл до приблизительно 50 нг/мл. В альтернативном варианте осуществления PDGF-C используется в концентрации приблизительно 50 нг/мл.
В одном из вариантов осуществления PDGF-D используется в концентрации от приблизительно 5 нг/мл до приблизительно 500 нг/мл. В альтернативном варианте осуществления PDGF-D используется в концентрации от приблизительно 5 нг/мл до приблизительно 50 нг/мл. В альтернативном варианте осуществления PDGF-D используется в концентрации приблизительно 50 нг/мл.
В одном из вариантов осуществления VEGF используется в концентрации от приблизительно 5 нг/мл до приблизительно 500 нг/мл. В альтернативном варианте осуществления VEGF используется в концентрации от приблизительно 5 нг/мл до приблизительно 50 нг/мл. В альтернативном варианте осуществления VEGF используется в концентрации приблизительно 50 нг/мл.
В одном из вариантов осуществления мусцимол используется в концентрации от приблизительно 1 мкМ до приблизительно 200 мкМ. В альтернативном варианте осуществления мусцимол используется в концентрации от приблизительно 1 мкМ до приблизительно 20 мкМ. В альтернативном варианте осуществления мусцимол используется в концентрации приблизительно 20 мкМ.
В одном из вариантов осуществления PD98059 используется в концентрации от приблизительно 0,1 мкМ до приблизительно 10 мкМ. В альтернативном варианте осуществления PD98059 используется в концентрации от приблизительно 0,1 мкМ до приблизительно 1 мкМ. В альтернативном варианте осуществления PD98059 используется в концентрации приблизительно 1 мкМ.
В одном из вариантов осуществления LY294002 используется в концентрации от приблизительно 0,25 мкМ до приблизительно 25 мкМ. В альтернативном варианте осуществления LY294002 используется в концентрации от приблизительно 0,25 мкМ до приблизительно 2,5 мкМ. В альтернативном варианте осуществления LY294002 используется в концентрации приблизительно 2,5 мкМ.
В одном из вариантов осуществления U0124 используется в концентрации от приблизительно 0,1 мкМ до приблизительно 10 мкМ. В альтернативном варианте осуществления U0124 используется в концентрации от приблизительно 0,1 мкММ до приблизительно 1 мкМ. В альтернативном варианте осуществления U0124 используется в концентрации приблизительно 1 мкМ.
В одном из вариантов осуществления U0126 используется в концентрации от приблизительно 0,1 мкМ до приблизительно 10 мкМ. В альтернативном варианте осуществления U0126 используется в концентрации от приблизительно 0,1 мкМ до приблизительно 1 мкМ. В альтернативном варианте осуществления U0126 используется в концентрации приблизительно 1 мкМ.
В одном из вариантов осуществления бутират натрия используется в концентрации от приблизительно 0,05 мкМ до приблизительно 5 мкМ. В альтернативном варианте осуществления бутират натрия используется в концентрации от приблизительно 0,05 мкМ до приблизительно 0,5 мкМ. В альтернативном варианте осуществления бутират натрия используется в концентрации приблизительно 0,5 мкМ.
В альтернативном варианте осуществления указанный по меньшей мере один другой фактор выбран из группы, включающей: анилин-пиридинотриазин, циклический анилин-пиридинотриазин, N-{[1-(фенилметил)азепан-4-ил]метил}-2-пиридин-3-илацетамид, 4-{[4-(4-{[2-(пиридин-2-иламино)этил]амино}-1,3,5-триазин-2-ил)пиридин-2-ил]окси}бутан-1-ол, 3-({3-[4-({2-[метил(пиридин-2-ил)амино]этил}амино)-1,3,5-триазин-2-ил]пиридин-2-ил}амино)пропан-1-ол, N~4~-[2-(3-фторфенил)этил]-N~2~-[3-(4-метилпиперазин-1-ил)пропил]пиридо[2,3-d]пиримидин-2,4-диамин, 1-метил-N-[(4-пиридин-3-ил-2-{[3-(трифторметил)фенил]амино}-1,3-тиазол-5-ил)метил]пиперидин-4-карбоксамид, 1,1-диметилэтил {2-[4-({5-[3-(3-гидроксипропил)фенил]-4H-1,2,4-триазол-3-ил}амино)фенил]этил}карбамат, 1,1-диметилэтил {[3-({5-[5-(3-гидроксипропил)-2-(метилокси)фенил]-1,3-оксазол-2-ил}амино)фенил]метил}карбамат, 1-({5-[6-({4-[(4-метилпиперазин-1-ил)сульфонил]фенил}амино)пиразин-2-ил]тиофен-2-ил}метил)пиперидин-4-ол, 1-({4-[6-({4-[(4-метилпиперазин-1-ил)сульфонил]фенил}амино)пиразин-2-ил]тиофен-2-ил}метил)пиперидин-4-карбоксамид и 2-{[4-(1-метилэтил)фенил]амино}-N-(2-тиофен-2-илэтил)-7,8-дигидропиридо[4,3-d]пиримидин-6(5H)-карбоксамид.
Соединения, составляющие предмет настоящего изобретения
Настоящее изобретение предлагает соединения, способные дифференцировать плюрипотентные стволовые клетки в клетки, экспрессирующие маркеры, характерные для линии сформированной эндодермы.
В одном из вариантов осуществления соединение, способное дифференцировать плюрипотентные стволовые клетки в клетки, экспрессирующие маркеры, характерные для линии сформированной эндодермы, представляет собой анилин-пиридинотриазин формулы (1):
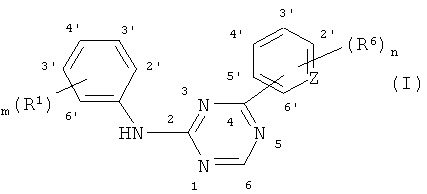 Формула (1)
Формула (1)
N-оксидные формы, фармацевтически приемлемые аддитивные соли и стереохимические изомерные формы, где:
m представляет собой целое число от 1 до 4; n представляет собой целое число от 1 до 4; Z представляет собой атом N или C;
R1 и R8 каждый независимо представляет собой водород, Het14, цианогруппу, галоген, гидроксигруппу, C1-6алкоксигруппу, C1-6алкил, моно-или ди(C1-4алкил)аминокарбонил, моно- или ди(C1-4алкил)аминосульфонил, галогензамещенную C1-6алкоксигруппу, или R1 представляет собой C1-6алкил, замещенный одним или, если возможно, двумя и более заместителями, выбираемыми из гидроксигруппы или галогена;
R2 и R9 каждый независимо представляет собой водород, C1-4алкил, C2-4алкенил, Het3, Het4-C1-4алкил, Het5-C1-4алкилкарбонил, моно- или ди(C1-4алкил)амино-C1-4алкилкарбонил или -фенил, необязательно замещенный одним или, если возможно, двумя и более заместителями, выбираемыми из водорода, гидроксигруппы, аминогруппы или C1-4алкилоксигруппы;
R3 и R7 каждый независимо представляет собой водород, C1-4алкил, Het6, Het7-C1-4алкил, C2-4алкенилкарбонил, необязательно замещенный Het8-C1-4алкиламинокарбонилом, C2-4алкенилсульфонил, C1-4алкилокси-C1-4алкил- или фенил, необязательно замещенный одним или, если возможно, двумя и более заместителями, выбираемыми из водорода, гидроксигруппы, аминогруппы или C1-4алкилоксигруппы;
R4, R5, R6 и R10 каждый независимо представляет собой водород или C1-4алкил, необязательно замещенный гидроксигруппой, Het9 или C1-4алкилоксигруппой;
Het1 и Het2 каждый независимо представляет собой гетероцикл, выбираемый из пирролидинила, пиперидинила, пиперазинила, пиридинила, пиримидинила, пиразинила, имидазолидинила или пиразолидинила, где указанные Het1 и Het2 необязательно замещены аминогруппой, гидроксигруппой, C1-4алкилом, гидрокси-C1-4алкилом, фенилом, фенил-C1-4алкилом, C1-4алкилокси-C1-4алкил-моно- или ди(C1-4алкил)амино- или аминокарбонильной группой;
Het3 и Het6 каждый независимо представляет собой гетероцикл, выбираемый из пирролидинила или пиперидинила, где указанные Het3 и Het6 необязательно замещены одним или, если возможно, двумя и более заместителями, выбираемыми из C1-4алкила, C3-6циклоалкила, гидрокси-C1-4алкила, C1-4алкилокси-C1-4алкила или полигидрокси-C1-4алкила;
Het4, Het7 и Het9 каждый независимо представляет собой гетероцикл, выбираемый из морфолинила, пирролидинила, пиперазинила или пиперидинила, где упомянутые Het4, Het7 и Het9 необязательно замещены одним или, если возможно, двумя и более заместителями, выбираемыми из C1-4алкила, C3-6циклоалкила, гидрокси-C1-4алкила, C1-4алкилокси-C1-4алкила или полигидрокси-C1-4алкила;
Het5 представляет собой гетероцикл, выбираемый из морфолинила, пирролидинила, пиперазинила или пипендинила, где упомянутый Het5 необязательно замещен одним или, если возможно, двумя и более заместителями, выбираемыми из C1-4алкила, C3-6циклоалкила, гидрокси-C1-4алкила, C1-4алкилокси-C1-4алкила или полигидрокси-C1-4алкила;
Het10, Het11 и Het13 каждый независимо представляет собой гетероцикл, выбираемый из пирролидинила, пиперидинила, пиперазинила, пиридинила, пиримидинила, пиразинила, имидазолидинила или пиразолидинила, где указанные Het10, Het11 и Het13 необязательно замещены аминогруппой, гидроксигруппой, C1-4алкилом, гидрокси-C1-4алкилом, фенилом, фенил-C1-4алкилом, C1-4алкилокси-C1-4алкилом, аминокарбонилом или моно- или ди(C1-4алкил)аминогруппой;
Het12 представляет собой гетероцикл, выбираемый из пирролидинила, пиперидинила, пиперазинила, пиридинила, пиримидинила, пиразинил, имидазолидинила или пиразолидинила, где указанный Het12 необязательно замещен аминогруппой, гидроксигруппой, C1-4алкилом, гидрокси-C1-4алкилом, фенилом, фенил-C1-4алкилом, C1-4алкилокси-C1-4алкилом; моно- или ди(C1-4алкил)амино- или аминокарбонильной группой;
Het14 представляет собой гетероцикл, выбираемый из морфолинила, пирролидинила, пиперазинила, имидазолила, пирролила, 2,3,4-триазапирролила, 1,2,3-триазолила, пиразолила или пиперидинила, где указанный Het14 необязательно замещен одним или, если возможно, двумя и более заместителями, выбираемыми из C1-4алкила, C3-6циклоалкила, гидрокси-C1-4алкила, C1-4алкокси-C1-4алкила или полигидрокси-C1-4алкила; в частности, Het14 представляет собой гетероцикл, выбираемый из морфолинила, пирролидинила, пирролила, 2,3,4-триазапирролила, пиперазинила или пиперидинила, где упомянутый Het14 необязательно замещен одним или, если возможно, двумя и более заместителями, выбираемыми из C1-4алкила, C3-6циклоалкила, гидрокси-C1-4алкила, C1-4алкокси-C1-4алкила или полигидрокси-C1-4алкила; более конкретно, Het14 представляет собой гетероцикл, выбираемый из морфолинила, пирролидинила, пиперазинила или пиперидинила, где упомянутый Het14 необязательно замещен одним или, если возможно, двумя и более заместителями, выбираемыми из C1-4алкила, C3-6циклоалкила, гидрокси-C1-4алкила, C1-4алкилокси-C1-4алкила или полигидрокси-C1-4алкила.
В одном из вариантов осуществления анилин-пиридинотриазин представляет собой соединение формулы (1).
В одном из вариантов осуществления анилин-пиридинотриазин представляет собой соединение формулы (2).
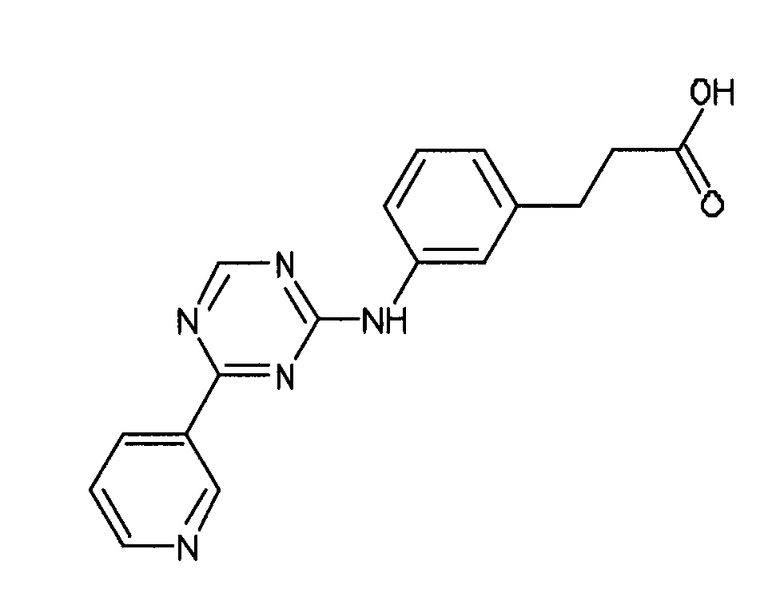
Формула (2): 3-{3-[(4-пиридин-3-ил-1,3,5-триазин-2-ил)амино]фенил}пропановая кислота, именуемая в настоящем документе «Соединение 1».
В одном из вариантов осуществления анилин-пиридинотриазин представляет собой соединение формулы (3).
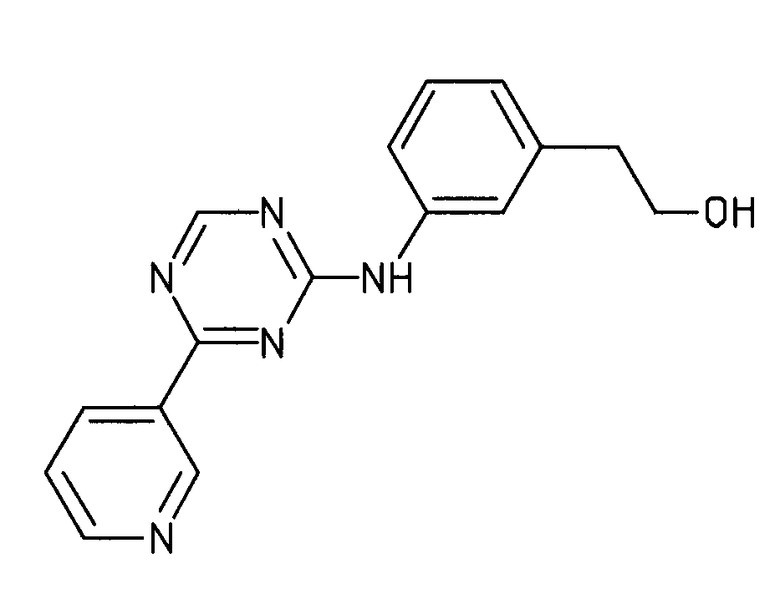
Формула (3): 2-{3-[(4-пиридин-3-ил-1,3,5-триазин-2-ил)амино]фенил}этанол, именуемый в настоящем документе «Соединение 2».
В одном из вариантов осуществления анилин-пиридинотриазин представляет собой соединение формулы (4).
Формула (4): 1,1-диметилэтил {2-[3-({4-[2-(3-гидроксипроп-1-ин-1-ил)пиридин-4-ил]-1,3,5-триазин-2-ил}амино)фенил]этил}карбамат, именуемый в настоящем документе «Соединение 3».
В одном из вариантов осуществления анилин-пиридинотриазин представляет собой соединение формулы (5).
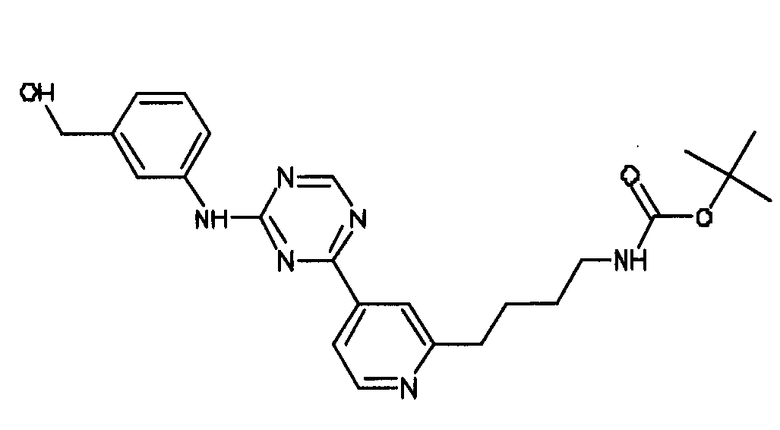
Формула (5): 1,1-диметилэтил {4-[4-(4-{[3-(гидроксиметил)фенил]амино}-1,3,5-триазин-2-ил)пиридин-2-ил]бутил}карбамат, именуемый в настоящем документе «Соединение 4».
В одном из вариантов осуществления анилин-пиридинотриазин представляет собой соединение формулы (6).
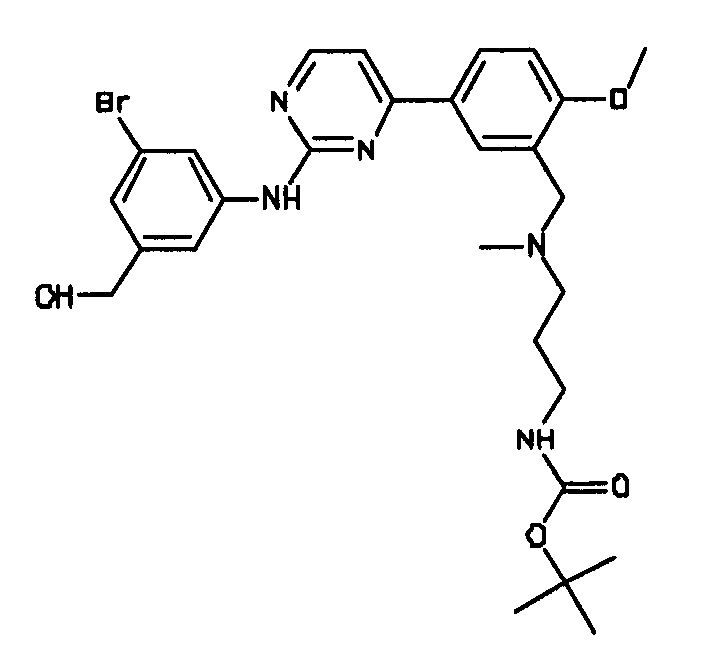
Формула (6): 1,1-диметилэтил {3-[{[5-(2-{[3-бром-5-(гидроксиметил)фенил]амино}пиримидин-4-ил)-2-(метилокси)фенил]метил}(метил)амино]пропил}карбамат, именуемый в настоящем документе «Соединение 5».
В одном из вариантов осуществления анилин-пиридинотриазин представляет собой соединение формулы (7).
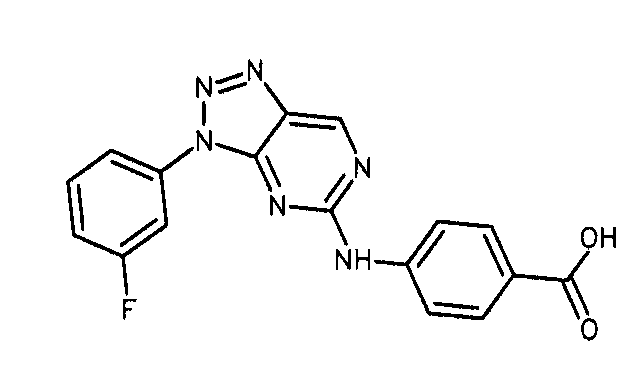
Формула (7): 4-{[3-(3-фторфенил)-3H-[1,2,3]триазоло[4,5-d]пиримидин-5-ил]амино}бензойная кислота, именуемая в настоящем документе «Соединение 6».
В одном из вариантов осуществления анилин-пиридинотриазин представляет собой соединение формулы (8).
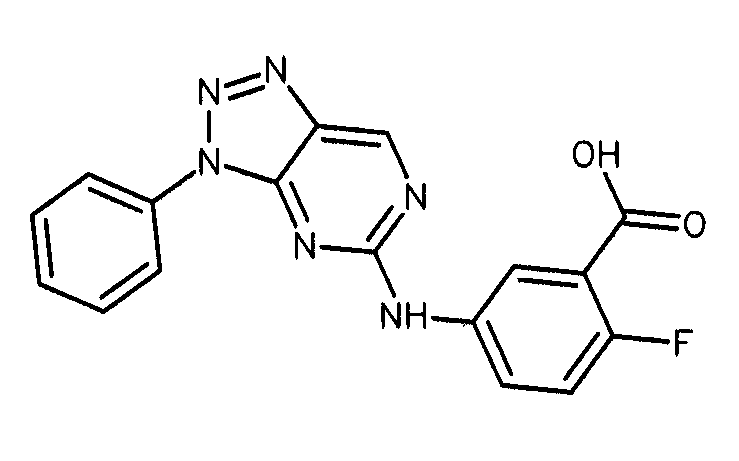
Формула (8): 2-фтор-5-[(3-фенил-3H-[1,2,3]триазоло[4,5-d]пиримидин-5-ил)амино]бензойная кислота, именуемая в настоящем документе «Соединение 7».
В одном из вариантов осуществления анилин-пиридинотриазин представляет собой соединение формулы (9).
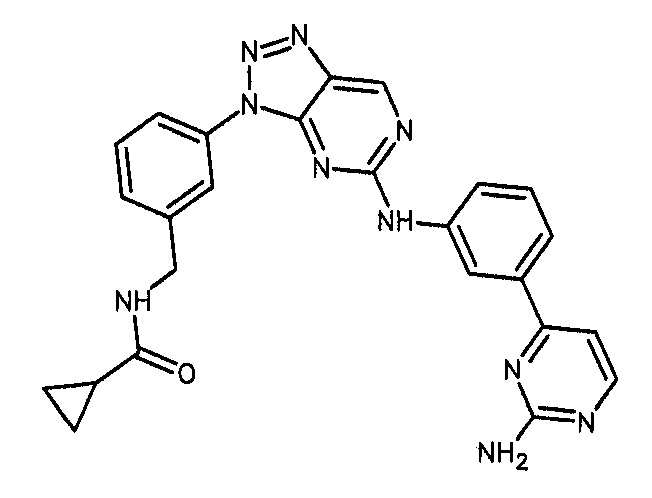
Формула (9): N-{[3-(5-{[3-(2-аминопиримидин-4-ил)фенил]амино}-3H-[1,2,3]триазоло[4,5-d]пиримидин-3-ил)фенил]метил}циклопропанкарбоксамид, именуемый в настоящем документе «Соединение 8».
В одном из вариантов осуществления анилин-пиридинотриазин представляет собой соединение формулы (10).
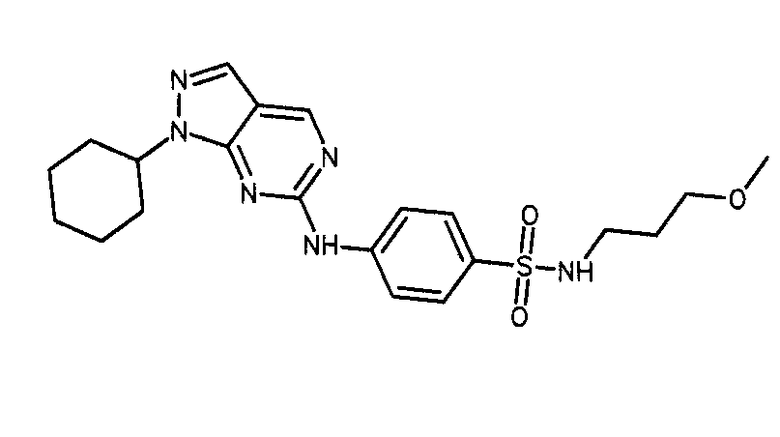
Формула (10): 4-[(1-циклогексил-1H-пиразоло[3,4-d]пиримидин-6-ил)амино]-N-[3-(метилокси)пропил]бензолсульфонамид, именуемый в настоящем документе «Соединение 9».
В одном из вариантов осуществления анилин-пиридинотриазин представляет собой соединение формулы (11).
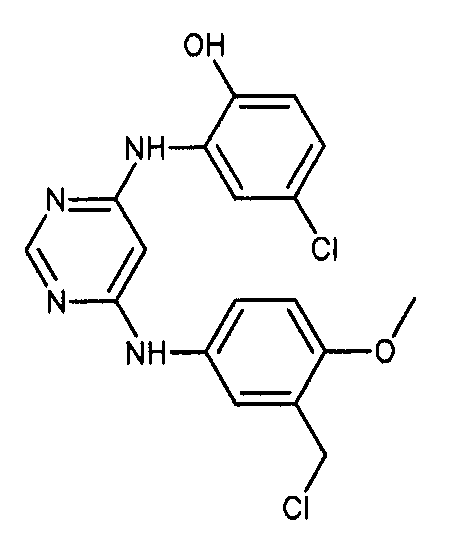
Формула (11): 4-хлор-2-[(6-{[3-(хлорметил)-4-метоксифенил]амино}пиримидин-4-ил)амино]фенол, именуемый в настоящем документе «Соединение 10».
В одном из вариантов осуществления анилин-пиридинотриазин представляет собой соединение формулы (12).
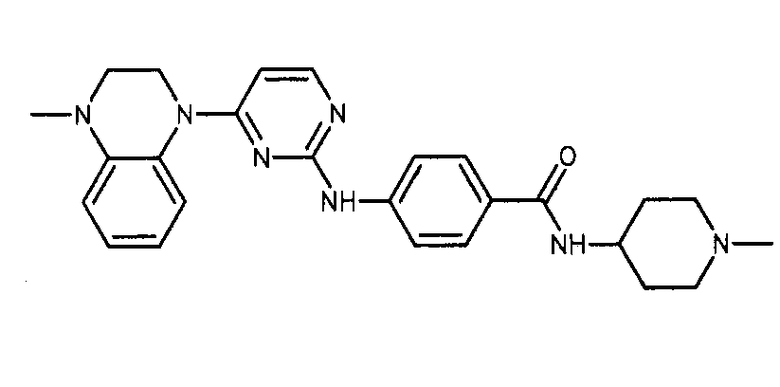
Формула (12): 4-{[4-(4-метил-3,4-дигидрохиноксалин-1(2H)-ил)пиримидин-2-ил]амино}-N-(1-метилпиперидин-4-ил)бензамид, именуемый в настоящем документе «Соединение 11».
В одном из вариантов осуществления анилин-пиридинотриазин представляет собой соединение формулы (13).
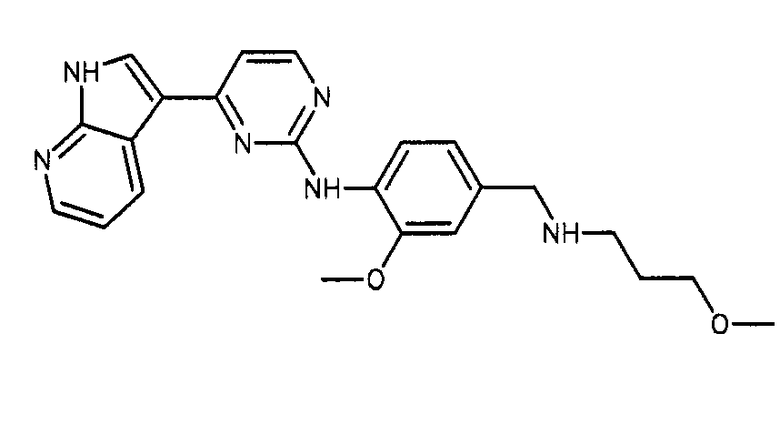
Формула (13): N-(2-метокси-4-{[(3-метоксипропил)амино]метил}фенил)-4-(1H-пирроло[2,3-b]пиридин-3-ил)пиримидин-2-амин, именуемый в настоящем документе «Соединение 12».
В одном из вариантов осуществления соединение, способное дифференцировать плюрипотентные стволовые клетки в клетки, экспрессирующие маркеры, характерные для линии сформированной эндодермы, представляет собой циклический анилин-пиридинотриазин формулы (14):
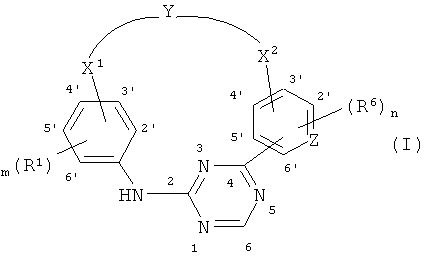
Формула (14)
N-оксидные формы, фармацевтически приемлемые аддитивные соли и стереохимические изомерные формы, где:
m представляет собой целое число от 1 до 4; n представляет собой целое число от 1 до 4; Z представляет собой атом N или C;
Y представляет собой -NR2-C1-6алкил-CO-NR4-, -C1-4алкил-NR9-C1-4алкил-, C1-6алкил-CO-Het10-, -Het11-CO-C1-6алкил-, -Het12-C1-6алкил-, -CO-Het13-C1-6алкил-, -CO-NR10-C1-6алкил-, -Het1-C1-6алкил-CO-NR5- или Het2-CO-NR6-, где -C1-6алкильное звено в группе -NR2-C1-6алкил-CO-NR4- или -Het1-C1-6алкил-CO-NR5 необязательно замещено одним или, если возможно, двумя и более заместителями, выбираемыми из гидроксигруппы, метоксигруппы, аминокарбонила, галогена, фенила, индолила, метилсульфида, тиола, гидроксифенила, цианофенила, аминогруппы и гидроксикарбонила;
X1 представляет собой прямую связь, C1-4алкил, C1-4алкоксигруппу, C1-4алкил-CO-, C2-4алкенил, C2-4алкинил или C1-4алкил-NR3-, где указанный C1-4алкил или C2-4алкенил необязательно замещен одним или, если возможно, двумя и более заместителями-галогенами;
X2 представляет собой прямую связь, C1-4алкил, C1-4алкоксигруппу, C1-4алкил-CO-, C2-4алкенил, C2-4алкинил или C1-4алкил-NR7-, где указанный C1-4алкил или C2-4алкенил необязательно замещен одним или, если возможно, двумя и более заместителями-галогенами;
R1 и R8 каждый независимо представляет собой водород, Het14, цианогруппу, галоген, гидроксигруппу, C1-6алкоксигруппу, C1-6алкил, моно-или ди(C1-4алкил)аминокарбонил, моно- или ди(C1-4алкил)аминосульфонил, галогензамещенную C1-6алкоксигруппу, или R1 представляет собой C1-6алкил, замещенный одним или, если возможно, двумя и более заместителями, выбираемыми из гидроксигруппы или галогена;
R2 и R9 каждый независимо представляет собой водород, C1-4алкил, C2-4алкенил, Het3, Het4-C1-4алкил, Het5-C1-4алкилкарбонил, моно- или ди(C1-4алкил)амино-C1-4алкилкарбонил или фенил, необязательно замещенный одним или, если возможно, двумя и более заместителями, выбираемыми из водорода, гидроксигруппы, аминогруппы или C1-4алкилоксигруппы;
R3 и R7 каждый независимо представляет собой водород, C1-4алкил, Het6, Het7-C1-4алкил, C2-4алкенилкарбонил, необязательно замещенный Het8-C1-4алкиламинокарбонилом, C2-4алкенилсульфонил, C1-4алкилокси-C1-4алкил или фенил, необязательно замещенный одним или, если возможно, двумя и более заместителями, выбираемыми из водорода, гидроксигруппы, аминогруппы или C1-4алкилоксигруппы;
R4, R5, R6 и R10 каждый независимо представляет собой водород или C1-4алкил, необязательно замещенный гидроксигруппой, Het9 или C1-4алкилоксигруппой;
Het1 и Het2 каждый независимо представляет собой гетероцикл, выбираемый из пирролидинила, пиперидинила, пиперазинила, пиридинила, пиримидинила, пиразинила, имидазолидинила или пиразолидинила, где указанные Het1 и Het2 необязательно замещены аминогруппой, гидроксигруппой, C1-4алкилом, гидрокси-C1-4алкилом, фенилом, фенил-C1-4алкилом, C1-4алкилокси-C1-4алкил-моно- или ди(C1-4алкил)аминогруппой или аминокарбонильной группой;
Het3 и Het6 каждый независимо представляет собой гетероцикл, выбираемый из пирролидинила или пиперидинила, где указанные Het3 и Het6 необязательно замещены одним или, если возможно, двумя и более заместителями, выбираемыми из C1-4алкила, C3-6циклоалкила, гидрокси-C1-4алкила, C1-4алкилокси-C1-4алкила или полигидрокси-C1-4алкила;
Het4, Het7 и Het9 каждый независимо представляет собой гетероцикл, выбираемый из морфолинила, пирролидинила, пиперазинила или пиперидинила, где указанные Het4, Het7 и Het9 необязательно замещены одним или, если возможно, двумя и более заместителями, выбираемыми из C1-4алкила, C3-6циклоалкила, гидрокси-C1-4алкила, C1-4алкилокси-C1-4алкила или полигидрокси-C1-4алкила;
Het5 представляет собой гетероцикл, выбираемый из морфолинила, пирролидинила, пиперазинила или пипендинила, где указанный Het5 необязательно замещен одним или, если возможно, двумя и более заместителями, выбираемыми из C1-4алкила, C3-6циклоалкила, гидрокси-C1-4алкила, C1-4алкилокси-C1-4алкила или полигидрокси-C1-4алкила;
Het10, Het11 и Het13 каждый независимо представляет собой гетероцикл, выбираемый из пирролидинила, пиперидинила, пиперазинила, пиридинила, пиримидинила, пиразинила, имидазолидинила или пиразолидинила, где указанные Het10, Het11 и Het13 необязательно замещены аминогруппой, гидроксигруппой, C1-4алкилом, гидрокси-C1-4алкилом, фенилом, фенил-C1-4алкилом, C1-4алкилокси-C1-4алкилом, аминокарбонилом или моно- или ди(C1-4алкил)аминогруппой;
Het12 представляет собой гетероцикл, выбираемый из пирролидинила, пиперидинила, пиперазинила, пиридинила, пиримидинила, пиразинил, имидазолидинила или пиразолидинила, где указанный Het12 необязательно замещен аминогруппой, гидроксигруппой, C1-4алкилом, гидрокси-C1-4алкилом, фенилом, фенил-C1-4алкилом, C1-4алкилокси-C1-4алкилом; моно- или ди(C1-4алкил)амином или аминокарбонилом;
Het14 представляет собой гетероцикл, выбираемый из морфолинила, пирролидинила, пиперазинила;,имидазолила, пирролила, 2,3,4-триазапирролила, 1,2,3-триазолила, пиразолила или пиперидинила, где указанный Het14 необязательно замещен одним или, если возможно, двумя и более заместителями, выбираемыми из C1-4алкила, C3-6циклоалкила, гидрокси-C1-4алкила, C1-4алкокси-C1-4алкила или полигидрокси-C1-4алкила; в частности, Het14 представляет собой гетероцикл, выбираемый из морфолинила, пирролидинила, пирролила, 2,3,4-триазапирролила, пиперазинила или пиперидинила, где указанный Het14 необязательно замещен одним или, если возможно, двумя и более заместителями, выбираемыми из C1-4алкила, C3-6циклоалкила, гидрокси-C1-4алкила, C1-4алкокси-C1-4алкила или полигидрокси-C1-4алкила; более конкретно, Het14 представляет собой гетероцикл, выбираемый из морфолинила, пирролидинила, пиперазинила или пиперидинила, где указанный Het14 необязательно замещен одним или, если возможно, двумя и более заместителями, выбираемыми из C1-4алкила, C3-6циклоалкила, гидрокси-C1-4алкила, C1-4алкилокси-C1-4алкила или полигидрокси-C1-4алкила.
Соединения формулы (7) раскрыты в публикации WO2007/003525, принадлежащей Janssen Pharmaceutica N.V.
В одном из вариантов осуществления циклический анилин-пиридинотриазин представляет собой соединение формулы (14).
В одном из вариантов осуществления циклический анилин-пиридинотриазин представляет собой соединение формулы (15).
Формула (15): 1,8,10,12,17,19,23,27,33-нонаазапентацикло[25.2.2.1~3,7~.1~9,13~.1~14,18~]тетратриаконта-3(34),4,6,9(33),10,12,14(32),15,17-нонаен-24-он, именуемый в настоящем документе «Соединение 13».
В одном из вариантов осуществления циклический анилин-пиридинотриазин представляет собой соединение формулы (16).
Формула (16): 10-хлор-14-этил-3,5,7,14,17,22,27-гептаазатетрацикло[19.3.1.1~2,6~.1~8,12~]гептакоза-1(25),2(27),3,5,8(26),9,11,21,23-нонаен-16-он, именуемый в настоящем документе «Соединение 14».
В одном из вариантов осуществления циклический анилин-пиридинотриазин представляет собой соединение формулы (17).
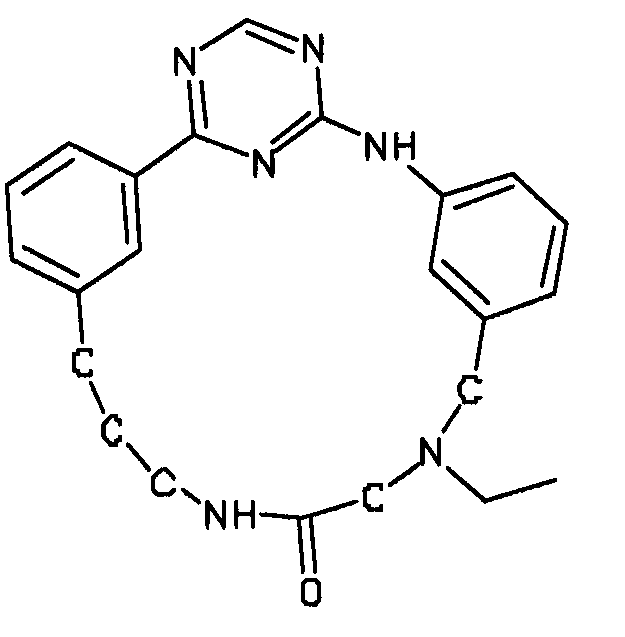
Формула (17): 14-этил-3,5,7,14,17,27-гексаазатетрацикло[19.3.1.1~2,6~.1~8,12~]гептакоза-1(25),2(27),3,5,8(26),9,11,21,23-нонаен-16-он, именуемый в настоящем документе «Соединение 15».
В одном из вариантов осуществления циклический анилин-пиридинотриазин представляет собой соединение формулы (18).
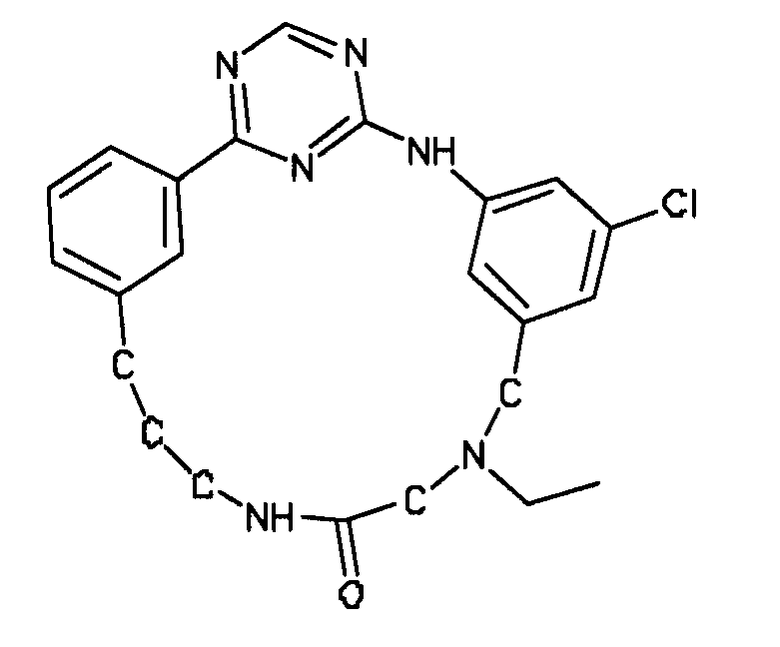
Формула (18): 10-хлор-14-этил-3,5,7,14,17,27-гексаазатетрацикло[19.3.1.1~2,6~.1~8,12~]гептакоза-1(25),2(27),3,5,8(26),9,11,21,23-нонаен-16-он, именуемый в настоящем документе «Соединение 16».
В одном из вариантов осуществления циклический анилин-пиридинотриазин представляет собой соединение формулы (19).
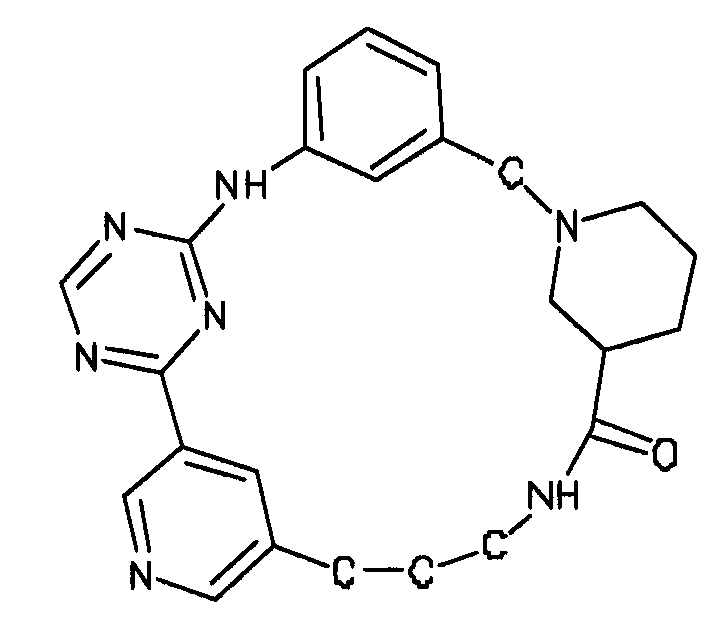
Формула (19): 3,5,7,14,20,26,31-гептаазапентацикло[22.3.1.1~2,6~.1~8,12~.1~14,18~]гентриаконта-1(28),2(31),3,5,8(30),9,11,24,26-нонаен-19-он, именуемый в настоящем документе «Соединение 17».
В одном из вариантов осуществления циклический анилин-пиридинотриазин представляет собой соединение формулы (20).
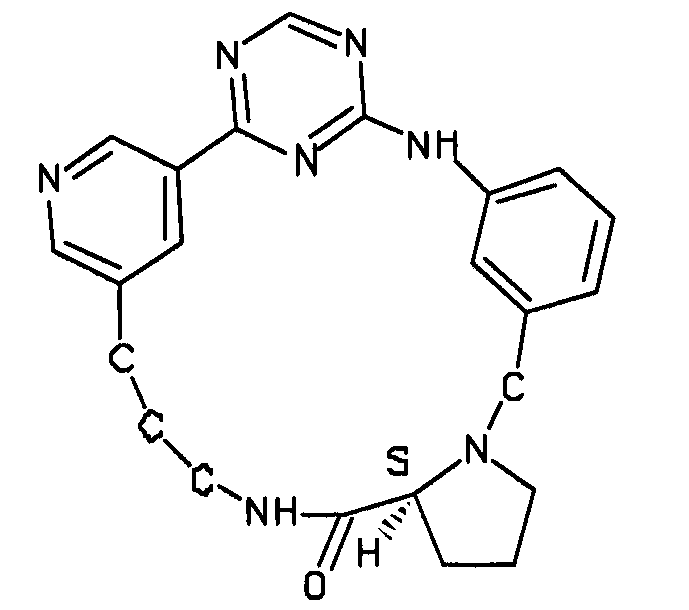
Формула (20): (18S)-3,5,7,14,20,26,30-гептаазапентацикло[22.3.1.1~2,6~.1~8,12~.0~14,18~]триаконта-1(28),2(30),3,5,8(29),9,11,24,26-нонаен-19-он, именуемый в настоящем документе «Соединение 18».
В одном из вариантов осуществления циклический анилин-пиридинотриазин представляет собой соединение формулы (21).
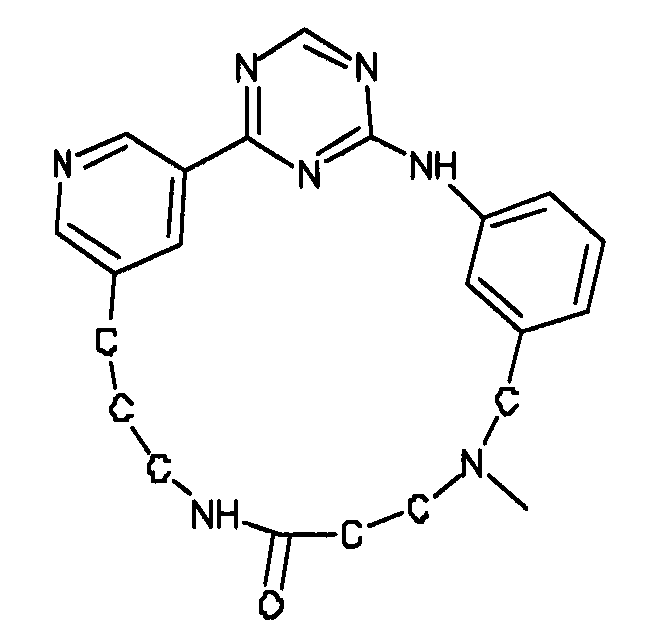
Формула (21): 14-метил-3,5,7,14,18,24,28-гептаазатетрацикло[20.3.1.1~2,6~.1~8,12~]октакоза-1(26),2(28),3,5,8(27),9,11,22,24-нонаен-17-он, именуемый в настоящем документе «Соединение 19».
В одном из вариантов осуществления циклический анилин-пиридинотриазин представляет собой соединение формулы (22).
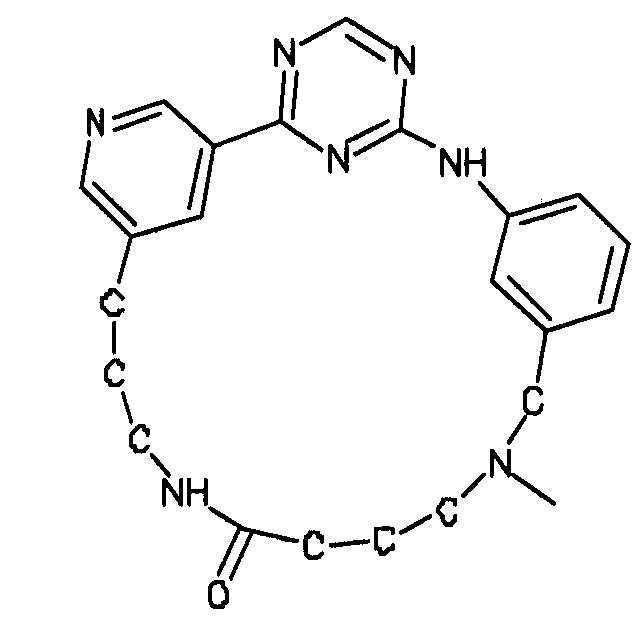
Формула (22): 14-метил-3,5,7,14,19,25,29-гептаазатетрацикло[21.3.1.1~2,6~.1~8,12~]нонакоза-1(27),2(29),3,5,8(28),9,11,23,25-нонаен-18-он, именуемый в настоящем документе «Соединение 20».
В одном из вариантов осуществления циклический анилин-пиридинотриазин представляет собой соединение формулы (23).
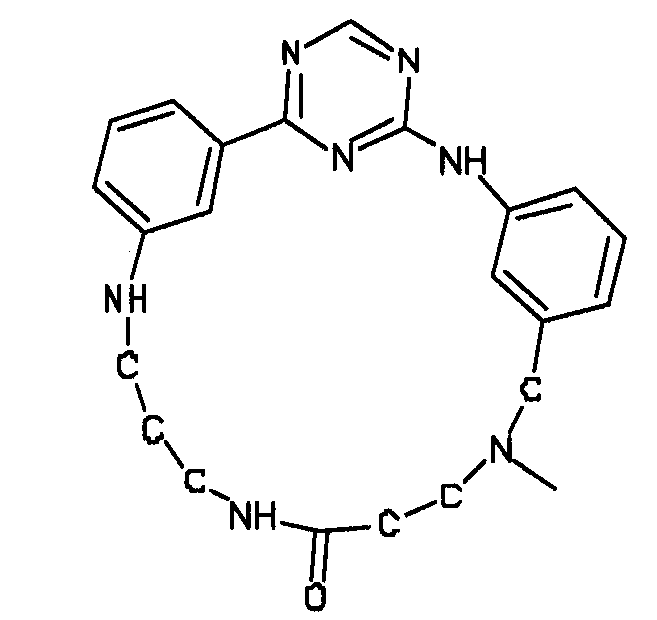
Формула (23): 14-метил-3,5,7,14,18,22,29-гептаазатетрацикло[21.3.1.1~2,6~.1~8,12~]нонакоза-1(27),2(29),3,5,8(28),9,11,23,25-нонаен-17-он, именуемый в настоящем документе «Соединение 21».
В одном из вариантов осуществления циклический анилин-пиридинотриазин представляет собой соединение формулы (24).
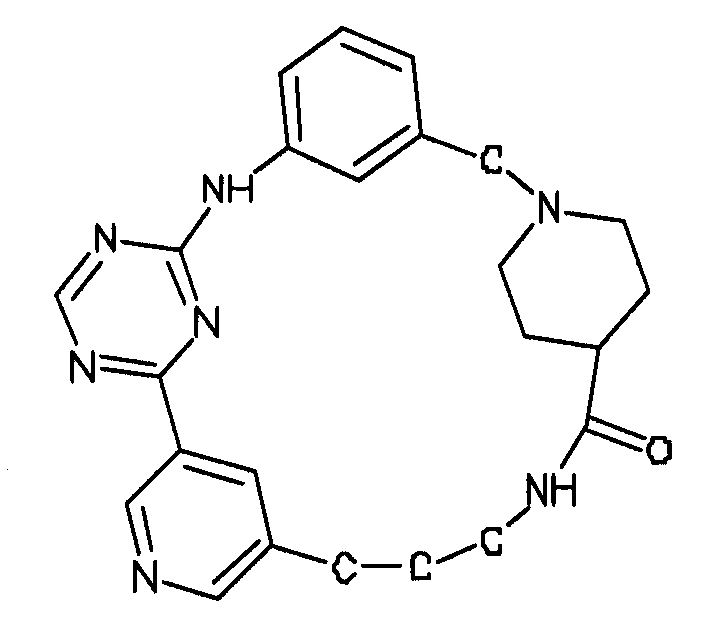
Формула (24): 1,8,10,12,16,22,30-гептаазапентацикло[22.2.2.1~3,7~.1~9,13~.1~14,18~]гентриаконта-3(31),4,6,9(30),10,12,14(29),15,17-нонаен-23-он, именуемый в настоящем документе «Соединение 22».
В одном из вариантов осуществления циклический анилин-пиридинотриазин представляет собой соединение формулы (25).
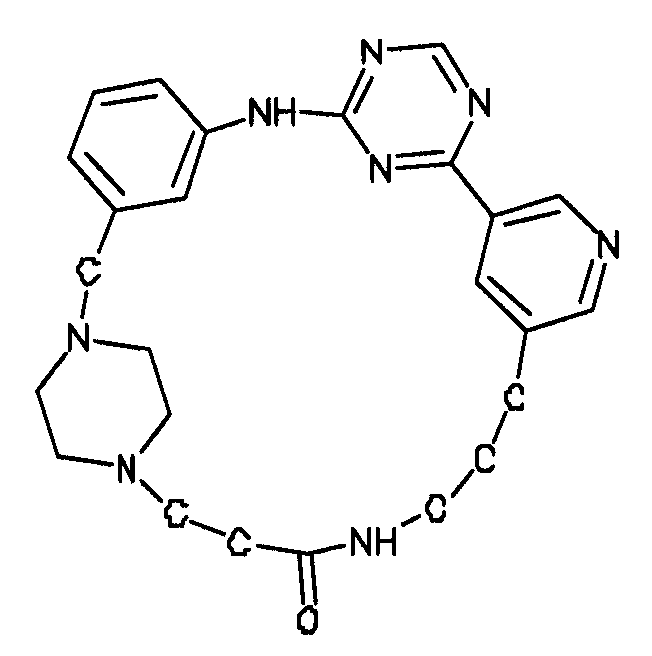
Формула (25): 1,8,10,12,16,22,26,32-октаазапентацикло[24.2.2.1~3,7~.1~9,13~.1~14,18~]тритриаконта-3(33),4,6,9(32),10,12,14(31),15,17-нонаен-23-он, именуемый в настоящем документе «Соединение 23».
В одном из вариантов осуществления циклический анилин-пиридинотриазин представляет собой соединение формулы (26).
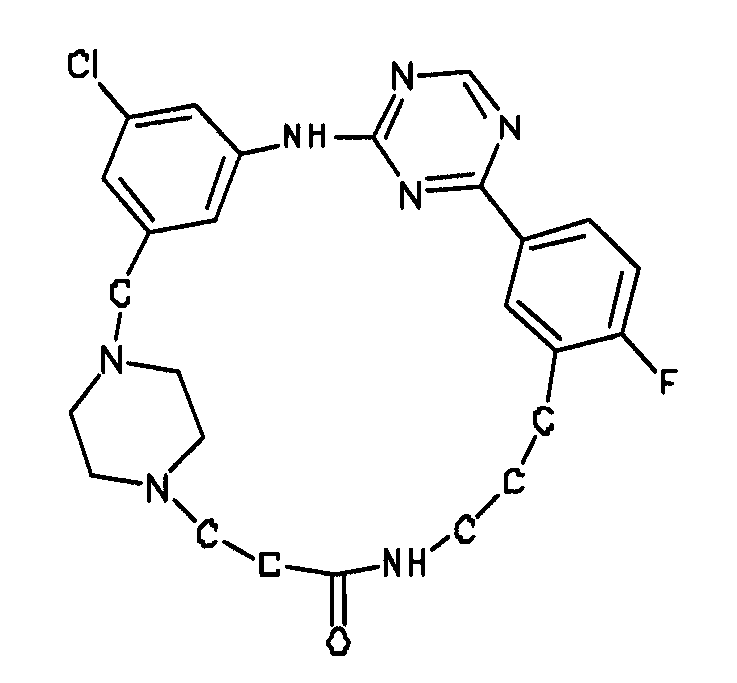
Формула (26): 5-хлор-17-фтор-1,8,10,12,22,26,32-гептаазапентацикло[24.2.2.1~3,7~.1~9,13~.1~14,18~]тритриаконта-3(33),4,6,9(32),10,12,14(31),15,17-нонаен-23-он, именуемый в настоящем документе «Соединение 24».
В одном из вариантов осуществления циклический анилин-пиридинотриазин представляет собой соединение формулы (27).
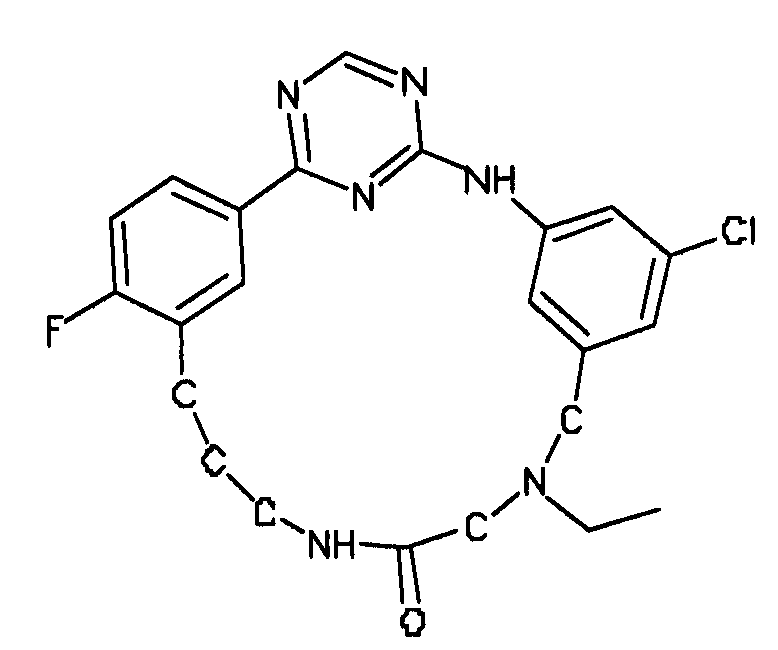
Формула (27): 10-хлор-14-этил-22-фтор-3,5,7,14,17,27-гексаазатетрацикло[19.3.1.1~2,6~.1~8,12~]гептакоза-1(25),2(27),3,5,8(26),9,11,21,23-нонаен-16-он, именуемый в настоящем документе «Соединение 25».
В одном из вариантов осуществления циклический анилин-пиридинотриазин представляет собой соединение формулы (28).
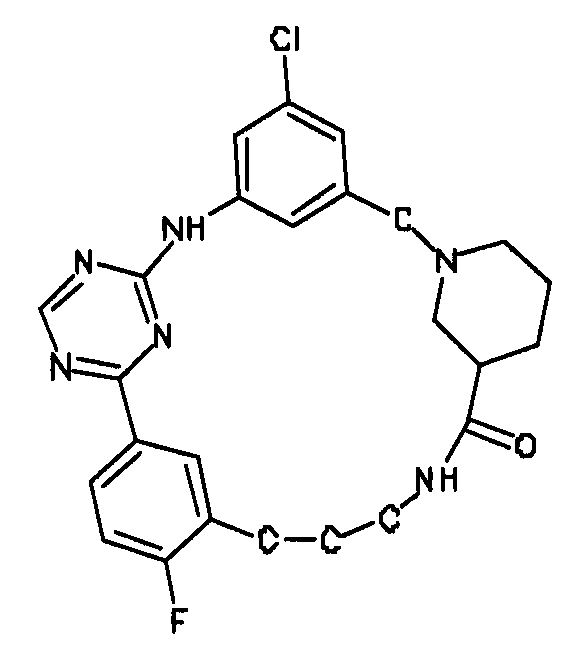
Формула (28): 10-хлор-25-фтор-3,5,7,14,20,31-гексаазапентацикло[22.3.1.1~2,6~.1~8,12~.1~14,18~]гентриаконта-1(28),2(31),3,5,8(30),9,11,24,26-нонаен-19-он, именуемый в настоящем документе «Соединение 26».
В одном из вариантов осуществления циклический анилин-пиридинотриазин представляет собой соединение формулы (29).
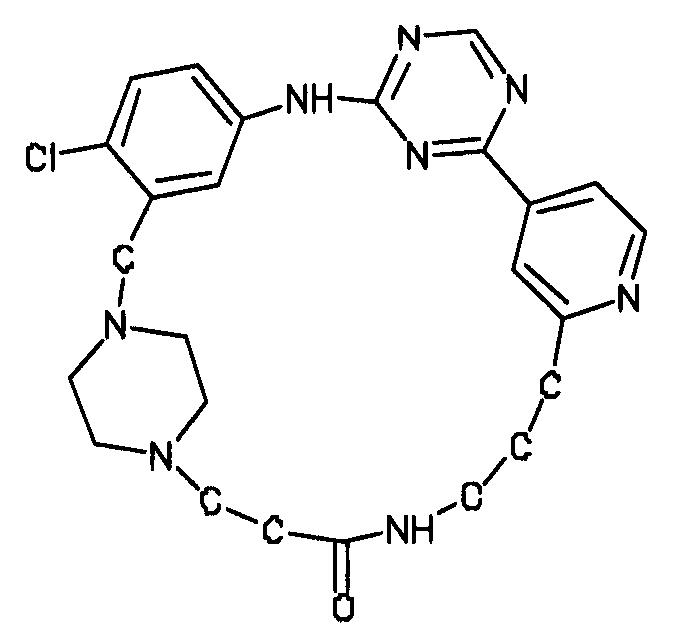
Формула (29): 4-хлор-1,8,10,12,17,22,26,32-октаазапентацикло[24.2.2.1~3,7~.1~9,13~.1~14,18~]тритриаконта-3(33),4,6,9(32),10,12,14(31),15,17-нонаен-23-он, именуемый в настоящем документе «Соединение 27».
В одном из вариантов осуществления циклический анилин-пиридинотриазин представляет собой соединение формулы (30).
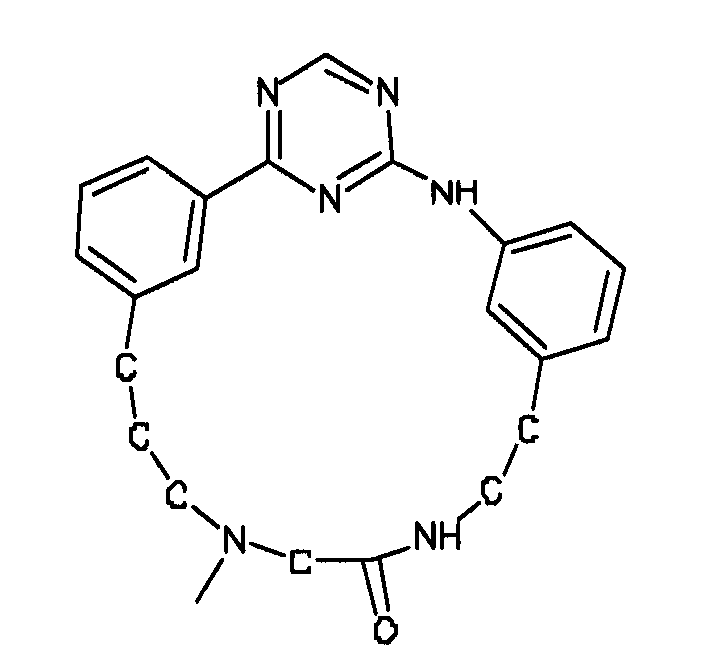
Формула (30): 18-метил-3,5,7,15,18,28-гексаазатетрацикло[20.3.1.1~2,6~.1~8,12~]октакоза-1(26),2(28),3,5,8(27),9,11,22,24-нонаен-16-он, именуемый в настоящем документе «Соединение 28».
В одном из вариантов осуществления циклический анилин-пиридинотриазин представляет собой соединение формулы (31).
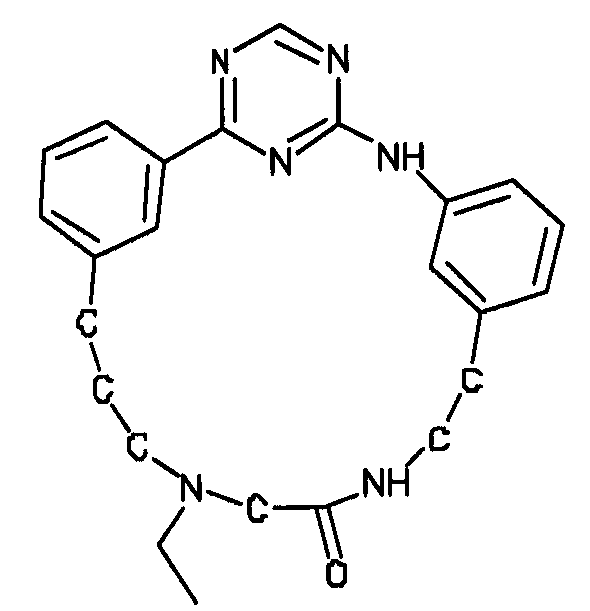
Формула (31): 18-этил-3,5,7,15,18,28-гексаазатетрацикло[20.3.1.1~2,6~.1~8,12~]октакоза-1(26),2(28),3,5,8(27),9,11,22,24-нонаен-16-он, именуемый в настоящем документе «Соединение 29».
В одном из вариантов осуществления циклический анилин-пиридинотриазин представляет собой соединение формулы (32).
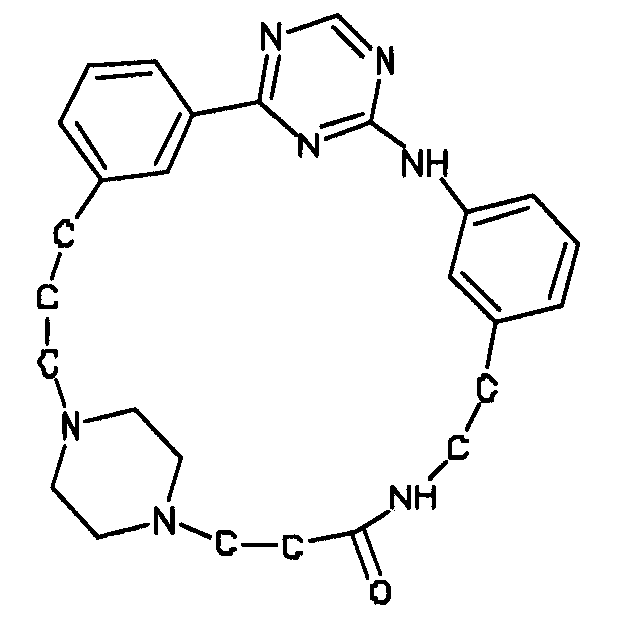
Формула (32): 1,8,10,12,17,19,23,27,33-нонаазапентацикло[25.2.2.1~3,7~.1~9,13~.1~14,18~]тетратриаконта-3(34),4,6,9(33),10,12,14(32),15,17-нонаен-24-он, именуемый в настоящем документе «Соединение 30».
В одном из вариантов осуществления циклический анилин-пиридинотриазин представляет собой соединение формулы (33).
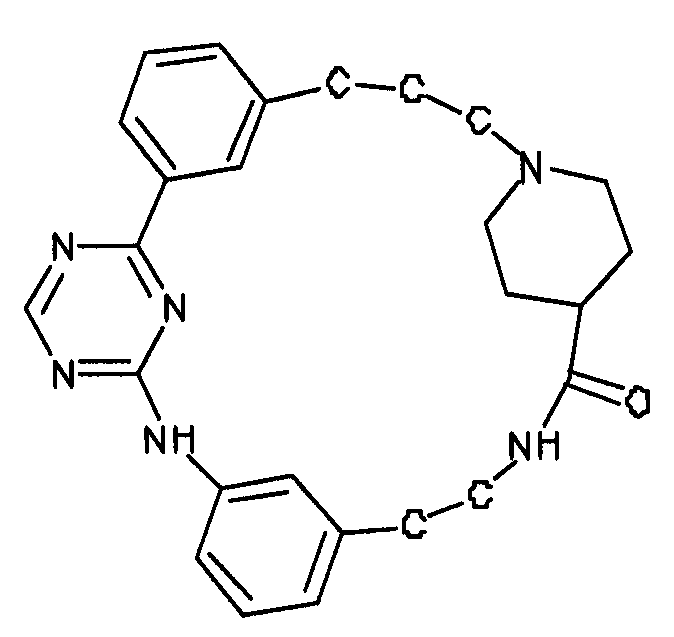
Формула (33): 1,11,13,15,23,31-гексаазапентацикло[23.2.2.1~5,9~.1~10,14~.1~16,20~]дотриаконта-5(32),6,8,10(31),11,13,16(30),17,19-нонаен-24-он, именуемый в настоящем документе «Соединение 31».
В одном из вариантов осуществления циклический анилин-пиридинотриазин представляет собой соединение формулы (34).
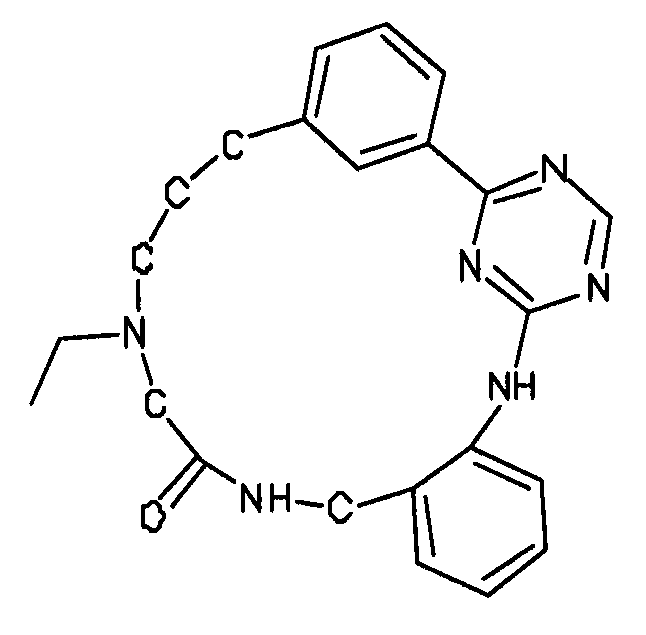
Формула (34): 15-этил-13,14,15,16,18,19-гексагидро-1H-6,2-(азено)-7,11-(метено)-1,3,5,15,18-бензопентаазациклогеникозин-17(12H)-он, именуемый в настоящем документе «Соединение 32».
В одном из вариантов осуществления циклический анилин-пиридинотриазин представляет собой соединение формулы (35).
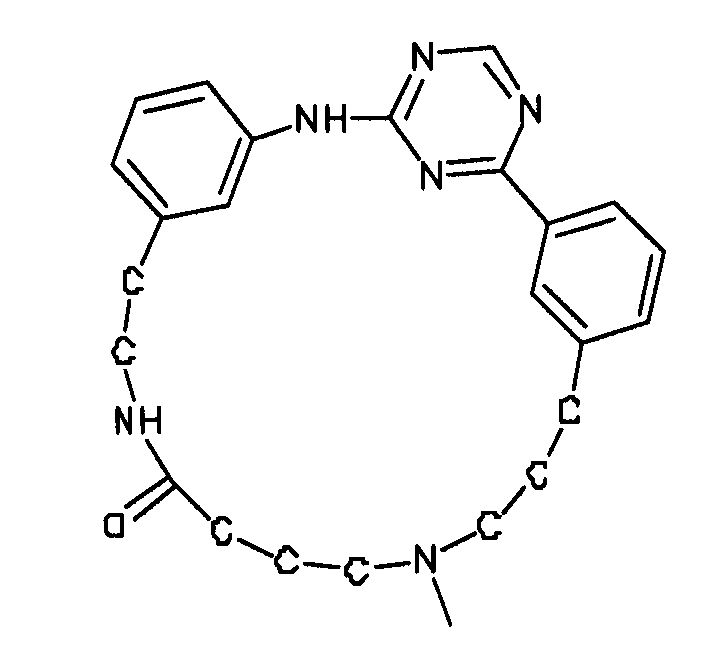
Формула (35): 20-метил-3,5,7,15,20,30-гексаазатетрацикло[22.3.1.1~2,6~.1~8,12~]триаконта-1(28),2(30),3,5,8(29),9,11,24,26-нонаен-16-он, именуемый в настоящем документе «Соединение 33».
В одном из вариантов осуществления циклический анилин-пиридинотриазин представляет собой соединение формулы (36).
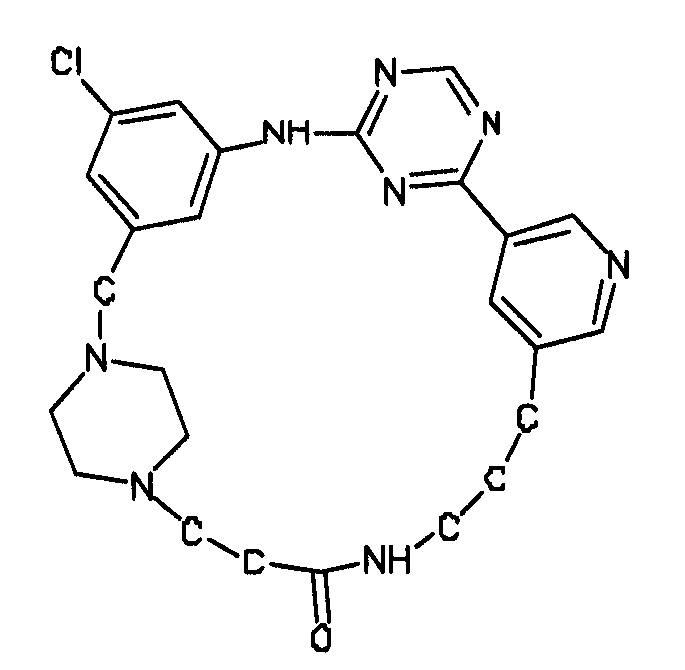
Формула (36): 5-хлор-1,8,10,12,16,22,26,32-октаазапентацикло[24.2.2.1~3,7~.1~9,13~.1~14,18~]тритриаконта-3(33),4,6,9(32),10,12,14(31),15,17-нонаен-23-он, именуемый в настоящем документе «Соединение 34».
В одном из вариантов осуществления циклический анилин-пиридинотриазин представляет собой соединение формулы (37).
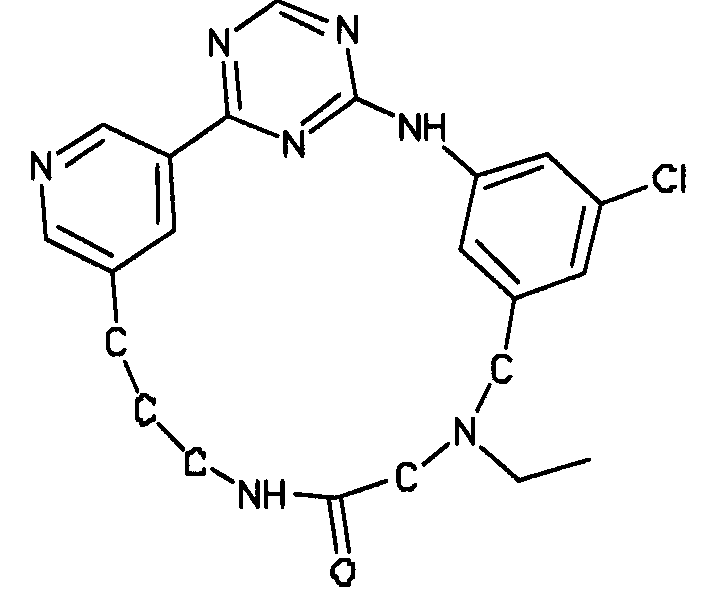
Формула (37): 10-хлор-14-этил-3,5,7,14,17,23,27-гептаазатетрацикло[19.3.1.1~2,6~.1~8,12~]гептакоза-1(25),2(27),3,5,8(26),9,11,21,23-нонаен-16-он, именуемый в настоящем документе «Соединение 35».
В одном из вариантов осуществления циклический анилин-пиридинотриазин представляет собой соединение формулы (38).
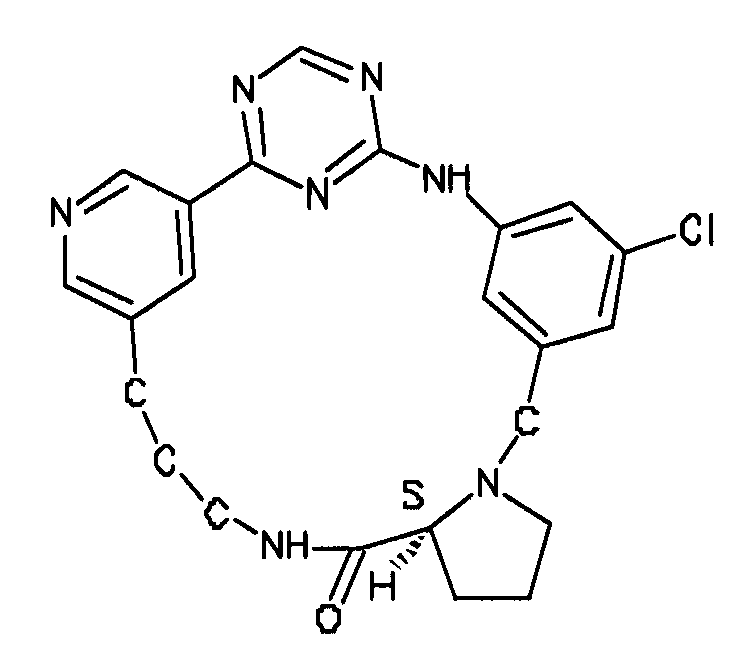
Формула (38): (18S)-10-хлор-3,5,7,14,20,26,30-гептаазапентацикло[22.3.1.1~2,6~.1~8,12~.0~14,18~]триаконта-1(28),2(30),3,5,8(29),9,11,24,26-нонаен-19-он, именуемый в настоящем документе «Соединение 36».
В одном из вариантов осуществления циклический анилин-пиридинотриазин представляет собой соединение формулы (39).
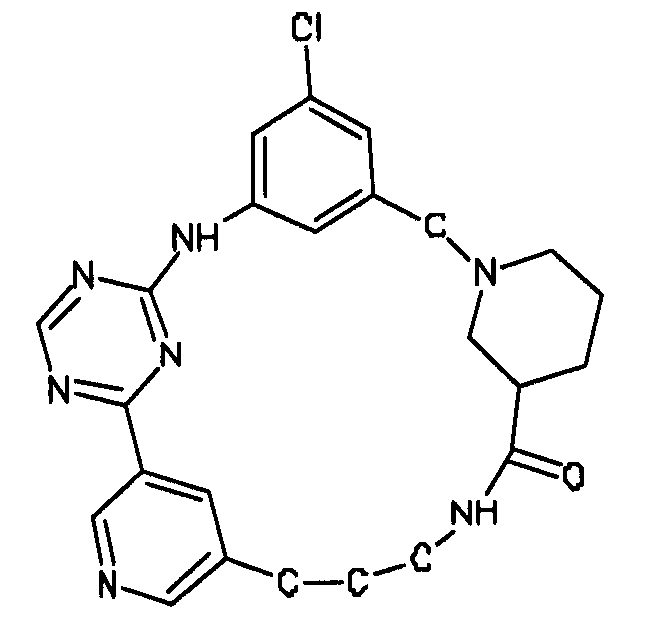
Формула (39): 10-хлор-3,5,7,14,20,26,31-гептаазапентацикло[22.3.1.1~2,6~.1~8,12~.1~14,18~]гентриаконта-1(28),2(31),3,5,8(30),9,11,24,26-нонаен-19-он, именуемый в настоящем документе «Соединение 37».
В одном из вариантов осуществления циклический анилин-пиридинотриазин представляет собой соединение формулы (40).
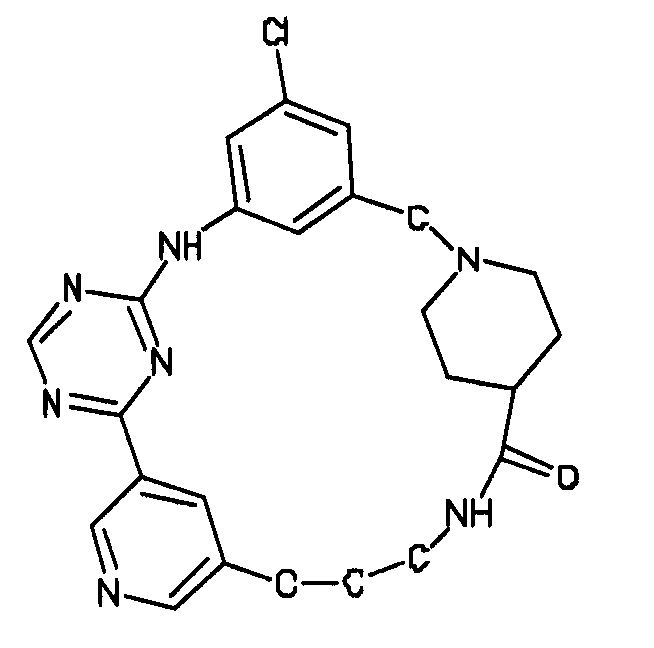
Формула (40): 5-хлор-1,8,10,12,16,22,30-гептаазапентацикло[22.2.2.1~3,7~.1~9,13~.1~14,18~]гентриаконта-3(31),4,6,9(30),10,12,14(29),15,17-нонаен-23-он, именуемый в настоящем документе «Соединение 38».
В одном из вариантов осуществления циклический анилин-пиридинотриазин представляет собой соединение формулы (41).
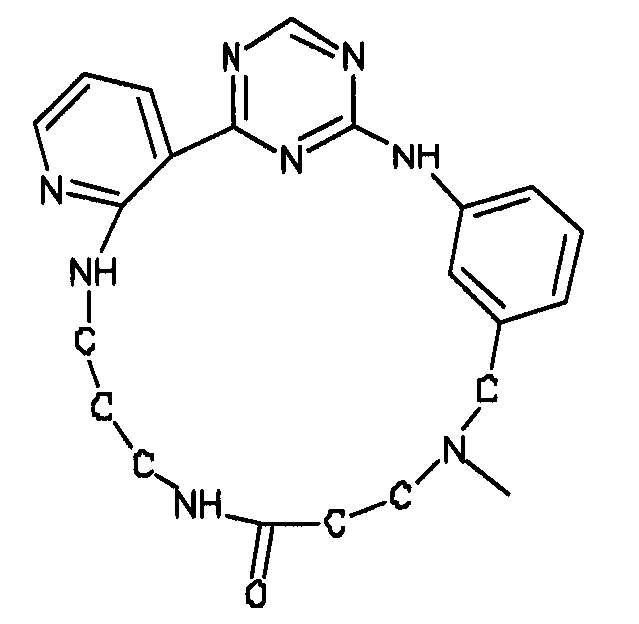
Формула (41): 9-метил-2,3,4,5,7,8,9,10-октагидро-16H-17,21-(азено)-11,15-(метено)пиридо[3,2-g][1,3,5,9,13,17]гексаазациклотрикозин-6(1H)-он, именуемый в настоящем документе «Соединение 39».
В одном из вариантов осуществления циклический анилин-пиридинотриазин представляет собой соединение формулы (42).
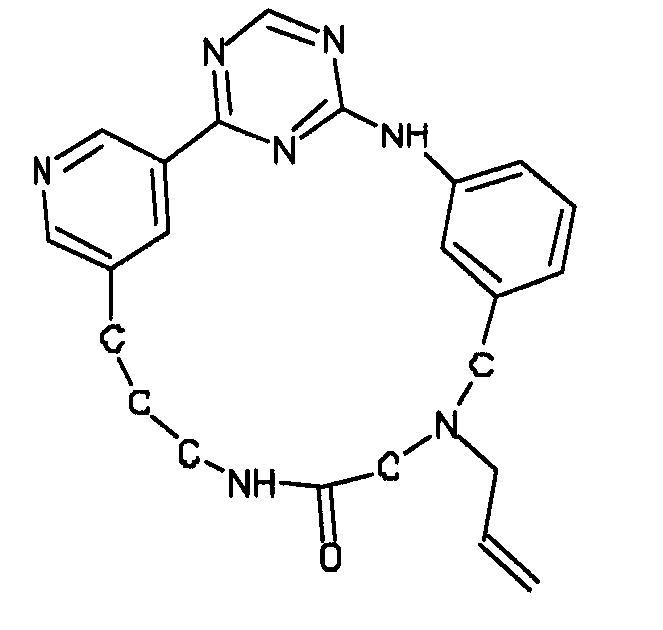
Формула (42): 14-проп-2-ен-1-ил-3,5,7,14,17,23,27-гептаазатетрацикло[19.3.1.1~2,6~.1~8,12~]гептакоза-1(25),2(27),3,5,8(26),9,11,21,23-нонаен-16-он, именуемый в настоящем документе «Соединение 40».
В одном из вариантов осуществления циклический анилин-пиридинотриазин представляет собой соединение формулы (43).
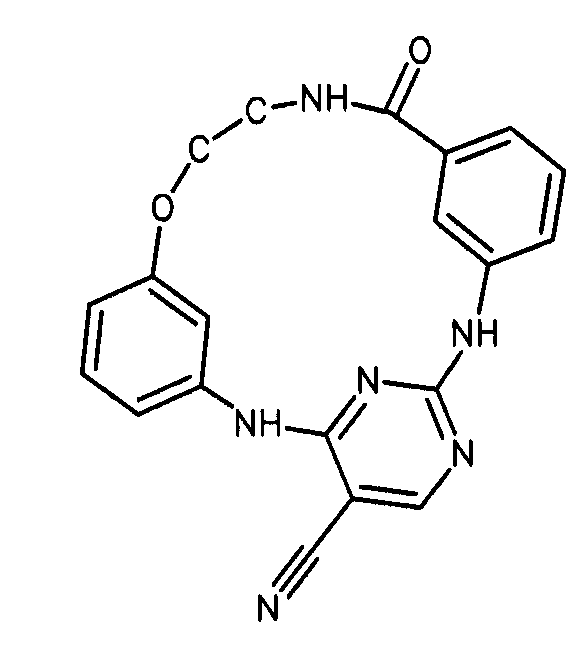
Формула (43): 18-оксо-14-окса-2,4,8,17,25-пентаазатетрацикло[17.3.1.1~3,7~.1~9,13~]пентакоза-1(23),3(25),4,6,9(24),10,12,19,21-нонаен-6-карбонитрил, именуемый в настоящем документе «Соединение 41».
В одном из вариантов осуществления циклический анилин-пиридинотриазин представляет собой соединение формулы (44).
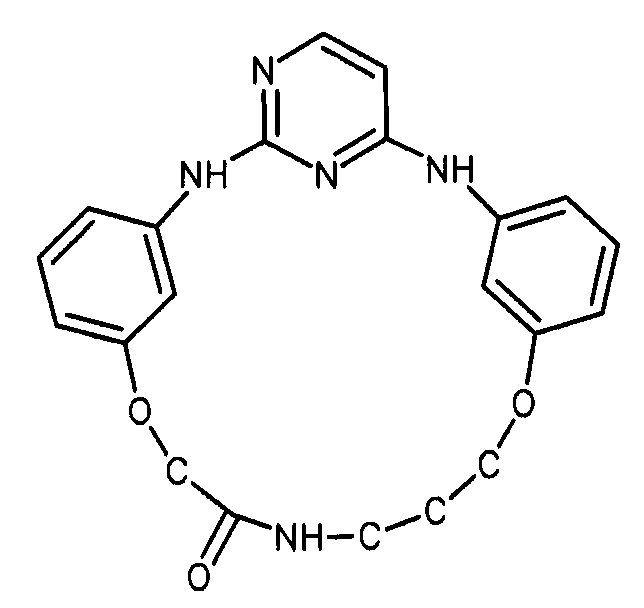
Формула (44): 14,21-диокса-2,4,8,18,28-пентаазатетрацикло[20.3.1.1~3,7~.1~9,13~]октакоза-1(26),3(28),4,6,9(27),10,12,22,24-нонаен-19-он, именуемый в настоящем документе «Соединение 42».
В одном из вариантов осуществления циклический анилин-пиридинотриазин представляет собой соединение формулы (45).
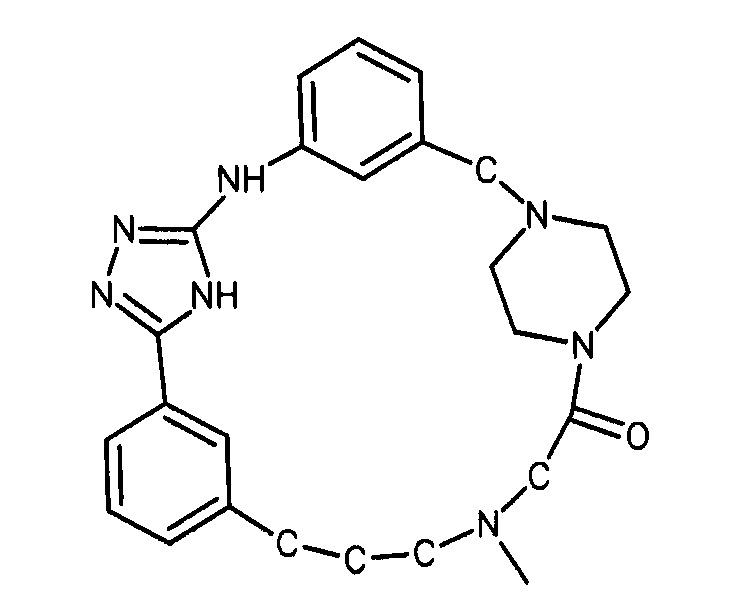
Формула (45): 21-метил-1,8,10,11,21,24,30-гептаазапентацикло[22.2.2.1~3,7~.1~9,12~.1~13,17~]гентриаконта-3(31),4,6,9,11,13(29),14,16-октаен-23-он, именуемый в настоящем документе «Соединение 43».
В одном из вариантов осуществления циклический анилин-пиридинотриазин представляет собой соединение формулы (46).
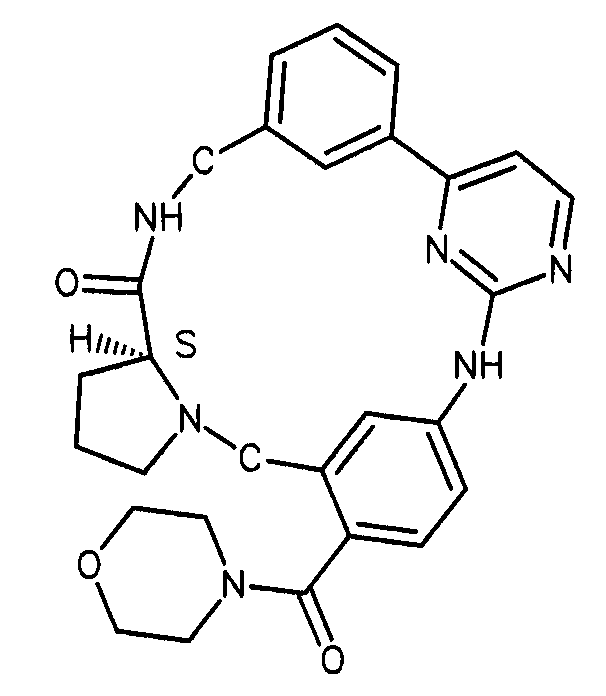
Формула (46): (18S)-11-(морфолин-4-илкарбонил)-5,7,14,20,28-пентаазапентацикло[20.3.1.1~2,6~.1~8,12~.0~14,18~]октакоза-1(26),2(28),3,5,8(27),9,11,22,24-нонаен-19-он, именуемый в настоящем документе «Соединение 44».
В одном из вариантов осуществления циклический анилин-пиридинотриазин представляет собой соединение формулы (47).
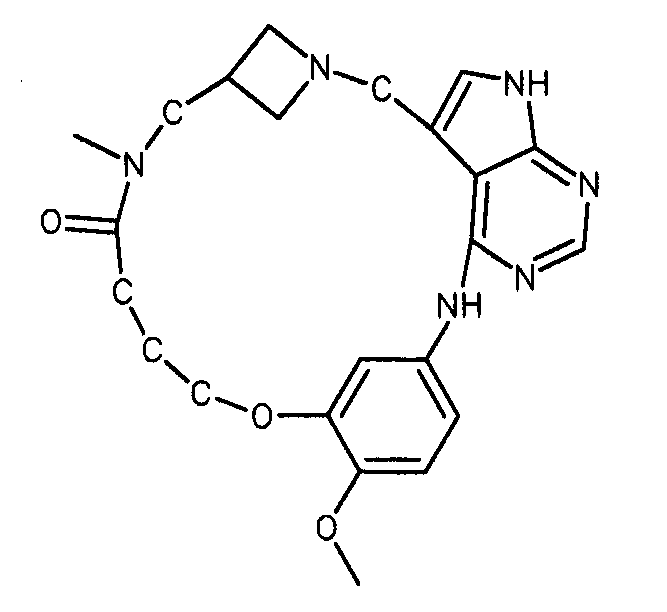
Формула (47): 10-метокси-17-метил-2,14,15,17,18,19,20,22-октагидро-6H-19,21-метано-7,11-(метено)-12-окса-2,3,5,6,17,21-гексаазациклоикоза[1,2,3-cd]инден-16(13H)-он, именуемый в настоящем документе «Соединение 45».
В одном из вариантов осуществления указанный по меньшей мере один другой фактор представляет собой соединение формулы (48):
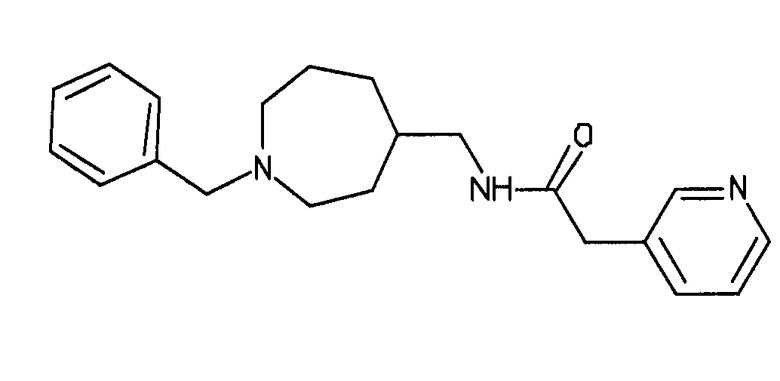
Формула (48). N-{[1-(фенилметил)азепан-4-ил]метил}-2-пиридин-3-илацетамид, именуемый в настоящем документе «Соединение 46».
В одном из вариантов осуществления указанный по меньшей мере один другой фактор представляет собой соединение формулы (49):
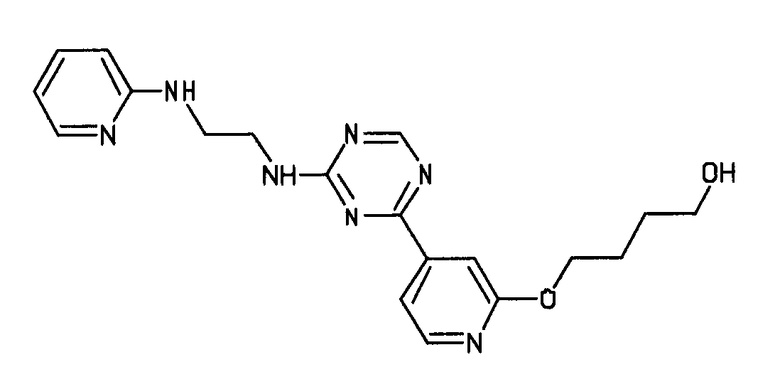
Формула (49). 4-{[4-(4-{[2-(пиридин-2-иламино)этил]амино}-1,3,5-триазин-2-ил)пиридин-2-ил]окси}бутан-1-ол, именуемый в настоящем документе «Соединение 47».
В одном из вариантов осуществления указанный по меньшей мере один другой фактор представляет собой соединение формулы (50):
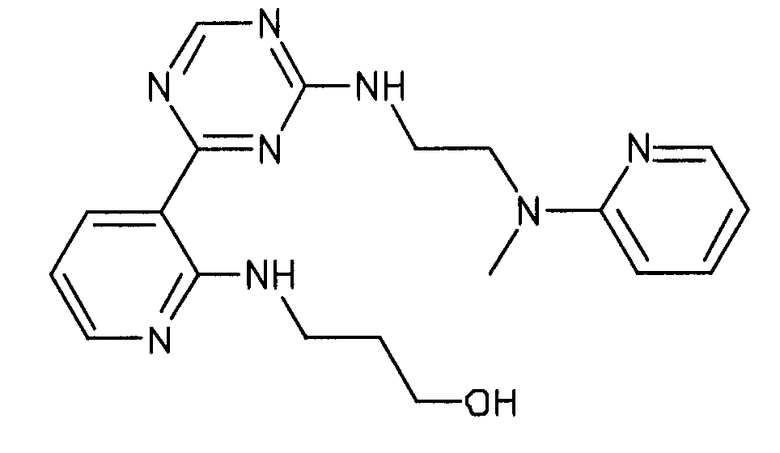
Формула (50). 3-({3-[4-({2-[метил(пиридин-2-ил)амино]этил}амино)-1,3,5-триазин-2-ил]пиридин-2-ил}амино)пропан-1-ол, именуемый в настоящем документе «Соединение 48».
В одном из вариантов осуществления указанный по меньшей мере один другой фактор представляет собой соединение формулы (51):
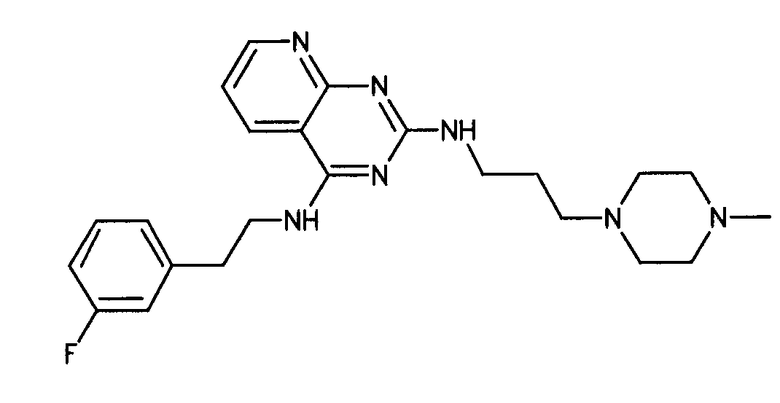
Формула (51). N~4~-[2-(3-фторфенил)этил]-N~2~-[3-(4-метилпиперазин-1-ил)пропил]пиридо[2,3-d]пиримидин-2,4-диамин, именуемый в настоящем документе «Соединение 49».
В одном из вариантов осуществления указанный по меньшей мере один другой фактор представляет собой соединение формулы (52):
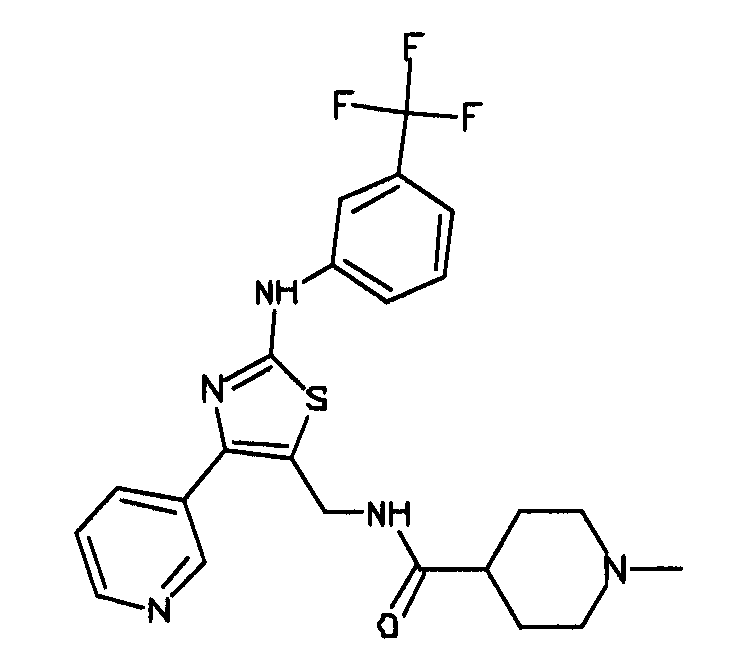
Формула (52). 1-метил-N-[(4-пиридин-3-ил-2-{[3-(трифторметил)фенил]амино}-1,3-тиазол-5-ил)метил]пиперидин-4-карбоксамид, именуемый в настоящем документе «Соединение 50».
В одном из вариантов осуществления указанный по меньшей мере один другой фактор представляет собой соединение формулы (53):
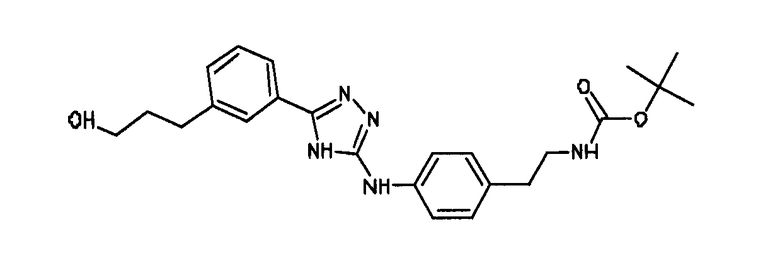
Формула (53). 1,1-диметилэтил {2-[4-({5-[3-(3-гидроксипропил)фенил]-4H-1,2,4-триазол-3-ил}амино)фенил]этил}карбамат, именуемый в настоящем документе «Соединение 51».
В одном из вариантов осуществления указанный по меньшей мере один другой фактор представляет собой соединение формулы (54):
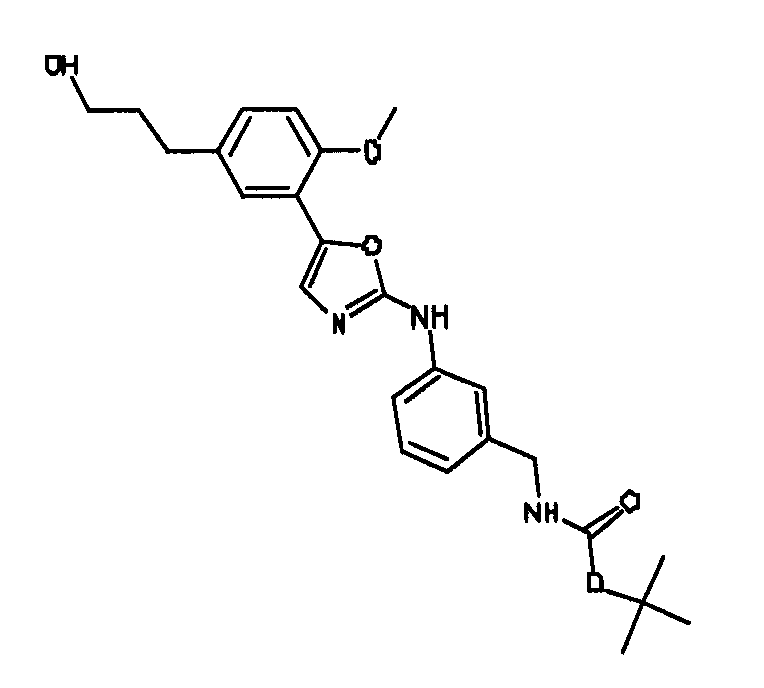
Формула (54). 1,1-диметилэтил {[3-({5-[5-(3-гидроксипропил)-2-(метилокси)фенил]-1,3-оксазол-2-ил}амино)фенил]метил}карбамат, именуемый в настоящем документе «Соединение 52».
В одном из вариантов осуществления указанный по меньшей мере один другой фактор представляет собой соединение формулы (55):
Формула (55). 1-({5-[6-({4-[(4-метилпиперазин-1-ил)сульфонил]фенил}амино)пиразин-2-ил]тиофен-2-ил}метил)пиперидин-4-ол, именуемый в настоящем документе «Соединение 53».
В одном из вариантов осуществления указанный по меньшей мере один другой фактор представляет собой соединение формулы (56):
Формула (56). 1-({4-[6-({4-[(4-метилпиперазин-1-ил)сульфонил]фенил}амино)пиразин-2-ил]тиофен-2-ил}метил)пиперидин-4-карбоксамид, именуемый в настоящем документе «Соединение 54».
В одном из вариантов осуществления указанный по меньшей мере один другой фактор представляет собой соединение формулы (57):
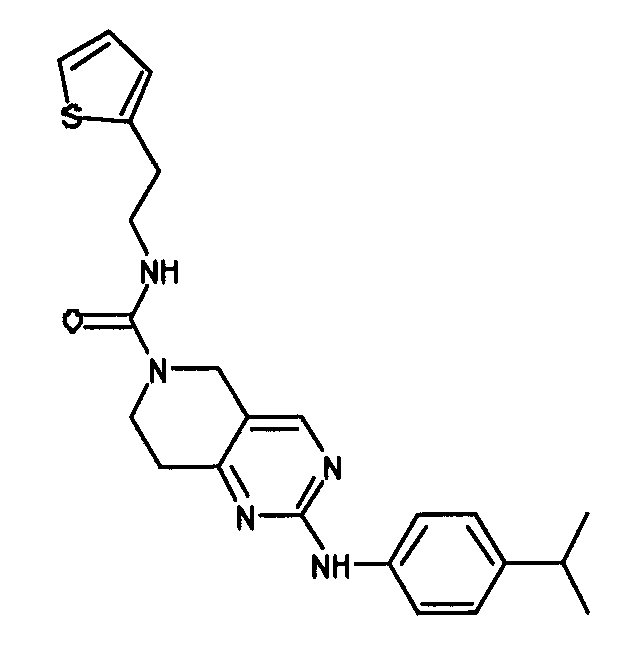
Формула (57). 2-{[4-(1-метилэтил)фенил]амино}-N-(2-тиофен-2-илэтил)-7,8-дигидропиридо[4,3-d]пиримидин-6(5H)-карбоксамид, именуемый в настоящем документе «Соединение 55».
В одном из вариантов осуществления указанный по меньшей мере один другой фактор представляет собой соединение формулы (58):
Формула (58). 6-[(2-{[4-(2,4-дихлорфенил)-5-(4-метил-1H-имидазол-2-ил)пиримидин-2-ил]амино}этил)амино]пиридин-3-карбонитрил, именуемый в настоящем документе «Соединение 56».
В одном из вариантов осуществления указанный по меньшей мере один другой фактор представляет собой соединение формулы (59):
Формула (59). 4-(6-{[(3-хлорфенил)метил]амино}имидазо[1,2-b]пиридазин-3-ил)-N-[2-(диметиламино)этил]бензамид, именуемый в настоящем документе «Соединение 57».
Обнаружение клеток, экспрессирующих маркеры, характерные для линии сформированной эндодермы
Образование клеток, экспрессирующих маркеры, характерные для линии сформированной эндодермы, может быть обнаружено путем проверки на наличие соответствующих маркеров до и после выполнения конкретного протокола. Плюрипотентные стволовые клетки, как правило, не экспрессируют такие маркеры. Таким образом, дифференцирование плюрипотентных клеток определяется по началу экспрессии таких маркеров.
Эффективность дифференцирования может быть определена путем обработки популяции клеток агентом (например, антителом), специфически распознающим белковый маркер, экспрессированный клетками, экспрессирующими маркеры, характерные для линии сформированной эндодермы.
Способы оценки экспрессии маркеров белков и нуклеиновых кислот в культивированных или выделенных клетках являются стандартными для данной области. К подобным способам относятся количественная полимеразная цепная реакция с обратной транскрипцией (ОТ-ПЦР), Нозерн-блоттинг, гибридизация in situ (см., например, Current Protocols in Molecular Biology (Ausubel et al., eds. 2001 supplement)) и иммунологические анализы, такие как иммуногистохимический анализ среза материала, Вестерн-блоттинг, а для доступных в интактных клетках маркеров - также способ проточной цитометрии (FACS) (см., например, Harlow and Lane, Using Antibodies: A Laboratory Manual, New York: Cold Spring Harbor Laboratory Press (1998)).
Например, характеристики плюрипотентных стволовых клеток хорошо известны специалистам в данной области, и продолжается выявление дополнительных характеристик плюрипотентных стволовых клеток. К маркерам плюрипотентных стволовых клеток относится, например, экспрессия одного или нескольких из следующих факторов: ABCG2, крипто, FOXD3, коннексин43, коннексин45, OCT4, SOX2, Nanog, hTERT, UTF-1, ZFP42, SSEA-3, SSEA-4, Tra1-60 или Tra1-81.
После обработки плюрипотентных стволовых клеток с применением способов, составляющих предмет настоящего изобретения, дифференцированные клетки могут быть выделены путем воздействия на популяцию клеток агентом (например, антителом), специфически распознающим белковый маркер, например CXCR4, экспрессируемый клетками, экспрессирующими маркеры, характерные для линии сформированной эндодермы.
Образование клеток, экспрессирующих маркеры, характерные для линии панкреатической эндодермы
Клетки, экспрессирующие маркеры, характерные для линии сформированной эндодермы, могут быть дифференцированы в клетки, экспрессирующие маркеры, характерные для линии панкреатической эндодермы, с использованием любого способа, известного специалистам в данной области, или с использованием способа, предложенного в настоящем изобретении.
Например, клетки, экспрессирующие маркеры, характерные для линии сформированной эндодермы, могут быть дифференцированы в клетки, экспрессирующие маркеры, характерные для линии панкреатической эндодермы, в соответствии со способами, описанными в публикации D'Amour et al, Nature Biotechnology 24, 1392-1401 (2006).
Например, клетки, экспрессирующие маркеры, характерные для линии сформированной эндодермы, далее дифференцируются в клетки, экспрессирующие маркеры, характерные для линии панкреатической эндодермы, путем обработки клеток, экспрессирующих маркеры, характерные для линии сформированной эндодермы, фактором роста фибробластов и ингибитором сигнального пути Hedgehog KAAD-циклопамином с последующим удалением содержащей фактор роста фибробластов и KAAD-циклопамин среды и культивированием клеток в среде, содержащей ретиноевую кислоту, фактор роста фибробластов и KAAD-циклопамин. Пример использования данного способа приведен в публикации Nature Biotechnology 24, 1392-1401 (2006).
В одном из аспектов настоящего изобретения клетки, экспрессирующие маркеры, характерные для линии сформированной эндодермы, далее дифференцируются в клетки, экспрессирующие маркеры, характерные для линии панкреатической эндодермы, путем обработки клеток, экспрессирующих маркеры, характерные для линии сформированной эндодермы, ретиноевой кислотой и по меньшей мере одним фактором роста фибробластов в течение периода времени, в соответствии со способами, описанными в заявке на патент США № 11/736908, принадлежащей LifeScan, Inc.
В одном из аспектов настоящего изобретения клетки, экспрессирующие маркеры, характерные для линии сформированной эндодермы, далее дифференцируются в клетки, экспрессирующие маркеры, характерные для линии панкреатической эндодермы, путем обработки клеток, экспрессирующих маркеры, характерные для линии сформированной эндодермы, ретиноевой кислотой и по меньшей мере одним фактором роста фибробластов в течение периода времени, в соответствии со способами, описанными в заявке на патент США № 11/779311, принадлежащей LifeScan, Inc.
В одном из аспектов настоящего изобретения клетки, экспрессирующие маркеры, характерные для линии сформированной эндодермы, далее дифференцируются в клетки, экспрессирующие маркеры, характерные для линии панкреатической эндодермы, путем обработки клеток, экспрессирующих маркеры, характерные для линии сформированной эндодермы, в соответствии со способами, описанными в заявке на патент США № 60/990529.
Клетки, экспрессирующие маркеры, характерные для линии сформированной эндодермы, могут быть обработаны по меньшей мере еще одним дополнительным фактором, который может стимулировать образование клеток, экспрессирующих маркеры, характерные для линии панкреатической эндодермы. В качестве альтернативы по меньшей мере еще один дополнительный фактор может стимулировать пролиферацию клеток, экспрессирующих маркеры, характерные для линии панкреатической эндодермы, полученных в соответствии со способами, составляющими предмет настоящего изобретения. Кроме того, по меньшей мере еще один дополнительный фактор может стимулировать способность клеток, экспрессирующих маркеры, характерные для линии панкреатической эндодермы, полученных в соответствии со способами, составляющими предмет настоящего изобретения, образовывать другие типы клеток или повышать эффективность дополнительных этапов дифференцирования.
По меньшей мере еще одним дополнительным фактором может быть, например, никотинамид, члены семейства TGF-β, включая TGF-β1, 2 и 3, сывороточный альбумин, члены семейства факторов роста фибробластов, тромбоцитарные факторы роста-AA и -BB, плазма, богатая тромбоцитами, инсулиноподобный фактор роста (IGF-I, II), фактор роста и дифференцирования (например, GDF-5, -6, -8, -10, 11), глюкагон-подобный пептид-I и II (GLP-I и II), миметические антитела GLP-1 и GLP-2, экзендин-4, ретиноевая кислота, паратиреоидный гормон, инсулин, прогестерон, апротинин, гидрокортизон, этаноламин, бета-меркаптоэтанол, эпидермальный фактор роста (EGF), гастрин I и II, хелатирующие агенты на основе меди, например триэтиленпентамин, форсколин, бутират натрия, активин, бетацеллюлин, ITS, Noggin, фактор роста нейритов, Nodal, вальпроевая кислота, трихостатинА, натрий бутират, фактор роста гепатоцитов (HGF), сфингозин 1, VEGF, MG132 (EMD, Калифорния, США), добавки N2 и B27 (Gibco, Калифорния, США), стероидный алкалоид, например циклопамин (EMD, Калифорния, США), фактор роста кератиноцитов (KGF), белки семейства Dickkopf, экстракт бычьего гипофиза, белок, ассоциированный с неогенезом островков (INGAP), Indian Hedgehog, Sonic Hedgehog, протеосомальные ингибиторы, ингибиторы сигнального пути Notch, ингибиторы Sonic Hedgehog и их сочетания.
По меньшей мере еще один дополнительный фактор может быть взят из кондиционированной среды, полученной от линий панкреатических клеток, таких как PANC-1 (ATCC No: CRL-1469), CAPAN-1 (ATCC No: HTB-79), BxPC-3 (ATCC No: CRL-1687), HPAF-II (ATCC No: CRL-1997), линий клеток печени, например, HepG2 (ATCC No: HTB-8065), линий клеток кишечника, например, FHs 74 (ATCC No: CCL-241).
Обнаружение клеток, экспрессирующих маркеры, характерные для линии панкреатической эндодермы
Маркеры, характерные для линии панкреатической эндодермы, хорошо известны специалистам в данной области, и дополнительные маркеры, характерные для линии панкреатической эндодермы, продолжают выявляться. Такие маркеры могут использоваться для подтверждения того, что клетки, прошедшие обработку в соответствии с настоящим изобретением, дифференцировались и приобрели характерные особенности линии панкреатической эндодермы. К маркерам, характерным для линии панкреатической эндодермы, относится экспрессия одного или нескольких факторов транскрипции, таких как, например, Hlxb9, PTF-1a, PDX-1, HNF-6, HNF-1beta.
Эффективность дифференцирования может быть определена путем обработки популяции клеток агентом (например, антителом), специфически распознающим белковый маркер, экспрессированный клетками, экспрессирующими маркеры, характерные для линии панкреатической эндодермы.
Способы оценки экспрессии маркеров белков и нуклеиновых кислот в культивированных или выделенных клетках являются стандартными для данной области. К подобным способам относятся количественная полимеразная цепная реакция с обратной транскрипцией (ОТ-ПЦР), Нозерн-блоттинг, гибридизация in situ (см., например, Current Protocols in Molecular Biology (Ausubel et al., eds. 2001 supplement)) и иммунологические анализы, такие как иммуногистохимический анализ среза материала, Вестерн-блоттинг, а для доступных в интактных клетках маркеров - также способ проточной цитометрии (FACS) (см., например, Harlow and Lane, Using Antibodies: A Laboratory Manual, New York: Cold Spring Harbor Laboratory Press (1998)).
Образование клеток, экспрессирующих маркеры, характерные для линии панкреатических эндокринных клеток
Клетки, экспрессирующие маркеры, характерные для линии панкреатической эндодермы, могут быть дифференцированы в клетки, экспрессирующие маркеры, характерные для линии панкреатических эндокринных клеток, с использованием любого способа, известного специалистам в данной области, или с использованием способа, раскрываемого в настоящем изобретении.
Например, клетки, экспрессирующие маркеры, характерные для линии панкреатической эндодермы, могут быть дифференцированы в клетки, экспрессирующие маркеры, характерные для линии панкреатических эндокринных клеток, в соответствии со способами, описанными в публикации D'Amour et al, Nature Biotechnology 24, 1392-1401 (2006).
Например, клетки, экспрессирующие маркеры, характерные для линии панкреатической эндодермы, далее дифференцируются в клетки, экспрессирующие маркеры, характерные для линии панкреатических эндокринных клеток, путем культивирования клеток, экспрессирующих маркеры, характерные для линии панкреатической эндодермы, в среде, содержащей DAPT и экзендин-4, с последующим удалением содержащей DAPT и экзендин-4 среды и культивированием клеток в среде, содержащей экзендин-1, IGF-1 и HGF. Пример использования данного способа приведен в публикации Nature Biotechnology 24, 1392-1401 (2006).
Например, клетки, экспрессирующие маркеры, характерные для линии панкреатической эндодермы, далее дифференцируются в клетки, экспрессирующие маркеры, характерные для линии панкреатических эндокринных клеток, путем культивирования клеток, экспрессирующих маркеры, характерные для линии панкреатической эндодермы, в среде, содержащей экзендин-4, с последующим удалением содержащей экзендин-4 среды и культивированием клеток в среде, содержащей экзендин-1, IGF-1 и HGF. Пример использования данного способа приведен в публикации D' Amour et al, Nature Biotechnology, 2006.
Например, клетки, экспрессирующие маркеры, характерные для линии панкреатической эндодермы, далее дифференцируются в клетки, экспрессирующие маркеры, характерные для линии панкреатических эндокринных клеток, путем культивирования клеток, экспрессирующих маркеры, характерные для линии панкреатической эндодермы, в среде, содержащей DAPT и экзендин-4. Пример использования данного способа приведен в публикации D' Amour et al, Nature Biotechnology, 2006.
Например, клетки, экспрессирующие маркеры, характерные для линии панкреатической эндодермы, далее дифференцируются в клетки, экспрессирующие маркеры, характерные для линии панкреатических эндокринных клеток, путем культивирования клеток, экспрессирующих маркеры, характерные для линии панкреатической эндодермы, в среде, содержащей экзендин-4. Пример использования данного способа приведен в публикации D' Amour et al, Nature Biotechnology, 2006.
В одном из аспектов настоящего изобретения клетки, экспрессирующие маркеры, характерные для линии панкреатической эндодермы, далее дифференцируются в клетки, экспрессирующие маркеры, характерные для линии панкреатических эндокринных клеток, путем обработки клеток, экспрессирующих маркеры, характерные для линии панкреатической эндодермы, фактором, ингибирующим сигнальный путь Notch, в соответствии со способами, описанными в заявке на патент США № 11/736908, принадлежащей LifeScan, Inc.
В одном из аспектов настоящего изобретения клетки, экспрессирующие маркеры, характерные для линии панкреатической эндодермы, далее дифференцируются в клетки, экспрессирующие маркеры, характерные для линии панкреатических эндокринных клеток, путем обработки клеток, экспрессирующих маркеры, характерные для линии панкреатической эндодермы, фактором, ингибирующим сигнальный путь Notch, в соответствии со способами, описанными в заявке на патент США № 11/779311, принадлежащей LifeScan, Inc.
В одном из аспектов настоящего изобретения клетки, экспрессирующие маркеры, характерные для линии панкреатической эндодермы, далее дифференцируются в клетки, экспрессирующие маркеры, характерные для линии панкреатических эндокринных клеток, путем обработки клеток, экспрессирующих маркеры, характерные для линии панкреатической эндодермы, фактором, ингибирующим сигнальный путь Notch, в соответствии со способами, описанными в заявке на патент США № 60/953178, принадлежащей LifeScan, Inc.
В одном из аспектов настоящего изобретения клетки, экспрессирующие маркеры, характерные для линии панкреатической эндодермы, дифференцируются в клетки, экспрессирующие маркеры, характерные для линии панкреатических эндокринных клеток, путем обработки клеток, экспрессирующих маркеры, характерные для линии панкреатической эндодермы, в соответствии со способами, описанными в заявке на патент США № 60/990529.
Клетки, экспрессирующие маркеры, характерные для линии панкреатической эндодермы, могут быть обработаны по меньшей мере еще одним дополнительным фактором, который может стимулировать образование клеток, экспрессирующих маркеры, характерные для линии панкреатических эндокринных клеток. В качестве альтернативы по меньшей мере еще один дополнительный фактор может стимулировать пролиферацию клеток, экспрессирующих маркеры, характерные для линии панкреатических эндокринных клеток, полученных в соответствии со способами, составляющими предмет настоящего изобретения. Кроме того, по меньшей мере еще один дополнительный фактор может стимулировать способность клеток, экспрессирующих маркеры, характерные для линии панкреатических эндокринных клеток, полученных в соответствии со способами, составляющими предмет настоящего изобретения, образовывать другие типы клеток или повышать эффективность дополнительных этапов дифференцирования.
По меньшей мере еще одним дополнительным фактором может быть, например, никотинамид, члены семейства TGF-β, включая TGF-β1, 2 и 3, сывороточный альбумин, члены семейства факторов роста фибробластов, тромбоцитарные факторы роста-AA и -BB, плазма, богатая тромбоцитами, инсулиноподобный фактор роста (IGF-I, II), фактор роста и дифференцирования (например, GDF-5, -6, -8, -10, 11), глюкагон-подобный пептид-I и II (GLP-I и II), миметические антитела GLP-1 и GLP-2, экзендин-4, ретиноевая кислота, паратиреоидный гормон, инсулин, прогестерон, апротинин, гидрокортизон, этаноламин, бета-меркаптоэтанол, эпидермальный фактор роста (EGF), гастрин I и II, хелатирующие агенты на основе меди, например, триэтиленпентамин, форсколин, бутират натрия, активин, бетацеллюлин, ITS, Noggin, фактор роста нейритов, Nodal, вальпроевая кислота, трихостатинА, натрий бутират, фактор роста гепатоцитов (HGF), сфингозин 1, VEGF, MG132 (EMD, Калифорния, США), добавки N2 и B27 (Gibco, Калифорния, США), стероидный алкалоид, например, циклопамин (EMD, Калифорния, США), фактор роста кератиноцитов (KGF), белки семейства Dickkopf, экстракт бычьего гипофиза, белок, ассоциированный с неогенезом островков (INGAP), Indian Hedgehog, Sonic Hedgehog, протеосомальные ингибиторы, ингибиторы сигнального пути Notch, ингибиторы Sonic Hedgehog и их сочетания.
По меньшей мере еще один дополнительный фактор может быть взят из кондиционированной среды, полученной от линий панкреатических клеток, таких как PANC-1 (ATCC No: CRL-1469), CAPAN-1 (ATCC No: HTB-79), BxPC-3 (ATCC No: CRL-1687), HPAF-II (ATCC No: CRL-1997), линий клеток печени, например, HepG2 (ATCC No: HTB-8065), линий клеток кишечника, например, FHs 74 (ATCC No: CCL-241).
Обнаружение клеток, экспрессирующих маркеры, характерные для линии панкреатических эндокринных клеток
Маркеры, характерные для линии панкреатических эндокринных клеток, хорошо известны специалистам в данной области, и продолжается выявление дополнительных маркеров, характерных для линии панкреатических эндокринных клеток. Такие маркеры могут использоваться для подтверждения того, что клетки, прошедшие обработку в соответствии с настоящим изобретением, дифференцировались и приобрели характерные особенности линии панкреатических эндокринных клеток. К маркерам, характерным для линии панкреатических эндокринных клеток, относится экспрессия одного или нескольких факторов транскрипции, таких как, например, NGN3, NEURO или ISL1.
Маркеры, характерные для клеток β-клеточной линии дифференцирования, хорошо известны специалистам в данной области, и продолжается выявление дополнительных маркеров, характерных для β-клеточной линии дифференцирования. Такие маркеры могут использоваться для подтверждения того, что клетки, прошедшие обработку в соответствии с настоящим изобретением, дифференцировались и приобрели свойства, характерные для β-клеточной линии дифференцирования. К специфичным характеристикам β -клеточной линии дифференцирования относится экспрессия одного или нескольких факторов транскрипции, таких как, например, PDX1, NKX2.2, NKX6.1, ISL1, PAX6, PAX4, NEUROD, HNF1 beta, HNF6, HNF3 beta или MAFA. Такие факторы транскрипции широко используются в данной области для идентификации эндокринных клеток. См., например, обзор Edlund (Nature Reviews Genetics 3: 524-632 (2002)).
Эффективность дифференцирования может быть определена путем обработки популяции клеток агентом (например, антителом), специфически распознающим белковый маркер, экспрессированный клетками, экспрессирующими маркеры, характерные для линии панкреатических эндокринных клеток. В качестве альтернативы эффективность дифференцирования может быть определена путем обработки популяции клеток агентом (например, антителом), специфически распознающим белковый маркер, экспрессированный клетками, экспрессирующими маркеры, характерные для β-клеточной линии дифференцирования.
Способы оценки экспрессии маркеров белков и нуклеиновых кислот в культивированных или выделенных клетках являются стандартными для данной области. К подобным способам относятся количественная полимеразная цепная реакция с обратной транскрипцией (ОТ-ПЦР), Нозерн-блоттинг, гибридизация in situ (см., например, Current Protocols in Molecular Biology (Ausubel et al., eds. 2001 supplement)) и иммунологические анализы, такие как иммуногистохимический анализ среза материала, Вестерн-блоттинг, а для доступных в интактных клетках маркеров - также способ проточной цитометрии (FACS) (см., например, Harlow and Lane, Using Antibodies: A Laboratory Manual, New York: Cold Spring Harbor Laboratory Press (1998)).
В одном из аспектов настоящего изобретения эффективность дифференцирования определялась путем измерения процентной доли инсулин-положительных клеток в данной клеточной культуре после обработки. В одном из вариантов осуществления способы, составляющие предмет настоящего изобретения, давали приблизительно 100% инсулин-положительных клеток в данной культуре. В альтернативном варианте осуществления способы, составляющие предмет настоящего изобретения, давали приблизительно 90% инсулин-положительных клеток в данной культуре. В альтернативном варианте осуществления способы, составляющие предмет настоящего изобретения, давали приблизительно 80% инсулин-положительных клеток в данной культуре. В альтернативном варианте осуществления способы, составляющие предмет настоящего изобретения, давали приблизительно 70% инсулин-положительных клеток в данной культуре. В альтернативном варианте осуществления способы, составляющие предмет настоящего изобретения, давали приблизительно 60% инсулин-положительных клеток в данной культуре. В альтернативном варианте осуществления способы, составляющие предмет настоящего изобретения, давали приблизительно 50% инсулин-положительных клеток в данной культуре. В альтернативном варианте осуществления способы, составляющие предмет настоящего изобретения, давали приблизительно 40% инсулин-положительных клеток в данной культуре. В альтернативном варианте осуществления способы, составляющие предмет настоящего изобретения, давали приблизительно 30% инсулин-положительных клеток в данной культуре. В альтернативном варианте осуществления способы, составляющие предмет настоящего изобретения, давали приблизительно 20% инсулин-положительных клеток в данной культуре. В альтернативном варианте осуществления способы, составляющие предмет настоящего изобретения, давали приблизительно 10% инсулин-положительных клеток в данной культуре. В альтернативном варианте осуществления способы, составляющие предмет настоящего изобретения, давали приблизительно 5% инсулин-положительных клеток в данной культуре.
В одном из аспектов настоящего изобретения эффективность дифференцирования определялась путем измерения стимулированной глюкозой секреции инсулина, определявшейся по количеству С-пептида, секретируемого клетками. В одном из вариантов осуществления клетки, полученные в соответствии со способами, составляющими предмет настоящего изобретения, производили приблизительно 1000 нг C-пептида/пг ДНК. В альтернативном варианте осуществления клетки, полученные в соответствии со способами, составляющими предмет настоящего изобретения, производили приблизительно 900 нг C-пептида/пг ДНК. В альтернативном варианте осуществления клетки, полученные в соответствии со способами, составляющими предмет изобретения, производили приблизительно 800 нг C-пептида/пг ДНК. В альтернативном варианте осуществления клетки, полученные в соответствии со способами, составляющими предмет настоящего изобретения, производили приблизительно 700 нг C-пептида/пг ДНК. В альтернативном варианте осуществления клетки, полученные в соответствии со способами, составляющими предмет настоящего изобретения, производили приблизительно 600 нг C-пептида/пг ДНК. В альтернативном варианте осуществления клетки, полученные в соответствии со способами, составляющими предмет настоящего изобретения, производили приблизительно 500 нг C-пептида/пг ДНК. В альтернативном варианте осуществления клетки, полученные в соответствии со способами, составляющими предмет настоящего изобретения, производили приблизительно 400 нг C-пептида/пг ДНК. В альтернативном варианте осуществления клетки, полученные в соответствии со способами, составляющими предмет настоящего изобретения, производили приблизительно 500 нг C-пептида/пг ДНК. В альтернативном варианте осуществления клетки, полученные в соответствии со способами, составляющими предмет настоящего изобретения, производили приблизительно 400 нг C-пептида/пг ДНК. В альтернативном варианте осуществления клетки, полученные в соответствии со способами, составляющими предмет настоящего изобретения, производили приблизительно 300 нг C-пептида/пг ДНК. В альтернативном варианте осуществления клетки, полученные в соответствии со способами, составляющими предмет настоящего изобретения, производили приблизительно 200 нг C-пептида/пг ДНК. В альтернативном варианте осуществления клетки, полученные в соответствии со способами, составляющими предмет настоящего изобретения, производили приблизительно 100 нг C-пептида/пг ДНК. В альтернативном варианте осуществления клетки, полученные в соответствии со способами, составляющими предмет настоящего изобретения, производили приблизительно 90 нг C-пептида/пг ДНК. В альтернативном варианте осуществления клетки, полученные в соответствии со способами, составляющими предмет настоящего изобретения, производили приблизительно 80 нг C-пептида/пг ДНК. В альтернативном варианте осуществления клетки, полученные в соответствии со способами, составляющими предмет настоящего изобретения, производили приблизительно 70 нг C-пептида/пг ДНК. В альтернативном варианте осуществления клетки, полученные в соответствии со способами, составляющими предмет настоящего изобретения, производили приблизительно 60 нг C-пептида/пг ДНК. В альтернативном варианте осуществления клетки, полученные в соответствии со способами, составляющими предмет настоящего изобретения, производили приблизительно 50 нг C-пептида/пг ДНК. В альтернативном варианте осуществления клетки, полученные в соответствии со способами, составляющими предмет настоящего изобретения, производили приблизительно 40 нг C-пептида/пг ДНК. В альтернативном варианте осуществления клетки, полученные в соответствии со способами, составляющими предмет настоящего изобретения, производили приблизительно 30 нг C-пептида/пг ДНК. В альтернативном варианте осуществления клетки, полученные в соответствии со способами, составляющими предмет настоящего изобретения, производили приблизительно 20 нг C-пептида/пг ДНК. В альтернативном варианте осуществления клетки, полученные в соответствии со способами, составляющими предмет настоящего изобретения, производили приблизительно 10 нг C-пептида/пг ДНК.
Способы лечения
В одном из аспектов настоящего изобретения предлагается способ лечения пациентов, страдающих от диабета 1 типа или подвергающихся риску развития этого заболевания. Такой способ включает культивирование плюрипотентных стволовых клеток, дифференцирование плюрипотентных стволовых клеток in vitro в β-клеточную линию дифференцирования и имплантирование клеток β-клеточной линии дифференцирования пациенту.
В еще одном аспекте настоящего изобретения предлагается способ лечения пациентов, страдающих от диабета 2 типа или подвергающихся риску развития этого заболевания. Такой способ включает культивирование плюрипотентных стволовых клеток, дифференцирование культивированных клеток in vitro в β-клеточную линию дифференцирования и имплантирование β-клеточной линии дифференцирования пациенту.
При необходимости, пациент дополнительно может получать лечение фармацевтическими средствами или биологически активными веществами, улучшающими выживаемость и функционирование трансплантированных клеток. К таким агентам могут относиться, например, инсулин, члены семейства TGF-β, включая TGF-β 1, 2 и 3, костные морфогенетические белки (BMP-2, -3, -4, -5, -6, -7, -11, -12 и -13), факторы роста фибробластов-1 и -2, тромбоцитарный фактор роста-AA и -BB, плазма, богатая тромбоцитами, инсулинподобный фактор роста (IGF-I, II), фактор роста и дифференцирования (GDF-5, -6, -7, -8, -10, -15), фактор роста из клеток эндотелия сосудов (VEGF), плейотропин, эндотелин. К другим фармацевтическим средствам могут относиться, например, никотинамид, глюкагон-подобный пептид-I (GLP-1) и II, миметические антитела GLP-1 и -2, экзендин-4, ретиноевая кислота, паратиреоидный гормон, ингибиторы MAPK, такие, например, как описаны в заявке на патент США № 2004/0209901 and заявке на патент США № 2004/0132729.
Плюрипотентные стволовые клетки могут быть дифференцированы в инсулин-продуцирующие клетки перед трансплантацией реципиенту. В конкретном варианте реализации плюрипотентные стволовые клетки являются полностью дифференцированными в β-клетки перед трансплантацией реципиенту. В качестве альтернативы плюрипотентные стволовые клетки могут быть трансплантированы реципиенту в недифференцированном или частично дифференцированном состоянии. Дальнейшее дифференцирование может происходить в организме реципиента.
Клетки сформированной эндодермы или, в качестве альтернативы, клетки панкреатической эндодермы, или, в качестве альтернативы, β-клетки могут имплантироваться в форме дисперсных клеток или клеток, образовавших кластеры, которые инфузионно могут вводиться в воротную вену печени. В качестве альтернативы клетки могут вводиться в биологически-совместимых разрушающихся полимерных вспомогательных материалах, в пористых неразрушающихся приспособлениях или в инкапсулированном виде для защиты от иммунного ответа организма-хозяина. Клетки могут имплантироваться в подходящее место организма реципиента. К таким местам для имплантации относятся, например, печень, естественная поджелудочная железа, пространство под почечной капсулой, сальник, брюшная полость, субсерозное пространство, кишечник, желудок или подкожный карман.
Для стимуляции дальнейшего дифференцирования, выживания и активности имплантированных клеток предварительно, одновременно или после имплантации клеток могут вводиться дополнительные факторы, такие как факторы роста, антиоксиданты или противовоспалительные агенты. В некоторых вариантах осуществления факторы роста применяются для дифференцирования введенных клеток in vivo. Такие факторы могут секретироваться эндогенными клетками и воздействовать на введенные клетки in situ. Дифференцирование имплантированных клеток можно индуцировать любым сочетанием эндогенных и введенных экзогенно факторов роста, известных специалистам в данной области.
Число клеток, используемое при имплантации, зависит от ряда различных факторов, в том числе от состояния пациента и его реакции на лечение, и может быть определено специалистом в данной области.
В одном из аспектов настоящего изобретения предлагается способ лечения пациентов, страдающих от диабета или подвергающихся риску развития этого заболевания. Такой способ включает культивирование плюрипотентных стволовых клеток, дифференцирование культивированных стволовых клеток in vitro в β-клеточную линию дифференцирования и заключение клеток в опорный материал с трехмерной структурой. Клетки могут поддерживаться in vitro на этом опорном материале до имплантации пациенту. В качестве альтернативы опорный материал, содержащий клетки, может напрямую имплантироваться в организм пациента без дополнительного культивирования in vitro. В опорный материал может быть заключено по меньшей мере одно фармацевтическое средство, улучшающее выживание и функционирование трансплантированных клеток.
К опорным материалам, соответствующим целям настоящего изобретения, относятся тканевые шаблоны, каналы, перегородки и резервуары, применяемые для восстановления тканей. В частности, соответствуют целям практического применения способов настоящего изобретения синтетические и природные материалы в форме пены, губок, гелей, гидрогелей, тканных и нетканых структур, применяемых in vitro и in vivo для реконструкции и регенерации биологической ткани, а также для доставки хемотаксических агентов, индуцирующих рост ткани. См., например, материалы, содержащиеся в патентах США №№ 5770417, 6022743, 5567612, 5759830, 6626950, 6534084, 6306424, 6365149, 6599323, 6656488, заявке на патент США № 2004/0062753 A1, патентах США №№ 4557264 и 6333029.
Для создания опорного материала с включенным фармацевтическим средством такое средство можно смешать с раствором полимера до изготовления опорной структуры. В качестве альтернативы фармацевтическое средство можно нанести на изготовленную опорную структуру, предпочтительно в присутствии фармацевтического носителя. Фармацевтическое средство может быть в виде жидкости, мелкодисперсного твердого вещества или иметь любую другую подходящую физическую форму. В качестве альтернативы опорный материал может содержать инертные наполнители для изменения скорости высвобождения фармацевтического средства. В альтернативном варианте осуществления опорный материал содержит по меньшей мере одно фармацевтическое средство, являющееся противовоспалительным средством, например, средства, описанные в патенте США № 6509369.
Опорный материал может содержать по меньшей мере одно фармацевтическое средство, являющееся антиапоптозным средством, как например, средства, описанные в патенте США № 6793945.
Опорный материал дополнительно может содержать по меньшей мере одно фармацевтическое средство, являющееся ингибитором фиброза, как например, средства, описанные в патенте США № 6331298.
Опорный материал дополнительно может содержать по меньшей мере одно фармацевтическое средство, являющееся стимулятором ангиогенеза, как например, средства, описанные в заявках на патенте США №№ 2004/0220393 и 2004/0209901.
Опорный материал дополнительно может содержать по меньшей мере одно фармацевтическое средство, являющееся иммуносупрессором, как например, средства, описанные в заявке на патенте США № 2004/0171623.
Опорный материал дополнительно может содержать по меньшей мере одно фармацевтическое средство, представляющее собой фактор роста, например, члены семейства TGF-β, включая TGF-β1, 2 и 3, костные морфогенетические белки (BMP-2, -3, -4, -5, -6, -7, -11, -12 и -13), факторы роста фибробластов-1 и -2, тромбоцитарный фактор роста-AA и -BB, плазма, богатая тромбоцитами, инсулинподобный фактор роста (IGF-I, II), фактор роста и дифференцирования (GDF-5, -6, -8, -10, -15), фактор роста из клеток эндотелия сосудов (VEGF), плейотропин, эндотелин. Другие фармацевтические средства могут включать, например, никотинамид, индуцированный гипоксией фактор 1-alpha, глюкагон-подобный пептид-I (GLP-1), миметические антитела GLP-1 и GLP-2, и II, экзендин-4, Nodal, Noggin, NGF, ретиноевую кислоту, паратиреоидный гормон, тенасцин-C, тропоэластин, пептиды тромбинового происхождения, кателицидины, дефензины, ламинин, биологические пептиды, содержащие связывающие клетки и гепарин домены протеинов адгезивного внеклеточного матрикса, такие как фибронектин и витронектин, ингибиторы MAPK, такие как средства, описанные в заявках на патенте США №№ 2004/0209901 и 2004/0132729.
Включение клеток, составляющих предмет настоящего изобретения, в опорный каркас может осуществляться путем простого нанесения клеток на опорный каркас. Клетки могут входить внутрь каркаса путем простой диффузии (J. Pediatr. Surg. 23 (1 Pt 2): 3-9 (1988)). Было разработано несколько других подходов для повышения эффективности посева клеток. Например, для посева хондроцитов на каркасы из полигликолевой кислоты используются центрифужные пробирки (Biotechnol. Prog. 14(2): 193-202 (1998)). Другой подход к посеву клеток представляет собой использование центрифугирования, создающего минимальный стресс для высеиваемых клеток и повышающий эффективность посева. Например, в публикации Yang et al. разработан способ посева клеток (J. Biomed. Mater. Res. 55(3): 379-86 (2001)), названный «центрифужной иммобилизацией клеток (CCI)».
Настоящее изобретение далее иллюстрируется, помимо прочего, следующими примерами.
ПРИМЕРЫ
Для ясности описания, а не в ограничение изобретения, приведенное ниже подробное описание изобретения разделено на следующие подразделы, описывающие или иллюстрирующие определенные особенности, варианты осуществления или области применения настоящего изобретения.
Пример 1
Культура эмбриональных стволовых клеток человека
Эмбриональные стволовые клетки человека линий H1, H7 и H9 получили из института WiCell Research Institute, Inc. (Мадисон, Висконсин) и культивировали в соответствии с инструкциями, предоставленными указанным выше институтом. Эмбриональные стволовые клетки человека также высеивали на планшеты, покрытые слоем препарата MATRIGEL™ с пониженным содержанием факторов роста в концентрации 1:30 (BD Biosciences; № по кат. 356231), и культивировали в кондиционированной среде MEF с добавлением 8 нг/мл bFGF (R&D Systems; № по кат. 233-FB). Культивированные на препарате MATRIGEL™ клетки обычным образом пассировали в виде кластеров с использованием коллагеназы IV (Invitrogen/GIBCO; № по кат. 17104-019), диспазы (Invitrogen; № по кат. 17105-041) или ферментативной смеси Liberase CI (Roche; № по кат. 11814435001). В некоторых случаях клетки пассировали в виде отдельных клеток с использованием препарата ACCUTASE (Sigma; № по кат. A6964).
Эмбриональные стволовые клетки человека, использованные в этих примерах, поддерживали в недифференцированном плюрипотентном состоянии со средним интервалом между последовательными пассированиями четыре дня. При пассировании культуры клеток обрабатывали раствором коллагеназы (1 или 10 мг/мл; Sigma-Aldrich) в течение 10-30 минут при температуре 37єC, а затем осторожно соскоблили кончиком пипетки, получив кластеры клеток. Полученным кластерам дали осесть под собственным весом и затем промыли их для удаления остатков коллагеназы. Кластеры клеток разделили в соотношении 1:3 для стандартного поддерживающего культивирования или в соотношении 1:1 для последующего анализа. Все линии эмбриональных стволовых клеток человека поддерживали при номерах пассажа менее 50 и периодически проверяли на нормальный кариотипический фенотип и отсутствие примесей микоплазмы.
Пример 2
Биологический анализ на образование клеток, экспрессирующих маркеры, характерные для линии сформированной эндодермы
Активин A является важным медиатором дифференцирования для широкого спектра типов клеток, включая дифференцирование эмбриональных стволовых клеток в линию сформированной эндодермы. Если эмбриональные стволовые клетки человека обрабатывают сочетанием активина A и Wnt3a, то увеличивается экспрессия генов, характерных для линии сформированной эндодермы. Биологический анализ, с помощью которого измеряется такое дифференцирование в эмбриональных стволовых клетках человека, для скрининга адаптировали к 96-луночным планшетам в миниатюрном формате. Проверку достоверности осуществляли путем обработки полученными из коммерческих источников рекомбинантными активином A и Wnt3a и измерения уровня экспрессии белков фактора транскрипции SOX17, который считается характерным маркером сформированной эндодермы.
Анализ живых клеток. Коротко говоря, кластеры эмбриональных стволовых клеток человека линии H1 выращивали на покрытых препаратом MATRIGELTM с пониженным содержанием факторов роста (Invitrogen; № по кат. 356231) носителях для культивирования тканей. Клетки пассировали путем обработки коллагеназой (Invitrogen; № по кат. 17104-019), затем осторожно соскоблили, промыли для удаления остатков фермента и высеяли в соотношении 1:1 (по площади поверхности) на покрытые препаратом MATRIGELTM с пониженным содержанием факторов роста 96-луночные черные планшеты (Packard ViewPlates; Perkin Elmer; № по кат. 6005182). Клеткам дали возможность прикрепиться (кластерами), после чего в течение 1-3 дней воспроизводили логарифмическую фазу роста, во время которой клетки ежедневно питали, добавляя в каждую лунку 100 мкл кондиционированной мышиными эмбриональными фибробластами (MEF) среды с добавлением 8 нг/мл bFGF (R&D Systems; № по кат. 233-FB).
Анализ начали с двукратной промывки лунок каждого планшета раствором PBS (Invitrogen; № по кат. 14190) с последующим добавлением в каждую лунку аликвоты (100 мкл) тестируемого образца в основной среде DMEM:F12 (Invitrogen; № по кат. 11330-032). Каждый тест выполняли в трех повторностях. В течение всего четырехдневного периода питание вводили через день, для чего среду из каждой лунки отсасывали и заменяли тестируемыми образцами. В 1 и 2 дни анализа тестируемые образцы, добавленные в лунки, разбавили средой DMEM:F12 с добавлением 0,5% FCS (HyClone; № по кат. SH30070.03) и 20 нг/мл Wnt3a (R&D Systems; № по кат. 1324-WN). На 3 и 4 день анализа тестируемые образцы, добавленные в лунки, разбавили средой DMEM:F12 с добавлением 2% FCS, без Wnt3a. Положительные контрольные образцы в течение всего периода анализа содержали рекомбинантый человеческий активин A (PeproTech; № по кат. 120-14) в концентрации 100 нг/мл и Wnt3a (20 нг/мл) в 1 и 2 дни. Отрицательные контрольные образцы не обрабатывали ни активином А, ни Wnt3a.
Одновременный многопараметрический анализ. По окончании четырехдневного периода культивирования аналитические планшеты дважды промыли раствором PBS (Invitrogen; № по кат. 14190), зафиксировали 4% параформальдегидом (Alexis Biochemical; № по кат. ALX-350-011) при комнатной температуре в течение 20 минут, затем трижды промыли раствором PBS и пермеабилизировали, обработав раствором 0,5% Triton X-100 (Sigma; № по кат. T8760-2) в течение 20 минут при комнатной температуре. Затем клетки снова трижды промыли раствором PBS и заблокировали раствором 4% куриной сыворотки (Invitrogen; № по кат. 16110082) в растворе PBS в течение 30 минут при комнатной температуре. Первичные антитела (козий анти-человеческий SOX17; R&D Systems; № по кат. AF1924) разбавили в соотношении 1:100 в 4% куриной сыворотке и добавили в каждую лунку на один час при комнатной температуре. Сопряженные с флюорофором Alexa Fluor 488 вторичные антитела (куриный анти-козий IgG; Molecular Probes; № по кат. AZ1467) разбавили в соотношении 1:200 в растворе PBS и добавили в каждую лунку после трехкратной промывки раствором PBS. Для подсчета окрашенных ядер на десять минут при комнатной температуре добавили 4 мкг/мл Hoechst 33342 (Invitrogen; № по кат. H3570). Затем планшеты один раз промыли раствором PBS и оставили в 100 мкл раствора PBS на лунку для томографии.
Томографию проводили на анализаторе IN Cell Analyzer 1000 (GE Healthcare) с применением дихроичного зеркала 51008bs для клеток, окрашенных красителями Hoechst 33342 и Alexa Fluor 488. Времена экспозиции оптимизировали с использованием лунок с положительными контрольными образцами и необработанных лунок с отрицательными контрольными образцами, окрашенных только вторичными антителами. Для компенсации возможных потерь клеток в ходе процедур анализа и последующего окрашивания для каждой лунки снимали по 15 проекций. Общее число клеток и общую интенсивность сигнала SOX17 измеряли для каждой лунки с помощью программного пакета IN Cell Developer Toolbox 1.7 (GE Healthcare). Сегментацию ядер определяли по ступеням шкалы серых тонов (исходный интервал 100-300) и размерам ядра. Для каждой повторной совокупности данных рассчитали средние значение и стандартные отклонения. Общий уровень экспрессии белка SOX17 выражается в виде общей интенсивности или интегральной интенсивности, определяемой как произведение общей интенсивности флюоресценции клетки на площадь клетки. Фон исключили на основании критериев соответствия ступеням шкалы серых тонов в диапазоне от 200 до 3500. Показатели общей интенсивности нормировали путем деления общей интенсивности для каждой лунки на среднюю общую интенсивность для положительного контрольного образца. Для каждого набора повторностей рассчитали нормированные данные для средних значений и их стандартных отклонений.
На фиг. 1 представлено подтверждение результатов скринингового анализа: тестирование кривой двукратных разведений коммерческого активина A (PeproTech) с измерением числа клеток (фиг. 1A) и интенсивности сигнала SOX17 (фиг. 1B). Оптимальный эффект активина А на индукцию экспрессии SOX17, в целом, наблюдали в диапазоне концентраций 100-200 нг/мл, а значение EC50 составило 30-50 нг/мл. При отсутствии обработки Wnt3a в 1 и 2 дни анализа измеримой экспрессии SOX17 не наблюдали (фиг. 1B, белые столбцы). При отсутствии активина A экспрессия SOX17 также не наблюдалась (фиг. 1B).
Пример 3
Первичный скрининг. Эффект соединений, составляющих предмет настоящего изобретения, на дифференцирование эмбриональных стволовых клеток человека в клетки, экспрессирующие маркеры, характерные для линии сформированной эндодермы, в отсутствие активина А
Дифференцирование плюрипотентных стволовых клеток в клетки, экспрессирующие маркеры, характерные для линии сформированной эндодермы, опосредуется рядом взаимодействий лиганд-рецептор, которые, в свою очередь, активируют рецепторные киназы, что ведет к фосфорилированию и транслокации в ядро субстратов, которые находятся дальше по каскаду, в конечном счете приводя к регуляции экспрессии определенных генов-мишеней. Для оптимальной активации таких сигнальных каскадов в некоторых типах клеток может потребоваться ингибирование исходно активированных путей противоположного действия. В других случаях одну или несколько сигнальных молекул могут частично заменить дублирующие сигнальные пути с участием альтернативных членов большого семейства киназ. В других случаях канонические и неканонические сигнальные пути могут расходиться при воздействии разных инициирующих стимулов, но вести к сходному функциональному результату.
Одним из подходов к идентификации новых мишеней и способов, влияющих на специфические клеточные ответы, является клеточный функциональный скрининг. Один из наиболее эффективных подходов связан с серией повторяющихся скрининговых анализов, при которых итоги или положительные результаты одного исследования используются в последующем исследовании. В качестве альтернативы несколько различных переменных величин комбинируют друг с другом (например, факторы роста и ингибиторы киназ), выявляя новые эффекты на дифференцирование клеток. В данном случае было проведено тестирование библиотеки низкомолекулярных соединений, состоящей из анилин-пиридинотриазинов, циклических анилин-пиридинотриазинов и промежуточных продуктов их синтеза, на предмет наличия свойств, важных для дифференцирования эмбриональных стволовых клеток человека в сформированную эндодерму, в частности, на предмет наличия эффектов, связанных с сохранением или увеличением числа клеток по окончании «первого» этапа дифференцирования в среде с низким содержанием сыворотки и в отсутствие фактора роста активина А.
Скрининговый анализ
Посев клеток для анализа. Коротко говоря, кластеры эмбриональных стволовых клеток человека H1 выращивали на покрытых препаратом MATRIGELTM с пониженным содержанием факторов роста (Invitrogen; № по кат. 356231) носителях для культивирования тканей. Клетки пассировали путем обработки коллагеназой (Invitrogen; № по кат. 17104-019), затем осторожно соскоблили, промыли для удаления остатков фермента и высеяли в соотношении 1:1 (по площади поверхности) на покрытые препаратом MATRIGELTM с пониженным содержанием факторов роста 96-луночные черные планшеты (Packard ViewPlates; PerkinElmer; № по кат. 6005182) в объеме 100 мкл на лунку. Клеткам дали возможность прикрепиться (кластерами), после чего в течение 1-3 дней воспроизводили логарифмическую фазу роста, во время которой клетки ежедневно питали, используя кондиционированную среду MEF с добавлением 8 нг/мл bFGF (R&D Systems; № по кат. 233-FB). В течение всего периода анализа планшеты держали при температуре 37єC в атмосфере 5% CO2 в увлажняемом боксе.
Подготовка соединений и анализ. Тестируемые соединения предоставили в виде исходных растворов концентрации 5 мМ на 96-луночных планшетах, растворили в 100% DMSO (Sigma; № по кат. D2650) и хранили при температуре -80єC. Соединения из такой библиотеки затем разбавили до промежуточной концентрации 0,2 мМ в 50 мМ HEPES (Invitrogen; № по кат. 15630-080), 20% DMSO и хранили при температуре 4єC. Каждый тест выполняли в трех повторностях. В течение всего четырехдневного периода анализа питание вводили через день. Первичный скрининговый анализ начали с отсасывания культуральной среды из каждой лунки с последующей трехкратной промывкой лунки раствором PBS (Invitrogen; № по кат. 14190) для удаления остатков факторов роста и сыворотки. В 1 день анализа в лунки добавили тестируемые образцы объемом 200 мкл, содержащие основную среду DMEM:F12 (Invitrogen; № по кат. 11330-032) с добавлением 0,5% FCS (HyClone; № по кат. SH30070.03), 20 нг/мл Wnt3a (R&D Systems; № по кат. 1324-WN) и 2,5 мкМ тестируемого соединения. На 3 день анализа в лунки добавили тестируемые образцы объемом 200 мкл, содержащие основную среду DMEM:F12 с добавлением 2% FCS и 2,5 мкМ тестируемого соединения, без Wnt3a. Положительные контрольные образцы содержали ту же основную среду с добавлением FCS, за исключением того, что вместо тестируемого соединения в течение всего четырехдневного периода анализа среда содержала 100 нг/мл рекомбинантного человеческого активина A (PeproTech; № по кат. 120-14), с добавлением 20 нг/мл Wnt3a в 1 и 2 дни. Отрицательные контрольные образцы содержали основную среду DMEM:F12 с добавлением FCS, которая не содержала активина А, но в дни 1 и 2 к ней добавляли Wnt3a.
Одновременный многопараметрический анализ. По окончании четырехдневного периода культивирования аналитические планшеты дважды промыли раствором PBS (Invitrogen; № по кат. 14190), зафиксировали 4% параформальдегидом (Alexis Biochemical; № по кат. ALX-350-011) при комнатной температуре в течение 20 минут, затем трижды промыли раствором PBS и пермеабилизировали, обработав раствором 0,5% Triton X-100 (Sigma; № по кат. T8760-2) в течение 20 минут при комнатной температуре. Затем клетки снова трижды промыли раствором PBS и заблокировали раствором 4% куриной сыворотки (Invitrogen; № по кат. 16110082) в PBS в течение 30 минут при комнатной температуре. Первичные антитела (козий анти-человеческий SOX17; R&D Systems; № по кат. AF1924) разбавили в соотношении 1:100 в 4% куриной сыворотке и добавили в каждую лунку на один час при комнатной температуре. Сопряженные с флюорофором Alexa Fluor 488 вторичные антитела (куриный анти-козий IgG; Molecular Probes; № по кат. AZ1467) разбавили в соотношении 1:200 в растворе PBS и добавили к каждому образцу после трехкратной промывки раствором PBS. Для подсчета окрашенных ядер на десять минут при комнатной температуре добавили 4 мкг/мл Hoechst 33342 (Invitrogen; № по кат. H3570). Затем планшеты один раз промыли раствором PBS и оставили в 100 мкл раствора PBS на лунку для томографии.
Томографию проводили на анализаторе IN Cell Analyzer 1000 (GE Healthcare) с применением дихроичного зеркала 51008bs для клеток, окрашенных красителями Hoechst 33342 и Alexa Fluor 488. Времена экспозиции оптимизировали с использованием лунок с положительными контрольными образцами и необработанных лунок с отрицательными контрольными образцами, окрашенных только вторичными антителами. Для компенсации возможных потерь клеток в ходе процедур анализа и последующего окрашивания для каждой лунки снимали по 15 проекций. Общее число клеток и общую интенсивность сигнала SOX17 измеряли для каждой лунки с помощью программного пакета IN Cell Developer Toolbox 1.7 (GE Healthcare). Сегментацию ядер определяли по ступеням шкалы серых тонов (исходный интервал 100-300) и размерам ядра. Для каждой повторной совокупности данных рассчитали средние значение и стандартные отклонения. Общий уровень экспрессии белка SOX17 выражается в виде общей интенсивности или интегральной интенсивности, определяемой как произведение общей интенсивности флюоресценции клетки на площадь клетки. Фон исключили на основании критериев соответствия ступеням шкалы серых тонов в диапазоне от 200 до 3500. Показатели общей интенсивности нормировали путем деления общей интенсивности для каждой лунки на среднюю общую интенсивность для положительного контрольного образца. Для каждого набора повторностей рассчитали нормированные данные для средних значений и стандартных отклонений.
В Таблице 1 представлены результаты первичного скринингового анализа соединений, демонстрирующие эффект этих соединений на дифференцирование эмбриональных стволовых клеток человека в клетки, экспрессирующие маркеры, характерные для линии сформированной эндодермы, в отсутствие активина А. Результаты содержат количественные показатели числа клеток и интенсивности сигнала SOX17, где соответствующие точки усреднены по трем лункам и проанализированы по каждому параметру с применением идентичных проекций для каждой лунки. Экспрессия фактора транскрипции SOX17 считается показателем дифференцирования в линию сформированной эндодермы. Результаты первичного скрининга были получены с восьми 96-луночных планшетов. Вариабельность результатов между планшетами была снижена благодаря включению в каждый планшет лунок с положительными и отрицательными контрольными образцами. Результаты нормированы и выражены в процентах от положительного контрольного образца. Акцент делался на сохранении или увеличении числа клеток в конце анализа.
В Таблице 2 приводится список тех 27 соединений, с результатами их анализа, полученными в ходе первичного скрининга, где число клеток сохранялось или превышало положительный контрольный образец, несмотря на отсутствие при проведении анализа активина А.
В некоторых случаях экспрессия SOX17 индуцировалась в отсутствие активина A (например, циклические анилин-пиридинотриазины, обозначенные как Соединение 35 и Соединение 22).
Соединения, представленные в Таблице 2, были отобраны для проведения дальнейшего анализа эффекта на дифференцирование эмбриональных стволовых клеток человека в клетки, экспрессирующие маркеры, характерные для линии сформированной эндодермы, в отсутствие активина A.
Пример 4
Вторичный скрининговый анализ. Эффект соединений, составляющих предмет настоящего изобретения, на дифференцирование эмбриональных стволовых клеток человека в клетки, экспрессирующие маркеры, характерные для линии сформированной эндодермы, с использованием EGF/FGF4, в отсутствие активина А
На титрационной кривой для активина A при постоянном количестве Wnt3a видны по меньшей мере два эффекта, проявляющихся при дифференцировании клеток в сформированную эндодерму: 1) сохранение числа клеток или предотвращение потери клеток; и 2) индукция маркеров сформированной эндодермы, например, индукция экспрессии SOX17 (Пример 2). При проведении первичного скрининга (Пример 3) были установлены соединения, способные сохранять на сходном уровне или увеличивать число клеток при проведении анализа в сравнении с добавлением только активина A/Wnt3a. Для оценки эффектов сочетаний установленных соединений с другими факторами роста, в частности EGF и FGF4, на образование сформированной эндодермы был произведен вторичный скрининговый анализ.
Посев клеток для анализа. Кластеры эмбриональных стволовых клеток человека линии H1 выращивали на покрытых препаратом MATRIGELTM с пониженным содержанием факторов роста (Invitrogen; № по кат. 356231) носителях для культивирования тканей. Клетки пассировали путем обработки коллагеназой (Invitrogen; № по кат. 17104-019), затем осторожно соскоблили, промыли для удаления остатков фермента и высеяли в соотношении 1:1 (по площади поверхности) на покрытые препаратом MATRIGELTM с пониженным содержанием факторов роста 96-луночные черные планшеты (Packard ViewPlates; PerkinElmer; № по кат. 6005182) в объеме 100 мкл на лунку. Клеткам дали возможность прикрепиться (кластерами), после чего в течение 1-3 дней воспроизводили логарифмическую фазу роста, во время которой клетки ежедневно питали, используя кондиционированную среду MEF с добавлением 8 нг/мл bFGF (R&D Systems; № по кат. 233-FB). В течение всего периода анализа планшеты держали при температуре 37єC в атмосфере 5% CO2 в увлажняемом боксе.
Подготовка соединений и факторов роста. Исходные концентрации EGF (R&D Systems; № по кат. 236-EG) и FGF4 (R&D Systems; № по кат. 235-F4) составляли 250 нг/мл, оба фактора роста растворили в PBS с 0,1% BSA (Sigma; № по кат. A7888). Соединения предоставили в виде исходных растворов концентрации 5 мМ на 96-луночных планшетах, растворили в 100% DMSO (Sigma; № по кат. D2650) и хранили при температуре -80єC. Затем соединения разбавили до промежуточной концентрации 0,2 мМ в 50 мМ HEPES (Invitrogen; № по кат. 15630-080), 20% DMSO и хранили при температуре 4єC. Все факторы роста и ингибиторы поместили в 96-луночный полипропиленовый планшет с глубокими лунками, в начале анализа разбавили до получения 5-кратных промежуточных растворов в основной среде DMEM:F12 и хранили при температуре 4єC.
При вторичном скрининговом анализе тесты выполняли в трех повторностях. В течение всего четырехдневного периода анализа питание вводили через день. Анализ начали с отсасывания культуральной среды из каждой лунки с последующей трехкратной промывкой лунки раствором PBS для удаления остатков факторов роста и сыворотки. Затем в лунки добавили тестируемые образцы объемом 80 мкл, содержащие основную среду DMEM:F12 (Invitrogen; № по кат. 11330-032) с добавлением 0,625% FCS (HyClone; № по кат. SH30070.03), 25 нг/мл Wnt3a (R&D Systems), 3,125 мкМ соединения и 20 мкл 5-кратного раствора факторов роста с получением следующих конечных концентраций: 0,5% FCS, 20 нг/мл Wnt3a, 2,5 мкМ соединения, 50 нг/мл EGF и 50 нг/мл FGF4. Лунки с положительными контрольными образцами (100 мкл на лунку) содержали ту же основную среду с добавлением 0,5% FCS, 20 нг/мл Wnt3a и 100 нг/мл активина A. Лунки с отрицательными контрольными образцами (100 мкл на лунку) содержали ту же основную среду с добавлением 0,5% FCS и 20 нг/мл Wnt3a, без активина A.
На 3 день среду из лунок отсосали и добавили 80 мкл основной среды DMEM:F12 с добавлением 2,5% FCS (HyClone), 3,125 мкМ соединения и 20 мкл 5-кратного раствора факторов роста в лунку, получив конечные концентрации 2% FCS, 2,5 мкМ соединения (без Wnt3a), 50 нг/мл EGF и FGF4. Лунки с положительными контрольными образцами (100 мкл на лунку) содержали ту же основную среду с добавлением 2% FCS и 100 нг/мл активина A, без Wnt3a. Лунки с отрицательными контрольными образцами (100 мкл на лунку) содержали ту же основную среду с добавлением 2% FCS, без активина A и Wnt3a.
Одновременный многопараметрический анализ. По окончании четырехдневного периода культивирования аналитические планшеты дважды промыли раствором PBS, зафиксировали 4% параформальдегидом (Alexis Biochemical; № по кат. ALX-350-011) при комнатной температуре в течение 20 минут, затем трижды промыли раствором PBS и пермеабилизировали, обработав раствором 0,5% Triton X-100 (Sigma; № по кат. T8760-2) в течение 20 минут при комнатной температуре. Затем клетки снова трижды промыли раствором PBS и заблокировали раствором 4% куриной сыворотки (Invitrogen; № по кат. 16110082) в PBS в течение 30 минут при комнатной температуре. Первичные антитела (козий анти-человеческий SOX17; R&D Systems; № по кат. AF1924) разбавили в соотношении 1:100 в 4% куриной сыворотке и добавили в каждую лунку на один час при комнатной температуре. Сопряженные с флюорофором Alexa Fluor 488 вторичные антитела (куриный анти-козий IgG; Molecular Probes; № по кат. AZ1467) разбавили в соотношении 1:200 в PBS и добавили к каждому образцу после трехкратной промывки раствором PBS. Для подсчета окрашенных ядер на десять минут при комнатной температуре добавили 4 мкг/мл Hoechst 33342 (Invitrogen; № по кат. H3570). Затем планшеты один раз промыли раствором PBS и оставили в 100 мкл раствора PBS на лунку для томографии.
Томографию проводили на анализаторе IN Cell Analyzer 1000 (GE Healthcare) с применением дихроичного зеркала 51008bs для клеток, окрашенных красителями Hoechst 33342 и Alexa Fluor 488. Времена экспозиции оптимизировали с использованием лунок с положительными контрольными образцами и необработанных лунок с отрицательными контрольными образцами, окрашенных только вторичными антителами. Для компенсации возможных потерь клеток в ходе процедур анализа и последующего окрашивания для каждой лунки снимали по 15 проекций. Общее число клеток и общую интенсивность сигнала SOX17 измеряли для каждой лунки с помощью программного пакета IN Cell Developer Toolbox 1.7 (GE Healthcare). Сегментацию ядер определяли по ступеням шкалы серых тонов (исходный интервал 100-300) и размерам ядра. Для каждой повторной совокупности данных рассчитали средние значение и стандартные отклонения. Общий уровень экспрессии белка SOX17 выражается в виде общей интенсивности или интегральной интенсивности, определяемой как произведение общей интенсивности флюоресценции клетки на площадь клетки. Фон исключили на основании критериев соответствия ступеням шкалы серых тонов в диапазоне от 200 до 3500. Показатели общей интенсивности нормировали путем деления общей интенсивности для каждой лунки на среднюю общую интенсивность для положительного контрольного образца. Для каждого набора повторностей рассчитали нормированные данные для средних значений и стандартных отклонений.
В Таблице 3A представлены результаты исследования эффектов сочетания двух факторов роста, EGF и FGF 4 (по 50 нг/мл), с анилин-пиридинотриазиновыми соединениями, представленными в Таблице 2, на дифференцирование эмбриональных стволовых клеток человека в клетки, экспрессирующие маркеры, характерные для линии сформированной эндодермы, в отсутствие активина A. Результаты отсортированы в порядке снижения эффектов на экспрессию SOX17. Хотя эффекты данных соединений на экспрессию SOX17 были сочтены слабыми по сравнению с положительными контрольными образцами (активин A/Wnt3a), ответные реакции на некоторые из этих соединений были сочтены достоверными. Например, часть этих соединений, как оказалось, обладает уникальными свойствами в плане сохранения большого числа клеток в лунке в ходе проведения анализа, которые могут быть связаны либо с предотвращением апоптоза, либо с регуляцией клеточного цикла. Кроме того, такие соединения синергично взаимодействовали с EGF и FGF4, обеспечивая умеренный уровень дифференцирования в сформированную эндодерму, оцениваемый по экспрессии SOX17. Наиболее эффективные соединения перечислены в Таблице 3B. Другие соединения, протестированные в сочетании с EGF и FGF4 в данном анализе, оказались неэффективными в плане индукции экспрессии SOX17, но могли сохранять число клеток (например, Соединение 90: число клеток 85%; экспрессия SOX17 2%).
Пример 5
Эффект соединений, составляющих предмет настоящего изобретения, в сочетании с другими факторами на дифференцирование эмбриональных стволовых клеток человека в клетки, экспрессирующие маркеры, характерные для линии сформированной эндодермы, в отсутствие активина А
Вторичный анализ проводился с целью оценить эффект соединений, составляющих предмет настоящего изобретения, в сочетании с другими отдельными факторами роста или соединениями, которые, по данным из литературных источников, регулируют дифференцирование в сформированную эндодерму.
Посев клеток для анализа. Кластеры эмбриональных стволовых клеток человека линии H1 выращивали на покрытых препаратом MATRIGELTM с пониженным содержанием факторов роста (Invitrogen; № по кат. 356231) носителях для культивирования тканей. Клетки пассировали путем обработки коллагеназой (Invitrogen; № по кат. 17104-019), затем осторожно соскоблили, промыли для удаления остатков фермента и высеяли в соотношении 1:1 (по площади поверхности) на покрытые препаратом MATRIGELTM с пониженным содержанием факторов роста 96-луночные черные планшеты (Packard ViewPlates; PerkinElmer; № по кат. 6005182) в объеме 100 мкл на лунку. Клеткам дали возможность прикрепиться (кластерами), после чего в течение 1-3 дней воспроизводили логарифмическую фазу роста, во время которой клетки ежедневно питали, используя кондиционированную среду MEF с добавлением 8 нг/мл bFGF (R&D Systems; № по кат. 233-FB). В течение всего периода анализа планшеты держали при температуре 37єC в атмосфере 5% CO2 в увлажняемом боксе.
Подготовка соединений и факторов роста. В R&D Systems были приобретены исходные растворы следующих факторов роста: EGF (№ по кат. 236-EG), FGF4 (№ по кат. 235-F4), PDGF-A (№ по кат. 221-AA), PDGF-B (№ по кат. 220-BB), PDGF-C (№ по кат. 1687-CC), PDGF-D (№ по кат. 1159-SB), PDGF-A/B (№ по кат. 222-AB), VEGF (№ по кат. 293-VE), BMP-1 (№ по кат. 1927-ZN), BMP-2 (№ по кат. 355-BM), BMP-4 (№ по кат. 314-BP), BMP-6 (№ по кат. 507-BP), BMP-7 (№ по кат. 222-AB), BMP-2/7 (№ по кат. 3229-BM). Для тестирования были приобретены также следующие реагенты: BMP-7 (Sigma; № по кат. B1434), LY294002 (Cayman; № по кат. 70920), PD98059, U0126, U0124 (EMD Biosciences; № по кат. 453710), мусцимол (Tocris; № по кат. 0289), бикукулин (Tocris; № по кат. 0130), бутират натрия (Sigma; № по кат. B5887). Все факторы роста растворили в PBS с добавлением 0,1% BSA (Sigma; № по кат. A7888) и хранили в замороженном состоянии при температуре -80єC. Низкомолекулярные вещества растворили в 100% DMSO (Sigma; № по кат. D2650) и хранили в замороженном состоянии при температуре -80єC. Соединения предоставили в виде исходных растворов концентрации 5 мМ на 96-луночных планшетах, растворили в 100% DMSO и хранили при температуре -80єC. Соединения, составляющие предмет настоящего изобретения, далее разбавили до промежуточной концентрации 0,2 мМ в 50 мМ HEPES (Invitrogen; № по кат. 15630-080), 20% DMSO и хранили при температуре 4єC. Все факторы роста и ингибиторы поместили в 96-луночный полипропиленовый планшет с глубокими лунками, в начале анализа разбавили до получения 5-кратных промежуточных растворов в основной среде DMEM:F12 и хранили при температуре 4єC.
При вторичном скрининговом анализе тесты выполняли в трех повторностях. В течение всего четырехдневного периода анализа питание вводили через день. Анализ начали с отсасывания культуральной среды из каждой лунки с последующей трехкратной промывкой лунки раствором PBS для удаления остатков факторов роста и сыворотки. Затем в лунки добавили тестируемые образцы объемом 80 мкл, содержащие основную среду DMEM:F12 (Invitrogen; № по кат. 11330-032) с добавлением 0,625% FCS (HyClone; № по кат. SH30070.03), 25 нг/мл Wnt3a (R&D Systems), 3,125 мкМ соединения и 20 мкл 5-кратного раствора факторов роста или низкомолекулярных веществ с получением следующих конечных концентраций: 0,5% FCS, 20 нг/мл Wnt3a, 2,5 мкМ соединения. Остальные факторы роста тестировали при конечной концентрации для анализа 50 нг/мл (EGF, FGF4, PDGF-A, PDGF-B, PDGF-C, PDGF-D, PDGF-A/B, VEGF, BMP-1, BMP-2, BMP-4, BMP-6, BMP-7, BMP-2/7). Конечные концентрации для анализа низкомолекулярных веществ были следующими: мусцимол (20 мкМ), PD98059 (1 мкМ), LY294002 (2,5 мкМ), U0124 (1 мкМ), U0126 (1 мкМ), бутират натрия (0,5 мМ). Лунки с положительными контрольными образцами (100 мкл на лунку) содержали ту же основную среду с добавлением 0,5% FCS, 20 нг/мл Wnt3a и 100 нг/мл активина A. Лунки с отрицательными контрольными образцами (100 мкл на лунку) содержали ту же основную среду с добавлением 0,5% FCS и 20 нг/мл Wnt3a, без активина A.
На 3 день среду из лунок отсосали и добавили 80 мкл основной среды DMEM:F12 с добавлением 2,5% FCS (HyClone), 3,125 мкМ циклического анилин-пиридинотриазинового соединения и 20 мкл 5-кратного раствора факторов роста или низкомолекулярных веществ, получив следующие конечные концентрации: 2% FCS и 2,5 мкМ соединения (без Wnt3a), а также приведенные в описании 1 дня концентрации остальных факторов роста и низкомолекулярных веществ. Лунки с положительными контрольными образцами (100 мкл на лунку) содержали ту же основную среду с добавлением 2% FCS и 100 нг/мл активина A, без Wnt3a. Лунки с отрицательными контрольными образцами (100 мкл на лунку) содержали ту же основную среду с добавлением 2% FCS, без активина A и Wnt3a.
Одновременный многопараметрический анализ. По окончании четырехдневного периода культивирования аналитические планшеты дважды промыли раствором PBS, зафиксировали 4% параформальдегидом (Alexis Biochemical; № по кат. ALX-350-011) при комнатной температуре в течение 20 минут, затем трижды промыли раствором PBS и пермеабилизировали, обработав раствором 0,5% Triton X-100 (Sigma; № по кат. T8760-2) в течение 20 минут при комнатной температуре. Затем клетки снова трижды промыли раствором PBS и заблокировали раствором 4% куриной сыворотки (Invitrogen; № по кат. 16110082) в PBS в течение 30 минут при комнатной температуре. Первичные антитела (козий анти-человеческий SOX17; R&D Systems; № по кат. AF1924) разбавили в соотношении 1:100 в 4% куриной сыворотке и добавили в каждую лунку на один час при комнатной температуре. Сопряженные с флюорофором Alexa Fluor 488 вторичные антитела (куриный анти-козий IgG; Molecular Probes; № по кат. AZ1467) разбавили в соотношении 1:200 в PBS и добавили к каждому образцу после трехкратной промывки раствором PBS. Для подсчета окрашенных ядер на десять минут при комнатной температуре добавили 4 мкг/мл Hoechst 33342 (Invitrogen; № по кат. H3570). Затем планшеты один раз промыли раствором PBS и оставили в 100 мкл раствора PBS на лунку для томографии.
Томографию проводили на анализаторе IN Cell Analyzer 1000 (GE Healthcare) с применением дихроичного зеркала 51008bs для клеток, окрашенных красителями Hoechst 33342 и Alexa Fluor 488. Времена экспозиции оптимизировали с использованием лунок с положительными контрольными образцами и необработанных лунок с отрицательными контрольными образцами, окрашенных только вторичными антителами. Для компенсации возможных потерь клеток в ходе процедур анализа и последующего окрашивания для каждой лунки снимали по 15 проекций. Общее число клеток и общую интенсивность сигнала SOX17 измеряли для каждой лунки с помощью программного пакета IN Cell Developer Toolbox 1.7 (GE Healthcare). Сегментацию ядер определяли по ступеням шкалы серых тонов (исходный интервал 100-300) и размерам ядра. Для каждой повторной совокупности данных рассчитали средние значение и стандартные отклонения. Общий уровень экспрессии белка SOX17 выражается в виде общей интенсивности или интегральной интенсивности, определяемой как произведение общей интенсивности флюоресценции клетки на площадь клетки. Фон исключили на основании критериев соответствия ступеням шкалы серых тонов в диапазоне от 200 до 3500. Показатели общей интенсивности нормировали путем деления общей интенсивности для каждой лунки на среднюю общую интенсивность для положительного контрольного образца. Для каждого набора повторностей рассчитали нормированные данные для средних значений и стандартных отклонений.
В Таблице 4 представлены результаты дифференцирования эмбриональных стволовых клеток человека в линию сформированной эндодермы после обработки соединениями, составляющими предмет настоящего изобретения, в сочетании с отдельными факторами роста или другими низкомолекулярными веществами. В целом, члены семейства BMP (BMP-1, BMP-2, BMP-4, BMP-6, BMP-7, BMP-2/7) ингибировали или оказывали незначительное влияние на экспрессию SOX17. Аналогичные результаты были получены и для протестированных низкомолекулярных ингибиторов ферментов (LY294002, PD98059, U0126, U0124, бутират натрия). Однако некоторые члены семейства PDGF (PDGF-A, -AB, -C и -D) обеспечивали увеличение экспрессии SOX17 (10-25% от эффекта контрольного сочетания активина A/Wnt3a). Другие факторы роста, которые вызывали сходное увеличение экспрессии SOX17, включают EGF (34%), VEGF (18%) и FGF4 (17%), но FGF4 не обеспечивал сохранение числа клеток. Низкомолекулярное вещество мусцимол (агонист рецептора GABAA) в сочетании с Соединением 35 также обеспечивал умеренное увеличение экспрессии SOX17; антагонист GABAA бикукулин не оказывал влияния на экспрессию SOX17. EGF, FGF4, PDGF-A, PDGF-B, PDGF-AB, PDGF-C, PDGF-D и мусцимол были отобраны для дополнительного анализа эффекта на дифференцирование клеток в сформированную эндодерму.
Пример 6
Эффект соединений, составляющих предмет настоящего изобретения, в сочетании с другими факторами на дифференцирование эмбриональных стволовых клеток человека в клетки, экспрессирующие маркеры, характерные для линии сформированной эндодермы, в отсутствие активина А
Вторичный анализ проводился с целью оценить эффект сочетаний различных соединений с другими отдельными агентами на дифференцирование клеток в сформированную эндодерму. Другие агенты, выбранные для данного скринингового анализа, как было показано ранее в тесте с Соединением 17 и как указано в Таблице 5, вызывали умеренное увеличение образования сформированной эндодермы. В рамках данного скринингового анализа исследовался более широкий спектр соединений в сочетании с данными агентами. Анализ проводился либо путем сравнения отдельных пар, либо при сочетании нескольких компонентов.
Посев клеток для анализа. Кластеры эмбриональных стволовых клеток человека линии H1 выращивали на покрытых препаратом MATRIGELTM с пониженным содержанием факторов роста (Invitrogen; № по кат. 356231) носителях для культивирования тканей. Клетки пассировали путем обработки коллагеназой (Invitrogen; № по кат. 17104-019), затем осторожно соскоблили, промыли для удаления остатков фермента и высеяли в соотношении 1:1 (по площади поверхности) на покрытые препаратом MATRIGELTM с пониженным содержанием факторов роста 96-луночные черные планшеты (Packard ViewPlates; PerkinElmer; № по кат. 6005182) в объеме 100 мкл на лунку. Клеткам дали возможность прикрепиться (кластерами), после чего в течение 1-3 дней воспроизводили логарифмическую фазу роста, во время которой клетки ежедневно питали, кондиционированную среду MEF с добавлением 8 нг/мл bFGF (R&D Systems; № по кат. 233-FB). В течение всего периода анализа планшеты держали при температуре 37°C в атмосфере 5% CO2 в увлажняемом боксе.
Подготовка соединений и факторов роста. В R&D Systems были приобретены исходные растворы следующих факторов роста: EGF (№ по кат. 236-EG), FGF4 (№ по кат. 235-F4), PDGF-A (№ по кат. 221-AA), PDGF-D (№ по кат. 1159-SB), PDGF-A/B (№ по кат. 222-AB) и VEGF (№ по кат. 293-VE). Мусцимол был приобретен в Tocris (№ по кат. 0289). Все факторы роста растворили в PBS с добавлением 0,1% BSA (Sigma; № по кат. A7888) и хранили в замороженном состоянии при температуре -80°C. Мусцимол растворили в 100% DMSO (Sigma; № по кат. D2650) и хранили в замороженном состоянии при температуре -80°C. Соединения предоставили в виде исходных растворов концентрации 5 мМ на 96-луночных планшетах, растворили в 100% DMSO и хранили при температуре -80°C. Затем соединения разбавили до промежуточной концентрации 0,2 мМ в 50 мМ HEPES (Invitrogen; № по кат. 15630-080), 20% DMSO и хранили при температуре 4°C. Все факторы роста и ингибиторы поместили в 96-луночный полипропиленовый планшет с глубокими лунками, в начале анализа разбавили до получения 5-кратных промежуточных растворов в основной среде DMEM:F12 и хранили при температуре 4°C.
При вторичном скрининговом анализе тесты выполняли в трех повторностях. В течение всего четырехдневного периода анализа питание вводили через день. Анализ начали с отсасывания культуральной среды из каждой лунки с последующей трехкратной промывкой лунки раствором PBS для удаления остатков факторов роста и сыворотки. Затем в лунки добавили тестируемые образцы объемом 80 мкл, содержащие основную среду DMEM:F12 (Invitrogen; № по кат. 11330-032) с добавлением 0,625% FCS (HyClone; № по кат. SH30070.03), 25 нг/мл Wnt3a (R&D Systems), 3,125 мкМ соединения и 20 мкл 5-кратного раствора факторов роста или низкомолекулярных веществ с получением следующих конечных концентраций: 0,5% FCS, 20 нг/мл Wnt3a, 2,5 мкМ соединения. Остальные факторы роста тестировали в конечной концентрации для анализа, равной 50 нг/мл (EGF, FGF4, PDGF-A, PDGF-A/B, VEGF). Конечная концентрация для анализа мусцимола составила 20 мкМ. Лунки с положительными контрольными образцами (100 мкл на лунку) содержали ту же основную среду с добавлением 0,5% FCS, 20 нг/мл Wnt3a и 100 нг/мл активина A. Лунки с отрицательными контрольными образцами (100 мкл на лунку) содержали ту же основную среду с добавлением 0,5% FCS и 20 нг/мл Wnt3a, без активина A.
На 3 день среду из лунок отсосали и добавили 80 мкл основной среды DMEM:F12 с добавлением 2,5% FCS (HyClone), 3,125 мкМ соединения и 20 мкл 5-кратного раствора факторов роста или низкомолекулярных веществ, получив следующие конечные концентрации: 2% FCS и 2,5 мкМ соединения (без Wnt3a), а также приведенные в описании 1 дня концентрации остальных факторов роста и низкомолекулярных веществ. Лунки с положительными контрольными образцами (100 мкл на лунку) содержали ту же основную среду с добавлением 2% FCS и 100 нг/мл активина A, без Wnt3a. Лунки с отрицательными контрольными образцами (100 мкл на лунку) содержали ту же основную среду с добавлением 2% FCS, без активина A и Wnt3a.
Одновременный многопараметрический анализ. По окончании четырехдневного периода культивирования аналитические планшеты дважды промыли раствором PBS, зафиксировали 4% параформальдегидом (Alexis Biochemical; № по кат. ALX-350-011) при комнатной температуре в течение 20 минут, затем трижды промыли раствором PBS и пермеабилизировали, обработав раствором 0,5% Triton X-100 (Sigma; № по кат. T8760-2) в течение 20 минут при комнатной температуре. Затем клетки снова трижды промыли раствором PBS и заблокировали раствором 4% куриной сыворотки (Invitrogen; № по кат. 16110082) в PBS в течение 30 минут при комнатной температуре. Первичные антитела (козий анти-человеческий SOX17; R&D Systems; № по кат. AF1924) разбавили в соотношении 1:100 в 4% куриной сыворотке и добавили в каждую лунку на один час при комнатной температуре. Сопряженные с флюорофором Alexa Fluor 488 вторичные антитела (куриный анти-козий IgG; Molecular Probes; № по кат. AZ1467) разбавили в соотношении 1:200 в PBS и добавили к каждому образцу после трехкратной промывки раствором PBS. Для подсчета окрашенных ядер на десять минут при комнатной температуре добавили 4 мкг/мл Hoechst 33342 (Invitrogen; № по кат. H3570). Затем планшеты один раз промыли раствором PBS и оставили в 100 мкл раствора PBS на лунку для томографии.
Томографию проводили на анализаторе IN Cell Analyzer 1000 (GE Healthcare) с применением дихроичного зеркала 51008bs для клеток, окрашенных красителями Hoechst 33342 и Alexa Fluor 488. Времена экспозиции оптимизировали с использованием лунок с положительными контрольными образцами и необработанных лунок с отрицательными контрольными образцами, окрашенных только вторичными антителами. Для компенсации возможных потерь клеток в ходе процедур анализа и последующего окрашивания для каждой лунки снимали по 15 проекций. Общее число клеток и общую интенсивность сигнала SOX17 измеряли для каждой лунки с помощью программного пакета IN Cell Developer Toolbox 1.7 (GE Healthcare). Сегментацию ядер определяли по ступеням шкалы серых тонов (исходный интервал 100-300) и размерам ядра. Для каждой повторной совокупности данных рассчитали средние значение и стандартные отклонения. Общий уровень экспрессии белка SOX17 выражается в виде общей интенсивности или интегральной интенсивности, определяемой как произведение общей интенсивности флюоресценции клетки на площадь клетки. Фон исключили на основании критериев соответствия ступеням шкалы серых тонов в диапазоне от 200 до 3500. Показатели общей интенсивности нормировали путем деления общей интенсивности для каждой лунки на среднюю общую интенсивность для положительного контрольного образца. Для каждого набора повторностей рассчитали нормированные данные для средних значений и стандартных отклонений.
В Таблице 5 представлены соединения, результаты тестирования которых на образование сформированной эндодермы в различных сочетаниях с факторами роста и мусцимолом, без активина А, были признаны положительными (Таблица 2). Некоторые соединения в сочетаниях со всеми использованными факторами роста имели минимальный или слабый эффект на экспрессию SOX17. Однако некоторые соединения оказались способны индуцировать существенную экспрессию SOX17 в сочетании с некоторыми, хотя и не всеми факторами роста. В частности, одно из соединений, Соединение 34, показало существенную синергическую ответную реакцию со всеми испытанными факторами роста, и обеспечивало в данном эксперименте увеличение как числа клеток, так и экспрессии SOX17: Соединение 39 в сочетании с 1) EGF+FGF4=77% от ответной реакции положительного контрольного образца; или 2) EGF+FGF4+PDGF-AB=68% от ответной реакции положительного контрольного образца; или 3) EGF+FGF4+PDGF-A+VEGF=31% от ответной реакции положительного контрольного образца.
Пример 7
Эффект Соединения 34 в сочетании с другими факторами на дифференцирование эмбриональных стволовых клеток человека в клетки, экспрессирующие маркеры, характерные для линии сформированной эндодермы, в отсутствие активина А
В данном примере была проведена работа по выявлению минимально необходимого числа факторов роста, которые в сочетании с наиболее эффективным анилин-пиридинотриазиновым соединением, Соединением 34, оказывали бы устойчивое влияние на экспрессию SOX17 в отсутствие активина A. Также в данном примере был проанализирован новый ростовой фактор, GDF-8. GDF-8, известный также как миостатин, является членом семейства TGF-β и, как было показано, использует активиновые рецепторы типа II и рецепторы TGF-β типа I (ALK4/5), индуцируя фосфорилирование SMAD 2/3.
Посев клеток для анализа. Кластеры эмбриональных стволовых клеток человека линии H1 выращивали на покрытых препаратом MATRIGELTM с пониженным содержанием факторов роста (Invitrogen; № по кат. 356231) носителях для культивирования тканей. Клетки пассировали путем обработки коллагеназой (Invitrogen; № по кат. 17104-019), затем осторожно соскоблили, промыли для удаления остатков фермента и высеяли в соотношении 1:1 (по площади поверхности) на покрытые препаратом MATRIGELTM с пониженным содержанием факторов роста 96-луночные черные планшеты (Packard ViewPlates; PerkinElmer; № по кат. 6005182) в объеме 100 мкл на лунку. Клеткам дали возможность прикрепиться (кластерами), после чего в течение 1-3 дней воспроизводили логарифмическую фазу роста, во время которой клетки ежедневно питали, используя кондиционированную среду MEF с добавлением 8 нг/мл bFGF (R&D Systems; № по кат. 233-FB). В течение всего периода анализа планшеты держали при температуре 37єC в атмосфере 5% CO2 в увлажняемом боксе.
Подготовка соединений и факторов роста. В R&D Systems были приобретены исходные растворы следующих факторов роста: EGF (№ по кат. 236-EG), FGF4 (№ по кат. 235-F4), PDGF-A (№ по кат. 221-AA), PDGF-D (№ по кат. 1159-SB), PDGF-A/B (№ по кат. 222-AB), VEGF (№ по кат. 293-VE) и GDF-8 (№ по кат. 788-G8). Мусцимол был приобретен в Tocris (№ по кат. 0289). Все факторы роста растворили в PBS с добавлением 0,1% BSA (Sigma; № по кат. A7888) и хранили в замороженном состоянии при температуре -80єC. Мусцимол растворили в 100% DMSO (Sigma; № по кат. D2650) и хранили в замороженном состоянии при температуре -80єC. Циклические анилин-пиридинотриазиновые соединения предоставили в виде исходных растворов концентрации 5 мМ на 96-луночных планшетах, растворили в 100% DMSO и хранили при температуре
-80єC. Соединение 34 затем разбавили до промежуточной концентрации 0,2 мМ в 50 мМ HEPES (Invitrogen; № по кат. 15630-080), 20% DMSO и хранили при температуре 4єC. Все факторы роста и ингибиторы поместили в 96-луночный полипропиленовый планшет с глубокими лунками, в начале анализа разбавили до получения 5-кратных промежуточных растворов в основной среде DMEM:F12 и хранили при температуре 4єC.
При вторичном скрининговом анализе тесты выполняли в трех повторностях. В течение всего четырехдневного периода анализа питание вводили через день. Анализ начали с отсасывания культуральной среды из каждой лунки с последующей трехкратной промывкой лунки раствором PBS для удаления остатков факторов роста и сыворотки. Затем в лунки добавили тестируемые образцы объемом 80 мкл, содержащие основную среду DMEM:F12 (Invitrogen; № по кат. 11330-032) с добавлением 0,625% FCS (HyClone; № по кат. SH30070.03), 25 нг/мл Wnt3a (R&D Systems), 3,125 мкМ Соединения 27 и 20 мкл 5-кратного раствора факторов роста или низкомолекулярных веществ с получением следующих конечных концентраций: 0,5% FCS, 20 нг/мл Wnt3a и 2,5 мкМ Соединения 34. Все остальные факторы роста тестировали в конечной концентрации для анализа, равной 50 нг/мл (EGF, FGF4, PDGF-A, PDGF-A/B, VEGF), за исключением GDF-8, который использовался в концентрации 25 нг/мл. Конечная концентрация для анализа мусцимола составила 20 мкМ. Лунки с положительными контрольными образцами (100 мкл на лунку) содержали ту же основную среду с добавлением 0,5% FCS, 20 нг/мл Wnt3a и 100 нг/мл активина A. Лунки с отрицательными контрольными образцами (100 мкл на лунку) содержали ту же основную среду с добавлением 0,5% FCS и 20 нг/мл Wnt3a, без активина A.
На 3 день среду из лунок отсосали и добавили 80 мкл основной среды DMEM:F12 с добавлением 2,5% FCS (HyClone), 3,125 мкМ Соединения 34 и 20 мкл 5-кратного раствора факторов роста или низкомолекулярных веществ, получив следующие конечные концентрации: 2% FCS и 2,5 мкМ Соединения 34 (без Wnt3a), а также приведенные в описании 1 дня концентрации остальных факторов роста и низкомолекулярных веществ. Лунки с положительными контрольными образцами (100 мкл на лунку) содержали ту же основную среду с добавлением 2% FCS и 100 нг/мл активина A, без Wnt3a. Лунки с отрицательными контрольными образцами (100 мкл на лунку) содержали ту же основную среду с добавлением 2% FCS, без активина A и Wnt3a.
Одновременный многопараметрический анализ. По окончании четырехдневного периода культивирования аналитические планшеты дважды промыли раствором PBS, зафиксировали 4% параформальдегидом (Alexis Biochemical; № по кат. ALX-350-011) при комнатной температуре в течение 20 минут, затем трижды промыли раствором PBS и пермеабилизировали, обработав раствором 0,5% Triton X-100 (Sigma; № по кат. T8760-2) в течение 20 минут при комнатной температуре. Затем клетки снова трижды промыли раствором PBS и заблокировали раствором 4% куриной сыворотки (Invitrogen; № по кат. 16110082) в PBS в течение 30 минут при комнатной температуре. Первичные антитела (козий анти-человеческий SOX17; R&D Systems; № по кат. AF1924) разбавили в соотношении 1:100 в 4% куриной сыворотке и добавили в каждую лунку на один час при комнатной температуре. Сопряженные с флюорофором Alexa Fluor 488 вторичные антитела (куриный анти-козий IgG; Molecular Probes; № по кат. AZ1467) разбавили в соотношении 1:200 в PBS и добавили к каждому образцу после трехкратной промывки раствором PBS. Для подсчета окрашенных ядер на десять минут при комнатной температуре добавили 4 мкг/мл Hoechst 33342 (Invitrogen; № по кат. H3570). Затем планшеты один раз промыли раствором PBS и оставили в 100 мкл раствора PBS на лунку для томографии.
Томографию проводили на анализаторе IN Cell Analyzer 1000 (GE Healthcare) с применением дихроичного зеркала 51008bs для клеток, окрашенных красителями Hoechst 33342 и Alexa Fluor 488. Времена экспозиции оптимизировали с использованием лунок с положительными контрольными образцами и необработанных лунок с отрицательными контрольными образцами, окрашенных только вторичными антителами. Для компенсации возможных потерь клеток в ходе процедур анализа и последующего окрашивания для каждой лунки снимали по 15 проекций. Общее число клеток и общую интенсивность сигнала SOX17 измеряли для каждой лунки с помощью программного пакета IN Cell Developer Toolbox 1.7 (GE Healthcare). Сегментацию ядер определяли по ступеням шкалы серых тонов (исходный интервал 100-300) и размерам ядра. Для каждой повторной совокупности данных рассчитали средние значение и стандартные отклонения. Общий уровень экспрессии белка SOX17 выражается в виде общей интенсивности или интегральной интенсивности, определяемой как произведение общей интенсивности флюоресценции клетки на площадь клетки. Фон исключили на основании критериев соответствия ступеням шкалы серых тонов в диапазоне от 200 до 3500. Показатели общей интенсивности нормировали путем деления общей интенсивности для каждой лунки на среднюю общую интенсивность для положительного контрольного образца. Для каждого набора повторностей рассчитали нормированные данные для средних значений и стандартных отклонений.
Результаты данного анализа представлены в Таблице 6. Во всех случаях сочетание Соединения 34 с фактором GDF-8 давало существенное увеличение экспрессии SOX17. Более того, сочетания GDF-8, Wnt3a и Соединения 34 оказалось достаточно для получения уровня экспрессии SOX17 (88% от контрольного образца), сопоставимого с наблюдаемым при обработке 100 нг/мл активина A/Wnt3a. Представляется, что фактор роста GDF-8 может служить заменой активину A при дифференцировании эмбриональных стволовых клеток человека в сформированную эндодерму.
Пример 8
Дополнительный скрининговый анализ соединений, способных дифференцировать плюрипотентные стволовые клетки в клетки, экспрессирующие маркеры, характерные для линии сформированной эндодермы
На основе структуры выявленных к настоящему моменту соединений, показавших положительные результаты, для проведения биологического анализа на образование сформированной эндодермы был проведен поиск сходных соединений. Субструктурный поиск позволил найти соединения для скринингового анализа. Параметры скринингового анализа основывались на сочетании факторов, которые показали оптимальные результаты в предыдущих анализах, в частности, сочетание EGF, FGF, PDGF-A,VEGF, PDGF-D, мусцимола и GDF-8 с низкомолекулярным соединением.
Посев клеток для анализа. Коротко говоря, кластеры эмбриональных стволовых клеток человека линии H1 выращивали на покрытых препаратом MATRIGELTM с пониженным содержанием факторов роста (Invitrogen; № по кат. 356231) носителях для культивирования тканей. Клетки пассировали путем обработки коллагеназой (Invitrogen; № по кат. 17104-019), затем осторожно соскоблили, промыли для удаления остатков фермента и высеяли в соотношении 1:1 (по площади поверхности) на покрытые препаратом MATRIGELTM с пониженным содержанием факторов роста 96-луночные черные планшеты (Packard ViewPlates; PerkinElmer; № по кат. 6005182) в объеме 100 мкл на лунку. Клеткам дали возможность прикрепиться (кластерами), после чего в течение 1-3 дней воспроизводили логарифмическую фазу роста, во время которой клетки ежедневно питали, используя кондиционированную среду MEF с добавлением 8 нг/мл bFGF (R&D Systems; № по кат. 233-FB). В течение всего периода анализа планшеты держали при температуре 37єC в атмосфере 5% CO2 в увлажняемом боксе.
Подготовка соединений и факторов роста. В R&D Systems были приобретены исходные растворы следующих факторов роста: EGF (№ по кат. 236-EG), FGF4 (№ по кат. 235-F4), PDGF-A (№ по кат. 221-AA), PDGF-D (№ по кат. 1159-SB), PDGF-A/B (№ по кат. 222-AB), VEGF (№ по кат. 293-VE) и GDF-8 (№ по кат. 788-G8). Мусцимол был приобретен в Tocris (№ по кат. 0289). Скрининговый анализ производился с использованием библиотеки соединений, которые предоставили в виде исходных растворов концентрации 5 мМ на 96-луночных планшетах, растворили в 100% DMSO (Sigma; № по кат. D2650) и хранили при температуре -80єC. Затем соединения разбавили до промежуточной концентрации 0,2 мМ в 50 мМ HEPES (Invitrogen; № по кат. 15630-080), 20% DMSO и хранили при температуре 4єC. Каждый тест выполняли в отдельных лунках. В течение всего четырехдневного периода анализа питание вводили через день. Первичный скрининговый анализ начали с отсасывания культуральной среды из каждой лунки с последующей трехкратной промывкой лунки раствором PBS (Invitrogen; № по кат. 14190) для удаления остатков факторов роста и сыворотки. В 1 день анализа в лунки добавили тестируемые образцы объемом 200 мкл, содержащие основную среду DMEM:F12 (Invitrogen; № по кат. 11330-032) с добавлением 0,5% FCS (HyClone; № по кат. SH30070.03), 20 нг/мл Wnt3a (R&D Systems; № по кат. 1324-WN) и 2,5 мкМ соединения. Все остальные факторы роста тестировали в конечной концентрации для анализа, равной 50 нг/мл (EGF, FGF4, PDGF-A, PDGF-A/B, VEGF), за исключением GDF-8, который использовался в концентрации 25 нг/мл. Конечная концентрация для анализа мусцимола составила 20 мкМ. Положительные контрольные образцы содержали ту же основную среду с добавлением 0,5% FCS, 20 нг/мл Wnt3a и 100 нг/мл рекомбинантного человеческого активина A (PeproTech; № по кат. 120-14). Отрицательные контрольные образцы содержали основную среду DMEM:F12 с добавлением 0,5% FCS и 20 нг/мл Wnt3a. На 3 день анализа в лунки добавили тестируемые образцы объемом 200 мкл, содержащие основную среду DMEM:F12 с добавлением 2% FCS и 2,5 мкМ соединения, без Wnt3a. Все остальные факторы роста тестировали в конечной концентрации для анализа 50 нг/мл (EGF, FGF4, PDGF-A, PDGF-A/B, VEGF), за исключением GDF-8, который использовался в концентрации 25 нг/мл. Конечная концентрация для анализа мусцимола составила 20 мкМ. Положительные контрольные образцы содержали ту же основную среду с добавлением 2% FCS и 100 нг/мл рекомбинантного человеческого активина A (PeproTech; № по кат. 120-14). Отрицательные контрольные образцы содержали основную среду DMEM:F12 с добавлением 2% FCS. Положительные контрольные образцы содержали ту же основную среду с FCS, за исключением того, что вместо анилин-пиридинотриазинового соединения в течение всего четырехдневного периода анализа среда содержала 100 нг/мл рекомбинантного человеческого активина A (PeproTech; № по кат. 120-14) с добавлением в 1 и 2 дни Wnt3a (20 нг/мл). Отрицательные контрольные образцы содержали основную среду DMEM:F12 с FCS, без активина А, но с добавлением в 1 и 2 дни Wnt3a.
Одновременный многопараметрический анализ. По окончании четырехдневного периода культивирования аналитические планшеты дважды промыли раствором PBS (Invitrogen; № по кат. 14190), зафиксировали 4% параформальдегидом (Alexis Biochemical; № по кат. ALX-350-011) при комнатной температуре в течение 20 минут, затем трижды промыли раствором PBS и пермеабилизировали, обработав раствором 0,5% Triton X-100 (Sigma; № по кат. T8760-2) в течение 20 минут при комнатной температуре. Затем клетки снова трижды промыли раствором PBS и заблокировали раствором 4% куриной сыворотки (Invitrogen; № по кат. 16110082) в PBS в течение 30 минут при комнатной температуре. Первичные антитела (козий анти-человеческий SOX17; R&D Systems; № по кат. AF1924) разбавили в соотношении 1:100 в 4% куриной сыворотке и добавили в каждую лунку на один час при комнатной температуре. Сопряженные с флюорофором Alexa Fluor 488 вторичные антитела (куриный анти-козий IgG; Molecular Probes; № по кат. AZ1467) разбавили в соотношении 1:200 в PBS и добавили к каждому образцу после трехкратной промывки раствором PBS. Для подсчета окрашенных ядер на десять минут при комнатной температуре добавили 4 мкг/мл Hoechst 33342 (Invitrogen; № по кат. H3570). Затем планшеты один раз промыли раствором PBS и оставили в 100 мкл раствора PBS на лунку для томографии.
Томографию проводили на анализаторе IN Cell Analyzer 1000 (GE Healthcare) с применением дихроичного зеркала 51008bs для клеток, окрашенных красителями Hoechst 33342 и Alexa Fluor 488. Времена экспозиции оптимизировали с использованием лунок с положительными контрольными образцами и необработанных лунок с отрицательными контрольными образцами, окрашенных только вторичными антителами. Для компенсации возможных потерь клеток в ходе процедур анализа и последующего окрашивания для каждой лунки снимали по 15 проекций. Общее число клеток и общую интенсивность сигнала SOX17 измеряли для каждой лунки с помощью программного пакета IN Cell Developer Toolbox 1.7 (GE Healthcare). Сегментацию ядер определяли по ступеням шкалы серых тонов (исходный интервал 100-300) и размерам ядра. Для каждой повторной совокупности данных рассчитали средние значение и стандартные отклонения. Общий уровень экспрессии белка Sox17 выражается в виде общей интенсивности или интегральной интенсивности, определяемой как произведение общей интенсивности флюоресценции клетки на площадь клетки. Фон исключили на основании критериев соответствия ступеням шкалы серых тонов в диапазоне от 200 до 3500. Показатели общей интенсивности нормировали путем деления общей интенсивности для каждой лунки на среднюю общую интенсивность для положительного контрольного образца. Для каждого набора повторностей рассчитали нормированные данные для средних значений и стандартных отклонений.
В Таблице 7 приводятся данные тестирования GDF-8 и сочетания факторов роста/агонистов (EGF, FGF, PDGF-A, VEGF, PDGF-D, мусцимол) с новым набором анилин-пиридинотриазиновых соединений. Результаты, полученные в одном эксперименте на двух аналитических планшетах, ранжированы по ответной реакции в виде экспрессии SOX17 (в процентах от положительного контрольного образца - обработки активином A и Wnt3a). Были выявлены дополнительные соединения, проявившие значительную синергическую активность с набором факторов роста/агонистов. Такие соединения были эффективны как в отношении сохранения числа клеток, так и в отношении экспрессии SOX17 в ходе дифференцирования эмбриональных стволовых клеток человека в отсутствие активина A. Список показавших положительные результаты соединений с активностью более 25% от положительного контрольного образца приведен в Таблице 8.
Следует отметить, что в библиотеке аналогов были продублированы четыре соединения, показавшие положительные результаты, выявленные при проведении первичного скринингового анализа (Таблица 2). Два из этих соединений снова показали положительные результаты при скрининговом анализе аналогов (Соединение 34 и Соединение 35; обведены рамкой в Таблице 8); одно соединение показало слабый положительный результат, а для одного соединения эффект не повторился.
Пример 9
Эффект соединений, составляющих предмет настоящего изобретения, на дифференцирование эмбриональных стволовых клеток человека в клетки, экспрессирующие маркеры, характерные для линии сформированной эндодермы, в присутствие низких концентраций активина А
Было важно установить, могут ли соединения, показавшие положительные результаты в описанном выше биологическом анализе на образование сформированной эндодермы, синергично взаимодействовать с очень низкими дозами активина A. Первоначальная оценка проводилась с использованием приведенного в Таблице 3B краткого перечня циклических анилин-пиридинотриазиновых соединений, показавших положительные результаты.
Посев клеток для анализа. Кластеры эмбриональных стволовых клеток человека линии H1 выращивали на покрытых препаратом MATRIGELTM с пониженным содержанием факторов роста (Invitrogen; № по кат. 356231) носителях для культивирования тканей. Клетки пассировали путем обработки коллагеназой (Invitrogen; № по кат. 17104-019), затем осторожно соскоблили, промыли для удаления остатков фермента и высеяли в соотношении 1:1 (по площади поверхности) на покрытые препаратом MATRIGELTM с пониженным содержанием факторов роста 96-луночные черные планшеты (Packard ViewPlates; PerkinElmer; № по кат. 6005182) в объеме 100 мкл на лунку. Клеткам дали возможность прикрепиться (кластерами), после чего в течение 1-3 дней воспроизводили логарифмическую фазу роста, во время которой клетки ежедневно питали, используя кондиционированную среду MEF с добавлением 8 нг/мл bFGF (R&D Systems; № по кат. 233-FB). В течение всего периода анализа планшеты держали при температуре 37єC в атмосфере 5% CO2 в увлажняемом боксе.
Подготовка соединений и факторов роста. В R&D Systems были приобретены исходные растворы следующих факторов роста: EGF (№ по кат. 236-EG), FGF4 (№ по кат. 235-F4), PDGF-A (№ по кат. 221-AA), PDGF-D (№ по кат. 1159-SB), PDGF-A/B (№ по кат. 222-AB), VEGF (№ по кат. 293-VE) и GDF-8 (№ по кат. 788-G8). Активин A был приобретен в PeproTech (№ по кат.). Мусцимол был приобретен в Tocris (№ по кат. 0289). Все факторы роста растворили в PBS с добавлением 0,1% BSA (Sigma; № по кат. A7888) и хранили в замороженном состоянии при температуре -80єC. Мусцимол растворили в 100% DMSO (Sigma; № по кат. D2650) и хранили в замороженном состоянии при температуре -80єC. Соединения предоставили в виде исходных растворов концентрации 5 мМ на 96-луночных планшетах, растворили в 100% DMSO и хранили при температуре -80єC. Соединения затем разбавили до промежуточной концентрации 0,2 мМ в 50 мМ HEPES (Invitrogen; № по кат. 15630-080), 20% DMSO и хранили при температуре 4єC. Все факторы роста и ингибиторы поместили в 96-луночный полипропиленовый планшет с глубокими лунками, в начале анализа разбавили до получения 5-кратных промежуточных растворов в основной среде DMEM:F12 и хранили при температуре 4єC.
При вторичном скрининговом анализе тесты выполняли в трех повторностях. В течение всего четырехдневного периода анализа питание вводили через день. Анализ начали с отсасывания культуральной среды из каждой лунки с последующей трехкратной промывкой лунки раствором PBS для удаления остатков факторов роста и сыворотки. Затем в лунки добавили тестируемые образцы объемом 80 мкл, содержащие основную среду DMEM:F12 (Invitrogen; № по кат. 11330-032) с добавлением 0,625% FCS (HyClone; № по кат. SH30070.03), 25 нг/мл Wnt3a (R&D Systems), 12,5 нг/мл активина А, 3,125 мкМ соединения и 20 мкл 5-кратного раствора факторов роста или низкомолекулярных веществ с получением следующих конечных концентраций: 0,5% FCS, 20 нг/мл Wnt3a, 10 нг/мл активина А и 2,5 мкМ соединения. Все остальные факторы роста тестировали в конечной концентрации для анализа, равной 50 нг/мл (EGF, FGF4, PDGF-A, PDGF-A/B, VEGF), за исключением GDF-8, который использовался в концентрации 25 нг/мл. Конечная концентрация для анализа мусцимола составила 20 мкМ. Лунки с положительными контрольными образцами (100 мкл на лунку) содержали ту же основную среду с добавлением 0,5% FCS, 20 нг/мл Wnt3a и 10 нг/мл (низкая доза) или 100 нг/мл (высокая доза) активина A. Лунки с отрицательными контрольными образцами (100 мкл на лунку) содержали ту же основную среду с добавлением 0,5% FCS и 20 нг/мл Wnt3a, без активина A.
На 3 день среду из лунок отсосали и добавили 80 мкл основной среды DMEM:F12 с добавлением 2,5% FCS (HyClone), 12,5 нг/мл активина А, 3,125 мкМ соединения и 20 мкл 5-кратного раствора факторов роста или низкомолекулярных веществ, получив следующие конечные концентрации: 2% FCS, 10 нг/мл активина А и 2,5 мкМ соединения (без Wnt3a), а также приведенные в описании 1 дня концентрации остальных факторов роста и низкомолекулярных веществ. Лунки с положительными контрольными образцами (100 мкл на лунку) содержали ту же основную среду с добавлением 2% FCS и 10 нг/мл или 100 нг/мл активина A, без Wnt3a. Лунки с отрицательными контрольными образцами (100 мкл на лунку) содержали ту же основную среду с добавлением 2% FCS, без активина A и Wnt3a.
Одновременный многопараметрический анализ. По окончании четырехдневного периода культивирования аналитические планшеты дважды промыли раствором PBS, зафиксировали 4% параформальдегидом (Alexis Biochemical; № по кат. ALX-350-011) при комнатной температуре в течение 20 минут, затем трижды промыли раствором PBS и пермеабилизировали, обработав раствором 0,5% Triton X-100 (Sigma; № по кат. T8760-2) в течение 20 минут при комнатной температуре. Затем клетки снова трижды промыли раствором PBS и заблокировали раствором 4% куриной сыворотки (Invitrogen; № по кат. 16110082) в PBS в течение 30 минут при комнатной температуре. Первичные антитела (козий анти-человеческий SOX17; R&D Systems; № по кат. AF1924) разбавили в соотношении 1:100 в 4% куриной сыворотке и добавили в каждую лунку на один час при комнатной температуре. Сопряженные с флюорофором Alexa Fluor 488 вторичные антитела (куриный анти-козий IgG; Molecular Probes; № по кат. AZ1467) разбавили в соотношении 1:200 в PBS и добавили к каждому образцу после трехкратной промывки раствором PBS. Для подсчета окрашенных ядер на десять минут при комнатной температуре добавили 4 мкг/мл Hoechst 33342 (Invitrogen; № по кат. H3570). Затем планшеты один раз промыли раствором PBS и оставили в 100 мкл раствора PBS на лунку для томографии.
Томографию проводили на анализаторе IN Cell Analyzer 1000 (GE Healthcare) с применением дихроичного зеркала 51008bs для клеток, окрашенных красителями Hoechst 33342 и Alexa Fluor 488. Времена экспозиции оптимизировали с использованием лунок с положительными контрольными образцами и необработанных лунок с отрицательными контрольными образцами, окрашенных только вторичными антителами. Для компенсации возможных потерь клеток в ходе процедур анализа и последующего окрашивания для каждой лунки снимали по 15 проекций. Общее число клеток и общую интенсивность сигнала SOX17 измеряли для каждой лунки с помощью программного пакета IN Cell Developer Toolbox 1.7 (GE Healthcare). Сегментацию ядер определяли по ступеням шкалы серых тонов (исходный интервал 100-300) и размерам ядра. Для каждой повторной совокупности данных рассчитали средние значение и стандартные отклонения. Общий уровень экспрессии белка SOX17 выражается в виде общей интенсивности или интегральной интенсивности, определяемой как произведение общей интенсивности флюоресценции клетки на площадь клетки. Фон исключили на основании критериев соответствия ступеням шкалы серых тонов в диапазоне от 200 до 3500. Показатели общей интенсивности нормировали путем деления общей интенсивности для каждой лунки на среднюю общую интенсивность для положительного контрольного образца. Для каждого набора повторностей рассчитали нормированные данные для средних значений и стандартных отклонений.
В Таблице 9 представлены результаты анализа различных соединений и различных сочетаний факторов роста с низкими дозами активина А. Некоторые соединения продемонстрировали синергичные ответные реакции с различными факторами роста. В других случаях синергичные эффекты были более умеренными, но значительными по сравнению с контрольным образцом с низкой дозой активина А. Другие соединения оказались неактивными по сравнению с контрольным образцом с низкой дозой активина А.
Пример 10
Эффект соединений, составляющих предмет настоящего изобретения, на дифференцирование одиночных эмбриональных стволовых клеток человека в клетки, экспрессирующие маркеры, характерные для линии сформированной эндодермы, в отсутствие активина А
Циклические анилин-пиридинотриазиновые соединения также были протестированы в формате скринингового анализа, при котором клетки диспергировали в ходе ферментативной обработки и высеивали монослоем для проведения анализа. В анализ также были внесены изменения с целью устранения сыворотки, в которой могут содержаться факторы роста, хотя и в низких дозах. Для этой цели основная среда была изменена, а сыворотку заменили на бычий сывороточный альбумин (BSA), не содержащий жирных кислот. Анализ укоротили с четырех до трех дней, временной интервал получения результатов был более узким. Наконец, в анализ были включены два ростовых фактора, EGF и FGF4, которые ранее показали достоверный, но недостаточно оптимальный эффект на дифференцирование клеток в сформированную эндодерму в отсутствие активина A.
Скрининговый анализ
Посев клеток для анализа. Коротко говоря, кластеры эмбриональных стволовых клеток человека линии H1 выращивали на покрытых препаратом MATRIGELTM с пониженным содержанием факторов роста (Invitrogen; № по кат. 356231) носителях для культивирования тканей. Культуры обработали препаратом Accutase (Sigma; № по кат. A6964) объемом, эквивалентным 10 мл на 10 см2 площади поверхности, в течение 5 минут при температуре 37єC, затем культуры осторожно ресуспендировали, осадили центрифугированием и ресуспендировали для подсчета в кондиционированной среде MEF. При посеве для анализа клетки наносили с плотностью 50000 клеток на см2 на покрытые препаратом MATRIGELTM с пониженным содержанием факторов роста 96-луночные черные планшеты (Packard ViewPlates; № по кат. 6005182) в объеме 100 мкл на лунку. Клеткам дали возможность прикрепиться, после чего в течение 1-3 дней воспроизводили логарифмическую фазу роста, во время которой клетки ежедневно питали, используя кондиционированную среду MEF с добавлением 8 нг/мл bFGF (R&D Systems; № по кат. 233-FB). В течение всего периода анализа планшеты держали при температуре 37єC в атмосфере 5% CO2 в увлажняемом боксе.
Подготовка соединений и анализ. Исходные растворы EGF и FGF4 поместили в 96-луночный полипропиленовый планшет (Corning, Inc.; № по кат. 3960). Соединение 22 предоставили в виде исходного раствора концентрации 5 мМ, растворили в 100% DMSO (Sigma; № по кат. D2650) и хранили при температуре -80oC. Анализ начали с отсасывания культуральной среды из каждой лунки с последующей трехкратной промывкой лунки раствором PBS для удаления остатков факторов роста и сыворотки. Затем в лунки добавили тестируемые образцы объемом 80 мкл, содержащие основную среду RPMI-1640 (Invitrogen; № по кат. 22400-089) с добавлением 2,5% BSA, не содержащего жирных кислот (MP Biomedicals LLC; № по кат. 152401), 10 нг/мл bFGF (PeproTech Inc; № по кат. 100-18B), 25 нг/мл Wnt3a (R&D Systems; № по кат. 1324-WN), 3,125 мкМ Соединения 22 и 20 мкл 5-кратного раствора факторов роста с получением следующих конечных концентраций: 2% BSA, не содержащего жирных кислот, 8 нг/мл bFGF(PeproTech Inc; № по кат. 100-18B), 20 нг/мл Wnt3a и 2,5 мкМ Соединения 22. Лунки с положительными контрольными образцами содержали ту же основную среду с добавлением 2% BSA, не содержащего жирных кислот, 8 нг/мл bFGF, 20 нг/мл Wnt3a и 100 нг/мл рекомбинантного человеческого активина A (PeproTech; № по кат. 120-14). Лунки с отрицательными контрольными образцами содержали ту же основную среду с добавлением 2% BSA, не содержащего жирных кислот, 8 нг/мл bFGF, 20 нг/мл Wnt3a, но без обработки активином A.
На 2 день анализа среду из лунок снова отсосали и добавили среду в объеме 80 мкл на лунку, содержащую основную среду RPMI-1640 с добавлением 2,5% BSA, не содержащего жирных кислот, 10 нг/мл bFGF, 3,125 мкМ Соединения 22 и 20 мкл 5-кратного раствора факторов роста с получением следующих конечных концентраций: 2% BSA, не содержащего жирных кислот, 8 нг/мл bFGF и 2,5 мкМ Соединения 22. Лунки с положительными контрольными образцами содержали ту же основную среду с добавлением 2% BSA, не содержащего жирных кислот, 8 нг/мл bFGF и 100 нг/мл рекомбинантного человеческого активина A. Лунки с отрицательными контрольными образцами содержали ту же основную среду с добавлением 2% BSA, не содержащего жирных кислот, и 8 нг/мл bFGF, но без обработки активином A.
Одновременный многопараметрический анализ. По окончании четырехдневного периода культивирования аналитические планшеты дважды промыли раствором PBS, зафиксировали 4% параформальдегидом (Alexis Biochemical; № по кат. ALX-350-011) при комнатной температуре в течение 20 минут, затем трижды промыли раствором PBS и пермеабилизировали, обработав раствором 0,5% Triton X-100 (Sigma; № по кат. T8760-2) в течение 20 минут при комнатной температуре. Затем клетки снова трижды промыли раствором PBS и заблокировали раствором 4% куриной сыворотки (Invitrogen; № по кат. 16110082) в PBS в течение 30 минут при комнатной температуре. Первичные антитела (козий анти-человеческий SOX17; R&D Systems; № по кат. AF1924) разбавили в соотношении 1:100 в 4% куриной сыворотке и добавили в каждую лунку на один час при комнатной температуре. Сопряженные с флюорофором Alexa Fluor 488 вторичные антитела (куриный анти-козий IgG; Molecular Probes; № по кат. AZ1467) разбавили в соотношении 1:200 в PBS и добавили к каждому образцу после трехкратной промывки раствором PBS. Для подсчета окрашенных ядер на десять минут при комнатной температуре добавили 4 мкг/мл Hoechst 33342 (Invitrogen; № по кат. H3570). Затем планшеты один раз промыли раствором PBS и оставили в 100 мкл раствора PBS на лунку для томографии.
Томографию проводили на анализаторе IN Cell Analyzer 1000 (GE Healthcare) с применением дихроичного зеркала 51008bs для клеток, окрашенных красителями Hoechst 33342 и Alexa Fluor 488. Времена экспозиции оптимизировали с использованием лунок с положительными контрольными образцами и необработанных лунок с отрицательными контрольными образцами, окрашенных только вторичными антителами. Для компенсации возможных потерь клеток в ходе процедур анализа и последующего окрашивания для каждой лунки снимали по 15 проекций. Общее число клеток и общую интенсивность сигнала SOX17 измеряли для каждой лунки с помощью программного пакета IN Cell Developer Toolbox 1.7 (GE Healthcare). Сегментацию ядер определяли по ступеням шкалы серых тонов (исходный интервал 100-300) и размерам ядра. Для каждой повторной совокупности данных рассчитали средние значение и стандартные отклонения. Общий уровень экспрессии белка SOX17 выражается в виде общей интенсивности или интегральной интенсивности, определяемой как произведение общей интенсивности флюоресценции клетки на площадь клетки. Фон исключили на основании критериев соответствия ступеням шкалы серых тонов в диапазоне от 200 до 3500. Показатели общей интенсивности нормировали путем деления общей интенсивности для каждой лунки на среднюю общую интенсивность для положительного контрольного образца. Для каждого набора повторностей рассчитали нормированные данные для средних значений и стандартных отклонений.
В Таблице 10 представлены результаты анализа с Соединением 34. Контрольные образцы, содержащие EGF и (или) FGF4, без Соединения 34 имели низкую экспрессию SOX17. Добавление Соединения 34 существенно повысило экспрессию SOX17.
Пример 11
Сравнение способности активина A и GDF-8 дифференцировать эмбриональные стволовые клетки человека в клетки, экспрессирующие маркеры, характерные для линии сформированной эндодермы
В предыдущем примере было показано, что GDF-8 может заменить активин A при дифференцировании эмбриональных стволовых клеток человека в клетки, экспрессирующие маркеры, характерные для линии сформированной эндодермы. Было важно определить относительную эффективность GDF-8 и активина A при дифференцировании эмбриональных стволовых клеток человека в клетки, экспрессирующие маркеры, характерные для линии сформированной эндодермы. Был проведен анализ зависимости доза-эффект при дифференцировании эмбриональных стволовых клеток с применением эквивалентных концентраций каждого из факторов роста, а полученные результаты были подвергнуты сравнению.
Подготовка клеток к анализу. Исходные культуры эмбриональных стволовых клеток человека (эмбриональные стволовые клетки человека линии H1) поддерживали в недифференцированном плюрипотентном состоянии на чашках, покрытых препаратом MATRIGEL с пониженным содержанием факторов роста, в кондиционированной среде MEF и пассировали в среднем каждые четыре дня. При пассировании клетки обрабатывали раствором диспазы 1 мг/мл (Invitrogen, № по кат. 17105-041) в течение 5-7 минут при 37єC с последующей промывкой монослоя кондиционированной культуральной средой MEF, затем осторожно соскоблили для получения кластеров клеток. Кластеры центрифугировали на низкой скорости для получения осадка клеток и удаления остатков диспазы. Кластеры клеток разделили в соотношении 1:3 или 1:4 для стандартного поддерживающего культивирования или в соотношении 1:1 для немедленного анализа. Все линии эмбриональных стволовых клеток человека поддерживали при номерах пассажа менее 50 и периодически проверяли на нормальный кариотипический фенотип и отсутствие примесей микоплазмы.
Кластеры клеток, предназначенные для анализа, равномерно ресуспендировали в кондиционированной среде MEF с добавлением 8 нг/мл bFGF и высеяли на покрытые препаратом MATRIGELTM с пониженным содержанием факторов роста 96-луночные планшеты Packard VIEWPLATES (PerkinElmer; № по кат. 6005182) в объеме 100 мкл на лунку. Для первоначального посева и размножения использовали кондиционированную среду MEF с добавлением 8 нг/мл bFGF. Ежедневное питание осуществляли путем отсасывания отработанной культуральной среды из каждой лунки и ее замены тем же объемом свежей среды. В течение всего периода анализа планшеты держали при температуре 37єC в атмосфере 5% CO2 в увлажняемом боксе.
Анализ. Анализ начали с отсасывания культуральной среды из всех лунок и добавления аликвот (100 мкл) тестируемой среды. Каждый тест выполнялся в четырех повторностях. В течение всего трехдневного периода питание вводили в 1 и 2 дни, для чего среду из каждой лунки отсасывали и заменяли свежей тестируемой средой. Для приготовления тестируемой среды, содержащей различные концентрации активина А (PeproTech; № по кат. 120-14) или GDF-8 (R&D Systems, № по кат. 788-G8), использовали два 12-канальных полипропиленовых лотка (Argos technologies, Inc, № по кат. B3135). В каждом лотке каналы с номерами со 2 по 12 содержали 1 мл анализируемой среды, содержащей RPMI-1640 (Invitrogen; № по кат. 22400) с добавлением 2% фракции V бычьего альбумина, не содержащего жирных кислот (FAF BSA) (MP Biomedicals, Inc; № по кат. 152401), 8 нг/мл bFGF (PeproTech Inc.; № по кат. 100-18B) и 20 нг/мл Wnt3a (R&D Systems; № по кат. 1324-WN/CF) добавляемого в день 1 и не добавляемого в дни 2 и 3. В каждом лотке канал с номером 1 содержал 1600 нг/мл активина A или 1600 нг/мл GDF-8, разведенных в той же анализируемой среде. Один миллилитр среды переносили из канала с номером 1 в канал с номером 2 и хорошо перемешивали. С помощью нового наконечника пипетки переносили один миллилитр среды из канала с номером 2 в канал с номером 3 и тщательно перемешивали. Процедуру последовательно повторяли в каждом лотке до канала с номером 11. В канале с номером 12 каждого лотка содержалась среда без активина А или GDF-8. В результате получалась серия двукратных тестовых разведений, где концентрации активина A или GDF-8 изменялись от 1,6 нг/мл до 1600 нг/мл, которые и добавляли в соответствующие лунки для анализа.
Одновременный многопараметрический анализ. По окончании трехдневного периода культивирования аналитические планшеты один раз промыли раствором PBS (Invitrogen; № по кат. 14190), зафиксировали 4% параформальдегидом (Alexis Biochemical; № по кат. ALX-350-011) при комнатной температуре в течение 20 минут, затем трижды промыли раствором PBS и пермеабилизировали, обработав раствором 0,5% Triton X-100 (Sigma; № по кат. T8760-2) в течение 20 минут при комнатной температуре. Затем клетки снова трижды промыли раствором PBS и заблокировали раствором 4% куриной сыворотки (Invitrogen; № по кат. 16110082) в PBS в течение 30 минут при комнатной температуре. Первичные антитела (козий анти-человеческий SOX17; R&D Systems; № по кат. AF1924) разбавили в соотношении 1:100 в 4% куриной сыворотке и добавили в каждую лунку на два часа при комнатной температуре. Сопряженные с флюорофором Alexa Fluor 488 вторичные антитела (куриный анти-козий IgG; Invitrogen; № по кат. A21467) разбавили в соотношении 1:200 в растворе PBS и добавили в каждую лунку после трехкратной промывки клеток раствором PBS. Для подсчета окрашенных ядер на пятнадцать минут при комнатной температуре добавили 5 мкг/мл Hoechst 33342 (Invitrogen; № по кат. H3570). Затем планшеты один раз промыли раствором PBS и оставили в 100 мкл раствора PBS на лунку для томографии.
Томографию проводили на анализаторе IN Cell Analyzer 1000 (GE Healthcare) с применением дихроичного зеркала 51008bs для клеток, окрашенных красителями Hoechst 33342 и Alexa Fluor 488. Для каждой лунки снимали по 25 проекций. Общую интенсивность сигнала SOX17 измеряли для каждой лунки с помощью программного пакета IN Cell Developer Toolbox 1.7 (GE Healthcare). Сегментацию ядер определяли по ступеням шкалы серых тонов (исходный интервал 100-300) и размерам ядра. Для каждой четырехкратно повторенной совокупности данных рассчитали средние значения и стандартные отклонения. Общий уровень экспрессии белка SOX17 выражается в виде общей интенсивности или интегральной интенсивности, определяемой как произведение общей интенсивности флюоресценции клетки на площадь клетки. Фон исключили на основании критериев соответствия ступеням шкалы серых тонов в диапазоне от 200 до 4500. Показатели общей интенсивности сигнала SOX17 были рассчитаны с помощью программы GraphPad Prism 4.02 (GraphPad Software, Inc., Lo Jolla, Калифорния, США). Данные нормировали, для чего наименьшее и наибольшее значения в каждой совокупности данных приняли за 0% и 100%, соответственно. В Таблице 11 представлены нормированные значения для всех совокупностей данных для активина A и GDF-8. На фиг. 2 изображены две сигмоидальные кривые доза-эффект, полученные на основании нормированных значений из Таблицы 11. Значения R2, показывающие коэффициенты аппроксимации, были вычислены с использованием программы GraphPad Prism и составили 0,9944 для активина A и 0,9964 для GDF-8. С использованием программы GraphPad Prism были вычислены значения EC50 для каждого из факторов роста, которые составили 13,9 нг/мл для активина A и 184,8 нг/мл для GDF-8. Эти данные показывают, что GDF-8 менее эффективен, чем активин A в отношении индукции дифференцирования эмбриональных стволовых клеток человека в клетки, экспрессирующие маркеры, характерные для линии сформированной эндодермы. Тем не менее, в определенных концентрациях GDF-8 может заменить активин A и обеспечить образование эквивалентной популяции клеток сформированной эндодермы, показателем чего является экспрессия SOX17.
Пример 12
Клетки, экспрессирующие маркеры, характерные для линии сформированной эндодермы, полученные с использованием способов, составляющих предмет настоящего изобретения, способны далее дифференцироваться в клетки, экспрессирующие маркеры, характерные для линии панкреатических эндокринных клеток
Параллельные популяции эмбриональных стволовых клеток человека дифференцировали в клетки, экспрессирующие маркеры, характерные для линии сформированной эндодермы, с использованием GDF-8 в сочетании с Соединением 34 или Соединением 56. Далее к обработанным клеткам применили протокол поэтапного дифференцирования для стимуляции их дифференцирования в линию панкреатической эндодермы и линию панкреатических эндокринных клеток. В течение всего периода поэтапного дифференцирования для сравнения присутствовал параллельный контрольный образец, состоящий из клеток, обработанных активином A и Wnt3a. На каждой стадии дифференцирования отбирались образцы для определения наличия белковых и мРНК-биомаркеров, соответствующих различным стадиям дифференцирования.
Подготовка клеток к анализу. Исходные культуры эмбриональных стволовых клеток человека (эмбриональные стволовые клетки человека линии H1) поддерживали в недифференцированном плюрипотентном состоянии на чашках, покрытых препаратом MATRIGELTM с пониженным содержанием факторов роста, в кондиционированной среде MEF и пассировали в среднем каждые четыре дня. При пассировании клетки обрабатывали раствором диспазы 1 мг/мл (Invitrogen; № по кат. 17105-041) в течение 5-7 минут при 37єC с последующей промывкой монослоя кондиционированной культуральной средой MEF, затем осторожно соскоблили для получения кластеров клеток. Кластеры центрифугировали на низкой скорости для получения осадка клеток и удаления остатков диспазы. Кластеры клеток разделили в соотношении 1:3 или 1:4 для стандартного поддерживающего культивирования или в соотношении 1:1 для немедленного анализа. Все линии эмбриональных стволовых клеток человека поддерживали при номерах пассажа менее 50 и периодически проверяли на нормальный кариотип и отсутствие микоплазмы.
Кластеры клеток равномерно ресуспендировали в кондиционированной среде MEF с добавлением 8 нг/мл bFGF и высеяли на покрытые препаратом MATRIGELTM с пониженным содержанием факторов роста 24-луночные культуральные планшеты с черными стенками (Arctic White; № по кат. AWLS-303012) в объеме 0,5 мл на лунку. Ежедневное питание осуществляли путем отсасывания отработанной культуральной среды из каждой лунки и ее замены тем же объемом свежей среды. В течение всего периода анализа планшеты держали при температуре 37єC в атмосфере 5% CO2.
Анализ. Анализ начали с отсасывания культуральной среды из всех лунок и добавления аликвот (0,5 мл) тестируемой среды. На первом этапе дифференцирования тесты проводили в течение трехдневного периода. Питание вводили ежедневно, для чего среду из каждой лунки отсасывали и заменяли свежей тестируемой средой. В 1 день анализа в соответствующие лунки добавили 100 нг/мл активина A (PeproTech; № по кат. 120-14) или 200 нг/мл GDF-8 (R&D Systems, № по кат. 788-G8), причем все факторы роста разбавили в среде RPMI-1640 (Invitrogen; № по кат. 22400) с добавлением 1% фракции V бычьего альбумина, не содержащего жирных кислот (FAF BSA) (MP Biomedicals, Inc; № по кат. 152401), 1% Probumin (Millipore; № по кат. 81-068-3) и 20 нг/мл Wnt3a (R&D Systems; № по кат. 1324-WN/CF). На 2 день анализа 100 нг/мл активина A или 200 нг/мл GDF-8 разбавили в среде RPMI-1640 с добавлением 2% FAF BSA, без Wnt3a. В некоторых тестируемых образцах с GDF-8 Wnt3a заменяли Соединением 34 или Соединением 56 в концентрации 2,5 мкМ, и Соединение 34 или Соединение 56 добавляли ежедневно в течение всех трех дней дифференцирования клеток в сформированную эндодерму. По завершении первого этапа дифференцирования клетки из некоторых лунок собрали с целью определения методом проточной цитометрии уровня CXCR4, маркера образования сформированной эндодермы. Клетки из других лунок собрали для определения методом ОТ-ПЦР других маркеров дифференцирования.
После завершения первого этапа дифференцирования дублирующиеся наборы параллельных лунок каждой из экспериментальных групп подвергли дальнейшему поэтапному дифференцированию. Важно отметить, что после первого этапа дифференцирования все лунки с клетками, подвергаемыми дальнейшему культивированию и дифференцированию, получали одинаковую обработку. Ниже представлено описание протокола, использованного для продолжения дифференцирования.
Второй этап протокола дифференцирования занял два дня. Питание клеткам вводили ежедневно, для чего среду из всех лунок отсасывали и заменяли свежей аликвотой (0,5 мл) среды DMEM:F12 (Invitrogen; № по кат. 11330-032) с добавлением 2% фракции V бычьего альбумина, не содержащего жирных кислот (FAF BSA) (MP Biomedicals, Inc; № по кат. 152401), 50 нг/мл FGF7 (PeproTech; № по кат. 100-19) и 250 нМ циклопамина (Calbiochem; № по кат. 239804).
Третий этап протокола дифференцирования занял четыре дня. Питание клеткам вводили ежедневно, для чего среду из всех лунок отсасывали и заменяли свежей аликвотой (0,5 мл) среды DMEM с высоким содержанием глюкозы (Invitrogen; № по кат. 10569) с добавлением 1% B27 (Invitrogen; № по кат. 17504-044), 50 нг/мл FGF7, 100 нг/мл Noggin (R&D Systems; № по кат. 3344-NG), 250 нМ KAAD-циклопамина (Calbiochem; № по кат. 239804) и 2 мкМ полностью транс-ретиноевой кислоты (RA) (Sigma-Aldrich; № по кат. R2625). По завершении третьего этапа дифференцирования клетки из некоторых лунок собрали с целью определения методом ОТ-ПЦР маркеров дифференцирования. Другие лунки подвергли одновременному многопараметрическому анализу на предмет определения уровня экспрессии белка Pdx1, фактора транскрипции, характерного для панкреатической эндодермы, и Cdx2, фактора транскрипции, характерного для интестинальной эндодермы.
Четвертый этап протокола дифференцирования занял четыре дня. Питание клеткам вводили ежедневно, для чего среду из всех лунок отсасывали и заменяли свежей аликвотой (0,5 мл) среды DMEM с высоким содержанием глюкозы с добавлением 1% B27, 100 нг/мл Noggin, 100 нг/мл Netrin-4, 1 мкМ DAPT (EMD Biosciences; № по кат. 565770) и 1 мкМ ингибитора Alk 5 (Axxora; № по кат. ALX-270-445). По завершении четвертого этапа дифференцирования клетки из некоторых лунок собрали с целью определения методом ОТ-ПЦР маркеров дифференцирования. Другие лунки подвергли одновременному многопараметрическому анализу на предмет определения уровня экспрессии белка PDX1.
Пятый этап протокола дифференцирования выполнялся в течение семи дней в среде DMEM с высоким содержанием глюкозы с добавлением 1% B27 и 1 мкМ ингибитора Alk 5. Ежедневно среду из всех лунок отсасывали и заменяли свежей аликвотой (0,5 мл). По завершении пятого этапа дифференцирования клетки из некоторых лунок собрали с целью определения методом ОТ-ПЦР маркеров дифференцирования. Другие лунки подвергли одновременному многопараметрическому анализу на предмет определения уровня экспрессии инсулина и глюкагона.
Шестой этап протокола дифференцирования выполнялся в течение семи дней в среде DMEM с высоким содержанием глюкозы с добавлением 1% B27. Среду во всех лунках через день отсасывали и заменяли свежей аликвотой (0,5 мл). По завершении шестого этапа дифференцирования клетки из некоторых лунок собрали с целью определения методом ОТ-ПЦР маркеров дифференцирования.
Анализ методом FACS. Клетки для анализа методом FACS заблокировали 0,5% раствором человеческого гамма-глобулина (Sigma; № по кат. G-4386), разведенным в соотношении 1:5 в PBS (Invitrogen; № по кат. 14040-133) и буфере для окрашивания BD FACS - BSA (BD; № по кат. 554657), в течение 15 минут при температуре 4єC. Затем клетки окрасили антителами к CD9 PE (BD; № по кат. 555372), CD99 PE (Caltag; № по кат. MHCD9904) и CXCR4 APC (R&D Systems; № по кат. FAB173A) в течение 30 минут при 4єC. После серии промывок в буфере для окрашивания BD FACS клетки окрасили для оценки жизнеспособности красителем 7-AAD (BD; № по кат. 559925) и проанализировали на биоанализаторе BD FACSArray. Для получения процентной доли положительных клеток использовали мышиные изотипические контрольные антитела типа IgG1K к PE и APC.
Анализ методом ОТ-ПЦР. Образцы РНК очистили путем связывания с силикагелевой мембраной (Rneasy Mini Kit, Qiagen, Калифорния, США) в присутствии этанолсодержащего высокосолевого буферного раствора, а затем промыли для удаления примесей. Затем РНК очищали далее, используя набор TURBO DNA-free (Ambion, Inc.), элюировав в воду РНК высокого качества. Выход и чистоту полученной РНК оценивали по измеряемому на спектрофотометре поглощению на длинах волн 260 и 280 нм. Копии кДНК получили с очищенной РНК, используя набор высокой емкости для получения кДНК из архивного материала компании (ABI, Калифорния, США).
Если не указано иное, все реагенты были приобретены в Applied Biosystems. Реакции ПЦР в режиме реального времени проводили с использованием системы для определения последовательности ABI PRISM® 7900 Sequence Detection System. Использовали набор TAQMAN® UNIVERSAL PCR MASTER MIX® (ABI, Калифорния, США) с 20 нг обратно транскрибированной РНК в общем объеме реакционной смеси 20 мкл. Каждый образец кДНК анализировали дважды для коррекции погрешности пипетирования. Праймеры и меченные красителем FAM зонды TAQMAN использовали в концентрациях 200 нМ. Уровень экспрессии для каждого целевого гена нормировали по эндогенному контрольному образцу, глицеральдегид-3-фосфатдегидрогеназе человека (ГАФДГ), ранее разработанному Applied Biosystems. Праймеры и наборы зондов перечислены в Таблице 12. После начальной инкубации при температуре 50°C в течение 2 мин с последующей инкубацией при температуре 95°C в течение 10 минут с образцами провели 40 двухстадийных циклов, включающих стадию денатурации при температуре 95°C продолжительностью 15 секунд и следующую за ней стадию отжига/роста при температуре 60°C продолжительностью 1 минута. Анализ данных проводили с использованием программного пакета GENEAMP®7000 Sequence Detection System. Для каждого комплекта праймеров и зондов определяли значение порогового цикла (Ct) как номер цикла, при котором интенсивность флюоресценции достигала определенного значения в середине экспоненциальной области амплификации. Уровни относительной экспрессии генов вычисляли методом сравнения Ct. Коротко говоря, для каждого образца кДНК значение Ct эндогенного контрольного образца вычитали из значения Ct целевого гена и получали величину дельта Ct (ΔCt). Нормированное количество целевого гена рассчитывали как 2-ΔCt, принимая, что амплификация имеет 100% эффективность. Окончательные данные были выражены относительно калибровочного образца.
Одновременный многопараметрический анализ. По окончании периода культивирования аналитические планшеты один раз промыли раствором PBS (Invitrogen; № по кат. 14190), зафиксировали 4% параформальдегидом (Alexis Biochemical; № по кат. ALX-350-011) при комнатной температуре в течение 20 минут, затем трижды промыли раствором PBS и пермеабилизировали, обработав раствором 0,5% Triton X-100 (Sigma; № по кат. T8760-2) в течение 20 минут при комнатной температуре. Затем клетки снова трижды промыли раствором PBS и заблокировали раствором 4% куриной сыворотки (Invitrogen; № по кат. 16110082) в PBS в течение 30 минут при комнатной температуре. Первичные антитела (козий анти-человеческий SOX17; R&D Systems; № по кат. AF1924) разбавили в соотношении 1:100 в 4% куриной сыворотке и добавили в каждую лунку на два часа при комнатной температуре. Сопряженные с флюорофором Alexa Fluor 488 вторичные антитела (куриный анти-козий IgG; Invitrogen; № по кат. A21467) разбавили в соотношении 1:200 в растворе PBS и добавили в каждую лунку после трехкратной промывки клеток раствором PBS. Для подсчета окрашенных ядер на пятнадцать минут при комнатной температуре добавили 5 мкг/мл Hoechst 33342 (Invitrogen; № по кат. H3570). Затем планшеты один раз промыли раствором PBS и оставили в 100 мкл раствора PBS на лунку для томографии. Другие первичные антитела, использованные в анализе, включали: разведенные 1:100 мышиные антитела к человеческому CDX2 (Invitrogen; № по кат. 397800), разведенные 1:100 козьи антитела к человеческому Pdx1 (Santa Cruz Biotechnology; № по кат. SC-14664), разведенные 1:200 кроличьи антитела к человеческому инсулину (Cell Signaling; № по кат. C27C9) и разведенные 1:1500 мышиные антитела к человеческому глюкагону (Sigma-Aldrich; № по кат. G2654). В анализе использовались следующие вторичные антитела: разведенные 1:400 куриные антитела к мышиному IgG Alexa Fluor 647 (Invitrogen; № по кат. A-21463), разведенные 1:200 ослиные антитела к козьему IgG Alexa Fluor 488 (Invitrogen; № по кат. A11055), разведенные 1:1000 куриные антитела к кроличьему IgG Alexa Fluor 647 (Invitrogen; № по кат. A21443) и разведенные 1:1000 куриные антитела к мышиному IgG Alexa Fluor 488 (Invitrogen; № по кат. A21200).
Томографию проводили на анализаторе IN Cell Analyzer 1000 (GE Healthcare) с применением дихроичного зеркала 51008bs для клеток, окрашенных красителями Hoechst 33342 и Alexa Fluor 488. Для каждой лунки снимали по 25 проекций. Общую интенсивность сигнала измеряли для каждой лунки с помощью программного пакета IN Cell Developer Toolbox 1.7 (GE Healthcare). Сегментацию ядер определяли по ступеням шкалы серых тонов (исходный интервал 100-300) и размерам ядра. Для каждой повторной совокупности данных рассчитали средние значение и стандартные отклонения. Общий уровень экспрессии белка выражается в виде общей интенсивности или интегральной интенсивности, определяемой как произведение общей интенсивности флюоресценции клетки на площадь клетки. Фон исключили на основании критериев соответствия ступеням шкалы серых тонов в диапазоне от 200 до 4500. Показатели общей интенсивности нормировали путем деления общей интенсивности для каждой лунки на среднюю общую интенсивность для положительного контрольного образца.
Результаты ПЦР-анализа клеток, собранных на каждом из этапов дифференцирования, на характерные маркеры представлены в Таблице 13. В образцах, обработанных GDF-8 и Wnt3a, или GDF-8 и либо Соединением 34, либо Соединением 56, наблюдался сходный, а иногда повышенный уровень экспрессии маркеров, ассоциирующихся с дифференцированием в эндодерму и эндокринные клетки.
На фиг. 3 представлены результаты анализа методом FACS, демонстрирующие экспрессию маркера сформированной эндодермы, CXCR4, после первого этапа дифференцирования. Обработка эмбриональных стволовых клеток человека GDF-8 и Wnt3a привела к образованию процентной доли CXCR4-положительных клеток, сходной с наблюдаемой при обработке активином A и Wnt3a. Аналогично, обработка эмбриональных стволовых клеток человека GDF-8 и низкомолекулярным соединением (Соединение 34 или Соединение 56) также привела к образованию сходной или несколько большей процентной доли CXCR4-положительных клеток. На фиг. 4 представлены результаты одновременного многопараметрического анализа на предмет нормированной экспрессии белка SOX17 в эмбриональных стволовых клетках человека после трех дней дифференцирования в сформированную эндодерму. Уровни экспрессии в группах, прошедших обработку GDF-8 и Wnt3a или GDF-8 и низкомолекулярным соединением, были сходными с экспрессией при обработке активином A и Wnt3a.
На фиг. 5 представлены результаты одновременного многопараметрического анализа на предмет нормированной экспрессии белков Pdx1 и Cdx2 в эмбриональных стволовых клетках человека после третьего этапа дифференцирования в панкреатическую эндодерму. В группах, прошедших обработку GDF-8 и Wnt3a или GDF-8 и соединением, составляющим предмет настоящего изобретения, уровни экспрессии белков PDX1 и CDX2 были эквивалентными. В некоторых экспериментальных группах число клеток, сохраняющихся после дифференцирования, снижалось, что увеличивало долю клеток, экспрессирующих PDX1. Также были получены сходные результаты, демонстрирующие эквивалентную нормированную экспрессию PDX1 во всех экспериментальных группах после четвертого этапа дифференцирования, что показано на фиг. 6. На фиг. 7 показаны нормированные уровни экспрессии белков инсулина и глюкагона, демонстрирующие эквивалентную экспрессию этих белков в группах, обработанных активином A и обработанных GDF-8.
В совокупности эти результаты показывают, что GDF-8 в сочетании с Wnt3a или Соединением 34, или Соединением 56 может заменить активин A при дифференцировании клеток в сформированную эндодерму и при дальнейшем дифференцировании в панкреатическую эндодерму и эндокринные клетки.
Пример 13
Образование клеток, экспрессирующих маркеры, характерные для линии сформированной эндодермы, с использованием других членов семейства белков GDF
Было важно установить, можно ли путем обработки эмбриональных стволовых клеток человека другими членами семейства GDF получить клетки, экспрессирующие маркеры, характерные для линии сформированной эндодермы. С целью определения способности членов семейства белков GDF обеспечивать дифференцирование эмбриональных стволовых клеток человека в клетки, экспрессирующие маркеры, характерные для линии сформированной эндодермы, на эмбриональных стволовых клетках человека была протестирована комбинация Wnt3a с Соединением 34 или Соединением 56 в сочетании с шестью разными факторами роста семейства GDF (GDF-3, GDF-5, GDF-8, GDF-10, GDF-11 и GDF-15). Для сравнения использовался параллельный контрольный образец, состоящий из клеток, обработанных активином A и Wnt3a.
Подготовка клеток к анализу. Исходные культуры эмбриональных стволовых клеток человека (эмбриональные стволовые клетки человека линии H1) поддерживали в недифференцированном плюрипотентном состоянии на чашках, покрытых препаратом MATRIGELTM с пониженным содержанием факторов роста (BD Biosciences; № по кат. 356231), в кондиционированной среде MEF и пассировали в среднем каждые четыре дня. При пассировании клетки обрабатывали раствором диспазы 1 мг/мл (Invitrogen; № по кат. 17105-041) в течение 5-7 минут при 37єC с последующей промывкой монослоя кондиционированной культуральной средой MEF, затем осторожно соскоблили для получения кластеров клеток. Кластеры центрифугировали на низкой скорости для получения осадка клеток и удаления остатков диспазы. Кластеры клеток разделили в соотношении 1:3 или 1:4 для стандартного поддерживающего культивирования или в соотношении 1:1 для немедленного анализа. Все линии эмбриональных стволовых клеток человека поддерживали при номерах пассажа менее 50 и периодически проверяли на нормальный кариотип и отсутствие микоплазмы.
Кластеры клеток равномерно ресуспендировали в кондиционированной среде MEF с добавлением 8 нг/мл bFGF и высеяли на покрытые препаратом MATRIGELTM с пониженным содержанием факторов роста 96-луночные планшеты Packard VIEWPLATES (PerkinElmer; № по кат. 6005182) в объеме 0,1 мл на лунку. Ежедневное питание осуществляли путем отсасывания отработанной культуральной среды из каждой лунки и ее замены тем же объемом свежей среды. В течение всего периода анализа планшеты держали при температуре 37єC в атмосфере 5% CO2.
Анализ. Анализ начали с отсасывания культуральной среды из всех лунок и добавления аликвот (100 мкл) тестируемой среды. Каждый тест выполнялся в трех повторностях. В течение всего четырехдневного периода питание вводили в 1 и 3 дни, для чего среду из каждой лунки отсасывали и заменяли свежей тестируемой средой. Для тестирования были получены следующие члены семейства белков GDF: GDF-3 (PeproTech; № по кат. 120-22); GDF-5 (DePuy Orthopaedics, Inc., компания Johnson & Johnson); GDF-8 (R&D Systems; № по кат. 788-G8); GDF-10 (R&D Systems; № по кат. 1543-BP); GDF11 (PeproTech; № по кат. 120-11); GDF-15 (R&D Systems; № по кат. 957-GD). В 1 день анализа во все лунки добавили аликвоту (80 мкл) основной среды DMEM:F12 (Invitrogen; № по кат. 11330-032) с добавлением 0,5% эмбриональной бычьей сыворотки (FBS) (Hyclone; № по кат. SH30070.03). Для тестирования активина А и различных GDF в сочетании либо с Wnt3a, либо с Соединением 34 или Соединением 56 была приготовлена серия из пяти различных контрольных или экспериментальных образцов. Эти образцы добавляли аликвотами объемом 20 мкл (с 5-кратной концентрацией) в соответствующие лунки, получая в каждой лунке среду, обладающую указанными параметрами, объемом 100 мкл. В первом наборе контрольных образцов тестировались следующие среды: 1) без добавок (т.е. без добавления факторов роста или низкомолекулярных соединений); 2) 100 нг/мл активина A (PeproTech; № по кат. 120-14) в сочетании с 20 нг/мл Wnt3a (R&D Systems; № по кат. 1324-WN/CF); 3) только 20 нг/мл Wnt3a; 4) только Соединение 34 (2,5 мкМ) без факторов роста и низкомолекулярных соединений; 5) только Соединение 56 (2,5 мкМ) без факторов роста и низкомолекулярных соединений. Во втором наборе тестируемых образцов тестировались следующие среды в сочетании со 100 нг/мл GDF3: 1) без добавок (т.е. только GDF-3); 2) 20 нг/мл Wnt3a; 3) 20нг/мл Wnt3a и Соединение 34 (2,5 мкМ); 4) Соединение 34 (2,5 мкМ); 5) Соединение 56 (2,5 мкМ); и 6) 20 нг/мл Wnt3a и Соединение 56 (2,5 мкМ). В третьем наборе тестируемых образцов каждая из шести упомянутых сред сочеталась со 100 нг/мл GDF-5. В четвертом наборе тестируемых образцов каждая из шести упомянутых сред сочеталась со 100 нг/мл GDF-8. В пятом наборе тестируемых образцов каждая из шести упомянутых сред сочеталась со 100 нг/мл GDF-10. В шестом наборе тестируемых образцов каждая из шести упомянутых сред сочеталась со 100 нг/мл GDF-11. В седьмом наборе тестируемых образцов каждая из шести упомянутых сред сочеталась со 100 нг/мл GDF-15. На третий день анализа во все лунки с тестируемыми образцами добавили 100 нг/мл активина A или 100 нг/мл соответствующего фактора роста GDF, без Wnt3a или Соединений 34 или 56, разведенные в среде DMEM:F12 с добавлением 2% FBS.
Одновременный многопараметрический анализ. По окончании периода культивирования аналитические планшеты один раз промыли раствором PBS (Invitrogen; № по кат. 14190), зафиксировали 4% параформальдегидом (Alexis Biochemical; № по кат. ALX-350-011) при комнатной температуре в течение 20 минут, затем трижды промыли раствором PBS и пермеабилизировали, обработав раствором 0,5% Triton X-100 (Sigma; № по кат. T8760-2) в течение 20 минут при комнатной температуре. Затем клетки снова трижды промыли раствором PBS и заблокировали раствором 4% куриной сыворотки (Invitrogen; № по кат. 16110082) в PBS в течение 30 минут при комнатной температуре. Первичные антитела (козий анти-человеческий SOX17; R&D Systems; № по кат. AF1924) разбавили в соотношении 1:100 в 4% куриной сыворотке и добавили в каждую лунку на два часа при комнатной температуре. Сопряженные с флюорофором Alexa Fluor 488 вторичные антитела (куриный анти-козий IgG; Invitrogen; № по кат. A21467) разбавили в соотношении 1:200 в растворе PBS и добавили в каждую лунку после трехкратной промывки клеток раствором PBS. Для подсчета окрашенных ядер на пятнадцать минут при комнатной температуре добавили 5 мкг/мл Hoechst 33342 (Invitrogen; № по кат. H3570). Затем планшеты один раз промыли раствором PBS и оставили в 100 мкл раствора PBS на лунку для томографии.
Томографию проводили на анализаторе IN Cell Analyzer 1000 (GE Healthcare) с применением дихроичного зеркала 51008bs для клеток, окрашенных красителями Hoechst 33342 и Alexa Fluor 488. Для каждой лунки снимали по 25 проекций. Общую интенсивность сигнала измеряли для каждой лунки с помощью программного пакета IN Cell Developer Toolbox 1.7 (GE Healthcare). Сегментацию ядер определяли по ступеням шкалы серых тонов (исходный интервал 100-300) и размерам ядра. Для каждой повторной совокупности данных рассчитали средние значение и стандартные отклонения. Общий уровень экспрессии белка выражается в виде общей интенсивности или интегральной интенсивности, определяемой как произведение общей интенсивности флюоресценции клетки на площадь клетки. Фон исключили на основании критериев соответствия ступеням шкалы серых тонов в диапазоне от 200 до 4500. Показатели общей интенсивности нормировали путем деления общей интенсивности для каждой лунки на среднюю общую интенсивность для положительного контрольного образца.
На фиг. 8 представлены результаты одновременного многопараметрического анализа на предмет экспрессии белка SOX17 в эмбриональных стволовых клетках человека после четырех дней дифференцирования в сформированную эндодерму. Во всех случаях результаты нормированы к положительному контрольному образцу - обработке активином A и Wnt3a. Как видно из фиг. 8A, только положительный контрольный образец индуцировал значительную экспрессию SOX17, а обработка только Wnt3a или только Соединением 34 или Соединением 56 не индуцировала экспрессию SOX17. На фиг. 8, части с B по G, представлены нормированные уровни экспрессии SOX17 в клетках, прошедших соответствующую обработку, при которой активин А заменялся на фактор роста GDF. GDF-3 (фиг. 8B) и GDF-5 (фиг. 8C) индуцировали слабую экспрессию SOX17 и только в тех образцах, где присутствовали соединения, составляющие предмет настоящего изобретения. GDF10 (фиг. 8D), GDF11 (фиг. 8E) и GDF15 (фиг. 8G) индуцировали значительную экспрессию SOX17, уровень которой был выше, чем при обработке GDF3 или 5, но ниже, чем при обработке активном A и Wnt3a. В целом, экспрессия SOX17 была пренебрежимо мала, если GDF-10, GDF-11 или GDF-15 сочетались с Wnt3a, но повышалась, если они сочетались с одним из соединений, составляющих предмет изобретения, в частности, в сочетании с Соединением 34. На фиг. 8D представлены результаты для экспериментальных групп, обработанных GDF-8, где видно, что GDF-8 в сочетании с Соединением 34 или Соединением 56 приводил к существенной индукции экспрессии SOX17, превышающей результаты, наблюдаемые в положительном контрольном образце с активином A/Wnt3a. В некоторых случаях присутствие Соединения 34 или Соединения 56 в сочетании с фактором роста GDF также приводило к увеличению числа клеток при дифференцировании.
В совокупности эти результаты показывают, что фактор GDF-8, используемый в комбинации с Соединением 34 или Соединением 56, превосходит все прочие протестированные факторы семейства GDF и может заменить активин А при дифференцировании клеток в сформированную эндодерму.
Пример 14
Образование клеток, экспрессирующих маркеры, характерные для линии сформированной эндодермы, с использованием других членов суперсемейства белков TGF
Было важно установить, можно ли путем обработки эмбриональных стволовых клеток человека другими членами суперсемейства TGF улучшить образование клеток, экспрессирующих маркеры, характерные для линии сформированной эндодермы. С целью определения способности членов суперсемейства белков TGF обеспечивать дифференцирование эмбриональных стволовых клеток человека в клетки, экспрессирующие маркеры, характерные для линии сформированной эндодермы, на эмбриональных стволовых клетках человека была протестирована комбинация Соединения 34 и Wnt3a в сочетании с одним из факторов TGFβ-1, BMP2, BMP3 или BMP4. Параллельно тестировали два коммерческих варианта GDF-8, выпущенных разными производителями, в сочетании с Wnt3a с целью определения их способности обеспечивать дифференцирование эмбриональных стволовых клеток человека в клетки, экспрессирующие маркеры, характерные для линии сформированной эндодермы. Для сравнения использовался положительный контрольный образец - обработка активином A и Wnt3a.
Подготовка клеток к анализу. Исходные культуры эмбриональных стволовых клеток человека (эмбриональные стволовые клетки человека линии H1) поддерживали в недифференцированном плюрипотентном состоянии на чашках, покрытых препаратом MATRIGELTM с пониженным содержанием факторов роста (BD Biosciences; № по кат. 356231), в кондиционированной среде MEF и пассировали в среднем каждые четыре дня. При пассировании клетки обрабатывали раствором диспазы 1 мг/мл (Invitrogen; № по кат. 17105-041) в течение 5-7 минут при 37єC с последующей промывкой монослоя кондиционированной культуральной средой MEF, затем осторожно соскоблили для получения кластеров клеток. Кластеры центрифугировали на низкой скорости для получения осадка клеток и удаления остатков диспазы. Кластеры клеток разделили в соотношении 1:3 или 1:4 для стандартного поддерживающего культивирования или в соотношении 1:1 для немедленного анализа. Все линии эмбриональных стволовых клеток человека поддерживали при номерах пассажа менее 50 и периодически проверяли на нормальный кариотип и отсутствие микоплазмы.
Кластеры клеток равномерно ресуспендировали в кондиционированной среде MEF с добавлением 8 нг/мл bFGF и высеяли на покрытые препаратом MATRIGELTM с пониженным содержанием факторов роста 96-луночные планшеты Packard VIEWPLATES (PerkinElmer; № по кат. 6005182) в объеме 0,1 мл на лунку. Ежедневное питание осуществляли путем отсасывания отработанной культуральной среды из каждой лунки и ее замены тем же объемом свежей среды. В течение всего периода анализа планшеты держали при температуре 37єC в атмосфере 5% CO2.
Анализ. Анализ начали с отсасывания культуральной среды из всех лунок и добавления аликвот (100 мкл) тестируемой среды. Каждый тест выполнялся в трех повторностях. В течение всего трехдневного периода питание вводили в 1 и 2 дни, для чего среду из каждой лунки отсасывали и заменяли свежей тестируемой средой. Для тестирования были получены следующие белковые факторы роста: BMP-2 (R&D Systems; № по кат. 355-BM); BMP-3 (R&D Systems; № по кат. 113-BP); BMP-4 (R&D Systems; № по кат. 314-BP); TGFβ-1 (R&D Systems; № по кат. 240-B); GDF-8 (PeproTech; № по кат. 120-00); GDF-8 (Shenandoah; № по кат. 100-22) и активин A (PeproTech; № по кат. 120-14). В 1 день анализа в каждую лунку добавили 80 мкл питательной среды [RPMI-1640 (Invitrogen; № по кат. 22400) с добавлением 2,5% фракции V бычьего альбумина, не содержащего жирных кислот (FAF BSA) (MP Biomedicals, Inc; № по кат. 152401) и 10 нг/мл bFGF]. В некоторых лунках в питательную среду добавили 25 нг/мл Wnt3a (R&D Systems; № по кат. 1324-WN/CF) с получением конечной концентрации для анализа, равной 20 нг/мл. В некоторых лунках в питательную среду добавили активин А, получив конечную концентрацию для анализа, равную 100 нг/мл. В некоторых лунках в питательную среду добавили 3,125 мкМ Соединения 34, получив конечную концентрацию для анализа, равную 2,5 мкМ. Также в соответствующие лунки с тестируемыми образцами была введена раститровка доз дополнительных факторов роста (5-кратная концентрация, разведение в RPMI-1640) с получением итогового объема 100 мкл в каждой лунке для всех видов обработки. На 2 день анализа были исключены Wnt3a и Соединение 34. Во все лунки добавили 80 мкл питательной среды [RPMI-1640 с добавлением 2,5% FAF BSA и 10 нг/мл bFGF] и 20 мкл должным образом разведенного фактора роста (5-кратная концентрация, разведение в RPMI-1640). Для сравнения использовались следующие контрольные образцы: 1) без добавления факторов роста; 2) только Wnt3a; и 3) активин A с Wnt3a. Каждый коммерческий GDF-8 тестировался в комбинации с Wnt3a. Каждый из факторов роста BMP, а также TGFβ-1 тестировались в сочетании с Wnt3a, с Соединением 34, а также в сочетании как с Wnt3a, так и с Соединением 34.
Одновременный многопараметрический анализ. По окончании периода культивирования аналитические планшеты один раз промыли раствором PBS (Invitrogen; № по кат. 14190), зафиксировали 4% параформальдегидом (Alexis Biochemical; № по кат. ALX-350-011) при комнатной температуре в течение 20 минут, затем трижды промыли раствором PBS и пермеабилизировали, обработав раствором 0,5% Triton X-100 (Sigma; № по кат. T8760-2) в течение 20 минут при комнатной температуре. Затем клетки снова трижды промыли раствором PBS и заблокировали раствором 4% куриной сыворотки (Invitrogen; № по кат. 16110082) в PBS в течение 30 минут при комнатной температуре. Первичные антитела (козий анти-человеческий SOX17; R&D Systems; № по кат. AF1924) разбавили в соотношении 1:100 в 4% куриной сыворотке и добавили в каждую лунку на два часа при комнатной температуре. Сопряженные с флюорофором Alexa Fluor 488 вторичные антитела (куриный анти-козий IgG; Invitrogen; № по кат. A21467) разбавили в соотношении 1:200 в растворе PBS и добавили в каждую лунку после трехкратной промывки клеток раствором PBS. Для подсчета окрашенных ядер на пятнадцать минут при комнатной температуре добавили 5 мкг/мл Hoechst 33342 (Invitrogen; № по кат. H3570). Затем планшеты один раз промыли раствором PBS и оставили в 100 мкл раствора PBS на лунку для томографии.
Томографию проводили на анализаторе IN Cell Analyzer 1000 (GE Healthcare) с применением дихроичного зеркала 51008bs для клеток, окрашенных красителями Hoechst 33342 и Alexa Fluor 488. Для каждой лунки снимали по 25 проекций. Общую интенсивность сигнала измеряли для каждой лунки с помощью программного пакета IN Cell Developer Toolbox 1.7 (GE Healthcare). Сегментацию ядер определяли по ступеням шкалы серых тонов (исходный интервал 100-300) и размерам ядра. Для каждой повторной совокупности данных рассчитали средние значение и стандартные отклонения. Общий уровень экспрессии белка выражается в виде общей интенсивности или интегральной интенсивности, определяемой как произведение общей интенсивности флюоресценции клетки на площадь клетки. Фон исключили на основании критериев соответствия ступеням шкалы серых тонов в диапазоне от 200 до 4500. Показатели общей интенсивности нормировали путем деления общей интенсивности для каждой лунки на среднюю общую интенсивность для положительного контрольного образца.
На фиг. 9 представлены результаты одновременного многопараметрического анализа на предмет экспрессии белка SOX17 в эмбриональных стволовых клетках человека после трех дней дифференцирования в сформированную эндодерму. Во всех случаях результаты нормированы к положительному контрольному образцу - обработке активином A с Wnt3a. Результаты, представленные на фиг. 9A, показывают, что при обработке только питательной средой или только Wnt3a экспрессия SOX17 не индуцируется; надежная экспрессия SOX17 индуцировалась только при добавлении активина А. На фиг. 9, части B и C, представлены результаты по каждому из коммерческих вариантов GDF-8, где демонстрируются различия эффективности препаратов разных производителей. В клетках, обработанных GDF-8 в сочетании с Wnt3a, наблюдалась существенная индукция экспрессии SOX17, хотя и меньшая, чем при использовании активина А. На фиг. 9, части D, E, F и G, представлены результаты дифференцирования клеток в сформированную эндодерму под действием BMP2, BMP3, BMP4 и TGFβ-1, с раститровкой доз каждого из факторов роста, в сочетании с Wnt3a или Соединением 34, или же в сочетании как с Wnt3a, так и с Соединением 34. Хотя некоторые виды обработки достоверно влияли на число клеток в конце анализа (например, BMP2 и BMP4), индукция экспрессии SOX17 под действием этих факторов роста и сочетаний была слабой или пренебрежимо малой в сравнении с обработкой только Wnt3a.
Пример 15
Определение оптимальной дозы выбранных соединений, составляющих предмет настоящего изобретения, для образования клеток, экспрессирующих маркеры, характерные для линии сформированной эндодермы
Было важно определить оптимальные рабочие концентрации Соединения 181, Соединения 180, Соединения 19, Соединения 202, Соединения 40 и Соединения 34, способствующие образованию клеток, экспрессирующих маркеры, характерные для линии сформированной эндодермы. Параллельно проводили непосредственное сравнение раститровок всех соединений в сочетании с активином А или GDF-8 в тесте на образование сформированной эндодермы. Наконец, в ходе анализа была проверена продолжительность воздействия каждого соединения, также в сочетании с активином А или GDF-8, для чего соединение добавлялось только в 1 день анализа или в течение всех трех дней образования сформированной эндодермы.
Подготовка клеток к анализу. Исходные культуры эмбриональных стволовых клеток человека (эмбриональные стволовые клетки человека линии H1) поддерживали в недифференцированном плюрипотентном состоянии на чашках, покрытых препаратом MATRIGELTM с пониженным содержанием факторов роста (BD Biosciences; № по кат. 356231), в кондиционированной среде MEF с добавлением 8 нг/мл bFGF (PeproTech Inc.; № по кат. 100-18B) и пассировали в среднем каждые четыре дня. При пассировании клетки обрабатывали раствором диспазы 1 мг/мл (Invitrogen; № по кат. 17105-041) в течение 5-7 минут при 37єC с последующей промывкой монослоя кондиционированной культуральной средой MEF, затем осторожно соскоблили для получения кластеров клеток. Кластеры центрифугировали на низкой скорости для получения осадка клеток и удаления остатков диспазы. Кластеры клеток разделили в соотношении 1:3 или 1:4 для стандартного поддерживающего культивирования или в соотношении 1:1 для немедленного анализа. Все линии эмбриональных стволовых клеток человека поддерживали при номерах пассажа менее 50 и периодически проверяли на нормальный кариотип и отсутствие микоплазмы.
Кластеры клеток равномерно ресуспендировали в кондиционированной среде MEF с добавлением 8 нг/мл bFGF и высеяли на покрытые препаратом MATRIGELTM с пониженным содержанием факторов роста 96-луночные планшеты Packard VIEWPLATES (PerkinElmer; № по кат. 6005182) в объеме 0,1 мл на лунку. Ежедневное питание осуществляли путем отсасывания отработанной культуральной среды из каждой лунки и ее замены тем же объемом свежей среды. В течение всего периода анализа планшеты держали при температуре 37єC в атмосфере 5% CO2.
Анализ. Анализ начали с отсасывания культуральной среды из всех лунок и добавления аликвот (100 мкл) тестируемой среды. Каждый тест выполнялся в четырех повторностях. В течение всего четырехдневного периода питание вводили ежедневно, для чего среду из каждой лунки отсасывали и заменяли свежей тестируемой средой. В каждую лунку добавили 80 мкл питающей среды [RPMI-1640 (Invitrogen; № по кат. 22400) с добавлением 2,5% фракции V бычьего альбумина, не содержащего жирных кислот (FAF BSA) (MP Biomedicals, Inc; № по кат. 152401), 10 нг/мл bFGF, дополнительные факторы роста (1,25-кратная концентрация)] и 20 мкл тестируемого соединения (5-кратная концентрация, разведение в RPMI-1640) с получением итогового объема 100 мкл в каждой лунке. Тестируемыми соединениями в данном анализе были шесть соединений, составляющих предмет настоящего изобретения: Соединение 181, Соединение 180, Соединение 19, Соединение 202, Соединение 40 и Соединение 34, а также BIO, коммерческий ингибитор GSK3i (EMD Chemicals, Inc.; № по кат. 361550). В 1 день анализа в лунки добавили различные контрольные или экспериментальные среды. Использовались следующие контрольные среды с указанными конечными концентрациями для анализа: 1) только питающая среда; 2) только 20 нг/мл Wnt3a (R&D Systems; № по кат. 1324-WN/CF); 3) 100 нг/мл активина A (PeproTech; № по кат. 120-14); 4) 100 нг/мл активина A и 20 нг/мл Wnt3a; 5) 100 нг/мл GDF-8 (R&D Systems, № по кат. 788-G8); 6) 100 нг/мл GDF-8 и 20 нг/мл Wnt3a. Тестируемые соединения последовательно разбавляли вдвое для получения итоговых концентраций в диапазоне от 78 нМ до 10 мкМ. В экспериментальных образцах последовательность разведений каждого из соединений сочеталась со 100 нг/мл активина A или 100 нг/мл GDF-8, и в обоих случаях обработка не включала Wnt3a. Во 2 и 3 дни анализа в некоторые лунки добавили 20 нг/мл Wnt3a или разведенного тестируемого соединения в сочетании либо с активином А, либо с GDF-8. В другие лунки во 2 и 3 дни добавили активин А или GDF-8, но без Wnt3a или разведенного тестируемого соединения.
Одновременный многопараметрический анализ. По окончании периода культивирования аналитические планшеты один раз промыли раствором PBS (Invitrogen; № по кат. 14190), зафиксировали 4% параформальдегидом (Alexis Biochemical; № по кат. ALX-350-011) при комнатной температуре в течение 20 минут, затем трижды промыли раствором PBS и пермеабилизировали, обработав раствором 0,5% Triton X-100 (Sigma; № по кат. T8760-2) в течение 20 минут при комнатной температуре. Затем клетки снова трижды промыли раствором PBS и заблокировали раствором 4% куриной сыворотки (Invitrogen; № по кат. 16110082) в PBS в течение 30 минут при комнатной температуре. Первичные антитела (козий анти-человеческий SOX17; R&D Systems; № по кат. AF1924) разбавили в соотношении 1:100 в 4% куриной сыворотке и добавили в каждую лунку на два часа при комнатной температуре. Сопряженные с флюорофором Alexa Fluor 488 вторичные антитела (куриный анти-козий IgG; Invitrogen; № по кат. A21467) разбавили в соотношении 1:200 в растворе PBS и добавили в каждую лунку после трехкратной промывки клеток раствором PBS. Для подсчета окрашенных ядер на пятнадцать минут при комнатной температуре добавили 5 мкг/мл Hoechst 33342 (Invitrogen; № по кат. H3570). Затем планшеты один раз промыли раствором PBS и оставили в 100 мкл раствора PBS на лунку для томографии.
Томографию проводили на анализаторе IN Cell Analyzer 1000 (GE Healthcare) с применением дихроичного зеркала 51008bs для клеток, окрашенных красителями Hoechst 33342 и Alexa Fluor 488. Для каждой лунки снимали по 25 проекций. Общую интенсивность сигнала измеряли для каждой лунки с помощью программного пакета IN Cell Developer Toolbox 1.7 (GE Healthcare). Сегментацию ядер определяли по ступеням шкалы серых тонов (исходный интервал 100-300) и размерам ядра. Для каждой повторной совокупности данных рассчитали средние значение и стандартные отклонения. Общий уровень экспрессии белка выражается в виде общей интенсивности или интегральной интенсивности, определяемой как произведение общей интенсивности флюоресценции клетки на площадь клетки. Фон исключили на основании критериев соответствия ступеням шкалы серых тонов в диапазоне от 200 до 4500. Показатели общей интенсивности нормировали путем деления общей интенсивности для каждой лунки на среднюю общую интенсивность для положительного контрольного образца.
Результаты. На фиг. 10-14 представлены результаты одновременного многопараметрического анализа на предмет экспрессии SOX17, а на фиг. 15-19 представлено итоговое число клеток по завершении анализа. На фиг. 10 представлены результаты измерения экспрессии SOX 17 при контрольных вариантах обработки активином A или GDF-8, как отдельно, так и в сочетании с Wnt3a. При обработке активином А наблюдалась значительно более высокая экспрессия SOX17, чем при обработке GDF-8. Аналогично, как видно на фиг. 15, обработка активином A давала в конце анализа большее число клеток, чем обработка GDF-8, независимо от того, присутствовал Wnt3a в течение одного или трех дней анализа. Добавление Соединения 181, Соединения 180, Соединения 19, Соединения 202, Соединения 40 или Соединения 34 при обработке активином A не вызывало увеличения экспрессии SOX 17 (фиг. 11-12) или числа клеток (фиг. 17-18), независимо от того, присутствовало ли соединение в течение одного дня в начале анализа или в течение трех дней. Однако добавление Соединения 181, Соединения 180, Соединения 19, Соединения 202, Соединения 40 или Соединения 34 в сочетании с GDF-8 значительно увеличивало экспрессию SOX17 (фиг. 13-14), а также увеличивало число клеток в конце анализа (фиг. 18-19). При использовании Соединения 181, Соединения 180, Соединения 19, Соединения 202, Соединения 40 или Соединения 34 в сочетании с GDF-8 увеличение экспрессии SOX17 и числа клеток во многих случаях было эквивалентно результатам, наблюдаемым при обработке активином A. Улучшение дифференцирования было очевидным на раститровке доз многих соединений (в сочетании с GDF-8), хотя в наивысших концентрациях иногда наблюдалась токсичность. В большинстве случаев оптимальные положительные эффекты обработки соединением и GDF-8 были очевидны только при однодневном воздействии соединения в начале анализа. В некоторых случаях присутствие соединения на всем протяжении анализа не приводило к вредным эффектам или вызывало небольшой положительный эффект. На основании совокупности представленных результатов был определен оптимальный рабочий диапазон концентраций для каждого соединения в сочетании с обработкой GDF8. Результаты были специфичными для конкретных соединений, но в целом для данного анализа находились в диапазоне 1-10 мкМ.
Пример 16
Клетки, экспрессирующие маркеры, характерные для линии сформированной эндодермы, полученные с использованием способов, составляющих предмет настоящего изобретения, способны далее дифференцироваться в клетки, экспрессирующие маркеры, характерные для линии панкреатических эндокринных клеток
При дифференцировании клеток в сформированную эндодерму были протестированы дополнительные низкомолекулярные соединения в сочетании с GDF-8. К этим соединениям относился коммерческий ингибитор GSK3, а также соединения, составляющие предмет настоящего изобретения. К клеткам, обработанным GDF-8 в сочетании с различными низкомолекулярными соединениями, применялся протокол поэтапного дифференцирования. Эффективность дифференцирования определялась по экспрессии генов биомаркеров, характерных для линии панкреатической эндодермы или линии панкреатических эндокринных клеток. В течение всего периода поэтапного дифференцирования для сравнения присутствовал параллельный контрольный образец, состоящий из клеток, обработанных активином A и Wnt3a.
Подготовка клеток к анализу. Исходные культуры эмбриональных стволовых клеток человека (эмбриональные стволовые клетки человека линии H1) поддерживали в недифференцированном плюрипотентном состоянии на чашках, покрытых препаратом MATRIGELTM с пониженным содержанием факторов роста (BD Biosciences; № по кат. 356231), в кондиционированной среде MEF и пассировали в среднем каждые четыре дня. При пассировании клетки обрабатывали раствором диспазы 1 мг/мл (Invitrogen, № по кат. 17105-041) в течение 5-7 минут при 37єC с последующей промывкой монослоя кондиционированной культуральной средой MEF, затем осторожно соскоблили для получения кластеров клеток. Кластеры центрифугировали на низкой скорости для получения осадка клеток и удаления остатков диспазы. Кластеры клеток разделили в соотношении 1:3 или 1:4 для стандартного поддерживающего культивирования или в соотношении 1:1 для немедленного анализа. Все линии эмбриональных стволовых клеток человека поддерживали при номерах пассажа менее 50 и периодически проверяли на нормальный кариотип и отсутствие микоплазмы.
Кластеры клеток равномерно ресуспендировали в кондиционированной среде MEF с добавлением 8 нг/мл bFGF и высеяли на покрытые препаратом MATRIGEL с пониженным содержанием факторов роста 24-луночные культуральные планшеты с черными стенками (Arctic White; № по кат. AWLS-303012) в объеме 0,5 мл на лунку. Ежедневное питание осуществляли путем отсасывания отработанной культуральной среды из каждой лунки и ее замены тем же объемом свежей среды. В течение всего периода анализа планшеты держали при температуре 37єC в атмосфере 5% CO2.
Анализ. Анализ начали с отсасывания культуральной среды из всех лунок и добавления аликвот (0,5 мл) тестируемой среды. На первом этапе дифференцирования тесты проводили в течение трехдневного периода. Питание вводили ежедневно, для чего среду из каждой лунки отсасывали и заменяли свежей тестируемой средой. В 1 день анализа в соответствующие лунки добавили 100 нг/мл активина A (PeproTech; № по кат. 120-14) или 100 нг/мл GDF-8 (R&D Systems, № по кат. 788-G8), причем все факторы роста разбавили в среде RPMI-1640 (Invitrogen; № по кат. 22400) с добавлением 2% фракции V бычьего альбумина, не содержащего жирных кислот (FAF BSA) (Proliant Inc. № по кат. SKU 68700), и 20 нг/мл Wnt3a (R&D Systems; № по кат. 1324-WN/CF). На 2 день анализа 100 нг/мл активина A или 100 нг/мл GDF-8 разбавили в среде RPMI-1640 с добавлением 2% FAF BSA, без Wnt3a. В некоторых тестируемых образцах с GDF-8 Wnt3a заменяли низкомолекулярным соединением, которое добавляли только в 1 день дифференцирования в сформированную эндодерму. Такие низкомолекулярные соединения включали: Соединение 19 (концентрация для анализа 2,5 мкМ), Соединение 202 (концентрация для анализа 2,5 мкМ), Соединение 40 (концентрация для анализа 2,5 мкМ) или коммерческий ингибитор GSK3 BIO (концентрация для анализа 0,5 мкМ) (EMD Chemicals, Inc.; № по кат. 361550). По завершении первого этапа дифференцирования клетки из некоторых лунок собрали с целью определения методом проточной цитометрии уровня CXCR4, маркера образования сформированной эндодермы. Клетки из других лунок собрали для определения методом ОТ-ПЦР других маркеров дифференцирования.
После завершения первого этапа дифференцирования в сформированную эндодерму дублирующиеся наборы параллельных лунок каждой из экспериментальных групп подвергли дальнейшему поэтапному дифференцированию. Важно отметить, что после первого этапа дифференцирования все лунки с клетками, подвергаемыми дальнейшему культивированию и дифференцированию, получали одинаковую обработку. Ниже представлено описание протокола, использованного для продолжения дифференцирования.
Второй этап протокола дифференцирования занял два дня. Питание клеткам вводили ежедневно, для чего среду из всех лунок отсасывали и заменяли свежей аликвотой (0,5 мл) среды DMEM:F12 (Invitrogen; № по кат. 11330-032) с добавлением 2% FAF BSA, 50 нг/мл FGF7 (PeproTech; № по кат. 100-19) и 250 нМ циклопамина-KAAD (Calbiochem; № по кат. 239804).
Третий этап протокола дифференцирования занял семь дней. Питание клеткам вводили ежедневно, для чего среду из всех лунок отсасывали и заменяли свежей аликвотой (0,5 мл) среды DMEM с высоким содержанием глюкозы (Invitrogen; № по кат. 10569) с добавлением 0,1% Albumax (Invitrogen; № по кат. 11020-021), 0,5-кратной смеси инсулин-трансферрин-селен (ITS-X; Invitrogen; № по кат. 51500056), 50 нг/мл FGF7, 100 нг/мл Noggin (R&D Systems; № по кат. 3344-NG), 250 нМ KAAD-циклопамина и 2 мкМ полностью транс-ретиноевой кислоты (RA) (Sigma-Aldrich; № по кат. R2625). По завершении третьего этапа дифференцирования клетки из некоторых лунок собрали с целью определения методом ОТ-ПЦР маркеров дифференцирования. Другие лунки подвергли одновременному многопараметрическому анализу на предмет определения уровня экспрессии белка Pdx1 и Cdx2.
Четвертый этап протокола дифференцирования занял три дня. Питание клеткам вводили ежедневно, для чего среду из всех лунок отсасывали и заменяли свежей аликвотой (0,5 мл) среды DMEM с высоким содержанием глюкозы с добавлением 0,1% Albumax, 0,5-кратной смеси инсулин-трансферрин-селен, 100 нг/мл Noggin и 1 мкМ ингибитора Alk 5 (Axxora; № по кат. ALX-270-445). По завершении четвертого этапа дифференцирования клетки из некоторых лунок собрали с целью определения методом ОТ-ПЦР маркеров дифференцирования. Другие лунки подвергли одновременному многопараметрическому анализу на предмет определения уровня экспрессии белка Pdx1.
Пятый этап протокола дифференцирования выполнялся в течение семи дней в среде DMEM с высоким содержанием глюкозы с добавлением 0,1% Albumax, 0,5-кратной смеси инсулин-трансферрин-селен и 1 мкМ ингибитора Alk 5. Ежедневно среду из всех лунок отсасывали и заменяли свежей аликвотой (0,5 мл). По завершении пятого этапа дифференцирования клетки из некоторых лунок собрали с целью определения методом ОТ-ПЦР маркеров дифференцирования. Другие лунки подвергли одновременному многопараметрическому анализу на предмет определения уровня экспрессии инсулина и глюкагона.
Анализ методом FACS. Клетки для анализа методом FACS заблокировали 0,5% раствором человеческого гамма-глобулина (Sigma; № по кат. G-4386), разведенным в соотношении 1:5 в PBS (Invitrogen; № по кат. 14040-133) и буфере для окрашивания BD FACS - BSA (BD; № по кат. 554657), в течение 15 минут при температуре 4єC. Затем клетки окрасили антителами к CD9 PE (BD; № по кат. 555372), CD99 PE (Caltag; № по кат. MHCD9904) и CXCR4 APC (R&D Systems; № по кат. FAB173A) в течение 30 минут при 4єC. После серии промывок в буфере для окрашивания BD FACS клетки окрасили для оценки жизнеспособности красителем 7-AAD (BD; № по кат. 559925) и проанализировали на биоанализаторе BD FACSArray. Для получения процентной доли положительных клеток использовали мышиные изотипические контрольные антитела типа IgG1K к PE и APC.
Анализ методом ОТ-ПЦР. Образцы РНК очистили путем связывания с силикагелевой мембраной (Rneasy Mini Kit, Qiagen, Калифорния, США) в присутствии этанолсодержащего высокосолевого буферного раствора, а затем промыли для удаления примесей. Затем РНК очищали далее, используя набор TURBO DNA-free (Ambion, Inc.), элюировав в воду РНК высокого качества. Выход и чистоту полученной РНК оценивали по измеряемому на спектрофотометре поглощению на длинах волн 260 и 280 нм. Копии кДНК получили с очищенной РНК, используя набор высокой емкости для получения кДНК из архивного материала компании (ABI, Калифорния, США).
Если не указано иное, все реагенты были приобретены в Applied Biosystems. Реакции ПЦР в режиме реального времени проводили с использованием системы для определения последовательности ABI PRISM® 7900 Sequence Detection System. Использовали набор TAQMAN® UNIVERSAL PCR MASTER MIX® (ABI, Калифорния, США) с 20 нг обратно транскрибированной РНК в общем объеме реакционной смеси 20 мкл. Каждый образец кДНК анализировали дважды для коррекции погрешности пипетирования. Праймеры и меченные красителем FAM зонды TAQMAN использовали в концентрациях 200 нМ. Уровень экспрессии для каждого целевого гена нормировали по эндогенному контрольному образцу, глицеральдегид-3-фосфатдегидрогеназе человека (ГАФДГ), ранее разработанному Applied Biosystems. Праймеры и наборы зондов перечислены в Таблице 12. После начальной инкубации при температуре 50°C в течение 2 минут с последующей инкубацией при температуре 95°C в течение 10 минут с образцами провели 40 двухстадийных циклов, включающих стадию денатурации при температуре 95°C продолжительностью 15 секунд и следующую за ней стадию отжига/роста при температуре 60°C продолжительностью 1 минута. Анализ данных проводили с использованием программного пакета GENEAMP®7000 Sequence Detection System. Для каждого комплекта праймеров и зондов определяли значение порогового цикла (Ct) как номер цикла, при котором интенсивность флюоресценции достигала определенного значения в середине экспоненциальной области амплификации. Уровни относительной экспрессии генов вычисляли методом сравнения Ct. Коротко говоря, для каждого образца кДНК значение Ct эндогенного контрольного образца вычитали из значения Ct целевого гена и получали величину дельта Ct (ΔCt). Нормированное количество целевого гена рассчитывали как 2-ΔCt, принимая, что амплификация имеет 100% эффективность. Окончательные данные были выражены относительно калибровочного образца.
Одновременный многопараметрический анализ. По окончании периода культивирования аналитические планшеты один раз промыли раствором PBS (Invitrogen; № по кат. 14190), зафиксировали 4% параформальдегидом (Alexis Biochemical; № по кат. ALX-350-011) при комнатной температуре в течение 20 минут, затем трижды промыли раствором PBS и пермеабилизировали, обработав раствором 0,5% Triton X-100 (Sigma; № по кат. T8760-2) в течение 20 минут при комнатной температуре. Затем клетки снова трижды промыли раствором PBS и заблокировали раствором 4% куриной сыворотки (Invitrogen; № по кат. 16110082) в PBS в течение 30 минут при комнатной температуре. Первичные антитела (козий анти-человеческий SOX17; R&D Systems; № по кат. AF1924) разбавили в соотношении 1:100 в 4% куриной сыворотке и добавили в каждую лунку на два часа при комнатной температуре. Сопряженные с флюорофором Alexa Fluor 488 вторичные антитела (куриный анти-козий IgG; Invitrogen; № по кат. A21467) разбавили в соотношении 1:200 в растворе PBS и добавили в каждую лунку после трехкратной промывки клеток раствором PBS. Для подсчета окрашенных ядер на пятнадцать минут при комнатной температуре добавили 5 мкг/мл Hoechst 33342 (Invitrogen; № по кат. H3570). Затем планшеты один раз промыли раствором PBS и оставили в 100 мкл раствора PBS на лунку для томографии. Другие первичные антитела, использованные в анализе, включали: разведенные 1:200 кроличьи антитела к человеческому инсулину (Cell Signaling; № по кат. C27C9) и разведенные 1:1500 мышиные антитела к человеческому глюкагону (Sigma-Aldrich; № по кат. G2654). В анализе использовались следующие вторичные антитела: разведенные 1:1000 куриные антитела к кроличьему IgG Alexa Fluor 647 (Invitrogen; № по кат. A21443) и разведенные 1:1000 куриные антитела к мышиному IgG Alexa Fluor 488 (Invitrogen; № по кат. A21200).
Томографию проводили на анализаторе IN Cell Analyzer 1000 (GE Healthcare) с применением дихроичного зеркала 51008bs для клеток, окрашенных красителями Hoechst 33342 и Alexa Fluor 488. Для каждой лунки снимали по 25 проекций. Общую интенсивность сигнала измеряли для каждой лунки с помощью программного пакета IN Cell Developer Toolbox 1.7 (GE Healthcare). Сегментацию ядер определяли по ступеням шкалы серых тонов (исходный интервал 100-300) и размерам ядра. Для каждой повторной совокупности данных рассчитали средние значение и стандартные отклонения. Общий уровень экспрессии белка выражается в виде общей интенсивности или интегральной интенсивности, определяемой как произведение общей интенсивности флюоресценции клетки на площадь клетки. Фон исключили на основании критериев соответствия ступеням шкалы серых тонов в диапазоне от 200 до 4500. Показатели общей интенсивности нормировали путем деления общей интенсивности для каждой лунки на среднюю общую интенсивность для положительного контрольного образца.
Результаты ПЦР-анализа клеток, собранных на каждом из этапов дифференцирования, на характерные маркеры представлены в Таблице 14. В образцах, обработанных GDF-8 и Wnt3a или GDF-8 и низкомолекулярным соединением, наблюдался сходный уровень экспрессии маркеров, ассоциирующихся с дифференцированием в эндодерму и эндокринные клетки.
На фиг. 20, часть А, представлены результаты анализа методом FACS, демонстрирующие экспрессию маркера сформированной эндодермы, CXCR4, после первого этапа дифференцирования. Обработка эмбриональных стволовых клеток человека GDF-8 и Wnt3a привела к образованию процентной доли CXCR4-положительных клеток, сходной с наблюдаемой при обработке активином A и Wnt3a. Обработка эмбриональных стволовых клеток человека GDF-8 и соединением, составляющим предмет настоящего изобретения (Соединением 19, Соединением 202, Соединением 40 или GSK3-ингибитором IX BIO) также привела к образованию сходной или несколько большей процентной доли CXC4-положительных клеток. На фиг. 20, часть B, представлены результаты одновременного многопараметрического анализа на предмет нормированной экспрессии белка SOX17 в эмбриональных стволовых клетках человека после трех дней дифференцирования в сформированную эндодерму. В некоторых случаях при обработке GDF-8 число клеток после завершения первого этапа дифференцирования уменьшилось. Однако обработка GDF-8 в сочетании с Wnt3a или низкомолекулярными ингибиторами, очевидно, индуцировала экспрессию SOX17, маркера сформированной эндодермы. В одном случае при обработке GDF-8 и Соединением 40 число клеток в культуре и экспрессия SOX17 были эквивалентны наблюдаемым при обработке активином A и Wnt3a.
На фиг. 20, часть C, представлены результаты одновременного многопараметрического анализа на предмет относительного числа клеток, выделенных из культур, которые подвергались обработке вплоть до пятого этапа дифференцирования. Как и ранее, в конце первого этапа некоторые виды обработки привели к уменьшению числа клеток относительно обработки активином A и Wnt3a. Такое уменьшение числа клеток наблюдалось, в частности, в группах с обработкой GDF-8 с GSK3-ингибитором BIO, а также при обработке GDF-8 с Соединением 19. В других группах с GDF-8 число клеток было сходным с наблюдаемым при обработке активином A и Wnt3a. На фиг. 20, части D-F, показаны нормированные уровни белков инсулина и глюкагона, а также их соотношение в каждой из экспериментальных групп. Каждый из вариантов обработки GDF-8 позволял получить уровни инсулина и глюкагона, сходные с наблюдаемыми при обработке активином А и Wnt3a. Этот результат показывает, что GDF-8 в сочетании с Wnt3a или низкомолекулярным соединением может заменить активин А в ходе дифференцирования клеток в сформированную эндодерму и последующего дифференцирования в панкреатическую эндодерму и эндокринные клетки.
Пример 17
Клетки, экспрессирующие маркеры, характерные для линии сформированной эндодермы, полученные с использованием GDF-8 и соединения, составляющего предмет настоящего изобретения, способны далее дифференцироваться в клетки, экспрессирующие маркеры, характерные для линии панкреатических эндокринных клеток
При дифференцировании клеток в сформированную эндодерму были протестированы дополнительные низкомолекулярные соединения в сочетании с GDF-8 и активином А. К этим соединениям относился коммерческий ингибитор GSK3, а также соединения, составляющие предмет настоящего изобретения. К клеткам, обработанным GDF-8 в сочетании с различными низкомолекулярными соединениями, применялся протокол поэтапного дифференцирования. Эффективность дифференцирования определялась по экспрессии генов биомаркеров, характерных для линии панкреатической эндодермы и линии панкреатических эндокринных клеток. В течение всего периода поэтапного дифференцирования для сравнения присутствовал параллельный контрольный образец, состоящий из клеток, обработанных активином A и Wnt3a.
Подготовка клеток к анализу. Исходные культуры эмбриональных стволовых клеток человека (эмбриональные стволовые клетки человека линии H1) поддерживали в недифференцированном плюрипотентном состоянии на чашках, покрытых препаратом MATRIGELTM с пониженным содержанием факторов роста (BD Biosciences; № по кат. 356231), в кондиционированной среде MEF с добавлением 8 нг/мл bFGF (PeproTech Inc.; № по кат. 100-18B) и пассировали в среднем каждые четыре дня. При пассировании клетки обрабатывали раствором диспазы 1 мг/мл (Invitrogen; № по кат. 17105-041) в течение 5-7 минут при 37єC с последующей промывкой монослоя кондиционированной культуральной средой MEF, затем осторожно соскоблили для получения кластеров клеток. Кластеры центрифугировали на низкой скорости для получения осадка клеток и удаления остатков диспазы. Кластеры клеток разделили в соотношении 1:3 или 1:4 для стандартного поддерживающего культивирования или в соотношении 1:1 для немедленного анализа. Все линии эмбриональных стволовых клеток человека поддерживали при номерах пассажа менее 50 и периодически проверяли на нормальный кариотип и отсутствие микоплазмы.
Кластеры клеток равномерно ресуспендировали в кондиционированной среде MEF с добавлением 8 нг/мл bFGF и высеяли на покрытые препаратом MATRIGELTM с пониженным содержанием факторов роста 24-луночные культуральные планшеты с черными стенками (Arctic White; № по кат. AWLS-303012) в объеме 0,5 мл на лунку. Ежедневное питание осуществляли путем отсасывания отработанной культуральной среды из каждой лунки и ее замены тем же объемом свежей среды. В течение всего периода анализа планшеты держали при температуре 37єC в атмосфере 5% CO2.
Анализ. Анализ начали с отсасывания культуральной среды из всех лунок и добавления аликвот (0,5 мл) тестируемой среды. На первом этапе дифференцирования тесты проводили в течение трехдневного периода. Питание вводили ежедневно, для чего среду из каждой лунки отсасывали и заменяли свежей тестируемой средой. В 1 день анализа в соответствующие лунки добавили 100 нг/мл активина A (PeproTech; № по кат. 120-14) или 100 нг/мл GDF-8 (R&D Systems, № по кат. 788-G8), причем все факторы роста разбавили в среде RPMI-1640 (Invitrogen; № по кат. 22400) с добавлением 2% фракции V бычьего альбумина, не содержащего жирных кислот (FAF BSA) (MP Biomedicals, Inc; № по кат. 152401). В некоторые образцы также добавили 20 нг/мл Wnt3a (R&D Systems; № по кат. 1324-WN/CF). На 2 день анализа 100нг/мл активина A или 100 нг/мл GDF-8 разбавили в среде RPMI-1640 с добавлением 2% FAF BSA, без Wnt3a. В некоторых тестируемых образцах с GDF-8 Wnt3a заменяли низкомолекулярным соединением в заданной концентрации, которое добавляли только в 1 день дифференцирования в сформированную эндодерму. Такие низкомолекулярные соединения включали: Соединение 181 (концентрация для анализа 1,25 мкМ), Соединение 180 (концентрация для анализа 2,5 мкМ), Соединение 19 (концентрация для анализа 10 мкМ), Соединение 202 (концентрация для анализа 2,5 мкМ), Соединение 40 (концентрация для анализа 5 мкМ), Соединение 34 (концентрация для анализа 2,5 мкМ), Соединение 206 (концентрация для анализа 2,5 мкМ) и коммерческий ингибитор GSK3 IX BIO (концентрация для анализа 10 мкМ) (EMD Chemicals, Inc.; № по кат. 361550). По завершении первого этапа дифференцирования клетки из некоторых лунок собрали с целью определения методом проточной цитометрии уровня CXCR4, маркера образования сформированной эндодермы. Клетки из других лунок собрали для определения методом ОТ-ПЦР других маркеров дифференцирования.
После завершения первого этапа дифференцирования в сформированную эндодерму дублирующиеся наборы параллельных лунок каждой из экспериментальных групп подвергли дальнейшему поэтапному дифференцированию. Важно отметить, что после первого этапа дифференцирования все лунки с клетками, подвергаемыми дальнейшему культивированию и дифференцированию, получали одинаковую обработку. Ниже представлено описание протокола, использованного для продолжения дифференцирования.
Второй этап протокола дифференцирования занял два дня. Питание клеткам вводили ежедневно, для чего среду из всех лунок отсасывали и заменяли свежей аликвотой (0,5 мл) среды DMEM:F12 (Invitrogen; № по кат. 11330-032) с добавлением 2% FAF BSA, 50 нг/мл FGF7 (PeproTech; № по кат. 100-19) и 250 нМ циклопамина-KAAD (Calbiochem; № по кат. 239804).
Третий этап протокола дифференцирования занял четыре дня. Питание клеткам вводили ежедневно, для чего среду из всех лунок отсасывали и заменяли свежей аликвотой (0,5 мл) среды DMEM с высоким содержанием глюкозы (Invitrogen; № по кат. 10569) с добавлением 0,1% Albumax (Invitrogen; № по кат. 11020-021), 0,5-кратной смеси инсулин-трансферрин-селен (ITS-X; Invitrogen; № по кат. 51500056), 50 нг/мл FGF7, 100 нг/мл Noggin (R&D Systems; № по кат. 3344-NG), 250 нМ KAAD-циклопамина и 2 мкМ полностью транс-ретиноевой кислоты (RA) (Sigma-Aldrich; № по кат. R2625). По завершении третьего этапа дифференцирования клетки из некоторых лунок собрали с целью определения методом ОТ-ПЦР маркеров дифференцирования.
Четвертый этап протокола дифференцирования занял три дня. Питание клеткам вводили ежедневно, для чего среду из всех лунок отсасывали и заменяли свежей аликвотой (0,5 мл) среды DMEM с высоким содержанием глюкозы с добавлением 0,1% Albumax, 0,5-кратной смеси инсулин-трансферрин-селен, 100 нг/мл Noggin и 1 мкМ ингибитора Alk 5 (Axxora; № по кат. ALX-270-445). По завершении четвертого этапа дифференцирования клетки из некоторых лунок собрали с целью определения методом ОТ-ПЦР маркеров дифференцирования.
Пятый этап протокола дифференцирования выполнялся в течение семи дней в среде DMEM с высоким содержанием глюкозы с добавлением 0,1% Albumax, 0,5-кратной смеси инсулин-трансферрин-селен и 1 мкМ ингибитора Alk 5. Ежедневно среду из всех лунок отсасывали и заменяли свежей аликвотой (0,5 мл). По завершении пятого этапа дифференцирования клетки из некоторых лунок собрали с целью определения методом ОТ-ПЦР маркеров дифференцирования. Другие лунки подвергли одновременному многопараметрическому анализу на предмет определения уровня экспрессии инсулина и глюкагона.
Анализ методом FACS. Клетки для анализа методом FACS заблокировали 0,5% раствором человеческого гамма-глобулина (Sigma; № по кат. G-4386), разведенным в соотношении 1:5 в PBS (Invitrogen; № по кат. 14040-133) и буфере для окрашивания BD FACS-BSA (BD; № по кат. 554657), в течение 15 минут при температуре 4єC. Затем клетки окрасили антителами к CD9 PE (BD; № по кат. 555372), CD99 PE (Caltag; № по кат. MHCD9904) и CXCR4 APC (R&D Systems; № по кат. FAB173A) в течение 30 минут при 4єC. После серии промывок в буфере для окрашивания BD FACS клетки окрасили для оценки жизнеспособности красителем 7-AAD (BD; № по кат. 559925) и проанализировали на биоанализаторе BD FACSArray. Для получения процентной доли положительных клеток использовали мышиные изотипические контрольные антитела типа IgG1K к PE и APC.
Анализ методом ОТ-ПЦР. Образцы РНК очистили путем связывания с силикагелевой мембраной (Rneasy Mini Kit, Qiagen, Калифорния, США) в присутствии этанолсодержащего высокосолевого буферного раствора, а затем промыли для удаления примесей. Затем РНК очищали далее, используя набор TURBO DNA-free (Ambion, Inc.), элюировав в воду РНК высокого качества. Выход и чистоту полученной РНК оценивали по измеряемому на спектрофотометре поглощению на длинах волн 260 и 280 нм. Копии кДНК получили с очищенной РНК, используя набор высокой емкости для получения кДНК из архивного материала компании (ABI, Калифорния, США).
Если не указано иное, все реагенты были приобретены в Applied Biosystems. Реакции ПЦР в режиме реального времени проводили с использованием системы для определения последовательности ABI PRISM® 7900 Sequence Detection System. Использовали набор TAQMAN® UNIVERSAL PCR MASTER MIX® (ABI, Калифорния, США) с 20 нг обратно транскрибированной РНК в общем объеме реакционной смеси 20 мкл. Каждый образец кДНК анализировали дважды для коррекции погрешности пипетирования. Праймеры и меченные красителем FAM зонды TAQMAN использовали в концентрациях 200 нМ. Уровень экспрессии для каждого целевого гена нормировали по эндогенному контрольному образцу, глицеральдегид-3-фосфатдегидрогеназе человека (ГАФДГ), ранее разработанному Applied Biosystems. Праймеры и наборы зондов перечислены в Таблице 12. После начальной инкубации при 50°C в течение 2 мин с последующей инкубацией при 95°C в течение 10 мин с образцами провели 40 двухстадийных циклов, включающих стадию денатурации при температуре 95°C продолжительностью 15 секунд и следующую за ней стадию отжига/роста при температуре 60°C продолжительностью 1 минута. Анализ данных проводили с использованием программного пакета GENEAMP®7000 Sequence Detection System. Для каждого комплекта праймеров и зондов определяли значение порогового цикла (Ct) как номер цикла, при котором интенсивность флюоресценции достигала определенного значения в середине экспоненциальной области амплификации. Уровни относительной экспрессии генов вычисляли методом сравнения Ct. Коротко говоря, для каждого образца кДНК значение Ct эндогенного контрольного образца вычитали из значения Ct целевого гена и получали величину дельта Ct (ΔCt). Нормированное количество целевого гена рассчитывали как 2-ΔCt, принимая, что амплификация имеет 100% эффективность. Окончательные данные были выражены относительно калибровочного образца.
Одновременный многопараметрический анализ. По окончании периода культивирования аналитические планшеты один раз промыли раствором PBS (Invitrogen; № по кат. 14190), зафиксировали 4% параформальдегидом (Alexis Biochemical; № по кат. ALX-350-011) при комнатной температуре в течение 20 минут, затем трижды промыли раствором PBS и пермеабилизировали, обработав раствором 0,5% Triton X-100 (Sigma; № по кат. T8760-2) в течение 20 минут при комнатной температуре. Затем клетки снова трижды промыли раствором PBS и заблокировали раствором 4% куриной сыворотки (Invitrogen; № по кат. 16110082) в PBS в течение 30 минут при комнатной температуре. Первичные антитела (козий анти-человеческий SOX17; R&D Systems; № по кат. AF1924) разбавили в соотношении 1:100 в 4% куриной сыворотке и добавили в каждую лунку на два часа при комнатной температуре. Сопряженные с флюорофором Alexa Fluor 488 вторичные антитела (куриный анти-козий IgG; Invitrogen; № по кат. A21467) разбавили в соотношении 1:200 в растворе PBS и добавили в каждую лунку после трехкратной промывки клеток раствором PBS. Для подсчета окрашенных ядер на пятнадцать минут при комнатной температуре добавили 5 мкг/мл Hoechst 33342 (Invitrogen; № по кат. H3570). Затем планшеты один раз промыли раствором PBS и оставили в 100 мкл раствора PBS на лунку для томографии. Другие первичные антитела, использованные в анализе, включали: разведенные 1:100 мышиные антитела к человеческому CDX2 (Invitrogen; № по кат. 397800), разведенные 1:100 козьи антитела к человеческому Pdx1 (Santa Cruz Biotechnology; № по кат. SC-14664), разведенные 1:200 кроличьи антитела к человеческому инсулину (Cell Signaling; № по кат. C27C9) и разведенные 1:1500 мышиные антитела к человеческому глюкагону (Sigma-Aldrich; № по кат. G2654). В анализе использовались следующие вторичные антитела: разведенные 1:400 куриные антитела к мышиному IgG Alexa Fluor 647 (Invitrogen; № по кат. A-21463), разведенные 1:200 ослиные антитела к козьему IgG Alexa Fluor 488 (Invitrogen; № по кат. A11055), разведенные 1:1000 куриные антитела к кроличьему IgG Alexa Fluor 647 (Invitrogen; № по кат. A21443) и разведенные 1:1000 куриные антитела к мышиному IgG Alexa Fluor 488 (Invitrogen; № по кат. A21200).
Томографию проводили на анализаторе IN Cell Analyzer 1000 (GE Healthcare) с применением дихроичного зеркала 51008bs для клеток, окрашенных красителями Hoechst 33342 и Alexa Fluor 488. Для каждой лунки снимали по 25 проекций. Общую интенсивность сигнала измеряли для каждой лунки с помощью программного пакета IN Cell Developer Toolbox 1.7 (GE Healthcare). Сегментацию ядер определяли по ступеням шкалы серых тонов (исходный интервал 100-300) и размерам ядра. Для каждой повторной совокупности данных рассчитали средние значение и стандартные отклонения. Общий уровень экспрессии белка выражается в виде общей интенсивности или интегральной интенсивности, определяемой как произведение общей интенсивности флюоресценции клетки на площадь клетки. Фон исключили на основании критериев соответствия ступеням шкалы серых тонов в диапазоне от 200 до 4500. Показатели общей интенсивности нормировали путем деления общей интенсивности для каждой лунки на среднюю общую интенсивность для положительного контрольного образца.
Результаты. Результаты анализа экспрессии характерных маркеров в клетках, собранных на каждом из этапов дифференцирования, представлены на фиг. 21 и в Таблице 15. На фиг. 21A и B представлены полученные методом проточной цитометрии данные об экспрессии CXCR4 на первом этапе дифференцирования клеток в сформированную эндодерму при различных способах обработки. На фиг. 21A показан эффект обработки различными соединениями в сочетании с активином А на экспрессию CXCR4. На фиг. 21B показан эффект обработки различными соединениями в сочетании с GDF-8 на экспрессию CXCR4. Соединения, составляющие предмет настоящего изобретения, в сочетании с активином А не усиливали экспрессию CXCR4. Однако все протестированные в данном примере соединения, составляющие предмет настоящего изобретения, усиливали экспрессию CXCR4 в сочетании с GDF-8.
На фиг. 21C и 21D представлены полученные методом ОТ-ПЦР нормированные данные по различным маркерам дифференцирования для использованных на первом этапе протокола видов обработки, в которых применялись некоторые соединения, составляющие предмет настоящего изобретения, в сочетании с активином А (фиг. 21C) или в сочетании с GDF-8 (фиг. 21D). Аналогичные полученные методом ОТ-ПЦР нормированные данные анализировали на третьем этапе протокола дифференцирования (фиг. 21E и 21F), в конце четвертого этапа протокола дифференцирования (фиг. 21G и 21H) и в конце пятого этапа протокола дифференцирования (фиг. 21I и 21J). Применяемые на первом этапе дифференцирования виды обработки, в которых соединение, составляющее предмет настоящего изобретения, сочеталось с GDF-8, улучшали экспрессию различных маркеров эндодермальных и панкреатических клеток в сравнении с обработкой только GDF-8 (фиг. 21F, 21H и 21J). Те виды обработки, в которых соединения, составляющие предмет настоящего изобретения, сочетались с активином А, незначительно увеличивали или не увеличивали вовсе экспрессию маркеров в сравнении с обработкой одним активином А или активином А в сочетании с Wnt3a (фиг. 21E, 21G и 21I). В Таблице 15 приводятся сравнительные значения CT по дополнительным генным маркерам в конце каждого этапа дифференцирования, где сравниваются варианты обработки на первом этапе дифференцирования, в которых активин А или GDF-8 сочетались или не сочетались с соединениями, составляющими предмет настоящего изобретения. В конце пятого этапа дифференцирования был проведен одновременный многопараметрический анализ для измерения числа клеток (фиг. 21K и 21M) и экспрессии белков инсулина и глюкагона (фиг. 21L и 21N). Обработка GDF-8 на первом этапе дифференцирования, как отдельная, так и в сочетании с соединением, составляющим предмет настоящего изобретения, вызывала экспрессию инсулина и глюкагона в конце пятого этапа дифференцирования. Это показывает, что GDF-8 способен заменить активин A на начальном этапе дифференцирования в сформированную эндодерму и в дальнейшем привести к образованию панкреатических гормональных клеток. В совокупности эти данные демонстрируют, что добавление любого из соответствующих низкомолекулярных соединений в сочетании с активином А минимально влияет на маркеры дифференцирования. Однако добавление низкомолекулярного соединения в сочетании с GDF-8 оказывает существенно более сильное влияние на немедленное дифференцирование клеток в сформированную эндодерму в конце первого этапа дифференцирования, а также на маркеры дальнейшего дифференцирования в конце третьего, четвертого и пятого этапов. В пределах набора низкомолекулярных соединений наблюдалась вариабельность, вероятно связанная с концентрацией соединения, использованного в анализе, и (или) с механизмом действия.
Пример 18
Клетки, экспрессирующие маркеры, характерные для линии сформированной эндодермы, полученные с использованием GDF-8 и соединения, составляющего предмет настоящего изобретения, способны выделять С-пептид после трансплантации грызунам
Было важно установить, способны ли клетки, экспрессирующие маркеры, характерные для линии панкреатической эндодермы, полученные in vitro путем обработки GDF-8 и низкомолекулярным соединением, образовывать in vivo функциональные эндокринные клетки. Изучение трансплантации in vivo было проведено с целью сравнения клеток, дифференцированных путем обработки активином A и Wnt3a, с клетками, дифференцированными путем обработки GDF-8 и низкомолекулярными соединениями.
Подготовка клеток. Кластеры эмбриональных стволовых клеток человека линии H1 выращивали на покрытых препаратом MATRIGELTM с пониженным содержанием факторов роста (Invitrogen; № по кат. 356231) носителях для культивирования тканей и пассировали в среднем каждые четыре дня. Для первоначального посева и размножения использовали кондиционированную среду MEF с добавлением 8 нг/мл bFGF. Все линии эмбриональных стволовых клеток человека поддерживали при номерах пассажа менее 50 и периодически проверяли на нормальный кариотип и отсутствие примесей микоплазмы.
При пассировании клетки обрабатывали раствором диспазы 1 мг/мл (Invitrogen; № по кат. 17105-041) в течение 5-7 минут при 37єC с последующей промывкой монослоя клеток кондиционированной средой MEF, а затем осторожно соскоблили для получения кластеров клеток. Кластеры клеток центрифугировали на низкой скорости в кондиционированной среде MEF для удаления остатков диспазы, а затем равномерно ресуспендировали в кондиционированной среде MEF с добавлением 8 нг/мл bFGF (PeproTech Inc.; № по кат. 100-18B) и высеяли на покрытые препаратом MATRIGEL с пониженным содержанием факторов роста (BD Biosciences; № по кат. 356231) 6-луночные планшеты (Nunc; № по кат. 140685) в соотношении 1:3 в объеме 2,5 мл на лунку. Ежедневное питание осуществляли путем отсасывания отработанной культуральной среды из каждой лунки и ее замены тем же объемом свежей среды. В течение всего периода культивирования планшеты держали при температуре 37єC в атмосфере 5% CO2.
Дифференцирование клеток. Процесс дифференцирования запустили через три дня после посева клеток на 6-луночные планшеты, покрытые препаратом MATRIGELTM с пониженным содержанием факторов роста. Для дифференцирования in vitro эмбриональных стволовых клеток человека линии H1 в клетки, экспрессирующие маркеры, характерные для линии панкреатической эндодермы, использовали протокол, включающий четыре этапа. На первом этапе, который занял три дня, образовались клетки сформированной эндодермы. В 1 день первого этапа дифференцирование инициировали путем отсасывания отработанной культуральной среды и добавления аналогичного объема основной среды RPMI-1640 (Invitrogen; № по кат. 22400) с добавлением 2% фракции V бычьего альбумина, не содержащего жирных кислот (FAF BSA) (Proliant Biologicals; № по кат. SKU 68700), и 8 нг/мл bFGF. Клетки первой экспериментальной группы обработали 100 нг/мл активина А (PeproTech; № по кат. 120-14) и 20 нг/мл Wnt3a (R&D Systems; № по кат. 1324-WN/CF). Клетки второй экспериментальной группы обработали 100 нг/мл GDF-8 (R&D Systems; № по кат. 788-G8) и 2,5 мкМ Соединения 40. Клетки третьей экспериментальной группы обработали 100 нг/мл GDF-8 (R&D Systems; № по кат. 788-G8) и 2,5 мкМ Соединения 202. На 2 и 3 день первого этапа дифференцирования к клеткам всех экспериментальных групп добавили среду RPMI-1640, содержащую 2% FAF BSA, 8 нг/мл bFGF и либо 100 нг/мл активина A (экспериментальная группа 1), либо 100 нг/мл GDF-8 (экспериментальные группы 2 и 3), без добавления Wnt3a или соединения, составляющего предмет настоящего изобретения. В конце третьего дня культивирования из каждой экспериментальной группы отобрали одну лунку для анализа методом FASC.
Второй этап протокола дифференцирования занял три дня. К клеткам всех экспериментальных групп добавили среду DMEM:F12 (Invitrogen; № по кат. 11330-032) с добавлением 2% FAF BSA и 50 нг/мл FGF7 (PeproTech; № по кат. 100-19).
Третий этап протокола дифференцирования занял четыре дня. К клеткам всех экспериментальных групп ежедневно добавляли среду DMEM с высоким содержанием глюкозы (Invitrogen; № по кат. 10569) с добавлением 1% B27 (Invitrogen; № по кат. 17504-044), 50 нг/мл FGF7, 100 нг/мл Noggin (R&D Systems; № по кат. 3344-NG), 250 нМ KAAD-циклопамина (Calbiochem; № по кат. 239804) и 2 мкМ полностью транс-ретиноевой кислоты (RA) (Sigma-Aldrich; № по кат. R2625).
Четвертый этап протокола дифференцирования занял три дня. К клеткам всех экспериментальных групп в течение первых двух дней ежедневно добавляли среду DMEM с высоким содержанием глюкозы с добавлением 1% B27, 100 нг/мл Noggin и 1 мкМ ингибитора ALK5 (Axxora; № по кат. ALX-270-445). На третий день клетки отделили от субстрата с помощью наконечника на 20 мкл (Rainin; № по кат. RT-L10F) и клеточного скребка (Corning; № по кат. 3008) и перенесли в пробирку на 50 мл. Затем клеткам дали возможность осесть под собственным весом, и удалили супернатант, не затрагивая осадок клеток. Клетки ресуспендировали в среде DMEM с высоким содержанием глюкозы с добавлением 1% B27, 100 нг/мл Noggin и 1 мкМ ингибитора ALK5, а затем культивировали в течение ночи в 6-луночных планшетах со сверхнизкой связывающей способностью Costar (Corning Inc., № по кат. 3471). На следующий день клетки отобрали и подсчитали их число во взвешенной культуре. Для трансплантации клеток использовали аликвоты 10Ч106 клеток на мышь. Для анализа методом ОТ-ПЦР отобрали аликвоты 0,5Ч106 клеток.
На фиг. 22, часть A, представлены результаты анализа методом проточной цитометрии клеток сформированной эндодермы, образовавшихся в конце первого этапа в каждой из экспериментальных групп. При обработке активином A и Wnt3a или при обработке GDF-8 и соединением, составляющим предмет настоящего изобретения, в конце первого этапа уровень экспрессии CXCR4 в клетках оказался сходным (более 85%). Можно предположить, что в каждой их экспериментальных групп образовались сходные популяции клеток сформированной эндодермы.
На фиг. 22, часть B, представлены результаты анализа клеток экспериментальных групп методом ОТ-ПЦР в конце четвертого этапа протокола дифференцирования. Клетки, дифференцированные в панкреатическую эндодерму (PE) с помощью активина A и Wnt3a, или с помощью GDF-8 и Соединения 40, или с помощью GDF-8 и Соединения 202, имели эквивалентные уровни экспрессии маркеров, характерных для линии панкреатической эндодермы: CDX2, MAFA, NGN3, NKX6.1, PDX1 and Ptf1 alpha. Эти результаты дают основание предполагать, что протокол дифференцирования с использованием GDF-8 и низкомолекулярного соединения был столь же эффективен для получения популяции клеток-предшественников панкреатический эндодермы.
Трансплантация мышам эмбриональных стволовых клеток человека, обработанных в соответствии со способами, составляющими предмет настоящего изобретения. Мыши с тяжелым комбинированным иммунодефицитом, вызванным врожденным отсутствием клеток-киллеров (C.B-Igh-1b/GbmsTac-Prkdc scid -Lyst bg N7), возрастом от пяти до шести недель были приобретены в Taconic Farms. Мышей содержали в клетках-микроизоляторах со свободным доступом к стерилизованной пище и воде. В ходе подготовки к хирургической операции мыши были помечены идентификационной меткой, нанесенной на ухо, была измерена масса их тела, а также содержание глюкозы в крови с помощью ручного глюкометра (LifeScan; One Touch). Для анестезии использовали смесь изофлурана и кислорода. Шерсть на месте операции выстригли малыми ножницами, предназначенными для животных. Перед операцией мышам подкожно ввели 0,1 мг/кг Buprenex. Для подготовки участок проведения операции обработали 70% изопропиловым спиртом, 10% повидон-йодом и 70% изопропиловым спиртом, затем с левой стороны произвели боковой разрез кожи и мышечного слоя. Открыли левую почку, которую поддерживали во влажном состоянии путем смачивания 0,9% раствором хлорида натрия. Для проникновения через почечную капсулу использовали катетер для внутривенных введений 24GЧѕ дюйма, после чего иглу удалили. Затем катетер продвинули под почечной капсулой к дистальному концу почки. В ходе предоперационной подготовки мышей клетки, предназначенные для трансплантации, отцентрифугировали в 1,5 мл микроцентрифужной пробирке, большую часть супернатанта удалили, оставив достаточное количество среды для отбора осажденных клеток. Клетки отобрали с помощью наконечника с объемным вытеснением Rainin Pos-D для пипеток, после чего пипетку перевернули, чтобы клетки могли осесть под собственным весом. Излишки среды выдавили, получив препарат уплотненных клеток для трансплантации. При трансплантации наконечник Pos-D плотно вставили во втулку катетера, и выдавили клетки из пипетки через катетер, введя их под почечной капсулой в дистальную часть почки. Просвет катетера промыли небольшим объемом культуральной среды для введения оставшихся клеток, после чего катетер извлекли. Почечную капсулу загерметизировали, используя низкотемпературное прижигание, после чего почку вернули в ее первоначальное анатомическое положение. Мышцы сшили непрерывным швом с помощью викриловой нити 5-0, а кожу стянули скобами для ран. Мышей вывели из наркоза и дали возможность полностью восстановиться. После операции мышам подкожно ввели 1,0 мг/кг Metacam.
После трансплантации мышей взвешивали раз в неделю, и дважды в неделю брали анализ крови для определения уровня глюкозы. На разных сроках после трансплантации мышам внутривенно ввели 3 г/кг глюкозы. Через 60 минут после введения глюкозы через ретроорбитальный синус отобрали кровь, поместив ее в микроцентрифужные пробирки, содержащие небольшое количество гепарина. Кровь центрифугировали, плазму собрали во вторую микроцентрифужную пробирку, заморозили на сухом льду и хранили при -80єC до проведения анализа на человеческий С-пептид. Концентрацию человеческого C-пептида определяли с помощью диагностического сверхчувствительного набора для твердофазного иммуноферментного анализа (ELISA) Mercodia/ALPCO C-peptide ELISA в соответствии с инструкциями изготовителя.
На фиг. 23 представлены полученные методом ELISA результаты определения человеческого С-пептида у мышей, которым были трансплантированы клетки из каждой из экспериментальных групп. Через четыре недели после трансплантации человеческий С-пептид не обнаруживался в крови мышей, получавших клетки из любых экспериментальных групп. Через 8 недель после трансплантации определимый уровень С-пептида был обнаружен у одной из двух мышей, получавших клетки, обработанные активином A и Wnt3a; у одной из трех мышей, получавших клетки, обработанные GDF-8 и Соединением 40; и у двух из трех мышей, получавших клетки, обработанные GDF-8 и Соединением 202. Эти результаты дают основание предполагать, что при использовании протокола дифференцирования с GDF-8 и низкомолекулярным соединением можно получать равноценную популяцию клеток-предшественников эндокринных клеток и что эти клетки в дальнейшем созревают in vivo в реагирующие на глюкозу инсулин-секретирующие клетки.
Пример 19
Клетки, экспрессирующие маркеры, характерные для линии сформированной эндодермы, полученные с использованием GDF-8, способны выделять С-пептид после трансплантации грызунам
Было важно продемонстрировать, что клетки, дифференцированные с помощью GDF-8 в отсутствие активина А, также могут далее дифференцироваться в популяцию эндокринных клеток, способных секретировать человеческий С-пептид на модели трансплантации in vivo грызунам.
Подготовка клеток. Кластеры эмбриональных стволовых клеток человека линии H1 выращивали на покрытых препаратом MATRIGELTM с пониженным содержанием факторов роста (Invitrogen; № по кат. 356231) носителях для культивирования тканей и пассировали в среднем каждые четыре дня. Для первоначального посева и размножения использовали кондиционированную среду MEF с добавлением 8 нг/мл bFGF. Все линии эмбриональных стволовых клеток человека поддерживали при номерах пассажа менее 50 и периодически проверяли на нормальный кариотип и отсутствие примесей микоплазмы.
При пассировании клетки обрабатывали раствором диспазы 1 мг/мл (Invitrogen; № по кат. 17105-041) в течение 5-7 минут при 37єC с последующей промывкой монослоя клеток кондиционированной средой MEF, а затем осторожно соскоблили для получения кластеров клеток. Кластеры клеток центрифугировали на низкой скорости в кондиционированной среде MEF для удаления остатков диспазы, а затем равномерно ресуспендировали в кондиционированной среде MEF с добавлением 8 нг/мл bFGF (PeproTech Inc.; № по кат. 100-18B) и высеяли на покрытые препаратом MATRIGELTM с пониженным содержанием факторов роста (BD Biosciences; № по кат. 356231) 6-луночные планшеты (Nunc; № по кат. 140685) в соотношении 1:3 в объеме 2,5 мл на лунку. Ежедневное питание осуществляли путем отсасывания отработанной культуральной среды из каждой лунки и ее замены тем же объемом свежей среды. В течение всего периода культивирования планшеты содержали при температуре 37єC в атмосфере 5% CO2.
Дифференцирование клеток. Процесс дифференцирования запустили через три дня после посева клеток на 6-луночные планшеты. Для дифференцирования in vitro эмбриональных стволовых клеток человека в клетки, экспрессирующие маркеры, характерные для линии панкреатической эндодермы, использовали протокол, включающий четыре этапа. На первом этапе, который занял три дня, образовались клетки сформированной эндодермы. В 1 день первого этапа дифференцирование инициировали путем отсасывания отработанной культуральной среды и добавления аналогичного объема базальной среды RPMI-1640 (Invitrogen; № по кат. 22400) с добавлением 2% фракции V бычьего альбумина, не содержащего жирных кислот (FAF BSA) (Proliant Biologicals; № по кат. SKU 68700), и 8 нг/мл bFGF. Параллельные наборы клеток первой экспериментальной группы обработали 100 нг/мл GDF-8 (R&D Systems; № по кат. 788-G8) и 20 нг/мл Wnt3a (R&D Systems; № по кат. 1324-WN/CF). Параллельные наборы клеток второй экспериментальной группы обработали 100 нг/мл GDF-8 и 2,5 мкМ Соединения 40. На 2 и 3 день первого этапа дифференцирования к клеткам всех экспериментальных групп добавили среду RPMI-1640, содержащую 2% FAF BSA, 8 нг/мл bFGF и 100 нг/мл GDF-8, без добавления Wnt3a или Соединения 40. В конце 3 дня культивирования из каждой экспериментальной группы отобрали одну лунку для анализа методом FASC.
Второй этап протокола дифференцирования занял три дня. К клеткам всех экспериментальных групп добавили среду DMEM:F12 (Invitrogen; № по кат. 11330-032) с добавлением 2% FAF BSA и 50 нг/мл FGF7 (PeproTech; № по кат. 100-19).
Третий этап протокола дифференцирования занял четыре дня. К клеткам всех экспериментальных групп ежедневно добавляли среду DMEM с высоким содержанием глюкозы (Invitrogen; № по кат. 10569) с добавлением 1% B27 (Invitrogen; № по кат. 17504-044), 50 нг/мл FGF7, 100 нг/мл Noggin (R&D Systems; № по кат. 3344-NG), 250 нМ KAAD-циклопамина (Calbiochem; № по кат. 239804) и 2 мкМ полностью транс-ретиноевой кислоты (RA) (Sigma-Aldrich; № по кат. R2625).
Четвертый этап протокола дифференцирования занял три дня. К клеткам всех экспериментальных групп ежедневно добавляли среду DMEM с высоким содержанием глюкозы с добавлением 1% B27, 100 нг/мл Noggin, 1 мкМ ингибитора ALK5 (Axxora; № по кат. ALX-270-445), а также 100 нг/мл GDF-8 (R&D Systems; № по кат. 788-G8) в течение первых двух дней. На 3 день четвертого этапа клетки собрали из 6-луночных планшета с помощью наконечника на 20 мкл (Rainin; № по кат. RT-L10F) и клеточного скребка (Corning; № по кат. 3008) и перенесли в пробирку на 50 мл. Затем клеткам дали возможность осесть под собственным весом, и удалили супернатант, не затрагивая осадок клеток. Клетки ресуспендировали в среде DMEM с высоким содержанием глюкозы с добавлением 1% B27, 100 нг/мл Noggin и 1 мкМ ингибитора ALK5, а затем культивировали в течение ночи в 6-луночных планшетах со сверхнизкой связывающей способностью Costar (Corning Inc., № по кат. 3471). На следующий день клетки отобрали и подсчитали их число во взвешенной культуре. Для трансплантации клеток использовали аликвоты 10Ч106 клеток на мышь. Для анализа методом ОТ-ПЦР отобрали аликвоты 0,5Ч106 клеток.
На фиг. 24А представлены результаты анализа методом проточной цитометрии клеток сформированной эндодермы, образовавшихся в конце первого этапа в каждой из экспериментальных групп. При обработке GDF-8 и Wnt3a или при обработке GDF-8 и Соединением 40 в конце первого этапа уровень экспрессии CXCR4 в клетках оказался сходным. Можно предположить, что в каждой из экспериментальных групп образовались сходные и жизнеспособные популяции клеток сформированной эндодермы. Результаты дублирующихся обработок хорошо согласовывались. На фиг. 24B представлены результаты анализа методом ОТ-ПЦР клеток перед трансплантацией в конце четвертого этапа протокола дифференцирования. Клетки, дифференцированные в панкреатическую эндодерму (PE) с помощью GDF-8 и Wnt3a или с помощью GDF-8 и Соединения 40, имели эквивалентные уровни экспрессии маркеров, характерных для линии панкреатической эндодермы: CDX2, MafA, Ngn3, NKX6.1, Pdx-1 и Ptf1A. Эти результаты показывают, что протокол дифференцирования с использованием GDF-8 и Wnt3a или GDF-8 и соединения, составляющего предмет настоящего изобретения, был эффективен для получения популяции клеток-предшественников панкреатический эндодермы. Протокол дифференцирования был применен к двум независимым, но идентичным экспериментальным группам. Полученные методом ОТ-ПЦР результаты для дублирующихся экспериментальных групп хорошо согласовывались.
Трансплантация мышам эмбриональных стволовых клеток человека. Мыши с тяжелым комбинированным иммунодефицитом, вызванным врожденным отсутствием клеток-киллеров (C.B-Igh-1b/GbmsTac-Prkdc scid -Lyst bg N7), возрастом от пяти до шести недель были приобретены в Taconic Farms. Мышей содержали в клетках-микроизоляторах со свободным доступом к стерилизованной пище и воде. В ходе подготовки к хирургической операции мыши были помечены идентификационной меткой, нанесенной на ухо, была измерена масса их тела, а также содержание глюкозы в крови с помощью ручного глюкометра (LifeScan; One Touch). Для анестезии использовали смесь изофлурана и кислорода. Шерсть на месте операции выстригли малыми ножницами, предназначенными для животных. Перед операцией мышам подкожно ввели 0,1 мг/кг Buprenex. Для подготовки участок проведения операции обработали 70% изопропиловым спиртом, 10% повидон-йодом и 70% изопропиловым спиртом, затем с левой стороны произвели боковой разрез кожи и мышечного слоя. Открыли левую почку, которую поддерживали во влажном состоянии путем смачивания 0,9% раствором хлорида натрия. Для проникновения через почечную капсулу использовали катетер для внутривенных введений 24GЧѕ дюйма, после чего иглу удалили. Затем катетер продвинули под почечной капсулой к дистальному концу почки. В ходе предоперационной подготовки мышей клетки, предназначенные для трансплантации, отцентрифугировали в 1,5 мл микроцентрифужной пробирке, большую часть супернатанта удалили, оставив достаточное количество среды для отбора осажденных клеток. Клетки отобрали с помощью наконечника с объемным вытеснением Rainin Pos-D для пипеток, после чего пипетку перевернули, чтобы клетки могли осесть под собственным весом. Излишки среды выдавили, получив препарат уплотненных клеток для трансплантации. При трансплантации наконечник Pos-D плотно вставили во втулку катетера, и выдавили клетки из пипетки через катетер, введя их под почечной капсулой в дистальную часть почки. Просвет катетера промыли небольшим объемом культуральной среды для введения оставшихся клеток, после чего катетер извлекли. Почечную капсулу загерметизировали, используя низкотемпературное прижигание, после чего почку вернули в ее первоначальное анатомическое положение. Мышцы сшили непрерывным швом с помощью викриловой нити 5-0, а кожу стянули скобами для ран. Мышей вывели из наркоза и дали возможность полностью восстановиться. После операции мышам подкожно ввели 1,0 мг/кг Metacam.
После трансплантации мышей взвешивали раз в неделю, и дважды в неделю брали анализ крови для определения уровня глюкозы. На разных сроках после трансплантации мышам внутривенно ввели 3 г/кг глюкозы. Через 60 минут после введения глюкозы через ретроорбитальный синус отобрали кровь, поместив ее в микроцентрифужные пробирки, содержащие небольшое количество гепарина. Кровь центрифугировали, плазму собрали во вторую микроцентрифужную пробирку, заморозили на сухом льду и хранили при -80єC до проведения анализа на человеческий С-пептид. Концентрацию человеческого C-пептида определяли с помощью диагностического сверхчувствительного набора для твердофазного иммуноферментного анализа (ELISA) Mercodia/ALPCO C-peptide ELISA в соответствии с инструкциями изготовителя. На фиг. 29C и D представлены полученные методом ELISA результаты определения человеческого С-пептида у мышей, которым были трансплантированы клетки из каждой из экспериментальных групп. При всех видах обработки через 8 недель после трансплантации определялись сходные уровни человеческого С-пептида, что указывает на то, что при использовании протоколов дифференцирования с применением сочетания GDF-8 и Wnt3a или GDF-8 и соединения, составляющего предмет настоящего изобретения, можно получать эквивалентные популяции клеток-предшественников эндокринных клеток.
Пример 20
Оценка способности ингибиторов CDK, GSK3 и TRK дифференцировать эмбриональные стволовые клетки человека в клетки, экспрессирующие маркеры, характерные для линии сформированной эндодермы
Была проведена оценка 14 патентованных низкомолекулярных соединений, известных своей специфичностью к сигнальным путям CDK, GSK3 и (или) TRK, на предмет их способности дифференцировать эмбриональные стволовые клетки человека в клетки, экспрессирующие маркеры, характерные для линии сформированной эндодермы.
Посев клеток для анализа. Коротко говоря, кластеры эмбриональных стволовых клеток человека линии H1 выращивали на покрытых препаратом MATRIGELTM с пониженным содержанием факторов роста (Invitrogen; № по кат. 356231) носителях для культивирования тканей. Клетки пассировали с использованием коллагеназы (Invitrogen; №356231 по кат. 17104-019), затем осторожно соскоблили, промыли для удаления остатков фермента и высеяли в соотношении 1:1 (по площади поверхности) на покрытые препаратом MATRIGELTM с пониженным содержанием факторов роста (BD Biosciences; № по кат. 356231) 96-луночные черные планшеты (Packard ViewPlates; PerkinElmer; № по кат. 6005182) в объеме 100 мкл на лунку. Клеткам дали возможность прикрепиться, после чего в течение 1-3 дней воспроизводили логарифмическую фазу роста, во время которой клетки ежедневно питали, используя кондиционированную среду MEF с добавлением 8 нг/мл bFGF (R&D Systems; № по кат. 233-FB). В течение всего периода анализа планшеты держали при температуре 37єC в атмосфере 5% CO2 в увлажняемом боксе.
Подготовка соединений и анализ. Скрининговый анализ проводился с использованием соединений, описанных в Таблице 16. Кроме того, в качестве положительного контрольного образца использовалось Соединение 34, описанное в предыдущих примерах. Соединения предоставили в виде исходных растворов концентрации 5 мМ на 96-луночных планшетах, растворили в 100% DMSO (Sigma; № по кат. D2650) и хранили при температуре -80єC. Затем соединения из такой библиотеки разбавили до промежуточной концентрации 0,2 мМ в 50 мМ HEPES (Invitrogen; № по кат. 15630-080), 20% DMSO и хранили при температуре 4єC. Каждый тест выполнялся в трех повторностях. В течение всего четырехдневного периода анализа питание вводили через день. Анализ начали с отсасывания культуральной среды из каждой лунки с последующей трехкратной промывкой лунки раствором PBS (Invitrogen; № по кат. 14190) для удаления остатков факторов роста. В 1 день проведения анализа в лунки добавили тестируемые образцы объемом 200 мкл, содержащие основную среду DMEM:F12 (Invitrogen; № по кат. 11330-032) с добавлением 0,5% FCS (HyClone; № по кат. SH30070.03), 100 нг/мл GDF-8 (R&D Systems № по кат. 788-G8) и 2,5 мкМ соединения. Параллельный набор тестируемых образцов подвергался аналогичной обработке, за исключением того, что в среде отсутствовал GDF-8. В 3 день анализа в лунки добавили тестируемые образцы объемом 100 мкл, содержащие основную среду DMEM:F12 с добавлением 2% FCS и 100 нг/мл GDF-8 (R&D Systems, № по кат. 788-G8). GDF-8 не добавлялся в те образцы, которые не подвергались обработке GDF-8 в 1 день анализа. Положительные контрольные образцы в течение четырех дней анализа содержали ту же основную среду с добавлением FCS и 100 нг/мл рекомбинантного человеческого активина A (PeproTech; № по кат. 120-14), к которой в 1 и 2 дни добавляли 20нг/мл Wnt3a. Отрицательные контрольные образцы содержали основную среду DMEM:F12 с добавлением FCS.
Одновременный многопараметрический анализ. По окончании четырехдневного периода культивирования аналитические планшеты дважды промыли раствором PBS (Invitrogen; № по кат. 14190), зафиксировали 4% параформальдегидом (Alexis Biochemical; № по кат. ALX-350-011) при комнатной температуре в течение 20 минут, затем трижды промыли раствором PBS и пермеабилизировали, обработав раствором 0,5% Triton X-100 (Sigma; № по кат. T8760-2) в течение 20 минут при комнатной температуре. Затем клетки снова трижды промыли раствором PBS и заблокировали раствором 4% куриной сыворотки (Invitrogen; № по кат. 16110082) в PBS в течение 30 минут при комнатной температуре. Первичные антитела (козий анти-человеческий SOX17; R&D Systems; № по кат. AF1924) разбавили в соотношении 1:100 в 4% куриной сыворотке и добавили в каждую лунку на один час при комнатной температуре. Сопряженные с флюорофором Alexa Fluor 488 вторичные антитела (куриный анти-козий IgG; Molecular Probes; № по кат. AZ1467) разбавили в соотношении 1:200 в PBS и добавили к каждому образцу после трехкратной промывки раствором PBS. Для подсчета окрашенных ядер на десять минут при комнатной температуре добавили 4 мкг/мл Hoechst 33342 (Invitrogen; № по кат. H3570). Затем планшеты один раз промыли раствором PBS и оставили в 100 мкл раствора PBS на лунку для томографии.
Томографию проводили на анализаторе IN Cell Analyzer 1000 (GE Healthcare) с применением дихроичного зеркала 51008bs для клеток, окрашенных красителями Hoechst 33342 и Alexa Fluor 488. Времена экспозиции оптимизировали с использованием лунок с положительными контрольными образцами и необработанных лунок с отрицательными контрольными образцами, окрашенных только вторичными антителами. Для компенсации возможных потерь клеток в ходе процедур анализа и последующего окрашивания для каждой лунки снимали по 15 проекций. Общее число клеток и общую интенсивность сигнала SOX17 измеряли для каждой лунки с помощью программного пакета IN Cell Developer Toolbox 1.7 (GE Healthcare). Сегментацию ядер определяли по ступеням шкалы серых тонов (исходный интервал 100-300) и размерам ядра. Для каждой повторной совокупности данных рассчитали средние значение и стандартные отклонения. Общий уровень экспрессии белка SOX17 выражается в виде общей интенсивности или интегральной интенсивности, определяемой как произведение общей интенсивности флюоресценции клетки на площадь клетки. Фон исключили на основании критериев соответствия ступеням шкалы серых тонов в диапазоне от 200 до 3500. Получали усредненные данные по трем лункам. Была вычислена процентная доля обработанных лунок относительно положительного контрольного образца.
Результаты этого скринингового анализа приведены в Таблице 17. Ни одно из низкомолекулярных соединений в течение четырехдневного процесса дифференцирования не индуцировало существенной экспрессии SOX17 в отсутствие GDF-8. Соединение 34 использовалось в качестве экспериментального контрольного образца и индуцировало существенную экспрессию SOX17 в присутствии GDF-8, уровень которой был эквивалентен наблюдаемым в положительном контрольном образце с использованием активина A и Wnt3a. Остальные протестированные в данном примере соединения, составляющие предмет настоящего изобретения, продемонстрировали в отношении экспрессии SOX17 активность в диапазоне от слабой до умеренной. Стоит отметить, что наблюдаемое в данном наборе влияние соединений на дифференцирование было связано с селективностью ко всем трем ферментативным сигнальным путям, что затрудняет определение точного механизма действия.
Пример 21
Скрининговый анализ аналогов соединений, составляющих предмет настоящего изобретения, способных участвовать в образовании клеток, экспрессирующих маркеры, характерные для линии сформированной эндодермы
На основе структуры соединений, составляющих предмет настоящего изобретения, был произведен поиск аналогов, в результате которого было обнаружено 118 аналогов. В ходе первичного скринингового анализа было выявлено, что некоторые аналоги способны индуцировать дифференцирование клеток в сформированную эндодерму в отсутствие активна А, но в сочетании с другими факторами роста. Было важно установить, способны ли эти аналоги индуцировать дифференцирование клеток в сформированную эндодерму в сочетании только с GDF-8.
Посев клеток для анализа. Коротко говоря, кластеры эмбриональных стволовых клеток человека линии H1 выращивали на покрытых препаратом MATRIGELTM с пониженным содержанием факторов роста (Invitrogen; № по кат. 356231) носителях для культивирования тканей. Клетки пассировали с использованием коллагеназы (Invitrogen; №356231 по кат. 17104-019), затем осторожно соскоблили, промыли для удаления остатков фермента и высеяли в соотношении 1:1 (по площади поверхности) на покрытые препаратом MATRIGELTM с пониженным содержанием факторов роста (BD Biosciences; № по кат. 356231) 96-луночные черные планшеты (Packard ViewPlates; PerkinElmer; № по кат. 6005182) в объеме 100 мкл на лунку. Клеткам дали возможность прикрепиться, после чего в течение 1-3 дней воспроизводили логарифмическую фазу роста, во время которой клетки ежедневно питали, используя кондиционированную среду MEF с добавлением 8 нг/мл bFGF (R&D Systems; № по кат. 233-FB). В течение всего периода анализа планшеты держали при температуре 37єC в атмосфере 5% CO2 в увлажняемом боксе.
Подготовка соединений и анализ. Скрининговый анализ проводился с использованием библиотеки соединений-аналогов. Соединения из библиотеки предоставили в виде исходных растворов концентрации 5 мМ на 96-луночных планшетах, растворили в 100% DMSO (Sigma; № по кат. D2650) и хранили при температуре -80oC. Затем соединения из такой библиотеки разбавили до промежуточной концентрации 0,2 мМ в 50 мМ HEPES (Invitrogen; № по кат. 15630-080), 20% DMSO и хранили при температуре 4єC. Каждый тест выполнялся в трех повторностях. В течение всего четырехдневного периода анализа питание вводили через день. Анализы начали с отсасывания культуральной среды из каждой лунки с последующей трехкратной промывкой лунки раствором PBS (Invitrogen; № по кат. 14190) для удаления остатков факторов роста. В 1 день проведения анализа в лунки добавили тестируемые образцы объемом 200 мкл, содержащие основную среду DMEM:F12 (Invitrogen; № по кат. 11330-032) с добавлением 0,5% FCS (HyClone; № по кат. SH30070.03), 200 нг/мл GDF-8 (R&D Systems № по кат. 788-G8) и 2,5 мкМ соединения. В 3 день анализа в лунки добавили тестируемые образцы объемом 100 мкл, содержащие основную среду DMEM:F12 с добавлением 2% FCS и 200 нг/мл GDF-8 (R&D Systems, № по кат. 788-G8). Положительные контрольные образцы в течение четырех дней анализа содержали ту же основную среду с добавлением FCS и 100 нг/мл рекомбинантного человеческого активина A (PeproTech; № по кат. 120-14), к которой в 1 и 2 дни добавляли 20нг/мл Wnt3a.Отрицательные контрольные образцы содержали основную среду DMEM:F12 с добавлением FCS, без активина А, но с добавлением в 1 и 2 дни Wnt3a.
Одновременный многопараметрический анализ. По окончании четырехдневного периода культивирования аналитические планшеты дважды промыли раствором PBS (Invitrogen; № по кат. 14190), зафиксировали 4% параформальдегидом (Alexis Biochemical; № по кат. ALX-350-011) при комнатной температуре в течение 20 минут, затем трижды промыли раствором PBS и пермеабилизировали, обработав раствором 0,5% Triton X-100 (Sigma; № по кат. T8760-2) в течение 20 минут при комнатной температуре. Затем клетки снова трижды промыли раствором PBS и заблокировали раствором 4% куриной сыворотки (Invitrogen; № по кат. 16110082) в PBS в течение 30 минут при комнатной температуре. Первичные антитела (козий анти-человеческий SOX17; R&D Systems; № по кат. AF1924) разбавили в соотношении 1:100 в 4% куриной сыворотке и добавили в каждую лунку на один час при комнатной температуре. Сопряженные с флюорофором Alexa Fluor 488 вторичные антитела (куриный анти-козий IgG; Molecular Probes; № по кат. AZ1467) разбавили в соотношении 1:200 в PBS и добавили к каждому образцу после трехкратной промывки раствором PBS. Для подсчета окрашенных ядер на десять минут при комнатной температуре добавили 4 мкг/мл Hoechst 33342 (Invitrogen; № по кат. H3570). Затем планшеты один раз промыли раствором PBS и оставили в 100 мкл раствора PBS на лунку для томографии.
Томографию проводили на анализаторе IN Cell Analyzer 1000 (GE Healthcare) с применением дихроичного зеркала 51008bs для клеток, окрашенных красителями Hoechst 33342 и Alexa Fluor 488. Времена экспозиции оптимизировали с использованием лунок с положительными контрольными образцами и необработанных лунок с отрицательными контрольными образцами, окрашенных только вторичными антителами. Для компенсации возможных потерь клеток в ходе процедур анализа и последующего окрашивания для каждой лунки снимали по 15 проекций. Общее число клеток и общую интенсивность сигнала SOX17 измеряли для каждой лунки с помощью программного пакета IN Cell Developer Toolbox 1.7 (GE Healthcare). Сегментацию ядер определяли по ступеням шкалы серых тонов (исходный интервал 100-300) и размерам ядра. Для каждой повторной совокупности данных рассчитали средние значение и стандартные отклонения. Общий уровень экспрессии белка SOX17 выражается в виде общей интенсивности или интегральной интенсивности, определяемой как произведение общей интенсивности флюоресценции клетки на площадь клетки. Фон исключили на основании критериев соответствия ступеням шкалы серых тонов в диапазоне от 200 до 3500. Показатели общей интенсивности нормировали путем деления общей интенсивности для каждой лунки на среднюю общую интенсивность для положительного контрольного образца. Для каждого набора повторностей рассчитали нормированные данные для средних значений и стандартных отклонений.
Результаты скринингового анализа, полученные на четырех аналитических планшетах в ходе одного эксперимента, представлены в Таблице 18. Соединения ранжированы по ответной реакции в виде экспрессии SOX17, в процентах от положительного контрольного образца - обработки активином A и Wnt3a. В Таблице 19 представлен список из 12 новых аналогов, показавших в ходе анализа положительные результаты.
Пример 22
Эмбриональные стволовые клетки человека, выращенные на микроносителях, могут быть дифференцированы в клетки, экспрессирующие маркеры, характерные для линии сформированной эндодермы, в соответствии со способами, составляющими предмет настоящего изобретения
Для дифференцирования и производства большого числа эндокринных клеток в промышленных условиях было важно показать, что эмбриональные стволовые клетки человека могут быть выращены и дифференцированы в сформированную эндодерму на гранулах микроносителя с использованием способов, составляющих предмет настоящего изобретения.
Подготовка клеток к анализу и дифференцированию. Клетки линии H1 p49C3 обычным методом выращивали на гранулах Cytodex3 (GE Healthcare Life Sciences, Нью-Джерси, США) во вращающейся колбе объемом 125 мл в соответствии со способами, описанными в заявке на патент США № 61/116447. Через семь дней клетки и гранулы перенесли на 6-луночный планшет (производитель; № по кат. XXX) в соотношении 30 см2 площади поверхности гранул на лунку, и планшет установили на качающуюся платформу. Клетки на гранулах, находящиеся в экспериментальной лунке с положительным контрольным образцом (обозначенной «AA/Wnt3a»), дифференцировали путем добавления 100 нг/мл активина A (PeproTech; № по кат. 120-14) и 20 нг/мл Wnt3a (R&D Systems; № по кат. 1324-WN/CF) в течение двух дней с последующим добавлением 100 нг/мл активина A и 8 нг/мл bFGF (PeproTech Inc.; № по кат. 100-18B) в течение одного дня в среде RPMI-1640 (Invitrogen; № по кат. 22400), содержащей 2% BSA без жирных кислот (MP Biomedicals, Inc; № по кат. 152401), в объеме 2 мл на лунку. В экспериментальную лунку с отрицательным контрольным образцом (обозначенную «CMP») в течение трех дней и в отсутствие других факторов роста добавляли Соединение 34 в конечной концентрации 2,5 мкМ в среде RPMI-1640, содержащей 2% BSA без жирных кислот, в объеме 2 мл на лунку. В третью экспериментальную лунку (обозначенную «CMP+8») в течение трех дней добавляли Соединение 34 в концентрации 2,5 мкМ и 50 нг/мл GDF-8 (R&D Systems, № по кат. 788-G8) в среде RPMI-1640, содержащей 2% BSA без жирных кислот, в объеме 2 мл на лунку. В четвертую экспериментальную лунку (обозначенную «CMP+8+D») в течение трех дней добавляли Соединение 34 в концентрации 2,5 мкМ, 50нг/мл GDF-8 и 50 нг/мл PDGF-D в среде RPMI-1640, содержащей 2% BSA без жирных кислот, в объеме 2 мл на лунку. В пятую экспериментальную лунку (обозначенную «CMP+8+D+V») в течение трех дней добавляли Соединение 34 в концентрации 2,5 мкМ, 50 нг/мл GDF-8,50 нг/мл PDGF-D и 50 нг/мл VEGF в среде RPMI-1640, содержащей 2% BSA без жирных кислот, в объеме 2 мл на лунку. В шестую экспериментальную лунку (обозначенную «CMP+8+D+V+M») в течение трех дней добавляли Соединение 34 в концентрации 2,5 мкМ, 50 нг/мл GDF-8, 50 нг/мл PDGF-D, 50 нг/мл VEGF и 20 нг/мл мусцимола в среде RPMI-1640, содержащей 2% BSA без жирных кислот, в объеме 2 мл на лунку. Замену сред и соединений производили ежедневно.
После завершения обработки и культивирования клетки собрали с гранул в соответствии со способами, описанными в заявке на патент США № 61/116447. Собранные клетки подсчитали и проанализировали методом проточной цитометрии в соответствии с описанными выше способами.
Результаты представлены на фиг. 25. Как видно в части A, во всех экспериментальных группах, подвергавшихся дифференцированию, было получено сходное число клеток. Как видно в части B, клетки, обработанные только Соединением 34, не дифференцировались в CXCR4-положительные клетки. В положительном контрольном образце, в котором в ходе дифференцирования добавлялись активин А и Wnt3a, 68% клеток полученной клеточной популяции экспрессировали CXCR4. При добавлении Соединения 34 в сочетании с различными факторами роста экспрессия CXCR4 индуцировалась в среднем в 50% клеток. Стоит отметить, что при обработке Соединением 34 в сочетании с одним фактором роста, GDF-8, или в сочетании с несколькими факторами роста, в числе которых был GDF-8, уровни экспрессии CXCR4 были эквивалентными. Это доказывает, что сочетание Соединения 34 и по меньшей мере GDF-8 может заменить сочетание активина A и Wnt3a при стимуляции дифференцирования клеток в сформированную эндодерму. Этот пример также показывает, что такая процедура обработки эффективна для клеток, выращиваемых и дифференцируемых на гранулах микроносителя.
Пример 23
Соединения, составляющие предмет настоящего изобретения, в сочетании с GDF-8 усиливают пролиферацию клеток
В предыдущем примере было показано, что GDF-8 может заменить активин A при дифференцировании эмбриональных стволовых клеток человека в клетки, экспрессирующие маркеры, характерные для линии сформированной эндодермы. Было важно определить относительную эффективность GDF-8 и активина A в отношении образования сформированной эндодермы. Был проведен анализ зависимости доза-эффект при дифференцировании эмбриональных стволовых клеток человека с применением эквивалентных концентраций каждого из факторов роста, а полученные результаты были подвергнуты сравнению.
Соединения, составляющие предмет настоящего изобретения, использованные в сочетании с GDF-8 при дифференцировании клеток в сформированную эндодерму, были исследованы на их способность индуцировать пролиферацию клеток. Результаты сравнивались с обработкой активином А или только GDF-8.
Подготовка клеток к анализу. Исходные культуры эмбриональных стволовых клеток человека (эмбриональные стволовые клетки человека линии H1) поддерживали в недифференцированном плюрипотентном состоянии на чашках, покрытых препаратом MATRIGELTM с пониженным содержанием факторов роста (BD Biosciences; № по кат. 356231), в кондиционированной среде MEF и пассировали в среднем каждые четыре дня. При пассировании клетки обрабатывали раствором диспазы 1 мг/мл (Invitrogen, № по кат. 17105-041) в течение 5-7 минут при 37єC с последующей промывкой монослоя кондиционированной культуральной средой MEF, затем осторожно соскоблили для получения кластеров клеток. Кластеры центрифугировали на низкой скорости для получения осадка клеток и удаления остатков диспазы. Кластеры клеток разделили в соотношении 1:3 или 1:4 для стандартного поддерживающего культивирования или в соотношении 1:1 для немедленного анализа. Все линии эмбриональных стволовых клеток человека поддерживали при номерах пассажа менее 50 и периодически проверяли на нормальный кариотипический фенотип и отсутствие примесей микоплазмы.
Кластеры клеток, предназначенные для анализа, равномерно ресуспендировали в кондиционированной среде MEF с добавлением 8 нг/мл bFGF и высеяли на покрытые препаратом MATRIGELTM с пониженным содержанием факторов роста 96-луночные планшеты Packard VIEWPLATES (PerkinElmer; № по кат. 6005182) в объеме 100 мкл на лунку. Для первоначального посева и размножения использовали кондиционированную среду MEF с добавлением 8 нг/мл bFGF. Ежедневное питание осуществляли путем отсасывания отработанной культуральной среды из каждой лунки и ее замены тем же объемом свежей среды. В лунки с фоновыми образцами на каждой аналитической планшете клетки не высеивали, но лунки обрабатывали в ходе анализа основной средой. В течение всего периода анализа планшеты держали при температуре 37єC в атмосфере 5% CO2 в увлажняемом боксе.
Анализ. Анализ начали с отсасывания культуральной среды из всех лунок и добавления готовых аликвот (100 мкл) тестируемой среды. Каждый тест выполнялся в трех повторностях. В течение всего трехдневного периода питание вводили ежедневно, для чего среду из каждой лунки отсасывали и заменяли свежей тестируемой средой. Параллельно проводили несколько идентичных анализов для оценки через 24, 48 и 72 часа.
В 1 день анализа во все лунки, содержащие клетки, добавили аликвоту (80 мкл) среды RPMI-1640 (Invitrogen; № по кат. 22400) с добавлением 2,5% фракции V бычьего альбумина, не содержащего жирных кислот (FAF BSA; итоговая концентрация 2%) (Proliant Inc. № по кат. SKU 68700). Различные контрольные и тестируемые образцы для добавления в соответствующие лунки готовили в 5-кратной концентрации (20 мкл на лунку). Использовали следующие контрольные среды (с указанными итоговыми концентрациями факторов роста): 1) основная среда с 2% FAF BSA; 2) 100 нг/мл активина A (PeproTech; № по кат. 120-14) и 8 нг/мл bFGF (PeproTech; № по кат. 100-18B); 3) 100 нг/мл активина A, 8 нг/мл bFGF и 20 нг/мл Wnt3a (R&D Systems; № по кат. 1324-WN/CF); 4) 100 нг/мл GDF-8 (R&D Systems, № по кат. 788-G8) и 8 нг/мл bFGF; 5) GDF-8, 8 нг/мл bFGF и 20 нг/мл Wnt3a. Клетки из еще одного набора контрольных лунок в течение всего периода анализа обрабатывали кондиционированной средой MEF. В некоторых контрольных образцах, где использовался GDF-8, Wnt3a заменяли на соединение, составляющее предмет настоящего изобретения. Для получения экспериментальных образцов восемь различных соединений последовательно разбавляли вдвое для получения трех различных доз, к которым потом добавили 100 нг/мл GDF-8 и 8 нг/мл bFGF. Такими низкомолекулярными соединениями были следующие патентованные соединения: Соединение 181, Соединение 180, Соединение 19, Соединение 202, Соединение 40, Соединение 34, Соединение 56 и коммерческий ингибитор GSK3 BIO (EMD Chemicals, Inc.; № по кат. 361550). Во 2 и 3 дни анализа из всех контрольных и экспериментальных лунок среду отсосали и заменили на новую, которая была идентична предыдущей, за исключением того, что из некоторых контрольных лунок был убран Wnt3a.
MTS-анализ. Через 24, 48 или 72 часа культивирования один из наборов аналитических планшетов подвергался MTS-анализу (Promega; № по кат. G3581) в соответствии с инструкциями изготовителя. Коротко говоря, в каждую лунку добавили 20 мкл реактива MTS, планшеты инкубировали при 37єC в атмосфере 5% CO2 в течение четырех часов, после чего сняли показания оптической плотности на длине волны 490 нм (OD490). При статистических расчетах вычитали значения фона (т.е. значения для экспериментальных лунок без клеток) и определяли средние значения по трем повторностям, а также стандартную ошибку среднего.
MTS-анализ является оценкой метаболической активности клеток по ферментативному восстановлению тетразолия в формазан. При измерении в одной временной точке MTS-анализ может служить сравнительным показателем жизнеспособности клеток. Если MTS-анализ проводится параллельно в последовательных временных точках, он может дать дополнительную информацию об увеличении метаболической активности клеток, которая, в свою очередь, может коррелировать с пролиферацией клеток в каждой из экспериментальных групп. На фиг. 26, часть A, приводятся показатели оптической плотности (OD490) во всех контрольных лунках за трехдневный период анализа. Изменения OD490 в клетках, обработанных кондиционированной средой, были небольшими, что указывает на сохранение числа клеток в данной экспериментальной группе. Напротив, у клеток, культивированных в основной среде без факторов роста (без обработки), значение OD490 равномерно уменьшалось, что коррелировало с уменьшением числа клеток со временем. Обработка активином А при дифференцировании, как в сочетании с Wnt3a, так и без него, приводила к постепенному увеличению OD490, что указывает на значительное увеличение популяции клеток со временем. Обработка GDF-8 в отсутствие Wnt3a приводила к уменьшению OD490 относительно обработки активном A; этот эффект был заметен в 1 день и сохранялся в течение всех трех дней культивирования. Добавление Wnt3a к GDF-8 приводило к восстановлению и увеличению OD490 к 3 дню культивирования.
На фиг. 26, части с B по I, показаны результаты MTS-анализа групп, обработанных низкомолекулярным ингибитором в сочетании с GDF-8. Значения OD490 для групп, обработанных соединением, составляющим предмет настоящего изобретения, и GDF-8, были равными или большими, чем при обработке активином A. Во всех случаях оптимальная концентрация каждого из низкомолекулярных соединений в сочетании с GDF-8 вызывала увеличение значений OD490 в течение трехдневного периода анализа относительно обработки одним GDF-8. Эти результаты дают основание предполагать, что соединения, составляющие предмет настоящего изобретения, важны для индукции пролиферации и роста клеточной популяции в ходе дифференцирования клеток в сформированную эндодерму.
Пример 24
Эмбриональные стволовые клетки человека, выращенные на микроносителях, могут быть дифференцированы в клетки-предшественники эндокринных клеток в соответствии со способами, составляющими предмет настоящего изобретения
Для дифференцирования и производства большего числа эндокринных клеток в промышленных условиях было важно показать, что эмбриональные стволовые клетки человека могут быть выращены и дифференцированы в клетки-предшественники эндокринных клеток на гранулах микроносителя с использованием протокола, не включающего активин А.
Подготовка клеток к анализу и дифференцированию. Клетки линии H1 p45 cells выращивали на гранулах Cytodex3 (GE Healthcare; № по кат. 17-0485-01) на 6-луночном планшете со сверхнизкой связывающей способностью (Costar; № по кат. 3471), помещенном на платформу, качающуюся со скоростью 1 оборот за 10 секунд (Vari Mix, Thermo Scientific, № по кат. М79735). Кондиционированную среду MEF меняли ежедневно в течение шести дней. Затем среду заменяли на следующие виды обработки в целях инициации дифференцирования клеток в эндодерму. Клетки на гранулах, находящиеся в экспериментальной лунке с положительным контрольным образцом (обозначенной «AA+Wnt»), дифференцировали путем добавления 100 нг/мл активина A (PeproTech; № по кат. 120-14), 8 нг/мл bFGF (PeproTech Inc.; № по кат. 100-18B) и 20 нг/мл Wnt3a (R&D Systems; № по кат. 1324-WN/CF) в течение одного дня с последующим добавлением 100 нг/мл активина A и 8 нг/мл bFGF (PeproTech Inc.; № по кат. 100-18B) в течение двух дней в среде RPMI-1640 (Invitrogen; № по кат.: 22400), содержащей 2% BSA без жирных кислот (Proliant Biomedicals, Inc; № SKU 68700), в объеме 2 мл на лунку. Во вторую экспериментальную лунку (обозначенную «GDF-8+MCX») в течение одного дня добавили Соединение 202 в концентрации 2,5 мкМ, 200 нг/мл GDF-8 (R&D Systems, № по кат. 788-G8) и 8нг/мл bFGF с последующим добавлением в течение двух дней 200 нг/мл GDF-8 и 8 нг/мл bFGF в среде RPMI-1640, содержащей 2% BSA без жирных кислот (2 мл на лунку). В третью экспериментальную лунку (обозначенную «GDF-8+Wnt») в течение одного дня добавили 200 нг/мл GDF-8, 20нг/мл Wnt3a и 8 нг/мл bFGF с последующим добавлением в течение двух дней 200 нг/мл GDF-8 и 8 нг/мл bFGF в среде RPMI-1640, содержащей 2% BSA без жирных кислот (2 мл на лунку). Замену сред и соединений производили ежедневно.
После завершения обработки и культивирования клетки собрали и подсчитали для определения выхода клеток и для проведения анализа методом проточной цитометрии. При всех трех видах обработки были зафиксированы высокие уровни CXCR4 и CD99 (фиг. 27A). Число клеток в образцах варьировалось (фиг. 27B). Более низкое, чем в других группах, число клеток было обнаружено в образцах, обработанных GDF-8, а также на стадии сформированной эндодермы и на четвертой стадии. Такие результаты дают основание предполагать, что соединения, составляющие предмет настоящего изобретения, могут увеличивать пролиферацию клеток в ходе дифференцирования.
В конце третьей стадии в клетках экспрессируются эндодермальные гены PDX1, HNF4 alpha и CDX2 (фиг. 27C, D). Обработка клеток GDF-8 и соединением, составляющим предмет настоящего изобретения, в течение первой стадии дифференцирования приводит к лучшей экспрессии Pdx1, чем в контрольном образце. В конце четвертой стадии эндодермальные гены активируются еще больше (фиг. 27E, F). Эти результаты позволяют сделать вывод, что GDF-8 в сочетании с Соединением 202 может заменить сочетание активина A и Wnt3a при дифференцировании сформированной эндодермы с образованием панкреатической эндодермы.
Публикации, цитируемые в настоящем документе, в силу ссылки на них полностью включаются в настоящий документ. Хотя различные аспекты настоящего изобретения иллюстрируются выше ссылками на примеры и предпочтительные варианты осуществления, подразумевается, что область изобретения определяется не упомянутым выше описанием, а следующими ниже пунктами формулы изобретения, составленными в соответствии с принципами патентного законодательства.
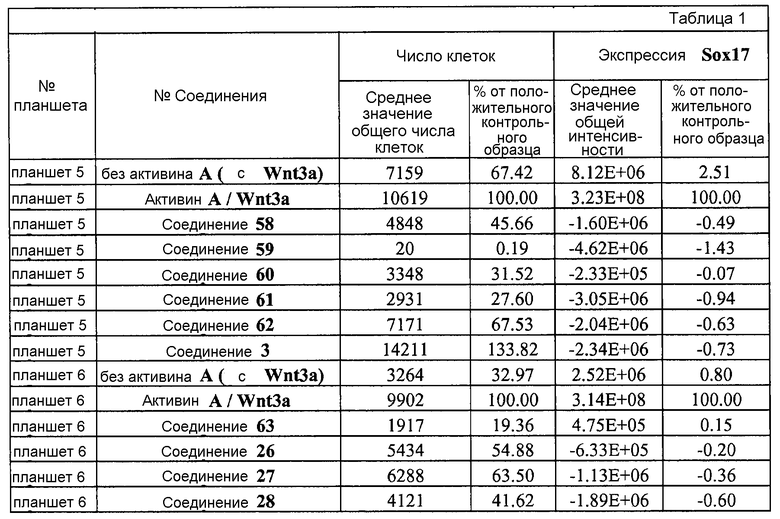
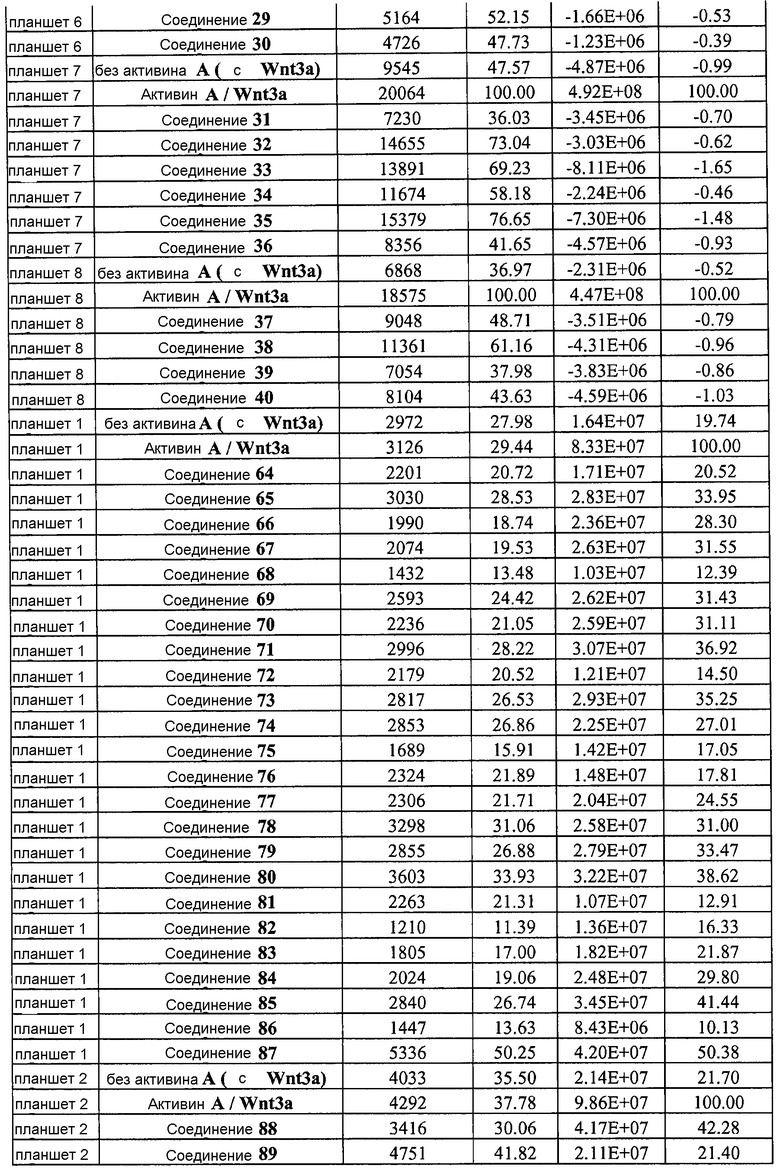
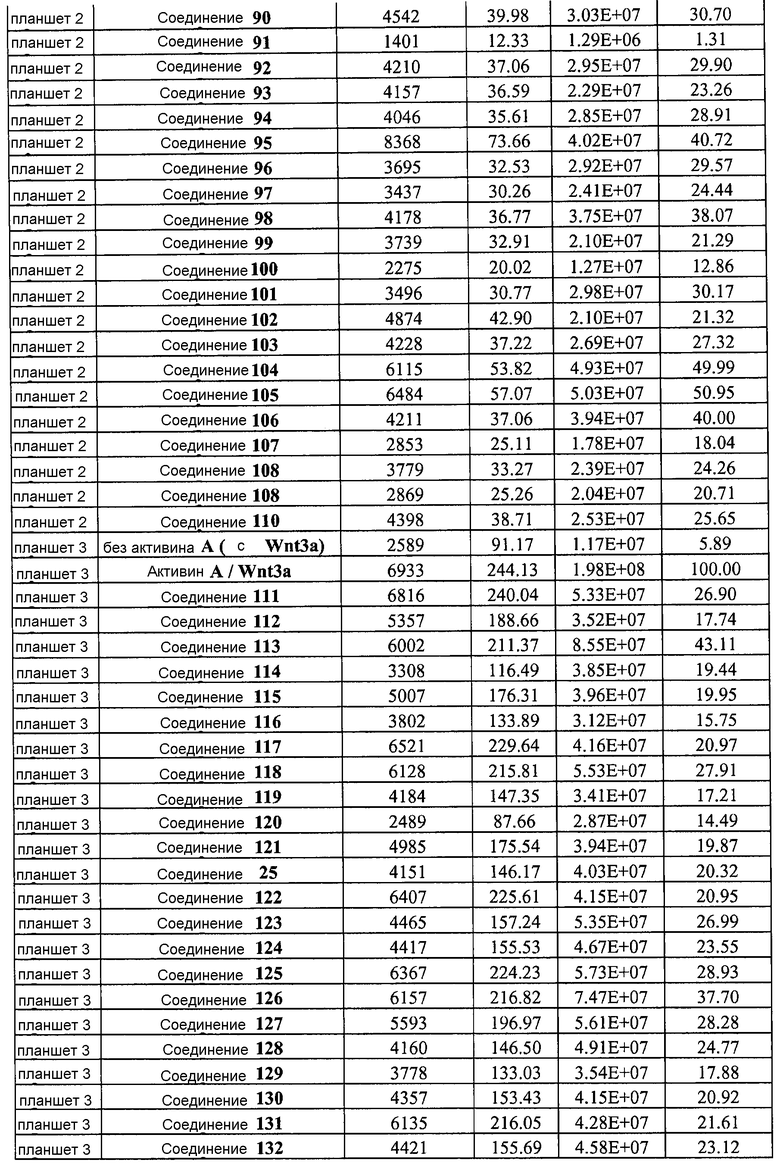
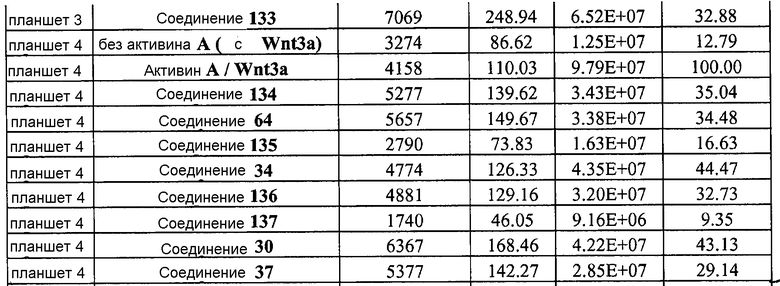
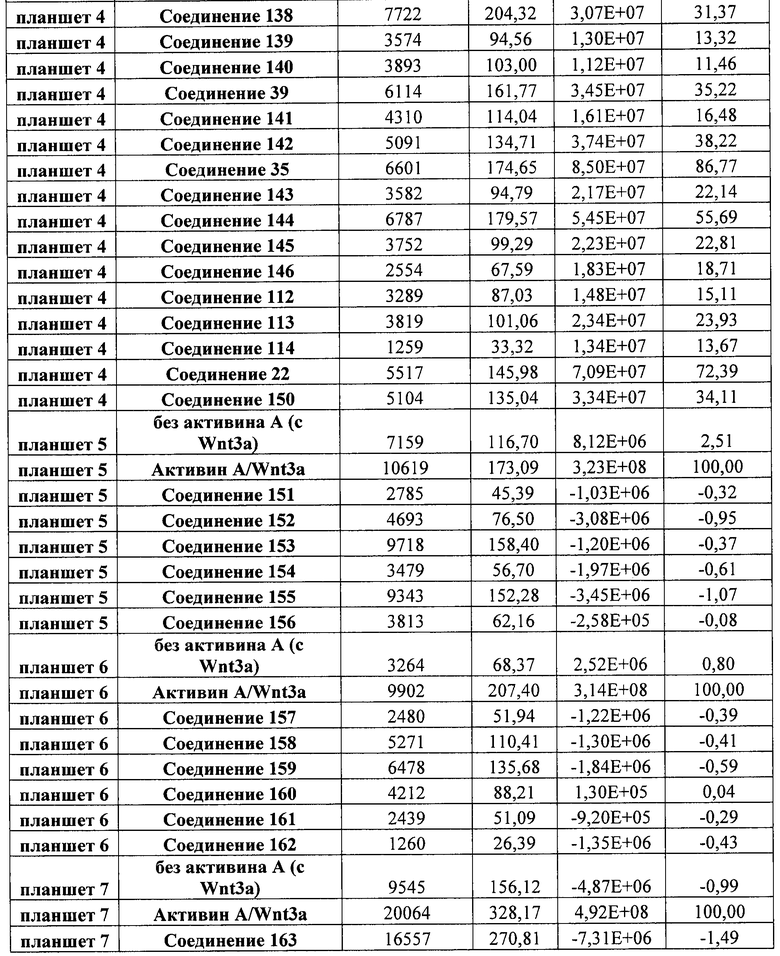
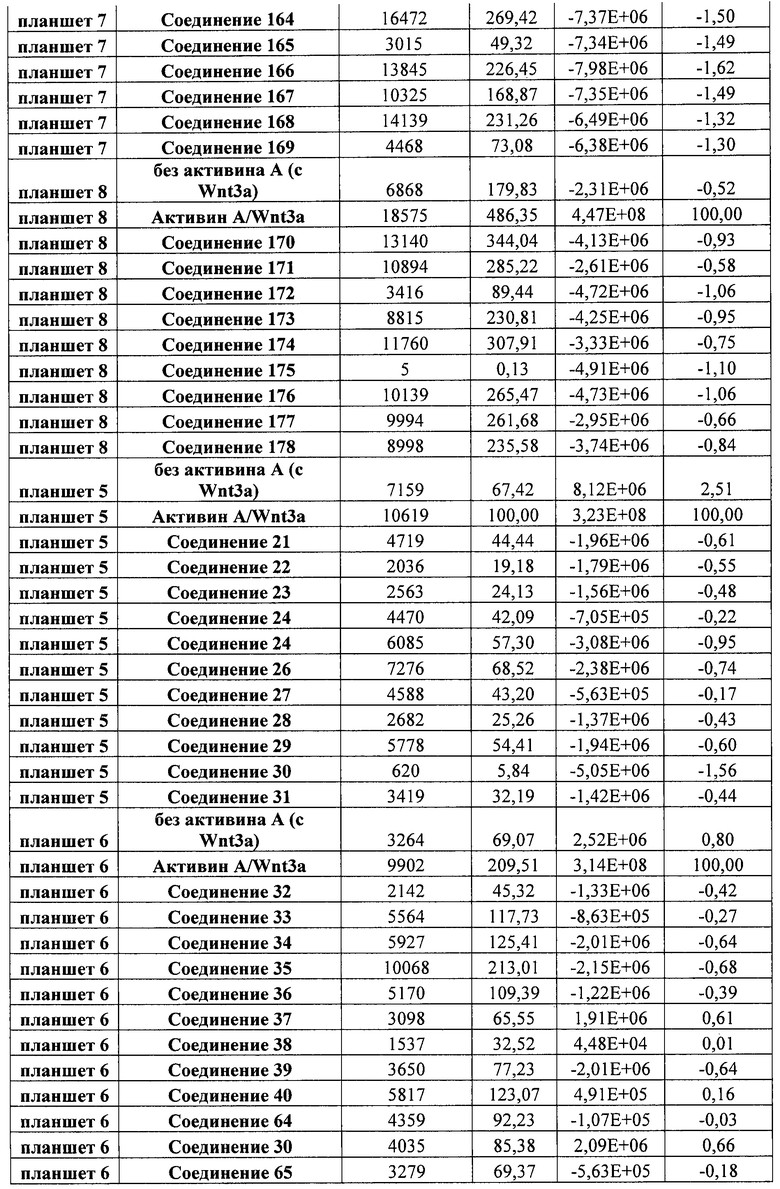
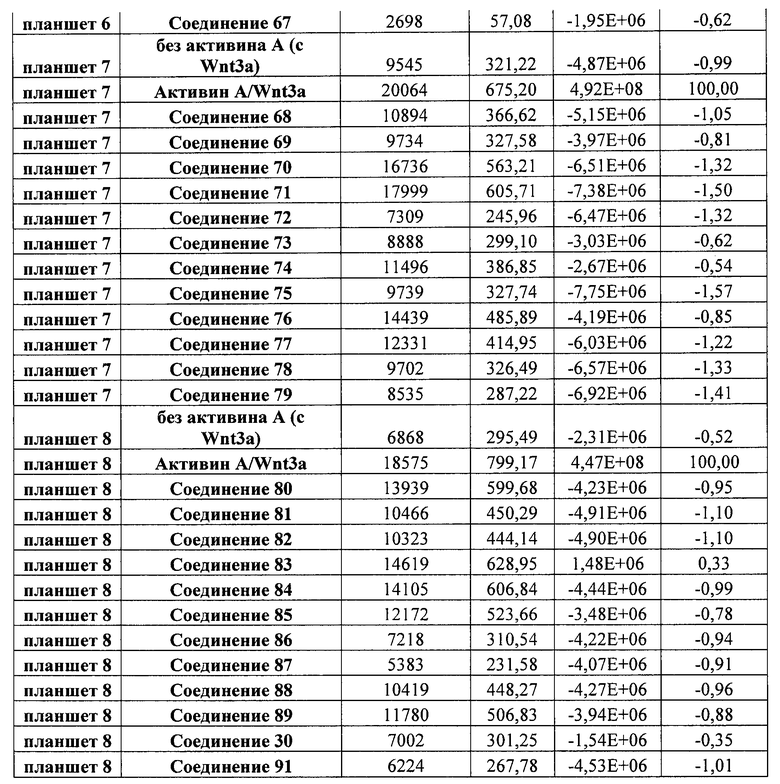
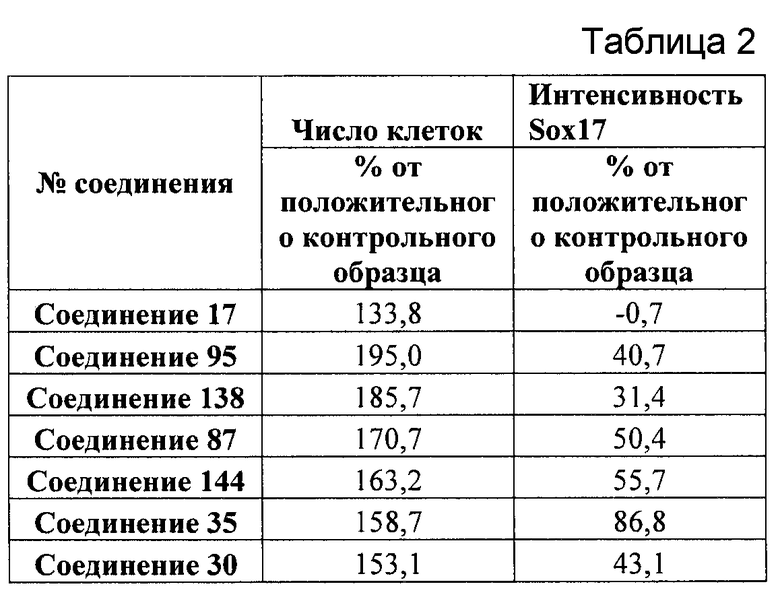
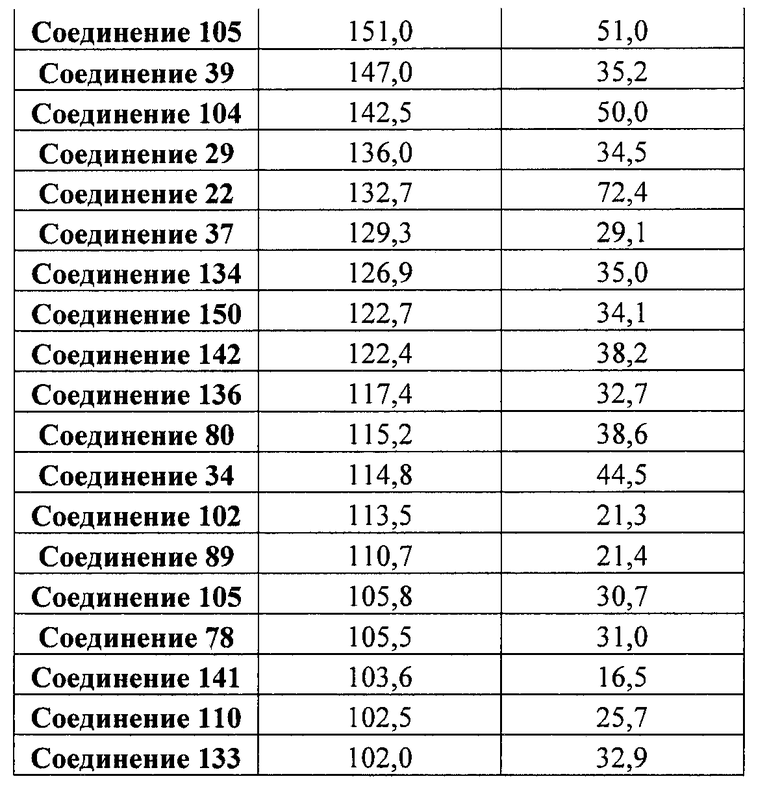
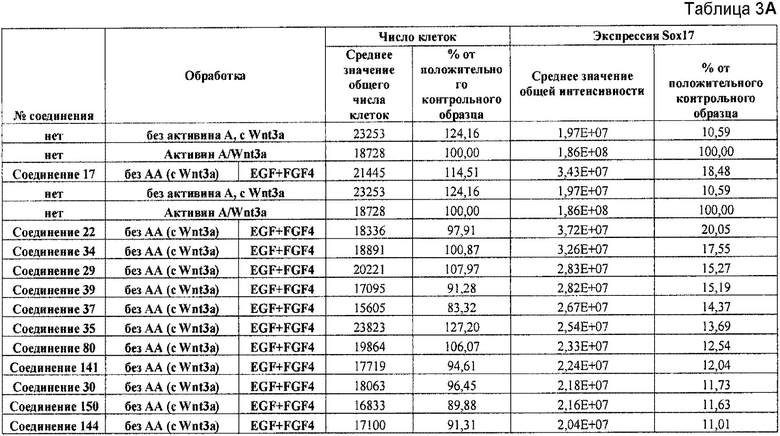
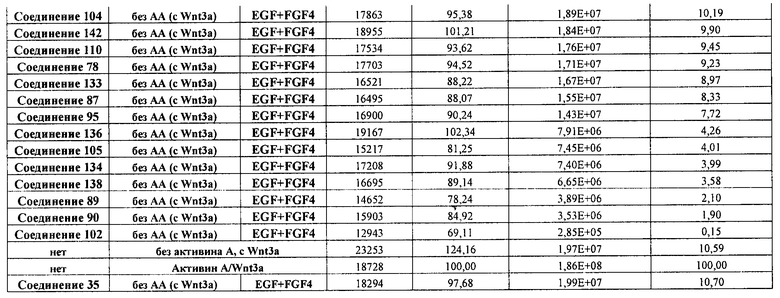
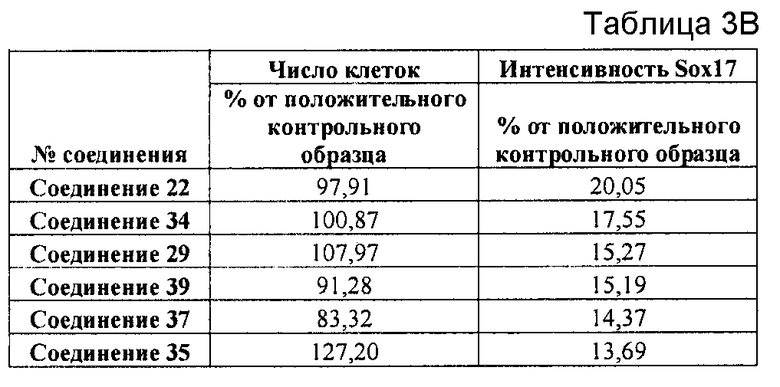
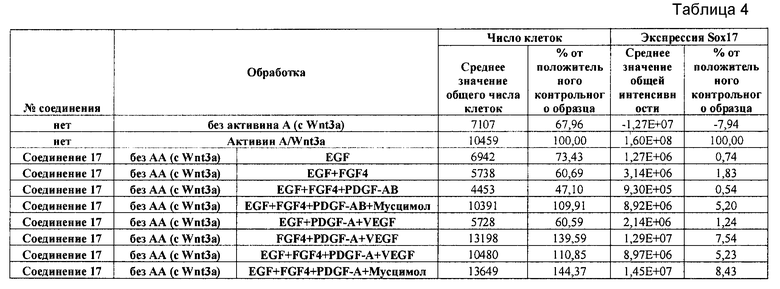
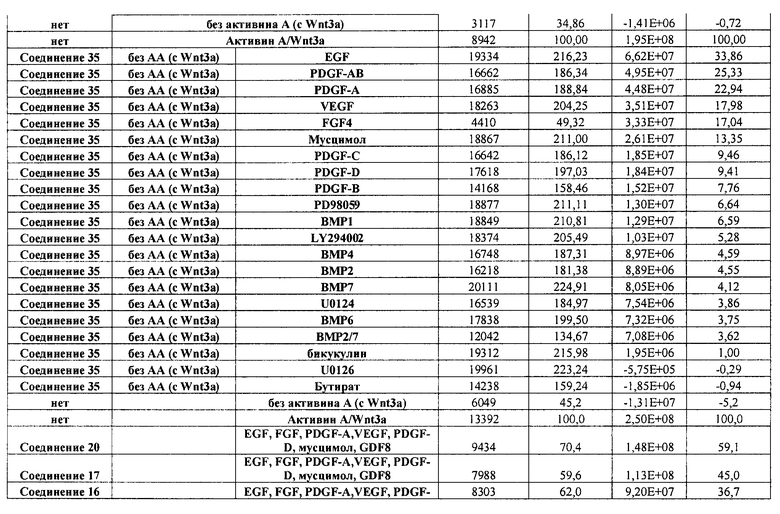
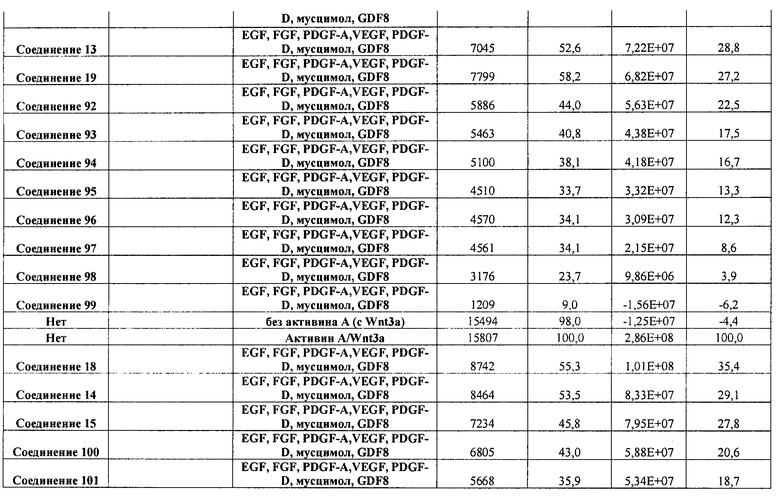
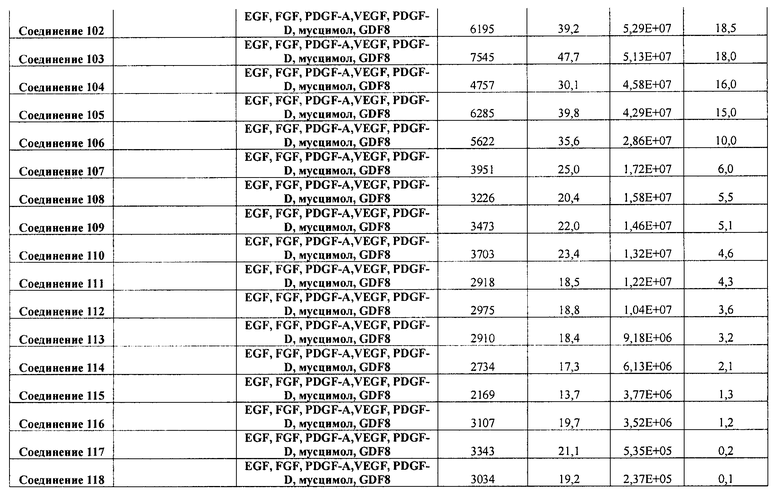

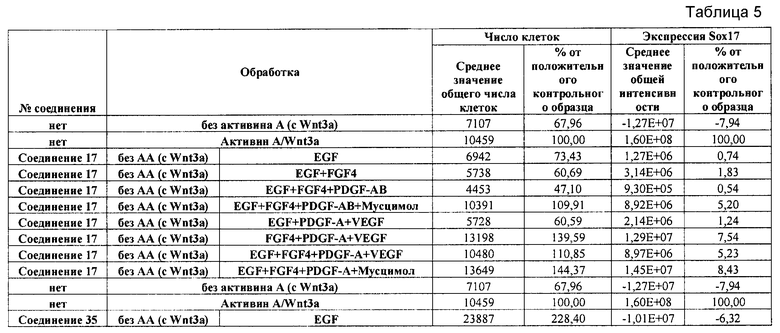
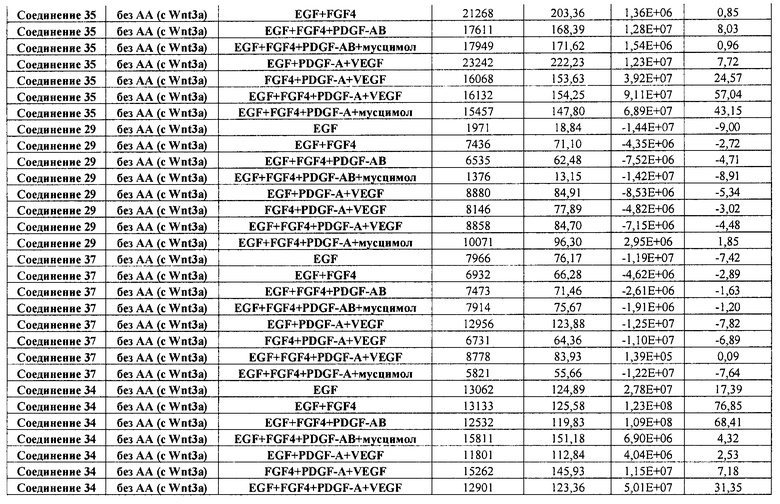
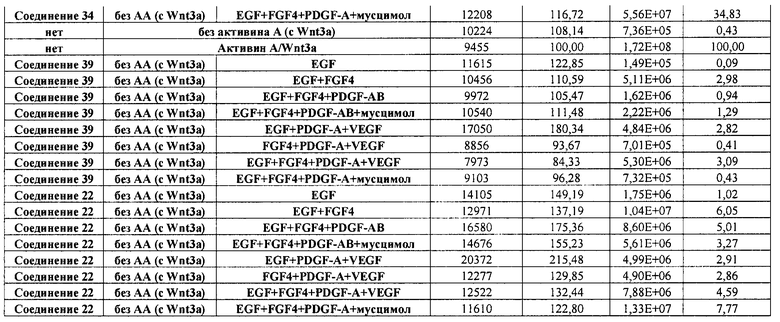
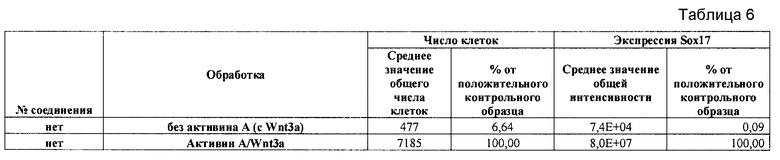
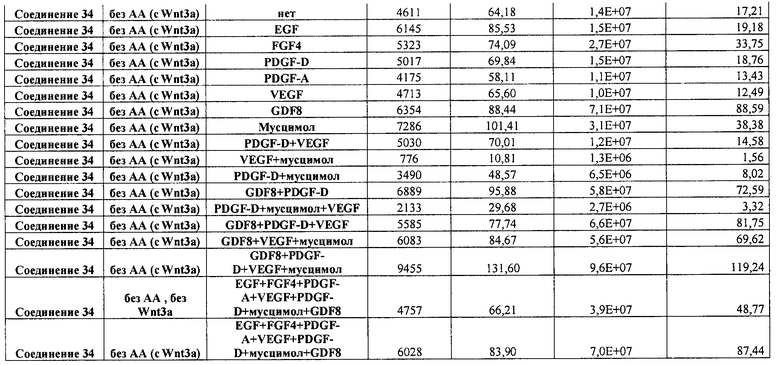
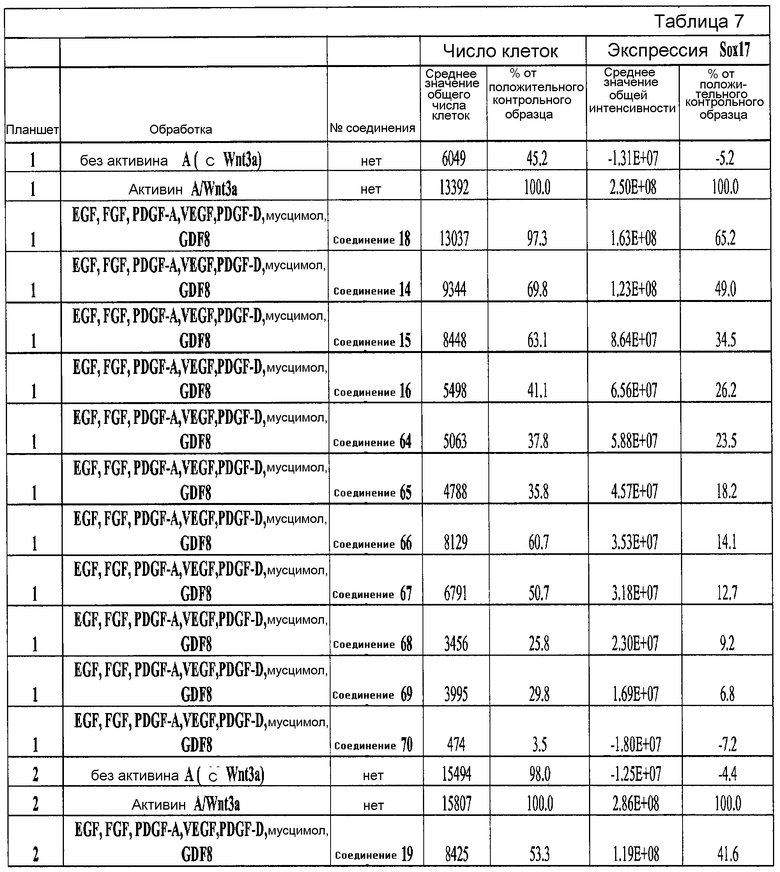
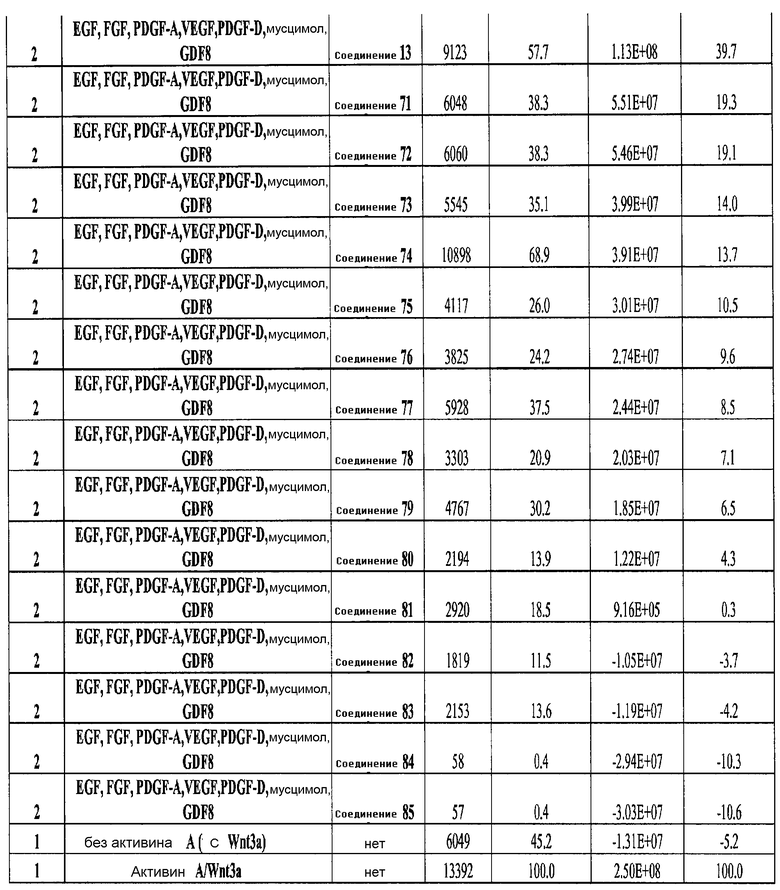
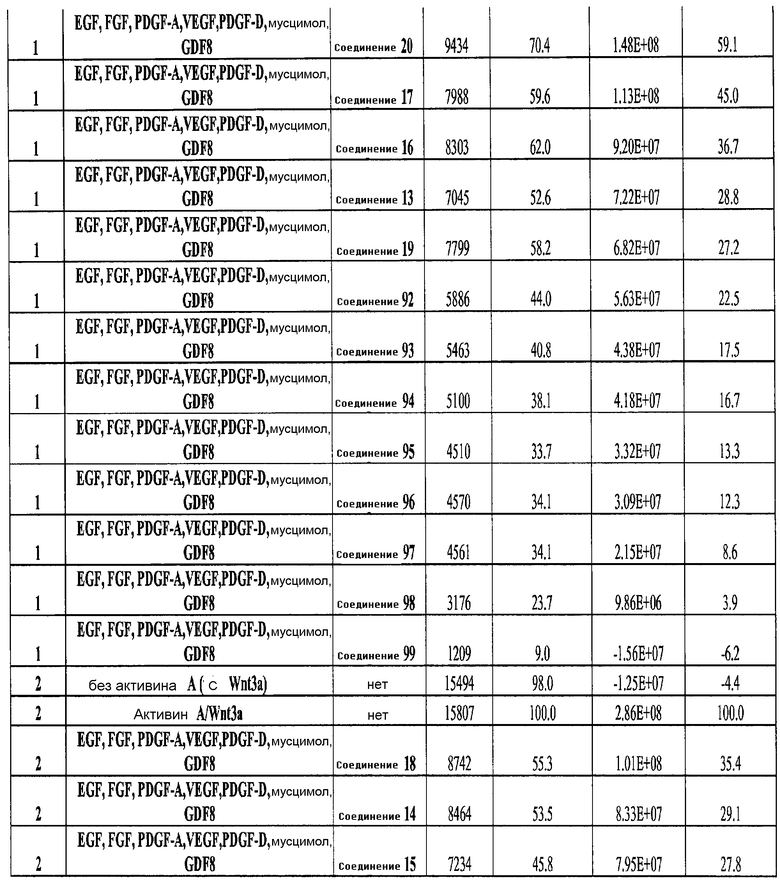
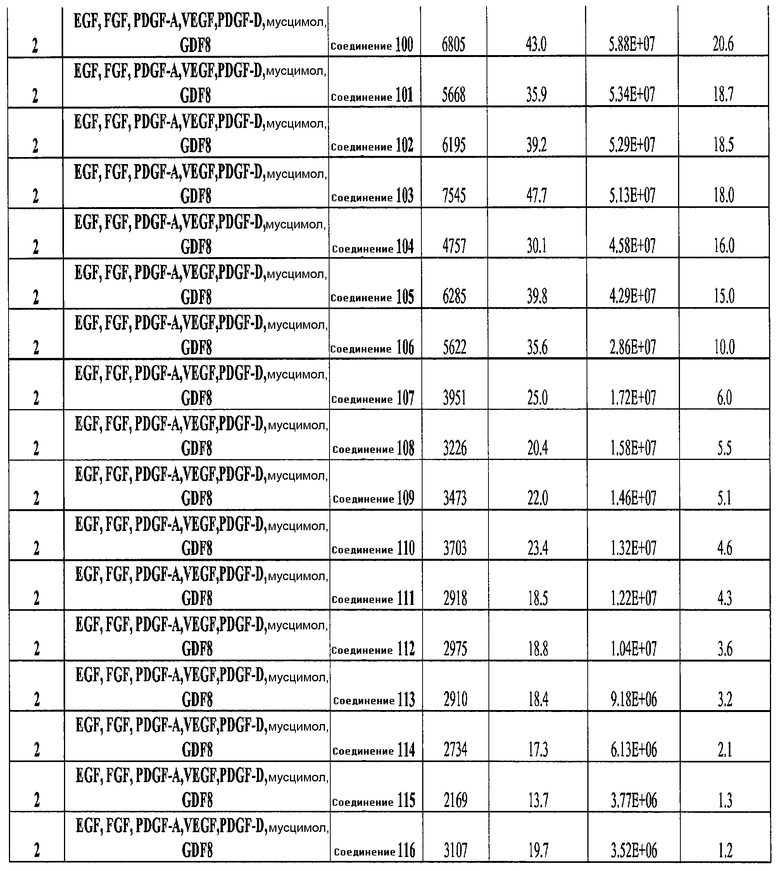
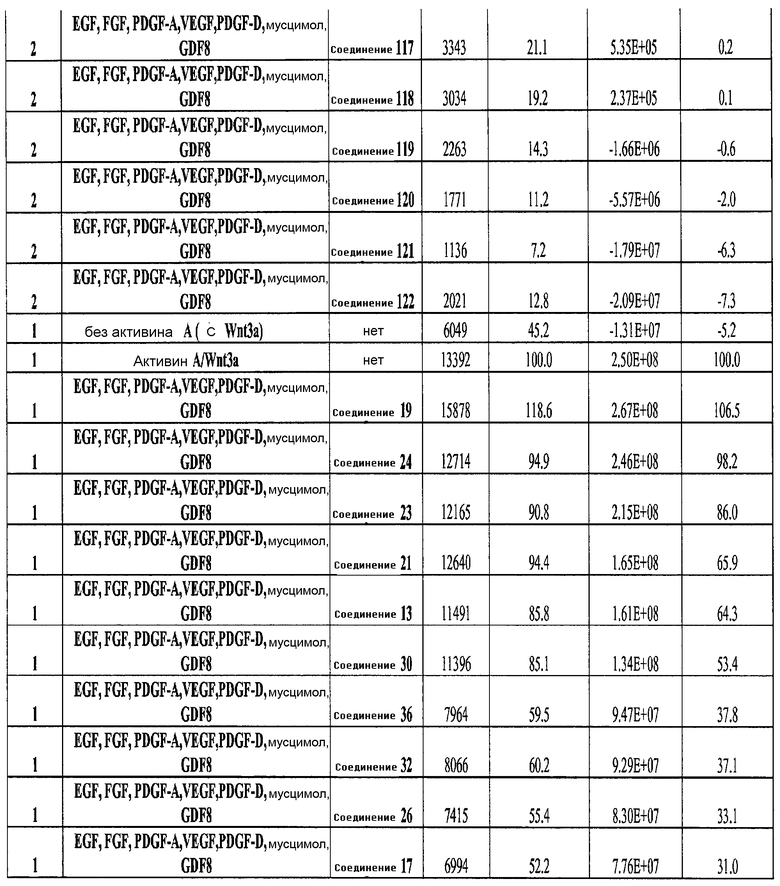
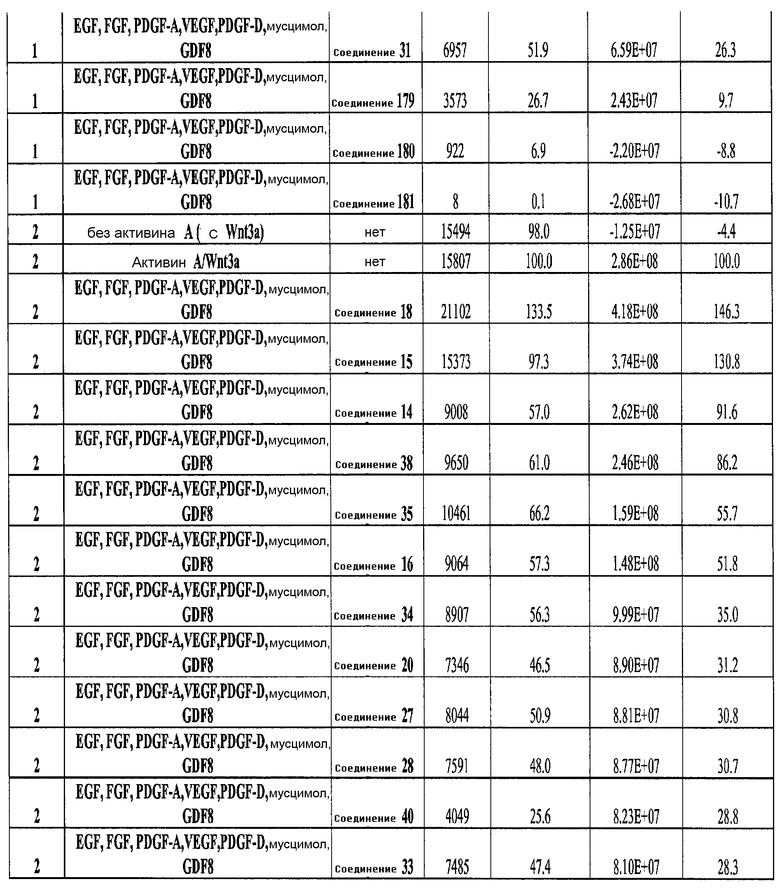
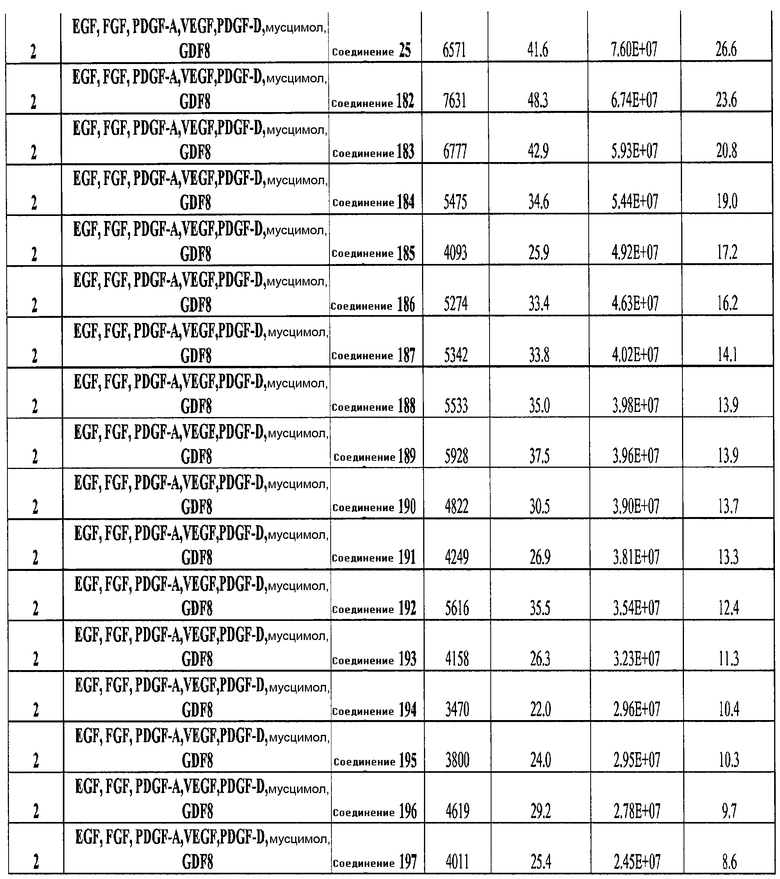

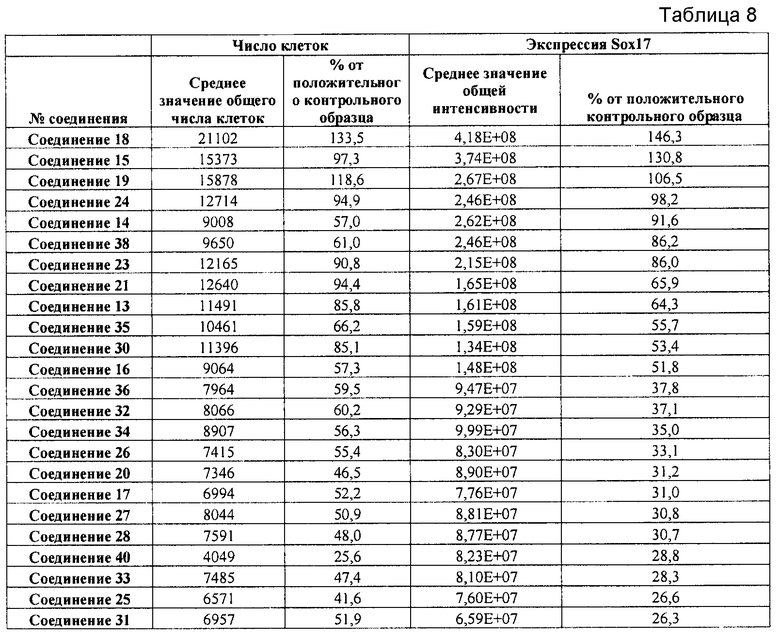
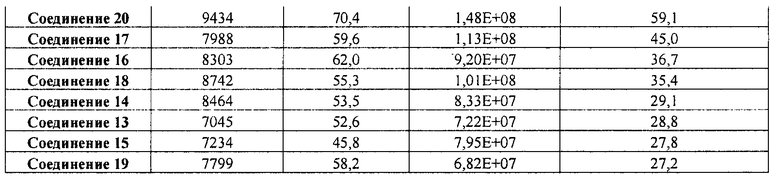
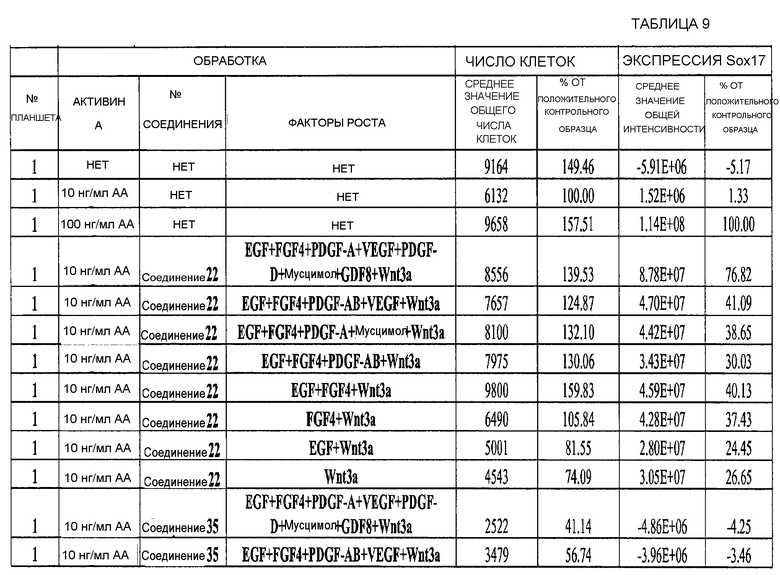
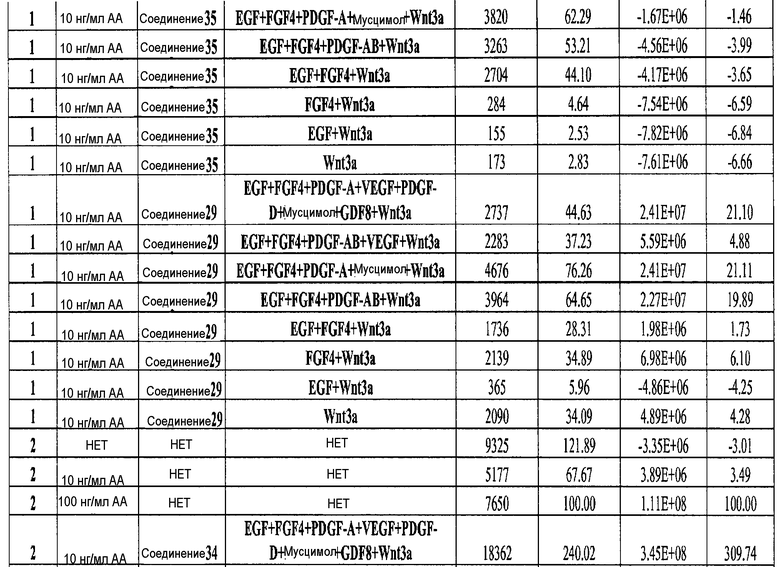

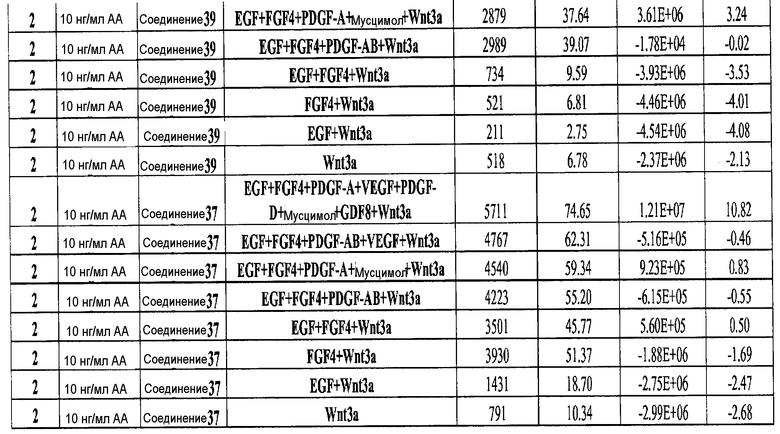
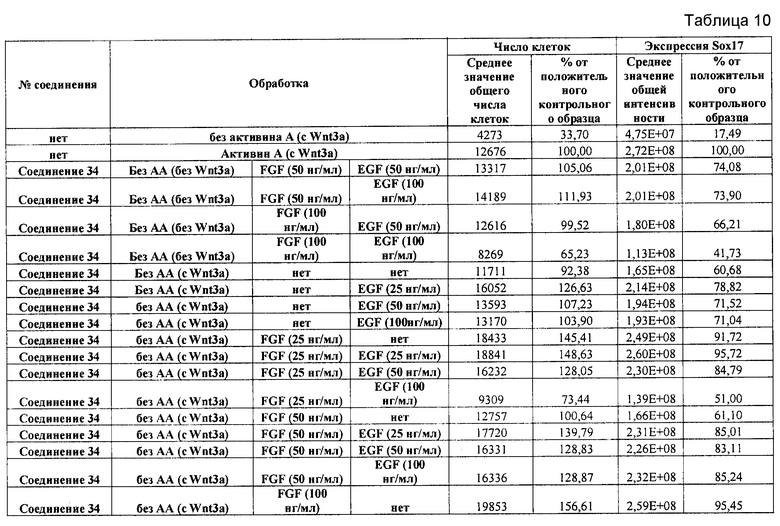
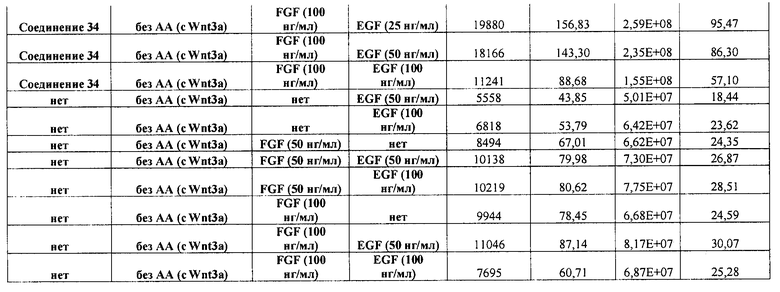
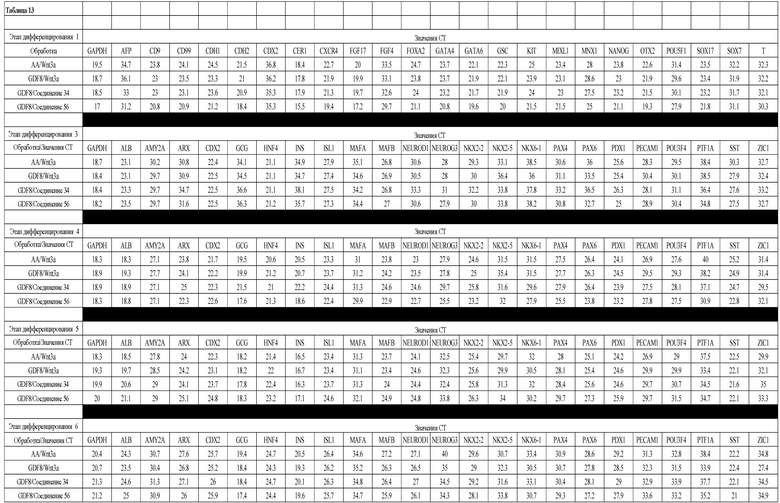
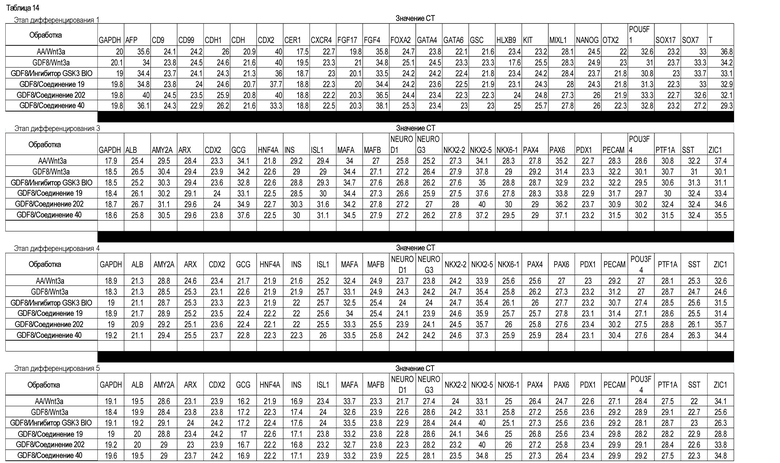
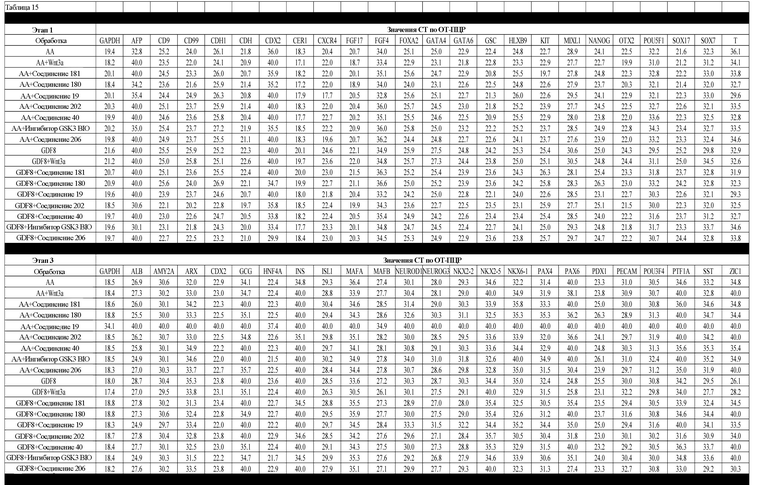
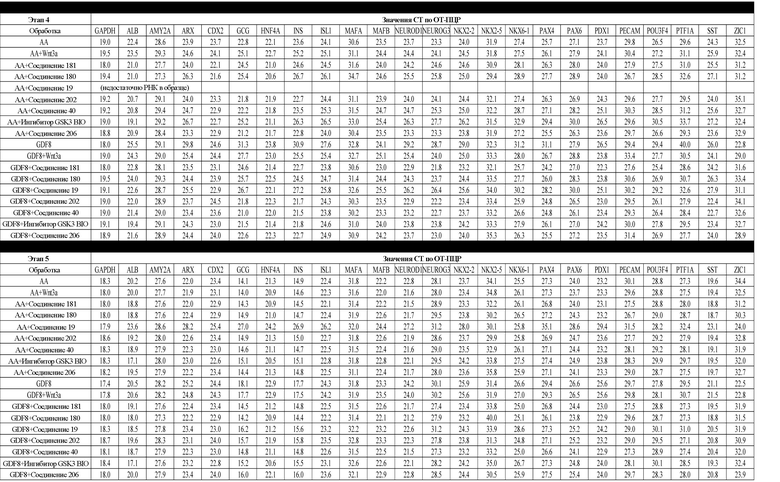
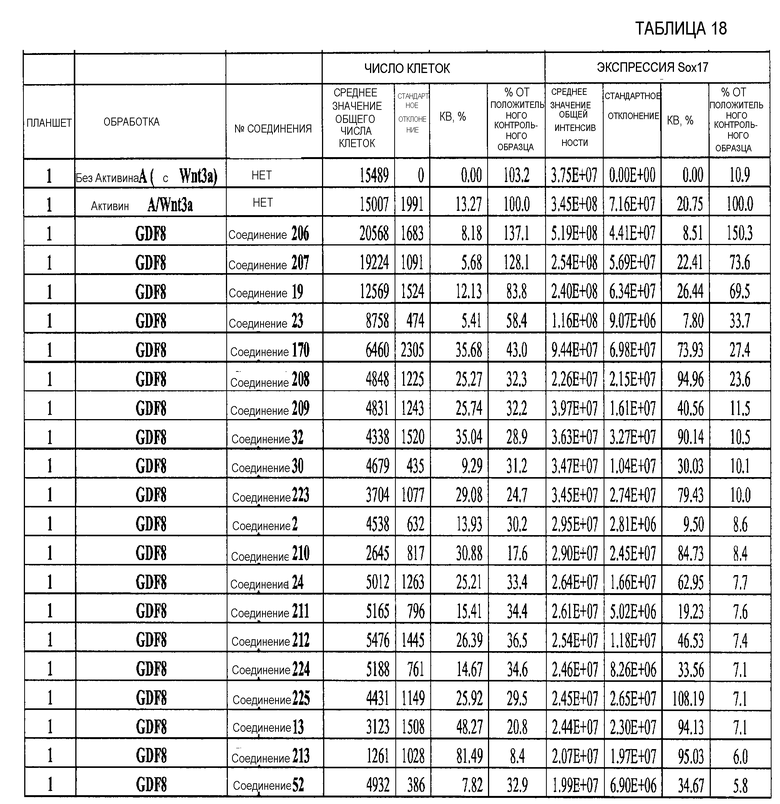
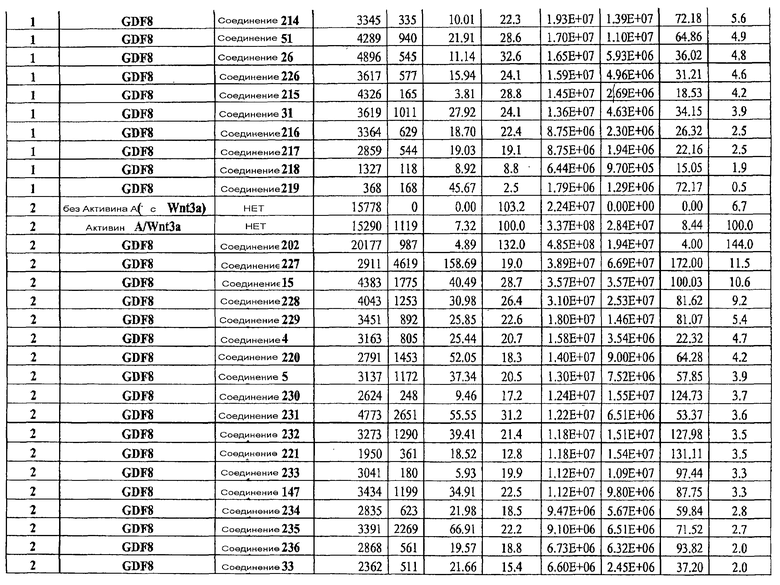
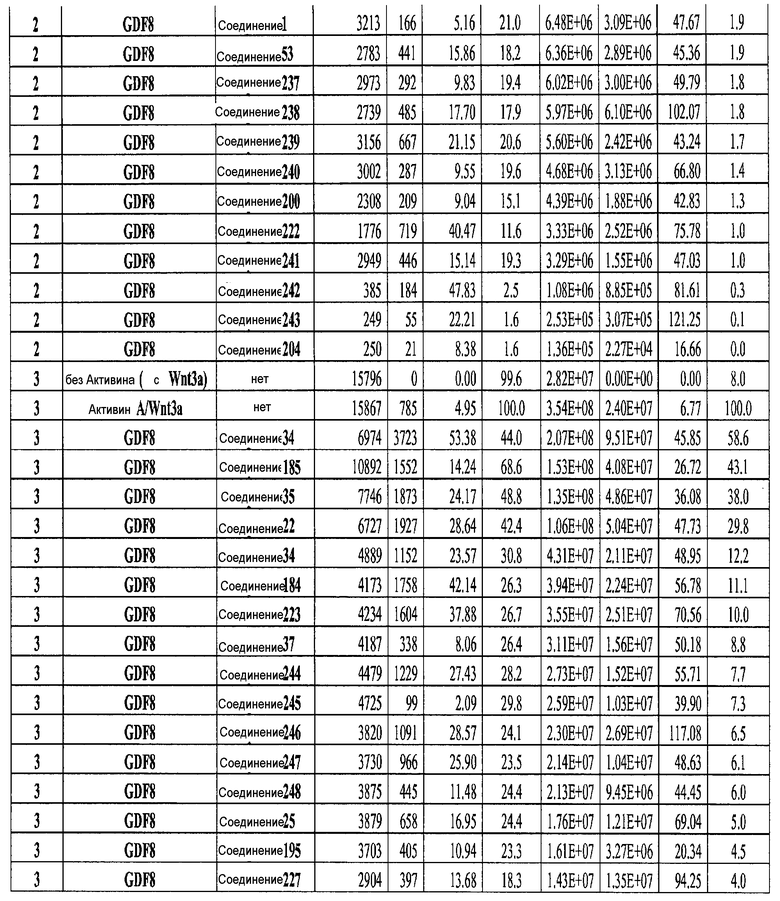
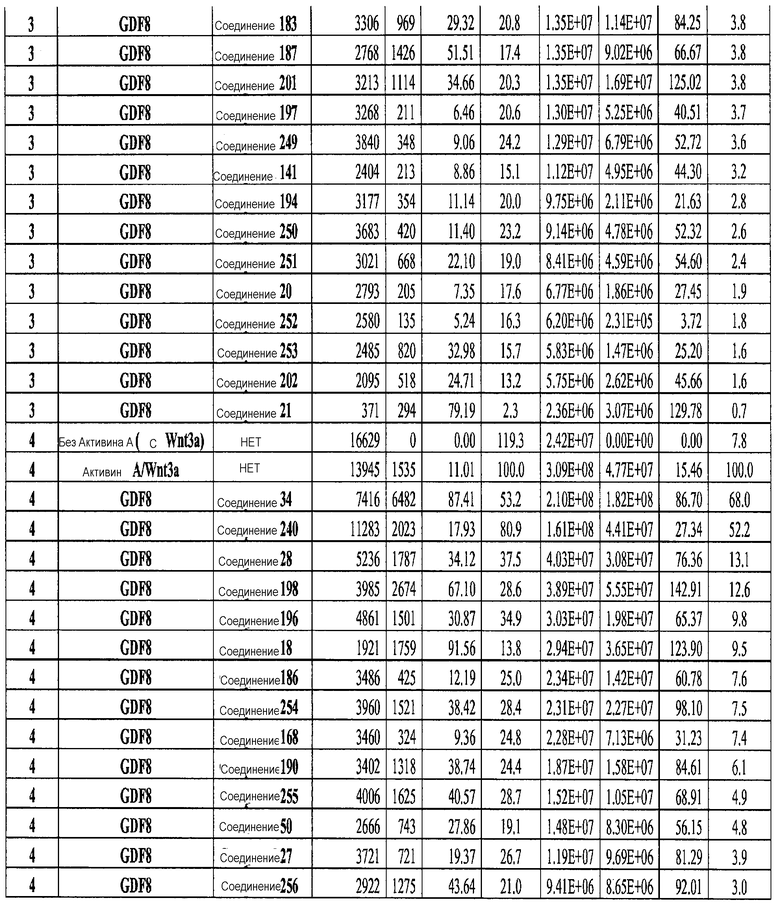
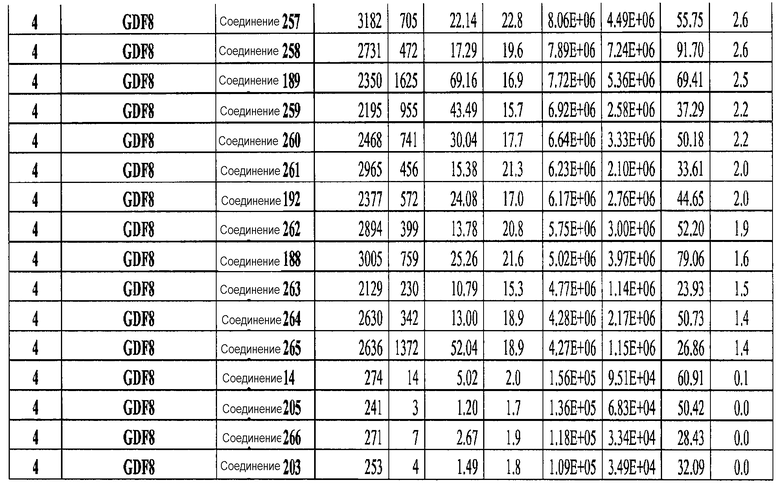
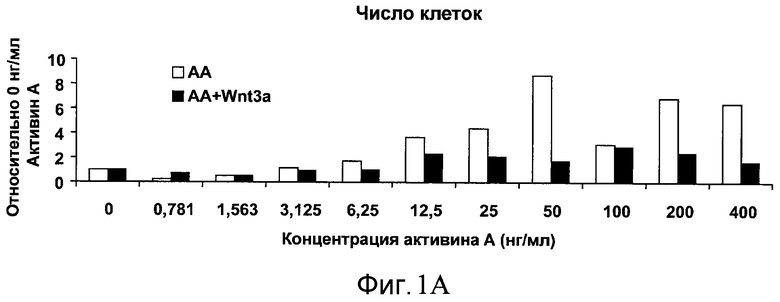
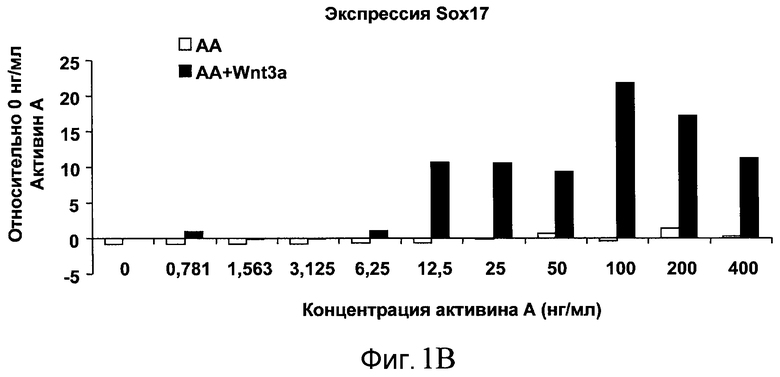
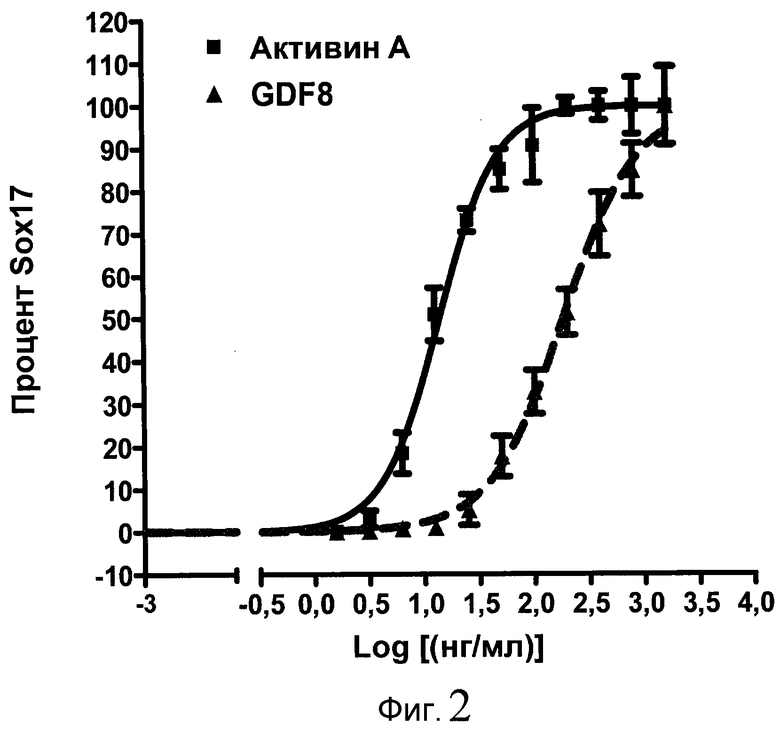
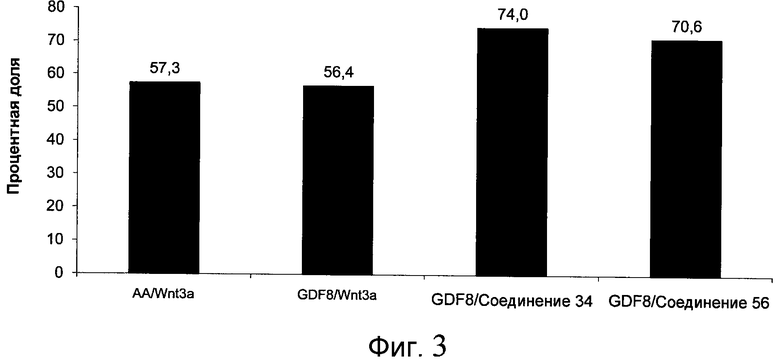
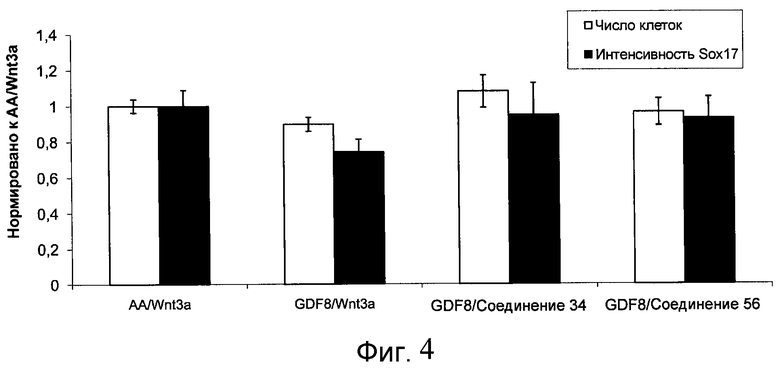
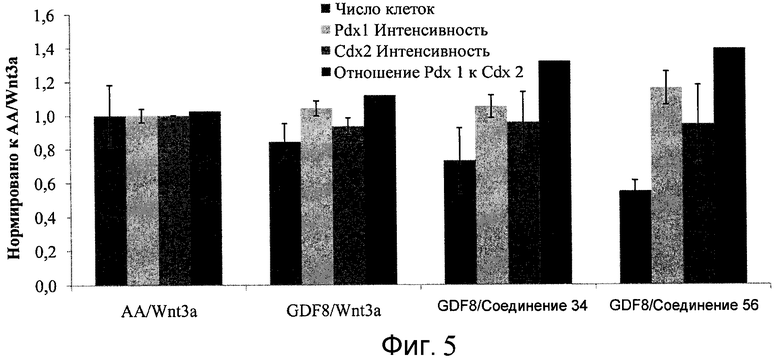
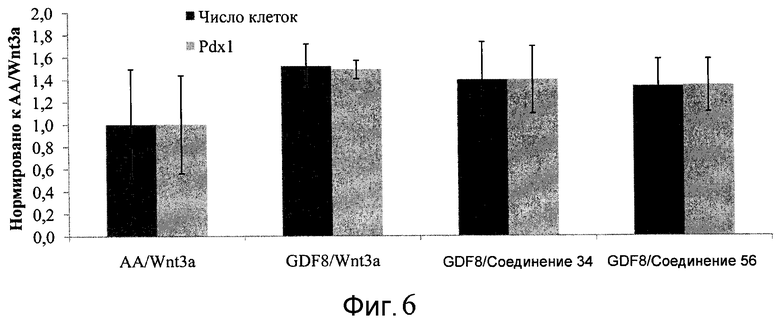
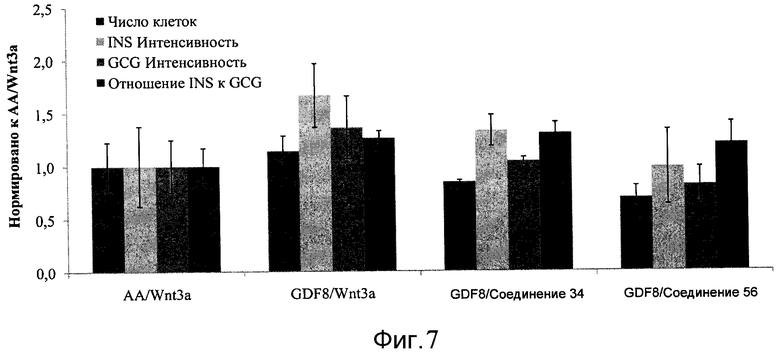
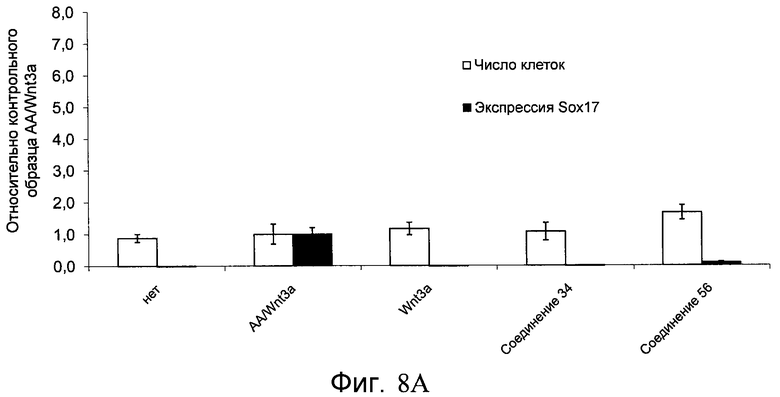
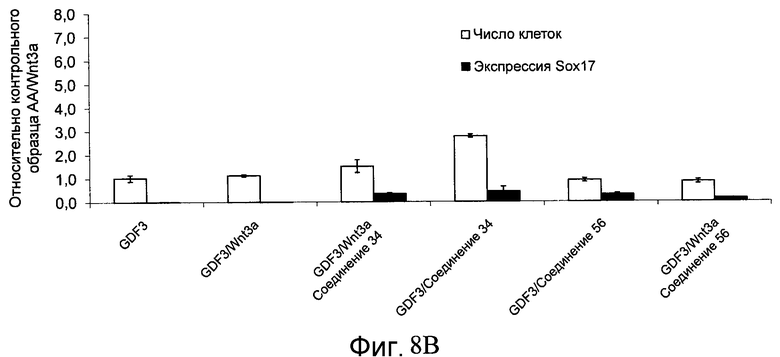
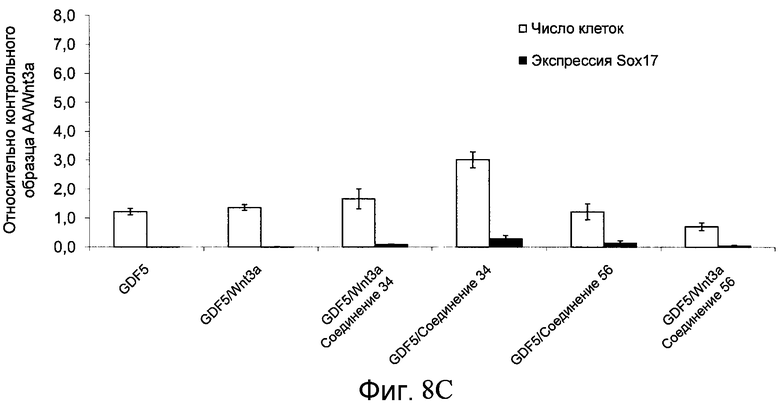
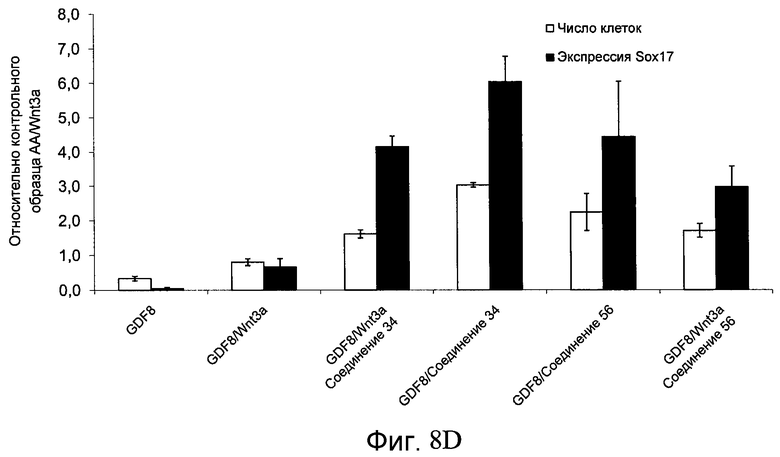
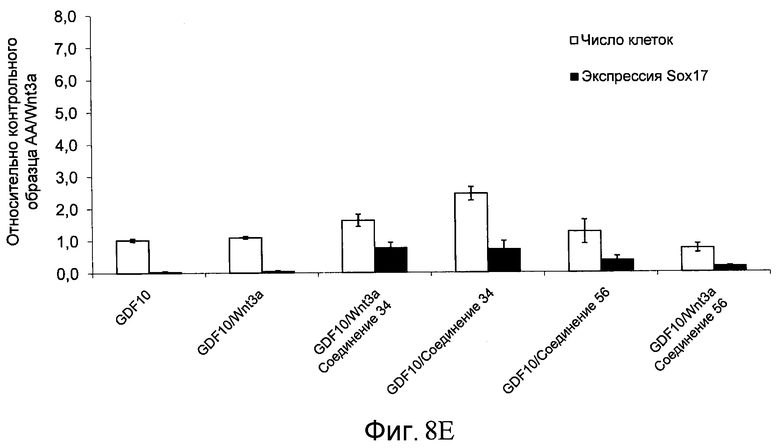
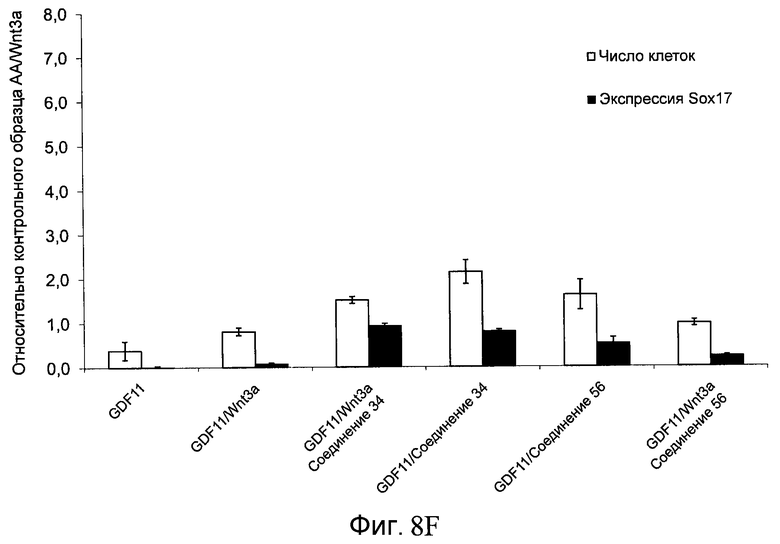
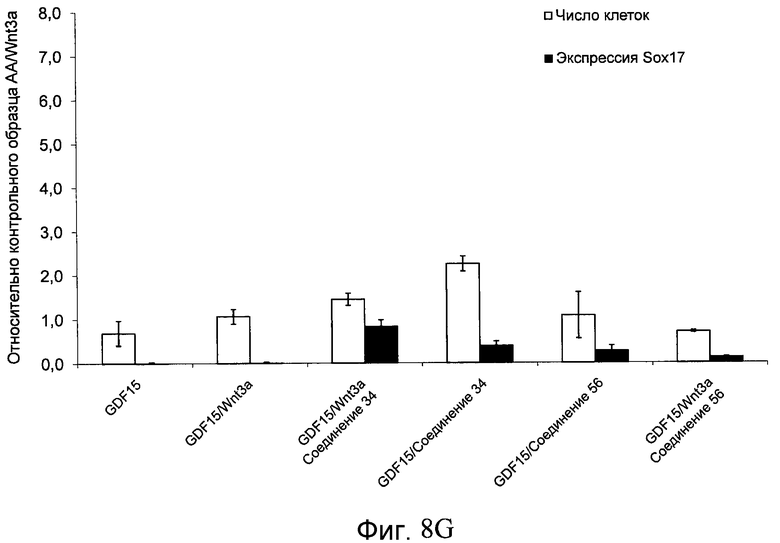
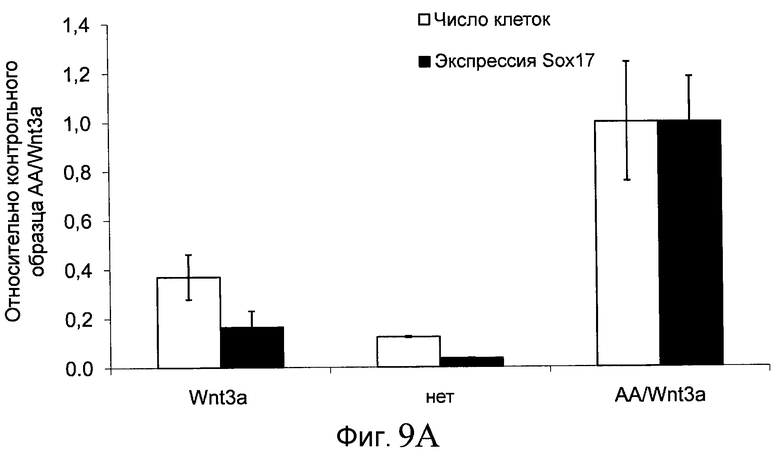
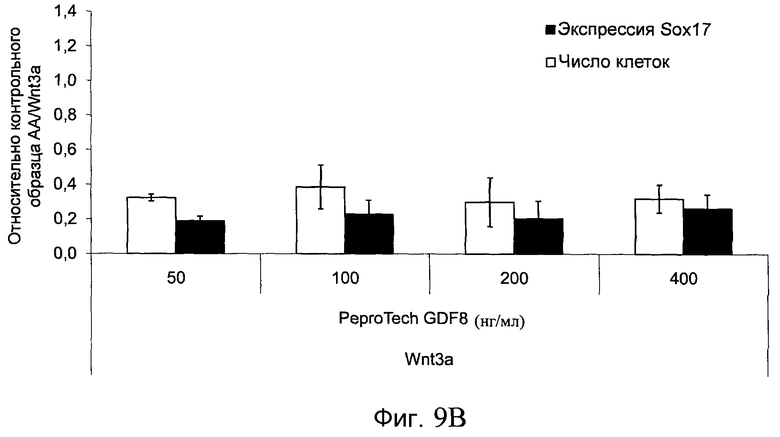
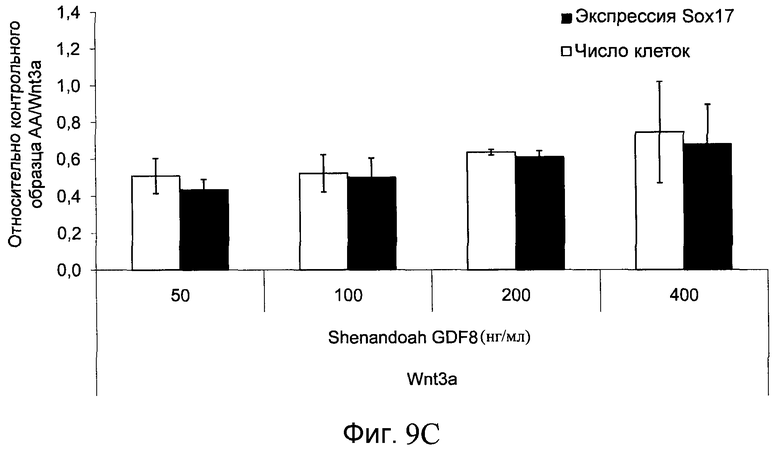
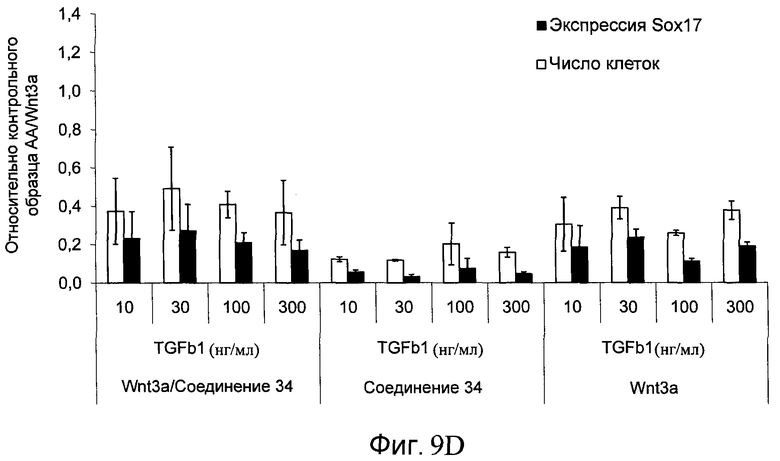
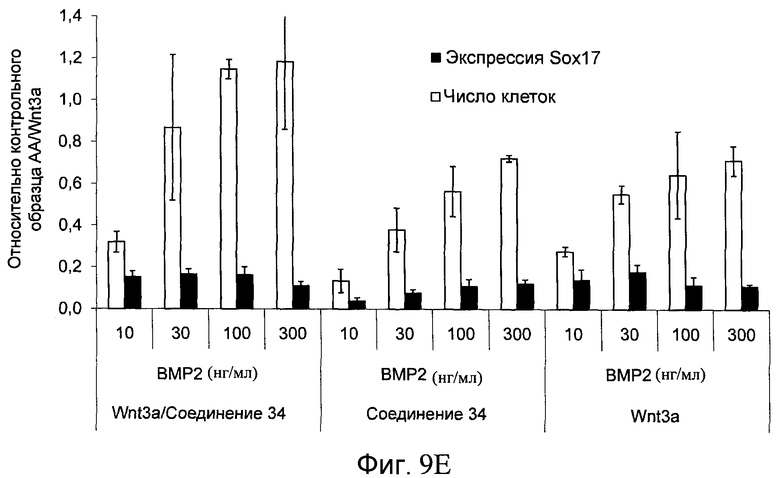
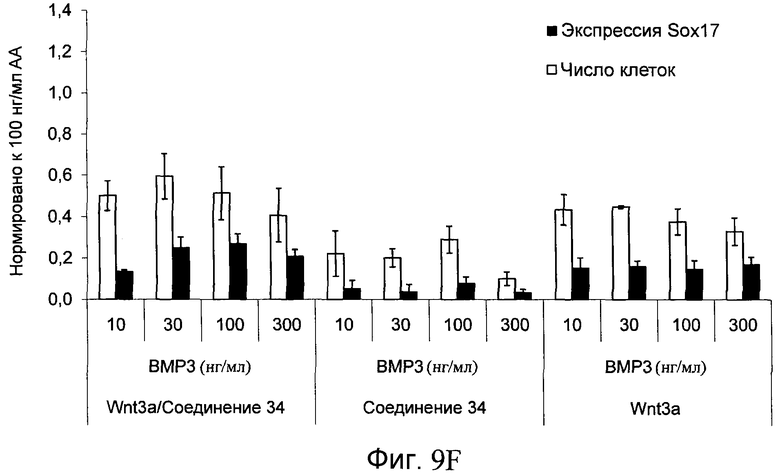
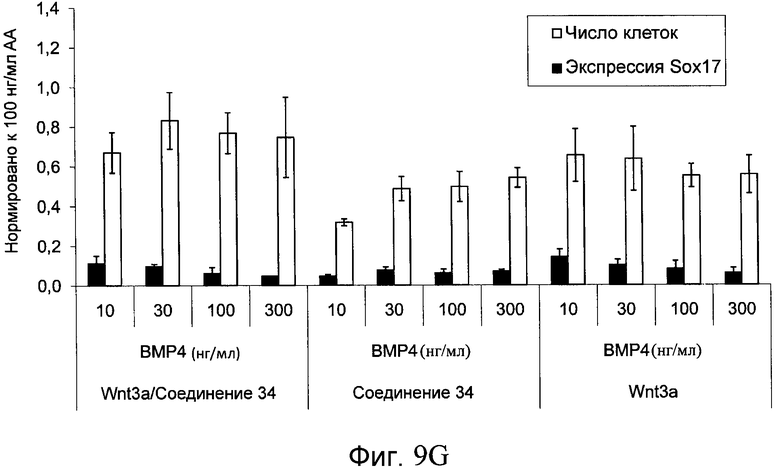
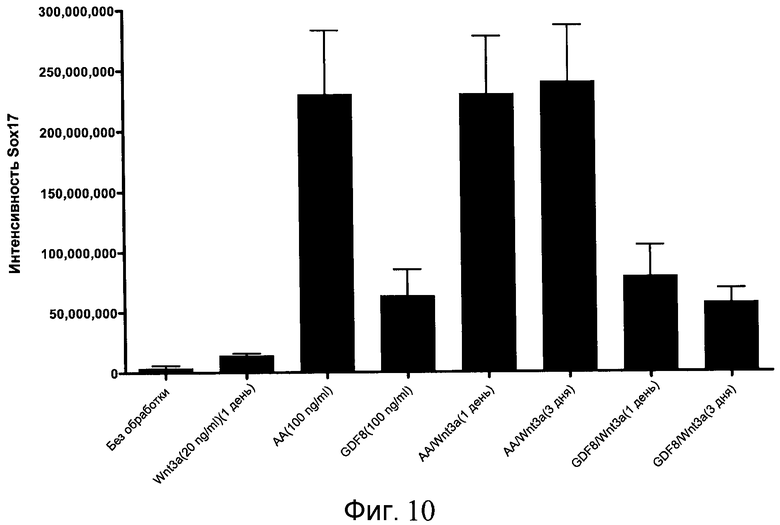
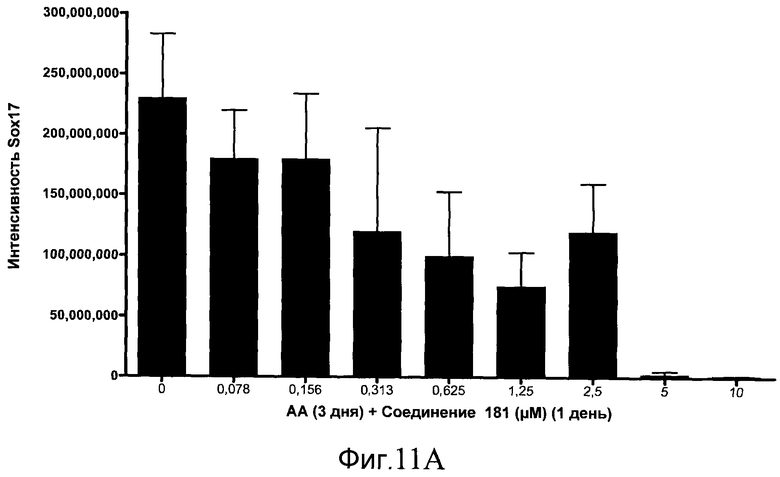
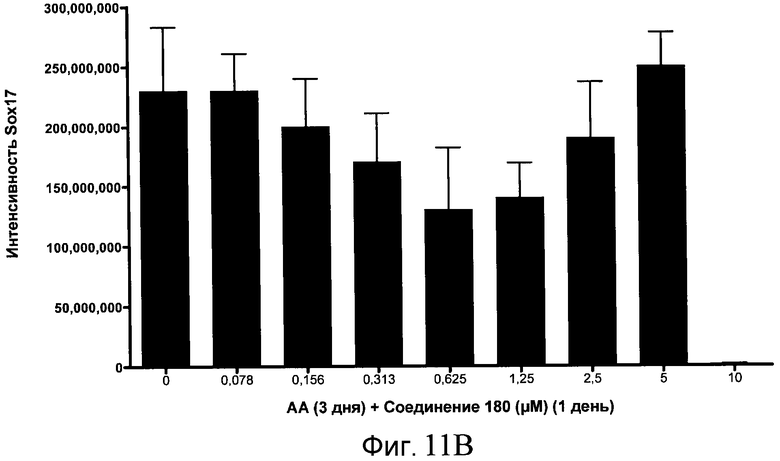
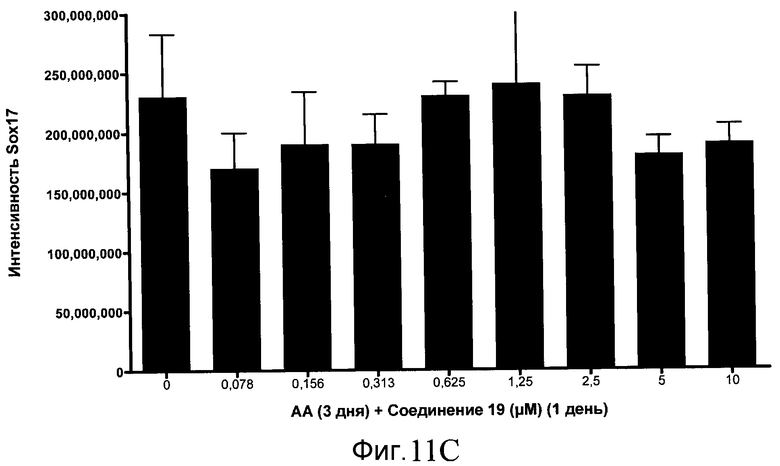
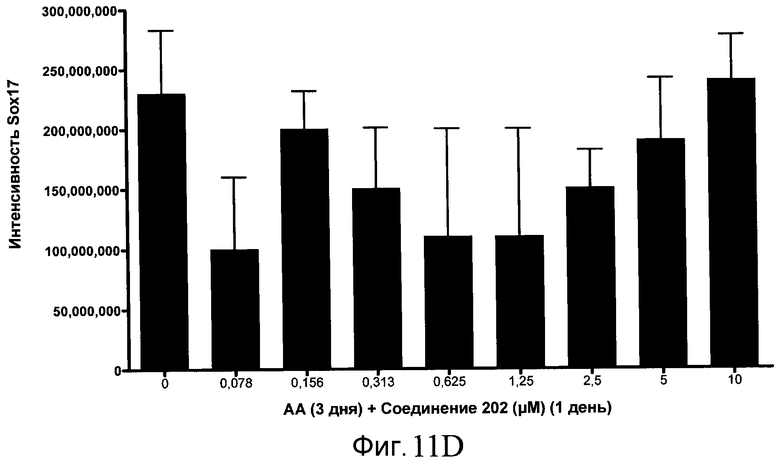
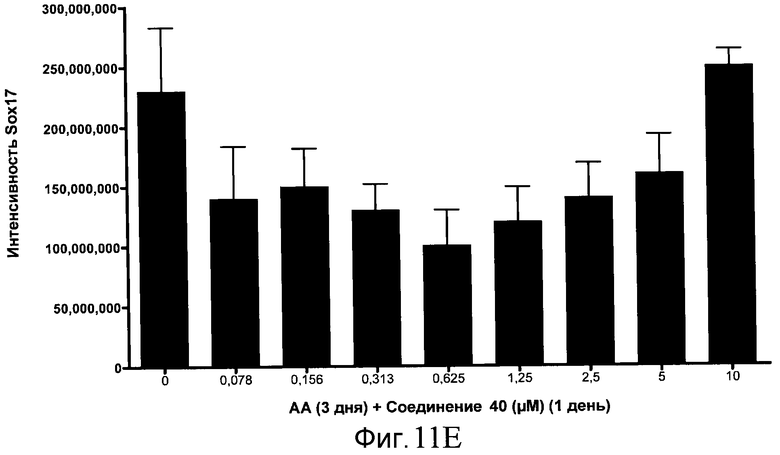
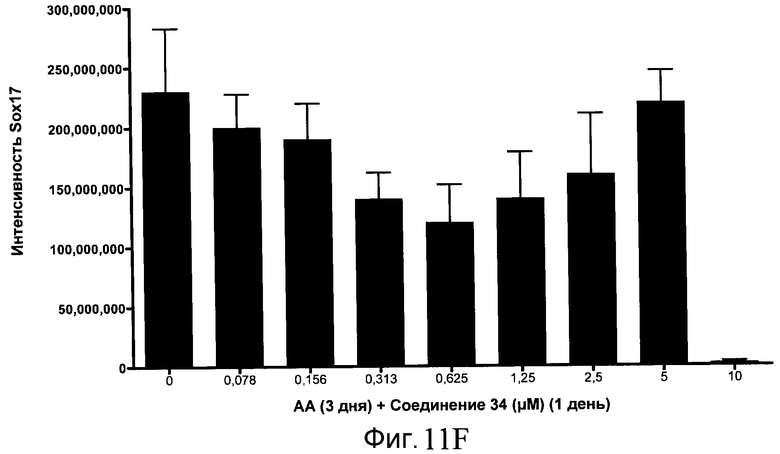
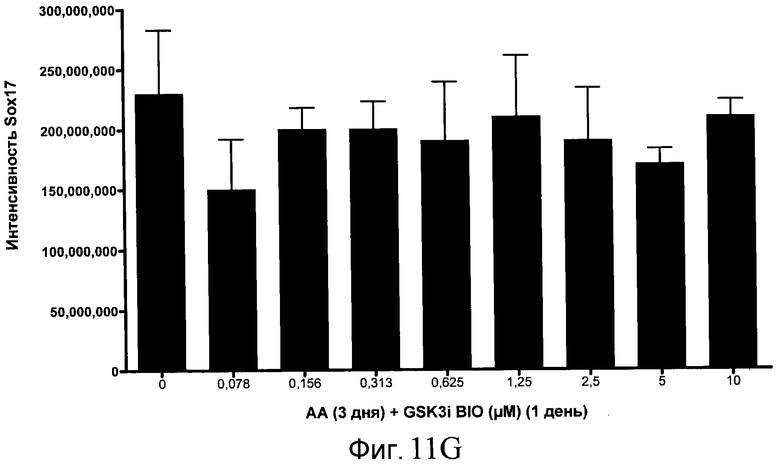
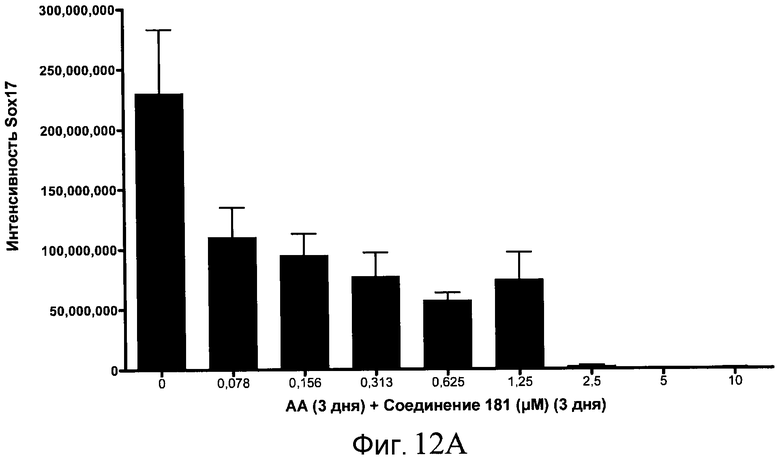
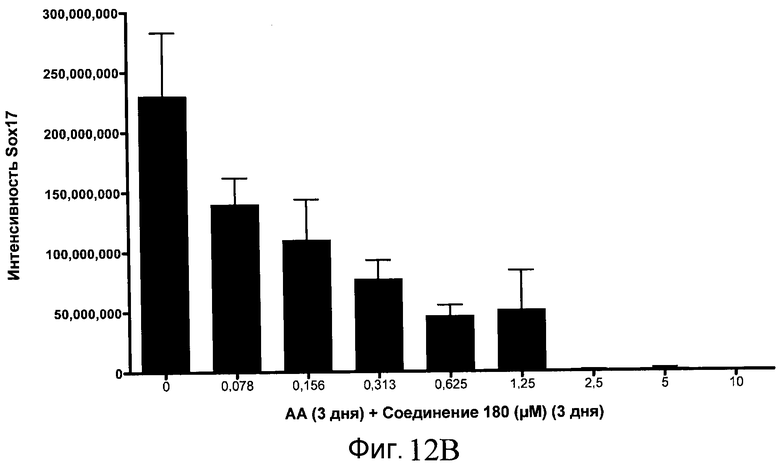
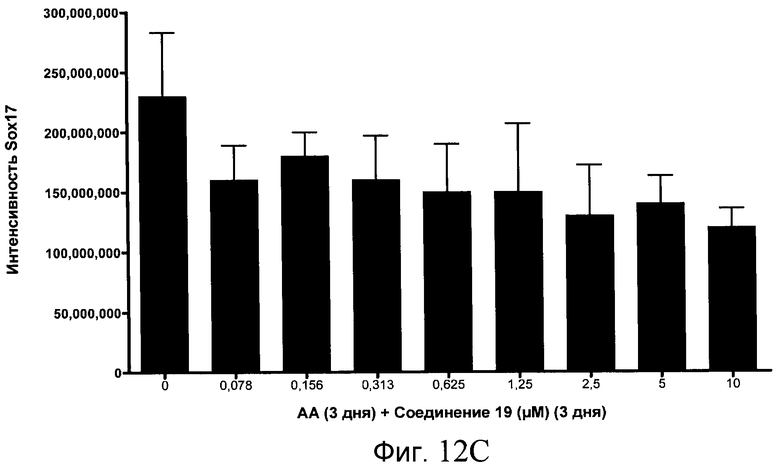
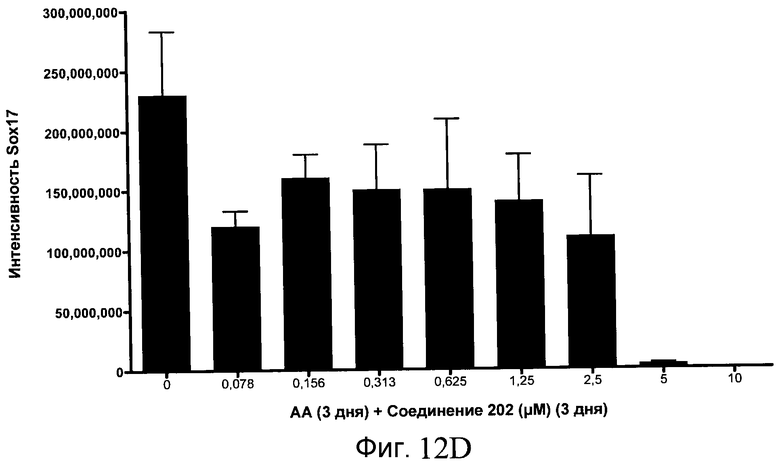
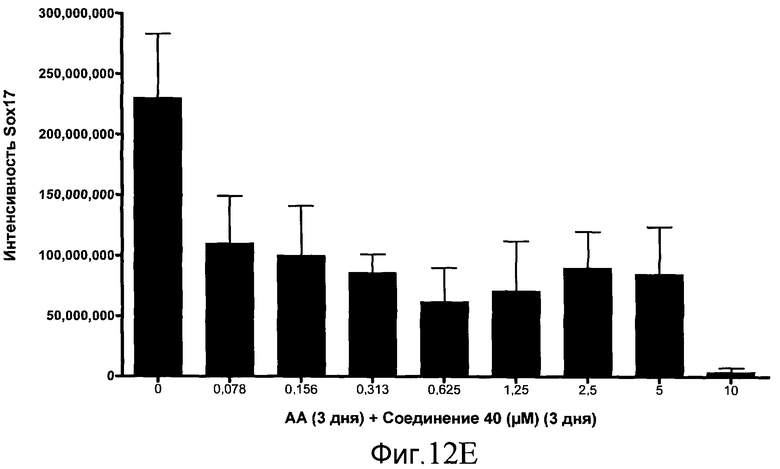
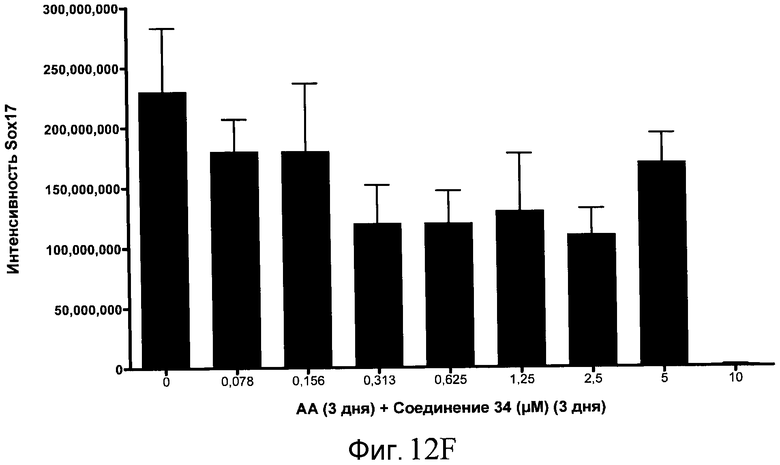
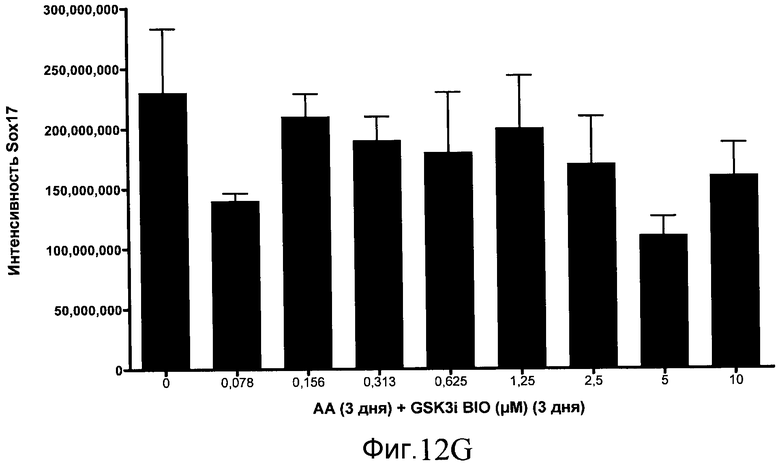
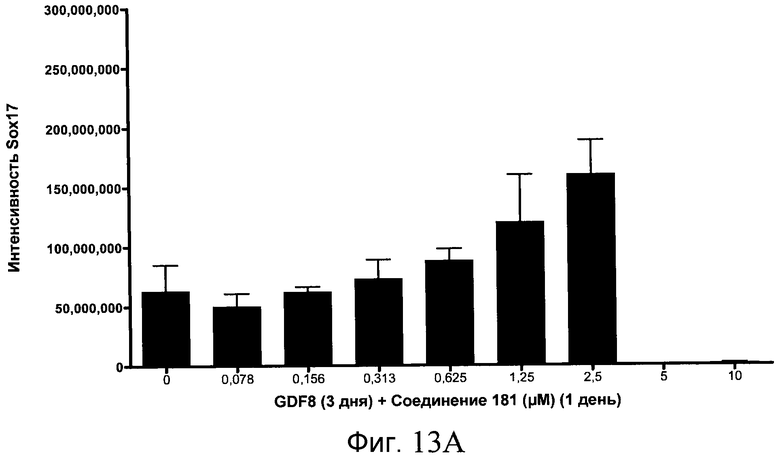
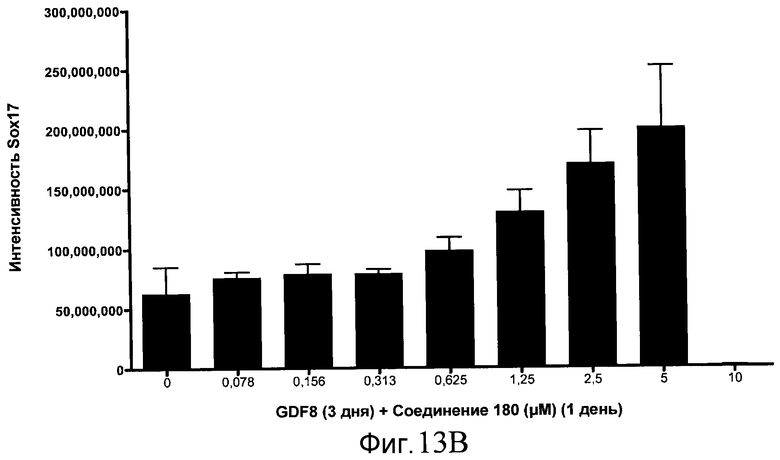
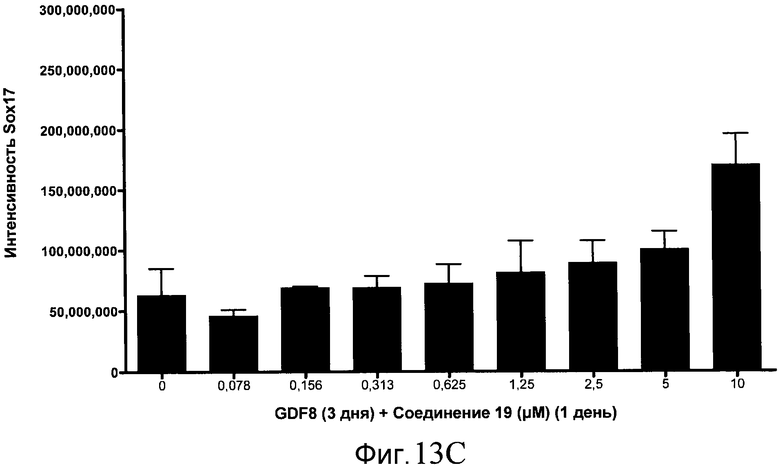
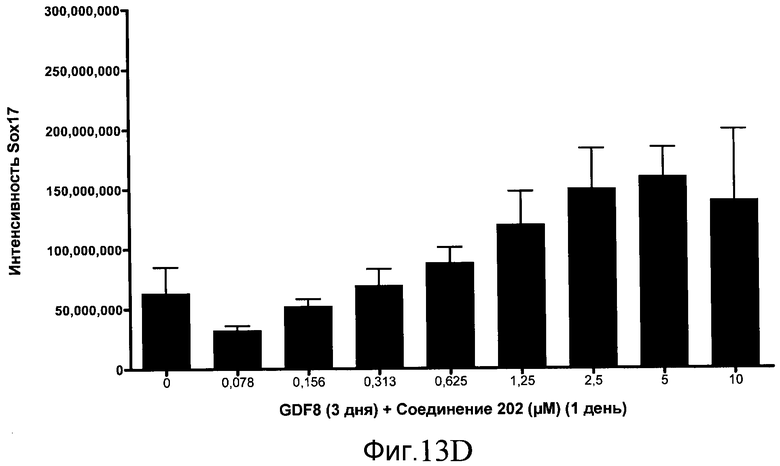
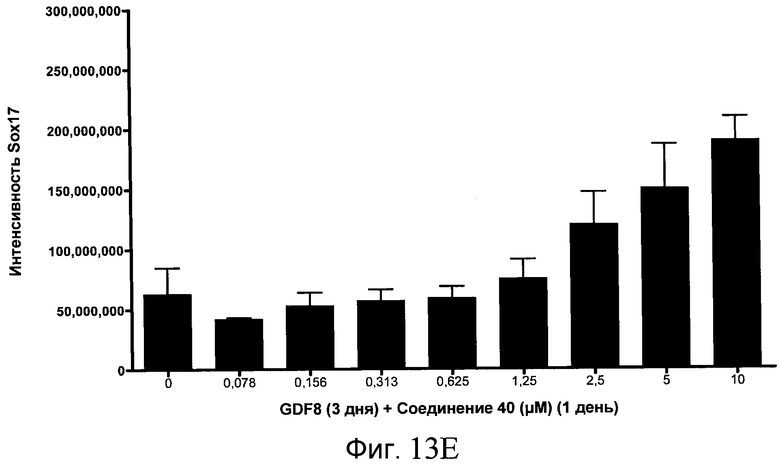
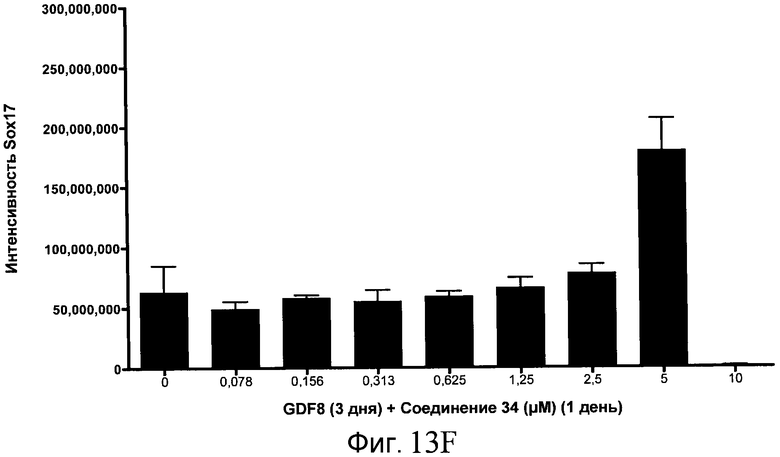
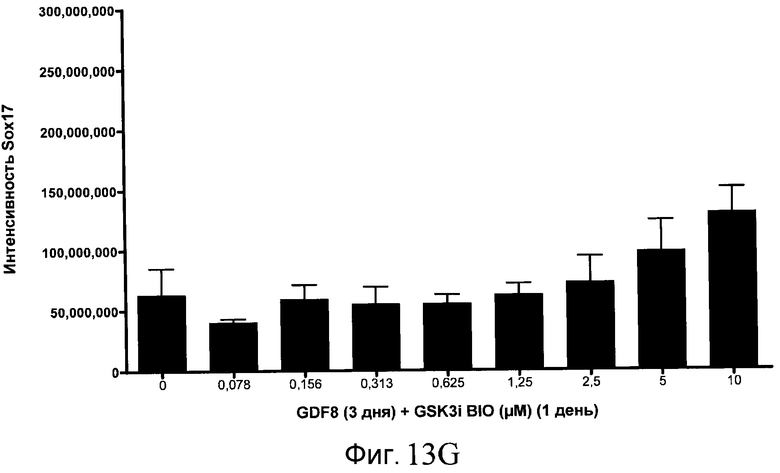
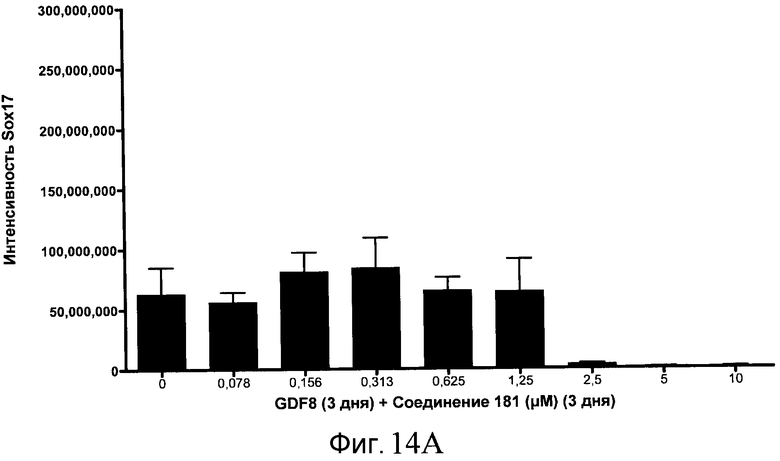
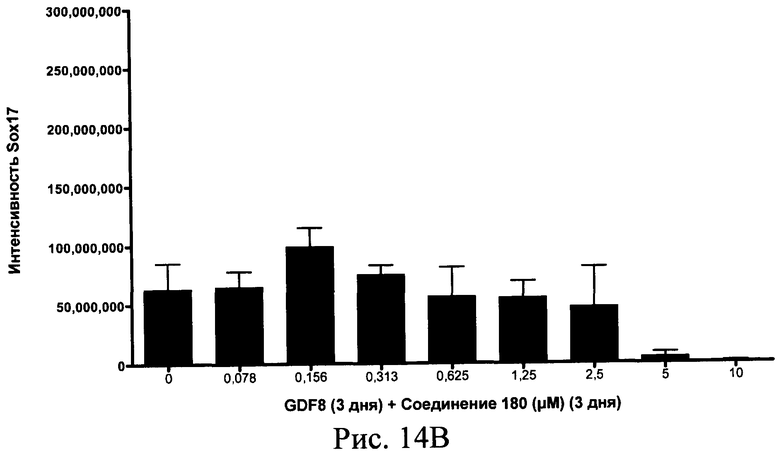
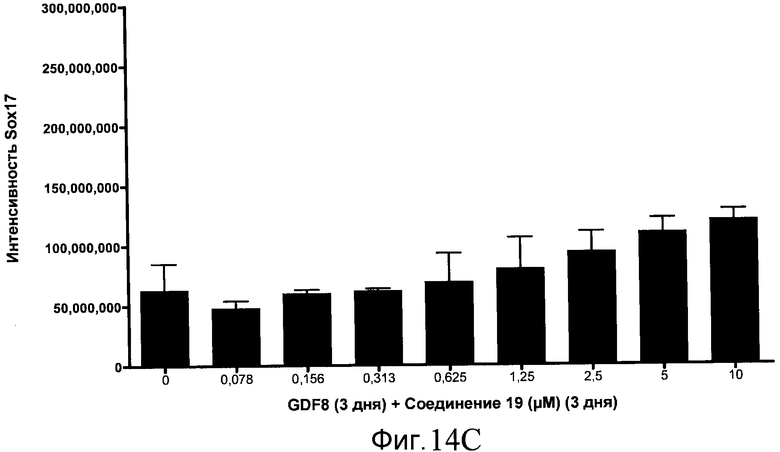
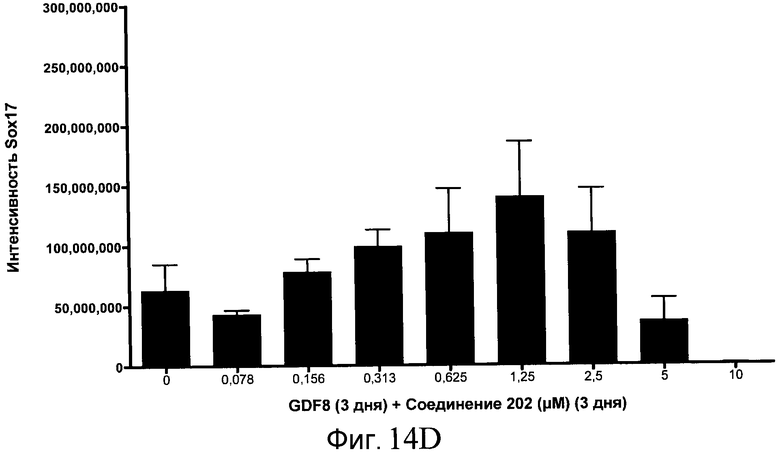
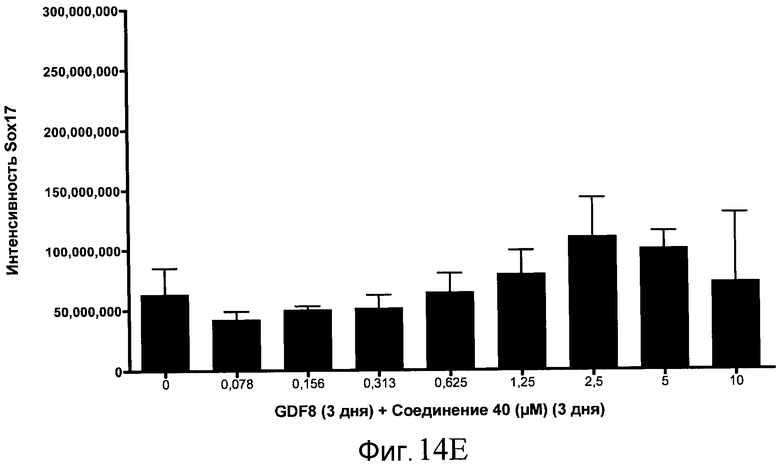
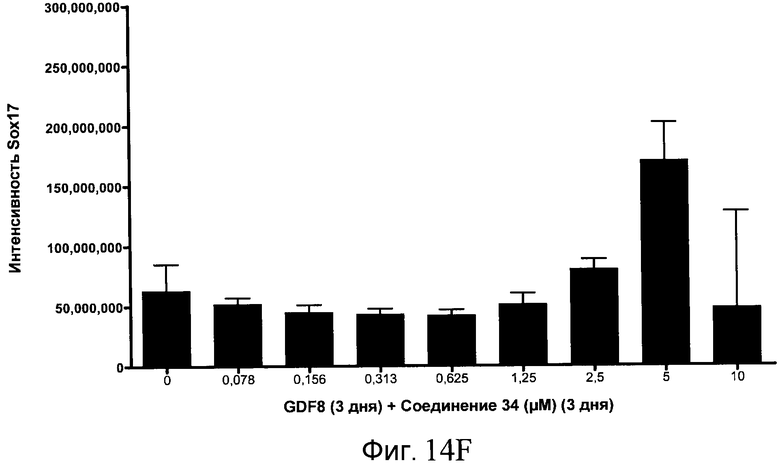
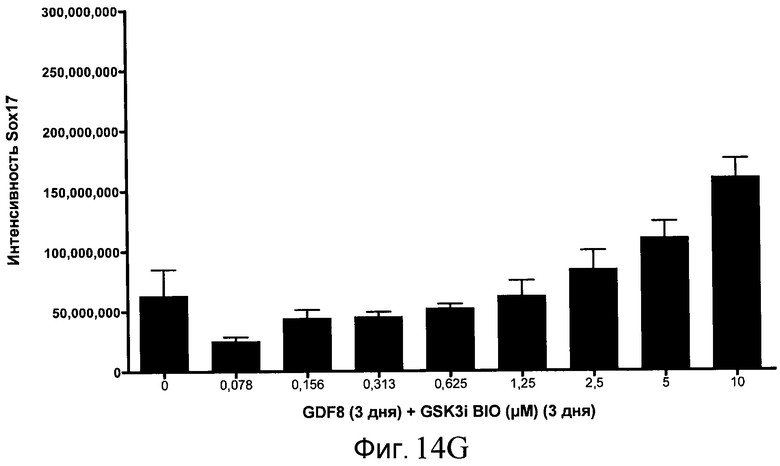
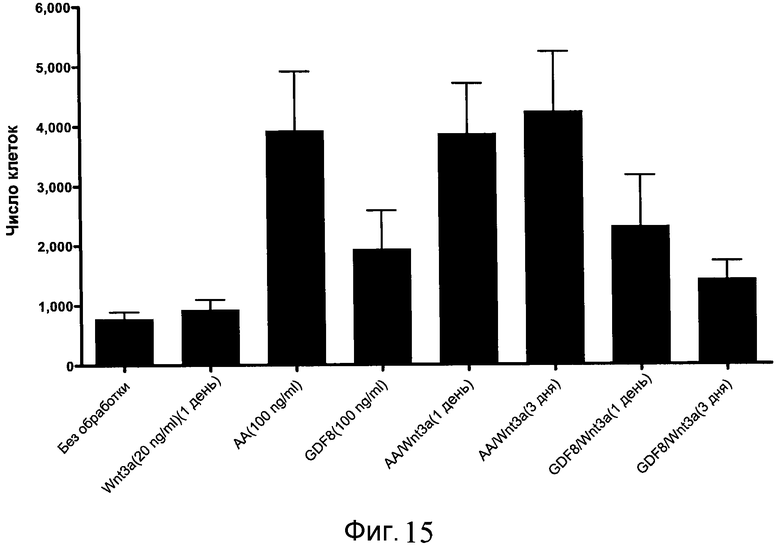
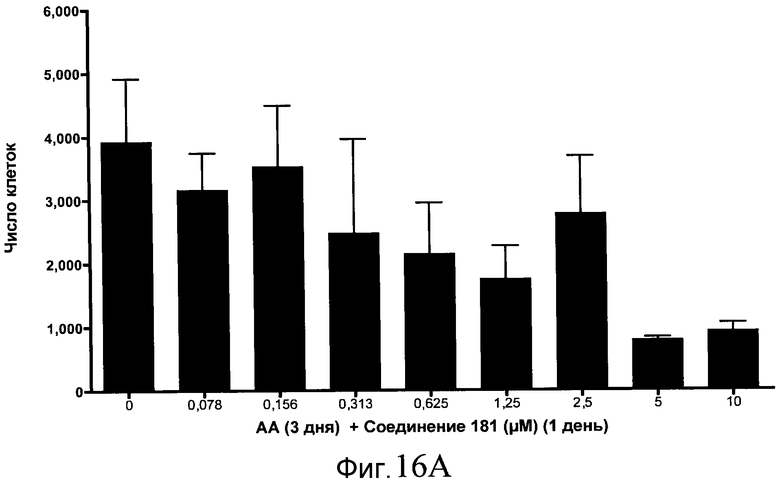
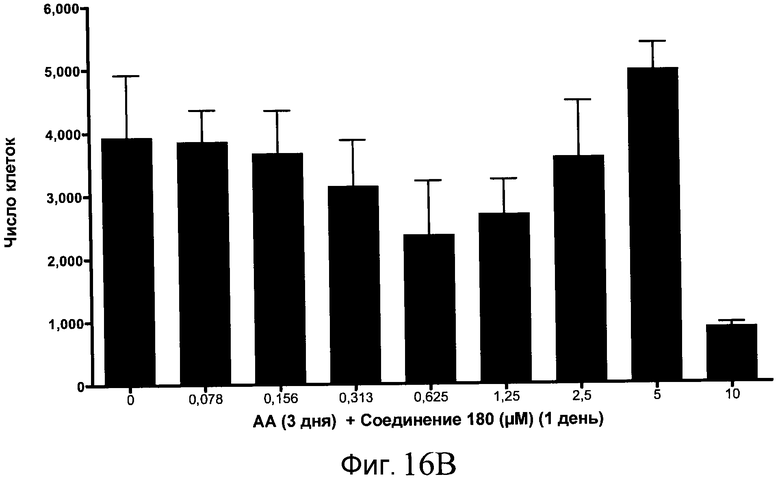
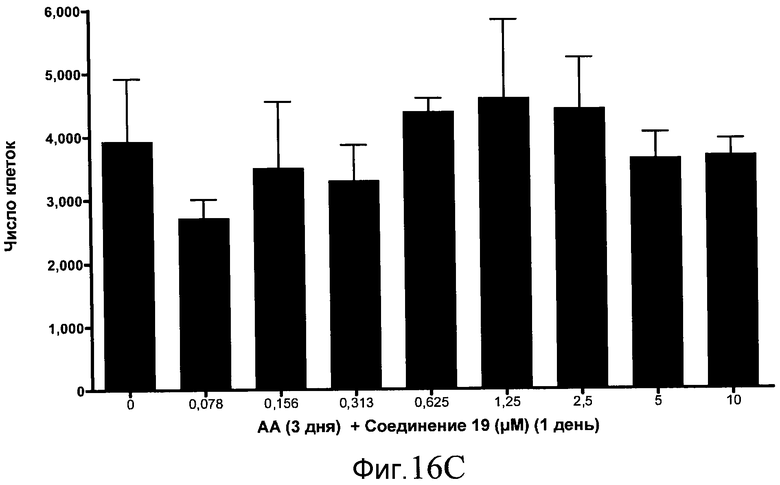
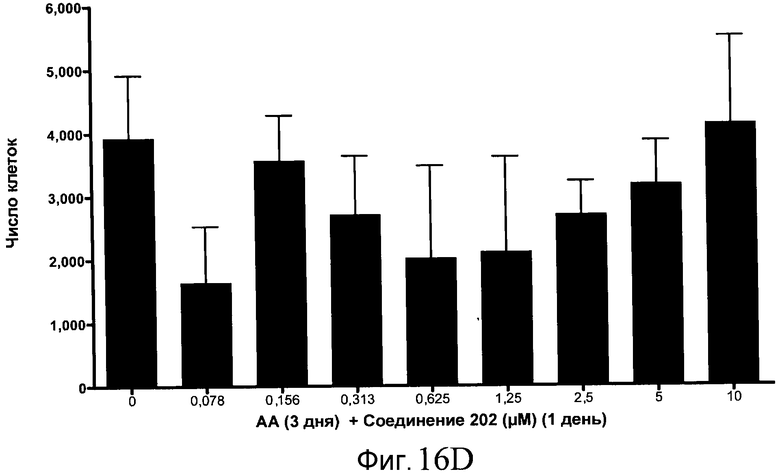
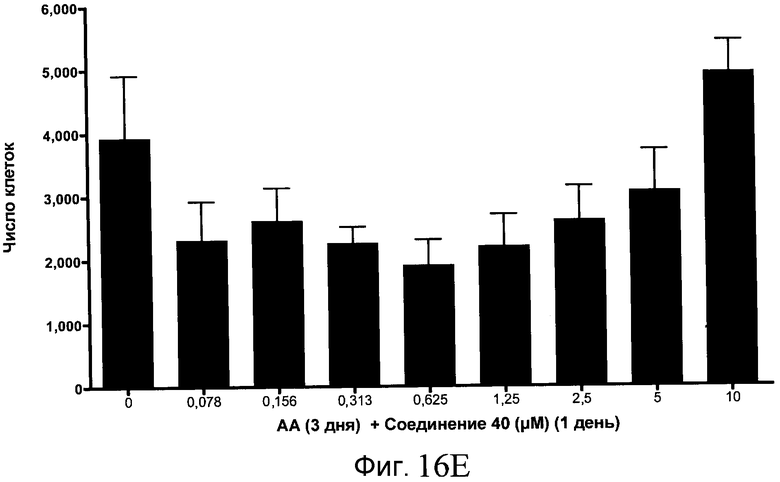
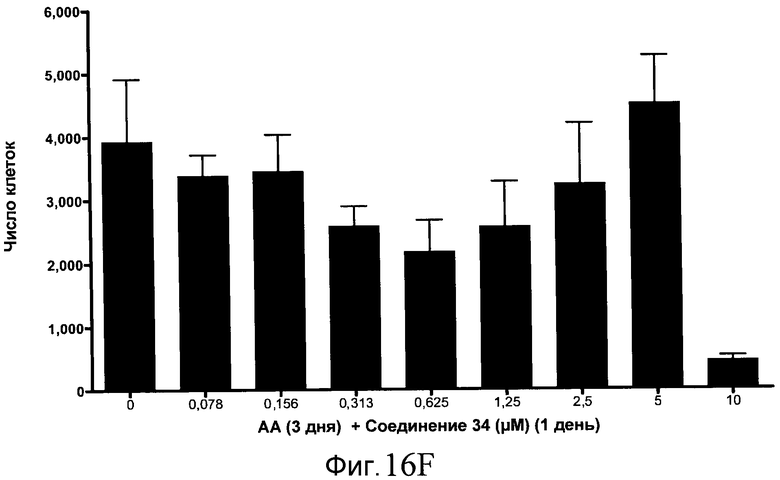
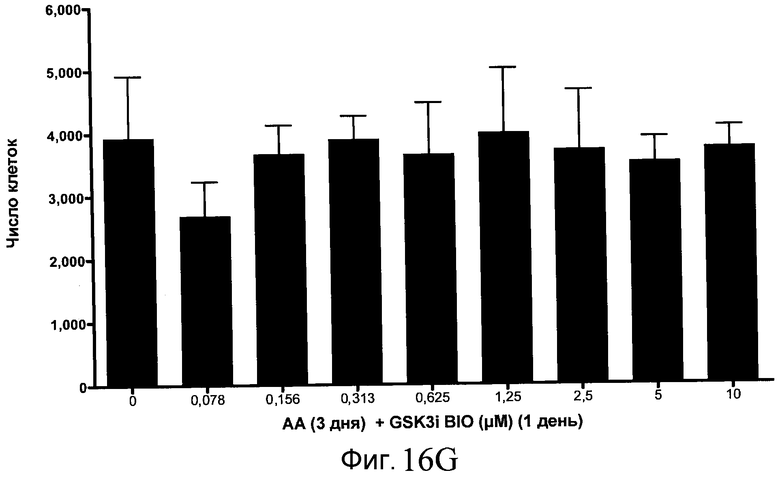
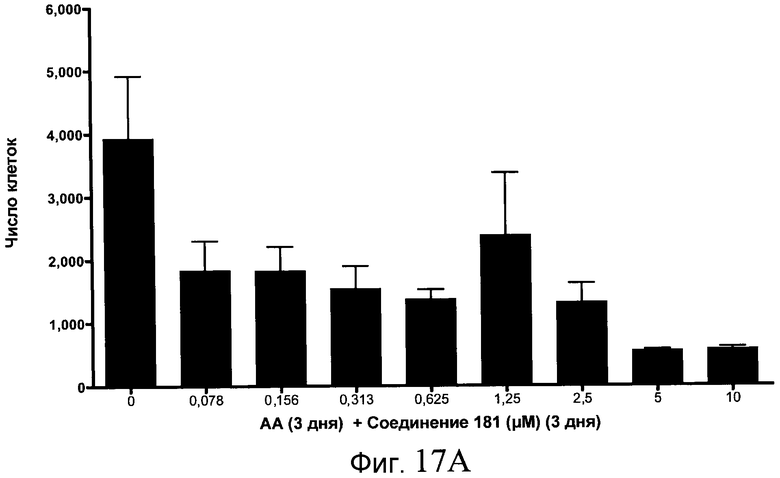
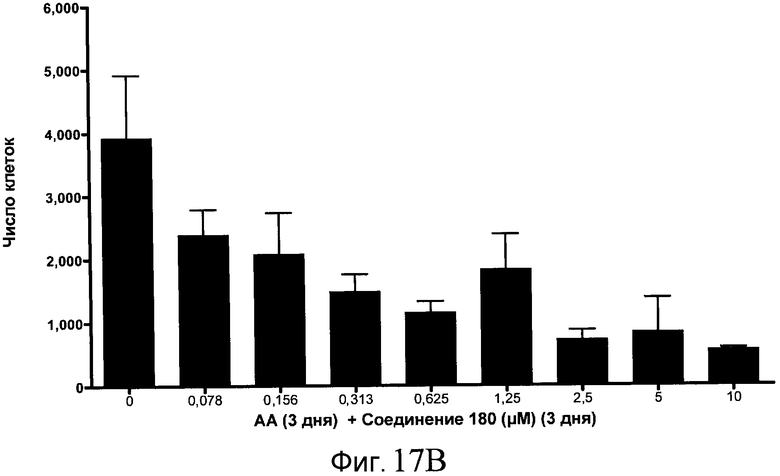
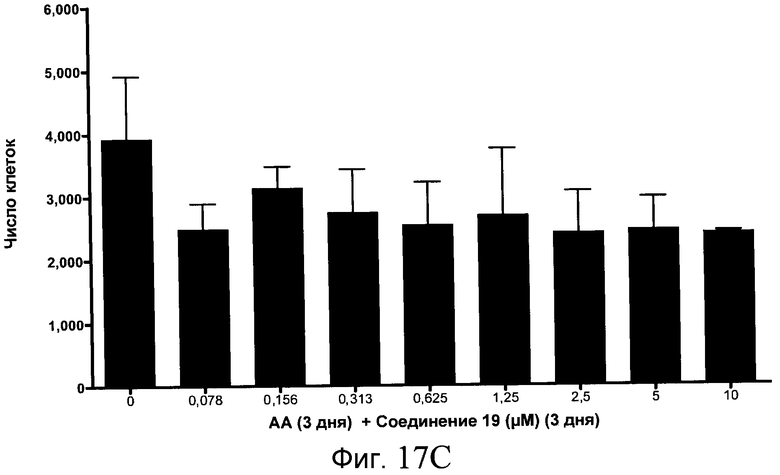
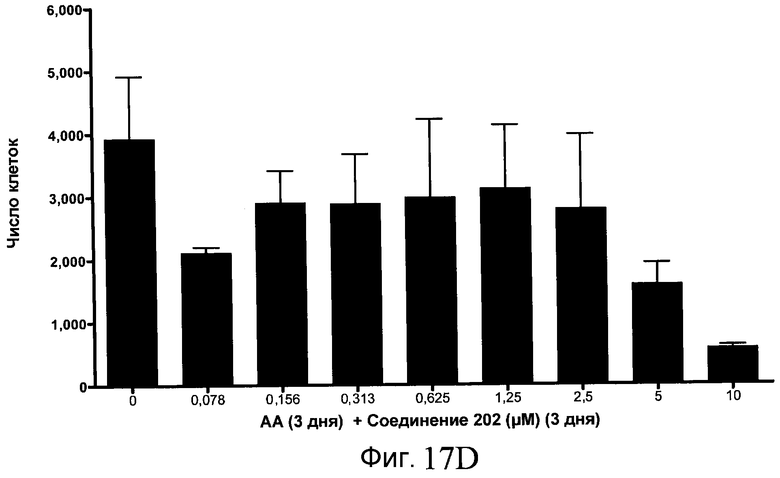
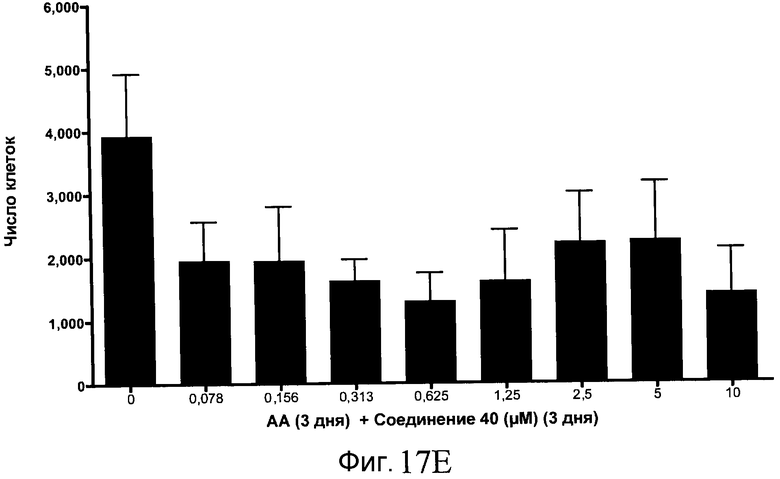
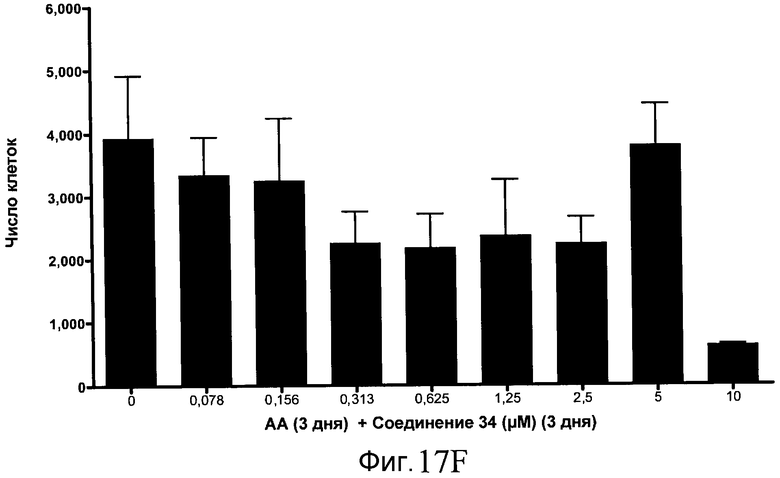
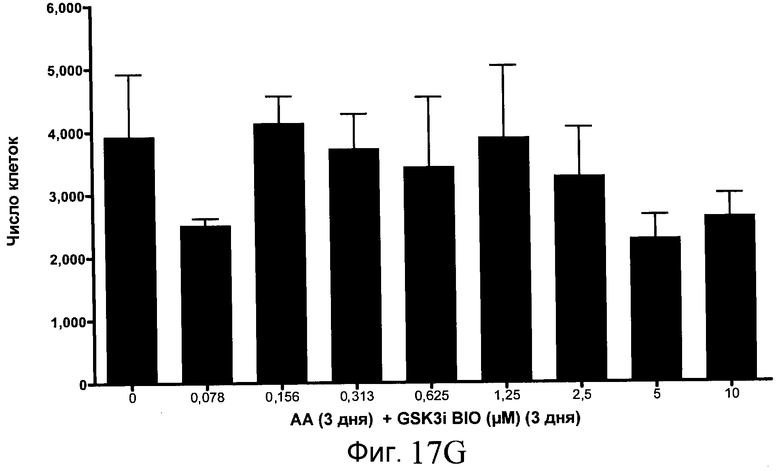
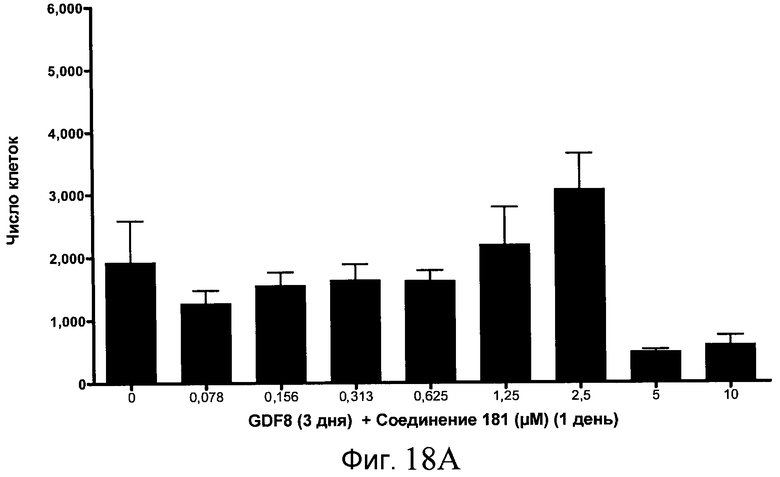
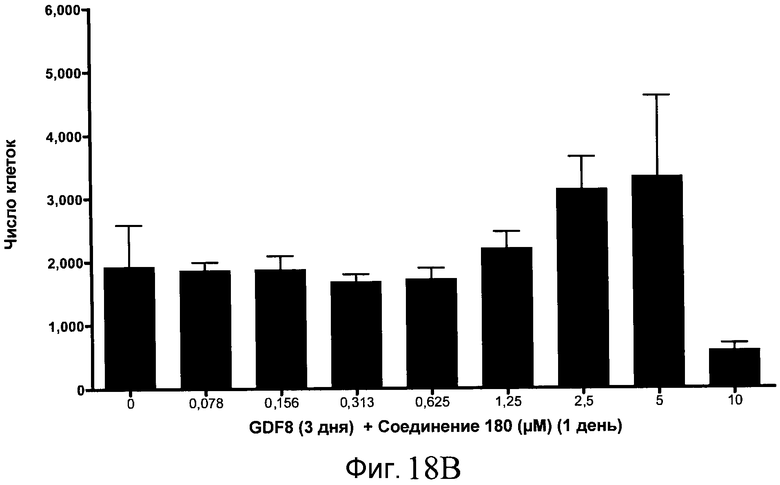
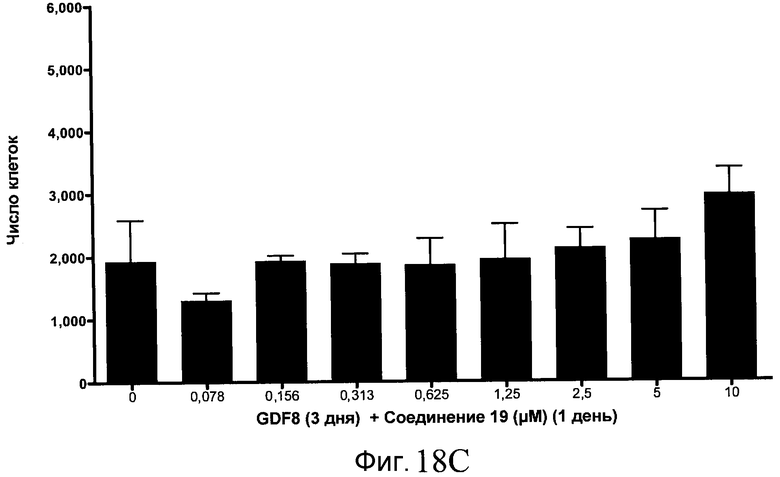
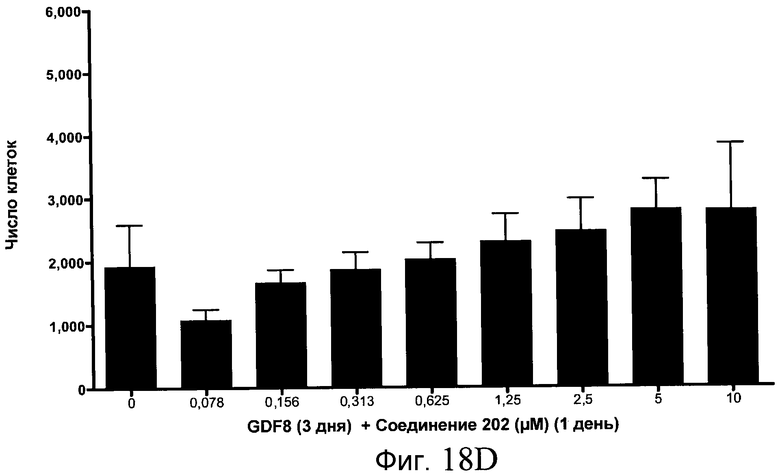
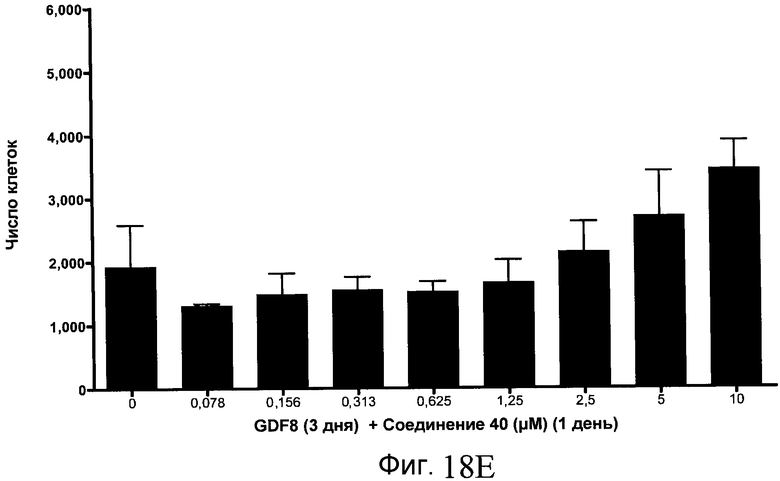
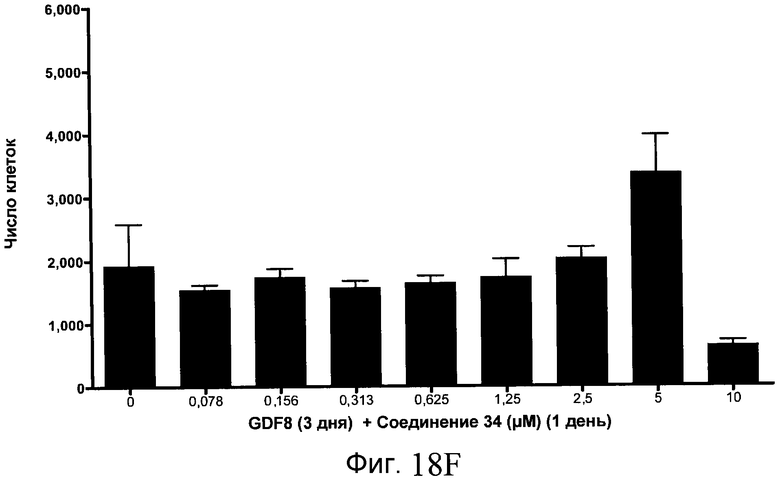
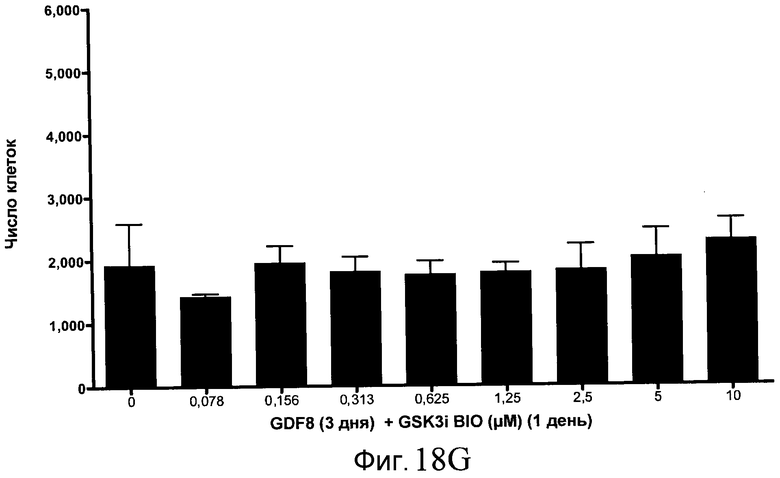
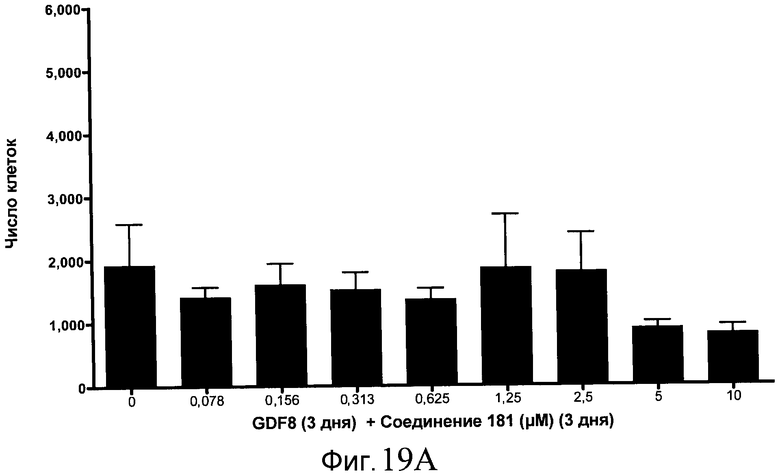
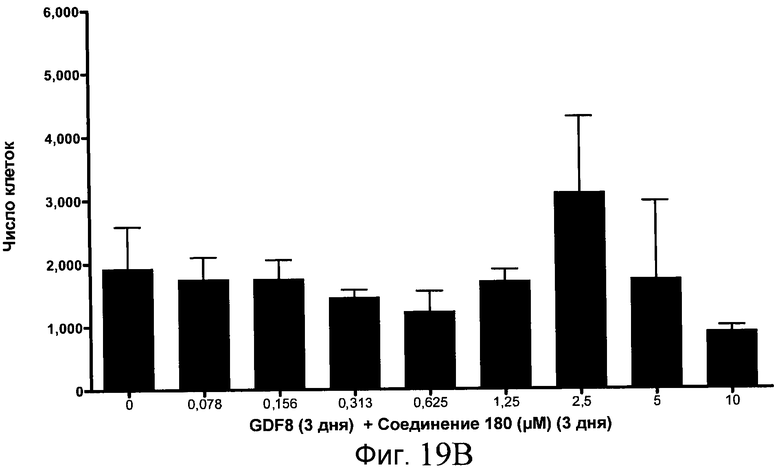
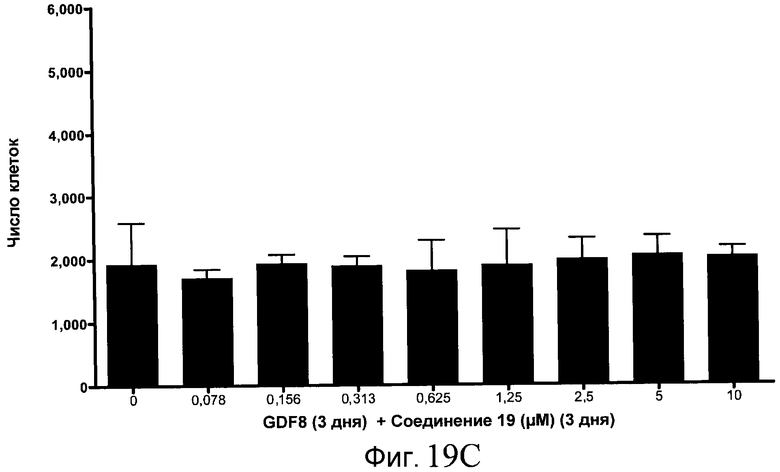
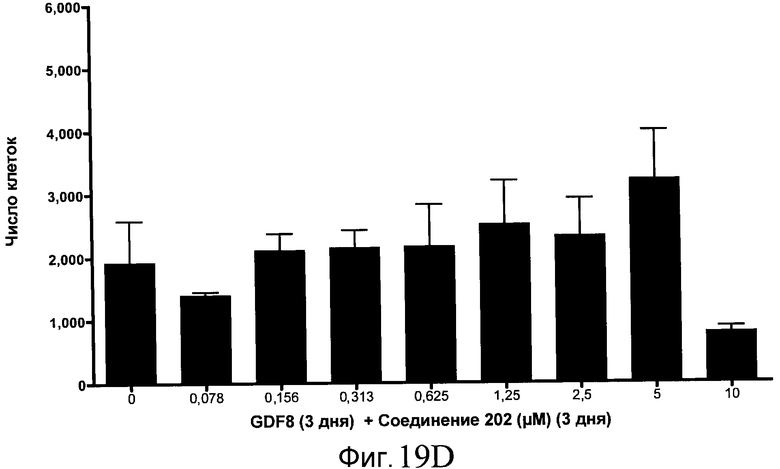
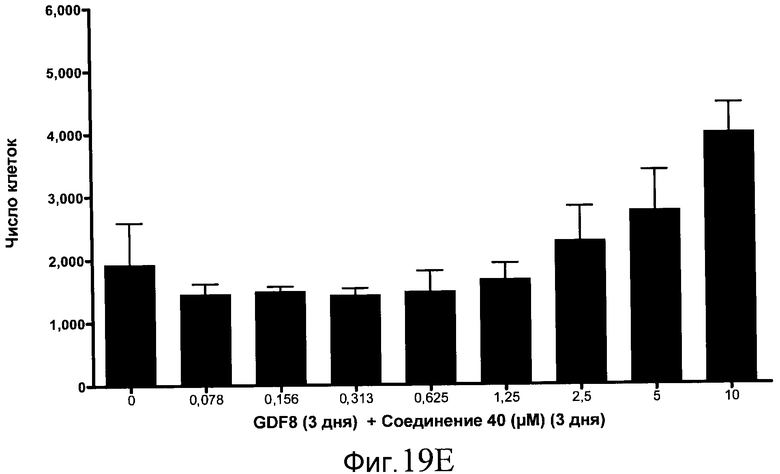
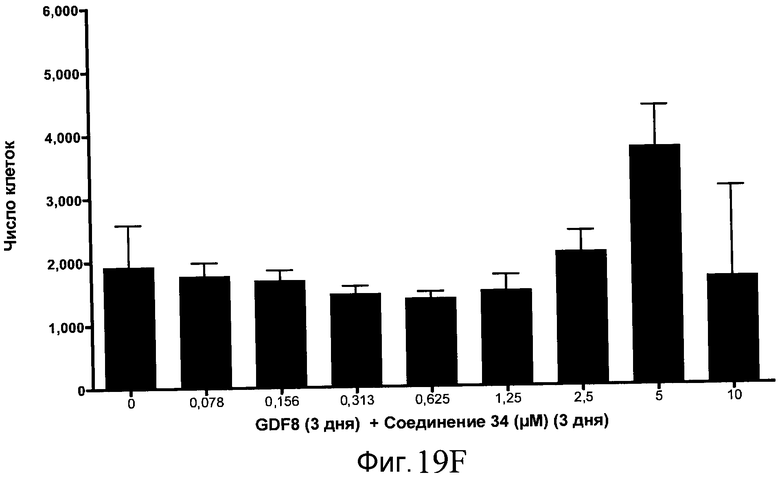
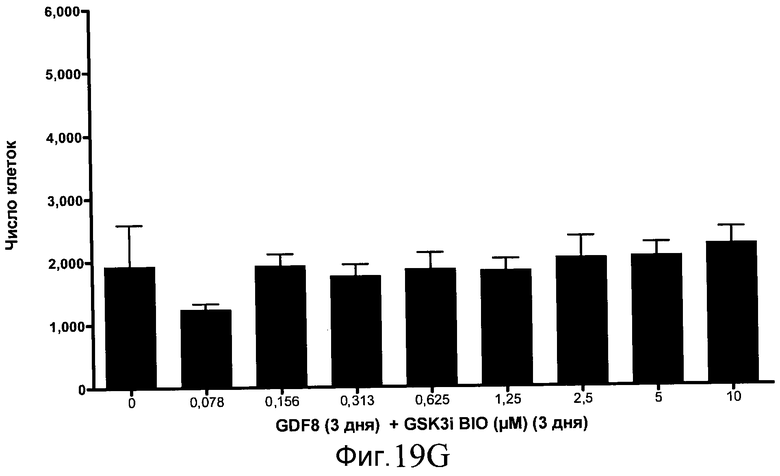
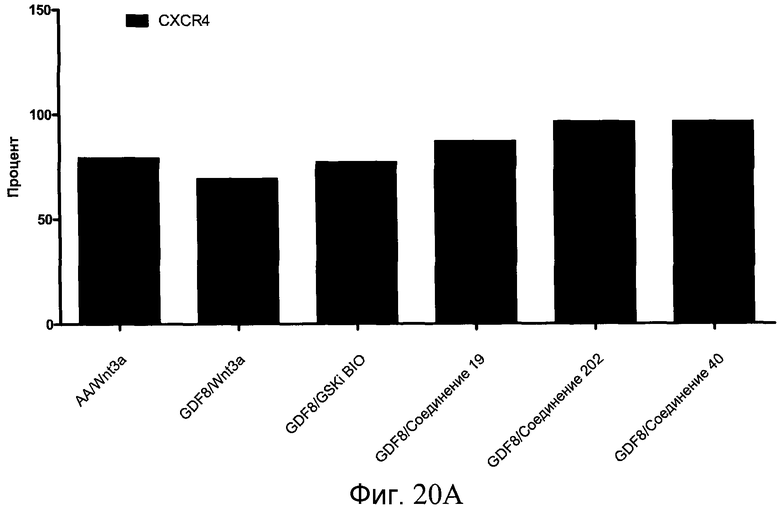
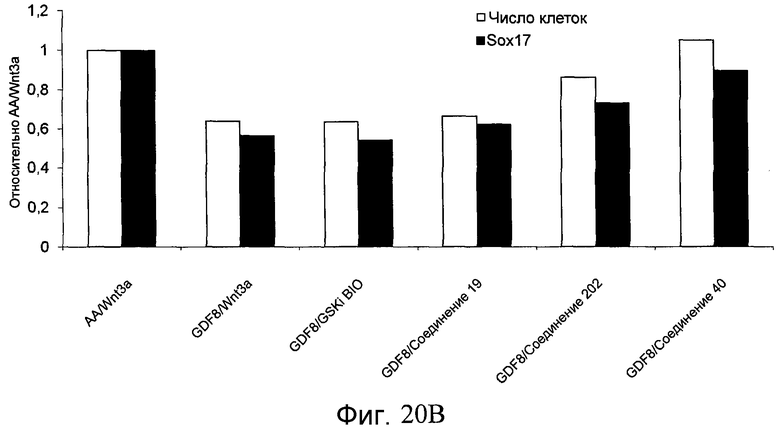
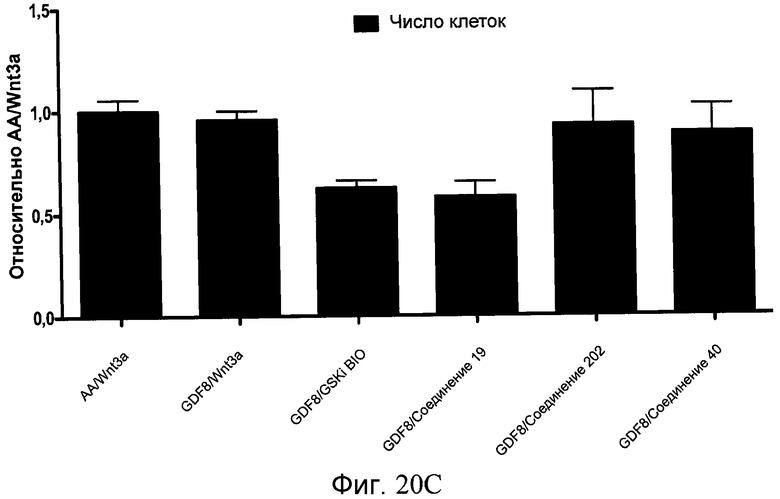
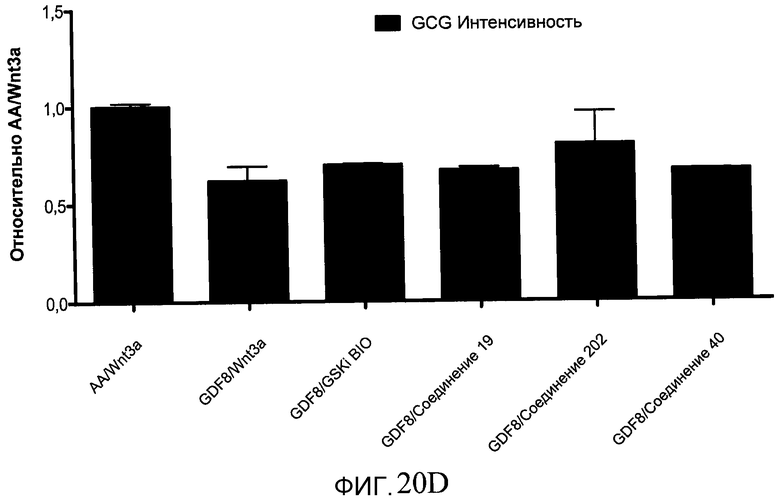
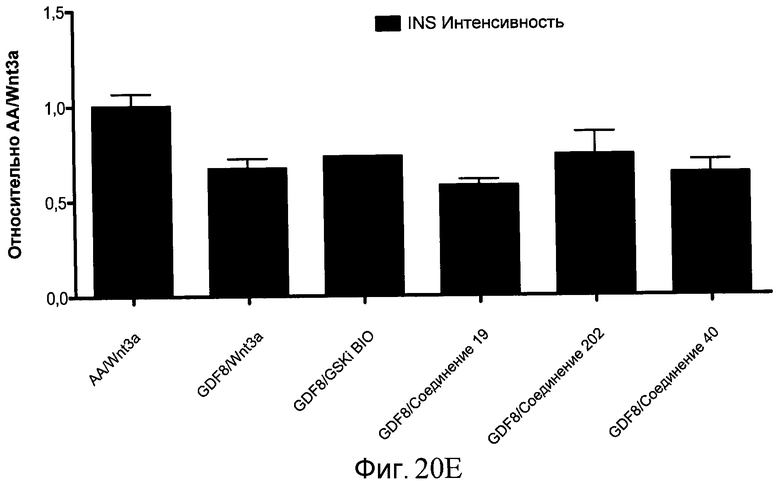
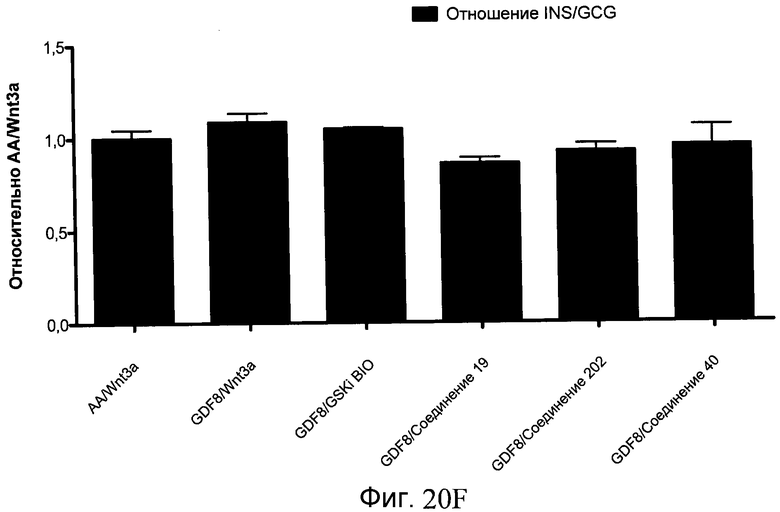
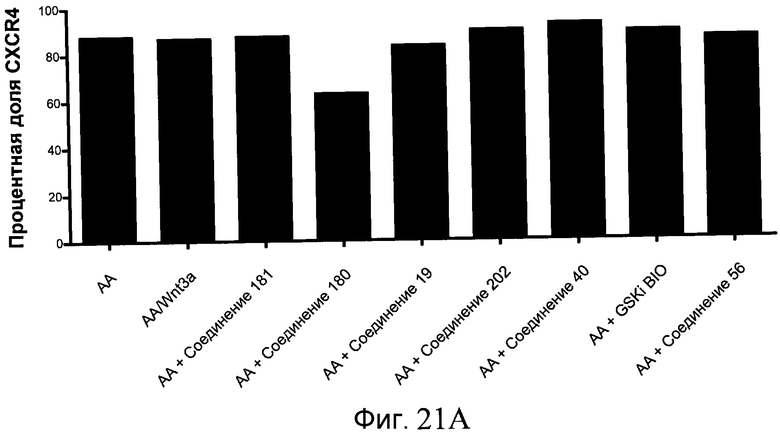
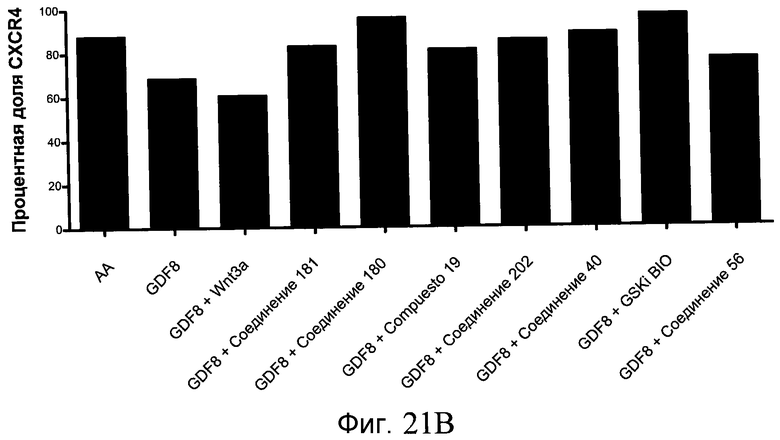
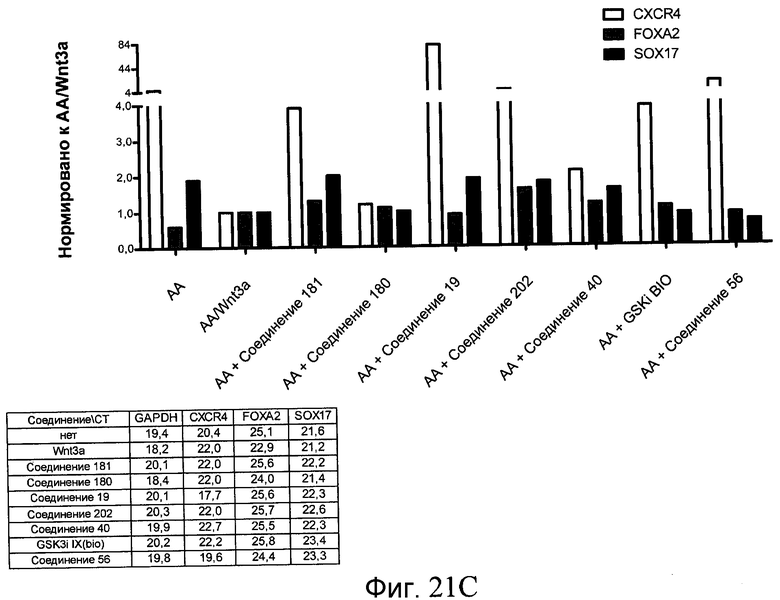
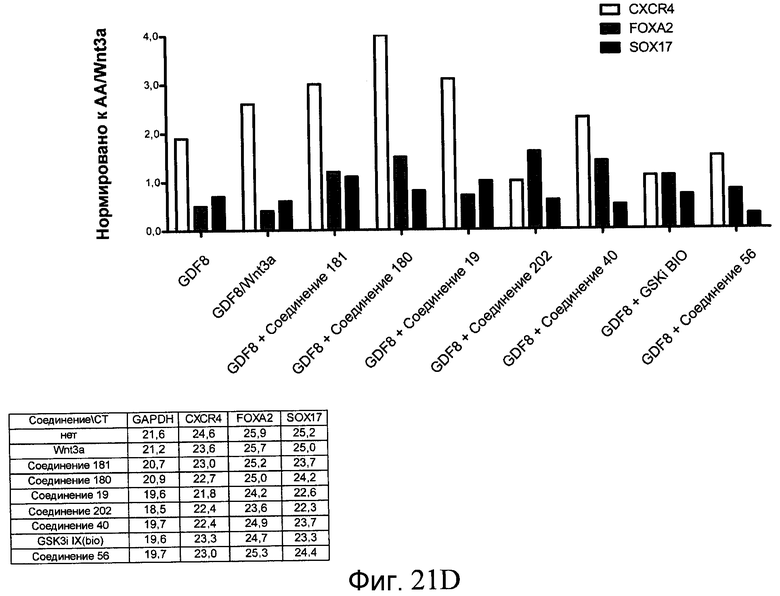
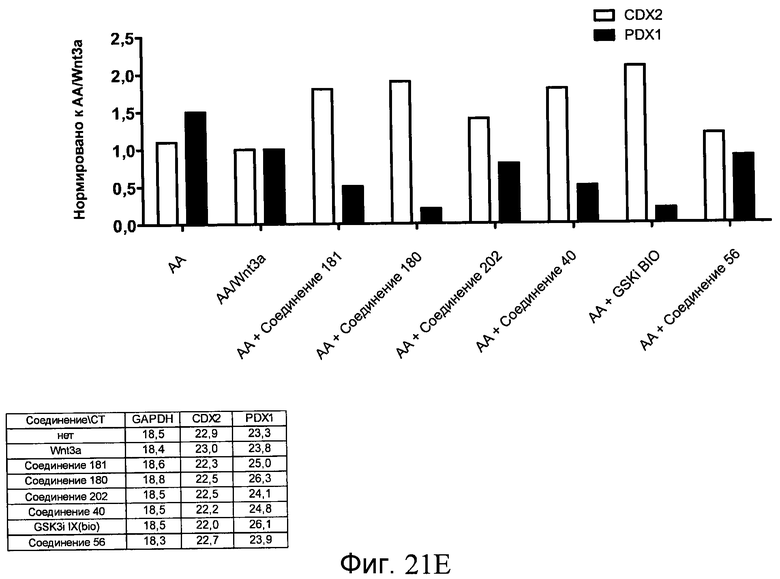
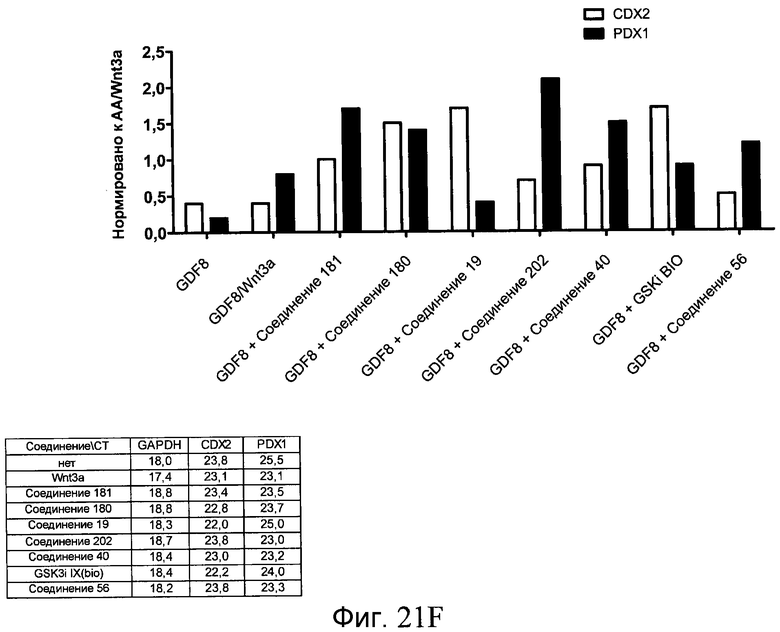
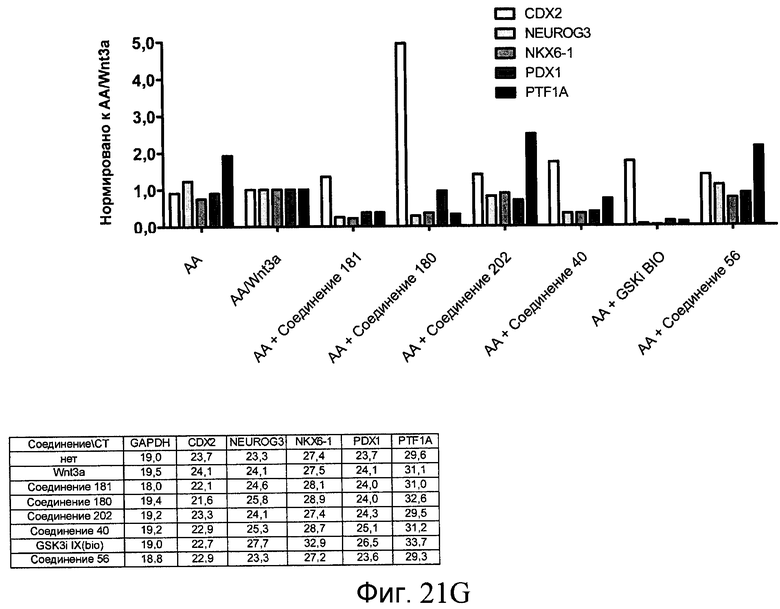
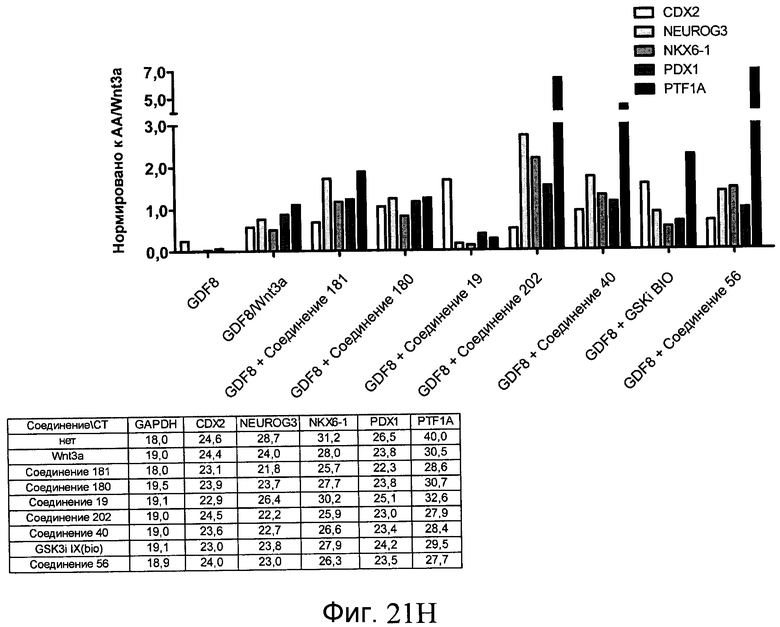
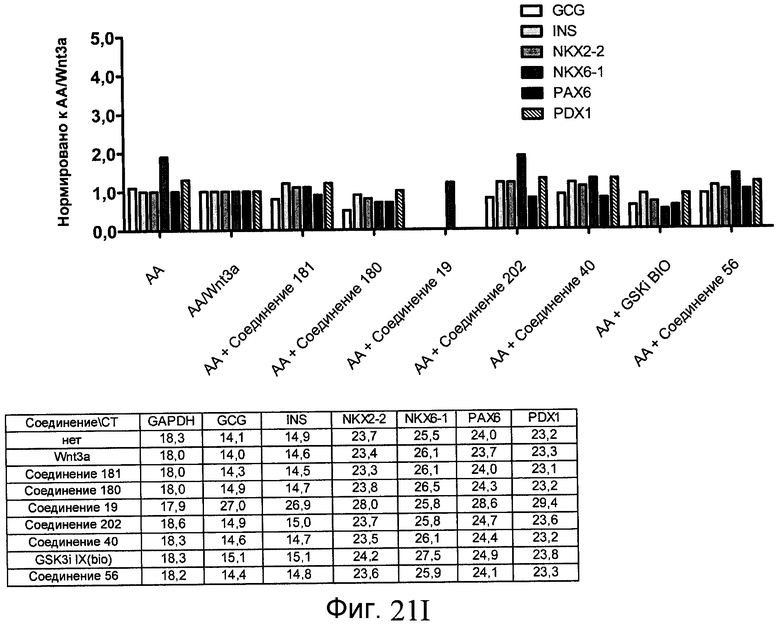
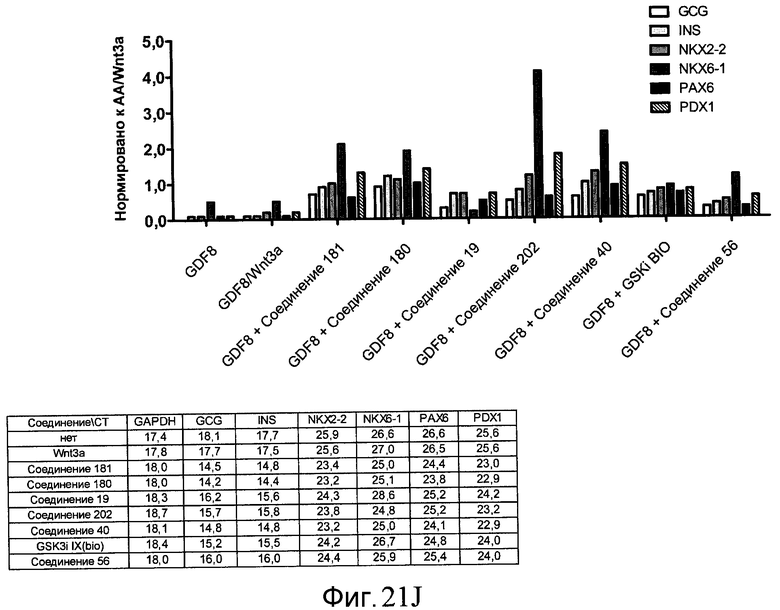
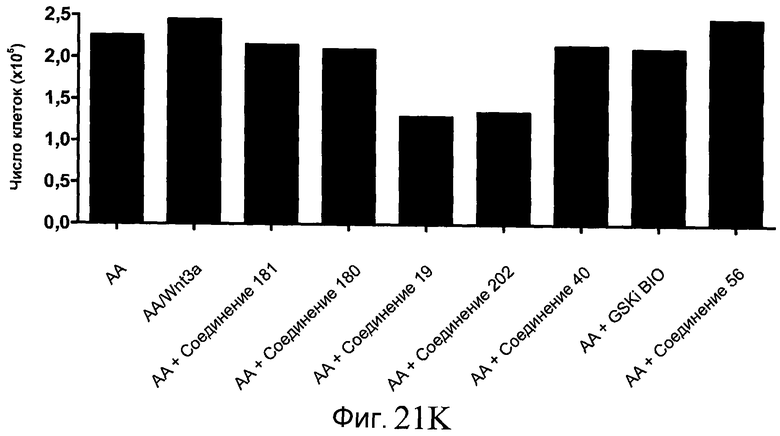
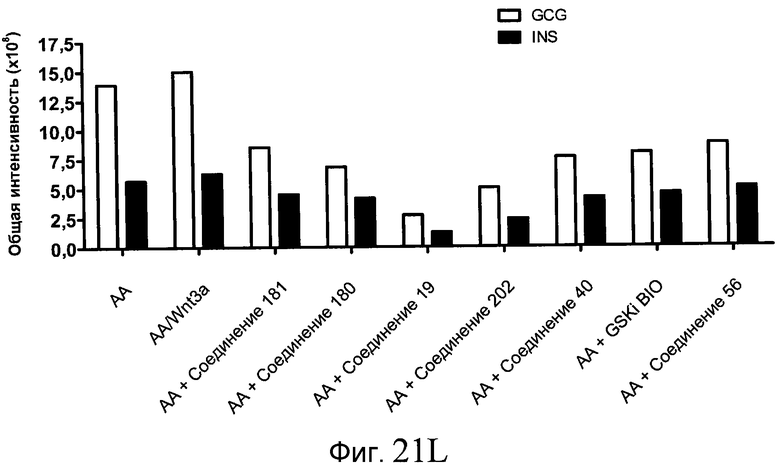
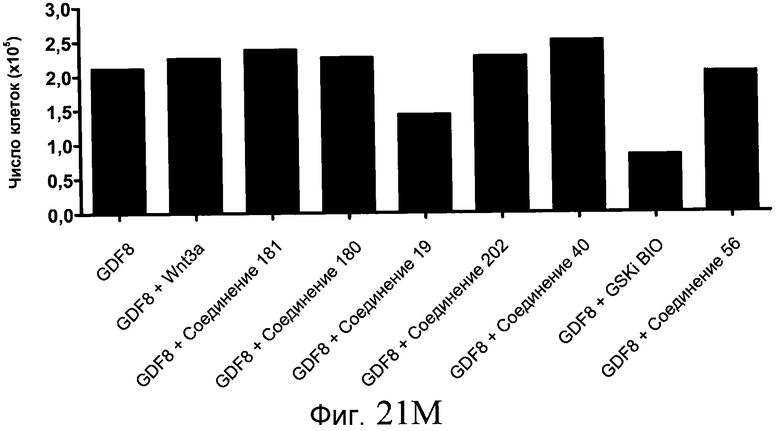
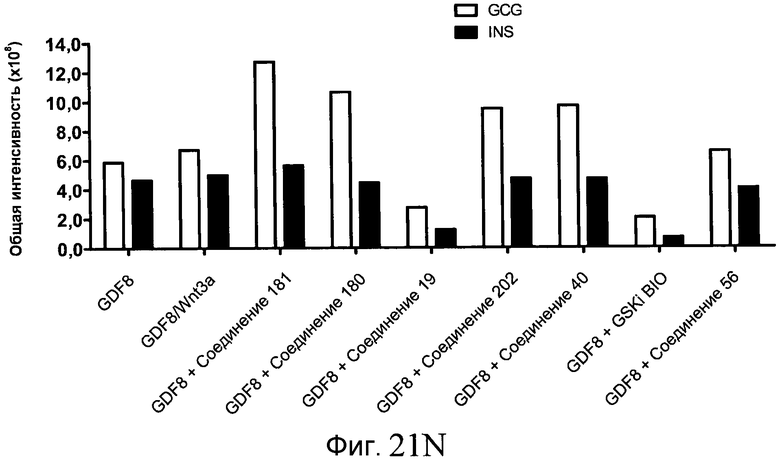
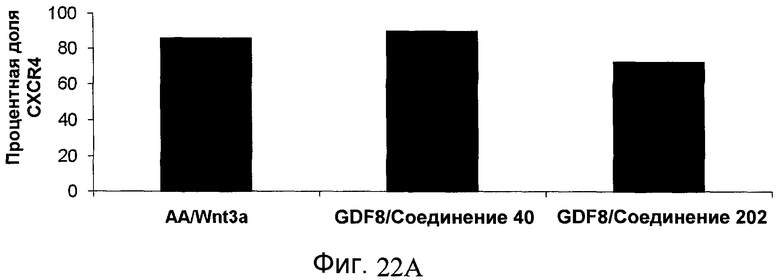
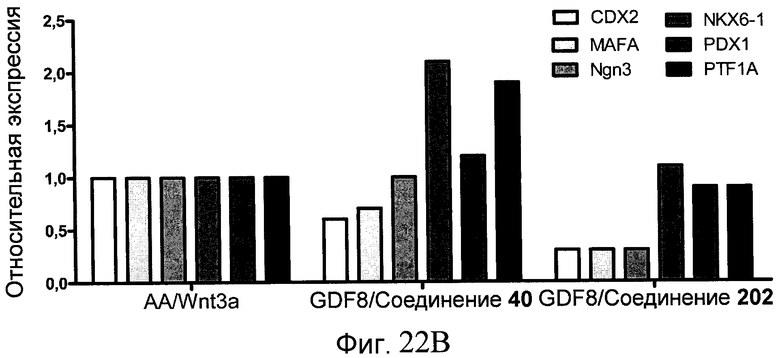
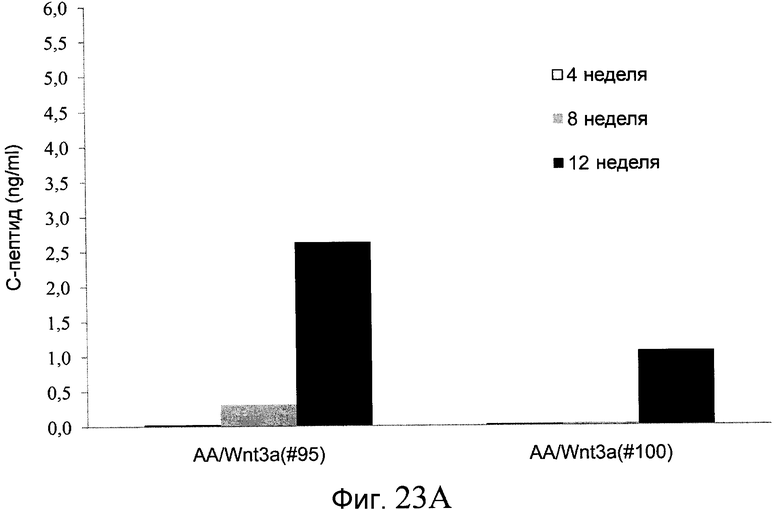
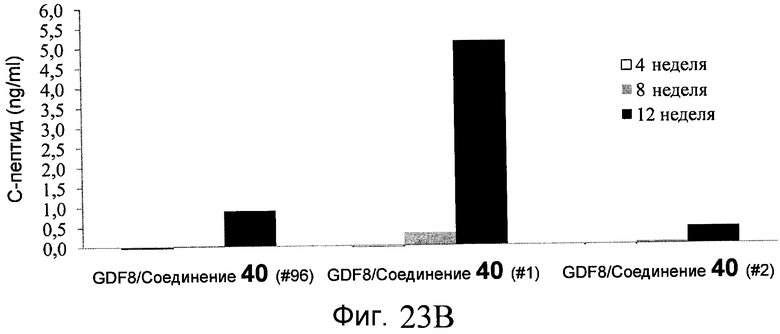
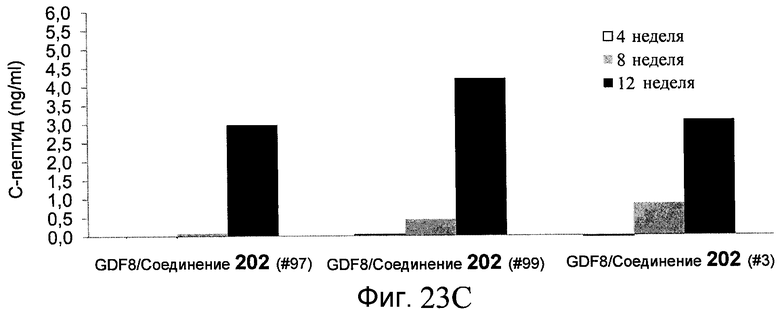
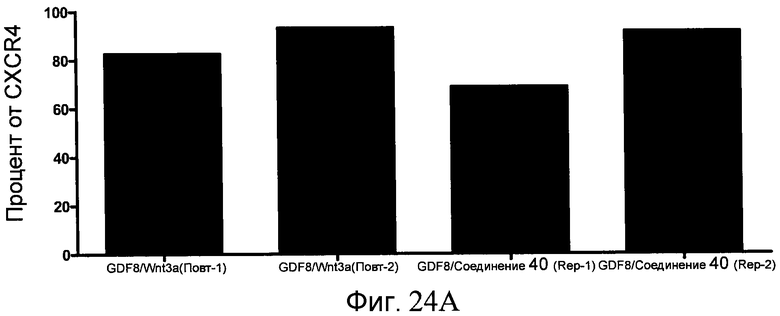
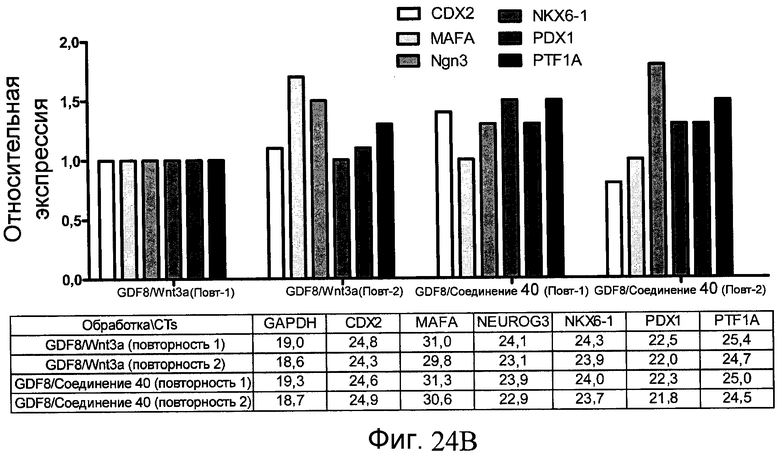
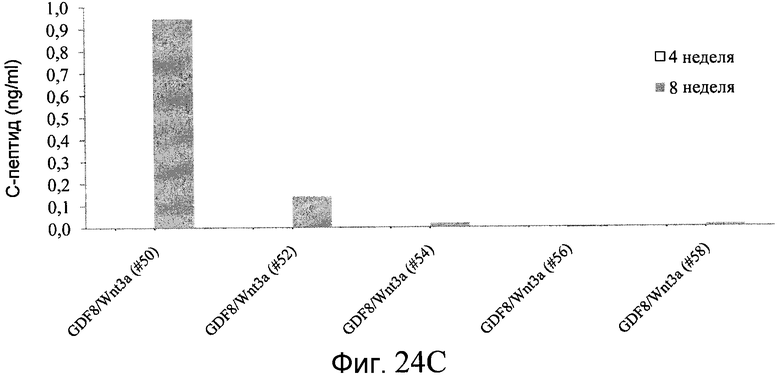
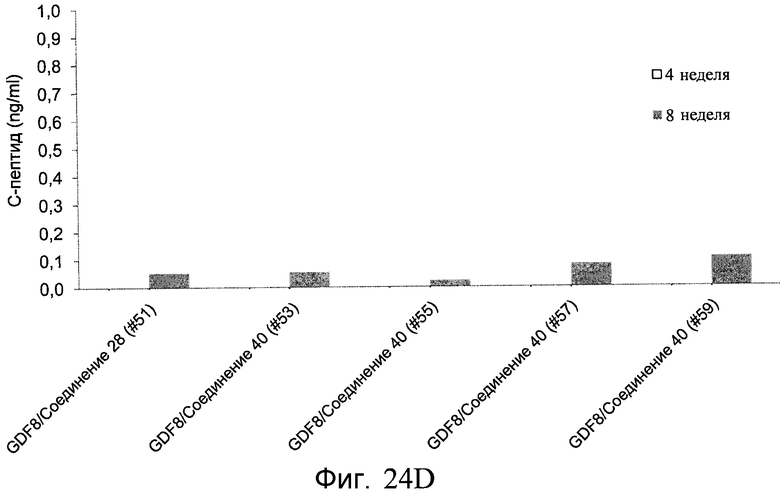
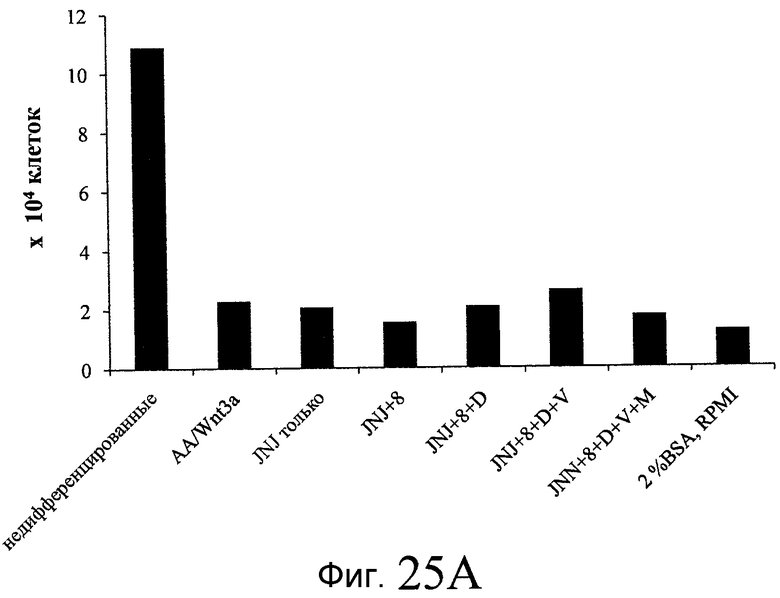
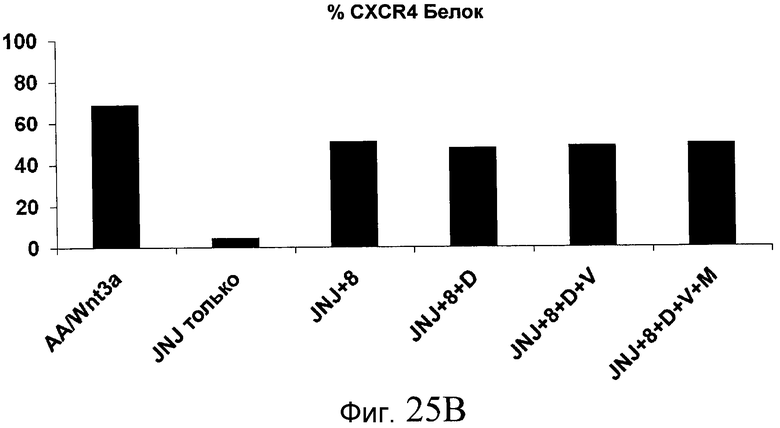
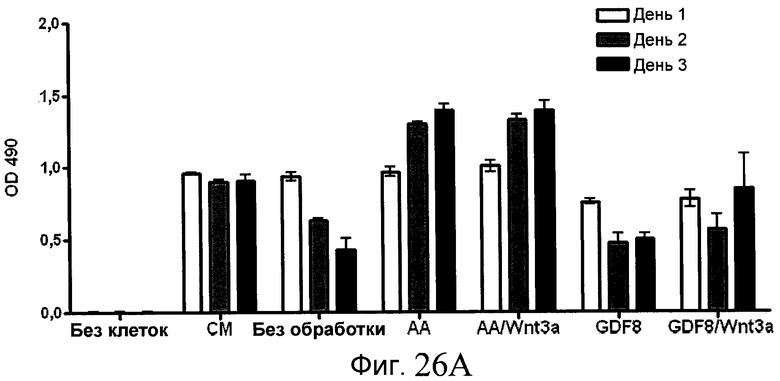
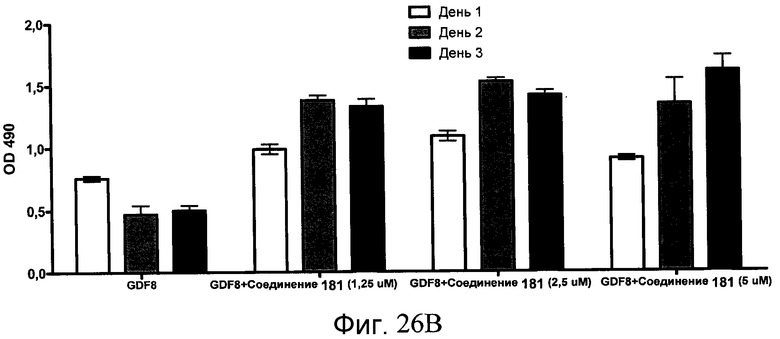
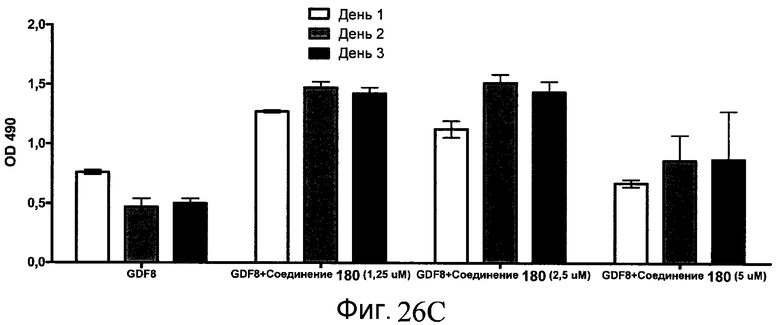
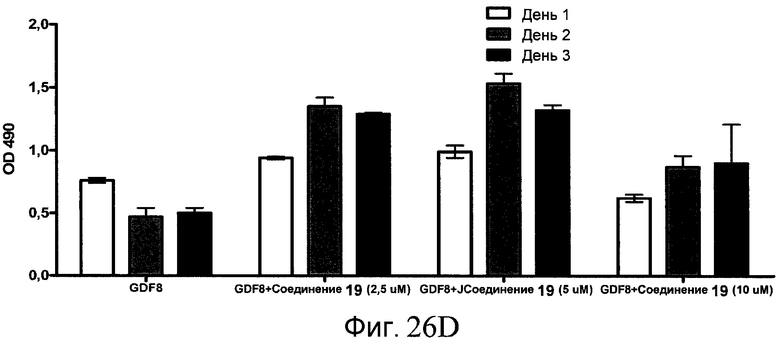
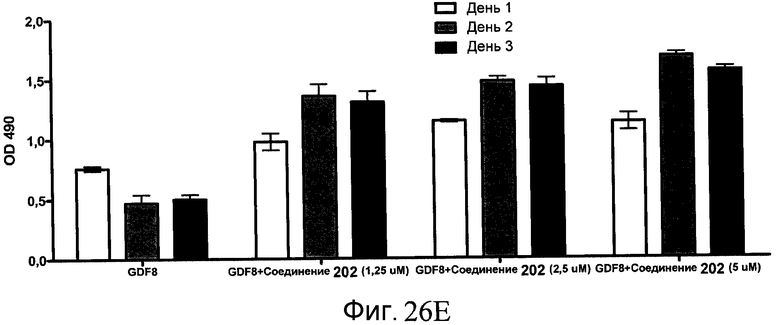
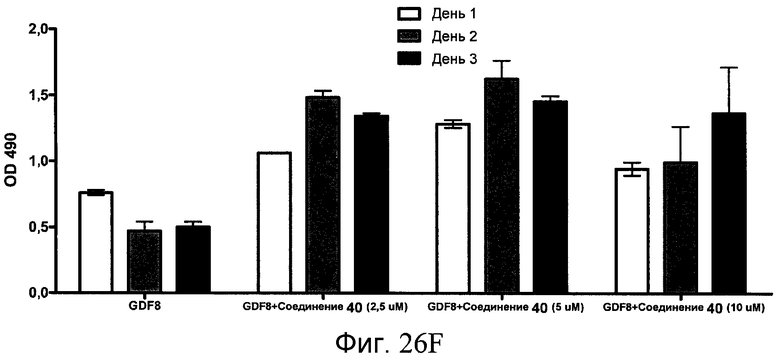
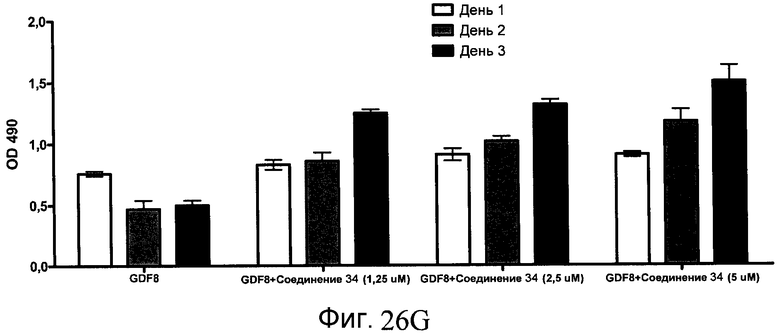
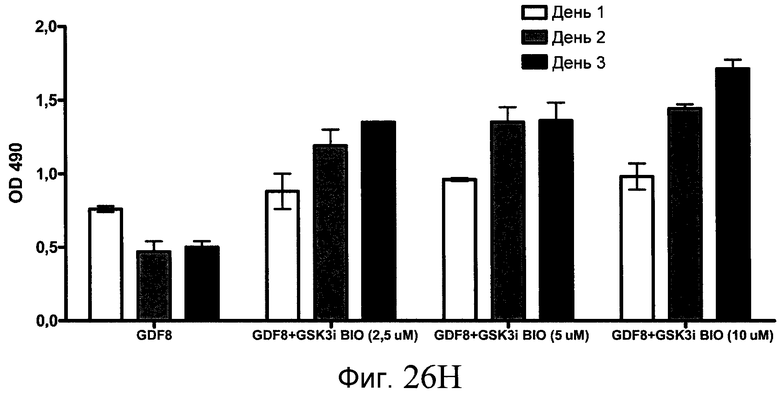
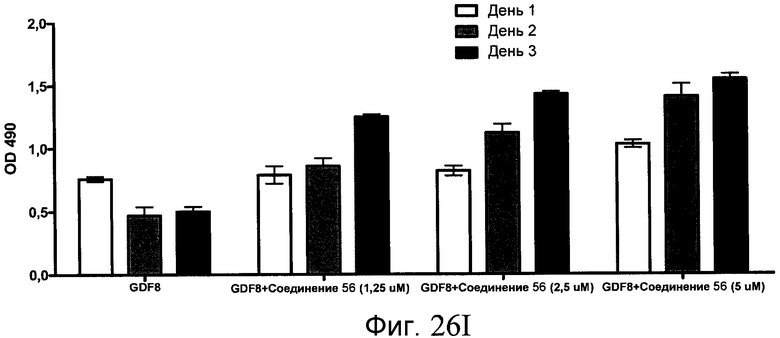
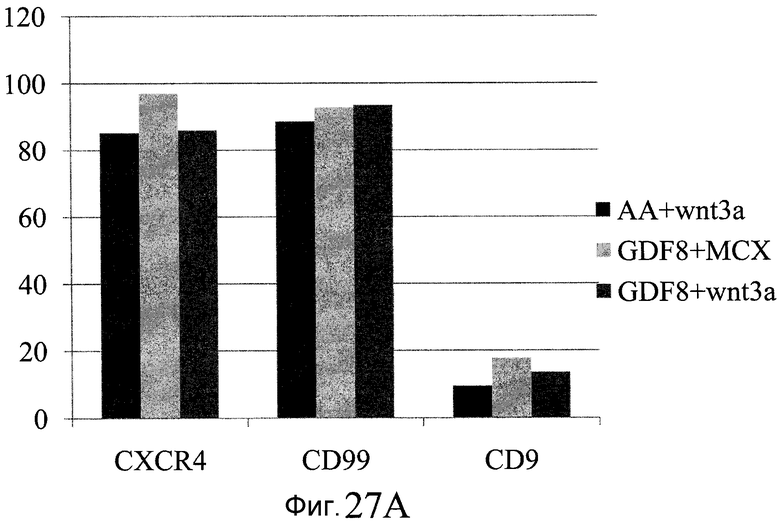
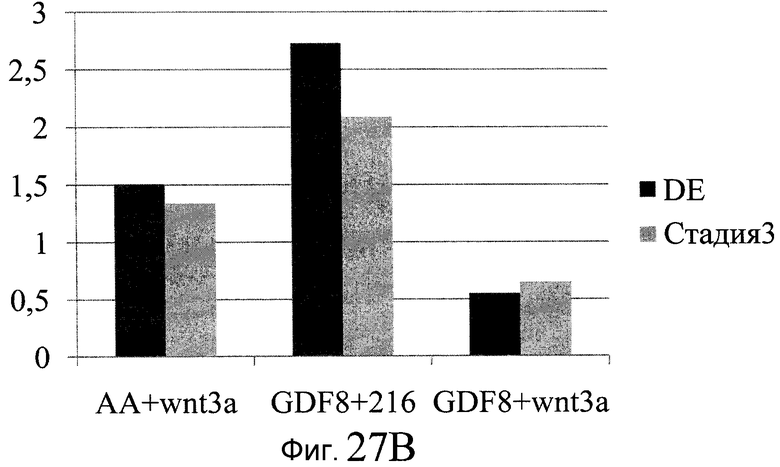
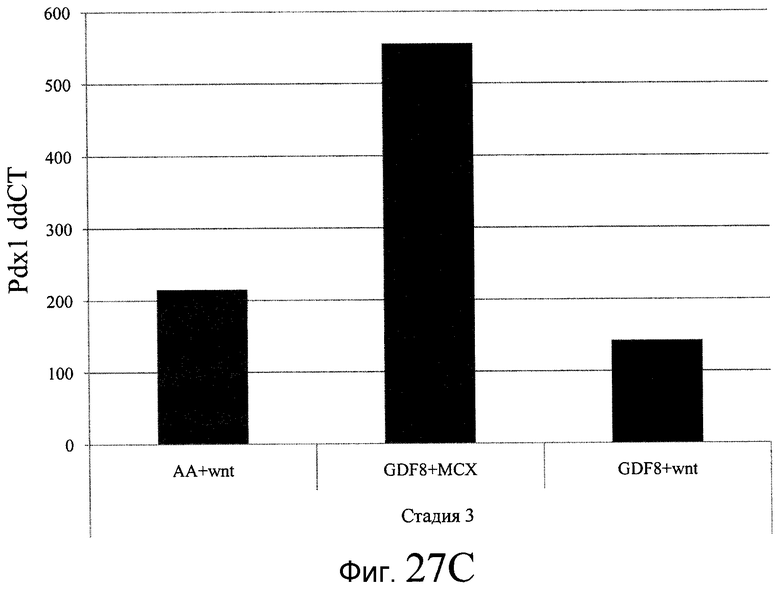

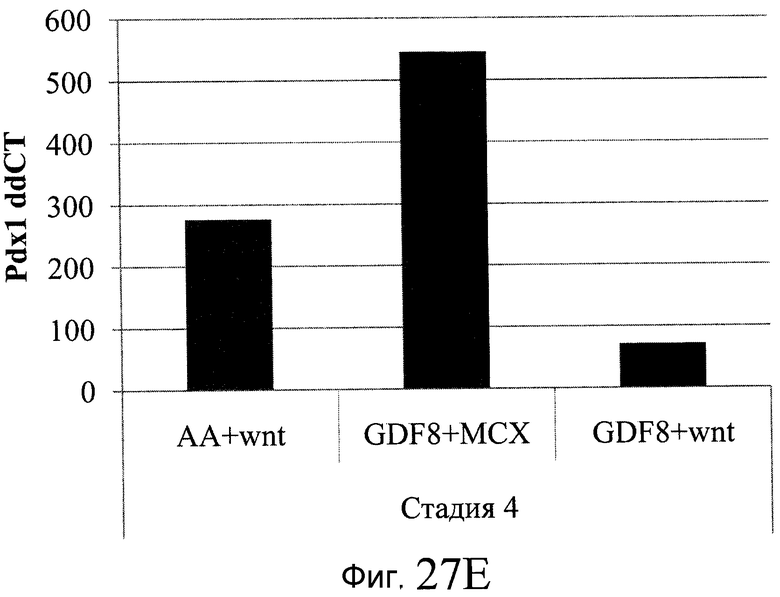

Изобретение относится к области медицины, биотехнологии и клеточных технологий. Способ дифференцирования плюрипотентных стволовых клеток, представляющих собой линию клеток человека, в клетки, экспрессирующие маркеры, характерные для линии сформированной эндодермы, включает обработку плюрипотентных стволовых клеток средой, отличающейся тем, что она не содержит активин А и содержит GDF-8, в течение периода времени, достаточного для того, чтобы плюрипотентные стволовые клетки дифференцировались в клетки, экспрессирующие маркеры, характерные для линии сформированной эндодермы. Изобретение может быть использовано в медицине для целей трансплантации. 13 з.п. ф-лы, 19 табл., 27 ил., 24 пр.
1. Способ дифференцирования плюрипотентных стволовых клеток, представляющих собой линию клеток человека, в клетки, экспрессирующие маркеры, характерные для линии сформированной эндодермы, включающий обработку плюрипотентных стволовых клеток средой, отличающейся тем, что она не содержит активин А и содержит GDF-8, в течение периода времени, достаточного для того, чтобы плюрипотентные стволовые клетки дифференцировались в клетки, экспрессирующие маркеры, характерные для линии сформированной эндодермы.
2. Способ по п.1, в котором среда, отличающаяся тем, что она не содержит активин А, также содержит по меньшей мере одно другое соединение, выбранное из группы, состоящей из: EGF, FGF4, PDGF-A, PDGF-B, PDGF-C, PDGF-D, VEGF, мусцимол, PD98059, LY294002, U0124, U0126 и бутират натрия.
3. Способ по п.1, где среда, отличающаяся тем, что она не содержит активин А, также содержит по меньшей мере одно другое соединение, выбранное из группы, состоящей из: анилин-пиридинотриазина, циклического анилин-пиридинотриазина,
N-{[1-(фенилметил)азелан-4-ил]метил}-2-пиридин-3-илацетамид, 4-{[4-(4-{[2-(пиридин-2-иламино)этил]амино}-1,3,5-триазин-2-ил)пиридин-2-ил]окси}бутан-1-ол, 3-({3-[4-({2-[метил(пиридин-2-ил)амино]этил}амино)-1,3,5-триазин-2-ил]пиридин-2-ил}амино)пропан-1-ол, N~4~-[2-(3-фторфенил)этил]-N~2~-[3-(4-метилпиперазин-1-ил)пропил]пиридо[2,3-d]пиримидин-2,4-диамин, 1-метил-N-[(4-пиридин-3-ил-2-{[3-(трифторметил)фенил]амино}-1,3-триазол-5-ил)метил]пиперидин-4-карбоксамид, 1,1-диметилэтил {2-[4-({5-[3-(3-гидроксипропил)фенил]-4H-1,2,4-триазол-3-ил}амино)фенил]этил}карбамат, 1,1-диметилэтил {[3-({5-[5-(3-гидроксипропил)-2-(метилокси)фенил]-1,3-оксазол-2-ил}амино)фенил]метил}карбамат, 1-({5-[6-({4-[(4-метилпиперазин-1-ил)сульфонил]фенил}амино)пиразин-2-ил]тиофен-2-ил}метил)пиперидин-4-ол, 1-({4-[6-({4-[(4-метилпиперазин-1-ил)сульфонил]фенил}амино)пиразин-2-ил]тиофен-2-ил}метил)пиперидин-4-карбоксамид и 2-{[4-(1-метилэтил)фенил]амино}-N-(2-тиофен-2-илэтил)-7,8-дигидропиридо[4,3-d]пиримидин-6(5H)-карбоксамид.
4. Способ по п. 3, где соединение выбрано из группы, состоящей из: анилин-пиридинотриазина и циклического анилин-пиридинотриазина.
5. Способ по п. 1, где GDF-8 используется при концентрации от 5 нг/мл до 500 нг/мл.
6. Способ по п.1, где GDF-8 используется при концентрации от 5 нг/мл до 25 нг/мл.
7. Способ по п. 1, где GDF-8 используется при концентрации 25 нг/мл.
8. Способ по любому из пп. 2-7, где EGF, FGF4, PDGF-A, PDGF-B, PDGF-C, PDGF-D и/или VEGF используются при концентрации от 5 нг/мл до 500 нг/мл.
9. Способ по любому из пп. 2-7, где EGF, FGF4, PDGF-A, PDGF-B, PDGF-C, PDGF-D и/или VEGF используются при концентрации от 5 нг/мл до 50 нг/мл.
10. Способ по любому из пп. 2-7, где EGF, FGF4, PDGF-A, PDGF-B, PDGF-C, PDGF-D и/или VEGF используются при концентрации 50 нг/мл.
11. Способ по любому из пп. 2-7, где мусцимол используется при концентрации от 1 мкМ до 200 мкМ, предпочтительно при концентрации 20 мкМ.
12. Способ по любому из пп. 2-7, где PD98059, U0124 и/или U0126 используются при концентрации от 0,1 мкМ до 10 мкМ, предпочтительно при концентрации 1 мкМ.
13. Способ по любому из пп. 2-7, где LY294002 используется при концентрации от 0,25 мкМ до 25 мкМ, предпочтительно при концентрации 2,5 мкМ.
14. Способ по любому из пп. 2-7, где бутират натрия используется при концентрации от 0,05 мкМ до 5 мкМ, предпочтительно при концентрации 0,5 мкМ.
| WO2008048647 A, 24.04.2008 | |||
| Кассета для образца для теплофизических испытаний влажных материалов | 1987 |
|
SU1516925A1 |
| БЕССЫВОРОТОЧНАЯ СРЕДА ДЛЯ КУЛЬТУРЫ КЛЕТОК, ИСПОЛЬЗУЕМЫХ ДЛЯ РЕКОНСТРУКЦИИ КОСТНЫХ И ХРЯЩЕВЫХ СЕГМЕНТОВ (ВАРИАНТЫ) | 1999 |
|
RU2272839C2 |
| RU 2005131581 A, 27.04.2006 | |||

