РОДСТВЕННАЯ ЗАЯВКА
По настоящей заявке испрашивается приоритет в соответствии с предварительной заявкой на патент США 61/625994, поданной 18 апреля 2012.
УРОВЕНЬ ТЕХНИКИ
Вирусы ответственны за многие инфекционные заболевания животных, включая, в частности, млекопитающих и человека. В отличие от инфекций, вызванных бактериями, для профилактики и лечения вирусных инфекций эффективным является относительно небольшое число агентов. Биология вирусных заболеваний в настоящее время хорошо изучена, включая транскрипцию, трансляцию и репликацию вирусного генома. В РНК-содержащих вирусах важным ферментом является РНК-зависимая РНК-полимераза, которая отвечает за репликацию вирусного генома. РНК-зависимая РНК-полимераза является важным белком, кодируемым в геномах всех РНК-содержащих вирусов, не имеющих ДНК-стадии, которые имеют отрицательно-полярную РНК. Фермент катализирует синтез РНК-нити, комплементарной данной матричной РНК. Поскольку репликация вируса зависит от РНК-полимеразы, этот фермент является перспективной мишенью в разработке новых противовирусных соединений.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Изобретение относится к соединениям формулы I, включая их фармацевтически приемлемые соли, для применения в ингибировании вирусной РНК-полимеразной активности или вирусной репликации и при лечении вирусных инфекций. Соединения характеризуются, кроме того, благоприятной фармакокинетикой активного фармацевтического ингредиента, особенно в сочетании с энтеральным введением, включая, в частности, пероральное введение. Изобретение также относится к фармацевтическим композициям, включающим одно или несколько соединений формулы I или их фармацевтически приемлемые соли, а также к способам получения вышеуказанных соединений. Также предложены способы ингибирования вирусной РНК-полимеразной активности, вирусной репликации и лечения вирусных инфекций.
КРАТКОЕ ОПИСАНИЕ ФИГУР
На фигуре 1 представлен график, показывающий фармакокинетику соединения 12i в плазме крови после перорального введения однократной дозы соединения 12i (треугольники, контроль) и соединения 30f (круги, исследуемое соединение) крысам. N=4 в каждой группе.
На фигуре 2 представлен график, иллюстрирующий действие соединения 12i (соед. 1) на выживание хомяков, инфицированных вирусом желтой лихорадки. **Р<0,01 по сравнению с плацебо. ***Р<0,001 по сравнению с плацебо. dpi - дней после инфекции.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Аспектом настоящего изобретения является соединение, представленное формулой I, или его фармацевтически приемлемая соль:
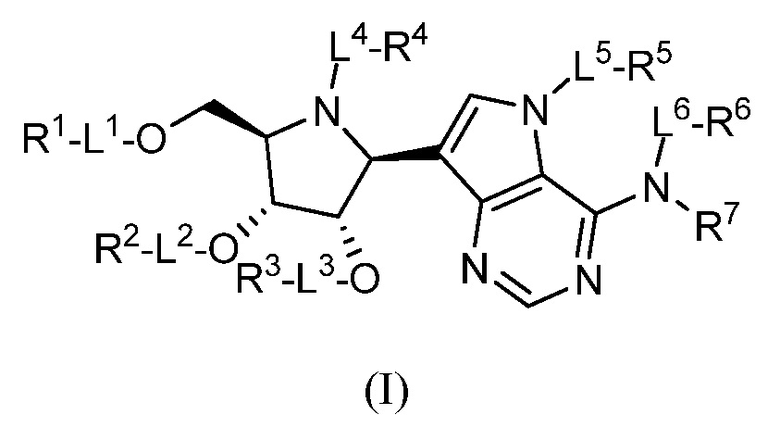
где:
L1, L2, L3, L4, L5 и L6, каждый, независимо, представляют собой связь или линкер -C(R0)2-O-;
R0, в каждом случае независимо, представляет собой H или (C1-C6)алкил;
R1, R2 и R3, каждый, независимо, выбраны из группы, включающей H, аминоацил, аминотионил, ацил, R10OC(O)-, фосфорил и аминофосфорил; или R1 и R2, взятые вместе, или R2 и R3, взятые вместе, выбраны из группы, включающей карбонил, тиокарбонил, фосфорил и (C1-C6)алкилфосфорил;
R4, R5 и R6, каждый, независимо, выбраны из группы, включающей H, ацил, фосфорил, алкилтио, R10OC(O)- и аминоалкил;
R7 представляет собой H; или R6, R7 и атом азота, к которому они присоединены, взятые вместе, представляют собой -N=CR20R21;
R10, в каждом случае независимо, выбран из группы, включающей H, (C1-C6)алкил, арил, гетероарил, аралкил и гетероаралкил;
R20 и R21, каждый, независимо, выбраны из группы, включающей H, алкил, амино, арил, гетероарил, аралкил и гетероаралкил;
при условии, что соединение, представленное формулой I, не представляет собой
В некоторых вариантах осуществления изобретения соединение формулы I представляет собой соединение, представленное формулой IA, или его фармацевтически приемлемую соль:
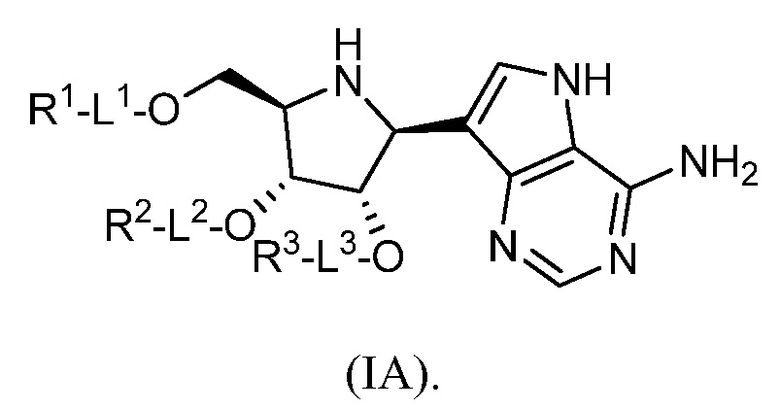
В некоторых вариантах осуществления изобретения, в соответствии с любым из вышеуказанных, L1-R1 представляет собой H.
В некоторых вариантах осуществления изобретения, в соответствии с любым из вышеуказанных, L2-R2 представляет собой H.
В некоторых вариантах осуществления изобретения, в соответствии с любым из вышеуказанных, L3-R3 представляет собой H.
Альтернативно, в некоторых вариантах осуществления изобретения, в соответствии с любым из вышеуказанных, L2-R2 и L3-R3 являются одинаковыми.
В некоторых вариантах осуществления изобретения, в соответствии с любым из вышеуказанных, каждый из L2-R2 и L3-R3 представляет собой H.
Альтернативно, в некоторых вариантах осуществления изобретения, в соответствии с любым из вышеуказанных, L1-R1 и L3-R3 являются одинаковыми.
В некоторых вариантах осуществления изобретения, в соответствии с любым из вышеуказанных, L1-R1 и L2-R2 являются одинаковыми.
В некоторых вариантах осуществления изобретения, в соответствии с любым из вышеуказанных, каждый из L1-R1 и L2-R2 представляет собой H.
В некоторых вариантах осуществления изобретения, в соответствии с любым из вышеуказанных, каждый из L1-R1 и L3-R3 представляет собой H.
Альтернативно, в некоторых вариантах осуществления изобретения, в соответствии с любым из вышеуказанных, L1-R1, L2-R2 и L3-R3 являются одинаковыми; и никто из L1-R1, L2-R2 и L3-R3 не представляет собой H.
В некоторых вариантах осуществления изобретения, в соответствии с любым из вышеуказанных, в каждом случае независимо, аминоацил представляет собой -C(=O)CH(NH2)(CH2)nCHR30R31, где n обозначает 0 или 1; и R30 и R31 каждый, независимо, выбраны из группы, включающей H, (C1-C6)алкил, арил, гетероарил, аралкил и гетероаралкил.
В некоторых вариантах осуществления изобретения R30 и R31, каждый, независимо, выбраны из группы, включающей H и (C1-C6)алкил.
В некоторых вариантах осуществления изобретения R30 и R31, каждый, независимо, представляют собой (C1-C6)алкил.
В некоторых вариантах осуществления изобретения, n обозначает 0; и R30 и R31 каждый, независимо, представляют собой метил.
В некоторых вариантах осуществления изобретения, в соответствии с любым из вышеуказанных, в каждом случае независимо, аминотионил представляет собой -C(=S)CH(NH2)(CH2)nCHR30R31, где n обозначает 0 или 1; и R30 и R31 каждый, независимо, выбраны из группы, включающей H, (C1-C6)алкил, арил, гетероарил, аралкил и гетероаралкил.
В некоторых вариантах осуществления изобретения R30 и R31 каждый, независимо, выбраны из группы, включающей H и (C1-C6)алкил.
В некоторых вариантах осуществления изобретения R30 и R31 каждый, независимо, представляют собой (C1-C6)алкил.
В некоторых вариантах осуществления изобретения, n обозначает 0; и R30 и R31 каждый, независимо, представляют собой метил.
В некоторых вариантах осуществления изобретения, в соответствии с любым из вышеуказанных, в каждом случае независимо, ацил представляет собой -C(=O)R40, где R40 выбран из группы, включающей H, (C1-C6)алкил, арил, гетероарил, аралкил и гетероаралкил.
В некоторых вариантах осуществления изобретения R40 представляет собой H.
В некоторых вариантах осуществления изобретения R40 представляет собой (C1-C6)алкил.
В некоторых вариантах осуществления изобретения, в соответствии с любым из вышеуказанных, в каждом случае независимо, R10 представляет собой H.
Альтернативно, в некоторых вариантах осуществления изобретения, в соответствии с любым из вышеуказанных, в каждом случае независимо, R10 представляет собой (C1-C6)алкил.
В некоторых вариантах осуществления изобретения, в соответствии с любым из вышеуказанных, в каждом случае независимо, аминофосфорил представляет собой -P(=O)(OR50)NR51R52, где
R50 выбран из группы, включающей H, (C1-C6)алкил, арил, арилалкил, гетероарил, гетероаралкил и -(CH2)mSC(=O)C(CH3)2CH2OH;
m обозначает 1 или 2;
R51 представляет собой H или (C1-C6)алкил; и
R52 выбран из группы, включающей H, (C1-C6)алкил, арил, аралкил, гетероарил, гетероаралкил и -CR60R61C(=O)OR62, где
R60 и R61 каждый, независимо, представляют собой H или (C1-C6)алкил; и
R62 выбран из группы, включающей H, (C1-C6)алкил, арил, аралкил, гетероарил, гетероаралкил.
В некоторых вариантах осуществления изобретения, в соответствии с любым из вышеуказанных, R50 представляет собой H.
В некоторых вариантах осуществления изобретения, в соответствии с любым из вышеуказанных, R50 представляет собой арил.
В некоторых вариантах осуществления изобретения, в соответствии с любым из вышеуказанных, R50 представляет собой -(CH2)mSC(=O)C(CH3)2CH2OH.
В некоторых вариантах осуществления изобретения, в соответствии с любым из вышеуказанных, m обозначает 2.
В некоторых вариантах осуществления изобретения, в соответствии с любым из вышеуказанных, R51 представляет собой H.
В некоторых вариантах осуществления изобретения, в соответствии с любым из вышеуказанных, R52 представляет собой аралкил.
Альтернативно, в некоторых вариантах осуществления изобретения, в соответствии с любым из вышеуказанных, R52 представляет собой -CR60R61C(=O)OR62.
В некоторых вариантах осуществления изобретения, в соответствии с любым из вышеуказанных, R60 представляет собой H; R61 представляет собой (C1-C6)алкил; и R62 представляет собой (C1-C6)алкил.
В некоторых вариантах осуществления изобретения соединение формулы I представляет собой соединение, представленное формулой IB, или его фармацевтически приемлемую соль:
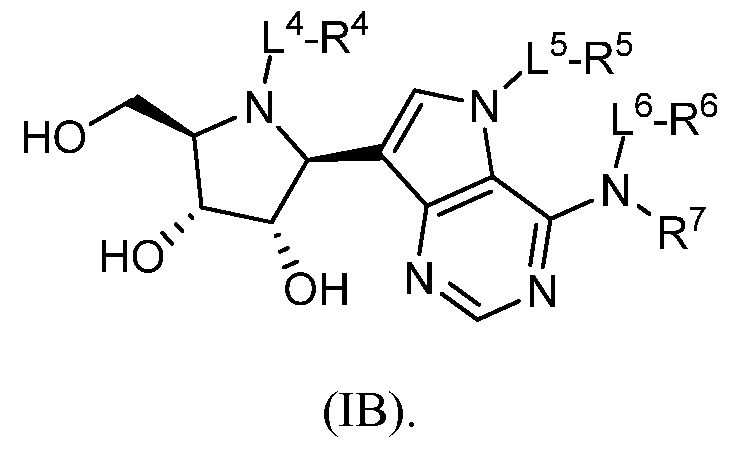
В некоторых вариантах осуществления изобретения, в соответствии с любым из вышеуказанных, R7 представляет собой H; каждый из L4, L5 и L6 представляет собой связь; и каждый из любых двух из R4, R5 и R6 представляет собой H.
В некоторых вариантах осуществления изобретения, в соответствии с любым из вышеуказанных, каждый из R4 и R5 представляет собой H.
Альтернативно, в некоторых вариантах осуществления изобретения, в соответствии с любым из вышеуказанных, каждый из R5 и R6 представляет собой H.
Альтернативно, в некоторых вариантах осуществления изобретения, в соответствии с любым из вышеуказанных, каждый из R4 и R6 представляет собой H.
В некоторых вариантах осуществления изобретения, в соответствии с любым из вышеуказанных, R10 из группы R10OC(O)- в определениях R4, R5 и R6 представляет собой H или (C1-C6)алкил.
В некоторых вариантах осуществления изобретения, в соответствии с любым из вышеуказанных, аминоалкил в определениях R4, R5 и R6 представляет собой -CH2N(CH3)2.
В некоторых вариантах осуществления изобретения, каждый из L4, L5 и L6 представляет собой связь; и R6, R7 и атом азота, к которому они присоединены, взятые вместе, представляют собой -N=CR20R21.
В некоторых вариантах осуществления изобретения, в соответствии с любым из вышеуказанных, R20 представляет собой H и R21 представляет собой амино.
В некоторых вариантах осуществления изобретения, в соответствии с любым из вышеуказанных, каждый из R4 и R5 представляет собой H.
Альтернативно, в некоторых вариантах осуществления R7 представляет собой Н; по меньшей мере, один из L4, L5 и L6, представляет собой линкер -C(R0)2-O-; и любой из R4, R5 или R6, связанный по меньшей мере с одним линкером -C(R0)2-O-, представляет собой фосфорил.
Определения
Термин "алкил", используемый в настоящем документе, является термином, используемым в данной области техники, и обозначает насыщенные алифатические группы, включая алкильные группы с прямой цепью, алкильные группы с разветвленной цепью, циклоалкильные (алициклические) группы, алкилзамещенные циклоалкильные группы и циклоалкилзамещенные алкильные группы. В некоторых вариантах осуществления алкил с линейной цепью или разветвленной цепью имеет в своем скелете около 30 или менее атомов углерода (например, С1-С30 для линейной цепи, С3-С30 для разветвленной цепи), и, альтернативно, около 20 или менее атомов углерода. Подобным образом, циклоалкилы содержат от около 3 до около 10 атомов углерода в своей кольцевой структуре, и, альтернативно, около 5, 6 или 7 атомов углерода в кольцевой структуре.
Термин "амино" является термином, используемым в данной области техники, и относится как к незамещенным, так и замещенным аминам, например, группе, которая может быть представлена общей формулой:
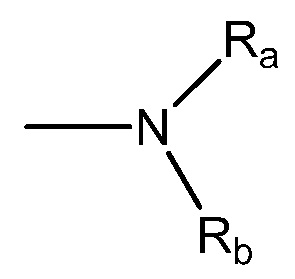 и
и 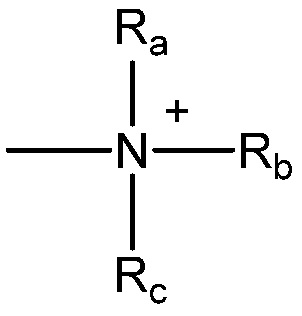 ,
,
где Ra, Rb и Rc, каждый, независимо, представляют собой водород, алкил, алкенил, -(CH2)x-Rd, или Ra и Rb, взяты вместе с атомом N, к которому они присоединены, образуют гетероцикл, содержащий от 4 до 8 атомов в кольцевой структуре; Rd представляет арил, циклоалкил, циклоалкенил, гетероциклил или полициклил; и x обозначает ноль или целое число в диапазоне от 1 до 8. В некоторых вариантах осуществления только один из Ra или Rb может представлять собой карбонил, например, Ra, Rb и азот, взятые вместе, не образуют имид. В других вариантах осуществления, Ra и Rb (и, необязательно, RC), каждый, независимо, представляют собой водород, алкил, алкенил или -(CH2)x-Rd.
Термин "ацил" является термином, используемым в данной области техники, и относится к любой группе или радикалу вида RCO-, где R представляет собой любую органическую группу, например, алкил, арил, гетероарил, аралкил и гетероаралкил. Типичные ацильные группы включают ацетил, бензоил и малонил.
Термин "аминоалкил", используемый в настоящем документе, относится к алкильной группе, замещенной одной или несколькими аминогруппами.
Термин “аминоацил” является термином, используемым в данной области техники, и относится к ацильной группе, замещенной одной или несколькими аминогруппами.
Термин "аминотионил", используемый в настоящем документе, относится к аналогу аминоацила, в котором O в RC(O)- заменен на серу и, соответственно, имеет вид RC(S)-.
Термин “фосфорил” является термином, используемым в данной области техники, и может обычно быть представлен формулой:
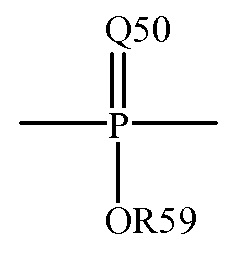
где Q50 представляет собой S или O, и R59 представляет собой водород, низший алкил или арил; например, -P(O)(OMe)- или -P(O)(OH)2. Когда она используется для замещения, например, алкила, фосфорильная группа фосфорилалкила может быть представлена общими формулами:
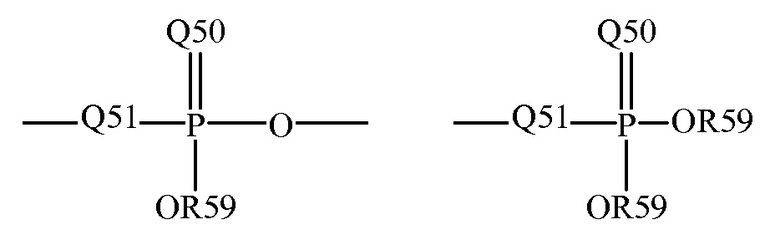
где Q50 и R59, каждый независимо, определены выше, и Q51 представляет собой О, S или N; например, -O-P(O)(OH)OMe или -NH-P(O)(OH)2. Когда Q50 представляет собой S, фосфорильный фрагмент представляет собой "фосфоротиоат".
Термин "аминофосфорил" используемый в настоящем документе, относится к фосфорильной группе, замещенной по меньшей мере одной аминогруппой, как определено в настоящем документе; например, -P(O)(OH)NMe2.
Термин “карбонил”, используемый в настоящем документе, относится к -C(O)-.
Термин “тиокарбонил”, используемый в настоящем документе, относится к -C(S)-.
Термин “алкилфосфорил”, используемый в настоящем документе, относится к фосфорильной группе, замещенной по меньшей мере одной алкильной группой, как определено в настоящем документе; например, -P(O)(OH)Me.
Термин “алкилтио”, используемый в настоящем документе, относится к алкил-S-.
Термин “арил” является термином, используемым в данной области техники, и относится к моноциклическим, бициклическим и полициклическим ароматическим углеводородным группам, например, бензол, нафталин, антрацен и пирен. Ароматическое кольцо может быть замещено в одном или нескольких положениях кольца одним или несколькими заместителями, такими как галоген, азид, алкил, аралкил, алкенил, алкинил, циклоалкил, гидроксил, алкоксил, амино, нитро, сульфгидрил, имино, амидо, фосфонат, фосфинат, карбонил, карбоксил, силил, простой эфир, алкилтио, сульфонил, сульфонамидо, кетон, альдегид, сложный эфир, гетероциклил, ароматические или гетероароматические группы, фторалкил (такой как трифторметил), циано или тому подобное. Термин "арил" также включает полициклические кольцевые системы, содержащие два или более циклических колец, в которых два или более атомов углерода являются общими для двух смежных колец (кольца представляют собой "конденсированные кольца"), где по меньшей мере одно из колец является ароматическим углеводородом, например, другие циклические кольца могут быть циклоалкилами, циклоалкенилами, циклоалкинилами, арилами, гетероарилами и/или гетероциклилами.
Термин "гетероатом" является термином, используемым в данной области техники, и включает атом любого элемента, отличный от атома углерода или водорода. Иллюстративные гетероатомы включают бор, азот, кислород, фосфор, серу и селен и, альтернативно, кислород, азот или серу.
Термин "гетероарил" является термином, используемым в данной области техники, и относится к моноциклическим, бициклическим и полициклическим ароматическим группам, имеющим один или несколько гетероатомов в структуре кольца, например, пиррол, фуран, тиофен, имидазол, оксазол, тиазол, триазол, пиразол, пиридин, пиразин, пиридазин и пиримидин и тому подобное. "Гетероарил" может быть замещен в одном или нескольких положениях кольца одним или несколькими заместителями, такими как галоген, азид, алкил, аралкил, алкенил, алкинил, циклоалкил, гидроксил, алкокси, амино, нитро, сульфгидрил, имино, амидо, фосфонат, фосфинат, карбонил, карбоксил, силил, простой эфир, алкилтио, сульфонил, сульфонамидо, кетон, альдегид, сложный эфир, гетероциклил, ароматические или гетероароматические остатки, фторалкил (такой как трифторметил), циано или тому подобное. Термин "гетероарил" также включает полициклические кольцевые системы, имеющие два или более циклических кольца, в которых два или более атомов углерода являются общими для двух смежных колец (кольца представляют собой "конденсированные кольца"), где по меньшей мере одно из колец является ароматической группой, содержащей один или несколько гетероатомов в структуре кольца, например, другие циклические кольца могут быть циклоалкилами, циклоалкенилами, циклоалкинилами, арилами, гетероарилами и/или гетероциклилами.
Термин “аралкил” является термином, используемым в данной области техники, и относится к алкильной группе, замещенной арильной группой.
Термин “гетероаралкил” является термином, используемым в данной области техники, и относится к алкильной группе, замещенной гетероарильной группой.
Некоторые соединения, содержащиеся в композициях по настоящему изобретению, могут существовать в определенных геометрических или стереоизомерных формах. Кроме того, соединения по настоящему изобретению также могут быть оптически активными. Настоящее изобретение рассматривает все такие соединения, в том числе цис- и транс-изомеры, (R)- и (S)-энантиомеры, диастереоизомеры, (D)-изомеры, (L)-изомеры, их рацемические смеси и другие их смеси, как включенные в объем настоящего изобретения. В заместителе, таком как алкильная группа, могут иметься дополнительные асимметрические атомы углерода. Все такие изомеры, а также их смеси, включены в настоящее изобретение.
Если требуется, например, конкретный энантиомер соединения по настоящему изобретению, он может быть получен путем асимметрического синтеза, либо путем взаимодействия с хиральным вспомогательным соединением, когда полученную смесь диастереомеров разделяют, и вспомогательную группу отщепляют с получением желаемых чистых энантиомеров. С другой стороны, когда молекула содержит основную функциональную группу, такую как амино, или кислотную функциональную группу, такую как карбоксил, диастереомерные соли образуются с соответствующей оптически-активной кислотой или основанием, с последующим разделением образованных таким образом диастереоизомеров путем фракционной кристаллизации или хроматографическими методами, хорошо известными в этой области техники, и последующей регенерацией чистых энантиомеров.
Следует иметь в виду, что "замещение" или "замещенный с помощью" включает предполагаемое условие, при котором осуществляется замещение в соответствии с допустимой валентностью замещенного атома и заместителя, и что замещение приводит к стабильному соединению, например, которое не подвергается спонтанно трансформации, такой как перегруппировка, циклизация, отщепление или иная реакция.
Термин "замещенный" также включает все допустимые заместители органических соединений. В широком аспекте допустимые заместители включают ациклические и циклические, разветвленные и неразветвленные, карбоциклические и гетероциклические, ароматические и неароматические заместители органических соединений. Иллюстративные заместители включают, например, те, которые описаны в настоящем документе выше. Количество допустимых заместителей может быть равно одному или их может быть несколько, и они могут быть одинаковыми или разными для соответствующих органических соединений. Для целей настоящего изобретения гетероатомы, такие как азот, могут иметь водородные заместители и/или любые допустимые заместители органических соединений, описанные в настоящем документе, которые соответствуют валентностям гетероатомов. Объем настоящего изобретения не ограничивают каким-либо образом допустимыми заместителями органических соединений.
Для целей настоящего изобретения химические элементы идентифицируются в соответствии с Периодической таблицей элементов, CAS version, Handbook of Chemistry and Physics, 67th Ed., 1986-87, внутри обложки.
Другие химические термины используются в настоящем документе в соответствии с их обычным употреблением в данной области, как проиллюстрировано, например, в словаре The McGraw-Hill Dictionary of Chemical Terms (ed. Parker, S., 1985), McGraw-Hill, San Francisco, включенном в настоящий документ посредством ссылки). Если не определено иначе, все технические и научные термины, используемые в настоящем документе, имеют те же значения, которые обычно подразумевает специалист с обычной квалификацией в области, к которой относится настоящее изобретение.
Термин "защитная группа", используемый в настоящем документе, относится к временным заместителям, которые защищают потенциально реакционноспособные функциональные группы от нежелательных химических преобразований. Примеры таких защитных групп включают сложноэфирные группы карбоновых кислот и бороновых кислот, группы эфиров спиртов, а также группы ацеталей и кеталей альдегидов и кетонов. Например, фраза «N-концевая защитная группа» или «амино-защитная группа», используемая в настоящем документе, относится к различным амино-защитным гpуппам, которые могут быть использованы для защиты N-конца аминокислоты или пептида от нежелательных реакций в ходе процедур синтеза. Примеры подходящих групп включают ацильные защитные группы, такие как, для иллюстрации, формил, дансил, ацетил, бензоил, трифторацетил, сукцинил и метоксисукцинил; ароматические, как, например, бензилоксикарбонил (Cbz); и алифатические уретановые защитные группы, такие как трет-бутоксикарбонил (Boc) или 9-флуоренилметоксикарбонил (Fmoc).
Термин "амино-защитная группа" или "N-концевая защитная группа" относится к таким группам, которые предназначены для защиты α-N-конца аминокислоты или пептида, или, в противном случае, для защиты аминогруппы аминокислоты или пептида от нежелательных реакций в процессе синтеза. Обычно используемые N-защитные группы описаны в обзоре Greene, Protective Groups In Organic Synthesis, (John Wiley & Sons, New York (1981)), который включен в настоящее описание посредством ссылки. Кроме того, защитные группы могут быть использованы в качестве пролекарств, которые легко расщепляются in vivo, например, путем ферментативного гидролиза, с высвобождением биологически активного исходного вещества. α-N-защитные группы включают группы низшего алканоила, такие как формил, ацетил ("Ac"), пропионил, пивалоил, трет-бутилацетил и тому подобное; другие ацильные группы включают 2-хлорацетил, 2-бромацетил, трифторацетил, трихлорацетил, фталил, о-нитрофеноксиацетил, хлорбутирил, бензоил, 4-хлорбензоил, 4-бромбензоил, 4-нитробензоил и тому подобное; сульфонильные группы, такие как бензолсульфонил, п-толуолсульфонил и тому подобное; образующие карбамат группы, такие как бензилоксикарбонил, п-хлорбензилоксикарбонил, п-метоксибензилоксикарбонил, п-нитробензилоксикарбонил, 2-нитробензилоксикарбонил, п-бромбензилоксикарбонил, 3,4-диметоксибензилоксикарбонил, 3,5-диметоксибензилоксикарбонил, 2,4-диметоксибензилоксикарбонил, 4-этоксибензилоксикарбонил, 2-нитро-4,5-диметоксибензилоксикарбонил, 3,4,5-триметоксибензилоксикарбонил, 1-(п-бифенилил)-1-метилэтоксикарбонил, α,α-диметил-3,5-диметоксибензилоксикарбонил, бензгидрилоксикарбонил, трет-бутилоксикарбонил, диизопропилметоксикарбонил, изопропилоксикарбонил, этоксикарбонил, метоксикарбонил, аллилоксикарбонил, 2,2,2-трихлорэтоксикарбонил, феноксикарбонил, 4-нитрофеноксикарбонил, флуоренил-9-метоксикарбонил, циклопентилоксикарбонил, адамантилоксикарбонил, циклогексилоксикарбонил, фенилтиокарбонил и тому подобное; арилалкильные группы, такие как бензил, трифенилметил, бензилоксиметил, 9-флуоренилметилоксикарбонил (Fmoc) и тому подобное, и силильные группы, такие как триметилсилил и тому подобные. Еще другие примеры включают теил, сукцинил, метоксисукцинил, суберил, адипил, азелаил, дансил, бензилоксикарбонил, метоксиазелалил, метоксиадипил, метоксисуберил и 2,4-динитрофенил.
Термин "карбокси-защитная группа" или "С-концевая защитная группа" относится к защищающей группу карбоновой кислоты сложноэфирной или амидной группе, используемых для блокирования или защиты функциональной группы карбоновой кислоты, в то время как выполняются реакции, охватывающие другие функциональные участки соединения. Карбоксизащитные группы описаны в обзоре Greene, Protective Groups in Organic Synthesis pp. 152-186 (1981), который включен в настоящем документе посредством ссылки. Кроме того, карбокси-защитная группа может быть использована в качестве пролекарства, тем самым карбокси-защитная группа может быть легко отщеплена in vivo, например, путем ферментативного гидролиза, с высвобождением биологически активного исходного вещества. Такие карбокси-защитные группы хорошо известны специалистам в данной области техники, широко применяются в защите карбоксильных групп в области пенициллина и цефалоспорина, как описано в патенте США № 3840556 и 3719667, описания которых, таким образом, включены в настоящий документ посредством ссылки. Типичными карбокси-защитными группами являются низший алкил C1-C8 (например, метил, этил или трет-бутил и тому подобное); арилалкил, такой как фенетил или бензил, и их замещенные производные, например алкоксибензильная или нитробензильная группы и тому подобное; арилалкенил, такой как фенилэтенил и тому подобное; арил и его замещенные производные, такие как 5-инданил и тому подобное; диалкиламиноалкил, такой как диметиламиноэтил и т.п.); алканоилоксиалкильные группы, такие как ацетоксиметил, бутирилоксиметил, валерилоксиметил, изобутирилоксиметил, изовалерилоксиметил, 1-(пропионилокси)-1-этил, 1-(пивалоилокси)-1-этил, 1-метил-1-(пропионилокси)-1-этил, пивалоилоксиметил, пропионилоксиметил и тому подобное; циклоалканоилоксиалкильные группы, такие как циклопропилкарбонилоксиметил, циклобутилкарбонилоксиметил, циклопентилкарбонилоксиметил, циклогексилкарбонилоксиметил и тому подобное; ароилоксиалкил, такой как бензоилоксиметил, бензоилоксиэтил и тому подобное; арилалкилкарбонилоксиалкил, такой как бензилкарбонилоксиметил, 2-бензилкарбонилоксиэтил и тому подобное; алкоксикарбонилалкил или циклоалкилоксикарбонилалкил, такой как метоксикарбонилметил, циклогексилоксикарбонилметил, 1-метоксикарбонил-1-этил и тому подобное; алкоксикарбонилоксиалкил или циклоалкилоксикарбонилоксиалкил, такой как метоксикарбонилоксиметил, трет-бутилоксикарбонилоксиметил, 1-этоксикарбонилокси-1-этил, 1-циклогексилоксикарбонилокси-1-этил и тому подобное; арилоксикарбонилоксиалкил, такой как 2-(феноксикарбонилокси)этил, 2-(5-инданилоксикарбонилокси)этил и тому подобное; алкоксиалкилкарбонилоксиалкил, такой как 2-(1-метокси-2-метилпропан-2-оилокси)этил и тому подобное; арилалкилоксикарбонилоксиалкил, такой как 2-(бензилоксикарбонилокси)этил и тому подобное; арилалкенилоксикарбонилоксиалкил, такой как 2-(3-фенилпропен-2-илоксикарбонилокси)этил и тому подобное; алкоксикарбониламиноалкил, такой как трет-бутилоксикарбониламинометил и тому подобное; алкиламинокарбониламиноалкил, такой как метиламинокарбониламинометил и тому подобное; алканоиламиноалкил, такой как ацетиламинометил и тому подобное; гетероциклилкарбонилоксиалкил, такой как 4-метилпиперазинилкарбонилоксиметил и тому подобное; диалкиламинокарбонилалкил, такой как диметиламинокарбонилметил, диэтиламинокарбонилметил и тому подобное; (5-(низший алкил)-2-оксо-1,3-диоксолен-4-ил)алкил, такой как (5-трет-бутил-2-оксо-1,3-диоксолен-4-ил)метил и тому подобное; и (5-фенил-2-оксо-1,3-диоксолен-4-ил)алкил, такой как (5-фенил-2-оксо-1,3-диоксолен-4-ил)метил и тому подобное. Типичные амидные карбокси-защитные группы представляют собой группы аминокарбонила и низшего алкиламинокарбонила. Например, аспарагиновая кислота может быть защищена на α-С-конце с помощью подвижной для кислоты группы (например, трет-бутил) и защищена на β-С-конце с помощью подвижной при гидрировании группы (например, бензил), потом защитную группу селективно удаляют в процессе синтеза. Как указано выше, карбокси-защитная группа может также представлять собой сложноэфирную группу, содержащую низший алкил, циклоалкил или арилалкил, например, метиловый эфир, этиловый эфир, пропиловый эфир, изопропиловый эфир, бутиловый эфир, втор-бутиловый эфир, амиловый эфир, изоамиловый эфир, октиловый эфир, циклогексиловый эфир, фенилэтиловый эфир и т.п., или алканоилоксиалкиловый эфир, циклоалканоилоксиалкиловый эфир, ароилоксиалкиловый эфир или арилалкилкарбонилоксиалкиловый эфир.
Термин “аминокислота” является термином, используемым в данной области техники, и относится к альфа- и бета-аминокарбоновым кислотам, включая так называемые природные альфа-аминокислоты и неприродные аминокислоты. Природные альфа-аминокислоты, в частности, включают аланин (Ala), аргинин (Arg), аспарагин (Asn), аспарагиновую кислоту (Asp), цистеин (Cys), глутаминовую кислоту (Glu), глутамин (Gln), глицин (Gly), гистидин (His), изолейцин (Ile), лейцин (Leu), лизин (Lys), метионин (Met), орнитин (Orn), фенилаланин (Phe), пролин (Pro), селеноцистеин, серин (Ser), таурин, треонин (Thr), триптофан (Trp), тирозин (Tyr) и валин (Val). Полярные встречающиеся в природе альфа-аминокислоты включают аргинин, аспарагин, аспарагиновую кислоту, цистеин, глутаминовую кислоту, глутамин, гистидин, лизин, орнитин, серин, треонин и тирозин. Неполярные встречающиеся в природе альфа-аминокислоты включают аланин, глицин, изолейцин, лейцин, метионин, фенилаланин, пролин, триптофан и валин.
Не встречающиеся в природе аминокислоты включают, но не ограничиваются ими, D-аминокислоты (то есть, аминокислота противоположной хиральности по отношению к встречающейся в природе форме), N-α-метил аминокислоты, C-α-метил аминокислоты, β-метил аминокислоты, β-аланин (β-Ala), норвалин (Nva), норлейцин (Nle), 4-аминомасляную кислоту (γ-Abu), 2-аминоизомасляную кислоту (Aib), 6-аминокапроновую кислоту (ε-Ahx), орнитин (Orn), гидроксипролин (Hyp), саркозин, цитруллин, цистеиновую кислоту, циклогексилаланин, α-амино изомасляную кислоту, трет-бутилглицин, трет-бутилаланин, 3-аминопропионовую кислоту, 2,3-диаминопропионовую кислоту (2,3-diaP), D- или L-фенилглицин, D- или L-2-нафтилаланин (2-Nal), 1,2,3,4-тетрагидроизохинолин-3-карбоновую кислоту (Tic), D- или L-2-тиенилаланин (Thi), D- или L-3-тиенилаланин, D- или L-1-, 2-, 3- или 4-пиренилаланин, D- или L-(2-пиридинил)аланин, D- или L-(3-пиридинил)аланин, D- или L-(2-пиразинил)аланин, D- или L-(4-изопропил)-фенилглицин, D-(трифторметил)-фенилглицин, D (трифторметил)-фенилаланин, D-п-фторфенилаланин, D- или L-п-бифенилаланин, D- или L-п-метоксибифенилаланин, метионин сульфоксид (MSO) и гомоаргинин (Хар). Другие примеры включают D- или L-2-индол(алкил)аланины и D- или L-алкилаланины, где алкил замещен или незамещен метилом, этилом, пропилом, гексилом, бутилом, пентилом, изопропилом, изобутилом или изопентилом, и фосфоно- или сульфатированные (например, -SO3H) не карбоксилатные аминокислоты.
Другие примеры неприродных амино кислот включают 3-(2-хлорфенил)-аланин, 3-хлор-фенилаланин, 4-хлор-фенилаланин, 2-фтор-фенилаланин, 3-фтор-фенилаланин, 4-фтор-фенилаланин, 2-бром-фенилаланин, 3-бром-фенилаланин, 4-бром-фенилаланин, гомофенилаланин, 2-метил-фенилаланин, 3-метил-фенилаланин, 4-метил-фенилаланин, 2,4-диметил-фенилаланин, 2-нитро-фенилаланин, 3-нитро-фенилаланин, 4-нитро-фенилаланин, 2,4-динитро-фенилаланин, 1,2,3,4-тетрагидроизохинолин-3-карбоновую кислоту, 1,2,3,4-тетрагидроноргарман-3-карбоновую кислоту, 1-нафтилаланин, 2-нафтилаланин, пентафторфенилаланин, 2,4-дихлор-фенилаланин, 3,4-дихлор-фенилаланин, 3,4-дифтор-фенилаланин, 3,5-дифтор-фенилаланин, 2,4,5-трифтор-фенилаланин, 2-трифторметил-фенилаланин, 3-трифторметил-фенилаланин, 4-трифторметил-фенилаланин, 2-циано-фенилаланин, 3-циано-фенилаланин, 4-циано-фенилаланин, 2-йод-фенилаланин, 3-йод-фенилаланин, 4-йод-фенилаланин, 4-метоксифенилаланин, 2-аминометил-фенилаланин, 3-аминометил-фенилаланин, 4-аминометил-фенилаланин, 2-карбамоил-фенилаланин, 3-карбамоил-фенилаланин, 4-карбамоил-фенилаланин, м-тирозин, 4-амино-фенилаланин, стирилаланин, 2-амино-5-фенил-пентановую кислоту, 9-антрилаланин, 4-трет-бутил-фенилаланин, 3,3-дифенилаланин, 4,4'-дифенилаланин, бензоилфенилаланин, α-метил-фенилаланин, α-метил-4-фтор-фенилаланин, 4-тиазолилаланин, 3-бензотиенилаланин, 2-тиенилаланин, 2-(5-бромтиенил)-аланин, 3-тиенилаланин, 2-фурилаланин, 2-пиридилаланин, 3-пиридилаланин, 4-пиридилаланин, 2,3-диаминопропионовую кислоту, 2,4-диаминобутановую кислоту, аллилглицин, 2-амино-4-бром-4-пентеновую кислоту, пропаргилглицин, 4-аминоциклопент-2-енкарбоновую кислоту, 3-аминоциклопентанкарбоновую кислоту, 7-амино-гептановую кислоту, дипропилглицин, пипеколиновую кислоту, азетидин-3-карбоновую кислоту, циклопропилглицин, циклопропилаланин, 2-метокси-фенилглицин, 2-тиенилглицин, 3-тиенилглицин, α-бензил-пролин, α-(2-фтор-бензил)-пролин, α-(3-фтор-бензил)-пролин, α-(4-фтор-бензил)-пролин, α-(2-хлор-бензил)-пролин, α-(3-хлор-бензил)-пролин, α-(4-хлор-бензил)-пролин, α-(2-бром-бензил)-пролин, α-(3-бром-бензил)-пролин, α-(4-бром-бензил)-пролин, α-фенетил-пролин, α-(2-метил-бензил)-пролин, α-(3-метил-бензил)-пролин, α-(4-метил-бензил)-пролин, α-(2-нитро-бензил)-пролин, α-(3-нитро-бензил)-пролин, α-(4-нитро-бензил)-пролин, α-(1-нафталенилметил)-пролин, α-(2-нафталенилметил)-пролин, α-(2,4-дихлор-бензил)-пролин, α-(3,4-дихлор-бензил)-пролин, α-(3,4-дифтор-бензил)-пролин, α-(2-трифторметил-бензил)-пролин, α-(3-трифторметил-бензил)-пролин, α-(4-трифторметил-бензил)-пролин, α-(2-циано-бензил)-пролин, α-(3-циано-бензил)-пролин, α-(4-циано-бензил)-пролин, α-(2-йод-бензил)-пролин, α-(3-йод-бензил)-пролин, α-(4-йод-бензил)-пролин, α-(3-фенил-аллил)-пролин, α-(3-фенил-пропил)-пролин, α-(4-трет-бутил-бензил)-пролин, α-бензгидрил-пролин, α-(4-бифенилметил)-пролин, α-(4-тиазолилметил)-пролин, α-(3-бензо[b]тиофенилметил)-пролин, α-(2-тиофенилметил)-пролин, α-(5-бром-2-тиофенилметил)-пролин, α-(3-тиофенилметил)-пролин, α-(2-фуранилметил)-пролин, α-(2-пиридинилметил)-пролин, α-(3-пиридинилметил)-пролин, α-(4-пиридинилметил)-пролин, α-аллил-пролин, α-пропинил-пролин, γ-бензил-пролин, γ-(2-фтор-бензил)-пролин, γ-(3-фтор-бензил)-пролин, γ-(4-фтор-бензил)-пролин, γ-(2-хлор-бензил)-пролин, γ-(3-хлор-бензил)-пролин, γ-(4-хлор-бензил)-пролин, γ-(2-бром-бензил)-пролин, γ-(3-бром-бензил)-пролин, γ-(4-бром-бензил)-пролин, γ-(2-метил-бензил)-пролин, γ-(3-метил-бензил)-пролин, γ-(4-метил-бензил)-пролин, γ-(2-нитро-бензил)-пролин, γ-(3-нитро-бензил)-пролин, γ-(4-нитро-бензил)-пролин, γ-(1-нафталенилметил)-пролин, γ-(2-нафталенилметил)-пролин, γ-(2,4-дихлор-бензил)-пролин, γ-(3,4-дихлор-бензил)-пролин, γ-(3,4-дифтор-бензил)-пролин, γ-(2-трифторметил-бензил)-пролин, γ-(3-трифторметил-бензил)-пролин, γ-(4-трифторметил-бензил)-пролин, γ-(2-циано-бензил)-пролин, γ-(3-циано-бензил)-пролин, γ-(4-циано-бензил)-пролин, γ-(2-йод-бензил)-пролин, γ-(3-йод-бензил)-пролин, γ-(4-йод-бензил)-пролин, γ-(3-фенил-аллил-бензил)-пролин, γ-(3-фенил-пропил-бензил)-пролин, γ-(4-трет-бутил-бензил)-пролин, γ-бензгидрил-пролин, γ-(4-бифенилметил)-пролин, γ-(4-тиазолилметил)-пролин, γ-(3-бензотиоиенилметил)-пролин, γ-(2-тиенилметил)-пролин, γ-(3-тиенилметил)-пролин, γ-(2-фуранилметил)-пролин, γ-(2-пиридинилметил)-пролин, γ-(3-пиридинилметил)-пролин, γ-(4-пиридинилметил)-пролин, γ-аллил-пролин, γ-пропинил-пролин, транс-4-фенил-пирролидин-3-карбоновую кислоту, транс-4-(2-фтор-фенил)-пирролидин-3-карбоновую кислоту, транс-4-(3-фтор-фенил)-пирролидин-3-карбоновую кислоту, транс-4-(4-фтор-фенил)-пирролидин-3-карбоновую кислоту, транс-4-(2-хлор-фенил)-пирролидин-3-карбоновую кислоту, транс-4-(3-хлор-фенил)-пирролидин-3-карбоновую кислоту, транс-4-(4-хлор-фенил)-пирролидин-3-карбоновую кислоту, транс-4-(2-бром-фенил)-пирролидин-3-карбоновую кислоту, транс-4-(3-бром-фенил)-пирролидин-3-карбоновую кислоту, транс-4-(4-бром-фенил)-пирролидин-3-карбоновую кислоту, транс-4-(2-метил-фенил)-пирролидин-3-карбоновую кислоту, транс-4-(3-метил-фенил)-пирролидин-3-карбоновую кислоту, транс-4-(4-метил-фенил)-пирролидин-3-карбоновую кислоту, транс-4-(2-нитро-фенил)-пирролидин-3-карбоновую кислоту, транс-4-(3-нитро-фенил)-пирролидин-3-карбоновую кислоту, транс-4-(4-нитро-фенил)-пирролидин-3-карбоновую кислоту, транс-4-(1-нафтил)-пирролидин-3-карбоновую кислоту, транс-4-(2-нафтил)-пирролидин-3-карбоновую кислоту, транс-4-(2,5-дихлор-фенил)-пирролидин-3-карбоновую кислоту, транс-4-(2,3-дихлор-фенил)-пирролидин-3-карбоновую кислоту, транс-4-(2-трифторметил-фенил)-пирролидин-3-карбоновую кислоту, транс-4-(3-трифторметил-фенил)-пирролидин-3-карбоновую кислоту, транс-4-(4-трифторметил-фенил)-пирролидин-3-карбоновую кислоту, транс-4-(2-циано-фенил)-пирролидин-3-карбоновую кислоту, транс-4-(3-циано-фенил)-пирролидин-3-карбоновую кислоту, транс-4-(4-циано-фенил)-пирролидин-3-карбоновую кислоту, транс-4-(2-метокси-фенил)-пирролидин-3-карбоновую кислоту, транс-4-(3-метокси-фенил)-пирролидин-3-карбоновую кислоту, транс-4-(4-метокси-фенил)-пирролидин-3-карбоновую кислоту, транс-4-(2-гидрокси-фенил)-пирролидин-3-карбоновую кислоту, транс-4-(3-гидрокси-фенил)-пирролидин-3-карбоновую кислоту, транс-4-(4-гидрокси-фенил)-пирролидин-3-карбоновую кислоту, транс-4-(2,3-диметокси-фенил)-пирролидин-3-карбоновую кислоту, транс-4-(3,4-диметокси-фенил)-пирролидин-3-карбоновую кислоту, транс-4-(3,5-диметокси-фенил)-пирролидин-3-карбоновую кислоту, транс-4-(2-пиридинил)-пирролидин-3-карбоновую кислоту, транс-4-(3-пиридинил)-пирролидин-3-карбоновую кислоту, транс-4-(6-метокси-3-пиридинил)-пирролидин-3-карбоновую кислоту, транс-4-(4-пиридинил)-пирролидин-3-карбоновую кислоту, транс-4-(2-тиенил)-пирролидин-3-карбоновую кислоту, транс-4-(3-тиенил)-пирролидин-3-карбоновую кислоту, транс-4-(2-фуранил)-пирролидин-3-карбоновую кислоту, транс-4-изопропил-пирролидин-3-карбоновую кислоту, 4-фосфонометил-фенилаланин, бензил-фосфотреонин, (1'-амино-2-фенил-этил)оксиран, (1'-амино-2-циклогексил-этил)оксиран, (1'-амино-2-[3-бром-фенил]этил)оксиран, (1'-амино-2-[4-(бензилокси)фенил]этил)оксиран, (1'-амино-2-[3,5-дифтор-фенил]этил)оксиран, (1'-амино-2-[4-карбамоил-фенил]этил)оксиран, (1'-амино-2-[бензилокси-этил])оксиран, (1'-амино-2-[4-нитро-фенил]этил)оксиран, (1'-амино-3-фенил-пропил)оксиран, (1'-амино-3-фенил-пропил)оксиран и/или соли и/или их производные с защитными группами.
Бета-аминокислоты включают, без ограничения, бета-аланин (3-аминопропановой кислоты).
Термин "соединение по настоящему изобретению", используемый в настоящем документе, означает соединение формулы I и его фармацевтически приемлемые соли.
Термин "фармацевтически приемлемая соль", используемый в настоящем документе, включает соли, полученные из неорганических или органических кислот, включающих, например, хлористоводородную, бромистоводородную, серную, азотную, хлорную, фосфорную, муравьиную, уксусную, молочную, малеиновую, фумаровую, янтарную, винную, гликолевую, салициловую, лимонную, метансульфоновую, бензолсульфоновую, бензойную, малоновую, трифторуксусную, трихлоруксусную, нафталин-2-сульфоновую и другие кислоты. Фармацевтически приемлемые соли могут включать формы, в которых соотношение молекул, содержащих соль, не равно 1: 1. Например, соль может содержать более чем одну неорганическую или органическую молекулу кислоты на молекулу основания, например, две молекулы соляной кислоты на молекулу соединения формулы I. В качестве другого примера, соль может содержать менее чем одну неорганическую или органическую молекулу кислоты на молекулу основания, например, две молекулы соединения формулы I на одну молекулу винной кислоты.
Термины "носитель" и "фармацевтически приемлемый носитель", используемые в настоящем описании, относятся к разбавителю, адъюванту, наполнителю или носителю, с которым соединение вводят или получают для введения. Неограничивающие примеры таких фармацевтически приемлемых носителей включают жидкости, такие как вода, физиологический раствор и масла; и твердые вещества, такие как аравийская камедь, желатин, крахмальная паста, тальк, кератин, коллоидный диоксид кремния, мочевина и тому подобное. Кроме того, могут быть использованы вспомогательные вещества, стабилизаторы, загустители, смазывающие вещества, ароматизаторы и красители. Другие примеры подходящих фармацевтических носителей описаны в Remington Remington's Pharmaceutical Sciences под редакцией E.W. Martin, который включен в настоящем документе в качестве ссылки полностью.
Термин «лечить», используемый в настоящем документе, означает предотвратить, остановить или замедлить развитие заболевания или состояния, или устранить заболевания или состояния у субъекта. В одном варианте осуществления «лечить» означает остановку или замедление развития заболевания или состояния, или устранить заболевания или состояния у субъекта. В одном варианте осуществления, "лечить" означает уменьшить по меньшей мере одно объективное проявление заболевания или состояния.
Термин "эффективное количество", используемый в настоящем документе, относится к количеству, которое достаточно, чтобы вызвать желаемый биологический эффект.
Термин "ингибировать", используемый в настоящем документе, означает снижение на объективно измеряемое количество или степень. В различных вариантах осуществления "ингибировать" означает уменьшение по меньшей мере на 5, 10, 20, 30, 40, 50, 60, 70, 80, 90 или 95 процентов по сравнению с соответствующим контролем. В одном варианте осуществления "ингибировать" означает снижение на 100 процентов, то есть, остановить или устранить.
Термин "субъект", используемый в настоящем документе, относится к млекопитающему. В различных вариантах осуществления субъектом является мышь, крыса, кролик, кошка, собака, свинья, овца, лошадь, корова или примат, не относящийся к человеческому роду. В одном варианте осуществления, субъектом является человек.
В некоторых вариантах осуществления изобретения, соединение, представленное формулой I, выбрано из группы, включающей:
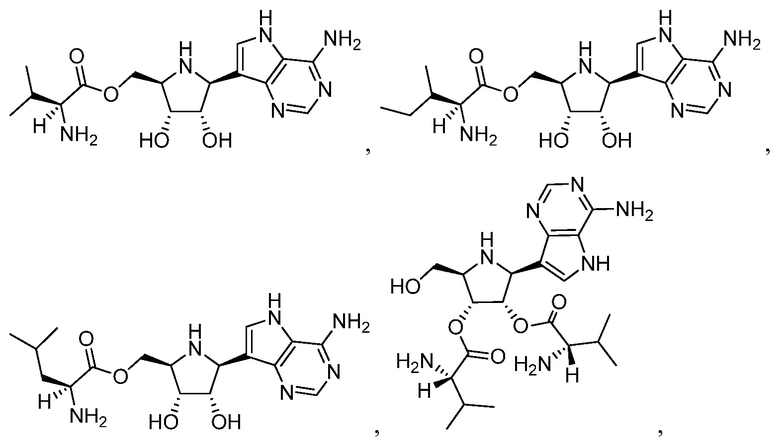
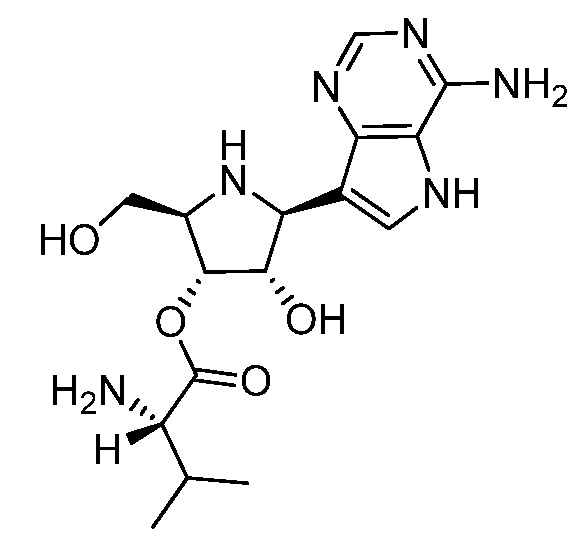 и
и 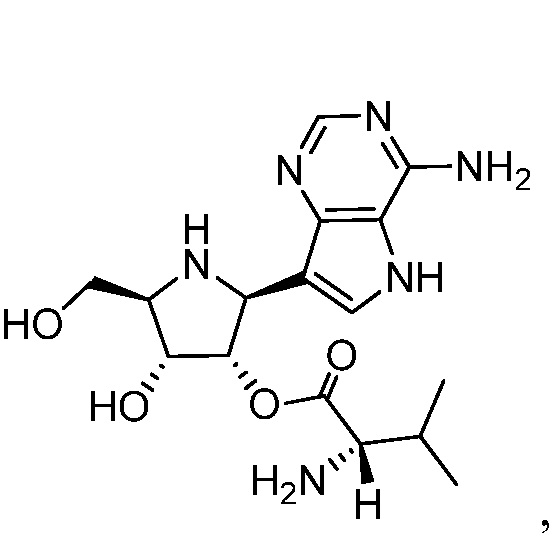
и их фармацевтически приемлемые соли.
В некоторых вариантах осуществления изобретения, соединение, представленное формулой I выбрано из группы, включающей
(S)-((2R,3R,4S,5S)-5-(4-амино-5H-пирроло[3,2-d]пиримидин-7-ил)-3,4-дигидроксипирролидин-2-ил)метил 2-амино-3-метилбутаноат;
(2S,3S)-((2R,3R,4S,5S)-5-(4-амино-5H-пирроло[3,2-d]пиримидин-7-ил)-3,4-дигидроксипирролидин-2-ил)метил 2-амино-3-метилпентаноат;
(S)-((2R,3R,4S,5S)-5-(4-амино-5H-пирроло[3,2-d]пиримидин-7-ил)-3,4-дигидроксипирролидин-2-ил)метил 2-амино-4-метилпентаноат;
(2S,2'S)-(2S,3S,4R,5R)-2-(4-амино-5H-пирроло[3,2-d]пиримидин-7-ил)-5-(гидроксиметил)пирролидин-3,4-диил бис(2-амино-3-метилбутаноат);
(S)-(2R,3R,4S,5S)-5-(4-амино-5H-пирроло[3,2-d]пиримидин-7-ил)-4-гидрокси-2-(гидроксиметил)пирролидин-3-ил 2-амино-3-метилбутаноат;
(S)-(2S,3S,4R,5R)-2-(4-амино-5H-пирроло[3,2-d]пиримидин-7-ил)-4-гидрокси-5-(гидроксиметил)пирролидин-3-ил 2-амино-3-метилбутаноат; и
их фармацевтически приемлемые соли.
Общие способы получения соединений по изобретению:
Гетероциклы и гетероарилы могут быть получены известными способами, как описано в литературе (a. Ring system handbook, published by American Chemical Society edition 1993 и последующие издания. b. The Chemistry of Heterocyclic Compounds; Weissberger, A., Ed.; Wiley: New York, 1962. c. Nesynov, E. P.; Grekov, A. P. The chemistry of 1,3,4-oxadiazole derivatives. Russ. Chem. Rev. 1964, 33, 508-515. d. Advances in Heterocyclic Chemistry; Katritzky, A. R., Boulton, A. J., Eds.; Academic Press: New York, 1966. e. Comprehensive Heterocyclic Chemistry; Potts, K. T., Ed.; Pergamon Press: Oxford, 1984. f. Eloy, F. A review of the chemistry of 1,2,4-oxadiazoles. Fortschr. Chem. Forsch. 1965, 4, pp 807-876. g. Adv. Heterocycl. Chem. 1976. h. Comprehensive Heterocyclic Chemistry; Potts, K. T., Ed.; Pergamon Press: Oxford, 1984. i. Chem. Rev. 1961 61, 87-127. j. 1,2,4-Triazoles; John Wiley & Sons: New York, 1981; Vol 37). Функциональные группы в процессе синтеза может должны быть защищены, а затем удалены. Примеры подходящих защитных групп можно найти в обзоре Protective Groups in Organic Synthesis, fourth edition, edited by Greene and Wuts.
Типичные способы, которые могут быть использованы для получения соединений по изобретению и промежуточных соединений, используемых для их получения, показаны на следующих схемах.
Схема 1
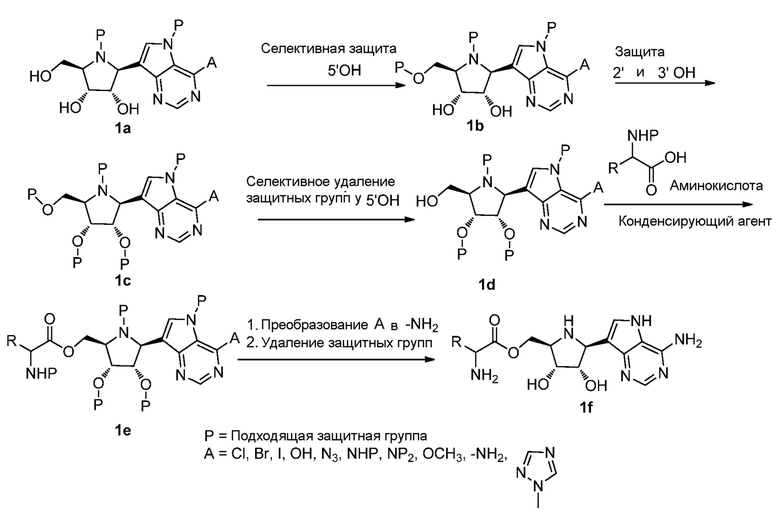
Схема 2
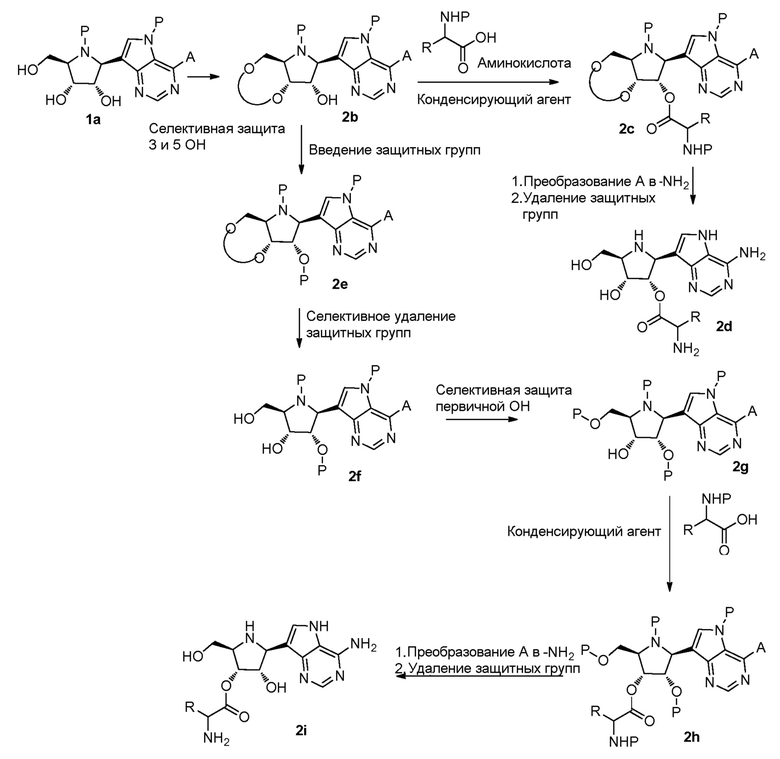
Схема 3
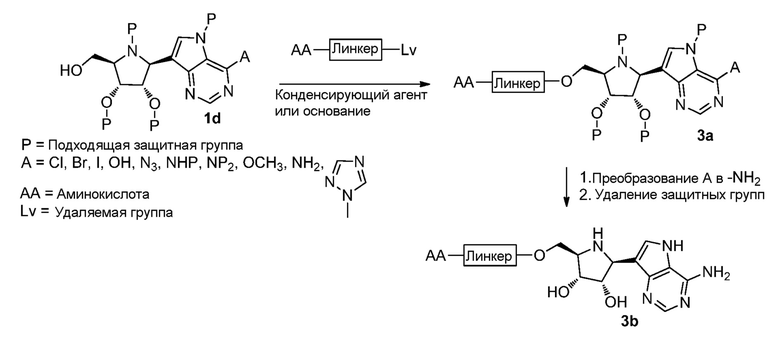
Схема 4
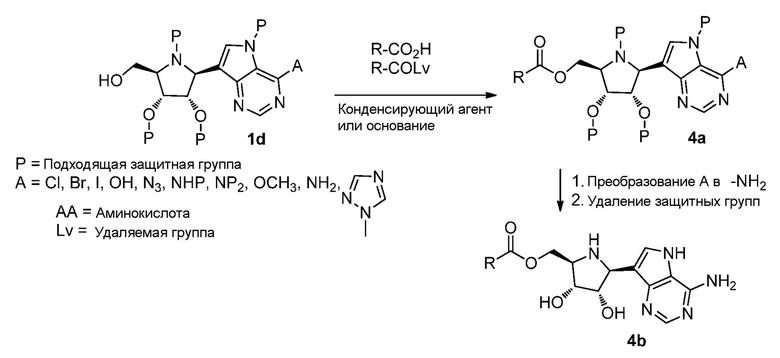
Схема 5
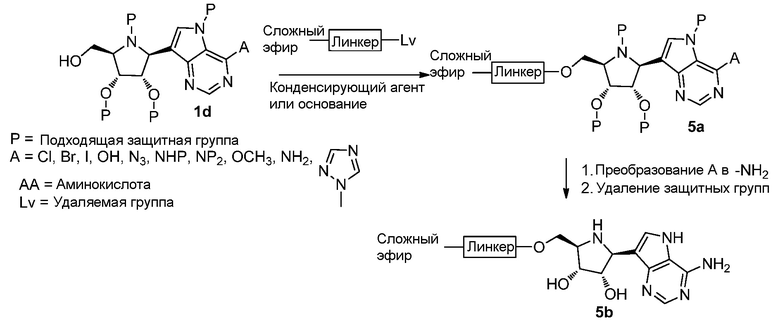
Схема 6
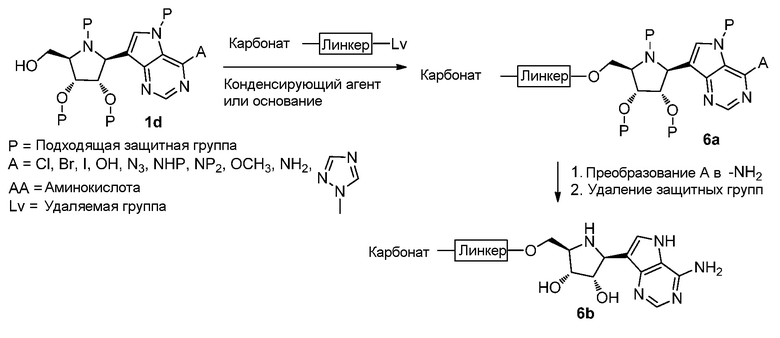
Схема 7
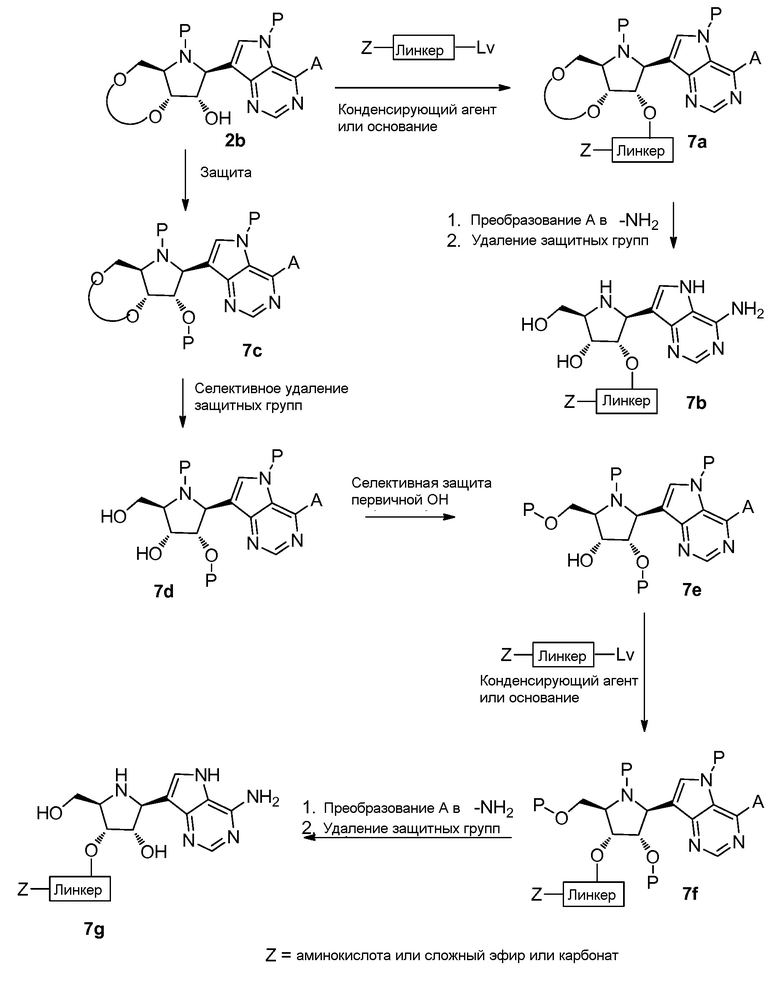
Схема 8
Ссылки к схеме 8:
1. WO 2011/123586 A1 (включено в качестве ссылки).
2. WO 2010/135520 A1 (включено в качестве ссылки).
3. WO 2009/069095 A2 (включено в качестве ссылки).
4. WO 2009/029729 A1 (включено в качестве ссылки).
5. WO 2008/082601 A2 (включено в качестве ссылки).
6. WO 2007/022073 A2 (включено в качестве ссылки).
7. Hecker, Scott J.; Reddy, K. Raja; van Poelje, Paul D.; Sun, Zhili; Huang, Wenjian; Varkhedkar, Vaibhav; Reddy, M. Venkat; Fujitaki, James M.; Olsen, David B.; Koeplinger, Kenneth A.; Boyer, Serge H.; Linemeyer, David L.; MacCoss, Malcolm; Erion, Mark D; Journal of Medicinal Chemistry (2007), 50(16), 3891-3896.
8. Yadava, Virendra Singh; Asian Journal of Chemistry (2005), 17(4), 2857-2859.
9. U.S. Pat. Appl. Publ. 2005/0182252 A1 (включено в качестве ссылки).
10. U.S. Pat. Appl. Publ. 2005/0070556 A1 (включено в качестве ссылки).
11. Reitz, Allen B.; Goodman, Michael G.; Pope, Barbara L.; Argentieri, Dennis C.; Bell, Stanley C.; Burr, Levelle E.; Chourmouzis, Erika; Come, Jon; Goodman, Jacquelyn H.; Klaubert, Dieter H.; Maryanoff, Bruce E.; McDonnell, Mark E.; Rampulla, Marianne S.; Schott, Mary R.; Chen, Robert; Journal of Medicinal Chemistry (1994), 37(21), 3561-78.
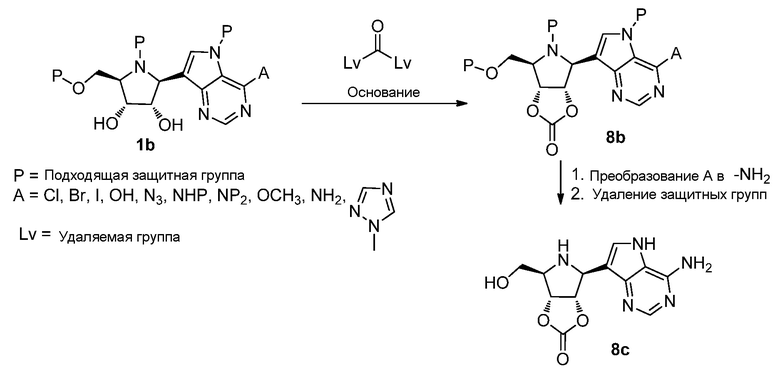
Схема 9
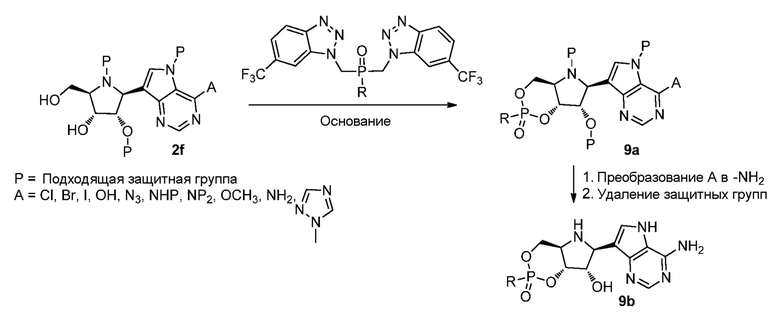
Ссылки к схеме 9
1. Roelen, H. C. P. F.; De Vroom, E.; Wang, A. H. J.; Van der Marel, G. A.; Van Boom, J. H; Nucleosides & Nucleotides (1992), 11(1), 141-56.
2. Kaji, Akira. (Japan) (1988), 5 pp. CODEN: JKXXAF JP 63135399 A 19880607 Patent written in Japanese. Application: JP 1986-282021 19861128.
Схема 10
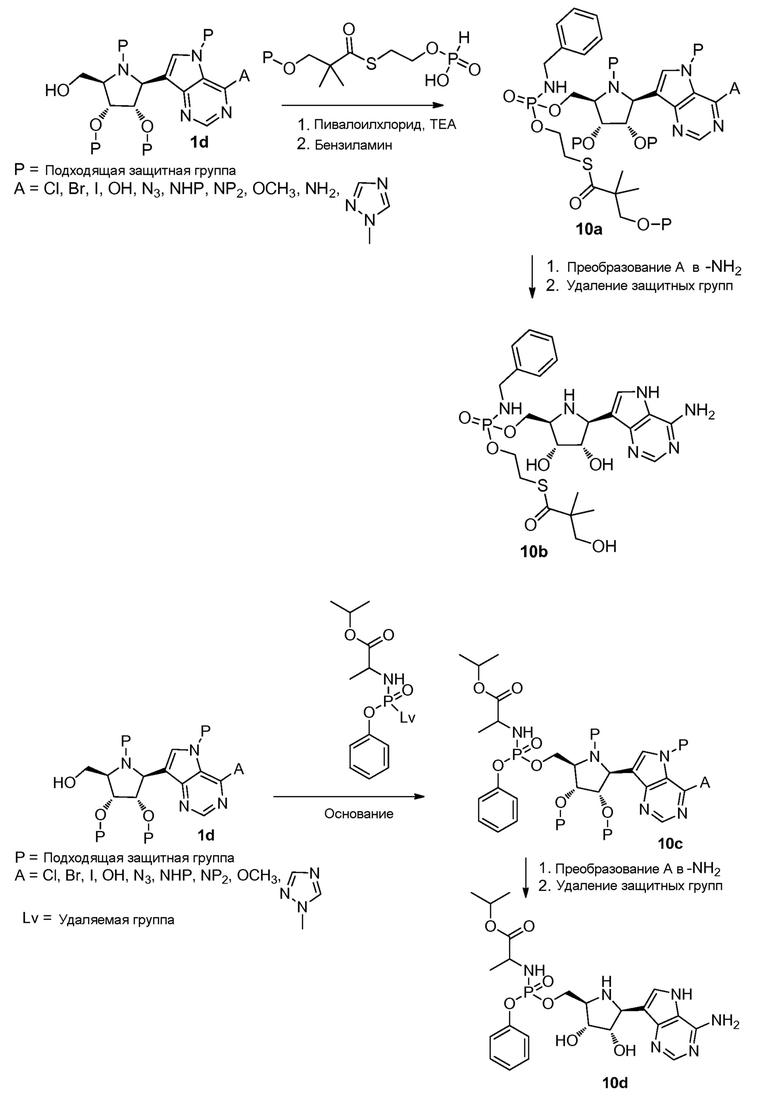
Ссылки к схеме 10:
1. U.S. Pat. Appl. Publ. 2010/0203015 A1 (включено в качестве ссылки).
2. WO 2009/132123 A1 (включено в качестве ссылки).
3. Hatton, Wilfried; Hunault, Julie; Egorov, Maxim; Len, Christophe; Pipelier, Muriel; Blot, Virginie; Silvestre, Virginie; Fargeas, Valerie; Ane, Adjou; McBrayer, Tami; Detorio, Mervi; Cho, Jong-Hyun; Bourgougnon, Nathalie; Dubreuil, Didier; Schinazi, Raymond F.; Lebreton, Jacques; European Journal of Organic Chemistry (2011), 2011(36), 7390-7399.
4. Zhang, Hong-wang; Zhou, Longhu; Coats, Steven J.; McBrayer, Tamara R.; Tharnish, Phillip M.; Bondada, Lavanya; Detorio, Mervi; Amichai, Sarah A.; Johns, Melissa D.; Whitaker, Tony; Schinazi, Raymond F; Bioorganic & Medicinal Chemistry Letters (2011), 21(22), 6788-6792.
5. Ross, Bruce S.; Ganapati Reddy, P.; Zhang, Hai-Ren; Rachakonda, Suguna; Sofia, Michael J; Journal of Organic Chemistry (2011), 76(20), 8311-8319.
6. McGuigan, Christopher; Madela, Karolina; Aljarah, Mohamed; Gilles, Arnaud; Battina, Srinivas K.; Ramamurty, Changalvala V. S.; Srinivas Rao, C.; Vernachio, John; Hutchins, Jeff; Hall, Andrea; Kolykhalov, Alexander; Henson, Geoffrey; Chamberlain, Stanley; Bioorganic & Medicinal Chemistry Letters (2011), 21(19), 6007-6012.
7. Cho, Jong Hyun; Amblard, Franck; Coats, Steven J.; Schinazi, Raymond F; Tetrahedron (2011), 67(30), 5487-5493.
8. WO 2010/135520 A1 (включено в качестве ссылки).
9. Perlikova, Pavla; Pohl, Radek; Votruba, Ivan; Shih, Robert; Birkus, Gabriel; Cihlar, Tomas; Hocek, Michal; Bioorganic & Medicinal Chemistry (2011), 19(1), 229-242.
10. WO 2010/108135 A1 (включено в качестве ссылки).
11. WO 2010/130726 A1 (включено в качестве ссылки).
12. WO 2010/030858 A1 (включено в качестве ссылки).
13. WO 2010/108140 A1 (включено в качестве ссылки).
14. WO 2010/026153 A1 (включено в качестве ссылки).
15. WO 2010/081082 A2 (включено в качестве ссылки).
16. McGuigan, Christopher; Madela, Karolina; Aljarah, Mohamed; Gilles, Arnaud; Brancale, Andrea; Zonta, Nicola; Chamberlain, Stanley; Vernachio, John; Hutchins, Jeff; Hall, Andrea; Ames, Brenda; Gorovits, Elena; Ganguly, Babita; Kolykhalov, Alexander; Wang, Jin; Muhammad, Jerry; Patti, Joseph M.; Henson, Geoffrey; Bioorganic & Medicinal Chemistry Letters (2010), 20(16), 4850-4854.
17. Derudas, Marco; Brancale, Andrea; Naesens, Lieve; Neyts, Johan; Balzarini, Jan; McGuigan, Christopher; Bioorganic & Medicinal Chemistry (2010), 18(7), 2748-2755.
18. Mehellou, Youcef; Valente, Rocco; Mottram, Huw; Walsby, Elisabeth; Mills, Kenneth I.; Balzarini, Jan; McGuigan, Christopher; Bioorganic & Medicinal Chemistry (2010), 18(7), 2439-2446.
19. McGuigan, Christopher; Gilles, Arnaud; Madela, Karolina; Aljarah, Mohamed; Holl, Sabrina; Jones, Sarah; Vernachio, John; Hutchins, Jeff; Ames, Brenda; Bryant, K. Dawn; Gorovits, Elena; Ganguly, Babita; Hunley, Damound; Hall, Andrea; Kolykhalov, Alexander; Liu, Yule; Muhammad, Jerry; Raja, Nicholas; Walters, Robin; Wang, Jin; Chamberlain, Stanley; Henson, Geoffrey; Journal of Medicinal Chemistry (2010), 53(13), 4949-4957.
20. Leisvuori, Anna; Aiba, Yuichiro; Loennberg, Tuomas; Poijaervi-Virta, Paeivi; Blatt, Laurence; Beigelman, Leo; Loennberg, Harri; или ganic & Biomolecular Chemistry (2010), 8(9), 2131-2141.
21. Mehellou, Youcef; Balzarini, Jan; McGuigan, Christopher; Antiviral Chemistry & Chemotherapy (2010), 20(4), 153-160.
22. Rondla, Ramu; Coats, Steven J.; McBrayer, Tamara R.; Grier, Jason; Johns, Melissa; Tharnish, Phillip M.; Whitaker, Tony; Zhou, Longhu; Schinazi, Raymond F; Antiviral Chemistry & Chemotherapy (2009), 20(2), 99-106.
23. WO 2008/121941 A1 (включено в качестве ссылки).
24. WO 2009/086192 A1 (включено в качестве ссылки).
25. McGuigan, Christopher; Kelleher, Mary Rose; Perrone, Plinio; Mulready, Sinead; Luoni, Giovanna; Daverio, Felice; Rajyaguru, Sonal; Le Pogam, Sophie; Najera, Isabel; Martin, Joseph A.; Klumpp, Klaus; Smith, David B; Bioorganic & Medicinal Chemistry Letters (2009), 19(15), 4250-4254.
26. McGuigan, Christopher; Perrone, Plinio; Madela, Karolina; Neyts, Johan; Bioorganic & Medicinal Chemistry Letters (2009), 19(15), 4316-4320.
Схема 11
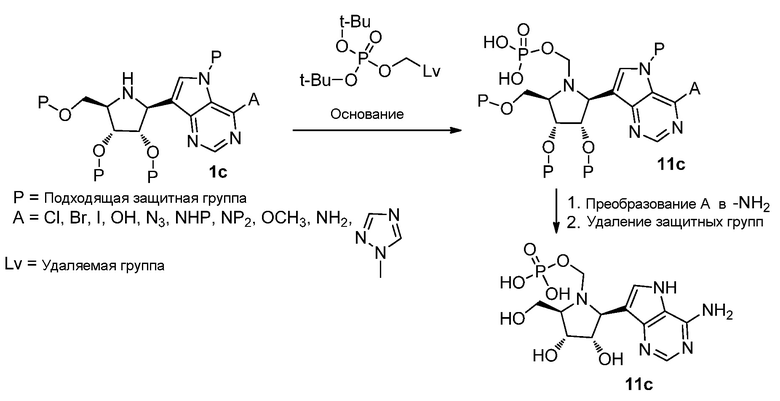
Ссылки к схеме 11:
1. WO 2011/150016 A1 (включено в качестве ссылки).
2. WO 2010/150761 A1 (включено в качестве ссылки).
3. WO 2011/068899 A1 (включено в качестве ссылки).
4. WO 2011/084849 A1 (включено в качестве ссылки).
5. U.S. Pat. Appl. Publ. 2011/0166128 A1 (включено в качестве ссылки).
6. WO 2011/084846 A1 (включено в качестве ссылки).
7. CN 102060874 A.
8. WO 2011/031979 A1 (включено в качестве ссылки).
9. WO 2011/002999 A1 (включено в качестве ссылки).
10. WO 2010/064735 A1 (включено в качестве ссылки).
11. WO 2010/036638 A2 (включено в качестве ссылки).
12. WO 2010/079443 A1 (включено в качестве ссылки).
13. WO 2010/010017 A1 (включено в качестве ссылки).
14. WO 2010/093789 A2 (включено в качестве ссылки).
Схема 12
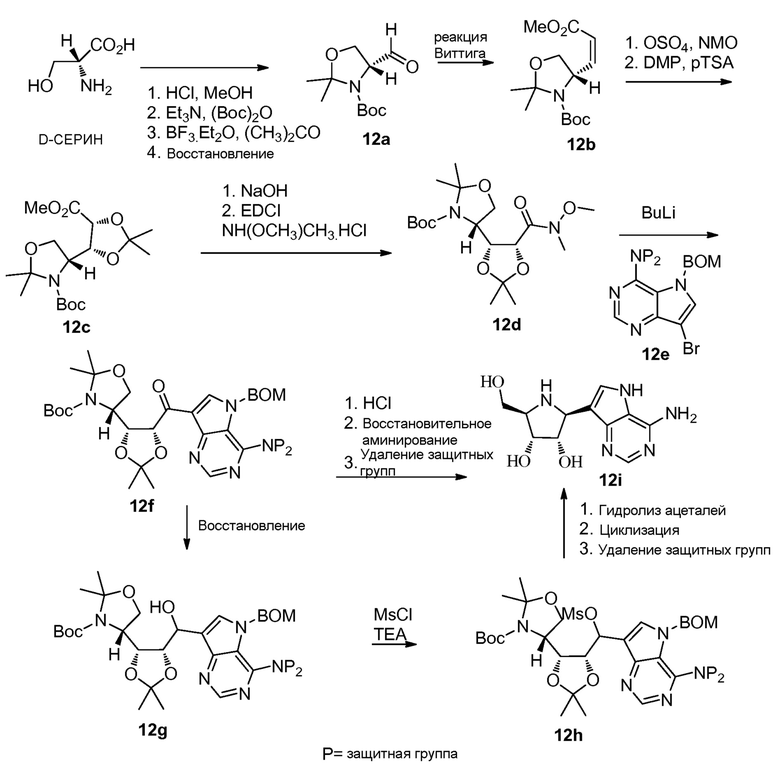
Ссылки для соединения альдегида Гарнера 12a:
1. Upadhyay, Puspesh K.; Kumar, Pradeep; Synthesis (2010), (18), 3063-3066.
2. U.S. Pat. Appl. Publ. 2010/0152098 A1 (включено в качестве ссылки).
3. Badarau, Eduard; Suzenet, Franck; Finaru, Adriana-Luminita; Guillaumet, Gerald; European Journal of Organic Chemistry (2009), (21), 3619-3627.
4. Belanger, Dominique; Tong, Xia; Soumare, Sadia; Dory, Yves L.; Zhao, Yue; Chemistry-A European Journal (2009), 15(17), 4428-4436.
5. Osada, Satoshi; Ishimaru, Takako; Kawasaki, Hiroshi; Kodama, Hiroaki; Heterocycles (2006), 67(1), 421-431.
6. Xin, Cong; Liao, Qing-Jiang; Yao, Zhu-Jun; Journal of Organic Chemistry (2004), 69(16), 5314-5321.
7. Dondoni, Alessandro; Perrone, Daniela; Organic Syntheses (2000), 77 64-77.
8. Campbell, Andrew D.; Raynham, Tony M.; Taylor, Richard J. K; Synthesis (1998), (12), 1707-1709.
Ссылки для реакции Виттига, связанной с соединением 12b:
1. Ma, Zhiqiang; Lu, Jianming; Wang, Xiao; Chen, Chuo; Chemical Communications (Cambridge, United Kingdom) (2011), 47(1), 427-429.
2. Spangenberg, Thomas; Schoenfelder, Angele; Breit, Bernhard; Mann, Andre; European Journal of Organic Chemistry (2010), (31), 6005-6018.
3. Osman, Sami; Albert, Brian J.; Wang, Yanping; Li, Miaosheng; Czaicki, Nancy L.; Koide, Kazunori; Chemistry--A European Journal (2011), 17(3), 895-904.
4. Passiniemi, Mikko; Koskinen, Ari M. P; Synthesis (2010), (16), 2816-2822.
5. Thander, Latibuddin; Sarkar, Kaushik; Chattopadhyay, Shital K; Tetrahedron: Asymmetry (2009), 20(11), 1213-1216.
6. Chiou, Wen-Hua; Schoenfelder, Angele; Mann, Andre; Ojima, Iwao; Pure and Applied Chemistry (2008), 80(5), 1019-1024.
7. Ribes, Celia; Falomir, Eva; Carda, Miguel; Marco, J. Alberto; Journal of Organic Chemistry (2008), 73(19), 7779-7782.
8. Mochizuki, Akiyoshi; Naito, Hiroyuki; Nakamoto, Yumi; Uoto, Kouichi; Ohta, Toshiharu; Heterocycles (2008), 75(7), 1659-1671.
9. Spangenberg, Thomas; Schoenfelder, Angele; Breit, Bernhard; Mann, Andre; organic Letters (2007), 9(20), 3881-3884.
10. Lebel, Helene; Ladjel, Chehla; organometallics (2008), 27(11), 2676-2678.
11. Liu, Fa; Hu, Tai-Shan; Yao, Zhu-Jun; Tetrahedron (2005), 61(21), 4971-4981.
12. Shigeki Sano, Tomoka Takehisa, Shiho Ogawa, Kenji yokoyama and Yoshimitsu Nagao Chem. Pharm. Bull. 50 (9) 1300-1302 (2002).
13. Raghavan, Sadagopan; Rajender, A.; Joseph, Suju C.; Rasheed, M. Abdul; Ravi Kumar, K; Tetrahedron: Asymmetry (2004), 15(2), 365-379.
Ссылки по дигидроксилированию для соединений, относящихся к 12c:
1. Dondoni, Alessandro; Merino, Pedro; Perrone, Daniela; Tetrahedron (1993), 49(14), 2939-56.
2. Ribes, Celia; Falomir, Eva; Carda, Miguel; Marco, J. Alberto; Journal of Organic Chemistry (2008), 73(19), 7779-7782.
3. Upadhyay, Puspesh K.; Kumar, Pradeep; Synthesis (2010), (18), 3063-3066.
Ссылка, относящаяся к синтезу основания 12e:
Bambuch, Vitezslav; Otmar, Miroslav; Pohl, Radek; Masojidkova, Milena; Holy, Antonin; Tetrahedron (2007), 63(7), 1589-1601.
Схема 13
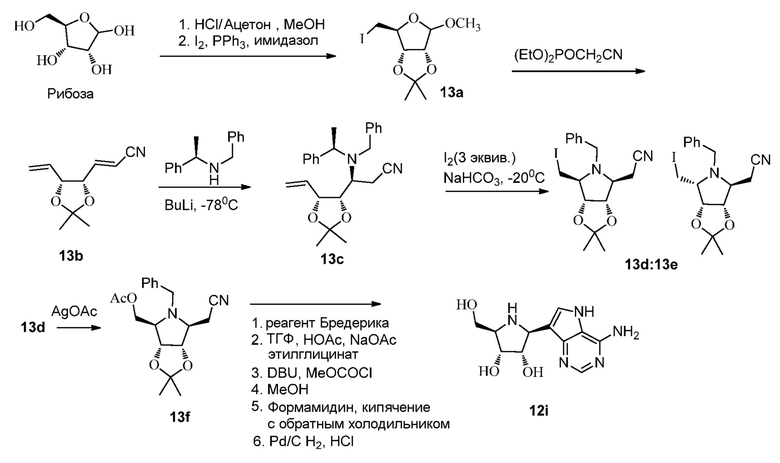
Ссылки для получения соединения 13a:
1. Mishra, Girija Prasad; Rao, Batchu Venkateswara; Tetrahedron: Asymmetry (2011), 22(7), 812-817.
2. Brock, E. Anne; Davies, Stephen G.; Lee, James A.; Roberts, Paul M.; Thomson, James E; Organic Letters (2011), 13(7), 1594-1597.
3. WO 2010/085377 A2 (включено в качестве ссылки).
4. Yadav, J. S.; Reddy, P. Narayana; Reddy, B. V. Subba; Synlett (2010), (3), 457-461.
5. Song, Kai; Zheng, Guo-jun; Huaxue Shiji (2010), 32(2), 171-172.
6. Prabhakar, Peddikotla; Rajaram, Singanaboina; Reddy, Dorigondla Kumar; Shekar, Vanam; Venkateswarlu, Yenamandra; Tetrahedron: Asymmetry (2010), 21(2), 216-221.
7. CN 101182342 A.
8. Baird, Lynton J.; Timmer, Mattie S. M.; Teesdale-Spittle, Paul H.; Harvey, Joanne E; Journal of Organic Chemistry (2009), 74(6), 2271-2277.
9. Wang, Xiang-cheng; Wang, Gang; Qu, Gang-lian; Huaxue Shijie (2008), 49(4), 226-228.
10. Ivanova, N. A.; Valiullina, Z. R.; Shitikova, O. V.; Miftakhov, M. S; Russian Journal of Organic Chemistry (2007), 43(5), 742-746.
11. Braga, Fernanda Gambogi; Coimbra, Elaine Soares; Matos, Magnum de Oliveira; Lino Carmo, Arturene Maria; Cancio, Marisa Damato; da Silva, Adilson David; European Journal of Medicinal Chemistry (2007), 42(4), 530-537.
12. Wender, Paul A.; Bi, F. Christopher; Buschmann, Nicole; Gosselin, Francis; Kan, Cindy; Kee, Jung-Min; Ohmura, Hirofumi; organic Letters (2006), 8(23), 5373-5376.
13. Fei, Xiangshu; Wang, Ji-Quan; Miller, Kathy D.; Sledge, George W.; Hutchins, Gary D.; Zheng, Qi-Huang; Nuclear Medicine and Biology (2004), 31(8), 1033-1041.
14. Abdel-Rahman, Adel A.-H.; Abdel-Megied, Ahmed E.-S.; Goda, Adel E.-S.; Zeid, Ibrahim F.; El Ashry, El Sayed H; Nucleosides, Nucleotides & Nucleic Acids (2003), 22(11), 2027-2038.
15. Palmer, Andreas M.; Jager, Volker; European Journal of Organic Chemistry (2001), (7), 1293-1308.
16. Paquette, Leo A.; Bailey, Simon; Journal of Organic Chemistry (1995), 60(24), 7849-56.
17. Classon, Bjoern; Liu, Zhengchun; Samuelsson, Bertil; Journal of Organic Chemistry (1988), 53(26), 6126-30.
18. Kissman, Henry M.; Baker, B. R; Journal of the American Chemical Society (1957), 79 5534-40.
Ссылки по циклизациям, относящимся к получению соединений типа 13d:
1. Davies, Stephen G.; Durbin, Matthew J.; Goddard, Euan C.; Kelly, Peter M.; Kurosawa, Wataru; Lee, James A.; Nicholson, Rebecca L.; Price, Paul D.; Roberts, Paul M.; Russell, Angela J.; Scott, Philip M.; Smith, Andrew D; Organic & Biomolecular Chemistry (2009), 7(4), 761-776.
2. Davies, Stephen G.; Nicholson, Rebecca L.; Price, Paul D.; Roberts, Paul M.; Russell, Angela J.; Savory, Edward D.; Smith, Andrew D.; Thomson, James E; Tetrahedron: Asymmetry (2009), 20(6-8), 758-772.
3. Davies, Stephen G.; Nicholson, Rebecca L.; Price, Paul D.; Roberts, Paul. M.; Smith, Andrew D; Synlett (2004), (5), 901-903.
4. Brock, E. Anne; Davies, Stephen G.; Lee, James A.; Roberts, Paul M.; Thomson, James E; Organic Letters (2011), 13(7), 1594-1597.
5. Gary B. Evans, Richard H. Furneaux, Andrzej Lewandowicz, Vern L. Schramm, and Peter C. Tyler, Journal of Medicinal Chemistry (2003), 46, 3412-3423.
Схема 14
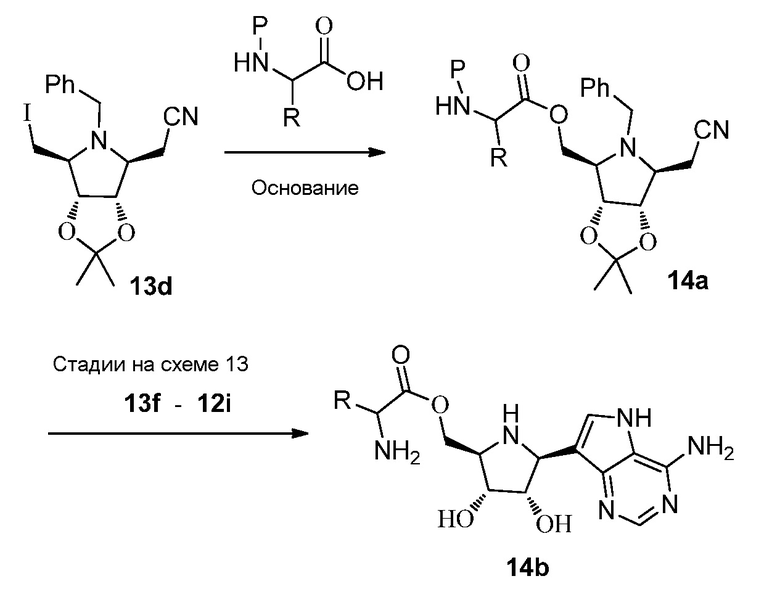
Схема 15
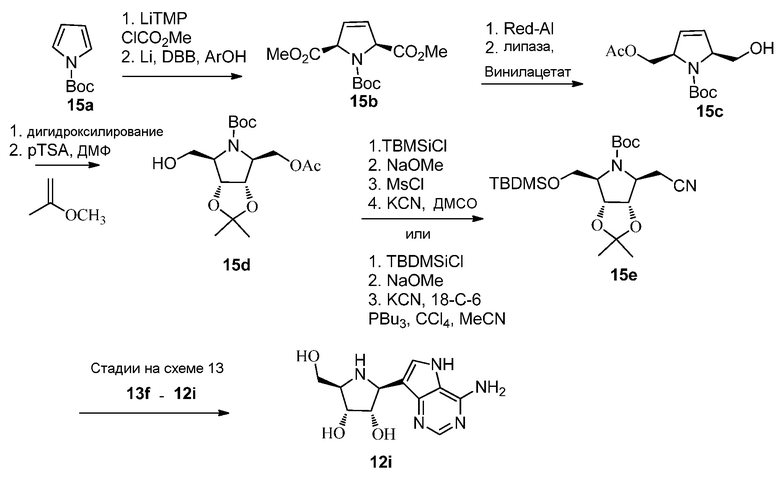
Ссылки к схеме 15:
1. Chenevert, Robert; Jacques, Frederic; Giguere, Pascall; Dasser, Mohammed; Tetrahedron: Asymmetry (2008), 19(11), 1333-1338.
2. Donohoe, Timothy J.; Thomas, Rhian E.; Cheeseman, Matthew D.; Rigby, Caroline L.; Bhalay, Gurdip; Linney, Ian D; organic Letters (2008), 10(16), 3615-3618.
3. Hanessian, Stephen; Therrien, Eric; Warrier, Jayakumar S.; Charron, Guillaume; Heterocycles (2006), 70 461-476.
4. Hamada, Yasumasa; Kawai, Akiyoshi; Kohno, Yasushi; Hara, Osamu; Shioiri, Takayuki; Journal of the American Chemical Society (1989), 111(4), 1524-5. (для преобразования спирта к циано).
Схема 16A
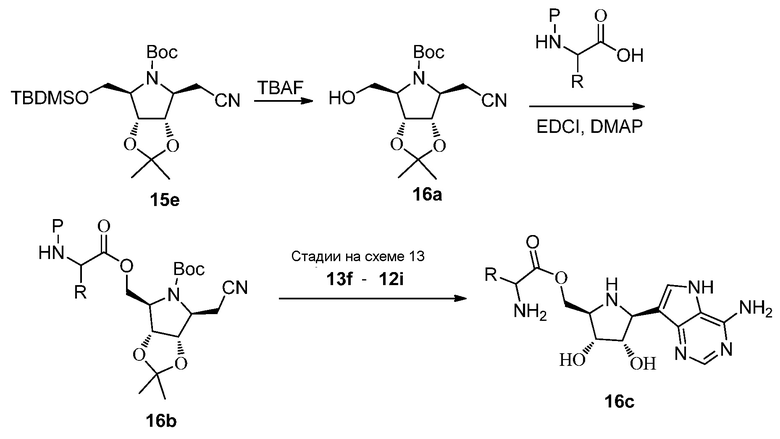
Схема 16B
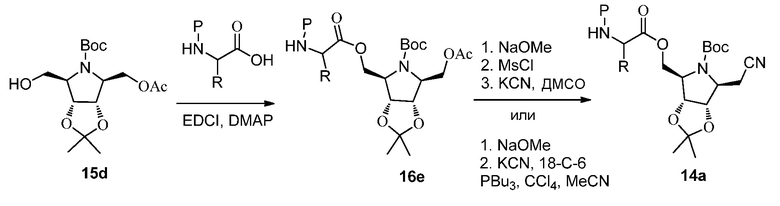
Схема 17
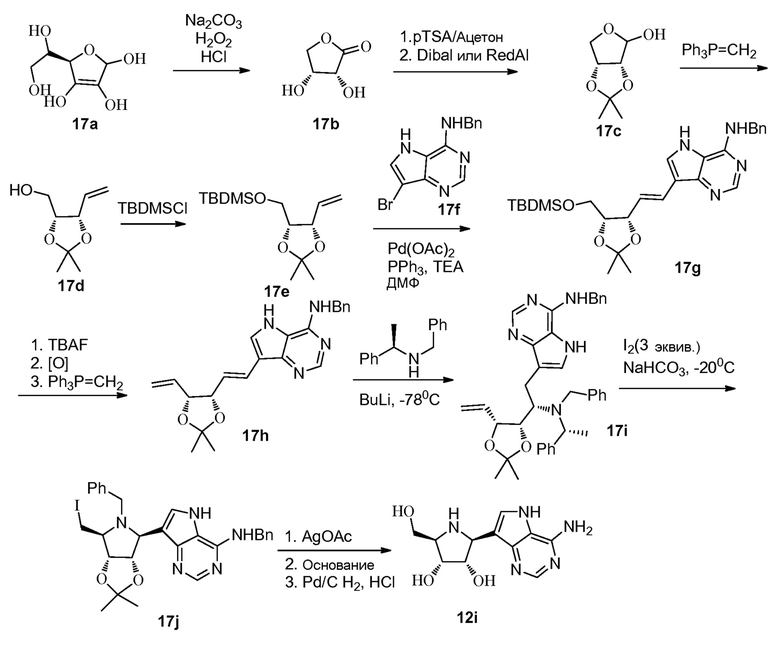
Ссылки к схеме 17:
1. Nikolaos G. Argyropoulos and Vassiliki C. Sarli; Tetrahedron Letters 45 (2004) 4237-4240.
2. Yuji Matsuya, Sho-ichi Takayanagi, and Hideo Nemoto; Chemistry A European Journal 2008, 14, 5275-5281.
3. Hyo-Joong; Ricardo, Alonso; Illangkoon, Heshan I.; Kim, Myong Jung; Carrigan, Matthew A.; Frye, Fabianne; Benner, Steven A; Journal of the American Chemical Society (2011), 133(24), 9457-9468.
4. Paudyal, Mahesh P.; Rath, Nigam P.; Spilling, Christopher D; organic Letters (2010), 12(13), 2954-2957.
5. Scarpi, Dina; Occhiato, Ernesto G.; Guarna, Antonio. Dipartimento di Chimica Organica 'U. Schiff; Tetrahedron: Asymmetry (2009), 20(3), 340-350.
6. WO 2008/108508 A1 (включено в качестве ссылки).
7. WO 2008/010776 A1 (включено в качестве ссылки).
8. U.S. Pat. Appl. Publ. 2007/0265333 A1 (включено в качестве ссылки).
9. Vu, Nguyen Quang; Chai, Christina L. L.; Lim, Kok Peng; Chia, Sze Chen; Chen, Anqi; Tetrahedron (2007), 63(30), 7053-7058.
10. WO 99/21858 A1 (включено в качестве ссылки).
11. Bambuch, Vitezslav; Otmar, Miroslav; Pohl, Radek; Masojidkova, Milena; Holy, Antonin; Tetrahedron (2007), 63(7), 1589-1601.
Схема 18
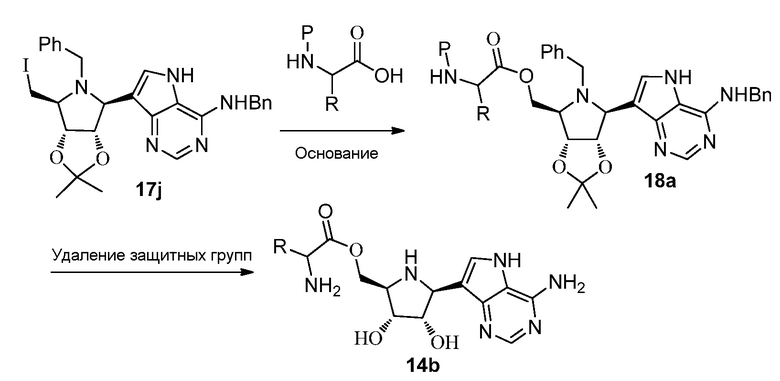
Схема 19
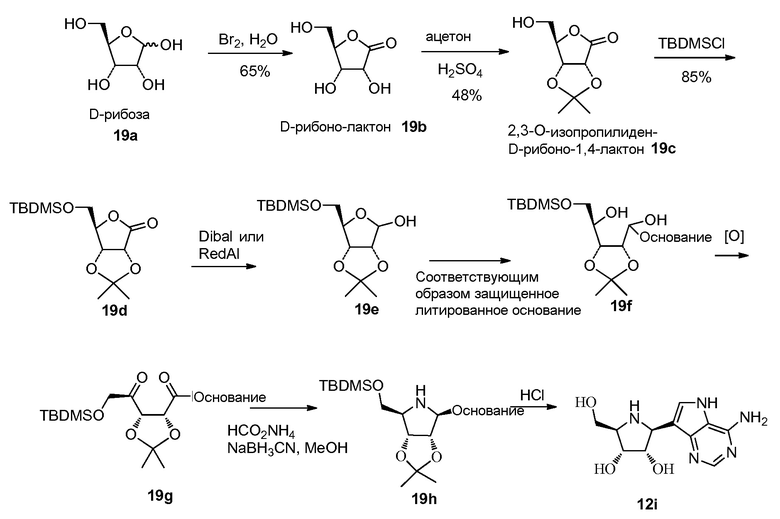
Ссылка к схеме 19:
Yokoyama, Masataka; Akiba, Takahiro; Ochiai, Yoshie; Momotake, Atsuya; Togo, Hideo; Journal of Organic Chemistry (1996), 61(17), 6079-6082.
Схема 20
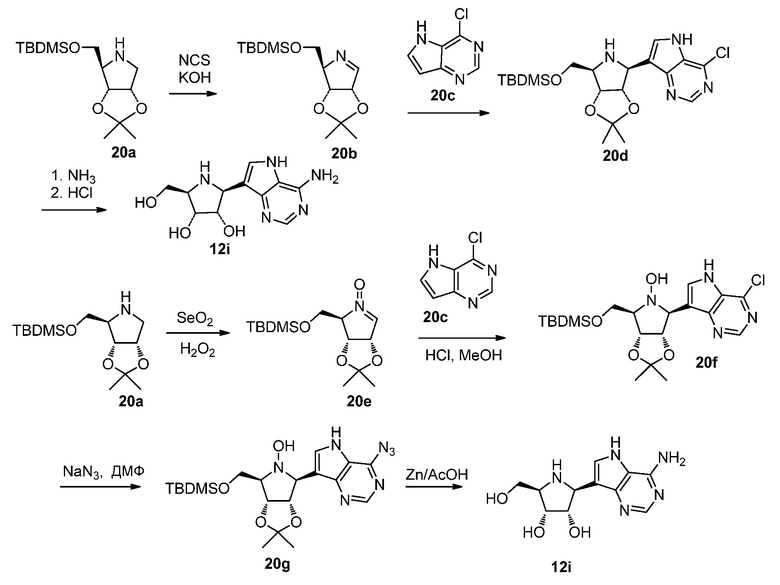
Ссылки к схеме 20:
1. Su, Jia-Kun; Jia, Yue-Mei; He, Ruirui; Rui, Pei-Xin; Han, Nanyin; He, Xihui; Xiang, Junfeng; Chen, Xin; Zhu, Jinghua; Yu, Chu-Yi; Synlett (2010), (11), 1609-1616.
2. Li, Xiao-Liu; Qin, Zhan-bin; Wang, Rui; Chen, Hua; Zhang, Ping-Zhu; Tetrahedron (2011), 67(10), 1792-1798.
Схема 21
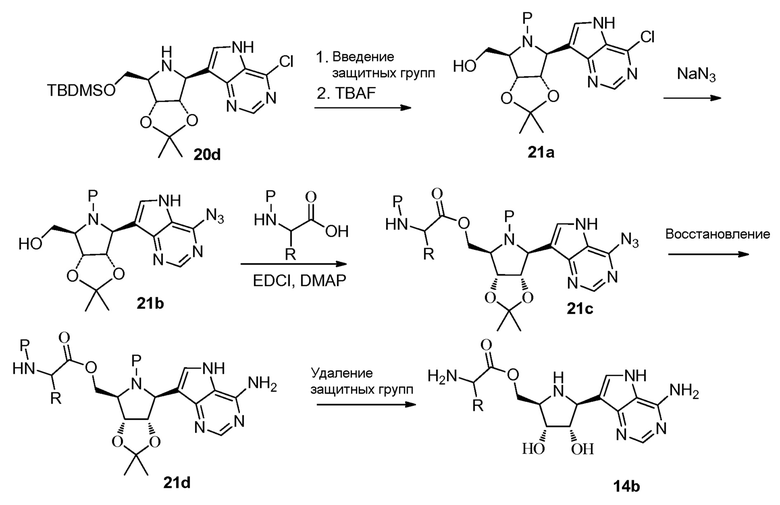
Схема 22
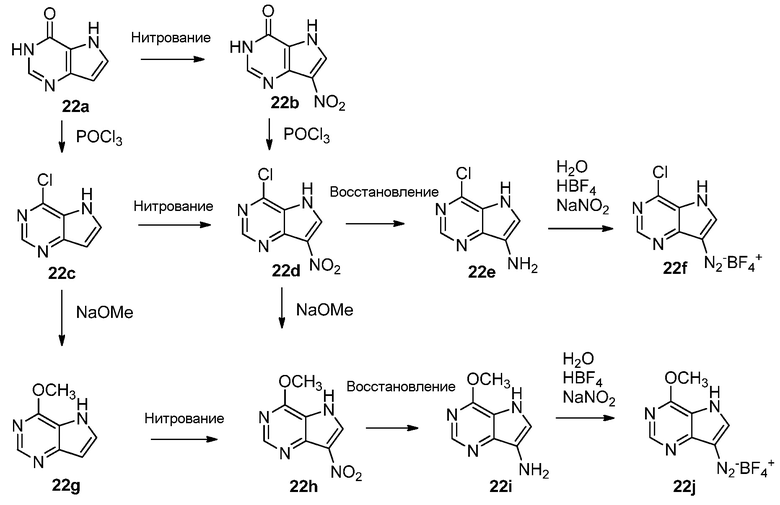
Ссылка к 7-нитро-3H-пирроло[3,2-d]пиримидин-4(5H)-ону:
1. WO 2008/063669 A1 (включено в качестве ссылки).
2. U.S. Pat. Appl. Publ. 2007/0155738 A1 (включено в качестве ссылки).
Схема 23
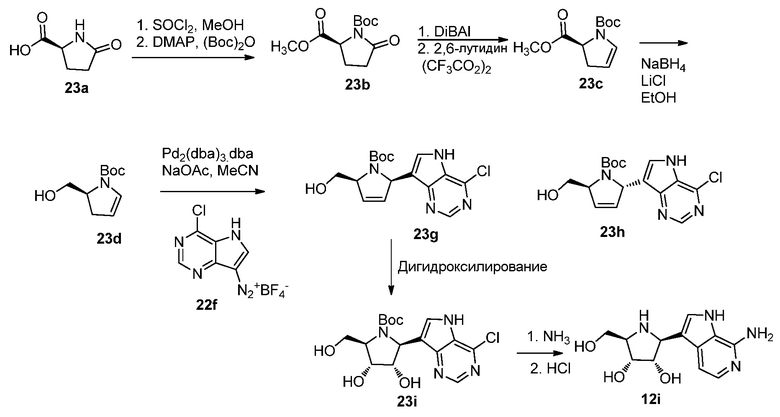
Ссылки по способам получения (S)-1-трет-бутил 2-метил 5-оксопирролидин-1,2-дикарбоксилата (23c):
1. U.S. Pat. Appl. Publ. 2011/0237636 A1 (включено в качестве ссылки).
2. WO 2011/015537 A1 (включено в качестве ссылки).
Ссылки по способам получения (S)-трет-бутил 2-(гидроксиметил)-2,3-дигидро-1H-пиррол-1-карбоксилат (23d):
1. Oliveira, Denilson F.; Miranda, Paulo C. M. L.; Correia, Carlos R. D; Journal of Organic Chemistry (1999), 64(18), 6646-6652.
2. Schumacher, Kelly K.; Jiang, Jianjun; Joullie, Madeleine M; Tetrahedron: Asymmetry (1998), 9(1), 47-53.
3. Dormoy, Jean Robert; Castro, Bertrand; Chappuis, Georges; Fritschi, Ulrich Stefan; Grogg, Peter; Angewandte Chemie (1980), 92(9), 761.
4. Woo, Grace H. C.; Kim, Se-Ho; Wipf, Peter; Tetrahedron (2006), 62(45), 10507-10517.
5. Moro, Angelica Venturini; Rodrigues dos Santos, Marcelo; Correia, Carlos Roque D; European Journal of Organic Chemistry (2011), 2011(36), 7259-7270.
Ссылка по реакции конденсации Хека, относящиеся к получению соединений 23g и 23h:
Severino, Elias A.; Costenaro, Edson R.; Garcia, Ariel L. L.; Correia, Carlos Roque D; organic Letters (2003), 5(3), 305-308.
Схема 24
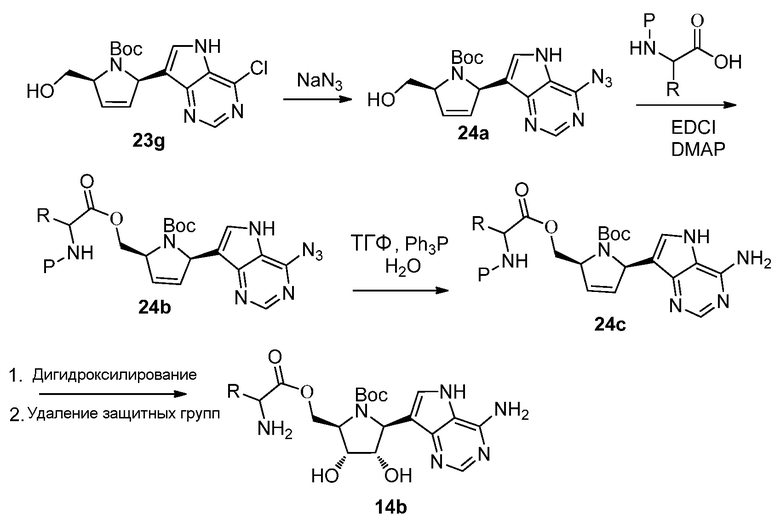
Схема 25
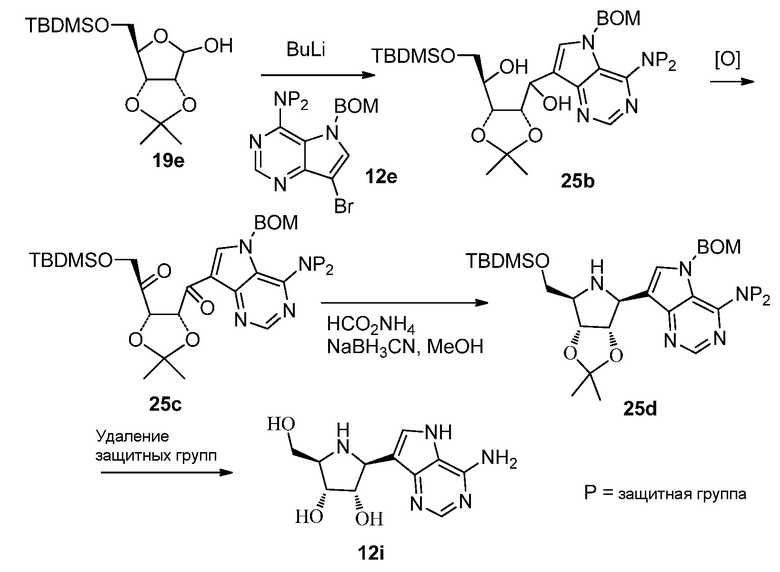
Схема 26
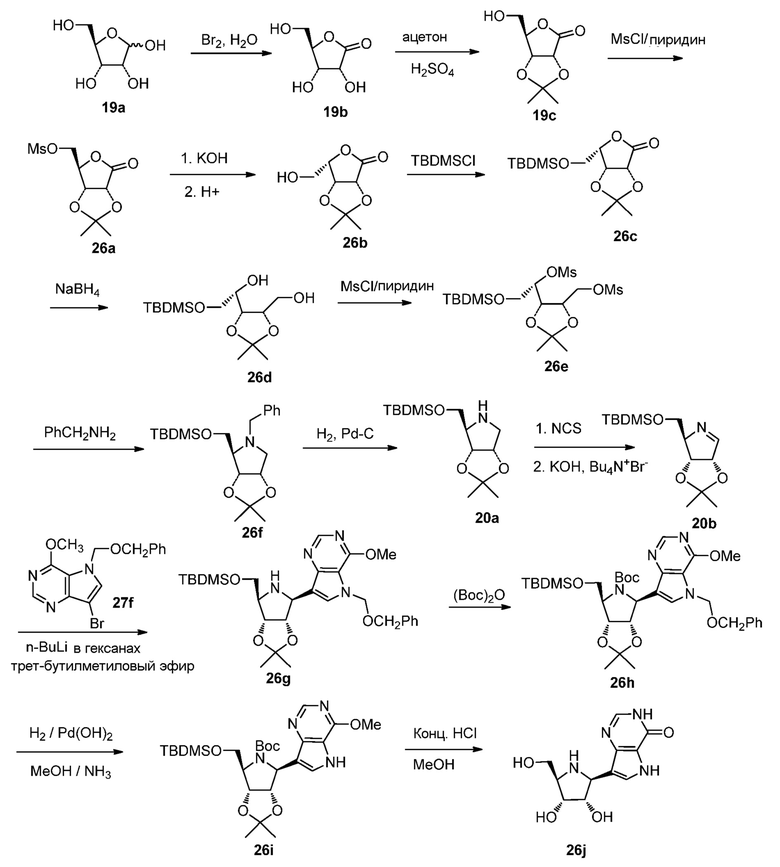
Схема 27
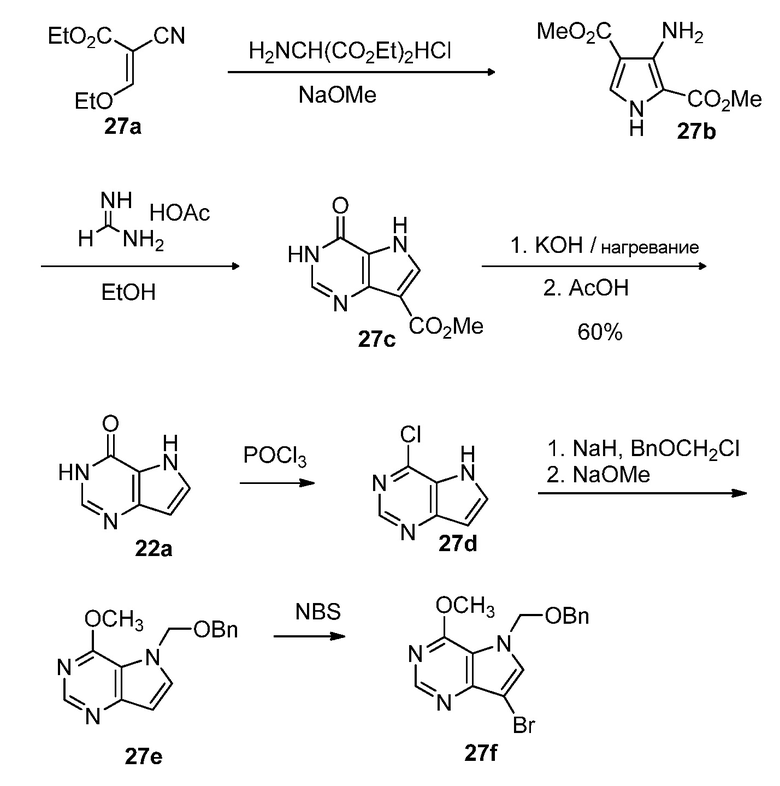
Схема 28
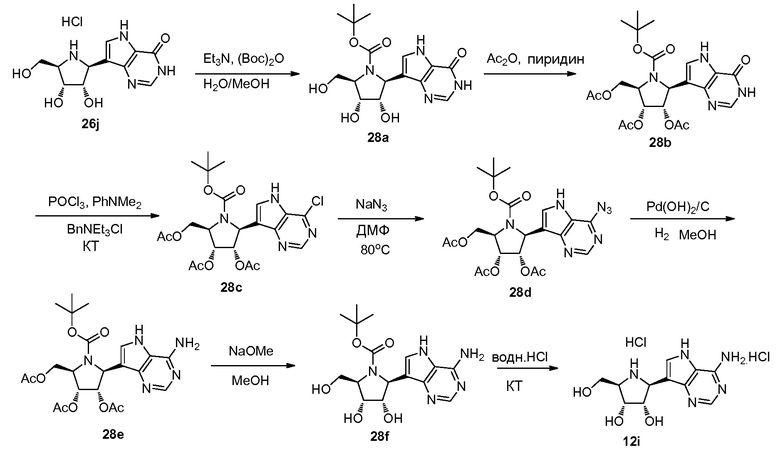
Схема 29
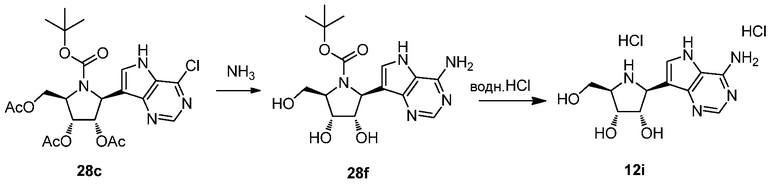
Схема 30
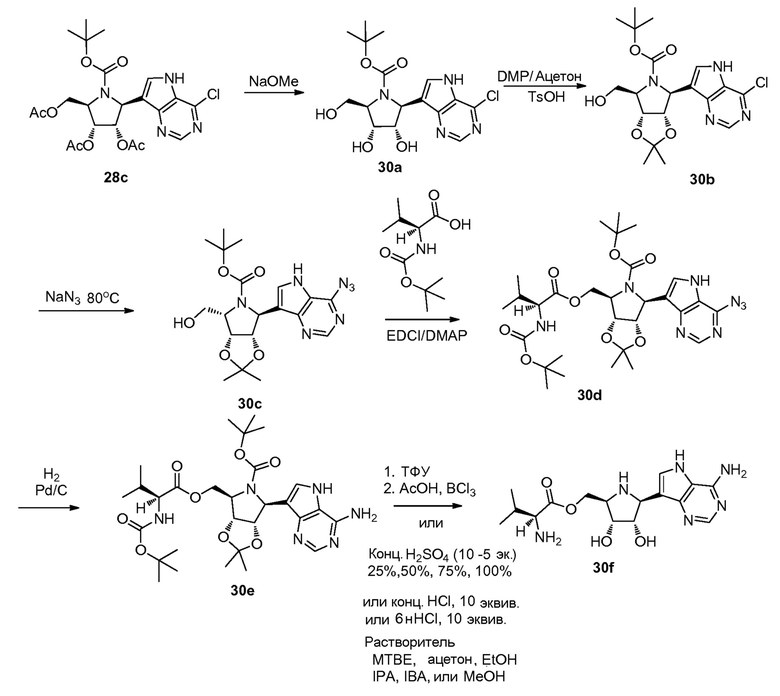
Схема 31
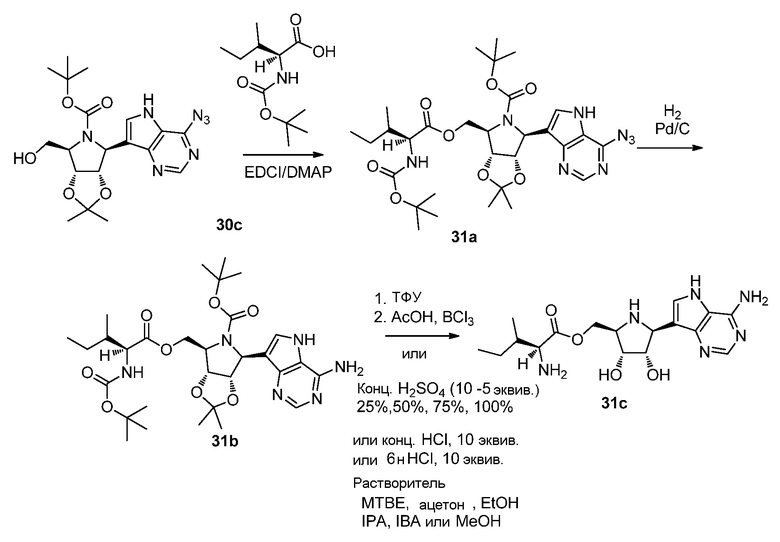
Схема 32
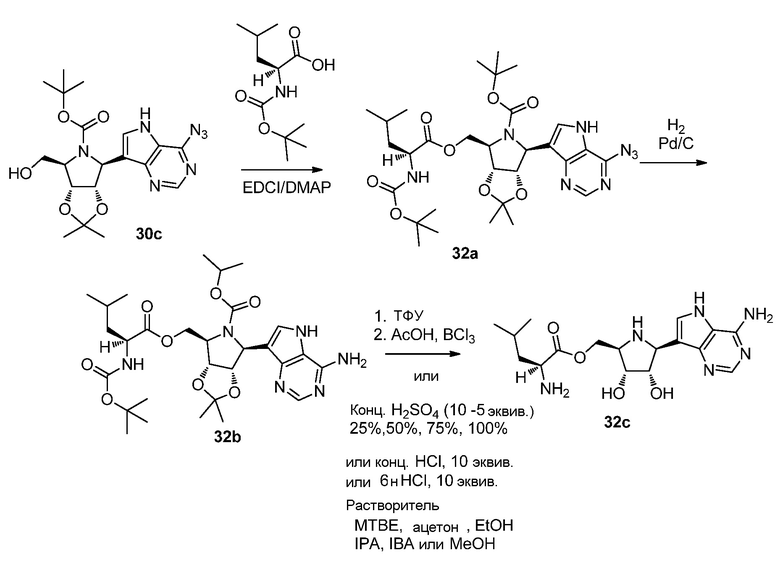
Схема 33
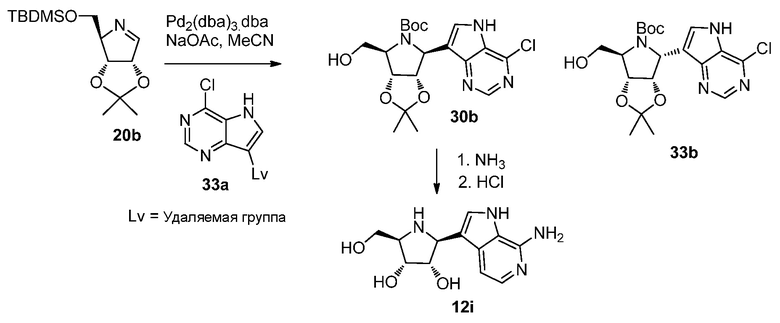
Схема 34
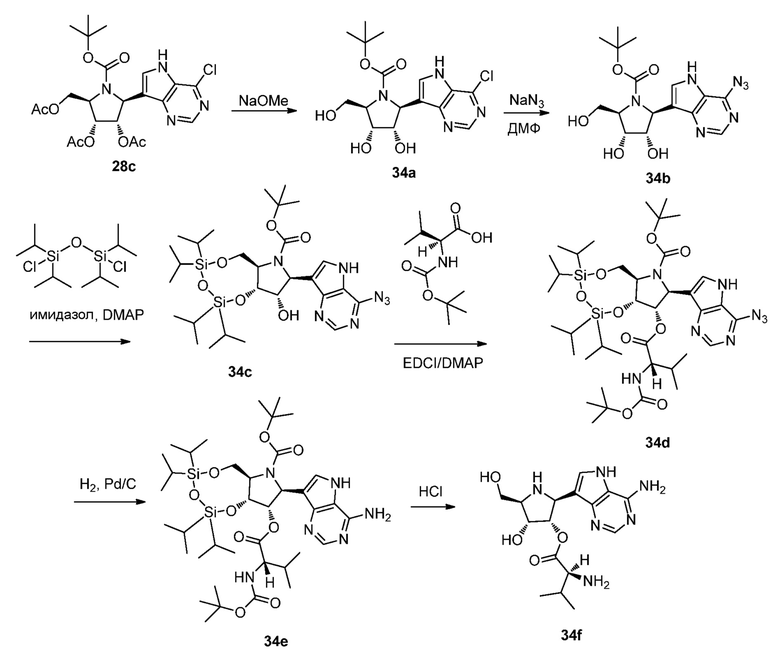
Схема 35
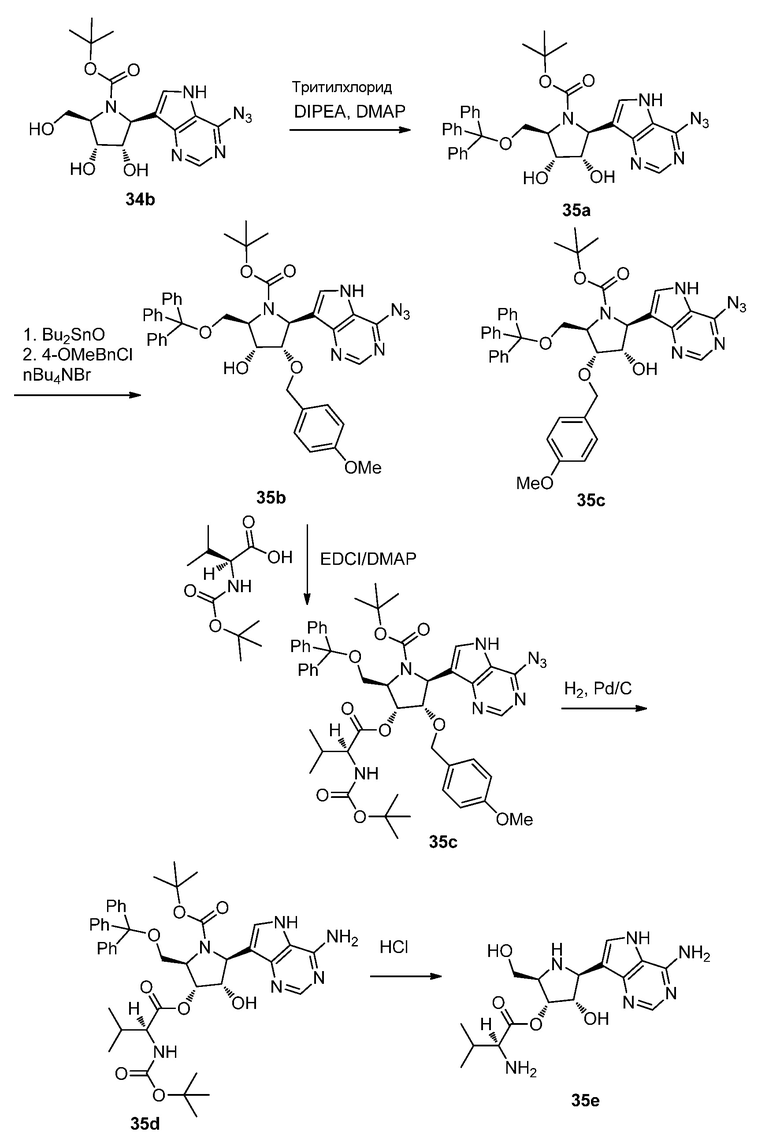
Схема 36
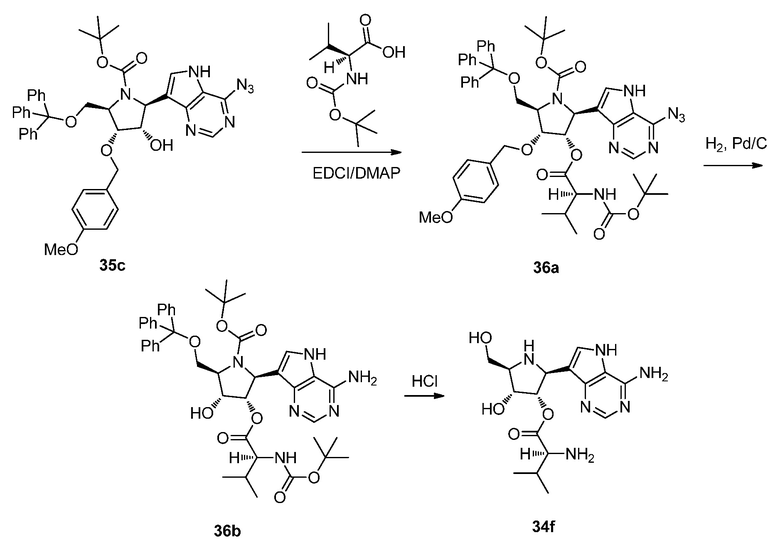
Схема 37
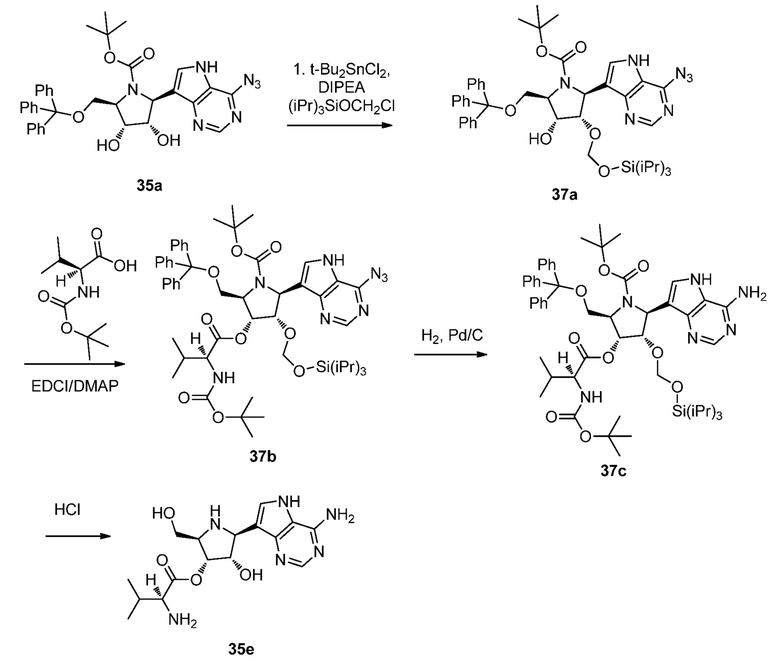
Схема 38
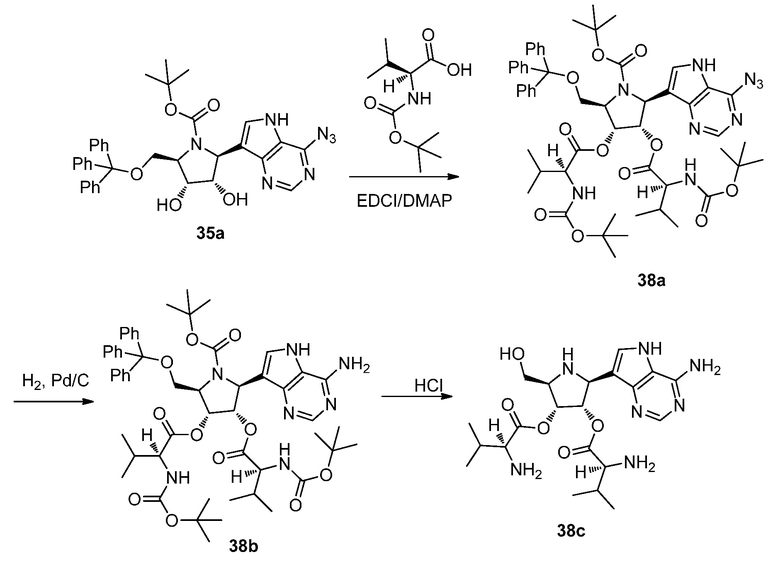
Схема 39A
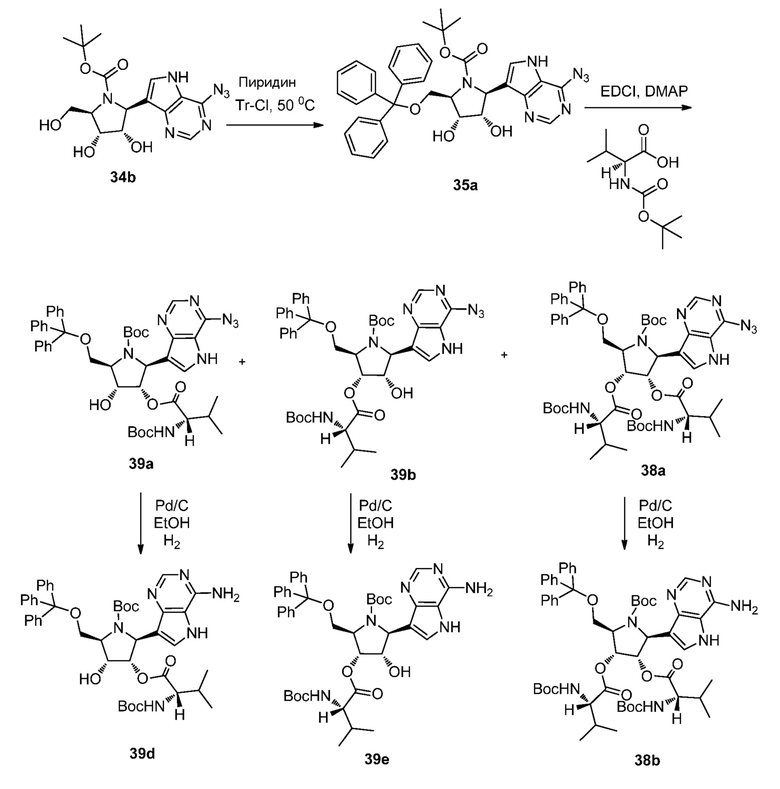
Схема 39B
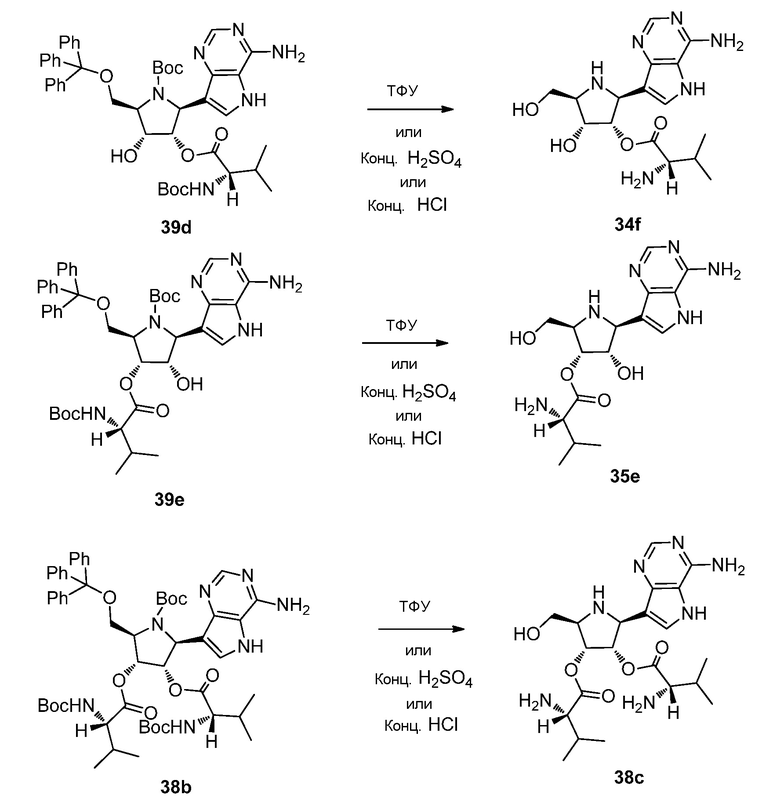
Схема 40
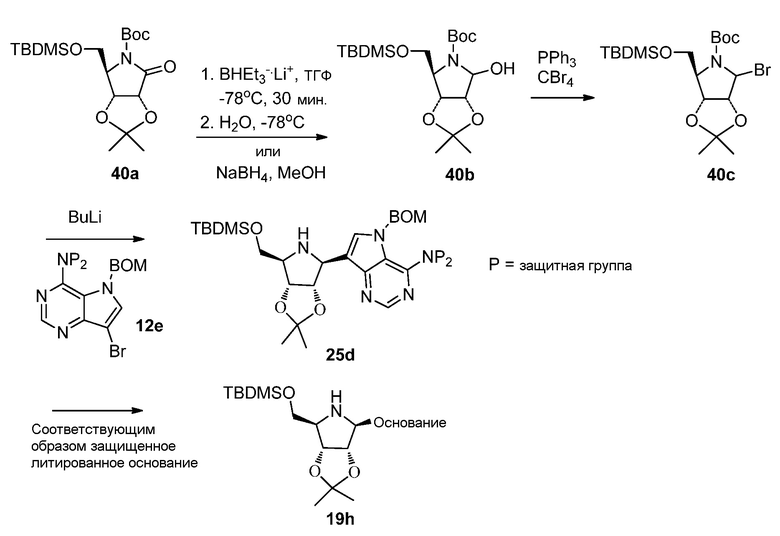
Ссылки для получения (3aR,4R,6aR)-трет-бутил 4-(((трет-бутилдиметилсилил)окси)метил)-2,2-диметил-6-оксодигидро-3aH-[1,3]диоксоло[4,5-c]пиррол-5(4H)-карбоксилата (40a):
1. Malladi, Venkata L. A.; Sobczak, Adam J.; Meyer, Tiffany M.; Pei, Dehua; Wnuk, Stanislaw F; Bioorganic & Medicinal Chemistry (2011), 19(18), 5507-5519.
2. Fiaux, Helene; Kuntz, Douglas A.; Hoffman, Daniela; Janzer, Robert C.; Gerber-Lemaire, Sandrine; Rose, David R.; Juillerat-Jeanneret; Bioorganic & Medicinal Chemistry (2008), 16(15), 7337-7346.
3. Yokoyama, Masataka; Ikenogami, Taku; Togo, Hideo. Inage-ku, Yayoi-cho; Perkin 1 (2000), (13), 2067-2071.
4. Zanardi, Franca; Battistini, Lucia; Nespi, Marika; Rassu, Gloria; Spanu, Pietro; Cornia, Mara; Casiraghi, Giovanni; Tetrahedron: Asymmetry (1996), 7(4), 1167-1180.
Ссылки по восстановлению лактона (40a) до лактола (40b):
1. Malladi, Venkata L. A.; Sobczak, Adam J.; Meyer, Tiffany M.; Pei, Dehua; Wnuk, Stanislaw F; Bioorganic & Medicinal Chemistry (2011), 19(18), 5507-5519.
2. Wang, Xiao-Ling; Huang, Wen-Feng; Lei, Xin-Sheng; Wei, Bang-Guo; Lin, Guo-Qiang; Tetrahedron (2011), 67(26), 4919-4923.
3. Liu, Xue-Kui; Qiu, Shi; Xiang, Yong-Gang; Ruan, Yuan-Ping; Zheng, Xiao; Huang, Pei-Qiang; Journal of Organic Chemistry (2011), 76(12), 4952-4963.
4. Hulme, Alison N.; Montgomery, Charles H; Tetrahedron Letters (2003), 44(41), 7649-7653.
Ссылки для преобразования лактола в соединение брома (40c):
1. Reddy, P. Ganapati; Chun, Byoung-Kwon; Zhang, Hai-Ren; Rachakonda, Suguna; Ross, Bruce S.; Sofia, Michael J; Journal of Organic Chemistry (2011), 76(10), 3782-3790.
2. Chatterjee, Abhishek; Hazra, Amrita B.; Abdelwahed, Sameh; Hilmey, David G.; Begley, Tadhg P; Angewandte Chemie, International Edition (2010), 49(46), 8653-8656.
3. WO 2010075549 A2 (включено в качестве ссылки).
4. WO 2010075517 A2 (включено в качестве ссылки).
5. WO 2009152095 A2 (включено в качестве ссылки).
6. Castro, Bertrand R. Ecole Nationale Superieure de Chimie de Montpellier, Montpellier, Fr. Organic Reactions (Hoboken, NJ, United States) (1983), 29 Publisher: John Wiley & Sons, Inc.
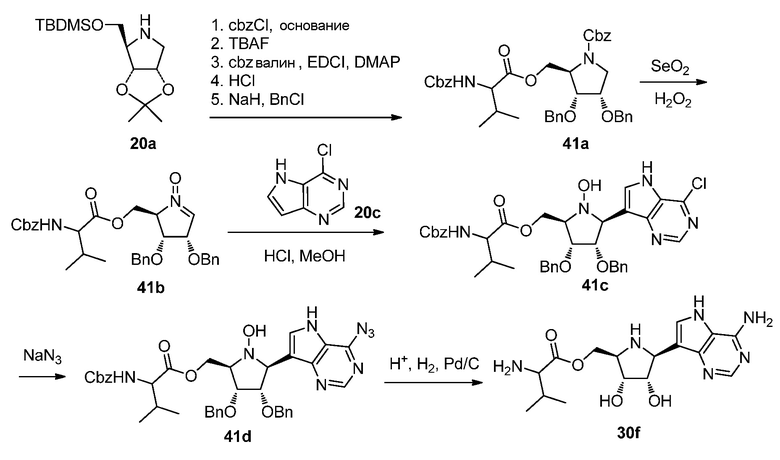
Схема 41
В варианте осуществления изобретения соединение формулы I выбрано из группы, включающей
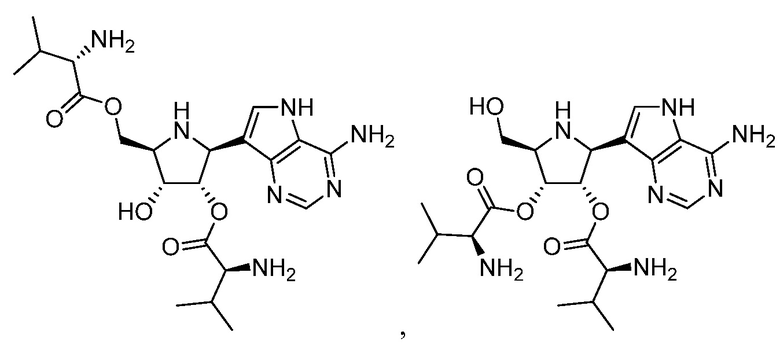 и
и
и их фармацевтически приемлемых солей.
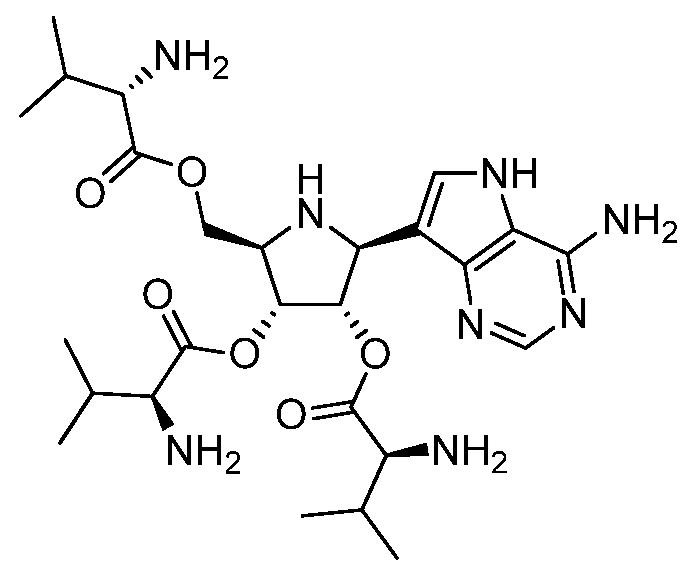
В одном варианте осуществления изобретения соединение формулы I представляет собой 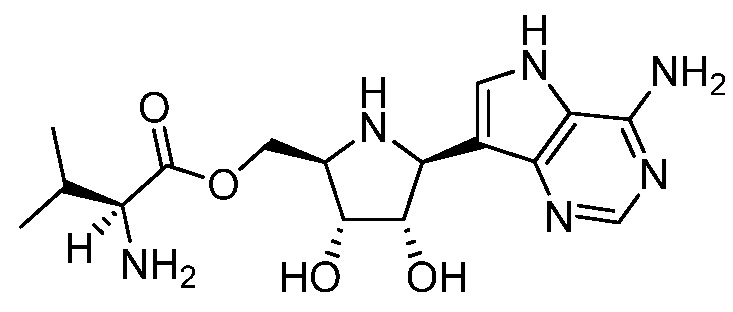 или его фармацевтически приемлемую соль.
или его фармацевтически приемлемую соль.
В одном варианте осуществления изобретения соединение формулы I выбрано из группы, включающей
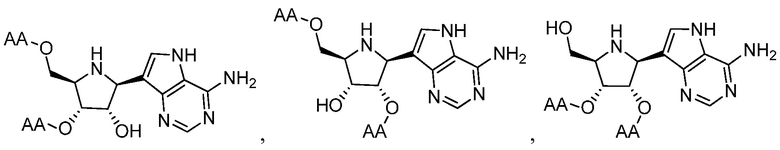
и 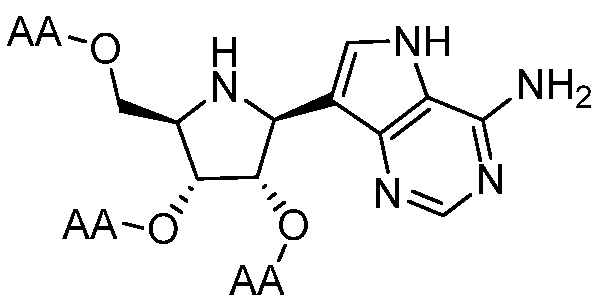 ,
,
и их фармацевтически приемлемые соли, где в каждом случае “AA” представляет собой аминоацильную группу амино кислоты, например, аланил, лейцил, метионил или валинил.
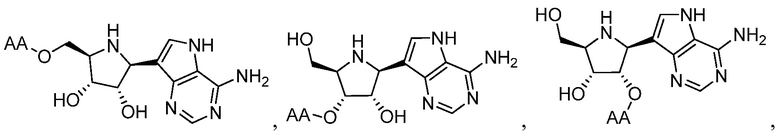
В одном варианте осуществления изобретения соединение формулы I выбрано из группы, включающей 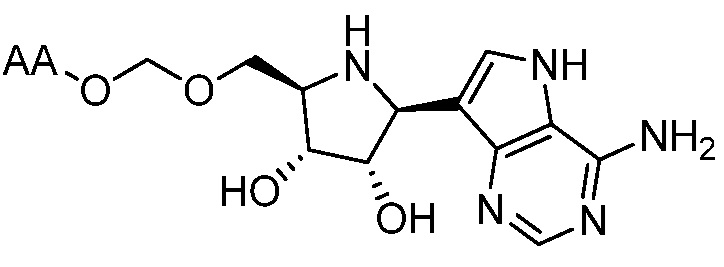 и
и 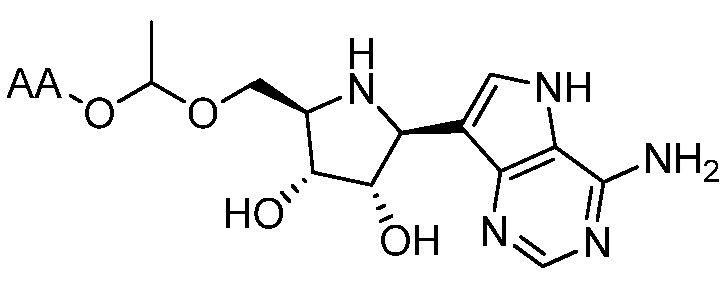 и их фармацевтически приемлемые соли, где в каждом случае “AA” представляет собой аминоацильную группу амино кислоты, например, аланил, лейцил, метионил или валинил.
и их фармацевтически приемлемые соли, где в каждом случае “AA” представляет собой аминоацильную группу амино кислоты, например, аланил, лейцил, метионил или валинил.
В одном варианте осуществления изобретения соединение формулы I выбрано из группы, включающей 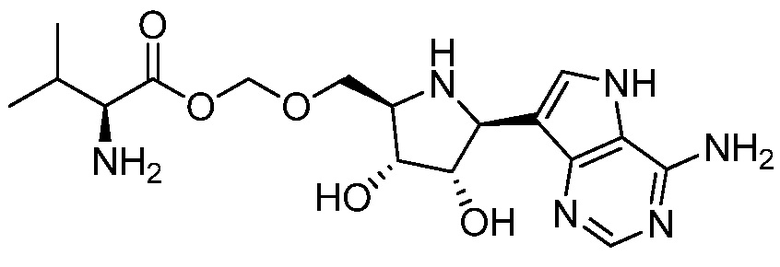 и
и 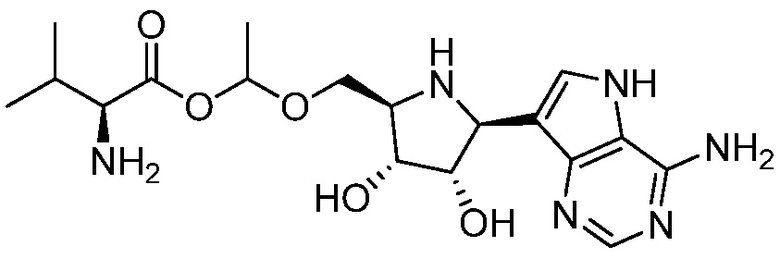 и их фармацевтически приемлемые соли.
и их фармацевтически приемлемые соли.
В одном варианте осуществления изобретения соединение формулы I выбрано из группы, включающей
 и
и 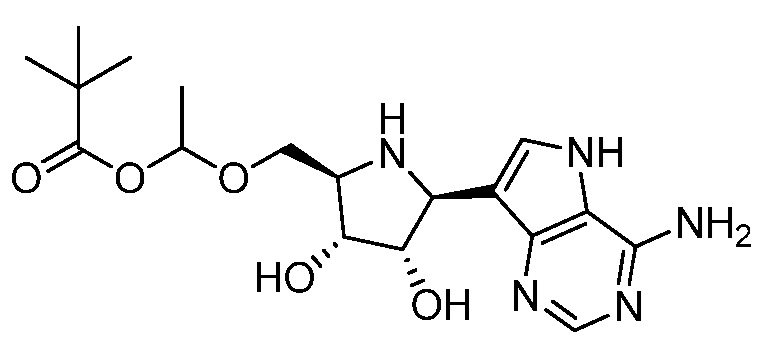 и их фармацевтически приемлемые соли.
и их фармацевтически приемлемые соли.
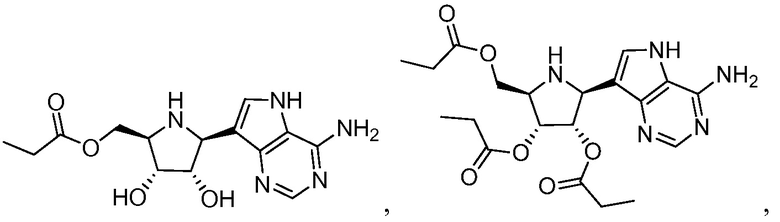
В одном варианте осуществления изобретения соединение формулы I выбрано из группы, включающей 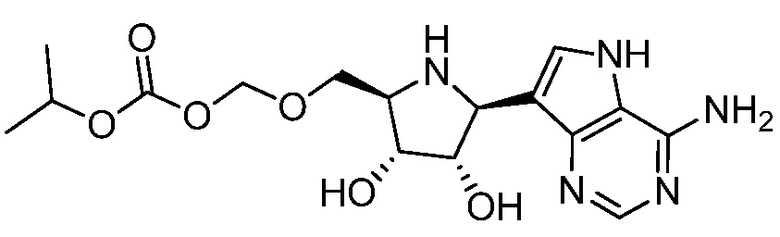 и
и 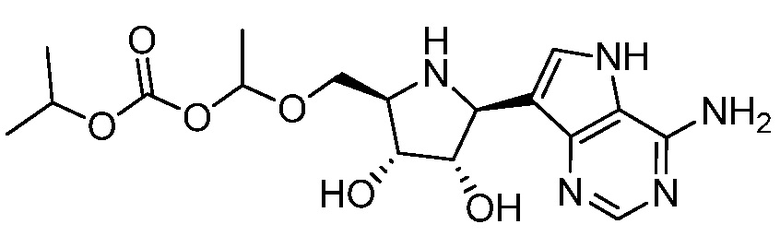 и их фармацевтически приемлемые соли.
и их фармацевтически приемлемые соли.
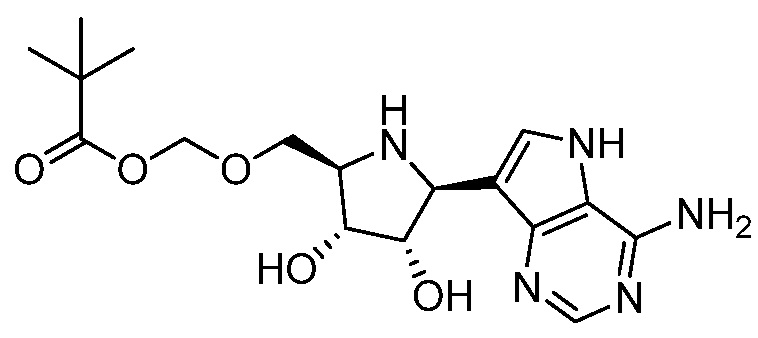
В одном варианте осуществления изобретения соединение формулы I представляет собой 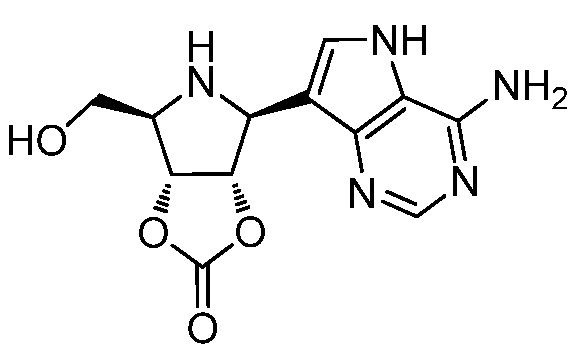 или его фармацевтически приемлемую соль.
или его фармацевтически приемлемую соль.
В одном варианте осуществления изобретения соединение формулы I представляет собой 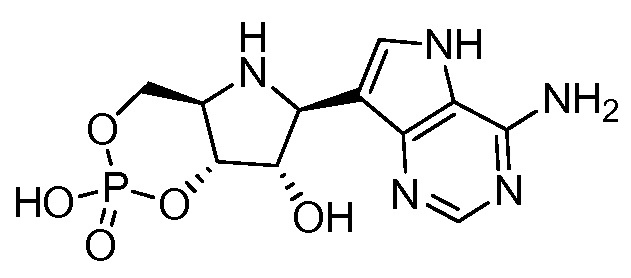 или его фармацевтически приемлемую соль.
или его фармацевтически приемлемую соль.
В одном варианте осуществления изобретения соединение формулы I выбрано из группы, включающей 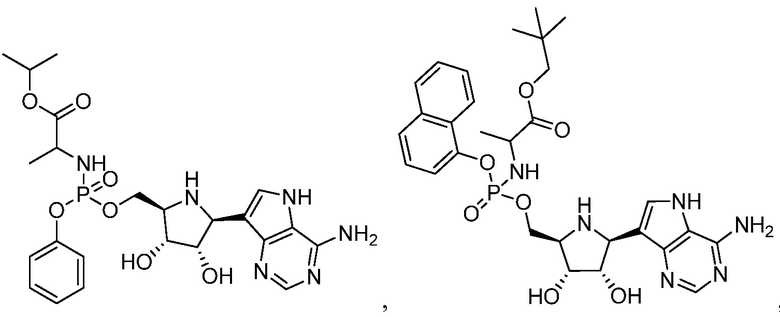 и
и 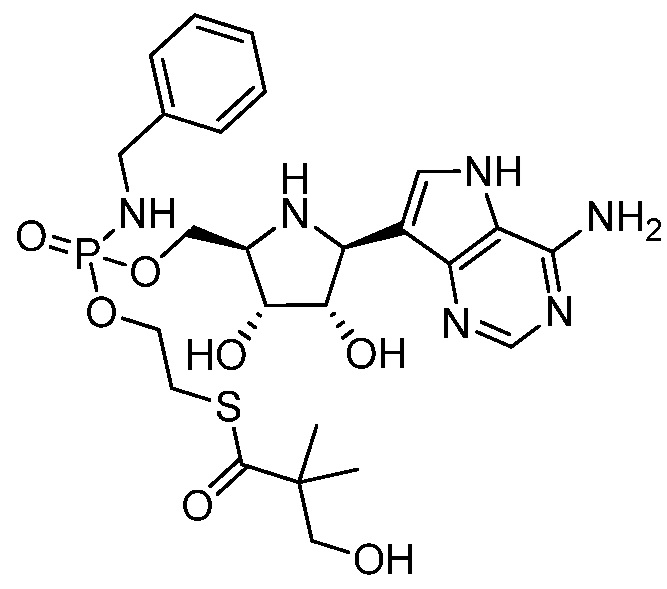 и их фармацевтически приемлемые соли.
и их фармацевтически приемлемые соли.
В одном варианте осуществления изобретения соединение формулы I выбрано из группы, включающей 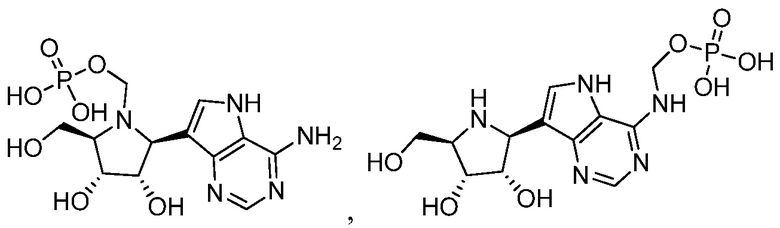 и
и 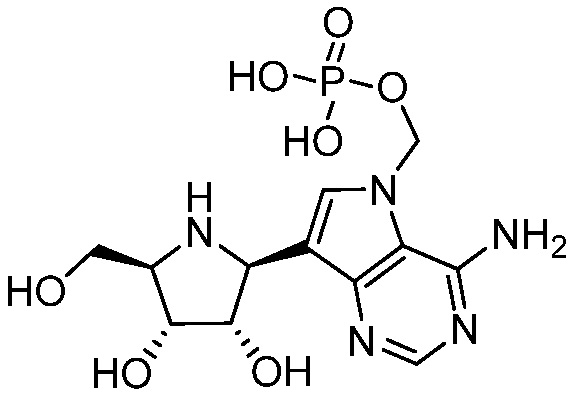 и их фармацевтически приемлемые соли.
и их фармацевтически приемлемые соли.
В одном варианте осуществления изобретения соединение формулы I выбрано из группы, включающей 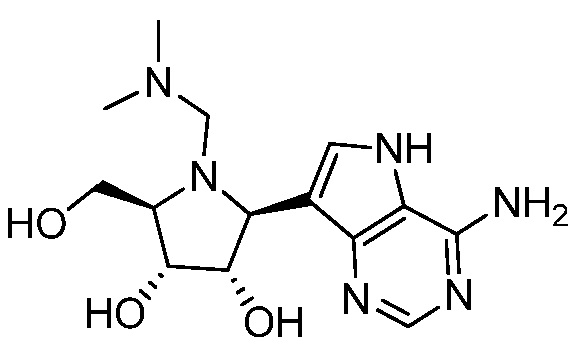 и
и 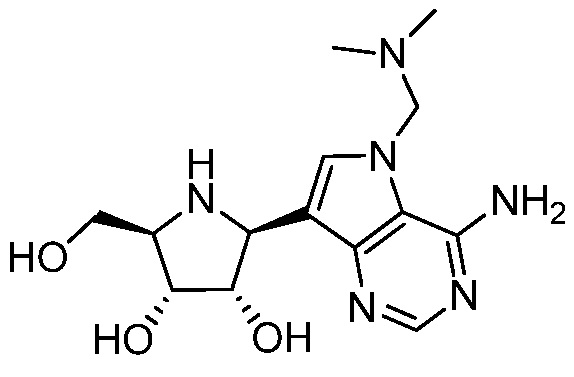 и их фармацевтически приемлемые соли.
и их фармацевтически приемлемые соли.
В одном варианте осуществления изобретения соединение формулы I представляет собой 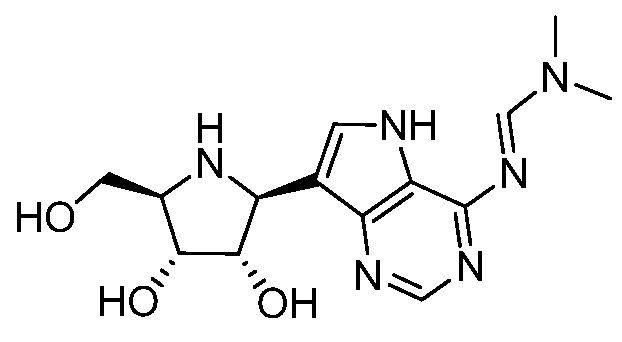 или его фармацевтически приемлемую соль.
или его фармацевтически приемлемую соль.
Аспектом настоящего изобретения является фармацевтическая композиция, содержащая соединение по изобретению или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель.
Аспектом настоящего изобретения является способ получения фармацевтической композиции. Способ включает стадию объединения соединения по изобретению или его фармацевтически приемлемой соли с фармацевтически приемлемым носителем.
Соединения по изобретению могут быть использованы для ингибирования активности полимеразы нуклеиновой кислоты некоторых вирусов. Соединения по изобретению могут быть использованы для ингибирования вирусной репликации или лечения вирусных инфекций.
РНК-вирусы у животных классифицируются на три отдельные группы в зависимости от их генома и способа репликации (и числовые группы на основе более старой классификации по Балтимору):
Двухцепочечные (ds) РНК-вирусы (группа III по классификации Балтимора) содержат от одного до десятка различных молекул РНК, каждая из которых кодирует один или несколько вирусных белков. Примеры дцРНК вирусов включают риовирусы.
Положительно-полярные одноцепочечные (оц) РНК-вирусы (группа IV по классификации Балтимор) имеют свой геном, используемый непосредственно, как если бы это были мРНК, продуцируя один белок, который модифицируется белками хозяина и вируса с образованием различных белков, необходимых для репликации. Один из них включает РНК-зависимую РНК-полимеразу, которая копирует вирусную РНК с формированием двухцепочечной репликативной формы, которая в свою очередь контролирует образование новых вирионов. Примеры положительно-полярных оцРНК-вирусов включают тогавирус, флавивирус, кальцивирус, коронавирус, пикорнавирус и тогавирус.
Отрицательно-полярные оцРНК-вирусы (группа V по классификации Балтимор) должны иметь свой геном, копируемый РНК-полимеразой с формированием положительно-полярной РНК. Это означает, что вирус должен “принести” с собой фермент - РНК-зависимую РНК-полимеразу. Затем положительно-полярная молекула РНК выполняет функцию вирусной мРНК, которая транслируется в белки рибосомами хозяина. Полученный белок идет на прямой синтез новых вирионов, например, капсидных белков и РНК-репликазы, используемых для продуцирования новых молекул отрицательно-полярных РНК. Отрицательно-полярные оцРНК-вирусы включают борнавирус, филовирус, ортомиксовирус, парамиксовирус, рабдовирус, аренавирус и буньявирус.
Ретровирусы (группа VI по классификации Балтимор) имеют одноцепочечный РНК-геном, но обычно не рассматриваются в качестве РНК-вирусов, поскольку для репликации они используют посредников ДНК. Обратная транскриптаза, вирусный фермент, который выходит из самого вируса, после того, как вирус теряет свою оболочку, и преобразует вирусную РНК в комплементарную нить ДНК, которая копируется для продукции двухцепочечной молекулы вирусной ДНК. После того, как ДНК интегрируется, экспрессия кодируемых генов может привести к образованию новых вирионов. Ретровирусы включают, без ограничения, ВИЧ-1 и ВИЧ-2.
Аспектом настоящего изобретения является способ ингибирования активности вирусной полимеразы нуклеиновой кислоты вируса. Способ включает стадию контактирования вирусной полимеразы нуклеиновой кислоты вируса с эффективным количеством соединения по изобретению или его фармацевтически приемлемой соли.
В одном варианте осуществления изобретения вирусная полимераза нуклеиновой кислоты представляет собой ДНК-полимеразу.
В одном варианте осуществления вирусная полимераза нуклеиновой кислоты представляет собой РНК-полимеразу.
В одном варианте осуществления изобретения вирус выбран из группы, состоящей из РНК-содержащих вирусов.
В одном варианте осуществления изобретения вирус выбран из группы, состоящей из ортомиксовируса, парамиксовируса, аренавируса, буньявируса, флавивируса, филовируса, тогавируса, пикорнавируса и коронавируса.
В одном варианте осуществления изобретения вирус выбран из группы, состоящей из аденовируса, риновируса, вируса гепатита А, вируса гепатита С, вируса полиомиелита, вируса кори, вируса Эбола, вируса Коксаки, вируса лихорадки западного Нила, вируса оспы, вируса желтой лихорадки, вируса лихорадки денге, вируса гриппа А, вируса гриппа В, вируса Ласса, вируса лимфоцитарного хориоменингита, вируса Хунин, вируса мачупо, вируса гуанарито, хантавируса, вируса лихорадки долины Рифт, вируса Ла-Кросс, вируса калифорнийского энцефалита, вируса Конго-Крым, вируса Марбург, вируса японского энцефалита, вируса болезни Кьясанурского Леса, вируса венесуэльского энцефалита лошадей, вируса восточного энцефалита лошадей, вируса западного энцефалита лошадей, вируса тяжелого острого респираторного синдрома (ТОРС), вируса парагриппа, респираторного синтициального вируса, вируса Пунта-Торо, вируса Такарибе и вируса Пичинде.
В одном варианте осуществления изобретения вирус выбран из группы, включающей аденовирус, вирус лихорадки денге, вирус Эбола, вирус Марбург, вирус гриппа А, вирус гриппа В, вирус Хунин, вирус кори, вирус парагриппа, вирус Пичинде, вирус Пунта-Торо, вирус респираторный синтициальный, риновирус, вирус лихорадки долины Рифт, вирус тяжелого острого респираторного синдрома, вирус Такарибе, вирус венесуэльского энцефалита лошадей, вирус лихорадки Западного Нила и вирус желтой лихорадки.
В одном варианте осуществления изобретения вирус выбран из группы, включающей вирус Эбола, вирус желтой лихорадки, вирус Марбург, вирус гриппа А и вирус гриппа В.
Аспектом настоящего изобретения является способ ингибирования репликации вируса. Способ включает стадию контактирования вируса с эффективным количеством соединения по изобретению или его фармацевтически приемлемой соли таким образом, чтобы ингибировать репликацию вируса.
В одном варианте осуществления изобретения вирус выбран из группы, включающей РНК-вирусы.
В одном варианте осуществления изобретения вирус выбран из группы, включающей ортомиксовирус, парамиксовирус, аренавирус, буньявирус, флавивирус, филовирус, тогавирус, пикорнавирус и коронавирус.
В одном варианте осуществления изобретения вирус выбран из группы, включающей аденовирус, риновирус, вирус гепатита А, вирус гепатита С, вирус полиомиелита, вирус кори, вирус Эбола, вирус Коксаки, вирус лихорадки западного Нила, вирус оспы, вирус желтой лихорадки, вирус лихорадки денге, вирус гриппа А, вирус гриппа В, вирус Ласса, вирус лимфоцитарного хориоменингита, вирус Хунин, вирус мачупо, вирус Гуанарито, хантавирус, вирус лихорадки долины Рифт, вирус Ла-кросс, вирус калифорнийского энцефалита, вирус Конго-Крым, вирус Марбург, вирус японского энцефалита, вирус болезни Кьясанурского Леса, вирус венесуэльского энцефалита лошадей, вирус восточного лошадиного энцефалита, вирус западного лошадиного энцефалита, вирус тяжелого острого респираторного синдрома (SARS), вирус парагриппа, вирус респираторный синтициальный, вирус Пунта-Торо, вирус Такарибе и вирус Пичинде.
В одном варианте осуществления изобретения вирус выбран из группы, включающей аденовирус, вирус лихорадки денге, вирус Эбола, вирус Марбург, вирус гриппа А, вирус гриппа В, вирус Хунин, вирус кори, вирус парагриппа, вирус Пичинде, вирус Пунта-Торо, вирус респираторный синтициальный, риновирус, вирус лихорадки долины Рифт, вирус тяжелого острого респираторного синдрома, вирус Такарибе, вирус венесуэльского энцефалита лошадей, вирус лихорадки Западного Нила и вирус желтой лихорадки.
В одном варианте осуществления изобретения вирус выбран из группы, включающей вирус Эбола, вирус желтой лихорадки, вирус Марбург, вирус гриппа А и вирус гриппа В.
Аспектом настоящего изобретения является способ лечения вирусной инфекции у субъекта. Способ включает в себя стадию введения субъекту, нуждающемуся в таком лечении, эффективного количества соединения по изобретению или его фармацевтически приемлемой соли.
В одном варианте осуществления изобретения вирус выбран из группы, включающей РНК-вирусы.
В одном варианте осуществления изобретения вирус выбран из группы, включающей ортомиксовирус, парамиксовирус, аренавирус, буньявирус, флавивирус, филовирус, тогавирус, пикорнавирус и коронавирус.
В одном варианте осуществления изобретения вирус выбран из группы, включающей аденовирус, риновирус, вирус гепатита А, вирус гепатита С, вирус полиомиелита, вирус кори, вирус Эбола, вирус Коксаки, вирус лихорадки Западного Нила, вирус оспы, вирус желтой лихорадки, вирус лихорадки денге, вирус гриппа А, вирус гриппа В, вирус Ласса, вирус лимфоцитарного хориоменингита, вирус Хунин, вирус мачупо, вирус Гуанарито, хантавирус, вирус Лихорадки долины Рифт, вирус Ла-кросс, вирус калифорнийского энцефалита, вирус Конго-Крым, вирус Марбург, вирус японского энцефалита, вирус болезни Кьясанурского Леса, вирус венесуэльского энцефалита лошадей, вирус восточного лошадиного энцефалита, вирус западного лошадиного энцефалита, вирус тяжелого острого респираторного синдрома (SARS), вирус парагриппа, вирус респираторный синтициальный, вирус Пунта-Торо, вирус Такарибе и вирус Пичинде.
В одном варианте осуществления изобретения вирус выбран из группы, включающей аденовирус, вирус лихорадки денге, вирус Эбола, вирус Марбург, вирус гриппа А, вирус гриппа В, вирус Хунин, вирус кори, вирус парагриппа, вирус Пичинде, вирус Пунта-Торо, вирус респираторный синтициальный, риновирус, вирус Лихорадки долины Рифт, Вирус тяжелого острого респираторного синдрома, вирус Такарибе, вирус венесуэльского энцефалита лошадей, вирус Лихорадки Западного Нила и вирус желтой лихорадки.
В одном варианте осуществления изобретения вирус выбран из группы, включающей Вирус Эбола, вирус желтой лихорадки, вирус Марбург, вирус гриппа А и вирус гриппа В.
Соединения по изобретению могут быть получены в виде фармацевтических композиций и введены млекопитающему-хозяину, такому как человек-пациент, в различных формах, адаптированных для выбранного способа введения, например, перорального или парентерального, внутривенного, внутрибрюшинного, внутримышечного, местного или подкожного введения.
Таким образом, соединения по настоящему изобретению могут быть введены системно, например, перорально, в сочетании с фармацевтически приемлемым носителем, таким как инертный разбавитель или усвояемый пищевой носитель. Они могут быть заключены в твердые или мягкие желатиновые капсулы, спрессованы в таблетки, или могут быть объединены непосредственно с пищевыми продуктами диеты пациента. Для перорального терапевтического введения активное соединение может быть объединено с одним или несколькими наполнителями и использованы в форме таблеток для приема внутрь, буккальных таблеток, пастилок, капсул, эликсиров, суспензий, сиропов, облаток и тому подобное. Такие композиции и препараты должны содержать по меньшей мере 0,1% активного соединения. Процентное содержание композиций и препаратов может, конечно, изменяться, и обычно составляет от около 2 до около 60% от массы заданной стандартной лекарственной формы. Количество активного соединения в таких терапевтически эффективных композициях является таким, которое обеспечивает получение эффективного уровня дозы.
Таблетки, пастилки, пилюли, капсулы и т.п. могут также содержать следующие разбавители и носители: связующие, такие как трагакантовая камедь, гуммиарабик, кукурузный крахмал или желатин; вспомогательные вещества, такие как дикальций фосфат; разрыхляющий агент, такой как кукурузный крахмал, картофельный крахмал, альгиновая кислота и тому подобное; и смазывающее вещество, такое как стеарат магния; и может быть добавлен подсластитель, такой как сахароза, фруктоза, лактоза или аспартам, или ароматизатор, такой как мята перечная, масло грушанки или вишневый ароматизатор. Когда стандартная лекарственная форма представляет собой капсулу, она может содержать, помимо веществ указанного выше типа, жидкий носитель, такой как растительное масло или полиэтиленгликоль. Другие различные вещества могут присутствовать в качестве покрытий или для изменения физической формы твердой стандартной лекарственной формы. Например, таблетки, пилюли или капсулы могут быть покрыты желатином, воском, шеллаком или сахаром и тому подобным. Сироп или эликсир могут содержать активное соединение, сахарозу или фруктозу в качестве подслащивающего агента, метил- и пропилпарабены в качестве консервантов, краситель и ароматизатор, такой как вишневый или апельсиновый ароматические добавки. Конечно, любое вещество, используемое в получении любой стандартной лекарственной формы, должно быть фармацевтически приемлемым и по существу нетоксичным в используемых количествах. Кроме того, активное соединение может быть включено в препараты и устройства с замедленным высвобождением.
Активное соединение может также быть введено внутривенно или внутрибрюшинно путем инфузии или инъекции. Растворы активного соединения или его соли могут быть приготовлены в воде или физиологически приемлемом водном растворе, необязательно, в смеси с нетоксичным поверхностно-активным веществом. Дисперсии могут быть получены в глицерине, жидких полиэтиленгликолях, триацетине, а также в их смесях и в маслах. При обычных условиях хранения и использования эти препараты содержат консервант для предотвращения роста микроорганизмов.
Фармацевтические лекарственные формы, подходящие для инъекции или инфузии, могут включать стерильные водные дисперсии или растворы или стерильные порошки, содержащие активный ингредиент, который адаптирован для экстемпорального поучения стерильных растворов или дисперсий, и, необязательно, инкапсулирован в липосомы. Во всех случаях, конечные лекарственные формы должны быть стерильными, жидкими и стабильными в условиях производства и хранения. Жидкий носитель или наполнитель может представлять собой растворитель или жидкую дисперсионную среду, включающую, например, воду, этанол и полиол (например, глицерин, пропиленгликоль, жидкие полиэтиленгликоли и тому подобное), растительные масла, нетоксичные эфиры глицерина и их подходящие смеси. Собственная текучесть может поддерживаться, например, с помощью образования липосом, путем сохранения требуемого размера частиц в случае дисперсий или с помощью использования поверхностно-активных веществ. Предотвращение действия микроорганизмов может быть осуществлено с помощью разных антибактериальных и противогрибковых агентов, например, парабенов, хлорбутанола, фенола, сорбиновой кислоты, тимеросала и т.п. Во многих случаях будет предпочтительным включить изотонические агенты, например, сахара, буфер или хлорид натрия. Пролонгированную абсорбцию инъецируемых композиций можно осуществить путем использования в композициях задерживающих абсорбцию агентов, например, моностеарата алюминия и желатина.
Стерильные инъекционные растворы получают путем введения активного соединения в требуемом количестве в соответствующий растворитель с различными другими ингредиентами, перечисленными выше, при необходимости, с последующей стерилизацией путем фильтрации. В случае стерильных порошков для приготовления стерильных инъекционных растворов способы приготовления включают методы вакуумной сушки и лиофилизации, которые позволяют получить порошок активного ингредиента дополнительно к любому дополнительно необходимому ингредиенту, присутствующему в предварительно стерильно отфильтрованных растворах.
Для местного введения соединения по настоящему изобретению могут применяться в чистой форме, например, когда они находится в жидкой форме. Однако, в целом, желательно наносить их на кожу в виде композиций или препаратов в сочетании с дерматологически приемлемым носителем, который может быть твердым или жидким.
Эффективные твердые носители включают высокодисперсные твердые вещества, такие как тальк, глину, микрокристаллическую целлюлозу, кремнезем, глинозем и т.п. Эффективные жидкие носители включают воду, спирты или гликоли, или водно-спиртовые/гликольные смеси, в которых соединения по настоящему изобретению могут растворяться или диспергироваться на эффективных уровнях, необязательно, с добавлением нетоксичных поверхностно-активных веществ. Адъюванты, такие как ароматизаторы и дополнительные антимикробные агенты, могут быть добавлены для оптимизации свойств для заданного применения. Полученные жидкие композиции могут наноситься на гигроскопические прокладки, которые используются для пропитки повязок и других перевязочных материалов, или распыляться на пораженную область с помощью дозирующих или аэрозольных распылителей.
Загустители, такие как синтетические полимеры, жирные кислоты, соли жирных кислот и сложные эфиры, жирные спирты, модифицированная целлюлоза или модифицированные минеральные вещества, также могут использоваться с жидкими носителями для образования легко намазывающихся паст, гелей, мазей, мыла и тому подобного, для непосредственного нанесения на кожу потребителя.
Примеры эффективных дерматологических композиций, которые могут быть использованы для нанесения соединений формулы I на кожу, известны из уровня техники; например, см. Jacquet et al. (патент США № 4608392; включенный в настоящий документ посредством ссылки), Geria (патент США № 4992478; включенный в настоящий документ посредством ссылки), Smith et al. (патент США № 4559157; включенный в настоящий документ посредством ссылки) и Wortzman (патент США № 4820508; включенный в настоящий документ посредством ссылки).
Эффективные дозы соединений по изобретению могут быть определены путем сравнения их in vitro и in vivo активности на животных моделях. Методы экстраполяции эффективных доз у мышей и других животных на человека известны из уровня техники; например, смотри патент США № 4938949.
Количество соединения или его активной соли или его производного, необходимое для использования в лечении, будет изменяться в зависимости не только от конкретного выбранного соединения или соли, но также от пути введения, природы заболевания, подлежащего лечению, возраста и состояния пациента, и будет в конечном счете находиться на рассмотрении лечащего терапевта или клинициста.
Однако обычно подходящая доза будет в диапазоне от около 0,5 до около 100 мг/кг массы тела реципиента в день, например, от приблизительно 3 до приблизительно 90 мг/кг массы тела в день, от около 6 до около 75 мг на килограмм массы тела в день, от приблизительно 10 до приблизительно 60 мг/кг/день или от приблизительно 15 до приблизительно 50 мг/кг/день.
Соединения по изобретению обычно могут быть получены в стандартной лекарственной форме; например, содержащей от 5 до 1000 мг, от 10 до 750 мг или от 50 до 500 мг активного ингредиента на стандартную лекарственную форму. В одном варианте осуществления изобретение относится к композиции, содержащей соединение по изобретению, полученной в такой стандартной лекарственной форме. Необходимая доза обычно может быть представлена в виде однократной дозы или в виде разделенных доз, которые вводят через соответствующие промежутки времени, например, в виде двух, трех, четырех или более субдоз в сутки. Сама субдоза может быть дополнительно разделена, например, на ряд отдельных введений с небольшими промежутками, такие как множественные ингаляции из инсуффлятора или путем нанесения множества капель в глаза.
Соединения по настоящему изобретению также могут быть введены в сочетании с другими терапевтическими средствами, например, другими агентами, которые являются эффективными для лечения вирусной инфекции.
Настоящее изобретение также относится к набору, содержащему соединение по изобретению или его фармацевтически приемлемую соль; по меньшей мере одно другое терапевтическое средство, упаковочный материал и инструкции по введению соединения по настоящему изобретению или его фармацевтически приемлемой соли и другого терапевтического средства или средств млекопитающему для лечения вирусной инфекции у млекопитающего. В одном варианте осуществления изобретения млекопитающее является человеком.
Далее изобретение будет проиллюстрировано следующими неограничивающими объем изобретения примерами.
ПРИМЕРЫ
Пример 1: (2S,3S,4R,5R)-2-(4-амино-5H-пирроло[3,2-d]пиримидин-7-ил)-5-(гидроксиметил)пирролидин-3,4-диол дигидрохлорид (12i)
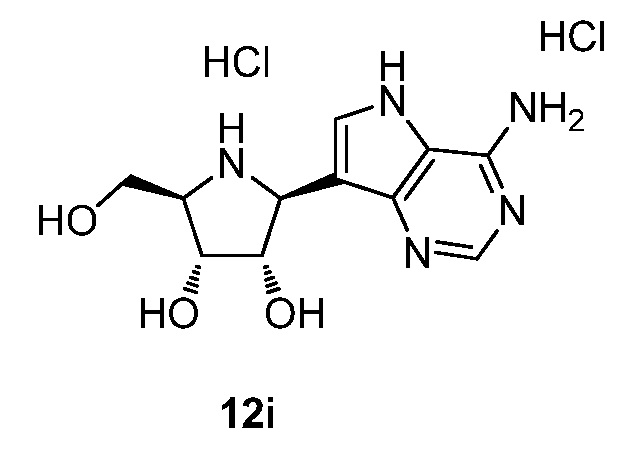
(2S,3S,4R,5R)-трет-бутил 2-(4-амино-5H-пирроло[3,2-d]пиримидин-7-ил)-3,4-дигидрокси-5-(гидроксиметил)пирролидин-1-карбоксилат (28f) обрабатывали следующим образом тремя партиями.
Партия 1. (28f) растворяли в водн. HCl (1658,8 ммоль, 118 мл конц. HCl и 293 мл воды).
Партия 2. (28f) растворяли в водн. HCl (239,6 ммоль, 169 мл конц. HCl и 421 мл воды).
Партия 3. (28f) растворяли в водн. HCl (263,5 ммоль, 186 мл конц. HCl и 468 мл воды).
Реакционные смеси перемешивали при комнатной температуре в течение 30 мин (сильное выделение газа CO2), и затем каждую партию концентрировали в вакууме досуха (80-90°C). Партии 2 и 3 объединяли с получением 226 г влажного прозрачного желтого продукта. Партия 1 давала 91,4 г продукта темного сероватого цвета. Кристаллизацию осуществляли следующим образом: Для партий 2 и 3 влажного продукта: 226 мл воды добавляли к продукту, затем нагревали до температуре 50°C, в этот момент медленно добавляли горячий этанол до начала кристаллизации. Смесь выдерживали при температуре 50°C в течение 10 минут, затем давали достичь температуры 25°C при энергичном перемешивании, далее фильтровали с получением порошка светло-желтого цвета (2S,3S,4R,5R)-2-(4-амино-5H-пирроло[3,2-d]пиримидин-7-ил)-5-(гидроксиметил)пирролидин-3,4-диола (12i) (88 г, 52%). Партию 1 очищали таким же образом с получением 33,0 г (59%) продукта светло-сероватого цвета. Общий выход был 121,0 г (53,5%) после высушивания при температуре 55°C в высоком вакууме. Маточный раствор после перекристаллизации партии 1 и 2 вновь обрабатывали с получением 15,0 г порошкообразного продукта светло-желтоватого цвета (12i); Т.пл.: 238°C. 1H ЯМР (300 МГц, ДМСО-d6) δ 14,60 (с, 1H), 13,25 (с, 1H), 10,23 (с, 1H), 9,13 (с, 2H), 8,84 (с, 1H), 8,63 (с, 1H), 8,11 (д, J=3,1 Гц, 1H), 5,55 (с, 2H), 4,78 (д, J=4,4 Гц, 1H), 4,44 (дд, J=8,8, 5,0 Гц, 1H), 4,14-4,02 (м, 1H), 3,73 (д, J=5,1 Гц, 2H), 3,52 (с, 1H); 1H ЯМР (300 МГц, D2O) δ 8,33 (с, 1H), 7,94 (с, 1H), 4,90 (д, J=8,9 Гц, 1H), 4,65 (с, 1H), 4,37 (дд, J=4,8, 3,4 Гц, 1H), 3,89 (с, 1H), 3,88 (с, 1H), 3,81 (дд, J=8,1, 4,5 Гц, 1H); MS (ES+) 266,3 (M+1); Оптическое вращение -52,69; (H2O, C=1,15); Анализ: Вычислено для C11H15N5O3⋅2HC⋅0,25H2O: C, 38,55; H, 5,15; Cl, 20,44; N, 20,69; Найдено: C, 38,67; H, 5,05; Cl, 20,45; N, 20,42.
Альтернативный способ получения дигидрохлорида (2S,3S,4R,5R)-2-(4-амино-5H-пирроло[3,2-d]пиримидин-7-ил)-5-(гидроксиметил)пирролидин-3,4-диола (12i) исходя из (2R,3R,4S,5S)-2-(ацетоксиметил)-1-(трет-бутоксикарбонил)-5-(4-хлор-5H-пирроло[3,2-d]пиримидин-7-ил)пирролидин-3,4-диил диацетата (28c).
В прозрачный раствор диацетата (2R,3R,4S,5S)-2-(ацетоксиметил)-1-(третбутоксикарбонил)-5-(4-хлор-5H-пирроло[3,2-d]пиримидин-7-ил)-пирролидин-3,4-диила (28c) (40 г, 78,29 ммоль) в этаноле (400 мл) выливали аммиак (35% объем относительно этанола) при температуре -50°C. Охлажденный раствор осторожно выливали в автоклав и нагревали в течение 16 ч при температуре 100-105°С. Проводили анализ ТСХ, чтобы быть уверенным в завершение реакции. Смесь оставляли охлаждаться до комнатной температуры. Растворитель отгоняли с получением (2S,3S,4R,5R)-трет-бутил 2-(4-амино-5H-пирроло[3,2-d]пиримидин-7-ил)-3,4-дигидрокси-5-(гидроксиметил)пирролидин-1-карбоксилата (28f) 38 г в виде темно-коричневой клейкой массы.
К перемешиваемому раствору трет-бутил (2S,3R,4S,5S)-5-(гидроксиметил)-2-(4-амино-5H-пирроло[3,2-d]пиримидин-7-ил)-пирролидин-3,4-дигидрокси карбоксилата (28f) (292 г, 799,16 ммоль) в деионизированной воде (584 мл) добавляли конц. HCl (423 мл). Полученный прозрачный раствор перемешивали в течение 30 мин при комнатной температуре. Затем его концентрировали досуха (водяная баня 80-90°C) с получением влажного твердого вещества желтого цвета. Влажный осадок затем разбавляли деионизированной водой (475 мл) и давали нагреться до температуры 70°C с получением прозрачного раствора и охлаждали до температуры 50°C. Медленно добавляли горячий этанол (1,6 л) с получением частичного осадка. Смесь перемешивали в течение 10 мин при температуре 60°C. Смеси давали охладиться до комнатной температуры и охлаждали до температуры 10°C и перемешивали в течение 1 часа при той же температуре. Полученное твердое вещество собирали путем фильтрации, сушили при температуре 55-60°C до достижения постоянной массы с получением дигидрохлорида (2S,3S,4R,5R)-2-(4-амино-5H-пирроло[3,2-d]пиримидин-7-ил)-5-(гидроксиметил)пирролидин-3,4-диола (12i) (65 г) в виде твердого вещества от бледно-желтого до не совсем белого цвета; Т.пл.: 255,5°C. 1H ЯМР (300 МГц, ДМСО-d6) δ 14,64 (с, 1H), 13,19 (с, 1H), 10,20 (с, 1H), 9,11 (с, 2H), 8,83 (с, 1H), 8,64 (с, 1H), 8,11 (д, J=3,1 Гц, 1H), 5,99-5,20 (ушир. с, 2H), 4,78 (с, 1H), 4,43 (дд, J=8,9, 4,9 Гц, 1H), 4,11 (т, J=4,2 Гц, 1H), 3,73 (д, J=5,1 Гц, 2H), 3,51 (с, 2H); MS (ES+) 266,1 (M+1); Оптическое вращение -51,74 (H2O,C=0,545); Анализ: Вычислено для C11H15N5O3·2HCl·0,25H2O: C, 38,55; H, 5,15; Cl, 20,69; N, 20,44; Найдено: C, 38,51; H, 5,11; Cl, 20,57; N, 20,31.
Получение (2R,3R,4S,5S)-2-(ацетоксиметил)-1-(трет-бутоксикарбонил)-5-(4-хлор-5H-пирроло[3,2-d]пиримидин-7-ил)пирролидин-3,4-диил диацетата (28c) и трет-бутил (2S,3R,4S,5S)-5-(гидроксиметил)-2-(4-амино-5H-пирроло[3,2-d]пиримидин-7-ил)-пирролидин-3,4-дигидрокси карбоксилата (28f).
Стадия 1: Получение D-Рибоно лактона (19b)
В 22-литровую трехгорлую колбу, снабженную механической мешалкой, 1 литровой выравнивающей давление капельной воронкой и эффективным холодильником, помещали D-рибозу (19a) (2,0 кг, 13,33 моль), твердый бикарбонат натрия (2,24 кг, 26,66 моль) и воду (12 л). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч, за это время большая часть твердого вещества исчезала. Реакционный сосуд помещали на ледяную баню с внутренней температурой, поддерживаемой при 5±1°C. В капельную воронку помещали бром (710 мл, 13,86 моль), и в перемешиваемый водный раствор энергично добавляли бром со скоростью приблизительно 5 мл/мин, чтобы температура поддерживалась в интервале 5-10°C. Когда добавление заканчивали (примерно 2,5 ч), полученный раствор оранжевого цвета перемешивали еще дополнительно 3 ч. В реакционную смесь добавляли твердый гидросульфит натрия (~75 г) небольшими партиями, пока оранжевый цвет полностью не исчезал. Прозрачный водный раствор переносили в 20 л перегонную колбу и выпаривали досуха на роторном испарителе (80°C, 10 мм рт.ст.) в течение 4 ч, получая полутвердый остаток. К остатку добавляли этиловый спирт (~4 л) и перемешивали при температуре 40°C в течение 1 ч. Смесь охлаждали и фильтровали через воронку, чтобы удалить большую часть нерастворимой неорганической соли. Твердый остаток промывали этиловым спиртом (1 л). Фильтрат переносили в 20-литровую перегонную колбу и концентрировали досуха на роторном испарителе (50°C, 10 мм рт.ст.) с получением твердого остатка. К этому остатку добавляли этиловый спирт (~3 л), и взвесь перемешивали при комнатной температуре в течение 12 ч. Твердое вещество собирали путем фильтрации и промывали этиловым спиртом (750 мл). Продукт D-Рибоно лактон (19b) сушили в вакуумной печи при температуре 40°C (0,1 мм рт.ст.). Выход 1,28 кг (65%); Т. пл. 77-80°C; 1H ЯМР (D2O) δ 4,72 (д, 1 H), 4,57 (т, 1 H), 4,42 (д, 1 H), 3,80 (м, 2 H).
Стадия 2: Получение 2,3-O-изопропилиден D-Рибоно-1,4-лактона (19c)
В 50-литровый реакционный сосуд с рубашкой помещали D-рибоно-1,4-лактон (19b) (3,0 кг, 20,27 моль) и 30 л ацетона со степенью чистоты, соответствующей стандарту ACS. Реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Внутреннюю температуру реакционного сосуда понижали до температуры 10°C, и в реакционную смесь медленно добавляли конц. серную кислоту (49 мл). После добавления серной кислоты внутренней температуре реакционной смеси давали медленно нагреться. Реакционную смесь перемешивали при этой температуре в течение 2,5-3 ч. Реакцию отслеживали с помощью ТСХ (ТСХ; 9:1, метиленхлорид:метиловый спирт, Rf=0,75). Реакционную смесь нейтрализовали путем добавления твердого бикарбоната натрия (~500 г) до нейтрального pH. Реакционную смесь фильтровали через воронку. Твердый остаток, содержащий неорганические соли, промывали ацетоном (3 л). Фильтрат переносили в 20-литровую перегонную колбу и упаривали досуха (50°C, 10 мм рт.ст.) с получением полутвердого соединения. Остаток обрабатывали этилацетатом (3 л) и перемешивали при комнатной температуре в течение 4 ч на роторном испарителе. Твердое вещество 2,3-O-изопропилиден D-Рибоно-1,4-лактон (19c) собирали путем фильтрации и сушили в вакуумной печи в течение 16 ч при температуре 40°C (0,1 мм рт.ст.). Выход: 1,819 кг (48%); т.пл. 136-140°C; 1H ЯМР (CDCl3) δ 4,8 (дд, 2 H), 4,6 (с, 1 H), 3,85 (дд, 2 H), 1,5 (с, 3 H), 1,4 (с, 3 H).
Стадия 3: Получение 2,3-O-изопропилиден 5-O-метансульфонил D Рибоно-1,4-лактона (26a)
Раствор 2,3-O-изопропилиден D-Рибоно-1,4-лактона (19c) (4,3 кг, 22,96 моль) в пиридине со степенью чистоты, соответствующей стандарту ACS, (20 л) перемешивали в 50 л реакционном сосуде при комнатной температуре в течение 15 мин до завершения растворения. Внутреннюю температуру реакционного сосуда понижали до -15°C, затем медленно добавляли метан сульфонилхлорид (1,96 л, 25,26 моль) в течение периода времени в 2 ч. Внутреннюю температуру поддерживали при 0-5°C. Реакцию перемешивали при температуре 0°C в течение ~2 ч в инертной атмосфере до тех пор, пока анализ ТСХ не показывал отсутствие исходного материала (ТСХ; 7:3 этилацетат:гексан, Rf=0,85). После завершения реакции добавляли DCM (10 л) и экстрагировали с помощью 3н HCl (4 раза, pH=3), [Повторная экстракция водного слоя с помощью DCM (5 л) каждый раз] с последующей быстрой промывкой насыщенным водным раствором NaHCO3. Органическую фракцию сушили над сульфатом натрия, фильтровали и упаривали до сиропа. Выход: 4,89 кг (80%). Продукт 2,3-O-изопропилиден 5-O-метансульфонил D Рибоно-1,4-лактон (26a) использовали на следующей стадии без какой-либо дополнительной очистки; 1H ЯМР (CDCl3) δ 4,8 (м, 3H), 4,5 (м, 2H), 3,08 (с, 3H), 1,5 (с, 3H), 1,4 (с, 3H).
Стадия 4: Получение 2,3-O-изопропилиден L Ликсоно-1,4-лактона (26b)
К 2,3-O-изопропилиден 5-O-метансульфонил D Рибоно-1,4-лактону (26a) (3,04 кг, 11,37 моль) добавляли воду (10 л), затем медленно добавляли твердый KOH (1,83 кг, 32,77 моль). (Внимание: Соединение переходит в раствор по мере добавления твердого KOH. Реакция является экзотермической, когда добавляется KOH, поэтому реакционный сосуд необходимо поместить на баню со льдом). После завершения добавления KOH температура реакции повышалась до 45°C. Реакционную смесь перемешивали при ~ комнатной температуре (комн. темп.) в течение 3 ч. Раствор вновь охлаждали на ледяной бане и затем подкисляли до pH=3 (точно) с использованием конц. раствора HCl. Реакционную смесь упаривали с получением твердого остатка коричневого цвета. Остаток перемешивали два раза с кипящим ацетоном (~5 л) в течение 1 ч, и органические продукты декантировали. Оставшиеся соли затем растворяли в минимальном количестве воды и pH доводили до 3 с использованием конц. HCl (~200 мл). Водный раствор концентрировали, и твердый остаток экстрагировали ацетоном (~5 л). Органический слой сушили, фильтровали и упаривали с получением белых игл 2,3-O-изопропилиден L Ликсоно-1,4-лактона (26b). Кристаллизация может быть проведена в горячем ацетоне. Выход: 1,60 кг (75%); 1H ЯМР (D2O) δ 5,00 (м, 2H), 3,8 (м, 3H), 1,5 (с, 3H), 1,4 (с, 3H).
Стадия 5: Получение 2,3-O-изопропилиден 5-O-третбутилдиметилсилил L Ликсоно-1,4-лактона (26c)
В 22-литровую 3-горлую колбу, снабженную механической мешалкой, помещали 2,3-O-изопропилиден L Ликсоно-1,4-лактон (26b) (2,0 кг, 10,63 моль), DMAP (~25 г), имидазол (1,60 кг, 23,40 моль, 2,2 эквив.) и перемешивали в ДМФ со степенью чистоты, соответствующей стандарту ACS (8 л), в течение 1 ч. Температуры реакции понижали до температуры 0°C с использованием ледяной бани. В реакционную смесь медленно добавляли TBDMSCl (2,08 кг, 13,81 моль, 1,3 эквив.) в течение периода времени в 2 ч. Реакционную смесь перемешивали при комнатной температуре в инертной атмосфере в течение 14 ч. После завершения реакции, согласно данным анализа ТСХ (7:3, EtOAc:гексан, Rf=0,80), реакционную смесь выливали в ледяную воду и экстрагировали с помощью EtOAc (~2). Органический слой отделяли, сушили и фильтровали с получением маслянистого остатка. Реакционный сосуд, содержащий продукт, помещали на ледяную баню, затем добавляли гексан (~3 л). Соединение хорошо кристаллизовалось в гексане. Фильтрование кристаллов и промывка кристаллов минимальным количеством гексана и помещение продукта в вакуумную печь при температуре 40°C в течение ночи с получением 2,3-O-изопропилиден 5-O-третбутилдиметилсилил L Ликсоно-1,4-лактона (26c) 3,01 кг (93%); 1H ЯМР (CDCl3) δ 4,8 (с, 2H), 4,5 (м, 1H), 3,9 (м, 2H), 1,5 (с, 3H), 1,4 (с, 3H), 0,9 (с, 9H), 0,0 (с, 6H).
Стадия 6: Получение 2-(трет-бутилдиметилсиланокси)-1-(5-гидроксиметил-2,2-диметил-[1,3]диоксолано-4-ил)-этанола (26d)
Раствор 2,3-O-изопропилиден 5-O-третбутилдиметилсилил L Ликсоно-1,4-лактона (26c) (3,00 кг, 9,93 моль) в ТГФ:MeOH (9:1 об./об. смесь, 15 л) перемешивали при комнатной температуре в течение 0,5 ч до видимого завершения растворения. Внутреннюю температуру реакционного сосуда понижали до температуры -5°C. Небольшими порциями добавляли боргидрид натрия (751 г, 19,86 моль, 2 экв.) таким образом, чтобы температура не превышала 15-17°C. Добавление реагента завершали в течение периода времени в 1 ч. Реакционной смеси давали достичь комнатной температуры в течение периода времени в 3 ч и затем продолжали перемешивание при данной температуре в течение 18 ч. Реакционную смесь отслеживали с помощью ТСХ (3:7, этилацетат:гексан, Rf=0,15). После завершения реакции раствор разбавляли EtOAc (5 л) и промывали 1н раствором HCl (2 раза). Органический слой промывали водой, сушили и упаривали с получением маслянистого остатка. К данной смеси добавляли ~3 л гексана и охлаждали перегонную колбу на ледяной бане. Кристаллы выпадали из раствора. Фильтровали кристаллы и промывали с помощью ~250 мл гексана. Сушили в вакуумной печи при температуре 40°C в течение 24 ч с получением 2-(трет-бутилдиметилсиланокси)-1-(5-гидроксиметил-2,2-диметил-[1,3]диоксолано-4-ил)-этанола (26d). Выход: 2,32 кг (77%); 1H ЯМР (CDCl3) δ 4,2 (м, 2H), 3,7 (м, 5H), 1,5 (с, 3H), 1,4 (с, 3H), 0,9 (с, 9H), 0,0 (с, 6H).
Стадия 7: Получение 2-(третбутилдиметилсилилокси)-1-1(5-метансульфонилоксиметил-2,2-диметил-[1,3]диоксолан-4-ил)-этиловый эфир метансульфоновой кислоты (26e)
В 500 мл 3-горлую колбу помещали сухой пиридин (20 мл), каталитическое количество DMAP, затем добавляли метан сульфонил хлорид (4,98 мл, 64,4 ммоль, 4,0 экв.) при температуре 0°C. В реакционный сосуд медленно добавляли 2-(трет-бутилдиметилсиланокси)-1-(5-гидроксиметил-2,2-диметил-[1,3]диоксолано-4-ил)-этанол (26d) (5,0 г, 16,3 ммоль), растворенный в сухом пиридине (20 мл). Реакцию перемешивали в инертной атмосфере в течение 4 ч при данной температуре. (ТСХ; 1:9 этилацетат:гексан, Rf=0,85). После завершения реакции добавляли 1 мл воды и 100 мл EtOAc и перемешивали. Экстрагировали органический слой водой, сушили и упаривали с получением сиропа 2-(трет-бутилдиметилсилилокси)-1-1(5-метансульфонилоксиметил-2,2-диметил-[1,3]диоксолан-4-ил)-этилового эфира метансульфоновой кислота (26e). Выход: 8,7 г (90%). Сырой продукт использовали на следующей стадии без какой-либо дополнительной очистки.
Стадия 8: Получение 5-O-третбутилдиметилсилил-1,4-N-бензилимино-2,3-O-изопропилиден-D-рибитола (26f)
К 2-(третбутилдиметилсилилокси)-1-1(5-метансульфонилоксиметил-2,2-диметил-[1,3]диоксолан-4-ил)-этиловому эфиру метансульфоновой кислоты (26e) (8,6 г) добавляли неразбавленный бензиламин (10 мл), и реакционную смесь нагревали при температуре 70°C в течение 48 ч. Данные ТСХ (4:1 гексан:EtOAc, Rf=0,68) показывали, что реакция завершалась. Реакционную смесь охлаждали, и в реакционную смесь добавляли насыщенный солевой раствор. Реакционную смесь экстрагировали дихлорметаном, промывали водой, сушили и упаривали с получением сиропа, который содержал большое количество аминового реагента. Остаток обрабатывали толуолом и к нему добавляли кусочки сухого льда для высаживания соли. Твердое вещество фильтровали, и фильтрат упаривали с получением желаемого продукта 5-O-третбутилдиметилсилил-1,4-N-бензилимино-2,3-O-изопропилиден-D-рибитола (26f) (5,6 г, 92%). Данный продукт использовали напрямую на следующей стадии без какой-либо дополнительной очистки. 1H ЯМР (CDCl3) δ 7,2-7,4 (м, 5H), 4,65 (м, 1H), 4,55 (дд, 1H), 4,0 (д, 1H), 3,6-3,8 (м, 3H), 3,1 (дд, 1H), 3,0 (м, 1H), 2,75 (дд, 1H), 1,5 (с, 3H), 1,34 (с, 3H), 0,9 (с, 9H), 0,0 (с, 6H).
Стадия 9: Получение 5-O-третбутилдиметилсилил-1,4-имино-2,3-O-изопропилиден-D-рибитола (20a)
К 5-O-третбутилдиметилсилил-1,4-N-бензилимино-2,3-O-изопропилиден-D-рибитолу (26f) (5,93 г, 15,74 ммоль) в EtOH (15 мл) добавляли Pd/C (50 мг), и реакционную смесь гидрировали при давлении 80 фунтов на квадратный дюйм в течение 5 ч или до тех пор, пока анализ ТСХ (3:2, гексан:EtOAc, Rf=0,18) не показывал завершение реакции. Реакционную смесь фильтровали через слой из целита, и слой Целита промывали EtOH (25 мл). Фильтрат пропускали через фильтр Миллипор (0,25 мкм) для удаления следов катализатора и упаривали с получением 5-O-третбутилдиметилсилил-1,4-имино-2,3-O-изопропилиден-D-рибитола (20a) в виде сиропа. Выход: 3,5 г (75%-стадии); 1H ЯМР (CDCl3) δ 4,65 (м, 2H), 3,60 (дд, 2H), 3,24 (т, 1H), 3,00 (д, 2H), 1,5 (с, 3H), 1,34 (с, 3H), 0,9 (с, 9H), 0,0 (с, 6H).
Стадия 10: Получение (3aR,4R,6aS)-4-(((трет-бутилдиметилсилил)окси)метил)-2,2-диметил-4,6a-дигидро-3aH-[1,3]диоксоло[4,5-c]пиррола (20b)
Раствор 5-O-третбутилдиметилсилил-1,4-имино-2,3-O-изопропилиден-D-рибитола (20a) (94 г, 327 ммоль) в толуоле (470 мл) добавляли к суспензии N-хлорсукцинимида (54,6 г, 408,8 ммоль) в толуоле (470 мл) при температуре от 17 до 23°C в течение периода времени от 60 до 90 минут. Реакционную смесь перемешивали при температуре от 17 до 23°C в течение 1 часа, охлаждали до температуры от -3 до 3°C и перемешивали в течение дополнительного часа. Побочный сукцинимидный продукт удаляли путем фильтрации, и отфильтрованный раствор сразу помещали в 60%-ый раствор гидроксида калия (458 г, 8175 ммоль в 305 мл воды), содержащий бромид тетрабутиламмония (10,53 г, 32,7 ммоль). Реакционную смесь перемешивали при температуре от -5 до 5°C в течение 17 ч. Затем в двухфазную смесь добавляли воду (700 мл) для растворения неорганических осадков, и раствор продукта в толуоле промывали аммонийацетатным буфером (pH ~4,5), буферированным насыщенным солевым раствором (700 мл) и стабилизировали триэтиламином, затем сушили циркуляцией над сульфатом магния в реакторе. Высушенный раствор, содержащий (3aR,4R,6aS)-4-(((трет-бутилдиметилсилил)окси)метил)-2,2-диметил-4,6a-дигидро-3aH-[1,3]диоксоло[4,5-c]пиррол (20b) в толуоле, использовали непосредственно на следующей стадии.
Стадия 11: Получение 1S-5-O-трет-бутилдиметилсилил-1,4-дидеокси-1-C-[(4-метоксипирроло[3,2-d]пиримидин-9-N-(бензилоксометил)-7-ил)]-1,4-имино-2,3-O-изопропилидин-D-рибитола (26g)
6-Метокси-N-(бензилоксиметил)-9-деазагипоксантин (27f) (271,0 г, 0,775 моль) в атмосфере N2 добавляли в 22 л 3-горлую круглодонную колбу, содержащую безводный анизол (1,7 л). Данную смесь аккуратно нагревали до тех пор, пока смесь не становилась гомогенной (≈45°C). Смесь охлаждали до температуры окружающей среды и добавляли безводный эфир (2,9 л). Реакционную колбу помещали на охлаждающую баню и охлаждали до температуры -70°C с использованием бани сухой лед/ацетон. При температуре ≈-20°C бромид начинал осаждаться в виде мелкого белого твердого вещества. К суспензии добавляли н-BuLi (1,6н, 486 мл, 0,778 моль) в течение 1,2 ч с помощью капельной воронки таким образом, чтобы внутренняя температура поддерживалась <-50°C. После последнего добавления анализ ТСХ (30% EtOAc/гексан) показывал наличие <2% остаточного бромида. С помощью капельной воронки в течение 15 минут добавляли (3aR,4R,6aS)-4-(((трет-бутилдиметилсилил)окси)метил)-2,2-диметил-4,6a-дигидро-3aH-[1,3]диоксоло[4,5-c]пиррол (20b) (183 г, 0,642 моль в толуоле, поддерживая внутреннюю температуру ниже -50°C. Реакционная смесь была бледно-янтарного цвета. Реакционную колбу отставляли с охлаждающей бани и давали нагреться. Реакционной смеси давали нагреться до температуры -2°C, и анализ ТСХ (40% EtOAc/гексан, визуализированный с помощью реагента Эрлиха) показывал отсутствие (3aR,4R,6aS)-4-(((трет-бутилдиметилсилил)окси)метил)-2,2-диметил-4,6a-дигидро-3aH-[1,3]диоксоло[4,5-c]пиррола (20b). Реакцию гасили H2O (2 л) и экстрагировали с помощью эфира (2 × 2 л). Объединенные органические слои сушили (MgSO4) и концентрировали в вакууме (для удаления анизола был использован высокий вакуум при температуре 60°C) с получением сырого масла твердого цвета 1S-5-O-трет-бутилдиметилсилил-1,4-дидеокси-1-C-[(4-метоксипирроло[3,2-d]пиримидин-9-N-(бензилоксометил)-7-ил)]-1,4-имино-2,3-O-изопропилидин-D-рибитола (26g), которое было подходящим для использования на следующей стадии. Выход 284 г (79%). Небольшое количество (5 г) смеси сырого продукта очищали путем флеш-хроматографии (силикагель, элюируя смесью 0-40% этилацетат в гексане) с получением 1S-5-O-трет-бутилдиметилсилил-1,4-дидеокси-1-C-[(4-метоксипирроло[3,2-d]пиримидин-9-N-(бензилоксометил)-7-ил)]-1,4-имино-2,3-O-изопропилидин-D-рибитола (26g) в виде сиропа оранжевого цвета (3,4 г); 1H ЯМР (ДМСО-d6) δ 0,02 (с, 3H), 0,03 (с, 3H), 0,8 (с, 9H), 1,25 (с, 3H), 1,48 (с, 3H), 3,11-3,20 (м, 1H), 3,60-3,71 (м, 2H), 4,05 (с, 3H), 4,26 (д, 1H, J=4,7 Гц), 4,49 (с, 2H), 4,52-4,56 (м, 1H), 4,81-4,85 (м, 1H), 5,71 (с, 2H), 7,21-7,32 (м, 5H), 7,80 (с, 1H), 8,40 (с, 1H); 13C ЯМР (CDCl3) δ -5,46, -5,43, 18,30, 25,53, 25,88, 27,63, 53,43, 61,59, 62,54, 66,14, 70,14, 76,93, 82,32, 86,40, 114,43, 116,22, 116,56, 127,67, 127,93, 128,43, 130,55, 136,93, 149,61, 149,82, 156,16; ИК 3420, 1610 см-1; MS (ES+) m/z 555,3; Анализ: Вычислено для C29H42N4O5Si: C, 62,79; H, 7,63; N, 10,10; Найдено: C, 62,95; H, 7,59; N, 9,95.
Стадия 12: Получение 1S-N-трет-бутоксикарбонил-5-O-трет-бутилдиметилсилил-1,4-дидеокси-1-C-[(4-метоксипирроло[3,2-d]пиримидин-9-N-(бензилоксометил)-7-ил)]-1,4-имино-2,3-O-изопропилидин-D-рибитола (26h)
Сырой 1S-5-O-трет-бутилдиметилсилил-1,4-дидеокси-1-C-[(4-метоксипирроло[3,2-d]пиримидин-9-N-(бензилоксометил)-7-ил)]-1,4-имино-2,3-O-изопропилидин-D-рибитол (26g) (275 г, 0,496 моль обрабатывали CH2Cl2 (1,4 л) и охлаждали до температуры 5°C на бане лед/вода. К этой охлажденной смеси добавляли Boc2O (168,5 г, 0,772 моль 4 порциями таким образом, чтобы температура реакционной смеси поддерживалась <10°C. Через 30 мин анализ ТСХ (40% этилацетат/гексан) показывал отсутствие оставшегося исходного материала. Сырую смесь абсорбировали на SiO2 (700 г) и очищали с помощью флеш-хроматографии (силикагель 1,5 кг, элюируя смесью 10% этилацетат в гексане). Соответствующие фракции объединяли и концентрировали в вакууме с получением 1S-N-трет-бутоксикарбонил-5-O-трет-бутилдиметилсилил-1,4-дидеокси-1-C-[(4-метоксипирроло[3,2-d]пиримидин-9-N-(бензилоксометил)-7-ил)]-1,4-имино-2,3-O-изопропилидин-D-рибитола (26h) (272 г, 84%) в виде сиропа желтого цвета; 1H ЯМР (CDCl3) δ 0,02 (с, 3H), 0,03 (с, 3H), 0,82 (с, 9H), 1,31-1,58 (м, 15H) 2,05-2,09 (м, 1H); 3,58-3,80 (м, 2H), 4,08 (с, 3H), 4,17-4,32 (м, 1H), 4,44 (с, 2H), 4,84-5,71 (м, 4H), 7,19-7,33 (м, 5H), 7,46 (с, 1H), 8,51 (с, 1H); 13C ЯМР (CDCl3) δ -5,31, -5,20, 14,10, 14,20, 18,32, 21,01, 22,64, 25,56, 25,93, 27,46, 28,46, 31,58, 53,44, 60,34, 62,48, 70,08, 76,96, 79,84, 111,69, 115,89, 127,67, 127,93, 128,43, 136,90, 148,62, 149,90, 154,38, 156,19; ИК 1692, 1608 см-1; MS (ES+) m/z 655,3; Анализ: Вычислено для C34H50N4O7Si: C, 62,43; H, 7,65; N, 8,56; Найдено: C, 62,79; H, 7,89; N, 8,47.
Стадия 13: Получение (3aR,4R,6S,6aS)-трет-бутил 4-(((трет-бутилдиметилсилил)окси)метил)-6-(4-метокси-5H-пирроло[3,2-d]пиримидин-7-ил)-2,2-диметилдигидро-3aH-[1,3]диоксоло[4,5-c]пиррол-5(4H)-карбоксилата (26i)
Гидроксид палладия на угле (120 г, 50% влажности) помещали в 2 л-овую коническую колбу. Метанол (7,60 кг) отвешивали в 20 л барабан с полиэтиленовым вкладышем. 0,80 кг данного метанола использовали для перенесения в коническую колбу, содержащую катализатор гидроксид палладия и коническую колбу взбалтывали до образования гомогенной смеси. Эту суспензию затем выливали в 20-литровый сосуд для гидрирования, который был продут азотом. Оставшийся гидроксид палладия на угле смывали из конической колбы в сосуд для гидрирования метанолом (25 мл). 1S-N-трет-бутоксикарбонил-5-O-трет-бутилдиметилсилил-1,4-дидеокси-1-C-[(4-метоксипирроло[3,2-d]пиримидин-9-N-(бензилоксометил)-7-ил)]-1,4-имино-2,3-O-изопропилидин-D-рибитол (26h) (380 г) помещали в 10-литровый барабан с полиэтиленовым вкладышем, затем 1,32 кг метанола из 20-литрового барабана с полиэтиленовым вкладышем. Указанный метанольный раствор 1S-N-трет-бутоксикарбонил-5-O-трет-бутилдиметилсилил-1,4-дидеокси-1-C-[(4-метоксипирроло[3,2-d]пиримидин-9-N-(бензилоксометил)-7-ил)]-1,4-имино-2,3-O-изопропилидин-D-рибитола (26h) помещали в 20-литровый сосуд для гидрирования. Остаток метанола в 20-литровом барабане с полиэтиленовым вкладышем помещали в сосуд, посредством 10-литрового барабана с полиэтиленовым вкладышем, путем споласкивания. Раствор аммиака в метаноле (7,0 M, 0,68 кг) отмеряли в 10-литровый барабан с полиэтиленовым вкладышем и переносили в сосуд для гидрирования. В сосуд подавали газообразный водород под давлением 5 бар, и содержимое нагревали при температуре 35°C при перемешивании. Эти условия реакции поддерживали в течение 20 ч, при добавлении водорода по мере необходимости. По прошествии этого времени анализ ВЭЖХ показывал, что оставалось приблизительно 2% исходного материала, что позволяло предположить, что реакция по существу завершилась. Содержимое сосуда переносили в 20-литровый барабан с полиэтиленовым вкладышем, затем фильтровали через слой из целита. При осуществлении этого процесса проводили продувку фильтровальной воронки азотом, и для промывки слоя на фильтре использовали метанол (1,50 кг). Фильтрат и промывки переносили в роторный испаритель и концентрировали при пониженном давлении до массы в 0,48 кг. В колбу роторного испарителя добавляли метанол (2,50 кг), и раствор концентрировали до постоянной массы (0,340 кг, приблизительно количественный выход). Добавляли дополнительное количество метанола (1,0 кг) к продукту (3aR,4R,6S,6aS)-трет-бутил 4-(((трет-бутилдиметилсилил)окси)метил)-6-(4-метокси-5H-пирроло[3,2-d]пиримидин-7-ил)-2,2-диметилдигидро-3aH-[1,3]диоксоло[4,5-c]пиррол-5(4H)-карбоксилату (26i), чтобы приготовить раствор для использования на следующей стадии.
Стадия 14: Получение гидрохлорида 7-((2S,3S,4R,5R)-3,4-дигидрокси-5-(гидроксиметил)пирролидин-2-ил)-3H-пирроло[3,2-d]пиримидин-4(5H)-она (26j)
Раствор (3aR,4R,6S,6aS)-трет-бутил 4-(((трет-бутилдиметилсилил)окси)метил)-6-(4-метокси-5H-пирроло[3,2-d]пиримидин-7-ил)-2,2-диметилдигидро-3aH-[1,3]диоксоло[4,5-c]пиррол-5(4H)-карбоксилата (26i) разбавляли метанолом до достижения общего объема 2,5 л и помещали в 5-литровую многогорлую круглодонную колбу, снабженную механической мешалкой, обратным холодильником и внутренним термометром. Раствор нагревали с использованием масляной бани и одновременно в течение 40 минут добавляли концентрированную соляную кислоту (37%, 2,18 л или 2,62 кг) (в течение этого времени внутренняя температура повышалась от 43°C до 58°C). Нагревание продолжали в течение еще 6 ч с поднятием внутренней температуры до 68°C, в этой точке раствору давали охладиться до комнатной температуры и перемешивали в течение последующих 15 ч. Раствор коричневого цвета концентрировали на роторном испарителе до объема 1,5-2,0 л, затем добавляли воду (0,5 л). Суспензию переносили обратно в 5 л колбу и нагревали для повторного растворения твердых веществ. После добавления дополнительного количества воды (0,50 л) нагревание доводили до 50°C. Добавляли активированный уголь (95 г), и суспензию перемешивали при температуре 50°C в течение 1 ч. Активированный уголь удаляли путем фильтрации через слой из целита, промывая водой (приблизительно 1,0 л). Фильтрат и промывки, теперь частично обесцвеченные, концентрировали на роторном испарителе до объема 0,95 л. Раствор комнатной температуры переносили в 10 л-овую колбу и охлаждали на ледяной бане при перемешивании. В раствор по частям добавляли этанол (7,90 л), вызывая кристаллизацию продукта. При перемешивании еще через последующие 2 ч перемешивания внутреннюю температуру понижали до 5°C. Твердый продукт собирали путем фильтрации в атмосфере азота и промывали предварительно охлажденным этанолом (3 × 250 мл). Продукт собирали без промывки на фильтровальной воронке в течение 30 минут, затем переносили в лоток для сушки. Продукт сушили в печи при температуре 70°C в течение ночи с получением гидрохлорида 7-((2S,3S,4R,5R)-3,4-дигидрокси-5-(гидроксиметил)пирролидин-2-ил)-3H-пирроло[3,2-d]пиримидин-4(5H)-она (26j) в виде твердого вещества не совсем белого цвета (101,2 г, 58%). Гидрохлорид 7-((2S,3S,4R,5R)-3,4-дигидрокси-5-(гидроксиметил)пирролидин-2-ил)-3H-пирроло[3,2-d]пиримидин-4(5H)-она (26j) (101,2 г) помещали в 20-литровый сосуд с рубашкой. Добавляли воду (1,52 л), и суспензию перемешивали до растворения твердых веществ. Добавляли концентрированную соляную кислоту (37%, 63,6 мл), и раствор перемешивали при температуре 25°C. После гомогенизации раствор переносили в барабан с полиэтиленовым вкладышем, и сосуд промывали чистой водой (506 мл). В качестве стадии очистки раствор гидрохлорида 7-((2S,3S,4R,5R)-3,4-дигидрокси-5-(гидроксиметил)пирролидин-2-ил)-3H-пирроло[3,2-d]пиримидин-4(5H)-она (26j) фильтровали через фильтровальную бумагу на полипропиленовой фильтровальной воронке и затем опять возвращали в сосуд. Промывки опять фильтровали таким же образом, затем возвращали обратно в сосуд. Раствор перемешивали при температуре приблизительно 15°C в течение 45 минут. К перемешиваемому раствору в течение 15 минут добавляли этанол (1,0 л). Для вызывания кристаллизации добавляли затравочные кристаллы (2,0 г) гидрохлорида 7-((2S,3S,4R,5R)-3,4-дигидрокси-5-(гидроксиметил)пирролидин-2-ил)-3H-пирроло[3,2-d]пиримидин-4(5H)-она (26j). Через 70 минут добавляли этанол (1,0 л), и суспензию перемешивали при температуре 15°C в течение еще 19,5 ч. К суспензии добавляли дополнительное количество этанола (8,0 л), и перемешивание при температуре 15°C продолжали в течение еще 5 ч. Температуру рубашки устанавливали равной 0°C, и перемешивание продолжали в течение дополнительных 2 ч. После этого суспензию перемещали в барабан с полиэтиленовым вкладышем и фильтровали через фильтровальную бумагу на полипропиленовой фильтровальной воронке. Слой на фильтре промывали охлажденным этанолом (1,0 л затем 0,5 л) и собирали без промывки на фильтровальной воронке в течение 30 минут. Твердое вещество затем переносили на лоток для сушки и сушили в печи при температуре 70°C в течение ночи с получением гидрохлорида 7-((2S,3S,4R,5R)-3,4-дигидрокси-5-(гидроксиметил)пирролидин-2-ил)-3H-пирроло[3,2-d]пиримидин-4(5H)-она (26j) в виде твердого вещества не совсем белого цвета (176,9 г, 87%-ый выход).
Стадия 15: Получение (2R,3R,4S,5S)-трет-бутил 3,4-дигидрокси-2-(гидроксиметил)-5-(4-оксо-4,5-дигидро-3H-пирроло[3,2-d]пиримидин-7-ил)пирролидин-1-карбоксилата (28a)
К суспензии 7-((2S,3S,4R,5R)-3,4-дигидрокси-5-(гидроксиметил)пирролидин-2-ил)-3H-пирроло[3,2-d]пиримидин-4(5H)-она (26j) (446,19 г, 1,47 моль) в смеси вода:метанол (1:1, 10,4 л) добавляли триэтиламин (621 мл, 4,42 моль, 3,0 экв.) при комнатной температуре, затем (Boc)2O (987 г, 4,53 моль, 3,1 экв.). Реакционная смесь становилась прозрачным окрашенным раствором после добавления (Boc)2O с легким повышением внутренней температуры от 28°C до 33°C. Раствор начал проявлять некоторое замутнение после 1 часа перемешивания. Раствор перемешивали при комнатной температуре в течение ночи. Твердый продукт собирали путем фильтрации и промывали водой (5,0 л), сушили в высоком вакууме при температуре 50°C с получением (2R,3R,4S,5S)-трет-бутил 3,4-дигидрокси-2-(гидроксиметил)-5-(4-оксо-4,5-дигидро-3H-пирроло[3,2-d]пиримидин-7-ил)пирролидин-1-карбоксилата (28a) (482 г, 89%) в виде твердого вещества не совсем белого цвета; 1H ЯМР (300 МГц, ДМСО-d6) δ 11,92 (с, 2H), 7,81 (с, 1H), 7,32 (д, J=22,7 Гц, 1H), 5,73-5,20 (м, 1H), 5,05-4,91 (м, 1H), 4,87-4,76 (м, 1H), 4,74-4,49 (м, 1H), 4,33-4,17 (м, 1H), 4,09-3,86 (м, 2H), 3,64-3,48 (м, 2H), 1,39-1,00 (м, 9H); MS (ES+) 755,1 (2M+Na), (ES-) 731,7 (2M-1); Анализ: Вычислено для C16H22N4O6: C, 52,45; H, 6,05; N, 15,29; Найдено: C, 52,24; H, 6,02; N, 15,05.
Стадия 16: Получение (2R,3R,4S,5S)-2-(ацетоксиметил)-1-(трет-бутоксикарбонил)-5-(4-оксо-4,5-дигидро-3H-пирроло[3,2-d]пиримидин-7-ил)пирролидин-3,4-диил диацетата (28b)
К суспензии (2R,3R,4S,5S)-трет-бутил 3,4-дигидрокси-2-(гидроксиметил)-5-(4-оксо-4,5-дигидро-3H-пирроло[3,2-d]пиримидин-7-ил)пирролидин-1-карбоксилата (28a) (482 г, 1,32 моль, 1,0 эквив.) в пиридине (740 мл, 9,21 моль, 7 эквив.) при комнатной температуре добавляли DMAP (3,22 г, 26,32 ммоль, 0,02 эквив.) и уксусный ангидрид (435 мл, 4,61 ммоль, 3,5 экв.). Внутренняя температура начинала повышаться по мере добавления уксусного ангидрида, поэтому требовалось охлаждение баней лед-вода. После полного добавления ангидрида температура повышалась до 67°C, затем понижалась до комнатной температуры. Баню лед-вода удаляли после достижения температуры реакционной смеси до 25°C. Суспензия не давала прозрачный раствор, но просветление суспензии наблюдалось. Реакционную смесь перемешивали при комнатной температуре в течение 14 ч с получением непрозрачного раствора. Обработанная аликвота показывала, что исходного материала больше не оставалось, и в анализе ТСХ имелось только два основных пятна (9:1 хлороформ:метанол), МС показывал два основных пика при (493,0, M+1) для продукта и тетраацетилированного продукта (M+1=535). Реакционную смесь разбавляли 3,0 л хлороформа, перемешивали в течение 10 минут, затем добавляли 2,0 л деионизированной воды. На поверхности раздела фаз вода-органика образовывался восковой белый неизвестный продукт. Этот неизвестный продукт после проведения разделения фаз оставался в водной фазе. Органическую фазу отделяли и промывали снова 2,0 л воды. Объединенные водные слои снова экстрагировали с помощью 1,0 л хлороформа. Объединенные органические фазы промывали водным 2,0н раствором HCl (2 × 1,0 л), водой (2 × 1,0 л), насыщенным бикарбонатом натрия (2 × 1,0 л) и насыщенным солевым раствором (2 × 1,0 л). Органический слой сушили над MgSO4, фильтровали и концентрировали досуха в вакууме и на водяной бане с температурой 50-55°C. Вакуум переключали на высокий вакуум масляного насоса до прекращения видимой перегонки с получением плотного сиропообразного продукта. Круглодонную колбу оставляли в высоком вакууме масляного насоса в течение 14 ч, чтобы свести к минимуму остатки пиридина. Была получена комбинация твердой пены, которая превращалась в хороший белый твердый и плотный остаток (2R,3R,4S,5S)-2-(ацетоксиметил)-1-(трет-бутоксикарбонил)-5-(4-оксо-4,5-дигидро-3H-пирроло[3,2-d]пиримидин-7-ил)пирролидин-3,4-диил диацетата (28b) (715, 110% выход). Этот процент отражал количество тетраацетилированного соединения. Продукт был достаточно чистым, чтобы использоваться на следующей стадии. Аналитический образец получали путем очистки смеси с помощью колоночной флэш-хроматографией (силикагель, элюируя смесью 0-100% (9:1) этилацетат/метанол в гексане) с получением (2R,3R,4S,5S)-2-(ацетоксиметил)-1-(трет-бутоксикарбонил)-5-(4-оксо-4,5-дигидро-3H-пирроло[3,2-d]пиримидин-7-ил)пирролидин-3,4-диил диацетата (28b) в виде твердого вещества белого цвета; 1H ЯМР (300 МГц, ДМСО-d6) δ 12,13 (с, 1H, D2O способный к обмену протонами), 11,98 (с, 1H, D2O способный к обмену), 7,82 (с, 1H), 7,29 (с, 1H), 5,76 (с, 1H), 5,37 (т, J=4,5 Гц, 1H), 4,99 (с, 1H), 4,55 (дд, J=11,3, 6,6 Гц, 1H), 4,34 (д, J=8,3 Гц, 1H), 4,03 (кв., J=7,1 Гц, 1H), 2,01 (д, J=12,6 Гц, 9H), 1,23 (дд, J=39,9, 32,8 Гц, 9H); MS (ES+) 493,0 (M+1); (ES-) 526,7 (M+Cl); Анализ: Вычислено для C22H28N4O9: C, 53,65; H, 5,73; N, 11,38; Найдено: C, 53,18; H, 5,89; N, 11,10.
Стадия 17: Получение (2R,3R,4S,5S)-2-(ацетоксиметил)-1-(трет-бутоксикарбонил)-5-(4-хлор-5H-пирроло[3,2-d]пиримидин-7-ил)пирролидин-3,4-диил диацетата (28c)
К раствору (2R,3R,4S,5S)-2-(ацетоксиметил)-1-(трет-бутоксикарбонил)-5-(4-оксо-4,5-дигидро-3H-пирроло[3,2-d]пиримидин-7-ил)пирролидин-3,4-диил диацетата (28b) (622 г, 1,26 моль, 1,0 экв.) в ацетонитриле (2,75 л) при комнатной температуре добавляли бензилтриэтиламмоний хлорид (575 г, 2,5 моль, 2,0 экв.), диметиланилин (240 мл, 1,9 моль, 1,5 экв.), затем POCl3 (706 мл, 7,58 моль, 6,0 экв.). Был получен прозрачный раствор светло-желтого цвета. Реакционную смесь медленно нагревали до температуры 80°C и выдерживали при данной температура в течение 10 минут. Анализ ТСХ в 9:1 хлороформ:метанол показывал, что реакция завершалась на >98%. Темный гомогенный раствор охлаждали до температуры 50,0°C и концентрировали в вакууме (водяная баня 70-73°C) для удаления POCl3; остаток помещали в высокий вакуум масляного насоса до тех пор, пока отгонки больше не наблюдалось. Остаток растворяли в 3,0 л хлороформа и быстро осторожно промывали водным насыщенным бикарбонатом натрия до достижения нейтрального pH. Органический слой отделяли, промывали водой (2 л), насыщенным солевым раствором (2 л), сушили над MgSO4, фильтровали и концентрировали в вакууме досуха (водяная баня с температурой 50-53°C). Темный продукт (2R,3R,4S,5S)-2-(ацетоксиметил)-1-(трет-бутоксикарбонил)-5-(4-хлор-5H-пирроло[3,2-d]пиримидин-7-ил)пирролидин-3,4-диил диацетата (28c) использовали непосредственно на следующей стадии без очистки. Аналитический образец получали с помощью очистки 0,5 г, используя флеш-хроматографию на колонке (силикагель 12 г, элюируя смесью от 0 до 50% этилацетат/метанол (9:1) в гексане), полученный соответствующий продукт растворяли в смеси эфир/гексан, оставляли в течение ночи, образовавшиеся кристаллы (301 мг) собирали путем фильтрации с получением (2R,3R,4S,5S)-2-(ацетоксиметил)-1-(трет-бутоксикарбонил)-5-(4-хлор-5H-пирроло[3,2-d]пиримидин-7-ил)пирролидин-3,4-диил диацетата (28c) в виде твердого вещества белого цвета; 1H ЯМР (300 МГц, ДМСО-d6) δ 12,55 (с, 1H, D2O способный к обмену), 8,65 (с, 1H), 7,87 (ушир. с, 1H), 5,79 (ушир. с, 1H), 5,44 (т, J=4,0 Гц, 1H), 5,10 (ушир. с, 1H), 4,56 (дд, J=11,5, 6,8 Гц, 1H), 4,38 (дд, J=11,4, 4,1 Гц, 1H), 4,08 (ушир. с, 1H), 2,07 (с, 3H), 2,00 (с, 6H), 1,38 (с, 4H), 1,13 (с, 5H); MS (ES+) 510,865 (M+1), (ES-) 508,717 (M-1); Анализ: Вычислено для C22H27ClN4O8: C, 51,72; H, 5,33; Cl, 6,94; N, 10,97; Найдено: C, 51,91; H, 5,32; Cl, 6,76; N, 10,90.
Стадия 18: Получение (2R,3R,4S,5S)-2-(ацетоксиметил)-5-(4-азидо-5H-пирроло[3,2-d]пиримидин-7-ил)-1-(трет-бутоксикарбонил)пирролидин-3,4-диил диацетата (28d)
К раствору (2R,3R,4S,5S)-2-(ацетоксиметил)-1-(трет-бутоксикарбонил)-5-(4-хлор-5H-пирроло[3,2-d]пиримидин-7-ил)пирролидин-3,4-диил диацетата (28c) (622 г, 1,26 моль, 1 экв.) в ДМФ (1,5 л) добавляли азид натрия (411 г, 6,32 моль, 5 эквив.) и нагревали при перемешивании при температуре 60°C в течение 10 ч, за это время реакцию завершалась (ТСХ в смеси 9:1 хлороформ:метанол и в смеси 1:1 гексан:этилацетат). Реакцию охлаждали до температуры 25°C, выливали на лед (2 л) и экстрагировали с помощью хлороформа (2 × 1 л). Объединенные хлороформные слои промывали водой (2 × 2 л), насыщенным солевым раствором (2 л), сушили, фильтровали и концентрировали в вакууме (водяная баня 70-80°C) с получением черного шлама. Очистку этого шлама осуществляли путем колоночной хроматографии (987 г черного шлама, 8 × 30 дюймовая колонка, ½ наполненная силикагелем, профиль элюирования гексан:этилацетат; 9:1 (40,0 л); 7:3 (20,0 л); 6:4 (20,0 л); 1:1 (20 л); 4:6 (20,0 л) и 2:8 (20,0 л). Соответствующие фракции объединяли и концентрировали в вакууме (водяная баня 50,0°C) с получением (2R,3R,4S,5S)-2-(ацетоксиметил)-5-(4-азидо-5H-пирроло[3,2-d]пиримидин-7-ил)-1-(трет-бутоксикарбонил)пирролидин-3,4-диил диацетата (28d) (407,05 г, 62,3% выход за две стадии) в виде плотного подобного меду продукта красноватого цвета. Аналитический образец получали путем очистки смеси с помощью флеш-хроматографии на колонке (0-100% этилацетат в гексане) с получением (2R,3R,4S,5S)-2-(ацетоксиметил)-5-(4-азидо-5H-пирроло[3,2-d]пиримидин-7-ил)-1-(трет-бутоксикарбонил)пирролидин-3,4-диил диацетата (28d) в виде твердого вещества оранжевого цвета. 1H ЯМР (300 МГц, ДМСО-d6) δ 13,08 (д, J=155,6 Гц, 1H, D2O возможный обмен протонов), 9,86 (с, 1H), 7,61 (д, J=76,8 Гц, 1H), 5,78 (т, J=4,5 Гц, 1H), 5,41 (т, J=4,3 Гц, 1H), 5,21 (с, 1H), 4,55 (дд, J=11,4, 6,4 Гц, 1H), 4,41 (дд, J=11,4, 3,9 Гц, 1H), 4,07 (д, J=16,5 Гц, 1H), 2,06 (с, 3H), 2,01 (д, J=9,9 Гц, 6H), 1,23 (дд, J=39,8, 32,7 Гц, 9H); MS (ES+) 518,0 (M+1), 540 (M+23); (ES-) 516,4 (M-1); Анализ: Вычислено для C22H27N7O8: C, 51,06; H, 5,26; N, 18,95; Найдено: C, 50,97; H, 5,30; N, 18,62.
Стадия 19: Получение (2R,3R,4S,5S)-2-(ацетоксиметил)-5-(4-амино-5H-пирроло[3,2-d]пиримидин-7-ил)-1-(трет-бутоксикарбонил) пирролидин-3,4-диил диацетата (28e)
(2R,3R,4S,5S)-2-(ацетоксиметил)-5-(4-азидо-5H-пирроло[3,2-d]пиримидин-7-ил)-1-(трет-бутоксикарбонил)пирролидин-3,4-диил диацетат (28d) восстанавливали тремя различными партиями следующим образом.
Партия 1: В 2,0-литровый гидрогенизатор Парра с тефлоновой вставкой помещали (2R,3R,4S,5S)-2-(ацетоксиметил)-5-(4-азидо-5H-пирроло[3,2-d]пиримидин-7-ил)-1-(трет-бутоксикарбонил)пирролидин-3,4-диил диацетат (28d) (108,01 г, 300 ммоль в метаноле, 800 мл), Pd(OH)2 (21,6 г, 20% масс/масс).
Партия 2: В 2,0-литровый гидрогенизатор Парра с тефлоновой вставкой помещали (2R,3R,4S,5S)-2-(ацетоксиметил)-5-(4-азидо-5H-пирроло[3,2-d]пиримидин-7-ил)-1-(трет-бутоксикарбонил)пирролидин-3,4-диил диацетат (28d) (140,70 г, 271,9 ммоль в метаноле, 1,0 л), Pd(OH)2 (28,14 г, 20% масс/масс).
Партия 3: В 2,0-литровый гидрогенизатор Парра с тефлоновой вставкой помещали (2R,3R,4S,5S)-2-(ацетоксиметил)-5-(4-азидо-5H-пирроло[3,2-d]пиримидин-7-ил)-1-(трет-бутоксикарбонил)пирролидин-3,4-диил диацетат (28d) (140,7 г, 271,9 ммоль в метаноле, 1,0 л), Pd(OH)2 (28,14 г, 20% масс/масс).
Реакционные смеси гидрировали под давлением 150 фунтов на квадратный дюйм в течение 15-18 ч. Реакционную смесь фильтровали через целит для удаления катализатора. Фильтрат концентрировали в вакууме (водяная баня 60-70°C) до постоянного веса с получением продукта темного цвета (2R,3R,4S,5S)-2-(ацетоксиметил)-5-(4-амино-5H-пирроло[3,2-d]пиримидин-7-ил)-1-(трет-бутоксикарбонил)пирролидин-3,4-диил диацетата (28e) (328,8 г, 89%). Продукт был достаточно чистым, чтобы использовать непосредственно на следующей стадии. Аналитический образец получали путем очистки смеси с использованием флеш-хроматографии на колонке (0-10% метанол в хлороформе). 1H ЯМР (300 МГц, ДМСО-d6) δ 11,06 (с, 1H), 8,12 (с, 1H), 7,49 (с, 1H), 6,94 (с, 2H), 5,86 (с, 1H), 5,44 (т, J=4,2 Гц, 1H), 5,02 (с, 1H), 4,56 (дд, J=11,3, 6,9 Гц, 1H), 4,40 (дд, J=11,3, 4,2 Гц, 1H), 4,16-3,98 (м, 1H), 2,09-1,94 (м, 9H), 1,48-1,14 (м, 9H); MS (ES+) 492,1 (M+1); (ES-) 526,4 (M+Cl); Анализ: Вычислено для C22H29N5O8, 1,25H2O: C, 51,41; H, 6,18; N, 13,62; Найдено: C, 51,24; H, 5,92; N, 13,33.
Стадия 20: Получение (2S,3S,4R,5R)-трет-бутил 2-(4-амино-5H-пирроло[3,2-d]пиримидин-7-ил)-3,4-дигидрокси-5-(гидроксиметил)пирролидин-1-карбоксилата (28f)
Партия 1. К (2R,3R,4S,5S)-2-(ацетоксиметил)-5-(4-амино-5H-пирроло[3,2-d]пиримидин-7-ил)-1-(трет-бутоксикарбонил)пирролидин-3,4-диил диацетату (28e) (81,5 г, 165,8 ммоль) добавляли безводный метанол (370 мл), затем добавляли NaOMe (метоксид натрия, 25 масс.% раствор в метаноле, 4,49 г, 20,76 ммоль) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре до получения данных анализа ТСХ (хлороформ:метанол 9:1), показывающих, что весь исходный материал прореагировал.
Партия 2. К (2R,3R,4S,5S)-2-(ацетоксиметил)-5-(4-амино-5H-пирроло[3,2-d]пиримидин-7-ил)-1-(трет-бутоксикарбонил)пирролидин-3,4-диил диацетату (28e) (117,8 г, 239,6 ммоль), добавляли безводный метанол (530 мл), затем добавляли NaOMe (метоксид натрия, 25 масс.% раствор в метаноле, 6,58 г, 30,45 ммоль) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре до получения данных анализа ТСХ (хлороформ:метанол 9:1), показывающих, что весь исходный материал прореагировал.
Партия 3. К (2R,3R,4S,5S)-2-(ацетоксиметил)-5-(4-амино-5H-пирроло[3,2-d]пиримидин-7-ил)-1-(трет-бутоксикарбонил)пирролидин-3,4-диил диацетату (28e) (129,5 г, 263,5 ммоль) добавляли безводный метанол (584 мл), затем добавляли NaOMe (метоксид натрия, 25 масс.% раствор в метаноле, 6,99 г, 32,35 ммоль) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре до получения данных анализа ТСХ (хлороформ:метанол 9:1), показывающих, что весь исходный материал прореагировал (7-8 ч).
Вышеуказанные растворы концентрировали (водяная баня 65-75°C) с получением (2S,3S,4R,5R)-трет-бутил 2-(4-амино-5H-пирроло[3,2-d]пиримидин-7-ил)-3,4-дигидрокси-5-(гидроксиметил)пирролидин-1-карбоксилата (28f), который был достаточно чистым, чтобы использовать непосредственно на следующей стадии. Аналитический образец получали путем очистки смеси с использованием флеш-хроматографию на колонке (0-10% метанол в хлороформе). 1H ЯМР (300 МГц, ДМСО-d6) δ 10,77 (с, 1H), 8,01 (с, 1H), 7,40 (с, 1H), 6,82 (с, 3H), 5,04-4,91 (м, 1H), 4,87-4,74 (м, 1H), 4,56-4,35 (м, 2H), 4,04-3,90 (м, 2H), 3,72-3,63 (м, 1H), 3,59-3,41 (м, 1H), 1,15 (2 c, 9H); MS (ES+) 366,1 (M+1); (ES-) 400,3 (M+Cl); Анализ: Вычислено для C16H23N5O5·0,25H2O: C, 51,33; H, 6,46; N, 18,71; Найдено: C, 51,04; H, 6,43; N, 18,48.
Получение метокси-N-(бензилоксиметил)-9-бром-9-деазагипоксантина (27f)
Стадия 1: Получение диметил 3-амино-1H-пиррол-2,4-дикарбоксилата (27b)
К раствору диэтил аминомалоната (370,4 г, 1,75 моль) в метаноле (3,6 л) при комнатной температуре одной порцией добавляли 5,4 M раствор NaOMe (975 мл, 5,25 моль) (реакционная смесь была светло-коричневой по цвету). В реакционную смесь добавляли тремя порциями этил (этоксиметилен)цианоацетат (27a) (296 г, 1,75 моль) (в процессе добавления наблюдалось незначительно изменение температуры, ~1°C, цвет реакционной смеси менялся от светло-коричневого до темно-коричневого). Реакционную смесь нагревали при температуре кипения с обратным холодильником в течение 48 ч (проводили ТСХ анализ, 50% этилацетат в гексане, для установления исчезновения исходного материала). Реакционную смесь нейтрализовали путем добавления AcOH (210 мл, 3,5 моль) до pH 6. Реакционную смесь концентрировали в вакууме с получением остатка коричневого цвета. Остаток обрабатывали водой (3 л), фильтровали, промывали водой (500 мл) и гексаном. Сушили на воздухе в течение 48 ч и в вакуумной печи при температуре 60°C с получением диметил 3-амино-1H-пиррол-2,4-дикарбоксилата (27b) 287 г (83%) в виде твердого вещества коричневого цвета. Продукт использовали как таковой на следующей стадии.
Стадия 2: Получение 3H,5H-пирроло[3,2-d]пиримидин-4-она (27c)
Смесь диметил 3-амино-1H-пиррол-2,4-дикарбоксилата (27b) (286 г, 1,44 моль) и формамидин ацетата (451 г, 4,33 моль) в этаноле (2,8 л, 2 мл/ммоль) нагревали при кипячении с обратным холодильником в течение ночи. Реакционная смесь сначала на была гомогенной, но после пары часов кипячения казалась гомогенной и темно-коричневой по цвету (перемешивание становилось затруднительным, поскольку из раствора начинал выпадать твердый продукт). ТСХ анализ аликвоты (50% этилацетат в гексане) свидетельствовал, что присутствовало еще некоторое количество непрореагировавшего материала. Реакционную смесь продолжали нагревать при кипячении с обратным холодильником в течение дополнительных 24 ч и охлаждали до комнатной температуры. Полученное твердое вещество собирали путем фильтрации, промывали водой и гексаном и сушили в вакууме с получением 3H,5H-пирроло[3,2-d]пиримидин-4-она (27c) (223 г, 80%) в виде твердого вещества светло-желтого цвета. Продукт использовали как таковой без очистки.
Стадия 3: Получение 3H,5H-пирроло[3,2-d]пиримидин-4-она (22a)
Смесь 3H,5H-пирроло[3,2-d]пиримидин-4-она (27c) (130,4 г, 0,675 моль) в 2н KOH (1,35 л, 2,7 моль) нагревали при осторожном кипячении в течение 40 ч. Реакционную смесь охлаждали до температуры 60°C и осторожно нейтрализовали с помощью ледяной уксусной кислоты (162 мл, 2,7 моль) до pH 6 (наблюдалось пенообразование из-за декарбоксилирования, и цвет реакционной смеси был черным). Реакционную смесь охлаждали до комнатной температуры, и полученное твердое вещество собирали путем фильтрации, промывали водой (2 × 250 мл) сушили на воздухе и сушили в высоком вакууме над P2O5 с получением продукта в виде твердого вещества темновато-серого цвета (145 г, 159%). Анализ ЯМР продукта указывал на большое количество уксусной кислоты или ее соли, поэтому выход был выше, анализ ТСХ показывал чистый продукт плюс немного продукта на старте с использованием смA-80 в качестве системы растворителей). Продукт обрабатывали водой (400 мл) и нейтрализовали насыщенным водным раствором NaHCO3 до закипания смеси и pH имел значения в области 7-8. Твердый продукт темновато-серого цвета собирали путем фильтрации и промывали водой с получением после сушки на воздухе в течение 48 ч 67,62 г (74%) продукта. Продукт затем сушили в вакууме при температуре кипения этанола с получением 3H,5H-пирроло[3,2-d]пиримидин-4-она (22a) в виде порошка темновато-серого цвета; Т.пл. аналитически чистого образца >250°C; 1H ЯМР (360 МГц, ДМСО-d6) δ 12,05 (с, D2O способный к обмену, 1H), 11,82 (с, D2O способный к обмену, 1H), 7,77 (с, 1H), 7,36 (с, 1H), 6,35 (с, 1H). 13C-ЯМР (ДМСО-d6) 153,88, 144,80, 141,66, 127,51, 117,92, 103,10; ИК (KBr) 3107, 3030 и 1674 см-1; MS (ES+) 136,2 (M+1); Анализ: Вычислено для C6H5N3O: C, 53,33; H, 3,73; N, 31,10; Найдено: C, 53,38; H, 3,77; N, 31,11.
Стадия 4: Получение 4-хлорпирроло[3,2-d]пиримидина (27d)
К образцу 3H,5H-пирроло[3,2-d]пиримидин-4-она (22a) (31,08 г, 230 ммоль) в атмосфере N2 добавляли оксихлорид фосфора (60 мл, 644 моль, 2,8 экв.). Смесь нагревали при кипячении с обратным холодильником в течение 1 ч, в течение этого времени реакционная смесь становилась гомогенной. Реакционную смесь охлаждали на бане лед-вода и затем выливали на дробленый лед (775 мл) с перемешиванием. pH водного раствора медленно устанавливали равным ~pH 8 с помощью концентрированного NH4OH (225 мл), при этом охлаждение смеси продолжали. Полученный осадок собирали с помощью вакуумной фильтрации и промывали водой. Твердое вещество переносили в лоток для сушки и сушили в вакууме при температуре 110°C с получением 4-хлорпирроло[3,2-d]пиримидина (27d) (31,48 г, 89%) в виде твердого вещества темно-серого цвета. Аналитический образец был получен с помощью колоночной хроматографии (силикагель, EtOAc-гексан, 35:65) с последующим упариванием соответствующих фракций. Обработка твердого вещества EtOAc-MeOH давала 4-хлорпирроло[3,2-d]пиримидин (27d) в виде твердого вещества не совсем белого цвета, т.пл. >150°C (разл.); 1H ЯМР (ДМСО-d6) δ 12,43 (с, D2O способный к обмену, 1H), 8,61 (с, 1H), 7,97 (дд, J=2,8, 2,8 Гц; D2O обмен разрушает до d, 1H), 6,72 (дд, J=1,7, 3,5 Гц; D2O обмен разрушает до d, 1H). 13C-ЯМР (ДМСО-d6) 151,30, 149,58, 142,12, 134,83, 124,32, 102,70; ИК (чистый) 3128, 3078, 2979, 1621 см-1; MS (ES+) 154,01 (100%, M+1) и 156,01 (33%); Анализ: Вычислено для C6H4N3Cl: C, 46,93; H, 2,63; N, 27,36; Cl, 23,09; Найдено: C, 47,10; H, 2,79; N, 27,15; Cl, 22,93.
Стадия 5: Получение 6-метокси-N-(бензилоксиметил)-9-деазагипоксантина (27e)
В суспензию предварительно промытого NaH (20 г, 500 ммоль, 1,25 экв., 60%-ая масляная дисперсия, промытая гексаном 2 раза) в безводном ТГФ (1,0 л), охлажденную до температуры 4°C, порциями осторожно добавляли твердый 4-хлорпирроло[3,2-d]пиримидин (27d) (61,4 г, 400 ммоль) при перемешивании в атмосфере N2 в течение 10-15 мин, таким образом, чтобы контролировать выделение газа H2. Спустя примерно час выделение газа прекращалось, и по каплям добавляли бензиловый хлорметиловый эфир (61 мл, 440 ммоль, 1,1 экв.) в течение 45 мин при температуре 4°C (наблюдалось дополнительное выделение газа). Полученной смеси давали нагреться до комнатной температуры и перемешивали в течение 1 ч. Реакционную смесь охлаждали до температуры 4°C и осторожно гасили метоксидом натрия (93 мл, 5,4 M раствор в метаноле, 500 ммоль). Смеси давали нагреться до комнатной температуры в течение ночи и нейтрализовали с помощью ледяной уксусной кислоты (30 мл, 500 ммоль) до pH 6. Смесь концентрировали, и остаток обрабатывали водой (2 × 400 мл). Водный слой декантировали, и остаток сушили в вакууме. Остаток обрабатывали этилацетатом (250 мл) и кипятили с обратным холодильником и фильтровали через складчатый бумажный фильтр. Остаток кипятили с этилацетатом (2 × 100 мл) и фильтровали (оставшийся остаток являлся нежелательным соединением и не двигался в ТСХ анализе 50% этилацетат в гексане). Объединенные фильтраты концентрировали в вакууме до 250 мл и выдерживали в холодильнике в течение ночи. Полученные кристаллы коричневого цвета собирали путем фильтрации, промывали ледяной смесью этилацетат/гексан (2 × 100 мл) и сушили в вакууме с получением 6-метокси-N-(бензилоксиметил)-9-деазагипоксантина (27e) (46,64 г, 43%) в виде твердого вещества оранжево-коричневого цвета. Аналитический образец получали путем перекристаллизации из этилацетата; т.пл. 123-127°C; 1H ЯМР (ДМСО-d6) δ 8,44 (с, 1H), 7,86 (д, J=3,1 Гц, 1 H), 7,31-7,22 (м, 5 H), 6,62 (д, J=3,6 Гц, 1 H), 5,75 (с, 2 H), 4,49 (с, 2 H), 4,05 (с, 3 H); 13C-ЯМР (ДМСО-d6) 156,11, 151,59, 150,09, 137,82, 134,80, 128,53, 127,87, 127,77, 114,99, 103,08, 77,55, 69,95, 53,73; ИК (KBr) 1602 см-1; MS (ES+) 269,97 (M+1); Анализ: Вычислено для C15H15N3O2: C, 66,90; H, 5,61; N, 15,60; Найдено: C, 67,09; H, 5,60; N, 15,60.
Стадия 6: Получение 6-метокси-N-(бензилоксиметил)-9-бром-9-деазагипоксантина (27f)
К раствору 6-метокси-N-(бензилоксиметил)-9-деазагипоксантина (27e) (59,81 г, 222 ммоль) в дихлорметане (225 мл) в атмосфере N2, охлажденному до температуры 4°C (гомогенная реакционная смесь), порциями добавляли NBS (40,3 г, 224 моль, 1,01 экв.) в течение 30 мин, так что температура реакции оставалась ниже 15°C. Смесь перемешивали при температуре 0°C в течение 15 мин и оставляли нагреваться до комнатной температуры в течение 15 мин (ТСХ анализ 50% этилацетат в гексане). Реакционную смесь фильтровали в вакууме для удаления нерастворимого сукцинимида. Фильтрат промывали водой (2 × 250 мл) и насыщенным солевым раствором (200 мл), сушили (Na2SO4), фильтровали и концентрировали в вакууме с получением продукта в виде твердого вещества светло-желтого цвета. Твердое вещество растворяли путем кипячения в этилацетате (200 мл) и разбавляли гексаном (200 мл). Раствор кипятили с обратным холодильником и фильтровали горячим очень быстро (чтобы избежать кристаллизации твердого вещества). Фильтрат затем кипятили, и порциями по 200 мл добавляли гексан (полный объем гексана 1600 мл). Горячий раствор декантировали, при необходимости, для удаления нерастворимых остатков (продукт растворим в горячем 10%-ом этилацетате в гексане). Горячему фильтрату давали охладиться до комнатной температуры и затем выдерживали в холодильнике в течение ночи. Полученное твердое вещество собирали путем фильтрования и промывали гексаном и сушили в вакууме при комнатной температуре с получением 6-метокси-N-(бензилоксиметил)-9-бром-9-деазагипоксантина (27f) (59,6 г, 77%) в виде твердого вещества светло-желтого цвета: Т.пл. 103-108°C; 1H ЯМР (ДМСО-d6) δ 8,51 (с, 1H), 8,12 (с, 1H), 7,31-7,22 (м, 5H), 5,74 (с, 2H), 4,52 (с, 2H), 4,07 (с, 3H). 13C-ЯМР (ДМСО-d6) 156,19, 150,66, 148,14, 137,59, 133,45, 128,38, 127,80, 127,67, 115,02, 90,90, 77,79, 70,25, 54,07; ИК (KBr) 3078, 1602, 1542 см-1; MS (ES+) 348,27 (100%), 350,28 (98%); Анализ: Вычислено для C15H14N3O2Br: C, 51,74; H, 4,05; N, 12,07; Найдено: C, 51,72; H, 4,04; N, 12,06.
Пример 2: Гидрохлорид (S)-((2R,3R,4S,5S)-5-(4-амино-5H-пирроло[3,2-d]пиримидин-7-ил)-3,4-дигидроксипирролидин-2-ил)метил 2-амино-3-метилбутаноата (30f)
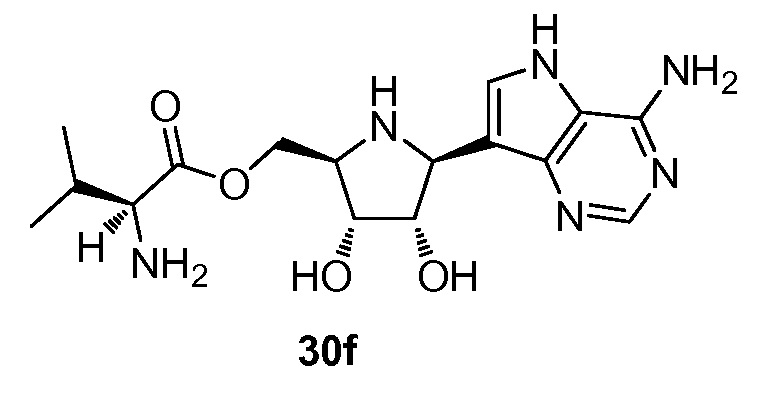
Способ A:
Раствор (3aS,4S,6R,6aR)-трет-бутил 4-(4-амино-5H-пирроло[3,2-d]пиримидин-7-ил)-6-(((2-((трет-бутоксикарбонил)амино)-3-метилбутаноил)окси)метил)-2,2-диметилдигидро-3aH-[1,3]диоксоло[4,5-c]пиррол-5(4H)-карбоксилата (30e) (600 мг, 1 ммоль) в ТФУ (10 мл) перемешивали при комнатной температуре в течение 1 ч и концентрировали в вакууме досуха. Остаток растворяли в 10 мл AcOH и добавляли раствор BCl3 (3,6 мл, 3,6 ммоль, 1 M в дихлорметане), перемешивали при комнатной температуре в течение 4 мин и гасили водой (5 мл). Реакционную смесь концентрировали досуха. Остаток сушили в холодильнике с получением гидрохлорида (S)-((2R,3R,4S,5S)-5-(4-амино-5H-пирроло[3,2-d]пиримидин-7-ил)-3,4-дигидроксипирролидин-2-ил)метил 2-амино-3-метилбутаноата (30f) (400 мг, 76%) в виде твердого вещества белого цвета; 1H ЯМР (300 МГц, ДМСО-d6/D2O) δ 8,64 (с, 1H), 8,21 (с, 1H), 4,83 (д, J=8,4 Гц, 1H), 4,63-4,49 (м, 3H), 4,27-4,19 (м, 1H), 3,94 (д, J=4,8 Гц, 1H), 3,82-3,70 (м, 1H), 2,33-2,18 (м, 1H), 0,99 (д, J=6,9 Гц, 6H); MS (ES+) 365,1 (M+1); Анализ: Вычислено для C16H27Cl3N6O4,3HCl,2,5H2O: C, 37,17; H, 6,07; Cl, 20,16; N, 16,09; Найдено: C, 37,04; H, 6,22; Cl, 20,50; N, 16,20.
Способ B:
К раствору (3aS,4S,6R,6aR)-трет-бутил 4-(4-амино-5H-пирроло[3,2-d]пиримидин-7-ил)-6-(((2-((трет-бутоксикарбонил)амино)-3-метилбутаноил)окси)метил)-2,2-диметилдигидро-3aH-[1,3]диоксоло[4,5-c]пиррол-5(4H)-карбоксилата (30e) (0,151 г, 0,25 ммоль) в ацетоне (2 мл) добавляли конц. серную кислоту (18н, 0,139 мл, 2,5 ммоль) и перемешивали при комнатной температуре в течение ночи. Реакционную смесь декантировали, и к остатку добавляли кипящий ацетон (10 мл) и охлаждали до комнатной температуры. Полученное твердое вещество собирали путем фильтрации с получением сульфата ((2R,3R,4S,5S)-5-(4-амино-5H-пирроло[3,2-d]пиримидин-7-ил)-3,4-дигидроксипирролидин-2-ил)метил 2-амино-3-метилбутаноата (30f) в виде твердого вещества белого цвета; 1H ЯМР (300 МГц, D2O) δ 8,42 (с, 1H), 8,04 (с, 1H), 5,05 (д, J=8,1 Гц, 1H), 4,79 (д, J=4,9 Гц, 1H), 4,62 (дд, J=12,6, 7,5 Гц, 1H), 4,55 (т, J=5,2 Гц, 1H), 4,20-4,08 (м, 3H), 2,45-2,28 (м, 1H), 1,06 (т, J=7,3 Гц, 6H).
Способ C:
К раствору (3aS,4S,6R,6aR)-трет-бутил 4-(4-амино-5H-пирроло[3,2-d]пиримидин-7-ил)-6-(((2-((трет-бутоксикарбонил)амино)-3-метилбутаноил)окси)метил)-2,2-диметилдигидро-3aH-[1,3]диоксоло[4,5-c]пиррол-5(4H)-карбоксилата (30e) (0,302 г, 0,5 ммоль) в MTBE (2,5 мл) добавляли воду (0,046 мл) и конц. серную кислоту (0,138 мл, 5,00 ммоль), затем через 15 мин MTBE (2,5 мл) и перемешивали при комнатной температуре в течение 4 ч. Декантировали TBDME, добавляли воду (0,5 мл) и перемешивали для растворения твердого вещества, затем добавляли этанол (9,5 мл) и энергично перемешивали в течение 2 ч. Полученное тонкоизмельченное твердое вещество собирали путем фильтрации, промывали этанолом с получением сульфата ((2R,3R,4S,5S)-5-(4-амино-5H-пирроло[3,2-d]пиримидин-7-ил)-3,4-дигидроксипирролидин-2-ил)метил 2-амино-3-метилбутаноата (30f) (0,288 г, 103% выход) в виде твердого вещества белого цвета; 1H ЯМР (300 МГц, ДМСО-d6/D2O) δ 8,26 (с, 1H), 7,81 (с, 1H), 4,67 (д, J=6,7 Гц, 1H), 4,59-4,41 (м, 3H), 4,27 (т, J=5,6 Гц, 1H), 3,91 (д, J=4,6 Гц, 1H), 3,81-3,69 (м, 1H), 2,28-2,10 (м, 1H), 0,97 (д, J=6,9 Гц, 6H).
Способ D:
К раствору (3aS,4S,6R,6aR)-трет-бутил 4-(4-амино-5H-пирроло[3,2-d]пиримидин-7-ил)-6-(((2-((трет-бутоксикарбонил)амино)-3-метилбутаноил)окси)метил)-2,2-диметилдигидро-3aH-[1,3]диоксоло[4,5-c]пиррол-5(4H)-карбоксилата (30e) (0,302 г, 0,5 ммоль) в MTBE (2,5 мл) добавляли воду (0,138 мл) и конц. серную кислоту (0,138 мл, 5,00 ммоль), затем через 15 мин MTBE (2,5 мл) и перемешивали при комнатной температуре в течение 4 ч. Декантировали TBDME, добавляли этанол (9,5 мл) и перемешивали в течение 2 ч, собирали твердое вещество путем фильтрации, сушили в вакууме, собирали твердое вещество с получением продукта белого цвета сульфата ((2R,3R,4S,5S)-5-(4-амино-5H-пирроло[3,2-d]пиримидин-7-ил)-3,4-дигидроксипирролидин-2-ил)метил 2-амино-3-метилбутаноата (30f) (0,160 г, 0,285 ммоль, 57,1% выход).
Получение (3aS,4S,6R,6aR)-трет-бутил 4-(4-амино-5H-пирроло[3,2-d]пиримидин-7-ил)-6-(((2-((трет-бутоксикарбонил)амино)-3-метилбутаноил)окси)метил)-2,2-диметилдигидро-3aH-[1,3]диоксоло[4,5-c]пиррол-5(4H)-карбоксилата (30e)
Стадия 1: Получение (2S,3S,4R,5R)-трет-бутил 2-(4-хлор-5H-пирроло[3,2-d]пиримидин-7-ил)-3,4-дигидрокси-5-(гидроксиметил)пирролидин-1-карбоксилата (30a)
Раствор (2R,3R,4S,5S)-2-(ацетоксиметил)-1-(трет-бутоксикарбонил)-5-(4-хлор-5H-пирроло[3,2-d]пиримидин-7-ил)пирролидин-3,4-диил диацетата (28c) (25,8 г, 50,5 ммоль) растворяли в метаноле (200 мл) и при комнатной температуре добавляли метоксид натрия 25 масс.% в метаноле (3,6 мл, 16,66 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь концентрировали досуха и очищали на колонке 600 г с получением (2S,3S,4R,5S)-трет-бутил 2-(4-хлор-5H-пирроло[3,2-d]пиримидин-7-ил)-3,4-дигидрокси-5-(гидроксиметил)пирролидин-1-карбоксилата (30a) (17,7 г, 46 ммоль, 91% выход) в виде бесцветной пены; 1H ЯМР (300 МГц, ДМСО-d6) δ 12,34 (с, 1H), 8,62 (с, 1H), 7,94 (с, 1H), 5,40-5,02 (м, 2H), 4,96-4,70 (м, 2H), 4,41-4,25 (м, 1H), 4,13-3,93 (м, 2H), 3,69-3,51 (м, 2H), 1,35 (с, 3H), 1,01 (с, 6H); MS (ES+) 384,9 (M+1), 792,6 (2M+Na); (ES-) 382,6 (M-1).
Стадия 2: Получение (3aS,4S,6R,6aR)-трет-бутил 4-(4-хлор-5H-пирроло[3,2-d]пиримидин-7-ил)-6-(гидроксиметил)-2,2-диметилдигидро-3aH-[1,3]диоксоло[4,5-c]пиррол-5(4H)-карбоксилата (30b)
К раствору (2S,3S,4R,5R)-трет-бутил 2-(4-хлор-5H-пирроло[3,2-d]пиримидин-7-ил)-3,4-дигидрокси-5-(гидроксиметил)пирролидин-1-карбоксилата (30a) (16,3 г, 42,4 ммоль) в ацетоне (400 мл) добавляли 2,2-диметоксипропан (11,17 мл, 89 ммоль) и гидрат 4-метилбензолсульфоновой кислоты (0,41 г, 2,12 ммоль). Реакцию перемешивали при комнатной температуре в течение ночи. Реакционную смесь гасили TEA (590 мкл, 4,24 ммоль) и концентрировали досуха. Остаток очищали путем флеш-хроматографии на колонке (силикагель 500 г) с получением (3aS,4S,6R,6aR)-трет-бутил 4-(4-хлор-5H-пирроло[3,2-d]пиримидин-7-ил)-6-(гидроксиметил)-2,2-диметилдигидро-3aH-[1,3]диоксоло[4,5-c]пиррол-5(4H)-карбоксилата (30b) (10,7 г, 25,2 ммоль, 59,5% выход) в виде бесцветной пены; 1H ЯМР (300 МГц, ДМСО-d6) δ 12,45 (с, 1H), 8,67 (с, 1H), 7,81 (с, 1H), 5,09 (д, J=36,7 Гц, 3H), 4,82 (д, J=5,7 Гц, 1H), 4,00 (с, 1H), 3,53 (с, 1H), 3,34 (с, 1H), 1,47 (с, 3H), 1,40 (ушир. с, 4H), 1,29 (ушир. с, 4H), 1,20 (ушир. с, 4H); MS (ES+) 426,9 (M+1); 422,6 (M-1).
Стадия 3: Получение (3aS,4S,6S,6aR)-трет-бутил 4-(4-азидо-5H-пирроло[3,2-d]пиримидин-7-ил)-6-(гидроксиметил)-2,2-диметилдигидро-3aH-[1,3]диоксоло[4,5-c]пиррол-5(4H)-карбоксилата (30c)
К раствору (3aS,4S,6R,6aR)-трет-бутил 4-(4-хлор-5H-пирроло[3,2-d]пиримидин-7-ил)-6-(гидроксиметил)-2,2-диметилдигидро-3aH-[1,3]диоксоло[4,5-c]пиррол-5(4H)-карбоксилата (30b) (5,1 г, 12 ммоль) в ДМФ (30 мл) добавляли азид натрия (3,9 г, 60 ммоль), полученный раствор перемешивали при температуре 80°C в течение 4 ч. Реакционную смесь концентрировали в вакууме для удаления большей части ДМФ, и полученный остаток растворяли в хлороформе. Органический слой промывали водой, сушили над MgSO4 и концентрировали в вакууме с получением (3aS,4S,6R,6aR)-трет-бутил 4-(4-азидо-5H-пирроло[3,2-d]пиримидин-7-ил)-6-(гидроксиметил)-2,2-диметилдигидро-3aH-[1,3]диоксоло[4,5-c]пиррол-5(4H)-карбоксилата (30c) (5 г, 97%); 1H ЯМР (300 МГц, ДМСО-d6) δ 13,22 (ушир. с, 1H), 9,87 (с, 1H), 7,69-7,47 (м, 1H), 5,28 (м, 1H), 5,05 (м, 2H), 4,81 (д, J=5,9, 1H), 4,06-3,91 (м, 1H), 3,57 (м, 1H), 3,51-3,38 (м, 1H), 1,48 (с, 3H), 1,41-1,23 (ушир. с, 9H), 1,30 (с, 3H); MS (ES+) 454 (M+Na), 863,1 (2M+1), 885,2 (2M+Na); (ES-) 429,7 (M-1).
Стадия 4: Получение (3aS,4S,6R,6aR)-трет-бутил 4-(4-азидо-5H-пирроло[3,2-d]пиримидин-7-ил)-6-((((S)-2-((трет-бутоксикарбонил)амино)-3-метилбутаноил)окси)метил)-2,2-диметилдигидро-3aH-[1,3]диоксоло[4,5-c]пиррол-5(4H)-карбоксилата (30d)
К раствору (3aS,4S,6R,6aR)-трет-бутил 4-(4-азидо-5H-пирроло[3,2-d]пиримидин-7-ил)-6-(гидроксиметил)-2,2-диметилдигидро-3aH-[1,3]диоксоло[4,5-c]пиррол-5(4H)-карбоксилата (30c) (1,088 г, 2,5 ммоль) и (S)-2-((трет-бутоксикарбонил)амино)-3-метилбутановой кислоты (L-Boc валин, 0,543 г, 2,5 ммоль) в ДМФ (20 мл) добавляли EDCI (1,198 г, 6,25 ммоль) и DMAP (92 мг, 0,75 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 10 дней и гасили водой (60 мл), экстрагировали с помощью этилацетата (3 × 50 мл). Органические слои объединяли, промывали водой (2 × 50 мл), насыщенным солевым раствором, сушили и концентрировали в вакууме. Полученный остаток очищали дважды путем флеш-хроматографии на колонке с получением (3aS,4S,6R,6aR)-трет-бутил 4-(4-азидо-5H-пирроло[3,2-d]пиримидин-7-ил)-6-((((S)-2-((трет-бутоксикарбонил)амино)-3-метилбутаноил)окси)метил)-2,2-диметилдигидро-3aH-[1,3]диоксоло[4,5-c]пиррол-5(4H)-карбоксилата (30d) (0,75 г, 47%) в виде твердого вещества белого цвета; 1H ЯМР (300 МГц, ДМСО-d6) δ 13,30 (с, 1H), 9,88 (с, 1H), 7,60 (с, 1H), 7,10 (с, 1H), 5,34 (с, 1H), 5,20 (дд, J=5,7, 1,5 Гц, 1H), 4,82 (д, J=5,8 Гц, 1H), 4,34-4,14 (м, 2H), 3,80 (дд, J=8,0, 5,9 Гц, 1H), 3,34 (с, 1H), 1,97-1,84 (м, 1H), 1,47 (с, 3H), 1,43-1,31(м, 21H), 0,80 (дд, J=6,9, 5,2 Гц, 6H); MS (ES-) 629,1 (M-1); ИК (KBr) 2315 см-1.
Стадия 5: Получение (3aS,4S,6R,6aR)-трет-бутил 4-(4-амино-5H-пирроло[3,2-d]пиримидин-7-ил)-6-((((S)-2-((трет-бутоксикарбонил)амино)-3-метилбутаноил)окси)метил)-2,2-диметилдигидро-3aH-[1,3]диоксоло[4,5-c]пиррол-5(4H)-карбоксилата (30e)
К раствору (3aS,4S,6R,6aR)-трет-бутил 4-(4-азидо-5H-пирроло[3,2-d]пиримидин-7-ил)-6-((((S)-2-((трет-бутоксикарбонил)амино)-3-метилбутаноил)окси)метил)-2,2-диметилдигидро-3aH-[1,3]диоксоло[4,5-c]пиррол-5(4H)-карбоксилата (30d) (0,72 г, 1,19 ммоль) в метаноле (20 мл) добавляли Pd/C (200 мг, 5 масс.% на C) и гидрировали в атмосфере водорода в течение 2 ч. Катализатор удаляли путем фильтрации через целит, и фильтрат концентрировали в вакууме. Полученный остаток очищали на колонке с получением (3aS,4S,6R,6aR)-трет-бутил 4-(4-амино-5H-пирроло[3,2-d]пиримидин-7-ил)-6-((((S)-2-((трет-бутоксикарбонил)амино)-3-метилбутаноил)окси)метил)-2,2-диметилдигидро-3aH-[1,3]диоксоло[4,5-c]пиррол-5(4H)-карбоксилата (30e) (600 мг, 83%) в виде твердого вещества белого цвета. 1H ЯМР (300 МГц, ДМСО-d6) δ 11,97 (с, 1H), 10,87 (с, 1H), 8,09 (с, 1H), 7,36 (с, 1H), 6,78 (с, 2H), 5,30-5,22 (м, 1H), 5,19-5,07 (м, 1H), 4,88 (д, J=5,9 Гц, 1H), 4,18-4,07 (м, 2H), 3,87-3,79 (м, 1H), 3,44 (квд, J=7,0, 5,1 Гц, 1H), 2,01-1,92 (м, 1H), 1,44-1,32 (м, 21H), 1,28 (с, 3H), 0,82 (д, J=6,7 Гц, 6H); MS (ES+) 605,1 (M+1).
Пример 3: Гидрохлорид (2S,3S)-((2R,3R,4S,5S)-5-(4-амино-5H-пирроло[3,2-d]пиримидин-7-ил)-3,4-дигидроксипирролидин-2-ил)метил 2-амино-3-метилпентаноата (31c)
Раствор (3aS,4S,6R,6aR)-трет-бутил 4-(4-амино-5H-пирроло[3,2-d]пиримидин-7-ил)-6-((((2S,3S)-2-((трет-бутоксикарбонил)амино)-3-метилпентаноил)окси)метил)-2,2-диметилдигидро-3aH-[1,3]диоксоло[4,5-c]пиррол-5(4H)-карбоксилата (31b) (0,398 г, 0,643 ммоль) в трифторуксусной кислоте (10 мл) перемешивали при комнатной температуре в течение 1 ч и концентрировали в вакууме досуха. Остаток обрабатывали толуолом (20 мл), концентрировали в вакууме досуха. Полученный остаток растворяли в AcOH (10 мл), и к этому раствору добавляли раствор трихлорборана (2,32 мл, 2,32 ммоль), перемешивали при комнатной температуре в течение 4 мин и гасили водой (5 мл). Реакционную смесь концентрировали в вакууме досуха. Полученное твердое смолистое вещество растворяли в воде (5 мл) и фильтровали. Фильтрат сушили вымораживанием с получением (2S,3S)-((2R,3R,4S,5S)-5-(4-амино-5H-пирроло[3,2-d]пиримидин-7-ил)-3,4-дигидроксипирролидин-2-ил)метил 2-амино-3-метилпентаноата (31c) (0,275 г, 88% выход) в виде твердого вещества белого цвета.
1H ЯМР (300 МГц, D2O) δ 8,24 (с, 1H), 7,87 (с, 1H), 4,89 (д, J=8,0 Гц, 1H), 4,61-4,43 (м, 3H), 4,41 (т, J=5,0 Гц, 1H), 4,04 (д, J=3,9 Гц, 1H), 3,98 (дт, J=6,7, 4,2 Гц, 1H), 2,00-1,88 (м, 1H), 1,41-1,10 (м, 2H), 0,88 (д, J=7,0 Гц, 3H), 0,79 (т, J=7,4 Гц, 3H); 1H ЯМР (300 МГц, ДМСО-d6/D2O) δ 8,61 (с, 1H), 8,17 (с, 1H), 4,85 (д, J=8,3 Гц, 1H), 4,62 (дд, J=12,2, 4,2 Гц, 1H), 4,57-4,46 (м, 2H), 4,24 (т, J=5,0 Гц, 1H), 4,01 (д, J=4,1 Гц, 1H), 2,01-1,94 (м, 1H), 1,56-1,39 (м, 1H), 1,36-1,22 (м, 1H), 0,96 (д, J=6,9 Гц, 3H), 0,90 (т, J=7,3 Гц, 3H); MS (ES+) 379,1 (M+1), (ES-) 412,5 (M+Cl); ВЭЖХ [Restek Pinnacle DB C18, 150 × 4,6 мм, 5 мкм, Скорость потока: 1,0 мл в минуту при температуре 40°C. "A" буфер = Растворяли 4,3 г моногидрата натриевой соли 1-октансульфоновой кислоты в 900 мл воды со степенью чистоты для ВЭЖХ. Добавляли 10 мл уксусной кислоты и 100 мл ацетонитрила. "B" буфер = Растворяли 4,3 г моногидрата натриевой соли 1-октансульфоновой кислоты в 600 мл воды со степенью чистоты для ВЭЖХ. Добавляли 10 мл уксусной кислоты и 400 мл ацетонитрила, УФ поглощение=260 нМ; (A:B, 85/15 (0 мин) до A:B 0/100 (25 мин) до A:B 0/100 (40 мин) до A:B 85/15 (50 мин)) Rt=22,79 (97,26%)]; Анализ: Вычислено для C17H26N6O4⋅3HCl⋅2,25H2O⋅2B(OH)3: C, 31,84; H, 6,21; Cl, 16,59; N, 13,11; Найдено: C, 31,99; H, 6,13; Cl, 16,33; N, 12,80.
Получение (3aS,4S,6R,6aR)-трет-бутил 4-(4-амино-5H-пирроло[3,2-d]пиримидин-7-ил)-6-((((2S,3S)-2-((трет-бутоксикарбонил)амино)-3-метилпентаноил)окси)метил)-2,2-диметилдигидро-3aH-[1,3]диоксоло[4,5-c]пиррол-5(4H)-карбоксилата (31b)
Стадия 1: Получение (3aS,4S,6R,6aR)-трет-бутил 4-(4-азидо-5H-пирроло[3,2-d]пиримидин-7-ил)-6-((((2S,3S)-2-((трет-бутоксикарбонил)амино)-3-метилпентаноил)окси)метил)-2,2-диметилдигидро-3aH-[1,3]диоксоло[4,5-c]пиррол-5(4H)-карбоксилата (31a)
К раствору (3aS,4S,6R,6aR)-трет-бутил 4-(4-азидо-5H-пирроло[3,2-d]пиримидин-7-ил)-6-(гидроксиметил)-2,2-диметилдигидро-3aH-[1,3]диоксоло[4,5-c]пиррол-5(4H)-карбоксилата (30c) (1,079 г, 2,5 ммоль), (2S,3S)-2-((трет-бутоксикарбонил)амино)-3-метилпентановой кислоты (Boc-L-изолейцин) (0,578 г, 2,5 ммоль) в ДМФ (20 мл) добавляли гидрохлорид N1-((этилимино)метилен)-N3,N3-диметилпропан-1,3-диамина (EDCI, 1,20 г, 6,25 ммоль) и N,N-диметилпиридин-4-амин (DMAP, 0,092 г, 0,75 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 5 дней, гасили 1н водн. HCl (5,00 мл) и водой (60 мл). Реакционную смесь экстрагировали с помощью этилацетата (3 × 50 мл). Органические слои объединяли, промывали водой (2 × 25 мл), насыщенным солевым раствором, сушили и концентрировали в вакууме. Полученный остаток очищали путем флеш-хроматографии на колонке (силикагель, 25 г, элюируя смесью этилацетата в гексане 0-100%) с получением (3aS,4S,6R,6aR)-трет-бутил 4-(4-азидо-5H-пирроло[3,2-d]пиримидин-7-ил)-6-((((2S,3S)-2-((трет-бутоксикарбонил)амино)-3-метилпентаноил)окси)метил)-2,2-диметилдигидро-3aH-[1,3]диоксоло[4,5-c]пиррол-5(4H)-карбоксилата (31a) (0,683 г, 42% выход) в виде твердого вещества белого цвета; 1H ЯМР (300 МГц, ДМСО-d6) δ 13,29 (с, 1H, D2O способный к обмену), 9,86 (с, 1H), 7,59 (с, 1H), 7,11 (д, J=7,0 Гц, 1H), 5,32 (с, 1H), 5,20 (д, J=5,8 Гц, 1H), 4,81 (д, J=5,7 Гц, 1H), 4,28 (ушир. с, 2H), 4,08-3,93 (м, 1H), 3,90-3,77 (м, 1H), 1,62 (с, 1H), 1,52-1,21 (м, 26H), 0,84-0,66 (м, 6H); MS (ES+) 645,2 (M+1), 667,2 (M+Na), (ES-) 643,1 (M-1).
Стадия 2: Получение (3aS,4S,6R,6aR)-трет-бутил 4-(4-амино-5H-пирроло[3,2-d]пиримидин-7-ил)-6-((((2S,3S)-2-((трет-бутоксикарбонил)амино)-3-метилпентаноил)окси)метил)-2,2-диметилдигидро-3aH-[1,3]диоксоло[4,5-c]пиррол-5(4H)-карбоксилата (31b)
К раствору (3aS,4S,6R,6aR)-трет-бутил 4-(4-азидо-5H-пирроло[3,2-d]пиримидин-7-ил)-6-((((2S,3S)-2-((трет-бутоксикарбонил)амино)-3-метилпентаноил)окси)метил)-2,2-диметилдигидро-3aH-[1,3]диоксоло[4,5-c]пиррол-5(4H)-карбоксилата (31a) (0,625 г, 0,969 ммоль) в метаноле (20 мл) добавляли (10%) палладий на угле (206 мг) и гидрировали под давлением 60 фунтов на квадратный дюйм в течение 3,5 ч. ТСХ анализ показывал (этилацетат/метанол (9:1) в гексане 1:1), что реакция завершалась. Катализатор удаляли путем фильтрации через целит, и фильтрат концентрировали в вакууме. Полученный остаток очищали путем флеш-хроматографии на колонке (силикагель 12 г, элюируя смесью этилацетат/метанол (9:1) в гексане 0-100%) с получением (3aS,4S,6R,6aR)-трет-бутил 4-(4-амино-5H-пирроло[3,2-d]пиримидин-7-ил)-6-((((2S,3S)-2-((трет-бутоксикарбонил)амино)-3-метилпентаноил)окси)метил)-2,2-диметилдигидро-3aH-[1,3]диоксоло[4,5-c]пиррол-5(4H)-карбоксилата (31b) (0,418 г, 70% выход) в виде твердого вещества белого цвета; 1H ЯМР (300 МГц, ДМСО-d6) δ 10,86 (ушир. с, 1H, D2O способный к обмену), 8,09 (с, 1H), 7,36 (д, J=2,8 Гц, 1H), 7,14 (с, 1H), 6,77 (с, 2H), 5,25 (с, 1H), 5,14 (ушир. с, 1H), 4,88 (д, J=5,9 Гц, 1H), 4,18 (с, 1H), 4,05 (с, 3H), 3,90 (с, 1H), 1,68 (ушир. с, 1H), 1,42 (с, 3H), 1,38 (с, 18H), 1,28 (с, 3H), 1,17 (с, 1H), 0,78 (м, 6H); MS (ES+) 619,2 (M+1), (ES-) 653,2 (M+Cl); Анализ: Вычислено для C30H46N6O8·0,25H2O: C, 57,82; H, 7,52; N, 13,48, Найдено: C, 57,56; H, 7,42; N, 13,40.
Пример 4: Гидрохлорид (S)-((2R,3R,4S,5S)-5-(4-амино-5H-пирроло[3,2-d]пиримидин-7-ил)-3,4-дигидроксипирролидин-2-ил)метил 2-амино-4-метилпентаноата (32c)
Раствор (3aS,4S,6R,6aR)-трет-бутил 4-(4-амино-5H-пирроло[3,2-d]пиримидин-7-ил)-6-((((S)-2-((трет-бутоксикарбонил)амино)-4-метилпентаноил)окси)метил)-2,2-диметилдигидро-3aH-[1,3]диоксоло[4,5-c]пиррол-5(4H)-карбоксилата (32b) (0,243 г, 0,393 ммоль) в трифторуксусной кислоте (10 мл) перемешивали при комнатной температуре в течение 1 ч и концентрировали в вакууме досуха. Остаток обрабатывали толуолом (20 мл) и концентрировали в вакууме досуха. Остаток растворяли в AcOH (10 мл) и добавляли раствор трихлорборана (1,4 мл, 1,4 ммоль), перемешивали при комнатной температуре в течение 4 мин и гасили водой (5 мл). Реакционную смесь концентрировали досуха. Твердое смолистое вещество растворяли водой (5 мл) и фильтровали. Фильтрат сушили вымораживанием с получением твердого вещества (201 мг). Твердое вещество растворяли в 0,37 мл воды и мягко нагревали до образования прозрачного раствора, опять добавляли 0,13 мл воды, затем разбавляли 9,0 мл 2-пропанола (IPA), затем добавляли 0,25 мл воды (теперь общий объем был 0,75 мл), раствор декантировали для удаления нерастворимой массы. На данной стадии раствор был прозрачным, нагревали и добавляли 5,5 мл IPA, раствор становился мутным и его оставляли стоять в течение 1 ч. Полученное твердое вещество собирали путем фильтрации с получением гидрохлорида (S)-((2R,3R,4S,5S)-5-(4-амино-5H-пирроло[3,2-d]пиримидин-7-ил)-3,4-дигидроксипирролидин-2-ил)метил 2-амино-4-метилпентаноата (32c) (73 мг, 49% выход) в виде твердого вещества белого цвета; 1H ЯМР (300 МГц, ДМСО-d6) δ 8,47 (с, 1H), 8,03 (с, 1H), 4,77 (д, J=7,5 Гц, 1H), 4,60-4,45 (м, 3H), 4,29 (т, J=5,0 Гц, 1H), 4,03 (т, J=6,4 Гц, 1H), 3,80-3,71 (м, 1H), 1,83-1,58 (м, 3H), 0,90 (д, J=5,2 Гц, 6H); MS (ES+) 379,1 (M+1), (ES-) 412,7 (M+Cl); ВЭЖХ [Restek Pinnacle DB C18, 150 × 4,6 мм, 5 мкм колонка, Скорость потока: 1,0 мл в минуту при температуре 40°C. "A" буфер = растворение 4,3 г моногидрата натриевой соли 1-октансульфоновой кислоты в 900 мл воды со степенью чистоты для ВЭЖХ. Добавляли 10 мл уксусной кислоты и 100 мл ацетонитрила. "B" буфер = растворение 4,3 г моногидрата натриевой соли 1-октансульфоновой кислоты в 600 мл воды со степенью чистоты для ВЭЖХ. Добавляли 10 мл уксусной кислоты и 400 мл ацетонитрила, УФ поглощение=260 нМ; (A:B, 85/15 (0 мин) до A:B 0/100 (25 мин) до A:B 0/100 (40 мин) до A:B 85/15 (50 мин)) Rt=22,79 (96,3027%)]; Анализ: Вычислено для C17H26N6O4 2,75 H2O 2,5HCl: C, 39,33; H, 6,60; Cl, 17,07; N, 16,19; Найдено: C, 39,04; H, 6,32; Cl, 17,46; N, 15,96.
Получение (3aS,4S,6R,6aR)-трет-бутил 4-(4-амино-5H-пирроло[3,2-d]пиримидин-7-ил)-6-((((S)-2-((трет-бутоксикарбонил)амино)-4-метилпентаноил)окси)метил)-2,2-диметилдигидро-3aH-[1,3]диоксоло[4,5-c]пиррол-5(4H)-карбоксилата (32b)
Стадия 1: получение (3aS,4S,6R,6aR)-трет-бутил 4-(4-азидо-5H-пирроло[3,2-d]пиримидин-7-ил)-6-((((S)-2-((трет-бутоксикарбонил)амино)-4-метилпентаноил)окси)метил)-2,2-диметилдигидро-3aH-[1,3]диоксоло[4,5-c]пиррол-5(4H)-карбоксилата (32a)
К раствору (3aS,4S,6R,6aR)-трет-бутил 4-(4-азидо-5H-пирроло[3,2-d]пиримидин-7-ил)-6-(гидроксиметил)-2,2-диметилдигидро-3aH-[1,3]диоксоло[4,5-c]пиррол-5(4H)-карбоксилата (30c) (1,088 г, 2,52 ммоль) и (S)-2-((трет-бутоксикарбонил)амино)-4-метилпентановой кислоты (Boc-L-лейцин) (0,583 г, 2,52 ммоль) в ДМФ (20 мл) добавляли гидрохлорид N1-((этилимино)метилен)-N3,N3-диметилпропан-1,3-диамина (EDCI, 1,21 г, 6,30 ммоль) и N,N-диметилпиридин-4-амин (DMAP, 0,092 г, 0,757 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 96 ч, гасили водой (60 мл) и экстрагировали с помощью EtOAc (3 × 50 мл). Объединенные органические слои промывали водой (2 × 50 мл), насыщенным солевым раствором (50 мл), сушили над MgSO4 и концентрировали досуха в вакууме. Полученный остаток очищали путем флеш-хроматографии на колонке (силикагель 25 г) с получением (3aS,4S,6R,6aR)-трет-бутил 4-(4-азидо-5H-пирроло[3,2-d]пиримидин-7-ил)-6-((((S)-2-((трет-бутоксикарбонил)амино)-4-метилпентаноил)окси)метил)-2,2-диметилдигидро-3aH-[1,3]диоксоло[4,5-c]пиррол-5(4H)-карбоксилата (32a) (633 мг, 39% выход) в виде пены белого цвета; 1H ЯМР (300 МГц, ДМСО-d6) δ 13,28 (с, 1H), 9,86 (с, 1H), 7,56 (с, 1H), 7,19 (д, J=7,4 Гц, 1H), 5,31 (с, 1H), 5,16 (д, J=6,8 Гц, 1H), 4,80 (д, J=5,8 Гц, 1H), 4,23 (с, 2H), 3,86 (т, J=11,4 Гц, 1H), 1,39 (м, 27H), 0,86 (дд, J=11,7, 6,0 Гц, 1H), 0,75 (дд, J=13,6, 6,5 Гц, 6H); MS (ES+) 645,19(M+1), 667,17 (M+Na); (ES-) 643,20(M-1), 679,18 (M+Cl).
Стадия 2: (3aS,4S,6R,6aR)-трет-бутил 4-(4-амино-5H-пирроло[3,2-d]пиримидин-7-ил)-6-((((S)-2-((трет-бутоксикарбонил)амино)-4-метилпентаноил)окси)метил)-2,2-диметилдигидро-3aH-[1,3]диоксоло[4,5-c]пиррол-5(4H)-карбоксилат (32b)
К раствору (3aS,4S,6R,6aR)-трет-бутил 4-(4-азидо-5H-пирроло[3,2-d]пиримидин-7-ил)-6-((((S)-2-((трет-бутоксикарбонил)амино)-4-метилпентаноил)окси)метил)-2,2-диметилдигидро-3aH-[1,3]диоксоло[4,5-c]пиррол-5(4H)-карбоксилата (32a) (584 мг, 0,91 ммоль) в метаноле (20 мл) добавляли 10%-ый палладий на угле (193 мг) и гидрировали при температуре 50 фунтов на квадратный дюйм в течение 2 ч. Катализатор фильтровали через слой из целита, и фильтрат концентрировали в вакууме. Полученный остаток очищали путем флеш-хроматографии на колонке (силикагель, 4 г) с получением (3aS,4S,6R,6aR)-трет-бутил 4-(4-амино-5H-пирроло[3,2-d]пиримидин-7-ил)-6-((((S)-2-((трет-бутоксикарбонил)амино)-4-метилпентаноил)окси)метил)-2,2-диметилдигидро-3aH-[1,3]диоксоло[4,5-c]пиррол-5(4H)-карбоксилата (32b) (300 мг, 53,5% выход) в виде твердого вещества белого цвета; 1H ЯМР (300 МГц, ДМСО-d6) δ 10,86 (с, 1H, способный к обмену), 8,09 (д, J=5,0 Гц, 1H), 7,34 (д, J=2,4 Гц, 1H), 7,22 (с, 1H), 6,78 (с, 2H, способный к обмену), 5,23 (д, J=5,3 Гц, 1H), 5,14 (с, 1H), 4,87 (д, J=5,7 Гц, 1H), 4,22-4,05 (м, 3H), 3,99-3,86 (м, 1H), 3,17 (д, J=5,2 Гц, 1H), 1,55 (дд, J=12,5, 6,0 Гц, 1H), 1,46-1,34 (м, 22H), 1,28 (с, 3H), 0,81 (дд, J=9,1, 6,7 Гц, 6H); MS (ES+) 619,13(M+1), 642,15 (M+Na); (ES-) 617,18(M-1), 653,27 (M+Cl); Анализ: Вычислено для C30H44N8O8·0,5H2O: C, 57,40; H, 7,55; N, 13,39; Найдено: C, 57,46; H, 7,53; N, 13,13.
Пример 5: (2S,2'S)-(2S,3S,4R,5R)-2-(4-амино-5H-пирроло[3,2-d]пиримидин-7-ил)-5-(гидроксиметил)пирролидин-3,4-диил бис(2-амино-3-метилбутаноат) (38c)
К раствору (2S,2'S)-(2S,3S,4R,5R)-2-(4-амино-5H-пирроло[3,2-d]пиримидин-7-ил)-1-(трет-бутоксикарбонил)-5-((тритилокси)метил)пирролидин-3,4-диил бис(2-((трет-бутоксикарбонил)амино)-3-метилбутаноата) (38b) (715 мг, 0,711 ммоль) в ацетоне (25 мл) добавляли 9 M серную кислоту (0,395 мл, 3,55 ммоль) и перемешивали при комнатной температуре в течение ночи. Ацетоновый слой декантировали, и остаток обрабатывали ацетоном и декантировали (3 раза). Полученный остаток очищали путем флеш-хроматографии на колонке (силикагель 12 г, элюируя смесью 0-100% смA-50 в смA-80) с получением (2S,2'S)-(2S,3S,4R,5R)-2-(4-амино-5H-пирроло[3,2-d]пиримидин-7-ил)-5-(гидроксиметил)пирролидин-3,4-диил бис(2-амино-3-метилбутаноата) (38c) (188 мг, 57%) в виде твердого вещества белого цвета; 1H ЯМР (300 МГц, ДМСО-d6) δ 10,84 (ушир. с, 1H, D2O способный к обмену), 8,06 (с, 1H), 7,51 (д, J=2,3 Гц, 1H), 6,77 (с, 2H, D2O способный к обмену), 5,33 (дд, J=7,6, 5,6 Гц, 1H), 5,24 (дд, J=5,7, 3,8 Гц, 1H), 4,39 (д, J=7,6 Гц, 1H), 3,64-3,51 (м, 2H), 3,17 (дд, J=4,5, 2,9 Гц, 2H), 3,06 (д, J=4,9 Гц, 1H), 2,01-1,88 (м, 1H), 1,87-1,75 (м, 1H), 0,91 (д, J=6,8 Гц, 3H), 0,84 (д, J=6,7 Гц, 3H), 0,79 (д, J=6,8 Гц, 3H), 0,73 (д, J=6,8 Гц, 3H); MS (ES+) 928,2 (2M+1); (ES-) 462,0 (M-1), 925,1 (2M-1).
Получение (2S,2'S)-(2S,3S,4R,5R)-2-(4-амино-5H-пирроло[3,2-d]пиримидин-7-ил)-1-(трет-бутоксикарбонил)-5-((тритилокси)метил)пирролидин-3,4-диил бис(2-((трет-бутоксикарбонил)амино)-3-метилбутаноата) (38b)
Стадия 1: (2S,3S,4R,5R)-трет-бутил 2-(4-хлор-5H-пирроло[3,2-d]пиримидин-7-ил)-3,4-дигидрокси-5-(гидроксиметил)пирролидин-1-карбоксилат (34a)
К раствору (2R,3R,4S,5S)-2-(ацетоксиметил)-1-(трет-бутоксикарбонил)-5-(4-хлор-5H-пирроло[3,2-d]пиримидин-7-ил)пирролидин-3,4-диил диацетата (28c) (25,8 г, 50,5 ммоль) в метаноле (200 мл) при комнатной температуре добавляли метоксид натрия 25 масс.% в метаноле (3,6 мл, 16,66 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь концентрировали в вакууме досуха и очищали путем флеш-хроматографии на колонке (силикагель 600 г) с получением (2S,3S,4R,5R)-трет-бутил 2-(4-хлор-5H-пирроло[3,2-d]пиримидин-7-ил)-3,4-дигидрокси-5-(гидроксиметил)пирролидин-1-карбоксилата (34a) (17,7 г, 91% выход) в виде бесцветной пены; 1H ЯМР (300 МГц, ДМСО-d6) δ 12,34 (с, 1H), 8,62 (с, 1H), 7,94 (с, 1H), 5,40-5,02 (м, 2H), 4,96-4,70 (м, 2H), 4,41-4,25 (м, 1H), 4,13-3,93 (м, 2H), 3,69-3,51 (м, 2H), 1,35 (с, 3H), 1,01 (с, 6H); MS (ES+) 384,9 (M+1), 792,6 (2M+Na); (ES-) 382,6 (M-1).
Стадия 2: Получение (2S,3S,4R,5R)-трет-бутил 2-(4-азидо-5H-пирроло[3,2-d]пиримидин-7-ил)-3,4-дигидрокси-5-(гидроксиметил)пирролидин-1-карбоксилата (34b)
К раствору (2S,3S,4R,5R)-трет-бутил 2-(4-хлор-5H-пирроло[3,2-d]пиримидин-7-ил)-3,4-дигидрокси-5-(гидроксиметил)пирролидин-1-карбоксилата (34a) (4 г, 10,39 ммоль) в ДМФ (80 мл) добавляли азид натрия (3,38 г, 52,0 ммоль) и нагревали с перемешиванием при температуре 80°C в течение 10 ч. Реакцию охлаждали до температуры 25°C, выливали на лед и экстрагировали с помощью этилацетата. Этилацетатный слой отделяли, промывали водой, насыщенным солевым раствором, сушили, фильтровали и концентрировали в вакууме досуха (водяная баня 50°C). Полученный сырой остаток очищали путем флеш-хроматографии на колонке (силикагель 120 г, элюируя смесью метанол в хлороформе 0-100%) с получением (2S,3S,4R,5R)-трет-бутил 2-(4-азидо-5H-пирроло[3,2-d]пиримидин-7-ил)-3,4-дигидрокси-5-(гидроксиметил)пирролидин-1-карбоксилата (34b) (1,28 г, 31% выход) в виде твердого вещества белого цвета; 1H ЯМР (300 МГц, ДМСО-d6) δ 13,05 (ушир. с, 1H, D2O способный к обмену), 9,84 (с, 1H), 7,81 (м, 1H), 5,13 (м, 1H), 5,05-4,83 (м, 3H, D2O способный к обмену), 4,24 (м, 1H), 4,09 (м, 1H), 4,03 (м, 1H), 3,59 (м, 2H), 1,38 (с, 4H для Boc) и 1,05 (с, 5H для Boc); MS (ES+) 782,8 (2M+1), (ES-) 389,6 (M-1).
Стадия 3: Получение (2S,3S,4R,5R)-трет-бутил 2-(4-азидо-5H-пирроло[3,2-d]пиримидин-7-ил)-3,4-дигидрокси-5-((тритилокси)метил)пирролидин-1-карбоксилата (35a)
К раствору (2S,3S,4R,5R)-трет-бутил 2-(4-азидо-5H-пирроло[3,2-d]пиримидин-7-ил)-3,4-дигидрокси-5-(гидроксиметил)пирролидин-1-карбоксилата (34b) (1,0 г, 2,55 ммоль) в пиридине (5,0 мл, 62,06 ммоль) добавляли хлортрифенилметан (0,85 г, 3,07 ммоль). Полученную смесь перемешивали при температуре 50°C в течение 4 ч, в это время реакция подходила к завершению (ТСХ в смеси 9:1 хлороформ:метанол). Реакционную смесь охлаждали до температуры 25°C, выливали в ледяную воду (80 мл) и экстрагировали с помощью этилацетата (100 мл, 2 × 60 мл). Органические слои объединяли, промывали водой, насыщенным солевым раствором, сушили, фильтровали и концентрировали в вакууме с получением твердого вещества не совсем белого цвета. Твердое вещество обрабатывали 5% EtOAc в н-гексане и собирали путем фильтрации с получением (2S,3S,4R,5R)-трет-бутил 2-(4-азидо-5H-пирроло[3,2-d]пиримидин-7-ил)-3,4-дигидрокси-5-((тритилокси)метил)пирролидин-1-карбоксилата (35a) (1,47 г, 90,74% выход) в виде твердого вещества бледно-желтого цвета; 1H ЯМР (300 МГц, ДМСО-d6, 370 K) δ 12,82 (с, 1H), 9,48 (с, 1H), 7,44 (с, 1H), 7,36 (д, J=8,0 Гц, 5H), 7,29-7,19 (м,9H), 4,96 (д, J=4,6 Гц, 1H), 4,72 (д, J=5,3 Гц, 1H), 4,59 (д, J=5,0 Гц, 1H), 4,54-4,46 (м, 1H), 4,38-4,30 (м, 1H), 4,08-3,99 (м, 1H), 3,93-3,84 (м, 1H), 3,46 (дд, J=9,1, 6,4 Гц, 1H), 3,37 (дд, J=9,2, 4,2 Гц, 1H), 1,19 (с, 9H); MS (ES+) 655,85 (M+Na), (ES-) 632,55 (M-1). ИК (KBr) 2133 см-1.
Стадия 4: Получение (2S,3S,4R,5R)-трет-бутил 2-(4-азидо-5H-пирроло[3,2-d]пиримидин-7-ил)-3-(((S)-2-((трет-бутоксикарбонил)амино)-3-метилбутаноил)окси)-4-гидрокси-5-((тритилокси)метил)пирролидин-1-карбоксилата (39a); (2S,3S,4R,5R)-трет-бутил 2-(4-азидо-5H-пирроло[3,2-d]пиримидин-7-ил)-4-(((S)-2-((трет-бутоксикарбонил)амино)-3-метилбутаноил)окси)-3-гидрокси-5-((тритилокси)метил)пирролидин-1-карбоксилата (39b) и (2S,2'S)-(2S,3S,4R,5R)-2-(4-азидо-5H-пирроло[3,2-d]пиримидин-7-ил)-1-(трет-бутоксикарбонил)-5-((тритилокси)метил)пирролидин-3,4-диил бис(2-((трет-бутоксикарбонил)амино)-3-метилбутаноата) (38a)
Способ 1:
К раствору (2S,3S,4R,5R)-трет-бутил 2-(4-азидо-5H-пирроло[3,2-d]пиримидин-7-ил)-3,4-дигидрокси-5-((тритилокси)метил)пирролидин-1-карбоксилата (35a) (1 г, 1,578 ммоль) и (S)-2-(трет-бутоксикарбониламино)-3-метилбутановой кислоты (L-Boc валин 0,343 г, 1,58 ммоль) в ДМФ (10 мл) при комнатной температуре добавляли гидрохлорид N1-((этилимино)метилен)-N3,N3-диметилпропан-1,3-диамина (EDCI, 0,756 г, 3,95 ммоль) и N,N-диметилпиридин-4-амин (DMAP, 0,193 г, 1,578 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. ТСХ анализ (10% хлороформ в метаноле) показывал небольшое количество непрореагировавшего (2S,3S,4R,5R)-трет-бутил 2-(4-азидо-5H-пирроло[3,2-d]пиримидин-7-ил)-3,4-дигидрокси-5-((тритилокси)метил)пирролидин-1-карбоксилата (35a). Реакцию гасили водой (50 мл) и экстрагировали с помощью этилацетата (3 × 100 мл). Органические слои объединяли, промывали водой, насыщенным солевым раствором (100 мл), сушили, фильтровали и концентрировали в вакууме. Полученный сырой остаток очищали путем флеш-хроматографии на колонке (силикагель 40 г) с получением: 2-(4-азидо-5H-пирроло[3,2-d]пиримидин-7-ил)-3,4-дигидрокси-5-((тритилокси)метил)пирролидин-1-карбоксилата (35a) (196 мг, 19,6%) в виде твердого вещества белого цвета;
(2S,3S,4R,5R)-трет-бутил 2-(4-азидо-5H-пирроло[3,2-d]пиримидин-7-ил)-3-(((S)-2-((трет-бутоксикарбонил)амино)-3-метилбутаноил)окси)-4-гидрокси-5-((тритилокси)метил)пирролидин-1-карбоксилат (39a) (511 мг, 38,9%) в виде твердого вещества белого цвета; 1H ЯМР (300 МГц, ДМСО-d6, 370K) δ 12,86 (с, 1H), 9,39 (с, 1H), 7,47 (с, 1H), 7,38 (д, J=7,9 Гц, 6H), 7,31-7,18 (м, 9H), 6,57 (д, J=7,3 Гц, 1H), 5,46 (с, 1H), 5,05 (д, J=6,2 Гц, 1H), 4,94 (д, J=6,3 Гц, 1H), 4,78-4,69 (м, 1H),4,13-4,05 (м, 2H), 3,53-3,36 (м, 2H), 2,19-2,04 (м, 1H), 1,42 (с, 9H), 1,17 (с, 9H), 0,92 (т, J=6,7 Гц, 6H); ИК (KBr) 2133 см-1; MS (ES-) 831,1 (M-1);
(2S,3S,4R,5R)-трет-бутил 2-(4-азидо-5H-пирроло[3,2-d]пиримидин-7-ил)-4-(((S)-2-((трет-бутоксикарбонил)амино)-3-метилбутаноил)окси)-3-гидрокси-5-((тритилокси)метил)пирролидин-1-карбоксилат (39b) (250 мг, 19%) в виде твердого вещества белого цвета; 1H ЯМР (300 МГц, ДМСО-d6, 370K) δ 12,84 (с, 1H), 9,49 (с, 1H), 7,51 (с, 1H), 7,36-7,28 (м, 6H), 7,25-7,17 (м, 9H), 6,48 (д, J=7,6 Гц, 1H), 5,81-5,71 (м, 1H), 5,22 (д, J=4,2 Гц, 1H), 5,00 (д, J=6,0 Гц, 1H), 4,71 (т, J=8,1 Гц, 1H), 3,91 (м, 2H), 3,56-3,32 (м, 2H), 2,10-2,00 (м, 1H),1,32 (с, 9H), 1,22 (с, 9H), 0,88 (дд, J=6,6, 3,4 Гц, 6H); ИК (KBr) 2134 см-1; MS (ES-) 831,1(M-1); и
(2S,2'S)-(2S,3S,4R,5R)-2-(4-азидо-5H-пирроло[3,2-d]пиримидин-7-ил)-1-(трет-бутоксикарбонил)-5-((тритилокси)метил)пирролидин-3,4-диил бис(2-((трет-бутоксикарбонил)амино)-3-метилбутаноат) (38a) (18 мг, 1,1%) в виде твердого вещества белого цвета; 1H ЯМР (300 МГц, ДМСО, 370K) δ 12,94 (с, 1H, N-H), 9,38 (с, 1H), 7,54 (с, 1H), 7,38-7,14 (м, 15H), 6,51 (с, 1H, N-H), 6,37 (с, 1H, N-H), 5,97 (д, J=17,2 Гц, 1H), 5,76 (с, 1H), 5,22 (т, J=11,3 Гц, 1H), 4,19-3,98 (м, 2H), 3,91 (д, J=5,3 Гц, 1H), 3,55 (д, J=18,8 Гц, 1H), 3,35 (м, 1H), 2,06 (м, 2H), 1,37 (с, 9H), 1,24 (с, 9H), 1,21 (с, 9H), 0,94-0,78 (м, 12H).
Способ 2:
К раствору (2S,3S,4R,5R)-трет-бутил 2-(4-азидо-5H-пирроло[3,2-d]пиримидин-7-ил)-3,4-дигидрокси-5-((тритилокси)метил)пирролидин-1-карбоксилата (35a) (1,0 г, 1,58 ммоль) и (S)-2-(трет-бутоксикарбониламино)-3-метилбутановой кислоты (L-Boc валин, 0,720 г, 3,31 ммоль) в ДМФ (10 мл) при комнатной температуре добавляли гидрохлорид N1-((этилимино)метилен)-N3,N3-диметилпропан-1,3-диамина (EDCI, 0,756 г, 3,95 ммоль) и N,N-диметилпиридин-4-амин (DMAP, 0,193 г, 1,578 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи и гасили водой (30 мл). Реакционную смесь экстрагировали с помощью этилацетата (3 × 60 мл). Органические слои объединяли, промывали водой, насыщенным солевым раствором (50 мл), сушили, фильтровали и концентрировали в вакууме. Полученный сырой остаток очищали путем флеш-хроматографии на колонке (силикагель 25 г, элюируя смесью этилацетат в гексане 0-50%) с получением (2S,2'S)-(2S,3S,4R,5R)-2-(4-азидо-5H-пирроло[3,2-d]пиримидин-7-ил)-1-(трет-бутоксикарбонил)-5-((тритилокси)метил)пирролидин-3,4-диил бис(2-((трет-бутоксикарбонил)амино)-3-метилбутаноата) (38a) (670 мг, 41,2% выход) в виде пены белого цвета, плюс смесь, содержащая (2S,3S,4R,5R)-трет-бутил 2-(4-азидо-5H-пирроло[3,2-d]пиримидин-7-ил)-3-(((S)-2-((трет-бутоксикарбонил)амино)-3-метилбутаноил)окси)-4-гидрокси-5-((тритилокси)метил)пирролидин-1-карбоксилат (39a) и (2S,3S,4R,5R)-трет-бутил 2-(4-азидо-5H-пирроло[3,2-d]пиримидин-7-ил)-4-(((S)-2-((трет-бутоксикарбонил)амино)-3-метилбутаноил)окси)-3-гидрокси-5-((тритилокси)метил)пирролидин-1-карбоксилат (39b) (610 мг, 47,9%) в виде пены белого цвета; MS (ES-) 831,5 (M-1).
Стадия 5: Получение (2S,2'S)-(2S,3S,4R,5R)-2-(4-амино-5H-пирроло[3,2-d]пиримидин-7-ил)-1-(трет-бутоксикарбонил)-5-((тритилокси)метил)пирролидин-3,4-диил бис(2-((трет-бутоксикарбонил)амино)-3-метилбутаноата) (38b)
К раствору (2S,2'S)-(2S,3S,4R,5R)-2-(4-азидо-5H-пирроло[3,2-d]пиримидин-7-ил)-1-(трет-бутоксикарбонил)-5-((тритилокси)метил)пирролидин-3,4-диил бис(2-((трет-бутоксикарбонил)амино)-3-метилбутаноата) (38a) (964 мг, 0,934 ммоль) в этаноле (25 мл) добавляли Pd/C (10%) (150 мг) и гидрировали под давлением 50 фунтов на квадратный дюйм в течение ночи. Катализатор удаляли путем фильтрации через слой из целита, и фильтрат концентрировали в вакууме. Полученный остаток очищали путем флеш-хроматографии на колонке (силикагель 4 г, элюируя смесью (этилацетат/метанол, 9:1) в гексане, 0-100%) с получением (2S,2'S)-(2S,3S,4R,5R)-2-(4-амино-5H-пирроло[3,2-d]пиримидин-7-ил)-1-(трет-бутоксикарбонил)-5-((тритилокси)метил)пирролидин-3,4-диил бис(2-((трет-бутоксикарбонил)амино)-3-метилбутаноата) (38b) (765 мг, 81% выход) в виде твердого вещества белого цвета; 1H ЯМР (300 МГц, ДМСО-d6, 370K) δ 10,61 (с, 1H, N-H), 7,86 (д, J=2,2 Гц, 1H), 7,37-7,17 (м, 16H), 6,38 (м, 2H, N-H), 6,30 (с, 2H, N-H), 6,16 (т, J=5,1 Гц, 1H), 5,87 (т, J=3,8 Гц, 1H), 5,06 (д, J=5,9 Гц, 1H), 4,12-4,00 (м, 2H), 3,98-3,86 (м, 1H), 3,78 (дд, J=9,7, 6,9 Гц, 1H), 3,20 (м, 1H), 2,05 (м, 2H), 1,40 (с, 9H), 1,31 (с, 9H), 1,23 (д, J=2,0 Гц, 9H), 0,90-0,80 (м, 12H); Анализ: Вычислено для C55H71N7O11.H2O: C, 64,50; H, 7,18; N, 9,57; Найдено: C, 64,36; H, 7,11; N, 9,38.
Пример 6: Получение (S)-(2R,3R,4S,5S)-5-(4-амино-5H-пирроло[3,2-d]пиримидин-7-ил)-4-гидрокси-2-(гидроксиметил)пирролидин-3-ил 2-амино-3-метилбутаноата (35e) и (S)-(2S,3S,4R,5R)-2-(4-амино-5H-пирроло[3,2-d]пиримидин-7-ил)-4-гидрокси-5-(гидроксиметил)пирролидин-3-ил 2-амино-3-метилбутаноата (34f)
Способ 1:
Исходя из (2S,3S,4R,5R)-трет-бутил 2-(4-амино-5H-пирроло[3,2-d]пиримидин-7-ил)-3-(((S)-2-((трет-бутоксикарбонил)амино)-3-метилбутаноил)окси)-4-гидрокси-5-((тритилокси)метил)пирролидин-1-карбоксилата (39d) и (2S,3S,4R,5R)-трет-бутил 2-(4-амино-5H-пирроло[3,2-d]пиримидин-7-ил)-4-(((S)-2-((трет-бутоксикарбонил)амино)-3-метилбутаноил)окси)-3-гидрокси-5-((тритилокси)метил)пирролидин-1-карбоксилата (39e)
К раствору (2S,3S,4R,5R)-трет-бутил 2-(4-амино-5H-пирроло[3,2-d]пиримидин-7-ил)-3-(((S)-2-((трет-бутоксикарбонил)амино)-3-метилбутаноил)окси)-4-гидрокси-5-((тритилокси)метил)пирролидин-1-карбоксилата (39d) и (2S,3S,4R,5R)-трет-бутил 2-(4-амино-5H-пирроло[3,2-d]пиримидин-7-ил)-4-(((S)-2-((трет-бутоксикарбонил)амино)-3-метилбутаноил)окси)-3-гидрокси-5-((тритилокси)метил)пирролидин-1-карбоксилата (39e) (744 мг, 0,922 ммоль) в ацетоне (15 мл) добавляли 9 M серную кислоту (0,512 мл, 4,61 ммоль) и перемешивали при комнатной температуре в течение ночи. Растворитель декантировали, и твердое вещество белого цвета промывали ацетоном и перемешивали в течение 30 мин, затем вновь декантировали. Тот же самый способ повторяли 3-4 раза, полученное твердое вещество собирали путем фильтрации, промывали ацетоном, сушили в вакууме при температуре 35°C с получением смеси (S)-(2R,3R,4S,5S)-5-(4-амино-5H-пирроло[3,2-d]пиримидин-7-ил)-4-гидрокси-2-(гидроксиметил)пирролидин-3-ил 2-амино-3-метилбутаноата (35e) и (S)-(2S,3S,4R,5R)-2-(4-амино-5H-пирроло[3,2-d]пиримидин-7-ил)-4-гидрокси-5-(гидроксиметил)пирролидин-3-ил 2-амино-3-метилбутаноата (34f) в виде сульфатной соли (500 мг, 1,081 ммоль, 97% выход) в виде твердого вещества белого цвета. Очистка с использованием флеш-хроматографии на колонке (233 мг образца смеси, силикагель, элюируя смесью 0-100% СМA-50 в СМA-80) давала смесь (S)-(2R,3R,4S,5S)-5-(4-амино-5H-пирроло[3,2-d]пиримидин-7-ил)-4-гидрокси-2-(гидроксиметил)пирролидин-3-ил 2-амино-3-метилбутаноата (35e) и (S)-(2S,3S,4R,5R)-2-(4-амино-5H-пирроло[3,2-d]пиримидин-7-ил)-4-гидрокси-5-(гидроксиметил)пирролидин-3-ил 2-амино-3-метилбутаноата (34f) (74 мг, 48%) в виде твердого вещества белого цвета; 1H ЯМР (300 МГц, ДМСО-d6) δ [8,08 (с, 0,65H), 8,07 (с, 0,35H) 1H], [7,49 (с, 0,35H), 7,48 (с, 0,65H) 1H], [5,09 (т, J=6,4 Гц, 0,35H), 5,01 (дд, J=5,7, 3,7 Гц, 0,65H) 1H], [4,35 (д, J=6,7 Гц, 0,35H), 4,21 (д, J=5,6 Гц, 0,35H), 4,18 (д, J=5,7 Гц, 0,65H), 4,14-4,09 (м, 1,65H) 3H], [3,63-3,48 (м, 1H) 1H], [3,25 (д, J=5,1 Гц, 0,7H), 3,20-3,10 (м, 1,3H) 2H], [2,05-1,82 (м, 1H)], [0,93 (д, J=6,8 Гц, 1,95H), 0,88 (д, J=6,8 Гц, 1,95H), 0,81 (д, J=6,9 Гц, 1,05H), 0,77 (д, J=6,8 Гц, 1,05H) 6H]; MS (ES+) 365,0 (M+1).
Способ 2:
Исходя из (6aR,8S,9S,9aR)-трет-бутил 8-(4-амино-5H-пирроло[3,2-d]пиримидин-7-ил)-9-(((S)-2-((трет-бутоксикарбонил)амино)-3-метилбутаноил)окси)-2,2,4,4-тетраизопропилтетрагидро-[1,3,5,2,4]триоксадисилоцино[7,6-b]пиррол-7(8H)-карбоксилата (34e)
К перемешиваемому раствору (6aR,8S,9S,9aR)-трет-бутил 8-(4-амино-5H-пирроло[3,2-d]пиримидин-7-ил)-9-(((S)-2-((трет-бутоксикарбонил)амино)-3-метилбутаноил)окси)-2,2,4,4-тетраизопропилтетрагидро-[1,3,5,2,4]триоксадисилоцино[7,6-b]пиррол-7(8H)-карбоксилата (34e) (0,843 г, 1,04 ммоль) в ацетоне (10 мл) при комнатной температуре добавляли конц. серную кислоту (50% раствор в воде, 1,16 мл, 10,44 ммоль) и перемешивали в течение 18 ч. Реакционную смесь разбавляли ацетоном (30 мл) и перемешивали. Ацетон декантировали, и эту операцию повторяли два раза. Отделяющееся твердое вещество собирали путем фильтрации, сушили в вакууме с получением смеси (S)-(2R,3R,4S,5S)-5-(4-амино-5H-пирроло[3,2-d]пиримидин-7-ил)-4-гидрокси-2-(гидроксиметил)пирролидин-3-ил 2-амино-3-метилбутаноата (35e) и сульфатной соли (S)-(2S,3S,4R,5R)-2-(4-амино-5H-пирроло[3,2-d]пиримидин-7-ил)-4-гидрокси-5-(гидроксиметил)пирролидин-3-ил 2-амино-3-метилбутаноата (34f) (0,4 г, 68%) в виде твердого вещества белого цвета. Твердое вещество очищали путем флеш-хроматографии на колонке (силикагель 4 г, элюируя смесью 0-100% СМA-50 в СМA-80) с получением смеси (S)-(2R,3R,4S,5S)-5-(4-амино-5H-пирроло[3,2-d]пиримидин-7-ил)-4-гидрокси-2-(гидроксиметил)пирролидин-3-ил 2-амино-3-метилбутаноата (35e) и (S)-(2S,3S,4R,5R)-2-(4-амино-5H-пирроло[3,2-d]пиримидин-7-ил)-4-гидрокси-5-(гидроксиметил)пирролидин-3-ил 2-амино-3-метилбутаноата (34f); ЯМР анализ показывал смесь соединения 35e и 34f; MS (ES+) 365,1 (M+1), (ES-) 362,9 (M-1).
Получение (2S,3S,4R,5R)-трет-бутил 2-(4-амино-5H-пирроло[3,2-d]пиримидин-7-ил)-3-(((S)-2-((трет-бутоксикарбонил)амино)-3-метилбутаноил)окси)-4-гидрокси-5-((тритилокси)метил)пирролидин-1-карбоксилата (39d) и (2S,3S,4R,5R)-трет-бутил 2-(4-амино-5H-пирроло[3,2-d]пиримидин-7-ил)-4-(((S)-2-((трет-бутоксикарбонил)амино)-3-метилбутаноил)окси)-3-гидрокси-5-((тритилокси)метил)пирролидин-1-карбоксилата (39e)
Способ 1:
Исходя из (2S,3S,4R,5R)-трет-бутил 2-(4-азидо-5H-пирроло[3,2-d]пиримидин-7-ил)-3-(((S)-2-((трет-бутоксикарбонил)амино)-3-метилбутаноил)окси)-4-гидрокси-5-((тритилокси)метил)пирролидин-1-карбоксилата (39a)
К раствору (2S,3S,4R,5R)-трет-бутил 2-(4-азидо-5H-пирроло[3,2-d]пиримидин-7-ил)-3-(((S)-2-((трет-бутоксикарбонил)амино)-3-метилбутаноил)окси)-4-гидрокси-5-((тритилокси)метил)пирролидин-1-карбоксилата (39a) (1,319 г, 1,584 ммоль) в этаноле (50 мл) добавляли 10% Pd/C (200 мг) и гидрировали под давлением 50 фунтов на квадратный дюйм в течение 8 ч. Катализатор удаляли путем фильтрации реакционной смеси через слой из целита. Фильтрат концентрировали в вакууме, и полученный остаток очищали путем флеш-хроматографии на колонке с получением смеси 3:2 (анализ путем ЯМР) (2S,3S,4R,5R)-трет-бутил 2-(4-амино-5H-пирроло[3,2-d]пиримидин-7-ил)-3-(((S)-2-((трет-бутоксикарбонил)амино)-3-метилбутаноил)окси)-4-гидрокси-5-((тритилокси)метил)пирролидин-1-карбоксилата (39d) и (2S,3S,4R,5R)-трет-бутил 2-(4-амино-5H-пирроло[3,2-d]пиримидин-7-ил)-4-(((S)-2-((трет-бутоксикарбонил)амино)-3-метилбутаноил)окси)-3-гидрокси-5-((тритилокси)метил)пирролидин-1-карбоксилата (39e) (945 мг, 1,171 ммоль, 74,0% выход) в виде твердого вещества белого цвета; MS (ES+) 806,9 (M+1); (ES-) 805,0 (M-1), 841,2 (M+Cl).
Способ 2:
Исходя из (2S,3S,4R,5R)-трет-бутил 2-(4-азидо-5H-пирроло[3,2-d]пиримидин-7-ил)-4-(((S)-2-((трет-бутоксикарбонил)амино)-3-метилбутаноил)окси)-3-гидрокси-5-((тритилокси)метил)пирролидин-1-карбоксилата (39b)
К раствору (2S,3S,4R,5R)-трет-бутил 2-(4-азидо-5H-пирроло[3,2-d]пиримидин-7-ил)-4-(((S)-2-((трет-бутоксикарбонил)амино)-3-метилбутаноил)окси)-3-гидрокси-5-((тритилокси)метил)пирролидин-1-карбоксилата (39b) (634 мг, 0,761 ммоль) в этаноле (25 мл) добавляли 10% Pd/C (100 мг) и гидрировали под давлением 50 фунтов на квадратный дюйм в течение 8 ч. Катализатор удаляли путем фильтрации реакционной смеси через слой из целита. Фильтрат концентрировали в вакууме, и полученный остаток очищали путем флеш-хроматографии на колонке с получением смеси 3:2 смесь (2S,3S,4R,5R)-трет-бутил 2-(4-амино-5H-пирроло[3,2-d]пиримидин-7-ил)-3-(((S)-2-((трет-бутоксикарбонил)амино)-3-метилбутаноил)окси)-4-гидрокси-5-((тритилокси)метил)пирролидин-1-карбоксилата (39d) и (2S,3S,4R,5R)-трет-бутил 2-(4-амино-5H-пирроло[3,2-d]пиримидин-7-ил)-4-(((S)-2-((трет-бутоксикарбонил)амино)-3-метилбутаноил)окси)-3-гидрокси-5-((тритилокси)метил)пирролидин-1-карбоксилата (39e) (474 мг, 0,587 ммоль, 77% выход) в виде твердого вещества белого цвета; ЯМР-спектр соответствовал продукту, полученному с использованием способа для получения соединения 39a; MS (ES+) 806,9 (M+1); (ES-) 805,7 (M-1).
Получение (6aR,8S,9S,9aR)-трет-бутил 8-(4-амино-5H-пирроло[3,2-d]пиримидин-7-ил)-9-(((S)-2-((трет-бутоксикарбонил)амино)-3-метилбутаноил)окси)-2,2,4,4-тетраизопропилтетрагидро-[1,3,5,2,4]триоксадисилоцино[7,6-b]пиррол-7(8H)-карбоксилата (34e)
Стадия 1: Получение (6aR,8S,9S,9aR)-трет-бутил 8-(4-азидо-5H-пирроло[3,2-d]пиримидин-7-ил)-9-гидрокси-2,2,4,4-тетраизопропилтетрагидро-[1,3,5,2,4]триоксадисилоцино[7,6-b]пиррол-7(8H)-карбоксилата (34c)
К перемешиваемому раствору (2S,3S,4R,5R)-трет-бутил 2-(4-азидо-5H-пирроло[3,2-d]пиримидин-7-ил)-3,4-дигидрокси-5-(гидроксиметил)пирролидин-1-карбоксилата (34b) (5 г, 12,78 ммоль) в ДМФ (25 мл) добавляли N,N-диметилпиридин-4-амин (DMAP, 0,078 г, 0,639 ммоль), 1H-имидазол (3,48 г, 51,1 ммоль) и 1,3-дихлор-1,1,3,3-тетраизопропилдисилоксан (4,63 мл, 14,05 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи и разбавляли водой (300 мл). Выделившееся твердое вещество собирали путем фильтрации и промывали водой. Твердое вещество очищали путем флеш-хроматографии на колонке (силикагель, элюируя смесью этилацетата в гексане от 0 до 35%) с получением (6aR,8S,9S,9aR)-трет-бутил 8-(4-азидо-5H-пирроло[3,2-d]пиримидин-7-ил)-9-гидрокси-2,2,4,4-тетраизопропилтетрагидро-[1,3,5,2,4]триоксадисилоцино[7,6-b]пиррол-7(8H)-карбоксилата (34c) (5,02 г, 62,0% выход) в виде твердого вещества белого цвета; 1H ЯМР (300 МГц, ДМСО-d6) δ 13,26 (с, 1H, D2O способный к обмену), 9,85 (с, 1H), 7,49 (с, 1H), 5,52 (д, J=3,2 Гц, 1H, D2O способный к обмену), 5,10 (с, 1H), 4,63-4,24 (м, 2H), 4,18-3,82 (м, 1H), 3,67 (дт, J=8,2, 2,9 Гц, 1H), 1,37 (д, J=45,1 Гц, 10H), 1,08-0,99 (м, 14H), 0,86 (м, 14H); MS (ES+) 633,9 (M+1); (ES-) 632,2 (M-1); Анализ: Вычислено для C28H47N7O6Si2: C, 53,05; H, 7,47; N, 15,47; Найдено: C, 53,00; H, 7,55; N, 15,15.
Стадия 2: Получение (6aR,8S,9S,9aR)-трет-бутил 8-(4-азидо-5H-пирроло[3,2-d]пиримидин-7-ил)-9-(((S)-2-((трет-бутоксикарбонил)амино)-3-метилбутаноил)окси)-2,2,4,4-тетраизопропилтетрагидро-[1,3,5,2,4]триоксадисилоцино[7,6-b]пиррол-7(8H)-карбоксилата (34d)
К перемешиваемому раствору (6aR,8S,9S,9aR)-трет-бутил 8-(4-азидо-5H-пирроло[3,2-d]пиримидин-7-ил)-9-гидрокси-2,2,4,4-тетраизопропилтетрагидро-[1,3,5,2,4]триоксадисилоцино[7,6-b]пиррол-7(8H)-карбоксилата (34c) (2 г, 3,16 ммоль) в ДМФ (20 мл) добавляли (S)-2-(трет-бутоксикарбониламино)-3-метилбутановую кислоту (L-Boc Валин, 1,03 г, 4,73 ммоль) и охлаждали до температуры 0°C. При температуре 0°C добавляли гидрохлорид N1-((этилимино)метилен)-N3,N3-диметилпропан-1,3-диамина (EDCI, 1,51 г, 7,89 ммоль) и N,N-диметилпиридин-4-амин (DMAP, 0,385 г, 3,16 ммоль), и в течение ночи давали реакционной смеси вернуться к комнатной температуре. Реакционную смесь разбавляли водой (100 мл) и экстрагировали с помощью этилацетата (2 × 100 мл). Объединенные этилацетатные слои промывали водой (2 × 50 мл), насыщенным солевым раствором (50 мл), сушили, фильтровали и концентрировали в вакууме. Полученный остаток очищали путем флеш-хроматографии на колонке (силикагель 80 г, элюируя смесью этилацетата в гексане от 0 до 35%) с получением (6aR,8S,9S,9aR)-трет-бутил 8-(4-азидо-5H-пирроло[3,2-d]пиримидин-7-ил)-9-(((S)-2-((трет-бутоксикарбонил)амино)-3-метилбутаноил)окси)-2,2,4,4-тетраизопропилтетрагидро-[1,3,5,2,4]триоксадисилоцино[7,6-b]пиррол-7(8H)-карбоксилата (34d) (2,1 г, 80%) в виде твердого вещества белого цвета; 1H ЯМР (300 МГц, ДМСО-d6) δ 13,40 (с, 1H, D2O способный к обмену), 9,85 (д, J=5,7 Гц, 1H), 7,57 (с, 1H), 7,23 (д, J=8,2 Гц, 1H), 5,84 (д, J=3,7 Гц, 1H), 5,24 (д, J=10,7 Гц, 1H), 4,82-4,58 (м, 1H), 4,41 (д, J=12,4 Гц, 1H), 4,03-3,93 (м, 2H), 3,67 (с, 1H), 2,08 (дт, J=13,5, 6,8 Гц, 1H), 1,49-1,33 (м, 18H), 1,15-0,67 (м, 35H); MS (ES+) 856,0 (M+Na), 832,4 (M-1). Анализ: Вычислено для C38H64N8O9Si2: C, 54,77; H, 7,74; N, 13,45; Найдено: C, 54,86; H, 7,78; N, 13,13.
Стадия 3: Получение (6aR,8S,9S,9aR)-трет-бутил 8-(4-амино-5H-пирроло[3,2-d]пиримидин-7-ил)-9-(((S)-2-((трет-бутоксикарбонил)амино)-3-метилбутаноил)окси)-2,2,4,4-тетраизопропилтетрагидро-[1,3,5,2,4]триоксадисилоцино[7,6-b]пиррол-7(8H)-карбоксилата (34e)
К суспензии палладия на угле (10%, 0,262 г) в этаноле (50 мл) добавляли (6aR,8S,9S,9aR)-трет-бутил 8-(4-азидо-5H-пирроло[3,2-d]пиримидин-7-ил)-9-(((S)-2-((трет-бутоксикарбонил)амино)-3-метилбутаноил)окси)-2,2,4,4-тетраизопропилтетрагидро-[1,3,5,2,4]триоксадисилоцино[7,6-b]пиррол-7(8H)-карбоксилат (34d) (2,05 г, 2,46 ммоль) и гидрировали под давлением 60 фунтов на квадратный дюйм в течение 12 ч. Катализатор удаляли путем фильтрации через слой из целита, и фильтрат концентрировали в вакууме. Полученный остаток очищали путем флеш-хроматографии на колонке (силикагель 25 г, элюируя смесью смA 80 в хлороформе 0 до 100%) с получением (6aR,8S,9S,9aR)-трет-бутил 8-(4-амино-5H-пирроло[3,2-d]пиримидин-7-ил)-9-(((S)-2-((трет-бутоксикарбонил)амино)-3-метилбутаноил)окси)-2,2,4,4-тетраизопропилтетрагидро-[1,3,5,2,4]триоксадисилоцино[7,6-b]пиррол-7(8H)-карбоксилата (34e) (1,9 г, 96% выход) в виде бесцветного твердого продукта; 1H ЯМР (300 МГц, ДМСО-d6) δ 10,94 (с, 1H, D2O способный к обмену), 8,05 (с, 1H), 7,46-7,30 (м, 1H), 7,17 (д, J=8,0 Гц, 1H), 6,82 (с, 2H, D2O способный к обмену), 5,86 (д, J=4,0 Гц, 1H), 5,16 (д, J=25,0 Гц, 1H, D2O способный к обмену), 5,00 (д, J=8,2 Гц, 1H), 4,17 (дд, J=12,2, 5,1 Гц, 1H), 3,98-3,88 (м, 1H), 3,61 (с, 1H), 2,16-1,92 (м, 1H), 1,41 (ушир. с, 18H), 1,01-0,83 (м, 35H); MS (ES+) 806,921 (M+1), 830,1 (M+Na), (ES-) 805,289 (M-1), 842,0 (M+Cl).
Пример 7: Фармакокинетика соединения 30f после перорального введения крысам
Здоровых 8-10-недельных самцов крыс линии Sprague-Dawley случайным образом разделяли на контрольную и экспериментальную группы, по N=4 в группе. Всех животных помещали в клетки и кормили в соответствии со стандартными процедурами. В день эксперимента всех животных изолировали в метаболических клетках и кормили приблизительно за 15 часов до введения дозы. Через два часа после введения дозы с контрольным или экспериментальным агентом, кормление животных возобновляли. Водой обеспечивали в неограниченном количестве. Непосредственно перед введением контрольное соединение дигидрохлорид (2S,3S,4R,5R)-2-(4-амино-5H-пирроло[3,2-d]пиримидин-7-ил)-5-(гидроксиметил)пирролидин-3,4-диола (12i) растворяли в воде с получением концентрации в 1 мг/мл. Аналогичным образом экспериментальное соединение 30f растворяли в воде с получением эквивалентной концентрации (1 мг/мл в отношении соединения 12i). После того, как каждое животное было взвешено, всем животным контрольной группы вводили через желудочный зонд 10 мг/кг массы тела соединения 12i в момент времени 0, а всем экспериментальным животным вводили 10 мг/кг веса тела соединения 30f через желудочный зонд в момент времени 0. Серийные образцы крови получали в момент времени 0, 15 мин, 30 мин, 1 ч, 2 ч, 4 ч, 8 ч, 12 ч и 24 ч. Все образцы помещали в микроцентрифужные пробирки и центрифугировали при 14000 оборотов в минуту в течение 3 минут. Плазму из каждой пробирки удаляли и переносили в предварительно меченные микроцентрифужные трубки и помещали на сухой лед до тех пор, пока образцы не были перенесены в морозильник для хранения при -80°C перед проведением анализа. Затем отдельные образцы анализировали на соединение 12i. Данные концентрации плазмы в зависимости от времени анализировали с помощью некомпартментных методов с использованием программного обеспечения WinNonlin. Получали фармакокинетические параметры Tmax, Cmax, T½, AUC(0-last), AUC(0-inf), MRT(0-inf) и строили графики концентраций в плазме и печени в зависимости от времени. Результаты представлены на фиг.1.
Как показано на фиг.1, экспериментальные и контрольные группы демонстрировали явные различия в фармакокинетике. В то время как значение Tmax было одинаковым для обеих групп (0,5 ч), значение Cmax для экспериментальной группы было 527 нг/мл, при этом значение Cmax в контрольной группе составляло только 123 нг/мл, и AUC(0-inf) для экспериментальной группы составляло 1076 нг⋅ч, а значение AUC(0-inf) в контрольной группе составляло только 219 нг⋅ч. На основании этих результатов было показано, что соединение 30f имеет приблизительно в четыре раза большую биодоступность, чем соединение 12i, и эстеразы плазмы быстро гидролизуют соединение 30f в соединение 12i.
Пример 8: Влияние вирусного ингибитора РНК-полимеразы (соединение 12i) на репликацию вируса кори в клетках почки африканской зеленой мартышки
Материалы и методы: клетки Vero-76 (клетки почки африканской зеленой мартышки) получали из Американской коллекции типовых культур (АТСС, Manassas, VA). По стандартной методике клетки помещали в минимальную питательную среду (MEM с 0,15% NaHCO3; Hyclone Laboratories, Logan, UT, USA) с добавлением 5% фетальной бычьей сыворотки (FBS, Hyclone). При оценке соединений сыворотку сводили до конечной концентрации 2,5%, и к аналитической среде добавляли гентамицин до конечной концентрации 50 мкг/мл. Штамм вируса кори (MV), Chicago, получали из Центров по контролю и профилактике заболеваний (Atlanta, GA).
Способы противовирусных исследований:
Исследование ингибирования цитопатического эффекта (визуальное исследование)
Клетки высевали на 96-луночные плоскодонные планшеты для тканевых культур (Corning Glass Works, Corning, NY), 0,2 мл/лунку, при соответствующей концентрации клеток, и инкубировали в течение ночи при 37°C для образования клеточного монослоя. После образования монослоя питательную среду декантировали, и в каждую лунку добавляли различные разведения исследуемого соединения (3 лунки/раствор, 0,1 мл/лунку). Среду разведения соединения добавляли в контрольные лунки с вирусом и клетками (0,1 мл/лунку). Вирус, разведенный в исследуемой среде, добавляли в лунки с исследуемым соединением (3 лунки/раствор соединения) и в контрольные лунки с вирусом (6 лунок) при 0,1 мл/лунку. Вирус (вирусная MOI=0,001) добавляли примерно через 5 минут после введения соединения. Исследуемую среду без вируса добавляли во все лунки для контроля токсичности (2 лунки/разведение каждого исследуемого соединения) и в контрольные лунки с клетками (6 лунок) при концентрации 0,1 мл/лунку. Планшеты инкубировали при 37°C в увлажненном инкубаторе в атмосфере с 5% CO2 и 95% воздуха, пока контрольные лунки с вирусом не показывали значения достаточного цитопатического эффекта (СРЕ) (80-100% клеточная деструкция). Он достигался, в зависимости от вируса, на 4-11 день после контакта вируса с клетками. Затем клетки исследовали под микроскопом на предмет CPE, который оценивали в баллах от 0 (нормальные клетки) до 4 (максимальный, 100%) CPE. Клетки в лунках контроля токсичности наблюдали под микроскопом на предмет морфологических изменений, присущих цитотоксичности. Эту цитотоксичность (клеточная деструкция и/или изменение морфологии) также оценивали при 100% токсичности, 80% цитотоксичности), 60% цитотоксичности, 40% цитотоксичности, 20% цитотоксичности и 0 (нормальные клетки). С помощью регрессионного анализа данных вирусного CPE и данных контроля токсичности, соответственно, вычисляли 50%-ую эффективную дозу (ЕС50) и 50%-ую цитотоксическую дозу (IC50). Для каждого исследуемого соединения вычисляли индекс селективности (SI) по следующей формуле: SI=CC50/EC50.
Исследование ингибирования CPE методом поглощения нейтрального красного (NR)
Поглощение NR было выбрано в качестве количественного метода поглощения красителя для оценки противовирусных препаратов, основанного на данных, полученных Smee et al (Virol. Methods 2002, 106: 71-79; включено в настоящий документ посредством ссылки полностью). Этот анализ был выполнен с использованием аналогичных аналитических планшетов для исследования ингибирования СРЕ, описанных выше, для контроля ингибирующей активности и цитотоксичности, наблюдаемой визуально. Анализ с использованием NR проводили модифицированным методом Cavenaugh et al. (Invest. New Drugs 1990, 8:347-354; включено в настоящий документ посредством ссылки полностью), как описано Barnard et al. ((Antiviral Chem. Chernother. 2001, 12:220-231; включено в настоящий документ посредством ссылки полностью). Вкратце, среду удаляли из каждой лунки планшета для оценки CPE из анализа ингибирования CPE, в каждую лунку планшета добавляли 0,034% NR, и планшет инкубировали в темноте в течение 2 ч при 37°C. Затем раствор NR удаляли из лунок. После промывки (иногда клетки откреплялись от планшета, приводя к ложно низкому нейтральному красному) и аспирации досуха оставшийся краситель экстрагировали в темноте в течение 30 мин при комнатной температуре из клеток с использованием абсолютного этанола, содержащего цитратный буфер Sorenson. Поглощение при 540 нм/405 нм считывали с помощью микропланшетного ридера (Opsys MR™, Dynex Technologies, Chantilly, VA, USA). Значения абсорбции выражали в процентах необработанных контролей, а значения EC50, CC50 и SI вычисляли способом, описанным выше.
Исследование снижения урожая вируса:
Исследование снижения урожая вируса выполняли с использованием анализа 50%-ой инфекционной дозы для клеточной культуры (CCID50), по существу, как описано ранее (Antimicrob. Agents Chemother. 1992, 3:1837-1842; включено в настоящий документ посредством ссылки полностью). Вкратце, супернатанты из каждой лунки серийно разводили в трех лунках 96-луночных планшетов, содержащих клетки Vero-76. Планшеты инкубировали в течение 6 дней, а затем проверяли на вирус-индуцированный CPE. Количественное определение титров вирусного урожая вычисляли методом с определением конечных точек Рида-Менча (Am. J. Hyg. 1938, 27:493-498; включено в настоящий документ посредством ссылки полностью). Для оценки концентрации, необходимой для ингибирования вирусного урожая на 90% или снижения титра вируса на один log10, вычисляли значение EC90 путем линейной регрессии.
Результаты и обсуждение:
Вирус кори эффективно подавляется соединением 12i (Таблица 1). Значения EC50 относительно вируса кори составляли 0,6 и 1,4 мкг/мл при использовании визуального анализа и анализа NR, соответственно. Соединение не обладало какой-либо цитотоксичностью как при визуальном анализе, так и анализе NR (IC50>100). Таким образом, селективные индексы, по обоим анализам, предполагают, что соединение 12i было весьма активно в отношении вируса кори (MV). Мощную ингибирующую активность в отношении MV подтверждали с помощью анализа снижения урожая вируса при EC90=0,36 мкг/мл, что составляет снижение титра вируса, продуцируемого в инфицированных клетках, на один log10.
Выводы:
Соединение 12i продемонстрировало мощную и селективную ингибирующую активность. По результатам исследования снижения урожая вируса соединение 12i также является мощным ингибитором MV (EC90=0,37 мкг/мл). Таким образом, было установлено, что соединение 12i является мощным ингибитором многих РНК-вирусов, и высказано предположение, что соединение 12i является ингибитором широкого спектра действия некоторых РНК-содержащих вирусов, что подтверждает дополнительный in vitro и in vivo анализ.
Влияние ингибитора полимеразы (соединение 12i) на репликацию различных вирусов
(клетки A-549)
(клетки Vero)
CA/04/2009
(Пандемический вирус HIN1)
Brisbane/10/2007
VN/I203/2004 гибрид
(на остове H1N1)
(клетки Vero)
(клетки MA-104)
(клетки Vero)
(клетки Vero 76)
(клетки MA-104)
(HeLa Ohio-1 клетки)
(клетки Vero 76)
(клетки Vero 76)
(клетки Vero)
(клетки Vero 76)
(Клетки Vero)
(клетки Vero 76)
Пример 9: Эффекты ингибитора вирусной РНК-полимеразы (соединение 12i) на репликацию различных РНК-вирусов
Материалы и методы
Клетки и вирус
Клетки почки африканской зеленой мартышки (MA-104) получали от фирмы Whitaker MA Bioproducts, Walkersville, MD, USA). Все клетки Vero (клетки почки африканской зеленой мартышки, клетки карциномы гортани человека (А-549), клетки Мадин-Дарби почек собаки получали из Американской коллекции типовых культур (АТСС, Manassas, VA). Клетки A-549 культивировали в минимальной эссенциальной среде Дульбекко (DMEM) с добавлением 0,15% NaHCO3 (Hyclone Laboratories, Logan, UT, USA) и 10% фетальной бычьей сыворотки (FBS, Hyclone). Остальные клетки по стандартной методике помещали в минимальную поддерживающую среду (MEM с 0,15% NaHCO3; Hyclone Laboratories, Logan, UT, USA) с добавлением 5% фетальной бычьей сыворотки (FBS, Hyclone).
При оценке соединений сыворотку сводили до конечной концентрации 2,5%, и к аналитической среде добавляли гентамицин до конечной концентрации 50 мкг/мл. Аналитическую среду для исследований гриппа, состоящую из MEM без сыворотки, 0,18% NaHCO3, 20 мкг трипсина/мл, 2,0 мкг ЭДТА/мл и 50 мкг гентамицина/мл.
Для оценки токсичности в активно растущих клетках цитотоксичность оценивали путем определения общего числа клеток, что показывает анализ поглощения NR после 3-дневного воздействия нескольких концентраций соединения. Для количественного роста клеток за 72 ч в присутствии или в отсутствие лекарственного средства на планшеты высевали 1 × 103 клеток MDCK и через 4 ч (оставляли все клетки прикрепляться к лункам планшета) подвергали воздействию отобранных концентраций лекарственного средства в MEM или MEM. Для NR анализа через 72 ч планшеты обрабатывали описанным выше способом. Значения оптической плотности выражали в процентах от необработанных контролей и с помощью регрессионного анализа вычисляли значения CC50.
Вирус Денге 2 (DV-2), штамм New Guinea C, респираторный синтициальный вирус (RSV) A2, риновирус 2 (RV-2), штамм HOP, вирус Tacaribe (TCV), штамм TRVL 11573, вирус венесуэльского энцефалита лошадей (VEE) и вирус желтой лихорадки (YFV), штамм 17D, все были приобретены из Американской Коллекции Типовых Культур (ATCC; Manassas, VA). Все вирусы гриппа, вирус Measles (MV), штамм Chicago, коронавирус SARS (SARS-CoV), штамм Urbani и вирус Лихорадки Западного Нила (WNV), прототипный изолят New York 1999, обозначающий штамм 996625, получали из Центров по контролю и профилактике заболеваний (Atlanta, GA). Вирус Пунта-Торо (PTV), штамм Adames получали от доктора Dominique Pifat из Медицинского научно-исследовательского института по инфекционным болезням армии США (U. S. Army Medical Research Institute for Infectious Diseases), Ft. Detrick (Frederick, MD). Вакцинный штамм вируса Лихорадки лолины Рифт (RVFV), MP-12 и вакцинный штамм вируса Junin (JUNV), Candid 1, были любезно предоставлены доктором Robert Tesh (World Reference Center for Emerging and Viruses and Arboviruses, University of Texas Medical Branch, Galveston, TX). Вирус Пичинде (PICV), штамм An 4763, был предоставлен доктором David Gangemi (Clemson University, Clemson, South Carolina). Вирус парагриппа тип 3 (PIV-3), штамм 14702/5/95, был получен от Jacquelin Boivin (госпиталь St. Justin, Montreal, Canada). Аденовирус (AV-1) тип 1, штамм Chicago/95, был выделен из промывной жидкости трахеи пациента детского возраста и предоставлен M.F. Smaron (Department of Medicine, University of Chicago, Chicago IL).
Процедура исследования противовирусного средства:
Исследование ингибирования цитопатического эффекта (визуальное исследование)
Клетки высевали на 96-луночные плоскодонные планшеты для тканевых культур (Corning Glass Works, Corning, NY), 0,2 мл/лунку, при соответствующей концентрации клеток, и инкубировали в течение ночи при 37°C для образования клеточного монослоя. После образования монослоя питательную среду декантировали и в каждую лунку добавляли различные разведения исследуемого соединения (3 лунки/раствор, 0,1 мл/лунку). Среду разведения соединения добавляли в контрольные лунки с вирусом и клетками (0,1 мл/лунку). Вирус, разведенный в исследуемой среде, добавляли в лунки с исследуемым соединением (3 лунки/раствор соединения) и в контрольные лунки с вирусом (6 лунок) при 0,1 мл/лунку. Вирус (вирусная MOI=0,001) добавляли примерно через 5 минут после введения соединения. Исследуемую среду без вируса добавляли во все лунки контроля токсичности (2 лунки/разведение каждого исследуемого соединения) и в контрольные лунки с клетками (6 лунок) при концентрации 0,1 мл/лунку. Планшеты инкубировали при 37°C в увлажненном инкубаторе в атмосфере с 5% CO2 и 95% воздуха, пока контрольные лунки с вирусом не показали значения достаточного цитопатического эффекта (СРЕ) (80-100% клеточная деструкция). Он достигался, в зависимости от вируса, на 4-11 день после контакта вируса с клетками. Затем клетки исследовали под микроскопом на предмет CPE, который оценивали в баллах от 0 (нормальные клетки) до 4 (максимальный, 100%) CPE. Клетки в лунках контроля токсичности наблюдали под микроскопом на предмет морфологических изменений, присущих цитотоксичности. Эту цитотоксичность (клеточная деструкция и/или изменение морфологии) также оценивали при 100% токсичности, 80% цитотоксичности), 60% цитотоксичности, 40% цитотоксичности, 20% цитотоксичности и 0 (нормальные клетки). С помощью регрессионного анализа данных вирусного CPE и данных контроля токсичности, соответственно, вычисляли 50%-ую эффективную дозу (ЕС50) и 50%-ую цитотоксическую дозу (IC50). Для каждого исследуемого соединения вычисляли индекс селективности (SI) по следующей формуле: SI=CC50/EC50.
Исследование ингибирования CPE и цитотоксичности соединения методом поглощения красителя нейтрального красного (NR)
Поглощение NR было выбрано в качестве количественного метода поглощения красителя для оценки противовирусных препаратов, основанного на данных, полученных Smee et al (см. выше). Этот анализ был выполнен с использованием аналогичных аналитических планшетов для исследования ингибирования СРЕ, описанных выше, для контроля ингибирующей активности и цитотоксичности, наблюдаемой визуально. Анализ с использованием NR проводили модифицированным методом Cavenaugh et al. (см. выше), как описано Barnard et al. Вкратце, среду удаляли из каждой лунки планшета для оценки CPE из анализа ингибирования CPE, в каждую лунку планшета добавляли 0,034% NR и планшет в темноте инкубировали в течение 2 ч при 37°C. Затем раствор NR удаляли из лунок. После промывки (иногда клетки открепляются от планшета, приводя к ложно низкому нейтральному красному) и аспирации досуха оставшийся краситель экстрагируют в течение 30 мин при комнатной температуре в темноте из клеток с использованием абсолютного этанола, содержащего цитратный буфер Sorenson. Поглощение при 540 нм/405 нм считывали с помощью микропланшетного ридера (Opsys MR™, Dynex Technologies, Chantilly, VA, USA). Значения абсорбции выражали в процентах необработанных контролей, а значения EC50, CC50 и SI вычисляли способом, описанным выше.
Другими вирусами, которые, как полагают, в значительной степени ингибируются соединением 12i (SI>10), были DV-2 (ЕС50=15, 13 мкг/мл), JUNV (ЕС50=29, 16 мкг/мл), YFV (EC50=8,3, 8,3 мкг/мл) (Таблица 1). Следующие вирусы незначительно ингибировались соединением 12i (3<SI<10): PIV-3 (EC50=7,1, 10 мкг/мл), SARS-коронавирус (ЕС50=14, 16 мкг/мл), PicV (ЕС50=61, 28 мкг/мл) и RVFV (ЕС50=75, 64 мкг/мл). Соединение 12i было исследовано в отношении штаммов подгруппы вируса гриппа (таблица 2) и продемонстрировало широкий спектр противогриппозной активности в отношении множества штаммов.
Широкий спектр противогриппозной активности соединения 12i.
(Пандемический H1N1)
(H3N2)
(H5N1)
(H1N1)
(H1N1-H274Y)
(H3N2)
Выводы
Соединение 12i продемонстрировало высокую активность в отношении всех исследуемых вирусов гриппа. Соединение 12i оказалось мощным ингибитором репликации вируса гриппа и предполагалось, что соединение 12i эффективно в качестве ингибитора широкого спектра действия в отношении некоторых РНК-содержащих вирусов, включая все вирусы гриппа.
Пример 10: In vitro противовирусная активность соединения 12i
Противовирусную активность соединения 12i исследовали in vitro на нескольких вирусах на предмет противовирусной активности. Значения ЕС50 изменяли в пределах от примерно 10 мкг/мл до примерно >300 мкг/мл в отношении Марбург (филовирус), Junin Candid 1 (аренавирус), вируса Пичинде (аренавирус), Chikungunya 181/25 (тогавирус) и Vaccinia NYCBH (поксвирус).
Пример 11: Синергетическая противовирусная активность соединения 12i и ингибитора нейраминидазы в клетках MDCK
Клетки Мадин-Дарби почек собак (MDCK) инфицировали вирусом гриппа подтип H3N2 (A/Victoria/3/75) и обрабатывали различными сочетаниями соединения 12i и перамивиром в течение 72 ч. Цитопатический эффект определяли с использованием анализа поглощения нейтрального красного красителя. Данные представлены в таблице 3.
Выраженное в процентах ингибирование цитопатического эффекта в грипп-инфицированных клетках
Экспериментальные данные оценивали с помощью трехмерного анализа с использованием программного обеспечения Mac Synergy II™ (Prichard and Shipman, 1990, 1990; включено в настоящий документ посредством ссылки полностью). Программа вычисляет теоретические аддитивные взаимодействия на основе кривых доза-ответ отдельных лекарственных препаратов. Расчетную аддитивную поверхность, которая представляет предполагаемые аддитивные взаимодействия, затем вычитают из экспериментальной поверхности, чтобы выявить области больших (синергия) или меньших (антагонизм), чем ожидалось, взаимодействий. Сочетание перамивира и соединения 12i в исследованиях клеточной культуры продемонстрировало синергический противовирусный эффект с объемом синергии, равным 92% мкM2 единица %.
Пример 12: Эффективность внутримышечной (IM) инъекции соединения 12i в мышиной модели гриппа
Мышей BALB/C 6-8 недельного возраста адаптировали к вирусу H3N2 (A/Victoria/3/75). Ежедневно, в течение 5 дней, начиная за 1 ч до инфицирования, вводили дозы 0, 30, 100 и 300 мг/кг/день путем внутримышечной (IM) инъекции. N=50 животных. За всеми животными наблюдали в течение 16 дней. Предельные значения включали летальность, средний срок до гибели и потерю веса животного.
Эффекты соединения 12i (IM) в мышиной модели вируса гриппа представлены в таблице 4. Соединение 12i, введенное внутримышечно, улучшило выживаемость и потерю веса у мышей, инфицированных вирусом гриппа.
Соединение 12i (IM) в мышиной модели вируса гриппа - H3N2 A/Vic/3/75
(мг/кг/день)
(Среднее значение ± стандартная погрешность)
(грамм ± стандартная погрешность)
День 8
неинфицированный
**P<0,001 по сравнению с группой, обработанной носителем (критерий Стьюдента)
Пример 13: Эффективность перорального введения соединения 12i в мышиной модели гриппа
Мышей BALB/C 6-8 недельного возраста адаптировали к вирусу H3N2 (A/Victoria/3/75). Дозы 0, 30, 100 и 300 мг/кг/сут, один раз в день, и 100 мг/кг/сут, два раза в день, вводили перорально. N=60 животных. За всеми животными наблюдали в течение 16 дней. Предельные значения включали летальность, средний срок до гибели и потерю веса животного. Эффекты вводимого перорально соединения 12i в отношении потери веса у мышей, инфицированных вирусом гриппа H3N2/Vic/3/75, представлены в таблице 5. Вводимое перорально соединение 12i улучшало выживаемость и потерю веса у мышей, инфицированных вирусом гриппа.
Соединение 12i (перорально) в мышиной модели вируса гриппа - H3N2 A/Vic/3/75
(мг/кг/день)
ежедневно
(грамм ± стандартная погрешность)
День 9
**P<0,001 по сравнению с носитель-инфицированной группой (критерий Стьюдента)
Пример 14: Фармакокинетические исследования на мышах
Самкам мышей BALB/с (N=30) перорально вводили соединение 12i в дозе 100 мг/кг. У мышей отбирали кровь из ретро-орбитального синуса в момент времени t=0,17, 0,5, 1,0, 3, 6 и 24 ч (5 мышей в каждой временной точке), центрифугировали и плазму хранили при -80°C. Уровни лекарственного средства в плазме измеряли методом LC/MS/MS (метод жидкостной хроматографии/тандемной масс-спектрометрии).
Уровни соединения 12i в плазме мышей после перорального введения приведены в таблице 6.
Уровни соединения 12i в плазме мышей после перорального введения
(Среднее значение ± стандартная погрешность)
Пример 15: Исследование профилактики вируса Эбола у мышей
Соединение 12i вводили внутрибрюшинно, подкожно и перорально (300 мг/кг/день, дважды в день) 8-12-недельным мышам C57BL/6 (N=10 на группу, 4 группы: одна группа, обработанная солевым раствором, и три группы, обработанные лекарственным средством). Восемь дней лечения, начиная за 4 ч до инфицирования. Адаптированную к мышам дозу вируса Эбола (Заир) вводили внутрибрюшинно. Смертность и вес контролировали в течение 14 дней после инфицирования.
Все обработанные солевым раствором мыши, инфицированные вирусом Эбола, погибли в день 8. Все мыши, обработанные внутрибрюшинно или внутримышечно соединением 12i, выжили в конце исследования (14 день). Восемьдесят процентов мышей, перорально обработанных соединением 12i, выжило в конце исследования (14 день).
Обработанные солевым раствором мыши, инфицированные вирусом Эбола, показали общую потерю веса до дня 8 (все контрольные мыши были мертвы в день 8). Мыши, обработанные внутрибрюшинно или внутримышечно соединением 12i, сохранили более 95% исходного веса в день 12. Мыши, обработанные перорально соединением 12i, сохраняли больше, чем 80% исходного веса в день 12. Все мыши, обработанные лекарственным средством, продолжали набирать вес после дня 12.
Пример 16: Исследование профилактики вируса Эбола у мышей
Соединение 12i вводили внутримышечно и перорально 8-12 недельным мышам C57BL/6. Субъекты исследования были разделены на 6 групп (N=10 на группу). Группа 1 была контрольной группой, которой вводили физиологический раствор, группе 2 вводили дозу соединения 12i, составляющую 150 мг/кг (перорально, дважды в сутки); группе 3 вводили дозу соединения 12i, составляющую 250 мг/кг (перорально, дважды в сутки); группе 4 вводили дозу соединения 12i, составляющую 150 мг/кг (внутримышечно, дважды в сутки). Группу 5 составляли неинфицированные мыши, обработанные физиологическим раствором (перорально, дважды в сутки), и группу 6 составляли неинфицированные мыши, обработанные соединением 12i в дозе 250 мг/кг (перорально, дважды в сутки). Лечение выполняли в течение девяти дней, начиная за 4 ч до инфицирования. Адаптированную к мышам дозу вируса Эбола (Заир) вводили внутрибрюшинно (1000 КОЕ). Смертность и вес контролировали в течение 14 дней после инфицирования.
Все обработанные солевым раствором мыши, инфицированные вирусом Эбола, были мертвы на 8 день. Все мыши, которым внутримышечно вводили соединение 12i, выжили в конце исследования, указывая на то, что внутримышечная доза соединения 12i обеспечила полную защиту. Восемьдесят процентов мышей или больше, получавших перорально соединение 12i, выжило в конце исследования.
Обработанные солевым раствором мыши, инфицированные вирусом Эбола, показали общую потерю веса до 7-го дня (все контрольные мыши погибли в день 8). Мыши, внутримышечно обработанные соединением 12i, показывали увеличение веса аналогично контрольной неинфицированной группе в день 11. Мыши, перорально обработанные соединением 12i, показывали обратимое снижение веса и сохраняли более 100% исходного веса в день 11.
Пример 17: Исследование вируса желтой лихорадки (YFV) на сирийских хомячках с временными интервалами
Вирус желтой лихорадки (штамм Jimenez) вводили внутрибрюшинно самкам сирийских золотистых хомячков (99 г) при 20 CCID50 на одного хомячка (~6,25 × LD50). Группы были разделены следующим образом: 1) соединение 12i вводили, начиная за 4 ч до инфицирования (N=15); 2) соединение 12i вводили, начиная с 1 дня после инфицирования (N=10); 3) соединение 12i вводили, начиная со 2-го дня после инфицирования (N=10); 4) соединение 12i вводили с 3-го дня после инфицирования (N=10); 5) соединение 12i вводили с 4-го дня после инфицирования (N=10); 6) рибавирин вводили, начиная за 4 ч (N=10); 7) физиологический носитель, начиная за 4 ч (N=16); 8) неинфицированные хомячки, обработанные соединением 12i, начиная за 4 ч (N=3); 9) неинфицированные хомячки, обработанные носителем, солевым раствором, начиная за 4 ч (N=3); и 10) неинфицированные необработанные нормальные контроли (N=3). Доза для лечения составляла 100 мг/кг внутрибрюшинно, дважды в сутки в течение 7 дней. Конечными критериями исследования были: смертность на 21 дней, вес, измеренный в дни 0, 3, 5 и 6; титры сыворотки и вируса в печени (день 4, соединение 12i за 4 ч, и носитель за 4 ч), и ALT и AST на 6-й день.
Результаты показали повышенную выживаемость для соединения 12i с задержкой лечения по сравнению с плацебо (фиг.2). Выживаемость хомячков, инфицированных YFV и обработанных соединением 12i два раза в день в течение 7 дней, начиная с различных моментов времени после инфицирования вирусом, обозначается (***p<0,001, **Р<0,1, по сравнению с плацебо). Выживаемость составляла 100% для соединения 12i, начиная c момента до инфицирования, и задержкой лечения не более 3-х дней после инфицирования. Выживаемость составляла 80% для соединения 12i, начиная с 4 дня после инфицирования, что указывает на значительное улучшение по сравнению с плацебо в группах с задержкой лечения. В отличие от этого рибавирин обеспечивает 90% выживаемость, начиная с момента до инфицирования, и носитель обеспечивает 12,5% выживаемости, начиная с момента перед инфицированием. Большинство случаев гибели происходило в течение 10 дней после инфицирования. Выживших животных повторно инфицируют YFV на 21 день после инфицирования.
Хомячки, инфицированные YFV и обработанные соединением 12i в течение между моментом до инфицирования и 4 днями после инфицирования, показывали увеличение веса по сравнению с плацебо и рибавирином, введенным до инфицирования.
Пример 18: Исследование воздействия соединения 12i на вирус Марбург
Соединение 12i вводили внутримышечно 10-12-недельным мышам BALB/с, инфицированным (внутрибрюшинно) 1000 БОЕ мышь-адаптированным MARV-Ravn. Мышей разбивали на 10 групп (N=10 на группу). Режимы дозирования, способы введения и дозы приведены в таблице 7. Соединение 12i растворяли в 0,9% солевом растворе перед введением, и жизнеспособность и вес контролировали в течение 14 дней после инфицирования.
План исследования для профилактики и лечения с помощью соединения 12i вирусной инфекции Марбург
Доза
(мг/кг)
Доза
(мг/кг/день)
-1-4 ч,
Дни 1-8 PI
Обработка группы 6, инициированная через 4 ч после инфицирования в день 0.
PI=после инфицирования.
Выживаемость, выраженная в процентах, в 10 группах на 12 день настоящего исследования представлена в Таблице 8. Выживаемость мышей, обработанных только носителем (0,9% солевой раствор), составляла 60% на 7 день и 30% на 8-12 дней. Соединение 12i показывало увеличение выживаемости по меньшей мере на 90% на 7-й день, и по меньшей мере на 80% на 8-12 дни при всех дозах.
Выраженная в процентах выживаемость при профилактике и лечении с помощью соединения 12i вирусной инфекции Марбург
(150 мг/кг)
(50 мг/кг)
(15 мг/кг)
(5 мг/кг)
(150 мг/кг)
+ 4 ч
(150 мг/кг) + 24 ч
(150 мг/кг) + 48 ч
(150 мг/кг)
+ 72 ч
(150 мг/кг)
+ 96 ч
Пример 19: Фармацевтические лекарственные формы
Далее представлены типичные фармацевтические лекарственные формы, содержащие соединение по изобретению ("соединение X"), для терапевтического или профилактического применения у людей.
Приведенные выше соединения могут быть получены обычными способами, хорошо известными в фармацевтической области.
Все публикации, патенты и патентные документы включены в данный документ посредством ссылки, как если бы каждый из этих документов в отдельности был включен посредством ссылки. Изобретение описано со ссылкой на различные конкретные и предпочтительные варианты осуществления и методы. Тем не менее, следует понимать, что могут быть осуществлены многие изменения и модификации, оставаясь при этом в пределах сущности и объема настоящего изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| СОЕДИНЕНИЯ И КОМПОЗИЦИИ, ПРИМЕНЯЕМЫЕ ДЛЯ ЛЕЧЕНИЯ РАССТРОЙСТВ, СВЯЗАННЫХ С NTRK | 2016 |
|
RU2744974C2 |
| СПОСОБЫ И КОМПОЗИЦИИ ДЛЯ ИНГИБИРОВАНИЯ ПОЛИМЕРАЗЫ | 2011 |
|
RU2718690C2 |
| ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ КАРБОНУКЛЕОЗИДА, ПРИМЕНЯЕМЫЕ В КАЧЕСТВЕ ПРОТИВОРАКОВЫХ АГЕНТОВ | 2017 |
|
RU2712944C1 |
| ИНГИБИТОРЫ RMT5 | 2019 |
|
RU2814198C2 |
| НОВОЕ ПИРРОЛОПИРИМИДИНОВОЕ СОЕДИНЕНИЕ ИЛИ ЕГО СОЛЬ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ЕЕ, В ЧАСТНОСТИ, АГЕНТ ДЛЯ ПРЕДОТВРАЩЕНИЯ И/ИЛИ ЛЕЧЕНИЯ ОПУХОЛЕЙ, И ТОМУ ПОДОБНОЕ, НА ОСНОВЕ ИНГИБИТОРНОГО ВОЗДЕЙСТВИЯ НА NAE | 2015 |
|
RU2658008C2 |
| ИНГИБИТОРЫ БЕТА-ЛАКТАМАЗ | 2009 |
|
RU2445314C9 |
| УСИЛИТЕЛЬ ПРОТИВООПУХОЛЕВОГО ВОЗДЕЙСТВИЯ, СОДЕРЖАЩИЙ СОЕДИНЕНИЕ ПИРРОЛОПИРИМИДИНА | 2016 |
|
RU2710380C1 |
| АГОНИСТЫ РЕЦЕПТОРА МЕЛАНОКОРТИНА | 2007 |
|
RU2411240C2 |
| БИАРИЛЬНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ БЕЛОК-БЕЛКОВОГО ВЗАИМОДЕЙСТВИЯ YAP/TAZ-TEAD | 2021 |
|
RU2830596C1 |
| 3,5-ДИЗАМЕЩЕННОЕ АЛКИНИЛБЕНЗОЛЬНОЕ СОЕДИНЕНИЕ И ЕГО СОЛЬ | 2013 |
|
RU2576384C1 |
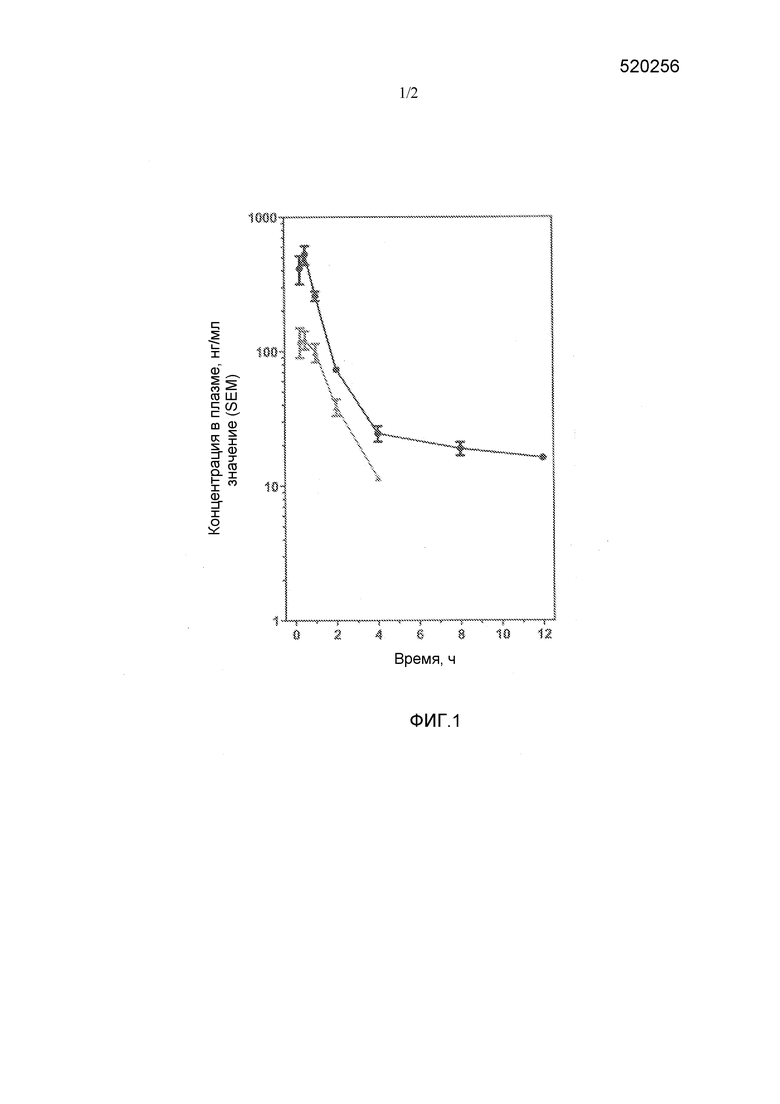
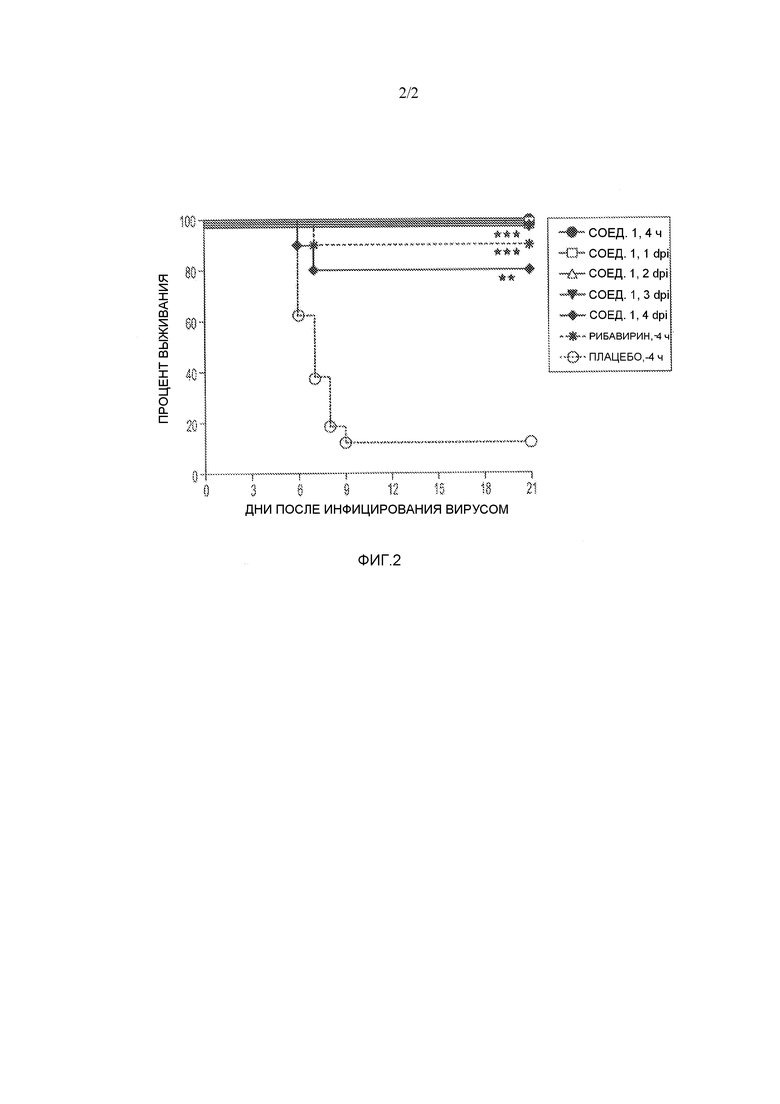
Изобретение относится к новым соединениям формулы (I), которые обладают свойствами ингибитора активности РНК полимеразы вируса и репликации вирусов РНК-содержащих вирусов, таких как ортомиксовируса, парамиксовируса, аренавируса, буньявируса, флавивируса, филовируса, тогавируса, пикорнавируса и коронавируса, аденовируса, риновируса, вируса гепатита А, вируса гепатита С, вируса полиомиелита, вируса кори, вируса Эбола, вируса Коксаки, вируса лихорадки Западного Нила, вируса оспы, вируса желтой лихорадки, вируса лихорадки Денге, вируса гриппа А, вируса гриппа В, вируса Ласса, вируса лимфоцитарного хориоменингита, вируса Хунин и др. В формуле (I)
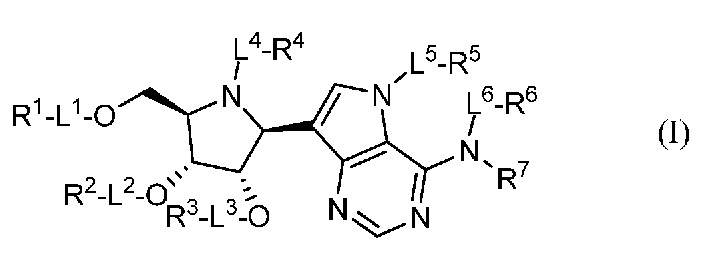
L1, L2 и L3 каждый независимо представляет собой связь или линкер -C(R0)2-O-; L4, L5 и L6 каждый представляет собой связь; R0, в каждом случае независимо, представляет собой H или (C1-C6)алкил; R1, R2 и R3 каждый независимо выбран из группы, состоящей из H; аминоацила, где в каждом случае независимо аминоацил представляет собой -C(=O)CH(NH2)(CH2)nCHR30R31, где n обозначает 0 или 1; и R30 и R31 каждый независимо представляет собой H или (C1-C6)алкил; ацила, где в каждом случае независимо ацил представляет собой -C(=O)R40, где R40 представляет собой (C1-C6)алкил; R10OC(O)-;фосфорила; и аминофосфорила, где в каждом случае независимо аминофосфорил представляет собой -P(=O)(OR50)NR51R52, где R50 выбран из группы, состоящей из фенила, нафтила и -(CH2)mSC(=O)C(CH3)2CH2OH, m обозначает 1 или 2, R51 представляет собой H или (C1-C6)алкил, и R52 выбран из группы, состоящей из H, (C1-C6)алкила, бензила и -CR60R61C(=O)OR62, где R60 и R61 каждый независимо представляет собой H или (C1-C6)алкил; и R62 представляет собой H или (C1-C6)алкил; или R1 и R2, взятые вместе, или R2 и R3, взятые вместе, могут быть выбраны из группы, состоящей из карбонила, фосфорила и (C1-C6)алкилфосфорила; R4, R5 и R6 представляют собой H; R7 представляет собой H; R10, в каждом случае независимо, представляет собой H или (C1-C6)алкил. 4 н. и 26 з.п. ф-лы, 2 ил., 8 табл., 19 пр.
1. Соединение, представленное формулой (I), или его фармацевтически приемлемая соль
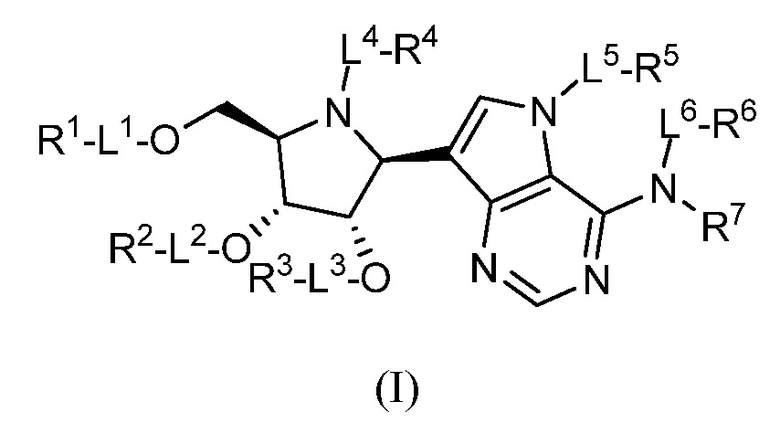
где:
L1, L2 и L3 каждый независимо представляет собой связь или линкер -C(R0)2-O-;
L4, L5 и L6 каждый представляет собой связь;
R0, в каждом случае независимо, представляет собой H или (C1-C6)алкил;
R1, R2 и R3 каждый независимо выбран из группы, состоящей из:
H;
аминоацила, где в каждом случае независимо аминоацил представляет собой -C(=O)CH(NH2)(CH2)nCHR30R31, где n обозначает 0 или 1; и R30 и R31 каждый независимо представляет собой H или (C1-C6)алкил;
ацила, где в каждом случае независимо ацил представляет собой -C(=O)R40, где R40 представляет собой (C1-C6)алкил;
R10OC(O)-;
фосфорила; и
аминофосфорила, где в каждом случае независимо аминофосфорил представляет собой -P(=O)(OR50)NR51R52, где:
R50 выбран из группы, состоящей из фенила, нафтила и -(CH2)mSC(=O)C(CH3)2CH2OH,
m обозначает 1 или 2,
R51 представляет собой H или (C1-C6)алкил, и
R52 выбран из группы, состоящей из H, (C1-C6)алкила, бензила и -CR60R61C(=O)OR62, где
R60 и R61 каждый независимо представляет собой H или (C1-C6)алкил; и
R62 представляет собой H или (C1-C6)алкил;
или R1 и R2, взятые вместе, или R2 и R3, взятые вместе, могут быть выбраны из группы, состоящей из карбонила, фосфорила и (C1-C6)алкилфосфорила;
R4, R5 и R6 представляют собой H;
R7 представляет собой H;
R10, в каждом случае независимо, представляет собой H или (C1-C6)алкил;
при условии, что соединение, представленное формулой (I), не является
2. Соединение по п.1, где L1-R1 и L2-R2 являются одинаковыми.
3. Соединение по п.2, где каждый из L1-R1 и L2-R2 представляет собой H.
4. Соединение по п.2, где L3-R3 представляет собой H.
5. Соединение по п.1, где L2-R2 и L3-R3 являются одинаковыми.
6. Соединение по п.5, где каждый из L2-R2 и L3-R3 представляет собой H.
7. Соединение по п.5, где L1-R1 представляет собой H.
8. Соединение по п.1, где L1-R1 и L3-R3 являются одинаковыми.
9. Соединение по п.8, где каждый из L1-R1 и L3-R3 представляет собой H.
10. Соединение по п.8, где L2-R2 представляет собой H.
11. Соединение по п.1, где L1-R1, L2-R2 и L3-R3 являются одинаковыми.
12. Соединение по любому из пп.1-11, где R30 и R31 каждый независимо представляет собой (C1-C6)алкил.
13. Соединение по п.12, где n обозначает 0; и R30 и R31 каждый независимо представляет собой метил.
14. Соединение по любому из пп.1-13, где в каждом случае независимо R10 представляет собой H.
15. Соединение по любому из пп.1-13, где в каждом случае независимо, R10 представляет собой (C1-C6)алкил.
16. Соединение по любому из пп.1-15, где R50 представляет собой -(CH2)mSC(=O)C(CH3)2CH2OH.
17. Соединение по п.16, где m обозначает 2.
18. Соединение по любому из пп.1-17, где R51 представляет собой H.
19. Соединение по любому из пп.1-18, где R52 представляет собой -CR60R61C(=O)OR62.
20. Соединение по п.19, где R60 представляет собой H; R61 представляет собой (C1-C6)алкил; и R62 представляет собой (C1-C6)алкил.
21. Соединение по п.1, выбранное из группы, состоящей из
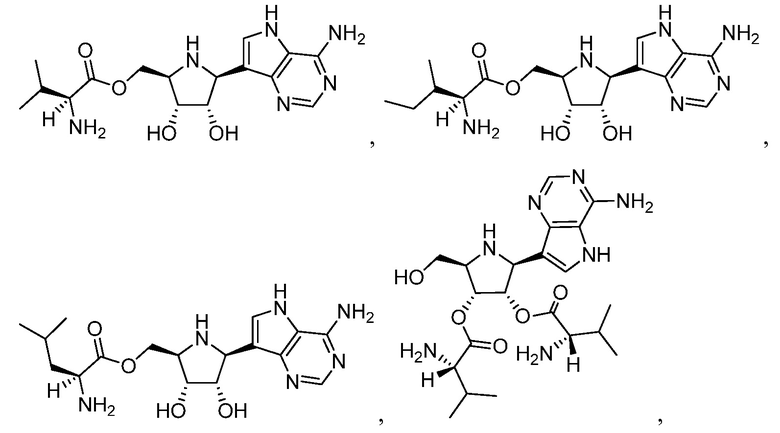
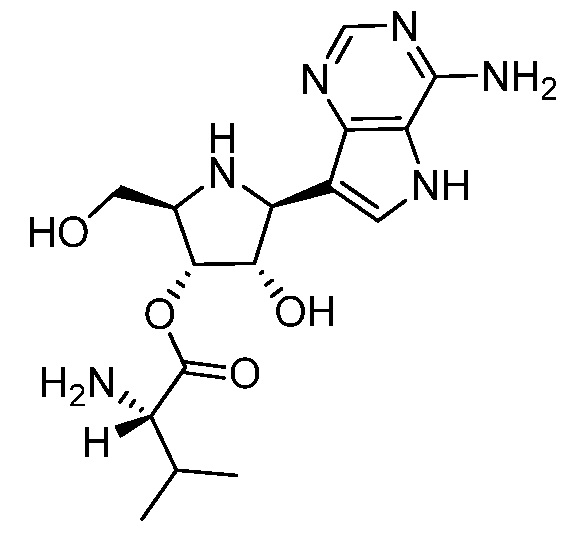 и
и 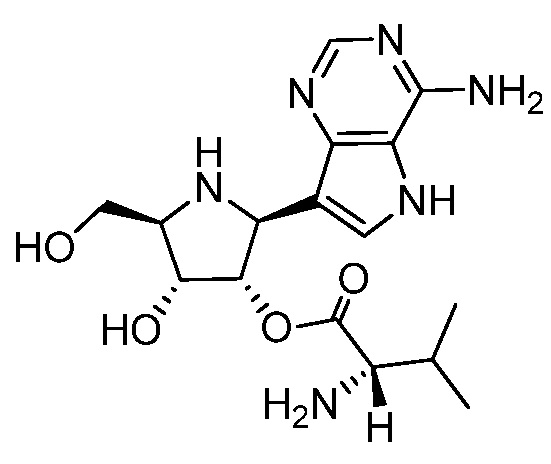 ,
,
и их фармацевтически приемлемые соли.
22. Соединение по п.1, представленное 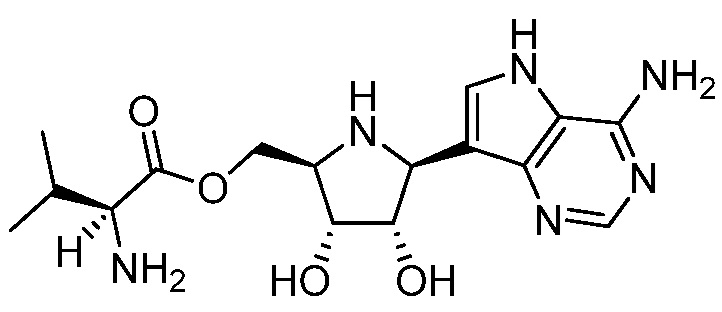 ,
,
или его фармацевтически приемлемая соль.
23. Фармацевтическая композиция, обладающая способностью ингибировать репликацию вируса, содержащая соединение по любому из пп.1-22 или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель.
24. Применение соединения по любому из пп.1-22 или его фармацевтически приемлемой соли при изготовлении лекарственного средства для ингибирования репликации вируса.
25. Применение соединения по любому из пп.1-22 или его фармацевтически приемлемой соли при изготовлении лекарственного средства для лечения вирусной инфекции.
26. Применение по п.24 или 25, в котором вирус выбран из группы, состоящей из РНК-содержащих вирусов.
27. Применение по п.26, в котором вирус выбран из группы, состоящей из ортомиксовируса, парамиксовируса, аренавируса, буньявируса, флавивируса, филовируса, тогавируса, пикорнавируса и коронавируса.
28. Применение по п.24 или 25, в котором вирус выбран из группы, состоящей из аденовируса, риновируса, вируса гепатита А, вируса гепатита С, вируса полиомиелита, вируса кори, вируса Эбола, вируса Коксаки, вируса лихорадки Западного Нила, вируса оспы, вируса желтой лихорадки, вируса лихорадки Денге, вируса гриппа А, вируса гриппа В, вируса Ласса, вируса лимфоцитарного хориоменингита, вируса Хунин, вируса мачупо, вируса Гуанарито, хантавируса, вируса лихорадки долины Рифт, вируса Ла-кросс, вируса калифорнийского энцефалита, вируса Конго-Крым, вируса Марбург, вируса японского энцефалита, вируса болезни Кьясанурского леса, вируса венесуэльского энцефалита лошадей, вируса восточного лошадиного энцефалита, вируса западного лошадиного энцефалита, вируса тяжелого острого респираторного синдрома (SARS), вируса парагриппа, респираторного синтициального вируса, вируса Пунта-Торо, вируса Такарибе и вируса Пичинде.
29. Применение по п.28, в котором вирус выбран из группы, состоящей из аденовируса, вируса лихорадки Денге, вируса Марбург, вируса гриппа А, вируса гриппа В, вируса Хунин, вируса кори, вируса парагриппа, вируса Пичинде, вируса Пунта-Торо, респираторного синтициального вируса, риновируса, вируса лихорадки долины Рифт, вируса SARS, вируса Такарибе, вируса венесуэльского энцефалита лошадей, вируса лихорадки западного Нила и вируса желтой лихорадки.
30. Применение по п.28, в котором вирус выбран из группы, состоящей из вируса Эбола, вируса желтой лихорадки, вируса Марбург, вируса гриппа А и вируса гриппа В.
| US 2007099942 A1, 03.05.2007 & US 7560434 B2, 14.07.2009 | |||
| US 2009298863 A1, 03.12.2009 | |||
| US 2010233120 A1, 16.09.2010 | |||
| US 2009253648 A1, 08.10.2009 & EA 200701850 A1, 28.02.2014 & EA 200701849 А1, 28.02.2008 | |||
| US 2010143300 A1, 10.06.2010 | |||
| Прибор для поверки законов центробежной силы | 1927 |
|
SU5774A1 |
| US 6066722 A, 23.05.2000 | |||
| WO 03080620 A1, 02.10.2003 | |||
| WO 2007069924 A1, 21.06.2007 | |||
| Прибор лабораторного типа для сортирования зерна по длине | 1929 |
|
SU20931A1 |

