Область техники, к которой относится изобретение
Настоящее изобретение относится к соединению в качестве антагониста аденозинового рецептора А2а, его стереоизомерам, его фармацевтически приемлемым солям, его медицинскому применению и содержащей его фармацевтической композиции.
Уровень техники
Аденозин относится к множеству биологически активных модификаторов сердечно-сосудистой и нервной систем, которые регулируют различные функции посредством взаимодействия со специфическими рецепторами клеточной поверхности. Кроме того, аденозин представляет собой иммунодепрессивный метаболит, который в большом количестве продуцируется в микроокружении опухоли, накапливается в опухолях, способствуя пролиферации опухолей, а также служит посредником в избегании опухолью иммунной системы, придавая резистентность к иммунной системе и т. д. Микроокружение опухоли является одним из важных регуляторов иммунных функций, влияющих на прогрессирование рака и его метастазирование. В микроокружении опухоли, высокая концентрация аденозина ингибирует ответы противоопухолевых цитотоксических лимфоцитов, и Т-клетки ингибируют их действие и экспрессируют аденозиновый рецептор А2а (А2аR), который блокирует удаление опухолей иммунитетом.
Аденозиновый рецептор A2a является одним из рецепторов A1, A2a, A2b и A3, которые представляют собой четыре подтипа рецептора, сопряженного с G-белком (GPCR), и широко распространен в тканях человека и сверхэкспрессируется в полосатом теле головного мозга, иммунных клетках, селезенке, тимусе, лейкоцитах, тромбоцитах, нейронах GABA-типа, обонятельных луковицах и подобных. В то же время, рецептор аденозина А2а также экспрессируется в других частях тела, таких как сердце, легкие, кровеносные сосуды, головной мозг и подобные, и проявляет высокую аффинность к аденозину. Рецептор A2b также широко экспрессируется, но в основном на низком уровне и менее чувствителен к аденозину. Рецептор А2а был лекарственной мишенью для лечения болезни Паркинсона, и недавно сообщалось, что он является многообещающей мишенью для иммунотерапии рака. (J. Med. Chem. 2020, 63, 21, 12196-12212)
По сути, иммунные клетки больных раком развивают резистентность к раковым антигенам и, таким образом, могут распознавать раковые клетки, но функционально ингибируются, что не позволяет эффективно уничтожать раковые клетки. Ключом к иммунотерапии является пробуждение иммунных клеток, которые стали резистентными, и побуждение их стать активированными иммунными клетками для уничтожения раковых клеток. Такая иммунотерапия включает цитокиновые терапевтические агенты, такие как гамма-интерферон, IL-2 и т.д., противораковые вакцины с использованием дендритных клеток, продукты клеточной терапии с использованием Т-клеток, иммунные контрольные точки блокирования иммунодепрессивных белков, и подобные.
Белки иммунных контрольных точек представляют собой белки клеточной мембраны, которые ингибируют дифференциацию, пролиферацию и активность иммунных клеток. Это говорит о том, что белки иммунных контрольных точек могут быть хорошей мишенью для лечения рака. Действительно, на нескольких моделях рака у животных было подтверждено, что блокирование CTLA4, PD1 и PDL1 антителами ингибирует рост рака и увеличивает выживаемость. Такой терапевтический эффект основан на механизме, посредством которого блокируется ингибирующий сигнал белков иммунных контрольных точек и, таким образом, активируются онкоспецифические Т-клетки. На основании результатов испытаний на животных было разработано и проведено множество клинических испытаний, и известно, что показан гораздо более высокий терапевтический эффект, чем у обычных противораковых лекарственных средств (Leone and Emens, Journal for ImmunoTherapy of Cancer (2018) 6:57).
Известно, что антагонист рецептора А2а может ингибировать ключевой путь иммунодепрессии в микроокружении опухоли при некоторых видах рака. Было обнаружено, что аденозин более широко распространен в микроокружении опухоли при некоторых видах рака, в отличие от других нормальных тканей, и было объявлено, что такой сверхэкспрессированный аденозин ослабляет основную иммунную систему, сосредоточенную на Т-клетках (Cancer Cell, 2015 Apr 13:27(4): 435-436).
Согласно недавнему исследованию, среди аденозиновых рецепторов, рецептор А2а особенно известен как основной фактор, влияющий на сверхэкспрессию аденозина в микроокружении опухоли некоторых видов рака, и, таким образом, сообщалось, что селективное и правильное блокирование этого рецептора может создать большую синергию в анти-PD-1 иммунотерапии (Cancer Immunol Res; 3 (5) May 2015; 506-517).
Как таковое, блокирование аденозинового сигнального пути рецептора А2а может снизить ингибирующее действие на иммунную систему и усилить иммунные функции Т-клеток, и, таким образом, антагонист аденозинового рецептора А2а представляет собой многообещающий отрицательный механизм, способный ингибировать рост опухоли.
Соответственно, авторы настоящего изобретения изобрели новую структуру соединения в качестве антагониста рецептора А2а, который селективно ингибирует аденозиновый рецептор А2а, и использовали его для ингибирования или лечения заболеваний, связанных с аденозиновым рецептором А2а, тем самым завершив настоящее изобретение.
Ссылки известного уровня техники
Не-патентные документы
J. Med. Chem. 2020, 63, 21, 12196-12212
Leone and Emens Journal for ImmunoTherapy of Cancer (2018) 6:57
Cancer Cell, 2015 Apr 13: 27 (4), 435-436
Cancer Immunol Res; 3 (5); 506-517
Описание изобретения
Техническая проблема
Целью настоящего изобретения является предложение соединения в качестве антагониста рецептора А2а, его стереоизомеров или его фармацевтически приемлемых солей.
Другой целью настоящего изобретения является предложение фармацевтической композиции, включающей соединение в качестве антагониста рецептора А2а, его стереоизомеры или его фармацевтически приемлемые соли.
Еще одной целью настоящего изобретения является предложение композиции для лечения или профилактики заболеваний, связанных с аденозиновым рецептором А2а, содержащей соединение в качестве антагониста рецептора А2а, его стереоизомеры или его фармацевтически приемлемые соли.
Еще одной целью настоящего изобретения является предложение композиции для лечения или профилактики рака или воспалительных заболеваний, включающей соединение в качестве антагониста рецептора А2а, его стереоизомеры или его фармацевтически приемлемые соли.
Еще одной целью настоящего изобретения является предложение способа лечения или профилактики заболеваний, связанных с аденозиновым рецептором А2а, включающего введение терапевтически эффективного количества указанного соединения или фармацевтической композиции, включающей это соединение.
Еще одной целью настоящего изобретения является предложение применения для лечения или профилактики заболеваний, связанных с аденозиновым рецептором А2а, или применения указанного соединения для приготовления лекарственного средства.
Техническое решение проблемы
Далее настоящее изобретение будет описано более подробно. Другими словами, все комбинации различных элементов, описанные в настоящем изобретении, попадают в объем настоящего изобретения. Кроме того, нельзя рассматривать объем настоящего изобретения как ограниченный конкретным описанием, приведенным ниже.
Соединения, представленные формулой 1
Согласно целям, соединения, предложенные в настоящем изобретении, могут быть показаны в (1) - (6) ниже.
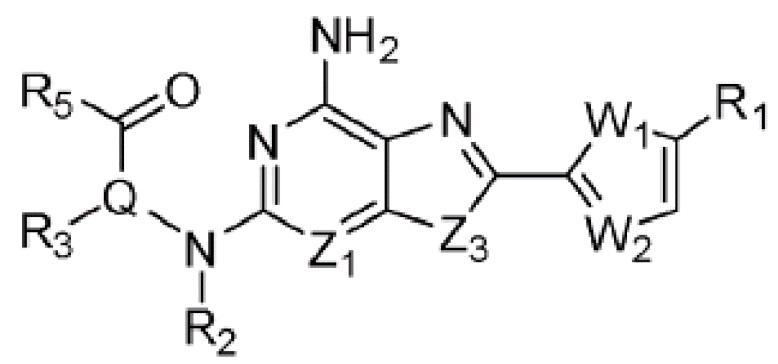
Настоящее изобретение может предложить соединение, представленное формулой 1 ниже, его стереоизомеры или его фармацевтически приемлемые соли:
(1) Соединение, представленное формулой 1, его стереоизомеры или его фармацевтически приемлемые соли:
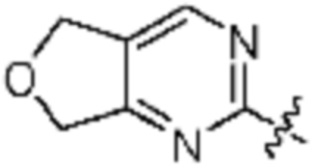
[Формула 1]
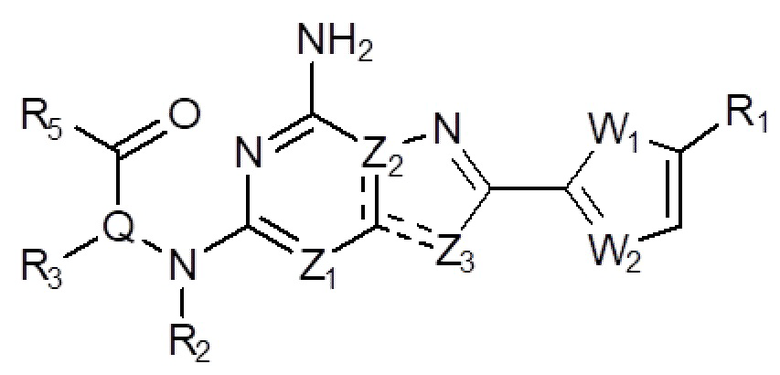
в формуле 1,
W1 представляет собой О или S;
W2 представляет собой N или CH;
Z1 представляет собой СH или N;
Z2 представляет собой С или N;
Z3 представляет собой N, O или S;
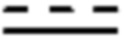 и
и 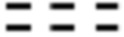 каждый независимо представляет одинарную связь или двойную связь (если представляет собой двойную связь, представляет собой одинарную связь, и если представляет собой одинарную связь, представляет собой двойную связь);
каждый независимо представляет одинарную связь или двойную связь (если представляет собой двойную связь, представляет собой одинарную связь, и если представляет собой одинарную связь, представляет собой двойную связь);
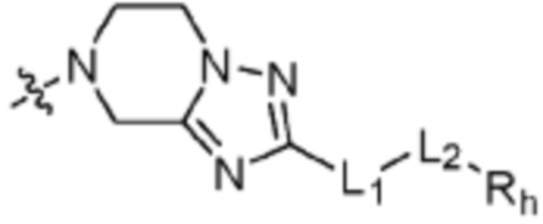
Q представляет собой С-R4 или N;
R1 представляет собой Н или -CH3;
R2 представляет собой Н или C1-C5 алкил, R3 представляет собой Н или -La-Ra, или R2 и R3 связаны с образованием кольца,
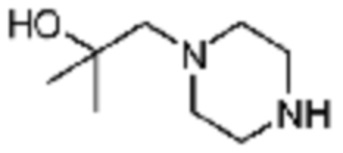
где La представляет собой одинарную связь или C1-C3 алкилен, Ra представляет собой С1-C5 алкил, C3-C6 циклоалкил, 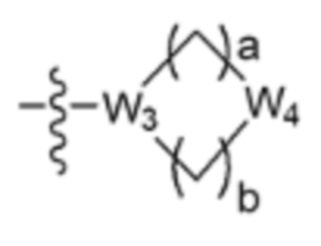 (a и b каждый независимо равен 1 или 2, W3 представляет собой СH или N, W4 представляет собой СH2 или O, где если W3 представляет собой СH, то W4 не является CH2), фенилом или -фенилен-O-бензилом, и если Ra представляет собой С1-C5 алкил или фенил, то по меньшей мере один из H может быть замещен -OH или C1-C5 алкокси;
(a и b каждый независимо равен 1 или 2, W3 представляет собой СH или N, W4 представляет собой СH2 или O, где если W3 представляет собой СH, то W4 не является CH2), фенилом или -фенилен-O-бензилом, и если Ra представляет собой С1-C5 алкил или фенил, то по меньшей мере один из H может быть замещен -OH или C1-C5 алкокси;
кольцо, образованное связыванием R2 и R3 представляет собой 4-6-членный N-содержащий гетероциклоалкил (где по меньшей мере каждый H из N-содержащего гетероциклоалкила может быть независимо замещен C1-C5 алкилом или OH), или 6-8-членный N-содержащий спирогетероциклоалкил;
R4 представляет собой Н или C1-C5 алкил;
R5 представляет собой
-NH-(CH2)y-Rb (где y равен любому целом числу из 1-3, и Rb представляет собой 5-6-членный гетероциклоалкил, включающий любой из O и N);
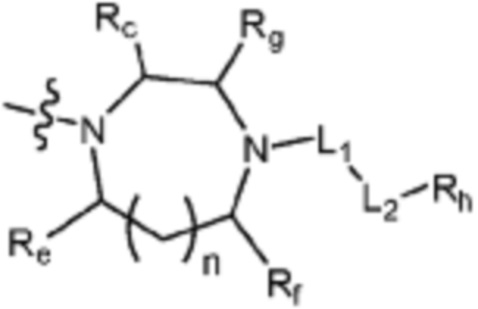
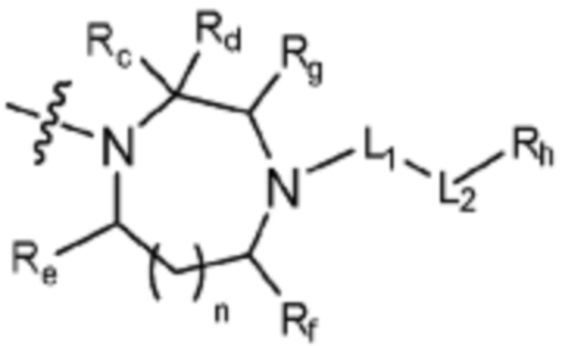 (где n равен 0 или 1, и Rc, Rd, Re, Rf и Rg каждый независимо представляет собой H или C1-C5 алкил, но два, выбранных из Rc, Rd, Re, Rf и Rg, могут быть связаны с образованием CH2 или CH2-CH2);
(где n равен 0 или 1, и Rc, Rd, Re, Rf и Rg каждый независимо представляет собой H или C1-C5 алкил, но два, выбранных из Rc, Rd, Re, Rf и Rg, могут быть связаны с образованием CH2 или CH2-CH2);
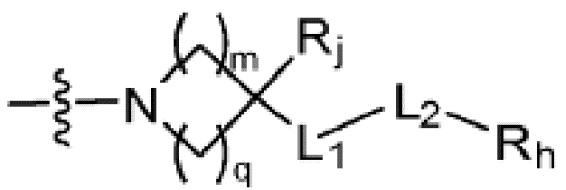 (где m и q каждый независимо равен любому целому числу из 0-3, m и q не могут быть 0 одновременно, и Rj представляет собой Н или галоген);
(где m и q каждый независимо равен любому целому числу из 0-3, m и q не могут быть 0 одновременно, и Rj представляет собой Н или галоген);
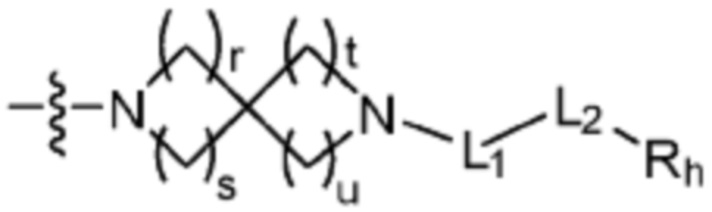 (где r, s, t и u каждый независимо равен 1 или 2);
(где r, s, t и u каждый независимо равен 1 или 2);
 ;
;
 ; или
; или
 ;
;
в вышеуказанном R5,
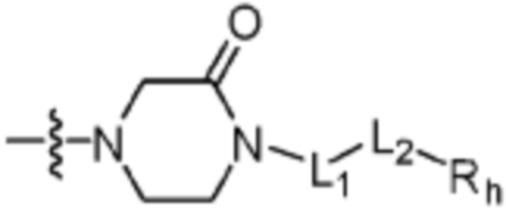
L1 представляет собой одинарную связь или C1-C3 алкилен;
L2 представляет собой одинарную связь, -C(=O)-, -C(=O)NH-, -C(=O)-N(C1-C5 алкил)-, -C(=O)-NH(C1-C5 алкилен)-, -S(=O)2- или -S(=O)2-(C1-C3 алкилен)-;
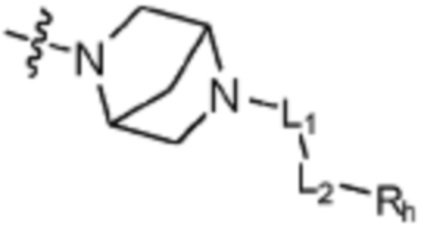
Rh представляет собой Н, C1-C5 алкил, C1-C5 алкокси, C1-C5 галогеналкил, галоген, C3-C6 циклоалкил, фенокси, фенил, -(C1-C5 алкилен)-фенил, -фенилен-O-(C1-C5 алкил), -фенилен-C(=O), -фенилен-пиперазинил, 4-6-членный гетероциклоалкил, включающий 1-3 гетероатома из по меньшей мере одного, выбранного из N, O и S, 5-10-членный гетероарил, включающий 1-3 гетероатома из по меньшей мере одного, выбранного из N, O, и S,  ,
,  ,
,  или -NR6R7;
или -NR6R7;
R6 и R7 каждый независимо представляет собой C1-C5 алкил или C1-C5 галогеналкил; и
по меньшей мере один H из Rh каждый может быть независимо замещен C1-C5 алкилом, C1-C5 алкокси, C1-C5 галогеналкилом, OH или галогеном.
В вышеуказанной формуле 1, если Z2 представляет собой С, то и не могут быть двойной связью одновременно, и не могут быть одинарной связью одновременно.
В вышеуказанной формуле 1, 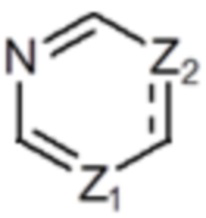 может быть выражен как
может быть выражен как 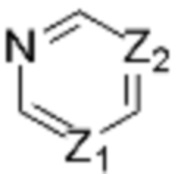 , если представляет собой двойную связь, но может явно рассматриваться как по существу то же самое, что и структура, выраженная как
, если представляет собой двойную связь, но может явно рассматриваться как по существу то же самое, что и структура, выраженная как 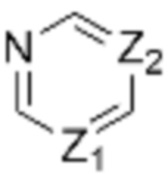 , принимая во внимание определение резонансной структуры.
, принимая во внимание определение резонансной структуры.
В вышеуказанной формуле 1, если Z2 представляет собой С, соединения, представленные формулами 1a и 1b ниже, могут означать по существу одно и то же соединение.
[Формула 1a]
[Формула 1b]
В вышеуказанной формуле 1, соединение, где R2 и R3 связаны с образованием кольца, может быть представлено формулой 1c ниже.
[Формула 1c]
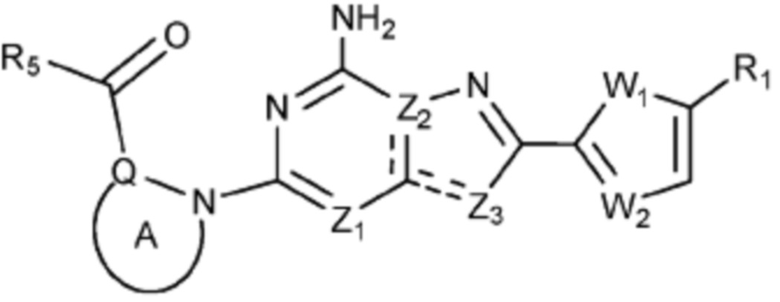
В вышеуказанной формуле 1c, Q представляет C-R4 или N, и кольцо, включающее Q и N, представляет 4-6-членный N-содержащий гетероциклоалкил (где по меньшей мере каждый H из N-содержащего гетероциклоалкила может быть независимо замещен C1-C5 алкилом или OH) или 6-8-членный N-содержащий спирогетероциклоалкил.
В настоящем изобретении, «алкил» может означать насыщенную углеводородную группу с прямой или разветвленной цепью, если не указано иное, и, например, «C1-C5 алкил» может включать метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, трет-бутил, изобутил, н-пентил, втор-пентил, трет-пентил, изопентил, втор-изопентил, нео-пентил и т. д.
В настоящем изобретении, «алкилен» может означать двухвалентную функциональную группу, полученную из определенного выше алкила (включая как неразветвленный, так и разветвленный), если не указано иное, и, например, «C1-C3 алкилен» может включать метилен (-CH2-), этилен(-CH2CH2-), н-пропилен(-CH2CH2CH2-), изопропилен(-CH(CH3)-CH2-) и т.д.
В настоящем изобретении, «гетеро» может относиться к гетероатому или гетероатомной группе (то есть атомной группе, содержащей гетероатом), если не указано иное, и может означать, например, такие атомы, как кислород (О), азот (N), сера (S) и/или подобные, и атомную группу, содержащую такой гетероатом.
В настоящем изобретении, «гетероарил» может означать гетероцикл, в котором по меньшей мере один атом углерода ароматической функциональной группы замещен гетероатомом, если не указано иное, и гетероатом может представлять собой O, N или S. Например, гетероарил может включать фурил, пирролил, пиразолил, пиридил, пиримидил, имидазолил, триазолил, триазинил, пиридазинил, пиразинил или подобный, но не ограничен ими.
В настоящем изобретении, «гетероциклоалкил» может означать циклическую алкильную группу, в которой по меньшей мере один атом углерода, образующий кольцо, замещен гетероатомом, если не указано иное. Гетероатомом может представлять собой, например, O, N или S. Например, гетероциклоалкил может включать пиперидинил, морфолинил, тиаморфолинил, пирролидинил, имидазолидинил, оксиранил, оксетанил, тетрагидрофуранил, тетрагидропиранил и т.д., но не ограничен ими.
В настоящем изобретении, «спирогетероциклоалкил» может представлять собой двойное кольцо, включающее два кольца, имеющие только один атом углерода, в котором по меньшей мере одно из двух колец включает гетероатом. Гетероатомом может быть, например, O, N или S. Когда одно из двух колец имеет форму с x углами, и другое имеет форму с y углами (где каждый из x и y представляет собой целое число 3 или более), его можно назвать (x+y-1)-членным спирогетероциклоалкилом. Например, спирогетероциклоалкил может представлять собой 7-членный 5-азаспиро[2,4]гептанил.
В настоящем изобретении, «галогеналкил» может означать функциональную группу, в которой по меньшей мере один водород замещен галогеном в алкильной группе, определенной выше. Примеры галогеналкила могут включать CF3, CF2H, CH2F, CH2CH2F, CH2CF3, C(CH3)2CF3 и т.д.
В настоящем изобретении, «галоген» может представлять собой F, Cl, Br или I, если не указано иное.
(2) Соединение, представленное формулой 1, его стереоизомеры или его фармацевтически приемлемые соли согласно указанному выше (1):
В формуле 1,
W1, W2, Z1, Z2, Z3, Q, R1, R2, R3, R4, и каждый является таким, как определено выше,
если W1 представляет собой О, то W2 представляет собой СH;
если W1 представляет собой S, то W2 представляет собой N;
R5 представляет собой
-NH-(CH2)y-Rb (где y равен любому целому числу из 1-3, и Rb представляет собой 5-6-членный гетероциклоалкил, включающий O);
 ,
,  ,
, 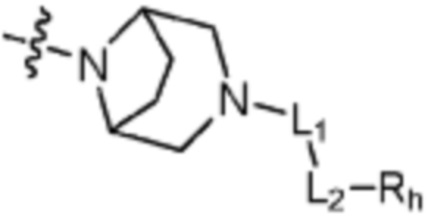 ,
, 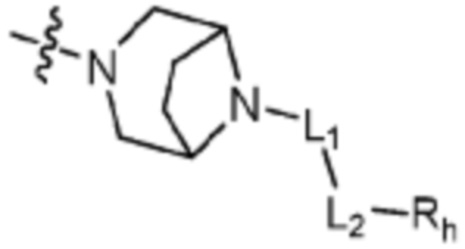 ,
,  (где n равен 0 или 1, Rc, Re, Rf и Rg каждый независимо представляет собой H или C1-C5 алкил) или
(где n равен 0 или 1, Rc, Re, Rf и Rg каждый независимо представляет собой H или C1-C5 алкил) или 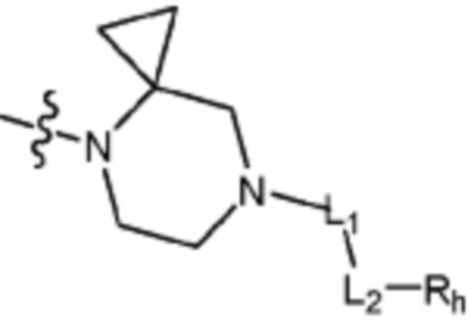 ;
;
(где m и q каждый независимо равен любому целому числу из 0-3, m и q не могут быть 0 одновременно, и Rj представляет собой Н или галоген);
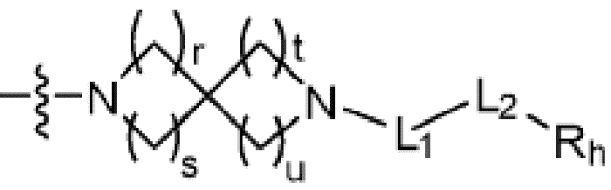 (где r, s, t и u каждый независимо равен 1 или 2);
(где r, s, t и u каждый независимо равен 1 или 2);
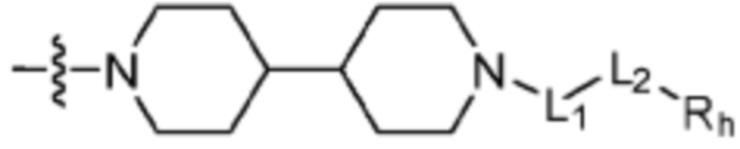 ;
;
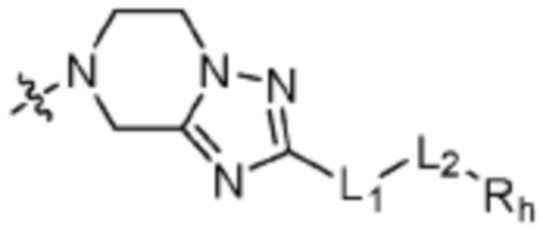 или
или
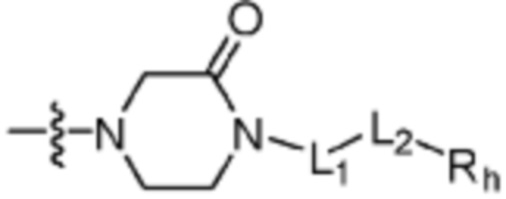 ;
;
в вышеуказанном R5, L1, L2 и Rh каждый является таким, как определено выше.
(3) Соединение, представленное формулой 1, его стереоизомеры или его фармацевтически приемлемые соли согласно указанному выше (1) или (2):
В формуле 1, W1, W2, Z1, Z2, Z3, и каждый является таким, как определено выше;
Q представляет собой С-R4;
R1 и R2 каждый представляет собой H;
R3 представляет собой Н или -La-Ra (где La представляет собой одинарную связь или C1-C3 алкилен; Ra представляет собой С1-C5 алкил, C3-C6 циклоалкил, 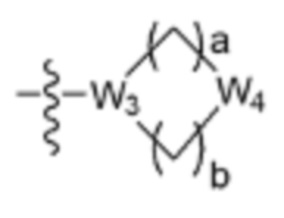 (a и b каждый независимо равен 1 или 2, W3 представляет собой СH или N, W4 представляет собой СH2 или O, где если W3 представляет собой СH, то W4 не является CH2), фенил или -фенилен-O-бензил, и если Ra представляет собой С1-C5 алкил или фенил, то по меньшей мере один из H может быть замещен -OH или C1-C5 алкокси;
(a и b каждый независимо равен 1 или 2, W3 представляет собой СH или N, W4 представляет собой СH2 или O, где если W3 представляет собой СH, то W4 не является CH2), фенил или -фенилен-O-бензил, и если Ra представляет собой С1-C5 алкил или фенил, то по меньшей мере один из H может быть замещен -OH или C1-C5 алкокси;
R4 представляет собой Н или C1-C5 алкил;
R5 представляет собой
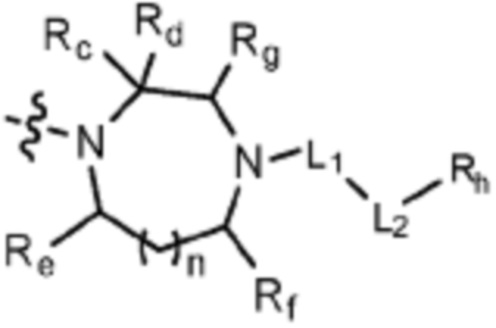 (где n равен 0 или 1, и Rc, Rd, Re, Rf и Rg каждый независимо представляет собой H или C1-C5 алкил, но два, выбранных из Rc, Rd, Re, Rf и Rg могут быть связаны с образованием CH2 или CH2-CH2);
(где n равен 0 или 1, и Rc, Rd, Re, Rf и Rg каждый независимо представляет собой H или C1-C5 алкил, но два, выбранных из Rc, Rd, Re, Rf и Rg могут быть связаны с образованием CH2 или CH2-CH2);
(где m и q каждый независимо равен любому целому числу из 0-3, и Rj представляет собой Н или галоген);
(где r, s, t и u каждый независимо равен 1 или 2);
 или
или  ;
;
в вышеуказанном R5,
L1 представляет собой одинарную связь или C1-C3 алкилен;
L2 представляет собой одинарную связь, -C(=O)- или -S(=O)2-;
Rh представляет собой Н, C1-C5 алкил, C1-C5 алкокси, C1-C5 галогеналкил, C3-C6 циклоалкил, фенокси, фенил, 5- или 6-членный гетероциклоалкил, включающий 1-3 гетероатома из по меньшей мере одного, выбранного из N и O, или 5- или 6-членный гетероарил включающий 1-3 гетероатома из по меньшей мере одного, выбранного из N и S; и
по меньшей мере один H из Rh каждый может быть независимо замещен C1-C5 алкокси, C1-C5 галогеналкилом, OH или галогеном.
(4) Соединение, представленное формулой 1, его стереоизомеры или его фармацевтически приемлемые соли по любому из вышеуказанных (1), (2), и (3):
В формуле 1, W1, W2, Z1, Z2, Z3, R1, и каждый является таким, как определено выше,
Q представляет собой С-R4 или N;
R2 и R3 связаны друг с другом с образованием 4-6-членного N-содержащего гетероциклоалкила (где по меньшей мере каждый H из N-содержащего гетероциклоалкила может быть независимо замещен C1-C5 алкилом или OH), или 6-8-членный N-содержащий спирогетероциклоалкил;
R4 представляет собой Н или C1-C5 алкил;
R5 представляет собой
-NH-(CH2)y-Rb (где y равен любому целому числу из 1-3, и Rb представляет собой 5-6-членный гетероциклоалкил, включающий O);
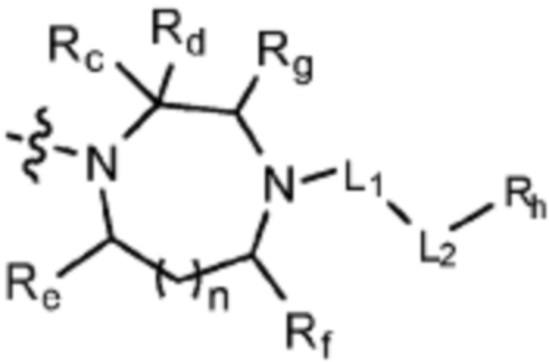 (где n равен 0 или 1, и Rc, Rd, Re, Rf и Rg каждый независимо представляет собой H или C1-C5 алкил, но два, выбранных из Rc, Rd, Re, Rf и Rg могут быть связаны с образованием CH2 или CH2-CH2);
(где n равен 0 или 1, и Rc, Rd, Re, Rf и Rg каждый независимо представляет собой H или C1-C5 алкил, но два, выбранных из Rc, Rd, Re, Rf и Rg могут быть связаны с образованием CH2 или CH2-CH2);
(где m и q каждый независимо равен любому целому числу из 0-3, m и q не могут быть 0 одновременно, и Rj представляет собой Н или галоген);
(где r, s, t и u каждый независимо равен 1 или 2);
 ;
;
 или
или
 ;
;
в вышеуказанном R5,
L1 представляет собой одинарную связь или C1-C3 алкилен;
L2 представляет собой одинарную связь, -C(=O)-, -C(=O)NH-, -C(=O)-N(C1-C5 алкил)-, -C(=O)-NH(C1-C5 алкилен)-, -S(=O)2- или -S(=O)2-(C1-C3 алкилен)-;
Rh представляет собой Н, C1-C5 алкил, C1-C5 алкокси, C1-C5 галогеналкил, галоген, C3-C6 циклоалкил, фенокси, фенил, -(C1-C3 алкилен)-фенил, -фенилен-O-(C1-C5 алкил), -фенилен-C(=O)-, -фенилен-пиперазинил, 4-6-членный гетероциклоалкил, включающий 1-3 гетероатома из по меньшей мере одного, выбранного из N, O и S, 5-10-членный гетероарил, включающий 1-3 гетероатома из по меньшей мере одного, выбранного из N, O, и S,  ,
,  ,
,  или -NR6R7;
или -NR6R7;
R6 и R7 каждый независимо представляет собой C1-C5 алкил или C1-C5 галогеналкил; и
по меньшей мере каждый H из Rh может быть независимо замещен C1-C5 алкилом, C1-C5 алкокси, C1-C5 галогеналкилом, OH или галогеном.
(5) Соединение, представленное формулой 1, его стереоизомеры или его фармацевтически приемлемые соли по любому из вышеуказанных (1), (2), (3), и (4), где соединение может представлять собой по меньшей мере одно соединение, выбранное из соединений, показанных в таблице 1 ниже.
[Таблица 1]
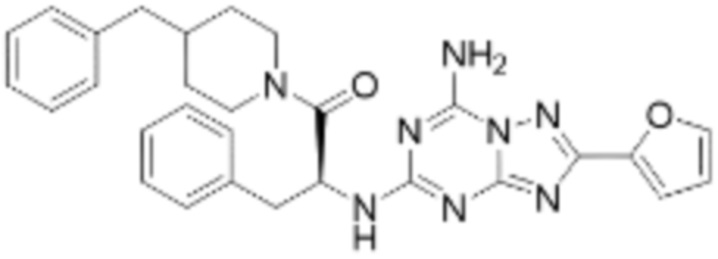
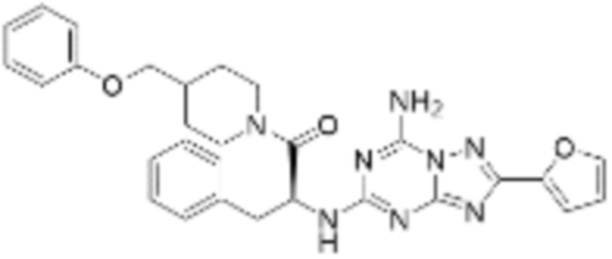
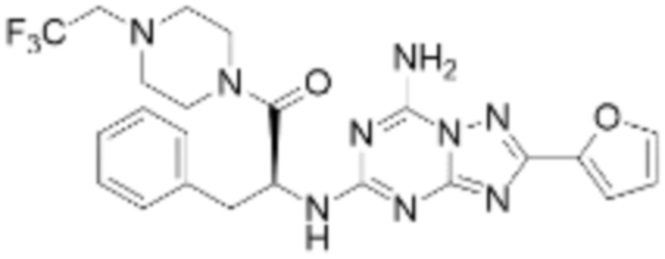
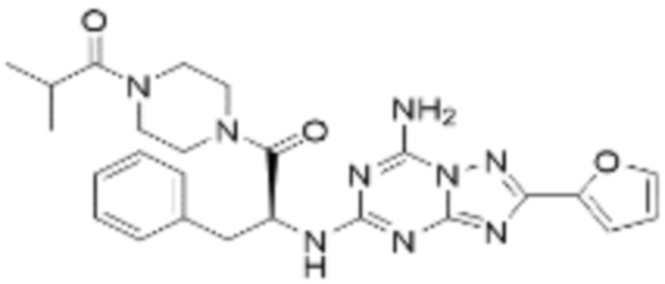
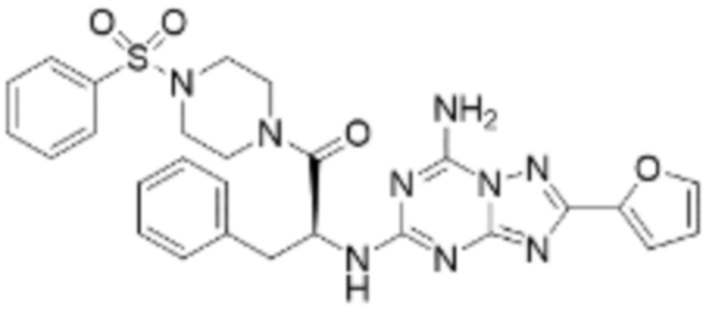
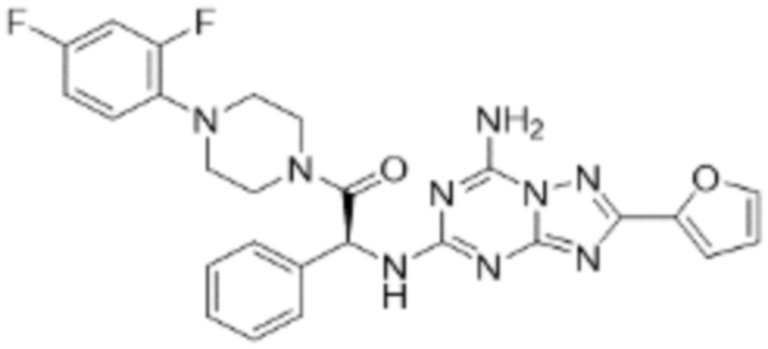
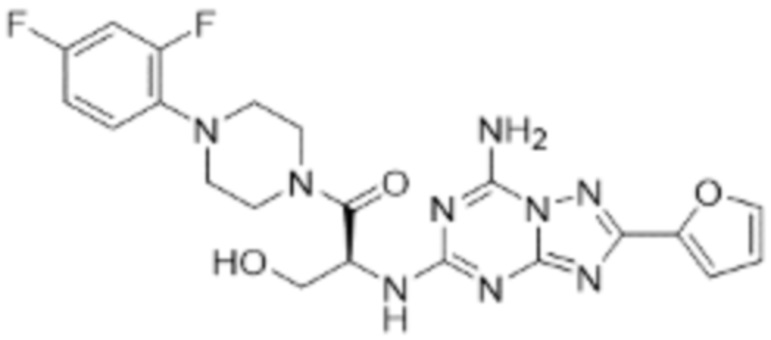
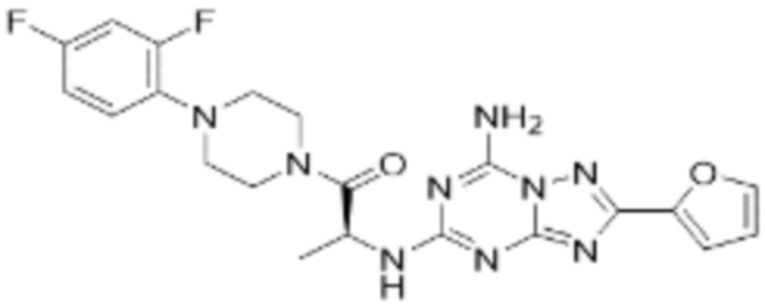
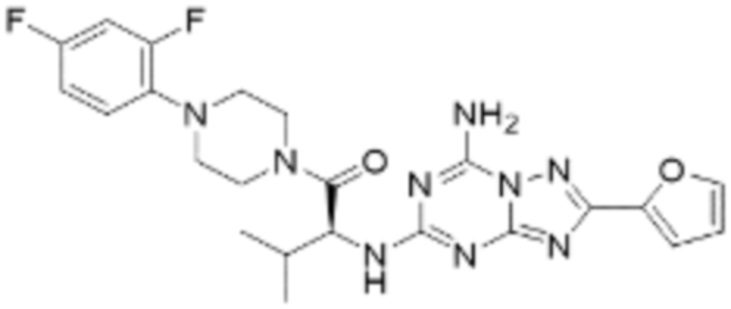
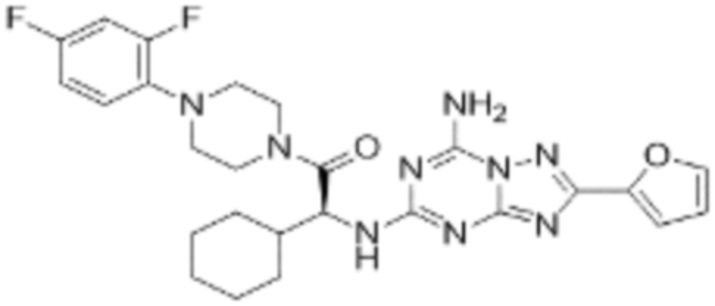
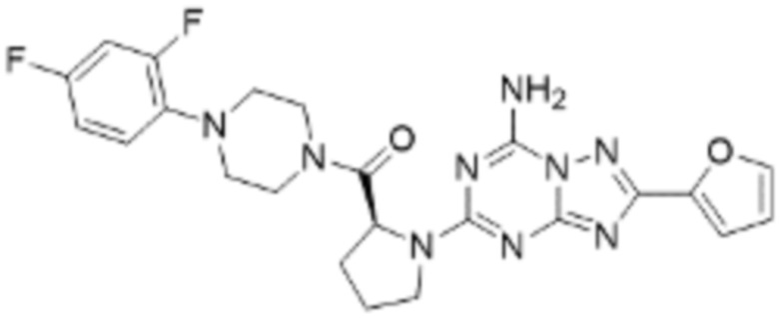
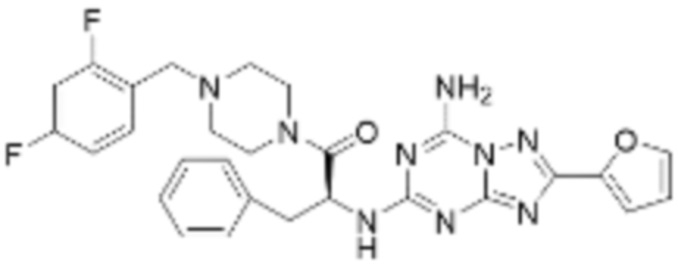
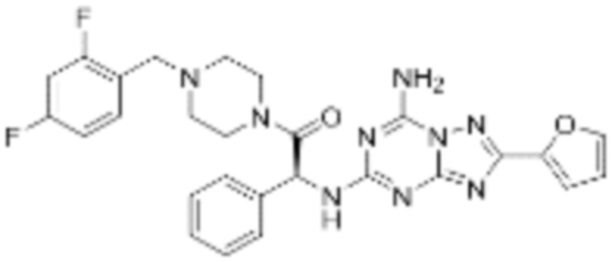
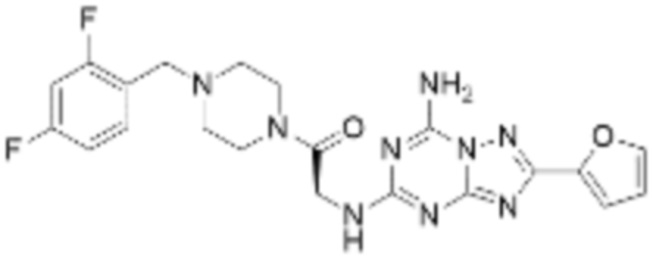
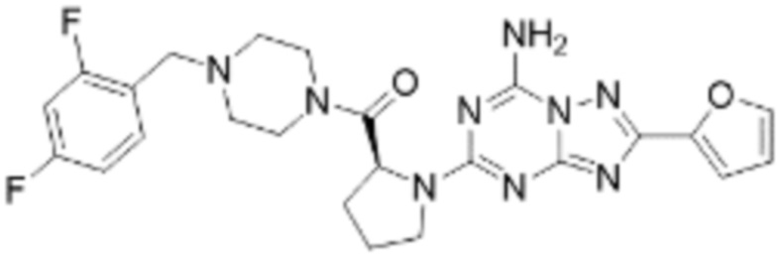
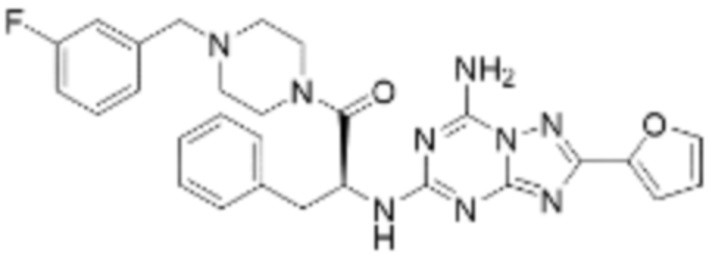
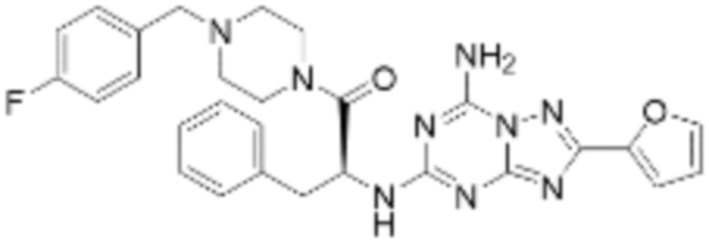
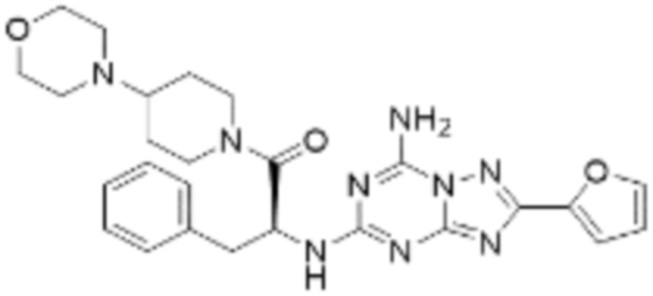
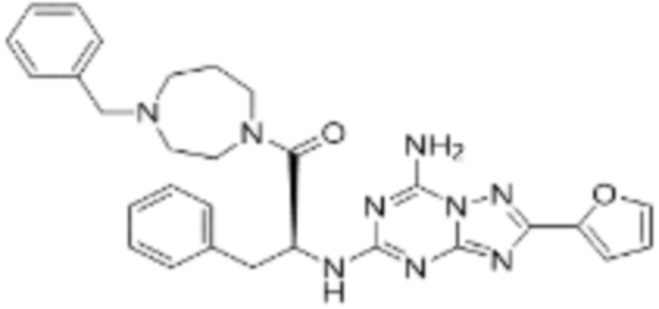
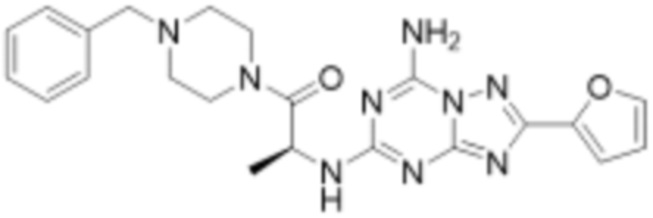
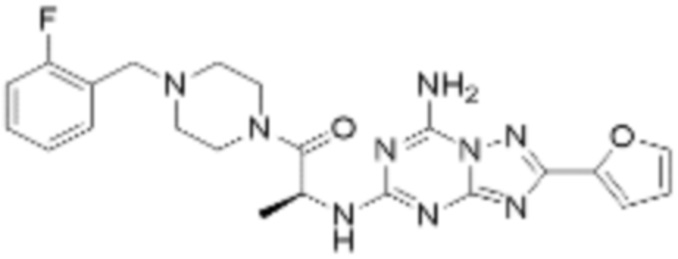
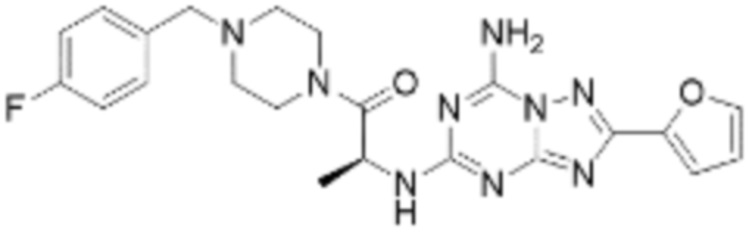
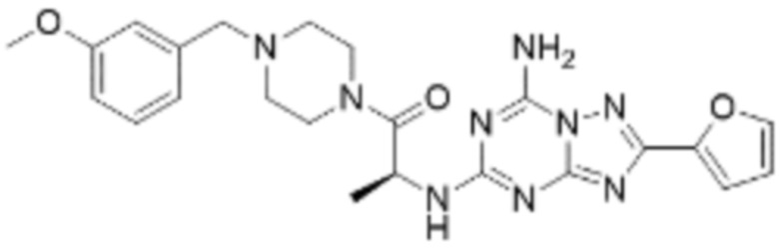
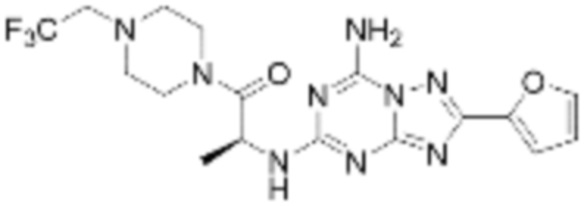
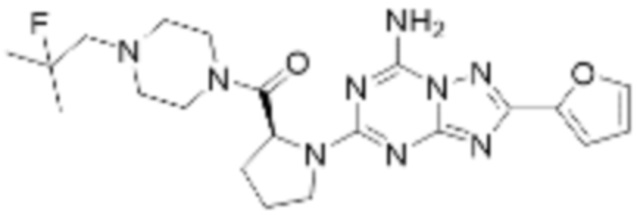
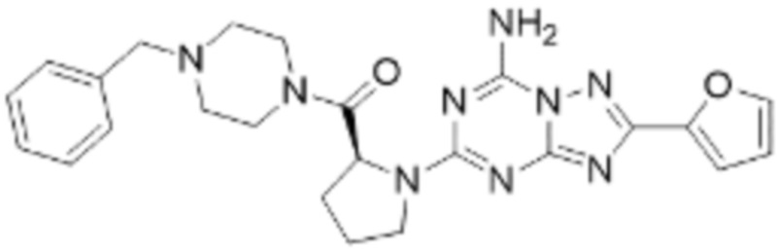
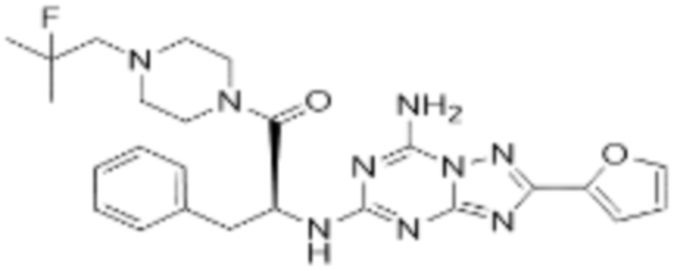
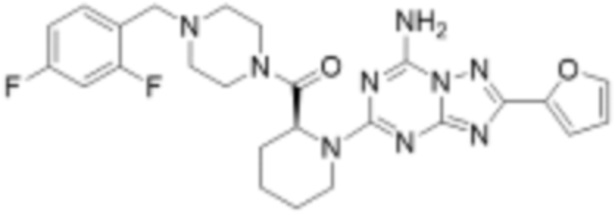
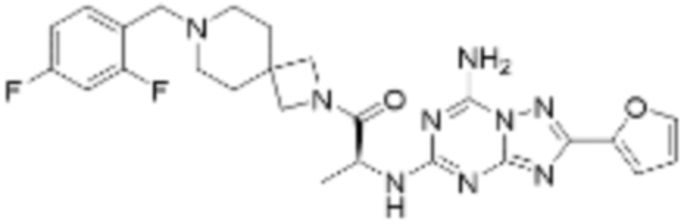
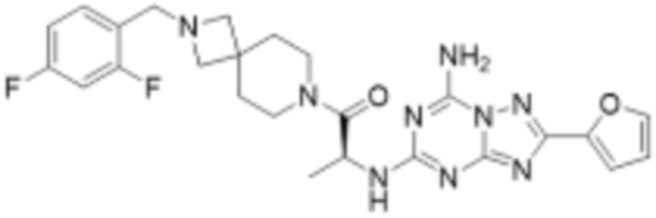
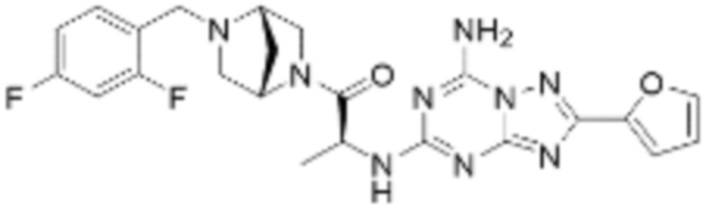
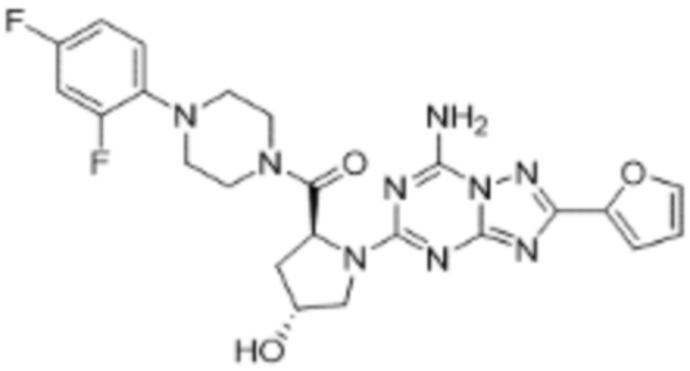
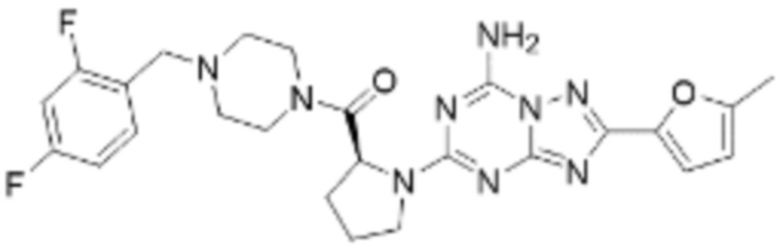
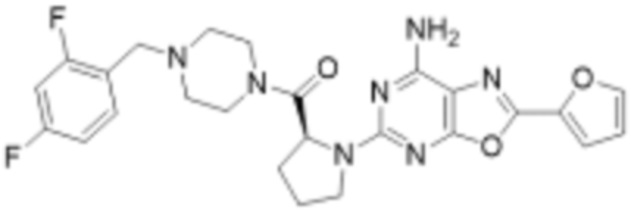
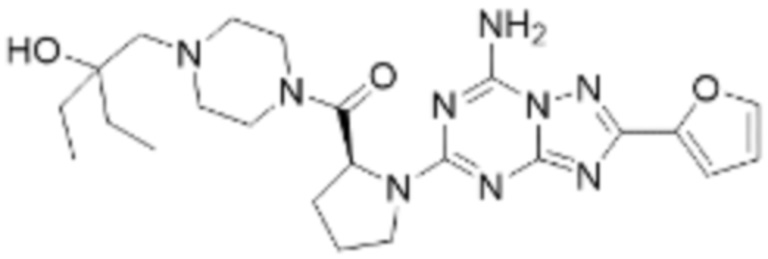
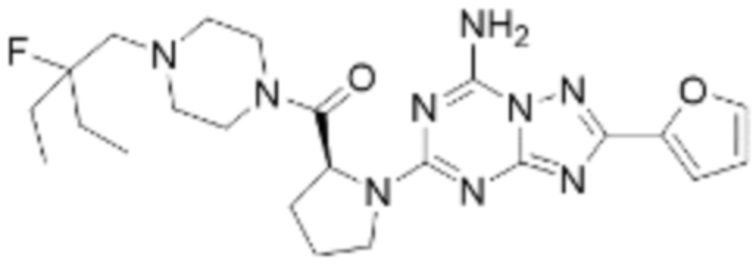
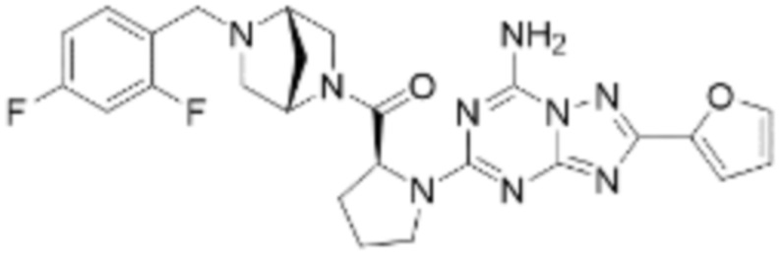
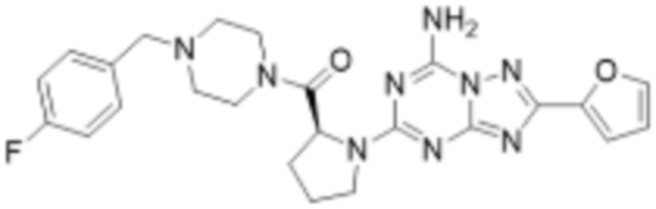
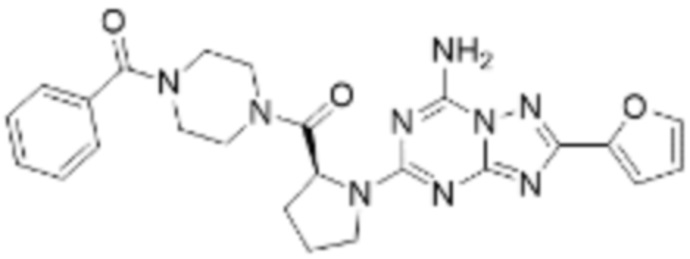
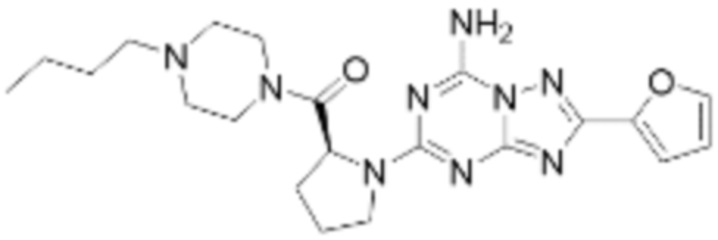
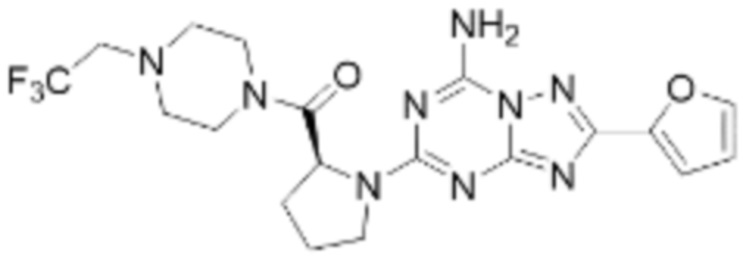
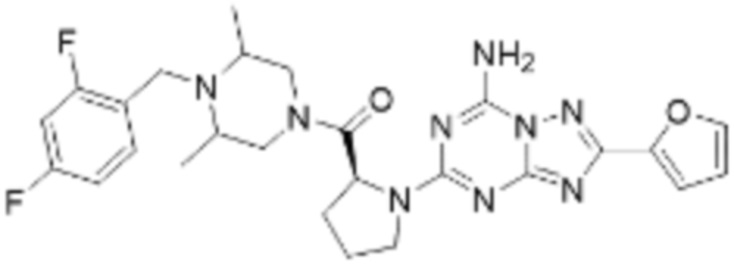
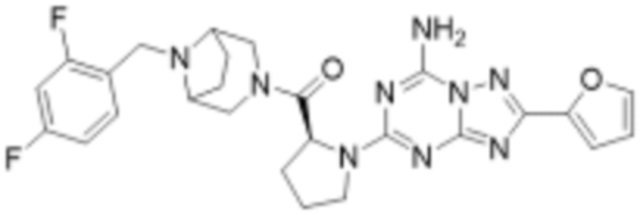
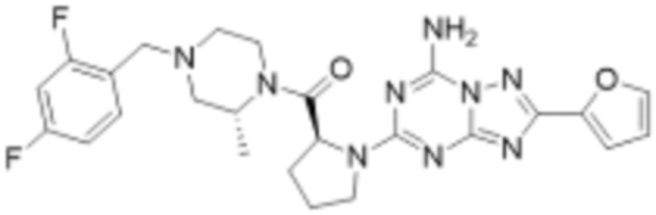
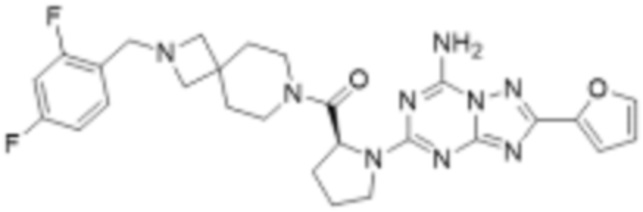
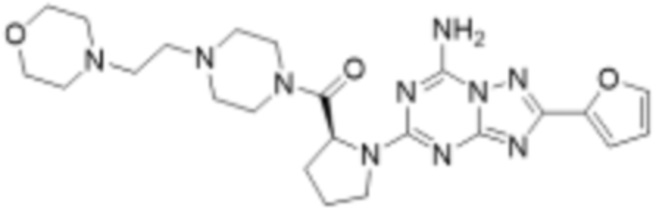
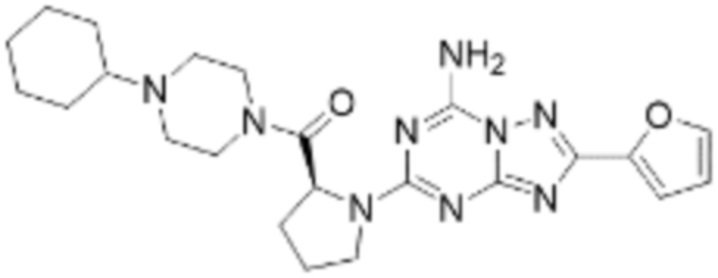
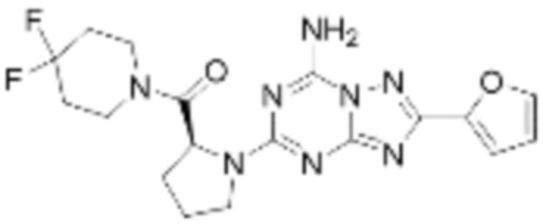
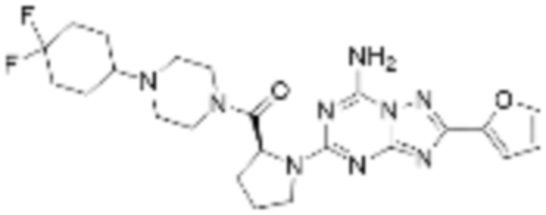
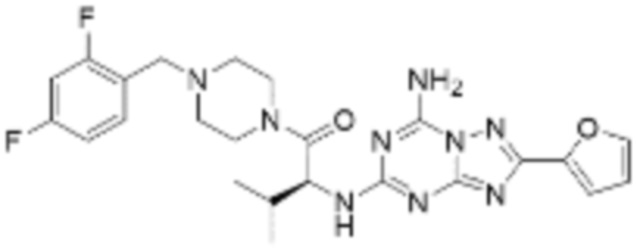
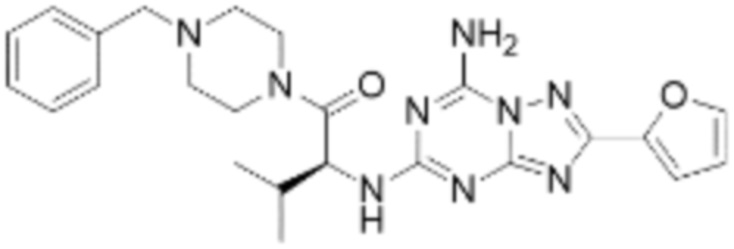
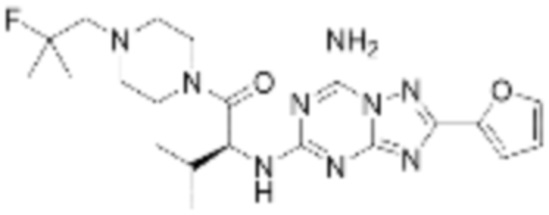
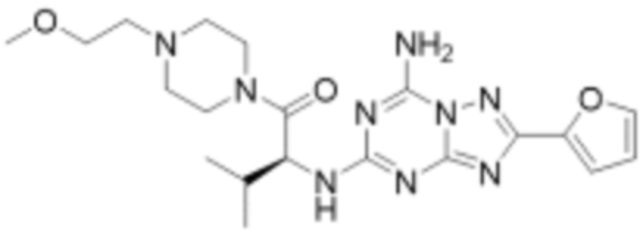
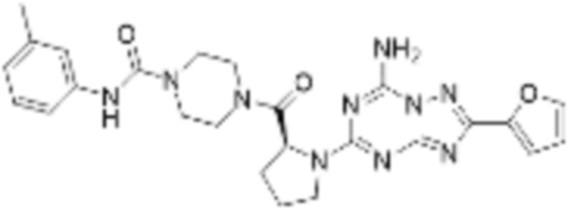
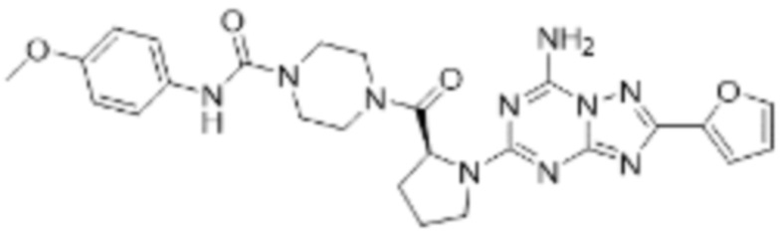
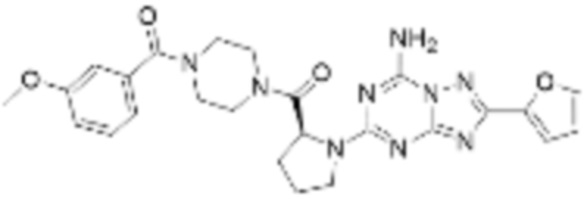
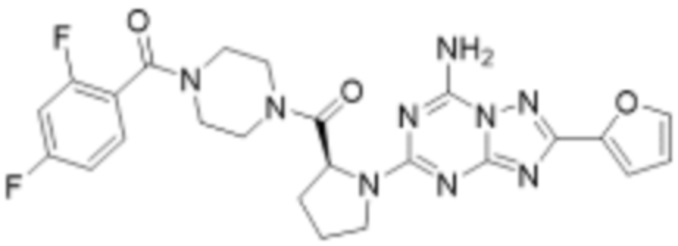
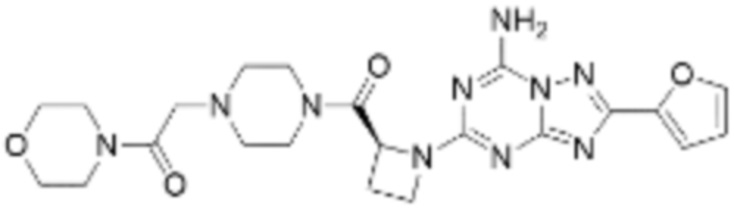
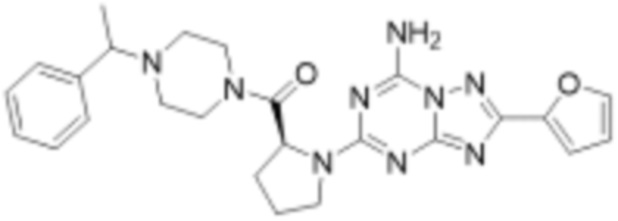
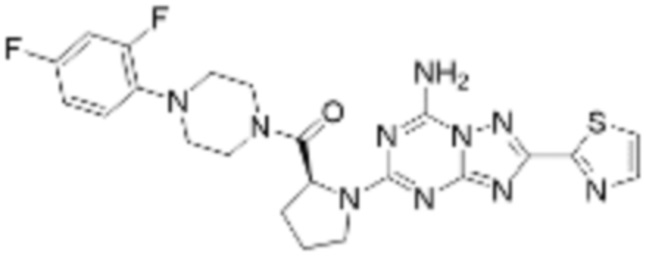
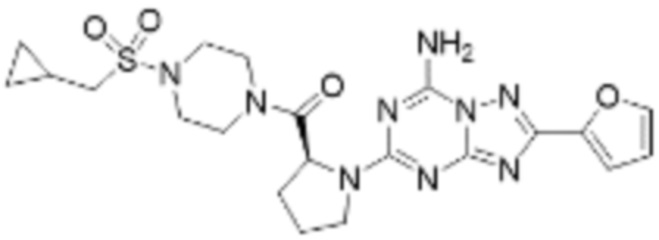
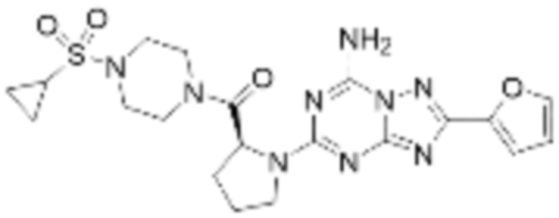
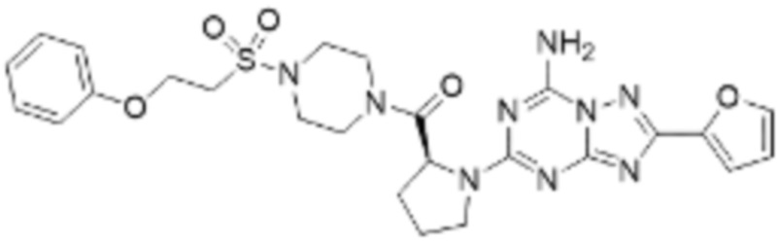
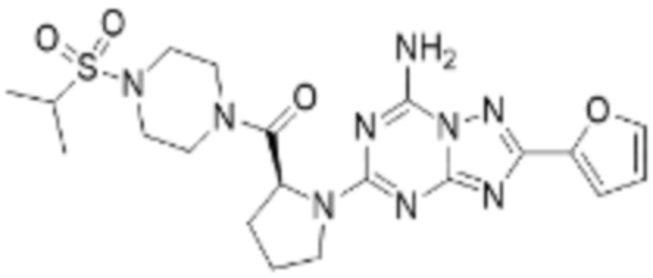
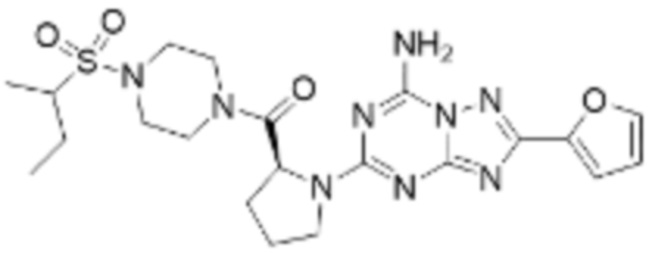
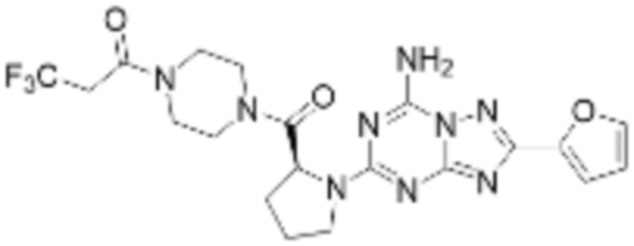
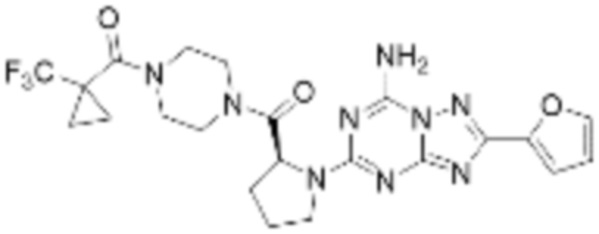
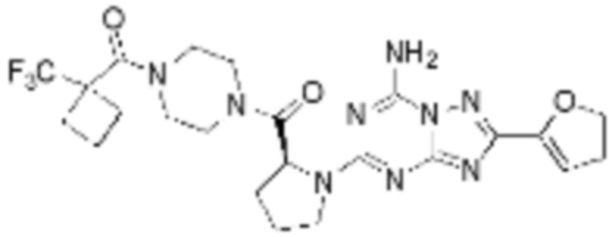
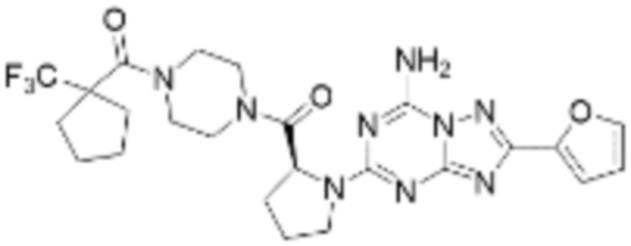

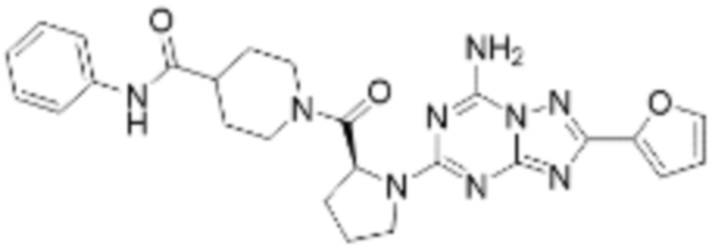
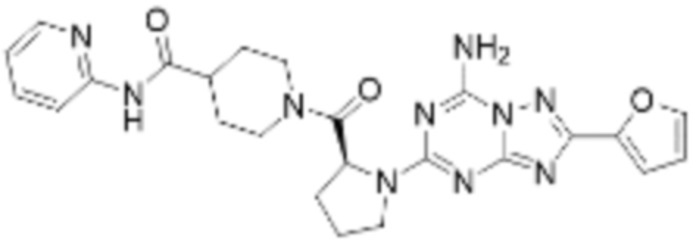
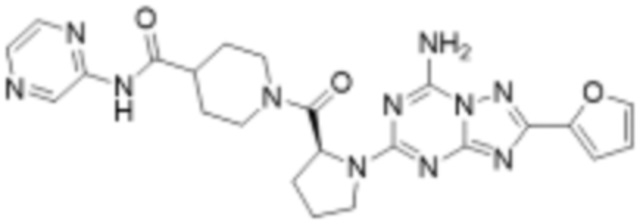
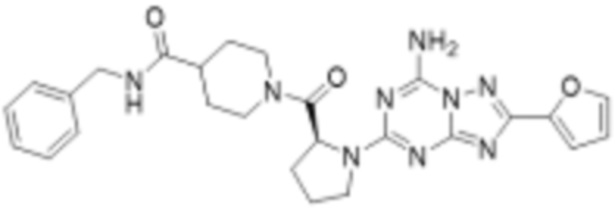
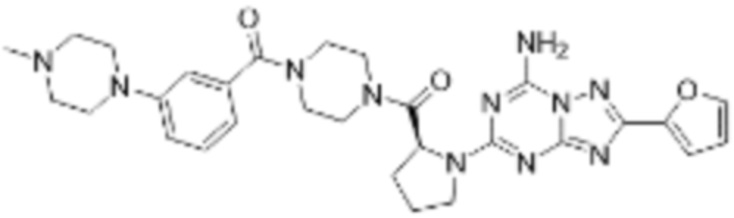
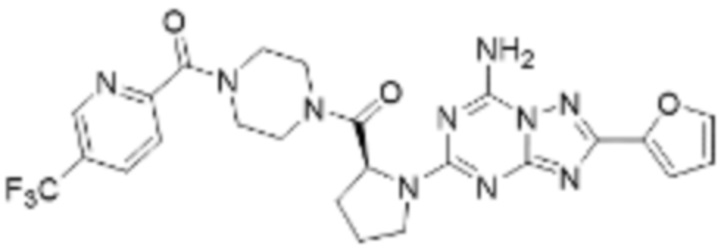
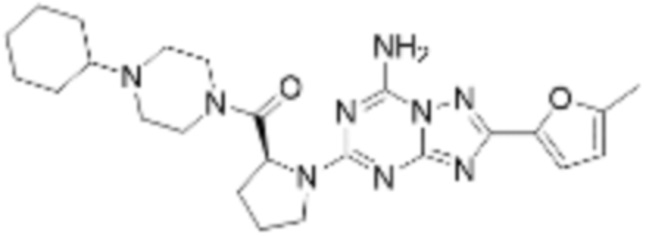
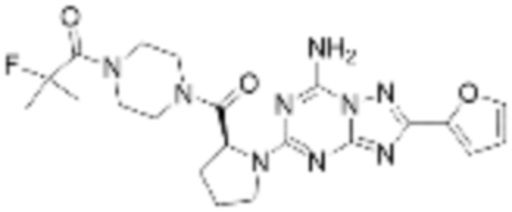
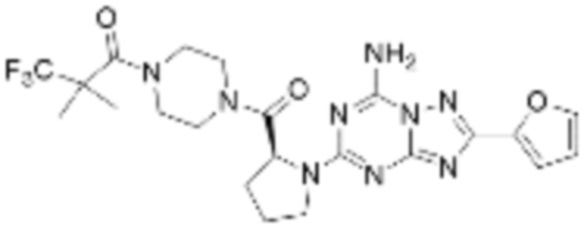
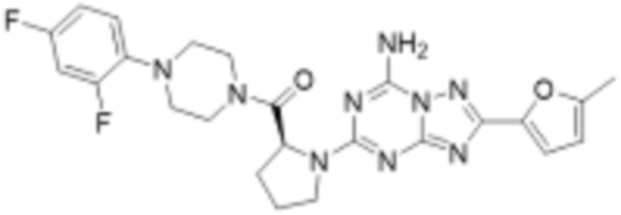
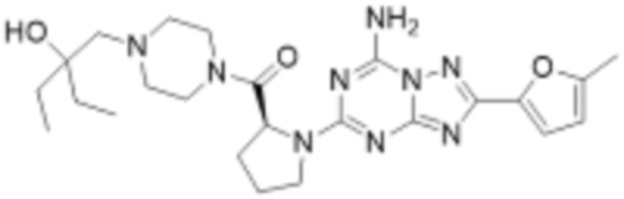
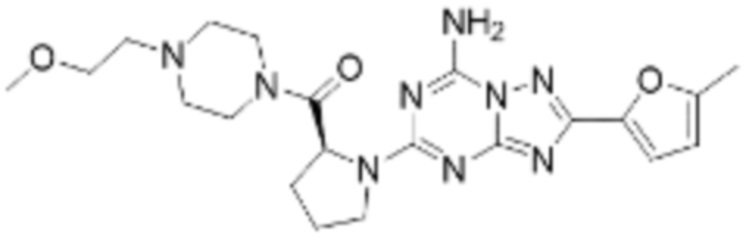
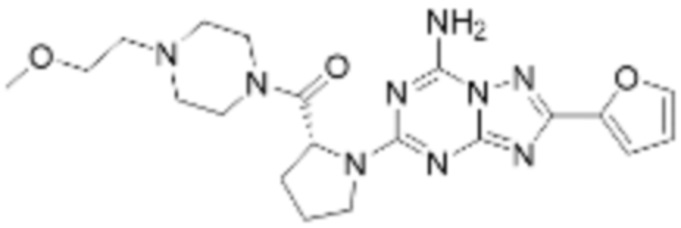
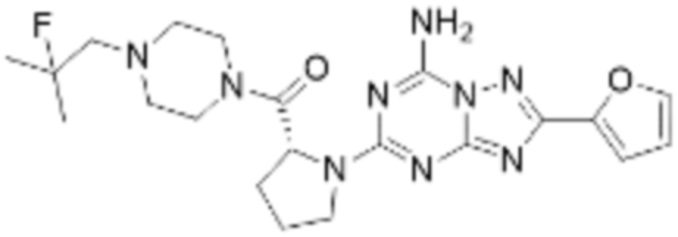
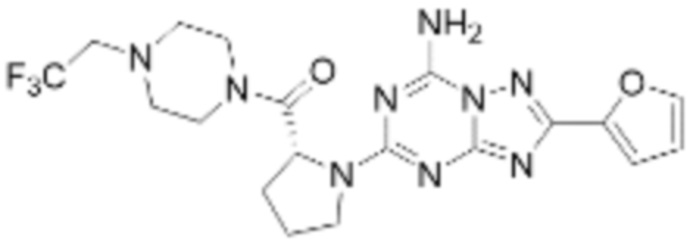
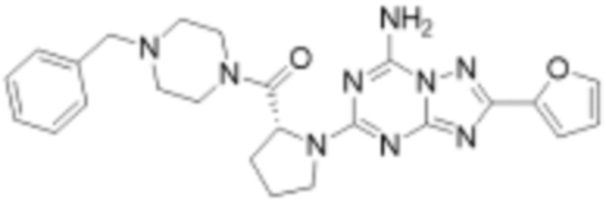

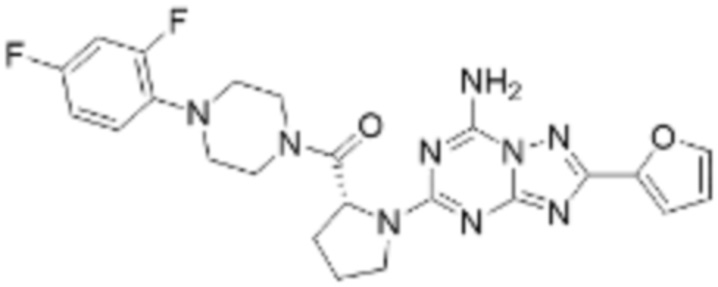
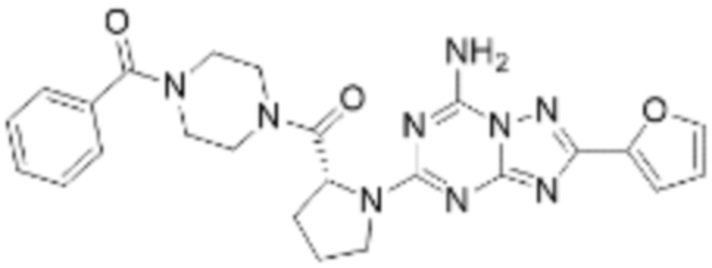
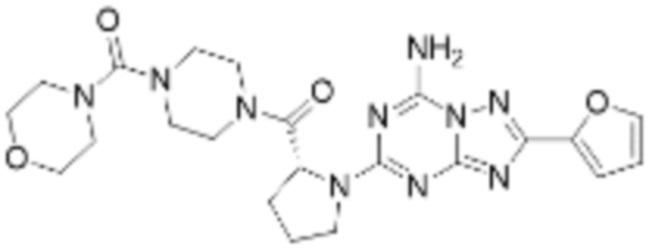
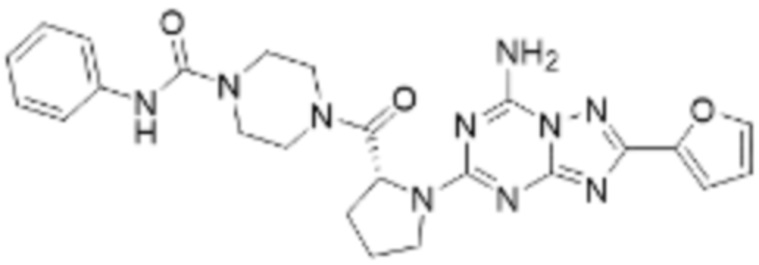
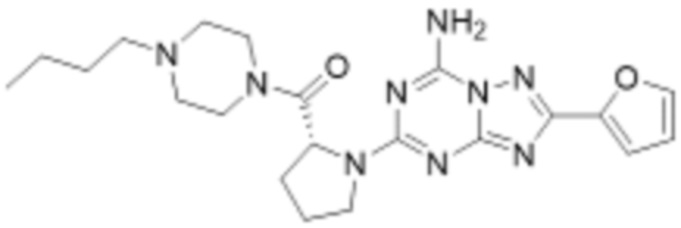
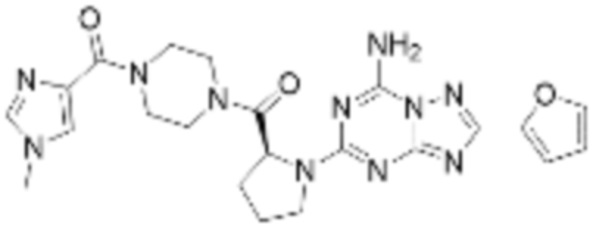
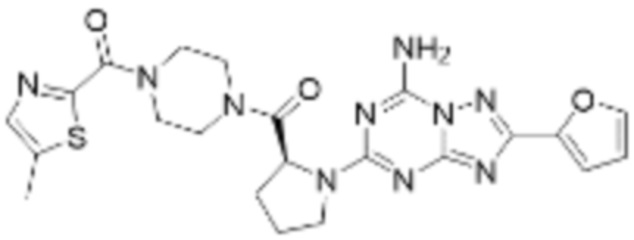
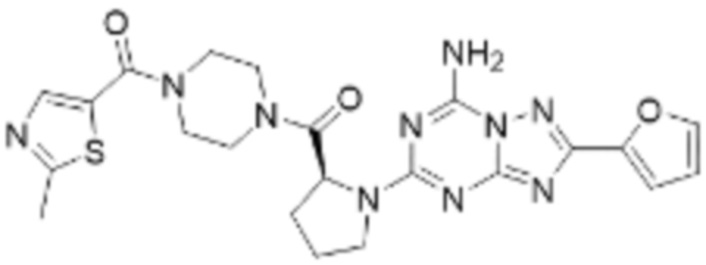
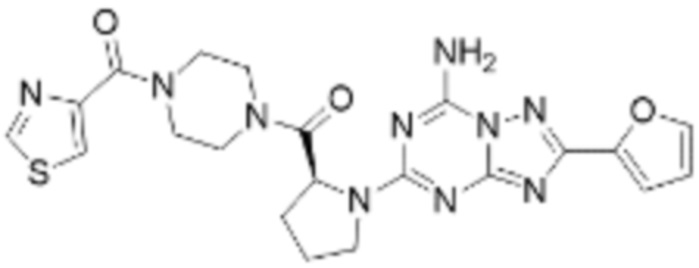
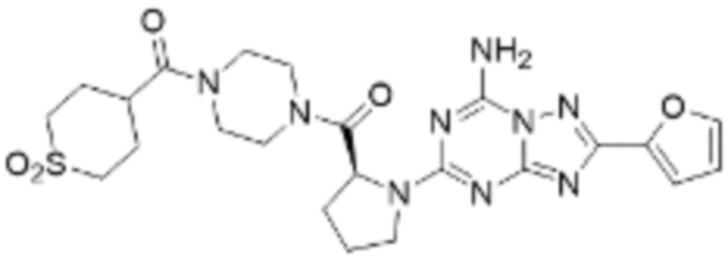
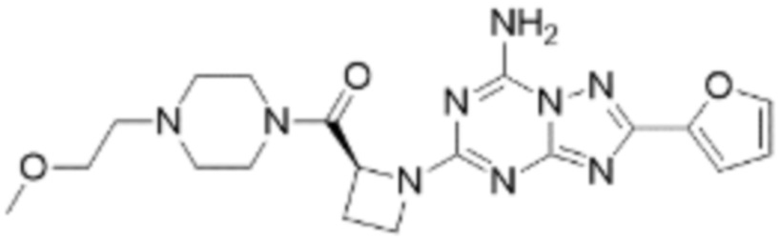
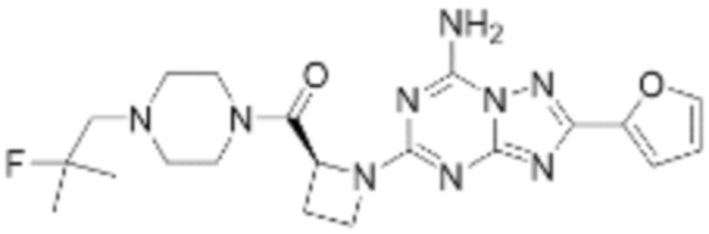
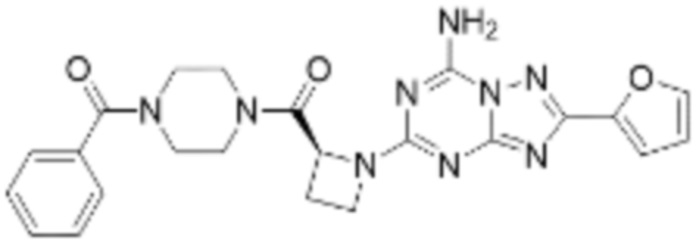
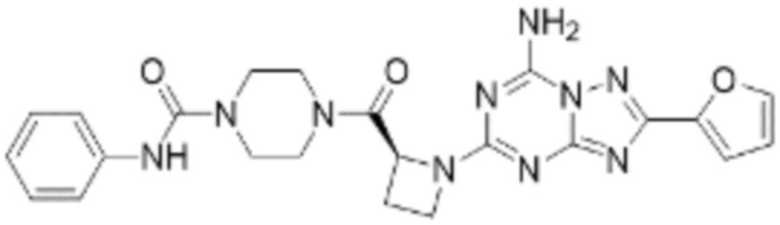
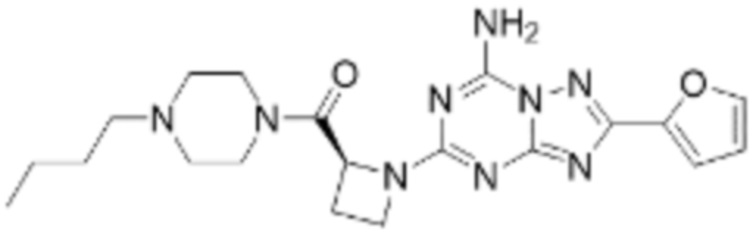
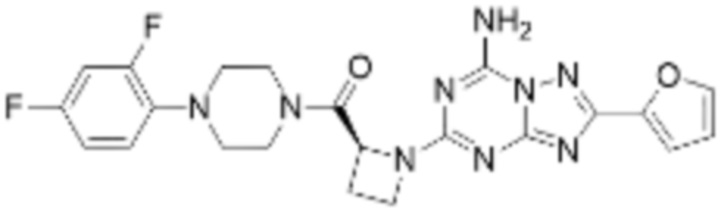
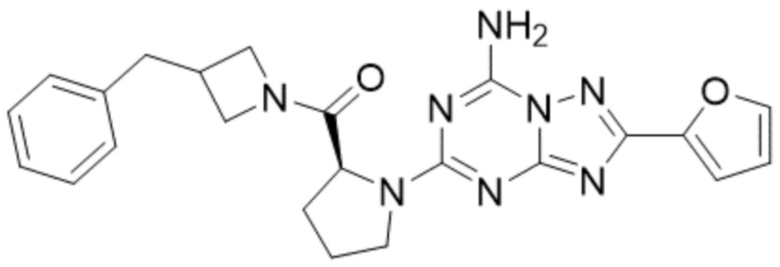
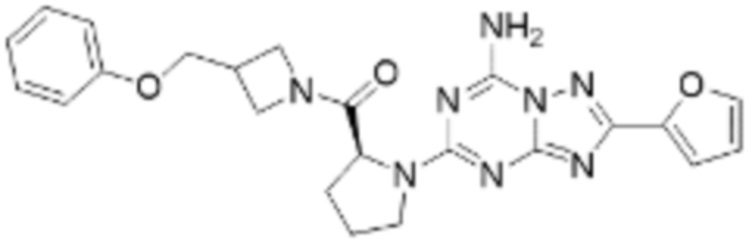
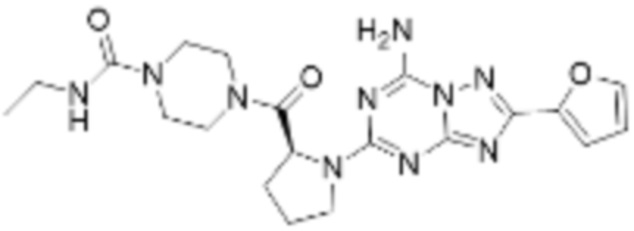
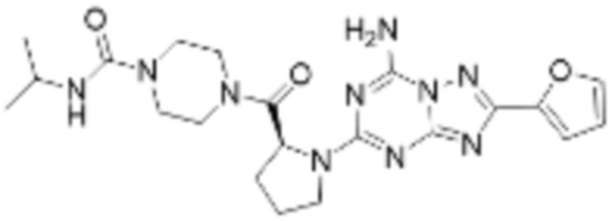
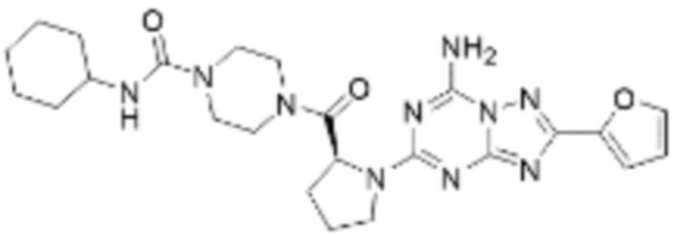

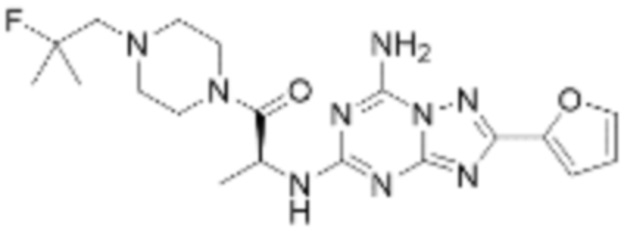
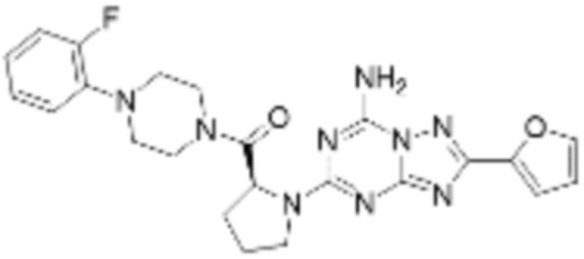
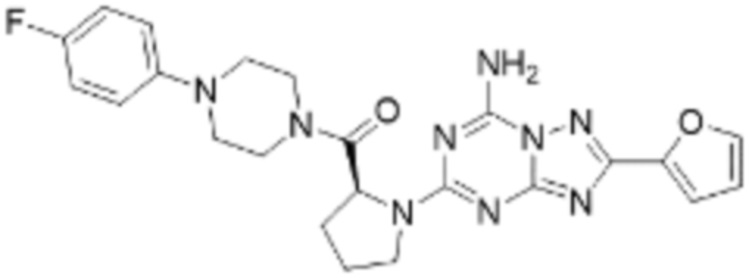
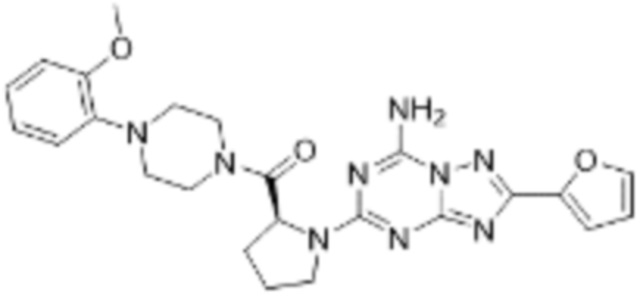
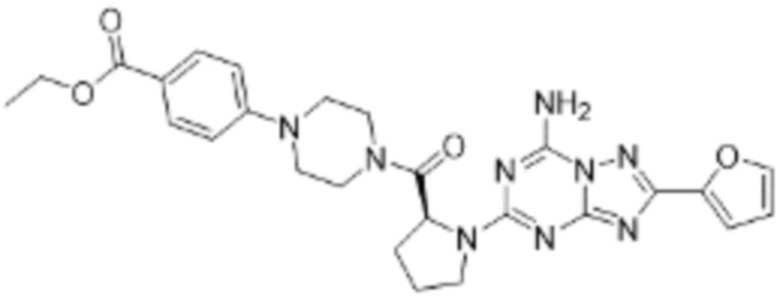
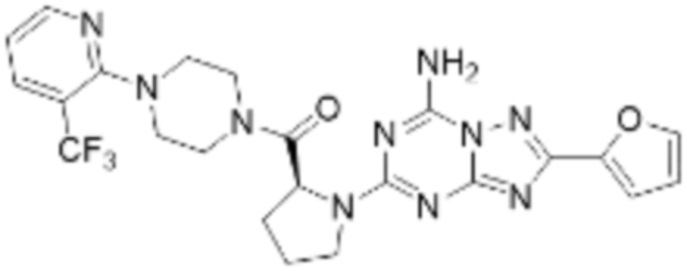
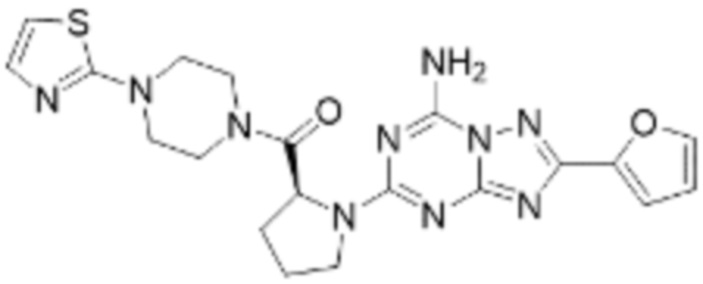
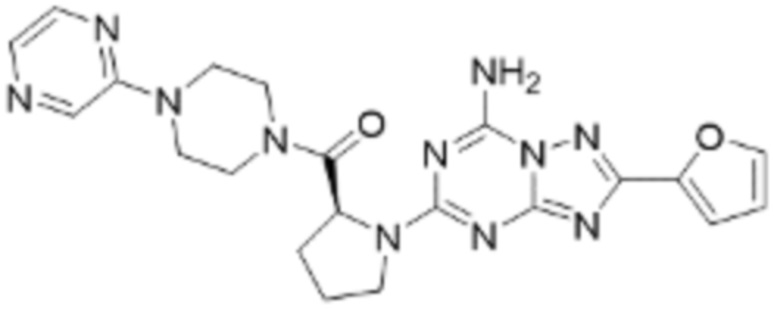
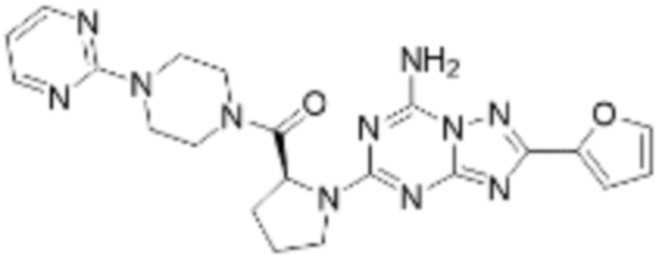
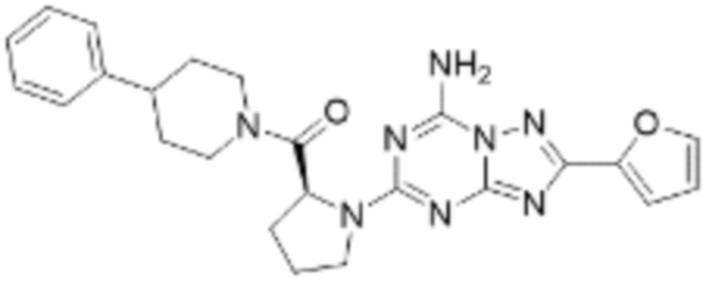
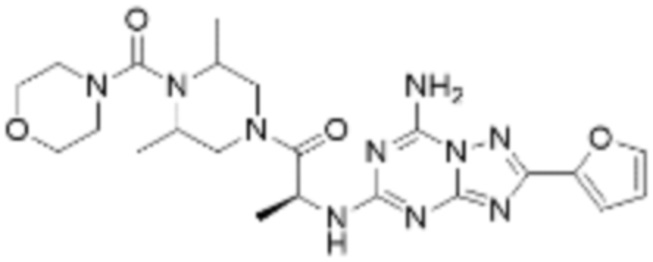
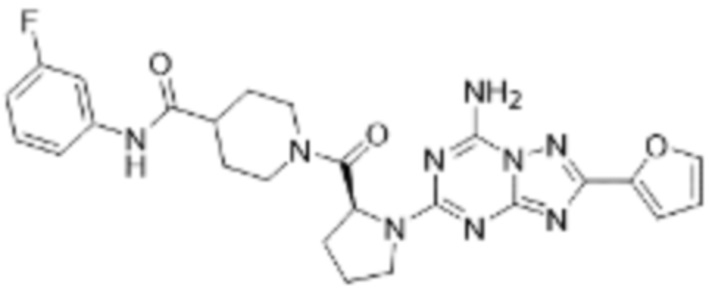
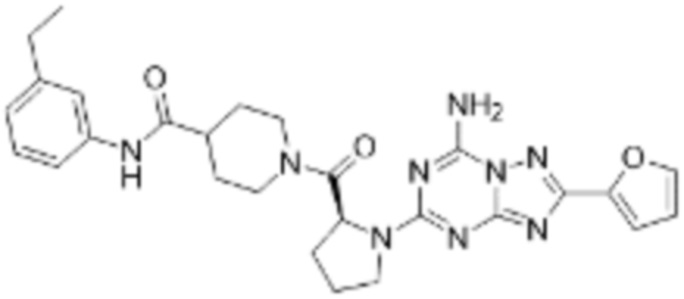
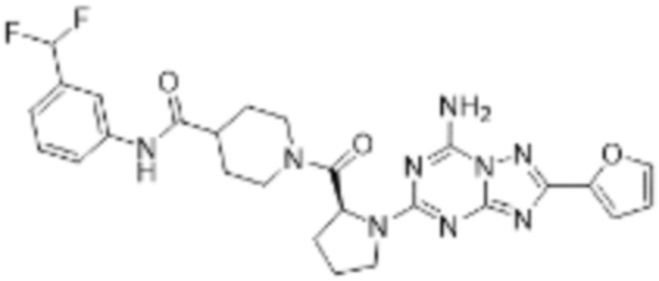
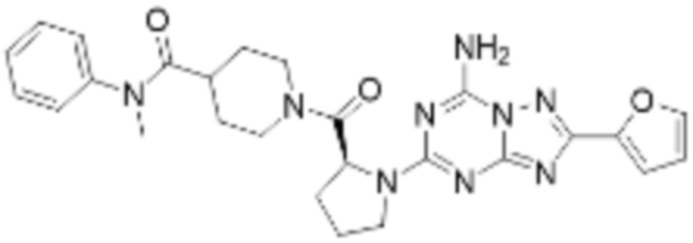
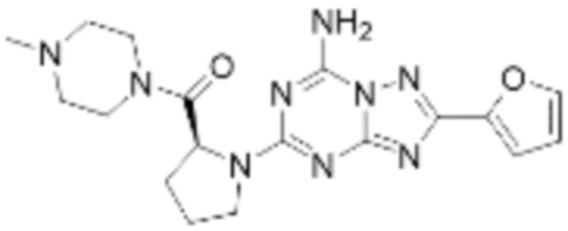
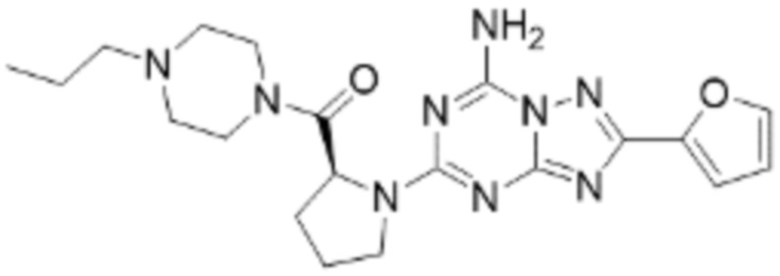
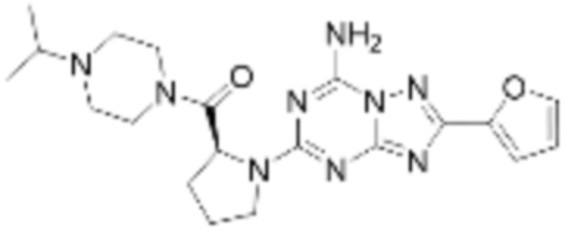
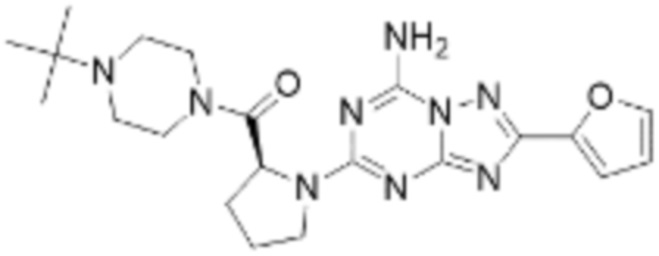
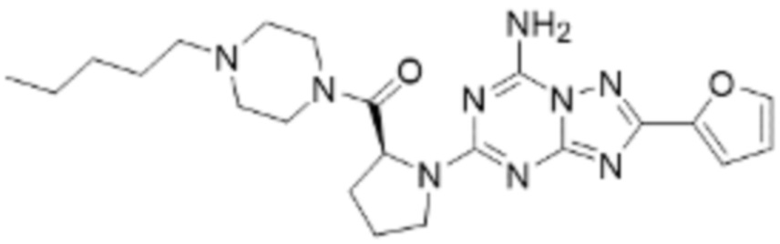
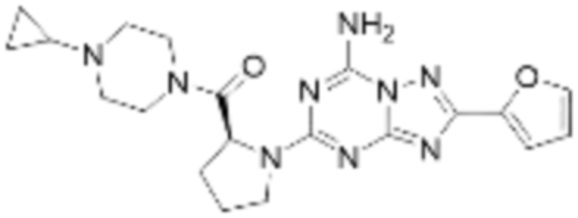
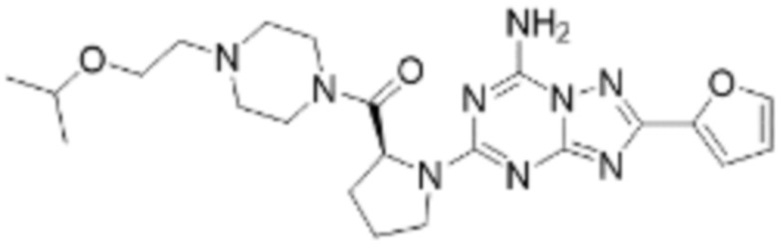
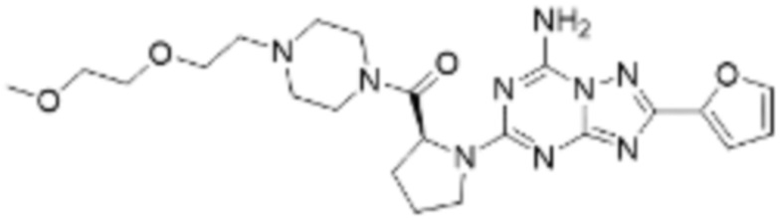
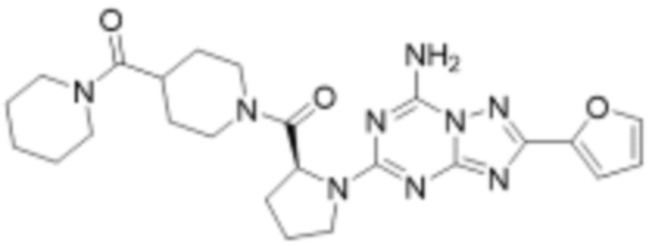
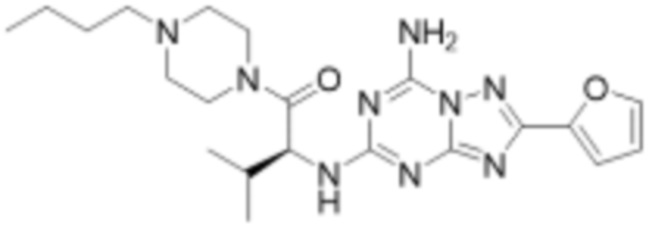
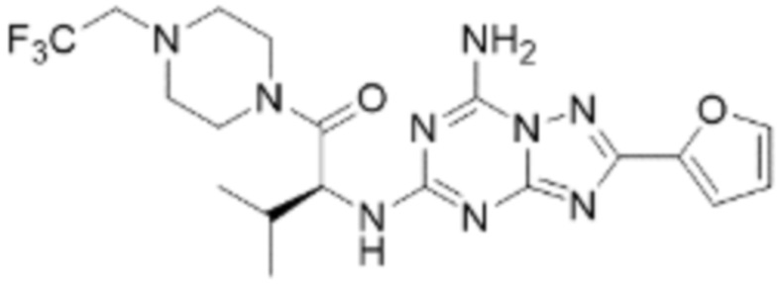
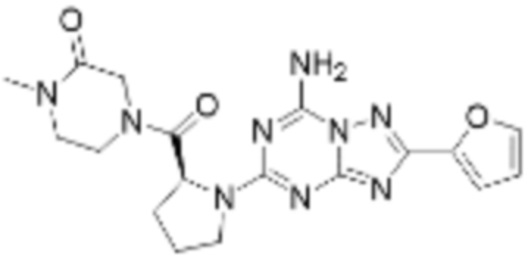
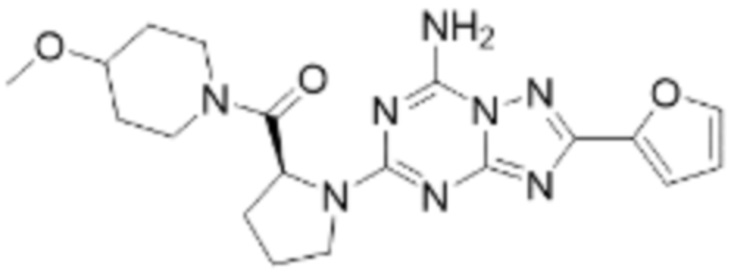
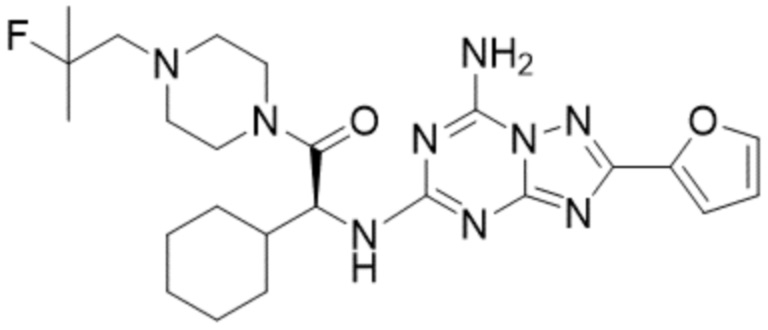
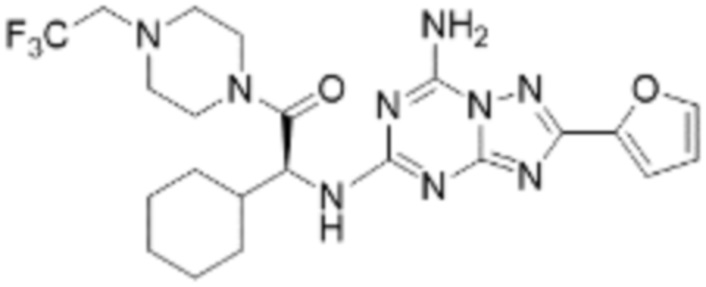
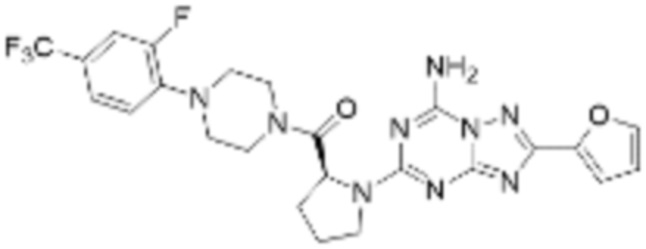
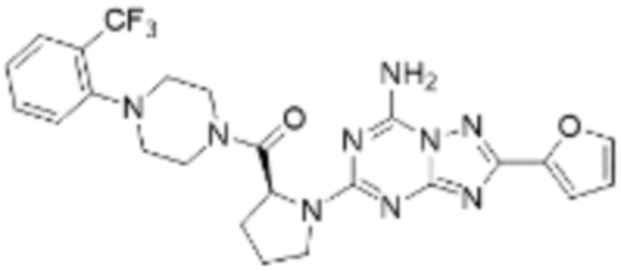
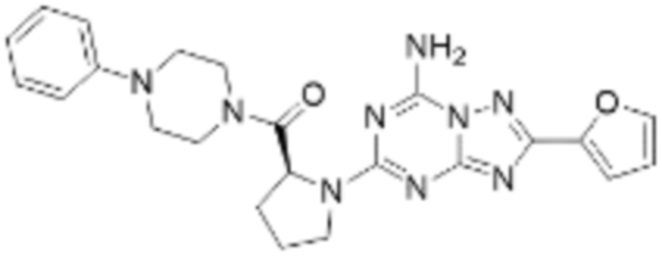
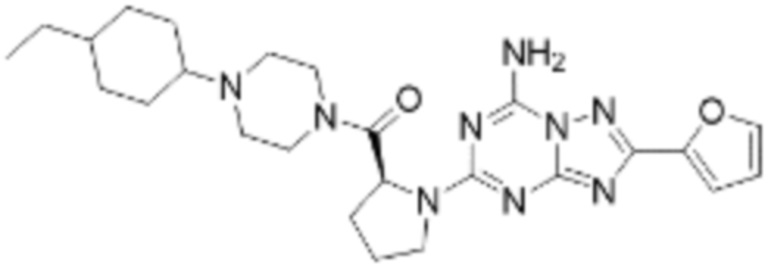
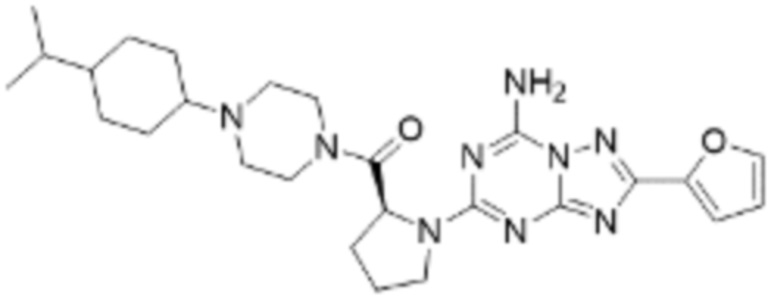
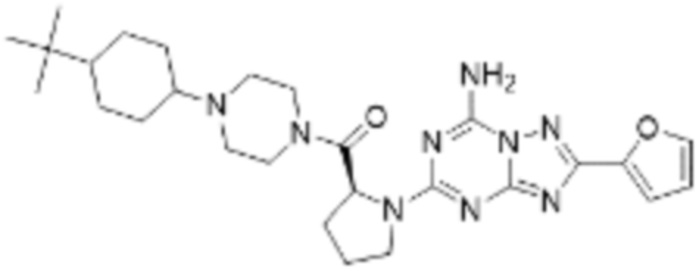
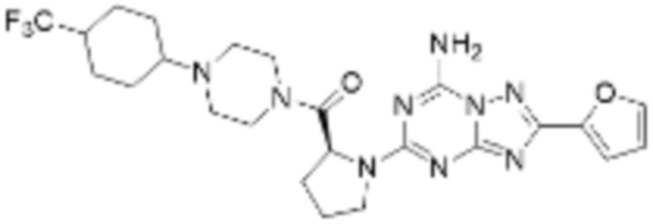
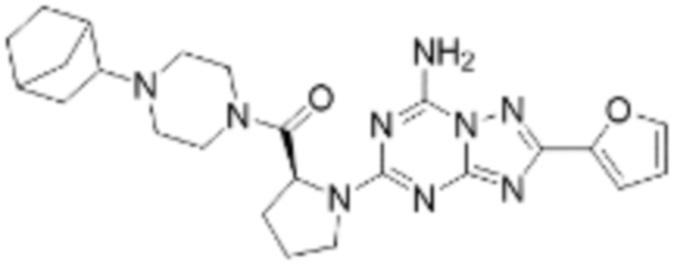
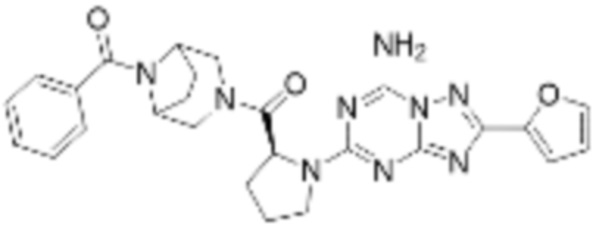
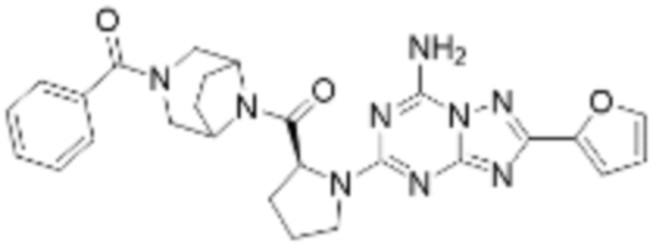
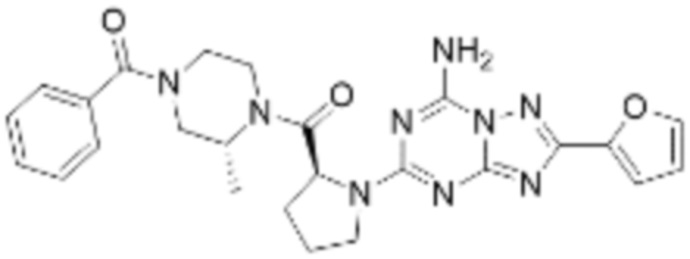
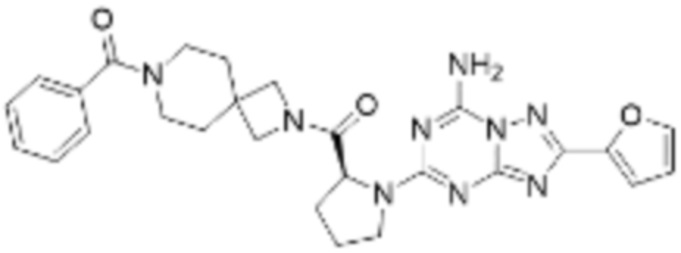
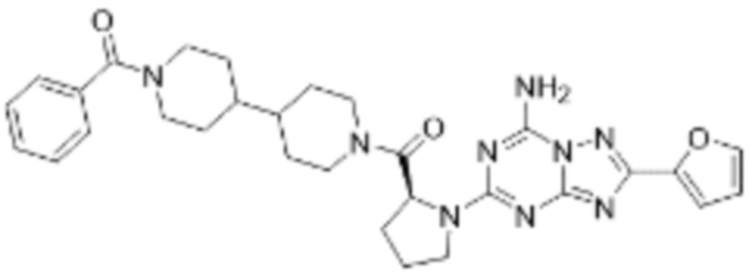
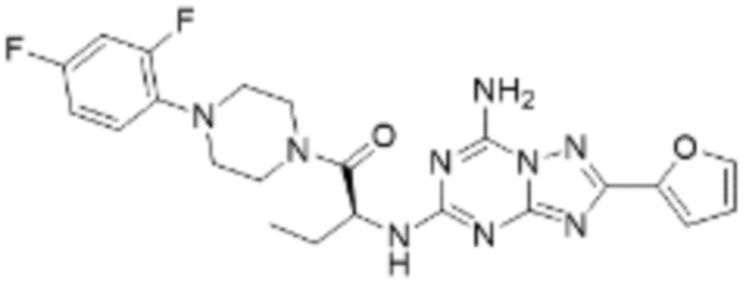
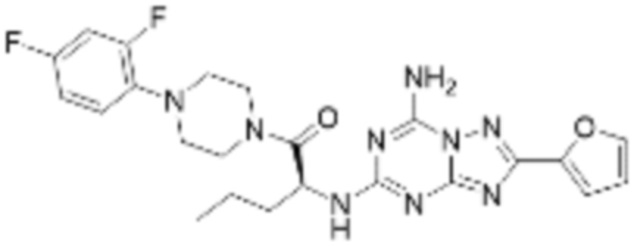
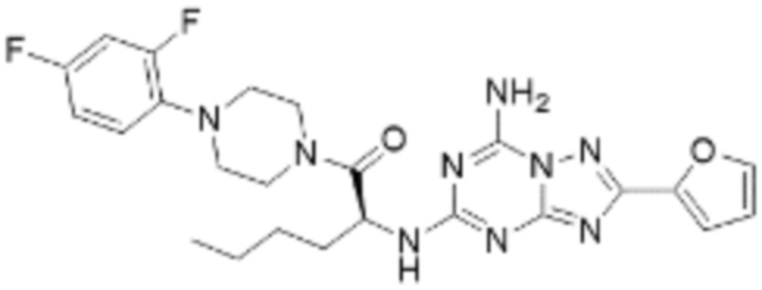
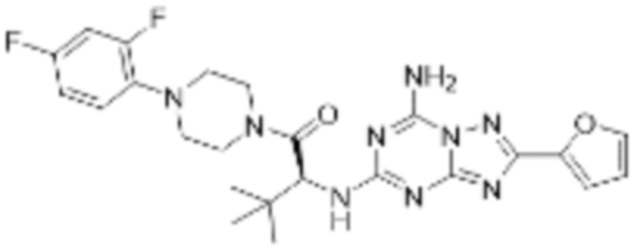
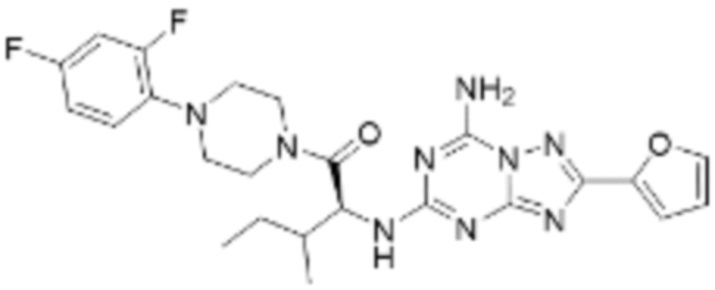
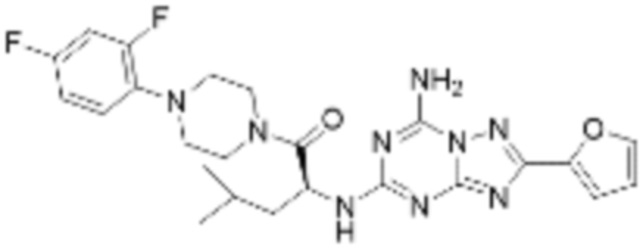
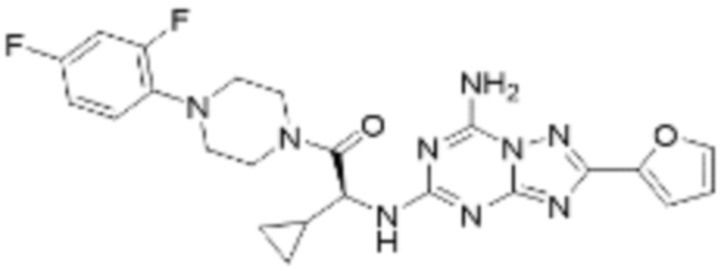
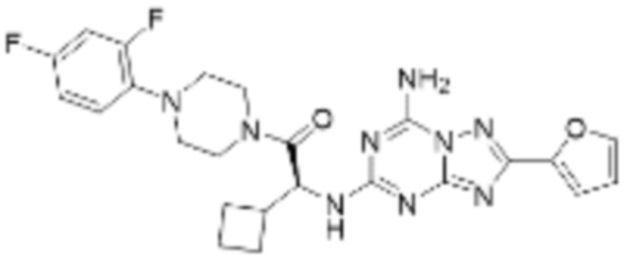
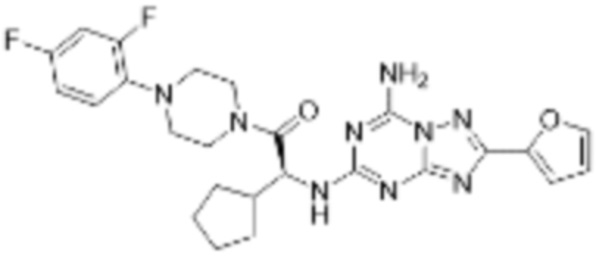
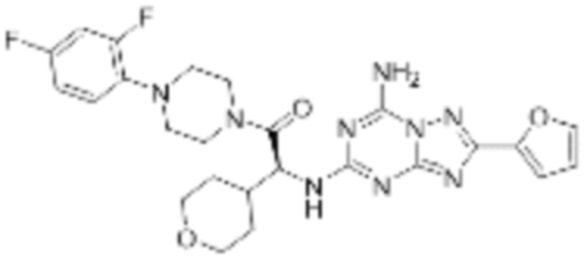
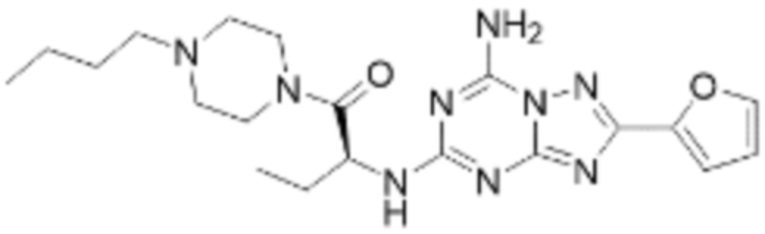
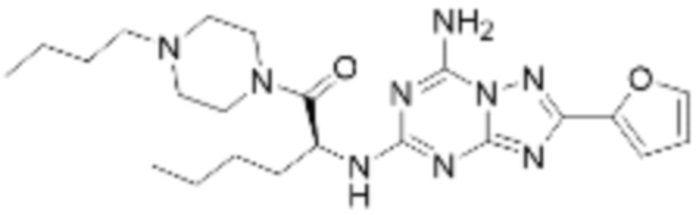
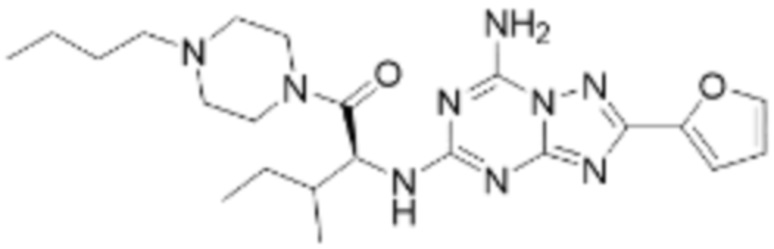
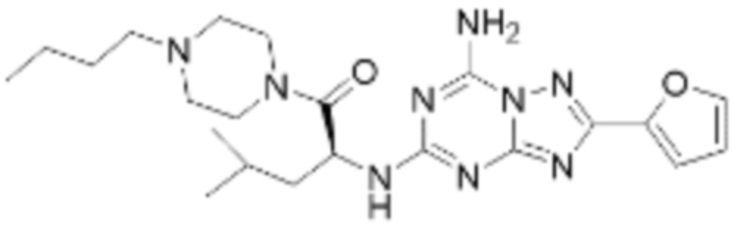
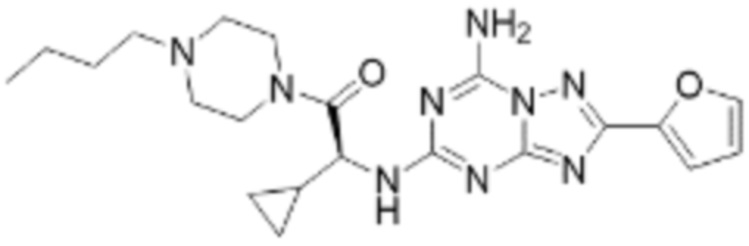
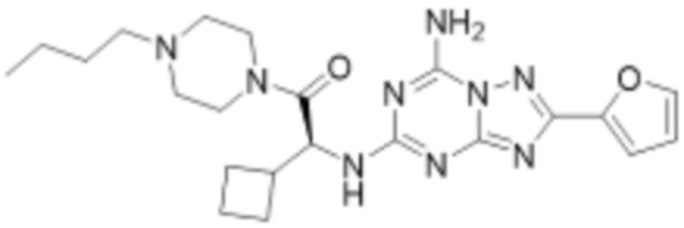
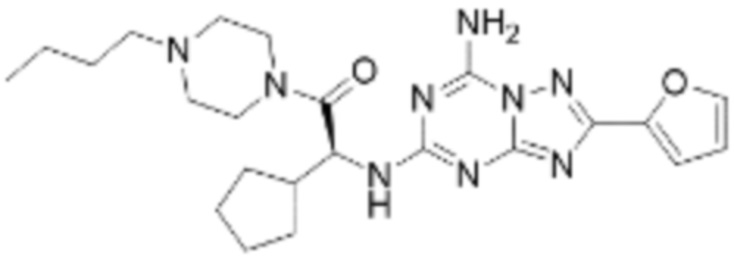
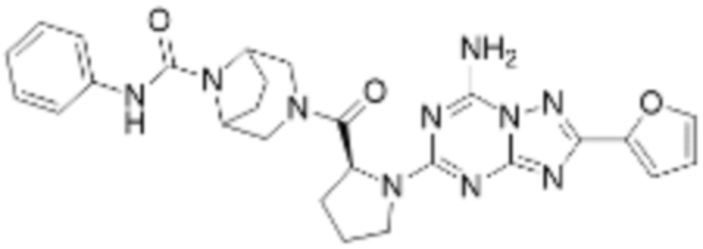
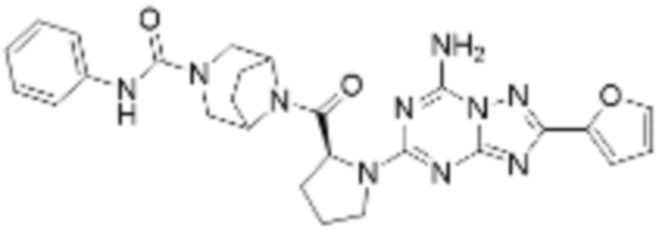
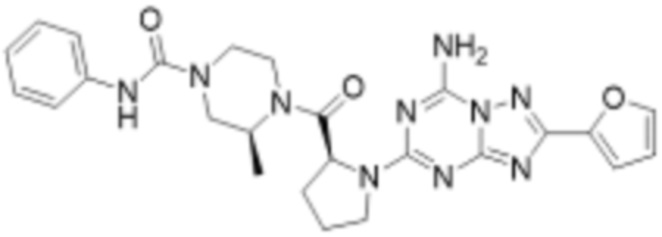
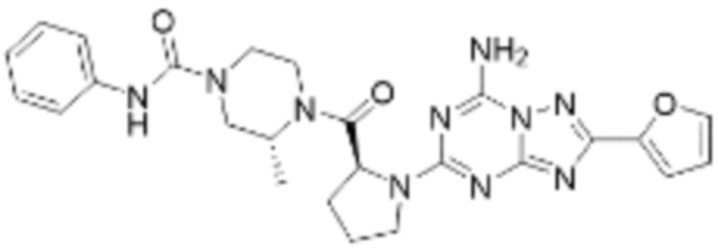
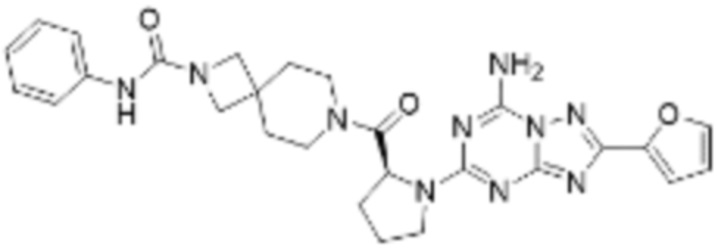
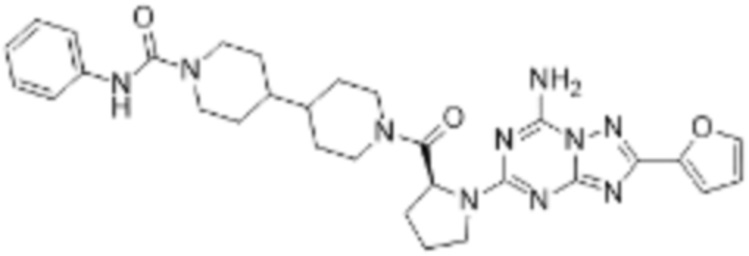
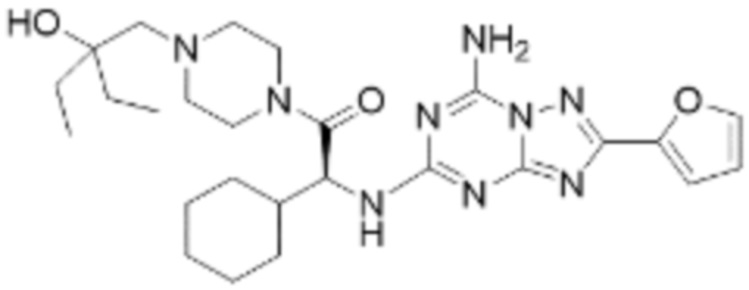
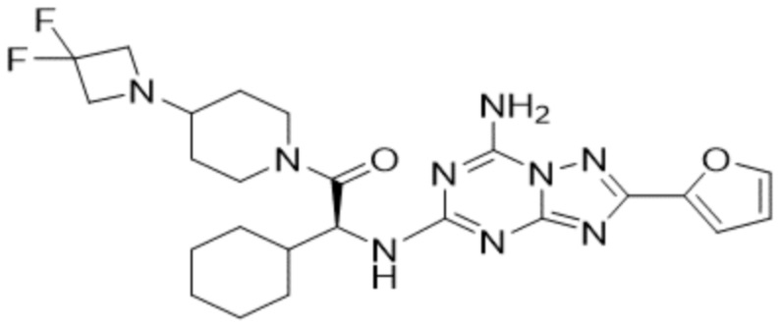
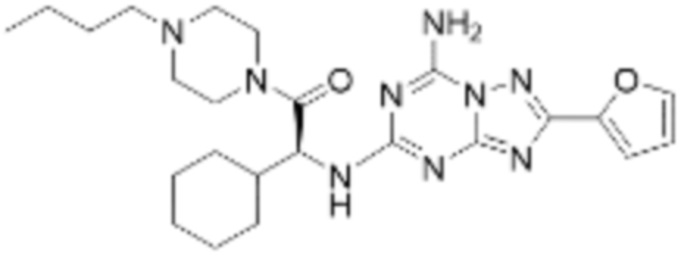
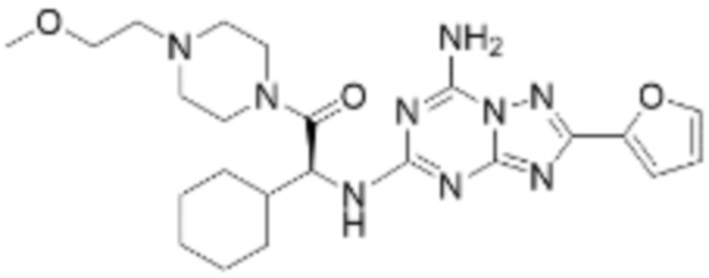
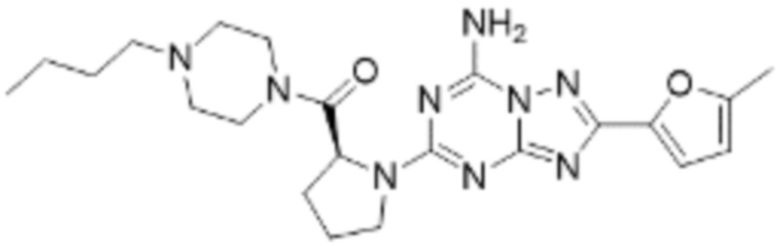
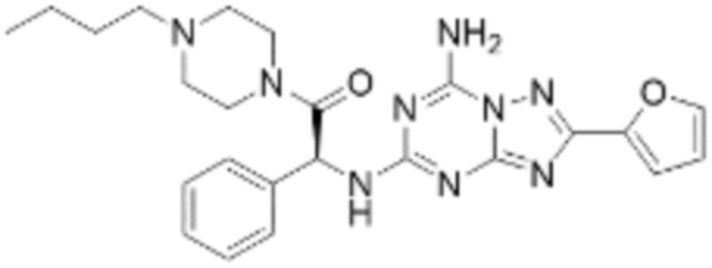
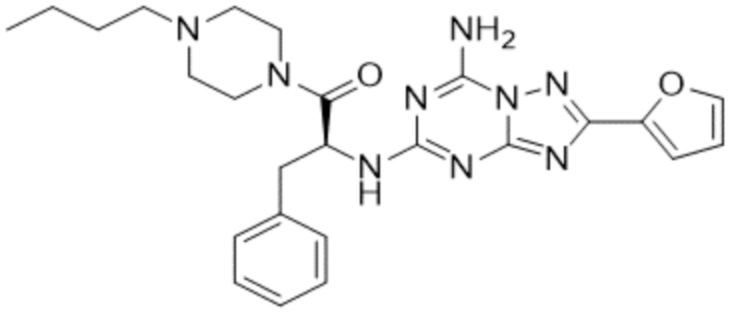
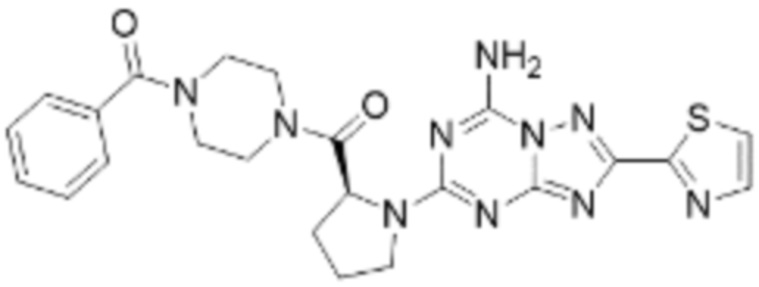
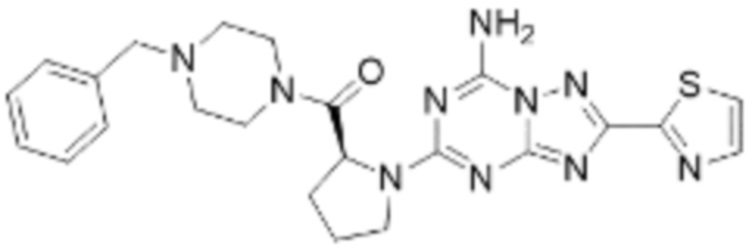
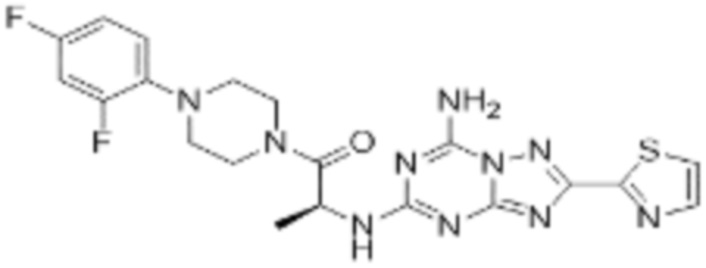
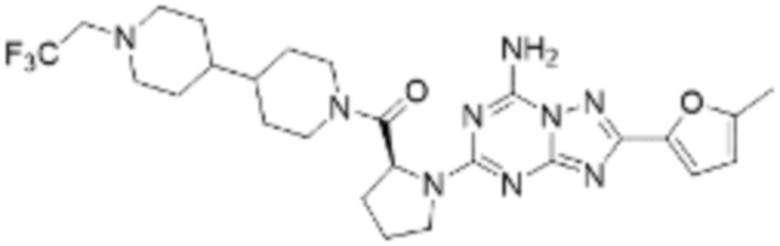
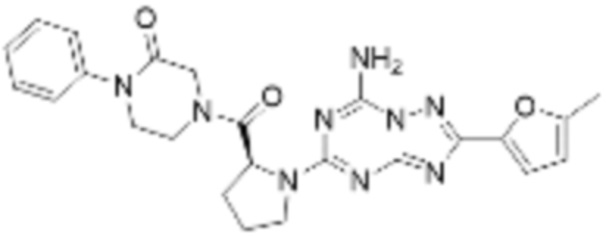
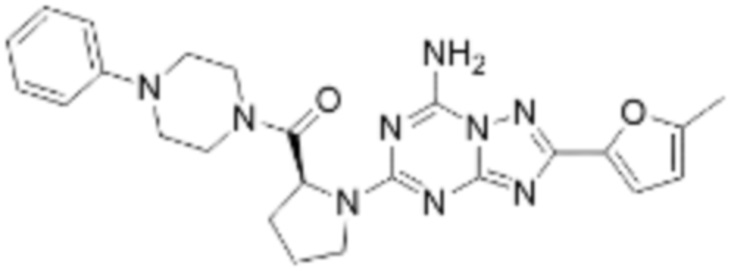
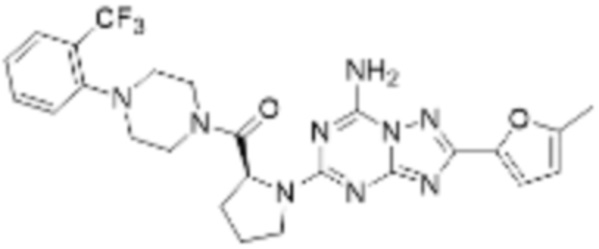
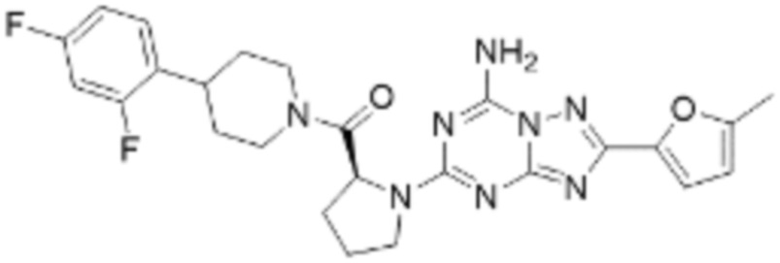
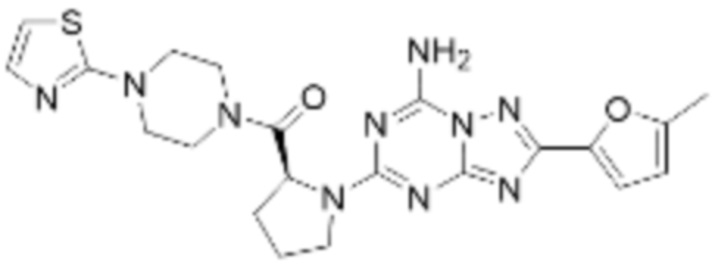
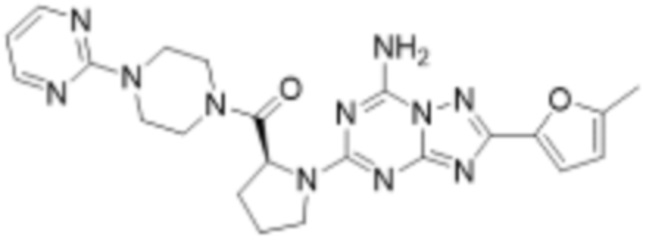
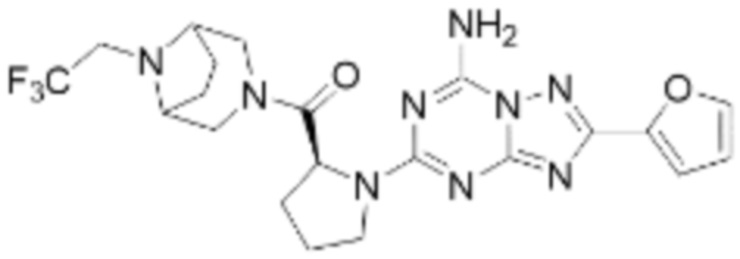
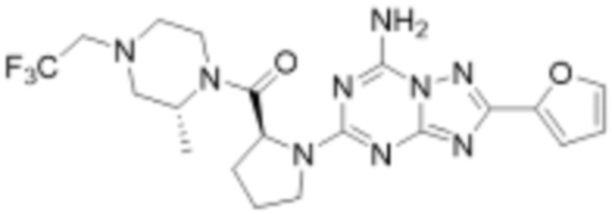
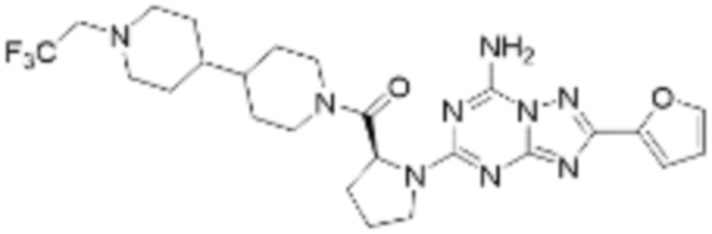
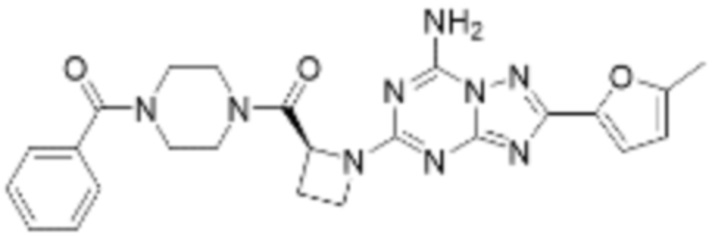
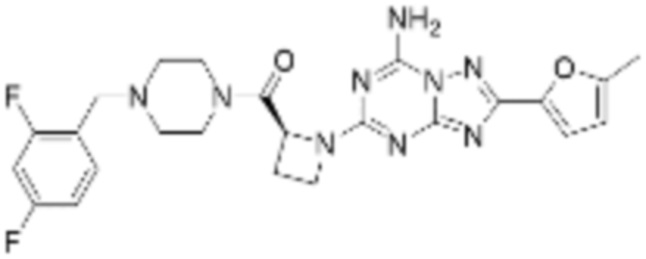
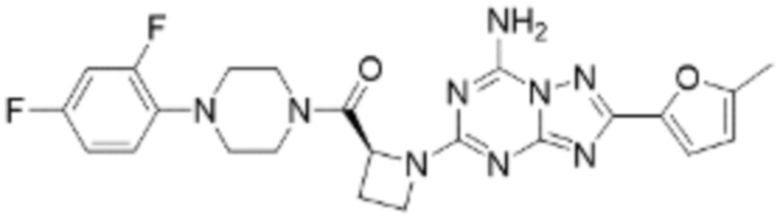
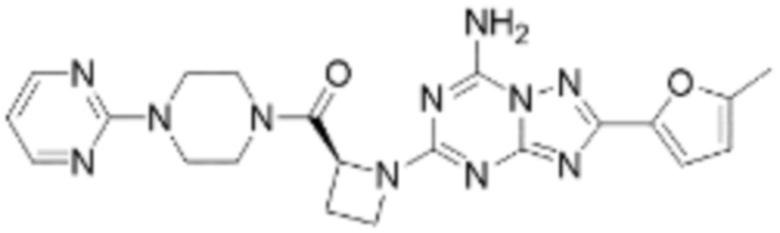
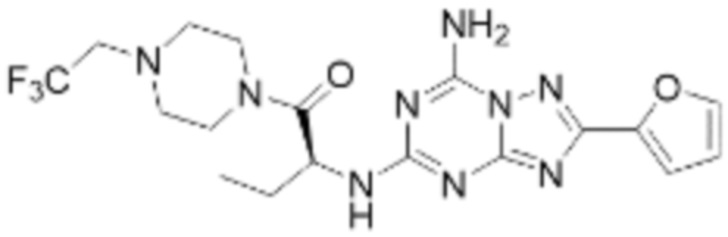
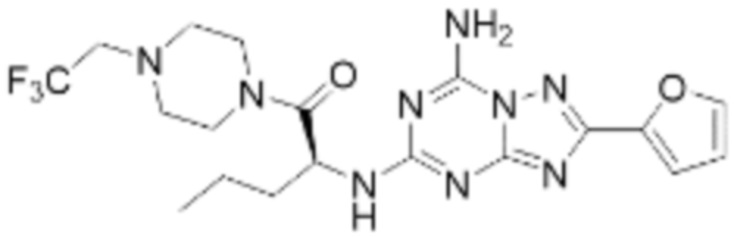
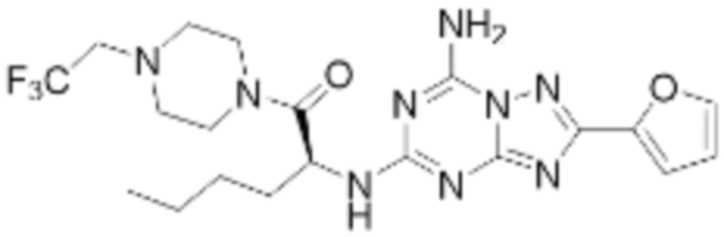
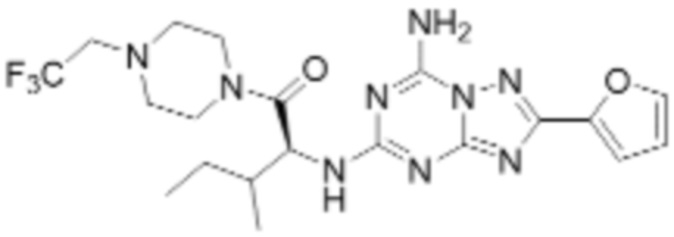
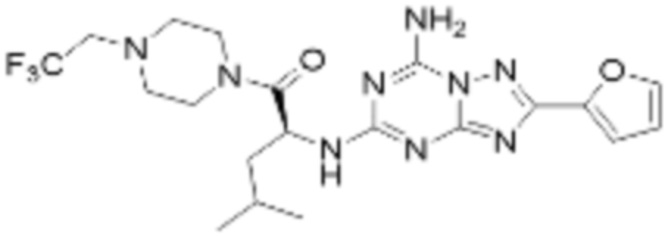
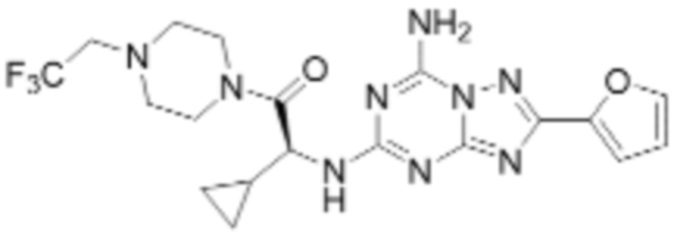
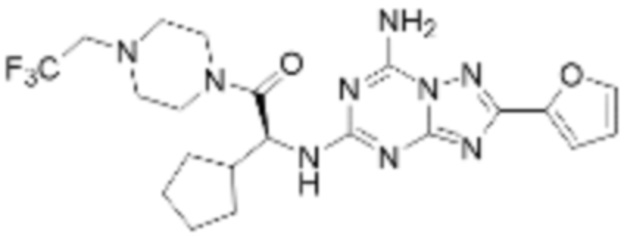
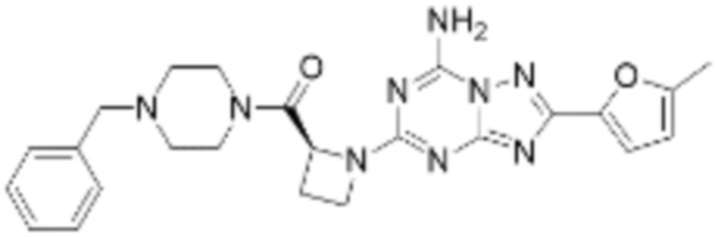
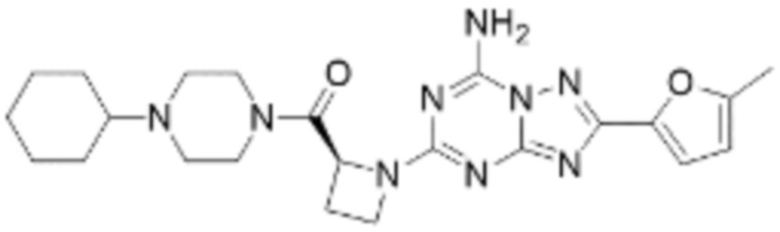
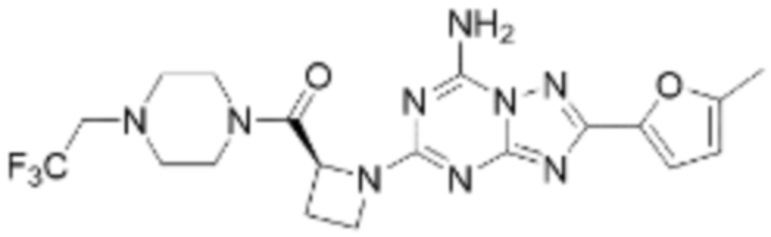
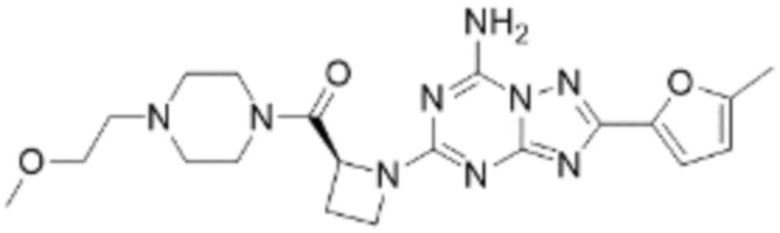
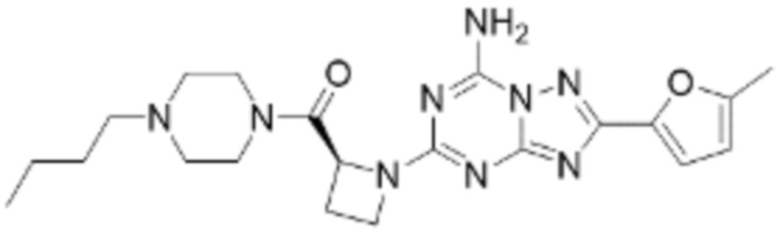
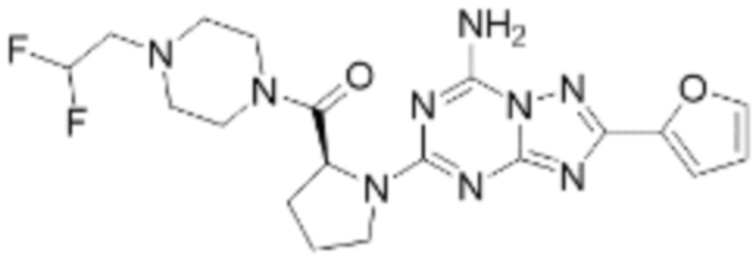
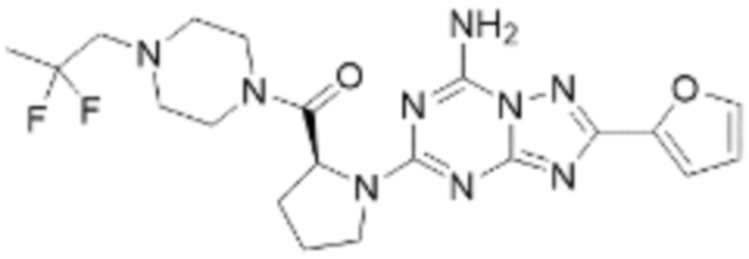
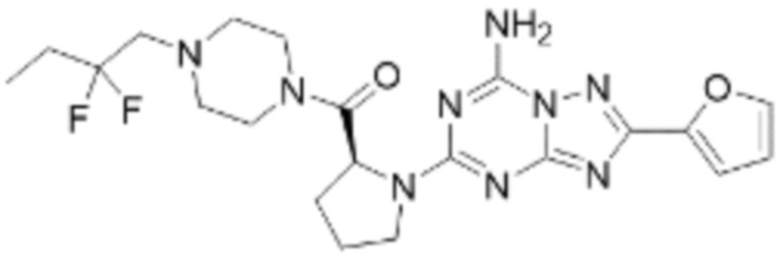
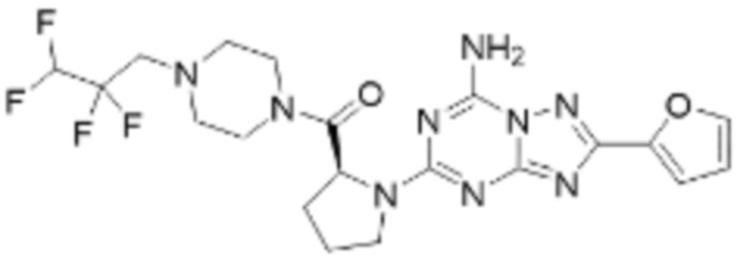
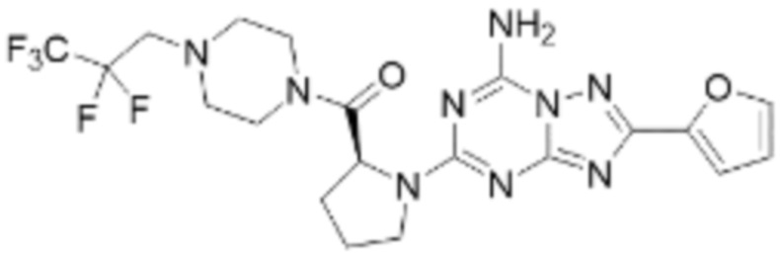
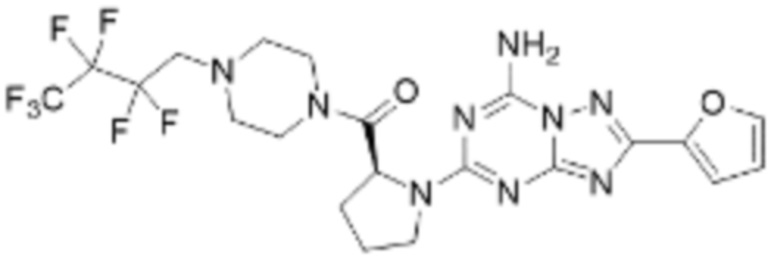
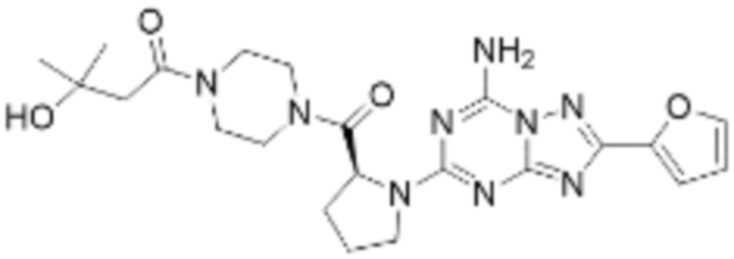
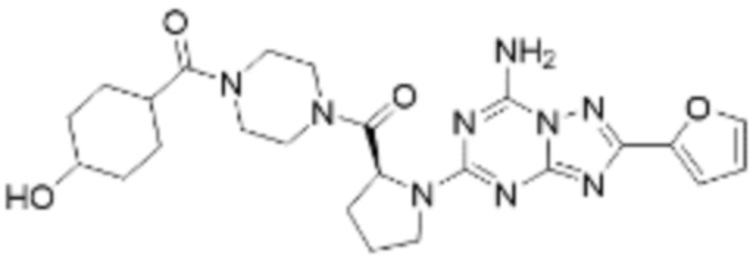
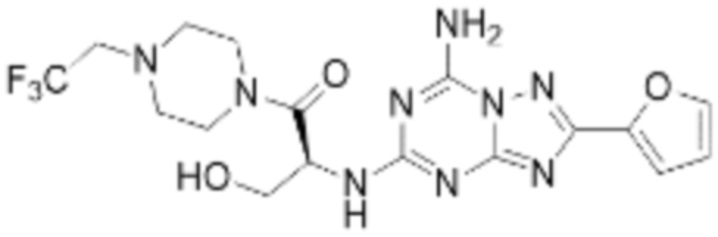
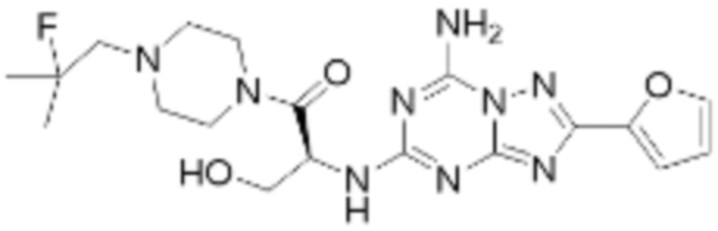
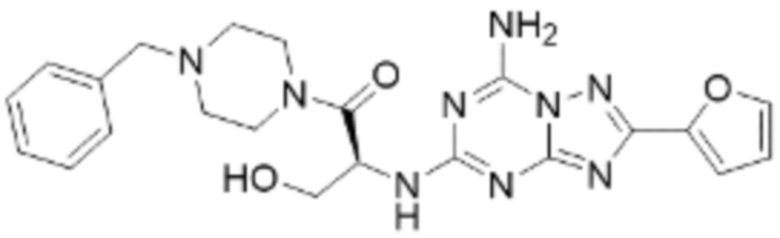
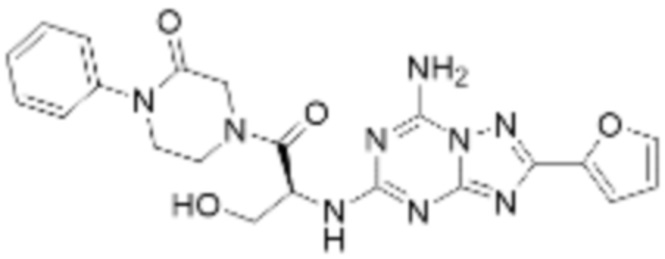
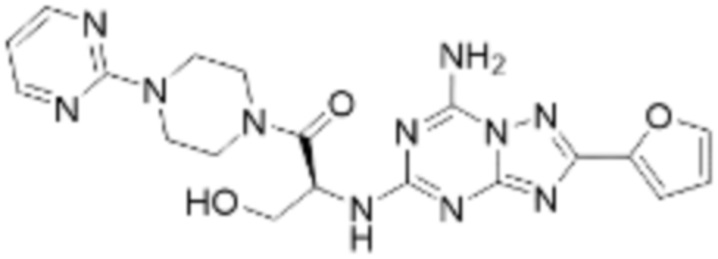
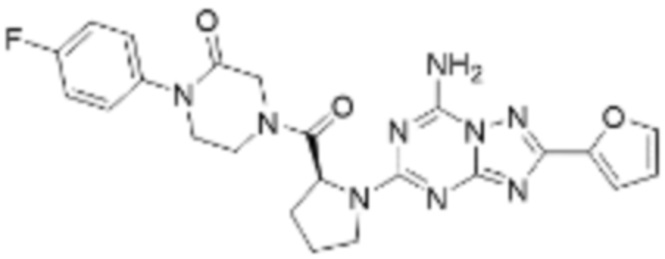
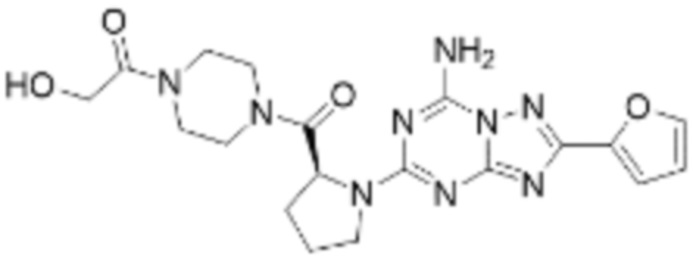
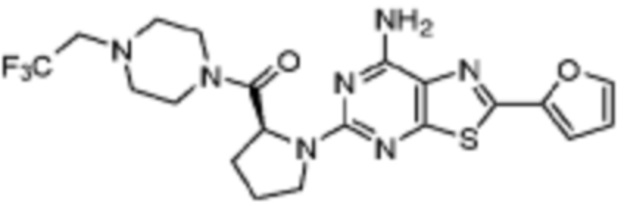
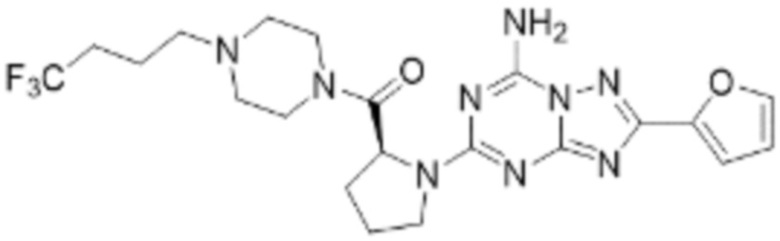
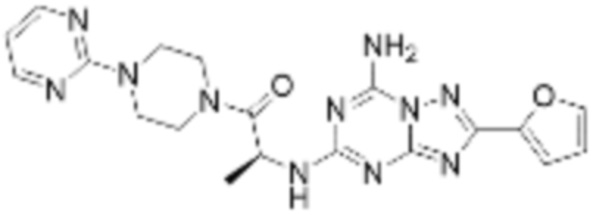
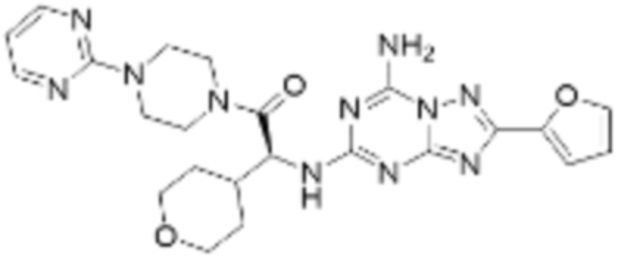
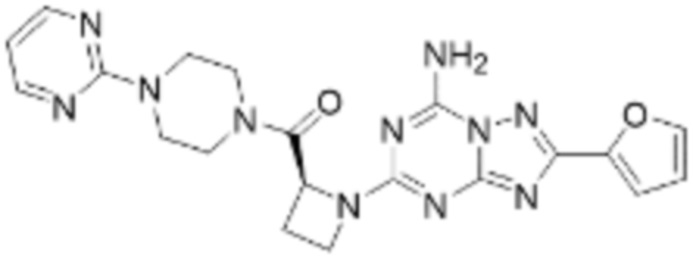
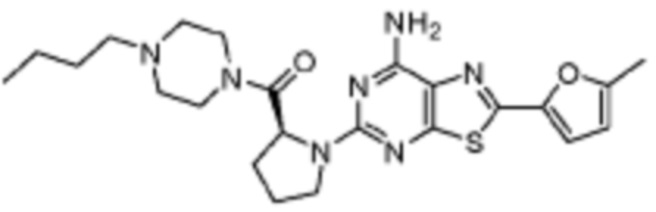
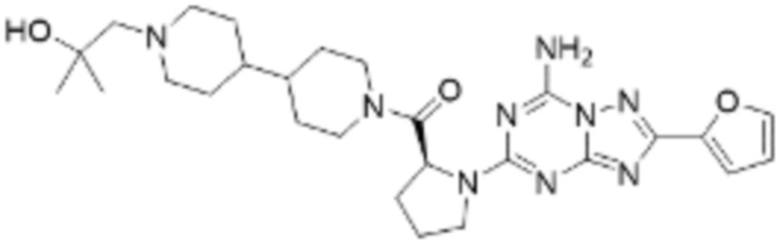
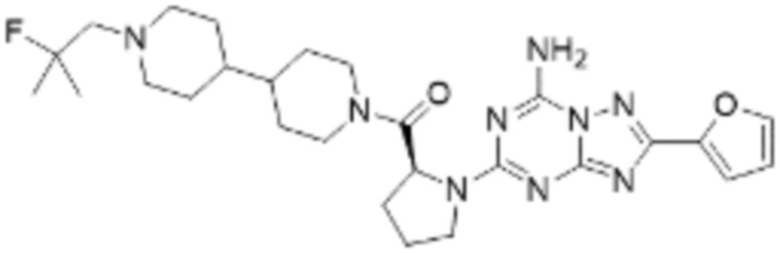
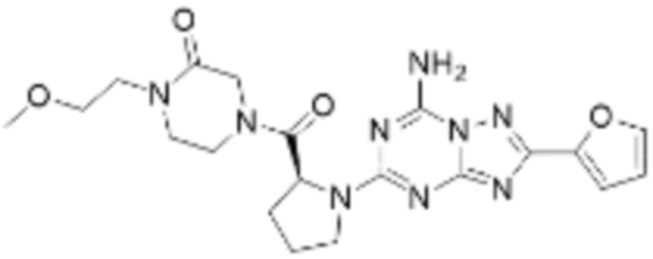
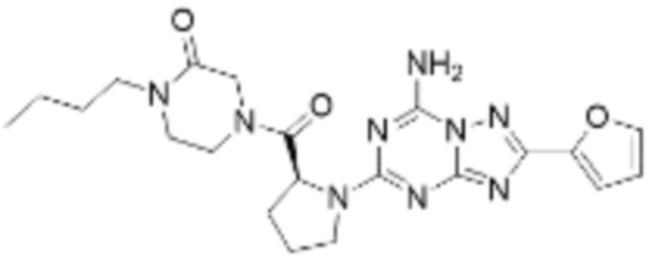
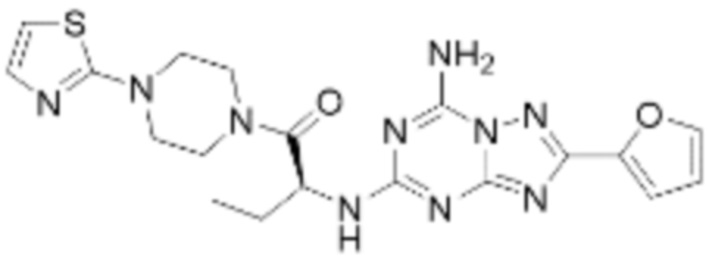
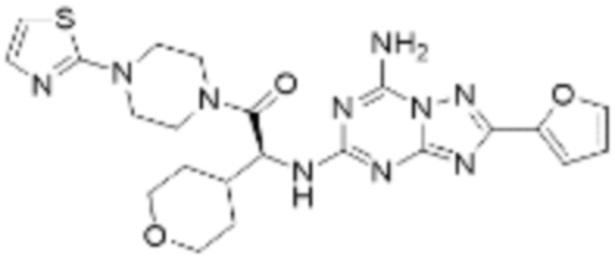
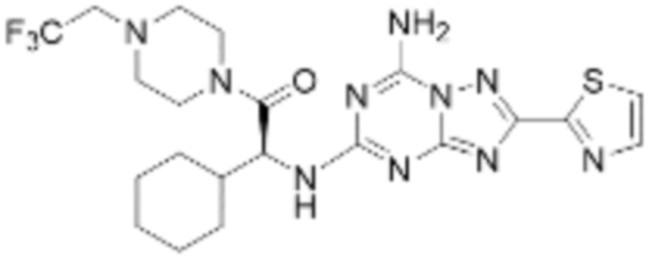
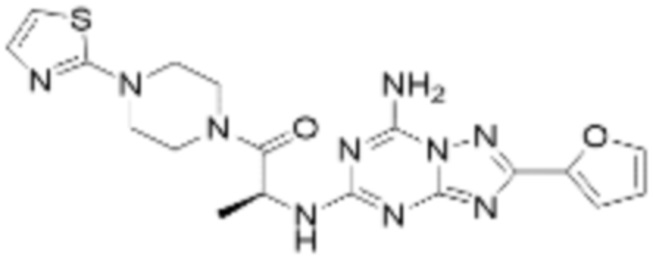
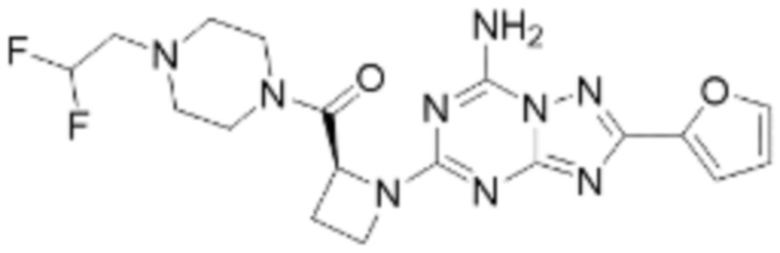
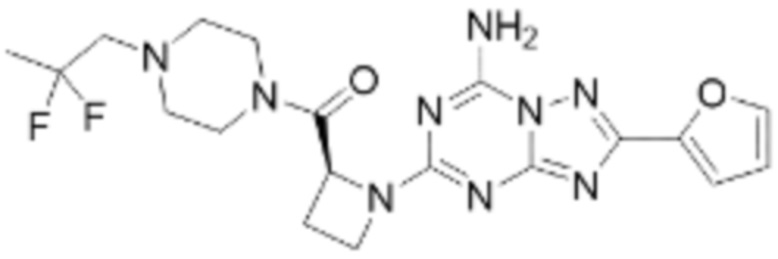
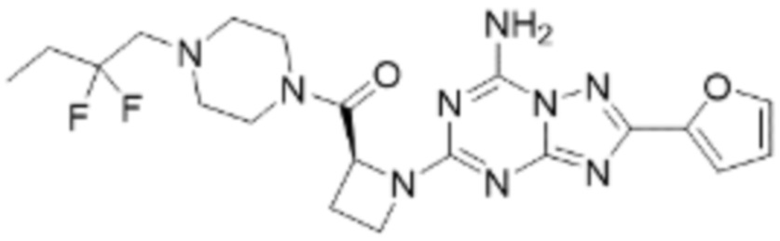
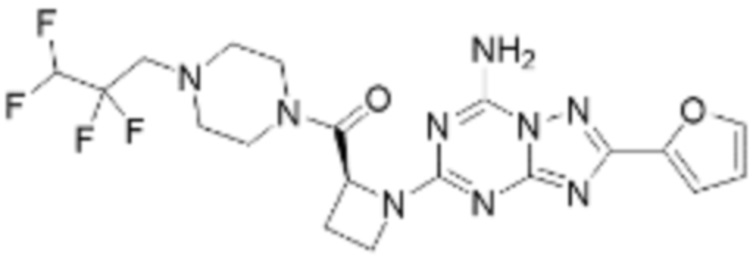
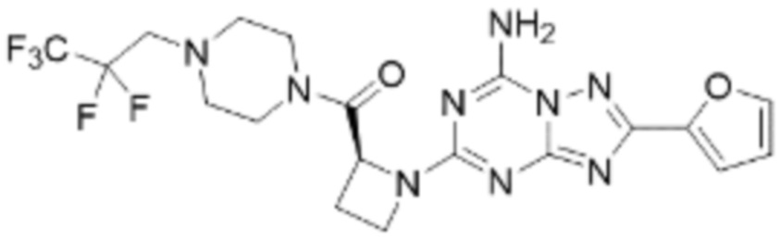
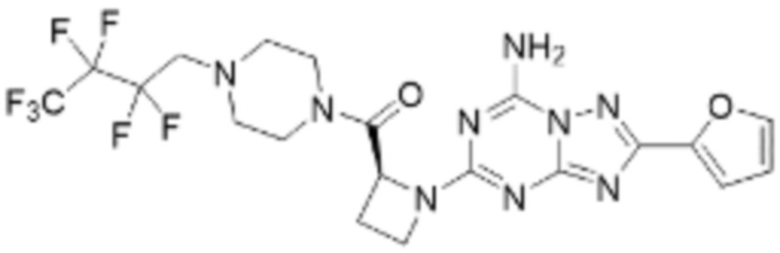
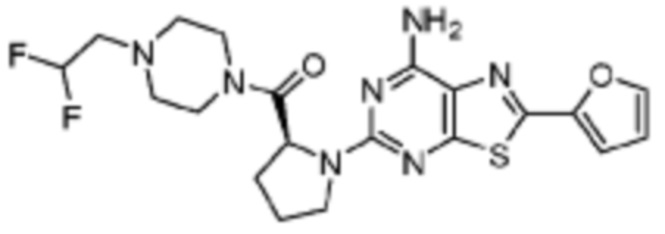
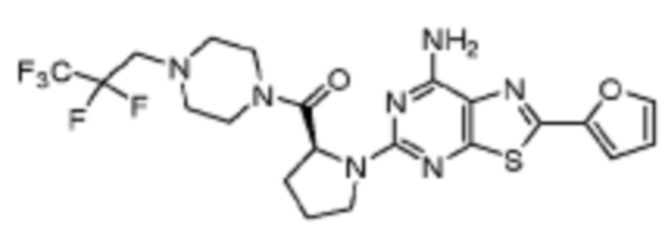
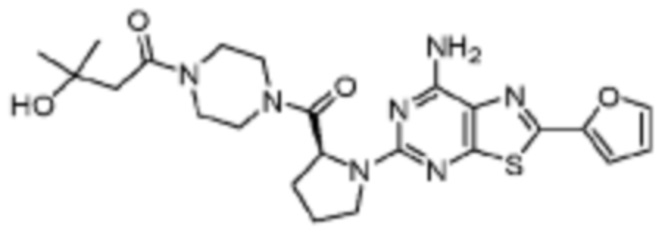
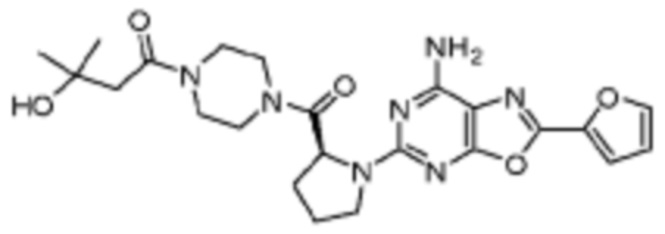
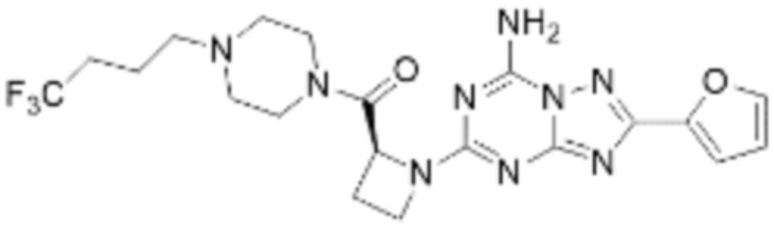
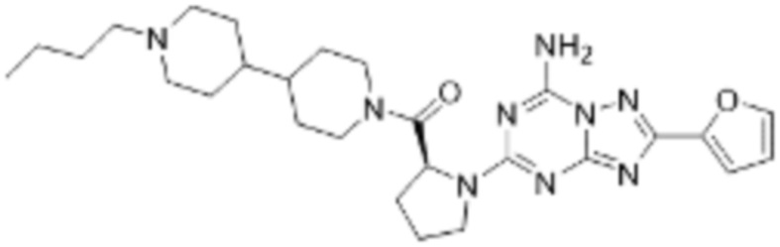
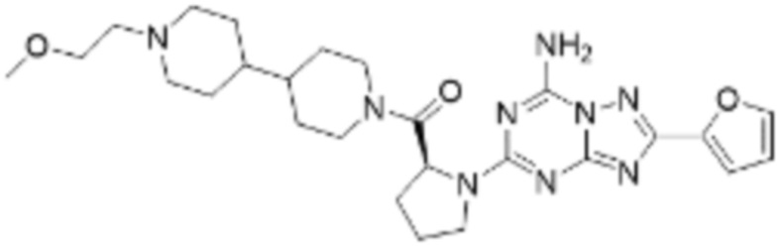
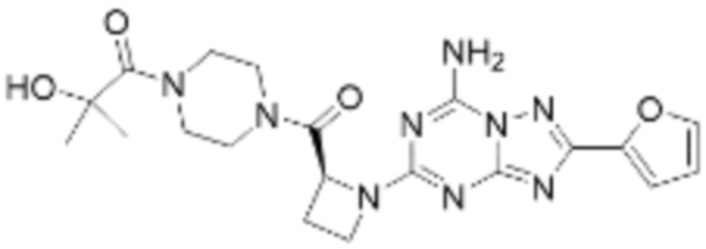
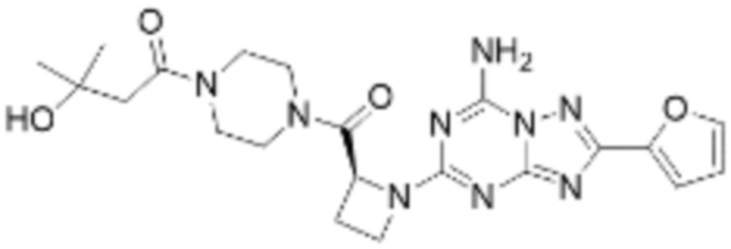
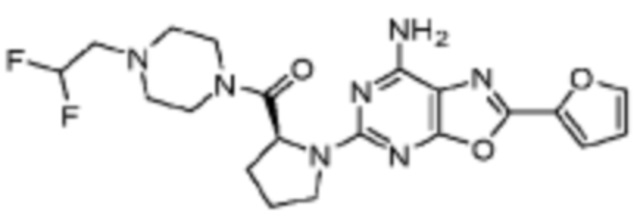
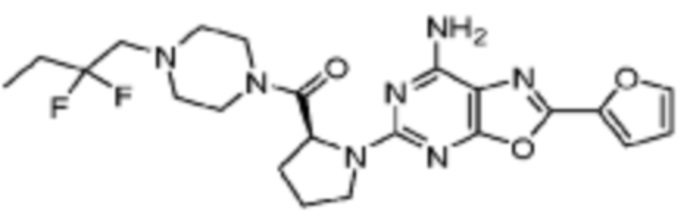
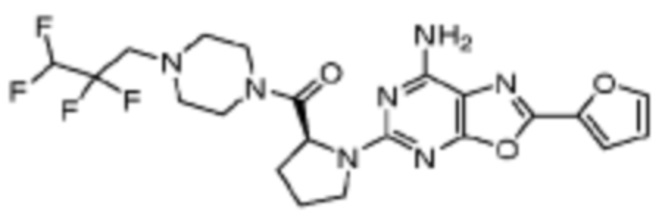
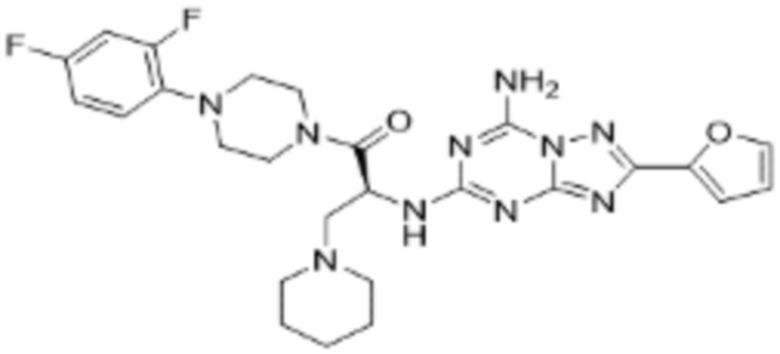
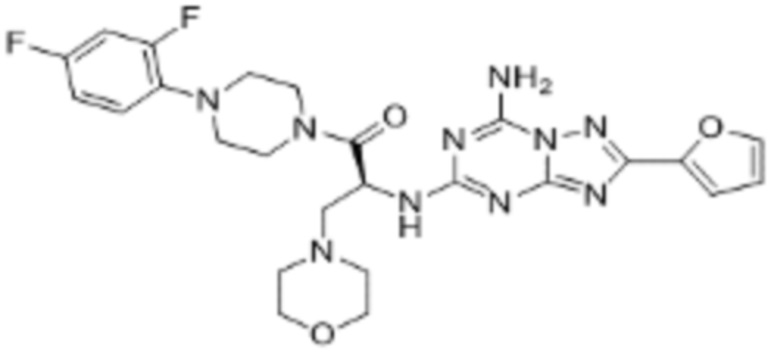
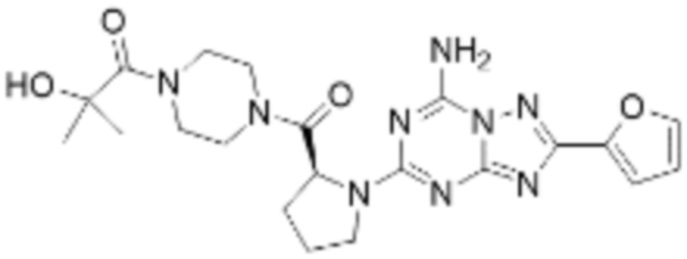
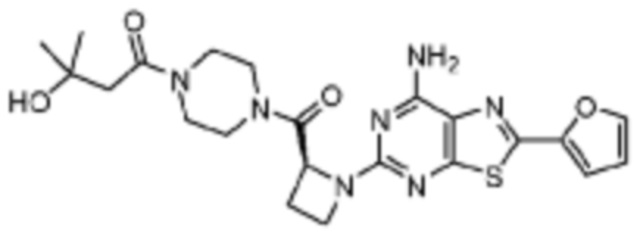
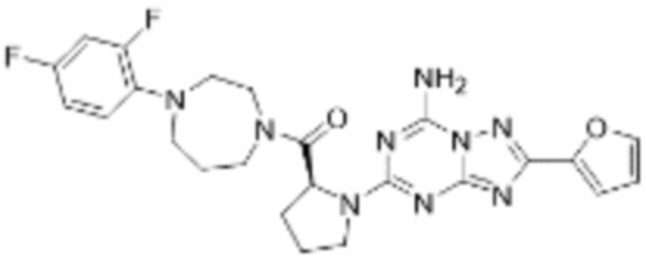
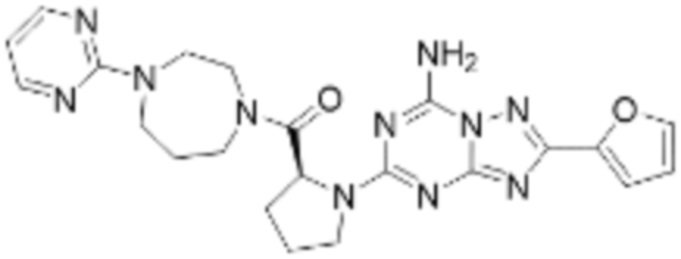
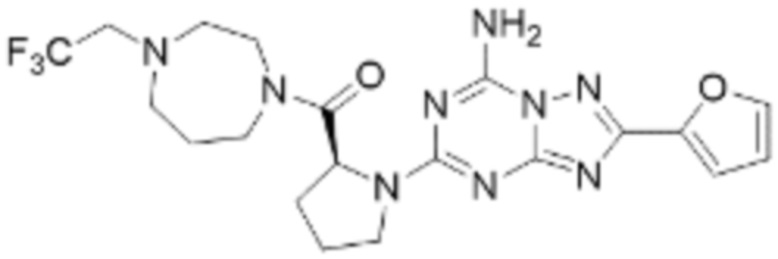
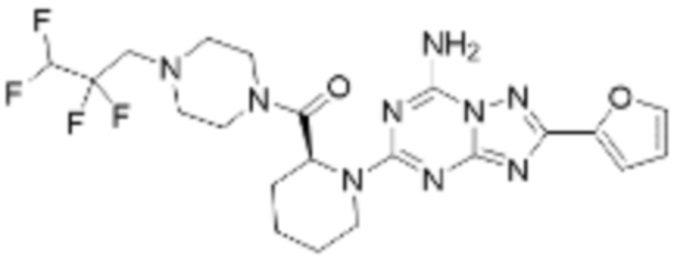
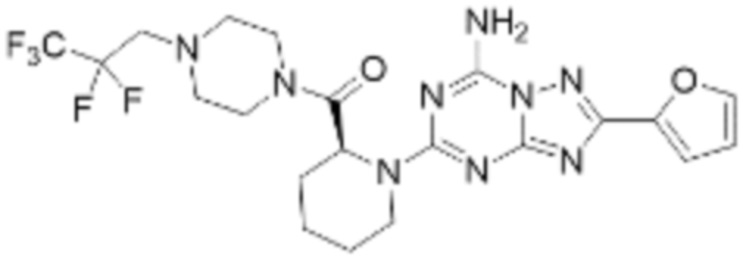
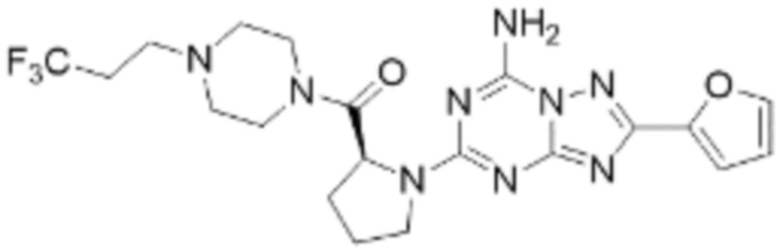
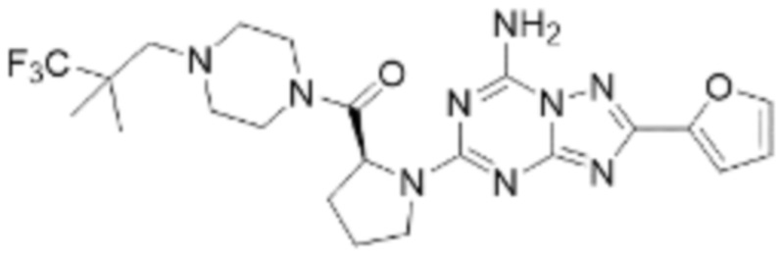
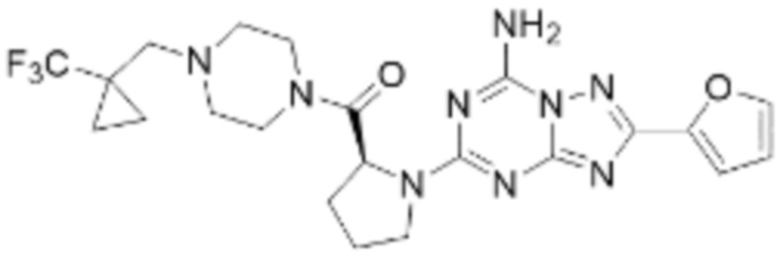
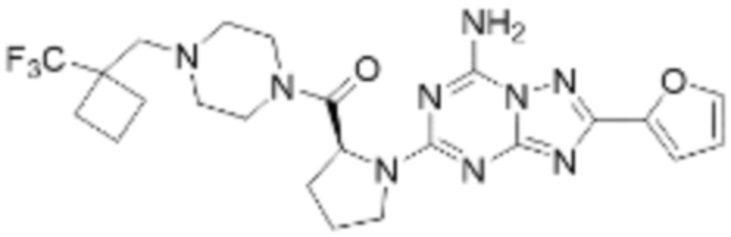
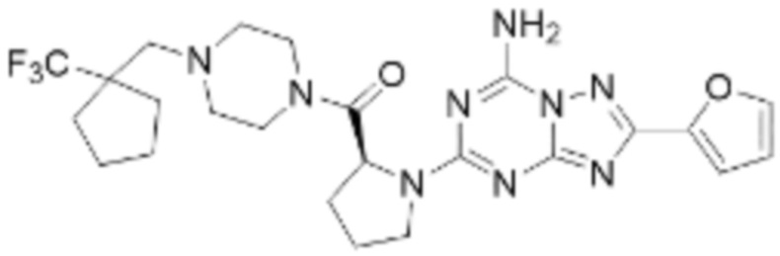
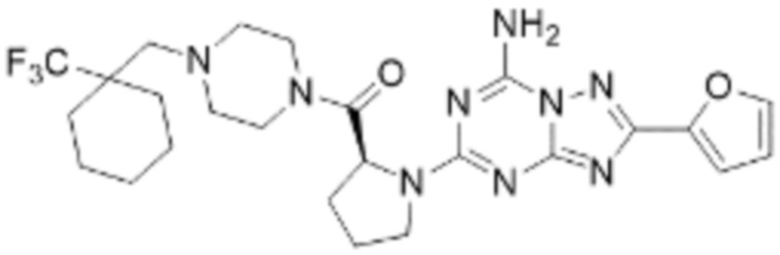
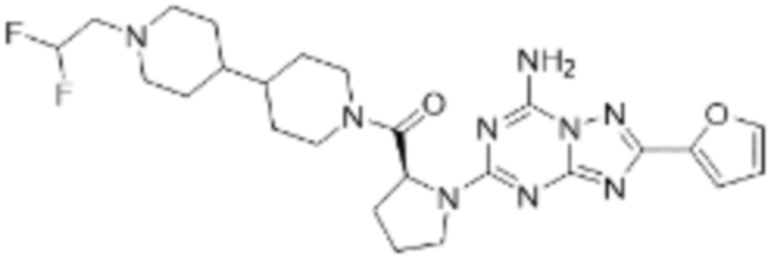
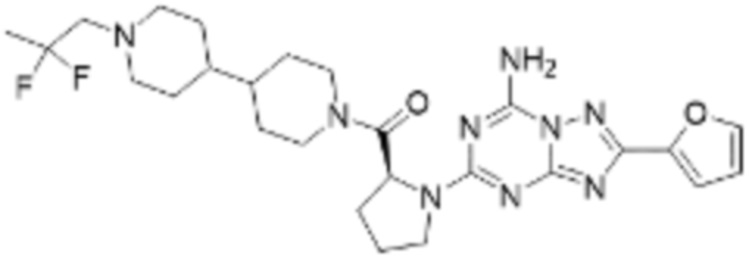
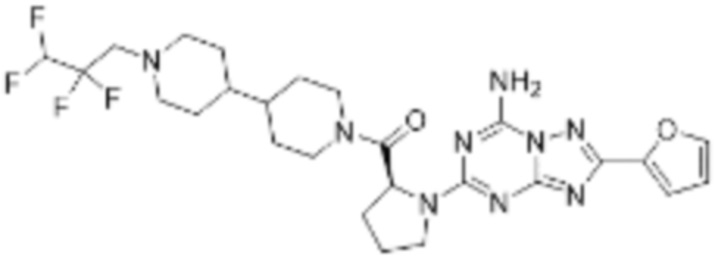
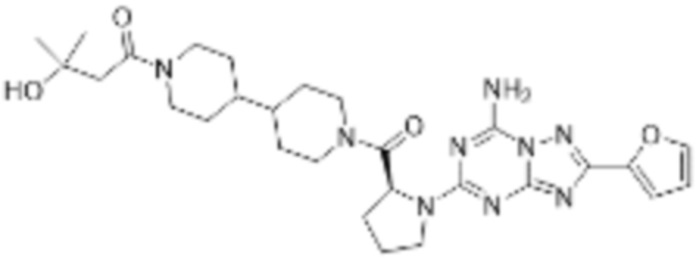
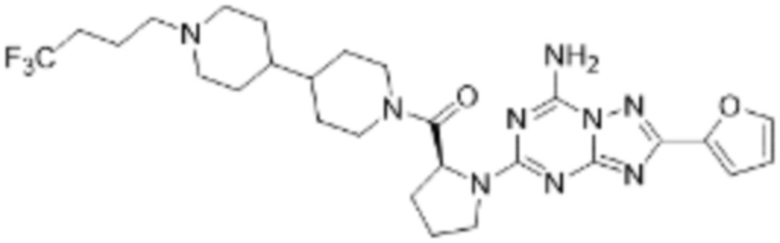
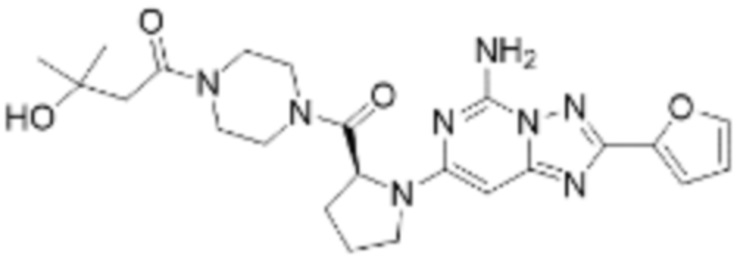
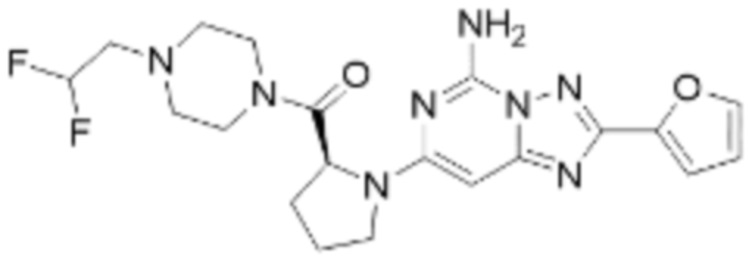
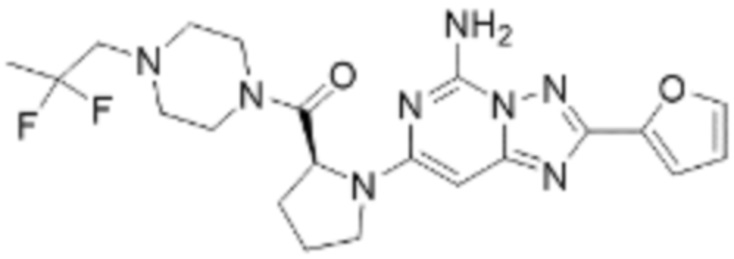
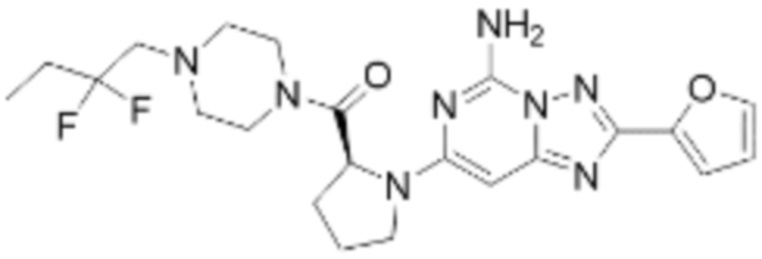
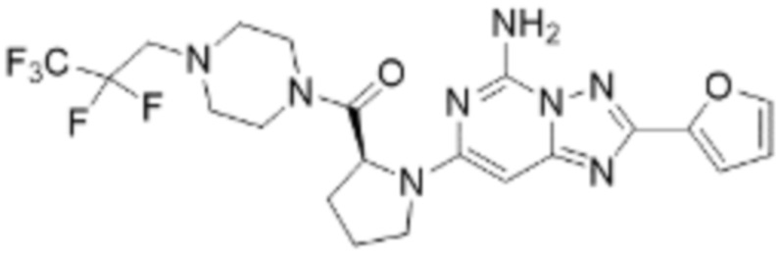
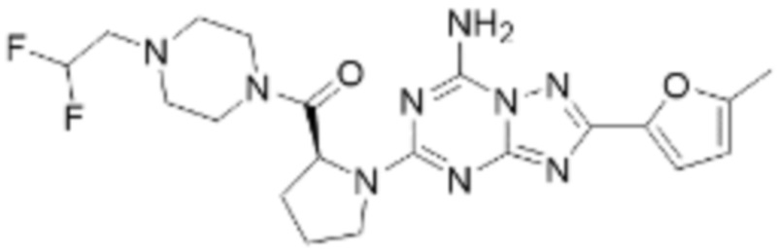
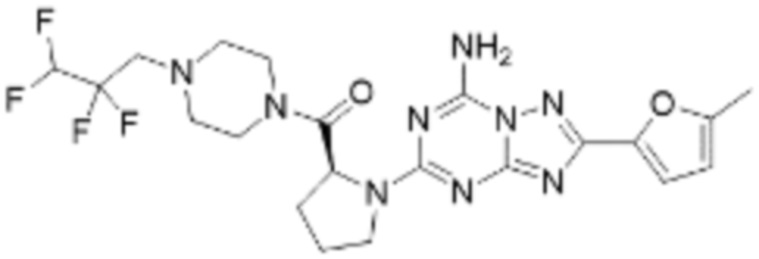
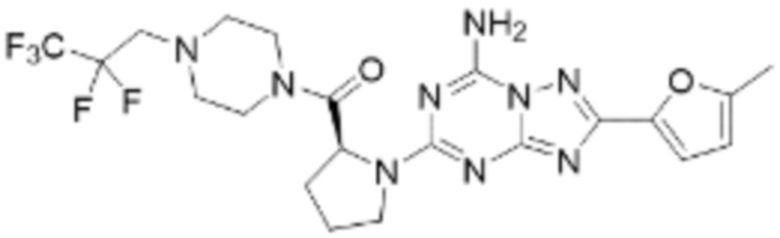
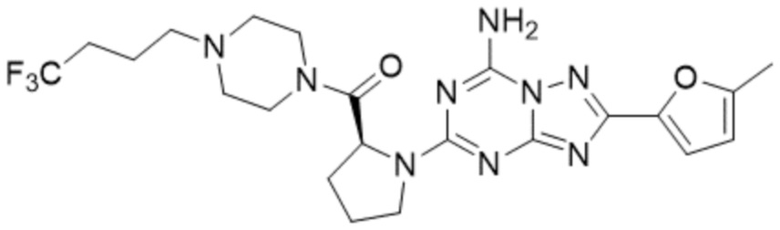
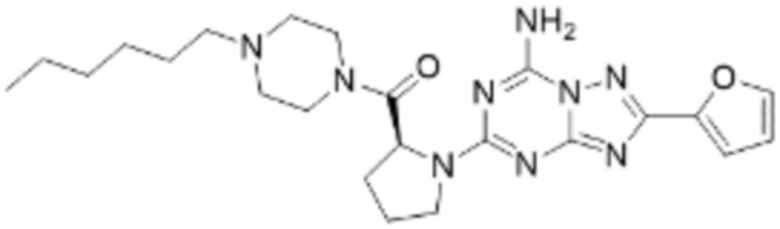
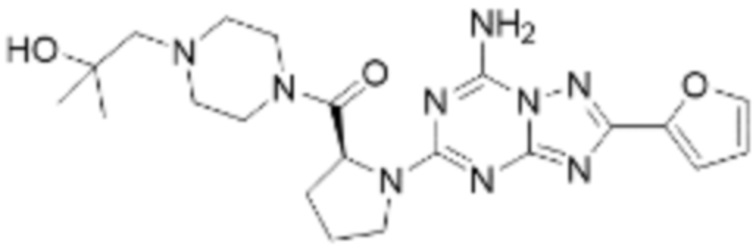
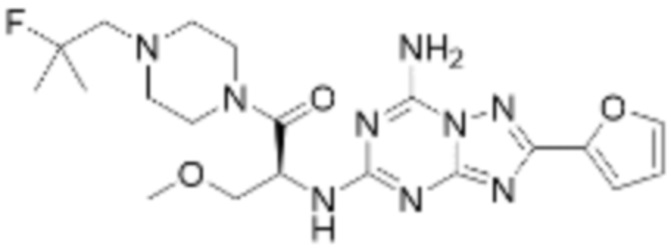
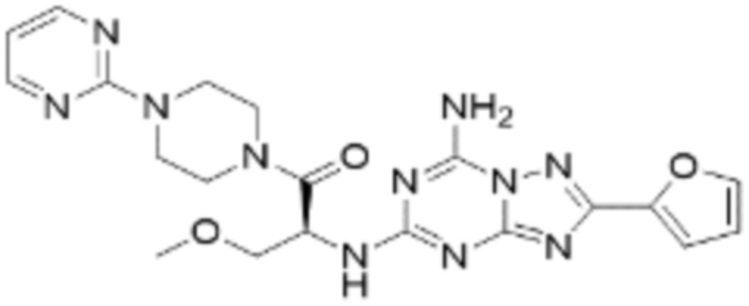
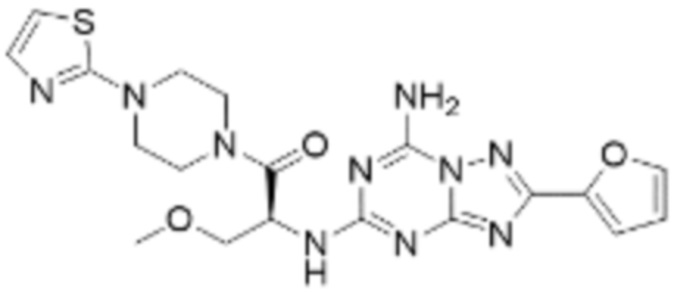
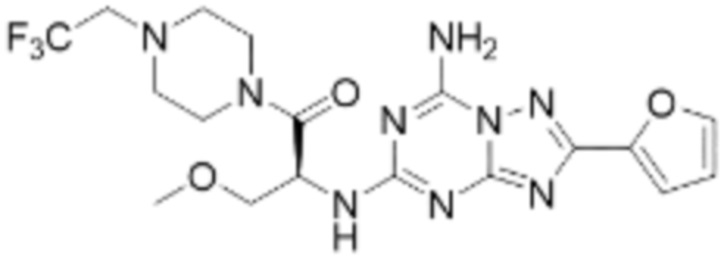
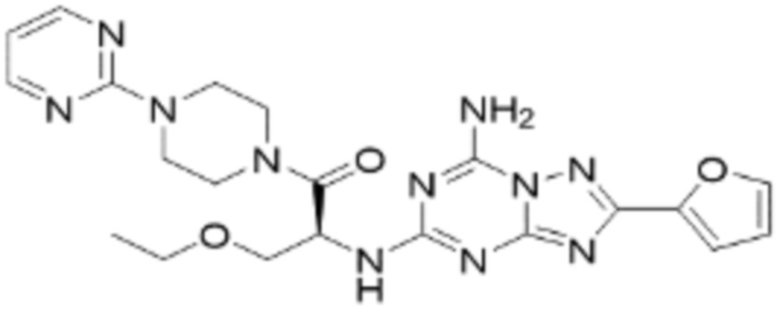
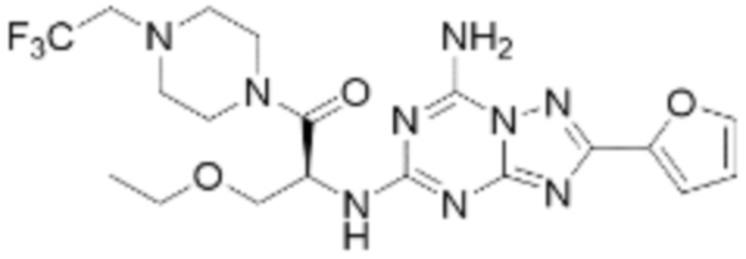
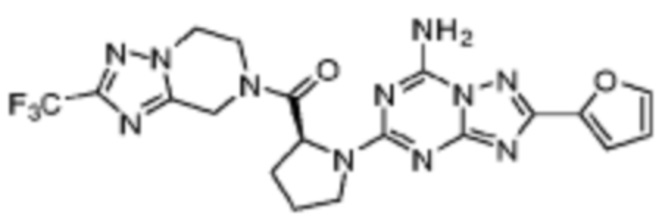
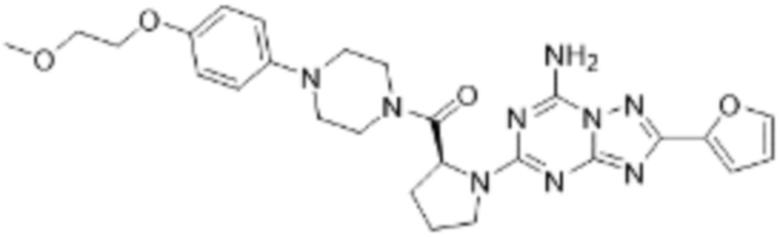
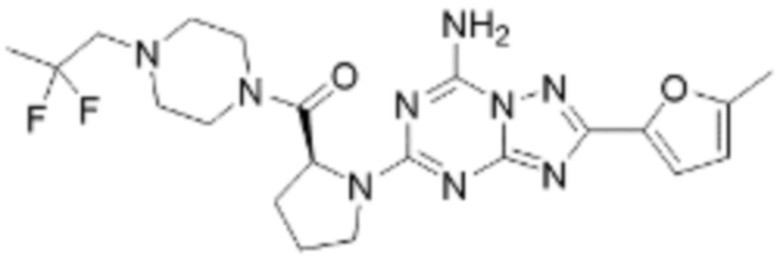
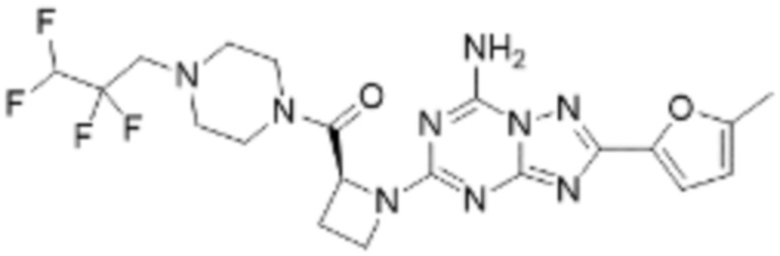
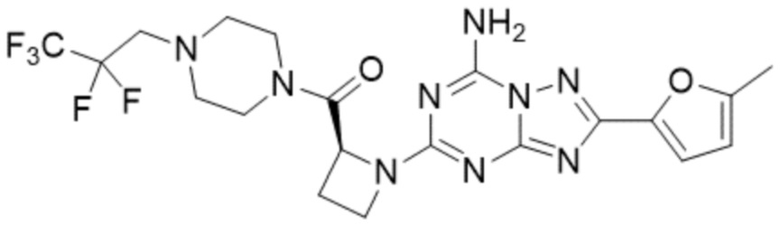
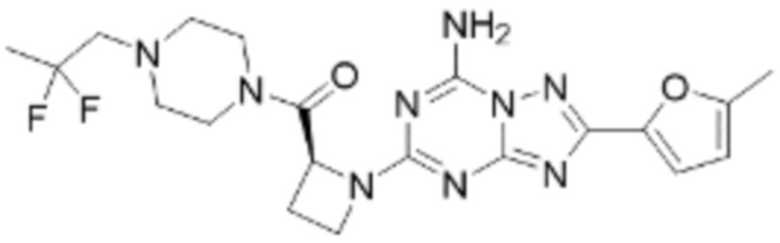
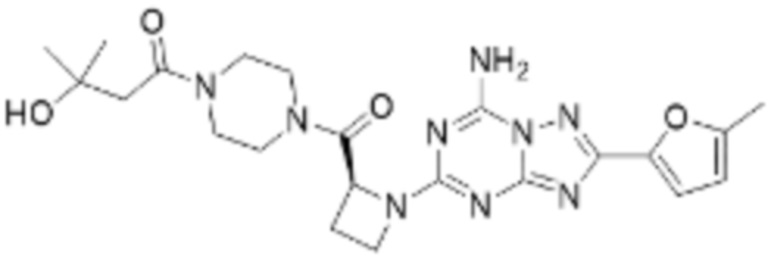
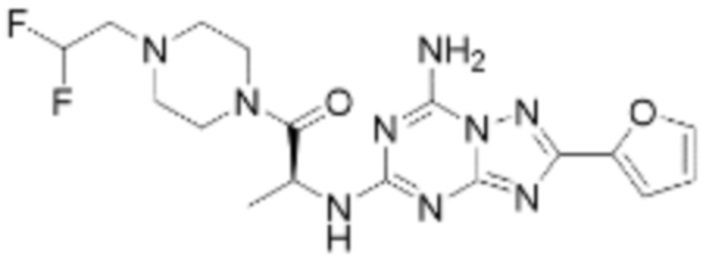
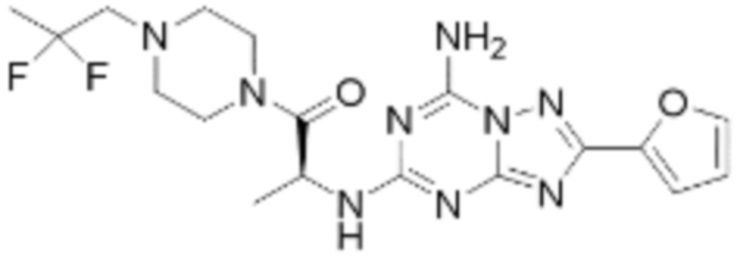
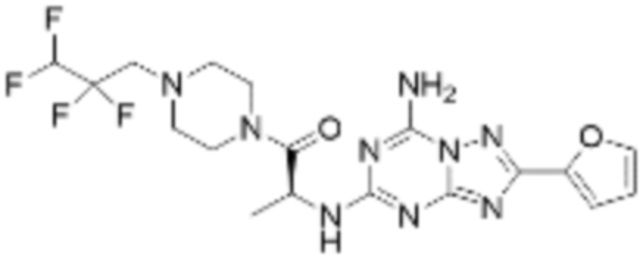
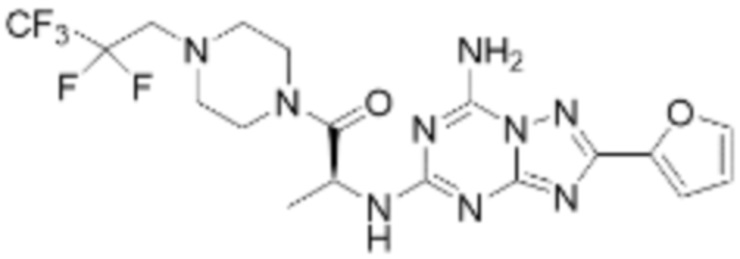
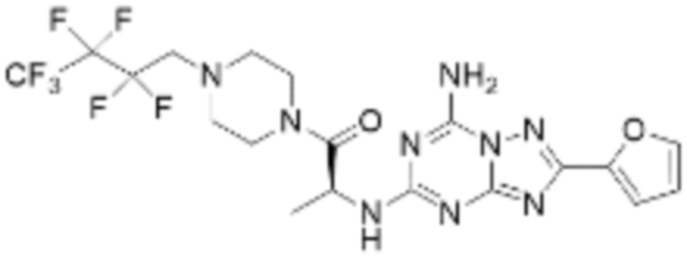
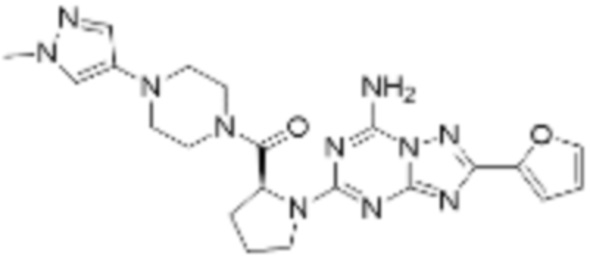
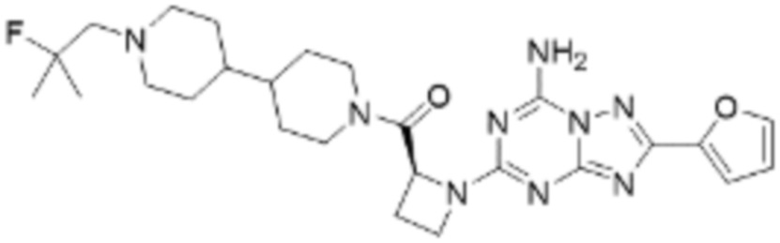
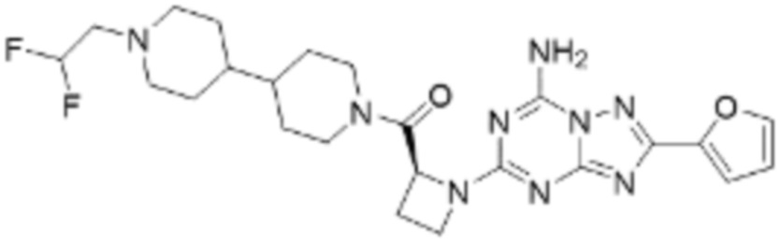
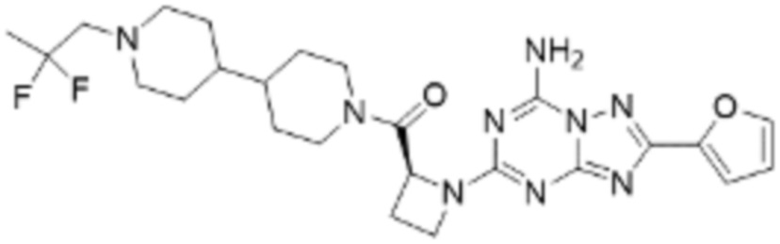
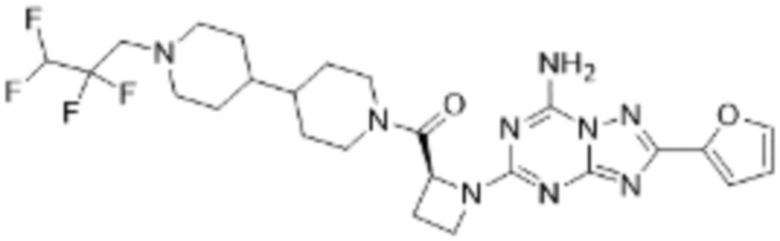
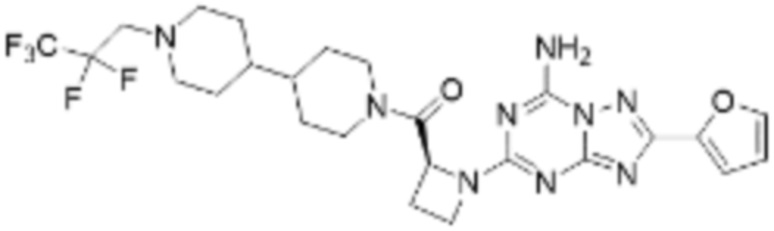
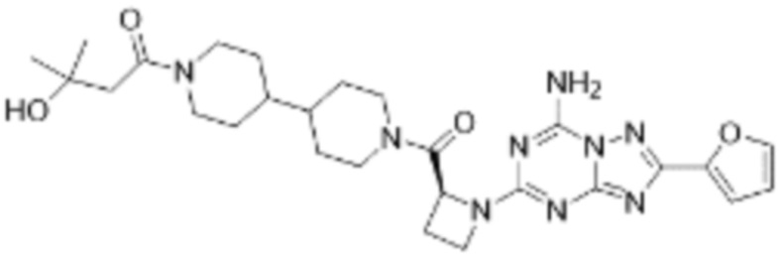
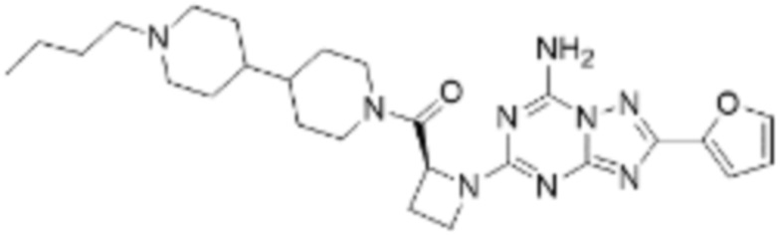
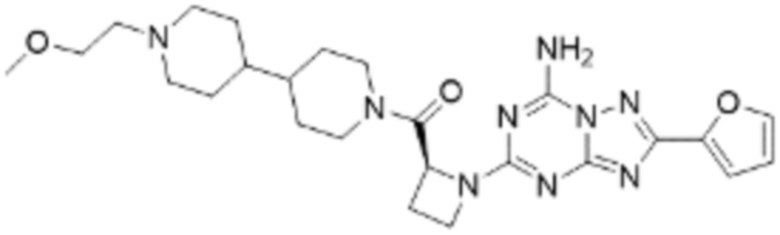
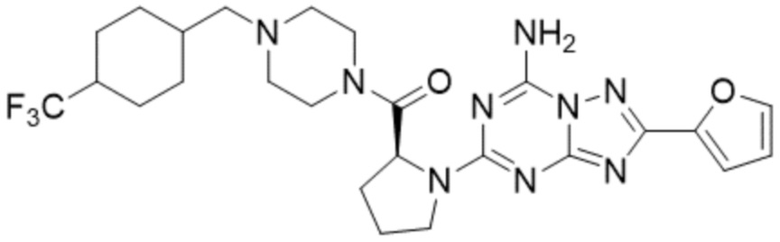
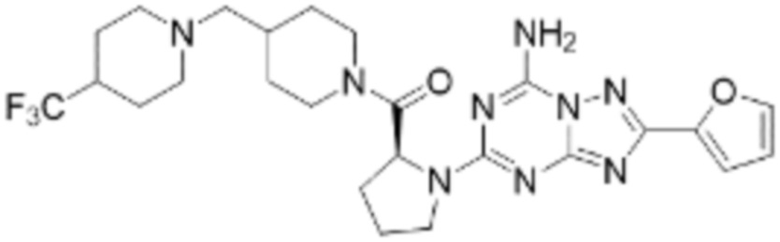
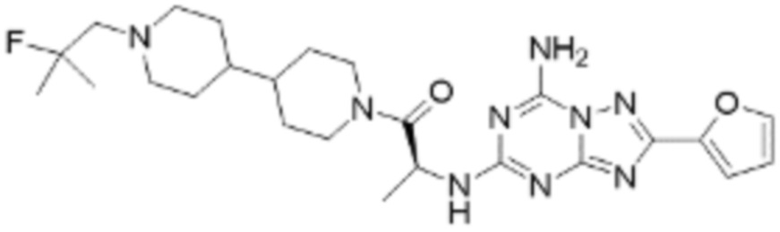
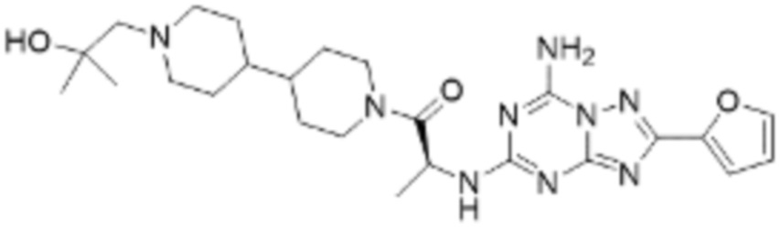
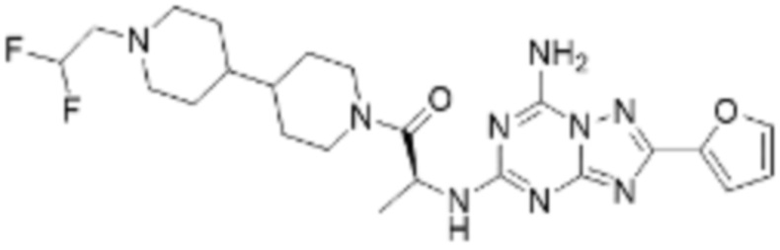
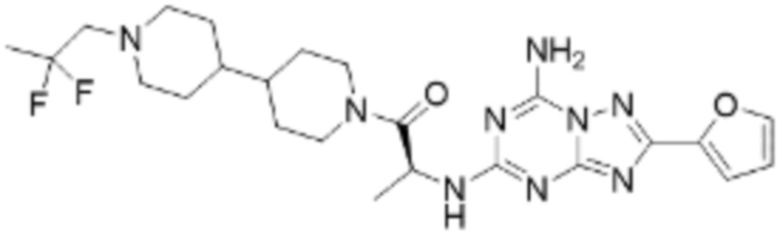
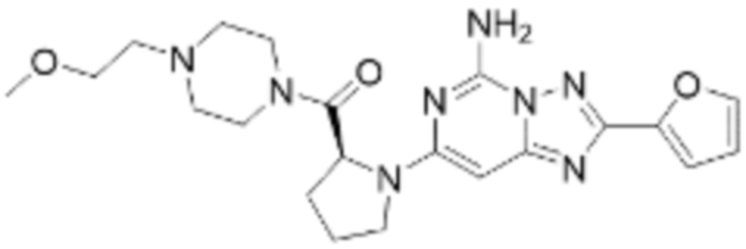
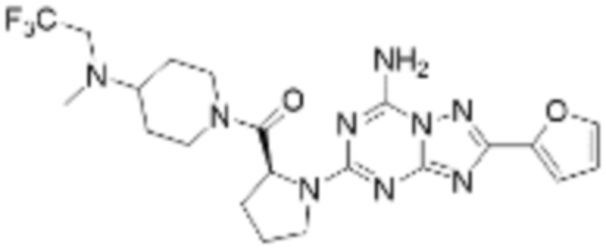
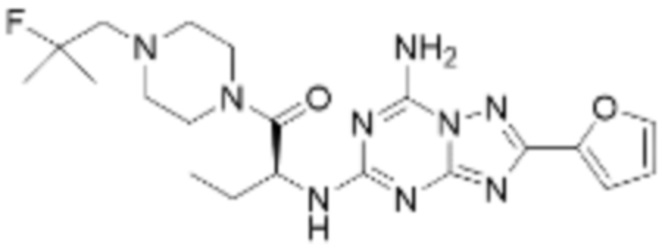
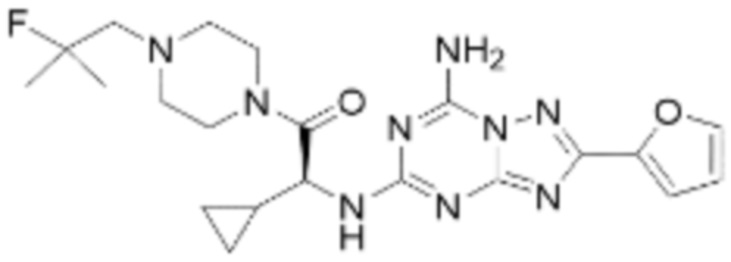
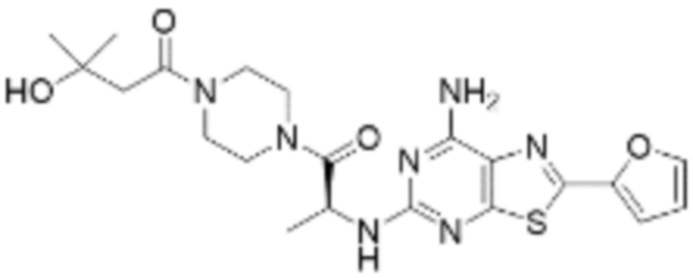
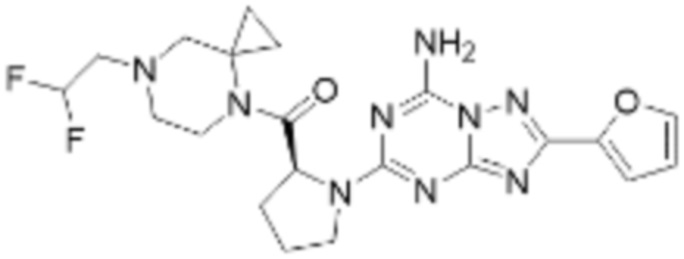
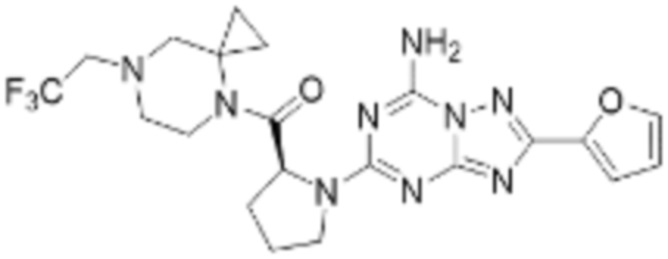
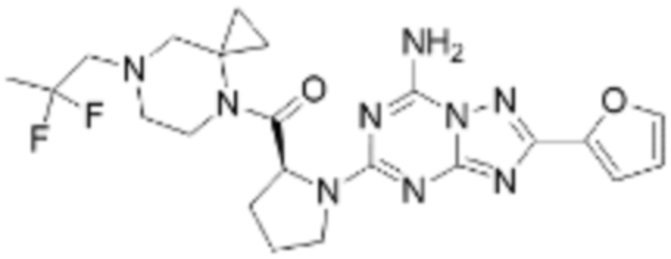
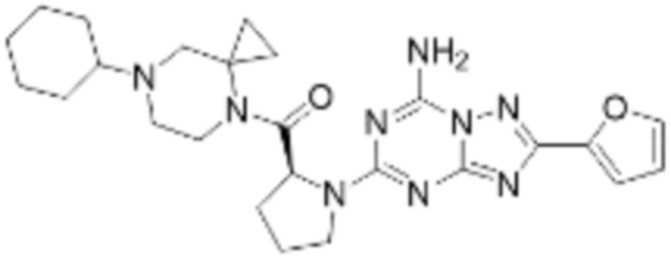
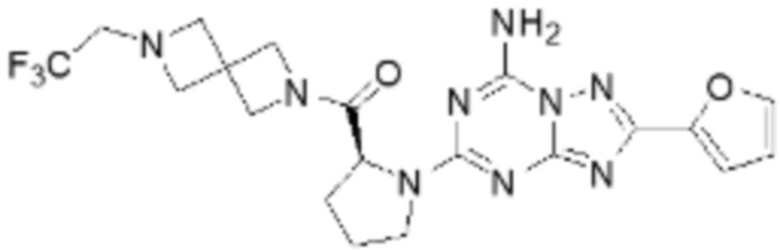
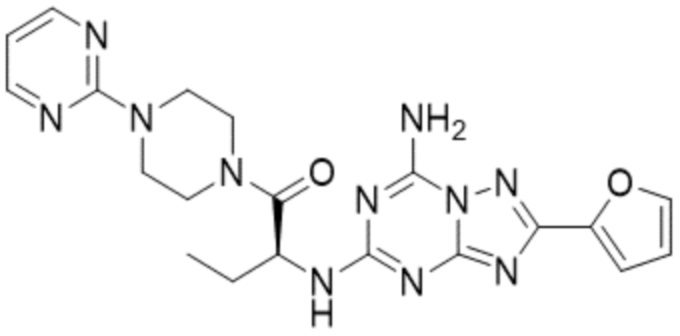
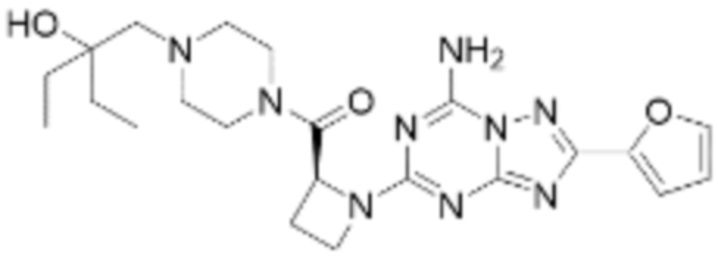
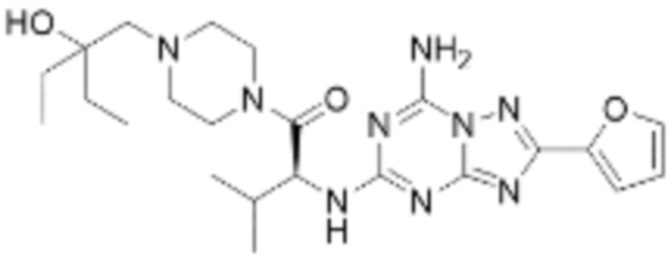
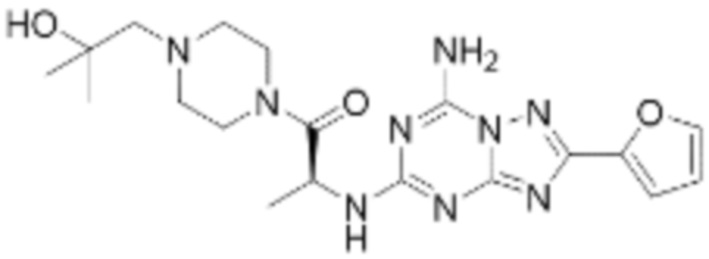
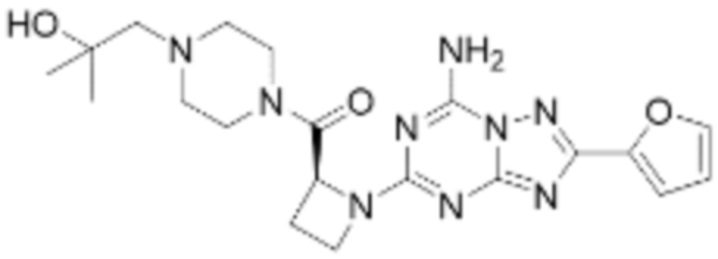
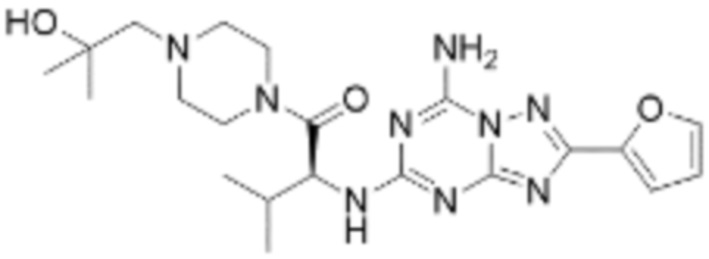
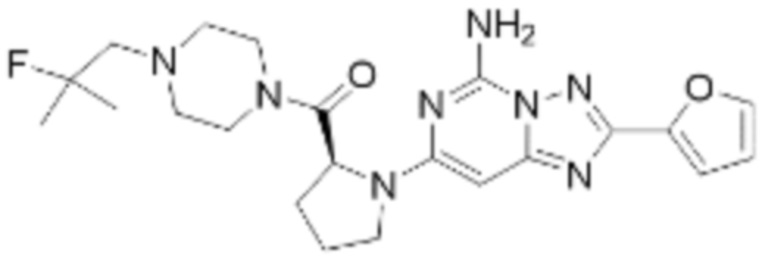
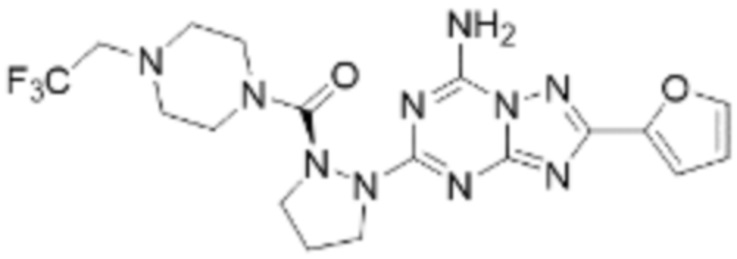
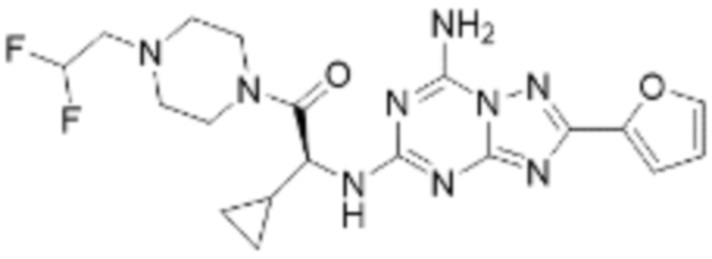
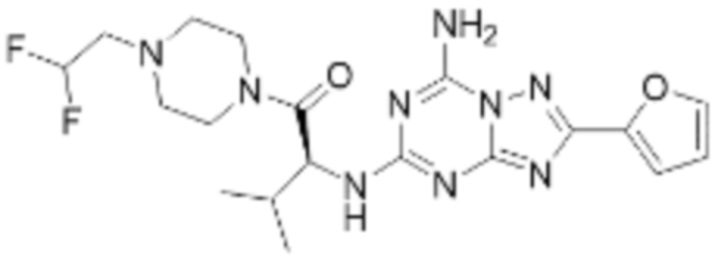
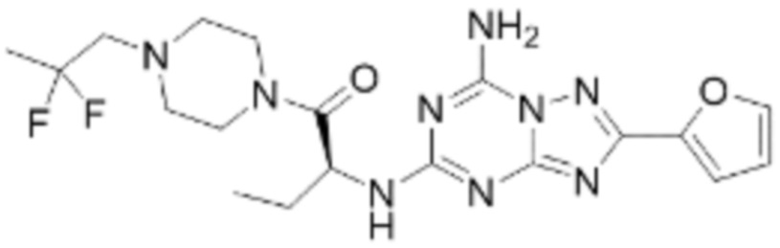
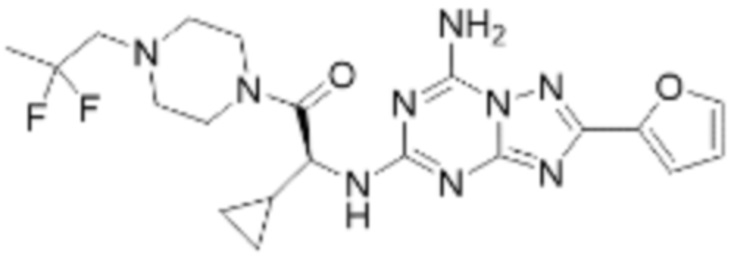
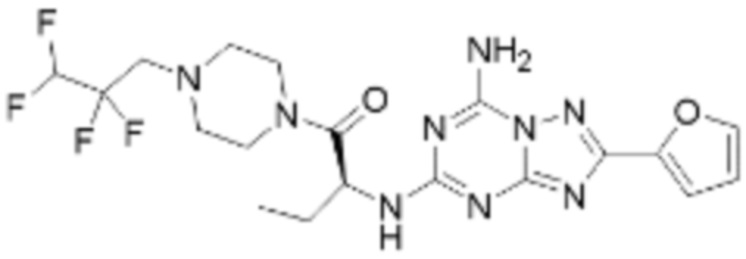
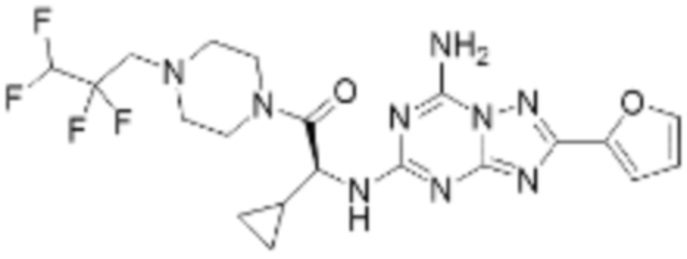
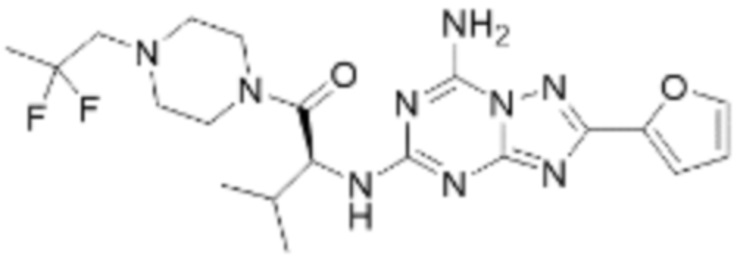
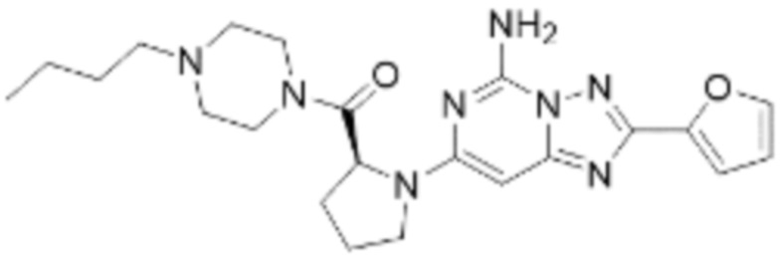
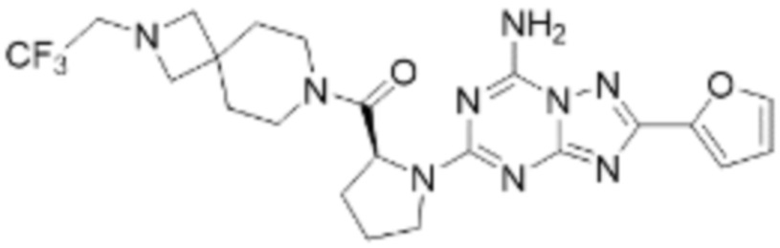
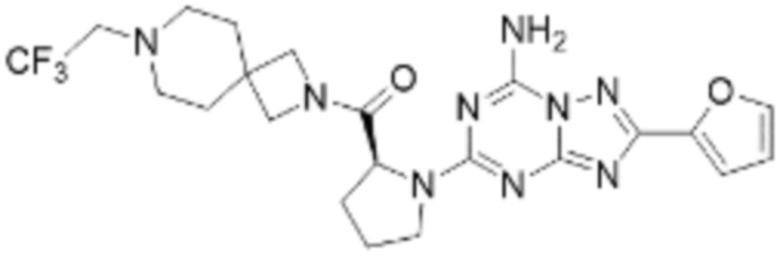
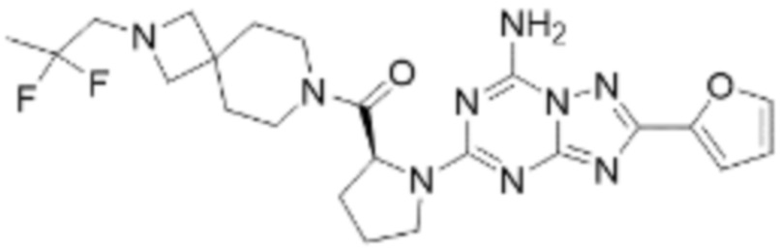
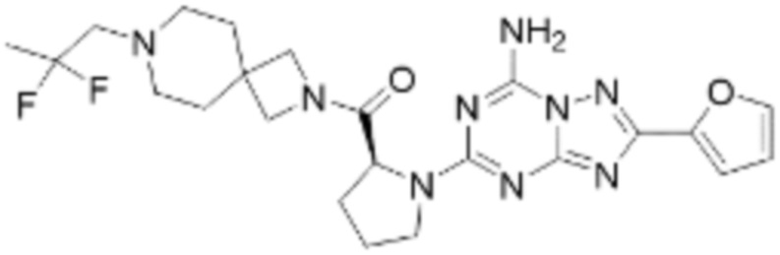
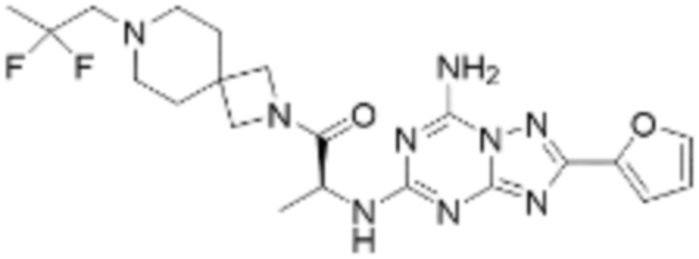
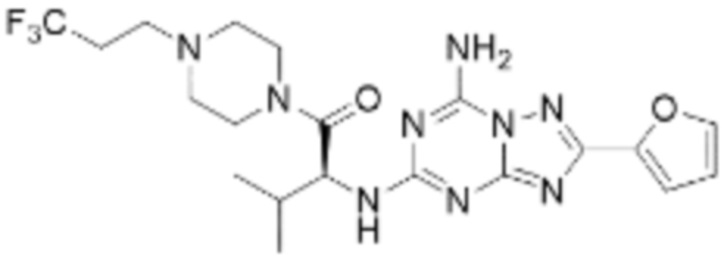
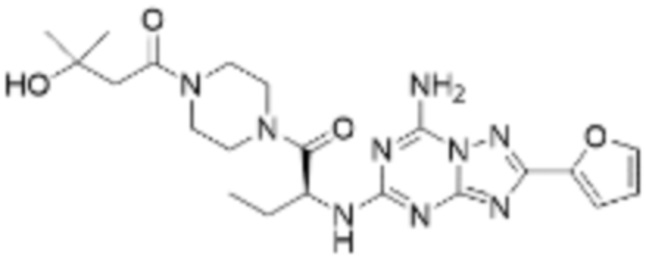
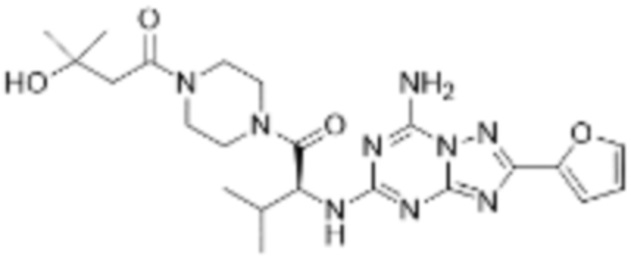
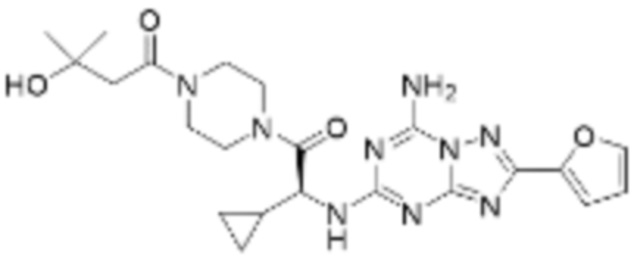
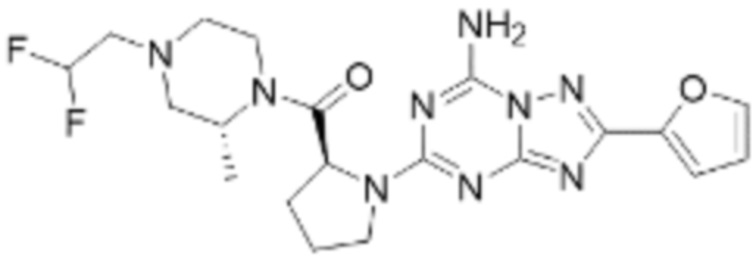
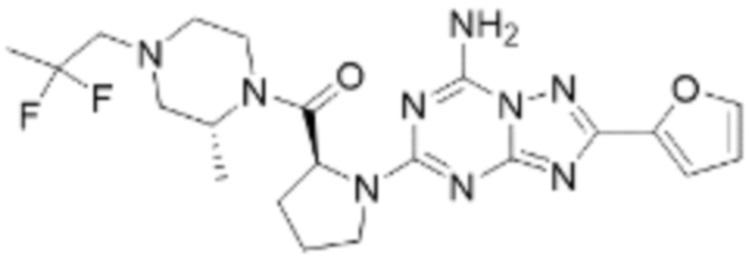
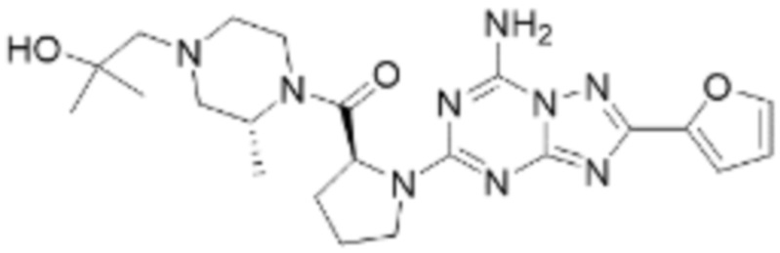
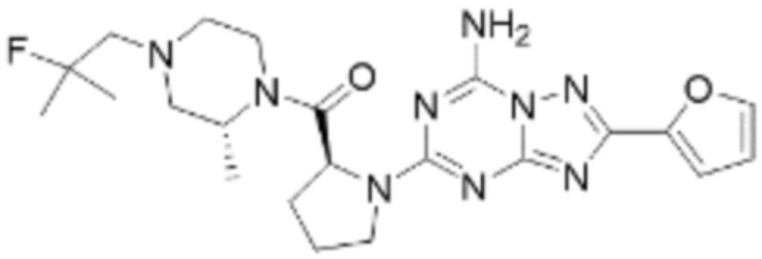
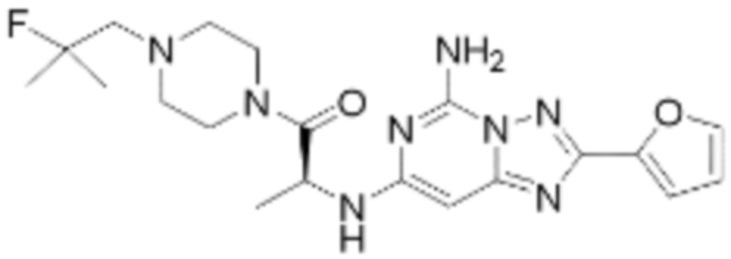
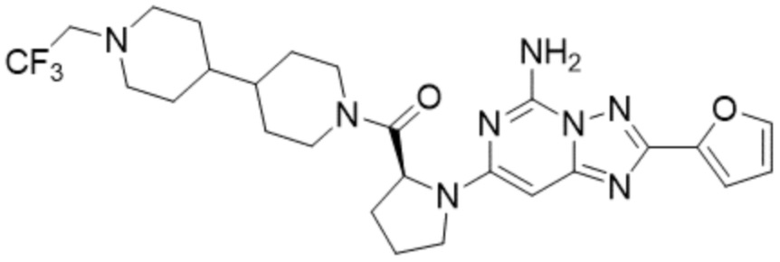
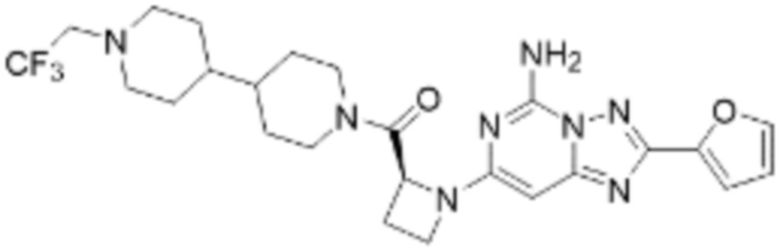
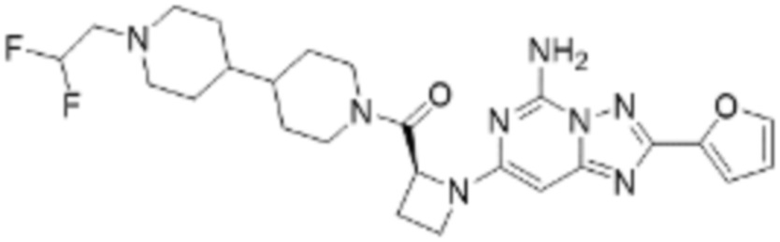
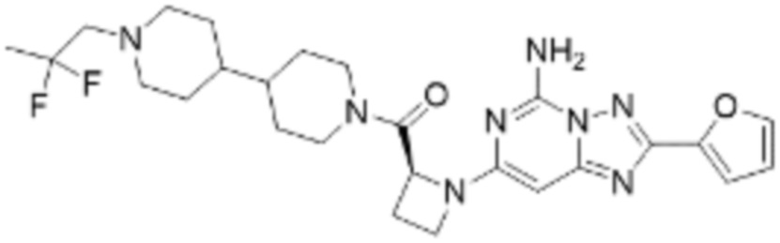
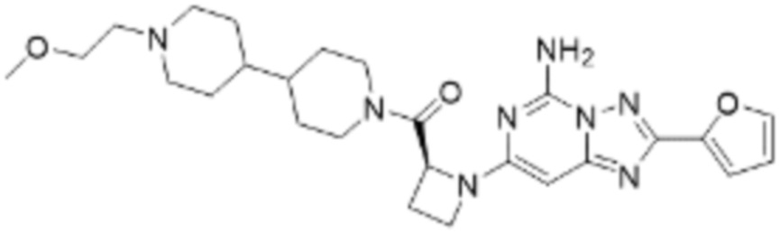
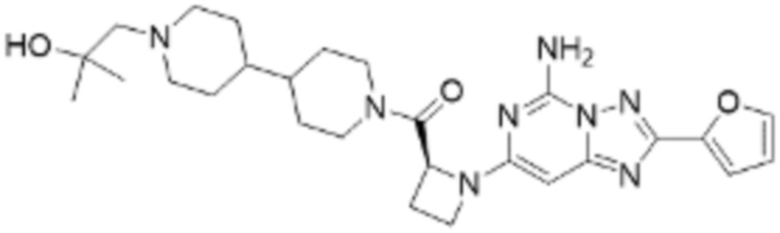
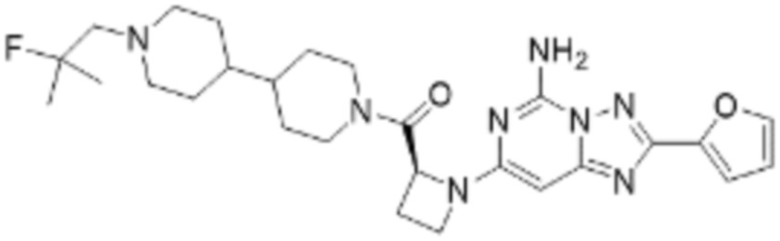
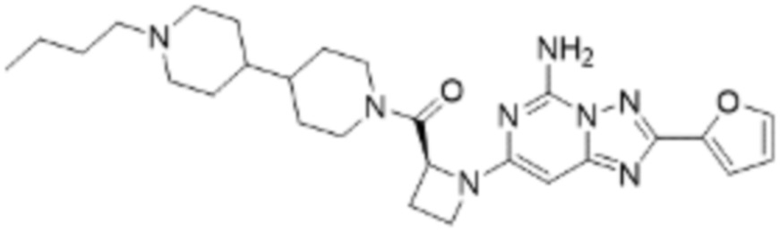
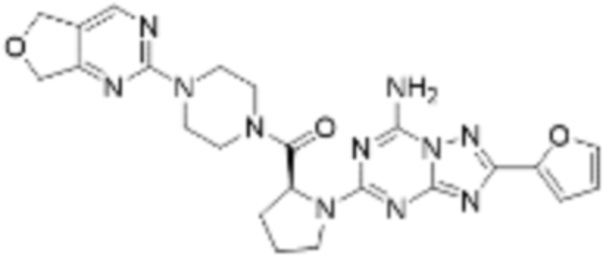
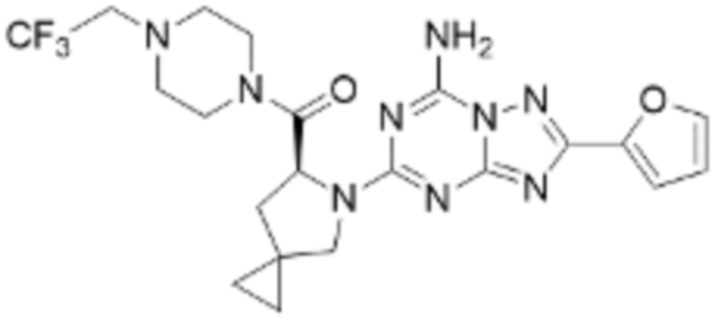
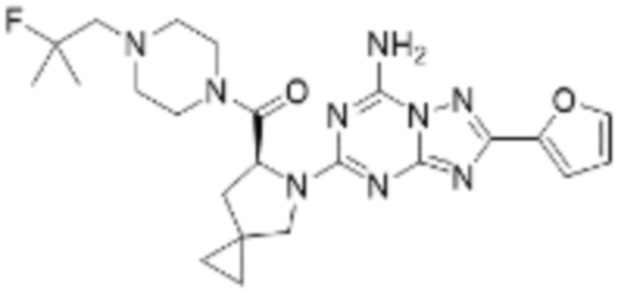
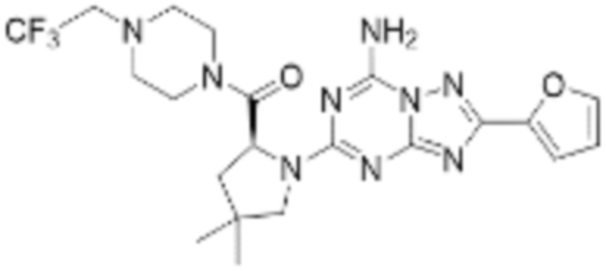
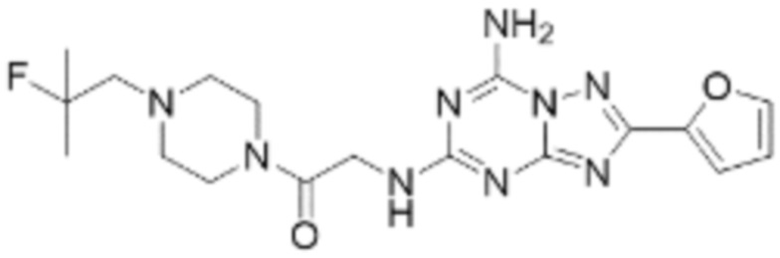
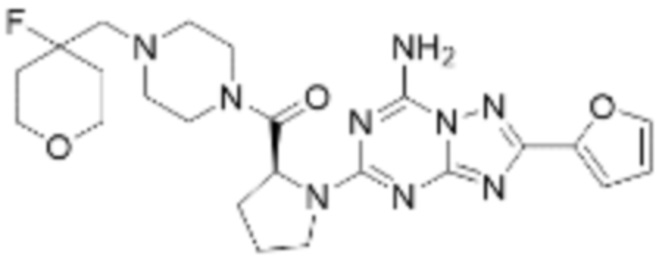
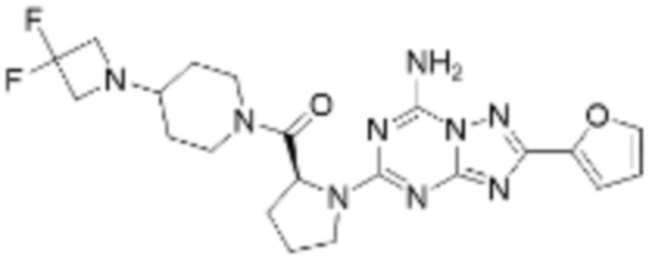
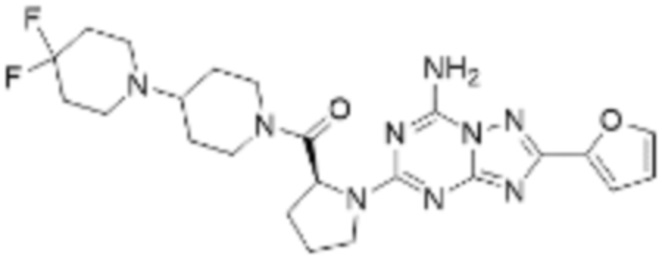
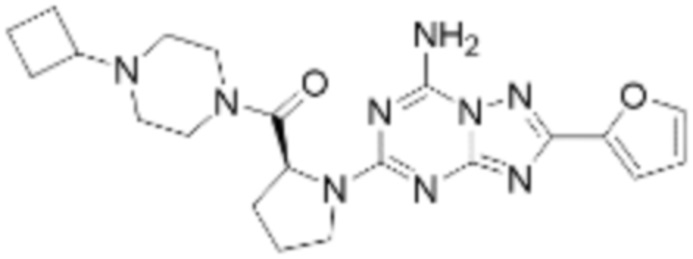
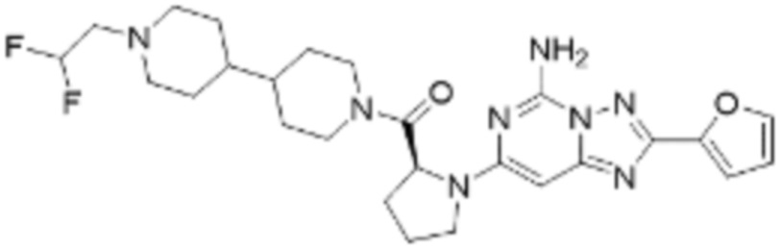
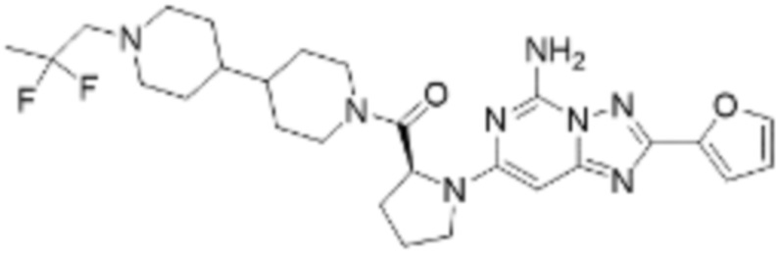
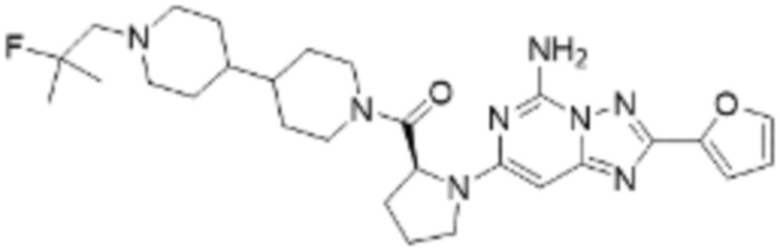
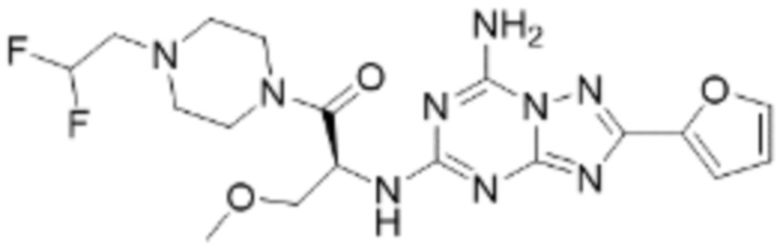
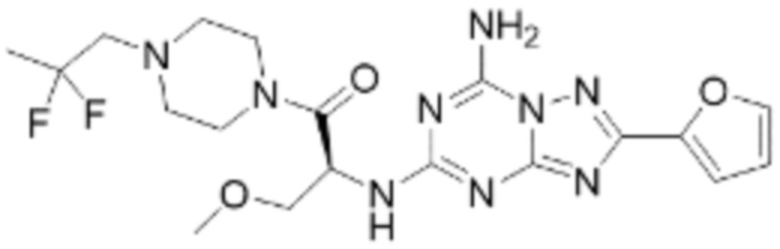
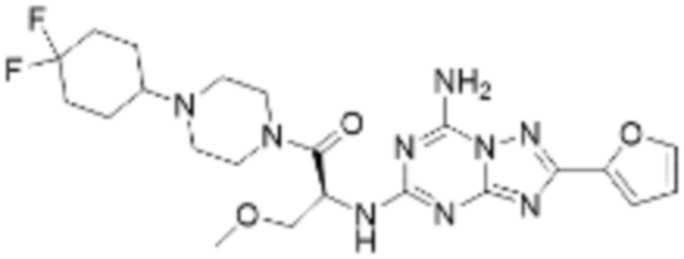
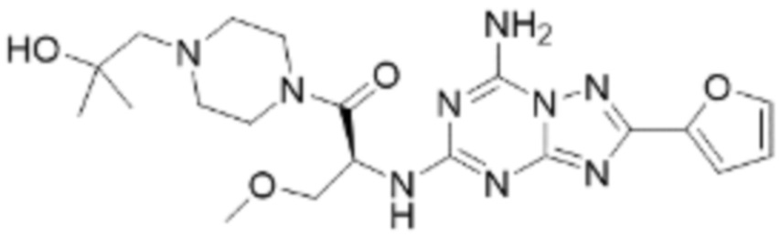
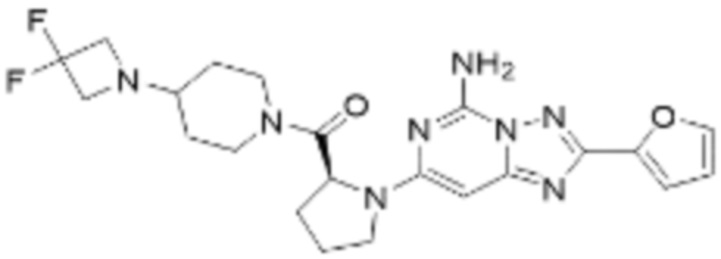
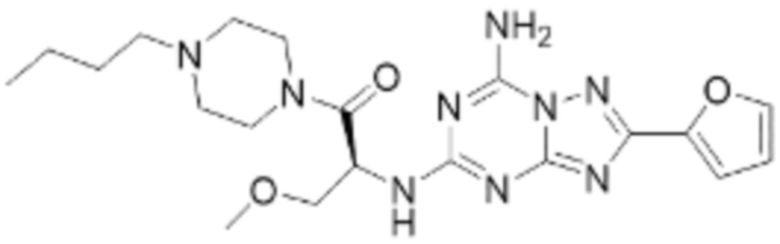
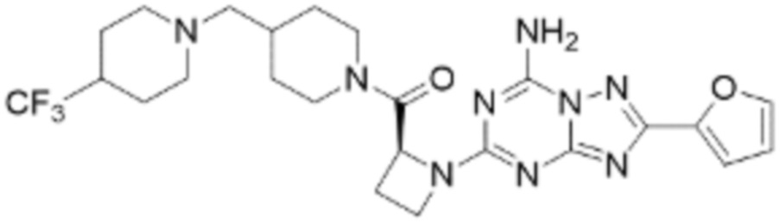
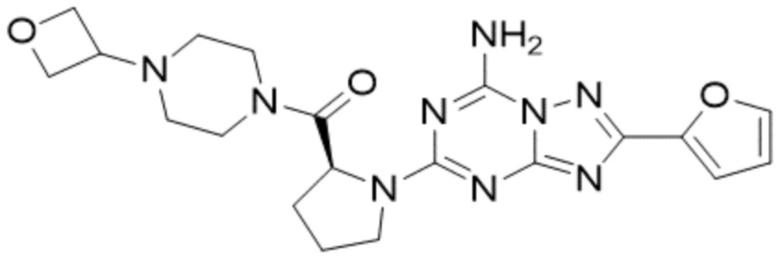
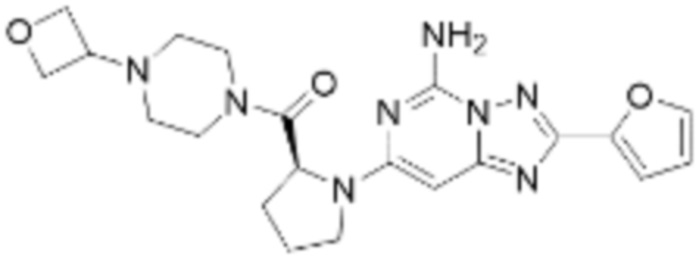
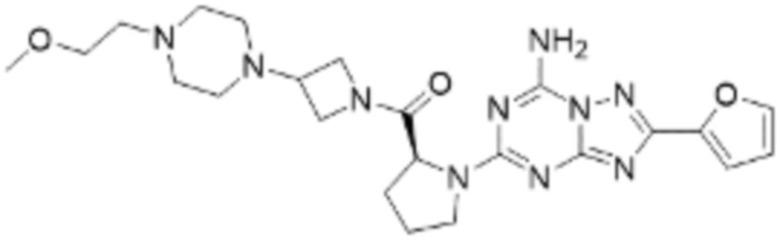
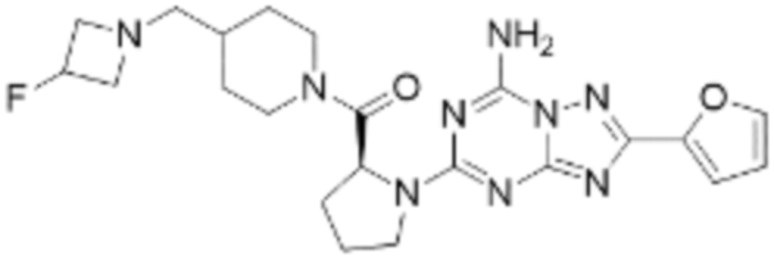
(6) Соединение, представленное формулой 1, его стереоизомеры или его фармацевтически приемлемые соли по любому из вышеуказанных (1), (2), (3), (4) и (5), где соединение, представленное приведенной выше формулой 1, его стереоизомеры или его фармацевтически приемлемые соли могут включать примеры соединений 25, 26, 48, 90, 111, 223, 224, 294, 303, 353 или 371.
В одном варианте осуществления, соединение, представленное приведенной выше формулой 1, его стереоизомеры или его фармацевтически приемлемые соли могут включать примеры соединений 4, 6, 10, 11, 13, 14, 17, 18, 32, 77, 123, 149, 150, 163. , 164, 165, 166, 167 или 169.
Настоящее изобретение может предложить соединение в качестве антагониста рецептора А2а, его стереоизомеры или его фармацевтически приемлемые соли могут представлять собой по меньшей мере одно соединение, выбранное из соединений, показанных в таблице 1 выше.
В настоящем изобретении, «фармацевтически приемлемые соли» могут означать соли, традиционно используемые в фармацевтической промышленности, например, соли неорганических ионов, полученные из кальция, калия, натрия, магния и подобных; соли неорганических кислот, полученные из хлористоводородной кислоты, азотной кислоты, фосфорной кислоты, бромноватой кислоты, йодноватой кислоты, перхлорной кислоты, винной кислоты, серной кислоты и подобных; соли органических кислот, полученные из уксусной кислоты, трифторуксусной кислоты, лимонной кислоты, малеиновой кислоты, янтарной кислоты, щавелевой кислоты, бензойной кислоты, фумаровой кислоты, миндальной кислоты, пропионовой кислоты, молочной кислоты, гликолевой кислоты, глюконовой кислоты, галактуроновой кислоты, глутаминовой кислоты, глутаровой кислоты, глюкуроновой кислоты, аспарагиновой кислоты, аскорбиновой кислоты, угольной кислоты, ванилиновой кислоты, иодистоводородной кислоты и т.д.; соли сульфоновой кислоты, полученные из метансульфоновой кислоты, этансульфоновой кислоты, бензолсульфоновой кислоты, п-толуолсульфоновой кислоты, нафталинсульфоновой кислоты и подобных; соли аминокислот, полученные из глицина, аргинина, лизина и т.п.; соли амина, полученные из триметиламина, триэтиламина, аммиака, пиридина, пиколина и т.п.; и подобные, но типы солей, подразумеваемые в настоящем изобретении, не ограничены перечисленными солями.
В настоящем изобретении, «стереоизомер» может включать диастереомер и оптический изомер (энантиомер), где оптический изомер может включать не только энантиомер, но также смесь энантиомера и даже рацемат. Такой изомер может быть выделен путем расщепления согласно известному уровню техники, например, с помощью колоночной хроматографии, ВЭЖХ или подобной. Альтернативно, стереоизомер каждого соединения, представленного формулой 1, может быть стереоспецифически синтезирован с использованием известного набора оптически чистых исходных материалов и/или реагентов.
В настоящем изобретении, соединение в качестве антагониста рецептора А2а может быть таким же, как список соединений в данном описании, но также включать фармацевтически приемлемое меченное изотопом соединение, где по меньшей мере одно соединение может быть замещено атомом, имеющим тот же атомный номер, но имеющим атомную массу или массовое число, отличающиеся от атомной массы или массового числа, преобладающих в природе. Примеры изотопов, которые могут быть включены в соединение по настоящему изобретению, могут включать: 2H, 3H, изотопы водорода; 11С, 13С, 14С, изотопы углерода; 36Cl, изотоп хлора; 18F, изотоп фтора; 123I, 125I, изотопы йода; 13N, 15N, изотопы азота; 15О, 17О, 18О, изотопы кислорода; 32P, изотоп фосфора; 35S, изотоп серы; и подобные. Определенное меченое изотопом соединение по настоящему изобретению, например соединение с включенными радиоактивными изотопами, может быть полезно при изучении лекарственных средств и/или распределения субстратов в тканях (например, в анализах). Радиоактивный изотоп тритий, то есть 3H, и углерод-14, то есть 14C, могут быть полезны ввиду простоты включения и средств немедленного обнаружения. Замещение более тяжелыми изотопами, например, замещение водорода (1H) на дейтерий (2H), может оказывать превосходный терапевтический эффект при заболеваниях за счет повышения метаболической стабильности, например, увеличения периода полужизни in vivo или снижения дозировки. Замещение позитронно-эмиссионными изотопами, например, 11C, 15F, 18F, 15O, 13N и т. д., может быть полезно при изучении позитронно-эмиссионной томографии (ПЭТ) для изучения занятости субстрата рецептором. Меченое изотопом соединение по настоящему изобретению обычно можно получить обычными методами, известными специалистам в данной области техники, методами, аналогичными тем, которые описаны в формулах реакций и/или примерах и примерах получения, описанных в настоящем описании, с использованием подходящего изотопно-меченого реагента вместо не меченого реагента, как используется в настоящем описании. Соединения, представленные формулой 1, и соединения, примеры которых приведены в настоящем описании, могут включать меченные изотопами соединения этих соединений, такие как, но не ограниченные ими, соединения, включающие дейтерированные и тритированные изотопы и все другие изотопы, обсуждавшиеся выше.
Способ получения соединения, представленного формулой 1
Настоящее изобретение может предложить способ получения соединения, представленного формулой 1, его стереоизомеров или его фармацевтически приемлемых солей.
Соединение, представленное формулой 1, его стереоизомеры или его фармацевтически приемлемые соли могут быть получены в соответствии с любым способом реакции формул 1-10, который может быть модифицирован до уровня, очевидного для специалистов в данной области техники.
В формулах реакций 1-10, приведенных ниже, каждый из R1 - R7, Z1 - Z3, W1 - W4, Q, La, Ra - Rh, a, b, m, n, q, r, t, s, u, y, L1 и L2 может быть по существу таким же, как определено в формуле 1, если не указано иное. «PG» может означать защитную группу и может включать трет-бутилоксикарбонил (Boc), бензилоксикарбонил (Cbz) тому подобную.
[Формула реакции 1]
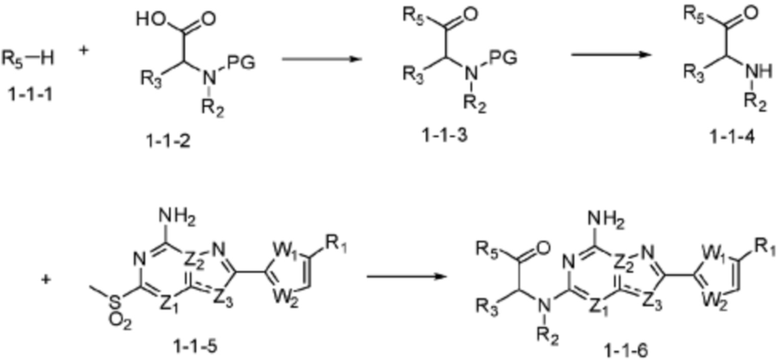
Согласно указанному выше формула реакции 1, соединение формулы 1-1-1 (R5-H) может быть подвергнуто реакции с формулой 1-1-2 с получением соединения формулы 1-1-3, после чего защитная группа (PG) может быть удалена из него с получением соединения формулы 1-1-4, которое затем может быть подвергнуто реакции замещения с соединением формулы 1-1-5, тем самым получая соединение формулы 1-1-6.
В настоящем изобретении, примеры соединений, полученных тем же способом, как показано в вышеуказанной формуле реакции 1 могут включать примеры соединений 1-3, 7-16, 19, 20, 26-29, 37, 40, 41, 48, 55, 57-60, 65, 67, 78-81, 84, 87, 90-98, 107-110, 112-114, 118-128, 134-151, 162-178, 187-195, 199-203, 207-222, 231-236, 238-243, 246-251, 264, 281, 300-310, 321, 336, 338, 339, 346, 349-352, 361, 367, 375, 385-399, 401 или подобные.
[Формула реакции 2]
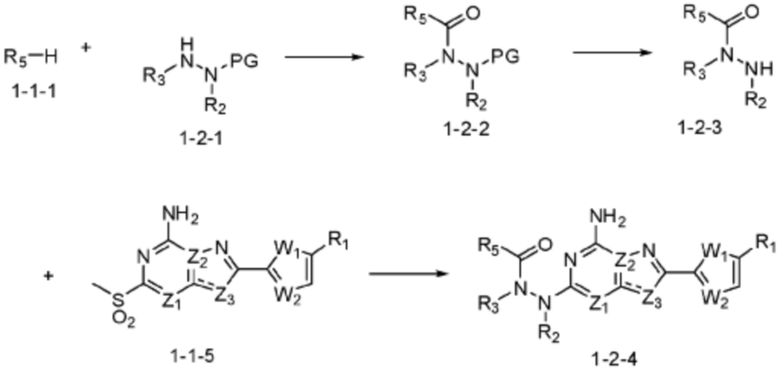
Вышеуказанная формула реакции 2 может показывать способ синтеза соединения пиразолидин-1-карбоксамида, где соединение формулы 1-1-1 и формулы 1-2-1 могут быть подвергнуты реакции с получением соединения формулы 1-2-2, после чего защитная группа может быть удалена из него с получением соединения формулы 1-2-3. После этого, соединение формулы 1-2-4 может быть получено реакцией замещения с соединением формулы 1-1-5.
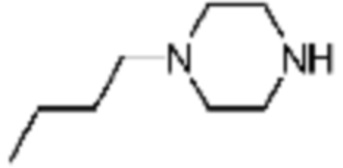
В настоящем изобретении, соединение, полученное тем же способом, который показан в вышеуказанной формул реакции 2 может включать пример соединения 353, и т.д.
[Формула реакции 3]
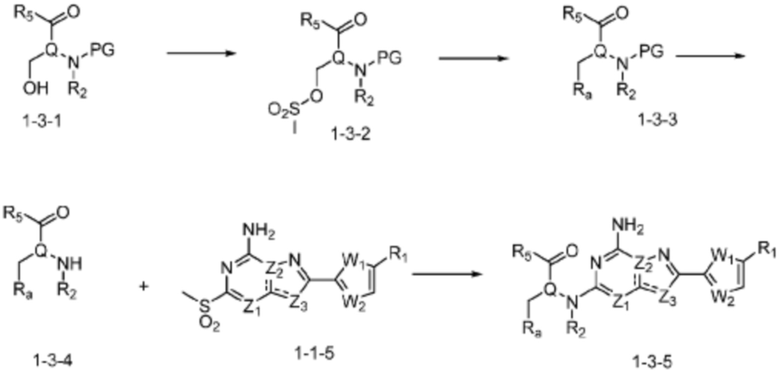
(В вышеуказанной формуле реакции 3, Ra может представлять .)
Согласно вышеуказанной формуле реакции 3, соединение формулы 1-3-1 может быть подвергнуто реакции метансульфонилирования с получением соединения формулы 1-3-2, которое затем может быть подвергнуто реакции замещения с получением соединения формулы 1-3-3, после чего защитная группа может быть удалена из него с получением соединения формулы 1-3-4. После этого, соединение формулы 1-3-5 может быть получено реакцией замещения с соединением формулы 1-1-5.
В настоящем изобретении, соединения, полученные тем же способом, который показан в вышеуказанной формул реакции 3, могут включать примеры соединений 272, 273, и т.д.
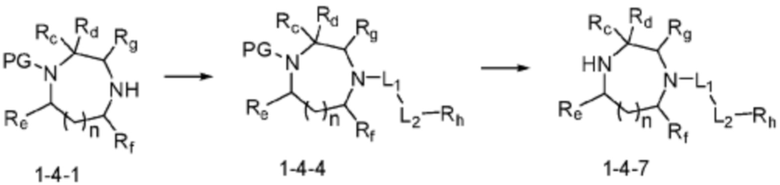
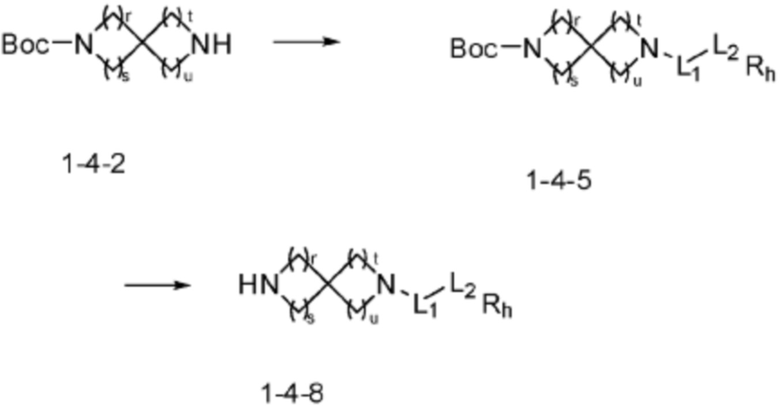
Соединение формулы 1-1-1 представленное R5-H в каждой из вышеуказанных формул реакции 1 и 2 может быть получено по способам, описанным в формулах реакции 4a - 4c ниже и формулах реакции 5a, 5b, и 6-8 ниже. Другими словами, соединение формулы 1-1, представленное R5-H в каждой из вышеуказанных формул реакции 1 и 2, может представлять собой соединения формул 1-4-7, 1-4-8, или 1-4-9 ниже, или может представлять собой соединения формул 1-5-5, 1-5-6, 1-6-3, 1-7-4, 1-7-5, или 1-8-3.
[Формула реакции 4a]
[Формула реакции 4b]
[Формула реакции 4c]
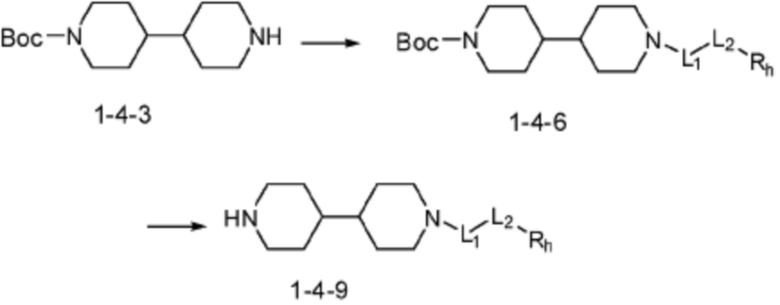
Согласно формулам реакции 4a - 4c, заместитель (-L1-L2-Rh) может быть введен в соединение формулы 1-4-1, 1-4-2 или 1-4-3 с получением соединения формулы 1-4-4, 1-4-5 или 1-4-6, после чего защитная группа (PG, Boc) может быть удалена из него с получением соединения формулы 1-4-7, 1-4-8 или 1-4-9 в качестве соединения формулы 1-1-1(R5-H).
В настоящем изобретении, примеры соединений, которые могут быть синтезированы согласно способу, показанному в вышеуказанной формуле реакции 1 или 2 с применением R5-H, полученного тем же способом, как показано в вышеуказанных формулах реакции 4a - 4c, могут включать примеры соединений 30-32, 42, 44, 49-52, 56, 73-77, 85, 86, 88, 129, 152-161, 179-184, 185, 196, 197, 204-206, 223-230, 237, 244, 252-263, 265-271, 274-280, 287-299, 311-320, 323-329, 333-335, 337, 340-345, 347, 348, 354-360, 362-366, 368-373, 376-381, 383, 384, 392-394, 396-398, 403, 404 или подобные.
Заместитель (-L1-L2-Rh), включенный в R5 в каждой из вышеуказанных формул реакции 4a - 4c, может быть введен в кольцо, включающее N с применением реакции сочетания с соединением, имеющим соединение галогенида, такое как хлорангидрид, оксалилхлорид, сульфонилхлорид, карбонилхлорид и т.д., или уходящую группу, такую как o-толуолсульфонилфторид, и т.д., реакции Бухвальда-Хартвига, реакции размыкания кольца через амидное сочетание и эпоксидный гидролиз, реакции восстановительного амидирования, и т.д. Например, введение может быть осуществлено в кольцо, содержащее N, реакцией алкилирования или арилирования с применением соединения галогенида, имеющего структуру X-L1-L2-Rh (где X представляет собой галоген).
[Формула реакции 5a]
[Формула реакции 5b]
(каждый Rx в вышеуказанных формулах реакции 5a и 5b независимо может представлять собой -C1-C7 алкилен-, и алкилен из Rx может означать двухвалетный заместитель прямого или разветвленного алкила.)
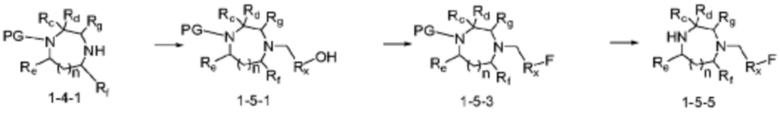
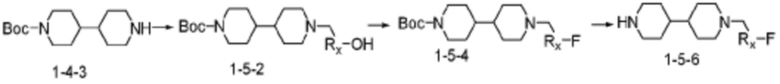
Согласно вышеуказанным формулам реакции 5a и 5b, заместитель может быть введен в соединение формулы 1-4-1 или 1-4-3 с получением гидроксисоединения формулы 1-5-1 или 1-5-2, которое может быть затем подвергнуто реакции фторирования с получением соединения формулы 1-5-3 или 1-5-4, после чего защитная группа (PG, Boc) может быть удалена из него с получением соединения формулы 1-5-5 или 1-5-6 в качестве соединения формулы 1-1-1 (R5-H).
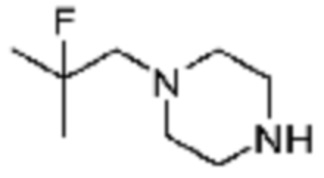
В настоящем изобретении, примеры соединений с R5, замещенным как получено тем же способом, который показан в вышеуказанной формуле реакции 5a или 5b, могут включать примеры соединений 43, 245, 322, 332, 374, 382, 395 или подобные.
[Формула реакции 6]
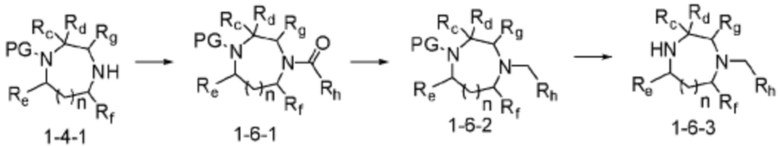
Согласно вышеуказанной формуле реакции 6, заместитель может быть введен в соединение формулы 1-4-1 с получением соединения амида формулы 1-6-1, которое может быть затем подвергнуто реакции восстановления с получением соединения формулы 1-6-2. Защитная группа может быть удалена с соединения формулы 1-6-2 с получением соединения формулы 1-6-3 в качестве соединения формулы 1-1-1 (R5-H).
В настоящем изобретении, примеры соединений, которые могут быть синтезированы способом из вышеуказанной формулы реакции 1 или 2 с применением R5-H, полученного тем же способом, который показан в вышеуказанной формуле реакции 6, могут включать примеры соединений 282-286, 330, 389 или подобные.
[Формула реакции 7]
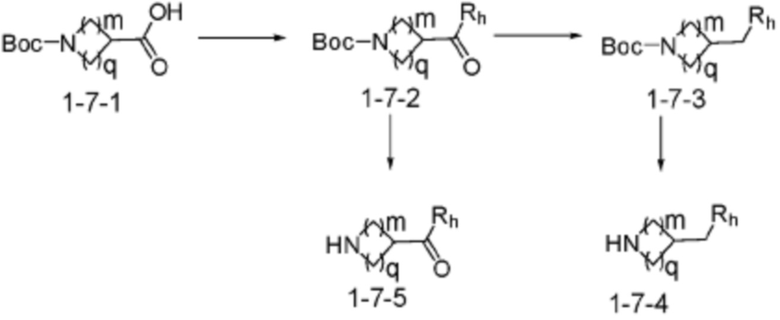
Согласно вышеуказанной формуле реакции 7, амид может быть введен в соединение формулы 1-7-1 с получением соединения формулы 1-7-2, которое затем может быть подвергнуто реакции восстановления с получением соединения формулы 1-7-3. Защитная группа может быть удалена из соединения формулы 1-7-3 с получением соединения формулы 1-7-4 в качестве соединения формулы 1-1-1 (R5-H). И, защитная группа может быть удалена из соединения формулы 1-7-2 as с получением соединения формулы 1-7-5 в качестве соединения формулы 1-1-1 (R5-H).
В настоящем изобретении, примеры соединений, которые могут быть синтезированы способом из вышеуказанной формулы реакции 1 или 2 с применением R5-H, полученного тем же способом, который показан в вышеуказанной формуле реакции 7, могут включать примеры соединений 131 t-o 133, 331, 402, 406 или подобные.
[Формула реакции 8]
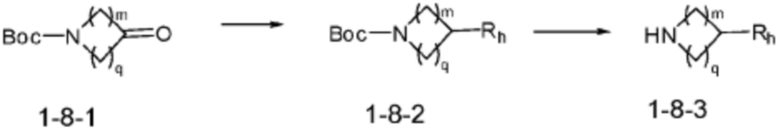
Согласно вышеуказанной формуле реакции 8, соединение формулы 1-8-2 может быть получено реакцией восстановительного аминирования с соединением формулы 1-8-1. Защитная группа (Boc) может быть удалена из соединения формулы 1-8-2 с получением соединения формулы 1-8-3.
В настоящем изобретении, примеры соединений, которые могут быть синтезированы способом из вышеуказанной формулы реакции 1 или 2 с применением R5-H, полученного тем же способом, который показан в вышеуказанной формуле реакции 8, могут включать примеры соединений 186, 390, 391, 400, 405 или подобные.
[Формула реакции 9]
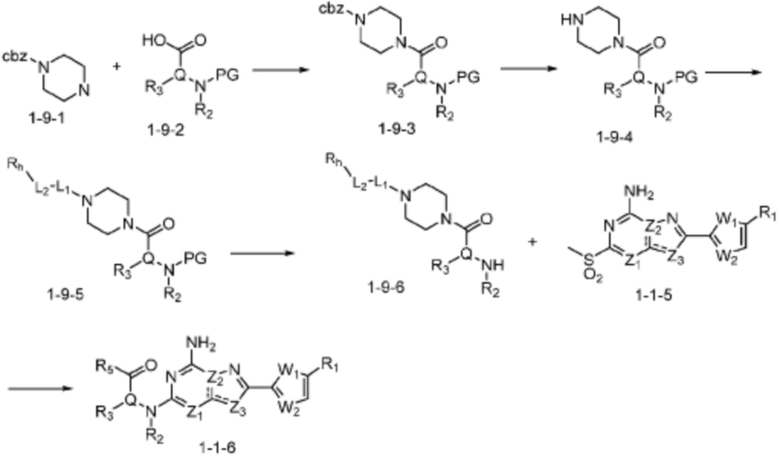
В вышеуказанной формуле реакции 9, соединение формулы 1-9-1 и формулы 1-9-2 могут быть подвергнуты реакции с получением соединения формулы 1-9-3, после чего защитная группа (Cbz) может быть удалена из N пиперазина с получением соединения формулы 1-9-4. Заместитель может быть введен в соединение формулы 1-9-4 с получением соединения формулы 1-9-5, после чего защитная группа (PG) может быть удалена из N амина с получением соединения формулы 1-9-6. После этого, соединение формулы 1-1-6 может быть получено реакцией замещения с соединением формулы 1-1-5.
В настоящем изобретении, соединения, полученные вышеуказанной формулой реакции 9, могут включать примеры соединений 4-6, 17, 18, 21-25, 45-47, 61-64, 66, 68-72, 82, 89, 99-106, 111, 115-117, и т.д.
[Формула реакции 10]
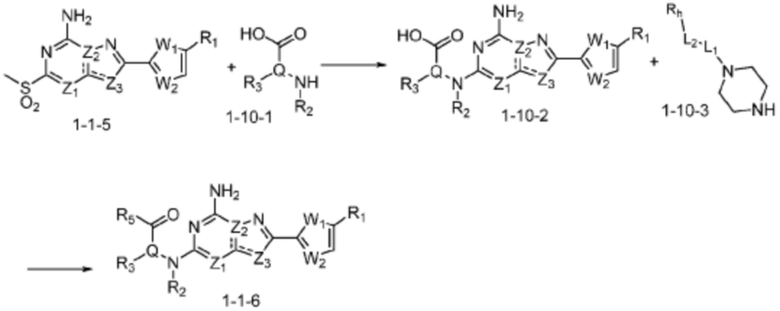
В вышеуказанной формуле реакции 10, соединение формулы 1-10-1 может быть введено в соединение формулы 1-1-5 с получением соединения формулы 1-10-2,после чего соединение формулы 1-10-2 и соединение формулы 1-10-3 могут быть подвергнуты реакции, тем самым получая соединение формулы 1-1-6.
В настоящем изобретении, соединения, полученные вышеуказанной формулой реакции 10 могут включать примеры соединений 53, 54, и т.д.
Композиция, включающая соединение, представленное формулой 1, ее применение и терапевтический способ с ее использованием
Настоящее изобретение может предложить фармацевтическую композицию, включающую соединение, представленное вышеуказанной формулой 1, соединения, примеры которых приведены в настоящем описании, их стереоизомеры или их фармацевтически приемлемые соли в качестве активного ингредиента.
Кроме того, настоящее изобретение может предложить фармацевтическую композицию для лечения или профилактики заболеваний, связанных с рецептором А2а, включающую соединение, представленное вышеуказанной формулой 1, соединения, примеры которых приведены в настоящем описании, их стереоизомеры или их фармацевтически приемлемые соли в качестве активного ингредиента.
Заболевания, связанные с рецептором А2а, могут представлять собой рак или воспалительные заболевания.
Рак может быть по меньшей мере одним из рака легких, рака желудка, рака яичников, рака предстательной железы, рака пищевода, рака желудочно-кишечного тракта, рака поджелудочной железы, колоректального рака, рака почки, рака яичка, рака мочевого пузыря, рака молочной железы, рака матки, рака шейки матки, рака головы и шеи, рака крови, рака костей, рака печени, рака щитовидной железы, рака кожи, лимфомы, лейкоза, миеломы, саркомы и вирус-ассоциированного рака.
Воспалительным заболеванием может быть по меньшей мере одно заболевание, выбранное из ревматоидного артрита, рассеянного склероза, болезни Крона, язвенного колита, заболевания «трансплантат против хозяина», системной красной волчанки, синдрома токсического шока, остеоартрита и инсулинозависимого диабета.
Для введения, фармацевтическая композиция по настоящему изобретению может дополнительно включать по меньшей мере один тип фармацевтически приемлемого носителя в дополнение к соединению, представленному вышеуказанной формулой 1, его стереоизомерам или его фармацевтически приемлемым солям. Фармацевтически приемлемый носитель, используемый в настоящем документе, может включать солевой раствор, стерилизованную воду, раствор Рингера, забуференный солевой раствор, раствор декстрозы, раствор мальтодекстрина, глицерин, этанол и смесь по меньшей мере одного их ингредиента, и с добавлением других обычных добавок, таких как антиоксиданты, буферные растворы, бактериостатические агенты и т. д., при необходимости. Кроме того, разбавители, диспергирующие агенты, поверхностно-активные вещества, связующие агенты и смазывающие агенты могут быть дополнительно добавлены для составления инъекционных дозированных форм, таких как водные растворы, суспензии, эмульсии и т.д., пилюль, капсул, гранул или таблеток. Таким образом, композиция по настоящему изобретению может представлять собой пластыри, жидкие лекарственные средства, пилюли, капсулы, гранулы, таблетки, суппозитории и т.д. Препараты могут быть приготовлены в соответствии с обычным способом, используемым для составления в данной области техники, или способом, описанным в Remington's Pharmaceutical Science (latest edition), Mack Publishing Company, Easton PA, и композиция может быть составлена в различные препараты в зависимости от каждого заболевания или ингредиента.
Композиция по настоящему изобретению может вводиться перорально или парентерально (например, применяться внутривенно, подкожно, внутрибрюшинно или местно) в соответствии с целевым способом, при котором ее дозировка может варьироваться в диапазоне, зависящем от веса, возраста, пола, состояния здоровья и диеты пациента, времени введения, способа введения, скорости выведения, тяжести заболевания и подобных. Соединение, представленное формулой 1 по настоящему изобретению, можно вводить один или несколько раз в день, разделив суточную дозу соединения, но не обязательно ограничиваясь этим.
В дополнение к соединению, представленному вышеприведенной формулой 1, соединению, приведенному в качестве примера в настоящем описании, его стереоизомерам или его фармацевтически приемлемым солям, фармацевтическая композиция по настоящему изобретению может дополнительно включать по меньшей мере один ингредиент, который может демонстрировать такие же или подобные медицинские эффекты или может обеспечить синергию медицинских эффектов в комбинации.
Настоящее изобретение может предложить способ лечения или профилактики заболеваний, связанных с аденозиновым рецептором А2а, включающий введение терапевтически эффективного количества соединения, представленного вышеприведенной формулой 1, соединения, приведенного в качестве примера в настоящем описании, его стереоизомеров или его фармацевтически приемлемых солей; или фармацевтической композиции, включающей его в качестве эффективного ингредиента, субъекту, нуждающемуся в этом.
Используемый в настоящем документе термин «терапевтически эффективное количество» может относиться к количеству соединения, соединения, представленного в качестве примера в настоящем описании, его стереоизомеров или его фармацевтически приемлемых солей, которые эффективны при лечении или профилактике заболеваний, связанных с аденозиновым рецептором А2а. Заболевания, связанные с аденозиновым рецептором А2а, могут представлять собой рак или воспалительные заболевания.
В настоящем изобретении, термин «субъект» может относиться к млекопитающим, включая человека, и термин «введение» может относиться к предоставлению заранее определенного материала субъекту любым подходящим способом. Специалистам в данной области техники очевидно, что терапевтически эффективная дозировка и количество введения эффективного ингредиента по настоящему изобретению может варьироваться в зависимости от желаемого эффекта.
В настоящем изобретении термин «профилактика» может относиться к задержке возникновения заболевания, нарушения или состояния. Если возникновение заболевания, нарушения или состояния задерживается на ожидаемый период времени, профилактику можно считать завершенной.
В настоящем изобретении, термин «лечение» может относиться к лечению, которое частично или полностью уменьшает, улучшает, облегчает, подавляет или задерживает возникновение определенного заболевания, нарушения и/или состояния, уменьшает его тяжесть или снижает частоту возникновения по меньшей мере одного его симптома или свойства.
Настоящее изобретение также может предложить применение соединения, представленного формулой 1, соединения, приведенного в качестве примера в настоящем описании, его стереоизомеров или его фармацевтически приемлемых солей; или фармацевтической композиции, включающей его в качестве эффективного ингредиента для лечения или профилактики заболеваний, связанных с аденозиновым рецептором А2а. Заболевания, связанные с аденозиновым рецептором А2а, могут представлять собой рак или воспалительные заболевания.
Настоящее изобретение также может предложить применение соединения, представленного формулой 1, соединения, приведенного в качестве примера в настоящем описании, его стереоизомеров или его фармацевтически приемлемых солей; или фармацевтической композиции, включающей его в качестве эффективного ингредиента, при приготовлении лекарственного средства для лечения или профилактики заболеваний, связанных с аденозиновым рецептором А2а. Заболевания, связанные с аденозиновым рецептором А2а, могут представлять собой рак или воспалительные заболевания.
Объекты, упомянутые в композиции, терапевтическом способе и применении по настоящему изобретению, применимы в равной степени, если не противоречат друг другу.
Полезные эффекты изобретения
Соединение по настоящему изобретению, его стереоизомеры или его фармацевтически приемлемые соли могут проявлять эффективную антагонистическую активность в отношении аденозиновых рецепторов А2а и могут быть успешно использованы для лечения или профилактики заболеваний, связанных с аденозиновым рецептором А2а.
Принцип изобретения
Далее настоящее изобретение будет описано более подробно с помощью примеров получения и типовых примеров. Однако следующие примеры получения и типовые примеры представлены с целью иллюстрации настоящего изобретения, и, таким образом, настоящее изобретение не ограничивается примерами получения и типовыми примерами.
Получение соединения, представленного формулой 1
Каждое из соединений по настоящему изобретению синтезируют следующим образом.
Чтобы получить соединения по настоящему изобретению, каждое из реакционных соединений, используемых в каждой реакции, приобретают у Sigma Aldrich (название компании) и т. д. или синтезируют с использованием способа органического синтеза, очевидного для специалистов в области химии, и используют без отдельного процесса очистки. Соединения из каждого примера идентифицируют с помощью 1H-ЯМР (Bruker, Avance II 400) и анализа по массам (Waters, SQD2).
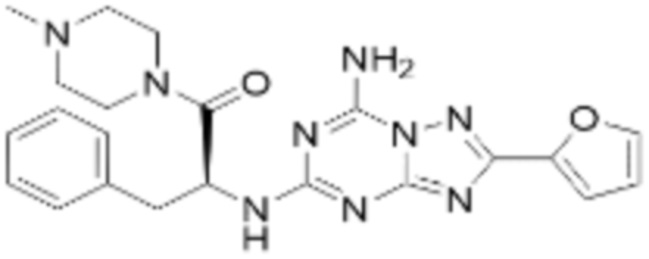
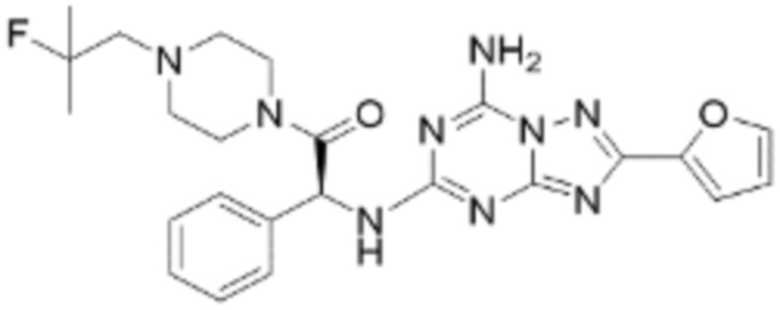
Пример 1: Синтез соединения 1, (S)-2-((7-амино-2-(фуран-2-ил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-5-ил)амино)-1-(4-метилпиперазин-1-ил)-3-фенилпропан-1-она
[Стадия 1] Синтез трет-бутил (S)-(1-(4-метилпиперазин-1-ил)-1-оксо-3-фенилпропан-2-ил)карбамат
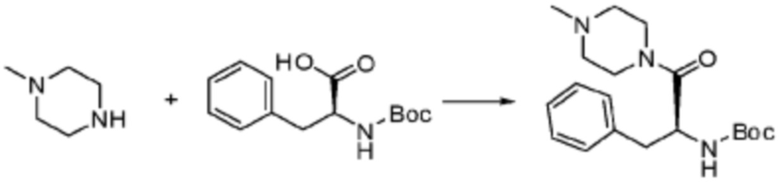
(Трет-бутоксикарбонил)-L-фенилаланин (10,000 г, 37,692 ммоль), 1-метилпиперазин (8,390 мл, 75,384 ммоль), [диметиламино(триазоло[4,5-b]пиридин-3-илокси)метилиден]-диметилазания, гексафторфосфат (28,664 г, 75,384 ммоль) и N, N-диизопропилэтиламин (13,130 мл, 75,384 ммоль) растворяют в N, N-диметилформамиде (200 мл) при комнатной температуре, и полученный раствор перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего воду выливают в полученный концентрат и органический слой экстрагируют этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют, и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 120 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (10,000 г, 76,4%) в виде белого твердого вещества пенистого типа.
[Стадия 2] Синтез (S)-2-амино-1-(4-метилпиперазин-1-ил)-3-фенилпропан-1-она
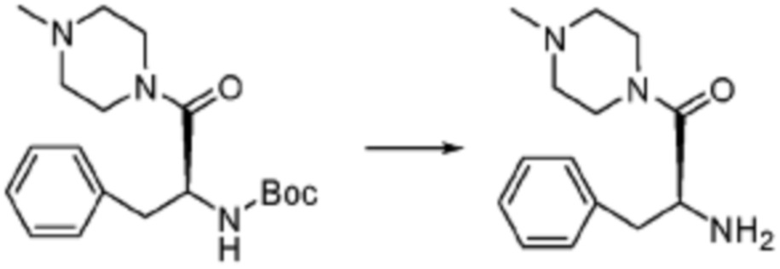
Трет-бутил (S)-(1-(4-метилпиперазин-1-ил)-1-оксо-3-фенилпропан-2-ил)карбамат (9,000 г, 25,902 ммоль), полученный на стадии 1, и 2,2,2-трифторуксусную кислоту (9,911 мл, 129,511 ммоль) растворяют в метаноле (100 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Водный раствор 2N-гидроксида натрия выливают в реакционную смесь, и органический слой экстрагируют этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют, и концентрируют при пониженном давлении. Полученный продукт используют без дополнительного процесса очистки (указанное в заголовке соединение, 3,500 г, 54,6%, белое твердое вещество).
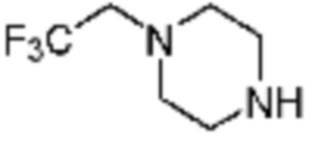
[Стадия 3] Синтез (S)-2-((7-амино-2-(фуран-2-ил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-5-ил)амино)-1-(4-метилпиперазин-1-ил)-3-фенилпропан-1-она
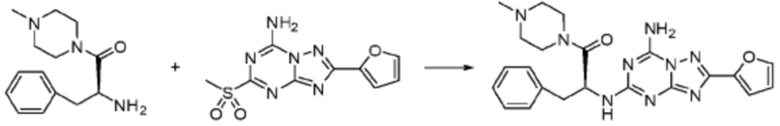
2-(Фуран-2-ил)-5-(метилсульфонил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амин (3,500 г, 12,488 ммоль), полученный на стадии 2, (S)-2-амино-1-(4-метилпиперазин-1-ил)-3-фенилпропан-1-он (3,089 г, 12,488 ммоль) и триэтиламин (3,481 мл, 24,977 ммоль) растворяют в диметилсульфоксиде (60 мл) при комнатной температуре, и полученный раствор перемешивают при той же температуре в течение 18 часов. Воду выливают в реакционную смесь и органический слой экстрагируют этилацетатом. Органический слой промывают водой, дегидратируют безводным сульфатом магния, фильтруют, и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-8%) и концентрируют с получением указанного в заголовке соединения (3,000 г, 53,7%) в виде белого твердого вещества.
1H ЯМР (400 MГц, Хлороформ-d) δ 9,34 (с, 1H), 8,47-8,36 (м, 1H), 7,62 (с, 1H), 7,31-7,08 (м, 6H), 6,59 (с, 1H), 6,30-6,25 (м, 1H), 5,72-5,65 (м, 1H), 3,81-3,38 (м, 4H), 3,11-3,05 (м, 2H), 2,39-2,31 (м, 2H), 2,21 (с, 3H), 2,17-2,13 (м, 1H), 1,83-1,79 (м, 1H), 1,33 (с, 1H), 1,28 (с, 1H); МСНР (ЭР) m/z 488,4
Примеры 2, 3, 13, 19, 20, 28 и 191
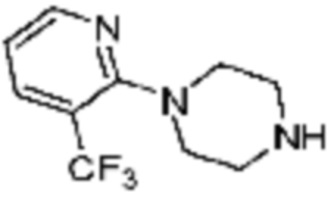
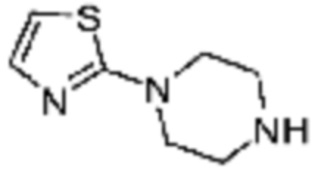
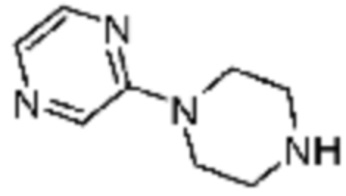
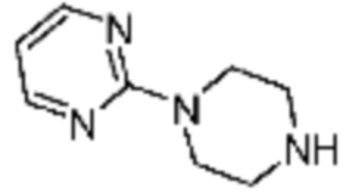
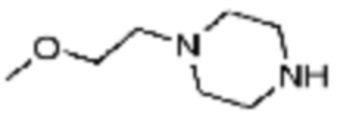
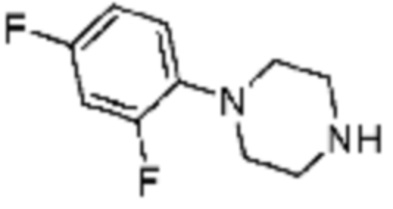
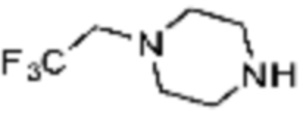
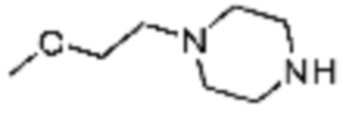
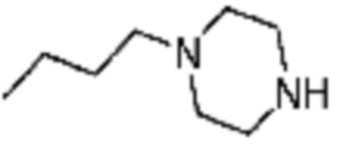
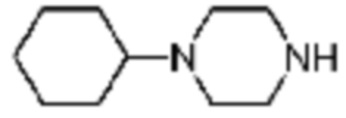
Примеры соединений 2, 3, 13, 19, 20, 28 и 191 получают по существу тем же способом синтеза, как и способ синтеза из примера соединения 1 за исключением применения соединений из следующей таблицы вместо 1-метилпиперазина в качестве R5-H из вышеуказанной формулы реакции 1 на стадии 1 способа синтеза из примера 1.
[Таблица 2]
№
№
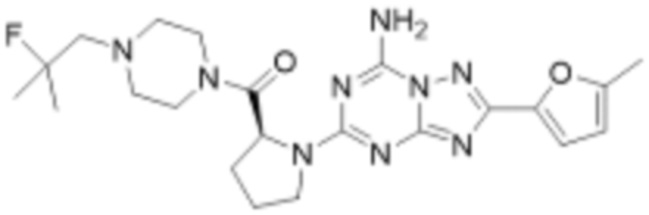
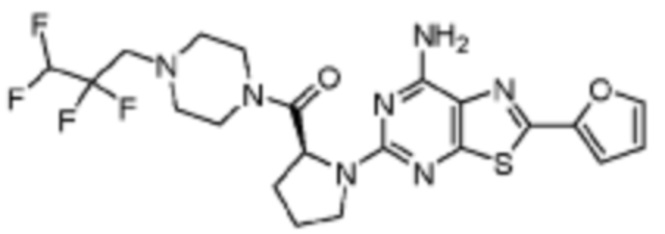
Пример 26: Синтез соединения 26, (S)-(1-(7-амино-2-(фуран-2-ил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-5-ил)пирролидин-2-ил)(4-(2-фтор-2-метилпропил)пиперазин-1-ил)метанона
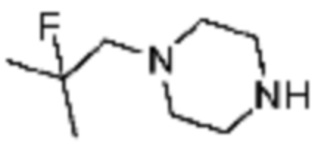
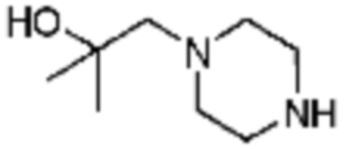
[Стадия 1] Синтез трет-бутил (S)-2-(4-(2-фтор-2-метилпропил)пиперазин-1-карбонил)пирролидин-1-карбоксилата
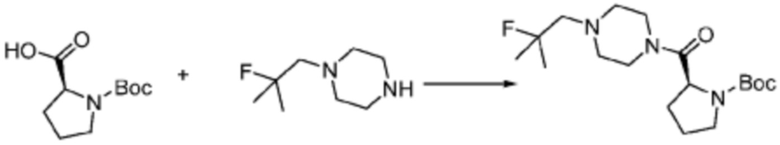
(Трет-бутоксикарбонил)-L-пролин (2,000 г, 9,292 ммоль), 1-(2-фтор-2-метилпропил)пиперазин (2,978 г, 18,583 ммоль), [диметиламино(триазоло[4,5-b]пиридин-3-илокси)метилиден]-диметилазания; гексафторфосфат (7,066 г, 18,583 ммоль) и N, N-диизопропилэтиламин (6,474 мл, 37,166 ммоль) растворяют в N, N-диметилформамиде (20 мл) при комнатной температуре, и полученный раствор перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего насыщенный водный раствор хлорида аммония выливают в полученный концентрат и органический слой экстрагируют этилацетатом. Органический слой промывают насыщенным водным раствором гидрокарбоната натрия, дегидратируют безводным сульфатом натрия, фильтруют, и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 24 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением указанного в заголовке соединения (2,000 г, 60,2%) в виде желтого твердого вещества.
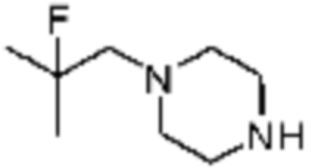
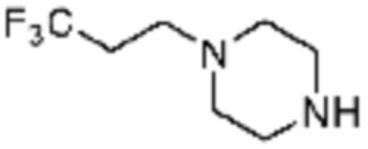
[Стадия 2] Синтез (S)-1-(2-фтор-2-метилпропил)-4-пропилпиперазина
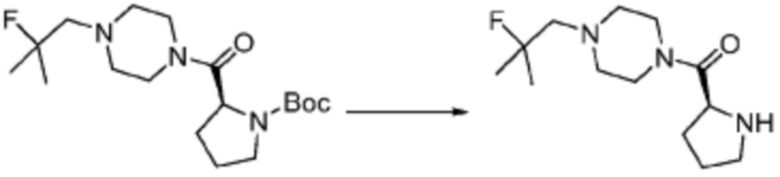
Трет-бутил (S)-2-(4-(2-фтор-2-метилпропил)пиперазин-1-карбонил)пирролидин-1-карбоксилат (0,200 г, 0,559 ммоль), полученный на стадии 1, и хлористоводородную кислоту (4,00 M раствор в диоксане, 0,699 мл, 2,797 ммоль) растворяют в дихлорметане (5 мл)/метанол (1 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный продукт используют без дополнительного процесса очистки (указанное в заголовке соединение, 0,140 г, 97,2%,коричневое твердое вещество).
[Стадия 3] Синтез (S)-(1-(7-амино-2-(фуран-2-ил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-5-ил)пирролидин-2-ил)(4-(2-фтор-2-метилпропил)пиперазин-1-ил)метанона
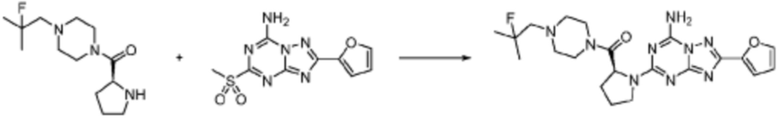
2-(фуран-2-ил)-5-(метилсульфонил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амин (0,050 г, 0,178 ммоль), полученный на стадии 2, (S)-1-(2-фтор-2-метилпропил)-4-пропилпиперазин (0,046 г, 0,178 ммоль) и триэтиламин (0,050 мл, 0,357 ммоль) растворяют в диметилсульфоксиде (1 мл) при комнатной температуре, после чего полученный раствор перемешивают в течение 18 часов при той же температуре. Воду выливают в реакционную смесь, и затем органический слой экстрагируют дихлорметаном, фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора из него, и концентрируют при пониженном давлении. Полученный концентрат очищают хроматографией (SiO2 тарелка, 20×20x1 мм; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,013 г, 15,3%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, Хлороформ-d) δ 7,60-7,51 (м, 1H), 7,24-7,13 (м, 1H), 6,54 (ддд, J=11,3, 3,4, 1,8 Гц, 1H), 6,30 (с, 1H), 6,14 (с, 1H), 5,13-4,83 (м, 1H), 3,95-3,39 (м, 6H), 2,72 (с, 1H), 2,57-2,38 (м, 4H), 2,29 (ддд, J=13,8, 9,7, 5,3 Гц, 1H), 2,16 (дт, J=12,7, 7,7 Гц, 1H), 1,47-1,33 (м, 6H). МСНР (ЭР) m/z 458,4 (M++ 1).
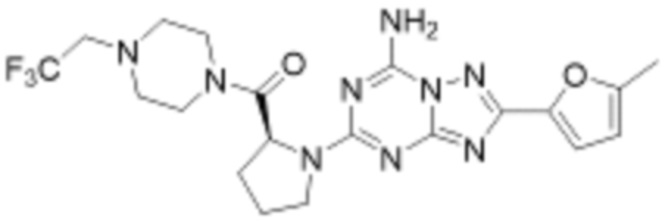
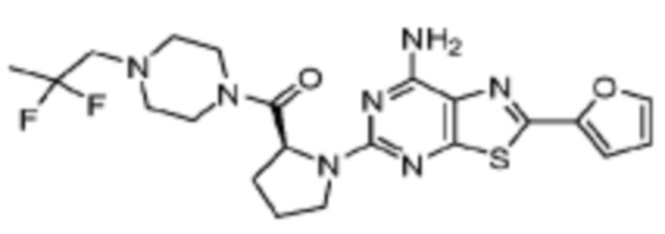
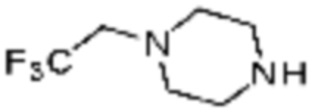
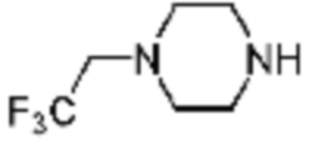
Пример 48: Синтез соединения 48, (S)-(1-(7-амино-2-(фуран-2-ил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-5-ил)пирролидин-2-ил)(4-(2,2,2-трифторэтил)пиперазин-1-ил)метанона
[Стадия 1] Синтез трет-бутил (S)-2-(4-(2,2,2-трифторэтил)пиперазин-1-карбонил)пирролидин-1-карбоксилата
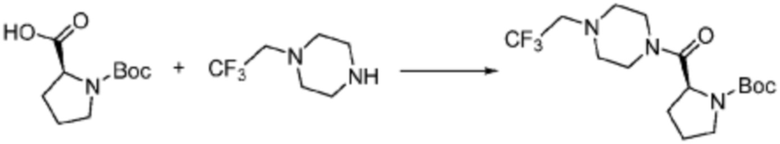
(Трет-бутоксикарбонил)-L-пролин (1,000 г, 4,646 ммоль), 1-(2,2,2-трифторэтил)пиперазин (0,781 г, 4,646 ммоль), [диметиламино(триазоло[4,5-b]пиридин-3-илокси)метилиден]-диметилазания; гексафторфосфат (3,533 г, 9,292 ммоль) и N, N-диизопропилэтиламин (1,618 мл, 9,292 ммоль) растворяют в N, N-диметилформамиде (20 мл) при комнатной температуре, и полученный раствор перемешивают при той же температуре в течение 18 часов. Воду выливают в реакционную смесь, и органический слой экстрагируют этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют, и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением указанного в заголовке соединения (0,700 г, 41,2%) в виде желтого твердого вещества.
[Стадия 2] Синтез (S)-1-пропил-4-(2,2,2-трифторэтил)пиперазина
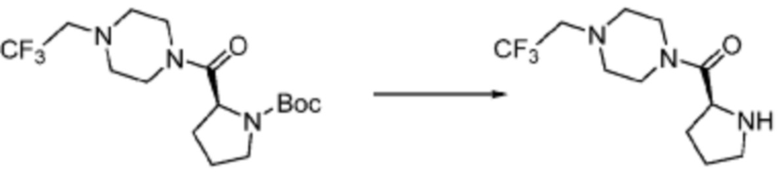
Трет-бутил (S)-2-(4-(2,2,2-трифторэтил)пиперазин-1-карбонил)пирролидин-1-карбоксилат (0,200 г, 0,547 ммоль), полученный на стадии 1, и хлористоводородную кислоту (4,00 M раствор в диоксане, 1,368 мл, 5,473 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный продукт используют без дополнительного процесса очистки (указанное в заголовке соединение, 0,140 г, 96,4%, белое твердое вещество).
[Стадия 3] Синтез (S)-(1-(7-амино-2-(фуран-2-ил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-5-ил)пирролидин-2-ил)(4-(2,2,2-трифторэтил)пиперазин-1-ил)метанона
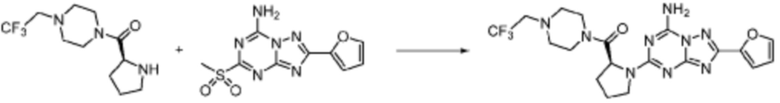
(S)-1-Пропил-4-(2,2,2-трифторэтил)пиперазин (0,050 г, 0,188 ммоль), полученный на стадии 2, 2-(фуран-2-ил)-5-(метилсульфонил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амин (0,053 г, 0,188 ммоль) и триэтиламин (0,053 мл, 0,377 ммоль) растворяют в диметилсульфоксиде (1 мл) при комнатной температуре, после чего полученный раствор перемешивают в течение 18 часов при той же температуре. Воду выливают в реакционную смесь, и затем органический слой экстрагируют дихлорметаном, фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора из него, и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,015 г, 16,5%) в виде белого твердого вещества.
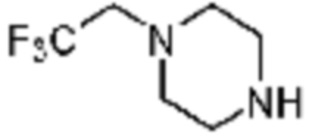
1H ЯМР (400 MГц, Хлороформ-d) δ 7,61-7,54 (м, 1H), 7,27-7,13 (м, 1H), 6,56 (ддд, J=5,2, 3,4, 1,8 Гц, 1H), 5,95 (д, J=74,2 Гц, 2H), 5,02 (ддд, J=63,5, 8,5, 3,0 Гц, 1H), 3,99-3,71 (м, 4H), 3,67-3,61 (м, 2H), 3,09 (п, J=9,5, 9,1 Гц, 3H), 2,80-2,60 (м, 3H), 2,38-2,08 (м, 2H), 2,06-1,97 (м, 2H). МСНР (ЭР) m/z 466,5 (M++ 1).
Пример 90: Синтез соединения 90, (S)-(1-(7-амино-2-(5-метилфуран-2-ил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-5-ил)пирролидин-2-ил)(4-(2-фтор-2-метилпропил)пиперазин-1-ил)метанона
[Стадия 1] Синтез (S)-(1-(7-амино-2-(5-метилфуран-2-ил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-5-ил)пирролидин-2-ил)(4-(2-фтор-2-метилпропил)пиперазин-1-ил)метанона
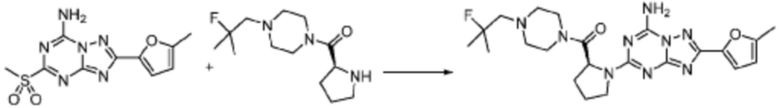
(S)-1-(2-Фтор-2-метилпропил)-4-пропилпиперазин (0,087 г, 0,340 ммоль), полученный синтезом со стадии 2 Примера 26, 2-(5-метилфуран-2-ил)-5-(метилсульфонил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амин (0,050 г, 0,170 ммоль) и триэтиламин (0,047 мл, 0,340 ммоль) растворяют в диметилсульфоксиде (5 мл) при комнатной температуре, и полученный раствор перемешивают при той же температуре в течение 18 часов. Воду выливают в реакционную смесь, и органический слой экстрагируют этилацетатом. Органический слой промывают водой, дегидратируют безводным сульфатом натрия, фильтруют, и концентрируют при пониженном давлении. Полученный концентрат очищают хроматографией (SiO2 тарелка, 20×20x1 мм; метанол/дихлорметан=5%) и концентрируют с получением указанного в заголовке соединения (0,035 г, 43,7%) в виде белого твердого вещества.
1H ЯМР (400 MГц, DMSO-d6) δ 8,27 (м, 2H), 6,94 (ддд, J=3,2, 2,2, 0,6 Гц, 1H), 6,29 (ддд, J=3,3, 1,7, 1,1 Гц, 1H), 5,02-4,92 (м, 1H), 3,72-3,39 (м, 6H), 2,72 (м, 1H), 2,61-2,52 (м, 5H), 2,48-2,18 (м, 4H), 2,04-1,71 (м, 3H), 1,39-1,26 (м, 6H); МСНР (ЭР) m/z 472,6 (M++ 1).
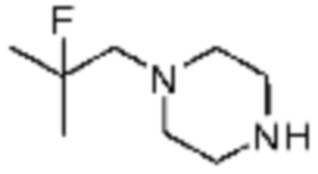
Примеры 12, 16, 27, 55, 78-81, 113, 114, 120-128, 134-142, 145, 146, 149, 150, 151, 236, 239, 246, 247, 265, 266, 281, 301, 302, 309, 310 и 321
Примеры соединений 12, 16, 27, 55, 78-81, 113, 114, 120-128, 134-142, 145, 146, 149, 150, 151, 236, 239, 246, 247, 265, 266, 281, 301, 302, 309, 310 и 321 получают по существу тем же способом синтеза, как и способ синтеза из примера соединения 26 за исключением применения соединений из следующей таблицы вместо 1-(2-фтор-2-метилпропил)пиперазина в качестве R5-H из вышеуказанной формулы реакции 1 на стадии 1 способа синтеза из примера соединения 26.
[Таблица 3]
№
№
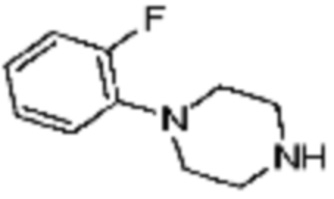
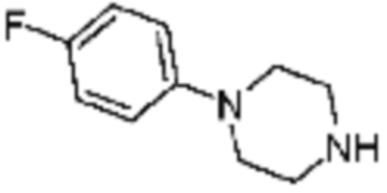
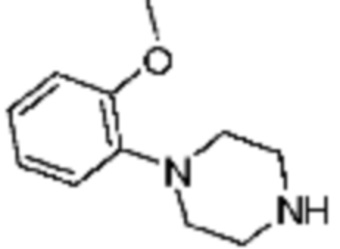
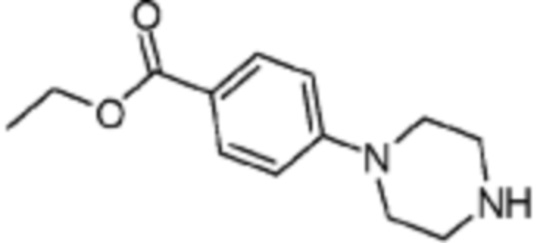
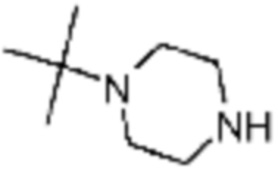
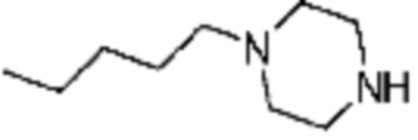
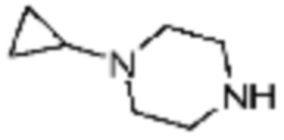
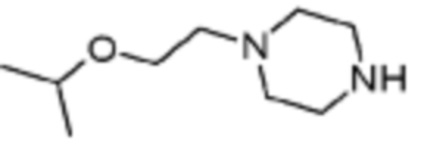
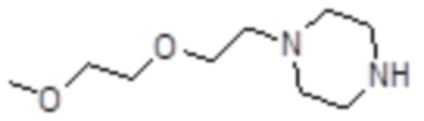
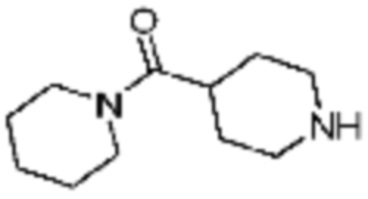
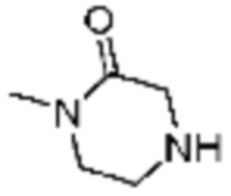
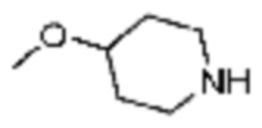
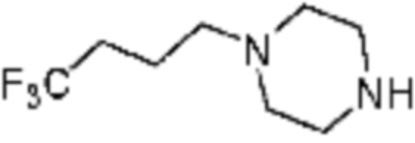
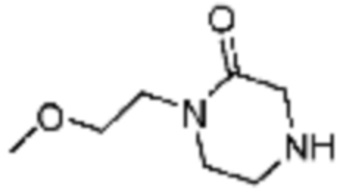
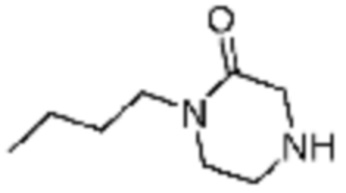
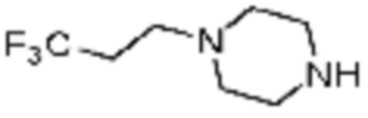
Пример 15: Синтез соединения 15
Пример соединения 15 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 16 за исключением применения (трет-бутоксикарбонил)глицина вместо (трет-бутоксикарбонил)-L-пролина.
Пример 37: Синтез соединения 37
Пример соединения 37 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 12 за исключением применения (2S,4R)-1-(трет-бутоксикарбонил)-4-гидроксипирролидин-2-карбоновой кислоты вместо (трет-бутоксикарбонил)-L-пролина.
Пример 385: Синтез соединения 385
Пример соединения 385 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 48 за исключением применения (S)-5-(трет-бутоксикарбонил)-5-азаспиро[2,4]гептан-6-карбоновой кислоты вместо (трет-бутоксикарбонил)-L-пролина.
Пример 386: Синтез соединения 386
Пример соединения 386 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 26 за исключением применения (S)-5-(трет-бутоксикарбонил)-5-азаспиро[2,4]гептан-6-карбоновой кислоты вместо (трет-бутоксикарбонил)-L-пролина.
Пример 387: Синтез соединения 387
Пример соединения 387 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 48 за исключением применения (S)-1-(трет-бутоксикарбонил)-4,4-диметилпирролидин-2-карбоновой кислоты вместо (трет-бутоксикарбонил)-L-пролина.
Пример 388: Синтез соединения 388
Пример соединения 388 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 26 за исключением применения (трет-бутоксикарбонил)глицина вместо (трет-бутоксикарбонил)-L-пролина.
Примеры 65, 107, 108, 109, 110, 112, 242, 264 и 350
Пример соединения 108 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 26 за исключением применения (S)-1-(трет-бутоксикарбонил)азетидин-2-карбоновой кислоты вместо (трет-бутоксикарбонил)-L-пролина.
В дополнение, примеры соединений 65, 107, 109, 110, 112, 242, 264 и 350 получают по существу тем же способом синтеза, как и способ синтеза из примера соединения 26 за исключением применения соединений из следующей таблицы вместо 1-(2-фтор-2-метилпропил)пиперазина в качестве R5-H из формулы реакции 1.
[Таблица 4]
№
№
Пример 29: Синтез соединения 29
Пример соединения 29 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 16 за исключением применения (S)-1-(трет-бутоксикарбонил)пиперидин-2-карбоновой кислоты вместо (трет-бутоксикарбонил)-L-пролина.
Примеры 92, 93, 94, 95, 96, 97 и 98
Примеры соединений 92, 93, 94, 95, 96, 97 и 98 получают по существу тем же способом синтеза, что и способ синтеза из Примера соединения 26 за исключением применения соединений из следующей таблицы вместо 1-(2-фтор-2-метилпропил)пиперазина в качестве R5-H из формулы реакции 1 и с применением трет-бутоксикарбонил-D-пролина вместо (трет-бутоксикарбонил)-L-пролина.
[Таблица 5]
№
№
Пример 7: Синтез соединения 7
Пример соединения 7 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 12 за исключением применения (S)-2-((трет-бутоксикарбонил)амино)-2-фенилуксусной кислоты вместо (трет-бутоксикарбонил)-L-пролина.
Пример 14: Синтез соединения 14
Пример соединения 14 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 16 за исключением применения (S)-2-((трет-бутоксикарбонил)амино)-2-фенилуксусной кислоты вместо (трет-бутоксикарбонил)-L-пролина.
Пример 190: Синтез соединения 190
Пример соединения 190 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 191 за исключением применения (S)-2-((трет-бутоксикарбонил)амино)-2-фенилуксусной кислоты вместо (трет-бутоксикарбонил)-L-фенилаланина.
Пример 195: Синтез соединения 195
Пример соединения 195 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 26 за исключением применения (S)-2-((трет-бутоксикарбонил)амино)-2-фенилуксусной кислоты вместо (трет-бутоксикарбонил)-L-пролина.
Примеры 171, 241 и 249
Пример соединения 171 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 12 за исключением применения (S)-2-((трет-бутоксикарбонил)амино)-2-(тетрагидро-2H-пиран-4-ил)уксусной кислоты вместо (S)-2-((трет-бутоксикарбонил)амино)-2-фенилуксусной кислоты.
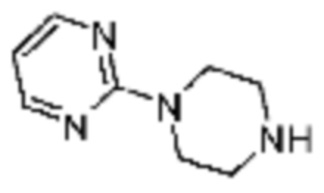
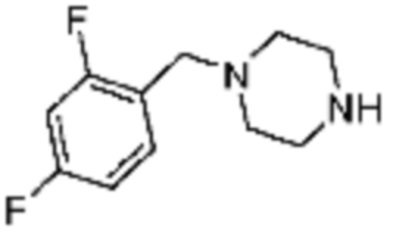
В дополнение, примеры соединений 241 и 249 получают по существу тем же способом синтеза, как и способ синтеза из примера соединения 171 за исключением применения соединений из следующей таблицы вместо 1-(2,4-дифторфенил)пиперазин в качестве R5-H.
[Таблица 6]
№
№
Примеры 8 и 231-235
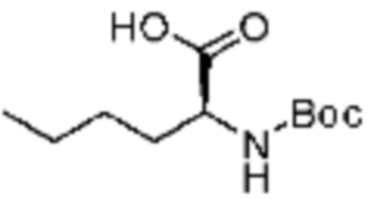
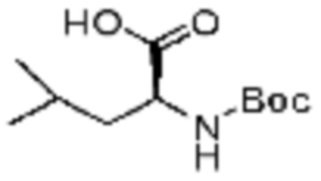
Пример соединения 8 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 12 за исключением применения (трет-бутоксикарбонил)-L-серина вместо (S)-2-((трет-бутоксикарбонил)амино)-2-фенилуксусной кислоты.
В дополнение, примеры соединений 231-235 получают по существу тем же способом синтеза, как и способ синтеза из примера соединения 8 за исключением применения соединений из следующей таблицы вместо 1-(2,4-дифторфенил)пиперазина в качестве R5-H.
[Таблица 7]
№
№
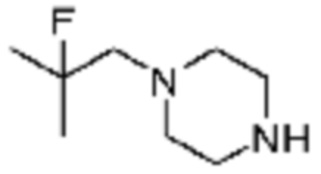
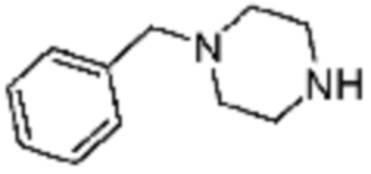
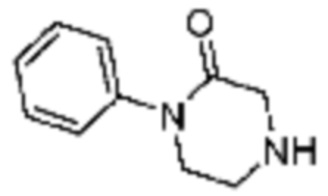
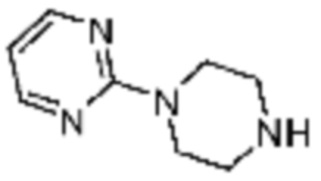
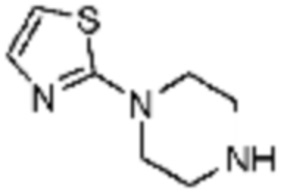
Примеры 9, 118, 119, 240, 251 и 349
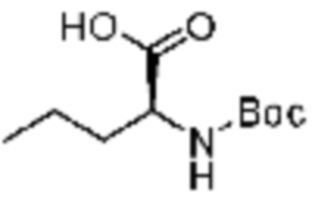
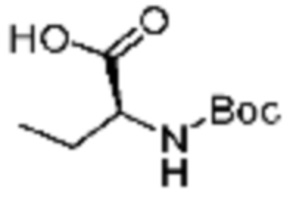
Пример соединения 9 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 8 за исключением применения (трет-бутоксикарбонил)-L-аланина вместо (трет-бутоксикарбонил)-L-серина.
В дополнение, примеры соединений 118, 119, 240, 251 и 349 получают по существу тем же способом синтеза, как и способ синтеза из примера соединения 9 за исключением применения соединений из следующей таблицы вместо 1-(2,4-дифторфенил)пиперазина в качестве R5-H.
[Таблица 8]
№
№
Примеры 10, 57-60, 143, 144, 351 и 367
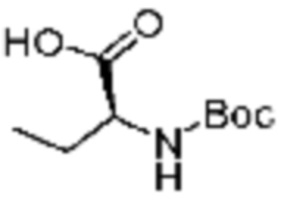
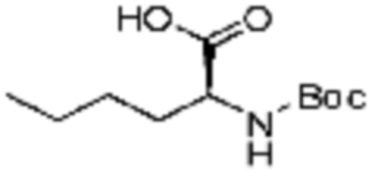
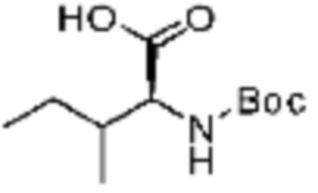
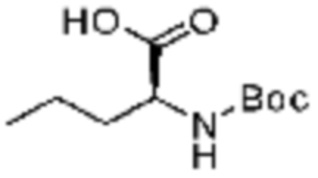
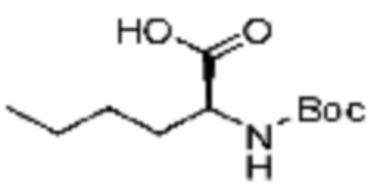
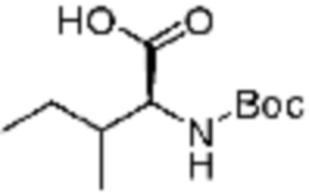
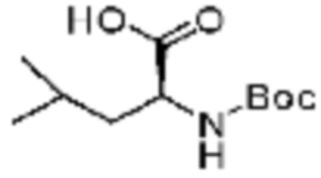
Пример соединения 10 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 8 за исключением применения (трет-бутоксикарбонил)-L-валина вместо (трет-бутоксикарбонил)-L-серина. В дополнение, примеры соединений 57-60, 143, 144, 351 и 367 получают по существу тем же способом синтеза, как и способ синтеза из примера соединения 10 за исключением применения соединений из следующей таблицы вместо 1-(2,4-дифторфенил)пиперазина в качестве R5-H.
[Таблица 9]
№
№
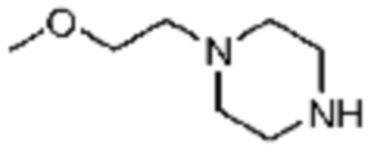
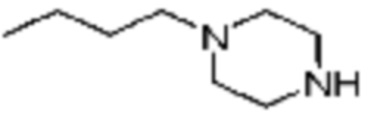
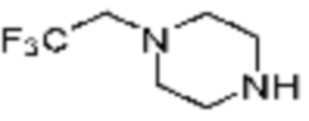
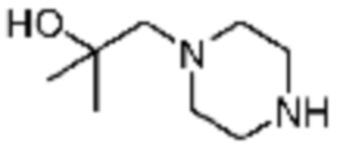
Примеры 162-167
Примеры соединений 162-167 получают по существу тем же способом синтеза, как и способ синтеза из примера соединения 9 за исключением применения соединений из следующей таблицы в качестве N-защищенной аминокислоты вместо (трет-бутоксикарбонил)-L-аланина.
[Таблица 10]
№
№
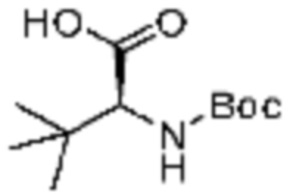
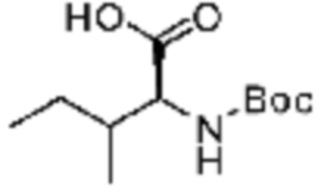
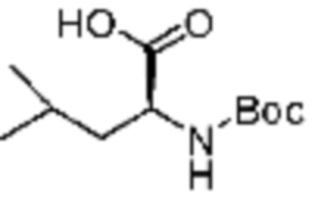
Примеры 172-175
Примеры соединений 172-175 получают по существу тем же способом синтеза, как и способ синтеза из примера соединения 143 за исключением применения соединений из следующей таблицы в качестве N-защищенной аминокислоты вместо (трет-бутоксикарбонил)-L-валина.
[Таблица 11]
№
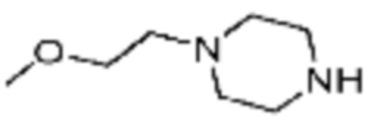
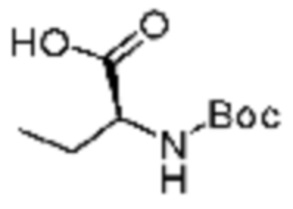
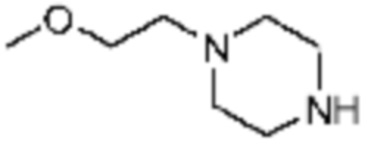
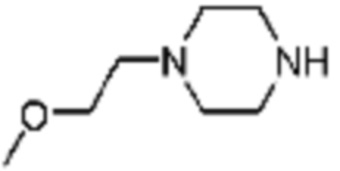
№
Примеры 211-215
Примеры соединений 211-215 получают по существу тем же способом синтеза, как и способ синтеза из примера соединения 144 за исключением применения соединений из следующей таблицы в качестве N-защищенной аминокислоты вместо (трет-бутоксикарбонил)-L-валина.
[Таблица 12]
№
№
Примеры 248, 338 и 346
Пример соединения 338 получают по существу тем же способом синтеза, как и способ синтеза из Примера соединения 240 за исключением применения (S)-2-((трет-бутоксикарбонил)амино)бутановой кислоты вместо (трет-бутоксикарбонил)-L-аланина.
В дополнение, примеры соединений 248 и 346 получают по существу тем же способом синтеза, как и способ синтеза из примера соединения 338 за исключением применения соединений из следующей таблицы вместо 1-(2-фтор-2-метилпропил)пиперазина в качестве R5-H.
[Таблица 13]
№
№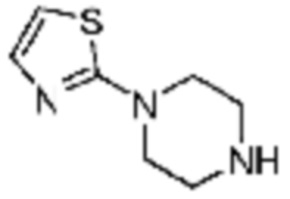
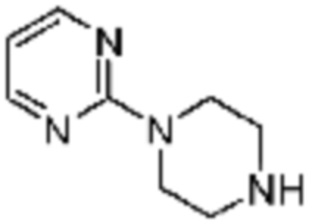
Примеры 11, 147, 148, 187 и 188
Пример соединения 11 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 12 за исключением применения (S)-2-((трет-бутоксикарбонил)амино)-2-циклогексилуксусной кислоты вместо (S)-2-((трет-бутоксикарбонил)амино)-2-фенилуксусной кислоты.
В дополнение, примеры соединений 147, 148, 187 и 188 получают по существу тем же способом синтеза, как и способ синтеза из примера соединения 11 за исключением применения соединений из следующей таблицы вместо 1-(2,4-дифторфенил)пиперазина в качестве R5-H.
[Таблица 14]
№
№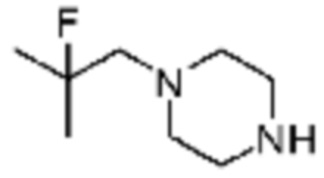
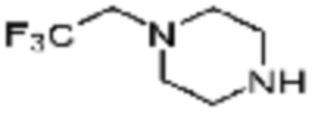
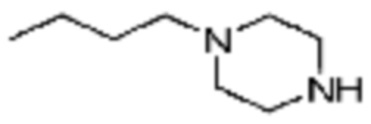
Примеры 168, 176, 216 и 339
Пример соединения 168 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 11 за исключением применения (S)-2-((трет-бутоксикарбонил)амино)-2-циклопропилуксусной кислоты вместо (S)-2-((трет-бутоксикарбонил)амино)-2-циклогексилуксусной кислоты.
Примеры соединений 176, 216 и 339 получают по существу тем же способом синтеза, как и способ синтеза из примера соединения 168 за исключением применения соединений из следующей таблицы вместо 1-(2,4-дифторфенил)пиперазина в качестве R5-H.
[Таблица 15]
№
№
Примеры 169 и 177
Пример соединения 169 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 11 за исключением применения (S)-2-((трет-бутоксикарбонил)амино)-2-циклобутилуксусной кислоты вместо (S)-2-((трет-бутоксикарбонил)амино)-2-циклогексилуксусной кислоты.
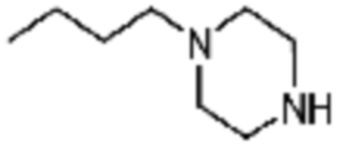
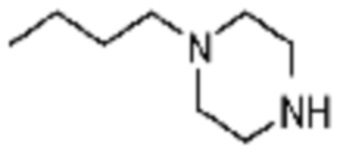
В дополнение, пример соединения 177 получают по существу тем же способом синтеза, как и способ синтеза из Примера соединения 169 за исключением применения 1-бутилпиперазина вместо 1-(2,4-дифторфенил)пиперазина.
Примеры 170, 178 и 217
Пример соединения 170 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 11 за исключением применения (S)-2-((трет-бутоксикарбонил)амино)-2-циклопентилуксусной кислоты вместо (S)-2-((трет-бутоксикарбонил)амино)-2-циклогексилуксусной кислоты.
В дополнение, примеры соединений 178 и 217 каждый синтезируют по существу тем же способом синтеза, как и способ синтеза из Примера соединения 170 за исключением применения соединений из следующей таблицы вместо 1-(2,4-дифторфенил)пиперазина в качестве R5-H.
[Таблица 16]
№
№
Примеры 40, 87, 89, 91, 189, 198, 199, 200, 201, 202, 203 и 300
Примеры соединений 40, 87, 89, 91, 189, 198, 199, 200, 201, 202, 203 и 300 каждый синтезируют по существу тем же способом синтеза, как и способ синтеза из Примера соединения 90 за исключением применения соединений из следующей таблицы вместо 1-(2-фтор-2-метилпропил)пиперазина в качестве R5-H.
[Таблица 17]
№
№
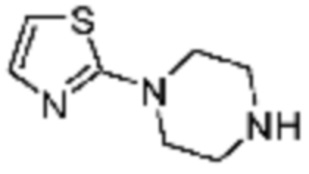
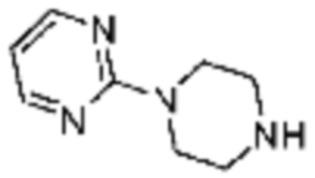
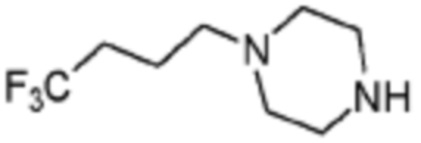
Примеры 207-210 и 218-222
Примеры соединений 207-210 и 218-222 каждый синтезируют по существу тем же способом синтеза, как и способ синтеза из Примера соединения 90 за исключением применения соединений из следующей таблицы вместо 1-(2-фтор-2-метилпропил)пиперазина в качестве R5-H и с применением (S)-1-(трет-бутоксикарбонил)азетидин-2-карбоновой кислоты вместо (трет-бутоксикарбонил)-L-пролина.
[Таблица 18]
№
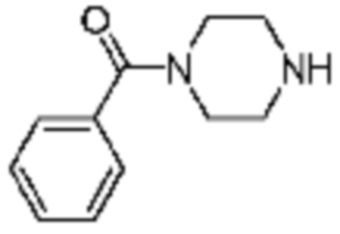
№
Пример 303: Синтез соединения 303, (S)-2-((7-амино-2-(фуран-2-ил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-5-ил)амино)-1-(4-(2-фтор-2-метилпропил)пиперазин-1-ил)-3-метоксипропан-1-она
[Стадия 1] трет-бутил (S)-(1-(4-(2-фтор-2-метилпропил)пиперазин-1-ил)-3-метокси-1-оксопропан-2-ил)карбамат
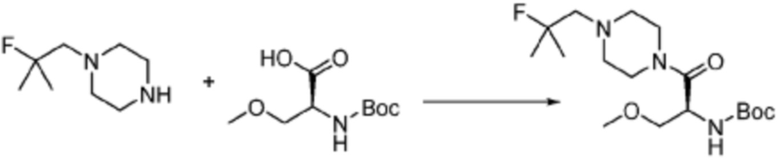
1-(2-Фтор-2-метилпропил)пиперазин (0,500 г, 3,120 ммоль), N-(трет-бутоксикарбонил)-O-метил-L-серин (1,368 г, 6,241 ммоль), гексафторфосфат 3-оксида 1-[бис(диметиламино)метилен]-1H-1,2,3-триазоло[4,5-b]пиридиния (ГАТУ, 2,373 г, 6,241 ммоль) и N, N-диизопропилэтиламин (2,717 мл, 15,602 ммоль) растворяют в дихлорметане (15 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего насыщенный водный раствор гидрокарбоната натрия выливают в полученный концентрат и органический слой экстрагируют этилацетатом. Органический слой промывают насыщенным водным раствором гидрокарбоната натрия, дегидратируют безводным сульфатом магния, фильтруют, и концентрируют при пониженном давлении. Полученный продукт используют без дополнительного процесса очистки (указанное в заголовке соединение, 1,000 г, 88,7%, коричневое масло).
[Стадия 2] (S)-2-амино-1-(4-(2-фтор-2-метилпропил)пиперазин-1-ил)-3-метоксипропан-1-он
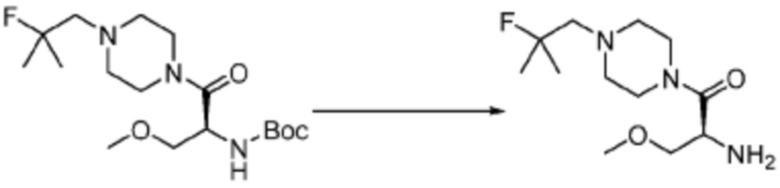
Трет-бутил (S)-(1-(4-(2-фтор-2-метилпропил)пиперазин-1-ил)-3-метокси-1-оксопропан-2-ил)карбамат (0,200 г, 0,553 ммоль), полученный на стадии 1, и хлороводород (4,00 M раствор в диоксане, 1,383 мл, 5,533 ммоль) растворяют в дихлорметане (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный продукт используют без дополнительного процесса очистки (указанное в заголовке соединение, 0,100 г, 69,2%, коричневое масло).
[Стадия 3] (S)-2-((7-амино-2-(фуран-2-ил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-5-ил)амино)-1-(4-(2-фтор-2-метилпропил)пиперазин-1-ил)-3-метоксипропан-1-он
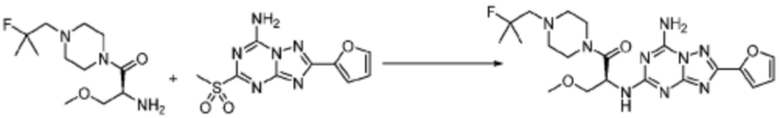
(S)-2-Амино-1-(4-(2-фтор-2-метилпропил)пиперазин-1-ил)-3-метоксипропан-1-он (0,100 г, 0,383 ммоль), полученный на стадии 2, 2-(фуран-2-ил)-5-(метилсульфонил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амин (0,054 г, 0,191 ммоль) и гидрокарбонат натрия (0,096 г, 1,148 ммоль) растворяют в ацетонитриле (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при 70°C в течение 18 часов до завершения реакции путем понижения температуры до комнатной температуры. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/этилацетат=0-10%) и концентрируют с получением указанного в заголовке соединения (0,015 г, 8,5%) в виде белого твердого вещества.
1H ЯМР (400 MГц, ДМСО-d6) δ 8,32 (с, 2H), 7,88 (с, 1H), 7,46 (дд, J=53,9, 8,2 Гц, 1H), 7,08 (д, J=3,2 Гц, 1H), 6,68 (дд, J=3,2, 1,7 Гц, 1H), 5,09 (дд, J=14,2, 6,3 Гц, 1H), 3,73-3,39 (м, 6H), 3,27 (с, 3H), 2,62-2,34 (м, 6H), 1,32 (д, J=21,5 Гц, 6H); МСНР (ЭР) m/z 462,5 (M++ 1).
Примеры 304, 305, 306, 399 и 401
Примеры соединений 304, 305, 306, 399 и 401 получают по существу тем же способом синтеза, как и способ синтеза из примера соединения 303 за исключением применения соединений из следующей таблицы вместо 1-(2-фтор-2-метилпропил)пиперазина в качестве R5-H из вышеуказанной формулы реакции 1 на стадии 1.
[Таблица 19]
№
№
Пример 307: Синтез соединения 307
Пример соединения 307 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 304 за исключением применения N-(трет-бутоксикарбонил)-O-этил-L-серина вместо N-(трет-бутоксикарбонил)-O-метил-L-серина.
Пример 308: Синтез соединения 308
Пример соединения 308 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 306 за исключением применения N-(трет-бутоксикарбонил)-O-этил-L-серина вместо N-(трет-бутоксикарбонил)-O-метил-L-серина.
Пример 352: Синтез соединения 352
Пример соединения 352 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 26 за исключением применения 2-(фуран-2-ил)-7-(метилсульфонил)-[1,2,4]триазоло[1,5-c]пиримидин-5-амина вместо 2-(фуран-2-ил)-5-(метилсульфонил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амина со стадии 3.
Примеры 336 и 361: Синтез соединений 336 и 361
Примеры соединений 336 и 361 каждый синтезируют по существу тем же способом синтеза, как и способ синтеза из Примера соединения 352 за исключением применения 1-(2-метоксиэтил)пиперазина и 1-бутилпиперазина, соответственно, вместо 1-(2,4-дифторфенил)пиперазина в качестве R5-H.
Пример 375: Синтез соединения 375
Пример соединения 375 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 119 за исключением применения 2-(фуран-2-ил)-7-(метилсульфонил)-[1,2,4]триазоло[1,5-c]пиримидин-5-амина вместо 2-(фуран-2-ил)-5-(метилсульфонил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амина.
Пример 41: Синтез соединения 41
Пример соединения 41 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 16 за исключением применения 2-(фуран-2-ил)-5-(метилсульфонил)-3a,7a-дигидрооксазоло[5,4-d]пиримидин-7-амина вместо 2-(фуран-2-ил)-5-(метилсульфонил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амина.
Пример 238: Синтез соединения 238
Пример соединения 238 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 48 за исключением применения 2-(фуран-2-ил)-5-(метилсульфонил)-3a,7a-дигидротиазоло[5,4-d]пиримидин-7-амина вместо 2-(фуран-2-ил)-5-(метилсульфонил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амина.
Пример 243: Синтез соединения 243
Пример соединения 243 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 189 за исключением применения 2-(фуран-2-ил)-5-(метилсульфонил)-3a,7a-дигидротиазоло[5,4-d]пиримидин-7-амина вместо 2-(фуран-2-ил)-5-(метилсульфонил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амина.
Пример 192: Синтез соединения 192
Пример соединения 192 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 48 за исключением применения фенил (пиперазин-1-ил)метанона вместо 1-(2,2,2-трифторэтил)пиперазина и с применением 5-(метилсульфонил)-2-(тиазол-2-ил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амина вместо 2-(фуран-2-ил)-5-(метилсульфонил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амина.
Пример 193: Синтез соединения 193
Пример соединения 193 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 48 за исключением применения 1-бензилпиперазина вместо 1-(2,2,2-трифторэтил)пиперазина и с применением 5-(метилсульфонил)-2-(тиазол-2-ил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амина вместо 2-(фуран-2-ил)-5-(метилсульфонил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амина.
Пример 194: Синтез соединения 194
Пример соединения 194 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 9 за исключением применения 5-(метилсульфонил)-2-(тиазол-2-ил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амина вместо 2-(фуран-2-ил)-5-(метилсульфонил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амина.
Пример 250: Синтез соединения 250
Пример соединения 250 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 148 за исключением применения 5-(метилсульфонил)-2-(тиазол-2-ил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амина вместо 2-(фуран-2-ил)-5-(метилсульфонил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амина.
Пример 67: Синтез соединения 67
Пример соединения 67 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 12 за исключением применения 5-(метилсульфонил)-2-(тиазол-2-ил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амина вместо 2-(фуран-2-ил)-5-(метилсульфонил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амина.
Пример 84: Синтез соединения 84
Пример соединения 84 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 139 за исключением применения 2-(5-метилфуран-2-ил)-5-(метилсульфонил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амина вместо 2-(фуран-2-ил)-5-(метилсульфонил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амина, и с применением 1-циклогексилпиперазина вместо 1-циклопропилпиперазина.
Аналитические данные каждого из соединений, полученных как описано выше, показаны в таблице ниже.
[Таблица 20]
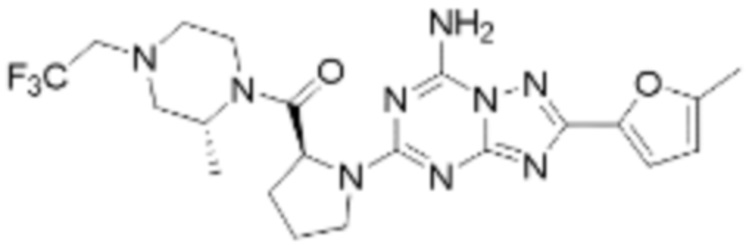
Пример 353: Синтез соединения 353, (2-(7-амино-2-(фуран-2-ил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-5-ил)пиразолидин-1-ил)(4-(2,2,2-трифторэтил)пиперазин-1-ил)метанона
[Стадия 1] трет-бутил 2-(4-(2,2,2-трифторэтил)пиперазин-1-карбонил)пиразолидин-1-карбоксилат

Трет-бутил пиразолидин-1-карбоксилат (1,000 г, 5,806 ммоль), 1,1′-карбонилдиимидазол (КДИ, 1,412 г, 8,709 ммоль) и N, N-диизопропилэтиламин (3,034 мл, 17,419 ммоль) растворяют в дихлорметане (20 мл) при комнатной температуре, после чего 1-(2,2,2-трифторэтил)пиперазин (1,953 г, 11,612 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего насыщенный водный раствор гидрокарбоната натрия выливают в полученный концентрат и органический слой экстрагируют этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют, и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-100%) и концентрируют с получением указанного в заголовке соединения (0,500 г, 23,5%) в виде белого твердого вещества пенистого типа.
[Стадия 2] Синтез пиразолидин-1-ил(4-(2,2,2-трифторэтил)пиперазин-1-ил)метанон

Трет-бутил 2-(4-(2,2,2-трифторэтил)пиперазин-1-карбонил)пиразолидин-1-карбоксилат (0,400 г, 1,092 ммоль), полученный на стадии 1, и хлористоводородную кислоту (4,00 M раствор в диоксане, 2,729 мл, 10,917 ммоль) растворяют в дихлорметане (5 мл)/метаноле (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный продукт используют без дополнительного процесса очистки (указанное в заголовке соединение, 0,250 г, 86,0%, коричневое масло).
[Стадия 3] Синтез (2-(7-амино-2-(фуран-2-ил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-5-ил)пиразолидин-1-ил)(4-(2,2,2-трифторэтил)пиперазин-1-ил)метанон
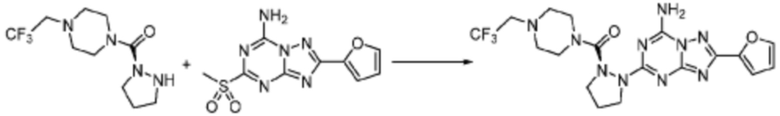
Пиразолидин-1-ил(4-(2,2,2-трифторэтил)пиперазин-1-ил)метанон (0,200 г, 0,751 ммоль), полученный на стадии 2, 2-(фуран-2-ил)-5-(метилсульфонил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амин (0,105 г, 0,376 ммоль) и гидрокарбонат натрия (0,189 г, 2,253 ммоль) растворяют в ацетонитриле (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при 70°C в течение 18 часов до завершения реакции путем понижения температуры до комнатной температуры. Реакционную смесь очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/этилацетат=0-10%) и концентрируют с получением продукта, после чего полученный продукт снова очищают хроматографией (SiO2 тарелка, 20×20x1 мм; метанол/дихлорметан=10%) и концентрируют с получением указанного в заголовке соединения (0,020 г, 5,7%) в виде белого твердого вещества.
1H ЯМР (400 MГц, ДМСО-d6) δ 8,40 (м, 2H), 7,88 (дд, J=1,7, 0,8 Гц, 1H), 7,08 (дд, J=3,4, 0,7 Гц, 1H), 6,69 (дд, J=3,4, 1,8 Гц, 1H), 3,73 (т, J=7,1 Гц, 2H), 3,43 (м, 6H), 3,30-3,10 (м, 2H), 2,73-2,58 (м, 4H), 2,12-1,94 (м, 2H); МСНР (ЭР) m/z 467,19 (M++ 1).
Пример 272: Синтез соединения 272, (2-(7-амино-2-(фуран-2-ил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-5-ил)пиразолидин-1-ил)(4-(2,2,2-трифторэтил)пиперазин-1-ил)метанона
[Стадия 1] Синтез трет-бутил (S)-(1-(4-(2,4-дифторфенил)пиперазин-1-ил)-3-гидрокси-1-оксопропан-2-ил)карбамата
1-(2,4-дифторфенил)пиперазин (10,000 г, 50,449 ммоль), (трет-бутоксикарбонил)-L-серин (20,705 г, 100,898 ммоль), гексафторфосфат 3-оксида 1-[бис(диметиламино)метилен]-1H-1,2,3-триазоло[4,5-b]пиридиния (ГАТУ, 38,364 г, 100,898 ммоль) и N, N-диизопропилэтиламин (43,936 мл, 252,245 ммоль) растворяют в дихлорметане (500 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего насыщенный водный раствор хлорида аммония выливают в полученный концентрат и органический слой экстрагируют этилацетатом. Органический слой промывают насыщенным водным раствором гидрокарбоната натрия, дегидратируют безводным сульфатом магния, фильтруют, и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 120 г картридж; этилацетат/гексан=0-45%) и концентрируют с получением указанного в заголовке соединения (18,000 г, 92,6%) в виде белого твердого вещества пенистого типа.
[Стадия 2] Синтез (S)-2-((трет-бутоксикарбонил)амино)-3-(4-(2,4-дифторфенил)пиперазин-1-ил)-3-оксопропилметансульфоната
Трет-бутил (S)-(1-(4-(2,4-дифторфенил)пиперазин-1-ил)-3-гидрокси-1-оксопропан-2-ил)карбамат (18,000 г, 46,704 ммоль), полученный на стадии 1, метансульфонилхлорид (3,976 мл, 51,374 ммоль) и триэтиламин (13,019 мл, 93,407 ммоль) растворяют в дихлорметане (500 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор хлорида аммония выливают в реакционную смесь, и органический слой экстрагируют дихлорметаном. Органический слой промывают насыщенным водным раствором гидрокарбоната натрия, дегидратируют безводным сульфатом магния, фильтруют, и концентрируют при пониженном давлении. Полученный продукт используют без дополнительного процесса очистки (указанное в заголовке соединение, 20,000 г, 92,4%, желтое масло).
[Стадия 3] Синтез трет-бутил (S)-(1-(4-(2,4-дифторфенил)пиперазин-1-ил)-1-оксо-3-(пиперидин-1-ил)пропан-2-ил)карбамата
(S)-2-((Трет-бутоксикарбонил)амино)-3-(4-(2,4-дифторфенил)пиперазин-1-ил)-3-оксопропилметансульфонат (1,000 г, 2,157 ммоль), полученный на стадии 2, карбонат цезия (1,406 г, 4,315 ммоль) и пиперидин (0,426 мл, 4,315 ммоль) растворяют в ацетонитриле (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при 90°C в течение 18 часов до завершения реакции путем понижения температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, и органический слой экстрагируют этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют, и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-40%) и концентрируют с получением указанного в заголовке соединения (0,450 г, 46,1%) в виде бесцветного масла.
[Стадия 4] Синтез (S)-2-амино-1-(4-(2,4-дифторфенил)пиперазин-1-ил)-3-(пиперидин-1-ил)пропан-1-она
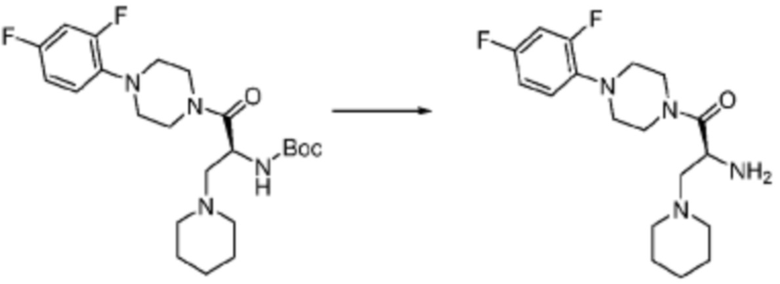
Трет-бутил (S)-(1-(4-(2,4-дифторфенил)пиперазин-1-ил)-1-оксо-3-(пиперидин-1-ил)пропан-2-1)карбамат (0,200 г, 0,442 ммоль), полученный на стадии 3, и хлористоводородную кислоту (4,00 M раствор в диоксане, 1,105 мл, 4,419 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный продукт используют без дополнительного процесса очистки (указанное в заголовке соединение, 0,100 г, 64,2%, коричневое масло).
[Стадия 5] Синтез (S)-2-((7-амино-2-(фуран-2-ил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-5-ил)амино)-1-(4-(2,4-дифторфенил)пиперазин-1-ил)-3-(пиперидин-1-ил)пропан-1-она
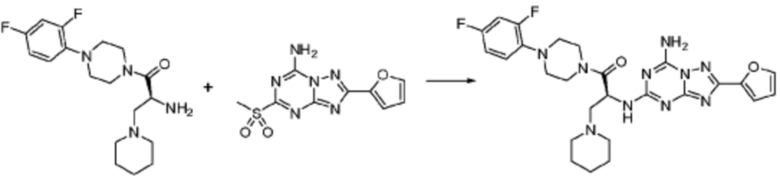
(S)-2-Амино-1-(4-(2,4-дифторфенил)пиперазин-1-ил)-3-(пиперидин-1-ил)пропан-1-он (0,100 г, 0,284 ммоль), полученный на стадии 4, 2-(фуран-2-ил)-5-(метилсульфонил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амин (0,080 г, 0,284 ммоль) и гидрокарбонат натрия (0,048 г, 0,567 ммоль) растворяют в ацетонитриле (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при 70°C в течение 18 часов до завершения реакции путем понижения температуры до комнатной температуры. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный концентрат очищают хроматографией (SiO2 тарелка, 20×20x1 мм; метанол/этилацетат=0-10%) и концентрируют с получением указанного в заголовке соединения (0,020 г, 12,8%) в виде желтого твердого вещества.
1H ЯМР (400 MГц, ДМСО-d6) δ 8,31 (м, 2H), 7,87 (с, 1H), 7,45 (дд, J=37,2, 7,0 Гц, 1H), 7,26-6,89 (м, 4H), 6,68 (дд, J=3,3, 1,8 Гц, 1H), 5,12-4,95 (м, 1H), 3,65 (м, 5H), 3,22 (м, 1H), 3,08-2,78 (м, 4H), 2,68-2,56 (м, 2H), 2,37 (м, 2H), 1,42 (м, 4H), 1,35 (м, 2H).
Пример 273: Синтез соединения 273
Пример соединения 273 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 272 с применением (S)-2-((трет-бутоксикарбонил)амино)-3-(4-(2,4-дифторфенил)пиперазин-1-ил)-3-оксопропилметансульфоната, полученного на стадии 2, за исключением применения морфолина вместо пиперидина.
1H ЯМР (400 MГц, ДМСО-d6) δ 8,47-8,00 (м, 2H), 7,87 (с, 1H), 7,50 (дд, J=32,6, 7,7 Гц, 1H), 7,29-6,92 (м, 4H), 6,68 (дд, J=3,2, 1,8 Гц, 1H), 5,11-4,95 (м, 1H), 3,95-3,58 (м, 4H), 3,55 (д, J=13,7 Гц, 4H), 3,30-3,13 (м, 2H), 3,11-2,79 (м, 4H), 2,70-2,48 (м, 4H).
Пример 44: Синтез соединения 44, ((S)-1-(7-амино-2-(фуран-2-ил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-5-ил)пирролидин-2-ил)((1S,4S)-5-(2,4-дифторбензил)-2,5-диазабицикло[2,2,1]гептан-2-ил)метанона
[Стадия 1] Синтез трет-бутил (1S,4S)-5-(2,4-дифторбензил)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилата
Трет-бутил (1S,4S)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилат (10,000 г, 50,436 ммоль), 1-(бромметил)-2,4-дифторбензол (6,406 мл, 50,436 ммоль) и карбонат калия (27,882 г, 201,745 ммоль) растворяют в N, N-диметилформамиде (80 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Воду выливают в реакционную смесь и органический слой экстрагируют этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют, и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением указанного в заголовке соединения (16,000 г, 97,8%) в виде белого твердого вещества.
[Стадия 2] Синтез (1S,4S)-2-(2,4-дифторбензил)-2,5-диазабицикло[2,2,1]гептана

Трет-бутил (1S,4S)-5-(2,4-дифторбензил)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилат (12,000 г, 36,995 ммоль), полученный на стадии 1, и хлористоводородную кислоту (4,00 M раствор в диоксане, 92,487 мл, 369,948 ммоль) растворяют в дихлорметане (100 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный продукт используют без дополнительного процесса очистки (указанное в заголовке соединение, 8,000 г, 96,4%, белое твердое вещество).
[Стадия 3] Синтез трет-бутил (S)-2-((1S,4S)-5-(2,4-дифторбензил)-2,5-диазабицикло[2,2,1]гептан-2-карбонил)пирролидин-1-карбоксилата
(1S,4S)-2-(2,4-Дифторбензил)-2,5-диазабицикло[2,2,1]гептан (0,500 г, 2,230 ммоль), полученный на стадии 2, (трет-бутоксикарбонил)-L-пролин (0,960 г, 4,459 ммоль), [диметиламино(триазоло[4,5-b]пиридин-3-илокси)метилиден]-диметилазания; гексафторфосфат (1,696 г, 4,459 ммоль) и N, N-диизопропилэтиламин (0,777 мл, 4,459 ммоль) растворяют в N, N-диметилформамиде (15 мл) при комнатной температуре, и полученный раствор перемешивают при той же температуре в течение 18 часов. Воду выливают в реакционную смесь и органический слой экстрагируют этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют, и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,200 г, 21,3%) в виде белого твердого вещества.
[Стадия 4] Синтез (1S,4S)-2-(L-пропил)-5-(2,4-дифторбензил)-2,5-диазабицикло[2,2,1]гептана
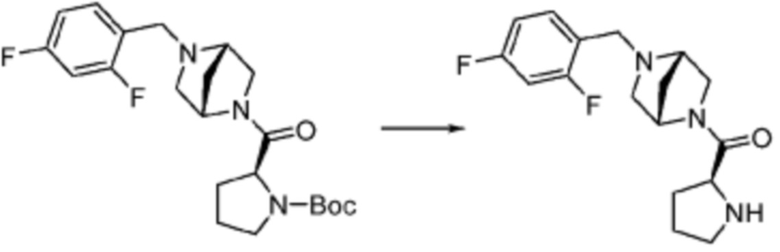
Трет-бутил (S)-2-((1S,4S)-5-(2,4-дифторбензил)-2,5-диазабицикло[2,2,1]гептан-2-карбонил)пирролидин-1-карбоксилат (0,200 г, 0,475 ммоль), полученный на стадии 3, и хлористоводородную кислоту (4,00 M раствор в диоксане, 1,186 мл, 4,745 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный продукт используют без дополнительного процесса очистки (указанное в заголовке соединение, 0,150 г, 98,4%, белое твердое вещество).
[Стадия 5] Синтез ((S)-1-(7-амино-2-(фуран-2-ил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-5-ил)пирролидин-2-ил)((1S,4S)-5-(2,4-дифторбензил)-2,5-диазабицикло[2,2,1]гептан-2-ил)метанона
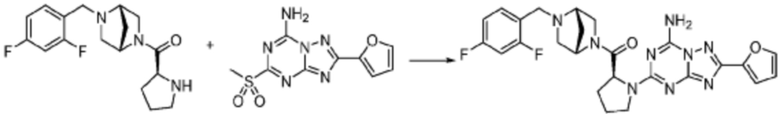
(1S,4S)-2-(L-Пропил)-5-(2,4-дифторбензил)-2,5-диазабицикло[2,2,1]гептан (0,200 г, 0,622 ммоль), полученный на стадии 4, 2-(фуран-2-ил)-5-(метилсульфонил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амин (0,087 г, 0,311 ммоль) и триэтиламин (0,173 мл, 1,245 ммоль) растворяют в диметилсульфоксиде (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Воду выливают в реакционную смесь, и органический слой экстрагируют этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют, и концентрируют при пониженном давлении. Полученный концентрат очищают хроматографией (SiO2 тарелка, 20×20x1 мм; метанол/дихлорметан=10%) и концентрируют с получением указанного в заголовке соединения (0,040 г, 12,3%) в виде белого твердого вещества.
1H ЯМР (400 MГц, ДМСО-d6) δ 8,33 (м, 1H), 7,90-7,44 (м, 2H), 7,18 (м, 1H), 7,12-6,87 (м, 2H), 6,67 (м, 1H), 4,66 (м, 2H), 4,13-3,37 (м, 7H), 3,29-2,74 (м, 2H), 2,47-2,20 (м, 2H), 2,14-1,64 (м, 5H); МСНР (ЭР) m/z 522,4 (M++ 1).
Пример 49, 50, 51 и 52
Примеры соединений 49, 50, 51 и 52 каждый синтезируют по существу тем же способом синтеза, как и способ синтеза из Примера соединения 44 за исключением применения соединений из следующей таблицы вместо трет-бутил (1S,4S)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилата на стадии 1.
[Таблица 21]
№
№
Примеры 30 и 31
Примеры соединений 30 и 31 каждый синтезируют по существу тем же способом синтеза, как и способ синтеза из Примера соединения 44 за исключением применения соединений из следующей таблицы вместо трет-бутил (1S,4S)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилата на стадии 1 и с применением (трет-бутоксикарбонил)-L-аланина вместо (трет-бутоксикарбонил)-L-пролина на стадии 3.
[Таблица 22]
№
№
Пример 32: Синтез соединения 32
Пример соединения 32 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 44 за исключением применения (трет-бутоксикарбонил)-L-аланина вместо (трет-бутоксикарбонил)-L-пролина на стадии 3.
Аналитические данные каждого из соединений, полученных как описано выше, показаны в таблице ниже.
[Таблица 23]
Пример 157: Синтез соединения 157, (3-((7-амино-2-(фуран-2-ил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-5-ил)-L-пропил)-3,8-диазабицикло[3,2,1]октан-8-ил)(фенил)метанона
[Стадия 1] Синтез трет-бутил 8-бензоил-3,8-диазабицикло[3,2,1]октан-3-карбоксилата
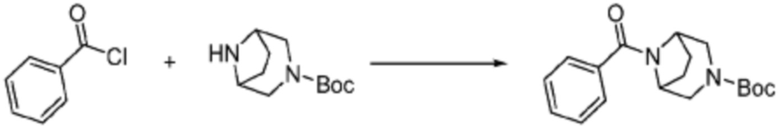
Трет-бутил 3,8-диазабицикло[3,2,1]октан-3-карбоксилат (0,319 г, 1,503 ммоль), бензоилхлорид (0,175 мл, 1,503 ммоль) и триэтиламин (0,314 мл, 2,254 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Водный раствор N-гидрокарбонат натрия выливают в реакционную смесь, и затем органический слой экстрагируют дихлорметаном, фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора из него, и концентрируют при пониженном давлении. Полученный продукт используют без дополнительного процесса очистки (указанное в заголовке соединение, 0,474 г, 99,7%, светло-желтое масло).
[Стадия 2] Синтез (3,8-диазабицикло[3,2,1]октан-8-ил)(фенил)метанона гидрохлорида

Трет-бутил 8-бензоил-3,8-диазабицикло[3,2,1]октан-3-карбоксилат (0,474 г, 1,498 ммоль), полученный на стадии 1, и хлористоводородную кислоту (4,00 M раствор в 1,4-диоксане, 1,498 мл, 5,992 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный продукт используют без дополнительного процесса очистки (указанное в заголовке соединение, 0,378 г, 99,8%, светло-желтое масло).
[Стадия 3] Синтез трет-бутил (2S)-2-(8-бензоил-3,8-диазабицикло[3,2,1]октан-3-карбонил)пирролидин-1-карбоксилата
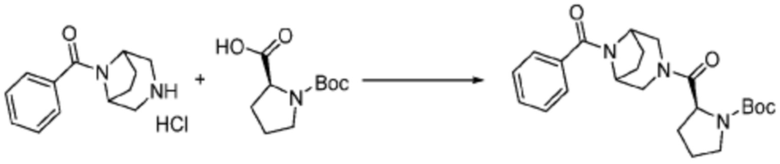
(3,8-Диазабицикло[3,2,1]октан-8-ил)(фенил)метанон гидрохлорид (0,378 г, 1,496 ммоль), полученный на стадии 2, (трет-бутоксикарбонил)-L-пролин (0,322 г, 1,496 ммоль), 2,4,6-трипропил-1,3,5,2,4,6-триоксатрифосфинеин 2,4,6-триоксид (T3P, 50,00% раствор в EtOAc, 1,334 мл, 2,243 ммоль) и N, N-диизопропилэтиламин (0,782 мл, 4,487 ммоль) растворяют в дихлорметане (8 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Водный раствор N-гидрокарбоната натрия выливают в реакционную смесь, и затем органический слой экстрагируют дихлорметаном, фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора из него, и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; дихлорметан/метанол=0-5%) и концентрируют с получением указанного в заголовке соединения (0,424 г, 68,6%) в виде прозрачного масла.
[Стадия 4] Синтез (3-(L-пропил)-3,8-диазабицикло[3,2,1]октан-8-ил)(фенил)метанона гидрохлорида

Трет-бутил (2S)-2-(8-бензоил-3,8-диазабицикло[3,2,1]октан-3-карбонил)пирролидин-1-карбоксилат (0,424 г, 1,025 ммоль), полученный на стадии 3, и хлороводород (4,00 M раствор в 1,4-диоксане, 1,025 мл, 4,101 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный продукт используют без дополнительного процесса очистки (указанное в заголовке соединение, 0,358 г, 99,8%, светло-желтое масло).
[Стадия 5] Синтез (3-((7-амино-2-(фуран-2-ил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-5-ил)-L-пропил)-3,8-диазабицикло[3,2,1]октан-8-ил)(фенил)метанона
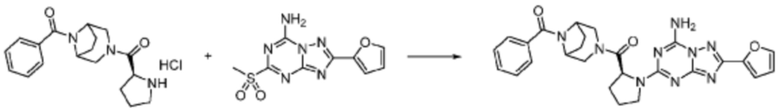
(3-(L-Пропил)-3,8-диазабицикло[3,2,1]октан-8-ил)(фенил)метанон гидрохлорид (0,358 г, 1,023 ммоль), полученный на стадии 4, 2-(фуран-2-ил)-5-(метилсульфонил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амин (0,287 г, 1,023 ммоль) и гидрокарбонат натрия (0,258 г, 3,070 ммоль) растворяют в циклопентилметиловом эфире (ЦПМЭ, 5 мл) при комнатной температуре, после чего полученный раствор перемешивают при 50°C в течение 18 часов до завершения реакции путем понижения температуры до комнатной температуры. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; дихлорметан/метанол=0-5%) и концентрируют с получением указанного в заголовке соединения (0,309 г, 58,9%) в виде белого твердого вещества.
1H ЯМР (400 MГц, Хлороформ-d) δ 7,69-7,33 (м, 5H), 7,24-7,09 (м, 1H), 6,51 (тд, J=3,7, 1,7 Гц, 1H), 6,31 (с, 1H), 5,18-4,69 (м, 2H), 4,16 (дд, J=78,6, 54,6 Гц, 2H), 3,92-3,49 (м, 3H), 3,42-2,78 (м, 1H), 2,61 (с, 1H), 2,53-2,26 (м, 1H), 2,26-2,09 (м, 2H), 2,00 (дп, J=17,5, 7,3, 6,1 Гц, 3H), 1,92-1,83 (м, 1H), 1,74 (дд, J=15,6, 8,1 Гц, 1H). МСНР (ЭР) m/z 514,5(M++ 1).
Примеры 158, 159, 160 и 161
Примеры соединений 158-161 каждый синтезируют по существу тем же способом синтеза, как и способ синтеза из Примера соединения 157 за исключением применения соединений из следующей таблицы вместо трет-бутил 3,8-диазабицикло[3,2,1]октан-3-карбоксилата на стадии 1 в качестве исходного материала.
[Таблица 24]
№
№
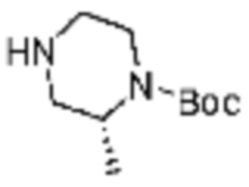
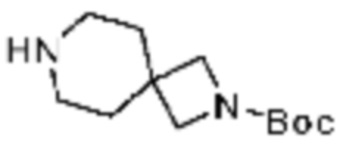
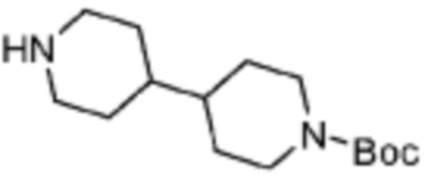
Аналитические данные каждого из соединений, полученных как описано выше, показаны в таблице ниже.
[Таблица 25]
Пример 277: Синтез соединения 277, (S)-(1-(7-амино-2-(фуран-2-ил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-5-ил)пирролидин-2-ил)(4-(пиримидин-2-ил)-1,4-диазепан-1-ил)метанона
[Стадия 1] Синтез трет-бутил 4-(пиримидин-2-ил)-1,4-диазепан-1-карбоксилата
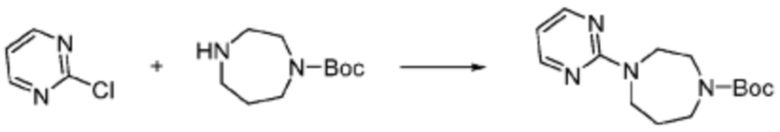
2-Хлорпиримидин (0,229 г, 1,999 ммоль), трет-бутил 1,4-диазепан-1-карбоксилат (0,400 г, 1,999 ммоль) и карбонат калия (0,829 г, 5,998 ммоль) растворяют в ацетонитриле (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при 80°C в течение 18 часов до завершения реакции путем понижения температуры до комнатной температуры. Воду выливают в реакционную смесь и органический слой экстрагируют дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют, и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; дихлорметан/метанол=0-30%) и концентрируют с получением указанного в заголовке соединения (0,556 г, 99,9%) в виде светло-желтой жидкости.
[Стадия 2] Синтез 1-(пиримидин-2-ил)-1,4-диазепана гидрохлорида

Трет-бутил 4-(пиримидин-2-ил)-1,4-диазепан-1-карбоксилат (0,556 г, 1,997 ммоль), полученный на стадии 1, и хлористоводородную кислоту (4,00 M раствор в 1,4-диоксане, 1,997 мл, 7,990 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение пяти часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный продукт используют без дополнительного процесса очистки (указанное в заголовке соединение, 0,215 г, 50,1%, белое твердое вещество).
[Стадия 3] Синтез трет-бутил (S)-2-(4-(пиримидин-2-ил)-1,4-диазепан-1-карбонил)пирролидин-1-карбоксилата
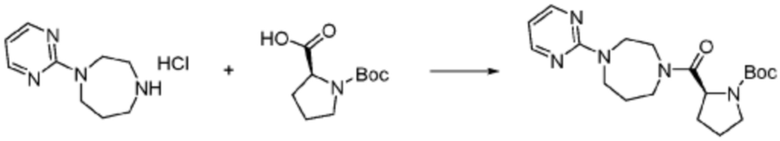
1-(Пиримидин-2-ил)-1,4-диазепан гидрохлорид (0,214 г, 0,997 ммоль), полученный на стадии 2, (трет-бутоксикарбонил)-L-пролин (0,215 г, 0,997 ммоль), 2,4,6-трипропил-1,3,5,2,4,6-триоксатрифосфинан 2,4,6-триоксид (T3P, 50,00% раствор в EtOAc, 0,914 мл, 1,495 ммоль) и N, N-диизопропилэтиламин (0,694 мл, 3,987 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Водный раствор N-гидрокарбонат натрия выливают в реакционную смесь, и затем органический слой экстрагируют дихлорметаном, фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора из него, и концентрируют при пониженном давлении. Полученный продукт используют без дополнительного процесса очистки (указанное в заголовке соединение, 0,3750 г, 100,0%, светло-желтая жидкость).
[Стадия 4] Синтез (S)-1-пропил-4-(пиримидин-2-ил)-1,4-диазепана гидрохлорида
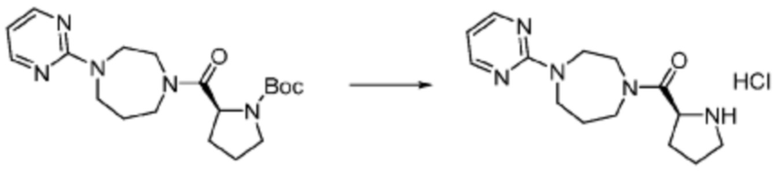
Трет-бутил (S)-2-(4-(пиримидин-2-ил)-1,4-диазепан-1-карбонил)пирролидин-1-карбоксилат (0,375 г, 0,999 ммоль), полученный на стадии 3, и хлористоводородную кислоту (4,00 M раствор в 1,4-диоксане, 0,999 мл, 3,995 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный продукт используют без дополнительного процесса очистки (указанное в заголовке соединение, 0,311 г, 99,9%, белое твердое вещество).
[Стадия 5] Синтез (S)-(1-(7-амино-2-(фуран-2-ил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-5-ил)пирролидин-2-ил)(4-(пиримидин-2-ил)-1,4-диазепан-1-ил)метанона
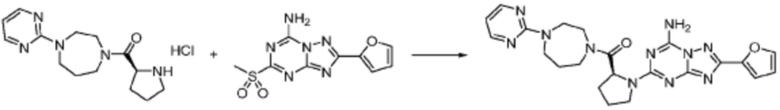
(S)-1-Пропил-4-(пиримидин-2-ил)-1,4-диазепан гидрохлорид (0,311 г, 0,997 ммоль), полученный на стадии 4, 2-(фуран-2-ил)-5-(метилсульфонил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амин (0,280 г, 0,997 ммоль) и гидрокарбонат натрия (0,251 г, 2,992 ммоль) растворяют в ацетонитриле (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при 70°C в течение 18 часов до завершения реакции путем понижения температуры до комнатной температуры. Реакционную смесь фильтруют через слой целита для удаления из нее твердого вещества, после чего растворитель удаляют из полученного фильтрата без твердого вещества при пониженном давлении. Затем, полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; дихлорметан/метанол= 0-30%) и концентрируют с получением указанного в заголовке соединения (0,109 г, 23,0%) в виде белого твердого вещества.
1H ЯМР (400 MГц, ДМСО-d6) δ 7,86 (дд, J=1,7, 0,8 Гц, 1H), 7,46 (шс, 2H), 7,06 (дд, J=3,4, 0,6 Гц, 1H), 6,67 (дд, J=3,4, 1,8 Гц, 1H), 5,59 (шс, 1H), 4,96 (шс, 1H), 3,79-3,38 (м, 5H), 2,79 (т, J=14,1 Гц, 3H), 2,64-2,36 (м, 4H), 2,24 (с, 1H), 2,06-1,77 (м, 3H), 1,66 (т, J=19,1 Гц, 3H); МСНР (ЭР) m/z 461,5 (M++ 1).
Примеры 328, 329 и 384
Примеры соединений 328, 329 и 384 каждый синтезируют по существу тем же способом синтеза, как и способ синтеза из Примера соединения 277 за исключением применения исходного материала 1 из таблицы ниже вместо 2-хлорпиримидина на стадии 1, с применением исходного материала 2 из таблицы ниже вместо трет-бутил 1,4-диазепан-1-карбоксилата, и с применением (S)-1-(трет-бутоксикарбонил)азетидин-2-карбоновой кислоты вместо (трет-бутоксикарбонил)-L-пролина на стадии 3.
[Таблица 26]
№
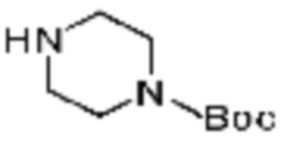
Пример 265: Синтез соединения 265
Пример соединения 265 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 328 за исключением применения (трет-бутоксикарбонил)-L-пролина вместо (S)-1-(трет-бутоксикарбонил)азетидин-2-карбоновой кислоты.
Пример 266: Синтез соединения 266
Пример соединения 266 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 329 за исключением применения (трет-бутоксикарбонил)-L-пролина вместо (S)-1-(трет-бутоксикарбонил)азетидин-2-карбоновой кислоты.
Пример 380: Синтез соединения 380
Пример соединения 380 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 329 за исключением применения 2-(фуран-2-ил)-7-(метилсульфонил)-[1,2,4]триазоло[1,5-c]пиримидин-5-амина вместо 2-(фуран-2-ил)-5-(метилсульфонил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амина.
Пример 383: Синтез соединения 383
Пример соединения 383 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 328 за исключением применения 2-(фуран-2-ил)-7-(метилсульфонил)-[1,2,4]триазоло[1,5-c]пиримидин-5-амина вместо 2-(фуран-2-ил)-5-(метилсульфонил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амина.
Аналитические данные каждого из соединений, полученных как описано выше, показаны в таблице ниже.
[Таблица 27]
№
Пример 229: Синтез соединения 229, 1-(4-((7-амино-2-(фуран-2-ил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-5-ил)-L-пропил)пиперазин-1-ил)-3-гидрокси-3-метилбутан-1-она
[Стадия 1] Синтез трет-бутил 4-(3-гидрокси-3-метилбутаноил)пиперазин-1-карбоксилата
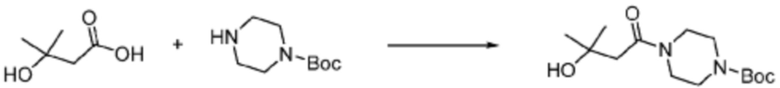
3-Гидрокси-3-метилбутановую кислоту (0,315 мл, 2,500 ммоль), трет-бутил пиперазин-1-карбоксилат (0,466 г, 2,500 ммоль), 1-этил-3-(3-диметиламинопропил)карбодиимид гидрохлорид (ЭДК-HCl, 0,959 г, 5,000 ммоль), 1H-бензо[d][1,2,3]триазол-1-ол (HOBt, 0,338 г, 2,500 ммоль) и N, N-диизопропилэтиламин (1,306 мл, 7,500 ммоль) растворяют в дихлорметане (8 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор хлорида аммония выливают в реакционную смесь, и затем органический слой экстрагируют дихлорметаном, фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора из него, и концентрируют при пониженном давлении. Полученный продукт используют без дополнительного процесса очистки (указанное в заголовке соединение, 0,715 г, 99,9%, белое твердое вещество).
[Стадия 2] Синтез 3-гидрокси-3-метил-1-(пиперазин-1-ил)бутан-1-она гидрохлорида
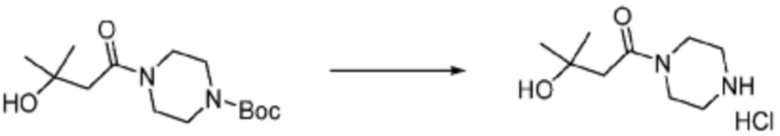
Трет-бутил 4-(3-гидрокси-3-метилбутаноил)пиперазин-1-карбоксилат (0,715 г, 2,497 ммоль), полученный на стадии 1, и хлороводород (4,00 M раствор в 1,4-диоксане, 3,121 мл, 12,484 ммоль) растворяют в дихлорметане (6 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный продукт используют без дополнительного процесса очистки (указанное в заголовке соединение, 0,555 г, 99,8%, белое твердое вещество).
[Стадия 3] Синтез трет-бутил (S)-2-(4-(3-гидрокси-3-метилбутаноил)пиперазин-1-карбонил)пирролидин-1-карбоксилата
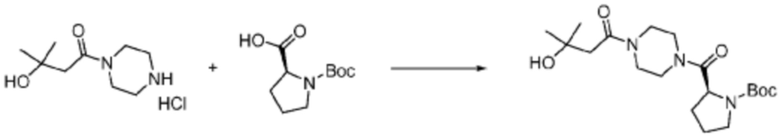
3-Гидрокси-3-метил-1-(пиперазин-1-ил)бутан-1-он гидрохлорид (0,555 г, 2,492 ммоль), полученный на стадии 2, (трет-бутоксикарбонил)-L-пролин (0,536 г, 2,492 ммоль), 1-этил-3-(3-диметиламинопропил)карбодиимид гидрохлорид (ЭДК-HCl, 0,955 г, 4,984 ммоль), 1H-бензо[d][1,2,3]триазол-1-ол (HOBt, 0,337 г, 2,492 ммоль) и N, N-диизопропилэтиламин (1,302 мл, 7,476 ммоль) растворяют в дихлорметане (8 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор хлорида аммония выливают в реакционную смесь, и затем органический слой экстрагируют дихлорметаном, фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора из него, и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 24 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,890 г, 93,1%) в виде бесцветного масла.
[Стадия 4] Синтез (S)-3-гидрокси-3-метил-1-(4-пропилпиперазин-1-ил)бутан-1-она гидрохлорида
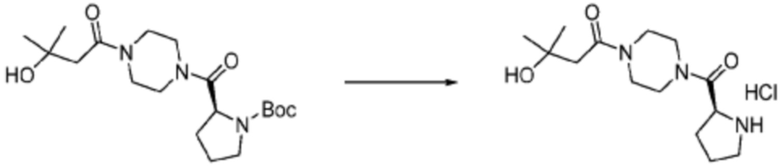
Трет-бутил (S)-2-(4-(3-гидрокси-3-метилбутаноил)пиперазин-1-карбонил)пирролидин-1-карбоксилат (0,890 г, 2,321 ммоль), полученный на стадии 3, и хлороводород (4,00 M раствор в 1,4-диоксане, 2,321 мл, 9,283 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при 40°C в течение двух часов до завершения реакции путем понижения температуры до комнатной температуры. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный продукт используют без дополнительного процесса очистки (указанное в заголовке соединение, 0,740 г, 99,7%, белое твердое вещество).
[Стадия 5] Синтез 1-(4-((7-амино-2-(фуран-2-ил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-5-ил)-L-пропил)пиперазин-1-ил)-3-гидрокси-3-метилбутан-1-она
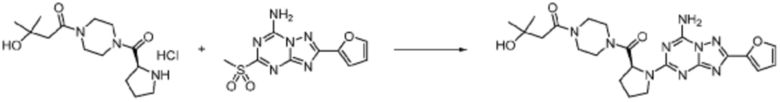
(S)-3-Гидрокси-3-метил-1-(4-пропилпиперазин-1-ил)бутан-1-он гидрохлорид (0,150 г, 0,469 ммоль), полученный на стадии 4, 2-(фуран-2-ил)-5-(метилсульфонил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амин (0,131 г, 0,469 ммоль) и гидрокарбонат натрия (0,079 г, 0,938 ммоль) растворяют в ацетонитриле (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при 50°C в течение 18 часов до завершения реакции путем понижения температуры до комнатной температуры. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением продукта, после чего полученный продукт снова очищают хроматографией (SiO2 тарелка, 20×20x1 мм; метанол/дихлорметан=10%) и концентрируют с получением указанного в заголовке соединения (0,117 г, 51,8%) в виде белого твердого вещества.
1H ЯМР (400 MГц, ДМСО-d6) δ 8,63-8,00 (м, 2H), 7,91-7,84 (м, 1H), 7,10-7,01 (м, 1H), 6,72-6,64 (м, 1H), 5,09-4,94 (м, 1H), 4,88-4,72 (м, 1H), 3,89-3,38 (м, 10H), 2,61-2,53 (м, 2H), 2,35-2,17 (м, 1H), 1,98-1,77 (м, 3H), 1,27-1,13 (м, 6H); МСНР (ЭР) m/z 484,3 (M++ 1).
Примеры 230 и 237
Примеры соединений 230 и 237 каждый синтезируют по существу тем же способом синтеза, как и способ синтеза из Примера соединения 229 за исключением применения исходных материалов из следующей таблицы вместо 3-гидрокси-3-метилбутановой кислоты на стадии 1.
[Таблица 28]
№
№
Пример 262: Синтез соединения 262
Пример соединения 262 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 268 за исключением применения 2-(фуран-2-ил)-5-(метилсульфонил)-3a,7a-дигидротиазоло[5,4-d]пиримидин-7-амина вместо 2-(фуран-2-ил)-5-(метилсульфонил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амина на стадии 5.
Пример 263: Синтез соединения 263
Пример соединения 263 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 229 за исключением применения 2-(фуран-2-ил)-5-(метилсульфонил)-3a,7a-дигидрооксазоло[5,4-d]пиримидин-7-амина вместо 2-(фуран-2-ил)-5-(метилсульфонил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амина на стадии 5.
Пример 267: Синтез соединения 267
Пример соединения 267 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 229 за исключением применения 2-гидрокси-2-метилпропановой кислоты вместо 3-гидрокси-3-метилбутановой кислоты на стадии 1, и с применением (S)-1-(трет-бутоксикарбонил)азетидин-2-карбоновой кислоты вместо (трет-бутоксикарбонил)-L-пролина на стадии 3.
Пример 268: Синтез соединения 268
Пример соединения 268 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 229 за исключением применения (S)-1-(трет-бутоксикарбонил)азетидин-2-карбоновой кислоты вместо (трет-бутоксикарбонил)-L-пролина на стадии 3.
Пример 274: Синтез соединения 274
Пример соединения 274 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 229 за исключением применения 2-гидрокси-2-метилпропановой кислоты вместо 3-гидрокси-3-метилбутановой кислоты на стадии 1.
Пример 275: Синтез соединения 275
Пример соединения 275 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 268 за исключением применения 2-(фуран-2-ил)-5-(метилсульфонил)-3a,7a-дигидротиазоло[5,4-d]пиримидин-7-амина вместо 2-(фуран-2-ил)-5-(метилсульфонил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амина на стадии 5.
Пример 290: Синтез соединения 290
Пример соединения 290 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 229 за исключением применения трет-бутил 4,4'-бипиперидин-1-карбоксилата вместо трет-бутил пиперазин-1-карбоксилата на стадии 1.
Пример 327: Синтез соединения 327
Пример соединения 327 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 290 за исключением применения (S)-1-(трет-бутоксикарбонил)азетидин-2-карбоновой кислоты вместо (трет-бутоксикарбонил)-L-пролина.
Пример 292: Синтез соединения 292
Пример соединения 292 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 229 за исключением применения 2-(фуран-2-ил)-7-(метилсульфонил)-[1,2,4]триазоло[1,5-c]пиримидин-5-амина вместо 2-(фуран-2-ил)-5-(метилсульфонил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амина на стадии 5.
Пример 315: Синтез соединения 315
Пример соединения 315 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 268 за исключением применения 2-(метилфуран-2-ил)-5-(метилсульфонил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амина вместо 2-(фуран-2-ил)-5-(метилсульфонил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амина на стадии 5.
Пример 340: Синтез соединения 340
Пример соединения 340 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 262 за исключением применения (трет-бутоксикарбонил)-L-аланина вместо (трет-бутоксикарбонил)-L-пролина.
Примеры 368, 369 и 370
Примеры соединений 368, 369 и 370 каждый синтезируют по существу тем же способом синтеза, как и способ синтеза из Примера соединения 229 за исключением применения соединений из следующей таблицы в качестве N-защищенной аминокислоты вместо (трет-бутоксикарбонил)-L-пролина на стадии 3.
[Таблица 29]
№
№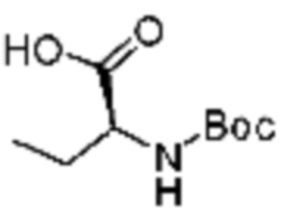
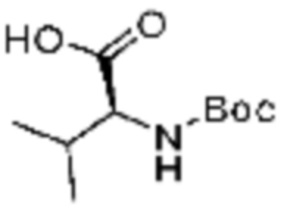
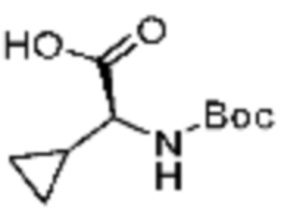
Аналитические данные каждого из соединений, полученных как описано выше, показаны в таблице ниже.
[Таблица 30]
Пример 276: Синтез соединения 276, (S)-(1-(7-амино-2-(фуран-2-ил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-5-ил)пирролидин-2-ил)(4-(2,4-дифторфенил)-1,4-диазепан-1-ил)метанона
[Стадия 1] Синтез трет-бутил 4-(2,4-дифторфенил)-1,4-диазепан-1-карбоксилата
1-Бром-2,4-дифторбензол (0,386 г, 2,000 ммоль), трет-бутил 1,4-диазепан-1-карбоксилат (0,401 г, 2,000 ммоль), хлор(2-дициклогексилфосфино-2′,6′-диизопропил-1,1′-дифенил)[2-(2′-амино-1,1′-дифенил)]палладий (II, 0,155 г, 0,200 ммоль) и карбонат цезия (1,955 г, 6,000 ммоль) растворяют в тетрагидрофуране (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при 80°C в течение 18 часов до завершения реакции путем понижения температуры до комнатной температуры. Воду выливают в реакционную смесь и органический слой экстрагируют дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют, и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; дихлорметан/метанол=0-30%) и концентрируют с получением указанного в заголовке соединения (0,624 г, 99,9%) в виде светло-желтой жидкости.
[Стадия 2] Синтез 1-(2,4-дифторфенил)-1,4-диазепана гидрохлорида
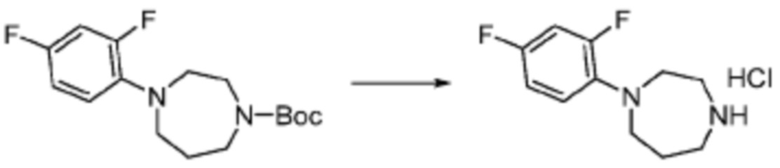
Трет-бутил 4-(2,4-дифторфенил)-1,4-диазепан-1-карбоксилат (0,624 г, 1,998 ммоль), полученный на стадии 1, и хлористоводородную кислоту (4,00 M раствор в 1,4-диоксане, 1,998 мл, 7,991 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение пяти часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный продукт используют без дополнительного процесса очистки (указанное в заголовке соединение, 0,248 г, 49,9%, белое твердое вещество).
[Стадия 3] Синтез трет-бутил (S)-2-(4-(2,4-дифторфенил)-1,4-диазепан-1-карбонил)пирролидин-1-карбоксилат
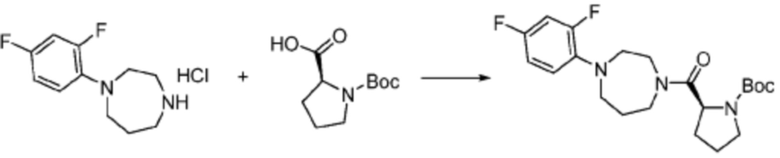
1-(2,4-Дифторфенил)-1,4-диазепан гидрохлорид (0,497 г, 1,998 ммоль), полученный на стадии 2, (трет-бутоксикарбонил)-L-пролин (0,430 г, 1,998 ммоль), 2,4,6-трипропил-1,3,5,2,4,6-триоксатрифосфинан 2,4,6-триоксид (T3P, 50,00% раствор в EtOAc, 1,833 мл, 2,998 ммоль) и N, N-диизопропилэтиламин (1,392 мл, 7,994 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Водный раствор N-гидрокарбоната натрия выливают в реакционную смесь, и затем органический слой экстрагируют дихлорметаном, фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора из него, и концентрируют при пониженном давлении. Полученный продукт используют без дополнительного процесса очистки (указанное в заголовке соединение, 0,409 г, 50,0%, светло-желтая жидкость).
[Стадия 4] Синтез (S)-1-(2,4-дифторфенил)-4-пропил-1,4-диазепана гидрохлорида
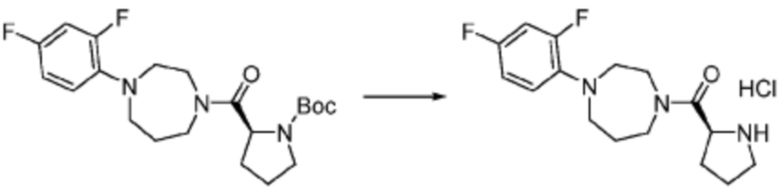
Трет-бутил (S)-2-(4-(2,4-дифторфенил)-1,4-диазепан-1-карбонил)пирролидин-1-карбоксилат (0,409 г, 0,999 ммоль), полученный на стадии 3, и хлористоводородную кислоту (4,00 M раствор в 1,4-диоксане, 0,999 мл, 3,995 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный продукт используют без дополнительного процесса очистки (указанное в заголовке соединение, 0,345 г, 99,9%, белое твердое вещество).
[Стадия 5] Синтез (S)-(1-(7-амино-2-(фуран-2-ил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-5-ил)пирролидин-2-ил)(4-(2,4-дифторфенил)-1,4-диазепан-1-ил)метанона
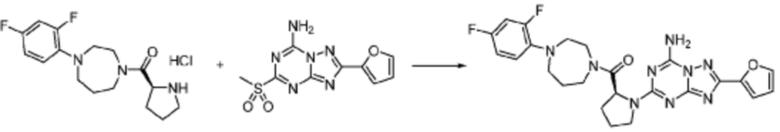
(S)-1-(2,4-Дифторфенил)-4-пропил-1,4-диазепан гидрохлорид (0,345 г, 0,998 ммоль), полученный на стадии 4, 2-(фуран-2-ил)-5-(метилсульфонил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амин (0,280 г, 0,998 ммоль) и (S)-(1-(7-амино-2-(фуран-2-ил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-5-ил)пирролидин-2-ил)(4-(2,4-дифторфенил)-1,4-диазепан-1-ил)метанон (1,525 г, 2,993 ммоль) растворяют в ацетонитриле (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при 70°C в течение 18 часов до завершения реакции путем понижения температуры до комнатной температуры. Реакционную смесь фильтруют через слой целита для удаления из нее твердого вещества, после чего растворитель удаляют из полученного фильтрата без твердого вещества при пониженном давлении. Затем, полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; дихлорметан/метанол= 0-30%) и концентрируют с получением указанного в заголовке соединения (0,011 г, 2,1%) в виде белого твердого вещества.
1H ЯМР (400 MГц, ДМСО-d6) δ 8,54-8,06 (м, 2H), 7,87 (с, 1H), 7,22-6,83 (м, 4H), 6,68 (ддд, J=5,2, 3,5, 1,6 Гц, 1H), 5,03-4,91 (м, 1H), 3,87-3,37 (м, 10H), 2,34-2,09 (м, 2H), 1,97-1,78 (м, 4H); МСНР (ЭР) m/z 510,5 (M++ 1).
Пример 42: Синтез соединения 42, (S)-(1-(7-амино-2-(фуран-2-ил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-5-ил)пирролидин-2-ил)(4-(2-этил-2-гидроксибутил)пиперазин-1-ил)метанона
[Стадия 1] Синтез трет-бутил 4-(2-этил-2-гидроксибутил)пиперазин-1-карбоксилата
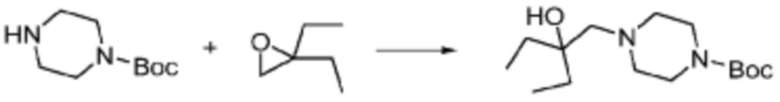
Трет-бутил пиперазин-1-карбоксилат (2,000 г, 10,738 ммоль), 2,2-диэтилоксиран (2,151 г, 21,475 ммоль) и карбонат калия (5,936 г, 42,951 ммоль) смешивают в этаноле (10 мл)/воде (2 мл) при комнатной температуре, облучают микроволнами, и нагревают при 110°C в течение одного часа до завершения реакции путем понижения температуры до комнатной температуры. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего воду выливают в полученный концентрат и органический слой экстрагируют этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют, и концентрируют при пониженном давлении. Полученный продукт используют без дополнительного процесса очистки (указанное в заголовке соединение, 2,800 г, 91,0%, белое твердое вещество).
[Стадия 2] Синтез 3-(пиперазин-1-илметил)пентан-3-ола
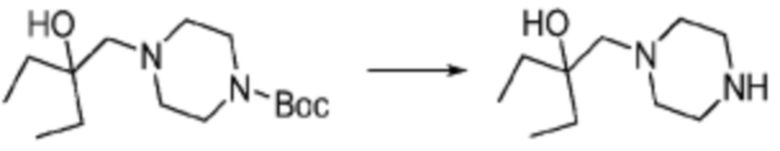
Трет-бутил 4-(2-этил-2-гидроксибутил)пиперазин-1-карбоксилат (0,600 г, 2,095 ммоль), полученный на стадии 1, и хлористоводородную кислоту (4,00 M раствор в диоксане, 5,237 мл, 20,948 ммоль) растворяют в дихлорметане (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный продукт используют без дополнительного процесса очистки (указанное в заголовке соединение, 0,300 г, 76,9%, белое твердое вещество).
[Стадия 3] Синтез трет-бутил (S)-2-(4-(2-этил-2-гидроксибутил)пиперазин-1-карбонил)пирролидин-1-карбоксилата
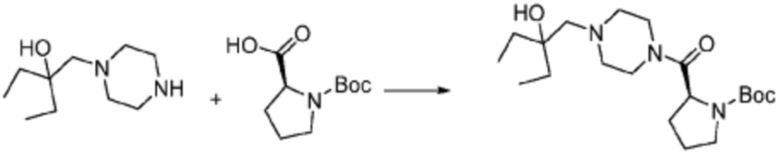
3-(Пиперазин-1-илметил)пентан-3-ол (0,500 г, 2,684 ммоль), полученный на стадии 2, (трет-бутоксикарбонил)-L-пролин (1,155 г, 5,368 ммоль), [диметиламино(триазоло[4,5-b]пиридин-3-илокси)метилиден]-диметилазания; гексафторфосфат (2,041 г, 5,368 ммоль) и N, N-диизопропилэтиламин (0,935 мл, 5,368 ммоль) растворяют в N, N-диметилформамиде (20 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Воду выливают в реакционную смесь и органический слой экстрагируют этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют, и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,200 г, 19,4%) в виде белого твердого вещества.
[Стадия 4] Синтез (S)-1-(2-этил-2-гидроксибутил)-4-пропилпиперазина
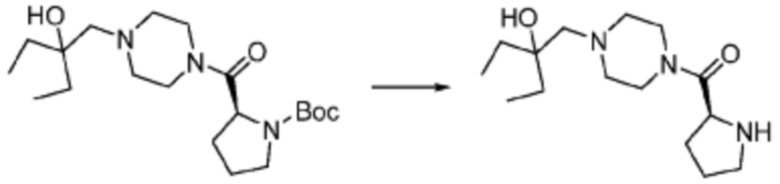
Трет-бутил (S)-2-(4-(2-этил-2-гидроксибутил)пиперазин-1-карбонил)пирролидин-1-карбоксилат (0,200 г, 0,501 ммоль), полученный на стадии 3, и хлористоводородную кислоту (4,00 M раствор в диоксане, 1,252 мл, 5,006 ммоль) растворяют в дихлорметане (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный продукт используют без дополнительного процесса очистки (указанное в заголовке соединение, 0,140 г, 93,4%, белое твердое вещество).
[Стадия 5] Синтез (S)-(1-(7-амино-2-(фуран-2-ил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-5-ил)пирролидин-2-ил)(4-(2-этил-2-гидроксибутил)пиперазин-1-ил)метанона
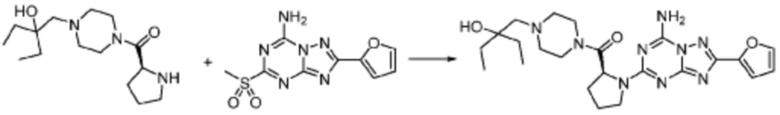
(S)-1-(2-Этил-2-гидроксибутил)-4-пропилпиперазин (0,200 г, 0,706 ммоль), полученный на стадии 4, 2-(фуран-2-ил)-5-(метилсульфонил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амин (0,099 г, 0,353 ммоль) и триэтиламин (0,197 мл, 1,411 ммоль) растворяют в диметилсульфоксиде (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Воду выливают в реакционную смесь и органический слой экстрагируют этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют, и концентрируют при пониженном давлении. Полученный концентрат очищают хроматографией (SiO2 тарелка, 20×20x1 мм; метанол/дихлорметан=10%) и концентрируют с получением указанного в заголовке соединения (0,006 г, 1,8%) в виде белого твердого вещества.
1H ЯМР (400 MГц, ДМСО-d6) δ 8,54-8,06 (м, 2H), 7,87 (с, 1H), 7,06 (д, J=3,1 Гц, 1H), 6,68 (д, J=3,3 Гц, 1H), 5,03-4,91 (м, 1H), 3,92-3,79 (м, 1H), 3,72-3,39 (м, 6H), 2,89-2,55 (м, 2H), 2,43-2,14 (м, 5H), 2,01-1,75 (м, 3H), 1,57-1,26 (м, 4H), 0,80 (т, J=7,0 Гц, 6H); МСНР (ЭР) m/z 484,4 (M++ 1).
Пример 88: Синтез соединения 88
Пример соединения 88 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 42 за исключением применения 2-(5-метилфуран-2-ил)-5-(метилсульфонил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амина вместо 2-(фуран-2-ил)-5-(метилсульфонил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амина на стадии 5.
Пример 185: Синтез соединения 185
Пример соединения 185 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 42 за исключением применения (S)-2-((трет-бутоксикарбонил)амино)-2-циклогексилуксусной кислоты вместо (трет-бутоксикарбонил)-L-пролина на стадии 3.
Пример 347: Синтез соединения 347
Пример соединения 347 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 42 за исключением применения (S)-1-(трет-бутоксикарбонил)азетидин-2-карбоновой кислоты вместо (трет-бутоксикарбонил)-L-пролина на стадии 3.
Пример 348: Синтез соединения 348
Пример соединения 348 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 42 за исключением применения (трет-бутоксикарбонил)-L-валина вместо (трет-бутоксикарбонил)-L-пролина на стадии 3.
Пример 244: Синтез соединения 244
Пример соединения 244 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 42 за исключением применения трет-бутил [4,4'-бипиперидин]-1-карбоксилата вместо трет-бутил пиперазин-1-карбоксилата на стадии 1 и с применением 2,2-диметилоксирана вместо 2,2-диэтилоксирана.
Пример 333: Синтез соединения 333
Пример соединения 333 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 244 за исключением применения (трет-бутоксикарбонил)-L-аланина вместо (трет-бутоксикарбонил)-L-пролина.
Пример 373: Синтез соединения 373
Пример соединения 373 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 244 за исключением применения трет-бутил (R)-2-метилпиперазин-1-карбоксилата вместо трет-бутил пиперазин-1-карбоксилата.
Пример 381: Синтез соединения 381
Пример соединения 333 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 244 за исключением применения (S)-1-(трет-бутоксикарбонил)азетидин-2-карбоновой кислоты вместо (трет-бутоксикарбонил)-L-пролина.
Аналитические данные каждого из соединений, полученных как описано выше, показаны в таблице ниже.
[Таблица 31]
№
Пример 223: Синтез соединения 223, (S)-(1-(7-амино-2-(фуран-2-ил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-5-ил)пирролидин-2-ил)(4-(2,2-дифторэтил)пиперазин-1-ил)метанона
[Стадия 1] Синтез трет-бутил 4-(2,2-дифторэтил)пиперазин-1-карбоксилата
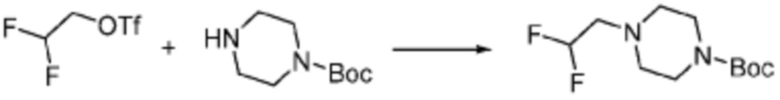
2,2-Дифторэтил трифторметансульфонат (95,00% раствор 0,279 мл, 1,899 ммоль), трет-бутил пиперазин-1-карбоксилат (0,354 г, 1,899 ммоль) и N, N-диизопропилэтиламин (0,331 мл, 1,899 ммоль) растворяют в тетрагидрофуране (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего водный раствор хлорида N-аммония выливают в полученный концентрат, и затем органический слой экстрагируют дихлорметаном, фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора из него, и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; дихлорметан/метанол=0-20%), и концентрируют с получением указанного в заголовке соединения (0,411 г, 86,1%) в виде белого твердого вещества.
[Стадия 2] Синтез 1-(2,2-дифторэтил)пиперазина гидрохлорида

Трет-бутил 4-(2,2-дифторэтил)пиперазин-1-карбоксилат (0,250 г, 0,999 ммоль), полученный на стадии 1, и хлористоводородную кислоту (4,00 M раствор в 1,4-диоксане, 0,999 мл, 3,995 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный продукт используют без дополнительного процесса очистки (указанное в заголовке соединение, 0,186 г, 99,8%, белое твердое вещество).
[Стадия 3] Синтез трет-бутил (S)-2-(4-(2,2-дифторэтил)пиперазин-1-карбонил)пирролидин-1-карбоксилата
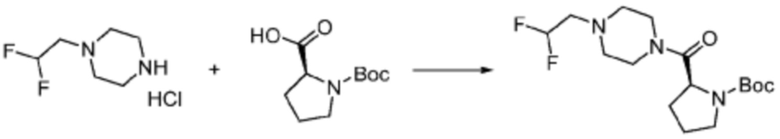
1-(2,2-Дифторэтил)пиперазин гидрохлорид (3,614 г, 19,365 ммоль), полученный на стадии 2, (трет-бутоксикарбонил)-L-пролин (4,168 г, 19,365 ммоль), 2,4,6-трипропил-1,3,5,2,4,6-триоксатрифосфинан 2,4,6-триоксид (T3P, 50,00% раствор в EtOAc, 17,767 мл, 29,048 ммоль) и N, N-диизопропилэтиламин (10,119 мл, 58,095 ммоль) растворяют в дихлорметане (130 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Водный раствор N-гидрокарбонат натрия выливают в реакционную смесь и органический слой экстрагируют дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют, и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; дихлорметан/метанол) и концентрируют с получением указанного в заголовке соединения (5,360 г, 79,7%) в виде желтой жидкости.
[Стадия 4] Синтез (S)-1-(2,2-дифторэтил)-4-пропилпиперазина гидрохлорида
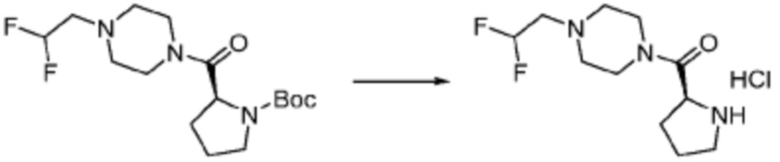
Трет-бутил (S)-2-(4-(2,2-дифторэтил)пиперазин-1-карбонил)пирролидин-1-карбоксилат (5,360 г, 15,428 ммоль), полученный на стадии 3, и хлороводород (4,00 M раствор в 1,4-диоксане, 15,428 мл, 61,714 ммоль) растворяют в дихлорметане (65 мл) при комнатной температуре, после чего полученный раствор перемешивают при 40°C в течение пяти часов до завершения реакции путем понижения температуры до комнатной температуры. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный продукт используют без дополнительного процесса очистки (указанное в заголовке соединение, 4,377 г, 100,0%, белое твердое вещество).
[Стадия 5] Синтез (S)-(1-(7-амино-2-(фуран-2-ил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-5-ил)пирролидин-2-ил)(4-(2,2-дифторэтил)пиперазин-1-ил)метанона
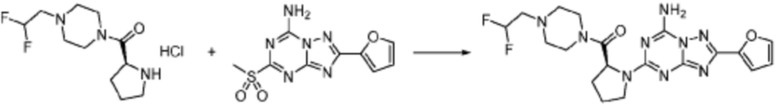
(S)-1-(2,2-Дифторэтил)-4-пропилпиперазин гидрохлорид (4,377 г, 15,426 ммоль), полученный на стадии 4, 2-(фуран-2-ил)-5-(метилсульфонил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амин (4,323 г, 15,426 ммоль) и гидрокарбонат натрия (3,887 г, 46,277 ммоль) растворяют в ацетонитриле (90 мл) при комнатной температуре, после чего полученный раствор перемешивают при 80°C в течение 18 часов до завершения реакции путем понижения температуры до комнатной температуры. Реакционную смесь фильтруют через слой целита для удаления из нее твердого вещества, после чего растворитель удаляют из полученного фильтрата без твердого вещества при пониженном давлении. Затем, полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; дихлорметан/метанол= 0-20%) и концентрируют с получением указанного в заголовке соединения (4,240 г, 61,4%) в виде светло-желтого твердого вещества.
1H ЯМР (400 MГц, ДМСО-d6) δ 8,33 (д, J=126,9 Гц, 2H), 7,90-7,84 (м, 1H), 7,06 (ддд, J=7,7, 3,4, 0,9 Гц, 1H), 6,68 (дт, J=3,4, 1,6 Гц, 1H), 6,40-6,02 (м, 1H), 4,99 (ддд, J=12,5, 8,6, 3,1 Гц, 1H), 4,12 (кв, J=5,3 Гц, 1H), 3,70-3,49 (м, 5H), 3,17 (д, J=5,1 Гц, 2H), 2,82 (тдд, J=17,4, 10,3, 4,2 Гц, 3H), 2,58 (д, J=9,2 Гц, 1H), 2,48-2,37 (м, 1H), 2,26 (ддд, J=12,2, 8,1, 3,8 Гц, 1H), 1,96-1,79 (м, 3H). МСНР (ЭР) m/z 448,3 (M++ 1).
Пример 224: Синтез соединения 224, (S)-(1-(7-амино-2-(фуран-2-ил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-5-ил)пирролидин-2-ил)(4-(2,2-дифторпропил)пиперазин-1-ил)метанона
[Стадия 1] Синтез трет-бутил 4-(2,2-дифторпропил)пиперазин-1-карбоксилата
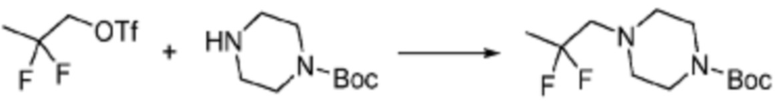
2,2-Дифторпропил трифторметансульфонат (0,456 г, 1,999 ммоль), трет-бутил пиперазин-1-карбоксилат (0,372 г, 1,999 ммоль) и N, N-диизопропилэтиламин (0,348 мл, 1,999 ммоль) растворяют в тетрагидрофуране (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего водный раствор хлорида N-аммония выливают в полученный концентрат, и затем органический слой экстрагируют дихлорметаном, фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора из него, и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; дихлорметан/метанол=0-20%) и концентрируют с получением указанного в заголовке соединения (0,423 г, 80,1%) в виде белого твердого вещества.
[Стадия 2] Синтез 1-(2,2-дифторпропил)пиперазина гидрохлорида

Трет-бутил 4-(2,2-дифторпропил)пиперазин-1-карбоксилат (0,264 г, 0,999 ммоль), полученный на стадии 1, и хлористоводородную кислоту (4,00 M раствор в 1,4-диоксане, 0,999 мл, 3,995 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный продукт используют без дополнительного процесса очистки (указанное в заголовке соединение, 0,200 г, 99,8%, белое твердое вещество).
[Стадия 3] Синтез трет-бутил (S)-2-(4-(2,2-дифторпропил)пиперазин-1-карбонил)пирролидин-1-карбоксилата
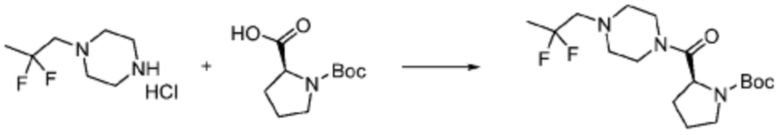
1-(2,2-Дифторпропил)пиперазин гидрохлорид (6,000 г, 29,901 ммоль), полученный на стадии 2, (трет-бутоксикарбонил)-L-пролин (6,436 г, 29,901 ммоль), 2,4,6-трипропил-1,3,5,2,4,6-триоксатрифосфинан 2,4,6-триоксид (T3P, 50,00% раствор в EtOAc, 27,434 мл, 44,852 ммоль) и N, N-диизопропилэтиламин (15,624 мл, 89,704 ммоль) растворяют в дихлорметане (150 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего насыщенный водный раствор N-гидрокарбонат натрия выливают в полученный концентрат и органический слой экстрагируют дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют, и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; дихлорметан/метанол=0-30%) и концентрируют с получением указанного в заголовке соединения (8,030 г, 74,3%) в виде светло-желтой жидкости.
[Стадия 4] Синтез (S)-1-(2,2-дифторпропил)-4-пропилпиперазина гидрохлорида
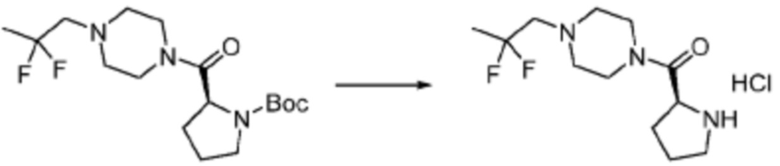
Трет-бутил (S)-2-(4-(2,2-дифторпропил)пиперазин-1-карбонил)пирролидин-1-карбоксилат (8,030 г, 22,217 ммоль), полученный на стадии 3, и хлористоводородную кислоту (4,00 M раствор в 1,4-диоксане, 22,217 мл, 88,869 ммоль) растворяют в дихлорметане (100 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный продукт используют без дополнительного процесса очистки (указанное в заголовке соединение, 6,615 г, 100,0%, светло-желтое твердое вещество).
[Стадия 5] Синтез (S)-(1-(7-амино-2-(фуран-2-ил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-5-ил)пирролидин-2-ил)(4-(2,2-дифторпропил)пиперазин-1-ил)метанона
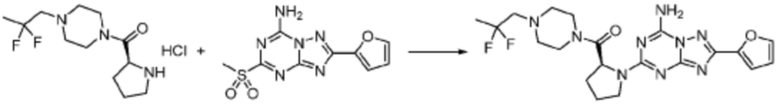
(S)-1-(2,2-Дифторпропил)-4-пропилпиперазин гидрохлорид (5,000 г, 16,791 ммоль), полученный на стадии 4, 2-(фуран-2-ил)-5-(метилсульфонил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амин (4,706 г, 16,791 ммоль) и гидрокарбонат натрия (4,232 г, 50,374 ммоль) растворяют в ацетонитриле (95 мл) при комнатной температуре, после чего полученный раствор перемешивают при 70°C в течение 18 часов до завершения реакции путем понижения температуры до комнатной температуры. Реакционную смесь фильтруют через слой целита для удаления из нее твердого вещества, после чего растворитель удаляют из полученного фильтрата без твердого вещества при пониженном давлении. Затем, полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; дихлорметан/метанол= 0-30%) и концентрируют с получением указанного в заголовке соединения (2,920 г, 37,7%) в виде белого твердого вещества.
1H ЯМР (400 MГц, ДМСО-d6) δ 8,33 (д, J=124,7 Гц, 2H), 7,88 (с, 1H), 7,06 (с, 1H), 6,68 (с, 1H), 4,98 (т, J=10,6 Гц, 1H), 3,63 (т, J=9,8 Гц, 5H), 3,17 (д, J=5,0 Гц, 1H), 2,80 (тд, J=13,9, 7,5 Гц, 3H), 2,60 (с, 1H), 2,46-2,19 (м, 2H), 2,00-1,77 (м, 3H), 1,66 (т, J=19,1 Гц, 3H). МСНР (ЭР) m/z 462,4 (M++ 1).
Пример 294: Синтез соединения 294, (S)-(1-(5-амино-2-(фуран-2-ил)-[1,2,4]триазоло[1,5-c]пиримидин-7-ил)пирролидин-2-ил)(4-(2,2-дифторпропил)пиперазин-1-ил)метанона
[Стадия 1] Синтез трет-бутил 4-(2,2-дифторпропил)пиперазин-1-карбоксилата
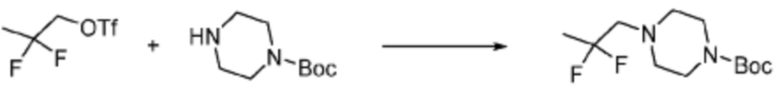
Трет-бутил пиперазин-1-карбоксилат (0,559 г, 3,000 ммоль), 2,2-дифторпропил трифторметансульфонат (0,447 мл, 3,000 ммоль) и карбонат калия (0,829 г, 6,000 ммоль) растворяют в ацетонитриле (8 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего воду выливают в полученный концентрат, и затем органический слой экстрагируют дихлорметаном, фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора из него, и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=1%) и концентрируют с получением указанного в заголовке соединения (0,790 г, 99,6%) в виде белого твердого вещества.
[Стадия 2] Синтез 1-(2,2-дифторпропил)пиперазина гидрохлорида

Трет-бутил 4-(2,2-дифторпропил)пиперазин-1-карбоксилат (0,790 г, 2,989 ммоль), полученный на стадии 1, и хлороводород (4,00 M раствор в 1,4-диоксане, 5,978 мл, 23,910 ммоль) растворяют в дихлорметане (6 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный продукт используют без дополнительного процесса очистки (указанное в заголовке соединение, 0,599 г, 99,9%, белое твердое вещество).
[Стадия 3] Синтез трет-бутил (S)-2-(4-(2,2-дифторпропил)пиперазин-1-карбонил)пирролидин-1-карбоксилата
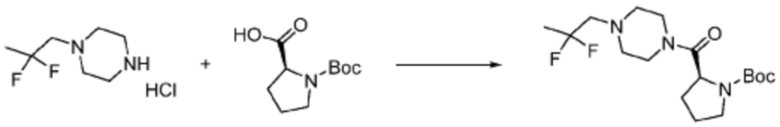
1-(2,2-Дифторпропил)пиперазин гидрохлорид (0,201 г, 1,000 ммоль), полученный на стадии 2, (трет-бутоксикарбонил)-L-пролин (0,215 г, 1,000 ммоль), триэтиламин (0,348 мл, 2,500 ммоль) и 2,4,6-трипропил-1,3,5,2,4,6-триоксатрифосфинан 2,4,6-триоксид (T3P, 50,00% раствор в EtOAc, 0,707 мл, 1,200 ммоль) растворяют в дихлорметане (4 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, и затем органический слой экстрагируют дихлорметаном, фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора из него, и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=1,5%) и концентрируют с получением указанного в заголовке соединения (0,360 г, 99,6%) в виде бесцветного масла.
[Стадия 4] Синтез (S)-1-(2,2-дифторпропил)-4-пропилпиперазина гидрохлорида

Трет-бутил (S)-2-(4-(2,2-дифторпропил)пиперазин-1-карбонил)пирролидин-1-карбоксилат (0,360 г, 0,996 ммоль), полученный на стадии 3, и хлороводород (4,00 M раствор в 1,4-диоксане, 1,494 мл, 5,976 ммоль) растворяют в дихлорметане (3 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный продукт используют без дополнительного процесса очистки (указанное в заголовке соединение, 0,227 г, 76,4%, белое твердое вещество).
[Стадия 5] Синтез (S)-(1-(5-амино-2-(фуран-2-ил)-[1,2,4]триазоло[1,5-c]пиримидин-7-ил)пирролидин-2-ил)(4-(2,2-дифторпропил)пиперазин-1-ил)метанона
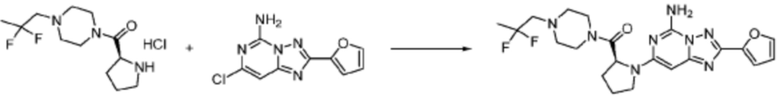
(S)-1-(2,2-Дифторпропил)-4-пропилпиперазин гидрохлорид (0,227 г, 0,761 ммоль), полученный на стадии 4, 7-хлор-2-(фуран-2-ил)-[1,2,4]триазоло[1,5-c]пиримидин-5-амин (0,179 г, 0,761 ммоль) и гидрокарбонат натрия (0,192 г, 2,284 ммоль) растворяют в N, N-диметилформамиде (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при 100°C в течение 18 часов до завершения реакции путем понижения температуры до комнатной температуры. Воду выливают в реакционную смесь, и затем органический слой экстрагируют дихлорметаном, фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора из него, и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением продукта, после чего полученный продукт снова очищают хроматографией (SiO2, 12 г картридж; ацетон/дихлорметан=10-100%) и концентрируют с получением указанного в заголовке соединения (0,055 г, 15,8%) в виде светло-желтого твердого вещества.
1H ЯМР (400 MГц, ДМСО-d6) δ 7,86 (дд, J=1,7, 0,8 Гц, 1H), 7,46 (шс, 2H), 7,06 (дд, J=3,4, 0,6 Гц, 1H), 6,67 (дд, J=3,4, 1,8 Гц, 1H), 5,59 (шс, 1H), 4,96 (шс, 1H), 3,79-3,38 (м, 5H), 2,79 (т, J=14,1 Гц, 3H), 2,64-2,36 (м, 4H), 2,24 (с, 1H), 2,06-1,77 (м, 3H), 1,66 (т, J=19,1 Гц, 3H); МСНР (ЭР) m/z 461,5 (M++ 1).
Пример 371: Синтез соединения 371, ((S)-1-(7-амино-2-(фуран-2-ил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-5-ил)пирролидин-2-ил)((R)-4-(2,2-дифторэтил)-2-метилпиперазин-1-ил)метанона
[Стадия 1] Синтез трет-бутил (R)-4-(2,2-дифторэтил)-2-метилпиперазин-1-карбоксилата
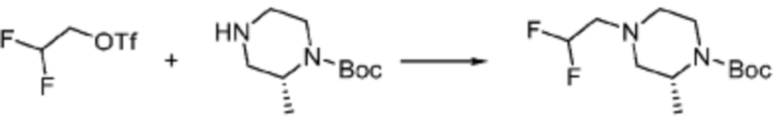
Трет-бутил (R)-2-метилпиперазин-1-карбоксилат (0,507 г, 2,531 ммоль), 2,2-дифторэтил трифторметансульфонат (0,542 г, 2,531 ммоль) и карбонат калия (1,050 г, 7,594 ммоль) растворяют в ацетонитриле (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Реакционную смесь фильтруют через пластиковый фильтр для удаления из нее твердого вещества, после чего растворитель удаляют из полученного фильтрата без твердого вещества при пониженном давлении, и затем полученный продукт используют без дополнительного процесса очистки (указанное в заголовке соединение, 0,668 г, 99,8%, прозрачная жидкость).
[Стадия 2] Синтез (R)-1-(2,2-дифторэтил)-3-метилпиперазина гидрохлорида

Трет-бутил (R)-4-(2,2-дифторэтил)-2-метилпиперазин-1-карбоксилат (0,668 г, 2,527 ммоль), полученный на стадии 1, и хлористоводородную кислоту (4,00 M раствор в 1,4-диоксане, 2,527 мл, 10,109 ммоль) растворяют в дихлорметане (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный продукт используют без дополнительного процесса очистки (указанное в заголовке соединение, 0,507 г, 100,0%, белое твердое вещество).
[Стадия 3] Синтез трет-бутил (S)-2-((R)-4-(2,2-дифторэтил)-2-метилпиперазин-1-карбонил)пирролидин-1-карбоксилата
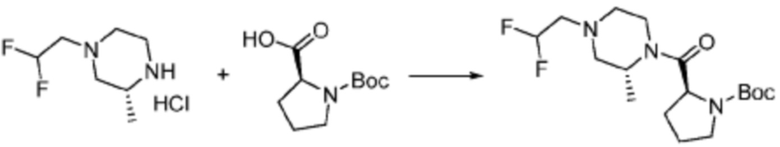
(R)-1-(2,2-Дифторэтил)-3-метилпиперазин гидрохлорид (0,507 г, 2,527 ммоль), полученный на стадии 2, (трет-бутоксикарбонил)-L-пролин (0,544 г, 2,527 ммоль), 2,4,6-трипропил-1,3,5,2,4,6-триоксатрифосфинан 2,4,6-триоксид (T3P, 50,00% раствор в EtOAc, 2,232 мл, 3,790 ммоль) и N, N-диизопропилэтиламин (1,320 мл, 7,580 ммоль) растворяют в дихлорметане (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре. Водный раствор N-гидрокарбонат натрия выливают в реакционную смесь и органический слой экстрагируют дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют, и концентрируют при пониженном давлении. Полученный продукт используют без дополнительного процесса очистки (указанное в заголовке соединение, 1,028 г, 98,9%, светло-желтая жидкость).
[Стадия 4] Синтез (R)-1-(L-пропил)-4-(2,2-дифторэтил)-2-метилпиперазина гидрохлорида
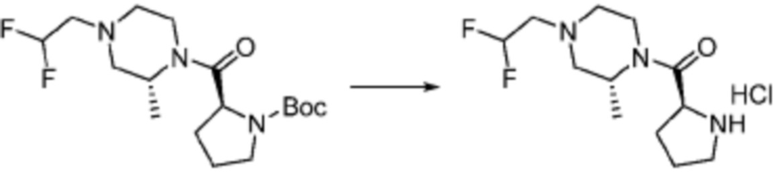
Трет-бутил (S)-2-((R)-4-(2,2-дифторэтил)-2-метилпиперазин-1-карбонил)пирролидин-1-карбоксилат (0,903 г, 2,498 ммоль), полученный на стадии 3, и хлористоводородную кислоту (4,00 M раствор в 1,4-диоксане, 2,498 мл, 9,994 ммоль) растворяют в дихлорметане (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный продукт используют без дополнительного процесса очистки (указанное в заголовке соединение, 0,743 г, 99,9%, белое твердое вещество).
[Стадия 5] ((S)-1-(7-амино-2-(фуран-2-ил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-5-ил)пирролидин-2-ил)((R)-4-(2,2-дифторэтил)-2-метилпиперазин-1-ил)метанон
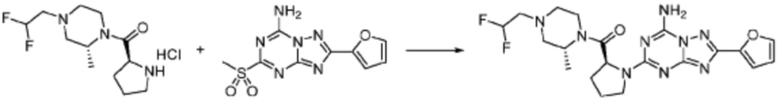
(R)-1-(L-Пропил)-4-(2,2-дифторэтил)-2-метилпиперазин гидрохлорид (0,743 г, 2,495 ммоль), полученный на стадии 4, 2-(фуран-2-ил)-5-(метилсульфонил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амин (0,699 г, 2,495 ммоль) и гидрокарбонат натрия (0,629 г, 7,486 ммоль) растворяют в ацетонитриле (15 мл) при комнатной температуре, после чего полученный раствор перемешивают при 75°C в течение 18 часов до завершения реакции путем понижения температуры до комнатной температуры. Реакционную смесь фильтруют через слой целита для удаления из нее твердого вещества, после чего растворитель удаляют из полученного фильтрата без твердого вещества при пониженном давлении. Затем, полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; дихлорметан/метанол= 0-30%) и концентрируют с получением указанного в заголовке соединения (0,509 г, 44,2%) в виде белого твердого вещества.
1H ЯМР (400 MГц, ДМСО-d6) δ 8,56-7,93 (м, 2H), 7,90-7,83 (м, 1H), 7,06 (дд, J=8,5, 3,3 Гц, 1H), 6,67 (ддд, J=6,4, 3,3, 1,6 Гц, 1H), 6,39-5,95 (м, 1H), 4,96 (дддд, J=35,1, 26,4, 8,5, 2,6 Гц, 1H), 4,33 (д, J=100,5 Гц, 1H), 4,15-3,76 (м, 1H), 3,70-3,53 (м, 2 H), 2,96-2,64 (м, 5H), 2,48-2,00 (м, 3H), 1,98-1,69 (м, 3H), 1,49 (дд, J=15,8, 6,6 Гц, 2H), 1,13 (д, J=6,8 Гц, 1H). МСНР (ЭР) m/z 462,4 (M++ 1).
Пример 258: Синтез соединения 258
Пример соединения 258 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 223 за исключением применения 2-(фуран-2-ил)-5-(метилсульфонил)-3a,7a-дигидротиазоло[5,4-d]пиримидин-7-амина вместо 2-(фуран-2-ил)-5-(метилсульфонил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амина в способе синтеза Примера соединения 223.
Пример 259: Синтез соединения 259
Пример соединения 259 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 224 за исключением применения 2-(фуран-2-ил)-5-(метилсульфонил)-3a,7a-дигидротиазоло[5,4-d]пиримидин-7-амина вместо 2-(фуран-2-ил)-5-(метилсульфонил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амина в способе синтеза Примера соединения 224.
Пример 260: Синтез соединения 260
Пример соединения 260 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 223 за исключением применения 1,1,2,2-тетрафтор-3-((трифторметил)сульфонил)пропана вместо 2,2-дифторэтил трифторметансульфоната и с применением 2-(фуран-2-ил)-5-(метилсульфонил)-3a,7a-дигидротиазоло[5,4-d]пиримидин-7-амина вместо 2-(фуран-2-ил)-5-(метилсульфонил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амина в способе синтеза Примера соединения 223.
Пример 261: Синтез соединения 261
Пример соединения 261 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 223 за исключением применения 1,1,1,2,2-пентафтор-3-((трифторметил)сульфонил)пропана вместо 2,2-дифторэтил трифторметансульфоната и с применением 2-(фуран-2-ил)-5-(метилсульфонил)-3a,7a-дигидротиазоло[5,4-d]пиримидин-7-амина вместо 2-(фуран-2-ил)-5-(метилсульфонил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амина в способе синтеза Примера соединения 223.
Пример 269: Синтез соединения 269
Пример соединения 269 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 223 за исключением применения 2-(фуран-2-ил)-5-(метилсульфонил)-3a,7a-дигидрооксазоло[5,4-d]пиримидин-7-амина вместо 2-(фуран-2-ил)-5-(метилсульфонил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амина в способе синтеза Примера соединения 223.
Пример 270: Синтез соединения 270
Пример соединения 270 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 223 за исключением применения 2,2-дифтор-1-((трифторметил)сульфонил)бутана вместо 2,2-дифторэтил трифторметансульфоната и с применением 2-(фуран-2-ил)-5-(метилсульфонил)-3a,7a-дигидрооксазоло[5,4-d]пиримидин-7-амина вместо 2-(фуран-2-ил)-5-(метилсульфонил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амина в способе синтеза Примера соединения 223.
Пример 271: Синтез соединения 271
Пример соединения 271 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 223 за исключением применения 1,1,2,2-тетрафтор-3-((трифторметил)сульфонил)пропана вместо 2,2-дифторэтил трифторметансульфоната и с применением 2-(фуран-2-ил)-5-(метилсульфонил)-3a,7a-дигидрооксазоло[5,4-d]пиримидин-7-амина вместо 2-(фуран-2-ил)-5-(метилсульфонил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амина в способе синтеза Примера соединения 223.
Пример 293: Синтез соединения 293
Пример соединения 293 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 223 за исключением применения 2-(фуран-2-ил)-7-(метилсульфонил)-[1,2,4]триазоло[1,5-c]пиримидин-5-амина вместо 2-(фуран-2-ил)-5-(метилсульфонил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амина в способе синтеза Примера соединения 223.
Пример 295: Синтез соединения 295
Пример соединения 295 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 223 за исключением применения 2,2-дифтор-1-((трифторметил)сульфонил)бутана вместо 2,2-дифторэтил трифторметансульфоната и с применением 2-(фуран-2-ил)-7-(метилсульфонил)-[1,2,4]триазоло[1,5-c]пиримидин-5-амина вместо 2-(фуран-2-ил)-5-(метилсульфонил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амина в способе синтеза Примера соединения 223.
Пример 296: Синтез соединения 296
Пример соединения 296 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 223 за исключением применения 1,1,1,2,2-пентафтор-3-((трифторметил)сульфонил)пропана вместо 2,2-дифторэтил трифторметансульфоната и с применением 2-(фуран-2-ил)-7-(метилсульфонил)-[1,2,4]триазоло[1,5-c]пиримидин-5-амина вместо 2-(фуран-2-ил)-5-(метилсульфонил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амина в способе синтеза Примера соединения 223.
Примеры 316, 317, 318 и 319
Примеры соединений 316, 317, 318 и 319 каждый синтезируют по существу тем же способом синтеза, что и каждый способ синтеза за исключением применения (трет-бутоксикарбонил)-L-аланина вместо (трет-бутоксикарбонил)-L-пролина и с применением 2-(фуран-2-ил)-5-(метилсульфонил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амина вместо 2-(фуран-2-ил)-5-(метилсульфонил)-3a,7a-дигидротиазоло[5,4-d]пиримидин-7-амина в каждом способе синтеза Примеров соединений 258, 259, 260 и 261.
Пример 320: Синтез соединения 320
Пример соединения 320 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 223 за исключением применения 1,1,1,2,2,3,3-гептафтор-4-((трифторметил)сульфонил)бутана вместо 2,2-дифторэтил трифторметансульфоната и с применением (трет-бутоксикарбонил)-L-аланина вместо (трет-бутоксикарбонил)-L-пролина в способе синтеза Примера соединения 223.
Примеры 323, 324, 325 и 326
Примеры соединений 323, 324, 325 и 326 каждый синтезируют по существу тем же способом синтеза, что и каждый способ синтеза за исключением применения (S)-1-(трет-бутоксикарбонил)азетидин-2-карбоновой кислоты вместо (трет-бутоксикарбонил)-L-аланин и с применением трет-бутил [4,4'-бипиперидин]-1-карбоксилата вместо трет-бутил пиперазин-1-карбоксилат в способе синтеза Примеров соединений 316, 317, 318 и 319.
Примеры 334 и 335
Примеры соединений 334 и 335 синтезируют по существу тем же способом синтеза, что и в каждом способе синтеза за исключением применения трет-бутил [4,4'-бипиперидин]-1-карбоксилата вместо трет-бутил пиперазин-1-карбоксилата в каждом способе синтеза Примеров соединений 316 и 317.
Примеры 252, 253, 255, 256 и 257
Примеры соединений 252, 253, 255, 256 и 257 каждый синтезируют по существу тем же способом синтеза, что и каждый способ синтеза за исключением применения (S)-1-(трет-бутоксикарбонил)азетидин-2-карбоновой кислоты вместо (трет-бутоксикарбонил)-L-аланина в способе синтеза Примеров соединений 316, 317, 318 и 319.
Пример 254: Синтез соединения 254
Пример соединения 254 синтезируют по существу тем же способом синтеза за исключением применения (S)-1-(трет-бутоксикарбонил)азетидин-2-карбоновой кислоты вместо трет-бутоксикарбонил-D-пролина в способе синтеза Примера соединения 225.
Примеры 204, 205, 206, 225, 226, 227, 228, 278, 362, 363, 364, 365, 287, 288, 289, 337, 341, 342, 343 и 345
Примеры соединений 204, 205, 206, 225, 226, 227, 228, 278, 362, 363, 364, 365, 287, 288, 289, 337, 341, 342, 343 и 345 каждый синтезируют по существу тем же способом синтеза, как и способ синтеза из Примера соединения 224 за исключением применения исходного материала 1 из таблицы ниже вместо трет-бутил пиперазин-1-карбоксилата на стадии 1 в способе синтеза Примера соединения 224 и с применением исходного материала 2 из таблицы ниже вместо дифторпропилтрифторметансульфоната.
[Таблица 32]
№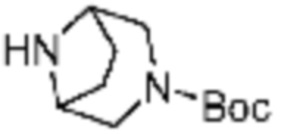
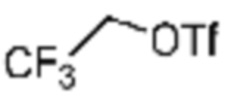
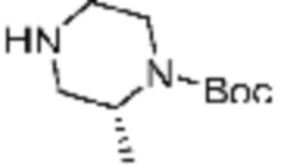
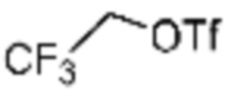
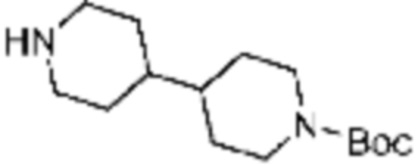
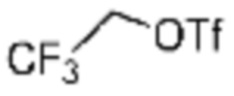
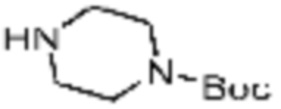
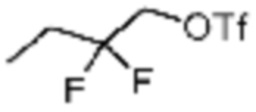
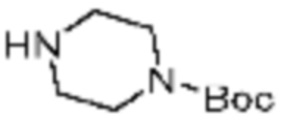
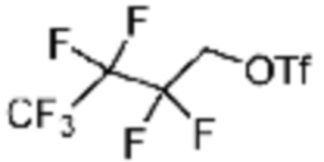
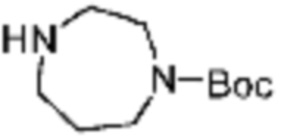
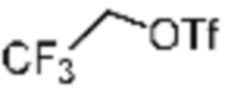
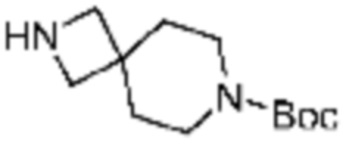

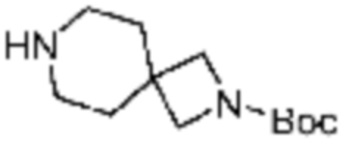
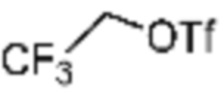
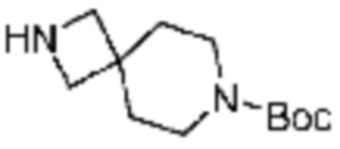
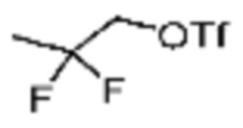
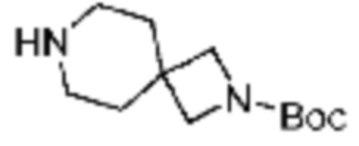
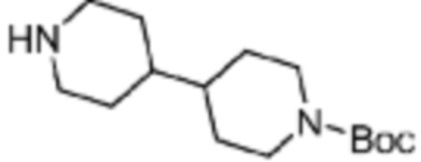
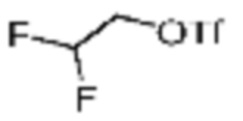
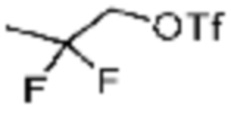
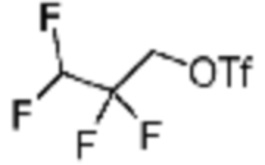
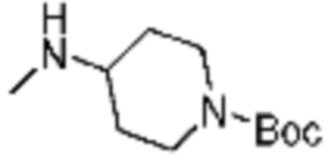
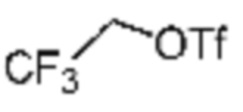
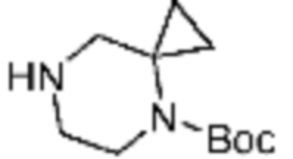
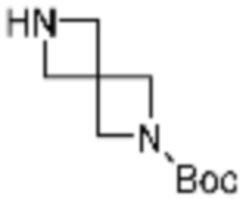
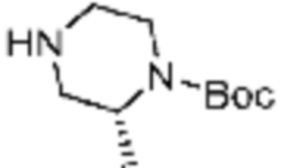
Пример 196: Синтез соединения 196
Пример соединения 196 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 205 за исключением применения 2-(метилфуран-2-ил)-5-(метилсульфонил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амина вместо 2-(фуран-2-ил)-5-(метилсульфонил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амина в способе синтеза Примера соединения 205.
Пример 197: Синтез соединения 197
Пример соединения 197 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 196 за исключением применения трет-бутил [4,4'-бипиперидин]-1-карбоксилата вместо трет-бутил (R)-2-метилпиперазин-1-карбоксилата.
Пример 279: Синтез соединения 279
Пример соединения 279 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 223 за исключением применения 1,1,2,2-тетрафтор-3-((трифторметил)сульфонил)пропана вместо 2,2-дифторэтил трифторметансульфоната и с применением (S)-1-(трет-бутоксикарбонил)пиперидин-2-карбоновой кислоты вместо (трет-бутоксикарбонил)-L-пролина.
Пример 280: Синтез соединения 280
Пример соединения 280 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 223 за исключением применения 1,1,1,2,2-пентафтор-3-((трифторметил)сульфонил)пропана вместо 2,2-дифторэтил трифторметансульфоната и с применением (S)-1-(трет-бутоксикарбонил)пиперидин-2-карбоновой кислоты вместо (трет-бутоксикарбонил)-L-пролина.
Пример 297: Синтез соединения 297
Пример соединения 297 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 223 за исключением применения 2-(метилфуран-2-ил)-5-(метилсульфонил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амина вместо 2-(фуран-2-ил)-5-(метилсульфонил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амина.
Пример 298: Синтез соединения 298
Пример соединения 298 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 223 за исключением применения 1,1,2,2-тетрафтор-3-((трифторметил)сульфонил)пропана вместо 2,2-дифторэтил трифторметансульфоната и с применением 2-(метилфуран-2-ил)-5-(метилсульфонил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амина вместо 2-(фуран-2-ил)-5-(метилсульфонил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амина.
Пример 299: Синтез соединения 299
Пример соединения 299 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 223 за исключением применения 1,1,1,2,2-пентафтор-3-((трифторметил)сульфонил)пропана вместо 2,2-дифторэтил трифторметансульфоната и с применением 2-(метилфуран-2-ил)-5-(метилсульфонил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амина вместо 2-(фуран-2-ил)-5-(метилсульфонил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амина.
Пример 311: Синтез соединения 311
Пример соединения 311 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 223 за исключением применения 2-(метилфуран-2-ил)-5-(метилсульфонил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амина вместо 2-(фуран-2-ил)-5-(метилсульфонил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амина.
Пример 312: Синтез соединения 312
Пример соединения 312 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 298 за исключением применения (S)-1-(трет-бутоксикарбонил)азетидин-2-карбоновой кислоты вместо трет-бутоксикарбонил-D-пролина.
Пример 313: Синтез соединения 313
Пример соединения 313 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 299 за исключением применения (S)-1-(трет-бутоксикарбонил)азетидин-2-карбоновой кислоты вместо трет-бутоксикарбонил-D-пролина.
Пример 314: Синтез соединения 314
Пример соединения 314 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 311 за исключением применения (S)-1-(трет-бутоксикарбонил)азетидин-2-карбоновой кислоты вместо трет-бутоксикарбонил-D-пролина.
Примеры 354, 355, 356, 357, 358, 359 и 360
Примеры соединений 354, 355, 356, 357, 358, 359 и 360 получают по существу тем же способом синтеза, как и способ синтеза из примера соединения 223 за исключением применения исходного материала 1 из таблицы ниже вместо 2,2-дифторэтилтрифторметансульфоната на стадии 1 и с применением Boc-защищенных аминокислот из таблицы ниже вместо (трет-бутоксикарбонил)-L-пролина.
[Таблица 33]
№
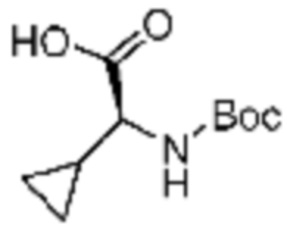
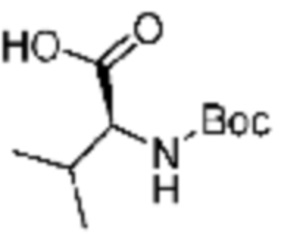
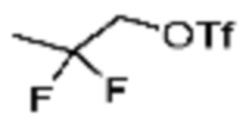
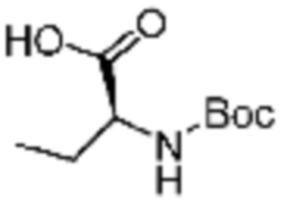
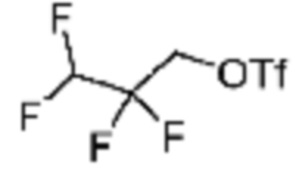
Примеры 376, 377, 378, 379, 393 и 394
Примеры соединений 376, 377, 378, 379, 393 и 394 каждый синтезируют по существу тем же способом синтеза, как и способ синтеза из Примера соединения 223 за исключением применения 2,2-дифторэтилтрифторметансульфонат на стадии 1 или исходного материала 1 из таблицы ниже, с применением (трет-бутоксикарбонил)-L-пролина или Boc-защищенной аминокислоты из таблицы ниже, и с применением 2-(фуран-2-ил)-7-(метилсульфонил)-[1,2,4]триазоло[1,5-c]пиримидин-5-амина вместо 2-(фуран-2-ил)-5-(метилсульфонил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амина.
[Таблица 34]
№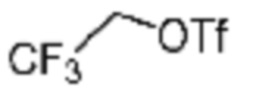
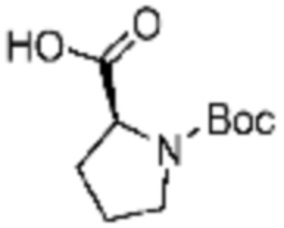
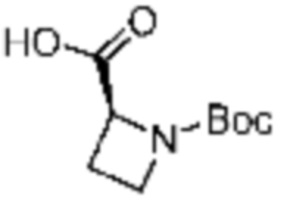
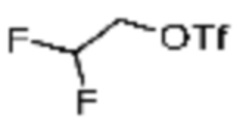
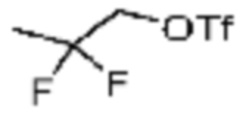
Пример 366: Синтез соединения 366
Пример соединения 366 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 365 за исключением применения (трет-бутоксикарбонил)-L-аланина вместо трет-бутоксикарбонил)-L-пролина.
Пример 396: Синтез соединения 396
Пример соединения 396 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 223 за исключением применения N-(трет-бутоксикарбонил)-O-метил-L-серина вместо (трет-бутоксикарбонил)-L-пролина.
Пример 397: Синтез соединения 397
Пример соединения 397 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 224 за исключением применения N-(трет-бутоксикарбонил)-O-метил-L-серина вместо (трет-бутоксикарбонил)-L-пролина.
Аналитические данные каждого из соединений, полученных как описано выше, показаны в таблице ниже.
[Таблица 35]
Пример 73: Синтез соединения 73, 1-(4-((7-амино-2-(фуран-2-ил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-5-ил)-L-пропил)пиперазин-1-ил)-3,3,3-трифторпропан-1-она
[Стадия 1] Синтез трет-бутил 4-(3,3,3-трифторпропаноил)пиперазин-1-карбоксилата
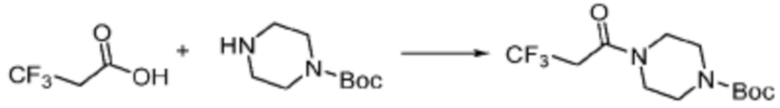
3,3,3-Трифторпропановую кислоту (0,500 г, 3,905 ммоль) и N, N-диметилформамид (0,003 мл, 0,039 ммоль) растворяют в дихлорметане (10 мл) при комнатной температуре, после чего оксалилдихлорид (0,335 мл, 3,905 ммоль) добавляют в полученный раствор и перемешивают при той же температуре в течение одного часа. К полученной смеси добавляют раствор, полученный растворением трет-бутил пиперазин-1-карбоксилата (0,636 г, 3,413 ммоль) и триэтиламина (0,951 мл, 6,826 ммоль) в дихлорметане (10 мл) при комнатной температуре, и перемешивают при той же температуре в течение одного часа. Воду выливают в реакционную смесь и органический слой экстрагируют дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида аммония, дегидратируют безводным сульфатом магния, фильтруют, и концентрируют при пониженном давлении. Полученный продукт используют без дополнительного процесса очистки (указанное в заголовке соединение, 1,000 г, 98,9%, белое твердое вещество).
[Стадия 2] Синтез 3,3,3-трифтор-1-(пиперазин-1-ил)пропан-1-она
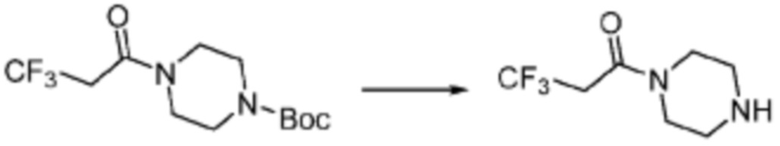
Трет-бутил 4-(3,3,3-трифторпропаноил)пиперазин-1-карбоксилат (1,000 г, 3,375 ммоль), полученный на стадии 1, и хлористоводородную кислоту (0,615 г, 16,875 ммоль) растворяют в дихлорметане (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный продукт используют без дополнительного процесса очистки (указанное в заголовке соединение, 0,500 г, 75,5%, белое твердое вещество).
[Стадия 3] Синтез трет-бутил (S)-2-(4-(3,3,3-трифторпропаноил)пиперазин-1-карбонил)пирролидин-1-карбоксилата
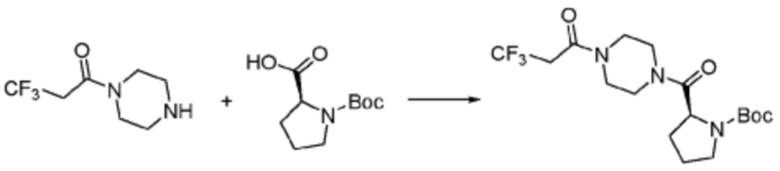
3,3,3-Трифтор-1-(пиперазин-1-ил)пропан-1-он (0,500 г, 2,549 ммоль), полученный на стадии 2, (трет-бутоксикарбонил)-L-пролин (1,097 г, 5,098 ммоль), [диметиламино(триазоло[4,5-b]пиридин-3-илокси)метилиден]-диметилазания; гексафторфосфат (1,938 г, 5,098 ммоль) и N, N-диизопропилэтиламин (1,776 мл, 10,195 ммоль) растворяют в N, N-диметилформамиде (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор хлорида аммония выливают в реакционную смесь, и органический слой экстрагируют этилацетатом. Органический слой промывают насыщенным водным раствором гидрокарбоната натрия, дегидратируют безводным сульфатом натрия, фильтруют, и концентрируют при пониженном давлении. Полученный продукт используют без дополнительного процесса очистки (указанное в заголовке соединение, 0,500 г, 49,9%, коричневое масло).
[Стадия 4] (S)-3,3,3-трифтор-1-(4-пропилпиперазин-1-ил)пропан-1-он
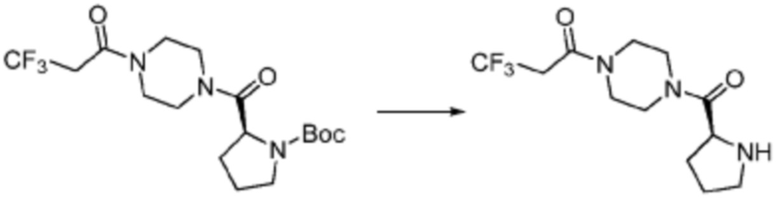
Трет-бутил (S)-2-(4-(3,3,3-трифторпропаноил)пиперазин-1-карбонил)пирролидин-1-карбоксилат (0,200 г, 0,508 ммоль), полученный на стадии 3, и хлористоводородную кислоту (0,093 г, 2,542 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный продукт используют без дополнительного процесса очистки (указанное в заголовке соединение, 0,120 г, 80,5%, коричневое твердое вещество).
[Стадия 5] 1-(4-((7-амино-2-(фуран-2-ил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-5-ил)-L-пропил)пиперазин-1-ил)-3,3,3-трифторпропан-1-он
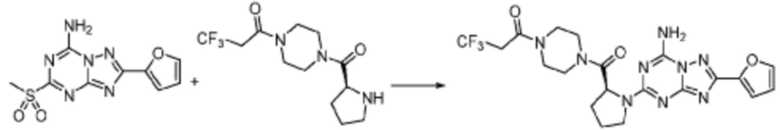
2-(Фуран-2-ил)-5-(метилсульфонил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амин (0,050 г, 0,178 ммоль), полученный на стадии 4, (S)-3,3,3-трифтор-1-(4-пропилпиперазин-1-ил)пропан-1-он (0,105 г, 0,357 ммоль) и триэтиламин (0,099 мл, 0,714 ммоль) растворяют в диметилсульфоксиде (5 мл) при комнатной температуре, и полученный раствор перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный концентрат очищают хроматографией (SiO2 тарелка, 20×20x1 мм; метанол/этилацетат=10%) и концентрируют с получением указанного в заголовке соединения (0,020 г, 22,7%) в виде желтого твердого вещества.
1H ЯМР (400 MГц, ДМСО-d6) δ 8,64-8,09 (м, 1H), 7,87 (ддд, J=5,5, 1,8, 0,8 Гц, 1H), 7,09-7,01 (м, 1H), 6,68 (ддд, J=5,6, 3,4, 1,8 Гц, 1H), 5,09-4,92 (м, 1H), 3,88-3,39 (м, 12H), 2,36-2,19 (м, 1H), 2,04-1,78 (м, 3H); МСНР (ЭР) m/z 494,5 (M++ 1).
Примеры 74-77, 85 и 86
Примеры соединений 74-77, 85 и 86 каждый синтезируют по существу тем же способом синтеза, как и способ синтеза из Примера соединения 73 за исключением применения исходных материалов, показанных в таблице ниже, вместо 3,3,3-трифторпропановой кислоты на стадии 1.
[Таблица 36]
№
№
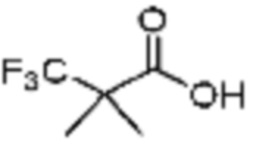
Аналитические данные каждого из соединений, полученных как описано выше, показаны в таблице ниже.
[Таблица 37]
№
Пример 56: Синтез соединения 56, (S)-(1-(7-амино-2-(фуран-2-ил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-5-ил)пирролидин-2-ил)(4-(4,4-дифторциклогексил)пиперазин-1-ил)метанон
[Стадия 1] Синтез трет-бутил 4-(4,4-дифторциклогексил)пиперазин-1-карбоксилат
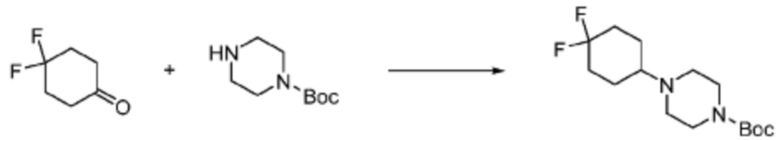
4,4-Дифторциклогексан-1-он (0,402 г, 3,000 ммоль), трет-бутил пиперазин-1-карбоксилат (0,559 г, 3,000 ммоль) и триацетоксиборгидрид натрия (0,954 г, 4,500 ммоль) растворяют в дихлорметане (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение двух часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, и затем органический слой экстрагируют дихлорметаном, фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора из него, и концентрируют при пониженном давлении. Полученный продукт используют без дополнительного процесса очистки (указанное в заголовке соединение, 0,910 г, 99,7%, светло-желтое масло).
[Стадия 2] Синтез 1-(4,4-дифторциклогексил)пиперазина гидрохлорида

Трет-бутил 4-(4,4-дифторциклогексил)пиперазин-1-карбоксилат (0,910 г, 2,990 ммоль), полученный на стадии 1, и хлороводород (4,00 M раствор в 1,4-диоксане, 2,990 мл, 11,959 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение пяти часов. Выпавшее в осадок твердое вещество фильтруют, промывают дихлорметаном и сушат с получением указанного в заголовке соединения (0,710 г, 98,7%) в виде белого твердого вещества.
[Стадия 3] Синтез трет-бутил (S)-2-(4-(4,4-дифторциклогексил)пиперазин-1-карбонил)пирролидин-1-карбоксилат
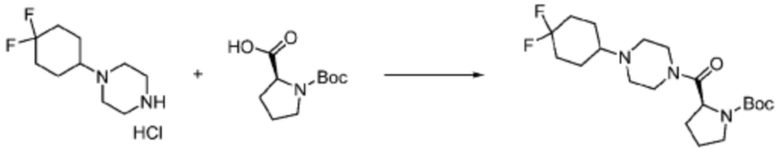
1-(4,4-Дифторциклогексил)пиперазин гидрохлорид (0,710 г, 2,949 ммоль), полученный на стадии 2, (трет-бутоксикарбонил)-L-пролин (0,698 г, 3,244 ммоль), 1-этил-3-(3-диметиламинопропил)карбодиимид гидрохлорид (ЭДК-HCl, 1,131 г, 5,899 ммоль), 1H-бензо[d][1,2,3]триазол-1-ол (HOBt, 0,399 г, 2,949 ммоль) и N, N-диизопропилэтиламин (1,541 мл, 8,848 ммоль) растворяют в дихлорметане (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Воду выливают в реакционную смесь, и затем органический слой экстрагируют дихлорметаном, фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора из него, и концентрируют при пониженном давлении. Полученный продукт используют без дополнительного процесса очистки (указанное в заголовке соединение, 1,180 г, 99,6%, светло-желтое масло).
[Стадия 4] Синтез (S)-1-(4,4-дифторциклогексил)-4-пропилпиперазина гидрохлорида

Трет-бутил (S)-2-(4-(4,4-дифторциклогексил)пиперазин-1-карбонил)пирролидин-1-карбоксилат (1,180 г, 2,939 ммоль), полученный на стадии 3, и хлороводород (4,00 M раствор в 1,4-диоксане, 2,939 мл, 11,756 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение трех часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный продукт используют без дополнительного процесса очистки (указанное в заголовке соединение, 0,990 г, 99,7%, светло-желтое твердое вещество).
[Стадия 5] Синтез (S)-(1-(7-амино-2-(фуран-2-ил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-5-ил)пирролидин-2-ил)(4-(4,4-дифторциклогексил)пиперазин-1-ил)метанона
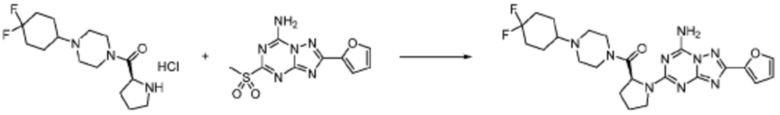
(S)-1-(4,4-Дифторциклогексил)-4-пропилпиперазин гидрохлорид (0,338 г, 1,000 ммоль), полученный на стадии 4, 2-(фуран-2-ил)-5-(метилсульфонил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амин (0,280 г, 1,000 ммоль) и триэтиламин (0,418 мл, 3,000 ммоль) растворяют в N, N-диметилформамиде (3 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Воду выливают в реакционную смесь, и затем органический слой экстрагируют дихлорметаном, фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора из него, и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением продукта, после чего дихлорметан (2 мл) вводят в полученный продукт, перемешивают для фильтрования выпавшего в осадок твердого вещества, промывают гексаном и сушат с получением указанного в заголовке соединения (0,131 г, 26,0%) в виде белого твердого вещества.
1H ЯМР (400 MГц, ДМСО-d6) δ 8,61-7,99 (м, 2H), 7,92-7,82 (м, 1H), 7,10-6,98 (м, 1H), 6,73-6,61 (м, 1H), 5,16-4,91 (м, 1H), 3,99-3,53 (м, 5H), 3,51-3,36 (м, 2H), 2,95-2,65 (м, 1H), 2,44-2,15 (м, 3H), 2,15-1,70 (м, 9H), 1,66-1,44 (м, 2H), 1,39-1,16 (м, 1H); МСНР (ЭР) m/z 502,6 (M++ 1).
Примеры 152-156, 392 и 403
Примеры соединений 152-156, 392 и 403 каждый синтезируют по существу тем же способом синтеза, как и способ синтеза из Примера соединения 56 за исключением применения исходных материалов, показанных в таблице ниже вместо 4,4-дифторциклогексан-1-она на стадии 1 в способе синтеза Примера соединения 56.
[Таблица 38]
№
№
Пример 344: Синтез соединения 344
Пример соединения 344 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 56 за исключением применения циклогексанона вместо 4,4-дифторциклогексан-1-она на стадии 1 и с применением трет-бутил 4,7-диазаспиро[2,5]октан-4-карбоксилата вместо трет-бутил пиперазин-1-карбоксилата.
Пример 398: Синтез соединения 398
Пример соединения 398 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 56 за исключением применения N-(трет-бутоксикарбонил)-O-метил-L-серина вместо (трет-бутоксикарбонил)-L-пролина.
Пример 404: Синтез соединения 404
Пример соединения 404 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 403 за исключением применения 2-(фуран-2-ил)-7-(метилсульфонил)-[1,2,4]триазоло[1,5-c]пиримидин-5-амина вместо 2-(фуран-2-ил)-5-(метилсульфонил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амина.
Пример 291: Синтез соединения 291
Пример соединения 291 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 56 за исключением применения 4,4,4-трифторбутанала вместо 4,4-дифторциклогексан-1-она на стадии 1 и с применением трет-бутил [4,4'-бипиперидин]-1-карбоксилата вместо трет-бутил пиперазин-1-карбоксилата.
Аналитические данные каждого из соединений, полученных как описано выше, показаны в таблице ниже.
[Таблица 39]
№
Пример 129: Синтез соединения 129, (2S)-2-((7-амино-2-(фуран-2-ил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-5-ил)амино)-1-(3,5-диметил-4-(морфолин-4-карбонил)пиперазин-1-ил)пропан-1-она
[Стадия 1] Синтез трет-бутил 3,5-диметил-4-(морфолин-4-карбонил)пиперазин-1-карбоксилата
Трет-бутил 3,5-диметилпиперазин-1-карбоксилат (0,321 г, 1,500 ммоль), N, N-диизопропилэтиламин (0,261 мл, 1,500 ммоль) и морфолин-4-карбонилхлорид (0,175 мл, 1,500 ммоль) растворяют в ацетонитриле (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, и затем органический слой экстрагируют дихлорметаном, фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора из него, и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,205 г, 41,7%) в виде белого твердого вещества.
[Стадия 2] Синтез (2,6-диметилпиперазин-1-ил)(морфолино)метанона гидрохлорида

Трет-бутил 3,5-диметил-4-(морфолин-4-карбонил)пиперазин-1-карбоксилат (0,205 г, 0,626 ммоль), полученный на стадии 1, и хлороводород (4,00 M раствор в 1,4-диоксане, 0,626 мл, 2,504 ммоль) растворяют в дихлорметане (3 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный продукт используют без дополнительного процесса очистки (указанное в заголовке соединение, 0,165 г, 99,9%, белое твердое вещество).
[Стадия 3] Синтез трет-бутил ((2S)-1-(3,5-диметил-4-(морфолин-4-карбонил)пиперазин-1-ил)-1-оксопропан-2-ил)карбамата
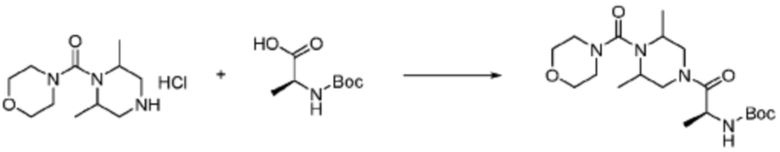
(2,6-Диметилпиперазин-1-ил)(морфолино)метанон гидрохлорид (0,165 г, 0,626 ммоль), полученный на стадии 2, (трет-бутоксикарбонил)-L-аланин (0,130 г, 0,688 ммоль), триэтиламин (0,218 мл, 1,564 ммоль) и 2,4,6-трипропил-1,3,5,2,4,6-триоксатрифосфинан 2,4,6-триоксид (T3P, 50,00% раствор в EtOAc, 0,558 мл, 0,938 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при 40°C в течение 18 часов до завершения реакции путем понижения температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, и затем органический слой экстрагируют дихлорметаном, фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора из него, и затем концентрируют при пониженном давлении. Полученный продукт используют без дополнительного процесса очистки (указанное в заголовке соединение, 0,249 г, 99,9%, светло-желтое масло).
[Стадия 4] Синтез (2S)-2-амино-1-(3,5-диметил-4-(морфолин-4-карбонил)пиперазин-1-ил)пропан-1-она гидрохлорида

Трет-бутил ((2S)-1-(3,5-диметил-4-(морфолин-4-карбонил)пиперазин-1-ил)-1-оксопропан-2-ил)карбамат (0,249 г, 0,625 ммоль), полученный на стадии 3, и хлороводород (4,00 M раствор в 1,4-диоксане, 0,625 мл, 2,499 ммоль) растворяют в дихлорметане (3 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный продукт используют без дополнительного процесса очистки (указанное в заголовке соединение, 0,209 г, 99,9%, белое твердое вещество).
[Стадия 5] Синтез (2S)-2-((7-амино-2-(фуран-2-ил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-5-ил)амино)-1-(3,5-диметил-4-(морфолин-4-карбонил)пиперазин-1-ил)пропан-1-она
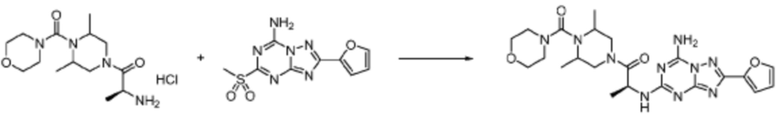
(2S)-2-Амино-1-(3,5-диметил-4-(морфолин-4-карбонил)пиперазин-1-ил)пропан-1-он гидрохлорид (0,209 г, 0,624 ммоль), полученный на стадии 4, 2-(фуран-2-ил)-5-(метилсульфонил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амин (0,175 г, 0,624 ммоль) и гидрокарбонат натрия (0,210 г, 2,497 ммоль) растворяют в циклопентилметиловом эфире (ЦПМЭ, 4 мл) при комнатной температуре, после чего полученный раствор перемешивают при 50°C в течение 18 часов до завершения реакции путем понижения температуры до комнатной температуры. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан= 0-5%) и концентрируют с получением указанного в заголовке соединения (0,069 г, 22,0%) в виде белого твердого вещества.
1H ЯМР (400 MГц, ДМСО-d6) δ 8,49-7,98 (м, 2H), 7,90-7,82 (м, 1H), 7,77-7,42 (м, 1H), 7,06 (дд, J=13,0, 3,5 Гц, 1H), 6,71-6,63 (м, 1H), 5,04-4,75 (м, 1H), 3,89-3,36 (м, 11H), 3,29-2,88 (м, 3H), 1,36-1,24 (м, 3H), 1,22-0,89 (м, 6H); МСНР (ЭР) m/z 499,6 (M++ 1).
Пример 179: Синтез соединения 179
Пример соединения 179 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 129 за исключением применения (трет-бутоксикарбонил)-L-пролина вместо (трет-бутоксикарбонил)-L-аланина и с применением трет-бутил 3,8-диазабицикло[3,2,1]октан-3-карбоксилата и хлорида фенилкарбаминовой кислоты вместо трет-бутил 3,5-диметилпиперазин-1-карбоксилата и морфолин-4-карбонилхлорида, соответственно.
Примеры 180-184
Примеры соединений 180-184 каждый синтезируют по существу тем же способом синтеза, как и способ синтеза из Примера соединения 179 за исключением применения соединений из следующей таблицы вместо трет-бутил 3,8-диазабицикло[3,2,1]октан-3-карбоксилата в качестве исходного материала.
[Таблица 40]
№
№
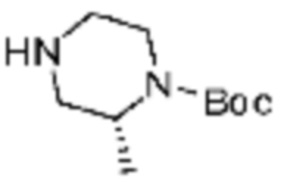
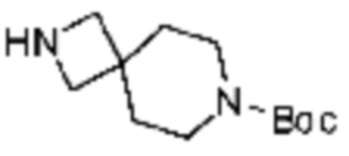
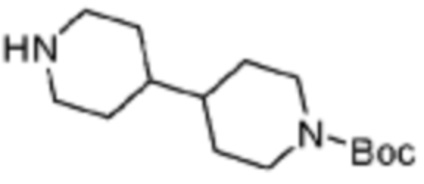
Аналитические данные каждого из соединений, полученных как описано выше, показаны в таблице ниже.
[Таблица 41]
№
Пример 43: Синтез соединения 43, (S)-(1-(7-амино-2-(фуран-2-ил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-5-ил)пирролидин-2-ил)(4-(2-этил-2-фторбутил)пиперазин-1-ил)метанона
[Стадия 1] Синтез трет-бутил 4-(2-этил-2-фторбутил)пиперазин-1-карбоксилата

Трет-бутил 4-(2-этил-2-гидроксибутил)пиперазин-1-карбоксилат (3,000 г, 10,474 ммоль) растворяют в дихлорметане (300 мл), после чего туда добавляют DAST (2,076 мл, 15,711 ммоль) при 0°C, затем перемешивают при той же температуре в течение 30 минут, и затем перемешивают при комнатной температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, и органический слой экстрагируют дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют, и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=0-25%) и концентрируют с получением указанного в заголовке соединения (0,500 г, 16,6%) в виде бесцветного масла.
[Стадия 2] Синтез 1-(2-этил-2-фторбутил)пиперазина
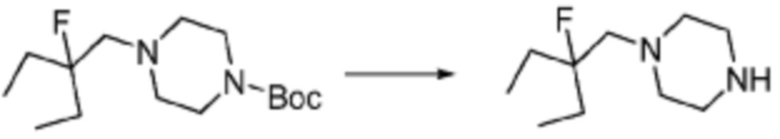
Трет-бутил 4-(2-этил-2-фторбутил)пиперазин-1-карбоксилат (0,500 г, 1,734 ммоль), полученный на стадии 1, и хлористоводородную кислоту (4,00 M раствор в диоксане, 4,334 мл, 17,336 ммоль) растворяют в дихлорметане (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный продукт используют без дополнительного процесса очистки (указанное в заголовке соединение, 0,320 г, 98,0%, коричневое твердое вещество).
[Стадия 3] Трет-бутил (S)-2-(4-(2-этил-2-фторбутил)пиперазин-1-карбонил)пирролидин-1-карбоксилата
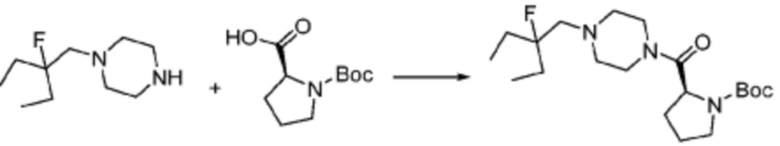
1-(2-Этил-2-фторбутил)пиперазин (0,500 г, 2,655 ммоль), полученный на стадии 2, (трет-бутоксикарбонил)-L-пролин (1,143 г, 5,311 ммоль), [диметиламино(триазоло[4,5-b]пиридин-3-илокси)метилиден]-диметилазания; гексафторфосфат (2,019 г, 5,311 ммоль) и N, N-диизопропилэтиламин (0,925 мл, 5,311 ммоль) растворяют в N, N-диметилформамиде (15 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Воду выливают в реакционную смесь и органический слой экстрагируют этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют, и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,200 г, 19,5%) в виде белого твердого вещества.
[Стадия 4] Синтез (S)-1-(2-этил-2-фторбутил)-4-пропилпиперазина
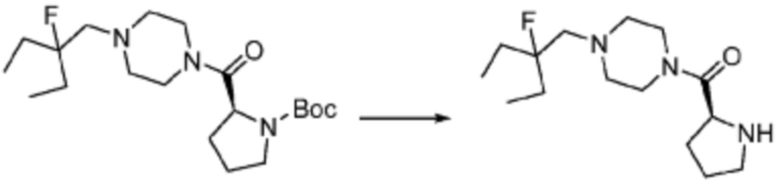
Трет-бутил (S)-2-(4-(2-этил-2-фторбутил)пиперазин-1-карбонил)пирролидин-1-карбоксилат (0,200 г, 0,519 ммоль), полученный на стадии 3, и хлористоводородную кислоту (4,00 M раствор в диоксане, 1,297 мл, 5,188 ммоль) растворяют в дихлорметане (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный продукт используют без дополнительного процесса очистки (указанное в заголовке соединение, 0,140 г, 94,6%, белое твердое вещество).
[Стадия 5] (S)-(1-(7-амино-2-(фуран-2-ил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-5-ил)пирролидин-2-ил)(4-(2-этил-2-фторбутил)пиперазин-1-ил)метанон
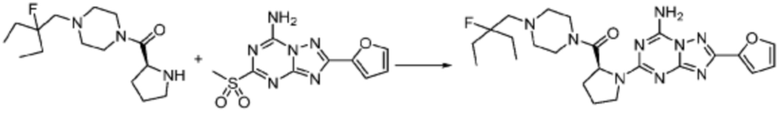
(S)-1-(2-Этил-2-фторбутил)-4-пропилпиперазин (0,150 г, 0,526 ммоль), полученный на стадии 4, 2-(фуран-2-ил)-5-(метилсульфонил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амин (0,074 г, 0,263 ммоль) и триэтиламин (0,147 мл, 1,051 ммоль) растворяют в диметилсульфоксиде (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Воду выливают в реакционную смесь и органический слой экстрагируют этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют, и концентрируют при пониженном давлении. Полученный концентрат очищают хроматографией (SiO2 тарелка, 20×20x1 мм; метанол/дихлорметан=10%) и концентрируют с получением указанного в заголовке соединения (0,005 г, 2,0%) в виде белого твердого вещества.
1H ЯМР (400 MГц, ДМСО-d6) δ 8,64-8,08 (м, 2H), 7,87 (с, 1H), 7,09-7,01 (м, 1H), 6,68 (с, 1H), 5,05-4,92 (м, 1H), 3,77-3,36 (м, 6H), 2,81-2,21 (м, 6H), 2,03-1,58 (м, 8H), 0,91-0,79 (м, 6H); МСНР (ЭР) m/z 486,5 (M++ 1).
Пример 245: Синтез соединения 245
Пример соединения 245 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 43 за исключением применения трет-бутил 1'-(2-этил-2-гидроксибутил)-[4,4'-бипиперидин]-1-карбоксилата вместо трет-бутил 4-(2-этил-2-гидроксибутил)пиперазин-1-карбоксилата на стадии 1.
Примеры 322 и 332
Примеры соединений 322 и 332 каждый синтезируют по существу тем же способом синтеза, как и способ синтеза из Примера соединения 245 за исключением применения (S)-1-(трет-бутоксикарбонил)азетидин-2-карбоновой кислоты и (трет-бутоксикарбонил)-L-аланина, соответственно, вместо (трет-бутоксикарбонил)-L-пролина.
Пример 374: Синтез соединения 374
Пример соединения 374 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 43 за исключением применения трет-бутил (R)-4-(2-гидрокси-2-метилпропил)-2-метилпиперазин-1-карбоксилата вместо трет-бутил 4-(2-этил-2-гидроксибутил)пиперазин-1-карбоксилата на стадии 1.
Пример 382: Синтез соединения 382
Пример соединения 382 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 322 за исключением применения 2-(фуран-2-ил)-7-(метилсульфонил)-[1,2,4]триазоло[1,5-c]пиримидин-5-амина вместо 2-(фуран-2-ил)-5-(метилсульфонил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амина.
Пример 395: Синтез соединения 395
Пример соединения 395 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 245 за исключением применения 2-(фуран-2-ил)-7-(метилсульфонил)-[1,2,4]триазоло[1,5-c]пиримидин-5-амина вместо 2-(фуран-2-ил)-5-(метилсульфонил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амина.
Аналитические данные каждого из соединений, полученных как описано выше, показаны в таблице ниже.
[Таблица 42]
№
Пример 282: Синтез соединения 282
[Стадия 1] Синтез трет-бутил 4-(3,3,3-трифтор-2,2-диметилпропаноил)пиперазин-1-карбоксилата
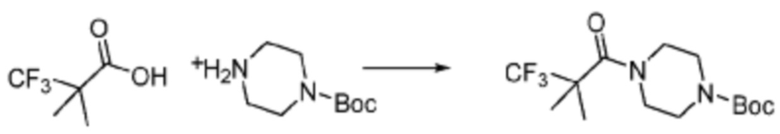
3,3,3-Трифтор-2,2-диметилпропановую кислоту (1,000 г, 6,406 ммоль), оксалилхлорид (0,550 мл, 6,406 ммоль) и N, N-диметилформамид (0,049 мл, 0,641 ммоль) растворяют в дихлорметане (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. К полученной смеси добавляют раствор, полученный растворением трет-бутил пиперазин-1-карбоксилата (1,067 г, 5,729 ммоль) и триэтиламина (1,597 мл, 11,458 ммоль) в дихлорметане (5 мл) при комнатной температуре, и перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего насыщенный водный раствор хлорида аммония выливают в полученный концентрат и органический слой экстрагируют этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют, и концентрируют при пониженном давлении. Полученный продукт используют без дополнительного процесса очистки (указанное в заголовке соединение, 1,800 г, 96,9%, белое масло).
[Стадия 2] Синтез трет-бутил 4-((1-(трифторметил)циклобутил)метил)пиперазин-1-карбоксилата

Трет-бутил 4-(1-(трифторметил)циклобутан-1-карбонил)пиперазин-1-карбоксилат (2,000 г, 6,166 ммоль), полученный на стадии 2, трифторборан (45,00% раствор в Et2O, 4,647 мл, 30,832 ммоль) и боргидрид натрия (0,467 г, 12,333 ммоль) растворяют в тетрагидрофуране (20 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего Насыщенный водный раствор гидрокарбоната натрия выливают в полученный концентрат, и органический слой экстрагируют этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют, и концентрируют при пониженном давлении. Полученный продукт используют без дополнительного процесса очистки (указанное в заголовке соединение, 1,500 г, 78,4%, коричневое масло).
[Стадия 3] 1-(3,3,3-трифтор-2,2-диметилпропил)пиперазин

Трет-бутил 4-(3,3,3-трифтор-2,2-диметилпропил)пиперазин-1-карбоксилат (1,500 г, 4,833 ммоль), полученный на стадии 2, и хлористоводородную кислоту (4,00 M раствор в диоксане, 12,083 мл, 48,331 ммоль) растворяют в дихлорметане (20 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный продукт используют без дополнительного процесса очистки (указанное в заголовке соединение, 0,900 г, 88,6%, коричневое масло).
[Стадия 4] Трет-бутил (S)-2-(4-(3,3,3-трифтор-2,2-диметилпропил)пиперазин-1-карбонил)пирролидин-1-карбоксилат
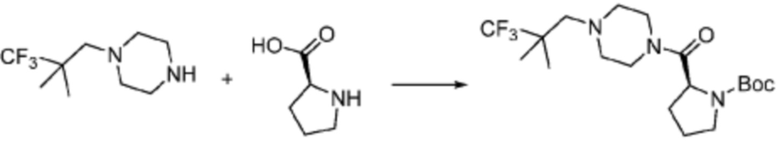
1-(3,3,3-Трифтор-2,2-диметилпропил)пиперазин (1,000 г, 4,756 ммоль), полученный на стадии 3, L-пролин (1,095 г, 9,513 ммоль), гексафторфосфат 3-оксида 1-[бис(диметиламино)метилен]-1H-1,2,3-триазоло[4,5-b]пиридиния (ГАТУ, 3,617 г, 9,513 ммоль) и N, N-диизопропилэтиламин (4,142 мл, 23,782 ммоль) растворяют в дихлорметане (20 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего Насыщенный водный раствор гидрокарбоната натрия выливают в полученный концентрат, и органический слой экстрагируют этилацетатом. Органический слой промывают насыщенным водным раствором гидрокарбоната натрия, дегидратируют безводным сульфатом магния, фильтруют, и концентрируют при пониженном давлении. Полученный продукт используют без дополнительного процесса очистки (указанное в заголовке соединение, 1,500 г, 77,4%, коричневое масло).
[Стадия 5] (S)-1-пропил-4-(3,3,3-трифтор-2,2-диметилпропил)пиперазин
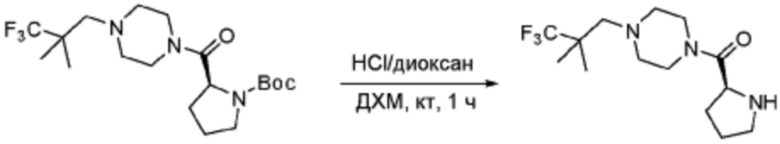
Трет-бутил (S)-2-(4-(3,3,3-трифтор-2,2-диметилпропил)пиперазин-1-карбонил)пирролидин-1-карбоксилат (0,300 г, 0,736 ммоль), полученный на стадии 5, и хлористоводородную кислоту (0,268 г, 7,362 ммоль) растворяют в дихлорметане (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный продукт используют без дополнительного процесса очистки (указанное в заголовке соединение, 0,200 г, 88,4%, коричневое масло).
[Стадия 6] (S)-(1-(7-амино-2-(фуран-2-ил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-5-ил)пирролидин-2-ил)(4-(3,3,3-трифтор-2,2-диметилпропил)пиперазин-1-ил)метанон 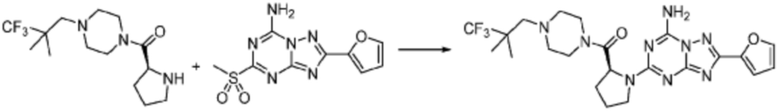
(S)-1-Пропил-4-(3,3,3-трифтор-2,2-диметилпропил)пиперазин (0,200 г, 0,651 ммоль), полученный на стадии 5, 2-(фуран-2-ил)-5-(метилсульфонил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амин (0,182 г, 0,651 ммоль) и гидрокарбонат натрия (0,164 г, 1,952 ммоль) растворяют в ацетонитриле (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при 70°C в течение 18 часов до завершения реакции путем понижения температуры до комнатной температуры. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан=0-100%) и концентрируют с получением указанного в заголовке соединения (0,010 г, 3,0%) в виде белого твердого вещества.
1H ЯМР (400 MГц, ДМСО-d6) δ 8,30 (м, 2H), 7,87 (ддд, J=2,6, 1,8, 0,8 Гц, 1H), 7,06 (ддд, J=6,5, 3,4, 0,8 Гц, 1H), 6,69-6,61 (м, 1H), 5,06-4,90 (м, 1H), 3,76-3,40 (м, 6H), 2,77 (м, 1H), 2,60-2,34 (м, 5H), 2,26 (м, 1H), 1,96-1,75 (м, 3H), 1,11 (м, 6H); МСНР (ЭР) m/z 508,5 (M++ 1).
Примеры 283, 284, 285, 286, 330 и 389
Примеры соединений 283, 284, 285, 286, 330 и 389 каждый синтезируют по существу тем же способом синтеза, как и способ синтеза из Примера соединения 282 за исключением применения соединений из следующей таблицы вместо 3,3,3-трифтор-2,2-диметилпропановой кислоты в качестве исходного материала.
[Таблица 43]
№
№
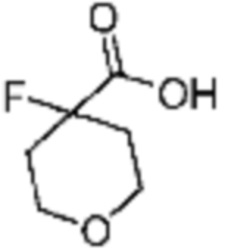
Аналитические данные каждого из соединений, полученных как описано выше, показаны в таблице ниже.
[Таблица 44]
№
Пример 130: Синтез соединения 130, 1-((7-амино-2-(фуран-2-ил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-5-ил)-L-пропил)-N-(3-фторфенил)пиперидин-4-карбоксамида
[Стадия 1] Синтез трет-бутил 4-((3-фторфенил)карбамоил)пиперидин-1-карбоксилата
1-(Трет-бутоксикарбонил)пиперидин-4-карбоновую кислоту (0,229 г, 1,000 ммоль), 3-фторанилин (0,096 мл, 1,000 ммоль), 2,4,6-трипропил-1,3,5,2,4,6-триоксатрифосфинан 2,4,6-триоксид (T3P, 50,00% раствор в EtOAc, 0,892 мл, 1,500 ммоль) и N, N-диизопропилэтиламин (0,523 мл, 3,000 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, и затем органический слой экстрагируют дихлорметаном, фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора из него, и концентрируют при пониженном давлении. Полученный продукт используют без дополнительного процесса очистки (указанное в заголовке соединение, 0,322 г, 99,9%, светло-желтое масло).
[Стадия 2] Синтез N-(3-фторфенил)пиперидин-4-карбоксамида гидрохлорида

Трет-бутил 4-((3-фторфенил)карбамоил)пиперидин-1-карбоксилат (0,322 г, 0,999 ммоль), полученный на стадии 1, и хлороводород (4,00 M раствор в 1,4-диоксане, 0,999 мл, 3,995 ммоль) растворяют в дихлорметане (4 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный продукт используют без дополнительного процесса очистки (указанное в заголовке соединение, 0,258 г, 99,8%, светло-желтое масло).
[Стадия 3] Синтез трет-бутил (S)-2-(4-((3-фторфенил)карбамоил)пиперидин-1-карбонил)пирролидин-1-карбоксилата
N-(3-фторфенил)пиперидин-4-карбоксамид гидрохлорид (0,258 г, 0,997 ммоль), полученный на стадии 2, (трет-бутоксикарбонил)-L-пролин (0,215 г, 0,997 ммоль), 2,4,6-трипропил-1,3,5,2,4,6-триоксатрифосфинан 2,4,6-триоксид (T3P, 50,00% раствор в EtOAc, 0,889 мл, 1,496 ммоль) и N, N-диизопропилэтиламин (0,521 мл, 2,992 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, и затем органический слой экстрагируют дихлорметаном, фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора из него, и концентрируют при пониженном давлении. Полученный продукт используют без дополнительного процесса очистки (указанное в заголовке соединение, 0,410 г, 98,0%, светло-желтое масло).
[Стадия 4] Синтез (S)-N-(3-фторфенил)-1-пропилпиперидин-4-карбоксамида гидрохлорида

Трет-бутил (S)-2-(4-((3-фторфенил)карбамоил)пиперидин-1-карбонил)пирролидин-1-карбоксилат (0,419 г, 0,999 ммоль), полученный на стадии 3, и хлороводород (4,00 M раствор в 1,4-диоксане, 0,999 мл, 3,995 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный продукт используют без дополнительного процесса очистки (указанное в заголовке соединение, 0,350 г, 98,5%, светло-желтое масло).
[Стадия 5] Синтез 1-((7-амино-2-(фуран-2-ил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-5-ил)-L-пропил)-N-(3-фторфенил)пиперидин-4-карбоксамида
(S)-N-(3-фторфенил)-1-пропилпиперидин-4-карбоксамид гидрохлорид (0,356 г, 1,000 ммоль), полученный на стадии 4, 2-(фуран-2-ил)-5-(метилсульфонил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амин (0,280 г, 1,000 ммоль) и гидрокарбонат натрия (0,252 г, 3,001 ммоль) растворяют в циклопентилметиловом эфире (ЦПМЭ, 5 мл) при комнатной температуре, после чего полученный раствор перемешивают при 50°C в течение 18 часов до завершения реакции путем понижения температуры до комнатной температуры. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего насыщенный водный раствор хлорида аммония выливают в полученный концентрат, и затем органический слой экстрагируют дихлорметаном, фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора из него, и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; дихлорметан/метанол=0-5%) и концентрируют с получением указанного в заголовке соединения (0,109 г, 21,0%) в виде белого твердого вещества.
1H ЯМР (400 MГц, ДМСО-d6) δ 10,46-10,07 (м, 1H), 8,27 (д, J=95,0 Гц, 1H), 7,89-7,81 (м, 1H), 7,67 (дд, J=40,6, 11,7 Гц, 1H), 7,33 (д, J=8,0 Гц, 2H), 7,05 (дд, J=9,5, 3,4 Гц, 1H), 6,95-6,83 (м, 1H), 6,68 (тд, J=3,4, 1,8 Гц, 1H), 5,12-4,92 (м, 1H), 4,44-4,28 (м, 1H), 4,21-4,01 (м, 1H), 3,71-3,56 (м, 2H), 3,30-3,12 (м, 1H), 2,82-2,58 (м, 2H), 2,39-2,13 (м, 1H), 1,98-1,77 (м, 5H), 1,68-1,39 (м, 2H). МСНР (ЭР) m/z 521,5 (M++ 1).
Примеры 131, 132 и 133
Примеры соединений 131, 132 и 133 каждый синтезируют по существу тем же способом синтеза, как и способ синтеза из Примера соединения 130 за исключением применения соединений из следующей таблицы вместо 3-фторанилина в качестве исходного материала.
[Таблица 45]
№
№
Аналитические данные каждого из соединений, полученных как описано выше, показаны в таблице ниже.
[Таблица 46]
№
Пример 331: Синтез соединения 331, (2-(7-амино-2-(фуран-2-ил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-5-ил)пиразолидин-1-ил)(4-(2,2,2-трифторэтил)пиперазин-1-ил)метанона
[Стадия 1] Синтез трет-бутил 4-(4-(трифторметил)пиперидин-1-карбонил)пиперидин-1-карбоксилата
1-(Трет-бутоксикарбонил)пиперидин-4-карбоновую кислоту (3,000 г, 13,084 ммоль), оксалилдихлорид (1,122 мл, 13,084 ммоль) и N, N-диметилформамид (0,050 мл, 0,654 ммоль) растворяют в дихлорметане (30 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение одного часа. 4-(трифторметил)пиперидин (1,236 г, 8,074 ммоль) и триэтиламин (1,688 мл, 12,110 ммоль) добавляют в полученный раствор, растворенный в дихлорметане (5 мл), при комнатной температуре и перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего воду выливают в полученный концентрат и органический слой экстрагируют этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют, и концентрируют при пониженном давлении. Полученный продукт используют без дополнительного процесса очистки (указанное в заголовке соединение, 1,500 г, 102,0%, белое твердое вещество).
[Стадия 2] Синтез трет-бутил 4-((4-(трифторметил)пиперидин-1-ил)метил)пиперидин-1-карбоксилата
Трет-бутил 4-(4-(трифторметил)пиперидин-1-карбонил)пиперидин-1-карбоксилат (1,500 г, 4,116 ммоль), полученный на стадии 1, трифторборан (45,00% раствор в Et2O, 0,457 мл, 20,581 ммоль) и боргидрид натрия (0,311 г, 8,232 ммоль) растворяют в тетрагидрофуране (20 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего насыщенный водный раствор гидрокарбоната натрия выливают в полученный концентрат, и органический слой экстрагируют этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют, и концентрируют при пониженном давлении. Полученный продукт используют без дополнительного процесса очистки (указанное в заголовке соединение, 0,500 г, 34,7%, белое твердое вещество).
[Стадия 3] Синтез 1-(пиперидин-4-илметил)-4-(трифторметил)пиперидина

Трет-бутил 4-((4-(трифторметил)пиперидин-1-ил)метил)пиперидин-1-карбоксилат (0,500 г, 1,427 ммоль), полученный на стадии 2, и хлористоводородную кислоту (4,00 M раствор в диоксане, 1,784 мл, 7,134 ммоль) растворяют в дихлорметане (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный продукт используют без дополнительного процесса очистки (указанное в заголовке соединение, 0,300 г, 84,0%, белое твердое вещество).
[Стадия 4] Синтез трет-бутил (S)-2-(4-((4-(трифторметил)пиперидин-1-ил)метил)пиперидин-1-карбонил)пирролидин-1-карбоксилата
1-(Пиперидин-4-илметил)-4-(трифторметил)пиперидин (0,300 г, 1,199 ммоль), полученный на стадии 3, (трет-бутоксикарбонил)-L-пролин (0,516 г, 2,397 ммоль), гексафторфосфат 3-оксида 1-[бис(диметиламино)метилен]-1H-1,2,3-триазоло[4,5-b]пиридиния (ГАТУ, 0,911 г, 2,397 ммоль) и N, N-диизопропилэтиламин (1,044 мл, 5,993 ммоль) растворяют в дихлорметане (30 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего водный раствор N-гидрокарбонат натрия выливают в полученный концентрат, и органический слой экстрагируют этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют, и концентрируют при пониженном давлении. Полученный продукт используют без дополнительного процесса очистки (указанное в заголовке соединение, 0,500 г, 93,2%, коричневое масло).
[Стадия 5] Синтез (S)-1-пропил-4-((4-(трифторметил)пиперидин-1-ил)метил)пиперидина
Трет-бутил (S)-2-(4-((4-(трифторметил)пиперидин-1-ил)метил)пиперидин-1-карбонил)пирролидин-1-карбоксилат (0,300 г, 0,670 ммоль), полученный на стадии 4, и хлористоводородную кислоту (4,00 M раствор в диоксане, 1,676 мл, 6,703 ммоль) растворяют в дихлорметане (20 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный продукт используют без дополнительного процесса очистки (указанное в заголовке соединение, 0,200 г, 85,9%, коричневое масло).
[Стадия 6] Синтез (S)-(1-(7-амино-2-(фуран-2-ил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-5-ил)пирролидин-2-ил)(4-((4-(трифторметил)пиперидин-1-ил)метил)пиперидин-1-ил)метанона
(S)-1-Пропил-4-((4-(трифторметил)пиперидин-1-ил)метил)пиперидин (0,300 г, 0,863 ммоль), полученный на стадии 5, 2-(фуран-2-ил)-5-(метилсульфонил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амин (0,060 г, 0,216 ммоль) и гидрокарбонат натрия (0,218 г, 2,590 ммоль) растворяют в ацетонитриле (20 мл) при комнатной температуре, после чего полученный раствор перемешивают при 70°C в течение 18 часов до завершения реакции путем понижения температуры до комнатной температуры. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный концентрат очищают хроматографией (SiO2 тарелка, 20×20x1 мм; метанол/дихлорметан=10%) и концентрируют с получением указанного в заголовке соединения (0,005 г, 1,1%) в виде белого твердого вещества.
1H ЯМР (400 MГц, ДМСО-d6) δ 8,56-8,03 (м, 2H), 7,87 (д, J=1,7 Гц, 1H), 7,09-6,95 (м, 1H), 6,68 (с, 1H), 5,01 (м, 1H), 4,41-3,87 (м, 2H), 3,73-3,44 (м, 4H), 3,24-2,85 (м, 6H), 2,38-2,02 (м, 4H), 2,01-1,57 (м, 8H), 1,48 (с, 1H), 1,10-0,75 (м, 2H); МСНР (ЭР) m/z 548,6 (M++ 1).
Пример 402: Синтез соединения 402
Пример соединения 402 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 331 за исключением применения (S)-1-(трет-бутоксикарбонил)азетидин-2-карбоновой кислоты вместо (трет-бутоксикарбонил)-L-пролина.
Пример 406: Синтез соединения 406
Пример соединения 406 получают по существу тем же способом синтеза, как и способ синтеза из Примера соединения 331 за исключением применения 3-фторазетидина вместо 4-(трифторметил)пиперидина.
Аналитические данные каждого из соединений, полученных как описано выше, показаны в таблице ниже.
[Таблица 47]
№
Пример 186: Синтез соединения 186, (S)-2-((7-амино-2-(фуран-2-ил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-5-ил)амино)-2-циклогексил-1-(4-(3,3-дифторазетидин-1-ил)пиперидин-1-ил)этан-1-она
[Стадия 1] Синтез трет-бутил 4-(3,3-дифторазетидин-1-ил)пиперидин-1-карбоксилата
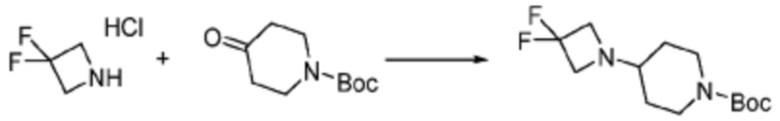
Трет-бутил 4-оксопиперидин-1-карбоксилат (0,500 г, 2,509 ммоль), 3,3-дифторазетидин (0,325 г, 2,509 ммоль), триацетоксиборгидрид натрия (0,798 г, 3,764 ммоль) и триэтиламин (0,350 мл, 2,509 ммоль) растворяют в дихлорметане (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Водный раствор N-гидрокарбонат натрия выливают в реакционную смесь, и затем органический слой экстрагируют дихлорметаном, фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора из него, и концентрируют при пониженном давлении. Полученный продукт используют без дополнительного процесса очистки (указанное в заголовке соединение, 0,653 г, 94,2%, прозрачное масло).
[Стадия 2] Синтез 4-(3,3-дифторазетидин-1-ил)пиперидина гидрохлорида

Трет-бутил 4-(3,3-дифторазетидин-1-ил)пиперидин-1-карбоксилат (0,653 г, 2,363 ммоль), полученный на стадии 1, и хлороводород (4,00 M раствор в 1,4-диоксане, 2,363 мл, 9,452 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный продукт используют без дополнительного процесса очистки (указанное в заголовке соединение, 0,502 г, 99,9%, белое твердое вещество).
[Стадия 3] Синтез трет-бутил (S)-(1-циклогексил-2-(4-(3,3-дифторазетидин-1-ил)пиперидин-1-ил)-2-оксоэтил)карбамата
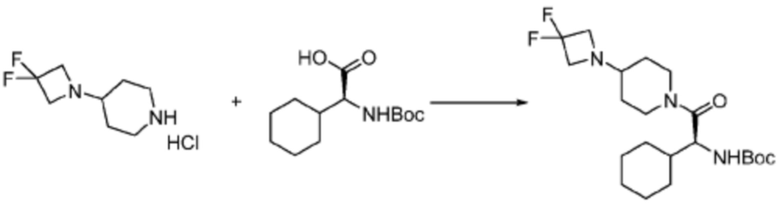
4-(3,3-Дифторазетидин-1-ил)пиперидин гидрохлорид (0,258 г, 1,213 ммоль), полученный на стадии 2, (S)-2-((трет-бутоксикарбонил)амино)-2-циклогексилуксусную кислоту (0,312 г, 1,213 ммоль), 2,4,6-трипропил-1,3,5,2,4,6-триоксатрифосфинан 2,4,6-триоксид (T3P, 50,00% раствор в EtOAc, 1,082 мл, 1,820 ммоль) и N, N-диизопропилэтиламин (0,634 мл, 3,639 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Водный раствор N-гидрокарбонат натрия выливают в реакционную смесь, и затем органический слой экстрагируют дихлорметаном, фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора из него, и концентрируют при пониженном давлении. Полученный продукт используют без дополнительного процесса очистки (указанное в заголовке соединение, 0,415 г, 82,3%, прозрачное масло).
[Стадия 4] Синтез (S)-2-амино-2-циклогексил-1-(4-(3,3-дифторазетидин-1-ил)пиперидин-1-ил)этан-1-он гидрохлорида
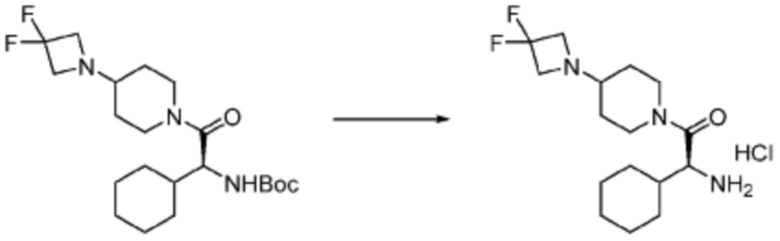
Трет-бутил (S)-(1-циклогексил-2-(4-(3,3-дифторазетидин-1-ил)пиперидин-1-ил)-2-оксоэтил)карбамат (0,415 г, 0,999 ммоль), полученный на стадии 3, и хлороводород (4,00 M раствор в 1,4-диоксане, 0,999 мл, 3,995 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный продукт используют без дополнительного процесса очистки (указанное в заголовке соединение, 0,351 г, 99,9%, светло-желтое твердое вещество).
[Стадия 5] Синтез (S)-2-((7-амино-2-(фуран-2-ил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-5-ил)амино)-2-циклогексил-1-(4-(3,3-дифторазетидин-1-ил)пиперидин-1-ил)этан-1-она
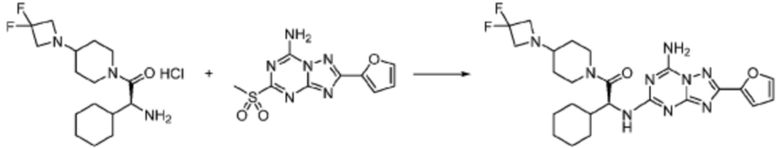
(S)-2-Амино-2-циклогексил-1-(4-(3,3-дифторазетидин-1-ил)пиперидин-1-ил)этан-1-он гидрохлорид (0,351 г, 0,998 ммоль), полученный на стадии 4, 2-(фуран-2-ил)-5-(метилсульфонил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амин (0,280 г, 0,998 ммоль) и гидрокарбонат натрия (0,251 г, 2,993 ммоль) растворяют в ацетонитриле (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при 70°C в течение 18 часов до завершения реакции путем понижения температуры до комнатной температуры. Реакционную смесь фильтруют через пластиковый фильтр для удаления из нее твердого вещества, после чего растворитель удаляют из полученного фильтрата без твердого вещества при пониженном давлении, и затем полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; дихлорметан/метанол=0-10%) и концентрируют с получением продукта, после чего полученный продукт снова очищают хроматографией (SiO2 тарелка, 20×20x1 мм; дихлорметан/метанол) и концентрируют с получением указанного в заголовке соединения (0,041 г, 7,9%) в виде белого твердого вещества.
1H ЯМР (400 MГц, Хлороформ-d) δ 9,71 (с, 1H), 8,47 (дд, J=23,4, 9,4 Гц, 1H), 7,57 (дд, J=3,1, 1,7 Гц, 1H), 7,23-7,07 (м, 1H), 6,54 (дт, J=3,8, 2,0 Гц, 1H), 6,28 (с, 1H), 5,23-5,05 (м, 1H), 4,41-4,05 (м, 2H), 3,66-3,44 (м, 5H), 3,22-3,09 (м, 1H), 2,51-2,35 (м, 1H), 1,96-1,52 (м, 9H), 1,21-0,89 (м, 6H). МСНР (ЭР) m/z 516,6(M++ 1).
Пример 390: Синтез соединения 390
Пример соединения 390 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 186 за исключением применения (трет-бутоксикарбонил)-L-пролина вместо (S)-2-((трет-бутоксикарбонил)амино)-2-циклогексилуксусной кислоты.
Пример 391: Синтез соединения 391
Пример соединения 391 получают по существу тем же способом синтеза, как и способ синтеза из Примера соединения 390 за исключением применения 4,4-дифторпиперидина вместо 3,3-дифторазетидина.
Пример 400: Синтез соединения 400
Пример соединения 400 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 390 за исключением применения 2-(фуран-2-ил)-7-(метилсульфонил)-[1,2,4]триазоло[1,5-c]пиримидин-5-амина вместо 2-(фуран-2-ил)-5-(метилсульфонил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амина.
Пример 405: Синтез соединения 405
Пример соединения 405 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 390 за исключением применения 1-(2-метоксиэтил)пиперазина вместо 3,3-дифторазетидина и с применением трет-бутил 3-оксоазетидин-1-карбоксилата вместо трет-бутил 4-оксопиперидин-1-карбоксилата.
Аналитические данные каждого из соединений, полученных как описано выше, показаны в таблице ниже.
[Таблица 48]
Пример 25: Синтез соединения 25, (S)-2-((7-амино-2-(фуран-2-ил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-5-ил)амино)-1-(4-(2,2,2-трифторэтил)пиперазин-1-ил)пропан-1-она
[Стадия 1] Синтез бензил 4-((трет-бутоксикарбонил)-L-аланил)пиперазин-1-карбоксилата
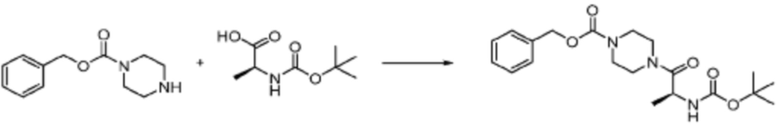
Бензилпиперазин-1-карбоксилат (1,000 г, 4,540 ммоль), (трет-бутоксикарбонил)-L-аланин (0,859 г, 4,540 ммоль), 1-этил-3-(3-диметиламинопропил)карбодиимид гидрохлорид (ЭДК-HCl, 2,611 г, 13,620 ммоль), 1H-бензо[d][1,2,3]триазол-1-ол (HOBt, 1,840 г, 13,620 ммоль) и N, N-диизопропилэтиламин (3,954 мл, 22,699 ммоль) растворяют в дихлорметане (30 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Воду выливают в реакционную смесь и органический слой экстрагируют дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют, и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=0-10%) и концентрируют с получением указанного в заголовке соединения (1,200 г, 67,5%) в виде бесцветного масла.
[Стадия 2] Синтез трет-бутил (S)-(1-оксо-1-(пиперазин-1-ил)пропан-2-ил)карбамата
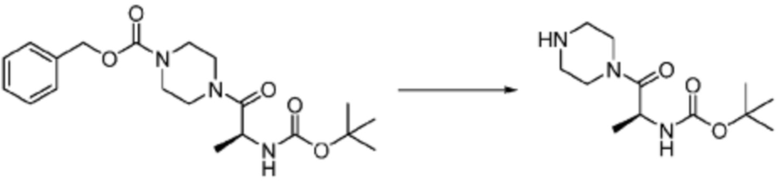
Бензил 4-((трет-бутоксикарбонил)-L-аланил)пиперазин-1-карбоксилат (1,200 г, 3,065 ммоль), полученный на стадии 1, растворяют в метаноле (20 мл) и перемешивают при комнатной температуре, после чего Pd/C (0,100 мг) медленно добавляют в полученный раствор при той же температуре и перемешивают в течение 18 часов в присутствии баллона водорода, присоединенного к нему, при той же температуре. Реакционную смесь фильтруют через слой целита для удаления из нее твердого вещества, после чего растворитель удаляют из полученного фильтрата при пониженном давлении, и затем полученный продукт используют без дополнительного процесса очистки (указанное в заголовке соединение 0,746 г, 94,6%, бесцветное масло).
[Стадия 3] Синтез трет-бутил (S)-(1-оксо-1-(4-(2,2,2-трифторэтил)пиперазин-1-ил)пропан-2-ил)карбамата
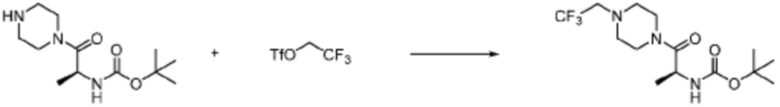
Трет-бутил (S)-(1-оксо-1-(пиперазин-1-ил)пропан-2-ил)карбамат (0,080 г, 0,311 ммоль), полученный на стадии 2, 2,2,2-трифторэтилтрифторметансульфонат (0,072 г, 0,311 ммоль) и карбонат калия (0,086 г, 0,622 ммоль) растворяют в ацетонитриле (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Реакционную смесь фильтруют через пластиковый фильтр для удаления из нее твердого вещества, после чего растворитель удаляют из полученного фильтрата при пониженном давлении, и затем полученный продукт используют без дополнительного процесса очистки (указанное в заголовке соединение, 0,065 г, 61,6%, светло-коричневое масло).
[Стадия 4] Синтез (S)-2-амино-1-(4-(2,2,2-трифторэтил)пиперазин-1-ил)пропан-1-она

Трет-бутил (S)-(1-оксо-1-(4-(2,2,2-трифторэтил)пиперазин-1-ил)пропан-2-ил)карбамат (0,065 г, 0,192 ммоль), полученный на стадии 3, и трифторуксусную кислоту (0,147 мл, 1,915 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, и затем органический слой экстрагируют дихлорметаном, фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора из него, и концентрируют при пониженном давлении. Полученный продукт используют без дополнительного процесса очистки (указанное в заголовке соединение, 0,042 г, 91,7%, желтое масло).
[Стадия 5] Синтез (S)-2-((7-амино-2-(фуран-2-ил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-5-ил)амино)-1-(4-(2,2,2-трифторэтил)пиперазин-1-ил)пропан-1-он
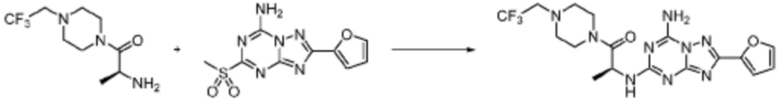
2-(фуран-2-ил)-5-(метилсульфонил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амин (0,042 г, 0,150 ммоль), полученный на стадии 4, (S)-2-амино-1-(4-(2,2,2-трифторэтил)пиперазин-1-ил)пропан-1-он (0,036 г, 0,150 ммоль) и триэтиламин (0,042 мл, 0,300 ммоль) растворяют в диметилсульфоксиде (1 мл) при комнатной температуре, и полученный раствор перемешивают при той же температуре в течение 18 часов. Воду выливают в реакционную смесь, и затем органический слой экстрагируют дихлорметаном, фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора из него, и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,010 г, 15,3%) в виде белого твердого вещества.
1H ЯМР (400 MГц, Хлороформ-d) δ 8,40 (с, 1H), 8,02 (д, J=8,6 Гц, 1H), 7,61 (с, 1H), 7,21 (с, 1H), 6,59 (с, 1H), 5,39-5,31 (м, 1H), 4,05-3,97 (м, 1H), 3,93-3,85 (м, 1H), 3,74-3,67 (м, 1H), 3,59-3,50 (м, 1H), 3,15-2,98 (м, 2H), 2,93-2,81 (м, 2H), 2,78-2,74 (м, 1H), 2,70-2,62 (м, 1H), 1,44 (д, J=6,8 Гц, 3H); МСНР (ЭР) m/z 440,4 (M++ 1).
Пример 4: Синтез соединения 4
Пример соединения 4 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 25 за исключением применения (трет-бутоксикарбонил)-L-фенилаланина вместо (трет-бутоксикарбонил)-L-аланина.
1H ЯМР (400 MГц, Хлороформ-d) δ 9,64 (с, 1H), 8,69 (д, J=9,2 Гц, 1H), 7,62 (с, 1H), 7,32-7,07 (м, 5H), 6,59 (с, 1H), 6,32 (с, 1H), 5,63 (кв, J=8,4 Гц, 1H), 3,81-3,71 (м, 1H), 3,68-3,60 (м, 1H), 3,57-3,48 (м, 2H), 3,09 (д, J=7,7 Гц, 2H), 2,91 (кв, J=9,0, 8,5 Гц, 2H), 2,70-2,64 (м, 1H), 2,64-2,54 (м, 1H), 2,41 (т, J=9,3 Гц, 1H), 2,04 (т, J=9,6 Гц, 1H); МСНР (ЭР) m/z 516,3 (M++ 1).
Пример 64: Синтез соединения 64, (4-((7-амино-2-(фуран-2-ил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-5-ил)-L-пропил)пиперазин-1-ил)(2,4-дифторфенил)метанона
[Стадия 1] Синтез трет-бутил (S)-2-(пиперазин-1-карбонил)пирролидин-1-карбоксилата
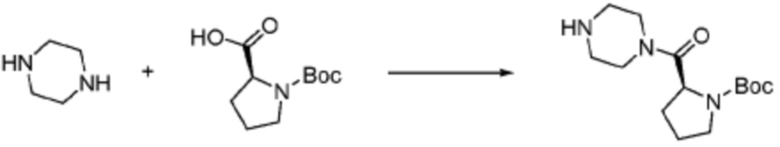
(Трет-бутоксикарбонил)-L-пролин (10,763 г, 50,000 ммоль), пиперазин (12,920 г, 150,000 ммоль), 1-этил-3-(3-диметиламинопропил)карбодиимид гидрохлорид (ЭДК-HCl, 19,170 г, 100,000 ммоль), 1H-бензо[d][1,2,3]триазол-1-ол (HOBt, 7,432 г, 55,000 ммоль) и N, N-диизопропилэтиламин (26,127 мл, 150,000 ммоль) растворяют в дихлорметане (200 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор хлорида аммония выливают в реакционную смесь, и органический слой экстрагируют дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют, и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 80 г картридж; метанол/дихлорметан=0-15%) и концентрируют с получением указанного в заголовке соединения (6,718 г, 47,4%) в виде белого твердого вещества.
[Стадия 2] Синтез трет-бутил (S)-2-(4-(2,4-дифторбензоил)пиперазин-1-карбонил)пирролидин-1-карбоксилата
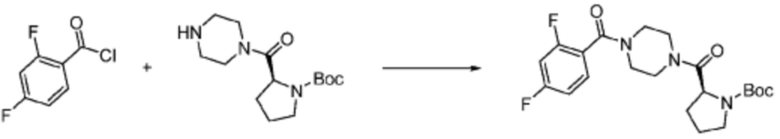
Трет-бутил (S)-2-(пиперазин-1-карбонил)пирролидин-1-карбоксилат (0,283 г, 1,000 ммоль), полученный на стадии 1, и 2,4-дифторбензоилхлорид (0,124 мл, 1,000 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный продукт используют без дополнительного процесса очистки (указанное в заголовке соединение, 0,420 г, 99,2%, светло-желтое масло).
[Стадия 3] Синтез (S)-(2,4-дифторфенил)(4-пропилпиперазин-1-ил)метанона гидрохлорида

Трет-бутил (S)-2-(4-(2,4-дифторбензоил)пиперазин-1-карбонил)пирролидин-1-карбоксилат (0,420 г, 0,992 ммоль), полученный на стадии 2, и хлороводород (4,00 M раствор в 1,4-диоксане, 0,992 мл, 3,967 ммоль) растворяют в дихлорметане (3 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение трех часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный продукт используют без дополнительного процесса очистки (указанное в заголовке соединение, 0,356 г, 99,8%, белое твердое вещество).
[Стадия 4] Синтез (4-((7-амино-2-(фуран-2-ил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-5-ил)-L-пропил)пиперазин-1-ил)(2,4-дифторфенил)метанона
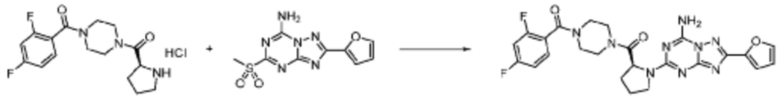
(S)-(2,4-Дифторфенил)(4-пропилпиперазин-1-ил)метанон гидрохлорид (0,356 г, 0,989 ммоль), полученный на стадии 3, 2-(фуран-2-ил)-5-(метилсульфонил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амин (0,277 г, 0,989 ммоль) и триэтиламин (0,414 мл, 2,968 ммоль) растворяют в N, N-диметилформамиде (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан= 0-5%) и концентрируют с получением указанного в заголовке соединения (0,227 г, 43,7%) в виде светло-желтого твердого вещества.
1H ЯМР (400 MГц, ДМСО-d6) δ 8,75-8,05 (м, 2H), 7,97-7,80 (м, 1H), 7,74-7,48 (м, 1H), 7,47-7,33 (м, 1H), 7,34-7,14 (м, 1H), 7,13-7,00 (м, 1H), 6,74-6,61 (м, 1H), 5,11-4,88 (м, 1H), 4,03-3,38 (м, 10H), 2,38-2,13 (м, 1H), 2,03-1,77 (м, 3H); МСНР (ЭР) m/z 524,5 (M++ 1).
Пример 5: Синтез соединения 5
Пример соединения 5 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 64 за исключением применения (трет-бутоксикарбонил)-L-фенилаланина вместо (трет-бутоксикарбонил)-L-пролина и с применением изобутурилхлорида вместо 2,4-дифторбензоилхлорида.
Примеры 46 и 63
Примеры соединений 46 и 63 получают по существу тем же способом синтеза, что и способ синтеза из Примера соединения 64 за исключением применения бензоилхлорида и 3-гидроксибензоилхлорида, соответственно, вместо 2,4-дифторбензоилхлорида.
Пример 99: Синтез соединения 99
Пример соединения 99 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 64 за исключением применения (трет-бутоксикарбонил)-D-пролина вместо (трет-бутоксикарбонил)-L-пролина и с применением морфолин-4-карбонилхлорида вместо 2,4-дифторбензоилхлорида.
Аналитические данные каждого из соединений, полученных как описано выше, показаны в таблице ниже.
[Таблица 49]
№
Пример 71: Синтез соединения 71, (S)-(1-(7-амино-2-(фуран-2-ил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-5-ил)пирролидин-2-ил)(4-(изопропилсульфонил)пиперазин-1-ил)метанона
[Стадия 1] Синтез трет-бутил (S)-2-(4-(изопропилсульфонил)пиперазин-1-карбонил)пирролидин-1-карбоксилата
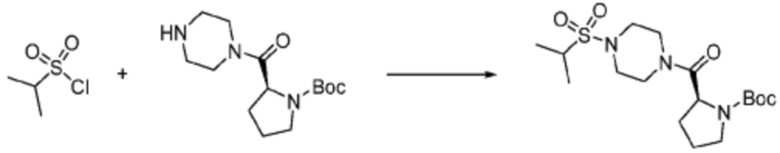
Трет-бутил (S)-2-(пиперазин-1-карбонил)пирролидин-1-карбоксилат (0,283 г, 1,000 ммоль), полученный на стадии 1 Примера 64, пропан-2-сульфонилхлорид (0,226 мл, 2,000 ммоль) и карбонат калия (0,415 г, 3,000 ммоль) растворяют в ацетонитриле (8 мл) при комнатной температуре, после чего полученный раствор перемешивают при 50°C в течение 18 часов, и затем реакцию завершают снижением температуры до комнатной температуры. Воду выливают в реакционную смесь, и затем органический слой экстрагируют дихлорметаном, фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора из него, и концентрируют при пониженном давлении. Полученный продукт используют без дополнительного процесса очистки (указанное в заголовке соединение, 0,770 г, 98,8%, светло-желтое масло).
[Стадия 2] Синтез (S)-1-(изопропилсульфонил)-4-пропилпиперазина гидрохлорида

Трет-бутил (S)-2-(4-(изопропилсульфонил)пиперазин-1-карбонил)пирролидин-1-карбоксилат (0,770 г, 1,977 ммоль), полученный на стадии 1, и хлороводород (4,00 M раствор в 1,4-диоксане, 1,977 мл, 7,907 ммоль) растворяют в дихлорметане (4 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный продукт используют без дополнительного процесса очистки (указанное в заголовке соединение, 0,640 г, 99,4%, светло-желтое масло).
[Стадия 3] Синтез (S)-(1-(7-амино-2-(фуран-2-ил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-5-ил)пирролидин-2-ил)(4-(изопропилсульфонил)пиперазин-1-ил)метанона
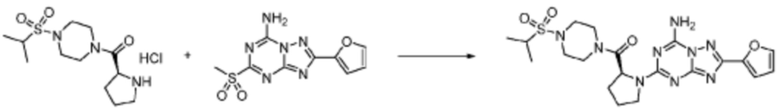
2-(Фуран-2-ил)-5-(метилсульфонил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амин (0,400 г, 1,427 ммоль), полученный на стадии 2, (S)-1-(изопропилсульфонил)-4-пропилпиперазин гидрохлорид (0,651 г, 1,998 ммоль) и гидрокарбонат натрия (0,360 г, 4,282 ммоль) растворяют в циклопентилметиловом эфире (ЦПМЭ, 10 мл) при комнатной температуре, после чего полученный раствор перемешивают при 70°C в течение 18 часов до завершения реакции путем понижения температуры до комнатной температуры. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан= 0-5%) и концентрируют с получением указанного в заголовке соединения (0,068 г, 9,7%) в виде белого твердого вещества.
1H ЯМР (400 MГц, ДМСО-d6) δ 8,70-8,09 (м, 2H), 7,90-7,84 (м, 1H), 7,10-6,98 (м, 1H), 6,75-6,63 (м, 1H), 5,09-4,92 (м, 1H), 3,97-3,53 (м, 6H), 3,53-3,38 (м, 3H), 3,31-3,11 (м, 2H), 2,32-2,16 (м, 1H), 2,05-1,78 (м, 3H), 1,36-1,18 (м, 6H); МСНР (ЭР) m/z 490,6 (M++ 1).
Пример 6: Синтез соединения 6
Пример соединения 6 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 71 за исключением применения бензолсульфонилхлорида вместо пропан-2-сульфонилхлорида и с применением (трет-бутоксикарбонил)-L-фенилаланина вместо (трет-бутоксикарбонил)-L-пролина.
Примеры 68, 69, 70 и 72
Примеры соединений 68, 69, 70 и 72 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 71 за исключением применения соединений из следующей таблицы вместо пропан-2-сульфонилхлорида в качестве исходного материала.
[Таблица 50]
№
№
Аналитические данные каждого из соединений, полученных как описано выше, показаны в таблице ниже.
[Таблица 51]
№
Пример 111: Синтез соединения 111, (S)-(1-(7-амино-2-(фуран-2-ил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-5-ил)азетидин-2-ил)(4-бутилпиперазин-1-ил)метанона
[Стадия 1] Синтез трет-бутил (S)-2-(пиперазин-1-карбонил)азетидин-1-карбоксилата
(S)-1-(Трет-бутоксикарбонил)азетидин-2-карбоновую кислоту (2,012 г, 10,000 ммоль), пиперазин (2,584 г, 30,000 ммоль) и 2,4,6-трипропил-1,3,5,2,4,6-триоксатрифосфинан 2,4,6-триоксид (T3P, 50,00% раствор в EtOAc, 8,918 мл, 15,000 ммоль) растворяют в дихлорметане (40 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, и органический слой экстрагируют дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют, и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,508 г, 18,9%) в виде бесцветного масла.
[Стадия 2] Синтез трет-бутил (S)-2-(4-бутилпиперазин-1-карбонил)азетидин-1-карбоксилата
Трет-бутил (S)-2-(пиперазин-1-карбонил)азетидин-1-карбоксилат (0,150 г, 0,557 ммоль), полученный на стадии 1, N, N-диизопропилэтиламин (0,097 мл, 0,557 ммоль) и 1-бромбутан (0,060 мл, 0,557 ммоль) растворяют в ацетонитриле (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Воду выливают в реакционную смесь, и затем органический слой экстрагируют этилацетатом, фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора из него, и концентрируют при пониженном давлении. Полученный продукт используют без дополнительного процесса очистки (указанное в заголовке соединение, 0,181 г, 99,9%, бесцветное масло).
[Стадия 3] Синтез (S)-азетидин-2-ил(4-бутилпиперазин-1-ил)метанона гидрохлорида

Трет-бутил (S)-2-(4-бутилпиперазин-1-карбонил)азетидин-1-карбоксилат (0,181 г, 0,556 ммоль), полученный на стадии 2, и хлороводород (4,00 M раствор в 1,4-диоксане, 0,556 мл, 2,225 ммоль) растворяют в дихлорметане (3 мл) при комнатной температуре, после чего полученный раствор перемешивают при 40°C в течение четырех часов до завершения реакции путем понижения температуры до комнатной температуры. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный продукт используют без дополнительного процесса очистки (указанное в заголовке соединение, 0,145 г, 99,6%, белое твердое вещество).
[Стадия 4] Синтез (S)-(1-(7-амино-2-(фуран-2-ил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-5-ил)азетидин-2-ил)(4-бутилпиперазин-1-ил)метанона
(S)-Азетидин-2-ил(4-бутилпиперазин-1-ил)метанон гидрохлорид (0,145 г, 0,554 ммоль), полученный на стадии 3, 2-(фуран-2-ил)-5-(метилсульфонил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амин (0,155 г, 0,554 ммоль) и гидрокарбонат натрия (0,140 г, 1,662 ммоль) растворяют в циклопентилметиловом эфире (ЦПМЭ, 4 мл) при комнатной температуре, после чего полученный раствор перемешивают при 50°C в течение 18 часов до завершения реакции путем понижения температуры до комнатной температуры. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан= 0-10%) и концентрируют с получением указанного в заголовке соединения (0,135 г, 57,5%) в виде белого твердого вещества.
1H ЯМР (400 MГц, ДМСО-d6) δ 8,78-8,01 (м, 2H), 7,92-7,82 (м, 1H), 7,12-6,99 (м, 1H), 6,73-6,62 (м, 1H), 5,20 (дд, 1H), 4,10-3,89 (м, 2H), 3,86-3,14 (м, 5H), 2,74-2,55 (м, 1H), 2,46-2,02 (м, 6H), 1,50-1,35 (м, 2H), 1,35-1,19 (м, 2H), 0,88 (т, J=7,3 Гц, 3H); МСНР (ЭР) m/z 426,6 (M++ 1).
Примеры 17 и 18
Примеры соединений 17 и 18 каждый синтезируют по существу тем же способом синтеза, как и способ синтеза из Примера соединения 111 за исключением применения трет-бутил (S)-(1-оксо-3-фенил-1-(пиперазин-1-ил)пропан-2-ил)карбамата вместо трет-бутил (S)-2-(пиперазин-1-карбонил)азетидин-1-карбоксилата и с применением 1-(бромметил)-3-фторбензола и 1-(бромметил)-4-фторбензола, соответственно, вместо 1-бромбутана.
Примеры 21, 22, 23 и 24
Примеры соединений 21, 22, 23 и 24 каждый синтезируют по существу тем же способом синтеза, как и способ синтеза из Примера соединения 111 за исключением применения трет-бутил (S)-(1-оксо-1-(пиперазин-1-ил)пропан-2-ил)карбамата вместо трет-бутил (S)-2-(пиперазин-1-карбонил)азетидин-1-карбоксилата и с применением соединений из следующей таблицы вместо 1-бромбутана в качестве исходного материала.
[Таблица 52]
№
№
Пример 45: Синтез соединения 45
Пример соединения 45 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 18 за исключением применения трет-бутил (S)-2-(пиперазин-1-карбонил)пирролидин-1-карбоксилата вместо трет-бутил (S)-(1-оксо-3-фенил-1-(пиперазин-1-ил)пропан-2-ил)карбамата.
Пример 47: Синтез соединения 47
Пример соединения 47 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 111 за исключением применения трет-бутил (S)-2-(пиперазин-1-карбонил)пирролидин-1-карбоксилата вместо трет-бутил (S)-2-(пиперазин-1-карбонил)азетидин-1-карбоксилата.
Пример 66: Синтез соединения 66
Пример соединения 66 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 111 за исключением применения трет-бутил (S)-2-(пиперазин-1-карбонил)пирролидин-1-карбоксилата вместо трет-бутил (S)-2-(пиперазин-1-карбонил)азетидин-1-карбоксилата, и с применением (1-бромэтил)бензола вместо 1-бромбутана.
Пример 101: Синтез соединения 101
Пример соединения 101 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 111 за исключением применения трет-бутил (R)-2-(пиперазин-1-карбонил)пирролидин-1-карбоксилата вместо трет-бутил (S)-2-(пиперазин-1-карбонил)азетидин-1-карбоксилата.
Аналитические данные каждого из соединений, полученных как описано выше, показаны в таблице ниже.
[Таблица 53]
№
Пример 61: Синтез соединения 61, 4-((7-амино-2-(фуран-2-ил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-5-ил)-L-пропил)-N-(м-толил)пиперазин-1-карбоксамида
[Стадия 1] Трет-бутил (S)-2-(4-(м-толилкарбамоил)пиперазин-1-карбонил)пирролидин-1-карбоксилата
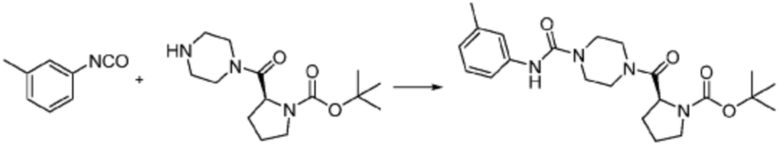
Трет-бутил (S)-2-(пиперазин-1-карбонил)пирролидин-1-карбоксилат (0,300 г, 1,059 ммоль), полученный на стадии 1 Примера 64, и 1-изоцианато-3-метилбензол (0,141 г, 1,059 ммоль) растворяют в диэтиловом эфире (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Воду выливают в реакционную смесь, и затем органический слой экстрагируют дихлорметаном, фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора из него, и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,198 г, 44,9%) в виде белого твердого вещества.
[Стадия 2] Синтез (S)-4-пропил-N-(м-толил)пиперазин-1-карбоксамида
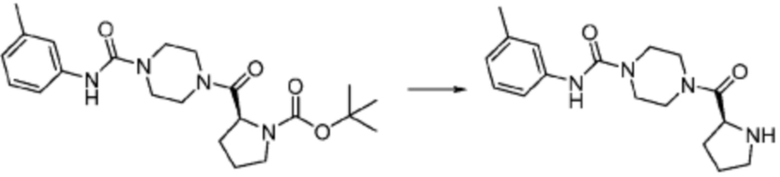
Трет-бутил (S)-2-(4-(м-толилкарбамоил)пиперазин-1-карбонил)пирролидин-1-карбоксилат (0,198 г, 0,475 ммоль), полученный на стадии 1, и хлористоводородную кислоту (4,00 M раствор в диоксане, 0,594 мл, 2,377 ммоль) смешивают, после чего полученную смесь перемешивают при комнатной температуре и перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, и затем органический слой экстрагируют дихлорметаном, фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора из него, и концентрируют при пониженном давлении. Полученный продукт используют без дополнительного процесса очистки (указанное в заголовке соединение, 0,098 г, 65,2%, светло-коричневое масло).
[Стадия 3] Синтез 4-((7-амино-2-(фуран-2-ил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-5-ил)-L-пропил)-N-(м-толил)пиперазин-1-карбоксамида
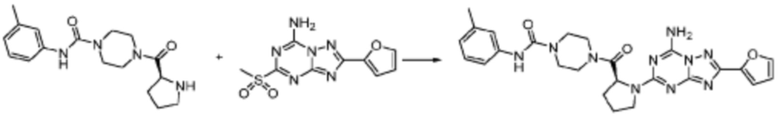
2-(Фуран-2-ил)-5-(метилсульфонил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амин (0,050 г, 0,178 ммоль), полученный на стадии 2, (S)-4-пропил-N-(м-толил)пиперазин-1-карбоксамид (0,056 г, 0,178 ммоль) и триэтиламин (0,050 мл, 0,357 ммоль) растворяют в диметилсульфоксиде (1 мл) при комнатной температуре, после чего полученный раствор перемешивают в течение 18 часов при той же температуре. Воду выливают в реакционную смесь, и затем органический слой экстрагируют дихлорметаном, фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора из него, и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением указанного в заголовке соединения (0,017 г, 18,4%) в виде белого твердого вещества.
1H ЯМР (400 MГц, Хлороформ-d) δ 7,58-7,45 (м, 1H), 7,27-7,20 (м, 1H), 7,21-7,11 (м, 3H), 7,08-6,91 (м, 1H), 6,90-6,83 (м, 1H), 6,55-6,47 (м, 1H), 6,10 (с, 0H), 5,03-4,81 (м, 1H), 3,97-3,36 (м, 10H), 2,32 (д, J=8,8 Гц, 4H), 2,28-2,11 (м, 1H), 2,08-1,85 (м, 2H); МСНР (ЭР) m/z 517,2 (M++ 1).
Пример 100: Синтез соединения 100
Пример соединения 100 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 61 за исключением применения трет-бутил (R)-2-(пиперазин-1-карбонил)пирролидин-1-карбоксилата вместо трет-бутил (S)-2-(пиперазин-1-карбонил)пирролидин-1-карбоксилата, и с применением изоцианатобензола вместо 1-изоцианато-3-метилбензола.
Примеры 62, 115, 116 и 117
Примеры соединений 62, 115, 116 и 117 каждый синтезируют по существу тем же способом синтеза, как и способ синтеза из Примера соединения 61 за исключением применения соединений из следующей таблицы вместо 1-изоцианато-3-метилбензола в качестве исходного материала.
[Таблица 54]
№
№
Аналитические данные каждого из соединений, полученных как описано выше, показаны в таблице ниже.
[Таблица 55]
№
Пример 82: Синтез соединения 82, (4-((7-амино-2-(фуран-2-ил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-5-ил)-L-пропил)пиперазин-1-ил)(3-(4-метилпиперазин-1-ил)фенил)метанона
[Стадия 1] Синтез трет-бутил (S)-2-(4-(3-(4-метилпиперазин-1-ил)бензоил)пиперазин-1-карбонил)пирролидин-1-карбоксилата

Трет-бутил (S)-2-(пиперазин-1-карбонил)пирролидин-1-карбоксилат (0,150 г, 0,529 ммоль), полученный на стадии 1 Примера 64, 3-(4-метилпиперазин-1-ил)бензойную кислоту (0,117 г, 0,529 ммоль), триэтиламин (0,221 мл, 1,588 ммоль) и 2,4,6-трипропил-1,3,5,2,4,6-триоксатрифосфинан 2,4,6-триоксид (T3P, 50,00% раствор в EtOAc, 0,945 мл, 1,588 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение трех часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, и затем органический слой экстрагируют дихлорметаном, фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора из него, и концентрируют при пониженном давлении. Полученный продукт используют без дополнительного процесса очистки (указанное в заголовке соединение, 0,250 г, 97,3%, светло-желтое масло).
[Стадия 2] Синтез (S)-(3-(4-метилпиперазин-1-ил)фенил)(4-пропилпиперазин-1-ил)метанона гидрохлорида

Трет-бутил (S)-2-(4-(3-(4-метилпиперазин-1-ил)бензоил)пиперазин-1-карбонил)пирролидин-1-карбоксилат (0,250 г, 0,515 ммоль), полученный на стадии 1, и хлороводород (4,00 M раствор в 1,4-диоксане, 0,515 мл, 2,059 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при 40°C в течение двух часов до завершения реакции путем понижения температуры до комнатной температуры. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный продукт используют без дополнительного процесса очистки (указанное в заголовке соединение, 0,210 г, 96,7%, светло-желтое твердое вещество).
[Стадия 3] Синтез (4-((7-амино-2-(фуран-2-ил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-5-ил)-L-пропил)пиперазин-1-ил)(3-(4-метилпиперазин-1-ил)фенил)метанона
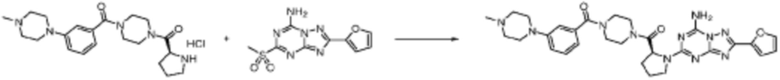
(S)-(3-(4-Метилпиперазин-1-ил)фенил)(4-пропилпиперазин-1-ил)метанон гидрохлорид (0,210 г, 0,498 ммоль), полученный на стадии 2, 2-(фуран-2-ил)-5-(метилсульфонил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амин (0,126 г, 0,448 ммоль) и гидрокарбонат натрия (0,125 г, 1,493 ммоль) растворяют в циклопентилметиловом эфире (ЦПМЭ, 3 мл) при комнатной температуре, после чего полученный раствор перемешивают при 70°C в течение 18 часов до завершения реакции путем понижения температуры до комнатной температуры. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан= 0-5%) и концентрируют с получением указанного в заголовке соединения (0,042 г, 14,5%) в виде белого твердого вещества.
1H ЯМР (400 MГц, ДМСО-d6) δ 8,60-8,09 (м, 2H), 7,90-7,85 (м, 1H), 7,36-7,23 (м, 1H), 7,10-7,01 (м, 2H), 6,99-6,89 (м, 1H), 6,82 (с, 1H), 6,73-6,65 (м, 1H), 5,00 (с, 1H), 4,04-3,48 (м, 10H), 3,26-2,93 (м, 6H), 2,38-2,14 (м, 4H), 2,03-1,77 (м, 3H), 1,17 (т, J=7,3 Гц, 2H); МСНР (ЭР) m/z 586,6 (M++ 1).
Примеры 83, 102, 103, 104, 105 и 106
Примеры соединений 83, 102, 103, 104, 105 и 106 каждый синтезируют по существу тем же способом синтеза, как и способ синтеза из Примера соединения 82 за исключением применения исходных материалов ниже вместо трет-бутил (S)-2-(пиперазин-1-карбонил)пирролидин-1-карбоксилата в способе синтеза Примера соединения 82.
[Таблица 56]
№
№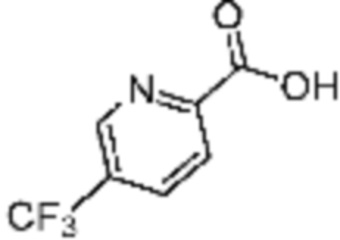
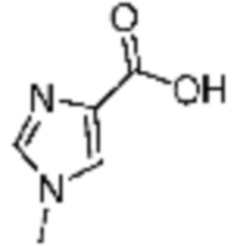
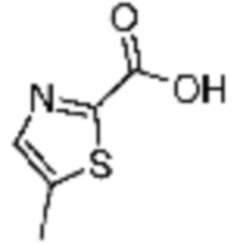
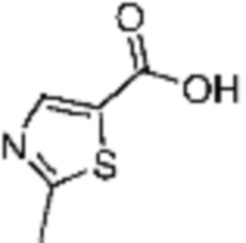
Аналитические данные каждого из соединений, полученных как описано выше, показаны в таблице ниже.
[Таблица 57]
№
Пример 53: Синтез соединения 53, 4-(2-(4-((7-амино-2-(фуран-2-ил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-5-ил)-L-пропил)пиперазин-1-ил)этил)морфолина
[Стадия 1] Синтез (7-амино-2-(фуран-2-ил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-5-ил)-L-пролина
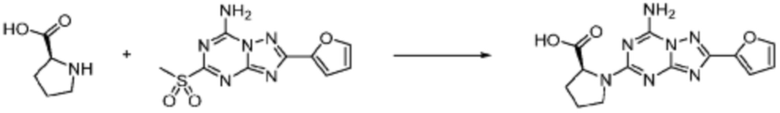
L-Пролин (1,151 г, 10,000 ммоль), 2-(фуран-2-ил)-5-(метилсульфонил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-7-амин (2,803 г, 10,000 ммоль) и триэтиламин (2,788 мл, 20,000 ммоль) растворяют в N, N-диметилформамиде (20 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Полученный продукт используют без дополнительного процесса очистки (указанное в заголовке соединение, 3,151 г, 99,9%, светло-коричневое масло).
[Стадия 2] Синтез 4-(2-(4-((7-амино-2-(фуран-2-ил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-5-ил)-L-пропил)пиперазин-1-ил)этил)морфолина
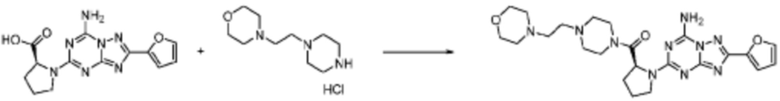
(7-Амино-2-(фуран-2-ил)-[1,2,4]триазоло[1,5-a][1,3,5]триазин-5-ил)-L-пролин (0,315 г, 1,000 ммоль), полученный на стадии 1, 4-(2-(пиперазин-1-ил)этил)морфолин гидрохлорид (0,354 г, 1,500 ммоль), 2,4,6-трипропил-1,3,5,2,4,6-триоксатрифосфинан 2,4,6-триоксид (T3P, 50,00% раствор в ДМФ, 1,908 мл, 3,000 ммоль) и триэтиламин (0,279 мл, 2,000 ммоль) растворяют в N, N-диметилформамиде (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Воду выливают в реакционную смесь, и затем органический слой экстрагируют дихлорметаном, фильтруют через пластиковый фильтр для удаления твердого остатка и слоя водного раствора из него, и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением указанного в заголовке соединения (0,010 г, 2,0%) в виде белого твердого вещества.
1H ЯМР (400 MГц, ДМСО-d6) δ 8,61-8,00 (м, 2H), 7,92-7,80 (м, 1H), 7,11-6,98 (м, 1H), 6,74-6,60 (м, 1H), 5,09-4,90 (м, 1H), 3,76-3,46 (м, 10H), 2,79-2,63 (м, 1H), 2,49-2,36 (м, 9H), 2,35-2,13 (м, 3H), 1,99-1,79 (м, 3H); МСНР (ЭР) m/z 497,5 (M++ 1).
Пример 54: Синтез соединения 54
Пример соединения 54 синтезируют по существу тем же способом синтеза, что и способ синтеза Примера соединения 53 за исключением применения 1-циклогексилпиперазина вместо 4-(2-(пиперазин-1-ил)этил)морфолина гидрохлорида.
1H ЯМР (400 MГц, ДМСО-d6) δ 8,64-8,01 (м, 2H), 7,92-7,82 (м, 1H), 7,11-6,98 (м, 1H), 6,78-6,60 (м, 1H), 5,08-4,91 (м, 1H), 3,77-3,44 (м, 5H), 3,30-3,08 (м, 1H), 2,84-2,64 (м, 1H), 2,45-2,17 (м, 4H), 2,04-1,66 (м, 7H), 1,59 (д, J=12,4 Гц, 1H), 1,35-1,00 (м, 6H); МСНР (ЭР) m/z 466,5 (M++ 1).
Протокол измерения и анализа активности соединения по настоящему изобретению
Экспериментальный Пример 1. Оценка аффинности связывания рецептора A2a
Аффинность связывания соединений из Примеров настоящего изобретения с аденозиновым рецептором А2а человека оценивают путем поручения поиска новых лекарственных средств SB в Великобритании.
Тест на связывание радиолиганда проводят с использованием [3H]-NECA (5'-N-[аденин-2,8-3H]-этилкарбоксамидоаденозина) и аденозиновой мембраны A2a. В качестве аденозиновой мембраны А2а используют клеточную мембрану, полученную из клеток HEK-293, трансфицированных аденозиновым рецептором А2а человека. Мембрану, используемую для тестирования, получают путем инкубации с радиолигандом до достижения равновесия. Для отделения мембраны, связанной с радиолигандом, несвязанный радиолиганд отделяют с помощью фильтрующего материала Packard Harverster и стеклянных фильтрующих пластин.
10 мкл тестируемого соединения, растворенного в буфере для связывания (50 мМ Tris, 10 мМ MgCl2, 1 мМ ЭДТК, pH 7,4) и 20 мкл [3H]-NECA (конечная концентрация 37 нМ) или эталонного ингибитора, смешивают с 20 мкл мембраны A2a в несвязанном 96-луночном планшете и инкубируют при комнатной температуре в течение одного часа. Перед фильтрованием, 96-луночный планшет для сбора покрывают 0,33% полиэтиленимином на 30 минут, и затем промывают аналитическим буфером. Реакцию связывания переносят на фильтровальную пластинку и трижды промывают промывочным буфером. Затем чашку сушат, добавляют сцинтиллятор и подсчитывают радиоактивность на сцинтилляционном счетчике (Topcount NXT, Packard).
Аффинность связывания IC50 (мкМ) с аденозиновым рецептором А2а человека, полученная в соответствии с описанным выше экспериментальным способом, показана в таблице ниже.
[Таблица 58]
Из приведенных выше результатов можно подтвердить, что соединения по типовому варианту осуществления настоящего изобретения обладают очень хорошей аффинностью связывания с аденозиновым рецептором А2а человека.
Экспериментальный пример 2. Тест на кинетическую растворимость
Растворимость соединения настоящего изобретения измеряют для оценки физических свойств. Реагенты и планшеты, использованные в эксперименте, представлены в таблице ниже.
(Фосфатно-солевой буфер, PBS)
(без пепсина, pH 1,0-1,4)
- Ricca Chemical Company 7108-16_500 мл
(кишечный сок, имитирующий состояние натощак (FASSIF), pH 6,6-7,0))
- FaSSIF буферный концентрат 215 г_ Biorelevant_ FASBUF01
- Микропланшет, 96-лунок, ПС, с F-образным кристально-прозрачным дном_greiner bio-one_655101
Каждое из соединений по типовому варианту осуществления настоящего изобретения растворяют в диметилсульфоксиде для приготовления растворов с различными концентрациями (500, 370, 250, 125, 62,5, 31,25, 15,62, 7,81, 3,90, 1,95 мкМ). После отбора 10 мкл раствора и смешивания со 190 мкл приготовленного раствора (буферный раствор, искусственный желудочный сок или искусственный кишечный сок) через час измеряют количество выпавшего осадка при каждой концентрации с помощью нефелометра (NEPHELOstar (BMG LABTECH)) для оценки растворимости.
Аппарат представляет собой прибор для измерения растворимости методом нефелометрии. После растворения соединения в различных концентрациях в желаемом растворителе, когда через раствор пропускают лазер, растворимость можно измерить на основании размера канала рассеяния нерастворимых частиц.
В результате подтверждено, что соединения по настоящему изобретению демонстрируют превосходную растворимость в нейтральных условиях и в условиях желудочного и кишечного сока в пищеварительном тракте, соответственно.
Хотя настоящее изобретение было подробно описано выше, специалистам в данной области техники очевидно, что такие подробные описания приведены только для иллюстрации типовых вариантов осуществления, но не ограничивают объем настоящего изобретения. Таким образом, следует понимать, что по существу объем настоящего изобретения определяется прилагаемой формулой изобретения и ее эквивалентами.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРА ГИСТОНДЕАЦЕТИЛАЗЫ 6 И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ИХ | 2021 |
|
RU2817736C1 |
| СОЕДИНЕНИЯ, ЯВЛЯЮЩИЕСЯ АНТАГОНИСТАМИ А3-АДЕНОЗИНОВОГО РЕЦЕПТОРА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ МЕДИЦИНСКОЕ ПРИМЕНЕНИЕ | 2017 |
|
RU2737157C2 |
| ИНГИБИТОРНЫЕ СОЕДИНЕНИЯ | 2013 |
|
RU2673079C2 |
| ИНГИБИТОРЫ МУТАЦИИ HER2 | 2022 |
|
RU2834124C2 |
| ПИРАЗОЛЬНЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ МОДУЛЯТОРОВ FSHR И ИХ ПРИМЕНЕНИЕ | 2014 |
|
RU2663898C2 |
| ЦИКЛОАЛКИЛНИТРИЛПИРАЗОЛОПИРИДОНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ЯНУС-КИНАЗЫ | 2014 |
|
RU2655380C2 |
| Соединения формул (I) и (A), фармацевтическая композиция, лекарственное средство, применение и способ получения соединения формулы (I) | 2018 |
|
RU2822758C2 |
| БИЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ И ЕГО ПРИМЕНЕНИЕ | 2020 |
|
RU2831535C1 |
| ПРОИЗВОДНЫЕ ИМИДАЗО[1,2-A]ПИРИДИНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ | 2017 |
|
RU2725147C1 |
| СПОСОБ ПОЛУЧЕНИЯ 3-ГЕТАРИЛ-7-АМИНОКУМАРИНОВ | 2022 |
|
RU2802966C1 |
Изобретение относится к соединению, представленному формулой 1
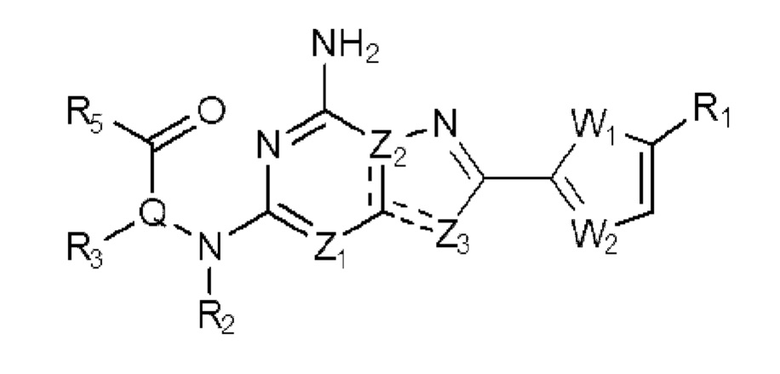 ,
,
где если W1 представляет собой О, то W2 представляет собой СН; если W1 представляет собой S, то W2 представляет собой N; Z1 представляет собой СН или N; Z2 представляет собой С или N; Z3 представляет собой N, О или S;  и
и  - каждый независимо представляет одинарную связь или двойную связь (если
- каждый независимо представляет одинарную связь или двойную связь (если  представляет собой двойную связь,
представляет собой двойную связь,  представляет собой одинарную связь и, если
представляет собой одинарную связь и, если  представляет собой одинарную связь,
представляет собой одинарную связь,  представляет собой двойную связь); Q представляет собой C-R4 или N; R1 представляет собой Н или -СН3; R2 представляет собой Н, R3 представляет собой Н или -La-Ra или R2 и R3 связаны с образованием кольца, где La представляет собой одинарную связь или С1-С3 алкилен, Ra представляет собой С1-С5 алкил, С3-С6 циклоалкил,
представляет собой двойную связь); Q представляет собой C-R4 или N; R1 представляет собой Н или -СН3; R2 представляет собой Н, R3 представляет собой Н или -La-Ra или R2 и R3 связаны с образованием кольца, где La представляет собой одинарную связь или С1-С3 алкилен, Ra представляет собой С1-С5 алкил, С3-С6 циклоалкил, 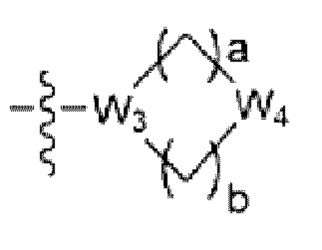 (а и b - каждый независимо равен 2, W3 представляет собой СН или N, W4 представляет собой СН2 или О, где если W3 представляет собой СН, то W4 не является СН2) или фенил, и если Ra представляет собой С1-С5 алкил или фенил, то по меньшей мере один из Н может быть замещен -ОН или С1-С5 алкокси; кольцо, образованное связыванием R2 и R3, представляет собой 4-6-членный N-содержащий гетероциклоалкил (где по меньшей мере каждый Н из N-содержащего гетероциклоалкила может быть независимо замещен С1-С5 алкилом или ОН) или 7-членный N-содержащий спирогетероциклоалкил; R4 представляет собой Н или С1-С5 алкил; R5 представляет собой
(а и b - каждый независимо равен 2, W3 представляет собой СН или N, W4 представляет собой СН2 или О, где если W3 представляет собой СН, то W4 не является СН2) или фенил, и если Ra представляет собой С1-С5 алкил или фенил, то по меньшей мере один из Н может быть замещен -ОН или С1-С5 алкокси; кольцо, образованное связыванием R2 и R3, представляет собой 4-6-членный N-содержащий гетероциклоалкил (где по меньшей мере каждый Н из N-содержащего гетероциклоалкила может быть независимо замещен С1-С5 алкилом или ОН) или 7-членный N-содержащий спирогетероциклоалкил; R4 представляет собой Н или С1-С5 алкил; R5 представляет собой
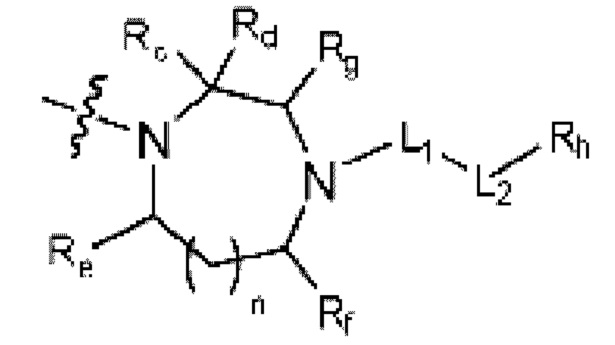
(где n равен 0 или 1 и Rc, Rd, Re, Rf и Rg - каждый независимо представляет собой Н или С1-С5 алкил, но два, выбранных из Rc, Rd, Re, Rf и Rg, могут быть связаны с образованием СН2 или СН2-СН2);
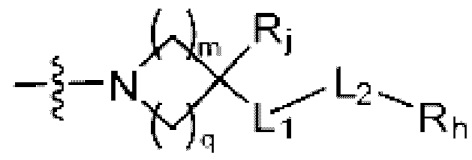
(где m и q - каждый независимо равен 1 или 2 и R3 представляет собой Н или галоген);
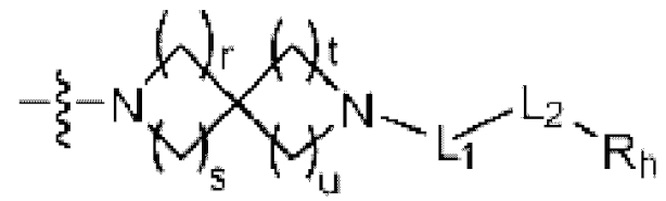
(где r, s, t и u - каждый независимо равен 1 или 2);
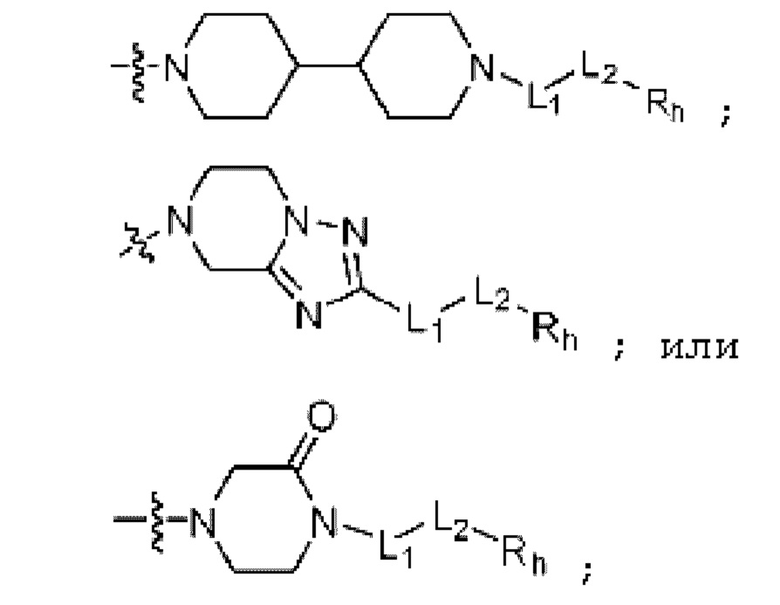
в вышеуказанном R5 L1 представляет собой одинарную связь или C1-С3 алкилен; L2 представляет собой одинарную связь, -С(=O)-, -С(=O)NH-, -С(=O)-N(С1-С5 алкил)-, -С(=O)-NH(С1-С5 алкилен)-, -S(=O)2- или -S(=O)2-(C1-С3 алкилен)-; Rh представляет собой Н, С1-С5 алкил, С1-С5 алкокси, С1-С5 галогеналкил, галоген, С3-С6 циклоалкил, фенокси, фенил, -(С1-С5 алкилен)-фенил, -фенилен-O-(С1-С5 алкил), -фенилен-С(=O), фенилен-пиперазинил, 4-6-членный гетероциклоалкил, включающий 1-3 гетероатома из по меньшей мере одного, выбранного из N, О и S, 5-10-членный гетероарил, включающий 1-3 гетероатома из по меньшей мере одного, выбранного из N, О и S,
 ,
,
или -NR6R7; R6 и R7 - каждый независимо представляет собой С1-С5 алкил или С1-С5 галогеналкил; и по меньшей мере один Н из Rh каждый может быть независимо замещен С1-С5 алкилом, С1-С5 алкокси, С1-С5 галогеналкилом, ОН или галогеном, а также к фармацевтическим композициям и применению соединений в качестве антагониста аденозинового рецептора A2a. Технический результат: получены новые соединения, которые могут быть использованы в качестве антагониста аденозинового рецептора A2a. 4 н. и 8 з.п. ф-лы, 58 табл., 54 пр.
1. Соединение, представленное формулой 1, его стереоизомеры или его фармацевтически приемлемые соли:
[Формула 1]
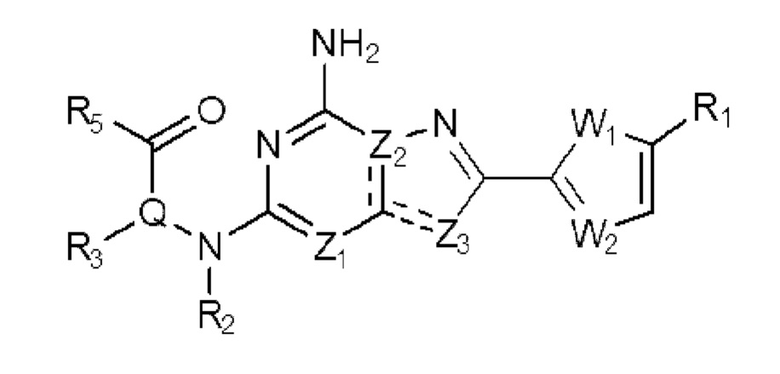 ,
,
где если W1 представляет собой О, то W2 представляет собой СН;
если W1 представляет собой S, то W2 представляет собой N;
Z1 представляет собой СН или N;
Z2 представляет собой С или N;
Z3 представляет собой N, О или S;
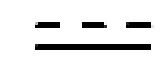 и
и  - каждый независимо представляет одинарную связь или двойную связь (если
- каждый независимо представляет одинарную связь или двойную связь (если  представляет собой двойную связь,
представляет собой двойную связь,  представляет собой одинарную связь и, если
представляет собой одинарную связь и, если  представляет собой одинарную связь,
представляет собой одинарную связь,  представляет собой двойную связь);
представляет собой двойную связь);
Q представляет собой C-R4 или N;
R1 представляет собой Н или -СН3;
R2 представляет собой Н, R3 представляет собой Н или -La-Ra или R2 и R3 связаны с образованием кольца,
где La представляет собой одинарную связь или С1-С3 алкилен, Ra представляет собой С1-С5 алкил, С3-С6 циклоалкил,
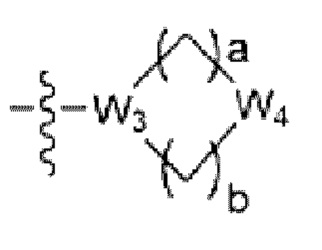
(а и b - каждый независимо равен 2, W3 представляет собой СН или N, W4 представляет собой СН2 или О, где если W3 представляет собой СН, то W4 не является СН2) или фенил, и если Ra представляет собой С1-С5 алкил или фенил, то по меньшей мере один из Н может быть замещен -ОН или С1-С5 алкокси;
кольцо, образованное связыванием R2 и R3, представляет собой 4-6-членный N-содержащий гетероциклоалкил (где по меньшей мере каждый Н из N-содержащего гетероциклоалкила может быть независимо замещен С1-С5 алкилом или ОН) или 7-членный N-содержащий спирогетероциклоалкил;
R4 представляет собой Н или С1-С5 алкил;
R5 представляет собой
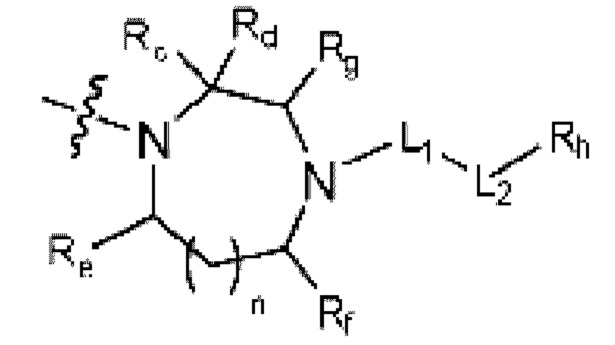
(где n равен 0 или 1 и Rc, Rd, Re, Rf и Rg - каждый независимо представляет собой Н или С1-С5 алкил, но два, выбранных из Rc, Rd, Re, Rf и Rg, могут быть связаны с образованием СН2 или СН2-СН2);
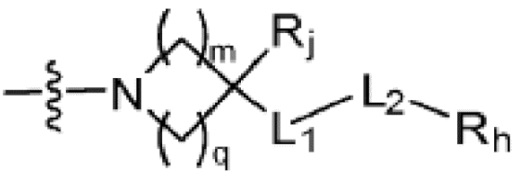
(где m и q - каждый независимо равен 1 или 2 и R3 представляет собой Н или галоген);
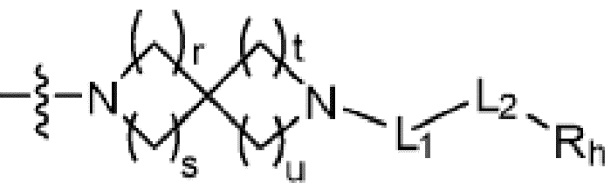
(где r, s, t и u - каждый независимо равен 1 или 2);
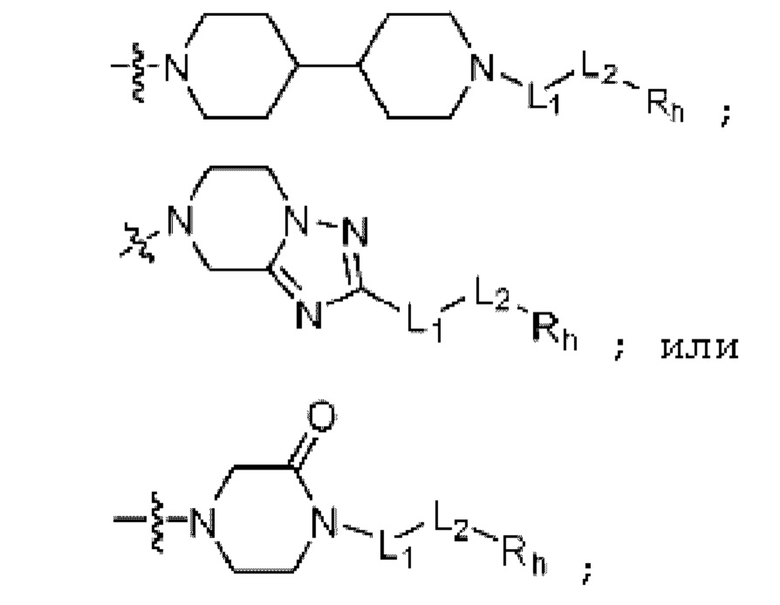
в вышеуказанном R5
L1 представляет собой одинарную связь или C1-С3 алкилен;
L2 представляет собой одинарную связь, -С(=O)-, -С(=O)NH-, - С(=O)-N (С1-С5 алкил)-, -С(=O) -NH(С1-С5 алкилен)-, -S(=O)2- или -S(=O)2-(C1-С3 алкилен)-;
Rh представляет собой Н, С1-С5 алкил, С1-С5 алкокси, С1-С5 галогеналкил, галоген, С3-С6 циклоалкил, фенокси, фенил, -(С1-С5 алкилен)-фенил, -фенилен-O-(С1-С5 алкил), -фенилен-С(=O), фенилен-пиперазинил, 4-6-членный гетероциклоалкил, включающий 1-3 гетероатома из по меньшей мере одного, выбранного из N, О и S, 5-10-членный гетероарил, включающий 1-3 гетероатома из по меньшей мере одного, выбранного из N, О, и S,  ,
,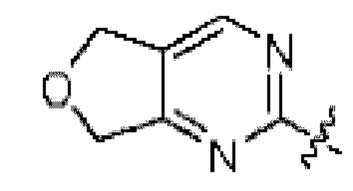 или -NR6R7;
или -NR6R7;
R6 и R7 - каждый независимо представляет собой С1-С5 алкил или С1-С5 галогеналкил; и
по меньшей мере один Н из Rh каждый может быть независимо замещен С1-С5 алкилом, С1-С5 алкокси, С1-С5 галогеналкилом, ОН или галогеном.
2. Соединение, представленное формулой 1, его стереоизомеры или его фармацевтически приемлемые соли по п. 1,
где в формуле 1
W1, W2, Z1, Z2, Z3, Q, R1, R2, R3, R4,  и
и  каждый является таким, как определено в п. 1,
каждый является таким, как определено в п. 1,
R5 представляет собой
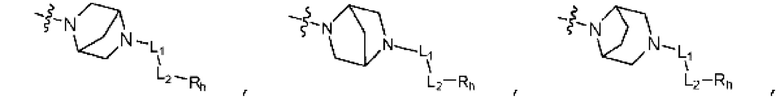
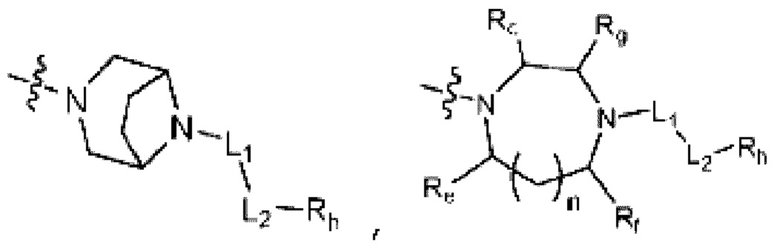
(где n равен 0 или 1, Rc, Re, Rf и Rg - каждый независимо представляет собой Н или С1-С5 алкил) или
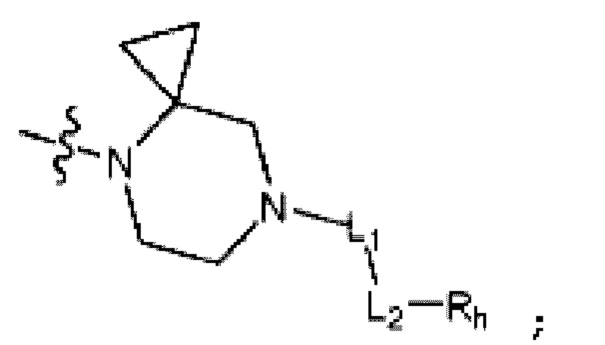
(где m и q - каждый независимо равен 1 или 2 и R3 представляет собой Н или галоген);
(где r, s, t и u - каждый независимо равен 1 или 2);
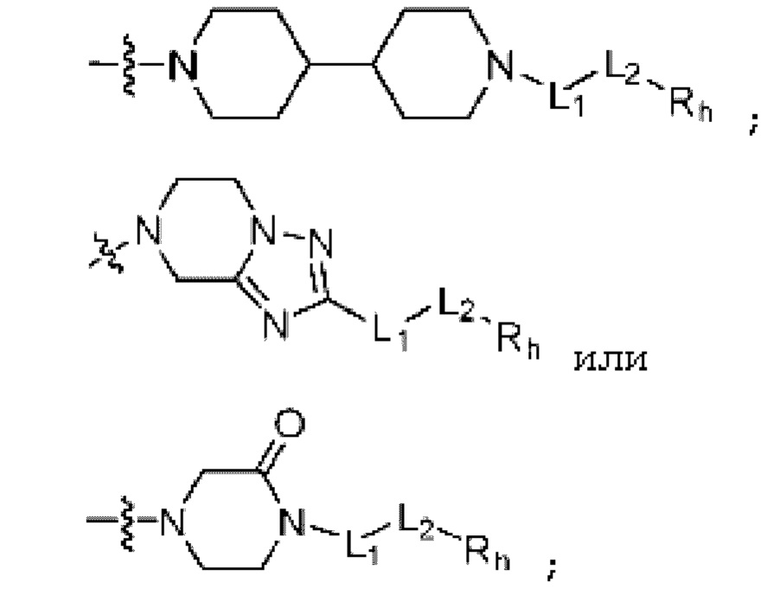
в вышеуказанном R5
L1, L2 и Rh каждый является таким, как определено выше.
3. Соединение, представленное формулой 1, его стереоизомеры или его фармацевтически приемлемые соли по п. 1,
где в формуле 1
W1, W2, Z1, Z2, Z3,  и
и  каждый является таким, как определено в п. 1;
каждый является таким, как определено в п. 1;
Q представляет собой C-R4;
R1 и R2 каждый представляет собой Н;
R3 представляет собой Н или -La-Ra, где La представляет собой одинарную связь или C1-С3 алкилен; Ra представляет собой С1-С5 алкил, С3-С6 циклоалкил, 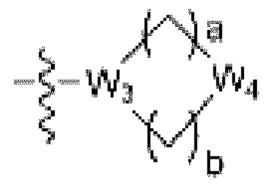 (а и b - каждый независимо равен 2, W3 представляет собой СН или N, W4 представляет собой СН2 или О, где если W3 представляет собой СН, то W4 не является СН2) или фенил, и если Ra представляет собой С1-С5 алкил или фенил, то по меньшей мере один из Н может быть замещен -ОН или С1-С5 алкокси;
(а и b - каждый независимо равен 2, W3 представляет собой СН или N, W4 представляет собой СН2 или О, где если W3 представляет собой СН, то W4 не является СН2) или фенил, и если Ra представляет собой С1-С5 алкил или фенил, то по меньшей мере один из Н может быть замещен -ОН или С1-С5 алкокси;
R4 представляет собой Н или С1-С5 алкил;
R5 представляет собой
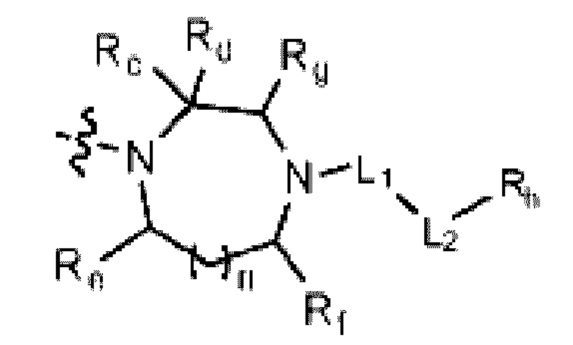
(где n равен 0 или 1 и Rc, Rd, Re, Rf и Rg - каждый независимо представляет собой Н или С1-С5 алкил, но два, выбранных из Rc, Rd, Re, Rf и Rg, могут быть связаны с образованием CH2 или CH2-CH2);
(где m и q - каждый независимо равен 1 или 2 и R3 представляет собой Н или галоген);
(где r, s, t и u - каждый независимо равен 1 или 2);
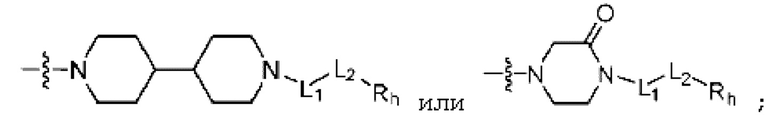
в вышеуказанном R5
L1 представляет собой одинарную связь или C1-С3 алкилен;
L2 представляет собой одинарную связь, -С(=O)- или -S(=O)2-;
Rh представляет собой Н, С1-С5 алкил, С1-С5 алкокси, С1-С5 галогеналкил, С3-С6 циклоалкил, фенокси, фенил, 5- или 6-членный гетероциклоалкил, включающий 1-3 гетероатома из по меньшей мере одного, выбранного из N и О, или 5- или 6-членный гетероарил, включающий 1-3 гетероатома из по меньшей мере одного, выбранного из N и S; и
по меньшей мере один Н из Rh каждый может быть независимо замещен С1-С5 алкокси, С1-С5 галогеналкилом, ОН или галогеном.
4. Соединение, представленное формулой 1, его стереоизомеры или его фармацевтически приемлемые соли по п. 1,
где в формуле 1
W1, W2, Z1, Z2, Z3, R1,  и
и  каждый является таким, как определено в п. 1,
каждый является таким, как определено в п. 1,
Q представляет собой C-R4 или N;
R2 и R3 связаны друг с другом с образованием 4-6-членного N-содержащего гетероциклоалкила (где по меньшей мере каждый Н из N-содержащего гетероциклоалкила может быть независимо замещен С1-С5 алкилом или ОН) или 7-членного N-содержащего спирогетероциклоалкила;
R4 представляет собой Н или С1-С5 алкил;
R5 представляет собой
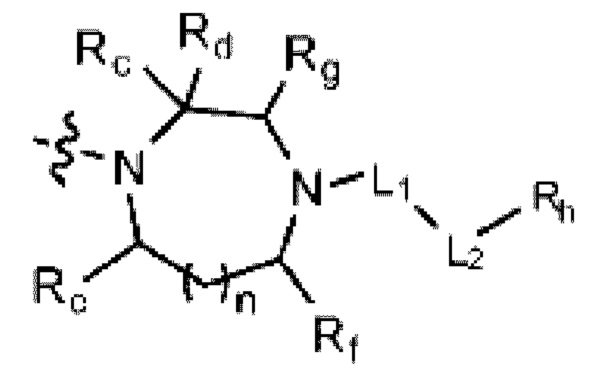
(где n равен 0 или 1 и Rc, Rd, Re, Rf и Rg - каждый независимо представляет собой Н или С1-С5 алкил, но два, выбранных из Rc, Rd, Re, Rf и Rg, могут быть связаны с образованием СН2 или СН2-СН2);
(где m и q - каждый независимо равен 1 ил 2 и R3 представляет собой Н или галоген);
(где r, s, t и u - каждый независимо равен 1 или 2);
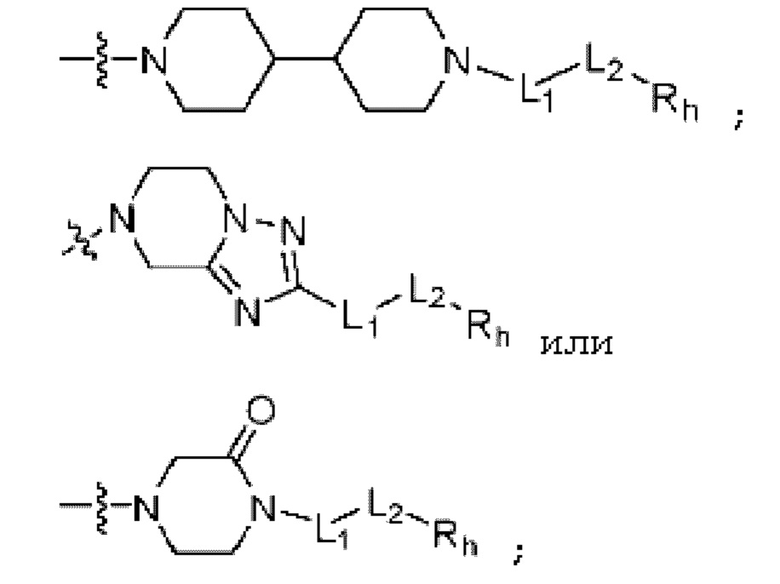
в вышеуказанном R5
L1 представляет собой одинарную связь или C1-С3 алкилен;
L2 представляет собой одинарную связь, -С(=O)-, -С(=O)NH-, -С(=O)-N(С1-С5 алкил)-, -С(=O)-NH(С1-С5 алкилен)-, -S(=O)2- или -S(=O)2-(C1-С3 алкилен)-;
Rh представляет собой Н, С1-С5 алкил, С1-С5 алкокси, С1-С5 галогеналкил, галоген, С3-С6 циклоалкил, фенокси, фенил, -(C1-С3 алкилен)-фенил, -фенилен-O-(С1-С5 алкил), -фенилен-С (=O)-, фенилен-пиперазинил, 4-6-членный гетероциклоалкил, включающий 1-3 гетероатома из по меньшей мере одного, выбранного из N, О и S, 5-10-членный гетероарил, включающий 1-3 гетероатома из по меньшей мере одного, выбранного из N, О, и S,  ,
,  или -NR6R7;
или -NR6R7;
R6 и R7 - каждый независимо представляет собой С1-С5 алкил или С1-С5 галогеналкил; и
по меньшей мере каждый Н из Rh может быть независимо замещен С1-С5 алкилом, С1-С5 алкокси, С1-С5 галогеналкилом, ОН или галогеном.
5. Соединение, его стереоизомеры или его фармацевтически приемлемые соли, где соединение может представлять собой одно соединение, выбранное из соединений ниже:
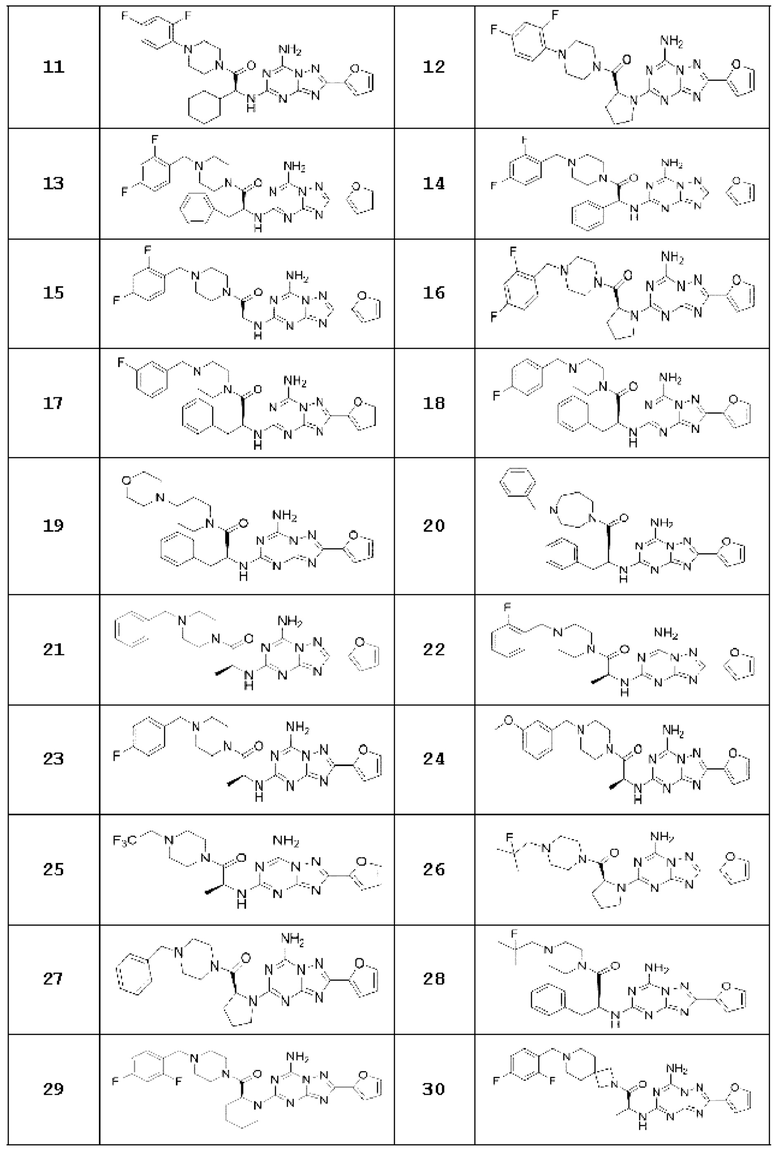
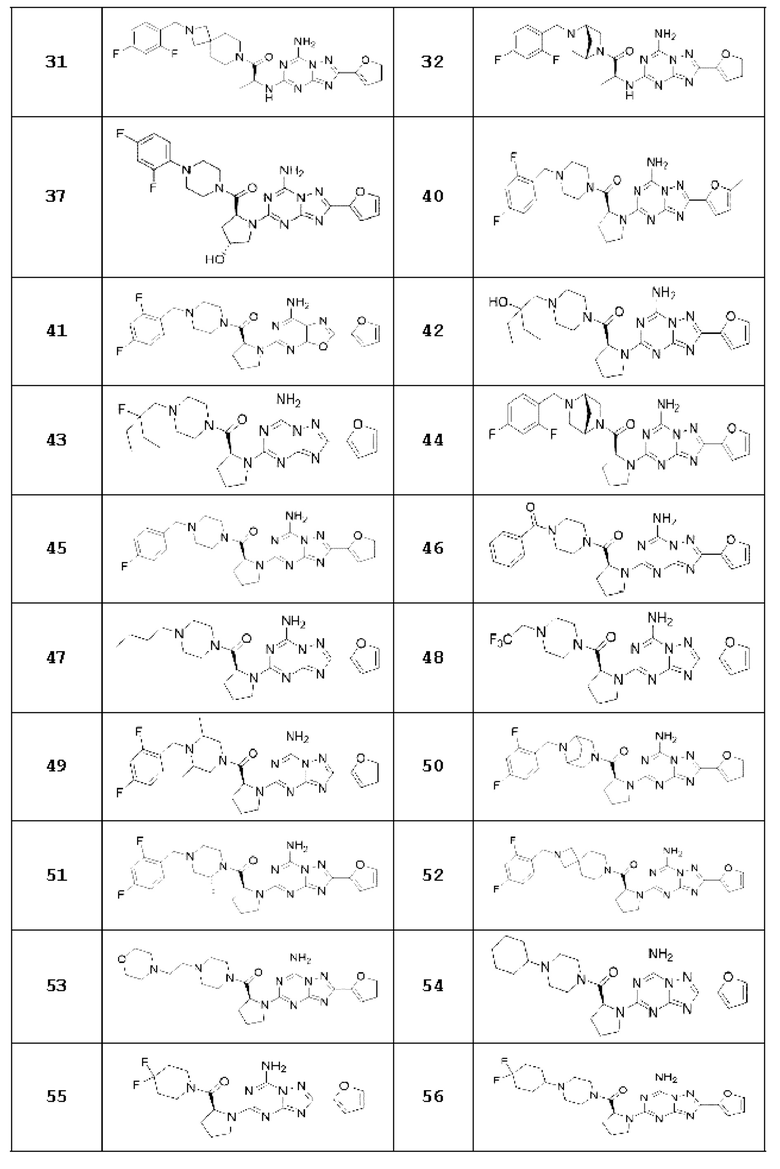
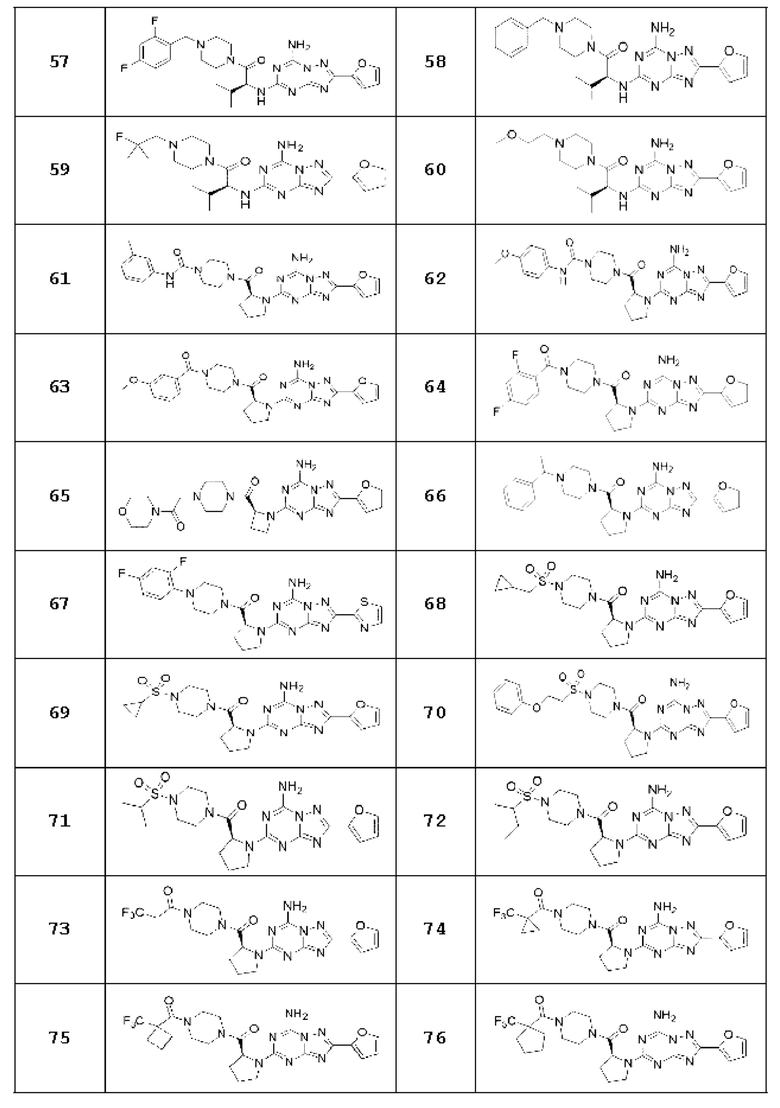
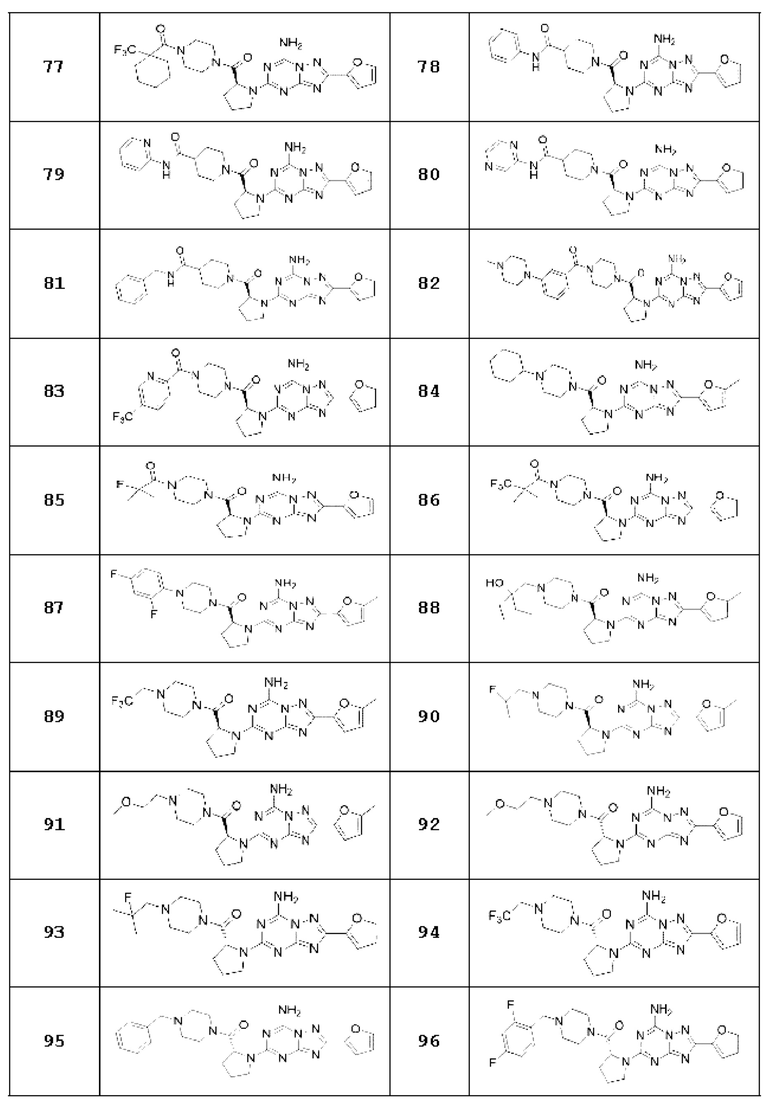
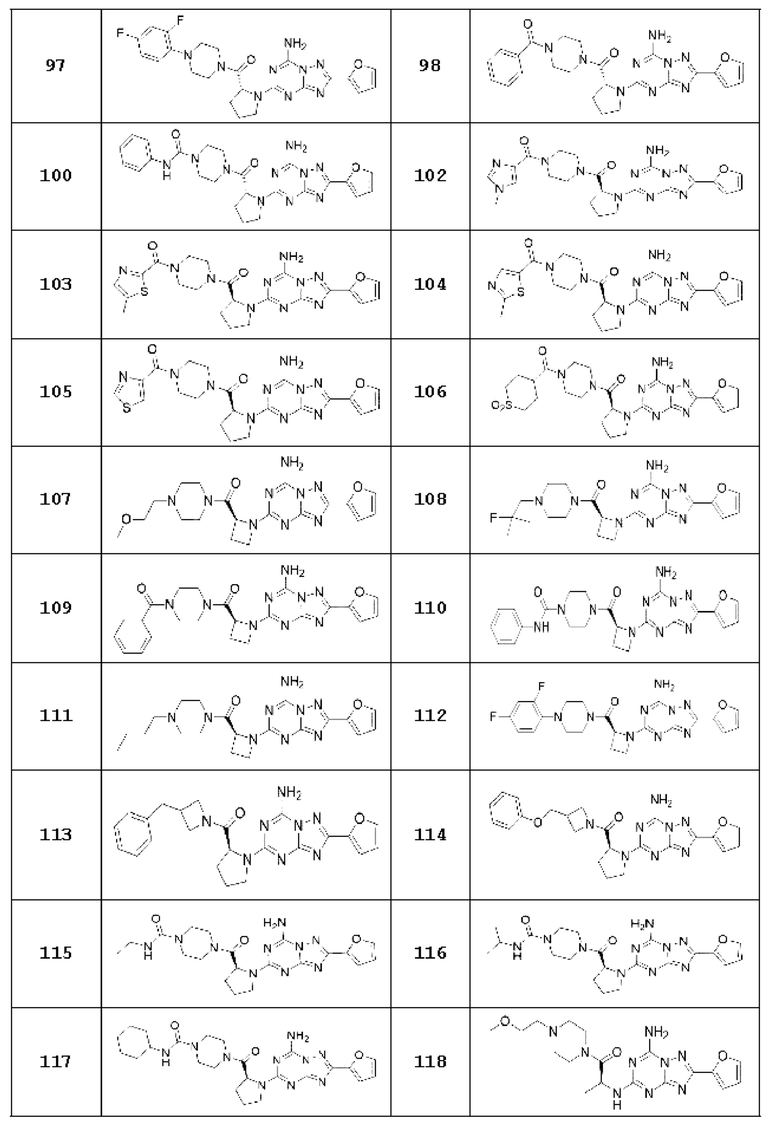
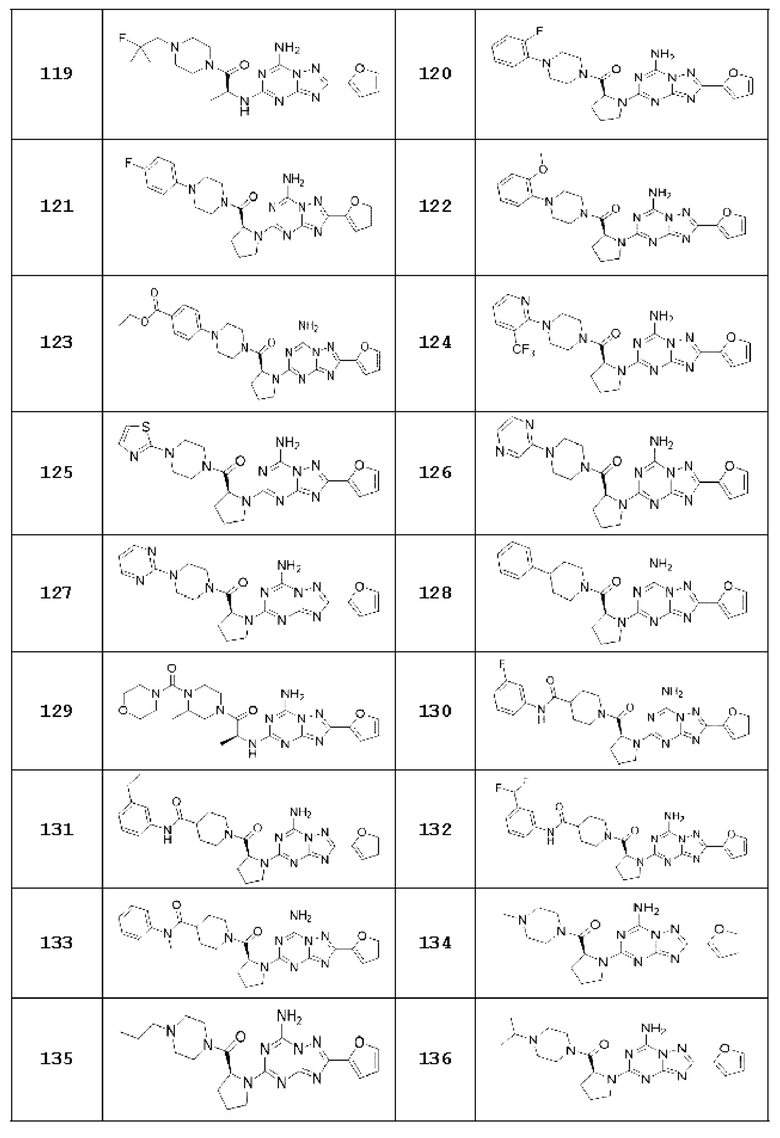
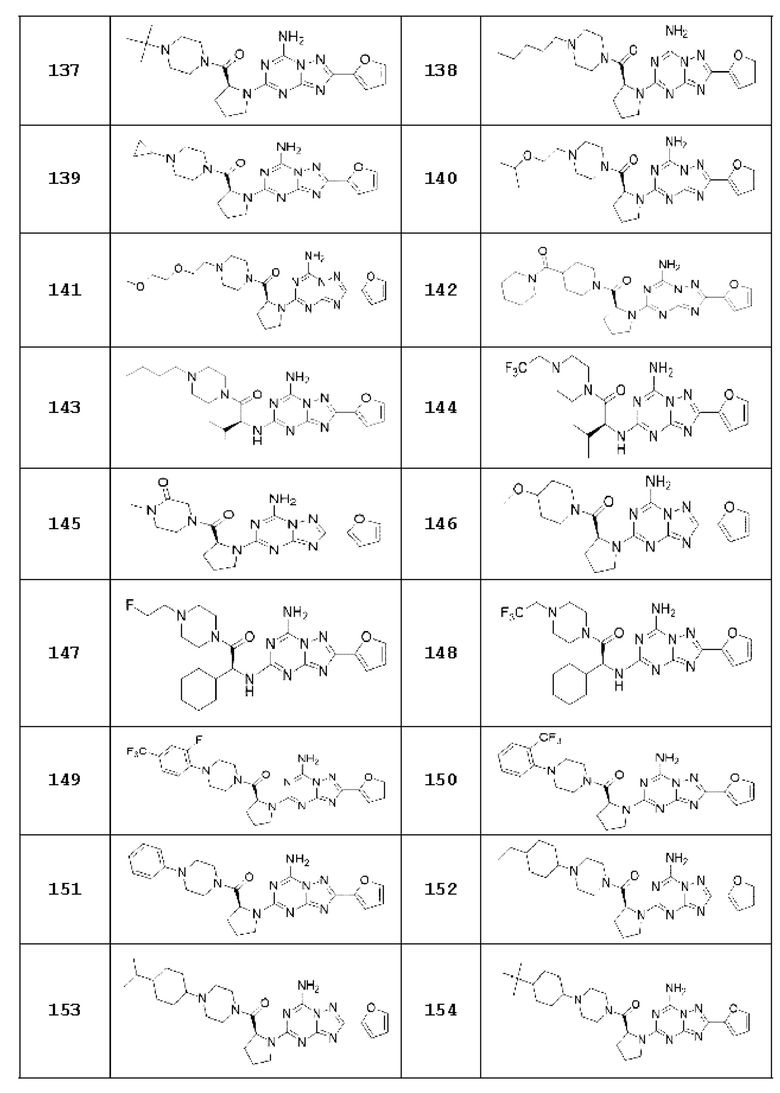
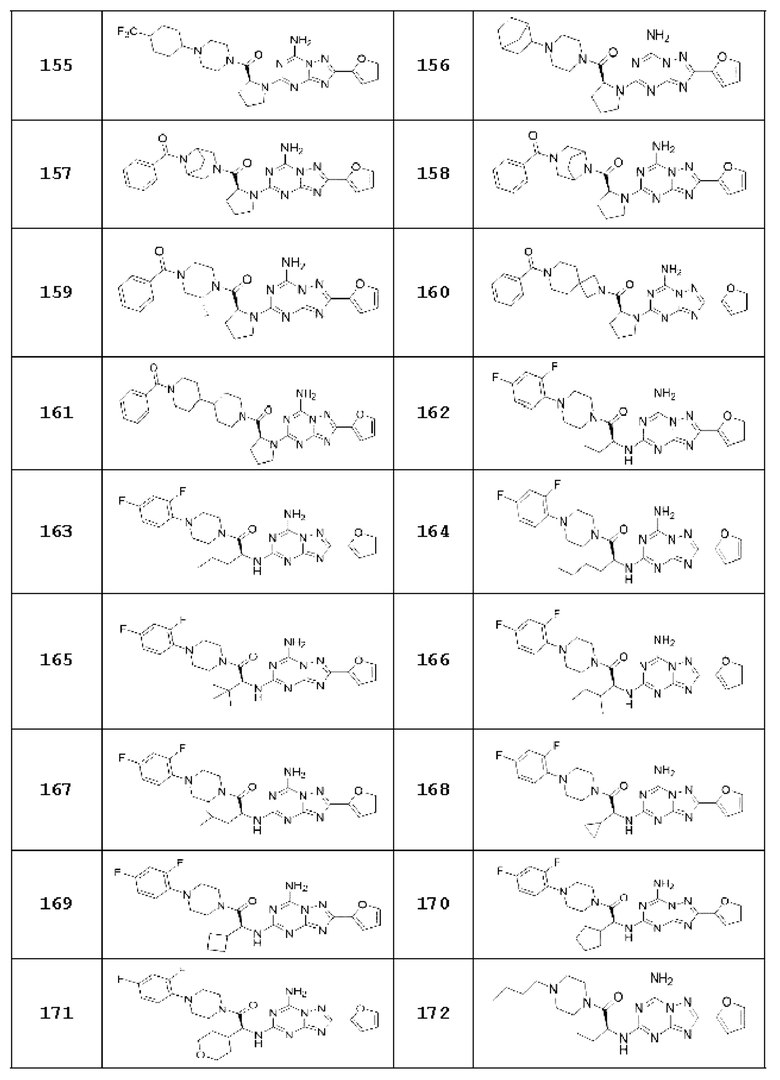
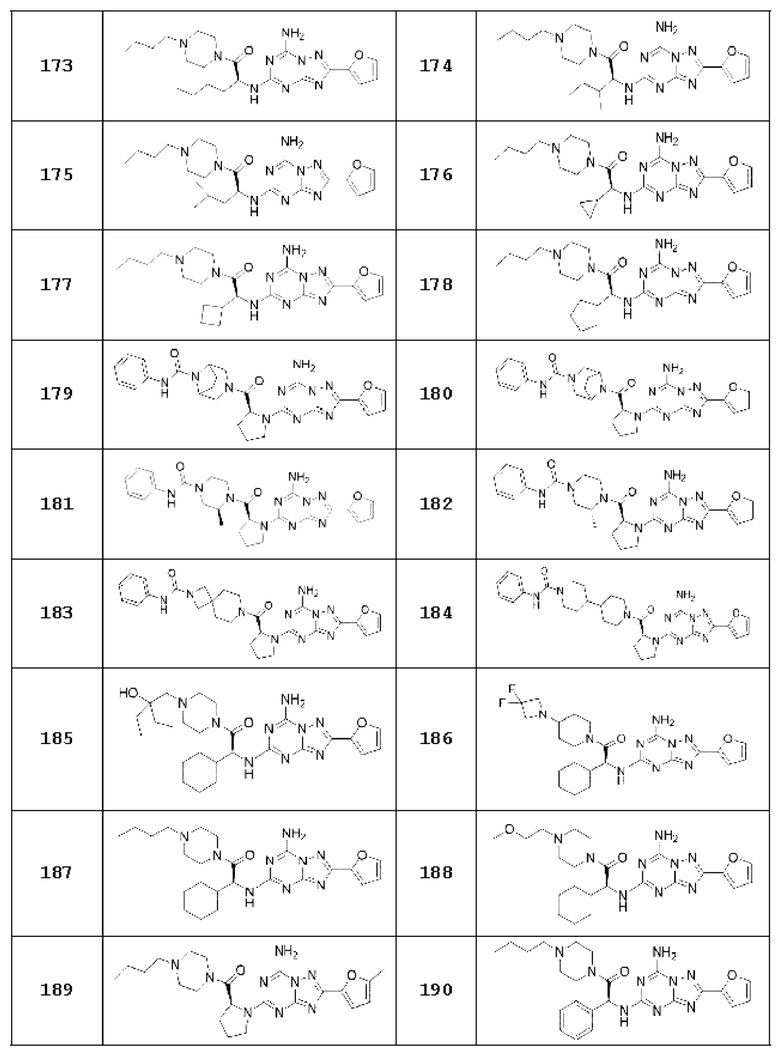
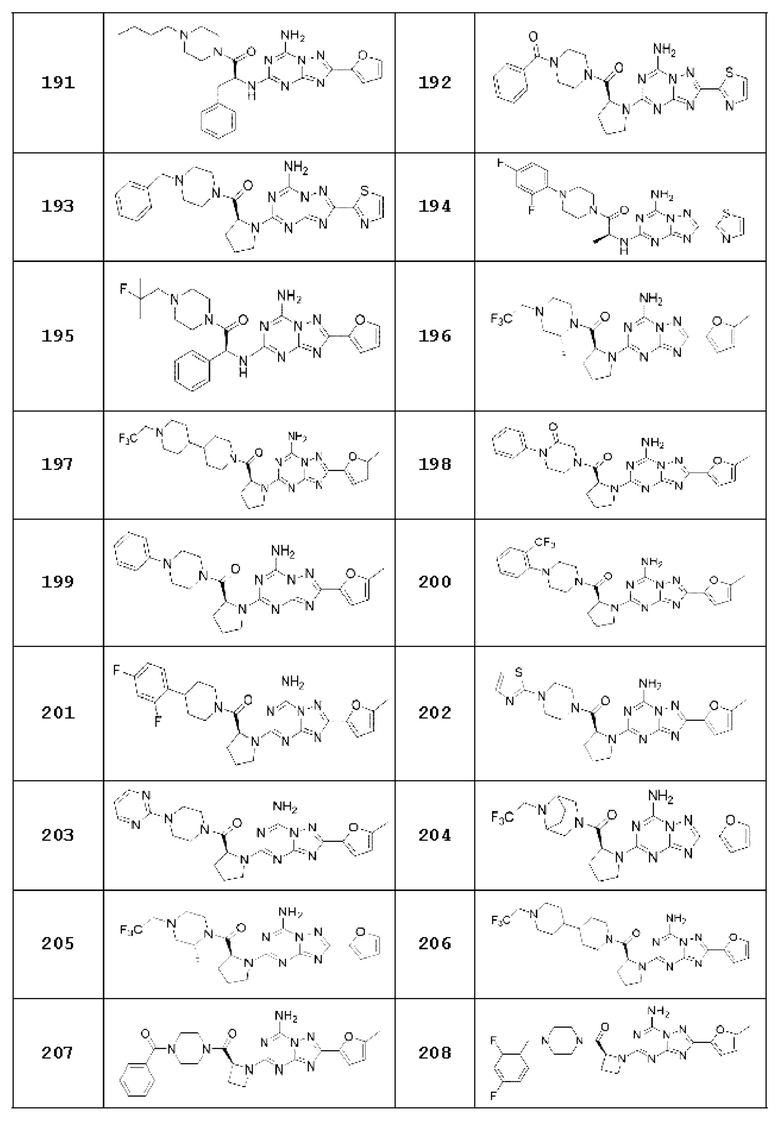
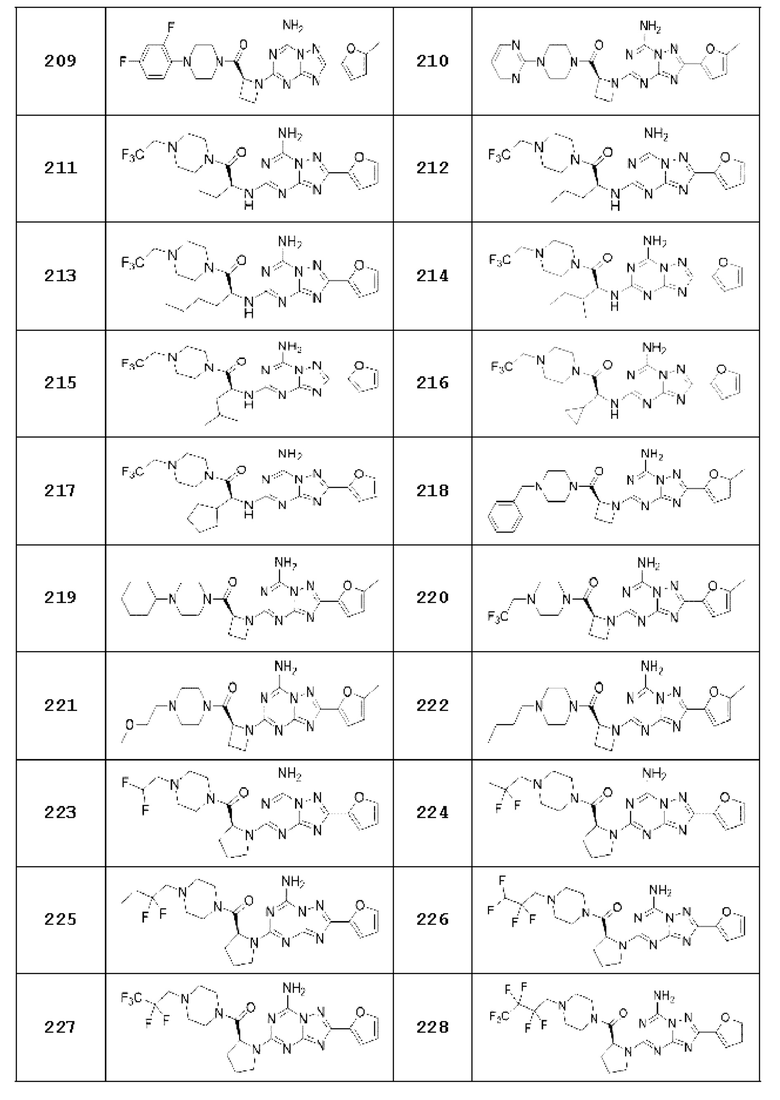
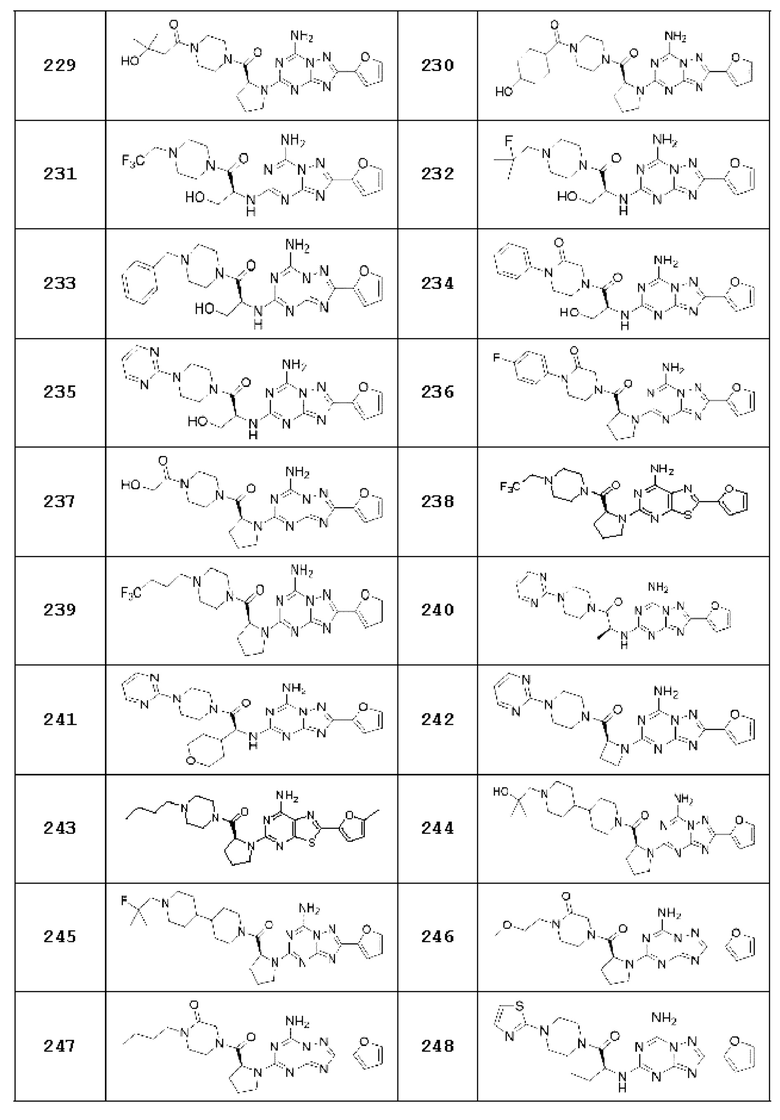
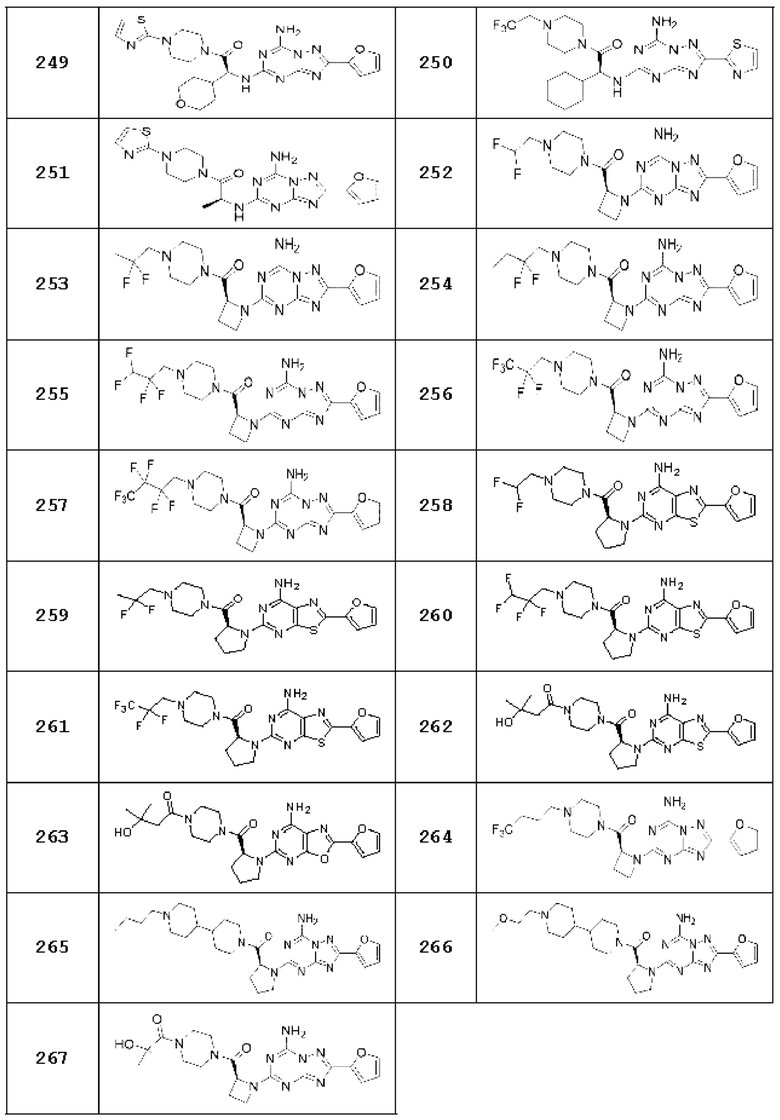
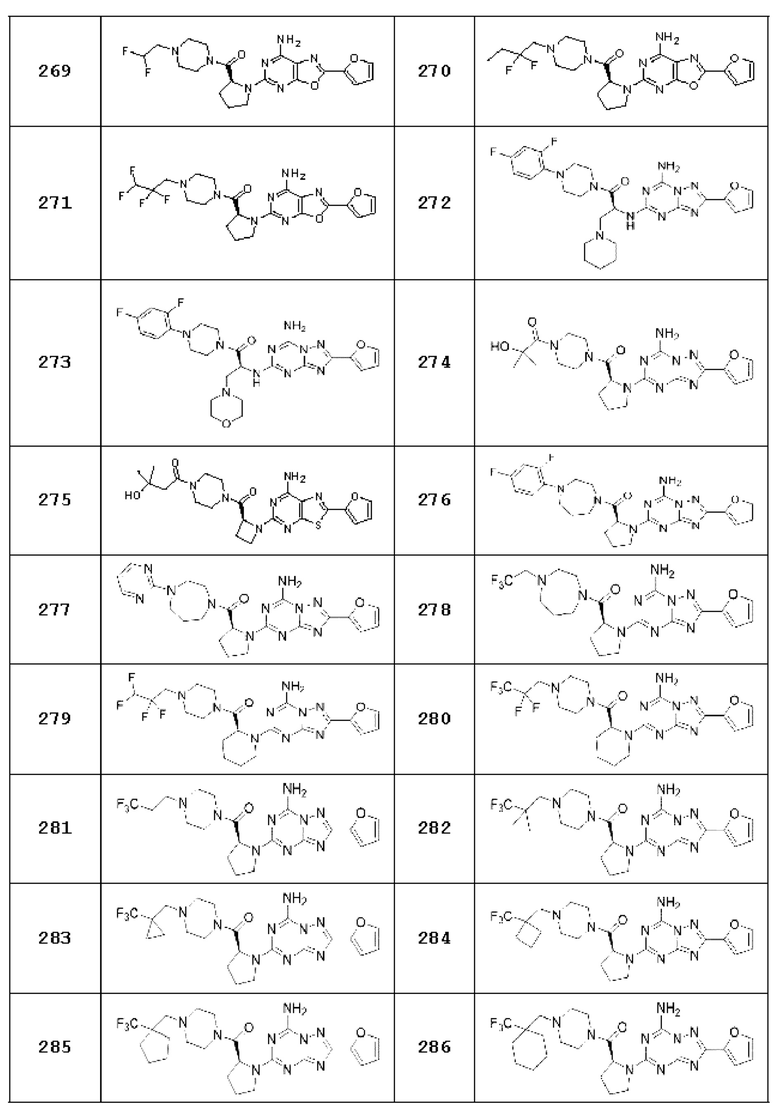
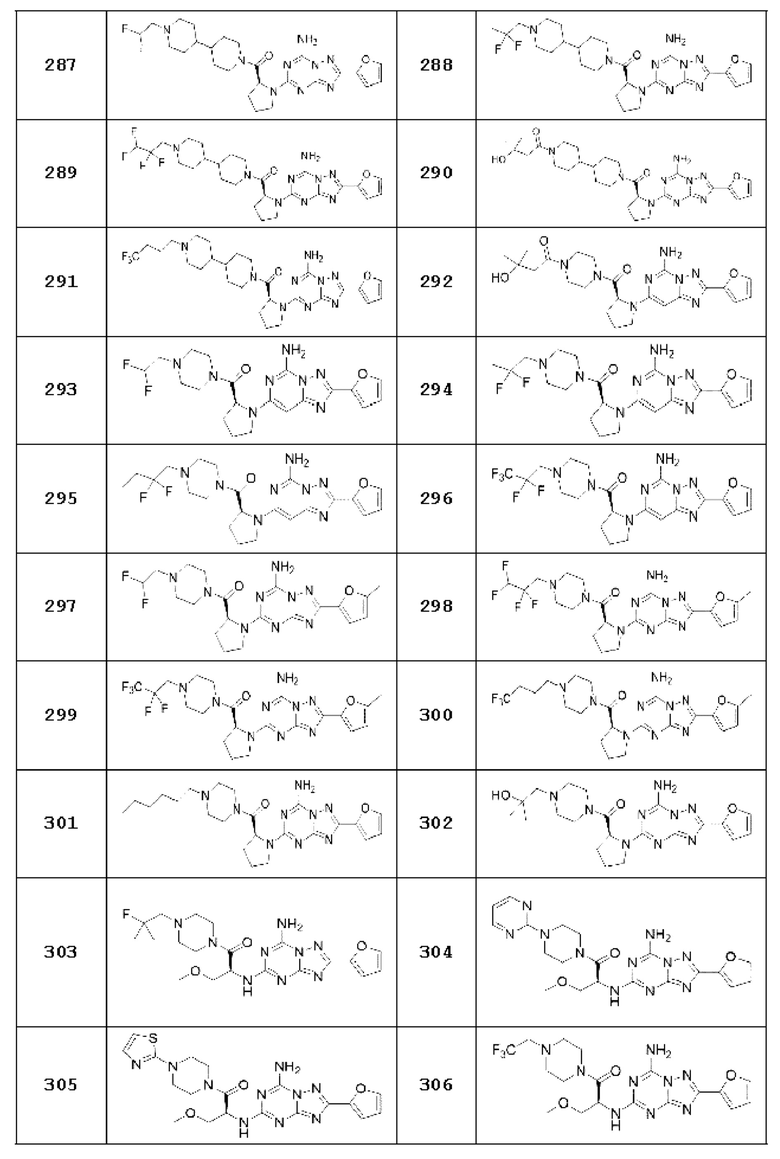
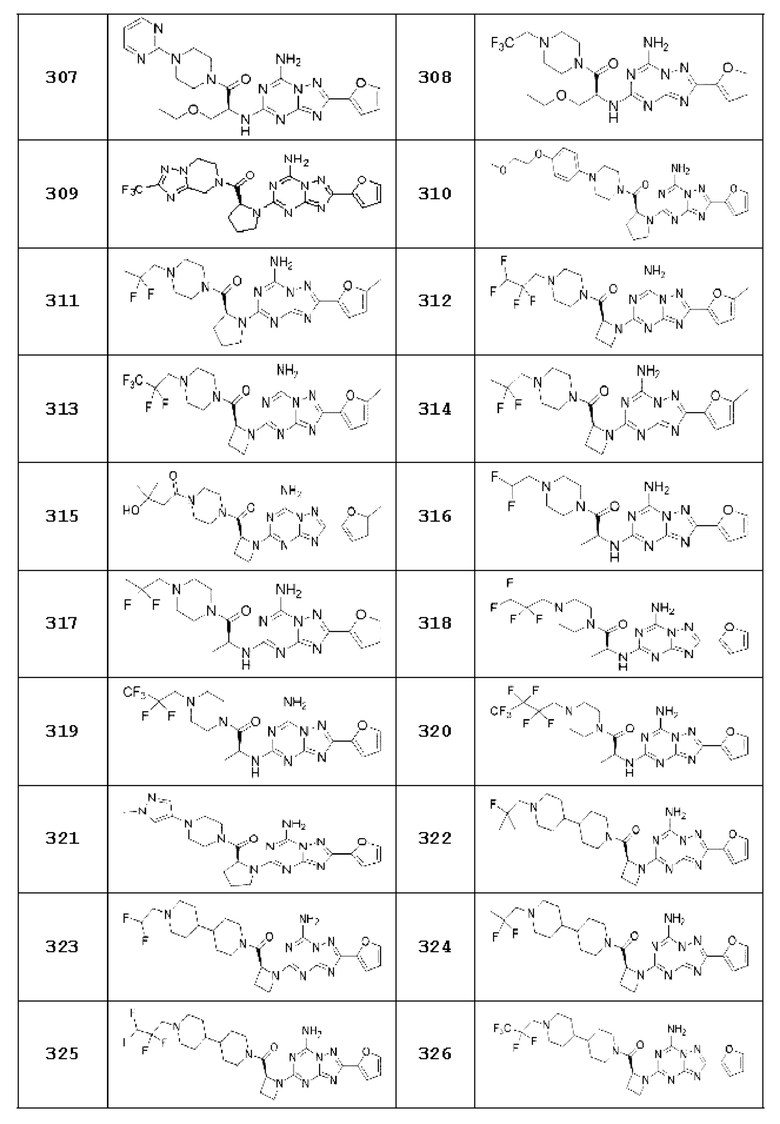
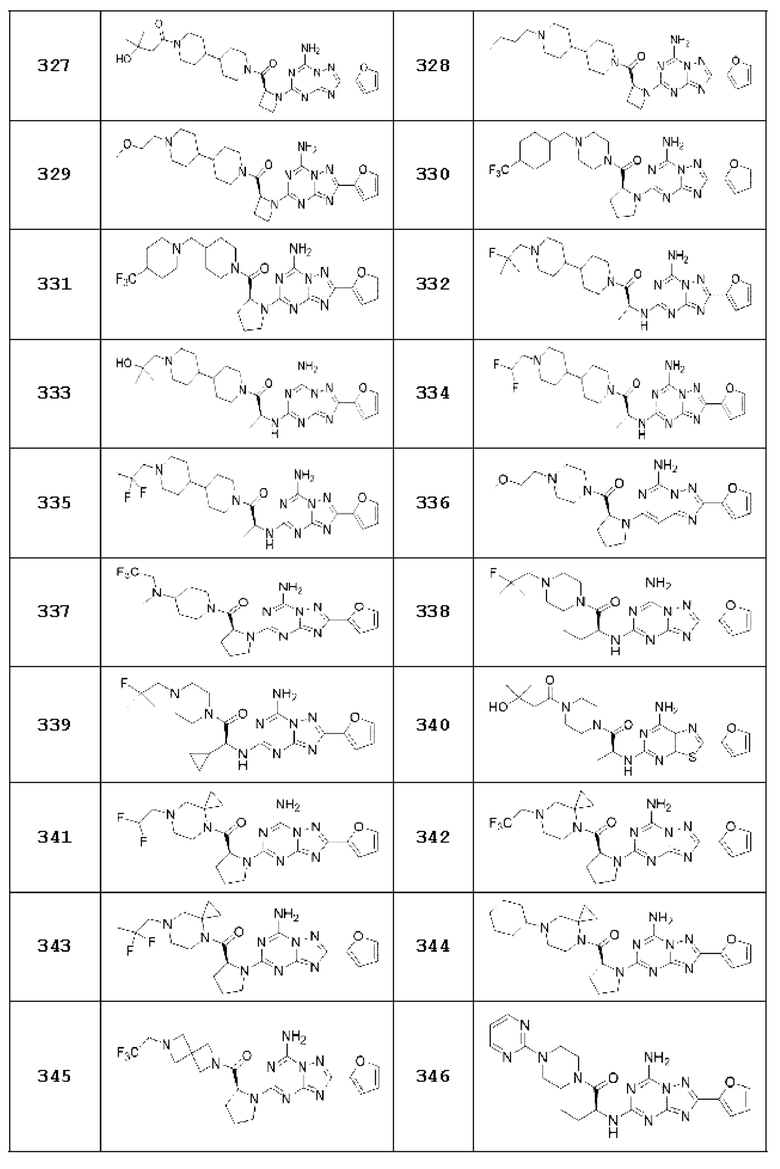
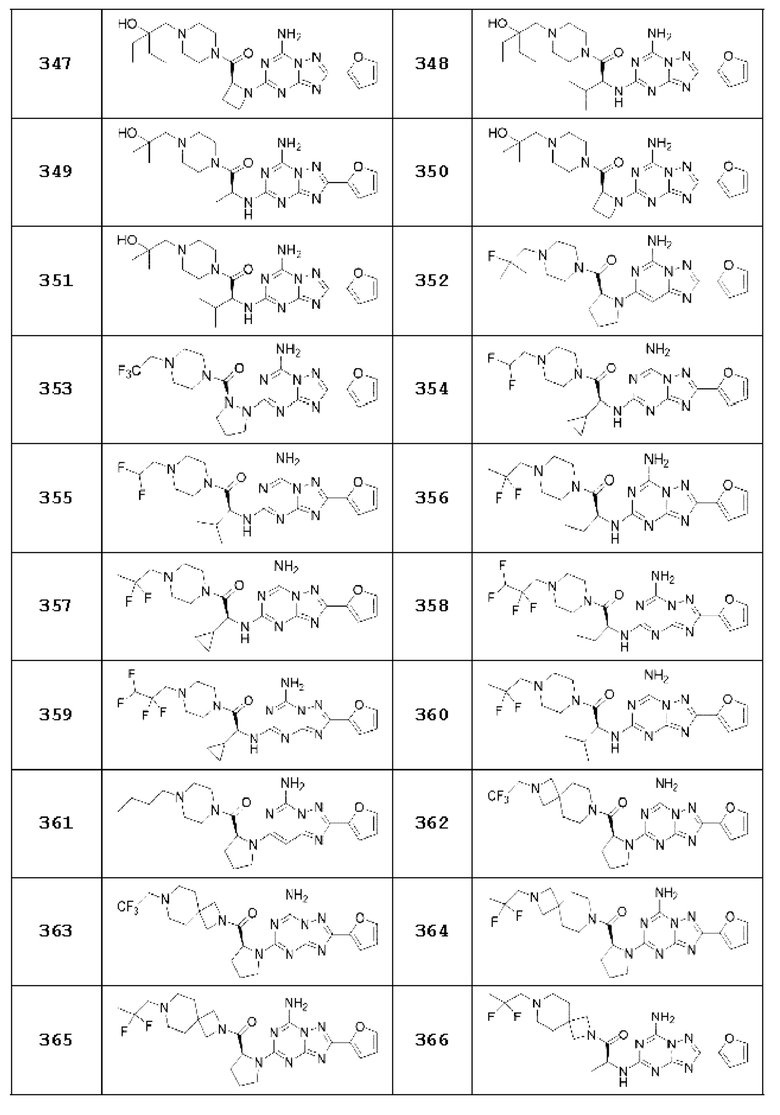
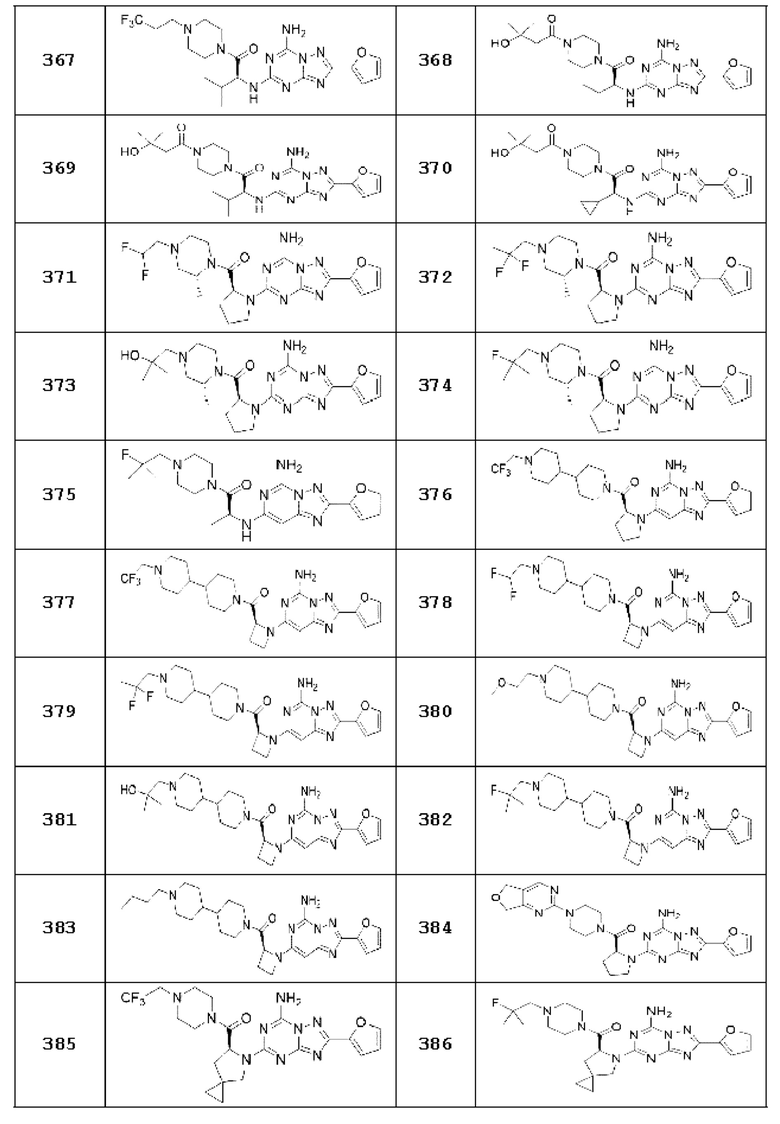
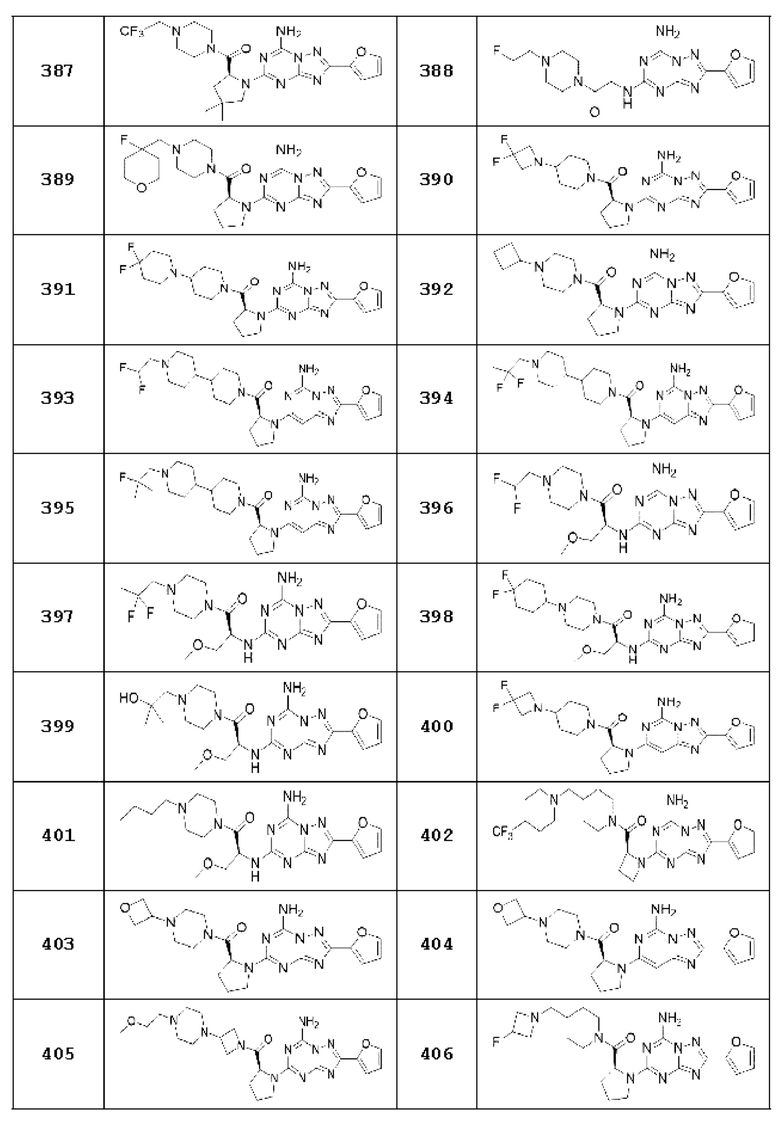
6. Соединение, его стереоизомеры или его фармацевтически приемлемые соли по п. 5, отличающееся тем, что соединение представляет собой одно соединение, выбранное из группы, состоящей из соединений ниже:
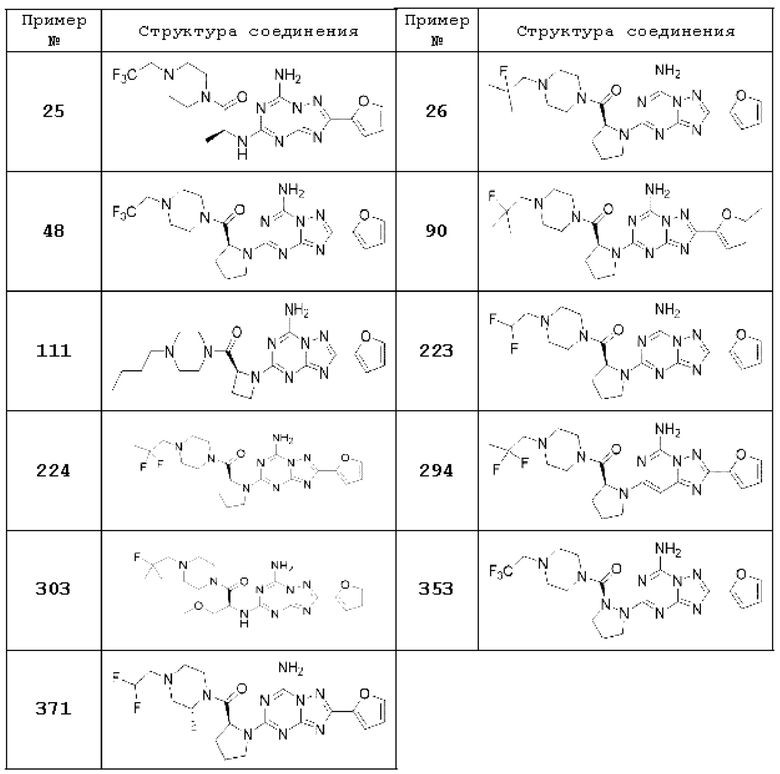
7. Фармацевтическая композиция, обладающая активностью антагониста рецептора аденозина А2а, содержащая эффективное количество соединения по любому из пп. 1-6, его стереоизомеров или его фармацевтически приемлемых солей в качестве активного ингредиента.
8. Фармацевтическая композиция по п. 7, отличающаяся тем, что фармацевтическая композиция предназначена для лечения или профилактики заболеваний, ассоциированных с аденозиновым рецептором А2а.
9. Фармацевтическая композиция по п. 8, отличающаяся тем, что заболевания, ассоциированные с аденозиновым рецептором А2а, представляют собой рак или воспалительные заболевания.
10. Фармацевтическая композиция по п. 9, отличающаяся тем, что рак представляет собой по меньшей мере один, выбранный из рака легких, рака желудка, рака яичников, рака предстательной железы, рака пищевода, рака желудочно-кишечного тракта, рака поджелудочной железы, колоректального рака, рака почки, рака яичка, рака мочевого пузыря, рака молочной железы, рака матки, рака шейки матки, рака головы и шеи, рака крови, рака костей, рака печени, рака щитовидной железы, рака кожи, лимфомы, лейкоза, миеломы, саркомы и вирус-ассоциированного рака.
11. Фармацевтическая композиция по п. 9, отличающаяся тем, что воспалительное заболевание представляет собой по меньшей мере одно, выбранное из ревматоидного артрита, рассеянного склероза, болезни Крона, язвенного колита, заболевания «трансплантат против хозяина», системной красной волчанки, синдрома токсического шока, остеоартрита и инсулинозависимого диабета.
12. Применение соединения по любому из пп. 1-6, его стереоизомеров или его фармацевтически приемлемых солей для приготовления лекарственного средства для лечения или профилактики заболеваний, ассоциированных с аденозиновым рецептором А2а.
| WO 03048163 A1, 12.06.2003 | |||
| CN 112028891 A, 04.12.2020 | |||
| US 2003162742 A1, 28.08.2003 | |||
| WO 03068776 A1, 21.08.2003 | |||
| ПРОИЗВОДНЫЕ ТРИАЗОЛО [4,5-d]ПИРИМИДИНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ АНТАГОНИСТОВ ПУРИНЕРГИЧЕСКОГО РЕЦЕПТОРА | 2002 |
|
RU2317084C2 |

