Область техники, к которой относится изобретение
Настоящее изобретение относится к новому триазолопиридиновому соединению, обладающему ингибирующим действием в отношении пролилгидроксилазы (здесь и далее в этом документе также называемой «PHD») и способностью индуцировать выработку эритропоэтина (здесь и далее в этом документе также называемого «EPO»). Настоящее изобретение также относится к ингибитору пролилгидроксилазы (здесь и далее в этом документе также называемому «ингибитором PHD») и к агенту, индуцирующему выработку эритропоэтина (здесь и далее в этом документе также называемому «агентом, индуцирующим выработку EPO»).
Уровень техники
EPO представляет собой состоящий из 165 аминокислот гормон, способствующий росту красных клеток крови. EPO в основном вырабатывается в почках и частично в печени, и его выработка увеличивается в условиях сниженного содержания кислорода.
Анемия относится к состоянию, характеризующемуся низким содержанием красных клеток крови и гемоглобина в крови. Ее симптомы проистекают из кислородного голодания вследствие сниженного числа красных клеток крови или изменений в динамике кровотока вследствие увеличенной частоты дыхания и частоты сердцебиения для компенсации кислородного голодания и включают «общее ощущение слабости», «быструю утомляемость», «одышку», «учащенное сердцебиение», «тяжесть в голове», «головокружение», «плохой цвет лица», «скованность в плечах», «трудность в пробуждении утром» и т.п.
Причину анемии, как правило, разделяют на низкую выработку, повышенное разрушение и повышенную потерю красных клеток крови, и анемия включает анемию вследствие нарушения кроветворения в костном мозге, анемию вследствие дефицита железа, витамина B12 или фолиевой кислоты, кровотечения во время несчастного случая или операции, анемию, ассоциированную с хроническим воспалением (аутоиммунные заболевания, злокачественная опухоль, хронические трансмиссивные заболевания, плазмоклеточная дискразия и т.д.), анемию, ассоциированную с эндокринными заболеваниями (гипотиреоз, аутоиммунный полигландулярный синдром, сахарный диабет типа IA, дисфункциональное маточное кровотечение и т.д.), анемию, ассоциированную с хронической сердечной недостаточностью, анемию, ассоциированную с язвой, анемию, ассоциированную с заболеваниями печени, старческую анемию, лекарственную анемию, почечную анемию (анемию, ассоциированную с почечной недостаточностью), анемию, ассоциированную с химиотерапией, и т.п.
В 1989 году препарат рекомбинантного EPO человека был одобрен Управлением по контролю качества продовольствия и медикаментов США (FDA) для применения при почечной анемии, анемии, ассоциированной с AZT-терапией у пациентов с ВИЧ, анемии, ассоциированной с химиотерапией у пациентов со злокачественной опухолью, или для снижения объема переливаемой крови для пациентов, перенесших операцию. Более того, его применение распространялось на анемию недоношенных и т.п.
Почечную анемию лечат стимуляторами эритропоэза (ESA). Почечная анемия, главным образом, вызвана снижением выработки EPO в интерстициальных клетках на периферии почечных канальцев почки. Существует применение, при котором рекомбинантный эритропоэтин человека весьма часто используется в качестве дополнения к EPO. Рекомбинантный эритропоэтин человека разительно уменьшил число пациентов, нуждающихся в периодическом переливании крови, улучшил различные симптомы, ассоциированные с анемией, и внес значительный вклад в улучшение ADL (повседневной активности) и QOL (качества жизни). С другой стороны, будучи биологическим препаратом, он дорог и требует высоких медицинских затрат. Кроме того, он обладает коротким периодом полувыведения из крови и требует внутривенного введения 2-3-раза в неделю через диализный контур у пациентов на гемодиализе. Таким образом, уменьшение частоты введения желательно для предупреждения медицинских осложнений, а также с точки зрения объема врачебной практики и медицинских отходов. Более того, для пациентов на перитонеальном диализе и пациентов с почечной недостаточностью в предиализный период, для которых использовалось подкожное введение, предоставляющее более продолжительный период действия, все же необходимо однократное введение в неделю или две недели. В этом случае пациентам часто требуется посещение больницы только для введения рекомбинантного эритропоэтина человека, что создает нагрузку на пациентов.
Более того, путем модификации EPO добавлением новой цепи сахара или цепи PEG был разработан лекарственный препарат EPO длительного действия, обладающий пролонгированным периодом полувыведения из крови при внутривенной инъекции или подкожной инъекции. Тем не менее, поскольку были разработаны лишь инъецируемые препараты, для предупреждения медицинских осложнений и уменьшения нагрузки на пациентов желательна разработка перорально вводимого ESA.
Более того, предполагается, что перорально вводимый ESA будет применим для более широкого диапазона видов терапии не только почечной анемии, но также анемии, вызванной различными причинами.
В качестве типичной молекулы, способствующей транскрипции EPO, может быть упомянут индуцируемый гипоксией фактор (здесь и далее в этом документе также называемый «HIF»). HIF представляет собой белок, состоящий из гетеродимера, содержащего регулируемую кислородом α-субъединицу и неизменно экспрессируемую β-субъединицу, где пролин в α-субъединице гидроксилируется пролилгидроксилазой (PHD) в присутствии кислорода, и полученная в результате α-субъединица связывается с белком фон Хиппеля-Линдау (VHL) и убиквитинируется. Однако поскольку в условиях сниженного содержания кислорода α-субъединица не подвергается гидроксилированию PHD, она не убиквитинируется, а связывается с внутриядерным гипоксия-респонсивным элементом (HRE) для промотирования транскрипции EPO содержащегося ниже гена HIF. Поэтому ингибирование активности PHD приводит к предотвращению убиквитинации HIF и его стабилизации. Следовательно, усиливается выработка EPO.
Примеры заболеваний, которые, как предполагается, подлежат улучшению путем ингибирования PHD для стабилизации HIF, включают ишемические болезни сердца (стенокардия, инфаркт миокарда и т.д.), ишемические цереброваскулярные нарушения (инсульт, церебральная эмболия, преходящие церебральные ишемические атаки и т.д.), виды хронической почечной недостаточности (ишемическая нефропатия, тубуло-интерстициальные нарушения почек и т.д.), диабетические осложнения (диабетические язвы и т.д.), когнитивные нарушения (деменция, болезнь Альцгеймера, болезнь Паркинсона, болезнь Гентингтона и т.д.) и т.п.
КРАТКОЕ ОПИСАНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Задачи, подлежащие решению настоящим изобретением
Из результатов, полученных в ранее проведенных исследованиях, было выяснено, что лекарственный препарат, ингибирующий пролилгидроксилазу (PHD), способствует выработке эритропоэтина (EPO) и является эффективным для профилактики или лечения различных заболеваний и патологий (нарушений), вызванных сниженной выработкой EPO, в частности, для лечения анемии.
Соответственно, настоящее изобретение направлено на обеспечение лекарственным препаратом, обладающим ингибирующим действием в отношении пролилгидроксилазы (PHD). Кроме того, настоящее изобретение направлено на обеспечение лекарственным препаратом, обладающим способностью индуцировать выработку EPO.
Средства решения задач
Авторы настоящего изобретения обнаружили соединение, обладающее ингибирующим действием в отношении пролилгидроксилазы (PHD) и способностью индуцировать выработку EPO, и оформили настоящее изобретение.
Более конкретно настоящее изобретение относится к следующему.
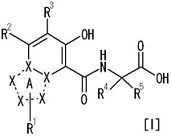
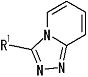
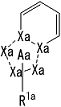
[1] Соединение, представленное следующей формулой [I] (в дальнейшем также называемое «соединением по настоящему изобретению»), или его фармацевтически приемлемая соль, или его сольват:
 ,
,
в котором
частичная структурная формула:
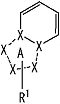
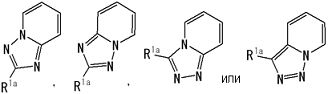
представляет собой группу, представленную любой из следующих формул:
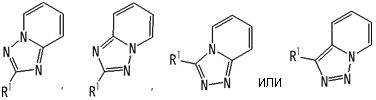 ;
;
R1 представляет собой
(1) атом водорода,
(2) C1-6алкильную группу,
(3) C6-14арильную группу,
(4) C3-8циклоалкильную группу,
(5) C6-14арил-C1-6алкильную группу или
(6) C3-8 циклоалкил-C1-6алкильную группу;
R2 представляет собой
(1) атом водорода,
(2) C1-10алкильную группу,
(3) C6-14арильную группу, необязательно замещенную одинаковыми или различными 1-5 заместителями, выбранными из следующей группы B,
(4) C3-8циклоалкильную группу, необязательно замещенную одинаковыми или различными 1-5 заместителями, выбранными из следующей группы B,
(5) C3-8циклоалкенильную группу, необязательно замещенную одинаковыми или различными 1-5 заместителями, выбранными из следующей группы B,
(6) гетероарильную группу, необязательно замещенную одинаковыми или различными 1-5 заместителями, выбранными из следующей группы B (в которой гетероарил содержит, кроме атома углерода, 1-6 гетероатомов, выбранных из атома азота, атома кислорода и атома серы),
(7) C6-14арил-C1-6алкильную группу (в которой C6-14арил необязательно замещен одинаковыми или различными 1-5 заместителями, выбранными из следующей группы B) или
(8) C3-8циклоалкил-C1-6алкильную группу (в которой C3-8циклоалкил необязательно замещен одинаковыми или различными 1-5 заместителями, выбранными из следующей группы B);
R3 представляет собой
(1) атом водорода,
(2) атом галогена,
(3) C1-6алкильную группу,
(4) C6-14арильную группу,
(5) C3-8циклоалкильную группу или
(6) C6-14арил-C1-6алкильную группу; и
каждый из R4 и R5 независимо представляет собой
(1) атом водорода или
(2) C1-6алкильную группу,
группа B:
(a) атом галогена,
(b) C1-6алкильная группа,
(c) C3-8циклоалкильная группа,
(d) цианогруппа и
(e) галоген-C1-6алкильная группа.
[2] Соединение, описанное в вышеупомянутом пункте [1], в котором частичная структурная формула:
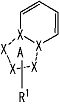
представляет собой группу, представленную следующей формулой
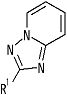 ,
,
или его фармацевтически приемлемая соль, или его сольват.
[3] Соединение, описанное в вышеупомянутом пункте [1], в котором частичная структурная формула:
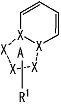
представляет собой группу, представленную следующей формулой
 ,
,
или его фармацевтически приемлемая соль, или его сольват.
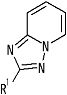
[4] Соединение, описанное в вышеупомянутом пункте [1], в котором частичная структурная формула:
представляет собой группу, представленную следующей формулой
 ,
,
или его фармацевтически приемлемая соль, или его сольват.
[5] Соединение, описанное в вышеупомянутом пункте [1], в котором частичная структурная формула:
представляет собой группу, представленную следующей формулой
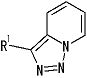 ,
,
или его фармацевтически приемлемая соль, или его сольват.
[6] Соединение, описанное в любом из вышеупомянутых пунктов [1]-[5], в котором R4 и R5, оба, представляют собой атомы водорода, или его фармацевтически приемлемая соль, или его сольват.
[7] Соединение, описанное в любом из вышеупомянутых пунктов [1]-[5], в котором R3 представляет собой атом водорода, или его фармацевтически приемлемая соль, или его сольват.
[8] Соединение, описанное в любом из вышеупомянутых пунктов [1]-[5], в котором R1 представляет собой атом водорода, или его фармацевтически приемлемая соль, или его сольват.
[9] Соединение, описанное в любом из вышеупомянутых пунктов [1]-[5], в котором R2 представляет собой
(1) C1-10алкильную группу,
(2) C6-14арильную группу, необязательно замещенную одинаковыми или различными 1-5 заместителями, выбранными из вышеупомянутой группы B,
(3) C6-14арил-C1-6алкильную группу (в которой C6-14арил необязательно замещен одинаковыми или различными 1-5 заместителями, выбранными из вышеупомянутой группы B) или
(4) C3-8циклоалкил-C1-6алкильную группу (в которой C3-8циклоалкил необязательно замещен одинаковыми или различными 1-5 заместителями, выбранными из вышеупомянутой группы B),
или его фармацевтически приемлемая соль, или его сольват.
[10] Соединение, описанное в вышеупомянутом пункте [2], в котором R4 и R5, оба, представляют собой атомы водорода, или его фармацевтически приемлемая соль, или его сольват.
[11] Соединение, описанное в вышеупомянутом пункте [10], в котором R3 представляет собой атом водорода, или его фармацевтически приемлемая соль, или его сольват.
[12] Соединение, описанное в вышеупомянутом пункте [11], в котором R1 представляет собой атом водорода, или его фармацевтически приемлемая соль, или его сольват.
[13] Соединение, описанное в вышеупомянутом пункте [12], в котором R2 представляет собой
(1) C1-10алкильную группу или
(2) C6-14арил-C1-6алкильную группу (в которой C6-14арил необязательно замещен одинаковыми или различными 1-5 заместителями, выбранными из вышеупомянутой группы B),
или его фармацевтически приемлемая соль, или его сольват.
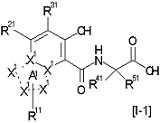
[14] Соединение, представленное следующей формулой [I-1], или его фармацевтически приемлемая соль, или его сольват:
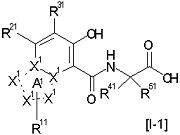
где частичная структурная формула:
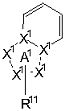
представляет собой группу, представленную любой из следующих формул:
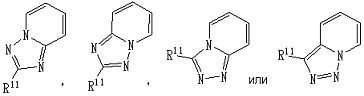 ;
;
R11 представляет собой
(1) атом водорода,
(2) C1-6алкильную группу,
(3) фенильную группу,
(4) C3-8циклоалкильную группу,
(5) фенил-C1-6алкильную группу или
(6) C3-8циклоалкил-C1-6алкильную группу;
R21 представляет собой
(1) атом водорода,
(2) C1-10алкильную группу,
(3) фенильную группу, необязательно замещенную одинаковыми или различными 1-5 заместителями, выбранными из следующей группы B,
(4) C3-8циклоалкильную группу,
(5) C3-8циклоалкенильную группу,
(6) тиенильную группу, необязательно замещенную одинаковыми или различными 1-5 заместителями, выбранными из следующей группы B,
(7) фенил-C1-6алкильную группу (в которой фенил необязательно замещен одинаковыми или различными 1-5 заместителями, выбранными из следующей группы B) или
(8) C3-8циклоалкил-C1-6алкильную группу;
R31 представляет собой
(1) атом водорода,
(2) атом галогена,
(3) C1-6алкильную группу,
(4) фенильную группу,
(5) C3-8циклоалкильную группу или
(6) фенил-C1-6алкильную группу; и
каждый из R41 и R51 независимо представляет собой
(1) атом водорода или
(2) C1-6алкильную группу;
группа B:
(a) атом галогена,
(b) C1-6алкильная группа,
(c) C3-8циклоалкильная группа,
(d) цианогруппа и
(e) галоген-C1-6алкильная группа.
[15] Соединение, представленное следующей формулой:
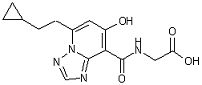 ,
,
или его фармацевтически приемлемая соль, или его сольват.
[16] Соединение, представленное следующей формулой:
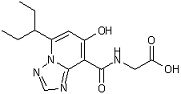 ,
,
или его фармацевтически приемлемая соль, или его сольват.
[17] Соединение, представленное следующей формулой:
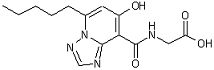 ,
,
или его фармацевтически приемлемая соль, или его сольват.
[18] Соединение, представленное следующей формулой:
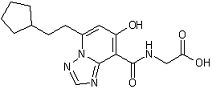 ,
,
или его фармацевтически приемлемая соль, или его сольват.
[19] Соединение, представленное следующей формулой:
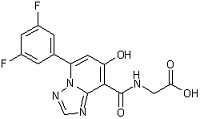 ,
,
или его фармацевтически приемлемая соль, или его сольват.
[20] Соединение, представленное следующей формулой:
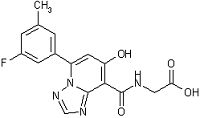 ,
,
или его фармацевтически приемлемая соль, или его сольват.
[21] Соединение, представленное следующей формулой:
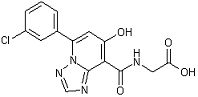 ,
,
или его фармацевтически приемлемая соль, или его сольват.
[22] Соединение, представленное следующей формулой:
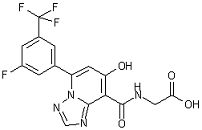 ,
,
или его фармацевтически приемлемая соль, или его сольват.
[23] Соединение, представленное следующей формулой:
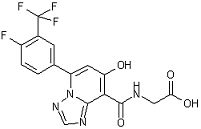 ,
,
или его фармацевтически приемлемая соль, или его сольват.
[24] Соединение, представленное следующей формулой:
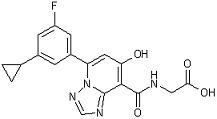 ,
,
или его фармацевтически приемлемая соль, или его сольват.
[25] Соединение, представленное следующей формулой:
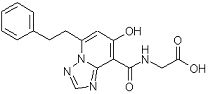 ,
,
или его фармацевтически приемлемая соль, или его сольват.
[26] Фармацевтическая композиция, содержащая соединение, описанное в любом из вышеупомянутых пунктов [1]-[25], или его фармацевтически приемлемую соль, или его сольват, и фармацевтически приемлемый носитель (в дальнейшем также называемый «фармацевтической композицией по настоящему изобретению»).
[27] Ингибитор пролилгидроксилазы, содержащий соединение, описанное в любом из вышеупомянутых пунктов [1]-[25], или его фармацевтически приемлемую соль, или его сольват.
[28] Агент, индуцирующий выработку эритропоэтина, содержащий соединение, описанное в любом из вышеупомянутых пунктов [1]-[25], или его фармацевтически приемлемую соль, или его сольват.
[29] Терапевтическое средство против анемии, содержащее соединение, описанное в любом из вышеупомянутых пунктов [1]-[25], или его фармацевтически приемлемую соль, или его сольват.
[30] Терапевтическое средство против почечной анемии, содержащее соединение, описанное в любом из вышеупомянутых пунктов [1]-[25], или его фармацевтически приемлемую соль, или его сольват.
[31] Применение соединения, описанного в любом из вышеупомянутых пунктов [1]-[25], или его фармацевтически приемлемой соли, или его сольвата для получения ингибитора пролилгидроксилазы.
[32] Применение соединения, описанного в любом из вышеупомянутых пунктов [1]-[25], или его фармацевтически приемлемой соли, или его сольвата для получения агента, индуцирующего выработку эритропоэтина.
[33] Применение соединения, описанного в любом из вышеупомянутых пунктов [1]-[25], или его фармацевтически приемлемой соли, или его сольвата для получения терапевтического средства против анемии.
[34] Применение соединения, описанного в любом из вышеупомянутых пунктов [1]-[25], или его фармацевтически приемлемой соли, или его сольвата для получения терапевтического средства против почечной анемии.
[35] Способ ингибирования пролилгидроксилазы, включающий введение млекопитающему эффективного количества соединения, описанного в любом из вышеупомянутых пунктов [1]-[25], или его фармацевтически приемлемой соли, или его сольвата.
[36] Способ индукции выработки эритропоэтина, включающий введение млекопитающему эффективного количества соединения, описанного в любом из вышеупомянутых пунктов [1]-[25], или его фармацевтически приемлемой соли, или его сольвата.
[37] Способ лечения анемии, включающий введение млекопитающему эффективного количества соединения, описанного в любом из вышеупомянутых пунктов [1]-[25], или его фармацевтически приемлемой соли, или его сольвата.
[38] Способ лечения почечной анемии, включающий введение млекопитающему эффективного количества соединения, описанного в любом из вышеупомянутых пунктов [1]-[25], или его фармацевтически приемлемой соли, или его сольвата.
[39] Упаковка для коммерческого использования, включающая фармацевтическую композицию, описанную в вышеупомянутом пункте [26], и связанный с ней печатный материал, причем в печатном материале указывается, что фармацевтическая композиция может или должна быть использована для лечения или профилактики заболевания, выбранного из анемии и почечной анемии.
[40] Набор, включающий фармацевтическую композицию, описанную в вышеупомянутом пункте [26], и связанный с ней печатный материал, причем в печатном материале указывается, что фармацевтическая композиция может или должна быть использована для лечения или профилактики заболевания, выбранного из анемии и почечной анемии.
Вариант осуществления для применения настоящего изобретения на практике
Определение каждого заместителя или каждого фрагмента, подлежащего применению в настоящем описании, является следующим.
«Атом галогена» представляет собой атом фтора, атом хлора, атом брома или атом йода.
«C1-10алкильная группа» представляет собой алкильную группу с неразветвленной или разветвленной цепью, содержащую число атомов углерода от 1 до 10, предпочтительно алкильную группу с неразветвленной или разветвленной цепью, содержащую число атомов углерода от 1 до 7. Например, могут быть упомянуты метильная группа, этильная группа, пропильная группа, изопропильная группа, бутильная группа, изобутильная группа, втор-бутильная группа, трет-бутильная группа, пентильная группа, изопентильная группа, трет-пентильная группа, 1-этилпропильная группа, неопентильная группа, гексильная группа, 2-этилбутильная группа, 3,3-диметилбутильная группа, 3,3-диметилпентильная группа, гептильная группа, октильная группа, нонильная группа, децильная группа и т.п.
«C1-6алкильная группа» представляет собой алкильную группу с неразветвленной или разветвленной цепью, содержащую число атомов углерода от 1 до 6, предпочтительно алкильную группу с неразветвленной или разветвленной цепью, содержащую число атомов углерода от 1 до 3. Например, могут быть упомянуты такие группы, как представленные в качестве примера вышеупомянутой «C1-10алкильной группы» и содержащие число атомов углерода от 1 до 6.
«C1-3алкильная группа» представляет собой алкильную группу с неразветвленной или разветвленной цепью, содержащую число атомов углерода от 1 до 3. Например, могут быть упомянуты такие группы, как представленные в качестве примера вышеупомянутой алкильной группы и содержащие число атомов углерода от 1 до 3.
«C6-14арильная группа» представляет собой ароматическую углеводородную группу, содержащую число атомов углерода от 6 до 14. Например, могут быть упомянуты фенильная группа, нафтильная группа, антрильная группа, инденильная группа, азуленильная группа, фторенильная группа, фенантрильная группа, пенталенильная группа и т.п., причем предпочтение отдается фенильной группе.
«C3-8циклоалкильная группа» представляет собой насыщенную циклоалкильную группу, содержащую число атомов углерода от 3 до 8, предпочтительно от 3 до 5, и, например, могут быть упомянуты циклопропильная группа, циклобутильная группа, циклопентильная группа, циклогексильная группа, циклогептильная группа, циклооктильная группа и т.п.
«C3-5циклоалкильная группа» представляет собой насыщенную циклоалкильную группу, содержащую число атомов углерода от 3 до 5. Например, могут быть упомянуты такие группы, как представленные в качестве примера вышеупомянутой «C3-8циклоалкильной группы» и содержащие число атомов углерода от 3 до 5.
«C6-14арил-C1-6алкильная группа» представляет собой C6-14арил-C1-6алкильную группу, в которой ее C6-14арильный фрагмент представляет собой определенную выше «C6-14арильную группу», а ее C1-6алкильный фрагмент представляет собой определенную выше «C1-6алкильную группу», причем предпочтение отдается C6-14арил-C1-6алкильной группе, в которой C1-6алкильный фрагмент представляет собой C1-6алкильную группу с неразветвленной цепью. Примеры C6-14арил-C1-6алкильной группы включают фенилметильную группу, фенилэтильную группу, фенилпропильную группу, фенилбутильную группу, фенилпентильную группу, фенилгексильную группу, нафтилметильную группу, нафтилэтильную группу, нафтилпропильную группу, нафтилбутильную группу, нафтилпентильную группу, нафтилгексильную группу, антрилметильную группу, инденилметильную группу, азуленилметильную группу, фторенилметильную группу, фенантрилметильную группу, пенталенилметильную группу и т.п.
«C3-8циклоалкил-C1-6алкильная группа» представляет собой C3-8циклоалкил-C1-6алкильную группу, в которой ее C3-8циклоалкильный фрагмент представляет собой определенную выше «C3-8циклоалкильную группу», а ее C1-6алкильный фрагмент представляет собой определенную выше «C1-6алкильную группу». Ее примеры включают циклопропилметильную группу, циклопропилэтильную группу, циклопропилпропильную группу, циклопропилбутильную группу, циклопропилпентильную группу, циклопропилгексильную группу, циклобутилметильную группу, циклобутилэтильную группу, циклобутилпропильную группу, циклобутилбутильную группу, циклобутилпентильную группу, циклобутилгексильную группу, циклопентилметильную группу, циклопентилэтильную группу, циклопентилпропильную группу, циклопентилбутильную группу, циклопентилпентильную группу, циклопентилгексильную группу, циклогексилметильную группу, циклогексилэтильную группу, циклогексилпропильную группу, циклогексилбутильную группу, циклогексилпентильную группу, циклогексилгексильную группу, циклогептилметильную группу, циклогептилэтильную группу, циклогептилпропильную группу, циклогептилбутильную группу, циклогептилпентильную группу, циклогептилгексильную группу, циклооктилметильную группу, циклооктилэтильную группу, циклооктилпропильную группу, циклооктилбутильную группу, циклооктилпентильную группу, циклооктилгексильную группу и т.п.
«C3-8циклоалкенильная группа» представляет собой циклоалкенильную группу, содержащую число атомов углерода от 3 до 8 и содержащую по меньшей мере одну, предпочтительно 1 или 2, двойные связи. Например, могут быть упомянуты циклопропенильная группа, циклобутенильная группа, циклопентенильная группа, циклопентадиенильная группа, циклогексенильная группа, циклогексадиенильная группа (2,4-циклогексадиен-1-ильная группа, 2,5-циклогексадиен-1-ильная группа и т.д.), циклогептенильная группа, циклооктенильная группа и т.п.
«Гетероарильная группа» представляет собой ароматический гетероцикл, содержащий помимо атома углерода в качестве кольцевого атома 1-6 гетероатомов, выбранных из атома азота, атома кислорода и атома серы, где число кольцевых атомов равно 3-14, включая моноцикл и конденсированное кольцо.
«Моноциклическая гетероарильная группа» представляет собой моноциклическую гетероарильную группу, предпочтительно содержащую 1-4 гетероатома, и, например, могут быть упомянуты тиенильная группа (например, тиофен-2-ил, тиофен-3-ил), фурильная группа (например, фуран-2-ил, фуран-3-ил и т.д.), пирролильная группа (например, 2-пирролин-1-ильная группа, 3-пирролин-3-ил и т.д.), оксазолильная группа (например, оксазол-2-ил, оксазол-4-ил, оксазол-5-ил и т.д.), изоксазолильная группа (например, изоксазол-3-ил, изоксазол-4-ил, изоксазол-5-ил и т.д.), тиазолильная группа (например, тиазол-2-ил, тиазол-4-ил, тиазол-5-ил и т.д.), изотиазолильная группа (например, изотиазол-3-ил, изотиазол-4-ил, изотиазол-5-ил и т.д.), имидазолильная группа (например, имидазол-1-ил, 1H-имидазол-2-ил, 1H-имидазол-4-ил и т.д.), пиразолильная группа (например, пиразол-1-ил, 1H-пиразол-3-ил, 2H-пиразол-3-ил, 1H-пиразол-4-ил и т.д.), оксадиазолильная группа (например, 1,3,4-оксадиазол-2-ил, 1,2,3-оксадиазол-4-ил, 1,2,3-оксадиазол-5-ил, 1,2,4-оксадиазол-3-ил, 1,2,4-оксадиазол-5-ил, 1,2,5-оксадиазол-3-ил и т.д.), тиадиазолильная группа (например, 1,3,4-тиадиазол-2-ил, 1,2,3-тиадиазол-4-ил, 1,2,3-тиадиазол-5-ил, 1,2,4-тиадиазол-3-ил, 1,2,4-тиадиазол-5-ил, 1,2,5-тиадиазол-3-ил и т.д.), триазолильная группа (например, 1,2,4-триазол-3-ил, 1,2,4-триазол-1-ил, 1,2,3-триазол-1-ил, 1,2,3-триазол-2-ил, 1,3,4-триазол-1-ил и т.д.), тетразолильная группа (например, тетразол-1-ил, тетразол-2-ил, 1H-тетразол-5-ил, 2H-тетразол-5-ил и т.д.), пиридильная группа (например, пиридин-2-ил, пиридин-3-ил, пиридин-4-ил и т.д.), пиримидинильная группа (например, пиримидин-2-ил, пиримидин-4-ил, пиримидин-5-ил и т.д.), пиридазинильная группа (например, пиридазин-3-ил, пиридазин-4-ил и т.д.), пиразинильная группа (например, пиразин-2-ил и т.д.), триазинильная группа (например, 1,3,5-триазин-2-ил и т.д.) и т.п.
Примеры «конденсированной гетероарильной группы» включают хинолильную группу, изохинолильную группу, хиназолинильную группу, хиноксалильную группу, фталазинильную группу, циннолинильную группу, нафтиридинильную группу, индолильную группу, бензимидазолильную группу, индолинильную группу, бензофуранильную группу, бензотиенильную группу, бензоксазолильную группу, бензотиазолильную группу, бензодиоксинильную группу, бензотиазолильную группу, тетрагидрохинолильную группу, дигидробензофуранильную группу, дигидробензотиенильную группу, дигидробензодиоксинильную группу, инденотиазолильную группу, тетрагидробензотиазолильную группу, 5,7-дигидропирроло[3,4-d]пиримидинильную группу, 6,7-дигидро-5H-циклопентапиримидинильную группу, имидазо[2,1-b]тиазолильную группу, птеридинильную группу, пуринильную группу и т.п.
«Галоген-C1-6алкильная группа» представляет собой определенную выше «C1-6алкильную группу», которая замещена одинаковыми или различными 1-5 атомами галогена, и, например, могут быть упомянуты хлорметил, фторметил, дифторметил, трифторметил, бромметил, хлорэтил, фторэтил, бромэтил, хлорпропил, фторпропил, бромпропил и т.п.
«Группа B» включает следующие замещающие группы (a)-(e):
(a) определенный выше «атом галогена»,
(b) определенную выше «C1-6алкильную группу»,
(c) определенную выше «C3-8циклоалкильную группу»,
(d) цианогруппу и
(e) определенную выше «галоген-C1-6алкильную группу».
«C6-14арильная группа, необязательно замещенная одинаковыми или различными 1-5 заместителями, выбранными из группы B» представляет собой определенную выше «C6-14арильную группу», которая необязательно замещена одинаковыми или различными 1-5 заместителями, и включает незамещенную C6-14арильную группу. Заместители являются одинаковыми или различными и выбраны из определенной выше «группы B».
«C3-8циклоалкильная группа, необязательно замещенная одинаковыми или различными 1-5 заместителями, выбранными из группы B» представляет собой определенную выше «C3-8циклоалкильную группу», которая необязательно замещена одинаковыми или различными 1-5 заместителями, и включает незамещенную C3-8циклоалкильную группу. Заместители являются одинаковыми или различными и выбраны из определенной выше «группы B».
«C3-8циклоалкенильная группа, необязательно замещенная одинаковыми или различными 1-5 заместителями, выбранными из группы B» представляет собой определенную выше «C3-8циклоалкенильную группу», которая необязательно замещена одинаковыми или различными 1-5 заместителями, и включает незамещенную C3-8циклоалкенильную группу. Заместители являются одинаковыми или различными и выбраны из определенной выше «группы B».
«Гетероарильная группа, необязательно замещенная одинаковыми или различными 1-5 заместителями, выбранными из группы B» представляет собой определенную выше «гетероарильную группу», которая необязательно замещена одинаковыми или различными 1-5 заместителями, и включает незамещенную гетероарильную группу. Заместители являются одинаковыми или различными и выбраны из определенной выше «группы B».
В вышеупомянутой формуле [I] предпочтительные группы являются такими, как описано ниже.
Частичная структурная формула:
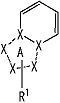
представляет собой группу, представленную любой из следующих формул:
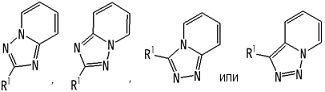 .
.
В качестве частичной структурной формулы предпочтительными являются группы, представленные
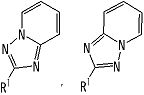
и т.п.
В качестве частичной структурной формулы более предпочтительной является группа, представленная
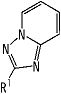 .
.
R1 представляет собой
(1) атом водорода,
(2) C1-6алкильную группу,
(3) C6-14арильную группу,
(4) C3-8циклоалкильную группу,
(5) C6-14арил-C1-6алкильную группу или
(6) C3-8циклоалкил-C1-6алкильную группу.
R1 предпочтительно представляет собой
(1) атом водорода,
(2) C1-3алкильную группу (например, метильную),
(3) C6-14арильную группу (например, фенильную),
(4) C3-5циклоалкильную группу (например, циклопропильную),
(5) C6-14арил (например, фенил)-C1-3алкильную (предпочтительно C1-3алкильную с неразветвленной цепью, например, этильную) группу,
(6) C3-8циклоалкил (например, циклогексил)-C1-3алкильную (например, этильную) группу или т.п.
R1 более предпочтительно представляет собой атом водорода.
R2 представляет собой
(1) атом водорода,
(2) C1-10алкильную группу,
(3) C6-14арильную группу, необязательно замещенную одинаковыми или различными 1-5 заместителями, выбранными из вышеупомянутой группы B,
(4) C3-8циклоалкильную группу, необязательно замещенную одинаковыми или различными 1-5 заместителями, выбранными из вышеупомянутой группы B,
(5) C3-8циклоалкенильную группу, необязательно замещенную одинаковыми или различными 1-5 заместителями, выбранными из вышеупомянутой группы B,
(6) гетероарильную группу, необязательно замещенную одинаковыми или различными 1-5 заместителями, выбранными из вышеупомянутой группы B (в которой помимо атома углерода гетероарил содержит 1-6 гетероатомов, выбранных из атома азота, атома кислорода и атома серы),
(7) C6-14арил-C1-6алкильную группу (в которой C6-14арил необязательно замещен одинаковыми или различными 1-5 заместителями, выбранными из вышеупомянутой группы B) или
(8) C3-8циклоалкил-C1-6алкильную группу (в которой C3-8циклоалкил необязательно замещен одинаковыми или различными 1-5 заместителями, выбранными из вышеупомянутой группы B).
R2 предпочтительно представляет собой
(1) атом водорода,
(2) C1-10алкильную группу,
(3) C6-14арильную группу, необязательно замещенную одинаковыми или различными 1-5 заместителями, выбранными из вышеупомянутой группы B,
(4) C3-8циклоалкильную группу (например, циклопентильную, циклогексильную, циклогептильную),
(5) C3-8циклоалкенильную группу (например, циклогексенильную),
(6) гетероарильную группу (предпочтительно, моноциклическую гетероарильную группу, например, тиенильную) необязательно замещенную одинаковыми или различными 1-5 (например, 1) заместителями, выбранными из вышеупомянутой группы B
(например, (a) атом галогена (например, атом хлора), и
(b) C1-6алкильная группа (например, метильная))
(в которой помимо атома углерода гетероарил содержит 1-6 (например, 1-4) гетероатомов, выбранных из атома азота, атома кислорода и атома серы (например, атома серы)),
(7) C6-14арил-C1-6алкильную группу (в которой C6-14арил необязательно замещен одинаковыми или различными 1-5 заместителями, выбранными из вышеупомянутой группы B) или
(8) C3-8циклоалкил-C1-3алкильную группу (в которой C3-8циклоалкил необязательно замещен одинаковыми или различными 1-5 заместителями, выбранными из вышеупомянутой группы B).
R2 более предпочтительно представляет собой
(1) C1-10алкильную группу (например, этильную, пропильную, изопропильную, бутильную, изобутильную, пентильную, изопентильную, трет-пентильную, гексильную, 1-этилпропильную, 2-этилбутильную, 3,3-диметилбутильную, 3,3-диметилпентильную),
(2) C6-14арильную группу (например, фенильную), необязательно замещенную одинаковыми или различными 1-5 (например, 1-3) заместителями, выбранными из вышеупомянутой группы B
(например, (a) атом галогена (например, атом хлора, атом фтора),
(b) C1-3алкильная группа (например, метильная),
(c) C3-5циклоалкильная группа (например, циклопропильная),
(d) цианогруппа и
(e) галоген-C1-3алкильная группа (например, трифторметильная)),
(3) C6-14арил (например, фенил)-C1-6алкильную (предпочтительно C1-6алкильную с неразветвленной цепью, например, метильную, этильную, пропильную) группу,
(C6-14арил необязательно замещен одинаковыми или различными 1-5 (например, 1-3) заместителями, выбранными из вышеупомянутой группы B
(например, (a) атом галогена (например, атом хлора, атом фтора),
(b) C3-8циклоалкильная группа (например, циклопропильная) и
(c) галоген-C1-3алкильная группа (например, трифторметильная))), или
(4) C3-8циклоалкил (например, циклопропил, циклобутил, циклопентил, циклогексил)-C1-3алкильную (например, метильную, этильную) группу
(C3-8циклоалкил необязательно замещен одинаковыми или различными 1-5 заместителями, выбранными из вышеупомянутой группы B).
R2 еще более предпочтительно представляет собой
(1) C1-6алкильную группу (например, бутильную, пентильную, 1-этилпропильную),
(2) фенил, необязательно замещенный одинаковыми или различными 1-3 заместителями, выбранными из
(a) атома галогена (например, атома хлора, атома фтора),
(b) C1-3алкильной группы (например, метильной),
(c) C3-5циклоалкильной группы (например, циклопропильной) и
(d) галоген-C1-3алкильной группы (например, трифторметильной),
(3) фенилэтил или
(4) циклопентилэтил.
R2 особенно предпочтительно представляет собой бутил, фенилэтил или 4-фтор-3-трифторметилфенил.
В другом варианте осуществления по настоящему изобретению R2 предпочтительно представляет собой
(1) C1-10алкильную группу или
(2) C6-14арил-C1-6алкильную группу (в которой C6-14арил необязательно замещен одинаковыми или различными 1-5 заместителями, выбранными из вышеупомянутой группы B).
R3 представляет собой
(1) атом водорода,
(2) атом галогена,
(3) C1-6алкильную группу,
(4) C6-14арильную группу,
(5) C3-8циклоалкильную группу или
(6) C6-14арил-C1-6алкильную группу.
R3 предпочтительно представляет собой
(1) атом водорода,
(2) атом галогена (например, атом хлора),
(3) C1-6алкильную группу (например, этильную, пентильную),
(4) C6-14арильную группу (например, фенильную) или
(5) C6-14арил (например, фенил)-C1-6алкильную (предпочтительно C1-6алкильную с неразветвленной цепью, например, этильную) группу.
R3 более предпочтительно представляет собой атом водорода.
Каждый из R4 и R5 независимо представляет собой
(1) атом водорода или
(2) C1-6алкильную группу.
Каждый из R4 и R5 предпочтительно независимо представляет собой
(1) атом водорода или
(2) C1-3алкильную группу (например, метильную).
Более предпочтительно, R4 и R5, оба, представляют собой атомы водорода.
В формуле [I] более предпочтительным является соединение, представленное следующей формулой [Ia]
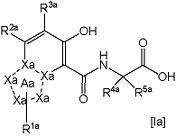 ,
,
в которой
частичная структурная формула:
представляет собой группу, представленную в виде
 ;
;
R1a представляет собой
(1) атом водорода,
(2) C1-3алкильную группу (например, метильную),
(3) C6-14арильную группу (например, фенильную),
(4) C3-5циклоалкильную группу (например, циклопропильную),
(5) C6-14арил (например, фенил)-C1-3алкильную (предпочтительно, C1-3алкильную с неразветвленной цепью, например, этильную) группу или
(6) C3-8циклоалкил (например, циклогексил)-C1-3алкильную (например, этильную) группу;
R2a представляет собой
(1) атом водорода,
(2) C1-10алкильную группу,
(3) C6-14арильную группу, необязательно замещенную одинаковыми или различными 1-5 заместителями, выбранными из вышеупомянутой группы B,
(4) C3-8циклоалкильную группу (например, циклопентильную, циклогексильную, циклогептильную),
(5) C3-8циклоалкенильную группу (например, циклогексенильную),
(6) гетероарильную группу (предпочтительно моноциклическую гетероарильную группу, например, тиенильную), необязательно замещенную одинаковыми или различными 1-5 (например, 1) заместителями, выбранными из вышеупомянутой группы B
(например, (a) атом галогена (например, атом хлора) и
(b) C1-6алкильная группа (например, метильная))
(в которой помимо атома углерода гетероарил содержит 1-6 (например, 1-4) гетероатомов, выбранных из атома азота, атома кислорода и атома серы (например, атом серы)),
(7) C6-14арил-C1-6алкильную группу (в которой C6-14арил необязательно замещен одинаковыми или различными 1-5 заместителями, выбранными из вышеупомянутой группы B) или
(8) C3-8циклоалкил-C1-3алкильную группу (в которой C3-8циклоалкил необязательно замещен одинаковыми или различными 1-5 заместителями, выбранными из вышеупомянутой группы B);
R3a представляет собой
(1) атом водорода,
(2) атом галогена (например, атом хлора),
(3) C1-6алкильную группу (например, этильную, пентильную),
(4) C6-14арильную группу (например, фенильную) или
(5) C6-14арил (например, фенил)-C1-6алкильную (предпочтительно C1-6алкильную с неразветвленной цепью, например, этильную) группу; и
каждый из R4a и R5a независимо представляет собой
(1) атом водорода или
(2) C1-3алкильную группу (например, метильную).
В качестве соединения по настоящему изобретению более предпочтительным является соединение, представленное вышеупомянутой формулой [Ia], в которой
R1a представляет собой атом водорода;
R2a представляет собой
(1) C1-10алкильную группу (например, этильную, пропильную, изопропильную, бутил, изобутильную, пентильную, изопентильную, трет-пентильную, гексильную, 1-этилпропильную, 2-этилбутильную, 3,3-диметилбутильную, 3,3-диметилпентильную),
(2) C6-14арильную группу (например, фенильную), необязательно замещенную одинаковыми или различными 1-5 (например, 1-3) заместителями, выбранными из вышеупомянутой группы B
(например, (a) атом галогена (например, атом хлора, атом фтора),
(b) C1-3алкильная группа (например, метильная),
(c) C3-5циклоалкильная группа (например, циклопропильная),
(d) цианогруппа и
(e) галоген-C1-3алкильная группа (например, трифторметильная)),
(3) C6-14арил (например, фенил)-C1-6алкильную (предпочтительно, C1-6алкильную с неразветвленной цепью, например, метильную, этильную, пропильную) группу
(C6-14арил, необязательно замещенный одинаковыми или различными 1-5 (например, 1-3) заместителями, выбранными из вышеупомянутой группы B
(например, (a) атом галогена (например, атом хлора, атом фтора),
(b) C3-8циклоалкильная группа (например, циклопропильная) и
(c) галоген-C1-6алкильная группа (например, трифторметильная))) или
(4) C3-8циклоалкил (например, циклопропил, циклобутил, циклопентил, циклогексил)-C1-3алкильную (например, метильную, этильную) группу
(C3-8циклоалкил, необязательно замещенный одинаковыми или различными 1-5 заместителями, выбранными из вышеупомянутой группы B);
R3a представляет собой атом водорода; и
R4a и R5a, оба, представляют собой атомы водорода.
В другом варианте осуществления по настоящему изобретению из соединений, представленных формулой [I], более предпочтительным является соединение, представленное следующей формулой [I-1]:
 ,
,
в которой
частичная структурная формула:
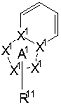
представляет собой группу, представленную любой из следующих формул:
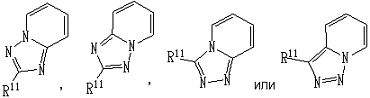 ;
;
R11 представляет собой
(1) атом водорода,
(2) C1-6алкильную группу (например, метильную),
(3) фенильную группу,
(4) C3-8циклоалкильную группу (например, циклопропильную),
(5) фенил-C1-6алкильную группу или
(6) C3-8циклоалкил-C1-6алкильную группу;
R21 представляет собой
(1) атом водорода,
(2) C1-10алкильную группу (например, этильную, н-пропильную, изопропильную, н-бутильную, изобутильную, н-пентильную, н-гексильную, 1-этилпропильную, 3-метилбутильную, 2,2-диметилпропильную, 3,3-диметилбутильную, 2-этилбутильную, 3,3-диметилпентильную),
(3) фенильную группу, необязательно замещенную одинаковыми или различными 1-5 заместителями (например, атомом фтора, атомом хлора, метилом, циано, циклопропилом, трифторметилом), выбранными из вышеупомянутой группы B,
(4) C3-8циклоалкильную группу (например, циклопентильную, циклогексильную, циклогептильную),
(5) C3-8циклоалкенильную группу (например, циклогексенильную),
(6) тиенильную группу, необязательно замещенную одинаковыми или различными 1-5 заместителями (например, атомом хлора, метилом), выбранными из вышеупомянутой группы B,
(7) фенил-C1-6алкильную группу (например, фенилметильную, фенилэтильную, фенилпропильную) (в которой фенил необязательно замещен одинаковыми или различными 1-5 заместителями (например, атомом фтора, атомом хлора, циклопропилом, трифторметилом), выбранными из вышеупомянутой группы B) или
(8) C3-8циклоалкил-C1-6алкильную группу (например, циклобутилметильную, циклопентилметильную, циклогексилметильную, циклопропилэтильную, циклобутилэтильную, циклопентилэтильную, циклогексилэтильную);
R31 представляет собой
(1) атом водорода,
(2) атом галогена (например, атом хлора),
(3) C1-6алкильную группу (например, этильную, н-пентильную),
(4) фенильную группу,
(5) C3-8циклоалкильную группу или
(6) фенил-C1-6алкильную группу (например, фенилэтильную); и
каждый из R41 и R51 независимо представляет собой
(1) атом водорода или
(2) C1-6алкильную группу (например, метильную).
Из соединений, представленных формулой [I-1], соединение, в котором
R11 представляет собой
(1) атом водорода,
(2) C1-6алкильную группу (например, метильную),
(3) фенильную группу или
(4) C3-8циклоалкильную группу (например, циклопропильную);
R21 представляет собой
(1) атом водорода,
(2) C1-10алкильную группу (например, этильную, н-пропильную, изопропильную, н-бутильную, изобутильную, н-пентильную, н-гексильную, 1-этилпропильную, 3-метилбутильную, 2,2-диметилпропильную, 3,3-диметилбутильную, 2-этилбутильную, 3,3-диметилпентильную),
(3) фенильную группу, необязательно замещенную одинаковыми или различными 1-5 заместителями (например, атомом фтора, атомом хлора, метилом, циано, циклопропилом, трифторметилом), выбранными из вышеупомянутой группы B,
(4) C3-8циклоалкильную группу (например, циклопентильную, циклогексильную, циклогептильную),
(5) C3-8циклоалкенильную группу (например, циклогексенильную),
(6) тиенильную группу, необязательно замещенную одинаковыми или различными 1-5 заместителями (например, атомом хлора, метилом), выбранными из вышеупомянутой группы B,
(7) фенил-C1-6алкильную группу (например, фенилметильную, фенилэтильную, фенилпропильную) (в которой фенил необязательно замещен одинаковыми или различными 1-5 заместителями (например, атомом фтора, атомом хлора, циклопропилом, трифторметилом), выбранными из вышеупомянутой группы B) или
(8) C3-8циклоалкил-C1-6алкильную группу (например, циклобутилметил, циклопентилметильную, циклогексилметильную, циклопропилэтильную, циклобутилэтильную, циклопентилэтильную, циклогексилэтильную);
R31 представляет собой
(1) атом водорода,
(2) атом галогена (например, атом хлора),
(3) C1-6алкильную группу (например, этильную, н-пентильную),
(4) фенильную группу или
(6) фенил-C1-6алкильную группу (например, фенилэтильную);
каждый из R41 и R51 независимо представляет собой
(1) атом водорода или
(2) C1-6алкильную группу (например, метильную),
является предпочтительным,
соединение, где
R11 представляет собой атом водорода, метил, фенил или циклопропил;
R21 представляет собой атом водорода; этил, н-пропил, изопропил, н-бутил, изобутил, н-пентил, н-гексил, 1-этилпропил, 3-метилбутил, 2,2-диметилпропил, 3,3-диметилбутил, 2-этилбутил, 3,3-диметилпентил; фенил, необязательно замещенный одинаковыми или различными 1-5 заместителями, выбранными из атома фтора, атома хлора, метила, циано, циклопропила и трифторметила; циклопентил, циклогексил, циклогептил; циклогексенил; тиенил, необязательно замещенный одинаковыми или различными 1-5 заместителями, выбранными из атома хлора и метила; фенилметил, фенилэтил, фенилпропил (фенил, необязательно замещенный одинаковыми или различными 1-5 заместителями, выбранными из атома фтора, атома хлора, циклопропила и трифторметила); циклобутилметил, циклопентилметил, циклогексилметил, циклопропилэтил, циклобутилэтил, циклопентилэтил или циклогексилэтил;
R31 представляет собой атом водорода, атом хлора, этил, н-пентил, фенил или фенилэтил; и
каждый R41 и R51 независимо представляет собой атом водорода или метил,
является более предпочтительным.
В качестве соединения по настоящему изобретению, или его фармацевтически приемлемой соли, или его сольвата соединения, описанные в примерах 1-122, являются предпочтительными, соединения, описанные в примерах 1, 2, 21, 31, 40, 44, 47, 52, 60, 74, 79, 116, 118, 119, 120, 121 и 122 являются особенно предпочтительными.
Фармацевтически приемлемой солью соединения, представленного формулой [I], может быть любая соль при условии, что она образует нетоксическую соль с соединением по настоящему изобретению. Ее примеры включают соли с неорганическими кислотами, соли с органическими кислотами, соли с неорганическими основаниями, соли с органическими основаниями, соли с аминокислотами и т.п.
Примеры солей с неорганической кислотой включают соль с соляной кислотой, азотной кислотой, серной кислотой, фосфорной кислотой, бромистоводородной кислотой и т.п.
Примеры солей с органической кислотой включают соли со щавелевой кислотой, малеиновой кислотой, лимонной кислотой, фумаровой кислотой, молочной кислотой, яблочной кислотой, янтарной кислотой, винной кислотой, уксусной кислотой, трифторуксусной кислотой, глюконовой кислотой, аскорбиновой кислотой, метансульфоновой кислотой, бензолсульфоновой кислотой, пара-толуолсульфоновой кислотой и т.п.
Примеры соли с неорганическим основанием включают натриевую соль, калиевую соль, кальциевую соль, магниевую соль, аммониевую соль и т.п.
Примеры солей с органическим основанием включают метиламин, диэтиламин, триметиламин, триэтиламин, этаноламин, диэтаноламин, триэтаноламин, этилендиамин, трис(гидроксиметил)метиламин, дициклогексиламин, N,N'-дибензилэтилендиамин, гуанидин, пиридин, пиколин, холин, цинхонин, меглумин и т.п.
Примеры соли с аминокислотой включают соли с лизином, аргинином, аспарагиновой кислотой, глутаминовой кислотой и т.п.
Каждая соль может быть получена путем осуществления взаимодействия соединения, представленного формулой [I], с неорганическим основанием, органическим основанием, неорганической кислотой, органической кислотой или аминокислотой в соответствии с известным способом.
«Сольват» представляет собой соединение, представленное формулой [I], или его фармацевтически приемлемую соль, где молекула растворителя является координированной, и также включает гидраты. В качестве сольвата фармацевтически приемлемый сольват является предпочтительным и включает, например, гидрат, этанолат, диметилсульфоксидат и т.п. соединения, представленного формулой [I], или его фармацевтически приемлемой соли. Его конкретные примеры включают гемигидрат, моногидрат, дигидрат и моноэтанолат соединения, представленного формулой [I], моногидрат натриевой соли, 2/3 этанолата дигидрохлорида и т.п. соединения, представленного формулой [I].
Сольват соединения по настоящему изобретению или его фармацевтически приемлемой соли может быть получен в соответствии со способом, известным per se.
Кроме того, соединение, представленное формулой [1], или его фармацевтически приемлемая соль, или его сольват, имеет различные изомеры. Например, E-форма и Z-форма присутствуют в качестве геометрических изомеров при наличии асимметрического атома углерода, энантиомер и диастереомер присутствуют в качестве стереоизомеров на его основе, и при наличии осевой хиральности присутствуют стереоизомеры на ее основе. Более того, также могут присутствовать таутомеры. Соответственно, настоящее изобретение охватывает все эти изомеры и их смеси.
Кроме того, соединение по настоящему изобретению, или его фармацевтически приемлемая соль, или его сольват могут быть помечены изотопом (например, 3H, 14C, 35S и т.д.).
В качестве соединения, представленного формулой [I], или его фармацевтически приемлемой соли, или его сольвата предпочтительным является соединение, представленное формулой [I], или его фармацевтически приемлемая соль, или его сольват, каждый из которых по существу очищен. Более предпочтительным является соединение, представленное формулой [I], или его фармацевтически приемлемая соль, или его сольват, каждый из которых очищен до чистоты, приемлемой для фармацевтического продукта.
В настоящем изобретении пролекарство соединения, представленного формулой [I], также может быть применимо в качестве лекарственного препарата. «Пролекарство» представляет собой производное соединения по настоящему изобретению, содержащее химически или метаболически разлагаемую группу, которое после введения в организм восстанавливается до исходного соединения путем, например, гидролиза, сольволиза или распада при физиологических условиях, и проявляет свойственную ему эффективность. Оно включает нековалентный комплекс и соль. Пролекарство применимо, например, для улучшения всасывания при пероральном введении или целевого воздействия на целевой фрагмент.
В соединении по настоящему изобретению примеры модифицированного фрагмента включают высоко реакционноспособную функциональную группу, такую как гидроксильная группа, карбоксильная группа, аминогруппа и т.п.
Конкретные примеры гидроксилмодифицирующей группы включают ацетильную группу, пропионильную группу, изобутирильную группу, пивалоильную группу, пальмитоильную группу, бензоильную группу, 4-метилбензоильную группу, диметилкарбамоильную группу, диметиламинометилкарбонильную группу, сульфогруппу, аланильную группу, фумарильную группу и т.п. Кроме того, может быть упомянута натриевая соль 3-карбоксибензоильной группы, 2-карбоксиэтилкарбонильной группы и т.п.
Конкретные примеры карбоксилмодифицирующей группы включают метильную группу, этильную группу, пропильную группу, изопропильную группу, бутильную группу, изобутильную группу, трет-бутильную группу, пивалоилоксиметильную группу, карбоксиметильную группу, диметиламинометильную группу, 1-(ацетилокси)этильную группу, 1-(этоксикарбонилокси)этильную группу, 1-(изопропилоксикарбонилокси)этильную группу, 1-(циклогексилоксикарбонилокси)этильную группу, (5-метил-2-оксо-1,3-диоксол-4-ил)метильную группу, бензильную группу, фенильную группу, орто-толильную группу, морфолиноэтильную группу, N,N-диэтилкарбамоилметильную группу, фталидильную группу и т.п.
Конкретные примеры аминомодифицирующей группы включают трет-бутильную группу, докозаноильную группу, пивалоилоксиметильную группу, аланильную группу, гексилкарбамоильную группу, пентилкарбамоильную группу, 3-метилтио-1-(ацетиламино)пропилкарбонильную группу, 1-сульфо-1-(3-этокси-4-гидроксифенил)метильную группу, (5-метил-2-оксо-1,3-диоксол-4-ил)метильную группу, (5-метил-2-оксо-1,3-диоксол-4-ил)метоксикарбонильную группу, тетрагидрофуранильную группу, пирролидилметильную группу и т.п.
Примеры «фармацевтической композиции» включают пероральные препараты, такие как таблетка, капсула, гранула, порошок, пастилка, сироп, эмульсия, суспензия и т.п., и парентеральные средства, такие как препарат для наружного применения, суппозиторий, инъекционный препарат, глазные капли, назальный препарат, легочный препарат и т.п.
Фармацевтическую композицию по настоящему изобретению приготавливают в соответствии со способом, известным в области фармацевтических препаратов, путем смешивания, при необходимости, соединения, представленного формулой [I], или его фармацевтически приемлемой соли, или его сольвата с подходящим количеством по меньшей мере одного типа фармацевтически приемлемого носителя и т.п. Хотя содержание соединения, представленного формулой [I], или его фармацевтически приемлемой соли, или его сольвата в фармацевтической композиции варьирует в зависимости от лекарственной формы, дозы и т.п., оно составляет, например, от 0,1 до 100% масс. от всей композиции.
Примеры «фармацевтически приемлемого носителя» включают различные органические или неорганические вещества-носители, обычно применяемые в качестве используемых в приготовлении веществ, например, наполнитель, разрыхлитель, связующее вещество, скользящее вещество, смазку и т.п. для твердых препаратов, и растворитель, солюбилизатор, способствующее суспендированию средство, изотоническое средство, буферное вещество, успокаивающее средство и т.п. для жидких препаратов. Более того, при необходимости применяют добавки, такие как консервант, антиоксидант, краситель, подсластитель и т.п.
Примеры «наполнителя» включают лактозу, сахарозу, D-маннит, D-сорбит, кукурузный крахмал, декстрин, микрокристаллическую целлюлозу, кристаллическую целлюлозу, кармеллозу, кармеллозу кальция, натрия карбоксиметилкрахмал, гидроксипропилцеллюлозу с низкой степенью замещения, аравийскую камедь и т.п.
Примеры «разрыхлителя» включают кармеллозу, кармеллозу кальция, кармеллозу натрия, натрия карбоксиметилкрахмал, кроскармеллозу натрия, кросповидон, гидроксипропилцеллюлозу с низкой степенью замещения, гидроксипропилметилцеллюлозу, кристаллическую целлюлозу и т.п.
Примеры «связующего вещества» включают гидроксипропилцеллюлозу, гидроксипропилметилцеллюлозу, повидон, кристаллическую целлюлозу, сахарозу, декстрин, крахмал, желатин, кармеллозу натрия, аравийскую камедь и т.п.
Примеры «вещества, способствующего скольжению» включают слабую безводную кремниевую кислоту, стеарат магния и т.п.
Примеры «смазки» включают стеарат магния, стеарат кальция, тальк и т.п.
Примеры «растворителя» включают очищенную воду, этанол, пропиленгликоль, макрогол, кунжутное масло, кукурузное масло, оливковое масло и т.п.
Примеры «солюбилизаторов» включают пропиленгликоль, D-маннит, бензилбензоат, этанол, триэтаноламин, карбонат натрия, цитрат натрия и т.п.
Примеры «способствующего суспендированию средства» включают бензалкония хлорид, кармеллозу, гидроксипропилцеллюлозу, пропиленгликоль, повидон, метилцеллюлозу, глицеролмоностеарат и т.п.
Примеры «изотонического средства» включают глюкозу, D-сорбит, хлорид натрия, D-маннит и т.п.
Примеры «буферного вещества» включают гидрофосфат натрия, ацетат натрия, карбонат натрия, цитрат натрия и т.п.
Примеры «успокаивающего средства» включают бензиловый спирт и т.п.
Примеры «консерванта» включают этилпарагидроксибензоат, хлорбутанол, бензиловый спирт, дегидроацетат натрия, сорбиновую кислоту и т.п.
Примеры «антиоксиданта» включают сульфит натрия, аскорбиновую кислоту и т.п.
Примеры «красителя» включают пищевые красители (например, пищевой краситель красный №2 или 3, пищевой краситель желтый №4 или 5 и т.п.), β-каротин и т.п.
Примеры «подсластителя» включают сахарин натрия, дикалия глицирризинат, аспартам и т.п.
Соединение по настоящему изобретению, или его фармацевтически приемлемая соль, или его сольват обладают способностью индуцировать выработку EPO вследствие ингибирующего действия в отношении пролилгидроксилазы (PHD) и могут быть использованы для профилактики или лечения различных заболеваний и патологий (нарушений), вызванных сниженной выработкой EPO.
В качестве различных заболеваний и патологий (нарушений), вызванных сниженной выработкой EPO, может быть упомянута анемия и т.п.
В общем, анемия включает анемию вследствие нарушения кроветворения в костном мозге, анемию вследствие дефицита железа, витамина B12 или фолиевой кислоты, кровотечения во время несчастного случая или операции, анемию, ассоциированную с хроническим воспалением (аутоиммунные заболевания, злокачественная опухоль, хронические трансмиссивные заболевания, плазмоклеточная дискразия и т.д.), анемию, ассоциированную с эндокринными заболеваниями (гипотиреоз, аутоиммунный полигландулярный синдром, сахарный диабет типа IA, дисфункциональное маточное кровотечение и т.д.), анемию, ассоциированную с хронической сердечной недостаточностью, анемию, ассоциированную с язвой, анемию, ассоциированную с заболеваниями печени, старческую анемию, лекарственную анемию, почечную анемию (анемию, ассоциированную с почечной недостаточностью), анемию, ассоциированную с химиотерапией, и т.п.
Примеры заболеваний, которые, как предполагается, подлежат улучшению путем ингибирования PHD для стабилизации HIF, включают ишемические болезни сердца (стенокардия, инфаркт миокарда и т.д.), ишемические цереброваскулярные нарушения (инсульт, церебральная эмболия, преходящие церебральные ишемические атаки и т.д.), виды хронической почечной недостаточности (ишемическая нефропатия, тубуло-интерстициальные нарушения почек и т.д.), диабетические осложнения (диабетические язвы и т.д.), когнитивные нарушения (деменция, болезнь Альцгеймера, болезнь Паркинсона, болезнь Гентингтона и т.д.) и т.п.
Ингибитор пролилгидроксилазы (PHD) и агент, индуцирующий выработку EPO, по настоящему изобретению предпочтительно используют в качестве средства для лечения анемии, более предпочтительно в качестве средства для лечения почечной анемии.
Фармацевтическая композиция по настоящему изобретению может быть введена перорально или парентерально (например, местное, ректальное, внутривенное введение и т.п.) человеку, а также отличным от человека млекопитающим (например, мышь, крыса, хомяк, морская свинка, кролик, кошка, собака, свинья, корова, лошадь, овца, обезьяна и т.п.). Доза варьирует в зависимости от субъекта введения, заболевания, симптома, лекарственной формы, пути введения и т.п. Например, суточная доза для перорального введения взрослому пациенту (масса тела приблизительно 60 кг) находится, как правило, в пределах от приблизительно 1 мг до 1 г, из расчета соединения по настоящему изобретению в качестве активного ингредиента. Это количество может быть введено однократно или несколькими порциями.
Поскольку соединение по настоящему изобретению, или его фармацевтически приемлемая соль, или его сольват ингибирует PHD и индуцирует выработку EPO, оно может быть использовано в качестве активного ингредиента терапевтического средства или профилактического средства против анемии.
«Ингибировать PHD» означает специфически ингибировать работу пролилгидроксилазы и устранить или ослабить ее активность. Например, это означает специфически ингибировать работу пролилгидроксилазы в условиях нижеупомянутого Тестового примера 1. «Ингибировать PHD» предпочтительно означает ингибировать PHD человека. В качестве «ингибитора PHD» предпочтительным является «ингибитор PHD человека».
«Индуцировать выработку EPO» означает, что выработка эритропоэтина в почках и т.д. усилена. Например, это означает, что выработка эритропоэтина индуцирована в условиях нижеупомянутого Тестового примера 2. «Индуцировать выработку EPO» предпочтительно означает «индуцировать выработку EPO человека». «Агент, индуцирующий выработку EPO» предпочтительно представляет собой «агент, индуцирующий выработку EPO человека».
Вышеупомянутое соединение, представленное формулой [I], или его фармацевтически приемлемая соль, или его сольват могут быть использованы в сочетании с одним или множеством других лекарственных средств (называемых здесь или далее в этом документе сопутствующим лекарством) в соответствии со способом, обычно используемым в области медицины (называемым здесь или далее в этом документе комплексным применением).
Продолжительность введения вышеупомянутого соединения, представленного формулой [I], или его фармацевтически приемлемой соли, или его сольвата и сопутствующего лекарства не ограничена, и они могут быть введены субъекту введения в виде комбинированного препарата, или оба препарата могут быть введены одновременно или через заданные интервалы. Кроме того, фармацевтическая композиция по настоящему соединению и сопутствующее лекарственное средство могут быть использованы в качестве лекарственного средства в форме набора. Доза сопутствующего лекарственного средства аналогична дозе, используемой в клинике, и может быть подходящим образом выбрана в соответствии с субъектом введения, заболеванием, симптомом, лекарственной формой, путем введения, временем введения, сочетанием и т.п. Форма введения сопутствующего лекарства особо не ограничена, и требует лишь ее сочетания с соединением по настоящему изобретению или с его фармацевтически приемлемой солью, или с его сольватом.
Примеры сопутствующего лекарственного средства включают средство для лечения и/или профилактики анемии или т.п., и могут быть использованы в сочетании с соединением по настоящему изобретению.
Примеры «терапевтического средства и/или профилактического средства против анемии» включают железистый цитрат, сульфат железа и т.п.
В качестве PHD могут быть упомянуты PHD2 и PHD3.
Далее конкретно пояснены способы получения соединения по настоящему изобретению, или его фармацевтически приемлемой соли, или его сольвата. Однако нет необходимости говорить, что настоящее изобретение не ограничивается такими способами получения. Для получения соединения по настоящему изобретению, или его фармацевтически приемлемой соли, или его сольвата порядок реакций может быть соответствующим образом изменен. Реакция может быть начата со стадии или фрагмента замещения, который представляется целесообразным.
Кроме того, между соответствующими стадиями может быть включена стадия соответствующего преобразования заместителя (преобразования или дальнейшей модификации заместителя). При наличии реакционноспособной функциональной группы соответствующим образом может проводиться защита и снятие защитных групп. Более того, при необходимости, для способствования протеканию реакции может быть использован реагент, отличный от реагента, приведенного в качестве примера. Кроме того, исходное соединение, способ получения которого не описан, является либо коммерчески доступным, либо может быть легко получено путем комбинации известных реакций синтеза.
Соединение, полученное на каждой стадии, может быть очищено общепринятым способом, таким как дистилляция, перекристаллизация, колоночная хроматография и т.п. В некоторых случаях соединение может быть применено на следующей стадии без выделения и очистки.
В следующем способе получения «комнатная температура» означает 1-40°C.
Способ получения I-1
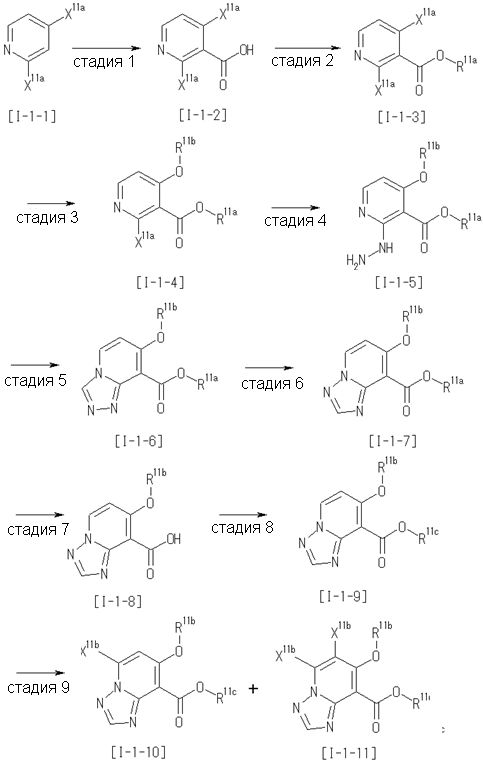
где каждый R11a и R11c представляет собой карбоксилзащитную группу, такую как метильная группа, этильная группа, бензильная группа, трет-бутильная группа и т.п., R11b представляет собой гидроксилзащитную группу, такую как ацетильная группа, бензильная группа, метильная группа, этильная группа, изопропильная группа, триметилсилильная группа, триэтилсилильная группа, трет-бутилдиметилсилильная группа, триизопропилсилильная группа, трет-бутилдифенилсилильная группа и т.п., каждый из X11a и X11b представляет собой атом галогена, такой как атом хлора, атом брома, атом йода, атом фтора и т.п., уходящую группу, такую как пара-толуолсульфонилоксигруппа, метансульфонилоксигруппа, трифторметансульфонилоксигруппа и т.п.
Стадия 1
Соединение [I-1-2] может быть получено путем металлирования соединения [I-1-1] в соответствии с общепринятым способом и введения карбоксильной группы с применением диоксида углерода. Металлирование выполняют путем осуществления взаимодействия с металлоорганическим реагентом, таким как н-бутиллитий, втор-бутиллитий, диизопропиламид лития, бис(триметилсилил)амид лития, бис(триметилсилил)амид натрия, бис(триметилсилил)амид калия, амид лития, амид натрия и т.п., в условиях низкой температуры в гексане, бензоле, толуоле, тетрагидрофуране, диэтиловом эфире, 1,4-диоксане и т.п. отдельно или в их смешанном растворителе, за которым следует осуществление взаимодействия с диоксидом углерода с получением соединения [I-1-2].
Стадия 2
Соединение [I-1-3] может быть получено путем введения защитной группы в карбоксильную группу соединения [I-1-2] в соответствии с общепринятым способом. Например, если защитной группой является трет-бутильная группа, то соединение [I-1-3] может быть получено путем осуществления взаимодействия с трет-бутил-2,2,2-трихлорацетимидатом в условиях от низкой температуры до нагревания в присутствии кислоты, такой как пара-толуолсульфоновая кислота, метансульфоновая кислота, трифторид бора, трихлорид бора, трибромид бора, трихлорид алюминия, хлороводород, бромоводород, фосфорная кислота, серная кислота, уксусная кислота, трифторуксусная кислота и т.п., в гексане, хлороформе, метиленхлориде, этилацетате, толуоле, 1,4-диоксане, тетрагидрофуране, 1,2-диметоксиэтане, диметилсульфоксиде, N,N-диметилформамиде, ацетонитриле и т.п. отдельно или в их смешанном растворителе.
Стадия 3
Соединение [I-1-4] может быть получено путем введения гидроксильной группы, защищенной защитной группой, представленной R11b, в соединение [I-1-3] в соответствии с общепринятым способом. Например, при введении гидроксильной группы, защищенной бензильной группой, соединение [I-1-3] подвергают взаимодействию с бензильным спиртом в условиях от низкой температуре до нагревания в присутствии основания, такого как триэтиламин, трет-бутоксид калия, карбонат калия, гидрид натрия, н-бутиллитий, диизопропиламид лития и т.п., в гексане, диметилсульфоксиде, N,N-диметилформамиде, ацетонитриле, тетрагидрофуране, 1,4-диоксане, 1,2-диметоксиэтане, толуоле и т.п. отдельно или в их смешанном растворителе, посредством чего может быть получено соединение [I-1-4].
Стадия 4
Соединение [I-1-5] может быть получено путем осуществления взаимодействия соединения [I-1-4] с моногидратом гидразина в условиях от низкой температуры до нагревания в хлороформе, толуоле, 1,4-диоксане, тетрагидрофуране, 1,2-диметоксиэтане, метаноле, этаноле, 2-пропаноле, диметилсульфоксиде, N,N-диметилформамиде, ацетонитриле, воде и т.п. отдельно или в их смешанном растворителе.
Стадия 5
Соединение [I-1-6] может быть получено путем осуществления взаимодействия соединения [I-1-5] с ортоэфирным соединением, таким как триметилортоформиат, триэтилортоформиат и т.п., или с муравьиной кислотой в условиях от низкой температуры до нагревания в присутствии кислоты, такой как пара-толуолсульфоновая кислота, метансульфоновая кислота, трифторид бора, трихлорид бора, трибромид бора, хлороводород, бромоводород, фосфорная кислота, серная кислота и т.п., в гексане, хлороформе, метиленхлориде, этилацетате, толуоле, 1,4-диоксане, тетрагидрофуране, 1,2-диметоксиэтане, метаноле, этаноле, 2-пропаноле, диметилсульфоксиде, N,N-диметилформамиде, ацетонитриле и т.п. отдельно или в смешанном растворителе или без растворителя.
Стадия 6
Соединение [I-1-7] может быть получено путем проведения реакции эндоциклической перегруппировки соединения [I-1-6] в условиях от комнатной температуры до нагревания в присутствии основания, такого как гидроксид натрия, морфолин, пиперидин, пирролидин и т.п. в гексане, хлороформе, метиленхлориде, этилацетате, толуоле, 1,2-диметоксиэтане, 1,4-диоксане, тетрагидрофуране, 1,2-диметоксиэтане, метаноле, этаноле, 2-пропаноле, диметилсульфоксиде, N,N-диметилформамиде, N,N-диметилацетамиде, ацетонитриле и т.п. отдельно или в их смешанном растворителе.
Стадия 7
Соединение [I-1-8] может быть получено путем удаления карбоксилзащитной группы соединения [I-1-7] в соответствии с общепринятым способом. Например, если R11a представляет собой трет-бутильную группу, то соединение [I-1-8] может быть получено путем осуществления взаимодействия с кислотой, такой как пара-толуолсульфоновая кислота, метансульфоновая кислота, трифторид бора, трихлорид бора, трибромид бора, трихлорид алюминия, хлороводород, бромоводород, фосфорная кислота, серная кислота, уксусная кислота, трифторуксусная кислота и т.п., в условиях от низкой температуры до нагревания в гексане, хлороформе, метиленхлориде, этилацетате, толуоле, 1,2-диметоксиэтане, 1,4-диоксане, тетрагидрофуране, метаноле, этаноле, 2-пропаноле, диметилсульфоксиде, N,N-диметилформамиде, N,N-диметилацетамиде, ацетонитриле, воде и т.п. отдельно или в их смешанном растворителе. Если R11a представляет собой метильную группу, этильную группу или трет-бутильную группу, то соединение [I-1-8] может быть получено путем гидролиза соединения [I-1-7] в условиях от низкой температуры до нагревания в присутствии основания, такого как гидроксид натрия, гидроксид калия, карбонат калия, карбонат натрия, гидроксид лития и т.п., в смешанном растворителе, состоящем из воды и растворителя, такого как метанол, этанол, 2-пропанол, тетрагидрофуран, 1,4-диоксан, 1,2-диметоксиэтан, N,N-диметилформамид, ацетонитрил и т.п.
Стадия 8
Соединение [I-1-9] может быть получено путем введения защитной группы в карбоксильную группу соединения [I-1-8] в соответствии с общепринятым способом. Например, если защитной группой является этильная группа, то соединение [I-1-9] может быть получено путем осуществления взаимодействия соединения [I-1-8] с N,N-диметилформамиддиэтилацеталем в условиях от низкой температуры до нагревания в хлороформе, метиленхлориде, этилацетате, толуоле, 1,4-диоксане, тетрагидрофуране, 1,2-диметоксиэтане, метаноле, этаноле, 2-пропаноле, диметилсульфоксиде, N,N-диметилформамиде, ацетонитриле и т.п. отдельно или в их смешанном растворителе.
Стадия 7 и стадия 8 могут быть пропущены. В этом случае R11a=R11c.
Стадия 9
Соединение [I-1-10] может быть получено путем введения уходящей группы в пиридиновое кольцо соединения [I-1-9] в соответствии с общепринятым способом. Может быть получено двузамещенное соединение [I-1-11]. Если уходящей группой является атом йода, то для проведения металлирования соединение [I-1―10] и соединение [I-1-11] могут быть получены путем осуществления взаимодействия с металлоорганическим реагентом, таким как н-бутиллитий, втор-бутиллитий, диизопропиламид лития, бис(триметилсилил)амид лития, бис(триметилсилил)амид натрия, бис(триметилсилил)амид калия, амид лития, амид натрия и т.п., в условиях низкой температуры в гексане, толуоле, 1,2-диметоксиэтане, диэтиловом эфире, 1,4-диоксане, тетрагидрофуране и т.п. отдельно или в их смешанном растворителе, с последующим осуществлением взаимодействия с йодом.
Способ получения I-2
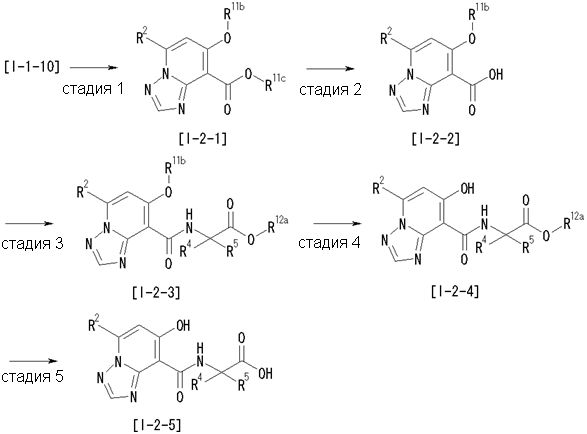 ,
,
где R12a представляет собой карбоксилзащитную группу, такую как метильная группа, этильная группа, бензильная группа, трет-бутильная группа и т.п., а значения других обозначений определены выше. Даже если R2 в [I-2-1]-[I-2-4] является отличным от определенных заместителей, он может быть использован, поскольку определенный заместитель может быть, в конечном итоге, получен путем осуществления соответствующего преобразования заместителя.
Стадия 1
Соединение [I-2-1] может быть получено путем введения заместителя R2 или его предшественника в соединение [I-1-10] в соответствии с общепринятым способом. Например, если R2 представляет собой бутильную группу, то соединение [I-2-1] может быть получено путем осуществления взаимодействия соединения [I-1-10] с бутилборной кислотой в условиях от комнатной температуры до нагревания в присутствии палладиевого катализатора, такого как дихлорид [1,1-бис(дифенилфосфино)ферроцен]палладия(II), тетракис(трифенилфосфин)палладий, дихлорид бис(трифенилфосфин)палладия(II), ацетаттрифенилфосфин палладия и т.п., и основания, такого как ацетат калия, карбонат калия, гидрокарбонат калия, гидрокарбонат натрия, фосфат калия, триэтиламин, диизопропилэтиламин, гидрофосфат натрия, карбонат цезия и т.п., путем добавления при необходимости соли серебра, такой как карбонат серебра, нитрат серебра, оксид серебра(I) и т.п., в гексане, N,N-диметилформамиде, N,N-диметилацетамиде, ацетонитриле, 1,2-диметоксиэтане, тетрагидрофуране, 1,4-диоксане, толуоле, воде и т.п. отдельно или в их смешанном растворителе.
Стадия 2
Соединение [I-2-2] может быть получено путем удаления карбоксилзащитной группы соединения [I-2-1] таким же образом, как в способе получения I-1, стадия 7.
Стадия 3
Соединение [I-2-3] может быть получено путем конденсации соединения [I-2-2] с производным глицина, представленным формулой H2NC(R4)(R5)COOR12a, в соответствии с общепринятым способом. Например, соединение [I-2-3] может быть получено путем конденсации соединения [I-2-2] с производным глицина, представленным формулой H2NC(R4)(R5)COOR12a, в условиях от низкой температуры до нагревания в присутствии агента конденсации, такого как дициклогексилкарбодиимид, 1,1'-карбонилдиимидазол, 1-этил-3-(3-диметиламинопропил)карбодиимид или его соль, дифенилфосфорилазид и т.п., и, при необходимости, N-гидроксисукцинимида, 1-гидроксибензотриазола, диметиламинопиридина и т.п., и, при необходимости, добавления основания, такого как карбонат калия, гидрокарбонат натрия, карбонат цезия, триэтиламин, диизопропилэтиламин, морфолин, пиридин и т.п., в растворителе, таком как N,N-диметилформамид, ацетонитрил, тетрагидрофуран, хлороформ, этилацетат, метиленхлорид, толуол и т.п.
Стадия 4
Соединение [I-2-4] может быть получено путем удаления гидроксилзащитной группы R11b соединения [I-2-3] в соответствии с общепринятым способом. Например, если R11b представляет собой бензильную группу, то соединение [I-2-4] может быть получено путем гидрирования в условиях от комнатной температуры до нагревания в атмосфере водорода в условиях от нормального давления до повышенного давления в присутствии катализатора, такого как палладированный уголь, гидроксид палладия, оксид платины, платина-углерод, никель Ренея и т.п., в гексане, метаноле, этаноле, 2-пропаноле, тетрагидрофуране, N,N-диметилформамиде, N,N-диметилацетамиде, этилацетате, уксусной кислоте, воде и т.п. отдельно или в их смешанном растворителе.
Стадия 5
Соединение [I-2-5] может быть получено таким же образом, как в способе получения I-1, стадия 7, путем удаления карбоксилзащитной группы соединения [I-2-4].
Способ получения I-3
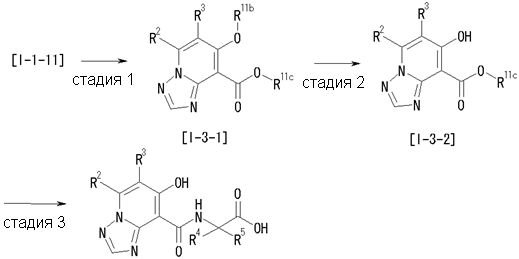
где даже если R2 и R3 в [I-3-1] и [I-3-2] являются отличными от определенных заместителей, они могут быть использованы, поскольку определенные заместители могут быть, в конечном итоге, получены путем соответствующего преобразования заместителя, а значения других символов определены выше.
Стадия 1
Соединение [I-3-1] может быть получено путем введения заместителей R2 и R3 или их предшественника в соединение [I-1-11] таким же путем, как в способе получения I-2, стадия 1. Например, если в качестве предшественника R2 и R3 вводят алкенильную группу, то соединение [I-3-1] может быть получено путем осуществления взаимодействия соединения [I-1-11] с алкенилбороновой кислотой таким же путем, как в способе получения I-2, стадия 1.
Стадия 2
Соединение [I-3-2] может быть получено путем снятия защитных групп с R11b соединения [I-3-1] таким же путем, как в способе получения I-2, стадия 4.
Стадия 3
Соединение [I-3-3] может быть получено путем осуществления взаимодействия соединения [I-3-2] с производным глицина, представленным формулой H2NC(R4)(R5)COOH. Например, соединение [I-3-3] может быть получено путем осуществления взаимодействия соединения [I-3-2] с натриевой солью производного глицина в условиях от комнатной температуры до нагревания в гексане, хлороформе, метиленхлориде, толуоле, 1,4-диоксане, тетрагидрофуране, 1,2-диметоксиэтане, метаноле, этаноле, 2-пропаноле, 2-метоксиэтаноле, диметилсульфоксиде, N,N-диметилформамиде, ацетонитриле, воде и т.п. отдельно или в их смешанном растворителе.
Способ получения I-4
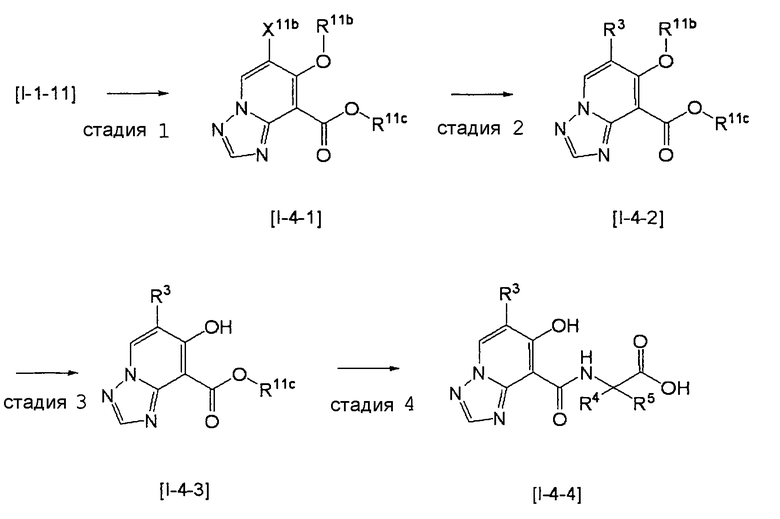 ,
,
где даже если R3 в [I-4-2] и [I-4-3] является отличным от определенных заместителей, он может быть использован, поскольку определенные заместители могут быть, в конечном итоге, получены путем соответствующего преобразования заместителя, а значения других символов определены выше.
Стадия 1
Соединение [I-4-1] может быть получено путем перемешивания соединения [I-1-11] в условиях от низкой температуры до нагревания в присутствии палладиевого катализатора, такого как дихлорид [1,1-бис(дифенилфосфино)ферроцен]палладия(II), тетракис(трифенилфосфин)палладий, дихлорид бис(трифенилфосфин)палладия(II), ацетаттрифенилфосфин палладия и т.п., и восстановителя, такого как гидрид три-н-бутилолова и т.п., в гексане, хлороформе, метиленхлориде, этилацетате, бензоле, толуоле, 1,2-диметоксиэтане, 1,4-диоксане, тетрагидрофуране, диэтиловом эфире, ацетонитриле, воде и т.п. отдельно или в их смешанном растворителе.
Стадия 2
Соединение [I-4-2] может быть получено путем замещения X11b соединения [I-4-1] заместителем R3 или его предшественником таким же путем, как в способе получения I-2, стадия 1.
Стадия 3
Соединение [I-4-3] может быть получено путем снятия защитных групп с R11b соединения [I-4-2] таким же путем, как в способе получения I-2, стадия 4.
Стадия 4
Соединение [I-4-4] может быть получено путем осуществления взаимодействия соединения [I-4-3] с производным глицина, представленным формулой H2NC(R4)(R5)COOH, или с его солью с металлом таким же путем, как в способе получения I-3, стадия 3.
Способ получения I-5
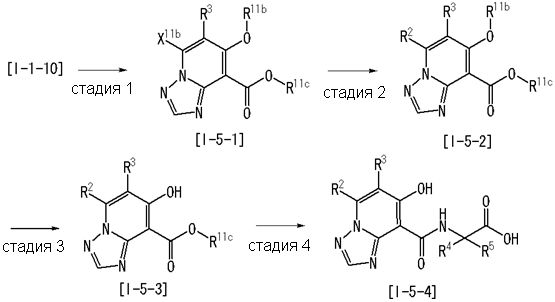 ,
,
где значение каждого обозначения определено выше.
Стадия 1
Соединение [I-5-1] может быть получено путем введения заместителя R3 в соединение [I-1-10] в соответствии с общепринятым способом. Например, если R3 представляет собой группу хлора, то соединение [I-5-1] может быть получено путем осуществления взаимодействия соединения [I-1-10] с хлорирующим агентом, таким как гексахлорэтан и т.п., в условиях низкой температуры в присутствии металлоорганического реагента, такого как н-бутиллитий, дисилазидгексаметил лития, бис(триметилсилил)амид натрия, гексаметилдисилазид калия, диизопропиламид лития, трет-бутоксид и т.п., в гексане, бензоле, толуоле, тетрагидрофуране, диэтиловом эфире, 1,4-диоксане и т.п. отдельно или в их смешанном растворителе.
Стадия 2
Соединение [I-5-2] может быть получено путем замещения заместителя X11b соединения [I-5-1] заместителем R2 или его предшественником таким же путем, как в способе получения I-2, стадия 1.
Стадия 3
Соединение [I-5-3] может быть получено путем снятия защитных групп с R11b соединения [I-5-2] таким же путем, как в способе получения I-2, стадия 4.
Стадия 4
Соединение [I-5-4] может быть получено путем осуществления взаимодействия соединения [I-5-3] с производным глицина, представленным формулой H2NC(R4)(R5)COOH, или с его солью с металлом таким же путем, как в способе получения I-3, стадия 3.
Способ получения I-6
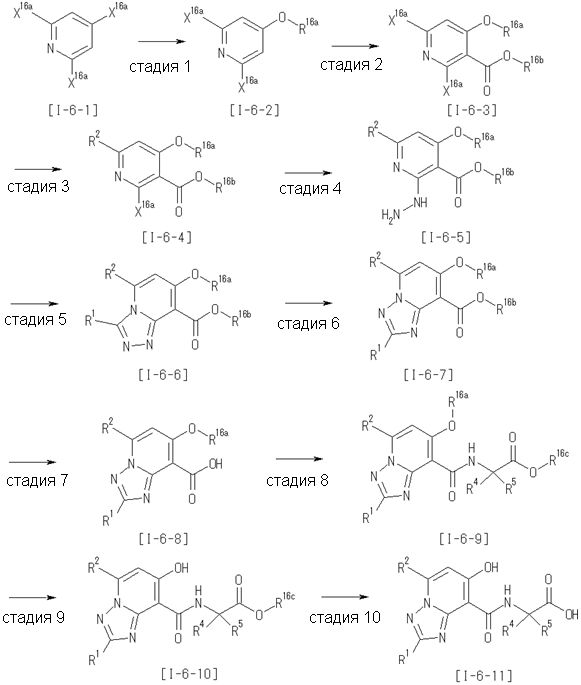 ,
,
где R16a представляет собой гидроксилзащитную группу, такую как ацетильная группа, бензильная группа, метильная группа, этильная группа, изопропильная группа, триметилсилильная группа, триэтилсилильная группа, трет-бутилдиметилсилильная группа, триизопропилсилильная группа, трет-бутилдифенилсилильная группа и т.п., каждый из R16b и R16c представляет собой карбоксилзащитную группу, такую как метильная группа, этильная группа, бензильная группа, трет-бутильная группа и т.п., X16a представляет собой атом галогена, такой как атом фтора, атом хлора, атом брома, атом йода и т.п., или уходящую группу, такую как пара-толуолсульфонилоксигруппа, метансульфонилоксигруппа, трифторметансульфонилоксигруппа и т.п., а значения других обозначений определены выше. Даже если R2 в [1-6-4]-[1-6-10] являются отличными от определенных заместителей, они могут быть использованы, поскольку определенные заместители могут быть, в конечном итоге, получены путем соответствующего преобразования заместителя.
Стадия 1
Соединение [I-6-2] может быть получено путем введения гидроксильной группы, защищенной защитной группой R16a, в соединение [I-6-1] таким же путем, как в способе получения I-1, стадия 3.
Стадия 2
Соединение [I-6-3] может быть получено путем введения карбоксильной группы, защищенной защитной группой R16b, в соединение [I-6-2] таким же путем, как в способе получения I-1, стадия 1.
Стадия 3
Соединение [I-6-4] может быть получено путем введения заместителя R2 или его предшественника в соединение [I-6-3] таким же путем, как в способе получения I-2, стадия 1.
Стадия 4
Соединение [I-6-5] может быть получено из соединения [I-6-4] таким же путем, как в способе получения I-1, стадия 4.
Стадия 5
Соединение [I-6-6] может быть получено из соединения [I-6-5] таким же путем, как в способе получения I-1, стадия 5.
Стадия 6
Соединение [I-6-7] может быть получено из соединения [I-6-6] таким же путем, как в способе получения I-1, стадия 6.
Стадия 7
Соединение [I-6-8] может быть получено путем удаления карбоксилзащитной группы соединения [I-6-7] таким же путем, как в способе получения I-1, стадия 7.
Стадия 8
Соединение [I-6-9] может быть получено путем конденсации соединения [I-6-8] с производным глицина, представленным формулой H2NC(R4)(R5)COOR16c, таким же путем, как в способе получения I-2, стадия 3.
Стадия 9
Соединение [I-6-10] может быть получено путем удаления гидроксилзащитной группы R16a соединения [I-6-9] таким же путем, как в способе получения I-2, стадия 4.
Стадия 10
Соединение [I-6-11] может быть получено путем удаления карбоксилзащитной группы соединения [I-6-10] таким же путем, как в способе получения I-1, стадия 7.
Способ получения I-7
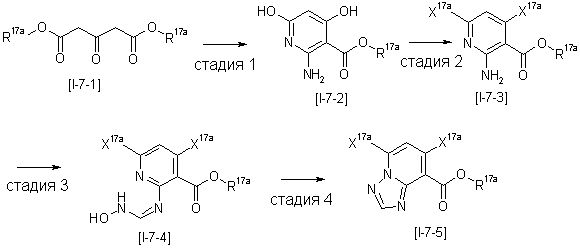 ,
,
где R17a представляет собой карбоксилзащитную группу, такую как метильная группа, этильная группа, бензильная группа, трет-бутильная группа и т.п., X17a представляет собой атом галогена, такой как атом фтора, атом хлора, атом брома, атом йода и т.п., или уходящую группу, такую как пара-толуолсульфонилоксигруппа, метансульфонилоксигруппа, трифторметансульфонилоксигруппа и т.п.
Стадия 1
Соединение [I-7-2] может быть получено путем осуществления взаимодействия соединения [I-7-1] с цианамидом в присутствии металлоорганического реагента, такого как ацетилацетонат никеля(II) и т.п., в условиях от низкой температуры до нагревания в гексане, этилацетате, хлороформе, метиленхлориде, толуоле, 1,4-диоксане, тетрагидрофуране, 1,2-диметоксиэтане, метаноле, этаноле, 2-пропаноле, диметилсульфоксиде, N,N-диметилформамиде, N-метил-2-пирролидоне, ацетонитриле, воде и т.п. отдельно или в их смешанном растворителе.
Стадия 2
Соединение [I-7-3] может быть получено путем преобразования гидроксильной группы соединения [I-7-2] в уходящую группу в соответствии с общепринятым способом. Например, если уходящей группой X17a является атом хлора, то соединение [I-7-3] может быть получено путем хлорирования соединения [I-7-2] тионилхлоридом, оксалилхлоридом, трифосгеном, пентахлоридом фосфора, оксихлоридом фосфора и т.п. в условиях от низкой температуры до нагревания в гексане, этилацетате, ацетоне, хлороформе, метиленхлориде, толуоле, 1,4-диоксане, тетрагидрофуране, 1,2-диметоксиэтане, метаноле, этаноле, 2-пропаноле, диметилсульфоксиде, N,N-диметилформамиде, 2-пирролидоне, ацетонитриле и т.п. отдельно, или в их смешанном растворителе, или без растворителя в присутствии, при необходимости, основания, такого как триэтиламин, пиридин, 4-(диметиламино)пиридин, N-метилморфолин, диизопропилэтиламин, тетраметилэтилендиамин и т.п., и, при необходимости, N,N-диметилформамида.
Стадия 3
Соединение [I-7-4] может быть получено путем осуществления взаимодействия соединения [I-7-3] с N,N-диметилформамиддиалкилацеталем в условиях от низкой температуры до нагревания в этилацетате, хлороформе, толуоле, 1,4-диоксане, тетрагидрофуране, 1,2-диметоксиэтане, метаноле, этаноле, 2-пропаноле, диметилсульфоксиде, N,N-диметилформамиде, ацетонитриле и т.п. отдельно или в их смешанном растворителе, а затем с гидроксиламином или его гидрохлоридом.
Стадия 4
Соединение [I-7-5] может быть путем дегидратации соединения [I-7-4] с применением полифосфорной кислоты, тионилхлорида, оксихлорида фосфора, пара-толуолсульфонилхлорида, уксусного ангидрида, ацетилхлорида, трифторуксусного ангидрида и т.п. в условиях от низкой температуры до высокой температуры в гексане, этилацетате, ацетоне, хлороформе, толуоле, 1,4-диоксане, тетрагидрофуране, 1,2-диметоксиэтане, диметилсульфоксиде, N,N-диметилформамиде, ацетонитриле и т.п. отдельно или в их смешанном растворителе.
Способ получения I-8
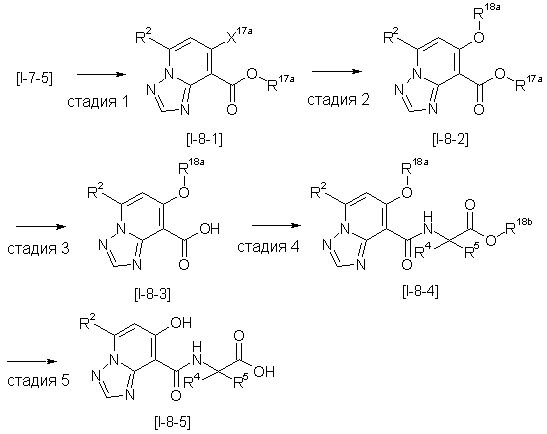 ,
,
где R18a представляет собой гидроксилзащитную группу, такую как ацетильная группа, бензильная группа, метильная группа, этильная группа, изопропильная группа, триметилсилильная группа, триэтилсилильная группа, трет-бутилдиметилсилильная группа, триизопропилсилильная группа, трет-бутилдифенилсилильная группа и т.п., R18b представляет собой карбоксилзащитную группу, такую как метильная группа, этильная группа, бензильная группа, трет-бутильная группа и т.п., а значения других обозначений определены выше.
Стадия 1
Соединение [I-8-1] может быть получено путем введения заместителя R2 или его предшественника в соединение [I-7-5] в соответствии с общепринятым способом таким же путем, как в способе получения I-2, стадия 1.
Стадия 2
Соединение [I-8-2] может быть получено путем введения гидроксильной группы, защищенной защитной группой, представленной формулой R18a, в соединение [I-8-1]. Например, если вводят гидроксильную группу, защищенную метильной группой, то соединение [I-8-2] может быть получено путем осуществления взаимодействия соединения [I-8-1] с метоксидом натрия в условиях от низкой температуры до нагревания в гексане, диметилсульфоксиде, N,N-диметилформамиде, ацетонитриле, тетрагидрофуране, 1,4-диоксане, 1,2-диметоксиэтане, толуоле, метаноле, воде и т.п. отдельно или в их смешанном растворителе, или с основанием, таким как триэтиламин, трет-бутоксид калия, метоксид натрия, карбонат калия, гидрид натрия, н-бутиллитий, диизопропиламид лития и т.п., в условиях от низкой температуры до нагревания в метаноле отдельно или в смешанном растворителе с гексаном, диметилсульфоксидом, N,N-диметилформамидом, ацетонитрилом, тетрагидрофураном, 1,4-диоксаном, 1,2-диметоксиэтаном, толуолом и т.п.
Стадия 3
Соединение [I-8-3] может быть получено путем удаления карбоксилзащитной группы соединения [I-8-2] таким же путем, как в способе получения I-1, стадия 7.
Стадия 4
Соединение [I-8-4] может быть получено путем осуществления конденсации соединения [I-8-3] с производным глицина, представленным формулой H2NC(R4)(R5)COOR18b, таким же путем, как в способе получения I-2, стадия 3.
Стадия 5
Соединение [I-8-5] может быть получено путем удаления гидроксилзащитной группы R18a и карбоксилзащитной группы R18b соединения [I-8-4] в соответствии с общепринятым способом. Например, если R18a представляет собой метильную группу, а R18b представляет собой трет-бутильную группу, то соединение [I-8-5] может быть получено путем перемешивания соединения [I-8-4] в условиях от комнатной температуры до нагревания в присутствии кислоты, такой как пара-толуолсульфоновая кислота, метансульфоновая кислота, трифторид бора, комплекс трифторида бора с диэтиловым эфиром, трихлорид бора, трибромид бора, хлороводород, бромоводород, фосфорная кислота, серная кислота, уксусная кислота, трифторуксусная кислота и т.п., в гексане, этилацетате, ацетоне, хлороформе, метиленхлориде, этилацетате, толуоле, 1,2-диметоксиэтане, 1,4-диоксане, тетрагидрофуране, 1,2-диметоксиэтане, метаноле, этаноле, изопропаноле, диметилсульфоксиде, N,N-диметилформамиде, N,N-диметилацетамиде, ацетонитриле, уксусной кислоте, воде и т.п. отдельно или в их смешанном растворителе.
Способ получения I-9
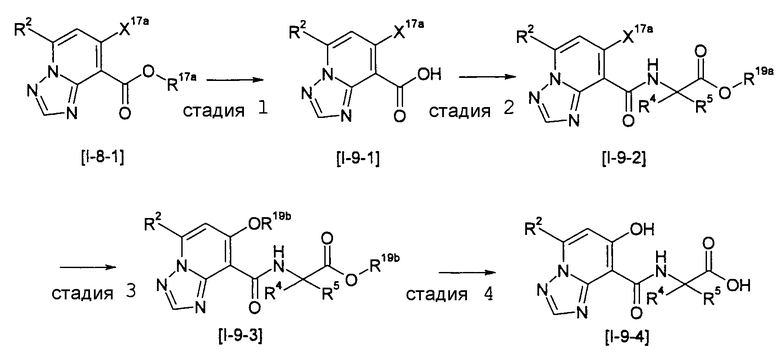 ,
,
где R19a представляет собой карбоксилзащитную группу, такую как метильная группа, этильная группа, бензильная группа, трет-бутильная группа и т.п., R19b представляет собой металл, образующий соль с карбоновой кислотой или фенолом, такой как литий, натрий, кальций и т.д., а значения других обозначений определены выше.
Стадия 1
Соединение [I-9-1] может быть получено путем удаления карбоксилзащитной группы соединения [I-8-1] таким же путем, как в способе получения I-1, стадия 7.
Стадия 2
Соединение [I-9-2] может быть получено путем конденсации соединения [I-9-1] с производным глицина, представленным формулой H2NC(R4)(R5)COOR19a, таким же путем, как в способе получения I-2, стадия 3.
Стадия 3
Соединение [I-9-3] может быть получено путем осуществления взаимодействия соединения [I-9-2] с основанием. Например, если R19b представляет собой натрий, то соединение [I-9-3] может быть получено путем осуществления взаимодействия соединения [I-9-2] с гидроксидом натрия в условиях от комнатной температуры до нагревания в диметилсульфоксиде, N,N-диметилформамиде, диметилацетамиде, ацетонитриле, тетрагидрофуране, 1,4-диоксане, 1,2-диметоксиэтане, толуоле, метаноле, этаноле, 2-метоксиэтаноле, 2-этоксиэтаноле, воде и т.п. отдельно или в их смешанном растворителе.
Стадия 4
Соединение [I-9-4] может быть получено путем осуществления взаимодействия соединения [I-9-3] с кислотой, такой как уксусная кислота, пара-толуолсульфоновая кислота, метансульфоновая кислота, трифторуксусная кислота, хлороводород, бромоводород, фосфорная кислота, серная кислота и т.п., в условиях от низкой температуры до нагревания в диметилсульфоксиде, N,N-диметилформамиде, ацетоне, ацетонитриле, тетрагидрофуране, 1,4-диоксане, 1,2-диметоксиэтане, толуоле, метаноле, этаноле, 2-метоксиэтаноле, 2-этоксиэтаноле, воде и т.п. отдельно или в их смешанном растворителе.
Способ получения II-1
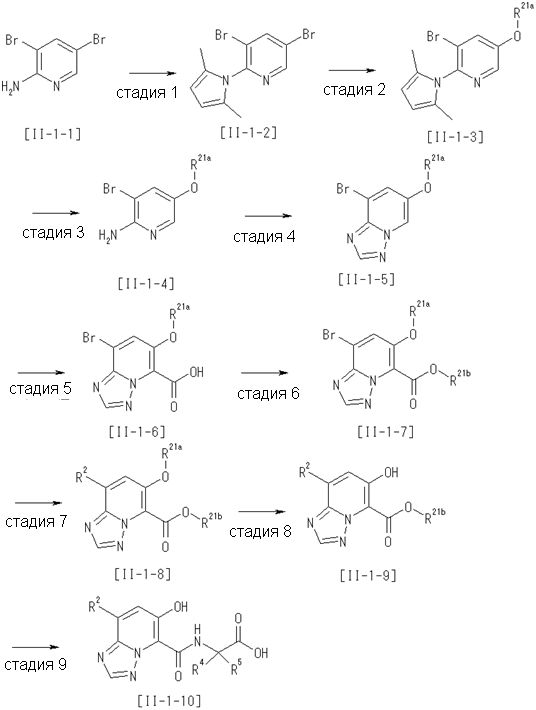 ,
,
где R21a представляет собой гидроксилзащитную группу, такую как бензильная группа, ацетильная группа, метильная группа, этильная группа, изопропильная группа, триметилсилильная группа, триэтилсилильная группа, трет-бутилдиметилсилильная группа, триизопропилсилильная группа, трет-бутилдифенилсилильная группа и т.п., R21b представляет собой карбоксилзащитную группу, такую как метильная группа, этильная группа, бензильная группа, трет-бутильная группа и т.п., а значения других обозначений определены выше. Даже если R2 в [II-1-8]-[II-1-9] являются отличными от определенных заместителей, они могут быть использованы, поскольку определенные заместители могут быть, в конечном итоге, получены путем соответствующего преобразования заместителя.
Стадия 1
Соединение [II-1-2] может быть получено путем осуществления взаимодействия соединения [II-1-1] с 2,5-гександионом в условиях от низкой температуры до нагревания в присутствии кислоты, такой как пара-толуолсульфоновая кислота, метансульфоновая кислота, трифторид бора, трихлорид бора, трибромид бора, трихлорид алюминия, хлороводород, бромоводород, фосфорная кислота, серная кислота, сульфамовая кислота, уксусная кислота, трифторуксусная кислота и т.п., в гексане, хлороформе, метиленхлориде, этилацетате, метаноле, этаноле, 2-пропаноле, толуоле, 1,4-диоксане, тетрагидрофуране, 1,2-диметоксиэтане, диметилсульфоксиде, N,N-диметилформамиде, ацетонитриле, воде и т.п. отдельно или в их смешанном растворителе.
Стадия 2
Соединение [II-1-3] может быть получено путем введения гидроксильной группы, защищенной защитной группой, представленной формулой R21a, в соединение [II-1-2] таким же путем, как в способе получения I-1, стадия 3.
Стадия 3
Соединение [II-1-4] может быть получено путем перемешивания соединения [II-1-3] в условиях от низкой температуры до нагревания в присутствии основания, такого как триэтиламин, трет-бутоксид калия, карбонат калия, гидрид натрия, диизопропиламид лития и т.п., и хлорида гидроксиламмония в метаноле, этаноле, 2-пропаноле, диметилсульфоксиде, N,N-диметилформамиде, ацетонитриле, воде и т.п. отдельно или в их смешанном растворителе.
Стадия 4
Соединение [II-1-4] приводят во взаимодействие с N,N-диметилформамиддиметилацеталем в условиях от комнатной температуры до нагревания в этилацетате, хлороформе, толуоле, 1,4-диоксане, тетрагидрофуране, 1,2-диметоксиэтане, метаноле, этаноле, 2-пропаноле, диметилсульфоксиде, N,N-диметилформамиде, ацетонитриле и т.п. отдельно или в их смешанном растворителе с получением соединения, которое подвергают взаимодействию с гидроксиламином или его солью в присутствии основания, такого как триэтиламин, диизопропилэтиламин, морфолин, пиридин и т.п., в условиях от низкой температуры до высокой температуры в гексане, этилацетате, ацетоне, хлороформе, толуоле, 1,4-диоксане, тетрагидрофуране, 1,2-диметоксиэтане, метаноле, этаноле, 2-пропаноле, диметилсульфоксиде, N,N-диметилформамиде, ацетонитриле и т.п. отдельно или в их смешанном растворителе, и подвергают взаимодействию с полифосфорной кислотой или гидроксиламин-O-сульфоновой кислотой, посредством чего может быть получено соединение [II-1-5].
Стадия 5
Соединение [II-1-6] может быть получено путем введения карбоксильной группы в соединение [II-1-5] таким же путем, как в способе получения I-1, стадия 1.
Стадия 6
Соединение [II-1-7] может быть получено путем введения защитной группы R21b в карбоксильную группу соединения [II-1-6] в соответствии с общепринятым способом. Например, если R21b представляет собой этильную группу, то соединение [II-1-7] может быть получено путем осуществления взаимодействия соединения [II-1-6] с N,N-диметилформамиддиэтилацеталем в условиях от комнатной температуры до нагревания в гексане, хлороформе, метиленхлориде, этилацетате, толуоле, 1,4-диоксане, тетрагидрофуране, 1,2-диметоксиэтане, диметилсульфоксиде, N,N-диметилформамиде, ацетонитриле и т.п. отдельно или в их смешанном растворителе.
Стадия 7
Соединение [II-1-8] может быть получено путем введения заместителя R2 или его предшественника в соединение [II-1-7] в соответствии с общепринятым способом. Например, при введении трет-бутилацетиленовой группы соединение [II-1-8] может быть получено путем осуществления взаимодействия соединения [II-1-7] с трет-бутилацетиленом в условиях от комнатной температуры до нагревания в присутствии палладиевого катализатора, такого как дихлорид [1,1-бис(дифенилфосфино)ферроцен]палладия(II), тетракис(трифенилфосфин)палладий, дихлорид бис(трифенилфосфин)палладия(II), ацетаттрифенилфосфин палладия и т.п., основания, такого как ацетат калия, карбонат калия, гидрокарбонат калия, гидрокарбонат натрия, фосфат калия, триэтиламин, диизопропилэтиламин, гидрофосфат натрия, карбонат цезия и т.п., и йодида меди в гексане, N,N-диметилформамиде, N,N-диметилацетамиде, ацетонитриле, 1,2-диметоксиэтане, тетрагидрофуране, 1,4-диоксане, толуоле, воде и т.п. отдельно или в их смешанном растворителе.
Стадия 8
Соединение [II-1-9] может быть получено путем снятия защитных групп с гидроксилзащитной группы R21a соединения [II-1-8] таким же путем, как в способе получения I-2, стадия 4.
Стадия 9
Соединение [II-1-10] может быть получено из соединения [II-1-9] таким же путем, как в способе получения I-3, стадия 3.
В данном способе получения был описан способ получения, когда R1 представляет собой атом водорода. Если R1 представляет собой вышеупомянутый и отличный от атома водорода заместитель, то на стадии 4 и стадии 5 вместо N,N-диметилформамиддиметилацеталя может быть применен N,N-диметилформамиддиметилацеталь, замещенный желаемым заместителем, а последующее может проводиться способом, подобным способу, описанному в данном способе получения.
Способ получения III-1
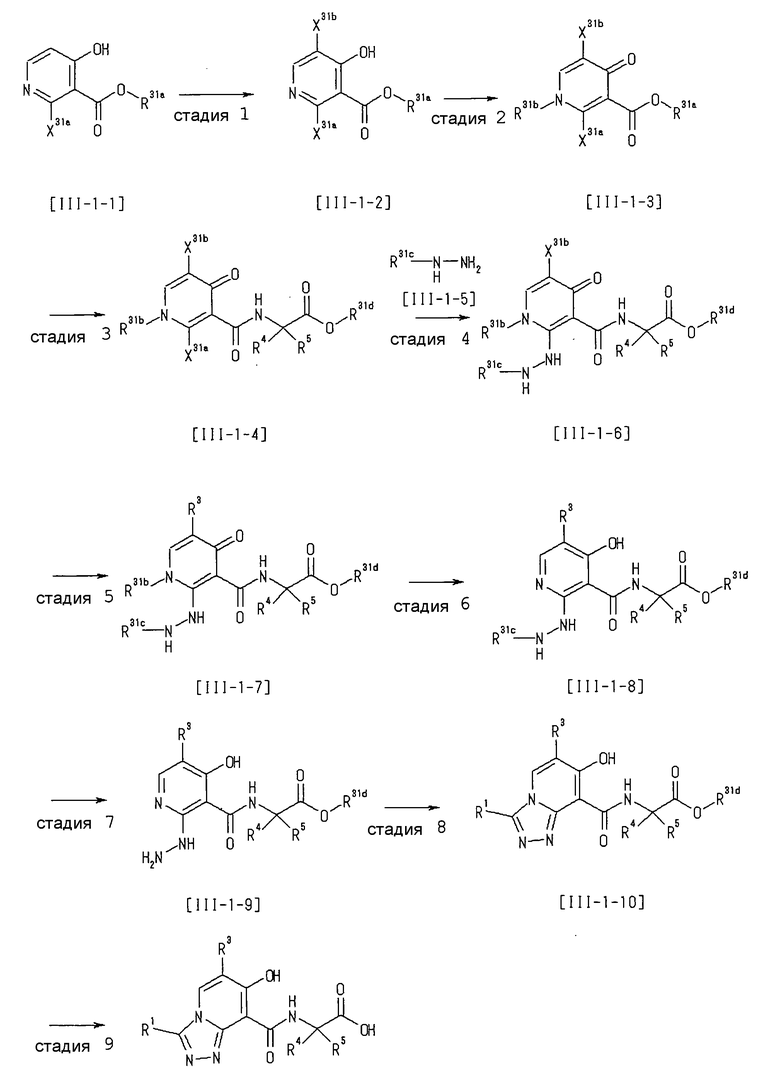 ,
,
где каждый из R31a и R31d представляет собой карбоксилзащитную группу, такую как метильная группа, этильная группа, бензильная группа, трет-бутильная группа и т.п., каждый из R31b и R31c представляет собой аминозащитную группу, такую как бензилоксикарбонильная группа, трет-бутоксикарбонильная группа, бензильная группа и т.п., каждый из X31a и X31b представляет собой атом галогена, такой как атом фтора, атом хлора, атом брома, атом йода и т.п., уходящую группу, такую как пара-толуолсульфонилоксигруппа, метансульфонилоксигруппа, трифторметансульфонилоксигруппа и т.п., а значения других обозначений определены выше.
Стадия 1
Соединение [III-1-2] может быть получено путем введения в соответствии с общепринятым способом уходящей группы X31b в соединение [III-1-1], полученное снятием защитной группы с R11b соединения [I-1-4] таким же путем, как в способе получения I-2, стадия 4. Например, если X31b представляет собой атом брома, то соединение [III-1-2] может быть получено путем осуществления взаимодействия соединения [III-1-1] с бромом или N-бромсукцинимидом в условиях от низкой температуры до нагревания в гексане, хлороформе, метиленхлориде, этилацетате, толуоле, тетрагидрофуране, 1,4-диоксане, ацетонитриле, воде и т.п. отдельно или в их смешанном растворителе.
Стадия 2
Соединение [III-1-3] может быть получено путем введения R31b в соединение [III-1-2] в соответствии с общепринятым способом. Например, если R31b представляет собой бензильную группу, то соединение [III-1-3] может быть получено путем осуществления взаимодействия соединения [III-1-2] с бензилхлоридом или бензилбромидом в присутствии основания, такого как карбонат калия, трет-бутоксид калия, гидрид натрия, карбонат цезия и т.п. в этилацетате, хлороформе, толуоле, 1,4-диоксане, тетрагидрофуране, 1,2-диметоксиэтане, диметилсульфоксиде, N,N-диметилформамиде, ацетонитриле, воде и т.п. отдельно или в их смешанном растворителе.
Стадия 3 и стадия 4
[III-1-6] может быть получено путем снятия защитной группы R31a соединения [III-1-3] таким же путем, как в способе получения I-1, стадия 7, преобразования соединения в хлорангидрид в соответствии с общепринятым способом, осуществления взаимодействия хлорангидрида с производным глицина, представленным формулой H2NC(R4)(R5)COOR31d, в присутствии основания, такого как триэтиламин, диизопропилэтиламин, пиридин и т.п., в условиях от низкой температуры до нагревания в гексане, хлороформе, метиленхлориде, этилацетате, толуоле, тетрагидрофуране и т.п. отдельно или в их смешанном растворителе с получением соединения [III-1-4], и осуществления взаимодействия этого соединения с соединением [III-1-5] в условиях от низкой температуры до нагревания в присутствии основания, такого как триэтиламин, диизопропилэтиламин, пиридин и т.п., в гексане, хлороформе, метиленхлориде, этилацетате, толуоле, тетрагидрофуране, 1,4-диоксане и т.п. отдельно или в их смешанном растворителе.
Стадия 5
Соединение [III-1-7] может быть получено из соединения [III-1-6] таким же путем, как в способе получения I-2, стадия 1.
Стадия 6
Соединение [III-1-8] может быть получено путем снятия защитных групп R31b соединения [III-1-7] таким же путем, как в способе получения I-2, стадия 4.
Стадия 7
Соединение [III-1-9] может быть получено путем удаления аминозащитной группы R31c соединения [III-1-8] в соответствии с общепринятым способом. Например, если R31c представляет собой трет-бутоксикарбонильную группу, то соединение [III-1-9] может быть получено путем перемешивания в условиях от низкой температуры до комнатной температуры в присутствии кислоты, такой как хлороводород, серная кислота, бромоводород, фосфорная кислота, уксусная кислота, трифторуксусная кислота и т.п. в гексане, хлороформе, метиленхлориде, этилацетате, толуоле, метаноле, этаноле, 2-пропаноле, тетрагидрофуране, 1,4-диоксане, ацетонитриле, воде и т.п. отдельно или в их смешанном растворителе.
Стадия 8
Соединение [III-1-10] может быть получено из соединения [III-1-9] таким же путем, как в способе получения I-1, стадия 5.
Стадия 9
Соединение [III-1-11] может быть получено путем удаления карбоксилзащитной группы соединения [III-1-10] таким же путем, как в способе получения I-1, стадия 7.
Способ получения III-2
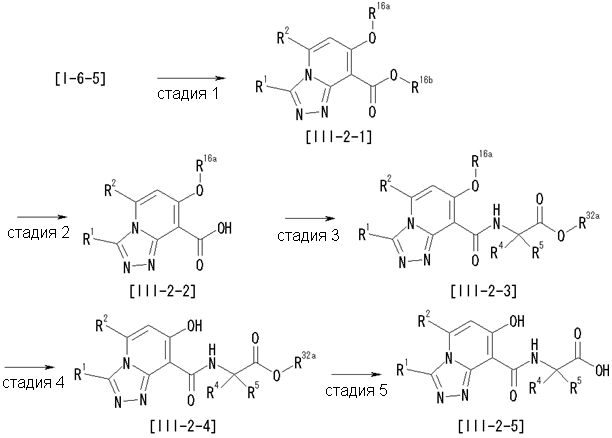 ,
,
где R32a представляет собой карбоксилзащитную группу, такую как метильная группа, этильная группа, бензильная группа, трет-бутильная группа и т.п., а значения других обозначений определены выше.
Стадия 1
Соединение [III-2-1] может быть получено из соединения [I-6-5] таким же путем, как в способе получения I-1, стадия 5.
Стадия 2
Соединение [III-2-2] может быть получено путем удаления карбоксилзащитной группы соединения [III-2-1] таким же путем, как в способе получения I-1, стадия 7.
Стадия 3
Соединение [III-2-3] может быть получено путем конденсации соединения [III-2-2] с производным глицина, представленным формулой H2NC(R4)(R5)COOR32a, таким же путем, как в способе получения I-2, стадия 3.
Стадия 4
Соединение [III-2-4] может быть получено путем снятия защитных групп R16a соединения [III-2-3] таким же путем, как в способе получения I-2, стадия 4.
Стадия 5
Соединение [III-2-5] может быть получено путем удаления карбоксилзащитной группы соединения [III-2-4] таким же путем, как в способе получения I-1, стадия 7.
Способ получения IV-1
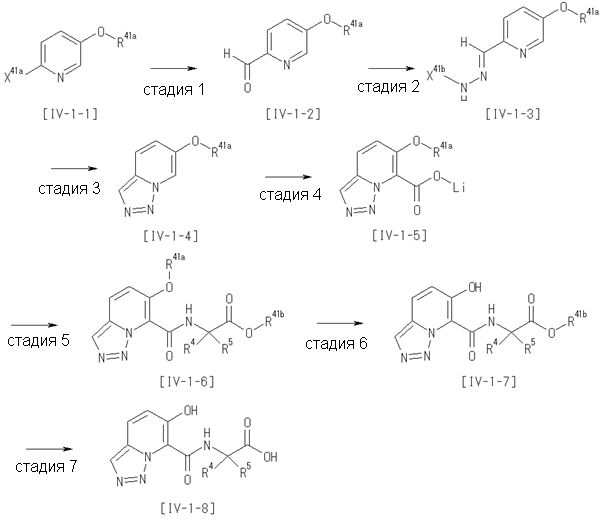 ,
,
где R41a представляет собой гидроксилзащитную группу, такую как ацетильная группа, бензильная группа, метильная группа, этильная группа, изопропильная группа, триметилсилильная группа, триэтилсилильная группа, трет-бутилдиметилсилильная группа, триизопропилсилильная группа, трет-бутилдифенилсилильная группа и т.п., R41b представляет собой карбоксилзащитную группу, такую как метильная группа, этильная группа, бензильная группа, трет-бутильная группа и т.п., каждый из X41a и X41b представляет собой атом галогена, такой как атом хлора, атом брома, атом йода и т.п., уходящую группу, такую как пара-толуолсульфонилоксигруппа, метансульфонилоксигруппа, трифторметансульфонилоксигруппа, пара-толуолсульфонильная группа, метансульфонильная группа и т.п., а значения других обозначений определены выше.
Стадия 1
Соединение [IV-1-2] может быть получено путем преобразования уходящей группы X41a соединения [IV-1-1] в формильную группу в соответствии с общепринятым способом. Соединение [IV-1-2] может быть получено путем осуществления взаимодействия соединения [IV-1-1] с металлоорганическим реагентом, таким как н-бутиллитий, втор-бутиллитий, диизопропиламид лития, бис(триметилсилил)амид лития, бис(триметилсилил)амид натрия, бис(триметилсилил)амид калия, амид лития, амид натрия и т.п. в условиях низкой температуры в гексане, бензоле, толуоле, тетрагидрофуране, диэтиловом эфире, 1,4-диоксане и т.п. отдельно или в их смешанном растворителе, а затем с N,N-диметилформамидом.
Стадия 2 и стадия 3
Соединение [IV-1-4] может быть получено путем осуществления взаимодействия соединения [IV-1-2] с гидразином, содержащим уходящую группу X41b, в условиях от комнатной температуры до нагревания в этилацетате, хлороформе, толуоле, 1,4-диоксане, тетрагидрофуране, 1,2-диметоксиэтане, метаноле, этаноле, изопропаноле, диметилсульфоксиде, N,N-диметилформамиде, ацетонитриле и т.п. отдельно или в их смешанном растворителе, а затем путем добавления основания, такого как морфолин, пиперидин, пирролидин и т.п., и перемешивания смеси.
Стадия 4
Соединение [IV-1-5] может быть получено из соединения [IV-1-4] таким же путем, как в способе получения I-1, стадия 1.
Стадия 5
Соединение [IV-1-6] может быть получено путем конденсации соединения [IV-1-5] с производным глицина таким же путем, как в способе получения I-2, стадия 3.
Стадия 6
Соединение [IV-1-7] может быть получено путем снятия защитных групп R41a соединения [IV-1-6] таким же путем, как в способе получения I-2, стадия 4.
Стадия 7
Соединение [IV-1-8] может быть получено путем удаления карбоксилзащитной группы соединения [IV-1-7] таким же путем, как в способе получения I-1, стадия 7.
В данном способе получения был описан способ получения, когда R1 представляет собой атом водорода. Если R1 представляет собой вышеупомянутый и отличный от атома водорода заместитель, то на стадии 1 и стадии 2 вместо N,N-диметилформамида может быть применен N,N-диметилформамид, замещенный желаемым заместителем, а последующее может быть проведено способом, подобным способу, описанному в данном способе получения.
Примеры
Далее посредством Примеров конкретно поясняется способ получения соединения по настоящему изобретению, или его фармацевтически приемлемой соли, или его сольвата. Тем не менее, настоящее изобретение не ограничивается Примерами.
Пример 1
Получение гидрохлорида {[5-(4-фтор-3-трифторметилфенил)-7-гидрокси[1,2,4]триазоло[1,5-a]пиридин-8-карбонил]амино}уксусной кислоты
Стадия 1-1
В токе азота смешивали диизопропиламин (198 мл) и тетрагидрофуран (1000 мл) и при охлаждении на бане со смесью сухой лед/гексан по каплям добавляли н-бутиллитий (2,76M, 500 мл). После перемешивания на бане со смесью сухой лед/гексан в течение 1 ч по каплям добавляли 2,4-дихлорпиридин. После перемешивания при охлаждении на бане со смесью сухой лед/гексан в течение 1 ч продували углекислым газом до прекращения повышения температуры, предупреждая снижение температуры ниже -60°C. Продували углекислым газом еще в течение 30 мин при охлаждении на бане со смесью сухой лед/гексан, и по каплям добавляли 4 н. соляную кислоту (1000 мл). Водный слой дважды экстрагировали этилацетатом (1000 мл, 500 мл). Органические слои объединяли, сушили над сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления. Полученное твердое вещество суспендировали в гексане с получением соединения (243 г, 96%), описанного в представленной выше схеме.
1H-ЯМР (ДМСО-D6) δ: 7,74 (1H, д, J=5,6 Гц), 8,47 (1H, д, J=5,6 Гц), 14,53 (1H, ушир. c).
Стадия 1-2
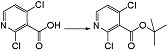
Смешивали соединение (234 г), полученное на стадии 1-1, и тетрагидрофуран (1200 мл), и добавляли комплекс трифторида бора/диэтилового эфира (8 мл). Затем при охлаждении на льду по каплям добавляли трет-бутил-2,2,2-трихлорацетимидат (361 мл). К этой реакционной смеси добавляли насыщенный водный раствор гидрокарбоната натрия (1200 мл) и воду (1200 мл) и экстрагировали водный слой этилацетатом (1200 мл). Органический слой промывали насыщенным солевым раствором, сушили над сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления. К полученному остатку добавляли гексан (1800 мл). Нерастворимый материал отфильтровывали, и концентрировали фильтрат в условиях пониженного давления с получением соединения (326 г), описанного в представленной выше схеме, в виде неочищенного продукта.
1H-ЯМР (CDCl3) δ: 1,63 (9H, с), 7,31 (1H, д, J=5,2 Гц), 8,31 (1H, д, J=5,2 Гц).
Стадия 1-3
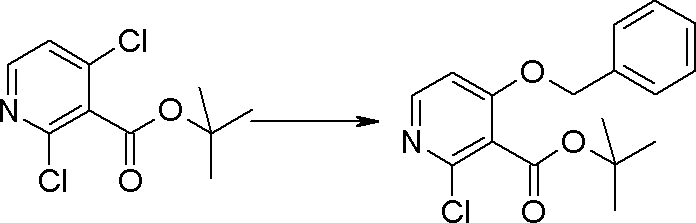
Под струей азота при охлаждении на льду смешивали гидрид натрия (60% суспензия в масле, 58 г) и N,N-диметилформамид (1000 мл). К смеси добавляли соединение (326 г), полученное на стадии 1-2, растворяли в N,N-диметилформамиде (300 мл) и добавляли к ним. К этой смеси добавляли смесь бензилового спирта (136 мл) и N,N-диметилформамида (200 мл). После перемешивания при охлаждении на льду в течение 15 мин добавляли гидрид натрия (60% суспензия в масле, 5,2 г). После перемешивания при охлаждении на льду еще в течение 20 мин добавляли воду (3000 мл), выпавшее в осадок твердое вещество фильтровали и сушили фильтрат в условиях пониженного давления при 50°C в течение ночи. Твердое вещество очищали по методу колоночной хроматографии (элюент: гексан/этилацетат = 10/1 - только этилацетат). Полученное твердое вещество затем суспендировали в гексане с получением целевого продукта. В то же время фильтрат концентрировали, очищали колоночной хроматографией и суспендировали в гексане с получением целевого продукта. Целевые продукты объединяли с получением соединения (334 г, выход 83%), описанного в представленной выше схеме.
1H-ЯМР (CDCl3) δ: 1,55 (9H, с), 5,17 (2H, с), 6,83 (1H, д, J=6,0 Гц), 7,32-7,42 (5H, м), 8,24 (1H, д, J=6,0 Гц).
Стадия 1-4
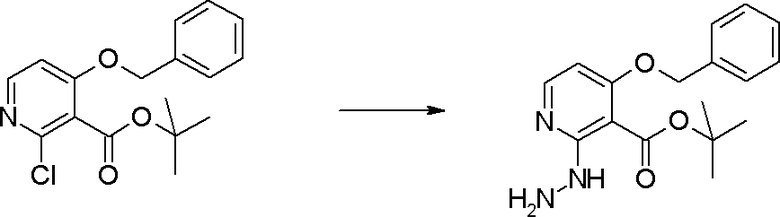
Смешивали соединение (167 г), полученное на стадии 1-3, моногидрат гидразина (127 мл) и 1,4-диоксан (1200 мл), и перемешивали реакционную смесь при 94°C в течение 17 ч. После охлаждения до комнатной температуры добавляли этилацетат (1700 мл), и последовательно промывали смесь насыщенным водным раствором гидрокарбоната натрия (500 мл)/водой (500 мл), насыщенным водным раствором гидрокарбоната натрия (250 мл)/водой (250 мл) и насыщенным водным раствором гидрокарбоната натрия (200 мл)/водой (200 мл). Органический слой сушили над сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления. Путем двукратного повторения операции получали соединение (266 г), описанное в представленной выше схеме, в виде неочищенного продукта.
1H-ЯМР (CDCl3) δ: 1,42 (9H, с), 3,98 (2H, ушир. c), 5,09 (2H, с), 6,32 (1H, д, J=5,6 Гц), 7,28-7,45 (5H, м), 7,96 (1H, ушир. c), 8,08 (1H, д, J=5,6 Гц).
Стадия 1-5
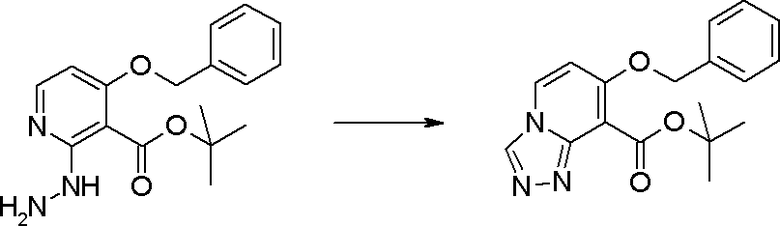
Смешивали соединение (266 г), полученное способом, аналогичным описанному на стадии 1-4, и триметилортоформиат (1000 мл), добавляли моногидрат пара-толуолсуфоновой кислоты (80 г) и перемешивали смесь при 56°C в течение 1 ч. Реакционную смесь концентрировали в условиях пониженного давления, и суспендировали полученный остаток в гексане/этилацетате (2/1). Затем остаток суспендировали в насыщенном водном растворе гидрокарбоната натрия/воде (1/1) с получением соединения (209 г, 76%), описанного в представленной выше схеме.
1H-ЯМР (ДМСО-D6) δ: 1,49 (9H, с), 5,36 (2H, с), 7,22 (1H, д, J=7,6 Гц), 7,32-7,50 (5H, м), 8,62 (1H, д, J=7,6 Гц), 9,13 (1H, с).
Стадия 1-6
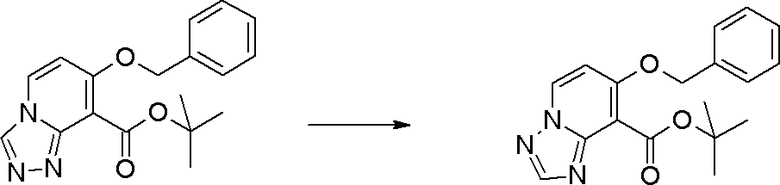
Смешивали соединение (200 г), полученное на стадии 1-5, и этилацетат (600 мл), добавляли морфолин (160 мл) и перемешивали смесь при 74°C в течение 3 ч. Смесь оставляли охлаждаться до комнатной температуры и добавляли воду (600 мл). Водный слой экстрагировали этилацетатом (400 мл), органические слои объединяли и последовательно промывали 5% водным раствором гидросульфата калия (600 мл) и насыщенным солевым раствором. Органический слой сушили над сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления с получением соединения (194 г, 97%), описанного в представленной выше схеме.
1H-ЯМР (CDCl3) δ: 1,59 (9H, с), 5,28 (2H, с), 6,85 (1H, д, J=7,6 Гц), 7,33-7,46 (5H, м), 8,29 (1H, с), 8,50 (1H, д, J=7,6 Гц).
Стадия 1-7

Под струей азота и при охлаждении на бане со смесью сухой лед/гексан смешивали соединение (194 г), полученное на стадии 1-6, и тетрагидрофуран (600 мл), и по каплям добавляли раствор йода (151 г) в тетрагидрофуране (500 мл). К этой смеси по каплям добавляли 1,6M бис(триметилсилил)амид лития (788 мл), не допуская снижения температуры ниже -60°C. После перемешивания при охлаждении на бане со смесью сухой лед/гексан в течение 2 ч по каплям добавляли 4 н. соляную кислоту в этилацетате (315 мл), не допуская снижения температуры ниже -60°C. К этой реакционной смеси добавляли сульфит натрия (76 г), насыщенный водный раствор хлорида аммония (1000 мл), воду (800 мл) и гексан/этилацетат (1/1, 1000 мл). Органический слой последовательно промывали насыщенным водным раствором гидрокарбоната натрия (500 мл) и насыщенным солевым раствором (800 мл), сушили над сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления с получением неочищенного продукта. Неочищенный продукт суспендировали в гексане с получением соединения (188 г, 70%), описанного в представленной выше схеме.
1H-ЯМР (ДМСО-D6) δ: 1,46 (9H, с), 5,39 (2H, с), 7,33-7,51 (5H, м), 7,87 (1H, с), 8,43 (1H, с).
Стадия 1-8
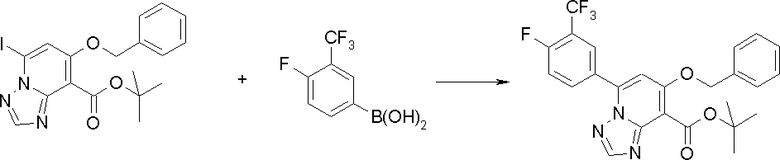
Смешивали соединение (60 г), полученное на стадии 1-7, 4-фтор-3-(трифторметил)фенилбороновую кислоту (29 г), комплекс дихлорида [1,1'-бис(дифенилфосфино)ферроцен]палладия(II) и дихлорметана (1/1, 5,4 г), фосфат калия (113 г) и 1,2-диметоксиэтан (600 мл) и перемешивали смесь при 80°C в течение 1 ч. К реакционной смеси добавляли воду и экстрагировали смесь этилацетатом. Органический слой промывали насыщенным солевым раствором, сушили над безводным сульфатом магния, фильтровали и концентрировали в условиях пониженного давления. Полученное твердое вещество очищали по методу колоночной хроматографии (элюент: хлороформ/этилацетат = 10/1) с получением неочищенного продукта, описанного в представленной выше схеме соединения. Этот продукт суспендировали в диизопропиловом эфире/гексане (1/1, 500 мл) с получением соединения (45 г, 70%), описанного в представленной выше схеме.
1H-ЯМР (CDCl3) δ: 1,61 (9H, с), 5,33 (2H, с), 6,88 (1H, с), 7,35-7,48 (6H, м), 8,04 (1H, дд, J=6,7, 2,1 Гц), 8,11-8,15 (1H, м), 8,31 (1H, с).
Стадия 1-9
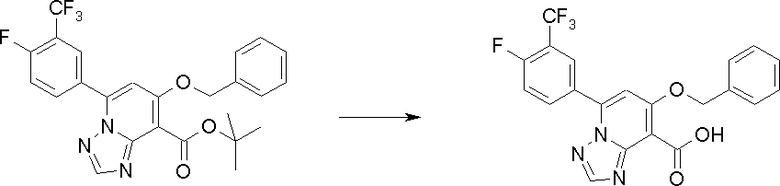
Смешивали соединение (45 г), полученное на стадии 1-8, и 1,4-диоксан (450 мл) и при комнатной температуре добавляли 4 н. водный раствор гидроксида натрия (116 мл). После перемешивания при 100°C в течение 17 ч реакционную смесь концентрировали в условиях пониженного давления. К смеси добавляли воду (450 мл), при охлаждении на льду нейтрализовали смесь добавлением 6 н. соляной кислоты (77 мл) и собирали выпавшее в осадок твердое вещество путем фильтрования с получением соединения (43 г), описанного в представленной выше схеме.
1H-ЯМР (ДМСО-D6) δ: 5,51 (2H, с), 7,34-7,38 (1H, м), 7,41-7,45 (2H, м), 7,50-7,52 (2H, м), 7,63 (1H, с), 7,80 (1H, дд, J=10,5, 8,9 Гц), 8,40-8,48 (2H, м), 8,48 (1H, с).
Стадия 1-10
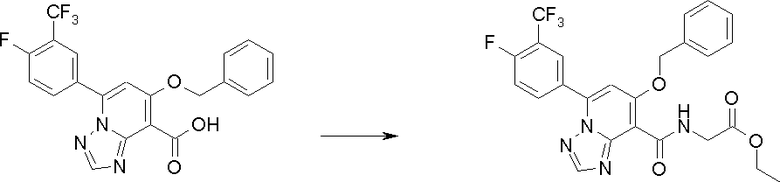
Смешивали соединение (43 г), полученное на стадии 1-9, гидрохлорид сложного этилового эфира глицина (15 г), гидрат 1-гидроксибензотриазола (17 г) и N,N-диметилформамид (430 мл) и при комнатной температуре добавляли триэтиламин (15 мл) и гидрохлорид 1-этил-3-(3-диметиламинопропил)карбодиимида (21 г). После перемешивания при комнатной температуре в течение 1 ч добавляли воду (860 мл) и насыщенный водный раствор гидрокарбоната натрия (215 мл) и собирали выпавшее в осадок твердое вещество путем фильтрования с получением соединения (43 г, 84%), описанного в представленной выше схеме.
1H-ЯМР (CDCl3) δ: 1,33 (3H, т, J=7,1 Гц), 4,28 (2H, кв., J=7,1 Гц), 4,35 (2H, д, J=4,8 Гц), 5,47 (2H, с), 6,95 (1H, с), 7,32-7,43 (4H, м), 7,54 (2H, д, J=7,3 Гц), 8,01 (1H, дд, J=6,4, 2,0 Гц), 8,10-8,14 (1H, м), 8,31 (1H, с), 9,72 (1H, т, J=4,8 Гц).
Стадия 1-11

Смешивали соединение (43 г), полученное на стадии 1-10, и трифторуксусную кислоту (430 мл) и после перемешивания при 80°C в течение 6 ч концентрировали реакционную смесь в условиях пониженного давления. К остатку добавляли метанол (86 мл) и воду (430 мл), перемешивали смесь при комнатной температуре в течение 30 мин и собирали выпавшее в осадок твердое вещество путем фильтрования. Твердое вещество очищали по методу колоночной хроматографии (элюент: хлороформ/этилацетат = 10/1) с получением соединения (28 г, 79%), описанного в представленной выше схеме.
1H-ЯМР (CDCl3) δ: 1,34 (3H, т, J=7,3 Гц), 4,30 (2H, кв., J=7,3 Гц), 4,33 (2H, д, J=5,2 Гц), 6,87 (1H, с), 7,41 (1H, дд, J=9,7, 8,9 Гц), 8,16-8,20 (1H, м), 8,24 (1H, дд, J=6,9, 2,4 Гц), 8,26 (1H, с), 10,15 (1H, т, J=5,2 Гц), 14,13 (1H, с).
Стадия 1-12
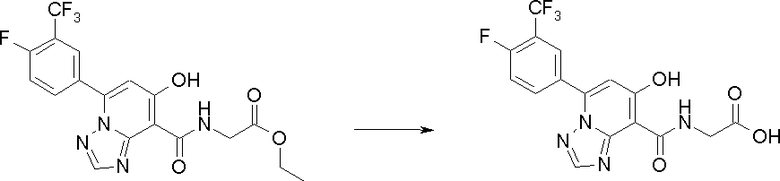
Смешивали соединение (27 г), полученное на стадии 1-11, и 2-пропанол (540 мл), и при комнатной температуре добавляли 4 н. водный раствор гидроксида лития (64 мл). После перемешивания при 70°C в течение 1 ч добавляли 6 н. соляную кислоту (43 мл). Смесь оставляли постепенно охлаждаться при перемешивании, и при 37°C в осадок выпадали кристаллические вещества. Добавляли воду (270 мл) и собирали кристаллические вещества путем фильтрования с получением соединения (22 г, 87%), описанного в представленной выше схеме.
1H-ЯМР (ДМСО-D6) δ: 4,24 (2H, д, J=5,6 Гц), 7,30 (1H, с), 7,77 (1H, дд, J=10,5, 9,3 Гц), 8,36-8,40 (1H, м), 8,47 (1H, д, J=6,9 Гц), 8,60 (1H, с), 9,97 (1H, ушир. c), 14,38 (1H, ушир. c).
Полученное соединение преобразовывали до гидрохлорида в соответствии с общепринятым способом с получением соединения Примера 1.
1H-ЯМР (ДМСО-D6) δ: 4,25 (д, 2H, J=5,6 Гц), 7,31 (c, 1H), 7,73-7,82 (м, 1H), 8,34-8,43 (м, 1H), 8,43-8,51 (м, 1H), 8,61 (c, 1H), 9,99 (т, 1H, J=5,6 Гц).
Пример 2
Получение гидрохлорида [(7-гидрокси-5-фенетил[1,2,4]триазоло[1,5-a]пиридин-8-карбонил)амино]уксусной кислоты
Стадия 2-1
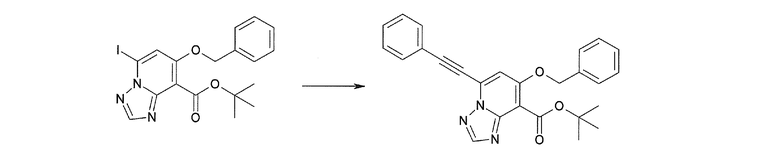
Смешивали соединение (5,00 г), полученное на стадии 1-7, толуол (35 мл) и фенилацетилен (1,34 мл) и при охлаждении на льду последовательно добавляли бис(трифенилфосфин)палладия дихлорид (0,233 г), йодид меди (0,063 г) и триэтиламин (1,85 мл). После перемешивания при комнатной температуре в течение 2 ч к реакционной смеси добавляли 5% водный аммиак (35 мл). Органический слой затем последовательно промывали 5% водным аммиаком, насыщенным водным раствором хлорида аммония и насыщенным солевым раствором и сушили над безводным сульфатом магния. После фильтрования фильтрат концентрировали в условиях пониженного давления и очищали полученный остаток по методу колоночной хроматографии (элюент: гексан/этилацетат = 3/1-1/1). Полученное соединение суспендировали в гексане с получением соединения (3,84 г, 82%), описанного в представленной выше схеме.
1H-ЯМР (ДМСО-D6) δ: 1,48 (9H, с), 5,42 (2H, с), 7,36 (1H, тт, J=7,1, 1,8 Гц),7,40-7,45 (2H, м), 7,48 (2H, дт, J=7,0, 1,9 Гц), 7,51-7,58 (3H, м), 7,72 (2H, дд, J=7,7, 1,6 Гц), 7,78 (1H, с), 8,49 (1H, с).
Стадия 2-2
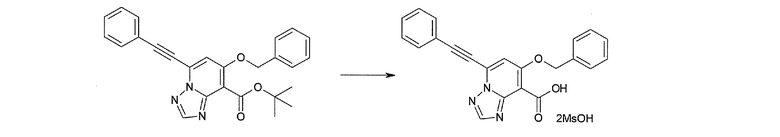
Смешивали соединение (3,84 г), полученное на стадии 2-1, толуол (29 мл) и этилацетат (9,5 мл), и при перемешивании при комнатной температуре по каплям в течение 10 мин добавляли смесь метансульфоновой кислоты (2,34 мл) и этилацетата (2,34 мл). После перемешивания при комнатной температуре в течение 3 ч к реакционной смеси добавляли этилацетат (9,5 мл) и собирали твердое вещество путем фильтрования с получением соединения (4,94 г, 98%), описанного в представленной выше схеме.
1H-ЯМР (ДМСО-D6) δ: 2,38 (6H, с), 5,48 (2H, с), 7,37 (1H, тт, J=7,2, 1,7 Гц), 7,41-7,45 (2H, м), 7,48-7,52 (2H, м), 7,53-7,62 (3H, м), 7,71-7,75 (2H, м), 7,86 (1H, с), 8,67 (1H, с).
Стадия 2-3
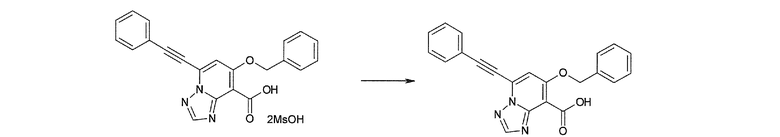
При комнатной температуре смешивали соединение (4,94 г), полученное на стадии 2-2, и N,N-диметилформамид (30 мл) и при 0°C по каплям в течение 10 мин добавляли воду (50 мл). Выпавшее в осадок твердое вещество собирали путем фильтрования с получением соединения (3,20 г, 98%), описанного в представленной выше схеме.
1H-ЯМР (ДМСО-D6) δ: 5,45 (2H, с), 7,36 (1H, тт, J=7,4, 2,1 Гц), 7,43 (2H, т, J=7,3 Гц), 7,49 (2H, д, J=7,5 Гц), 7,52-7,60 (3H, м), 7,72 (2H, дд, J=6,7, 1,9 Гц), 7,78 (1H, с), 8,51 (1H, с), 13,59 (1H, с).
Стадия 2-4
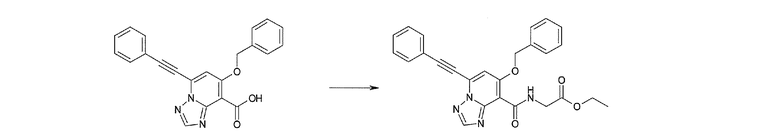
Осуществляли взаимодействие соединения (3,20 г), полученного на стадии 2-3, с гидрохлоридом сложного этилового эфира глицина (1,33 г) способом, аналогичным описанному в Примере 1, стадия 1-10, с получением соединения (3,38 г, 81%), описанного в представленной выше схеме.
1H-ЯМР (ДМСО-D6) δ: 1,21 (3H, т, J=7,1 Гц), 4,10 (2H, д, J=5,7 Гц), 4,13 (2H, кв., J=7,5 Гц), 5,44 (2H, с), 7,34 (1H, тт, J=7,2, 1,7 Гц), 7,38-7,43 (2H, м), 7,52-7,58 (5H, м), 7,71-7,74 (2H, м), 7,75 (1H, с), 8,52 (1H, с), 9,18 (1H, т, J=5,8 Гц).
Стадия 2-5
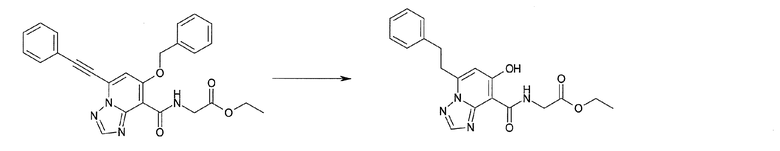
К раствору соединения (3,38 г), полученного на стадии 2-4, в тетрагидрофуране (34 мл) и метаноле (17 мл) добавляли 5% палладированный уголь (0,34 г) и перемешивали смесь в атмосфере водорода и в условиях нормального давления в течение 4 ч. Реакционную смесь фильтровали через целит и концентрировали в условиях пониженного давления. Полученный остаток очищали по методу колоночной хроматографии (элюент: хлороформ/метанол = 20/0-20/1) и суспендировали в гексане/диизопропиловом эфире (1/1) с получением соединения (2,29 г, 83%), описанного в представленной выше схеме.
1H-ЯМР (ДМСО-D6) δ: 1,23 (3H, т, J=7,2 Гц), 3,12 (2H, т, J=7,8 Гц), 3,41 (2H, т, J=7,9 Гц), 4,17 (2H, кв., J=7,1 Гц), 4,29 (2H, д, J=5,7 Гц), 6,82 (1H, с), 7,18-7,32 (5H, м), 8,58 (1H, с), 9,87 (1H, т, J=5,6 Гц), 14,12 (1H, с).
Стадия 2-6
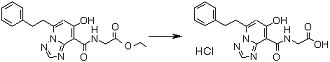
Соединение (2,28 г), полученное на стадии 2-5, гидролизовали способом, аналогичным описанному на стадии 1-12, и преобразовывали полученное соединение до гидрохлорида в соответствии с общепринятым способом с получением указанного в заголовке соединения (2,16 г).
1H-ЯМР (ДМСО-D6) δ: 3,12 (т, 2H, J=7,8 Гц), 3,41 (т, 2H, J=7,8 Гц), 4,21 (д, 2H, J=5,6 Гц), 6,81 (c, 1H), 7,14-7,33 (м, 5H), 8,60 (c, 1H), 9,85 (т, 1H, J=5,6 Гц).
Пример 3
Получение гидрохлорида [(5-бутил-7-гидрокси[1,2,4]триазоло[1,5-a]пиридин-8-карбонил)амино]уксусной кислоты
Стадия 3-1
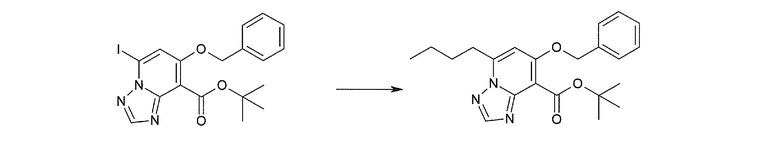
Смешивали соединение (0,2 г), полученное на стадии 1-7, комплекс дихлорида [1,1'-бис(дифенилфосфино)ферроцен]палладия(II) и дихлорметана (1/1, 0,011 г), бутилбороновую кислоту (0,050 г), оксид серебра(I) (0,12 г), карбонат калия (0,15 г) и тетрагидрофуран (1,6 мл), и перемешивали смесь при 80°C в течение 40 ч. Нерастворимый материал отфильтровывали, и концентрировали фильтрат в условиях пониженного давления. Остаток очищали по методу колоночной хроматографии (элюент: гексан/этилацетат = 8/2-6/4) с получением соединения (0,13 г, 77%), описанного в представленной выше схеме.
1H-ЯМР (ДМСО-D6) δ: 0,91 (т, 3H, J=7,7 Гц), 1,28-1,39 (м, 2H), 1,48 (c, 9H), 1,71-1,81 (м, 2H), 3,11 (т, 2H, J=7,7 Гц), 5,38 (c, 2H), 7,23 (c, 1H), 7,32-7,51 (м, 5H), 8,39 (c, 1H).
Стадия 3-2
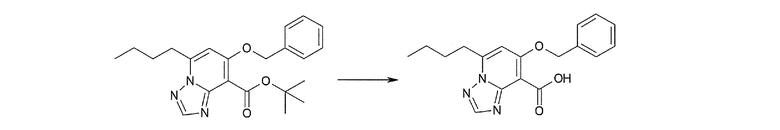
Карбоксилзащитную группу соединения (53,7 г), полученного на стадии 3-1, удаляли способом, аналогичным описанному на стадии 2-2, с получением карбоновой кислоты в виде смеси (69,8 г, 80%) с метансульфоновой кислотой (290% мол.). Смесь обрабатывали способом, аналогичным описанному на стадии 2-3, с получением соединения, описанного в представленной выше схеме, в виде смеси (42,8 г, 99%) с метансульфоновой кислотой (50% мол.).
1H-ЯМР (ДМСО-D6) δ: 0,91 (т, 3H, J=7,7 Гц), 1,29-1,40 (м, 2H), 1,71-1,80 (м, 2H), 2,34 (c, 1,5H), 3,14 (т, 2H, J=7,7 Гц), 5,47 (c, 2H), 7,32-7,45 (м, 4H), 7,48-7,53 (м, 2H), 8,73 (c, 1H).
Стадия 3-3
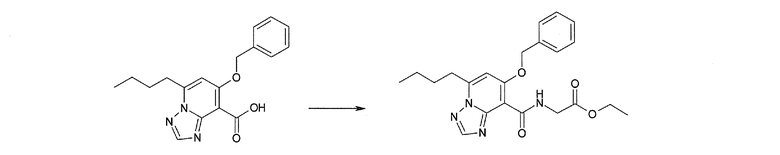
Способом, аналогичным описанному на стадии 1-10, осуществляли взаимодействие соединения (42,8 г), полученного на стадии 3-2, с гидрохлоридом сложного этилового эфира глицина (19,3 г) с получением соединения (43,5 г, 92%), описанного в представленной выше схеме.
1H-ЯМР (CDCl3) δ: 0,94 (т, 3H, J=7,5 Гц), 1,31 (т, 3H, J=7,1 Гц), 1,34-1,43 (м, 2H), 1,70-1,79 (м, 2H), 3,11 (т, 2H, J=7,5 Гц), 4,26 (кв., 2H, J=7,1 Гц), 4,33 (д, 2H, J=5,2 Гц), 5,41 (c, 2H), 6,69 (c, 1H), 7,29-7,55 (м, 5H), 8,28 (c, 1H), 9,77 (т, 1H, J=5,2 Гц).
Стадия 3-4
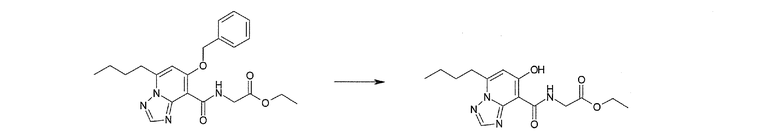
Способом, аналогичным описанному на стадии 2-5, из соединения (7,2 г), полученного на стадии 3-3, получали соединение (5,0 г, 90%), описанное в представленной выше схеме.
1H-ЯМР (ДМСО-D6) δ: 0,93 (т, 3H, J=7,5 Гц), 1,22 (т, 3H, J=7,3 Гц), 1,33-1,44 (м, 2H), 1,71-1,81 (м, 2H), 3,09 (т, 2H, J=7,5 Гц), 4,16 (кв., 2H, J=7,3 Гц), 4,29 (д, 2H, J=5,6 Гц), 6,85 (c, 1H), 8,54 (c, 1H), 9,88 (ушир. с, 1H), 14,14 (c, 1H).
Стадия 3-5

Способом, аналогичным описанному на стадии 1-12, из соединения (4,94 г), полученного на стадии 3-4, получали соединение (4,56 г), описанное в представленной выше схеме.
1H-ЯМР (ДМСО-D6) δ: 0,93 (т, 3H, J=7,5 Гц), 1,33-1,44 (м, 2H), 1,71-1,80 (м, 2H), 3,10 (т, 2H, J=7,5 Гц), 4,20 (д, 2H, J=5,2 Гц), 6,85 (c, 1H), 8,55 (c, 1H), 9,84 (ушир. с, 1H), 14,26 (ушир. с, 1H).
Стадия 3-6

Смешивали соединение (4,56 г), полученное на стадии 3-5, и 4 н. раствор соляной кислоты в этилацетате (91 мл), и перемешивали смесь при комнатной температуре в течение 1,5 ч. Реакционную суспензию концентрировали в условиях пониженного давления, добавляли к остатку гексан (100 мл), и дважды концентрировали смесь в условиях пониженного давления. К остатку добавляли смешанный раствор диэтилового эфира/гексана (1/2, 100 мл), и перемешивали смесь в течение 30 мин. Твердое вещество собирали путем фильтрования с получением указанного в заголовке соединения (4,88 г, 96%).
1H-ЯМР (ДМСО-D6) δ: 0,93 (т, 3H, J=7,5 Гц), 1,34-1,44 (м, 2H), 1,71-1,80 (м, 2H), 3,10 (т, 2H, J=7,5 Гц), 4,21 (д, 2H, J=5,6 Гц), 6,86 (c, 1H), 8,57 (c, 1H), 9,83 (т, 1H, J=5,6 Гц).
Пример 4
Получение гидрохлорида [(5,6-диэтил-7-гидрокси[1,2,4]триазоло[1,5-a]пиридин-8-карбонил)амино]уксусной кислоты
Стадия 4-1
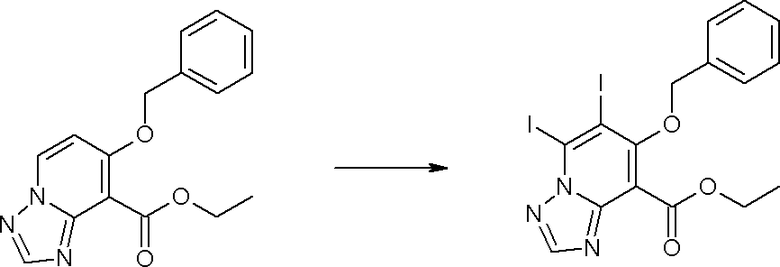
Под струей азота смешивали соединение (3,98 г), полученное способом, аналогичным описанному на стадии 1-6, и тетрагидрофуран (40 мл), и при охлаждении сухим льдом/денатурированным этанолом по каплям добавляли раствор йода (3,4 г) в тетрагидрофуране (16 мл). К этой смеси по каплям в течение 10 мин добавляли 1M бис(триметилсилил)амид лития (26,8 мл). После перемешивания как есть в течение 2,5 ч, реакционную смесь вливали при охлаждении на льду в смесь насыщенного водного раствора хлорида аммония (40 мл) и воды (40 мл). К этой смеси добавляли сульфит натрия (1,7 г), и отделяли от смеси органический слой. Органический слой концентрировали в условиях пониженного давления, полученный остаток объединяли с водным слоем, и дважды экстрагировали смесь этилацетатом. Органический слой промывали насыщенным солевым раствором (70 мл), сушили над сульфатом натрия, отфильтровывали сульфат натрия, и концентрировали фильтрат в условиях пониженного давления. Полученный остаток очищали по метолу колоночной хроматографии. Полученный продукт очистки перекристаллизовывали из гептана/хлороформа с получением соединения (0,295 г, 4%), описанного в представленной выше схеме.
1H-ЯМР (CDCl3) δ: 1,38 (т, 3H, J=7,1 Гц), 4,50 (кв., 2H, J=7,1 Гц), 5,22 (c, 2H), 7,36-7,56 (м, 5H), 8,33 (c, 1H).
Стадия 4-2
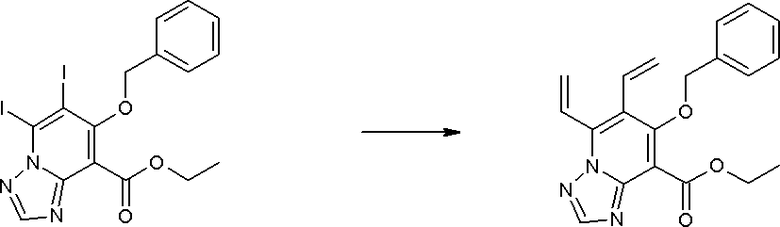
Способом, аналогичным описанному в Примере 1, стадия 1-8, из соединения (0,1 г), полученного на стадии 4-1, получали соединение (0,019 г, 30%), описанное в представленной выше схеме.
1H-ЯМР (CDCl3) δ: 1,38 (т, 3H, J=7,1 Гц), 4,50 (кв., 2H, J=7,1 Гц), 5,09 (c, 2H), 5,73 (дд, 1H, J=17,7, 1,4 Гц), 5,76 (дд, 1H, J=11,5, 1,4 Гц), 6,05 (дд, 1H, J=11,5, 1,4 Гц), 6,77 (дд, 1H, J=17,7, 11,7 Гц), 6,87 (дд, 1H, J=17,7, 1,2 Гц), 7,14 (дд, 1H, J=17,7, 11,5 Гц), 7,35-7,44 (м, 5H), 8,37 (c, 1H).
Стадия 4-3
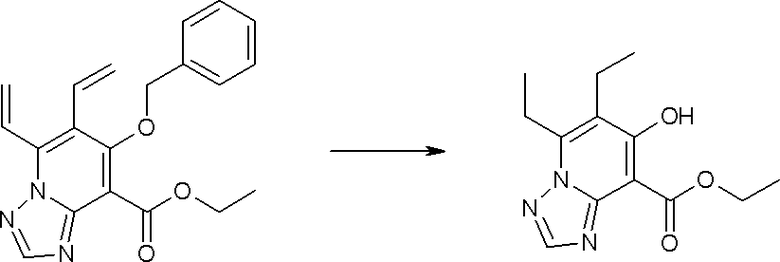
Способом, аналогичным описанному в Примере 2, стадия 2-5, из соединения (0,036 г), полученного на стадии 4-2, получали соединение (0,020 г, 75%), описанное в представленной выше схеме.
1H-ЯМР (CDCl3) δ: 1,23 (т, 3H, J=7,5 Гц), 1,36 (т, 3H, J=7,5 Гц), 1,52 (т, 3H, J=7,3 Гц), 2,78 (кв., 2H, J=7,5 Гц), 3,25 (кв., 2H, J=7,5 Гц), 4,64 (кв., 2H, J=7,3 Гц), 8,26 (c, 1H), 13,10 (c, 1H).
Стадия 4-4
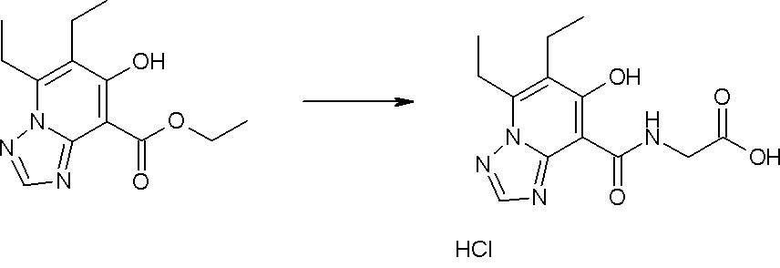
Смешивали соединение (0,020 г), полученное на стадии 4-3, и 2-метоксиэтанол (2 мл), и добавляли натриевую соль глицина (0,030 г). После перемешивания при 130°C в течение 1,5 ч смесь охлаждали до комнатной температуры. К реакционной смеси добавляли 1 н. соляную кислоту (0,34 мл) и воду (10 мл), и перемешивали смесь. Осадок собирали путем фильтрования и сушили в условиях пониженного давления. К полученному твердому веществу добавляли этилацетат (1 мл) и 4 н. соляную кислоту в этилацетате (0,1 мл), и перемешивали смесь при комнатной температуре в течение 20 мин. Твердое вещество собирали путем фильтрования с получением указанного в заголовке соединения (0,052 мг, 67%).
1H-ЯМР (ДМСО-D6) δ: 1,15 (т, 3H, J=7,5 Гц), 1,29 (т, 3H, J=7,5 Гц), 2,72 (кв., 2H, J=7,5 Гц), 3,20 (кв., 2H, J=7,6 Гц), 4,21 (д, 2H, J=5,6 Гц), 8,52 (c, 1H), 9,95 (т, 1H, J=5,6 Гц).
Пример 5
Получение гидрохлорида [(7-гидрокси-6-фенетил[1,2,4]триазоло[1,5-a]пиридин-8-карбонил)амино]уксусной кислоты
Стадия 5-1
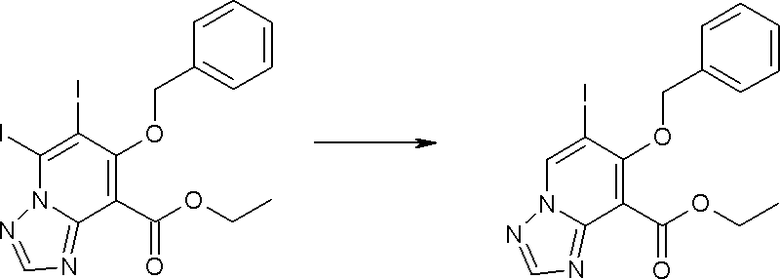
Смешивали соединение (1,31 г), полученное способом, аналогичным описанному в Примере 4, стадия 4-1, тетракис(трифенилфосфин)палладий(0) (0,137 г) и тетрагидрофуран (15 мл), и при охлаждении на льду добавляли гидрид три-н-бутилолова (0,7 мл). После перемешивания при охлаждении на льду в течение 20 мин смесь перемешивали при комнатной температуре в течение 20 мин и концентрировали в условиях пониженного давления. Полученный остаток очищали по методу колоночной хроматографии (элюент: гексан/этилацетат = 10/0-1/1) с получением соединения (0,44 г, 44%), описанного в представленной выше схеме.
1H-ЯМР (CDCl3) δ: 1,39 (т, 3H, J=7,2 Гц), 4,51 (кв., 2H, J=7,2 Гц), 5,23 (c, 2H), 7,36-7,56 (м, 5H), 8,31 (c, 1H), 8,99 (c, 1H).
Стадия 5-2
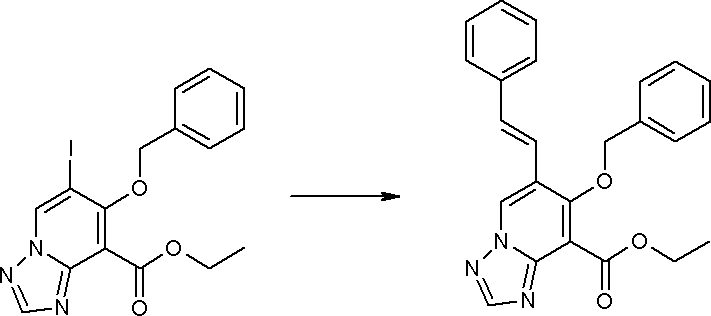
Способом, аналогичным описанному в Примере 1, стадия 1-8, из соединения (0,070 г), полученного на стадии 5-1, получали соединение (0,078 г, 109%), описанное в представленной выше схеме.
1H-ЯМР (CDCl3) δ: 1,43 (т, 3H, J=7,1 Гц), 4,54 (кв., 2H, J=7,1 Гц), 5,17 (c, 2H), 7,13 (д, 1H, J=9,3 Гц), 7,29-7,45 (м, 11H), 8,34 (c, 1H), 8,80 (c, 1H).
Стадия 5-3
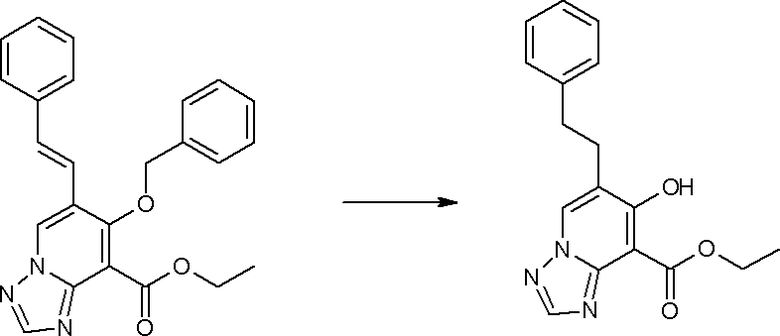
Способом, аналогичным описанному в Примере 2, стадия 2-5, из соединения (0,070 г), полученного на стадии 5-2, получали соединение (0,037 г, 72%), описанное в представленной выше схеме.
1H-ЯМР (CDCl3) δ: 1,54 (т, 3H, J=7,2 Гц), 3,00 (c, 4H), 4,66 (кв., 2H, J=7,2 Гц), 7,16-7,31 (м, 5H), 8,20 (c, 1H), 8,23 (c, 1H), 13,17 (c, 1H).
Соединение, полученное на этой стадии, преобразовывали до гидрохлорида способом, аналогичным описанному в Примере 4, стадия 4-4, и в соответствии с общепринятым способом с получением указанного в заголовке соединения.
1H-ЯМР (ДМСО-D6) δ: 2,93 (c, 4H), 4,22 (д, 2H, J=5,7 Гц), 7,19 (тт, 1H, J=7,1, 1,8 Гц), 7,23-7,31 (м, 4H), 8,50 (c, 1H), 8,78 (c, 1H), 9,97 (c, 1H).
Пример 6
Получение гидрохлорида [(5-бутил-6-хлор-7-гидрокси[1,2,4]триазоло[1,5-a]пиридин-8-карбонил)амино]уксусной кислоты
Стадия 6-1
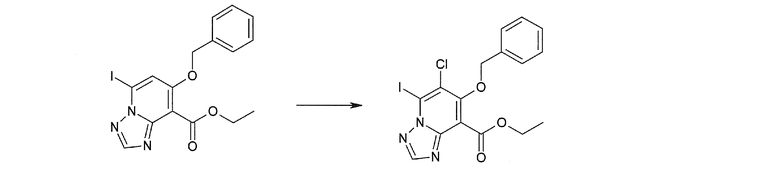
Смешивали соединение (200 мг), полученное способом, аналогичным описанному в Примере 4, стадия 4-1, гексахлорэтан (224 мг) и тетрагидрофуран (4,0 мл), и при -78°C добавляли бис(триметилсилил)амид лития (0,473 мл). После перемешивания при -78°C в течение 2 ч смесь медленно нагревали до -40°C. После этого смесь по каплям добавляли к насыщенному водному раствору гидрокарбоната натрия и добавляли этилацетат. Органический слой отделяли от смеси и дважды экстрагировали водный слой этилацетатом. Органические слои объединяли, дважды промывали насыщенным солевым раствором, сушили над сульфатом натрия и концентрировали в условиях пониженного давления. Концентрированный остаток очищали по методу колоночной хроматографии (элюент: гексан/этилацетат = 3/1-2/1) с получением неочищенного продукта (180 мг), содержащего описанное в представленной выше схеме соединение в качестве главного компонента.
Стадия 6-2
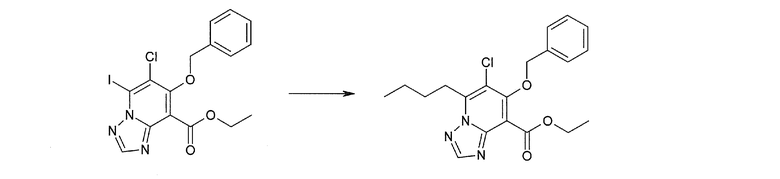
Способом, аналогичным описанному в Примере 3, стадия 3-1, из соединения (180 мг), полученного на стадии 6-1, получали соединение (71 мг), описанное в представленной выше схеме.
1H-ЯМР (ДМСО-D6) δ: 0,94 (3H, т, J=7,5 Гц), 1,28 (3H, т, J=7,1 Гц), 1,37-1,47 (2H, м), 1,68-1,75 (2H, м), 3,32-3,37 (2H, м), 4,37 (2H, кв., J=7,1 Гц), 5,19 (2H, с), 7,37-7,52 (5H, м), 8,57 (1H, с).
Стадия 6-3
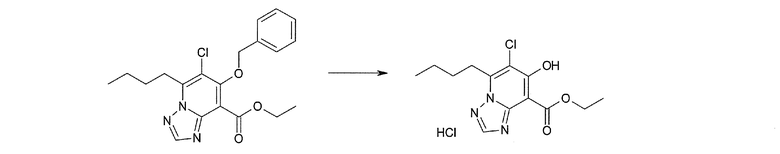
Смешивали соединение (70 мг), полученное на стадии 6-2, и трифторуксусную кислоту (1 мл) и перемешивали смесь при комнатной температуре в течение 3 ч. К этой реакционной смеси добавляли хлороформ и концентрировали смесь в условиях пониженного давления. К концентрированному остатку добавляли 4 н. соляную кислоту в этилацетате (1 мл) и перемешивали смесь. Добавляли гексан (3 мл) и перемешивали смесь. Твердое вещество собирали путем фильтрования с получением соединения (50 мг, 83%), описанного в представленной выше схеме.
1H-ЯМР (ДМСО-D6) δ: 0,92 (3H, т, J=7,3 Гц), 1,31 (3H, т, J=7,1 Гц), 1,37-1,45 (2H, м), 1,62-1,69 (2H, м), 3,17 (2H, т, J=7,7 Гц), 4,35 (2H, кв., J=7,1 Гц), 8,69 (1H, с).
Стадия 6-4
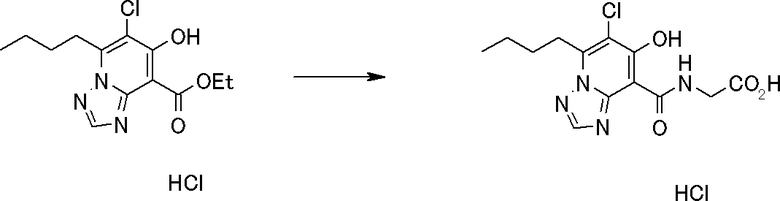
Способом, аналогичным описанному в Примере 4, стадия 4-4, из соединения (50 мг), полученного на стадии 6-3, получали соединение (48 мг, 88%), описанное в представленной выше схеме.
1H-ЯМР (ДМСО-D6) δ: 0,93 (т, 3H, J=7,3 Гц), 1,36-1,47 (м, 2H), 1,64-1,72 (м, 2H), 3,15-3,28 (м, 2H), 4,15 (д, 2H, J=2,8 Гц), 8,74 (ушир. с, 1H), 10,20 (ушир. с, 1H).
Пример 7
Получение гидрохлорида [(7-гидрокси-2-метил-5-фенетил[1,2,4]триазоло[1,5-a]пиридин-8-карбонил)амино]уксусной кислоты
Стадия 7-1
Смешивали 2,4,6-трихлорпиридин (50 г) и N,N-диметилформамид (400 мл) и при охлаждении на льду порциями добавляли гидрид натрия (60% суспензия в масле, 11,5 г). К этой смеси при охлаждении на льду по каплям в течение 40 мин добавляли бензиловый спирт (28 мл) и перемешивали смесь при той же температуре в течение 3 ч. При охлаждении на льду по каплям добавляли воду (550 мл) и собирали полученное твердое вещество путем фильтрования с получением соединения (50 г, 72%), описанного в представленной выше схеме.
1H-ЯМР (CDCl3) δ: 5,11 (c, 2H), 6,86 (c, 2H), 7,36-7,46 (м, 5H).
Стадия 7-2
При охлаждении на бане с сухим льдом/ацетоном смешивали тетрагидрофуран (250 мл) и н-бутиллитий (1,65M, 119 мл) и по каплям в течение 30 мин добавляли раствор соединение (50 г), полученного на стадии 7-1, в тетрагидрофуране (110 мл). К этой смеси по каплям в течение 1 ч добавляли раствор ди-трет-бутилдикарбоната (45 мл) в тетрагидрофуране (100 мл) и перемешивали смесь при той же температуре в течение 30 мин. Для гашения реакции добавляли воду (450 мл), для разделения органического слоя добавляли этилацетат (200 мл) и промывали органический слой водой (200 мл) и насыщенным солевым раствором (200 мл). Органический слой концентрировали в условиях пониженного давления, очищали остаток по методу колоночной хроматографии (элюент: гексан/этилацетат = 50/0-11/1), а затем суспендировали полученное твердое вещество в гексане (100 мл) с получением соединения (15,7 г, 31%), описанного в представленной выше схеме.
1H-ЯМР (CDCl3) δ: 1,53 (c, 9H), 5,11 (c, 2H), 6,86 (c, 1H), 7,32-7,46 (м, 5H).
Стадия 7-3
Смешивали соединение (15 г), полученное на стадии 7-2, и 1,4-диоксан (150 мл), добавляли карбонат калия (18 г), фенилвинилбороновую кислоту (7,1 г), комплекс дихлорида [1,1'-бис(дифенилфосфино)ферроцен]палладия(II) и дихлорметана (1/1, 1,8 г) и воду (45 мл) и перемешивали смесь при 80°C в течение 1,5 ч при нагревании. Добавляли фенилвинилбороновую кислоту (0,64 г) и перемешивали смесь в течение 1,5 ч. Смесь охлаждали до комнатной температуры, и для разделения органического слоя добавляли воду, этилацетат и насыщенный солевой раствор. Органический слой концентрировали в условиях пониженного давления и очищали полученный остаток по методу колоночной хроматографии (элюент: хлороформ). К полученному твердому веществу добавляли изопропиловый спирт (100 мл) и суспендировали смесь при 70°C в течение 0,5 ч при охлаждении на льду с получением соединения (11,5 г, 62%), описанного в представленной выше схеме.
1H-ЯМР (CDCl3) δ: 1,55 (c, 9H), 5,21 (c, 2H), 6,87 (c, 1H), 7,01 (д, 1H, J=15,9 Гц), 7,30-7,44 (м, 8H), 7,56 (д, 2H, J=7,1 Гц), 7,65 (д, 1H, J=16,1 Гц).
Стадия 7-4
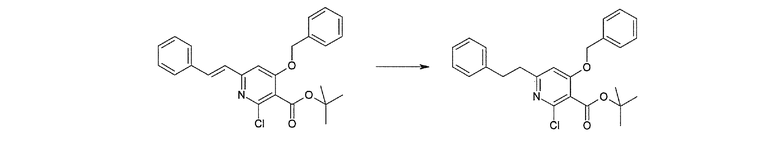
Смешивали соединение (11,5 г), полученное на стадии 7-3, этилацетат (120 мл) и 2% платинированный уголь (2,5 г) и перемешивали смесь в атмосфере водорода (3,8 кгс/см2) при комнатной температуре в течение 23 ч. Реакционную смесь фильтровали через целит и концентрировали фильтрат в условиях пониженного давления с получением соединения (11,5 г, 100%), описанного в представленной выше схеме.
1H-ЯМР (CDCl3) δ: 1,55 (c, 9H), 3,02 (ушир. с, 4H), 5,05 (c, 2H), 6,55 (c, 1H), 7,16-7,41 (м, 10H).
Стадия 7-5
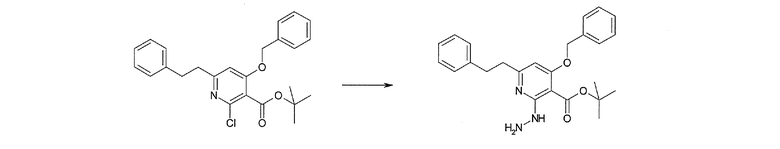
Способом, аналогичным описанному в Примере 1, стадия 1-4, из соединения (11,5 г), полученного на стадии 7-4, получали соединение (11,9 г, 104%), описанное в представленной выше схеме.
1H-ЯМР (CDCl3) δ: 1,39 (c, 9H), 2,92 (т, 2H, J=8,2 Гц), 3,03 (т, 2H, J=8,2 Гц), 4,05 (ушир. с, 2H), 5,00 (c, 2H), 6,10 (c, 1H), 7,16-7,43 (м, 10H), 8,10 (c, 1H).
Стадия 7-6
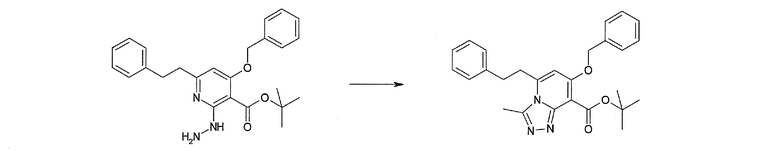
Смешивали соединение (126 мг), полученное на стадии 7-5, моногидрат пара-толуолсульфоновой кислоты (50 мг) и триметилортоформиат (1 мл), добавляли толуол (1 мл) и нагревали смесь при 60°C в течение 1 ч. Реакционную смесь при комнатной температуре по каплям добавляли к насыщенному водному раствору гидрокарбоната натрия и добавляли этилацетат для разделения органического слоя. Органический слой концентрировали в условиях пониженного давления и очищали концентрированный остаток по методу тонкослойной хроматографии (элюент: хлороформ/метанол = 9/1) с получением соединения (32 мг, 24%), описанного в представленной выше схеме.
1H-ЯМР (CD3OD) δ: 1,51 (9H, с), 2,93 (3H, с), 3,06 (2H, т, J=7,6 Гц), 3,52 (2H, т, J=7,6 Гц), 5,16 (2H, с), 6,72 (1H, с), 7,14-7,48 (10H, м).
Стадия 7-7
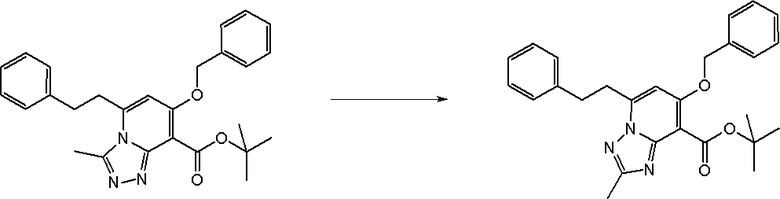
Способом, аналогичным описанному в Примере 1, стадия 1-6, из соединения (55 мг), полученного на стадии 7-6, получали соединение (29 мг, 53%), описанное в представленной выше схеме.
1H-ЯМР (CD3OD) δ: 1,49 (9H, с), 2,52 (3H, с), 3,13 (2H, т, J=7,6 Гц), 3,42 (2H, т, J=7,7 Гц), 5,16 (2H, с), 6,83 (1H, с), 7,15-7,19 (3H, м), 7,24-7,28 (2H, м), 7,32-7,44 (5H, м).
Стадия 7-8
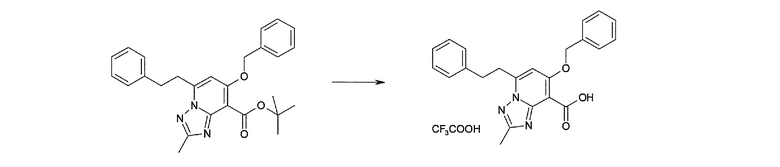
Смешивали соединение (54 мг), полученное на стадии 7-7, и хлороформ (0,5 мл), добавляли трифторуксусную кислоту (0,22 мл) и перемешивали смесь при комнатной температуре в течение 2 ч. Реакционную смесь концентрировали в условиях пониженного давления и подвергали остаток азеотропной перегонке с толуолом с получением неочищенного продукта (69 мг), содержащего соединение, описанное в представленной выше схеме, в качестве главного компонента.
Соединение, полученное на этой стадии, обрабатывали способом, аналогичным описанному в Примере 1, стадии 1-10-1-12, и преобразовывали полученное соединение до гидрохлорида общепринятым способом с получением указанного в заголовке соединения.
1H-ЯМР (ДМСО-D6) δ: 2,52 (c, 3H), 3,10 (т, 2H, J=7,8 Гц), 3,35 (т, 2H, J=7,8 Гц), 4,19 (д, 2H, J=5,7 Гц), 6,69 (c, 1H), 7,18-7,31 (м, 5H), 9,81 (т, 1H, J=5,5 Гц).
Пример 8
Получение гидрохлорида {[8-(3,3-диметилбутил)-6-гидрокси[1,2,4]триазоло[1,5-a]пиридин-5-карбонил]амино}уксусной кислоты
Стадия 8-1
2-Амино-3,5-дибромпиридин (50,4 г), 2,5-гександион (23,5 г) и моногидрат пара-толуолсульфоновой кислоты (2,7 г) растворяли в толуоле (300 мл) и нагревали смесь с обратным холодильником в течение 5 ч, удаляя при этом воду. Смесь оставляли охлаждаться до комнатной температуры, добавляли этилацетат и последовательно промывали смесь насыщенным водным раствором гидрокарбоната натрия, водой и насыщенным солевым раствором (каждым один раз). Органический слой сушили над сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления с получением неочищенного продукта (66,6 г), описанного в представленной выше схеме соединения.
1H-ЯМР (CDCl3) δ: 2,00 (6H, с), 5,92 (2H, с), 8,22 (1H, д, J=2,4 Гц), 8,63 (1H, д, J=2,4 Гц).
Стадия 8-2
Способом, аналогичным описанному в Примере 1, стадия 1-3, из соединения (66,6 г), полученного на стадии 8-1, получали соединение (58,4 г, 82%), описанное в представленной выше схеме.
1H-ЯМР (CDCl3) δ: 1,99 (6H, с), 5,15 (2H, с), 5,89 (2H, с), 7,37-7,48 (5H, м), 7,64 (1H, д, J=2,8 Гц), 8,31 (1H, д, J=2,8 Гц).
Стадия 8-3
Смешивали соединение (58,4 г), полученное на стадии 8-2, хлорид гидроксиламмония (233 г), этанол (600 мл) и воду (350 мл). К этой смеси при комнатной температуре по каплям добавляли триэтиламин (46 мл) и этанол (100 мл) и нагревали смесь с обратным холодильником на бане с температурой 95°C в течение 89 ч. Реакционную смесь охлаждали на льду и последовательно добавляли 50% водный раствор гидроксида натрия (96 мл), 8 н. водный раствор гидроксида натрия (134 мл) и насыщенный водный раствор гидрокарбоната натрия (50 мл). При комнатной температуре добавляли воду (1800 мл), перемешивали смесь в течение 1 ч и собирали выпавшее в осадок твердое вещество путем фильтрования. Выпавшее в осадок твердое вещество сушили в условиях пониженного давления в течение ночи и суспендировали неочищенный продукт (49 г) в 2-пропаноле (120 мл) с получением соединения (34,3 г, 75%), описанного в представленной выше схеме.
1H-ЯМР (CDCl3) δ: 4,60 (2H, ушир. c), 5,00 (2H, с), 7,31-7,40 (5H, м), 7,41 (1H, д, J=2,8 Гц), 7,83 (1H, д, J=2,8 Гц).
Стадия 8-4
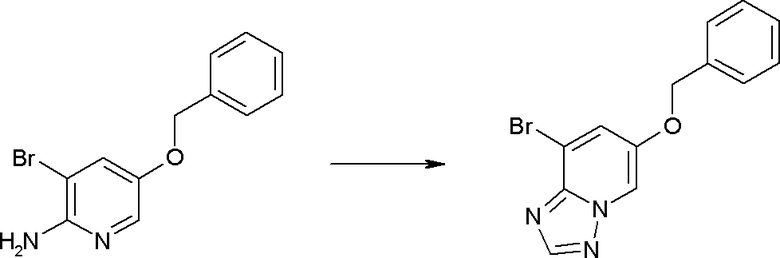
Смешивали соединение (7,25 г), полученное на стадии 8-3, N,N-диметилформамид (11 мл) и диметилацеталь N,N-диметилформамида (11 мл) и перемешивали смесь при нагревании при 130°C в течение 15 мин. После перемешивания при комнатной температуре в течение 20 мин смесь концентрировали в условиях пониженного давления. К полученному остатку добавляли метанол (58 мл) и пиридин (4,2 мл), и при охлаждении на льду добавляли к смеси гидроксиламин-O-сульфоновую кислоту (4,1 г). После перемешивания при комнатной температуре в течение ночи при охлаждении на льду добавляли воду (29 мл) и насыщенный водный раствор гидрокарбоната натрия (58 мл) и перемешивали смесь при комнатной температуре в течение 1 ч. Полученное твердое вещество собирали путем фильтрования с получением соединения (6,13 г, 78%), описанного в представленной выше схеме.
1H-ЯМР (CDCl3) δ: 5,09 (c, 2H), 7,36-7,45 (м, 5H), 7,66 (д, 1H, J=2,0 Гц), 8,19 (д, 1H, J=2,0 Гц), 8,29 (c, 1H).
Стадия 8-5
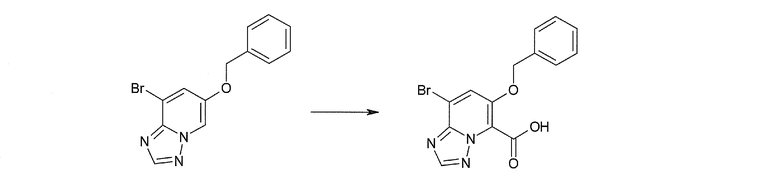
Способом, аналогичным описанному в Примере 1, стадия 1-1, из соединения (10 г), полученного на стадии 8-4, получали соединение (5,67 г, 49%), описанное в представленной выше схеме.
1H-ЯМР (ДМСО-D6) δ: 5,33 (c, 2H), 7,31-7,49 (м, 5H), 8,37 (c, 1H), 8,56 (c, 1H).
Стадия 8-6
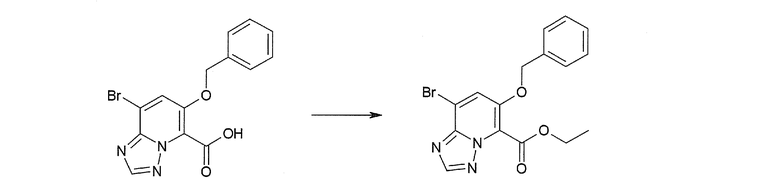
К соединению (5,68 г), полученному на стадии 8-5, добавляли толуол (57 мл) и при 80°C тремя порциями добавляли диэтилацеталь N,N-диметилформамида (4,5 мл). После завершения реакции смесь концентрировали в условиях пониженного давления и очищали полученный остаток по методу колоночной хроматографии (элюент: хлороформ/этилацетат = 10/1-4/1). К полученному соединению добавляли гексан и суспендировали выпавшее в осадок твердое вещество с получением соединения (4,68 г, 69%), описанного в представленной выше схеме.
1H-ЯМР (CDCl3) δ: 1,39 (т, 3H, J=7,1 Гц), 4,53 (кв., 2H, J=7,1 Гц), 5,21 (c, 2H), 7,35-7,43 (м, 5H), 7,72 (c, 1H), 8,38 (c, 1H).
Стадия 8-7
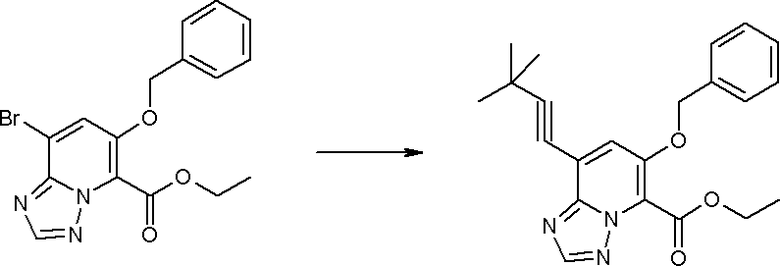
В бутыль с винтовой крышкой добавляли соединение (100 мг), полученное на стадии 8-6, трет-бутилацетилен (0,1 мл), дихлорид бис(трифенилфосфин)палладия(II) (18 мг) и йодид меди(I) (5 мг). К этой смеси добавляли тетрагидрофуран (0,4 мл) и триэтиламин (0,8 мл) и плотно закрывали бутыль. Смесь перемешивали при комнатной температуре в течение 1 ч, пропускали через небольшое количество силикагеля и концентрировали в условиях пониженного давления. Остаток дважды очищали по методу тонкослойной хроматографии (элюент: гексан/этилацетат = 2/1) с получением соединения (85 мг, 85%), описанного в представленной выше схеме.
Стадия 8-8
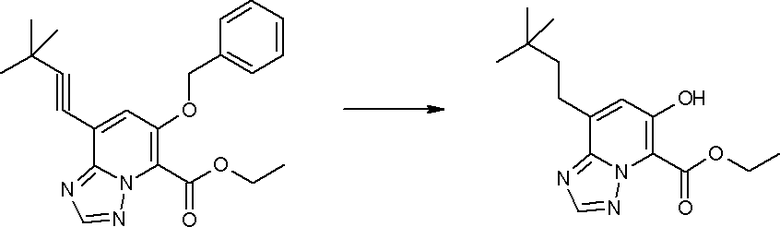
Способом, аналогичным описанному в Примере 2, стадия 2-5, из соединения (85 мг), полученного на стадии 8-7, получали соединение (62 мг, 94%), описанное в представленной выше схеме.
Способом, аналогичным описанному в Примере 4, стадия 4-4, указанное в заголовке соединение получали из соединения, полученного на этой стадии.
1H-ЯМР δ: 0,98 (c, 9H), 1,57-1,66 (м, 2H), 2,89-2,99 (м, 2H), 4,25 (д, 2H, J=5,4 Гц), 7,42 (c, 1H), 8,64 (c, 1H), 10,42 (т, 1H, J=5,4 Гц), 13,28 (c, 1H).
Пример 9
Получение [(7-гидрокси-6-фенил[1,2,4]триазоло[4,3-a]пиридин-8-карбонил)амино]уксусной кислоты
Стадия 9-1
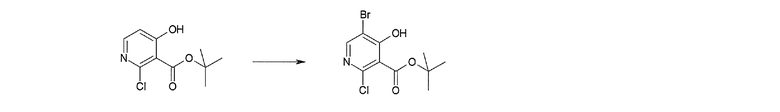
Гидроксилзащитную группу соединения, полученного в Примере 1, стадия 1-3, удаляли способом, аналогичным описанному в Примере 2, стадия 2-5. К раствору полученного соединения (3,30 г) в хлороформе (30 мл) при охлаждении на льду добавляли N-бромсукцинимид (2,82 г). После перемешивания при комнатной температуре в течение 5 ч реакционную смесь разделяли путем добавления насыщенного водного раствора гидрокарбоната натрия (20 мл). Органический слой затем промывали водным раствором сульфита натрия и насыщенным солевым раствором и сушили над безводным сульфатом магния. После фильтрования смесь концентрировали в условиях пониженного давления с получением соединения (5,37 г), описанного в представленной выше схеме, в виде неочищенного продукта.
1H-ЯМР (CDCl3) δ: 1,66 (9H, с), 8,39 (1H, с), 12,77 (1H, с).
Стадия 9-2
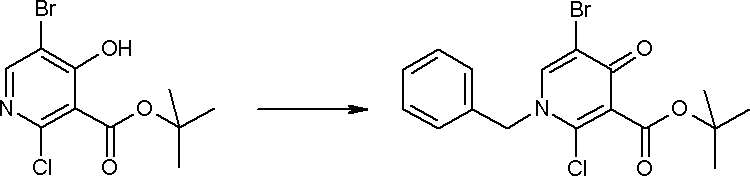
К раствору соединения (5,37 г), полученного на стадии 9-1, в N,N-диметилформамиде при охлаждении на льду добавляли карбонат калия (2,19 г) и бензилбромид (1,88 мл) и перемешивали смесь при комнатной температуре в течение 20 ч. Реакционную смесь разделяли путем добавления воды (50 мл) и этилацетата (50 мл). Органический слой затем промывали насыщенным солевым раствором, сушили над безводным сульфатом магния и концентрировали в условиях пониженного давления. Остаток очищали по методу колоночной хроматографии (элюент: гексан/этилацетат = 3/1-1/2) с получением соединения (3,73 г, 65% в 2 этапа), описанного в представленной выше схеме.
1H-ЯМР (CDCl3) δ: 1,58 (9H, с), 5,21 (2H, с), 7,19-7,21 (2H, м), 7,37-7,46 (3H, м), 7,76 (1H, с).
Стадия 9-3
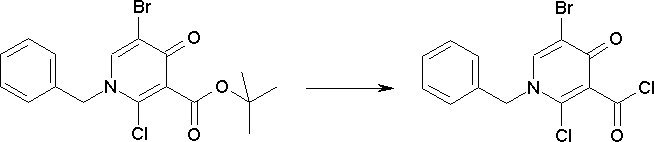
К соединению (610 мг), полученному на стадии 9-2, при охлаждении на льду добавляли трифторуксусную кислоту (180 мл), и перемешивали смесь при комнатной температуре в течение 2 ч. Реакционную смесь разбавляли хлороформом (6 мл) и концентрировали в условиях пониженного давления. К остатку снова добавляли хлороформ (6 мл), добавляли тионилхлорид (0,18 мл) и N,N-диметилформамид (одна капля) и нагревали смесь при 70°C в течение 30 мин. Полученный раствор концентрировали в условиях пониженного давления с получением соединения (540 мг, 98%), описанного в представленной выше схеме.
Стадия 9-4
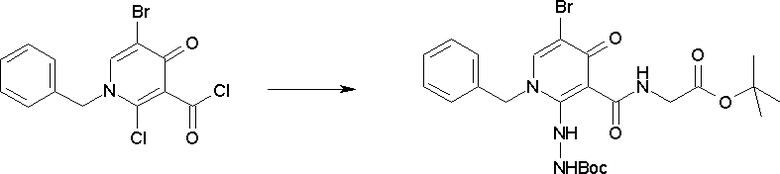
К раствору соединения (540 мг), полученного на стадии 9-3, в тетрагидрофуране (6 мл) при охлаждении на льду добавляли диизопропилэтиламин (0,29 мл) и сложный трет-бутиловый эфир глицина (0,21 мл) и перемешивали смесь в течение 2 ч. К реакционной смеси добавляли диизопропилэтиламин (0,29 мл) и трет-бутилкарбазат и нагревали смесь при 70°C в течение 3 ч. Реакционную смесь концентрировали в условиях пониженного давления и очищали остаток по методу колоночной хроматографии (элюент: гексан/этилацетат = 3/1-3/2) с получением соединения (790 мг, 92%), описанного в представленной выше схеме.
1H-ЯМР (CDCl3) δ: 1,30 (9H, с), 1,48 (9H, с), 4,05 (2H, д, J=5,5 Гц), 5,24 (2H, с), 6,27 (1H, с), 7,14 (2H, т, J=4,1 Гц), 7,34-7,41 (3H, м), 7,47 (1H, с), 11,50 (1H, т, J=5,4 Гц), 11,83 (1H, с).
Стадия 9-5
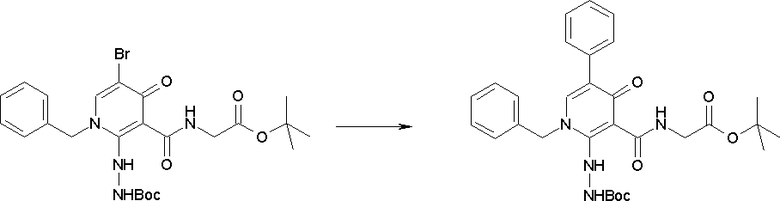
Способом, аналогичным описанному в Примере 1, стадия 1-8, из соединения (330 мг), полученного на стадии 9-4, получали соединение (280 г, 84%), описанное в представленной выше схеме.
1H-ЯМР (CDCl3) δ: 1,31 (9H, с), 1,48 (9H, с), 4,06 (2H, д, J=5,5 Гц), 5,30 (2H, с), 6,16 (1H, с), 7,17 (2H, д, J=7,4 Гц), 7,18 (1H, с), 7,28-7,40 (6H, м), 7,43-7,46 (2H, м), 11,79 (1H, с), 11,85 (1H, т, J=5,4 Гц).
Стадия 9-6
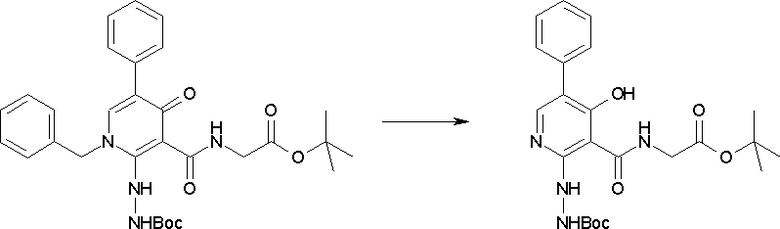
Способом, аналогичным описанному в Примере 2, стадия 2-5, из соединения (120 мг), полученное на стадии 9-5, получали соединение (86 мг, 87%), описанное в представленной выше схеме.
1H-ЯМР (CDCl3) δ: 1,45 (9H, с), 1,49 (9H, с), 4,15 (2H, с), 7,06-7,29 (8H, м), 10,40 (1H, с), 10,95 (1H, с).
Стадия 9-7
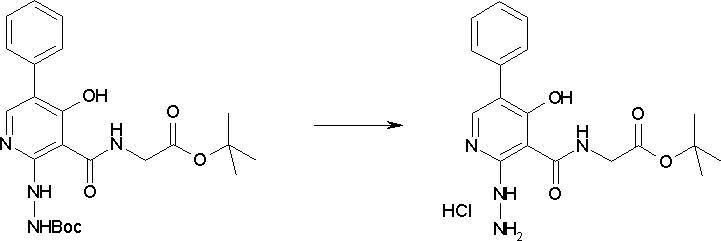
К раствору соединения (110 мг), полученного на стадии 9-6, в хлороформе (0,3 мл) добавляли 4 н. раствор соляной кислоты в диоксане (1 мл) и перемешивали смесь при охлаждении на льду в течение 2 ч. Реакционную смесь концентрировали в условиях пониженного давления с получением соединения (84 мг, 87%), описанного в представленной выше схеме.
1H-ЯМР (CD3OD) δ: 1,49 (9H, с), 4,04 (2H, с), 7,36-7,47 (5H, м), 7,57 (1H, с).
Стадия 9-8
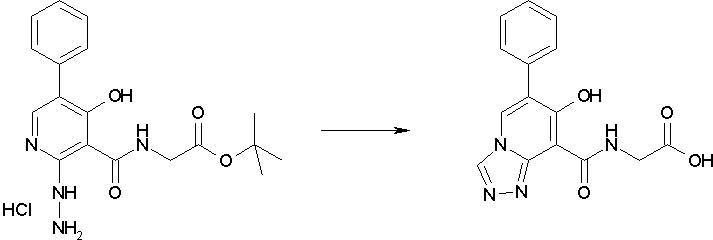
К соединению (41 мг), полученному на стадии 9-7, добавляли триметилортоформиат (0,41 мл) и нагревали смесь при 100°C в течение 30 мин. Реакционную смесь концентрировали в условиях пониженного давления и растворяли остаток в хлороформе (1 мл). Добавляли трифторуксусную кислоту (2 мл) и перемешивали смесь при комнатной температуре в течение 3 ч. Реакционную смесь концентрировали в условиях пониженного давления, добавляли 4 н. раствор соляной кислоты в диоксане (1 мл) и перемешивали смесь при комнатной температуре в течение 30 мин. Реакционную смесь концентрировали в условиях пониженного давления и суспендировали остаток в воде с получением указанного в заголовке соединения (29 мг, 89%).
1H-ЯМР (ДМСО-D6) δ: 4,06 (2H, д, J=5,5 Гц), 7,39-7,46 (3H, м), 7,60 (2H, дд, J=8,3, 1,4 Гц), 8,39 (1H, с), 8,86 (1H, с), 10,50 (1H, т, J=5,7 Гц), 12,59 (1H, с), 13,75 (1H, с).
Пример 10
Получение гидрохлорида [(7-гидрокси[1,2,4]триазоло[4,3-a]пиридин-8-карбонил)амино]уксусной кислоты
Стадия 10-1
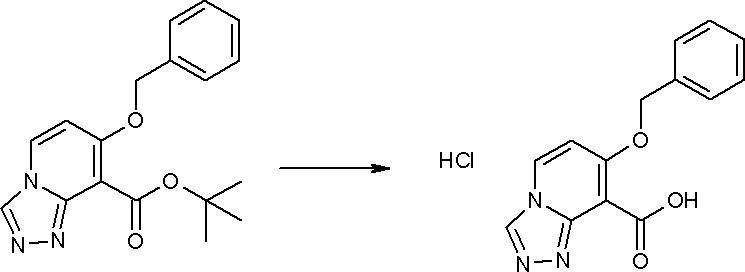
Способом, аналогичным реакции снятия карбоксилзащитной группы в Примере 9, стадия 9-8, из соединения (0,050 г), полученного в Примере 1, стадия 1-5, получали неочищенный продукт, содержащий описанное в представленной выше схеме соединение в качестве главного компонента.
1H-ЯМР (ДМСО-D6) δ: 5,60 (c, 2H), 7,35-7,55 (м, 5H), 7,69 (д, 1H, J=7,7 Гц), 9,03 (д, 1H, J=7,7 Гц), 9,48 (c, 1H).
Стадия 10-2
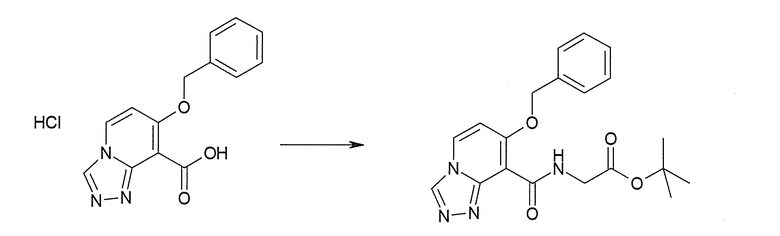
Способом, аналогичным описанному в Примере 1, стадия 1-10, из соединения, полученного на стадии 10-1, получали соединение (0,024 г, 41%), описанное в представленной выше схеме.
1H-ЯМР (CDCl3) δ: 1,51 (c, 9H), 4,23 (д, 2H, J=5,6 Гц), 5,35 (c, 2H), 6,81 (д, 1H, J=7,7 Гц), 7,26-7,51 (м, 5H), 8,10 (д, 1H, J=7,7 Гц), 8,67 (c, 1H), 9,66 (ушир. с, 1H).
Соединение, полученное на этой стадии, подвергали снятию гидроксилзащитной группы и карбоксилзащитной группы способами, аналогичными описанным выше, и преобразовывали полученное соединение до гидрохлорида общепринятым способом с получением указанного в заголовке соединения.
1H-ЯМР (ДМСО-D6) δ: 4,07 (c, 2H), 6,65 (д, 1H, J=7,7 Гц), 8,32 (д, 1H, J=7,7 Гц), 8,99 (c, 1H), 10,09 (c, 1H).
Пример 11
Получение [(6-гидрокси[1,2,3]триазоло[1,5-a]пиридин-7-карбонил)амино]уксусной кислоты
Стадия 11-1
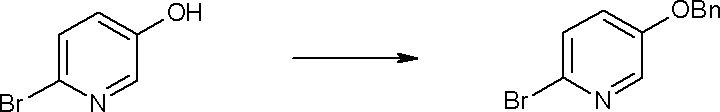
Смешивали 2-бром-5-гидроксипиридин (5,8 г), N,N-диметилформамид (58 мл) и карбонат калия (5,1 г), при охлаждении на льду добавляли бензилбромид (4,4 мл) и перемешивали смесь при комнатной температуре в течение 13 ч. К реакционной смеси добавляли этилацетат (58 мл) и воду (87 мл), органический слой отделяли от смеси и последовательно промывали дважды водой (60 мл, 30 мл) и насыщенным солевым раствором (30 мл). Органический слой сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали в условиях пониженного давления. Полученный остаток очищали по методу колоночной хроматографии (элюент: гексан/этилацетат = 10/1-7/1) с получением соединения (7,4 г, 83%), описанного в представленной выше схеме.
1H-ЯМР (CDCl3) δ: 5,09 (c, 2H), 7,15 (дд, 1H, J=8,1, 3,2 Гц), 7,33-7,38 (м, 1H), 7,36 (д, 1H, J=8,1 Гц), 7,40-7,41 (м, 4H), 8,13 (д, 1H, J=3,2 Гц).
Стадия 11-2
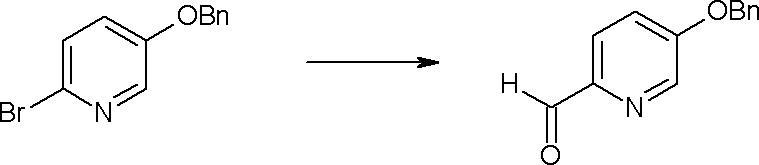
К н-бутиллитию (1,54 мол./л раствор в гексане, 25 мл) при -78°C по каплям в течение 7 мин добавляли раствор соединения (1 г), полученного на стадии 11-1, в толуоле (4 мл). Реакционную смесь перемешивали при -78°C в течение 50 мин, и по каплям добавляли раствор N,N-диметилформамида (0,352 мл) в толуоле (4 мл). Реакционную смесь дополнительно перемешивали при -78°C в течение 1 ч, при -10°C добавляли воду (6 мл), и перемешивали смесь при комнатной температуре в течение 2 ч. Органический слой и водный слой разделяли, водный слой дважды экстрагировали толуолом (5 мл). Органические слои объединяли и промывали насыщенным солевым раствором (10 мл). Органический слой сушили над безводным сульфатом магния, и смесь фильтровали. Фильтрат концентрировали в условиях пониженного давления, и очищали полученный остаток по методу колоночной хроматографии (элюент: гексан/этилацетат = 10/1-7/1) с получением соединения (641 мг, 79%), описанного в представленной выше схеме.
1H-ЯМР (CDCl3) δ: 5,21 (c, 2H), 7,35-7,45 (м, 6H), 7,95 (д, 1H, J=8,9 Гц), 8,51 (д, 1H, J=2,8 Гц), 9,99 (c, 1H).
Стадия 11-3
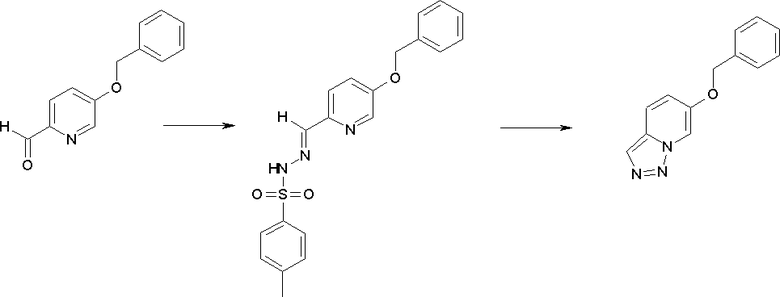
Смешивали соединение (637 мг), полученное на стадии 11-2, метанол (6,4 мл) и тозилгидразид (574 мг) и нагревали смесь с обратным холодильником в течение 10 мин. Реакционную смесь концентрировали в условиях пониженного давления, добавляли морфолин (6,4 мл) и нагревали смесь с обратным холодильником в течение 30 мин. Реакционную смесь концентрировали в условиях пониженного давления и добавляли этилацетат (12 мл), 2M водный раствор карбоната натрия (6 мл) и воду (5 мл). Затем добавляли тетрагидрофуран (6 мл) и разделяли органический слой. Водный слой дважды экстрагировали этилацетатом (5 мл), органические слои объединяли и промывали насыщенным солевым раствором (10 мл). Органический слой сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали в условиях пониженного давления и суспендировали полученное твердое вещество в диизопропиловом эфире с получением соединения (566 мг, 84%), описанного в представленной выше схеме.
1H-ЯМР (CDCl3) δ: 5,12 (c, 2H), 7,11 (дд, 1H, J=9,7, 2,0 Гц), 7,36-7,47 (м, 5H), 7,61 (дд, 1H, J=9,7, 0,8 Гц), 7,99 (д, 1H, J=0,8 Гц), 8,34 (д, 1H, J=2,0 Гц).
Стадия 11-4
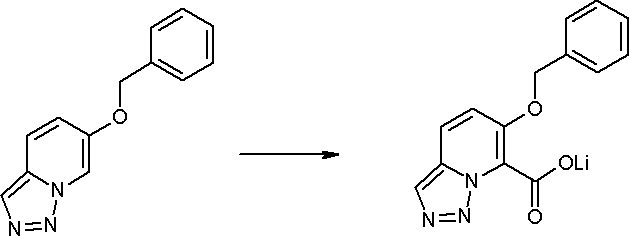
Смешивали соединение (200 мг), полученное на стадии 11-3, и тетрагидрофуран (2 мл) и при -40°C добавляли диизопропиламид лития (2M раствор в тетрагидрофуране, гептане, этилбензоле, 0,45 мл). После перемешивания при -40°C в течение 1 ч добавляли сухой лед и оставляли смесь нагреваться до комнатной температуры при перемешивании в течение 1 ч. После этого добавляли метанол (2 мл) и концентрировали реакционную смесь в условиях пониженного давления с получением соединения, описанного в представленной выше схеме, в виде неочищенного продукта. Этот продукт использовали непосредственно на следующей стадии.
Стадия 11-5
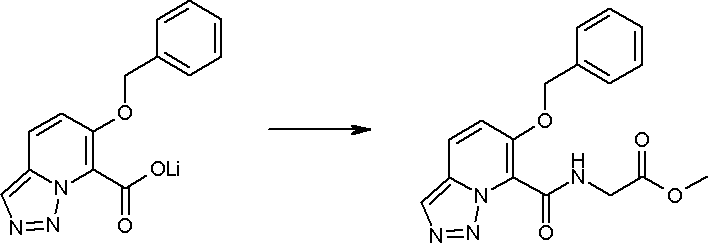
Способом, аналогичным описанному в Примере 1, стадия 1-10, из неочищенного продукта, полученного на стадии 11-4, получали соединение (85 мг, 28% (в 2 этапа)), описанное в представленной выше схеме.
1H-ЯМР (CDCl3) δ: 3,80 (c, 3H), 4,38 (д, 2H, J=5,2 Гц), 5,32 (c, 2H), 7,24 (д, 1H, J=9,7 Гц), 7,31-7,41 (м, 3H), 7,46-7,49 (м, 2H), 7,75 (д, 1H, J=9,7 Гц), 8,11 (c, 1H), 8,79 (ушир. с, 1H).
Стадия 11-6
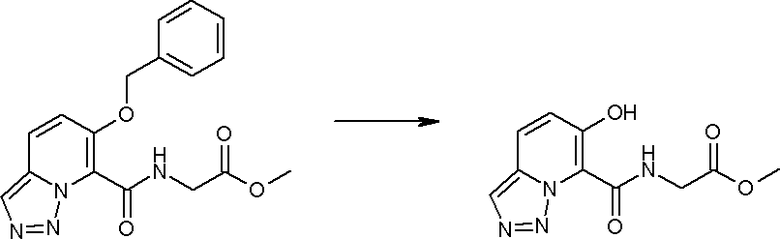
Способом, аналогичным описанному в Примере 2, стадия 2-5, из соединения (72 мг), полученного на стадии 11-5, получали соединение (40 мг, 76%), описанное в представленной выше схеме.
1H-ЯМР (CDCl3) δ: 3,83 (c, 3H), 4,37 (д, 2H, J=6,0 Гц), 7,17 (д, 1H, J=9,7 Гц), 7,80 (д, 1H, J=9,7 Гц), 8,12 (c, 1H), 10,57 (ушир. с, 1H), 13,56 (c, 1H).
Соединение, полученное на этой стадии, подвергали удалению карбоксильной группы способом, аналогичным описанному выше, с получением указанного в заголовке соединения.
1H-ЯМР (ДМСО-D6) δ: 4,28 (д, 2H, J=5,2 Гц), 7,31 (д, 1H, J=9,7 Гц), 8,20 (д, 1H, J=9,7 Гц), 8,39 (c, 1H), 10,36 (т, 1H, J=5,2 Гц), 13,82 (c, 1H).
Пример 116
Получение [(7-гидрокси-5-фенетил[1,2,4]триазоло[1,5-a]пиридин-8-карбонил)амино]уксусной кислоты
Стадия 116-1
Смешивали цианамид (1,4 г) и 1,4-диоксан (20 мл) и добавляли диметил-1,3-ацетондикарбоксилат (2,0 г) и ацетилацетонат никеля(II) (0,30 г). Смесь нагревали с обратным холодильником в течение 16 ч. Смесь охлаждали до комнатной температуры, перемешивали в течение 1 ч и собирали полученное твердое вещество путем фильтрования. К полученному твердому веществу добавляли метанол (6,0 мл) и перемешивали смесь при комнатной температуре в течение 1,5 ч. Твердое вещество собирали путем фильтрования с получением соединения (1,4 г, 64%), описанного в представленной выше схеме.
1H-ЯМР (ДМСО-D6) δ: 3,80 (c, 3H), 4,92 (c, 1H), 7,20 (c, 2H), 10,29 (c, 1H), 11,51 (c, 1H).
Стадия 116-2
Смешивали соединение (30 г), полученное на стадии 116-1, оксихлорид фосфора (150 мл) и N,N-диизопропилэтиламин (30 мл) и перемешивали смесь при комнатной температуре в течение 3 суток. Реакционную смесь концентрировали и трижды подвергали азеотропной перегонке с толуолом. При охлаждении на льду добавляли метанол (30 мл) и воду (150 мл) и перемешивали смесь при комнатной температуре в течение 1 ч. Полученное твердое вещество собирали путем фильтрования и объединяли с твердым веществом, полученным из фильтрата. Добавляли метанол (50 мл) и перемешивали смесь при комнатной температуре в течение 1 ч. Твердое вещество собирали путем фильтрования с получением первичных кристаллических веществ. Фильтрат концентрировали, добавляли к остатку метанол (10 мл) и собирали полученное твердое вещество путем фильтрования с получением вторичного кристаллического вещества. Первичное кристаллическое вещество и вторичное кристаллическое вещество объединяли с получением соединения (21 г, 59%), описанного в представленной выше схеме.
1H-ЯМР (ДМСО-D6) δ: 3,84 (c, 3H), 6,84 (c, 1H), 7,16 (ушир. с, 2H).
Стадия 116-3
Смешивали соединение (2,2 г), полученное на стадии 116-2, и 2-пропанол (31 мл) и добавляли диметилацеталь N,N-диметилформамида (2,9 мл). Смесь нагревали с обратным холодильником в течение 30 мин. Реакционную смесь охлаждали до комнатной температуры, добавляли гидрохлорид гидроксиламина (1,4 г) и перемешивали смесь при комнатной температуре в течение 30 мин. Реакционную смесь концентрировали приблизительно до половины объема и добавляли воду (40 мл) и 2-пропанол (6,6 мл). Смесь перемешивали при комнатной температуре в течение 30 мин и собирали полученное твердое вещество путем фильтрования с получением соединения (2,2 г, 84%), описанного в представленной выше схеме.
1H-ЯМР (ДМСО-D6) δ: 3,92 (c, 3H), 7,37 (c, 1H), 7,85 (д, 1H, J=9,5 Гц), 10,12 (д, 1H, J=9,5 Гц), 10,83 (c, 1H).
Стадия 116-4
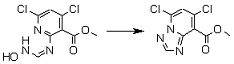
Смешивали соединение (0,66 г), полученное на стадии 116-3, и тетрагидрофуран (6,6 мл) и добавляли трифторуксусный ангидрид (0,37 мл). Смесь нагревали с обратным холодильником в течение 22 ч. Реакционную смесь охлаждали до комнатной температуры, добавляли этилацетат и насыщенный водный раствор гидрокарбоната натрия и разделяли органический слой. Органический слой промывали насыщенным солевым раствором и сушили над сульфатом натрия. После фильтрования фильтрат концентрировали в условиях пониженного давления и очищали полученный остаток по методу колоночной хроматографии (элюент: хлороформ/этилацетат = 4/1) с получением соединения (0,32 г, 51%), описанного в представленной выше схеме.
1H-ЯМР (ДМСО-D6) δ: 3,99 (c, 3H), 7,92 (c, 1H), 8,71 (c, 1H).
Стадия 116-5
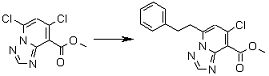
Смешивали соединение (1,0 г), полученное на стадии 116-4, фенетилбороновую кислоту (1,2 г), карбонат калия (1,7 г), комплекс дихлорида [1,1'-бис(дифенилфосфино)ферроцен]палладия(II) и дихлорметана (1:1) (0,083 г), циклопентилметиловый эфир (6,0 мл) и воду (0,15 мл) и перемешивали смесь при нагревании при 50°C в течение 6 ч. Реакционную смесь охлаждали до комнатной температуры и дважды промывали органический слой 3% водным раствором диэтилентриамина (10 мл) и насыщенным солевым раствором (5 мл). Добавляли сульфат натрия и улавливающий металл силикагель (1 г), смесь перемешивали при комнатной температуре в течение 1 ч и фильтровали через воронку Кириямы, наполненную силикагелем (1 г). Фильтрат концентрировали в условиях пониженного давления с получением соединения (1,87 г), описанного в представленной выше схеме, в виде грубо очищенного продукта.
1H-ЯМР (CDCl3) δ: 3,17 (т, 2H, J=7,8 Гц), 3,48 (т, 2H, J=7,8 Гц), 4,08 (c, 3H), 6,84 (c, 1H), 7,18-7,28 (м, 5H), 8,42 (c, 1H).
Стадия 116-6
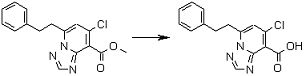
Смешивали соединение (1,87 г), полученное на стадии 116-5, и тетрагидрофуран и при охлаждении на льду добавляли 4 н. водный раствор гидроксида натрия (4,0 мл). После перемешивания при комнатной температуре в течение 2 ч реакционную смесь нейтрализовали добавлением концентрированной соляной кислоты (1,4 мл) при охлаждении на льду. К суспензии добавляли этанол (5 мл) и воду (1,3 мл) и перемешивали смесь в течение 1 ч. Твердое вещество собирали путем фильтрования с получением соединения (0,847 г, 69%), описанного в представленной выше схеме.
1H-ЯМР (ДМСО-D6) δ: 3,13 (т, 2H, J=7,8 Гц), 3,45 (т, 2H, J=7,8 Гц), 7,23-7,31 (м, 5H), 8,65 (c, 1H), 14,19 (c, 1H).
Стадия 116-7
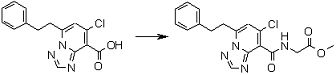
Смешивали соединение (0,200 г), полученное на стадии 116-6, ацетонитрил (1,0 мл), моногидрат 1-гидроксибензотриазола (0,122 г) и гидрохлорид сложного метилового эфира глицина (0,100 г) и при охлаждении на льду добавляли триэтиламин (0,111 мл) и гидрохлорид 1-этил-3-(3-диметиламинопропил)карбодиимида (0,153 г). Смесь перемешивали при комнатной температуре в течение 2 ч. К реакционной смеси добавляли насыщенный водный раствор гидрокарбоната натрия (2 мл) и собирали выпавшее в осадок твердое вещество путем фильтрования с получением соединения (0,194 г, 78%), описанного в представленной выше схеме.
1H-ЯМР (CDCl3) δ: 3,18 (т, 2H, J=7,8 Гц), 3,48 (т, 2H, J=7,8 Гц), 3,81 (c, 3H), 4,34 (д, 2H, J=5,2 Гц), 6,92 (c, 1H), 7,15-7,33 (м, 5H), 8,42 (c, 1H), 9,90 (c, 1H).
Стадия 116-8
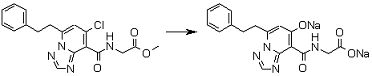
Смешивали соединение (0,150 г), полученное на стадии 116-7, 2-этоксиэтанол (0,75 мл) и 8 н. водный раствор гидроксида натрия (0,23 мл) и перемешивали смесь при 90°C в течение 18 ч. К этой смеси добавляли этанол (0,75 мл) и воду (0,23 мл) и перемешивали смесь при комнатной температуре в течение 1 ч. Твердое вещество собирали путем фильтрования с получением грубо очищенного продукта, описанного в представленной выше схеме соединения (0,19 г).
1H-ЯМР (ДМСО-D6) δ: 2,99-3,11 (м, 4H), 3,58 (д, 2H, J=4,4 Гц), 6,01 (c, 1H), 7,20-7,27 (м, 5H), 7,86 (c, 1H), 11,23 (т, 1H, J=4,4 Гц).
Стадия 116-9
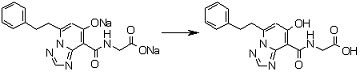
Смешивали соединение (0,19 г), полученное на стадии 116-8, и воду (0,63 мл), и нагревали смесь до 50°C. Добавляли ацетон (0,78 мл) и 6 н. соляную кислоту (0,2 мл) и перемешивали смесь при той же температуре в течение 1 ч. После перемешивания при охлаждении на льду в течение 1 ч собирали твердое вещество путем фильтрования с получением соединения (0,11 г, 80%), описанного в представленной выше схеме.
1H-ЯМР (ДМСО-D6) δ: 3,12 (т, 2H, J=7,9 Гц), 3,40 (т, 3H, J=7,9 Гц), 4,22 (д, 2H, J=5,2 Гц), 6,79 (c, 1H), 7,21-7,29 (м, 5H), 8,58 (c, 1H), 9,84 (т, 1H, J=5,2 Гц), 12,97 (c, 1H), 14,22 (c, 1H).
Стадия 116-10
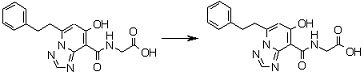
Смешивали соединение (0,050 г), полученное на стадии 116-9, и метанол (3 мл) и нагревали смесь до 60°C. Раствор охлаждали до комнатной температуры и перемешивали в течение суток. Твердое вещество собирали путем фильтрования с получением указанного в заголовке соединения (0,031 г, 61%).
1H-ЯМР (ДМСО-D6) δ: 3,12 (т, 2H, J=7,9 Гц), 3,40 (т, 3H, J=7,9 Гц), 4,22 (д, 2H, J=5,2 Гц), 6,79 (c, 1H), 7,21-7,29 (м, 5H), 8,58 (c, 1H), 9,84 (т, 1H, J=5,2 Гц), 12,97 (c, 1H), 14,22 (c, 1H).
Пример 117
Получение [(5-бутил-7-гидрокси[1,2,4]триазоло[1,5-a]пиридин-8-карбонил)амино]уксусной кислоты
Стадия 117-1
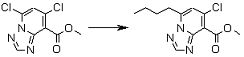
Смешивали соединение (0,050 г), полученное на стадии 116-4, бутилбороновую кислоту (0,042 г), оксид серебра(I) (0,071 г), карбонат калия (0,084 г), комплекс дихлорида [1,1'-бис(дифенилфосфино)ферроцен]палладия(II) и дихлорметана (1/1, 0,008 г) и тетрагидрофуран (1,0 мл) и смесь нагревали с обратным холодильником в течение 10 ч. Нерастворимый материал отфильтровывали через целит и добавляли насыщенный водный раствор гидрокарбоната натрия и этилацетат. Органический слой отделяли от смеси. Органический слой дважды промывали насыщенным водным раствором гидрокарбоната натрия и насыщенным солевым раствором, сушили над сульфатом натрия и фильтровали. Фильтрат концентрировали в условиях пониженного давления и очищали полученный остаток по методу тонкослойной хроматографии (элюент: гексан/этилацетат = 1/1) с получением соединения (0,046 г, 77%), описанного в представленной выше схеме.
1H-ЯМР (CDCl3) δ: 1,00 (т, 3H, J=7,4 Гц), 1,45-1,53 (м, 2H), 1,79-1,87 (м, 2H), 3,18 (т, 2H, J=7,9 Гц), 4,08 (c, 3H), 6,92 (c, 1H), 8,38 (c, 1H).
Стадия 117-2
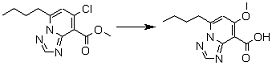
Смешивали соединение (0,043 г), полученное на стадии 117-1, и метанол (0,22 мл), и добавляли 28% раствор метоксида натрия в метаноле (0,014 мл). Смесь перемешивали при комнатной температуре в течение 4 ч. Добавляли воду (0,22 мл) и перемешивали смесь при комнатной температуре в течение 1 ч. К реакционной смеси добавляли 1 н. соляную кислоту (0,16 мл) и собирали полученное твердое вещество путем фильтрования с получением соединения (0,026 г, 66%), описанного в представленной выше схеме.
1H-ЯМР (CDCl3) δ: 1,01 (т, 3H, J=7,3 Гц), 1,46-1,57 (м, 2H), 1,82-1,90 (м, 2H), 3,21 (т, 2H, J=7,9 Гц), 4,15 (c, 3H), 6,77 (c, 1H), 8,28 (c, 1H).
Стадия 117-3
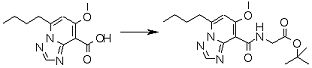
Смешивали соединение (0,025 г), полученное на стадии 117-2, и N,N-диметилформамид (0,50 мл) и добавляли моногидрат 1-гидроксибензотриазола (0,016 г), сложный трет-бутиловый эфир глицина (0,015 мл) и гидрохлорид 1-этил-3-(3-диметиламинопропил)карбодиимида (0,020 г). Смесь перемешивали при комнатной температуре в течение 1,5 ч. При охлаждении на льду к реакционной смеси добавляли 5% водный раствор гидрокарбоната натрия и воду и собирали полученное твердое вещество путем фильтрования с получением соединения (0,033 г, 94%), описанного в представленной выше схеме.
1H-ЯМР (CDCl3) δ: 1,00 (т, 4H, J=7,3 Гц), 1,51 (c, 9H), 1,80-1,90 (м, 2H), 3,18 (т, 2H, J=7,9 Гц), 4,21 (д, 2H, J=5,2 Гц), 6,72 (c, 1H), 8,29 (c, 1H), 9,72 (т, 1H, J=4,2 Гц).
Стадия 117-4
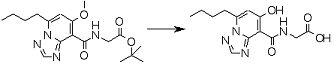
Смешивали соединение (0,030 г), полученное на стадии 117-3, и 25% раствор бромоводорода в уксусной кислоте (0,60 мл) и нагревали смесь с обратным холодильником в течение 3 ч. Реакционную смесь концентрировали в условиях пониженного давления. К полученному остатку при охлаждении на льду добавляли воду (0,60 мл) и 4 н. водный раствор гидроксида натрия (0,23 мл). Затем при охлаждении на льду добавляли 2 н. соляную кислоту (0,23 мл) и собирали полученное твердое вещество путем фильтрования с получением соединения (0,010 г, 42%), описанного в представленной выше схеме.
1H-ЯМР (ДМСО-D6) δ: 0,93 (т, 3H, J=7,5 Гц), 1,33-1,44 (м, 2H), 1,71-1,80 (м, 2H), 3,10 (т, 2H, J=7,5 Гц), 4,20 (д, 2H, J=5,2 Гц), 6,85 (c, 1H), 8,55 (c, 1H), 9,84 (ушир. с, 1H), 14,26 (ушир. с, 1H).
Стадия 117-5
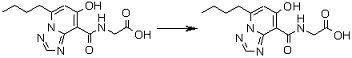
Смешивали соединение (0,100 г), полученное на стадии 117-4, и метилэтилкетон (1,0 мл) и нагревали до 80°C. К раствору добавляли гептан (1,0 мл) и перемешивали смесь при комнатной температуре в течение ночи. Твердое вещество собирали путем фильтрования с получением указанного в заголовке соединения (0,089 г, 89%).
1H-ЯМР (ДМСО-D6) δ: 0,93 (т, 3H, J=7,5 Гц), 1,33-1,44 (м, 2H), 1,71-1,80 (м, 2H), 3,10 (т, 2H, J=7,5 Гц), 4,20 (д, 2H, J=5,2 Гц), 6,85 (c, 1H), 8,55 (c, 1H), 9,84 (ушир. с, 1H), 14,26 (ушир. с, 1H).
Соединения Примеров 12-115 и Примеров 118-122, представленные в последующих Таблицах 3-24, получали тем же способом, что и в упомянутых выше Примерах 1-11, Примере 116 или 117, или, при необходимости, другими общепринятыми способами.
Структурные формулы и свойства соединений Примеров 1-122 представлены в последующих таблицах 1-24.
(M-H)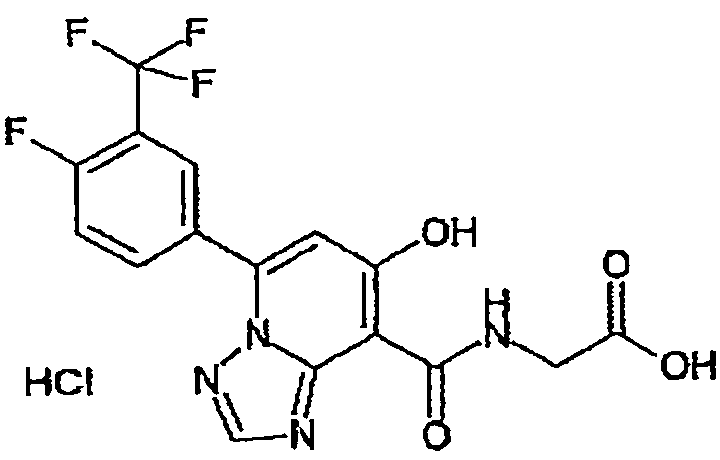
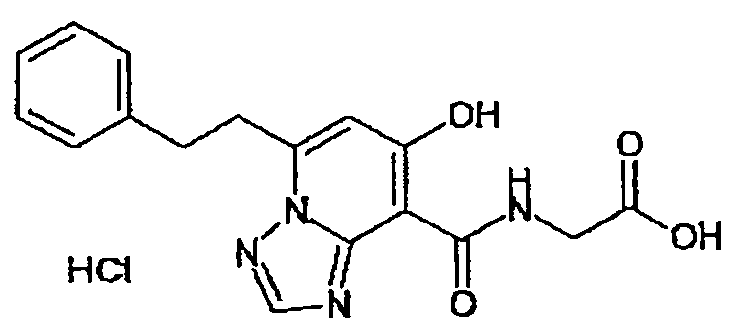
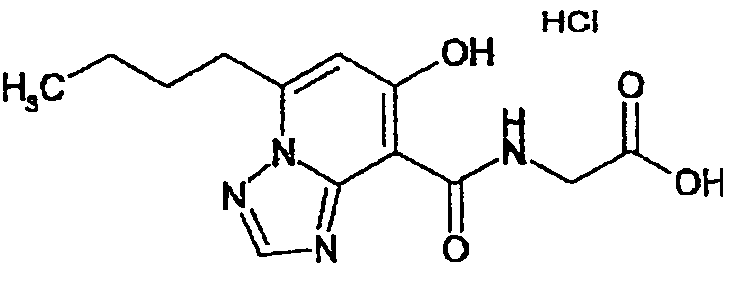
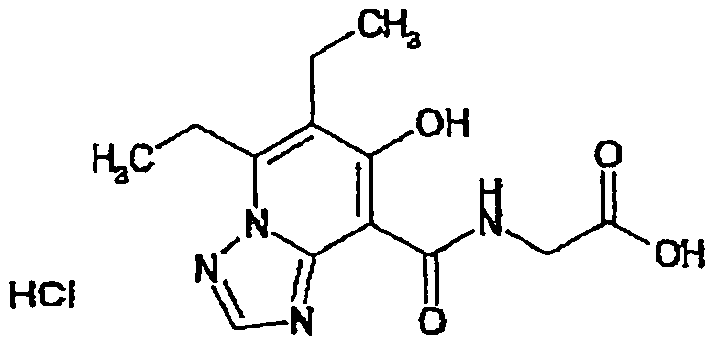
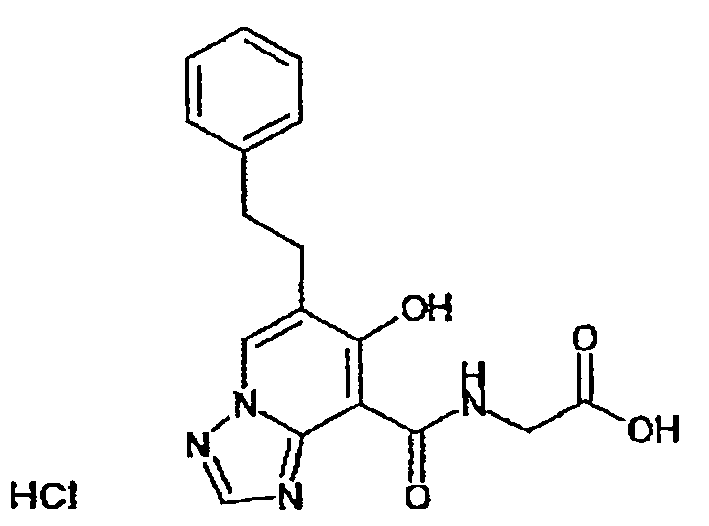
(M-H)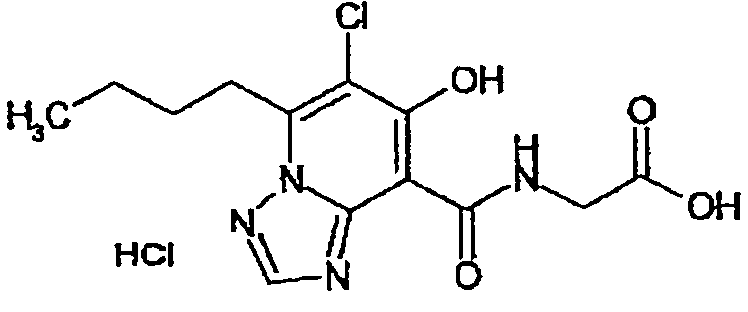
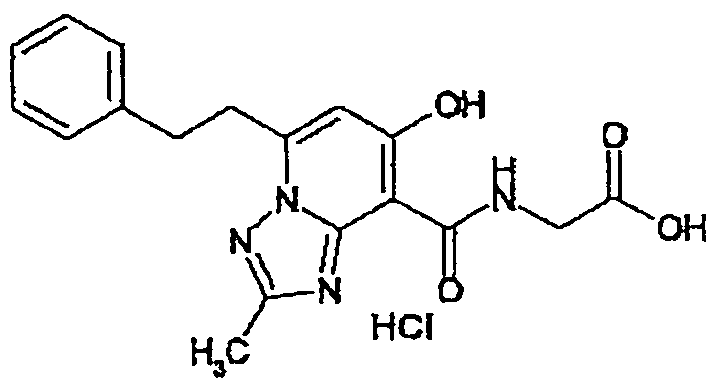
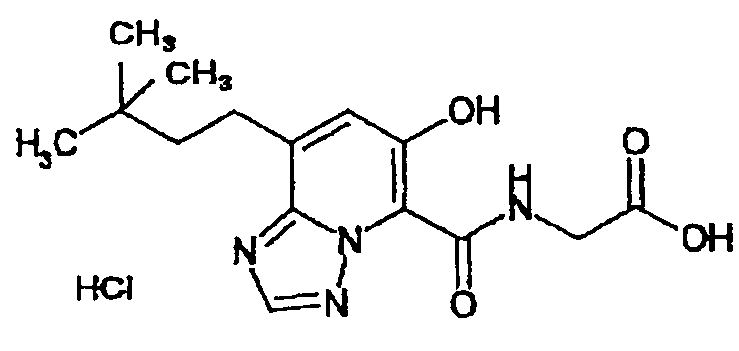
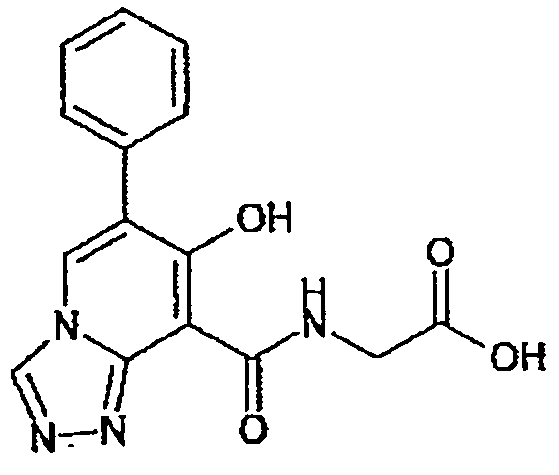
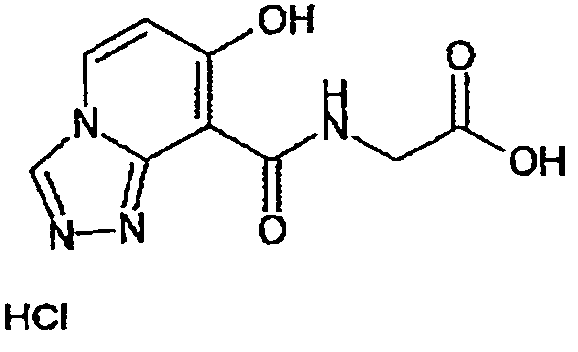
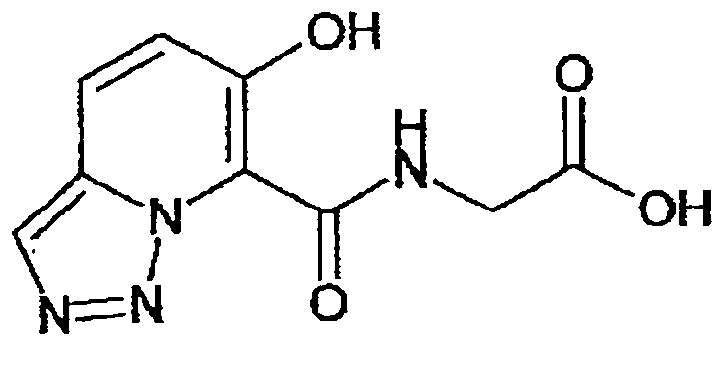
(M-H)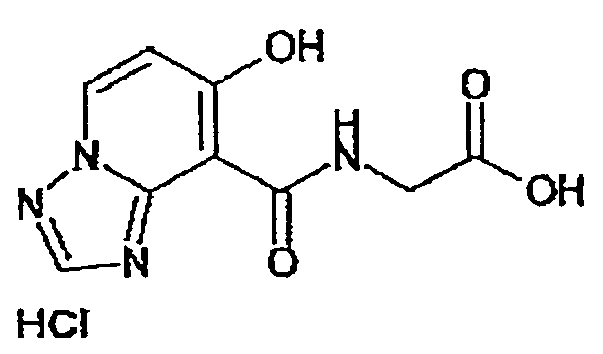
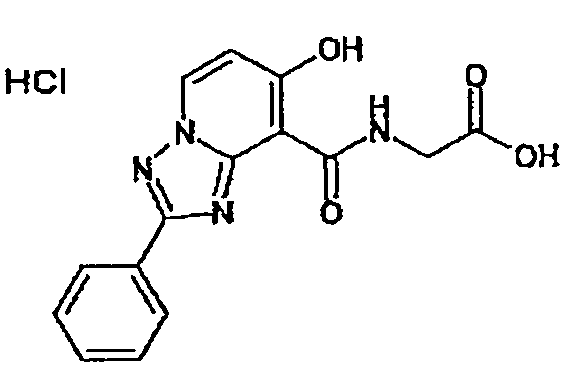
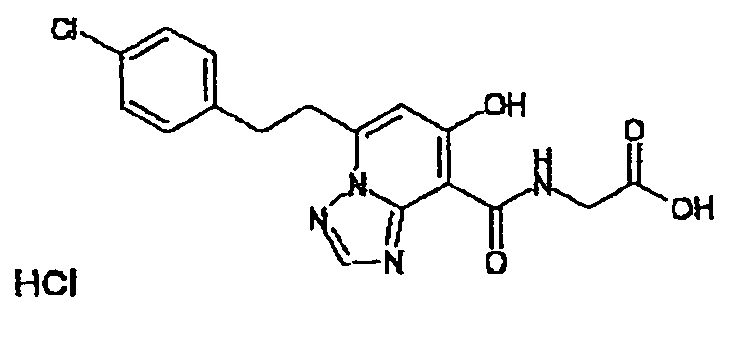
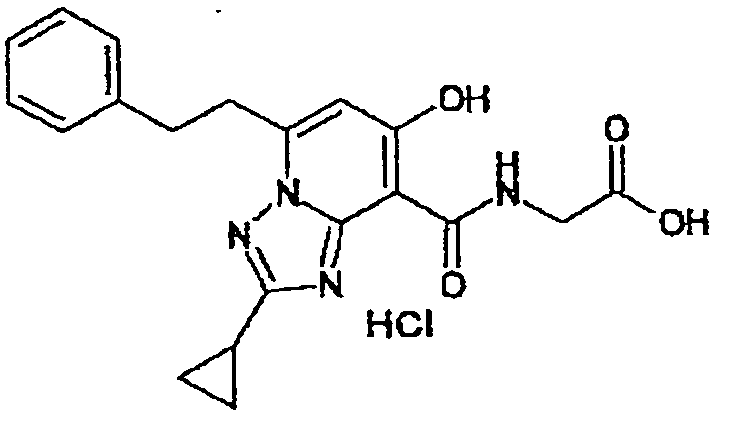
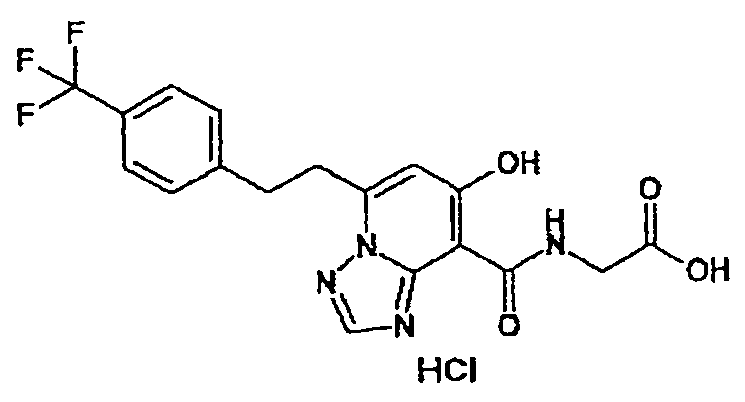
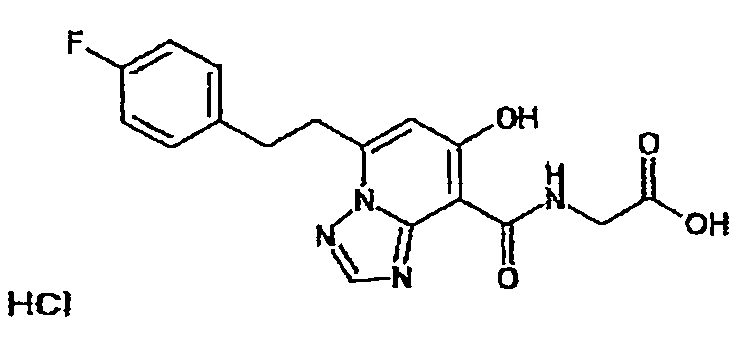
(M-H)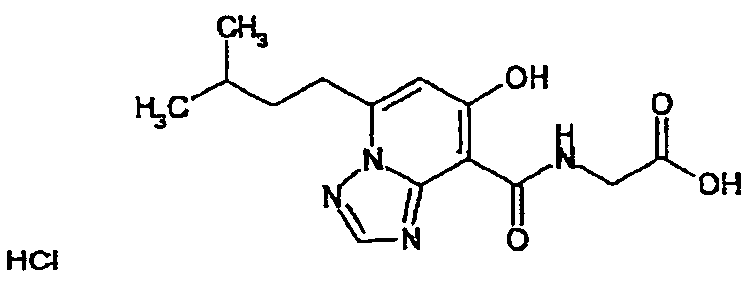
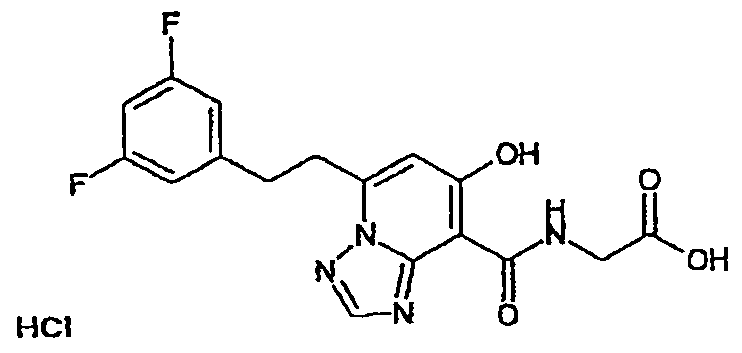
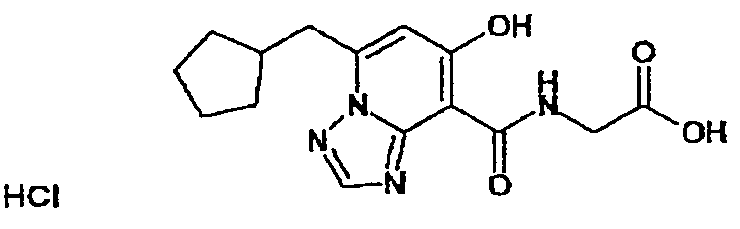
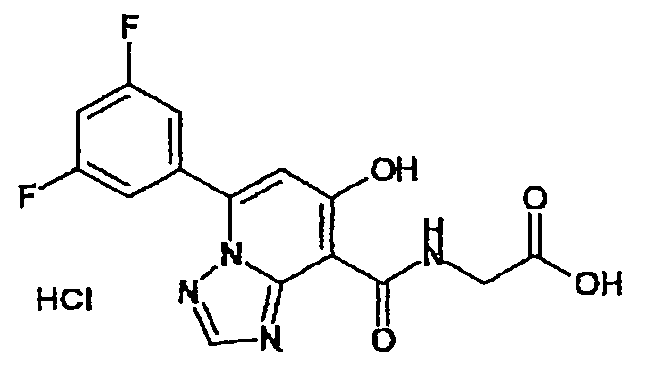
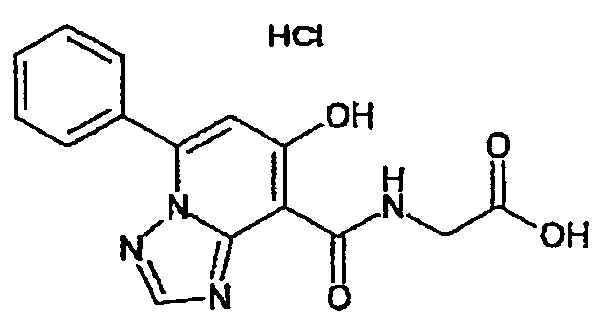
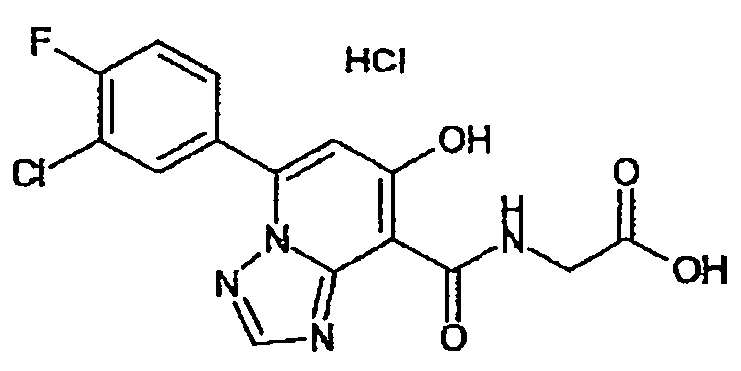
(M-H)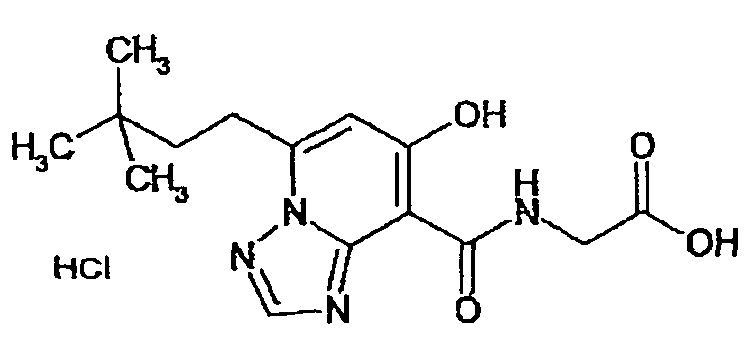
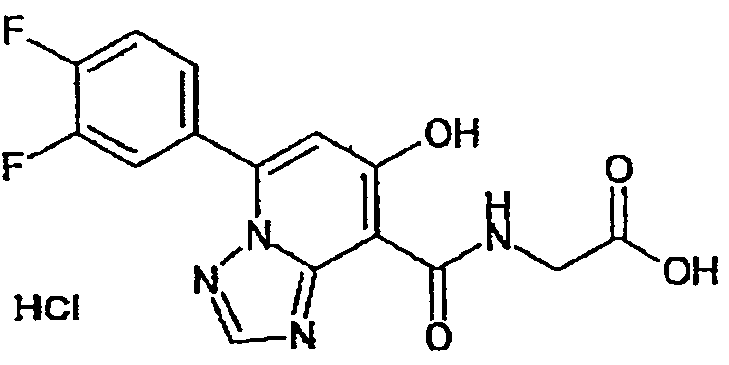
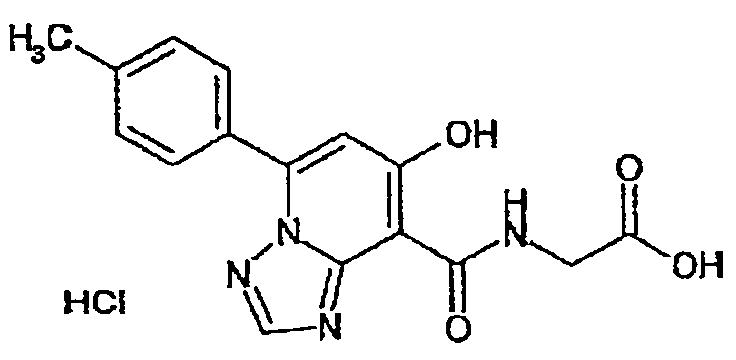
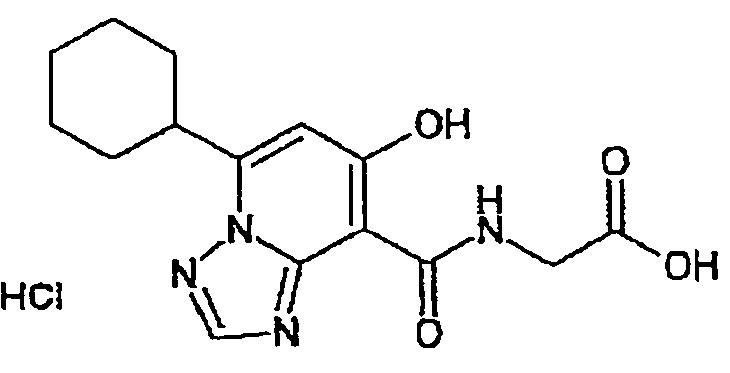
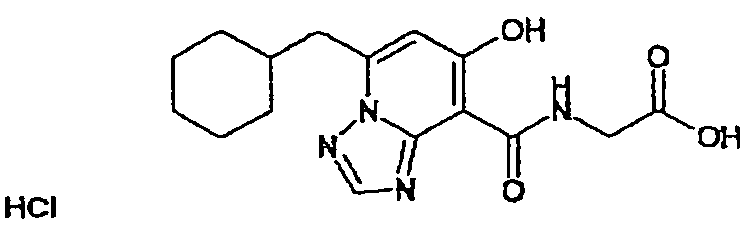
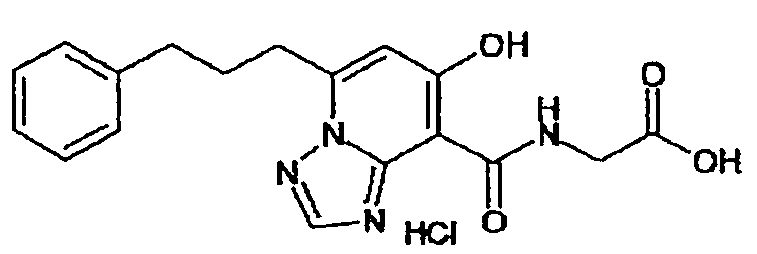
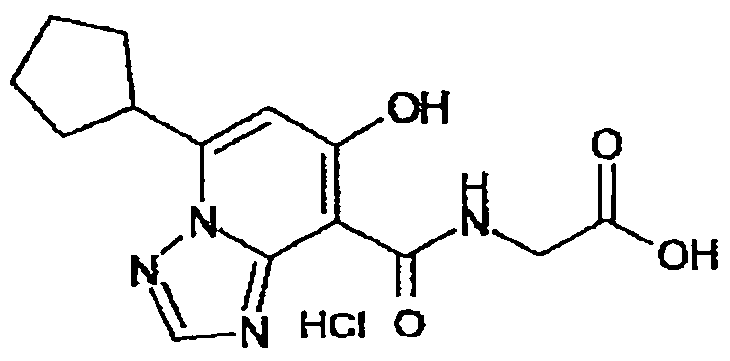
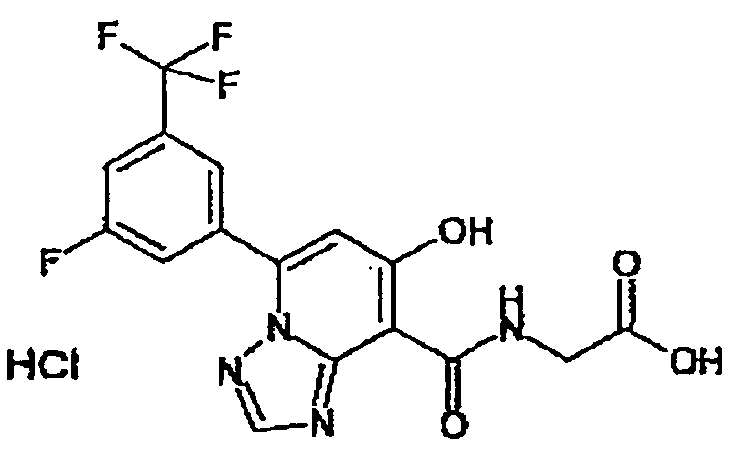
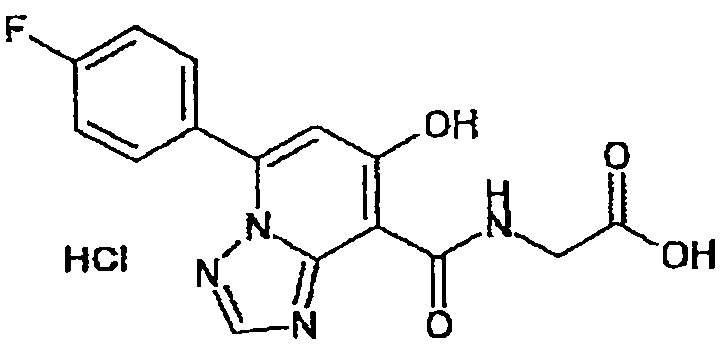
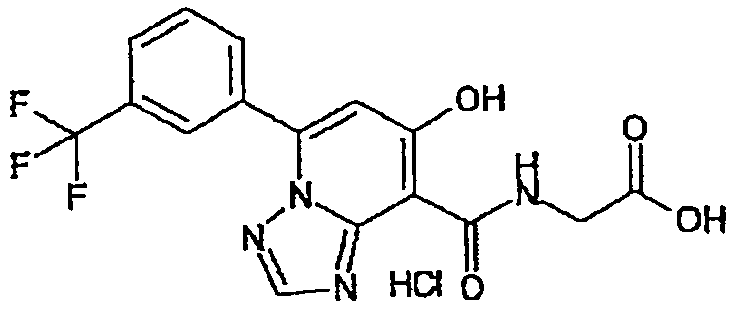
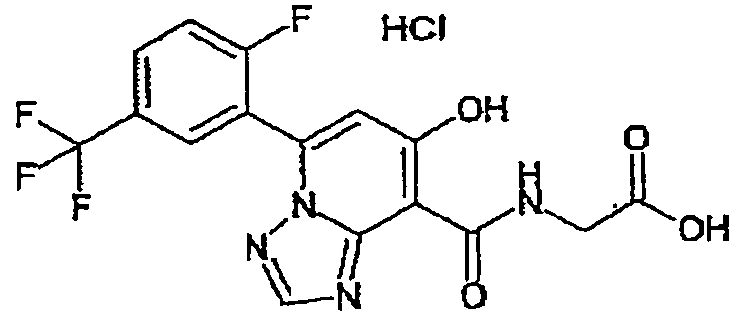
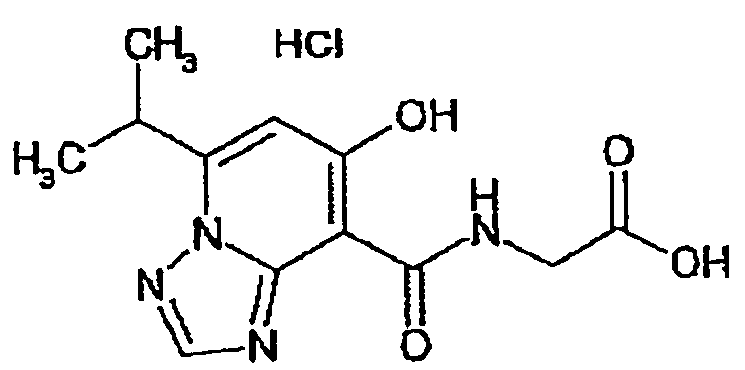
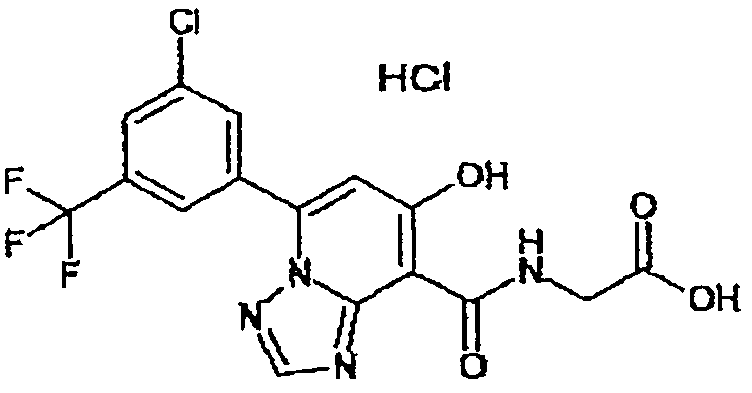
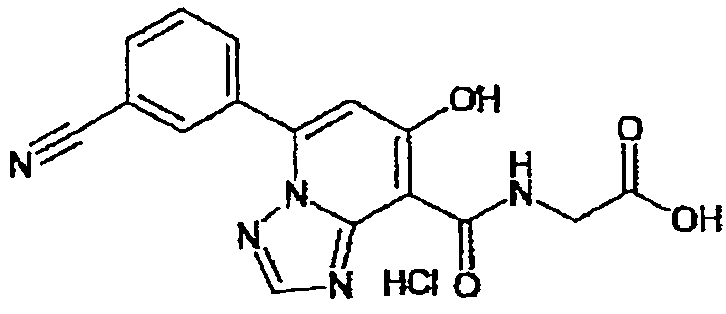
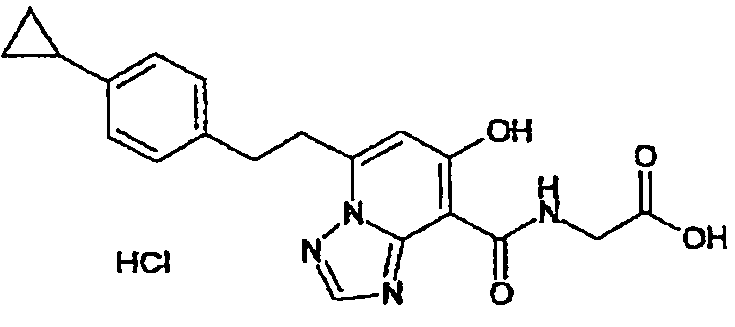
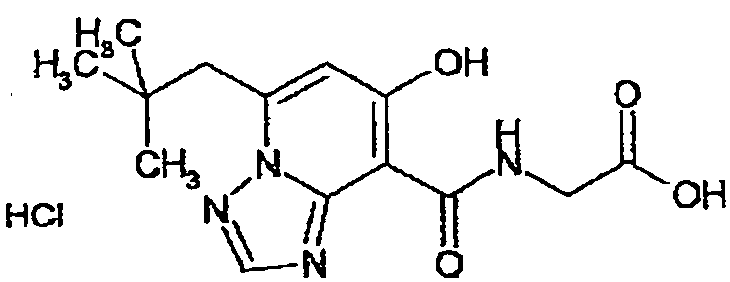
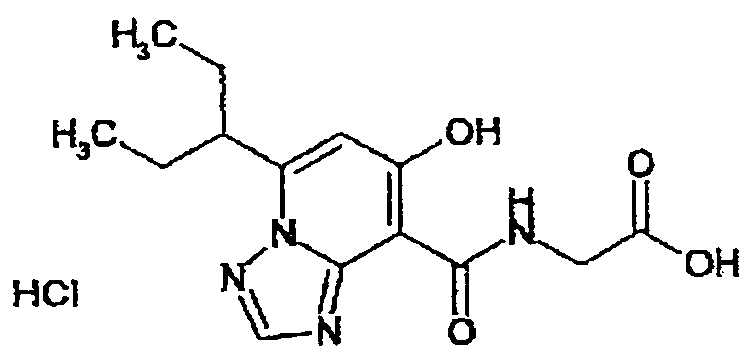
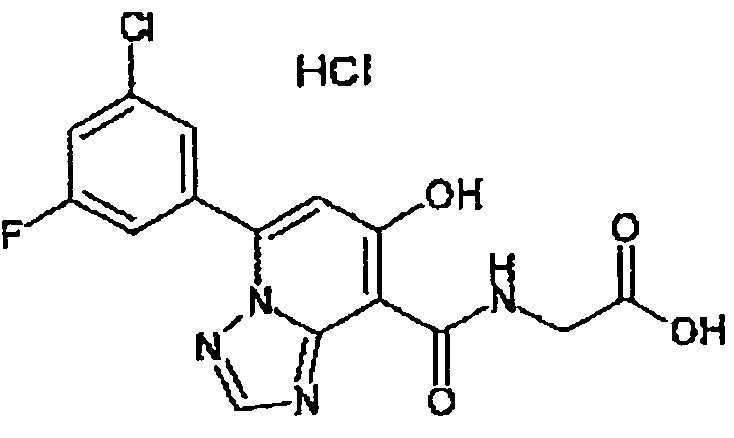
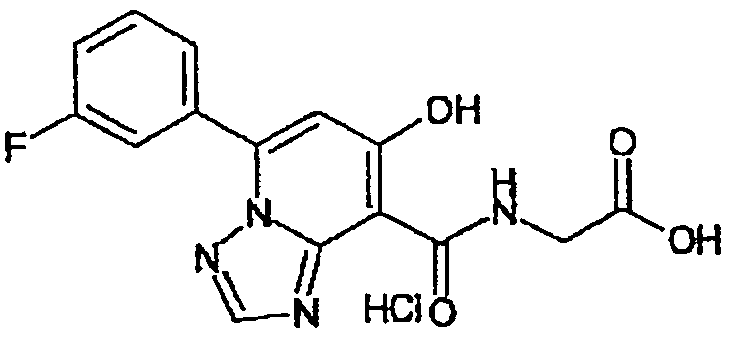
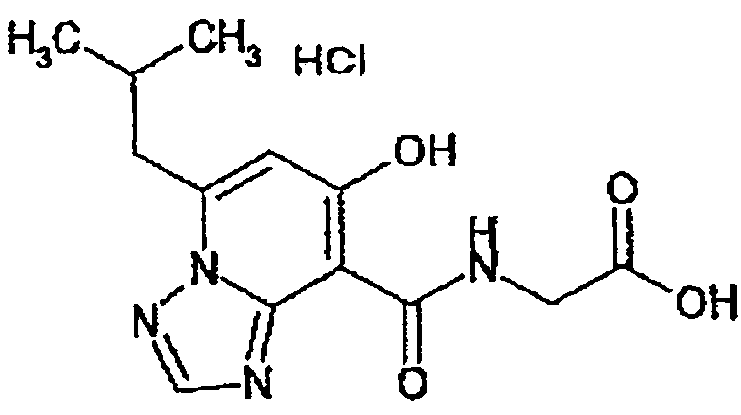
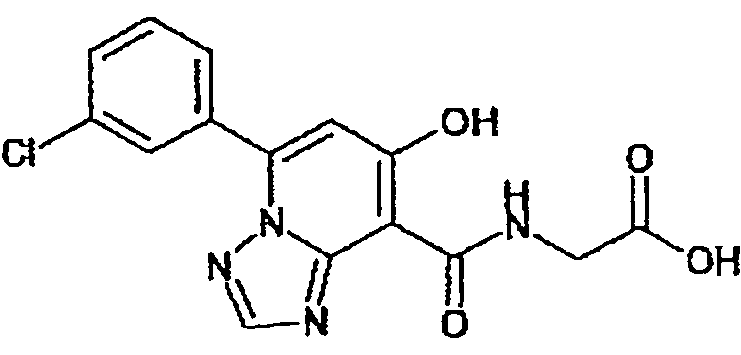
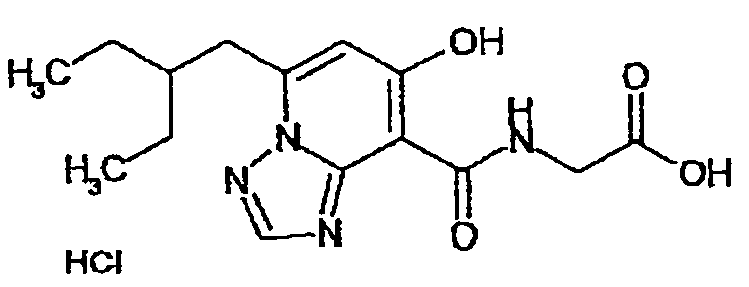
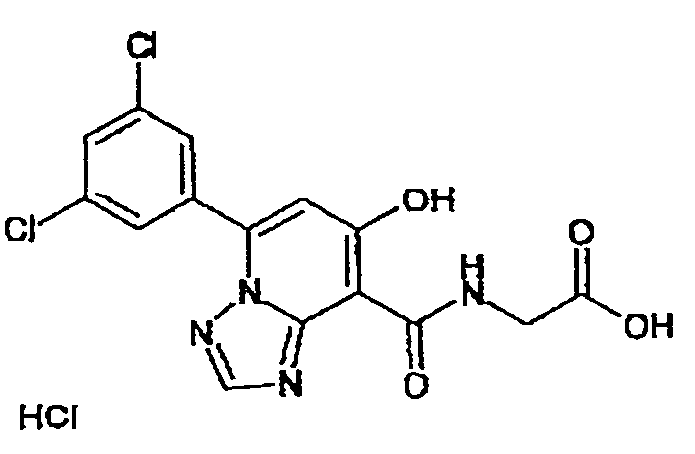
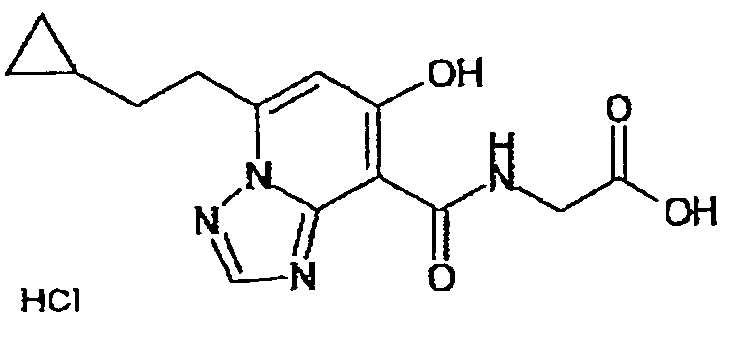
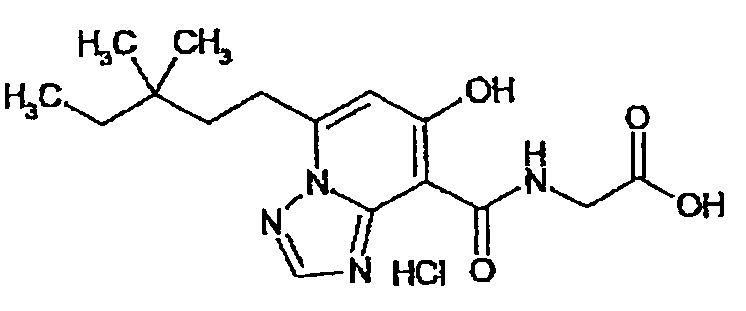
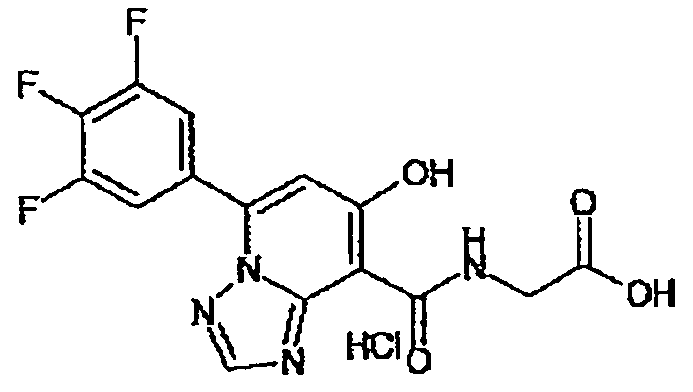
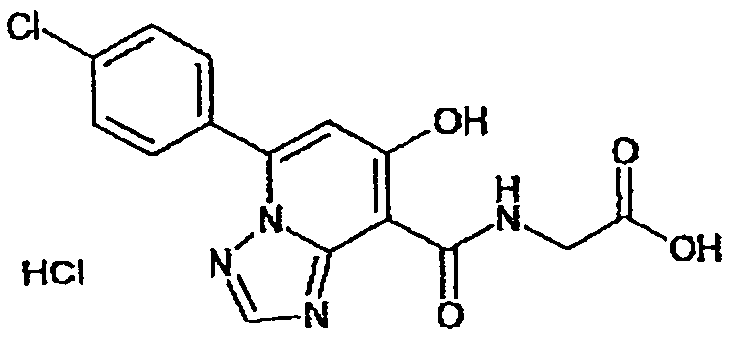
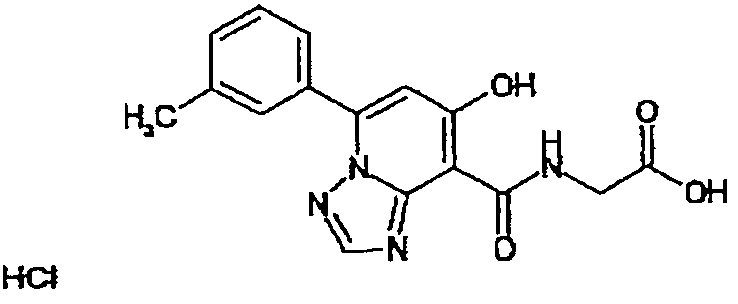
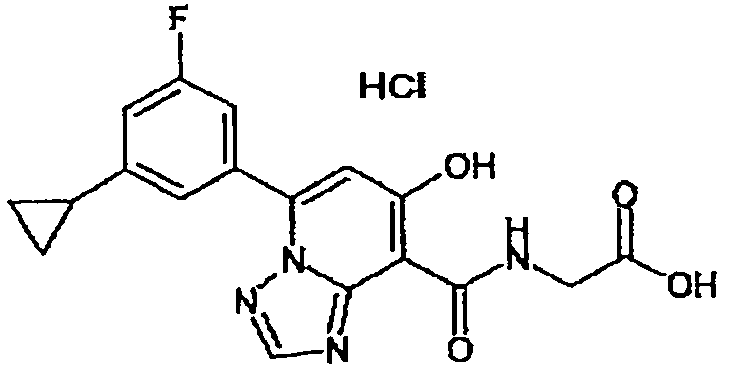
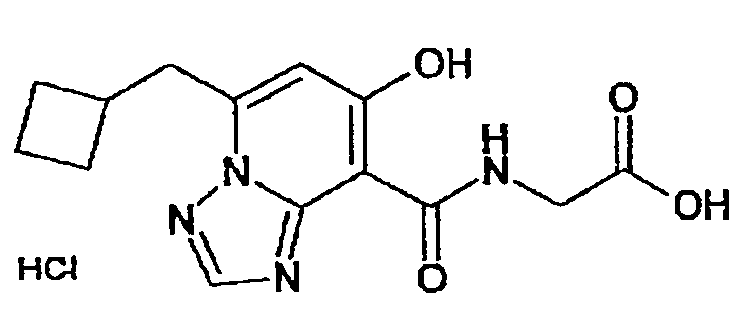
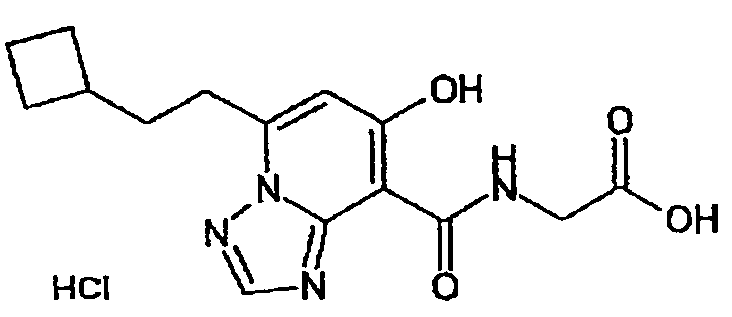
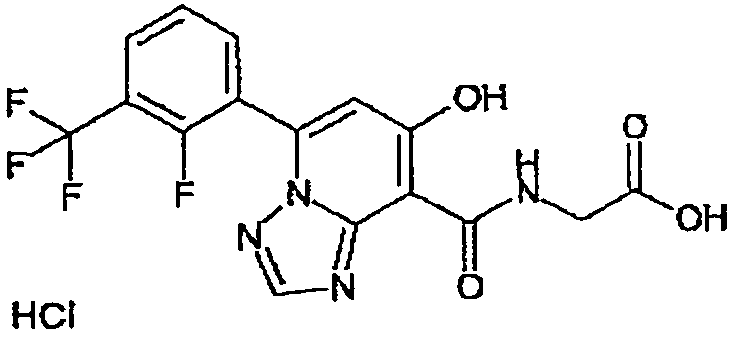
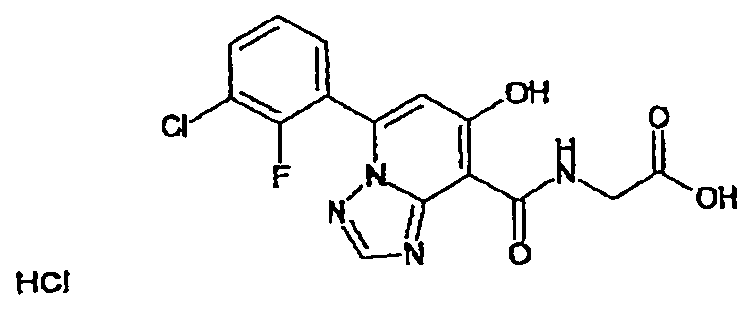
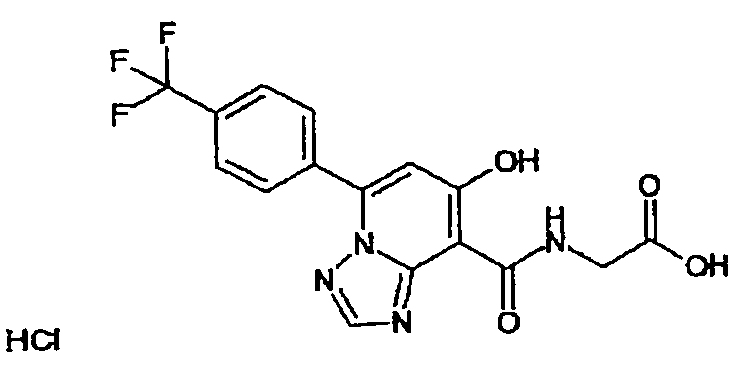
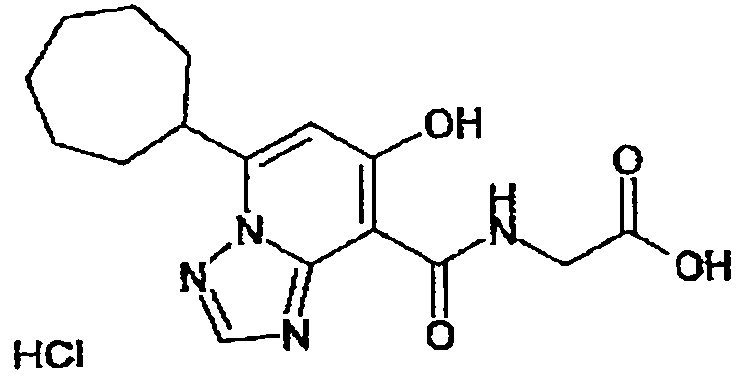
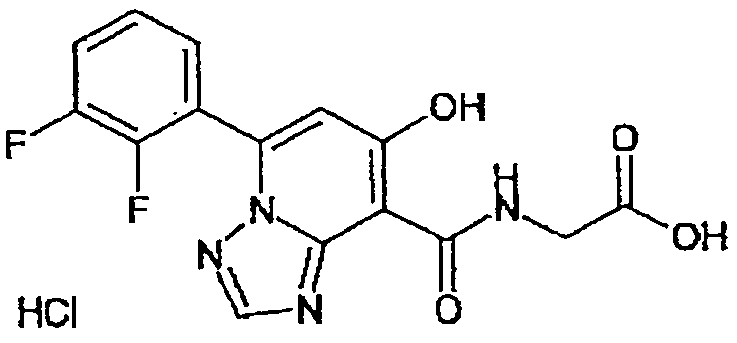
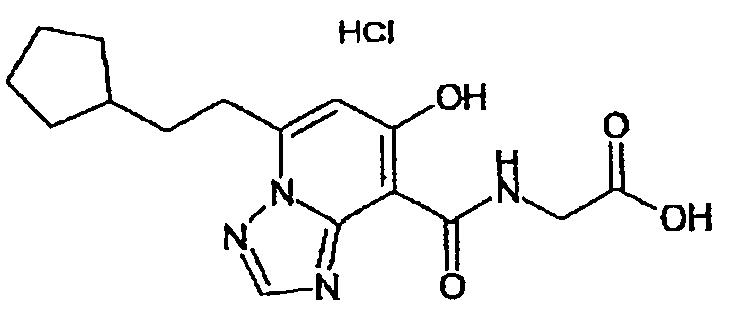
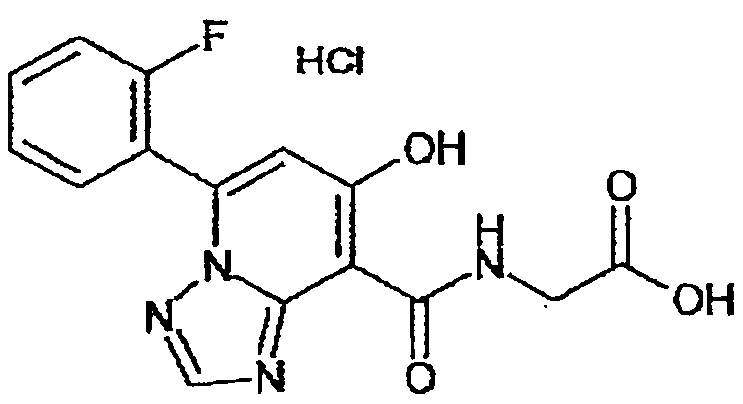
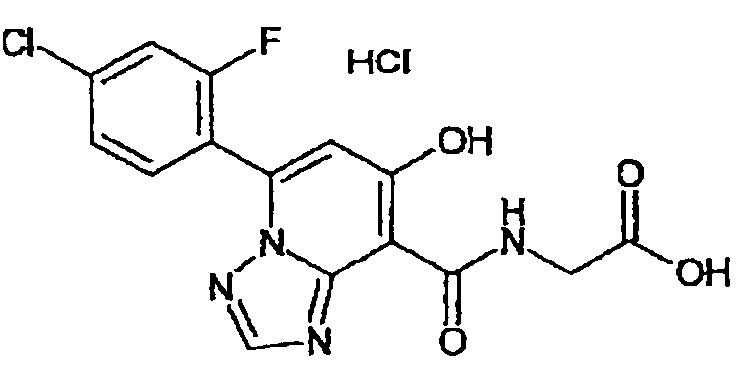
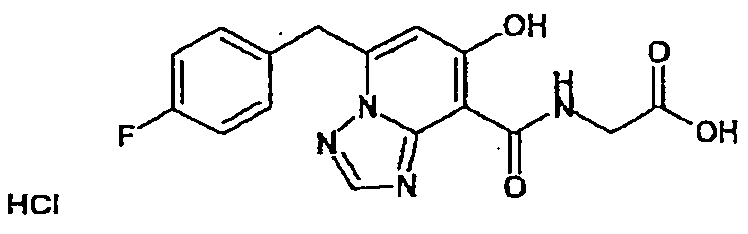
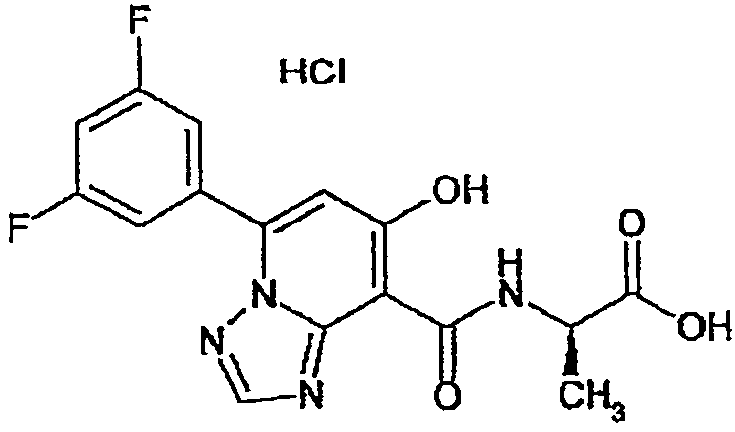
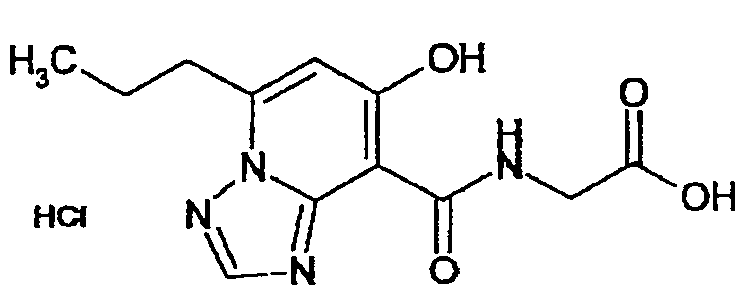
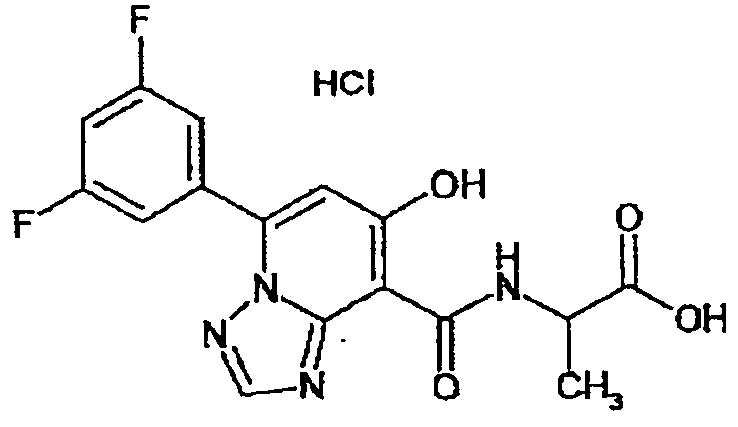
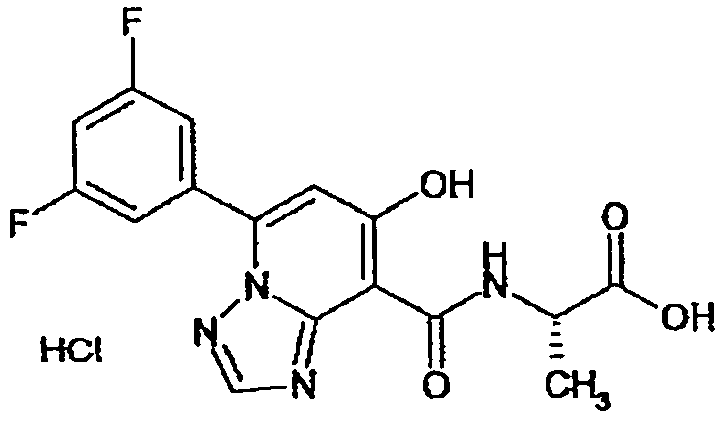
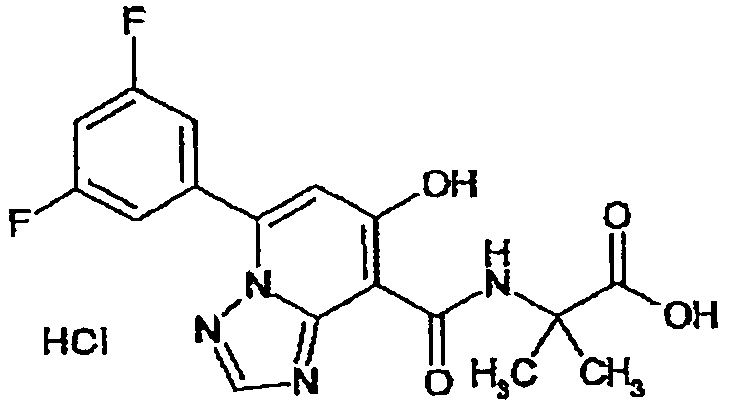
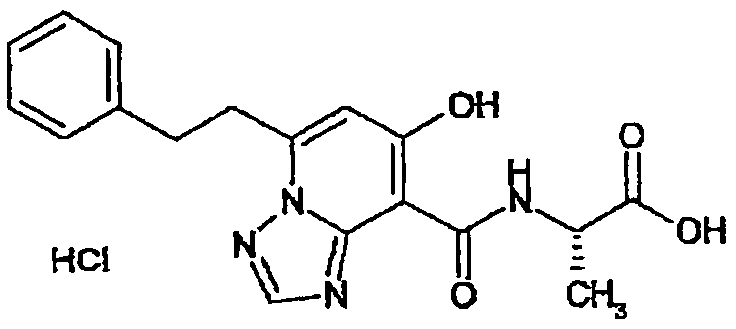
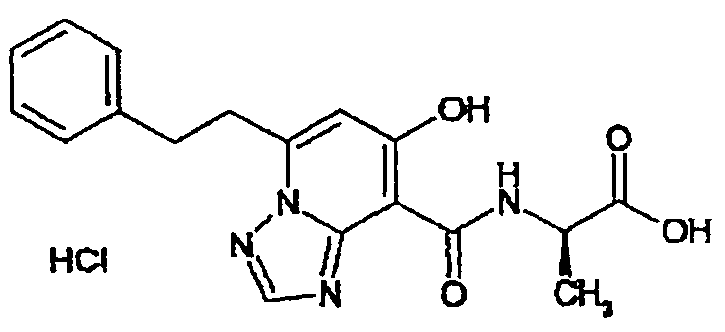
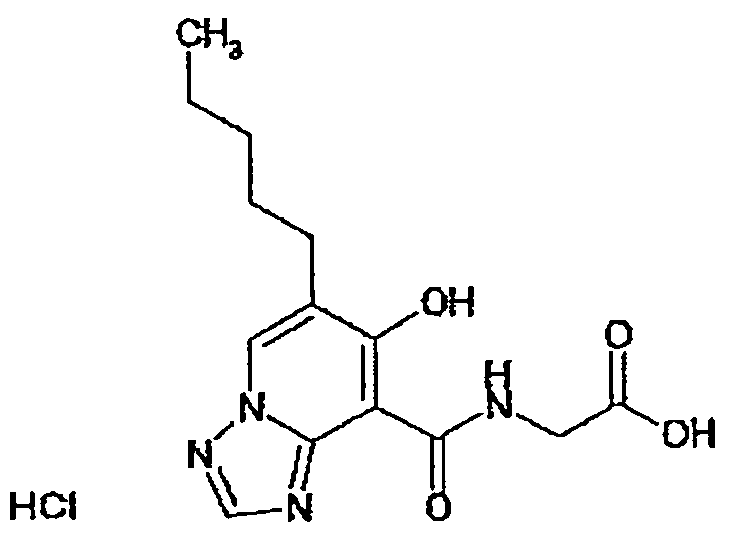
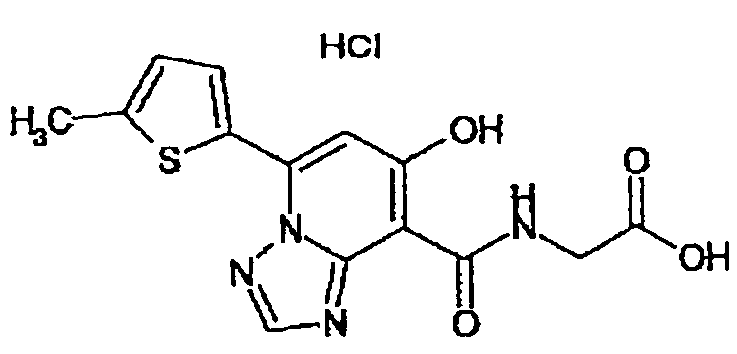
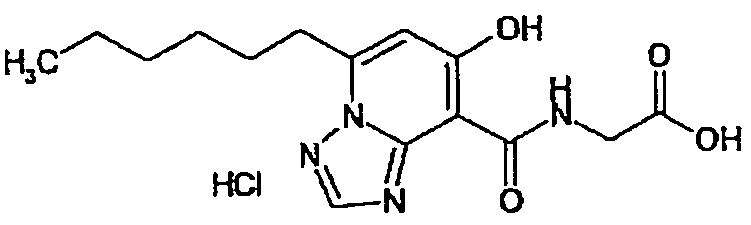
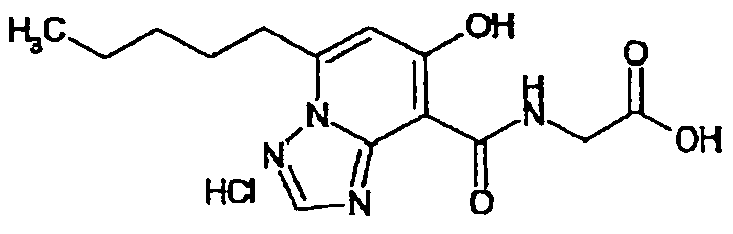
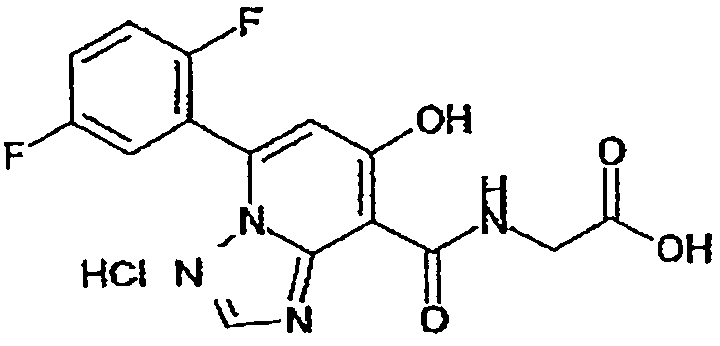
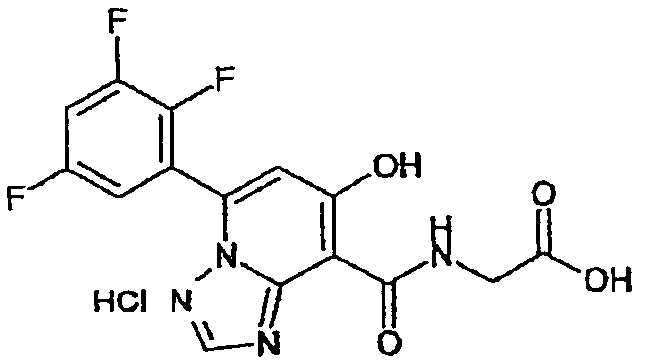
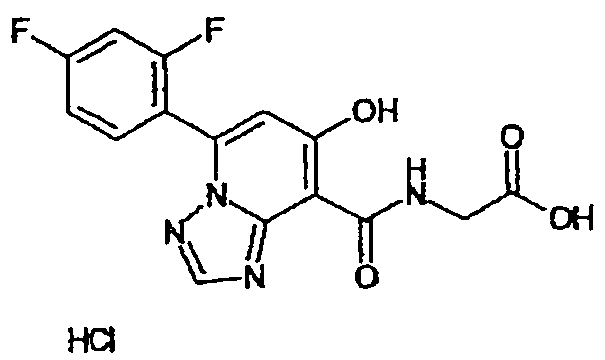
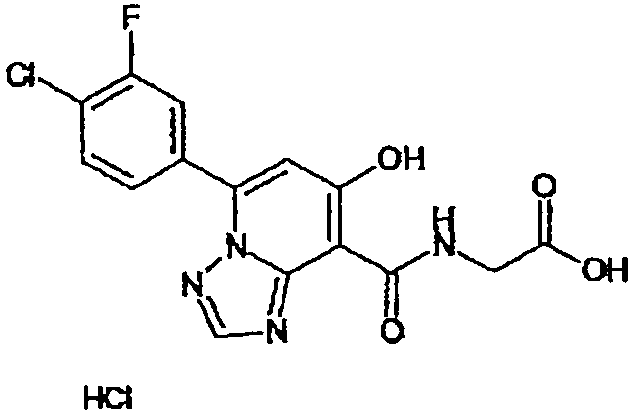
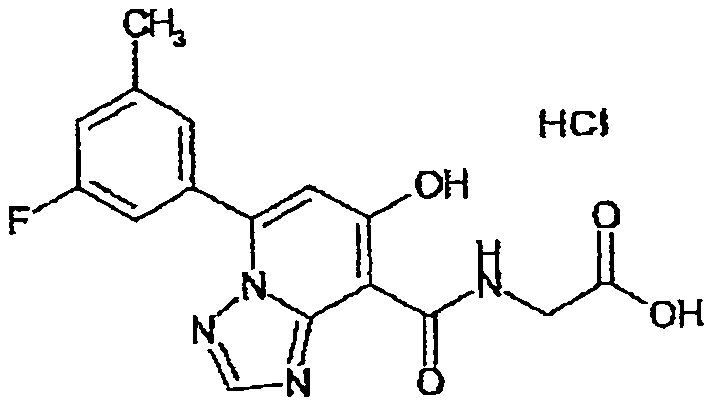
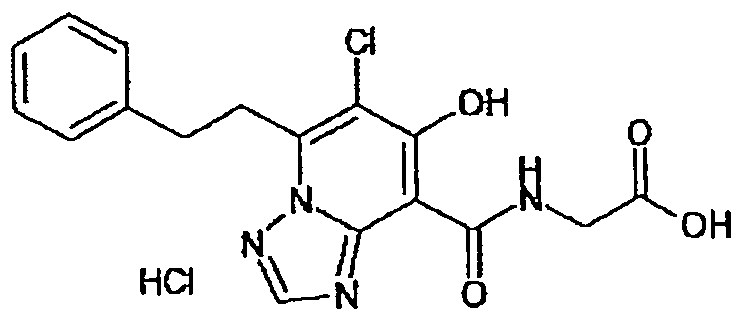
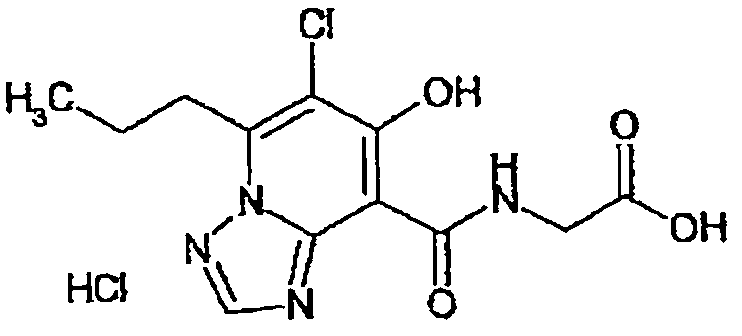
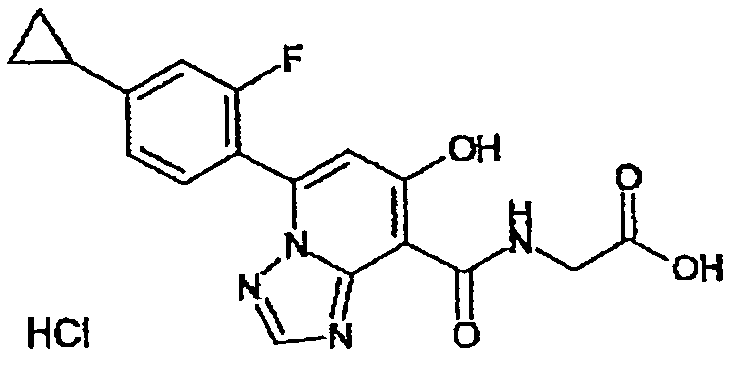
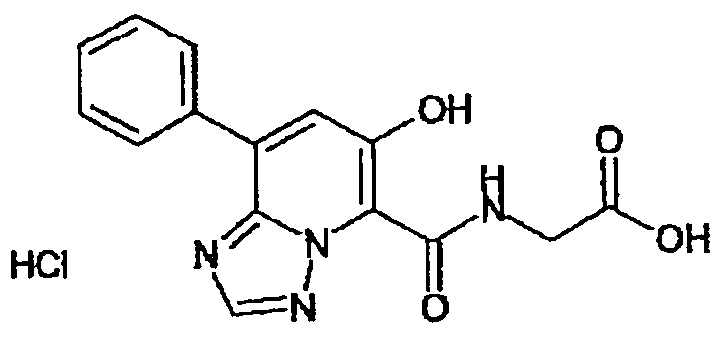
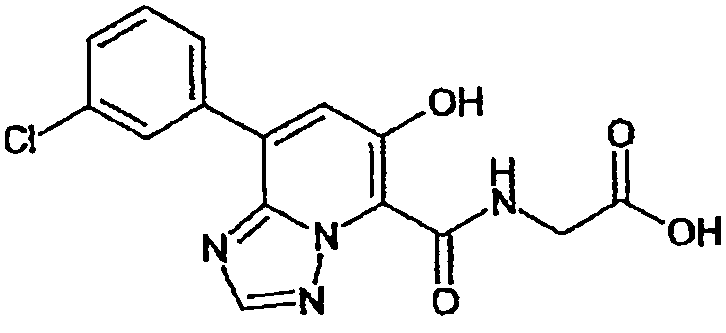
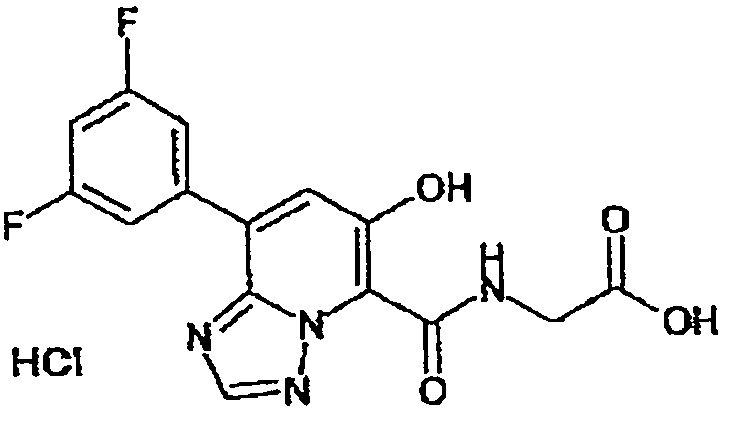
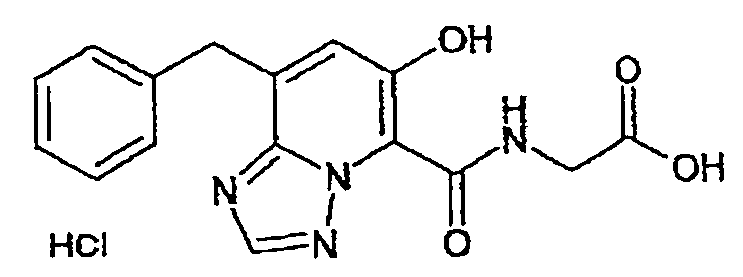
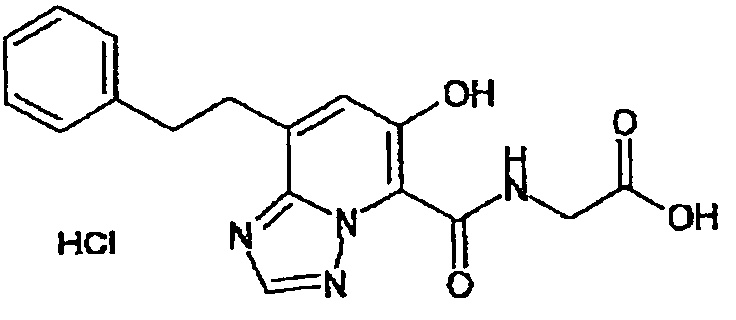
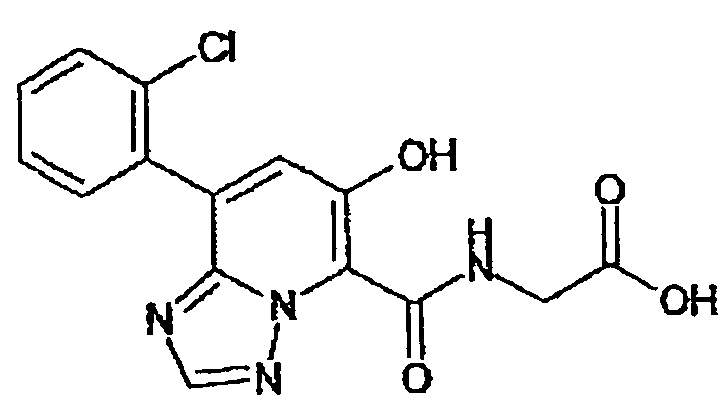
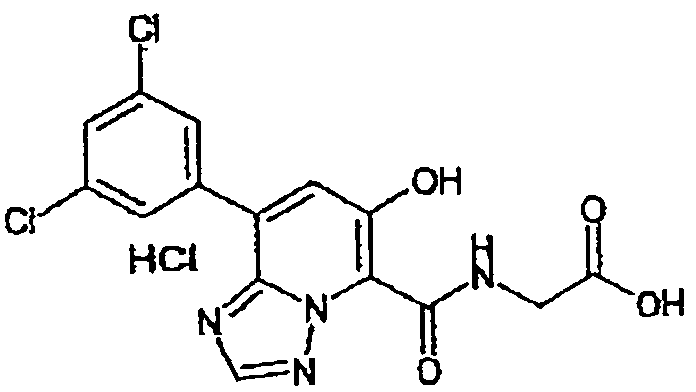
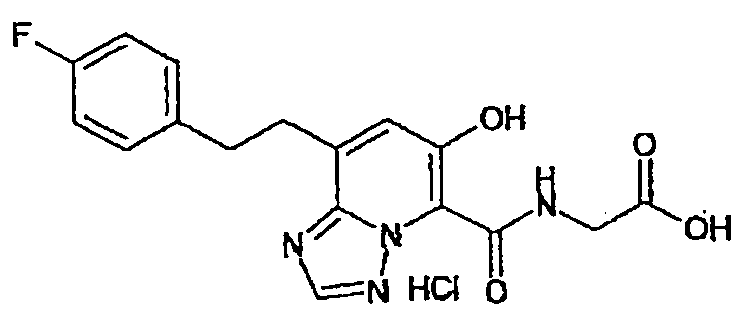
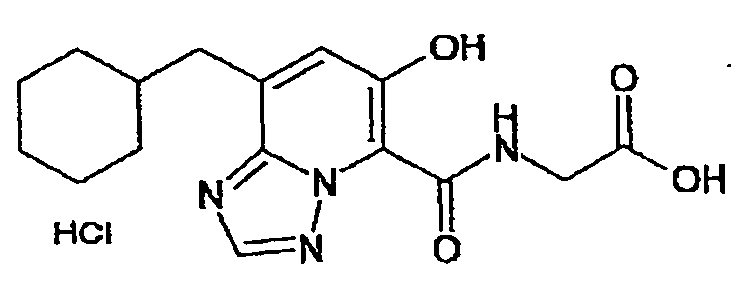
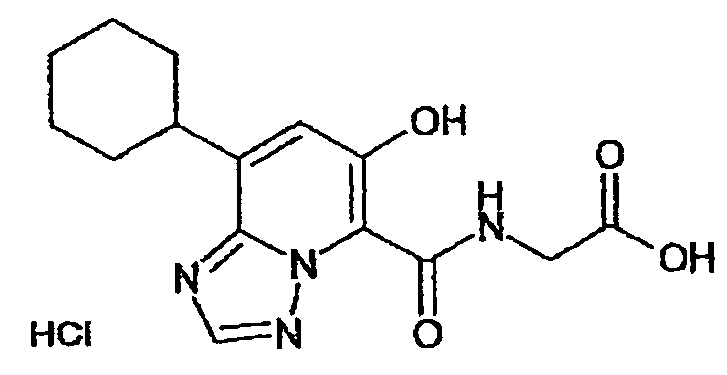
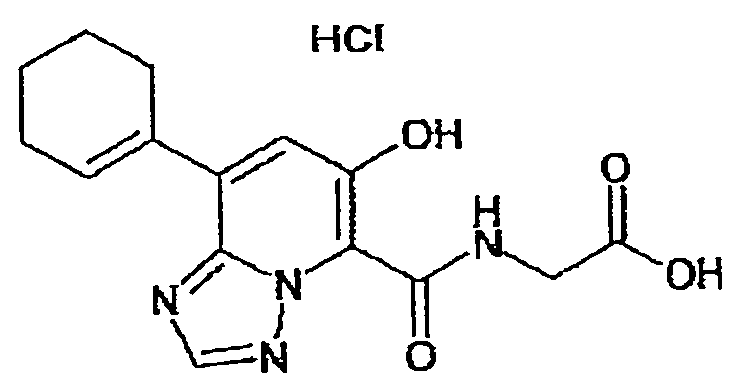
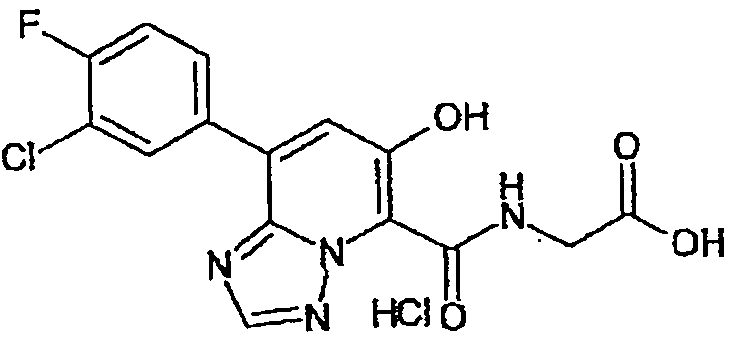
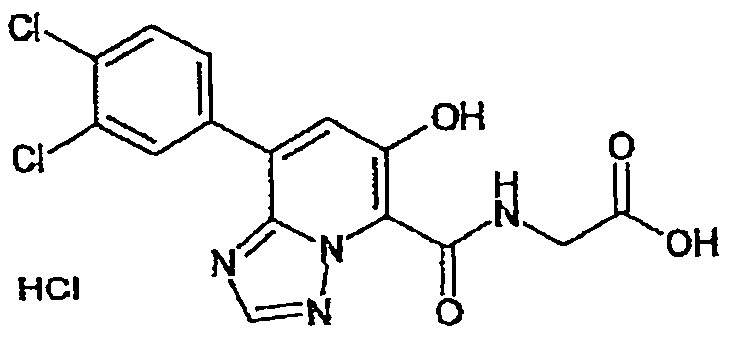
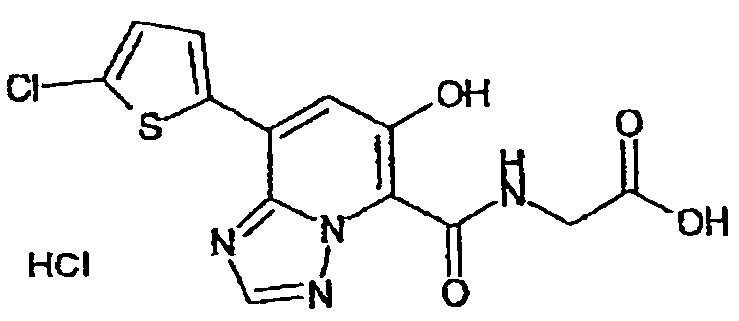
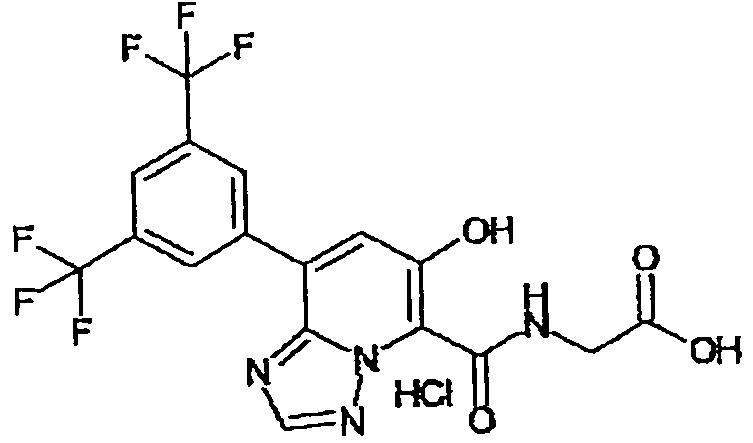
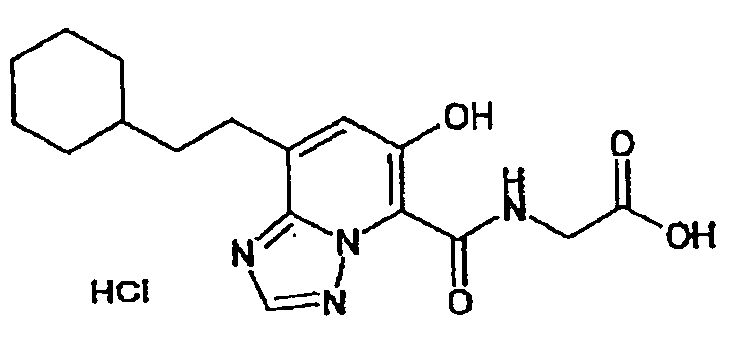
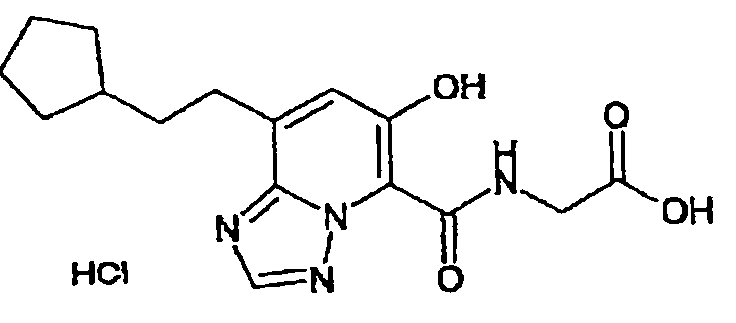
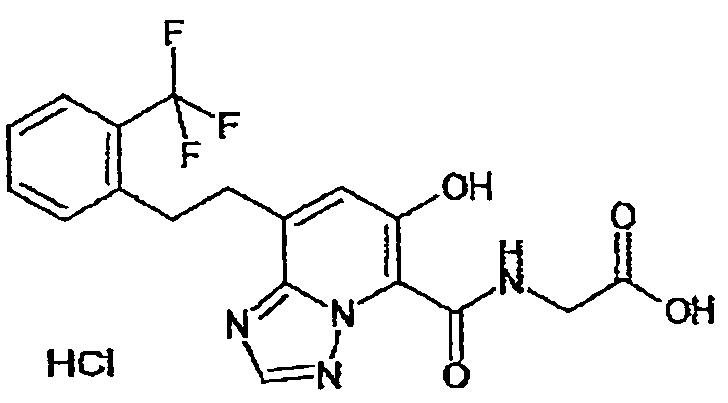
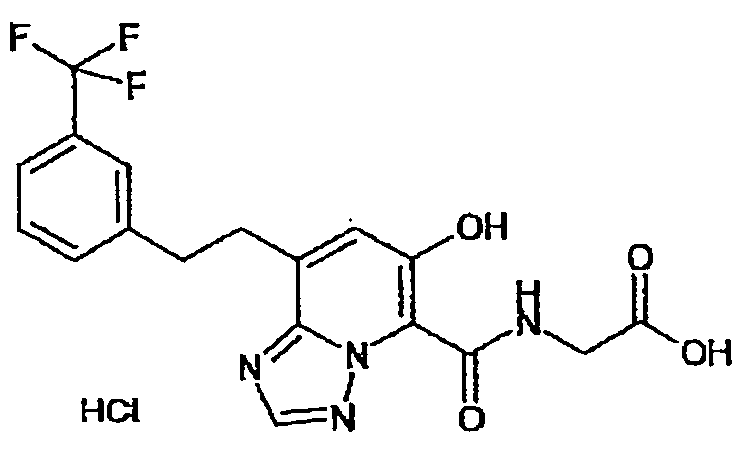
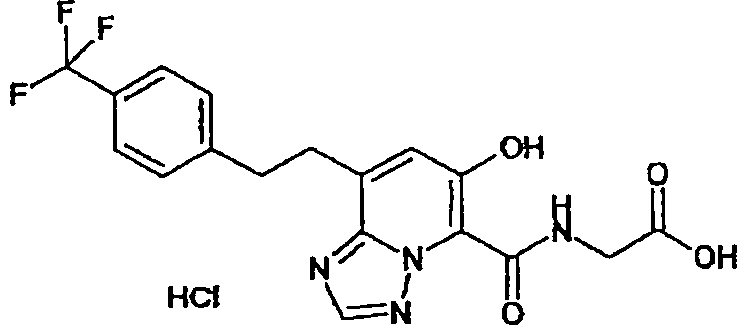
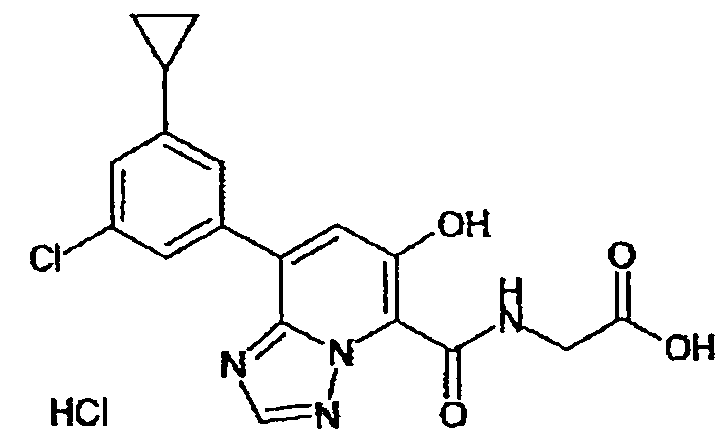
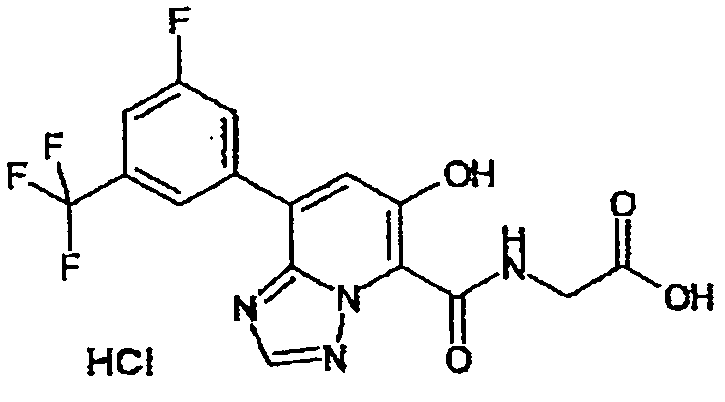
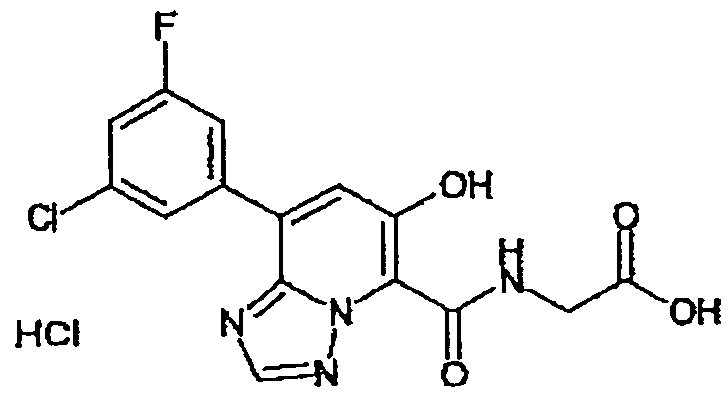
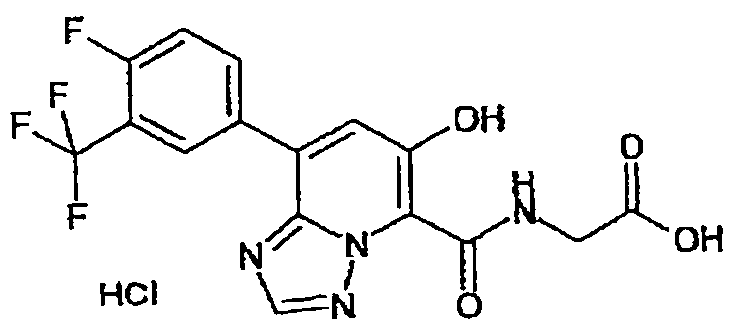
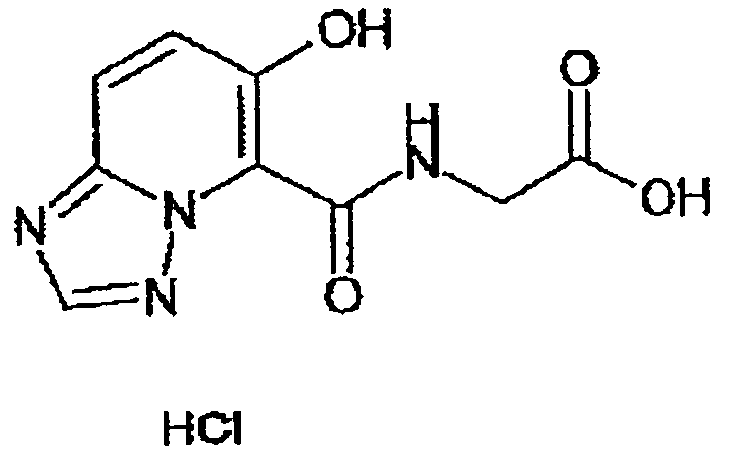
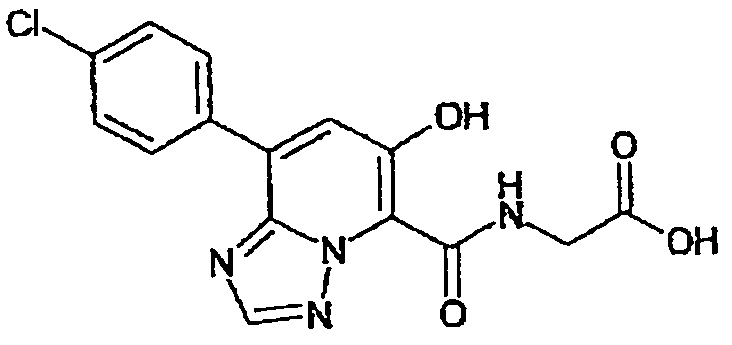
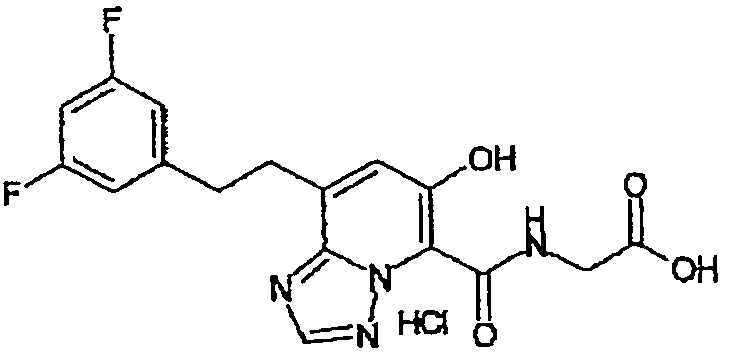
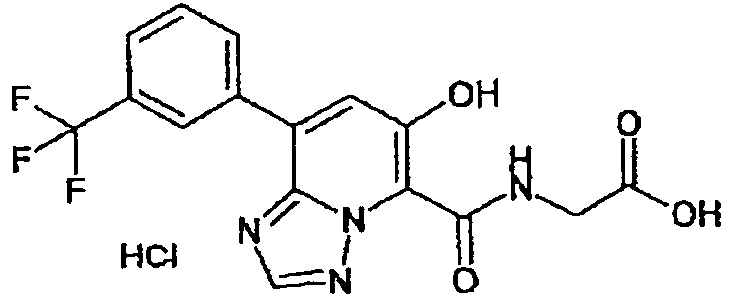
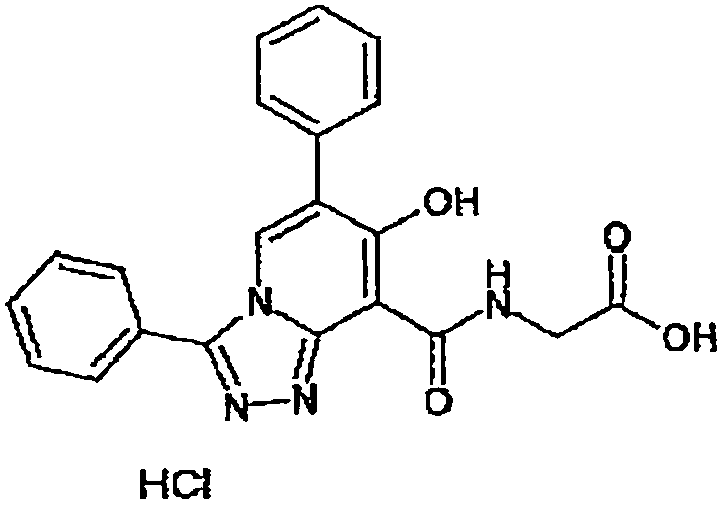
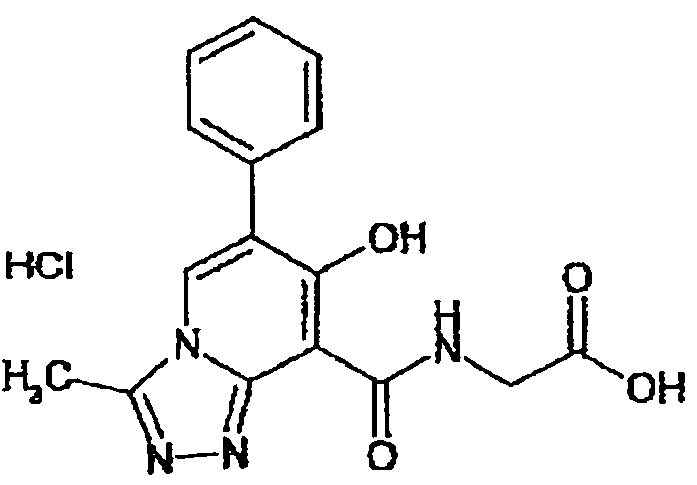
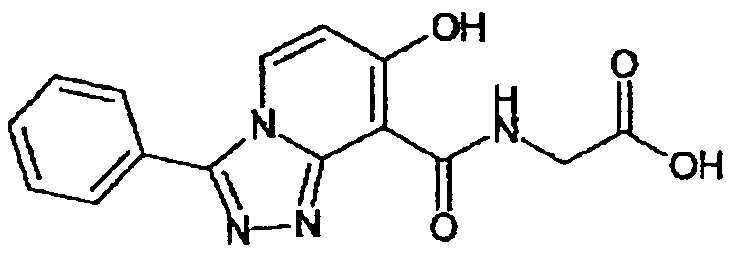
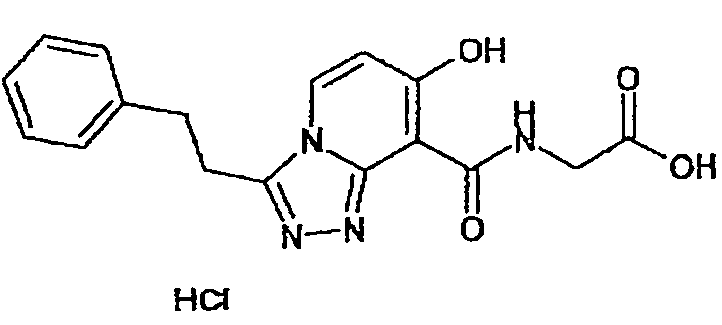
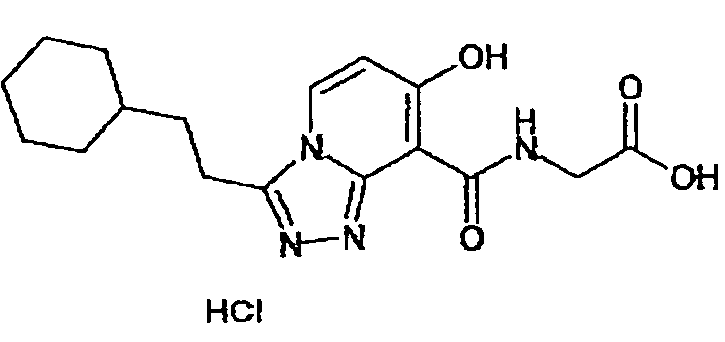
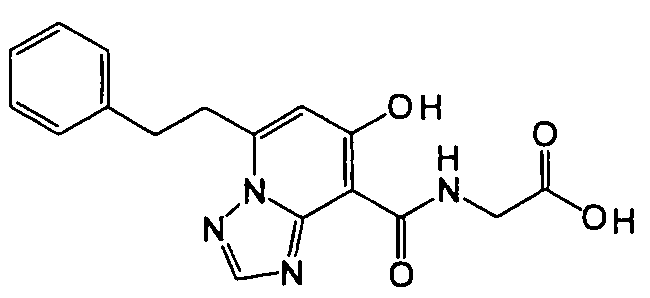
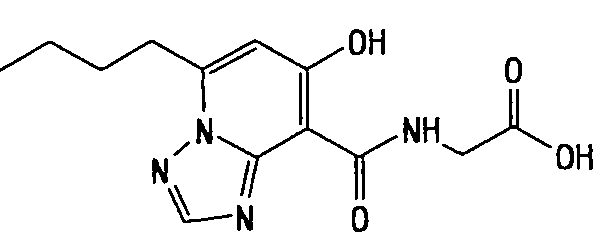
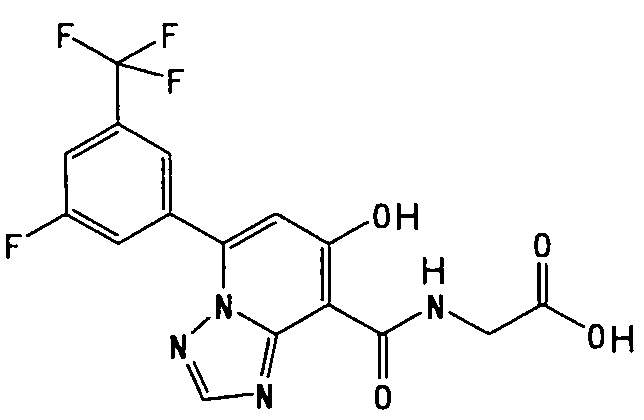
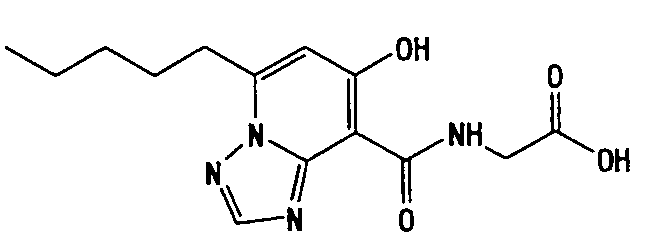
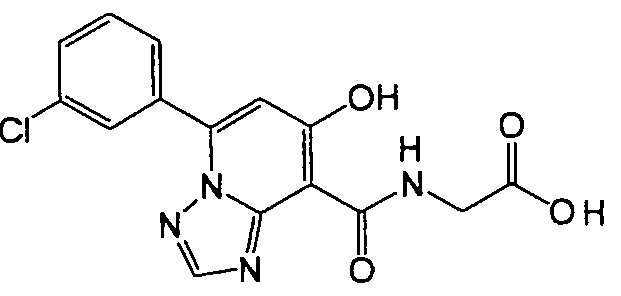
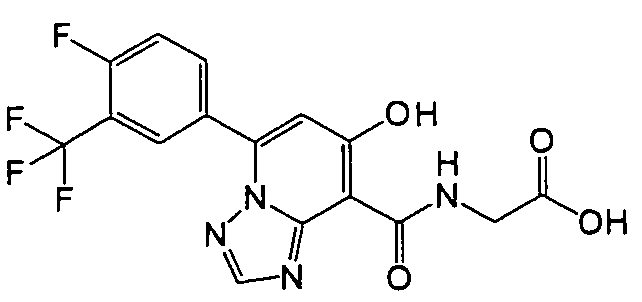
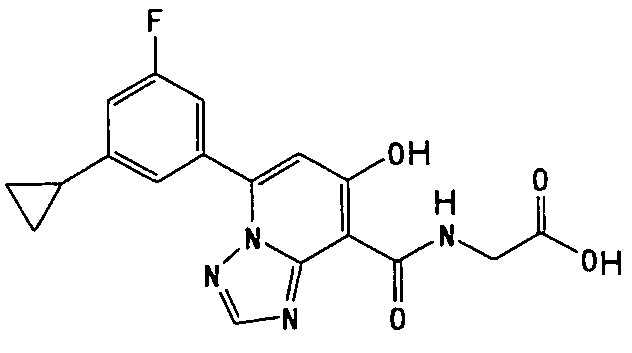
Примеры приготовления состава по настоящему изобретению включают следующие приготовления состава. Тем не менее, настоящее изобретение не ограничивают такими примерами приготовления состава.
Компоненты 1), 2), 3) и 4) смешивают и заключают в желатиновую капсулу.
Общее количество компонентов 1), 2), 3) и 30 г компонента 4) замешивают с водой, высушивают в условиях вакуума и просеивают. Просеянный порошок смешивают с 14 г компонента 4) и 1 г компонента 5) и таблетируют смесь при помощи таблетирующей машины. Таким образом, получают 1000 таблеток, содержащих 10 мг соединения Примера 1 на таблетку.
Далее описаны методы оценки ингибирующей активности в отношении человеческого PHD и индукции выработки EPO соединения по настоящему изобретению, или его фармацевтически приемлемой соли, или его сольвата.
Тестовый пример 1
Измерение ингибирующей активности в отношении PHD человека
i) Экспрессия и очистка PHD2 человека
PHD2 человека экспрессировали в клетках насекомых (клетки Sf9). Осуществляли вставку FLAG-последовательности в N-концевую часть трансляционного участка зарегистрированной последовательности PHD2 человека (NM_022051), последовательность вводили в вектор pVL1393 и подтверждали последовательность. Вектор и бакуловирус котрансфицировали в Sf9 и изолировали в Sf9 бакуловирус, экспрессирующий PHD2 человека. С использованием вируса получали клетки, экспрессирующие PHD2 человека. После выращивания клеток при 27°C в течение 72 ч добавляли лизирующий раствор с различным содержанием ингибиторов протеаз и разрушали клетки ультразвуком. Клеточный лизат наносили на колонку, заполненную аффинным гелем ANTI-FLAG M2 Affinity Gel Freezer Safe (SIGMA), промывали, элюировали и собирали PHD2 человека с добавленной N-концевой FLAG-последовательностью. По методу вестерн-блоттинга с использованием анти-FLAG антител и анти-PHD2 антител подтверждали, что продукт очистки представляет собой PHD2 человека.
ii) Экспрессия и очистка комплекса VBC
Комплекс VBC (VHL/элонгин B/элонгин C) экспрессировали в Escherichia coli (BL21(DE3)). Осуществляли вставку GST-слияния в N-концевую часть трансляционного участка зарегистрированной последовательности VHL человека (NM_000551). Осуществляли вставку FLAG-последовательности в N-концевую часть трансляционного участка зарегистрированной последовательности элонгина B человека (NM_207013), последовательность вводили в вектор pETDuet-1 и подтверждали последовательность. Осуществляли вставку His-последовательности в N-концевую часть трансляционного участка зарегистрированной последовательности элонгина C человека (NM_005648), последовательность вводили в вектор pETDuet-1 и подтверждали последовательность. После трансфекции указанных векторов экспрессии в Escherichia coli (BL21(DE3)), Escherichia coli выращивали при 37°C в среде, содержащей IPTG. Собранные Escherichia coli разрушали ультразвуком и наносили на колонку, заполненную Ni-NTA superflow (QIAGEN), промывали, элюировали и собирали продукт. Элюаты наносили на колонку, заполненную глутатионом Sepharose 4B, промывали, элюировали и собирали продукт. По методу вестерн-блоттинга с использованием анти-GST антител·анти-FLAG антител и анти-His антител подтверждали, что продукт очистки представляет собой VHL/элонгин В/элонгин C человека.
iii) Связывающая активность комплекса VBC
На покрытом стрептавидином планшете измеряли связывающую активность комплекса VBC, полученного в вышеупомянутом пункте ii), в отношении к 19 остаткам меченного биотином неполного пептида (HIF-1α-C19) на основе последовательности HIF-1α или меченного биотином неполного пептида (HIF-1α-C19 (Hyp)), где пролиновый остаток в упомянутой последовательности гидроксилирован. Для обнаружения проводили ELISA с использованием анти-GST-антител и подтверждали связывание комплекса VBC только с гидроксилированным неполным пептидом HIF-1α.
iv) Измерение ингибирующей активности в отношении PHD человека
Что касается ферментативной активности PHD2 человека, то гидроксилирование пролинового остатка, содержащегося в 19 остатках неполного пептида на основе последовательности HIF-1α, в качестве субстрата измеряли по методу TR-FRET (флуоресцентного резонансного переноса энергии с временным разрешением).
Фермент и субстрат, каждый, разбавляли 50 мМ трис-гидрохлоридным буфером (pH 7,5), содержащим 50 мкм сульфата железа, 120 мМ NaCl, 0,1% БСА, 0,1 мМ аскорбиновой кислоты, 10 мкМ 2-оксоглутаровой кислоты, 0,2 мМ CHAPS и разбавляли тестируемое соединение диметилсульфоксидом (DMSO).
Тестируемое соединение и раствор субстрата добавляли в 96-луночный планшет. Реакцию инициировали добавлением к реакционной системе раствора фермента PHD2 человека (конечная концентрация 1 нМ). После инкубации при 25°C в течение 30 мин добавляли стоп-раствор, содержащий EDTA и добавляли раствор комплекса VBC, содержащий европий (Eu) и Xlent, и определяли содержание гидроксилированного пролинового остатка по методу флуоресцентной спектроскопии с временным разрешением. Флуоресценцию с временным разрешением измеряли в каждой лунке и, исходя из значений в лунках без добавления фермента и без добавления тестируемого соединения, вычисляли ингибирующую активность (%) тестируемого соединения в отношении PHD человека. Ингибирующая активность каждого соединения в отношении PHD человека представлена в виде значений IC50 (мкМ) или значений ингибирующей активности (%) в отношении PHD человека при 30 мкМ в последующих Таблицах 25-29. В указанных Таблицах значения, состоящие исключительно из чисел, представляют значения IC50 (мкМ), а значения, содержащие %, представляют собой значения ингибирующей активности (%) в отношении PHD человека при 30 мкМ.
Тестовый пример 2
Активность в отношении выработки EPO человека
Активность тестируемого соединения в отношении выработки EPO человека измеряли с использованием Hep3B (ATCC), созданных из клеточной линии, полученной из человеческой печени.
Клетки Hep3B выращивали в модифицированной среде Игла, содержащей 10% эмбриональной бычьей сыворотки, и разбавляли тестируемое соединение диметилсульфоксидом (DMSO).
Клетки Hep3B выращивали в 96-луночном планшете и спустя 24 ч добавляли тестируемое соединение в каждой концентрации. После инкубации при 37°C в течение 24 ч отбирали культуральную надосадочную жидкость. Концентрацию EPO человека, выработанного в культуральной надосадочной жидкости, измеряли с использованием набора для EPO-ELISA человека (производства StemCell Technologies, 01630) в соответствии с описанием производителя, и рассчитывали активность тестируемого соединения в отношении выработки EPO человека (%), исходя из максимального значения выработки в указанных условиях и значения выработки без добавления тестируемого соединения. Активность каждого тестируемого соединения в отношении выработки EPO человека представлена в последующих Таблицах 30-34 в виде значений IC50 (мкМ) или значений активности тестируемого соединения в отношении выработки EPO человека (%) при 30 мкМ. В указанных Таблицах значения, состоящие исключительно из чисел, представляют значения IC50 (мкМ), а таковые, содержащие %, представляют собой значения активности тестируемого соединения в отношении выработки EPO человека (%) при 30 мкМ.
Как видно из упомянутых выше результатов, соединение по настоящему изобретению, или его фармацевтически приемлемая соль, или его сольват обладают ингибирующей активностью в отношении PHD человека и способностью к выработке EPO человека.
Промышленная применимость
Соединение по настоящему изобретению, или его фармацевтически приемлемая соль, или его сольват ингибируют связывание HIF и PHD исходя из его ингибирующей активности в отношении PHD, и стабилизируют HIF, что позволяет способствовать выработке EPO.
Таким образом, соединение по настоящему изобретению, или его фармацевтически приемлемая соль, или его сольват могут представлять собой лекарственный препарат, эффективный для профилактики или лечения различных заболеваний и патологий (нарушений), вызванных сниженной выработкой EPO, и могут быть эффективно использованы для лечения анемии.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИТОРЫ ПРОЛИЛГИДРОКСИЛАЗЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2007 |
|
RU2429226C9 |
| АМИНОТРИАЗОЛОПИРИДИНЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗ | 2009 |
|
RU2552642C2 |
| ПРОИЗВОДНОЕ 1,2,4-ТРИАЗОЛОНА | 2011 |
|
RU2566754C2 |
| СПОСОБЫ И КОМПОЗИЦИИ ДЛЯ НАПРАВЛЕННОЙ ДЕГРАДАЦИИ БЕЛКА | 2020 |
|
RU2820673C2 |
| АЗОТСОДЕРЖАЩИЕ АРОМАТИЧЕСКИЕ ПРОИЗВОДНЫЕ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ИХ СОДЕРЖАЩАЯ, СПОСОБ ЛЕЧЕНИЯ И ПРИМЕНЕНИЕ | 2003 |
|
RU2310651C2 |
| ПРОИЗВОДНЫЕ ПИРИДИНА В КАЧЕСТВЕ ИНГИБИТОРОВ ДИПЕПТИДИЛПЕПТИДАЗЫ IV | 2004 |
|
RU2353617C2 |
| СУЛЬФОНАМИДНОЕ ПРОИЗВОДНОЕ И ЕГО МЕДИЦИНСКОЕ ПРИМЕНЕНИЕ | 2013 |
|
RU2607081C2 |
| СОЕДИНЕНИЯ АЗАЛАКТАМА В КАЧЕСТВЕ ИНГИБИТОРОВ HPK1 | 2019 |
|
RU2801140C2 |
| ГЕТЕРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ | 2018 |
|
RU2791533C2 |
| СОЕДИНЕНИЕ В КАЧЕСТВЕ АНТАГОНИСТА АДЕНОЗИНОВОГО РЕЦЕПТОРА A2a И СОДЕРЖАЩАЯ ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2022 |
|
RU2840068C2 |
Изобретение относится к области органической химии, а именно к триазолопиридиновому соединению общей формулы [I] или к его фармацевтически приемлемой соли, где частичная структурная формула: 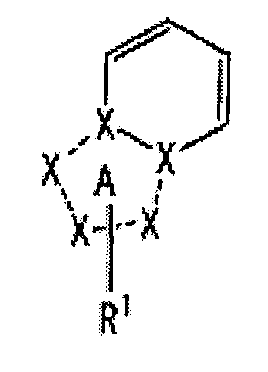 представляет собой группу, представленную любой из следующих формул:
представляет собой группу, представленную любой из следующих формул: 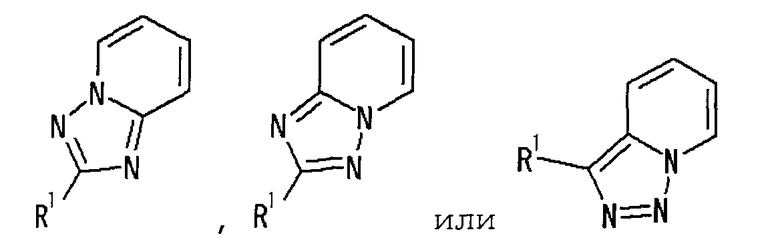 , R1 представляет собой (1) атом водорода, (2) C1-6алкильную группу, (3) фенильную группу или (4) С3-8циклоалкильную группу; R2 представляет собой (1) атом водорода, (2) C1-10алкильную группу, (3) фенильную группу, необязательно замещенную одинаковыми или различными 1-3 заместителями, выбранными из следующей группы В, (4) С3-8циклоалкильную группу, (5) С3-8циклоалкенильную группу, (6) тиенильную группу, необязательно замещенную 1 заместителем, выбранным из галогена или C1-6алкильной группы, (7) фенил-С1-6алкильную группу (в которой фенил необязательно замещен одинаковыми или различными 1-2 заместителями, выбранными из галогена, С3-8циклоалкильной или галоген-С1-6алкильной группы) или (8) С3-8циклоалкил-С1-6акильную группу; R3 представляет собой (1) атом водорода, (2) атом галогена, (3) С1-6алкильную группу, (4) фенильную группу или (6) фенил-С1-6алкильную группу; и каждый из R4 и R5 представляют собой оба атомы водорода или группа В: (a) атом галогена, (b) С1-6алкильная группа, (c) С3-8циклоалкильная группа, (d) цианогруппа и (e) галоген-С1-6алкильная группа. Также изобретение относится к конкретным соединениям, фармацевтической композиции на основе соединения формулы [I] и применению соединения формулы [I]. Технический результат: получены новые триазолопиридиновые соединения, обладающие ингибирующим действием в отношении пролилгидроксилазы и способностью индуцировать выработку эритропоэтина. 21 н. и 9 з.п. ф-лы, 34 табл.
, R1 представляет собой (1) атом водорода, (2) C1-6алкильную группу, (3) фенильную группу или (4) С3-8циклоалкильную группу; R2 представляет собой (1) атом водорода, (2) C1-10алкильную группу, (3) фенильную группу, необязательно замещенную одинаковыми или различными 1-3 заместителями, выбранными из следующей группы В, (4) С3-8циклоалкильную группу, (5) С3-8циклоалкенильную группу, (6) тиенильную группу, необязательно замещенную 1 заместителем, выбранным из галогена или C1-6алкильной группы, (7) фенил-С1-6алкильную группу (в которой фенил необязательно замещен одинаковыми или различными 1-2 заместителями, выбранными из галогена, С3-8циклоалкильной или галоген-С1-6алкильной группы) или (8) С3-8циклоалкил-С1-6акильную группу; R3 представляет собой (1) атом водорода, (2) атом галогена, (3) С1-6алкильную группу, (4) фенильную группу или (6) фенил-С1-6алкильную группу; и каждый из R4 и R5 представляют собой оба атомы водорода или группа В: (a) атом галогена, (b) С1-6алкильная группа, (c) С3-8циклоалкильная группа, (d) цианогруппа и (e) галоген-С1-6алкильная группа. Также изобретение относится к конкретным соединениям, фармацевтической композиции на основе соединения формулы [I] и применению соединения формулы [I]. Технический результат: получены новые триазолопиридиновые соединения, обладающие ингибирующим действием в отношении пролилгидроксилазы и способностью индуцировать выработку эритропоэтина. 21 н. и 9 з.п. ф-лы, 34 табл.
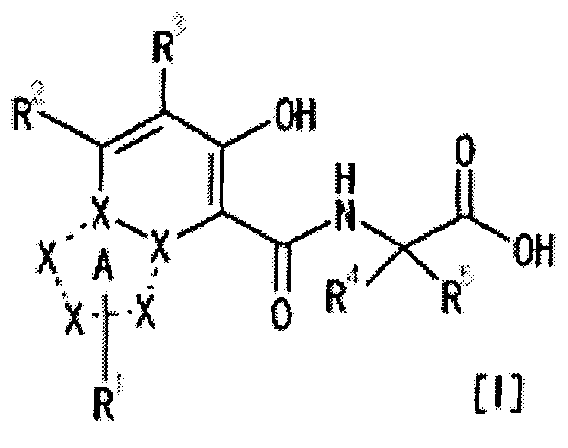
1. Соединение, представленное следующей формулой [I], или его фармацевтически приемлемая соль:
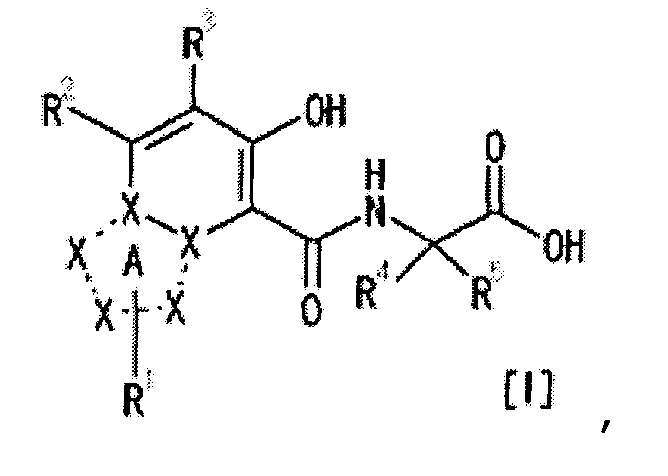
в котором
частичная структурная формула:
представляет собой группу, представленную любой из следующих формул:
R1 представляет собой
(1) атом водорода,
(2) C1-6алкильную группу,
(3) фенильную группу или
(4) С3-8циклоалкильную группу;
R2 представляет собой
(1) атом водорода,
(2) C1-10алкильную группу,
(3) фенильную группу, необязательно замещенную одинаковыми или различными 1-3 заместителями, выбранными из следующей группы
В,
(4) С3-8циклоалкильную группу,
(5) С3-8циклоалкенильную группу,
(6) тиенильную группу, необязательно замещенную 1 заместителем, выбранным из галогена или C1-6алкильной группы,
(7) фенил-С1-6алкильную группу (в которой фенил необязательно замещен одинаковыми или различными 1-2 заместителями, выбранными из галогена, С3-8циклоалкильной или галоген-С1-6алкильной группы) или
(8) С3-8циклоалкил-С1-6акильную группу;
R3 представляет собой
(1) атом водорода,
(2) атом галогена,
(3) С1-6алкильную группу,
(4) фенильную группу или
(6) фенил-С1-6алкильную группу; и
каждый из R4 и R5 представляют собой оба атомы водорода или
группа В:
(a) атом галогена,
(b) С1-6алкильная группа,
(c) С3-8циклоалкильная группа,
(d) цианогруппа и
(e) галоген-С1-6алкильная группа.
2. Соединение по п.1, в котором частичная структурная формула:
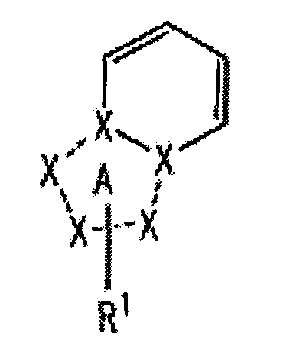
представляет собой группу, представленную следующей формулой
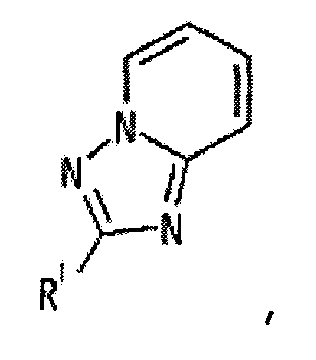
или его фармацевтически приемлемая соль.
3. Соединение по п.1, в котором частичная структурная формула:
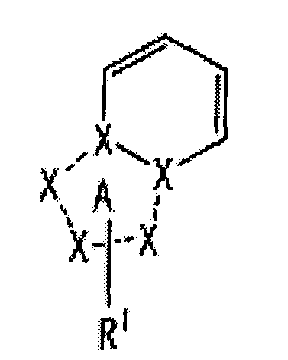
представляет собой группу, представленную следующей формулой
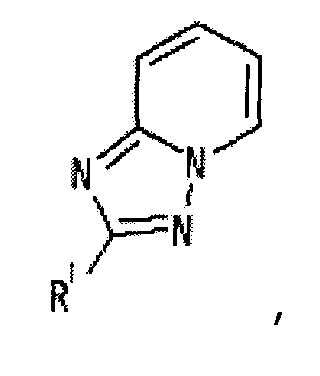
или его фармацевтически приемлемая соль.
4. Соединение по п.1, в котором частичная структурная формула:
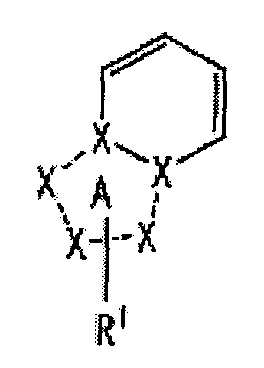
представляет собой группу, представленную следующей формулой
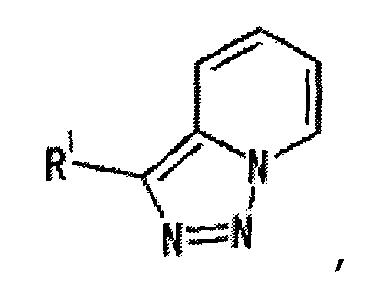
или его фармацевтически приемлемая соль.
5. Соединение по любому из пп.1-4, в котором R3 представляет собой атом водорода, или его фармацевтически приемлемая соль.
6. Соединение по любому из пп.1-5, в котором R1 представляет собой атом водорода, или его фармацевтически приемлемая соль.
7. Соединение по любому из пп.1-5, в котором R2 представляет собой
(1) C1-10алкильную группу,
(2) фенильную группу, необязательно замещенную одинаковыми или различными 1-3 заместителями, выбранными из вышеупомянутой группы В,
(3) фенил-С1-6алкильную группу (в которой фенил необязательно замещен одинаковыми или различными 1-2 заместителями, выбранными из галогена, С3-8циклоалкильной или галоген-С1-6алкильной группы) или
(4) С3-8циклоалкил-С1-6алкильную группу,
или его фармацевтически приемлемая соль.
8. Соединение по п.2, в котором R3 представляет собой атом водорода, или его фармацевтически приемлемая соль.
9. Соединение по п.8, в котором R1 представляет собой атом водорода, или его фармацевтически приемлемая соль.
10. Соединение по п.9, в котором R2 представляет собой
(1) C1-10алкильную группу или
(2) фенил-С1-6алкильную группу (в которой фенил необязательно замещен одинаковыми или различными 1-2 заместителями, выбранными из галогена, С3-8циклоалкильной или галоген-С1-6алкильной группы),
или его фармацевтически приемлемая соль.
11. Соединение, представленное следующей формулой:
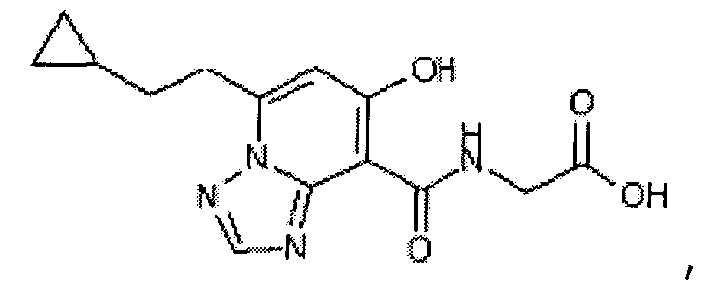
или его фармацевтически приемлемая соль.
12. Соединение, представленное следующей формулой:
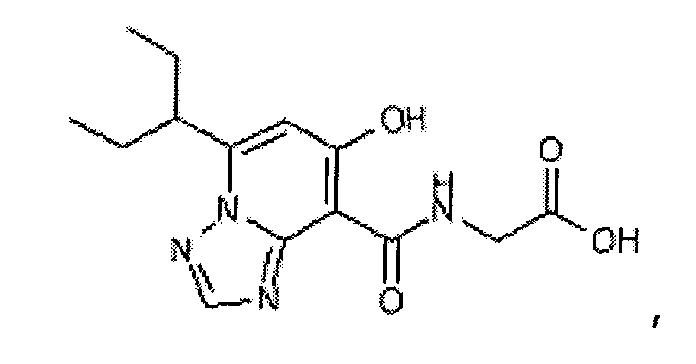
или его фармацевтически приемлемая соль.
13. Соединение, представленное следующей формулой:
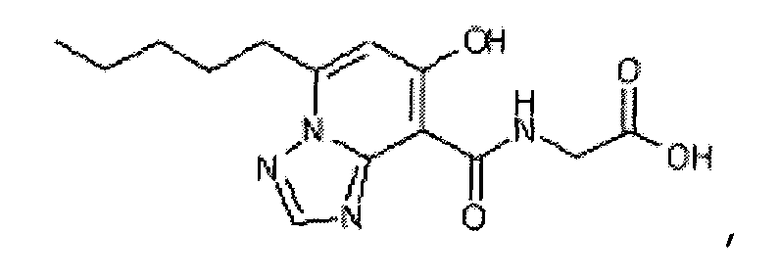
или его фармацевтически приемлемая соль.
14. Соединение, представленное следующей формулой:
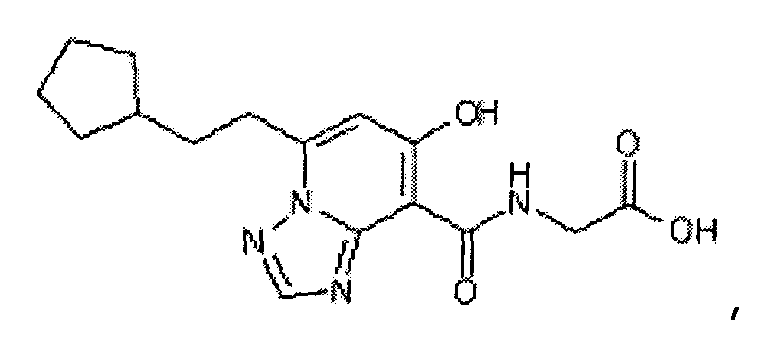
или его фармацевтически приемлемая соль.
15. Соединение, представленное следующей формулой:
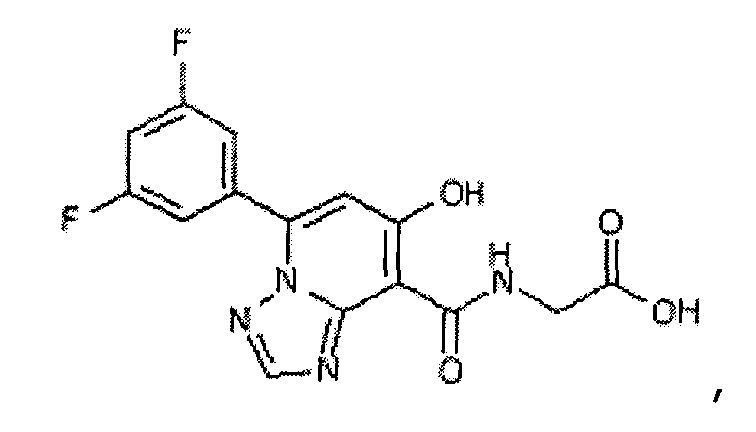
или его фармацевтически приемлемая соль.
16. Соединение, представленное следующей формулой:
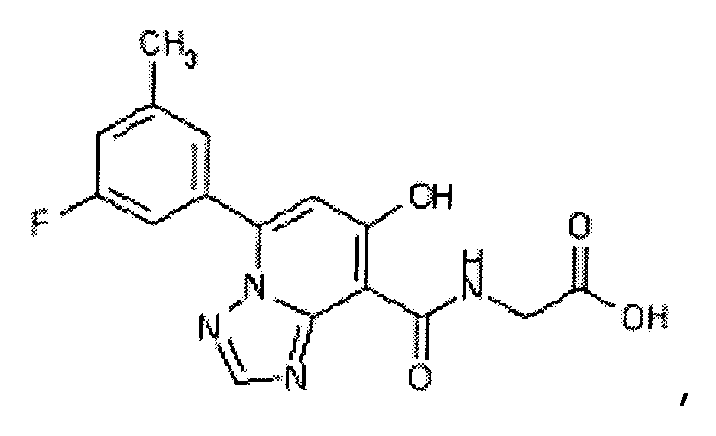
или его фармацевтически приемлемая соль.
17. Соединение, представленное следующей формулой:
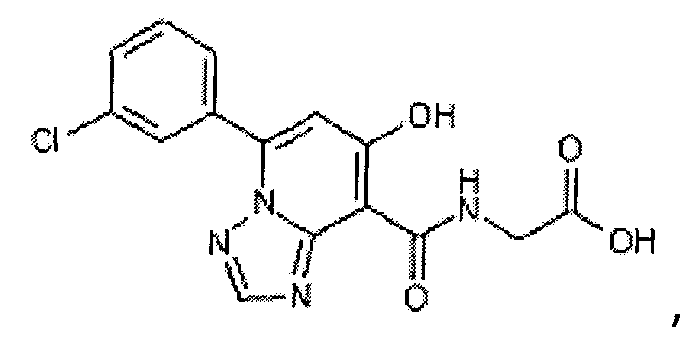
или его фармацевтически приемлемая соль.
18. Соединение, представленное следующей формулой:
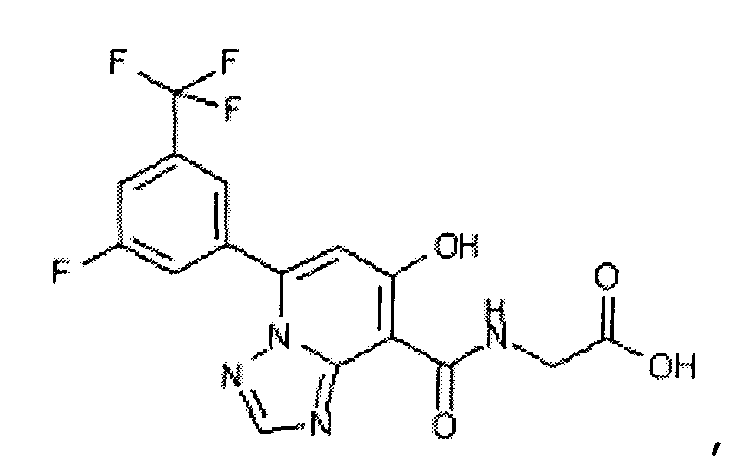
или его фармацевтически приемлемая соль.
19. Соединение, представленное следующей формулой:
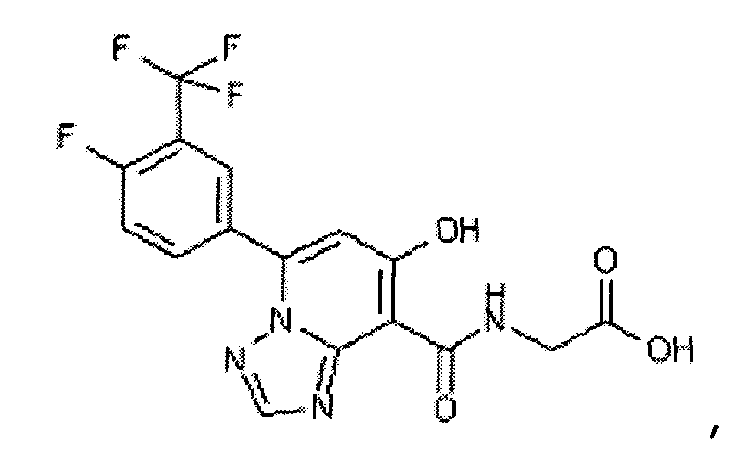
или его фармацевтически приемлемая соль.
20. Соединение, представленное следующей формулой:
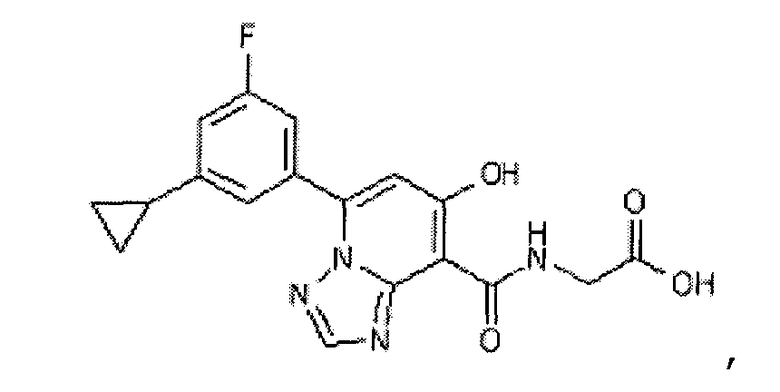
или его фармацевтически приемлемая соль.
21. Соединение, представленное следующей формулой:
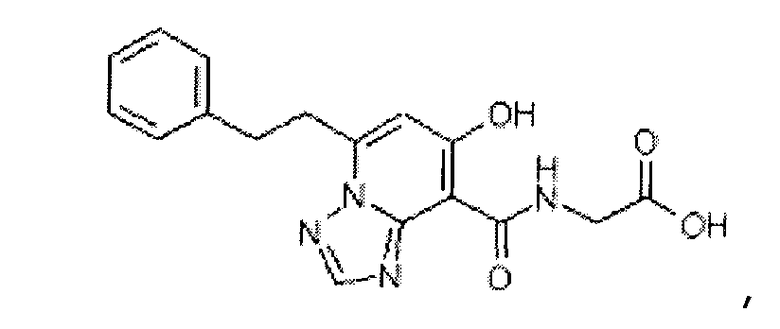
или его фармацевтически приемлемая соль.
22. Фармацевтическая композиция для лечения анемии или почечной анемии, содержащая соединение по любому из пп.1-21 или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель.
23. Применение соединения, представленного формулой (I) по любому из пп.1-10, или его фармацевтически приемлемой соли в качестве ингибитора пролилгидроксилазы.
24. Применение соединения по любому из пп.11-21 или его фармацевтически приемлемой соли в качестве ингибитора пролилгидроксилазы.
25. Применение соединения, представленного формулой (I) по любому из пп.1-10, или его фармацевтически приемлемой соли в качестве агента, индуцирующего выработку эритропоэтина.
26. Применение соединения по любому из пп.11-21 или его фармацевтически приемлемой соли в качестве агента, индуцирующего выработку эритропоэтина.
27. Применение соединения, представленного формулой (I) по любому из пп.1-10, или его фармацевтически приемлемой соли для лечения анемии.
28. Применение соединения по любому из пп.11-21 или его фармацевтически приемлемой соли для лечения анемии.
29. Применение соединения, представленного формулой (I) по любому из пп.1-10, или его фармацевтически приемлемой соли для лечения почечной анемии.
30. Применение соединения по любому из пп.11-21 или его фармацевтически приемлемой соли для лечения почечной анемии.
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |
| Электронно-лучевой осциллограф | 1980 |
|
SU911340A1 |
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |

