Область техники.
Настоящее изобретение относится к области функционализированных наночастиц, их приготовлению и применению.
Уровень техники, к которой относится изобретение.
Известно, что некоторые органические молекулы способны адсорбироваться на поверхностях твердых неорганических материалов, и это свойство широко используется в такой степени, что сформировались целые классы технологически важных соединений, таких как диспергенты и «смачивающие агенты».
Некоторые из этих молекул не только адсорбируются упомянутой поверхностью, но также способствуют образованию компактных структур, которые могут глубоко изменить ее свойства.
Типичными примерами органических молекул упомянутого выше типа (называемых далее связующими) являются простые монофункционализированные алифатические соединения, такие как тиолы, додецилфосфат натрия, бромид цетилтриметиламмония, различные алифатические фосфаты и фосфоновые, карбоксильные и гидроксамовые кислоты.
Взаимодействие обычно происходит между единственной функциональной группой и металлической неорганической поверхностью, оставляя, таким образом, свободной простую алифатическую цепочку, которая никаким образом не способна взаимодействовать с другими функциональными молекулами.
Сродство между органическими молекулами и поверхностями зависит от химической природы каждого из них: эти взаимодействия были изучены для некоторых очень хорошо известных случаев, однако полного понимания сродства разных связующих веществ с поверхностями наночастиц все еще нет, вопрос обсуждается в академических кругах, поскольку результаты часто являются противоречивыми.
Известно также, что наночастицы являются материалами с размерами менее 500 нм или, согласно некоторым авторам, менее 100 нм, которые могут образовывать устойчивую дисперсию в жидкостях, если между отдельными частицами существует потенциал отталкивания. В дисперсии не наблюдается никакого осаждения, поскольку присущее частицам сообщаемое им температурой движение препятствует их осаждению под действием тяжести. Потенциал взаимодействия между двумя частицами прежде всего зависит от состояния поверхности наночастицы и он может меняться в результате адсорбции или химического связывания с другими молекулярными или ионными частицами, присутствующими в растворе.
Известны некоторые комплексы, образованные наночастицами и монофункциональными связующими указанного выше типа [см., например, Aronoff Y.G. et al., J. Am. Chem. Soc. 1997, 119, 259-262; Heimer T.A., d'Arcangelis et al., Langmuir, 2002, 18, 5205-5212; Yee C. et al., Langmuir, 1999, 15, 7111-7115; Folkers et al., Langmuir, 1995, 11, 813-824], но они обладают различными недостатками.
Не говоря о скудости изученных материалов и связующих, указанные выше продукты нерастворимы в водно-спиртовой среде, что является очень важным условием для биомедицинских и фармакологических применений. Наряду с этим остающаяся свободной простая алифатическая цепочка абсолютно не способна взаимодействовать с функциональными группами, обычно присутствующими в биоактивных молекулах.
С учетом сказанного выше, безусловно, важно иметь комплексы, образуемые наночастицами и функциональными связующими, которые сделали бы их пригодными для разных желаемых целей, устранив указанные выше недостатки.
Краткое раскрытие изобретения.
Устойчивые комплексы могут быть получены при связывания наночастиц различных типов оксидов переходных металлов с моно- и бифункциональными соединениями.
Краткое описание чертежей.
Фиг.1 схематически иллюстрирует способы приготовления комплексов, образованных наночастицами с описанными выше бифункциональными связующими, и последующие реакции этих комплексов с биополимерами, молекулами (циклодекстринами, антителами и т.д.) и белками.
Фиг.2а и 2b показывают дзета-потенциал суспензии в этаноле до и после функционализации.
Фиг.3а и 3b показывают дзета-потенциал суспензии в воде до и после функционализации.
Детальное описание изобретения.
В настоящей работе неожиданным образом было обнаружено, что бифункциональные соединения способны связываться с наночастицами, образованными различными типами оксидов переходных металлов и металлами, образуя устойчивые комплексы.
В указанных бифункциональных связующих дополнительная функциональная группа (которая не взаимодействует с неорганической металлической поверхностью) приводит к изменениям в растворимости наночастицы в жидкой среде, делая наночастицу пригодной для процессов производства разных типов новых материалов (некоторых типов гидрофильных пластиков, волокон); эта группа делает также возможными взаимодействия с разными сложными веществами, такими как биополимеры, циклодекстрины, антитела и лекарственные препараты, для применения в фармацевтической и диагностической области.
Кроме того, применение бифункциональных соединений позволяет получать комплексы наночастиц со связующими, в которых достигается полное и компактное покрытие наночастицы без значительного изменения определяемых ею свойств (например, магнитных или оптических свойств).
Среди других преимуществ следует обратить внимание на то, что благодаря полному покрытию поверхности, получаемому при использовании указанных связующих веществ, наночастицы являются нетоксичными.
Согласно настоящему изобретению выражение «бифункциональные соединения» подразумевает тиолы, карбоксильные кислоты, гидроксамовые кислоты, эфиры фосфорных кислот и их соли, имеющие алифатическую цепочку, которая имеет вторую функциональную группу в концевом положении (называемым ω-положением).
Вторую функциональную группу преимущественно выбирают из группы, состоящей из ОН, NH2, СООН, COOR3, где R3 определено ниже.
Более конкретно, бифункциональные соединения согласно настоящему изобретению являются соединениями общей формулы:
Rl-(CH2)n-R2
в которой n обозначает целое число от 2 до 20,
R1 выбирают из группы, состоящей из ОН, NH2, СООН,
R2 выбирают из CONHOH, PO(OH)2, PO(OH)(OR3), СООН, SH,
R3 обозначает щелочной металл, преимущественно К, Na или Li, или защитную органическую группу.
Описанные выше бифункциональные соединения известны или могут быть получены согласно известным способам.
Способ получения, как правило, предполагает начало синтеза исходя из простого бифункционального соединения, имеющегося в продаже (например, карбоновые кислоты или ω-функционализированные спирты), защиту функциональной группы в ω-положении и, наконец, активацию карбоксильной (или спиртовой) функции для последующего введения гидроксамовой или фосфорнокислотной функциональности.
Согласно настоящему изобретению термин «наночастицы» подразумевает частицы с размерами от 1 до 200 нм.
Особо предпочтительными согласно изобретению являются наночастицы, состоящие из металлов и оксидов металлов, принадлежащих к ряду переходных металлов, в частности из соединений общей формулы MIIMIII 2O4, где MII=Co, Ni, Fe, Mn и MIII=FeIII, Со, Al; оксиды Fe2O3 типа магхемита; в частности феррит кобальта CoFe2O4, магнетит FeFe2O4, магхемит γ-Fe2O3; металлические частицы, состоящие из металлических Fe0 и Со0 и их сплавов, даже с благородными металлами.
Комплексы наночастиц и связующих получают, вводя описанные выше моно- или бифункциональные производные в реакцию с описанными выше наночастицами таким образом, чтобы полностью покрыть их свободную поверхность.
Процесс приготовления осуществляется введением дисперсии наночастиц в органическом растворителе (например, этиленгликоле) в реакцию с предпочтительным связующим при перемешивании в течение нескольких часов при пониженной температуре.
Продукт после этого осаждают (например, с помощью ацетона), центрифугируют, отделяют и при необходимости очищают путем повторного диспергирования в подходящем растворителе и повторного осаждения. Степень покрытия и степень протекания реакции оценивают, используя разные экспериментальные методы, включая термогравиметрию DSC-TG, ИК-спектроскопию с Фурье-преобразованием (FT-ИК), элементный анализ и метод динамического светорассеяния (МДС, DLS).
Было также определено влияние поверхностной функционализации на магнитные свойства продукта.
Полученные таким образом функционализированные наночастицы могут быть использованы в процессах, которые требуют особого гидрофильно-гидрофобного поведения, в частности, в производстве пластиков (например, полиэтилена или полиэфира) или синтетических волокон (например, нейлона) и натуральных волокон (например, хлопчатобумажных).
Наночастицы, обработанные бифункциональными связующими, могут быть дополнительно модифицированы по функциональной группе определенными реакционноспособными молекулами (например, циклодекстринами, фолевой кислотой, антителами и лекарственными препаратами), белками или полимерами (например, полиамидоамином), с целью того, чтобы соединить свойства частицы (магнетизм) со свойствами этих молекул или этих полимеров (биосовместимость, невидимость для иммунной системы), или этих белков.
Магнитные свойства могут быть использованы для конструирования контрастных агентов общего действия или селективных контрастных агентов, для анализа методом магнитного резонанса или, в сочетании с лекарствами, для образования транспортирующих систем, высвобождение которых контролируется нагревом частиц за счет гипертермического эффекта.
В целом можно утверждать, что для сборки комплекса наночастица/бифункциональное связующее, который мы далее будем называть функционализированной наночастицей: указанными выше молекулой, полимером или белком, могут соблюдаться следующие критерии.
a) Функционализированные наночастицы, которые содержат амины в качестве наружных (внешних) функциональных групп, могут быть связаны с указанными выше молекулами, полимерами или белками, которые могут содержать одну из следующих функциональных групп: карбоновые кислоты, альдегиды и акриламиды.
b) Функционализированные наночастицы, которые содержат карбоновые кислоты в качестве внешних функциональных групп, могут быть связаны с биополимерами, белками или молекулами (циклодекстринами, фолиевой кислотой, антителами и лекарственными препаратами), которые, в свою очередь, могут содержать одну из следующих функциональных групп: спирты, амины и тиолы.
c) Функционализированные наночастицы, которые содержат оксигидрильные группы в качестве внешних функциональных групп, могут быть связаны с биополимерами, белками или молекулами (циклодекстринами, фолиевой кислотой, антителами и лекарственными препаратами), которые, в свою очередь, могут содержать одну из следующих функциональных групп: карбоновые кислоты.
Как можно убедиться, соединения, образуемые комплексами наночастица/бифункциональное связующее и описанными выше функциональными молекулами, могут быть получены различными препаративными способами.
Способы.
Способ А.
Функционализация наночастицы простыми бифункциональными связующими, такими как, например, ω-гидрокси-, ω-карбокси-, ω-аминокарбоновые кислоты; ω-гидрокси-, ω-карбокси-, ω-аминогидроксамовые кислоты; ω-гидрокси-, ω-карбокси-, ω-аминофосфорные кислоты; ω-гидрокси-, ω-карбокси- и ω-аминотиолы. Последующее связывание бифункционализированных частиц с молекулами, белками или полимерами - через бифункциональные связующие.
Способ В.
Закрепление молекул, полимеров или модифицированных белков с помощью связующих на функционализированных наночастицах путем обмена связующих.
Способ С.
Идентичен способу А за исключением того, что функционализация наночастицы осуществляется смесями бифункционализированных связующих.
Способ D.
Идентичен способу В за исключением того, что функционализация наночастицы осуществляется смесями бифункционализированных связующих.
Способ Е.
Прямая функционализация наночастицы молекулами, полимерами или белками, предварительно связанными с подходящим бифункциональным связующим.
Способ F.
Функционализация наночастицы смесями, содержащими молекулы, полимеры или белки, уже связанные с подходящим бифункциональным связующим и каким-либо иным бифункциональным связующим.
Чтобы лучше проиллюстрировать изобретение, ниже приводятся некоторые конкретные примеры получения связующих, комплексов и их последующей функционализации.
Пример 1.
Синтез 12-амино-N-гидроксидодеканамида.

а) Синтез 12-амино(трет-бутоксикарбонил)додекановой кислоты.
В 250-мл двугорлой колбе фирмы Sovirel с магнитным перемешивателем, снабженной перфорированной мембраной и краном для аргона, растворяют в диоксане (20 мл) продажную 12-аминододекановую кислоту (5,2 г, 25,8 ммоль) и добавляют Boc2O (6,5 мл, 28 ммоль). Систему доводят до 0°С и медленно прибавляют по каплям 2 н. NaOH (13,2 мл). Раствору дают возможность прореагировать в условиях нагревания с обратным холодильником в течение 24 час, добавляют дистиллированную воду (60 мл) и экстрагируют Et2O (2×30 мл). Водную фазу подкисляют лимонной кислотой (25 вес.%) до рН 5, экстрагируют EtOAc (3×50 мл), объединяют полученные фракции, обезвоживают с помощью MgSO4 и упаривают на роторном испарителе Rotavapor и с высоковакуумным насосом. Получают 6,0 г 12-амино(трет-бутоксикарбонил)додекановой кислоты (выход 73%).
Т.пл. 80-82°С.
Данные спектроскопии:
ИК: 3385, 2919, 2853, 1727, 1688, 1520, 1469, 1365, 1246, 1172, 946.
1Н-ЯМР (400 МГц, CD3OD): 1,36 (с, 9Н), 1,40-1,60 (м, 18Н), 2,35 (т, J=7,0 Гц, 2Н), 3,00 (т, J=6,6 Гц, 2Н), 4,80 (с, ушир., 1Н).
13С-ЯМР (100,2 МГц, CD3OD): 24,9; 26,7; 27,7; 29,1; 29,3; 29,4 (2СН2); 29,48; 29,5; 29,8; 33,8; 40,2; 78,6; 157,3; 176,4.
Масс-спектр: 315 (М+).
b) Синтез дициклогексиламмониевой соли 12-амино-(трет-бутоксикарбонил)додекановой кислоты.
Дициклогексиламин (3,92 мл, 19,7 ммоль) добавляют к суспензии 12-амино-(трет-бутоксикарбонил)додекановой кислоты (5,8 г, 18,4 ммоль) в метаноле (20 мл). Полученную суспензию перемешивают 10 мин при комнатной температуре, удаляют в вакууме растворитель и получают 9,1 г продукта (выход 100%) в виде порошкообразного белого твердого вещества, который используют далее без какой-либо очистки.
c) Синтез трет-бутилового эфира 12-(бензилоксиамино)-12-оксододецилкарбаминовой кислоты.
Дициклогексиламмониевую соль 12-амино-(трет-бутоксикарбонил)додекановой кислоты (9,1 г, 18,4 ммоль) помещают в 100-мл двугорлую колбу фирмы Sovirel с магнитным перемешивателем, снабженную перфорированной мембраной и краном для аргона, и добавляют пиридин (1,50 мл, 15,2 ммоль) и дихлорметан (18 мл).
Добавляют с помощью шприца тиенилхлорид (22,1 ммоль, 1,62 мл) и оставляют смесь реагировать в течение 5 мин при комнатной температуре. Одновременно в другую двугорлую колбу помещают навеску гидрохлорида бензилоксиламина (2,9 г, 18,4 ммоль) и добавляют 4-диметиламинопиридин (DIMAP, 3,6 г, 3,0 ммоль) и дихлорметан (36 мл). Полученный раствор прибавляют по каплям с помощью шприца в первую колбу и полученную смесь перемешивают 1 час при комнатной температуре. Растворитель удаляют на роторном испарителе Rotavapor и проводят очистку с помощью колоночной хроматографии на силикагеле (элюент: этилацетат/петролейный эфир, 1:1), что приводит к выделению 3,8 г (выход 50%) продукта в виде желто-белого твердого вещества.
Т.пл.68-73°С.
Данные спектроскопии:
ИК: 3346, 3298, 2922, 2851, 1682, 1657, 1540, 1356, 1269, 1254, 1171.
1Н-ЯМР (400 МГц, CDCl3): 1,05-1,10 (м, 16Н), 1,40 (с, 9Н), 1,40-1,55 (м, 2Н), 2,00 (с, ушир., 2Н), 3,00-3,10 (м, 2Н), 4,80 (с, ушир., 1Н), 4,90 (с, 2Н), 7,25-7,35 (м, 5Н), 9,25 (с, ушир., 1Н).
13С-ЯМР (75,3 МГц, CDCl3): 25,2, 26,4, 28,1, 28,8, 28,9, 29,1, 29,2, 29,7, 32,7, 40,3, 77,5, 78,6, 128,0, 128,7 (2ArCH), 135,3, 155,8, 170,1.
Масс-спектр: 420 (М+).
d) Синтез 12-амино-N-(бензилокси)додеканамида.
В одногорлую колбу, содержащую трет-бутиловый эфир 12-(бензилоксиамино)-12-оксододецилкарбаминовой кислоты (3,14 г, 7,5 ммоль), добавляют в инертной атмосфере хлороформ (30 мл). Медленно прибавляют по каплям трифторуксусную кислоту (5,6 мл, 7,5 ммоль) и перемешивают смесь 1 час при комнатной температуре. Растворитель удаляют на роторном испарителе Rotavapor и добавляют концентрированный аммиак до рН 9. Добавляют дистиллированную воду (30 мл) и хлороформ (30 мл). Экстракцию проводят хлороформом (3×25 мл), органическую фазу обезвоживают сульфатом магния, фильтруют и удаляют растворитель, получая 2,0 г (выход 85%) продукта в виде желтоватого твердого вещества.
Т.пл.=76-78°С.
Данные спектроскопии:
ИК: 3357, 3225, 2907, 2841, 1657, 1553, 1369, 1203, 1057.
1Н-ЯМР (400 МГц, CDCl3): 1,00-1,40 (м, 16Н), 1,45-1,55 (с, ушир., 2Н), 2,00 (с, ушир., 2Н), 2,45 (с, ушир., 2Н), 4,80-5,00 (м, ушир., 5Н), 7,20-7,40 (м, 5Н).
13С-ЯМР (75,3 МГц, CDCl3): 25,3, 26,5, 28,9, 29,0, 29 1, 29,2, 29,24, 32,7, 32,9, 41,5, 77,5, 128,2, 129,0 (2ArCH), 135,7, 170,7.
Масс-спектр: 320 (М+).
e) Синтез 12-амино-N-гидроксидодеканамида.
Проводят гидрирование с помощью водорода в реакторе Парра. В реактор помещают 120 мг Pd/C, 12-амино-N-(бензилокси)додеканамид (1,0 г, 2,4 ммоль) и этанол (40 мл). Желательно вначале подогреть продукт в этаноле до 50°С в колбе Эрленмейера. Гидрирование длится 30 час, после чего проводят фильтрацию на пористой мембране со слоем целлита, промывая мембрану несколько раз этанолом. Раствор упаривают на роторном испарителе Rotavapor и на высоковакуумном насосе, получая 12-амино-N-гидроксидодеканамид в виде белого твердого вещества (500 мг, выход 66%).
T.пл. 112-116°С.
Данные спектроскопии:
ИК: 3247, 2973, 2856, 1712, 1635, 1465, 1207, 1155, 1041.
1Н-ЯМР (400 МГц, CDCl3): 1, 10-1,60 (м, 18Н), 2,0 (т, ушир., 2Н), 2,70-2,75 (м, 4Н), 6,80 (с, ушир., 1Н), 7,40 (с, ушир., 1H).
13С-ЯМР (75,3 МГц, CDCl3): интервал СН2 25,9-33,0, 41,8, 169,8.
Масс-спектр: 230 (М+).
Согласно тому же синтетическому протоколу из 12-гидроксидодекановой кислоты может быть получен N-12-дигидроксидодеканамид.

Пример 2.
Синтез однозамещенного 12-аминододецилфосфоната калия.

a) Синтез трет-бутилового эфира 12-гидроксидодецилкарбаминовой кислоты.
В 100-мл двугорлую колбу с обратным холодильником и магнитным перемешивателем, помещенную под статический слой азота, помещают навеску 12-аминододекан-1-ола гидрохлорида (3,34 г, 14,1 ммоль) и добавляют пиридин (40 мл), 'Pr2NEt (2,45 мл, 14,1 ммоль) и Вос2О (3,24 мл, 14,1 ммоль). Смесь перемешивают 60 час при 70°С, упаривают на роторном испарителе Rotavapor и на высоковакуумном насосе и очищают продукт с помощью колоночной хроматографии на силикагеле, используя в качестве элюента смесь петролейный эфир/этилацетат, 1:1. Выделяют 3,1 г трет-бутилового эфира 12-гидроксидодецилкарбаминовой кислоты в виде белого твердого вещества с выходом 73%.
Т.пл. 78°С.
Данные спектроскопии:
ИК: 3424, 3370, 2920, 2852, 1686, 1523, 1172, 1058.
1Н-ЯМР (400 МГц, CDCl3): 1,20-1,30 (с, ушир., 20Н), 1,40 (с, ушир., 9Н), 3,15 (bis, 2Н), 3,6 (т, J=8,5 Гц, 2Н), 4,4 (с, ушир., 1Н).
13С-ЯМР (75,3 МГц, CDCl3): 24,8, 26,7, 27,6, 29,0, 29,2 (2СН2), 29,5, 29,6, 29,7, 29,73, 33,7, 40,1, 78,9, 157,1.
Масс-спектр: 301 (М+).
b) Синтез трет-бутилового эфира 12-бромдодецилкарбаминовой кислоты.
В 250-мл двугорлой колбе с обратным холодильником и магнитным перемешивателем, помещенной под статический слой азота, трет-бутиловый эфир 12-гидроксидодецилкарбаминовой кислоты (3,07 г, 10,2 ммоль) растворяют в дихлорметане (75 мл) и добавляют PPh3 (2,94 г, 11,2 ммоль) и NBS (2,42 г, 10,7 ммоль). Смесь перемешивают при кипячении 24 часа, упаривают на роторном испарителе Rotavapor и продукт очищают с помощью колоночной хроматографии на силикагеле, используя в качестве элюента смесь петролейный эфир/этилацетат, 3:1. Выделяют 2,9 г трет-бутилового эфира 12-бромдодецилкарбаминовой кислоты в виде легкоплавкого белого твердого вещества с выходом 78%.
Т.пл. 42-44°С.
Данные спектроскопии:
ИК: 3421, 3366, 2924, 2853, 1688, 1521, 1170, 1061.
1Н-ЯМР (400 МГц, CDCl3): 1,10-1,20 (с, ушир., 20Н), 1,35 (с, ушир., 9Н), 3,05 (с, ушир., 2Н), 3,60 (т, J=6,0 Гц, 2Н), 4,80 (с, ушир., 1H).
13С-ЯМР (100,4 МГц, CDCl3): 26,4, 27,8, 28,1, 28,4, 28,9, 29 1, 29,15, 29,2, 29,7, 32,5, 33,4, 40,2, 78,2, 155,2.
Масс-спектр: 363 (М+).
c) Синтез трет-бутилового эфира 12-(диэтоксифосфорил)додецилкарбаминовой кислоты.
В 250-мл одногорлую колбу с обратным холодильником помещают навеску трет-бутилового эфира 12-бромдодецилкарбаминовой кислоты (2,39 г, 6,6 ммоль) и добавляют триэтилфосфат (2,25 мл, 13,1 ммоль). Реакционную смесь доводят до 150°С и перемешивают под статическим слоем азота. Через 18 час одногорлую колбу подсоединяют к высоковакуумному насосу для удаления летучих продуктов, а получаемое густое масло помещают непосредственно на силикагель хроматографической колонки и элюируют смесью этилацетат/петролейный эфир, 1:1, выделяя 0,4 г трет-бутилового эфира 12-(диэтоксифосфорил)додецилкарбаминовой кислоты (выход 14%) в виде бесцветного масла.
Данные спектроскопии:
ИК: 3420, 3371, 2922, 2850, 1687, 1218, 1060.
1Н-ЯМР (400 МГц, CDCl3): 1,20-1,45 (м+т, J=7,0 Гц, 35Н), 1,55-1,60 (bm, 2Н), 3,05 (кв, ушир., 2Н), 3,90-4,15 (м, 4Н).
13С-ЯМР (75,3 МГц, CDCl3): 15,6,24,9-29,8 (10CH2+t-Bu), 40,0, 61,2, 65,2, 78,3, 155,6.
Масс-спектр: 421 (М+).
d) Однозамещенный 12-аминододецилфосфонат калия.
В одногорлую колбу с обратным холодильником помещают навеску трет-бутилового эфира 12-(диэтоксифосфорил)додецилкарбаминовой кислоты (0,35 г, 8,3 ммоль) и добавляют концентрированную HCl (1,5 мл). Доводят температуру до 100°С и перемешивают смесь под статическим слоем азота. Через 18 час смесь упаривают с помощью высоковакуумного насоса, получая светло-коричневое резиноподобное твердое вещество.
Данные спектроскопии:
ИК: 3431, 2900, 2841, 1631, 1470, 1172, 1045, 952.
1Н-ЯМР (400 МГц, CDCl3): уширенные сигналы: (1,0-1,80, м), с, ушир., 2,80, с, ушир., 3,40.
13С-ЯМР (100,4 МГц, CDCl3): 23,0-28,8 (перекрывающиеся сигналы), 31,2, 33,4.
Масс-спектр: 265 (М+).
Пример 3
Синтез однозамещенного 12-гидроксидодецилфосфоната калия.

а) Синтез 12-бромдодецилбензоата.
В 250-мл двугорлую колбу под статическим слоем азота помещают навеску 12-бромдодеканола (5,0 г, 18,9 ммоль), добавляют пиридин (25 мл) и доводят смесь до 0°С с помощью внешней бани со льдом и солью. Медленно прибавляют по каплям бензоилхлорид и, после завершения прибавления, удаляют ледяную баню и перемешивают смесь при комнатной температуре. Через 18 час добавляют этилацетат (100 мл) и дистиллированную воду (100 мл). Органическую фазу трижды промывают дистиллированной водой (3×50 мл) и обезвоживают над безводным сульфатом натрия, после чего фильтруют под вакуумом и удаляют растворитель на роторном испарителе Rotavapor и на высоковакуумном насосе. Продукт очищают с помощью колоночной хроматографии на силикагеле, используя в качестве элюента смесь петролейный эфир/этиловый эфир, 5:1. Выделяют 4,5 г 12-бромдодецилбензоата в виде бесцветного масла с выходом 65%.
В альтернативном варианте через 18 час к реакционной смеси добавляют этилацетат (100 мл) и смесь после этого промывают насыщенным водным раствором сульфата меди (3×80 мл) с целью удаления пиридина. В этом случае выход повышается до 90% без колоночной хроматографии и продукт непосредственно используют на последующей стадии.
Данные спектроскопии:
ИК: 2926, 2853, 1716, 1269, 1109.
1Н ЯМР (400 МГц, CDCl3): 1,10-1,60 (м, 16 Н), 1,60-1,80 (м, 4Н), 3,55 (т, J=6,8 Гц, 2Н), 4,25 (т, J=6,8 Гц, 2Н), 7,30-7,35 (м, 3Н), 8,00-8,05 (м, 2Н) м.д.
13С ЯМР (75,3 МГц, CDCl3): 25,8, 26,6, 28,5, 28,6-29,3 (6СН2), 32,4, 44,8, 64,8, 128,0, 129,3, 130,3, 132,5, 166,3.
Масс-спектр: 369 (М+).
b) Синтез 1,2-диэтоксифосфорилбензоата
В одногорлую колбу с обратным холодильником помещают навеску 1,2-бромдодецилбензоата (425 г, 11,5 ммоль) и добавляют триэтилфосфит (4,11 мл, 24 ммоль). Реакционную смесь доводят до 150°С и перемешивают под статическим слоем азота. Через 24 часа одногорлую колбу подсоединяют к высоковакуумному насосу для удаления летучих продуктов, а получаемое густое масло помещают непосредственно на силикагель хроматографической колонки и элюируют смесью этилацетат/петролейный эфир, 1:1, выделяя 4,0 г (выход 82%) 1,2-диэтоксифосфорилбензоата в виде бесцветного масла.
Данные спектроскопии:
ИК: 3663, 3425, 2927, 2844, 1721, 1218, 1064.
1Н-ЯМР (400 МГц, CDCl3): 1,30 (т, J=7,0 Гц, 6Н), 1,40-1,80 (м, 22Н), 3,95-4,05 (м, 4Н), 4,25 (т, J=6,0 Гц, 2Н), 7,40-7,65 (м, 3Н), 8,00-8,05 (м, 2Н) м.д.
13С-ЯМР (75.3 МГц, CDCl3): 16,0, 22,6, 24,2-34,1 (10CH2), 61,0, 65,3, 128,2, 129,4, 131,4, 167,1.
Масс-спектр: 426 (М+).
c) Синтез однозамещенного 1,2-гидроксидодецилфосфоната калия.
В одногорлую колбу с обратным холодильником помещают навеску 12-диэтоксифосфорилбензоата (4,0 г, 9,3 ммоль) и добавляют концентрированную HCl (10 мл). Доводят смесь до температуры 100°С и перемешивают ее под статическим слоем азота. Через 72 часа добавляют этилацетат (80 мл) и дистиллированную воду (40 мл). Производят разделение в делительной воронке и экстрагируют воду еще три раза этилацетатом (3×50 мл). Объединенные органические фазы промывают насыщенным раствором NaCl, сушат безводным сульфатом натрия и упаривают на роторном испарителе Rotavapor и на высоковакуумном насосе. Проводят колоночную хроматографию на силикагеле, используя в качестве элюента смесь петролейный эфир/этилацетат, 1:1. Вначале выделяют побочно образовавшуюся бензойную кислоту и затем, при замене элюента на чистый метанол, выделяют в качестве продукта 12-бензилоксидодецилфосфорную кислоту. Продолжая хроматографию на колонке, выделяют также продукт полного гидролиза - 12-гидроксидодецилфосфорную кислоту. Последние два продукта (примерно 2,0 г) хранят вместе и используют на следующей стадии.
Два выделенных продукта помещают в одногорлую колбу с обратным холодильником и добавляют метанол (50 мл), дистиллированную воду (20 мл) и карбонат калия (13 ммоль, 1,8 г). Смесь доводят до 50°С и перемешивают 18 час под статическим слоем азота. Метанол удаляют на роторном испарителе Rotavapor и трижды экстрагируют остаток этиловым эфиром (3×20 мл) с целью удаления образовавшегося побочно метилбензоата. К водному раствору добавляют 10%-ную HCl до кислого рН. После выпадения в осадок твердого белого вещества воду удаляют на роторном испарителе Rotavapor и на высоковакуумном насосе. Полученное твердое вещество растворяют в метаноле и декантируют, удаляя тем самым хлорид калия.
Т.пл. 270-279°С.
Данные спектроскопии:
ИК: 3357, 2917, 2850, 1467, 1233, 1162, 1010, 936.
1Н-ЯМР (400 МГц, D2O): 1,10-1 90 (м, 22Н), 3,40 (с, ушир., 2Н).
13С-ЯМР (75,3 МГц, D2O): 24,5, 25,3, 29,0-29,3 (7СН2), 30,5, 31,7, 61,9.
Масс-спектр: 266 (М+).
Полученную таким образом фосфорную кислоту обрабатывают эквимолярным количеством КОН и нагревают в метаноле с целью получения соответствующей калиевой соли. Получают (из 12-диэтоксифосфорилбензоата) 1,3 г калиевой соли 12-гидроксидодецилфосфоната, выход 57%, в виде порошкообразного белого твердого вещества.
Т.пл. 336-348°С.
Данные спектроскопии:
ИК: 3308, 2918, 2851, 2364, 1651, 1553, 1399, 1082, 977, 831.
1Н-ЯМР (400 МГц, CD3OD): 1,20-1,85 (м, 22Н), 3,50 (т, J=6,8 Гц, 2Н).
13С-ЯМР (75,3 МГц, CD3OD): 22,9, 25,7, 29,1-29,5 (7СН2), 30,7 (д, 1=12 Гц), 61,8.
Масс-спектр: 265 (М-), 39 (К+).
Пример 4.
Синтез однозамещенного 13-этокси-13-оксатридецилфосфоната калия.

а) Синтез этил-12-гидроксидодеканоата.
В 100-мл двугорлую колбу с обратным холодильником и магнитным перемешивателем в статическом токе азота помещают навеску 12-гидроксидодекановой кислоты (5,0 г, 23,2 ммоль) и добавляют этанол (20 мл) и ацетилхлорид (1,62 ммоль, 0,09 мл, 0,1 экв.). Смесь перемешивают 24 часа при кипячении, упаривают на роторном испарителе Rotavapor и на высоковакуумном насосе и очищают продукт с помощью колоночной хроматографии на силикагеле, используя в качестве элюента смесь петролейный эфир/этилацетат, 5:4. Выделяют 3,30 г этил-12-гидроксидодеканоата в виде светло-желтоватого масла с выходом 96%.
Данные спектроскопии:
ИК:. 3662, 2926, 2853, 1731.
1Н-ЯМР (400 МГц, CDCl3): 1,05-1,25 (м, 17Н), 1,40-1,60 (м, 4Н), 2,17 (т, J=7,2 Гц, 2Н), 2,34 (с, 1Н), 3,49 (т, J=6,8 Гц, 2Н), 4,01 (1, J=7,2 Гц, 2Н).
13С-ЯМР (75,3 МГц, CDCl3): 14,0, 24,7, 25,6, 28,9, 29,0, 29,2, 29,2, 29,3, 29,4, 32,6, 34,2, 59,98, 62,6,173,8.
Масс-спектр: 234 (М+).
b) Синтез этил-12-бромдодеканоата.
В 100-мл двухгорлой колбе с обратным холодильником и магнитным перемешивателем под статическим слоем азота растворяют этил-12-гидроксидодеканоат (1,65 г, 6,7 ммоль) в дихлорметане (20 мл) и добавляют PPh3 (1,93 г, 7,4 ммоль) и NBS (N-бромсукцинимид) (1,6 г, 7,0 ммоль). Смесь перемешивают при кипячении 24 часа, упаривают на роторном испарителе Rotavapor и продукт очищают с помощью колоночной хроматографии на силикагеле, используя в качестве элюента смесь петролейный эфир/этилацетат, 5:1. Выделяют 1,92 г (выход 92%) этил-12-бромдодеканоата в виде светло-желтоватого масла.
Данные спектроскопии:
ИК: 2926, 2853, 1731.
1Н-ЯМР (400 МГц, CDCl3): 1,20-1,45 (м, 15Н), 1,55-1,65 (м, 4Н), 1,80-1,90 (м, 2Н), 2,30 (т, J=7,0 Гц, 2Н), 3,40 (т, J=7,l Гц, 2Н), 4,10 (1, J=7,2 Гц, 2Н).
13С-ЯМР (75,3 МГц, CDCl3): 14,2, 24,9, 28,1, 28,7, 29,1, 29,3 (2СН2), 29,4, 32,8, 33,9, 34,3, 61,1, 173,8.
Масс-спектр: 296 (М+).
c) Синтез этилового эфира 13-(диэтоксифосфорил)тридекановой кислоты.
В одногорлую колбу с обратным холодильником помещают навеску этил-12-бромдодеканоата (1,8 г, 7,37 ммоль) и добавляют триэтилфосфит (2,6 мл, 15 ммоль). Реакционную смесь доводят до 150°С и перемешивают под статическим слоем азота. Через 24 часа подсоединяют идущий от горла колбы шланг к высоковакуумному насосу с целью удаления летучих продуктов, а полученное в результате этого густое масло непосредственно хроматографируют на колонке с силикагелем, используя в качестве элюента смесь этилацетат/петролейный эфир, 1:1, и выделяют 2,5 г (выход 94%) этилового эфира 13-(диэтоксифосфорил)тридекановой кислоты в виде бесцветного масла.
Данные спектроскопии:
ИК: 3684, 3445, 2978, 2853, 1730, 1216, 1058.
1Н-ЯМР (400 МГц, CDCl3): 1,05-1,15 (м, 25Н), 1,40-1,80 (м, 4Н), 2,0-2,1 (м, 2Н), 4,00 (с, ушир., 6Н).
13С-ЯМР (75,3 МГц, CDCl3): 14,0, 15,9, 16,2 (д, J=5,6 Гц), 22,1, 22,2, 24,7, 26,2, 26,8, 29,0, 29,1, 29,2, 30,3 (д, J=16,1 Гц), 34,1, 59,9, 61,11 (д, J=6,4 Гц), 63,7 (д, J=5,6 Гц), 173,6.
Масс-спектр: 364 (М+).
d) Синтез однозамещенного 13-этокси-13-оксатридецилфосфоната.
В одногорлую колбу с обратным холодильником помещают навеску этилового эфира 13-(диэтоксифосфорил)тридекановой кислоты (1,3 г, 3,6 ммоль) и добавляют концентрированную HCl (2 мл). Температуру смеси доводят до 100°С и перемешивают смесь под статическим слоем азота. Через 6 дней смесь упаривают на высоковакуумном насосе, получая липкое белое твердое вещество. Анализ 1Н-ЯМР показывает все еще присутствующую сложноэфирную группу. Добавляют КОН (460 мг в 20 мл смеси вода/метанол, 1:1) и вновь полученную смесь перемешивают в течение ночи при комнатной температуре. Утром смесь высушивают и экстрагируют этилацетатом возможные органические примеси. Водную фазу упаривают, добавляют к полученному липкому белому твердому веществу 10 мл метанола и кипятят смесь с обратным холодильником в течение 5 мин. Раствор отделяют с помощью пипетки, а твердый белый остаток сушат в высоком вакууме и характеризуют с помощью спектроскопии. Получают 800 мг (выход 62%)) продукта в виде порошкообразного белого твердого вещества.
Т.пл. 350-360°С.
Данные спектроскопии:
ИК: 3411 (ушир.): 2922, 2848, 1649, 1566, 1410, 1041, 977.
1Н-ЯМР (400 МГц, D2O): 1,00-1,40 (м, 20Н), 2,0 (т, J=7,6 Гц, 2Н).
13С-ЯМР (100,3 МГц, D2O): 23,5, 24,4, 26,1, 28,7, 28,9, 31,3, 37,87 (только различимые сигналы для СН2).
Масс-спектр (m/z): 278/2=139 (М+).
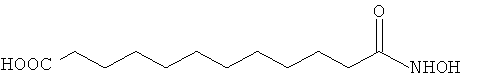
Следуя тому же протоколу синтеза, можно получить 12-гидроксиламино-12-оксододекановую кислоту.
Комплексы наночастица/бифункциональное связующее.
Пример 5.
Синтез этилового эфира 12-гидроксиламино-12-оксододекановой кислоты.
Синтез этил-12-гидроксидодеканоата.
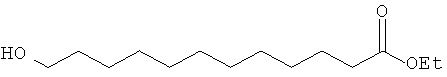
В двугорлую колбу с обратным холодильником вводят при перемешивании в токе азота 12-гидроксидодекановую кислоту (5,0 г, 23,2 ммоль), этанол (20 ммоль) и ацетилхлорид (0,9 мл, 1,62 ммоль) и кипятят смесь в течение 24 час. Затем раствор упаривают на роторном испарителе в высоком вакууме и очищают сырой продукт с помощью колоночной хроматографии на силикагеле, используя в качестве элюента смесь петролейный эфир/этилацетат, 5:4. Выделяют 5,45 г (выход 96%) желаемого продукта в виде бледного желтого масла.
Данные спектроскопии:
Синтез 12-этокси-12-оксододекановой кислоты.

В одногорлой колбе, снабженной перфорированной мембраной, к перйодной кислоте (5,13 г, 22,5 ммоль) добавляют в токе аргона при перемешивании ацетонитрил (80 мл) и через 15 мин доводят температуру до 0°С. В этих условиях в колбу прибавляют по каплям раствор этил-12-гидроксидодеканоата (5; 2,5 г, 10,2 ммоль) и пиридин-хлорохромат (РСС, 44 мг, 0,20 ммоль) в ацетонитриле (20 мл). После добавления реакцию ведут в течение 24 час при комнатной температуре. Реакцию останавливают добавлением этилацетата (100 мл). Реакционный раствор промывают раствором (1:1) дистиллированная вода/рассол (2×50 мл), насыщенным водным раствором бисульфита натрия (NaHSO3; 2×25 мл) рассолом (2×50 мл). Органическую фазу обезвоживают сульфатом натрия и фильтруют под вакуумом. Растворитель отгоняют и продукт сушат в высоком вакууме, получая 2,45 г белого твердого вещества. Продукт очищают с помощью колоночной хроматографии с использованием силикагеля, в качестве элюента используют смесь этилацетат/петролейный эфир, 3:1. Получают 2,1 г (выход 80%) целевого продукта в виде белого твердого вещества. Реакцию проводят согласно Hunsen, М. Synthesis, 2005, 2487-2490.
Данные спектроскопии:
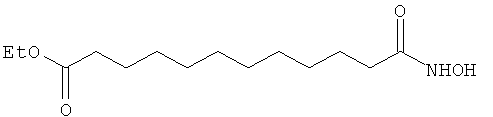
Синтез этилового эфира 12-гидроксиламино-12-оксододекановой кислоты.
В одногорлой колбе, оборудованной обратным холодильником, растворяют при перемешивании в токе аргона 12-этокси-12-оксододекановую кислоту (13; 1,5 г, 5,8 ммоль) в хлороформе (20 мл). Прибавляют по каплям тионилхлорид (SOCl2; 0,64 мл, 8,8 ммоль) и проводят реакцию при кипячении в течение 3 час. Затем смесь охлаждают до комнатной температуры и удаляют растворитель при высоком вакууме. Полученный продукт растворяют в дихлорметане (20 мл), смешивают при комнатной температуре и при перемешивании с раствором гидроксиламина-гидрохлорида (0,61 г, 8,8 ммоль) в пиридине (10 мл) и оставляют в тех же условия реагировать на 12 час. Весь растворитель удаляют в высоком вакууме, оставшийся продукт растворяют в этилацетате (50 мл) и промывают дистиллированной водой (3×20 мл). Органическую фазу обезвоживают безводным сульфатом натрия и фильтруют под вакуумом. После отгонки растворителя и высушивания при высоком вакууме получают 1,3 г (выход 82%) продукта в виде желтого твердого вещества.
Данные спектроскопии:
Пример 6.
Комплексы наночастиц феррит кобальта/12-гидроксидодецилфосфоновая кислота (диаграмма продукта 1.2).
10 г дисперсии в диэтиленгликоле, содержащей 3 вес.% наночастиц, например, феррита кобальта с диаметром 5 нм, добавляют к 0,3 г 12-гидроксидодецилфосфоновой кислоты после ее растворения в 20 г слегка нагретого этанола. Смесь перемешивают 2 часа при комнатной температуре. После этого осаждают ацетоном образец, центрифугируют и отделяют. Образец повторно диспергируют в этаноле и вновь осаждают, центрифугируют и отделяют с целью удаления примесей. Влажный образец может после этого быть вновь диспергированным в желаемом растворителе.
Пример 7.
Комплексы наночастиц феррит кобальта/12-амино-N-гидроксидодеканамид (диаграмма продукта 1.2).
10 г дисперсии в диэтиленгликоле, содержащей 3 вес.% наночастиц, например, феррита кобальта с диаметром 5 нм, добавляют к 0,21 г 12-амино-N-гидроксидодеканамида после его растворения в 20 г кипящей воды, смесь перемешивают 2 часа при комнатной температуре. Затем образец осаждают ацетоном, центрифугируют и отделяют. Образец повторно диспергируют в этаноле и вновь осаждают, центрифугируют и отделяют с целью удаления примесей. Влажный образец может после этого быть вновь диспергированным в желаемом растворителе.
Комплексы неорганических наночастиц полимер/функциональная молекула.
Пример 8.
Синтез функционализированных соединений наночастиц с полиамидоамином (РАА), состоящим из этилендиаминодиуксусной кислоты-бисакрилоилпиперазина - диаграмма продукта 1.2.1.
10 г водной дисперсии, содержащей 0,1 вес.% наночастиц, например, феррита кобальта с диаметром 5 нм, функционализированных гидроксамовой 12-аминододекановой кислотой, добавляют к 10 г раствора, содержащего 0,02 г полимера. Значение рН доводят до 8 добавлением нескольких капель триэтиленамина. Раствор перемешивают 2 дня в темноте при 25°С. Полученный продукт затем фильтруют с использованием фильтрационной системы Amicon для удаления непрореагировавшего полимера. После этого продукт может быть оставлен в растворе или высушен для проведения анализа с целью его характеристики.
Пример 9.
Синтез соединения функционализированная наночастица/циклодекстрин.
a) Последовательность операций для прямой фиксации циклодекстрина на «привитом» продукте (диаграмма продукта 1.2.1).
10 г дисперсии в диэтиленгликоле, содержащей 0,1 вес.% наночастиц, например, феррита кобальта с диаметром 5 нм, добавляют к этанольному раствору, содержащему 0,21 г гидроксамовой 12-гидроксидодекановой кислоты после ее растворения в 20 г слегка нагретого этанола. Смесь перемешивают 1 час при 60°С. После этого осаждают ацетоном образец, центрифугируют и отделяют. Полученное твердое вещество повторно диспергируют в этаноле и вновь осаждают, центрифугируют и отделяют с целью удаления примесей. Влажный образец может после этого быть вновь диспергированным в ДМФ (15 мл). Добавляют дициклогексилкарбодиимид (DCC, 2 г) и 4-диметиламинопиридин (DMAP, 0,2 г) и охлаждают полученную смесь до 0°С. α-Циклодекстринкарбоновую кислоту (6-дезокси-6-карбокси-α-циклодекстрин, 1 г) суспендируют в ДМФ (25 мл). Суспензию охлаждают до 0°С и медленно прибавляют к реакционной смеси. Перемешивают смесь 48 час при комнатной температуре. Раствор вливают в ацетон (100 мл), отделяют образовавшийся осадок и сушат его в высоком вакууме. Сырой продукт может быть дополнительно очищен на Sephadex СМ-25.
b) Последовательность операций для прямой фиксации циклодекстрина на функциональном связующем с последующей прививкой на феррите кобальта (диаграмма продукта 1.4).
К раствору 6-дезокси-6-карбокси-α-декстрина (1 г, 0,87 ммоль) в H2O/EtOH, 1:1 (20 мл) добавляют DCC (197 мг, 0,96 ммоль), DMAP (12 мг, 0,087 ммоль, 10% каталитическое количество) и гидроксамовую 12-гидроксидодекановую кислоту (0,2 г, 0,87 ммоль). Реакционную смесь перемешивают 72 часа при комнатной температуре. Сырой продукт очищают на Sephadex СМ-25, получая 360 мг (30%) циклодекстрина, связанного с гидроксамовой 12-гидроксидодекановой кислотой.
200 мг полученного продукта растворяют в 20 мл 96%-ного этанола и добавляют к 10 мл дисперсии в диэтиленгликоле, содержащей 0,1 вес.% наночастиц феррита кобальта с диаметром 5 нм. Смесь перемешивают 2 часа при комнатной температуре. После этого осаждают ацетоном образец, центрифугируют и отделяют. Образец повторно диспергируют в этаноле и вновь осаждают, центрифугируют и отделяют с целью удаления примесей. Образец может быть затем вновь диспергирован в желаемом растворителе.
| название | год | авторы | номер документа |
|---|---|---|---|
| СОЕДИНЕНИЯ, ОКАЗЫВАЮЩИЕ ВОЗБУЖДАЮЩЕЕ ДЕЙСТВИЕ НА РЕЦЕПТОР АКТИВАТОРА ПРОЛИФЕРАЦИИ ПЕРОКСИСОМ ПОДТИПА Б, СПОСОБ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ УКАЗАННЫХ СОЕДИНЕНИЙ | 2009 |
|
RU2522450C2 |
| СПОСОБ СИНТЕЗА ФТАЛАСЦИДИНА И ПРОМЕЖУТОЧНЫХ ПРОДУКТОВ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ | 2001 |
|
RU2267492C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПАКЛИТАКСЕЛА | 2001 |
|
RU2276147C2 |
| СОЕДИНЕНИЯ С ГЕТЕРОАРОМАТИЧЕСКИМИ КОЛЬЦАМИ В КАЧЕСТВЕ ИНГИБИТОРОВ RET-КИНАЗ И ИХ ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ | 2021 |
|
RU2829729C1 |
| СПОСОБЫ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ БИЦИКЛО [3.1.0]ГЕКСАНА И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДЛЯ ЭТОЙ ЦЕЛИ | 2004 |
|
RU2388747C2 |
| Соединение диарилтиогидантоина в качестве антагониста андрогенового рецептора | 2018 |
|
RU2804108C9 |
| ИНГИБИТОР НЕКРОЗА КЛЕТОК, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2019 |
|
RU2793918C2 |
| 2,3,5-ТРИЗАМЕЩЕННЫЕ ПИРРОЛЫ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ЛЕКАРСТВЕННОЕ СРЕДСТВО НА ИХ ОСНОВЕ, СПОСОБ ПОЛУЧЕНИЯ СОЕДИНЕНИЙ (ВАРИАНТЫ) | 2010 |
|
RU2549885C2 |
| НОВЫЙ СПОСОБ СИНТЕЗА АГОМЕЛАТИНА | 2014 |
|
RU2683279C1 |
| УСОВЕРШЕНСТВОВАННЫЕ КОНЪЮГАТЫ N4 ХЕЛАТООБРАЗУЮЩИХ АГЕНТОВ | 2005 |
|
RU2360701C2 |



Изобретение относится к устойчивым комплексам, состоящим из оксидов металлов, железа, кобальта или их сплавов в форме наночастиц и бифункциональных соединений, где бифункциональные соединения выбирают из тиолов, карбоновых кислот, гидроксамовых кислот, эфиров фосфорных кислот или их солей, имеющих алифатическую цепочку, содержащую вторую функциональную группу в конечном положении ω, которые могут использоваться в некоторых новых гидрофильных пластиках и волокнах, а также к способу получения указанных комплексов. Способ состоит в том, что дисперсию указанных наночастиц вводят в реакцию в органическом растворителе с подходящим связующим, перемешивают смесь в течение нескольких часов при низкой температуре и затем осаждают полученный продукт, который затем отделяют центрифугированием и который может быть очищен путем повторного диспергирования в подходящем растворителе и повторного осаждения. Получены новые комплексы с улучшенной растворимостью в водно-спиртовой среде. 3 н. и 10 з.п. ф-лы, 3 ил.
1. Устойчивые комплексы, состоящие из оксидов металлов, железа, кобальта или их сплавов в форме наночастиц и бифункциональных соединений, где бифункциональные соединения выбирают из группы, состоящей из: тиолов, карбоновых кислот, гидроксамовых кислот, эфиров фосфорных кислот или их солей, имеющих алифатическую цепочку, содержащую вторую функциональную группу в конечном положении ω.
2. Комплексы по п.1, где указанные оксиды металлов в форме наночастиц являются соединениями формулы:
MIIMIII 2O4,
где MII=Со, Ni, FeII, Zn, Mn и
MIII=FeIII, Со, Al.
3. Комплексы по п.2, где указанные оксиды являются оксидами Fe2O3 типа магхемита.
4. Комплексы по п.3, где указанные оксиды выбирают из группы, состоящей из: феррита кобальта CoFe2O4, магнетита FeFe2O4, магхемита Fe2O3.
5. Комплексы по п.4, где указанную вторую функциональную группу выбирают из группы, состоящей из: ОН, NH2, СООН, COOR3, где R3 обозначает щелочной металл или защитную органическую группу.
6. Комплексы по п.5, где указанные бифункциональные соединения имеют общую формулу (II):
R1-(CH2)n-R2,
в которой: n обозначает целое число от 2 до 20,
R1 выбирают из группы, состоящей из ОН, NH2, СООН,
R2 выбирают из: CONHOH, РО(ОН)2, РО(ОН)(OR3), СООН, SH,
R3 обозначает щелочной металл или защитную органическую группу.
7. Комплексы по п.6, где указанный щелочной металл выбирают из группы, состоящей из K, Na или Li.
8. Комплексы по пп.1-7, состоящие из:
наночастица феррит кобальта/12-гидроксидодецилфосфоновая кислота;
наночастица феррит кобальта/12-амино-N-гидроксидодеканамид;
наночастица/функционализированная полиамидоамином (РАА), образуемым этилендиаминодиуксусной кислотой-бисакрилоилпиперазином.
9. Способ приготовления комплексов по пп.1-8, в котором дисперсию указанных наночастиц вводят в реакцию в органическом растворителе с подходящим связующим, перемешивают смесь в течение нескольких часов при низкой температуре и затем осаждают полученный продукт, который затем отделяют центрифугированием и который может быть очищен путем повторного диспергирования в подходящем растворителе и повторного осаждения.
10. Соединения, состоящие из комплексов по пп.1-8, имеющие бифункциональное производное, где внешняя функциональная группа указанного бифункционального производного соединена с молекулами, белками или полимерами.
11. Соединения по п.10, в которых указанные молекулы выбирают из: циклодекстринов, фолиевой кислоты, антител, полиамидоамина.
12. Соединение по п.11, состоящее из соединения феррита кобальта/12-гидроксидодецилфосфоновой кислоты и карбоксиметилированного циклодекстрина.
13. Соединение по п.11, состоящее из феррита кобальта/кислоты феррита кобальта/12-амино-N-гидроксидодеканамида и карбоксиметилированного циклодекстрина.
| С.YEE ЕТ AL | |||
| «Self-assembled monolayers of alkanesulfonic and phosphonic acids on amorphous iron oxide nanoparticles», Langmuir, 1999, 15, 7111-7115 | |||
| G | |||
| KATABY ET AL | |||
| «Coating a bola-amphiphile on amorphous iron nanoparticles», J.Mater, chem., 1999, 9, 1501-1506 | |||
| O.ROZENFELD ET AL | |||
| «Self-assembled monolayer coatings on amorphous |

