Область техники
Изобретение относится к медицине, а именно биотехнологии, органической химии, молекулярной биологии и биохимии, а именно настоящее изобретение относится к получению высокоспецифичного ингибитора протеазы коронавируса Mpro SARS-CoV-2 в виде синтетического олигопептида, а также применению данного препарата для ингибирования вирусных протеаз.
Уровень техники
В настоящее время в качестве одного из эффективных способов лечения заболевания вызванного SARS-CoV-2 рассматривают низкомолекулярные вещества прямого действия, препятствующие размножению вируса в клетке хозяина. Большой РНК-геном SARS-CoV-2 содержит две открытые рамки считывания и кодирует два полипептида, которые далее процессируются 3С-подобной протеазой (основная протеаза, Mpro или 3CLpro) и папаин-подобной цистеиновой протеазой (PLpro), что приводит к образованию всего набора вирусных белков. Обе протеазы необходимы для жизненного цикла вируса, что делает их привлекательными мишенями для терапевтического воздействия (Cannalire R. et al. J. Med. Chem., 2020; Rut W. et al. Sci. Adv., 2020; Ullrich S. et al. Bioorg. Med. Chem. Lett., 2020). Каталитически активная форма M9™ представляет собой гомодимер с сайтом связывания субстрата в виде протяженного кармана, который обеспечивает связывание 5-ти целевых аминокислотных остатков (Dai W. et al. Science, 2020; Zhang L. et al. Science, 2020), Mpro имеет высокую специфичность по отношению к последовательности -Y-Z-Leu-Gln-X, где X представляет собой небольшой аминокислотный остаток, чаще всего Ser, Ala или Gly, Y является гидрофобным остатком, a Z-аминокислотный остаток, направленный из кармана активного центра в раствор. Каталитическая диада Cys145-His41 в активном центре участвует в гидролизе целевой пептидной связи, что приводит к формированию 11 из 13 отдельных вирусных белков (Gadlage MJ. et al. J. Virol., 2010). Такая субстратная специфичность не свойственна известным протеазам человека, поэтому можно предположить, что потенциальные ингибиторы Mpro за счет высокой селективности не будут обладать побочными эффектами (Zhang L. et al. Science, 2020). Следует отметить, что на сегодняшний день среди наиболее перспективных для использования противовирусных препаратов SARS-CoV-2 известно несколько лекарственных препаратов, относящихся к семейству ингибиторов цистеиновых протеаз. Как правило, в этих соединениях присутствуют реакционноспособные группы, необходимые для образования ковалентной связи между молекулой ингибитора и остатком Cys145 в активном центре фермента.
Среди ингибиторов 3С-подобных протеаз известны соединения боцепревир и телапревир-противовирусные соединения, которые были разработаны как ингибиторы протеазы NS3 гепатита С, и одобрены для безопасного применения у людей (Lang L. Gastroenterology. 2007; Rotella D.P. Expert Opin. DrugDiscov. 2013).
Соединение GC-376 было разработано как ингибитор вирусных протеаз 3С-подобного семейства и первоначально использовалось для лечения инфекционного перитонита кошек в качестве противовирусного соединения против таких вирусов как MERS-CoV, FIPV и норовирус (Kim Y. et al. J. Virol., 2012; Kim Y. et al. PLoS Pathog., 2016; Pedersen N. et al. J. Feline Med. Surg., 2018).
Недостатком данных ингибиторов является их невысокая селективность по отношению к протеазе коронавируса SARS-CoV-2.
Одно из наиболее перспективных соединений-ингибиторов протеазы SARS-CoV-2, PF-00835231, было первоначально разработано в ответ на предыдущую эпидемию коронавируса в 2003 году в качестве ингибитора Mpro SARS-CoV (Hoffman R.L. et al. J. Med. Chem., 2020). Было показано (Boras В. et al. bioRxiv., 2021; de Vries M. et al. J. Virol, 2021), что PF-00835231 обладает противовирусной активностью против SARS-CoV-2 с лучшей на сегодняшний день эффективностью среди всех опубликованных низкомолекулярных соединений (KI ~ 6 нМ).
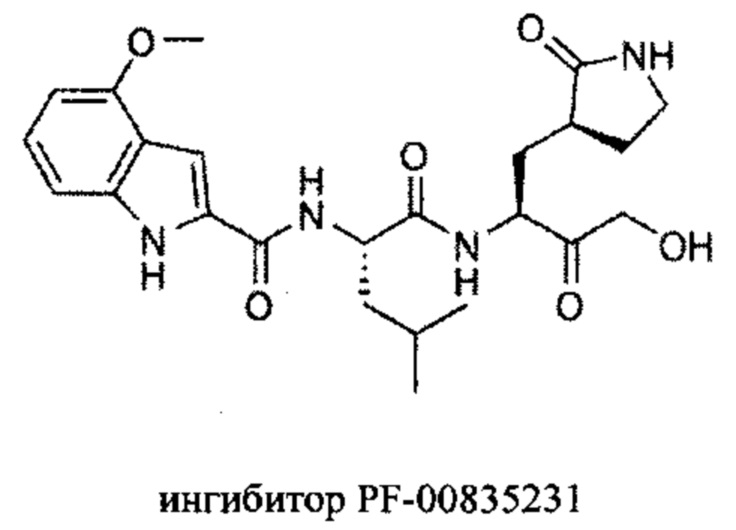
Недостатком соединения PF-00835231 являются неблагоприятные фармакокинетические свойства, такие как биодоступность и растворимость. Поэтому на основе ингибитора PF-00835231 были разработаны модифицированные варианты, обладающие лучшей биодоступностью при введении в организм, например PF-07304814 (de Vries М. et al. J. Virol., 2021).
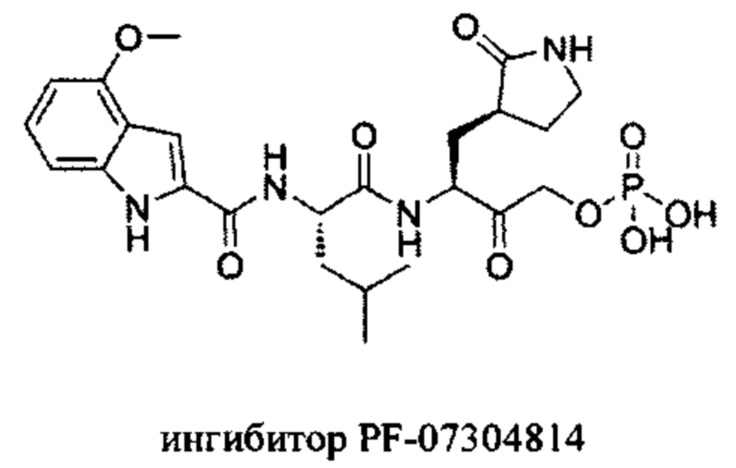
Однако, добавление высокогидрофильной фосфатной группы в составе PF-07304814 приводит к значительному снижению способности всасывания в кишечнике и, как следствие, потере эффективности при пероральном введении (de Vries М. et al. J. Virol., 2021).
Еще одним известным ингибитором протеазы коронавируса SARS-CoV-2 является соединение PF-07321332, входящий в состав препарата «Паксловид».
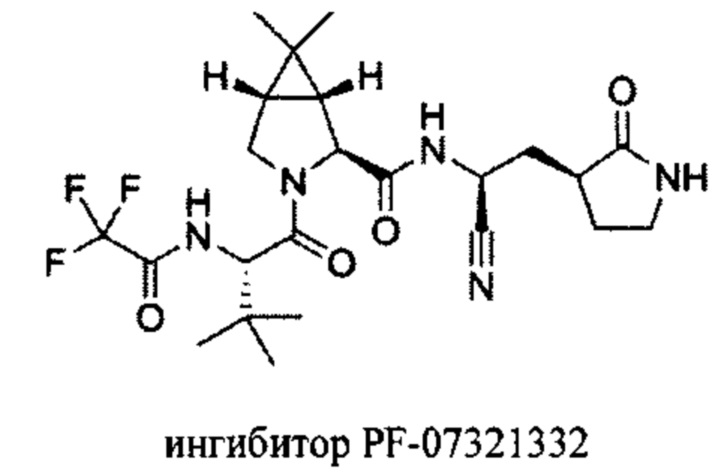
PF-07321332 имеет умеренную растворимость около 1 мг/мл, а результаты клинических испытаний показали, что биодоступность этого соединения при пероральном приеме составляет примерно 50%, что является хорошим показателем для лекарственного средства (Owen, D.R. et al. Science, 2021). Клинические исследования показали, что препарат снижает риск госпитализации больных на 89%.
Недостатком известных ингибиторов протеазы коронавируса SARS-CoV-2 является то, что эти соединения труднодоступны в связи со сложностью синтеза и их последующего выделения из реакционной смеси. Таким образом, в настоящее время существует необходимость в разработке российского высокоспецифичного ингибитора протеазы коронавируса Мрго SARS-CoV-2.
Сущность изобретения
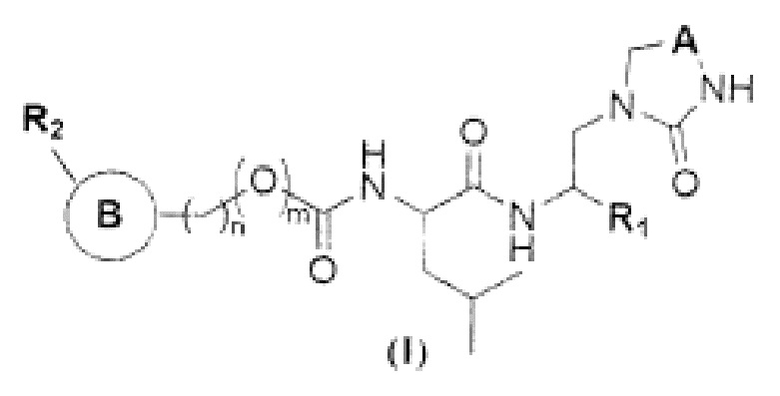
Настоящее изобретение относится к соединению общей формулы (I) в качестве ингибитора основной протеазы коронавируса SARS-CoV2:
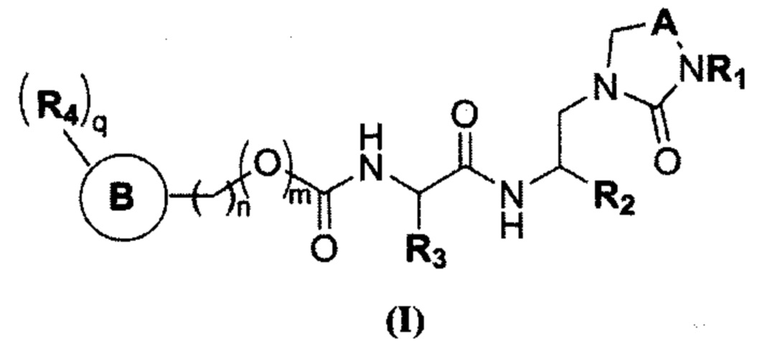
или его фармацевтически приемлемой соли, где:
R1 представляет собой Н или C1-С3 алкил;
R2 представляет собой -СН(SO3H)ОН, -CN или -XYR5, где
X представляет собой -С(О)- или -СНСН-,
Y отсутствует или представляет собой -О-, -С(О)- или -СН2О-,
R5 представляет собой водород, C1-С6 алкил, мезил, алкинил, С3-С10 циклоалкил, -РО3Н2, O-(C1-C6 алкил), незамещенный или C1-С6 алкил или С3-С6 циклоалкил замещенный амин, тиазолил, бензотиазолил, фенил, пиридил, фурил, причем R5 необязательно замещен одним или более гидроксилами или галогенами или O-(C1-C6 алкил), или незамещенной или C1-С6 алкил или С3-С6 циклоалкил замещенным амином, выбранными независимо;
R3 представляет собой C1-С6 алкил, 2-метил-1-пропил, 2-метил-2-пропил, 2,2диметил-1-пропил, С3-С6 циклоалкил, бензил, причем R3 необязательно замещен одним и более гидроксилами или галогенами или O-(C1-C6 алкил), выбранными независимо;
m равно 0 или 1;
n равно 0, 1 или 2;
А представляет собой -СН2- или -С(О)-;
кольцо В или отсутствует, или представляет собой фенил, индолил, 5-членный гетероарил, содержащий 1 или 2 гетероатома, независимо выбранных из N, О и S;
q равно от 1 до 5;
каждый R4-независимо выбраны из водорода, галогена, гидроксила, C1-С6 алкила, O-(C1-C6 алкила), незамещенного или C1-С6 алкил или С3-С6 циклоалкил замещенного амина, фенила, пиридила, фурила, причем R4 необязательно замещен одним или более гидроксилами или галогенами или O-(C1-C6 алкил), или незамещенной или C1-С6 алкил или С3-С6 циклоалкил замещенным амином, выбранными независимо.
Другой аспект изобретения относится к применению описываемого соединения для специфического ингибирования основной протеазы коронавируса Mpro SARS-CoV2.
Краткое описание чертежей
На фиг.1 представлена схема рационального поиска новых ингибиторов основной протеазы коронавируса на основании структуры активного центра фермента. Отдельные карманы в активном центре фермента обозначены S0-S4, заместители в составе соединения общей формулой (I), которые обеспечивают необходимые специфические взаимодействия с ферментом в данных карманах обозначены R и R1-R4.
На фиг.2 представлена общая схема разработки синтеза целевых соединений общей формулы (I), состоящей из четырех этапов-разработка синтеза неприродных аминоксилотных мономеров; синтез Вос-(третбутоксикарбонил-) и Z- (бензилоксикарбонил-) защищенных дипептидов; сборка трипептидных фрагментов; модификация активной группы в С-концевой аминокислоте и получение ковалентных ингибиторов.
На фиг.3 представлены кинетические флуоресцентные кривые, характеризующие процесс гидролиза FRET-субстрата (30 мкМ) в присутствии Mpro (150 нМ) и различных концентраций ингибитора (от 0 до 20 мкМ).
На фиг.4 представлена зависимость начальной скорости гидролиза субстрата (15, 20, 30, 35 мкМ) от концентрации ингибитора при постоянной концентрации фермента (150 нМ).
Подробное описание изобретения
Задачей настоящего изобретения является разработка нового ингибитора протеазы коронавируса Mpro SARS-CoV-2 в виде глубоко модифицированного олигопептида, а также установление возможности его применения в качестве ингибитора протеазы коронавируса Mpro SARS-CoV-2.
Технический результат изобретения заключается в создании эффективного противовирусного средства, активного в качестве высокоспецифичного ингибитора протеазы коронавируса Mpro SARS-CoV-2. Указанный технический результат достигается тем, что создано соединение вышеуказанной общей формулы (I), которое пригодно в качестве ингибитора основной протеазы коронавируса SARS-CoV-2. На основании структуры активного центра основной протеазы Mpro коронавируса SARS-CoV-2 проведен молекулярный дизайн соединения, представленного общей формулой (I). Для этого карман активного центра был параметризован и определены структурные полости, которые отвечают за образование потенциальных контактов с молекулой-ингибитором (фиг.1). Поскольку у каждой части молекулы-ингибитора должно быть структурное соответствие с конкретной полостью фермента, то возможно провести дизайн таких соединений, структурные характеристики которых будут соответствовать пространственной топологии кармана активного центра, что в итоге позволит данным молекулам эффективно связываться в активном центре фермента.
На основании анализа структуры активного центра (фиг.1), содержащего различные ингибиторы, можно заключить, что ингибитор протеазы должен включать:
1) жесткий и объемный остов от N-концевого до второго заместителя R;
2) гидрофобный второй заместитель R2;
3) может иметь ароматический или алифатический заместитель R1;
4) может иметь заместитель R3, формирующий дополнительные водородные связи в активном центре;
5) может иметь заместитель R4, обеспечивающий ковалентное присоединение к остатку Cys145 активного центра.
Выбор соединения, представленного общей формулой (I), был основан на максимальном соответствии структуры ингибитора и активного центра фермента, позволяющим образовать наибольшее число межмолекулярных контактов, которые должны обеспечить высокую эффективность связывания и высокую селективность по отношению к вирусной протеазе. Одним из самых важных видов контактов, которые возникают между ингибитором и ферментом, являются водородные связи, увеличивающие сродство ингибитора к ферменту. Согласно данным рентгеноструктурного анализа, наиболее эффективные ингибиторы, в том числе PF-00835231, PF-07321332 и GC-376, образуют с ферментом не менее шести водородных связей: одну-за счет защитной группы на N-конце, одну-за счет остова первого аминокислотного остатка, одну-за счет остова второго аминокислотного остатка, две-за счет боковой цепи второго аминокислотного остатка, одну-за счет защитной группы на С-конце. Совокупность заместителей, входящих в соединение с общей формулой (I), приводит к образованию не менее шести водородных связей с аминокислотными остатками активного центра протеазы Mpro независимо от комбинации заместителей.
Для предсказания ингибирующей способности соединения, представленного общей формулой (I), с помощью компьютерного моделирования методом молекулярной динамики была определена эффективность связывания с ферментом ингибиторов, содержащих двадцать одну комбинацию заместителей (таблица 1). В качестве эталонов для сравнения были выбраны известные ингибиторы протеазы: соединения PF-00835231, PF-07321332 и GC-373 (активная форма GC-376). Для каждого из этих трех соединений в базе данных кристаллических структур PDB имеются структуры комплекса ингибитора с протеазой Mpro коронавируса SARS-CoV-2, которые также были использованы для определения эффективности связывания методом молекулярной динамики.
В качестве начальной структуры для моделирования комплекса протеазы Mpro с соединением, представленным общей формулой (I), на примере двадцать одной комбинации заместителей (таблица 1) использовали кристаллическую структуру комплекс основной протеазы SARS-CoV-2 с ингибитором PF-00835231 (PDB ID: 6ХНМ). Молекулы соединения, представленного общей формулой (I), располагали на месте PF-00835231 в том же положении, что было возможно благодаря структурной схожести этих соединений. После получения начальных комплексов с новым соединением проводили МД-моделирование длительностью не менее 300 не. Моделирование комплексов с известными ингибиторами также длилось не менее 300 не. Далее из полученных молекулярно-динамических траекторий выбирали последние 5 не для вычисления энергии связывания ингибитора с протеазой методом MM/PBSA (molecular mechanics / Poisson-Boltzmann surface area).
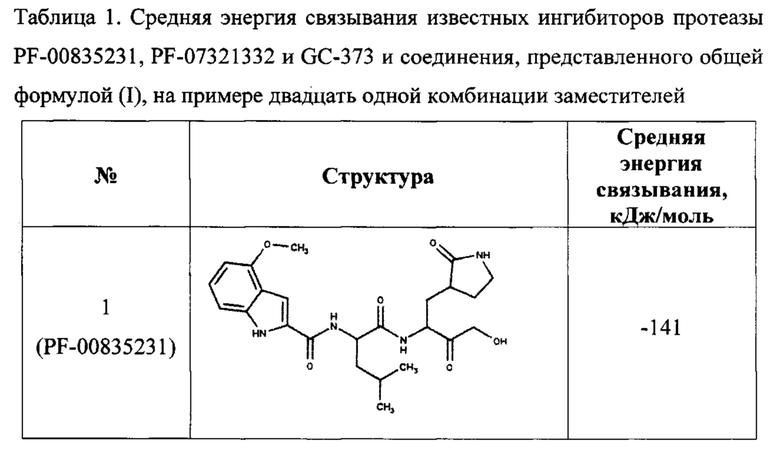
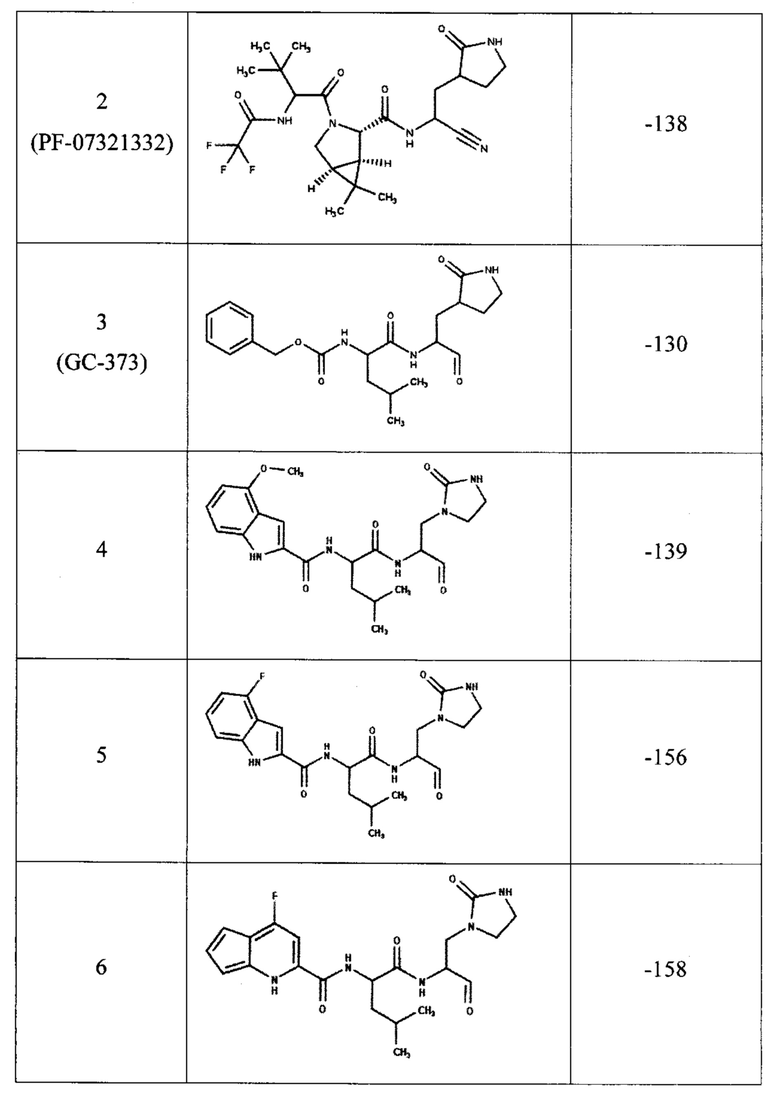
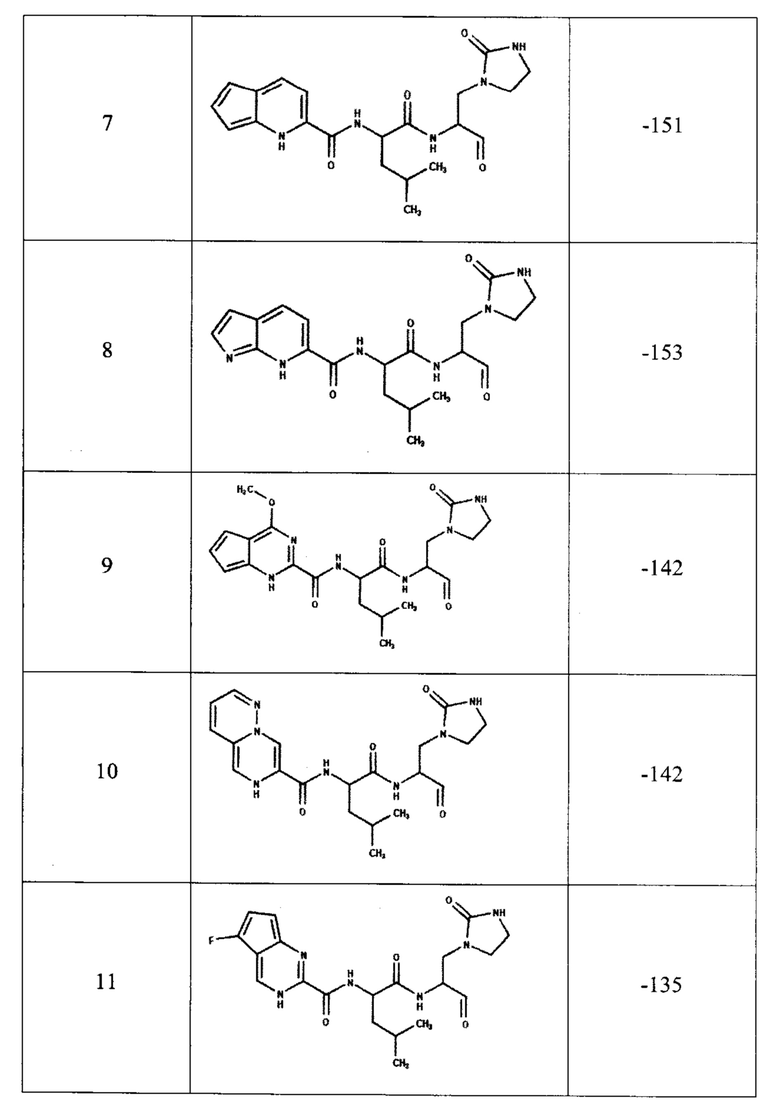
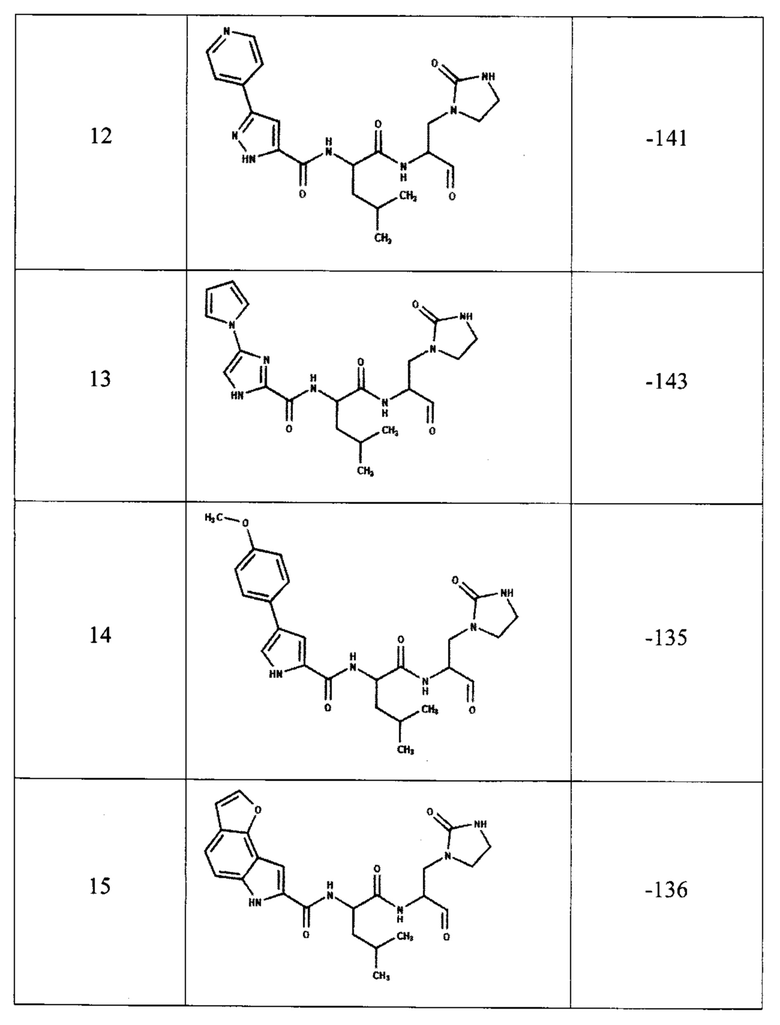
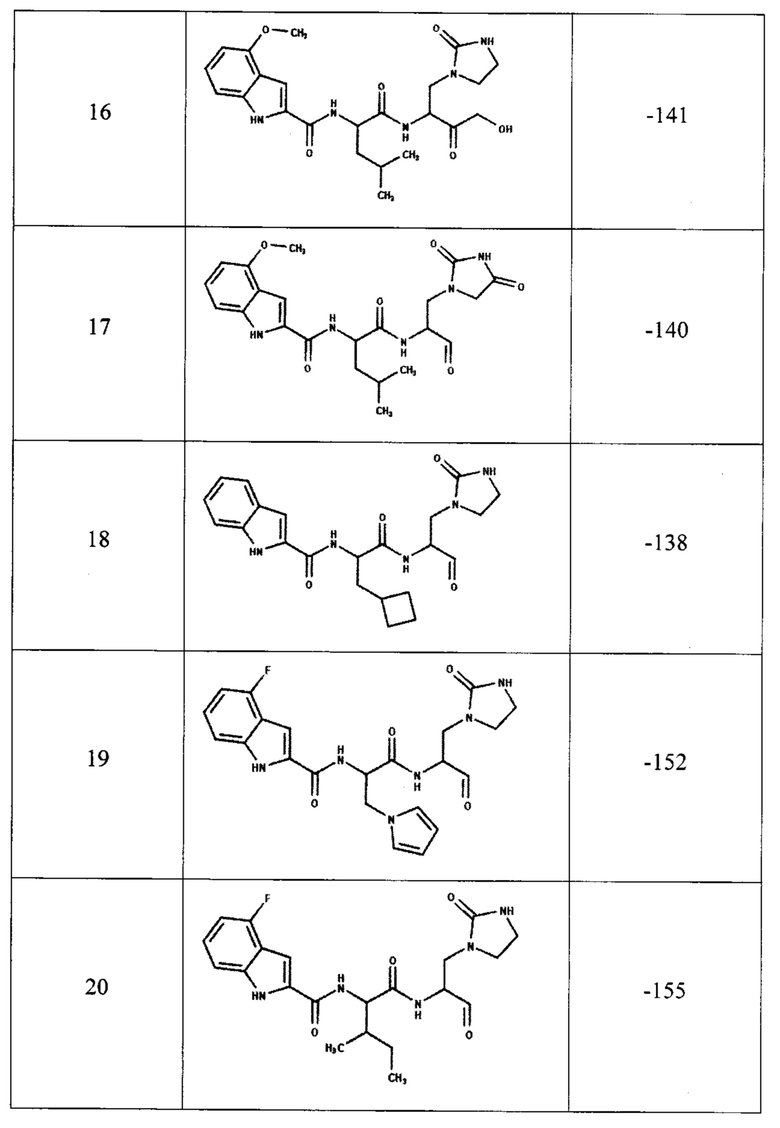
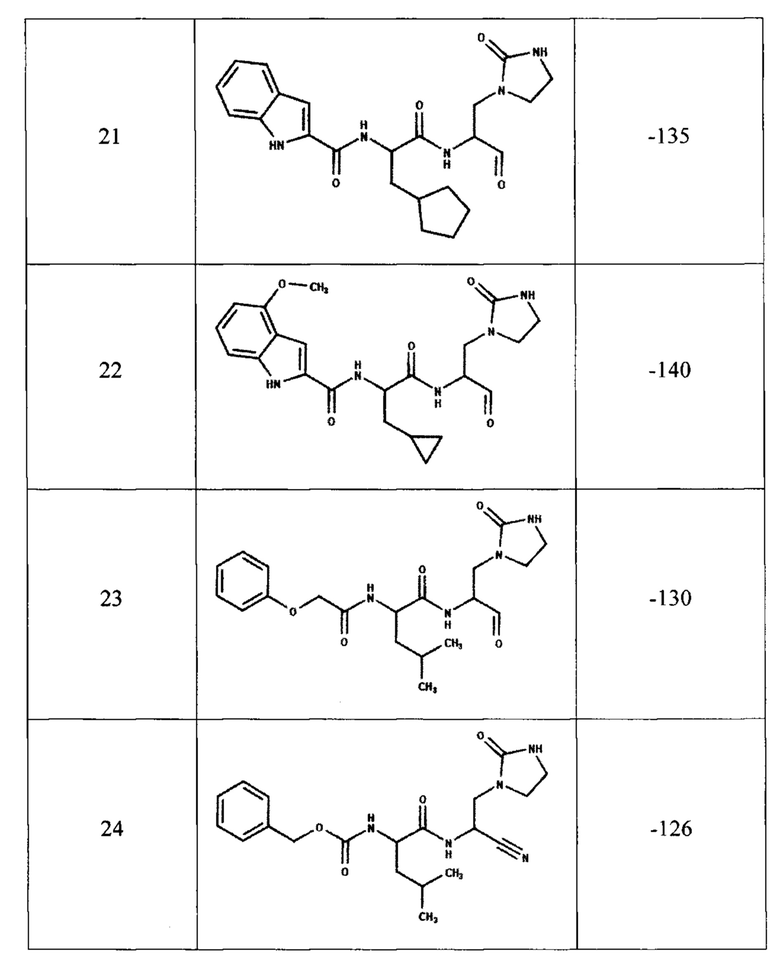
Согласно полученным расчетам, соединения-аналоги PF-00835231 (номера с 4 по 22) имеют энергии связывания, близкие к энергии связывания PF-00835231 в активном центре протеазы, что подтверждает способность соединения, представленного общей формулой (I), эффективно связываться с ферментом. Кроме того, некоторые комбинации заместителей (№5, 6, 7, 8, 19, 20) приводят к значительному уменьшению энергии связывания, что свидетельствует о повышении их сродства к активному центру протеазы. При этом, соединения-аналоги GC-373 (№23, 24) также имеют энергии связывания, сравнимые с ингибитором GC-373. Следует отметить, что результаты МД-моделирования, полученные на примере 21 комбинации заместителей соединения, представленного общей формулой (I), подтвердили образование как минимум 6 водородных связей между ингибитором и активным центром протеазы Mpro.
Предлагаемые соединения общей формулы (I) получают из Nα-(трет-бутоксикарбонил)-аспарагина или N-карбоксибензил-аспарагина, а также N-карбоксибензил-лейцина в 8-15 стадий методами пептидного синтеза с применением перегруппировки Гофмана, ацилирования, нуклеофильного замещения, восстановительного аминирования, этерификации, омыления, восстановления боргидридом натрия, окисления по Сверну или аналогичным методикам, гидрирования на палладии и др. Целевое соединение общей формулы (I) состоит из трех принципиальных фрагментов, соединенных пептидными связями (фиг.2): I с синим ароматическим Ar, II лейцин (консервативный фрагмент) и крайний справа-III необычная аминокислота с гетероциклическим заместителем (зеленый Het).
ЭКПЕРИМЕНТАЛЬНАЯ ЧАСТЬ.
Получение основных промежуточных соединений.
Получение трет-бутил 3-амино-2-((трет-бутоксикарбонил)амино)пропаноата. (Вос-Вар-CtBu).
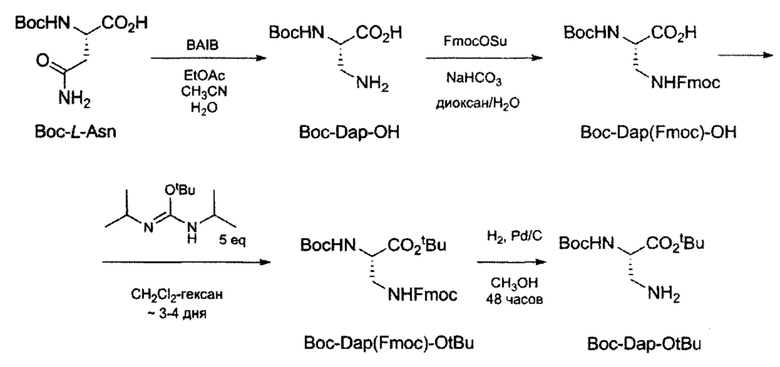
Схема 1. Синтез Boc-Dap-OtBu (трет-бутил 3-амино-2-((трет-бутоксикарбонил)амино)пропаноата) из (трет-бутоксикарбонил)-L-аспарагина (Boc-L-Asn).
Стадия 1: Синтез 3-амино-2(((трет-бутоксикарбонил))амино)пропионовой кислоты (Boc-Dap-OH).
(Трет-бутоксикарбонил)-L-аспарагин (2,015 г, 8,685 ммоль, 1 экв) суспендировали смесью EtOAc (15 мл) CH3CN (15 мл) и H2O (12 мл), а затем охлаждали на ледяной бане до 0°С в течение 15 мин. К полученной белой суспензии при перемешивании и 0°С медленно присыпали (диацетоксийодо)бензол (3,352 г, 10,409 ммоль, 1,2 экв). Реакционную смесь перемешивали 30 мин на ледяной бане, а затем 24 ч при комнатной температуре. Белую суспензию 3 ч выдерживали в холодильнике, а затем отделяли белый осадок на фильтре Шотта. Полученный осадок промывали хол. Et2O (40 мл), а затем сушили на ротационном испарителе. В результате получали продукт Boc-Dap-OH (1,389 г, выход 79%);
Rf=0.51 (EtOAc-MeOH 1:1);
1Н ЯМР (300 МГц, DMSO-d6) δ 6.17 (д, J=5.6 Гц, 1Н), 3.62-3.56 (м, 1Н), 3.00 (дд, J=11.8, 4.9 Гц, 1Н), 2,72 (дд, J=11.8,1.9 Гц, 1H), 1.38 (с, 9Н).
Стадия 2: Синтез 3-((((9Н-флуорен-9-ил)окси)карбонил)амино)-2-((трет-бутоксикарбонил)амино)пропановой кислоты (Boc-Dap(Fmoc)-OH).
Смесь 3 -амино-2-((трет-бутоксикарбонил)амино)пропионовой кислоты (Boc-Dap-OH) (3,138 г, 15,365 ммоль, 1 экв.), NaHCO3 (2,589 г, 30,730 ммоль, 2 экв.) и FmocOSu (6,215 18,438 ммоль, 1,2 экв.) суспендировали в смеси диоксана (70 мл) и НгО (70 мл). Полученную белую суспензию перемешивали 2 ч при комнатной температуре до исчезновения пятна исходного соединения (ТСХ-контроль в системе EtOAc:СН3ОН (1:1)). Затем реакционную смесь промывали Et20 (20 мл). Водную фазу подкисляли водн. 10% лимонной кислоты до рН=3, при подкислении выпадал осадок. Полученный водный раствор с осадком экстрагировали CH2Cl2 (3×70 мл). Органическую фазу промывали Н2О (20 мл), затем сушили над безв. Na2SO4 и упаривали на ротационном испарителе. Из полученного бесцветного масла методом перекристаллизации из смеси CH2Cl2-гексан (1:1) выделяли белый осадок, который затем отфильтровывали, промывали хол. гексаном, сушили на ротационном испарителе. В результате получали продукт Вос-Dap(Fmoc)-OH: бел. крист.(6,356 г, выход 97%);
Rf=0,71 (EtOAc-МеОН 1:1);
1Н ЯМР (300 МГц, CDCl3) δ 7.76 (д, J=7.4 Гц, 2Н), 7.57 (д, J=7.4 Гц, 2Н), 7.40 (т, J=7.4 Гц, 2Н), 7.31 (т, J=7.4 Гц, 2Н), 5.85 (с, 1Н), 5.46 (с, 1Н), 4.41 (д, J=7.0 Гц, 2Н), 4.34-4.26 (м, 1Н), 4.20 (т, J=6.8 Гц, 1H), 3.69-3.53 (м, 1Н), 1.45 (с, 9Н);
13С ЯМР (75 МГц, CDCl3) δ 173.39, 156.44, 143.66, 141.26, 127.74, 127.09, 125.07, 119.99, 80.97, 67.27, 67.05, 54.53, 47.01, 28.28.
Стадия 3: Синтез трет-бутил 3-((((9Н-флуорен-9-ил)окси)карбонил)амино)-2-((трет-бутокси-карбонил)амино)пропионата (Boc-Dap(Fmoc)-OtBu). 3-((((9Н-флуорен-9-ил)окси)карбонил)амино)-2-((трет-бутоксикарбонил)амино)пропановую кислоту (Boc-Dap(Fmoc)-OH) (0,497 г, 1,167 ммоль, 1 экв.) суспендировали в смеси сух. CH2Cl2-гексан (5:12) (25,5 мл). При перемешивании на ледяной бане к полученной белой суспензии прикапывали трет-бутил-N,N'-диизопропилкарбамидат (1,662 мл, 5,833 ммоль 5 экв.), получали прозрачный раствор, который 15 мин выдерживали при перемешивании на ледяной бане, а затем-4 суток при комнатной температуре, из прозрачного раствора выпадал белый осадок (ТСХ контроль этилацетат-гексан 1:1). К реакционной смеси добавляли гексан (25 мл) и выдерживали ее в холодильнике 1 ч. Осадок отделяли фильтрованием через целит, промывая хол. гексаном (20 мл). Супернатант упаривали на ротационном испарителе до бесцветного масла, из которого методом колоночной хроматографии (EtOAc-гексан 1:9 → 3:7) выделяли продукт Boc-Dap(Fmoc)-OtBu (0,375 г, выход 67%), бел. стеклующаяся пена;
Rf=0,75 (EtOAc-гексан 1:1);
1Н ЯМР (300 МГц, CDCl3) δ 7.76 (д, J=7.4 Гц, 2Н), 7.58 (д, J=7.4 Гц, 2Н), 7.40 (т, J=7.4 Гц, 2Н), 7.31 (т, J=7.4, Гц, 2Н), 5.40 (с, 1H), 5.25 (с, 1Н), 4.37 (д, J=6.6 Гц, 2Н), 4.32-4.25 (м, 1H), 4.20 (т, J=7.0 Гц, 1H), 3.64-3.54 (м, 2Н), 1.46 (с, 9Н), 1.45 (с, 9Н);
13С ЯМР (75 МГц, CDCl3) δ 169.53, 156.52, 143.87, 143.82, 141.27, 127.70, 127.06, 125.08, 119.97, 82.91, 80.18, 66.98, 54.43, 47.13, 43.43, 28.30, 27.92; ЖХ-МС (ESI) m/z: вычислено для C27H35N2O6+ ([М+Н]+) 483.2, найдено 483.2.
Стадия 4: Синтез трет-бутил 3-амино-2-((трет-бутоксикарбонил)амино)пропаноата (Boc-Dap-OtBu).
К раствору 3-((((9Н-флуорен-9-ил)окси)карбонил)амино)-2-((трет-бутокси-карбонил)амино)пропионата (Boc-Dap(Fmoc)OtBu) (1,176 г, 2,437 ммоль) в 5 мл СН3ОН добавляли 10 мас. % Pd/C (101 мг, 3,5 моль %). Воздух удаляли с помощью водоструйного насоса до закипания раствора. Затем реакционную смесь перемешивали в атмосфере Н2 (1 атм) 2 суток. (ТСХ-контроль этилацетат:гексан 1:1). Pd/C удаляли центрифугированием, полученный супернатант упаривали на ротационном испарителе до бесцветного масла, из которого методом колоночной хроматографии (EtOAc-гексан 1:1 → CH2Cl2-МеОН 9:1) выделяли продукт Boc-Dap-OtBu: бесцветное масло (551 мг, выход 87%);
Rf=0,17 (CH2Cl2-МеОН 9:1);
1Н ЯМР (300 МГц, CDCl3) δ 5.32 (д, J=6.0 Гц, 1H), 4.30-4.01 (м, 1Н), 3.09 -2.91 (м, 2Н), 1.47 (с, 9Н), 1.44 (с, 9Н), 1.40 (с, 1Н);
ЖХ-МС (ESI) m/z: вычислено для C12H25N2O4+ ([М+Н]+) 261.17, найдено 261.18.
Получение метил 3-амино-2-(((бензилокси)карбонил)амино)пропаноат гидрохлорида (Cbz-Dap-OMe).
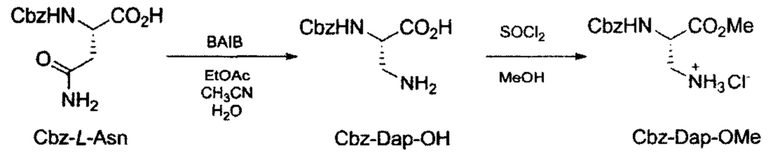
Схема 2. Синтез Cbz-Dap-OMe (метил 3-амино-2-(((бензилокси)карбонил)амино)пропаноат гидрохлорида) из N-карбоксибензил-L-аспарагина (Cbz-Z-Asn).
Стадия 1: Синтез 3-амино-2-(((бензилокси)карбонил)амино)пропановой кислоты (Cbz-Dap-OH).
Навеску N-карбоксибензил-L-аспарагина (2.0 г, 7.5 ммоль, 1 экв.) растворяли в смеси EtOAc (20 мл), MeCN (20 мл) и воды (10 мл), полученный раствор охлаждали до 0°С, после чего в смесь добавляли PhI(ОАс)2 (2.66 г, 8.3 ммоль, 1.1 экв.) и оставляли перемешиваться на ночь при комнатной температуре. Наутро наблюдали обильное выпадение осадка. Реакционную смесь выдерживали в холодильнике в течение 4 часов, после чего осадок отфильтровывали и сушили в вакууме до полного исчезновения запаха йодбензола, получая продукт Cbz-Dap-OH (1.64 г, выход 92%), бел. хлопьевидн. крист. Продукт использовали в следующей стадии без доп.очистки.
Стадия 2: Синтез метил 3-амино-2-(((бензилокси)карбонил)амино)пропаноат гидрохлорида (Cbz-Dap-OMe). 3-Амино-2-(((бензилокси)карбонил)амино)пропановую кислоту (396 мг, 1.66 ммоль, 1 экв.) суспензировали в метаноле (7.5 мл) и охлаждали на ледяной бане, после чего по каплям добавляли SOCl2 (133 мкл, 1.83 ммоль, 1.1 экв.) и оставляли при перемешивании при комнатной температуре. Спустя сутки реакционную смесь упаривали и сухой остаток промывали небольшим объемом Et2O, после высушивания получали продукт Cbz-Dap-ОМе (406 мг, выход 85%), бел. крист.;
1Н ЯМР (300 МГц, DMSO-d6) δ 8.38 (с, 3Н), 7.95 (д, J=8.17 Гц, 1Н), 7.43 -7.26 (м, 5Н), 5.06 (с, 2Н), 4.46 (м, 1Н), 3.67 (с, 3Н), 3.21 (м, 1Н), 3.07 (м, 1H); 13С ЯМР (75 MHz, DMSO-d6) δ 170.3, 156.5, 137.1, 128.8, 128.3, 128.2, 66.3, 53.0, 52.2.
Получение метил 2-амино-3-(2,4-диоксоимидазолидин-1-ил) пропионата (II).
Схема 3. Синтез метил 2-амино-3-(2,4-диоксоимидазолидин-1-ил) пропионата (II) из Boc-Dap-OtBu (трет-бутил 3-амино-2-((трет-бутоксикарбонил)амино)пропаноата).
Стадия 1: Синтез трет-бутил 2-((трет-бутоксикарбонил)амино)-3-((2-метокси-2-оксоэтил)амино)пропаноата (P1).
К раствору соединения трет-бутил 3-амино-2-((трет-бутоксикарбонил)амино)пропаноата (Boc-Dap-OtBu) (0,19 г, 1,904 ммоль) в 4 мл безводного ДМФА в атмосфере аргона прибавляли гидрокабонат натрия (2,40 г, 28,56 ммоль) и иодид калия (0,35 г, 2,09 ммоль). К суспензии при интенсивном перемешивании добавляли раствор метилбромоацетата (0,15 г, 0,95 ммоль) в 1 мл безводного ДМФА. Реакционную массу выдерживали при перемешивании в течение суток в атмосфере аргона при комнатной температуре. Далее ДМФА упаривали при пониженном давлении, к сухому остатку добавляли 5 мл ТГФ, полученную суспензию отфильтровывали. Из фильтрата методом колоночной хроматографии (SiO2, EtOAc-Hexane, 1:1, Rf=0,3 5) выделяли продукт Р1 (0,26 г, выход 81%) в виде бесцветного масла;
1Н ЯМР (300 МГц, CDCl3) δ 5.39 (д, J=7.8 Гц, 1Н), 4.22 (м, 1Н), 3.72 (с, 3Н), 3.49-3.31 (м, 2Н), 3.08-2.85 (м, 2Н), 1.47 (с, 9Н), 1.44 (с, 9Н).
Стадия 2: Синтез трет-бутил 3-((2-амино-2-оксоэтил)(этоксикарбонил)амино)-2-((трет-бутоксикарбонил)амино)пропаноата (Р2).
К раствору трет-бутил 2-((трет-бутоксикарбонил)амино)-3-((2-метокси-2-оксоэтил)амино)пропаноата (0,32 г, 0,98 ммоль) в 20 мл ТГФ добавляли этилхлорформиат (0,21 г, 1,95 ммоль) и триэтиламин (0,11 г, 1,07 ммоль), после чего наблюдали быстрое выпадение мелкого осадка. Реакционную массу перемешивали при комнатной температуре 20 минут, после чего растворитель отгоняли в вакууме, остаток перерастворяли в метанольном растворе аммиака (20 мл). Реакционную смесь выдерживали при перемешивании при комнатной температуре в течение 48 ч, после чего упаривали, из остатка методом колоночной хроматографии (SiO2, EtOAc, Rf=0,4) выделяли продукт Р2 (0,24 г, выход 64%);
1Н ЯМР (300 МГц, CDCl2) δ 6.81 (уш. с, 1Н), 6.34 (с, 0.5Н), 5.41 (с, 1H), 4.37 (с, 1Н), 4.12 (кв, J=7.1 Гц, 2Н), 3.94 (с, 2Н), 3.65 (с, 2Н), 1.45 (д, J=11.7 Гц, 18Н), 1.25 (т, J=7.1 Гц, 3Н).
Стадия 3: Синтез 2-((трет-бутоксикарбонил)амино)-3-(2,4-диоксоимидазолидин-1-ил)пропионовой кислоты (Р3).
К раствору трет-бутил 3-((2-амино-2-оксоэтил)(этоксикарбонил)амино)-2-((трет-бутоксикарбонил)амино)пропаноата (0,20 г, 0,52 ммоль) в 9,5 мл этанола добавляли 2,1 мл 2 N КОН. Реакционную смесь выдерживали при перемешивании при 45°С 3 часа, после чего ночь при комнатной температуре. На следующий день рН раствора доводили до 6,5 ледяной уксусной кислотой, после чего растворители упаривали, из сухого остатка методом колоночной хроматографии (SiO2, ацетон-вода, 95:5, Rf=0,3) выделяли продукт Р3 (0,12 г, выход 77%) в виде белых кристаллов; 1Н ЯМР(300МГц, DMSO-d6) δ 10.62 (уш. с, 1Н), 6.18 (уш. с, 1Н), 4.12-3.76 (м, 3Н), 3.56 (дд, J=13.8, 4.1 Гц, 1Н), 3.44-3.30 (м, 1H), 1.35 (с, 9Н).
Стадия 4: Синтез метил 2-амино-3-(2,4-диоксоимидазолидин-1-ил)пропаноата (II).
К раствору 2-((трет-бутоксикарбонил)амино)-3-(2,4-диоксоимидазолидин-1-ил)пропионовой кислоты (0.17 ммоль) в метаноле добавляли 10% раствор тионилхлорида в метаноле (10 экв.). Реакционную смесь выдерживали при перемешивании при нагревании до 50°С 2 часа, затем разбавляли толуолом, упаривали под вакуумом, повторяли два раза. Полученный продукт II (0.17 ммоль, количеств.) использовали далее без дополнительной очистки;
1Н ЯМР (700 МГц, MeOD) δ 4.40-4.35 (м, 1H), 4.08 (д, J=17.1 Гц, 1Н), 3.98-3.92 (м, 2Н), 3.89 (с, 3Н), 3.88-3.84 (м, 1H);
13С ЯМР (176 МГц, MeOD) δ 173.17,169.07,160.20, 54.12, 53.22, 53.21,49.85, 43.97.
Получение метил 2-амино-3-(2-оксоимидазолидин-1-ил)пропаноата (I2).
Схема 3. Синтез 2-амино-3-(2-оксоимидазолидин-1-ил)пропаноата (I2) из ди-трет-бутил-карбамата и Cbz-Dap-OMe (метил 3-амино-2-(((бензилокси)карбонил)амино)пропаноат гидрохлорида).
Стадия 1. Синтез трет-бутил (2-гидроксиэтил)карбамата (Р5). Навеску дя-трет-бутия дикарбоната (14.28 г, 66 ммоль) растворяли в 5 мл свежеперегнанного ДХМ, после чего смесь охлаждали на льду. К смеси при перемешивании в течение одной минуты прикапывали предварительно высушенный в вакууме этаноламин (4.0 г, 66 ммоль), после чего смесь снимали со льда и оставляли при перемешивании. Смесь мутнела, а через несколько минут снова становилась прозрачной. Через 1 час из реакционной массы отгоняли дихлорметан, из полученного вязкого прозрачного масла методом флэш- хроматографии (гексан : изопропанол = 4:1) выделяли продукт Р5 (10.28 г, выход 97%): бцв. масло;
1Н ЯМР (300 МГц, CDCl3) δ 4.97 (с, 1Н), 3.79-3.63 (м, 2Н), 3.38-3.18 (м, 2Н), 2.49 (с, 1Н), 1.46 (с, 9Н).
Стадия 2. Синтез трет-бутил (2-оксоэтил)карбамата (Р6). К смеси диметилсульфоксида (1.91 мл, 27.0 ммоль, 1.4 экв.) и 20 мл сухого дихлорметана при -78°С прикапывали оксалил хлорид (1.98 мл, 23.1 ммоль, 1.2 экв.) в течение 5 минут. Затем реакционную смесь перемешивали 10 минут при -78°С, после чего прикапывали раствор трет-бутил (2-гидроксиэтил)карбамата (Р5) (3.10 г, 19 ммоль, 1.0 экв.) в 10 мл сухого ДХМ в течение 5 минут при постоянном перемешивании. Через 20 минут к смеси прикапывали триэтиламин (8.02 мл, 57.8 ммоль, 3 экв.) в течение 5 минут, после чего смесь отогревали до 0°С в течение 15 минут, затем разбавляли 50 мл ДХМ и 50 мл смеси насыщенного раствора NH4Cl и льда в отношении 1:1. Органическую фазу сушили над безводным Na2SO4 и упаривали при пониженном давлении при температуре 15-20°С. Полученное желтое масло подвергали хроматографии, аналогичной описанной в методике 1, получая продукт трет-бутил (2-оксоэтил)карбамат (Р6) (2.10 г, выход 70%), бцв. масло, хранится при -80°С. В следующую стадию продукт вводили без дополнительной очистки.
Стадия 3. Синтез метил 2-(((бензилокси)карбонил)амино)-3-((трет-бутоксикарбонил)(2-((трет-бутоксикарбонил)амино)этил)амино)пропаноата (Р7).
В реакционную смесь, содержащую неочищенный альдегид Р6 (3.89 ммоль), добавляли метил 3-амино-2-(((бензилокси)карбонил)амино)пропаноат гидрохлорид (Cbz-Dap-OMe) (0.800 г, 2.78 ммоль, 1 экв.) и NaBH(OAc)3 (1.178 г, 5.56 ммоль, 2 экв.), после чего оставляли при перемешивании на ночь. Затем к реакционной смеси прибавляли 4М р-р K2CO3 в объеме, равным объему реакционной смеси. Органическую фазу отделяли, упаривали и прибавляли ди-трет-бутил дикарбонат (3 экв.) в 15 мл ДХМ, после чего оставляли на сутки. Затем реакционную смесь упаривали и подвергали прямофазной градиентной хроматографии, градиент с последовательной элюцией 1 CV 20%, 1 CV 30%, 4 CV 40% этилацетат в гексане (ТСХ контроль фракций этилацетат-гексан 2:3, Rf=0.30, нингидрин), получали Р7 (0.565 г, выход 41%);
1Н ЯМР (300 МГц, CDCl3) δ 7.33-7.27 (м, 5Н), 6.04-5.41 (м, 1Н), 5.07 (с, 2Н), 4.98-4.45 (м, 2Н), 3.71 (с, 3Н), 3.69-3.42 (м, 1H), 3.34-3.12 (м, 5Н), 1.48-1.38 (м, 18Н);
13С ЯМР (75 МГц, CDCl3) δ 171.0,156.1,155.9, 136.3,128.5,128.1, 128.0, 80.8, 79.3, 77.5, 77.1, 76.7, 66.9, 53.9, 52.6, 48.9, 47.9, 39.2, 28.4, 28.2.
Стадия 4. Синтез метил 2-(((бензилокси)карбонил)амино)-3-(2-оксоимидазолидин-1-ил)пропаноата (Р8).
Метил 2-(((бензилокси)карбонил)амино)-3-((трет-бутоксикарбонил)(2-((трет-бутоксикарбонил)амино)этил)амино)пропаноат (Р7) (0.784 г, 1.58 ммоль, 1 экв) растворяли в 20% растворе трифторуксусной кислоты в сухом ДХМ (20 мл), после чего оставляли при перемешивании в течение 2 часов. Окончание удаления защит отслеживали при помощи ЖХ-МС. По окончании к смеси приливали толуол (20 мл) и упаривали при пониженном давлении и температуре 40°С. Процедуру повторяли еще два раза, после чего полученное желтое масло растворяли в сухом ДХМ (50 мл) и добавляли триэтиламин (1.4 мл, 6 экв) и карбонилдиимидазол (256 мг, 1.58 ммоль, 1 экв). Смесь оставляли на 5 часов при перемешивании при комнатной температуре, после чего упаривали досуха при пониженном давлении. Из полученного вязкого остатка методом колоночной хроматографии (CHCl3 -метанол 97.5:2.5, Rf=0.27) выделяли продукт Р8 (361 мг, выход 70%), бцв. масло;
1Н ЯМР (300 МГц, CDCl3) δ 7.44-7.28 (м, 5Н), 6.04 (д, J=7.8 Гц, 1Н), 5.10 (с, 2Н), 4.82 (с, 1Н), 4.55-4.41 (м, 1Н), 3.75 (с, 3Н), 3.69-3.24 (м, 6Н).
Стадия 5. Синтез метил 2-амино-3-(2-оксоимидазолидин-1-ил)пропаноата (I2).
К раствору соединения метил 2-(((бензилокси)карбонил)амино)-3-(2-оксоимидазолидин-1-ил)пропаноата (Р8) (272 мг, 0.85 ммоль) в метаноле (5 мл) добавляли палладий на активированном угле (45 мг, 5 мол. %) и затем выдерживали при перемешивании в атмосфере водорода в течение 4 часов (ТСХ контроль хлороформ-метанол 9:1). Затем реакционную смесь фильтровали через целит, промывая метанолом (5 мл), после чего фильтрат упарили при пониженном давлении. Методом флэш-хроматографии выделяли продукт 12 (156 мг, выход 98%), бел. пена;
1Н ЯМР (300 МГц, CD3OD) δ 4.25 (дд, J=6.0, 4.4 Гц, 1H), 3.88 (с, 3Н), 3.79-3.40 (м, 6Н),3.36 (с, 1Н);
13С ЯМР (75 МГц, CD3OD) 5 168.58, 164.15, 52.42, 52.21, 46.29, 44.62, 37.97.
Получение целевых ди- и трипептидов.
Общие методы:
Метод А. Карбоновую кислоту (1 экв.) и амин (1 экв., если не указано другое) растворяли в безводном диметилформамиде. Реакционную смесь охлаждали до 0°С, добавляли 1.6 экв. HBTU (N,N,N',N'-тетраметил-O-(1H-бензотриазол-1-ил)уроний гексафторфосфат) и 5 экв. DIPEA (диизопропилэтиламин). Реакционную смесь выдерживали при перемешивании при 0°С 1 час, затем 12 часов при -20°С, затем разбавляли водой, экстрагировали в этилацетат, промывали NaClнac. Органический слой высушивали над безводным сульфатом натрия, упаривали под вакуумом. Метод Б. К раствору исходного соединения в дихлорметане (0.5 мл на 10 мг) добавляли TFA (0.5 мл на 10 мг). Через 2 часа реакционную смесь разбавляли толуолом и упаривали, повторяли два раза. Полученный продукт использовали в следующей стадии без дополнительной очистки. Метод В. К раствору исходного соединения в ТГФ (100 мкл растворителя на 1 мг) добавляли 10 экв. гидроксида лития (1М раствор в воде). Перемешивали при комнатной температуре в течение 1 часа. рН раствора доводили до 7 с помощью 1М HCl. Упаривали под вакуумом. Получение 2-(2-(((бензилокси)карбонил)амино)-4-метилпентанамидо)-3-(2-оксоимидазолидин-1-ил)пропановой кислоты.
Стадия 1. Синтез метил 2-(2-(((бензилокси)карбонил)амино)-4-метилпентанамидо)-3-(2-оксоимидазолидин-1-ил)пропаноата. К раствору 2,5-диоксопирролидин-1-ил ((бензилокси)карбонил)лейцината (0.08 ммоль, 1 экв.) в дихлорметане добавляли метил 2-амино-3-(2-оксоимидазолидин-1-ил)пропаноат (0.08 ммоль, 1 экв.) и триэтиламин (0.4 ммоль, 5 экв.). Реакционную смесь выдерживали при перемешивании при комнатной температуре 12 часов, далее упаривали. Из сухого остатка методом колоночной хроматографии (SiO2; хлороформ-этанол от 99/1 до 95/5) получали продукт метил 2-(2-(((бензилокси)карбонил)амино)-4-метилпентанамидо)-3-(2-оксоимидазолидин-1-ил)пропаноат (0.05 ммоль, выход 61%);
1Н ЯМР (300 МГц, ацетон-d6) δ 7.45-7.28(м, 5Н), 5.09 (с, 2Н), 4.64-4.54 (м, 1H), 4.28-4.19 (м, 1H), 3.69 (с, 1H), 3.56-3.41 (м, 4Н), 3.33 (т, J=1.1 Гц, 2Н), 1.81-1.56 (м, 3Н), 0.93 (дд, J=6.5, 4.3 Гц, 6Н).
Стадия 2. Синтез 2-(2-(((бензилокси)карбонил)амино)-4-метилпентанамидо)-3-(2-оксоимидазолидин-1-ил)пропановой кислоты.
Из метил 2-(2-(((бензилокси)карбонил)амино)-4-метилпентанамидо)-3-(2-оксоимидазолидин-1-ил)пропаноата (0.09 ммоль) методом В. Очистка методом ОФХ на приборе PuriFlash. Элюент А: вода, элюент Б-ацетонитрил. Градиент от 10% до 100% элюента Б. Получали продукт 2-(2-(((Бензилокси)карбонил)амино)-4-метилпентанамидо)-3-(2-оксоимидазолидин-1-ил)пропановую кислоту (0.046 ммоль, выход 51%);
1Н ЯМР (700 МГц, MeOD) δ 7.39 (д, J=7.1 Гц, 2Н), 7.36 (д, J=7.3 Гц, 2Н), 7.31 (т, J=7.0 Гц, 1Н),5.14(д, J=15.3 Гц, 1Н), 5.10 (д, J=15.3 Гц, 1Н), 4.52-4.47 (м, 1Н), 4.22-4.17 (м, 1H), 3.63 (кв, J=7.0 Гц, 2Н), 3.59-3.57 (м, 1Н), 3.53-3.46 (м, 1H), 3.35-3.33 (м, 2Н), 1.74-1.68 (м, 1H), 1.65-1.60 (м, 1Н), 1.59-1.54 (м, 1H), 0.97 (д, J=6.5 Гц, 3Н), 0.94 (д, J=6.5 Гц, 3Н);
13С ЯМР (175 МГц, MeOD) δ 157.09, 136.85, 128.09, 127.59, 127.35, 66.33, 56.93, 53.84, 52.86, 45.39, 40.56, 37.83, 24.52, 22.07, 20.36; ЖХ-МС (ESI) m/z: вычислено для C20H29N4O6+ ([М+Н]+) 421.21, найдено 421.31.
Получение 2-(2-(((бензилокси)карбонил)амино)-4-метилпентанамидо)-3-(2,4-ди-оксоимидазолидин-1-ил)пропановой кислоты.
Стадия 1. Синтез метил 2-(2-(((бензилокси)карбонил)амино)-4-метилпентанамидо)-3-(2,4-ди-оксоимидазолидин-1-ил)пропаноата. К раствору 2,5-диоксопирролидин-1-ил ((бензилоксикарбонил)лейцината (0.28 ммоль, 1 экв.) в дихлорметане добавляли метил 2-амино-3-(2,4-диоксоимидазолидин-1-ил)пропаноат (0.28 ммоль, 1 экв.) и триэтиламин (1.4 ммоль, 5 экв.). Реакционную смесь выдерживали при перемешивании при комнатной температуре 12 часов, далее упаривали. Из сухого остатка методом колоночной хроматографии (хлороформ-этанол от 99/1 до 95/5) выделяли продукт метил 2-(2-(((бензилокси)карбонил)амино)-4-метилпентанамидо)-3-(2,4-ди-оксоимидазолидин-1-ил)пропаноат (0.14 ммоль, выход 50%);
1Н ЯМР (300 МГц, ацетон-d6) δ 7.44-7.28 (м, 5Н), 5.08 (с, 2Н), 4.81-4.72 (м, 1Н), 4.32-4.20 (м, 1Н), 4.15-3.95 (м, 3Н), 3.79- 3.57 (м, 4Н), 1.87-1.62 (м, 3Н), 0.97-0.87 (м, 6Н).
Стадия 2. Синтез 2-(2-(((бензилокси)карбонил)амино)-4-метилпентанамидо)-3-(2,4-диоксоимидазолидин-1-ил)пропановой кислоты. Из метил 2-(2-(((бензилокси)карбонил)амино)-4-метилпентанамидо)-3-(2,4-ди-оксоимидазолидин-1-ил)пропаноата (0.033 ммоль) методом В. Очистка методом ОФХ на приборе PuriFlash. Элюент А: вода, элюент Б -ацетонитрил. Градиент от 10% до 100% элюента Б. Получали продукт 2-(2-(((бензилокси)карбонил)амино)-4-метилпентанамидо)-3-(2,4-диоксоимидазолидин-1-ил)пропановую кислоту (0.011 ммоль, выход 33%);
1Н ЯМР (300 МГц, MeOD) δ 7.50-7.15 (м, 5Н), 5.09 (д, J=7.1 Гц, 2Н), 4.66 (с, 1Н), 4.28-3.95 (м, 3Н), 3.75 (с, 3Н), 1.80-1.47 (м, 3Н), 1.07-0.81 (м, 6Н); ЖХ-МС (ESI) m/z: вычислено для C20H27N4O7+ ([М+Н]+) 435.19, найдено 435.22.
Получение билдинг-блока (4-фтор-1Н-индол-2-карбонил)лейцина.
Стадия 1. Синтез трет-бутил (4-фтор-1Н-индол-2-карбонил)лейцината.
Получали из 4-фтор-1Н-индол-2-карбоновой кислоты (0.22 ммоль) и трет-бутил лейцинат гидрохлорида (0.22 ммоль) методом А. Методом колоночной хроматографии (этилацетат-гексан 1:9 -> 3:7) выделяли трет-бутил (4-фтор-1Н-индол-2-карбонил)лейцинат (0.14 ммоль, выход 64%);
1Н ЯМР (300 МГц, CDCl3) δ 9.57 (с, 1Н), 6.85-6.72 (м, 3Н), 4.75 (тд, J=8.4, 5.2 Гц, 1Н), 1.82-1.62 (м, 3Н), 1.50 (с, 9Н), 0.99 (д, J=4.7 Гц, 6Н). Стадия 2. Синтез (4-фтор-1H-индол-2-карбонил)лейцина. Получали из трет-бутил (4-фтор-1H-индол-2-карбонил)лейцината (0.017 ммоль) методом Б. В следующей стадии продукт использовали без дополнительной очистки. Получение 2-(2-(2-(4-фтор-1H-индол-2-карбоксамидо)-4-метилпентанамидо)-3-(2-оксоимидазолидин-1-ил)пропановой кислоты. Стадия 1. Синтез метил 2-(2-(2-(4-фтор-1H-индол-2-карбоксамидо)-4-метилпентанамидо)-3-(2-оксоимидазолидин-1-ил)пропаноата.
Получали путем конденсации (4-фтор-1H-индол-2-карбонил)лейцина (0.017 ммоль) с метил 2-амино-3-(2-оксоимидазолидин-1-ил)пропаноатом (0.017 ммоль) по методу А. Очистка методом ОФХ на приборе PuriFlash. Элюент А: вода, элюент Б-ацетонитрил. Градиент от 10% до 100% элюента Б. Получали продукт метил 2-(2-(2-(4-фтор-1H-индол-2-карбоксамидо)-4-метилпентанамидо)-3-(2-оксоимидазолидин-1-ил)пропаноат (0.009 ммоль, выход 51%);
1Н ЯМР (800 МГц, MeOD) δ 7.30 (д, J=0.9 Гц, 1Н), 7.28 (д, J=8.3 Гц, 1H), 7.24-7.18 (м, 1Н), 6.77 (дд, J=10.5, 7.7 Гц, 1Н), 4.74 (дд, J=8.1, 5.1 Гц, 1Н), 4.72-4.69 (м, 1Н), 3.75 (с, 3Н), 3.64-3.45 (м, 6Н), 1.84-1.71 (м, 3Н), 1.04 (д, J=6.5 Гц, 3Н), 1.01 (д, J=6.3 Гц, 3Н).
Стадия 2. Синтез 2-(2-(2-(4-фтор-1H-индол-2-карбоксамидо)-4-метилпентанамидо)-3 -(2-оксоимидазолидин-1-ил)пропановой кислоты. Получали из метил 2-(2-(2-(4-фтор-1H-индол-2-карбоксамидо)-4-метилпентанамидо)-3-(2-оксоимидазолидин-1-ил)пропаноата (0.009 ммоль) методом С.Очистка ОФ-ВЭЖХ (Transgenomic). Элюент А: вода, элюент Б -ацетонитрил. Градиент от 10% до 100% элюента Б. Получали продукт 2-(2-(2-(4-фтор-1H-индол-2-карбоксамидо)-4-метилпентанамидо)-3-(2-оксоимидазолидин-1-ил)пропановую кислоту (0.002 ммоль, выход 23%); 1Н ЯМР (600 МГц, MeOD) 8 8.47-8.39 (м, 1H), 8.27 (д, J=8.6 Гц, 1Н), 7.30-7.24 (м, 2Н), 7.22-7.15 (м, 1Н), 6.78-6.72 (м, 1Н), 4.80-4.74 (м, 1Н), 4.72 -4.69 (м, 1Н), 3.63-3.49 (м, 4Н), 3.42-3.35 (м, 2Н), 1.84-1.72 (м, 3Н), 1.02 (т, J=5.8 Hz, 3Н), 0.99 (т, J=4.7 Hz, 3Н);
ЖХ-МС (ESI) m/z: вычислено для C21H27FN5O5+ ([М+Н]+) 448.20, найдено 448.33.
Получение 3-(2,4-диоксоимидазолидин-1-ил)-2-(2-(2-(4-фтор-1H-индол-2-карбоксамидо)-4-метилпентанамидо)пропановой кислоты.
Стадия 1. Синтез метил 3-(2,4-диоксоимидазолидин-1-ил)-2-(2-(4-фтор-1H- индол-2-карбоксамидо)-4-метилпентанамидо)пропаноата.
Получали путем конденсации (4-фтор-1H-индол-2-карбонил)лейцина (0.017 ммоль) с метил 2-амино-3-(2,4-диоксоимидазолидин-1-ил)пропаноатом (0.017 ммоль) по методу А. Очистка методом ОФХ на приборе PuriFlash. Элюент А: вода, элюент Б-ацетонитрил. Градиент от 10% до 100% элюента Б. Получали продукт метил 3-(2,4-диоксоимидазолидин-1-ил)-2-(2-(4-фтор-1H-индол-2-карбоксамидо)-4-метилпентанамидо)пропаноат (0.005 ммоль, выход 29%);
1Н ЯМР (700 МГц, MeOD) δ 7.32-7.24 (м, 2Н), 7.19 (ddd, J=11.0, 8.0, 3.9 Гц, 1H), 6.76 (дд, J=10.5, 7.7 Гц, 1Н), 4.70-4.62 (м, 1H), 4.20-3.99 (м, 2Н), 3.84-3.71 (м, 5Н),3.63 (кв, J=7.0 Hz, 1H), 1.86-1.68 (м, 3Н), 1.06-0.98 (м, 6Н).
Стадия 2. Синтез 3-(2,4-диоксоимидазолидин-1-ил)-2-(2-(2-(4-фтор-1H-индол-2-карбоксамидо)-4-метилпентанамидо)пропановой кислоты. Получали из метил 3-(2,4-диоксоимидазолидин-1-ил)-2-(2-(4-фтор-1H-индол-2-карбоксамидо)-4-метилпентанамидо)пропаноата (0.005 ммоль) методом С.Очистка ОФ-ВЭЖХ (Transgenomic). Элюент А: вода, элюент Б -ацетонитрил. Градиент от 10% до 100% элюента Б. Получали продукт 3-(2,4-диоксоимидазолидин-1-ил)-2-(2-(2-(4-фтор-1H-индол-2-карбоксамидо)-4-метилпентанамидо)пропановую кислоту (0.002 ммоль, выход 40%);
1Н ЯМР (600 МГц, MeOD) δ 11.25 (д, J=27.7 Гц, 1Н), 10.62 (д, J=18.1 Гц, 1Н), 8.47-8.41 (м, 1Н), 8.41-8.35 (м, 1Н), 7.30-7.24 (м, 2Н), 7.18 (тд, J=8.1, 5.1 Гц, 1Н), 6.75 (дд, J=10.5, 8.0 Гц, 1Н), 4.85-4.82 (м, 1Н), 4.15-3.98 (м, 3Н), 3.80-3.69 (м, 2Н), 1.84-1.68 (м, 3Н), 1.02 (дд, J=9.0, 6.4 Гц, 3Н), 0.99 (дд, J=6.4, 5.0 Гц, 3Н);
ЖХ-МС (ESI) m/z: вычислено для C21H25FN5O6+ ([М+Н]+) 462.18, найдено 462.22.
Получение билдинг-блока (4-(4-метоксифенил)-1Н-пиррол-2-карбонил)лейцина.
Стадия 1. Синтез трет-бутил (4-(4-метоксифенил)-1Н-пиррол-2-карбонил)лейцината.
Получали из 4-(4-метоксифенил)-1Н-пиррол-2-карбоновой кислоты (0,07 ммоль) и трет-бутил лейцинат гидрохлорида (0,09 ммоль, 1,3 экв.) методом А. Очистка методом ОФХ на приборе PuriFlash. Элюент А: вода, элюент Б -ацетонитрил. Градиент от 10% до 100% элюента Б. Получали продукт трет-бутил (4-(4-метоксифенил)-1H-пиррол-2-карбонил)лейцинат (0.03 ммоль, выход 33%). В следующей стадии продукт использовали без дополнительной очистки.
Стадия 2. Синтез (4-(4-метоксифенил)-1H-пиррол-2-карбонил)лейцина. Получали из трет-бутил (4-(4-метоксифенил)-1H-пиррол-2-карбонил)лейцината (0.028 ммоль) методом В. В следующей стадии продукт использовали без дополнительной очистки.
1Н ЯМР (300 МГц, MeOD) δ 7.47 (д, J=8.7 Hz, 2Н), 7.29-7.15 (м, 3Н), 6.90 (д, J=8.7 Гц, 1Н), 4.71-4.53 (м, 1H), 3.80 (с, 3Н), 1.72 (дт, J=11.9, 8.6 Гц, 3Н), 0.99 (ддд, J=8.8, 6.1, 3.2 Гц, 6Н).
Получение 2-(2-(2-(4-(4-Метоксифенил)-1H-пиррол-2-карбоксамидо)-4-метилпентанамидо)-3-(2-оксоимидазолидин-1-ил)пропановой кислоты. Стадия 1. Синтез метил 2-(2-(4-(4-(4-метоксифенил)-1Н-пиррол-2-карбоксамидо)-4-метил-пентанамидо)-3-(2-оксоимидазолидин-1-ил)пропаноата.
Получали путем конденсации (4-(4-метоксифенил)-1H-пиррол-2-карбонил)лейцина (0.012 ммоль) с метил 2-амино-3-(2-оксоимидазолидин-1-ил)пропаноатом (0.018 ммоль, 1.5 экв.) по методу А. Очистка методом ОФХ на приборе PuriFlash. Элюент А: вода, элюент Б -ацетонитрил. Градиент от 10% до 100% элюента Б. Получали продукт метил 2-(2-(4-(4-(4-метоксифенил)-1H-пиррол-2-карбоксамидо)-4-метил-пентанамидо)-3-(2-оксоимидазолидин-1-ил)пропаноат (0.006 ммоль, выход 50%);
1Н ЯМР (600 МГц, MeOD) δ 7.61 (d, J=8.7 Гц, 2Н), 7.35 (т, J=1.4 Гц, 1Н), 7.32 (дд, J=3.0, 1.7 Гц, 1Н), 7.04 (д, J=8.8 Гц, 2Н), 4.85 (дд, J=7.9, 5.2 Гц, 1Н), 4.82-4.73 (м, 1Н), 3.94 (с, 3Н), 3.87 (д, J=2.2 Гц, 3Н), 3.74-3.57 (м, 6Н), 1.89-1.81 (м, 3Н), 1.18-1.06 (м, 6Н).
Стадия 2. Синтез 2-(2-(2-(4-(4-метоксифенил)-1H-пиррол-2-карбоксамидо)-4-метилпентанамидо)-3-(2-оксоимидазолидин-1-ил)пропановой кислоты. Получали из метил 2-(2-(4-(4-(4-метоксифенил)-1H-пиррол-2-карбоксамидо)-4-метил-пентанамидо)-3-(2-оксоимидазолидин-1-ил)пропаноата (0.006 ммоль) методом С.Очистка ОФ-ВЭЖХ (Transgenomic). Элюент А: вода, элюент Б-ацетонитрил. Градиент от 10% до 100% элюента Б. Получали продукт 2-(2-(2-(4-(4-метоксифенил)-1H-пиррол-2-карбоксамидо)-4-метилпентанамидо)-3 -(2-оксоимидазолидин-1-ил)пропановую кислоту (0.002 ммоль, выход 33%);
1Н ЯМР (700 МГц, MeOD) δ 11.00 (д, У=11.3 Гц, 2Н), 8.20 (дд, 7=13.6, 8.1 Гц, 1Н), 8.00 (дд, J=18.9, 8.2 Гц, 1H), 7.48 (с, 2Н), 7.22-7.16 (м, 2Н), 6.91 (с, 2Н), 4.85-4.72(м, 2Н), 3.81 (с, 3Н), 3.66-3.40 (м, 6Н), 1.78-1.68 (м, 3Н), 1.01 (т, J=5.9 Гц, 3Н), 0.98 (дд, J=6.5, 3.0 Гц, 3Н);
ЖХ-МС (ESI) m/z: вычислено для C24H32N5O6+ ([М+Н]+) 486.24, найдено 486.47.
Получение 3-(2,4-диоксоимидазолидин-1-ил)-2-(2-(2-(4-(4-метоксифенил)-1H-пиррол-2-карбоксамидо)-4-метилпентанамидо)пропановой кислоты. Стадия 1. Синтез метил 3-(2,4-диоксоимидазолидин-1-ил)-2-(2-(4-(4-метоксифенил)-1H-пиррол-2-карбоксамидо)-4-метилпентанамидо)пропаноата.
Получено путем конденсации (4-(4-метоксифенил)-1H-пиррол-2-карбонил)лейцина (0.05 ммоль) с метил 2-амино-3-(2,4-диоксоимидазолидин-1-ил) пропаноатом (0,08 ммоль, 1,5 экв.) по методу А. Аминокислоту, HBTU и DIPEA добавляли в 2 порции с промежутком 2 часа. Очистка методом ОФХ на приборе PuriFlash. Градиент от 10% до 100% элюента Б. Элюент А: вода, элюент Б-ацетонитрил. Получали продукт метил 3-(2,4-диоксоимидазолидин-1-ил)-2-(2-(4-(4-метоксифенил)-1Н-пиррол-2-карбоксамидо)-4-метилпентанамидо)пропаноат (0.018 ммоль, выход 36%);
ЖХ-МС (ESI) m/z: вычислено для C25H32N5O7+ ([М+Н]+) 514.23, найдено 514.55.
Стадия 2. Синтез 3-(2,4-диоксоимидазолидин-1-ил)-2-(2-(2-(4-(4-метоксифенил)-1Н-пиррол-2-карбоксамидо)-4-метилпентанамидо)пропановой кислоты.
Получали из метил 3-(2,4-диоксоимидазолидин-1-ил)-2-(2-(4-(4-метоксифенил)-1Н-пиррол-2-карбоксамидо)-4-метилпентанамидо)пропаноата (0.018 ммоль) методом С.Очистка ОФ-ВЭЖХ (Transgenomic). Элюент А: вода, элюент Б-ацетонитрил. Градиент от 10% до 100% элюента Б. Получали продукт 3-(2,4-диоксоимидазолидин-1-ил)-2-(2-(2-(4-(4-метоксифенил)-1Н-пиррол-2-карбоксамидо)-4-метилпентанамидо)пропановую кислоту (0.005 ммоль, выход 30%); 1Н ЯМР (700 МГц, MeOD) δ 11.04 (с, 1Н), 8.15 (с, 1H), 7.96 (с, 1H), 7.44 (д,7=9.8 Hz, 2Н), 7.21 (д, J=13.6 Гц, 2Н), 6.93-6.82 (м, 2Н), 4.17-3.94 (м, 1H), 3.86-3.69 (м, 5Н), 3.79 (м, 3Н), 1.82-1.59 (м, 3Н), 0.99 (д, J=6.6 Гц, 3Н), 0.94(d, J=6.6 Гц,3Н);
ЖХ-МС (ESI) m/z: вычислено для C24H30N5O7+ ([М+Н]+) 500.21, найдено 500.29.
Получение билдинг-блока (3-(пиридин-4-ил)-1Н-пиразол-5-карбонил)лейцина.
Стадия 1. Синтез трет-бутил (3-(пиридин-4-ил)-1Н-пиразол-5-карбонил)лейцината.
Получали из 3-(пиридин-4-ил)-1Н-пиразол-5-карбоновой кислоты (0.50 ммоль) и трет-бутил лейцинат гидрохлорида (0.50 ммоль) методом А. Методом колоночной хроматографии (хлороформ-этанол 99:1 -> 90:10) выделяли продукт трет-бутил (3-(пиридин-4-ил)-1Н-пиразол-5-карбонил)лейцинат (0.42 ммоль, выход 84%);
1Н ЯМР (300 МГц, ДМСО-d6) δ 8.76-8.55 (м, 2Н), 7.94 (с, 2Н), 7.73 (д, J=3.6 Гц, 1Н), 4.37 (с, 1Н), 1.84-1.51 (м, 3Н), 1.41 (д, 7=1.7 Гц, 9Н), 0.98 -0.84 (м, 6Н).
Стадия 2. Синтез (3-(пиридин-4-ил)-1H-пиразол-5-карбонил)лейцина. Получали из трет-бутил (3-(пиридин-4-ил)-1Н-пиразол-5-карбонил)лейцината (0.27 ммоль) методом В. Полученный продукт (3-(пиридин-4-ил)-1Н-пиразол-5-карбонил)лейцин использовали в следующей стадии без дополнительной очистки.
1Н ЯМР (700 МГц, MeOD) δ 8.68 (с, 2Н), 7.97 (с, 2Н), 7.44 (с, 1H), 4.70 (дд, J=10.3, 3.9 Гц, 1Н), 1.90-1.69 (м, 3Н), 1.02 (дд, J=14.6, 5.3 Гц, 6Н). Получение 2-(4-метил-2-(3-(пиридин-4-ил)- 1H-пиразол-5-карбоксамидо)пентанамидо)-3-(2-оксоимидазолидин-1-ил)пропановой кислоты.
Стадия 1. Синтез метил 2-(4-метил-2-(3-(пиридин-4-ил)-1H-пиразол-5-карбоксамидо)пентанамидо)-3-(2-оксоимидазолидин-1-ил)пропаноата.
Получали путем конденсации (3-(пиридин-4-ил)-1H-пиразол-5-карбонил)лейцина (0.08 ммоль) с метил 2-амино-3-(2-оксоимидазолидин-1-ил) пропаноатом (0.09 ммоль, 1,1 экв.) по методу А. Очистка методом ОФХ на приборе PuriFlash. Элюент А: вода, элюент Б-ацетонитрил. Градиент от 10% до 100% элюента Б. Получали продукт метил 2-(4-метил-2-(3-(пиридин-4-ил)-1H-пиразол-5-карбоксамидо)пентан-амидо)-3-(2-оксоимидазолидин-1-ил)пропаноат (0.01 ммоль, выход 13%);
ЖХ-МС (ESI) m/z: вычислено для C22H30N7O5+ ([М+Н]+) 472.23, найдено 472.34.
Стадия 2. Синтез 2-(4-метил-2-(3-(пиридин-4-ил)-1H-пиразол-5-карбоксамидо)пентанамидо)-3-(2-оксоимидазолидин-1-ил)пропановой кислоты.
Получали из метил 2-(4-метил-2-(3-(пиридин-4-ил)-1Н-пиразол-5-карбоксамидо)пентанамидо)-3-(2-оксоимидазолидин-1-ил)пропаноата (0.005 ммоль) методом С.Элюент А: вода, элюент Б-ацетонитрил. Очистка методом ОФХ на приборе PuriFlash. Градиент от 10% до 100% элюента Б. Получали продукт 2-(4-метил-2-(3-(пиридин-4-ил)-1H-пиразол-5-карбоксамидо)пентанамидо)-3-(2-оксоимидазолидин-1-ил)пропановую кислоту (0.001 ммоль, выход 14%);
1Н ЯМР (700 МГц, MeOD) δ 8.64 (с, 1Н), 7.78 (с, 1Н), 4.73-4.69 (м, 1Н), 4.54 (с, 1Н), 3.68-3.66 (м, 1Н), 3.60-3.57 (м, 2Н), 3.55-3.48 (м, 3Н), 3.35 (с, 2Н), 1.79-1.73 (м, 1Н), 1.01 (д, J=6.1 Гц, 3Н), 0.99 (д, J=5.4 Гц, 3Н);
13С ЯМР (175 МГц, MeOD) δ 149.2, 51.92, 45.65, 45.20, 37.83, 24.72, 22.10, 20.50;
ЖХ-МС (ESI) m/z: вычислено для C21H28N7O5+ ([М+Н]+) 458.22, найдено 458.43.
Получение 3-(2,4-диоксоимидазолидин-1-ил)-2-(4-метил-2-(3-(пиридин-4-ил)-1H-пиразол-5-карбоксамидо)пентанамидо)пропановой кислоты. Стадия 1. Синтез метил 2-(2-((трет-бутоксикарбонил)амино)-4-метилпентанамидо)-3-(2,4-диоксоимидазолидин-1-ил)пропаноата. К раствору 2,5-диоксопирролидин-1-ил (трет-бутоксикарбонил)лейцината (1 экв., 0.11 ммоль) и метил 2-амино-3-(2,4-диоксоимидазолидин-1-ил)пропаноата (1 экв., 0.11 ммоль) в дихлорметане добавляли триэтиламин (5 экв., 0.55 ммоль). Реакционную смесь выдерживали при перемешивании при комнатной температуре в течение 12 часов. После упаривания проводили очистку методом обращенно-фазовой хроматографии на приборе PuriFlash. Элюент А: вода, элюент Б-ацетонитрил. Градиент от 20% до 100% элюента Б. Получали метил 2-(2-((трет-бутоксикарбонил)амино)-4-метилпентанамидо)-3-(2,4-диоксоимидазолидин-1-ил)пропаноат (0.047 ммоль, выход 43%);
1Н ЯМР (300 МГц, CDCl3) δ5 9.15 (с, 1Н), 7.49 (д, J=8.5 Гц, 1Н), 5.12 (д, J=9.6 Гц, 1Н), 4.91-4.62 (м, 1Н), 4.17 (с, 1Н), 4.14-3.78 (м, 3Н), 3.77 (д, J=2.0 Гц, 3Н), 3.61 (дд, J=14.5, 4.8 Гц, 1H), 1.75-1.46 (м, 3Н), 1.42 (с, 9Н), 0.98-0.85 (м, 6Н).
Стадия 2. Синтез метил 2-(2-амино-4-метилпентанамидо)-3-(2,4-диоксоимидазолидин- 1-ил) пропаноата.
Из метил 2-(2-((трет-бутоксикарбонил)амино)-4-метилпентанамидо)-3-(2,4-диоксоимидазолидин-1-ил)пропаноата (0.036 ммоль) методом В получали метил 2-(2-амино-4-метилпентанамидо)-3-(2,4-диоксоимидазолидин-1-ил) пропаноат, который использовали в следующей стадии без дополнительной очистки.
Стадия 3. Синтез метил 3-(2,4-диоксоимидазолидин-1-ил)-2-(4-метил-2-(3-(пиридин-4-ил)-1H-пиразол-5-карбоксамидо)пентанамидо)пропаноата.
Получали путем конденсации метил 2-(2-амино-4-метилпентанамидо)-3-(2,4-диоксоимидазолидин-1-ил) пропаноата (0.032 ммоль) с метил 2-амино-3-(2,4-диоксоимидазолидин-1-ил) пропаноатом (0.032 ммоль) по методу А.
Очистка методом ОФХ на приборе PuriFlash. Элюент А: вода, элюент Б-ацетонитрил. Градиент от 10% до 100% элюента Б. Получали метил 3-(2,4-диоксоимидазолидин-1-ил)-2-(4-метил-2-(3-(пиридин-4-ил)-1H-пиразол-5-карбоксамидо)пентанамидо)пропаноат (0.027 ммоль, выход 84%);
1Н ЯМР (300 МГц, Ацетон-d6) 5 8.64 (д, J=5.7 Гц, 2Н), 7.92-7.83 (м, 1H), 7.44-7.32 (м, 2Н), 4.86-4.64 (м, 1Н), 4.11-4.01 (м, 1Н), 4.01-3.87 (м, 2Н), 3.85-3.64 (м, 5Н), 1.87-1.66 (м, 2Н), 1.00-0.82 (м, 6Н).
Стадия 4. Синтез 3-(2,4-диоксоимидазолидин-1-ил)-2-(4-метил-2-(3-(пиридин-4-ил)-1H-пиразол-5-карбоксамидо)пентанамидо)пропановой кислоты.
Получали из метил 3-(2,4-диоксоимидазолидин-1-ил)-2-(4-метил-2-(3-(пиридин-4-ил)-1Н-пиразол-5-карбоксамидо)пентанамидо)пропаноата (0.02 ммоль) методом С.Очистка методом ОФХ на приборе PuriFlash. Элюент А: вода, элюент Б-ацетонитрил. Градиент от 10% до 100% элюента Б. Получали 3-(2,4-диоксоимидазолидин-1-ил)-2-(4-метил-2-(3-(пиридин-4-ил)-1Н-пиразол-5-карбоксамидо)пентанамидо)пропановую кислоту (0.005 ммоль, выход 23%);
1Н ЯМР (700 МГц, MeOD) δ 8.61 (с, 2Н), 7.83 (с, 2Н), 7.38 (с, 1Н), 4.72-4.64 (м, 1Н), 4.53-4.43 (м, 1Н), 4.15 (дд, J=40.7, 17.5 Гц, 1Н), 4.03 (дд, J=17.5, 11.2 Гц, 1Н), 3.79-3.69 (м, 2Н), 1.75 (д, J=7.1 Гц, 3Н), 1.01 (т,./=5.6 Гц, 3Н), 0.99 (дд, J=6.1, 4.0 Гц, 3Н);
ЖХ-МС (ESI) m/z: вычислено для C21H26N7O6+([М+Н]+) 472.19, [М+Н] 472.27.
Получение бензил (4-метил-1-оксо-1-((1-оксо-3-(2-оксоимидазолидин-1-ил)пропан-2-ил)амино)пентан-2-ил)карбамата.
Стадия 1. Синтез бензил (1-((1-гидрокси-3 -(2-оксоимидазолидин-1-ил)пропан-2-ил)амино)-4-метил-1-оксопентан-2-ил)карбамата. К раствору метил 2-(2-(((бензилокси)карбонил)амино)-4-метилпентанамидо)-3-(2-оксоимидазолидин-1-ил)пропаноата (1.26 г, 2.90 ммоль, 1 экв.) в ТГФ (110 мл) прибавляли боргидрид натрия (656 мг, 17.34 ммоль, 6 экв.), а затем метанол (4 мл) при перемешивании на ледяной бане. Реакционную смесь 1 час перемешивали при комнатной температуре, а затем прибавляли нас. раствор NH4Cl (30 мл). При добавлении водного раствора NH4Cl бурно выделялся газ. Реакционную смесь экстрагировали этилацетатом (2×100 мл, 1×50 мл). Органическую фазу отделяли, промывали нас. раствором NaCl (100 мл), далее сушили над безв. Na2SO4 и упаривали на ротационном испарителе. Из полученной белой пены методом колоночной хроматографии (SiO2; CHCl3-СН3ОН 2/98 → 5/96) выделяли продукт бензил (1-((1-гидрокси-3-(2-оксоимидазолидин-1-ил)пропан-2-ил)амино)-4-метил-1-оксопентан-2-ил)карбамат (1.13 г, выход 96%);
Rf=0.44 (CHCl3-МеОН 9:1);
1Н ЯМР (700 МГц, CDCl3) δ 7.36-7.26 (м, 5Н), 7.11 (д, J=8.8 Гц, 1H), 5.67 (д, J=8.4 Гц, 1Н), 5.08 (м, 2Н), 4.25-4.19 (м, 1Н), 4.17-4.12 (м, 1H), 4.12-4.05 (м, 1Н), 3.67-3.18 (м, 8Н), 1.70-1.58 (м,2Н), 1.49 (ддд, J=14.3, 9.8, 5.0 Гц, 1Н), 0.98-0.85 (м 6Н);
13С ЯМР (176 МГц, CDCl3) δ 172.95, 163.92, 156.29, 136.53, 128.64, 128.26, 128.15,67.08,62.11,54.01,49.30,45.99,43.65,42.09,38.52,24.90,23.16.21.99; ЖХ-МС (ESI) m/z: вычислено для C20H31N4O5 ([М+Н]+) 407.22, найдено 407.25.
Стадия 2. Синтез бензил (4-метил-1-оксо-1-((1-оксо-3-(2-оксоимидазолидин-1-ил)пропан-2-ил)амино)пентан-2-ил)карбамата. Py⋅SO3 (159 мг, 0.49 ммоль, 4 экв.) и ДМСО (44 мкл, 0.62 ммоль, 5 экв.) растворяли в CH2Cl2 (1 мл), через 15 минут к полученному раствору прибавляли бензил (1-((1-гидрокси-3-(2-оксоимидазолидин-1-ил)пропан-2-ил)амино)-4-метил-1-оксопентан-2-ил)карбамат (50 мг, 0.12 ммоль, 1 экв.), триэтиламин (102 мкл, 0.74 ммоль, 6 экв.). Реакционную смесь выдерживали при перемешивании 1,5 ч при комнатной температуре, затем прибавляли Py⋅SO3 (119 мг, 0.37 ммоль, 3 экв.), ДМСО (33 мкл, 0.47 ммоль, 3.75 экв.), триэтиламин (77 мкл, 0.56 ммоль, 6 экв.) и перемешивали еще 1 ч, затем к реакционной смеси прибавляли нас. раствор NH4Cl (3×1 мл). Органическую фазу отделяли, сушили над безв. Na2SO4, концентрировали на ротационном испарителе. Из полученного желт, масла методом колоночной хроматографии (SiO2; CHCl3-CH3OH 2/98 → 5/95) выделяли продукт бензил (4-метил-1 -оксо-1 -((1 -оксо-3-(2-оксоимидазолидин-1-ил)пропан-2-ил)амино)пентан-2-ил)карбамат (9 мг, выход 18%);
Rf=0.52 (CHCl3-МеОН 9:1);
1Н ЯМР (700 МГц, ацетон-d6) δ 9.56 (с, 1Н), 7.99 (д, J=6.8 Гц, 1Н), 7.38 (д, J=7.5 Гц, 2Н), 7.35 (т, J=7.5 Гц, 2Н), 7.30 (т, J=7.5 Гц, 1H), 6.50-6.50 (м, 1Н), 5.66-5.36 (м, 1Н), 5.09 (с, 2Н), 4.58-4.34 (м, 1Н), 4.31-4.14 (м, 1Н), 3.71-3.38 (м, 4Н), 3.36-3.26 (м, 2Н), 1.79-1.72 (м, 1H), 1.70-1.53 (м, 2Н), 0.95-0.89 (м, 6Н);
13С ЯМР (МГц, ацетон-d6) δ 200.35, 173.96, 163.92, 157.21, 138.08, 129.29, 128.70, 128.67, 66.93, 59.56, 54.58, 47.14, 43.83, 42.03, 38.94, 25.55, 23.49, 21.99;
ЖХ-МС (ESI) m/z: вычислено для C20H29N4O5 ([М+Н]+) 405.21, найдено 405.24.
Получение бензил (1-((1-(2,4-диоксоимидазолидин-1-ил)-3-оксопропан-2-ил)амино)-4-метил-1-оксопентан-2-ил)карбамата.
Стадия 1. Синтез бензил (1-((1-(2,4-диоксоимидазолидин-1-ил)-3-гидроксипропан-2-ил)амино)-4-метил-1-оксопентан-2-ил)карбамата. К раствору метил 2-(2-(((бензилокси)карбонил)амино)-4-метилпентанамидо)-3-(2-оксоимидазолидин-1-ил)пропаноата (48 мг, 0.11 ммоль, 1 экв.) в тетрагидрофуране (2 мл), при до 0°С по частям добавляли NaBH4 (24 мг, 0.64 ммоль, 6 экв.) и метанола (150 мкл). Спустя 30 минут реакционную смесь отогревали до комнатной температуры и оставляли при перемешивании 1.5 часа. Затем добавляли 5 мл насыщенного раствора хлорида аммония, экстрагировали этилацетатом (3×10 мл). Органический слой сушили над безводным сульфатом натрия и упаривали под вакуумом на роторном испарителе. Затем методом колоночной хроматографии на силикагеле (хлороформ/этанол 99/1-90/10) выделяли бензил (1-((1-(2,4-диоксоимидазолидин-1-ил)-3 -гидроксипропан-2-ил)амино)-4-метил-1 -оксопентан-2-ил)карбамат (20 мг, выход 44%);
Rf=0.10 (CHCl3-EtOH 95:5);
1Н ЯМР (300 МГц, CDCl3) δ 7.33-7.27 (м, 5Н), 5.83-5.68 (м, 1Н), 5.09 (д, J=12.0 Гц, 1H), 5.02 (д, J=12.2 Гц, 1H), 4.25-4.07 (м, 4Н), 4.07-3.75 (м, 2Н), 3.66-3.50 (м, 3Н), 3.39-3.20 (м, 1Н), 1.65-1.37 (м, 3Н), 0.89 (д, J=5.6 Гц, 6Н).
Стадия 2. Синтез бензил (1-((1-(2,4-диоксоимидазолидин-1-ил)-3-оксопропан-2-ил)амино)-4-метил-1-оксопентан-2-ил)карбамата. К смеси дихлорметана (200 мкл) и ДМСО (21 мкл, 0.29 ммоль, 4.2 экв.) при -78°С в атмосфере аргона прикапывали оксалилхлорид (22 мкл, 0.26 ммоль, 3.6 экв.). Реакционную смесь выдерживали при перемешивании 15 минут, затем прикапывали раствор бензил (1-((1-(2,4-диоксоимидазолидин-1-ил)-3-гидроксипропан-2-ил)амино)-4-метил-1 -оксопентан-2-ил)карбамата (3 0 мг, 0.071 ммоль) в дихлорметане (0.5 мл). Через 20 минут добавляли триэтиламин (79 мкл, 0.57 ммоль, 8 экв.). Далее реакционную смесь отогревали до 0°С, разбавляли дихлорметаном (2 мл) и добавили нас. раствор хлорида аммония (2 мл). Экстрагировали дихлорметаном (3×5 мл). Органический слой сушили над сульфатом натрия, концентрировали на роторном испарителе. Из сухого остатка методом колоночной хроматографии на силикагеле (хлороформ-этанол 98/2-90/10) выделяли продукт бензил (1-((1-(2,4-диоксоимидазолидин-1-ил)-3-оксопропан-2-ил)амино)-4-метил-1-оксопентан-2-ил)карбамат (7 мг, выход 23%);
Rf=0.35 (CHCl3-EtOH 95:5);
ЖХ-МС (ESI) m/z: вычислено для C20H27N4O6+ ([М+Н]+) 419.19, найдено 419.27.
Получение бензил (1-((1-циано-2-(2-оксоимидазолидин-1-ил)этил)амино)-4-метил-1-оксопентан-2-ил)карбамата.
К метил 2-(2-(((бензилокси)карбонил)амино)-4-метилпентанамидо)-3-(2-оксоимидазолидин-1-ил)пропаноату (50 мг, 0.12 ммоль, 1 экв.) добавляли раствор аммиака в метаноле (3 мл). Через 12 часов реакционную смесь концентрировали на роторном испарителе. Сухой остаток перерастворяли в этилацетате, растворитель упаривали под вакуумом. Полученный продукт вводили в следующую стадию без выделения. К раствору бензил (1-((1-амино-1-оксо-3-(2-оксоимидазолидин-1-ил)пропан-2-ил)амино)-4-метил-1-оксопентан-2-ил)карбамата в смеси пиридин-дихлорметан (1 мл, 1:1 v:v) добавляли 1H-имидазол (18 мг, 0.26 ммоль, 2.6 экв.), реакционную смесь охлаждали до -35°С, далее прикапывали оксихлорид фосфора (53 мкл, 0.57 ммоль, 5.7 экв.). Через 2 часа добавляли еще, далее через 30 минут еще оксихлорида фосфора (18 мкл, 2 экв., 0.2 ммоль) до полной конверсии. Реакционную смесь отогревали до 0°С, добавляли 1М HCl (3 мл), экстрагировали дихлорметаном (3×10 мл). Органический слой осушали над безводным сульфатом натрия и упаривали под вакуумом на роторном испарителе. Затем из сухого остатка методом колоночной хроматографии на силикагеле (этилацетат) получали продукт бензил (1-((1-циано-2-(2-оксоимидазолидин-1-ил)этил)амино)-4-метил-1-оксопентан-2-ил)карбамат (21 мг, выход 53%);
Rf=0.27 (этилацетат);
1Н ЯМР (300 МГц, ацетон-d6) δ 8.39 (д, J=7.8 Hz, 1Н), 7.42-7.35 (м, 4Н), 7.35-7.26 (м, 1H), 6.59 (д, J=8.3 Гц, 1Н), 5.79 (с, 1Н), 5.09 (с, 1Н), 5.08 (с, 1Н), 5.02 (тт, J=6.3, 3.7 Гц, 1H), 4.23 (к, J=7.9 Гц, 1Н), 3.66-3.48 (м, 4Н), 3.47-3.35 (м, 2Н), 1.73 (дк, J=12.8, 6.5 Гц, 1H), 1.62 (дд, J=8.1, 5.1 Гц, 2Н), 0.93 (д, J=5.1 Гц, 3Н), 0.91 (д, J=5.3 Гц, 3Н);
13С ЯМР (75 МГц, ацетон-d6) δ172.54, 162.88, 156.26, 137.29, 128.32, 127.74, 117.68,66.02,53.50,45.96,45.92,45.55,40.81,40.45,40.33,38.13,38.04,24.51, 22.49, 20.93;
ЖХ-МС (ESI) m/z: вычислено для C20H28N5O4+ ([М+Н]+) 402.21, найдено 402.33.
Ингибирование основной протеазы Mpro коронавируса SARS-CoV-2 (KI) различными концентрациями соединения вышеуказанной общей формулы (I).
Анализ эффективности ингибирования соединения в стационарных условиях позволил определить тип и константу ингибирования KI. Кинетический анализ проводили с использованием FRET-субстрата, Dabcyl-KTSAVLQ↓SGGFRKM-E(Edans)-NH2 (BPS Bioscience, США), содержащий сайт расщепления основной протеазой (обозначен стрелкой в приведенной последовательности). Исследуемый ингибитор растворяли в ДМСО (конечная концентрация 5,0 мМ). Для определения константы ингибирования фермента (150 нМ) в реакционном буфере (20 мМ Трис-HCl, рН 7,5, 100 мМ NaCl, 1,0 мМ ЭДТА и 1,0 мМ ДТТ) инкубировали с различными концентрациями тестируемого соединения (или ДМСО) в течение 10 мин при 4°С. Реакцию инициировали добавлением FRET субстрата (15-35 мкМ) в реакционном буфере, который расщепляется Mpro с образованием продукта, содержащего свободную группу Edans. Кинетические кривые были получены с помощью планшетного флуориметра Thermo Scientific Varioscan. Длина волны возбуждения составляла 350 нм, интенсивность флуоресценции детектировали на длине волны 460 нм. Время реакции составляло 1 ч при 25°С. Начальную скорость рассчитывали с помощью линейной регрессии для первых 10 минут кинетических кривых процесса. Тип ингибирования и константу ингибирования определяли из зависимости начальной скорости от концентрации субстрата и ингибитора в двойных обратных координатах. На фиг.3 представлены кинетические флуоресцентные кривые, характеризующие процесс гидролиза FRET-субстрата (30 мкМ) в присутствии Mpro в присутствии ингибитора. Из фиг.3 видно, что описываемое соединение оказывает выраженное ингибирующее действие на основной протеазы Mpro коронавируса SARS-CoV-2 при микромолярных концентрациях.
Из зависимости начальной скорости гидролиза от концентрации ингибитора (фиг.4) была рассчитана константа ингибирования KI = 0,92 мкМ. Таким образом, предлагаемое соединение оказывает специфическое ингибирующее действие на фермент Mpro и, являясь доступным российским аналогом известных ингибиторов, расширяет арсенал специфических ингибиторов данного фермента и может с успехом применяться в медицине.
| название | год | авторы | номер документа |
|---|---|---|---|
| Адамантилсодержащие производные 1,2,4-триазола и 1,3,4-тиадиазола, имеющие монотерпеноидные фрагменты, используемые в качестве ингибиторов фермента тирозил-ДНК-фосфодиэстеразы 1 | 2020 |
|
RU2761880C1 |
| Способ получения (6R,7S,7aS)-6-((R)-1-(3,5-бис(трифторметил)фенил)этокси)-7-(4-фторфенил)гексагидро-3Н-пирролизин-3-она | 2022 |
|
RU2789599C1 |
| Способ получения замещенных 3-арилпирролов | 2024 |
|
RU2831117C1 |
| Способ получения эфиров пиперидин-4,4-дикарбоновых кислот | 2021 |
|
RU2765464C1 |
| 3-(2-ОКСОИМИДАЗОЛИДИНИЛ-5)ИНДОЛЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2019 |
|
RU2720490C1 |
| Замещенные 4-арил-гексагидро-7Н-имидазоло[1,5-b][1,2]оксазин-7-оны и способ их получения | 2018 |
|
RU2670097C1 |
| Способ получения ацилоксизамещенных барбитуровых кислот | 2017 |
|
RU2649146C1 |
| Нуклеозидные производные 1,3-диаза-2-оксофеноксазина в качестве ингибиторов репликации герпесвирусов. | 2019 |
|
RU2731381C1 |
| СРЕДСТВО ДЛЯ ИНГИБИРОВАНИЯ ФЕРМЕНТА ТИРОЗИЛ-ДНК-ФОСФОДИЭСТЕРАЗЫ 1 ЧЕЛОВЕКА | 2015 |
|
RU2612875C1 |
| Способ получения трициклических органических дипероксидов | 2020 |
|
RU2752957C1 |
Изобретение относится к соединению формулы (I), где R1 представляет собой -СН(SO3H)ОН, -CN или -C(O)XR3, где X отсутствует или представляет собой -О- или -СН2О-, R3 представляет собой водород, C1-С6 алкил, -РО3Н2; m равно 0 или 1; n равно 0 или 1; А представляет собой -СН2- или -С(O)-; кольцо В представляет собой фенил, индолил, 5-членный гетероарил, содержащий 1 или 2 гетероатома, независимо выбранных из N; каждый R2 независимо выбран из водорода, фтора, фенила, пиридила, причем R2 необязательно замещен одним О-(C1-C6 алкил). Также изобретение относится к применению соединения формулы (I) для ингибирования основной протеазы коронавируса SARS-CoV-2. Технический результат – создание эффективного противовирусного средства, активного в качестве высокоспецифичного ингибитора протеазы коронавируса SARS-CoV-2. 2 н.п. ф-лы, 4 ил., 1 табл.
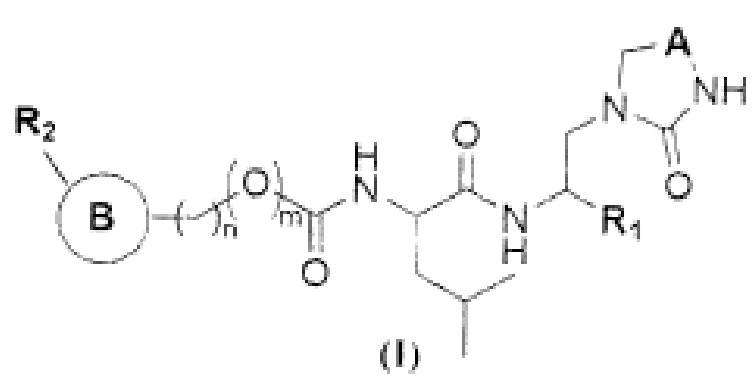
1. Соединение общей формулы (I):
 , где
, где
R1 представляет собой -СН(SO3H)ОН, -CN или -C(O)XR3, где
X отсутствует или представляет собой -О- или -СН2О-,
R3 представляет собой водород, C1-С6 алкил, -РО3Н2;
m равно 0 или 1;
n равно 0 или 1;
А представляет собой -СН2- или -С(O)-;
кольцо В представляет собой фенил, индолил, 5-членный гетероарил, содержащий 1 или 2 гетероатома, независимо выбранных из N;
каждый R2 независимо выбран из водорода, фтора, фенила, пиридила, причем R2 необязательно замещен одним О-(C1-C6 алкил).
2. Применение соединения по п. 1 для ингибирования основной протеазы коронавируса SARS-CoV-2.
| WO 2022040186 A1, 24.02.2022 | |||
| US 20230234984 A1, 27.07.2023 | |||
| WO 2021252644 A1, 16.12.2021 | |||
| Rotella, D.P | |||
| The discovery and development of boceprevir | |||
| Expert opinion on Drug Discovery, 2013, 8 (11), p.1439-1447 | |||
| Hoffman, R.L | |||
| et al | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |

