Настоящее изобретение относится к азотсодержащим ассоциативным молекулам, включающим, по меньшей мере, одно звено, позволяющее связываться молекулам друг с другом или с наполнителем за счет ковалентных связей, и включающим функциональную группу, способную взаимодействовать с полимером, содержащим ненасыщенные связи, с образованием ковалентной связи с указанным полимером.
В промышленности смеси полимеров с наполнителями используются очень часто. Для того чтобы эти смеси отличались хорошими свойствами, постоянно изыскивают средства для улучшения диспергирования носителей внутри полимеров. Одним из средств для достижения этого результата является использование сочетающих агентов, способных установить взаимодействия между полимером и наполнителем.
Агенты сочетания полимера с наполнителем, содержащие азотсодержащие биполярные группы, описаны в документах, опубликованных под номерами US7186845B2 и JP 2008208163.
Эти документы описывают модификацию полимеров, содержащих диеновые звенья, с помощью биполярных азотсодержащих соединений, включающих, кроме того, гетероцикл, причем указанный гетероцикл сам содержит атом азота, а также атом кислорода и/или серы.
Более конкретно, описанные соединения являются нитронами, несущими оксазолиновые, тиазолиновые функциональные группы, как, например, соединение (-(2-оксазолил)-фенил-N-метилнитрон)
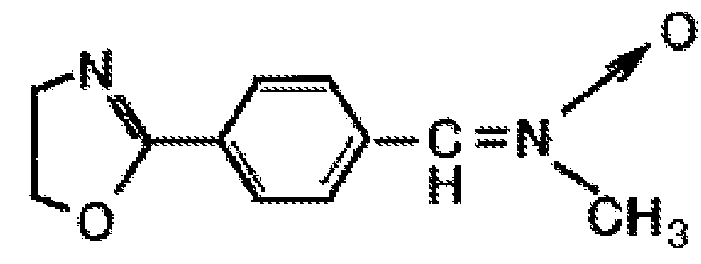
Когда диеновые полимеры вводят во взаимодействие с такого рода соединениями, то получаемые в результате полимеры несут оксазолиновые или тиазолиновые циклы.
Эти циклы, находящиеся на полимере, сами в свою очередь вступают в реакцию с поверхностными функциональными группами наполнителей (как сажа или двуокись кремния), с которыми смешиваются полимеры. Эта реакция ведет к установлению ковалентных связей между полимером, модифицированным сочетающим агентом и наполнителем в связи с раскрытием оксазолинового или тиазолинового цикла. Действительно, как было описано в документе US7186845B2, оксазолиновые и/или тиазолиновые циклы способны раскрываться в присутствии нуклеофила, который может, в качестве примера, находиться на поверхности наполнителя.
Однако возникновение таких ковалентных связей создает неудобства во время приготовления смесей, содержащих эти полимеры, модифицированные агентами сочетания с наполнителем. В частности, существование преждевременно образующихся ковалентных связей между полимером и наполнителями делает эти смеси очень вязкими в несшитом состоянии, что затрудняет любые операции, осуществляемые перед сшиванием (вулканизацией), с составами на основе каучука, в частности приготовление смесей из соответствующих компонентов и их формование; эти недостатки оказывают сильное влияние на производительность промышленного предприятия. Следовательно, желательно предложить новые молекулы, не имеющие вышеуказанных недостатков, т.е. молекулы, которые способны после взаимодействия с полимером и смешивания с наполнителем, не образовывать ковалентные связи с наполнителем и таким образом не вызывать слишком сильного увеличения вязкости смеси.
Объектом настоящего изобретения является соединение, включающее, по меньшей мере, одну группу Q и, по меньшей мере, одну группу А, связанные друг с другом, по меньшей мере и предпочтительно, одной «спейсерной» группой Sp, в котором:
- Q включает одну биполярную группу, содержащую, по меньшей мере и предпочтительно, один атом азота,
- А включает способную к ассоциации группу, содержащую, по меньшей мере, один атом азота,
- Sp является атомом или группой атомов, образующих связь между Q и А.
Полимер, привитый соединением, которое определено выше, смешивают с наполнителями, при этом полимер создает только лабильные связи с наполнителями, что позволяет обеспечить хорошее взаимодействие полимер - наполнитель, благоприятное для конечных свойств полимера, но без недостатков, которые могли бы возникнуть вследствие очень сильного взаимодействия между полимером и наполнителем.
Соединения согласно изобретению обеспечивают хорошее взаимодействие с наполнителями благодаря установлению лабильных связей между полимерными цепями и наполнителем и таким образом ограничивают технологические проблемы.
Под биполярной группой подразумевается функциональная группа, способная образовывать 1,3-биполярное присоединение к ненасыщенной углерод-углеродной связи.
Под «способной к ассоциации группой» подразумевают группы, способные ассоциироваться друг с другом посредством водородных, ионных и/или гидрофобных связей. В предпочтительном варианте осуществления изобретения речь идет о группах, способных ассоциироваться посредством водородных связей.
Когда способные к ассоциации группы способны связываться посредством водородных связей, то каждая способная к ассоциации группа содержит, по меньшей мере, один донорный участок и один акцепторный участок по отношению к водородной связи, и таким образом две одинаковые способные к ассоциации группы становятся взаимодополняющими и могут связываться друг с другом с образованием, по меньшей мере, двух водородных связей.
Способные к ассоциации группы согласно изобретению способны также связываться водородными, ионными и/или гидрофобными связями с функциональными группами, находящимися на наполнителях.
Соединения согласно изобретению, содержащие одну группу Q, одну «спейсерную» группу и одну способную к ассоциации группу, могут быть представлены, например, следующей формулой (Ia):
A-Sp-Q (Ia)
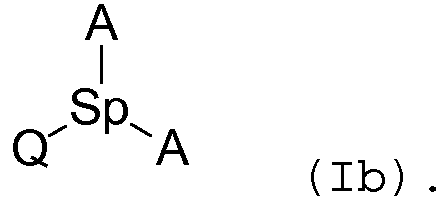
Соединения согласно изобретению, содержащие одну группу Q, одну «спейсерную» группу и две способные к ассоциации группы, могут быть представлены, например, следующей формулой (Ib):
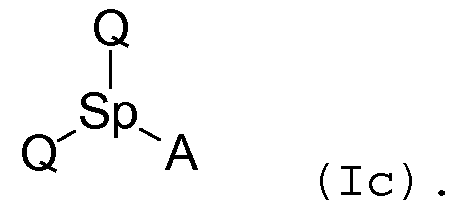
Аналогично, соединения согласно изобретению, содержащие две группы Q, одну «спейсерную» группу и одну способную к ассоциации группу, могут быть представлены, например, следующей формулой (Ic):
Используя такой же принцип, соединения согласно изобретению, содержащие две группы Q, одну «спейсерную» группу и две способные к ассоциации группы, могут быть представлены, например, следующей формулой (Id):
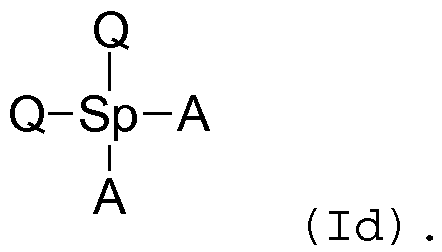
Предпочтительно, способную к ассоциации группу выбирают из имидазолидинильной, уреидной, бис-уреидной, уреидо-пиримидильной, триазолильной групп.
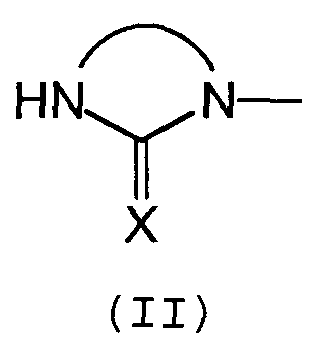
Предпочтительно, группа А отвечает одной из следующих формул (II)-(VI):
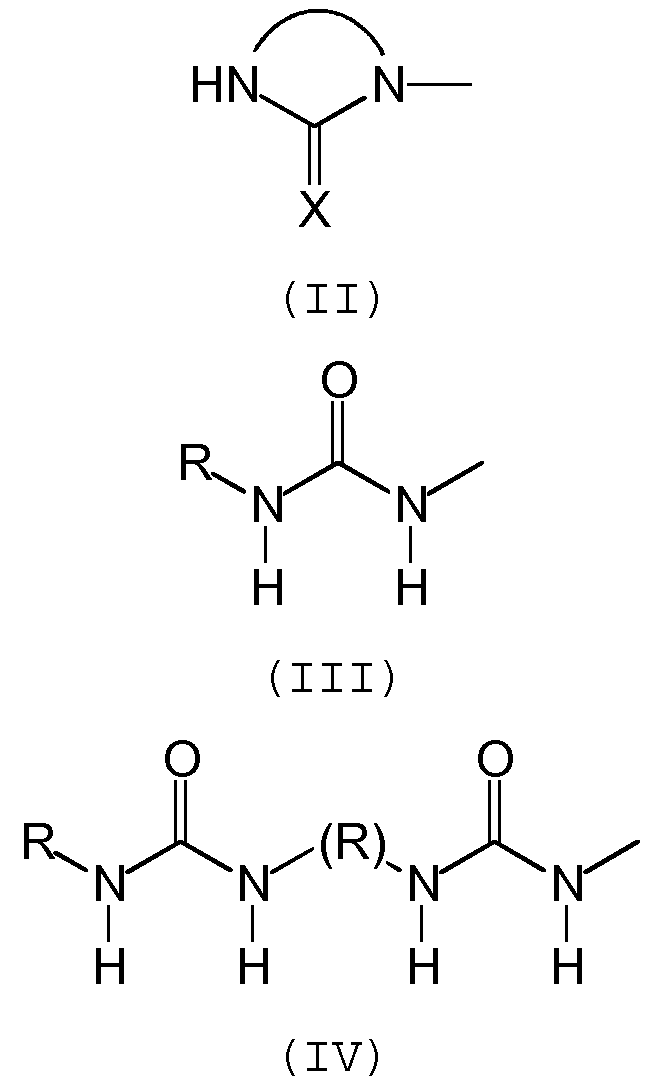
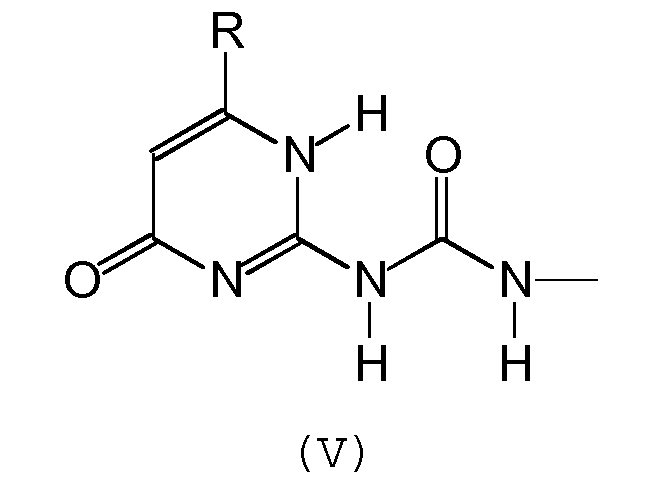
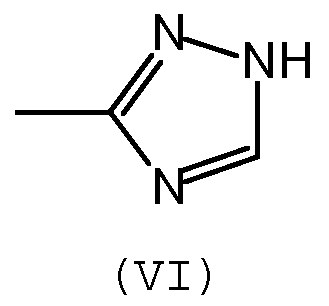
где:
- R обозначает углеводородную группу, которая может необязательно содержать гетероатомы,
- Х обозначает атом кислорода или серы, предпочтительно, атом кислорода.
Предпочтительно, группа А включает 5- или 6-членный азотсодержащий гетероцикл с двумя или тремя атомами азота, предпочтительно, с двумя атомами азота, и включает, по меньшей мере, одну карбонильную функциональную группу.
Еще более предпочтительно, группа А включает одну имидазолидинильную группу формулы (II).
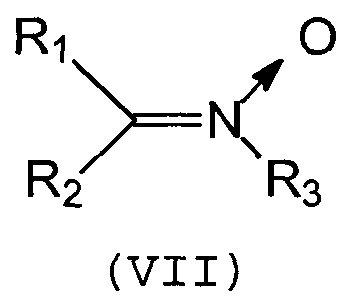
Группа Q способна связываться с полимерной цепью, содержащей, по меньшей мере, одну ненасыщенную связь, за счет образования ковалентной связи (прививка). Предпочтительно, группа Q включает нитрилоксидную, нитроновую или нитрилиминовую функциональную группу, которая может связываться с ненасыщенным полимером путем циклоприсоединения типа [3+2].
Предпочтительно, группа Q является группой следующей формулы (VII), (VIII) или (IX):
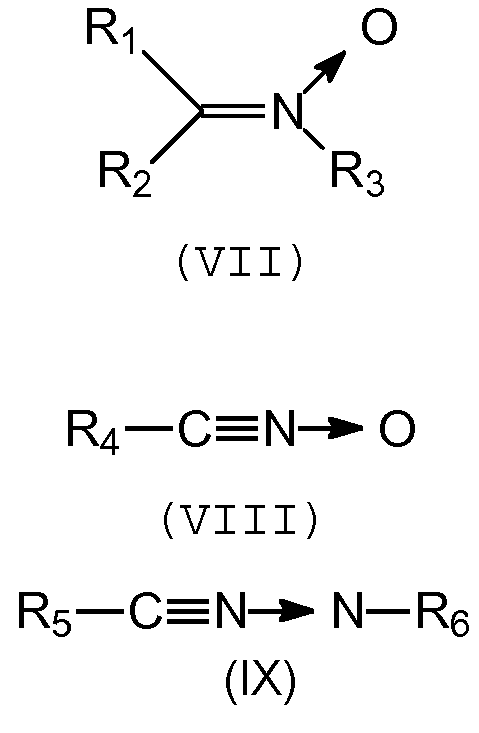 ,
,
в которых R1-R6 выбирают независимо из спейсерной группы Sp, атома водорода, С1-С20 алкила, линейного или разветвленного, С3-С20 циклоалкила, линейного или разветвленного, С6-С20 арила, линейного или разветвленного, и группы формулы (Х):
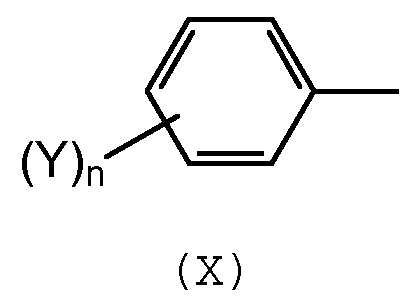
в которой n обозначает 1, 2, 3, 4 или 5 и каждый Y обозначает независимо спейсерную группу Sp, алкил или галоген.
«Спейсерная» группа Sp позволяет связаться с, по меньшей мере, одной группой Q и/или с, по меньшей мере, одной способной к ассоциации группой А, и поэтому может быть любой спейсерной группой известного типа. Однако «спейсерная» группа не должна или может в малой степени влиять на группу Q и способную к ассоциации группу соединения согласно изобретению.
Следовательно, указанная «спейсерная» группа рассматривается как группа, инертная по отношению к группе Q и способной к ассоциации группе. Под «спейсерной» группой, инертной по отношению к группе Q, подразумевают группу, которая не содержит алкенильной или алкинильной функциональной группы, способной вступать в реакцию с этой группой. Под «спейсерной» группой, инертной по отношению к способной к ассоциации группе, подразумевают группу, которая не содержит способных к ассоциации функциональных групп, таких, которые определены согласно изобретению.
«Спейсерная» группа, предпочтительно, представляет собой углеводородную цепь, линейную, разветвленную, циклическую, которая может содержать один или несколько ароматических радикалов и/или один или несколько гетероатомов. Указанная цепь может быть необязательно замещена, при условии, если заместители будут инертны по отношению к группе Q и способной к ассоциации группе.
Согласно предпочтительному варианту осуществления «спейсерная» группа является линейной или разветвленной С1-С24 алкильной цепью, предпочтительно, С1-С10 алкильной цепью, необязательно прерванной одним или несколькими атомами азота или кислорода, более предпочтительно, линейной С1-С6 алкильной цепью.
Предпочтительно, группа Q является группой формулы (XI):
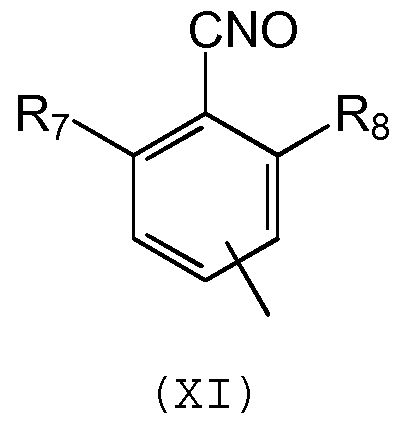
в которой R7 и R8 обозначают независимо С1-С5 алкил или галоген, предпочтительно, R7 и R8 обозначают независимо метил или атом хлора, а группа А является группой формулы (XII):
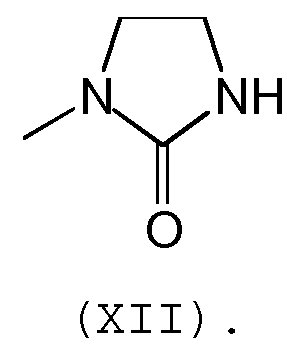
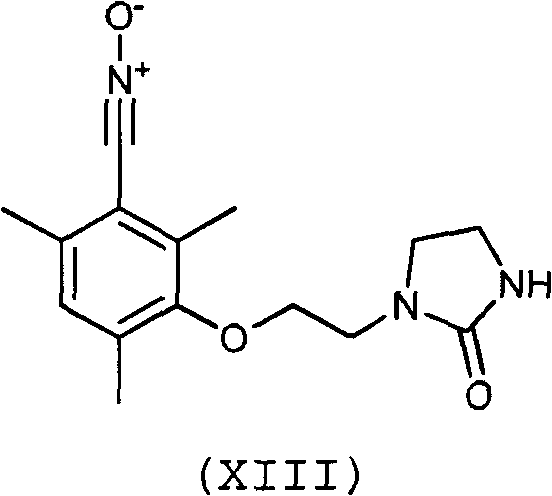
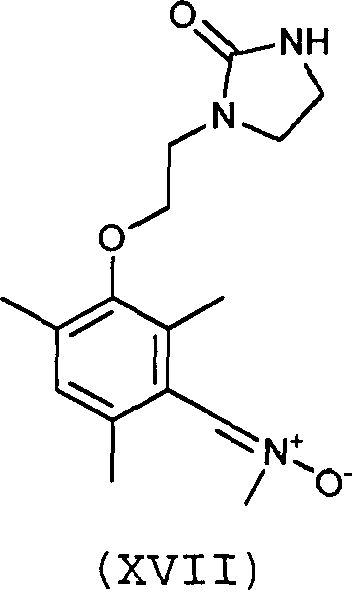
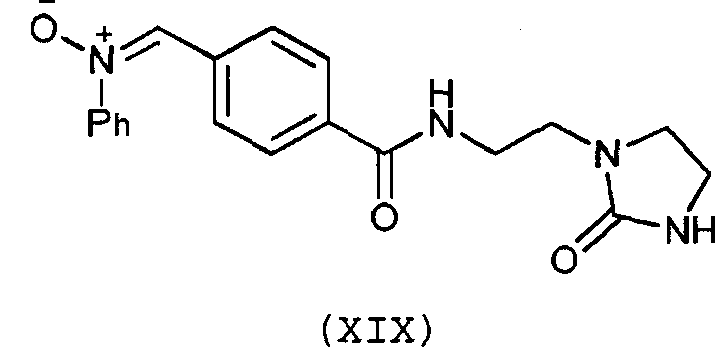
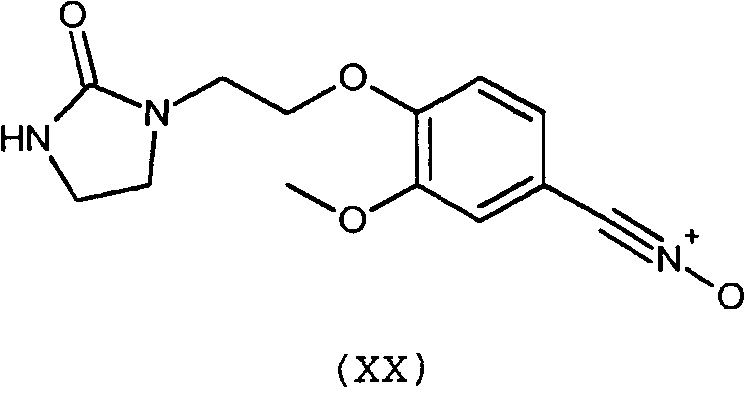
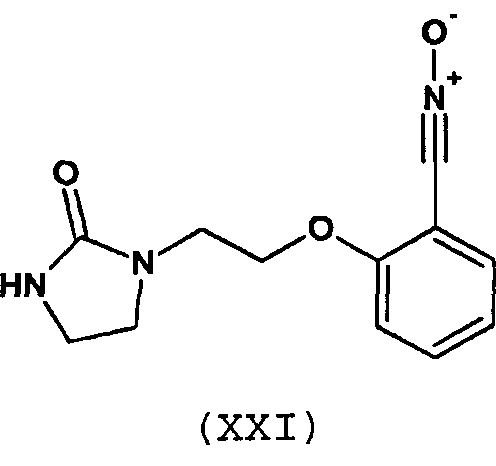
Предпочтительно, соединение согласно изобретению выбирают из соединений следующих формул (XIII)-(XXI):
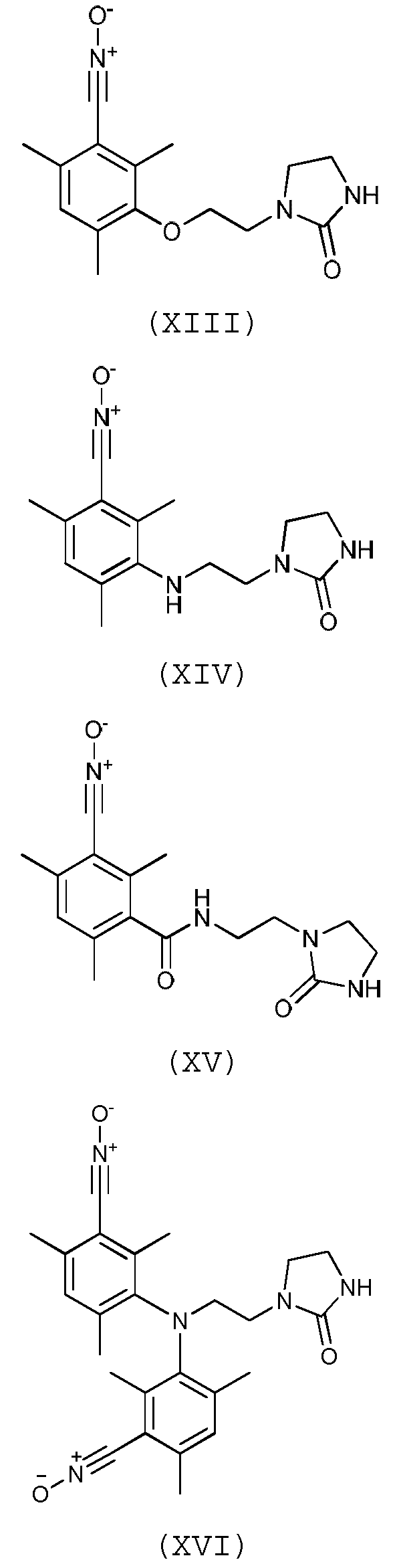
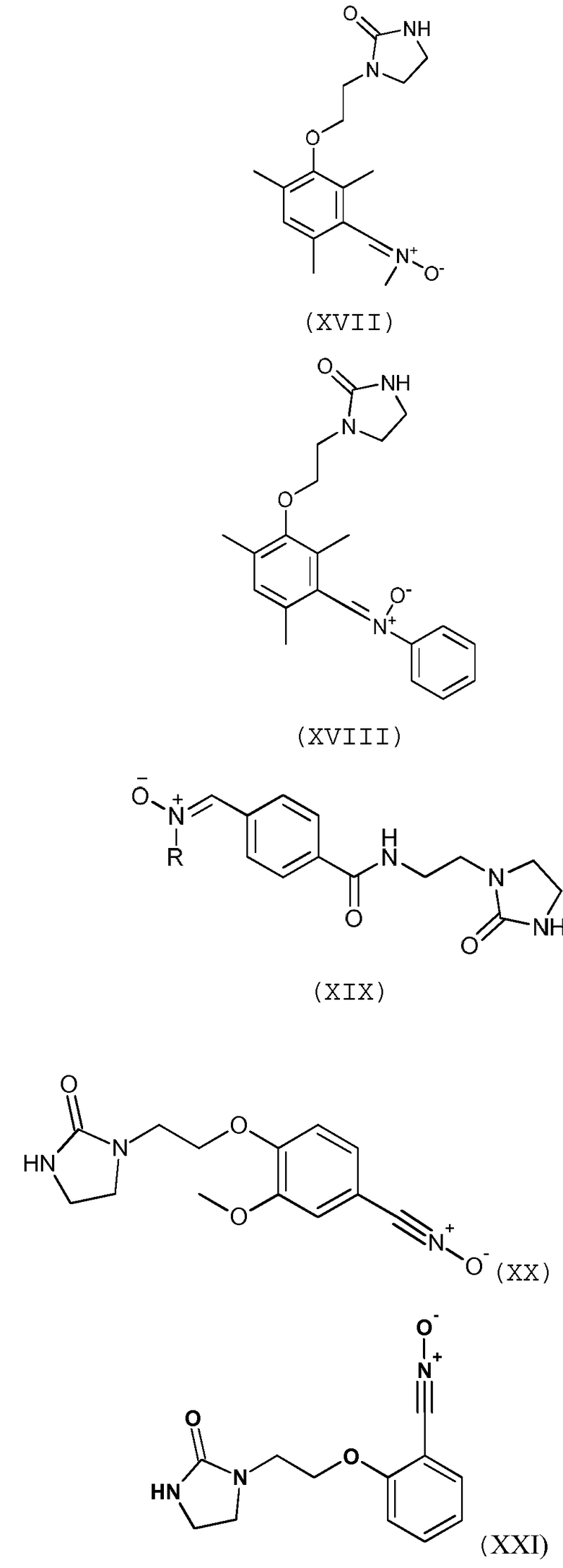
Согласно другому варианту осуществления изобретения, соединение согласно изобретению, предназначенное для прививки к полимеру, выбирают из соединений следующих формул (XXII)-(XXIII):
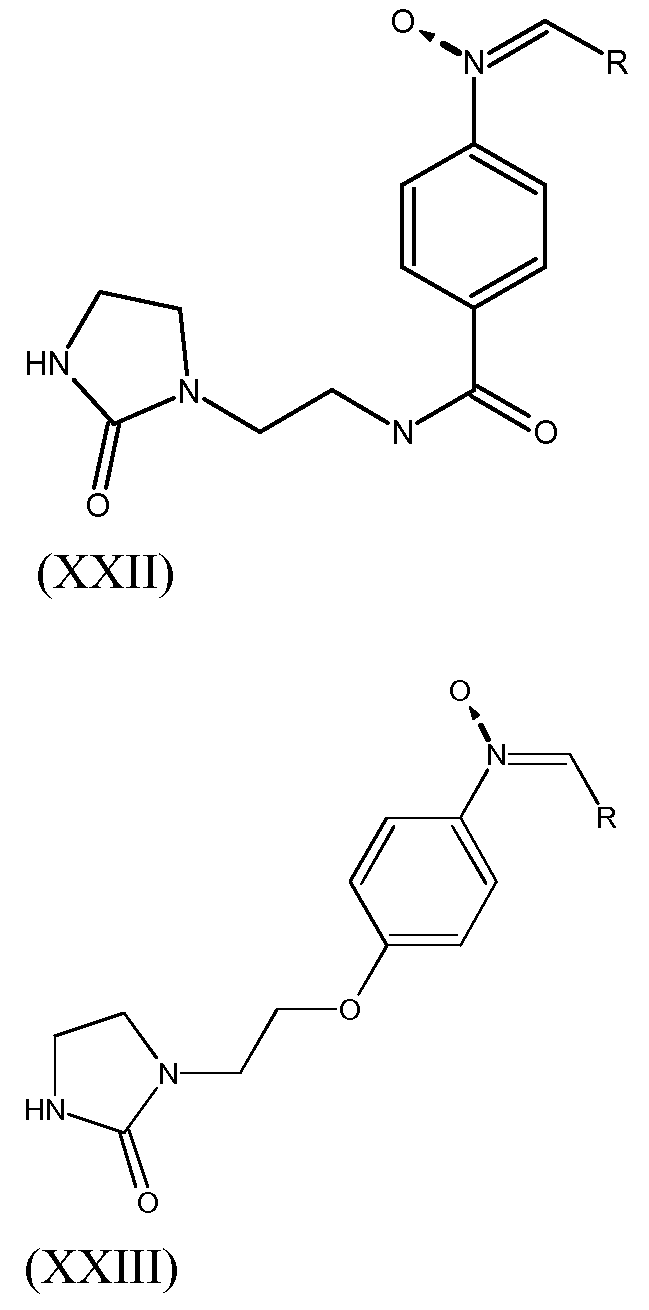
в которых R выбирают из спейсерной группы Sp, атома водорода, С1-С20 алкила, линейного или разветвленного, С3-С20 циклоалкила, линейного или разветвленного, С6-С20 арила, линейного или разветвленного, и группы формулы (Х):
 (Х)
(Х)
в которой n обозначает 1, 2, 3, 4 или 5 и каждый Y обозначает независимо спейсерную группу Sp, алкил или галоген.
Изобретение иллюстрируется дополнительно следующими неограничивающими примерами.
ПРИМЕРЫ ОСУЩЕСТВЛЕНИЯ
Структурный анализ и определение чистоты синтезированных молекул осуществляли с помощью ЯМР. Спектры регистрировали на спектрометре Avance 500 МГц BRUKER, снабженном «широкополосным» зондом BBIz-grad 5 мм. Для проведения количественной спектрометрии ПМР использовали единичную последовательность импульса в 30ºС с периодом повтора 3 секунды между каждой из 64 регистраций спектра. Образцы растворяли в дейтерированном диметилсульфоксиде (ДМСО). Этот же растворитель использовали для получения лок-сигнала. Калибровку осуществляли по сигналу протонов дейтерированного ДМСО с химическим сдвигом 2,44 м.д. относительно ТМС стандарта с химическим сдвигом 0 м.д. Спектр ЯМР 1Н совмещен с двумя анализами 2D HSQC 1H/13C и HMBC 1H/13C, позволяющими определять структуру молекул (см. таблицу сигналов). Количественный анализ в молях осуществляли на основе количественного спектра ЯМР 1D 1H.
Инфракрасный анализ позволил определить наличие нитрилоксидных групп в ароматическом соединении. Спектры снимали на спектрометре с Фурье-трансформацией VERTEX 70, оснащенном детектором DTGS. Спектры снимали на протяжении 32 сканирований в диапазоне частот 4000 см-1 - 400 см-1 с разрешением 2 см-1. Образцы готовили в форме таблеток с KBr в качестве носителя. Функциональная нитрилоксидная группа в ароматическом соединении характеризуется полосой поглощения 2295 см-1.
Спектрометрический анализ выполняли с прямым инжектированием образцов методом ионизации электрораспылением (ID/ESI). Анализы проводили с помощью спектрометра HCT Bruker (скорость потока 600 мкл/мин, давление газа-распылителя 10 psi, скорость газа-распылителя 4 л/мин).
Пример 1: Получение 1-(2-(3'-нитрилоксимезитил-1'-окси)этил)имидазолидин-2-она
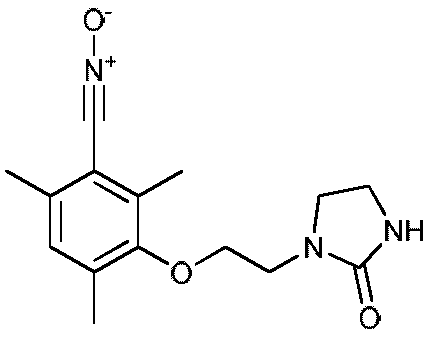
Это соединение может быть получено из мезитола гидроксиэтилимидазолидона согласно следующей схеме синтеза.
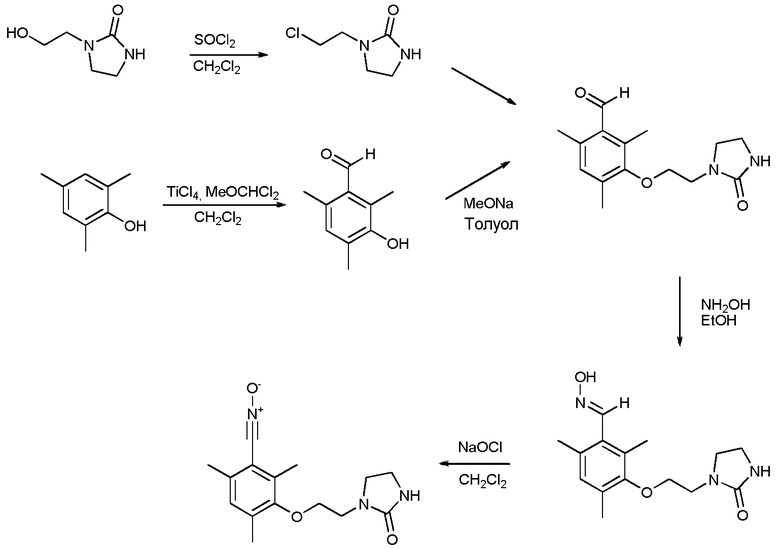
а) Получение 3-гидрокси-2,4,6-триметилбензальдегида
Это соединение может быть получено согласно процедуре, описанной в следующей статье: Yakubov, A.P.; Tayganov, D.V.; Belen'kii, L.I.; Krayushkin, M. M.; Bulletin of the Academy of Sciences of the USSR, Division of Chemical Science (English Translation); vol. 40; nb. 7.2; (1991); p. 1427-1432; Izvestiya Akademii Nauk SSSR, Seriya Khimicheskaya; nb. 7; (1991); р. 1609-1615.
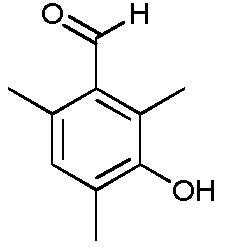
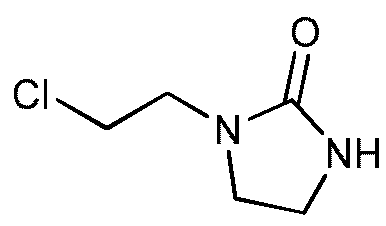
b) Получение 1-(2-хлорэтил)имидазолидин-2-она
Этот продукт описан в статье Nagarajan K., Arya V.P., Shah R. K.; Indian Journal of Chemistry, Section B: Organic Chemistry Including Medicinal Chemistry; 21; 10; 1982; 928-940.
К раствору 1-(2-гидроксиэтил)имидазолидин-2-она (50,0 г, 0,39 моль) в дихлорметане (250 мл) прибавляют по каплям при комнатной температуре тионилхлорид (34 мл, 0,47 моль) в течение 35 минут. К концу добавления температура реакционной среды составляет 35ºС. Реакционную среду выдерживают при температуре 35-40ºС в течение 2,5 часов. После выпаривания при пониженном давлении (Тбани 35ºС, 15-17 мбар) получают неочищенный продукт (67 г). Этот неочищенный продукт кристаллизуют в смеси ацетона и петролейного эфира (35 г на 950 мл ацетона и 820 мл петролейного эфира при -24ºС в течение 10-15 часов). Кристаллы отфильтровывают, промывают петролейным эфиром (2 раза по 40 мл), затем сушат в течение 10-15 часов при атмосферном давлении и комнатной температуре.
Получают твердое вещество белого цвета (33,3 г, выход 66%) с температурой плавления 93ºС.
Чистота продукта выше 97% (ЯМР 1Н).
Характеристика продукта с помощью спектров ЯМР 1Н и ЯМР 13С представлена в следующей таблице 1.
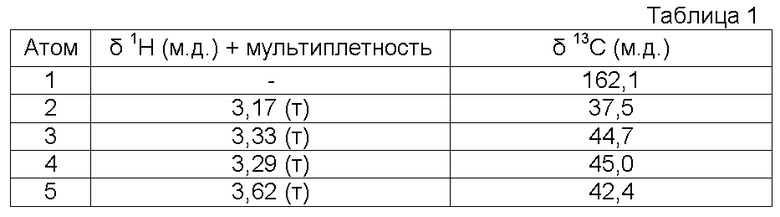
Используемый растворитель: ДМСО - Калибровка по сигналу ДМСО с химическим сдвигом 1Н в области 2,44 м.д., и с химическим сдвигом 13С в области 39,5 м.д.
с) Получение 2,4,6-триметил-3-(2-(2-оксоимидазолидин-1-ил)этокси)бензальдегида
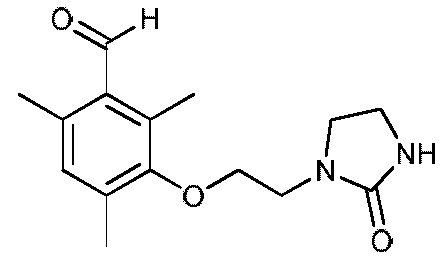
К раствору натрия (1,63 г, 0,071 моль) в метаноле (60 мл) прибавляют по каплям 3-гидрокси-2,4,6-триметилбензальдегид (11,90 г, 0,073 моль) в безводном толуоле (300 мл). Смесь кипятят с обратным холодильником, затем метанол отгоняют (собирают азеотропную смесь в объеме 80-90 мл). После возвращения к температуре 80-90ºС добавляют за один прием (2-хлорэтил)имидазолидин-2-он (10,45 г, 0,070 моль) к реакционной среде. После кипячения с обратным холодильником в течение 7 часов растворители выпаривают при пониженном давлении (Тбани 50ºС, 25 мбар). К полученной смеси прибавляют дихлорметан (150 мл) и воду (30 мл). Затем органический слой промывают 2 раза водой (20 мл). После высушивания над Na2SO4 дихлорметан выпаривают при пониженном давлении (Тбани 35ºС, 33 мбар). К полученной смеси (24 г) прибавляют петролейный эфир (3 порциями по 50 мл) и воду (50 мл) и полученный осадок отфильтровывают и промывают на фильтре водой (15 мл) и петролейным эфиром (2 раза по 15 мл).
Полученный продукт снова очищают путем промывки продукта, растворенного в дихлорметане (80 мл), 4%-ным раствором NaOH в воде (3 раза по 60 мл). После выпаривания растворителей при пониженном давлении продукт осаждают петролейным эфиром. Осадок отфильтровывают и сушат в течение 15-20 часов под атмосферным давлением и при комнатной температуре.
Получают твердый продукт белого цвета (8,55 г, выход 44%) с температурой плавления 139ºС.
Чистота продукта выше 94% (ЯМР 1Н).
Характеристика продуктов с помощью спектров ЯМР 1Н и 13С представлена в следующей таблице 2.
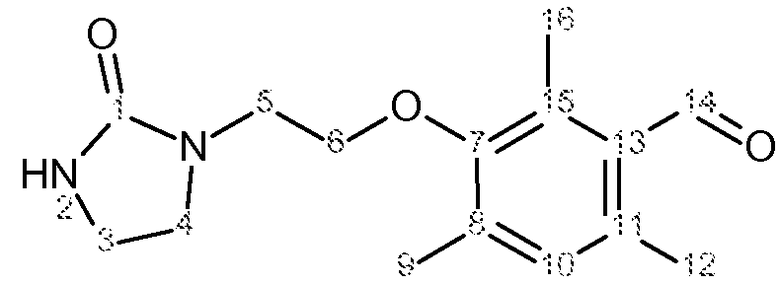
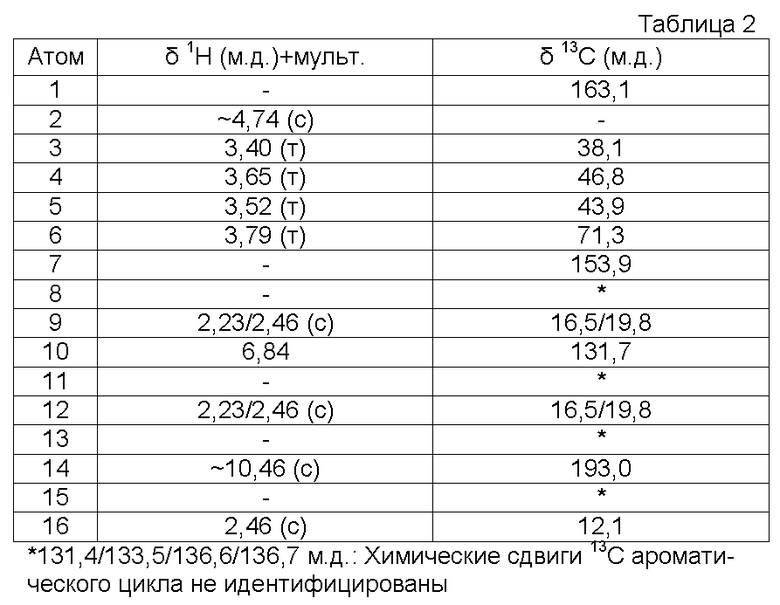
Используемый растворитель: CDCl3 - Калибровка по сигналу хлороформа с химическим сдвигом 1Н в области 7,2 м.д. и с химическим сдвигом 13С в области 77 м.д.
d) Получение 2,4,6-триметил-3-(2-(2-оксоимидазолидин-1-ил)этокси)бензальдегидоксима
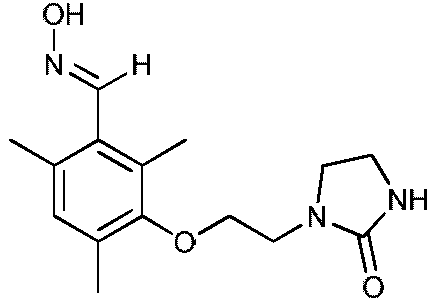
К раствору 2,4,6-триметил-3-(2-(2-оксоимидазолидин-1-ил)этокси)бензальдегида (7,90 г, 0,029 моль) в этаноле (70 мл), поддерживаемому при температуре 45ºС, прибавляют водный раствор гидроксиламина (2,83 г, 0,043 моль, 50%-ный раствор в воде) в этаноле (10 мл). Затем реакционную среду перемешивают в течение 2,5 часов при температуре между 50 и 55ºС. Растворитель выпаривают при пониженном давлении (Тбани 37ºС, 35 мбар). К полученному неочищенному продукту прибавляют петролейный эфир (80 мл). Полученный осадок отфильтровывают, промывают петролейным эфиром (2 раза по 20 мл) и сушат в течение 15-20 часов под атмосферным давлением и при комнатной температуре.
Получают твердый продукт белого цвета (7,82 г, выход 94%) с температурой плавления 165ºС.
Чистота продукта выше 84% (остальные 16% включают, в частности, 7 мол.% EtOH), согласно ЯМР 1Н.
Характеристика продукта с помощью спектров ЯМР 1Н и 13С представлена в следующей таблице 3.
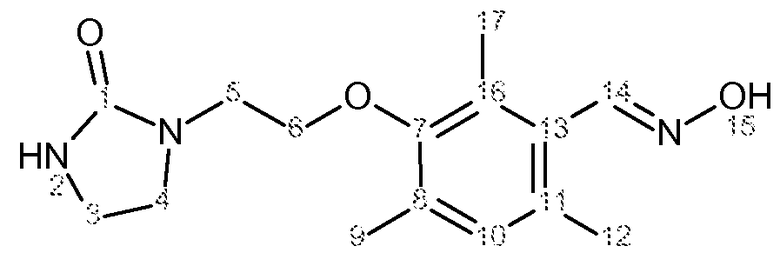
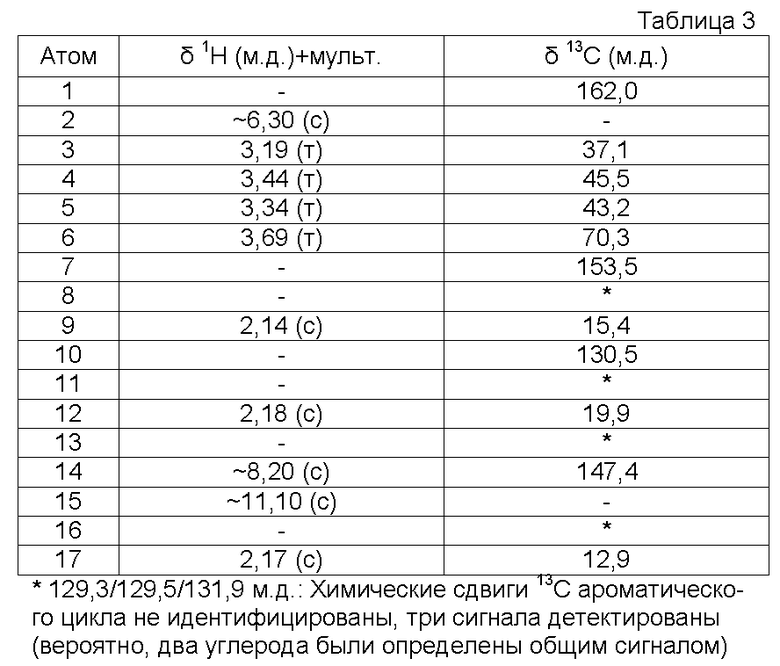
Используемый растворитель: ДМСО - Калибровка по сигналу ДМСО с химическим сдвигом 1Н в области 2,44 м.д. и с химическим сдвигом 13С в области 39,5 м.д.
е) Получение 2,4,6-триметил-3-(2-(2-оксоимидазолидин-1-ил)этокси)нитрилоксида, соединение согласно изобретению
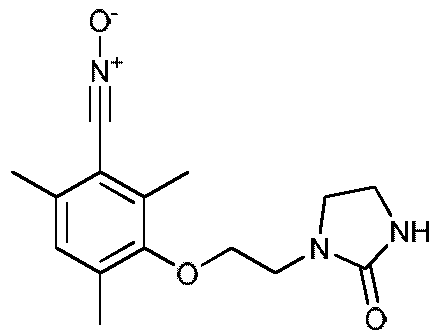
К раствору полученного выше оксима (6,00 г, 0,021 моль) в дихлорметане (250 мл) прибавляют по каплям при температуре 2ºС водный раствор NaOCl (4% активного хлора, 52 мл) в течение 5-7 минут. Температуру реакционной среды поддерживают при температуре между 0 и -4ºС. Затем реакционную среду перемешивают в течение 3 часов при температуре между 0 и 5ºС. Затем органический слой отделяют. Водный слой экстрагируют дихлорметаном (2 раза по 15 мл). Органические слои объединяют, затем промывают водой (2 раза по 20 мл), высушивают над Na2SO4. Объем растворителя снижают путем выпаривания при пониженном давлении (Тбани 22ºС, 220 мбар) до объема 50-60 мл. После этого прибавляют петролейный эфир (75 мл) и раствор выдерживают при температуре -18ºС в течение 10-15 часов. Полученный осадок отфильтровывают и промывают смесью этилацетата/петролейного эфира (1/2) (10 мл) и затем сушат в течение 10-15 часов под атмосферным давлением и при комнатной температуре.
Получают твердый продукт белого цвета (4,70 г, выход 79%) с температурой плавления 156ºС.
Чистота продукта выше 85% (ЯМР 1Н).
Характеристика продукта с помощью спектров ЯМР 1Н и 13С представлена в следующей таблице 4.
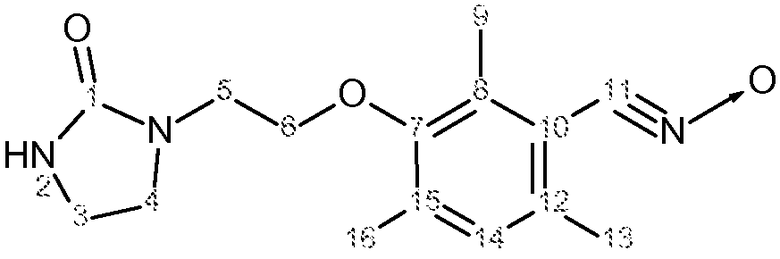
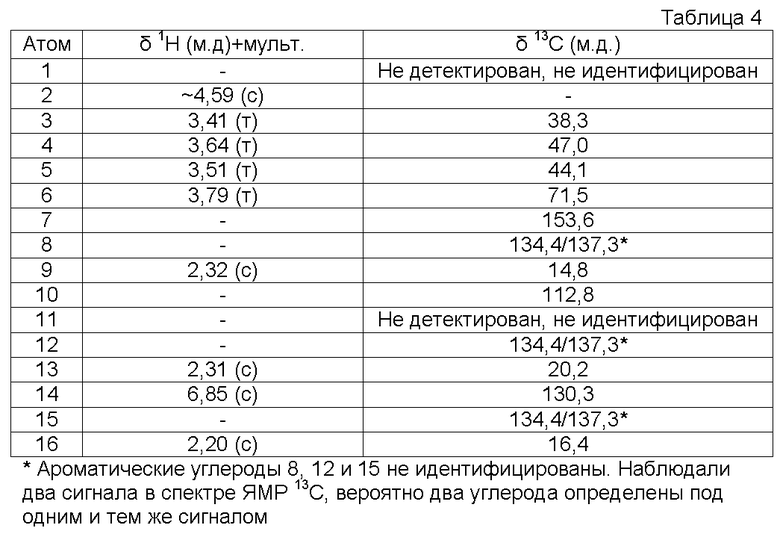
Функциональная группа -C≡N→O имела в ИК-спектре характеристическую полосу поглощения 2295 см-1.
Используемый растворитель: CDCl3 - Калибровка по сигналу хлороформа с химическим сдвигом 1Н в области 7,2 м.д., и с химическим сдвигом 13С в области 77 м.д.
Пример 2: Получение 2-[2-(2-оксоимидазолидин-1-ил)этокси]бензонитрилоксида
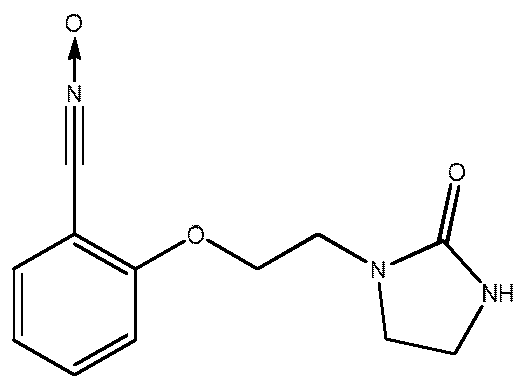
Это соединение может быть получено из салицилового альдегида и 2-хлорэтилимидазолидона согласно следующей схеме синтеза:
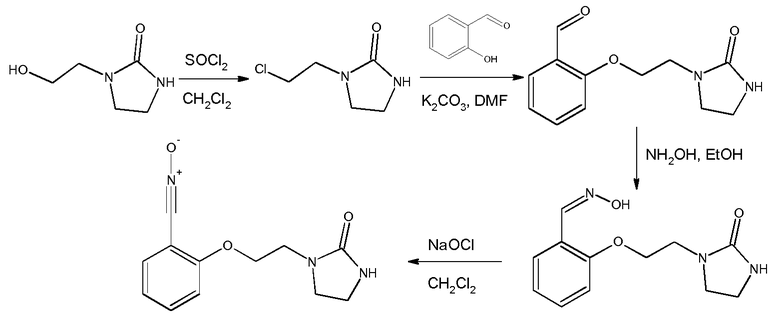
а) Получение 1-(2-хлорэтил)имидазолидин-2-она описано в примере 1
b) Получение 2-[2-(2-оксоимидазолидин-1-ил)этокси]бензальдегида
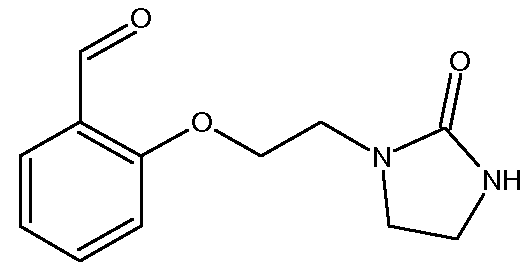
К раствору салицилового альдегида (22,0 г, 0,180 моль) в ДМФ (100 мл) прибавляют К2СО3 (87,1 г, 0,631 моль). Смесь перемешивают при 52ºС. После поддерживания этой температуры в течение 10 минут прибавляют порциями 1-(2-хлорэтил)имидазолидин-2-он (40,0 г, 0,270 моль, чистота >90%). Температуру смеси доводят до 90ºС (Тбани) в течение одного часа и эту температуру поддерживают в течение 5 часов. После возвращения к комнатной температуре смесь разбавляют водой (1,3 л) и продукт экстрагируют с помощью CH2Cl2 (500 мл, 5 раз по 100 мл). Органические слои объединяют, затем промывают водой (два раза по 50 мл) и выпаривают до получения сырого реакционного продукта 70-80 г (густая суспензия) (Тбани=40ºС). Сырой продукт реакции обрабатывают Et2O (120 мл) и суспензию перемешивают при комнатной температуре в течение 20 минут. Полученный осадок отфильтровывают и промывают смесью ДМФ/Et2O/Н2О (5 мл/20 мл/15 мл), затем с помощью Et2O (2 раза по 10 мл). Полученный твердый продукт сушат при комнатной температуре.
Получают твердый продукт (30,6 г, выход 73%) с температурой плавления 150ºС. Чистота продукта выше 84% (ЯМР 1Н).
Полученный 2-[2-(2-оксоимидазолидин-1-ил)этокси]бензальдегид сразу используют на следующей стадии без дополнительной очистки.
Характеристика продукта с помощью спектров ЯМР 1Н и 13С:
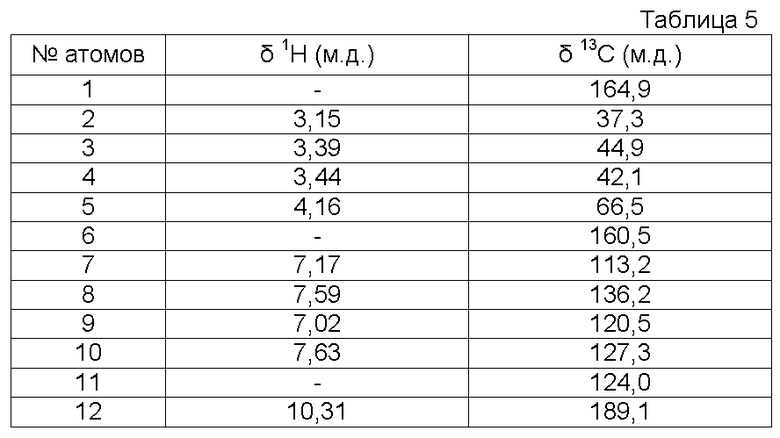
Используемый растворитель: ДМСО - Калибровка по сигналу ДМСО с химическим сдвигом 1Н в области 2,44 м.д. и с химическим сдвигом 13С в области 39,5 м.д.
с) Получение 2-[2-(2-оксоимидазолидин-1-ил)этокси]бензальдегидоксима
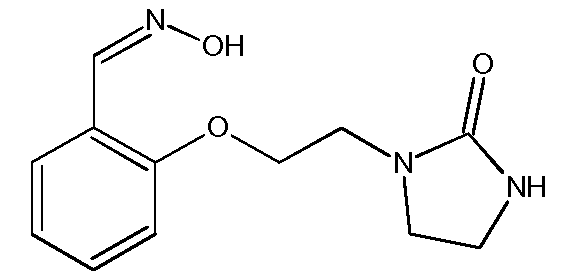
Раствор 2-[2-(2-оксоимидазолидин-1-ил)этокси]бензальдегида (10,0 г, 0,043 моль) в EtOH (100 мл) нагревают до 50ºС. При этой температуре добавляют раствор гидроксиламина (4,5 г, 0,068 моль, 50% раствор в воде, от компании Aldrich) в EtOH (10 мл). Затем реакционную среду перемешивают в течение 6 часов при температуре между 50ºС и 70ºС. Реакционную среду выпаривают при пониженном давлении (Тбани 45ºС, 65-70 мбар) до получения суспензии. Затем сырой продукт реакции обрабатывают водой (5 мл). Полученный раствор охлаждают до 5ºС и поддерживают эту температуру в течение 15 часов. Полученный осадок отфильтровывают и промывают на фильтре смесью EtOH/вода (2 мл/2 мл), затем смесью EtOH/петролейный эфир (1 мл/4 мл), затем петролейным эфиром (2 раза по 10 мл). Твердый продукт сушат под атмосферным давлением при комнатной температуре.
Получают твердый продукт белого цвета (9,25 г, выход 87%) с температурой плавления 88ºС.
Чистота продукта выше 99% (ЯМР 1Н).
Характеристика продукта с помощью спектров ЯМР 1Н и 13С:
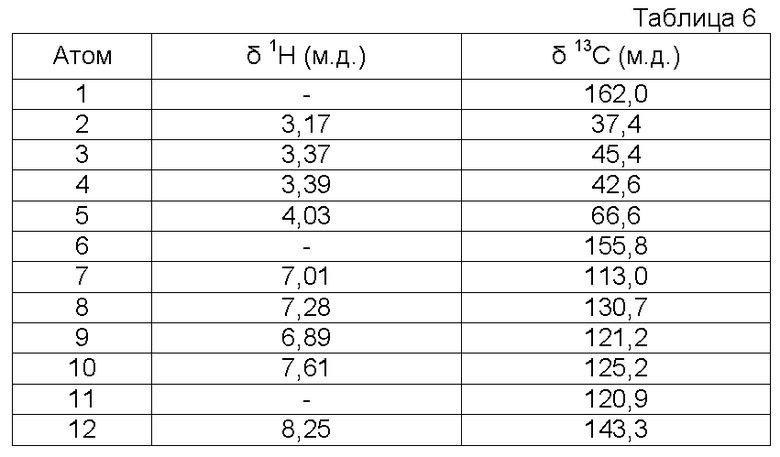
Используемый растворитель: ДМСО - Калибровка по сигналу ДМСО с химическим сдвигом 1Н в области 2,44 м.д. и с химическим сдвигом 13С в области 39,5 м.д.
d) Получение 2-[2-(2-оксоимидазолидин-1-ил)этокси]бензонитрилоксида
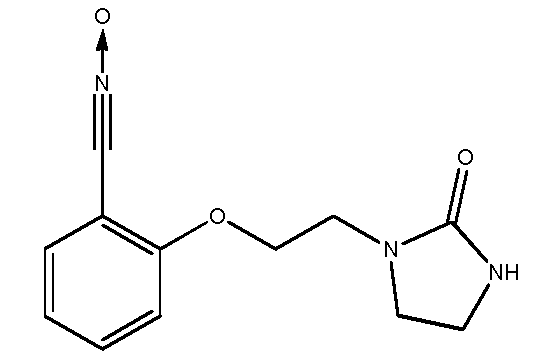
К суспензии 2-[2-(2-оксоимидазолидин-1-ил)этокси]бензальдегидоксима (20,2 г, 0,081 моль ) в CH2Cl2 (400 мл) при -1ºС прибавляют по каплям в течение 10 минут водный раствор NaOCl в воде (157 мл, Aldrich, >4% активного хлора). Затем реакционную среду перемешивают в течение 20 минут. Разделяют водные и органические слои и водный слой экстрагируют с помощью CH2Cl2 (2 раза по 75 мл). Объединенные органические слои промывают водой (3 раза по 10 мл) и высушивают над Na2SO4. Слои концентрируют до объема 100 мл при пониженном давлении и комнатной температуре. Добавляют 50 мл петролейного эфира. Раствор охлаждают до -18ºС (3 часа). Осадок отфильтровывают, промывают смесью CH2Cl2/петролейный эфир (5 мл/10 мл; затем 5 мл/20 мл; затем 0 мл/20 мл), затем сушат под атмосферным давлением и при комнатной температуре.
Получают твердый продукт (11,32 г, выход 57%) с температурой плавления 109-110ºС с разложением продукта.
Чистота продукта выше 94% (ЯМР 1Н).
Характеристика продукта с помощью спектров ЯМР 1Н и 13С:
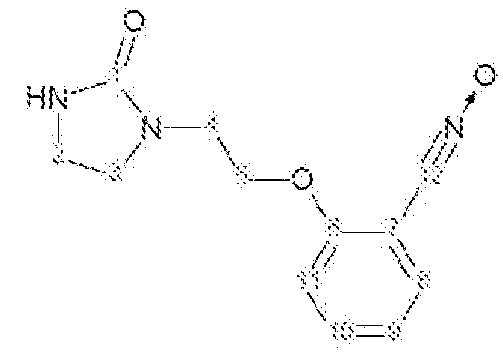
Используемый растворитель: ДМСО - Калибровка по сигналу ДМСО с химическим сдвигом 1Н в области 2,44 м.д. и с химическим сдвигом 13С в области 39,5 м.д.
Характеристика продукта с помощью инфракрасного спектра (таблетки с KBr)
ν (см-1): 2295 (функциональная группа Ar-C≡N→O).
Характеристика продукта с помощью масс-спектрометрии
C12H13N3O3, Mw=247,25 г/мол.
Образцы анализировали прямым внесением образца в масс-спектрометр с использованием метода ионизации электрораспылением (ID/ESI).
Приготовление образца
20 г образца растворяли в 2 мл ацетонитрила.
m/z: 270 ([M+Na]+), 517 ([2M+Na]+).
Пример 3: Получение 3-метокси-4-[2-(2-оксоимидазолидин-1-ил)этокси]бензонитрилоксида
Это соединение может быть получено из ванилина и 2-хлорэтилимидазолидона согласно следующей схеме синтеза:
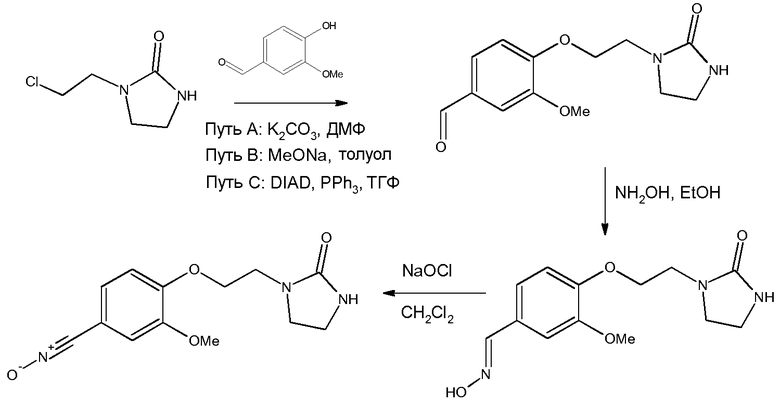
а) Получение 3-метокси-4-[2-(2-оксоимидазолидин-1-ил)этокси]бензальдегида
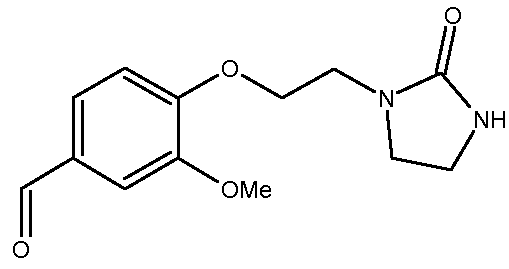
Путь А
Суспензию ванилина (30,0 г, 0,197 моль) и К2СО3 (95,4 г, 0,690 моль) в ДМФ (200 мл) нагревают при 50ºС в течение 15 минут. К этой суспензии прибавляют порциями 1-(2-хлорэтил)имидазолидин-2-он (44,0 г, 0,296 моль, чистота >90%) в ДМФ (30 мл). Реакционную среду нагревают до 90ºС (Тбани) и эту температуру поддерживают в течение примерно 4 часов. Реакционную среду доводят до комнатной температуры и добавляют воду (1,25 л). Продукт экстрагируют с помощью CH2Cl2 (400 мл, 4 раза по 100 мл). Объединенные органические слои промывают водой (60 мл) и концентрируют при пониженном давлении (14 мбар, 40ºС). Сырой продукт реакции разбавляют Et2O (100 мл) и суспензию перемешивают при комнатной температуре в течение 15-20 минут. Полученный осадок отфильтровывают, промывают Et2O (3 раза по 15 мл) и сушат при комнатной температуре.
Получают твердый продукт (31,2 г, выход 60%) с температурой плавления 130ºС.
Чистота продукта выше 92% (ЯМР 1Н).
Путь В
К раствору натрия (1,51 г, 0,066 моль) в СН3ОН (60 мл) прибавляют ванилин (10,0 г, 0,066 моль) в безводном толуоле (250 мл). Реакционную среду кипятят с обратным холодильником в инертной атмосфере, затем остаточный метанол отгоняют. После возвращения к температуре 80-90ºС суспензию 1-(2-хлорэтил)имидазолидин-2-она (9,28 г, 0,064 мол, чистота >95%) в толуоле (50 мл) вводят одной порцией в реакционную среду. После проведения реакции в течение 25 часов реакционную среду концентрируют при пониженном давлении (Тбани 50ºС, 30 мбар). Сырой продукт реакции обрабатывают CH2Cl2 (150 мл). Непрореагировавший ванилин удаляют экстрагированием 7%-ным водным раствором NaOH (5 раз по 30 мл). Объединенные органические слои промывают водой (4 раза по 50 мл), высушивают над Na2SO4 и выпаривают при пониженном давлении (Тбани 27ºС, 20 мбар). Сырой продукт реакции (4,81 г) разбавляют смесью петролейного эфира и EtOAc и полученный осадок отфильтровывают.
Получают твердый продукт (0,91 г, выход 6%) с температурой плавления 127ºС.
Чистота продукта выше 81% (ЯМР 1Н).
Путь С
Процедура проведения реакции Мицунобу описана, например, в следующих ссылках: Mitsunobu, О.; Yamada, Y. Bull. Chem. Soc. Japan 1967, 40, 2380-2382, The Use of Diethyl Azodicarboxylate and Triphenylphosphine in Synthesis and Transformation of Natural Products Mitsunobu, O. Synthesis 1981, 1-28, патент ЕР1149092 В1, 2003.
К раствору ванилина (5,02 г, 0,033 моль), безводного 1-(2-гидроксиэтил)имидазолидин-2-она (6,38 г, 0,049 мол, Aldrich) и PPh3 (13,1 г, 0,50 моль) в безводном ТГФ (300 мл) при 2°С прибавляют по каплям в течение 20 минут раствор диизопропилазодикарбоксилата (10,1 г, 0,050 мол, Aldrich) в безводном ТГФ (150 мл). Реакционную среду перемешивают в течение 14 часов при комнатной температуре, затем разбавляют водой (150 мл). Реакционную среду концентрируют при пониженном давлении (45 мбар, Тбани 28ºС). Водный слой экстрагируют с помощью EtOAc (3 раза по 200 мл). Объединенные органические слои промывают насыщенным водным раствором NaCl, затем концентрируют при пониженном давлении так, чтобы получить раствор в объеме 150 мл. Растворенный сырой продукт реакции очищают колоночной хроматографией (SiO2, элюент 1: EtOAc, элюент 2: EtOAC/EtOH=4/1, Rf продукта 0,36, Rf соединения Ph3PO 0,71 в смеси EtOAc: EtOH=5:1).
Получают твердый продукт (6,59 г, выход 76%) с температурой плавления 130ºС.
Чистота продукта выше 88% (ЯМР 1Н).
Полученный 3-метокси-4-[2-(2-оксоимидазолидин-1-ил)этокси]бензальдегид сразу используют на следующей стадии без дополнительного очищения.
Характеристика продукта с помощью спектров ЯМР 1Н и 13С:
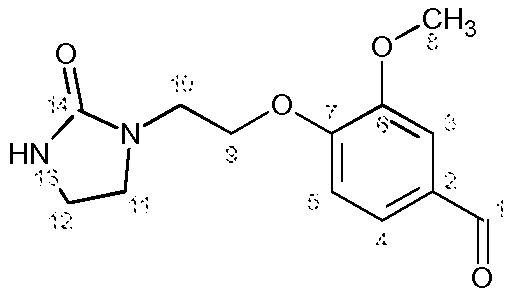
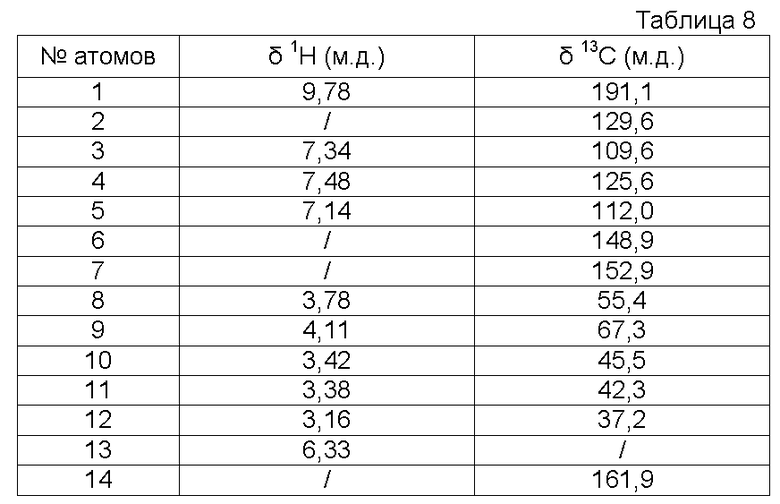
Используемый растворитель: ДМСО - Калибровка по сигналу ДМСО с химическим сдвигом 1Н в области 2,44 м.д. и с химическим сдвигом 13С в области 39,5 м.д.
b) Получение 3-метокси-4-[2-(2-оксоимидазолидин-1-ил)этокси]бензальдегидоксима
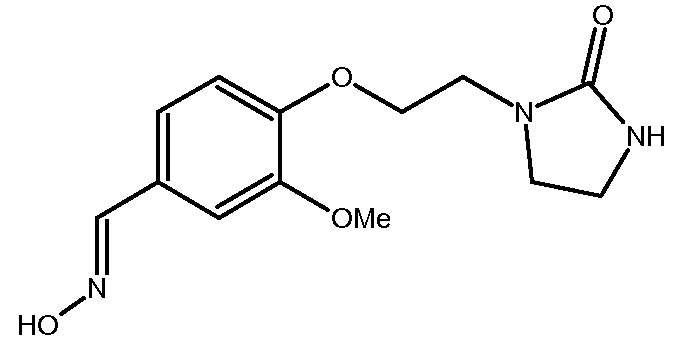
К раствору 3-метокси-4-[2-(2-оксоимидазолидин-1-ил)этокси]бензальдегида (25,6 г, 0,097 моль) в EtOH (250 мл) при 52ºС добавляют раствор гидроксиламина (10,2 г, 0,155 моль, 50% раствор в воде, Aldrich) в EtOH (20 мл). Затем реакционную среду перемешивают в течение 4,5 часов при температуре между 50 и 60ºС. Реакционную среду концентрируют при пониженном давлении (Тбани=42ºС, 60 мбар) до получения остатка объемом 70-80 мл. Полученный осадок отфильтровывают, промывают смесью EtOH/вода (2 раза 5 мл/15 мл) и сушат под атмосферным давлением и при комнатной температуре.
Получают твердый продукт белого цвета (22,14 г, выход 82%) с температурой плавления 189ºС.
Чистота продукта выше 88% (ЯМР 1Н).
Характеристика продукта с помощью спектров ЯМР 1Н и 13С:
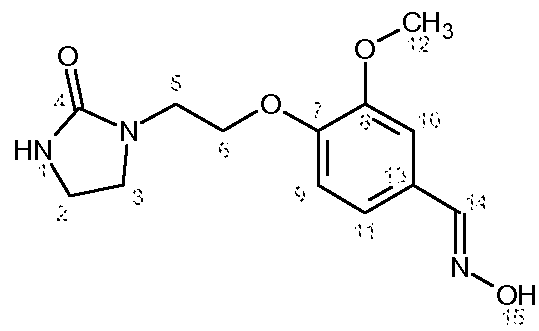
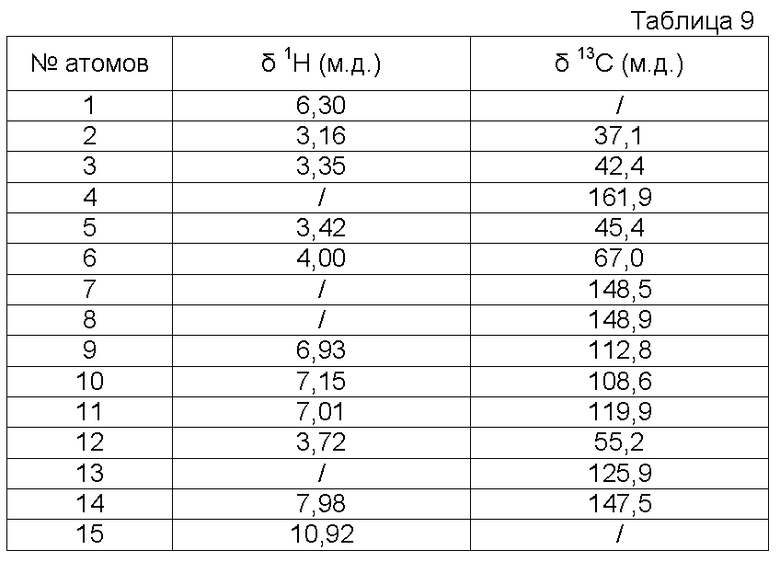
Используемый растворитель: ДМСО - Калибровка по сигналу ДМСО с химическим сдвигом 1Н в области 2,44 м.д. и с химическим сдвигом 13С в области 39,5 м.д.
с) Получение 3-метокси-4-[2-(2-оксоимидазолидин-1-ил)этокси]бензонитрилоксида
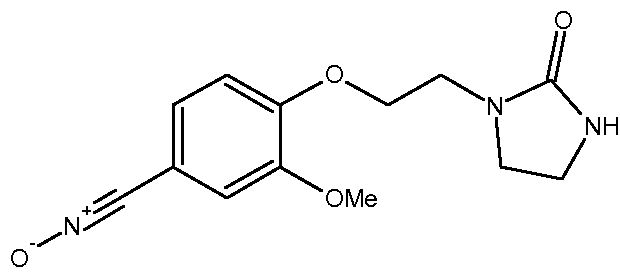
К суспензии 3-метокси-4-[2-(2-оксоимидазолидин-1-ил)этокси]бензальдегидоксима (21,7 г, 0,078 моль) в CH2Cl2 (950 мл) при -3ºС прибавляют по каплям водный раствор NaOCl в воде (161 мл, Aldrich, >4% активного хлора) в течение 10 минут. Затем реакционную среду перемешивают в течение 20 минут при 0ºС. Отделяют органический слой и водный слой экстрагируют с помощью CH2Cl2 (4 раза по 100 мл). Объединенные органические слои промывают водой (3 раза по 100 мл), высушивают над Na2SO4, затем концентрируют при пониженном давлении (Тбани 22ºС) до объема 200-220 мл. Полученный осадок отфильтровывают, промывают с помощью CH2Cl2 (2 раза по 10 мл) и сушат под атмосферным давлением и при комнатной температуре.
Получают твердый продукт (9,13 г, выход 42%) с температурой плавления 109-111ºС с разложением продукта.
Чистота продукта выше 80% (ЯМР 1Н). После перекристаллизации из EtOH чистота соединения выше 90% по массе.
Характеристика продукта с помощью спектров ЯМР 1Н и 13С:
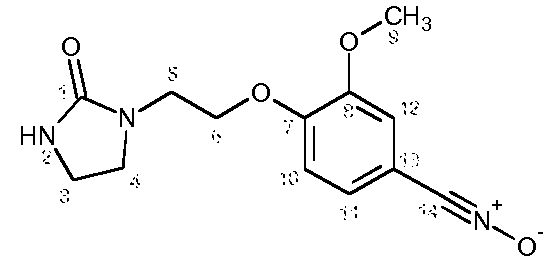
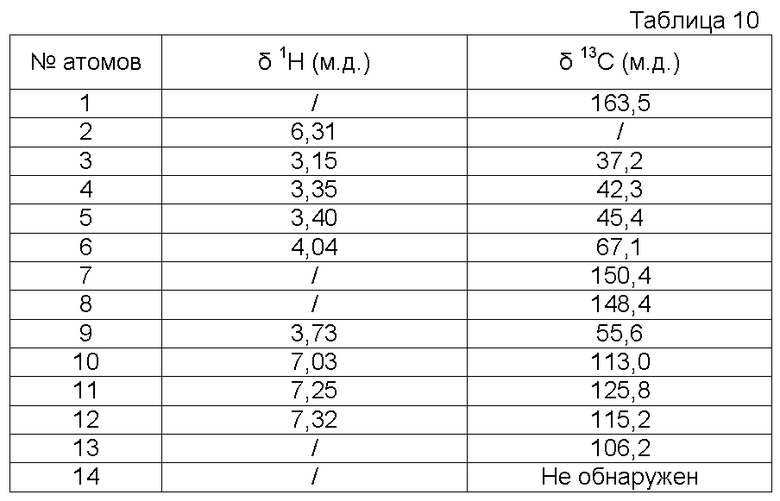
Используемый растворитель: ДМСО - Калибровка по сигналу ДМСО с химическим сдвигом 1Н в области 2,44 м.д. и с химическим сдвигом 13С в области 39,5 м.д.
Характеристика продукта с помощью инфракрасного спектра (таблетки с KBr)
ν (см-1): 2305 (функциональная группа Ar-C≡N→O).
Характеристика продукта с помощью масс-спектрометрии
C13H15N3O4, Mw=277,27 г/моль
Образцы анализировали прямым внесением образца в масс-спектрометр, используя метод ионизации электрораспылением (ID/ESI).
Приготовление образца
Примерно 20 мг образца растворяли в 25 мл метанола, затем разбавляли в соотношении 1/100 для анализа ID/ESI.
Режим положительных ионов
m/z: 300 ([M+Na]+), 577 ([2M+Na]+).
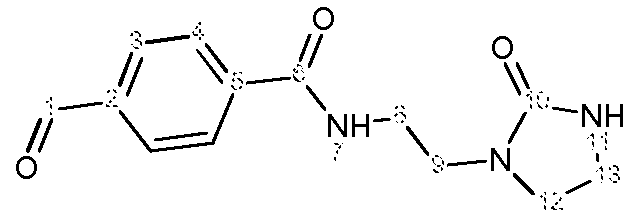
Пример 4: Получение (Z,E)-N-(4-(2-(2-оксоимидазолидин-1-ил)этилкарбамоил)бензилиден)анилиноксида
Соединение может быть получено из 4-формилбензойной кислоты и 2-аминоэтилимидазолидона согласно следующей схеме синтеза:
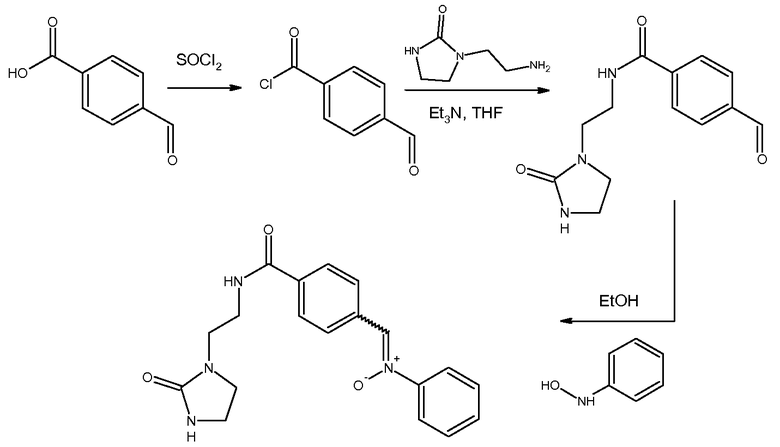
а) Получение 4-формилбензоилхлорида
Синтез этого соединения описан в следующих ссылках: JANSSEN PHARMACEUTICA N.V.; WO2007/53386; (2007); (A2). Температура плавления синтезированного формилбензоилхлорида соответствует данным, описанным в следующих ссылках: Graffner-Nordberg, Malin; Sjoedin, Karin; Tunek, Anders; Hallberg, Anders Chemical& Pharmaceutical Bulletin, 1998, vol.46, 4, h.591-601 et Kuhlmann; Alexander Inorganica Chemica Acta, 1979, vol.34, p.197, 207 et Simonis Chemische Berichte, 1912, vol.45, p.1586.
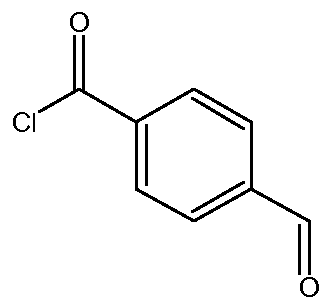
b) Получение 4-формил-N-[2-(2-оксоимидазолидин-1-ил)этил]бензамида
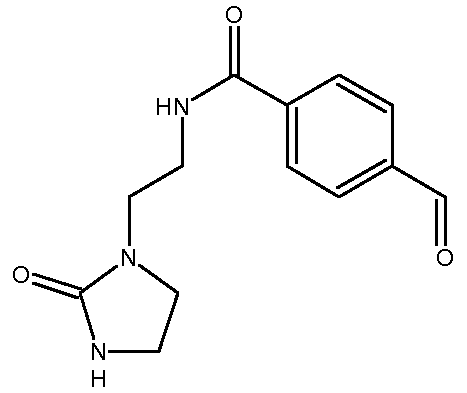
К суспензии 1-(2-аминоэтил)имидазолидин-2-она (12,6 г, 0,098 моль) и Et3N (19,8 г, 0,195 моль) в сухом ТГФ (300 мл) при -35°С прибавляют раствор 4-формилбензоилхлорида (16,5 г, 0,098 моль) в сухом ТГФ (100 мл) в течение 30 минут. Во время прибавления температуру реакционной среды поддерживают между -35ºС и -38ºС. Затем температуру реакционной среды медленно, в течение 4 часов, доводят до комнатной температуры. Полученный осадок (представляющий собой, главным образом, смесь целевого продукта с триэтиламина гидрохлоридом Et3N.HCl) отфильтровывают и промывают ТГФ (2 раза по 20 мл). Сырой продукт реакции солюбилизируют в водном растворе Na2CO3 (3,4 г, 0,032 мол в 40 мл воды). Целевой продукт экстрагируют несколько раз с помощью EtOAc (общий объем: 3,5 л). Объединенные органические слои высушивают над Na2SO4 и концентрируют при пониженном давлении (Тбани=40ºС).
Получают твердый продукт (5,53 г, выход 22%) с температурой плавления 138ºС.
Чистота продукта выше 81% (ЯМР 1Н). Этот продукт сразу используют на следующей стадии без дополнительной очистки.
Характеристика продукта с помощью спектров ЯМР 1Н и 13С:
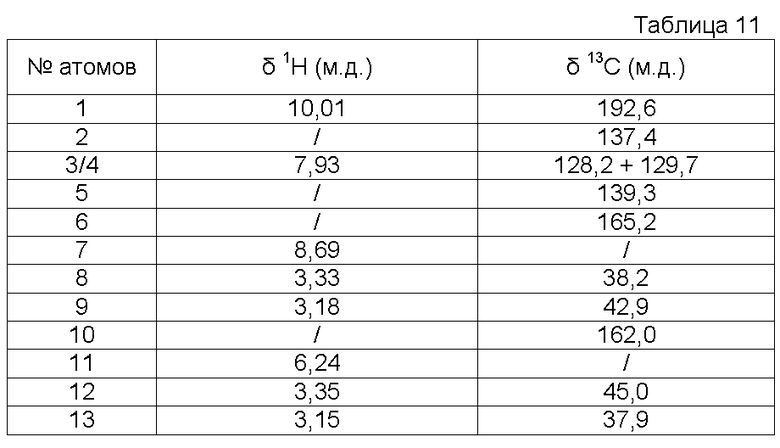
Используемый растворитель: ДМСО - Калибровка по сигналу ДМСО с химическим сдвигом 1Н в области 2,44 м.д. и с химическим сдвигом 13С в области 39,5 м.д.
с) Получение N-фенилгидроксиламина
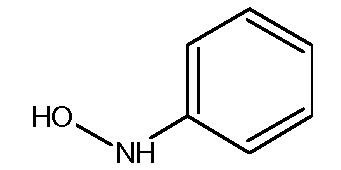
Синтез этого соединения из нитробензола описан в Organic Syntheses, Coll. Vol. p.445 (1941); Vol.4. p. 57 (1925).
d) Получение (Z,E)-N-(4-(2-(2-оксоимидазолидин-1-ил)этилкарбамоил)бензилиден)анилиноксида
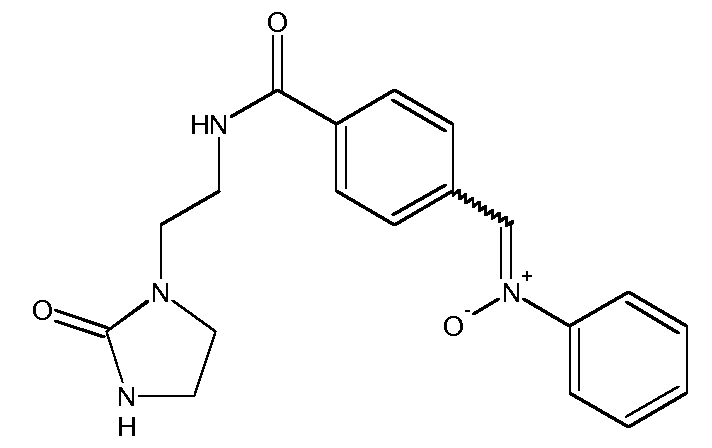
К раствору 4-формил-N-[2-(2-оксоимидазолидин-1-ил)этил]бензамида (5,3 г, 0,020 моль) в EtOH (50 мл) прибавляют раствор N-фенилгидроксиламина (2,21 г, 0,020 моль) в EtOH (10 мл). Реакционную смесь нагревают с обратным холодильником в течение 4 часов, затем охлаждают до комнатной температуры. Полученный осадок отфильтровывают, промывают EtOH (3 раза по 5 мл) и сушат при комнатной температуре на открытом воздухе.
Получают твердый продукт (4,65 г, выход 66%) с температурой плавления 209ºС.
Чистота продукта выше 92% (ЯМР 1Н).
Характеристика продукта с помощью спектров ЯМР 1Н и 13С:
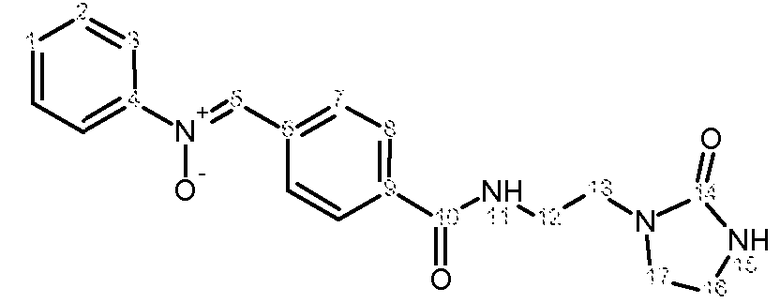
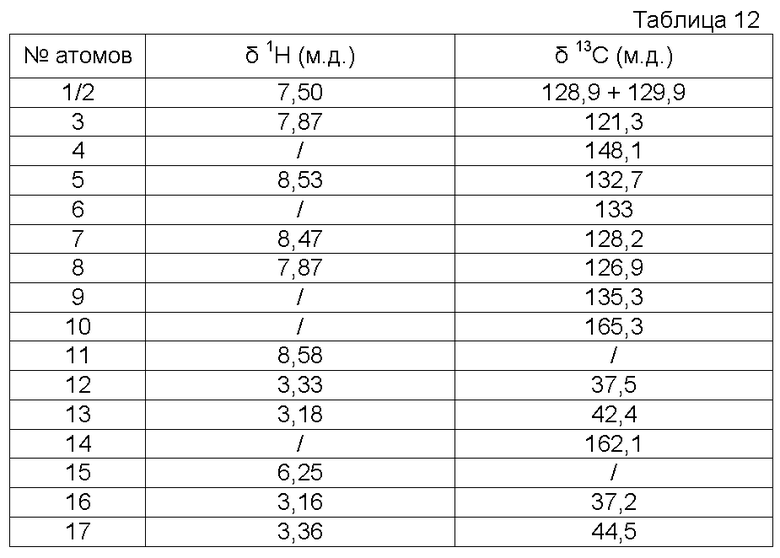
Используемый растворитель: ДМСО - Калибровка по сигналу ДМСО с химическим сдвигом 1Н в области 2,44 м.д. и с химическим сдвигом 13С в области 39,5 м.д.
Характеристика продукта с помощью масс-спектрометрии
C19H20N4O3, Mw=352,38 г/мол.
Образец анализировали прямым внесением образца в масс-спектрометр, используя метод ионизации электрораспылением (ID/ESI).
Приготовление образца
Примерно 20 мг образца растворяли в 0,5 мл ДМСО+24,5 мл метанола, затем разбавляли до 1/100 метанолом для анализа ID/ESI.
Режим положительных ионов
m/z: 375 ([M+Na]+), 727 ([2M+Na]+).
Режим отрицательных ионов
m/z: 351 ([M-Н]-), 703 ([2M-Н]-).
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ СИНТЕЗА МОЛЕКУЛ, СОДЕРЖАЩИХ ФУНКЦИОНАЛЬНУЮ ГРУППУ НИТРИЛОКСИДА | 2016 |
|
RU2724105C2 |
| СПОСОБ СИНТЕЗА АРОМАТИЧЕСКИХ ОКСИМОВ | 2016 |
|
RU2720240C2 |
| Полимер, несущий определенные боковые имидазолидинонные функциональные группы | 2018 |
|
RU2758218C2 |
| Полиароматическая молекула, имеющая нитрилоксидную функциональную группу | 2018 |
|
RU2742027C1 |
| СПОСОБ ПОЛУЧЕНИЯ ЗАМЕЩЕННЫХ 2,3,5,6-ТЕТРАОКСАБИЦИКЛО[2.2.1]ГЕПТАНОВ | 2012 |
|
RU2494102C1 |
| Спироконденсированные производные 2,3-дигидроиндола, их применение в офтальмологии | 2017 |
|
RU2712039C2 |
| ФУНКЦИОНАЛИЗИРОВАННЫЕ НАНОЧАСТИЦЫ, ИХ ПРИГОТОВЛЕНИЕ И ПРИМЕНЕНИЕ | 2007 |
|
RU2437890C2 |
| 3-(2-ОКСОИМИДАЗОЛИДИНИЛ-5)ИНДОЛЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2019 |
|
RU2720490C1 |
| СПОСОБ ДЕАЦЕТИЛИРОВАНИЯ α-АМИНОАЦЕТАЛЕЙ | 2008 |
|
RU2477270C2 |
| Ингибитор основной протеазы коронавируса SARS-CoV-2 | 2023 |
|
RU2840908C2 |
Изобретение относится к соединениям, содержащим, по меньшей мере, одну группу Q и, по меньшей мере, одну группу А, связанные друг с другом «спейсерной» группой Sp, и которые могут быть использованы в качестве сочетающих агентов, способных устанавливать взаимодействия между полимером и наполнителем. В указанных соединениях Q представляет собой нитрилоксидную или нитроновую группу формулы (VII) или (VIII):
 ,
,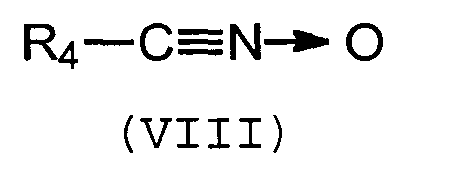
в которых R1-R4 выбирают независимо из атома водорода, С6 арила и группы формулы (X):
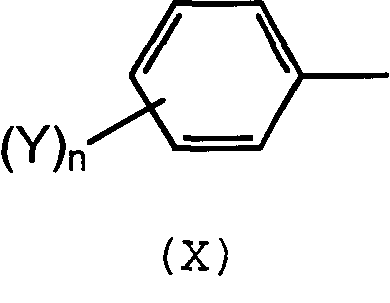
в которой n обозначает 1, 2, 3, 4 или 5, и каждый Y обозначает независимо спейсерную группу Sp или C1-C5 алкил, А соответствует формуле (II):
где Х обозначает атом кислорода, Sp представляет собой C1-C5 алкильную цепь, необязательно прерванную одним или несколькими атомами азота или кислорода, образующую связь между Q и А. 2 з.п. ф-лы, 12 табл., 4 пр.
1. Соединение, содержащее, по меньшей мере, одну группу Q и, по меньшей мере, одну группу А, связанные друг с другом, по меньшей мере и предпочтительно, «спейсерной» группой Sp, в котором:
Q представляет собой нитрилоксидную или нитроновую группу следующих формул (VII) или (VIII):
в которых R1-R4 выбирают независимо из атома водорода, С6 арила и группы формулы (X):
в которой n обозначает 1, 2, 3, 4 или 5, и каждый Y обозначает независимо спейсерную группу Sp или C1-C5 алкил.
- А соответствует формуле (II):
где
- Х обозначает атом кислорода,
- Sp представляет собой C1-C5 алкильную цепь, необязательно прерванную одним или несколькими атомами азота или кислорода, образующую связь между Q и А.
2. Соединение по п.1, отличающееся тем, что способная к ассоциации группа A выбрана из имидазолидинильной группы.
3. Соединение по п.1 или 2, отличающееся тем, что оно выбрано из соединений следующих формул (XIII), (XVII), (XIX)-(XXI):
| WO2010031956 A1, 25.03.2010 | |||
| US20060084730 A1, 20.04.2006 | |||
| US3884285 A, 20.05.1975 | |||
| US7534839 B2, 19.05.2009 | |||
| US20040010090 A1, 15.01.2004 | |||
| ПРИМЕНЕНИЕ АССОЦИАЦИИ СИЛИКОНОВЫХ СОЕДИНЕНИЙ В КАЧЕСТВЕ АГЕНТА СОЧЕТАНИЯ В КРЕМНЕЗЕМСОДЕРЖАЩИХ ЭЛАСТОМЕРНЫХ КОМПОЗИЦИЯХ | 1997 |
|
RU2177011C2 |

