Область техники, к которой относится изобретение
Настоящее изобретение относится к способу продуцирования аминокислот семейства аспартата, в частности, L-лизина, и их предшественников с использованием рекомбинантного микроорганизма. Более того, настоящее изобретение также относится к рекомбинантному микроорганизму, имеющему модифицированные пути, вовлеченные в синтез аминокислот семейства аспартата, по сравнению с исходным микроорганизмом, и к применению такого микроорганизма для продуцирования аминокислот семейства аспартата, в частности, L-лизина, и их предшественников.
Уровень техники
Аминокислоты и их производные являются важными предшественниками в фармацевтической промышленности, и их добавляют в широкий спектр пищевых продуктов и кормов в качестве добавок. Некоторые аминокислоты, такие как глутамат, лизин и треонин, продуцируют с использованием их природных путей биосинтеза. В природном биосинтезе аминокислот аминокислота аспартат служит в качестве предшественника для образования других аминокислот, таких как лизин, треонин, изолейцин и метионин.
Составляя на мировом рынке 900000 тонн/год, незаменимая аминокислота L-лизин является одним из наиболее важных продуктов биотехнологической ферментации (Kohl and Tauch, 2009). Ее, главным образом, применяют в качестве добавки в корм для животных (Anastassiadis, 2007). Дополнение таких кормовых материалов с богатым лизином источником обеспечивает оптимизированный рост, например, свиней или кур. Продолжающееся увеличение потребления белого мяса привело к увеличенной потребности в лизине в течение последних десятилетий.
Лизин можно продуцировать, например, способами ферментации. Было показано, что для этой цели особенно пригодны определенные микроорганизмы, такие как C. glutamicum. Ферментация в качестве способа промышленной продукции аминокислоты появилась с открытием секретирующей глутамат бактерии Corynebacterium glutamicum. В течение нескольких лет после ее открытия первые экскретирующие лизин мутанты C. glutamicum были применены для крупномасштабной продукции (Kinoshita, et al., 1961). Продолжаются исследования в направлении новых технологий для достижения высокоэффективной ферментации, включая оптимизацию процедуры ферментации, последующий процессинг, а также инженерию штаммов.
Штаммы классически конструировали путем повторяющегося подхода случайного мутагенеза с помощью УФ-облучения или химических мутагенов и последующей селекции штаммов. Ключом к успеху в то время было использование токсических аналогов лизина, таких как S-(2-аминоэтил)цистеин, для селекции штаммов, устойчивых по принципу обратной связи (Nakayama and Araki, 1973). Эти классические штаммы, как правило, имеют общие точечные мутации в гене аспартокиназы, который высвобождает кодированный фермент при ингибировании по принципу обратной связи лизином и треонином (Kalinowski, et al., 1991; Thierbach, et al., 1990). С помощью таких классически полученных штаммов достигают исключительных продукционных свойств, таких как выход конверсии вплоть до 50% и титр лизина·Cl 100 г·л-1 (Leuchtenberger, et al., 2005). Однако, как правило, это было связано с длительным временем ферментации, составлявшим 2-3 суток, ограничивающим продуктивность. Кроме того, ауксотрофия и слабая толерантность к стрессу вследствие нежелательных мутаций, которые накапливались в ходе разработки штамма (Ohnishi et al., 2002), далее отражают недостатки общепринятых штаммов для продукции. В последние годы, технология рекомбинантных ДНК и молекулярная биология дали начало новой эре конструирования-рациональной оптимизации штаммов путем метаболической инженерии (Ikeda, et al., 2006). Многие из этих исследований фокусировались на оптимизации процесса биосинтеза лизина путем прямой модификации ферментов этого пути. Освобождение аспартаткиназы от контроля по принципу обратной связи в настоящее время воспринимают как один из наиболее важных признаков промышленных продуцирующих штаммов. Помимо модификаций, касающихся регуляции пути, ключевым моментом инженерии штаммов является внутриклеточная активность определяющих скорость ферментов биосинтетического пути. Стратегии увеличения активности фермента в клетке вовлекают сверхэкспрессию с использованием более сильных промоторов, мутацию промоторных последовательностей или регуляторных областей выше гена, или увеличение числа копий кодирующего гена. Связанная с плазмидой сверхэкспрессия в этом контексте является пригодной для достижения более высокой активности фермента и более высоких выходов лизина (Eggeling, et al., 1998), однако может быть трудно применить ее в промышленном процессе.
Идентификация благоприятных мишеней помимо самого биосинтетического пути вскоре стала необходимой для устранения препятствий в предоставлении предшественников и кофакторов в целях создания конкурентоспособных продуцирующих штаммов. Однако это является более проблематичным и трудным, поскольку требуется понимание организма на системном уровне. В этом отношении, доступность последовательности генома Corynebacterium glutamicum стала важнейшим элементом для метаболической инженерии (Haberhauer, et al., 2001; Kalinowski, et al., 2003; Ohnishi, et al., 2002; Pompejus, et al., 2002). Она обеспечила основу для (i) выведения геномов путем сравнительного анализа последовательностей между классически полученными продуцирующими штаммами и штаммами дикого типа (Ohnishi, et al., 2002), (ii) детального реконструирования in silico метаболической сети C. glutamicum (Kjeldsen and Nielsen, 2009), включая подходы стехиометрического моделирования для анализа теоретической продуцирующей способности, а также вовлеченных метаболических путей (Kromer, et al., 2006; Wittmann and Becker, 2007), и (iii) открытия транскрипционных регуляторных сетей с помощью специфических мотивов последовательностей в геноме (Kohl and Tauch, 2009). Однако эти модели не применимы для предсказания активности метаболических путей in vivo, т.е. флуксома, в качестве ключевой характеристики для понимания систем и ориентирования в инженерии штаммов. Анализ потоков является центральным элементом в метаболической инженерии (Stephanopoulos, 1999), на что указывает впечатляющий прогресс в оценке метаболических потоков in vivo (Christensen and Nielsen, 2000; Christensen, et al., 2000; Frick and Wittmann, 2005; Van Dien, et al., 2006; Wittmann, 2007; Wittmann and Heinzle, 2002). Помимо понимания биологической системы, было показано, что 13C-анализ метаболических потоков является пригодным для характеризации штаммов и идентификации благоприятных мишеней для продукции лизина (Kiefer, et al., 2004; Wittmann and Heinzle, 2002). Вместе с дополнительными данными, полученными при определении активного набора генов (транскриптом) (Hayashi, et al., 2006) и белков (протеом) (Bendt, et al., 2003) и количественном определении уровней внутриклеточных метаболитов (метаболом) (Borner, et al., 2007) предоставляется широкий набор данных для глубокого понимания клеточной физиологии на общем уровне. Этот ориентированный на системы подход представляет собой превосходную платформу для метаболической инженерии (Lee, et al., 2005).
Основной продуцирующий лизин микроорганизм Corynebacterium glutamicum был открыт в 1950 в крупной скрининговой программе в Японии. Штаммы успешно оптимизировали для продуцирования лизина с использованием повторяющегося процесса случайного мутагенеза и скрининга в отношении усовершенствованных продукционных характеристик. Это привело к эффективным продуцирующим штаммам, а также привело к накоплению побочных мутаций, вызывающих снижение роста, низкую устойчивость к стрессу и увеличенные потребности в питательных веществах. Таким образом, достигнутая в настоящее время эффективность продуцирования является значительно более низкой, чем теоретическая способность, предсказанная для C. glutamicum. Прогресс в молекулярной биологии и системно-ориентированных инструментов для анализа метаболизма и регуляционного состояния клетки в настоящее время привел к сдвигу инженерии в направлении более точной и направленной метаболической инженерии оптимизированных систем. Она направлена на улучшенные сверхпродуцирующие штаммы с исключительными наборами благоприятных модификаций, проявляющих увеличенный продукционный выход, титр и продуктивность.
В прошлом были предприняты различные попытки для увеличения продуцирования L-лизина с использованием микроорганизмов. Основные попытки увеличить продуцирование, например, метионина или лизина путем активации и/или подавления экспрессии генов, вовлеченных в биосинтез метионина или лизина, описаны, например, в WO 02/10209, WO 2006/008097 и WO 2005/059093.
Центральная катаболическая сеть, ранее идентифицированная в C. glutamicum, включает путь гликолиза, пентозофосфатный путь (PPP), цикл трикарбоновых кислот (TCA) и глиоксалатный шунт.
WO 03/042389 относится к генетически модифицированному микроорганизму, в который введен ген G6PD (zwf) или в котором ген G6PD модифицирован, и к применению такого микроорганизма для получения представляющего интерес соединения, такого как аминокислота, особенно предпочтительно лизин.
WO 0220542 относится к способу ферментативного получения L-аминокислот, в частности L-лизина, в котором метаболические пути, которые снижают образование желаемой L-аминокислоты, по меньшей мере частично устранены, например с использованием усиленного гена gap2. Также приводится длинный перечень дополнительных генов, которые можно модифицировать.
В EP 0435132 описаны микроорганизмы рода Corynebacterium или Brevibacterium, которые содержат рекомбинантную ДНК и пригодны для получения аминокислот, особенно L-лизина. Последовательности ДНК происходят, в частности, из продуцирующих L-лизин штаммов рода Corynebacterium или Brevibacterium, предпочтительно из мутанта, полученного мутагенезом Corynebacterium glutamicum ATCC 13032 со сниженным ингибированием по принципу обратной связи аспартаткиназы.
В EP1067193 описаны продуцирующая L-лизин коринеформная бактерия (A) с амплифицированным геном pyc (пируваткарбоксилаза), в которой по меньшей мере один из дополнительных генов dapA (дигидропиколинатсинтаза), lysC (аспартаткиназа), lysE (экспортер-переносчик лизина) и/или dapB (дигидропиколинатредуктаза) амплифицирован, предпочтительно сверхэкспрессирован.
В EP0854189 описана коринеформная бактерия, содержащая аспартокиназу, в которой ингибирование по принципу обратной связи L-лизином и L-треонином существенно снижено, и содержащая усиленную последовательность ДНК, кодирующую дигидродипиколинатредуктазу, усиленную последовательность ДНК, кодирующую дигидропиколинатредуктазу, усиленную последовательность ДНК, кодирующую дигидропиколинатсинтазу, усиленную последовательность ДНК, кодирующую диаминопимелатдекарбоксилазу, и усиленную последовательность ДНК, кодирующую аспартатаминотрансферазу.
EP0857784 относится к ДНК, содержащей ген аспартокиназы, не подверженный ингибированию по принципу обратной связи L-лизином, и т.д., и содержащей ген декарбоксилазы диаминопимелиновой кислоты и обладающей улучшенной способностью коринеформной бактерии к продуцированию L-лизина.
В WO/2007/017526 описан способ получения аспартата и происходящих из него аминокислот, таких как лизин, треонин, изолейцин, метионин или гомосерин, с использованием микроорганизма с усиленной экспрессией изоцитратлиазы и/или малатсинтазы.
Описание изобретения
Техническая проблема
Основной задачей изобретения была оптимизация продукции лизина путем рациональной инженерии штамма. В качестве модельного микроорганизма использовали Corynebacterium glutamicum в системно-ориентированном подходе. Поскольку затраты на исходные материалы составляют основные затраты на продукцию при промышленном продуцировании лизина, оптимизация штамма нацелена на увеличение выхода лизина, титра и продуктивности. Центральная стратегия нацелена на технологии уровня техники для установления метаболического и регуляторного состояния C. glutamicum на системно-ориентированном уровне и использование полученного знания для идентификации генетических мишеней для оптимальной эффективности продуцирования. В C. glutamicum биосинтез лизина тесно связан с центральным метаболизмом вследствие потребности в предшественниках углерода и NADPH в качестве восстанавливающей силы. Вследствие этого исследование включало реакции центральных метаболических путей в качестве перспективных мишеней для оптимизации штамма. Стратегии были сфокусированы, главным образом, на метаболизме NADPH, цикле TCA, инженерии поступления оксалоацетата, а также на биосинтезе лизина. Сначала исследовали пригодность некоторых стратегий инженерии для улучшенной продукции лизина путем оценки мишеней в продуцирующих лизин штаммах. Штаммы по изобретению детально исследовали путем экспериментов по сравнительному культивированию, а также на уровне транскриптома, метаболома и флуксома для (i) более глубокого понимания клеточной физиологии, (ii) оценки пользы используемой стратегии в отношении промышленного применения и (iii) предоставления ценной информации для рационального моделирования продуцирующего штамма.
Более того, модификации путей биосинтеза, в частности, метаболизма NADPH, цикла TCA, инженерия поступления оксалоацетата в микроорганизмах в соответствии с настоящим изобретением важны не только для продуцирования L-лизина, но также важны для продуцирования других аминокислот семейства аспартата и их предшественников. Таким образом, также можно продуцировать другие представители семейства происходящих из аспартата аминокислот, такие как метионин, треонин или изолейцин, учитывая информацию, предоставленную в настоящем описании.
Эти и другие задачи, которые станут очевидными из последующего описания изобретения, решаются с помощью настоящего изобретения, как описано в независимых пунктах формулы изобретения. Зависимые пункты формулы изобретения относятся к предпочтительным вариантам осуществления.
Решение проблемы
Изобретение относится, в частности, к созданию микроорганизма, сверхпродуцирующего происходящие из аспартата аминокислоты, предпочтительно штаммов микроорганизмов, сверхпродуцирующих лизин, на основе C. glutamicum дикого типа, и к способам их применения. Такие штаммы были получены с помощью полезных модификаций, идентифицированных в этой работе. Специально полученная клеточная фабрика имеет высокий выход конверсии углерода, а также высокий выход в единицу времени, высокий конечный титр лизина, хорошие характеристики роста и предпочтительно также снижение образования побочных продуктов. Эти характеристики продуцирования обеспечивают быструю и эффективную конверсию предоставленных субстратов и, таким образом, экономичное продуцирование лизина.
Более того, продуцирование происходящих из аспартата аминокислот, предпочтительно L-лизина, увеличивается при использовании генетически модифицированных микроорганизмов, предпочтительно Corynebacterium, более предпочтительно C. glutamicum.
В предпочтительном способе продуцирования происходящих из аспартата аминокислот, более предпочтительно при продуцировании L-лизина, используют микроорганизм, например, с модифицированным биосинтезом лизина и модифицированным поступлением предшественника. Предпочтительно, микроорганизм представляет собой рекомбинантный микроорганизм. В особенно предпочтительном варианте осуществления микроорганизм может иметь модифицированную, например, увеличенную, активность гена и фермента глюкозо-6-фосфатдегидрогеназы. Более предпочтительно, модифицированный ген глюкозо-6-фосфатдегидрогеназы (zwf) содержит модифицированный промотор, предпочтительно замену промотора дикого типа гена zwf гетерологичным, т.е. неприродным промотором, более предпочтительно промотором супероксиддисмутазы (sod). Неприродный промотор может происходить из того же организма, но из другого гена, или из отличающегося вида. Еще более предпочтительно, микроорганизм также содержит точечную мутацию A243T в гене zwf. В следующем предпочтительном варианте осуществления, микроорганизм содержит модифицированный оперон tkt, где промотор дикого типа tkt-оперона заменен гетерологичным промотором, предпочтительно sod-промотором, что также обеспечивает увеличенную активность гена глюкозо-6-фосфатдегидрогеназы и фермента, соответственно. С использованием сильного sod-промотора, ген zwf также может сверхэкспрессироваться путем сверхэкспрессии оперона tkt, который содержит, среди прочего, ген транскетолазы (tkt), ген, кодирующий трансальдолазу (tal) и zwf.
В другом предпочтительном варианте осуществления микроорганизм, кроме того, также имеет увеличенную активность гена фруктозо-1,6-бисфосфатазы и фермента (fbp). Эта задача решается с помощью микроорганизма, который, в дополнение к указанным выше модификациям, содержит сильный гетерологичный промотор, например, sod-промотор или предпочтительно промотор eftu, заменяющий исходный промотор.
Также предпочтительно, чтобы микроорганизм, кроме того, содержал ослабленный ген изоцитратдегидрогеназы (icd) и обладал соответственно сниженной ферментативной активностью, предпочтительно путем замены исходного инициирующего кодона ATG инициирующим кодоном GTG.
В более предпочтительном варианте осуществления микроорганизм, кроме того, имеет увеличенную активность гена и фермента диаминопимелатдегидрогеназы. Этого можно достигать, например, путем сверхэкспрессии соответствующего гена, кодирующего диаминопимелатдегидрогеназу (ddh). Более предпочтительно, диаминопимелатдегидрогеназа сверхэкспрессируется микроорганизмом, который содержит по меньшей мере одну дополнительную копию гена ddh.
Более того, особенно предпочтительно, чтобы микроорганизм, в дополнение к описанным выше модификациям, содержал модифицированную аспартаткиназу (lysC) с увеличенной активностью гена и фермента, предпочтительно путем замены аминокислоты T311I.
Предпочтительные варианты осуществления относятся к микроорганизмам, содержащим, в дополнение к описанным выше модификациям, по меньшей мере одну из следующих модификаций: (i) сверхэкспрессию дигидропиколинатредуктазы (dapB), диаминопимелатдекарбоксилазы (lysA), (ii) модификацию гомосериндегидрогеназы путем введения мутантного варианта, содержащего аминокислотную замену V59A, (iii) сверхэкспрессию пируваткарбоксилазы, и (iv) делецию PEP-карбоксикиназы.
Микроорганизмом, который предпочтительно используют в способах согласно изобретению, является Corynebacterium glutamicum, предпочтительно производное C. glutamicum ATCC13032, более предпочтительно микроорганизм, депонированный согласно Будапештскому договору в Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH (DSMZ), Германия, 11 мая 2010 года, и который имеет номер доступа DSM 23586.
Преимущественные эффекты изобретения
Согласно настоящему изобретению, микроорганизмы по изобретению пригодны для применения в способе продуцирования происходящих из аспартата аминокислот, предпочтительно L-лизина. Дальнейшие предпочтительные аспекты изобретения могут быть установлены из представленного ниже подробного описания и примеров.
Клетки, используемые для способа продуцирования, могут быть прокариотами, низшими эукариотами, выделенными клетками растений, клетками дрожжей, выделенными клетками насекомых или выделенными клетками млекопитающих, в частности, клетками в клеточных культуральных системах. В контексте настоящего изобретения, термин "микроорганизм" используют для указанных типов клеток. Предпочтительным типом микроорганизмов для осуществления настоящего изобретения является Corynebacterium и особенно предпочтительно C. glutamicum.
Краткое описание фигур
Фиг.1: Стратегия ПЦР для конструирования вставки, проиллюстрированная для делеции гена путем частичной делеции последовательности открытой рамки считывания (ORF). Два отдельных фрагмента ДНК подвергали слиянию на трех стадиях ПЦР с использованием сайт-специфических праймеров (например, праймеров, обозначенных как P1, P2, P3 и P4).
Фиг.2: Встраивающийся вектор для трансформации pClik с селективными маркерами KanR и sacB, ORI для E. coli и множественным участком клонирования.
Фиг.3: Корреляция между оптической плотностью (OD660, Libra S11) и сухой массой клеток для C. glutamicum BS1 (закрашенные ромбы), C. glutamicum BS87 (закрашенные круги) и C. glutamicum BS205 (незакрашенные круги), для каждого в трех биологических повторах.
Фиг.4: Профиль культивирования BS1 C. glutamicum (lysCT311I) в ходе роста в стандартной минимальной среде с глюкозой в качестве единственного источника углерода (A) и характеристики роста и продуцирования (B). Линейная корреляция между образованием биомассы и лизина и потреблением глюкозы, соответственно, указывает на метаболическое устойчивое состояние.
Фиг.5: Подтверждение изотопного устойчивого состояния роста C. glutamicum BS13 на [1-13C] глюкозе (A) и эквимолярной смеси [U-13C] глюкозы и природно меченной глюкозы (B). Профили мечения аминокислот определяли при различных концентрациях сухой массы клеток (CDM) в процессе культивирования. Аминокислоты, иллюстративно представленные здесь, происходят из различных частей метаболической сети и включают аланин (закрашенный квадрат), фенилаланин (незакрашенный квадрат), валин (закрашенный круг), глицин (незакрашенный круг), глутамат (закрашенный треугольник), треонин (незакрашенный треугольник) и серин (закрашенный ромб). M0 (без мечения), M1 (с однократным мечением) и M2 (с двойным мечением) обозначают относительные доли соответствующих массовых изотопомеров.
Фиг.6: Схема эксперимента для количественного определения параметров потока в пируватном модуле Corynebacterium glutamicum с эквимолярной смесью [U-13C] глюкозы и природно меченной глюкозы. Относительное изменение распределения массовых изотопомеров аспартата (m/z 418) с варьирующим ΦPEPC, относительное изменение распределения массовых изотопомеров валина (m/z 288) с варьирующим ζPEPC/PEPCK, относительное изменение распределения массовых изотопомеров аланина (m/z 260) с варьирующим ζPC/MalE.
Фиг.7: Корреляция экспериментально определенных и смоделированных долей массовых изотопомеров аминокислот из клеточного белка и секретируемой трегалозы в процессе культивирования C. glutamicum BS1 на 99% [1-13C] глюкозе и 50% [U-13C] глюкозе. Данные включают экспериментальные данные ГХ-МС и величины, предсказанные путем решения математической модели, соответствующей оптимизированному набору потоков.
Фиг.8: Распределение потока углерода в пируватном модуле продуцирующих лизин C. glutamicum BS1 в процессе роста на глюкозе. Все потоки приведены в виде молярного процента удельной скорости потребления глюкозы qGlc=4,6 ммоль·г-1·час-1, которая принята за 100%. Ошибки отражают соответствующие 90% доверительные интервалы для различных потоков, полученных с помощью анализа Монте-Карло.
Фиг.9: Удельная активность изоцитратдегидрогеназы (ICD), фосфоглюкоизомеразы (PGI), пируватдегидрогеназы (PDH) и глюкозо-6-фосфатдегидрогеназы (G6PDH) в неочищенных клеточных экстрактах C. glutamicum BS87 и 6 мутантов после второго события рекомбинации в качестве возможных кандидатов для нуклеотидной замены. Данные отражают однократное измерение для клеток, растущих в комплексной среде.
Фиг.10: Удельная активность in vitro G6P-дегидрогеназы в штаммах C. glutamicum ATCC 13032, BS1 (lysCT311I), BS5 (Psodzwf) и BS6 (PsodzwfA243T) (A); удельная активность in vitro [%] белка дикого типа и варианта A243T при добавлении NADPH в качестве ингибитора (B). Данные соответствуют средним значениям для трех различных измерений в клеточных экстрактах клеток, выращенных в минимальной среде с глюкозой в качестве источника углерода, и соответствующим отклонениям.
Фиг.11: Внутриклеточные потоки и соответствующие 90% доверительные интервалы для C. glutamicum BS1, C. glutamicum BS5, C. glutamicum BS6 и C. glutamicum BS7 (сверху вниз), культивированных на [1-13C] глюкозе. Данные приведены в виде относительных величин (в %), нормализованных к уровню потребления глюкозы (таблица 15). Данные о потоках для эталонного штамма C. glutamicum BS1 взяты из (Becker, et al., 2005).
Фиг.12: Удельная активность глюкозо-6-фосфатдегидрогеназы (A), трансальдолазы (B) и транскетолазы (C) в ответ на сверхэкспрессию tkt-оперона с помощью промотора супероксиддисмутазы (sod).
Фиг.13: Удельная активность яблочного фермента в неочищенных клеточных экстрактах C. glutamicum WT, C. glutamicum BS1 (lysCT311I) и C. glutamicum BS6 (PsodzwfA243T), выращенных на глюкозе или фруктозе, соответственно. Определение проводили в трех повторах, и приведены соответствующие отклонения.
Фиг.14: Характеристики продуцирования различных продуцирующих лизин штаммов C. glutamicum в ходе периодического культивирования на глюкозе. Выходы определяли в виде наклона линейной наилучшей аппроксимации между образованием биомассы или лизина, соответственно, и потреблением субстрата в течение всего времени культивирования. Выходы определяли для трех биологических повторов.
Фиг.15: Баланс NADPH C. glutamicum BS1 (lysCT311I), вычисленный из потоков in vivo, при рассмотрении реакций PPP, изоцитратдегидрогеназы и яблочного фермента в качестве предоставляющих NADPH реакций, и образования биомассы и секреции лизина в качестве реакций с потреблением NADPH.
Фиг.16: Корреляция между отношением NADPH/NADP и удельной активностью яблочного фермента в C. glutamicum ATCC 13032 (треугольник), C. glutamicum BS1 (lysCT311I) (квадрат) и C. glutamicum BS6 (Psod zwf A243T) (круг).
Фиг.17: Удельная активность диаминопимелатдегидрогеназы в неочищенных клеточных экстрактах C. glutamicum BS1 (столбец в полоску) и C. glutamicum BS222 (серый столбец), выращенных в стандартной минимальной среде с использованием глюкозы в качестве единственного источника углерода.
Фиг.18: Удельная скорость роста C. glutamicum BS1 (lysCT311I) при различных концентрациях сульфата аммония.
Фиг.19: Выход лизина из C. glutamicum BS222, выращенных в минимальной среде при различных концентрациях сульфата аммония.
Фиг.20: Удельная активность диаминопимелатдегидрогеназы C. glutamicum BS1 и C. glutamicum BS222 при 2 г·л-1 и 15 г·л-1 сульфата аммония, соответственно.
Фиг.21: Количественные физиологические характеристики продуцирующих лизин C. glutamicum BS1 (A, B) и их производного с делецией pyk BS13 (C, D) в периодической культуре на глюкозе. Линейная корреляция между ростом, продукцией лизина и дигидроксиацетона (DHA) и потреблением глюкозы указывает на метаболическое устойчивое состояние в ходе культивирования.
Фиг.22: Распределение потока углерода in vivo при центральном метаболизме продуцирующих лизин C. glutamicum BS1 (верх) и их производного с дефицитом пируваткиназы C. glutamicum BS13 (низ) в ходе роста на глюкозе. Все потоки приведены в виде молярного процента средней удельной скорости потребления глюкозы qGlc=4,6 ммоль·г-1·ч-1 (для BS1) и 4,5 ммоль·г-1·ч-1 (для BS13), которая принята за 100%. Для обратимых метаболических реакций обратимость потока, кроме того, приведена в скобках и направление суммарного потока указано стрелкой.
Фиг.23: Распределение суммарного потока углерода в пируватном модуле продуцирующих лизин C. glutamicum BS1 (A) и их производного с дефицитом пируваткиназы C. glutamicum BS13 (B), культивированных на глюкозе. Истинные величины потока показаны толщиной соответствующих стрелок.
Фиг.24: Баланс NADPH для C. glutamicum BS1 (слева) и их производного с дефицитом пируваткиназы C. glutamicum BS13 (справа).
Фиг.25: Участок области инициирующего кодона после выравнивания последовательностей исходного штамма C. glutamicum BS87, двух мутантов после второго события рекомбинации со сниженной удельной активностью PDH и вектора для трансформации, использованного для конструирования штаммов.
Фиг.26: Удельная ферментативная активность пируватдегидрогеназы в выращенных на глюкозе C. glutamicum BS87 (столбец в полоску) и их aceEatt-производного C. glutamicum BS238 (белый столбец). Серый столбец указывает на удельную ферментативную активность мутанта по инициирующему кодону после непрерывного выращивания на протяжении 50 поколений в последовательных партиях. Величины определяли в трех повторах и соответствующие отклонения показаны планками погрешностей.
Фиг.27: Выход лизина и биомассы C. glutamicum BS87 (столбец в полоску) и C. glutamicum BS238 (серый столбец), определенный в минимальной среде для трех биологических повторов. Приведены соответствующие отклонения. Выходы определяли в виде наклона наилучшей линейной аппроксимации между образованием продукта и потреблением субстрата.
Фиг.28: Выравнивание области инициирующего кодона гена icd C. glutamicum BS87, вектора для трансформации для замены инициирующего кодона гена icd, и 4 клонов после второго рекомбинационного события.
Фиг.29: Удельная активность изоцитратдегидрогеназы в неочищенных клеточных экстрактах C. glutamicum BS87 (столбец в полоску) и C. glutamicum BS205 (icd att) (белый столбец), выращенных в минимальной среде с глюкозой в качестве единственного источника углерода. Серый столбец указывает на удельную активность ICD мутанта по инициирующему кодону icd att после выращивания на протяжении 50 поколений в последовательных партиях. Данные соответствуют средним величинам для трех параллелей и соответствующим отклонениям.
Фиг.30: Характеристики роста и продуцирования для продуцирующих лизин C. glutamicum BS87 (A, B) и C. glutamicum BS205 (icd att) (C, D) в ходе периодического культивирования на глюкозе. Линейная корреляция между продукцией биомассы и лизина и потреблением глюкозы, соответственно, указывает на метаболическое устойчивое состояние в ходе культивирования. Приведенные данные соответствуют величинам для трех биологических реплик.
Фиг.31: Потоки in vivo через цикл TCAa и пируваткарбоксилазуb для выращиваемых на глюкозе C. glutamicum BS87 и C. glutamicum BS205 (icd att). Ошибки отражают 90% доверительный интервал, полученный с помощью анализа Монте-Карло.
aПриведено в качестве входящего потока через цитратсинтазу.
bПриведено в качестве обобщенного суммарного потока через анаболическое карбоксилирование.
Фиг.32: Моделирование и конструирование специально полученного продуцирующего лизин штамма посредством инженерии путей. Модификации включают lysCT311I (1), 2×ddh (2), Dpck (3), Psod dapB (4), 2 lysA (5), Psod lysCT311I (6), hom V59A (7), pyc P458S (8), Psod pyc P458S (9), icd att (10), Peftu fbp (11) и Psod tkt (12), которые были успешно осуществлены для конструирования штаммов C. glutamicum BS1 (незакрашенный ромб), C. glutamicum BS222 (закрашенный ромб), C. glutamicum BS87 (закрашенный квадрат), C. glutamicum 205 (незакрашенный квадрат), C. glutamicum BS242 (закрашенный круг) и C. glutamicum 244 (незакрашенный круг).
Фиг.33: Выход лизина в генеалогии продуцентов лизина на основе дикого типа, протестированных во вращающихся флаконах в минимальной среде с 15 г·л-1 сульфата аммония и с глюкозой в качестве единственного источника углерода. Данные соответствуют средним величинам для трех биологических повторов с соответствующими отклонениями. Представленными штаммами являются C. glutamicum WT (ATCC 13032), C. glutamicum BS1 (lysCT311I) (1), C. glutamicum BS222 (ddh) (2), C. glutamicum BS87 (9), C. glutamicum BS205 (icd att) (10), C. glutamicum BS242 (Peftu fbp) (11) и C. glutamicum BS244 (Psod tkt) (12).
Фиг.34: Профиль культивирования продуцирующих лизин C. glutamicum BS244 в процессе периодической ферментации с подпиткой на комплексной среде на основе патоки. Концентрация сахара приведена в виде обобщенной концентрации глюкозы, фруктозы и сахарозы, соответственно.
Фиг.35: Характеристики двухфазного продуцирования лизина C. glutamicum BS244 в процессе периодической ферментации с подпиткой. Общая продукция лизина нанесена на график относительно общего потребления сахара.
Наилучшие способы осуществления изобретения
Ферменты и гены C. glutamicum
Ферменты и гены C. glutamicum, имеющие значение для настоящей работы, приведены в таблице 1 с соответствующим номером EC и систематическим названием гена согласно базе данных KEGG.
Систематическая характеризация ферментов и генов C. glutamicum согласно присвоенным кодам ферментов и номенклатуре генов базы данных KEGG
Как используют в контексте настоящего изобретения, формы единственного числа также включают соответствующие формы множественного числа, если контекст явно не указывает на иное. Таким образом, термин "микроорганизм" может включать более одного микроорганизма, а именно, два, три, четыре, пять и т.д. микроорганизмов этого типа.
Термин "приблизительно" в контексте числовой величины или диапазона параметров обозначает интервал точности, как понятно специалисту в данной области, который все еще обеспечивает технический эффект рассматриваемого признака. Термин, как правило, указывает на отклонение от указанной числовой величины +/- 10%, предпочтительно +/- 5%.
Термин "клетка-хозяин" для целей настоящего изобретения относится к любой выделенной клетке, которую обычно используют для экспрессии нуклеотидных последовательностей для продуцирования, например, полипептидов или продуктов тонкого органического синтеза. В частности, термин "клетка-хозяин" относится к клеточным культуральным системам прокариот, низших эукариот, клеток растений, клеток дрожжей или клеток млекопитающих.
Термин "микроорганизм" относится к прокариотам, низшим эукариотам, выделенным клеткам растений, клеткам дрожжей, выделенным клеткам насекомых или выделенным клеткам млекопитающих, в частности, к клеткам в клеточных культуральных системах. Микроорганизмы, пригодные для осуществления настоящего изобретения, включают дрожжи, такие как S. pombe или S. cerevisiae и Pichia pastoris. Культуральные системы клеток млекопитающих могут быть выбраны из группы, включающей, например клетки NIH T3, клетки CHO, клетки COS, клетки 293, клетки Jurkat и клетки HeLa. В контексте настоящего изобретения микроорганизм предпочтительно представляет собой прокариотическую клетку или клетку дрожжей. Предпочтительные микроорганизмы в контексте настоящего изобретения указаны ниже в разделе "Подробное описание". Особенно предпочтительными являются Corynebacteria, такие как C. glutamicum и их производные.
Термин "нативный" является синонимом для "дикого типа" и "встречающегося в природе". Микроорганизм "дикого типа" представляет собой, если не указано иное, распространенную встречающуюся в природе форму указанного микроорганизма. Как правило, микроорганизм дикого типа является нерекомбинантным микроорганизмом.
"Исходный" является синонимом "начальному". "Исходная" нуклеотидная последовательность или ферментативная активность представляет собой начальную точку для их модификации, например путем мутации или добавления ингибиторов. "Исходная" последовательность, фермент или микроорганизм лишены отличительного признака, которым обладают их "конечные" или "модифицированные" аналоги, и который указан в конкретном контексте. Термин "исходный" в контексте настоящего изобретения охватывает значение термина "нативный" и в предпочтительном аспекте является синонимом термина "нативный".
Любой микроорганизм дикого типа или мутантный (нерекомбинантный или рекомбинантный мутант) микроорганизм можно далее модифицировать нерекомбинантными (например, добавлением специфических ингибиторов фермента) или рекомбинантными способами, с получением микроорганизма, который отличается от исходного микроорганизма по меньшей мере одним физическим или химическим свойством.
В контексте настоящего изобретения исходный немодифицированный микроорганизм означают как "исходный микроорганизм" или "исходный (микроорганизм) штамм".
Любую модификацию активности выбранного гена или фермента в микроорганизме (например, количество нуклеиновых кислот или белков, находящихся в модифицированных клетках, или количество образовавшегося продукта, например, количество L-лизина) по сравнению с начальным штаммом с данным уровнем активности определяют путем сравнения активности в обоих микроорганизмах в сравнимых условиях с использованием стандартных способов молекулярной биологии, представленных в стандартных справочниках, или способов, конкретно описанных в разделе "Примеры".
Как правило, микроорганизмы согласно изобретению получают путем внесения генетических изменений в исходный микроорганизм, который не содержит указанного генетического изменения.
"Производное" штамма микроорганизма представляет собой штамм, который получен из его исходного штамма, например, путем классического мутагенеза и селекции или путем направленного мутагенеза.
Термин "нуклеотидная последовательность" или "последовательность нуклеиновой кислоты" для целей настоящего изобретения относится к любой молекуле нуклеиновой кислоты, которая кодирует полипептиды, такие как пептиды, белки и т.д. Эти молекулы нуклеиновых кислот могут быть получены из ДНК, РНК или их аналогов. Однако предпочтительными являются молекулы нуклеиновых кислот, полученные из ДНК.
"Рекомбинантный" в контексте настоящего изобретения означает "полученный способами генетической инженерии или являющийся результатом генетической инженерии". Таким образом, "рекомбинантный микроорганизм" включает по меньшей мере одну "рекомбинантную нуклеиновую кислоту или рекомбинантный белок". Рекомбинантный микроорганизм предпочтительно содержит экспрессирующий вектор или клонирующий вектор, или он модифицирован способами генетической инженерии, чтобы он содержал клонированную последовательность(и) нуклеиновой кислоты в эндогенном геноме клетки-хозяина.
"Гетерологичная" представляет собой любую нуклеиновую кислоту или полипептид/белок, введенные в клетку или организм способами генетической инженерии, относительно указанной клетки или организма и безотносительно от организма, из которого она происходит. Таким образом, ДНК, выделенная из микроорганизма и введенная в другой микроорганизм того же вида, представляет собой гетерологичную ДНК в отношении этого последнего генетически модифицированного микроорганизма в контексте настоящего изобретения, даже несмотря на то, что иногда в данной области для такого типа модификаций способами генетической инженерии используют термин "гомологичный". Однако предпочтительно термин "гетерологичный" относится к негомологичной нуклеиновой кислоте или полипептиду/белку в контексте настоящего изобретения. "Гетерологичные белок/нуклеиновая кислота" являются синонимом для рекомбинантных белка/нуклеиновой кислоты".
Термины "экспрессировать", "экспрессирующий", "экспрессированный" и "экспрессия" относятся к экспрессии продукта гена (например, биосинтетического фермента гена каскада) в организме-хозяине. Экспрессию можно проводить путем генетического изменения микроорганизма, который используют в качестве исходного организма. В некоторых вариантах осуществления микроорганизм может быть генетически изменен (например, способами генетической инженерии) для экспрессии продукта гена на увеличенном уровне относительно продукта гена, продуцируемого исходным микроорганизмом или сравнимым микроорганизмом, который не был изменен. Генетическое изменение включает, но не ограничивается ими, изменение или модификацию регуляторных последовательностей или участков, ассоциированных с экспрессией конкретного гена (например, путем добавления сильных промоторов, индуцибельных промоторов или множественных промоторов или путем удаления регуляторных последовательностей, так чтобы экспрессия была конститутивной), модификацию хромосомной области конкретного гена, изменение последовательностей нуклеиновых кислот, соседних с конкретным геном, таких как участок связывания рибосомы или терминатор транскрипции, увеличение числа копий конкретного гена, модификацию белков (например, регуляторных белков, супрессоров, энхансеров, активаторов транскрипции и т.п.), вовлеченных в транскрипцию конкретного гена и/или трансляцию конкретного продукта гена, или любые другие общепринятые средства для нарушения регуляции экспрессии конкретного гена с использованием стандартных способов в данной области (включая, но, не ограничиваясь ими, использование антисмысловых молекул нуклеиновых кислот, например, для блокирования экспрессии репрессорных белков).
"Консервативная аминокислотная замена" означает, что одна или несколько аминокислот в исходной аминокислотной последовательности заменены аминокислотами со сходными химическими свойствами, например, Val на Ala. Доля замещенных аминокислот по сравнению с исходной полипептидной последовательностью предпочтительно составляет от 0 до 30% всех аминокислот исходной аминокислотной последовательности, более предпочтительно от 0 до 15%, наиболее предпочтительно от 0 до 5%.
Консервативные аминокислотные замены предпочтительно являются заменами между представителями одной из следующих групп:
кислотные аминокислоты (аспарагиновая и глутаминовая кислота);
основные аминокислоты (лизин, аргинин, гистидин);
гидрофобные аминокислоты (лейцин, изолейцин, метионин, валин, аланин);
гидрофильные аминокислоты (серин, глицин, аланин, треонин);
аминокислоты, имеющие алифатические боковые цепи (глицин, аланин, валин, лейцин, изолейцин);
аминокислоты, имеющие алифатические гидроксильные боковые цепи (серин, треонин);
аминокислоты, имеющие аминосодержащие боковые цепи (аспарагин, глутамин);
аминокислоты, имеющие ароматические боковые цепи (фенилаланин, тирозин, триптофан);
аминокислоты, имеющие содержащие серу боковые цепи (цистеин, метионин).
Особенно предпочтительными консервативными аминокислотными заменами являются следующие:
Термин "выделенный" означает "отделенный или очищенный от организма, из которого он происходит". Более конкретно, выделенная клетка многоклеточного организма является отделенной или очищенной от организма, из которого она происходит. Она охватывает биохимически очищенные и рекомбинантно продуцированные клетки.
В контексте настоящего изобретения термин "происходящая из аспартата аминокислота" означает аминокислоты лизин, метионин, изолейцин и треонин, предпочтительно указанные аминокислоты находятся в L-конфигурации.
Как используют в данном описании, "предшественник" или "биохимический предшественник" аминокислоты представляет собой соединение, предшествующее ("расположенное выше") этой аминокислоте в биохимическом пути, ведущем к образованию указанной аминокислоты в микроорганизме по настоящему изобретению, особенно соединение, образующееся на последних нескольких стадиях указанного биохимического пути. В контексте настоящего изобретения "предшественник" аминокислоты лизина представляет собой любое промежуточное соединение, образовавшееся в процессе биохимического конвертирования аспартата в лизин в организме дикого типа in vivo.
"Промежуточное соединение" или "промежуточный продукт" означает соединение, которое временно или постоянно образуется в ходе химического или биохимического процесса, необязательно в прямо поддающейся аналитической детекции концентрации. Указанное промежуточное соединение может быть удалено из указанного химического процесса путем второй химической или биохимической реакции, в частности, путем последующего ферментативного превращения, как определено ниже в разделе "Подробное описание". Указанное последующее ферментативное превращение предпочтительно происходит в микроорганизме в соответствии с настоящим изобретением. В способе согласно этому предпочтительному аспекту микроорганизм содержит по меньшей мере один гетерологичный фермент, катализирующий стадию реакции при последующем конвертировании эндогенного промежуточного соединения в конечный продукт способа.
"Выход углерода" представляет собой найденное количество углерода (продукта) на потребленное количество углерода (источника углерода в ферментации, обычно сахара), т.е. отношение углерода продукта к углероду источника.
"Модификация" или "модифицированный", используемые в контексте настоящего изобретения, означают, что геном микроорганизмов по настоящему изобретению изменен с использованием способов, известных в данной области, таких как мутации, делеции, инсерции, вставки остатков нуклеиновых кислот путем гомологичной рекомбинации, замены промоторных последовательностей, дупликации генов и т.п.; соответствующие иллюстративные справочники приведены выше. В результате такой модификации, информация нуклеиновой кислоты в микроорганизмах по настоящему изобретению отличается, например, от микроорганизма дикого типа или уже модифицированного микроорганизма, используемого для дальнейшей модификации. Результат способов модификации может быть подтвержден с использованием способов, известных в данной области, например, способов на основе ПЦР, саузерн-блоттинга, нозерн-блоттинга, вестерн-блоттинга, расщепления ферментом рестрикции и визуализации амплифицированных и/или расщепленных фрагментов нуклеиновых кислот, измерения ферментативной активности по сравнению с исходным микроорганизмом, который использовали для дальнейшей модификации, и т.д.
В качестве примера для модификации в контексте настоящего изобретения "модифицированная глюкозо-6-фосфатдегидрогеназа" означает, что нуклеиновая кислота, кодирующая указанный ген (zwf), или оперон, содержащий указанный ген, изменены, т.е. последовательность нуклеиновой кислоты отличается от последовательности дикого типа или первоначально встречающейся последовательности нуклеиновой кислоты, кодирующей указанный фермент. Как следствие такой модификации, ферментативная активность обычно также отличается от исходного фермента при сравнении в сходных условиях и она может быть выражена в единицах на миллиграмм фермента (удельная активность) или в виде молекул субстрата, превращенных в минуту, на молекулу фермента. Предпочтительно, нуклеиновая кислота, кодирующая указанный выше фермент, изменена так, чтобы исходный промотор был заменен сильным гетерологичным промотором, таким как промотор супероксиддисмутазы (sod). Альтернативно или дополнительно к использованию сильного промотора, последовательность нуклеиновой кислоты, кодирующая указанный выше фермент, может быть подвергнута мутации. Предпочтительной модификацией является мутация, приводящая к аминокислотной последовательности, где остаток аланина (A) в положении 243 заменен остатком треонина (T). В следующем предпочтительном варианте осуществления промотор tkt-оперона заменен гетерологичным промотором, таким как приведенный выше sod-промотор.
"Активность G6PDH" в контексте настоящего изобретения означает любую ферментативную активность ICD, особенно любой каталитический эффект, проявляемый G6PDH. В частности, под "активностью G6PDH" понимают конвертирование изоцитрата в альфа-кетоглутарат. Активность G6PDH может быть выражена в виде единиц на миллиграмм фермента (удельная активность) или в виде молекул субстрата, конвертированных в минуту на молекулу фермента.
В контексте настоящего изобретения, когда активность модифицированного гена или фермента описана как "улучшенная", "увеличенная", "ослабленная", "пониженная", "сниженная", "уменьшенная", "пониженная" или "ингибированная" и т.д., это означает, что рассматриваемый ген транскрибируется и транслируется либо в более высокой, либо в более низкой степени (т.е. в более высоких или более низких количествах в единицу времени, например, в сутки, в час, в минуту и т.д.) по сравнению с исходным немодифицированным геном. Таким образом, ферментативная активность модифицированного продукта гена также является более высокой или более низкой, соответственно.
Активность генов или ферментов, которую можно модифицировать в микроорганизмах по изобретению, может быть определена, как правило, способами, аналогично описанным выше в отношении гена zwf и кодируемого им фермента. Конкретные способы определения соответствующих видов активности описаны в разделе "Примеры".
Подробное описание изобретения
Настоящее изобретение относится к получению модифицированного микроорганизма и его применению для продуцирования L-лизина. Ниже более подробно описаны предпочтительные модификации нуклеиновых кислот микроорганизмов по изобретению.
Штаммы микроорганизмов по изобретению были получены на основе подробных знаний о метаболизме C. glutamicum. В этом отношении, 13C-анализ метаболических потоков обеспечил ценную информацию об активности каскадов in vivo и (i) предсказал мишени для преодоления препятствий, а именно, в пентозофосфатном пути в качестве основного источника NADPH, а также в самом биосинтезе лизина, и (ii) идентифицировал реакции, такие как цикл TCA, которые прямо конкурируют с биосинтезом лизина. Для увеличения числа измеряемых потоков в C. glutamicum существующая модель для определения потоков была значительно расширена в контексте изобретения. Она была сфокусирована на сети реакций, соединяющих C3-метаболиты гликолиза и C4-метаболиты цикла TCA. В моделирующей части это включало разделение объединенных метаболитов пирувата и фосфоенолпирувата и вовлечение дополнительных фракций массовых изотопомеров для оценки потоков. Для экспериментальной схемы применяли комбинаторный подход экспериментов с применением радиоизотопных индикаторов с [1-13C] глюкозой и эквимолярной смесью естественным образом меченной и [U-13C] глюкозы. С использованием этого подхода можно было точно установить каждую реакцию, вовлеченную в анаплеротическое карбоксилирование или декарбоксилирование. Затем эту расширенную модель применяли для установления метаболических последствий удаления пируваткиназы из продуцирующих лизин C. glutamicum. Указанная модификация, предположительно увеличивающая продукцию лизина путем сдвига потока от пируваткиназы в направлении анаплеротического карбоксилирования и, таким образом, увеличивая поступление предшественника лизина оксалоацетата, компенсировалась in vivo с помощью метаболического обходного пути, обеспечиваемого PEP-карбоксилазой и яблочным ферментом, демонстрируя высокую гибкость C. glutamicum в отношении генетических нарушений.
Генетическая инженерия для оптимизации штаммов согласно настоящему изобретению включает модификацию связанных с лизином ключевых каскадов различными способами, включая, например, делецию генов, сверхэкспрессию путем замены промотора или адаптацию кодонов. Польза от проведенных изменений может быть подтверждена путем экспериментов с культивированием, ферментных анализов, а также анализов метаболома и флуксома.
Для замены инициирующего кодона было выявлено, что использование редкого инициирующего кодона GTG всегда приводило к более низкой удельной активности фермента в клетке по сравнению с экспрессией гена под контролем общего инициирующего кодона ATG. Таким образом, этот способ можно использовать для изменения удельной активности фермента, тем самым, дополняя экспериментальный набор инструментов, используемый в настоящее время для генетической инженерии. Альтернативно, редкий инициирующий кодон TTG можно использовать для замены более частых инициирующих кодонов.
В одном варианте осуществления настоящего изобретения увеличения поступления предшественника достигали путем подавления пируватдегидрогеназы (PDH), которая прямо конкурирует с пируваткарбоксилазой, главным ферментом, предоставляющим предшественник лизина оксалоацетат. Ослабления PDH в продуцирующих лизин C. glutamicum BS87 на основе дикого типа достигали путем замены инициирующего кодона ATG на GTG. Это приводило к снижению удельной активности PDH на 60% и увеличению выхода лизина на 17%. Оценка метаболических потоков показала, что улучшение было следствием эффективного перенаправления потока с пируватдегидрогеназы на пируваткарбоксилазу, что приводило к более эффективному поступлению оксалоацетата.
Как оказалось, модификация способами инженерии цикла TCA является эффективной альтернативой увеличению поступления предшественника в C. glutamicum. Замена инициирующего кодона (ATG→GTG) в гене icd, кодирующем изоцитратдегидрогеназу (ICD), увеличивала продуцирование лизина более чем на 40% вследствие сниженной на 70% удельной активности ICD. В настоящем изобретении C. glutamicum отвечали на это преднамеренно индуцированное препятствие путем перенаправления потока в сторону анаплеротического карбоксилирования.
Прямая модификация способами инженерии пути биосинтеза лизина посредством сверхэкспрессии гена ddh, кодирующего диаминопимелатдегидрогеназу, отчетливо показала, что польза от осуществленной модификации может быть еще более усилена с помощью надлежащих условий культивирования. В этом случае, улучшение является особенно выраженным при концентрации аммония в культуральной в среде в диапазоне приблизительно от 2 г·л-1 до 10 г·л-1 сульфата аммония, приводящей к приблизительно от 10% до приблизительно 50% увеличению выхода лизина, соответственно. В предпочтительном варианте осуществления способа по настоящему изобретению условия культивирования адаптируют соответствующим образом, т.е. концентрация аммония в культуральной среде предпочтительно находится в диапазоне приблизительно от 10 до 100 г·л-1, более предпочтительно в диапазоне приблизительно от 20 до 100 г·л-1, от 30 до 100 г·л-1, от 40 до 100 г·л-1, от 50 до 100 г·л-1, от 30 до 90 г·л-1, от 30 до 80 г·л-1, от 30 до 70 г·л-1, от 30 до 60 г·л-1, от 30 до 50 г·л-1, более предпочтительно от 40 до 90 г·л-1, от 40 до 80 г·л-1, от 40 до 70 г·л-1, от 40 до 60 г·л-1 или от 40 до 50 г·л-1.
Затем были проведены успешные исследования по метаболической инженерии самого биосинтетического пути лизина и поступления центрального предшественника оксалоацетата путем направленной оптимизации внутриклеточного поступления NADPH, требующегося в качестве кофактора для биосинтеза лизина в высоких количествах.
Увеличенного поступления NADPH для поддержания продуцирования лизина в C. glutamicum можно достигать путем увеличения потока через PPP с использованием сверхэкспрессии контролирующего скорость фермента глюкозо-6-фосфатдегидрогеназы (G6PDH), который кодируется геном zwf. Более того, фермент может быть усовершенствован проведением аминокислотной замены A243T, которая снижает его чувствительность к негативной регуляции метаболитами, образующимися в промежуточном метаболизме. Альтернативно, в предпочтительном способе по изобретению может быть сверхэкспрессирован оперон транскетолазы (tkt), так чтобы увеличивалась экспрессия гена zwf.
В генетическом фоне C. glutamicum lysCT311I, имеющих модифицированную улучшенную аспартаткиназу, которая более не подвергается ингибированию по принципу обратной связи, эти модификации приводили к 15% увеличению потока PPP, что значительно увеличивало продукцию лизина на вплоть до 40%.
Генетическая локализация гена zwf, кодирующего глюкозо-6-фосфатдегидрогеназу (G6PDH), в опероне транскетолазы альтернативно обеспечивает сверхэкспрессию G6PDH в комбинации с транскетолазой и трансальдолазой, образуя неокислительную часть PPP. Использование sod-промотора для контроля экспрессии приводило к значительно увеличенной активности G6PDH, транскетолазы и трансальдолазы, тем самым, устраняя потенциально предстоящие препятствия в PPP.
В дополнение к прямой модификации ферментов PPP, поступление NADPH далее увеличивали путем сверхэкспрессии фруктозо-1,6-бисфосфатазы (fbp). В комбинации со сверхэкспрессией и модификацией G6PDH, эти изменения увеличивали выход лизина на глюкозе на 70%. Значительное усовершенствование также было достигнуто для других, имеющих промышленное значение сахаров: фруктозы и сахарозы. Предпочтительно, штаммы, имеющие улучшенное поступление NADPH на одном или нескольких из указанных выше имеющих значение сахаров, имеют модифицированный ген lysC, например, lysCT311I.
В качестве дополнительного преимущества модификации способами инженерии PPP согласно настоящему изобретению, образование побочного продукта трегалозы снижалось вследствие снижения внутриклеточного уровня G6P. Подробное изучение метаболизма может далее обеспечить новое представление о метаболизме NADPH. До настоящего времени, источником NADPH при продуцировании лизина в C. glutamicum, главным образом, считали PPP и ICD. По сравнению с диким типом, продуцирующие лизин штаммы C. glutamicum проявляли увеличенную удельную активность NADPH-зависимого яблочного фермента (MalE). Делеция кодирующего malE гена в генетическом фоне исходных C. glutamicum lysCT311I в действительности снижала образование требующих NADPH продуктов: биомассы и лизина. Общее потребление восстановленного NADPH, таким образом, полностью совпадало с потоком in vivo через яблочный фермент, составляющим 15%, определенным 13C-анализом потоков. Это указывает на то, что C. glutamicum активируют яблочный фермент в условиях ограниченного NADPH для удовлетворения измененных физиологических потребностей.
Исходя из обширных знаний о метаболизме C. glutamicum и различных ключевых мишенях, идентифицированных для настоящего изобретения, в предпочтительном варианте осуществления настоящего изобретения был создан улучшенный продуцент лизина. Для этой цели особый набор благоприятных изменений был внесен в геном непродуцирующих C. glutamicum дикого типа ATCC 13032.
Сравнение эффективности продуцирования генеалогии созданных штаммов показало, что набор только из 5 модификаций согласно настоящему изобретению был особенно предпочтительным для увеличения продуцирования образующихся из аспартата аминокислот, в частности, продуцирования лизина. Эти модификации включали нарушение регуляции аспартаткиназы (lysC) ингибированием по принципу обратной связи, сверхэкспрессию диаминопимелатдегидрогеназы, ослабление icd, и модификацию способами инженерии метаболизма NADPH путем сверхэкспрессии фруктозо-1,6-бисфосфатазы и оперона транскетолазы. Рационально сконструированный штамм проявлял примечательные продукционные свойства, такие как конечный титр лизина HCl 120 г·л-1, который достигали в течение 30 ч, и выход конверсии вплоть до 55%. С такой эффективностью продуцирования сверхпродуцент лизина, полученный в контексте настоящего изобретения, является наилучшим продуцирующим штаммом на основе дикого типа, известным авторам настоящего изобретения. Достигнутый конечный титр лизина и выход конвертирования углерода лежит даже на уровне максимального предела, достигаемого с помощью используемых в промышленности продуцирующих штаммов, оптимизируемых в течение более 40 лет. Что касается выхода продукта в единицу времени, продуцент на основе дикого типа даже превосходит классические штаммы вследствие быстрого роста и, таким образом, сниженного времени ферментации. С такими продукционными свойствами этот штамм является высоко привлекательным для промышленного продуцирования.
Особенно предпочтительный штамм содержал 12 генетических модификаций. Таким образом, модификация биосинтеза лизина (нарушение регуляции по принципу обратной связи и сверхэкспресия аспартаткиназы, сверхэкспрессия диаминопимелатдегидрогеназы, дигидродипиколинатредуктазы и диаминопимелатдекарбоксилазы) и поступления предшественников (сверхэкспрессия и мутация пируваткарбоксилазы, делеция PEP-карбоксикиназы и мутация гомосериндегидрогеназы) была завершена с помощью других ключевых мишеней поступления предшественников (ослабление изоцитратдегидрогеназы) и метаболизма NADPH (сверхэкспрессия фруктозо-1,6-бисфосфатазы и оперона транскетолазы).
В предпочтительном аспекте настоящего изобретения способ продуцирования представляет собой ферментативный способ. Однако также предусмотрены другие способы биотехнологического получения химических соединений, включая продуцирование in vivo в растениях и не являющихся человеком животных.
Способ ферментативного продуцирования согласно настоящему изобретению может включать культивирование по меньшей мере одного, предпочтительно рекомбинантного, микроорганизма, содержащего описанные выше модификации.
В следующих предпочтительных аспектах изобретения микроорганизм, используемый в способе продуцирования, представляет собой рекомбинантный микроорганизм. Поскольку также предусматриваются другие способы биотехнологического получения химических соединений, включая продуцирование in vivo в растениях и не являющихся человеком животных, предпочтительным для выбора организмом также предпочтительно является рекомбинантный организм.
В предпочтительных вариантах осуществления настоящего изобретения, активность глюкозо-6-фосфатдегидрогеназы (zwf) в используемом микроорганизме является модифицированной, т.е. ген является сверхэкспрессированным. В контексте настоящего изобретения "сверхэкспрессия" или "увеличенная активность" означает, что исходная активность немодифицированного микроорганизма того же вида и с тем же генетическим фоном, является более низкой, предпочтительно по меньшей мере на 10%, 20%, 30%, 40%, 50%, 60% 70%, 80%, 90%, 95% более низкой. Напротив, активность гена или фермента в модифицированном микроорганизме увеличена предпочтительно по меньшей мере приблизительно на 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 95% или даже более. Принципы, сходные с принципами в отношении активности глюкозо-6-фосфатдегидрогеназы, также применимы к другим генам или ферментам, которые могут быть сверхэкспрессированы в контексте настоящего изобретения, т.е. к диаминопимелатдегидрогеназе (ddh), транскетолазе (tkt), трансальдолазе (tal), фруктозо-1,6-бисфосфатазе (fbp), аспартаткиназе (lysC), дигидропиколинатредуктазе (dapB), диаминопимелатдекарбоксилазе (lysA) или пируваткарбоксилазе (pyc).
Понятно, что не всегда желательно увеличивать активность указанных выше генов или ферментов, насколько это возможно. В некоторых случаях, может быть достаточным или желательным неполное снижение любых из уровней, указанных выше, а также промежуточные уровни, например, такие как 25%, 40%, 50% и т.д.
Более того, предпочтительными являются микроорганизмы согласно настоящему изобретению, обладающие сниженной ферментативной активностью, например, микроорганизмы, которые утратили их исходную активность ICD частично или полностью, по сравнению с исходным микроорганизмом того же вида и с тем же генетическим фоном. Предпочтительно, утрачивается приблизительно по меньшей мере 1%, по меньшей мере 2%, по меньшей мере 4%, по меньшей мере 6%, по меньшей мере 8%, по меньшей мере 10%, более предпочтительно по меньшей мере 20%, по меньшей мере 40%, по меньшей мере 60%, по меньшей мере 80%, по меньшей мере 90%, по меньшей мере 95% или вся исходная активность ICD. Степень снижения активности определяют по сравнению с уровнем активности эндогенной ICD в исходном микроорганизме в сравнимых условиях.
Аналогично сниженной активности изоцитратдегидрогеназы, можно модифицировать другие гены или ферменты, которые экспрессируются на более низком уровне в контексте настоящего изобретения, например, фосфоенолпируваткарбоксикиназу (pck) и, необязательно, также пируватдегидрогеназу, хотя это не является наиболее предпочтительным вариантом осуществления изобретения. Понятно, что не всегда желательно снижать активность этих генов или ферментов, насколько это возможно. В определенных случаях может быть достаточным или желательным неполное снижение любых из уровней, указанных выше, а также промежуточные уровни, например, такие как 25%, 40%, 50% и т.д.
В вариантах осуществления, где микроорганизм характеризуется полной или практически полной (т.е. 90% или более) утратой конкретной ферментативной активности, среду для культивирования микроорганизма, особенно среду, используемую в способах согласно настоящему изобретению, можно дополнять одним или несколькими незаменимыми соединениями, отсутствующими в микроорганизме вследствие подавления ферментативной активности. Например, когда подавляется активность ICD, среду можно дополнять глутаматом, поскольку он является недорогим, легкодоступным соединением.
Следующим примером фермента со сниженной активностью, используемого в контексте настоящего изобретения, является ген hom. Модификация этого гена более подробно описана ниже. Точечная мутация, например V59A, приводит к снижению активности и снижению потока в направлении синтеза треонина.
Увеличение или снижение ферментативной активности, соответственно, необходимое для настоящего изобретения, может быть либо эндогенным признаком микроорганизма, используемого в способе согласно настоящему изобретению, например, признаком вследствие спонтанной мутации, либо признаком вследствие любого способа, известного в данной области, для усиления, подавления или ингибирования ферментативной активности частично или полностью, особенно ферментативной активности in vivo. Увеличение или снижение ферментативной активности, соответственно, может происходить на любой стадии синтеза фермента и ферментативных реакций, на генетическом уровне, на уровне транскрипции, трансляции или реакции.
Увеличение или снижение ферментативной активности, соответственно, предпочтительно является результатом генетической инженерии. Для увеличения или снижения уровня экспрессии одного или нескольких эндогенного гена(ов) в клетке-хозяине и, тем самым, снижения количества и/или активности фермента в клетке-хозяине, в которой ген(ы)-мишень усилен или подавлен, соответственно, можно использовать любой способ, известный в данной области.
Для снижения экспрессии гена в микроорганизме, таком как E. coli или C. Glutamicum, или других клетках-хозяевах, таких как P. pastoris и A. niger, доступно множество технологий, таких как подходы нокаута генов, антисмысловая технология, технология РНКи и т.д. Можно удалять исходную копию соответствующего гена и/или заменять ее мутантной версией, демонстрирующей сниженную активность, в частности, сниженную удельную активность, или экспрессировать ее со слабого промотора. Альтернативно, можно заменять инициирующий кодон гена, промотор гена, вносить мутации случайным или направленным мутагенезом, разрушать ген или осуществлять нокаут гена. Более того, можно вносить дестабилизирующие элементы в мРНК или вносить генетические модификации, приводящие к повреждению участков связывания рибосом (RBS) на РНК. Наконец, в реакционную смесь можно добавлять специфические ингибиторы ICD.
Для активации (например, сверхэкспрессии) или усиления экспрессии гена в микроорганизме, таком как E. coli или C. Glutamicum, или других клетках-хозяевах, таких как P. pastoris и A. niger, существует множество технологий, например, можно использовать более сильные промоторы, амплификацию генов и т.д.
В одном предпочтительном аспекте "снижение экспрессии" означает ситуацию, в которой, если заменить эндогенную нуклеотидную последовательность, кодирующую полипептид, модифицированной нуклеотидной последовательностью, которая кодирует полипептид, по существу с той же аминокислотной последовательностью и/или функцией, в модифицированных клетках будет экспрессироваться сниженное количество кодируемого полипептида.
В следующем предпочтительном аспекте "снижение экспрессии" означает подавление экспрессии с помощью антисмысловой технологии или РНК-интерференции (когда это применимо, например, в культуре эукариотических клеток), чтобы препятствовать экспрессии генов. Эти технологии могут влиять на уровни мРНК и/или эффективность трансляции.
В следующем предпочтительном аспекте "снижение экспрессии" означает делецию или разрушение гена в комбинации с введением "слабого" гена, т.е. гена, кодирующего белок, ферментативная активность которого ниже, чем исходная, или путем встраивания гена в слабо экспрессирующийся участок, что приводит к меньшей ферментативной активности внутри клетки. Это можно осуществлять путем встраивания гена в хромосомной локус, с которого гены транскрибируются хуже, или путем введения мутантного или гетерологичного гена с более низкой удельной активностью или с меньшей эффективностью транскрипции, меньшей эффективностью трансляции или меньшей стабильностью в клетке. Встраивание такого мутантного гена можно проводить с использованием реплицирующейся плазмиды или встраивания в геном.
В следующем предпочтительном аспекте "снижение экспрессии" означает, что сниженная активность является результатом снижения уровней мРНК путем снижения транскрипции с кодируемого на хромосоме гена, предпочтительно путем мутации исходного промотора и замены исходного промотора ослабленной версией указанного промотора или более слабым гетерологичным промотором.
В следующем предпочтительном аспекте "снижение экспрессии" означает, что сниженная ферментативная активность является результатом мутации RBS, приводящей к сниженному связыванию рибосом с участком инициации трансляции и, таким образом, к сниженной трансляции мРНК. Мутация может представлять собой простую нуклеотидную замену, и/или также она может влиять на расположение в пространстве RBS относительно инициирующего кодона. Для обеспечения этих мутаций, можно получать библиотеку мутантов, содержащую набор мутантных RBS. Пригодный RBS можно отбирать, например, путем отбора по более низкой ферментативной активности. Затем исходный RBS можно заменять выбранным RBS.
В следующем предпочтительном аспекте "снижения экспрессии" достигают путем снижения уровней мРНК, уменьшая стабильность мРНК, например, изменяя вторичную структуру.
В следующем предпочтительном аспекте "снижения экспрессии" достигают с помощью регуляторов, например, регуляторов транскрипции.
В случае модифицированных нуклеотидных последовательностей, которые подлежат экспрессии в Corynebacterium и особенно предпочтительно в C. glutamicum для снижения количества гена или фермента, кодируемого указанной нуклеотидной последовательностью, по меньшей мере один, по меньшей мере два, по меньшей мере три, по меньшей мере четыре, по меньшей мере пять, по меньшей мере шесть, по меньшей мере seven, по меньшей мере восемь, по меньшей мере девять, по меньшей мере десять, предпочтительно по меньшей мере 1%, по меньшей мере 2%, по меньшей мере 4%, по меньшей мере 6%, по меньшей мере 8%, по меньшей мере 10%, более предпочтительно по меньшей мере 20%, по меньшей мере 40%, по меньшей мере 60%, по меньшей мере 80%, еще более предпочтительно по меньшей мере 90% или по меньшей мере 95% и наиболее предпочтительно все из кодонов немодифицированных нуклеотидных последовательностей можно заменять в модифицированной нуклеотидной последовательности менее часто используемыми кодонами для соответствующей аминокислоты. В еще более предпочтительном варианте осуществления указанное выше количество кодонов, подлежащее замене, относится к частым, очень частым и крайне частым или наиболее частым кодонам. В другом, особенно предпочтительном варианте осуществления описанные выше количества кодонов заменяют наименее часто встречающимися кодонами. Во всех из этих случаев эталонное использование кодонов основывается на использовании кодонов в Corynebacterium и предпочтительно C. Glutamicum, и предпочтительно на использовании кодонов имеющихся в большом количестве белков Corynebacterium и предпочтительно C. glutamicum. Также см. PCT/EP2007/061151 для подробного пояснения.
Как указано выше, настоящее изобретение относится не только к способу продуцирования лизина, но также к микроорганизмам и к применению микроорганизмов для продуцирования лизина. Термин "микроорганизм" для целей настоящего изобретения относится к любому не являющемуся человеком организму, который обычно используют для экспрессии нуклеотидных последовательностей для продуцирования L-лизина, в частности, микроорганизму, как определено выше, растениям, включая водоросли и мхи, дрожжам и не являющимся человеком животным. Организмы, помимо микроорганизмов, которые особенно пригодны для продуцирования L-лизина, могут представлять собой растения или части растений. Такие растения могут быть однодольными или двудольными, такими как однодольные и двудольные сельскохозяйственные растения, пищевые растения или кормовые растения. Примерами однодольных растений являются растения, принадлежащие роду avena (овес), triticum (пшеница), secale (рожь), hordeum (ячмень), oryza (рис), panicum, pennisetum, setaria, sorghum (просо), zea (маис) и т.п.
Двудольные сельскохозяйственные растения включают, среди прочих, хлопок, стручковые растения, такие как бобовые растения и, в частности, люцерна, соя, рапс, томат, сахарная свекла, декоративные растения, а также деревья. Кроме того, сельскохозяйственные растения могут включать фруктовые (в частности, яблони, груши, вишню, виноград, цитрусовые, ананас и банан), масличные пальмы, чайные кусты, деревья какао и деревья кофе, табак, сизаль, а также, что касается медицинских растений, раувольфию и дигиталис. Особенно предпочтительными являются зерна пшеницы, рожь, овес, ячмень, рис, маис и просо, сахарная свекла, рапс, соя, томат, картофель и табак. Другие сельскохозяйственные растения могут быть найдены в US 6137030.
Специалисту в данной области хорошо известно, что различные организмы и клетки, такие как микроорганизмы, растения и растительные клетки, животные и клетки животных, и т.д., отличаются с точки зрения количества и типа генов и белков в клетке, используемых в контексте настоящего изобретения. Даже в одном и том же организме, различные штаммы могут демонстрировать до некоторой степени гетерогетерогенный профиль экспрессии на уровне белка.
В случае, когда при осуществлении настоящего изобретения используют организм, отличающийся от микроорганизма, можно использовать способ неферментативного продуцирования.
В настоящем изобретении можно использовать любой микроорганизм, как определено выше. Предпочтительно, микроорганизмом является прокариотический организм. Особенно предпочтительными для осуществления настоящего изобретения являются микроорганизмы, выбранные из родов Corynebacterium и Brevibacterium, предпочтительно Corynebacterium, конкретно фокусируясь на Corynebacterium glutamicum, рода Escherichia, конкретно фокусируясь на Escherichia coli, рода Bacillus, в частности, Bacillus subtilis, рода Streptomyces и рода Aspergillus.
Предпочтительный вариант осуществления изобретения относится к применению микроорганизмов, которые выбраны из коринеформных бактерий, таких как бактерии рода Corynebacterium. Особенно предпочтительными являются виды Corynebacterium glutamicum, Corynebacterium acetoglutamicum, Corynebacterium acetoacidophilum, Corynebacterium callunae, Corynebacterium ammoniagenes, Corynebacterium thermoaminogenes, Corynebacterium melassecola и Corynebacterium effiziens. Другие предпочтительные варианты осуществления изобретения относятся к применению Brevibacteria и, в частности, видов Brevibacterium flavum, Brevibacterium lactofermentum и Brevibacterium divarecatum.
В предпочтительных вариантах осуществления изобретения микроорганизм может быть выбран из группы, состоящей из Corynebacterium glutamicum ATCC13032, C. acetoglutamicum ATCC15806, C. acetoacidophilum ATCC13870, Corynebacterium thermoaminogenes FERMBP-1539, Corynebacterium melassecola ATCC17965, Corynebacterium effiziens DSM 44547, Corynebacterium effiziens DSM 44549, Brevibacterium flavum ATCC14067, Brevibacterium lactoformentum ATCC13869, Brevibacterium divarecatum ATCC 14020, Corynebacterium glutamicum KFCC10065 и Corynebacterium glutamicum ATCC21608, а также штаммов, которые происходят из них, например, путем классического мутагенеза и селекции или путем направленного мутагенеза.
Другие предпочтительные штаммы C. glutamicum могут быть выбраны из группы, состоящей из ATCC13058, ATCC13059, ATCC13060, ATCC21492, ATCC21513, ATCC21526, ATCC21543, ATCC13287, ATCC21851, ATCC21253, ATCC21514, ATCC21516, ATCC21299, ATCC21300, ATCC39684, ATCC21488, ATCC21649, ATCC21650, ATCC19223, ATCC13869, ATCC21157, ATCC21158, ATCC21159, ATCC21355, ATCC31808, ATCC21674, ATCC21562, ATCC21563, ATCC21564, ATCC21565, ATCC21566, ATCC21567, ATCC21568, ATCC21569, ATCC21570, ATCC21571, ATCC21572, ATCC21573, ATCC21579, ATCC19049, ATCC19050, ATCC19051, ATCC19052, ATCC19053, ATCC19054, ATCC19055, ATCC19056, ATCC19057, ATCC19058, ATCC19059, ATCC19060, ATCC19185, ATCC13286, ATCC21515, ATCC21527, ATCC21544, ATCC21492, NRRLB8183, NRRLW8182, B12NRRLB12416, NRRLB12417, NRRLB12418 и NRRLB11476.
Сокращение KFCC означает Korean Federation of Culture Collection, ATCC означает American-Type Strain Culture Collection, и сокращение DSM означает Deutsche Sammlung von Mikroorganismen und Zellkulturen. Сокращение NRRL означает коллекцию культур ARS, Northern Regional Research Laboratory, Peorea, IL, США.
Такой штамм представляет собой, например, Corynebacterium glutamicum ATCC13032, и, особенно, его производные. Также можно предпочтительно использовать штаммы ATCC 13286, ATCC 13287, ATCC 21086, ATCC 21127, ATCC 21128, ATCC 21129, ATCC 21253, ATCC 21299, ATCC 21300, ATCC 21474, ATCC 21475, ATCC 21488, ATCC 21492, ATCC 21513, ATCC 21514, ATCC 21515, ATCC 21516, ATCC 21517, ATCC 21518, ATCC 21528, ATCC 21543, ATCC 21544, ATCC 21649, ATCC 21650, ATCC 21792, ATCC 21793, ATCC 21798, ATCC 21799, ATCC 21800, ATCC 21801, ATCC 700239, ATCC 21529, ATCC 21527, ATCC 31269 и ATCC 21526, которые известны тем, что они продуцируют лизин. Особенно предпочтительными являются штаммы Corynebacterium glutamicum, которые уже способны продуцировать L-лизин. Таким образом, особенно предпочтительными являются штаммы, происходящие из Corynebacterium glutamicum, имеющих устойчивую к механизму обратной связи аспартокиназу, и их производные. Эти предпочтительные штаммы охватывают штаммы, происходящие из Corynebacterium glutamicum ATCC13032, имеющие устойчивую к механизму обратной связи аспартокиназу, и, в частности, относятся к штаммам ATCC13032 lysCfbr и ATCC13286.
C. glutamicum ATCC13032 lysCfbr, ATCC13032 или ATCC13286 и их производные, имеющие устойчивую к механизму обратной связи аспартокиназу, являются особенно предпочтительными микроорганизмами в контексте настоящего изобретения. Также предпочтительными являются ATCC13032 lysCfbr или ATCC13286.
ATCC13032 lysCfbr можно получать, начиная с ATCC13032. Для получения такого продуцирующего лизин штамма в C. glutamicum ATCC13032 проводят аллельную замену в гене lysC дикого типа. Для этого в ген lysC вносят нуклеотидную замену, так чтобы полученный белок имел изолейцин в положении 311 вместо треонина. Подробно конструирование этого штамма описано в патентной заявке WO 2005/059093. Регистрационным номером гена lysC является P26512.
Можно использовать производные ATCC13032 lysCfbr, где активность ICD активность снижена путем замены ATG в качестве инициирующего кодона нуклеотидной последовательности, кодирующей изоцитратдегидрогеназу, предпочтительно путем замены ATG на GTG. Штамм, описанный в разделе "Примеры", где заменен инициирующий кодон icd, является особенно предпочтительным в контексте настоящего изобретения (т.е. штамм ICD ATG→GTG).
Для способов, которые можно использовать для модификации микроорганизмов, описанных в настоящем описании, см., например, Ausubel et al., (eds.), CURRENT PROTOCOLS IN MOLECULAR BIOLOGY, стр. с 8.2.8 по 8.2.13 (1990), Wosnick et al., Gene 60:115 (1987); Ausubel et al. (eds.), SHORT PROTOCOLS IN MOLECULAR BIOLOGY, 3rd Edition, стр. с 8-8 по 8-9, John Wiley & Sons, Inc. (1995), которые, тем самым, включены в настоящее описание посредством ссылки.
Молекулы ДНК генов, кодирующих ферменты клостридий, используемые в настоящем изобретении, которые рассмотрены выше, можно получать скринингом кДНК или генетических библиотек с помощью полинуклеотидных зондов, имеющих нуклеотидные последовательности, обратно транслированные с соответствующих аминокислотных последовательностей, или с помощью полинуклеотидных зондов, имеющих соответствующие нуклеотидные последовательности. Например, пригодную библиотеку можно получать путем получения геномной ДНК из штамма Clostridium subterminale SB4 (ATCC No. 29748) и конструирования библиотеки согласно стандартным способам. См., например, Ausubel et al. (eds.), SHORT PROTOCOLS IN MOLECULAR BIOLOGY, 3rd Edition, стр. с 2-1 по 2-13 и с 5-1 по 5-6 (John Wiley & Sons, Inc. 1995).
Альтернативно, гены можно получать путем синтеза молекул ДНК с использованием служащих взаимными затравками длинных олигонуклеотидов или ПЦР. См., например, Ausubel et al., (eds.), CURRENT PROTOCOLS IN MOLECULAR BIOLOGY, стр. с 8.2.8 по 8.2.13 (1990), Wosnick et al., Gene 60:115 (1987); Ausubel et al. (eds.), SHORT PROTOCOLS IN MOLECULAR BIOLOGY, 3rd Edition, стр. с 8-8 по 8-9, John Wiley & Sons, Inc. (1995) применительно к синтезу ДНК, которые включены в настоящее описание посредством ссылки.
Ферменты, приведенные в контексте настоящего изобретения, могут кодироваться последовательностью нуклеиновой кислоты, которая адаптирована для использования кодонов указанного микроорганизма, обладающего способностью продуцировать лизин.
Способ согласно настоящему изобретению, кроме того, может включать стадию выделения соединения-мишени (L-лизина) или его предшественника. Термин "извлечение" включает удаление, сбор, выделение или очистку соединения из культуральной среды. Извлечение соединения можно проводить согласно общепринятой методологии выделения или очистки, известной в данной области, включая, но, не ограничиваясь ими, обработку общепринятой смолой (например, анионо- или катионообменной смолой, неионной адсорбционной смолой и т.д.), обработку общепринятым адсорбентом (например, активированным углем, кремниевой кислотой, силикагелем, целлюлозой, оксидом алюминия и т.д.), изменение pH, экстракцию растворителем (например, общепринятым растворителем, таким как спирт, этилацетат, гексан и т.п.), дистилляцию, диализ, фильтрацию, концентрирование, кристаллизацию, перекристаллизацию, коррекцию pH, лиофилизацию и т.п. Например, соединение-мишень можно выделять из культуральной среды, сначала удаляя микроорганизмы. Оставшийся бульон затем пропускают через или над катионообменной смолой для удаления нежелательных катионов, и затем через или над анионообменной смолой для удаления нежелательных неорганических анионов и органических кислот.
Специалисту в данной области известно, как заменить, например, ген или эндогенную нуклеотидную последовательность, которая кодирует определенный полипептид, модифицированной нуклеотидной последовательностью. Этого можно достигать, например, путем внесения подходящей конструкции (плазмида без ориджина репликации, линейный фрагмент ДНК без ориджина репликации), путем электропорации, химической трансформации, конъюгации или других подходящих способов трансформации. После этого следует, например, гомологичная рекомбинация с использованием селективных маркеров, которые обеспечивают идентификацию только тех клеток, которые несут модифицированную нуклеотидную последовательность вместо эндогенной встречающейся в природе последовательности. Другие способы включают разрушение гена в эндогенном хромосомном локусе и эксперессию модифицированных последовательностей, например, с плазмид. Другие способы включают, например, транспозицию. Дополнительная информация в отношении векторов и клеток-хозяев, которые можно использовать, приведена ниже.
Как правило, специалисту в данной области известно моделирование конструкций, таких как векторы, для запуска экспрессии полипептида в микроорганизмах, таких как E. coli и C. glutamicum. Специалисту в данной области также хорошо известны условия культивирования микроорганизмов, таких как C. glutamicum и E. coli, а также способы сбора и очистки аминокислот и, в частности лизина, из приведенных выше микроорганизмов. Некоторые из этих аспектов дополнительно подробно описаны ниже.
Специалисту в данной области также известны способы, которые позволяют изменять исходную немодифицированную нуклеотидную последовательность на модифицированную нуклеотидную последовательность, кодирующую полипептиды с идентичной аминокислотной последовательностью, но отличающейся последовательностью нуклеиновой кислоты. Например, этого можно достигать способами мутагенеза на основе полимеразной цепной реакции, с помощью широко известных методик клонирования, химическим синтезом и т.д. Стандартные способы технологии рекомбинантных ДНК и молекулярной биологии описаны в различных публикациях, например, в Sambrook et al. (2001), Molecular Cloning: A Laboratory Manual, 3rd edition, Cold Spring Harbor Laboratory Press, или Ausubel et al. (Eds.) Current protocols in molecular biology (John Wiley & Sons, Inc. 2007). Ausubel et al., Current Protocols in Protein Science, (John Wiley & Sons, Inc. 2002). Ausubel et al. (eds.), SHORT PROTOCOLS IN MOLECULAR BIOLOGY, 3rd Edition (John Wiley & Sons, Inc. 1995). Способы конкретно для C. glutamicum описаны в Eggeling and Bott (eds.) Handbook of Corynebacterium (Taylor and Francis Group, 2005). Некоторые из этих способов представлены ниже и в разделе "Примеры".
Ниже описано и подробно представлено, как можно проводить генетические манипуляции в микроорганизмах, таких как Corynebacterium glutamicum.
Векторы и клетки-хозяева
Как используют в данном описании, термин "вектор" относится к молекуле нуклеиновой кислоты, способной транспортировать другую нуклеиновую кислоту, с которой она связана.
Одним из типов вектора является "плазмида", которая относится к замкнутой двухцепочечной петле ДНК, в которую можно лигировать дополнительные сегменты ДНК. Другим типом вектора является вирусный вектор, где в вирусный геном могут быть лигированы дополнительные сегменты ДНК.
Определенные векторы способны к автономной репликации в клетке-хозяине, в которую их вводят (например, бактериальные векторы, имеющие бактериальный ориджин репликации и эписомальные векторы млекопитающих). Другие векторы (например, неэписомальные векторы млекопитающих) встраиваются в геном клетки-хозяина при введении в клетку-хозяина и, таким образом, реплицируются вместе с геномом хозяина. Более того, определенные векторы способны регулировать экспрессию генов, с которыми они функционально связаны. Такие векторы называют в настоящем описании "экспрессирующими векторами".
Как правило, экспрессирующие векторы, пригодные в способах рекомбинантных ДНК, часто находятся в форме плазмид. В настоящем описании термины "плазмида" и "вектор" могут быть использованы взаимозаменяемо, поскольку плазмида является наиболее часто используемой формой вектора. Однако изобретение включает другие формы экспрессирующих векторов, такие как вирусные векторы (например, ретровирусы с дефектом репликации, аденовирусы и аденоассоциированные вирусы), которые выполняют эквивалентные функции.
Рекомбинантный экспрессирующий вектор, пригодный для получения рекомбинантного микроорганизма по изобретению, может включать гетерологичную нуклеиновую кислоту, как определено выше, в форме, пригодной для экспрессии соответствующей нуклеиновой кислоты в клетке-хозяине, что означает, что рекомбинантные экспрессирующие векторы включают одну или несколько регуляторных последовательностей, выбранных на основе клеток-хозяев, подлежащих использованию для экспрессии, которые функционально связаны с последовательностью нуклеиновой кислоты, подлежащей экспрессии.
В рекомбинантном экспрессирующем векторе "функционально связанный" означает, что представляющая интерес нуклеотидная последовательность связана с регуляторной последовательностью(ями) таким образом, чтобы обеспечивалась экспрессия нуклеотидной последовательности (например, в системе транскрипции/трансляции in vitro или в клетке-хозяине, когда вектор вводят в клетку-хозяина). Термин "регуляторная последовательность" включает промоторы, участки связывания репрессоров, участки связывания активаторов, энхансеры и другие элементы контроля экспрессии (например, терминаторы, сигналы полиаденилирования или другие элементы вторичной структуры мРНК). Такие регуляторные последовательности описаны, например, в Goeddel; Gene Expression Technology: Methods in Enzymology 185, Academic Press, San Diego, CA (1990). Регуляторные последовательности включают последовательности, которые обеспечивают конститутивную экспрессию нуклеотидной последовательности во многих типах клеток-хозяев и последовательности, которые обеспечивают экспрессию нуклеотидной последовательности только в определенных клетках-хозяевах. Предпочтительными регуляторными последовательностями являются, например, промоторы, такие как cos-, tac-, trp-, tet-, trp-, tet-, lpp-, lac-, lpp-lac-, lacIq-, T7-, T5-, T3-, gal-, trc-, ara-, SP6-, arny, SP02, e-Pp- или PL, SOD, EFTu, EFTs, GroEL, MetZ (последние пять из C. glutamicum), которые предпочтительно используют в бактериях. Дополнительными регуляторными последовательностями являются, например, промоторы из дрожжей и грибов, такие как ADC1, MFa, AC, P-60, CYC1, GAPDH, TEF, rp28, ADH, промоторы из растений, такие как CaMV/35S, SSU, OCS, lib4, usp, STLS1, B33, nos, или промоторы убиквитина или фазеолина. Также можно использовать искусственные промоторы. Специалисту в данной области будет понятно, что конструкция экспрессирующего вектора может завесить от таких факторов, как выбор клетки-хозяина, подлежащей трансформации, уровень экспрессии желаемого белка и т.д. Экспрессирующие векторы можно вводить в клетки-хозяева, чтобы, тем самым, продуцировать белки или пептиды, включая слитые белки или пептиды.
Можно использовать любой вектор, пригодный для запуска экспрессии модифицированной нуклеотидной последовательности в клетке-хозяине, предпочтительно в Corynebacterium и особенно предпочтительно в C. glutamicum. Такой вектор, например, может представлять собой плазмидный вектор, который автономно реплицируется в коринеформных бактериях. Примерами являются pZ1 (Menkel et al. (1989), Applied and Environmental Microbiology 64:549-554), pEKEx1 (Eikmanns et al. (1991), Gene 102:93-98), pHS2-1 (Sonnen et al. (1991), Gene 107:69-74). Эти векторы основаны на криптических плазмидах pHM1519, pBL1 или pGA1. Другими пригодными векторами являются pClik5MCS (WO 2005/059093) или векторы на основе pCG4 (US-A 4489160) или pNG2 (Serwold-Davis et al. (1990), FEMS Microbiology Letters 66:119-124), или pAG1 (US-A 5158891). Примеры других пригодных векторов могут быть найдены в Handbook of Corynebacterium, Chapter 23 (под редакцией Eggeling and Bott, ISBN 0-8493-1821-1, 2005).
Рекомбинантные экспрессирующие векторы можно конструировать для экспрессии конкретных нуклеотидных последовательностей в прокариотических или эукариотических клетках. Например, нуклеотидные последовательности можно экспрессировать в бактериальных клетках, таких как C. glutamicum и E. coli, в клетках насекомых (с использованием бакуловирусных экспрессирующих векторов), в клетках дрожжей и других грибов (см. Romanos, M.A. et al. (1992), Yeast 8:423-488; van den Hondel, C.A. M.J. J. et al. (1991), More Gene Manipulations in Fungi, J.W. Bennet & L.L. Lasure, eds., p. 396-428, Academic Press: San Diego; и van den Hondel, C. A. M. J. J. & Punt, P.J. (1991), Applied Molecular Genetics of Fungi, Peberdy, J.F. et al., eds., p. 1-28, Cambridge University Press: Cambridge), algae and multicellular plant cells (см. Schmidt, R. и Willmitzer, L. (1988) Plant Cell Rep.:583-586). Пригодные клетки-хозяева далее рассмотрены в Goeddel, Gene Expression Technology: Methods in Enzymology 185, Academic Press, San Diego, CA (1990). Альтернативно, рекомбинантный экспрессирующий вектор можно транскрибировать и транслировать in vitro, например, с использованием регуляторных последовательностей промотора T7 и полимеразы T7.
Экспрессию белков в прокариотах наиболее часто осуществляют с помощью векторов, содержащих конститутивные или индуцибельные промоторы, направляющие экспрессию либо слитых, либо неслитых белков.
Осуществляющие слияние векторы добавляют несколько аминокислот к белку, кодируемому ими, обычно на N-конец рекомбинантного белка, а также и на C-конец, или подвергаются слиянию с определенными областями в белках. Такие осуществляющие слияние векторы, как правило, служат для целей: 1) увеличения экспрессии рекомбинантного белка; 2) увлечения растворимости рекомбинантного белка; 3) способствования очистке рекомбинантного белка, действуя в качестве лиганда для аффинной очистки; и 4) предоставления "метки" для последующей детекции белка. Часто, в осуществляющие слияние экспрессирующие векторы встраивают участок протеолитического расщепления в точке соединения подвергаемой слиянию части и рекомбинантного белка для обеспечения отделения рекомбинантного белка от подвергаемой слиянию части после очистки слитого белка. Такие ферменты и распознаваемые ими последовательности включают фактор Xa, тромбин и энтерокиназу.
Конкретные, осуществляющие слияние экспрессирующие векторы включают pGEX (Pharmacia Biotech Inc; Smith, D.B. и Johnson, K.S. (1988) Gene 67:31-40), pMAL (New England Biolabs, Beverly, MA) и pRIT5 (Pharmacia, Piscataway, NJ), которые осуществляют слияние с глутатион-S-трансферазой (GST), связывающим мальтозу E белком или белком A, соответственно.
Примеры пригодных индуцибельных, не осуществляющих слияние экспрессирующих векторов E. coli включают pTrc (Amann et al., (1988) Gene 69: 301-315), pLG338, pACYC184, pBR322, pUC18, pUC19, pKC30, pRep4, pHS1, pHS2, pPLc236, pMBL24, pLG200, pUR290, pIN-III113-B1, egtll, pBdCl и pET lld (Studier et al., Gene Expression Technology: Methods in Enzymology 185, Academic Press, San Diego, California (1990) 60-89; и Pouwels et al., eds. (1985) Cloning Vectors. Elsevier: New York, ISBN 0 444 904018). Экспрессия гена-мишени с вектора pTrc основана на транскрипции РНК-полимеразы хозяина с гибридного слитого промотора trp-lac. Экспрессия гена-мишени с вектора pET lld основана на транскрипции со слитого промотора T7 gnlO-lac, опосредуемой коэкспрессируемой вирусной РНК-полимеразой (T7gnl). Эта вирусная полимераза предоставляется штаммами-хозяевами BL21 (DE3) или HMS174 (DE3) из резидентного профага X, обладающего геном T7gnl, под транскрипционным контролем промотора lacUV 5. Для трансформации других типов бактерий можно выбирать соответствующие векторы. Например, известно, что плазмиды pIJ101, pIJ364, pIJ702 и pIJ361 пригодны для трансформации Streptomyces, в то время как плазмиды pUB110, pC194 или pBD214 пригодны для трансформации видов Bacillus. Некоторые плазмиды, применимые для переноса генетической информации в Corynebacterium, включают pHM1519, pBL1, pSA77 или pAJ667 (Pouwels et al., eds. (1985) Cloning Vectors. Elsevier: New York ISBN 0 444 904018).
Примерами пригодных челночных векторов C. glutamicum и E. coli являются, например, pClik5aMCS (WO 2005/059093) или они могут быть найдены у Eikmanns et al. ((1991) Gene 102:93-8).
Примеры пригодных векторов для манипулирования Corynebacteria могут быть найдены в Handbook of Corynebacterium (под редакцией Eggeling and Bott, ISBN 0-8493-1821-1, 2005). Там можно найти перечень челночных векторов E. coli - C. glutamicum (таблица 23.1), перечень челночных экспрессирующих векторов E. coli - C. glutamicum (таблица 23.2), перечень векторов, которые можно использовать для встраивания ДНК в хромосому C. glutamicum (таблица 23.3), перечень экспрессирующих векторов для встраивания в хромосому C. glutamicum (таблица 23.4.), а также перечень векторов для сайт-специфического встраивания в хромосому C. glutamicum (таблица 23.6).
В другом варианте осуществления экспрессирующий вектор представляет собой экспрессирующий вектор дрожжей. Примеры векторов для экспрессии в дрожжах S. cerevisiae включают pYepSec1 (Baldari, et al., (1987) Embo J. 6:229-234), 2i, pAG-1, Yep6, Yep13, pEMBLYe23, pMFa (Kurjan and Herskowitz, (1982) Cell 30:933-943), pJRY88 (Schultz et al., (1987) Gene 54:113-123) и pYES2 (Invitrogen Corporation, San Diego, CA). Векторы и способы конструирования векторов, пригодных для применения в других грибах, таких как нитчатые грибы, включают векторы, подробно описанные в: van den Hondel, C. A. M. J. J. & Punt, P. J. (1991), Applied Molecular Genetics of Fungi, J.F. Peberdy et al., eds., p. 1-28, Cambridge University Press: Cambridge, и Pouwels et al., eds. (1985) Cloning Vectors. Elsevier: New York (ISBN 0 444 904018).
Для целей настоящего изобретения функциональную связь понимают как последовательное расположение промотора (включая участок связывания рибосом (RBS)), кодирующей последовательности, терминатора и, необязательно, дополнительных регуляторных элементов таким образом, чтобы каждый регуляторный элемент мог выполнять его функцию, согласно его назначению при экспрессии кодирующей последовательности.
В другом варианте осуществления гетерологичные нуклеотидные последовательности могут экспрессироваться в клетках одноклеточных растений (таких как водоросли) или в клетках высших растений (например, семенных растений, таких как сельскохозяйственные растения). Примеры экспрессирующих векторов растений включают векторы, подробно описанные в: Becker, D., Kemper, E., Schell, J. и Masterson, R. (1992) Plant Mol. Biol. 20:1195-1197; и Bevan, M.W. (1984) Nucl. Acid. Res. 12:8711-8721, и включают pLGV23, pGHlac+, pBIN19, pAK2004 и pDH51 (Pouwels et al., eds. (1985) Cloning Vectors. Elsevier: New York ISBN 0 444 904018).
Для других пригодных экспрессирующих систем как для прокариотических, так и для эукариотических клеток, см. главы 16 и 17 Sambrook, J. et al. Molecular Cloning: A Laboratory Manual. 3rd ed., Cold Spring Harbor Laboratory, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, 2003.
В другом варианте осуществления рекомбинантный экспрессирующий вектор способен направлять экспрессию нуклеиновой кислоты предпочтительно в конкретном типе клеток, например в клетках растений (например, для экспрессии нуклеиновой кислоты используют тканеспецифические регуляторные элементы). Тканеспецифические регуляторные элементы известны в данной области.
Другой аспект изобретения относится к организмам или клеткам-хозяевам, в которые введен рекомбинантный экспрессирующий вектор или нуклеиновая кислота. Полученная клетка или организм представляет собой рекомбинантную клетку или организм, соответственно. Понятно, что такие термины относится не только к конкретной рассматриваемой клетке, но также к потомкам или потенциальным потомкам такой клетки, когда потомок содержит рекомбинантную нуклеиновую кислоту. Поскольку в последующих поколениях могут происходить определенные модификации вследствие либо мутации, либо влияний окружающей среды, такие потомки, в действительности, могут быть не идентичны исходной клетке, но все еще входят в объем термина, как используют в данном описании, при условии, что потомки все еще экспрессируют или способны экспрессировать рекомбинантный белок.
ДНК вектор может быть введен в прокариотические или эукариотические клетки общепринятыми способами трансформации или трансфекции. Как используют в данном описании, термины "трансформация" и "трансфекция", "конъюгация" и "трансдукция" относятся к различным известным в данной области способам введения чужеродной нуклеиновой кислоты (например, линейной ДНК или РНК (например, линеаризованного вектора или конструкции гена отдельно без вектора) или нуклеиновой кислоты в форме вектора (например, плазмиды, фага, фасмиды, фагмиды, транспозона или другой ДНК)) в клетку-хозяина, включая совместное осаждение с фосфатом кальция или хлоридом кальция, опосредуемую DEAE-декстраном трансфекцию, липофекцию, естественную конкуренцию, опосредуемый конъюгацией с химическим веществом перенос или электропорацию. Пригодные способы для трансформации или трансфекции клеток-хозяев могут быть найдены в Sambrook, et al. (Molecular Cloning: A Laboratory Manual. 3rd ed., Cold Spring Harbor Laboratory, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, 2003), и в других лабораторных справочниках.
Для идентификации и селекции этих вставок, обычно в клетки-хозяева вместе с представляющим интерес геном вводят ген, который кодирует селективный маркер (например, устойчивость к антибиотикам). Предпочтительные селективные маркеры включают маркеры, которые придают устойчивость к лекарственным средствам, таким как G418, гигромицин, канамицин, тетрациклин, ампициллин и метотрексат. Нуклеиновую кислоту, кодирующую селективный маркер, можно вводить в клетку-хозяина в том же векторе, который кодирует указанный выше модифицированные нуклеотидные последовательности, или ее можно вводить в отдельном векторе. Клетки, стабильно трансфицированные введенной нуклеиновой кислотой, можно идентифицировать селекцией с помощью лекарственного средства (например, клетки, в которые включили ген селективного маркера, выживут, в то время как другие клетки погибнут).
Когда используют плазмиды без ориджина репликации и два различных маркера гена (например, pClik int sacB), также можно получать штаммы без маркеров, которые имеют часть вставки, встроенную в геном. Этого достигают путем двух последовательных событий гомологичной рекомбинации (также см. Becker et al., APPLIED AND ENVIRONMENTAL MICROBIOLOGY, 71 (12), p. 8587-8596; Eggeling and Bott (eds) Handbook of Corynebacterium (Taylor and Francis Group, 2005)). Последовательность плазмиды pClik int sacB может быть найдена в WO 2005/059093 в виде SEQ ID NO:24; в указанном источнике плазмиду называют pCIS.
В другом варианте осуществления можно получать рекомбинантные микроорганизмы, которые содержат подобранные системы, которые позволяют регулируемую экспрессию введенного гена. Например, включение нуклеотидной последовательности в вектор, помещая ее под контроль lac-оперона, позволяет экспрессию гена, только в присутствии IPTG. Такие регуляторные системы хорошо известны в данной области.
Выращивание Escherichia coli и Corynebacterium glutamicum -среда и условия культивирования
В одном варианте осуществления способ включает культивирование микроорганизма в пригодной среде для продуцирования лизина. В другом варианте осуществления способ дополнительно включает выделение лизина из среды или клетки-хозяина.
Специалисту в данной области известно культивирование распространенных микроорганизмов, таких как C. glutamicum и E. coli. Таким образом, ниже приведены общие указания для культивирования E. coli и C. glutamicum. Дополнительная информация может быть получена из стандартных справочников по культивированию E. coli и C. glutamicum.
Штаммы E. coli обычно выращивают в бульоне MB и LB, соответственно (Follettie et al. (1993) J. Bacteriol. 175:4096-4103). Минимальной средой для E. coli является M9 и модифицированная MCGC (Yoshihama et al. (1985) J. Bacteriol. 162:591-597), соответственно. Глюкозу можно добавлять в конечной концентрации 1%. Антибиотики можно добавлять в следующих количествах (микрограмм на миллилитр): ампициллин, 50; канамицин, 25; налидиксовая кислота, 25. Аминокислоты, витамины, и другие добавки можно добавлять в следующих количествах: метионин, 9,3 мМ; аргинин, 9,3 мМ; гистидин, 9,3 мМ; тиамин, 0,05 мМ. Клетки E. coli обычно выращивают при 37°С, соответственно.
Генетически модифицированные Corynebacteria, как правило, культивируют в синтетической или природной среде для роста. Ряд различных сред для роста Corynebacteria является как хорошо известным, так и легкодоступным (Liebl et al. (1989) Appl.Microbiol. Biotechnol., 32:205-210; von der Osten et al. (1998) Biotechnology Letters, 11:11-16; патент DE 4120867; Liebl (1992) "The Genus Corynebacterium", The Procaryotes, Volume II, Balows, A. et al., eds. Springer-Verlag). Также инструкции могут быть найдены в Handbook of Corynebacterium (edited by Eggeling и Bott, ISBN 0-8493-1821-1, 2005).
Эти среды состоят из одного или нескольких источников углерода, источников азота, неорганических солей, витаминов и микроэлементов. Предпочтительными источниками углерода являются сахара, такие как моно-, ди- или полисахариды. Например, в качестве источников углерода используют глюкозу, фруктозу, маннозу, галактозу, рибозу, сорбозу, лактозу, мальтозу, сахарозу, глицерин, раффинозу, крахмал или целлюлозу.
Также сахар можно добавлять в среду посредством комплексных соединений, таких как патока или другие побочные продукты рафинирования сахара. Также является преимущественным предоставление смесей различных источников углеродов. Другими возможными источниками углерода являются спирты и органические кислоты, такие как метанол, этанол, уксусная кислота или молочная кислота. Источники азота обычно представляют собой органические или неорганические азотсодержащие соединения, или материалы, которые содержат эти соединения. Иллюстративные источники азота включают газообразный аммиак или соли аммиака, такие как NH4Cl или (NH4)2SO4, NH4OH, нитраты, мочевину, аминокислоты или комплексные источники азота, такие как кукурузный сироп, соевая мука, соевый белок, дрожжевой экстракт, мясной экстракт и другие.
Неорганические соединения солей, которые могут быть включены в среду, включают хлоридные, фосфорные или сульфатные соли кальция, магния, натрия, кобальта, молибдена, калия, марганца, цинка, меди и железа. В среду можно добавлять хелатирующие соединения для поддержания ионов металлов в растворе. Особенно пригодные хелатирующие соединения включают дигидроксифенолы, такие как катехол или протокатеховая кислота, или органические кислоты, такие как лимонная кислота. Обычным для среды является содержание других факторов роста, таких как витамины или другие стимуляторы роста, примеры которых включают биотин, рибофлавин, тиамин, фолиевую кислоту, никотиновую кислоту, пантотенат и пиридоксин. Факторы роста и соли часто происходят из комплексных компонентов сред, таких как дрожжевой экстракт, патока, кукурузный сироп и прочие. Точная композиция соединений среды строго зависит от конкретного эксперимента, и ее индивидуально подбирают для каждого конкретного случая. Информация об оптимизации сред доступна в справочнике "Applied Microbiol. Physiology, A Practical Approach" (Eds. P.M. Rhodes, P.F. Stanbury, IRL Press (1997) pp. 53-73, ISBN 0 19 963577 3). Также можно выбирать среду для роста от коммерческих поставщиков, такую как standard 1 (Merck) или BHI (сердечно-мозговой экстракт, DIFCO) или другие.
Примеры предпочтительных сред в контексте настоящего изобретения описаны в разделе "Примеры" ниже.
Все компоненты среды должны быть стерилизованными либо нагреванием (20 мин при 1,5 бар (150 кПа) и 121°С), либо стерильной фильтрацией. Компоненты могут быть стерилизованы вместе или, если необходимо, по отдельности.
Все компоненты среды могут присутствовать в начале роста или необязательно их можно добавлять непрерывно или партиями. Условия культивирования определяют отдельно для каждого эксперимента.
Температура зависит от используемого микроорганизма, и обычно она должна находиться в диапазоне от 15°С до 45°С. Температуру можно поддерживать постоянной или можно изменять в ходе эксперимента. pH среды может находиться в диапазоне от 5 до 8,5, предпочтительно приблизительно 7,0, и его можно поддерживать добавлением в среду буферов. Иллюстративным буфером для этой цели является калий-фосфатный буфер. Можно использовать синтетические буферы, альтернативно или одновременно, такие как MOPS, HEPES, ACES и другие. Также можно поддерживать постоянное значение pH культуры путем добавления NaOH или NH4OH в процессе роста. Если используют комплексные компоненты среды, такие как дрожжевой экстракт, необходимость в дополнительных буферах может быть снижена благодаря тому факту, что многие комплексные соединения обладают высокой буферной емкостью. Если для культивирования микроорганизмов используют ферментер, pH также можно контролировать с использованием газообразного аммиака.
Время инкубации, как правило, находится в диапазоне от нескольких часов до нескольких суток. Это время выбирают для обеспечения накопления в бульоне максимального количества продукта. Описанные эксперименты по выращиванию можно проводить в различных емкостях, таких как микропланшеты для титрования, стеклянные пробирки, стеклянные колбы или стеклянные или металлические ферментеры различных размеров. Для скрининга большого количества клонов, микроорганизмы следует культивировать в микропланшетах для титрования, стеклянных пробирках или вращающихся флаконах либо с перегородками, либо без них. Предпочтительно используют 100-мл вращающиеся флаконы, заполненные на 10% (по объему) требуемой средой для роста. Флаконы следует вращать на вращающем устройстве (амплитуда 25 мм) с использованием диапазона скоростей 100-300 об./мин. Потери на испарение можно снижать путем поддержания влажной атмосферы; альтернативно, следует проводить математическую коррекцию потерь на испарение. Примеры предпочтительных условий культивирования описаны в разделе "Примеры" ниже.
Если тестируют генетически модифицированные клоны, также следует тестировать немодифицированный контрольный клон (например, исходный штамм) или контрольный клон, содержащий основную плазмиду без какой-либо вставки. Среду инокулируют до OD600 0,5-1,5 с использованием клеток, выращенных на планшетах с агаром, таких как планшеты CM (10 г/л глюкозы, 2,5 г/л NaCl, 2 г/л мочевины, 10 г/л полипептона, 5 г/л дрожжевого экстракта, 5 г/л мясного экстракта, 22 г/л агара, pH 6,8 с 2М NaOH), которые инкубировали при 30°С. Инокуляцию среды проводят либо путем введения суспензии в физиологическом растворе клеток C. glutamicum из планшетов CM, либо путем добавления жидкой предварительной культуры этих бактерий.
Количественное определение аминокислот и их промежуточных соединений
Количественное определение аминокислот и их промежуточных соединений можно проводить согласно любому описанному в литературе способу, известному специалисту в данной области. Далее указанное количественное определение проиллюстрировано посредством количественного определения метионина. Другие примеры количественного определения приведены в разделе "Примеры". Последние являются предпочтительными в контексте настоящего изобретения.
Анализ проводят ВЭЖХ (Agilent 1100, Agilent, Waldbronn, Германия) с защитной кассетой и 4-мкм колонкой Sinergi (Max-RP 80 Å 150*4,6 мм) (Phenomenex, Aschaffenburg, Германия). Перед инъекцией анализируемые образцы преобразуют в производные с использованием o-фтальдиальдегида (OPA) и меркаптоэтанола в качестве восстанавливающего агента (2-MCE). Кроме того, сульфгидрильные группы блокируют йодоуксусной кислотой. Разделение проводят при скорости потока 1 мл/мин с использованием 40 мМ NaH2PO4 (элюент A, значение pH=7,8, доведенное с помощью NaOH) в качестве полярной фазы и смеси метанол-вода (100/1) в качестве неполярной фазы (элюент B). Используют следующий градиент: начало 0% B; 39 мин 39% B; 70 мин 64% B; 100% B в течение 3,5 мин; 2 мин 0% B для уравновешивания. Преобразование в производные при комнатной температуре автоматизируют, как описано ниже. Первоначально 0,5 мкл 0,5% 2-MCE в бицине (0,5М, pH 8,5) смешивают с 0,5 мкл клеточного экстракта. Затем добавляют 1,5 мкл 50 мг/мл йодоуксусной кислоты в бицине (0,5М, pH 8,5), и затем добавляют 2,5 мкл бицинового буфера (0,5М, pH 8,5). Преобразование в производные проводят добавлением 0,5 мкл 10 мг/мл реагента OPA, растворенного в 1/45/54 об./об./об. 2-MCE/MeOH/бицин (0,5M, pH 8,5). Наконец, смесь разбавляют 32 мкл H2O. Между каждой из указанных выше стадий пипетирования имеется период ожидания 1 мин. Затем в колонку инъецируют общий объем 37,5 мкл. Результаты анализа могут быть существенно улучшены, если иглу автоматического пробоотборника периодически очищать в процессе (например в период ожидания) и после приготовления образца. Детекцию проводят с помощью флуоресцентного детектора (возбуждение 340 нм, испускание 450 нм, Agilent, Waldbronn, Германия). Для количественного определения в качестве внутреннего стандарта используют α-аминомасляную кислоту (ABA).
Протокол рекомбинации для C. glutamicum
Ниже описано, как можно конструировать штамм C. glutamicum с увеличенной эффективностью продуцирования продуктов тонкого органического синтеза с использованием конкретного протокола рекомбинации.
"Campbell in", как используют в данном описании, относится к трансформанту исходной клетки-хозяина, в котором целая замкнутая двухцепочечная молекула ДНК (например, плазмида на основе pClik int sacB) встроена в хромосому путем одного события гомологичной рекомбинации (событие "cross-in"), которое эффективно приводит к встраиванию линеаризованной версии указанной замкнутой молекулы ДНК в первую последовательность ДНК хромосомы, которая гомологична первой последовательности ДНК указанной замкнутой молекулы ДНК. "Campbelled in" относится к линеаризованной последовательности ДНК, которая встроилась в хромосому трансформанта "Campbell in". "Campbell in" содержит дупликации первой гомологичной последовательности ДНК, каждая копия которой включает и окружает копию точки кроссинговера при гомологичной рекомбинации. Название происходит от профессора Alan Campbell, который первым предложил этот тип рекомбинации.
"Campbell out", как используют в данном описании, относится к клетке, происходящей из трансформанта "Campbell in", в которой произошло второе событие гомологичной рекомбинации (событие "cross out") между второй последовательностью ДНК, которая содержится в линеаризованной встроенной ДНК на ДНК "Campbelled in", и второй последовательностью ДНК хромосомного происхождения, которая гомологична второй последовательности ДНК указанной линеаризованной вставки, причем второе событие рекомбинации приводит к делеции (удалению) части встроенной последовательности ДНК, но, важно, также приводит к тому, что часть (она может составлять только одно основание) встроенной ДНК "Campbelled in" остается в хромосоме, так что, по сравнению с исходной клеткой-хозяином, клетка "Campbell out" содержит одно или несколько преднамеренных изменений в хромосоме (например, замену одного основания, замену множества оснований, вставку гетерологичного гена или последовательности ДНК, вставку дополнительной копии или копий гомологичного гена или модифицированного гомологичного гена, или вставку последовательности ДНК, содержащей более одного из таких приведенных выше примеров).
Клетку или штамм "Campbell out" обычно, но не обязательно, получают путем контрселекции против гена, который содержится в части (часть, удаление которой является желательным) последовательности ДНК "Campbelled in", например, ген sacB Bacillus subtilis, который является летальным при экспрессии в клетке, которую выращивают в присутствии приблизительно 5%-10% сахарозы. Либо с контрселекцией, либо без нее, желаемую клетку "Campbell out" можно получить или идентифицировать путем скрининга на желаемую клетку с использованием любого поддающегося скринингу фенотипа, такого как, но, не ограничиваясь ими, морфология колоний, цвет колоний, присутствие или отсутствие устойчивости к антибиотику, присутствие или отсутствие данной последовательности ДНК в полимеразной цепной реакции, присутствие или отсутствие ауксотрофии, присутствие или отсутствие фермента, гибридизация нуклеиновых кислот колоний, скрининг антител и т.д. Термин "Campbell in" и "Campbell out" также можно использовать в качестве глаголов в различных временах для указания на способ или процесс, описанный выше.
Следует понимать, что события гомологичной рекомбинации, которые ведут к "Campbell in" или "Campbell out", могут происходить для диапазона оснований ДНК в гомологичной последовательности ДНК, и, поскольку гомологичные последовательности являются идентичными друг другу по меньшей мере в части этого диапазона, обычно невозможно точно указать, где произошло событие кроссинговера. Иными словами, невозможно точно указать, какая последовательность исходно была из встроенной ДНК, а какая исходно была из хромосомной ДНК. Более того, первая гомологичная последовательность ДНК и вторая гомологичная последовательность ДНК обычно разделены областью частичной негомологии, и именно эта область негомологии остается в хромосоме клетки "Campbell out".
Для практичности в C. glutamicum конкретные первая и вторая гомологичные последовательности ДНК имеют длину по меньшей мере приблизительно 200 пар оснований, и могут иметь длину вплоть до нескольких тысяч пар оснований, однако способ можно адаптировать для более коротких или более длинных последовательностей. Например, длина первой и второй гомологичных последовательностей может находиться в диапазоне приблизительно от 500 до 2000 оснований, и получению "Campbell out" из "Campbell in" способствует обеспечение первой и второй гомологичных последовательностей, имеющих приблизительно одну длину, предпочтительно с отличием менее чем 200 пар оснований и наиболее предпочтительно, более короткой из них, составляющей по меньшей мере 70% длины более длиной в парах оснований. "Способ Campbell In и Out" описан в WO 2007/012078 и Eggeling и Bott (eds) Handbook of Corynebacterium (Taylor and Francis Group, 2005), Chapter 23. Предпочтительные протоколы рекомбинации для C. glutamicum описаны в разделе "Примеры".
Настоящее изобретение более подробно описано с помощью следующих примеров. Следует понимать, что эти примеры представлены только для иллюстративных целей, и их не следует истолковывать как ограничивающие изобретение.
ПРИМЕРЫ
В следующих примерах использовали стандартные способы технологии рекомбинантных ДНК и молекулярной биологии, которые описаны в различных публикациях, например, Sambrook et al. (2001), Molecular Cloning: A Laboratory Manual, 3rd edition, Cold Spring Harbor Laboratory Press, или Ausubel et al. (2007), Current Protocols in Molecular Biology, Current Protocols in Protein Science, edition as of 2002, Wiley Interscience. Если не указано иное, все клетки, реагенты, устройства и наборы использовали согласно инструкциям изготовителей.
Материалы и способы
Бактериальные штаммы
Все штаммы C. glutamicum были получены из ATCC 13032 дикого типа (American Type Culture Collection, Manassas, VA, США) путем стабильной генетической модификации. Для амплификации вектора и метилирования ДНК использовали Escherichia coli DH5α и NM522, соответственно, приобретенные в Invitrogen (Karlsruhe, Германия). Все штаммы приведены в таблице 2.
Бактериальные штаммы, использованные в настоящей работе, для метаболической и генетической инженерии
C. glutamicum дикого типа ATCC 13032 получали в American Type Culture Collection (Manassas, США).
Праймеры и плазмиды
Трансформацию C. glutamicum проводили с помощью встраивающихся векторов pK19 или pClik (фиг.2) (Becker, et al., 2005), соответственно. Исходные векторы имеют размер 4 п.н. и содержат ориджин репликации (ORI) для E. coli, участок множественного клонирования (MCS), маркер устойчивости к канамицину (KanR) и открытую рамку считывания гена sacB, кодирующего левансахаразу B. subtilis. KanR и sacB используют качестве маркеров для положительной селекции по двум событиям рекомбинации (Jager, et al., 1995; Jager, et al., 1992).
Соответствующий фрагмент ДНК для генетической модификации C. glutamicum получали способом ПЦР, и затем встраивали в MCS исходного вектора через участки распознавания выбранных ферментов рестрикции. Последовательности плазмид и праймеров, использованные для конструирования штаммов, приведены в таблицах 3 и 4, соответственно. Для моделирования праймеров и разработки стратегии клонирования использовали программное обеспечение Vector NTI 10.0 (Invitrogen GmbH, Karlsruhe, Германия).
Плазмиды, использованные в настоящей работе
В таблице ниже приведены плазмиды, использованные для конструирования штамма C. glutamicum BS87.
Плазмиды, использованные для конструирования штамма C. glutamicum BS87
Нуклеотидные последовательности генов, приведенных в настоящем описании, также доступны через общедоступные базы данных, например www.ncbi.nlm.nih.gov.
Полный геном Corynebacterium glutamicum был секвенирован, и он доступен под номером доступа GenBank BA000036.3.
Реагенты
Дрожжевой экстракт, говяжий экстракт, триптон, пептон и агар приобретали от Difco Laboratories (Detroit, США). Все другие реагенты были аналитической категории, и они были получены от Sigma, Fluka или Merck. Меченую глюкозу приобретали в Campro Scientific (Veenendaal, The Netherlands). Ферменты рестрикции приобретали в Fermentas (St. Leon-Roth, Германия).
Состав среды
Комплексные среды
Среда LB
Культивирование E. coli на планшетах с агаром или в жидкой культуре проводили в среде LB (Луриа-Бертани). Она содержала на литр: 5 г дрожжевого экстракта, 10 г триптона и 5 г NaCl. Планшеты с агаром приготавливали добавлением 18 г·л-1 агара. Стерилизацию проводили автоклавированием в течение 20 минут при 121°С. При необходимости добавляли канамицин и тетрациклин до конечной концентрации 50 мкг.мл-1 или 12,5 мкг.мл-1, соответственно.
Среда SOC
Регенерацию E. coli DH5α и NM522 после трансформации посредством теплового шока проводили в обогащенной среде SOB (супероптимизированный бульон), состоящей из трех растворов. Раствор 1 содержал 20 г триптона, 5 г дрожжевого экстракта, 0,5 г NaCl и 10 мл KCl (250 мМ) в общем объеме 975 мл и был стерилизован автоклавированием. После охлаждения до комнатной температуры, добавляли 5 мл стерильного MgCl2 (2M) и 20 мл стерильной глюкозы (1M).
Среда CM
Первую предварительную культуру Corynebacterium glutamicum выращивали в среде CM. Для ее приготовления, 10 г пептона, 5 г говяжьего экстракта, 5 г дрожжевого экстракта и 2,5 г NaCl растворяли в 925 мл воды и автоклавировали в течение 20 минут при 121°С. Затем добавляли 25 мл глюкозы (400 г·л-1, стерилизованная фильтрацией, 0,2-мкм Ultrafree-MC, Millipore) и 50 мл мочевины (40 г·л-1, стерилизованная фильтрацией, 0,2 мкм Ultrafree-MC, Millipore). Чашки с агаром получали добавлением 18 г·л-1 агара. Для целей селекции в процессе конструирования штаммов приготавливали агар CMSac добавлением 100 г сахарозы перед автоклавированием. Агар CMKan содержал канамицин в конечной концентрации 50 мкг·мл-1. Канамицин добавляли из исходного раствора 50 мг·мл-1 после охлаждения автоклавированного агара до 50-60°С.
Среда BHI ++
Среду BHI++, используемую для выращивания клеток для электропорации, приготавливали растворением 37 г BHI (сердечно-мозговой экстракт) в 850 мл воды. После автоклавирования (20 мин, 121°С), добавляли 50 мл стерильного 2М (NH4)2SO4 и 100 мл стерильной 40% глюкозы.
Среда BHIS
Для регенерации клеток после электропорации C. glutamicum выращивали в течение 90 мин без селективного давления в среде BHIS. Один литр BHIS содержал 37 г BHI и 250 мл 2М сорбита, который стерилизовали фильтрацией и добавляли после автоклавирования.
Минимальные среды
Для физиологических исследований, включающих анализ метаболических потоков, использовали минимальную среду (таблица 5). Она состояла из различных растворов, которые комбинировали непосредственно перед применением.
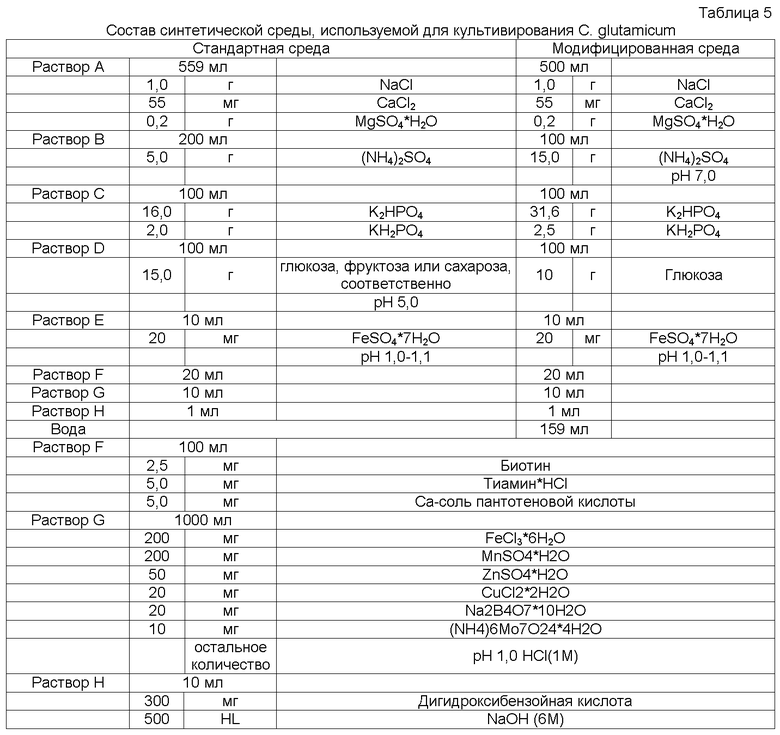
Растворы A-D и воду стерилизовали автоклавированием в течение 20 мин при 121°С. Растворы F-H стерилизовали фильтрацией (0,2-мкм Ultrafree-MC, Millipore). Минимальную среду модифицировали в выбранных экспериментах путем увеличения концентрации аммония и буферной емкости и снижения концентрации субстрата.
Среда для промышленного продуцирования
Периодическую ферментацию с подпиткой в 5-л ферментере Sartorius проводили в комплексной среде на основе патоки, которая была дополнена витаминами и микроэлементами. Состав среды для ферментации с подпиткой представлен в таблице 6. Для приготовления раствора подпитки, 200 г патоки и 800 г глюкозы*H2O растворяли в 1,8 л воды и стерилизовали автоклавированием в течение 20 мин при 121°С. Затем добавляли стерильный раствор 200 мл (NH4)2SO4 (400 г·л-1), 0,8 мл раствора E и 2 мл пеногасителя (пеногаситель Tego KS911, Goldschmidt GmbH, Essen, Германия).
Состав среды для ферментации с подпиткой на основе патоки, используемой для ферментации
Раствор E содержал 300 мг·л-1 биотина, 500 мг·л-1 тиамина*HCl, 2 г·л-1 Ca-соли пантотеновой кислоты и 600 мг·л-1 никотинамида. Раствор FeSO4-цитрат состоял из 20 г·л-1 FeSO4*7H2O и 18,14 г·л-1 цитрата. Растворы A-D и воду стерилизовали автоклавированием в течение 20 мин при 121°С. Раствор E стерилизовали фильтрацией (0,2-мкм Ultrafree-MC, Millipore).
Хранение штаммов
Один мл экспоненциально выращенных клеток из комплексной жидкой культуры (LB для E. coli и CM для C. glutamicum) смешивали с 1 мл стерильного 60% глицерина и хранили при -80°С.
Конструирование штаммов
Выделение нуклеиновых кислот
Выделение ДНК из C. glutamicum
Для выделения очищенной геномной ДНК клетки из чашек с агаром, инкубированные в течение 2 суток при 30°С, собирали с помощью стерильной петли для инокуляции и добавляли в 500 мкл стерильной воды. Разрушение клеток проводили в течение 1 минуты при 30 Гц в Ribolyzer (MM301, Retsch, Haan, Германия) после двукратного добавления стеклянных гранул на кончике шпателя (0,45-0,5 мм) и 700 мкл смеси фенол-хлороформ-изоамиловый спирт (Carl-Roth GmbH, Karlsruhe, Германия). После разделения водной и органической фаз (5 мин, 13000 об./мин., центрифуга 5415R, Eppendorf, Hamburg, Германия), ДНК из водной фазы осаждали добавлением 65 мкл ацетата натрия (3М, pH 5,5) и 1,3 мл этанола (100%) и стадией центрифугирования (10 мин, 13000 об./мин., центрифуга 5415R). Затем супернатант удаляли, и геномную ДНК растворяли в 100 мкл стерильной воды. Концентрацию ДНК определяли с помощью спектрофотометра NanoDrop ND-1000 (Thermo Fisher Scientific, Waltham, США), и геномную ДНК хранили при 4°С. Для быстрого выделения ДНК для целей скрининга в процессе конструирования штаммов, единичные колонии отбирали с помощью стерильной зубочистки и растворяли в 500 мкл стерильной воды, и проводили разрушение клеток, как описано выше. Клеточный дебрис удаляли центрифугированием, и 10 мкл супернатанта использовали для анализа способом ПЦР.
Выделение плазмиды из E. coli
Выделение плазмидной ДНК из Escherichia coli NM522 и DH5α проводили с использованием наборов для выделения ДНК HiSpeed Plasmid Midi Prep Kit (Qiagen, Hilden, Германия) и GFX Micro Plasmid Prep Kit (GE Healthcare, Munich, Германия), соответственно. Выделение плазмид с помощью набора GFX Micro Plasmid Prep проводили с 3 мл ночной культуры DH5α, выращенной в среде LBKan, как описано в руководстве. Для выделения плазмид из NM522 10 мл предварительной культуры (LBTet+Kan) инокулировали одной колонией из чашки с агаром и выращивали в течение 8 ч. Клетки из предварительной культуры использовали для инокуляции основной культуры, которую проводили в 50 мл LBTet+Kan, и выращивали в течение ночи. Сбор клеток и выделение плазмиды проводили согласно инструкции к набору HiSpeed Plasmid Midi Prep. Концентрацию ДНК определяли с помощью NanoDrop и выделенные плазмиды контролировали расщеплением двумя различными ферментами рестрикции и хранили при -20°С Все культивирования проводили во вращающихся флаконах с перегородками на вращающем устройстве (Multitron, Infors AG, Bottmingen, Швейцария) при 37°С и 230 об./мин. с диаметром вращения 5 см.
Полимеразная цепная реакция
Конструирование вставки и проверку штамма проводили способом ПЦР с использованием градиента mastercycler EP (Eppendorf, Hamburg, Германия). Для конструирования вставки и продуктов ПЦР, предназначенных для секвенирования, использовали PWO master (Roche Applied Science, Mannheim, Германия) с функцией точного считывания, в то время как проверку штамма проводили с помощью PCR master (Roche Applied Science, Mannheim, Germany), содержащего Taq-полимеразу. Получение желаемых фрагментов ДНК для конструирования вектора, индукцию делеций генов, внесение точечных мутаций, добавление искусственным образом участков распознавания ферментов рестрикции для направленного лигирования с векторами для трансформации, проводили общепринятыми способами. После каждой реакции продукты ПЦР очищали с использованием набора для очистки GFXTM PCR DNA и Gel Band purification (GE Healthcare, Munich, Германия). Амплификацию проводили при стандартном профиле температур, начиная с начальной стадии денатурации (5 мин при 95°С). За ней следовали 30 циклов денатурации в течение 0,5 мин при 95°С, отжига в течение 0,5 мин и элонгации в течение 1 мин при 72°С. После конечной стадии элонгации в течение 5 мин при 72°С, термоблок охлаждался до 15°С. Температуру отжига корректировали индивидуально согласно праймерам и вычисляли, в зависимости от их содержания GC в соответствии со следующим уравнением:
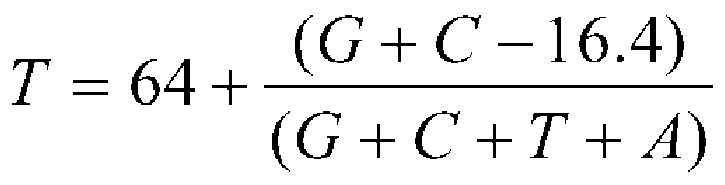 (2)
(2)
Реакции, как правило, проводили в общем объеме 50 мкл. Праймеры добавляли до конечной концентрации 400 нМ. Один мкл очищенной геномной ДНК или плазмидной ДНК добавляли в качестве матрицы. Для ПЦР колоний использовали ДНК-матрицу после быстрого выделения и объем увеличивали до 10 мкл. Отрицательный контроль анализировали без ДНК-матрицы.
Гель-электрофорез
Продукты полимеразной цепной реакции или ферментативного расщепления разделяли электрофорезом в 1% агарозном геле. Электрофорез проводили в 1×буфере TAE при 100-120 В в течение 1-1,5 ч (гель B1A легко сбрасываемый мини гель (easy cast mini gel) или система wide гелей D2, Thermo Scientific, Rochester, США, с Power Pack P25T, Biometra, Gottingen, Германия). Размер полос определяли с помощью маркерной ДНК с интервалом 1 п.н. (O' GeneRulerTM 1 kb DNA Ladder ready-to-use, Fermentas, St. Leon-Roth, Германия). Перед нанесением на гель образцы смешивали с 1/10 объема буфера для нанесения OrangeG. Состав буфера TAE и OrangeG представлены в таблице 7.
Состав буфера для нанесения на гель OrangeG и буфера TAE, используемых для гель-электрофореза
Гели окрашивали с помощью бани с красителем бромидом этидия (0,5 мкг.мл-1 бромида этидия). Детекцию ДНК проводили при УФ-облучении в Gel Ix Imager (Intas, Gottingen, Германия).
Ферментативное расщепление
Для конструирования вектора для трансформации, пустой вектор pClik и вставку, полученную способом осуществляющей слияние ПЦР, расщепляли двумя ферментами рестрикции. Реакцию расщепления вектора проводили в общем объеме 25 мкл, и реакционная смесь содержала 1 мкл каждого фермента, 2,5 мкл буфера для реакции (10×), 5 мкл плазмидной ДНК и 15,5 мкл воды. Расщепление проводили в течение ночи при 37°С или в течение 20 минут, когда использовали ферменты FastDigest (Fermentas, St. Leon-Roth, Германия). Затем фосфатный остаток из линеаризованного вектора удаляли обработкой щелочной фосфатазой (щелочная фосфатаза креветки (SAP), Fermentas) в течение 1 ч при комнатной температуре, чтобы избежать повторного лигирования вектора. Расщепление после выделения плазмиды из NM522 проводили без обработки SAP. Расщепление вставки проводили с помощью 25 мкл продукта осуществляющей слияние ПЦР, 2 мкл каждого фермента, 5 мкл 10× реакционного буфера и 16 мкл воды.
Лигирование ДНК
Для лигирования приготавливали две смеси с различными соотношениями вектор-вставка. Реакцию проводили с набором для лигирования RapidDNA ligation (Roche Applied Science, Mannheim, Германия), и реакционная смесь включала 0,5 мкл или 1 мкл линеаризованного вектора, 2 мкл ДНК-вставки, 2 мкл буфера для разведения, 10 мкл буфера для лигирования, 1 мкл лигазы T4 и 4 мкл или 4,5 мкл воды. Смеси инкубировали в течение 30 мин при комнатной температуре, и затем использовали для трансформации посредством теплового шока E. coli DH5α.
Трансформация
Приготовление компетентных посредством теплового шока E. coli (Inoue, et al., 1990)
Клетки выращивали в одной предварительной культуре и одной основной культуре при 23°С и 230 об./мин. на вращающем устройстве (Multitron, Infors). Предварительную культуру (5 мл среды LB, содержавшей 20 мМ MgCl2) инокулировали единичной колонией с чашки с агаром и выращивали в течение ночи. Для E. coli NM522, несущих плазмиду pTC, добавляли 12,5 мкг·мл-1 тетрациклина. Два мл ночной культуры использовали для инокуляции основной культуры, которую культивировали в 2-л вращающемся флаконе с перегородками с 250 мл среды. При OD600 0,4-0,6 клетки помещали на лед на 10 мин, затем переносили в предварительно охлажденные 50-мл пробирки falcon и центрифугировали в течение 10 мин (5000 об./мин. 4°С, центрифуга 5804R, Eppendorf, Hamburg, Германия). После стадии промывания 40 мл ледяного буфера TB (таблица 8), клетки разводили в 20 мл того же буфера и 1,5 мл ДМСО, помещали на 10 мин на лед и, наконец, разделяли на 220-мкл аликвоты в предварительно охлажденные 1,5-мл пробирки Eppendorf, которые затем подвергали шоковой заморозке на бане этанол-сухой лед. Компетентные клетки хранили при -80°С.
Состав буфера TB, использованного для получения компетентных посредством теплового шока E. coli
Трансформация посредством теплового шока
После размораживания на льду 50 мкл компетентных клеток E. coli смешивали с 3 мкл плазмидной ДНК и инкубировали в течение 30 мин на льду. Тепловой шок применяли в течение 45 с при 45°С в термоблоке (Thermomixer comfort, Eppendorf, Hamburg, Германия). Сразу после этого клетки помещали на лед на 2 минуты. Затем добавляли 900 мкл среды SOC и проводили регенерацию клеток без селекционного давления в течение 60 мин при 37°С в термосмесителе при 500 об./мин. (Thermomixer comfort, Eppendorf). Затем для целей селекции клетки высевали на чашки с LBKan или LBTet+Kan и инкубировали в течение 1 суток при 37°С. Единичные колонии отбирали. Выделение плазмиды и расщепление вектора проводили, как описано выше. Штаммы, содержавшие желаемый вектор, хранили при -80°C.
Получение электрокомпетентных C. Glutamicum
Клетки из исходных культур в глицерине распределяли на планшетах с агаром и инкубировали в течение 48 ч при 30°С. Первую предварительную культуру (5 мл среды BHI++) инокулировали одной колонией, выращивали в течение ночи при 30°С и 230 об./мин. во вращающем устройстве (диаметр вращения 5 см, Multitron, Infors AG, Bottmingen, Швейцария) и использовали в качестве посевного материала для основной культуры. Это осуществляли в 50 мл среды BHI++ и инокуляцию проводили при OD660 0,3. При OD660 1,2-1,5 клетки собирали (7500×g, 3 мин, 4°С в стерильных, предварительно охлажденных 50-мл пробирках falcon, промывали 4 раза ледяным 10% глицерином, и затем разводили в 4 мл глицерина на грамм сырой массы клеток (CWW). Суспензию клеток использовали для тестирования времени электропорации (tE). При необходимости (tE<8 мс) суспензию клеток далее разводили 10% глицерином.
Транфмормация посредством электропорации
Электропорацию C. glutamicum проводили с 200 мкл суспензии клеток и 2,5 мкг и 5 мкг плазмидной ДНК, соответственно при 2,5 кВ, 25 мкФ и 400 Ом в 2-мм кюветах для электропорации с BioRad gene pulser XcellTM (Biorad, Hercules, США). В качестве отрицательного контроля использовали реакционную смесь без ДНК. Сразу после электрического импульса добавляли 1 мл среды BHIS, клеточную суспензию переносили в пробирки eppendorf и инкубировали без селекционного давления в течение 90 мин (30°С, 500 об./мин., термосмеситель comfort, Eppendorf, Hamburg, Германия). Поскольку pClik и pK19 не могут реплицироваться в C. glutamicum, наследственный перенос вектора требует встраивания плазмидной ДНК в геном посредством гомологичной рекомбинации. Селекцию трансформантов после первого события рекомбинации проводили на чашках с агаром CMKan в течение 2 суток при 30°С. Единичные колонии из CMKan использовали для второй рекомбинации.
Вторая рекомбинация
Для модификации C. glutamicum без маркера, вектор, введенный электропорацией, удаляли путем второго события рекомбинации и селекции на сахарозе (Jager, et al., 1992). Для этой цели единичные колонии после первого события рекомбинации выращивали в течение ночи в 5 мл среды BHI++ (30°С, 230 об./мин.) и 100 мкл и 200 мкл, соответственно, клеток в разведении 1:1000 высевали на CMSac. Вследствие летальности экспрессии sacB в процессе роста C. glutamicum только на сахарозе, могут расти рекомбинантные штаммы, в которых плазмида, включающая sacB, была удалена посредством второго события гомологичной рекомбинации. Единичные колонии, выращенные на CMSac, отбирали зубочисткой и наносили штрихами на растровые чашки CMSac и CMKan. От десяти до пятнадцати случайным образом выбранных чувствительных к канамицину и толерантных к сахарозе клонов далее исследовали в отношении желаемой модификации.
Скрининг и проверка штамма
В зависимости от модификации, скрининг и проверка штамма включают анализ способом ПЦР, определение активности фермента или секвенирование. Скрининг замены промоторов и делеции генов обычно проводили способом ПЦР и проверяли путем определения удельной активности фермента. Однако скрининг для замены инициирующего кодона проводили с помощью тестов ферментативной активности, и перспективные клоны с измененной удельной активностью более подробно исследовали анализом последовательности.
Характеризация штамма
Периодическое культивирование в минимальной среде
Культивирование во вращающихся флаконах проводили при 30°С и 230 об./мин. на вращающем устройстве (диаметр вращения 5 см, Multitron, Infors AG, Bottmingen, Швейцария). Первые предварительные культуры (50 мл среды в 500-мл колбе с перегородками) инокулировали единичными колониями с чашек с агаром, культивированных в течение 2 суток, и инкубировали в течение 8 ч. Клетки собирали центрифугированием (8800×g, 2 мин, 4°С, промывали стерильным 0,9% NaCl и использовали в качестве инокулята для второго предварительного культивирования. Затем это осуществляли в 50 мл минимальной среды в 500 мл колбах с перегородками, содержавших источник углерода для последующей основной культуры. Культивирование основных культур проводили в трех повторах (25 мл в 250-мл колбах с перегородками или 200 мл в 2-л колбах с перегородками) с использованием клеток из второй предварительной культуры в качестве инокулята. Мониторинг растворенного кислорода в колбах проводили с помощью иммобилизованных сенсорных пятен, содержавших флуорофор с O2-зависимым временем затухания люминесценции (Wittmann, et al., 2003). Уровень растворенного кислорода составлял выше 20% насыщения в ходе каждого культивирования, чтобы обеспечить достаточное поступление кислорода к клеткам. Значение pH находилось в диапазоне 7,0±0,2 в процессе всего культивирования. Для экспериментов с использованием радиоактивных индикаторов, естественным образом меченную глюкозу заменяли 99% [1-13C] глюкозой, 99% [6-13C] глюкозой или эквимолярной смесью естественным образом меченной и 99% [U-13C] глюкозы. Ферментацию в минимальной среде проводили в 250-мл биореакторе (Meredos, Bovenden, Германия) с рабочим объемом 100 мл при 30±0,1°С pH 7,0±0,1 и 800 об./мин. Уровень растворенного кислорода составлял более 20% насыщения в процессе всего культивирования. Скорость аэрации поддерживали при 100 мл·мин-1 с помощью контроллера потока массы (Brooks Instruments, Veenendaal, Нидерланды). Состав газов для аэрации и отходящих газов измеряли в интерактивном режиме с помощью квадрупольного масс-спектрометра (Omnistar, Balzers, Vaduz, Лихтенштейн) с интервалами 2 мин.
Периодическая ферментации с подпиткой
Периодическую ферментацию с подпиткой в среде для продуцирования на основе патоки проводили в биореакторе Sartorius с контролирующим элементом Biostat B plus в 5-л емкости. Скорость аэрации была установлена на 2,5 л·мин-1 посредством встроенного контроллера потока газа. Для мониторинга pH использовали pH-электрод (Mettler Toledo, Giessen, Германия). Для поддержания pH постоянным на уровне 6,9, добавляли 25% NH4OH с помощью перистальтической насосной системы контролирующего элемента Biostat. Добавленный объем определяли гравиметрически (Lab Balance Cupis, Sartorius, Gottingen, Германия). Растворенный кислород определяли с использованием pO2-электрода (Mettler Toledo, Giessen, Германия) и поддерживали при насыщении 20% путем варьирования скорости мешалки. Такой контроль осуществляли с помощью программного обеспечения для контроля над процессами BaseLab (BASF SE, Ludwigshafen, Германия). Для калибровки pO2-электрода, среду, в свою очередь, аэрировали синтетическим воздухом и азотом, соответственно. Температуру доводили до 30°С с использованием охлаждающей рубашки. CO2 и O2 в отходящем газе анализировали с помощью масс-спектрометра. Мониторинг всех данных процесса проводили интерактивно и регистрировали с интервалом 5 мин с помощью программного обеспечения для контроля над процессами BaseLab. Подпитку начинали при сигнале pO2. Изменение растворенного кислорода со скоростью, превышающей 10% мин-1, активировало перистальтический насос для подпитки контролирующего элемента Biostat. Дозировку контролировали с помощью программного обеспечения для контроля над процессом посредством гравиметрически определенного количество добавленной подпитки.
Ферментацию начинали в объеме 1 л. Его инокулировали клетками, выращиваемыми в течение 6 ч при 30°С и 230 об./мин. на вращающем устройстве (диаметр вращения 5 см, Multitron, Infors AG, Bottmingen, Швейцария) в среде CM. Клетки из 600-мл предварительной культуры собирали центрифугированием (8800×g, 2 мин, RT), разводили 50 мл стерильного NaCl (0,9%) и использовали для инокуляции. Периодическую ферментацию с подпиткой начинали при оптической плотности 10. Фазу подпитки начинали через 9 ч и ферментацию проводили в течение всего 30 ч.
Общее описание поэтапного конструирования векторов и трансформации бактерий
Конструирование вектора для трансформации No 4 (pClik_lysC T311I ), несущего модифицированный ген аспартаткиназы
Вектор No 4 (pClik_lysCT311I), указанный в таблице 3 (выше) конструируют с использованием праймеров P1-P4 (SEQ ID NO:35-38) для внесения точечной мутации в генетический фон, например, C. glutamicum дикого типа ATCC 13032, путем аллельной замены.
Для получения штамма C. glutamicum, несущего точечную мутацию, приводящую к аминокислотной замене треонина изолейцином в положении 311 LysC-полипептида дикого типа (SEQ ID NO:2), амплифицировали ORF, кодирующую ген аспартаткиназы (lysC, SEQ ID NO:1).
Для этого выделенную геномную ДНК C. glutamicum ATCC 13032 подвергают амплификации способом ПЦР с использованием подходящих праймеров, вносящих модификацию нуклеиновой кислоты в положении 932 гена lysC дикого типа.
Аминокислотная последовательность и нуклеотидная последовательность модифицированного lysC/LysC представлены в SEQ ID NO:3 и 4, соответственно.
Более подробно, два отдельных фрагмента ДНК сначала амплифицируют способом ПЦР с использованием пары праймеров P1/P2 (ПЦР1) и P3/P4 (ПЦР2), соответственно. ПЦР1 и ПЦР2 используют для внесения точечной мутации в положение 932 в нуклеотидной последовательности гена lysC с помощью P2 и P3, соответственно. Праймеры P1 и P4 используют для внесения участков распознавания для ферментов рестрикции, например, для XhoI и SpeI.
В ПЦР1 получают фрагмент ДНК размером 1467 п.н., который содержит последовательность размером приблизительно 500 п.н. выше lysC и приблизительно 946 п.н. гена lysC, включая точечную мутацию в положении 932, и участок распознавания для XhoI.
В ПЦР2 получают фрагмент ДНК размером 449 п.н., который содержит часть гена lysC, включающую нуклеотидную замену в положении 932 гена lysC, последовательность размером приблизительно 150 п.н. ниже гена lysC с искусственно добавленным участком распознавания SpeI.
На следующей стадии, эти два фрагмента ДНК подвергают слиянию в ПЦР3 с использованием комбинации праймеров P1 и P4, а также очищенной ДНК, продуцированной в ПЦР1 и ПЦР2, соответственно, в качестве матриц. Полученный фрагмент ДНК имеет размер 1916 п.н. и содержит последовательность размером приблизительно 500 п.н. выше lysC, полную ORF lysC, включая точечную мутацию в положении 932 и последовательность размером приблизительно 150 п.н. ниже lysC, а также искусственно добавленные участки распознавания для XhoI и SpeI.
В альтернативном способе получают желаемую векторную конструкцию с использованием праймеров P1 и P4 и ДНК-матрицы, которая уже содержит желаемую нуклеотидную замену в гене lysC.
После стадий амплификации, продукт ПЦР расщепляют с использованием ферментов рестрикции XhoI и SpeI и встраивают в вектор pClik, который затем используют для трансформации E. coli DH5α. После культивирования трансформированных E. coli DH5α, вектор выделяют и используют для трансформации E. coli NM522. Этот штамм уже содержит плазмиду pTC (таблица 3, выше), экспрессирующий вектор для ДНК-метилтрансферазы C. glutamicum. Совместное существование обеих плазмид в E. coli NM522 приводит к амплификации правильно метилированного вектора для трансформации.
Плазмиду pClik_lysCT311I, содержащую правильно метилированный ген lysCT311I, используют для трансформации штаммов дикого типа ATCC 13032 с использованием электропорации. Первый и второй процессы рекомбинации проводят, как описано выше. Выбранные случайным образом штаммы исследуют с помощью анализа последовательности. Положительно идентифицированные рекомбинанты, ниже называемые штаммом 1, используют для последующих генетических модификаций.
Конструирование вектора для трансформации No 5 (pK19_2× ddh), содержащего две копии гена ddh, кодирующего диаминопимелатдегидрогеназу
Вектор No. 5 (см. таблицу 3) конструируют с использованием праймеров, обозначенных как P5-P8 (SEQ ID NO:39-42), для внесения второй копии гена ddh в генетический фон описанного выше штамма 1 путем аллельной замены.
Для амплификации гена ddh дикого типа (SEQ ID NO:5), кодирующего диаминопимелатдегидрогеназу (SEQ ID NO:6), выделенную геномную ДНК, например C. glutamicum ATCC 13032, подвергают амплификации способом ПЦР с использованием праймеров P5, P6, P7 и P8.
В ПЦР1 полную последовательность ddh, а также последовательность размером 245 п.н. выше гена ddh и последовательность размером 84 п.н. ниже гена dhh, амплифицируют с использованием P5 и P6. Полученный продукт ПЦР имеет размер 1305 п.н. и содержит участок распознавания для EcoRI, который вносится с помощью P5.
В ПЦР2 полную последовательность ddh амплифицируют вместе с последовательностью размером 245 п.н. выше гена ddh и последовательностью размером 84 п.н. ниже гена ddh с использованием праймеров P7 и P8. Полученный фрагмент ДНК имеет размер 1305 п.н. и содержит участок распознавания для SalI, который вносится с помощью P8.
На следующей стадии два очищенных продукта ПЦР, полученных в ПЦР1 и ПЦР2, соответственно, используют в качестве матриц и подвергают слиянию в ПЦР3 с использованием комбинации праймеров P5 и P8.
Полученный фрагмент ДНК имеет размер 2530 п.н. и содержит полные гены ddh, каждый из которых фланкирован вышележащей частью размером 245 п.н. и нижележащей частью размером 84 п.н., а также участки распознавания для EcoRI и SalI (см. SEQ ID NO:7)
После стадий амплификации, продукт ПЦР расщепляли с использованием ферментов рестрикции EcoRI и SalI и встраивали в вектор pK19, который использовали для трансформации E. coli DH5α. После стадии культивирования вектор выделяли и использовали для трансформации E. coli NM522, несущий плазмиду pTC, обеспечивающую амплификацию правильно метилированного вектора для трансформации.
Плазмиду pK19_2×ddh, содержащую правильно метилированные гены ddh, используют для трансформации описанного выше штамма 1 с использованием электропорации. Первый и второй процессы рекомбинации проводят, как описано выше. Выбранные случайным образом штаммы исследуют с помощью анализа ПЦР и путем определения ферментативной активности. Положительно идентифицированные рекомбинанты, ниже называемые штаммом 2, используют для последующих генетических модификаций.
Конструирование вектора для трансформации No 6 (pClik_Δpck), несущего частичную делецию PEP-карбоксикиназы
Для конструирования вектора для трансформации No 6 (pClik_Δpck; таблица 3, выше) использовали праймеры P9-P12 (SEQ ID NO:43-46) для внесения разрушенной формы гена pck (SEQ ID NO:10), кодирующей укороченную PEP-карбоксикиназу (SEQ ID NO:11) в генетический фон описанного выше штамма 2 путем аллельной замены.
Для внесения частичной делеции в ген pck (SEQ ID NO:8), кодирующий карбоксикиназу PEP (SEQ ID NO:9), геномную ДНК C. glutamicum ATCC 13032 подвергают амплификации способом ПЦР с использованием праймеров P9, P10, P11 и P12.
С использованием праймеров P9 и P10 амплифицируют фрагмент ДНК размером 420 т.п.н., содержащий нуклеотиды 111-509 гена pck, в ПЦР1. Также используют P10 для искусственного добавления 21 п.н., которые комплементарны 21 п.н. праймера P11. Полученная последовательность содержит природный участок распознания для BamHI.
В ПЦР2 амплифицируют фрагмент ДНК размером 417 п.н., который содержит нуклеотиды 1437-1833 гена pck с использованием пары праймеров P11 и P12. P11 используют для искусственного добавления 21 п.н., которые комплементарны 21 п.н. P10. Для искусственного добавления участка распознавания для BamHI используют P12.
На следующей стадии два очищенных фрагмента ДНК, полученных в ПЦР1 и ПЦР2, соответственно, подвергают слиянию в PCR3 с использованием комбинации праймеров P9 и P12. Полученный фрагмент ДНК имеет размер 807 п.н. и содержит нуклеотиды 111-509 и 1437-1833 гена pck, 21 дополнительную пару оснований, добавленных с помощью праймеров P11 и P12, а также два участка распознавания для BamHI.
После стадий амплификации, продукт ПЦР расщепляют с использованием фермента рестрикции BamHI и встраивают в вектор pClik, который используют для трансформации E. coli DH5α. Затем вектор выделяют из трансформированных E. coli DH5α и используют для трансформации E. coli NM522, как описано выше, приводящей к амплификации правильно метилированного вектора для трансформации. Правильно метилированную ДНК, находящуюся в плазмиде pClik_Δpck, трансформировали в указанный выше штамм 2 с использованием электропорации. Первый и второй процессы рекомбинации проводят, как описано выше. Выбранные случайным образом штаммы исследуют анализом способом ПЦР и саузерн-блоттингом. Положительно идентифицированные рекомбинанты, ниже называемые штаммом 3, используют для последующих генетических модификаций.
Конструирование вектора для трансформации No 7 (pK19_P sod dap B), содержащего модифицированную дигидродипиколинатредуктазу
Вектор для трансформации No 7 (pK19_Psod dapB; см таблицу 3, выше) получают с использованием праймеров, которые специфично связываются с sod-промотором, геном dapB (SEQ ID NO:12), кодирующем дигидродипиколинатредуктазу (SEQ ID NO:13), и выше гена dapB для внесения модификации промотора в генетический фон описанного выше штамма 3 путем аллельной замены.
Более подробно, для замены промотора в гене dapB геномную ДНК C. glutamicum ATCC 13032 подвергают амплификации способом ПЦР с использованием праймеров, которые специфично связываются с последовательностью sod-промотора, геном dapB и выше гена dapB, соответственно. Соответствующие последовательности праймеров, обозначенные как P13, P14, P15, P16, P17 и P18, представлены в SEQ ID NO:47-52.
В ПЦР1 амплифицируют фрагмент ДНК размером приблизительно 200 п.н., который содержит последовательность sod-промотора. Этого достигают с помощью P13 и P14, посредством которых на 3'-конец добавляют последовательность, перекрывающуюся с геном dapB.
В ПЦР2 амплифицируют часть гена dapB, включающую инициирующий кодон и 5'-часть гена. Этого достигают с помощью праймеров P15 и P16, искусственно добавляющих перекрывающуюся последовательность с sod-промотором на 5'-конце инициирующего кодона.
На следующей стадии два очищенных фрагмента ДНК, полученных в ПЦР1 и ПЦР2, соответственно, подвергают слиянию в ПЦР3 с последовательностями праймеров, специфичными к sod и dapB.
В ПЦР4 амплифицируют часть вышележащей последовательности гена dapB. Этого достигают с использованием праймеров P17 и P18, которые искусственно добавляют последовательность, перекрывающуюся с sod-промотором, на 3'-конце.
На следующей стадии очищенные фрагменты ДНК, полученные в ПЦР3 и ПЦР4, соответственно, подвергают слиянию с использованием праймеров P17 и P16. Эти праймеры фланкируют конечный фрагмент ДНК и искусственно добавляют участки распознавания для ферментов рестрикции, которые пригодны для лигирования вектор-вставка.
Для лигирования вектор-вставка продукт ПЦР и вектор разрезают с помощью выбранных ферментов рестрикции, затем лигируют и используют для трансформации E. coli DH5α. После стадии культивирования вектор выделяют и используют для трансформации E. coli NM522, содержащих плазмиду pTC, обеспечивающую амплификацию правильно метилированного вектора для трансформации.
Правильно метилированную плазмиду pK19_Psod dapB используют для трансформации описанного выше штамма 3 с использованием электропорации. Первый и второй процессы рекомбинации проводят, как описано выше. Выбранные случайным образом штаммы исследуют с помощью анализа способом ПЦР. Положительно идентифицированные рекомбинанты, ниже называемые штаммом 4, используют для последующих генетических модификаций.
Конструирование вектора для трансформации No 8 (pK19_2× lysA и argS), содержащего две копии диаминопимелатдекарбоксилазы
Вектор для трансформации No 8 (pK19_2× argSlysA; таблица 3, выше) получают с помощью праймеров, которые специфично связываются выше гена argS и с геном argS, кодирующим аргинил-тРНК-синтетазу, а также с геном lysA и ниже гена lysA с получением второй копии гена lysA вместе с последовательностью argS, которую встраивают в штамм 4 путем аллельной замены. Кластер генов argSlysA представлен в SEQ ID NO:15. Аминокислота LysA представлена в SEQ ID NO:17.
Для внесения второй копии генов lysA и argS, геномную ДНК, например, из C. glutamicum ATCC 13032, подвергают амплификации способом ПЦР. Для этого каждую копию кластера argSlysA конструируют с 3 отдельно амплифицированными фрагментами ДНК, охватывающими полный кластер. Эти фрагменты получают в отдельных ПЦР. Последовательности праймеров, используемые для этой цели, представляют собой P19, P20, P21, P22, P23, P24, P25 и P26 (SEQ ID NO:53-60).
В ПЦР1 с использованием праймеров P19 и P20 5'-часть гена argS амплифицируют вместе с приблизительно 500 п.н. выше гена argS. 5'-часть включает промоторную последовательность argS. Праймер P19 также используют для искусственного добавления участка распознавания BamHI. Праймер P20 расположен в природном участке распознавания для EcoRI. Полученный фрагмент ДНК имеет размер 1410 п.н.
С использованием праймеров P21 и P22 3'-часть гена argS вместе с 5'-частью гена lysA амплифицируют в ПЦР2. Праймер P21 расположен в природном участке распознавания для EcoRI. Праймер P22 расположен в природном участке распознавания для SalI. Полученный фрагмент ДНК имеет размер приблизительно 935 п.н.
В ПЦР3 3'-часть гена lysA амплифицируют вместе с приблизительно 80 п.н. нижележащей последовательности lysA с использованием праймеров P23 и P24. Праймер P23 расположен в природном участке распознавания для SalI. Праймер P24 используют для добавления участка распознавания для XbaI. Полученный фрагмент ДНК имеет размер приблизительно 1290 п.н. Затем фрагменты ДНК связывают посредством участков распознавания для EcoRI и SalI. Связанные фрагменты ДНК из ПЦР1, ПЦР2 и ПЦР3 представляют собой первую копию кластера argSlysA. На следующей стадии конструируют второй кластер argSlysA.
В ПЦР4 с использованием P25 и P20 5'-часть гена argS амплифицируют вместе с приблизительно 500 п.н. выше гена argS. Эта вышележащая часть включает промоторную последовательность argS. P25 используют для искусственного добавления участка распознавания для XbaI. P20 расположен на природном участке распознавания для EcoRI. Полученный фрагмент ДНК имеет размер приблизительно 1374 п.н.
В ПЦР5 с использованием праймеров P21 и P22 3'-часть гена argS амплифицируют вместе с 5'-частью гена lysA. P21 расположен на природном участке распознавания для EcoRI, P22 расположен на природном участке распознавания для SalI. Полученный фрагмент ДНК имеет размер приблизительно 935 п.н.
В PCR6 с использованием праймеров P23 и P26 3'-часть гена lysA амплифицируют вместе с приблизительно 70 п.н. нижележащей последовательности lysA. P23 расположен на природном участке распознавания для SalI. P26 используют для добавления участка распознавания для HindIII. Полученный фрагмент ДНК имеет размер приблизительно 1256 п.н.
Затем фрагменты ДНК связывают через участки распознавания для EcoRI и SalI. Связанные фрагменты ДНК из ПЦР4, ПЦР5 и ПЦР6 представляют собой вторую копию кластера argSlysA.
Копию 1 и копию 2 argSlysA, соответственно, связывают через участок распознавания для XbaI, который добавлен с помощью праймеров P24 и P25. Связанные копии (SEQ ID NO:16) встраивают в вектор для трансформации pK19 через участки распознавания BamHI и HindIII, которые добавлены с помощью праймеров P19 и P26, соответственно. Полученный вектор используют для трансформации E. coli DH5α. После стадии культивирования вектор выделяют и используют для трансформации E. coli NM522, несущих плазмиду pTC, обеспечивающую амплификацию правильно метилированного вектора для трансформации. Правильно метилированную конструкцию, находящуюся в плазмиде pK19_2×argSlysA, используют для трансформации штамма 4 с использованием электропорации. Первый и второй процессы рекомбинации проводят, как описано выше. Выбранные случайным образом штаммы исследуют анализом способом ПЦР. Положительно идентифицированные рекомбинанты, ниже называемые штаммом 5, использовали для последующих генетических модификаций.
Конструирование вектора для трансформации No 9 (pClik_P sod LysC ), несущего ген аспартаткиназы с sod-промотором
Конструирование вектора для трансформации No 9 (pClik_Psod lysC; таблица 3, выше) с использованием праймеров, которые специфично связывают sod-промотор, ген lysC и выше гена lysC, и внесение модификации промотора и одновременной замены инициирующего кодона с GTG на ATG проводили в генетическом фоне штамма 5 путем аллельной замены. Последовательность ДНК lysC, несущего модификацию инициирующего кодона, sod-промотор и замену T311I, представлена в SEQ ID NO:3.
Для замены промотора гена lysC геномную ДНК, например, C. glutamicum ATCC 13032, подвергают амплификации способом ПЦР с использованием праймеров, которые специфично связывают последовательность sod-промотора, гена lysC и выше гена lysC, соответственно. Соответствующие праймеры обозначают как P27, P28, P29, P30, P31 и P32 (SEQ ID NO:61-66).
В ПЦР1 амплифицируют фрагмент ДНК размером приблизительно 200 п.н., который содержит последовательность sod-промотора. Это осуществляют с использованием праймеров P27 и P28, посредством которых на 3'-конец искусственно добавляется последовательность, перекрывающаяся с геном lysC. P28 содержит желаемую нуклеотидную замену в положении инициирующего кодона lysC, так что обеспечивается замена инициирующего кодона с GTG на ATG.
В ПЦР2 амплифицируют часть гена lysC, включающую инициирующий кодон и 5'-часть гена. Это осуществляют с использованием праймеров P29 и P30, посредством которых на 5'-конец рядом с инициирующим кодоном, добавляют последовательность, перекрывающуюся с sod-промотором. Кроме того, P29 содержит желаемое нуклеотидное изменение для замены инициирующего кодона. На следующей стадии два полученных и очищенных фрагмента ДНК, продуцированные в ПЦР1 и ПЦР2, подвергают слиянию в PCR3 с помощью специфических к sod и lysC последовательностей праймеров, ранее используемых в ПЦР1 и ПЦР2, соответственно.
В ПЦР4 часть вышележащей последовательности гена lysC амплифицируют с помощью P31 и P32, посредством чего на 3'-конец искусственно добавляют последовательность, перекрывающуюся с sod-промотором.
На следующей стадии фрагменты ДНК из PCR3 и PCR4 подвергают слиянию с использованием очищенной ДНК из ПЦР3 и ПЦР4 в качестве матрицы, а также праймеров P31 и P30.
Концевые праймеры (P31 и P30) используют для искусственного добавления участков распознавания для ферментов рестрикции, которые пригодны для лигирования вектор-вставка. Для этого продукт ПЦР и вектор разрезают с помощью выбранных ферментов рестрикции, лигируют, b затем используют для трансформации E. coli DH5α. После стадии культивирования вектор выделяют и используют для трансформации E. coli NM522, несущего плазмиду pTC, обеспечивающую амплификацию правильно метилированного вектора для трансформации. Правильно метилированную конструкцию, несущую pClik_PsodlysC, используют для трансформации штамма 5 с использованием электропорации. Первый и второй процессы рекомбинации проводят, как описано выше. Выбранные случайным образом штаммы исследуют анализом способом ПЦР. Положительно идентифицированные рекомбинанты, ниже называемые штаммом 6, используют для последующих генетических модификаций.
Конструирование вектора для трансформации No 10 (pClik_hom V59A ), несущего мутантную гомосериндегидрогеназу
Вектор для трансформации No 10 (pClik_hom V59A, таблица 3, выше) конструируют с использованием праймеров P33-P36 (SEQ ID NO:67-70) для внесения точечной мутации в ген hom (SEQ ID NO:18) в генетический фон штамма 6 путем аллельной замены.
Для получения штамма C. glutamicum, несущего точечную мутацию, приводящую к аминокислотной замене с валина на аланин в положении 59 полипептида Hom (SEQ ID NO:19), амплифицируют ORF, кодирующую гомосериндегидрогеназу.
Для этого геномную ДНК, например, C. glutamicum ATCC 13032, подвергают амплификации способом ПЦР с использованием подходящих праймеров, приводящих к модификации нуклеиновой кислоты в положении 176 гена hom дикого типа. Этого достигают с использованием P33, P34, P35 и P36. Нуклеотидная последовательность и полипептидная последовательность модифицированного hom/Hom представлена в SEQ ID NO:20 и 21, соответственно.
Более подробно, два отдельных фрагмента ДНК сначала амплифицируют с использованием праймеров P33/P34 (ПЦР1) и P35/P36 (ПЦР2), соответственно.
ПЦР1 и ПЦР2, соответственно, используют для внесения точечной мутации в положение 176 в нуклеотидной последовательности гена hom с помощью праймеров 34 и 35.
Когда используют эту комбинацию праймеров, в ПЦР1 получают фрагмент ДНК размером 686 п.н., который содержит последовательность размером приблизительно 500 п.н. выше гена hom и приблизительно 186 п.н. ниже гена hom, включая точечную мутацию в положении 176.
В ПЦР2 получают фрагмент ДНК размером 1054 п.н., содержащий часть гена hom, которая содержит нуклеотидную замену в положении 176 гена hom.
На следующей стадии эти два фрагмента ДНК подвергают слиянию в PCR3 с использованием комбинации праймеров P33 и P36 и очищенной ДНК, полученной в ПЦР1 и ПЦР2 в качестве матриц, соответственно. Полученный фрагмент ДНК имеет размер 1718 п.н. и содержит последовательность размером приблизительно 500 п.н. выше hom и неполный ген hom размером приблизительно 1218 п.н., включающий точечную мутацию в положении 176.
Альтернативно, желаемую конструкцию получают с использованием праймеров P33 и P36 и матрицы ДНК, которая уже содержит желаемую нуклеотидную замену в гене hom. Когда используют праймеры P33-P36, амплифицированный фрагмент ДНК природным образом содержит участок для XmaI и NheI.
После стадий амплификации продукт ПЦР расщепляют с использованием ферментов рестрикции XmaI и NheI и полученный фрагмент встраивают в вектор pClik, который ранее был расщеплен посредством XmaI и SpeI. SpeI и NheI образуют совместимые липкие концы в процессе расщепления ДНК. Затем векторы используют для трансформации E. coli DH5α. После стадии культивирования вектор выделяют и используют для трансформации E. coli NM522, несущих плазмиду pTC, обеспечивающую амплификацию правильно метилированного вектора для трансформации. Правильно метилированный ген hom V59A, содержащийся в плазмиде pClik_hom V59A, используют для трансформации штамма 6 с использованием электропорации. Первый и второй процессы рекомбинации проводили, как описано выше. Случайным образом выбранные штаммы исследовали с помощью анализа последовательности. Положительно идентифицированные рекомбинанты, ниже называемые штаммом 7, использовали для последующих генетических модификаций.
Конструирование вектора для трансформации No 11 (pClik__pyc P458S ), несущего мутантную пируваткарбоксилазу
Вектор для трансформации No 11 (pClik_pyc P458S, таблица 3, выше) конструируют с помощью праймеров P37-P40, которые пригодны для внесения точечной мутации в ген pyc (SEQ ID NO:22) в генетический фон штамма 7 путем аллельной замены.
Для получения штамма C. glutamicum, несущего точечную мутацию, приводящую к аминокислотной замене пролина серином в положении 458 полипептида Pyc (SEQ ID NO:23), амплифицировали ORF, кодирующую пируваткарбоксилазу.
Для этого выделенную геномную ДНК, например, C. glutamicum ATCC 13032 подвергают амплификации способом ПЦР с использованием подходящих праймеров, которые приводят к модификации нуклеиновой кислоты в положении 1372 гена pyc дикого типа. Этого достигают с помощью праймеров, обозначенных как P37, P38, P39 и P40 (SEQ ID NO:71-74). Нуклеотидная последовательность и полипептидная последовательность модифицированного pyc/Pyc представлена в SEQ ID NO:24 и 25, соответственно.
Более подробно, продуцируют два отдельных фрагмента ДНК с использованием праймеров P37/P38 (ПЦР1) и P39/P40 (ПЦР2), соответственно. В этом случае, ПЦР1 и ПЦР2 используют для внесения точечной мутации в положение 1372 в нуклеотидную последовательность гена pyc с помощью P38 и P39, соответственно.
В ПЦР1 получают фрагмент ДНК размером 745 п.н., который содержит неполный ген pyc, включающий точечную мутацию в положении 1372 гена.
В ПЦР2 получают фрагмент ДНК размером 744 п.н., содержащий часть гена pyc, включающую нуклеотидную замену в положении 1372 гена pyc.
На следующей стадии эти два фрагмента ДНК, полученные в ПЦР1 и ПЦР2, соответственно, подвергают слиянию в ПЦР3 с использованием комбинации праймеров P37 и P40. Полученный фрагмент ДНК имеет размер 1461 т.п.н. и содержит неполную последовательность гена pyc, содержащую желаемую точечную мутацию в положении 1372 гена.
Альтернативно, желаемую конструкцию получают с использованием праймеров P37 и P40 и ДНК-матрицы, которая уже содержит желаемую нуклеотидную замену в гене pyc. Амплифицированный фрагмент ДНК можно встраивать в вектор pClik с помощью соответствующих участков распознавания для ферментов рестрикции, таких как MluI и SalI.
Затем вектор, несущий мутантную пируваткарбоксилазу, используют для трансформации E. coli DH5α. После стадии культивирования вектор выделяют и используют для трансформации E. coli NM522, несущих плазмиду pTC, обеспечивающую амплификацию правильно метилированного вектора для трансформации. Правильно метилированный ген pyc P458S, содержащийся в плазмиде pClik_pycP458S, используют для трансформации штамма 7 путем электропорации. Первый и второй процессы рекомбинации проводят, как описано выше. Выбранные случайным образом штаммы исследуют с помощью анализа последовательности. Положительно идентифицированные рекомбинанты, ниже называемые штаммом 8, используют для последующих генетических модификаций.
Конструирование вектора для трансформации No 12 (pClik_P sod pyc ), несущего пируваткарбоксилазу с sod-промотором
Вектор для трансформации No 12 (pClik_Psod pyc) конструируют с помощью праймеров, которые специфично связывают sod-промотор, ген pyc и выше гена pyc, для внесения замены промотора в генетический фон штамма 8 путем аллельной замены. Последовательность pyc с sod-промотором представлена в SEQ ID NO:24. Эта последовательность также содержит аминокислотную замену в положении 458, описанную выше.
Для замены промотора гена pyc геномную ДНК, например, C. glutamicum ATCC 13032 подвергали амплификации способом ПЦР с использованием праймеров, которые специфично связывают последовательность sod-промотора и гена pyc, а также выше гена pyc, соответственно. Соответствующие последовательности праймеров представлены последовательностях, кодирующих праймеры, обозначенные как P41, P42, P43, P44, P45 и P46 (SEQ ID NO:75-80).
В ПЦР1 амплифицируют фрагмент ДНК размером приблизительно 200 п.н., который содержит последовательность sod-промотора. Этого достигают с использованием P41 и P42, посредством которых на 3'-конец искусственно добавляют последовательность, перекрывающаяся с геном pyc.
В ПЦР2 амплифицируют часть гена pyc, включающую инициирующий кодон, и 5'-часть гена. Это осуществляют с использованием P43 и P44, посредством которых на 5'-конец рядом с инициирующим кодоном искусственно добавляется последовательность, перекрывающаяся с sod-промотором.
На следующей стадии два полученных фрагмента ДНК подвергают слиянию в ПЦР3 с последовательностями праймеров, специфичными к sod и pyc, использованными в ПЦР1 и ПЦР2, соответственно, и очищенной ДНК из PCR1 и ПЦР2.
В ПЦР4 амплифицируют часть вышележащей последовательности гена pyc. Это можно осуществлять с помощью праймеров P45 и P46, посредством которых на 3'-конец искусственно добавляется последовательность, перекрывающаяся с sod-промотором.
После этого фрагменты ДНК, полученные в ПЦР3 и ПЦР4, подвергают слиянию с использованием очищенной ДНК из ПЦР3 и ПЦР4 в качестве матриц и праймеров P45 и P44. Праймеры P45 и P44 используют для искусственного добавления участков распознавания для ферментов рестрикции, которые способствуют лигированию вектор-вставка. Для этого продукт ПЦР и вектор разрезают выбранными ферментами рестрикции, затем лигируют и используют для трансформации E. coli DH5α. После стадии культивирования вектор выделяют и используют для трансформации E. coli NM522, несущих плазмиду pTC, обеспечивающую амплификацию правильно метилированного вектора для трансформации. Правильно метилированную конструкцию, содержащуюся в плазмиде pClik_Psod pyc, используют для трансформации штамма 8 путем электропорации. Первый и второй процессы рекомбинации проводят, как описано выше. Выбранные случайным образом штаммы исследуют с помощью анализа способом ПЦР. Положительно идентифицированные рекомбинанты, ниже называемые штаммом 9, используют для последующих генетических модификаций.
Конструирование вектора для трансформации No 13 (pClik_icd A1G ), содержащего модифицированный ген изоцитратдегидрогеназы
Вектор для трансформации No 13 (pClik_icd A1G; таблица 3, выше) конструируют с помощью праймеров P47-P50 для внесения замены инициирующего кодона в генетический фон штамма 9 путем аллельной замены.
Для получения штамма C. glutamicum, несущего замену инициирующего кодона ATG→GTG в гене icd, кодирующем изоцитратдегидрогеназу (SEQ ID NO:27), амплифицируют ORF, кодирующую изоцитратдегидрогеназу (icd, SEQ ID NO:26).
Для этого ранее выделенную геномную ДНК C. glutamicum ATCC 13032 подвергают амплификации способом ПЦР с использованием подходящих праймеров, которые вносят модификацию нуклеиновой кислоты в положение инициирующего кодона гена icd. Используют праймеры P47, P48, P49 и P50 (SEQ ID NO:81-84). Нуклеотидная последовательность модифицированного гена icd представлена в SEQ ID NO:28.
Более подробно, два отдельных фрагмента ДНК сначала амплифицируют с использованием праймеров для ПЦР P47/P48 (ПЦР1) и P49/P50 (ПЦР2), соответственно. ПЦР1 и ПЦР2 используют для внесения точечной мутации в положение инициирующего кодона в нуклеотидной последовательности гена icd с использованием P48 и P49. P47 использовали для добавления участка распознавания XhoI, и P50 использовали для добавления участка распознавания MluI.
В ПЦР1 получали фрагмент ДНК размером 520 п.н., который содержал последовательность размером приблизительно 502 п.н. выше icd и последовательность размером приблизительно 18 п.н. гена icd, включающую точечную мутацию в положении инициирующего кодона, и участок распознавания XhoI.
В ПЦР2 получают фрагмент ДНК размером 520 п.н., содержащий часть гена icd, включающую нуклеотидную замену в положении инициирующего кодона гена icd, последовательность размером приблизительно 7 п.н. выше гена icd и участок распознавания MluI.
На следующей стадии два полученных фрагменты ДНК подвергают слиянию в ПЦР3 с использованием комбинации праймеров P47 и P50 и очищенной ДНК из ПЦР1 и ПЦР2, соответственно, в качестве матриц. Полученный фрагмент ДНК имеет размер 1014 п.н. и содержит последовательность размером приблизительно 500 п.н. выше icd, замену инициирующего кодона и приблизительно 500 п.н. гена icd, а также участки распознавания XhoI и MluI. После стадий амплификации продукт ПЦР расщепляют с использованием ферментов рестрикции XhoI и MluI и встраивают в вектор pClik, который затем используют для трансформации E. coli DH5α. После стадии культивирования вектор выделяют и используют для трансформации E. coli NM522, несущих плазмиду pTC, обеспечивающую амплификацию правильно метилированного вектора для трансформации.
Правильно метилированный ген icd A1G, содержащийся в плазмиде pClik_icd A1G, используют для трансформации штамма 9 с использованием электропорации. Первый и второй процессы рекомбинации проводят, как описано выше. Выбранные случайным образом штаммы исследуют с помощью анализа ферментативной активности и анализа последовательности. Положительно идентифицированные рекомбинанты, ниже называемые штаммом 10, используют для последующих генетических модификаций.
Конструирование вектора для трансформации No 14 (pClik_P eftu fbp ), содержащего фруктозо-1,6-бисфосфатазу с промотором eftu
Вектор No 14 (pClik_Peftu fbp); таблица 3, выше) конструируют с использованием праймеров, специфично связывающихся с промотором eftu, геном fbp и выше гена fbp. Вектор используют для внесения замены промотора в генетический фон описанного выше штамма 10 путем аллельной замены.
Для замены промотора гена fbp дикого типа (SEQ ID NO:29, демонстрирующая последовательность гена fbp и нативного промотора), кодирующего фруктозо-1,6-бисфосфатазу (SEQ ID NO:30), геномную ДНК C. glutamicum ATCC 13032 подвергают амплификации способом ПЦР. Используют праймеры, которые специфично связывают последовательность промотора eftu и гена fbp (нуклеотидная последовательность fbp, рекомбинированного с промотором eftu, представлена в SEQ ID NO:31), а также выше гена fbp соответственно. Соответствующие последовательности праймеров представлены в SEQ ID NO:85-90, кодирующих праймеры, обозначенные как P51, P52, P53, P54, P55 и P56.
В ПЦР1 с использованием P51 и P52 амплифицируют фрагмент ДНК размером приблизительно 200 п.н., который содержит последовательность промотора eftu. В ПЦР2 амплифицируют часть гена fbp, включающую инициирующий кодон и 5'-часть гена. Этого достигают с использованием P53 и P54, посредством которых на 5'-конец рядом с инициирующим кодоном искусственно добавляется последовательность, перекрывающаяся с промотором eftu.
На следующей стадии два очищенных фрагмента ДНК, полученных в ПЦР1 и ПЦР2, соответственно, подвергают слиянию в ПЦР3 со специфическими промоторами eftu и fbp, использованными в ПЦР1 и ПЦР2, соответственно.
В ПЦР4 амплифицируют часть вышележащей последовательности гена fbp. Этого достигают с помощью праймеров P55 и P56, посредством которых на 3'-конец искусственно добавляется последовательность, перекрывающаяся с промотором eftu.
На следующей стадии фрагменты ДНК из ПЦР3 и ПЦР4 подвергают слиянию с использованием праймеров P55 и P54, а также очищенной ДНК из ПЦР3 и ПЦР4 в качестве матрицы. Праймеры P55 и P54 также пригодны для добавления искусственных участков распознавания для ферментов рестрикции, таких как MluI и SalI, которые можно использовать для лигирования вектор-вставка. Для этого продукт ПЦР и вектор расщепляют выбранными ферментами рестрикции, затем лигируют и используют для трансформации E. coli DH5α. После стадии культивирования вектор выделяют и используют для трансформации E. coli NM522, содержащих плазмиду pTC, обеспечивающую амплификацию правильно метилированного вектора для трансформации. Затем правильно метилированную конструкцию, содержащуюся в плазмиде pClik_Peftu fbp используют для трансформации штамма 10 с использованием электропорации. Первый и второй процессы рекомбинации проводят, как описано выше. Выбранные случайным образом штаммы исследуют анализом способом ПЦР. Положительно идентифицированные рекомбинанты, ниже называемые штаммом 11, используют для последующих генетических модификаций.
Конструирование вектора для трансформации No 15 (pClik_P sod tkt ), содержащего оперон tkt и sod-промотор
Вектор для трансформации No 15 (pClik_Psod tkt) конструируют с использованием праймеров, которые специфично связываются с sod-промотором, геном tkt и выше гена tkt, обеспечивающих замену промотора в генетическом фоне штамма 11 путем аллельной замены.
Подробно, для осуществления замены промотора оперона tkt (SEQ ID NO:32, которая демонстрирует последовательность промотора tkt и гена tkt, который является первым геном в опероне tkt), геномную ДНК, например, C. glutamicum ATCC 13032 подвергают амплификации способом ПЦР с использованием праймеров, которые специфично связываются с последовательностью sod-промотора и геном tkt, который является первым геном оперона tkt и кодирует транскетолазу (SEQ ID NO:33), а также выше гена tkt, соответственно. Соответствующие последовательности праймеров представлены в последовательностях праймеров, обозначенных как P57, P58, P59, P60, P61 и P62 (SEQ ID NO:91-96).
В ПЦР1 амплифицируют фрагмент ДНК размером приблизительно 200 п.н., который содержит последовательность sod-промотора. Это осуществляют с использованием P57 и P58. Рекомбинация последовательности sod-промотора и гена tkt представлена в SEQ ID NO:34.
В ПЦР2 амплифицируют часть гена tkt, включая инициирующий кодон и 5'-часть гена. Это осуществляют с использованием P58 и P59, посредством которых на 5-конец рядом с инициирующим кодоном искусственно добавляется последовательность, перекрывающаяся с sod-промотором.
На следующей стадии два фрагмента ДНК, полученных в ПЦР1 и ПЦР2, очищали и использовали в качестве матриц в ПЦР3 для слияния обоих фрагментов со специфическими для sod и tkt последовательностями праймеров, использованных в ПЦР1 и ПЦР2.
В ПЦР4 амплифицируют часть вышележащей последовательности гена tkt. Это осуществляют с помощью праймеров P61 и P62, посредством которых на 3'-конец искусственно добавляют перекрывающуюся последовательность с sod-промотором.
На следующей стадии фрагменты ДНК из ПЦР3 и ПЦР4 подвергают слиянию с использованием очищенной ДНК, полученной в ПЦР3 и ПЦР4 в качестве матриц и P61 и P60 в качестве праймеров. P61 и P60 используют для искусственного добавления участков распознавания для ферментов рестрикции, таких как MluI и SalI, которые пригодны для лигирования вектор-вставка. Для этого продукт ПЦР и вектор разрезают выбранными ферментами рестрикции, затем лигируют и используют для трансформации E. coli DH5α. После стадии культивирования вектор выделяют и используют для трансформации E. coli NM522, содержащего плазмиду pTC, обеспечивая амплификацию правильно метилированного вектора для трансформации.
Затем правильно метилированную конструкцию, содержащуюся в плазмиде pClik_Psod tkt используют для трансформации штамма 11 с использованием электропорации. Первый и второй процессы рекомбинации проводят, как описано выше. Выбранные случайным образом штаммы исследуют анализом способом ПЦР. Положительно идентифицированные рекомбинанты далее называются штаммом 12.
Конструирование специализированного гиперпродуцента лизина
В предпочтительном варианте осуществления настоящего изобретения успешно использовали различные стратегии для рациональной оптимизации продуцирования лизина из C. glutamicum.
Используя в качестве примера исходные штаммы C. glutamicum BS1 и C. glutamicum BS87, исследовали пользу нескольких мишеней ключевых путей биосинтеза лизина, метаболизма NADPH, поступления предшественника и образования CO2. Имея задачу создать гиперпродуцент дикого типа, идентифицированные модификации комбинировали в одном штамме.
Исходя из C. glutamicum дикого типа ATCC 13032, осуществляли только полезные модификации для минимизации какого-либо вредоносного побочного эффекта, имеющегося в штаммах для промышленного продуцирования.
Конструирование штаммов и эффективность продуцирования
Первой стадией в направлении создания гиперпродуцента было освобождение аспартаткиназы от ингибирования по принципу отрицательной обратной связи лизином и треонином, путем введения аминокислотной замены T311I (Becker, et al., 2005). Это дополняли путем описанной выше сверхэкспрессии ddh. Единое внесение этих двух модификаций и дополнительная оптимизация среды создавала продуцент лизина, который уже продуцировал 125 ммоль·лизина (моль·глюкоза)-1.
Чтобы избежать других препятствий в пути биосинтеза лизина, в дополнение к ddh сверхэкспрессировали дигидродипиколинатредуктазу (dapB), аспартаткиназу (lysC) и диаминопимелатдекарбоксилазу (lysA). Все эти ферменты являются частью общего пути биосинтеза лизина. Таким образом, эффект на продуцирование лизина должен быть независимым от распределения потоков между ветвью сукцинилазы и дегидрогеназы.
Дальнейшую оптимизацию штамма проводили путем осуществления нескольких дополнительных генетических модификаций. Они включали замену природной гомосериндегидрогеназы мутантным вариантом, обладающим аминокислотной заменой V59A. Эта мутация увеличивает продуцирование лизина путем снижения потока углерода в направлении каскада треонина, который прямо конкурирует с биосинтезом лизина (Ohnishi et al., 2002). Для обеспечения достаточного поступления оксалоацетата для образования лизина, дополнительные модификации были сфокусированы на предоставлении предшественника. Это предполагало сверхэкспрессию и модификацию основного анаплеротического фермента пируваткарбоксилазы (Ohnishi, et al., 2002; Peters-Wendisch, et al., 2001) и делецию потребляющей OAA реакционной PEP-карбоксикиназы (Petersen, et al., 2001). Таким образом, полученный штамм C. glutamicum BS87 содержал 9 модификаций в биосинтезе лизина и поступлении предшественника (фиг.32).
Из сравнения с его более ранним штаммом-предшественником C. glutamicum BS222, обладающим только регуляторной точечной мутацией в гене lysC и второй копией гена ddh, становится очевидным, что дополнительные модификации в C. glutamicum BS87 обеспечивают только небольшое улучшение продуцирования лизина (фиг.18). Следовательно, стратегия инженерии, которая сфокусирована только на конкретной части метаболизма, не пригодна для создания эффективного продуцирующего штамма. Однако эта стратегия явно демонстрирует историю оптимизации штамма в направлении улучшенного продуцирования лизина путем метаболической инженерии. Таким образом, ключевые каскады цикла TCA и метаболизма NADPH в качестве основных реакций в концепции оптимизированного продуцента лизина были практически не приняты во внимание. В этом отношении, настоящая работа выявила генетические мишени, которые прекрасно дополняются предшествующими стратегиями для преодоления этих ограничений, тем самым, обеспечивая продвижение в направлении конструирования улучшенного продуцирующего штамма. Таким образом, следующая стадия в направлении создания предполагаемого гиперпродуцента, была сфокусирована на цикле TCA. Как описано выше, ослабление изоцитратдегидрогеназы (icd att) в генетическом фоне C. glutamicum BS87 значительно увеличивало продуцирование лизина путем эффективного перенаправления потока с цикла TCA в сторону анаплеротического карбоксилирования. Что касается продуцирования лизина, эта стратегия инженерии имеет преимущество вследствие увеличенного поступления оксалоацетата, а также сниженной потери углерода посредством образования CO2. Последующую модификацию способами инженерии метаболизма NADPH проводили с помощью двух различных стратегий. Сначала сверхэкспрессировали фермент глюконеогенеза фруктозо-1,6-бисфосфатазу, который, как показано выше, вносит значительный вклад в увеличенное поступление NADPH (Becker et al., 2005). Как показано выше, этот подход хорошо дополняется усиленной экспрессией глюкозо-6-фосфатдегидрогеназы. Для преодоления дальнейших ограничений в PPP вследствие ограниченной способности транскетолазы и трансальдолазы с самого начала, была осуществлена сверхэкспрессия G6PDH одновременно со сверхэкспрессией транскетолазы и трансальдолазы путем активации полного оперона tkt через sod-промотор, как описано выше. В ответ на замену промотора значительно увеличивалась удельная активность G6PDH, транскетолазы и трансальдолазы. Концепция стратегии инженерии проиллюстрирована на фиг.32. Польза от модификаций становится очевидной из сравнения выхода конверсии углерода в полученной генеалогии (фиг.18).
Начиная с C. glutamicum дикого типа ATCC 13032, последующее осуществление благоприятных модификаций приводило к гиперпродуценту лизина на основе штамма дикого типа. В настоящей работе впервые описан такой полный рациональный штамм, в котором учитываются все каскады. Важным было первоначальное внесение модификации в аспартаткиназу для преодоления ингибировании по принципу обратной связи этого ключевого регуляторного фермента. Значительная польза в виде практически 50% улучшения вследствие сверхэкспрессии ddh явно показала, что общая способность пути биосинтеза лизина в генетическом фоне C. glutamicum BS1 (lysC T311I) сильно ограничивала продуцирование лизина. Однако дальнейшая модификация каскада лизина, а также модификация способами инженерии поступления предшественника, которая была описана в предшествующих исследованиях (Ohnishi, et al., 2002; Peters-Wendisch, et al., 2001; Petersen, et al., 2001), имела, в данном случае, только умеренный успех. Это явно указывает на то, что существуют другие препятствия в центральном метаболизме C. glutamicum, которые ухудшают продуцирование лизина. Таким образом, в настоящем исследовании интерес в направлении дальнейшей модификации способами инженерии продуцирования лизина был смещен на другие части промежуточного метаболизма. Успех этой концепции отражался непосредственно ответом на ослабление изоцитратдегидрогеназы (icd att). Внесение единичной нуклеотидной замены в инициирующий кодон icd увеличило выход лизина с 141 ммоль·моль-1 в C. glutamicum BS87 вплоть до 200 ммоль·моль-1 в соответствующем мутанте icd, что отражает увеличение на 40%. Последующая сверхэкспрессия фруктозо-1,6-бисфосфатазы, которая, как ранее было показано, приводит к эффективному перенаправлению потоков в направлении предоставляющего NADPH PPP (Becker et al., 2005), далее усиливала продуцирование на 18% (фиг.18). Конечная внесенная модификация в описанную в настоящем описании генеалогию включала замену промотора оперона tkt. Это значительно увеличивало активность ферментов G6PDH, TKT и TAL, кодируемых этим опероном, что, в свою очередь, приводило к улучшению на 13% выхода лизина. Преимущество вследствие сверхэкспрессии fbp и оперона tkt явно демонстрирует, что недостаточное поступление NADPH строго ограничивает продуцирование лизина в C. glutamicum, и что стратегии инженерии, нацеленные на увеличение потока PPP, являются необходимыми для цели создания эффективного гиперпродуцирующего лизин штамма. Дополнительное преимущество усиленной экспрессии генов пентозофосфатного пути наблюдали в отношении поведения роста. По сравнению с его предшественником C. glutamicum BS242 (Peftu fbp), проявляющим удельную скорость роста 0,23 ч-1, мутант tkt рос со скоростью 0,32 ч-1. Более высокий выход конверсии углерода (YLys/S=26%) в комбинации с улучшенным поведением C. glutamicum BS244 увеличил эффективность продуцирования этого штамма, поскольку это обеспечивает эффективное продуцирование лизина за укороченное время ферментации. Путем осуществления последних трех модификаций достигали общего увеличения продуцирования лизина, составляющего почти 90%. Это является примечательной, а также неожиданной пользой вследствие только трех генетических изменений. Таким образом, можно заключить, что такие стратегии инженерии частично имеют преимущество перед ранее осуществленными мутациями, например, удалением препятствий в биосинтезе лизина.
Характеристики продуцирования исследуемых штаммов также тестировали при культивировании во вращающихся флаконах для экспоненциально растущих клеток в минимальной среде. В этих условиях наилучший продуцент C. glutamicum BS244 уже проявлял примечательный выход лизина 26%, хотя постановка эксперимента, включающая культивирование во вращающемся флаконе в минимальной среде при низких концентрациях субстрата, определенно ограничивает их продуктивность. Таким образом, было актуально исследовать эффективность продуцирования гиперпродуцента лизина в процессе ферментации с подпиткой на типичной промышленной среде для продуцирования, поскольку эта стратегия культивирования наиболее близка промышленному продуцированию. Более того, она позволяет более реалистичную оценку возможностей нового гиперпродуцента, особенно по сравнению классически получаемыми продуцирующими штаммами. Наилучший продуцент C. glutamicum BS244(Psod tkt) исследовали в промышленных условиях, включающих периодическую ферментацию с подпиткой на комплексной среде на основе патоки.
Эффективность продуцирования в промышленных условиях ферментации
Профиль культивирования периодической ферментации с подпиткой для C. glutamicum BS244 представлен на фиг.19. Продуцирование лизина начиналось рано и концентрация лизина в культуральном супернатанте постоянно увеличивалась в течение 30 ч вплоть до неожиданного высокого конечного титра 120 г·л-1. Основное увеличение, главным образом, достигалось на фазе подпитки, которую начинали сразу после потребления сахара, предоставленного в среде для периодической ферментации (100 г·л-1). В качестве сигнала для автоматизированной подпитки в ходе процесса использовали сигнал, основанный на растворенном кислороде (pO2). Насыщение O2 в ферментере контролировали скоростью мешалки и поддерживали постоянным на уровне 20%. Ограничение углерода в среде приводило к немедленному и быстрому увеличению pO2, которое активировало насос для подпитки, когда увеличение pO2 превышало 10% мин-1. Раствор для подпитки на основе патоки и глюкозы, дополнительно обогащали сульфатом аммония для обеспечения достаточного поступления аммония для продуцирования лизина. Посредством этой стратегии концентрацию сахара в фазе подпитки поддерживали на уровне ниже 10 г·л-1.
Концентрация биосмассы с течением времени, на которую указывала оптическая плотность, явно отличалась от концентрации лизина (фиг.34). В ходе стационарной фазы, оптическая плотность увеличивалась на коэффициент, равный пяти, в то время как на фазе подпитки наблюдали только пороговое увеличение. Более того, концентрация биомассы не увеличивалась до конца периода культивирования, но достигала максимума через 24 ч.
Более тщательное исследование характеристик продуцирования штамма C. glutamicum BS244 показало, что процесс ферментации может быть далее разделен. Как представлено на фиг.35, различные фазы могут отличаться, исходя из достигнутого выхода лизина. На фазе наилучшего продуцирования, обозначенную в данном случае фазой подпитки 2, гиперпродуцент лизина обладал выходом 55%. Другой характеристикой этой фазы является практически не изменяющаяся концентрация биомассы. Таким образом, потребленный сахар эффективно направляется на путь биосинтеза лизина. В начале процесса ферментации выход лизина был низким. Этот интервал, включающий стационарную фазу и фазу подпитки 1, может быть четко описан, как основная фаза роста, характеризующаяся обширным образованием биомассы (фиг.34). Интересно, что на данной фазе был достигнут выход лизина 25%, который хорошо соответствует выходу лизина, достигнутому в экспериментах с культивированием во вращающихся флаконах в ходе экспоненциальной фазы роста.
Гиперпродуцент лизина, созданный в данной работе, представляет собой наилучший продуцирующий штамм на основе дикого типа, описанный до настоящего времени. Учитывая конечный титр лизина, а также выход углерода и выход в единицу времени, он является явно лучшим, чем описанные рационально созданные штаммы (Ikeda et al., 2006). Имея конечный титр лизина 120 г·л-1 и выход конверсии углерода вплоть до 55%, этот штамм даже проявляет характеристики продуцирования, которые находятся на уровне максимального предела классически получаемых штаммов с описанными выходами углерода 40-50% (Leuchtenberger et al., 2005) и титрами лизина 80-120 г·л-1 (Anastassiadis, 2007). Однако такая эффективность продуцирования классических продуцентов, главным образом, связана с длительным временем ферментации вплоть до 100 ч (Anastassiadis, 2007), которое значительно ухудшает выход в единицу времени. Это, как правило, связано с плохим ростом, вызываемом рядом вредоносных вторичных мутаций, которые накапливаются в процессе разработки штамма вследствие неспецифического мутагенеза. Путем единственно осуществления исключительно набора только из 12 благоприятных модификаций, таких нежелательных побочных действий можно было избежать в продуцирующем штамме на основе дикого типа. Быстрый рост и, таким образом, укороченное время ферментации, составляющее только 30 ч, приводит к примечательно высокому выходу в единицу времени, составляющему 4 г·л-1·ч-1, который значительно превышает выход в единицу времени, составляющий 2,1 г·л-1·ч-1, достигаемый в ходе периодической ферментации с подпиткой классическими продуцентами (Hirao et al., 1989). Продуктивность, полученная в данном случае, может быть еще увеличена дополнительной модификацией способами генетической инженерии или усовершенствованием технологических действий. В этом отношении, особенно оптимизированная стратегия подпитки может значительно улучшить продуктивность (Hirao et al., 1989; Ikeda, 2003).
Гиперпродуцент лизина на основе дикого типа по настоящему изобретению демонстрирует возможность системной митаболической инженерии в качестве способа оптимизации штаммов. Штамм C. glutamicum BS244 является превосходным подтверждением концепции и подтверждением ценности полного рационального продуцирующего штамма, который является весьма привлекательным для промышленного применения.
Аналитические способы
Концентрация клеток
Взятие образцов
Для определения оптической плотности из вращающихся флаконов отбирали образцы объемом 1 мл в стерильных условиях с помощью пипетки и переносили их в пробирку eppendorf. Для гравиметрического определения концентрации клеток отбирали образцы объемом 10-15 мл с помощью стерильной одноразовой исследовательской пипетки (Sarstedt, Numbrecht, Германия) и переносили в предварительно высушенные 15-мл пробирки falcon. Точный объем культуры определяли на аналитических весах (CP225D, Sartorius, Gottingen, Германия). Первичные фракции и образцы во время культивирования в биореакторе отбирали с помощью шприца через трехходовой кран и затем переносили в пробирку falcon.
Количественное определение
Концентрацию клеток определяли фотометрически (Libra S11, Biochrome, Cambridge, Великобритания, и NovaspecII, Pharmacia Biotech, Little Chalfont, Великобритания) при 660 нм в 1,5-мл кюветах из полистирола (Plastibrand, Wertheim, Германия) с использованием воды в качестве эталона. При необходимости суспензию клеток разводили на аналитических весах (CP225D, Sartorius, Gottingen, Германия) до получения оптической плотности от 0,05 до 0,3. Все измерения проводили в двух повторах. Для получения корреляции между оптической плотностью и сухой массой клеток (CDM), концентрацию клеток, кроме того, определяли гравиметрически (CP225D, Sartorius, Gottingen, Германия). Для этого 15-мл пробирки falcon сушили до постоянной массы при 80°С и использовали для сбора клеток из 15 мл культурального бульона центрифугированием (10 мин, 9800×g, Biofuge stratos, Heraeus, Hanau, Германия). Супернатант осторожно удаляли, чтобы избежать потери клеточного материала. Клетки промывали три раза водой и затем сушили при 80°С до постоянной массы. Корреляцию определяли для трех штаммов, каждый с тремя биологическими повторами. Полученная корреляция между OD660 (Libra S11, Biochrome, Cambridge, Великобритания) и CDM представлена на фиг.3. Соответствующий коэффициент корреляции определяли путем линейной регрессии до CDM=0,255×OD (приведенный в граммах на литр).
Аминокислоты
Взятие образцов
Супернатант, используемый для количественного определения аминокислот, получали отделением биомассы на стадии центрифугирования (13000×g, 5 мин, 4°С для пробирок eppendorf, 8500×g, 5 мин, 4°С для пробирок falcon). Образцы периодической ферментации с подпиткой, кроме того, стерилизовали фильтрацией (0,2-мкм фильтр Minisart, Sartorius, Gottingen, Германия) для обеспечения полного удаления биомассы, поскольку ферментационный бульон обладал очень высокой концентрацией биомассы, которая не удалялась надлежащим образом центрифугированием.
Количественное определение
Для определения аминокислот после культивирования в минимальной среде, культуральный супернатант разводили 1:10 α-аминомасляной кислотой (237,18 мкм) в качестве внутреннего стандарта. Анализ проводили ВЭЖХ (Agilent Series 1100, Agilent Technology, Waldbronn, Германия). Протокол включал доколоночное получение производных с o-фтальдиальдегидом (OPA) и разделение на колонке C18 (Gemini5u, Phenomenex, Aschaffenburg, Германия) с градиентом элюента A (40 мМ NaH2PO4, pH 7,8) и элюента B (45% метанол, 45% ацетонитрил, 10% вода) при 40°С (Kromer et al., 2005). Разделение всех протеиногенных аминокислот осуществляли по временному профилю стандартного способа, описанного выше. В случаях, когда супернатант содержал только лизин, а также небольшие количества глутамата и глицина, градиент модифицировали для снижения времени измерения (таблица 9).
Профиль градиента для элюента A и элюента B для разделения и количественного определения методом ВЭЖХ на колонке Gemini5u (Phenomenex, Aschaffenburg, Германия)
Концентрацию лизина в образцах из периодической ферментации с подпиткой определяли в разведенном 1:20 супернатанте в виде лизин-HCl способом ВЭЖХ (Agilent Series 1100, Agilent Technology, Waldbronn, Германия) с использованием RI-детектора. Разделение проводили на колонке Ionospher 5C (100×3,0 мм; Chrompack, Engstingen, Германия) при 40°С и скорости потока 1,5 мл·мин-1. Подвижная фаза состояла из 1,80 мМ цитрата, 4,88 мМ этилендиамина и 20 мМ 2,3-дигидроксисукцината в 5% метаноле и 95% воде.
Сахара и органические кислоты
Взятие образцов
Супернатант, используемый для количественного определения сахаров и органических кислот, получали, как описано выше, с помощью стадии центрифугирования и фильтрации.
Количественное определение
Супернатант для культивирования в минимальной среде разводили 1:10 и использовали для определения субстратов: глюкозы и фруктозы, и побочных продуктов: трегалозы, глицерина, DHA, а также органических кислот, методом ВЭЖХ (Kontron Instruments, Neufahrn, Германия). Разделение проводили на колонке Aminex HPX-87H (300×7,8 мм; Bio-Rad, Hercules, США) при 45°С с 5 мМ H2SO4 в качестве подвижной фазы и скорости потока 0,5 мл·мин-1. Для детекции использовали индекс рефракции (сахара, глицерин) и УФ-поглощение при 210 нм (органические кислоты, DHA). Для количественного определения сахарозы температуру колонки снижали до 15°С. Концентрацию элюента увеличивали до 10 мМ H2SO4. Альтернативно, глюкозу количественно определяли с помощью биохимического анализатора (YSI 2700 Select, Kreienbaum, Langenfeld, Германия).
Концентрацию сахара и органических кислот в периодической ферментации с подпиткой определяли в неразбавленных образцах супернатанта методом ВЭЖХ (Agilent Series 1100, Agilent Technology, Waldbronn, германия). Использовали колонку Aminex HPX-87H (300×7,8 мм; Bio-Rad, Hercules, США) с предколонкой Cation H при 30°С с 5 мМ H2SO4 в качестве подвижной фазы при скорости потока 0,5 мл·мин-1. Концентрацию сахара в растворе для подпитки определяли в разведении 1:10. Для детекции использовали индекс рефракции.
Массовые изотопомеры в ГХ-МС
Взятие образцов
Клетки собирали на экспоненциальной фазе роста (13000 об./мин., Biofuge fresco, Heraeus, Hanau, Германия), промывали два раза водой и хранили при -20°С. Супернатант получали путем разделения клеток на стадии центрифугирования и хранили по отдельности при -20°С.
Количественное определение
Количество клеток, равное приблизительно 1 мг CDM, разводили 50 мкл HCl (6M) и белок клеток гидролизовали в течение 24 ч при 105°С. Гидролизаты нейтрализовали 6М NaOH, и клеточный дебрис удаляли фильтрацией (Ultrafree-MC centrifugal Filter Devices, Amicon, Bioseparations, Bedford, США). Затем определяли паттерн мечения методом ГХ-МС, как описано ранее (Wittmann et al., 2002). ГХ-МС состоял из газового хроматографа HP 6890 (Hewlett Packard, Paolo Alto, Californien) с колонкой HP-5-MS (5% фенилметилсилоксан-дифенилполисилоксан; 60 м×0,251 мм×0,25 мкм, Agilent, Waldbronn, Германия) и гелием в качестве газа-носителя. Детекцию осуществляли с помощью квадрупольного детектора MS 5973 (Agilent). Приготовление образцов включало лиофилизацию и преобразование в производное. Лиофилизированные гидролизаты растворяли в 50 мкл диметилформамида (ДМФА+0,1% пиридин) и 50 мкл N-метил-N-трет-бутилдиметилсилилтрифторацетамида (MBDSTFA, Macherey-Nagel, Duren, Германия) и затем инкубировали в течение 30 мин при 80°С. При необходимости соли удаляли центрифугированием перед измерением. Идентификацию аминокислот проводили по времени удерживания и паттерну фрагментации с использованием стандарта аминокислот. Аминокислоты, преобразованные в производное с MBDSTFA, показывают типичный паттерн фрагментации, включающий фрагменты масс [M-57], [M-85] и [M-159], причем [M-57] включает полный углеродный остов аминокислоты. Отделение аминокислотного остатка приводит к образованию фрагмента с массой 302, содержащего атомы углерода C1 и C2 соответствующей аминокислоты. Фрагменты масс, использованные для определения потока, приведены и описаны в таблице 10.
Распределение массовых изотопомеров трегалозы определяли в 50 мкл лиофилизированного супернатанта для культивирования. Преобразование в производное осуществляли с использованием 50 мкл метоксиламина (25 мг·мл-1 метоксиламин в пиридине) и 50 мкл BSTFA (Macherey-Nagel) в течение 30 мин при 80°С (Kiefer et al., 2004).
Массовые фрагменты протеиногенных аминокислот в качестве производных TBDMS, используемых для оценки потоков. Указанная масса относится к фрагменту M0, содержащему только немеченые массовые изотопомеры C, O, H, N, S и Si
Позиционные изотопомеры согласно ЯМР
Взятие образцов
Клетки выращивали, как описано выше, в двух предварительных культурах и одной основной культуре. Естественным образом меченную глюкозу заменяли [6-13C] глюкозой. Супернатант получали центрифугированием (13000 об./мин., Biofuge fresco, Heraeus, Hanau, Германия) и сушили в токе азота.
Количественное определение
Нестабильные протоны заменяли дейтерием лиофилизацией два раза в 3 мл 99,9% D2O (Eurisotop, Saint-Aubain Cedex, Франция), и конечный образец суспендировали в 600 мкл 100 мМ DCl (Eurisotop). Все спектры 1D и 2D ЯМР получали на спектрометре Avance 500 МГц (Bruker, Wissembourg, Cedex, Франция), оборудованном 5-мм z-градиентным зондом BBI. Данные получали и обрабатывали с использованием программного обеспечения TOPSPIN 1.3. Температура составляла 298°K. 1D протонные спектры регистрировали с использованием количественных условий. Для полного восстановления сигнала релаксационная задержка между сканированиями составляла 30 с и угол импульса составлял 30°. Ширина сектора составляла 5000 Гц и время получения данных составляло 1,6 с. Получали всего 16 сканограмм. Селективное возбуждение осуществляли с помощью сформированных импульсов. Диапазон возбуждения составлял 60 Гц, за исключением протонов, для которых он составлял 100 Гц вследствие большей ширины мультиплета. Последовательность DANTE-Z с 1D 13C-развязкой состояла из восьми последовательных сканограмм резонансных импульсов Inversion BURP (I-BURP), за которыми следовали восемь последовательных сканограмм нерезонансных импульсов I-BURP (Geen and Freeman, 1991). Этот цикл повторяли четыре раза. После селективного возбуждения использовали градиент продувки 0,7 Гс·мм-1. Последовательность DPFGSE с 1D 13C-развязкой состояла из двух импульсов REfocusing BURP (RE-BURP). Мощность градиента была установлена на 0,7 и 0,3 Гс·мм-1, соответственно. Развязку 13C ядер в ходе селективных импульсов проводили с использованием последовательности GARP4 с мощностью 3,8 кГц. Последовательность DANTE-Z с 13C-развязкой, Zero-Quantum Filtered TOCSY (DANTE-Z ZQF-TOCSY) получали встраиванием импульсного модуля с 13C-развязкой DANTE-Z в ранее описанную последовательность ZQFTOCSY (Massou et al., 2007). Импульс DANTE-Z с 13C-развязкой использовали с такими же параметрами, как указано выше. Перенос TOCSY осуществляли с использованием смешивающей последовательности DIPSI-2, применяемой в течение 20 мс с шириной поля 10 кГц. В конце периода смешивания намагниченность в плоскости подавляли квадратным градиентом продувки 2,6 Гс·мм-1. Перед получением данных использовали фильтр Zero-Quantum (Thrippleton and Keeler, 2003), состоящий из адиабатического импульса 180°CHIRP с 10% сглаживанием, шириной сектора 20 кГц, и длительностью 30 мс при максимальной мощности RF 330 Гц. В ходе адиабатического импульса использовали квадратный градиентный импульс 0,2 Гс·мм-1. Все изменения ЯМР проводили в институте Ingenierie des systemes biologiques et des procedes (INSA, Toulouse, Франция).
Внутриклеточные метаболиты
Взятие образцов
Для гашения метанолом 12 мл суспензии клеток (OD660=5) отбирали в пластмассовый шприц и прямо инъецировали в предварительно охлажденную пробирку falcon, заполненную 24 мл 60% метанола (-58°С 10 мМ HEPES, pH 7), сразу энергично перемешивали и центрифугировали в течение 5 мин (10000×g, -19°С Biofuge Stratos, Kendro Laboratory Products, Langenselbold, Германия). Взятие образцов проводили четыре раза параллельно для экстракции кислотой, а также щелочью.
Гашение всей культуры проводили путем переноса 10 мл культуры с клетками из фазы экспоненциального роста в стеклянные колбы, инкубированные в жидком азоте. Параллельно отбирали образцы супернатанта с разделением клеток фильтраций (размер пор 0,2 мкм, Sartorius, Gottingen, Германия) для учета метаболитов, находящихся в супернатанте (Bolten et al., 2007). Гашение метанолом осуществляли для взятия образцов нуклеотидов, в то время как образцы для гашения цельной культуры использовали для определения глюкозо-6-фосфата и фруктоза-6-фосфата, соответственно.
Экстракция
Окисленные нуклеотиды после гашения метанолом экстрагировали добавлением 900 мкл HClO4 (pH 1) к клеточному осадку, инкубацией в течение 7 мин при 55°С на водяной бане, с последующим помещением на лед для устранения нестабильности метаболитов. Образцы с перхлорной кислотой центрифугировали в течение 5 мин при 10000×g и 4°С для отделения клеточного дебриса. Нейтрализации супернатанта достигали осторожным добавлением 5М K2CO3 до достижения pH 7,0. Полученный осадок (KClO4) удаляли центрифугированием (5 мин при 13000×g). Экстракцию щелочью восстановленных нуклеотидов проводили путем добавления 900 мкл раствора KOH в этаноле (200 мл 1М K2HPO4+50 мл EtOH+20 мл 5М KOH, pH 12,4) к клеточному осадку и инкубации в течение 2 мин при 55°С на водяной бане, с последующей инкубацией в ледяной воде и конечной стадией центрифугирования для удаления клеточного дебриса (5 мин при 10000×g и 4°С). Метаболиты после гашения цельной культуры экстрагировали добавлением 10М HCl до конечного pH 1,2. Затем клетки размораживали и экстрагировали в течение 8 мин при 50°С на водяной бане, после сего следовала нейтрализация 10М KOH и удаление клеточного дебриса центрифугированием (15 мин, 10000×g при 4°С.
Количественное определение
Фосфорилированные нуклеотиды NADP и NADPH количественно определяли в 96-луночном планшете при 540 нм (iEMS Reader MF, Thermo Fisher Scientific, Waltham, MA, США) путем ферментативного циклического анализа (Bernofsky and Swan, 1973). Для измерения получали основную смесь для 110 реакций, содержащую 50 мМ Tris-HCl (pH 7,8), 250 мМ глюкозо-6-фосфат, 0,5 мМ тиазолил синий, 2 мМ этосульфат феназина и 100 мЕ·мл-1 глюкозо-6-фосфатдегидрогеназы. 25-мкм образец или стандартный раствор (NADP или NADPH, соответственно) предоставляли в планшете с лунками, и затем начинали реакцию добавлением 175 мкл основной смеси. Мониторинг изменения поглощения проводили в течение 10 минут. Количественное определение проводили с использованием калибровочной кривой с нуклеотидным стандартом.
Глюкозо-6-фосфат (G6P) и фруктозо-6-фосфат (F6P) определяли ферментативно в многостадийной реакции (Bergmeyer and Hohorst, 1970). Для измерения реакционную смесь с общим объемом 1 мл, содержащим 200 мМ Tris-HCl (pH 7,6), 0,2 мМ NADP, 5 мМ MgCl2 и 500 мкл образца инкубировали в течение 5 мин и определяли базовое поглощение при 340 нм (E0). Затем добавляли 5 мкл глюкозо-6-фосфатдегидрогеназы (34 Е·мл-1) и через 5 мин определяли E1. Конверсию F6P начинали добавлением 10 мкл PGI (35 Е·мл) и конечное поглощение при 340 нм измеряли через дополнительные 5 мин (E2). Положительный контроль анализировали с помощью стандартного раствора, содержавшего смесь G6P и F6P, отрицательный контроль анализировали с помощью воды. Учитывая коэффициент экстинкции NADPH ε340=6,22 л·ммоль-1·см-1 и фактор разведения DF образца, можно вычислить концентрации G6P и F6P с помощью следующих уравнений:
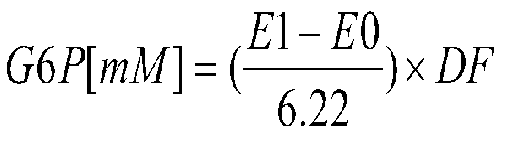 (3)
(3)
(4)
Активность фермента
Взятие образцов
На фазе экспоненциального роста клетки собирали центрифугированием (5 мин, 9800×g, Biofuge stratos, Heraeus, Hanau, Германия), промывали один раз буфером для разрушения и, наконец, суспендировали до 0,25 г CWW на мл. Разрушение проводили обработкой ультразвуком (MSE soniprep 150, Sanyo Europe, Munich, Германия) на бане с ледяной водой с пятью импульсами длительностью 15 с в 20 микрон или механически. Для этого суспензию клеток разделяли на аликвоты в количествах 750 мкл в 2-мл пробирки Eppendorf, содержавшие стеклянные гранулы. Разрушение проводили в ribolyser (MM301, Retsch, Haan, Германия) при 30 Гц (2×5 мин; перерыв в 5 минут между ними). Неочищенные экстракты клеток получали центрифугированием в течение 10 минут при 13000 об./мин. (Biofuge fresco, Heraeus, Hanau, Германия) для разрушения с помощью ribolyser или 2×30 мин при 8500 об./мин. (Biofuge stratos, Heraeus, Hanau, Германия) для разрушения ультразвуком. Содержание белка определяли способом Bradford (Bradford, 1976) с раствором реагента от BioRad (Quick Start Bradford Dye, BioRad, Hercules, США) и белковым стандартом BSA.
Количественное определение
Все измерения проводили в трех повторах в общем объеме 1 мл с интерактивным мониторингом изменения поглощения в спектрофотометре (Specord 40, Analytik Jena, Jena, Германия, или Helios alpha, Thermo Scientific, Waltham, США). Отрицательные контроли анализировали без субстрата или клеточного экстракта, соответственно. Активность приведена в качестве удельной ферментативной активности [Е·мг-1], причем 1Е=1 мкмоль·мин-1, при 30°С, и ее вычисляли из изменения поглощения [A мин-1] и удельного коэффициента экстинкции ε340=6,22 л·ммоль-1·см-1 для NAD(P)H. В качестве общей методики приготавливали основную смесь без субстрата и клеточного экстракта из исходных растворов для буфера реакции, MgCl2, кофакторов (NADP, NAD, NADH, ADP, TPP, CoA) и присоединяющих ферментов (LDH, G6PDH, PGI, рибоза-5-фосфатизомераза (RPI), рибулоза-5-фосфатэпимераза (RPE), TPI/GDH) и инкубировали в течение 10 мин при 30°С. Раствор субстрата и клеточный экстракт предоставляли в 1,5-мл кюветах из полистирола (Plastibrand, Wertheim, Германия). Реакцию начинали быстрым добавлением основной смеси.
Глюкозо-6-фосфатдегидрогеназа (G6PDH) (Moritz et al., 2000)
Разрушение клеток проводили в буфере, содержавшем 100 мМ Tris/HCl (pH 7,5), 10 мМ MgCl2 и 0,75 мМ DTT. DTT приготавливали свежим в виде 100-мМ исходного раствора и добавляли к суспензии клеток перед разрушением. Вследствие нестабильности фермента активность измеряли сразу после разрушения клеток. Реакционная смесь для количественного определения активности G6P-дегидрогеназы содержала 100 мМ Tris/HCl (pH 7,8), 200 мМ KCl, 1 мМ NADP, 10 мМ MgCl2, 5 мМ G6P и 50 мкл клеточного экстракта. Константы аффинности Михаэлеса-Ментена определяли, варьируя концентрации субстратов, G6P или NADP, соответственно. Для тестирования влияния эффекторов на активность, реакционную смесь, кроме того, дополняли ATP, PEP, FBP или NADPH в различных концентрациях.
Пируваткиназа (PYK) (Netzer et al., 2004)
Для определения активности пируваткиназы клетки разрушали в 100 мМ Tris/HCl (pH 7,5), содержавшим 10 мМ MgCl2. Активность определяли с использованием лактатдегидрогеназы (LDH) в качестве фермента для присоединения и интерактивного мониторинга A340. Анализ проводили при pH 7,0 с 100 мМ Tris/HCl, 15 мМ MgCl2, 1 мМ ADP, 0,25 мМ NADH, 5,5 Е LDH, 10 мМ фосфоенолпируватом (PEP) и 10 мкл неочищенного клеточного экстракта. Исходный раствор субстрата (1М PEP) доводили до pH 7,0 перед использованием.
Яблочный фермент (MalE) (Gourdon et al., 2000)
Буфер для разрушения для определения активности яблочного фермента содержал 100 мМ Tris/HCl (pH 7,8) и 200 мМ KCl. Активность MalE измеряли в том же буфере, дополнительно содержавшем 2 мМ MgCl2, 1 мМ NADP, 40 мМ малат и 50 мкл клеточного экстракта. Малат приготавливали в виде исходного раствора в концентрации 1 моль·л-1 и pH 7,8.
Изоцитратдегидрогеназа (ICD)
Разрушение клеток и измерение активности проводили в 100 мМ Tris/HCl (pH 7,8), содержавшем 10 мМ MgCl2. Реакционная смесь для определения активности фермента дополнительно содержала 1 мМ изоцитрат, 0,5 мМ NADP и 25 мкл неочищенного клеточного экстракта.
Пируватдегидрогеназа (PDH)
Разрушение проводили в 100 мМ Tris/HCl (pH 7,4), 10 мМ MgCl2 и 3 мМ цистеине (Blombach et al., 2007). Реакционная смесь для количественного определения активности пируватдегидрогеназы содержала 50 мМ Tris/HCl, 0,01 мМ CaCl2, 0,3 мМ TPP, 0,12 мМ CoA, 2 мМ NAD, 3 мМ цистеин, 5 мМ пируват и 50 мкл клеточного экстракта, и его проводили при pH 7,4 (Guest and Creaghan, 1974).
Фосфоглюкоизомераза (PGI) (Dominguez et al., 1998)
Для определения PGI клетки разрушали в 100 мМ Tris/HCl (pH 7,8), содержавшем 10 мМ MgCl2. Активность количественно определяли с использованием глюкозо-6-фосфатдегидрогеназы (G6PDH) в качестве фермента для присоединения и интерактивного мониторинга образования NADPH при 340 нм. Анализ проводили при pH 7,8 с 100 мМ Tris/HCl, 10 мМ MgCl2, 0,5 мМ NADP, 1,25 Е G6PDH, 4 мМ фруктоза-6-фосфатом и 10 мкл неочищенного клеточного экстракта.
Диаминопимелатдегидрогеназа (DDH) (Cremer et al., 1988)
Буфер для разрушения для определения активности DDH состоял из 100 мМ Tris/HCl и 10 мМ MgCl2, и его доводили до pH 7,8. Активность DDH измеряли в направлении окисления мезодиаминопимелата и одновременного образования NADPH. Для поддержания этого направления реакцию проводили при щелочном pH 10,5. Реакцию проводили с 200 мМ глицином/NaOH (pH 10,5), 10 мМ MgCl2, 2 мМ NADP, 4 мМ мезодиаминопимелатом и 50 мкл клеточного экстракта.
Фруктозо-1,6-бисфосфатаза (FBPase)
Определение активности FBPase in vitro было основано на протоколе Sugimoto и Shiio (Sugimoto and Shiio, 1989) с небольшими модификациями. Реакционная смесь содержала 100 мМ Tris/HCl, 10 мМ MgCl2, 0,5 мМ NADP, 1 мМ фруктозо-1,6-бисфосфат, 2 Е фосфоглюкоизомеразы, 1 Е глюкозо-6-фосфатдегидрогеназы и 50 мкл клеточного экстракта. Разрушение клеток проводили в 100-мМ буфере Tris/HCl при pH 7,8.
Транскетолаза (TKT)
Неочищенные клеточные экстракты приготавливали в 100 мМ Tris/HCl при pH 7,8. Определение активности TKT требовало использования нескольких ферментов для присоединения. RPI и RPE использовали в качестве ферментов для присоединения для предоставления субстратов рибулозо-5-фосфата и ксилулозо-5-фосфата из рибозо-5-фосфата (R5P), предоставленных в анализе. Интерактивный мониторинг при 340 нм обеспечивали путем дальнейшей конверсии глицеральдегид-3-фосфата в качестве продукта реакции TKT посредством триозофосфатизомеразы и глицерофосфатдегидрогеназы (TPI/GDH). В противоположность приведенной выше методике получали две различных основных смеси. Смесь A содержала буфер, TPI, RPI, RPE и NADH в общем объеме 650 мкл, смесь B содержала буфер, TPP, MgCl2 и клеточный экстракт в общем объеме 300 мкл. Обе из них предварительно нагревали в течение 10 мин при 30°С. Рибоза-5-фосфат предоставляли в кювете и реакцию начинали путем последовательного добавления смеси B и смеси A. Конечная реакционная смесь содержала 50 мМ Tris/HCl (pH 7,8), 0,5 мМ NADH, 1 Е RPI, 0,5 Е RPE, 1 Е TPI/GDH, 10 мМ MgCl2, 0,2 мМ TPP, 20 мМ R5P и 50 мкл неочищенного клеточного экстракта.
Трансальдолаза (TAL)
Разрушение клеток проводили в буфере Tris/HCl (100 мМ, pH 7,8). Реакцию определения трансальдолазы проводили в 50 мМ буфере Tris/HCl (pH 7,8), дополнительно содержавшем 1 мМ фруктозо-6-фосфат, 0,5 мМ NADH, 4 мМ эритрозо-4-фосфат, 6 Е TPI/GDH и 50 мкл неочищенного клеточного экстракта. TPI/GDH использовали в качестве присоединяющих ферментов для обеспечения интерактивного мониторинга при 340 нм.
Анализ метаболических потоков
Системный анализ [ 13 C] метаболических потоков
Метаболическая сеть для роста и продуцирования лизина C. glutamicum, выращенными на глюкозе, включала все центральные метаболические пути, т.е. гликолиз, PPP, цикл трикарбоновых кислот и анаплеротическое карбоксилирование и декарбоксилирование. Кроме того, применяли пути для биосинтеза лизина и различных побочных продуктов, а также анаболические пути промежуточных предшественников биомассе. Для синтеза глицина рассматривали два возможных пути, т.е. через серинальдолазу и через треонинальдолазу (Simic et al., 2002). Исходя из предшествующих результатов, глиоксалатный путь считали неактивным (Wittmann and Heinzle, 2002). В C. glutamicum, пируваткарбоксилаза, PEP-карбоксилаза, яблочный фермент и фосфоенолпируваткарбоксикиназа связывают гликолиз и цикл TCA путем взаимной конверсии метаболитов C3 и C4 (Wittmann and Becker, 2007). На основной модели, карбоксилирование и декарбоксилирование, соответственно, считали объединенными потоками. Однако в расширенном подходе все единичные ферменты рассматривали в качестве отдельных реакций. Для этой цели, PEP и пируват подразделяли на отдельные метаболические группы. Вычисление анаболической потребности в различных предшественниках было основано на данных о составе биомассы C. glutamicum, которые учитывали конкретные анаболические потребности для синтеза клеточной стенки на основе содержания диаминопимелата в клетке (Wittmann and de Graaf, 2005). Данные о мечении протеиногенных аминокислот и трегалозы и средние величины стехиометрических данных для трех параллельных культивирований объединяли для вычисления метаболических потоков. Набор потоков, которые дали минимальное отклонение между экспериментальными (Mi, экспер.) и стимулированными (Mi, вычисл.) фракциями массовых изотопомеров брали в качестве наилучшей оценки для внутриклеточного распределения потоков. Сеть была переопределена, так что был возможен подход наименьших квадратов. В качестве критерия ошибок использовали взвешенную сумму наименьших квадратов (Wittmann and Heinzle, 2002). Статистический анализ полученных потоков проводили с помощью подхода Монте-Карло (Wittmann and Heinzle, 2002). Из полученных данных вычисляли 90% доверительные интервалы для единичных параметров. Все метаболическое моделирование осуществляли на персональном компьютере с использованием Matlab 7.0 или Matlab 9.0 (Mathworks Inc.) (Wittmann and Heinzle, 2001a; Wittmann and Heinzle, 2001b; Wittmann and Heinzle, 2002). Соотношение распределения потоков (Φ) и обратимость потоков (ζ) определяли как относительный поток в одну из двух ветвей, и как отношение обратного или обменного потока к суммарному потоку в прямом направлении, соответственно (Wittmann and Heinzle, 2002). Для пируватного модуля с реакциями, катализируемыми фосфоенолпируваткарбоксилазой (PEPC), пируваткарбоксилазой (PC), фосфоенолпируваткарбоксикиназой (PEPCK) и яблочным ферментом (MAE) определения проводили следующим образом:
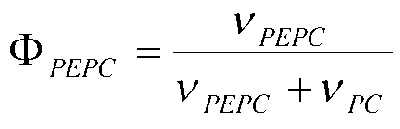 (5)
(5)
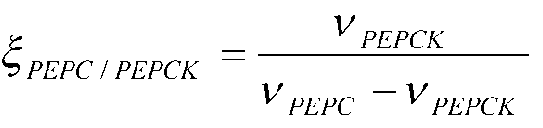 (6)
(6)
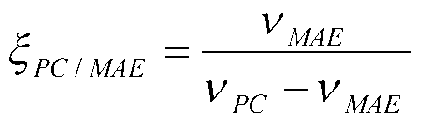 (7)
(7)
Кроме того, определяли соотношение расщепления для пути биосинтеза лизина из специфического 13C обогащения атомов углерода C? и C? из лизина и C? из ацетата из культурального супернатанта C. glutamicum, культивированных на [6-13C] глюкозе с помощью следующего уравнения:
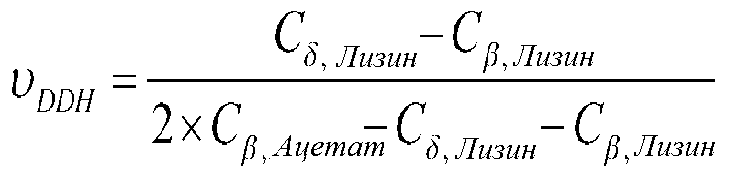 (8)
(8)
Оценка потоков на основе стехиометрии
Близкая корреляция относительного потока синтеза цитрата (νCIS) и относительного потока пируватдегидрогеназы (νPDH) с выходом лизина (YLys/S) и выходом биомассы (YX/S) обеспечила прочную основу для оценки потоков, исходя из стехиометрических данных.
Требуемой стехиометрической корреляции достигали с помощью параболоидной аппроксимации обширного набора данных от 18 независимых оценок потоков, и они обеспечивали вычисление νCIS и νPDH следующим образом:
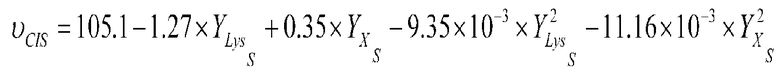 (9)
(9)
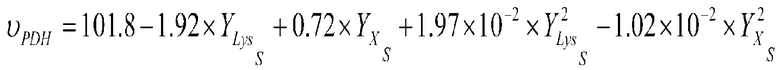 (10)
(10)
Аналогично, суммарный поток через анаплероз (νPYC) близко коррелировал со стехиометрическими характеристиками (Wittmann and Heinzle, 2002). Отсутствие образования побочного продукта цикла TCA, как наблюдали в данном случае, позволило вычисление νPYC через удаление углерода из цикла TCA для анаболизма и синтеза лизина:
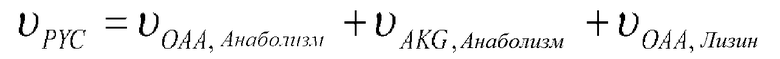 (11)
(11)
Анаболическую потребность в оксалоацетате (νOAA, анаболизм)) и α-кетоглутарате (νAKG, анаболизм) определяли из выхода биомассы и клеточного состава C. glutamicum, приведенного в таблице A.4 (Wittmann and de Graaf, 2005). Средние значения и доверительные интервалы для этих параметров потоков вычисляли из 100 статистически варьирующих величин для выхода биомассы, выхода лизина и анаболических потоков с использованием подхода Монте-Карло. Величины потоков статистически оценивали с помощью t-критерия с использованием программного обеспечения Origin (Microcal Origin 6.0).
Установление окислительно-восстановительного равновесия
Для большего понимания метаболизма кофакторов было установлено полное окислительно-восстановительное равновесие (уравнение 12), исходя из данных о потоках и метаболической сети C. glutamicum. Для вычисления рассматривали все возможные окислительно-восстановительные реакции в процессе окислительного фосфорилирования (Yang et al., 2006a).
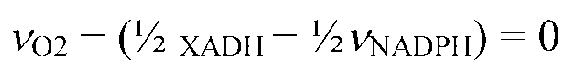 (12)
(12)
где νO2 обозначает поток потребления кислорода и νXADH и νNADPH представляют собой суммарное продуцирование NADH, FADH и NADPH, соответственно. Учитывая все имеющие значение реакции, νXADH и νNADPH можно вычислить из потоков метаболической сети C. glutamicum:
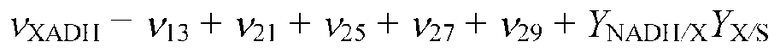 (13)
(13)
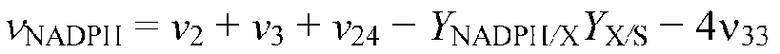 (14)
(14)
с анаболическим потреблением NADPH YNADPH/X=16,4 ммоль·г-1 (Wittmann and de Graaf, 2005) и анаболическим продуцированием NADH YNADH/X=3,2 ммоль·г-1 (Yang et al., 2006a). G6P-дегидрогеназу (ν2), 6-фосфоглюконатдегидрогеназу (ν3) и изоцитратдегидрогеназу (ν24) считают основными предоставляющими NADPH ферментами (Wittmann and de Graaf, 2005). Поток потребления кислорода вычисляли с помощью экспериментально определенного дыхательного коэффициента (RQ) и потока продуцирования диоксида углерода (νCO2):
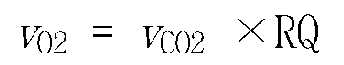 (15)
(15)
Для вычисления νCO2 учитывали все реакции продуцирования и потребления CO2 из центрального метаболизма углерода и из анаболизма, где
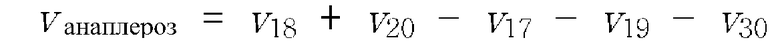 (16)
(16)
отражает анаплеротический суммарный поток, и анаболическое продуцирование CO2 для C. glutamicum представляет собой YCO2/X=1025 мкмоль·г-1 (Yang et al., 2006a).
 (17)
(17)
Анализ метаболических потоков продуцирующих лизин C. glutamicum
Установление потоков in vivo обеспечивает важную информацию о метаболическом ответе исследуемого организма на генетические неблагоприятные воздействия или неблагоприятные воздействия внешней среды. Это особенно важно для рациональной оптимизации штаммов, поскольку сложность живых клеток препятствует точному предсказанию влияния генетических модификаций.
Предпосылки
Анализ метаболических потоков в этой работе проводили в качестве стационарного анализа потоков с использованием информации о мечении, полученной для гидролизованного клеточного белка способом ГХ-МС. Этот подход требует метаболического и изотопного устойчивого состояния исследуемой культуры, где незаменим постоянный параметр культивирования, т.е. pH и pO2. Как проиллюстрировано на фиг.4A для штамма C. glutamicum BS1 (lysCT311I), выбранные условия культивирования поддерживали экспоненциальный рост C. glutamicum и поддержание pH, а также достаточное поступление кислорода в используемых вращающихся флаконах с перегородками вплоть до оптической плотности 10 (Novaspec III). Кроме того, присутствие метаболического устойчивого состояния обеспечивалось постоянным ростом и продуцирующим поведением исследованных штаммов (фиг.4B).
Как проиллюстрировано для различных протеиногенных аминокислот, из двух параллельных исследований с радиоактивными индикаторами с взятием образцов в различные моменты времени при культивировании, также паттерны 13C-мечения метаболитов оставались постоянными с течением времени (фиг.5). Это указывало на изотопное устойчивое состояние в ходе культивирования, так что полученные распределения потоков могли быть взяты в качестве типичных для всего периода культивирования.
Вычисление метаболических потоков было основано на минимизации отклонения между экспериментально измеренными и смоделированными фракциями массовых изотопомеров. Подход включал установление метаболического равновесия на каждой стадии с учетом стехиометрических данных по росту и образованию продукта для трех параллельных культивирований и анаболических потребностей в предшественниках биомассы (Wittmann and de Graaf, 2005).
Метаболические потоки C. glutamicum lysC T311 I
Активность метаболических путей C. glutamicum BS1 (lysCT311I) определяли из экспериментов по мечению с использованием [1-13C]глюкозы в качестве отслеживаемого субстрата. Модель потока включала все имеющие значение метаболические пути C. Glutamicum, т.е. пентозофосфатный путь, гликолиз, цикл TCA, анаплеротическое карбоксилирование и декарбоксилирование, а также биосинтез лизина. Исходя из предшествующих открытий, было предположено, что в процессе роста на глюкозе шунт глиоксиловой кислоты является неактивным (Wittmann and Heinzle, 2002). Пируват и PEP, и оксалоацетат, и малат, соответственно, считали общими группами. Таким образом, связывающие их реакции, т.е. пируваткарбоксилазу, PEP-карбоксилазу, яблочный фермент и PEP-карбоксикиназу, определяли в качестве общих потоков через карбоксилирование и декарбоксилирование, соответственно. Что касается полученной аппроксимации, было достигнуто превосходное соответствие между экспериментально определенными и вычисленными фракциями массовых изотопомеров (таблица 11).
Относительные фракции массовых изотопомеров аминокислот из клеточного белка и секретируемой трегалозы продуцирующих лизин C. glutamicum BS1 (lysCT311I), культивируемых на 99% [1-13C]глюкозе. Данные включают экспериментальные данные ГХ-МС (Эксп.) и величины, предсказанные решением математической модели, соответствующей оптимизированному набору потоков (Вычисл.)
Из полученной карты потоков становится очевидным, что активность центральных путей, особенно потоков через пентозофосфатный путь и гликолитический поток, была определена с очень высокой точностью.
По сравнению с диким типом (Kim et al., 2006; Kromer et al., 2008), освобождение аспартаткиназы от ингибирования по принципу обратной связи лизином и треонином в C. glutamicum приводило к значительному изменению потоков. Наиболее очевидным отличием определенно является увеличение на 8% потока лизина, которое является прямым следствием нарушения регуляции продуцирования лизина. Суммарный поток через анаплеротическое карбоксилирование был значительно увеличен для поступления достаточного количества оксалоацетата, требуемого для продуцирования лизина. Более того, мутант с нарушенным регулированием обладал измененными потоками через пентозофосфатный путь и цикл TCA. Вследствие сниженного образования биомассы в C. glutamicum BS1, меньше углерода удаляется из промежуточного метаболизма для удовлетворения анаболических потребностей. Использованный в данном случае подход для определения потоков полностью пригоден для установления таких глобальных изменений потоков при сравнимо низких усилиях. Таким образом, можно было эффективно применять постановку эксперимента, а также модель потоков, включающую информацию о мечении для 29 фракций массовых изотопомеров, для общепринятого 13C MFA для исследования эффекта генетических изменений или условий окружающей среды.
Расширенный анализ потоков
Однако в отдельных случаях, используемый выше подход для определения потоков не был достаточным. Установление полной метаболической сети в пируватном модуле, включающем пируваткарбоксилазу, PEP-карбоксилазу, яблочный фермент и PEP-карбоксикиназу, является значительно более детальным. Что касается продуцирования лизина в C. glutamicum, эти реакции имеют существенное значение. Таким образом, модель потоков была расширена для установления всех потоков через PEP-карбоксилазу (PEPC), пируваткарбоксилазу (PC) и яблочный фермент (MalE), а также PEP-карбоксикиназу (PEPCK).
Моделирование сети и постановка эксперимента
Первым этапом для адаптации модели было расширение метаболической сети путем рассмотрения PEP и пирувата в качестве отдельных групп и PC, PEPC, PEPCK и яблочного фермента в качестве отдельных ферментативных реакций. Для этого сценария описанный выше подход с использованием данных о мечении из единого исследования с радиоактивными индикаторами с [1-13C] глюкозой был значительно расширен, поскольку он не был способен полностью установить все эти потоки, а только общие потоки карбоксилирования и декарбоксилирования (Kim et al., 2006). Что касается центральных метаболических путей, [1-13C] глюкоза является ценной для установления верхней части метаболизма, в частности, окислительного пути PP, гликолиза и, в случае его наличия, пути Энтнера-Дудорфа, в то время как использование смесей [U-13C]глюкозы и немеченой глюкозы является особенно пригодным для установления потоков ниже PEP, особенно в пируватном модуле (Fischer et al., 2004; Wittmann and Heinzle, 2001b). Вследствие этого, такая экспериментальная стратегия была основана на двух параллельных исследованиях с радиоактивными индикаторами с (i) [1-13C] глюкозой и (ii) смесью [U-13C] глюкозы и естественным образом меченной глюкозы для объединения информации, доступной от анализа мечения ГХ-МС для метаболитов из различных отслеживаемых субстратов для анализа потоков. Поскольку известно, что более подробную информацию о вычислении потоков можно получить с помощью ГХ-МС посредством дополнительного анализа фрагментарных ионов, которые содержат только конкретные части углеродного скелета анализируемого соединения, кроме того, рассматривали ряд фрагментарных ионов из протеиногенных аминокислот, для которых ранее было показано, что они пригодны для анализа потоков в комплексных метаболических сетях прокариот (Klapa et al., 2003) и эукариот (Frick and Wittmann, 2005). В целом, здесь рассмотрено 197 различных фракций массовых изотопомеров для каждого штамма, в то время как исходный упрощенный подход только с одним экспериментом с радиоактивным индикатором и меньшим количеством измеренных фрагментов учитывал 29 фракций массовых изотопомеров. Расширенный подход тестировали с помощью компьютерных моделирующих исследований, чтобы увидеть, можно ли дополнительную рассматриваемую информацию о мечении использовать для определения дополнительных добавленных свободных потоков в пируватном модуле. Для этой цели чувствительность для доступных фракций массовых изотопомеров и представляющих интерес параметров потоков получали от частичных производных, как описано ранее (Wittmann and Heinzle, 2001b). На паттерны мечения ряда вновь рассматриваемых фрагментарных ионов влияло варьирование свободных потоков в пируватном модуле и, таким образом, они содержали ценную информацию для определения представляющих интерес параметров потоков. Это проиллюстрировано для исследования с [U-13C] глюкозой и немеченой глюкозой в качестве отслеживаемого субстрата и варьирования параметров свободных потоков ΦPEPC (распределение потоков между PEPC и PC), ζPEPC/PEPCK (обменный поток для PEPC и PEPCK) и ζPC/MalE (обменный поток для PC и яблочного фермента), которые сильно влияют на паттерн мечения анализируемых метаболитов (фиг.6).
Паттерны мечения в качестве единого вклада PEPC (ΦPEPC=100%), и высоко обратимых потоков в пируватном модуле (ζPEPC/PEPCK=ζPC/MalE=10) взяты в качестве эталонной точки и приняты за 100%. С использованием синтетических наборов данных о мечении было получено уникальное решение для всех трех потоков при множественной оценке параметров с рандомизированными исходными величинами, демонстрируя возможность наблюдения и возможность идентификации всех свободных потоков в сети и пригодность разработанного расширенного подхода. Применимость для биологической системы подтверждали с помощью штаммов C. glutamicum BS1 (lysCT311I) и C. glutamicum BS13 (lysCT311IDpyk).
Исследования с радиоактивными индикаторами и оценка параметров
Постоянный рост и характеристики продуцирования, а также изотопное устойчивое состояние, продемонстрированные выше, позволили определение потоков путем 13C обогащения протеиногенных аминокислот из клеточного белка. Метаболическое распределение потоков получали путем минимизации отклонения между экспериментально определенными и вычисленными фракциями массовых изотопомеров. Очевидно, что достигали превосходной аппроксимации, как проиллюстрировано для сопоставления паттерна мечения C. glutamicum BS 1 (фиг.7).
В модели метаболической сети, использованной в данном случае, малат и оксалоацетат рассматриваются в качестве одной группы, т.е. полностью уравновешенный обмен при мечении между двумя группами. Превосходная аппроксимация данных о мечении подтверждает присутствие обратимой внутренней конверсии между малатом и оксалоацетатом. Другие доказательства получены в исследованиях in vitro в C. glutamicum, демонстрирующих обратимое конвертирование малата и оксалоацетата посредством согласованного действия цитоплазматической (MDH) и мембраносвязанной малатдегидрогеназы (MQO) (Molenaar et al., 2000). Набор внутриклеточных потоков, которые дали минимальное отклонение между экспериментальным и смоделированным паттерном мечения, принимали в качестве наилучшей оценки для внутриклеточного распределения потоков. На фиг.8 представлено изображение комплексной сети в пируватном модуле и потоков in vivo, определенных для C. glutamicum BS1.
С использованием расширенной модели очень точно определили потоки in vivo через PEPC, PC, PEPCK и яблочный фермент, на что указывают узкие доверительные интервалы, полученные при статистической оценке. В то время как PC является основным анаплеротическим ферментом в C. glutamicum BS1, PEPC приводит только в небольшой степени к анаплеротическому карбоксилированию. Это очень хорошо соответствует предшествующим данным, что продукция лизина в C. glutamicum в значительной степени зависит от уровня экспрессии пируваткарбоксилазы (Peters-Wendisch et al., 2001), в то время как активность PEP-карбоксилазы необязательна для продуцирования лизина (Peters-Wendisch et al., 1993). Более того, расширенный подход показал значительный поток через яблочный фермент. Это обеспечило первое указание на то, что яблочный фермент служит в качестве источника NADPH в процессе роста на глюкозе, до настоящего времени эту роль постулировали только для выращиваемых на фруктозе C. glutamicum (Dominguez et al., 1998; Kiefer et al., 2004). Суммарные потоки через карбоксилирование и декарбоксилирование, соответственно, определенные с помощью расширенной модели, очень хорошо согласуются с общими потоками, полученными с помощью простой модели. Таким образом, дополнительные усилия, требуемые для полного установления этой комплексной сети, оправданы только в отдельных случаях, которые требуют точного определения одного из вовлеченных потоков.
Адаптация кодонов в качестве инструмента для метаболической инженерии
В последние годы метаболическая и генетическая инженерии появились в виде мощного подхода для продуцирования лизина в Corynebacterium glutamicum при получении улучшенных штаммов. Этот подход требует сложного набора инструментов для характеризации штаммов, таких как описанная выше методология установления видов активности центрального метаболического пути. Однако предполагаемые модификации для оптимизации штаммов также можно реализовывать с помощью генетических инструментов. В этом отношении направленное модулирование ферментативной активности в клетке является принципиальным для увеличения или снижения конверсии углерода с помощью выбранных метаболических путей. Генетические изменения, применяемые для этой цели, как правило, являются сильными, т.е. вызывают полный блок каскада путем делеции гена или многократную амплификацию посредством сверхэкспрессии генов на основе плазмид. Во многих случаях эти чрезмерные изменения приводят к неблагоприятным побочным эффектам. Делеция центральных гликолитических ферментов, например, фосфоглюкоизомеразы, вызывала тяжелый дефицит роста в C. glutamicum (Marx et al., 2003). Мутанты C. glutamicum с делецией пируватдегидрогеназы не могут расти на продуцирующих средах на основе сахаров и, кроме того, демонстрируют обширное образование побочных продуктов (Blombach et al., 2007). В настоящей работе приведенные выше инструменты, используемые в настоящее время для метаболической инженерии, дополнены более мягким подходом с использованием замены инициирующего кодона для изменения удельной активности фермента в C. glutamicum.
Использование инициирующего кодона в Corynebacterium glutamicum
В C. glutamicum на положение начала трансляции указывает один из инициирующих кодонов ATG, GTG или TTG. Для определения точного распределения инициирующих кодонов, всю геномную последовательность загружали из базы данных NCBI в виде файла FASTA и проводили в ней поиск точного количества A, G и T в качестве начального нуклеотида в положении инициирующего кодона с использованием Excel (MS Office, 2003). Точное количество A, G и T было определено как 2215, 627 и 215, соответственно, что соответствует относительной частоте 72%, 21% и 7%. Частота этих инициирующих кодонов немного отличается в центральном метаболизме C. glutamicum с 81% ATG, 15% GTG и 4% TTG.
Из генов центральных метаболических каскадов было выбрано три гена для замены ATG на GTG и один для замены природного GTG на ATG. Выбранные ферменты PGI, PDH, ICD и G6PDH расположены в ключевых точках ветвления. Перенаправление потоков в этих модулях, например пентозофосфатном пути (PPP), анаплеротическом карбоксилировании и цикле TCA, как правило, имеет высокое значение для направленного сверхпродуцирования лизина (Wittmann and Becker, 2007; Wittmann and Heinzle, 2002).
Модулирование ферментативной активности заменой инициирующего кодона
Замену инициирующего кодона осуществляли в генетическом фоне C. glutamicum BS87. Посредством двух событий гомологичной рекомбинации аллель дикого типа заменяли мутантным аллелем. Исходя из исходного протокола клонирования, полученные клоны после второго события рекомбинации могут либо вновь отражать исходный штамм, либо мутант по инициирующему кодону. Скрининг рекомбинантных штаммов, имеющих точечную мутацию в инициирующем кодоне, проводили путем определения удельной ферментативной активности ICD, PGI, PDH или G6PDH, соответственно. Как представлено на фиг.9, несколько клонов проявляли измененную активность фермента-мишени по сравнению с исходным штаммом BS87.
Эти клоны исследовали более подробно с помощью анализа последовательностей. Последовательности выбранных клонов и исходного штамма продемонстрировали 100% идентичность, за исключением нуклеотида инициирующего кодона. В данном случае, все штаммы, проявляющие измененную активность in vitro, также продемонстрировали нуклеотидную замену в инициирующем кодоне. После подтверждения нуклеотидной замены секвенированием определяли удельную активность в минимальной среде с использованием глюкозы в качестве единственного источника углерода (таблица 12).
Удельная активность изоцитратдегидрогеназы (ICD), фосфоглюкоизомеразы (PGI), пируватдегидрогеназы (PDH) и глюкозо-6-фосфатдегидрогеназы (G6PDH) в зависимости от инициирующего кодона в C. glutamicum. Величины соответствуют средним значением для трех биологических реплик с соответствующими отклонениями.
[мЕ·мг-1]
[мЕ·мг-1]
[мЕ·мг-1]
[мЕ·мг-1]
Выраженное воздействие на ферментативную активность было обнаружено для гена icd. В соответствующем мутанте удельная активность ICD была снижена на 70%. Замена начального положения трансляции в aceE и pgi приводила к снижению удельной активности пируватдегидрогеназы и фосфоглюкоизомеразы на 56% и 47%, соответственно. В случае G6PDH удельная активность увеличивалась на 20% вследствие использования инициирующего кодона ATG вместо GTG. Дальнейшая стратегия для трансляционного подавления активности ICD включала использование редких кодонов GGG для глицина и AUA для изолейцина. Для максимизации эффекта природные кодоны трех соседних остатков глицина заменяли на редкие варианты GGG. Они были расположены в положениях 570, 572. В случае изолейцина, осуществляли две замены кодонов в положениях 78 и 79. Несмотря на успешное осуществление в кодирующем гене icd, что было подтверждено секвенированием, воздействие на удельную ферментативную активность отсутствовало. Это могло быть связано с локализацией проводимых нуклеотидных замен, которые, как было обнаружено, участвует по меньшей мере в оптимизации кодонов в миксомицетах (Vervoort et al., 2000).
Замена кодонов в качестве способа регуляции экспрессии генов не является новым подходом. Однако такая стратегия до настоящего времени была сфокусирована, главным образом, на оптимизации кодонов в процессах, вовлекающих экспрессию гетерологичных генов. В данном случае, адаптация кодонов к их относительному появлению в штамме хозяине (Jo et al., 2007; Wiedemann and Boles, 2008) или, как недавно применялось, дополнение тРНК (Lee et al., 2009) приводили к улучшенной экспрессии. По сравнению с этими требующими времени и непрямыми способами, замена инициирующего кодона, проведенная в данной работе, очевидно является более простой и удобной, поскольку она сфокусирована только на одном кодоне. Результаты ухудшения кодонов, т.е. внесения GGG для глицина и AUA для изолейцина, далее подтверждает, что адаптация инициирующего кодона является лучшим способом для достижения успеха. Что касается метаболической инженерии, особенно интересной является возможность снижать экспрессию генов путем ухудшения инициации трансляции, поскольку подавление нежелательных путей не является тривиальным. До настоящего времени, поток углерода часто полностью блокировали делецией гена, которая во многих случаях приводит к тяжелому дефициту роста или нежелательной ауксотрофии (Blombach et al., 2007; Marx et al., 2003). Недавно связанную с плазмидой экспрессию антисмысловой РНК успешно применили для снижения активности 2-оксоглутаратдегидрогеназного комплекса (ODHC) в продуцирующих глутамат C. glutamicum (Kim et al., 2009). Однако этот подход требовал постоянного селекционного давления и, таким образом, не пригоден для процессов промышленного продуцирования (Kim et al., 2009). Замена инициирующего кодона обеспечивает возможность для направленного ослабления выбранных ферментов без таких мешающих эффектов. Вследствие высокой частоты инициирующего кодона ATG (72%) в C. glutamicum этот способ имеет высокий потенциал в качестве общего инструмента для метаболической инженерии. Два успешных примера такой стратегии оптимизации продуцирования лизина в C. glutamicum более подробно рассмотрены ниже.
Между тем как предыдущие две главы относились к ключевым способам в качестве прочной основы для характеризации штаммов и генетической инженерии, представленное ниже описание относится к различным стратегиям инженерии в направлении усовершенствованного продуцирования лизина посредством C. glutamicum. Ключевые пути для продуцирования лизина в C. glutamicum, т.е. метаболизм NADPH, биосинтез лизина, поступление предшественников и цикл TCA, были успешно изменены способами инженерии в направлении усовершенствованного продуцирования лизина.
Модификация способами инженерии метаболизма NADPH
Ввиду высокой потребности в пути биосинтеза лизина в кофакторе NADPH, метаболизм NADPH в C. glutamicum является одной из ключевых мишеней для рациональной инженерии. Таким образом, казалось перспективным усиление потока через пентозофосфатный путь, ранее идентифицированный как основной источник NADPH (Wittmann and Heinzle, 2002). Сначала этого достигали путем сверхэкспрессии и модификации ограничивающего скорость фермента глюкозо-6-фосфатдегидрогеназы, дополненных сверхэкспрессией гена fbp, кодирующего фруктозо-1,6-бисфосфатазу. В качестве альтернативного подхода увеличения активности глюкозо-6-фосфатдегидрогеназы достигали одновременно сверхэкспрессией трансальдолазы и транскетолазы путем замены промотора tkt-оперона, включающего, среди прочих, кодирующие гены zwf, tal и tkt. Более того, исследовали роль яблочного фермента в поступлении NADPH в продуцирующих лизин C. glutamicum, до настоящего времени только малопонятную.
Сверхэкспрессия и модификация глюкозо-6-фосфатдегидрогеназы
Сверхэкспрессию осуществляли путем геномной замены природного промотора гена zwf сильным промотором гена sod, кодирующего супероксиддисмутазу. Более того, в G6PDH вносили точечную мутацию A243T. Эта мутация была ранее идентифицирована путем сравнительного секвенирования между штаммом дикого типа и классически преобразованным продуцирующим штаммом, и было показано, что она приводит к увеличенному продуцированию лизина, несмотря на то, что точные метаболические последствия этой мутации до настоящего времени еще не установлены (Zelder et al., 2005). Затем тестировали, можно ли эти мишени дополнять сверхэкспрессией фруктозо-1,6-бисфосфатазы, которая, как показано ранее, значительно способствовала перенаправлению потока в сторону PPP (Becker et al., 2005). Все штаммы характеризовали в отношении роста клеток и продуцирования лизина. Исследования дополняли характеризацией G6P-дегидрогеназы дикого типа и мутантной G6P-дегидрогеназы для исследования последствий внесенной мутации A243T. Кроме того, анализировали внутриклеточные уровни метаболитов и метаболические потоки через центральный метаболизм углерода для установления метаболических последствий генетических модификаций и понимания общего клеточного поведения.
Активность in vitro
Сверхэкспрессия кодирующего гена zwf очевидно влияла на удельную активность G6P-дегидрогеназы in vitro (фиг.10A). В то время как штамм дикого типа и штамм BS1 (lysCT311I) продемонстрировали сходную активность, замена естественного промотора гена zwf sod-промотором, кодирующим супероксиддисмутазу, приводила к четырехкратному увеличению активности фермента. Это указывает на то, что использование sod-промотора приводит к увеличенному количеству G6P-дегидрогеназы в клетке. Точечная мутация A243T не оказывала существенного влияния на удельную активность G6P-дегидрогеназы, на что указывали сходные величины, наблюдаемые для BS5 (Psod zwf) и BS6 (Psod zwf A243T).
Кинетические свойства дикого типа и варианта A243T
Для понимания последствий точечной мутации в гене zwf, два варианта ферментов сравнивали в отношении их кинетического поведения. В различных аспектах мутантный фермент продемонстрировал улучшенные свойства. Они включали более высокую аффинность в отношении NADP, одному из его субстратов (таблица 15). Более того, можно было наблюдать более высокую устойчивость к ингибированию метаболитами центрального метаболизма. При добавлении 5 мМ ATP или PEP для мутантного фермента сохранялась значительно более высокая активность, в то время как несколько более слабого влияния достигали в случае добавления FBP. Комбинированное добавление всех трех метаболитов при физиологическом диапазоне концентраций от 1 до 2 мМ также показало, что точечная мутация A243T снижает регуляторный контроль G6P-дегидрогеназы на метаболическом уровне. Не наблюдали значимых отличий в отношении аффинности двух ферментов к субстрату G6P (таблица 13) и ингибирования посредством NADPH (фиг.10B).
Кинетическая характеризация дикого типа и варианта A243T G6P-дегидрогеназы из C. glutamicum: константы аффинности Михаэлиса-Ментен для G6P (KM,G6P) и NADP (KM,NADP) и ингибирование активности фермента посредством согласованного действия ATP, фосфоенолпирувата (PEP) и фруктозо-1,6-бисфосфата (FBP), добавленных в эквимолярных количествах. Данные соответствуют средним величинам для трех различных измерений с клеточными экстрактами из клеток, выращенных на минимальной среде с глюкозой в качестве источника углерода.
ATP
PEP
FBP
PEP, FBP
b
b [ATP]=[FBP]=[PEP]
Таким образом, польза мутации A243T в гене zwf является следствием улучшенного кинетического поведения кодируемой G6P-дегидрогеназы и не является результатом увеличенной удельной активности. Значительно более слабое ингибирование мутантного фермента наблюдали уже при концентрации 1 мМ, которая находится в пределах физиологического диапазона, найденного для протестированных метаболитов PEP, FBP и ATP в C. glutamicum (Moritz et al., 2002). Таким образом, кажется вероятным, что мутантный вариант фермента проявляет более высокую активность in vivo. Также боле высокая аффинность в отношении NADP может вносить вклад в этот эффект, поскольку истинные уровни NADP находятся в диапазоне 100 мкм (Moritz et al., 2002).
Образование продукта и метаболические потоки
Затем различные мутанты сравнивали в отношении характеристик роста и продуцирования (таблица 14 и 15). Для этого штаммы выращивали в стандартной минимальной среде, содержащей один из источников углерода: глюкозу, фруктозу или сахарозу, соответственно. Для устойчивого к регуляции по принципу обратной связи исходного штамма C. glutamicum BS1 выход лизина находился в диапазоне от 74 c-ммоль·c-моль-1 для клеток, выращенных на фруктозе и 85 c-ммоль·c-моль-1 для выращенных на глюкозе клеток (Becker et al., 2005).
Характеристики роста и продуцирования продуцирующих лизин C. glutamicum BS1 (lysCT311I), C. glutamicum BS5 (Psod
zwf), C. glutamicum BS6 (Psod
zwf
A243T) и C. glutamicum BS7 (Psod
fbp_zwf
A243T) на глюкозе, фруктозе и сахарозе. Приведенные данные представляют собой выход лизина (YLys/S) и выход биомассы (YX/S) и они отражают средние значения для трех параллельных экспериментов по культивированию.
[c-ммоль·c-моль-1]
[г·c-моль-1]
Сверхэкспрессия гена zwf посредством sod-промотора значительно увеличивала продуцирование лизина. Положительный эффект на продуцирование лизина в соответствующем штамме C. glutamicum BS5 наблюдали на всех протестированных источниках углерода, в то время как наибольшее увеличение, составляющее более 30%, было получено для выращенных на глюкозе клеток (таблица 16). C. glutamicum BS6, в которых ген zwf был, кроме того, модифицирован аминокислотной заменой A243T, продемонстрировали еще более высокое продуцирование лизина. Это было наиболее выражено на фруктозе и сахарозе в качестве источника углерода. Посредством дополнительной сверхэкспрессии гена fbp продуцирование лизина далее усиливали на всех источниках углерода. Существенное влияние внесенных модификаций становится очевидным из сравнения выхода лизина между C. glutamicum BS7, несущими все три мутации, и штаммом C. glutamicum BS1. Комбинированное внесение модификаций привело к общему увеличению выхода лизина, составляющему приблизительно 50%, на глюкозе и на фруктозе, в то время как на сахарозе увеличение составляло приблизительно 70%. С увеличением выхода лизина выход биомассы последовательно снижался на глюкозе и на сахарозе, в то время как он оставался практически постоянным на фруктозе (таблица 14). Что касается удельной скорости, потребление глюкозы оставалось практически неизмененным посредством генетических модификаций (таблица 15), так что наблюдаемые отличия между мутантами проявляют перераспределение потока углерода. В ответ на это, удельная скорость роста снижалась при увеличении продуцирования лизина.
Удельные скорости роста (μ), потребление глюкозы (qGlc) и продуцирование лизина (qLys) и дыхательный коэффициент (RQ) C. glutamicum BS1 (lysCT311I), C. glutamicum BS5 (Psod
zwf), C. glutamicum BS6 (Psod
zwf
A243T) и C. glutamicum BS7 (Psod
fbp_zwf
A243T), выращенных на глюкозе
[ч-1]
[ммоль·г-1·ч-1]
[ммоль·г-1·ч-1]
[моль·моль-1]
b n.d. = не определено
Для исследования того, как генетические модификации детально влияли на поток in vivo через PPP и другие метаболические пути C. glutamicum, проводили анализ 13C метаболических потоков для различных штаммов. Для этой цели, проводили исследования с радиоактивным индикатором на [1-13C]глюкозе. Полученные ключевые потоки центральных метаболических реакций в различных мутантах, выращенных на глюкозе, обобщенно представлены на фиг.11, причем отклонения, приведенные для каждого параметра потока, соответствуют 90% доверительным интервалам. Получили превосходное соответствие между смоделированными и измеренными паттернами мечения. Повторение двадцать раз оценки параметров со статистически варьирующими исходными величинами для свободных параметров потоков привело к идентичным результатам для каждого из штаммов, что подтвердило идентификацию абсолютного минимума.
Штаммы BS5 и BS6 показали сильно усиленный поток PPP по сравнению с исходным штаммом, в то время как гликолитический поток был существенно снижен. Таким образом, сверхэкспрессия гена zwf вызывала явное перенаправление потока в модуле G6P. Дополнительная сверхэкспрессия фруктозо-1,6-бисфосфатазы приводила к дальнейшему увеличению потока в PPP. Помимо PPP, также сильно усиливался поток через цикл TCA, другой важный путь поступления NADPH в C. glutamicum. В основном высокая активность каскадов предоставления NADPH: PPP и цикла TCA в различных мутантах zwf приводила к высокому поступлению NADPH, которое даже превышало истинную потребность для анаболизма и продуцирования лизина, и вызывало высокий кажущийся избыток NADPH во всех мутантах zwf. Более подробное исследование метаболизма кофактора для мутантов с модифицированным способами инженерии PPP проводили с помощью расширенного окислительно-восстановительного равновесия. Это учитывало все возможные реакции в процессе окислительного фосфорилирования, приводящего к окислению восстанавливающих эквивалентов (NADH, FADH и NADPH), которые образуются в катаболических и анаболических путях метаболизма углерода. Все мутанты показали существенный избыток как NADH/FADH, так и NADPH. Общее потребление кислорода не было достаточным для полного объяснения окисления всех образовавшихся восстанавливающих эквивалентов, т.е. окислительно-восстановительное равновесие было замкнутым, как можно было ожидать. Это является четким признаком дополнительных метаболических реакций, вносящих вклад в окисление кофакторов, которое более подробно рассмотрено ниже. Интересно, что более высокая потребность в оксалоацетате в качестве предшественника лизина в оптимизированных штаммах не отражалась путем увеличенного потока через анаплеротическое карбоксилирование (фиг.11). Это, вероятно, связано со сниженной потребностью в оксалоацетате для анаболизма вследствие уменьшенного выхода биомассы (таблица 14).
Посредством стратегии сверхэкспрессии и модификации глюкозо-6-фосфатдегидрогеназы, продуцирование лизина из C. glutamicum значительно увеличивалось на различных источниках углерода, включая имеющие промышленное значение источники углерода: глюкозу и сахарозу. Однако 4-кратное увеличение активности фермента in vitro отражалось только увеличением соответствующей активности in vivo в 1,4 раза, т.е. потока, что указывает на сильную метаболическую регуляцию G6P-дегидрогеназы in vivo (Moritz et al., 2000). При сравнении продуцирования лизина различными штаммами полученной генеалогии на глюкозе, можно было сразу определить, что сверхэкспрессия через sod-промотор была наиболее благоприятной, поскольку это приводило к наибольшему увеличению продуцирования, в то время как дополнительное внесение точечной мутации A243T не приводило к дальнейшему улучшению (таблица 14). Однако изменение порядка внесенных мутаций продемонстрировало полностью отличающуюся картину. Дополнительно сконструированный штамм BS3, содержащий только мутантный фермент с его природным промотором, проявлял высокий выход лизина, составляющий 120 c-ммоль·c-моль-1, который не увеличивался далее, когда ген zwf дополнительно сверхэкспрессировали с помощью sod-промотора. Это явно демонстрирует, что метаболический ответ на генетическое изменение зависит не только от внесенной модификации, но также в большой степени от генетического фона штамма-хозяина.
Образование побочных продуктов
Кроме увеличения продуцирования лизина способы метаболической инженерии также изменяли образование трегалозы, основного побочного продукта C. glutamicum при выращивании на глюкозе. Экскреция трегалозы является нежелательной при продуцировании лизина, поскольку это соединение не может захватываться клетками вследствие отсутствия соответствующей системы захвата. Все различные мутанты zwf C. glutamicum проявляли значительно сниженное образование трегалозы, которое является положительным побочным эффектом метаболической инженерии потока PPP. Как представлено в таблице 17, сверхэкспрессия гена zwf приводила к снижению на 53% образования трегалозы в штамме BS5 по сравнению с исходным штаммом BS1. В штамме BS6 продуцирование трегалозы было снижена даже на 67%. В C. glutamicum, трегалоза может образовываться посредством трех различных путей, во всех из которых используется G6P в качестве субстрата (Tzvetkov et al., 2003). Внутриклеточный анализ метаболитов показал, что сниженный поток трегалозы в штаммах BS5 и BS6 был связан со сниженной доступностью G6P (таблица 16).
Образование трегалозы и внутриклеточный уровень G6P и F6P в процессе культивирования продуцирующих лизин C. glutamicum BS1, C. glutamicum BS5, C. glutamicum BS6 и C. glutamicum BS7 на глюкозе. Выход трегалозы определяли в трех параллельных культивированиях. Внутриклеточный уровень G6P и F6P измеряли в двух независимых клеточных экстрактах, каждый из которых анализировали в двух повторах.
[c-ммоль·c-моль
-1
]
[мкмоль·г
-1
]
Ускоренное конвертирование G6P посредством PPP в результате более высокой активности G6PDH снижает образование трегалозы путем снижения доступности этого предшественника. Одновременная сверхэкспрессия гена fbp ухудшала эффект модификаций zwf, так что снижение образования трегалозы было очевидным, но в некоторой степени меньшим в тройном мутантном штамме (28%). Это должно быть следствием того, что эта модификация "толкает" углерод в PPP через совокупность G6P, в то время как G6P-дегидрогеназа "толкает" углерод из совокупности G6P на этот путь. Таким образом, совокупность G6P в этом мутанте была существенно более высокой. В ответ на это, совокупность F6P также была значительно более высокой в штамме BS7 (таблица 16).
Установление окислительно-восстановительного равновесия и метаболизм NADPH
Как показано с помощью анализа метаболических потоков, генетические манипуляции приводили к крайним изменениям метаболизма NADPH в клетке. Увеличенные потоки через PPP и цикл TCA приводили к значительно увеличенному поступлению NADPH. Увеличенный выход лизина, ассоциированный с этими изменениями потоков, демонстрирует, что поступление NADPH имеет определенно ограничивающую роль в проецировании лизина в периодических культурах C. glutamicum, хотя предположения для хемостатных культур отличаются (Sahm et al., 2000). В то время как исходный штамм BS1 проявляет небольшой очевидный дефицит NADPH (Kim et al., 2006), поступление NADPH в штаммы с модифицированным способами инженерии PPP значительно превышало их потребность (таблица 17). Полученный высокий кажущийся избыток NADPH в этих штаммах указывает на то, что проведенные изменения нельзя полностью использовать для продуцирования лизина, и что остается потенциал для дальнейшей оптимизации. Степень этого потенциала иллюстрируется тем фактом, что выход продукта наилучшего мутанта C. glutamicum BS7 может быть даже удвоен, если можно было направить весь избыток NADPH на путь лизина. Этот потенциал может быть в некоторой степени снижаться частичным окислением NADPH яблочным ферментом, действующим в направлении карбоксилирования. Неистраченный или направленный неправильно NADPH в мутантах zwf указывает на ограничения в других частях метаболической сети, которые появляются в ответ на модификацию способами инженерии потока PPP. Эти пределы могут быть ассоциированы с поступлением оксалоацетата, поскольку было показано, что в особенности увеличение анаплеротического потока в C. glutamicum приводит к увеличенному продуцированию лизина (Koffas et al., 2003; Peters-Wendisch et al., 2001). В действительности, анаплеротический суммарный поток оставался практически постоянным в штаммах, исследованных в данном случае, так что только снижение анаболической потребности в оксалоацетате обеспечило более высокий поток этого предшественника на путь лизина.
Высокий кажущийся избыток NADPH в мутантах PPP указывает на дополнительные реакции, которые потребляют NADPH. Возможные кандидаты включают образующую супероксид NAD(P)H-оксидазу в дыхательной цепи (Matsushita et al., 2001) или работу яблочного фермента в направлении карбоксилирования (de Graaf, 2000; Petersen et al., 2000). До настоящего времени не получено явных экспериментальных доказательств, подтверждающих роль любой из этих реакций. Однако было предположено, что яблочный фермент, действующий в направлении декарбоксилирования, может функционировать в качестве дополнительного источника NADPH в штаммах с кажущимся дефицитом NADPH (Dominguez et al., 1998). Для получения большего понимания метаболизма кофакторов в мутантах с модифицированным способами инженерии PPP, было установлено полное равновесие, учитывая все возможные окислительно-восстановительные реакции в процессе окислительного фосфорилирования (таблица 17).
Окислительно-восстановительное равновесие C. glutamicum BS5, C. glutamicum BS6 и C. glutamicum BS7 с модифицированным способами инженерии PPP, вычисленное из оцененных потоков (фиг.11), анаболического продуцирования CO2 и NADH, анаболической потребности в NADPH и дыхательного коэффициента (таблица 15)
Ни в одном из штаммов равновесие кофакторов не было замкнутым. Это указывает на присутствие дополнительного до настоящего времени не остановленного потока окисления [H], требуемого для замыкания равновесия. Во всех трех штаммах этот поток окисления [H] был практически настолько же высокими, как и кажущийся избыток NADPH. Существенный вклад NAD(P)H-оксидазы в дыхательной цепи в окисление NADPH может быть исключено, поскольку активность этого фермента в качестве части дыхательного пути может привести к потреблению кислорода и, таким образом, привести к замкнутому окислительно-восстановительному равновесию. Это принцип также справедлив для яблочного фермента. Окисление NADPH посредством активности яблочного фермента, действующего в направлении карбоксилирования, приводит к образованию малата, который конвертируется в оксалоацетат, тем самым образуя NADH. Полное окисление NADH в дыхательной цепи может, в свою очередь, быть связано с потреблением кислорода. Однако исследование удельной активности яблочного фермента в штаммах дикого типа и различных продуцентах лизина дало некоторое первое указание в направлении возрастающего физиологического значения яблочного фермента в продуцирующих лизин C. glutamicum (фиг.13). Таким образом, его физиологическая роль была исследована более подробно.
Сверхэкспрессия tkt-оперона
В C. glutamicum ген zwf, кодирующий глюкозо-6-фосфатдегидрогеназу, расположен на tkt-опероне вместе с генами opcA, кодирующим предполагаемую субъединицу G6PDH, tal, кодирующим трансальдолазу, tkt, кодирующим транскетолазу, и pgl, кодирующим 6-фосфоглюконолактоназу. Альтернативно описанному выше подходу, таким образом, сверхэкспрессию G6PDH можно осуществить путем усиленной экспрессии полного оперона, тем самым, активируя расширенный ферментативный набор пентозофосфатного пути.
Хотя глюкозо-6-фосфатдегидрогеназу считают контролирующим скорость ферментом PPP (Moritz et al., 2000), нельзя исключить, что в ответ на сверхэкспрессию zwf, в неокислительной части PPP появляются другие препятствия вследствие ограниченной способности транскетолазы и трансальдолазы. В этом отношении, было показано, что сверхэкспрессия tkt эффективно увеличивает конвертирование углерода с помощью неокислительного пути (Ikeda and Katsumata, 1999). Таким образом, комбинированная сверхэкспрессия ферментов окислительной и неокислительной части PPP предполагает эффективное увеличение потока через PPP и, таким образом, увеличенное поступление NADPH для продуцирования лизина.
Конструирование штамма и удельная активность фермента
Сверхэкспрессию обеспечивали путем аллельной замены промотора дикого типа tkt-оперона сильным sod-промотором. Замену промотора подтверждали анализом способом ПЦР. Для этого, сайт-специфический праймер, связывающийся с промотором, комбинировали с праймером, связывающимся с tkt-опероном. Этот подход приводил только к продуктам ПЦР из мутантов, обладающих заменой промотора в геноме. Для исследования эффекта осуществленной замены промоторов на представляющие интерес ферменты определяли удельную активность. В ответ на транскрипционную активацию tkt-оперона sod-промотором, существенно увеличивалась удельная активность глюкозо-6-фосфатдегидрогеназы, транскетолазы и трансальдолазы (фиг.12).
Базовый уровень экспрессии генов tkt-оперона очевидно контролируется общим промотором, замена которого более сильным sod-промотором увеличивала базовый уровень экспрессии, тем самым, увеличивая удельную активность кодируемых ферментов. Это явно упрощает одновременную сверхэкспрессию zwf, tkt и tal, поскольку она требует только одной замены промотора вместо множества генетических модификаций. Для достижения неограниченного потока углерода через пентозофосфатный путь, сверхэкспрессия целого tkt-оперона, таким образом, является наиболее перспективной и эффективной стратегией.
Функциональная роль яблочного фермента для метаболизма NADPH
Анализ метаболических потоков описанных выше штаммов показал, что ни один из исследованных штаммов не мог достигнуть замкнутого равновесия NADPH при рассмотрении только PPP и ICD в качестве предоставляющих NADPH и образования биомассы и лизина в качестве потребляющих NADPH реакций. Поскольку поступление NADPH является одной из основных проблем в отношении оптимизации штаммов, теперь было особенно интересно узнать, как штамм C. glutamicum BS1 (lysCT311I), который очевидно обладал дефицитом NADPH, мог полностью удовлетворить его потребность в NADPH. Потенциальным кандидатом является яблочный фермент, роль которого до настоящего времени мало понятна. Таким образом, его роль для метаболизма NADPH исследовали более подробно. Для этого кодирующий ген malE удаляли в генетическом фоне C. glutamicum BS1 и штамма C. glutamicum BS6 модифицированным способами инженерии PPP для исследования воздействия на характеристики роста и продуцирования.
Конструирование и подтверждение штаммов
Удаление яблочного фермента обеспечивали путем аллельной замены гена malE дикого типа укороченным фрагментом ДНК, лишенным 400 п.н. последовательности malE. Подтверждение проводили анализом способом ПЦР и определением удельной активности яблочного фермента. Активность MalE в мутантах с делецией была ниже предела детекции (<0,01 Е·мг-1).
Активность яблочного фермента
Удельная активность яблочного фермента строго зависела от генетического фона и пищевого статуса. По сравнению со штаммом дикого типа, продуцирующие лизин C. glutamicum BS1 проявляли значительно увеличенную активность яблочного фермента (фиг.13). В C. glutamicum BS1, кроме того, исследовали влияние условий роста на активность яблочного фермента. В данном случае, активность MalE была увеличена в растущих на фруктозе клетках по сравнению с клетками, культивированными на глюкозе (фиг.13).
Из предшествующих работ известно, что уровень экспрессии malE значительно варьирует, в зависимости от пищевого статуса (Dominguez et al., 1998; Hayashi et al., 2002; Polen et al., 2007). Наблюдаемая в данном случае различная активность in vitro, косвенно отражающая способность фермента и, таким образом, его уровень экспрессии, указывает на увеличенную экспрессию яблочного фермента вследствие генетических изменений.
Физиологический ответ на делецию malE
Для исследования физиологической роли яблочного фермента, сравнивали исходный штамм C. glutamicum BS1 и его производное DmalE, рассматривая рост и продуцирование лизина в стандартной минимальной среде. Как представлено на фиг.14, выход лизина и биомассы был снижен у мутанта с делецией. Однако на рост делеция malE влияла только незначительно. В штамме C. glutamicum BS6 с модифицированным способами инженерии NADPH, в котором было увеличено поступление NADPH через PPP, картина отличалась. Вследствие сверхэкспрессии и модификации глюкозо-6-фосфатдегидрогеназы поступление NADPH в этом штамме является значительно улучшенным по сравнению с C. glutamicum BS1, что приводило к изменению характеристик роста и продуцирования. Удаление яблочного фермента в этом штамме не оказывало какого-либо эффекта на образование лизина и биомассы (фиг.14).
Для дальнейшего исследования достигнутых последствий для метаболизма NADPH в C. glutamicum, вычисляли общую потребность в NADPH штаммов для образования биомассы и продуцирования лизина, учитывая анаболическую потребность в 16,4 ммоль NADPH (г сухой массы клеток)-1 и 4 моль NADPH (моль лизина)-1. Соотношение соответствующей потребности в NADPH для штамма C. glutamicum BS44 с делецией malE и его исходного штамма C. glutamicum BS1 составляло 0,93, показывая, что потребление этого кофактора было снижено в штамме с делецией. Образование биомассы и лизина в штамме C. glutamicum BS6 с модифицированным способами инженерии NADPH и соответствующем штамме с делецией malE было идентичным. Следовательно, оба штамма не отличались в отношении их потребности в NADPH для синтеза продукта, что приводило к соотношению их соответствующего потребления NADPH, равному 1,00. Полученный набор данных и 13C-анализ метаболических потоков с расширенной моделью потока явно показал, что яблочный фермент участвует в поступлении NADPH в продуцирующих лизин C. glutamicum BS1. По сравнению с диким типом, активность MalE была увеличена в продуценте лизина относительно увеличенной потребности в NADPH и делеция malE приводила к уменьшенному образованию потребляющих NADPH продуктов: лизина и биомассы. При выражении потребности в NADPH в качестве относительного потока [%], нормализованного к удельной скорости поглощения глюкозы, принятой за 100%, общий поток потребления NADPH в C. glutamicum BS1 был на 13% выше, чем общее потребление NADPH в его производном с дефицитом malE. Это отличие очень хорошо соответствует потоку in vivo, составляющему 15%, через яблочный фермент в C. glutamicum BS1, определенному в модели расширенного потока (фиг.20), и нулевому потоку через яблочный фермент в штамме с делецией. Из равновесия NADPH (фиг.15) становится очевидным, что этот поток in vivo через яблочный фермент требуется в продуцирующих лизин C. glutamicum BS1 для достижения замкнутого равновесия NADPH.
Однако для C. glutamicum дикого типа недавно было продемонстрировано, что в процессе роста на глюкозе PPP и цикл TCA отдельно поставляют достаточно NADPH для роста клеток (Kromer et al., 2008), объясняя таким образом, что делеция яблочного фермента в фоне дикого типа не влияет на характеристики роста на этом источнике углерода (Gourdon et al., 2000). C. glutamicum очевидно активирует яблочный фермент в условиях продуцирования лизина, чтобы удовлетворять увеличенной потребности в NADPH, которая отражается увеличенным уровнем экспрессии, а также увеличенной удельной активностью MalE. Основополагающая регуляторная стратегия для контроля экспрессии malE не была прояснена до настоящего времени. В данном случае, несколько исследований показали контроль экспрессии несколькими регуляторами. Например, существуют некоторые доказательства, что malE относится к регулону ramB (Gerstmeir et al., 2004), зависимость от пищевого статуса была описана (Dominguez et al., 1998; Hayashi et al., 2002; Polen et al., 2007) и недавно была открыта связь с общей регуляцией азота посредством amtR (Buchinger et al., 2009). Однако внутриклеточный сигнал все еще остается неясным. Вследствие очевидного вовлечения яблочного фермента в метаболизм NADPH в C. glutamicum, окислительно-восстановительный статус клетки считается пусковым фактором. Таким образом, соотношение NADPH/NADP было определено в качестве возможной сигнальной системы для окислительно-восстановительного состояния клетки.
Внутриклеточное соотношение NADPH/NADP в качестве сигнала для контроля экспрессии
Определение внутриклеточного соотношения NADPH/NADP показало существенные отличия между исследованными штаммами. В C. glutamicum BS1 соотношение NADPH/NADP составило 0,82±0,10. Отсутствие активности яблочного фермента в этом генетическом фоне не имело эффекта, что отражалось сходной величиной 0,76±0,10 в C. glutamicum BS44. Однако увеличенное поступление NADPH путем модулирования способами инженерии PPP явно влияло на соотношение NADPH/NADP. Его измеренная величина в C. glutamicum BS6 (1,45±0,40) была значительно более высокой по сравнению с исходным C. glutamicum BS1. C. glutamicum BS52 проявлял соотношение NADPH/NADP 1,76±0,20 и, таким образом, не отличался от его исходного штамма C. glutamicum BS6. Как представлено на фиг.16, активность MalE явно коррелирует с соотношением NADPH/NADP. C. glutamicum ATCC 13032, проявляющий наиболее высокое соотношение, равное 2,35 (Kromer et al., 2008), продемонстрировал наиболее низкую активность MalE.
Снижение соотношения NADPH/NADP, т.е. в качестве внутриклеточного сигнала для ограничения NADPH, по-видимому, запускает экспрессию malE, которая отражается увеличенной удельной активностью в исследованных продуцентах лизина. Предшествующие исследования показали, что в E. coli окислительно-восстановительное состояние, отражаемое доступностью NADPH в клетке, распознается как сигнал для регуляции экспрессии супероксиддисмутазы (Gardner and Fridovich, 1993). Вследствие увеличенного поступления NADPH в штамм с модифицированным способами инженерии PPP дополнительное поступление NADPH посредством яблочного фермента является необязательным, что объясняет сниженную активность MalE в этом штамме. Это предположение подтверждается ответом на делецию malE в этом штамме. Ни продуцирование лизина, ни образование биомассы не обеспечивается отсутствием активности яблочного фермента, показывая, что предоставление NADPH посредством PPP является достаточным для удовлетворения полной потребности в NADPH для этого штамма.
Полученные данные ясно показывают, что C. glutamicum очевидно адаптирован к различным клеточным потребностям посредством модулирования активности MalE. В условиях ограниченной доступности NADPH, например, при выращивании на фруктозе или продуцирование лизина, экспрессия malE увеличивается и представляет собой важный дополнительный источник NADPH в C. glutamicum. В этом отношении MalE поддерживает метаболическую гибкость и прочность, чтобы справиться с изменяющимися клеточными потребностями. Однако может ли яблочный фермент участвовать в установлении окислительно-восстановительного баланса в штаммах с модифицированным способами инженерии PPP с избыточным образованием NADPH, и как это происходит, остается неясным. Очевидным в этом отношении является то, что: удаление яблочного фермента не приводит к увеличенному продуцированию требующих NADPH продуктов: лизина и биомассы, которое можно было бы предположить, если malE был ответственен за окисление кажущегося избытка NADPH. Однако нельзя исключить, что лишенный malE штамм отвечал на общую перестройку метаболических потоков, включая потоки через PPP и цикл TCA, тем самым, снижая общее поступление NADPH. Изобретение может потребовать дальнейших исследований, вовлекающих анализ метаболических потоков с использованием расширенного подхода и определение полного окислительно-восстановительного и NADPH равновесия.
Модификация способами инженерии биосинтеза лизина
Что касается оптимизации штаммов, ферменты, образующие каскад биосинтеза лизина, являются ключевыми мишенями в метаболической инженерии. Однако различные пути образования лизина препятствуют моделированию штамма, поскольку расщепленный путь обеспечивает высокую гибкость. Эта работа была сфокусирована на диаминопимелатдегидрогеназе, составляющей дегидрогеназную ветвь пути лизина в C. glutamicum. Путем осуществления второй копии кодирующего гена ddh, диаминопимелатдегидрогеназу сверхэкспрессировали в генетическом фоне штамма C. glutamicum BS1 с нарушенной регуляцией по принципу обратной связи.
Влияние сверхэкспрессии ddh на удельную активность фермента
Обеспечение второй копии гена ddh привело к значительно увеличенной удельной активности DDH в неочищенных клеточных экстрактах C. glutamicum BS222 по сравнению с исходным штаммом BS1. Как представлено на фиг.17, мутант по ddh проявлял удельную активность 440 мЕ·мг-1, в то время как BS1 продемонстрировал удельную активность 210 мЕ·мг-1. Влияние на активность фермента отражает дополнительную копию гена диаминопимелатдегидрогеназы.
Влияние сверхэкспрессии ddh на рост и продуцирование
Чтобы увидеть, как удвоенная активность DDH влияла на продуцирование лизина, а также на поведение при выращивании, проводили эксперименты по культивированию в стандартной минимальной среде на глюкозе. Оба штамма проявляли хорошо сбалансированный рост и метаболическую стабильность на протяжении всего периода культивирования. Увеличенная активность DDH в штамме C. glutamicum BS222 приводила к увеличению выхода лизина, составляющему 23% (таблица 18).
Характеристики роста и продуцирования C. glutamicum BS1 и C. glutamicum BS222 в стандартной минимальной среде с глюкозой в качестве единственного источника углерода. Выход определяли в качестве наклона линейной наилучшей аппроксимации между потреблением глюкозы и образованием продукта.
Дополнительная копия гена также влияла на образование биомассы, а также на рост (таблица 19). Оба этих параметра были немного снижены в C. glutamicum BS222 по сравнению с его исходным штаммом. Он рос с удельной скоростью 0,38 ч-1, в то время как в мутанте по ddh μ была снижена до 0,31 ч-1.
В отличие от сукцинилазной ветви пути биосинтеза лизина, диаминопимелатдегидрогеназа прямо использует аммоний. Вследствие низкой аффинности DDH к (NH4)+ (Wehrmann et al., 1998), его концентрация в среде, вероятно, влияет на активность DDH in vivo и, таким образом, воздействие сверхэкспрессии ddh на продуцирование лизина. При дальнейшем культивировании во вращающихся флаконах, два штамма таким образом сравнивали в ходе роста при различных концентрациях сульфата аммония.
Ускорение роста посредством увеличенных концентраций сульфата аммония
Культивирование штамма C. glutamicum BS1 при различных концентрациях сульфата аммония показало четкую корреляцию между концентрацией и удельной скоростью роста. Как представлено на фиг.18, удельная скорость роста возрастала при увеличении доступности сульфата аммония.
При наиболее низкой концентрации, составляющей 2 г·л-1, C. glutamicum BS1 проявлял удельную скорость роста 0,32 ч-1 и достигал максимальной скорости роста 0,43 ч-1 при концентрации 15 г·л-1. В противоположность поведению роста, различная концентрация сульфата аммония в среде не влияла на образование лизина и биомассы C. glutamicum BS1 (таблица 19).
Выход лизина и биомассы в C. glutamicum BS1 (lysCT311I), выращенных в минимальной среде при различных концентрациях сульфата аммония.
Увеличение продуцирования лизина путем увеличенной доступности аммония
Картина для штамма C. glutamicum BS222 с модифицированным способами инженерии ddh полностью отличалась. В данном случае, продуцирование лизина чрезвычайно зависело от концентрации сульфата аммония в среде. Как представлено на фиг.19, выход лизина в BS222 последовательно увеличивался при увеличении доступности сульфата аммония. При концентрации 2 г·л-1 сульфата аммония, штамм проявлял выход лизина 89,4 ммоль·моль-1. Он возрастал вплоть до 125,2 ммоль·моль-1 при 10 г·л-1 сульфата аммония. Дальнейшее дополнение сульфатом аммония не приводило к дальнейшему улучшению продуцирования, указывая на то, что концентрация 10 г·л-1 является достаточной для насыщения модифицированной способами инженерии диаминопимелатдегидрогеназы, что согласуется с величиной km 34 мМ DDH в отношении аммония (Wehrmann et al., 1998).
В противоположность C. glutamicum BS1, на удельную скорость роста мутанта ddh не влияла концентрация сульфата аммония в среде. Штамм рос со средней скоростью роста 0,30 ч-1±0,01. Единственным исключением, в данном случае, было культивирование при 2 г·л-1 сульфата аммония, при котором скорость роста немного снижалась до 0,26 ч-1. Эта концентрация представляла собой концентрацию, которая также приводила к наихудшему росту исходного штамма BS1. Такое низкое количество аммония очевидно не поддерживает оптимальный рост, а ограничивает образование биомассы (Silberbach et al., 2005b). Это согласовывалось с данными, что при концентрации ниже 2,75 г·л-1 несколько генов, вовлеченных в ассимиляцию аммония, активируются для обеспечения приспособления к аммонию. Однако образование биомассы снижалось при концентрациях выше 10 г·л-1 сульфата аммония (таблица 20), что, вероятно, связано с чрезвычайно увеличивающимся выходом лизина.
Удельная скорость роста и выход биомассы в C. glutamicum BS222, выращенных в минимальной среде при различных концентрациях сульфата аммония.
С их двумя путями биосинтеза лизина C. glutamicum хорошо подготовлены для адаптации к варьирующей концентрации сульфата аммония. Путь дегидрогеназы с низкой аффинностью в отношении NH4 + становится активным при высоких концентрациях, в то время как путь сукцинилазы также обеспечивает рост при незначительной доступности аммония. Это также отражается в распределении потоков между этими двумя путями в процессе роста при различных концентрациях сульфата аммония (Eikmanns et al., 1993; Sonntag et al., 1993). Подавление dapD, кодирующего первый фермент ветви сукцинилазы общим регулятором азота amtR, в данном случае, подтверждает то, что путь потребления энергии имеет только небольшое значение, когда поступление аммония является высоким (Buchinger et al., 2009; Silberbach et al., 2005b). Это объясняет не изменяющийся выход лизина в C. glutamicum BS1. Его путь лизина, включающий ветвь сукцинилазы и дегидрогеназы, обладает определенной емкостью и варьирование сульфата аммония, в данном случае, приводит только к сдвигу потоков между этими двумя ветвями. Определение соотношения расщепления потоков с помощью анализа ЯМР показало, что при 5 г·л-1 сульфата аммония ветвь дегидрогеназы обеспечивает приблизительно 40% общего потока в направлении лизина. При такой концентрации сульфата аммония DDH не действует на его максимальном уровне вследствие низкой аффинности в отношении NH4 +, тем самым, ограничивая поток углерода через эту ветвь. Увеличение доступности NH4 + увеличивает активность DDH и, таким образом, ее вклад в общий поток лизина (Sonntag et al., 1993), но, в свою очередь, снижает экспрессию ферментов, образующих ветвь сукцинилазы (Buchinger et al., 2009), так что общий поток лизина не увеличивается. Однако в C. glutamicum BS222 общая емкость каскада лизина возрастает посредством сверхэкспрессии ddh. Вследствие низкой аффинности диаминопимелатдегидрогеназы в отношении NH4 +, эта емкость может быть полностью использована при более высоких концентрациях. В отличие от C. glutamicum BS1, в C. glutamicum BS222 используется эта увеличенная емкость пути лизина для продуцирования лизина, и они не отвечают ускорением роста. Важность ddh для продуцирования лизина в C. glutamicum при высоких концентрациях аммония далее подчеркивается экспериментами с делецией, проведенными ранее (Schrumpf et al., 1991). Делеция, в данном случае, приводила к сильно сниженной продукционной способности лизина.
Однако связанная с плазмидой сверхэкспрессия ddh не была преимущественной в отношении продуцирования лизина (Cremer et al., 1991), хотя обеспечивалось достаточное поступление аммония. В данном случае, следует отметить два возможных объяснения: в дополнение к зависимости от доступности аммония, соотношение для расщепления потоков в пути биосинтеза лизина также проявляет значительную зависимость от времени (Sonntag et al., 1993). При высоких концентрациях сульфата аммония, путь дегидрогеназы обеспечивает более 70% общего потока лизина в пределах первых 12 ч культивирования, что соответствует фазе культивирования, исследованной в настоящем исследовании. Однако на поздней стадии культивирования, исследованной в предшествующей работе, его вклад значительно снижается вплоть до 12% (Sonntag et al., 1993). Более того, сверхэкспрессию проводили в генетически не определенном продуценте. Его получали химическим мутагенезом и отбирали относительно аспартаткиназы с нарушенной регуляцией по принципу обратной связи (Menkel et al., 1989). Накопление дальнейших мутаций, которые преодолевают положительный эффект сверхэкспрессии ddh, таким образом, не может быть исключено. Из этих данных становится очевидным, что пользу стратегии модификации способами инженерии необходимо тщательно проверять, учитывая как генетический фон модифицированного штамма, так и условия тестирования в качестве важных переменных.
На удельную активность диаминопимелатдегидрогеназы не влияла концентрация сульфата аммония в культуральной среде. Оба штамма продемонстрировали идентичную удельную активность при выращивании при 2 г·л-1 или 15 г·л-1, соответственно (фиг.20). Это явно указывает на то, что наблюдаемые отличия в отношении продукционной способности лизина коррелируют с активностью DDH in vivo, которая поддерживается донированием NH4 + в среду и не приводит к измененным уровням сверхэкспрессии ddh в ответ на измененную концентрацию NH4 +.
До некоторой степени, на достигнутое улучшение при высокой концентрации аммония также может влиять уровень экспрессии lysA, кодирующего диаминопимелатдегидрогеназу. При низкой доступности аммония экспрессия lysA подавляется amtR, чтобы способствовать росту вместо образования лизина. Однако это подавление устраняется при увеличении концентраций сульфата аммония, тем самым, обеспечивая увеличение экспрессии lysA (Silberbach et al., 2005b) и, таким образом, более высокую емкость фермента, которая может поддерживать продуцирование лизина в штамме с модифицированным способами инженерии ddh.
Модификация способами инженерии поступления предшественника
Вследствие избытка поступления NADPH, описанные выше штаммы с модифицированным способом PPP имели существенный остаточный потенциал в отношении продуцирования лизина. В данном случае, синтез лизина, вероятно, ограничен недостаточным поступлением оксалоацетата, которое отражается неизмененным анаплеротическим суммарным потоком (фиг.24). Усовершенствования штамма в этом отношении достигали путем увеличения поступления OAA посредством сверхэкспрессии пируваткарбоксилазы или PEP-карбоксилазы (Peters-Wendisch et al., 2001; Sano et al., 1987). Стратегия, примененная в этой работе, была сфокусирована на новых мишенях в направлении улучшенного поступления OAA путем снижения конверсии углерода через метаболические пути, конкурирующие за пируват и фосфоенолпируват в качестве прямых углеродных предшественников OAA.
Метаболический ответ на удаление пируваткиназы
Пируваткиназа играет важную роль в контроле потоков промежуточного метаболизма. Она катализирует необратимую конверсию фосфоенолпирувата в пируват и находится под аллостерическим контролем ATP и AMP (Jetten et al., 1994b). В продуцирующих лизин C. glutamicum удаление пируваткиназы кажется перспективной стратегией для усовершенствования штаммов, поскольку в штаммах с дефицитом пируваткиназы требуемого эквимолярного соотношения двух предшественников лизина: оксалоацетата и пирувата, можно было достигнуть посредством согласованного действия системы фосфотрансферазы (PTS) и фосфоенолпируваткарбоксилазы (PEPC), причем сниженное количество углерода может утрачиваться в виде CO2 вследствие сниженного потока в цикл TCA. Однако в предшествующих исследованиях удаление пируваткиназы в продуцирующих лизин C. glutamicum не обеспечивало четкую картину, так что точные метаболические последствия не полностью понятны (Gubler et al., 1994; Ozaki and Shiio, 1969; Shiio et al., 1990). В данном случае, метаболический ответ C. glutamicum на удаление пируваткиназы детально исследовали на уровне потоков in vivo с использованием расширенной модели, описанной выше. Штамм C. glutamicum BS13 с дефицитом пируваткиназы конструировали путем делеции кодирующего гена pyk в генетическом фоне продуцента лизина C. glutamicum BS1 (lysCT311I).
Активность пируваткиназы
Мутант с делецией pyk C. glutamicum BS13 продемонстрировал полное отсутствие активности пируваткиназы in vitro (<0,6 мЕ·мг-1), в то время как исходный штамм C. glutamicum BS1 продемонстрировал сильную удельную активность, равную 1099 мЕ·мг-1, для этого фермента. Отсутствие активности PYK продемонстрировало, что в C. glutamicum BS13 не остается активности, подобной пируваткиназе. В штамме с делецией прямая конверсия PEP в пируват, таким образом, была ограничена до потребления глюкозы системой фосфотрансферазы.
Влияние делеции на характеристики роста и продуцирования
Для исследования количественных физиологических эффектов делеции пируваткиназы, продуцирующие лизин C. glutamicum BS1 и их производное с дефицитом пируваткиназы C. glutamicum BS13, выращивали в периодической культуре (фиг.21A-D).
Оба штамма проявляли относительно сходную удельную скорость роста, удельную скорость потребления глюкозы или выход биомассы. Однако выход и удельная скорость продуцирования лизина были немного ниже в C. glutamicum BS13 (таблица 21). Более того, отсутствие активности пируваткиназы индуцировало образование избытка метаболитов дигидроксиацетона (DHA) и глицерина. Образование трегалозы было немного снижено.
Характеристики роста и продуцирования лизина в C. glutamicum BS1 и BS13 в процессе периодического культивирования на глюкозе. Приведенные данные представляют собой выход биомассы (YX/S), выход лизина (YLys/S), выход глицерина (YGly/S), выход дигидроксиацетона (YDHA/S), выход трегалозы (YTre/S), а также удельную скорость роста (μ), удельную скорость потребления глюкозы (qGlc) и удельную скорость продуцирования лизина (qLys). Все величины представляют собой средние величины для трех параллельных экспериментов по культивированию. Выходы определяли в качестве наклона линейной наилучшей аппроксимации при нанесении на график образования продукта и потребления субстрата, как представлено на фиг.21, для трех биологических реплик.
Основные кинетические и стехиометрические параметры, включающие удельную скорость роста и выход биомассы, лизина и побочных продуктов, оставались постоянными на протяжении культивирования (фиг.21A-D). Это четко показывает, что оба штамма были в метаболическом устойчивом состоянии, позволяющем использование описанной выше модели расширенных потоков для установления метаболических потоков.
Ответ метаболических потоков на делецию пируваткиназы
Ответ продуцирующих лизин C. glutamicum на удаление пируваткиназы далее исследовали на уровне метаболических потоков углерода. Было интересно детально увидеть, как C. glutamicum BS13 могут компенсировать утрату этого центрального гликолитического гена и практически сохранить рост и характеристики продуцирования исходного штамма.
В данном случае, было необходимо разделение групп метаболитов PEP и пирувата и, таким образом, было необходимо установление всех потоков, вовлеченных в карбоксилирование и декарбоксилирование, так что проводили метаболический анализ потоков с увеличенной метаболической сетью и информацией о мечении из экспериментов с радиоактивным индикатором с 99% [1-13C] глюкозой и 50% [U-13C] глюкозой. В качестве прямого ответа на генетическую модификацию, общий поток конверсии с PEP в пируват значительно снижался с 138% до 100%, что отражало исключительную конверсию PEP в пируват системой фосфотрансферазы для потребления глюкозы. Распределение метаболических потоков на фиг.22 обнаруживает, что удаление пируваткиназы далее приводило к локальному перенаправлению потока в пируватном модуле для обхода ограниченного прямого конвертирования из PEP в пируват. Ответы в пентозофосфатном пути и цикле TCA были скорее слабыми. Более конкретно, два штамма значительно отличались по относительному потреблению PC и PEPC для анаплеротического поступления в цикл TCA. В то время как PC была основным анаплеротическим ферментом в C. glutamicum BS1, она была полностью неактивна в мутанте с делецией, в котором предпочтительно использовалась PEPC для этой цели. Поток через осуществляющие декарбоксилирование ферменты PEPCK и яблочный фермент продемонстрировал только небольшие отличия между двумя штаммами. В общем, делеция пируваткиназы приводила к существенному сдвигу анаплеротического суммарного потока от пирувата в направлении карбоксилирования PEP и обеспечивала метаболический обходной путь с PEP через оксалоацетат и малат в направлении пирувата, вовлекающий PEPC, малатдегидрогеназу и яблочный фермент (фиг.23). Этот обходной путь обеспечил достаточное поступление пирувата так, что поддерживался высокий поток через пируватдегидрогеназу и цикл TCA. В целом C. glutamicum BS13 смог компенсировать делецию генов путем локального перестроения потоков, вовлекающего гибкое использование их анаплеротических ферментов. Узкие доверительные интервалы указывают на то, что все потоки углерода были оценены с высокой точностью (фиг.22). Различия потоков, рассмотренные в данном случае, таким образом, четко связаны с делецией пируваткиназы. Поскольку удельная скорость потребления глюкозы была практически идентична для двух штаммов (таблица 22), все заключения, сделанные для относительных потоков, также были справедливы для абсолютных величин для потоков. Для обоих штаммов была достигнута превосходная аппроксимация между экспериментально определенными и смоделированными массовыми изотопомерами.
Конверсия оксалоацетата в малат в качестве части метаболического обходного пути происходит в обратном направлении суммарного потока цикла TCA, скорее образуя оксалоацетат из малата, и она может потребовать обратимого взаимного конвертирования этих двух метаболитов. Как указано выше, модель расширенных потоков, примененная в данном случае, в действительности учитывает малат и оксалоацетат в качестве единой группы, и превосходная аппроксимация данных о мечении служит в поддержку присутствия обратимого взаимного конвертирования между малатом и оксалоацетатом. Активация этого обходного пути является ключом для компенсации потери активности пируваткиназы и сохранения потока углерода через цикл TCA, а также через другие центральные пути, включающие PPP, гликолиз или анаболизм. Тот же метаболический обходной путь также активируется в E. coli с дефицитом пируваткиназы в процессе выращивания на глюкозе (Al Zaid Siddiquee et al., 2004; Emmerling et al., 2002) и очевидно отражает общую стратегию для микроорганизмов, обладающих обоими из PEPC и PC. Напротив, B. subtilis, лишенные PEPC, не могут расти на глюкозе в отсутствие пируваткиназы (Diesterhaft and Freese, 1973; Fry et al., 2000).
Как C. glutamicum BS1, так и мутант с делецией pyk проявляли существенный поток через яблочный фермент, демонстрируя важную роль этого фермента в гибкости метаболизма C. glutamicum. В мутанте с делецией яблочный фермент является частью созданного метаболического обходного пути и, таким образом, вносит вклад в устойчивость метаболизма к генетическим изменениям. Его поток, т.е. его активность in vivo в обоих штаммах, демонстрирует, что он также играет важную роль в гибкости метаболизма кофакторов в C. glutamicum и демонстрирует, как рассмотрено выше, важность этого фермента в качестве источника NADPH в продуцирующих лизин штаммах C. glutamicum. Как показано по равновесию NADPH (фиг.24), яблочный фермент требуется обоим штаммам для удовлетворения потребности клетки в NADPH и вносит существенный вклад в поступление этого кофактора. Глюкозо-6-фосфатдегидрогеназа и 6-фосфоглюконатдегидрогеназа (PPP), изоцитратдегидрогеназа (ICD) и яблочный фермент (MalE) рассматривали в качестве реакций для поступления NADPH и анаболизма со стехиометрической потребностью в 16,4 ммоль NADPH (г сухой массы клетки)-1 (Wittmann and de Graaf, 2005) и продуцирования лизина со стехиометрической потребностью в 4 моль NADPH (моль лизина)-1 в качестве потребляющих NADPH реакций. Это в значительной степени подтверждает заключения из исследований физиологической роли яблочного фермента.
Более низкий поток через яблочный фермент, а также через PPP в штамме с делецией пируваткиназы может быть связан с увеличенным потоком через изоцитратдегидрогеназу, приводящим к усиленному поступлению NADPH через цикл TCA. Совместную регуляцию путей поступления NADPH, уравновешивающих общее поступление, ранее наблюдали в продуцирующих лизин C. glutamicum (Wittmann and Heinzle, 2002).
Точный эффект делеции пируваткиназы на продуцирование лизина в C. glutamicum очевидно зависит от проецирующего штамма, а также от условий культивирования. В то же время, при делеции пируваткиназы продуцирование лизина было усилено в различных штаммах B. flavum (Ozaki and Shiio, 1969; Shiio et al., 1990), снижение продуцирования наблюдали в C. lactofermentum (Gubler et al., 1994) и, в некоторой степени, также в настоящей работе. Поскольку общее поступление NADPH не изменялось существенно, возможным объяснением может быть ограниченная доступность предшественника лизина - оксалоацетата. Физиологические характеристики и внутриклеточные потоки углерода в C. glutamicum BS1 и C. glutamicum BS13, исследованные в данном изобретении, указывают на возможные объяснения. В этом отношении, накопление дигидроксиацетона и глицерина, в частности, связанное с делецией пируваткиназы, кажется представляющим интерес. Эти избыточные метаболиты образуются C. glutamicum, когда поток, входящий в нижнюю гликолитическую цепь, превышает способность к реакциям нижележащего глицеральдегид-3-фосфата, например в ходе роста на фруктозе (Dominguez et al., 1998; Kiefer et al., 2004). В этих условиях препятствие приписывают глицеральдегид-3-фосфатдегидрогеназе, и оно обеспечивается неблагоприятным соотношением NAD/NADH, снижающим способность этого фермента. Однако наблюдаемые метаболические потоки для C. glutamicum BS1 и мутанта с делецией pyk не предполагают существенных изменений этого соотношения в штамме с дефицитом пируваткиназы, который может вызывать сниженную способность глицеральдегид-3-фосфатдегидрогеназы. Поскольку поток in vivo через этот фермент был также идентичен в двух штаммах, исследованных в данном изобретении, вклад глицеральдегид-3-фосфатдегидрогеназы в наблюдаемое ограничение с высокой вероятностью не проявится. Другие кандидаты являются более перспективными. Как показано, анаплеротический поток полностью сдвигается в сторону PEPC в мутанте с делецией. По сравнению с исходным штаммом, общий поток через этот фермент возрастает приблизительно в 2,5 раза, что может отражать максимальную способность, которой может обладать этот фермент в данных условиях. В этом отношении, штаммы C. glutamicum, ранее демонстрировавшие усиленное продуцирование лизина при делеции пируваткиназы, кроме того, содержали устойчивый к регуляции по принципу обратной связи вариант PEPC, нечувствительный к аллостерическому контролю аспартатом (Shiio et al., 1987), в то время как двойной мутант, лишенный пируваткиназы и PEPC, проявлял существенно нарушенную утилизацию глюкозы (Park et al., 1997). Также кажется возможным, что ограниченная способность яблочного фермента вовлечена в наблюдаемое ограничение. Активность яблочного фермента в C. glutamicum дикого типа с дефицитом пируваткиназы не является достаточной для обеспечения роста на ацетате или цитрате, однако рост может быть сохранен посредством сверхэкспрессии этого фермента (Netzer et al., 2004).
Ослабление пируватдегидрогеназы
Что касается продуцирования лизина в C. glutamicum, пируватдегидрогеназа прямо конкурирует с анаплеротическим ферментом пируваткарбоксилазой в качестве главного источника предшественника лизина - оксалоацетата (Peters-Wendisch et al., 2001). Было показано, что делеция aceE, кодирующего первую субъединицу этого центрального гликолитического гена, увеличивает продуцирование лизина в C. glutamicum. Однако эта делеция полностью блокировала поток углерода через цикл TCA и приводила к существенному дефициту роста и неспособности мутанта расти на типичной среде для продуцирования на основе сахара. Более того, отсутствие активности пируватдегидрогеназы индуцировало обширное образование побочных продуктов (Blombach et al., 2007). В этой работе экспрессию PDH осторожно подавляли путем замены инициирующих кодонов для снижения потока углерода через PDH и поступления в цикл TCA предшественника ацетил-CoA, таким образом, нацеливаясь на увеличенное продуцирование лизина без вредоносных побочных эффектов.
Конструирование и проверка штамма
Замену инициирующего кодона в гене aceE обеспечивали путем замены аллеля дикого типа на мутантный с использованием гомологичной рекомбинации. Модификацию осуществляли в генетическом фоне C. glutamicum BS87. Этот штамм уже содержал несколько модификаций в пути биосинтеза лизина, а также модифицированное способами инженерии поступление предшественников путем сверхэкспрессии и модификации пируваткарбоксилазы и делеции PEP-карбоксикиназы (таблица 1). Клоны после второй рекомбинации подвергали скринингу в отношении их удельной активности PDH, как описано выше (фиг.21). Дальнейший анализ мутантов секвенированием области инициирующего кодона aceE, содержащего участок размером приблизительно 1000 п.н., продемонстрировал 100% идентичность последовательности с диким типом, за исключением нуклеотида инициирующего кодона. Два мутанта со сниженной активностью PDH содержали нуклеотидную замену в инициирующем кодоне (фиг.25). Поскольку никакие другие мутации не встречались в процессе конструирования штамма, сниженная удельная активность PDH в мутантах может быть четко приписана внесенной точечной мутации. Дальнейшие эксперименты проводили с клоном 2, который обозначен как C. glutamicum BS238.
Влияние замены инициирующего кодона на удельную активность фермента
Удельную активность PDH в рекомбинантном штамме BS238 и исходном штамме BS87 определяли в минимальной среде с использованием глюкозы в качестве единственного источника углерода. Как представлено на фиг.26, использование редкого инициирующего кодона GTG вместо распространенного ATG приводило к существенно сниженной удельной активности пируватдегидрогеназы. Хотя исходный штамм C. glutamicum BS87 проявлял удельную активность PDH приблизительно 30 мЕ·мг-1, в C. glutamicum BS238 она составляла только 13 мЕ·мг-1.
Для успешного применения ослабления PDH заменой инициирующего кодона в промышленном процессе, стабильность внесенной точечной мутации является основной проблемой. Ее подтверждали в длительном эксперименте по культивированию, включавшем последовательный перенос экспоненциально растущих клеток в периодической культуре. Этот процесс не влиял на удельную активность PDH в мутантном C. glutamicum BS238. После 50 поколений мутантный штамм все еще проявлял сниженную удельную активность пируватдегидрогеназы (фиг.26), показывая, что модификация стабильно осуществлялась в геноме и не подвергалась обратному изменению.
Влияние на характеристики роста и продуцирования
Подавление активности PDH вызывало увеличение выхода лизина с 142 ммоль·моль-1 в BS87 вплоть до 165 ммоль·моль-1 в C. glutamicum BS238, что соответствует увеличению на 17%. На образование биомассы модифицированная активность фермента влияла мало (фиг.27).
Однако удельная скорость роста C. glutamicum BS238 (μ=0,25 ч-1) была немного снижена по сравнению с C. glutamicum BS87 (μ=0,33 ч-1). Рекомбинантный штамм не продемонстрировал ни аукстотрофии, ни значительных изменений в образование побочных продуктов, и продуцировались только небольшие количества трегалозы (<10 ммоль·моль-1). Выходы лизина и биомассы, а также удельная скорость роста были постоянными на протяжении всего культивирования, демонстрируя, что штаммы были в метаболическом устойчивом состоянии. Наблюдаемые отличия между двумя штаммами, таким образом, свойственны измененной последовательности инициирующего кодона и, таким образом, сниженной удельной активности пируватдегидрогеназы.
Ответ потоков на подавление пируватдегидрогеназы
Измененная физиология штамма C. glutamicum BS238 в ответ на сниженную удельную активность пируватдегидрогеназы также указывает не измененные потоки в пируватном модуле. Для более подробного исследования влияния модифицированной активности фермента на эти метаболические пути, оценивали поток PDH и суммарный поток через пируваткарбоксилазу в C. glutamicum BS238 и его исходном штамме. В данном случае, стехиометрическую корреляцию между выходом лизина, выходом биомассы и потоком PDH, полученными для 18 независимых экспериментов по 13C мечению с C. glutamicum, использовали в качестве основы для вычисления (фиг.13). Это выявило небольшое, но существенное снижение потока через модифицированный фермент пируватдегидрогеназу (таблица 23). Вместе с одновременным увеличением суммарного потока через пируваткарбоксилазу (таблица 22), это приводит к сдвигу потока от PDH к анаплерозу. Значимость наблюдаемых отличий подтверждали с помощью t-критерия, учитывая поток PDH и поток PYC (таблица 22).
Поток in vivo через пируватдегидрогеназу (nPDH) и общий суммарный поток через анаболическое карбоксилирование (nPYC) выращенных на глюкозе C. glutamicum BS87 (исходный) и C. glutamicum BS238 (aceEatt). Отклонение отражает 90% доверительный интервал, полученный анализом Монте-Карло.
Центральной целью подходов рациональной метаболической инженерии, нацеленных на усиленное продуцирование лизина, является идентификация и внесение исключительно благоприятных мутаций, которые не приводят к нежелательным вредоносным побочным эффектам (Ohnishi et al., 2002). Это обеспечивает улучшенные клеточные фабрики без нарушенного поведения роста или нарушенной устойчивости к стрессу (Park and Lee, 2008). Однако понятно, что в дополнение к предполагаемым благоприятным изменениям, многие из этих хорошо определенных направленных модификаций все еще приводят к неблагоприятным побочным эффектам, поскольку используемые генетические изменения, как правило, являются сильными. Подходящим примером является недавно описанная делеция пируватдегидрогеназы в продуцирующих лизин C. glutamicum, обеспечивающая увеличенное образование побочных продуктов и неспособность расти на имеющих промышленное значение средах на основе сахаров (Blombach et al., 2007). Это строго вызывает потребность в более постепенной модификации каскада, которая может уравновешивать положительные и отрицательные эффекты. Помимо предшествующей работы, 60% ослабление пируватдегидрогеназы усиливает продуцирование лизина, но все еще обеспечивает сбалансированный рост на имеющих промышленное значение сахарах. Более того, можно полностью избежать обширного образования побочных продуктов в пируватном модуле, обнаруженного для мутантов с делецией (Blombach et al., 2007). Оценка метаболических потоков в пируватном модуле показала, что польза от сниженной активности может быть четко приписана сдвигу потока от пируватдегидрогеназы в направлении анаплеротического карбоксилирования. Модифицированный штамм уже проявляет модифицированное способами инженерии поступление предшественников, включающее усиленную экспрессию пируваткарбоксилазы, а также ключевую мутацию P458S в гене pyc. Было показано, что обе мутации увеличивают продуцирование посредством усиленного поток анаплеротического карбоксилирования (Ohnishi et al., 2002; Peters-Wendisch et al., 2001). Эти модификации и, таким образом, увеличенная емкость пируваткарбоксилазы, могут участвовать в эффективном перенаправлении потоков, тем самым, увеличивая продуцирование лизина. Интересно отметить, что на 60% сниженной активности пируватдегидрогеназы достигали путем замены только одного нуклеотида, т.е. модификацией кодона инициации трансляции. Этот способ обеспечивает доступ к ферментам и путям, которые являются довольно чувствительными в отношении генетических нарушений и являются необходимыми для поддержания роста и жизнеспособности.
Модификация способами инженерии цикла TCA
В дополнение к метаболизму NADPH, поступлению предшественников и биосинтезу лизина, цикл TCA является перспективным метаболическим путем для модификации способами инженерии продуцирования лизина в C. glutamicum, что отражается наблюдаемой корреляцией между потоком лизина и потоком цикла TCA (Фиг.12) (Wittmann and Becker, 2007; Wittmann and Heinzle, 2002). Однако генетическая инженерия, нацеленная на сниженную конверсию углерода посредством этого каскада, не является тривиальной, поскольку цикл TCA необходим для роста клеток, что не позволяет полное блокирование путем делеции гена. В этой работе экспрессию icd, кодирующего изоцитратдегидрогеназу, подвергали рациональному ослаблению. В противоположность другим организмам, уровень экспрессии ICD в C. glutamicum не зависит ни от фазы роста, ни от условий роста (Eikmanns, 2005). При удельной активности приблизительно 1 Е·мг-1 (Eikmanns et al., 1995), ICD является наиболее высоко экспрессируемым ферментом цикла TCA и, таким образом, является перспективным кандидатом для подавления. Это было осуществлено, в данном случае, путем замены инициирующего кодона кодирующего гена icd. Модификация была осуществлена в генетическом фоне продуцента лизина C. glutamicum BS87. Затем исследовали эффект на активность фермента, характеристики продуцирования и поток цикла TCA.
Конструирование и проверка штамма
В двух событиях гомологичной рекомбинации аллель дикого типа изоцитратдегидрогеназы, содержащий инициирующий кодон ATG, заменяли мутантным аллелем начального кодона GTG. Скрининг проводили, как описано выше, посредством определения удельной активности фермента в клонах с увеличенным ростом после второго события рекомбинации (фиг.21). Проведенный впоследствии анализ последовательности клонов с существенно сниженной удельной активностью ICD явно указывает на эти клоны как на носители инициирующего кодона GTG (фиг.28).
Установление полной последовательности, вовлеченной в события гомологичной рекомбинации, обеспечили таким образом, чтобы в процессе конструирования штамма не происходили какие-либо другие мутации. Выравнивание последовательностей, в данном случае, выявило 100% идентичность между выбранными мутантами и исходным штаммом BS87 относительно полного участка из 1000 п.н., за исключением нуклеотидной замены инициирующего кодона. Наблюдаемое снижение ферментативной активности, таким образом, было прямым следствием использования редкого инициирующего кодона GTG.
Влияние замены инициирующего кодона на активность фермента
После проверки штамма секвенированием удельную активность ICD исходного штамма C. glutamicum BS87 и мутанта по инициирующему кодону C. glutamicum BS205 сравнивали в процессе роста на ферментативной среде. Как представлено на фиг.28, использование редкого инициирующего кодона GTG вместо общего ATG приводило к существенно сниженной удельной активности изоцитратдегидрогеназы. Хотя исходный штамм C. glutamicum BS87 проявлял удельную активность ICD приблизительно 1,3 Е·мг-1, в C. glutamicum BS205 она составляла только 0,3 Е·мг-1.
Сниженная активность ICD в штамме C. glutamicum BS205 может быть четко приписана замене инициирующего кодона и, таким образом, отражает трансляционное ослабление экспрессии генов. По сравнению с аллелем icd дикого типа, распознавание инициирующего кодона в мутанте по GTG ухудшено, что приводит к нарушению процесса инициации трансляции, вовлекающего связывание мРНК с рибосомой (O'Donnell and Janssen, 2001; Vellanoweth and Rabinowitz, 1992). Это существенно снижает частоту трансляции, а также стабильность мРНК, обе из которых приводят к снижению завершения образования функционально активного белка изоцитратдегидрогеназы и, таким образом, сниженной ферментативной активности. Как было подтверждено длительным культивированием с последовательными партиями, модификация и ее эффект на активность фермента были стабильными (фиг.29), что является важной предпосылкой в отношении промышленного применения.
Влияние на характеристики роста и продуцирования
Для исследования влияния измененной ферментативной активности на характеристики продуцирования, проводили эксперименты по культивированию для сравнения роста, продуцирования лизина и образования биомассы в различных штаммах в процессе роста в модифицированной минимальной среде. Путем подавления ICD, выход лизина в процессе роста на глюкозе увеличивался с 141 ммоль·моль-1 в BS87 вплоть до 200 ммоль·моль-1 в C. glutamicum BS205, что соответствовало увеличению, составляющему 42%. Влияние на образование биомассы было незначительным (таблица 23). Однако удельная скорость роста была снижена в мутантном штамме. В то время как исходный штамм BS87 рос с удельной скоростью 0,32 ч-1, BS205 проявлял удельную скорость роста 0,28 ч-1.
Характеристики продуцирования продуцирующих лизин C. glutamicum BS87 и C. glutamicum BS205 (icdatt) в ходе роста на глюкозе. Приведенные данные представляют собой выход биомассы (YX/S) и выход лизина (YLys/S). Выходы определяли для трех биологических повторений в качестве наклона линейной наилучшей аппроксимации при нанесении образования продукта против потребления субстрата (фиг.30).
Достаточное поступление кислорода препятствовало образованию побочных продуктов, индуцируемых недостатком кислорода (Inui et al., 2004). В обоих штаммах продуцировалось только небольшое количество трегалозы (<10 ммоль·моль-1). Выходы лизина и биомассы, а также удельная скорость роста были постоянными на протяжении всего культивирования, демонстрируя, что штаммы находились в метаболическом устойчивом состоянии (фиг.30). Наблюдаемые отличия между двумя штаммами, таким образом, четко свойственны измененной последовательности инициирующего кодона и, таким образом, сниженной удельной активности изоцитратдегидрогеназы.
Ответ метаболических потоков на подавление icd
Данные из определения активности фермента и сравнительных экспериментов по культивированию четко показали, что использование редкого инициирующего кодона GTG вместо ATG существенно изменяло клеточную физиологию штамма. Для более подробного исследования влияния активности модифицированного фермента на метаболические пути, поток через цикл TCA и суммарный поток через пируваткарбоксилазу оценивали в C. glutamicum BS87 и C. glutamicum BS205. Для этого устанавливали стехиометрическую корреляцию между выходом лизина, выходом биомассы и потоком цикла TCA (фиг.12). Входящий поток через цитратсинтазу снижался в мутанте по icd с одновременным увеличением суммарного потока через пируваткарбоксилазу (фиг.31). Полученный t-критерий явно показал значимое отличие между штаммами при рассмотрении потока цикла TCA (t=-10,1) и потока PYC (t=43,6), соответственно. В целом, эти изменения потоков приводили к перенаправлению потока от цикла TCA в направлении анаплероза.
Как уже предположено для ослабления PDH, сдвиг потока в направлении анаплеротического карбоксилирования, вероятно, поддерживается модифицированной пируваткарбоксилазой. Достигнутое в данном случае улучшение выхода лизина, составляющее 40%, значительно превышает увеличение вследствие замены инициирующего кодона в гене aceE. Это может отражать общее достигнутое снижение активности фермента, которое было наиболее выраженным для гена icd с результатом 70%. Несмотря на пользу сниженной активности фермента для продуцирования лизина, модификация не нарушала в высокой степени рост или конверсию углерода рассматриваемыми каскадами, что, тем самым, проявлялось образованием побочных продуктов. Оставшуюся высокую активность ICD, составляющую 300 мЕ·мг-1 в мутанте по инициирующему кодону оставляет возможность для продолжения оптимизации путем дальнейшего подавления. Отсутствие какого-либо образования побочного продукта в результате индуцированного препятствия в циклах TCA указывает на то, что метаболическая сеть в пируватном модуле C. glutamicum BS205 обладала способностью к удовлетворению еще большему ослаблению. Одной возможной стратегией могло быть внесение наиболее редкого варианта инициирующего кодона TTG. Помимо настоящего исследования, направленное подавление цикла TCA кажется также перспективным для различных других продуктов, поскольку высокая потеря углерода через образование CO2 в цикле TCA является, как правило, нежелательной с отношении выхода углерода для желаемого продукта. Обычными продуктами в этом отношении являются аминокислоты семейства аспартата или продуцирование диаминопентана, который прямо образуется из лизина декарбоксилированием. То же самое также справедливо для ослабления пируватдегидрогеназы, описанного выше.
Конструирование специализированного гиперпродуцента лизина
В предшествующих главах успешно использовали различные стратегии для рациональной оптимизации продуцирования лизина в C. glutamicum. Проиллюстрированные для исходных штаммов C. glutamicum BS1 и C. glutamicum BS87, исследовали пользу нескольких мишеней ключевых путей биосинтеза лизина, метаболизма NADPH, поступления предшественников и образования CO2. С целью создания гиперпродуцента на основе дикого типа в качестве представляющего интерес штамма для промышленного продуцирования, идентифицированные модификации теперь комбинировали в одном штамме. Эта работа была основана на C. glutamicum дикого типа ATCC 13032 для осуществления только благоприятных модификаций и для минимизации какого-либо вредоносного побочного действия, появляющегося в промышленных продуцирующих штаммах.
Конструирование штамма и эффективность продуцирования
Первым шагом в направлении создания гиперпродуцента было освобождение аспартаткиназы от ингибирования по принципу обратной связи лизином и треонином, внесение аминокислотной замены T311I (Becker et al., 2005). Это дополнялось описанной выше сверхэкспрессией ddh. Единое осуществление этих двух модификаций и дополнительная оптимизация среды обеспечивали продуцент лизина, который уже продуцировал 125 ммоль лизина (моль глюкозы)-1. Чтобы избежать других препятствий в пути биосинтеза лизина, в дополнение к ddh сверхэкспрессировали дигидродипиколинатредуктазу (dapB), аспартаткиназу (lysC) и диаминопимелатдекарбоксилазу (lysA). Все эти ферменты являются частью общего пути биосинтеза лизина. Таким образом, эффект на продуцирование лизина должен быть независимым от распределения потоков между ветвью сукцинилазы и дегидрогеназы. Дальнейшую оптимизацию штамма проводили путем осуществления нескольких дополнительных генетических модификаций, которые уже доказали свою ценность в последние годы. Они включали замену природной гомосериндегидрогеназы мутантным вариантом, обладающим аминокислотной заменой V59A. Эта мутация увеличивает продуцирование лизина путем снижения потока углерода в направлении каскада треонина, который прямо конкурирует с биосинтезом лизина (Ohnishi et al., 2002). Для обеспечения достаточного поступления оксалоацетата для образования лизина, дополнительные модификации были сфокусированы на предоставлении предшественника. Это предполагало сверхэкспрессию и модификацию основного анаплеротического фермента пируваткарбоксилазы (Ohnishi, et al., 2002; Peters-Wendisch, et al., 2001) и делецию потребляющей OAA реакционной PEP-карбоксикиназы (Petersen, et al., 2001). Таким образом, полученный штамм C. glutamicum BS87 содержал 9 модификаций в биосинтезе лизина и поступлении предшественника (фиг.32).
Из сравнения с его более ранним штаммом-предшественником C. glutamicum BS222, обладающим только регуляторной точечной мутацией в гене lysC и второй копии гена ddh, становится очевидным, что дополнительные модификации в C. glutamicum BS87 обеспечивают только небольшое улучшение продуцирования лизина (фиг.18). Следовательно, стратегия инженерии, которая сфокусирована только на конкретной части метаболизма, не пригодна для создания эффективного продуцирующего штамма. Однако история оптимизации штамма в направлении улучшенного продуцирования лизина путем метаболической инженерии, явно демонстрируется этой стратегией. Таким образом, ключевые каскады цикл TCA и метаболизм NADPH в качестве основных реакций в концепции оптимизированного продуцента лизина были практически не приняты во внимание. В этом отношении, настоящая работа выявила генетические мишени, которые прекрасно дополняются предшествующими стратегиями для преодоления этих ограничений, тем самым, обеспечив продвижение в направлении конструирования улучшенного продуцирующего штамма. Таким образом, следующая стадия в направлении создания предполагаемого гиперпродуцента, была сфокусирована на цикле TCA. Как описано выше, ослабление изоцитратдегидрогеназы (icd att) в генетическом фоне C. glutamicum BS87 значительно увеличивало продуцирование лизина путем эффективного перенаправления потока с цикла TCA в сторону анаплеротического карбоксилирования. Что касается продуцирования лизина, эта стратегия инженерии имеет преимущество вследствие увеличенного поступления оксалоацетата, а также сниженной потери углерода посредством образования CO2. Последующую модификацию способами инженерии метаболизма NADPH проводили с помощью двух различных стратегий. Сначала сверхэкспрессировали фермент глюконеогенеза фруктозо-1,6-бисфосфатазу, который, как ранее было показано, вносит значительный в клад в увеличенное поступление NADPH (Becker et al., 2005). Как показано выше, этот подход хорошо дополняется усиленной экспрессией глюкозо-6-фосфатдегидрогеназы. Для преодоления дальнейших ограничений в PPP вследствие ограниченной способности транскетолазы и трансальдолазы с самого начала, была осуществлена сверхэкспрессия G6PDH одновременно со сверхэкспрессией транскетолазы и трансальдолазы путем активации полного оперона tkt через sod-промотор, как описано выше. В ответ на замену промотора значительно увеличивалась удельная активность G6PDH, транскетолазы и трансальдолазы. Концепция стратегии инженерии проиллюстрирована на фиг.32. Польза от модификаций становится очевидной из сравнения выхода конверсии углерода в полученной генеалогии (фиг.18).
Начиная с C. glutamicum дикого типа ATCC 13032, последующее осуществление благоприятных модификаций приводило к гиперпродуценту лизина на основе штамма дикого типа. В настоящей работе впервые описан такой полный рациональный штамм, в котором учитываются все каскады. Важным было первоначальное внесение модификации в аспартаткиназу для преодоления ингибировании по принципу обратной связи этого ключевого регуляторного фермента. Значительная польза в виде практически 50% улучшения вследствие сверхэкспрессии ddh явно показала, что общая способность пути биосинтеза лизина в генетическом фоне C. glutamicum BS1 (lysCT311I) сильно ограничивала продуцирование лизина. Однако дальнейшая модификация каскада лизина, а также модификация способами инженерии поступления предшественника, которая была описана в предшествующих исследованиях (Ohnishi, et al., 2002; Peters-Wendisch, et al., 2001; Petersen, et al., 2001), имела в данном случае только умеренный успех. Это явно указывает на то, что существуют другие препятствия в центральном метаболизме C. glutamicum, которые ухудшают продуцирование лизина. Таким образом, в настоящем исследовании интерес в направлении дальнейшей модификации способами инженерии продуцирования лизина был смещен на другие части промежуточного метаболизма. Успех этой концепции отражался непосредственно ответом на ослабление изоцитратдегидрогеназы (icd att). Внесение единичной нуклеотидной замены в инициирующий кодон icd увеличило выход лизина с 141 ммоль·моль-1 в C. glutamicum BS87 вплоть до 200 ммоль·моль-1 в соответствующем мутанте icd, что отражает увеличение на 40%. Последующая сверхэкспрессия фруктозо-1,6-бисфосфатазы, которая, как ранее было показано, приводит к эффективному перенаправлению потоков в направлении предоставляющего NADPH PPP (Becker et al., 2005), далее усиливала продуцирование на 18% (фиг.18). Конечная внесенная модификация в описанную в настоящем описании генеалогию включала замену промотора оперона tkt. Это значительно увеличивало активность ферментов G6PDH, TKT и TAL, кодируемых этим опероном, что, в свою очередь, приводило к улучшению на 13% выхода лизина. Преимущество вследствие сверхэкспрессии fbp и оперона tkt явно демонстрирует, что недостаточное поступление NADPH строго ограничивает продуцирование лизина в C. glutamicum, и что стратегии инженерии, нацеленные на увеличение потока PPP, являются необходимыми для цели создания эффективного гиперпродуцирующего лизин штамма. Дополнительное преимущество усиленной экспрессии генов пентозофосфатного пути наблюдали в отношении поведения роста. По сравнению с его предшественником C. glutamicum BS242 (Peftufbp), проявляющим удельную скорость роста 0,23 ч-1, мутант tkt рос со скоростью 0,32 ч-1. Более высокий выход конверсии углерода (YLys/S=26%) в комбинации с улучшенным поведением C. glutamicum BS244 увеличил эффективность продуцирования этого штамма, поскольку это обеспечивает эффективное продуцирование лизина за укороченное время ферментации. Путем осуществления последних трех модификаций достигали общего увеличения продуцирования лизина, составляющего почти 90%. Это является примечательной, а также неожиданной пользой вследствие только трех генетических изменений. Таким образом, можно заключить, что эти стратегии инженерии частично имеют преимущество от ранее осуществленных мутаций, например, удаления препятствий в биосинтезе лизина.
Характеристики продуцирования исследуемых штаммов также тестировали при культивировании во вращающихся флаконах для экспоненциально растущих клеток в минимальной среде. В этих условиях наилучший продуцент C. glutamicum BS244 уже проявлял примечательный выход лизина 26%, хотя постановка эксперимента, включающая культивирование во вращающемся флаконе в минимальной среде при низких концентрациях субстрата, определенно ограничивает их продуктивность. Таким образом, было актуально исследовать эффективность продуцирования гиперпродуцента лизина в процессе ферментации с подпиткой на конкретной промышленной среде для продуцирования, поскольку эта стратегия культивирования наиболее близка промышленному продуцированию. Более того, она позволяет более реалистичную оценку возможностей нового гиперпродуцента, особенно по сравнению классически получаемыми продуцирующими штаммами. Наилучший продуцент C. glutamicum BS244(Psod tkt) исследовали в промышленных условиях, включающих периодическую ферментацию с подпиткой на комплексной среде на основе патоки.
Эффективность продуцирования в промышленных условиях ферментации
Профиль культивирования периодической ферментации с подпиткой для C. glutamicum BS244 представлен на фиг.19. Продуцирование лизина начиналось рано и концентрация лизина в культуральном супернатанте постоянно увеличивалась в течение 30 ч вплоть до неожиданного высокого конечного титра 120 г·л-1. Основное увеличение, главным образом, достигалось в фазу подпитки, которую начинали сразу после потребления сахара, предоставленного в среде для периодической ферментации (100 г·л-1). В качестве сигнала для автоматизированной подпитки в ходе процесса использовали сигнал, основанный на растворенном кислороде (pO2). Насыщение O2 в ферментере контролировали скоростью мешалки и поддерживали постоянным на уровне 20%. Ограничение углерода в среде приводило к немедленному и быстрому увеличению pO2 которое активировало насос для подпитки, когда увеличение pO2 превышало 10% мин-1. Раствор для подпитки на основе патоки и глюкозы, дополнительно обогащали сульфатом аммония для обеспечения достаточного поступления аммония для продуцирования лизина. Посредством этой стратегии концентрацию сахара в фазе подпитки поддерживали на уровне ниже 10 г·л-1.
Концентрация биосмассы с течением времени, на которую указывала оптическая плотность, явно отличалась от концентрации лизина (фиг.34). В ходе стационарной фазы, оптическая плотность увеличивалась на коэффициент, равный пяти, в то время как в фазу подпитки наблюдали только пороговое увеличение. Более того, концентрация биомассы не увеличивалась до конца периода культивирования, но достигала максимума через 24 ч.
Более тщательное исследование характеристик продуцирования штамма C. glutamicum BS244 показало, что процесс ферментации может быть далее разделен. Как представлено на фиг.35, различные фазы могут отличаться, исходя из достигнутого выхода лизина. На фазе наилучшего продуцирования, обозначенной здесь как фаза подпитки 2, гиперпродуцент лизина обладал выходом 55%. Другой характеристикой этой фазы является практически не изменяющаяся концентрация биомассы. Таким образом, потребленный сахар эффективно направляется на путь биосинтеза лизина. В начале процесса ферментации выход лизина был низким. Этот интервал, включающий стационарную фазу и фазу подпитки 1, может быть четко описан как основная фаза роста, характеризующаяся обширным образованием биомассы (фиг.34). Интересно, что на этой фазе был достигнут выход лизина 25%, который хорошо соответствует выходу лизина, достигаемому в экспериментах с культивированием во вращающихся флаконах в ходе экспоненциальной фазы роста.
Гиперпродуцент лизина, созданный в этой работе, представляет собой наилучший продуцирующий штамм на основе дикого типа, описанный до настоящего времени. Учитывая конечный титр лизина, а также выход углерода и выход в единицу времени, он является явно лучшим, чем описанные рационально созданные штаммы (Ikeda et al., 2006). Имея конечный титр лизина 120 г·л-1 и выход конверсии углерода вплоть до 55%, этот штамм даже проявляет характеристики продуцирования, которые находятся на уровне максимального предела классически получаемых штаммов с описанными выходами углерода 40-50% (Leuchtenberger et al., 2005) и титрами лизина 80-120 г·л-1 (Anastassiadis, 2007). Однако такая эффективность продуцирования классических продуцентов, главным образом, связана с длительным временем ферментации вплоть до 100 ч (Anastassiadis, 2007), которое значительно ухудшает выход в единицу времени. Это, как правило, связано с плохим ростом, вызываемым рядом вредоносных вторичных мутаций, которые накапливались в процессе разработки штамма вследствие неспецифического мутагенеза. Путем единственно осуществления исключительно набора только из 12 благоприятных модификаций, этих нежелательных побочных действий можно было избежать в продуцирующем штамме на основе дикого типа. Быстрый рост и, таким образом, укороченное время ферментации, составляющее только 30 ч, приводит к примечательно высокому выходу в единицу времени, составляющему 4 г·л-1·ч-1, который значительно превышает выход в единицу времени, составляющий 2,1 г·л-1·ч-1, достигаемый в ходе периодической ферментации с подпиткой классическими продуцентами (Hirao et al., 1989). Продуктивность, полученная в данном случае, может быть еще увеличена дополнительной модификацией способами генетической инженерии или усовершенствованием технологических действий. В этом отношении, особенно оптимизированная стратегия подпитки может значительно улучшить продуктивность (Hirao et al., 1989; Ikeda, 2003).
Гиперпродуцент лизина на основе дикого типа по настоящему изобретению демонстрирует возможность системной митаболической инженерии в качестве способа для оптимизации штаммов. Штамм C. glutamicum BS244 является превосходным подтверждением концепции и подтверждением ценности полного рационального продуцирующего штамма, который является высоко привлекательным для промышленного применения.
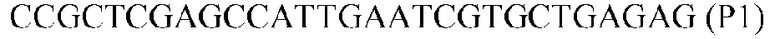
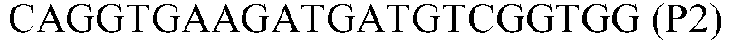
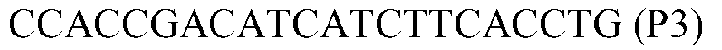
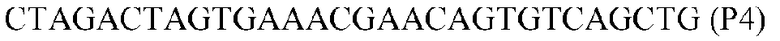
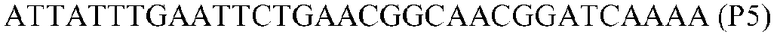
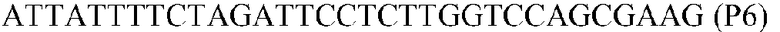
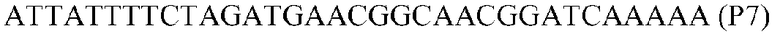
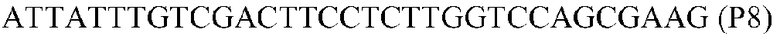
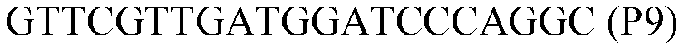
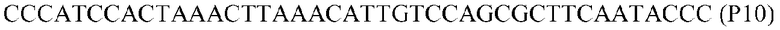
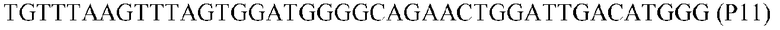
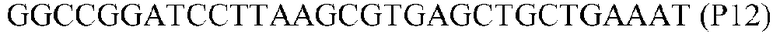
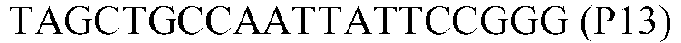
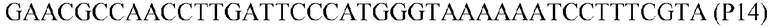
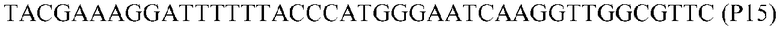
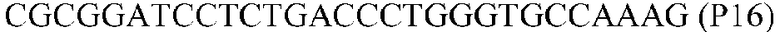
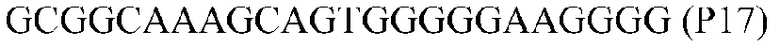
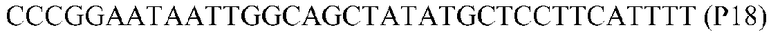
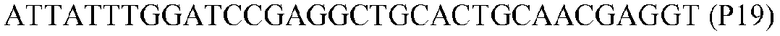


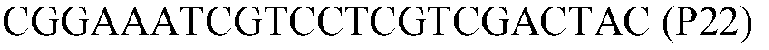
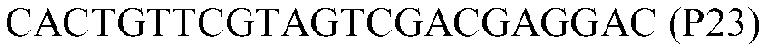
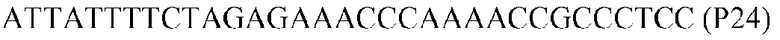
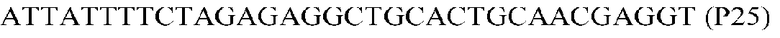
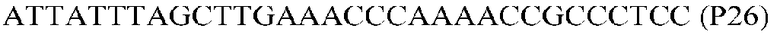

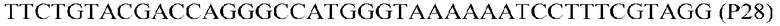
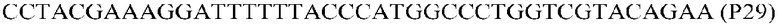
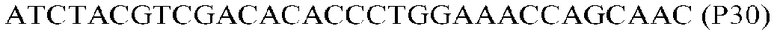

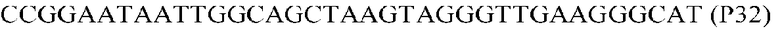
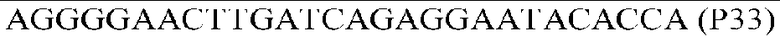

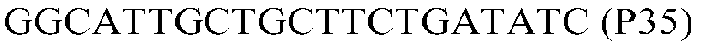
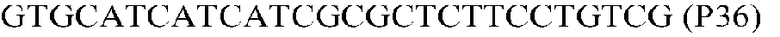
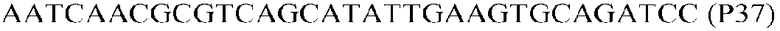
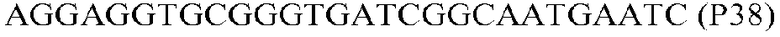
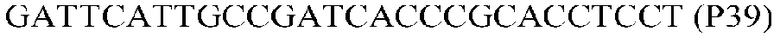
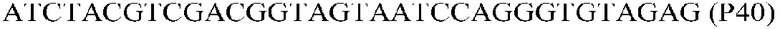
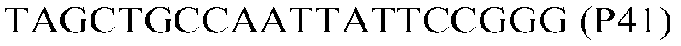
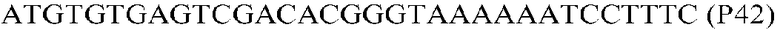
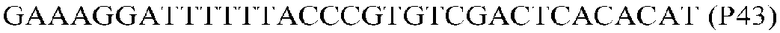
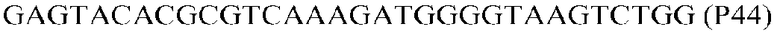
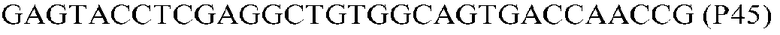
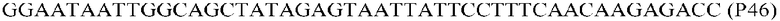
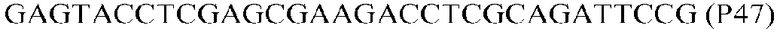
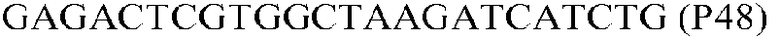
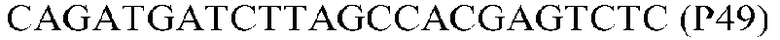
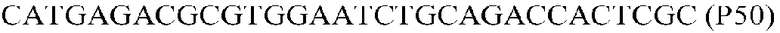

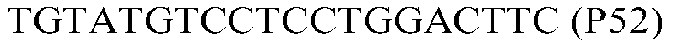
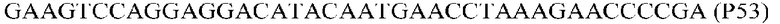
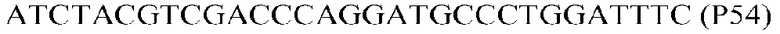
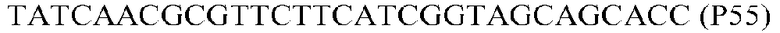
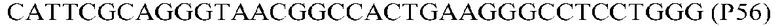
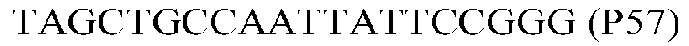
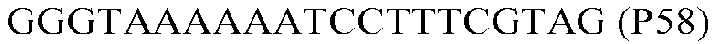
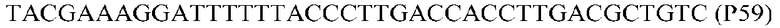
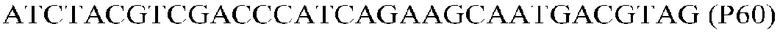
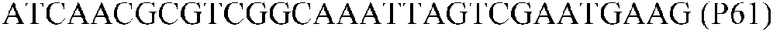
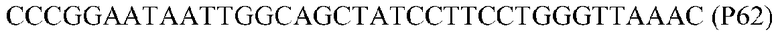
ОПРЕДЕЛЕНИЯ
Сокращения
2-OXO - 2-оксоглутарат
AA - Аминокислота
Ac-CoA - Ацетил-кофермент A
Ace - Ацетат
aceE - Ген, кодирующий субъединицу один комплекса пируватдегидрогеназы
ADP - Аденозиндифосфат
amtR - Ген, кодирующий общий регулятор азота
amyA - Ген, кодирующий α-амилазу
ara - Кластер генов для утилизации арабинозы
asd - Ген, кодирующий аспартатполуальдегиддегидрогеназу
argS - Ген, кодирующий аргинил-тРНК-синтетазу
ATCC - American type culture collection
ATP - Аденозинтрифосфат
BamH1 - Фермент рестрикции из Bacillus amyloli
п.н. - пара(ы) оснований (нуклеотидов)
BSA - Бычий сывороточный альбумин
BSTF - N,O-бис(триметилсилил)трифторацетамид
CDM - Сухая масса клеток
CIT - Цитрат
CoA - Кофермент A
CWW - Влажная масса клеток
dapA - Ген, кодирующий дигидродипиколинатсинтазу
dapB - Ген, кодирующий дигидродипиколинатредуктазу
dapC - Ген, кодирующий сукциниламинокетопимелаттрансаминазу
dapD - Ген, кодирующий тетрагидропиколинатсукцинилазу
dapE - Ген, кодирующий сукцинилдиаминопимелатдесукцинилазу
dapF - Ген, кодирующий диаминопиметалэпимеразу
ddh - Ген, кодирующий диаминопимелатдегидрогеназу
DDH - Диаминопимелатдегидрогеназа
DHA - Дигидроксиацетон
DHAP - Дигидроксиацетонфосфат
ДМСО - Диметилсульфоксид
DNase - Дезоксирибонуклеаза
DTT - Дитиотреитол
E4P - Эритроза-4-фосфат
eftu - Ген, кодирующий фактор элонгации tu
F1P - Фруктоза-1-фосфат
F6P - Фруктоза-6-фосфат
FBP - Фруктозо-1,6-бифосфат
fbp - Ген, кодирующий фруктозо-1,6-бифосфатазу
FBPase - Фруктозо-1,6-бифосфатаза
fbr - Устойчивый к регуляции по принципу обратной связи
Fru - Фруктоза
G6P - Глюкозо-6-фосфат
G6PDH - Глюкозо-6-фосфатдегидрогеназа
GAP - Глицеральдегид-3-фосфат
ГХ-МС - Газовая хроматография/масс-спектрометрия
GDH - Гицерофосфатдегидрогеназа
Glu - Глюкоза
gltA - Ген, кодирующий цитратсинтазу
Gly - Глицерин
GRAS - В целом признан безопасным
hom - Ген, кодирующий гомосериндегидрогеназу
ВЭЖХ - Высокоэффективная жидкостная хроматография
icd - Ген, кодирующий изоцитратдегидрогеназу
ICD - Изоцитратдегидрогеназа
Kan - Канамицин
Lac - Лактат
LDH - Лактатдегидрогеназа
Lys - Лизин
lysA - Ген, кодирующий диаминопимелатдекарбоксилазу
lysC - Ген, кодирующий аспартаткиназу (аспартокиназа)
lysE - Ген, кодирующий лизинпермеазу
malE - Ген, кодирующий яблочный фермент
MalE - Яблочный фермент
MHDSTFA - N-метил-N-трет-бутилдиметилсилилтрифторацетамид
MCS - Сайт множественного клонирования
MDV - Вектор распределения масс
MDH - Цитоплазматическая малатдегидрогеназа
MFA - Метаболический анализ потоков
MQO - Мембраносвязанная малатдегидрогеназа
NAD/NADH - Никотинамидадениндинуклеотид окисленный/восстановленный
NADP/NADPH - Никотинамидадениндинуклеотидфосфат окисленный/восстановленный
OAA - Оксалоацетат
OD - Оптическая плотность
ODHC - Комплекс 2-оксоглутаратдегидрогеназы
OPA - Ортофтальдиальдегид
opcA - Ген, кодирующий предполагаемую субъединицу G6PDH
ORF - Открытая рамка считывания
ORI - Ориджин репликации
PC - Пируваткарбоксилаза
pyc - Ген, кодирующий пируваткарбоксилазу
pck - Ген, кодирующий PEP-карбоксикиназу
PCR - Полимеразная цепная реакция
PDH - Комплекс пируватдегидрогеназы
PEP - Фосфоенолпируват
PEPC - Фосфоенолпируваткарбоксилаза
PEPCK - Фосфоенолпируваткарбоксикиназа
pgi - Ген, кодирующий фосфоглюкоизомеразу
PGI - Фосфоглюкоизомераза
PPP - Пентозофосфатный путь
PTS - Система фосфотрансферазы
Pwo - ДНК-полимераза из Pyrococcus woesei
pyk - Ген, кодирующий пируваткиназу
Pyr - Пируват
R5P - Рибозо-5-фосфат
RNase - Рибонуклеаза
RPE - Рибулозо-5-фосфатэпимераза
RPI - Рибоза-5-фосфатизомераза
RQ - Дыхательный коэффициент
S7P - Седогептулоза-7-фосфат
sacB - Ген, кодирующий левансукразу Bacillus subtilis
SAP - Щелочная фосфатаза креветок
sod - Ген, кодирующий супероксиддисмутазу
Suc - Сахароза
SUC - Сукцинат
tal - Ген, кодирующий трансальдолазу
TAL - Трансальдолаза
Taq - ДНК-полимераза из Thermus aquaticus
Цикл TCA - Цикл трикарбоновых кислот
Tet - Тетрациклин
tkt - Ген, кодирующий транскетолазу/оперон транскетолазы
TKT - Транскетолаза
TPI - Триозофосфатизомераза
TPP - Тиаминпирофосфат
SLS - Сумма наименьших квадратов
Tre - Трегалоза
об. в мин. - Обороты в минуту
XhoI - Фермент рестрикции из Xanthomonas holcicola
xyl - Кластер генов для утилизации ксилозы
Обозначения
C - Емкость [Ф]
c - Концентрация [моль·л-1] или [г·л-1]
м - Масса [г]
µ - Удельная скорость роста [ч-1]
n - Количество вещества [моль]
qs - Удельная скорость поглощения [ммоль·г-1·ч-1]
qp - Удельная скорость продуцирования [ммоль·г-1·ч-1]
R - Сопротивление [Ом]
T - Температура [°С]
t - Время [ч]
Е - Единицы [мкмоль·мин-1]
ν - Поток [%]
λ - Длина волны [нм]
YP/S - Выход продукта [моль·моль-1]
YX/S - Выход биомассы [г·моль-1]
Ссылки
Al Zaid Siddiquee, K., et al. (2004) Appl Microbiol Biotechnol 63, 407-17.
Amidon, T.E. and Liu, S. (2009). Biotechnol Adv 27, 542-50.
Anastassiadis, S. (2007). Recent Pat Biotechnol 1, 11-24.
Aristidou, A. and Penttila, M. (2000). Curr Opin Biotechnol 11, 187-98.
Bailey, J.E. (1991). Science 252, 1668-75.
Bathe, B., et al. (1996). Mol Gen Genet 252, 255-65.
Becker, J., et al. (2005). Appl Environ MicrobioF Vol. 71, 8587-8596.
Bellmann, A., et al. (2001). Microbiology 147, 1765-74.
Bendt, A.K., et al. (2003). Proteomics 3, 1637-46.
Bergmeyer, H.U. and Hohorst, H.-J. (1970). Methoden der enzymatischen Analyse, 2 edn. Chemie GmbH Weinheim.
Bernofsky, C. and Swan, M. (1973) Anal Biochem 53, 452-8.
Blombach, B., et al. (2007). Appl Microbiol Biotechnol 76, 615-23.
Bolten, C.J., et al. (2007). Anal Chem 79, 3843-9.
Bonnassie, S., et al., (1990). Nucleic Acids Res 18, 6421.
Borner, J., et al. (2007). Anal Biochem 361, 143-51.
Bradford, M.M. (1976). Anal Biochem 72, 248-54.
Buchinger, S., et al. (2009). J Biotechno 140, 68-74.
Chen, R. and Yang, H. (2000). Arch Biochem Biophys 383, 238-45.
Chen, Z., et al. (1998), Anal Biochem 259, 203-11.
Christensen, B. and Nielsen, J. (2000) Adv Biochem Eng Biotechnol 66, 209-31.
Christensen, B., et al. (2000). Appl Microbiol Biotechno l54, 212-7.
Cocaign-Bousquet, M. and Lindley, N.D. (1995). Enzyme Microb Technol 17, 260-267.
Cremer, J., et al. (1991). Appl Environ Microbiol 57 (6), 1746-1752.
Cremer, J., et al. (1988). J Gen Microbiol 134 (Pt 12), 3221-9.
de Graaf, A.A. (2000). in Schugerl, K. and Bellgard, K.H. (Eds), Bioreaction Engineering, Springer Verlag, pp. 506-555.
de Graaf, A.A., et al. (2000). J Biotechnol 77, 25-35.
Diesterhaft, M.D. and Freese, E. (1973). J Biol Chem 248, 6062-70.
Dominguez, H. and Lindley, N.D. (1996). Appl Environ Microbio l62 (10), 3878-3880.
Dominguez, H., et al. (1998). Eur J Biochem 254, 96-102.
Drysch, A., et al. (2003). Metab Eng 5, 96-107.
Drysch, A., et al. (2004). Biotechnol Bioeng 85, 497-505.
Eggeling, L. and Bott, M. (2005). Handbook of Corynebacterium glutamicum. CRC Press.
Eggeling, L., et al. (1998). Appl Microbiol Biotechno 149, 24-30.
Eikmanns, B.J. (2005). in Eggeling, L. and Bott, M. (Eds), Handbook of Corynebacterium glutamicum, CRC Press, pp. 241-276.
Eikmanns, B.J., et al. (1993). Antonie Van Leeuwenhoek 64, 145-63.
Eikmanns, B.J., et al. (1989). Mol Gen Genet 218, 330-9.
Eikmanns, B.J., et al. (1995). J Bacteriol 177, 774-82.
El Massaoudi, et al. (2003). Metab Eng 5, 86-95.
Emmerling, M., et al. (2002). J Bacteriol 184, 152-64.
Fischer, E., et al. (2004). Anal Biochem 325, 308-16.
Frick, O. and Wittmann, C. (2005). Microb Cell Fact 4, 30.
Fry, B., et al. (2000). Appl Environ Microbiol 66, 4045-9.
Gardner, P.R. and Fridovich, I. (1993). J Biol Chem 268, 12958-63.
Geen, H. and Freeman, R. (1991). J. Magn. Reson. 93, 93-141.
Georgi, T., et al. (2005). Metab Eng 7, 291-301.
Gerstmeir, R., et al. (2004). J Bacteriol 186, 2798-809.
Glanemann, C, et al. (2003). Appl Microbiol Biotechnol 61, 61-8.
Goodfellow, M., et al. (1976). J Gen Microbiol 96, 351-8.
Gourdon, P., et al. (2000). Appl Environ Microbiol 66, 2981-7.
Gubler, M., et al. (1994). Appl Environ Microbiol 60, 2494-500.
Guest, J.R. and Creaghan, I.T. (1974). J Gen Microbiol 81, 237-45.
Haberhauer, G., et al. (2001). Corynebacterium glutamicum genes encoding proteins involved in membrane synthesis and membrane transport.
Hartmann, M., et al. (2003). J Biotechnol l04, 199-211.
Hayashi, M., et al. (2002). Biosci Biotechnol Biochem 66, 1337-44.
Hayashi, M., et al. (2006). Biosci Biotechnol Biochem 70, 546-50.
Hermann, T., et al.. (2000). TElectrophoresis 21, 654-9.
Hirao, T., et al. (1989). Appl Microbiol Biotechnol 32, 269-273.
Holatko, J., et al. (2009). J Biotechnol 139, 203-10.
Hua, Q., et al. (2000). J Biosci Bioeng 90, 184-92.
Ihnen, E.D. and Demain, A.L. (1969). J Bacteriol 98, 1151-8.
Ikeda, M. (2003). Adv Biochem Eng Biotechnol 79, 1-35.
Ikeda, M. and Katsumata, R. (1992). Appl Environ Microbiol 58, 781-785.
Ikeda, M. and Katsumata, R. (1999). Appl Environ Microbiol 65, 2497-502.
Ikeda, M. and Nakagawa, S. (2003). Appl Microbiol Biotechnol 62, 99-109.
Ikeda, M., et al. (1994). Biosci Biotechnol Biochem 58, 674-8.
Ikeda, M., et al. (2006). J bid Microbiol Biotechnol 33, 610-5.
Inoue, H., et al. (1990). Gene 96, 23-8.
Inui, M., et al. (2004). J Mol Microbiol Biotechnol 7, 182-96.
Jager, W., et al. (1995). FEMS Microbiol Lett l26, 1-6.
Jager, W., et al. (1992). J Bacteriol 174, 5462-5.
Jetten, M., et al. (1994a). Appl Microbiol Biotechnol 41, 47-52.
Jetten, M. and Sinskey, A. J. (1993). FEMS Microbiol Lett 111, 183-188.
Jetten, M.S., et al. (1995). Appl Microbiol Biotechnol 43, 76-82.
Jetten, M.S., et al. (1994b). Appl Environ Microbiol 60, 2501-7.
Jetten, M.S. and Sinskey, A.J. (1995). Antonie Van Leeuwenhoek 67, 221-7.
Jo, S.J., et al. (2007). J Biosci Bioeng 104, 457-63.
Kalinowski, J., et al. (2003). J Biotechnol 104, 5-25.
Kalinowski, J., et al. (1991). Mol Microbiol 5, 1197-204.
Kawaguchi, H., et al. (2008). Appl Microbiol Biotechnol 77, 1053-62.
Kawaguchi, H., et al. (2006). Appl Environ Microbiol 72, 3418-28.
Kelle, R., et al. (2005). L-Lysine Production in Eggeling, L. and Bott, M. (Eds), Handbook of Corynebacterium glutamicum, CRC Press, pp. 465-488.
Kelleher, J.K. (2001). Metab Eng 3, 100-10.
Kiefer, P., et al. (2002). J Ind Microbiol Biotechnol 28, 338-43.
Kiefer, P., et al. (2004). Appl Environ Microbiol 70, 229-39.
Kim, H.M., et al. (2006). J Microbiol Biotechnol 16, 1174-1179.
Kim, J., et al. Appl Microbiol Biotechnol 81, 1097-106.
Kinoshita, S., et al. (1961). Method of producing L-lysine by fermentation in Office, U. S. P. (Ed).
Kinoshita, S., et al. (1957) J. Gen. Appl. Microbiol 3, 193-205.
Kircher, M. and Pfefferle, W. (2001). Chemosphere 43, 27-31.
Kjeldsen, K.R. and Nielsen, J. (2009). Biotechnol Bioeng l02, 583-97.
Klapa, M.I., et al. (2003). Eur J Biochem 270, 3525-42.
Koffas, M.A., et al. (2003). Metab Eng 5, 32-41.
Kohl, T.A. and Tauch, A. (2009). J Biotechnol.
Kromer, J.O., et al. (2008). Microbiology 154, 3917-30.
Kromer, J.O., et al. (2005). Anal Biochem 340, 171-3.
Kromer, J.O., et al. (2004). J Bacteriol 186, 1769-84.
Kromer, J.O., et al. (2006). Metab Eng 8.
Lange, C, et al. (2003). Appl Environ Microbiol 69, 2521-32.
Lee, S.F., et al. (2009). Microbiology.
Lee, S.Y., et al. (2005). Trends Biotechnol 23, 349-58.
Leuchtenberger, W., et al. (2005). Appl Microbiol Biotechnol 69, 1-8.
Liebl, W. (2005). Corynebacterium Taxonomy in Eggeling, L. and Bott, M. (Eds), Handbook of Corynebacterium glutamicum, CRC Press, pp. 9-34.
Liebl, W., et al. (1989). FEMS Microbiol Lett 53, 299-303.
Liebl, W., et al. (1991). Int J Syst Bacteriol 41, 255-60.
Malumbres, M. and Martin, J.F. (1996). FEMS Microbiol Lett 143, 103-14.
Marx, A., et al., (1996). Biotechnol Bioeng 49 (2), 111-129.
Marx, A., E et al., (1999). Metab Eng 1, 35-48.
Marx, A., et al., (2003). J Biotechnol 104, 185-97.
Marx, A., et al., (1997). Biotechnol Bioeng 56 (2), 168-180.
Massou, S., et al., (2007). Metab Eng 9, 252-7.
Matsushita, K., et al., (2001). N FEMS Microbiol Lett 204, 271-6.
Menkel, E., et al., (1989). Appl Environ Microbiol 55, 684-8.
Michal, G. (1999). Biochemical Pathways. John Wiley & Sons.
Mimitsuka, T., et al., (2007). Biosci Biotechnol Biochem 71, 2130-5.
Mizukami, T., et al., (1994). Biosci Biotechnol Biochem 58, 635-8.
Molenaar, D., et al., (2000). J Bacteriol 182, 6884-91.
Mollney, M., et al., (1999). Biotechnol Bioeng 66, 86-103.
Moon, M.W., et al., (2007). J Mol Microbiol Biotechnol 12, 43-50.
Moritz, B., et al., (2000). Eur J Biochem 261, 3442-52.
Moritz, B., et al., (2002). Metab Eng 4, 295-305.
Muffler, A., et al., (2002). J Biotechnol 98, 255-68.
Nakayama, K. and Araki, K. (1973). Process for producing L-lysine.
Nakayama, K., et al., (1978). Adv Exp Med Biol 105, 649-61.
Netzer, R., et al., (2004). Arch Microbiol 182, 354-63.
Noh, K. and Wiechert, W. (2006). Biotechnol Bioeng 94, 234-51.
O'Donnell, S.M. and Janssen, G.R. (2001). J Bacteriol 183, 1277-83.
O'Regan, M., et al., (1989). Gene 77, 237-51.
Ohnishi, J., et al., (2005). FEMS Microbiol Lett 242, 265-74.
Ohnishi, J., et al., (2002). Appl Microbiol Biotechnol 58, 217-23.
Ozaki, H. and Shiio, I. (1969). J Biochem (Tokyo) 66, 297-311.
Palsson, B. (2000). Nat Biotechnol 18, 1147-50.
Parche, S., et al., (2001). J Mol Microbiol Biotechnol 3, 423-8.
Park, J.H. and Lee, S.Y. (2008). Curr Opin Biotechnol 19, 454-60.
Park, S.M., (1997). Biotechnol Bioeng 55, 864-879.
Peters-Wendisch, et al., (1993). FEMS Microbiol Lett 112, 269-274.
Peters-Wendisch, P.G., et al., (1998). Microbiology 144 (Pt 4), 915-27.
Peters-Wendisch, P.G., et al., (2001). J Mol Microbiol Biotechnol 3, 295-300.
Petersen, S., et al., (2000). J Biol Chem 275, 35932-41.
Petersen, S., et al. (2001). Metab Eng 3, 344-61.
Pisabarro, A., et al. (1993). J Bacteriol 175, 2743-9.
Polen, T., et al. (2007). FEMS Microbiol Lett 273, 109-19.
Pompejus, M., et al. (2002). Corynebacterium glutamicum genes encoding metabolic pathway proteins, BASF AG (DE).
Riedel, C, et al. (2001). J Mol Microbiol Biotechnol 3, 573-83.
Rittmann, D., et al. (2008). Appl Environ Microbiol 74, 6216-22.
Roessner, U., et al. (2000). Plant J 23, 131-42.
Sahm, H., et al. (2000). Biol Chem 381, 899-910.
Sano, K., et al. (1987). Agric Biol Chem 51 (2), 597-599.
Sasaki, M., et al. (2009). Appl Microbiol Biotechnol.
Sauer, U. (2004). Curr Opin Biotechnol 15, 58-63.
Sauer, U. and Eikmanns, B.J. (2005). FEMS Microbiol Rev 29, 765-94.
Schaffer, S. and Burkovski, A. (2005). Proteomics in Eggeling, L. and Bott, M. (Eds), Handbook of Corynebacterium glutamicum, CRC Press, pp. 99-118.
Schaffer, S., et al. (2001). Electrophoresis 22, 4404-22.
Schilling, C.H., et al. (1999). Biotechnol Prog 15, 296-303.
Schmid, R., et al. (2000). FEMS Microbiol Lett 187, 83-8.
Schmidt, K., et al. (1999). Metab Eng 1, 166-79.
Schrumpf, B., et al. (1991). J Bacteriol 173, 4510-6.
Seibold, G., et al. (2006). J Biotechnol 124, 381-91.
Shiio, I., et al. (1987). Agric Biol Chem 51, 2485-2493.
Shiio, I., et al. (1990). Agric Biol Chem 54, 3275-3285.
Shinfuku, Y., et al. (2009). Microb Cell Fact 8, 43.
Silberbach, M., et al. (2005a). J Biotechnol.
Silberbach, M., et al. (2005b). Appl Environ Microbiol 71, 2391-402.
Simic, P., et al. (2002). Appl Environ Microbiol 68, 3321-7.
Sonntag, K., et al. (1993). Eur J Biochem 213, 1325-31.
Sonntag, K., et al. (1995). Appl Microbiol Biotechnol 44, 489-495.
Stephanopoulos, G. (1999). Metab Eng 1, 1-11.
Stephanopoulos, G. and Vallino, J.J. (1991). Science 252, 1675-81.
Sugimoto, S.-I. and Shiio, I. (1989). Agric Biol Chem 53 (5), 1261-1268.
Tauch, A., et al. (2002a). J Biotechnol 95, 25-38.
Tauch, A., et al. (2002b). Curr Microbiol 45, 362-7.
Tauch, A., et al. (1994). FEMS Microbiol Lett 123, 343-7.
Thierbach, G., et al. (1990). Appl Microbiol Biotechnol 32, 443-8.
Thrippleton, M.J. and Keeler, J. (2003). Angew Chem Int Ed Engl 42, 3938-41.
Tzvetkov, M., et al. (2003). Microbiology 149, 1659-73.
Udaka, S. (1960). J Bacteriol 79, 754-5.
Van Dien, S.J., et al. (2006). J Biosci Bioeng 102, 34-40.
Vasicova, P., et al. (1999). J Bacteriol 181, 6188-91.
Vellanoweth, R.L. and Rabinowitz, J.C. (1992). Mol Microbiol 6, 1105-14.
Vervoort, E.B., et al. (2000). Nucleic Acids Res 28, 2069-74.
Villas-Boas, S.G., et al. (2005). Mass Spectrom Rev 24, 613-46.
Vrljic, M., et al. (1996). Mol Microbiol 22, 815-26.
Wehrmann, A., et al. (1998). J Bacteriol 180, 3159-65.
Wendisch, V.F. (2003). J Biotechnol 104, 273-85.
Wendisch, V.F. (2006). J. Microbiol. Biotechnol. 16(7), 999-1009.
Wendisch, V.F., et al. (2006). J Biotechnol 124, 74-92.
Wendisch, V.F., et al. (2000). J Bacteriol 182, 3088-96.
Wiechert, W. (2001). Metab Eng 3, 195-206.
Wiechert, W. and Noh, K. (2005). Adv Biochem Eng Biotechnol 92, 145-72.
Wiedemann, B. and Boles, E. (2008) Appl Environ Microbiol 74, 2043-50.
Wittmann, C. (2002). Adv Biochem Eng Biotechnol 74, 39-64.
Wittmann, C. (2007). Microb Cell Fact 6, 6.
Wittmann, C. and Becker, J. (2007) in Wendisch, V. F. (Ed), Springer Berlin/Heidelberg pp. 39-70.
Wittmann, C. and de Graaf, A. (2005) in Eggeling, L. and Bott, M. (Eds), Handbook of Corynebacterium glutamicum, CRC Press, pp. 277-304.
Wittmann, C, et al. (2002). Anal Biochem 307, 379-82.
Wittmann, C. and Heinzle, E. (2001a). Eur J Biochem 268, 2441-55.
Wittmann, C. and Heinzle, E. (2001b). Metab Eng 3, 173-91.
Wittmann, C. and Heinzle, E. (2002). Appl Environ Microbiol 68, 5843-59.
Wittmann, C, et al. (2004a). Appl Environ Microbiol 70, 7277-87.
Wittmann, C, et al. (2004b). Biotechnol Bioeng 87, 1-6.
Wittmann, et al. (2003). Biotechnol Lett 25, 377-80.
Yang, T.H., et al. (2003). Rapid Commun Mass Spectrom 17, 2721-31.
Yang, T.H., et al. (2006a). Metab Eng 8, 432-46.
Yang, T.H., et al. (2006b). Metab Eng 8, 417-31.
Yokota, A. and Lindley, N.D. (2005) in Eggeling, L. and Bott, M. (Eds), Handbook of Corynebacterium glutamicum, CRC Press, pp. 215-240.
Zelder, O., et al. Genes coding for glucose 6-phosphate dehydrogenase proteins in Office, U. P. (Ed).





































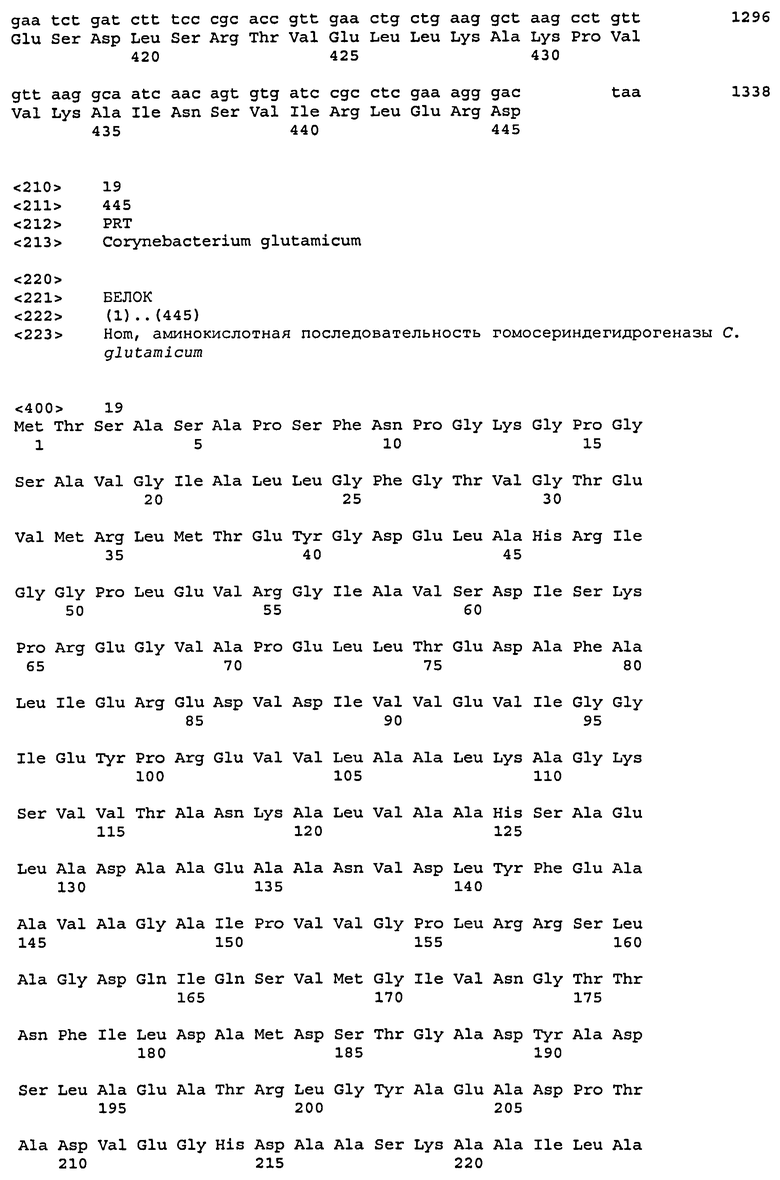
























































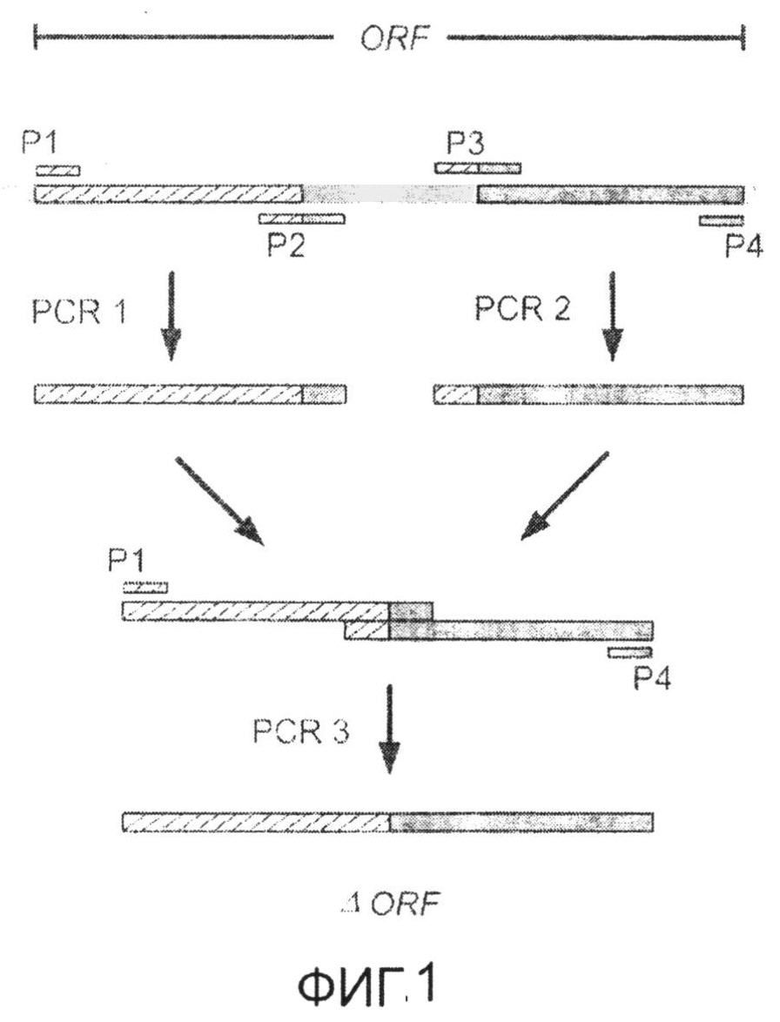
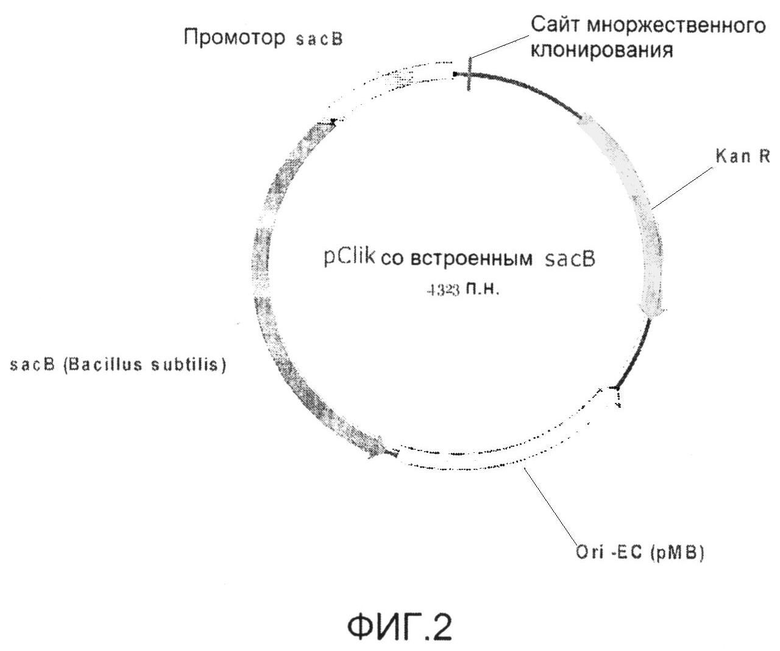
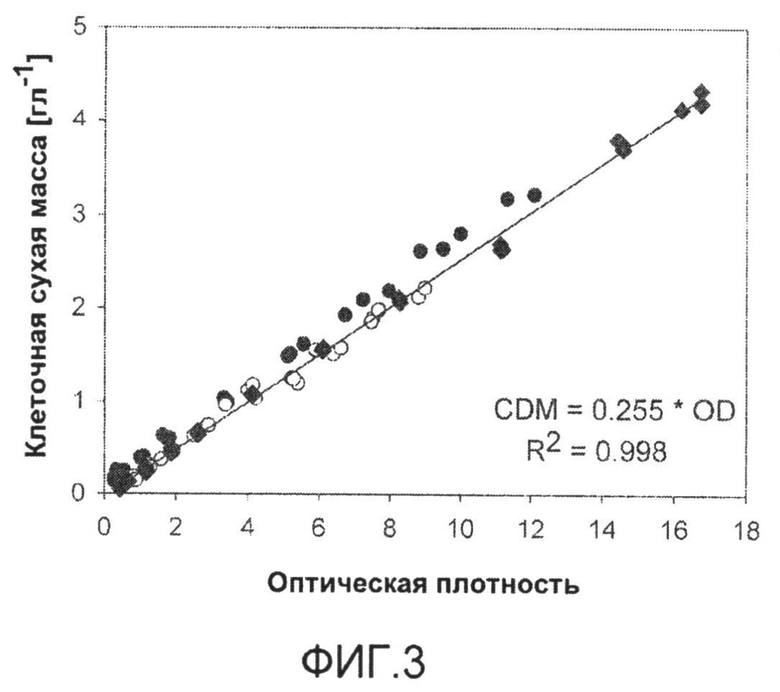
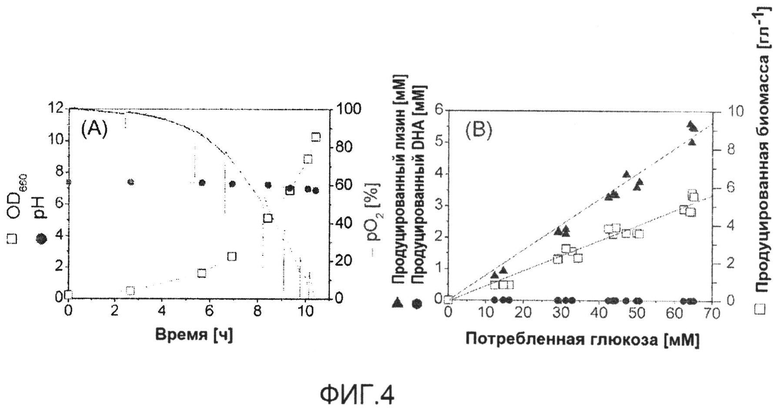
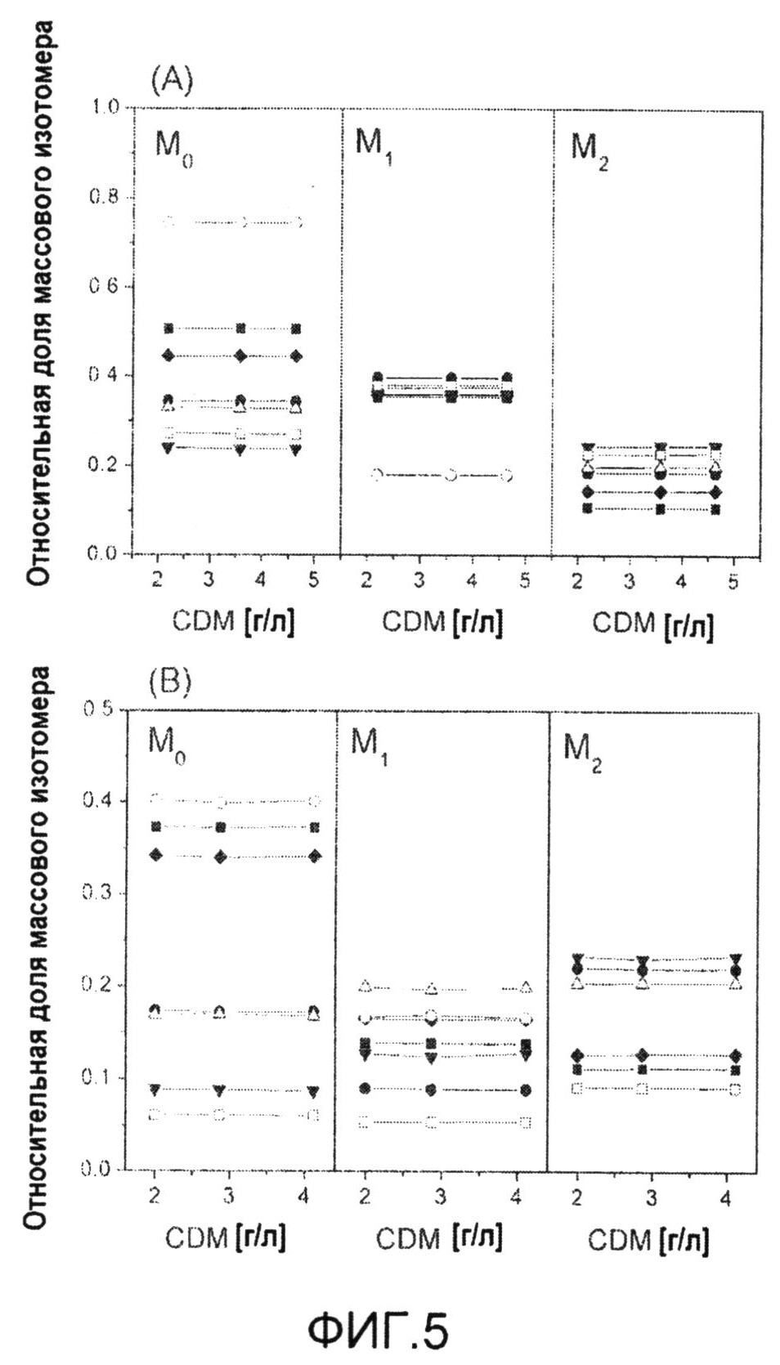
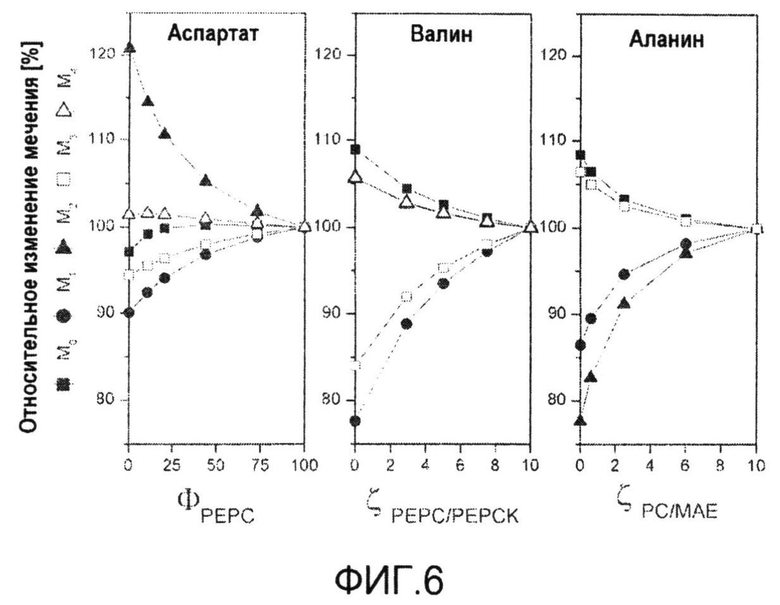
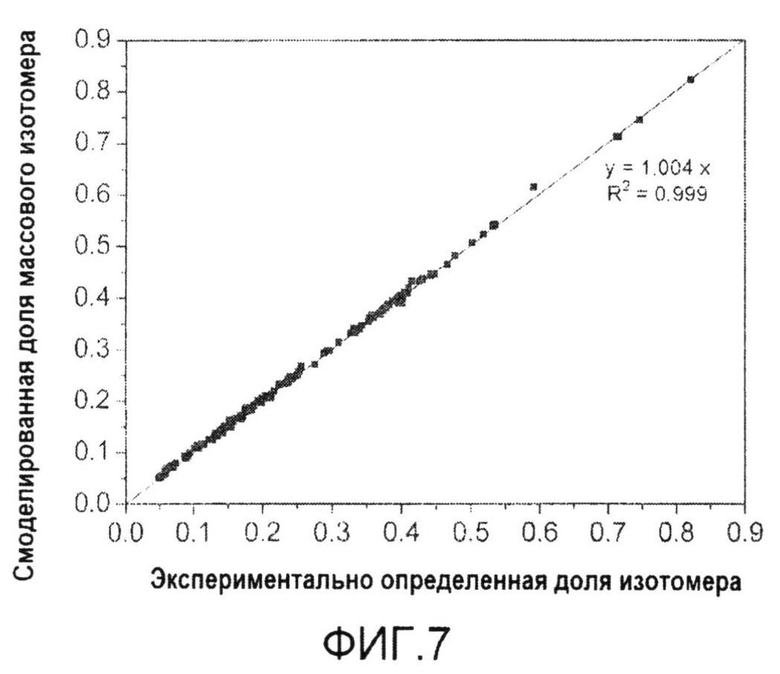
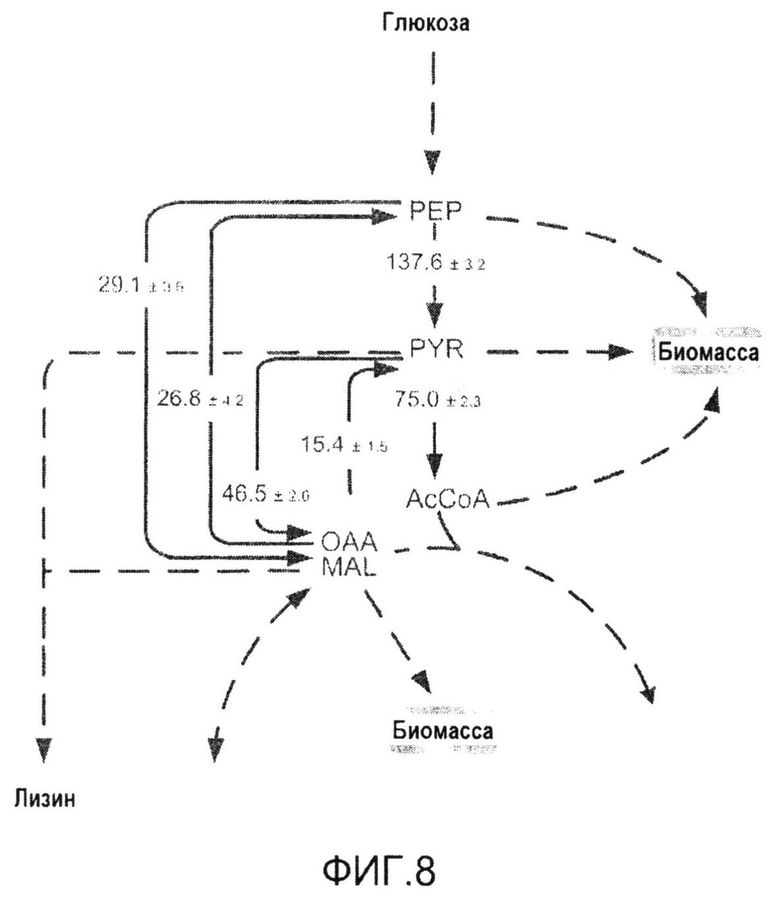
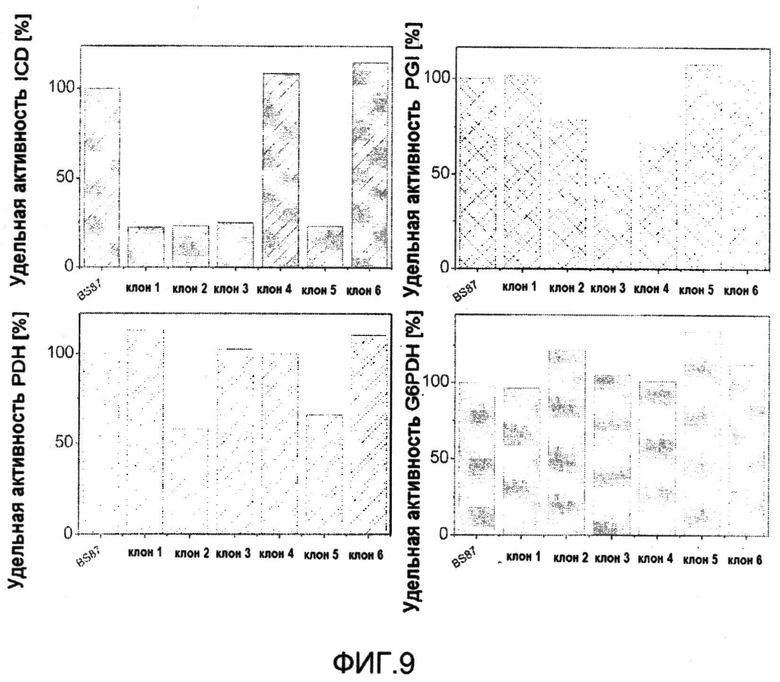

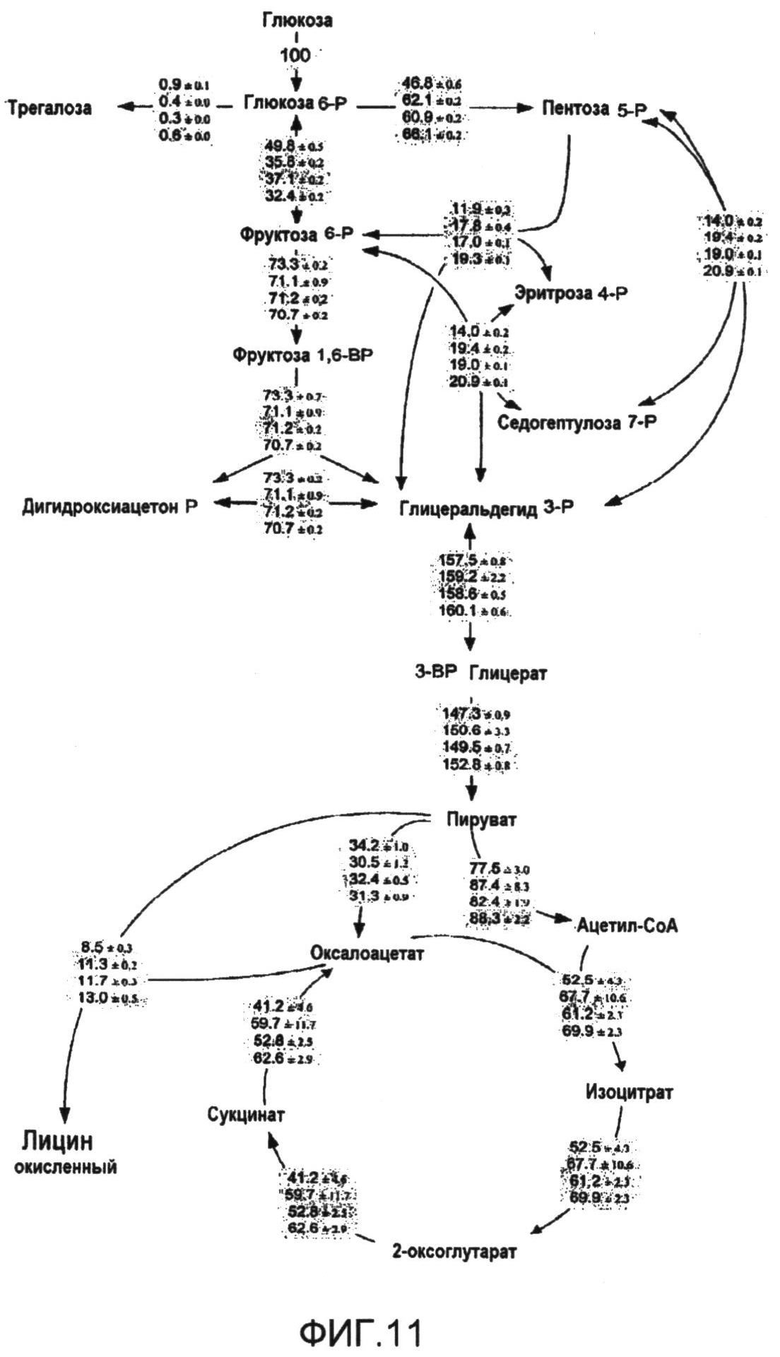
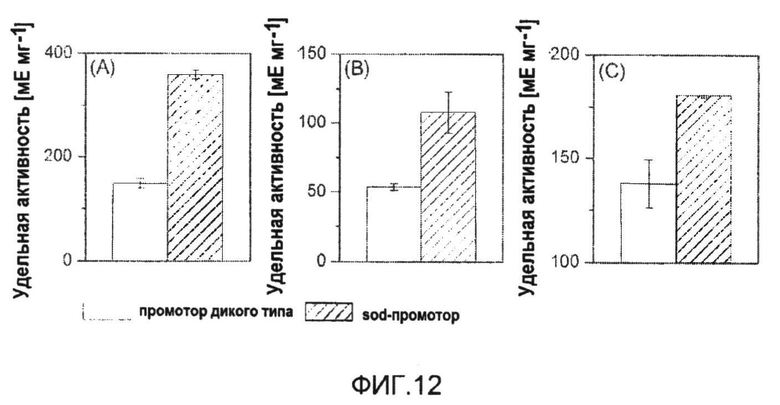
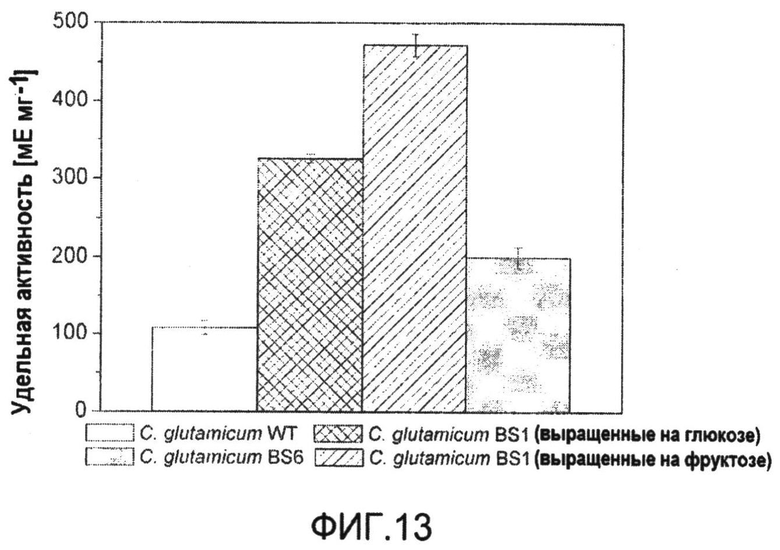
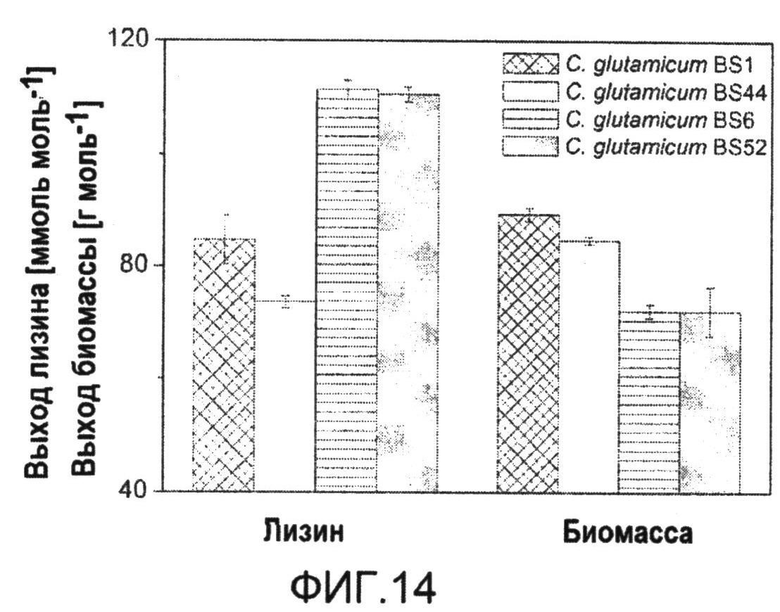
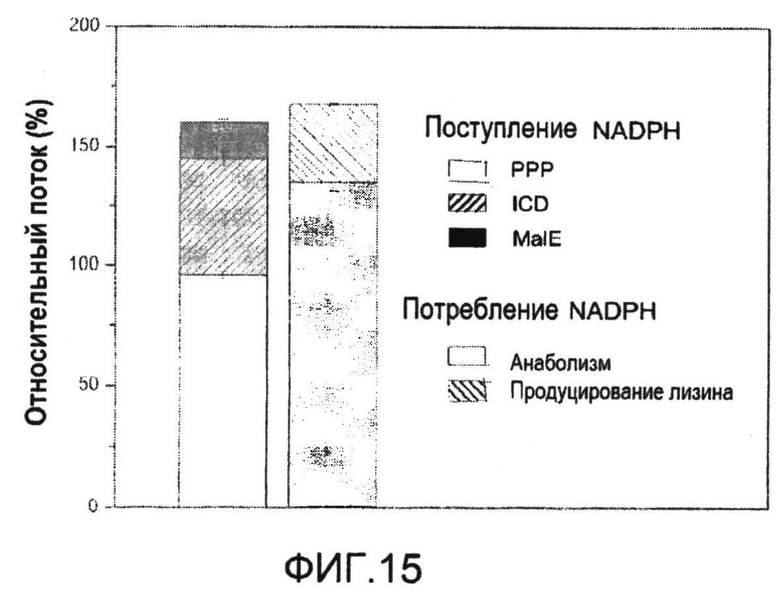
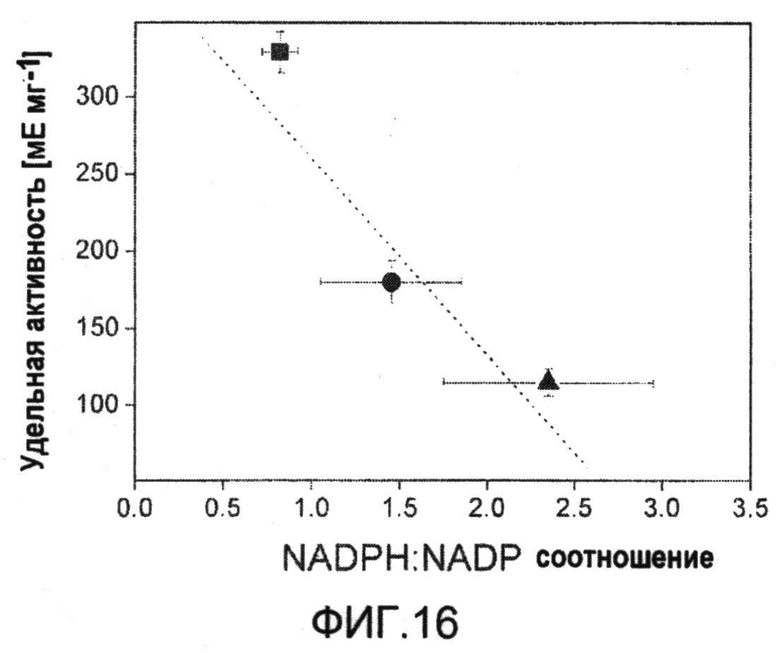
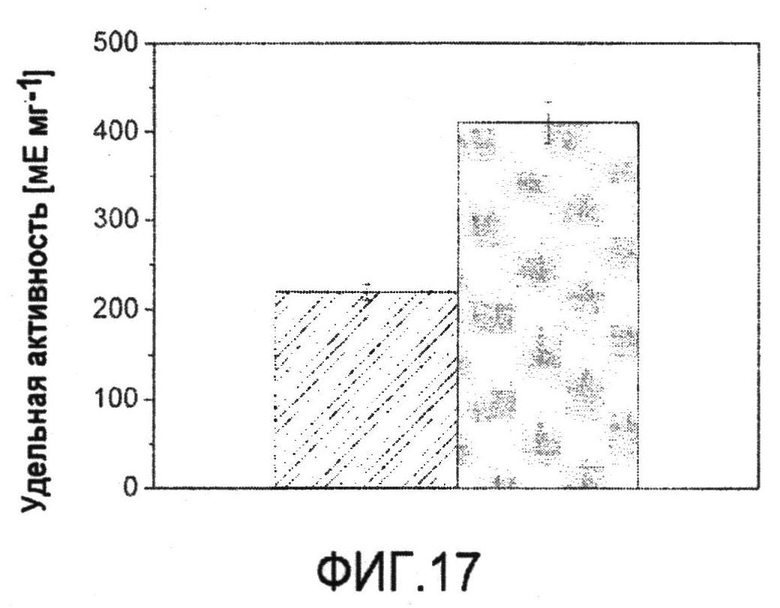
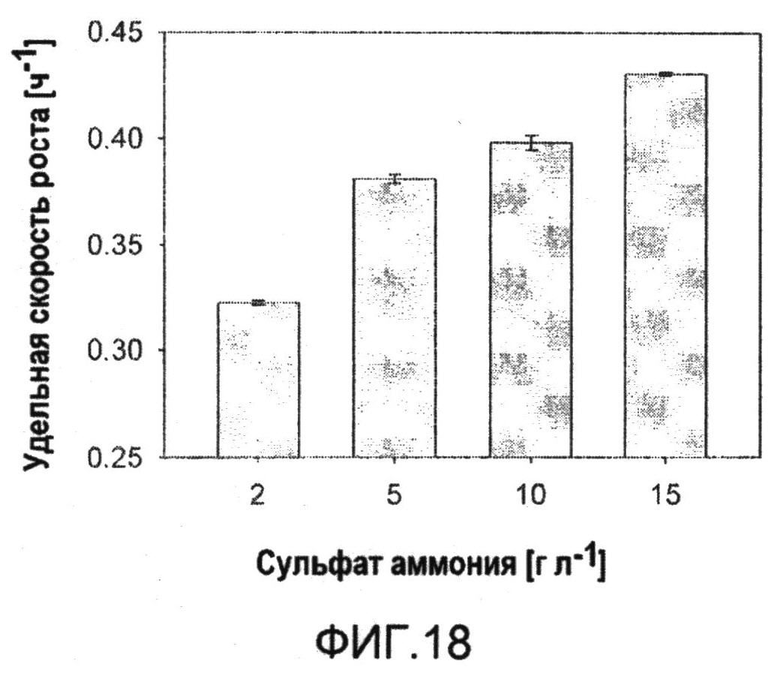
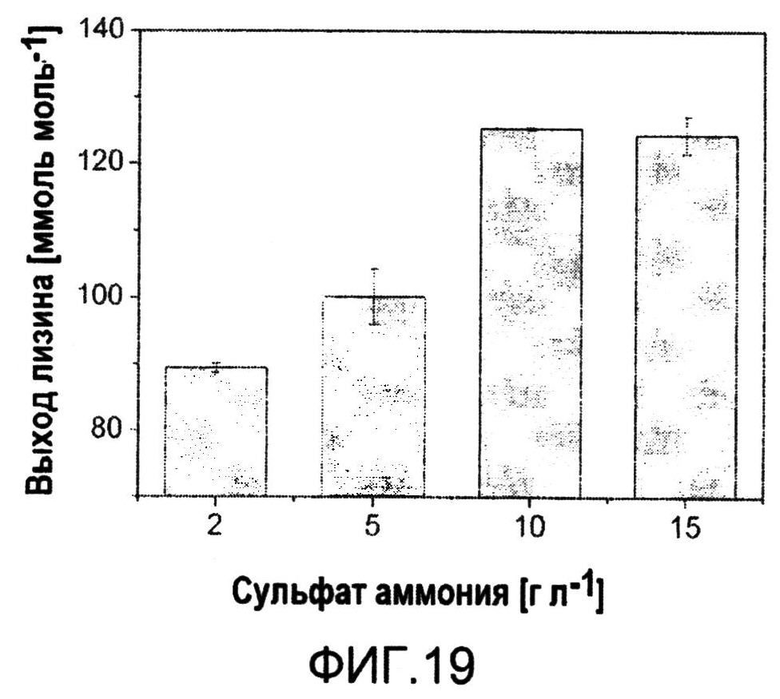
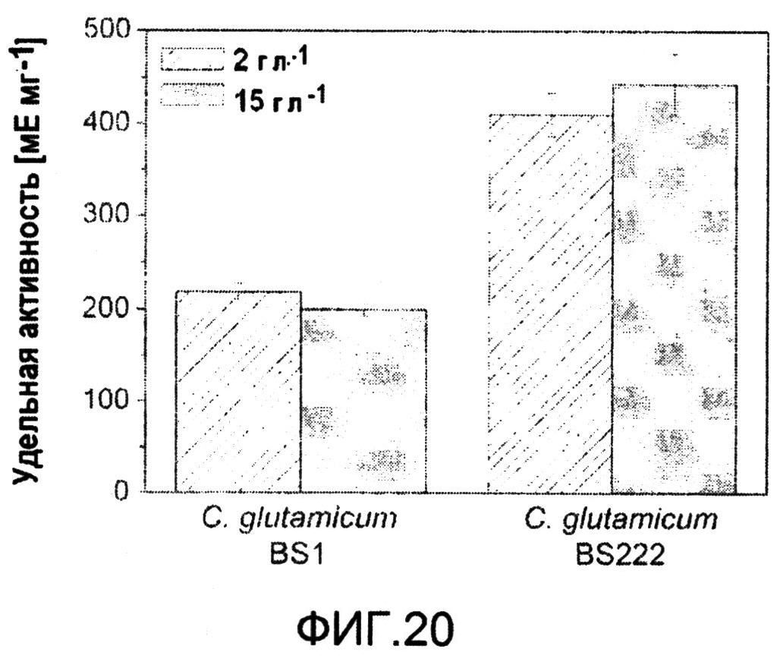
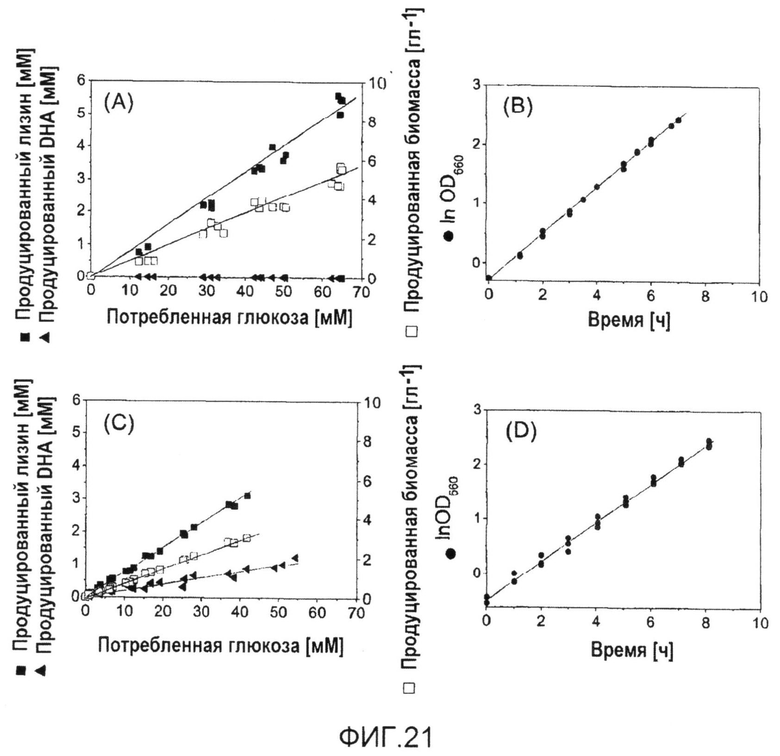
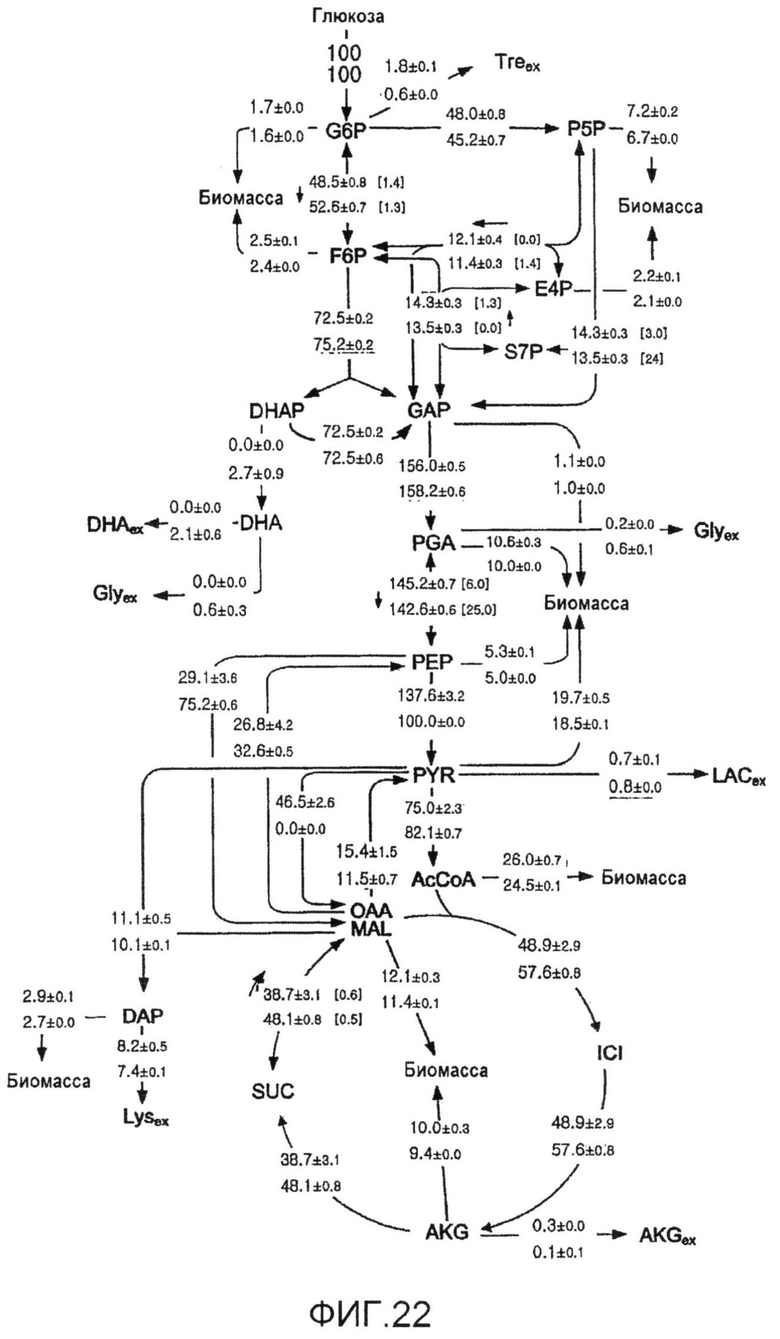
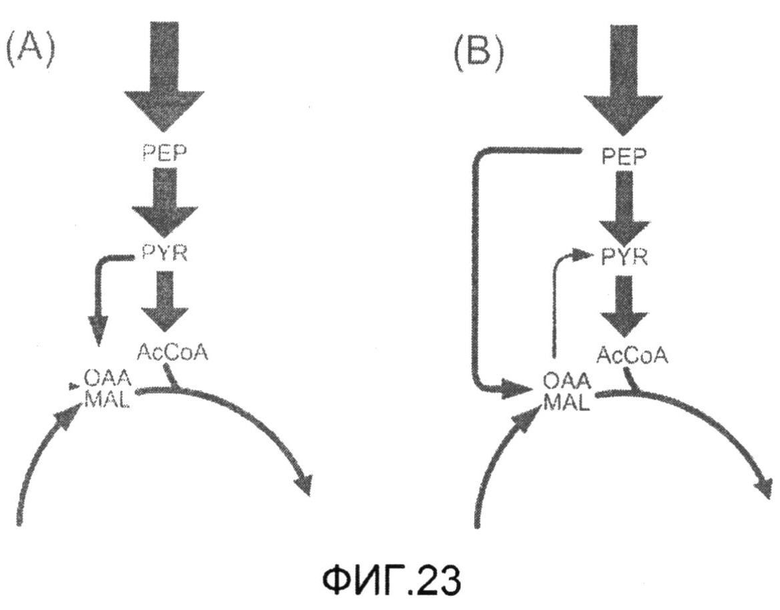
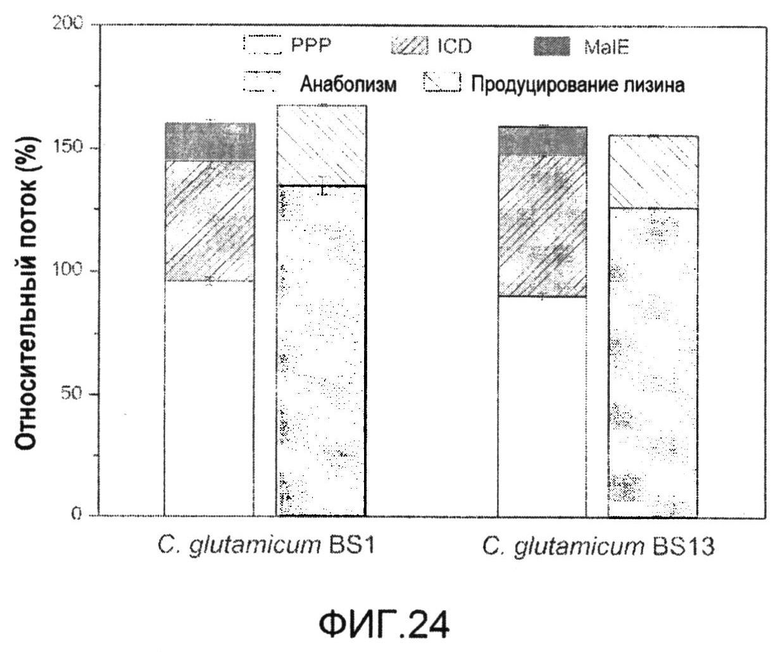
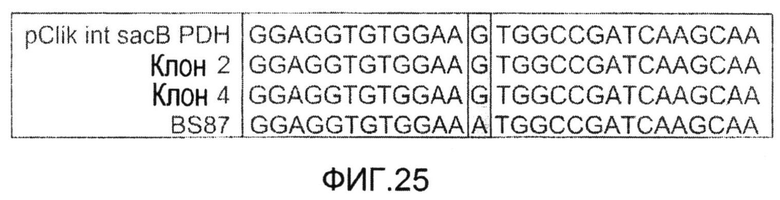
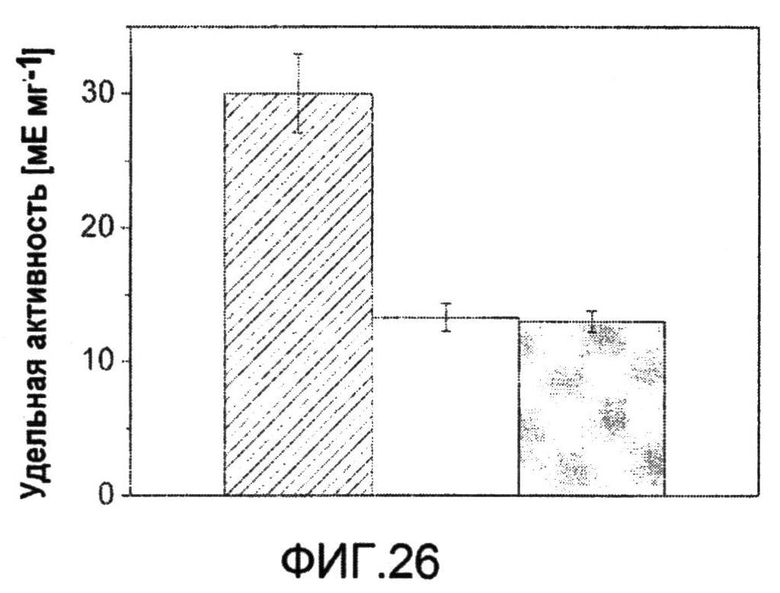
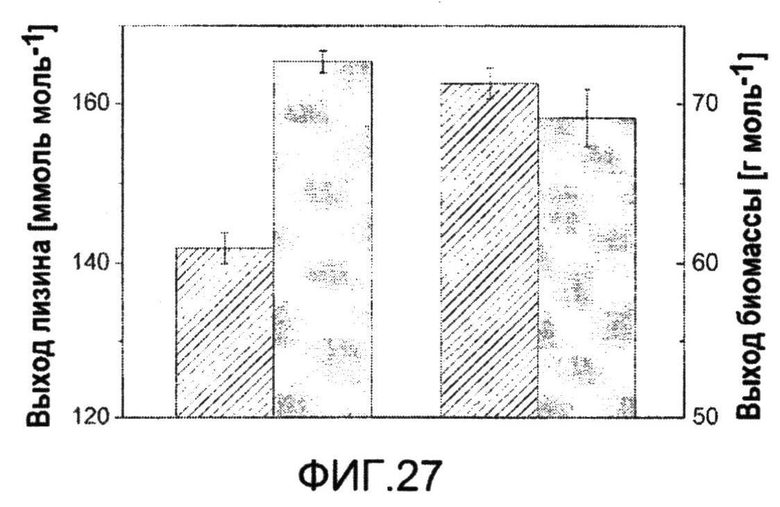
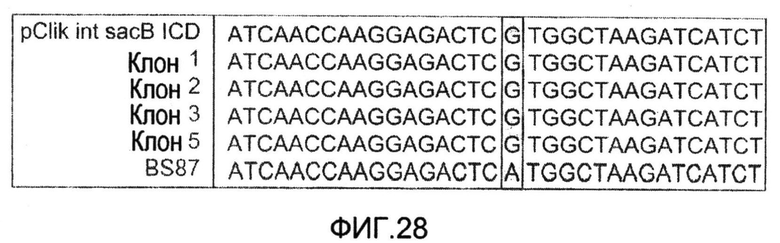
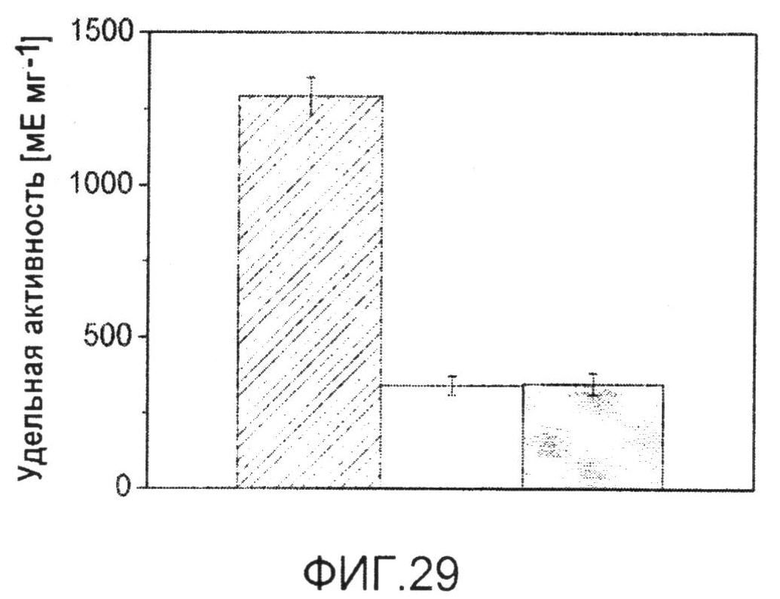
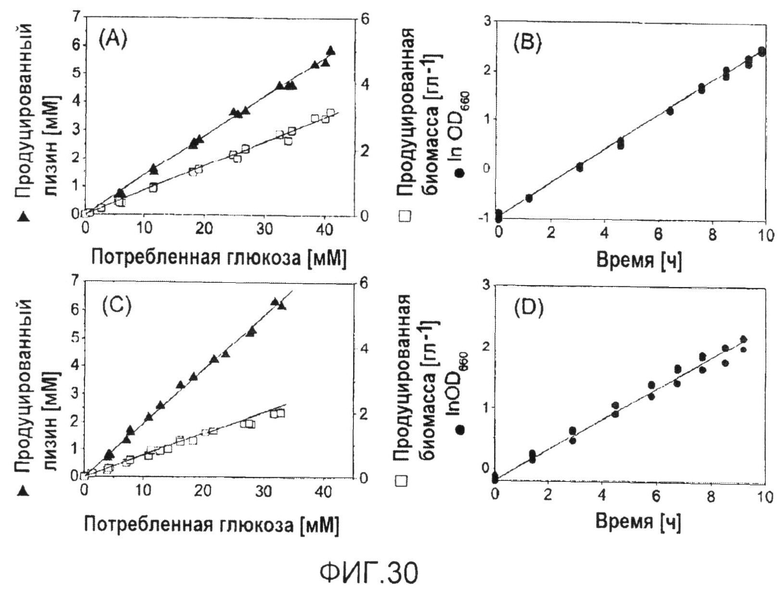
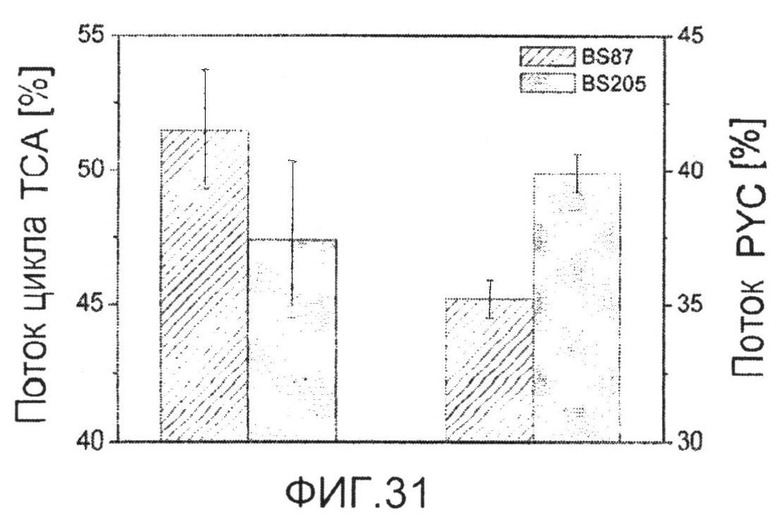
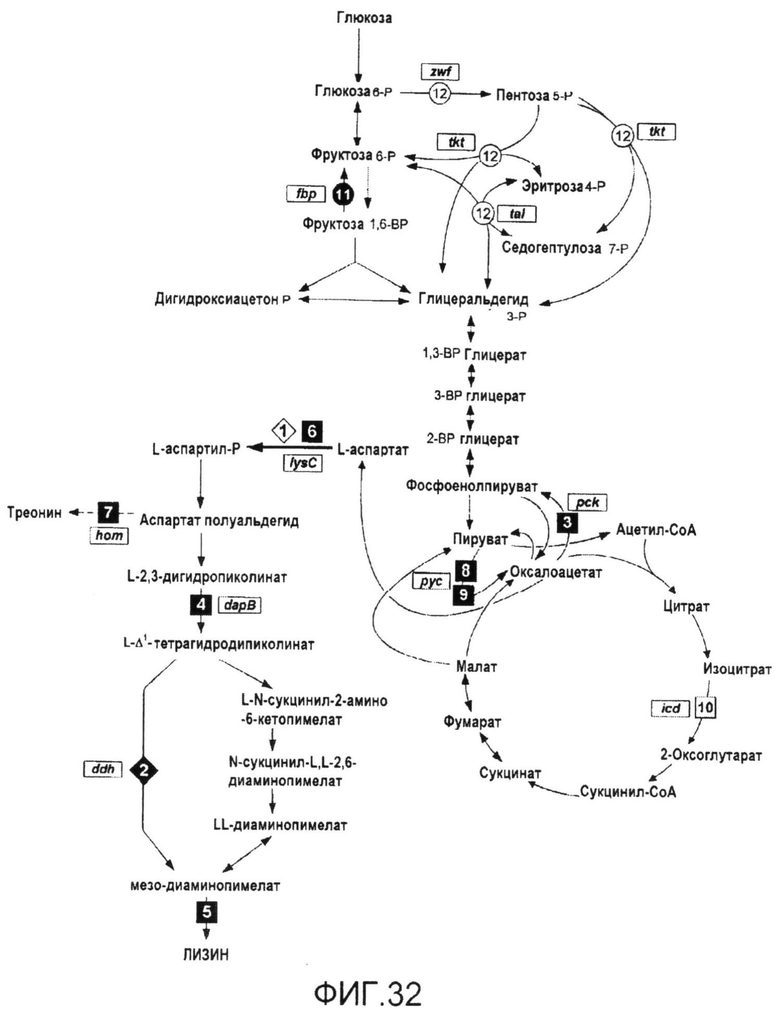
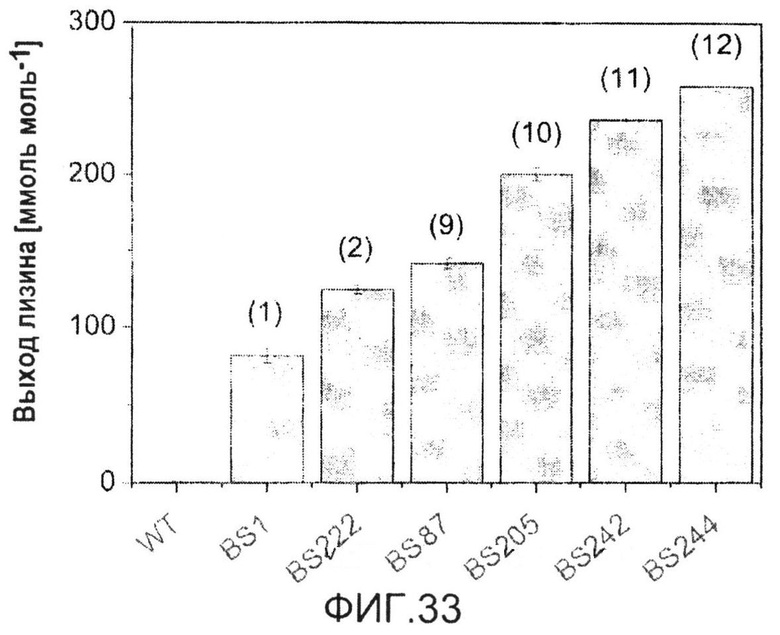
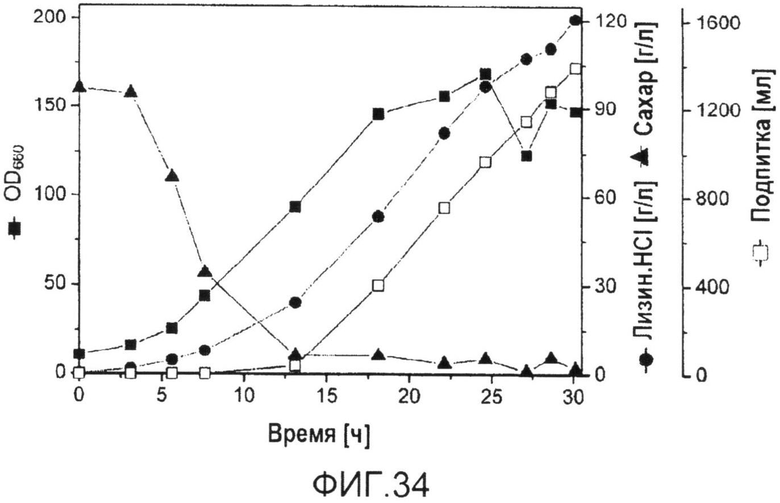
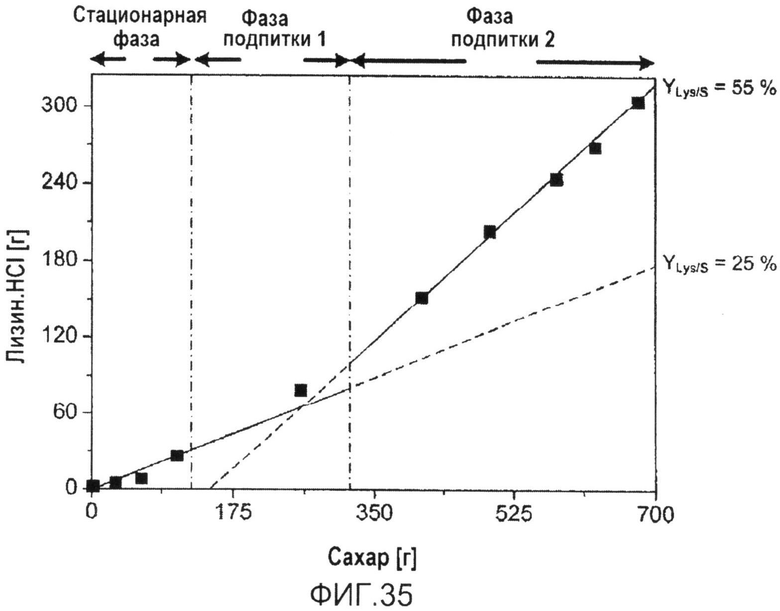
Изобретение относится к биотехнологии, в частности к способу продуцирования происходящих из аспартата аминокислот с использованием рекомбинантного микроорганизма, обладающего по меньшей мере следующими свойствами по сравнению с исходным микроорганизмом: (a) увеличенной активностью глюкозо-6-фосфатдегидрогеназы, (b) увеличенной активностью фруктозо-1,6-бифосфатазы, (c) ослабленной активностью изоцитратдегидрогеназы, (d) увеличенной активностью диаминопимелатдегидрогеназы и (e) увеличенной активностью аспартаткиназы, где микроорганизм представляет собой Corynebacterium. Изобретение позволяет продуцировать аминокислоты семейства аспартата, в частности L-лизин, с высокой степенью эффективности. 3 н. и 12 з.п. ф-лы, 35 ил., 20 табл., 1 пр.
1. Способ продуцирования происходящих из аспартата аминокислот, предпочтительно лизина, с использованием микроорганизма, обладающего по меньшей мере следующими свойствами по сравнению с исходным микроорганизмом:
(a) увеличенной активностью глюкозо-6-фосфатдегидрогеназы,
(b) увеличенной активностью фруктозо-1,6-бифосфатазы,
(c) ослабленной активностью изоцитратдегидрогеназы,
(d) увеличенной активностью диаминопимелатдегидрогеназы и
(e) увеличенной активностью аспартаткиназы,
где микроорганизм представляет собой Corynebacterium.
2. Способ по п.1, где указанный микроорганизм представляет собой рекомбинантный микроорганизм.
3. Способ по п.1, где
(a) увеличенная активность глюкозо-6-фосфатдегидрогеназы является следствием (i) сверхэкспрессии гена zwf, предпочтительно на основе замены природного промотора гена zwf гетерологичным промотором и/или (ii) замены аминокислотного остатка А243Т или (iii) замены природного промотора tkt-оперона гетерологичным промотором,
(b) увеличенная активность фруктозы-1,6 является следствием сверхэкспрессии гена fbp, предпочтительно на основе замены природного промотора гетерологичным промотором,
(c) ослабленная активность изоцитратдегидрогеназы основана на замене природного инициирующего кодона гена icd отличающимся инициирующим кодоном,
(d) увеличенная активность диаминопимелатдегидрогеназы основана на сверхэкспрессии гена ddh, предпочтительно путем умножения гена,
(e) увеличенная активность аспартаткиназы основана на сверхэкспрессии гена lysC, предпочтительно посредством модификации гена lysC, приводящей к аминокислотной замене T311I.
4. Способ по п.1, где гетерологичный промотор в признаках (а) и/или (b) выбран из промотора супероксиддисмутазы (sod) или промотора фактора элонгации tu (eftu).
5. Способ по п.1, где рекомбинантный микроорганизм дополнительно содержит по меньшей мере одну из следующих модификаций:
(f) сверхэкспрессию дигидродипиколинатредуктазы,
(g) сверхэкспрессию диаминопимелатдекарбоксилазы,
(h) сверхэкспрессию и мутацию пируваткарбоксилазы,
(i) делецию РЕР-карбоксикиназы и
(j) мутацию гомосериндегидрогеназы.
6. Способ по п.1, где микроорганизм депонирован как Corynebacterium glutamicum DSM 23586 в DSMZ.
7. Способ по п.1, где рекомбинантный микроорганизм представляет собой модифицированный С. glutamicum.
8. Способ по любому из пп. 1-7, дополнительно включающий стадии сбора и очистки происходящей из аспартата аминокислоты, предпочтительно лизина.
9. Рекомбинантный микроорганизм для получения происходящих из аспартата аминокислот, обладающий следующими характеристиками:
(а) увеличенной активностью глюкозо-6-фосфатдегидрогеназы,
(b) увеличенной активностью фруктозо-1,6-бифосфатазы,
(c) ослабленной активностью изоцитратдегидрогеназы,
(d) увеличенной активностью диаминопимелатдегидрогеназы и
(e) увеличенной активностью аспартаткиназы,
где микроорганизм представляет собой Corynebacterium.
10. Рекомбинантный микроорганизм по п.9, где
(a) увеличенная активность глюкозо-6-фосфатдегидрогеназы является следствием (i) сверхэкспрессии гена zwf, предпочтительно на основе замены природного промотора гена zwf гетерологичным промотором и/или (ii) замены аминокислотного остатка А243Т или (iii) замены природного промотора tkt-оперона гетерологичным промотором,
(b) увеличенная активность фруктозы-1,6 является следствием сверхэкспрессии гена fbp, предпочтительно на основе замены природного промотора гетерологичным промотором,
(c) ослабленная активность изоцитратдегидрогеназы основана на замене природного инициирующего кодона гена icd отличающимся инициирующим кодоном,
(d) увеличенная активность диаминопимелатдегидрогеназы основана на сверхэкспрессии гена ddh, предпочтительно путем умножения гена,
(e) увеличенная активность аспартаткиназы основана на сверхэкспрессии гена lysC, предпочтительно посредством модификации гена lysC, приводящей к аминокислотной замене T311I.
11. Микроорганизм по п.9, где гетерологичный промотор выбран из промотора супероксиддисмутазы (sod) или фактора элонгации tu (eftu).
12. Микроорганизм по п.9, который дополнительно содержит по меньшей мере одну из следующих модификаций:
(f) сверхэкспрессию дигидродипиколинатредуктазы,
(g) сверхэкспрессию диаминопимелатдекарбоксилазы,
(h) сверхэкспрессию и мутацию пируваткарбоксилазы,
(i) делецию РЕР-карбоксикиназы и
(j) мутацию гомосериндегидрогеназы.
13. Микроорганизм по п. 9, где рекомбинантный микроорганизм представляет собой модифицированный С. glutamicum.
14. Микроорганизм по п. 9, который депонирован как Corynebacterium glutamicum DSM 23586 в DSMZ.
15. Применение микроорганизма по любому одному из пп. 9-14 в способе продуцирования происходящей из аспартата аминокислоты, предпочтительно при продуцировании L-лизина.
| RU 2001107500 A , 20.03.2003 | |||
| US 20080032374 A1, 07.02.2008 | |||
| Устройство для испытания материалов на фрикционную усталость | 1985 |
|
SU1293560A1 |

