ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение направлено, в частности, на ариламидные соединения и способы их получения и применения.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Противомикробные пептиды (AMP) являются первой линией защиты против микробов для многих видов. AMP обычно представляют собой маленькие (12-80 аминокислот) катионные амфифилы. Существует два типа AMP, включающих рибосомно и нерибосомно синтезированные пептиды. Было идентифицировано свыше 700 AMP, и они имеют обычно α-спираль (магаинин и секропин) или обогащенные дисульфидом β-складчатые слои (бактенецин и дефензин). Хотя пептиды составлены из множества различных последовательностей, их физиологические свойства в высшей степени сходны. Они принимают амфифильную структуру с положительно заряженными группами, изолированными на одной стороне вторичной структуры, и гидрофобными группами на противоположной поверхности. У млекопитающих пептиды вырабатываются и секретируются в коже, на слизистых поверхностях и в нейтрофилах и действуют локально в ответ на инфекцию. Существуют общие физикохимические свойства, которые преимущественно отвечают за биологическую активность этих пептидов.
Некоторые противомикробные активности белков иммунной защиты были связаны с прямыми цитотоксическими действиями и модуляцией врожденной иммунной системы. Предполагается, что их прямые противомикробные активности включают как мембранные, так и немембранные эффекты. Противомикробные пептиды остаются эффективным средством защиты от бактериальной инфекции на протяжении времени эволюции, указывая на то, что их механизм действия препятствует бактериальным ответам, которые ведут к резистентности к токсическим веществам. Это предположение поддерживается прямыми экспериментальными данными, показывающими, что отсутствие значимой резистентности к действию противомикробных пептидов наблюдается после многократных серийных пассажей бактерий в присутствии сублетальных концентраций пептидов.
Существует крайняя необходимость в разработке новых противомикробных средств, которые атакуют новые мишени, ускользая от продуктов резистентности, которые ограничивают применимость многих антибиотиков. Кроме того, эти новые средствв должны проявлять свою противомикробную активность через механизмы, которые делают сопротивление бактерий не эффективным. Были разработаны серии непептидных аналогов, которые имеют множество преимуществ над пептидами по причине их маленького размера, которые увеличивают стабильность и улучшают распределение в тканях, и обладают возможностью тонко регулировать их физические свойства для оптимизации эффективности и безопасности. Было найдено, что серии ариламидных соединений, которые имитируют структурные свойства противомикробных пептидов, проявляют эффективные противомикробные активности и широкие индексы избирательности против клеток млекопитающих.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
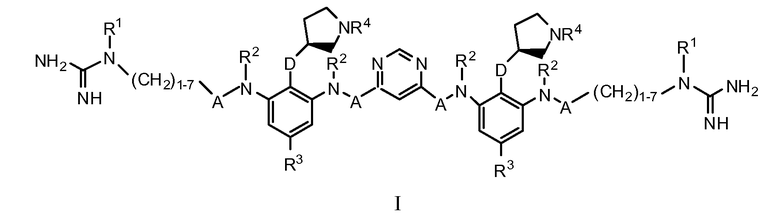
Настоящее изобретение предоставляет соединения Формулы I
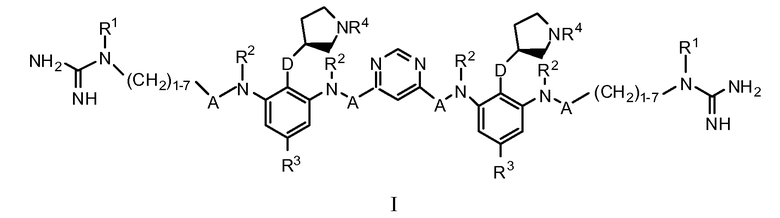 ,
,
где: каждый A представляет собой независимо -C=O, -C=S или -CH2; каждый D представляет собой независимо O или S; каждый R1 представляет собой независимо водород, C1-3алкил, C1-3алкокси, гало или галоС1-3алкил; каждый R2 представляет собой независимо водород, C1-3алкил, C1-3алкокси, гало или галоС1-3алкил; каждый R3 представляет собой независимо водород, C1-4алкил, C1-4алкокси, гало или галоС1-4алкил; и каждый R4 представляет собой независимо водород, C1-3алкил, C1-3алкокси, гало или галоС1-3алкил; или их фармацевтически приемлемую соль.
В некоторых вариантах осуществления изобретения каждый A представляет собой -C=O.
В некоторых вариантах осуществления изобретения каждый D представляет собой O.
В некоторых вариантах осуществления изобретения каждый R1 представляет собой независимо водород, метил, этил, метокси, этокси, гало или галоС1-3алкил. В некоторых вариантах осуществления изобретения каждый R1 представляет собой независимо водород, метил, метокси, гало или галоС1-3алкил. В некоторых вариантах осуществления изобретения каждый R1 представляет собой независимо водород, метил или метокси. В некоторых вариантах осуществления изобретения каждый R1 представляет собой водород.
В некоторых вариантах осуществления изобретения каждый R2 представляет собой независимо водород, метил, этил, метокси, этокси, гало или галоС1-3алкил. В некоторых вариантах осуществления изобретения каждый R2 представляет собой независимо водород, метил, метокси или гало. В некоторых вариантах осуществления изобретения каждый R2 представляет собой водород.
В некоторых вариантах осуществления изобретения каждый R3 представляет собой независимо водород, метил, этил, метокси, этокси, гало или галоС1-3алкил. В некоторых вариантах осуществления изобретения каждый R3 представляет собой независимо метил, метокси, гало или галоС1-3алкил. В некоторых вариантах осуществления изобретения каждый R3 представляет собой независимо гало или галоС1-3алкил. В некоторых вариантах осуществления изобретения каждый R3 представляет собой независимо галоС1-3алкил. В некоторых вариантах осуществления изобретения каждый R3 представляет собой трифторметил.
В некоторых вариантах осуществления изобретения каждый R4 представляет собой независимо водород, метил, этил, метокси, этокси или галоС1-3алкил. В некоторых вариантах осуществления изобретения каждый R4 представляет собой независимо водород, метил, метокси, гало или галоС1-3алкил. В некоторых вариантах осуществления изобретения каждый R4 представляет собой независимо водород, метил, метокси или гало. В некоторых вариантах осуществления изобретения каждый R4 представляет собой водород.
В некоторых вариантах осуществления изобретения каждый A представляет собой независимо -C=O, -C=S или -CH2; каждый D представляет собой независимо O или S; каждый R1 представляет собой независимо водород, метил, этил, метокси, этокси, гало, галометил или галоэтил; каждый R2 представляет собой независимо водород, метил, метокси, гало или галометил; каждый R3 представляет собой независимо C1-3алкил, C1-3алкокси, гало или галоалкил; и каждый R4 представляет собой независимо водород, метил, этил, метокси, этокси, гало, галометил или галоэтил.
В некоторых вариантах осуществления изобретения каждый A представляет собой независимо -C=O или -C=S; каждый D представляет собой независимо O или S; каждый R1 представляет собой независимо водород, метил, метокси, гало или галометил; каждый R2 представляет собой независимо водород, гало или галометил; каждый R3 представляет собой независимо метил, этил, метокси, этокси, гало, галометил или галоэтил; и каждый R4 представляет собой независимо водород, метил, этил, метокси, этокси, гало, галометил или галоэтил.
В некоторых вариантах осуществления изобретения каждый A представляет собой -C=O; каждый D представляет собой O; каждый R1 представляет собой независимо водород, гало или галометил; каждый R2 представляет собой независимо водород или гало; каждый R3 представляет собой независимо метил, метокси, гало или галометил; и каждый R4 представляет собой независимо водород, метил, метокси, гало или галометил.
В некоторых вариантах осуществления изобретения каждый A представляет собой -C=O; каждый D представляет собой O; каждый R1 представляет собой независимо водород или гало; каждый R2 представляет собой независимо водород или гало; каждый R3 представляет собой независимо метил, гало или галометил; и каждый R4 представляет собой независимо водород, метил, гало или галометил.
В некоторых вариантах осуществления изобретения каждый A представляет собой -C=O; каждый D представляет собой O; каждый R1 представляет собой независимо водород или гало; каждый R2 представляет собой независимо водород или гало; каждый R3 представляет собой независимо гало или галометил; и каждый R4 представляет собой независимо водород или гало.
В некоторых вариантах осуществления изобретения каждый A представляет собой -C=O; каждый D представляет собой O; каждый R1 представляет собой независимо водород или гало; каждый R2 представляет собой независимо водород или гало; каждый R3 представляет собой независимо метил, гало или галометил; и каждый R4 представляет собой независимо водород, метил, гало или галометил.
В некоторых вариантах осуществления изобретения соединение представляет собой
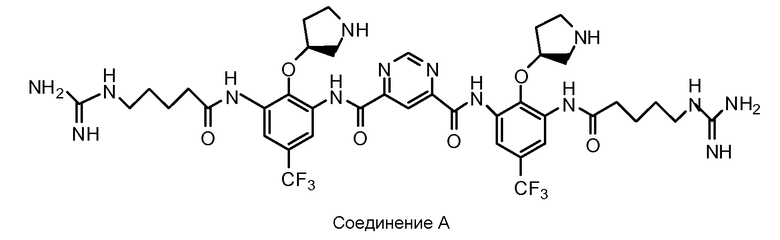
или его фармацевтически приемлемую соль.
Настоящее изобретение также предоставляет фармацевтические композиции, включающие одно или несколько соединений, описанных выше, или соль любого из соединений, описанных выше, и фармацевтически приемлемый носитель.
Настоящее изобретение также предоставляет препараты, включающие одно или несколько соединений, описанных выше, где препарат включает физиологический раствор, воду, раствор циклодекстрина или забуференный раствор при pH 3-9. В некоторых вариантах осуществления изобретения препарат представляет собой пероральный не абсорбируемый состав. В некоторых вариантах осуществления изобретения препарат включает эксципиент, выбранный из очищенной воды, пропиленгликоля, полиэтиленгликоля 400 (PEG 400), глицерина, DMA, этанола, бензилового спирта, лимонной кислоты/натрия цитрата (pH 3), лимонной кислоты/натрия цитрата (pH 5), трис(гидроксиметил)амино метан HCl (pH 7,0), 0,9% физиологического раствора и 1,2% физиологического раствора или любой их комбинации. В некоторых вариантах осуществления изобретения состав включает эксципиент, выбранный из пропиленгликоля, очищенной воды и глицерина. В некоторых вариантах осуществления изобретения препарат включает эксципиент, выбранный из 20% масса/объем пропиленгликоля в физиологическом растворе, 30% масса/объем пропиленгликоля в физиологическом растворе, 40% масса/объем пропиленгликоля в физиологическом растворе, 50% масса/объем пропиленгликоля в физиологическом растворе, 15% масса/объем пропиленгликоля в очищенной воде, 30% масса/объем пропиленгликоля в очищенной воде, 50% пропиленгликоля в очищенной воде, 30% масса/объем пропиленгликоля и 5 масса/объем этанола в очищенной воде, 15% масса/объем глицерина в очищенной воде, 30% масса/объем глицерина в очищенной воде, 50% масса/объем глицерина в очищенной воде, 20% масса/объем Клептозы в очищенной воде, 40% масса/объем Клептозы в очищенной воде и 25% масса/объем Каптизола в очищенной воде.
Настоящее изобретение также предоставляет способы получения Соединения A, включающие:
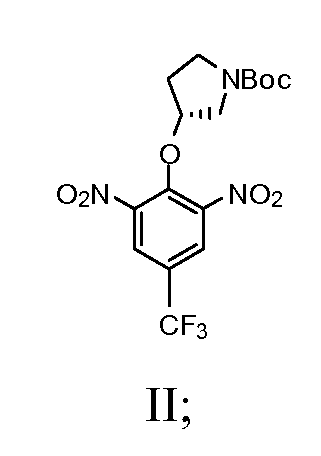
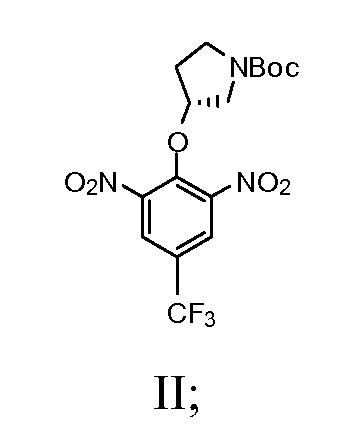
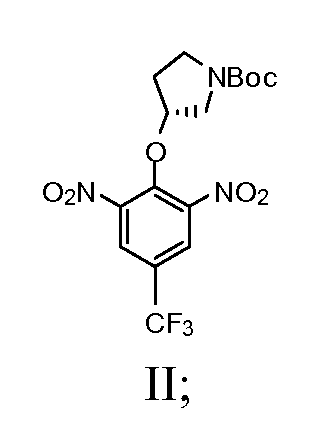
а) взаимодействие (R)-(-)-N-Boc-3-пирролидинола с сильным основанием с получением смеси; далее взаимодействие смеси с 2-хлор-5-(трифторметил)-1,3-динитробензолом с получением соединения, имеющего Формулу II
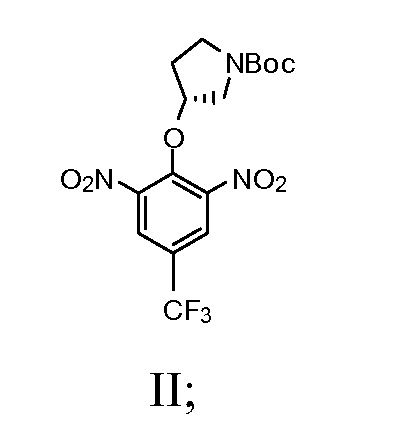
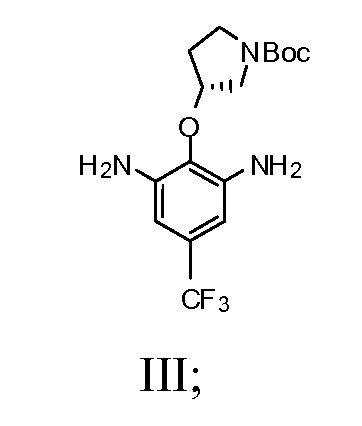
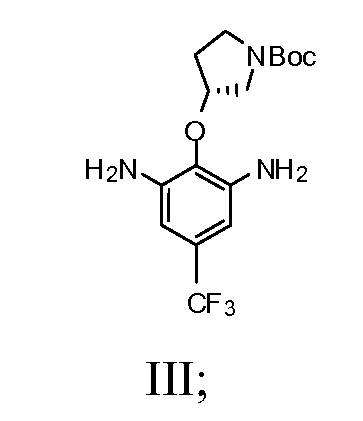
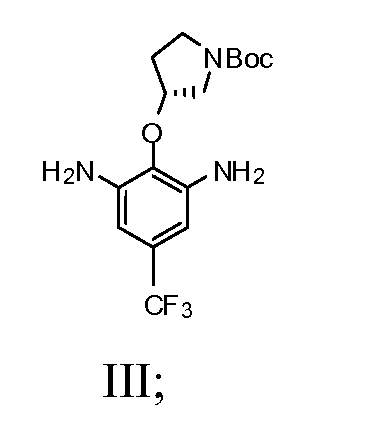
b) взаимодействие соединения Формулы II со спиртом и катализатором переходного металла в присутствии водорода с получением соединения Формулы III
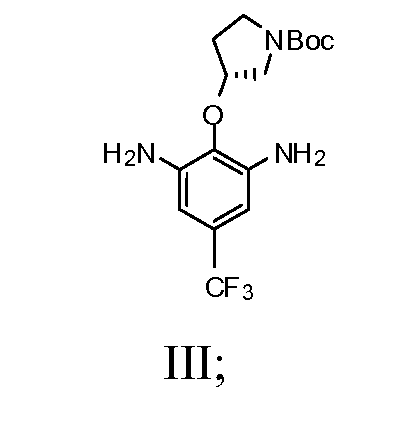
с) добавление соединения Формулы III и пиримидин-4,6-дикарбоновой кислоты в смесь 2-хлор-4,6-диметокси-1,3,5-триазина и N-метилморфолина с получением соединения Формулы IV
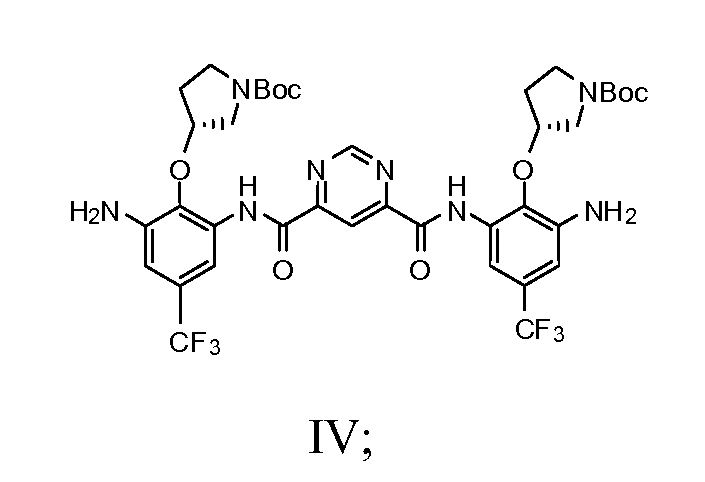
d) взаимодействие соединения Формулы IV с N-Boc-гуанидин масляной кислотой с получением соединения Формулы V
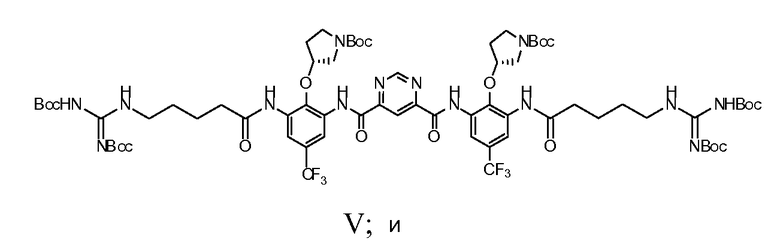
e) снятие защитных групп соединения Формулы V с получением Соединения A. В некоторых вариантах осуществления изобретения в а) сильное основание представляет собой NaH; и в b) катализатор переходного металла представляет собой Pd/C, и спирт представляет собой этанол.
Настоящее изобретение также предоставляет дополнительные способы получения Соединения A, включающие:
а) снятие защитных групп трет-бутилового эфира (R)-3-гидроксипирролидин-1-карбоновой кислоты и взаимодействие полученного соединения с 2-хлор-1,3-динитро-5-трифторметилбензолом с получением трет-бутилового эфира (R)-3-(2,6-динитро-4-трифторметилфенокси)пирролидин-1-карбоновой кислоты;
b) восстановление трет-бутилового эфира (R)-3-(2,6-динитро-4-трифторметилфенокси)пирролидин-1-карбоновой кислоты в присутствии спирта, катализатора переходного металла и водорода с получением трет-бутилового эфира (R)-3-(2,6-диамино-4-трифторметилфенокси)пирролидин-1-карбоновой кислоты;
c) сочетание трет-бутилового эфира (R)-3-(2,6-диамино-4-трифторметилфенокси)пирролидин-1-карбоновой кислоты с пиримидин-4,6-дикарбоновой кислотой в присутствии 1-[(3-(диметиламино)пропил)]-3-этилкарбодиимида гидрохлорида с получением бис-{[3-амино-2-((R)-1-(трет-бутоксикарбонил)пирролидин-3-илокси)-5-трифторметил-фенил]амида} пиримидин-4,6-дикарбоновой кислоты;
d) взаимодействие бис-{[3-амино-2-((R)-1-(трет-бутоксикарбонил)пирролидин-3-илокси)-5-трифторметил-фенил]амида} пиримидин-4,6-дикарбоновой кислоты с ({[(трет-бутоксикарбонил)амино][(трет-бутоксикарбонил)имино]метил}амино)пентановой кислотой в присутствии фосфора оксихлорида с получением бис-{[3-(5-({[(трет-бутоксикарбонил)амино][трет-бутоксикарбонил)имино]метил}амино)пентаноиламино)-2-((R)-1-(трет-бутоксикарбонил-пирролидин-3-илокси)-5-трифторметил-фенил]амида} пиримидин-4,6-дикарбоновой кислоты;
e) снятие защитных групп бис-{[3-(5-({[(трет-бутоксикарбонил)амино][трет-бутоксикарбонил)имино]метил}амино)пентаноиламино)-2-((R)-1-(трет-бутоксикарбонил)пирролидин-3-илокси)-5-трифторметил-фенил]амида} пиримидин-4,6-дикарбоновой кислоты с получением неочищенного бис-{[3-(5-гуанидино-пентаноиламино)-2-((R)-пирролидин-3-илокси)-5-трифторметил-фенил]амид}тетрагидрохлорида пиримидин-4,6-дикарбоновой кислоты; и
f) очистку неочищенного бис-{[3-(5-гуанидино-пентаноиламино)-2-((R)-пирролидин-3-илокси)-5-трифторметил-фенил]амид}тетрагидрохлорида пиримидин-4,6-дикарбоновой кислоты с помощью, например, хроматографии с обращенной фазой.
Настоящее изобретение также предоставляет другие дополнительные способы получения Соединения A, включающие:
а) снятие защитных групп трет-бутилового эфира (R)-3-гидроксипирролидин-1-карбоновой кислоты и далее взаимодействие полученного соединения с 2-хлор-1,3-динитро-5-трифторметилбензолом с получением трет-бутилового эфира (R)-3-(2,6-динитро-4-трифторметилфенокси)пирролидин-1-карбоновой кислоты;
b) восстановление трет-бутилового эфира (R)-3-(2,6-динитро-4-трифторметилфенокси)пирролидин-1-карбоновой кислоты в присутствии спирта, катализатора переходного металла и водорода с получением трет-бутилового эфира (R)-3-(2,6-диамино-4-трифторметилфенокси)пирролидин-1-карбоновой кислоты;
c) сочетание трет-бутилового эфира (R)-3-(2,6-диамино-4-трифторметилфенокси)-пирролидин-1-карбоновой кислоты с пиримидин-4,6-дикарбоновой кислотой в присутствии 1-[(3-(диметиламино)пропил)]-3-этилкарбодиимида гидрохлорида с получением бис-{[3-амино-2-((R)-1-(трет-бутоксикарбонил)пирролидин-3-илокси)-5-трифторметил-фенил]амида} пиримидин-4,6-дикарбоновой кислоты;
d) взаимодействие бис-{[3-амино-2-((R)-1-(трет-бутоксикарбонил)пирролидин-3-илокси)-5-трифторметил-фенил]амида} пиримидин-4,6-дикарбоновой кислоты с N-Cbz кислотой в присутствии тионила хлорида;
e) восстановление полученного соединения d) в присутствии спирта, катализатора переходного металла и водорода;
f) взаимодействие полученного соединения e) с ди-Boc пиразолом; и
g) снятие защитных групп полученного соединения f) с получением Соединения A.
Настоящее изобретение также предоставляет способы получения фармацевтически приемлемой соли Соединения A, включающие:
а) взаимодействие (R)-(-)-N-Boc-3-пирролидинола с сильным основанием с получением смеси; далее взаимодействие смеси с 2-хлор-5-(трифторметил)-1,3-динитробензолом с получением соединения, имеющего Формулу II
b) взаимодействие соединения Формулы II со спиртом и катализатором переходного металла в присутствии водорода с получением соединения Формулы III
c1) добавление соединения Формулы III и пиримидин-4,6-дикарбоновой кислоты в смесь 2-хлор-4,6-диметокси-1,3,5-триазина и N-метилморфолина с получением соединения Формулы IV

c2) добавление соединения Формулы III и пиримидин-4,6-дикарбоновой кислоты в смесь 1-этил-3-[3-(диметиламино)пропил]карбодиимида гидрохлорида (EDC1) и безводного пиридина с получением соединения Формулы IV
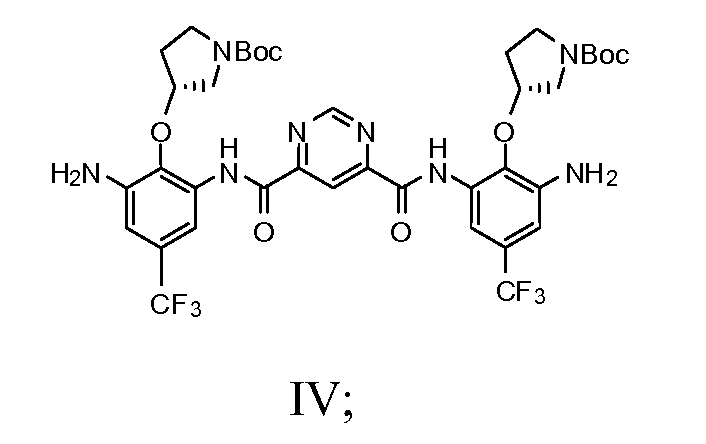
d) добавление соединения Формулы IV с N-Cbz кислотой в раствор, включающий безводный пиридин, диметиламинопропиламин и любой один из тионила хлорида, POCl3, (EtO)2POCl или оксалила хлорида с получением соединения Формулы Va
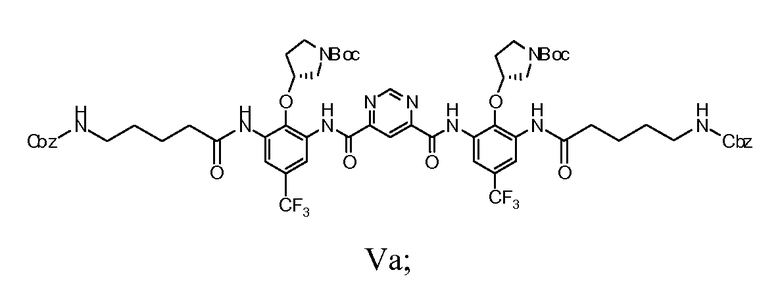
e) гидрогенолиз группы Cbz соединения Формулы Va с получением соединения Формулы VI
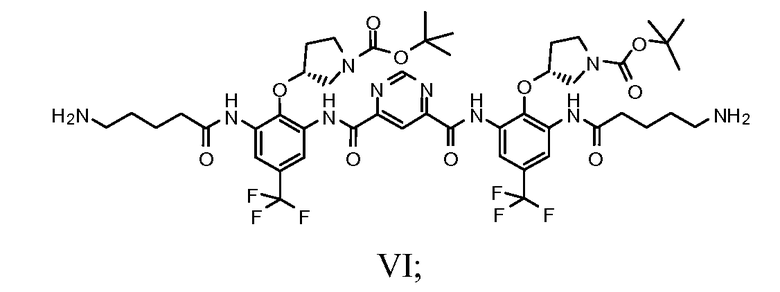
f) защиту соединения Формулы VI с получением соединения Формулы VII
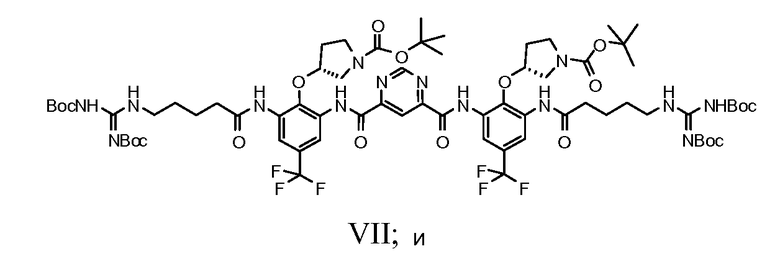
g) снятие защитных групп соединения Формулы VII с получением фармацевтически приемлемой соли Соединения A.
Настоящее изобретение также предоставляет способы ингибирования роста микроба, включающие контактирование микроба с любым из соединений, описанных выше, или его фармацевтически приемлемыми солями.
Настоящее изобретение также предоставляет способы лечения млекопитающего, страдающего микробной инфекцией, включающие введение млекопитающему, нуждающемуся в этом, противомикробного эффективного количества любого из соединений, описанных выше, или его фармацевтически приемлемых солей.
В некоторых вариантах осуществления изобретения микроб или микробная инфекция представляют собой грамотрицательный аэроб, грамположительный аэроб, грамотрицательный анаэроб, грамположительный анаэроб, микобактерию или дрожжи. В некоторых вариантах осуществления изобретения грамотрицательный аэроб представляет собой Escherichia coli, Citrobacter freundii, Citrobacter diverus, Citrobacter koseri, Enterobacter cloacae, Enterobacter faecalis, Klebsiella pneumonia, Klebsiella oxytoca, Morganella morganii, Providencia stuartii, Proteus vulgaris, Proteus mirabilis, Serratia marcescens, Acinetobacter haemolyticus, Acinetobacter junii, Acinetobacter lwoffii, Haemophilus influenzae, Stenotrophomonas maltophilia или Pseudomonas aeruginosa. В некоторых вариантах осуществления изобретения грамположительный аэроб представляет собой Enterococcus faecalis, Enterococcus faecium, Mycobacterium tuberculosis, Staphylococcus aureus, Staphylococcus pneumoniae, Staphylococcus epidermidis, Staphylococcus saprophyticus, Staphylococcus colmii, Staphylococcus sciuri, Staphylococcus warneri, Streptococcus agalactiae, Streptococcus pyogenes, Streptococcus anginosus, Streptococcus mitis или Streptococcus oralis. В некоторых вариантах осуществления изобретения грамотрицательный анаэроб представляет собой Bacteroides fragilis. В некоторых вариантах осуществления изобретения грамположительный анаэроб представляет собой Clostridium difficile или Clostridium perfringens. В некоторых вариантах осуществления изобретения микобактерия представляет собой Mycobacterium tuberculosis, Mycobacterium bovis, Mycobacterium africanum, Mycobacterium canetti или Mycobacterium microti. В некоторых вариантах осуществления изобретения дрожжи представляют собой Candida albicans или Candida krusei.
Настоящее изобретение также предоставляет любое из соединений, описанных выше, для лечения микробной инфекции.
Настоящее изобретение также предоставляет любое из соединений, описанных выше, или их фармацевтически приемлемых солей для использования при изготовлении лекарственного средства для лечения микробной инфекции.
Настоящее изобретение также предоставляет использование любого из соединений, описанных выше, или их фармацевтически приемлемых солей для ингибирования роста микроба.
Настоящее изобретение также предоставляет использование любого из соединений, описанных выше, или их фармацевтически приемлемых солей для лечения микробной инфекции млекопитающего.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
На фигуре 1A и фигуре 1B представлены результаты исследования время-эрадикация Соединения A против S. aureus ATCC27660 (фигура 1B представляет собой развернутый вид Фигуры 1A).
На фигуре 2 показана ассоциация пассажа S. aureus с норфлоксацином со значимым повышением значений MIC [минимальная ингибирующая концентрация] при пассаже 3 (4 двойных разведения) для MSSA и MRSA.
На фигуре 3 показана эффективность Соединения A против S. aureus на мышиной модели нагрузки на бедренную кость.
На фигуре 4 показана эффективность Соединения A по сравнению с ванкомицином против S. aureus на крысиной модели нагрузки на бедренную кость.
На фигуре 5 показана эффективность Соединения A против S. aureus на мышиной модели сепсиса.
ОПИСАНИЕ ВАРИАНТОВ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
Как использовано в данном описании, термин «примерно» означает ±5% описанного значения. Например, примерно 100 означает от 95 до 105.
Как использовано в данном описании. термины «C1-3алкил», «C1-4алкил» и «(CH2)1-7» означают насыщенные, моновалентные неразветвленные или разветвленные углеводородные цепи, содержащие от 1 до 3 углеродов, от 1 до 4 углеродов и от 1 до 7 углеродов соответственно. Примеры алкильных групп включают, но не ограничиваясь ими, (C1-C7)алкильные группы, такие как метил, этил, пропил, изопропил, 2-метил-1-пропил, 2-метил-2-пропил, 2-метил-1-бутил, 3-метил-1-бутил, 2-метил-3-бутил, 2-метил-1-пентил, 2,2-диметил-1-пропил, 3-метил-1-пентил, 4-метил-1-пентил, 2-метил-2-пентил, 3-метил-2-пентил, 4-метил-2-пентил, 2,2-диметил-1-бутил, 3,3-диметил-1-бутил, 2-этил-1-бутил, бутил, изобутил, трет-бутил, пентил, изопентил, неопентил, гексил и гептил. Алкильная группа может быть незамещенной или замещена одним или двумя приемлемыми заместителями.
Как использовано в данном описании, термины «C1-3алкокси» и «C1-4алкокси» означают -O-алкил с алкилом, определенным, как указано выше. Алкокси группа может быть незамещенной или замещена одним или двумя приемлемыми заместителями. Алкильная цепь алкоксигруппы содержит от 1 до 3 или от 1 до 4 атомов углерода в длину.
Как использовано в данном описании, термин «гало» означает галоген, такой как фтор, хлор, бром или йод.
Как использовано в данном описании, термины «галоС1-3алкил» и «галоС1-4алкил» означают алкильные группы, как определено выше, где один или несколько (т.е. 1, 2, 3, 4, 5, 6, 7, 8, 9 или 10) водородов замещаются на гало, как определено выше.
Как использовано в данном описании, «изолированный» означает, что соединения Формулы I выделяются из других компонентов или (а) природного источника, такого как клетка, такого как бактериальная культура, или (b) синтетической органической химической реакционной смеси, такой как используемая в общепринятых методиках, соединения Формулы I очищаются.
Как использовано в данном описании, термин «млекопитающее» означает грызуна (т.е. мышь, крыса или морская свинка), обезьяну, кошку, собаку, корову, лошадь, свинью или человека. В некоторых вариантах осуществления изобретения млекопитающим является человек.
Как использовано в данном описании, термин «микроб» означает бактерии, грибы, простейшие или вирус.
Как использовано в данном описании, фраза «фармацевтически приемлемая соль(и)» включает, но не ограничиваясь ими, соли кислых или основных групп.
Как использовано в данном описании, термин «очищенный» означает, что при изолировании изолят содержит по меньшей мере 90%, по меньшей мере 95%, по меньшей мере 98% или по меньшей мере 99% соединения Формулы I по массе изолята.
Как использовано в данном описании, фраза «приемлемый заместитель» означает группу, которая не отменяет синтетическую или фармацевтическую ценность соединений Формулы I или интермедиатов, используемых для их получения. Примеры приемлемых заместителей включают, но не ограничиваясь ими: (C1-C4)алкил, (C1-C4)алкенил, (C1-C4)алкинил, (C1-C4)алкокси, -CN, -OH, оксо, гало, -NO2, -CO2H, -NH2, -NH((C1-C4)алкил), -N((C1-C4)алкил)2, -CHO, -CO((C1-C4)алкил) и -CO2((C1-C4)алкил). Специалист в данной области может легко выбрать приемлемый заместитель на основе стабильности и фармакологической и синтетической активности соединения Формулы I.
Как использовано в данном описании, фраза «противомикробное эффективное количество» соединения, включающего Формулу I, оценивается противомикробной эффективностью соединения. В некоторых вариантах осуществления изобретения противомикробное эффективное количество ингибирует рост определенного микроба на по меньшей мере 10%, на по меньшей мере 20%, на по меньшей мере 30%, на по меньшей мере 40%, на по меньшей мере 50%, на по меньшей мере 60%, на по меньшей мере 70%, на по меньшей мере 80%, на по меньшей мере 90% или на по меньшей мере 95%. В некоторых вариантах осуществления изобретения противомикробное эффективное количество также означает «терапевтически эффективное количество», тем самым соединение снижает или устраняет по меньшей мере один неблагоприятный эффект микроба на млекопитающего.
Настоящее изобретение предоставляет соединения Формулы I
 ,
,
где:
каждый A представляет собой независимо -C=O, -C=S или -CH2;
каждый D представляет собой независимо O или S;
каждый R1 представляет собой независимо водород, C1-3алкил, C1-3алкокси, гало или галоС1-3алкил;
каждый R2 представляет собой независимо водород, C1-3алкил, C1-3алкокси, гало или галоС1-3алкил;
каждый R3 представляет собой независимо водород, C1-4алкил, C1-4алкокси, гало или галоС1-4алкил; и
каждый R4 представляет собой независимо водород, C1-3алкил, C1-3алкокси, гало или галоС1-3алкил;
или их фармацевтически приемлемую соль.
В некоторых вариантах осуществления изобретения по меньшей мере один A представляет собой -C=O. В некоторых вариантах осуществления изобретения каждый A представляет собой -C=O.
В некоторых вариантах осуществления изобретения по меньшей мере один D представляет собой O. В некоторых вариантах осуществления изобретения каждый D представляет собой O.
В некоторых вариантах осуществления изобретения каждый R1 представляет собой независимо водород, метил, этил, метокси, этокси, гало или галоС1-3алкил. В некоторых вариантах осуществления изобретения каждый R1 представляет собой независимо водород, метил, метокси, гало или галоС1-3алкил. В некоторых вариантах осуществления изобретения каждый R1 представляет собой независимо водород, метил или метокси. В некоторых вариантах осуществления изобретения по меньшей мере один R1 представляет собой водород. В некоторых вариантах осуществления изобретения каждый R1 представляет собой водород.
В некоторых вариантах осуществления изобретения каждый R2 представляет собой независимо водород, метил, этил, метокси, этокси, гало или галоС1-3алкил. В некоторых вариантах осуществления изобретения каждый R2 представляет собой независимо водород, метил, метокси или гало. В некоторых вариантах осуществления изобретения по меньшей мере один R2 представляет собой водород. В некоторых вариантах осуществления изобретения каждый R2 представляет собой водород.
В некоторых вариантах осуществления изобретения каждый R3 представляет собой независимо водород, метил, этил, метокси, этокси, гало или галоС1-3алкил. В некоторых вариантах осуществления изобретения каждый R3 представляет собой независимо метил, метокси, гало или галоС1-3алкил. В некоторых вариантах осуществления изобретения каждый R3 представляет собой независимо гало или галоС1-3алкил. В некоторых вариантах осуществления изобретения каждый R3 представляет собой независимо галоС1-3алкил. В некоторых вариантах осуществления изобретения по меньшей мере один R3 представляет собой трифторметил. В некоторых вариантах осуществления изобретения каждый R3 представляет собой трифторметил.
В некоторых вариантах осуществления изобретения каждый R4 представляет собой независимо водород, метил, этил, метокси, этокси или галоС1-3алкил. В некоторых вариантах осуществления изобретения каждый R4 представляет собой независимо водород, метил, метокси, гало или галоС1-3алкил. В некоторых вариантах осуществления изобретения каждый R4 представляет собой независимо водород, метил, метокси или гало. В некоторых вариантах осуществления изобретения по меньшей мере один R4 представляет собой водород. В некоторых вариантах осуществления изобретения каждый R4 представляет собой водород.
В некоторых вариантах осуществления изобретения каждый A представляет собой независимо -C=O или -C=S; каждый D представляет собой независимо O или S; каждый R1 представляет собой независимо водород, метил, этил, метокси, этокси, гало, галометил или галоэтил; каждый R2 представляет собой независимо водород, метил, метокси, гало или галометил; каждый R3 представляет собой независимо C1-3алкил, C1-3алкокси, гало или галоалкил; и каждый R4 представляет собой независимо водород, метил, этил, метокси, этокси, гало, галометил или галоэтил.
В некоторых вариантах осуществления изобретения каждый A представляет собой независимо -C=O или -C=S; каждый D представляет собой независимо O или S; каждый R1 представляет собой независимо водород, метил, метокси, гало или галометил; каждый R2 представляет собой независимо водород, гало или галометил; каждый R3 представляет собой независимо метил, этил, метокси, этокси, гало, галометил или галоэтил; и каждый R4 представляет собой независимо водород, метил, этил, метокси, этокси, гало, галометил или галоэтил.
В некоторых вариантах осуществления изобретения каждый A представляет собой -C=O; каждый D представляет собой O; каждый R1 представляет собой независимо водород, гало или галометил; каждый R2 представляет собой независимо водород или гало; каждый R3 представляет собой независимо метил, метокси, гало или галометил; и каждый R4 представляет собой независимо водород, метил, метокси, гало или галометил.
В некоторых вариантах осуществления изобретения каждый A представляет собой -C=O; каждый D представляет собой O; каждый R1 представляет собой независимо водород или гало; каждый R2 представляет собой независимо водород или гало; каждый R3 представляет собой независимо метил, гало или галометил; и каждый R4 представляет собой независимо водород, метил, гало или галометил.
В некоторых вариантах осуществления изобретения каждый A представляет собой -C=O; каждый D представляет собой O; каждый R1 представляет собой независимо водород или гало; каждый R2 представляет собой независимо водород или гало; каждый R3 представляет собой независимо гало или галометил; и каждый R4 представляет собой независимо водород или гало.
В некоторых вариантах осуществления изобретения каждый A представляет собой -C=O; каждый D представляет собой O; каждый R1 представляет собой независимо водород или гало; каждый R2 представляет собой независимо водород или гало; каждый R3 представляет собой независимо метил, гало или галометил; и каждый R4 представляет собой независимо водород, метил, гало или галометил.
В некоторых вариантах осуществления изобретения каждый A представляет собой -C=O; каждый D представляет собой O; каждый R1 представляет собой независимо водород или гало; каждый R2 представляет собой независимо водород или гало; каждый R3 представляет собой независимо гало или галометил; и каждый R4 представляет собой независимо водород, метил, гало или галометил.
В некоторых вариантах осуществления изобретения соединение представляет собой Соединение A
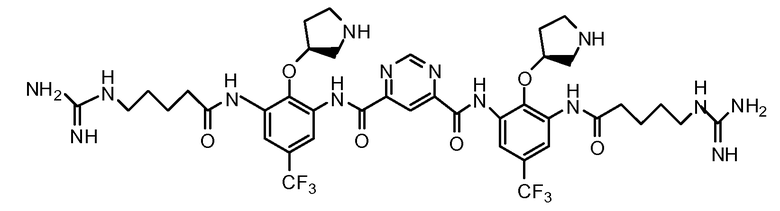
или его фармацевтически приемлемую соль.
Приемлемые примеры солей включают, например, соляную кислоту и трифторуксусную кислоту.
Соединения Формулы I могут содержать один или несколько хиральных центров и/или двойные связи и поэтому существуют как стереоизомеры, такие как изомеры с двойной связью (т.е. геометрические изомеры), энантиомеры или диастереомеры. В соответствии с изобретением химические структуры представлены в данном описании, и поэтому Соединения Формулы I охватывают все соответствующие энантиомеры и стереоизомеры соединения, а именно стереомерно правильную форму (например, геометрически правильная, энантиомерно правильная или диастереомерно правильная) и энантиомерные или стереоизомерные смеси. Энантиомерные или стереоизомерные смеси могут быть разрешены по своим компонентным энантиомерам или стереоизомерам с помощью хорошо известных методов, таких как газовая хроматография с хиральной фазой, высокоэффективная жидкостная хроматография с хиральной фазой, кристаллизация соединения в виде комплекса хиральных солей или кристаллизация соединения в хиральном растворителе. Энантиомеры и стереоизомеры могут быть также получены из стереомерно или энантиомерно чистых интермедиатов, реагентов и катализироваться с помощью хорошо известных методов асимметрического синтеза.
Соединения Формулы I далее включают гидраты и сольваты.
Соединения, содержащие аминную функцию, могут также образовывать N-оксиды. В данном описании приводится ссылка на соединение, которое содержит аминную функцию, также включая N-оксид. В случае если соединение содержит несколько аминных функций, один или более чем один атом азота может быть окислен с получением N-оксида. Примеры N-оксидов включают N-оксиды третичного амина или атом азота, содержащего азот гетероцикла. N-оксиды могут образовываться путем обработки соответствующим амином с окисляющим агентом, таким как водорода пероксид или перкислота (например, перкарбоновая кислота) (см. Advanced Organic Chemistry, by Jerry March, 4th Edition, Wiley Interscience).
В некоторых вариантах осуществления изобретения соединения Формулы I изолируются и/или очищаются.
Настоящее изобретение также предоставляет фармацевтические композиции, включающие одно или несколько соединений, описанных выше, или одну или несколько их солей, и фармацевтически приемлемый носитель.
Приемлемые композиции включают, но не ограничиваясь ими, пероральные не абсорбируемые композиции. Приемлемые композиции также включают, но не ограничиваясь ими, физиологический раствор, воду, растворы циклодекстрина и забуференные растворы с pH 3-9.
Соединения, описанные в данном описании, включая Соединение A или его фармацевтически приемлемые соли, могут быть получены с использованием многочисленных эксципиентов, включая, но не ограничиваясь ими, очищенную воду, пропиленгликоль, PEG 400, глицерин, DMA, этанол, бензиловый спирт, лимонную кислоту/натрия цитрат (pH 3), лимонную кислоту/натрия цитрат (pH 5), трис(гидроксиметил)амино метан HCl (pH 7,0), 0,9% физиологический раствор и 1,2% физиологический раствор и любую их комбинацию. В некоторых вариантах осуществления изобретения эксципиент выбран из пропиленгликоля, очищенной воды и глицерина.
В некоторых вариантах осуществления изобретения эксципиент представляет собой многокомпонентную систему, выбранную из 20% масса/объем пропиленгликоля в физиологическом растворе, 30% масса/объем пропиленгликоля в физиологическом растворе, 40% масса/объем пропиленгликоля в физиологическом растворе, 50% масса/объем пропиленгликоля в физиологическом растворе, 15% масса/объем пропиленгликоля в очищенной воде, 30% масса/объем пропиленгликоля в очищенной воде, 50% масса/объем пропиленгликоля в очищенной воде, 30% масса/объем пропиленгликоля и 5 масса/объем этанола в очищенной воде, 15% масса/объем глицерина в очищенной воде, 30% масса/объем глицерина в очищенной воде, 50% масса/объем глицерина в очищенной воде, 20% масса/объем Клептозы в очищенной воде, 40% масса/объем Клептозы в очищенной воде и 25% масса/объем Каптизола в очищенной воде. В некоторых вариантах осуществления изобретения эксципиент выбран из 50% масса/объем пропиленгликоля в очищенной воде, 15% масса/объем глицерина в очищенной воде, 20% масса/объем Клептозы в очищенной воде, 40% масса/объем Клептозы в очищенной воде и 25% масса/объем Каптизола в очищенной воде. В некоторых вариантах осуществления изобретения эксципиент выбран из 20% масса/объем Клептозы в очищенной воде, 20% масса/объем пропиленгликоля в очищенной воде и 15% масса/объем глицерина в очищенной воде.
В некоторых вариантах осуществления изобретения состав включает 50 мг/мл Соединения A в 20% масса/объем Клептозе в очищенной воде.
В некоторых вариантах осуществления изобретения состав может быть лиофилизирован в твердое вещество и восстановлен, например, водой перед использованием.
При введении млекопитающему (например, животному для ветеринарного применения или человеку для клинического применения) соединения Формулы I могут быть введены в изолированной форме. Альтернативно соединения Формулы I могут быть введены вместе с (т.е. как комбинированный состав или как отдельные составы) другими антибиотиками, такими как, например: 1) ингибиторы белкового синтеза, включая, но не ограничиваясь ими, амикацин, анизомицин, апрамицин, азитромицин, бластицидин S, брефелдин A, бутирозин, хлорамфеникол, хлортетрациклин, клиндамицин, клотримазол, циклогексимид, демеклоциклин, дибекацин, дигидрострептомицин, доксициклин, дурамицин, эметин, эритромицин, фузидовая кислота, G418, гентамицин, гельволовая кислота, гигромицин B, джозамицин, канамицин, кирромицин, линкомицин, меклоциклин, мепартрицин, мидекамицин, миноциклин, неомицин, нетилмицин, нитрофурантоин, ноурсетрицин, олеандомицин, окситетрациклин, паромомицин, пуромицин, рапамицин, рибостамицин, рифампицин, рифамицин, розамицин, сизомицин, спектиномицин, спирамицин, стрептомицин, тетрациклин, триамфеникол, тиострептон, тобрамицин, туникамицин, тилозин, виомицин и вирджиниамицин; 2) средства, препятствующие синтезу ДНК, включая, но не ограничиваясь ими, камптотецин, 10-деацетилбаккатин III, азацитидин, 7-аминоактиномицин D, 8-хинолинол, 9-дигидро-13-ацетилбаккатин III, акларубицин, актиномицин D, актиномицин I, актиномицин V, бафиломицин A1, блеомицин, капреомицин, хромомицин, циноксацин, ципрофлоксацин, цис-диаминплатина(II) дихлорид, кумермицин A1, L(+)-молочную кислоту, цитохалазин B, цитохалазин D, дакарбазин, даунорубицин, дистамицин A, доксорубицин, эхиномицин, энрофлоксацин, этопозид, флумехин, формицин, фумагиллин, ганцикловир глиотоксин, ломефлоксацин, метронидазол, митрамицин A, митомицин C, налидиксовую кислоту, нетропсин, нитрофурантоин, ногаламицин, нонактин, новобиоцин, офлоксацин, оксолиновую кислоту, паклитаксел, феназин, флеомицин, пипемидовую кислоту, ребескамицин, синефунгин, стрептонигрин, стрептозоцин, сукцинилсульфатиазол, сульфадиазин, сульфадиметоксин, сульфагуанидин чистый, сульфаметазин, сульфамонометоксин, сульфаниламид, сульфахиноксалин, сульфасалазин, сульфатиазол, триметоприм, туберцидин, 5-азацитидин, кордицепин и формицин A; 3) средства, препятствующие синтезу клеточной стенки, включая, но не ограничиваясь ими, (+)-6-аминопенициллановую кислоту, 7-аминодезацетоксицефалоспорановую кислоту, амоксициллин, ампициллин, азлоциллин, бацитрацин, карбенициллин, цефаклор, цефамандол, цефазолин, цефметазол, цефоперазон, цефотаксим, цефсулодин, цефтриаксон, цефалексин, цефалоспорин C, цефалотин, цефрадин, клоксациллин, D-циклосерин, диклоксациллин, D-пеницилламин, эконазол, этамбутол, лизостафин, моксалактам, нафциллин, никкомицин Z, нитрофурантоин, оксациллин, пенициллин, пенициллин G, фенетициллин, феноксиметилпенициллиновую кислоту, фосфомицин, пипемидовую кислоту, пиперациллин, ристомицин и ванкомицин; 4) средства, препятствующие проницаемости клеточной мембраны (ионофоры), включая, но не ограничиваясь ими, 2-меркаптопиридин, 4-бромкальцимицин A23187, аламетицин, амфотерицин B, кальцимицин A23187, хлоргексидин, клотримазол, колистин, эконазол, гидрокортизон, филипин, глиотоксин, грамицидин A, грамицидин C, иономицин, лазалоцид A, лономицин A, монензин, N-(6-аминогексил)-5-хлор-1-нафталенсульфонамид, наразин, нигерицин, низин, нонактин, нистатин, феназин, пимарицин, полимиксин B, DL-пеницилламин, полимиксин B, празиквантел, салиномицин, сурфактин и валиномицин; 5) ферментативные ингибиторы, включая, но не ограничиваясь ими, (+)-усниновую кислоту, (±)-миконазол, (S)-(+)-камптотецин, 1-дезоксиманноджиримицин, 2-гептил-4-гидроксихинолин N-оксид, кордицептин, 1,10-фенантролин, 6-диазо-5-оксо-L-норлейцин, 8-хинолинол, антимицин, антипаин, аскомицин, азасерин, бафиломицин, церуленин, хлорохин, циноксацин, ципрофлоксацин, мевастатин, конканамицин A, конканамицин C, кумермицин A1, L(+)-молочную кислоту, циклоспорин A, эконазол, энрофлоксацин, этопозид, флумехин, формицин A, фуразолидон, фузаровую кислоту, гелданамицин, глиотоксин, грамицидин A, грамицидин C, гербимицин A, индометацин, иргазан, ломефлоксацин, микофеноловую кислоту, миксотиазол, N-(6-аминогексил)-5-хлор-1-нафталенсульфонамид, налидиксовую кислоту, нетропсин, никлозамид, никкомицин, N-метил-1-дезоксиноджиримицин, ногаламицин, нонактин, новобиоцин, офлоксацин, олеандомицин, олигомицин, оксолиновую кислоту, пирицидин A, пипемидовую кислоту, радицикол, рапамицин, ребескамицин, синефунгин, стауроспорин, стигмателлин, сукцинилсульфатиазол, сульфадиазин, сульфадиметоксин, сульфагуанидин, сульфаметазин, сульфамонометоксин, сульфаниламид, сульфахиноксалин, сульфасалазин, сульфатиазол, триасцин C, триметоприм и винеомицин A1; и 6) мембранные модификаторы, включая, но не ограничиваясь ими, парацельзин.
В некоторых вариантах осуществления изобретения термин «фармацевтически приемлемый» означает утвержденный Агентством по регулированию Федерального или правительственного уровня или перечисленный в Фармакопеи США, или другой общепризнанной фармакопеи, используемой для животных, и конкретнее для людей. Термин «носитель» относится к разбавителю, адъюванту или эксципиенту, с которым вводится соединение Формулы I. Такие фармацевтические носители могут представлять собой жидкости, такие как вода и масла, включая масла нефтяного, животного, растительного или синтетического происхождения, такие как арахисовое масло, соевое масло, минеральное масло, кунжутное масло и тому подобное. Фармацевтическик носители могут также представлять собой физиологический раствор, аравийскую камедь, желатин, крахмальный клейстер, тальк, кератин, коллоидный кремний, мочевину и тому подобное. Дополнительно могут быть использованы вспомогательные, стабилизирующие, загущающие, смазывающие и красящие агенты. При введении человеку соединения Формулы I и фармацевтически приемлемые носители могут быть стерильными. Вода является приемлемым носителем, когда соединение Формулы I вводится внутривенно. Физиологические растворы и растворы водной декстрозы и глицерина могут быть также применены в качестве жидких носителей, особенно для инъецируемых растворов. Приемлемые фармацевтические носители также включают эксципиенты, такие как крахмал, глюкоза, лактоза, сахароза, желатин, солод, рис, мука, мел, силикатный гель, натрия стеарат, глицерол моностеарат, тальк, натрия хлорид, сухое обезжиренное молоко, глицерин, пропилен, гликоль, вода, этанол и тому подобное. Настоящие композиции, если желательно, могут также содержать небольшое количество увлажняющих или эмульгирующих агентов или забуферивающих pH агентов.
Композиции, описанные в данном описании, могут принимать форму раствора, суспензии, эмульсии, таблетки, пилюли, гранулы, капсулы, капсулы, содержащей жидкость, порошка, состава с замедленным высвобождением, суппозитория, аэрозоля, спрея или любую другую форму, приемлемую для использования. Примеры приемлемых фармацевтических носителей описаны в Remington's Pharmaceutical Sciense, A.R. Gennaro (Editor) Mack Publishing Co.
В одном из вариантов осуществления изобретения соединения Формулы I получают в соответствии с обычной процедурой как фармацевтическую композицию, адаптированную для введения людям. Обычно соединения Формулы I представляют собой растворы в стерильном изотоническом водном буфере. При необходимости соединения могут также включать солюбилизирующий агент. Композиции для внутривенного введения могут при необходимости включать местный анестетик, такой как лидокаин, для облегчения боли в месте инъекции. Обычно ингредиенты предоставляются раздельно или смешанные в стандартной лекарственной форме, например, в виде сухого лиофилизированного порошка или безводного концентрата в герметически закрытом контейнере, таком как ампула или саше, с указанием количества активного агента. В случае если соединение изобретения следует вводить путем инфузии, оно может быть приготовлено, например, с использованием флакона для инфузии, содержащего стерильную фармацевтически чистую воду для инфузии или физиологический раствор. В случае если соединение Формулы I вводится путем инъекции, может быть предоставлена ампула стерильной воды для инъекции или физиологического раствора так, чтобы ингредиенты могли быть смешаны до введения.
Соединения Формулы I и композиции, включающие то же самое, могут быть введены перорально. Соединения и композиции для пероральной доставки могут находиться в форме, например таблеток, пастилок, водных или масляных суспензий, гранул, порошков, эмульсий, капсул, сиропов или эликсиров. Перорально введенные композиции могут содержать один или несколько необязательных агентов, например, подслащивающие агенты, такие как фруктоза, аспартам или сахарин; ароматизирующие агенты, такие как мята перечная, масло гаультерии или вишни; окрашивающие агенты; и консервирующие агенты, с получением фармацевтически приятного на вкус препарата. Кроме того, когда имеется форма таблетки или пилюли, композиции могут быть покрыты для замедления распадаемости и абсорбции в желудочно-кишечном тракте, тем самым обеспечивая непрерывное действие в течение длительного периода времени. Избирательно проницаемые мембраны, окружающие осмотически активно перемещающееся соединение, также приемлемы для перорально вводимых соединений Формулы I. Пероральные композиции могут включать стандартные основы, такие как маннит, лактоза, крахмал, магния стеарат, натрия сахарин, целлюлоза, магния карбонат и т.д. Такие основы являются соответственно фармацевтически чистыми.
Фармацевтические композиции могут быть в стандартной лекарственной форме. В такой форме композиция может быть разделена на одноразовые дозы, содержащие соответствующее количество активного компонента. Стандартная лекарственная форма может находиться в виде упакованного препарата, упаковка содержит дискретные количества препаратов, например упакованные таблетки, капсулы и порошки во флаконах или ампулах. Стандартная лекарственная форма может также находиться в виде капсулы, саше или самой таблетки, или она может представлять собой соответствующее количество любой из этих упакованных форм.
Настоящее изобретение также предоставляет способы получения Соединения A, включающие:
1а) взаимодействие (R)-(-)-N-Boc-3-пирролидинола с сильным основанием с получением смеси; далее взаимодействие смеси с 2-хлор-5-(трифторметил)-1,3-динитробензолом с получением соединения, имеющего Формулу II
1b) взаимодействие соединения Формулы II со спиртом и катализатором переходного металла в присутствии водорода с получением соединения Формулы III
1c) добавление соединения Формулы III и пиримидин-4,6-дикарбоновой кислоты в смесь 2-хлор-4,6-диметокси-1,3,5-триазина и N-метилморфолина с получением соединения Формулы IV
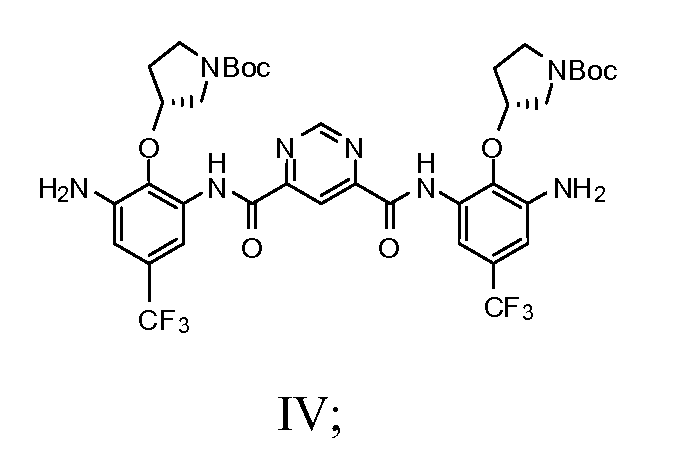
1d) взаимодействие соединения Формулы IV с N-Boc-гуанидином масляной кислоты с получением соединения Формулы V
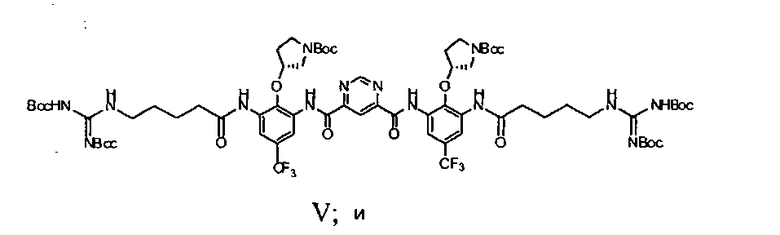
1e) снятие защитных групп соединения Формулы V с получением Соединения A.
В некоторых вариантах осуществления изобретения в a) сильное основание представляет собой NaH; и в b) катализатор переходного металла представляет собой Pd/C, а спирт представляет собой этанол. В частности, этот способ описан ниже боле детально в Примере 1.
Настоящее изобретение также предоставляет дополнительные способы получения Соединения A, включающие:
a) депротонирование трет-бутилового эфира (R)-3-гидроксипирролидин-1-карбоновой кислоты и взаимодействие полученного соединения с 2-хлор-1,3-динитро-5-трифторметилбензолом с получением трет-бутилового эфира (R)-3-(2,6-динитро-4-трифторметилфенокси)пирролидин-1-карбоновой кислоты;
b) восстановление трет-бутилового эфира (R)-3-(2,6-динитро-4-трифторметилфенокси)пирролидин-1-карбоновой кислоты в присутствии спирта, катализатора переходного металла и водорода с получением трет-бутилового эфира (R)-3-(2,6-диамино-4-трифторметилфенокси)пирролидин-1-карбоновой кислоты;
c) сочетание трет-бутилового эфира (R)-3-(2,6-диамино-4-трифторметилфенокси)пирролидин-1-карбоновой кислоты с пиримидин-4,6-дикарбоновой кислотой в присутствии 1-[(3-(диметиламино)пропил)]-3-этилкарбодиимида гидрохлорида с получением бис-{[3-амино-2-((R)-1-(трет-бутоксикарбонил)пирролидин-3-илокси)-5-трифторметил-фенил]амида} пиримидин-4,6-дикарбоновой кислоты;
d) взаимодействие бис-{[3-амино-2-((R)-1-(трет-бутоксикарбонил)пирролидин-3-илокси)-5-трифторметил-фенил]амида} пиримидин-4,6-дикарбоновой кислоты с ({[(трет-бутоксикарбонил)амино][(трет-бутоксикарбонил)имино]метил}амино)пентановой кислотой в присутствии фосфора оксихлорида с получением бис-{[3-(5-({[(трет-бутоксикарбонил)амино][трет-бутоксикарбонил)имино]метил}амино)пентаноиламино)-2-((R)-1-(трет-бутоксикарбонил)пирролидин-3-илокси)-5-трифторметил-фенил]амида} пиримидин-4,6-дикарбоновой кислоты;
е) снятие защитных групп бис-{[3-(5-({[(трет-бутоксикарбонил)амино][трет-бутоксикарбонил)имино]метил}амино)-пентаноиламино)-2-((R)-1-(трет-бутоксикарбонил)пирролидин-3-илокси)-5-трифторметил-фенил]амида} пиримидин-4,6-дикарбоновой кислоты с получением неочищенного бис-{[3-(5-гуанидино-пентаноиламино)-2-((R)-пирролидин-3-илокси)-5-трифторметил-фенил]амид}тетрагидрохлорида пиримидин-4,6-дикарбоновой кислоты; и
f) очистку неочищенного бис-{[3-(5-гуанидино-пентаноиламино)-2-((R)-пирролидин-3-илокси)-5-трифторметил-фенил]амид}тетрагидрохлорида пиримидин-4,6-дикарбоновой кислоты с помощью хроматографии с обращенной фазой.
В некоторых вариантах осуществления изобретения в b) катализатор переходного металла представляет собой Pd/C, и спирт представляет собой этанол. В частности, этот способ описан ниже более детально в Примере 2.
Настоящее изобретение также предоставляет другие дополнительные способы получения Соединения A, включающие:
a) депротонирование трет-бутилового эфира (R)-3-гидроксипирролидин-1-карбоновой кислоты и далее взаимодействие полученного соединения с 2-хлор-1,3-динитро-5-трифторметилбензолом с получением трет-бутилового эфира (R)-3-(2,6-динитро-4-трифторметилфенокси)пирролидин-1-карбоновой кислоты;
b) восстановление трет-бутилового эфира (R)-3-(2,6-динитро-4-трифторметилфенокси)пирролидин-1-карбоновой кислоты в присутствии спирта, катализатора переходного металла и водорода с получением трет-бутилового эфира (R)-3-(2,6-диамино-4-трифторметилфенокси)пирролидин-1-карбоновой кислоты;
c) сочетание трет-бутилового эфира (R)-3-(2,6-диамино-4-трифторметилфенокси)пирролидин-1-карбоновой кислоты с пиримидин-4,6-дикарбоновой кислотой в присутствии 1-[(3-(диметиламино)пропил)]-3-этилкарбодиимида гидрохлорида с получением бис-{[3-амино-2-((R)-1-(трет-бутоксикарбонил)пирролидин-3-илокси)-5-трифторметил-фенил]амида} пиримидин-4,6-дикарбоновой кислоты;
d) взаимодействие бис-{[3-амино-2-((R)-1-(трет-бутоксикарбонил)пирролидин-3-илокси)-5-трифторметил-фенил]амида} пиримидин-4,6-дикарбоновой кислоты с N-Cbz кислотой в присутствии тионила хлорида;
e) восстановление полученного соединения d) в присутствии спирта, катализатора переходного металла и водорода;
f) взаимодействие полученного соединения e) с ди-Boc пиразолом; и
g) снятие защитных групп полученного соединения f) с получением Соединения A.
В некоторых вариантах осуществления изобретения в b) и e) катализатор переходного металла представляет собой Pd/C, и спирт представляет собой этанол. В частности, этот способ описан ниже более детально в Примере 3.
Специалист в данной области сумеет заменить приемлемые реагенты на реагенты, перечисленные в способах, описанных в данном описании, с получением Соединения A, а также дополнительных соединений Формулы I.
Настоящее изобретение также предоставляет способы получения фармацевтически приемлемой соли Соединения A, включающие:
а) взаимодействие (R)-(-)-N-Boc-3-пирролидинола с сильным основанием с получением смеси; далее взаимодействие смеси с 2-хлор-5-(трифторметил)-1,3-динитробензолом с получением соединения, имеющего Формулу II
b) взаимодействие соединения Формулы II со спиртом и катализатором переходного металла в присутствии водорода с получением соединения Формулы III
c1) добавление соединения Формулы III и пиримидин-4,6-дикарбоновой кислоты в смесь 2-хлор-4,6-диметокси-1,3,5-триазина и N-метилморфолина с получением соединения Формулы IV

c2) добавление соединения Формулы III и пиримидин-4,6-дикарбоновой кислоты в смесь 1-этил-3-[3-(диметиламино)пропил]карбодиимида гидрохлорида (EDC1) и безводного пиридина с получением соединения Формулы IV
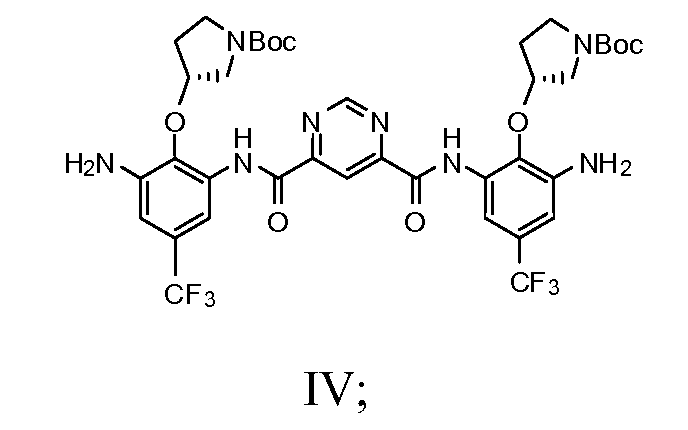
d) добавление соединения Формулы IV с N-Cbz кислотой в раствор, включающий безводный пиридин, диметиламинопропиламин и любой один из тионила хлорида, POCl3, (EtO)2POCl или оксалила хлорида с получением соединения Формулы Va
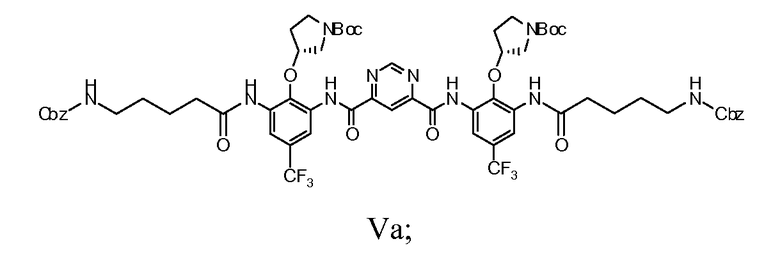
e) гидрогенолиз группы Cbz соединения Формулы Va с получением соединения Формулы VI
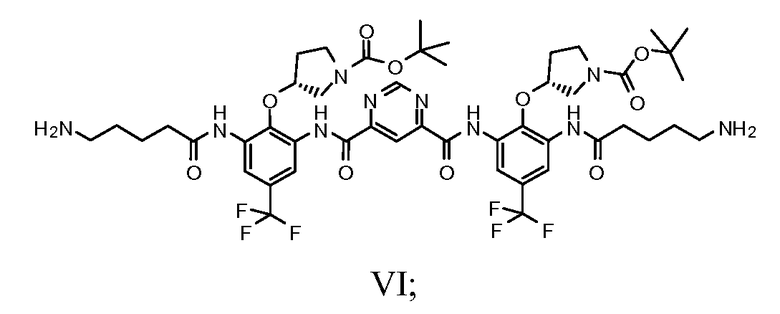
f) защиту соединения Формулы VI с получением соединения Формулы VII
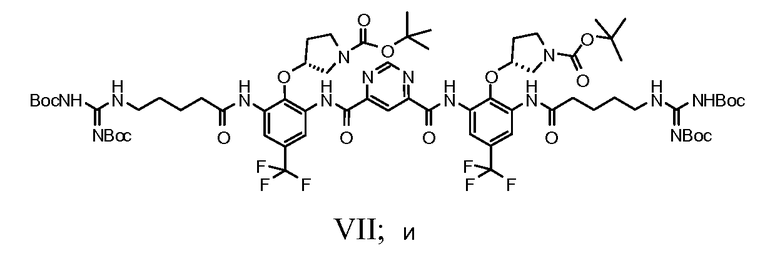
g) снятие защитных групп соединения Формулы VII с получением фармацевтически приемлемой соли Соединения A.
Получение соединений Формулы I может включать защиту или снятие защиты различных химических групп. Необходимость защиты и снятия защиты и выбор соответствующих защитных групп могут быть легко определены специалистом в данной области. Химия защитных групп может быть найдена, например, в работе T. W. Greene и P. G. M. Wuts, Protective Groups in Organic Synthesis, 3rd Ed. Wiley & Sons, Inc. New York (1999), которая включена в данное описании посредством ссылки во всей своей полноте.
Настоящее изобретение также предоставляет способы ингибирования роста микроба, включающие контактирование микроба с одним или несколькими соединениями, описанными выше, или их фармацевтически приемлемой солью. В некоторых вариантах осуществления изобретения соединение Формулы I может действовать как антисептическое средство для очищения поверхностей, таких как, например, в кухнях и ванных комнатах. В этих вариантах осуществления изобретения соединение Формулы I может быть получено для такого использования с помощью методик, хорошо известных специалисту в данной области.
Настоящее изобретение также предоставляет способы лечения млекопитающего, страдающего микробной инфекцией, включающие введение млекопитающему, нуждающемуся в этом, противомикробного эффективного количества любого из соединений, описанных выше, или его фармацевтически приемлемой соли. В некоторых вариантах осуществления изобретения млекопитающее может быть предварительно диагностировано на наличие микробной инфекции до лечения. В некоторых вариантах осуществления изобретения официальный диагноз не может быть сделан; в таких вариантах осуществления изобретения млекопитающее может подозреваться на наличие микробной инфекции, для которой лечение признано как желательное.
В одном варианте осуществления изобретения «лечение» или «обработка» относятся к ослаблению микробной инфекции или по меньшей мере ее одного заметного симптома; или к ослаблению по меньшей мере одного измеряемого физического параметра, не обязательно замечаемого пациентом; или к ингибированию прогрессирования микробной инфекции; или к задерживанию начала развития микробной инфекции.
В некоторых вариантах осуществления изобретения микроб представляет собой или микробная инфекция вызывается грамотрицательным аэробом, грамположительным аэробом, грамотрицательным анаэробом, грамположительным анаэробом или дрожжами. В некоторых вариантах осуществления изобретения грамотрицательный аэроб выбран из, но не ограничиваясь ими, Escherichia coli, Citrobacter freundii, Citrobacter diverus, Citrobacter koseri, Enterobacter cloacae, Enterobacter faecalis, Klebsiella pneumonia, Klebsiella oxytoca, Morganella morganii, Providencia stuartii, Proteus vulgaris, Proteus mirabilis, Serratia marcescens, Acinetobacter haemolyticus, Acinetobacter junii, Acinetobacter lwoffii, Haemophilus influenzae, Stenotrophomonas maltophilia и Pseudomonas aeruginosa. В некоторых вариантах осуществления изобретения грамположительный аэроб выбран из, но не ограничиваясь ими, Enterococcus faecalis, Enterococcus faecium, Mycobacterium tuberculosis, Staphylococcus aureus, Staphylococcus pneumoniae, Staphylococcus epidermidis, Staphylococcus saprophyticus, Staphylococcus colmii, Staphylococcus sciuri, Staphylococcus warneri, Streptococcus agalactiae, Streptococcus pyogenes, Streptococcus anginosus, Streptococcus mitis и Streptococcus oralis. В некоторых вариантах осуществления изобретения грамотрицательный анаэроб представляет собой Bacteroides fragilis. В некоторых вариантах осуществления изобретения грамположительный анаэроб представляет собой Clostridium difficile или Clostridium perfringens. В некоторых вариантах осуществления изобретения микобактерия представляет собой Mycobacterium tuberculosis, Mycobacterium bovis, Mycobacterium africanum, Mycobacterium canetti или Mycobacterium microti. В некоторых вариантах осуществления изобретения дрожжи выбраны из, но не ограничиваясь ими, Candida albicans и Candida krusei.
В некоторых вариантах осуществления изобретения микроб представляет собой резистентный к антибиотику штамм бактерий, такой как штамм бактерий, перечисленный в Примерах ниже.
Соединения Формулы I или их фармацевтически приемлемая соль и композиции, включающие их, могут быть введены различными способами, такими как, например, инфузия или болюсная инъекция, и могут быть введены вместе с другим биологически активным агентом, таким как другой антибиотик. Введение может быть системным или местным. Известны различные системы доставки, например инкапсулирование в липосомы, микрочастицы, микрокапсулы, капсулы, и т.д., и они могут быть использованы для введения соединения Формулы I. Пути введения включают, но не ограничиваясь ими, внутридермальный, внутримышечный, внутрибрюшинный, внутривенный, подкожный, интраназальный, эпидуральный, оральный, подъязычный, интраназальный, интрацеребральный, интравагинальный, трансдермальный, ректальный, легочный, путем ингаляции или топически, особенно в уши, нос, глаза или на кожу. В некоторых вариантах осуществления изобретения приемлемые пути введения включают внутривенный, топический или подкожный. Желательный путь введения остается на усмотрение практикующего врача и будет зависеть, частично, от локализации микробной инфекции и состояния здоровья млекопитающего или человека, которых подвергают лечению. В большинстве случаев введение может приводить к высвобождению соединений Формулы I в кровяное русло.
В некоторых вариантах осуществления изобретения может быть желательно применять одно или несколько соединений Формулы I или их фармацевтически приемлемую соль местно в область, нуждающуюся в лечении. Это может быть достигнуто, например, и без ограничения путем местной инфузии в процессе хирургической операции, топического нанесения, например, вместе с раневой повязкой после хирургической операции, путем инъекции, посредством катетера, посредством суппозитория или посредством имплантата, где имплантат представляет собой пористый, не пористый или гелеобразный материал, включая мембраны, такие как сиаластиковые мембраны или волокна.
Количество соединения Формулы I или его фармацевтически приемлемой соли, которое будет эффективно для лечения конкретной микробной инфекции, будет зависеть от природы нарушения или патологического состояния и может быть определено с помощью стандартной клинической методики. Дополнительно при необходимости могут быть применены анализы in vitro или in vivo, чтобы помочь идентифицировать оптимальный диапазон доз. Точная доза для применения в композициях будет также зависеть от пути введения и тяжести инфекции, и должно быть принято решение в соответствии с медицинской оценкой практикующего врача и состояния каждого пациента. Однако приемлемый диапазон доз для введения составляет обычно от примерно 0,001 миллиграмма до примерно 200 миллиграммов на килограмм массы тела. В некоторых вариантах осуществления изобретения доза составляет от примерно 0,01 миллиграмма до примерно 70 миллиграммов на килограмм массы тела, или от примерно 0,1 миллиграмма до примерно 50 миллиграммов на килограмм массы тела, или от примерно 0,5 миллиграммов до примерно 20 миллиграммов на килограмм массы тела, или от примерно 1 миллиграмма до примерно 10 миллиграммов на килограмм массы тела. В некоторых вариантах осуществления изобретения доза составляет примерно 5 миллиграммов на килограмм массы тела. Количество доз, описанных в данном описании, относится к общему введенному количеству; то есть, если вводится более чем одно соединение Формулы I, дозы соответствуют общему количеству введенных соединений Формулы I. Композиции могут содержать от 10% до 95% активного ингредиента по массе. Эффективные дозы могут быть экстраполированы по кривым доза-ответ, полученным in vitro или на животных модельных тест-системах. Такие животные модели и системы хорошо известны в данной области.
Настоящее изобретение также предоставляет одно или несколько соединений, описанных выше, или их фармацевтически приемлемую соль, или фармацевтическую композицию, включающую одно или несколько соединений, описанных выше, для лечения микробной инфекции.
Настоящее изобретение также предоставляет одно или несколько соединений, описанных выше, или их фармацевтически приемлемую соль, или фармацевтическую композицию, включающую одно или несколько соединений, описанных выше, для использования при изготовлении лекарственного средства для лечения микробной инфекции.
Настоящее изобретение также предоставляет применение одного или нескольких соединений, описанных выше, или их фармацевтически приемлемой соли, или фармацевтической композиции, включающей одно или несколько соединений, описанных выше, для ингибирования роста микроба.
Настоящее изобретение также предоставляет применение одного или нескольких соединений, описанных выше, или их фармацевтически приемлемой соли, или фармацевтической композиции, включающей одно или несколько соединений, описанных выше, для лечения микробной инфекции млекопитающего.
Для того чтобы изобретение, раскрытое в данном описании, могло быть лучше понято, ниже приводятся примеры. Следует понимать, что эти примеры служат только для иллюстративных целей и не толкуются как ограничивающие изобретение любым образом. В этих примерах выполнили реакции молекулярного клонирования и другие стандартные методики на основе рекомбинантной ДНК в соответствии с методами, описанными у Maniatis с сотр., Molecular cloning - A Laboratory Manual, 2nd ed., Cold Spring Harbor Press (1989), с использованием коммерчески пригодных реагентов, за исключением случаев, когда указано иное.
Примеры
Вкратце, результаты, полученные на основании примеров, приведенных ниже, указывают, что Соединение A является активным против Staphylococci spp. и других грамположительных и грамотрицательных организмов. Например, выполнен скрининг чувствительности против 150 изолятов S. aureus и отрицательных по коагулазе стафилококков с определенной антибактериальной чувствительностью к другим противомикробным агентам. В общем, были получены значения MIC90 от 0,5 до 2,0 мкг/мл при скрининге 150 организмов стафилококков, и отсутствовала чувствительность фенотипов к другим антибиотикам. Серийный пассаж метициллин-чувствительных (MSSA ATCC 29213) и резистентных (MRSA ATCC 33591) штаммов S. aureus при концентрациях 0,5x MIC в течение 17 пассажей не вызвал какого-либо изменения в значениях MIC. В общем, Соединение A являлось бактерицидным в отношении время-эрадикация в диапазоне от 30 минут до 6 часов.
Соединение A являлось эффективным in vivo на мышиной модели нагрузки на бедренную кость против MSSA 29213 и MRSA 33591 и на мышиной модели перитонита/сепсиса против MSSA 27660. На мышиной модели нагрузки на бедренную кость с использованием MSSA 27660 Соединение A обеспечивало снижение через 24 часа после инфицирования вплоть до 410 КОЕ/бедренная кость относительно необработанных инфицированных мышей в дозах, которые хорошо переносились в повторных исследованиях токсичности дозы. Таким образом, устойчивую эффективность против MSSA и MRSA наблюдали на мышиной модели нагрузки на бедренную кость и против MSSA на крысиной модели нагрузки на бедренную кость и мышиной модели перитонита. Соединение A являлось стабильным в присутствии плазмы и изолированных гепатоцитов множества видов.
Соединение A являлось в большей степени толерантным в исследованиях острой токсичности, когда вводилось посредством IV [внутривенной] инфузии. MTD [максимальная толерантная доза] (IV болюсное [струйное] введение) для Соединения A у мыши (30 мг/кг) являлась значимо более высокой, чем статичная эффективная доза на модели нагрузки на бедренную кость (2-4 мг/кг).
Соединение A в настоящее время находится в Фазе 1 клинических испытаний на человеке для разработки как IV пан-стафилококковый агент.
Пример 1: синтез Соединения A
Стадия 1:
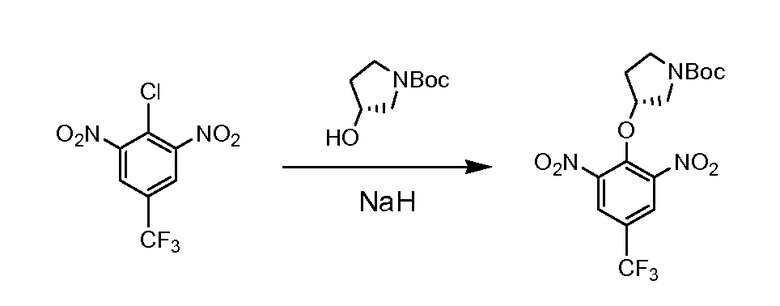
Натрия гидрид (1,12 г, 60% в минеральном масле, 28 мМ) добавляли по частям в безводный DMF (24 мл) раствор (R)-(-)-N-Boc-3-пирролидинола (5,0 г, 27,6 мМ) при комнатной температуре. Полученную смесь перемешивали дополнительно 15 минут. Эту смесь затем добавляли по каплям в DMF (20 мл) раствор 2-хлор-5-(трифторметил)-1,3-динитробензола (7,45 г, 27,6 мМ) при 0°C. Раствор темно-красного цвета перемешивали при комнатной температуре в течение 4 часов. Реакцию останавливали ледяной водой и экстрагировали этилацетатом. Органический слой промывали солевым раствором и водой и сушили над Na2SO4. После удаления растворителя остаток очищали с помощью колонны для отгона легких фракций (этилацетат/гексаны = ¼ объем/объем). Выход составил 54%.
Стадия 2:

(R)-трет-бутил-3-(4-трифторметил)-2,6-динитрофенокси)пирролидин-1-карбоксилат (4,84 г, 9,8 мМ) и Pd/C (0,78 г, 10% на углерод) и этанол (140 мл) помещали в колбу Парра. Смесь выпаривали под водородом три раза и перемешивали при 40 кПа водорода при комнатной температуре в течение ночи. Смесь фильтровали через целит. Осадок на фильтре промывали дважды этанолом (2×20 мл). Фильтрат выпаривали под вакуумом. Получили серовато-белое твердое вещество и использовали как таковое для последующей реакции. Выход составил 100%.
Стадия 3:
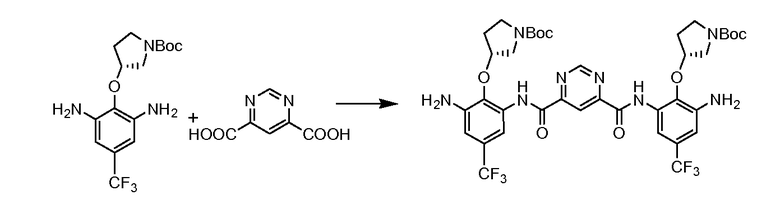
2-Хлор-4,6-диметокси-1,3,5-триазин (5,97 г, 34 мМ) перемешивали в безводном THF (200 мл). Добавляли N-метилморфолин (7,5 мл, 68 мМ). Полученную смесь перемешивали при комнатной температуре в течение 30 минут. Затем добавляли (R)-трет-бутил-3-(2,6-диамино-4-(трифторметил)фенокси)пирролидин-1-карбоксилат (10,84 г, 30 мМ) и пиримидин-4,6-дикарбоновую кислоту (2,48 г, 14,8 мМ). Смесь перемешивали при комнатной температуре в течение 24 часов. Растворитель выпаривали полностью в вакууме. Добавляли воду (250 мл) и смесь перемешивали в течение 4 часов. После фильтрования желтый осадок промывали водой (3×100 мл) и перемешивали в воде (250 мл) в течение 4 часов. Процедуру фильтрования и промывания повторяли дважды. Твердое вещество сушили на воздухе и перемешивали в дихлорметане (20 мл) в течение 30 минут с последующей ультразвуковой обработкой в течение 1 часа. После фильтрования желтый осадок на фильтре быстро промывали холодным дихлорметаном (2×10 мл). Продукт (10,0 г, выход: 79,1%) использовали как таковой для последующей реакции.
Стадия 4:
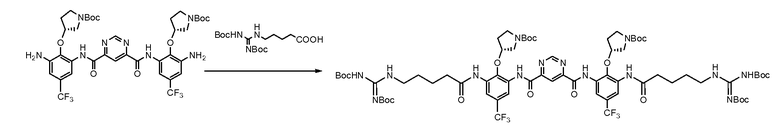
Исходный материал (6,5 г, 7,6 мМ), N-Boc-гуанидин масляной кислоты (10,9 г, 30,4 мМ) перемешивали в безводном пиридине (40 мл) при 0°C. POCl3 (2,78 мл, 30,4 мМ) в пиридине (4 мл) добавляли по каплям. Полученную смесь перемешивали при 0°C в течение 1,5 часов. Реакционную смесь выпаривали под вакуумом. Воду (140 мл) добавляли к остатку. Смесь экстрагировали с использованием этилацетата (260 мл). Органический слой промывали солевым раствором (100 мл) и сушили над Na2SO4. После выпаривания остаток очищали с использованием колонны (элюент: этилацетат/гексаны/дихлорметан = 1/1/1, объем/объем/объем, затем 2%~4% метанол в дихлорметане). Выход составил 29,1%. Rf был таким же, как в стандартном образце, который охарактеризовали с помощью ЯМР.
Стадия 5:
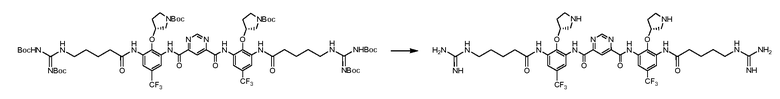
Исходный материал (3,4 г, 2,3 мМ) перемешивали в 4 н. HCl (34 мл) при комнатной температуре в течение ночи. Растворитель удаляли под вакуумом. Остаток титровали в эфире. Твердое вещество фильтровали и очищали с помощью C18 с обращенной фазой C18 колонны. В качестве продукта получили светло-желтое твердое вещество с чистотой 98% (ВЭЖХ); LC-MC (M+1): 937. Выход: 51%.
Пример 2: синтез Соединения A
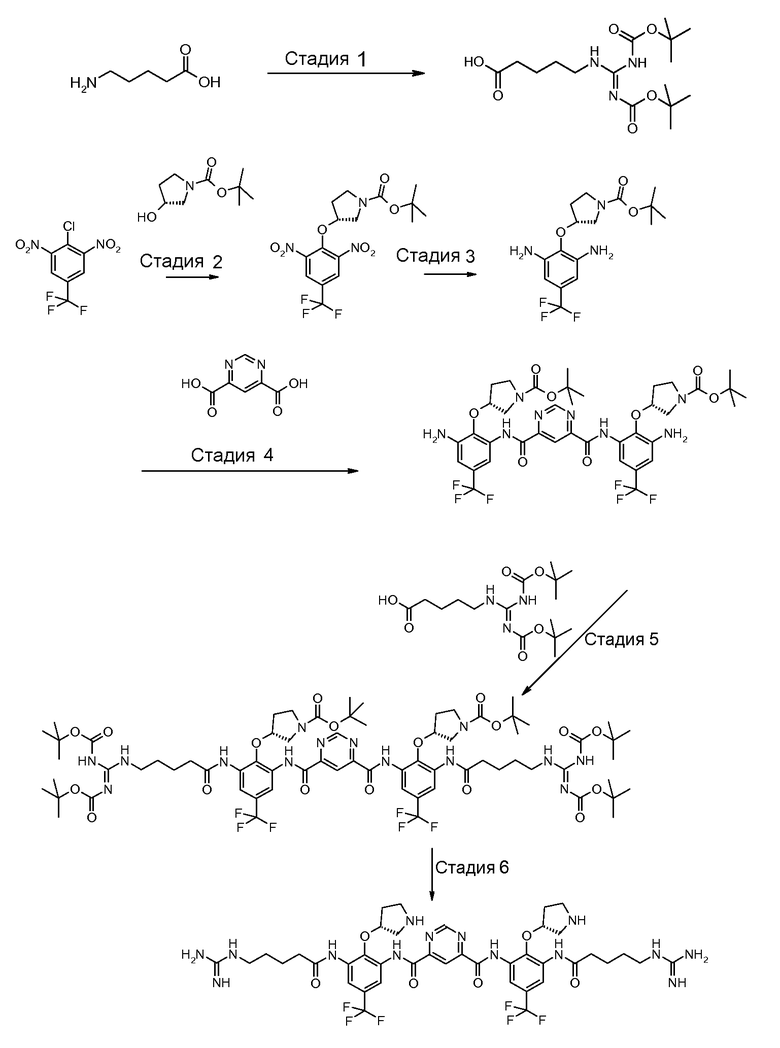
Стадия 1: трет-бутиловый эфир (R)-3-гидроксипирролидин-1-карбоновой кислоты депротонируют с использованием калия трет-бутоксида (KOtBU) в тетрагидрофуране (THF). Полученный анион взаимодействует с 2-хлор-1,3-динитро-5-трифторметилбензолом в трет-бутилметиловом эфире (MTBE)/THF. Когда реакция заканчивается, реакционную смесь гасят водой и разделяют с использованием добавочного MTBE. Органический слой промывают солевым раствором и водой и концентрируют на ротационном испарителе. Твердый концентрат повторно растворяют в метаноле и повторно осаждают с водой. Полученный осадок фильтруют и сушат с предоставлением трет-бутилового эфира (R)-3-(2,6-динитро-4-трифторметилфенокси)пирролидин-1-карбоновой кислоты, который может быть использован в следующей стадии без дальнейшей очистки.
Стадия 2: продукт стадии 1 растворяют в метаноле и гидрогенизируют при 100-200 кПа и 30-50°C в присутствии 10% Pd/C до тех пор, когда восстановление будет считаться законченным с помощью ВЭЖХ. Реакционную смесь фильтруют через цЦелит. Фильтрат концентрируют и сушат с предоставлением трет-бутилового эфира (R)-3-(2,6-диамино-4-трифторметилфенокси)пирролидин-1-карбоновой кислоты, который может быть использован в следующей стадии без дальнейшей очистки.
Стадия 3: продукт стадии 2 соединяют с пиримидин-4,6-дикарбоновой кислотой, в приблизительном отношении 2 М диамин:1 М диацид в присутствии 1-[(3-(диметиламино)пропил)]-3-этилкарбодиимида гидрохлорида (EDCI) в пиридине под инертной атмосферой при температуре окружающей среды. Когда реакция заканчивается, реакционную смесь разводят в воде. Полученный осадок выделяют и повторно растворяют в MTBE. Раствор MTBE промывают водой, 0,2 н. HCl и солевым раствором, сушат над безводным натрия сульфатом, выделяют и разводят в гептане. Полученный осадок изолируют посредством фильтрования и сушат с предоставлением бис-{[3-амино-2-((R)-1-(трет-бутоксикарбонил)пирролидин-3-илокси)-5-трифторметил-фенил]амида} пиримидин-4,6-дикарбоновой кислоты, который может быть использован в следующей стадии без дальнейшей очистки.
Стадия 4: продукт стадии 3 взаимодействует с 2,5-3 молярными эквивалентами ({[(трет-бутоксикарбонил)амино][(трет-бутоксикарбонил)имино]метил}амино)пентановой кислоты в пиридине в присутствии фосфора оксихлорида при температуре примерно от -5 до -10°C. Реакцию гасят водой при температуре 15°C. Супернатант выделяют из аморфного осадка, который повторно растворяют в MTBE, промывают водой и солевым раствором, сушат над безводным натрия сульфатом, выделяют и разводят в гептане. Полученный осадок изолируют посредством фильтрования и сушат с предоставлением бис-{[3-(5-({[(трет-бутоксикарбонил)амино][трет-бутоксикарбонил)имино]метил}амино)пентаноиламино)-2-((R)-1-(трет-бутоксикарбонил)пирролидин-3-илокси)-5-трифторметил-фенил]амида} пиримидин-4,6-дикарбоновой кислоты, который используется в следующей стадии без дальнейшей очистки.
Стадия 5: продукт стадии 4 лишают защитных групп (удаляя шесть трет-бутоксикарбонильных групп) с использованием 4М HCl/1,4-диоксан в муравьиной кислоте при температуре окружающей среды. Реакционную смесь разводят в 1,4-диоксане. Полученный осадок фильтруют, промывают в 1,4-диоксане и сушат с предоставлением неочищенного бис-{[3-(5-гуанидино-пентаноиламин)-2-((R)пирролидин-3-илокси)-5-трифторметил-фенил]-амид}тетрагидрохлорида пиримидин-4,6-дикарбоновой кислоты (неочищенное Соединение A). Неочищенный продукт далее очищают путем повторного осаждения из раствора метанола с использованием THF (50°C до температуры окружающей среды) и/или повторно осаждают из воды/раствора метанола с использованием THF при температуре окружающей среды.
Стадия 6 (хроматографическая очистка): конечная очистка Соединения A достигается путем хроматографии с обращенной фазой (ОФ-ВЭЖХ) с использованием фазы YMC ODS-AQ, 50 микрон, 120 ангстрем, суспензию упаковывали в колонку с динамическим осевым сжатием ProChrom. Мобильной фазой является градиент растворителя B в растворителе A, где растворитель A является водой с 0,05% трифторуксусной кислоты (TFA) и растворитель B является ацетонитрилом с 0,05% TFA. Фракции, содержащие очищенный продукт, концентрируют с помощью ротационного испарителя с предоставлением Соединения A как соли трифторацетата. Конечную форму хлористоводородной соли повторно получают путем пропускания водно/метанольного раствора соли трифторацетата через ионообменную колонку Dowex 1×2-400 (Cl-форма), собирают API-содержащий элюат, концентрируют и сушат.
Вещество нерасфасованной лекарственной формы Соединения A хранят при 2-8°C, защищая от света и воздуха, в желтых контейнерах HDPE или в сумках из двойного полиэтилена в картонном барабане.
Пример 3: синтез Соединения A
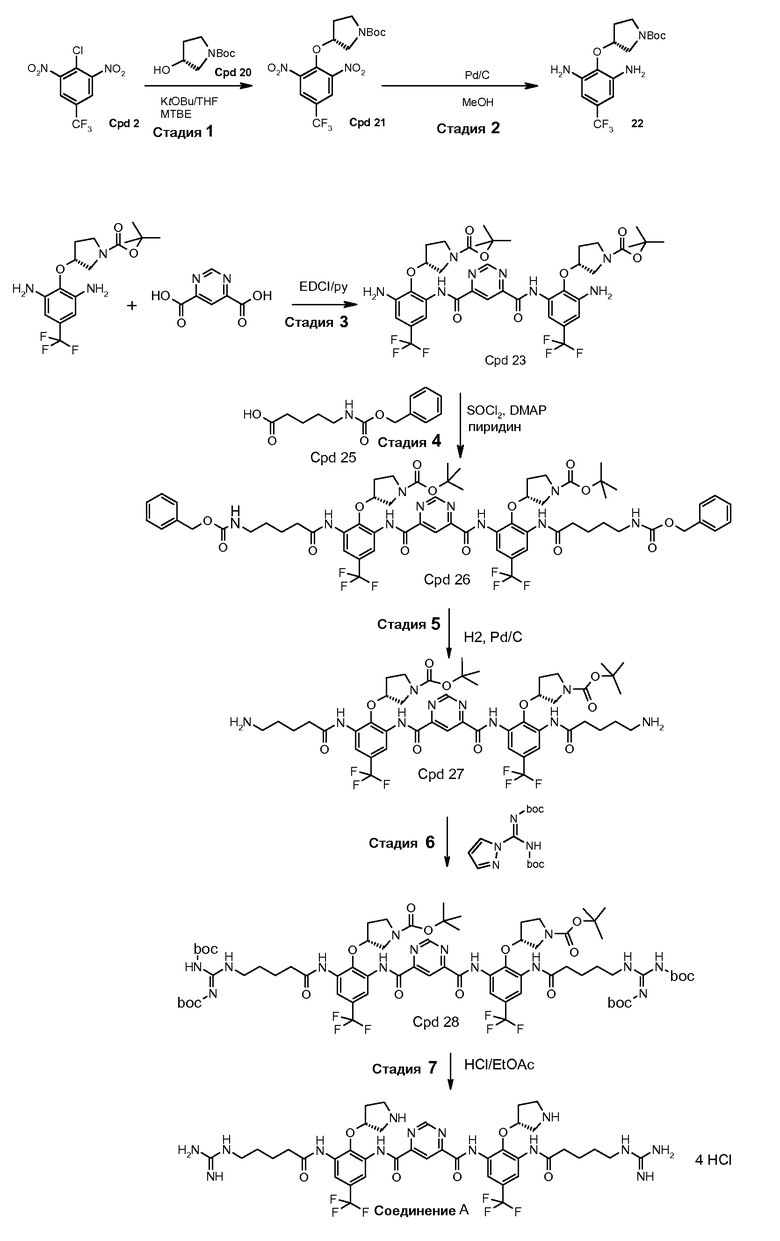
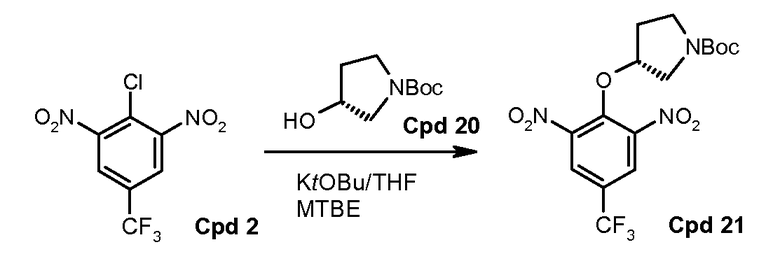
Стадия 1: трет-бутиловый эфир (R)-3-гидроксипирролидин-1-карбоновой кислоты (соединение 20) депротонируют с использованием калия трет-бутоксида (KOtBU) в тетрагидрофуране (THF). Полученный анион реагирует с 2-хлор-1,3-динитро-5-трифторметилбензолом (соединение 2) в трет-бутилметиловом эфире (MTBE)/THF. Когда реакция заканчивается, реакционную смесь гасят водой и разделяют с использованием добавочного MTBE. Органический слой промывают солевым раствором и водой и концентрируют на ротационном испарителе. Твердый концентрат повторно растворяют в метаноле и повторно осаждают с водой. Полученный осадок фильтруют и сушат с предоставлением трет-бутилового эфира (R)-3-(2,6-динитро-4-трифторметилфенокси)пирролидин-1-карбоновой кислоты, который может быть использован в следующей стадии без дальнейшей очистки. Эта реакция будет выполняться в масштабе с использованием 4,2 кг соединения 2.
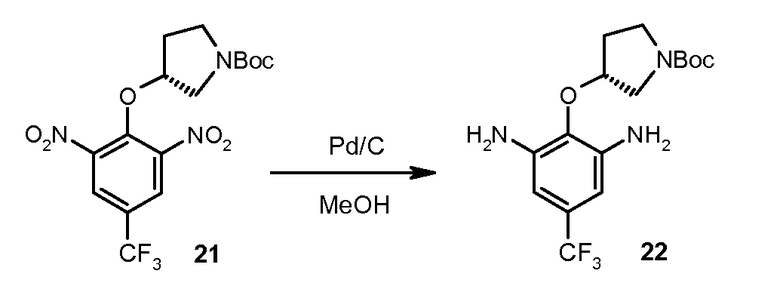
Стадия 2: соединение 21 растворяют в метаноле и гидрогенизируют при 100-200 кПа и 30-50°C в присутствии 10% Pd/C до тех пор, когда восстановление будет считаться законченным с помощью ВЭЖХ. Реакционную смесь фильтруют через целит. Фильтрат концентрируют и сушат с предоставлением трет-бутилового эфира (R)-3-(2,6-диамино-4-трифторметилфенокси)пирролидин-1-карбоновой кислоты (соединение 22) с ВЭЖХ чистотой 92,2%. Реакцию выполняют в четыре серии в масштабе 1,64 кг соединения 21 для каждой серии.
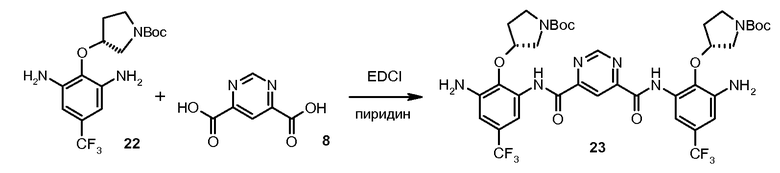
Стадия 3: Соединение 22 соединяют с пиримидин-4,6-дикарбоновой кислотой (соединение 8), в приблизительном отношении 2 М диамин:1 М диацид в присутствии 1-[(3-(диметиламино)пропил)]-3-этилкарбодиимида гидрохлорида (EDCI) в пиридине под инертной атмосферой при температуре окружающей среды. Когда реакция заканчивается, реакционную смесь разводят водой. Полученный осадок выделяют и повторно растворяют в MTBE. Раствор MTBE промывают водой, 0,2 н. HCl и солевым раствором, сушат над безводным натрия сульфатом, выделяют и разводят в гептане. Полученный осадок выделяют посредством фильтрования и сушат с предоставлением бис-{[3-амино-2-((R)-1-(трет-бутоксикарбонил)пирролидин-3-илокси)-5-трифторметил-фенил]амида} пиримидин-4,6-дикарбоновой кислоты (соединение 23), который может быть использован в следующей стадии без дальнейшей очистки. Реакцию выполняют в масштабе с использованием 3,15 кг соединения 22.
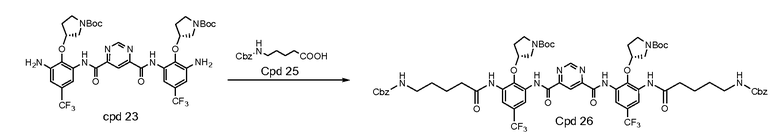
Стадия 4: раствор 3,66 г DMAP в 60 мл безводного пиридина охлаждали до 0°C в ванне со льдом. Медленно добавляли 3,60 г тионила хлорида. Полученный раствор перемешивали в течение 10 минут. Исходный материал N-Cbz кислота (7,53 г, 30 мМ), соединение (Cpd) 23 (8,54 г, 10 мМ) добавляли в раствор соответственно. Полученную смесь перемешивали при комнатной температуре (RT) в течение 4 часов. Добавляли воду (500 мл). После энергичного перемешивания смеси при комнатной температуре в течение 2 часов твердое вещество фильтровали и промывали 250 мл воды. Твердое вещество растворяли в этилацетате (300 мл). Органический слой промывали 10% раствором лимонной кислоты (100 мл) и солевым раствором (100 мл) и сушили над Na2SO4. После выпаривания остаток растворяли в 40 мл DCM, затем добавляли 250 мл гексана. Осадок собирали и сушили под вакуумом. Получили 13,20 г продукта с 95% чистотой. Выход: 100%.
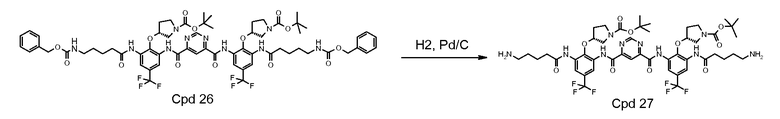
Стадия 5: соединение 26 (13,20 г) растворяли в MeOH с 2 эквив. 1 н. HCl и добавляли 1,0 г катализатора Pd/C (10%). Реакционную смесь вводили в гидрогенизатор Парра и встряхивали в течение 2 часов при 60 кПа водорода. Если LCMASS показывала отсутствие прогресса, добавляли еще 1,0 г катализатора. Реакционную смесь вводили в гидрогенизатор Парра и встряхивали в течение 3 часов при 60 кПа водорода. Смесь фильтровали через целит для удаления катализатора. Фильтрат концентрировали до сухого состояния на ротационном испарителе при 30°C. Получили 11,50 г продукта с 95% чистотой. Выход: 100%.
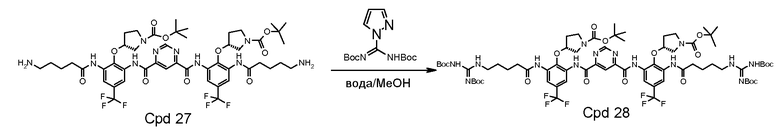
Стадия 6: соединение 27 (11,50 г, 10 мМ) растворяли в 60 мл метанола и DCM (1:1). Затем добавляли 4,04 г триэтиламина (40 мМ). Добавляли di-Boc пиразол 9,3 граммов (30 мМ) и полученную смесь перемешивали при комнатной температуре в течение 1 часа. После удаления 95% растворителя добавляли 300 мл воды и смесь энергично перемешивали в течение 2 часов. Твердое вещество фильтровали и промывали 300 мл воды. Твердое вещество растворяли в 300 мл этилацетата и сушили над Na2SO4. После выпаривания растворителя твердое вещество растворяли в 40 мл DCM, затем 500 мл гексана использовали для осаждения продукта. Твердое вещество собирали и сушили под вакуумом. Получили 13,0 граммов продукта с 85% выходом (90% чистота).
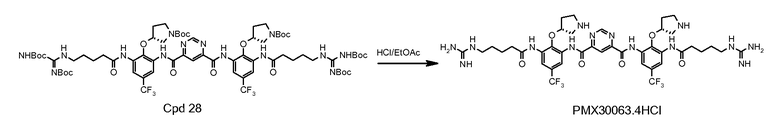
Стадия 7: соединение 28 (1,5 г) очищали на колонке с 80 г силикагеля с использованием градиента 10-88% EtOAc в DCM. Фракции с чистотой выше примерно 95% собирали, выпаривали под вакуумом и сушили. Выход составил 50-60%. Соединение 28 с 95% чистотой (0,3 г) растворяли и перемешивали в этилацетате (3 мл) при комнатной температуре (22°C) под аргоном. Газом HCl барботировали раствор в течение 20 минут. Цвет раствора изменялся на темно-желтый при проведении барботирования. Твердое вещество начинало дробиться в течение 15 минут. Раствор перемешивали при комнатной температуре еще 1 час. Дополнительно 4 мл этилацетата вводили в реакционную смесь по причине потери этилацетата. Смесь барботировали газом HCl в течение 10 минут. Смесь перемешивали в течение 2,5 часов. Одну треть смеси фильтровали и промывали этилацетатом. Две трети смеси перемешивали при комнатной температуре в течение ночи. Затем в смесь добавляли 30 мл этилацетата. После фильтрования осадок на фильтре промывали этилацетатом дважды (2×140 мл) и сушили. Твердое вещество погружали в этилацетат (8 мл) и хранили в морозильной камере. Реакционный процесс выполняли в течение 4 часов. Перемешивание в течение ночи не дало значительного изменения. Чистота конечного продукта составляла 98% с одной главной примесью 1,2%.
Пример 4: синтез Соединения A
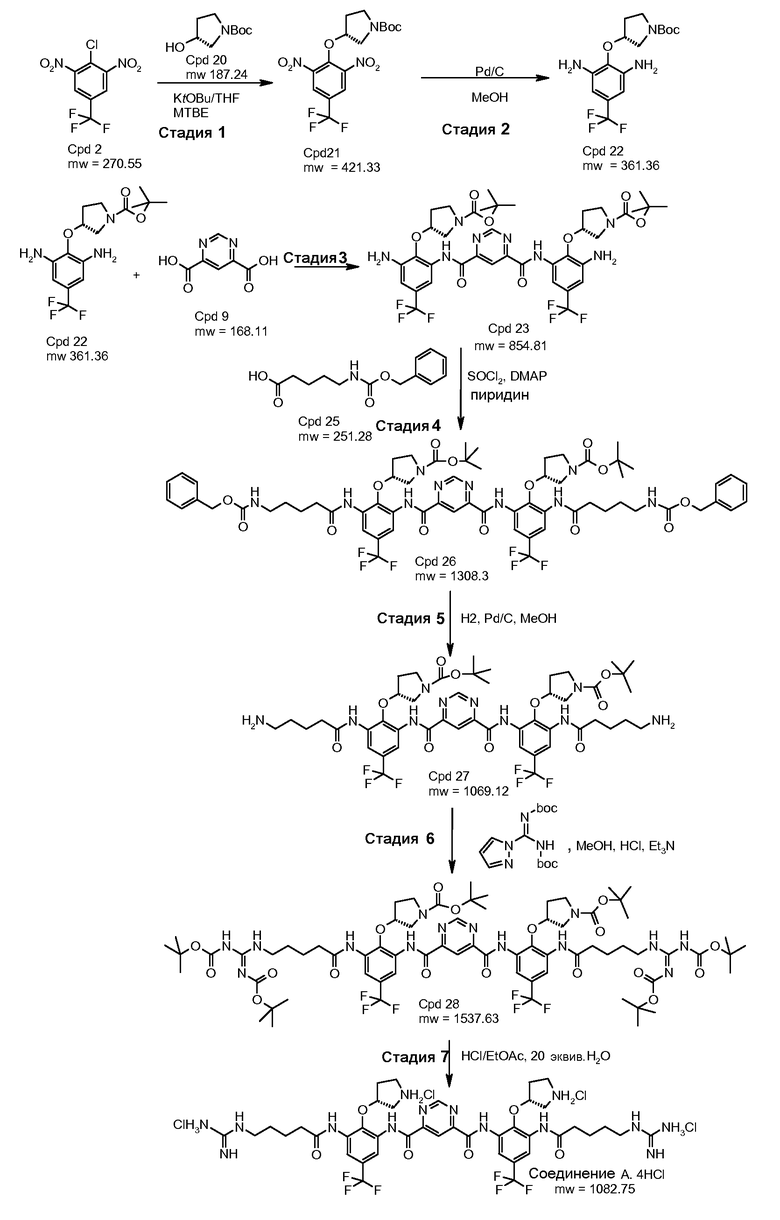
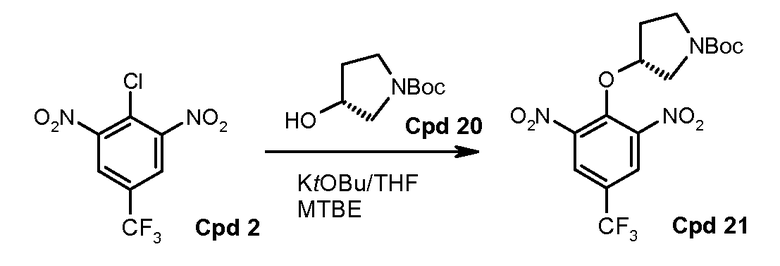
Стадия 1: в продутую азотом 4-горлую 12 л RBF добавляли 305,32 г соединения 2, 700 мл MTBF с перемешиванием и смесь охлаждали в ледяной/водной ванне. Соединение 20 (212,43 г) растворяли с калия трет-бутоксидом (1,31 л 1 М раствора в THF) с получением слегка мутной смеси. Эту смесь добавляли к раствору соединения 21 в RBF в течение 86 минут с перемешиванием, в то же время поддерживая внутреннюю температуру <9,0°C. Реакцию удаляли из холодной ванны через 30 минут и обеспечивали перемешивание при температуре окружающей среды в течение 15,5 часов. При перемешивании несколько маленьких кусочков льда добавляли для снижения температуры от 21,4°C до 18,4°C. Затем добавляли воду (1,5 л) MTBF (1,5 л) и смесь перемешивали в течение 10 минут. Смесь представляла собой расщепленную фазу и разделялась в 6-л делительной воронке. Водный слой повторно экстрагировали с MTBF (500 мл). Органические слои объединяли и промывали 2:1 вода/насыщенный солевой раствор (3×900 мл) и концентрировали в твердое вещество красновато/рыжеватого цвета при пониженном давлении. Это твердое вещество растворяли в 2,45 л MeOH и раствор переносили в 4-л колбу Эрленмейера. Затем добавляли воду (1 л) с перемешиванием частями с образованием густой суспензии. Смесь закрывали и помещали в холодильник (1-5°C) на 16 часов. Твердое вещество собирали путем фильтрования и сушили под вакуумом. Высушенный продукт представлял собой ярко-желтый порошок. Выход: 395,3 г, ВЭЖХ чистота 94%.
Калия трет-бутоксид может быть заменен, например, на любой алкоксид, натрия гидрид, калия гидрид или любое основание, которое может депротонировать гидроксил соединения 20. Соединение 2 может быть замещено в положении 2 любым галогенидом.
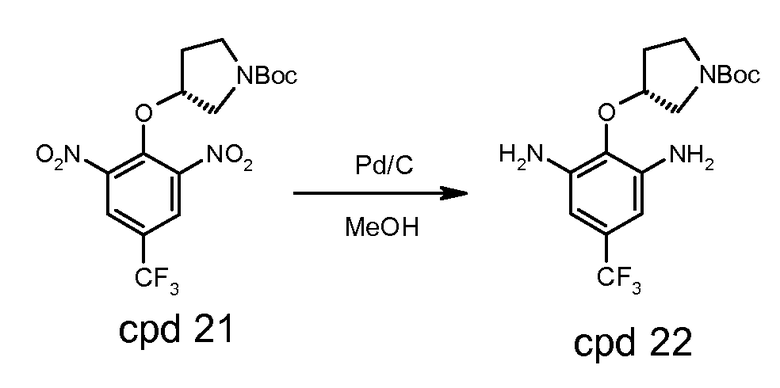
Стадия 2: в мешалку Парра из нержавеющей стали на 2 галлона добавляли катализатор Pd/C (10% масса/объем, 20 г), соединение 21 из стадии 1 (394,37 г) и затем осторожно с перемешиванием 2 л MeOH. Сосуд загружали водородом и дважды вентилировали. Смесь затем перемешивали, начиная при 82 кПа водорода; давление понижали до 0 кПа. Сосуд повторно наполняли до 62 кПа, 28 кПа и 36 кПа соответственно, каждый раз обеспечивая возвращение до 0 кПа (общее поглощение 208 кПа за 51 минуту). Внутренняя температура начиналась с 16°C, и смесь показала плавный, но быстрый экзотермический эффект с максимумом при 38°C. Внутренняя температура поддерживалась при 33-38°C. Сосуд подвергали давлению в 49 кПа с поглощением 23 кПа. Сосуд подвергали давлению в 90 кПа с поглощением 12 кПа в течение 1 часа. Сосуд подвергали давлению в 120 кПа с поглощением 82 кПа в течение 6 часов. Сосуд затем повторно подвергали давлению в 51 кПа с поглощением 37 кПа в течение 14,33 часов (общее поглощение водорода составило 362 кПа). Реактор демонтировали и смесь фильтровали через подушку целита 545, предварительно увлажненную MeOH (воронка Бюхнера 11 см диаметр). Реактор и подушку прополаскивали MeOH и из подушки откачивали фильтрат до состояния медленного стекания каплями и бесцветного фильтрата (~3,0 л общий объем). Фильтрат далее фильтровали через рифленый бумажный диск для удаления некоторого количества тонкоизмельченных темных порошков. Прозрачный фильтрат переносили в 5-л RBF и концентрировали до окрашенного в оранжевый/коричневый цвет густого масла, которое охлаждали до 3-4°C в течение ночи, за это время материал частично затвердевал/кристаллизовался. Материал нагревали и далее откачивали при пониженном давлении с получением твердого рыжевато/коричневого липкого/воскообразного жесткого твердого агломерата. В RBF добавляли гептаны (2×700 мл) и MTBF (700 мл). Смесь перемешивали с использованием верхнеприводной механической мешалки в течение 3,5 часов. Жидкий слой декантировали от твердого вещества, агломерат дробили на мелкие части, жидкость помещали обратно в RBF и суспензию энергично перемешивали в течение 16,75 часов. Затем оставленные мелкие части далее дробили с использованием конца стеклянного стержня мешалки и смесь энергично перемешивали в течение 70 минут. Суспензию фильтровали через воронку из тарированного фриттированного стекла с использованием фильтрата для полного переноса. Воронку закрывали и вакуумом высушивали при слабом нагревании (41°C) в течение 4 часов с предоставлением продукта в виде тусклого персикового/бежевого порошка. Выход: 270,60 г, ВЭЖХ чистота 98,5%.
Катализатор Pd/C может быть заменен, например, любым катализатором, приемлемым для гидрогенизации нитрогруппы.
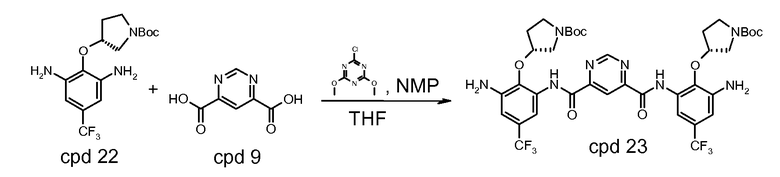
Стадия 3 (Опция 1): 2-хлор-4,6-диметокси-1,3,5-триазин (4,0 г) перемешивали с безводным THF (60 мл). Добавляли N-метилморфолин (4,4 г). Полученную смесь перемешивали при комнатной температуре в течение 30 минут. Затем добавляли соединение 22 (7,2 г) и пиримидин-4,6-дикарбоновую кислоту (1,68 г). Смесь перемешивали при комнатной температуре в течение 24 часов. Затем растворитель выпаривали полностью в вакууме. Добавляли воду (250 мл) и смесь перемешивали в течение 4 часов. После фильтрования желтый осадок на фильтре промывали водой (3×100 мл) и снова перемешивали в воде (250 мл) в течение 4 часов. Процедуру фильтрования и промывания повторяли дважды. Затем твердое вещество сушили на воздухе. Твердое вещество растворяли в 15 мл раствора DCM:гексан:ацетон (5:5:1). Смесь выдерживали при комнатной температуре в течение 2 дней. Твердое вещество фильтровали и промывали 10 мл раствора DCM:гексан (1:1) дважды. Процедуру рекристаллизации повторяли еще один раз с предоставлением светло-желтого твердого вещества. Выход: 70%, ВЭЖХ чистота 100%.
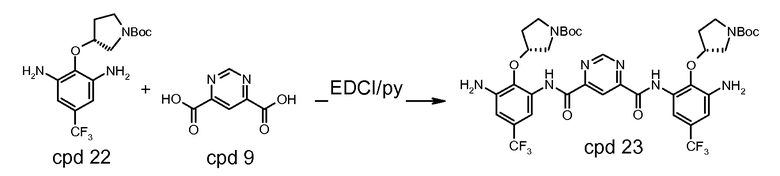
Стадия 3 (Опция 2): EDCI (6,0 г) перемешивали в безводном пиридине (60 мл). Затем добавляли соединение 22 (7,2 г) и соединение 9 (0,56 г). Смесь перемешивали при комнатной температуре в течение 2 часов. Затем добавляли другую часть пиримидин-4,6-дикарбоновой кислоты (0,56 г). После того как смесь перемешали при комнатной температуре еще 2 часа, добавляли третью часть пиримидин-4,6-дикарбоновой кислоты (0,56 г). Полученную смесь перемешивали при комнатной температуре в течение 24 часов. Затем растворитель полностью выпаривали в вакууме. Добавляли воду (250 мл) и смесь перемешивали в течение 4 часов. После фильтрования желтый осадок на фильтре промывали водой (3×100 мл) и перемешивали в воде (250 мл) снова 4 часа. Процедуру фильтрования и промывания повторяли дважды. Затем твердое вещество сушили на воздухе. Твердое вещество растворяли в 15 мл раствора DCM:гексан:ацетон (5:5:1). Смесь оставляли при комнатной температуре в течение 2 дней. Твердое вещество фильтровали и промывали 10 мл раствора DCM:гексан (1:1) дважды. Процедуру рекристаллизации повторяли еще один раз с предоставлением светло-желтого твердого вещества. Выход 70%, ВЭЖХ чистота 100%.
EDCI может быть заменен, например, любыми амидными связующими реагентами, которые продуцируют ангидрид кислоты или активированный сложный эфир, такой как CDI, DCC, HOBt, HOAt, POCl3.
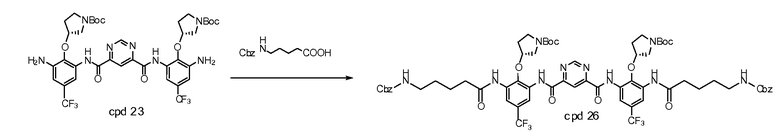
Стадия 4: раствор DMAP (3,66 г) в 60 мл безводного пиридина охлаждали до 0°C в ванне со льдом. Медленно добавляли тионил хлорид (3,60 г). Затем полученный раствор перемешивали в течение 10 минут. Исходный материал N-Cbz кислоту (7,53 г, 30 мМ), Cpd 5 (8,54 г, 10 мМ) добавляли в раствор соответственно. Полученную смесь перемешивали при комнатной температуре в течение 4 часов. Затем добавляли воду (500 мл). После смесь энергично перемешивали при комнатной температуре в течение 2 часов, твердое вещество фильтровали и промывали 250 мл воды. Твердое вещество растворяли в этилацетате (300 мл). Органический слой промывали 10% раствором лимонной кислоты (100 мл) и солевым раствором (100 мл) и сушили над Na2SO4. После выпаривания остаток растворяли в 40 мл DCM, затем добавляли 250 мл гексана. Осадок собирали и сушили под вакуумом. Получили 13,20 г продукта с 95% чистотой. Выход: 100%.
Тионил хлорид может быть заменен, например, POCl3, (EtO)2POCl или оксалил хлоридом.
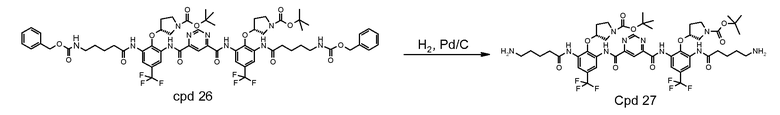
Стадия 5: соединение 26 (13,20 г) растворяли в MeOH с 2 эквив. 1 н. HCl и добавляли 1,0 грамм катализатора Pd/C (10%). Реакционную смесь вводили в гидрогенизатор Парра и встряхивали в течение 2 часов при 60 кПа водорода. Если LCMASS показывала отсутствие прогресса, добавляли еще 1,0 грамм катализатора. Реакционную смесь вводили в гидрогенизатор Парра и встряхивали в течение 3 часов при 60 кПа водорода. Смесь фильтровали через целит для удаления катализатора. Фильтрат концентрировали до сухого состояния на ротационном испарителе при 30°C. Получили 11,50 г продукта с 95% чистотой. Выход: 100%.
Катализатор Pd/C может быть заменен, например, на любой катализатор, приемлемый для гидрогенизации CBZ группы.
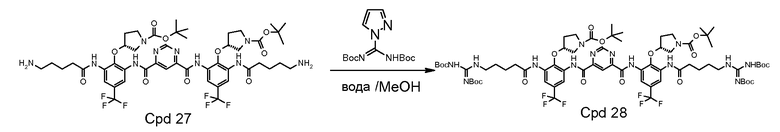
Стадия 6: соединение 27 (11,50 г, 10 мМ) растворяли в 60 мл метанола и DCM (1:1). Затем добавляли 4,04 г триэтиламина (40 мМ). Добавляли di-Boc пиразол 9,3 г (30 мМ) и полученную смесь перемешивали при комнатной температуре в течение 1 часа. После удаления 95% растворителя добавляли 300 мл воды и смесь энергично перемешивали в течение 2 часов. Твердое вещество фильтровали и промывали 300 мл воды. Твердое вещество растворяли в 300 мл этилацетата и сушили над Na2SO4. После выпаривания растворителя твердое вещество растворяли в 40 мл DCM, затем 500 мл гексана использовали для осаждения продукта. Твердое вещество собирали и сушили под вакуумом. Получили 13,0 грамма продукта с 85% выходом (90% чистота).
di-Boc пиразол может быть заменен, например, изомочевиной или di-Boc изомочевиной.
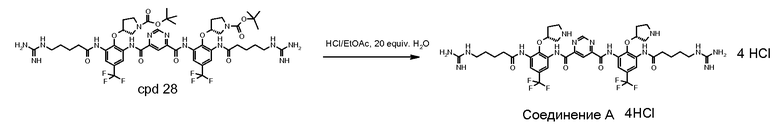
Стадия 7: соединение 28 (1,17 кг, 0,76 М) растворяли в 24 л EtOAc с последующим добавлением 281 мл воды. Газ HCl добавляли в раствор, тогда как температуру реакции поддерживали ниже 45°C путем регулирования скорости добавления. Общее время реакции составило 5 часов, из них 1,5 часа представляет собой время добавления HCl. ВЭЖХ показала, что исходный материал составляет менее чем 1%, и осажденный продукт собирали путем фильтрования под азотом. Твердое вещество прополаскивали с использованием EtOAc, титровали с MeOH/THF (1:1) и сушили под вакуумом. Выход 84%.
HCl/EtOAc может быть заменен, например, на HCl/диоксан.
Пример 5: противомикробная активность против грамположительных клинических изолятов (таблица 1A) и грамотрицательных клинических изолятов (таблица 1B)
Соединение A оценивали in vitro в соответствии с приведенными документами CLSI [Институт клинических и лабораторных стандартов], специфичными для организмов (аэробных, анаэробных или дрожжей), тестированных в этом исследовании. Ампициллин, цефтазидим, цефуроксим, ципрофлоксацин, линезолид и ванкомицин тестировали в одном ряду как агенты сравнения для аэробных бактерий; клиндамицин и метронидазол тестировали как агенты сравнения для анаэробов; фуконазол тестировали как агент сравнения для изолятов дрожжей. Базовые растворы Соединения A получали в диметилсульфоксиде (ДМСО). Ампициллин, цефтазидим, цефуроксим, ципрофлоксацин, линезолид, ванкомицин, метронидазол, клиндамицин и фуконазол получали каждый в соответствии с его руководством по производству.
Аэробы (M7-A7)1
Минимальные ингибирующие концентрации (MIC) в мкг/мл определяли в соответствии с руководством CLSI M7-A7 путем микроразведений в бульоне. Все аэробы тестировали с использованием бульонной среды Мюллера-Хинтона, за исключением Streptococcus spp., которые тестировали с использованием регулируемого по катионам бульона Мюллера-Хинтона с добавлением 2-5% лизированной лошадиной крови. Результаты показаны в таблице 1A и таблице 1B.
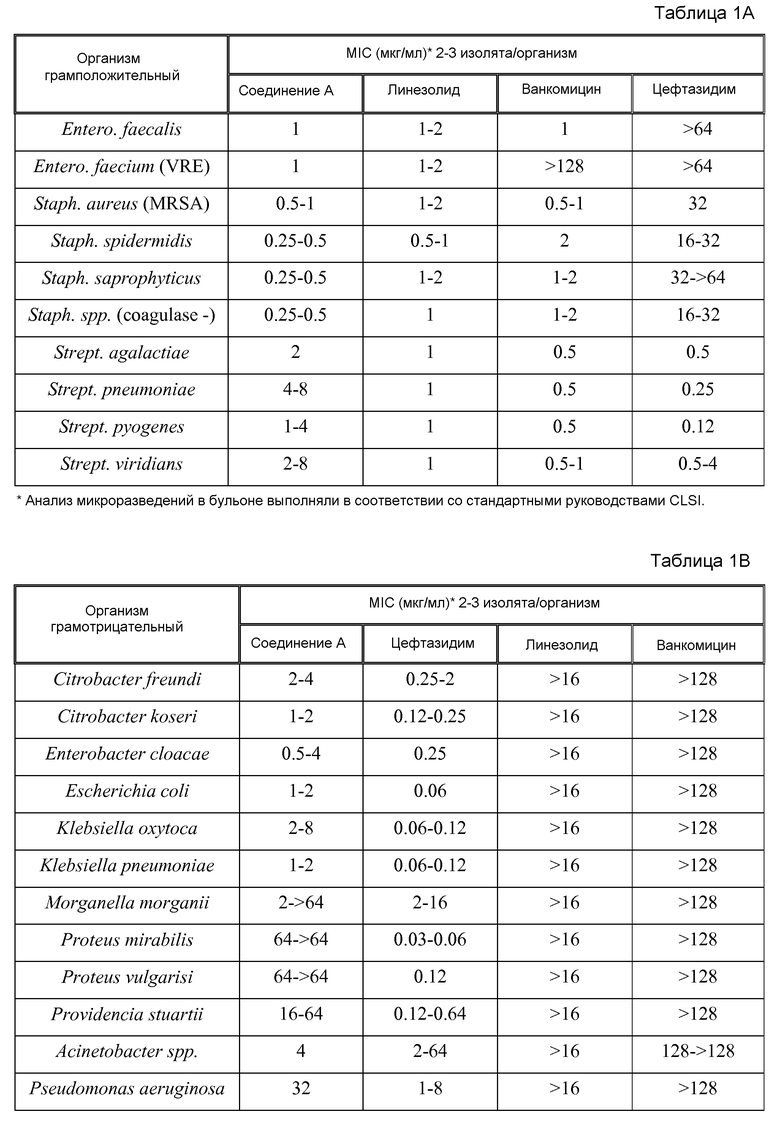

Соединение A проявило широкий спектр активности против грамположительных патогенов с S. aureus и отрицательных по коагулазе видов стафилококков, показывая наименьшие MIC. Соединение A являлось активным против грамотрицательных патогенов, но полный спектр активности был меньше, чем для грамположительных организмов.
Пример 6: MIC с видами стафилококков с определенными фенотипами резистентности
Оценку профилей чувствительности Соединения A против отобранных изолятов выполняли in vitro с помощью методики микроразведений в бульоне с использованием бульонной среды Мюллера-Хинтона в соответствии с документом CLSI M7-A7. Применили интерпретированные пределы чувствительности к антибиотику CLSI, когда применимость находится в соответствии с требованиями документа M100-S17 CLSI. Результаты показаны в таблице 2.
венная
чувств.
OXA-R
OXA-R
OXA-R
DAP-NS
OXA-R
OXA-R: оксациллин-резистентный; VRSA: ванкомицин-резистентный S. aureus; VISA: промежуточно чувствительный к ванкомицину S. aureus; LZD-NS: не чувствительный к линезолиду; DAP-NS: не чувствительный к даптомицину.
Соединение A являлось активным in vitro против всех изолятов S. aureus и отрицательных по коагулазе стафилококков, включая изоляты S. aureus с охарактеризованной резистентностью к даптомицину, линезолиду и ванкомицину (самое новое терапевтическое средство лечения резистентноЙ S. aureus, такое как MRSA). По отношению к изолятам S. aureus отсутствовали изменения в активности против резистентных изолятов относительно чувствительных изолятов. По отношению к отрицательным по коагулазе стафилококкам активность не изменялась вследствие резистентности к метициллину.
Пример 7: цитотоксичность и селективность
Цитотоксичность Соединения A оценивали в колориметрическом анализе с использованием трансформированной клеточной линии печени человека (HepG2, HB-8065) и эмбриональной клеточной линии мыши (клетки NIH/3T3, CRL-1658). С помощью этого анализа измеряли биовосстановление нового соединения тетразолия в растворимом продукте формазана посредством жизнеспособных клеток. Клетки HepG2 засевали в 96-луночные планшеты с плотностью 2×104 клетки/лунка в среду MEM с 10% фетальной бычьей сыворотки (FBS) на 24 часа до использования. Клетки NIH/3T3 засевали в 96-луночные планшеты с плотностью 2×104 клетки/лунка в среду DMEM с 10% телячьей сыворотки (BSC) на 24 часа до использования. Клеточные монослои прополаскивали бессывороточной средой и инкубировали в течение одного часа с Соединением A в бессывороточной среде. После инкубации среду заменяли на среду с добавлением сыворотки и живые клетки измеряли с использованием набора Cell Titer 96 Aqueous Non-Proliferation Assay (Promega, Мэдисон, шт. Висконсин). Значения EC50 определяли с использованием четырех параметров логистического уровнения: Y=нижний уровень+(верхний уровень-нижний уровень)/(1+10^)((LogEC50-X)*уровень пологого склона)).
Цитотоксичность Соединения A также оценивали в анализе гемолиза с использованием эритроцитов человека. Смешанную цельную кровь человека центрифугировали для выделения красных кровяных телец [эритроцитов] (RBC). Изолированные RBC прополаскивали и разводили в трис-забуференном физиологическом растворе (TBS-буфер, pH 7,4) с получением 0,22% базовой суспензии RBC. 5 мкл Соединения A базового раствора добавляли к 45 мкл суспензии RBC и инкубировали с подкачиванием в течение 1 часа при 37°C. После окончания времени инкубации образцы центрифугировали и 30 мкл супернатанта добавляли к 100 мкл воды. Результаты измерения OD414 снимали для концентрации гемоглобина. В качестве положительного контроля использовали пептид яда пчелы мелиттин. Значения EC50 определяли, как описано выше. Результаты показаны в таблице 3.
(EC50 мкг/мл)
(EC50/MIC)
Соединение A демонстрирует большую общую селективность.
Пример 8: время-эрадикация против S. aureus (ATCC 27660)
Исследования кривой время-эрадикация Соединения A против E coli ATCC25922, E coli D31 (лаб. штамм) и S. aureus ATCC27660 определяли в стандартном протоколе путем измерения времени, которое требуется для снижения первоначальных инокулятов на 3 log единицы. Три мл регулируемой катионом среды Мюллера-Хинтона инокулировали 20 мкл замороженного бактериального штамма и инкубировали при 37°C на качалке с платформой (250 об/мин) в течение ночи. Суспензию разводили примерно до 5×105 КОЕ/мл и обрабатывали 2x, 5x, 10x и 20×MIC (MIC=1 мкг/мл). Получили базовый раствор Соединения A 10 мг/мл в ДМСО. В определенные моменты времени бактерии отбирали и число жизнеспособных бактерий подсчитывали на чашках с MH агаром после 18 часов инкубации. Исследования по анализу кинетики кривой время-эрадикация Соединения A против S. aureus ATCC 27660 при концентрациях 2x MIC показали, что снижение на 3 log10 единицы в первоначальном инокуляте происходит за 5 часов. Отсутствие повторного роста наблюдали в культурах через 72 часа при концентрациях 1x MIC. См. фигуры 1A и 1B.
Пример 9: резистентность серийного пассажа в MSSA (ATCC 29213) и MRSA (ATCC 33591)
Замороженные бактериальные штаммы (20 мкл) S. aureus ATCC29213 или резистентного к метициллину S. aureus (MRSA ATCC 33591) инокулировали в 3 мл регулируемой катионом среды Мюллера-Хинтона и инкубировали при 37°C на качалке с платформой (250 об/мин) в течение ночи. Суспензию разводили примерно до 5×105 КОЕ/мл и инокулировали в полипропиленовый (Costar) 96-луночный круглодонный планшет (объемы 90 мкл). Соединение базовых растворов Соединения A и норфлоксацина (Sigma Aldrich, Сент-Луис, Миссури; Каталог #N9890) получали в ДМСО и делали серийные двукратные разведения соединения в 0,01% уксусной кислоте, 0,2% бычьем сывороточном альбумине прямо в лунках полипропиленового планшета по 10 мкл/лунка. Конечные концентрации Соединения A составили 50; 25; 12,5; 6,25; 3,13; 1,56; 0,78; 0,39; 0,19; 0,098; 0,049 и 0,024 мкг/мл. Диапазоны конечных концентраций норфлоксацина составили 100; 50; 25; 12,5; 6,25; 3,13; 1,56; 0,78; 0,39; 0,19; 0,098 и 0,049 мкг/мл. Концентрации ДМСО не превышали 1% в анализе. Все образцы вносили в трех повторностях. После 24 часов инкубации при 37°C клеточный рост оценивали по наблюдению присутствия «допустимого роста», определенного по CLSI как ≥2 мм сгусток или определенной мутности. MIC лунок определяли как наименьшую концентрацию, при которой не наблюдали приемлемого роста. Для серийного пассажа получали аликвоты 50 мкл из 2 из 3 повторных лунок при 0,5× MIC и смешивали с 900 мкл свежей регулируемой катионом среды Мюллера-Хинтона. Измеряли OD600 и клеточные суспензии инокулировали в полипропиленовый 96-луночные круглодонные планшеты (объемы 90 мкл) при примерно 5x105 КОЕ/мл. Десять мкл соединения базовых растворов предварительно добавляли в лунки для получения диапазона концентраций для каждого соединения, описанного выше. Все образцы вносили в трех повторностях. Лунки инкубировали в течение 24 часов при 37°C. Этот процесс повторяли для всех 17 пассажей и значения MIC регистрировали в каждом пассаже.
Пассаж S. aureus с норфлоксацином ассоциировался со значимым увеличением значений MIC в пассаже 3 (4 двойных разведения) для MSSA и MRSA, которые достигали 128-кратного и 64-кратного увеличения соответственно в пассаже 15. С другой стороны, отсутствовали изменения MIC для Соединения A против MSSA ATCC 29213 или MRSA ATCC 33591 в динамике полных 17 пассажей. См. фигуру 2.
Пример 10: in vitro метаболическая стабильность Соединения A - плазма крови
Смешанные образцы плазмы человека (смешанные по полу), крысы (смешанные по породе и полу) и собаки (смешанные по породе и полу) инкубировали с Соединением A (5 мкМ) при 37°C в течение 0 и 60 минут (дублирующие образцы). Инкубацию заканчивали добавлением ледяного растворителя для осаждения (ацетонитрил:ледяная уксусная кислота, 9:1 объем/объем). Супернатанты разводили равным объемом 0,1% муравьиной кислоты и анализировали с помощью ВЭЖХ-МС/МС. Стабильность плазмы представляли как % исходного соединения для 60 минут относительно количества исходного соединения для 0 минут. Результаты показаны в таблице 4.
В плазме человека, крысы и собаки имеется незначительное количество или отсутствует Соединение A после 1 часа инкубации при 37°C, указывая на высокую стабильность плазмы. Также имелось незначительное количество или отсутствовало Соединение A у людей, яванского макака и кроликов (данные не показаны).
Пример 11: эффективность Соединения A на мышиной модели нагрузки на бедренную кость
У самок 6-7-недельного возраста мышей CD-1 вызывали нейтропению циклофосфамидом (150 мг/кг, в/б) в дни 4 и 1 перед в/м инокуляцией S. aureus (ATCC 13709). Инокулят S. aureus получали путем переноса колоний из 18-20-часовых культур на триптическом соевом агаре (TSA) в стерильный PBS. Плотность регулировали до приблизительно 106 КОЕ/мл для спектрофотометра и концентрацию инокулята определяли с помощью метода разведений с подсчетом на чашке. Мышей инокулировали путем инъекции в каждую заднюю часть бедренной кости 0,1 мл инокулята. Соединение A давали отдельным группам мышей (4 самки/группа) посредством в/в болюсных доз 1 или 2 мг/кг/доза в 1 и 5, 1 и 9 или 1 и 13 часов после инокуляции, как показано в таблице 5. Отдельная контрольная группа из 4 мышей получала инокулят без обработки антибиотиком. Соединение A растворяли в 50%/50% объем/объем стерильном USP очищенная вода/PBS. Бедренные кости выделяли через 25 часов после инокуляции. Мышцу бедренной кости и костную ткань гомогенизировали, аликвоты серийных разведений помещали на TSA и инкубировали при 37°C в течение 20 часов, и получали число колоний для расчета КОЕ/бедренная кость. Параметры показаны в таблице 5.
доза)
(мл/кг)
(ч после
инокуляции)
бедренной
кости
(ч после
инокуляции)
Мышей
Контроль
A
A
A
A
A
A
Соединение A являлось более эффективным в снижении бактериальной популяции в инокулированных бедренных костях, когда вводилось 2 мг/кг/доза в 1 и 5 или в 1 и 9 часов после инокуляции. Снижение числа бактерий в этих 2 группах было на 3,96 и 3,93 log более низким соответственно, чем то же в инокулированной контрольной группе. См. фигуру 3.
Пример 12: эффективность против ванкомицина на крысиной модели нагрузки на бедренную кость
Для каждого эксперимента у самок крыс 8-9-недельного возраста с канюлированной бедренной веной Crl:CD(SD) вызывали нейтропению циклофосфамидом (150 мг/кг, в/б) в дни 4 и 1 перед в/м инокуляцией S. aureus (ATCC 13507). Суспензию S. aureus получали из колоний, полученных из ночной культуры, помещенной в PBS и отрегулированной приблизительно до 107 КОЕ/мл для спектрофотометра. Каждую крысу инъецировали 0,2 мл инокулята в мышцу бедренной кости правой задней конечности. Бедренные кости собирали через 25 часов после инокуляции и обрабатывали для определения КОЕ/бедренная кость. Соединение A давали в/в болюсной инъекцией в хвостовую вену или 1-часовой в/в инфузией, или 4-часовой в/в инфузией через канюлированную бедренную вену в различные временные интервалы после инокуляции. Отдельные контрольные группы инокуляции включали в каждый эксперимент, и группы ванкомицина включали как агенты сравнения в первый и второй эксперименты. Каждая группа, включая контроли и агент сравнения, состояла из 4 крыс.
Для Соединения A в/в болюсная инъекция (10 мг/кг/доза, 20 мг/кг общая доза) и 1 час в/в инфузий (10 мг/кг/доза, 20 мг/кг общая доза) снижали бактериальную нагрузку на 3,2 и 3,0 log соответственно по сравнению с инокулированными контролями. Снижение относительно уровней инокулята через 1 час после инфицирования составляло приблизительно от 2,2 до 2,0 log соответственно. Эффективность сравнивали с ванкомицином. См. фигуру 4.
Пример 13: эффективность Соединения A на мышиной модели сепсиса: инфекция S. aureus
Стерильный физиологический раствор, ванкомицин или Соединение A вводили в отдельные группы 8-недельных самок мышей CD-1 (8 мышей/группа) через 1 и 7 часов после в/б инъекций S. aureus (ATCC 13709, 5×107 КОЕ/мл в 5% муцине, 0,5 мл/мышь). Соединение A растворяли в 50%/50% объем/объем стерильном USP очищенная вода/TBS. Суспензию S. aureus получали из колоний, перенесенных из чашки с TSA на стерильный PBS. Аликвоту базовой суспензии добавляли в 5% муцин до конечной концентрации примерно 5×107 КОЕ/мл. Планирование исследования и дозы показаны в таблице 6. Мышей наблюдали в течение 6 дней после инокуляции до летального исхода.
(мг/кг/
доза)
доза
(мг/кг)
емого
соединения
(мл/кг)
инъекции
тестирумого
соединения
(ч после инокуляции)
мышей
контроль
Дозозависимую эффективность наблюдали с Соединением A, которая была сравнима с группой обработки ванкомицином. Все необработанные мыши погибли в первый день обработки. При дозах 2×5 и 2×10 мг/кг Соединения A полная защита достигалась с Соединением A. См. фигуру 5.
Пример 14: исследование острой токсичности - максимально толерантные дозы
Определения максимально толерантной дозы (MTD) получены в исследованиях восходящих/нисходящих доз у мышей и крыс. Соединение A вводили путем в/в болюсной инъекции в хвостовую вену мышей и крыс или путем в/в инфузии через катетер в бедренную вену крыс. В каждой дозе двум или трем животным вводили соединение и клинические признаки регистрировали через период от 4 до 7 дней.
В заключение исследования выполняли общую некропсию. Результаты показаны в таблице 7.
MTD для Соединения A у крысы была >24 мг/кг при введении путем в/в инфузии в течение 1 часа.
Пример 15: фармакокинетика Соединения A у крыс
Крысам Crl:CD(SD) вводили Соединения A путем в/в болюсной инъекции в указанных дозах. Плазму получали из образцов крови, взятых в 9 временных точках (n=3) в пределах 28 часов. Уровни соединения определяли с помощью ВЭЖХ-МС/МС. Все животные были оснащены двумя канюлями в яремной вене (JVC) по одной для введения дозы и забора крови. Каждый путь введения дозировали как N=3. Животных обеспечивали коммерческим рационом для грызунов и водой неограниченно. Каждая крыса получала дозированное болюсное введение через соответствующий путь введения в момент времени ноль в день введения дозы. Время сбора образцов крови показано в таблице 8.
Каждый образец крови собирали от крыс через JVC и помещали в охлажденные полипропиленовые пробирки, содержащие натрия ЭДТА в качестве антикоагулянта. Образцы центрифугировали при температуре 4°C и скорости 13000 об/мин в течение 5 минут. Образцы поддерживали охлажденными на всем протяжении обработки. Каждый образец плазмы затем переносили в меченые полипропиленовые пробирки, помещали на сухой лед и хранили в морозильной установке с поддержанием температуры от -60°C до -80°C.
Образцы исследуемой плазмы экстрагировали и анализировали с использованием ранее разработанного метода. Образцы для одной стандартной кривой и шесть повторов образцов качественного контроля в трех концентрациях экстрагировали с использованием ДМСО, содержащего 0,1% муравьиной кислоты. Образцы плазмы (50 мкл) добавляли к 150 мкл растворителя и центрифугировали. Супернатанты анализировали с помощью LC/MSMS [жидкостная хроматография с тандемной масс-спектрометрией] с использованием Perkin Elmer серий 200 микронасоса и масс-спектрометра с электрораспылением PE Sciex API4000. Стандартные кривые получали при концентрациях 10000, 5000, 1000, 500, 250, 100, 50 и 25 нг/мл. Образцы качественного контроля получали при концентрациях 5000, 500 и 50 нг/мл. Образцы для стандартной кривой и качественного контроля получали из независимо полученных базовых растворов. По меньшей мере 5/8 стандартов должны иметь точность в пределах ±15%, за исключением LLOQ, где ±20% приемлемы. Две трети партии QC должны иметь точность в пределах ±15% номинала, и по меньшей мере одна треть QC должна проходить каждый уровень для того, чтобы пробег считался принятым.
Индивидуальные концентрации плазмы против временных данных для Соединения A подвергали анализу без учета компартментов с использованием фармакокинетической программы WinNonlin v4.1. Концентрации плазмы ниже предела количественного анализа (25 нг/мл) оценивали по значению ноль для фармакокинетического анализа. Номинальные дозовые концентрации использовали во всех расчетах.
Результаты показаны ниже в таблице 9.
Полупериод существования в плазме Соединения A в плазме крысы было значимо большим, а значения клиренса ниже.
Пример 16: составы
Растворимость до насыщения Соединения A в различных эксципиентах исследовали при 25°C, и результаты представлены в таблице 10 (растворимость до насыщения Соединения A как чистой основы).
Категория
аминометан HCl (pH 7,0)
Предварительные исследования показали, что бензиловый спирт, этанол и DMA являлись плохими несущими растворами для Соединения A со значением растворимости до насыщения 1,83; 1,13 и 0,60 мг/мл соответственно. С другой стороны, хорошая растворимость до насыщения 90 мг/мл, 65 мг/мл и 53 мг/мл достигалась в пропиленгликоле, очищенной воде и глицерине, и они поэтому исследовались далее. Хорошие значения растворимости достигались при pH 3 и 7,4 со значениями 65,5 и 61,4 мг/мл соответственно. Растворимость, однако, являлась капельной при pH 5 со значением 12,1 мг/мл. Растворимость до насыщения Соединения A в 0,9% и 1,2% растворе натрия хлорида не могла быть определена, так как состав образовывал вязкий желтый гель.
Растворимость до насыщения Соединения A в различных многокомпонентных системах исследовали при 25°C, и результаты представлены в таблице 11 (растворимость до насыщения Соединения A как чистой основы)
*: состав желировался во время центрифугировании, но ожижался в процессе выдерживания.
Составы, которые проявили приемлемые результаты, включали 50% масса/объем пропиленгликоль и 15% масса/объем глицерин в очищенной воде со значением растворимости до насыщения при 25°C 74,7 мг/мл и 63,5 мг/мл соответственно. Хорошая растворимость до насыщения также достигалась с различными комплексообразующими агентами со значениями 79,7; 102,0 и 64,3 мг/мл для 20% масса/объем Клептозы, 40% масса/объем Клептозы и 25% масса/объем Каптизола соответственно. Результаты также показывали, что добавление Соединения A в 20-50% масса/объем пропиленгликоль в физиологическом растворе приводило к образованию вязкого желтого геля и, следовательно, не могло быть проанализировано с помощью УФ. Однако феномен гелеобразования зависел от концентрации и наблюдался в составах, когда концентрация Соединения A в составе достигала значения растворимости до насыщения лекарственного средства. Кроме того, процесс гелеобразования можно было легко обратить путем добавления небольшого объема состава эксципиента или нескольких капель этанола. Добавление 5% масса/объем этанола в состав не ингибировало феномен гелеобразования, но он все же являлся легко обратимым и мог быть проанализирован с помощью УФ-спектрофотометрии. После оценки предварительных данных скрининга эксципиентов выбрали три приемлемых состава для дальнейшей разработки состава. Эти составы представляют собой 20% масса/объем раствор Клептозы, 20% масса/объем пропиленгликоль в очищенной воде и 15% масса/объем глицерин в очищенной воде. Состав 50 мг/мл Соединения A в 20% масса/объем Клептозе выбрали в фазе I клинических испытаний. Кроме того, могут быть взяты аликвоты растворов Соединения A в воде, Клептозе или Манните и лиофилизированы в твердое вещество. Твердое вещество может быть восстановлено с применением воды перед использованием.
Различные модификации изобретения, в дополнение к тем, которые описаны в данном описании, будут очевидны специалистам в данной области из вышеизложенного описания. Такие модификации также находятся в пределах прилагаемой формулы изобретения. Эта заявка испрашивает приоритет предварительной заявки США серийный № 61/108595, поданной 27 октября 2008 г., которая приведена в данном описании в качестве ссылки во всей своей полноте. Настоящее изобретение поддерживалось фондами правительства США ((NIH Грант № AI4866 и 1R43AI058407), и правительство США может поэтому иметь определенные права на это изобретение.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ГЕТЕРОАРИЛПИРРОЛИДИНИЛ- И ПИПЕРИДИНИЛКЕТОНА | 2007 |
|
RU2479575C2 |
| ПРОИЗВОДНЫЕ ПИРРОЛИДИНА ДЛЯ ПРИМЕНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ КАТЕПСИНА | 2011 |
|
RU2548684C2 |
| Макроциклический ингибитор тирозинкиназы и его применение | 2019 |
|
RU2798231C2 |
| КОМПОЗИЦИИ И СПОСОБЫ ИНГИБИРОВАНИЯ ВИРУСНОЙ ПОЛИМЕРАЗЫ | 2013 |
|
RU2654482C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ МОДУЛЯЦИИ АУТОИММУННЫХ ЗАБОЛЕВАНИЙ, СПОСОБ ПОЛУЧЕНИЯ ЭТИХ СОЕДИНЕНИЙ | 2000 |
|
RU2255937C2 |
| ЗАМЕЩЕННЫЕ АЗЕТИДИНОНЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЛЕЧЕНИЯ | 1993 |
|
RU2148056C1 |
| ЗАМЕЩЕННОЕ ГЕТЕРОЦИКЛИЧЕСКОЕ КОНДЕНСИРОВАННОЕ ЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2020 |
|
RU2815814C1 |
| Регулятор на основе азотсодержащего гетероциклического производного, способ его получения и его применение | 2020 |
|
RU2829553C2 |
| ПРОИЗВОДНЫЕ 2-АМИНО-ПИРИМИДИНА В КАЧЕСТВЕ ИНГИБИТОРОВ JNK | 2011 |
|
RU2572247C2 |
| НОВЫЕ ДИАМИДЫ ПИРИМИДИН-4,6-ДИКАРБОНОВОЙ КИСЛОТЫ ДЛЯ СЕЛЕКТИВНОГО ИНГИБИРОВАНИЯ КОЛЛАГЕНАЗ | 2003 |
|
RU2344129C2 |
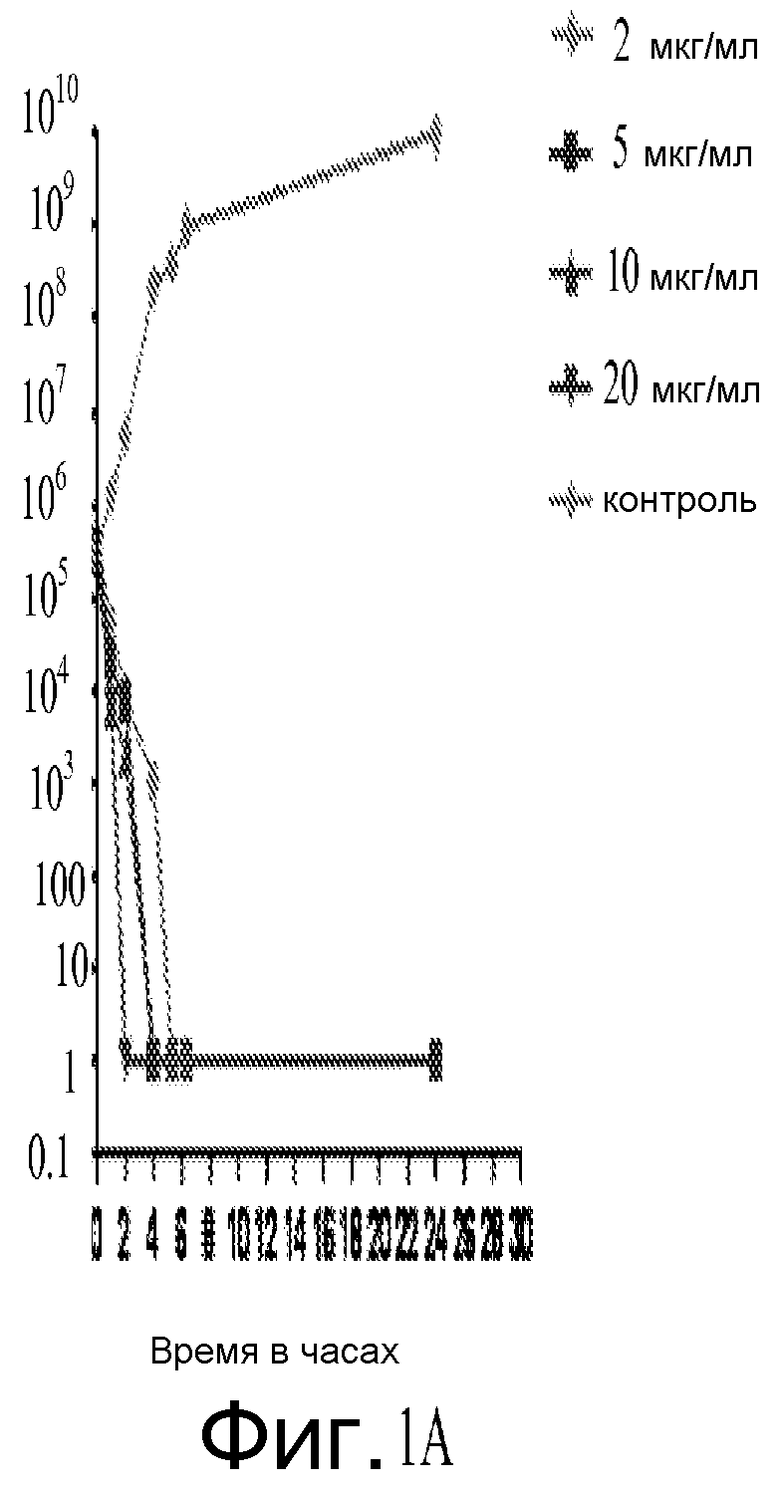
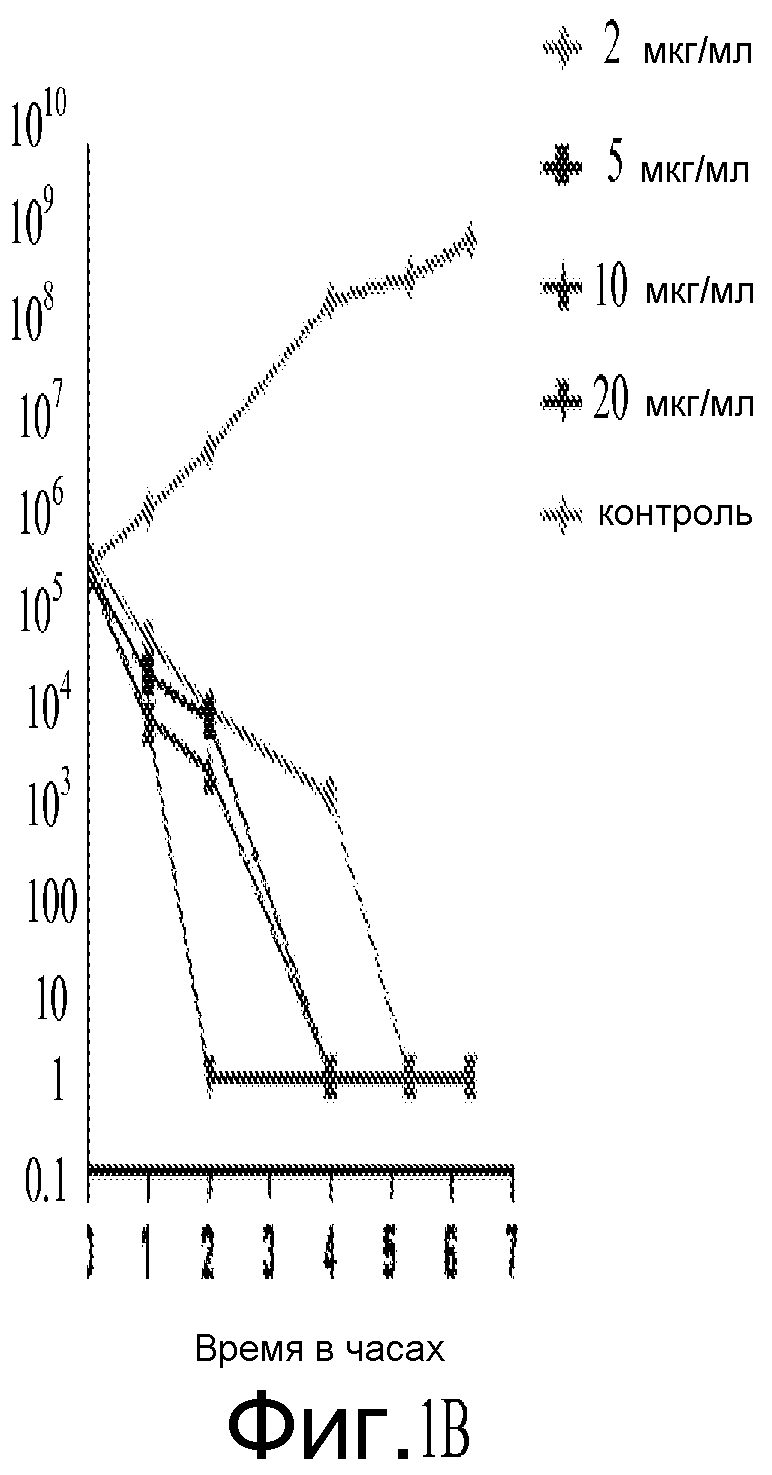
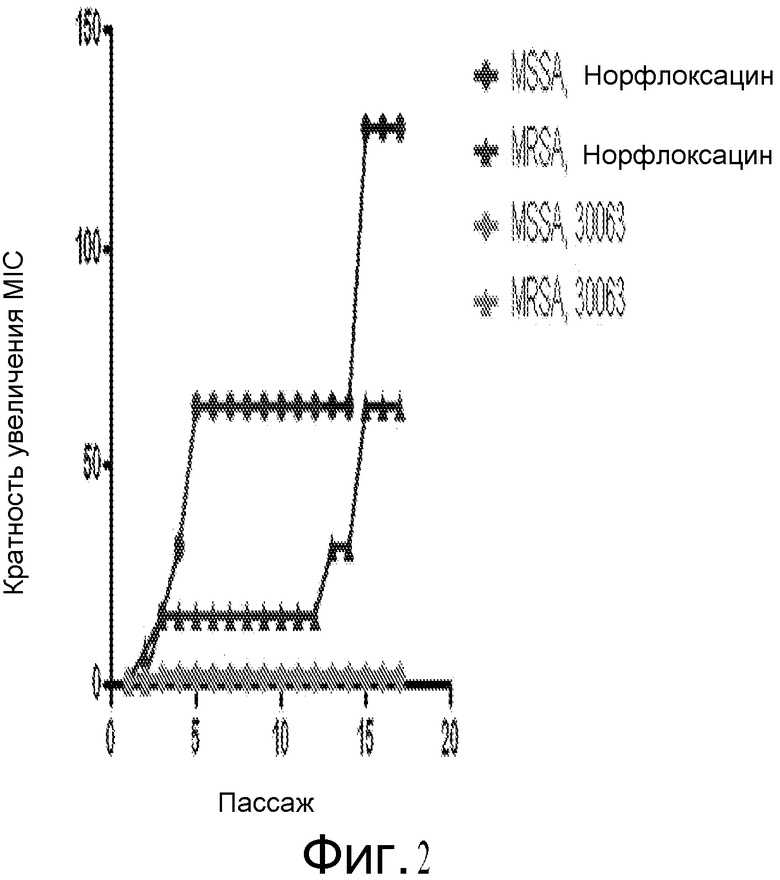
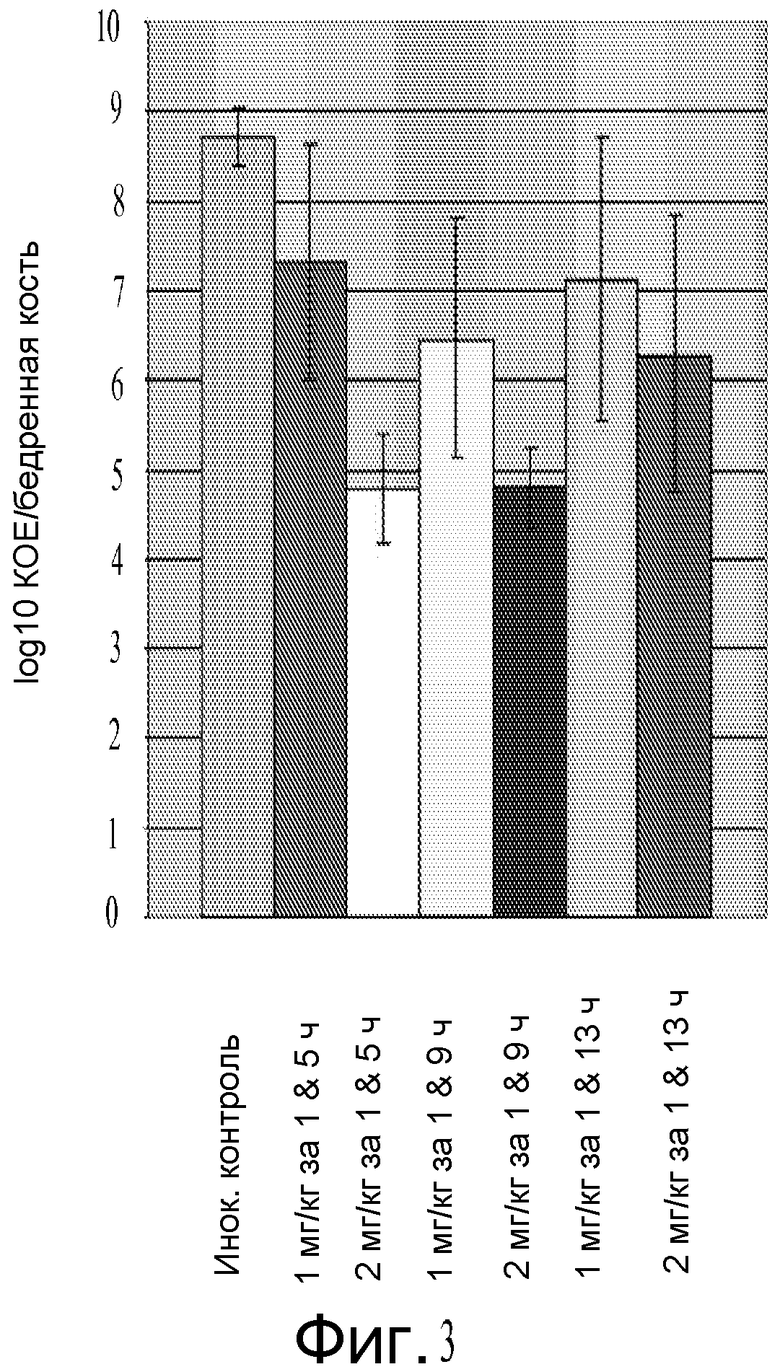
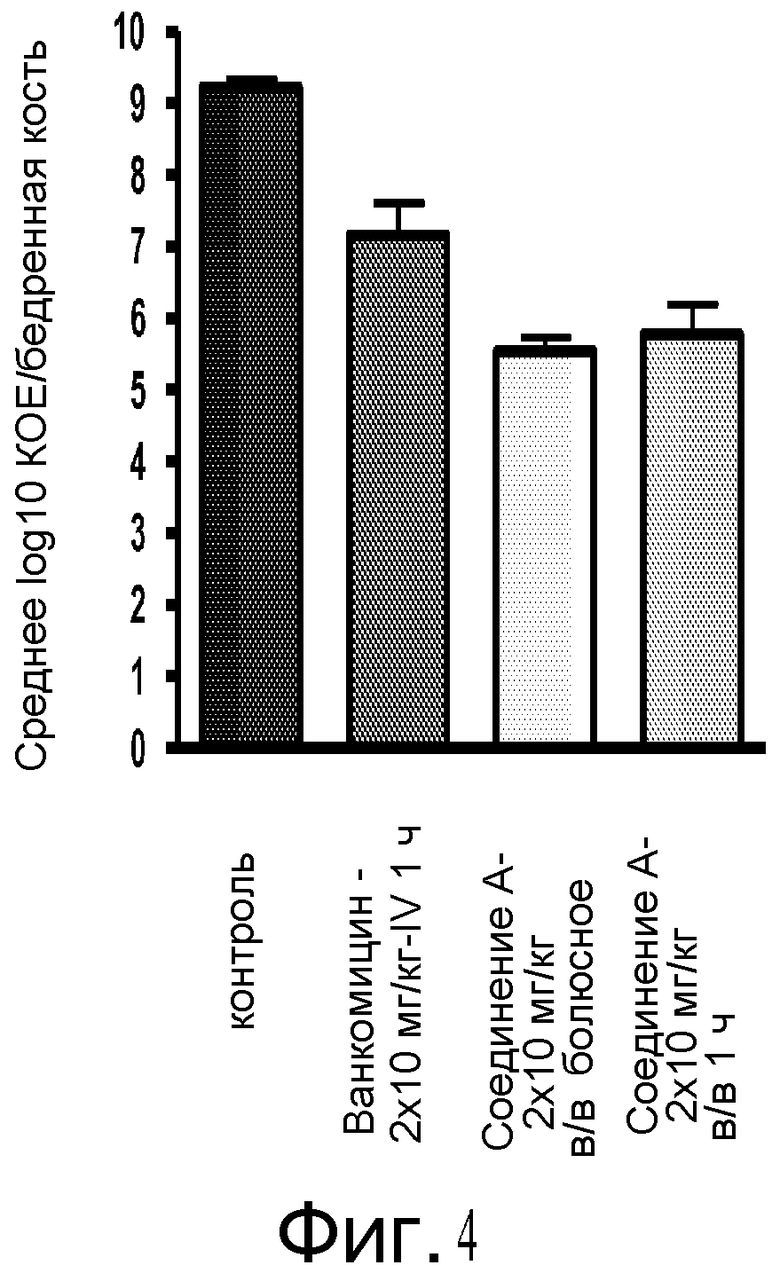
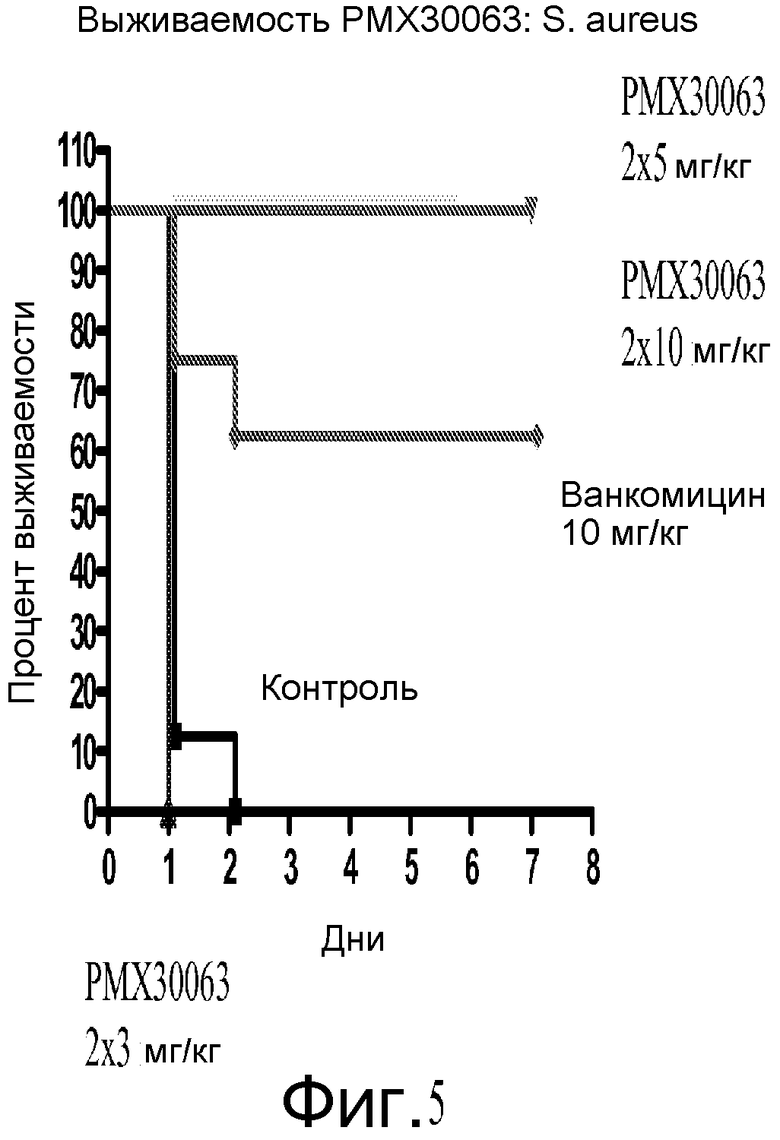
Настоящее изобретение относится к препарату, ингибирующему рост микробов, включающему ариламидное соединение в качестве активного соединения и клептозу или каптизол. Предложены способы получения препарата, применение и способ лечения микробных инфекций. 11 н. и 10 з.п. ф-лы, 5 ил., 11 табл., 16 пр.
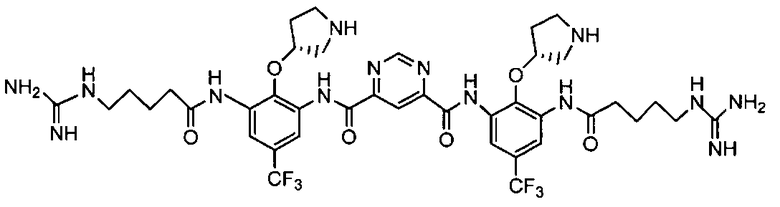
1. Способ получения препарата, включающего 50 мг/мл соединения следующей формулы:
где указанный способ включает:
а) взаимодействие (R)-(-)-N-Boc-3-пирролидинола с сильным основанием с получением смеси; и последующее взаимодействие смеси с 2-хлор-5-(трифторметил)-1,3-динитробензолом с получением соединения, имеющего Формулу II
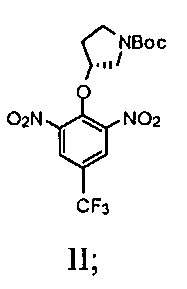
b) взаимодействие соединения Формулы II со спиртом и катализатором переходного металла в присутствии водорода с получением соединения Формулы III
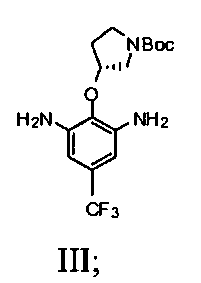
с) добавление соединения Формулы III и пиримидин-4,6-дикарбоновой кислоты в смесь 2-хлор-4,6-диметокси-1,3,5-триазина и N-метилморфолина с получением соединения Формулы IV
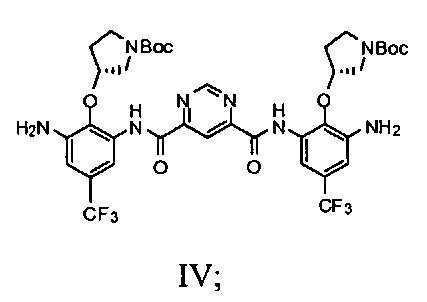
d) взаимодействие соединения Формулы IV с N-Boc-гуанидин масляной кислоты с получением соединения Формулы V
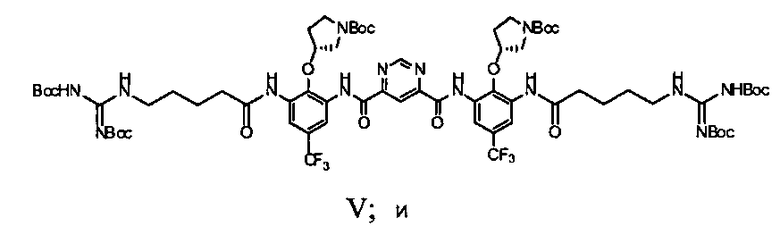
е) снятие защитных групп соединения Формулы V с получением соединения следующей формулы:
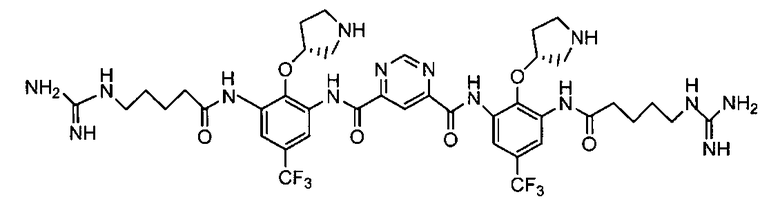
f) формулирование полученного соединения в количестве 50 мг/мл с 20% масса/объем β-циклодекстрином или 40% масса/объем β-циклодекстрином в воде.
2. Способ получения препарата, включающего 50 мг/мл соединения следующей формулы:
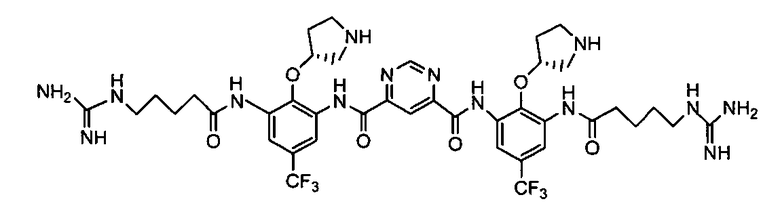
где указанный способ включает:
a) депротонирование трет-бутилового эфира (R)-3-гидроксипирролидин-1-карбоновой кислоты и взаимодействие полученного соединения с 2-хлор-1,3-динитро-5-трифторметилбензолом с получением трет-бутилового эфира (R)-3-(2,6-динитро-4-трифторметилфенокси)пирролидин-1-карбоновой кислоты;
b) восстановление трет-бутилового эфира (R)-3-(2,6-динитро-4-трифторметилфенокси)пирролидин-1-карбоновой кислоты в присутствии спирта, катализатора переходного металла и водорода с получением трет-бутилового эфира (R)-3-(2,6-диамино-4-трифторметилфенокси)пирролидин-1-карбоновой кислоты;
с) сочетание трет-бутилового эфира (R)-3-(2,6-диамино-4-трифторметилфенокси)пирролидин-1-карбоновой кислоты с пиримидин-4,6-дикарбоновой кислотой в присутствии 1-[(3-(диметиламино)пропил)]-3-этилкарбодиимида гидрохлорида с получением бис-{[3-амино-2-((R)-1-(трет-бутоксикарбонил)пирролидин-3-илокси)-5-трифторметил-фенил]амида}пиримидин-4,6-дикарбоновой кислоты;
d) взаимодействие бис-{[3-амино-2-((R)-1-(трет-бутоксикарбонил)пирролидин-3-илокси)-5-трифторметил-фенил]амида}пиримидин-4,6-дикарбоновой кислоты с ({[(трет-бутоксикарбонил)амино][(трет-бутоксикарбонил)имино]метил}амино)пентановой кислотой в присутствии фосфора оксихлорида с получением бис-{[3-(5-({[(трет-бутоксикарбонил)амино][трет-бутоксикарбонил)имино]метил}амино)пентаноиламино)-2-((R)-1-(трет-бутоксикарбонил)пирролидин-3-илокси)-5-трифторметил-фенил]амида}пиримидин-4,6-дикарбоновой кислоты;
e) снятие защитных групп бис-{[3-(5-({[(трет-бутоксикарбонил)амино][трет-бутоксикарбонил)имино]метил}амино)-пентаноиламино)-2-((R)-1-(трет-бутоксикарбонил)пирролидин-3-илокси)-5-трифторметил-фенил]амида} пиримидин-4,6-дикарбоновой кислоты с получением неочищенного бис-{[3-(5-гуанидино-пентаноиламино)-2-((R)-пирролидин-3-илокси)-5-трифторметил-фенил]амид}тетрагидрохлорида пиримидин-4,6-дикарбоновой кислоты; и
f) очистку неочищенного бис-{[3-(5-гуанидино-пентаноиламино)-2-((R)-пирролидин-3-илокси)-5-трифторметил-фенил]амид}тетрагидрохлорида пиримидин-4,6-дикарбоновой кислоты с помощью хроматографии с обращенной фазой;
g) формулирование полученного соединения в количестве 50 мг/мл с 20% масса/объем β-циклодекстрином или 40% масса/объем β-циклодекстрином в воде.
3. Способ получения препарата, включающего 50 мг/мл соединения следующей формулы:
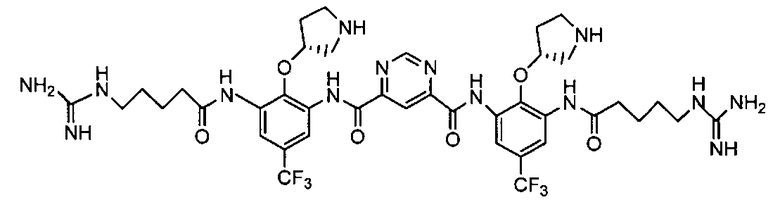
где указанный способ включает:
a) депротонирование трет-бутилового эфира (R)-3-гидроксипирролидин-1-карбоновой кислоты и последующее взаимодействие полученного соединения с 2-хлор-1,3-динитро-5-трифторметилбензолом с получением трет-бутилового эфира (R)-3-(2,6-динитро-4-трифторметилфенокси)пирролидин-1-карбоновой кислоты;
b) восстановление трет-бутилового эфира (R)-3-(2,6-динитро-4-трифторметилфенокси)пирролидин-1-карбоновой кислоты в присутствии спирта, катализатора переходного металла и водорода с получением трет-бутилового эфира (R)-3-(2,6-диамино-4-трифторметилфенокси)пирролидин-1-карбоновой кислоты;
c) сочетание трет-бутилового эфира (R)-3-(2,6-диамино-4-трифторметилфенокси)пирролидин-1-карбоновой кислоты с пиримидин-4,6-дикарбоновой кислотой в присутствии 1-[(3-(диметиламино)пропил)]-3-этилкарбодиимида гидрохлорида с получением бис-{[3-амино-2-((R)-1-(трет-бутоксикарбонил)пирролидин-3-илокси)-5-трифторметил-фенил]амида} пиримидин-4,6-дикарбоновой кислоты;
d) взаимодействие бис-{[3-амино-2-((R)-1-(трет-бутоксикарбонил)пирролидин-3-илокси)-5-трифторметил-фенил]амида} пиримидин-4,6-дикарбоновой кислоты с N-Cbz кислотой в присутствии тионила хлорида;
e) восстановление полученного соединения d) в присутствии спирта, катализатора переходного металла и водорода;
f) взаимодействие полученного соединения е) с ди-Boc пиразолом;
g) снятие защитных групп полученного соединения f) с получением соединения следующей формулы:
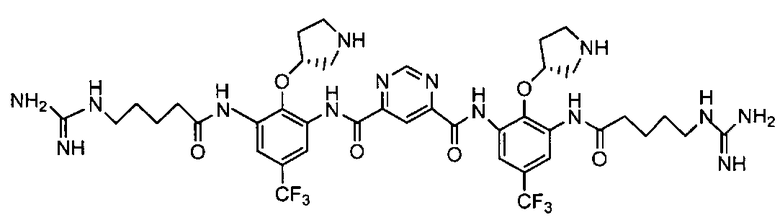
h) формулирование полученного соединения в количестве 50 мг/мл с 20% масса/объем β-циклодекстрином или 40% масса/объем β-циклодекстрином в воде.
4. Способ получения препарата, включающего 50 мг/мл соединения следующей формулы:
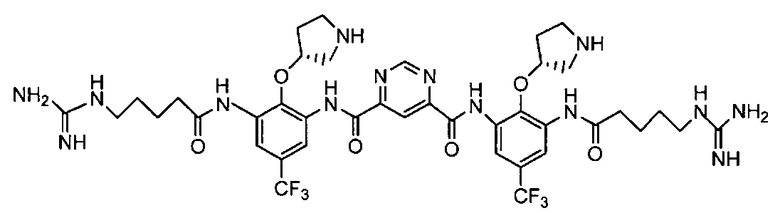
в форме фармацевтически приемлемой соли, где указанный способ включает:
а) взаимодействие (R)-(-)-N-Boc-3-пирролидинола с сильным основанием с получением смеси; последующее взаимодействие смеси с 2-хлор-5-(трифторметил)-1,3-динитробензолом с получением соединения, имеющего Формулу II

b) взаимодействие соединения Формулы II со спиртом и катализатором переходного металла в присутствии водорода с получением соединения Формулы III

c1) добавление соединения Формулы III и пиримидин-4,6-дикарбоновой кислоты в смесь 2-хлор-4,6-диметокси-1,3,5-триазина и N-метилморфолина с получением соединения Формулы IV
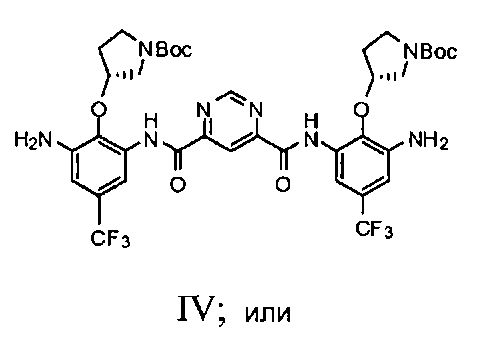
c2) добавление соединения Формулы III и пиримидин-4,6-дикарбоновой кислоты в смесь 1-этил-3-[3-(диметиламино)пропил]карбодиимида гидрохлорида (EDC1) и безводного пиридина с получением соединения Формулы IV
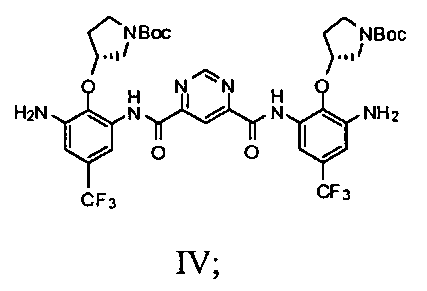
d) добавление соединения Формулы IV с N-Cbz кислотой в раствор, включающий безводный пиридин, диметиламинопропиламин и любой один из тионила хлорида, POCl3, (EtO)2POCl или оксалила хлорида с получением соединения Формулы Va

е) гидрогенолиз группы Cbz соединения Формулы Va с получением соединения Формулы VI
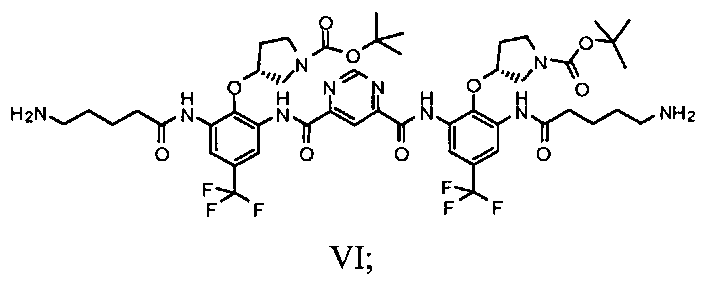
f) защиту соединения Формулы VI с получением соединения Формулы VII
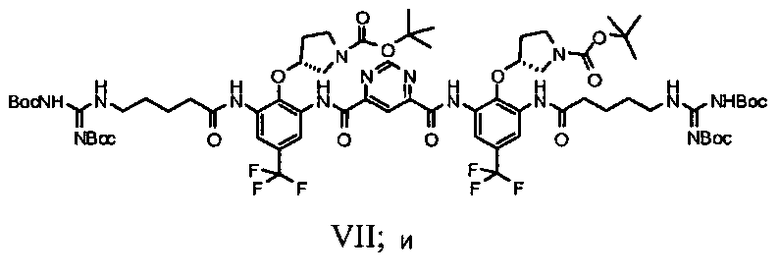
g) снятие защитных групп соединения Формулы VII с получением фармацевтически приемлемой соли соединения следующей формулы:
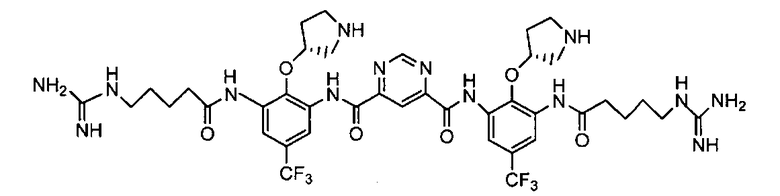
h) формулирование полученного в форме фармацевтически приемлемой соли соединения в количестве 50 мг/мл с 20% масса/объем β-циклодекстрином или 40% масса/объем β-циклодекстрином в воде.
5. Препарат, предназначенный для ингибирования роста микроба, включающий 50 мг/мл соединения следующей формулы:
или его фармацевтически приемлемую соль и 20% масса/объем β-циклодекстрин (Клептоза) в очищенной воде, 40% масса/объем β-циклодекстрин (Клептоза) в очищенной воде или 25% масса/объем Каптизол в очищенной воде.
6. Препарат по п.5, включающий соединение следующей формулы:
или его фармацевтически приемлемую соль в 20% масса/объем β-циклодекстрине (Клептозе) в очищенной воде или 40% масса/объем β-циклодекстрине (Клетозе) в очищенной воде.
7. Препарат по п.5 или 6, включающий соединение следующей формулы:
или его фармацевтически приемлемую соль в 20% масса/объем β-циклодекстрине (Клептозе) в очищенной воде.
8. Препарат, предназначенный для ингибирования роста микроба, полученный способом по любому из пп.1-4.
9. Способ ингибирования роста микроба, включающий контактирование микроба с препаратом по любому одному из пп.5-8.
10. Способ по п.9, где микробом является грамотрицательный аэроб, грамположительный аэроб, грамотрицательный анаэроб, грамположительный анаэроб или дрожжи.
11. Способ лечения субъекта, страдающего микробной инфекцией, включающий введение субъекту, нуждающемуся в этом, противомикробного эффективного количества препарата по любому одному из пп.5-8.
12. Способ по п.11, где микробной инфекцией является грамотрицательный аэроб, грамположительный аэроб, грамотрицательный анаэроб, грамположительный анаэроб или дрожжи.
13. Препарат по любому из пп.5-8, где микробом является грамотрицательный аэроб, грамположительный аэроб, грамотрицательный анаэроб, грамположительный анаэроб или дрожжи.
14. Препарат по любому из пп.5-8, предназначенный для лечения микробной инфекции.
15. Препарат по п.14, где микробной инфекцией является грамотрицательный аэроб, грамположительный аэроб, грамотрицательный анаэроб, грамположительный анаэроб или дрожжи.
16. Применение препарата по любому из пп.5-8 для получения лекарственного средства для лечения микробной инфекции.
17. Применение по п.16, где микробной инфекцией является грамотрицательный аэроб, грамположительный аэроб, грамотрицательный анаэроб, грамположительный анаэроб или дрожжи.
18. Применение препарата по любому из пп.5-8 для получения лекарственного средства для ингибирования роста микроба.
19. Применение по п.18, где микробом является грамотрицательный аэроб, грамположительный аэроб, грамотрицательный анаэроб, грамположительный анаэроб или дрожжи.
20. Применение препарата по любому из пп.5-8 для лечения микробной инфекции.
21. Применение по п.20, где микробной инфекцией является грамотрицательный аэроб, грамположительный аэроб, грамотрицательный анаэроб, грамположительный анаэроб или дрожжи.
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| Топчак-трактор для канатной вспашки | 1923 |
|
SU2002A1 |
| СПОСОБ ПОЛУЧЕНИЯ СИСТЕМЫ ДОСТАВКИ ВОДОНЕРАСТВОРИМЫХ И ПЛОХОРАСТВОРИМЫХ БИОЛОГИЧЕСКИ АКТИВНЫХ ВЕЩЕСТВ И ЛЕКАРСТВЕННАЯ ФОРМА НА ЕЕ ОСНОВЕ | 2006 |
|
RU2325151C2 |

