ОБЛАСТЬ ПРИМЕНЕНИЯ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к способу обработки плюрипотентных клеток, позволяющему эффективно размножать плюрипотентные клетки в культуре и дифференцировать их путем обработки ингибитором активности фермента GSK-3B.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Последние достижения в области заместительной клеточной терапии для лечения сахарного диабета I типа и нехватка островков Лангеранса для трансплантации заставили обратить внимание на разработку источников инсулинопродуцирующих клеток или β-клеток, которые могли бы использоваться для трансплантации. Одним из возможных подходов является генерация функциональных β-клеток из плюрипотентных клеток, таких как, например, эмбриональные стволовые клетки.
При эмбриональном развитии позвоночных плюрипотентные клетки дают начало группе клеток, формирующих три зародышевых листка (эктодерму, мезодерму и эндодерму) в ходе процесса, именуемого гаструляцией. Такие ткани, как, например, щитовидная железа, тимус, поджелудочная железа, кишечник и печень, будут развиваться из эндодермы через промежуточную стадию. Промежуточной стадией данного процесса является образование сформированной эндодермы. Клетки сформированной эндодермы экспрессируют ряд маркеров, таких как HNF-3 beta, GATA-4, Mixl1, CXCR4 и SOX-17.
Формирование поджелудочной железы происходит при дифференцировании сформированной эндодермы в панкреатическую эндодерму. Клетки панкреатической эндодермы экспрессируют ген панкреатическо-дуоденального гомеобокса, PDX-1. В отсутствие PDX-1 развитие поджелудочной железы не идет дальше формирования вентрального и дорсального зачатков. Таким образом, экспрессия PDX-1 характеризует критическую стадию органогенеза поджелудочной железы. Зрелая поджелудочная железа содержит, помимо других типов клеток, экзокринную ткань и эндокринную ткань. Экзокринная и эндокринная ткани образуются при дифференцировании панкреатической эндодермы.
Для получения достаточного количества клеточного материала для трансплантации необходим источник требуемого клеточного материала, который можно было бы эффективно размножать в культуре, а затем эффективно дифференцировать в требуемые ткани, например в функциональные β-клетки.
Известные на сегодняшний день способы культивации эмбриональных стволовых клеток человека достаточно сложны и требуют применения экзогенных факторов или культуральной среды химически определенного состава для поддержания пролиферации клеток без потери их плюрипотентности. Кроме того, дифференцирование эмбриональных стволовых клеток часто приводит к сокращению количества клеток, доступных для дальнейшего размножения в культуре.
В одном примере, в работе Cheon et al. (BioReprod DOI:10.1095/biolreprod.105.046870, October 19, 2005), описывается не содержащая питающих клеток и сыворотки культуральная система, в которой эмбриональные стволовые клетки поддерживаются в некондиционированной заменяющей сыворотку среде (SR) с добавлением различных факторов роста, способных запускать самообновление эмбриональных стволовых клеток.
В другом примере, US 20050233446, описывается среда с определенным составом, которая может быть использована при культивировании стволовых клеток, включая недифференцированные зародышевые стволовые клетки приматов. В растворе данная среда является существенно изотонической относительно культивируемых стволовых клеток. При культивировании клеток указанная среда содержит основную среду и количества bFGF, инсулина и аскорбиновой кислоты, достаточные для поддержания роста зародышевых стволовых клеток без их существенного дифференцирования.
В другом примере, WO 2005086845, описывается способ поддержания недифференцированных стволовых клеток, где упомянутый способ включает воздействие на стволовые клетки одним из членов семейства белков трансформирующего ростового фактора-бета (TGFβ), одним из членов семейства белков фактора роста фибробластов (FGF) или никотинамидом (NIC) в количестве, достаточном для поддержания клеток в недифференцированном состоянии в течение периода времени, достаточного для получения желаемого результата.
Известно, что ингибиторы гликогенсинтазы киназы-3 (GSK-3) стимулируют пролиферацию и размножение стволовых клеток взрослой особи. В одном примере, Tateishi et al. (Biochemical and Biophysical Research Communications (2007) 352: 635), показано, что ингибирование GSK-3 повышает эффективность роста и выживаемость сердечных стволовых клеток человека (hCSCs), выделенных из сердца новорожденного или взрослого человека и обладающих мезенхимальными признаками.
Например, в работе Rulifson et al. (PNAS 144, 6247-6252, (2007)) утверждается, что активация каскада передачи сигнала Wnt стимулирует пролиферацию островковых β-клеток.
В другом примере, WO 2007016485, утверждается, что добавление ингибиторов GSK-3 к культуре неэмбриональных стволовых клеток, включая мультипотентные клетки-предшественники взрослых особей, приводит к поддержанию плюрипотентного фенотипа в процессе размножения и позволяет получить более четко выраженный дифференцировочный отклик.
В другом примере, US 2006030042, используется способ ингибирования GSK-3 путем добавления либо Wnt, либо низкомолекулярного ингибитора активности фермента GSK-3 для поддержания эмбриональных стволовых клеток без использования слоя питающих клеток.
В другом примере, WO 2006026473, сообщается о добавлении ингибитора GSK-3B для стабилизации плюрипотентных клеток через транскрибционную активацию c-myc и стабилизации белка c-myc.
В другом примере, WO 2006100490, сообщается об использовании культуральной среды для стволовых клеток, содержащей ингибитор GSK-3 и агонист gp130, для поддержания самовозобновляющейся популяции плюрипотентных стволовых клеток, включая эмбриональные стволовые клетки мыши или человека.
В другом примере, Sato et al. (Nature Medicine (2004) 10:55-63), показано, что ингибирование GSK-3 специфическим фармакологическим препаратом может сохранять недифференцированный фенотип эмбриональных стволовых клеток и поддерживать уровень экспрессии специфичных для плюрипотентного состояния факторов транскрипции, таких как Oct-3/4, Rex-1 и Nanog.
В другом примере, Maurer et al. (Journal of Proteome Research (2007) 6:1198-1208), показано, что обработанные ингибитором GSK-3 нервные стволовые клетки взрослой особи демонстрируют более высокую эффективность нейронального дифференцирования, в особенности путем стимулирования транскрипции целевых генов β-катенина и торможения апоптоза.
В другом примере, Gregory et al. (Annals of the New York Academy of Sciences (2005) 1049:97-106), сообщается, что ингибиторы GSK-3B повышают эффективность остеогенеза in vitro.
В другом примере, Feng et al. (Biochemical and Biophysical Research Communcations (2004) 324:1333-1339), показано, что гемопоэтическое дифференцирование эмбриональных стволовых клеток связано с понижением эффективности каскада Wnt/β-катенин, где Wnt представляет собой естественный ингибитор GSK3.
Поэтому сохраняется значительная потребность в разработке способов обработки плюрипотентных стволовых клеток для их размножения с последующим использованием в клинических целях и при этом с сохранением их способности к дифференцированию в панкреатические эндокринные клетки, панкреатические экспрессирующие гормоны клетки или панкреатические секретирующие гормоны клетки.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение предлагает способ размножения и дифференцирования плюрипотентных клеток путем обработки упомянутых плюрипотентных клеток ингибитором активности фермента GSK-3B.
В одном из вариантов осуществления настоящее изобретение предлагает способ размножения и дифференцирования плюрипотентных клеток, включающий:
а. культивирование плюрипотентных клеток, и
b. обработку плюрипотентных клеток ингибитором активности фермента GSK-3B.
В одном из вариантов осуществления настоящего изобретения упомянутые плюрипотентные клетки дифференцируются в клетки, экспрессирующие маркеры, характерные для линии сформированной эндодермы.
Упомянутые плюрипотентные клетки могут представлять собой эмбриональные стволовые клетки человека или могут быть экспрессирующими маркеры плюрипотентности производными от эмбриональных стволовых клеток человека клетками в соответствии со способами, раскрытыми в публикации 60/913475.
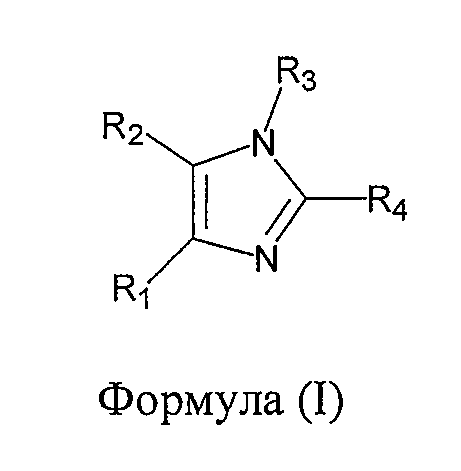
В одном из вариантов осуществления настоящего изобретения упомянутый ингибитор активности фермента GSK-3B представляет собой соединение формулы (I):

В одном из вариантов осуществления настоящего изобретения упомянутый ингибитор активности фермента GSK-3B представляет собой соединение формулы (II):

В одном из вариантов осуществления настоящего изобретения упомянутый ингибитор активности фермента GSK-3B представляет собой соединение формулы (III):

КРАТКОЕ ОПИСАНИЕ ФИГУР
На фиг. 1 показан эффект добавления соединения JNJ 17189731 в различной концентрации на численность клеток, определенную по числу наблюдаемых ядер (часть A), и на уровень экспрессии Sox-17, определенный по интенсивности иммунофлюоресцентной окраски (часть B). Приведенные результаты были получены для эмбриональных стволовых клеток человека линии H1 (белые столбики) или эмбриональных стволовых клеток человека линии H9 (черные столбики) на анализаторе IN Cell Analyzer 1000 (GE Healthcare).
На фиг. 2 показан эффект добавления соединения JNJ 17163796 в различной концентрации на численность клеток, определенную по числу наблюдаемых ядер (часть A), и на уровень экспрессии Sox-17, определенный по интенсивности иммунофлюоресцентной окраски (часть B). Приведенные результаты были получены для эмбриональных стволовых клеток человека линии H1 (белые столбики) или эмбриональных стволовых клеток человека линии H9 (черные столбики) на анализаторе IN Cell Analyzer 1000 (GE Healthcare).
На фиг. 3 показан эффект добавления соединения JNJ 17223375 в различной концентрации на численность клеток, определенную по числу наблюдаемых ядер (часть A), и на уровень экспрессии Sox-17, определенный по интенсивности иммунофлюоресцентной окраски (часть B). Приведенные результаты были получены для эмбриональных стволовых клеток человека линии H1 (белые столбики) или эмбриональных стволовых клеток человека линии H9 (черные столбики) на анализаторе IN Cell Analyzer 1000 (GE Healthcare).
На фиг. 4 показан эффект добавления соединения JNJ 18157698 в различной концентрации на численность клеток, определенную по числу наблюдаемых ядер (часть A), и на уровень экспрессии Sox-17, определенный по интенсивности иммунофлюоресцентной окраски (часть B). Приведенные результаты были получены для эмбриональных стволовых клеток человека линии H1 (белые столбики) или эмбриональных стволовых клеток человека линии H9 (черные столбики) на анализаторе IN Cell Analyzer 1000 (GE Healthcare).
На фиг. 5 показан эффект добавления соединения JNJ 26158015 в различной концентрации на численность клеток, определенную по числу наблюдаемых ядер (часть A), и на уровень экспрессии Sox-17, определенный по интенсивности иммунофлюоресцентной окраски (часть B). Приведенные результаты были получены для эмбриональных стволовых клеток человека линии H1 (белые столбики) или эмбриональных стволовых клеток человека линии H9 (черные столбики) на анализаторе IN Cell Analyzer 1000 (GE Healthcare).
На фиг. 6 показан эффект добавления соединения JNJ 26483197 в различной концентрации на численность клеток, определенную по числу наблюдаемых ядер (часть A), и на уровень экспрессии Sox-17, определенный по интенсивности иммунофлюоресцентной окраски (часть B). Приведенные результаты были получены для эмбриональных стволовых клеток человека линии H1 (белые столбики) или эмбриональных стволовых клеток человека линии H9 (черные столбики) на анализаторе IN Cell Analyzer 1000 (GE Healthcare).
На фиг. 7 показан эффект добавления соединения JNJ 26483249 в различной концентрации на численность клеток, определенную по числу наблюдаемых ядер (часть A), и на уровень экспрессии Sox-17, определенный по интенсивности иммунофлюоресцентной окраски (часть B). Приведенные результаты были получены для эмбриональных стволовых клеток человека линии H1 (белые столбики) или эмбриональных стволовых клеток человека линии H9 (черные столбики) на анализаторе IN Cell Analyzer 1000 (GE Healthcare).
На фиг. 8 показан эффект добавления соединения JNJ 10220067 в различной концентрации на численность клеток, определенную по числу наблюдаемых ядер (часть A), и на уровень экспрессии Sox-17, определенный по интенсивности иммунофлюоресцентной окраски (часть B). Приведенные результаты были получены для эмбриональных стволовых клеток человека линии H1 (белые столбики) или эмбриональных стволовых клеток человека линии H9 (черные столбики) на анализаторе IN Cell Analyzer 1000 (GE Healthcare).
На фиг. 9 приведены уровни экспрессии CXCR4 на поверхности клеток, определенные способами иммунофлюоресцентного окрашивания и проточной цитометрии, для клеток, обработанных указанными соединениями согласно способам, описанным в примере 8.
На фиг. 10 приведены уровни экспрессии CXCR4 (часть A), HNF-3 beta (часть B) и Sox-17 (часть C), определенные методом ПЦР в реальном времени, для клеток, обработанных указанными соединениями согласно способам, описанным в примере 8.
На фиг. 11 показан эффект добавления указанных соединений в различной концентрации на численность клеток, определенную по числу наблюдаемых ядер (часть A), и на уровень экспрессии Pdx-1, определенный по интенсивности иммунофлюоресцентной окраски (часть B). Измерения проводились на анализаторе IN Cell Analyzer 1000 (GE Healthcare). Клетки обрабатывали согласно способам, описанным в примере 9.
На фиг. 12 показан эффект добавления указанных соединений в различной концентрации на уровень экспрессии Pdx-1 (белые столбики) и HNF-6 (черные столбики), определенные методом ПЦР в реальном времени. Клетки обрабатывали согласно способам, описанным в примере 9.
На фиг. 13 показан эффект добавления указанных соединений в различной концентрации на численность клеток, определенную по числу наблюдаемых ядер (часть A), и на уровень экспрессии инсулина, определенный по интенсивности иммунофлюоресцентной окраски (часть B). Измерения проводили на анализаторе IN Cell Analyzer 1000 (GE Healthcare). Клетки обрабатывали согласно способам, описанным в примере 10.
На фиг. 14 показан эффект добавления указанных соединений в различной концентрации на уровень экспрессии Pdx-1 (белые столбики) и инсулина (черные столбики), определенные методом ПЦР в реальном времени. Клетки обрабатывали согласно способам, описанным в примере 10.
На фиг. 15 показан эффект добавления указанных соединений в различной концентрации на численность клеток, определенную по числу наблюдаемых ядер (часть A), и на уровень экспрессии инсулина, определенный по интенсивности иммунофлюоресцентной окраски (часть B). Измерения проводили на анализаторе IN Cell Analyzer 1000 (GE Healthcare). Клетки обрабатывали согласно способам, описанным в примере 11.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Для ясности описания, а не в ограничение изобретения, приведенное ниже подробное описание изобретения разделено на следующие подразделы, описывающие или иллюстрирующие определенные особенности, варианты осуществления или области применения настоящего изобретения.
Определения
Стволовые клетки представляют собой недифференцированные клетки, определяемые по их способности на уровне единичной клетки как самообновляться, так и дифференцироваться с образованием клеток-потомков, таких как самообновляющиеся клетки-предшественники, необновляющиеся клетки-предшественники и окончательно дифференцированные клетки. Стволовые клетки также характеризуются способностью дифференцироваться in vitro в функциональные клетки различных клеточных линий дифференцирования из нескольких зародышевых листков (эндодермы, мезодермы и эктодермы), а также после трансплантации давать начало тканям, происходящим от нескольких зародышевых листков, и вносить существенный вклад в формирование большинства, если не всех, тканей после инъекции в бластоцисты.
По потенциалу развития стволовые клетки классифицируются следующим образом: (1) тотипотентные, т.е. способные давать начало всем эмбриональным и внеэмбриональным типам клеток; (2) плюрипотентные, т.е. способные давать начало всем эмбриональным типам клеток; (3) мультипотентные, т.е. способные давать начало группе клеточных линий дифференцирования в пределах конкретной ткани, органа или физиологической системы (например, гематопоэтические стволовые клетки (HSC) могут давать таких потомков, как HSC (самообновление), олигопотентные предшественники, ограниченные клетками крови, и все типы клеток и клеточных элементов (таких как тромбоциты), являющиеся нормальными компонентами крови); (4) олигопотентные, т.е. способные давать начало более ограниченному набору клеточных линий дифференцирования, чем мультипотентные стволовые клетки; и (5) унипотентные, т.е. способные давать начало единственной клеточной линии дифференцирования (например, сперматогенные стволовые клетки).
Дифференцирование представляет собой процесс, при помощи которого неспециализированная («некоммитированная») или менее специализированная клетка приобретает свойства специализированной клетки, например нервной или мышечной клетки. Дифференцированная клетка или клетка с индуцированным дифференцированием представляет собой клетку, занявшую более специализированное («коммитированное») положение в линии дифференцирования клетки. Термин «коммитированная» применительно к процессу дифференцирования обозначает клетку, дошедшую в ходе процесса дифференцирования до стадии, от которой в нормальных условиях она продолжит дифференцироваться до определенного типа клеток или набора типов клеток и не сможет в нормальных условиях дифференцироваться в иной тип клеток или вернуться обратно к менее дифференцированному типу. Дедифференцированием называется процесс, в ходе которого клетка возвращается к менее специализированному (или коммитированному) положению в линии дифференцирования клетки. Используемый в настоящей заявке термин «линия дифференцирования клетки» определяет наследственность клетки, то есть определяет, из какой клетки произошла данная клетка и каким клеткам она может дать начало. В линии дифференцирования клетка помещается в наследственную схему развития и дифференцирования. Маркером, специфичным для линии дифференцирования, называется характерная особенность, специфически ассоциированная с фенотипом клеток конкретной линии дифференцирования, которая может использоваться для оценки дифференцирования некоммитированных клеток в клетки данной линии дифференцирования.
Используемый в настоящей заявке термин «β-клеточная линия дифференцирования» относится к клеткам, положительным по экспрессии гена транскрипционного фактора PDX-1 и как минимум одного из следующих транскрипционных факторов: NGN-3, Nkx2.2, Nkx6.1, NeuroD, Isl-1, HNF-3 beta, MAFA, Pax4 и Pax6. Клетки с экспрессией маркеров, характерных для β-клеточной линии дифференцирования, включают β-клетки.
Используемый в настоящей заявке термин «клетки, экспрессирующие маркеры, характерные для линии сформированной эндодермы» относится к клеткам с экспрессией по меньшей мере одного из следующих маркеров: SOX-17, GATA-4, HNF-3 beta, GSC, Cer1, Nodal, FGF8, Brachyury, гомеобоксный белок Mix-like, FGF4 CD48, эомезодермин (EOMES), DKK4, FGF17, GATA-6, CXCR4, C-Kit, CD99 и OTX2. Клетки, экспрессирующие маркеры, характерные для линии сформированной эндодермы, включают клетки-предшественники первичной полоски, клетки первичной полоски, клетки мезоэндодермы и клетки сформированной эндодермы.
Используемый в настоящей заявке термин «клетки с экспрессией маркеров, характерных для линии дифференцирования в клетки панкреатической эндодермы» относится к клеткам с экспрессией по меньшей мере одного из следующих маркеров: PDX-1, HNF-1 beta, PTF-1 alpha, HNF-6 и HB9. Клетки с экспрессией маркеров, характерных для линии дифференцирования в клетки панкреатической эндодермы, включают клетки панкреатической эндодермы.
Используемый в настоящей заявке термин «клетки с экспрессией маркеров, характерных для линии дифференцирования в панкреатические эндокринные клетки» относится к клеткам с экспрессией по меньшей мере одного из следующих маркеров: NGN-3, NeuroD, Islet-1, PDX-1, NKX6.1, Pax-4, Ngn-3 и PTF-1 alpha. Клетки с экспрессией маркеров, характерных для линии дифференцирования в панкреатические эндокринные клетки, включают панкреатические эндокринные клетки, панкреатические экспрессирующие гормоны клетки и панкреатические секретирующие гормоны клетки, а также клетки β-клеточной линии дифференцирования.
Используемый в настоящей заявке термин «сформированная эндодерма» относится к клеткам, обладающим характерными особенностями клеток, происходящих в ходе гаструляции от эпибласта, и формирующим желудочно-кишечный тракт и его производные. Клетки сформированной эндодермы экспрессируют следующие маркеры: HNF-3 beta, GATA-4, SOX-17, церберус, OTX2, гузекоид, C-Kit, CD99 и Mixl1.
Используемый в настоящей заявке термин «внеэмбриональная эндодерма» относится к популяции клеток, экспрессирующих по меньшей мере один из следующих маркеров: SOX-7, AFP и SPARC.
Используемый в настоящей заявке термин «маркеры» означает молекулы нуклеиновых кислот или полипептидов с дифференциальной экспрессией в интересующих клетках. В данном контексте под дифференциальной экспрессией подразумевается повышение уровня экспрессии для положительного маркера и понижение уровня экспрессии для отрицательного маркера. Поддающийся обнаружению уровень маркерной нуклеиновой кислоты или полипептида в интересующих клетках оказывается значительно выше или ниже по сравнению с другими клетками, что позволяет идентифицировать интересующую клетку и отличить ее от других клеток с помощью любого из множества известных в данной области способов.
Используемый в настоящей заявке термин «клетка мезэндодермы» относится к клеткам, экспрессирующим по меньшей мере один из следующих маркеров: CD48, эомезодермин (EOMES), SOX-17, DKK4, HNF-3 beta, GSC, FGF17, GATA-6.
Используемый в настоящей заявке термин «панкреатическая эндокринная клетка» или «панкреатическая экспрессирующая гормоны клетка» относится к клеткам, способным к экспрессии по меньшей мере одного из следующих гормонов: инсулин, глюкагон, соматостатин и панкреатический полипептид.
Используемый в настоящей заявке термин «панкреатическая секретирующая гормоны клетка» относится к клеткам, способным к секреции по меньшей мере одного из следующих гормонов: инсулин, глюкагон, соматостатин и панкреатический полипептид.
Используемый в настоящей заявке термин «клетка-предшественник клетки первичной полоски» относится к клеткам, экспрессирующим по меньшей мере один из следующих маркеров: Nodal и FGF8
Используемый в настоящей заявке термин «клетка первичной полоски» относится к клеткам, экспрессирующим по меньшей мере один из следующих маркеров: Brachyury, гомеобоксный белок Mix-like или FGF4.
В одном из вариантов осуществления настоящее изобретение предлагает способ размножения и дифференцирования плюрипотентных клеток, заключающийся в обработке упомянутых плюрипотентных клеток ингибитором активности фермента GSK-3B.
В одном из вариантов осуществления настоящее изобретение предлагает способ размножения и дифференцирования плюрипотентных клеток, включающий:
c. культивирование плюрипотентных клеток, и
d. обработку плюрипотентных клеток ингибитором активности фермента GSK-3B.
В одном из вариантов осуществления упомянутые плюрипотентные клетки дифференцируются в клетки, экспрессирующие маркеры, характерные для линии сформированной эндодермы.
Маркеры, характерные для линии сформированной эндодермы, выбраны из группы, включающей следующие маркеры: SOX17, GATA4, Hnf-3beta, GSC, Cer1, Nodal, FGF8, Brachyury, гомеобоксный белок Mix-like, FGF4 CD48, эомезодермин (EOMES), DKK4, FGF17, GATA6, CXCR4, C-Kit, CD99 и OTX2. В рамках настоящего изобретения возможно использование клеток - производных плюрипотентных клеток, которые экспрессируют по меньшей мере один из маркеров, характерных для линии сформированной эндодермы. В одном из аспектов настоящего изобретения клетка, экспрессирующая маркеры, характерные для линии сформированной эндодермы, представляет собой клетку-предшественник первичной полоски. В другом аспекте настоящего изобретения клетка с экспрессией маркеров, характерных для линии сформированной эндодермы, представляет собой мезэндодермальную клетку. В другом аспекте настоящего изобретения клетка с экспрессией маркеров, характерных для линии сформированной эндодермы, представляет собой клетку сформированной эндодермы.
Упомянутые плюрипотентные клетки могут быть обработаны ингибитором активности фермента GSK-3B в течение от приблизительно одного до приблизительно 72 часов. В качестве альтернативы упомянутые плюрипотентные клетки могут быть обработаны ингибитором активности фермента GSK-3B в течение от приблизительно 12 до приблизительно 48 часов. В качестве альтернативы упомянутые плюрипотентные клетки могут быть обработаны ингибитором активности фермента GSK-3B в течение приблизительно 48 часов.
В одном из вариантов осуществления настоящего изобретения упомянутый ингибитор активности фермента GSK-3B используется в концентрации от приблизительно 100 нМ до приблизительно 100 мкМ. В качестве альтернативы упомянутый ингибитор активности фермента GSK-3B используется в концентрации от приблизительно 1 мкМ до приблизительно 10 мкМ. В качестве альтернативы упомянутый ингибитор активности фермента GSK-3B используется в концентрации приблизительно 10 мкМ.
Соединения, пригодные для применения в способах, составляющих предмет настоящего изобретения
В одном из вариантов осуществления настоящего изобретения упомянутый ингибитор активности фермента GSK-3B представляет собой соединение формулы (I):
 ,
,
где:
R1 представляет собой фенил, замещенный фенил, где заместители для фенила выбраны из следующей группы заместителей: C1-5алкила, галогена, нитро, трифторметила и нитрила; или пиримидинила;
R2 представляет собой фенил, замещенный фенил, где заместители для фенила выбраны из следующей группы заместителей: C1-5алкила, галогена, нитро, трифторметила и нитрила; или пиримидинила, который необязательно несет в качестве заместителя C1-4алкил, и по меньшей мере один из заместителей R1 и R2 представляет собой пиримидинил;
R3 представляет собой водород, 2-(триметилсилил)этоксиметил, C1-5алкоксикарбонил, арилоксикарбонил, арил-C1-5алкилоксикарбонил, арил-C1-5алкил, замещенный арил-C1-5алкил, где упомянутые один или несколько заместителей для арильного фрагмента каждый независимо выбран из следующей группы заместителей: C1-5алкил, C1-5алкокси, галоген, амино, C1-5алкиламино и ди-C1-5алкиламино, фталимидо-C1-5алкил, амино-C1-5алкил, диаминоC1-5алкил, сукцинимидо-C1-5алкил, C1-5алкилкарбонил, арилкарбонил, C1-5алкилкарбонил-C1-5алкил и арилоксикарбонил-C1-5алкил;
R4 представляет собой фрагмент -(A)-(CH2)q-X;
A представляет собой винилен, этинилен или  ;
;
R5 выбран из следующей группы заместителей: водорода, C1-5алкила, фенила и фенил-C1-5алкила;
q равен 0-9;
X выбран из следующей группы заместителей: водорода, гидрокси, винила, замещенного винила, где один или несколько заместителей для винильной группы выбраны из следующей группы заместителей: фтора, брома, хлора и йода, этинила, замещенного этинила, где заместители для этинильной группы выбраны из следующей группы заместителей: фтора, брома, хлора и йода, C1-5алкила, замещенного C1-5алкила, где упомянутые один или несколько заместителей для алкильной группы выбраны из следующей группы заместителей: C1-5алкокси, тригалоалкила, фталимидо и амино, C3-7циклоалкила, C1-5алкокси, замещенного C1-5алкокси, где заместители для алкильной группы выбраны из следующей группы заместителей: фталимидо и амино, фталимидоокси, фенокси, замещенный фенокси, где упомянутые один или несколько заместителей для фенильной группы выбраны из следующей группы заместителей: C1-5алкила, галогена и C1-5алкокси, фенила, замещенного фенила, где упомянутые один или несколько заместителей для фенильной группы выбраны из следующей группы заместителей: C1-5алкила, галогена и C1-5алкокси, арил-C1-5алкила, замещенного арил-C1-5алкила, где упомянутые один или несколько заместителей для арильной группы выбраны из следующей группы заместителей: C1-5алкила, галогена и C1-5алкокси, арилокси-C1-5алкиламино, C1-5алкиламино, ди-C1-5алкиламино, нитрила, оксима, бензилоксиимино, C1-5алкилоксиимино, фталимидо, сукцинимидо, C1-5алкилкарбонилокси, фенилкарбонилокси, замещенного фенилкарбонилокси, где упомянутые один или несколько заместителей для фенильной группы выбраны из следующей группы заместителей: C1-5алкила, галогена и C1-5алкокси, фенил-C1-5алкилкарбонилокси, где упомянутые один или несколько заместителей для фенильной группы выбраны из следующей группы заместителей: C1-5алкила, галогена и C1-5алкокси, аминокарбонилокси, C1-5алкиламинокарбонилокси, ди-C1-5алкиламинокарбонилокси, C1-5алкоксикарбонилокси, замещенного C1-5алкоксикарбонилокси, где упомянутые один или несколько заместителей для алкильной группы выбраны из следующей группы заместителей: метила, этила, изопропила и гексила, феноксикарбонилокси, замещенного феноксикарбонилокси, где упомянутые один или несколько заместителей для фенильной группы выбраны из следующей группы заместителей: C1-5алкила, C1-5алкокси и галогена, C1-5алкилтио, замещенного C1-5алкилтио, где заместители для алкильной группы выбраны из следующей группы заместителей: гидрокси и фталимидо, C1-5алкилсульфонила, фенилсульфонила, замещенного фенилсульфонила, где упомянутые один или несколько заместителей для фенильной группы выбраны из следующей группы заместителей: брома, фтора, хлора, C1-5алкокси и трифторметила; с тем условием, что если A представляет собой  , q равен 0, и X представляет собой H, то R3 не может представлять собой 2-(триметилсилил)этоксиметил; а также их фармацевтически приемлемые соли.
, q равен 0, и X представляет собой H, то R3 не может представлять собой 2-(триметилсилил)этоксиметил; а также их фармацевтически приемлемые соли.
Пример варианта осуществления настоящего изобретения включает соединение формулы (I), где R1 представляет собой замещенный фенил и R2 представляет собой пиримидин-3-ил.
Пример варианта осуществления настоящего изобретения включает соединение формулы (I), где R1 представляет собой 4-фторфенил.
Пример варианта осуществления настоящего изобретения включает соединение формулы (I), где R3 представляет собой водород, арил-C1-5алкил или замещенный арил-C1-5алкил.
Пример варианта осуществления настоящего изобретения включает соединение формулы (I), где R3 представляет собой водород или фенил-C1-5алкил.
Пример варианта осуществления настоящего изобретения включает соединение формулы (I), где A представляет собой этинилен и q равен 0-5.
Пример варианта осуществления настоящего изобретения включает соединение формулы (I), где X представляет собой сукцинимидо, гидрокси, метил, фенил, C1-5алкилсульфонил, C3-6циклоалкил, C1-5алкилкарбонилокси, C1-5алкокси, фенилкарбонилокси, C1-5алкиламино, ди-C1-5алкиламино или нитрил.
Соединения формулы (I) раскрыты в выданном авторам настоящего изобретения патенте США за номером 6214830, полностью включенном в настоящую заявку путем ссылки.
Пример варианта осуществления настоящего изобретения включает соединение формулы (I), где упомянутое соединение выбрано из следующей группы соединений:
Пример варианта осуществления настоящего изобретения включает соединение формулы (I), где упомянутое соединение представляет собой соединение 5 формулы:
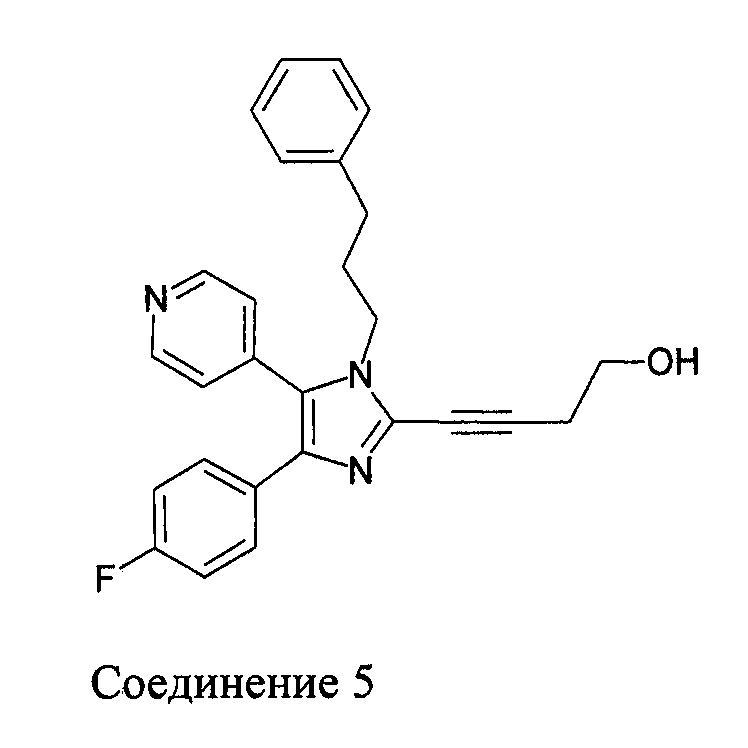
В одном из вариантов осуществления настоящего изобретения упомянутый ингибитор активности фермента GSK-3B представляет собой соединение формулы (II):
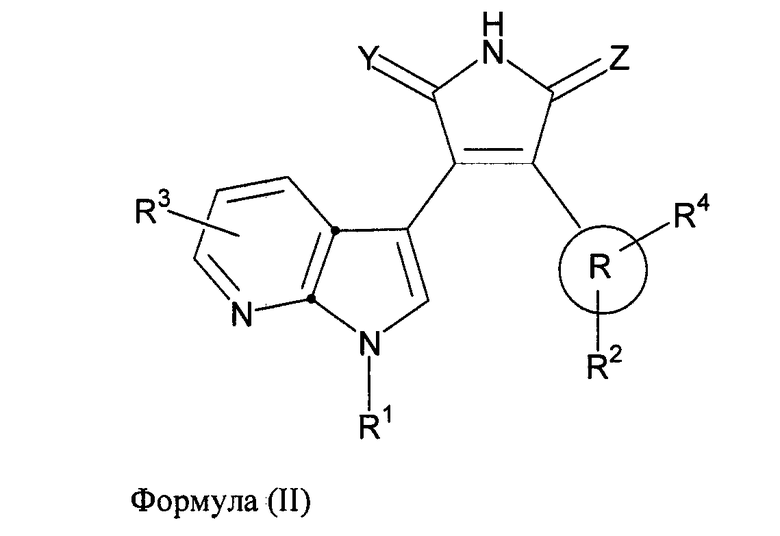 ,
,
где:
R выбран из следующей группы заместителей: Ra, -C1-8алкил-Ra, -C2-8алкенил-Ra, -C2-8алкинил-Ra и циано;
Ra выбран из следующей группы заместителей: циклоалкила, гетероциклила, арила и гетероарила;
R1 выбран из следующей группы заместителей: водорода, -C1-8алкил-R5, -C2-8алкенил-R5, -C2-8алкинил-R5, -C(O)-(C1-8)алкил-R9, -C(O)-арил-R8, -C(O)-O-(C1-8)алкил-R9, -C(O)-O-арил-R8, -C(O)-NH(C1-8алкил-R9), -C(O)-NH(арил-R8), -C(O)-N(C1-8алкил-R9)2, -SO2-(C1-8)алкил-R9, -SO2-арил-R8, -циклоалкил-R6, -гетероциклил-R6, -арил-R6 и -гетероарил-R6; где гетероциклил и гетероарил присоединены к атому азота в первом положении азаиндольного ядра через кольцевой атом углерода гетероциклильного или гетероарильного фрагмента;
R5 представляет собой от 1 до 2 заместителей, независимо выбранных из следующей группы заместителей: водорода, -O-(C1-8)алкила, -O-(C1-8)алкил-OH, -O-(C1-8)алкил-O-(C1-8)алкил, -O-(C1-8)алкил-NH2, -O-(C1-8)алкил-NH(C1-8алкил), -O-(C1-8)алкил-N(C1-8алкил)2, -O-(C1-8)алкил-S-(C1-8)алкил, -O-(C1-8)алкил-SO2-(C1-8)алкил, -O-(C1-8)алкил-SO2-NH2, -O-(C1-8)алкил-SO2-NH(C1-8алкил), -O-(C1-8)алкил-SO2-N(C1-8алкил)2, -O-C(O)H, -O-C(O)-(C1-8)алкил, -O-C(O)-NH2, -O-C(O)-NH(C1-8алкил), -O-C(O)-N(C1-8алкил)2, -O-(C1-8)алкил-C(O)H, -O-(C1-8)алкил-C(O)-(C1-8)алкил, -O-(C1-8)алкил-CO2H, -O-(C1-8)алкил-C(O)-O-(C1-8)алкил, -O-(C1-8)алкил-C(O)-NH2, -O-(C1-8)алкил-C(O)-NH(C1-8алкил), -O-(C1-8)алкил-C(O)-N(C1-8алкил)2, -C(O)H, -C(O)-(C1-8)алкил, -CO2H, -C(O)-O-(C1-8)алкил, -C(O)-NH2, -C(NH)-NH2, -C(O)-NH(C1-8алкил), -C(O)-N(C1-8алкил)2, -SH, -S-(C1-8)алкил, -S-(C1-8)алкил-S-(C1-8)алкил, -S-(C1-8)алкил-O-(C1-8)алкил, -S-(C1-8)алкил-O-(C1-8)алкил-OH, -S-(C1-8)алкил-O-(C1-8)алкил-NH2, -S-(C1-8)алкил-O-(C1-8)алкил-NH(C1-8алкил), -S-(C1-8)алкил-O-(C1-8)алкил-N(C1-8алкил)2, -S-(C1-8)алкил-NH(C1-8алкил), -SO2-(C1-8)алкил, -SO2-NH2, -SO2-NH(C1-8алкил), -SO2-N(C1-8алкил)2, -N-R7, циано, (гало)1-3, гидрокси, нитро, оксо, -циклоалкил-R6, -гетероциклил-R6, -арил-R6 и -гетероарил-R6;
R6 представляет собой от 1 до 4 заместителей, присоединенных к атому углерода или атому азота и независимо выбранных из следующей группы заместителей: водорода, -C1-8алкила, -C2-8алкенила, -C2-8алкинила, -C(O)H, -C(O)-(C1-8)алкила, -CO2H, -C(O)-O-(C1-8)алкила, -C(O)-NH2, -C(NH)-NH2, -C(O)-NH(C1-8алкила), -C(O)-N(C1-8)алкил)2, -SO2-(C1-8)алкила, -SO2-NH2, -SO2-NH(C1-8алкила), -SO2-N(C1-8алкил)2, -(C1-8)алкил-N-R7, -(C1-8)алкил-(гало)1-3, -(C1-8)алкил-OH, -арил-R8, -(C1-8)алкил-арил-R8 и -(C1-8)алкил-гетероарил-R8; с тем условием, что когда заместитель R6 присоединен к атому углерода, упомянутый заместитель R6 может также быть выбран из следующей группы заместителей: -C1-8алкокси, -(C1-8)алкокси-(гало)1-3, -SH, -S-(C1-8)алкила, -N-R7, циано, гало, гидрокси, нитро, оксо и -гетероарил-R8;
R7 представляет собой 2 заместителя, независимо выбранных из следующей группы заместителей: водорода, -C1-8алкила, -C2-8алкенила, -C2-8алкинила, -(C1-8)алкил-OH, -(C1-8)алкил-O-(C1-8)алкила, -(C1-8)алкил-NH2, -(C1-8)алкил-NH(C1-8алкил), -(C1-8)алкил-N(C1-8алкил)2, -(C1-8)алкил-S-(C1-8)алкила, -C(O)H, -C(O)-(C1-8)алкила, -C(O)-O-(C1-8)алкила, -C(O)-NH2, -C(O)-NH(C1-8алкил), -C(O)-N(C1-8алкил)2, -SO2-(C1-8)алкила, -SO2-NH2, -SO2-NH(C1-8алкил), -SO2-N(C1-8алкил)2, -C(N)-NH2, -циклоалкил-R8, -(C1-8)алкил-гетероциклил-R8, -арил-R8, -(C1-8)алкил-арил-R8 и -(C1-8)алкил-гетероарил-R8;
R8 представляет собой от 1 до 4 заместителей, присоединенных к атому углерода или атому азота и независимо выбранных из следующей группы заместителей: водорода, -C1-8алкила, -(C1-8)алкил-(гало)1-3 и -(C1-8)алкил-OH; с тем условием, что когда заместитель R8 присоединен к атому углерода, упомянутый заместитель R8 может также быть выбран из следующей группы заместителей: -C1-8алкокси, -NH2, -NH(C1-8алкил), -N(C1-8алкил)2, циано, гало, -(C1-8)алкокси-(гало)1-3, гидрокси и нитро;
R9 представляет собой от 1 до 2 заместителей, независимо выбранных из следующей группы заместителей: водорода, -C1-8алкокси, -NH2, -NH(C1-8алкил), -N(C1-8алкил)2, циано, (гало)1-3, гидрокси и нитро;
R2 представляет собой один заместитель, присоединенный к атому углерода или атому азота и выбранный из следующей группы заместителей: водорода, -C1-8алкил-R5, -C2-8алкенил-R5, -C2-8алкинил-R5, -C(O)H, -C(O)-(C1-8)алкил-R9, -C(O)-NH2, -C(O)-NH(C1-8алкил-R9), -C(O)-N(C1-8алкил-R9)2, -C(O)-NH(арил-R8), -C(O)-циклоалкил-R8, -C(O)-гетероциклил-R8, -C(O)-арил-R8, -C(O)-гетероарил-R8, -CO2H, -C(O)-O-(C1-8)алкил-R9, -C(O)-O-арил-R8, -SO2-(C1-8)алкил-R9, -SO2-арил-R8, -циклоалкил-R6, -арил-R6 и -(C1-8)алкил-N-R7; с тем условием, что когда заместитель R2 присоединен к атому углерода, упомянутый заместитель R2 может также выбираться из следующей группы заместителей: -C1-8алкокси-R5, -N-R7, циано, галогена, гидрокси, нитро, оксо, -гетероциклил-R6 и -гетероарил-R6;
R3 представляет собой от 1 до 3 заместителей, присоединенных к атому углерода и независимо выбранных из следующей группы заместителей: водорода, -C1-8алкил-R10, -C2-8алкенил-R10, -C2-8алкинил-R10, -C1-8алкокси-R10, -C(O)H, -C(O)-(C1-8)алкил-R9, -C(O)-NH2, -C(O)-NH(C1-8алкил-R9), -C(O)-N(C1-8алкил-R9)2, -C(O)-циклоалкил-R8, -C(O)-гетероциклил-R8, -C(O)-арил-R8, -C(O)-гетероарил-R8, -C(NH)-NH2, -CO2H, -C(O)-O-(C1-8)алкил-R9, -C(O)-O-арил-R8, -SO2-(C1-8)алкил-R9, -SO2-арил-R8, -N-R7, циано, галогена, гидрокси, нитро, -циклоалкил-R8, -гетероциклил-R8, -арил-R8 и -гетероарил-R8;
R4 представляет собой от 1 до 4 заместителей, присоединенных к атому углерода и независимо выбранных из следующей группы заместителей: водорода, -C1-8алкил-R10, -C2-8алкенил-R10, -C2-8алкинил-R10, -C1-8алкокси-R10, -C(O)H, -C(O)-(C1-8)алкил-R9, -C(O)-NH2, -C(O)-NH(C1-8алкил-R9), -C(O)-N(C1-8алкил-R9)2, -C(O)-циклоалкил-R8, -C(O)-гетероциклил-R8, -C(O)-арил-R8, -C(O)-гетероарил-R8, -C(NH)-NH2, -CO2H, -C(O)-O-(C1-8)алкил-R9, -C(O)-O-арил-R8, -SH, -S-(C1-8)алкил-R10, -SO2-(C1-8)алкил-R9, -SO2-арил-R8, -SO2-NH2, -SO2-NH(C1-8алкил-R9), -SO2-N(C1-8алкил-R9)2, -N-R7, циано, галогена, гидрокси, нитро, -циклоалкил-R8, -гетероциклил-R8, -арил-R8 и -гетероарил-R8;
R10 представляет собой от 1 до 2 заместителей, независимо выбранных из следующей группы заместителей: водорода, -NH2, -NH(C1-8алкил), -N(C1-8алкил)2, циано, (гало)1-3, гидрокси, нитро и оксо; и
Y и Z каждый независимо выбран из группы O, S, (H,OH) и (H,H); с тем условием, что один из Y и Z представляет собой атом O, а второй выбран из группы O, S, (H,OH) и (H,H); а также их фармацевтически приемлемые соли.
Варианты осуществления настоящего изобретения включают соединения формулы (II), где R выбран из следующей группы заместителей: Ra, -C1-4алкил-Ra, -C2-4алкенил-Ra, -C2-4алкинил-Ra и циано.
Варианты осуществления настоящего изобретения включают соединения формулы (II), где Ra выбран из следующей группы заместителей: гетероциклила, арила и гетероарила.
В одном из вариантов осуществления настоящего изобретения Ra выбран из следующей группы заместителей: дигидропиранила, фенила, нафтила, тиенила, пирролила, имидазолила, пиразолила, пиридинила, азаиндолила, индазолила, бензофурила, бензотиенила, дибензофурила и дибензотиенила.
Варианты осуществления настоящего изобретения включают соединения формулы (II), где R1 выбран из следующей группы заместителей: водорода, -C1-4алкил-R5, -C2-4алкенил-R5, -C2-4алкинил-R5, -C(O)-(C1-4)алкил-R9, -C(O)-арил-R8, -C(O)-O-(C1-4)алкил-R9, -C(O)-O-арил-R8, -C(O)-NH(C1-4алкил-R9), -C(O)-NH(арил-R8), -C(O)-N(C1-4алкил-R9)2, -SO2-(C1-4)алкил-R9, -SO2-арил-R8, -циклоалкил-R6, -гетероциклил-R6, -арил-R6 и -гетероарил-R6; где гетероциклил и гетероарил присоединены к атому азота в первом положении азаиндольного ядра через кольцевой атом углерода гетероциклильного или гетероарильного фрагмента.
В одном из вариантов осуществления настоящего изобретения R1 выбран из следующей группы заместителей: водорода, -C1-4алкил-R5, -арил-R6 и -гетероарил-R6; где гетероарил присоединен к атому азота в первом положении азаиндольного ядра через кольцевой атом углерода гетероарильного фрагмента.
В одном из вариантов осуществления настоящего изобретения R1 выбран из следующей группы заместителей: водорода, -C1-4алкил-R5 и -нафтил-R6.
Варианты осуществления настоящего изобретения включают соединения формулы (II), где R5 представляет собой от 1 до 2 заместителей, независимо выбранных из следующей группы заместителей: водорода, -O-(C1-4)алкила, -O-(C1-4)алкил-OH, -O-(C1-4)алкил-O-(C1-4)алкила, -O-(C1-4)алкил-NH2, -O-(C1-4)алкил-NH(C1-4алкил), -O-(C1-4)алкил-N(C1-4алкил)2, -O-(C1-4)алкил-S-(C1-4)алкил, -O-(C1-4)алкил-SO2-(C1-4)алкил, -O-(C1-4)алкил-SO2-NH2, -O-(C1-4)алкил-SO2-NH(C1-4алкил), -O-(C1-4)алкил-SO2-N(C1-4алкил)2, -O-C(O)H, -O-C(O)-(C1-4)алкила, -O-C(O)-NH2, -O-C(O)-NH(C1-4алкил), -O-C(O)-N(C1-4алкил)2, -O-(C1-4)алкил-C(O)H, -O-(C1-4)алкил-C(O)-(C1-4)алкил, -O-(C1-4)алкил-CO2H, -O-(C1-4)алкил-C(O)-O-(C1-4)алкил, -O-(C1-4)алкил-C(O)-NH2, -O-(C1-4)алкил-C(O)-NH(C1-4алкил), -O-(C1-4)алкил-C(O)-N(C1-4алкил)2, -C(O)H, -C(O)-(C1-4)алкила, -CO2H, -C(O)-O-(C1-4)алкила, -C(O)-NH2, -C(NH)-NH2, -C(O)-NH(C1-4алкил), -C(O)-N(C1-4алкил)2, -SH, -S-(C1-4)алкила, -S-(C1-4)алкил-S-(C1-4)алкил, -S-(C1-4)алкил-O-(C1-4)алкила, -S-(C1-4)алкил-O-(C1-4)алкил-OH, -S-(C1-4)алкил-O-(C1-4)алкил-NH2, -S-(C1-4)алкил-O-(C1-4)алкил-NH(C1-4алкил), -S-(C1-4)алкил-O-(C1-4)алкил-N(C1-4алкил)2, -S-(C1-4)алкил-NH(C1-4алкил), -SO2-(C1-4)алкил, -SO2-NH2, -SO2-NH(C1-4алкил), -SO2-N(C1-4алкил)2, -N-R7, циано, (гало)1-3, гидрокси, нитро, оксо, -циклоалкил-R6, -гетероциклил-R6, -арил-R6 и -гетероарил-R6.
В одном из вариантов осуществления настоящего изобретения R5 представляет собой от 1 до 2 заместителей, независимо выбранных из следующей группы заместителей: водорода, -O-(C1-4)алкила, -N-R7, гидрокси и -гетероарил-R6.
В одном из вариантов осуществления настоящего изобретения R5 представляет собой от 1 до 2 заместителей, независимо выбранных из следующей группы заместителей: водорода, -O-(C1-4)алкила, -N-R7, гидрокси, -имидазолил-R6, -триазолил-R6 и -тетразолил-R6.
Варианты осуществления настоящего изобретения включают соединения формулы (II), где R6 представляет собой от 1 до 4 заместителей, присоединенных к атому углерода или атому азота и независимо выбранных из следующей группы заместителей: водорода, -C1-4алкила, -C2-4алкенила, -C2-4алкинила, -C(O)H, -C(O)-(C1-4)алкила, -CO2H, -C(O)-O-(C1-4)алкила, -C(O)-NH2, -C(NH)-NH2, -C(O)-NH(C1-4алкил), -C(O)-N(C1-4)алкил)2, -SO2-(C1-4)алкила, -SO2-NH2, -SO2-NH(C1-4алкил), -SO2-N(C1-4алкил)2, -(C1-4)алкил-N-R7, -(C1-4)алкил-(гало)1-3, -(C1-4)алкил-OH, -арил-R8, -(C1-4)алкил-арил-R8 и -(C1-4)алкил-гетероарил-R8; с тем условием, что когда заместитель R6 присоединен к атому углерода, упомянутый заместитель R6 может также выбираться из следующей группы заместителей: -C1-4алкокси, -(C1-4)алкокси-(гало)1-3, -SH, -S-(C1-4)алкила, -N-R7, циано, гало, гидрокси, нитро, оксо и -гетероарил-R8.
В одном из вариантов осуществления настоящего изобретения R6 представляет собой водород.
Варианты осуществления настоящего изобретения включают соединения формулы (II), где R7 представляет собой 2 заместителя, независимо выбранных из следующей группы заместителей: водорода, -C1-4алкила, -C2-4алкенила, -C2-4алкинила, -(C1-4)алкил-OH, -(C1-4)алкил-O-(C1-4)алкила, -(C1-4)алкил-NH2, -(C1-4)алкил-NH(C1-4алкил), -(C1-4)алкил-N(C1-4алкил)2, -(C1-4)алкил-S-(C1-4)алкила, -C(O)H, -C(O)-(C1-4)алкила, -C(O)-O-(C1-4)алкила, -C(O)-NH2, -C(O)-NH(C1-4алкил), -C(O)-N(C1-4алкил)2, -SO2-(C1-4)алкила, -SO2-NH2, -SO2-NH(C1-4алкил), -SO2-N(C1-4алкил)2, -C(N)-NH2, -циклоалкил-R8, -(C1-4)алкил-гетероциклил-R8, -арил-R8, -(C1-4)алкил-арил-R8 и -(C1-4)алкил-гетероарил-R8.
В одном из вариантов осуществления настоящего изобретения R7 представляет собой 2 заместителя, независимо выбранных из следующей группы заместителей: водорода, -C1-4алкила, -C(O)H, -C(O)-(C1-4)алкила, -C(O)-O-(C1-4)алкила, -SO2-NH2, -SO2-NH(C1-4алкил) и -SO2-N(C1-4алкил)2.
Варианты осуществления настоящего изобретения включают соединения формулы (II), где R8 представляет собой от 1 до 4 заместителей, присоединенных к атому углерода или атому азота и независимо выбранных из следующей группы заместителей: водорода, -C1-4алкила, -(C1-4)алкил-(гало)1-3 и -(C1-4)алкил-OH; с тем условием, что когда заместитель R8 присоединен к атому углерода, упомянутый заместитель R8 может также выбираться из следующей группы заместителей: -C1-4алкокси, -NH2, -NH(C1-4алкил), -N(C1-4алкил)2, циано, гало, -(C1-4)алкокси-(гало)1-3, гидрокси и нитро.
В одном из вариантов осуществления настоящего изобретения R8 представляет собой водород.
Варианты осуществления настоящего изобретения включают соединения формулы (II), где R9 представляет собой от 1 до 2 заместителей, независимо выбранных из следующей группы заместителей: водорода, -C1-4алкокси, -NH2, -NH(C1-4алкил), -N(C1-4алкил)2, циано, (гало)1-3, гидрокси и нитро.
В одном из вариантов осуществления настоящего изобретения R9 представляет собой водород.
Варианты осуществления настоящего изобретения включают соединения формулы (II), где R2 представляет собой один заместитель, присоединенный к атому углерода или атому азота и выбранный из следующей группы заместителей: водорода, -C1-4алкил-R5, -C2-4алкенил-R5, -C2-4алкинил-R5, -C(O)H, -C(O)-(C1-4)алкил-R9, -C(O)-NH2, -C(O)-NH(C1-4алкил-R9), -C(O)-N(C1-4алкил-R9)2, -C(O)-NH(арил-R8), -C(O)-циклоалкил-R8, -C(O)-гетероциклил-R8, -C(O)-арил-R8, -C(O)-гетероарил-R8, -CO2H, -C(O)-O-(C1-4)алкил-R9, -C(O)-O-арил-R8, -SO2-(C1-4)алкил-R9, -SO2-арил-R8, -циклоалкил-R6, -арил-R6 и -(C1-4)алкил-N-R7; с тем условием, что когда заместитель R2 присоединен к атому углерода, упомянутый заместитель R2 может также выбираться из следующей группы заместителей: -C1-4алкокси-R5, -N-R7, циано, галогена, гидрокси, нитро, оксо, -гетероциклил-R6 и -гетероарил-R6.
В одном из вариантов осуществления настоящего изобретения R2 представляет собой один заместитель, присоединенный к атому углерода или атому азота и выбранный из следующей группы заместителей: водорода, -C1-4алкил-R5, -C2-4алкенил-R5, -C2-4алкинил-R5, -CO2H, -C(O)-O-(C1-4)алкил-R9, -циклоалкил-R6, -арил-R6 и -(C1-4)алкил-N-R7; с тем условием, что когда заместитель R2 присоединен к атому азота, не происходит образования четвертичной соли; и с тем условием, что когда заместитель R2 присоединен к атому углерода, упомянутый заместитель R2 может также выбираться из следующей группы заместителей: -C1-4алкокси-R5, -N-R7, циано, галогена, гидрокси, нитро, оксо, -гетероциклил-R6 и -гетероарил-R6.
В одном из вариантов осуществления настоящего изобретения R2 представляет собой один заместитель, присоединенный к атому углерода или атому азота и выбранный из следующей группы заместителей: водорода, -C1-4алкил-R5 и -арил-R6; с тем условием, что когда заместитель R2 присоединен к атому азота, не происходит образования четвертичной соли; и с тем условием, что когда заместитель R2 присоединен к атому углерода, упомянутый заместитель R2 может также выбираться из следующей группы заместителей: -N-R7, галогена, гидрокси и -гетероарил-R6.
Варианты осуществления настоящего изобретения включают соединения формулы (II), где R3 представляет собой от 1 до 3 заместителей, присоединенных к атому углерода и независимо выбранных из следующей группы заместителей: водорода, -C1-4алкил-R10, -C2-4алкенил-R10, -C2-4алкинил-R10, -C1-4алкокси-R10, -C(O)H, -C(O)-(C1-4)алкил-R9, -C(O)-NH2, -C(O)-NH(C1-4алкил-R9), -C(O)-N(C1-4алкил-R9)2, -C(O)-циклоалкил-R8, -C(O)-гетероциклил-R8, -C(O)-арил-R8, -C(O)-гетероарил-R8, -C(NH)-NH2, -CO2H, -C(O)-O-(C1-4)алкил-R9, -C(O)-O-арил-R8, -SO2-(C1-8)алкил-R9, -SO2-арил-R8, -N-R7, -(C1-4)алкил-N-R7, циано, галогена, гидрокси, нитро, -циклоалкил-R8, -гетероциклил-R8, -арил-R8 и -гетероарил-R8.
В одном из вариантов осуществления настоящего изобретения R3 представляет собой один заместитель, присоединенный к атому углерода и выбранный из следующей группы заместителей: водорода, -C1-4алкил-R10, -C2-4алкенил-R10, -C2-4алкинил-R10, -C1-4алкокси-R10, -C(O)H, -CO2H, -NH2, -NH(C1-4алкил), -N(C1-4алкил)2, циано, галогена, гидрокси и нитро.
В одном из вариантов осуществления настоящего изобретения R3 представляет собой один заместитель, присоединенный к атому углерода и выбранный из следующей группы заместителей: водорода, -C1-4алкил-R10, -NH2, -NH(C1-4алкил), -N(C1-4алкил)2, галогена и гидрокси.
Варианты осуществления настоящего изобретения включают соединения формулы (II), где R4 представляет собой от 1 до 4 заместителей, присоединенных к атому углерода и независимо выбранных из следующей группы заместителей: водорода, -C1-4алкил-R10, -C2-4алкенил-R10, -C2-4алкинил-R10, -C1-4алкокси-R10, -C(O)H, -C(O)-(C1-4)алкил-R9, -C(O)-NH2, -C(O)-NH(C1-4алкил-R9), -C(O)-N(C1-4алкил-R9)2, -C(O)-циклоалкил-R8, -C(O)-гетероциклил-R8, -C(O)-арил-R8, -C(O)-гетероарил-R8, -C(NH)-NH2, -CO2H, -C(O)-O-(C1-4)алкил-R9, -C(O)-O-арил-R8, -SH, -S-(C1-4)алкил-R10, -SO2-(C1-4)алкил-R9, -SO2-арил-R8, -SO2-NH2, -SO2-NH(C1-4алкил-R9), -SO2-N(C1-4алкил-R9)2, -N-R7, циано, галогена, гидрокси, нитро, -циклоалкил-R8, -гетероциклил-R8, -арил-R8 и -гетероарил-R8.
В одном из вариантов осуществления настоящего изобретения R4 представляет собой от 1 до 4 заместителей, присоединенных к атому углерода и независимо выбранных из следующей группы заместителей: водорода, -C1-4алкил-R10, -C2-4алкенил-R10, -C2-4алкинил-R10, -C1-4алкокси-R10, -C(O)H, -CO2H, -NH2, -NH(C1-4алкил), -N(C1-4алкил)2, циано, галогена, гидрокси, нитро, -циклоалкила, -гетероциклила, -арила и -гетероарила.
В одном из вариантов осуществления настоящего изобретения R4 представляет собой от 1 до 4 заместителей, присоединенных к атому углерода и независимо выбранных из следующей группы заместителей: водорода, C1-4алкил-R10, C1-4алкокси-R10, -NH2, -NH(C1-4алкил), -N(C1-4алкил)2, галогена и гидрокси.
В одном из вариантов осуществлении настоящего изобретения R4 представляет собой от 1 до 4 заместителей, присоединенных к атому углерода и независимо выбранных из следующей группы заместителей: водорода, C1-4алкил-R10, C1-4алкокси-R10, -NH2, -NH(C1-4алкил), -N(C1-4алкил)2, хлора, фтора и гидрокси.
Варианты осуществления настоящего изобретения включают соединения формулы (II), где R10 представляет собой от 1 до 2 заместителей, независимо выбранных из следующей группы заместителей: водорода, -NH2, -NH(C1-4алкил), -N(C1-4алкил)2, циано, (гало)1-3, гидрокси, нитро и оксо.
В одном из вариантов осуществления настоящего изобретения R10 представляет собой от 1 до 2 заместителей, независимо выбранных из следующей группы заместителей: водорода и (гало)1-3.
В одном из вариантов осуществления настоящего изобретения R10 представляет собой от 1 до 2 заместителей, независимо выбранных из следующей группы заместителей: водорода и (фтор)3.
Варианты осуществления настоящего изобретения включают соединения формулы (II), где Y и Z каждый независимо выбран из группы O, S, (H,OH) и (H,H); с тем условием, что один из Y и Z представляет собой атом O, а второй выбран из группы O, S, (H,OH) и (H,H).
В одном из вариантов осуществления настоящего изобретения Y и Z каждый независимо выбран из группы O и (H,H); с тем условием, что один из Y и Z представляет собой атом O, а второй выбран из группы O и (H,H).
В одном из вариантов осуществления настоящего изобретения Y и Z представляют собой атомы O.
Соединения формулы (II) раскрыты в выданном авторам настоящего изобретения патенте США за номером 7125878, полное раскрытие которого включено в настоящую заявку путем ссылки.
Пример варианта осуществления настоящего изобретения включает соединение формулы (II), где упомянутое соединение выбрано из следующей группы соединений:
Пример варианта осуществления настоящего изобретения включает соединение формулы (II), где упомянутое соединение выбрано из следующей группы соединений:

В одном из вариантов осуществления настоящего изобретения упомянутый ингибитор активности фермента GSK-3B представляет собой соединение формулы (III):
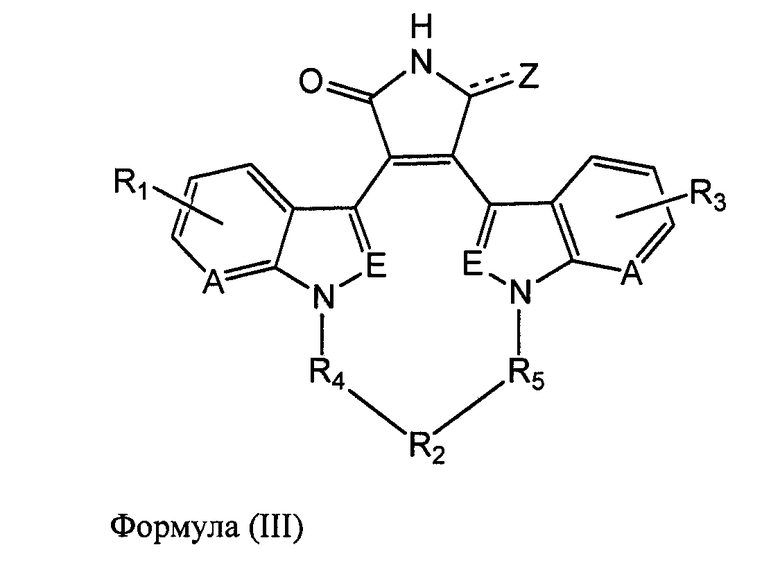 ,
,
где
фрагменты A и E каждый независимо выбран из фрагмента CH и атома азота; где  выбран из следующей группы: 1H-индола, 1H-пирроло[2,3-b]пиридина, 1H-пиразоло[3,4-b]пиридина и 1H-индазола;
выбран из следующей группы: 1H-индола, 1H-пирроло[2,3-b]пиридина, 1H-пиразоло[3,4-b]пиридина и 1H-индазола;
Z представляет собой атом O; в качестве альтернативы Z представляет собой два атома водорода, где каждый атом водорода присоединен через одинарную связь;
R4 и R5 каждый независимо выбран из следующей группы заместителей: C1-8алкила, C2-8алкенила и C2-8алкинила, необязательно несущие в качестве заместителей оксогруппы;
R2 выбран из следующей группы заместителей: -C1-8алкил-, -C2-8алкенил-, -C2-8алкинил-, -O-(C1-8)алкил-O-, -O-(C2-8)алкенил-O-, -O-(C2-8)алкинил-O-, -C(O)-(C1-8)алкил-C(O)- (где любая из вышеупомянутых алкильной, алкенильной и алкинильной мостиковых групп представляет собой линейную углеводородную цепь, необязательно несущую от одного до четырех заместителей, независимо выбранных из следующей группы заместителей: C1-8алкила, C1-8алкокси, C1-8алкокси(C1-8)алкила, карбоксила, карбоксил-(C1-8)алкила, -C(O)O-(C1-8)алкила, -C1-8алкил-C(O)O-(C1-8)алкила, амино (несущая заместитель, независимо выбранный из следующей группы заместителей: водорода и C1-4алкила), амино-(C1-8)алкила (где аминогруппа несет заместитель, независимо выбранный из следующей группы заместителей: водорода и C1-4алкил), галогена, (гало)1-3(C1-8)алкила, (гало)1-3(C1-8)алкокси, гидрокси, гидрокси-(C1-8)алкила и оксо; и где любая из вышеупомянутых алкильной, алкенильной и алкинильной мостиковых групп необязательно несет от одного до двух заместителей, независимо выбранных из следующей группы заместителей: гетероциклила, арила, гетероарила, гетероциклил-(C1-8)алкила, арил-(C1-8)алкила, гетероарил-(C1-8)алкила, спироциклоалкила и спирогетероциклила (где любой из вышеупомянутых циклоалкильного, гетероциклильного, арильного и гетероарильного заместителей необязательно несет от одного до четырех заместителей, независимо выбранных из следующей группы заместителей: C1-8алкила, C1-8алкокси, C1-8алкокси-(C1-8)алкила, карбоксила, карбоксил-(C1-8)алкила, амино (несущая заместитель, независимо выбранный из следующей группы заместителей: водорода и C1-4алкила), амино-(C1-8)алкила (где аминогруппа несет заместитель, независимо выбранный из следующей группы заместителей: водорода и C1-4алкила), галогена, (гало)1-3(C1-8)алкила, (гало)1-3(C1-8)алкокси, гидрокси и гидрокси-(C1-8)алкила; и где любой из вышеупомянутых гетероциклильных заместителей необязательно несет в качестве заместителей оксогруппы)), циклоалкила, гетероциклила, арила, гетероарила (где циклоалкил, гетероциклил, арил и гетероарил необязательно несут от одного до четырех заместителей, независимо выбранных из следующей группы заместителей: C1-8алкила, C1-8алкокси, C1-8алкокси-(C1-8)алкила, карбоксила, карбоксил-(C1-8)алкила, амино (несущая заместитель, независимо выбранный из следующей группы заместителей: водорода и C1-4алкила), амино-(C1-8)алкила (где аминогруппа несет заместитель, независимо выбранный из следующей группы заместителей: водорода и C1-4алкила), галогена, (гало)1-3(C1-8)алкила, (гало)1-3(C1-8)алкокси, гидрокси и гидрокси-(C1-8)алкила; и где гетероциклил необязательно несет в качестве заместителей оксогруппы), -(O-(CH2)1-6)0-5-O-, -O-(CH2)1-6-O-(CH2)1-6-O-, -O-(CH2)1-6-O-(CH2)1-6-O-(CH2)1-6-O-, -(O-(CH2)1-6)0-5-NR6-, -O-(CH2)1-6-NR6-(CH2)1-6-O-, -O-(CH2)1-6-O-(CH2)1-6-NR6-, -(O-(CH2)1-6)0-5-S-, -O-(CH2)1-6-S-(CH2)1-6-O-, -O-(CH2)1-6-O-(CH2)1-6-S-, -NR6-, -NR6-NR7-, -NR6-(CH2)1-6-NR7-, -NR6-(CH2)1-6-NR7-(CH2)1-6-NR8-, -NR6-C(O)-, -C(O)-NR6-, -C(O)-(CH2)0-6-NR6-(CH2)0-6-C(O)-, -NR6-(CH2)0-6-C(O)-(CH2)1-6-C(O)-(CH2)0-6-NR7-, -NR6-C(O)-NR7-, -NR6-C(NR7)-NR8-, -O-(CH2)1-6-NR6-(CH2)1-6-S-, -S-(CH2)1-6-NR6-(CH2)1-6-O-, -S-(CH2)1-6-NR6-(CH2)1-6-S-, -NR6-(CH2)1-6-S-(CH2)1-6-NR7- и -SO2- (где R6, R7 и R8 независимо выбираются из следующей группы заместителей: водорода, C1-8алкила, C1-8алкокси-(C1-8)алкила, карбоксил-(C1-8)алкила, амино-(C1-8)алкила (где аминогруппа несет заместитель, независимо выбранный из следующей группы заместителей: водорода и C1-4алкила), гидрокси-(C1-8)алкила, гетероциклил-(C1-8)алкила, арил-(C1-8)алкила и гетероарил-(C1-8)алкила (где вышеупомянутые гетероциклильные, арильные и гетероарильные заместители необязательно несут от одного до четырех заместителей, независимо выбранных из следующей группы заместителей: C1-8алкила, C1-8алкокси, C1-8алкокси-(C1-8)алкила, карбоксила, карбоксил-(C1-8)алкила, амино (несущая заместитель, независимо выбранный из следующей группы заместителей: водорода и C1-4алкила), амино-(C1-8)алкила (где аминогруппа несет заместитель, независимо выбранный из следующей группы заместителей: водорода и C1-4алкила), галогена, (гало)1-3(C1-8)алкила, (гало)1-3(C1-8)алкокси, гидрокси и гидрокси-(C1-8)алкила; и где гетероциклил необязательно несет в качестве заместителей оксогруппы)); с тем условием, что если в качестве A и E выбраны фрагменты CH, то заместитель R2 выбран из следующей группы заместителей: -C2-8алкинил-, -O-(C1-8)алкил-O-, -O-(C2-8)алкенил-O-, -O-(C2-8)алкинил-O-, -C(O)-(C1-8)алкил-C(O)- (где любая из вышеупомянутых алкильной, алкенильной и алкинильной мостиковых групп предсталяет собой линейную углеводородную цепь, необязательно несущую от одного до четырех заместителей, независимо выбранных из следующей группы заместителей: C1-8алкила, C1-8алкокси, C1-8алкокси-(C1-8)алкила, карбоксила, карбоксил-(C1-8)алкила, -C(O)O-(C1-8)алкила, -C1-8алкил-C(O)O-(C1-8)алкила, амино (несущая заместитель, независимо выбранный из следующей группы заместителей: водорода и C1-4алкила), амино-(C1-8)алкила (где аминогруппа несет заместитель, независимо выбранный из следующей группы заместителей: водорода и C1-4алкила), галогена, (гало)1-3(C1-8)алкила, (гало)1-3(C1-8)алкокси, гидрокси, гидрокси-(C1-8)алкила и оксо; и где любая из вышеупомянутых алкильной, алкенильной и алкинильной мостиковых групп необязательно несет от одного до двух заместителей, независимо выбранных из следующей группы заместителей: гетероциклила, арила, гетероарила, гетероциклил-(C1-8)алкила, арил-(C1-8)алкила, гетероарил-(C1-8)алкила, спироциклоалкила и спирогетероциклила (где любой из вышеупомянутых циклоалкильного, гетероциклильного, арильного и гетероарильного заместителей необязательно несет от одного до четырех заместителей, независимо выбранных из следующей группы заместителей: C1-8алкила, C1-8алкокси, C1-8алкокси-(C1-8)алкила, карбоксила, карбоксил-(C1-8)алкила, амино (несущая заместитель, независимо выбранный из следующей группы заместителей: водорода и C1-4алкила), амино-(C1-8)алкила (где аминогруппа несет заместитель, независимо выбранный из следующей группы заместителей: водорода и C1-4алкила), галогена, (гало)1-3(C1-8)алкила, (гало)1-3(C1-8)алкокси, гидрокси и гидрокси-(C1-8)алкила; и где любой из вышеупомянутых гетероциклильных заместителей необязательно несет в качестве заместителей оксогруппы)), циклоалкил (где циклоалкил необязательно несет от одного до четырех заместителей, независимо выбранных из следующей группы заместителей: C1-8алкила, C1-8алкокси, C1-8алкокси-(C1-8)алкила, карбоксила, карбоксил-(C1-8)алкила, амино (несущая заместитель, независимо выбранный из следующей группы заместителей: водорода и C1-4алкила), амино-(C1-8)алкила (где аминогруппа несет заместитель, независимо выбранный из следующей группы заместителей: водорода и C1-4алкила), галогена, (гало)1-3(C1-8)алкила, (гало)1-3(C1-8)алкокси, гидрокси и гидрокси-(C1-8)алкила), -(O-(CH2)1-6)1-5-O-, -O-(CH2)1-6-O-(CH2)1-6-O-, -O-(CH2)1-6-O-(CH2)1-6-O-(CH2)1-6-O-, -(O-(CH2)1-6)1-5-NR6-, -O-(CH2)1-6-NR6-(CH2)1-6-O-, -O-(CH2)1-6-O-(CH2)1-6-NR6-, -(O-(CH2)1-6)0-5-S-, -O-(CH2)1-6-S-(CH2)1-6-O-, -O-(CH2)1-6-O-(CH2)1-6-S-, -NR6-NR7-, -NR6-(CH2)1-6-NR7-, -NR6-(CH2)1-6-NR7-(CH2)1-6-NR8-, -NR9-C(O)-, -C(O)-NR9-, -C(O)-(CH2)0-6-NR6-(CH2)0-6-C(O)-, -NR6-(CH2)0-6-C(O)-(CH2)1-6-C(O)-(CH2)0-6-NR7-, -NR6-C(O)-NR7-, -NR6-C(NR7)-NR8-, -O-(CH2)1-6-NR6-(CH2)1-6-S-, -S-(CH2)1-6-NR6-(CH2)1-6-O-, -S-(CH2)1-6-NR6-(CH2)1-6-S- и -NR6-(CH2)1-6-S-(CH2)1-6-NR7- (где R6, R7 и R8 каждый независимо выбран из следующей группы заместителей: водорода, C1-8алкила, C1-8алкокси-(C1-8)алкила, карбоксил-(C1-8)алкила, амино-(C1-8)алкила (где аминогруппа несет заместитель, независимо выбранный из следующей группы заместителей: водорода и C1-4алкила), гидрокси-(C1-8)алкила, гетероциклил-(C1-8)алкила, арил-(C1-8)алкила и гетероарил-(C1-8)алкила (где вышеупомянутые гетероциклильные, арильные и гетероарильные заместители необязательно несут от одного до четырех заместителей, независимо выбранных из следующей группы заместителей: C1-8алкила, C1-8алкокси, C1-8алкокси-(C1-8)алкила, карбоксила, карбоксил-(C1-8)алкила, амино (несущая заместитель, независимо выбранный из следующей группы заместителей: водорода и C1-4алкила), амино-(C1-8)алкила (где аминогруппа несет заместитель, независимо выбранный из следующей группы заместителей: водорода и C1-4алкила), галогена, (гало)1-3(C1-8)алкила, (гало)1-3(C1-8)алкокси, гидрокси и гидрокси-(C1-8)алкила; и где гетероциклил необязательно дополнительно несет в качестве заместителей оксогруппы); и где R9 выбран из следующей группы заместителей: C1-8алкила, C1-8алкокси-(C1-8)алкила, карбоксил-(C1-8)алкила, амино-(C1-8)алкила (где аминогруппа несет заместитель, независимо выбранный из следующей группы заместителей: водорода и C1-4алкила), гидрокси-(C1-8)алкила, гетероциклил-(C1-8)алкила, арил-(C1-8)алкила и гетероарил-(C1-8)алкила (где вышеупомянутые гетероциклильные, арильные и гетероарильные заместители необязательно несут от одного до четырех заместителей, независимо выбранных из следующей группы заместителей: C1-8алкила, C1-8алкокси, C1-8алкокси-(C1-8)алкила, карбоксила, карбоксил-(C1-8)алкила, амино (несущая заместитель, независимо выбранный из следующей группы заместителей: C1-4алкила, амино-(C1-8)алкила (где аминогруппа несет заместитель, независимо выбранный из следующей группы заместителей: водорода и C1-4алкила), галогена, (гало)1-3(C1-8)алкила, (гало)1-3(C1-8)алкокси, гидрокси и гидрокси-(C1-8)алкила; и где гетероциклил необязательно несет в качестве заместителей оксогруппы)); и,
R1 и R3 каждый независимо выбран из следующей группы заместителей: водорода, C1-8алкила, C2-8алкенила, C2-8алкинила (где алкил, алкенил и алкинил необязательно несут заместитель, выбранный из следующей группы заместителей: C1-8алкокси, алкокси-(C1-8)алкила, карбоксила, карбоксил-(C1-8)алкила, амино (несущая заместитель, независимо выбранный из следующей группы заместителей: водорода и C1-4алкила), амино-(C1-8)алкила (где аминогруппа несет заместитель, независимо выбранный из следующей группы заместителей: водорода и C1-4алкила), (гало)1-3, (гало)1-3(C1-8)алкила, (гало)1-3(C1-8)алкокси, гидрокси, гидрокси-(C1-8)алкила и оксо), C1-8алкокси, C1-8алкоксикарбонила, (гало)1-3(C1-8)алкокси, C1-8алкилтио, арила, гетероарила (где арил и гетероарил необязательно несут заместитель, независимо выбранный из следующей группы заместителей: C1-8алкила, C1-8алкокси, алкокси-(C1-8)алкила, карбоксила, карбоксил-(C1-8)алкила, амино (несущая заместитель, независимо выбранный из следующей группы заместителей: водорода и C1-4алкила), амино-(C1-8)алкил (где аминогруппа несет заместитель, независимо выбранный из следующей группы заместителей: водорода и C1-4алкила), галогена, (гало)1-3(C1-8)алкила, (гало)1-3(C1-8)алкокси, гидрокси и гидрокси-(C1-8)алкила), амино (несущая заместитель, независимо выбранный из следующей группы заместителей: водорода и C1-4алкила), циано, галогена, гидрокси и нитро; а также их фармацевтически приемлемые соли.
В одном из вариантов осуществления настоящего изобретения соединение формулы (III) представляет собой соединение, выбираемое из следующей группы соединений:
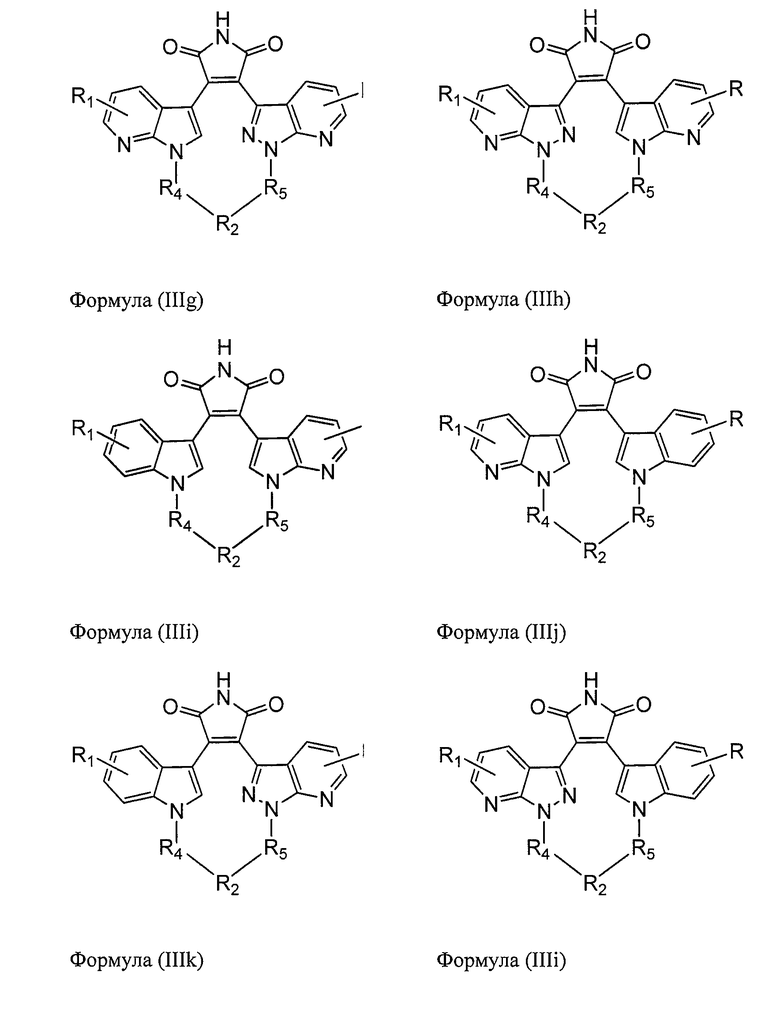
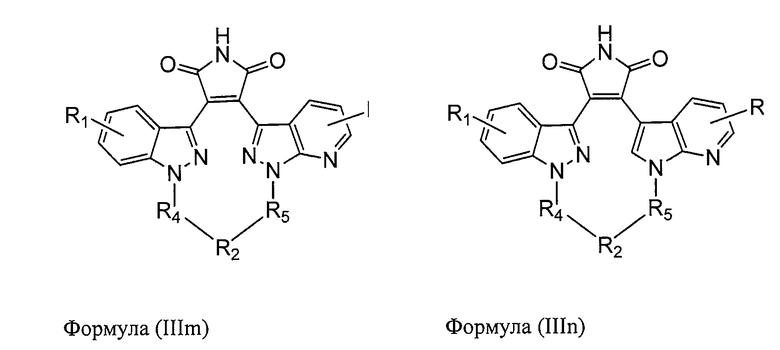 ,
,
где все остальные химические переменные выбраны в соответствии с вышеприведенным описанием; а также их фармацевтически приемлемые соли.
В одном из вариантов осуществления настоящего изобретения соединение формулы (III) представляет собой соединение, выбранное из следующей группы соединений:
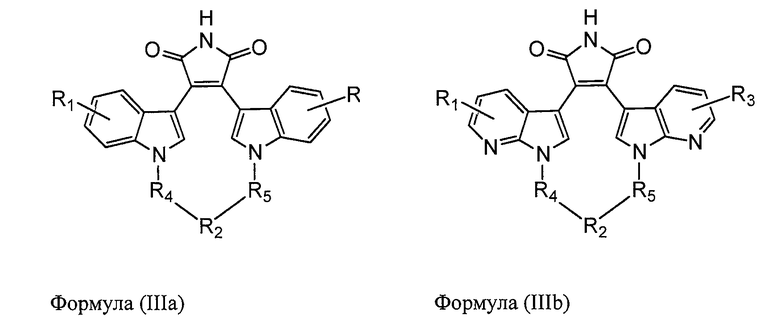
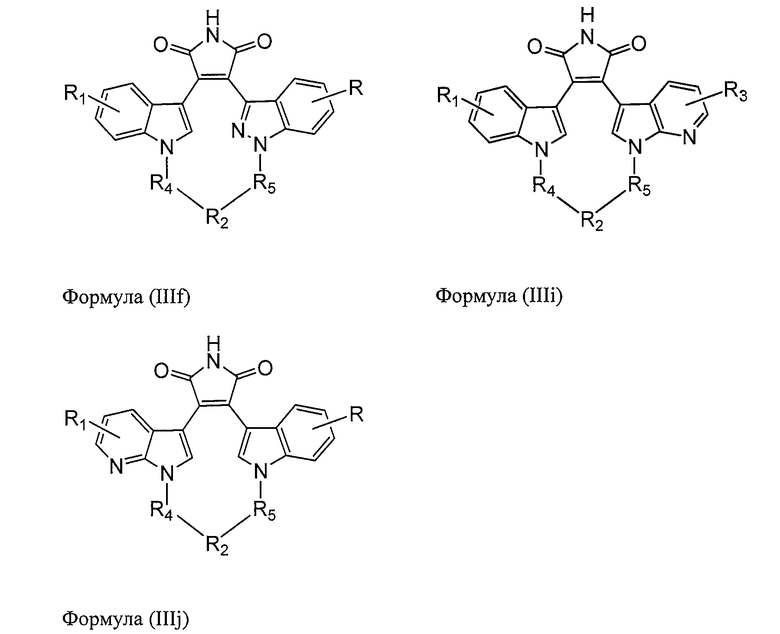 ,
,
где все остальные химические переменные выбраны в соответствии с вышеприведенным описанием; а также их фармацевтически приемлемые соли.
Соединения формулы (III) раскрыты в выданном авторам настоящего изобретения патенте США за номером 6828327, полностью включенном в настоящую заявку путем ссылки.
Пример варианта осуществления настоящего изобретения включает соединение формулы (III), где упомянутое соединение выбрано из следующей группы соединений:
Пример варианта осуществления настоящего изобретения включает соединение формулы (III), где упомянутое соединение выбрано из следующей группы соединений:
Другие примеры вариантов осуществления настоящего изобретения включают соединение, выбранное из следующей группы соединений:
Другие примеры вариантов осуществления настоящего изобретения включают соединение, выбранное из следующей группы соединений:



Клетки, пригодные для обработки, согласно способам, составляющим предмет настоящего изобретения
Плюрипотентные клетки, пригодные для использования в целях настоящего изобретения, экспрессируют по меньшей мере один из следующих маркеров плюрипотентности, выбранных из следующей группы: ABCG2, крипто, FoxD3, коннексин43, коннексин45, Oct4, SOX-2, Nanog, hTERT, UTF-1, ZFP42, SSEA-3, SSEA-4, Tra1-60 и Tra1-81.
В одном из вариантов осуществления настоящего изобретения упомянутые плюрипотентные клетки представляют собой эмбриональные стволовые клетки. В альтернативном варианте осуществления упомянутые плюрипотентные клетки представляют собой экспрессирующие маркеры плюрипотентности клетки, являющиеся производными от эмбриональных стволовых клеток человека. В одном из вариантов осуществления упомянутые эмбриональные стволовые клетки представляют собой клетки человека.
Выделение, размножение и культивирование эмбриональных стволовых клеток человека
Характеристика эмбриональных стволовых клеток человека: Эмбриональные стволовые клетки человека могут экспрессировать один или несколько стадий-специфичных эмбриональных антигенов (SSEA) 3 и 4, а также обнаруживаемые антителами маркеры, обозначаемые как Tra-1-60 и Tra-1-81 (Thomson et al., Science 282:1145, 1998). Дифференцирование эмбриональных стволовых клеток человека in vitro приводит к потере экспрессии SSEA-4, Tra-1-60 и Tra-1-81 (при ее наличии) и повышению экспрессии SSEA-1. Недифференцированные эмбриональные стволовые клетки человека, как правило, обладают щелочно-фосфатазной активностью, которую можно обнаружить путем фиксирования клеток 4% раствором параформальдегида с последующим проявлением с использованием красителя Vector Red в качестве субстрата, следуя рекомендациям производителя (Vector Laboratories, Бурлингем, Калифорния, США). Недифференцированные плюрипотентные стволовые клетки также, как правило, экспрессируют Oct-4 и TERT, обнаруживаемые методом ОТ-ПЦР.
Другим желательным фенотипическим свойством выращенных эмбриональных стволовых клеток человека является потенциал дифференцирования в клетки всех трех зародышевых листков: в эндодермальные, мезодермальные и эктодермальные ткани. Плюрипотентность эмбриональных стволовых клеток человека может быть подтверждена, например, путем инъекции клеток мышам с тяжелым комбинированным иммунодефицитом (SCID), фиксирования образующихся тератом с помощью 4% параформальдегида и их гистологического исследования для поиска клеточных типов, происходящих от трех зародышевых листков. В качестве альтернативы плюрипотентность можно определить по созданию эмбриоидных телец и анализа их на предмет присутствия маркеров, ассоциируемых с тремя зародышевыми листками.
Выращенные линии эмбриональных стволовых клеток человека могут быть кариотипированы с применением стандартного способа окрашивания с использованием красителя Гимза (G-banding) и сравнения с опубликованными кариотипами соответствующих видов приматов. Желательно получать клетки, имеющие «нормальный кариотип», т.е. эуплоидные клетки, в которых все человеческие хромосомы присутствуют и не имеют видимых изменений.
Источники эмбриональных стволовых клеток человека: К типам эмбриональных стволовых клеток человека, которые можно использовать, относятся устойчивые линии плюрипотентных клеток, получаемые из формируемой после вынашивания плода ткани, в том числе из преэмбриональной ткани (например, бластоциста), эмбриональной ткани или ткани плода, взятой в любой момент в ходе вынашивания, как правило, но не обязательно, до срока приблизительно 10-12 недель беременности. Неограничивающими настоящее изобретение примерами являются устойчивые линии эмбриональных стволовых клеток человека или эмбриональных зародышевых клеток человека, например, линии эмбриональных стволовых клеток человека H1, H7 и H9 (WiCell). Также возможно использование описываемых в настоящей заявке составов в ходе первоначального установления или стабилизации таких клеток, в этом случае исходными клетками являются первичные плюрипотентные клетки, взятые непосредственно из тканей-источников. Также соответствуют целям настоящего изобретения клетки, взятые из популяции плюрипотентных стволовых клеток, уже культивируемых в отсутствие питающих клеток. Также соответствуют целям настоящего изобретения клетки мутантных линий эмбриональных стволовых клеток человека, таких как, например, BG01v (BresaGen, Атенс, Джорджия, США).
В одном из вариантов осуществления эмбриональные стволовые клетки человека готовятся, как описано в следующих публикациях Thomson et al. (U.S. Pat. № 5843780; Science 282:1145, 1998; Curr. Top. Dev. Biol. 38:133 ff., 1998; Proc. Natl. Acad. Sci. U.S.A. 92:7844, 1995).
Культивирование эмбриональных стволовых клеток человека: В одном из вариантов осуществления настоящего изобретения эмбриональные стволовые клетки человека культивируют в культуральной системе, существенно свободной от питающих клеток, но, тем не менее, способной поддерживать пролиферацию эмбриональных стволовых клеток человека без существенного дифференцирования. Рост эмбриональных стволовых клеток человека в свободной от питающих клеток культуральной системе без дифференцирования поддерживается путем использования среды, кондиционированной посредством предварительного культивирования клеток иного типа. В качестве альтернативы рост эмбриональных стволовых клеток человека в свободной от питающих клеток культуральной системе без дифференцирования поддерживается путем использования среды с химически определенным составом.
В альтернативном варианте осуществления эмбриональные стволовые клетки человека исходно культивируются на слое питающих клеток, который поддерживает эмбриональные стволовые клетки человека в различных отношениях. Затем эмбриональные стволовые клетки человека переносятся в культуральную систему, существенно свободную от питающих клеток, но тем не менее способную поддерживать пролиферацию эмбриональных стволовых клеток человека без существенного дифференцирования.
Примеры соответствующей целям настоящего изобретения кондиционированной среды раскрыты в следующих публикациях: US 20020072117, US 6642048, WO 2005014799 и Xu et al. (Stem Cells 22:972-980, 2004).
Пример соответствующей целям настоящего изобретения среды с химически определенным составом можно найти в US 20070010011.
Соответствующая целям настоящего изобретения культуральная среда может быть приготовлена из следующих компонентов, таких как, например, модифицированная по способу Дульбекко среда Игла (DMEM), Gibco № 11965-092; модифицированная по способу Дульбекко нокаут-среда Игла (KO DMEM), Gibco № 10829-018; основная среда Хэма F12/50% DMEM; 200 мМ L-глутамина, Gibco № 15039-027; раствор заменимых аминокислот, Gibco 11140-050; β-меркаптоэтанол, Sigma № M7522; человеческий рекомбинантный основной фактор роста фибробластов (bFGF), Gibco № 13256-029.
В одном из вариантов осуществления эмбриональные стволовые клетки человека высевают на соответствующий культуральный субстрат, предварительно обработанный перед проведением обработки в соответствии со способами, составляющими предмет настоящего изобретения. В одном из вариантов осуществления упомянутая предварительная обработка состоит в нанесении компонента внеклеточного матрикса, такого как, например, полученного из базальной мембраны компонента или компонента, который может участвовать в лиганд-рецепторном взаимодействии с участием молекулы адгезивного слоя. В одном из вариантов осуществления упомянутый соответствующий культуральный субстрат представляет собой субстрат, обработанный препаратом MATRIGEL® (Becton Dickenson). MATRIGEL® представляет собой растворимый препарат из клеток опухоли Engelbreth-Holm-Swarm, который при комнатной температуре превращается в гель, образуя восстановленную базальную мембрану.
В качестве альтернативы в этих целях могут использоваться и другие компоненты внеклеточного матрикса и смеси компонентов. Последние могут включать ламинин, фибронектин, протеогликан, энтактин, гепарансульфат и т.д. как по отдельности, так и в различных сочетаниях.
Эмбриональные стволовые клетки человека высевают на субстрат с соответствующим распределением по поверхности и в присутствии среды, поддерживающей выживание, размножение и сохранение требуемых характеристик клеток. Все эти характеристики улучшаются при тщательном подходе к распределению клеток при посеве и могут быть определены специалистом в данной области.
Выделение, размножение и культивирование экспрессирующих маркеры плюрипотентности производных от эмбриональных стволовых клеток человека клеток
В одном из вариантов осуществления настоящего изобретения экспрессирующие маркеры плюрипотентности клетки получены из эмбриональных стволовых клеток человека способом, включающим:
а. культивирование эмбриональных стволовых клеток человека,
b. дифференцирование эмбриональных стволовых клеток человека в клетки, экспрессирующие маркеры, характерные для клеток сформированной эндодермы, и
c. изъятие клеток и последующее культивирование их в условиях гипоксии на культуральном субстрате, который не был предварительно обработан белком или внеклеточным матриксом.
В одном из вариантов осуществления настоящего изобретения экспрессирующие маркеры плюрипотентности клетки получены из эмбриональных стволовых клеток человека способом, включающим:
а. культивирование эмбриональных стволовых клеток человека, и
b. изъятие клеток и последующее культивирование их в условиях гипоксии на культуральном субстрате, который не был предварительно обработан белком или внеклеточным матриксом.
Культивирование клеток в условиях гипоксии на культуральном субстрате, который не был предварительно обработан белком или внеклеточным матриксом
В одном из вариантов осуществления настоящего изобретения клетки культивируют в условиях гипоксии на культуральном субстрате, на который не нанесен слой внеклеточного матрикса, в течение от приблизительно 1 до приблизительно 20 дней. В альтернативном варианте осуществления клетки культивируют в условиях гипоксии на культуральном субстрате, на который не нанесен слой внеклеточного матрикса, в течение от приблизительно 5 до приблизительно 20 дней. В альтернативном варианте осуществления клетки культивируют в условиях гипоксии на культуральном субстрате, на который не нанесен слой внеклеточного матрикса, в течение приблизительно 15 дней.
В одном из вариантов осуществления настоящего изобретения упомянутые условия гипоксии представляют собой содержание кислорода от приблизительно 1% O2 до приблизительно 20% O2. В альтернативном варианте осуществления упомянутые условия гипоксии представляют собой содержание кислорода от приблизительно 2% O2 до приблизительно 10% O2. В альтернативном варианте осуществления упомянутые условия гипоксии представляют собой содержание кислорода приблизительно 3% O2.
Клетки могут быть культивированы в условиях гипоксии на культуральном субстрате, который не был предварительно обработан белком или внеклеточным матриксом, в среде, содержащей сыворотку, Activin A и лиганд Wnt. В качестве альтернативы упомянутая среда может также содержать IGF-1.
Концентрация сыворотки в упомянутой культуральной среде может находиться в диапазоне от приблизительно 2% до приблизительно 5%. В альтернативном варианте осуществления концентрация сыворотки может составлять приблизительно 2%.
Activin A может использоваться в концентрации от приблизительно 1 пг/мл до приблизительно 100 мкг/мл. В альтернативном варианте осуществления концентрация может составлять от приблизительно 1 пг/мл до приблизительно 1 мкг/мл. В другом альтернативном варианте осуществления концентрация может составлять от приблизительно 1 пг/мл до приблизительно 100 нг/мл. В другом альтернативном варианте осуществления концентрация может составлять от приблизительно 50 нг/мл до приблизительно 100 нг/мл. В другом альтернативном варианте осуществления концентрация может составлять приблизительно 100 нг/мл.
Упомянутый лиганд Wnt может выбираться из следующей группы лигандов: Wnt-1, Wnt-3a, Wnt-5a и Wnt-7a. В одном из вариантов осуществления упомянутый лиганд Wnt представляет собой Wnt-1. В альтернативном варианте осуществления упомянутый лиганд Wnt представляет собой Wnt-3a.
Упомянутый лиганд Wnt может использоваться в концентрации от приблизительно 1 нг/мл до приблизительно 1000 нг/мл. В альтернативном варианте осуществления упомянутый лиганд Wnt может использоваться в концентрации от приблизительно 10 нг/мл до приблизительно 100 нг/мл. В одном из вариантов осуществления концентрация лиганда Wnt составляет приблизительно 20 нг/мл.
IGF-1 может использоваться в концентрации от приблизительно 1 нг/мл до приблизительно 100 нг/мл. В альтернативном варианте осуществления IGF-1 может использоваться в концентрации от приблизительно 10 нг/мл до приблизительно 100 нг/мл. В одном из вариантов осуществления концентрация IGF-1 составляет приблизительно 50 нг/мл.
Экспрессирующие маркеры плюрипотентности клетки, полученные способами, составляющими предмет настоящего изобретения, способны к размножению при культивировании в условиях гипоксии на культуральном субстрате, который не был предварительно обработан белком или внеклеточным матриксом.
Экспрессирующие маркеры плюрипотентности клетки, полученные способами, составляющими предмет настоящего изобретения, экспрессируют по меньшей мере один из следующих маркеров плюрипотентности, выбранных из следующей группы маркеров: ABCG2, крипто, FoxD3, коннексин43, коннексин45, Oct4, SOX-2, Nanog, hTERT, UTF-1, ZFP42, SSEA-3, SSEA-4, Tra1-60 и Tra1-81.
Дальнейшее дифференцирование клеток, экспрессирующих маркеры, характерные для линии сформированной эндодермы
Клетки, экспрессирующие маркеры, характерные для линии сформированной эндодермы, могут быть дифференцированы в клетки, экспрессирующие маркеры, характерные для линии панкреатической эндодермы, любым известным в данной области способом.
Например, клетки, экспрессирующие маркеры, характерные для линии сформированной эндодермы, могут быть дифференцированы в клетки, экспрессирующие маркеры, характерные для линии панкреатической эндодермы, в соответствии со способами, описанными в работе D'Amour et al., Nature Biotechnology 24, 1392-1401 (2006).
Например, клетки, экспрессирующие маркеры, характерные для линии сформированной эндодермы, далее дифференцируются в клетки, экспрессирующие маркеры, характерные для линии панкреатической эндодермы, путем обработки клеток, экспрессирующих маркеры, характерные для линии сформированной эндодермы, фактором роста фибробластов и KAAD-циклопамином с последующим удалением содержащей фактор роста фибробластов и KAAD-циклопамин среды и культивированием клеток в среде, содержащей ретиноевую кислоту, фактор роста фибробластов и KAAD-циклопамин. Пример описанного способа раскрыт в работе D' Amour et al., Nature Biotechnology, 24:1392-1401, (2006).
Маркеры, характерные для линии панкреатической эндодермы, выбираются из группы, включающей Pdx1, HNF-1beta, PTF1a, HNF-6, HB9 и PROX1. Для целей настоящего изобретения пригодны клетки с экспрессией по меньшей мере одного из маркеров, характерных для линии панкреатической эндодермы. В одном из аспектов настоящего изобретения клетка, экспрессирующая маркеры, характерные для линии панкреатической эндодермы, представляет собой клетку панкреатической эндодермы.
Дальнейшее дифференцирование клеток, экспрессирующих маркеры, характерные для линии панкреатической эндодермы
Клетки, экспрессирующие маркеры, характерные для линии панкреатической эндодермы, могут быть дифференцированы в клетки, экспрессирующие маркеры, характерные для линии панкреатических эндокринных клеток, любым известным в данной области способом.
Например, клетки, экспрессирующие маркеры, характерные для линии панкреатической эндодермы, могут быть дифференцированы в клетки, экспрессирующие маркеры, характерные для линии панкреатических эндокринных клеток, в соответствии со способами, описанными в работе D'Amour et al., Nature Biotechnology 24, 1392-1401 (2006).
Маркеры, характерные для линии панкреатических эндокринных клеток, выбираются из группы, включающей NGN-3, NeuroD, Islet-1, Pdx-1, NKX6.1, Pax-4, Ngn-3 и PTF-1 alpha. В одном из вариантов осуществления панкреатическая эндокринная клетка способна экспрессировать как минимум один из следующих гормонов: инсулин, глюкагон, соматостатин и панкреатический полипептид. Для целей настоящего изобретения пригодны клетки с экспрессией по меньшей мере одного из маркеров, характерных для линии панкреатических эндокринных клеток. В одном из аспектов настоящего изобретения клетка, экспрессирующая маркеры, характерные для линии панкреатических эндокринных клеток, представляет собой панкреатическую эндокринную клетку. Панкреатическая эндокринная клетка может представлять собой панкреатическую экспрессирующую гормоны клетку. В качестве альтернативы панкреатическая эндокринная клетка может представлять собой панкреатическую секретирующую гормоны клетку.
В одном из аспектов настоящего изобретения упомянутая панкреатическая эндокринная клетка представляет собой клетку, экспрессирующую маркеры, характерные для β-клеточной линии дифференцирования. Клетка, экспрессирующая маркеры, характерные для β-клеточной линии дифференцирования, экспрессирует Pdx1 и по меньшей мере один из следующих факторов транскрипции: NGN-3, Nkx2.2, Nkx6.1, NeuroD, Isl-1, HNF-3 beta, MAFA, Pax4 и Pax6. В одном из аспектов настоящего изобретения клетка, экспрессирующая маркеры, характерные для β-клеточной линии дифференцирования, представляет собой β-клетку.
Обнаружение клеток, экспрессирующих маркеры, характерные для линии сформированной эндодермы
Образование клеток, экспрессирующих маркеры, характерные для линии сформированной эндодермы, может быть обнаружено путем проверки на наличие соответствующих маркеров до и после выполнения конкретного протокола. Плюрипотентные стволовые клетки, как правило, не экспрессируют такие маркеры. Таким образом, дифференцирование плюрипотентных клеток определяется по началу экспрессии таких маркеров.
Эффективность дифференцирования может быть определена путем обработки популяции клеток агентом (например, антителом), специфически распознающим белковый маркер, экспрессированный клетками, экспрессирующими маркеры, характерные для линии сформированной эндодермы.
Способы оценки уровней экспрессии маркеров белков и нуклеиновых кислот в культивированных или выделенных клетках являются стандартными для данной области. К подобным способам относятся количественная полимеразная цепная реакция с обратной транскрипцией (ОТ-ПЦР), Нозерн-блоттинг, гибридизация in situ (см., например, Current Protocols in Molecular Biology (Ausubel et al., eds. 2001 supplement)), и иммунологические анализы, такие как иммуногистохимический анализ среза материала, Вестерн-блоттинг, а для доступных в интактных клетках маркеров - также способ проточной цитометрии (FACS) (см., например, Harlow and Lane, Using Antibodies: A Laboratory Manual, New York: Cold Spring Harbor Laboratory Press (1998)).
Примеры антител, используемых для обнаружения определенных белковых маркеров, приведены в таблице IA. Следует отметить, что существуют или могут быть легко созданы альтернативные антитела, связывающиеся с теми же маркерами, которые распознаются перечисленными в таблице IA антителами. Такие альтернативные антитела также можно применять для анализа экспрессии маркеров в клетках, выделенных в соответствии с настоящим изобретением.
Например, характеристики плюрипотентных стволовых клеток хорошо известны специалистам в данной области, и продолжается выявление дополнительных характеристик плюрипотентных стволовых клеток. К маркерам плюрипотентных стволовых клеток относится, например, экспрессия одного или нескольких из следующих факторов: ABCG2, крипто, FoxD3, коннексин43, коннексин45, Oct4, Sox2, Nanog, hTERT, UTF-1, ZFP42, SSEA-3, SSEA-4, Tra1-60, Tra1-81.
После обработки плюрипотентных стволовых клеток с применением способов, составляющих предмет настоящего изобретения, дифференцированные клетки могут быть выделены путем воздействия на популяцию клеток агентом (например, антителом), специфически распознающим белковый маркер, например, CXCR4, экспрессируемый клетками, экспрессирующими маркеры, характерные для линии сформированной эндодермы.
Обнаружение клеток, экспрессирующих маркеры, характерные для линии панкреатической эндодермы
Маркеры, характерные для линии панкреатической эндодермы, хорошо известны специалистам в данной области, и дополнительные маркеры, характерные для линии панкреатической эндодермы, продолжают выявляться. Такие маркеры могут использоваться для подтверждения того, что клетки, прошедшие обработку в соответствии с настоящим изобретением, дифференцировались и приобрели характерные особенности линии панкреатической эндодермы. К маркерам, характерным для линии панкреатической эндодермы, относится экспрессия одного или нескольких факторов транскрипции, таких как, например, Hlxb9, PTF-1a, PDX-1, HNF-6, HNF-1beta.
Эффективность дифференцирования может быть определена путем обработки популяции клеток агентом (например, антителом), специфически распознающим белковый маркер, экспрессированный клетками, экспрессирующими маркеры, характерные для линии панкреатической эндодермы.
Способы оценки экспрессии маркеров белков и нуклеиновых кислот в культивированных или выделенных клетках являются стандартными для данной области. К подобным способам относятся количественная полимеразная цепная реакция с обратной транскрипцией (ОТ-ПЦР), Нозерн-блоттинг, гибридизация in situ (см., например, Current Protocols in Molecular Biology (Ausubel et al., eds. 2001 supplement)), и иммунологические анализы, такие как иммуногистохимический анализ среза материала, Вестерн-блоттинг, а для доступных в интактных клетках маркеров - также способ проточной цитометрии (FACS) (см., например, Harlow and Lane, Using Antibodies: A Laboratory Manual, New York: Cold Spring Harbor Laboratory Press (1998)).
Примеры антител, используемых для обнаружения определенных белковых маркеров, приведены в таблице IA. Следует отметить, что существуют или могут быть легко созданы альтернативные антитела, связывающиеся с теми же маркерами, которые распознаются перечисленными в таблице IA антителами. Такие альтернативные антитела также можно применять для анализа экспрессии маркеров в клетках, выделенных в соответствии с настоящим изобретением.
Обнаружение клеток, экспрессирующих маркеры, характерные для линии панкреатических эндокринных клеток
Маркеры, характерные для линии панкреатических эндокринных клеток, хорошо известны специалистам в данной области, и продолжается выявление дополнительных маркеров, характерных для линии панкреатических эндокринных клеток. Такие маркеры могут использоваться для подтверждения того, что клетки, прошедшие обработку в соответствии с настоящим изобретением, дифференцировались и приобрели свойства, характерные для линии панкреатических эндокринных клеток. К маркерам, характерным для линии панкреатических эндокринных клеток, относится экспрессия одного или нескольких факторов транскрипции, таких как, например, NGN-3, NeuroD, Islet-1.
Маркеры, характерные для клеток β-клеточной линии дифференцирования, хорошо известны специалистам в данной области, и продолжается выявление дополнительных маркеров, характерных для β-клеточной линии дифференцирования. Такие маркеры могут использоваться для подтверждения того, что клетки, прошедшие обработку в соответствии с настоящим изобретением, дифференцировались и приобрели свойства, характерные β-клеточной линии дифференцирования. К специфичным характеристикам β-клеточной линии дифференцирования относится экспрессия одного или нескольких факторов транскрипции, таких как, например, Pdx1 (ген панкреатического и дуоденального гомеобокса-1), Nkx2.2, Nkx6.1, Isl1, Pax6, Pax4, NeuroD, Hnf1b, Hnf-6, Hnf-3beta и MafA, а также иных факторов. Такие факторы транскрипции широко используются в данной области для идентификации эндокринных клеток. См., например, обзор Edlund (Nature Reviews Genetics 3:524-632 (2002)).
Эффективность дифференцирования может быть определена путем обработки популяции клеток агентом (например, антителом), специфически распознающим белковый маркер, экспрессированный клетками, экспрессирующими маркеры, характерные для линии панкреатических эндокринных клеток. В качестве альтернативы эффективность дифференцирования может быть определена путем обработки популяции клеток агентом (например, антителом), специфически распознающим белковый маркер, экспрессированный клетками, экспрессирующими маркеры, характерные для β-клеточной линии дифференцирования.
Способы оценки уровней экспрессии маркеров белков и нуклеиновых кислот в культивированных или выделенных клетках являются стандартными для данной области. К подобным способам относятся количественная полимеразная цепная реакция с обратной транскрипцией (ОТ-ПЦР), Нозерн-блоттинг, гибридизация in situ (см., например, Current Protocols in Molecular Biology (Ausubel et al., eds. 2001 supplement)), и иммунологические анализы, такие как иммуногистохимический анализ среза материала, Вестерн-блоттинг, а для доступных в интактных клетках маркеров - также способ проточной цитометрии (FACS) (см., например, Harlow and Lane, Using Antibodies: A Laboratory Manual, New York: Cold Spring Harbor Laboratory Press (1998)).
Примеры антител, используемых для обнаружения определенных белковых маркеров, приведены в таблице IA. Следует отметить, что существуют или могут быть легко созданы альтернативные антитела, связывающиеся с теми же маркерами, которые распознаются перечисленными в таблице IA антителами. Такие альтернативные антитела также можно применять для анализа экспрессии маркеров в клетках, выделенных в соответствии с настоящим изобретением.
Настоящее изобретение далее иллюстрируют, помимо прочих, следующие примеры.
Пример 1
Культивирование эмбриональных стволовых клеток человека
Стволовые клетки представляют собой недифференцированные клетки, определяемые по их способности на уровне единичной клетки как самообновляться, так и дифференцироваться с образованием клеток-потомков, таких как самообновляющиеся клетки-предшественники, необновляющиеся клетки-предшественники и окончательно дифференцированные клетки. Стволовые клетки также характеризуются способностью дифференцироваться in vitro в функциональные клетки различных линий дифференцирования из нескольких зародышевых листков (эндодермы, мезодермы и эктодермы), а также после трансплантации давать начало тканям, происходящим от нескольких зародышевых листков, и вносить существенный вклад в формирование большинства, если не всех, тканей после инъекции в бластоцисты.
Линии эмбриональных стволовых клеток человека H1, H7 и H9 были получены из института WiCell Research Institute, Inc., (Мэдисон, Висконсин, США) и культивировались в соответствии с инструкциями, предоставленными указанным выше институтом. Коротко говоря, клетки культивировали на питающем слое из мышиных эмбриональных фибробластов (MEF) в среде для эмбриональных стволовых клеток, содержащей DMEM/F12 (Invitrogen/GIBCO) с добавлением 20% нокаут заменителя сыворотки, 100 нМ раствора заменимых аминокислот MEM, 0,5 мМ бетамеркаптоэтанола, 2мМ L-глутамина и 4 нг/мл основного фактора роста фибробластов человека (bFGF) (все производства компании Invitrogen/GIBCO). Мышиные эмбриональные фибробласты, выделенные из мышиных эмбрионов E13-13,5, были приобретены в Charles River. Мышиные эмбриональные фибробласты размножали в среде DMEM с добавлением 10% эмбриональной телячьей сыворотки (FBS) (Hyclone), 2 мМ глутамина и 100 мМ заменимых аминокислот MEM. Субконфлюэнтные культуры мышиных эмбриональных фибробластов обрабатывали 10 мкг/мл митомицина C (Sigma, Сент-Луис, Миссури, США) в течение 3 часов для остановки деления клеток, затем обработали трипсином и высеяли с плотностью 2×104/см2 на чашки, покрытые слоем 0,1% бычьего желатина. В качестве питающего слоя использовали мышиные эмбриональные фибробласты со второго по четвертый пассажи. Эмбриональные стволовые клетки человека, высеянные на питающий слой мышиных эмбриональных фибробластов, культивировали при температуре 37°C в атмосфере 5% CO2/ в инкубаторе с увлажнением для тканевых культур. При начале слияния культур (приблизительно через 5-7 дней после нанесения) эмбриональные стволовые клетки человека обработали раствором коллагеназы типа IV с концентрацией 1 мг/мл (Invitrogen/GIBCO) в течение 5-10 мин и затем осторожно соскоблили с поверхности при помощи 5-мл пипетки. Клетки осадили центрифугированием при 900 об/мин в течение 5 мин, осадок ресуспендировали и снова высеяли с соотношением клеток от 1:3 до 1:4 в свежую культуральную среду.
Параллельно эмбриональные стволовые клетки человека линий H1, H7 и H9 также высеяли на носителе, покрытом слоем препарата MATRIGEL™ с пониженным содержанием факторов роста (BD Biosciences), в разведении 1:30 и культивировали в кондиционируемой среде MEF с добавлением 8 нг/мл bFGF. Культивированные на препарате MATRIGEL™ клетки стандартно пассировали с использованием коллагеназы IV (Invitrogen/GIBCO), диспазы (BD Biosciences) или ферментативной смеси LIBERASE (Source). Некоторые культуры эмбриональных стволовых клеток человека инкубировали в условиях гипоксии (приблизительно 3% O2).
Пример 2
Получение и культивирование экспрессирующих маркеры плюрипотентности производных от эмбриональных стволовых клеток человека клеток
Клетки линий H1 и H9 эмбриональных стволовых клеток человека различных пассажей (пассажи 30-54) культивировали в условиях гипоксии (приблизительно 3% O2) в течение по меньшей мере трех пассажей. Клетки культивировали в кондиционированной среде MEF с добавлением 8 нг/мл bFGF и высеяли на покрытые препаратом MATRIGEL носители в соответствии с примером 1.
Затем клетки обрабатывали средой DMEM/F12 с добавлением 0,5% FBS, 20 нг/мл WNT-3a (Кат. № 1324-WN-002, R&D Systems, Миннесота, США) и 100 нг/мл Activin A (R&D Systems, Миннесота, США) в течение двух дней с последующей обработкой средой DMEM/F12 с добавлением 2% FBS и 100 нг/мл Activin A (AA) в течение еще 3-4 дней. Описанный протокол привел с существенному повышению уровней экспрессии маркеров сформированной эндодермы.
Затем клетки обработали раствором TrypLE™ Express (Invitrogen, Калифорния, США) в течение 5 минут. Выделенные клетки ресуспендировали в среде DMEM/F12 с добавлением 2% FBS, восстановили центрифугированием и подсчитали при помощи гемоцитометра. Выделенные клетки высеяли с плотностью 1000-10000 клеток/см2 в полистирольные культуральные сосуды (TCPS) и культивировали в среде DMEM-F12+2% FBS+100 нг/мл Activin A+20 нг/мл WNT-3A в условиях гипоксии (приблизительно 3% O2) при температуре 37°C в стандартном культуральном инкубаторе. Полистирольные культуральные сосуды не имели покрытия из препарата MATRIGEL или иных белков внеклеточного матрикса. Культуральную среду меняли ежедневно. Для некоторых культур в среду также добавили 10-50 нг/мл IGF-I (инсулиновый фактор роста-I, R&D Systems, Миннесота, США) или 1X ITS (инсулин, трансферрин и селен, Invitrogen, Калифорния, США). Для некоторых культур в основную среду (DM-F12+2% FBS) также добавили 0,1 мМ меркаптоэтанола (Invitrogen, Калифорния, США) и раствор заменимых аминокислот (1X, NEAA, Invitrogen, Калифорния, США).
Через 5-15 дней культивирования на носителях образовались хорошо определенные колонии клеток, окруженные большим количеством клеток увеличенного размера, которые находились в фазе старения. При степени слияния 50-60% полученные культуры пассировали путем обработки раствором TrypLE™ Express в течение 5 минут при комнатной температуре. Выделенные клетки ресуспендировали в среде DMEM-F12+2% FBS, восстановили центрифугированием, высеяли с плотностью 10000 клеток/см2 в полистирольные культуральные сосуды (TCPS) и культивировали в среде DMEM-F12+2% FBS+100 нг/мл Activin A+20 нг/мл WNT-3A+/-50 нг/мл IGF-I. Указанная среда будет далее именоваться «ростовая среда».
Пример 3
Получение экспрессирующих маркеры плюрипотентности производных из одноклеточной суспензии эмбриональных стволовых клеток человека клеток
Эмбриональные стволовые клетки человека линий H1 (P33) и H9 (P45) культивировали в условиях гипоксии (приблизительно 3% O2) в течение по меньшей мере трех пассажей. Клетки культивировали в кондиционированной среде MEF с добавлением 8 нг/мл bFGF и высеяли на покрытые препаратом MATRIGEL носители в соответствии с примером 1. При степени слияния приблизительно 60% полученные культуры обрабатывали раствором TrypLE™ Express (Invitrogen, Калифорния, США) в течение 5 минут. Выделенные клетки ресуспендировали в среде DMEM/F12 с добавлением 2% FBS, восстановили центрифугированием и подсчитали при помощи гемоцитометра. Выделенные клетки высеяли с плотностью 1000-10000 клеток/см2 в полистирольные культуральные сосуды (TCPS) и культивировали в среде DMEM-F12+2% FBS+100 нг/мл Activin A+20 нг/мл WNT-3A+50 нг/мл IGF-I+0,1 мМ меркаптоэтанол (Invitrogen, Калифорния, США) и растворе заменимых аминокислот (1X, NEAA, Invitrogen, Калифорния, США) в условиях гипоксии (приблизительно 3% O2) при температуре 37°C в стандартном культуральном инкубаторе. Полистирольные культуральные сосуды не имели покрытия из препарата MATRIGEL или иных белков внеклеточного матрикса. Культуральную среду меняли ежедневно. Клетки первого пассажа обозначаются P1.
Пример 4
Различные ростовые среды, пригодные для размножения экспрессирующих маркеры плюрипотентности производных от эмбриональных стволовых клеток человека клеток
Экспрессирующие маркеры плюрипотентности производные от эмбриональных стволовых клеток человека клетки успешно культивировали в средах перечисленного ниже состава в течение по меньшей мере 2-30 пассажей:
1. DM-F12+2% FBS+100 нг/мл AA+20 нг/мл WNT-3A
2. DM-F12+2% FBS+100 нг/мл AA+20 нг/мл WNT-3A+50 нг/мл IGF-I
3. DM-F12+2% FBS+100 нг/мл AA+20 нг/мл WNT-3A+10 нг/мл IGF-I
4. DM-F12+2% FBS+50 нг/мл AA+20 нг/мл WNT-3A+50 нг/мл IGF-I
5. DM-F12+2% FBS+50 нг/мл AA+10 нг/мл WNT-3A+50 нг/мл IGF-I
6. DM-F12+2% FBS+50 нг/мл AA+20 нг/мл WNT-3A+10 нг/мл IGF-I
7. DM-F12+2% FBS+100 нг/мл AA+10 нг/мл WNT-3A+10 нг/мл IGF-I
8. Среда определенного состава HEScGRO (Chemicon, Калифорния, США)
Основной компонент перечисленных выше сред может быть заменен на другие подобные среды, такие как RPMI, DMEM, CRML, Knockout ™DMEM и F12.
Пример 4
Эффект ингибиторов активности фермента GSK-3β на жизнеспособность экспрессирующих маркеры плюрипотентности клеток
Получение и поддержание экспрессирующих маркеры плюрипотентности клеток проводили, как описано в примере 2. Клетки выращивали в среде DMEM:F12 с добавлением 2% FCS (Invitrogen), 100 нг/мл Activin A, 20 нг/мл Wnt-3a и 50 нг/мл IGF(R&D Biosystems). Клетки высеяли с плотностью 10000 клеток/см2 в полистирольные флаконы типа Falcon и выращивали в монослойной культуре при температуре 37°C, в атмосфере 5% CO2 и пониженной концентрации кислорода. При достижении степени слияния 60-70% полученные клетки пассировали путем промывания монослоя раствором PBS и инкубирования с раствором TrypLE (Invitrogen) в течение 3-5 минут для открепления клеток и получения одноклеточной суспензии.
Скрининг проводили с использованием тестовых соединений из патентованной библиотеки малых молекул, отобранных по их способности ингибировать активность фермента GSK-3B. Соединения из этой библиотеки были предоставлены в виде исходных растворов концентрации 1 мМ на 96-луночных планшетах в 50 мМ HEPES, 30% DMSO. Для проведения анализа экспрессирующие маркеры плюрипотентности клетки промыли, подсчитали и высеяли в нормальной культуральной среде с плотностью 20000 клеток на лунку в 96-луночные планшеты, состоящие из непрозрачных лунок с прозрачным дном (Costar). Предварительные эксперименты показали, что указанная плотность посева дает оптимальное образование монослоя при культивировании в течение одной ночи. На следующий день культуральную среду удалили, монослои клеток трижды промыли раствором PBS и в лунки добавили 80 мкл аликвоты испытываемых соединений, разведенных в среде для проведения анализа до конечной концентрации 10 мкМ. На 2 день анализа из всех лунок удалили старую среду и заменили ее на свежую аликвоту испытываемых соединений, разведенных в среде для проведения анализа. Среда для проведения анализа на 1 и 2 день культивирования состояла из среды DMEM:F12 с добавлением 0,5% FCS и 100 нг/мл Activin A. На 3 и 4 день анализа из всех лунок удалили старую среду и заменили ее на среду DMEM:F12 с добавлением 0,5% FCS и 100 нг/мл Activin A (без испытываемых соединений). На 4 день анализа в каждую лунку добавили 15 мкл MTS (Promega), планшеты инкубировали при температуре 37°C в течение 1,5-4 часов, и затем измерили оптические плотности лунок на длине волны 490 нм с помощью анализатора SpectraMax (Molecular Devices). Для каждого дублирующего набора рассчитали статистические характеристики: среднее значение, стандартное отклонение и коэффициент вариации. Для каждой лунки с испытываемым образцом также рассчитали токсичность относительно положительного контрольного образца (лунки, обработанные Activin A и Wnt3a на 1 и 2 день культивирования).
Сводка всех результатов скрининга приведена в таблице II. В данном анализе экспрессирующие маркеры плюрипотентности клетки изначально высеяли в виде слитного монослоя, поэтому полученные результаты адекватно отражают степень токсичности за четырехдневный период культивирования. Полученные результаты представлены в виде процентной доли выживаемости клеток по отношению к контрольному образцу и демонстрируют различную токсичность некоторых соединений при использованных для скрининга концентрациях 10 мкМ. Большинство испытанных соединений показали минимальную токсичность или оказались нетоксичны в проведенном клеточном анализе.
Небольшой набор выбранных соединений повторно проверили в узком диапазоне дозировок с использованием экспрессирующих маркеры плюрипотентности клеток в анализе, аналогичном описанному выше. В таблице III приведены полученные в этом анализе результаты, свидетельствующие о наличии эффекта варьирования дозировки для испытанного набора токсичных и нетоксичных соединений.
Пример 5
Эффект ингибиторов активности фермента GSK-3β на дифференцирование и пролиферацию эмбриональных стволовых клеток человека, установленный с помощью одновременного многопараметрического анализа
Поддержание эмбриональных стволовых клеток человека (линия H9) проводили, как описано в примере 1. Колонии клеток поддерживали в недифференцированном плюрипотентном состоянии со средним интервалом четыре дня между последовательными пассированиями. При пассировании культуры клеток обрабатывали раствором коллагеназы (1 мг/мл; Sigma-Aldrich) в течение 10-30 минут при температуре 37°C, затем осторожно соскоблили кончиком пипетки, получив кластеры клеток. Полученным кластерам дали осесть под собственным весом и затем промыли их для удаления остатков коллагеназы. Кластеры клеток разделили в соотношении 1:3 для стандартного поддерживающего культивирования или в соотношении 1:1 для немедленного анализа. Использованные в анализе линии эмбриональных стволовых клеток человека поддерживали при номерах пассажа менее 50 и периодически проверяли на нормальный кариотипический фенотип и отсутствие примесей микоплазмы.
Используемые в анализе кластеры клеток однородно ресуспендировали в нормальной культуральной среде и высеяли на покрытые препаратом MATRIGEL 96-луночные планшеты типа Packard VIEWPLATE (PerkinElmer) в объемах 100 мкл/лунку. Для первоначального посева и восстановления использовали кондиционированную среду MEF с добавлением 8 нг/мл bFGF. Ежедневное питание осуществляли путем отсасывания отработанной культуральной среды из каждой лунки и ее замены тем же объемом свежей среды. В течение всего анализа планшеты держали при температуре 37°C в атмосфере 5% CO2 в увлажняемом боксе.
Скрининг проводили с использованием тестовых соединений из патентованной библиотеки малых молекул, отобранных по их способности ингибировать активность фермента GSK-3B. Соединения из этой библиотеки были предоставлены в виде исходных растворов концентрацией 1 мМ на 96-луночных планшетах в 50 мМ HEPES, 30% DMSO. Испытание соединений проводили в двух или трех повторностях. Первичный скрининговый анализ начали с отсасывания культуральной среды из каждой лунки с последующей трехкратной промывкой лунки раствором PBS для удаления остатков факторов роста и сыворотки. Затем в лунки добавили испытываемые образцы объемом от 80 до 100 мкл, содержащие основную среду DMEM:F12 (Invitrogen) с добавлением 0,5% FCS (HyClone) и 100 нг/мл Activin A (R&D Biosystems), плюс 10 мкМ испытываемого соединения. Лунки с положительными контрольными образцами содержали ту же основную среду с заменой испытываемого соединения на 10-20 нг/мл Wnt3a (R&D Biosystems). Лунки с отрицательными контрольными образцами содержали основную среду с добавлением 0,5% FCS и только Activin A (только AA) или в качестве альтернативы - 0,5% FCS, без Activin A или Wnt3a (без обработки). На 2 день анализа из лунок отсосали отработанную среду и заменили на идентичную по составу свежую среду. На 3 и 4 день из всех тестовых лунок удалили среду и заменили ее на среду DMEM:F12 с добавлением 2% FCS и 100 нг/мл Activin A (без испытываемого соединения или Wnt3a); лунки с параллельными отрицательными контрольными образцами поддерживали в основной среде DMEM:F12 с добавлением 2% FCS и Activin A (только AA) или в качестве альтернативы - 2% FCS, без Activin A (без обработки).
По окончании культивирования находящиеся в 96-луночных планшетах клетки зафиксировали 4% параформальдегидом при комнатной температуре в течение 20 минут, трижды промыли раствором PBS и затем пермеабилизовали, обработав раствором 0,5% Triton X-100 в течение 20 минут при комнатной температуре. В качестве альтернативы клетки зафиксировали ледяным 70% спиртом в течение ночи при температуре -20°C, трижды промыли раствором PBS и затем пермеабилизовали раствором 0,5% Triton X-100 в течение 5 минут при температуре 4°C. После фиксации и пермеабилизации клетки снова трижды промыли раствором PBS и заблокировали раствором 4% куриной сыворотки (Invitrogen) в растворе PBS в течение 30 минут при комнатной температуре. Первичные антитела (козий античеловеческий Sox17 и козий античеловеческий HNF-3beta; R&D Systems) развели 1:100 в 4% куриной сыворотке и добавили к клеткам на один час при комнатной температуре. Сопряженные с флюорофором Alexa Fluor 488 вторичные антитела (куриный антикозий IgG; Molecular Probes) развели 1:200 в растворе PBS и добавили к клеткам после их трехкратной промывки раствором PBS. Для контрастной окраски ядер добавили 5 мМ Draq5 (Alexis Biochemicals) на пять минут при комнатной температуре. Затем клетки один раз промыли раствором PBS и оставили в 100 мл раствора PBS/лунку для томографии.
Томографию клеток проводили на анализаторе IN Cell Analyzer 1000 (GE Healthcare), используя дихроичное зеркало 51008bs для клеток, окрашенных Draq5 и Alexa Fluor 488. Времена экспозиции оптимизировали с использованием лунок с положительными контрольными образцами и лунок с вторичными антителами только для необработанных отрицательных контрольных образцов. Для компенсации возможных потерь клеток в ходе процедур обработки и окрашивания для каждой лунки снимали по двенадцать проекций. Общее количество клеток и общую интенсивность клеток для Sox-17 и HNF-3beta измеряли с помощью программного пакета IN Cell Developer Toolbox 1.6 (GE Healthcare). Сегментацию ядер определяли по ступеням шкалы серых тонов (исходный интервал 100-300) и размерам ядра. Для повторностей рассчитали средние значения и их стандартные отклонения. Общий уровень экспрессии белка выражается в виде общей интенсивности или интегральной интенсивности, определяемой как произведение суммарной интенсивности флюоресценции клетки на площадь клетки. Фон исключили на основании критериев соответствия ступеням шкалы серых тонов в диапазоне от 300 до 3000 и форм-факторам от 0,4 и выше. Показатели общей интенсивности нормировали путем деления общей интенсивности для каждой лунки на среднюю общую интенсивность для положительного контрольного образца Wnt3a/Activin A. Для каждого набора повторностей рассчитали нормированные данные для средних значений и их стандартных отклонений.
В таблице IV приведена представительная сводка всех результатов проведенного скрининга. В таблице V приведен список положительных результатов проведенного скрининга. Сильно положительные результаты определяются как более чем или равные 120% от контрольных значений; умеренно положительные результаты определяются как находящиеся в диапазоне 60-120% от контрольных значений. В данном анализе значительное число соединений индуцировали пролиферативный отклик. Параллельно в данном анализе значительное число соединений индуцировали дифференцирование, измеряемое по уровню экспрессии белков факторов транскрипции Sox17 и Hnf-3b.
Пример 6
Эффект ингибиторов активности фермента GSK-3β на пролиферацию эмбриональных стволовых клеток человека, установленный с помощью спектрофотометра вертикального сканирования
Поддержание эмбриональных стволовых клеток человека (линии H9 или H1) проводили, как описано в примере 1. Колонии клеток поддерживали в недифференцированном плюрипотентном состоянии со средним интервалом четыре дня между последовательными пассированиями. При пассировании культуры клеток обрабатывали раствором коллагеназы (1 мг/мл; Sigma-Aldrich) в течение 10-30 минут при температуре 37°C, затем осторожно соскоблили кончиком пипетки, получив кластеры клеток. Полученным кластерам дали осесть под собственным весом и затем промыли их для удаления остатков коллагеназы. Кластеры клеток разделили в соотношении 1:3 площади монослоя для стандартного поддерживающего культивирования или в соотношении 1:1 для немедленного анализа. Использованные в этих примерах линии эмбриональных стволовых клеток человека поддерживали при номерах пассажа менее 50 и периодически проверяли на нормальный кариотипический фенотип и отсутствие примесей микоплазмы.
Используемые в анализе кластеры клеток однородно ресуспендировали в нормальной культуральной среде и высеяли на покрытые препаратом MATRIGEL 96-луночные планшеты типа Packard VIEWPLATE (PerkinElmer) в объемах 100 мкл/лунку. Для первоначального посева и восстановления использовали кондиционированную среду MEF с добавлением 8 нг/мл bFGF. Ежедневное питание осуществляли путем отсасывания отработанной культуральной среды из каждой лунки и ее замены тем же объемом свежей среды. В течение всего анализа планшеты держали при температуре 37°C в атмосфере 5% CO2 в увлажняемом боксе.
Первичный скрининговый анализ начали с отсасывания культуральной среды из каждой лунки с последующей трехкратной промывкой лунки раствором PBS для удаления остатков факторов роста и сыворотки. Затем в лунки добавили испытываемые образцы объемом от 80 до 100 мкл, содержащие основную среду DMEM:F12 (Invitrogen) с добавлением 0,5% FCS (HyClone) и 100 нг/мл Activin A (R&D Biosystems) и 10 мкМ испытываемого соединения. Лунки с положительными контрольными образцами содержали ту же основную среду с заменой испытываемого соединения на 10-20 нг/мл Wnt3a (R&D Biosystems). Лунки с отрицательными контрольными образцами содержали основную среду с добавлением 0,5% FCS, без Activin A или Wnt3a. Скрининг соединений проводили в трех повторностях. На 2 день анализа из лунок отсосали отработанную среду и заменили на идентичную по составу свежую среду. На 3 и 4 день из всех тестовых лунок удалили среду и заменили ее на среду DMEM:F12 с добавлением 2% FCS и 100 нг/мл Activin A, за исключением лунок с отрицательными контрольными образцами, которые поддерживали в основной среде DMEM:F12 с добавлением 2% FCS.
На 4 день анализа в каждую лунку добавили 15-20 мкл MTS (Promega) и инкубировали планшеты при температуре 37°C в течение 1,5-4 часов. Оптические плотности лунок на длине волны 490 нм измерили с помощью спектрофотометра вертикального сканирования Molecular Devices. Для каждого дублирующего набора рассчитали среднее значение, стандартное отклонение и коэффициент вариации. Результаты для каждой лунки с испытываемым образцом сравнили с результатом для положительного контрольного образца Activin A/Wnt3a и рассчитали степень пролиферации как процентную величину по отношению к контрольному образцу.
В таблице VI приведена представительная сводка всех результатов проведенного скрининга. В таблице VII приведен список положительных результатов проведенного скрининга. Сильно положительные результаты определяются как более чем или равные 120% от контрольных значений; умеренно положительные результаты определяются как находящиеся в диапазоне 60-120% от контрольных значений. В данном анализе значительное число соединений индуцировали пролиферативный отклик.
Пример 7
Эффект ингибиторов активности фермента GSK-3β на дифференцировку и пролиферацию эмбриональных стволовых клеток человека: титрование дозы эффективных соединений
Было важно подтвердить активность соединений, показавших положительный результат при первичном скрининге, и далее титрованием дозы проанализировать диапазон активности. Свежие образцы выбранного подмножества соединений, показавших положительный результат при первичном скрининге, получили в виде сухих порошков, растворили для получения свежих маточных растворов и разбавили для проведения вторичных подтверждающих анализов по оценке их воздействия на эмбриональные стволовые клетки человека.
Культивирование эмбриональных стволовых клеток человека двух линий (H1 и H9) проводили, как описано в примере 1. Колонии клеток поддерживали в недифференцированном плюрипотентном состоянии на покрытых препаратом MatrigelTM (Invitrogen) полистирольных носителях, используя для покрытия поверхности разведение 1:30 препарата MatrigelTM в среде DMEM:F12. Клетки разделили ферментативным пассированием со средним интервалом четыре дня между последовательными пассированиями. При пассировании монослой клеток обрабатывали раствором коллагеназы (1 мг/мл; Sigma-Aldrich) в течение 10-60 минут при температуре 37°C, затем осторожно соскоблили кончиком пипетки, получив кластеры клеток. Полученным кластерам дали осесть под собственным весом и затем промыли их для удаления остатков коллагеназы. Кластеры клеток разделили в соотношении 1:3 для стандартного поддерживающего культивирования или в соотношении 1:1 для немедленного анализа. Использованные в анализе линии эмбриональных стволовых клеток человека поддерживали при номерах пассажа менее 50 и периодически проверяли на нормальный кариотипический фенотип и отсутствие примесей микоплазмы.
Подготовка клеток для анализа: Используемые в анализе кластеры клеток линий H1 или H9 эмбриональных стволовых клеток человека однородно ресуспендировали в культуральной среде и высеяли на покрытые препаратом MatrigelTM 96-луночные планшеты типа Packard VIEWPLATE (PerkinElmer) в объемах 100 мкл/лунку. Для первоначального посева и размножения использовали кондиционированную среду MEF с добавлением 8 нг/мл bFGF. Ежедневное питание осуществляли путем отсасывания отработанной культуральной среды из каждой лунки и ее замены тем же объемом свежей среды. Перед началом анализа культуры размножали в течение от одного до трех дней после посева. В течение всего анализа планшеты держали при температуре 37°C в атмосфере 5% CO2 в увлажняемом боксе.
Подготовка соединений и среды для анализа: Для проведения дальнейших исследований и вторичных подтверждающих анализов использовали подмножество соединений, показавших положительный результат при первичном скрининге. Двадцать соединений, доступных в виде сухих порошков, растворили в DMSO до концентрации 10 мМ и до использования хранили с дессикатором при температуре -20°C. Непосредственно перед проведением анализа маточные растворы соединений разбавили 1:1000 до получения 10 мкМ раствора испытываемого соединения в основной среде DMEM:F12 (Invitrogen) с добавлением 0,5% FCS (HyClone) и 100 нг/мл Activin A (R&D Biosystems). Полученные растворы далее последовательно разбавили вдвое, получив для каждого соединения кривые разбавления из семи точек, также используя для разбавления основную среду DMEM:F12 с 0,5% FCS и 100 нг/мл Activin A.
Вторичный скрининговый анализ: Анализ начали с отсасывания культуральной среды из каждой лунки с монослоем клеток с последующей трехкратной промывкой лунки раствором PBS для удаления остатков факторов роста и сыворотки. Затем в лунки добавили испытываемые образцы объемом 100 мкл, содержащие основную среду с добавлением 0,5% FCS, варьируемую концентрацию испытываемых ингибиторов, 100 нг/мл Activin A, без Wnt3a. Лунки с положительными контрольными образцами содержали ту же основную среду с 0,5% FCS и 20 нг/мл Wnt3a (R&D Biosystems), без испытываемого соединения. Лунки с отрицательными контрольными образцами содержали ту же основную среду с 0,5% FCS, без Activin A, Wnt3a и испытываемого соединения. На 2 день анализа из лунок отсосали отработанную среду и лунки снова наполнили растворами с теми же концентрациями испытываемых соединений или контрольными растворами. На 3 и 4 день содержимое всех лунок отсосали и лунки наполнили средой DMEM:F12 с добавлением 2% FCS и 100 нг/мл Activin A, без тестовых соединений и Wnt3a. Лунки с параллельными отрицательными контрольными образцами на 3 и 4 день поддерживали в основной среде DMEM:F12 с добавлением 2% FCS.
Оценка результатов анализа: По окончании культивирования находящиеся в 96-луночных планшетах клетки дважды промыли раствором PBS, зафиксировали 4% параформальдегидом при комнатной температуре в течение 20 минут, трижды промыли раствором PBS и затем пермеабилизовали, обработав раствором 0,5% Triton X-100 в течение 20 минут при комнатной температуре. После фиксации и пермеабилизации клетки снова трижды промыли раствором PBS и заблокировали раствором 4% куриной сыворотки (Invitrogen) в растворе PBS в течение 30 минут при комнатной температуре. Первичные антитела (козий античеловеческий Sox17; R&D Systems) развели 1:100 в 4% куриной сыворотке и добавили к клеткам на один час при комнатной температуре. Сопряженные с флюорофором Alexa Fluor 488 вторичные антитела (куриный антикозий IgG; Molecular Probes) развели 1:200 в растворе PBS и добавили в каждую лунку к клеткам после их трехкратной промывки раствором PBS. Для контрастной окраски ядер добавили 2 мкг/мл красителя Hoechst 33342 (Invitrogen) на 10 минут при комнатной температуре. Затем клетки один раз промыли раствором PBS и оставили в 100 мкл раствора PBS/лунку для томографии.
Томографию клеток проводили на анализаторе IN Cell Analyzer 1000 (GE Healthcare), используя дихроичное зеркало 51008bs для клеток, окрашенных Hoechst 33342 и Alexa Fluor 488. Времена экспозиции оптимизировали с использованием лунок с положительными контрольными образцами и лунок, окрашенных только вторичными антителами, в качестве необработанных отрицательных контрольных образцов. Для компенсации возможных потерь клеток в ходе процедур обработки и окрашивания для каждой лунки снимали по 15 проекций. Общее количество клеток и общую интенсивность Sox-17 измеряли для каждой лунки с помощью программного пакета IN Cell Developer Toolbox 1.7 (GE Healthcare). Сегментацию ядер определяли по ступеням шкалы серых тонов (исходный интервал 100-300) и размерам ядра. Для каждой повторной совокупности данных рассчитали средние значение и стандартные отклонения. Общий уровень экспрессии белка Sox17 выражается в виде общей интенсивности или интегральной интенсивности, определяемой как произведение суммарной интенсивности флюоресценции клетки на площадь клетки. Фон исключили на основании критериев соответствия ступеням шкалы серых тонов в диапазоне от 300 до 3000 и форм-факторам от 0,4 и выше. Показатели общей интенсивности нормировали путем деления общей интенсивности для каждой лунки на среднюю общую интенсивность для положительного контрольного образца Wnt3a/Activin A. Для каждого набора повторностей рассчитали нормированные данные для средних значений и стандартных отклонений.
Результаты
Приведены результаты для восьми ингибиторов фермента GSK-3B, где титрованием в описанном вторичном анализе была подтверждена активность ингибитора и определена его эффективность. Приведенные данные демонстрируют эффект испытываемых соединений на численность клеток и интенсивность Sox17, где соответствующие точки получены усреднением по наборам повторностей и для каждого параметра извлечены из одних и тех же проекций для одних и тех же лунок. В приведенном примере уровень экспрессии Sox17 является индикатором дифференцирования в линию сформированной эндодермы. Результаты по количеству клеток и интенсивности Sox17, полученные для линии H1 эмбриональных стволовых клеток человека, приведены в таблицах VIII и IX, соответственно. Результаты для линии H9 эмбриональных стволовых клеток человека приведены в таблицах X и XI. Величины для положительных контрольных образцов нормированы на 1,000 для численности клеток и интенсивности Sox17. Величины для отрицательных контрольных образцов составили менее чем 0,388 для численности клеток и менее чем 0,065 для интенсивности Sox17 для обеих клеточных линий. Графическое изображение полученных данных, со сравнением двух линий эмбриональных стволовых клеток человека и включением титрования дозы для каждого соединения, приведено на фиг. 1-8. Численность клеток представлена в части A; интенсивность Sox17 представлена в части B каждого рисунка. Приведенные данные подтверждают, что каждое соединение может стимулировать пролиферацию эмбриональных стволовых клеток человека и их дифференцирование в клетки линии сформированной эндодермы, и указывают оптимальные диапазоны активности.
Пример 8
Эффект ингибиторов фермента GSK-3β на уровни экспрессии дополнительных маркеров, ассоциируемых с линией дифференцирования сформированной эндодермы
Было важно показать способность соединений, показавших положительный результат при первичном скрининге, в дополнение к фактору транскрипции Sox17 индуцировать также другие маркеры, характерные для дифференцирования по линии сформированной эндодермы. Выбранное подмножество соединений, показавших положительный результат при первичном скрининге, проверили на способность стимулировать экспрессию CXCR4, белка поверхностного рецептора и HNF-3 beta, фактора транскрипции, который также ассоциирован с дифференцированием по линии сформированной эндодермы.
Подготовка клеток для анализа: Используемые в анализе кластеры клеток линии H1 эмбриональных стволовых клеток человека однородно ресуспендировали в культуральной среде и высеяли на покрытые препаратом MATRIGELTM (в разбавлении 1:30) 6-луночные планшеты (Corning) в объемах 2 мл/лунку. Для первоначального посева и размножения использовали кондиционированную среду MEF с добавлением 8 нг/мл bFGF. Ежедневное питание осуществляли путем отсасывания отработанной культуральной среды из каждой лунки и ее замены тем же объемом свежей среды. Перед началом анализа культуры размножали в течение от одного до трех дней после посева. В течение всего анализа планшеты держали при температуре 37°C в атмосфере 5% CO2.
Подготовка соединений и среды для анализа: Для проведения дальнейших исследований и вторичных анализов использовали подмножество из семи соединений, показавших положительный результат при первичном скрининге. Чистые соединения растворили в DMSO до концентрации 10 мМ и до использования хранили с дессикатором при температуре -20°C. Непосредственно перед проведением анализа маточные растворы соединений разбавили до получения конечной концентрации в диапазоне от 1 мкМ до 5мкМ в основной среде DMEM:F12 (Invitrogen) с добавлением 0,5% FCS (HyClone) и 100 нг/мл Activin A (R&D Biosystems).
Анализ: Анализ начали с отсасывания культуральной среды из каждой лунки с монослоем клеток с последующей трехкратной промывкой лунки раствором PBS для удаления остатков факторов роста и сыворотки. Затем в лунки добавили испытываемые образцы объемом 2 мл, содержащие основную среду с добавлением 0,5% FCS, варьируемую концентрацию испытываемых ингибиторов с 100 нг/мл Activin A, без Wnt3a. Лунки с положительными контрольными образцами содержали ту же основную среду с 0,5% FCS, 100 нг/мл Activin A и 20 нг/мл Wnt3a (R&D Biosystems), без испытываемого соединения. Лунки с отрицательными контрольными образцами содержали ту же основную среду с 0,5% FCS, без Activin A, Wnt3a и испытываемого соединения. На 2 день анализа из лунок отсосали отработанную среду, и лунки снова наполнили растворами с теми же концентрациями испытываемых соединений или контрольными растворами. На 3 и 4 день содержимое всех лунок отсосали и лунки наполнили средой DMEM:F12 с добавлением 2% FCS и 100 нг/мл Activin A, без тестовых соединений и Wnt3a. Лунки с параллельными отрицательными контрольными образцами на 3 и 4 день поддерживали в основной среде DMEM:F12 с добавлением 2% FCS.
Оценка результатов анализа: По окончании культивирования монослои клеток промыли раствором PBS и собрали с культуральных планшетов путем инкубации с раствором TrypLE™ Express (Invitrogen) в течение 5 минут. Затем клетки ресуспендировали в кондиционированной среде MEF и разделили на пары равных образцов. Один набор образцов далее окрасили с помощью меченных различными флюоресцентными метками антител и анализировали способом проточной цитометрии (FACS). Второй параллельный набор образцов анализировали способом количественного ПЦР.
Клетки для цитометрического анализа промыли в растворе PBS и в течение 15 минут блокировали при температуре 4°C в 0, 125% растворе гамма-глобулина человека (Sigma, Кат. № G-4386), разведенного в растворе PBS и буфере для окрашивания BD FACS. Аликвоты клеток (приблизительно 105 клеток каждая) окрашивали в течение 30 минут при температуре 4°C антителами, непосредственно сопряженными с флюоресцентной меткой и специфичными по отношению к CD9 PE (BD № 555372), CD99 PE (Caltag № MHCD9904) или CXCR-4 APC (R&D Systems, Кат. № FAB173A). После ряда промывок в окрашивающем буфере BD FACS клетки окрасили 7-AAD (BD № 559925) для оценки их жизнеспособности и анализировали на анализаторе BD FACS Array (BD Biosciences), накапливая не менее 10000 событий. Для получения процентной доли положительных клеток использовали мышиные IgG1k изотипические контрольные антитела к PE и APC.
Клетки для количественного ПЦР обработали для выделения РНК, очистки и синтеза кДНК. Образцы РНК очистили путем связывания с силикагелевой мембраной (Rneasy Mini Kit, Qiagen, Калифорния, США) в присутствии этанолсодержащего высокосолевого буферного раствора, а затем промыли для удаления примесей. Затем РНК очищали далее, используя набор TURBO DNA-free (Ambion, Inc.), элюировав в воду РНК высокого качества. Выход и чистоту полученной РНК оценивали по измеряемому на спектрофотометре поглощению на длинах волн 260 и 280 нм. Копии кДНК получили с очищенной РНК, используя набор высокой емкости для получения кДНК из архивного материала компании Applied Biosystems, Inc. (ABI, Калифорния, США).
Если не указано иное, все реактивы для ПЦР-амплификации в реальном времени и количественного определения приобретались у компании ABI. Реакции ПЦР в реальном времени проводили с использованием системы для определения последовательности ABI PRISM 7900 Sequence Detection System. Использовали набор TAQMAN UNIVERSAL PCR MASTER MIX (ABI, Калифорния, США) с 20 нг обратно транскрибированной РНК при полном объеме реакционной смеси 20 мкл. Каждый образец кДНК анализировали дважды для коррекции погрешности пипетирования. Праймеры и меченные красителем FAM зонды TAQMAN использовали в концентрациях 200 нМ. Уровень экспрессии для каждого целевого гена нормировали по эндогенному контрольному образцу, глицеральдегид-3-фосфатдегидрогеназе человека (ГАФДГ), ранее разработанному компанией ABI. Использовали следующие комплекты праймеров и зондов: CXCR4 (Hs00237052), GAPDH (4310884E), HNF3b (Hs00232764), SOX17 (зондовая часть № 450025, прямая и обратная часть № 4304971).
После начальной инкубации при температуре 50°C в течение 2 мин с последующей инкубацией при температуре 95°C в течение 10 минут с образцами провели 40 двухстадийных циклов, включающих стадию денатурации при температуре 95°C продолжительностью 15 секунд и следующую за ней стадию отжига/роста при температуре 60°C продолжительностью 1 минута. Анализ данных проводили с использованием программного пакета GENEAMP 7000 Sequence Detection System. Для каждого комплекта праймеров и зондов определялось значение порогового цикла (Ct) как номер цикла, при котором интенсивность флюоресценции достигала определенного значения в середине экспоненциальной области амплификации. Уровни относительной экспрессии генов вычисляли методом сравнения Ct. Коротко говоря, для каждого образца кДНК значение Ct эндогенного контрольного образца вычитали из значения Ct целевого гена и получали величину дельта Ct (∆Ct). Нормированное количество целевого гена рассчитывали как 2-∆Ct, принимая, что амплификация имеет 100% эффективность. Окончательные данные были выражены относительно калибровочного образца.
Результаты
На фиг. 9 приведены результаты цитометрического анализа (FACS) процента положительных клеток с экспрессией поверхностного рецептора CXCR4 после обработки различными ингибиторами GSK3. Приведены данные для двух концентраций каждого соединения в диапазоне от 1 мкМ до 5 мкМ в сравнении с необработанной популяцией клеток (отрицательный контрольный образец) и клетками, обработанными Activin A и Wnt3 (положительный контрольный образец). На фиг. 10A, B и C приведены данные ПЦР в реальном времени для CXCR4, Sox17 и HNF3beta, которые также рассматриваются в качестве маркеров сформированной эндодермы. И цитометрический анализ, и анализ ПЦР в реальном времени свидетельствуют о значительном росте каждого из указанных маркеров в дифференцированных клетках относительно необработанных контрольных клеток. В ряде случаев уровни экспрессии указанных маркеров сформированной эндодермы оказались эквивалентны соответствующим уровням в положительных контрольных образцах, тем самым демонстрируя, что ингибитор GSK3 может заменить Wnt3a на этой стадии клеточной дифференцирования.
Пример 9
Эффект ингибиторов фермента GSK-3β на формирование панкреатической эндодермы
Было важно показать, что обработка ингибиторами GSK3β на стадии индуцирования сформированной эндодермы не препятствует дальнейшему дифференцированию клеток других типов, таких как, например, клетки панкреатической эндодермы. Выбранное подмножество соединений, показавших положительный результат при первичном скрининге, проверили на способность стимулировать экспрессию PDX1 и HNF6, ключевых факторов транскрипции, ассоциированных с дифференцированием по линии панкреатической эндодермы.
Поддержание эмбриональных стволовых клеток человека (линий H1 и H9) проводили, как описано в примере 1. Колонии клеток поддерживали в недифференцированном плюрипотентном состоянии со средним интервалом четыре дня между последовательными пассированиями. При пассировании культуры клеток обрабатывали раствором коллагеназы (1 мг/мл; Sigma-Aldrich) в течение 10-30 минут при температуре 37°C, затем осторожно соскоблили кончиком пипетки, получив кластеры клеток. Полученным кластерам дали осесть под собственным весом и затем промыли их для удаления остатков коллагеназы. Кластеры клеток разделили в соотношении 1:3 для стандартного поддерживающего культивирования или в соотношении 1:1 для немедленного анализа. Использованные в анализе линии эмбриональных стволовых клеток человека поддерживали при номерах пассажа менее 50 и периодически проверяли на нормальный кариотипический фенотип и отсутствие примесей микоплазмы.
Подготовка клеток для анализа: Используемые в анализе кластеры клеток линии H1 эмбриональных стволовых клеток человека однородно ресуспендировали в культуральной среде и высеяли на покрытые препаратом MATRIGELTM (в разбавлении 1:30) 24-луночные планшеты (черные лунки; Arctic White) в объеме 1 мл/лунку. Для первоначального посева и размножения использовали кондиционированную среду MEF с добавлением 8 нг/мл bFGF. Во втором эксперименте кластеры клеток линии H9 эмбриональных стволовых клеток человека высеяли на 96-луночные планшеты с нанесенным питающим слоем из мышиных эмбриональных фибропластов, инактивированных обработкой митомицином C (Sigma Chemical Co). Культуральная среда для эмбриональных стволовых клеток человека на монослоях мышиных эмбриональных фибробластов состояла из среды DMEM:F12 с 20% нокаут-заменителем сыворотки (Invitrogen) с добавлением минимально необходимых незаменимых аминокислот (Invitrogen), L-глутамина и 2-меркаптоэтанола. Ежедневное питание осуществляли путем отсасывания отработанной культуральной среды из каждой лунки и ее замены тем же объемом свежей среды. Перед началом анализа культуры размножали в течение от одного до трех дней после посева. В течение всего анализа планшеты держали при температуре 37°C в атмосфере 5% CO2.
Подготовка соединений и среды для анализа: Для проведения дальнейших исследований и вторичных анализов использовали подмножество из восьми соединений, показавших положительный результат при первичном скрининге. Чистые соединения растворили в DMSO до концентрации 10 мМ и до использования хранили с дессикатором при температуре -20°C. Непосредственно перед проведением анализа маточные растворы соединений разбавили до получения конечной концентрации в диапазоне от 1 мкМ до 5 мкМ в основной среде с добавками.
Анализ: В этом анализе ингибиторы GSK3 вводили только на 1 и 2 день стадии дифференцирования по линии сформированной эндодермы, заменяя ими Wnt3a. Анализ культур эмбриональных стволовых клеток на покрытых препаратом MATRIGELTM носителях начали, как описано в примерах 7 и 8 выше, с отсасывания культуральной среды из каждой лунки с монослоем клеток с последующей трехкратной промывкой лунки раствором PBS для удаления остатков факторов роста и сыворотки. Для дифференцирования в клетки сформированной эндодермы в лунки добавили испытываемые образцы (объемом 0,5 мл/лунку для 24-луночных планшетов, 100 мкл/лунку для 96-луночных планшетов), содержащие среду DMEM:F12 с добавлением 0,5% FCS, варьируемую концентрацию испытываемых ингибиторов, 100 нг/мл Activin A, без Wnt3a. Лунки с положительными контрольными образцами содержали ту же основную среду с 0,5% FCS, 100 нг/мл Activin A и 20 нг/мл Wnt3a (R&D Biosystems), без испытываемого соединения. Лунки с отрицательными контрольными образцами содержали ту же основную среду с 0,5% FCS, без Activin A, Wnt3a и испытываемого соединения. На 2 день анализа из лунок отсосали отработанную среду и лунки наполнили растворами с теми же концентрациями испытываемых соединений или контрольными растворами. На 3 и 4 день содержимое всех лунок отсосали и лунки наполнили средой DMEM:F12 с добавлением 2% FCS и 100 нг/мл Activin A, без тестовых соединений и Wnt3a. Лунки с параллельными отрицательными контрольными образцами на 3 и 4 день поддерживали в основной среде DMEM:F12 с добавлением 2% FCS. Для дифференцирования по линии панкреатической эндодермы клетки обрабатывали в течение трех дней, ежедневно внося основную среду DMEM:F12 с 2% FCS с добавлением 0,25 мкМ циклопамина 3-кето-N-(аминоэтил-аминокапроил-дигидроциннамоила (KAAD) (EMD Biosciences) и 20 нг/мл фактора роста фибробластов FGF7 (R&D Biosystems). Затем клетки обрабатывали в течение еще четырех дней, ежедневно внося основную среду DMEM:F12 с добавлением 1% B27 (Invitrogen), 0,25 мкМ KAAD-циклопамина, 2 мкМ ретиноевой кислоты (RA; Sigma-Aldrich) и 20 нг/мл FGF7. Лунки с параллельными отрицательными контрольными образцами все время поддерживали в основной среде DMEM:F12 с добавлением 2% FCS (стадия 2) или 1% B27 (стадия 3) без каких-либо других добавок.
Параллельно на питающих слоях мышиных эмбриональных фибробластов выращивали культуры клеток линии H9 эмбриональных стволовых клеток человека и дифференцировали их в клетки панкреатической эндодермы. Дифференцирование сформированной эндодермы провели путем культивирования клеток в среде, состоящей из основной среды RPMI-1640 (Invitrogen), не содержащей сыворотки на 1 день и содержащей 0,2% FCS на 2 и 3 дни, с добавлением различных концентраций испытываемых ингибиторов и 100 нг/мл Activin A. Лунки с положительными контрольными образцами содержали ту же основную среду (с сывороткой или без сыворотки) с 100 нг/мл Activin A и 20 нг/мл Wnt3a (R&D Biosystems), без испытываемого соединения. Лунки с отрицательными контрольными образцами содержали ту же основную среду (с сывороткой или без сыворотки), без Activin A, Wnt3a и испытываемого соединения. На 2 день анализа из лунок отсосали отработанную среду и лунки снова наполнили растворами с теми же концентрациями испытываемых соединений или контрольными растворами. На 3 день содержимое всех лунок отсосали и лунки наполнили средой RPMI-1640 с добавлением 2% FCS и 100 нг/мл Activin A, без тестовых соединений и Wnt3a. Лунки с параллельными отрицательными контрольными образцами на 3 день поддерживали в основной среде RPMI-1640 с добавлением 2% FCS. Клетки дифференцировали в клетки линии панкреатической эндодермы путем четырехдневной обработки, ежедневно внося основную среду RPMI-1640 с 2% FCS с добавлением 0,25 мкМ KAAD-циклопамина (EMD Biosciences) и 50 нг/мл фактора роста фибробластов FGF10 (R&D Biosystems). Затем клетки обрабатывали в течение еще трех дней, ежедневно внося основную среду RPMI-1640 с добавлением 1% B27 (Invitrogen), 0,25 мкМ KAAD-циклопамина, 2 мкМ ретиноевой кислоты (RA; Sigma-Aldrich) и 50 нг/мл FGF10. Лунки с параллельными отрицательными контрольными образцами все время поддерживали в основной среде RPMI-1640 с добавлением 2% FCS (стадия 2) или 1% B27 (стадия 3) без каких-либо других добавок.
Оценка результатов анализа: По окончании дифференцирования клетки проанализировали методом ПЦР в реальном времени на уровни экспрессии генов, как описано в примере 8 выше. Для флюоресцентного окрашивания, используемого в методе одновременного многопараметрического анализа, клетки в 96-луночных планшетах дважды промыли раствором PBS, зафиксировали 4% параформальдегидом при комнатной температуре в течение 20 минут, снова трижды промыли раствором PBS и затем пермеабилизовали, обработав раствором 0,5% Triton X-100 в течение 20 минут при комнатной температуре. После фиксации и пермеабилизации клетки снова трижды промыли раствором PBS и заблокировали раствором 4% куриной сыворотки (Invitrogen) в растворе PBS в течение 30 минут при комнатной температуре. Первичные антитела (козий античеловеческий Pdx1; Santa Cruz) развели 1:100 в 4% куриной сыворотке и добавили к клеткам на два часа при комнатной температуре. Сопряженные с флюорофором Alexa Fluor 488 вторичные антитела (куриный антикозий IgG; Molecular Probes) развели 1:200 в растворе PBS и добавили в каждую лунку после трехкратной промывки клеток раствором PBS. Для контрастной окраски ядер добавили 2 мкг/мл красителя Hoechst 33342 (Invitrogen) на 10 минут при комнатной температуре. Затем клетки один раз промыли раствором PBS и оставили в 100 мкл раствора PBS/лунку для томографии.
Томографию клеток проводили на анализаторе IN Cell Analyzer 1000 (GE Healthcare), используя дихроичное зеркало 51008bs для клеток, окрашенных Hoechst 33342 и Alexa Fluor 488. Времена экспозиции оптимизировали с использованием лунок с положительными контрольными образцами и лунок, окрашенных только вторичными антителами. Для компенсации возможных потерь клеток в ходе процедур обработки и окрашивания для каждой лунки снимали по 15 проекций. Общее количество клеток и общую интенсивность Pdx1 измеряли для каждой лунки с помощью программного пакета IN Cell Developer Toolbox 1.7 (GE Healthcare). Сегментацию ядер определяли по ступеням шкалы серых тонов (исходный интервал 100-300) и размерам ядра. Для каждой повторной совокупности данных рассчитали средние значения и стандартные отклонения. Общий уровень экспрессии белка Pdx1 выражается в виде общей интенсивности или интегральной интенсивности, определяемой как произведение суммарной интенсивности флюоресценции клетки на площадь клетки. Фон исключили на основании критериев соответствия ступеням шкалы серых тонов в диапазоне от 300 до 3000. Показатели общей интенсивности нормировали путем деления общей интенсивности для каждой лунки на среднюю общую интенсивность для положительного контрольного образца Wnt3a/Activin A. Для каждого набора повторностей рассчитали нормированные данные для средних значений и стандартных отклонений.
Клетки для количественного ПЦР лизировали в буфере RLT (Qiagen) и затем обработали для выделения РНК, очистки и синтеза кДНК. Образцы РНК очистили путем связывания с силикагелевой мембраной (Rneasy Mini Kit, Qiagen, Калифорния, США) в присутствии этанолсодержащего высокосолевого буферного раствора, а затем промыли для удаления примесей. Затем РНК очищали далее, используя набор TURBO DNA-free (Ambion, Inc.), элюировав в воду РНК высокого качества. Выход и чистоту полученной РНК оценивали по измеряемому на спектрофотометре поглощению на длинах волн 260 и 280 нм. Копии кДНК получили с очищенной РНК, используя набор высокой емкости для получения кДНК из архивного материала компании Applied Biosystems, Inc. (ABI, Калифорния, США).
Если не указано иное, все реактивы для ПЦР-амплификации в реальном времени и количественного определения приобретались у компании ABI. Реакции ПЦР в реальном времени проводили с использованием системы для определения последовательности ABI PRISM 7900 Sequence Detection System. Использовали набор TAQMAN UNIVERSAL PCR MASTER MIX с 20 нг обратно транскрибированной РНК при полном объеме реакционной смеси 20 мкл. Каждый образец кДНК анализировали дважды для коррекции погрешности пипетирования. Праймеры и меченные красителем FAM зонды TAQMAN использовали в концентрациях 200 нМ. Уровень экспрессии для каждого целевого гена нормировали по эндогенному контрольному образцу, глицеральдегид-3-фосфатдегидрогеназе человека (ГАФДГ), ранее разработанному компанией ABI. Использовали следующие комплекты праймеров и зондов: PDX1 (Hs00236830_m1), GAPDH (4310884E) и HNF6 (Hs00413554_m1).
После начальной инкубации при температуре 50°C в течение 2 мин с последующей инкубацией при температуре 95°C в течение 10 минут с образцами провели 40 двухстадийных циклов, включающих стадию денатурации при температуре 95°C продолжительностью 15 секунд и следующую за ней стадию отжига/роста при температуре 60°C продолжительностью 1 минута. Анализ данных проводили с использованием программного пакета GENEAMP 7000 Sequence Detection System. Для каждого комплекта праймеров и зондов определялось значение порогового цикла (Ct) как номер цикла, при котором интенсивность флюоресценции достигала определенного значения в середине экспоненциальной области амплификации. Уровни относительной экспрессии генов вычисляли методом сравнения Ct. Коротко говоря, для каждого образца кДНК значение Ct эндогенного контрольного образца вычитали из значения Ct целевого гена и получали величину дельта Ct (∆Ct). Нормированное количество целевого гена рассчитывали как 2-∆Ct, принимая, что амплификация имеет 100% эффективность. Окончательные данные были выражены относительно калибровочного образца.
Результаты
Результаты приведены для восьми ингибиторов фермента GSK-3β. Представленные на фиг. 11 результаты одновременного многопараметрического анализа демонстрируют эффект испытываемых соединений на численность клеток (часть A) и интенсивность Pdx1 (часть B) для линии H1 эмбриональных стволовых клеток человека, где соответствующие точки получены усреднением по наборам повторностей и для каждого параметра извлечены из одних и тех же проекций для одних и тех же лунок. Представленные на фиг. 12 результаты ПЦР в реальном времени демонстрируют эффект описываемых низкомолекулярных ингибиторов на индуцированную экспрессию двух факторов транскрипции, Pdx1 и HNF6. В приведенных примерах уровни экспрессии Pdx1 и HNF6 являются индикатором дифференцирования в линию панкреатической эндодермы. В проведенных анализах соединения-ингибиторы GSK3β могут заменить Wnt3a на ранних стадиях коммитирования клеток к линии дифференцирования; формируемые при этом клетки сохраняют способность к образованию панкреатической эндодермы в ходе последующих стадий дифференцирования.
Пример 10
Эффект ингибиторов фермента GSK-3β на формирование панкреатических эндокринных клеток
Было важно показать, что обработка ингибиторами GSK3 на стадии индуцирования сформированной эндодермы не препятствует дальнейшему дифференцированию клеток других типов, таких как, например, панкреатических эндокринных клеток или клеток, вырабатывающих инсулин. Выбранное подмножество соединений, показавших положительный результат при первичном скрининге, проверили на способность стимулировать экспрессию панкреатических гормонов.
Подготовка клеток для анализа: Клетки панкреатичекой эндодермы, полученные в соответствии с описанными в примере 9 способами (культивированные на 96-луночных и 24-луночных планшетах), затем обработали агентами, приводящими к дифференцированию клеток в панкреатические экспрессирующие гормоны клетки.
Анализ культур эмбриональных стволовых клеток человека линии H1 на покрытых препаратом MATRIGELTM носителях начали, как описано в примерах 7-9 выше, с отсасывания культуральной среды из каждой лунки с монослоем клеток с последующей трехкратной промывкой лунки раствором PBS для удаления остатков факторов роста и сыворотки. Для дифференцирования в клетки сформированной эндодермы в лунки добавили испытываемые образцы (объемом 0,5 мл/лунку для 24-луночных планшетов, 100 мкл/лунку для 96-луночных планшетов), содержащие среду с добавлением 0,5% FCS, варьируемую концентрацию испытываемых ингибиторов, 100 нг/мл Activin A, без Wnt3a. Лунки с положительными контрольными образцами содержали ту же основную среду с 0,5% FCS, 100 нг/мл Activin A и 20 нг/мл Wnt3a (R&D Biosystems), без испытываемого соединения. Лунки с отрицательными контрольными образцами содержали ту же основную среду с 0,5% FCS, без Activin A, Wnt3a и испытываемого соединения. На 2 день анализа из лунок отсосали отработанную среду и лунки снова наполнили растворами с теми же концентрациями испытываемых соединений или контрольными растворами. На 3, 4 и 5 день содержимое всех лунок отсосали и лунки наполнили средой DMEM:F12 с добавлением 2% FCS и 100 нг/мл Activin A, без тестовых соединений и Wnt3a. Лунки с параллельными отрицательными контрольными образцами на 3, 4 и 5 день поддерживали в основной среде DMEM:F12 с добавлением 2% FCS. Для дифференцирования по линии панкреатической эндодермы клетки обрабатывали в течение трех дней, ежедневно внося основную среду DMEM:F12 с 2% FCS с добавлением 0,25 мкМ KAAD-циклопамина (EMD Biosciences) и 20 нг/мл FGF7 (R&D Biosystems). Затем клетки обрабатывали в течение еще четырех дней, ежедневно внося основную среду DMEM:F12 с добавлением 1% B27 (Invitrogen), 0,25 мкМ KAAD-циклопамина, 2 мкМ ретиноевой кислоты (RA; Sigma-Aldrich) и 20 нг/мл FGF7. Лунки с параллельными отрицательными контрольными образцами на стадиях 2 и 3 все время поддерживали в основной среде DMEM:F12 с добавлением 2% FCS или 1% B27 без каких-либо других добавок. После формирования панкреатической эндодермы клетки обрабатывали далее в течение шести дней, ежедневно внося основную среду DMEM:F12 с добавлением 1% B27 с 1 мкМ DAPT (ингибитор гамма-секретазы: EMD Biosciences) и 50 нг/мл Exendin 4 (Sigma-Aldrich). Затем клетки обрабатывали в течение еще трех дней, ежедневно внося основную среду DMEM:F12 с добавлением 1% B27, 50 нг/мл Exendin 4, 50 нг/мл IGF (R&D Biosystems) и 50 нг/мл HGF (R&D Biosystems). Лунки с параллельными отрицательными контрольными образцами все время поддерживали в основной среде DMEM:F12 с добавлением 1% B27 без каких-либо других добавок.
Оценка результатов анализа: По окончании культивирования клетки обработали, как описано в примерах 7 и 8 выше, для анализа методами одновременного многопараметрического анализа или ПЦР в реальном времени.
Для флуоресцентного окрашивания, используемого в методе одновременного многопараметрического анализа, клетки в 96-луночных планшетах дважды промыли раствором PBS, зафиксировали 4% параформальдегидом при комнатной температуре в течение 20 минут, трижды промыли раствором PBS и затем пермеабилизовали, обработав раствором 0,5% Triton X-100 в течение 20 минут при комнатной температуре. После фиксации и пермеабилизации клетки снова трижды промыли раствором PBS и заблокировали раствором 4% куриной сыворотки (Invitrogen) в растворе PBS в течение 30 минут при комнатной температуре. Первичные антитела (морской свинки антисвиной инсулин, перекрестная активность с инсулином человека; DakoCytomation) развели 1:500 в 4% козьей сыворотке и добавили к клеткам на один час при комнатной температуре. Клетки трижды промыли раствором PBS и затем окрасили сопряженными с флюорофором Alexa Fluor 488 вторичными антителами (козий антиморской свинки IgG; Molecular Probes), разбавленными 1:100 в 4% козьей сыворотке. Для контрастной окраски ядер добавили 2 мкг/мл красителя Hoechst 33342 (Invitrogen) на 10 минут при комнатной температуре. Затем клетки один раз промыли раствором PBS и оставили в 100 мкл раствора PBS/лунку для томографии.
Томографию клеток проводили на анализаторе IN Cell Analyzer 1000 (GE Healthcare), используя дихроичное зеркало 51008bs для клеток, окрашенных Hoechst 33342 и Alexa Fluor 488. Времена экспозиции оптимизировали с использованием лунок с положительными контрольными образцами и лунок, окрашенных только вторичными антителами. Для компенсации возможных потерь клеток в ходе процедур обработки и окрашивания для каждой лунки снимали по 15 проекций. Общее количество клеток и общую интенсивность инсулина измеряли для каждой лунки с помощью программного пакета IN Cell Developer Toolbox 1.7 (GE Healthcare). Сегментацию ядер определяли по ступеням шкалы серых тонов (исходный интервал 100-300) и размерам ядра. Для каждой повторной совокупности данных рассчитали средние значения и стандартные отклонения. Общий уровень экспрессии белка инсулин выражается в виде общей интенсивности или интегральной интенсивности, определяемой как произведение суммарной интенсивности флюоресценции клетки на площадь клетки. Фон исключили на основании критериев соответствия ступеням шкалы серых тонов в диапазоне от 300 до 3000. Показатели общей интенсивности нормировали путем деления общей интенсивности для каждой лунки на среднюю общую интенсивность для положительного контрольного образца Wnt3a/Activin A. Для каждого набора трех повторностей рассчитали нормированные данные для средних значений и стандартных отклонений.
Клетки для количественного ПЦР лизировали в буфере RLT (Qiagen) и затем обработали для выделения РНК, очистки и синтеза кДНК. Образцы РНК очистили путем связывания с силикагелевой мембраной (Rneasy Mini Kit, Qiagen, CA) в присутствии этанолсодержащего высокосолевого буферного раствора, а затем промыли для удаления примесей. Затем РНК очищали далее, используя набор TURBO DNA-free (Ambion, Inc.), элюировав в воду РНК высокого качества. Выход и чистоту полученной РНК оценивали по измеряемому на спектрофотометре поглощению на длинах волн 260 и 280 нм. Копии кДНК получили с очищенной РНК, используя набор высокой емкости для получения кДНК из архивного материала компании Applied Biosystems, Inc. (ABI, Калифорния, США).
Если не указано иное, все реактивы для ПЦР-амплификации в реальном времени и количественного определения приобретались у компании ABI. Реакции ПЦР в реальном времени проводили с использованием системы для определения последовательности ABI PRISM®7900 Sequence Detection System. Использовали набор TAQMAN® UNIVERSAL PCR MASTER MIX® (ABI, Калифорния, США) с 20 нг обратно транскрибированной РНК при полном объеме реакционной смеси 20 мкл. Каждый образец кДНК анализировали дважды для коррекции погрешности пипетирования. Праймеры и меченные красителем FAM зонды TAQMAN®использовали в концентрациях 200 нМ. Уровень экспрессии для каждого целевого гена нормировали по эндогенному контрольному образцу, глицеральдегид-3-фосфатдегидрогеназе человека (ГАФДГ), ранее разработанному компанией ABI. Использовали следующие комплекты праймеров и зондов: PDX1 (Hs00236830_m1), Insulin (Hs00355773) и GAPDH (4310884E).
После начальной инкубации при температуре 50°C в течение 2 мин с последующей инкубацией при температуре 95°C в течение 10 минут с образцами провели 40 двухстадийных циклов, включающих стадию денатурации при температуре 95°C продолжительностью 15 секунд и следующую за ней стадию отжига/роста при температуре 60°C продолжительностью 1 минута. Анализ данных проводили с использованием программного пакета GENEAMP®7000 Sequence Detection System. Для каждого комплекта праймеров и зондов определялось значение порогового цикла Ct как номер цикла, при котором интенсивность флюоресценции достигала определенного значения в середине экспоненциальной области амплификации. Уровни относительной экспрессии генов вычисляли методом сравнения Ct. Коротко говоря, для каждого образца кДНК значение Ct эндогенного контрольного образца вычитали из значения Ct целевого гена и получали величину дельта Ct (∆Ct). Нормированное количество целевого гена рассчитывали как 2-∆Ct, принимая, что амплификация имеет 100% эффективность. Окончательные данные были выражены относительно калибровочного образца.
Результаты
Результаты приведены для восьми ингибиторов фермента GSK-3B. Представленные на фиг. 13 результаты одновременного многопараметрического анализа демонстрируют эффект испытываемых соединений на численность клеток (часть A) и интенсивность инсулина (часть B) для эмбриональных стволовых клеток человека линии H1, где соответствующие точки получены усреднением по наборам трех повторностей и для каждого параметра извлечены из одних и тех же проекций для одних и тех же лунок. Представленные на фиг. 14 результаты ПЦР в реальном времени демонстрируют эффект соединений для Pdx1 и инсулина. В приведенных примерах уровни экспрессии Pdx1 и инсулина являются индикатором дифференцирования в линию панкреатической эндодермы и выработки гормон-положительных клеток. В проведенных анализах соединения-селективные ингибиторы GSK3β могут заменить Wnt3a на ранних стадиях коммитирования клеток к линии дифференцирования и могут индуцировать и поддерживать формирование панкреатических бета-клеток в ходе последующих стадий дифференцирования, как показывают результаты и инсулинового иммуноокрашивания, и ПЦР в реальном времени.
Пример 11
Аддитивные эффекты ингибиторов фермента GSK-3β на формирование панкреатических эндокринных клеток
Было важно показать, что обработка ингибиторами GSK3β может улучшить дифференцирование панкреатических бета клеток, если ингибиторы добавляются на нескольких стадиях коммитирования клеток. Выбранное подмножество соединений, показавших положительный результат при первичном скрининге, проверили методом последовательного добавления на способность повышать уровень экспрессии инсулина, ассоциируемый с панкреатическими гормон-положительными клетками.
Подготовка клеток для анализа: Клетки панкреатичекой эндодермы, полученные в соответствии с описанными в примерах 9 и 10 способами (культивированные на 96-луночных планшетах), затем обработали агентами, приводящими к дифференцированию клеток в панкреатические экспрессирующие гормоны клетки.
Анализ культур эмбриональных стволовых клеток человека линии H1 на покрытых препаратом MATRIGELTM носителях начали, как описано в примерах 7-9 выше, с отсасывания культуральной среды из каждой лунки с монослоем клеток с последующей трехкратной промывкой лунки раствором PBS для удаления остатков факторов роста и сыворотки. Для дифференцирования в клетки сформированной эндодермы в лунки добавили испытываемые образцы (объемом 100 мкл/лунку для 96-луночных планшетов), содержащие среду с добавлением 0,5% FCS, варьируемую концентрацию испытываемых ингибиторов, 100 нг/мл Activin A, без Wnt3a. Лунки с положительными контрольными образцами содержали ту же основную среду с 0,5% FCS, 100 нг/мл Activin A и 20 нг/мл Wnt3a (R&D Biosystems), без испытываемого соединения. Лунки с отрицательными контрольными образцами содержали ту же основную среду с 0,5% FCS, без Activin A, Wnt3a и испытываемого соединения. На 2 день анализа из лунок отсосали отработанную среду и лунки снова наполнили растворами с теми же концентрациями испытываемых соединений или контрольными растворами. На 3, 4 и 5 день содержимое всех лунок отсосали и лунки наполнили средой DMEM:F12 с добавлением 2% FCS и 100 нг/мл Activin A, без тестовых соединений и Wnt3a. Лунки с параллельными отрицательными контрольными образцами на 3, 4 и 5 день поддерживали в основной среде DMEM:F12 с добавлением 2% FCS. Для дифференцирования по линии панкреатической эндодермы клетки обрабатывали в течение трех дней, ежедневно внося основную среду DMEM:F12 с 2% FCS с добавлением 0,25 мкМ KAAD-циклопамина (EMD Biosciences) и 20 нг/мл FGF7 (R&D Biosystems). Затем клетки обрабатывали в течение еще четырех дней, ежедневно внося основную среду DMEM:F12 с добавлением 1% B27 (Invitrogen), 0,25 мкМ KAAD-циклопамина, 2 мкМ ретиноевой кислоты (RA; Sigma-Aldrich) и 20 нг/мл FGF7. Лунки с параллельными отрицательными контрольными образцами все время поддерживали в основной среде DMEM:F12 с добавлением 2% FCS или 1% B27 без каких-либо других добавок. После формирования панкреатической эндодермы клетки обрабатывали далее в течение шести дней, ежедневно внося основную среду DMEM:F12 с добавлением 1% B27 с 1 мкМ DAPT (ингибитор гамма-секретазы: EMD Biosciences) и 50 нг/мл Exendin 4 (Sigma-Aldrich) и 1 мкМ ингибитора II TGFbeta R1 (ингибитор ALK5; EMD Biosciences). В течение данного шестидневного периода ингибиторы GSK3β добавляли в соответствующие лунки, используя те же концентрации, что и при предыдущей обработке для инициации дифференцирования. Затем клетки обрабатывали в течение еще трех дней, ежедневно внося основную среду DMEM:F12 с добавлением 1% B27, 50 нг/мл Exendin 4, 50 нг/мл IGF (R&D Biosystems) и 50 нг/мл HGF (R&D Biosystems), и 1 мкМ ингибитора II TGFbeta R1 (ингибитор ALK5; EMD Biosciences). В течение данного трехдневного периода ингибиторы GSK3β добавляли в соответствующие лунки, используя те же концентрации, что и при предыдущей обработке для инициации дифференцирования. Лунки с параллельными положительными контрольными образцами обрабатывали в присутствии или в отсутствии 20 нг/мл Wnt3a. Лунки с параллельными отрицательными контрольными образцами все время поддерживали в основной среде DMEM:F12 с добавлением 1% B27 без каких-либо других добавок.
Оценка результатов анализа: По окончании культивирования клетки обработали, как описано в примере 10 выше, для анализа методом одновременного многопараметрического анализа.
Для флуоресцентного окрашивания, используемого в методе одновременного многопараметрического анализа, клетки в 96-луночных планшетах дважды промыли раствором PBS, зафиксировали 4% параформальдегидом при комнатной температуре в течение 20 минут, трижды промыли раствором PBS и затем пермеабилизовали, обработав раствором 0,5% Triton X-100 в течение 20 минут при комнатной температуре. После фиксации и пермеабилизации клетки опять трижды промыли раствором PBS и заблокировали раствором 4% куриной сыворотки (Invitrogen) в растворе PBS в течение 30 минут при комнатной температуре. Первичные антитела (морской свинки антисвиной инсулин, перекрестная активность с инсулином человека; DakoCytomation) развели 1:500 в 4% козьей сыворотке и добавили к клеткам на один час при комнатной температуре. Клетки трижды промыли раствором PBS и затем окрасили сопряженными с флюорофором Alexa Fluor 488 вторичными антителами (козий антиморской свинки IgG; Molecular Probes), разбавленными 1:100 в 4% козьей сыворотке. Для контрастной окраски ядер добавили 2 мкг/мл красителя Hoechst 33342 (Invitrogen) на 10 минут при комнатной температуре. Затем клетки один раз промыли раствором PBS и оставили в 100 мкл раствора PBS/лунку для томографии.
Томографию клеток проводили на анализаторе IN Cell Analyzer 1000 (GE Healthcare), используя дихроичное зеркало 51008bs для клеток, окрашенных Hoechst 33342 и Alexa Fluor 488. Времена экспозиции оптимизировали с использованием лунок с положительными контрольными образцами и лунок, окрашенных только вторичными антителами. Для компенсации возможных потерь клеток в ходе процедур обработки и окрашивания для каждой лунки снимали по 15 проекций. Общее количество клеток и общую интенсивность инсулина измеряли с помощью программного пакета IN Cell Developer Toolbox 1.7 (GE Healthcare). Сегментацию ядер определяли по ступеням шкалы серых тонов (исходный интервал 100-300) и размерам ядра. Для каждой повторной совокупности данных рассчитали средние значения и стандартные отклонения. Общий уровень экспрессии белка инсулин выражается в виде общей интенсивности или интегральной интенсивности, определяемой как произведение суммарной интенсивности флюоресценции клетки на площадь клетки. Фон исключили на основании критериев соответствия ступеням шкалы серых тонов в диапазоне от 300 до 3000. Показатели общей интенсивности нормировали путем деления общей интенсивности для каждой лунки на среднюю общую интенсивность для положительного контрольного образца Wnt3a/Activin A. Для каждого набора трех повторностей рассчитали нормированные данные для средних значений и стандартных отклонений.
Результаты
Результаты приведены для восьми ингибиторов фермента GSK-3β. Представленные на фиг. 15 результаты одновременного многопараметрического анализа демонстрируют эффект испытываемых соединений на численность клеток (часть A) и интенсивность инсулина (часть B) для эмбриональных стволовых клеток человека линии H1, где соответствующие точки получены усреднением по наборам трех повторностей и для каждого параметра извлечены из одних и тех же проекций для одних и тех же лунок. В приведенном примере уровень экспрессии инсулина является индикатором дифференцирования в линию панкреатических гормон-положительных клеток. В проведенных анализах соединения-селективные ингибиторы GSK3β могут заменить Wnt3a на ранних стадиях коммитирования клеток к линии дифференцирования и, при добавлении на более поздних стадиях, стимулируют повышение уровня экспрессии инсулина по отношению к положительному контрольному образцу.
Публикации, цитируемые в настоящем документе, в силу ссылки на них полностью включаются в настоящий документ. Хотя различные аспекты настоящего изобретения иллюстрируются выше ссылками на примеры и предпочтительные варианты осуществления, подразумевается, что область изобретения определяется не упомянутым выше описанием, а следующими ниже пунктами формулы изобретения, составленными в соответствии с принципами патентного законодательства.
Список первичных антител, используемых для цитометрического анализа (FACS) и иммуноокрашивания
Список вторичных сопряженных антител, используемых для цитометрического анализа (FACS) и иммуноокрашивания
Эффекты ингибиторов активности фермента GSK-3B на жизнеспособность экспрессирующих маркеры плюрипотентности клеток
отклонение
Эффекты ингибиторов активности фермента GSK-3B на жизнеспособность экспрессирующих маркеры плюрипотентности клеток
данные
Эффекты ингибиторов активности фермента GSK-3B на дифференцировку и пролиферацию эмбриональных стволовых клеток человека
Эффекты ингибиторов активности фермента GSK-3B на дифференцировку и пролиферацию эмбриональных стволовых клеток человека
Эффекты ингибиторов активности фермента GSK-3B на пролиферацию эмбриональных стволовых клеток человека
отклонение
DMSO
DMSO
DMSO
DMSO
Эффекты ингибиторов активности фермента GSK-3B на пролиферацию эмбриональных стволовых клеток человека
Зависящие от дозы эффекты ингибиторов активности фермента GSK-3B на пролиферацию эмбриональных стволовых клеток человека линии H1
клеток
клеток
клеток
клеток
клеток
Зависящие от дозы эффекты ингибиторов активности фермента GSK-3B на дифференцирование эмбриональных стволовых клеток человека линии H1
Sox17
Зависящие от дозы эффекты ингибиторов активности фермента GSK-3B на пролиферацию эмбриональных стволовых клеток человека линии H9
Зависящие от дозы эффекты ингибиторов активности фермента GSK-3B на дифференцирование эмбриональных стволовых клеток человека линии H9
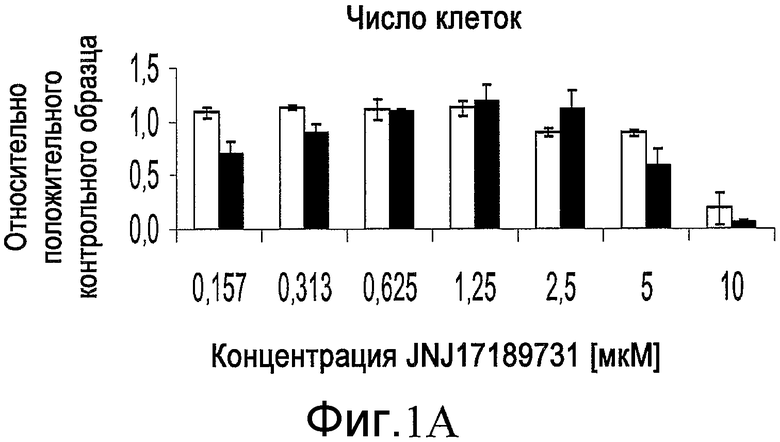


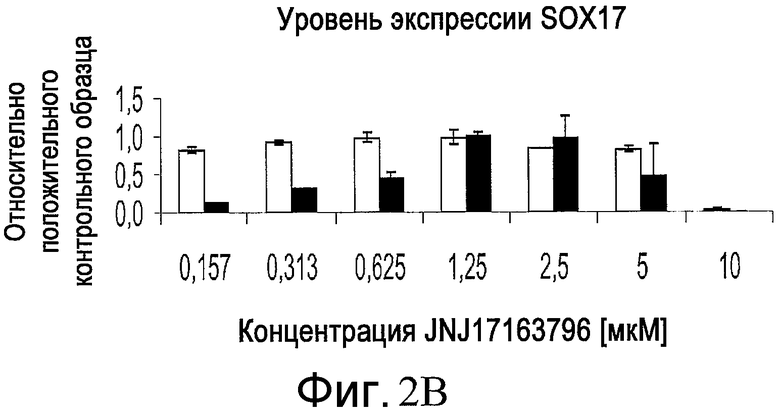




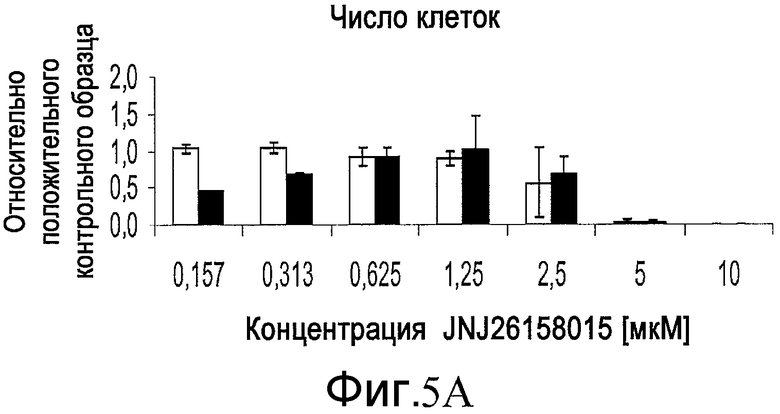

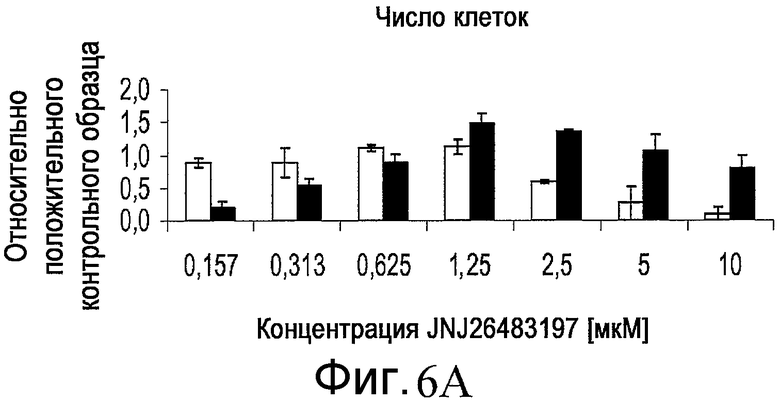
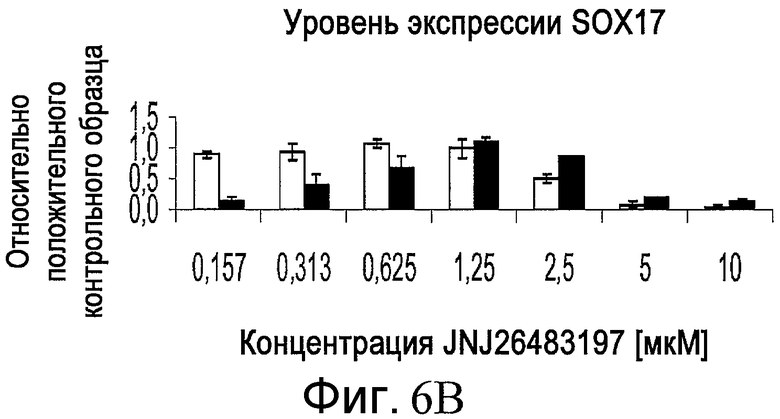


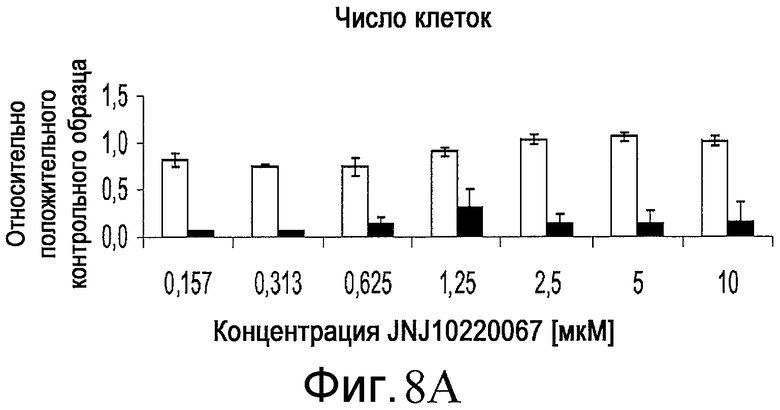





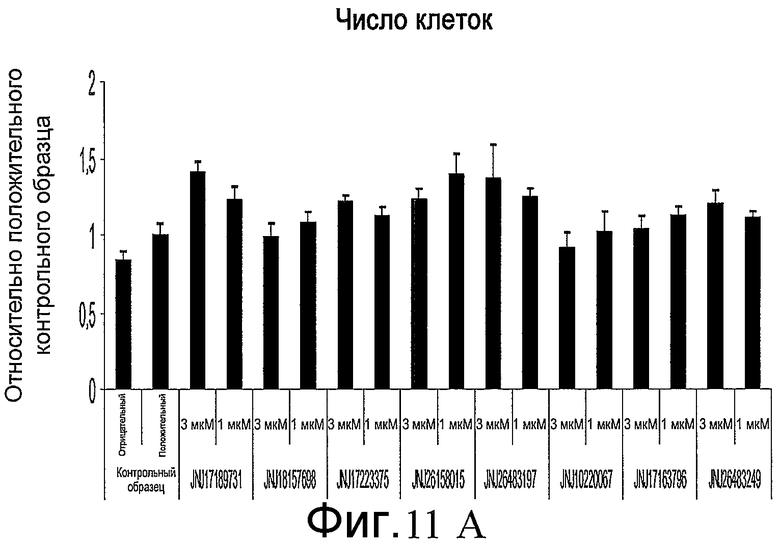
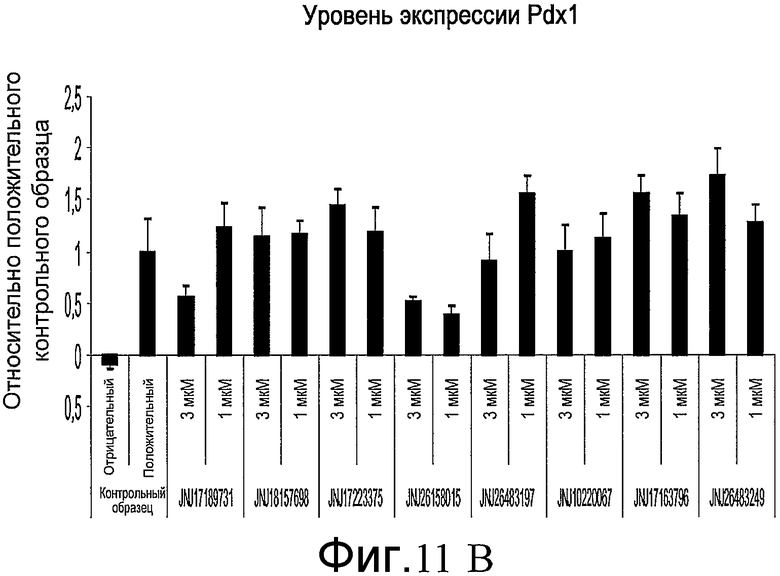
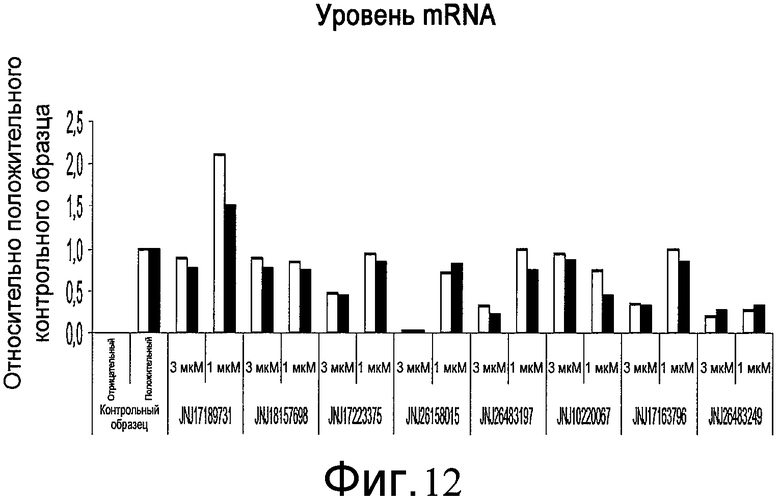
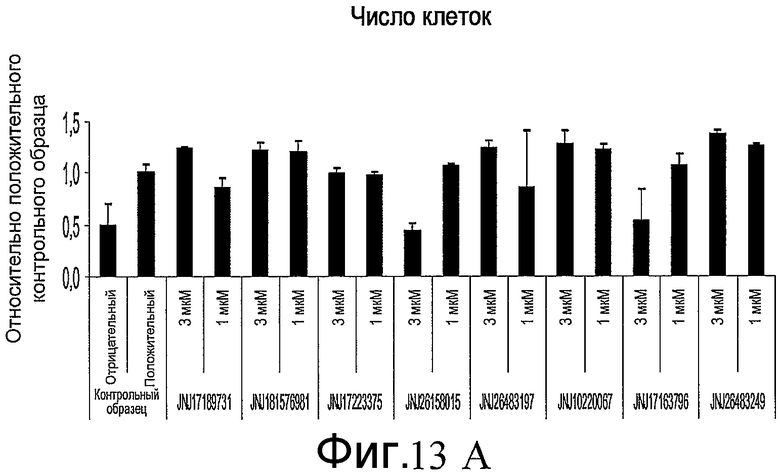

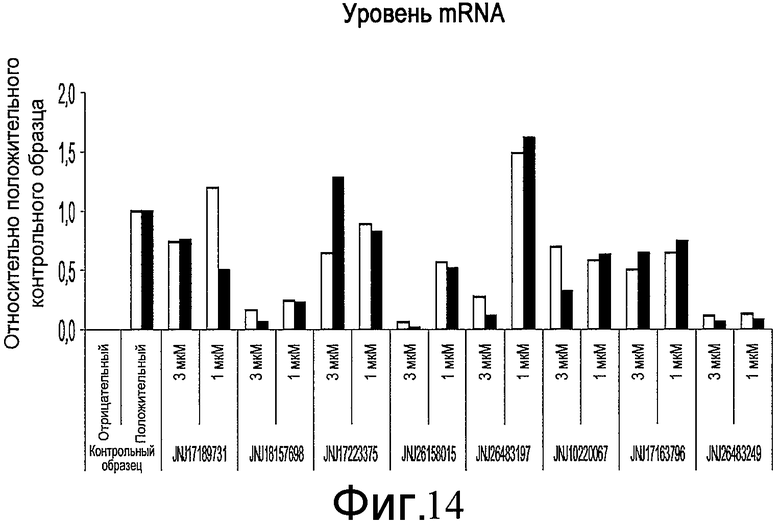

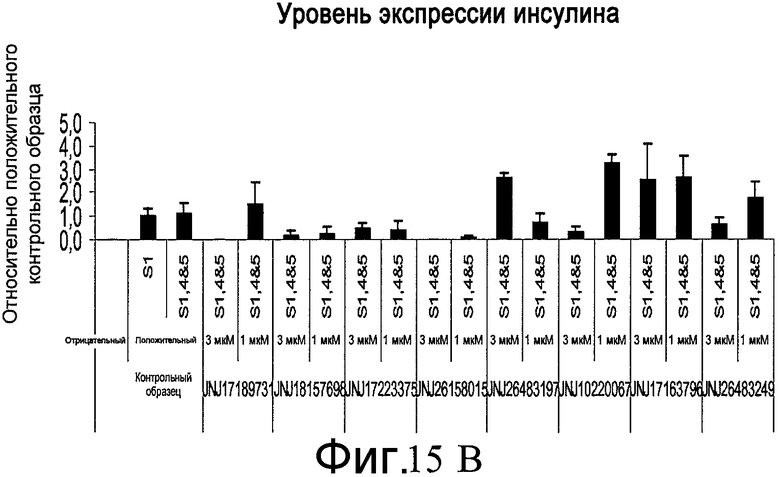
Настоящее изобретение касается способа размножения и дифференцирования плюрипотентных клеток. Описанный способ включает стадии: культивирование плюрипотентных клеток и обработку плюрипотентных клеток ингибитором активности фермента GSK-3B, где указанный ингибитор представляет собой 3-[1-(2-гидроксиэтил)-1H-индол-3-ил]-4-(1-пиридин-3-ил-1H-индол-3-ил)-пиррол-2,5-дион. Представленное изобретение позволяет получить достаточное количество клеточного материала с сохранением способности к дифференцированию для последующего использования в клинических целях. 9 з.п. ф-лы, 15 ил., 11 табл., 11 пр.
1. Способ размножения и дифференцирования плюрипотентных клеток, представляющих клеточную линию человека, включающий стадии:
a. культивирования плюрипотентных клеток, и
b. обработки плюрипотентных клеток ингибитором активности фермента GSK-3B, где указанный ингибитор представляет собой 3-[1-(2-гидроксиэтил)-1H-индол-3-ил]-4-(1-пиридин-3-ил-1H-индол-3-ил)-пиррол-2,5-дион.
2. Способ по п.1, в котором плюрипотентные клетки представляют собой клетки, экспрессирующие маркеры плюрипотентности эмбриональных стволовых клеток.
3. Способ по п.2, в котором клетки, экспрессирующие маркеры плюрипотентности, экспрессируют по меньшей мере один из маркеров, выбранных из группы, состоящей из: ABCG2, cripto, FoxD3, Connexin43, Connexin45, Oct4, SOX-2, Nanog, hTERT, UTF-1, ZFP42, SSEA-3, SSEA-4, Tral-60 и Tral-81.
4. Способ по п.1, в котором плюрипотентные клетки дифференцируются в клетки, экспрессирующие маркеры, характерные для линии дефинитивной эндодермы.
5. Способ по п.1, в котором плюрипотентные клетки обрабатываются ингибитором активности фермента GSK-3B в течение от 1 до 72 часов.
6. Способ по п.1, в котором плюрипотентные клетки обрабатываются ингибитором активности фермента GSK-3B в течение от 12 до 48 часов.
7. Способ по п.1, где плюрипотентные клетки обрабатываются ингибитором активности фермента GSK-3B в течение 48 часов.
8. Способ по п.1, в котором ингибитор GSK-3B используется в концентрации от 100 нМ до 100 мкМ.
9. Способ по п.1, в котором ингибитор GSK-3B используется в концентрации от 1 мкМ до 10 мкМ.
10. Способ по п.1, в котором ингибитор GSK-3B используется в концентрации 10 мкМ.
| US 20070254359 A1, 01.11.2007 | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| ДИФФЕРЕНЦИРОВКА ЭМБРИОНАЛЬНЫХ СТВОЛОВЫХ КЛЕТОК ЧЕЛОВЕКА | 2008 |
|
RU2465323C2 |

