ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Данное изобретение относится к замещенным соединениям пиразолоспирокетонов, действующим в качестве ингибиторов ацетил-КоА-карбоксилаз(ы), и их применению в лечении заболеваний, состояний или расстройств, модулируемых посредством ингибирования фермента(ов) ацетил-КоА-карбоксилаз(ы).
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Ацетил-КоА-карбоксилазы (АСС) представляют собой семейство ферментов, катализирующих получение малонил-КоА из ацетил-КоА, которые обнаружены у большинства видов и ассоциированы с синтезом и метаболизмом жирных кислот. У млекопитающих идентифицированы две изоформы фермента АСС. АСС1, уровни экспрессии которой высоки в липогенных тканях, таких как жировая и печеночная ткани, регулирует первую осуществляемую стадию в биосинтезе длинноцепочечных жирных кислот. Если ацетил-КоА не карбоксилируется с образованием малонил-КоА, он метаболизируется через цикл Кребса. АСС2, минорный компонент АСС в печени, но преобладающая изоформа в сердечной и скелетной мышце, катализирует образование малонил-КоА у цитозольной поверхности митохондрий и посредством этого регулирует утилизацию жирных кислот при β-окислении, ингибируя карнитин-пальмитоил трансферазу. Таким образом, непрерывное введение ингибитора АСС (ACC-I), повышая степень утилизации жирных кислот и предотвращая усиление синтеза жирных кислот de novo, также может опустошить депо триглицеридов (TG) в печеночной и жировой ткани у тучных субъектов посредством потребления пищи с высоким или низким содержанием жиров, что приводит к избирательному уменьшению содержания жира в организме.
Согласно исследованиям, проведенным Abu-Etheiga и др., можно предположить, что АСС2 играет существенную роль в регуляции окисления жирных кислот и, по существу, может представлять собой мишень в терапии ожирения и ассоциированных с ожирением заболеваний, таких как диабет 2 типа. См. Abu-Etheiga L., et al., ″Acetyl-CoA carboxylase 2 mutant mice are protected against obesity and diabetes induced by high-fat/high-carbohydrate diets″, PNAS, 100 (18), 10207-10212 (2003). См. также Choi C.S., et al., ″Continuous fat oxidation in acetyl-CoA carboxylase 2 knockout mice increases total energy expenditure, reduces fat mass, and improves insulin sensitivity″, PNAS, 104 (42), 16480-16485 (2007).
Становится все более очевидно, что накопление липидов в печени вызывает печеночную инсулинорезистентность и вносит вклад в патогенез диабета 2 типа. Salvage и др. продемонстрировали, что оба фермента АСС1 и АСС2 вовлечены в регуляцию окисления жиров в гепатоцитах, в то время как АСС1, доминантная изоформа в печени крыс, представляет собой единственный регулятор синтеза жирных кислот. Кроме того, согласно их модели одновременное уменьшение содержания обеих изоформ необходимо, чтобы существенно снизить уровни малонил-КоА в печени, усилить окисление жиров в состоянии сытости, уменьшить накопление липидов и улучшить действие инсулина in vivo. Таким образом, это указывает на то, что ингибиторы печеночных АСС1 и АСС2 могут быть полезны в лечении неалкогольной жировой болезни печени (NAFLD) и печеночной инсулинорезистентности. См. Savage D.B. et al., ″Reversal of diet-induced hepatic steatosis and hepatic insulin resistance by antisense oligonucleotide inhibitors of acetyl-CoA carboxylases 1 and 2″, J. Clin. Invest. doi: 10.1172/JCI27300. См. также Oh W. et al., ″Glucose and fat metabolism in adipose tissue of acetyl-CoA carboxylase 2 knockout mice″, PNAS, 102 (5), 1384-1389 (2005).
Таким образом, существует необходимость в лекарственных средствах, содержащих ингибиторы АСС1 и/или АСС2, для лечения ожирения и ассоциированных с ожирением заболеваний (таких как NAFLD и диабет 2 типа) посредством ингибирования синтеза жирных кислот и посредством усиления окисления жирных кислот.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
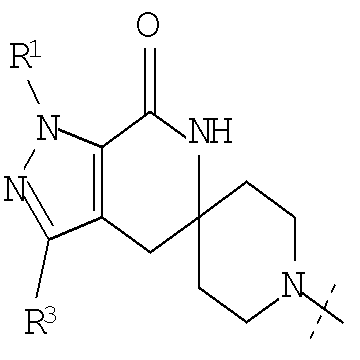
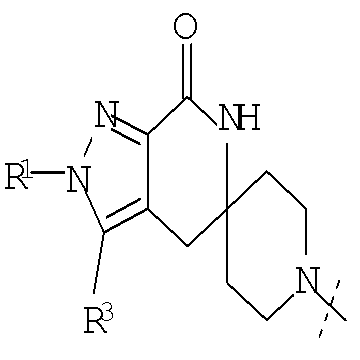
Настоящее изобретение относится к соединениям, имеющим структуру формулы (I),
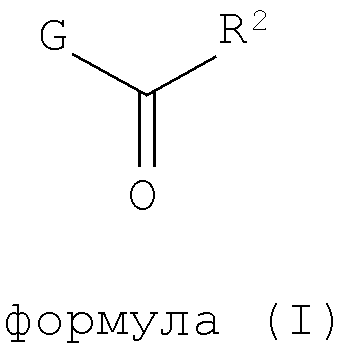
где G представляет собой
 или
или  ,
,
R1 представляет собой (С1-С6)алкил или (С3-С5)циклоалкил; R2 представляет собой фенил, нафтил, 5-12-членный гетероарил или 8-12-членный конденсированный гетероциклический арил; где каждая группа R2 возможно замещена одним-тремя заместителями, независимо выбранными из (С1-С3)алкила, (С1-С3)алкокси, галогено и CONH2; и R3 представляет собой водород или (С1-С3)алкил; или их фармацевтически приемлемой соли.
Предпочтительным воплощением настоящего изобретения являются соединения формулы (I), где R1 представляет собой изопропил или трет-бутил; или их фармацевтически приемлемые соли.
Другим предпочтительным воплощением настоящего изобретения являются соединения формулы (I), где R2 представляет собой бензоимидазолил, пирролопиридинил, пиразолопиридинил, индазолил, хинолинил или изохинолинил, причем указанный R2 возможно моно- или дизамещен независимо одним-двумя заместителями, независимо выбранными из (С1-С3)алкила, (С1-С3)алкокси и галогено; или их фармацевтически приемлемая соль. Еще одним другим предпочтительным воплощением настоящего изобретения являются соединения формулы (I), где R2 представляет собой индазолил, бензоимидазолил или 1H-пирроло[3,2-b]пиридинил, причем указанный R2 возможно замещен одной-двумя группами метил, метокси или хлоро; или их фармацевтически приемлемые соли.
Другим предпочтительным воплощением настоящего изобретения является соединение, выбранное из 1'-изопропил-1-(2-метил-1Н-бензо[d]имидазол-5-карбонил)-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(1'Н)-она; 1'-изопропил-1-(2-метил-2Н-индазол-5-карбонил)-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(1'Н)-она; 1'-изопропил-1-(1Н-пирроло[2,3-b]пиридин-5-карбонил)-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(1'Н)-она и 1'-изопропил-1-(1Н-пирроло[3,2-b]пиридин-6-карбонил)-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-c]пиридин]-7'(1'Н)-она; или его фармацевтически приемлемая соль.
Другим предпочтительным воплощением настоящего изобретения является соединение, выбранное из 1'-изопропил-1-(1-метил-1Н-индазол-5-карбонил)-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(1'Н)-она; 1-(4,8-диметоксихинолин-2-карбонил)-1'-изопропил-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(1'Н)-она; 1'-изопропил-1-(1Н-пирроло[3,2-b]пиридин-2-карбонил)-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(1'Н)-она и 1'-изопропил-1-(1Н-пиразоло[4,3-b]пиридин-6-карбонил)-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-c]пиридин]-7'(1'Н)-она; или его фармацевтически приемлемая соль.
Другим предпочтительным воплощением настоящего изобретения является соединение, выбранное из 1-(3,7-диметил-1Н-индазол-5-карбонил)-1'-изопропил-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(1'Н)-она; 1'-изопропил-1-(7-метил-1Н-индазол-5-карбонил)-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(1'Н)-она; 1-(1Н-индазол-5-карбонил)-1'-изопропил-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(1'Н)-она; 1'-трет-бутил-1-(1Н-индазол-5-карбонил)-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(1'Н)-она; 1'-трет-бутил-1-(7-метил-1Н-индазол-5-карбонил)-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(1'Н)-она и 1'-трет-бутил-1-(3,7-диметил-1Н-индазол-5-карбонил)-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(1'Н)-она; или его фармацевтически приемлемая соль.
Другим предпочтительным воплощением настоящего изобретения является соединение, выбранное из 1-(7-хлор-1Н-индазол-5-карбонил)-1'-изопропил-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(1'Н)-она; 1'-изопропил-1-(4-метокси-1Н-индазол-6-карбонил)-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(1'Н)-она; 1-(7-этил-1Н-индазол-5-карбонил)-1'-изопропил-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(1'Н)-она; 1-(3-этил-1Н-индазол-5-карбонил)-1'-изопропил-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(1'Н)-она и 1'-изопропил-1-(3-метил-1Н-индазол-5-карбонил)-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(1'Н)-она; или его фармацевтически приемлемая соль.
Другим предпочтительным воплощением настоящего изобретения является соединение, выбранное из 1-(1Н-индазол-5-карбонил)-2'-изопропил-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(2'Н)-она; 2'-трет-бутил-1-(1Н-индазол-5-карбонил)-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(2'Н)-она; 2'-изопропил-1-(7-метил-1Н-индазол-5-карбонил)-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(2'Н)-она; 1-(3,7-диметил-1Н-индазол-5-карбонил)-2'-изопропил-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(2'Н)-она; 2'-трет-бутил-1-(7-метил-1Н-индазол-5-карбонил)-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(2'Н)-она; 2'-трет-бутил-1-(3,7-диметил-1Н-индазол-5-карбонил)-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(2'Н)-она и 2'-изопропил-1-(2-метил-1Н-бензо[d]имидазол-5-карбонил)-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(2'Н)-она; или его фармацевтически приемлемая соль.
Другим предпочтительным воплощением настоящего изобретения является соединение, выбранное из 1'-изопропил-1-(хинолин-3-карбонил)-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(1'Н)-она; 1'-изопропил-1-(хинолин-6-карбонил)-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(1'Н)-она; 1'-изопропил-1-(изохинолин-6-карбонил)-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(1'Н)-она; 1'-изопропил-1-(изохинолин-7-карбонил)-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(1'Н)-она и 1'-изопропил-1-(хинолин-7-карбонил)-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(1'Н)-она; или его фармацевтически приемлемая соль.
Другим воплощением настоящего изобретения является соединение формулы (I),
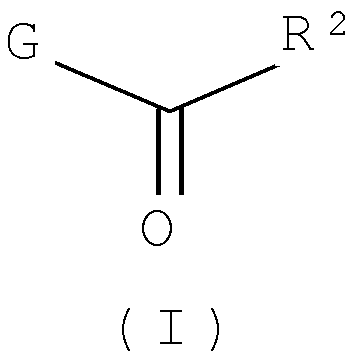
или его фармацевтически приемлемая соль; где
G представляет собой
 ,
, ,
,
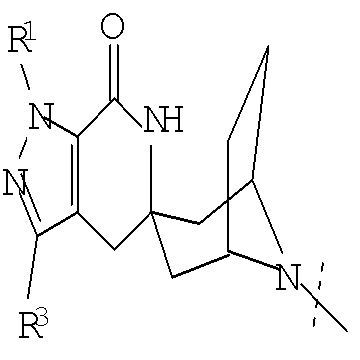 или
или 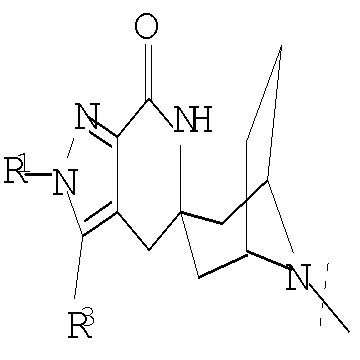 ;
;
R1 представляет собой (С1-С6)алкил или (С3-С5)циклоалкил; R2 представляет собой фенил; нафтил; 5-12-членный гетероарил или 8-12-членный конденсированный гетероциклический арил; где каждая группа R2 возможно замещена одним-тремя заместителями, независимо выбранными из (С1-С6)алкила, (С3-С7)циклоалкила, (С1-С6)алкокси, галогено, циано, CONR4R5, NR4R5 или 3-7-членного гетероциклила, где указанные (С1-С6)алкил, (С3-С7)циклоалкил или (С1-С6)алкокси возможно замещены 1-5 группами фторо; R3 представляет собой водород или (С1-С3)алкил; и R4 и R5 в каждом случае независимо выбраны из водорода, (С1-С6)алкила, (С3-С7)циклоалкила, (С1-С3)алкокси-(С1-С6)алкила или 3-7-членного гетероциклила; где указанные (С1-С6)алкил, (С3-С7)циклоалкил или (С1-С3)алкокси-(С1-С6)алкил возможно замещены 1-5 группами фторо.
Еще одним воплощением настоящего изобретения является соединение формулы (I), где G представляет собой
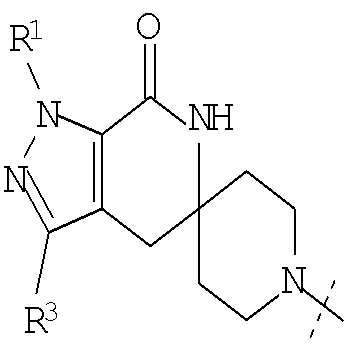 или
или 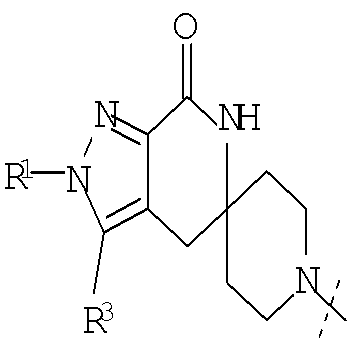 ; и
; и
R1 представляет собой изопропил или трет-бутил; или его фармацевтически приемлемая соль.
Другим воплощением настоящего изобретения является соединение формулы (I), где R2 представляет собой бензоимидазолил, бензотриазолил, пирролопиридинил, пиразолопиридинил, индолил, индазолил, хинолинил или изохинолинил, причем указанный R2 возможно замещен одним-двумя заместителями, независимо выбранными из (С1-С6)алкила, (С1-С6)алкокси, галогено или NR4R5, где указанные (С1-С6)алкил или (С1-С6)алкокси возможно замещен 1-5 группами фторо; или его фармацевтически приемлемая соль.
Другим воплощением настоящего изобретения является соединение формулы (I), где R2 представляет собой индазолил, индолил, бензоимидазолил или 1H-пирроло[3,2-b]пиридинил, причем указанный R2 возможно замещен независимо одной-двумя группами метил, метокси, NH2, NHCH3 или хлоро; или его фармацевтически приемлемая соль.
Еще одним воплощением настоящего изобретения является соединение формулы (I), где R2 представляет собой хинолинил или изохинолинил, причем указанный R2 возможно замещен независимо одной-двумя группами метил, метокси, NH2, NHCH3, NHCH2CH3, NHCH2CF3 или хлоро; или его фармацевтически приемлемая соль.
Предпочтительным воплощением настоящего изобретения является соединение, выбранное из группы, состоящей из 1-(3,7-диметил-1Н-индазол-5-карбонил)-1'-изопропил-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(1'Н)-она; 1-(3,7-диметил-1Н-индазол-5-карбонил)-1'-трет-бутил-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(1'Н)-она; 1-(3,7-диметил-1Н-индазол-5-карбонил)-2'-изопропил-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(1'Н)-она; 1-(6-метоксихинолин-3-карбонил)-2'-трет-бутил-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(1'Н)-она; 1-(1-метоксиизохинолин-7-карбонил)-2'-трет-бутит-4',6'-дигидро-спиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(1'Н)-она; 1-(3-хлор-7-метил-1Н-индазол-5-карбонил)-2'-трет-бутил-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(1'Н)-она; 1-(2-метоксихинолин-7-карбонил)-2'-трет-бутил-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(1'Н)-она; 1-(2-аминохинолин-7-карбонил)-2'-трет-бутил-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(1'Н)-она; 1-(5-метоксихинолин-3-карбонил)-2'-трет-бутил-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(1'Н)-она; 1-(2-амино-1Н-бензо[d]имидазол-5-карбонил)-2'-трет-бутил-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(1'Н)-она; 1-(2-(метиламино)хинолин-7-карбонил)-2'-трет-бутил-4',6'-дигидроспиро-[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(1'Н)-она; 1-(1-(метиламино)-изохинолин-7-карбонил)-2'-трет-бутил-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(1'Н)-она; 1-(3-хлор-1Н-индол-6-карбонил)-2'-трет-бутил-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(1'Н)-она; 1-(3-хлор-1Н-пирроло[3,2-b]пиридин-6-карбонил)-2'-трет-бутил-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(1'Н)-она; 1-(2-(метиламино)хинолин-7-карбонил)-1'-изопропил-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(1'Н)-она; 1-(1-(2,2,2-трифторэтиламино)хинолин-7-карбонил)-2'-трет-бутил-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(1'Н)-она и 1-(1-(этиламино)изохинолин-7-карбонил)-2'-трет-бутил-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(1'Н)-она; или его фармацевтически приемлемая соль.
Другой аспект настоящего изобретения относится к фармацевтической композиции, содержащей соединение формулы (I), описанное в любом из воплощений, или его фармацевтически приемлемую соль и фармацевтически приемлемый эксципиент, разбавитель или носитель. Предпочтительно, композиция содержит терапевтически эффективное количество соединения по настоящему изобретению. Композиция также может содержать по меньшей мере один дополнительный фармацевтический агент. Предпочтительные агенты включают антидиабетические средства и/или средства против ожирения.
В другом аспекте настоящее изобретение относится к способу лечения заболевания, состояния или расстройства, опосредованного ингибированием фермента(ов) ацетил-КоА-карбоксилаз(ы), у млекопитающего, включающему стадию введения млекопитающему, предпочтительно человеку, нуждающемуся в таком лечении, терапевтически эффективного количества соединения по настоящему изобретению или его фармацевтически приемлемой соли либо фармацевтической композиции на их основе.
Заболевания, расстройства или состояния, опосредованные ингибиторами ацетил-КоА-карбоксилаз, включают диабет II типа и диабет-ассоциированные заболевания, такие как неалкогольная жировая болезнь печени (NAFLD), печеночная инсулинорезистентность, гипергликемия, метаболический синдром, нарушенная толерантность к глюкозе, диабетическая нейропатия, диабетическая нефропатия, диабетическая ретинопатия, ожирение, дислипидемия, гипертензия, гиперинсулинемия и синдром инсулинорезистентности. Предпочтительные заболевания, расстройства или состояния включают диабет II типа, неалкогольную жировую болезнь печени (NAFLD), печеночную инсулинорезистентность, гипергликемию, нарушенную толерантность к глюкозе, ожирение и синдром инсулинорезистентности. Более предпочтительными являются диабет II типа, неалкогольная жировая болезнь печени (NAFLD), печеночная инсулинорезистентность, гипергликемия и ожирение. Наиболее предпочтительным заболеванием является диабет II типа.
Одно из предпочтительных воплощений относится к способу лечения (например, замедлению прогрессирования или задержке начала) диабета 2 типа и диабет-ассоциированных расстройств у животных, включающему стадию введения животному, нуждающемуся в таком лечении, терапевтически эффективного количества соединения по настоящему изобретению или его фармацевтически приемлемой соли либо композиции на его основе.
Другое предпочтительное воплощение относится к способу лечения ожирения и ассоциированных с ожирением расстройств у животных, включающему стадию введения животному, нуждающемуся в таком лечении, терапевтически эффективного количества соединения по настоящему изобретению или его фармацевтически приемлемой соли либо композиции на их основе.
Еще одно предпочтительное воплощение относится к способу лечения неалкогольной жировой болезни печени (NAFLD) или печеночной инсулинорезистентности у животных, включающему стадию введения животному, нуждающемуся в таком лечении, терапевтически эффективного количества соединения по настоящему изобретению или его фармацевтически приемлемой соли либо композиции на его основе.
Соединения по настоящему изобретению могут быть введены в комбинации с другими фармацевтическими агентами (в частности, средствами против ожирения и антидиабетическими средствами, изложенными в данном описании ниже). Комбинированную терапию можно назначать с введением в виде (а) единой фармацевтической композиции, содержащей соединение по настоящему изобретению, по меньшей мере один дополнительный фармацевтический агент, изложенный в данном описании, и фармацевтически приемлемый эксципиент, разбавитель или носитель; или (б) двух отдельных фармацевтических композиций, содержащих (1) первую композицию, содержащую соединение по настоящему изобретению и фармацевтически приемлемый эксципиент, разбавитель или носитель, и (2) вторую композицию, содержащую по меньшей мере один дополнительный фармацевтический агент, изложенный в данном описании, и фармацевтически приемлемый эксципиент, разбавитель или носитель. Эти фармацевтические композиции можно вводить одновременно или последовательно и в любом порядке.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Определения
Фраза ″терапевтически эффективное количество″ означает количество соединения по настоящему изобретению или его фармацевтически приемлемой соли, которое (1) лечит или предупреждает конкретное заболевание, состояние или расстройство, (2) ослабляет, улучшает или устраняет один или более симптомов конкретного заболевания, состояния или расстройства, или (3) предупреждает или задерживает начало одного или более симптомов конкретного заболевания, состояния или расстройства, изложенного в данном описании.
Термин ″животное″ относится к людям (мужского или женского пола), домашним животным (например, собакам, кошкам и лошадям), животным-источникам продовольствия, животным зоопарков, морским животным, птицам и другим подобным видам животных. Термин ″годные в пищу животные″ относится к животным-источникам продовольствия, таким как крупный рогатый скот, свиньи, овцы и домашняя птица.
Фраза ″фармацевтически приемлемый″ указывает на то, что данные вещество или композиция должны быть совместимы с точки зрения химии и/или токсикологии с другими ингредиентами, входящими в состав препарата, и/или млекопитающим, которого лечат им.
Термины ″подвергание лечению″, ″лечить″ или ″лечение″ включают как превентивное, т.е. профилактическое, так и паллиативное лечение.
Термин ″модулируемый″, или ″модулирование″, или ″модулируют(ет)″, как использовано в данном описании, если не указано иное, относится к ингибированию фермента(ов) ацетил-КоА-карбоксилаз(ы) (АСС) соединениями по настоящему изобретению.
Термин ″опосредуемый″, или ″опосредующий″, или ″опосредуют(ет)″, как использовано в данном описании, если не указано иное, относится к (1) лечению или предупреждению конкретного заболевания, состояния или расстройства, (2) ослаблению, улучшению или устранению одного или более симптомов конкретного заболевания, состояния или расстройства или (3) предупреждению или задержке начала одного или более симптомов конкретного заболевания, состояния или расстройства, изложенного в данном описании, посредством ингибирования фермента(ов) ацетил-КоА-карбоксилаз(ы) (АСС).
Термин ″соединения по настоящему изобретению″ (если конкретно не указано иное) относится к соединениям формулы (I) и любым фармацевтически приемлемым солям соединений, а также всем стереоизомерам (включая диастереоизомеры и энантиомеры), таутомерам, конформационным изомерам и меченным изотопами соединениям. Гидраты и сольваты соединений по настоящему изобретению рассматриваются как композиции по настоящему изобретению, в которых соединение ассоциировано с водой или растворителем, соответственно.
Термины ″(С1-С6)алкил″ и ″(С1-С3)алкил″ относятся к алкильным группам, содержащим конкретное количество атомов углерода от одного до шести или от одного до трех атомов углерода, соответственно, которые могут быть либо прямыми, либо разветвленными. Например, термин ″(С1-С3)алкил″ означает группу из одного-трех атомов углерода и включает в себя метил, этил, н-пропил и изопропил.
Термин ″(С3-С7)циклоалкил″ означает циклоалкильную группу из трех-семи атомов углерода и включает в себя циклопропил, циклобутил, циклопентил, циклогексил и циклогептил. Термин ″галогено″ означает фторо, хлоро, бромо или иодо. Термин ″(С6-С10)арил″ означает ароматическую карбоциклическую группу, состоящую из шести-десяти атомов углерода, такую как фенил или нафтил.
Термин ″5-12-членный гетероарил″ означает пяти-двенадцатичленную ароматическую группу, содержащую, по меньшей мере, один гетероатом, выбранный из азота, кислорода и серы. Как использовано в данном описании, место присоединения ″5-12-членной гетероарильной″ группы находится на атоме углерода этой группы. Группа ″5-12-членный гетероарил″ может быть бициклической. Предпочтительные воплощения бициклических гетероарилов включают, но не ограничиваются этим, радикалы следующих кольцевых систем:

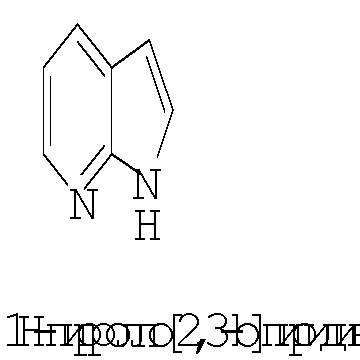
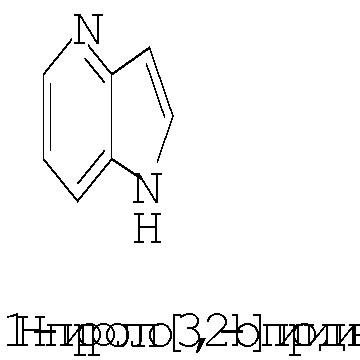
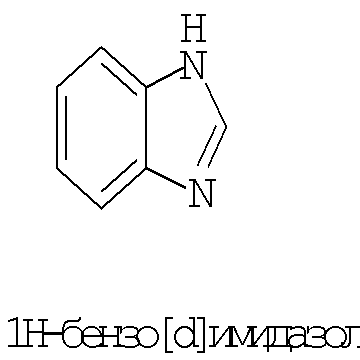
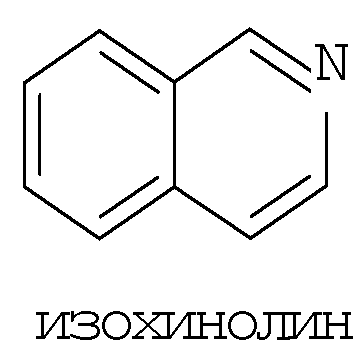
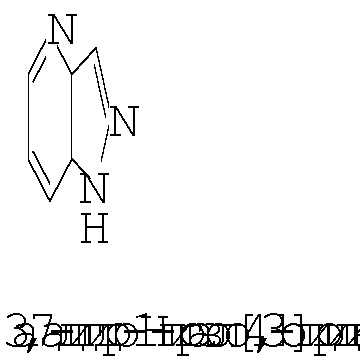
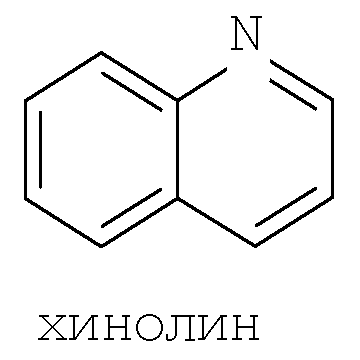
Термин ″8-12-членный конденсированный гетероциклический арил″ означает 8-12-членную кольцевую систему, в которой неароматическое гетероциклическое кольцо конденсировано с арильным кольцом. Как использовано в данном описании, место присоединения ″8-12-членной конденсированной гетероциклической арильной″ группы находится на атоме углерода этой группы. Термин ″3-7-членный гетероциклил″ означает трех-семи-членное насыщенное кольцо, где от одного до трех атомов являются гетероатомами, независимо выбранными из азота, кислорода и серы. Примеры ″3-7-членных гетероциклических″ групп включают, но не ограничиваются этим, такие группы, как азиридинил, азетидинил, пирролидинил, пиперидинил, оксиранил, оксетанил, тетрагидрофуранил, тетрагидро-2Н-пиранил, тетрагидро-2H-тиопиранил, пиперазинил, морфолинил и тиоморфолинил. Место присоединения ″3-7-членного гетероциклила″ может находиться на атоме углерода или азота, как будет целесообразно для конкретной группы.
В одном из воплощений соединение формулы I представляет собой N1-лактамное АСС-ингибирующее соединение, имеющее следующую структуру:
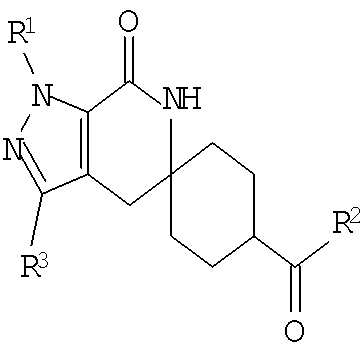 ,
,
где R1 представляет собой (С1-С6)алкил или (С3-С5)циклоалкил; R2 представляет собой фенил, нафтил, 5-12-членный гетероарил или 8-12-членный конденсированный гетероциклический арил; где каждая группа R2 возможно замещена одним-тремя заместителями, независимо выбранными из (С1-С3)алкила, (С1-С3)алкокси, галогено и CONH2; и R3 представляет собой водород или (С1-С3)алкил; или его фармацевтически приемлемую соль.
В одном из воплощений соединение формулы I представляет собой N2-лактамное АСС-ингибирующее соединение, имеющее следующую структуру:
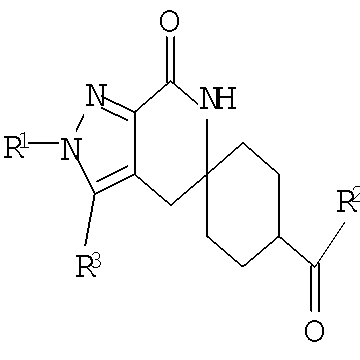 ,
,
где R1 представляет собой (С1-С6)алкил или (С3-С5)циклоалкил; R2 представляет собой фенил; нафтил; 5-12-членный гетероарил или 8-12-членный конденсированный гетероциклический арил; где каждая группа R2 возможно замещена одним-тремя заместителями, независимо выбранными из (С1-С3)алкила, (С1-С3)алкокси, галогено и CONH2; и R3 представляет собой водород или (С1-С3)алкил; или его фармацевтически приемлемую соль.
Соединения по настоящему изобретению могут быть синтезированы с использованием путей синтеза, включающих аналогичные хорошо известным в области химии, в частности, в свете приведенного в данном изобретении описания. Как правило, исходные вещества доступны из коммерческих источников, таких как Aldrich Chemicals (Milwaukee, WI), или их легко получить, используя способы, хорошо известные специалистам в данной области (например, получить способами, в целом описанными в Louis F. Fieser and Mary Fieser, Reagents for Organic Synthesis, v.1-19, Wiley, New York (1967-1999 ed.) или Beilsteins Handbuch der organischen Chemie, 4, Aufl. ed. Springer-Verlag, Berlin, включая приложения (также доступны через онлайн-базу данных Бейльштейна)).
В иллюстративных целях на приведенных ниже реакционных схемах представлены возможные пути синтеза соединений по настоящему изобретению, а также ключевых промежуточных соединений. Для более подробного описания отдельных реакционных стадий см. раздел Примеры, приведенный ниже. Специалистам в данной области техники будет очевидно, что для синтеза соединений по настоящему изобретению можно использовать другие пути синтеза. Несмотря на то, что на схемах приведены и ниже обсуждаются конкретные исходные вещества и реагенты, они легко могут быть заменены на другие исходные вещества и реагенты для обеспечения разнообразия производных и/или реакционных условий. Помимо этого, большинство соединений, полученных описанными ниже способами, могут быть модифицированы далее с учетом данного описания с использованием методов традиционной химии, хорошо известных специалистам в данной области.
При получении соединений по настоящему изобретению может потребоваться введение защиты функциональной группы (например, первичного или вторичного амина) в промежуточных соединениях. Необходимость в такой защите будет варьировать в зависимости от природы такой функциональной группы и условий осуществления способов получения. Подходящие амино-защитные группы (NH-Pg) включают ацетил, трифторацетил, трет-бутоксикарбонил (ВОС), бензилоксикарбонил (CBz) и 9-флуоренилметиленоксикарбонил (Fmoc). Аналогично, ″гидроксил-защитная группа″ относится к заместителю группы гидрокси, который блокирует или защищает функциональную гидроксигруппу. Подходящие гидроксил-защитные группы (O-Pg) включают, например аллил, ацетил, силил, бензил, пара-метоксибензил, тритил и тому подобные. Необходимость в такой защите легко определяется специалистом в данной области. Для общего описания защитных групп и их использования см. Т.W. Greene, Protective Groups in Organic Synthesis. John Wiley & Sons, New York, 1991.
На следующих далее реакционных схемах, от реакционной схемы I до реакционной схемы V, приведены репрезентативные методики, которые используются для получения соединений формулы (I). Следует понимать, что эти реакционные схемы не должны истолковываться как ограничивающие и что для получения соединений формулы (I) можно использовать разумные варианты представленных способов.
На реакционной схеме I приведены общие методики, которые могут быть использованы для получения N1-лактамных АСС-ингибирующих соединений по настоящему изобретению, имеющих формулу Ia, в которых R1 представляет собой (С1-С6)алкил или (С3-С5)циклоалкил, и R2 представляет собой фенил, нафтил, 5-12-членный гетероарил или 8-12-членный конденсированный гетероциклический арил; где каждая группа R2 возможно замещена одним-тремя заместителями, независимо выбранными из (С1-С3)алкила, (С1-С3)алкокси, галогено и CONH2.
РЕАКЦИОННАЯ СХЕМА 1
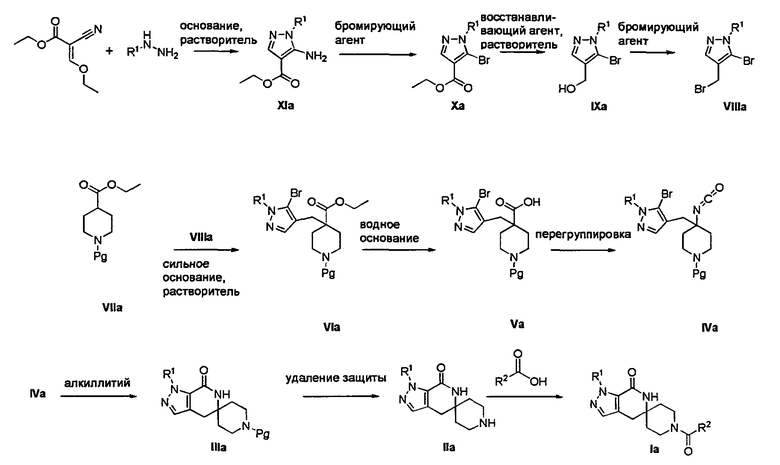
Согласно Схеме I соединение формулы XIa может быть образовано в результате приведения во взаимодействие метил-2-циано-3-этоксиакрилата с соответствующим алкилгидразином (R1NHNH2) в присутствии основания, такого как карбонат калия, и растворителя. Например, соединение формулы XIa может быть образовано в результате приведения во взаимодействие метил-2-циано-3-этоксиакрилата с соответствующим алкилгидразином (R1NHNH2) в присутствии основания, такого как карбонат калия (″К2СО3″), в этаноле с кипячением с обратным холодильником с получением желаемого циклизованного соединения, при температуре от примерно 20°С до примерно 80°С в течение примерно 2-24 часов.
Соединение формулы Xa может быть образовано в результате превращения ариламина формулы XIa в арилбромид с использованием нитрита, такого как изоамилнитрит, нитрит натрия или трет-бутилнитрит, и источника брома, такого как бромид меди(II) в ацетонитриле, с получением соединения формулы Xa, при температуре от примерно 20°С до примерно 80°С в течение от примерно 2 до примерно 18 часов.
Затем можно получить соединение формулы IXa путем обработки сложного эфира формулы Xa восстанавливающим агентом, таким как диизобутилалюминийгидрид (″DIBAL″) или алюмогидрид лития (″LAH″), в апротонном растворителе, таком как тетрагидрофуран (″THF″), толуол или диэтиловый эфир, при температуре от примерно 0°С до примерно 80°С в течение от примерно 1 до примерно 12 часов.
Соединение формулы VIa может быть образовано сначала в результате взаимодействия соединения формулы IXa с бромирующим агентом, таким как трибромид фосфора (″PBr3″) или смесь тетрабромида углерода и трифенилфосфина, при температуре от примерно -20°С до примерно 60°С в течение от примерно 30 до примерно 120 минут с образованием соединения формулы VIIIa. Затем соединение формулы VIIIa приводят во взаимодействие с защищенным производным пиперидина - соединением формулы VIIa в присутствии сильного основания, такого как бис(триметилсилил)амид лития (″LiHMDS″) или диизопропиламин лития (″LDA″), в апротонном растворителе, таком как THF, толуол или диэтиловый эфир, при температуре от примерно -78°С до примерно 20°С в течение от примерно 1 до примерно 18 часов. Группа Pg представляет собой соответствующую защитную группу амина и является предпочтительно N-трет-бутоксикарбонилом (″ВОС″) или карбобензилокси (″Cbz″).
Затем с соединения формулы VIa удаляют защиту путем гидролиза сложноэфирной группы в присутствии сильного водного основания, такого как гидроксид лития или гидроксид натрия, при температуре от примерно 0°С до примерно 100°С в течение от примерно 1 до примерно 18 часов с образованием содержащего группу карбоновой кислоты соединения формулы Va.
Затем может быть образовано изоцианатное соединение формулы IVa путем приведения во взаимодействие карбоновой кислоты формулы Va с дифенилфосфорилазидом (″DPPA″) в присутствии основания, такого как триэтиламин (″Et3N″) или диизопропилэтиламин, при температуре от примерно 60°С до примерно 120°С в течение от примерно 1 до примерно 12 часов.
Затем может быть образовано лактамное соединение формулы IIIa путем циклизации изоцианата (формула IVa) с использованием алкиллития, такого как н-бутиллитий (″н-BuLi″) или трет-бутиллитий (″f-BuLi″), при температуре от примерно -78°С до примерно 0°С в течение от примерно 5 до примерно 120 минут.
Затем с лактамного соединения формулы (IIIa) можно удалить защиту с получением свободного производного спиропиперидина формулы (IIa), используя стандартные методы в зависимости от того, какая защитная группа Pg была применена. Например, когда Pg представляет собой ВОС, то чтобы удалить группу ВОС, можно использовать стандартные условия удаления защиты с применением сильной кислоты, такой как 4 н соляная кислота в диоксане или трифторуксусная кислота в соответствующем растворителе, таком как дихлорметан. В том случае, когда Pg представляет собой Cbz, для удаления защиты можно использовать гидрирование над палладием на угле в этаноле или обработку с применением источника водорода, такого как формиат аммония или 1-метил-1,4-циклогексадиен в присутствии палладия на угле в этаноле или этилацетате.
Затем, чтобы получить соединение формулы (Ia), производное спиропиперидина формулы (IIa) можно подвергнуть ацилированию путем применения стандартных способов. Например, в таком случае соединение (Ia) может быть образовано с использованием стандартной реакции пептидного сочетания с желаемой карбоновой кислотой (R2CO2H). Например, сочетание промежуточного спиропиперидина (IIa) и карбоновой кислоты (R2CO2H) может быть осуществлено посредством образования активированного сложного эфира карбоновой кислоты, например, путем приведения в контакт карбоновой кислоты (R2CO2H) с реагентом пептидного сочетания, таким как гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (″HATU″) или гидрохлорид 1-этил-3-(3-диметиламинопропил)карбодиимида (″EDC·HCl″), в присутствии или в отсутствие активирующего агента, такого как гидроксибензотриазол (″HOBt″), и в присутствии подходящего основания, такого как N,N-диизопропилэтиламин (″DIEA″), триэтиламин или N-метилморфолин (″NMM″), в подходящем растворителе, таком как THF и/или DMF (диметилформамид), диметилацетамид (″DMA″) или дихлорметан, и затем приведения в контакт этого активированного эфира карбоновой кислоты с производным спиропиперидина (IIa) с образованием соединения формулы (Ia).
На реакционной схеме II приведены общие методики, которые могут быть использованы для получения N2-лактамных АСС-ингибирующих соединений по настоящему изобретению, имеющих формулу Ib, в которых R1 представляет собой (С1-С6)алкил или (С3-С5)циклоалкил, и R2 представляет собой фенил, нафтил, 5-12-членный гетероарил или 8-12-членный конденсированный гетероциклический арил; где каждая группа R2 возможно замещена одним-тремя заместителями, независимо выбранными из (С1-С3)алкила, (С1-С3)алкокси, галогено и CONH2.
РЕАКЦИОННАЯ СХЕМА II
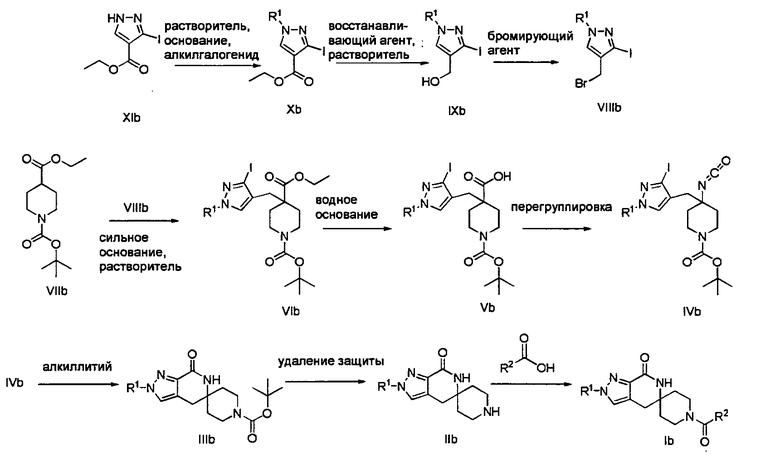
Согласно Схеме II алкилирование соединения пиразола формулы XIb до соединения формулы Х с использованием первичного или вторичного алкилгалогенида, такого как метилиодид, этилиодид, 1-бромпропан, 1-иодпропан, 2-бромпропан, 2-иодпропан, 1-иодбутан, 2-иодбутан, 1-иод-2-метилпропан или 1-(бромметил)циклопропан, может быть осуществлено в присутствии основания, такого как карбонат цезия (″Cs2CO3″) или карбонат калия (″K2CO3″), и растворителя, такого как диметилформамид (″DMF″), при температуре от примерно 20°С до примерно 100°С в течение от примерно 1 до примерно 12 часов.
Затем можно получить соединение формулы IXb путем обработки соединения формулы Xb восстанавливающим агентом, таким как DIBAL или LAH, в апротонном растворителе, таком как THF, толуол или диэтиловый эфир, при температуре от примерно -78°С до примерно 60°С в течение от примерно 1 до примерно 12 часов.
Соединение формулы VIb может быть образовано сначала в результате взаимодействия соединения формулы IXb с бромирующим агентом, таким как PBr3 или смесь тетрабромида углерода и трифенилфосфина, при температуре от примерно -20°С до примерно 60°С в течение от примерно 30 до примерно 120 минут с образованием соединения формулы VIIIb. Затем соединение формулы VIIIb приводят во взаимодействие с защищенным производным пиперидина - соединением формулы VIIb в присутствии сильного основания, такого как бис(триметилсилил)амид лития (″LiHMDS″) или диизопропиламин лития (″LDA″), в апротонном растворителе, таком как THF, толуол или диэтиловый эфир, при температуре от примерно -78°С до примерно 20°С в течение от примерно 1 до примерно 18 часов. Группа Pg представляет собой соответствующую защитную группа амина и предпочтительно BOC или Cbz.
Затем с соединения формулы VIb удаляют защиту путем гидролиза сложноэфирной группы в присутствии сильного водного основания, такого как гидроксид лития или гидроксид натрия, при температуре от примерно 0°С до примерно 100°С в течение от примерно 1 до примерно 18 часов с образованием содержащего группу карбоновой кислоты соединения формулы Vb. Затем может быть образовано изоцианатное соединение формулы IVb путем приведения во взаимодействие карбоновой кислоты формулы Vb с DPPA в присутствии основания, такого как EtaN или диизопропилэтиламин, при температуре от примерно 60°С до примерно 120°С в течение от примерно 1 до примерно 12 часов.
Затем может быть образовано лактамное соединение формулы 1Kb путем циклизации изоцианата (формула IVb) с использованием алкиллития, такого как н-BuLi или t-BuLi, при температуре от примерно -78°С до примерно 0°С в течение от примерно 5 до примерно 120 минут.
Затем с лактамного соединения формулы (IIIb) можно удалить защиту с получением свободного производного спиропиперидина формулы (IIb), используя стандартные методы в зависимости от того, какая защитная группа Pg была применена. Например, когда Pg представляет собой ВОС, то чтобы удалить группу ВОС, можно использовать стандартные условия удаления защиты с применением сильной кислоты, такой как 4 н соляная кислота в диоксане или трифторуксусная кислота в соответствующем растворителе, таком как дихлорметан. В том случае, когда Pg представляет собой Cbz, для удаления защиты можно использовать гидрирование над палладием на угле в этаноле или обработку с применением источника водорода, такого как формиат аммония или 1-метил-1,4-циклогексадиен, в присутствии палладия на угле в этаноле или этилацетате.
Затем, чтобы получить соединение формулы (Ib), производное спиропиперидина формулы (IIb) можно подвергнуть ацилированию путем применения стандартных способов. Например, в таком случае соединение (Ib) может быть образовано с использованием стандартной реакции пептидного сочетания с желаемой карбоновой кислотой (R2CO2H). Например, сочетание промежуточного спиропиперидина (IIb) и карбоновой кислоты (R2CO2H) может быть осуществлено посредством образования активированного сложного эфира карбоновой кислоты, например, путем приведения в контакт карбоновой кислоты (R2CO2H) с реагентом пептидного сочетания, таким как HATU или EDC·HCl, в присутствии или в отсутствие активирующего агента, такого как HOBt, и в присутствии подходящего основания, такого как DIEA, NMM, в подходящем растворителе, таком как THF и/или DMF, DMA или дихлорметан, и затем приведения в контакт этого активированного эфира карбоновой кислоты с производным спиропиперидина (IIb) с образованием соединения формулы (Ib).
На реакционной схеме III приведены общие методики, которые могут быть использованы для получения N2-лактамных АСС-ингибирующих соединений по настоящему изобретению, имеющих формулу Ic, в которых R1 представляет собой (С1-С6)алкил или (С3-С5)циклоалкил, и R2 представляет собой фенил, нафтил, 5-12-членный гетероарил или 8-12-членный конденсированный гетероциклический арил; где каждая группа R2 возможно замещена одним-тремя заместителями, независимо выбранными из (С1-С3)алкила, (С1-С3)алкокси, галогено и CONH2.
РЕАКЦИОННАЯ СХЕМА III
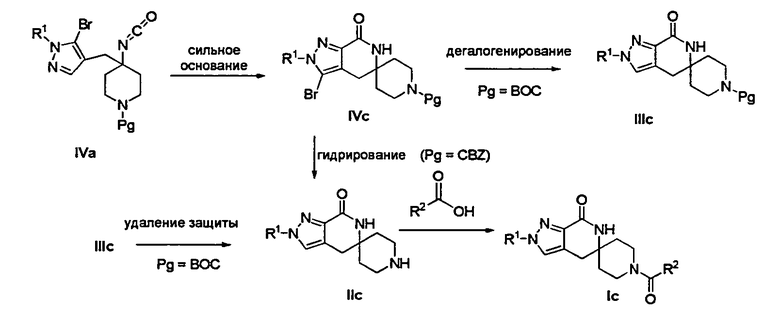
Лактамное соединение формулы IVc может быть образовано в результате циклизации изоцианата (формула IVa) с использованием сильного основания, такого как 2,2,6,6-тетраметилпиперидин лития (″LTMP″) или 2,2,6,6-тетраметилпиперидин магния, при температуре от примерно -78°С до примерно 0°С в течение от примерно 30 минут до примерно 6 часов.
Лактамное соединение формулы (IVc), где Pg представляет собой ВОС, затем может быть подвергнуто дегалогенированию с получением лактамного соединения формулы (IIIc) путем гидрирования в присутствии основания, такого как Et3N, над палладием на угле в этаноле или обработке с применением источника водорода, такого как формиат аммония или 1-метил-1,4-циклогексадиен, в присутствии основания, такого как Et3N, и палладия на угле в этаноле или этилацетате при температуре от примерно 20°С до примерно 100°С в течение от примерно 30 минут до примерно 6 часов.
Затем с лактамного соединения формулы (IIIc), где Pg представляет собой ВОС, можно удалить защиту с получением свободного производного спиропиперидина формулы (IIc), используя стандартные условия удаления защиты с применением сильной кислоты, такой как 4 н. соляная кислота в диоксане или трифторуксусная кислота в соответствующем растворителе, таком как дихлорметан, чтобы удалить группу ВОС.
Лактамное соединение формулы (IVc), где Pg представляет собой Cbz, может быть подвергнуто одновременно дегалогенированию и удалению защиты путем гидрирования над палладием на угле в этаноле или обработке с применением источника водорода, такого как формиат аммония или 1-метил-1,4-циклогексадиен, в присутствии палладия на угле в этаноле или этилацетате.
Затем, чтобы получить соединение формулы (Ic), производное спиропиперидина формулы (IIc) можно подвергнуть ацилированию путем применения стандартных способов. Например, в таком случае соединение (Ic) может быть образовано с использованием стандартной реакции пептидного сочетания с желаемой карбоновой кислотой (R2CO2H). Например, сочетание промежуточного спиропиперидина (IIc) и карбоновой кислоты (R2CO2H) может быть осуществлено посредством образования активированного сложного эфира карбоновой кислоты, например, путем приведения в контакт карбоновой кислоты (R2CO2H) с реагентом пептидного сочетания, таким как HATU или EDC·HCl, в присутствии или в отсутствие активирующего агента, такого как HOBt, и в присутствии подходящего основания, такого как DIEA, триэтиламин или NMM, в подходящем растворителе, таком как THF и/или DMF, DMA или дихлорметан, и затем приведения в контакт этого активированного эфира карбоновой кислоты с производным спиропиперидина (IIc) с образованием соединения формулы (Ic).
На реакционной схеме IV приведены общие методики, которые могут быть использованы для получения N2-лактамных АСС-ингибирующих соединений по настоящему изобретению, имеющих формулу Id, в которых R1 представляет собой (С1-С6)алкил или (С3-С5)циклоалкил, и R2 представляет собой фенил, нафтил, 5-12-членный гетероарил или 8-12-членный конденсированный гетероциклический арил; где каждая группа R2 возможно замещена одним-тремя заместителями, независимо выбранными из (С1-С3)алкила, (С1-С3)алкокси, галогено и CONH2.
РЕАКЦИОННАЯ СХЕМА IV
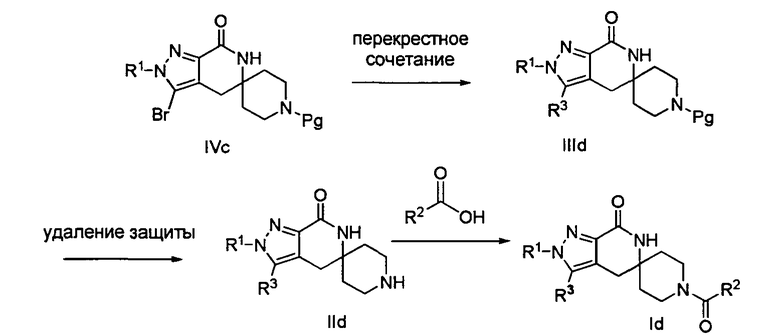
Лактамное соединение формулы IIId может быть образовано в результате палладий-катализируемого перекрестного сочетания бромида формулы IVc с алкил- или алкенил-трибутилстаннаном, таким как метил-три-н-бутилстаннан, винил-три-н-бутилстаннан или аллил-три-н-бутилстаннан, или триалкилбороксином, таким как триметилбороксин или тривинилбороксин, в присутствии палладиевого катализатора, такого как тетракис-(трифенилфосфин)палладий(0), или комбинации предкатализатора и лиганда, такой как ацетат палладия(II) и 2-дициклогексилфосфино-2',6'-диметоксибифенил (″SPhos″), и в присутствии или в отсутствие основания, такого как карбонат калия, в протонном растворителе, таком как этанол или трет-амиловый спирт, или апротонном растворителе, таком как тетрагидрофуран или диметилформамид, при температуре от примерно 20°С до примерно 100°С в течение от примерно 2 часов до примерно 18 часов или при температуре от примерно 100°С до примерно 150°С в течение от примерно 5 минут до примерно 60 минут при нагревании микроволнами. Если для введения группы R3 используют алкенилтриалкилстаннан или алкенилбороксин, то восстановление полученного олефина может быть осуществлено путем гидрирования над палладием на угле в этаноле или обработки с применением источника водорода, такого как формиат аммония или 1-метил-1,4-циклогексадиен, в присутствии палладия на угле в этаноле или этилацетате.
Затем с лактамного соединения формулы (IIId) можно удалить защиту с получением свободного производного спиропиперидина формулы (IId), используя стандартные методы в зависимости от того, какая защитная группа Pg была применена. Например, когда Pg представляет собой ВОС, то чтобы удалить группу ВОС, можно использовать стандартные условия удаления защиты с применением сильной кислоты, такой как 4 н. соляная кислота в диоксане или трифторуксусная кислота в соответствующем растворителе, таком как дихлорметан. В том случае, когда Pg представляет собой Cbz, для удаления защиты можно использовать гидрирование над палладием на угле в этаноле или обработку с применением источника водорода, такого как формиат аммония или 1-метил-1,4-циклогексадиен, в присутствии палладия на угле в этаноле или этилацетате.
Затем, чтобы получить соединение формулы (Id), производное спиропиперидина формулы (IId) можно подвергнуть ацилированию путем применения стандартных способов. Например, в таком случае соединение (Id) может быть образовано с использованием стандартной реакции пептидного сочетания с желаемой карбоновой кислотой (R2CO2H). Например, сочетание промежуточного спиропиперидина (IId) и карбоновой кислоты (R2CO2H) может быть осуществлено посредством образования активированного сложного эфира карбоновой кислоты, например, путем приведения в контакт карбоновой кислоты (R2CO2H) с реагентом пептидного сочетания, таким как HATU или EDC·HCl, в присутствии или в отсутствие активирующего агента, такого как HOBt, и в присутствии подходящего основания, такого как DIEA, триэтиламин или NMM, в подходящем растворителе, таком как THF и/или DMF, DMA или дихлорметан, и затем приведения в контакт этого активированного эфира карбоновой кислоты с производным спиропиперидина (IId) с образованием соединения формулы (Id).
На реакционной схеме V приведены общие методики, которые могут быть использованы для получения N2-лактамных АСС-ингибирующих соединений по настоящему изобретению, имеющих формулу Id, в которых R1 представляет собой (С1-С6)алкил или (С3-С5)циклоалкил, и R2 представляет собой фенил, нафтил, 5-12-членный гетероарил или 8-12-членный конденсированный гетероциклический арил; где каждая группа R2 возможно замещена одним-тремя заместителями, независимо выбранными из (С1-С3)алкила, (С1-С3)алкокси, галогено и CONH2.
РЕАКЦИОННАЯ СХЕМА V
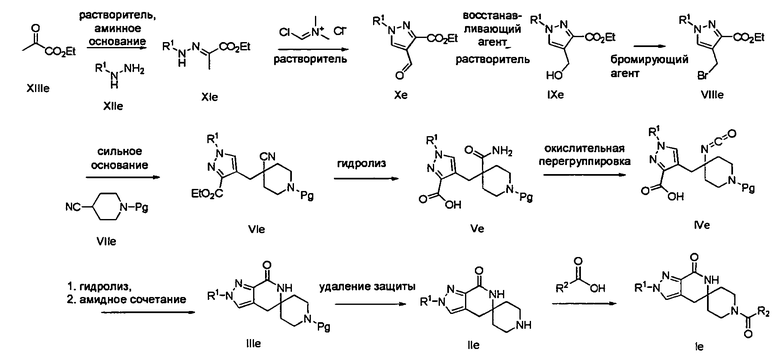
Согласно Схеме V соединение формулы XIe может быть получено в результате конденсации сложного кетоэфира - соединения формулы XIIIe с соответствующим гидрохлоридом алкилгидразина формулы XIIe, таким как гидрохлорид трет-бутилгидразина, в присутствии основания - третичного амина, такого как триэтиламин или N,N-диизопропилэтиламин, в полярном протонном растворителе, таком как этанол, при температуре от примерно 20°С до примерно 100°С в течение от примерно 1 до примерно 12 часов.
Соединение формулы Хе может быть получено путем обработки соединения формулы XIe (хлорметилен)диметилхлоридом аммония (солью Вильсмейера, Sigma-Aldrich, номер по кат. 280909) в непротонном растворителе, таком как диметилформамид или толуол или 1,2-дихлорэтан, при температуре от примерно 0°С до примерно 120°С в течение от примерно 1 до примерно 12 часов.
Соединение формулы IXe может быть получено путем обработки альдегида формулы Хе восстанавливающим агентом, таким как боргидрид натрия, в протонном растворителе, таком как метанол или этанол, при температуре от примерно 0°С до примерно 60°С в течение от примерно 1 до примерно 6 часов.
Соединение формулы VIe может быть образовано сначала в результате взаимодействия соединения формулы IXe с бромирующим агентом, таким как трибромид фосфора (″PBr3″) или смесь тетрабромида углерода и трифенилфосфина, при температуре от примерно -20°С до примерно 60°С в течение от примерно 30 до примерно 120 минут с образованием соединения формулы VIIIe. Затем соединение формулы VIIIe приводят во взаимодействие с защищенным производным 4-цианопиперидина - соединением формулы VIIe в присутствии сильного основания, такого как бис(триметилсилил)амид лития (″LiHMDS″) или диизопропиламин лития (″LDA″), в апротонном растворителе, таком как тетрагидрофуран (″THF″), толуол или диэтиловый эфир, при температуре от примерно -78°С до примерно 20°С в течение от примерно 1 до примерно 18 часов. Группа Pg представляет собой соответствующую защитную группа амина и предпочтительно N-трет-бутоксикарбонил (″BOC″) или карбобензилокси (″Cbz″).
Амидное соединение формулы Ve может быть получено в результате обработки нитрильного соединения формулы VIe в условиях гидролиза, как например, в присутствии водного, являющегося гидроксидом, основания, такого как гидроксид лития или гидроксид натрия, и растворителя, такого как метанол или этанол либо тетрагидрофуран, при температуре от примерно 20°С до примерно 100°С в течение примерно 1-12 часов. Альтернативно, можно использовать пероксидный комплекс, такой как гидроперит, в комбинации с водным являющимся, гидроксидом основанием, таким как гидроксид натрия, в растворителе, таком как метанол или этанол, при температуре от примерно 0°С до примерно 60°С в течение примерно 1-12 часов.
Перегруппировка амидного соединения формулы Ve в изоцианатное соединения формулы IVe может быть осуществлена в результате обработки таким реагентом, как (бис(трифторацетокси)иод)бензол, в присутствии неорганического основания, такого как бикарбонат натрия, в растворителе, таком как ацетонитрил, при температуре от примерно 20°С до примерно 60°С в течение примерно 1-6 часов.
Превращение изоцианатного соединения формулы IVe в лактамное соединение формулы IIIe сначала может быть осуществлено путем гидролиза изоцианата в присутствии водного являющегося гидроксидом основания, такого как гидроксид натрия или гидроксид лития, в растворителе, таком как метанол или тетрагидрофуран. Затем полученный амин можно обработать реагентом амидного сочетания, таким как 1-этил-3-(3-диметиламинопропил)карбодиимид или гексафторфосфат 2-(7-аза-1Н-бензотриазол-1-ил)-1,1,3,3-тетраметилурония, в присутствии являющегося алкиламином основания, такого как триэтиламин или N,N-диизопропилэтиламин, в растворителе, таком как дихлорметан или диметилформамид, при температуре от примерно 0°С до примерно 60°С в течение примерно 1-24 часов, с получением лактамного соединения формулы IIIe.
Затем с лактамного соединения формулы (IIIe) можно удалить защиту с получением свободного производного спиропиперидина формулы (IIe), используя стандартные методы в зависимости от того, какая защитная группа Pg была применена. Например, когда Pg представляет собой трет-бутилоксикарбонил (″ВОС″), то чтобы удалить группу ВОС, можно использовать стандартные условия удаления защиты с применением сильной кислоты, такой как 4 н. соляная кислота в диоксане или трифторуксусная кислота в соответствующем растворителе, таком как дихлорметан. В том случае, когда Pg представляет собой карбобензилокси (″Cbz″), для удаления защиты можно использовать гидрирование над палладием на угле в этаноле или обработку с применением источника водорода, такого как формиат аммония или 1-метил-1,4-циклогексадиен, в присутствии палладия на угле в этаноле или этилацетате.
Затем, чтобы получить соединение формулы (Ie), производное спиропиперидина формулы (IIe) можно подвергнуть ацилированию путем применения стандартных способов. Например, в таком случае соединение (Ie) может быть образовано с использованием стандартной реакции пептидного сочетания с желаемой карбоновой кислотой (R2CO2H). Например, сочетание промежуточного спиропиперидина (IIe) и карбоновой кислоты (R2CO2H) может быть осуществлено посредством образования активированного сложного эфира карбоновой кислоты, например, путем приведения в контакт карбоновой кислоты (R2CO2H) с реагентом пептидного сочетания, таким как HATU или EDC·HCl, в присутствии или в отсутствие активирующего агента, такого как гидроксибензотриазол (″HOBt″), и в присутствии подходящего основания, такого как DIEA, NMM, в подходящем растворителе, таком как THF и/или DMF, DMA или дихлорметан, и затем приведения в контакт этого активированного эфира карбоновой кислоты с производным спиропиперидина (IIe) с образованием соединения формулы (Ie).
Соединения по настоящему изобретению могут быть выделены и использованы в том виде, как они есть, или в форме своих фармацевтически приемлемых солей. Согласно настоящему изобретению соединения, имеющие многочисленные основные атомы азота, могут образовывать соли с различным числом эквивалентов (″экв.″) кислоты. Специалистам-практикам очевидно, что все такие соли включены в объем настоящего изобретения.
Фармацевтически приемлемые соли, как использовано в данном описании в отношении соединений по настоящему изобретению, включают фармацевтически приемлемые неорганические и органические соли соединения. Эти соли могут быть получены in situ в процессе окончательного выделения и очистки соединения или путем отдельного взаимодействия входящего в их состав соединения с подходящей органической или неорганической кислотой и выделения образовавшейся в результате этого соли. Репрезентативные соли включают, но не ограничиваются ими, соли гидробромид, гидрохлорид, гидроиодид, сульфат, бисульфат, нитрат, ацетат, трифторацетат, оксалат, безилат, пальмитат, памоат, малонат, стеарат, лаурат, малат, борат, бензоат, лактат, фосфат, гексафторфосфат, бензолсульфонат, тозилат, формиат, цитрат, малеат, фумарат, сукцинат, тартрат, нафтилат, мезилат, глюкогептонат, лактобионат и лаурилсульфонат, и тому подобное. Они также могут включать катионы щелочных и щелочноземельных металлов, таких как натрий, литий, калий, кальций, магний и тому подобное, а также нетоксичные катионы аммония, четвертичного аммония и аминов, включая аммоний, тетраметиламмоний, тетраэтиламмоний, метиламмоний, диметиламмоний, триметиламмоний, триэтиламмоний, этиламмоний и тому подобное, но не ограничиваются ими. Для дополнительных примеров см., например, Berge, et al., J. Pharm. Sci., 66, 1-19 (1977).
Соединения по настоящему изобретению могут существовать более чем в одной кристаллической форме. Полиморфы соединений формулы (I) и их солей (включая сольваты и гидраты) составляют часть данного изобретения и могут быть получены путем кристаллизации соединения по настоящему изобретению в различных условиях. Например, используя для перекристаллизации разные растворители или разные смеси растворителей; кристаллизацию при различных температурах; различные режимы охлаждения в процессе кристаллизации в диапазоне от очень быстрого до очень медленного охлаждения. Полиморфы также могут быть получены путем нагревания или плавления соединения по настоящему изобретению с последующим постепенным или быстрым охлаждением. Присутствие полиморфов можно определить посредством твердофазной спектроскопии ядерного магнитного резонанса (ЯМР), инфракрасной (ИК) спектроскопии, дифференциальной сканирующей калориметрии, дифракции рентгеновских лучей на порошке или других подобных методов.
Данное изобретение также включает меченые изотопом соединения, которые идентичны соединениям, описанным формулой (I), если не считать, что один или более атомов заменены атомами, имеющим атомную массу или массовое число, отличную(ое) от атомной массы или массового числа, обычно встречающихся в природе. Примеры изотопов, которые могут быть инкорпорированы в соединения по изобретению, включают изотопы водорода, углерода, азота, кислорода, серы, и фтора, такие как 2H, 3H, 13С, 14С, 15N, 18О, 17О, 35S, 36Cl, 125I, 129I и 18F, соответственно. Некоторые меченные изотопом соединения по настоящему изобретению, например те, в которые инкорпорированы такие радиоактивные изотопы, как 3H и 14С, применимы в анализах распределения лекарственного средства и/или субстрата в тканях. Изотопы трития (т.е. 3H) и углерода-14 (т.е. 14С) особенно предпочтительны ввиду простоты их получения и детекции. Кроме того, замещение на более тяжелые изотопы, такие как дейтерий (т.е. 2H), может давать некоторые терапевтические преимущества, обусловленные большей метаболической стабильностью, например, увеличенный in vivo период полувыведения или снижение дозировки, и поэтому может быть предпочтительным в некоторых обстоятельствах. В общем случае, меченные изотопом соединения по настоящему изобретению могут быть получены путем осуществления методик, описанных ниже на схемах и/или в разделе Примеры, с заменой не меченного изотопом реагента легко доступным меченным изотопом реагентом.
Соединения по настоящему изобретению могут содержать стереогенные центры. Эти соединения могут существовать в виде смесей энантиомеров или в виде чистых энантиомеров. В том случае, когда соединение включает стереогенный центр, соединения могут быть разделены на чистые энантиомеры методами, известными специалистам в данной области, например, путем образования диастереоизомерных солей, которые могут быть разделены, например, кристаллизацией; путем образования стереоизомерных производных или комплексов, которые могут быть разделены, например кристаллизацией, газожидкостной или жидкостной хроматографией; путем избирательного взаимодействия одного энантиомера со специфичным к энантиомеру реагентом, например, посредством ферментативной этерификации; или газожидкостной или жидкостной хроматографией в хиральной среде, например на хиральной подложке, такой как диоксид кремния с пришитым к нему хиральным лигандом или в присутствии хирального растворителя. Очевидно, что, если желаемый стереоизомер в результате осуществления одной из описанных выше методик разделения превращается в другую химическую разновидность, потребуется дополнительная стадия для высвобождения желаемой энантиомерной формы. Альтернативно, конкретные стереоизомеры можно синтезировать, используя оптически активное исходное вещество, применяя асимметрический синтез с использованием оптически активных реагентов, субстратов, катализаторов или растворителей либо превращая один стереоизомер в другой путем асимметрической трансформации.
Соединения по настоящему изобретению могут существовать в различных стабильных конформационных формах, которые могут поддаваться разделению. Благодаря ассиметрии крутильных колебаний, обусловленной ограничением вращения вокруг асимметричной одинарной связи, например, ввиду стерических затруднений или напряжения в кольце, можно разделить разные конформеры. Соединения по настоящему изобретению также включают каждый конформационный изомер соединений формулы 1 и их смеси.
Соединения по настоящему изобретению полезны для лечения заболеваний, состояний и/или расстройств, модулируемых посредством ингибирования фермента(ов) ацетил-КоА-карбоксилаз(ы) (в частности, АСС1 и АСС2). Другое воплощение настоящего изобретения относится к фармацевтической композиции, содержащей терапевтически эффективное количество соединения по настоящему изобретению и фармацевтически приемлемый эксципиент, разбавитель или носитель. Соединения по настоящему изобретению (в том числе композиции и способы, используемые в данном изобретении) также могут быть применены в изготовлении лекарственного средства для терапевтических применений, изложенных в данном описании.
Типичную композицию готовят путем смешивания соединения по настоящему изобретению и носителя, разбавителя или эксципиента. Подходящие носители, разбавители и эксципиенты хорошо известны специалистам в данной области техники и включают такие вещества, как углеводы, воски, растворимые и/или набухаемые в воде полимеры, гидрофильные или гидрофобные вещества, желатин, масла, растворители, воду и им подобное. Выбор конкретного используемого носителя, разбавителя или эксципиента будет зависеть от средства и назначения, по которому соединение по настоящему изобретению будет применяться. Как правило, растворители выбирают из растворителей, признанных специалистами в данной области безвредными (GRAS) для введения млекопитающему. Как правило, безвредными растворителями являются нетоксичные водные растворители, такие как вода и другие нетоксичные растворители, которые растворимы в воде или смешиваются с водой. Подходящие водные растворители включают воду, этанол, пропиленгликоль, полиэтиленгликоли (например, ПЭГ400, ПЭГ300) и т.д. и их смеси. Композиции также могут включать в себя одно или более из следующего: буферы, стабилизирующие агенты, поверхностно-активные вещества, увлажняющие агенты, смазывающие вещества, эмульгаторы, суспендирующие агенты, консерванты, антиоксиданты, придающие непрозрачность агенты, глиданты, технологические добавки, красители, подсластители, отдушки, корригенты и другие известные добавки для обеспечения наилучшей презентации лекарственного средства (т.е. соединения по настоящему изобретению или фармацевтической композиции на его основе) или содействия производству фармацевтического продукта (т.е. для применения в изготовлении лекарства).
Композиции могут быть изготовлены с использованием традиционных методик растворения и смешивания. Например, лекарственную субстанцию (то есть соединение по настоящему изобретению или стабилизированную форму соединения (например, комплекс с производным циклодекстрина или другим известным агентом комплексообразования)) растворяют в подходящем растворителе в присутствии одного или более эксципиентов, описанных выше. Скорость растворения слабо растворимых в воде соединений можно повысить путем применения диспергирования распылительной сушкой, такой как описанная Takeuchi H. и др. в ″Enhancement of the dissolution rate of a poorly water-soluble drug (tolbutamide) by a spray-drying solvent deposition method and disintegrants″ J. Pharm. Pharmacol., 39, 769-773 (1987); и в ЕР 0901786 В1 (US 2002/009494), включенного в данное описание посредством ссылки. Соединение по настоящему изобретению обычно вводят в состав фармацевтических лекарственных форм, обеспечивающих легко контролируемую дозировку лекарственного средства и дающих лучший продукт, с которым легко обращаться пациенту.
Фармацевтические композиции также включают сольваты и гидраты соединений по настоящему изобретению. Термин ″сольват″ относится к молекулярному комплексу соединения, представленного формулой (I) (включая его фармацевтически приемлемые соли), с одной или более чем одной молекулой растворителя. Молекулы таких растворителей представляют собой молекулы растворителей, обычно используемых в фармацевтической области, которые известны как безвредные для реципиента, например вода, этанол, этиленгликоль и тому подобное. Термин ″гидрат″ относится к комплексу, в) котором молекулой растворителя является вода. Сольваты и/или гидраты предпочтительно существуют в кристаллической форме. В качестве промежуточных сольватов при получении более желаемых сольватов могут быть использованы другие растворители, такие как метанол, метил-трет-бутиловый эфир, этилацетат, метилацетат, (S)-пропиленгликоль, (R)-пропиленгликоль, 1,4-бутин-диол и тому подобные.
Фармацевтическая композиция для применения может быть упакована различными способами в зависимости от способа, используемого для введения данного лекарства. В общем случае, изделие для распространения включает контейнер с размещенным в нем фармацевтическим препаратом в соответствующей форме. Подходящие контейнеры хорошо известны специалистам в данной области и включают такие материалы, как бутылки (пластиковые и стеклянные), саше, ампулы, пластиковые пакеты, металлические цилиндрические сосуды и тому подобное. Контейнер также может включать в себя блок, предохраняющий от неумелого обращения, для предупреждения неосторожного доступа к содержимому упаковки. Помимо этого, контейнер снабжен этикеткой с описанием содержимого контейнера. Этикетка также может содержать соответствующие предостережения.
Согласно настоящему изобретению также предложен способ лечения заболеваний, состояний и/или расстройств, модулируемых посредством ингибирования фермента(ов) ацетил-КоА-карбоксилаз(ы) у животного, включающий введение животному, нуждающемуся в таком лечении, терапевтически эффективного количества соединения по настоящему изобретению или фармацевтической композиции, содержащей эффективное количество соединения по настоящему изобретению и фармацевтически приемлемый эксципиент, разбавитель или носитель. Данный способ особенно полезен для лечения заболеваний, состояний и/или расстройств, на которые оказывает благоприятное воздействие ингибирование фермента(ов) ацетил-КоА-карбоксилаз(ы).
Один из аспектов настоящего изобретения относится к лечению ожирения и ассоциированных с ожирением расстройств (например, избыточного веса, прироста массы или поддержания веса).
Как правило, ожирение и избыточный вес определяют по индексу массы тела (BMI), который коррелирует с общим содержанием жира в организме и дает оценку относительного риска заболевания. BMI рассчитывают как массу в килограммах, деленную на рост в метрах в квадрате (кг/м2). Избыточный вес обычно определяют, когда BMI составляет 25-29,9 кг/м2, а ожирение обычно определяют, когда BMI составляет 30 кг/м2. См., например, National Heart, Lung, and Blood Institute, Clinical Guidelines on the Identification, Evaluation, and Treatment of Overweight and Obesity in Adults (Национальный институт сердца, легких и крови, Клиническое руководство по идентификации, оценке и лечению избыточного веса и ожирения у взрослых), The Evidence Report, Washington, DC: U.S. Department of Health and Human Services, NIH publication no. 98-4083 (1998)).
Другой аспект настоящего изобретения относится к лечению (например, задержке прогрессирования или начала) диабета или диабет-ассоциированных расстройств, включая диабет 1 типа (инсулинозависимый сахарный диабет, также обозначаемый как ″IDDM″) и 2 типа (инсулинонезависимый сахарный диабет, также обозначаемый как ″NIDDM″), нарушенную толерантность к глюкозе, инсулинорезистентность, гипергликемию и диабетические осложнения (такие как атеросклероз, коронарное заболевание сердца, инсульт, периферическое сосудистое заболевание, нефропатия, гипертензия, нейропатия и ретинопатия).
Еще в одном аспекте настоящего изобретения предложено лечение сопутствующих ожирению заболеваний, таких как метаболический синдром. Метаболический синдром включает такие заболевания, состояния или расстройства, как дислипидемия, гипертензия, инсулинорезистентность, диабет (например, диабет 2 типа), ишемическую болезнь сердца и сердечную недостаточность. Для более подробной информации о метаболическом синдроме см., например, Zimmet P.Z., et al., ″The Metabolic Syndrome: Perhaps an Etiologic Mystery but Far From a Myth - Where Does the International Diabetes Federation Stand?″, Diabetes & Endocrinology, 7(2), (2005); и Alberti K.G., et al., ″The Metabolic Syndrome - A New Worldwide Definition″, Lancet, 366, 1059-62 (2005). Предпочтительно, чтобы введение соединений по настоящему изобретению обеспечивало статистически значимое (р<0,05) уменьшение по меньшей мере одного фактора риска сердечнососудистых заболеваний, как например снижение содержания лептина, С-реактивного белка (CRP) и/или холестерина в плазме крови по сравнению с контролем, получающим препарат, не содержащий никакого лекарственного средства. Введение соединений по настоящему изобретению также может обеспечивать статистически значимое (р<0,05) уменьшение уровня глюкозы в сыворотке крови.
Еще в одном другом аспекте изобретения предложено лечение неалкогольной жировой болезни печени (NAFLD) и печеночной инсулинорезистентности.
Для нормального взрослого человека, имеющего массу тела примерно 100 кг, дозировка в диапазоне от примерно 0,001 мг до примерно 10 мг на килограмм массы тела обычно бывает достаточной, предпочтительно от примерно 0,01 мг/кг до примерно 5,0 мг/кг, более предпочтительно от примерно 0,01 мг/кг до примерно 1 мг/кг. Однако может потребоваться некоторая вариабельность в общем диапазоне дозировок в зависимости от возраста и массы подвергаемого лечению субъекта, планируемого способа введения, конкретного вводимого соединения и тому подобного. Определение диапазонов дозировок и оптимальных дозировок для конкретного пациента находится в пределах компетенции среднего специалиста в данной области, располагающего настоящим описанием. Также необходимо отметить, что соединения по настоящему изобретению могут быть использованы в форме препаратов с непрерывным высвобождением, регулируемым высвобождением и задержанным высвобождением, и такие формы также хорошо известны среднему специалисту в данной области.
Соединения по данному изобретению также могут быть использованы вместе с другими фармацевтическими агентами для лечения заболеваний, состояний и/или расстройств, изложенных в данном описании. Поэтому также предложены способы лечения, включающие введение соединений по настоящему изобретению в комбинации с другими фармацевтическими агентами. Подходящие фармацевтические агенты, которые могут быть использованы в комбинации с соединениями по настоящему изобретению, включают средства против ожирения (включая подавляющие аппетит средства), антидиабетические средства, антигипергликемические средства, снижающие содержание липидов агенты и антигипертензивные средства.
Подходящие снижающие содержание липидов агенты, которые могут быть объединены с соединениями по настоящему изобретению, включают, например, агенты, описанные в WO 2011005611 со страницы 30, строка 20, по страницу 31, строка 30. Снижающие содержание липидов агенты включают вещества, усиливающие экскрецию желчных кислот, ингибиторы HMG-KoA-редуктазы (3-гидрокси-3-метилглутарил-КоА-редуктазы), ингибиторы HMG-KoA-синтазы, ингибиторы всасывания холестерина, ингибиторы ацил-кофермент А-холестерол-ацилтрансферазы (АСАТ), ингибиторы СЕТР (белок-переносчик эфиров холестерина), ингибиторы скваленсинтетазы, агонисты PPARα (α-рецепторов, активируемых пролифератором пероксисом), модуляторы рецептора FXR (фарнезоидный Х-рецептор), модуляторы рецептора LXR (печеночный Х-рецептор), ингибиторы синтеза липопротеинов, ингибиторы ренин-ангиотензиновой системы, частичные агонисты PPARδ, ингибиторы обратного всасывания желчных кислот, агонисты PPARγ, ингибиторы синтеза триглицеридов, ингибиторы микросомального транспорта триглицеридов, модуляторы транскрипции, ингибиторы скваленэпоксидазы, индукторы рецепторов липопротеинов низкой плотности, ингибиторы агрегации тромбоцитов, ингибиторы 5-LO (5-липоксигеназа) или FLAP (белок, активирующий 5-липоксигеназу), связанный ниацином хром и другие агенты, влияющие на липидный состав.
Подходящие антигипертензивные средства, которые могут быть объединены с соединениями по настоящему изобретению, включают, например, средства, описанные в WO 2011005611 со страницы 31, строка 31, по страницу 32, строка 18. Антигипертензивные средства включают диуретики, бета-адренергические блокаторы, блокаторы кальциевых каналов, ингибиторы ангиотензин-превращающего фермента (АСЕ), ингибиторы нейтральных эндопептидаз, антагонисты рецепторов эндотелина, вазодилататоры, антагонисты рецепторов ангиотензина II, α/β-адреноблокаторы, альфа1-блокаторы, альфа2-агонисты, ингибиторы альдостерона, ингибиторы минералокортикоидных рецепторов, ингибиторы ренина и ангиопоэтин-2-связывающие вещества.
Подходящие антидиабетические средства включают ингибитор ацетил-КоА-карбоксилазы (АСС), например описанный в WO2009144554, WO2003072197, WO2009144555 и WO2008065508, ингибитор диацилглицерол-O-ацилтрансферазы 1 (DGAT-1), например описанный в WO09016462 или WO2010086820, AZD7687 или LCQ908, ингибитор диацилглицерол-O-ацилтрансферазы 2 (DGAT-2), ингибиторы моноацилглицерол-O-ацилтрансферазы, ингибитор фосфодиэстеразы(РОЕ10), активатор АМРК (АМФ-активируемая протеинкиназа), производные сульфонилмочевины (например, ацетогексамид, хлорпропамид, диабинез, глибенкламид, глипизид, глибурид, глимепирид, гликлазид, глипентид, гликвидон, глизоламид, толазамид и толбутамид), меглитинид, ингибитор α-амилазы (например, тендамистат, трестатин и AL-3688), ингибитор α-глюкозидгидролазы (например, акарбоза), ингибитор α-глюкозидазы (например, адипозин, камиглибоза, эмиглитат, миглитол, воглибоза, прадимицин-Q и салбостатин), агонист PPARγ (например, балаглитазон, циглитазон, дарглитазон, энглитазон, изаглитазон, пиоглитазон, розиглитазон и троглитазон), агонист PPARα/γ (например, CLX-0940, GW-1536, GW-1929, GW-2433, KRP-297, L-796449, LR-90, МК-0767 и SB-219994), бигуанид (например, метформин), модулятор глюкагоноподобного пептида-1 (GLP-1), такой как агонист (например, эксендин-3 и эксендин-4), лираглутид, албиглутид, эксенатид (Byetta®), албиглутид, таспоглутид, ликсисенатид, дулаглутид, семаглутид, NN-9924, ТТР-054, ингибитор протеинтирозинфосфатазы-1B (РТР-1В) (например, тродусквемин, экстракт из морских губок Hyrtios sp. и соединения, описанные в Zhang, S., et al., Drug Discovery Today, 12(9/10), 373-381 (2007)), ингибитор SIRT-1 (сиртуина-1) (например, ресвератрол, GSK2245840 или GSK184072), ингибитор дипептидилпептидазы IV (DPP-IV) (например, такой как в WO2005116014, ситаглиптин, вилдаглиптин, алоглиптин, дутоглиптин, линаглиптин и саксаглиптин), стимулятор секреции инсулина, ингибитор окисления жирных кислот, А2- антагонист, ингибитор c-Jun N-концевой киназы (JNK), активаторы глюкокиназы (GKa), например описанные в WO2010103437, WO2010103438, WO2010013161, WO2007122482, TTP-399, TTP-355, TTP-547, AZD1656, ARRY403, MK-0599, TAK-329, AZD5658 или GKM-001, инсулин, миметик инсулина, ингибитор гликогенфосфорилазы (например, GSK1362885), агонист рецептора VPAC2 (рецептора вазоактивного интестинального пептида 2), ингибиторы SGLT2 (натрий-зависимый переносчик глюкозы 2), например, описанные в Е.С.Chao et al. Nature Reviews Drug Discovery 9, 551-559 (July 2010), включая дапаглифлозин, канаглифлозин, BI-10733, тофоглифлозин (tofogimozin; CSG452), ASP-1941, THR1474, TS-071, ISIS388626 и LX4211, а также таковые из WO2010023594, модулятор рецептора глюкагона, например, описанный в Demong, D.E. et al. Annual Reports in Medicinal Chemistry 2008, 43, 119-137, модуляторы GPR119 (G-белок-связанный рецептор 119), в частности, агонисты, например, описанные в WO2010140092, WO2010128425, WO2010128414, WO2010106457, Jones, R.M. et al., Medicinal Chemistry 2009, 44, 149-170 (например, МВХ-2982, GSK1292263, APD597 и PSN821), производные или аналоги FGF21 (фактор роста фибробластов 21), например, описанные в Kharitonenkov, A. et al., Current Opinion in Investigational Drugs, 2009, 10(4) 359-364, модуляторы рецептора TGR5 (рецептор желчных кислот 5) (также называемого GPBAR1 (G-белок-связанный рецептор 1 желчных кислот)), в частности, агонисты, например, описанные в Zhong, M., Current Topics in Medicinal Chemistry, 2010, 10 (4), 386-396 и INT777, агонисты GPR40 (G-белок-связанный рецептор 40), например, описанные в Medina, J.C., Annual Reports in Medicinal Chemistry, 2008, 43, 75-85, включая ТАК-875, но этим не ограничиваясь, модуляторы GPR120 (G-белок-связанный рецептор 120), в частности, агонисты, активаторы высокоаффинного рецептора никотиновой кислоты (НМ74А) и ингибиторы SGLT1 (натрий-зависимый переносчик глюкозы 1), такие как GSK1614235. Дополнительный репрезентативный список антидиабетических средств, которые могут быть объединены с соединениями по настоящему изобретению, можно найти, например, в WO2011005611 со страницы 28, строка 35, по страницу 30, строка 19. Предпочтительными антидиабетическими средствами являются метформин и ингибиторы DPP-IV (например, ситаглиптин, вилдаглиптин, алоглиптин, дутоглиптин, линаглиптин и саксаглиптин). Другие антидиабетические средства могут включать ингибиторы или модуляторы ферментов карнитинпальмитоил-трансфераз, ингибиторы фруктозо-1,6-бифосфатазы, ингибиторы альдозоредуктазы, ингибиторы минералокортикоидных рецепторов, ингибиторы TORC2 (transducer of regulated CREB (cAMP responsive element binding protein) activity 2; белок, связывающийся с цАМФ-зависимым элементом 2), ингибиторы CCR2 (С-С-хемокиновый рецептор 2) и/или CCR5, ингибиторы изоформ РКС (протеинкиназа С) (например, РКСα, РКСβ, РКСγ), ингибиторы синтетазы жирных кислот, ингибиторы серинпальмитоилтрансферазы, модуляторы GPR81 (G-белок-связанный рецептор 81), GPR39 (G-белок-связанный рецептор 39), GPR43 (G-белок-связанный рецептор 43), GPR41 (G-белок-связанный рецептор 41), GPR105 (G-белок-связанный рецептор 105), Kv1.3 (калиевый канал подтип 1.3), ретинолсвязывающего белка 4, глюкокортикоидного рецептора, соматостатиновых рецепторов (например, SSTR1, SSTR2, SSTR3 и SSTR5), ингибиторы или модуляторы PDHK2 (киназа пируватдегидрогеназы 2) или PDHK4 (киназа пируватдегидрогеназы 4), ингибиторы МАР4К4 (митоген-активируемая киназа киназы 4 протеинкиназы 4), модуляторы семейства IL1 (интерлейкинов 1), включая IL1 бета, модуляторы РХРальфа (ретиноидный X-рецептор альфа). Помимо это, подходящие антидиабетические средства включают механизмы, перечисленные в Carpino P.A., Goodwin В. Expert Opin. Ther. Pat., 2010, 20 (12), 1627-51.
Подходящие средства против ожирения (некоторые из них также могут действовать в качестве антидиабетических средств) включают ингибиторы 11β-гидроксистероид-дегидрогеназы-1 (11β-HSD, тип 1), ингибитор стеароил-КоА-десатуразы-1 (SCD-1), агонисты MCR-4 (рецептор меланокортина-4), агонисты холецистокинина-А (ССК-А), ингибиторы обратного захвата моноаминов (такие как сибутрамин), симпатомиметические средства, β3-адренергические агонисты, дофаминовые агонисты (такие как бромкриптин), аналоги меланоцит-стимулирующего гормона, агонисты 5НТ2с рецепторов (серотониновых рецепторов типа 2 с), антагонисты меланин-концентрирующий гормона, лептин (белок OB; кодируется геном ожирения (obese gene)), аналоги лептина, агонисты лептина, антагонисты галанина, ингибиторы липаз (такие как тетрагидролипстатин, т.е. орлистат), анорексигенные средства (такие как агонист бомбезина), антагонисты нейропептида-Y (например, антагонисты NPY Y5, такие как велнеперит), пептид PYY3-36 (пептид тирозин-тирозин) (включая его аналоги), модулятор BRS3 (рецептор бомбезина, подтип 3), смешанные антагонисты подтипов опиодных рецепторов, тиромиметические средства, дегидроэпиандростерон или его аналог, агонисты или антагонисты глюкокортикоидов, антагонисты орексина, агонисты глюкагоноподобного пептида-1, цилиарные нейротрофические факторы (такие как Axokine™, поставляемый Regeneron Pharmaceuticals, Inc., Tarrytown, NY и Procter & Gamble Company, Cincinnati, ОН), ингибиторы агути-связанного белка (AGRP) человека, антагонисты или обратные агонисты гистаминового Н3-рецептора, агонисты нейромедина U, ингибиторы МТР/АроВ (микросомальный белок-переносчик триглицеридов/аполипопротеин В) (например, действующие избирательно в кишечнике ингибиторы МТР, такие как дирлотапид, JTT130, усистапид (Usistapide), SL×4090), опиоидный антагонист, модуляторы мюпиоидных рецепторов, включая, но не ограничиваясь этим, GSK1521498, ингибиторы MetAp2 (метионинаминопептидаза 2), включая, но не ограничиваясь этим, ZGN-433, агенты со смешанной модуляторной активностью в отношении 2-х или более рецепторов глюкагона, GIP (желудочный ингибирующий полипептид) и GLP1 (глюкогоноподобный пептид-1) рецепторов, такие как MAR-701 или ZP2929, ингибиторы переносчика норэпинефрина, антагонисты/обратные агонисты, агонисты/антагонисты грелина, оксинтомодулин и аналоги, ингибиторы обратного захвата моноаминов, такие как, но не ограничиваясь этим, тезофензин, антагонист орексина, комбинированные агенты (как например, бупропион плюс зонисамид, прамлинтид плюс метрелептин, бупропион плюс налтрексон, фентермин плюс топирамат) и тому подобное.
Предпочтительные средства против ожирения для применения в аспектах настоящего изобретения, относящихся к комбинациям, включают действующие избирательно в кишечнике ингибиторы МТР (например, дирлотапид, митратапид и имплитапид, R56918 (№ в CAS (от англ. Chemical Abstracts Service) 403987) и № в CAS 913541-47-6), агонисты ССКа (например, N-бензил-2-[4-(1Н-индол-3-илметил)-5-оксо-1-фенил-4,5-дигидро-2,3,6,10b-тетрааза-бензо[е]азулен-6-ил]-N-изопропил-ацетамид, описанный в публикации РСТ (от англ. Patent Cooperation Treaty) № WO 2005/116034 или публикации США №2005-0267100 А1), агонисты 5НТ2с (например, лоркасерин), агонист MCR4 (например, соединения, описанные в US 6818658), ингибиторы липаз (например, цетилистат), PYY3-36 (как использовано в данном описании, ″PYY3-36″ включает такие аналоги, как пегилированный PYY3-36, например, описанные в публикации US 2006/0178501), опиоидные антагонисты (например, налтрексон), олеоилэстрон (№ в CAS 180003-17-2), обинепитид (ТМ30338), прамлинтид (Symlin®), тезофензин (NS2330), лептин, бромкриптин, орлистат, AOD-9604 (№ в CAS 221231-10-3) и сибутрамин. Предпочтительно, соединения по настоящему изобретению и комбинированные терапии назначают в сочетании с физическими упражнениями и разумной диетой.
Все приведенные патенты и публикации США (включая все технические бюллетени, на которые сделаны ссылки в разделе Примеры) включены в данное описание посредством ссылки во всей своей полноте.
Примеры, приведенные в данном описании ниже, даются только в целях иллюстрации. Композиции, способы и различные параметры, отраженные в данном описании, предназначены только для пояснения различных аспектов и воплощений изобретения и никоим образом не предназначены для ограничения объема заявляемого изобретения.
Вещества, описанные ниже, были использованы в синтезе соединений, приведенных в последующих примерах.
Для получения соединений, описанных ниже в разделе Примеры, были использованы следующие имеющиеся в продаже исходные вещества: метил-3-иод-1Н-индазол-5-карбоксилат (Anichem LLC, North Brunswick, NJ), (1R,5S)-8-(трет-бутоксикарбонил)-8-азабицикло[3.2.1]октан-3-карбоновая кислота (AstaTech, Inc., Bristol, PA), 6-бромизохинолин-3-амин (Ark Pharm, Inc., Libertyville, IL), 3-гидрокси-1Н-индазол-5-карбоновая кислота (Aces Pharma, Inc., Branford, CT), этил-хинолин-7-карбоксилат (ASW MedChem, Inc., New Brunswick, NJ), 7-бромизохинолин-1(2Н)-он (Alfa Aesar, Ward Hill, MA), 3-оксо-2,3-дигидро-1Н-индазол-6-карбоновая кислота (ASW MedChem, Inc., New Brunswick, NJ), 5-бром-3-(трифторметил)-1Н-индазол (J&W PharmLab LLC., Levittown, PA), 6-бромизохинолин-1(2Н)-он (Anichem LLC, North Brunswick, NJ), метил-1H-пирроло[3,2-b]пиридин-6-карбоксилат (ACS Scientific Inc., Metuchen, NJ), 4-бром-2-фтор-N-метилбензамид (Oakwood Products, Inc., West Columbia, SC), 7-бром-3-хлоризохинолин (Allichem LLC, Baltimore, MD), 7-бромизохинолин-3-амин (Allichem LLC, Baltimore, MD), 6-бромизохинолин-3-ол (Ark Pharm, Inc., Libertyville, IL), 1Н-пирроло[2,3-b]пиридин-5-карбоновая кислота (ASDI Inc., Newark, DE), 1-хлоризохинолин-7-карбоновая кислота (American Custom Chemicals Corp., San Diego, CA), 3,7-диметил-1Н-индазол-5-карбоновая кислота (Annker Organics Co. Ltd., Wuhan, China), 7-метил-1Н-индазол-5-карбоновая кислота (J&W PharmLab LLC, Levittown, PA), 2-метил-2Н-индазол-5-карбоновая кислота (Bepharm Ltd., Shanghai, China), 1Н-пирроло[3,2-b]пиридин-6-карбоновая кислота (Sinova Inc., Bethesda, MD), 7-хлор-1Н-индазол-5-карбоновая кислота (Annker Organics Co. Ltd., Wuhan, China), 4-метокси-1Н-индазол-6-карбоновая кислота (ASW MedChem Inc., New Brunswick, NJ), 1-метил-1Н-индазол-5-карбоновая кислота (J&W PharmLab LLC, Levittown, PA), 7-этил-1Н-индазол-5-карбоновая кислота (Annker Organics Co. Ltd., Wuhan, China), 3-этил-1Н-индазол-5-карбоновая кислота (Allichem LLC, Baltimore, MD), 3-метил-1Н-индазол-5-карбоновая кислота (Ark Pharm Inc., Libertyville, IL), 1H-пирроло[3,2-b]пиридин-2-карбоновая кислота (Aces Pharma Inc., Branford, CT), хинолин-3-карбоновая кислота (Beta Pharma Inc., Branford, CT), хинолин-7-карбоновая кислота (Ark Pharm Inc., Libertyville, IL), изохинолин-6-карбоновая кислота (Ark Pharm Inc., Libertyville, IL), изохинолин-7-карбоновая кислота (Indofine Chemical Company Inc., Hillsborough, NJ), 6-метоксихинолин-3-карбоновая кислота (Princeton Biomolecular Research Inc., Monmouth Junction, NJ), 4-метокси-7-метил-1Н-индол-2-карбоновая кислота (Aurora Fine Chemicals LLC, San Diego, CA), 2-аминохинолин-6-карбоновая кислота (Princeton Biomolecular Research Inc., Monmouth Junction, NJ), 8-метоксихинолин-3-карбоновая кислота (BioBlocks Inc., San Diego, CA), 2-аминохинолин-7-карбоновая кислота (Princeton Biomolecular Research Inc., Monmouth Junction, NJ), 2-метил-1Н-бензо[d]имидазол-5-карбоновая кислота (Acros Organics, Geel, Belgium), 1Н-индазол-5-карбоновая кислота (Sigma Aldrich, St. Louis, МО), хинолин-6-карбоновая кислота (Acros Organics, Geel, Belgium), 6-метокси-2-нафтойная кислота (Sigma Aldrich, St. Louis, МО), 1Н-индазол-6-карбоновая кислота (Sigma Aldrich, St. Louis, МО), 1Н-бензо[d][1,2,3]триазол-5-карбоновая кислота (Sigma Aldrich, St. Louis, МО), 3,4-диамино-5-хлорбензойная кислота (Princeton BioMolecular Research, Inc., Monmouth Junction, NJ), 7-бром-1-хлоризохинолин (Alfa Aesar, Ward Hill, MA) 7-бромхинолин (Anichem LLC, North Brunswick, NJ).
С использованием опубликованных ранее способов были получены следующие карбоновые кислоты (которые использовали для получения соединений, описанных ниже в разделе Примеры): 3,7-диметил-1Н-индазол-5-карбоновая кислота (публикация РСТ № WO2009144554), 7-метил-1Н-индазол-5-карбоновая кислота (публикация РСТ № WO2009144554), 7-метокси-2-нафтойная кислота (публикация РСТ № WO2003018586), 5-метокси-2-нафтойная кислота (публикация РСТ № WO2003072578), 4,8-диметоксихинолин-2-карбоновая кислота (публикация РСТ № WO2007011809), 3-хлор-7-метил-1Н-индазол-5-карбоновая кислота (публикация РСТ № WO2009144554), 3-хлор-1Н-индазол-5-карбоновая кислота (публикация РСТ № WO2009144554), 8-метокси-2-нафтойная кислота (публикация РСТ № WO2003072578), 3-хлор-1Н-индол-5-карбоновая кислота (публикация РСТ № WO2008065508), 3-хлор-1Н-индол-6-карбоновая кислота (публикация РСТ № WO2008065508), 7-метокси-3-метил-1Н-индазол-5-карбоновая кислота (WO2009144554), 4,8-диметоксихинолин-2-карбоновая кислота (публикация РСТ № WO2007011809).
ПРИМЕРЫ
Составление названий соединений и промежуточных соединений, описанных ниже, выполняли, используя способ присвоения названий, приведенный в Chemdraw Ultra, версия 11.0.1 (CambridgeSoft Corp., Cambridge Massachusetts). Способ присвоения названий, приведенный в Chemdraw Ultra, версия 11.0.1, хорошо известен специалистам в данной области техники, и считается, что способ присвоения названий, приведенный в Chemdraw Ultra, версия 11.0.1, в целом согласуется с рекомендациями IUPAC (International Union for Pure ans Applied Chemistry; Международный союз теоретической и прикладной химии) по номенклатуре органических соединений и правилами индексации в CAS (химическая реферативная служба). Если не указано иное, все реактивы получали из коммерческих источников. Все ссылки, приведенные в данном описании ниже, включены посредством ссылки.
Флэш-хроматографию проводили способом, описанным в Still et al., J. Org. Chem., 1978, 43, 2923.
Все Biotage®-очистки, рассмотренные в данном описании, выполняли, используя колонки Biotage® SNAP, содержащие диоксид кремния KP-SIL (40-63 мкм, 60 ангстрем) (Biotage AB; Uppsala, Sweden).
Все Combiflash®-очистки, рассмотренные в данном описании, выполняли, используя систему CombiFlash® Companion system (Teledyne Isco; Lincoln, Nebraska), в которой применены упакованные диоксидом кремния колонки RediSep®.
Масс-спектры регистрировали на спектрометре (Micromass PIatform II) для микромасс от Waters (Waters Corp.; Milford, MA). Если не указано иное, масс-спектры регистрировали на спектрометре (Micromass Platform II) для микромасс от Waters (Milford, MA).
Химические сдвиги протонного ЯМР приведены в частях на миллион относительно тетраметилсилана, и их регистрировали на спектрометре Varian Unity 300, 400 или 500 МГц (мегагерц) (Varian Inc.; Palo Alto, CA). Химические сдвиги ЯМР приведены в частях на миллион относительно тетраметилсилана (для протона) или фтортрихлорметана (для фтора).
Времена удерживания для HPLC (высокоэффективная жидкостная хроматография) измеряли, используя следующие методы: метод А: колонка: Atlantis dC18 (4,6×50 мм; 5 мкм) от Waters; подвижная фаза А: 0,05%-ная TFA (трифторуксусная кислота) в воде (об./об.); подвижная фаза В: 0,05%-ная TFA в ацетонитриле (об./об.); градиент: линейный от 95% N5% В до 5% N95% В в течение 4,0 минут, выдерживание при 5% N95% В до 5,0 минут; скорость потока: 2,0 мл/минута. Метод В: колонка: XBridge C18 (4,6×50 мм; 5 мкм) от Waters; подвижная фаза А: 0,03%-ный NH4OH в воде (об./об.); подвижная фаза В: 0,03%-ный NH4OH в ацетонитриле (об./об.); градиент: линейный от 95% N5% В до 5% N95% В в течение 4,0 минут, выдерживание при 5% N95% В до 5,0 минут; скорость потока: 2,0 мл/минута.
Способы получения, описанные ниже, использовали в синтезе соединений, приведенных в следующих далее примерах.
Способы получения промежуточных соединений и исходных веществ
Промежуточные карбоновые кислоты приобретали путем закупок, получали, как описано ниже, получали, как описано в публикации РСТ № WO 2009/144554, получали с использованием способов получения, общеизвестных специалистам в данной области, или получали аналогично методам, описанным выше для других промежуточных карбоновых кислот.
Промежуточное соединение 1: гидрохлоридная соль 1'-трет-бутил-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(1'Н)-она
Стадия 1. Этил-5-бром-1-трет-бутил-1Н-пиразол-4-карбоксилат
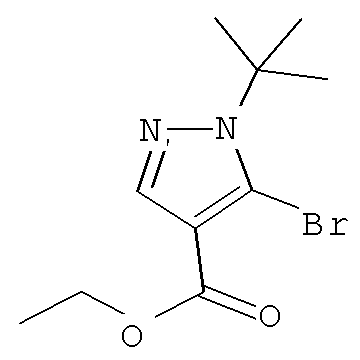
К раствору этил-5-амино-1-трет-бутил-1Н-пиразол-4-карбоксилата (674 мг; 3,19 ммоль; Li et al. J. Heterocycl. Chem., 2007, 44, 749) в ацетонитриле (20 мл) добавляли бромид меди(II) (720 мг; 3,19 ммоль) и изоамилнитрит (0,56 мл; 4,15 ммоль). Золотистую суспензию нагревали при 45°С в течение 2 часов и затем охлаждали до комнатной температуры, разбавляли этилацетатом (100 мл) и промывали насыщенным водным бикарбонатом натрия (50 мл), водой (50 мл) и рассолом (50 мл). Органическую фазу сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали флэш-хроматографией (5-40% этилацетат/гептаны; 10 г силикагеля), получая 685 мг этил-5-бром-1-трет-бутил-1Н-пиразол-4-карбоксилата в виде прозрачного масла. +APCl (химическая ионизация при атмосферном давлении) (М+Н) 275,0; 1H ЯМР (400 МГц, CDCl3, δ): 7.87 (s, 1Н), 4.32 (q, J=7,0 Гц, 2Н), 1.77 (s, 9H), 1.36 (t, J=7,1 Гц, 3Н).
Стадия 2: (5-бром-1-трет-бутил-1Н-пиразол-4-ил)метанол
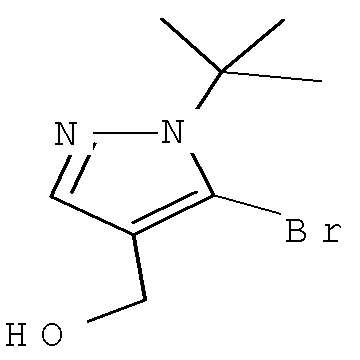
Раствор этил-5-бром-1-трет-бутил-1Н-пиразол-4-карбоксилата (685 мг; 2,49 ммоль) в THF (20 мл) охлаждали до -78°С и обрабатывали гидридом диизобутилалюминия (7,47 мл; 7,47 ммоль; 1 М THF), добавляя его по каплям. Смесь перемешивали при -78°С в течение 30 минут и затем нагревали до комнатной температуры в течение 18 часов. Смесь гасили этилацетатом (10 мл) и перемешивали в течение 15 минут. Затем смесь обрабатывали насыщенной водной сегнетовой солью (25 мл) и перемешивали в течение 1 часа при комнатной температуре. Смесь разбавляли этилацетатом (100 мл) и промывали водой (100 мл). Органический слой сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали флэш-хроматографией (градиент 10-80% этилацетат/гептан; 25 г силикагеля), получая 460 мг (5-бром-1-трет-бутил-1Н-пиразол-4-ил)метанола в виде прозрачного масла. +APCl (М+Н) 233,1; (М+2+Н) 235,1; 1H ЯМР (400 МГц, CDCl3, δ): 7.51 (s, 1H), 4.53 (d, 2Н), 1.74 (s, 9H), 1.55 (t, J=5,8 Гц, 1Н).
Стадия 3: 5-бром-4-(бромметил)-1-трет-бутил-1Н-пиразол
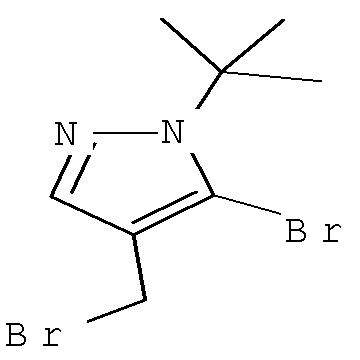
Раствор (5-бром-1-трет-бутил-1Н-пиразол-4-ил)метанола (460 мг; 1,97 ммоль) в дихлорметане (25 мл) охлаждали до 0°С и затем обрабатывали бромидом фосфора(III) (0,37 мл; 3,46 ммоль), добавляя его по каплям в течение 5 минут. Смесь перемешивали 30 минут при 0°С и затем 1 час при комнатной температуре. Смесь медленно гасили водой (50 мл), перемешивали 30 минут и затем экстрагировали этилацетатом (2×50 мл). Органическую фазу промывали насыщенным водным бикарбонатом натрия (50 мл), сушили над сульфатом натрия, фильтровали и концентрировали, получая 362 мг 5-бром-4-(бромметил)-1-трет-бутил-1Н-пиразола в виде прозрачного масла. 1H ЯМР (400 МГц, CDCl3, δ): 7.54 (s, 1H), 4.39 (s, 2H), 1.74 (s, 9H).
Стадия 4: 1-трет-бутил-4-этил-4-((5-бром-1-трет-бутил-1Н-пиразол-4-ил)метил)пиперидин-1,4-дикарбоксилат
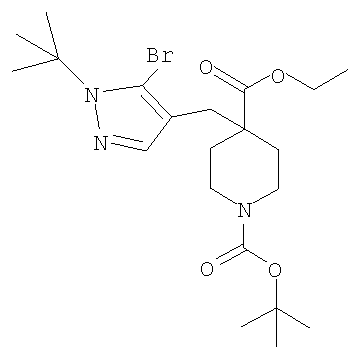
Раствор 1-трет-бутил-4-этил-пиперидин-1,4-дикарбоксилата (0,37 мл; 1,47 ммоль) в THF (15 мл) охлаждали до -78°С и затем обрабатывали бис(триметилсилил)амидом лития (1,48 мл; 1,48 ммоль; 1 М в толуоле), добавляя его по каплям. Реакционную смесь перемешивали 15 минут при -78°С, нагревали до 0°С в течение 30 минут и затем опять охлаждали до -78°С. Добавляли раствор 5-бром-4-(бромметил)-1-трет-бутил-1Н-пиразола (335 мг; 1,13 ммоль) в THF (10 мл), смесь перемешивали 1 час при -78°С и затем оставляли перемешиваться в течение 18 часов при комнатной температуре. Реакцию гасили насыщенным водным хлоридом аммония (20 мл), перемешивали 30 минут при комнатной температуре, разбавляли водой (50 мл) и экстрагировали этилацетатом (2×50 мл). Органические экстракты объединяли, сушили над сульфатом натрия, фильтровали и концентрировали. Полученный остаток очищали флэш-хроматографией (5-40% этилацетат/гептан; 25 г силикагеля), получая 256 мг 1-трет-бутил-4-этил-4-((5-бром-1-трет-бутил-1Н-пиразол-4-ил)метил)пиперидин-1,4-дикарбоксилата в виде прозрачного масла. +ESI (электрораспылительная ионизация) (М+Н) 474,2; (М+2+Н) 476,2; 1H ЯМР (400 МГц, CDCl3, δ): 7.20 (s, 1H), 4.16 (q, J=7,2 Гц, 2Н), 3.93 (br. s., 2H), 2.84 (m, 2H), 2.66 (s, 2H), 2.10 (d, J=12,5 Гц, 2Н), 1.72 (s, 9H), 1.45 (m, 1 1Н), 1.25 (t, J=7,1 Гц, 3Н).
Стадия 5: 4-((5-бром-1-трет-бутил-1Н-пиразол-4-ил)метил)-1-(трет-бутоксикарбонил)пиперидин-4-карбоновая кислота
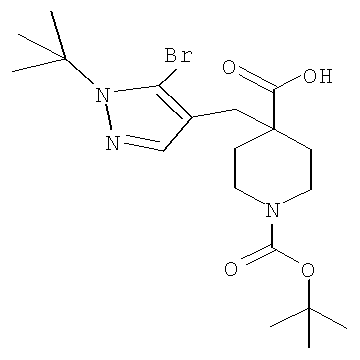
К раствору 1-трет-бутил-4-этил-4-((5-бром-1-трет-бутил-1Н-пиразол-4-ил)метил)пиперидин-1,4-дикарбоксилата (256 мг; 0,54 ммоль) в метаноле (15 мл) добавляли водный 2,5 M NaOH (5 мл) и полученную смесь нагревали при температуре дефлегмации в течение 18 часов. Смесь охлаждали до комнатной температуры и удаляли метанол при пониженном давлении. Полученную суспензию переносили в 25 мл воды, подкисляли водным 1 н. HCl и затем экстрагировали этилацетатом (2×50 мл). Объединенные органические экстракты сушили над сульфатом натрия, фильтровали и концентрировали, получая 241 мг 4-((5-бром-1-трет-бутил-1Н-пиразол-4-ил)метил)-1-(трет-бутоксикарбонил)пиперидин-4-карбоновой кислоты в виде бесцветного твердого вещества. +APCl (М+Н) 444,2; (М+2+Н) 446,2; 1H ЯМР (400 МГц, CDCl3, δ): 7.35 (s, 1H), 3.95 (br. s., 2H), 2.92 (br. s., 2H), 2.71 (s, 2H), 2.08 (d, J=12,9 Гц, 2H), 1.73 (s, 9H), 1.50 (m, 11H).
Стадия 6: трет-бутил-4-((5-бром-1-трет-бутил-1Н-пиразол-4-ил)метил)-4-изоцианатопиперидин-1-карбоксилат
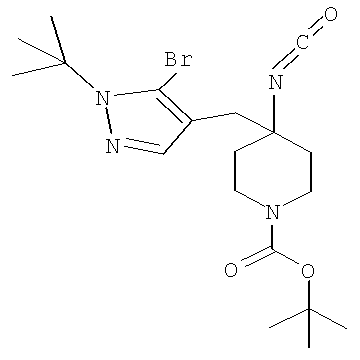
К раствору 4-((5-бром-1-трет-бутил-1Н-пиразол-4-ил)метил)-1-(трет-бутоксикарбонил)пиперидин-4-карбоновой кислоты (241 мг; 0,54 ммоль) в толуоле (10 мл) добавляли триэтиламин (91 мкл; 0,65 ммоль) и дифенилфосфорилазид (0,14 мл; 0,65 ммоль). Смесь нагревали при 120°С в течение 3 часов, реакционную смесь охлаждали и летучие вещества удаляли при пониженном давлении. Полученное масло очищали флэш-хроматографией (25 г диоксида кремния; градиент 7-60% этилацетат/гептан), получая 225 мг трет-бутил-4-((5-бром-1-трет-бутил-1Н-пиразол-4-ил)метил)-4-изоцианато-пиперидин-1-карбоксилата в виде прозрачного масла. +APCl (М+Н) 385,1; 1H-ЯМР (400 МГц, CDCl3, δ): 7.40 (s, 1H), 4.03 (br. s., 2H), 2.97 (br. t, J=12,3, 12,3 Гц, 2H), 2.70 (s, 2H), 1.74 (s, 9H), 1.67 (m, 2H), 1.62 (m, 2H), 1.46 (s, 9H).
Стадия 7: трет-бутил-1'-трет-бутил-7'-оксо-1',4',6',7'-тетрагидро-спиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-1-карбоксилат
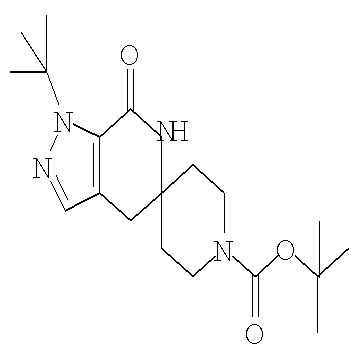
Раствор трет-бутил-4-((5-бром-1-трет-бутил-1Н-пиразол-4-ил)-метил)-4-изоцианатопиперидин-1-карбоксилата (225 мг; 0,51 ммоль) в THF (10 мл) охлаждали до -78°С и по каплям в течение 2 минут добавляли трет-бутиллитий (0,6 мл; 1,7 М в пентане). Смесь перемешивали 30 минут при -78°С, нагревали до 0°С и затем гасили насыщенным водным NH4Cl (20 мл). Смесь перемешивали 30 минут при комнатной температуре, разбавляли водой (25 мл) и затем экстрагировали этилацетатом (2×50 мл). Объединенные органические экстракты сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали флэш-хроматографией (12-100% этилацетат/гептан; 10 г силикагеля), получая 137 мг трет-бутил-1'-трет-бутил-7'-оксо-1',4',6',7'-тетрагидро-спиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-1-карбоксилата в виде бесцветного твердого вещества. +ESI (M-tBu) 307,2; 1H-ЯМР (400 МГц, DMSO-d6, δ): 7.74 (s, 1H), 7.30 (s, 1H), 3.51 (m, 2H), 3.20 (m, 2H), 2.79 (s, 2H), 1.64 (s, 9H), 1.56 (t, J=5,8 Гц, 4Н), 1.38 (s, 9H).
Стадия 8: гидрохлоридная соль 1'-трет-бутил-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(1'Н)-она
К раствору трет-бутил-1'-трет-бутил-7'-оксо-1',4',6',7'-тетрагидро-спиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-1-карбоксилата (137 мг; 0,39 ммоль) в этилацетате (4 мл) добавляли 4 н. HCl в диоксане (2 мл). После перемешивания в течение 1 часа при комнатной температуре летучие вещества удаляли при пониженном давлении и полученное бесцветное твердое вещество растирали в гептане (10 мл), получая 112 мг указанного в заголовке соединения в виде бесцветного твердого вещества. +APCl (М+Н) 263,3; 1H ЯМР (400 МГц, DMSO-d6, δ): 8.84 (m, 2H), 8.00 (s, 1H), 7.29 (s, 1H), 3.13 (d, J=6,1 Гц, 2H), 3.03 (br. s., 2H), 2.78 (s, 2H), 1.76 (m, 4H), 1.60 (s, 9H).
Промежуточное соединение 2: показанный ниже 1'-изопропил-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(1'Н)-он получали следующим образом.
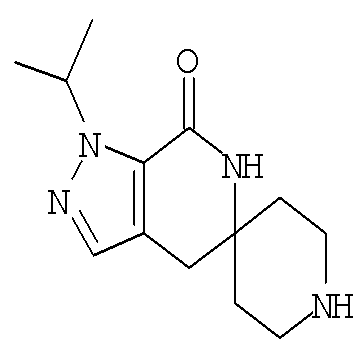
Стадия 1: 5-амино-1-изопропил-1Н-пиразол-4-карбоксилат

Смесь этил-2-циано-3-этоксиакрилата (84,4 г; 0,50 моль), гидрохлорида изопропилгидразина (55,2 г; 0,50 моль) и карбоната калия (68,8 г; 0,50 моль) в смеси 90%-ный этанол/метанол (1,5 л) нагревали при температуре дефлегмации в течение 16 часов. Затем растворитель удаляли в вакууме и добавляли воду и этилацетат. Смесь разделяли, органический слой сушили над сульфатом магния, фильтровали и растворитель удаляли в вакууме, получая этил-5-амино-1-изопропил-1Н-пиразол-4-карбоксилат (92,4 г; 94%). +ESI (М+Н) 198,1; 1H ЯМР (400 МГц, CDCl3, δ): 7.63 (s, 1H), 4.97 (br. s., 2H), 4.28 (q, 2H), 4.18 (m, 1H), 1.45 (d, 6H), 1.31 (t, 3H).
Стадия 2: 5-амино-1-изопропил-1Н-пиразол-4-карбоксилат
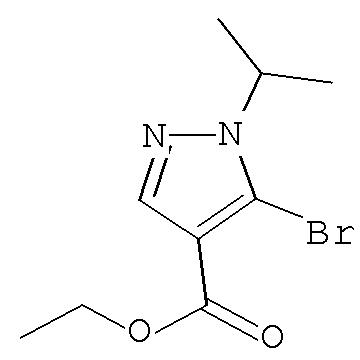
К смеси этил-5-амино-1-изопропил-1Н-пиразол-4-карбоксилата (107,5 г; 0,55 моль) в ацетонитриле (1 л) добавляли бромид меди(II) (182,6 г; 0,82 моль) при комнатной температуре в атмосфере аргона. Смесь нагревали до 50°С и по каплям добавляли изоамилнитрит (109,8 мл; 0,82 моль) (наблюдали экзотермический эффект и повышение температуры до 65°С). Реакционную смесь перемешивали при 50°С в течение 2 часов, затем смесь охлаждали до комнатной температуры, выливали в 2 М HCl, перемешивали в течение 15 минут и после этого дважды экстрагировали этилацетатом. Органические слои объединяли, промывали рассолом, затем насыщенным водным бикарбонатом натрия, сушили над сульфатом магния, фильтровали и растворитель удаляли в вакууме, получая этил-5-бром-1-изопропил-1Н-пиразол-4-карбоксилат (163 г; количественный выход), который использовали на следующей стадии без дополнительной очистки. 1H ЯМР (400 МГц, CDCl3, δ): 7.97 (s, 1Н), 4.77 (m, 1H), 4.28 (q, 2H), 1.35 (t, 3H), 0.90 (d, 6H).
Стадия 3: (5-бром-1-изопропил-1Н-пиразол-4-ил)метанол
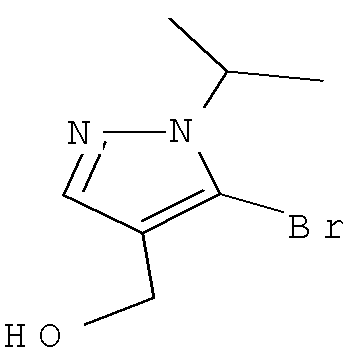
К раствору этил-5-бром-1-изопропил-1Н-пиразол-4-карбоксилата (163 г; 0,50 моль) в 2-метил-тетрагидрофуране (400 мл) в атмосфере аргона при 0°С добавляли боран-DMS (диметилсульфид) (140 мл; 1,50 моль) (выделение газа прекращалось после добавления 50 мл). Смесь перемешивали при комнатной температуре в течение 30 минут, далее нагревали до 70°С в течение 2 часов и затем до температуры дефлегмации в течение 17 часов. Добавляли дополнительную порцию боран-DMS (40 мл) и смесь перемешивали при температуре дефлегмации в течение еще 3 часов. Смесь охлаждали до комнатной температуры, затем постепенно с перемешиванием добавляли к охлажденному во льду метанолу (500 мл) в течение 30 минут. Смесь перемешивали при комнатной температуре в течение 30 минут, после чего добавляли 2 М водный гидроксид натрия (1,5 л). Слои разделяли и водный слой экстрагировали этилацетатом (2×500 мл). Органические слои объединяли, промывали рассолом (500 мл), сушили над сульфатом магния, фильтровали и растворитель удаляли в вакууме. Неочищенный продукт очищали флэш-хроматографией на сухой колонке (0-50% этилацетата в гептане), получая (5-бром-1-изопропил-1Н-пиразол-4-ил)метанол (70,8 г; 65% за две стадии). +ESI (М+Н) 220,9; 1H ЯМР (400 МГц, CDCl3, δ): 7.52 (s, 1H), 4.67 (m, 1H), 4.47 (s, 2H), 2.59 (br. s., 1H), 1.41 (s, 6H).
Стадия 4: 5-бром-4-(бромметил)-1-изопропил-1Н-пиразол
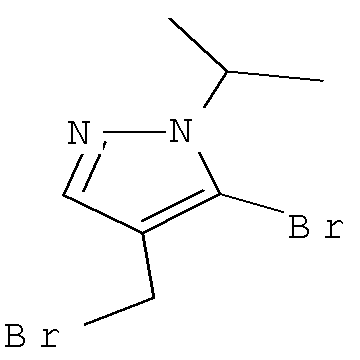
К перемешиваемому раствору (5-бром-1-изопропил-1Н-пиразол-4-ил)метанола (10,0 г; 45,7 ммоль) в дихлорметане (200 мл) добавляли PBr3 (6,5 мл; 68,5 ммоль) при 0°С. По завершении добавления смесь оставляли нагреваться до комнатной температуры и перемешивали в течение 3 часов. Смесь выливали в ледяную воду (300 мл), встряхивали, разделяли и после этого дважды промывали охлажденной до температуры замерзания водой (2×100 мл) и затем рассолом (100 мл), сушили над сульфатом натрия, фильтровали и растворитель удаляли в вакууме, получая 5-бром-4-(бромметил)-1-изопропил-1Н-пиразол (12,2 г; 95%). 1Н ЯМР (300 МГц, CDCl3, δ): 7.58 (s, 1H), 4.64 (m, 1H), 4.35 (s, 2H), 1.43 (d, 6H).
Стадия 5: 1-трет-бутил-4-этил-4-((5-бром-1-изопропил-1Н-пиразол-4-ил)метил)пиперидин-1,4-дикарбоксилат
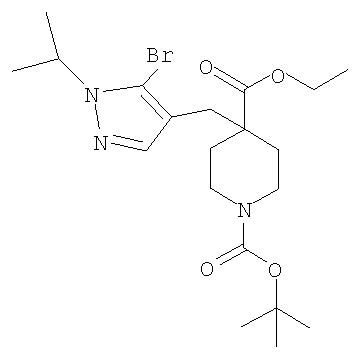
К перемешиваемому раствору 1-трет-бутил-4-этил-пиперидин-1,4-дикарбоксилата (14,5 г; 56,3 ммоль) в 2-метилтетрагидрофуране (120 мл) в атмосфере аргона по каплям добавляли 1 M LiHMDS в тетрагидрофуране (57 мл; 56,3 ммоль) при -78°С. Через 20 мин добавляли 5-бром-4-(бромметил)-1-изопропил-1Н-пиразол (12,2 г; 43,3 ммоль) в 2-метилтетрагидрофуране (10 мл). Смесь оставляли нагреваться до комнатной температуры и перемешивали в течение 18 часов. Смесь разбавляли водой (200 мл) и проводили разделение смеси. Органическую фазу промывали 10%-ным раствором лимонной кислоты (2×100 мл), затем рассолом (100 мл), сушили над сульфатом натрия, фильтровали и растворитель удаляли в вакууме. Неочищенный продукт очищали колоночной флэш-хроматографией (10-30% этилацетата в гептане), получая 1-трет-бутил-4-этил-4-((5-бром-1-изопропил-1Н-пиразол-4-ил)метил)пиперидин-1,4-дикарбоксилат (9,3 г). Из колонки также выделяли 7,1 г фракции, представляющей собой смесь исходного эфира и желаемого продукта. Ее перемешивали с 1 эквивалентом гидроксида натрия в смеси 90%-ный этанол/метанол в течение 2 часов при комнатной температуре. Растворитель удаляли в вакууме и добавляли этилацетат (100 мл). Смесь промывали 2 н. гидроксидом натрия (2×50 мл), затем рассолом (100 мл), сушили над сульфатом натрия, фильтровали и растворитель удаляли в вакууме, получая вторую порцию 1-трет-бутил-4-этил-4-((5-бром-1-изопропил-1Н-пиразол-4-ил)метил)пиперидин-1,4-дикарбоксилата (5,1 г). Объединенный выход составлял 14,4 г (72%). +ESI (М+Н) 404,0; 1H ЯМР (400 МГц, CDCl3, δ): 4.62 (m, 1Н), 4.12 (q, 2Н), 3.90 (br. s., 2H), 2.82 (m, 2H), 2.63 (s, 2H), 2.08 (d, 2H), 1.66 (m, 2H), 1.42 (s, 9H), 1.21 (t, 3H).
Стадия 6: 4-((5-бром-1-изопропил-1Н-пиразол-4-ил)метил)-1-(трет-бутоксикарбонил)пиперидин-4-карбоновая кислота
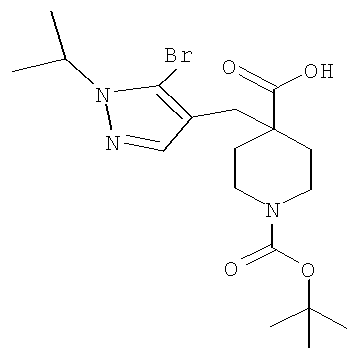
К раствору 1-трет-бутил-4-этил-4-((5-бром-1-изопропил-1Н-пиразол-4-ил)метил)пиперидин-1,4-дикарбоксилата (14,5 г; 31,6 ммоль) в метаноле (50 мл) добавляли гидроксид лития (1,52 г; 36,2 ммоль) и смесь перемешивали при 80°С в течение 18 часов. Добавляли дополнительную порцию гидроксида лития (2,55 г; 63,3 ммоль) и смесь нагревали при энергичном кипячении с обратным холодильником в течение 3 часов, охлаждали до комнатной температуры, растворитель удаляли в вакууме. Остаток промывали этилацетатом, фильтровали и фильтрат сохраняли. Твердые вещества растворяли в 2 н. водном гидроксиде натрия (40 мл) и затем подкисляли до pH 5 10%-ным раствором лимонной кислоты. Водный раствор экстрагировали этилацетатом (3×40 мл), органические экстракты объединяли, сушили над сульфатом магния, фильтровали и затем объединяли с исходным фильтратом. Растворитель из фильтрата удаляли при пониженном давлении и полученный остаток очищали колоночной флэш-хроматографией (этилацетат/гептаны), что позволило получить 4-((5-бром-1-изопропил-1Н-пиразол-4-ил)метил)-1-(трет-бутоксикарбонил)пиперидин-4-карбоновую кислоту (10,1 г; 74%) в виде бесцветного твердого вещества. +ESI (М+Н) 429,9; 1H ЯМР (300 МГц, CDCl3, δ): 7.41 (s, 1Н), 4.64 (m, 1Н), 3.94 (m, 2Н), 2.95 (m, 2H), 2.68 (m, 2H), 2.09 (m, 2H), 1.47 (m, 17H).
Стадия 7: трет-бутил-4-((5-бром-1-изопропил-1Н-пиразол-4-ил)метил)-4-изоцианатопиперидин-1-карбоксилат
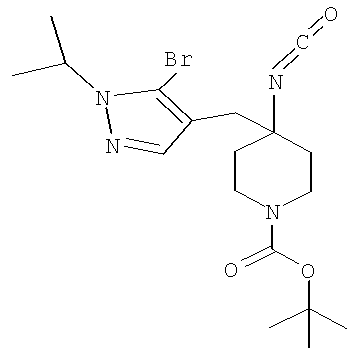
Смесь 4-((5-бром-1-изопропил-1Н-пиразол-4-ил)метил)-1-(трет-бутоксикарбонил)пиперидин-4-карбоновой кислоты (2,54 г; 5,9 ммоль), дифенилфосфорилазида (1,79 г; 6,5 ммоль) и триэтиламина (0,91 мл; 6,5 ммоль) в толуоле (15 мл) нагревали при температуре дефлегмации в течение 3 часов. Затем смесь охлаждали до комнатной температуры и растворитель удаляли в вакууме. Неочищенный продукт очищали колоночной хроматографией, получая трет-бутил-4-((5-бром-1-изопропил-1Н-пиразол-4-ил)метил)-4-изоцианатопиперидин-1-карбоксилат (2,8 г; 100%). 1H ЯМР (300 МГц, CDCl3, δ): 7.47 (s, 1Н), 4.68 (m, 1Н), 3.99 (m, 2H), 2.95 (m, 2H), 2.67 (s, 2H), 1.62 (m, 4H), 1.45 (m, 15H).
Стадия 8: трет-бутил-1'-изопропил-7'-оксо-1',4',6',7'-тетрагидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-1-карбоксилат
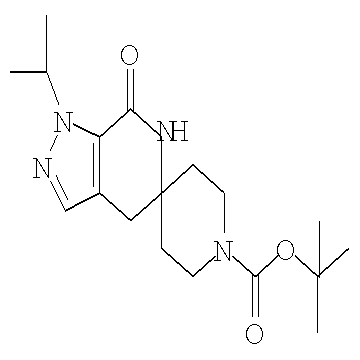
К смеси трет-бутил-4-((5-бром-1-изопропил-1Н-пиразол-4-ил)метил)-4-изоцианатопиперидин-1-карбоксилата (1,4 г; 3,3 ммоль) в 2-метил-тетрагидрофуране (10 мл) в атмосфере аргона при -78°С добавляли трет-бутиллитий (1,7 М в гексане, 4,3 мл; 7,2 ммоль). По завершении добавления смесь оставляли нагреваться до комнатной температуры и перемешивали в течение 18 часов. Смесь гасили водой (10 мл) и затем разбавляли этилацетатом (20 мл). Слои разделяли, органический слой промывали рассолом (10 мл), сушили над сульфатом натрия, фильтровали и растворитель удаляли в вакууме. Неочищенный продукт очищали колоночной флэш-хроматографией, получая трет-бутил-1'-изопропил-7'-оксо-1',4',6',7'-тетрагидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-1-карбоксилат (0,77 г; 67%). +ESI (М+Н) 374,1; 1H-ЯМР (300 МГц, CDCl3, δ): 7.34 (s, 1H), 6.35 (s, 1H), 5.45 (m, 1H), 3.57 (m, 2H), 3.42 (m, 2H), 2.79 (s, 2H), 1.70 (m, 4H), 1.45 (m, 15H).
Стадия 9: 1'-изопропил-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(1'Н)-он
К раствору трет-бутил-1'-изопропил-7'-оксо-1',4',6',7'-тетрагидро-спиро[пиперидин-4,5′-пиразоло[3,4-с]пиридин]-1-карбоксилата (100 мг; 0,29 ммоль) в 4 мл этилацетата добавляли 4 н. HCl в диоксане (2 мл). После перемешивания в течение 30 минут при комнатной температуре добавляли метанол (1 мл) и полученный раствор перемешивали в течение 5 часов при комнатной температуре. Летучие вещества удаляли при пониженном давлении и полученное бесцветное твердое вещество растирали в смеси 1:1 ацетонитрил/дихлорметан, получая 71 мг указанного в заголовке соединения в виде бесцветного твердого вещества. 1H ЯМР (400 МГц, DMSO-d6, δ): 8.72 (br. s., 2H), 8.05 (s, 1H), 7.37 (s, 1H), 5.36 (m, 1H), 3.15 (m, 2H), 3.05 (m, 2H), 2.78 (s, 2H), 1.78 (m, 4H), 1.33 (d, J=6,6 Гц, 6Н).
Промежуточное соединение 3: показанную ниже гидрохлоридную соль 2'-изопропил-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(2'Н)-она получали следующим образом.
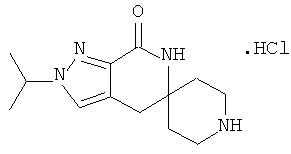
Стадия 1: этил-3-иод-1-изопропил-1Н-пиразол-4-карбоксилат
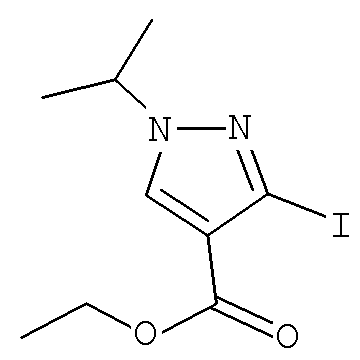
К раствору этил-3-иодпиразол-4-карбоксилата (1,58 г; 5,94 ммоль; Truong; et al., Bioorg. Med. Chem. Lett., 19, 4920 (2009)) в 20 мл N,N-диметилформамида добавляли карбонат цезия (3,87 г; 11,9 ммоль) и 2-иодпропан (0,89 мл; 8,90 ммоль). Смесь перемешивали 2 часа при 60°С и затем охлаждали до температуры окружающей среды. Реакционную смесь разбавляли 150 мл воды и экстрагировали диэтиловым эфиром (2×100 мл). Объединенные органические экстракты промывали 50 мл рассола, сушили над сульфатом натрия, фильтровали и концентрировали. Полученное масло очищали флэш-хроматографией (градиент 7-60% этилацетат/гептан; 50 г диоксида кремния), получая 340 мг этил-5-иод-1-изопропил-1Н-пиразол-4-карбоксилата в виде прозрачного масла, которое кристаллизовалось при стоянии, и 740 мг этил-3-иод-1-изопропил-1Н-пиразол-4-карбоксилата в виде прозрачного масла. Этил-5-иод-1-изопропил-1Н-пиразол-4-карбоксилат: +APCl (М+Н) 309,0; 1H ЯМР (400 МГц, CDCl3, δ): 8.05 (s, 1Н), 4.82 (spt, J=6,6 Гц, 1Н), 4.33 (q, J=7,2 Гц, 2Н), 1.50 (d, J=6,6 Гц, 6Н), 1.37 (t, J=7,1 Гц, 3Н). Этил-3-иод-1-изопропил-1Н-пиразол-4-карбоксилат: +APCl (М+Н) 309,0; 1H ЯМР (400 МГц, CDCl3, δ): 7.84 (s, 1Н), 4.52 (spt, J=6,7 Гц, 1Н), 4.32 (q, J=7,1 Гц, 2Н), 1.52 (d, J=6,6 Гц, 6Н), 1.37 (t, J=7,1 Гц, 3H).
Стадия 2: (3-иод-1-изопропил-1Н-пиразол-4-ил)метанол

Раствор этил-3-иод-1-изопропил-1Н-пиразол-4-карбоксилата (740 мг; 2,40 ммоль) в тетрагидрофуране (20 мл) охлаждали до -78°С и обрабатывали гидридом диизобутилалюминия (1,5 М в толуоле; 0,8 мл; 7,21 ммоль), добавляя его по каплям. Смесь перемешивали при -78°С в течение 1 ч и затем нагревали до комнатной температуры в течение 2 часов. Смесь гасили 10 мл этилацетата, перемешивали в течение 15 минут и затем обрабатывали 25 мл насыщенной водной сегнетовой соли. После перемешивания в течение еще 1 часа при комнатной температуре смесь разбавляли 50 мл этилацетата и промывали 100 мл воды. Водный слой дополнительно экстрагировали 50 мл этилацетата. Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали. Затем остаток очищали флэш-хроматографией (12-100% этилацетат/гептаны; 25 г силикагеля), получая 630 мг (3-иод-1-изопропил-1Н-пиразол-4-ил)метанола в виде прозрачного масла. +APCl (М+Н) 266,8; 1H ЯМР (400 МГц, CDCl3, δ): 7.37 (s, 1H), 4.49 (m, 3Н), 1.67 (t, J=5,9 Гц, 1Н), 1.50 (s, 6H).
Стадия 3: 4-(бромметил)-3-иод-1-изопропил-1Н-пиразол
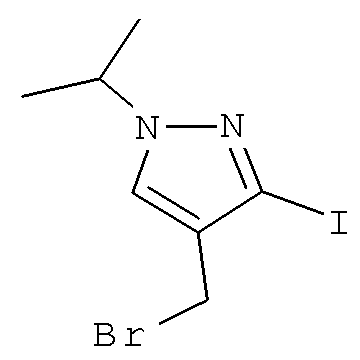
Раствор (3-иод-1-изопропил-1Н-пиразол-4-ил)метанола (0,63 г; 2,37 ммоль) в 20 мл дихлорметана охлаждали до 0°С. К раствору добавляли бромид фосфора(III) (0,67 мл; 7,10 ммоль) и смесь перемешивали 30 минут при 0°С, 1 час при комнатной температуре, затем гасили 50 мл воды и перемешивали в течение 15 минут при комнатной температуре. Смесь обрабатывали насыщенным водным бикарбонатом натрия и экстрагировали этилацетатом (2×50 мл). Объединенные органические слои промывали рассолом (50 мл), сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали флэш-хроматографией (10-80% этилацетат/гептаны; 25 г силикагеля), получая 400 мг 4-(бромметил)-3-иод-1-изопропил-1H-пиразола в виде бесцветного твердого вещества. +APCl (М+Н) 329,0; 1H ЯМР (400 МГц, CDCl3, δ): 7.42 (s, 1Н), 4.47 (spt, J=6,7 Гц, 1Н), 4.35 (s, 2H), 1.50 (d, J=6,6 Гц, 6H).
Стадия 4: 1-трет-бутил-4-этил-4-((3-иод-1-изопропил-1Н-пиразол-4-ил)метил)пиперидин-1,4-дикарбоксилат
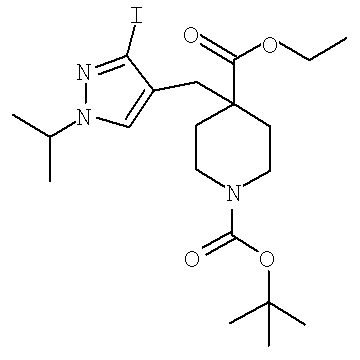
Раствор 1-трет-бутил-4-этил-пиперидин-1,4-дикарбоксилата (0,54 мл; 2,11 ммоль) в тетрагидрофуране (15 мл) в сухой 100 мл круглодонной колбе в атмосфере азота охлаждали до -78°С и затем обрабатывали бис(триметилсилил)амидом лития (1 М в толуоле; 2,13 мл; 2,13 ммоль). После перемешивания в течение 45 минут при -78°С добавляли 4-(бромметил)-3-иод-1-изопропил-1Н-пиразол (535 мг; 1,63 ммоль) в виде суспензии в 10 мл тетрагидрофурана. Смесь перемешивали в течение 1 часа при -78°С и затем оставляли перемешиваться в течение 18 часов при комнатной температуре. Реакционную смесь гасили 20 мл насыщенного водного хлорида аммония, перемешивали 30 минут при комнатной температуре, разбавляли 50 мл воды и затем экстрагировали этилацетатом (2×50 мл). Объединенные органические экстракты сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали флэш-хроматографией (10-80% этилацетат/гептаны; 25 г силикагеля), получая 1-трет-бутил-4-этил-4-((3-иод-1-изопропил-1Н-пиразол-4-ил)метил)пиперидин-1,4-дикарбоксилат (645 мг) в виде прозрачного масла. +ESI (M-tBu) 450,1; 1H ЯМР (400 МГц, CDCl3, δ): 7.02 (s, 3H), 4.44 (spt, J=6,6 Гц, 1Н), 4.17 (m, 2H), 3.92 (m, 2H), 2.86 (m, 2H), 2.62 (s, 2H), 2.08 (m, 2H), 1.46 (m, 17H), 1.25 (t, J=7,1 Гц, 3Н).
Стадия 5: 1-(трет-бутоксикарбонил)-4-((3-иод-1-изопропил-1Н-пиразол-4-ил)метил)пиперидин-4-карбоновая кислота
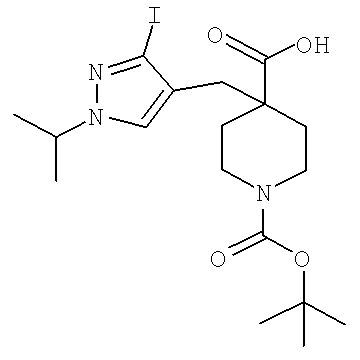
К раствору 1-трет-бутил-4-этил-4-((3-иод-1-изопропил-1Н-пиразол-4-ил)метил)пиперидин-1,4-дикарбоксилата (455 мг; 0,9 ммоль) в метаноле (20 мл) добавляли 2 н. NaOH (5 мл). После перемешивания в течение 18 часов при комнатной температуре метанол удаляли при пониженном давлении, полученную суспензию переносили в 20 мл воды, подкисляли 2 н. HCl и экстрагировали этилацетатом (2×30 мл). Объединенные органические экстракты сушили над сульфатом натрия, фильтровали и концентрировали, получая 1-(трет-бутоксикарбонил)-4-((3-иод-1-изопропил-1Н-пиразол-4-ил)метил)пиперидин-4-карбоновую кислоту (430 мг) в виде бесцветного твердого вещества. -APCl (М-Н) 476,1; 1H ЯМР (400 МГц, CDCl3, δ): 7.11 (s, 1H), 4.45 (dquin, J=13,4, 6,7 Гц, 1H), 3.95 (br. s., 2H), 2.91 (m, 2H), 2.69 (s, 2H), 2.08 (m, 2H), 1.47 (m, 8H).
Стадия 6: трет-бутил-4-((3-иод-1-изопропил-1Н-пиразол-4-ил)метил)-4-изоцианатопиперидин-1-карбоксилат
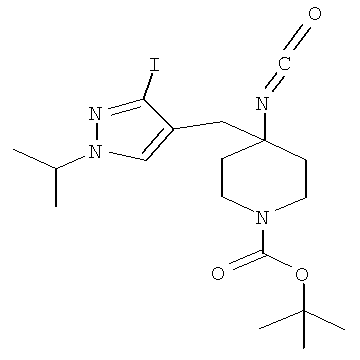
К раствору 1-(трет-бутоксикарбонил)-4-((3-иод-1-изопропил-1Н-пиразол-4-ил)метил)пиперидин-4-карбоновой кислоты (430 мг; 0,90 ммоль) в толуоле (10 мл) добавляли триэтиламин (0,15 мл; 1,08 ммоль) и дифенилфосфорилазид (0,24 мл; 1,08 ммоль). Смесь нагревали при 120°С в течение 3 часов, летучие вещества удаляли при пониженном давлении и полученное масло очищали флэш-хроматографией (7-60% этилацетат/гептаны; 25 г силикагеля), получая трет-бутил-4-((3-иод-1-изопропил-1Н-пиразол-4-ил)метил)-4-изоцианато-пиперидин-1-карбоксилат (280 мг) в виде прозрачного масла. (ФП-ИК (инфракрасная спектроскопия с Фурье-преобразованием)) (см-1): 2253; 1H ЯМР (400 МГц, CDCl3, δ): 7.27 (s, 1Н), 4.50 (m, 1Н), 4.03 (br. s., 2H), 2.97 (br. s., 2H), 2.65 (s, 2H), 1.65 (m, 4H), 1.50 (s, 6H), 1.47 (s, 9H).
Стадия 7: трет-бутил-2'-изопропил-7'-оксо-2',4',6',7'-тетрагидроспиро-[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-1-карбоксилат
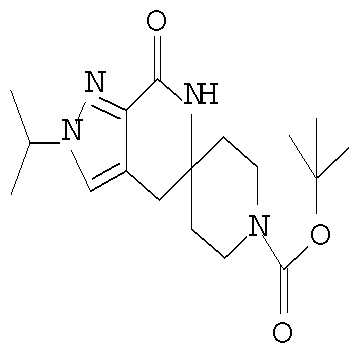
К раствору трет-бутил-4-((3-иод-1-изопропил-1Н-пиразол-4-ил)метил)-4-изоцианатопиперидин-1-карбоксилата (280 мг; 0,59 ммоль) в тетрагидрофуране (10 мл) при -78°С по каплям добавляли трет-бутиллитий (0,7 мл; 1,7М в пентане). После перемешивания в течение 30 минут при -78°С смесь нагревали до 0°С, гасили 20 мл насыщенного водного хлорида аммония и перемешивали в течение еще 30 минут при комнатной температуре. Реакционную смесь разбавляли 25 мл воды и экстрагировали этилацетатом (2×50 мл). Объединенные органические экстракты сушили над сульфатом натрия, фильтровали и концентрировали. Затем остаток очищали флэш-хроматографией (12-100% этилацетат/гептаны; 10 г силикагеля), получая трет-бутил-2'-изопропил-7'-оксо-2',4',6',7'-тетрагидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-1-карбоксилат (130 мг) в виде бесцветного твердого вещества. +ESI (М+Н) 349,1; 1H ЯМР (500 МГц, CDCl3, δ): 7.28 (s, 1Н), 5.78 (s, 1H), 4.57 (spt, J=6,6 Гц, 1Н), 3.59 (m, 2H), 3.37 (m, 2H), 2.82 (s, 2H), 1.74 (m, 4H), 1.55 (d, J=6,6 Гц, 6H), 1.47 (s, 9H).
Стадия 8: гидрохлорид 2'-изопропил-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(2'Н)-она
К раствору трет-бутил-2'-изопропил-7'-оксо-2',4',6',7'-тетрагидроспиро-[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-1-карбоксилата (130 мг; 0,37 ммоль) в этилацетате (5 мл) добавляли 4 М соляную кислоту (2 мл) в 1,4-диоксане. Реакционную смесь перемешивали 3 часа при комнатной температуре, летучие вещества удаляли при пониженном давлении и полученный остаток растирали в10 мл гептана. Твердое вещество сушили при пониженном давлении, получая гидрохлорид 2'-изопропил-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(2'Н)-она (105 мг) в виде беловатого твердого вещества. +ESI (М+Н) 249,1; 1H ЯМР (500 МГц, DMSO-d6, δ): 7.91 (s, 1H) 7.69 (s, 1H) 4.48-4.62 (m, 1H) 3.02-3.28 (m, 4H) 2.78 (s, 2H) 1.74-1.89 (m, 4H) 1.41 (d, J=6,59 Гц, 6Н).
Промежуточное соединение 4: показанную ниже гидрохлоридную соль 2'-трет-бутил-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(2'Н)-она получали следующим образом.
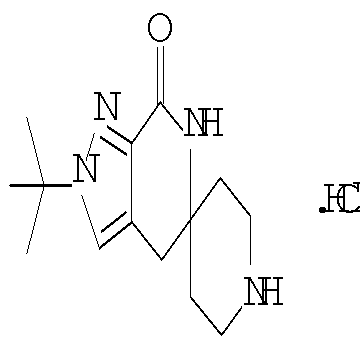
Стадия 1: (Е)-этил-2-(2-трет-бутилгидразоно)пропаноат
К раствору этилпирувата (20,22 г; 174,1 ммоль) в этаноле (150 мл) добавляли гидрохлорид трет-бутилгидразина (21,7 г; 174 ммоль) и N,N-диизопропилэтиламин (33,4 мл; 192 ммоль). После перемешивания при температуре дефлегмации в течение 18 часов реакционную смесь охлаждали и летучие вещества удаляли при пониженном давлении. Полученное золотистое масло переносили в 300 мл этилацетата и промывали 200 мл воды и 300 мл насыщенного водного бикарбоната натрия. Органический слой сушили над сульфатом натрия, фильтровали и концентрировали, получая (Е)-этил-2-(2-трет-бутилгидразоно)пропаноат (23,1 г) в виде прозрачного бледно-желтого масла. +APCl (М+Н) 187,3; 1H ЯМР (400 МГц, CDCl3, δ): 5.51 (br. s., 1H), 4.25 (q, J=7,2 Гц, 2H), 1.89 (s, 3H), 1.32 (t, J=7,1 Гц, 3Н), 1.28 (s, 9H).
Стадия 2: этил-1-трет-бутил-4-формил-1Н-пиразол-3-карбоксилат
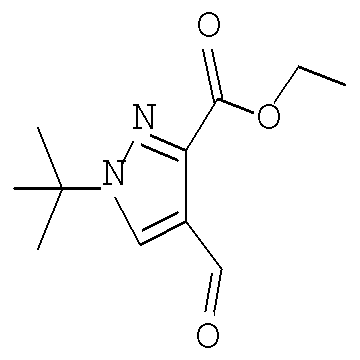
К желто-оранжевому раствору (Е)-этил-2-(2-трет-бутилгидразоно)-пропаноата (22,9 г; 123 ммоль) в толуоле (300 мл) добавляли в виде одной порции хлорид (хлорметилен)диметиламмония (соль Вильсмейера; 34,0 г; 252 ммоль). Суспензию перемешивали 3 часа при комнатной температуре с медленным превращением в двухфазную смесь толуола над густым оранжевым маслом. Реакционную смесь охлаждали до 0°С и медленно нейтрализовали насыщенным водным бикарбонатом натрия. Слои разделяли и водный слой дополнительно экстрагировали этилацетатом (2×200 мл). Органические слои объединяли, промывали 200 мл рассола, сушили над сульфатом натрия, фильтровали и концентрировали, получая этил-1-трет-бутил-4-формил-1Н-пиразол-3-карбоксилат (18,6 г) в виде коричневато-оранжевого масла, которое отвердевало при стоянии. +APCl (М+Н) 225,1; 1H ЯМР (400 МГц, CDCl3, δ): 10.37 (s, 1Н), 8.14 (s, 1Н), 4.48 (q, J=7,0 Гц, 2Н), 1.65 (s, 9H), 1.44 (t, 3H).
Стадия 3: этил-1-трет-бутил-4-(гидроксиметил)-1Н-пиразол-3-карбоксилат
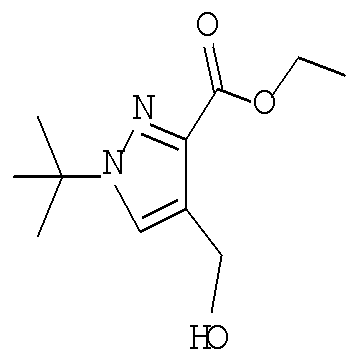
К раствору этил-1-трет-бутил-4-формил-1Н-пиразол-3-карбоксилата (2,87 г; 12,8 ммоль) в этаноле (50 мл) добавляли в виде одной порции боргидрид натрия (0,97 г; 25,6 ммоль). После перемешивания в течение 30 минут при комнатной температуре смесь гасили 1 н. водной соляной кислотой (100 мл), перемешивали в течение 15 минут и затем нейтрализовали насыщенным водным бикарбонатом натрия. Смесь экстрагировали этилацетатом (2×150 мл), затем объединенные органические экстракты сушили над сульфатом натрия, фильтровали и концентрировали, получая этил-1-трет-бутил-4-(гидроксиметил)-1Н-пиразол-3-карбоксилат (2,57 г) в виде прозрачного масла. +APCl (M+Na) 249,2; 1H ЯМР (400 МГц, CDCl3, δ): 7.49 (s, 1Н), 4.65 (d, J=6,8 Гц, 2Н), 4.43 (q, J=7,2 Гц, 2Н), 3.62 (t, J=6,9 Гц, 1Н), 1.59 (s, 9H), 1.41 (t, J=7,1 Гц, 3Н).
Стадия 4: этил-4-(бромметил)-1-трет-бутил-1Н-пиразол-3-карбоксилат
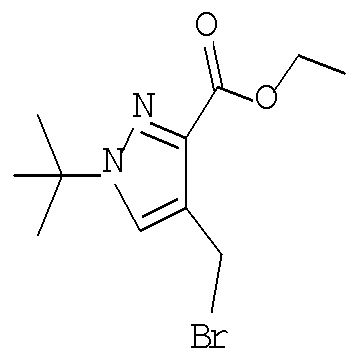
К раствору этил-1-трет-бутил-4-(гидроксиметил)-1Н-пиразол-3-карбоксилата (3,9 г; 17,24 ммоль) в дихлорметане (120 мл) при 0°С добавляли трибромид фосфора (4,91 мл; 51,7 ммоль) и полученную смесь перемешивали 30 минут при 0°С и затем 1 час при комнатной температуре. Смесь гасили 50 мл воды, нейтрализовали насыщенным водным бикарбонатом натрия, перемешивали 30 минут и затем экстрагировали дихлорметаном (2×150 мл). Объединенные органические экстракты промывали 100 мл рассола, сушили над сульфатом натрия, фильтровали и концентрировали. Полученный остаток очищали флэш-хроматографией (7-60% этилацетат/гептаны; 50 г силикагеля), получая этил-4-(бромметил)-1-трет-бутил-1Н-пиразол-3-карбоксилат (4,12 г) в виде прозрачного масла. +APCl (М+Н) 289,1; 1H ЯМР (400 МГц, CDCl3, δ): 7.61 (s, 1Н), 4.70 (s, 2Н), 4.41 (q, J=7,2 Гц, 2Н), 1.60 (s, 9H), 1.40 (t, 3H).
Стадия 5: трет-бутил-4-((1-трет-бутил-3-(этоксикарбонил)-1Н-пиразол-4-ил)метил)-4-цианопиперидин-1-карбоксилат

К раствору трет-бутил-4-цианопиперидин-1-карбоксилата (1,0 г; 4,76 ммоль) в тетрагидрофуране (20 мл) при -78°С добавляли бис(триметилсилил)амид лития (4,76 мл; 1 М в тетрагидрофуране). Смесь перемешивали 30 минут при -78°С, нагревали до 0°С в течение 30 минут и затем охлаждали до -78°С. Затем по каплям добавляли раствор этил-4-(бромметил)-1-трет-бутил-1Н-пиразол-3-карбоксилата (1,38 г; 4,76 ммоль) в тетрагидрофуране. После перемешивания в течение 30 минут при -78°С смесь оставляли нагреваться до комнатной температуры и перемешивали еще 18 часов. Реакционную смесь гасили насыщенным водным хлоридом аммония (50 мл), перемешивали в течение 30 минут, разбавляли водой (50 мл) и затем экстрагировали этилацетатом (2×50 мл). Органические экстракты объединяли, сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали флэш-хроматографией (7-60% этилацетат/гептаны; 100 г силикагеля), получая трет-бутил-4-((1-трет-бутил-3-(этоксикарбонил)-1Н-пиразол-4-ил)метил)-4-цианопиперидин-1-карбоксилат (455 мг) в виде прозрачного масла. +APCl (М+Н) 419,3; 1H ЯМР (500 МГц, CDCl3, δ): 7.68 (s, 1H), 4.40 (q, J=7,2 Гц, 2Н), 4.13 (br. s., 2H), 3.17 (s, 2H), 2.97 (br. s., 2H), 1.81 (d, J=13,2 Гц, 2H), 1.63 (s, 9H), 1.56 (m, 2H), 1.46 (s, 9H), 1.41 (t, J=7,2 Гц, 3Н).
Стадия 6: 4-((1-(трет-бутоксикарбонил)-4-карбамоилпиперидин-4-ил)метил)-1-трет-бутил-1Н-пиразол-3-карбоновая кислота
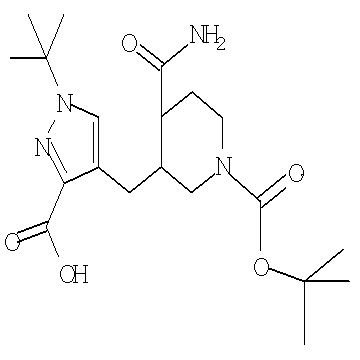
К раствору трет-бутил-4-((1-трет-бутил-3-(этоксикарбонил)-1Н-пиразол-4-ил)метил)-4-цианопиперидин-1-карбоксилата (455 мг; 1,09 ммоль) в метаноле (11 мл) при 0°С по каплям добавляли раствор гидроперита (1,05 г; 10,9 ммоль) в 1 М водном гидроксиде натрия (10,9 мл). После перемешивания в течение 18 часов при комнатной температуре летучие вещества удаляли при пониженном давлении и полученную суспензию переносили в воду (50 мл), подкисляли 2 н. водной соляной кислотой и экстрагировали этилацетатом (2×50 мл). Объединенные органические экстракты сушили над сульфатом натрия, фильтровали и концентрировали, получая 4-((1-(трет-бутоксикарбонил)-4-карбамоилпиперидин-4-ил)метил)-1-трет-бутил-1Н-пиразол-3-карбоновую кислоту (418 мг) в виде бесцветного твердого вещества.
-APCl (М-Н) 407,3; 1H ЯМР (400 МГц, DMSO-d6, δ): 12.33 (br. s., 1H), 7.47 (s, 1H), 7.11 (br. s., 1H), 6.99 (s, 1H), 3.59 (d, J=13,3 Гц, 2H), 2.89 (s, 2H), 2.77 (m, 2H), 1.84 (m, 2H), 1.44 (s, 9H), 1.31 (s, 9H), 1.16 (m, 2H).
Стадия 7: 4-((1-(трет-бутоксикарбонил)-4-изоцианатопиперидин-4-ил)метил)-1-трет-бутил-1Н-пиразол-3-карбоновая кислота
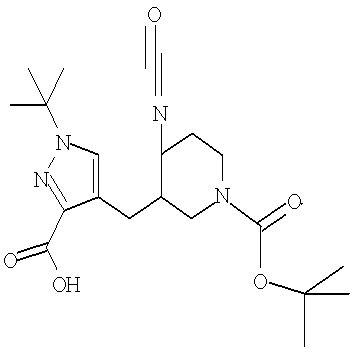
К суспензии 4-((1-(трет-бутоксикарбонил)-4-карбамоилпиперидин-4-ил)метил)-1-трет-бутил-1Н-пиразол-3-карбоновой кислоты (388 мг; 0,95 ммоль) в ацетонитриле (20 мл) добавляли бикарбонат натрия (319 мг; 3,80 ммоль) и бис(трифторацетокси)иодозобензол (632 мг; 1,42 ммоль). Смесь перемешивали 90 минут при комнатной температуре, разбавляли 50 мл воды, подкисляли 1 н. водной соляной кислотой и затем экстрагировали этилацетатом (2×50 мл). Объединенные органические экстракты сушили над сульфатом натрия, фильтровали и концентрировали. Полученный остаток очищали флэш-хроматографией (1-10% метанол/дихлорметан, 25 г силикагеля), получая 4-((1-(трет-бутоксикарбонил)-4-изоцианатопиперидин-4-ил)метил)-1-трет-бутил-1Н-пиразол-3-карбоновую кислоту (172 мг) в виде бесцветного твердого вещества. -APCl (М-Н) 405,4; 1H ЯМР (400 МГц, DMSO-d6, δ): 12.52 (br. s., 1H), 7.82 (s, 1H), 3.83 (br. s., 2H), 3.03 (s, 2H), 2.82 (br. s., 2H), 1.49 (m, 13H), 1.36 (m, 9H).
Стадия 8: трет-бутил-2'-трет-бутил-7'-оксо-2',4',6',7'-тетрагидроспиро-[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-1-карбоксилат
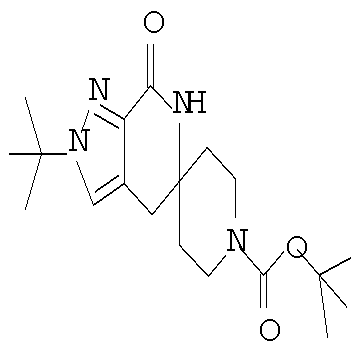
Раствор 4-((1-(трет-бутоксикарбонил)-4-изоцианатопиперидин-4-ил)метил)-1-трет-бутил-1Н-пиразол-3-карбоновой кислоты (180 мг; 0,44 ммоль) в тетрагидрофуране (5 мл) обрабатывали 2 н. водным гидроксидом натрия (0,664 мл; 1,33 ммоль). Смесь перемешивали в течение 3 часов при комнатной температуре, тетрагидрофуран и воду удаляли на роторном испарителе, полученное бесцветное твердое вещество суспендировали в ацетонитриле (10 мл) и затем концентрировали досуха. Еще дважды повторяли растирание в ацетонитриле (10 мл). Полученное бесцветное твердое вещество переносили в дихлорметан (10 мл) и обрабатывали гидрохлоридом (3-(диметиламино)пропил)-этилкарбодиимида (170 мг; 0,89 ммоль). Смесь перемешивали 18 часов при комнатной температуре, затем разбавляли дихлорметаном (50 мл) и промывали водой (30 мл). Органическую фазу сушили над сульфатом натрия, фильтровали и концентрировали. Затем остаток очищали флэш-хроматографией (30-100% этилацетат/гептаны, 10 г силикагеля), получая трет-бутил-2'-трет-бутил-7'-оксо-2',4',6',7'-тетрагидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-1-карбоксилат (70 мг) в виде бесцветного твердого вещества. +ESI (М+Н) 363,3; 1H ЯМР (400 МГц, DMSO-d6, δ): 7.68 (s, 1Н), 7.57 (s, 1Н), 3.47 (m, 2H), 3.20 (m, 2H), 2.73 (s, 2H), 1.53 (t, J=5,7 Гц, 4Н), 1.49 (s, 9H), 1.36 (s, 9H).
Стадия 9: указанное в заголовке соединение - гидрохлоридная соль 2'-трет-бутил-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(2'Н)-она
К раствору трет-бутил-2'-трет-бутил-7'-оксо-2'4',6',7'-тетрагидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-1-карбоксилата (70 мг; 0,19 ммоль) в этилацетате (5 мл) добавляли 4М соляную кислоту в 1,4-диоксане (2 мл) и смесь перемешивали в течение 3 часов при комнатной температуре. Летучие вещества удаляли при пониженном давлении, полученное бесцветное твердое вещество растирали в гептанах (10 мл) и сушили при пониженном давлении, получая гидрохлоридную соль 2'-трет-бутил-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(2'Н)-она (56 мг) в виде беловатого твердого вещества. +ESI (М+Н) 263.1; 1H ЯМР (500 МГц, DMSO-d6, δ): 8.72 (m, 2H), 7.92 (s, 1Н), 7.75 (s, 1Н), 3.20 (br. s, 2H), 3.09 (br. s., 2H), 2.78 (s, 2H), 1.79 (m, 4Н), 1.48 (s, 9H).
Промежуточное соединение 5: показанный ниже 2'-трет-пентил-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(2'Н)-он получали следующим образом.
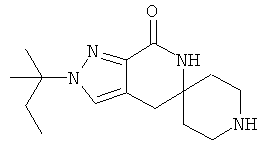
Стадия 1: этил-3-бром-1Н-пиразол-4-карбоксилат
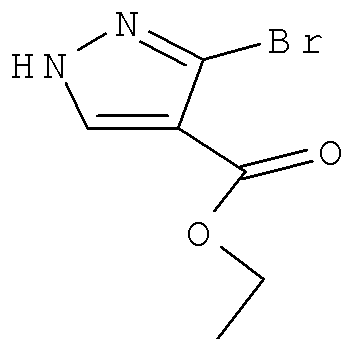
К раствору этил-3-амино-1Н-пиразол-4-карбоксилата (5,0 г; 32 ммоль) и бромида меди(II) (7,2 г; 32 ммоль) в ацетонитриле (65 мл) при (ГС медленно добавляли изоамилнитрит (12 мл; 86 ммоль). Реакционную смесь нагревали до 50°С и перемешивали в течение ночи. Реакционную смесь охлаждали до комнатной температуры и гасили 1 н. водной соляной кислотой (150 мл). Смесь экстрагировали этилацетатом (3×100 мл). Объединенные органические экстракты промывали водой, сушили над сульфатом натрия, фильтровали и концентрировали, получая указанное в заголовке соединение в виде коричневого масла, которое частично отвердевало под вакуумом в течение ночи (7,1 г; 100%). 1Н ЯМР (400 МГц, CDCl3, δ): 9.78 (br. s., 1Н), 8.10 (br. s., 1H), 4.33 (q, J=7,22 Гц, 2Н), 1.36 (m, 3H).
Стадия 2: (3-бром-1-трет-пентил-1Н-пиразол-4-ил)метанол
Концентрированную серную кислоту (0,45 мл; 4,8 ммоль) добавляли к смеси этил-3-бром-1Н-пиразол-4-карбоксилата (1,0 г; 4,6 ммоль) и трет-амилового спирта (3,0 мл; 27 ммоль). Реакционную смесь нагревали до 100°С в течение 2,5 часов. Затем реакционную смесь охлаждали до комнатной температуры и оставляли перемешиваться в течение ночи. Реакционную смесь разбавляли этилацетатом и промывали водой. Органический слой сушили над сульфатом натрия, фильтровали и концентрировали, получая этил-3-бром-1-трет-пентил-1Н-пиразол-4-карбоксилат (1,3 г) в виде неочищенного коричневого масла.
Этот неочищенный продукт (1,3 г) растворяли в тетрагидрофуране (24 мл) и охлаждали до -78°С. Медленно добавляли раствор гидрида диизобутилалюминия (1,5М в толуоле; 9,0 мл; 160 ммоль) и реакционную смесь перемешивали при -78°С в течение 1 часа. Затем реакционную смесь оставляли нагреваться до комнатной температуры и перемешивали в течение еще 2 часов. Реакционную смесь разбавляли этилацетатом (20 мл) и насыщенной водной сегнетовой солью (20 мл). Смесь перемешивали при комнатной температуре в течение ночи. Слои разделяли и водный слой экстрагировали этилацетатом. Объединенные органические экстракты промывали рассолом, сушили над сульфатом натрия, фильтровали и концентрировали. После очистки колоночной флэш-хроматографией (0-100% этилацетат/гептаны) получали указанное в заголовке соединение (685 мг; 62%) в виде бледно-желтого масла. 1Н ЯМР (400 МГц, CDCl3, δ): 7.45 (s, 1H), 4.51 (s, 2Н), 1.86 (q, J=7,41 Гц, 2Н), 1.66 (s, 1Н), 1.51 (s, 6H), 0.69 (m, 3H).
Стадия 3: 3-бром-4-(хлорметил)-1-трет-пентил-1Н-пиразол
Раствор (3-бром-1-трет-пентил-1Н-пиразол-4-ил)метанола (675 мг; 2,73 ммоль) в дихлорметане (10 мл) охлаждали до 0°С. Добавляли триэтиламин (0,53 мл; 3,8 ммоль) и метансульфонилхлорид (0,28 мл; 3,6 ммоль). Реакционную смесь перемешивали при 0°С в течение 15 минут, затем нагревали до комнатной температуры и перемешивали в течение 1,5 часов. Реакционную смесь разбавляли этилацетатом, промывали водой и рассолом, сушили над сульфатом натрия, фильтровали и концентрировали, получая указанное в заголовке соединение (725 мг, 100%) в виде прозрачного масла. 1H ЯМР (400 МГц, CDCl3, δ): 7.48 (s, 1H), 4.47 (s, 2Н), 1.86 (q, J=7,48 Гц, 2Н), 1.52 (s, 6H), 0.69 (m, 3H).
Стадия 4: 3-бром-4-(иодметил)-1-трет-пентил-1Н-пиразол
К раствору 3-бром-4-(хлорметил)-1-трет-пентил-1Н-пиразола (725 мг; 2,73 ммоль) в ацетоне (25 мл) добавляли иодид натрия (4,09 г; 27,3 ммоль). Реакционную смесь нагревали при температуре дефлегмации в течение 2 часов, затем охлаждали до комнатной температуры и перемешивали в течение ночи. Растворитель удаляли в вакууме и остаток распределяли между этилацетатом и водой. Органический слой промывали насыщенным водным тиосульфатом натрия и рассолом. Органические слои сушили над сульфатом натрия, фильтровали и концентрировали, получая указанное в заголовке соединение (824 мг, 85%) в виде коричневого масла. 1H ЯМР (400 МГц, CDCl3, δ): 7.47 (s, 1Н), 4.26 (s, 2Н), 1.83 (q, J=7,41 Гц, 2Н), 1.50 (s, 6H), 0.67 (t, J=7,51 Гц, 3Н).
Стадия 5: 2'-трет-пентил-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(2'Н)-он
Указанное в заголовке соединение получали способом, аналогичным описанному для промежуточного соединения 3 на Стадиях 4-8, используя 3-бром-4-(иодметил)-1-трет-пентил-1Н-пиразол. +ESI (М+Н) 277,3; 1H ЯМР (400 МГц, CD3OD, δ): 7.67 (s, 1Н), 3.22-3.37 (m, 4Н), 2.93 (s, 2Н), 1.92 (q, J=7,61 Гц, 2Н), 1.88-2.05 (m, 4Н), 1.57 (s, 6H), 0.67 (t, J=7,41 Гц, 3Н).
Промежуточное соединение 6: показанный ниже 2'-циклобутил-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(2'Н)-он получали, следующим образом.
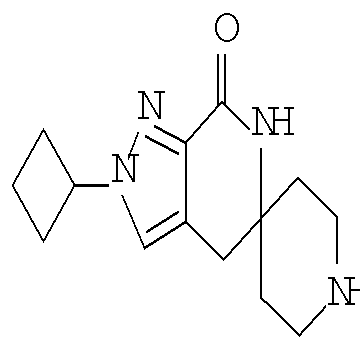
Стадия 1: этил-3-бром-1-циклобутил-1Н-пиразол-4-карбоксилат
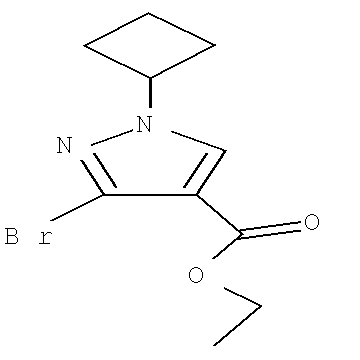
Смесь этил-3-бром-1Н-пиразол-4-карбоксилата (1,00 г; 4,56 ммоль), циклобутилбромида (0,65 мл; 6,9 ммоль) и карбоната цезия (2,97 г; 9,13 ммоль) в N,N-диметилформамиде (10 мл) нагревали до 60°С и перемешивали в течение ночи. Реакционную смесь охлаждали до комнатной температуры и распределяли между смесью 1:1 гептаны/этилацетат и водой. Водный слой вновь экстрагировали смесью 1:1 гептаны/этилацетат. Объединенные органические экстракты промывали рассолом, сушили над сульфатом натрия, фильтровали и концентрировали. После очистки колоночной флэш-хроматографией получали два региоизомерных продукта в виде бесцветного масла.
Этил-5-бром-1-циклобутил-1Н-пиразол-4-карбоксилат (230 мг; 18%): +ESI (М+Н+1) 275,1; 1H ЯМР (400 МГц, CDCl3, δ): 7.98 (s, 1H), 4.98 (m, 1H), 4.30 (q, J=7,02 Гц, 2H), 2.61-2.74 (m, 2H), 2.43 (m, 2H), 1.84-1.95 (m, 2H), 1.34 (m, 3Н).
Этил-3-бром-1-циклобутил-1Н-пиразол-4-карбоксилат (570 мг; 46%): +ESI (М+Н+1) 275,1; 1H ЯМР (400 МГц, CDCl3, δ): 7.87 (s, 1H), 4.69 (m, 1H), 4.29 (q, J=7,22 Гц, 2H), 2.41-2.61 (m, 4H), 1.78-1.98 (m, 2H), 1.34 (m, 3H).
Стадия 2: (3-бром-1-циклобутил-1Н-пиразол-4-ил)метанол

Раствор этил-3-бром-1-циклобутил-1Н-пиразол-4-карбоксилата (565 мг; 2,07 ммоль) в тетрагидрофуране (10 мл) охлаждали до -78°С. Медленно добавляли гидрид диизобутилалюминия (4,13 мл; 6,02 ммоль; 1,5 М в толуоле) и реакционную смесь перемешивали при -78°С в течение 1 часа. Затем реакционную смесь оставляли нагреваться до комнатной температуры и перемешивали в течение еще 2 часов. Реакционную смесь разбавляли этилацетатом (20 мл) и насыщенной водной сегнетовой солью (20 мл). Смесь перемешивали при комнатной температуре в течение ночи. Смесь еще раз разбавляли этилацетатом и промывали водой. Водный слой экстрагировали этилацетатом, объединенные органические экстракты сушили над сульфатом натрия, фильтровали и концентрировали, получая указанное в заголовке соединение (478 мг, 100%) в виде бесцветного масла. +APCl (М+Н+1) 233,1; 1H ЯМР (400 МГц, CDCl3, δ): 7.40 (s, 1H), 4.62-4.71 (m, 1H), 4.51 (s, 2H), 2.39-2.59 (m, 4H), 1.74-1.92 (m, 3H).
Стадия 3: 2'-циклобутил-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(2'Н)-он
Указанное в заголовке соединение получали способом, аналогичным описанному для промежуточного соединения 5, стадии 3-5, используя (3-бром-1-циклобутил-1Н-пиразол-4-ил)метанол. +ESI (М+Н) 261,3; 1H ЯМР (400 МГц, CD3OD, δ): 7.62 (s, 1H), 4.84-4.92 (m, 1H), 3.21-3.36 (m, 4H), 2.93 (s, 2H), 2.50-2.63 (m, 2H), 2.40-2.50 (m, 2H), 1.82-2.05 (m, 6H).
Промежуточное соединение 7: показанный ниже гидрохлорид 2'-трет-бутил-4',6'-дигидро-8-азаспиро[бицикло[3.2.1]октан-3,5'-пиразоло[3,4-с]пиридин]-7'(2'Н)-она получали следующим образом.
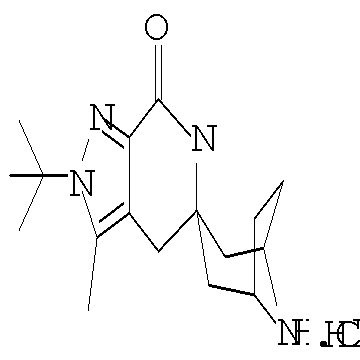
Стадия 1: этил-3-иод-1Н-пиразол-4-карбоксилат
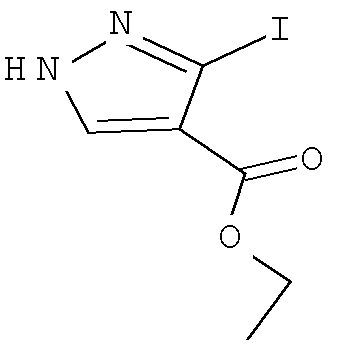
Этил-3-амино-1Н-пиразол-4-карбоксилат (860,0 мг; 5,54 ммоль) растворяли в уксусной кислоте (5 мл) и воде (5 мл). Добавляли иодид калия (921 мг; 5,54 ммоль) и смесь перемешивали до тех пор, пока не растворились все твердые вещества. Затем по каплям добавляли раствор нитрита натрия (386 мг; 5,54 ммоль) в воде (2 мл). Реакционную смесь перемешивали при комнатной температуре в течение 2 минут, после чего перемешивание затруднялось вследствие образования осадка. Добавляли дополнительное количество воды (5 мл) и реакционную смесь оставляли перемешиваться в течение ночи. Уксусную кислоту удаляли при пониженном давлении. Коричневый остаток переносили в насыщенный водный бикарбонат натрия и экстрагировали этилацетатом (2×50 мл). Объединенные органические экстракты промывали насыщенным водным тиосульфатом натрия (50 мл), сушили над сульфатом натрия, фильтровали и концентрировали. После очистки колоночной флэш-хроматографией (10-80% этилацетат/гептаны) получали указанное в заголовке соединение (863 мг, 59%) в виде белого твердого вещества. +APCl (М+Н) 267,2; 1H ЯМР (400 МГц, CDCl3, δ): 12.63 (br. s., 1Н), 8.13 (s, 1Н), 4.34 (q, J=7,0 Гц, 2Н), 1.38 (t, J=7,2 Гц, 3Н).
Стадия 2: этил-1-трет-бутил-3-иод-1Н-пиразол-4-карбоксилат
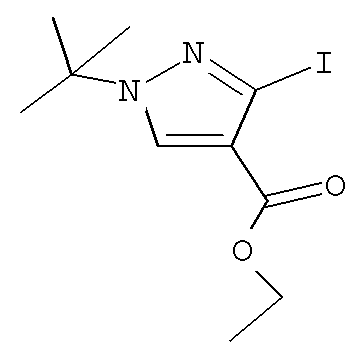
К раствору этил-3-иод-1Н-пиразол-4-карбоксилата (1,10 г; 3,91 ммоль) в трет-бутаноле (5 мл) добавляли серную кислоту (0,40 мл; 4,18 ммоль, 18 М). Реакционную смесь нагревали до 100°С и перемешивали в течение 3 часов. Реакционную смесь охлаждали до комнатной температуры и разбавляли этилацетатом (100 мл) и водой (25 мл). pH подводили до 8, используя насыщенный водный бикарбонат натрия. Слои разделяли, органические слои сушили над сульфатом натрия, фильтровали и концентрировали. После очистки колоночной флэш-хроматографией (7-60% этилацетат/гептаны) получали 2 региоизомерных продукта.
Этил-1-трет-бутил-5-иод-1Н-пиразол-4-карбоксилат: 1H ЯМР (400 МГц, CDCl3, δ): 7.91 (s, 1Н), 4.32 (q, J=7,2 Гц, 2Н), 1.83 (s, 9H), 1.37 (t, J=7,1 Гц, 3Н).
Этил-1-трет-бутил-3-иод-1Н-пиразол-4-карбоксилат (976 мг; 73%) в виде прозрачного масла: +APCl (М+Н) 323,3; 1H ЯМР (400 МГц, CDCl3, δ): 7.90 (s, 1H), 4.31 (q, J=7,0 Гц, 2H), 1.59 (s, 9H), 1.36 (t, J=7,1 Гц, 3Н).
Стадия 3: 1-трет-бутил-3-иод-4-(иодметил)-1Н-пиразол
Указанное в заголовке соединение получали способом, аналогичным описанному для промежуточного соединения 5, стадии 2-4, используя этил-1-трет-бутил-3-иод-1Н-пиразол-4-карбоксилат. 1H ЯМР (400 МГц, CDCl3, δ): 7.49 (s, 1H), 4.25 (s, 2H), 1.56 (s, 9H).
Стадия 4: (1R,5S)-8-трет-бутил-3-метил-8-азабицикло[3.2.1]октан-3,8-дикарбоксилат
К раствору (1R,5S)-8-(трет-бутоксикарбонил)-8-азабицикло[3.2.1]октан-3-карбоновой кислоты (500 мг; 1,96 ммоль) в N,N-диметилформамиде (5 мл) добавляли карбонат калия (541 мг; 3,92 ммоль) и метилиодид (0,18 мл; 2,94 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 18 часов. Реакционную смесь разбавляли этилацетатом (50 мл) и гептанами (50 мл) и затем промывали водой (100 мл) и рассолом (50 мл). Органические экстракты сушили над сульфатом натрия, фильтровали и концентрировали. После очистки колоночной флэш-хроматографией получали указанное в заголовке соединение (486 мг; 92%) в виде прозрачного масла. 1H ЯМР (400 МГц, CDCl3, δ): 4.17-4.29 (m, 2Н), 3.65 (s, 3Н), 2.75-2.86 (m, 1H), 1.93-2.01 (m, 2Н), 1.79-1.92 (m, 2Н), 1.67-1.76 (m, 2Н), 1.58-1.66 (m, 2Н), 1.45 (s, 9H).
Стадия 5: гидрохлорид 2'-трел1-бутил-4',6'-дигидро-8-азаспиро[бицикло[3.2.1]октан-3,5'-пиразоло[3,4-с]пиридин]-7'(2'Н)-она
Указанное в заголовке соединение получали способом, аналогичным описанному для промежуточного соединения 3, стадии 4-8, используя (1R,5S)-8-трет-бутил-3-метил-8-азабицикло[3.2.1]октан-3,8-дикарбоксилат и 1-трет-бутил-3-иод-4-(иодметил)-1Н-пиразол. +ESI (М+Н) 289,2; 1H ЯМР (400 МГц, CD3OD, δ): 7.69 (s, 1H), 4.03-4.10 (m, 2Н), 2.74 (s, 2Н), 2.39-2.46 (m, 2Н), 2.10-2.25 (m, 6H), 1.59 (s, 9H).
Промежуточное соединение 8: показанную ниже 1Н-пиразоло[4,3-b]пиридин-6-карбоновую кислоту получали следующим образом.

Стадия 1: диэтил-2-(5-бром-3-нитропиридин-2-ил)малонат
К суспензии гидрида натрия (5,08 г; 127 ммоль) в N,N-диметилформамиде (75 мл) добавляли диэтилмалонат (19,26 мл; 127 ммоль) при 0°С. Затем раствор перемешивали при температуре окружающей среды в течение 30 минут и по каплям добавляли раствор 5-бром-2-хлор-3-нитропиридина (30 г; 127 ммоль) в N,N-диметилформамиде (75 мл). Затем темно-коричневую смесь перемешивали при 100°С в течение 2 часов, после чего охлаждали до температуры окружающей среды и гасили насыщенным раствором хлорида аммония (500 мл) при 0°С. Смесь экстрагировали этилацетатом (3×500 мл), объединенные органические экстракты сушили над сульфатом магния и фильтровали. Растворитель удаляли в вакууме, получая темно-коричневое масло, которое очищали колоночной флэш-хроматографией (10% этилацетат/гексан), что позволило получить диэтил-2-(5-бром-3-нитропиридин-2-ил)малонат в виде коричневого твердого вещества (31,8 г; 69%). 1H ЯМР (400 МГц, CDCl3, δ): 8.86 (s, 1H), 8.61 (s, 1H), 5.44 (s, 1H), 4.29 (q, 4H), 1.27 (t, 6H).
Стадия 2: 5-бром-2-метил-3-нитропиридин
Смесь диэтил-2-(5-бром-3-нитропиридин-2-ил)малоната (31,8 г; 88 ммоль) в водной соляной кислоте (6 М; 1,4 л) перемешивали при температуре дефлегмации в течение 18 часов. Реакционную смесь охлаждали до температуры окружающей среды и добавляли очень медленно к насыщенному водному раствору бикарбоната натрия (4 л) при 0°С. Затем смесь экстрагировали дихлорметаном (7 л), сушили над сульфатом магния и растворитель удаляли в вакууме, получая 5-бром-2-метил-3-нитропиридин в виде оранжевого масла (13,8 г; 72%), которое отвердевало при стоянии. 1H ЯМР (300 МГц, CDCl3, δ): 8.78 (s, 1H), 8.43 (s, 1H), 2.79 (s, 3H).
Стадия 3: 6-бром-2-метилпиридин-3-амин
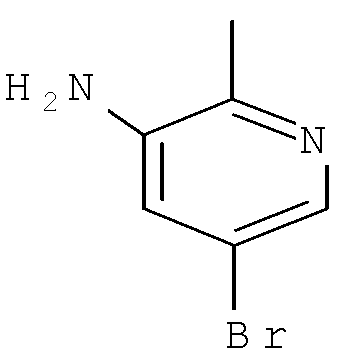
К раствору 5-бром-2-метил-3-нитропиридина (13,8 г; 63,9 ммоль) в промышленном денатурированном спирте (330 мл) при 40°С добавляли порошок железа (20 г) (порциями, чтобы избежать образования комков), затем концентрированную водную соляную кислоту (5 мл). Темно-коричневую смесь энергично перемешивали при температуре дефлегмации в течение 2 часов, затем охлаждали и фильтровали через Celite® (который промывали 1 л промышленного денатурированного спирта. Затем растворитель удаляли в вакууме, остаток переносили в этилацетат (200 мл), промывали насыщенным водным раствором бикарбоната натрия (200 мл), сушили над сульфатом магния и растворитель удаляли в вакууме, получая 5-бром-2-метилпиридин-3-амин в виде оранжевого твердого вещества (10,7 г; 90%). 1H ЯМР (400 МГц, CDCl3, δ): 7.91 (s, 1H), 7.00 (s, 1H), 3.75 (br. s., 2H), 2.25 (s, 3H).
Стадия 4: N-(5-бром-2-метилпиридин-3-ил)ацетамид
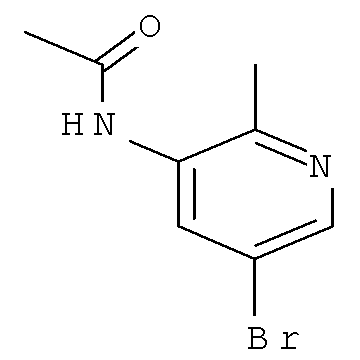
К раствору 5-бром-2-метилпиридин-3-амина (10,7 г; 57,5 ммоль) в дихлорметане (575 мл) добавляли уксусный ангидрид (12 мл; 126,5 ммоль) при 0°С, затем триэтиламин (22 мл; 158 ммоль). Смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение 18 часов и в этот момент дополнительно добавляли эквивалент уксусного ангидрида (6 мл; 63 ммоль). Смесь перемешивали при температуре окружающей среды в течение следующих 72 часов. Реакционную смесь гасили насыщенным водным раствором бикарбоната натрия (500 мл), органическую фазу промывали насыщенным водным хлоридом натрия (500 мл), сушили над сульфатом магния и концентрировали в вакууме, получая коричневое твердое вещество. Это твердое вещество растирали в 30%-ном этилацетате в гексанах, получая N-(5-бром-2-метилпиридин-3-ил)ацетамид в виде беловатого твердого вещества (8,28 г; 63%). 1H ЯМР (400 МГц, CD3OD, δ): 8.31 (s, 1H), 8.18 (s, 1H), 2.43 (s, 3H), 2.18 (s, 3H).
Стадия 5: 6-бром-1Н-пиразоло[4,3-b]пиридин
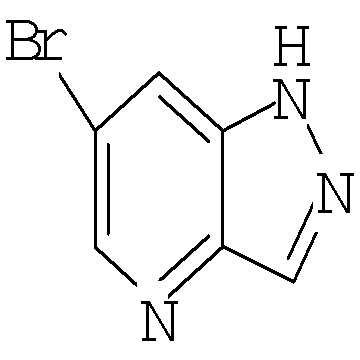
К раствору N-(5-бром-2-метилпиридин-3-ил)ацетамида (8,28 г; 36 ммоль) в хлороформе (550 мл) при температуре окружающей среды добавляли ацетат калия (4,32 г; 43,6 ммоль), уксусную кислоту (2,5 мл; 43,6 ммоль) и далее уксусный ангидрид (6,86 мл; 72,6 ммоль). Смесь перемешивали при температуре окружающей среды в течение 15 минут, затем нагревали до 40°С. Затем по каплям добавляли изоамилнитрит. Далее реакционную смесь перемешивали при 60°С в течение 48 часов. Реакционную смесь медленно выливали в насыщенный раствор бикарбоната натрия (500 мл) при 0°С. Органическую фазу сохраняли, а водную фазу экстрагировали дихлорметаном (500 мл). Затем объединенные органические фазы концентрировали до коричневого масла, которое растворяли в метаноле (500 мл). Добавляли при 0°С водный гидроксид натрия (2 М, 500 мл) и смесь перемешивали при температуре окружающей среды в течение 1 часа, после чего метанол удаляли в вакууме. Водную смесь затем экстрагировали этилацетатом (3×500 мл). Объединенные органические экстракты сушили над сульфатом магния, фильтровали и растворитель удаляли в вакууме, получая 6-бром-1Н-пиразоло[4,3-b]пиридин в виде светло-коричневого твердого вещества (5,5 г; 77%). 1H ЯМР (400 МГц, CD3OD, δ): 8.55 (s, 1H), 8.24 (s, 1H), 8.21 (s, 1H).
Стадия 6: метил-1Н-пиразоло[4,3-b]пиридин-6-карбоксилат
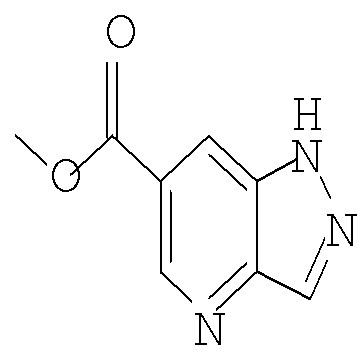
К раствору 6-бром-1Н-пиразоло[4,3-b]пиридина (5,5 г; 27,9 м моль) в метаноле (125 мл) и ацетонитриле (75 мл) добавляли триэтиламин (22 мл; 156 ммоль), 2,2'-бис(дифенилфосфино)-1,1'-бинафтил (1,98 г; 3,07 ммоль) и дихлорид палладия (1,23 г; 6,98 ммоль). Смесь помещали в атмосферу монооксида углерода под давлением 20 бар (2000 кПа), нагревали до 100°С и энергично перемешивали в течение 18 часов. Реакционную смесь охлаждали до температуры окружающей среды и фильтровали через Celite®, затем растворитель удаляли в вакууме, получая коричневое масло. Это неочищенное масло затем очищали колоночной флэш-хроматографией (50% этилацетат/гексаны), получая метил-1Н-пиразоло[4,3-b]пиридин-6-карбоксилат в виде бледно-желтого твердого вещества (4,52 г; 92%). 1H ЯМР (400 МГц, CDCl3, δ): 10.56 (s, 1Н), 9.23 (s, 1Н), 8.35 (s, 1Н), 8.40 (s, 1H), 4.01 (s, 3H).
Стадия 7: 1Н-пиразоло[4,3-b]пиридин-6-карбоновая кислота
К раствору метил-1Н-пиразоло[4,3-b]пиридин-6-карбоксилата (3,52 г; 20 ммоль) в метаноле (250 мл) и воде (190 мл) при 0°С по каплям добавляли водный гидроксид натрия (2 М; 64 мл; 128 ммоль). Затем суспензию оставляли нагреваться до температуры окружающей среды и перемешивали в течение 18 часов. Затем метанол удаляли в вакууме и водную смесь экстрагировали этилацетатом (250 мл). Водный слой подкисляли (до pH 5-6), используя 2 н. водную соляную кислоту (70 мл). Выпавшее в осадок кремовое твердое вещество осадок затем отфильтровывали и сушили в эксикаторе, получая указанное в заголовке соединение (0,675 г; 21%). 1H ЯМР (400 МГц, DMSO-d6, δ): 8.97 (s, 1Н), 8.45 (s, 1Н), 8.39 (s, 1Н).
Промежуточное соединение 9: показанную ниже 3-карбамоил-1Н-индазол-5-карбоновую кислоту получали следующим образом.
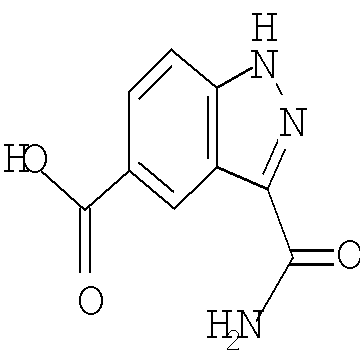
Стадия 1: метил-3-циано-1Н-индазол-5-карбоксилат
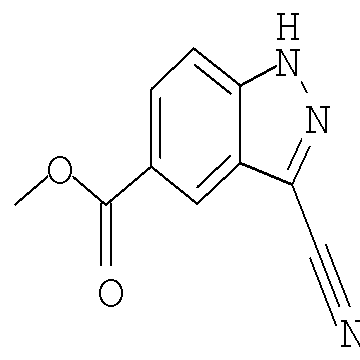
Метил-3-иод-1Н-индазол-5-карбоксилат (30,7 г; 102 ммоль), цианид цинка (20,3 г; 173 ммоль), цинковую пыль (4,05 г; 61,9 ммоль), [1,1'-бис(дифенил-фосфино)ферроцен]-дихлорпалладий(II) (комплекс с дихлорметаном; 12 г; 15 ммоль) и иодид меди(1) (19,7 г; 103 ммоль) объединяли в круглодонной колбе емкостью 1 л. Добавляли N,N-диметилацетамид (500 мл) и реакционную смесь продували азотом в течение 10 минут. Реакционную смесь нагревали до 120°С в течение 1 часа. Реакционную смесь охлаждали до комнатной температуры, разбавляли этилацетатом (1 л) и оставляли перемешиваться в течение 20 минут. Реакционную смесь фильтровали через набивку целита, промывая 500 мл этилацетата. Фильтрат добавляли к раствору насыщенного хлорида аммония и концентрированного гидроксида аммония (2 л) (полученному путем добавления гидроксида аммония к насыщенному водному раствору хлорида аммония до момента достижения pH 8) и этот двухфазный раствор энергично перемешивали в течение 1 часа. Полученную эмульсию фильтровали через небольшую набивку целита. Слои разделяли и водный слой дважды дополнительно экстрагировали этилацетатом (1100 мл), каждый раз фильтруя полученную эмульсию через целит. Объединенные органические слои промывали водой (2×900 мл) и рассолом (900 мл), сушили над сульфатом натрия, фильтровали и концентрировали. К неочищенному веществу добавляли метанол (100 мл) и смесь перемешивали в течение 20 минут. Полученный осадок отфильтровывали и промывали метанолом (10 мл). Фильтрат концентрировали, получая указанное в заголовке соединение (13,2 г; 65%) в виде твердого вещества. -ESI (М-Н) 200,0; 1H ЯМР (400 МГц, DMSO-d6, δ): 8.43-8.45 (m, 1Н), 8.05 (dd, J=8,8, 1,6 Гц, 1Н), 7.85 (dd, J=8,9, 0,9 Гц, 1Н), 3.88 (s, 3Н).
Стадия 2: 3-карбамоил-1Н-индазол-5-карбоновая кислота
Суспензию метил-3-циано-1Н-индазол-5-карбоксилата (50,0 г; 249 ммоль) в метаноле (1 л) охлаждали до 10°С. По каплям добавляли раствор гидроперита (241 г; 2,49 моль) в гидроксиде натрия (1 л; 2,5 н.) и воду (100 мл), поддерживая внутреннюю температуру ниже 25°С. По завершении добавления ледяную баню удаляли и реакционную смесь оставляли перемешиваться при комнатной температуре в течение 16 часов. Используя HPLC, обнаруживали присутствие небольшого количества непрореагировавшего исходного вещества. Реакционную смесь охлаждали до 15°С и порциями добавляли дополнительное количество гидроперита (50 г). Отмечали интенсивное выделение пузырьков газа. Реакционную смесь оставляли перемешиваться в течение еще 2 часов. Неочищенную реакционную смесь фильтровали для удаления присутствующих твердых веществ и фильтрат концентрировали удалением метанола. Оставшийся раствор охлаждали в ледяной бане и по каплям добавляли 6 н. соляную кислоту (420 мл) для подведения pH до 4. Раствор перемешивали в течение 20 минут и полученное рыжевато-коричневое твердое вещество собирали фильтрацией и сушили, получая 57,2 г неочищенного продукта. К этому неочищенному продукту добавляли ацетонитрил (700 мл) и дихлорметан (700 мл) и смесь перемешивали при комнатной температуре в течение 1 часа. Твердое вещество собирали фильтрацией, промывали смесью 1:1 ацетонитрил:дихлорметан (400 мл) и сушили, получая указанное в заголовке соединение (39,5 г; 77%) в виде рыжевато-коричневого твердого вещества. +ESI (М+Н) 206,1; 1H ЯМР (400 МГц, DMSO-d6, δ): 13.81 (s, 1Н), 12.85 (br. s., 1H), 8.82 (d, J=0,8 Гц, 1Н), 7.93 (dd, J=8,8, 1,6 Гц, 1Н), 7.79-7.85 (m, 1Н), 7.64 (d, J=8,6 Гц, 1Н), 7.44 (s, 1Н).
Промежуточное соединение 10: показанную ниже 3-циано-1Н-индазол-5-карбоновую кислоту получали следующим образом.
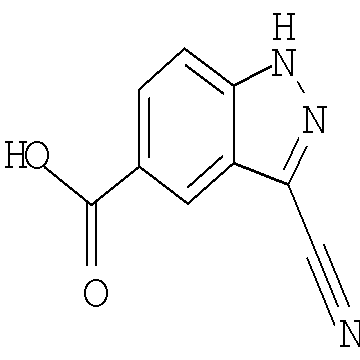
Метил-3-циано-1Н-индазол-5-карбоксилат (500 мг; 2,5 ммоль) растворяли в метаноле (12 мл) и добавляли 2 н. водный гидроксид лития (3,7 мл; 7 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь концентрировали с удалением метанола и остаток подкисляли до pH 4, используя 1 н. водную соляную кислоту. Полученный желтый осадок собирали фильтрацией, промывали водой и сушили в вакуумном сушильном шкафу, получая указанное в заголовке соединение (445 мг; 96%). -ESI (M-H) 186,4; 1Н ЯМР (400 МГц, DMSO-d6, δ): 13.17 (br. s., 1H), 8.42 (s, 1H), 8.05 (dd, J=8,8,1,6 Гц, 1H), 7.83 (d, 1H).
Промежуточное соединение 11: показанную ниже 3-циано-1Н-индазол-6-карбоновую кислоту получали следующим образом.
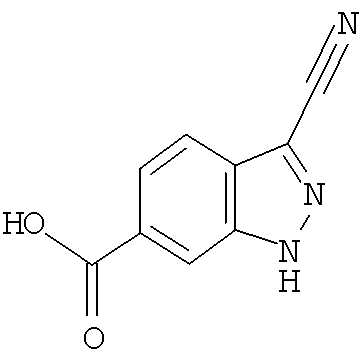
Стадия 1: метил-1Н-индазол-6-карбоксилат
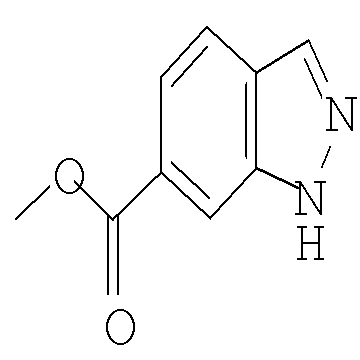
К раствору 1Н-индазол-6-карбоновой кислоты (3,00 г; 18,5 ммоль) в N,N-диметилформамиде (46 мл) добавляли карбонат натрия (2,06 г; 19,4 ммоль), затем по каплям добавляли иодметан (2,75 г; 1,21 мл; 19,4 ммоль). Смесь перемешивали при комнатной температуре в течение ночи. Смесь выливали в полунасыщенный бикарбонат натрия и трижды экстрагировали этилацетатом. Объединенные органические слои промывали рассолом, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме, что позволило получить коричневое масло. Этот остаток очищали колоночной флэш-хроматографией (12-100% этилацетат/гептаны), что позволило получить метил-1Н-индазол-6-карбоксилат в виде желтого твердого вещества (2,95 г; 90%). 1H ЯМР (400 МГц, CDCl3, δ): 10.40 (br. s., 1H), 8.26 (s, 1H), 8.13 (s, 1H), 7.84 (d, J=8,4 Гц, 1Н), 7.79 (d, J=8,4 Гц, 1Н), 3.96 (s, 3H).
Стадия 2: метил-3-иод-1Н-индазол-6-карбоксилат
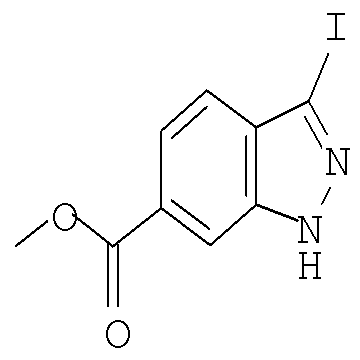
К раствору метил-1Н-индазол-6-карбоксилата (865 мг; 4,91 ммоль) в N,N-диметилформамиде (12 мл) добавляли гидроксид калия (840 мг; 3,05 ммоль), затем иод (1,54 г; 5,9 ммоль). Смесь перемешивали при комнатной температуре в течение 3 часов. Добавляли бисульфат натрия (30 мл 5%-ного водного) и смесь дважды экстрагировали этилацетатом. Объединенные органические слои промывали рассолом, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Остаток очищали колоночной флэш-хроматографией (5-65% этилацетат/гептаны), что позволило получить метил-3-иод-1Н-индазол-6-карбоксилат в виде бесцветного твердого вещества (1,16 г; 78%). 1H ЯМР (400 МГц, DMSO-d6, δ): 13.84 (s, 1H), 8.13 (s, 1H), 7.72 (d, J=8,4 Гц, 1H), 7.54 (d, J=8,6 Гц, 1H), 3.87 (s, 3H).
Стадия 3: метил-3-циано-1Н-индазол-6-карбоксилат
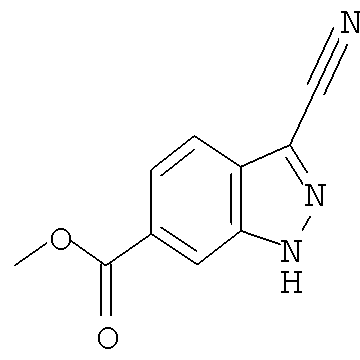
Смесь метил-3-иод-1Н-индазол-6-карбоксилата (3,0 г; 9,9 ммоль), цинковой пыли (400 мг; 6,11 ммоль), цианида цинка (2,0 г; 17,0 ммоль), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия(II) (комплекс с дихлорметаном; 1,15 г; 1,41 ммоль) и иодида меди(1) (1,90 г; 9,97 ммоль) в диметилацетамиде (55 мл) продували азотом в течение 15 минут. Смесь перемешивали при 120°С в течение 15 часов. Реакционную смесь охлаждали, разбавляли этилацетатом (250 мл) и фильтровали через целит, промывая этилацетатом (100 мл). К фильтрату добавляли ~400 мл насыщенного водного раствора хлорида аммония и концентрированного гидроксида аммония (полученного путем добавления гидроксида аммония к насыщенному водному раствору хлорида аммония до момента достижения pH 8). Смесь перемешивали в течение 1 часа. Затем проводили разделение слоев. Органический слой промывали водой и рассолом, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. К остатку добавляли метанол (40 мл) и смесь перемешивали в течение ночи. Смесь фильтровали и твердое вещество сушили в вакууме, получая метил-3-циано-1Н-индазол-6-карбоксилат в виде рыжевато-коричневого твердого вещества (1,47 г; 73%). 1H ЯМР (400 МГц, DMSO-d6, δ): 13.40 (br. s., 1H), 8.25 (s, 1H), 7.94 (d, J=8,6 Гц, 1Н), 7.83 (d, J=8,4 Гц, 1Н), 3.88 (s, 3H).
Стадия 4: 3-циано-1Н-индазол-6-карбоновая кислота
К раствору метил-3-циано-1Н-индазол-6-карбоксилата (1,47 г; 7,31 ммоль) в метаноле (36 мл) и тетрагидрофуране (20 мл) добавляли 2 н. водный гидроксид лития (16 мл; 32 ммоль). Реакционную смесь нагревали до 50°С в течение 72 часов. Реакционную смесь охлаждали до комнатной температуры и концентрировали. Остаток разбавляли водой и pH подводили до 4, используя 1 н. водную соляную кислоту. Полученный осадок отфильтровывали, промывали водой и сушили под вакуумом, получая указанное в заголовке соединение (500 мг; 37%) в виде рыжевато-коричневого твердого вещества. +ESI (М+Н) 188,2.
Промежуточное соединение 12: показанную ниже 1-метоксиизохинолин-7-карбоновую кислоту получали следующим образом.
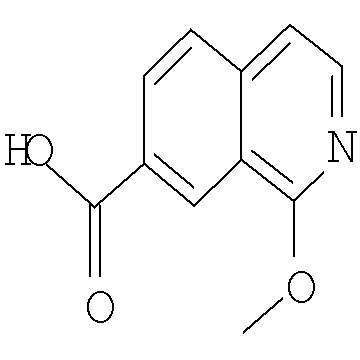
Стадия 1: 7-бром-1-метоксиизохинолин
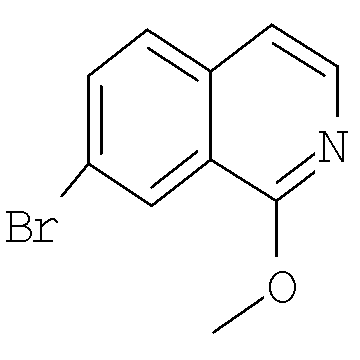
7-Бром-1-хлоризохинолин (570 мг; 2,4 ммоль) объединяли с метанолом (10 мл) и метилатом натрия (25 масс.%-ным в метаноле; 1,5 мл; 24 ммоль) во флаконе для микроволновой печи. Флакон герметично закрывали и нагревали до 130°С в течение 3 часов в микроволновой печи. Реакционную смесь концентрировали. Неочищенный остаток переносили в этилацетат и промывали водой и насыщенным водным бикарбонатом натрия. Водный слой дважды экстрагировали горячим этилацетатом. Объединенные органические экстракты сушили над сульфатом натрия, фильтровали и концентрировали, получая указанное в заголовке соединение (520 мг; 93%). +ESI (М+Н+1) 240,0; 1H ЯМР (400 МГц, DMSO-d6, δ): 8.25-8.28 (m, 1H), 8.04 (d, J=5,9 Гц, 1Н), 7.86-7.89 (m, 2Н), 7.40 (dd, J=6,0, 0,9 Гц, 1H), 4.03 (s, 3H).
Стадия 2: метил-1-метоксиизохинолин-7-карбоксилат
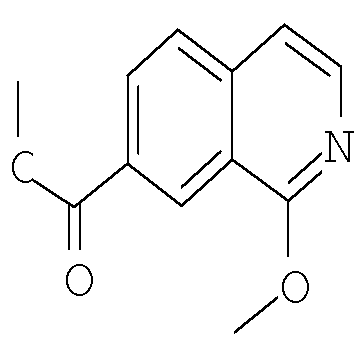
К раствору 7-бром-1-метоксиизохинолина (520 мг; 2,2 ммоль) в метаноле (30 мл) добавляли ацетат натрия (517 мг; 6,30 ммоль) и [1,1'-бис(дифенил-фосфино)ферроцен]-дихлорпалладий(II) (комплекс с дихлорметаном; 257 мг; 0,315 ммоль). Сосуд со смесью трижды вакуумировали и вновь заполняли азотом. Затем в реакционном сосуде создавали давление монооксида углерода до 25 фунт/кв. дюйм (172,37 кПа). Реакционную смесь нагревали до 70°С и встряхивали в течение 20 часов. Реакционную смесь фильтровали, промывали метанолом. Фильтрат концентрировали. Полученный остаток переносили в дихлорметан и оставшиеся твердые вещества отфильтровывали. Фильтрат концентрировали и очищали колоночной флэш-хроматографией (0-100% этилацетат/гептаны), получая указанное в заголовке соединение (443 мг; 93%) в виде белого твердого вещества. +ESI (М+Н) 218,1; 1H ЯМР (400 МГц, DMSO-d6, δ): 8.77 (d, J=0,8 Гц, 1Н), 8.20 (dd, J=8,6, 1,8 Гц, 1Н), 8.13 (d, J=5,9 Гц, 1Н), 8.00 (d, J=8,6 Гц, 1Н), 7.46 (d, J=5,9 Гц, 1Н), 4.08 (s, 3H), 3.90 (s, 3H).
Стадия 3: 1-метоксиизохинолин-7-карбоновая кислота
К раствору метил-1-метоксиизохинолин-7-карбоксилата (443 мг; 2,04 ммоль) в метаноле (10 мл) добавляли 2 н. водный гидроксид лития (20 мл). Реакционную смесь перемешивали при комнатной температуре в течение 24 часов. Реакционную смесь разбавляли 1 н. водной соляной кислотой и этилацетатом. Слои разделяли и водный слой еще дважды экстрагировали этилацетатом. Объединенные органические экстракты промывали рассолом, сушили над сульфатом натрия, фильтровали и концентрировали, что позволило получить указанное в заголовке соединение (414 мг; 100%) в виде твердого вещества. +ESI (М+Н) 204,1; 1H ЯМР (400 МГц, DMSO-d6, δ): 13.24 (s, 1H), 8.76 (d, J=0,8 Гц, 1Н), 8.18 (dd, J=8,6, 1,8 Гц, 1Н), 8.11 (d, J=5,9 Гц, 1H), 7.97 (d, J=8,4 Гц, 1Н), 7.45 (d, J=5,9 Гц, 1Н), 4.07 (s, 3H).
Промежуточное соединение 13: показанную ниже 3-аминоизохинолин-6-карбоновую кислоту получали следующим образом.
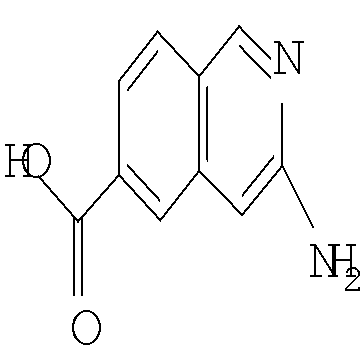
Указанное в заголовке соединение получали способом, аналогичным описанному для промежуточного соединения 12, стадии 2-3, используя 6-бромизохинолин-3-амин. +ESI (М+Н) 189,0; 1H ЯМР (400 МГц, DMSO-d6, δ): 13.15 (br. s., 1H), 8.94 (s, 1H), 8.20 (s, 1H), 7.91 (m, 1Н), 7.62-7.59 (m, 1H), 6.78 (s, 1H), 6.14 (s, 2H).
Промежуточное соединение 14: показанную ниже 3-амино-1Н-индазол-5-карбоновую кислоту получали следующим образом.
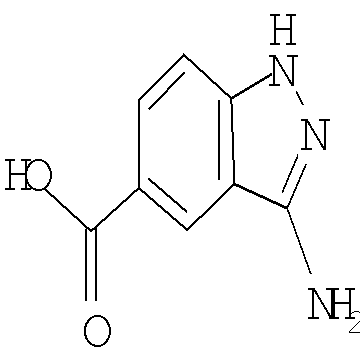
К раствору 3-циано-4-фторбензойной кислоты (980,0 мг; 5,94 ммоль) в этаноле (6 мл) добавляли гидрат гидразина (0,89 мл; 17,8 ммоль). Реакционную смесь нагревали при температуре дефлегмации в течение 3 часов. Реакционную смесь охлаждали до комнатной температуры и удаляли этанол при пониженном давлении. Полученное желтое масло переносили в воду (50 мл) и подщелачивали 1 н. водным гидроксидом натрия (5 мл). Этот раствор один раз промывали этилацетатом (25 мл). Водную фазу подкисляли до pH 3, используя 6 н. водную соляную кислоту, и оставляли перемешиваться при комнатной температуре в течение 1 часа. Полученный осадок собирали фильтрацией и сушили под вакуумом, получая указанное в заголовке соединение (612 мг; 48%) в виде розового твердого вещества. +ESI (М+Н) 178,1; 1H ЯМР (400 МГц, DMSO-d6, δ): 8.42-8.47 (m, 1H), 7.76 (dd, J=8,8, 1,6 Гц, 1H), 7.21 (d, J=8,8 Гц, 1H).
Промежуточное соединение 15: показанную ниже 3-амино-1Н-индазол-6-карбоновую кислоту получали следующим образом.
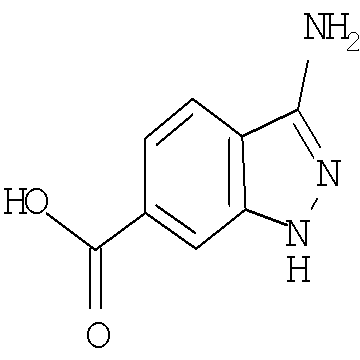
К раствору 4-циано-3-фторбензойной кислоты (500 мг; 3,0 ммоль) в н-бутаноле (9 мл) добавляли моногидрат гидразина (0,5 мл; 10 ммоль). Реакционную смесь нагревали до 110°С в течение ночи. Реакционную смесь охлаждали до комнатной температуры и осадок собирали фильтрацией. Затем твердое вещество растворяли в 1 н. водном гидроксиде натрия (2 мл) и экстрагировали этилацетатом (2×). Водный слой подкисляли до рН 4, используя 1 н. водную соляную кислоту. Полученный осадок собирали фильтрацией и сушили под вакуумом, получая указанное в заголовке соединение (140 мг; 26%) в виде красного твердого вещества. +ESI (М+Н) 178,2; 1H ЯМР (400 МГц, CD3OD, δ): 7.99-8.01 (m, 1H), 7.73 (dd, J=8,4, 0,8 Гц, 1Н), 7.61 (dd, J=8,5, 1,3 Гц, 1Н).
Промежуточное соединение 16: показанную ниже 2-метил-3-оксо-2,3-дигидро-1Н-индазол-5-карбоновую кислоту получали следующим образом.
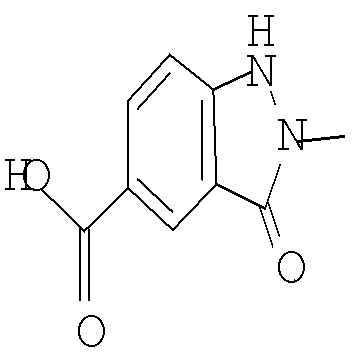
Стадия 1: метил-3-гидрокси-1Н-индазол-5-карбоксилат
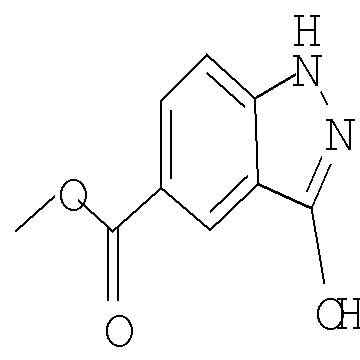
3-Гидрокси-1Н-индазол-5-карбоновую кислоту (1,5 г; 8,4 ммоль) суспендировали в метаноле (17 мл). Добавляли концентрированную соляную кислоту (3,11 мл; 101 ммоль) и реакционную смесь нагревали до 100°С в течение 6 часов. Реакционную смесь концентрировали, получая указанное в заголовке соединение (1,60 г; 99%). +ESI (М+Н) 193,1; 1H ЯМР (400 МГц, DMSO-d6, δ): 12.00 (br. s., 1H), 8.35 (s, 1H), 7.83 (dd, J=8,9, 1,7 Гц, 1Н), 7.33 (dd, J=8,9, 0,7 Гц, 1H), 3.82 (s, 3H).
Стадия 2: 1-этил-5-метил-3-гидрокси-1Н-индазол-1,5-дикарбоксилат
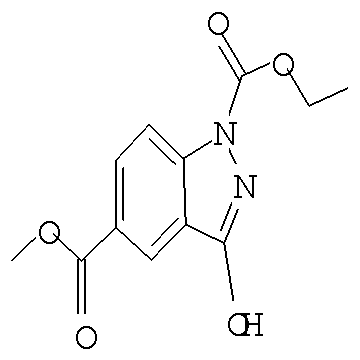
Метил-3-гидрокси-1Н-индазол-5-карбоксилат (1,60 г; 8,33 ммоль) суспендировали в пиридине (10 мл). Медленно добавляли этилхлорформиат (0,90 мл; 9,3 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Добавляли дополнительное количество этилхлорформиата (0,30 мл; 3,1 ммоль) и реакционную смесь перемешивали в течение следующих 30 минут. Реакционную смесь выливали в воду (65 мл) и охлаждали в холодильнике в течение 3 часов. Коричневое твердое вещество собирали фильтрацией, промывали водой и сушили под вакуумом, получая указанное в заголовке соединение (1,75 г; 80%). +ESI (М+Н) 265,1; 1H ЯМР (400 МГц, CDCl3, δ): 8.56 (s, 1H), 8.29 (d, J=7,8 Гц, 1H), 8.13 (br. s., 1H), 4.59 (q, J=7,0 Гц, 2H), 3.97 (s, 3H), 1.56 (t, J=7,0 Гц, 3Н).
Стадия 3: 1-этил-5-метил-2-метил-3-оксо-2,3-дигидро-1Н-индазол-1,5-дикарбоксилат
1-Этил-5-метил-3-гидрокси-1Н-индазол-1,5-дикарбоксилат (1,75 г; 6,62 ммоль) суспендировали в ацетоне (85 мл). Добавляли карбонат цезия (2,27 г; 6,95 ммоль) и метилиодид (1,3 мл; 20 ммоль) и реакционную смесь перемешивали при температуре дефлегмации в течение 22 часов. Реакционную смесь концентрировали досуха и остаток распределяли между дихлорметаном (60 мл) и водой (100 мл). Слои разделяли и водный слой еще раз экстрагировали дихлорметаном (60 мл). Объединенные органические экстракты сушили над сульфатом магния, фильтровали и концентрировали. После очистки колоночной флэш-хроматографией (7-60% этилацетат/гептаны) получали два региоизомерных продукта.
1-Этил-5-метил-3-метокси-1Н-индазол-1,5-дикарбоксилат (590 мг; 32%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, CDCl3, δ): 8.41 (dd, J=1,6, 0,8 Гц, 1H), 8.22 (dd, J=9,2, 3,5 Гц, 1H), 8.14 (d, J=9,2 Гц, 1H), 4.57 (q, J=7,1 Гц, 2H), 4.20 (s, 3H), 3.95 (s, 3H), 1.51 (t, J=7,1 Гц, 3Н).
1-Этил-5-метил-2-метил-3-оксо-2,3-дигидро-1Н-индазол-1,5-дикарбоксилат (699 мг; 38%) в виде желтого твердого вещества. +ESI (М+Н) 279,1; 1H ЯМР (400 МГц, CDCl3, δ): 8.56 (dd, J=1,8, 0,6 Гц, 1H), 8.30 (dd, J=8,8, 1,8 Гц, 1H), 7.93 (d, J=8,8 Гц, 1H), 4.50 (q, J=7,0 Гц, 2H), 3.94 (s, 3Н), 3.67 (s, 3H), 1.48 (t, J=7,1 Гц, 3Н).
Стадия 4: 2-метил-3-оксо-2,3-дигидро-1Н-индазол-5-карбоновая кислота
1-Этил-5-метил-2-метил-3-оксо-2,3-дигидро-1Н-индазол-1,5-дикарбоксилат (300 мг; 1,08 ммоль) растворяли в этаноле (4 мл). Добавляли гидроксид калия (485 мг; 8,62 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 1,5 часов. Данные LCMS (жидкостная хроматография в сочетании с масс-спектрометрией) показали, что реакция не завершена. Затем добавляли водный раствор гидроксида калия (10 мл; 10 ммоль; 1,0 М) и реакционную смесь нагревали до 65°С в течение 2 часов. Реакционную смесь охлаждали до комнатной температуры и концентрировали. Полученное оранжевое твердое вещество растворяли в воде и подкисляли 1 н. водной соляной кислотой. Осадок собирали фильтрацией и сушили под вакуумом, получая указанное в заголовке соединение (158 мг; 76%) в виде белого твердого вещества. +ESI (М+Н) 193,1; 1H ЯМР (400 МГц, DMSO-d6, δ): 12.75 (br. s., 1Н), 11.06 (s, 1H), 8.15 (s, 1H), 7.99 (dd, J=8,7, 1,5 Гц, 1Н), 7.28 (d, J=8,6 Гц, 1H), 3.37 (s, 3H).
Промежуточное соединение 17: показанную ниже 3-метокси-1Н-индазол-5-карбоновую кислоту получали следующим образом.
Указанное в заголовке соединение получали способом, аналогичным описанному для промежуточного соединения 16, используя 1-этил-5-метил-3-метокси-1Н-индазол-1,5-дикарбоксилат, региоизомерный продукт, полученный на стадии 3. +ESI (М+Н) 193,1; 1H ЯМР (400 МГц, DMSO-d6, δ): 12.65 (br. s., 1Н), 12.26 (s, 1Н), 8.18 (s, 1Н), 7.86 (dd, J=8,9, 1,5 Гц, 1Н), 7.38 (d, J=8,8 Гц, 1Н), 3.99 (s, 3H).
Промежуточное соединение 18: показанную ниже 7-метокси-1-(тетрагидро-2Н-пиран-2-ил)-1Н-индазол-5-карбоновую кислоту получали следующим образом.
Стадия 1: этил-7-метокси-1-(тетрагидро-2Н-пиран-2-ил)-1Н-индазол-5-карбоксилат

К смеси этил-7-гидрокси-1-(тетрагидро-2Н-пиран-2-ил)-1Н-индазол-5-карбоксилата (WO2009144554) (100 мг; 0,34 ммоль) и карбоната калия (95,1 мг; 0,68 ммоль) в N,N-диметилформамиде (1 мл) добавляли метилиодид (32 мкл; 0,51 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавляли водой и экстрагировали этилацетатом (4х). Объединенные органические слои промывали водой и рассолом, сушили над сульфатом натрия, фильтровали и концентрировали, получая указанное в заголовке соединение (105 мг, 100%) в виде желтого масла. +ESI (М+1-ТНР (тетрагидропиран)) 221,2; 1H ЯМР (400 МГц, CDCl3, δ): 8.07-8.10 (m, 2Н), 7.43 (d, J=0,98 Гц, 1Н), 6.24 (dd, J=10,24, 2,44 Гц, 1Н), 4.38 (q, J=7,15 Гц, 2Н), 4.08 (dt, J=11,56, 2,02 Гц, 1Н), 4.04 (s, 3H), 3.70-3.78 (m, 1Н), 2.54-2.66 (m, 1Н), 2.09-2.19 (m, 1Н), 2.01-2.08 (m, 1Н), 1.71-1.83 (m, 2Н), 1.55-1.64 (m, 1H), 1.41 (t, J=7,12 Гц, 3H).
Стадия 2: 7-метокси-1-(тетрагидро-2Н-пиран-2-ил)-1Н-индазол-5-карбоновая кислота
К раствору этил-7-метокси-1-(тетрагидро-2Н-пиран-2-ил)-1Н-индазол-5-карбоксилата (102 мг; 0,33 ммоль) в тетрагидрофуране (2 мл) добавляли 1 н. водный гидроксид лития (0,67 мл; 0,67 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Данные LCMS показали, что реакция не завершена. Добавляли дополнительное количество гидроксида лития (0,35 мл; 2 M; 0,7 ммоль) и реакционную смесь нагревали до 40°С в течение 1 часа. Затем реакционную смесь оставляли перемешиваться при комнатной температуре в течение 70 часов. Тетрагидрофуран удаляли в вакууме и остаток подкисляли до pH 4, используя 1 н. водную соляную кислоту. Раствор экстрагировали этилацетатом (3×). Объединенные органические экстракты промывали рассолом, сушили над сульфатом натрия, фильтровали и концентрировали, получая указанное в заголовке соединение (84 мг, 91%) в виде твердого вещества. (М+1-ТНР) 193,2; 1H ЯМР (400 МГц, CDCl3, δ): 8.18 (d, J=1,37 Гц, 1H), 8.12 (s, 1H), 7.46 (d, J=1,17 Гц, 1Н), 6.26 (dd, J=10,15, 2,54 Гц, 1Н), 4.07-4.12 (m, 1H), 4.06 (s, 3H), 3.65-3.81 (m, 1H), 2.54-2.72 (m, 1H), 2.10-2.22 (m, 1H), 2.01-2.10 (m, 1H), 1.71-1.85 (m, 2H), 1.57-1.67 (m, 1H).
Промежуточное соединение 19: показанную ниже 2-метоксихинолин-7-карбоновую кислоту получали следующим образом.
Стадия 1: 1-оксид 7-(этоксикарбонил)хинолина
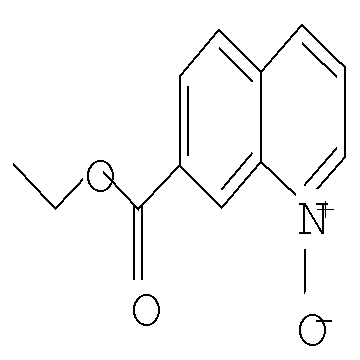
К раствору этил-хинолин-7-карбоксилата (1,02 г; 5,05 ммоль) в дихлорметане (20 мл) добавляли перуксусную кислоту (2,13 мл; 10,1 ммоль; 32 масс.%-ную в уксусной кислоте). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь распределяли между водой и дихлорметаном. Слои разделяли и водный слой экстрагировали дихлорметаном (4×). Объединенные органические экстракты промывали водой и рассолом, сушили над сульфатом натрия, фильтровали и концентрировали. Твердое вещество несколько раз концентрировали из гептанов и этилацетата, затем сушили под вакуумом, получая указанное в заголовке соединение (1,01 г; 92%) в виде желтого твердого вещества. +ESI (М+Н) 218,2; 1H ЯМР (400 МГц, CDCl3, δ): 9.40 (s, 1H), 8.65 (d, J=6,05 Гц, 1H), 8.27 (dd, J=8,58, 1,56 Гц, 1H), 7.95 (d, J=8,39 Гц, 1H), 7.82 (d, J=8,58 Гц, 1H), 7.42 (dd, J=8,49, 6,15 Гц, 1H), 4.47 (q, J=7,02 Гц, 2H), 1.45 (t, J=7,1 Гц, 3H).
Стадия 2: этил-2-метоксихинолин-7-карбоксилат
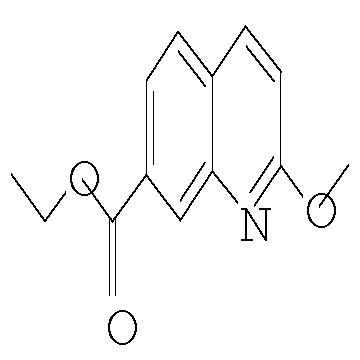
К раствору 1-оксида 7-(этоксикарбонил)хинолина (150 мг; 0,69 ммоль) и толуол-4-сульфонилхлорида (171 мг; 0,89 ммоль) в метаноле (5 мл) при 0°С добавляли триэтиламин (0,19 мл; 1,4 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Данные LCMS показали, что реакция не завершена. Добавляли дополнительное количество триэтиламина (0,05 мл) и реакционную смесь перемешивали в течение следующих 4 часов. Реакционную смесь концентрировали и остаток распределяли между этилацетатом и насыщенным водным карбонатом натрия. Слои разделяли и водный слой еще дважды экстрагировали этилацетатом. Объединенные органические экстракты промывали рассолом, сушили над сульфатом натрия, фильтровали и концентрировали. После очистки колоночной флэш-хроматографией (0-40% этилацетат/гептаны) получали указанное в заголовке соединение (130 мг; 81%) в виде бледно-желтого твердого вещества. +ESI (М+Н) 232,2; 1H ЯМР (400 МГц, CDCl3, δ): 8.49-8.60 (m, 1Н), 7.95-8.05 (m, 2H), 7.75 (d, J=8,19 Гц, 1Н), 6.98 (d, J=8,78 Гц, 1Н), 4.43 (q, J=7,22 Гц, 2H), 4.08 (s, 3H), 1.43 (t, J=7,12 Гц, 3Н).
Стадия 3: 2-метоксихинолин-7-карбоновая кислота
К раствору этил-2-метоксихинолин-7-карбоксилата (125 мг; 0,54 ммоль) в тетрагидрофуране (1,5 мл) добавляли 2 н. водный гидроксид лития (0,81 мл; 1,6 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 65 часов. Тетрагидрофуран удаляли в вакууме и остаток подкисляли до pH 4, используя 1 н. водную соляную кислоту. Смесь разбавляли водой, полученный осадок собирали фильтрацией и сушили под вакуумом, получая указанное в заголовке соединение (106 мг; 96%) в виде белого твердого вещества. +ESI (М+Н) 204,2; 1H ЯМР (400 МГц, CDCl3, δ): 8.64 (d, J=1,37 Гц, 1Н), 8.01-8.04 (m, 1Н), 8.01 (s, 1Н), 7.79 (d, J=8,58 Гц, 1Н), 7.01 (d, J=8,78 Гц, 1H), 4.09 (s, 3H).
Промежуточное соединение 20: показанную ниже 2-(метиламино)-хинолин-6-карбоновую кислоту получали следующим образом.
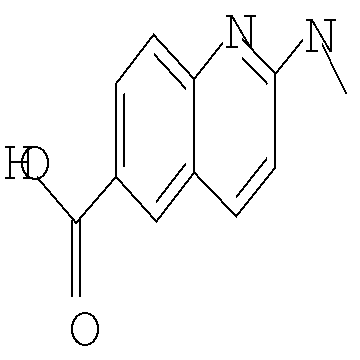
Стадия 1: этил-хинолин-6-карбоксилат
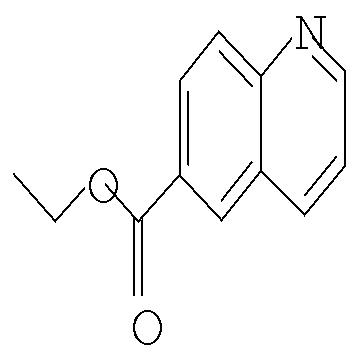
К раствору хинолин-6-карбоновой кислоты (2,8 г; 16 ммоль) в этаноле (100 мл) добавляли концентрированную серную кислоту (2 мл). Реакционную смесь нагревали до температуры дефлегмации в течение ночи. Растворитель выпаривали, получая коричневый остаток, который переносили в этилацетат (150 мл). Смесь промывали водой (2×30 мл), насыщенным водным бикарбонатом натрия (2×30 мл) и рассолом (2×30 мл). Органический слой сушили над сульфатом натрия, фильтровали и концентрировали до масла. После очистки колоночной флэш-хроматографией получали указанное в заголовке соединение (2,0 г; 81%) в виде коричневого твердого вещества.
Стадия 2:1-оксид 6-(этоксикарбонил)хинолина
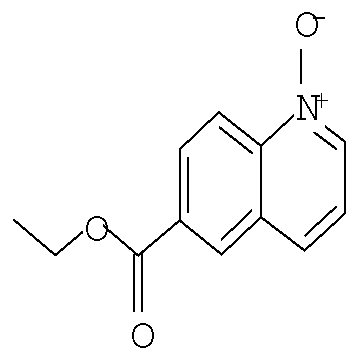
К этил-хинолин-6-карбоксилату (3,2 г; 16 ммоль) в дихлорметане (120 мл) порциями добавляли мета-хлорпероксибензойную кислоту (4,9 г; 0,024 моль). Реакционную смесь перемешивали при комнатной температуре в течение 4 часов. Реакционную смесь разбавляли дихлорметаном и промывали насыщенным водным карбонатом натрия (3×30 мл) и рассолом (2×40 мл). Органический слой сушили над сульфатом натрия, фильтровали и концентрировали. После очистки колоночной флэш-хроматографией получали указанное в заголовке соединение (2,45 г; 71%) в виде коричневого твердого вещества. 1H ЯМР (400 МГц, CDCl3, δ): 8.81-8.79 (d, 1H), 8.62 (s, 2H), 8.35-8.33 (d, 1H), 7.87-7.85 (d, 1H), 7.39 (s, 1H), 4.49-4.44 (q, 2H), 1.47-1.43 (t, 3Н).
Стадия 3: этил-2-(метиламино)хинолин-6-карбоксилат
Ангидрид трифторметансульфоновой кислоты (1,92 мл; 11,4 ммоль) по каплям при -70°С добавляли к раствору 1-оксида 6-(этоксикарбонил)хинолина (2,25 г; 10,4 ммоль) в дихлорметане (150 мл). Смесь перемешивали при -70°С в течение 5 минут. Затем по каплям добавляли раствор метиламина в тетрагидрофуране (31 мл; 62 ммоль, 2 М). Смесь перемешивали при -70°С в течение 5 минут. Реакцию гасили водой (20 мл). Слои разделяли и водный слой экстрагировали дихлорметаном (3×30 мл). Объединенные органические экстракты промывали рассолом, сушили над сульфатом натрия, фильтровали и концентрировали. После очистки колоночной флэш-хроматографией получали указанное в заголовке соединение (850 мг, 35%) в виде желтого твердого вещества. 1Н ЯМР (400 МГц, CDCl3, δ): 8.33 (d, 1H), 8.16-8.13 (m, 1H), 7.90-7.87 (d, 1H), 7.70-7.67 (d, 1H), 6.68 (d, 1H), 5.30 (br. s., 1H), 4.43-4.38 (q, 2H), 3.13-3.12 (d, 3H), 1.44-1.40 (m, 3H).
Стадия 4: 2-(метиламино)хинолин-6-карбоновая кислота Водный гидроксид натрия (4 мл; 8 ммоль; 2 н.) добавляли к раствору этил-2-(метиламино)хинолин-6-карбоксилата (850 мг; 3,7 ммоль) в этаноле (10 мл). Реакционную смесь нагревали до 50°С в течение ночи. Этанол удаляли в вакууме и остаток подкисляли до pH 5, используя 1 н. водную соляную кислоту. Полученный осадок собирали фильтрацией и сушили под вакуумом, получая указанное в заголовке соединение (710 мг, 96%) в виде желтого твердого вещества. 1Н ЯМР (400 МГц, DMSO-d6, δ): 8.26 (d, 1H), 7.96-7.93 (m, 2H), 7.50 (d, 1H), 7.43 (d, 1H), 6.81 (d, 1H), 2.91 (d, 3H).
Промежуточное соединение 21: показанную ниже 7-(трифторметил)-1Н-индазол-5-карбоновую кислоту получали следующим образом.
Стадия 1: 4-бром-2-метил-6-(трифторметил)анилин
К раствору 2-метил-6-(трифторметил)анилина (3,0 г; 17 ммоль) в ацетонитриле (85 мл) при комнатной температуре в течение 30 минут небольшими порциями добавляли N-бромсукцинимид (3,0 г; 17 ммоль). Реакционную смесь оставляли перемешиваться в течение 1 часа. Реакционную смесь выливали в смесь вода/рассол и экстрагировали этилацетатом (3×). Объединенные органические экстракты сушили над сульфатом магния, фильтровали и концентрировали. После очистки колоночной флэш-хроматографией (0-40% этилацетат/гептаны) получали указанное в заголовке соединение (4,13 г; 95%) в виде коричневого масла. 1H ЯМР (400 МГц, CDCl3, δ): 7.42 (d, J=2,34 Гц, 1H), 7.31 (s, 1H), 2.17 (s, 3H).
Стадия 2: 5-бром-7-(трифторметил)-1Н-индазол
К раствору 4-бром-2-метил-6-(трифторметил)анилина (3,3 г; 13 ммоль) в толуоле (65 мл) и ледяной уксусной кислоте (11,2 мл; 195 ммоль) порциями добавляли ацетат калия (10,2 г; 104 ммоль). Через 15 минут образовывалось большое количество осадка, затрудняющего перемешивание реакционной смеси. Реакционную смесь разбавляли уксусной кислотой (10 мл). Затем по каплям добавляли изоамилнитрит (1,92 мл; 14,3 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Добавляли дополнительное количество изоамилнитрита (0,5 мл; 3,7 ммоль) и реакционную смесь оставляли перемешиваться в течение 15 часов. Реакционную смесь разбавляли водой (100 мл) и перемешивали в течение 1,5 часов. Раствор распределяли между этилацетатом и насыщенным водным бикарбонатом натрия. Слои разделяли, органические слои промывали рассолом, сушили над сульфатом магния, фильтровали и концентрировали. После очистки колоночной флэш-хроматографией (5-50% этилацетат/гептаны) получали указанное в заголовке соединение (1,78 г; 52%) в виде желтого порошка. -ESI (М-Н+1) 264,9; 1H ЯМР (400 МГц, CDCl3, δ): 8.13 (s, 1H), 8.09-8.11 (m, 1H), 7.76 (dd, J=1,66, 0,88 Гц, 1Н).
Стадия 3: метил-7-(трифторметил)-1Н-индазол-5-карбоксилат
В герметично закрываемую пробирку добавляли [1,1'-бис(дифенилфосфино)ферроцен]-дихлорпалладий(II) (комплекс с дихлорметаном; 45,7 мг; 0,056 ммоль), 5-бром-7-(трифторметил)-1Н-индазол (100 мг; 0,38 ммоль), триэтиламин (105 мкл, 0,752 ммоль) и метанол (2 мл). Пробирку закрывали крышкой и в течение 5 минут осуществляли барботирование монооксидом углерода. Затем реакционную смесь нагревали до 70°С в течение 5 часов. Реакционную смесь охлаждали до комнатной температуры и оставляли перемешиваться в течение 2 суток. Реакционную смесь концентрировали и очищали колоночной флэш-хроматографией (0-50% этилацетат/гептаны), получая указанное в заголовке соединение (64 мг, 69%) в виде белого порошка. -ESI (М-Н) 243,1; 1H ЯМР (400 МГц, CDCl3, δ): 8.72 (s, 1H), 8.37 (s, 1H), 8.28 (s, 1H), 3.98 (s, 3Н).
Стадия 4: 7-(трифторметил)-1Н-индазол-5-карбоновая кислота
К раствору метил-7-(трифторметил)-1Н-индазол-5-карбоксилата (62 мг; 0,25 ммоль) в метаноле (2 мл) и тетрагидрофуране (2 мл) добавляли 1 н. водный гидроксид лития (0,76 мл; 0,76 ммоль). Реакционную смесь нагревали до 60°С в течение 17 часов. Реакционную смесь концентрировали, остаток разбавляли водой и подкисляли до pH 3, используя 1 н. водную соляную кислоту. Раствор экстрагировали дихлорметаном (3×). Объединенные органические экстракты промывали рассолом, сушили над сульфатом магния, фильтровали и концентрировали, получая указанное в заголовке соединение (17 мг, 29%) в виде беловатого порошка. +ESI (М+Н) 231,1.
Промежуточное соединение 22: показанную ниже 3-(метиламино)-изохинолин-6-карбоновую кислоту получали следующим образом.
Стадия 1: 6-бром-N-метилизохинолин-3-амин
К раствору 6-бромизохинолин-3-амина (50,0 мг; 2,6 ммоль) в N,N-диметилформамиде (10 мл) добавляли N,N-диметилформамида диметилацеталь (2 мл). Реакционный сосуд герметично закрывали и нагревали в микроволновой печи Biotage Smith Synthesizer до 110°С в течение 20 минут. Затем к реакционной смеси добавляли триацетоксиборгидрид натрия (59 мг; 0,28 ммоль). Флакон снова герметично закрывали и опять нагревали до 110°С в микроволновой печи Biotage Smith Synthesizer в течение 10 минут. Реакционную смесь концентрировали. Остаток растворяли в этилацетате (50 мл) и промывали рассолом (2×20 мл). Органические экстракты сушили над сульфатом натрия, фильтровали и концентрировали. После очистки колоночной флэш-хроматографией получали указанное в заголовке соединение (23 мг, 43%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, CDCl3, δ): 8.76 (s, 1Н), 7.74 (s, 1Н), 7.61 (d, 1Н), 7.28 (d, 1H), 6.40 (s, 1H), 5.09-5.07 (m, 1H), 2.97 (s, 3H).
Стадия 2: 3-(метиламино)изохинолин-6-карбоновая кислота
Метил-3-(метиламино)изохинолин-6-карбоксилат получали способом, аналогичным описанному на стадии 3 для промежуточного соединения 21, используя 6-бром-N-метилизохинолин-3-амин. К неочищенному веществу (580 мг; 2,7 ммоль) добавляли воду (5 мл), метанол (5 мл) и моногидрат гидроксида лития (300 мг; 7 ммоль). Смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь концентрировали и остаток подкисляли до pH 5, используя 1 н. водную соляную кислоту. Полученный остаток сушили под вакуумом и очищали обращенно-фазовой HPLC, получая указанное в заголовке соединение (512 мг, 89%) в виде белого твердого вещества. 1H ЯМР (400 МГц, CD3OD, δ): 8.81 (s, 1H), 8.23 (s, 1H), 7.80 (d, 1H), 7.72 (d, 1H), 6.70 (s, 1H), 2.93 (s, 3H).
Промежуточное соединение 23: показанную ниже 2-(метиламино)-хинолин-7-карбоновую кислоту получали следующим образом.
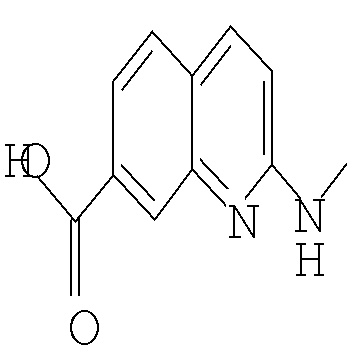
Указанное в заголовке соединение получали способом, аналогичным описанному на стадиях 3-4 для промежуточного соединения 20, используя 1-оксид 7-(этоксикарбонил)хинолина. 1H ЯМР (400 МГц, DMSO-d6, δ): 8.08 (s, 1H), 7.90 (d, 1H), 7.71-7.62 (m, 2H), 7.21 (s, 1H), 6.84 (d, 1H), 2.91 (d, 3H).
Промежуточное соединение 24: показанную ниже 5-метоксихинолин-3-карбоновую кислоту получали следующим образом.
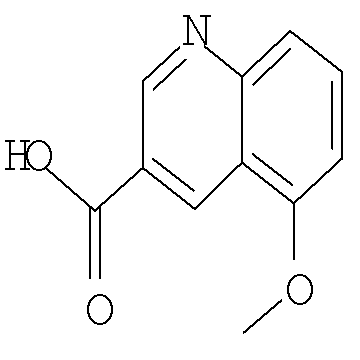
Метил-5-метоксихинолин-3-карбоксилат (Organic and Biomolecular Chemistry, 7(12), 2612-2618, 2009) омыляли, используя водный гидроксид лития. +ESI (М+Н) 203.9; 1H ЯМР (400 МГц, DMSO-d6, δ): 9.30 (d, 1H), 9.03 (d, 1H), 7.84-7.80 (m, 1H), 7.66 (d, 1H), 7.15 (d, 1H), 4.04 (s, 3H).
Промежуточное соединение 25: показанную ниже 2-(метиламино)-1Н-бензо[d]имидазол-5-карбоновую кислоту получали следующим образом.
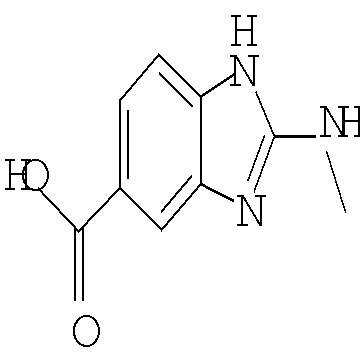
Стадия 1. Метил-2-(метиламино)-1Н-бензо[о]имидазол-5-карбоксилат

Смесь 3,4-диаминобензойной кислоты (15 г; 0,09 моль) и метилизотиоцианата (6,6 г; 0,09 моль) растворяли в тетрагидрофуране (90 мл). Реакционную смесь нагревали при температуре дефлегмации в течение 3 часов и затем концентрировали. Остаток выливали в ледяную воду. Полученный осадок отфильтровывали, промывали водой и сушили под вакуумом, получая метил-4-амино-3-(3-метилтиоуреидо)бензоат (12,0 г; 56%).
К этому твердому веществу (12 г; 0,05 моль) добавляли этанол (200 мл), затем метилиодид (35,5 г; 0,25 моль). Реакционную смесь нагревали до температуры дефлегмации и перемешивали в течение ночи. Реакционную смесь концентрировали и остаток подщелачивали гидроксидом аммония. Твердые вещества собирали фильтрацией и промывали водой. После очистки колоночной хроматографией (9-25% этилацетат/петролейный эфир) получали указанное в заголовке соединение (2,9 г; 28%) в виде желтого твердого вещества. 1H ЯМР (400 МГц, CDCl3, δ): 8.37 (s, 1H), 7.92-7.96 (m, 1H), 7.51 (d, J=8,4 Гц, 1H), 3.93 (s, 3Н), 2.81 (s, 3Н).
Стадия 2. 2-(метиламино)-1Н-бензо[d]имидазол-5-карбоновая кислота 3 н. водную соляную кислоту (14 мл; 42 ммоль) добавляли к метил-2-(метиламино)-1Н-бензо[d]имидазол-5-карбоксилату (2,9 г; 14 ммоль) и реакционную смесь перемешивали при температуре дефлегмации в течение ночи. Реакционную смесь концентрировали, получая указанное в заголовке соединение (2,4 г; 90%) в виде желтого твердого вещества. 1H ЯМР (400 МГц, CD3OD, δ): 7.96-8.00 (m, 2Н), 7.40 (d, J=8,4 Гц, 1H), 3.10 (s, 3Н).
Промежуточное соединение 26: показанную ниже 2-амино-1Н-бензо[d]имидазол-5-карбоновую кислоту получали следующим образом.
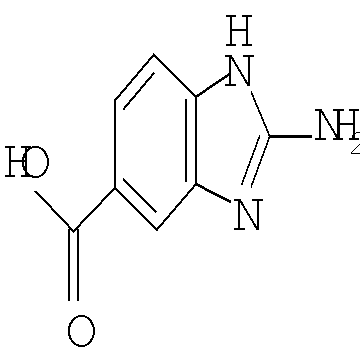
Раствор бромциана (5,0 мл; 5 М в ацетонитриле; 25 ммоль) добавляли к смеси метил-3,4-диаминобензоата (3,0 г; 18 ммоль) в воде (50 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи. К реакционной смеси добавляли водный аммиак (20 мл) и этилацетат (100 мл) и слои разделяли. Органические слои сушили над сульфатом натрия, фильтровали и концентрировали. К неочищенному остатку добавляли 2 н. водную соляную кислоту (18 мл; 36,0 ммоль) и смесь нагревали при температуре дефлегмации в течение ночи. Реакционную смесь концентрировали, получая указанное в заголовке соединение (2,90 г; 97%). 1H ЯМР (400 МГц, DMSO-d6, δ): 8.75 (s, 2H), 7.84 (s, 1H), 7.77 (dd, J=1,2, 8,4 Гц, 1H), 7.38 (d, J=8,4 Гц, 1H).
Промежуточное соединение 27: показанную ниже 1-(4-метоксибензил-амино)изохинолин-7-карбоновую кислоту получали следующим образом.
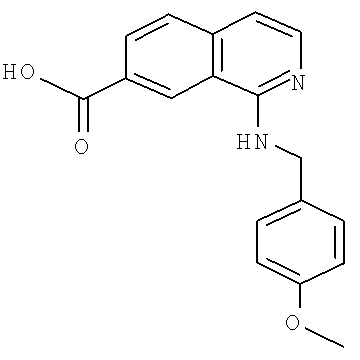
Стадия 1: 1-оксо-1,2-дигидроизохинолин-7-карбоновая кислота
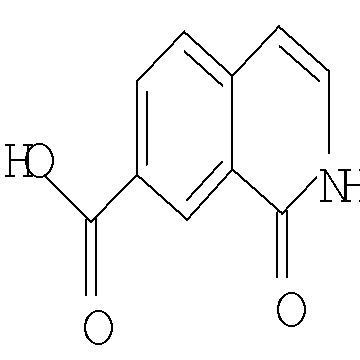
К суспензии 7-бромизохинолин-1(2Н)-она (70 г; 0,31 моль) в N,N-диметилформамиде (1 л) добавляли цианид меди (56 г; 0,63 моль). Реакционную смесь нагревали до 180°С в течение 2 часов. Реакционную смесь охлаждали до комнатной температуры и разбавляли водой (1 л). Раствор экстрагировали этилацетатом (3×). Органические экстракты сушили над сульфатом натрия, фильтровали и концентрировали, получая неочищенный 1-оксо-1,2-дигидроизохинолин-7-карбонитрил (37 г). Это неочищенное вещество переносили в этанол (500 мл) и добавляли 1 н. водный гидроксид натрия (400 мл). Смесь нагревали до температуры дефлегмации и перемешивали в течение 2 часов. Реакционную смесь охлаждали до комнатной температуры и pH подводили до ~2, используя 1 н. водную соляную кислоту. Твердые вещества собирали фильтрацией, промывали водой и сушили под вакуумом, получая указанное в заголовке соединение (35 г; 85%) в виде беловатого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6, δ): 13.15 (br. s., 1H), 11.49 (s, 1H), 8.75 (s, 1H), 8.17-8.14 (m, 1H), 7.75 (d, 1H), 7.34-7.29 (m, 1H), 6.62 (d, 1H).
Стадия 2: 1-хлоризохинолин-7-карбонилхлорид
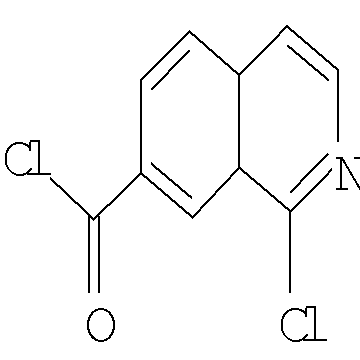
Оксихлорид фосфора (74 мл; 793 ммоль) добавляли к 1-оксо-1,2-дигидроизохинолин-7-карбоновой кислоте (3,0 г; 20 ммоль). Реакционную смесь нагревали до 90°С в течение 5 часов. Реакционную смесь концентрировали досуха. Вещество переносили в дихлорметан (250 мл) и насыщенный водный бикарбонат натрия (200 мл). Слои разделяли и водный слой еще раз экстрагировали дихлорметаном (100 мл). Объединенные органические экстракты сушили над сульфатом магния, фильтровали и концентрировали, получая указанное в заголовке соединение (3,0 г; 80%) в виде желтого твердого вещества. +ESI (М+Н) 227,1; 1H ЯМР (400 МГц, CDCl3, δ): 9.18-9.22 (m, 1H), 8.44 (d, J=5,7 Гц, 1Н), 8.32 (dd, J=8,8, 1,8 Гц, 1Н), 7.95 (d, J=8,8 Гц, 1Н), 7.68 (d, J=5,7 Гц, 1Н).
Стадия 3: этил-1-хлоризохинолин-7-карбоксилат
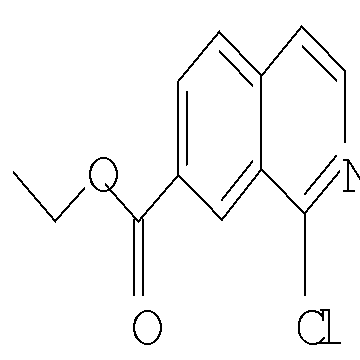
1-Хлоризохинолин-7-карбонилхлорид (3,02 г; 13,4 ммоль) растворяли в тетрагидрофуране (135 мл) и охлаждали до 0°С. Добавляли этанол (6,1 мл; -94 ммоль) и триэтиламин (2,05 мл; 14,7 ммоль). Реакционную смесь оставляли нагреваться до комнатной температуры и перемешивали в течение 2 часов. Реакционную смесь распределяли между этилацетатом (500 мл) и насыщенным водным бикарбонатом натрия (250 мл). Органический слой сушили над сульфатом магния, фильтровали и концентрировали, получая указанное в заголовке соединение (3,0 г; 96%) в виде желтого твердого вещества. +ESI (М+Н) 236,1; 1H ЯМР (400 МГц, CDCl3, δ): 9.06 (s, 1H), 8.30-8.39 (m, 2Н), 7.89 (d, J=8,6 Гц, 1H), 7.63 (d, J=5,7 Гц, 1H), 4.48 (q, J=7,1 Гц, 2Н), 1.46 (t, J=7,1 Гц, 3Н).
Стадия 4: этил-1-(4-метоксибензиламино)изохинолин-7-карбоксилат
К раствору этил-1-хлоризохинолин-7-карбоксилата (548 мг; 2,32 ммоль) в N,N-диметилформамиде (9,3 мл) добавляли 4-метокси-бензиламин (4,6 мл; 35 ммоль) и карбонат калия (5,14 г; 37,2 ммоль). Реакционную смесь нагревали до 70°С и перемешивали в течение ночи. Реакционную смесь охлаждали до комнатной температуры и разбавляли этилацетатом и водой. Слои разделяли и водный слой дважды экстрагировали этилацетатом. Объединенные органические экстракты промывали водой и рассолом, сушили над сульфатом магния, фильтровали и концентрировали. После очистки колоночной флэш-хроматографией (0-35% этилацетат/гептаны) получали указанное в заголовке соединение (430 мг; 55%) в виде зеленоватого масла. 1H ЯМР (400 МГц, CDCl3, δ): 8.49 (s, 1Н), 8.16 (dd, J=8,6, 1,6 Гц, 1Н), 8.09 (d, J=5,9 Гц, 1Н), 7.69 (d, J=8,6 Гц, 1H), 7.33-7.40 (m, 2Н), 6.96 (d, J=5,9 Гц, 1H), 6.87-6.93 (m, 2Н), 5.67 (br. s., 1Н), 4.76 (d, J=5,1 Гц, 2Н), 4.41 (q, J=7,2 Гц, 2Н), 3.81 (s, 3H), 1.37-1.43 (m, 3Н).
Стадия 5: 1-(4-метоксибензиламино)изохинолин-7-карбоновая кислота
К раствору этил-1-(4-метоксибензиламино)изохинолин-7-карбоксилата (430 мг; 1,28 ммоль) в метаноле (8,5 мл) добавляли 6 н. водный гидроксид натрия (1,1 мл; 6,4 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь концентрировали. Остаток переносили в воду и подкисляли 1 н. водной соляной кислотой до образования осадка. Твердое вещество собирали фильтрацией и сушили под вакуумом, получая указанное в заголовке соединение (328 мг, 83%) в виде желтого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6, δ): 8.92 (s, 1H), 8.30 (t, J=5,8 Гц, 1H), 8.06 (dd, J=8,4, 1,4 Гц, 1H), 7.88 (d, J=5,7 Гц, 1H), 7.69 (d, J=8,6 Гц, 1H), 7.24-7.31 (m, 2H), 6.88 (d, J=5,7 Гц, 1H), 6.79-6.85 (m, 2H), 4.62 (d, J=5,9 Гц, 2H), 3.67 (s, 3Н).
Промежуточное соединение 28: показанную ниже 3-метокси-1Н-индазол-6-карбоновую кислоту получали следующим образом.
Стадия 1: метил-3-гидрокси-1Н-индазол-6-карбоксилат
3-Оксо-2,3-дигидро-1Н-индазол-6-карбоновую кислоту (1,5 г; 8,4 ммоль) суспендировали в метаноле (17 мл). Добавляли концентрированную соляную кислоту (3,1 мл; 101 ммоль) и реакционную смесь нагревали до температуры дефлегмации в течение 24 часов. Реакционную смесь концентрировали, получая указанное в заголовке соединение (1,6 г; 100%). +ESI (М+Н) 193,1; 1H ЯМР (400 МГц, DMSO-d6, δ): 11.98 (br. s., 1H), 7.89 (s, 1H), 7.72 (d, J=8,6 Гц, 1H), 7.50 (dd, J=8,5, 1,3 Гц, 1H), 3.85 (s, 3Н).
Стадия 2: 1-этил-6-метил-3-гидрокси-1Н-индазол-1,6-дикарбоксилат
Метил-3-гидрокси-1Н-индазол-6-карбоксилат (1,6 г; 8,3 ммоль) суспендировали в пиридине (10 мл). Медленно добавляли этилхлорформиат (1,0 мл; 10 ммоль) и реакционную смесь оставляли перемешиваться при комнатной температуре в течение 2 часов. Реакционную смесь выливали в воду (65 мл) и охлаждали в холодильнике в течение 4 часов. Полученный коричневый осадок собирали фильтрацией, промывали водой и сушили под вакуумом, получая указанное в заголовке соединение (1,35 г; 61%) в виде бежевого твердого вещества. +ESI (М+Н) 265,0; 1H ЯМР (400 МГц, CDCl3, δ): 8.80 (d, J=6,0 Гц, 1Н), 8.01 (dd, J=8,2, 1,2 Гц, 1Н), 7.88 (d, J=8,6 Гц, 1Н), 4.60 (q, J=7,0 Гц, 2Н), 3.98 (s, 3H), 1.57 (t, J=7,1 Гц, 3Н).
Стадия 3: 1-этил-6-метил-3-метокси-1Н-индазол-1,6-дикарбоксилат
1-Этил-6-метил-3-гидрокси-1Н-индазол-1,6-дикарбоксилат (1,35 г; 5,11 ммоль) суспендировали в ацетоне (65 мл). Добавляли карбонат цезия (1,75 г; 5,36 ммоль) и метилиодид (1,0 мл; 15 ммоль) и реакционную смесь нагревали до температуры дефлегмации в течение 23 часов. Реакционную смесь концентрировали досуха. Остаток переносили в дихлорметан (100 мл) и воду (100 мл). Слои разделяли и водный слой еще раз экстрагировали дихлорметаном. Объединенные органические экстракты сушили над сульфатом магния, фильтровали и концентрировали. После очистки колоночной флэш-хроматографией получали два региоизомерных продукта.
1-Этил-6-метил-3-метокси-1Н-индазол-1,6-дикарбоксилат (444 мг, 31%) в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3, δ): 8.78 (s, 1H), 7.96 (dd, J=8,2, 1,4 Гц, 1H), 7.70 (dd, J=8,2,0,8 Гц, 1H), 4.57 (q, J=7,2 Гц, 2Н), 4.19 (s, 3Н), 3.96 (s, 3Н), 1.51 (t, J=7,1 Гц, 3Н).
1-Этил-6-метил-2-метил-3-оксо-2,3-дигидро-1Н-индазол-1,6-дикарбоксилат (514 мг, 36%) в виде желтого твердого вещества. 1H ЯМР (400 МГц, CDCl3, δ): 8.56 (s, 1Н), 8.00 (m, 1Н), 7.92 (d, J=8,6 Гц, 1Н), 4.49 (q, J=7,2 Гц, 2Н), 3.97 (s, 3Н), 3.69 (s, 3Н), 1.49 (t, J=7,1 Гц, 3Н).
Стадия 4: 3-метокси-1Н-индазол-6-карбоновая кислота
1-Этил-6-метил-3-метокси-1Н-индазол-1,6-дикарбоксилат (444 мг; 1,60 ммоль) суспендировали в этаноле (5 мл). Добавляли водный раствор гидроксида калия (16 мл, 16 ммоль, 1 M), реакционную смесь нагревали до 65°С и перемешивали в течение 1,5 часов. Реакционную смесь охлаждали до комнатной температуры и концентрировали. Остаток переносили в воду и раствор подкисляли 1 н. водной соляной кислотой до образования осадка. Твердое вещество собирали фильтрацией, промывали водой и сушили под вакуумом, получая указанное в заголовке соединение (232 мг, 76%) в виде оранжевого твердого вещества. +ESI (М+Н) 193,2; 1H ЯМР (400 МГц, DMSO-d6, δ): 12.22 (s, 1H), 7.90-7.94 (m, 1H), 7.64 (d, J=8,4 Гц, 1Н), 7.53 (dd, J=8,4, 1,4 Гц, 1Н), 3.99 (s, 3Н).
Промежуточное соединение 29: показанную ниже 3-(трифторметил)-1Н-индазол-5-карбоновую кислоту получали следующим образом.
Указанное в заголовке соединение получали способом, аналогичным описанному на стадиях 3-4 для промежуточного соединения 21, используя 5-бром-3-(трифторметил)-1Н-индазол. +ESI (М+Н) 231,1.
Промежуточное соединение 30: показанную ниже 1-(4-метоксибензил-амино)изохинолин-6-карбоновую кислоту получали следующим образом.
Стадия 1: 1-оксо-1,2-дигидроизохинолин-6-карбоновая кислота
В смеси 6-бромизохинолин-1(2Н)-она (30 г; 0,134 моль), триэтиламина (17,6 г; 0,174 моль), хлорида палладия(II) (0,24 г; 1,34 ммоль) и 2,2'-бис(дифенилфосфино)-1,1'-бинафтила (0,84 г; 1,34 ммоль) в метаноле (300 мл) создавали давление монооксида углерода до 2 МПа. Реакционную смесь нагревали до 100°С и перемешивали в течение 12 часов. Реакционную смесь фильтровали через целит и концентрировали. Остаток промывали водой и твердое вещество сушили под вакуумом, получая неочищенный метил-1-оксо-1,2-дигидроизохинолин-6-карбоксилат (23,8 г; 95,2%) в виде желтого твердого вещества. Твердое вещество разбавляли тетрагидрофураном (200 мл) и водой (200 мл). К этой смеси добавляли гидроксид лития (16,8 г; 0,4 моль) и реакционную смесь перемешивали при комнатной температуре в течение 4 часов. Реакционную смесь промывали этилацетатом (3×) и эти промывки отбрасывали. Водный слой подкисляли, используя 4 н. водную соляную кислоту, до pH 5. Полученный осадок собирали фильтрацией и сушили под вакуумом, получая указанное в заголовке соединение (11,3 г; 49%) в виде желтого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6, δ): 11.48 (s, 1H), 8.24 (d, 2Н), 7.93 (d, 1Н), 7.22 (d, 1Н), 6.68 (d, 1Н).
Стадия 2: 1-(4-метоксибензиламино)изохинолин-6-карбоновая кислота
Указанное в заголовке соединение получали способом, аналогичным описанному на стадиях 2-5 для промежуточного соединения 27, используя 1-оксо-1,2-дигидроизохинолин-6-карбоновую кислоту. +ESI (М+Н) 309,2; 1H ЯМР (400 МГц, CD3OD, δ): 8.37 (d, J=1,56 Гц, 1H), 8.34 (d, J=8,78 Гц, 1H), 8.12 (dd, J=8,68, 1,66 Гц, 1H), 7.67 (d, J=6,44 Гц, 1H), 7.29-7.36 (m, 2Н), 7.15 (d, J=6,24 Гц, 1H), 6.86-6.93 (m, 2Н), 4.73 (s, 2Н), 3.76 (s, 3Н).
Промежуточное соединение 31: показанную ниже 1-(метиламино)-изохинолин-7-карбоновую кислоту получали следующим образом.
Стадия 1: этил-1-(метиламино)изохинолин-7-карбоксилат

Раствор метиламина в тетрагидрофуране (30 мл, 60 ммоль, 2 M) добавляли к этил-1-хлоризохинолин-7-карбоксилату (полученному на стадии 3 для промежуточного соединения 27) (705 мг; 2,99 ммоль) в герметично закрываемой пробирке. Реакционную смесь нагревали до 60°С и перемешивали в течение ночи. Данные LCMS показали, что реакция не завершена. Добавляли дополнительное количество метиламина (10 мл; 20 ммоль; 2М в THF) и реакционную смесь нагревали до 60°С в течение следующих 18 часов. Реакционную смесь охлаждали до комнатной температуры и концентрировали. Остаток распределяли между водой и дихлорметаном. Органический слой сушили над сульфатом магния, фильтровали и концентрировали. После очистки колоночной флэш-хроматографией (25-65% этилацетат/гептаны) получали указанное в заголовке соединение (584 мг, 85%) в виде желтого масла, которое отвердевало при стоянии. +ESI (М+Н) 231,1; 1H ЯМР (400 МГц, DMSO-d6, δ): 8.83-8.94 (m, 1H), 8.07 (dd, J=8,58, 1,56 Гц, 1H), 7.99 (d, J=5,85 Гц, 1H), 7.89 (d, J=4,49 Гц, 1H), 7.77 (d, J=8,58 Гц, 1Н), 6.92 (d, J=5,07 Гц, 1Н), 4.38 (q, J=7,02 Гц, 2Н), 2.97 (d, J=4,49 Гц, 3Н), 1.38 (t, J=7,12 Гц, 3Н).
Стадия 2: 1-(метиламино)изохинолин-7-карбоновая кислота
Указанное в заголовке соединение получали способом, аналогичным описанному на стадии 3 для промежуточного соединения 19, используя этил-1-(метиламино)изохинолин-7-карбоксилат. +ESI (М+Н) 203,1; 1H ЯМР (400 МГц, DMSO-dg, δ): 13.03 (br. s., 1H), 8.87 (s, 1H), 8.06 (dd, J=8,51, 1,47 Гц, 1H), 7.97 (d, J=5,67 Гц, 1Н), 7.85 (d, J=4,50 Гц, 1Н), 7.75 (d, J=8,41 Гц, 1Н), 6.91 (d, J=5,87 Гц, 1H), 2.95 (d, J=4,50 Гц, 3Н).
Промежуточное соединение 32: показанную ниже 3-(трифторметил)-1Н-индазол-6-карбоновую кислоту получали следующим образом.
Стадия 1: 1-(4-бром-2-фторфенил)-2,2,2-трифторэтанол

К раствору 4-бром-2-фторбензальдегида (1,00 г; 4,93 ммоль) в тетрагидрофуране (50 мл) при 0°С по каплям в течение 5 минут добавляли триметилсилилтрифторметан (0,77 мл; 4,9 ммоль). Реакционную смесь перемешивали при 0°С в течение 10 минут. Затем медленно добавляли фторид тетрабутиламмония (0,49 мл; 0,49 ммоль; 1 М в тетрагидрофуране), реакционную смесь оставляли постепенно нагреваться до комнатной температуры и перемешивали в течение 3 суток. Реакционную смесь концентрировали и остаток переносили в дихлорметан. Раствор один раз промывали 1 н. водной соляной кислотой и один раз рассолом. Органические слои сушили над сульфатом магния, фильтровали и концентрировали. После очистки колоночной хроматографией (0-50% этилацетат/гептаны) получали указанное в заголовке соединение (1,0 г; 75%) в виде прозрачного масла. 1H ЯМР (400 МГц, CDCI3, δ): 7.48 (d, J=7,61 Гц, 1Н), 7.39 (d, J=1,76 Гц, 1Н), 7.29 (dd, J=9,56, 1,95 Гц, 1Н), 5.33-5.40 (m, 1Н), 2.70 (d, J=5,46 Гц, 1Н).
Стадия 2: 1-(4-бром-2-фторфенил)-2,2,2-трифторэтанон
К раствору 1-(4-бром-2-фторфенил)-2,2,2-трифторэтанола (1,09 г; 3,99 ммоль) в этилацетате (30 мл) добавляли 2-иодоксибензойную кислоту (2,28 г; 7,97 ммоль). Реакционную смесь нагревали до температуры дефлегмации в течение ночи. Реакционную смесь охлаждали до комнатной температуры и разбавляли гептанами (30 мл). Смесь фильтровали через целит и фильтрат концентрировали, получая указанное в заголовке соединение (1,03 г; 95%) в виде бледно-желтого масла. 1H ЯМР (400 МГц, CDCl3, δ): 7.44 (dd, J=10,15, 1,56 Гц, 1Н), 7.48 (m, 1Н), 7.76 (m, 1Н).
Стадия 3: 6-бром-3-(трифторметил)-1Н-индазол
Гидрат гидразина (3,5 мл; 45 ммоль) добавляли к раствору 1-(4-бром-2-фторфенил)-2,2,2-трифторэтанона (1,00 г; 3,69 ммоль) в 1-бутаноле (15 мл). Реакционную смесь нагревали до температуры дефлегмации в течение 5 часов, затем охлаждали до комнатной температуры и оставляли перемешиваться в течение ночи. Реакционную смесь разбавляли водой (50 мл) и экстрагировали этилацетатом (3×). Объединенные органические экстракты промывали рассолом, сушили над сульфатом магния, фильтровали и концентрировали. После очистки колоночной флэш-хроматографией (0-50% этилацетат/гептаны) получали указанное в заголовке соединение (310 мг, 32%) в виде беловатого твердого вещества. 1H ЯМР (400 МГц, CDCl3, δ): 7.42 (dd, J=8,58, 1,56 Гц, 1Н), 7.72 (d, J=8,58 Гц, 1Н), 7.75 (dd, J=1,56, 0,78 Гц, 1Н).
Стадия 4: 3-(трифторметил)-1Н-индазол-6-карбоновая кислота
Указанное в заголовке соединение получали способом, аналогичным описанному на стадиях 3-4 для промежуточного соединения 21, используя 6-бром-3-(трифторметил)-1Н-индазол. -ESI (М-Н) 229,1.
Промежуточное соединение 33: показанную ниже 2-метил-3-оксо-2,3-дигидро-1Н-индазол-6-карбоновую кислоту получали следующим образом.
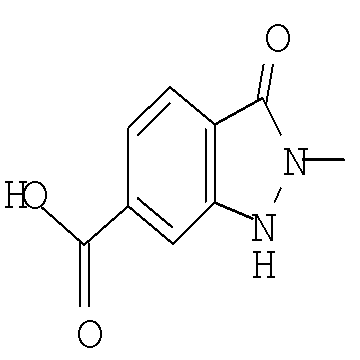
К суспензии 1-этил-6-метил-2-метил-3-оксо-2,3-дигидро-1Н-индазол-1,6-дикарбоксилата (полученного на стадии 3 для промежуточного соединения 28) (514 мг; 1,85 ммоль) в этаноле (6 мл) добавляли 1 н. водный гидроксид калия (18,5 мл; 18,5 ммоль). Реакционную смесь нагревали до 65°С в течение 1,5 часов. Реакционную смесь охлаждали до комнатной температуры и концентрировали досуха. Остаток переносили в воду и подкисляли 1 н. водной соляной кислотой до образования осадка. Твердое вещество собирали фильтрацией и сушили под вакуумом, получая указанное в заголовке соединение (196 мг, 55%) в виде коричневого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6, δ): 13.12 (br. s., 1Н), 10.61 (br. s., 1Н), 7.76 (s, 1H), 7.70 (d, J=8,2 Гц, 1Н), 7.60 (dd, J=8,2, 1,2 Гц, 1Н), 3.38 (s, 3H).
Промежуточное соединение 34: показанную ниже 3-хлор-1Н-пирроло[3,2-b]пиридин-6-карбоновую кислоту получали следующим образом.
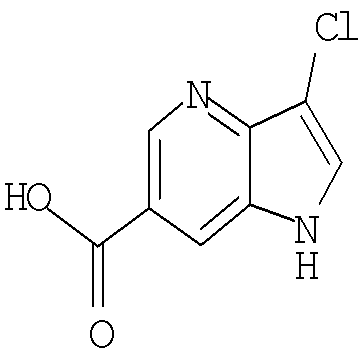
Стадия 1: метил-3-хлор-1Н-пирроло[3,2-b]пиридин-6-карбоксилат
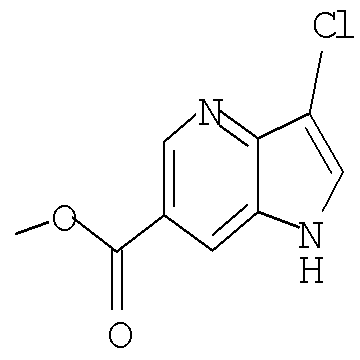
К раствору метил-1Н-пирроло[3,2-b]пиридин-6-карбоксилата (1,00 г; 5,68 ммоль) в N,N-диметилформамиде (15 мл) при 0°С добавляли N-хлорсукцинимид (895 мг; 5,96 ммоль). Реакционную смесь оставляли постепенно нагреваться до комнатной температуры и перемешивали в течение ночи. Реакционную смесь разбавляли водой (125 мл) и перемешивали в течение 20 минут. Полученное твердое вещество собирали фильтрацией, промывали водой и сушили под вакуумом, получая указанное в заголовке соединение (1,11 г; 93%) в виде оранжевого порошка. +ESI (М+Н) 211,0; 1H ЯМР (400 МГц, DMSO-d6, δ): 11.99 (br. s., 1H), 8.92 (d, J=2,0 Гц, 1Н), 8.31 (d, J=1,8 Гц, 1Н), 8.08 (d, J=3,1 Гц, 1Н), 3.88 (s, 3Н).
Стадия 2: 3-хлор-1Н-пирроло[3,2-b]пиридин-6-карбоновая кислота
Метил-3-хлор-1Н-пирроло[3,2-b]пиридин-6-карбоксилат (1,10 г; 5,22 ммоль) суспендировали в 1,4-диоксане (25 мл) и добавляли 6 н. водную соляную кислоту (8,7 мл). Реакционную смесь оставляли перемешиваться при комнатной температуре в течение ночи. Затем реакционную смесь концентрировали, получая указанное в заголовке соединение (1,2 г; 100%). +ESI (М+Н) 197,1; 1H ЯМР (400 МГц, DMSO-d6, δ): 12.50 (br. s., 1H), 8.92 (d, J=1,6 Гц, 1H), 8.46 (br. s., 1H), 8.19 (br. s., 1H).
Промежуточное соединение 35: показанную ниже 3-(метиламино)-1Н-индазол-6-карбоновую кислоту получали следующим образом.
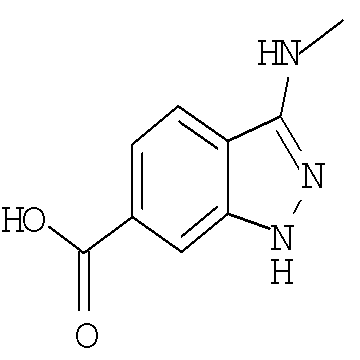
Стадия 1: 4-бром-2-фтор-N-метилбензотиоамид
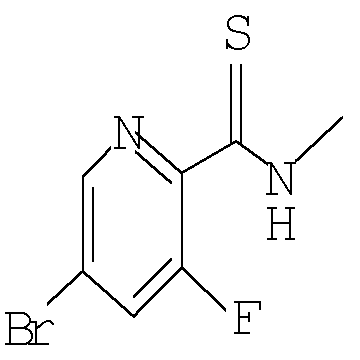
Смесь 4-бром-2-фтор-N-метилбензамида (500 мг; 2 ммоль) и реагента Лавессона (872 мг; 2,16 ммоль) в толуоле (10 мл) нагревали до 100°С и перемешивали в течение 4 часов. Реакционную смесь охлаждали до комнатной температуры, разбавляли толуолом и фильтровали. Фильтрат концентрировали и после очистки остатка колоночной флэш-хроматографией (0-20% этилацетат/гептаны) получали указанное в заголовке соединение (520 мг, 97%) в виде желтого твердого вещества. +ESI (М+Н+1) 250,1; 1H ЯМР (400 МГц, CDCl3, δ): 8.09 (t, J=8,58 Гц, 1Н), 8.03 (br. s., 1H), 7.35 (dd, J=8,19, 2,15 Гц, 1Н), 7.27 (dd, J=11,41, 1,85 Гц, 1Н), 3.36 (dd, J=4,88, 0,78 Гц, 3Н).
Стадия 2: 6-бром-N-метил-1Н-индазол-3-амин
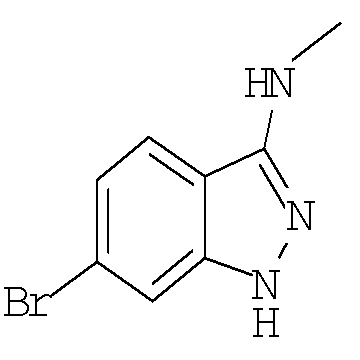
Безводный гидразин (0,25 мл; 8,1 ммоль) добавляли к раствору 4-бром-2-фтор-N-метилбензотиоамида (200 мг; 0,8 ммоль) в диметилсульфоксиде (2,5 мл). Реакционную смесь нагревали до 100°С и перемешивали в течение 2 часов. Реакционную смесь охлаждали до комнатной температуры и разбавляли этилацетатом и водой. Слои разделяли и водный слой экстрагировали этилацетатом (3×). Объединенные органические экстракты промывали насыщенным водным карбонатом натрия и рассолом, сушили над сульфатом натрия, фильтровали и концентрировали. После очистки колоночной флэш-хроматографией (20-100% этилацетат/гептаны) получали указанное в заголовке соединение (98 мг, 54%) в виде белого твердого вещества. +ESI (М+Н+1) 228,0; 1H ЯМР (400 МГц, CD3OD, δ): 7.52 (d, J=8,58 Гц, 1Н), 7.43 (s, 1Н), 7.04 (d, J=8,39 Гц, 1Н), 2.94 (s, 3Н).
Стадия 3: метил-3-(метиламино)-1Н-индазол-6-карбоксилат
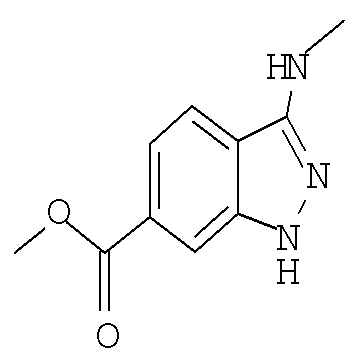
Указанное в заголовке соединение получали способом, аналогичным описанному на стадии 2 для промежуточного соединения 12, используя 6-бром-N-метил-1Н-индазол-3-амин. +ESI (М+Н) 206,2; 1H ЯМР (400 МГц, CD3OD, δ): 7.95 (t, J=1,17 Гц, 1Н), 7.67 (dd, J=8,39, 0,78 Гц, 1Н), 7.55 (dd, J=8,49, 1,27 Гц, 1Н), 3.90 (s, 3H), 2.96 (s, 3H).
Стадия 4: 3-(метиламино)-1Н-индазол-6-карбоновая кислота
К раствору метил-3-(метиламино)-1Н-индазол-6-карбоксилата (30,0 мг; 0,15 ммоль) в 1,4-диоксане (0,2 мл) добавляли 3 н. водную соляную кислоту (0,2 мл; 0,6 ммоль). Смесь нагревали до 100°С в течение 2 часов. Реакционную смесь концентрировали и сушили под вакуумом, получая указанное в заголовке соединение (33 мг, 99%) в виде рыжевато-коричневого твердого вещества. +ESI (М+Н) 192,1; 1H ЯМР (400 МГц, CD3OD, δ): 8.09 (s, 1H), 7.98 (dd, J=8,58, 0,78 Гц, 1H), 7.85 (dd, J=8,58, 1,37 Гц, 1H), 3.12 (s, 3Н).
Промежуточное соединение 36: показанную ниже 3-метоксиизохинолин-7-карбоновую кислоту получали следующим образом.
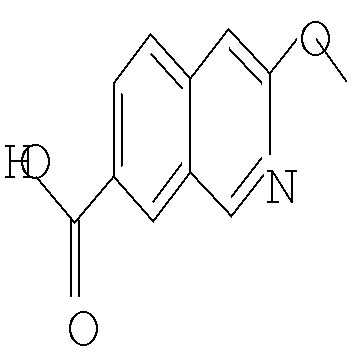
Стадия 1: 7-бром-3-метоксиизохинолин
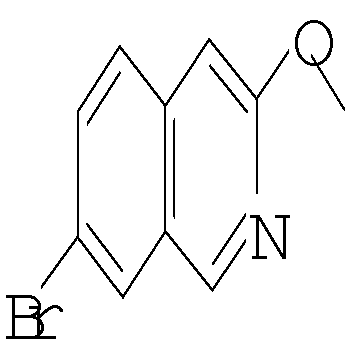
Смесь 7-бром-3-хлоризохинолина (100 мг; 0,4 ммоль) и метилата натрия (113 мг; 2,1 ммоль) в диглиме (1 мл) нагревали до 150°С в течение 1 часа. Реакционную смесь охлаждали до комнатной температуры и разбавляли толуолом и водой. Слои разделяли и водный слой экстрагировали толуолом (3×). Объединенные органические экстракты промывали водой и рассолом, сушили над сульфатом натрия, фильтровали и концентрировали до масла. Масло сушили под вакуумом в течение ночи, получая указанное в заголовке соединение (83 мг, 85%) в виде желтого твердого вещества. +ESI (М+Н+1) 240,1; 1H ЯМР (400 МГц, CDCl3, δ): 8.87 (s, 1H), 8.01-8.05 (m, 1H), 7.58-7.64 (m, 1H), 7.53-7.58 (m, 1H), 6.97 (s, 1H), 4.02 (s, 3Н).
Стадия 2: 3-метоксиизохинолин-7-карбоновая кислота
Указанное в заголовке соединение получали способом, аналогичным описанному на стадиях 3-4 для промежуточного соединения 21, используя 7-бром-3-метоксиизохинолин. +ESI (М+Н) 204,2; 1H ЯМР (400 МГц, CD3OD, δ): 9.08 (s, 1H), 8.71 (s, 1H), 8.14 (dd, J=8,78, 1,56 Гц, 1H), 7.83 (d, J=8,78 Гц, 1H), 7.17 (s, 1H), 4.02 (s, 3H).
Промежуточное соединение 37: показанную ниже 1-(метиламино)-изохинолин-6-карбоновую кислоту получали следующим образом.
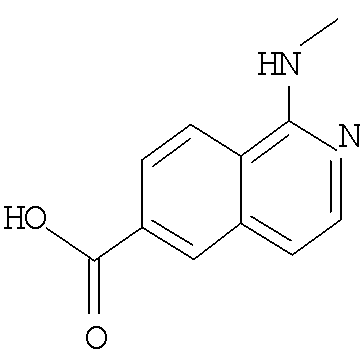
Стадия 1: этил-1-хлоризохинолин-6-карбоксилат

Указанное в заголовке соединение получали способом, аналогичным описанному на стадиях 1-3 для промежуточного соединения 27, используя 6-бромизохинолин-1(2Н)-он. 1H ЯМР (400 МГц, CDCl3, δ): 8.56 (d, J=1,6 Гц, 1Н), 8.38 (d, J=8,8 Гц, 1Н), 8.34 (d, J=5,7 Гц, 1Н), 8.25 (dd, J=8,8, 1,6 Гц, 1Н), 7.70 (d, J=6,0 Гц, 1Н), 4.47 (q, J=7,0 Гц, 2Н), 1.45 (t, J=7,1 Гц, 3Н).
Стадия 2: этил-1-(метиламино)изохинолин-6-карбоксилат
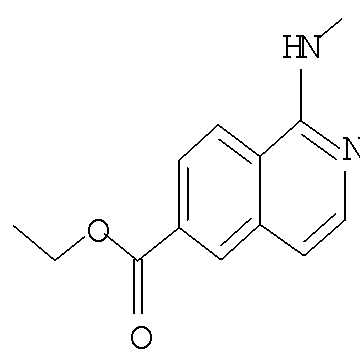
Указанное в заголовке соединение получали способом, аналогичным описанному на стадии 1 для промежуточного соединения 31, используя этил-1-хлоризохинолин-6-карбоксилат. +ESI (М+Н) 231,1; 1H ЯМР (400 МГц, CDCl3, δ): 8.39 (s, 1Н), 8.06-8.14 (m, 2Н), 8.00 (d, J=5,9 Гц, 1Н), 7.02 (d, J=6,0 Гц, 1Н), 4.44 (q, J=7,3 Гц, 2Н), 3.25 (d, J=4,7 Гц, 3Н), 1.43 (t, J=7,1 Гц, 3Н).
Стадия 3: 1-(метиламино)изохинолин-6-карбоновая кислота
К суспензии этил-1-(метиламино)изохинолин-6-карбоксилата (150 мг; 0,65 ммоль) в этаноле (2,5 мл) добавляли 1 н. водный гидроксид калия (6,5 мл; 6,5 ммоль). Реакционную смесь нагревали до 65°С в течение 1,5 часов. Реакционную смесь охлаждали до комнатной температуры и концентрировали досуха. Твердое вещество растворяли в воде, и раствор подкисляли, используя 1 н. HCl. Смесь концентрировали. Твердое вещество растворяли в воде (50 мл) и дважды экстрагировали 2-бутанолом (50 мл). Объединенные органические экстракты промывали рассолом (20 мл), сушили над сульфатом магния, фильтровали и концентрировали, получая указанное в заголовке соединение (95 мг, 72%) в виде белого твердого вещества. +ESI (М+Н) 203,2; 1H ЯМР (400 МГц, DMSO-d6, δ): 13.07 (br. s., 1Н), 10.25 (d, J=4,9 Гц, 1Н), 8.74 (d, J=8,8 Гц, 1Н), 8.51 (s, 1H), 8.15 (dd, J=8,6, 1,8 Гц, 1Н), 7.67 (d, J=6,8 Гц, 1Н), 7.35 (d, J=7,0 Гц, 1Н), 3.15 (d, J=4,7 Гц, 3Н).
Промежуточное соединение 38: 1-метоксиизохинолин-6-карбоновая кислота
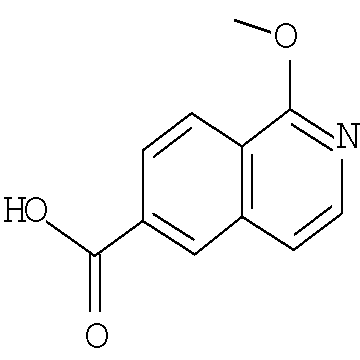
Раствор метилата натрия получали путем медленного добавления с перемешиванием металлического натрия (870 мг, 37 ммоль) к метанолу (25 мл). После того, как весь металлический натрий прореагировал, этот раствор добавляли к этил-1-хлоризохинолин-6-карбоксилату (440 мг; 1,9 ммоль). Полученную суспензию нагревали до температуры дефлегмации и перемешивали в течение 3 суток. Реакционную смесь охлаждали до комнатной температуры и концентрировали. Остаток распределяли между водой и этилацетатом. Слои разделяли и водный слой подкисляли 1 н. водной соляной кислотой до образования осадка. Твердое вещество собирали фильтрацией и сушили под вакуумом, получая указанное в заголовке соединение (294 мг, 78%) в виде белого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6, δ): 8.50 (d, J=1,2 Гц, 1Н), 8.23 (d, J=8,8 Гц, 1H), 8.02-8.10 (m, 2H), 7.54 (d, J=6,0 Гц, 1Н), 4.05 (s, 3Н).
Промежуточное соединение 39: показанную ниже 3-(метиламино)-1Н-индазол-5-карбоновую кислоту получали следующим образом.

Стадия 1: 5-бром-2-фтор-N-метилбензамид
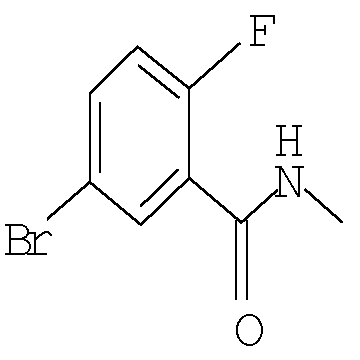
К смеси 5-бром-2-фторбензойной кислоты (200 мг; 0,91 ммоль) в дихлорметане (5 мл) добавляли оксалилхлорид (0,16 мл; 1,8 ммоль), затем 1 каплю N,N-диметилформамида. Реакционную смесь перемешивали при комнатной температуре в течение 1,5 часов. Реакционную смесь концентрировали, полученный остаток растворяли в дихлорметане (3 мл) и охлаждали до 0°С. Добавляли метиламин (2,3 мл; 5 ммоль; 2 М в тетрагидрофуране) и реакционную смесь оставляли перемешиваться при 0°С в течение 30 минут. Реакцию гасили водой и смесь концентрировали. Остаток разбавляли водой, полученные твердые вещества отфильтровывали, промывали водой и сушили под вакуумом, получая указанное в заголовке соединение (196,6 мг; 93%) в виде белого твердого вещества. +ESI (М+Н+1) 234,1; 1H ЯМР (400 МГц, CDCl3, δ); 8.22 (dd, J=6,83, 2,73 Гц, 1Н), 7.55 (ddd, J=8,68, 4,49, 2,63 Гц, 1Н), 7.00 (dd, J=11,32, 8,58 Гц, 1Н), 6.67 (br. s., 1H), 3.02 (dd, J=4,88, 1,17 Гц, 3Н).
Стадия 2: 5-бром-2-фтор-N-метилбензотиоамид
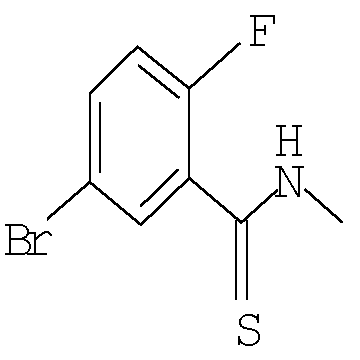
Смесь 5-бром-2-фтор-N-метилбензамида (500 мг, 2 ммоль) и реагента Лавессона (872 мг; 2,16 ммоль) в толуоле (10 мл) нагревали до 100°С и перемешивали в течение 3,5 часа. Реакционную смесь охлаждали до комнатной температуры, разбавляли толуолом и фильтровали. Фильтрат концентрировали и очищали колоночной флэш-хроматографией (0-20% этилацетат/гептаны), получая указанное в заголовке соединение (494 мг, 92%) в виде желтого масла, которое отвердевало при стоянии. +ESI (М+Н+1) 250,1; 1H ЯМР (400 МГц, CDCl3, δ): 8.20 (dd, J=6,93, 2,63 Гц, 1Н), 8.06 (br. s., 1Н), 7.47 (ddd, J=8,73, 4,44, 2,63 Гц, 1Н), 6.95 (dd, J=11,12, 8,78 Гц, 1Н), 3.32 (dd, J=4,88, 0,78 Гц, 3Н).
Стадия 3: 5-бром-N-метил-1Н-индазол-3-амин
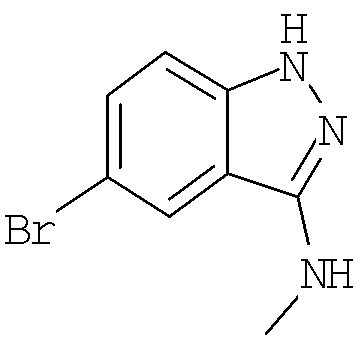
Смесь 5-бром-2-фтор-N-метилбензотиоамида (480 мг; 1,9 ммоль) и безводного гидразина (0,61 мл; 19 ммоль) в диметилсульфоксиде (6 мл) нагревали до 80°С и перемешивали в течение 1 часа. Температуру повышали до 100°С, и реакционную смесь перемешивали в течение 40 минут. Температуру повышали еще до 130°С, и реакционную смесь перемешивали в течение следующих 45 минут. Реакционную смесь охлаждали до комнатной температуры и разбавляли этилацетатом и рассолом. Слои разделяли и водный слой экстрагировали этилацетатом (4×). Объединенные органические экстракты промывали водой и рассолом, сушили над сульфатом натрия, фильтровали и концентрировали. После очистки колоночной флэш-хроматографией (20-70% этилацетат/гептаны) получали указанное в заголовке соединение (103 мг, 23%) в виде белого твердого вещества. +ESI (М+Н+1) 228,0; 1H ЯМР (400 МГц, CD3OD, δ): 7.78 (dd, J=1,85, 0,68 Гц, 1Н), 7.29-7.40 (m, 1H), 7.17 (dd, J=8,88, 0,68 Гц, 1Н), 2.94 (s, 3H).
Стадия 4: метил-3-(метиламино)-1Н-индазол-5-карбоксилат
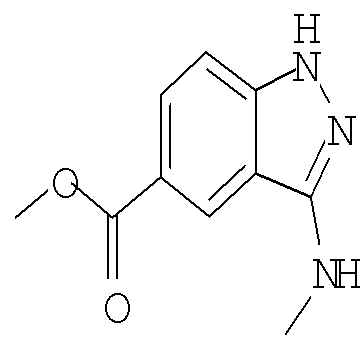
Указанное в заголовке соединение получали способом, аналогичным описанному на стадии 3 для промежуточного соединения 21, используя 5-бром-N-метил-1Н-индазол-3-амин. +ESI (М+Н) 206,2; 1H ЯМР (400 МГц, CD3OD, δ): 8.44 (dd, J=1,56, 0,78 Гц, 1Н), 7.92 (dd, J=8,78, 1,56 Гц, 1Н), 7.26 (dd, J=8,78, 0,78 Гц, 1Н), 3.88 (s, 3H), 2.96 (s, 3H).
Стадия 5: 3-(метиламино)-1Н-индазол-5-карбоновая кислота
Метил-3-(метиламино)-1Н-индазол-5-карбоксилат (60,0 мг; 0,29 ммоль) растворяли в 1,4-диоксане (0,5 мл). Добавляли 3 н. водную соляную кислоту (0,3 мл; 0,9 ммоль) и реакционную смесь нагревали до 100°С в течение 11,5 часов. Нагревание прекращали и реакционную смесь оставляли перемешиваться при комнатной температуре в течение ночи. Реакционную смесь концентрировали, получая указанное в заголовке соединение (63 мг, 95%) в виде рыжевато-коричневого твердого вещества. +ESI (М+Н) 192,1; 1H ЯМР (400 МГц, CD3OD, δ): 8.61 (d, J=0,78 Гц, 1Н), 8.19 (dd, J=8,80, 1,57 Гц, 1Н), 7.38 (d, J=8,80 Гц, 1Н), 3.02 (s, 3H).
Промежуточное соединение 40: показанную ниже 3-аминоизохинолин-7-карбоновую кислоту получали следующим образом.
Указанное в заголовке соединение получали способом, аналогичным описанному на стадиях 3-4 для промежуточного соединения 21, используя 7-бромизохинолин-3-амин. +ESI (М+Н) 189,2; 1H ЯМР (400 МГц, CD3OD, δ): 8.87 (s, 1Н), 8.52 (d, J=0,78 Гц, 1Н), 7.98 (dd, J=8,78, 1,76 Гц, 1Н), 7.54 (d, J=8,78 Гц, 1Н), 6.77 (s, 1Н).
Промежуточное соединение 41: показанную ниже 3-(метиламино)-изохинолин-7-карбоновую кислоту получали следующим образом.
Стадия 1: 7-бром-N-метилизохинолин-3-амин
Смесь 7-бром-3-хлоризохинолина (100 мг; 0,4 ммоль), гидрохлорида метиламина (139 мг; 2,06 ммоль) и карбоната калия (456 мг; 3,30 ммоль) в 1-метокси-2-(2-метоксиэтокси)этане (1 мл) нагревали до 150°С и перемешивали в течение 60 часов. Добавляли дополнительное количество гидрохлорида метиламина (100 мг; 1,5 ммоль) и карбоната калия (200 мг; 1,4 ммоль) и нагревание продолжали в течение следующих 40 часов. Реакционную смесь охлаждали до комнатной температуры и разбавляли водой. Смесь перемешивали в течение 30 минут. Полученное твердое вещество отфильтровывали, промывали водой и сушили под вакуумом. После очистки колоночной флэш-хроматографией (10-30% этилацетат/гептаны) получали указанное в заголовке соединение (82 мг) в виде бледно-желтого твердого вещества. -APCl (М-Н+1) 237,8; 1H ЯМР (400 МГц, CDCl3, δ): 8.70 (s, 1Н), 7.84 (d, J=1,95 Гц, 1Н), 7.48 (dd, J=8,97, 2,15 Гц, 1Н), 7.38 (d, J=8,97 Гц, 1Н), 6.39 (s, 1H), 2.92 (s, 3H).
Стадия 2: 3-(метиламино)изохинолин-7-карбоновая кислота
Указанное в заголовке соединение получали способом, аналогичным описанному на стадиях 3-4 для промежуточного соединения 21, используя 7-бром-N-метилизохинолин-3-амин. +ESI (М+Н) 203,1; 1H ЯМР (400 МГц, CD3OD, δ): 8.87 (s, 1H), 8.51 (s, 1H), 7.98 (dd, J=8,88, 1,66 Гц, 1Н), 7.58 (d, J=8,78 Гц, 1H), 6.60 (s, 1H), 2.93 (s, 3Н).
Промежуточное соединение 42: показанную ниже 3-хлор-1Н-пирроло[2,3-b]пиридин-5-карбоновую кислоту получали следующим образом.
Суспензию 1Н-пирроло[2,3-b]пиридин-5-карбоновой кислоты (250 мг; 1,5 ммоль) в N,N-диметилформамиде (5 мл) нагревали до 40°С. Добавляли N-хлорсукцинимид (243 мг; 1,62 ммоль) и смесь перемешивали при 55°С в течение 5 часов. Реакционную смесь охлаждали до комнатной температуры и оставляли перемешиваться в течение 2 суток. Смесь разбавляли водой (20 мл) и перемешивали в течение ночи. Полученное твердое вещество собирали фильтрацией и сушили, получая указанное в заголовке соединение (161 мг, 55%). +ESI (М+Н) 197,1; 1H ЯМР (400 МГц, DMSO-d6, δ): 13.08 (br. s., 1H), 12.39 (br. s., 1H), 8.86 (d, J=1,8 Гц, 1Н), 8.40 (d, J=1,2 Гц, 1Н), 7.84 (d, J=2,5 Гц, 1Н).
Промежуточное соединение 43: показанный ниже 6-бром-3-метоксиизохинолин получали следующим образом.
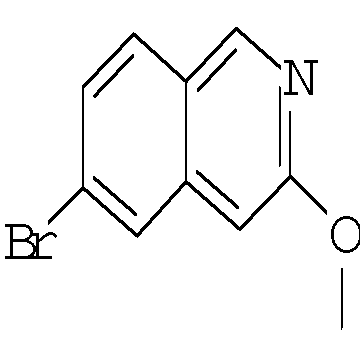
Смесь 6-бромизохинолин-3-ола (606 мг; 2,70 ммоль), карбоната серебра (1,5 г; 5,3 ммоль) и N,N-диметилформамида (12 мл) перемешивали при комнатной температуре в течение 16 минут. Добавляли метилиодид (186 мкл; 2,97 ммоль) и реакционную смесь оставляли перемешиваться в течение 18 часов. Реакционную смесь разбавляли метанолом и фильтровали через целит. Фильтрат концентрировали и очищали колоночной флэш-хроматографией, получая указанное в заголовке соединение (90 мг, 14%). +ESI (М+Н+1) 240,0; 1H ЯМР (400 МГц, CDCl3, δ): 8.91 (s, 1H), 7.86 (d, J=1,8 Гц, 1H), 7.73 (d, J=8,8 Гц, 1Н), 7.43 (dd, J=8,8, 1,8 Гц, 1H), 6.90 (s, 1H), 4.02 (s, 3H).
Промежуточное соединение 44: показанную ниже 2-хлорхинолин-7-карбоновую кислоту получали следующим образом.
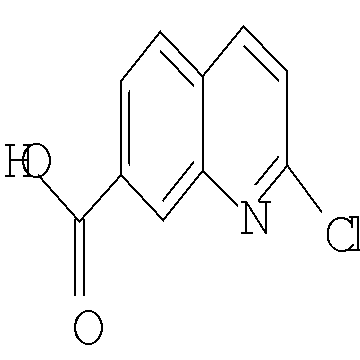
Стадия 1: этил-2-хлорхинолин-7-карбоксилат
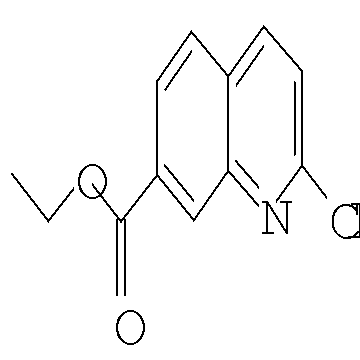
Оксихлорид фосфора (1,94 мл; 20,7 ммоль) добавляли к раствору 1-оксида 7-(этоксикарбонил)хинолина (450 мг; 2,07 ммоль) в дихлорметане (15 мл). Реакционную смесь нагревали до 50°С в течение 3 часов. Затем реакционную смесь охлаждали до комнатной температуры и медленно с перемешиванием выливали в 200 мл воды. Смесь оставляли перемешиваться в течение 1 часа и затем нейтрализовали, используя 1 н. водный гидроксид калия. Смесь экстрагировали дихлорметаном (3×). Экстракты промывали рассолом, сушили над сульфатом магния, фильтровали и концентрировали. После очистки колоночной хроматографией (0-20% этилацетат/гептаны) получали указанное в заголовке соединение (254 мг; 52%) в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3, δ): 8.70-8.79 (m, 1H), 8.13-8.18 (m, 2Н), 7.87 (d, J=8,39 Гц, 1H), 7.47 (d, J=8,58 Гц, 1H), 4.44 (q, J=7,02 Гц, 2Н), 1.43 (t, J=7,12 Гц, 3H).
Стадия 2: 2-хлорхинолин-7-карбоновая кислота
К раствору этил-2-хлорхинолин-7-карбоксилата (800 мг; 3,4 ммоль) в тетрагидрофуране (10 мл) добавляли 1 н. водный гидроксид лития (7 мл; 7 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь концентрировали, остаток разбавляли водой и подкисляли 1 н. водной соляной кислотой. Полученный осадок собирали фильтрацией и сушили под вакуумом, получая указанное в заголовке соединение (648 мг, 92%) в виде белого порошка. +ESI (М+Н) 208,1; 1H ЯМР (400 МГц, DMSO-d6, δ): 13.43 (s, 1H), 8.53 (d, J=8,7 Гц, 1H), 8.44-8.45 (m, 1H), 8.14 (d, J=8,4 Гц, 1Н), 8.07-8.11 (m, 1Н), 7.70 (d, J=8,5 Гц, 1Н).
Промежуточное соединение 45: показанную ниже 2-((2,2,2-трифторэтил)-амино)хинолин-7-карбоновую кислоту получали следующим образом.
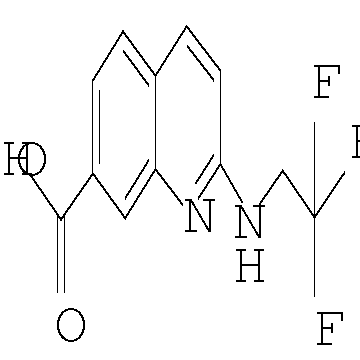
Стадия 1: метил-хинолин-7-карбоксилат
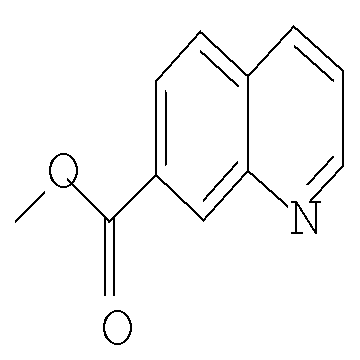
Указанное в заголовке соединение получали способом, аналогичным описанному на стадии 3 для промежуточного соединения 21, используя 7-бромхинолин в качестве исходного вещества.
Стадия 2: показанный ниже 1-оксид 7-(метоксикарбонил)хинолина получали следующим образом.

К раствору метил-хинолин-7-карбоксилата (17,8 г; 94,87 ммоль) в дихлорметане (315 мл) добавляли перуксусную кислоту (39,9 мл; 190 ммоль; 32%-ную в уксусной кислоте). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Добавляли перуксусную кислоту (10 мл; 48 ммоль; 32%-ную в уксусной кислоте) и смесь перемешивали в течение 5 ч. Реакционную смесь разбавляли насыщенным водным раствором бикарбоната натрия. Водную фазу экстрагировали в дихлорметан (2×1 л). Экстракты объединяли, сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении. После очистки флэш-хроматографией (2-15% метанола в дихлорметане) получали указанное в заголовке соединение (17,4 г; 90%) в виде желтого твердого вещества. 1H ЯМР (400 МГц, хлороформ-d, δ): 9.41 (1Н, s), 8.56 (1 H, dd, J=6,0, 0,8 Гц), 8.24 (1 H, dd, J=8,5, 1,7 Гц), 7.93 (1 H, d, J=8,6 Гц), 7.75 (1 H, d, J=8,6 Гц), 7.39 (1 H, dd, J=8,6, 6,0 Гц), 4.01 (3Н, s).
Стадия 3: показанный ниже метил-2-((2,2,2-трифторэтил)амино)хинолин-7-карбоксилат получали следующим образом.

К раствору 1-оксида 7-(метоксикарбонил)хинолина (200 мг; 0,984 ммоль) и 2,2,2-трифторэтиламина (292 мг; 0,295 ммоль) при 0°С порциями в течение 45 минут добавляли ангидрид 4-метилбензолсульфоновой кислоты (964 мг; 2,95 ммоль). Реакционную смесь оставляли нагреваться до комнатной температуры и перемешивали в течение ночи. Реакционную смесь разбавляли дихлорметаном и промывали насыщенным раствором хлорида аммония. Водный слой экстрагировали в дихлорметан (1×). Органические экстракты объединяли и промывали рассолом, сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении. После очистки флэш-хроматографией получали указанное в заголовке соединение (172 мг, 62%). +ESI (M+H) 285,1.
Стадия 4: 2-((2,2,2-трифторэтил)амино)хинолин-7-карбоновая кислота
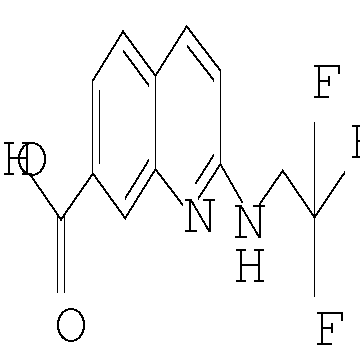
К раствору метил-2-((2,2,2-трифторэтил)амино)хинолин-7-карбоксилата (172 мг; 0,605 ммоль) в тетрагидрофуране (5 мл) при комнатной температуре добавляли водный гидроксид лития (1,82 мл; 1,82 ммоль; 1М раствор). Реакционную смесь перемешивали в течение 2,5 суток. Растворитель удаляли при пониженном давлении и остаток подкисляли 1 н. водной соляной кислотой. Полученный осадок отфильтровывали и сушили, получая указанное в заголовке соединение (65 мг, 40%) +ESI (М+Н) 271,1; 1H ЯМР (400 МГц, DMSO-d6, δ): 4.31-4.41 (m, 2Н), 7.01 (d, J=8,87 Гц, 1H), 7.69-7.80 (m, 2H), 8.06 (d, J=8,66 Гц, 1Н), 8.14 (s, 1H), 13.03 (bs, 1H).
Промежуточное соединение 46: показанную ниже 2-((2,2-дифторпропил)-амино)хинолин-7-карбоновую кислоту получали следующим образом.
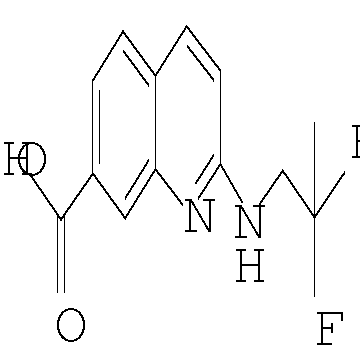
Указанное в заголовке соединение получали способом, аналогичным описанному для промежуточного соединения 45, используя 2,2-дифторэтиламин вместо 2,2,2-трифторэтиламина. +ESI (М+Н) 267,2; 1H ЯМР (400 МГц, DMSO-d6, δ): 1.63 (t, J=19,02 Гц, 3Н), 3.89-3.99 (m, 2H), 6.97 (d, J=8,97 Гц, 1Н), 7.54 (t, 1Н), 7.62-7.68 (m, 1H), 7.71 (d, J=8,19 Гц, 1Н), 7.96 (d, J=9,10 Гц, 1Н), 8.06-8.09 (m, 1Н), 12.95 (bs, 1Н).
Промежуточное соединение 47: показанную ниже 7-хлор-1Н-бензо[d][1,2,3]триазол-5-карбоновую кислоту получали следующим образом.
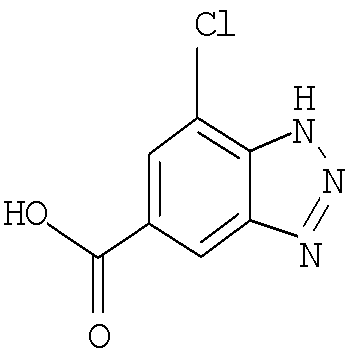
К раствору 3,4-диамино-5-хлорбензойной кислоты (125 мг; 0,67 ммоль) в концентрированной серной кислоте (0,45 мл) при 0°С добавляли воду (2 мл). Реакционную смесь перемешивали при 0°С в течение 1 ч. Смесь оставляли перемешиваться в течение ночи. Реакционную смесь разбавляли водой и полученный осадок отфильтровывали, получая указанное в заголовке соединение (124 мг, 94%) в виде коричневого твердого вещества. +APCl (М+Н) 198,0; 1H ЯМР (400 МГц, метанол-d4, δ): 8.53 (d, J=1,2 Гц, 1Н), 8.10 (d, J=1,0 Гц, 1Н).
Пример 1: 1'изопропил-1-(2-метил-1Н-бензо[d]имидазол-5-карбонил)-4'6'-гидроспиро[пиперидин-4,5'пиразоло[3,4-с]пиридин]-7'(1'Н)-он
К раствору 2-метил-1Н-бензо[d]имидазол-5-карбоновой кислоты (42 мг; 0,13 ммоль) в дихлорметане (2 мл) добавляли гидрохлоридную соль 1'-изопропил-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7' (1Н)-она (42 мг; 0,13 ммоль), триэтиламин (0,01 мл; 0,07 ммоль) и гексафторфосфат (1Н-7-азабензотриазол-1-ил)-1,1,3,3-тетраметилурония (54,8 мг; 0,144 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Смесь концентрировали в вакууме, полученные твердые вещества растворяли в этилацетате, промывали насыщенным бикарбонатом натрия и сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Остаток растворяли в диметилсульфоксиде (1 мл) и очищали обращенно-фазовой HPLC (колонка: Waters XBridge C18 19×100; 5 мкм; подвижная фаза А: 0,03% NH4OH в воде (об./об.); подвижная фаза В: 0,03% NH4OH в ацетонитриле (об./об.); градиент: 90% N10% В линейный до 0% N100% В в течение 8,5 мин, выдерживание при 0% N100% В в течение 10,0 мин; скорость потока: 25 мл/мин. +ESI (М+Н) 407,2; время удерживания при HPLC 1,74 минуты (метод А).
Пример 2: 1-(3,7-диметил-1Н-индазол-5-карбонил)-1'-изопропил-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(1'Н)-он
К раствору гидрохлоридной соли 1'-изопропил-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(1'Н)-она (промежуточного соединения 2; 430 мг; 1,3 ммоль) и 3,7-диметил-1Н-индазол-5-карбоновой кислоты (306 мг; 1,6 ммоль) в диметилформамиде (2 мл) добавляли триэтиламин (0,75 мл; 5,4 ммоль), 4-диметиламинопиридин (33 мг; 0,37 ммоль) и циклический ангидрид 1-пропанфосфоновой кислоты (0,52 мл; 1,74 ммоль, 50%-ный раствор в этилацетате) и реакционную смесь перемешивали в течение ночи при комнатной температуре. Реакционную смесь концентрировали в вакууме, переносили в этилацетат и промывали насыщенным водным бикарбонатом натрия. Органический слой сушили над сульфатом натрия, фильтровали и концентрировали до твердого вещества. Твердое вещество очищали колоночной флэш-хроматографией (0-15% метанол/дихлорметан), что позволило получить стеклообразное твердое вещество. Это стеклообразное твердое вещество перемешивали в этилацетате в течение 16 часов и полученное твердое вещество собирали вакуумной фильтрацией, что позволило получить желаемый продукт в виде белого твердого вещества (138 мг). +ESI (М+Н) 421,0; 1H ЯМР (400 МГц, CD3OD, δ): 7.65 (s, 1H), 7.42 (s, 1H), 7.21 (s, 1H), 5.50 (m, 1H), 3.95 (br. s., 1H), 3.50-3.62 (br. s., 3H), 2.97 (s, 2H), 2.56 (m, 6H), 1.83 (br. s., 4H), 1.44 (d, 6H).
Пример 3: 1'-изопропил-1-(2-метил-2Н-индазол-5-карбонил)-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(1'Н)-он
К раствору 2-метил-2Н-индазол-5-карбоновой кислоты (28 мг; 0,16 ммоль) в безводном диметилформамиде добавляли 1-этил-3-(3-диметиламинопропил)-карбодиимид (37 мг; 0,19 ммоль), 1-гидроксибензотриазол (26 мг; 0,19 ммоль) и N,N-диизопропилэтиламин (84 мкл; 0,48 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 10 минут, затем добавляли гидрохлорид 1'-изопропил-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(1'Н)-она (промежуточное соединение 2; 30 мг; 0,12 ммоль) и реакционную смесь перемешивали в течение 16 часов. Смесь выливали в охлажденную воду и полученный осадок собирали вакуумной фильтрацией. Полученное твердое вещество растирали в диэтиловом эфире, что позволило получить 1'-изопропил-1-(2-метил-2Н-индазол-5-карбонил)-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(1'Н)-он (25 мг). +ESI (М+Н) 407,3; 1H ЯМР (400 МГц, DMSO-d6, δ): 8.41 (s, 1H), 7.85 (s, 1H), 7.75 (s, 1H), 7.63 (d, 1H), 7.40 (s, 1H), 7.20 (s, 1H), 5.40 (m, 1H), 4.18 (s, 3H), 3.60 (br. s., 4H), 2.85 (s, 2H), 1.70 (br. s., 4H), 1.35 (d, 6H).
Соединения, приведенные ниже в Таблице 1, получали, используя методики, аналогичные описанным выше для синтеза соединений из Примеров 1-3, с применением соответствующих исходных веществ, имеющихся в продаже, полученных с использованием способов, хорошо известных специалистам в данной области, или полученных аналогично путям, описанным выше для других промежуточных соединений. Приведенные ниже соединения сначала были выделены в виде свободного основания, и для тестирования их можно превратить в фармацевтически приемлемую соль.
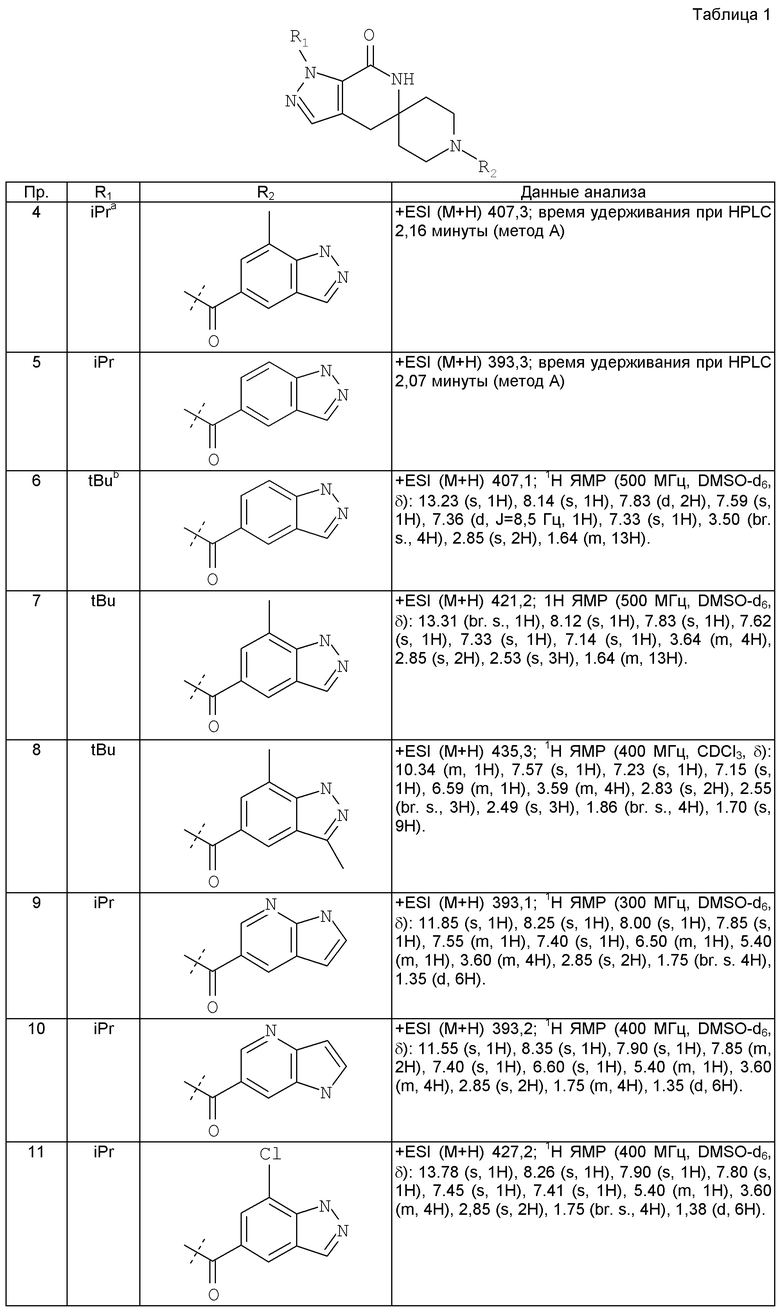

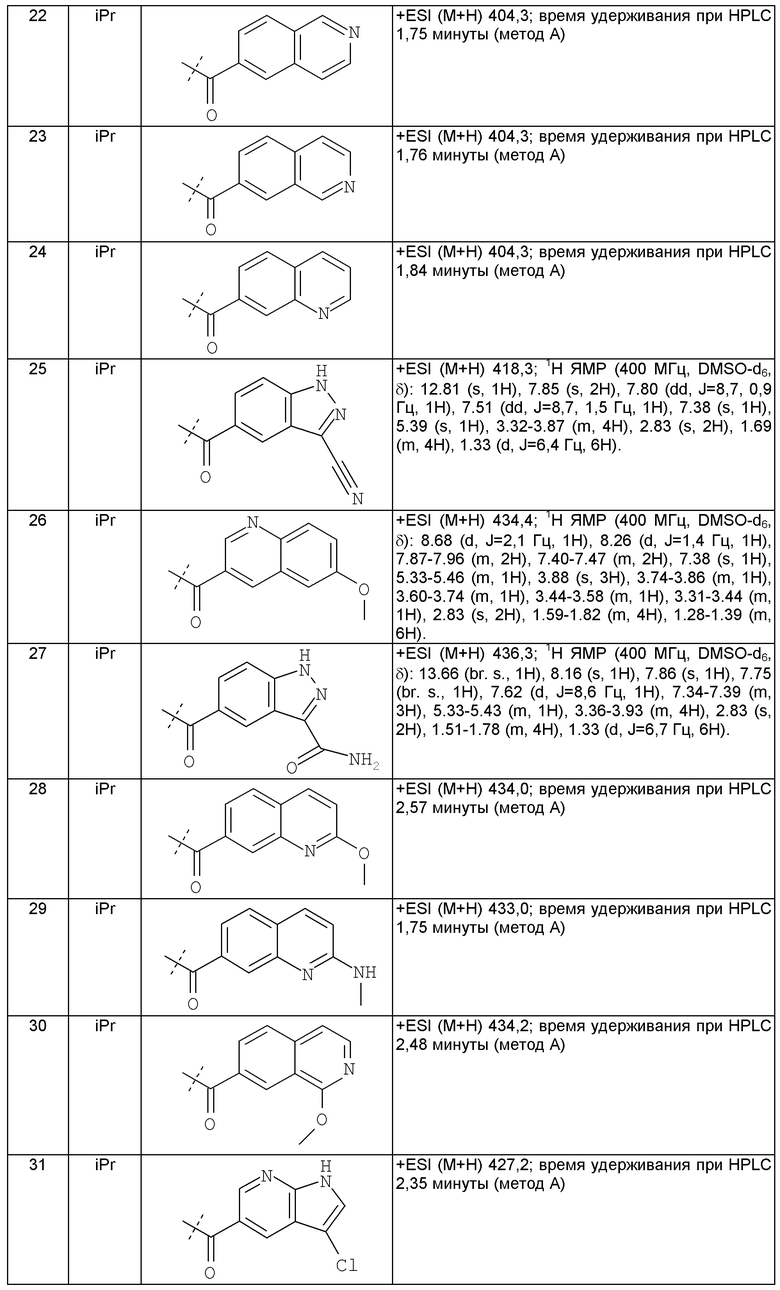
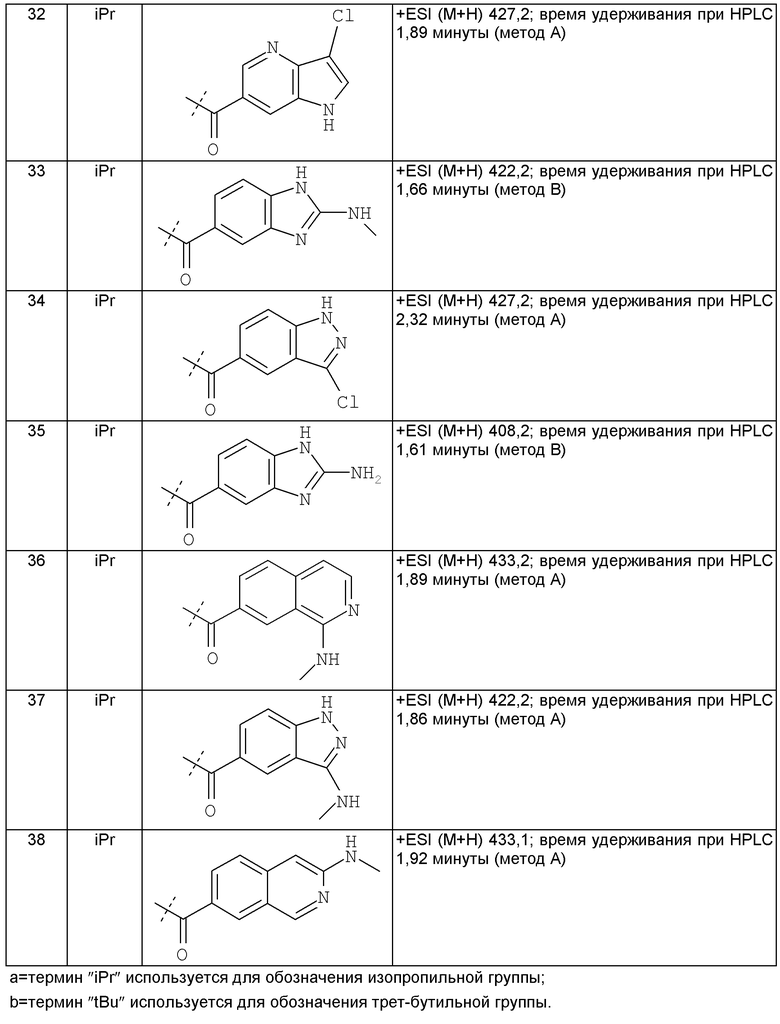
Соединения, приведенные ниже в Таблице 2, получали, используя методики, аналогичные описанным выше для синтеза соединений из Примеров 1-3, с применением соответствующих исходных веществ, имеющихся в продаже, полученных с использованием способов, хорошо известных специалистам в данной области, или полученных аналогично путям, описанным выше для других промежуточных соединений. Приведенные ниже соединения
сначала были выделены в виде свободного основания, и для тестирования их можно превратить в фармацевтически приемлемую соль.
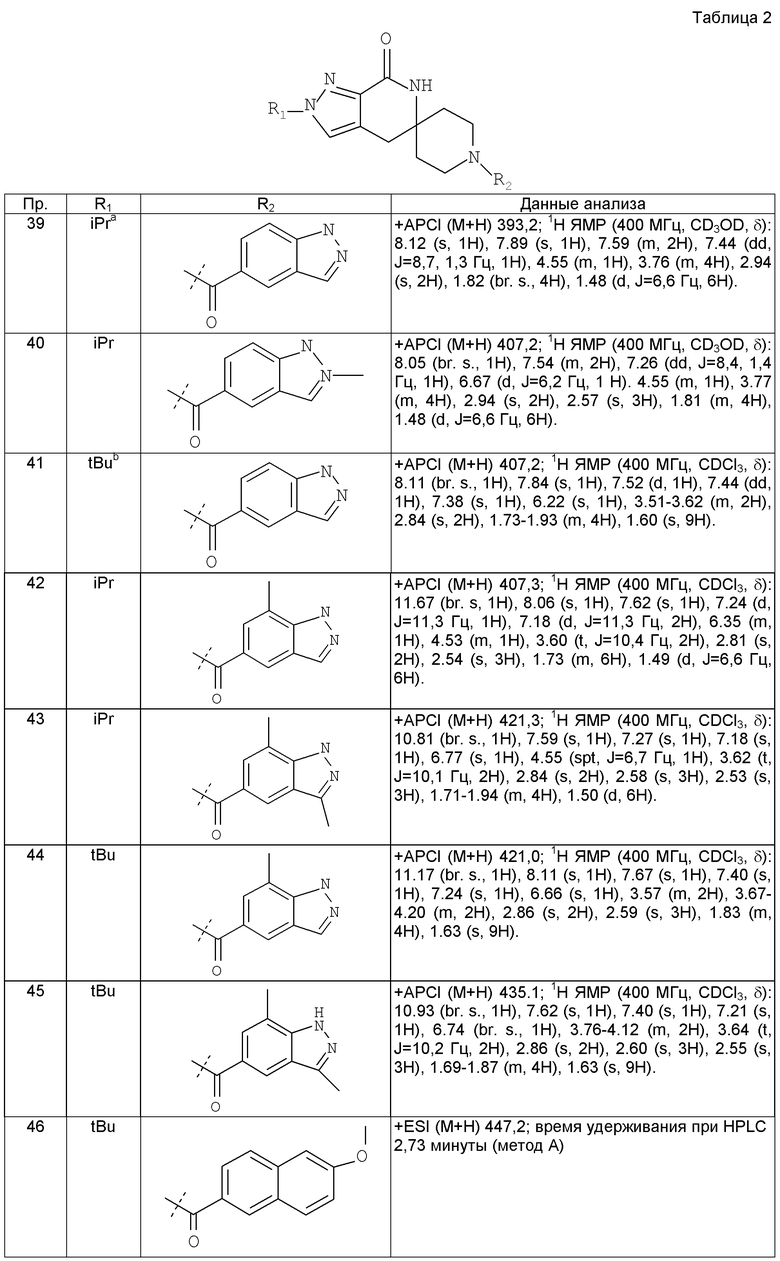
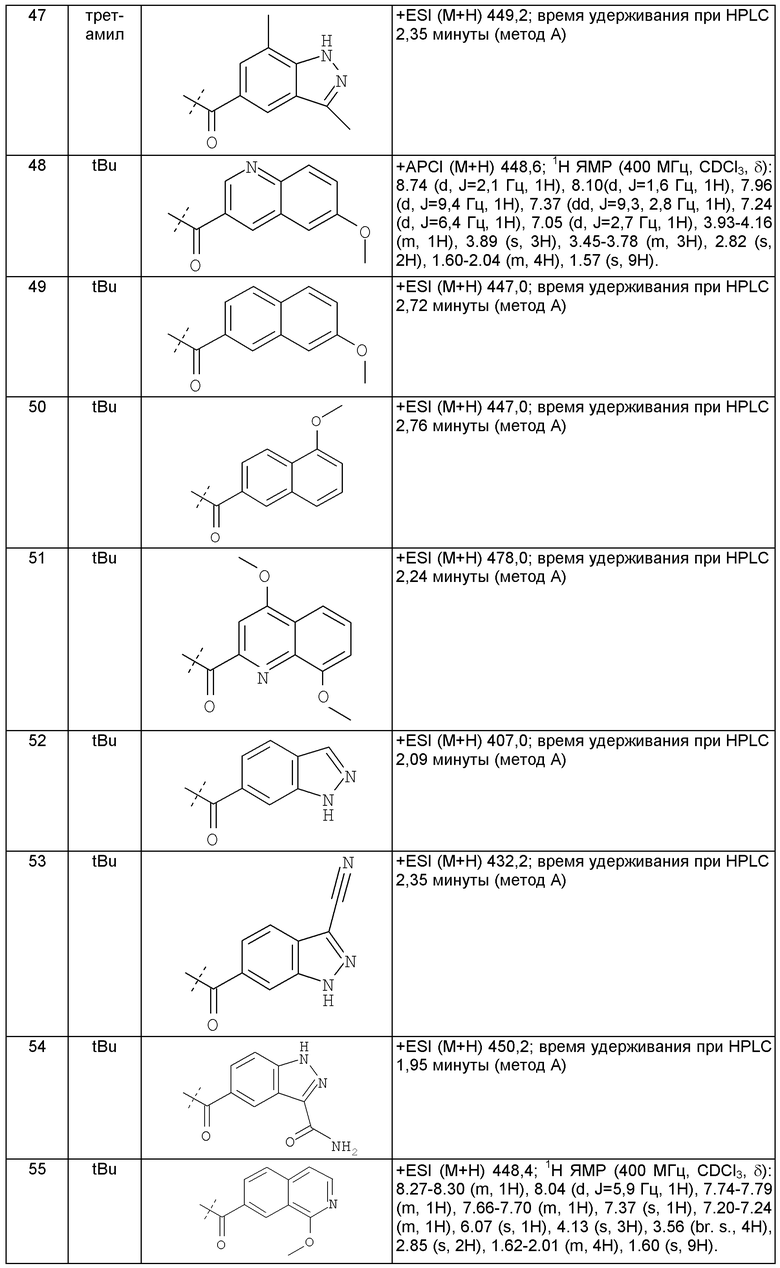
Пример 99: 2'-трет-бутил-1-(7-метокси-1Н-индазол-5-карбонил)-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(2'Н)-он
К смеси гидрохлоридной соли 2'-трет-бутил-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(2'Н)-она (промежуточного соединения 4; 25 мг; 0,075 ммоль) и 7-метокси-1-(тетрагидро-2Н-пиран-2-ил)-1Н-индазол-5-карбоновой кислоты (промежуточного соединения 18; 25 мг; 0,090 ммоль) в N,N-диметилформамиде (0,4 мл) добавляли триэтиламин (0,05 мл; 0,37 ммоль). Смесь перемешивали в течение 5 минут. Затем добавляли циклический ангидрид 1-пропанфосфоновой кислоты (0,09 мл; 0,1 ммоль; 50%-ный раствор в этилацетате) и реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавляли водой и экстрагировали этилацетатом (3×). Объединенные органические слои промывали рассолом, сушили над сульфатом натрия, фильтровали и концентрировали до желтого смолообразного вещества. К этому неочищенному веществу добавляли соляную кислоту (0,19 мл; 0,75 ммоль; 4 М в диоксане). Смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь концентрировали. После очистки обращенно-фазовой HPLC получали указанное в заголовке соединение (3,4 мг; 10%). +ESI (М+Н) 437,3; время удерживания при HPLC 2,12 минуты (метод А).
Пример 100: 1-(1-аминоизохинолин-7-карбонил)-2'-трет-бутил-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(2'Н)-он
Стадия 1: 2'-трет-бутил-1-(1-(4-метоксибензиламино)изохинолин-7-карбонил)-4',6'-дигидроспиро[пиперидин-4,5′-пиразоло[3,4-с]пиридин]-7'(2'Н)-он
Указанное в заголовке соединение получали способом, аналогичным описанному в Примере 3, используя гидрохлоридную соль 2'-трет-бутил-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(2'Н)-она (промежуточное соединение 4) и 1-(4-метоксибензиламино)изохинолин-7-карбоновую кислоту (промежуточное соединение 27). +ESI (М+Н) 553,5.
Стадия 2: 1-(1-аминоизохинолин-7-карбонил)-2'-трет-бутил-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(2'Н)-он
К раствору 2'-трет-бутил-1-(1-(4-метоксибензиламино)изохинолин-7-карбонил)-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(2'Н)-она (28 мг; 0,051 ммоль) в трифторуксусной кислоте (0,51 мл) добавляли анизол (8,3 мкл; 0,076 ммоль). Реакционную смесь нагревали до 65°C и перемешивали в течение 19 часов. Реакционную смесь концентрировали. После очистки обращенно-фазовой HPLC получали указанное в заголовке соединение (7,1 мг; 32%). +ESI (M+H) 433,2; время удерживания при HPLC 1,79 минуты (метод А).
Пример 101: 1-(1-аминоизохинолин-6-карбонил)-2'-трет-бутил-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(2'Н)-он
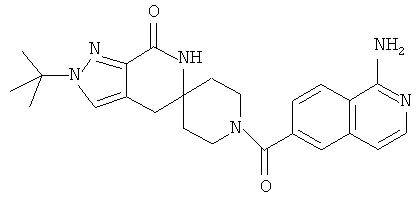
Указанное в заголовке соединение получали способом, аналогичным описанному для Примера 100, используя 1-(4-метоксибензиламино)изохинолин-6-карбоновую кислоту (промежуточное соединение 30) на стадии 1. +ESI (М+Н) 433,2; время удерживания при HPLC 1,82 минуты (метод А).
Пример 102: 2'-трет-бутил-1-(3-метоксиизохинолин-6-карбонил)-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(2'Н)-он
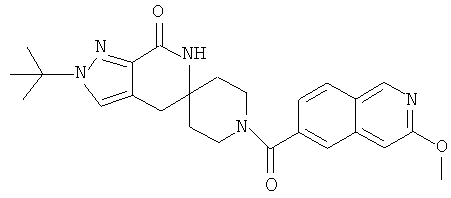
К раствору 6-бром-3-метоксиизохинолина (промежуточного соединения 43; 89,9 мг; 0,378 ммоль) в 1,4-диоксане (6 мл) добавляли гидрохлоридную соль 2'-трет-бутил-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(2'Н)-она (промежуточное соединение 4; 244 мг; 0,727 ммоль) и ацетат натрия (130 мг; 1,5 ммоль). Смесь барботировали газообразным азотом в течение 15 минут. Затем добавляли [1,1'-бис(дифенилфосфино)ферроцен]-дихлорпалладий(II) (комплекс с дихлорметаном; 102 мг; 0,125 ммоль), закрывали реакционный сосуд и барботировали газообразным монооксидом углерода в течение 5 минут. Затем реакционную смесь нагревали до 80°С в течение 18 часов. Реакционную смесь охлаждали до комнатной температуры и разбавляли этилацетатом. Смесь фильтровали через целит и фильтрат концентрировали. После очистки обращенно-фазовой HPLC получали указанное в заголовке соединение. +ESI (M+H) 448,1; время удерживания при HPLC 2,26 минуты (метод А).
Пример 103: 2'-трет-бутил-1-(1-(диметиламино)изохинолин-7-карбонил)-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(2'Н)-он
Стадия 1: 2'-трет-бутил-1-(1-хлоризохинолин-7-карбонил)-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(2'Н)-он
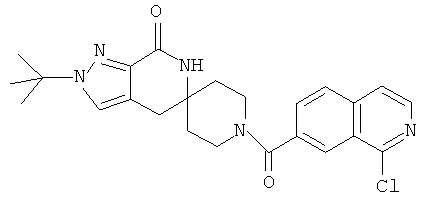
Указанное в заголовке соединение получали способом, аналогичным описанному для Примера 2, используя гидрохлоридную соль 2'-трет-бутил-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(2'Н)-она (промежуточное соединение 4) и 1-хлоризохинолин-7-карбоновую кислоту, и исключая 4-диметиламинопиридин. +ESI (M+H) 452,3; 1H ЯМР (400 МГц, CDCl3, δ): 8.37 (s, 1H), 8.32 (d, J=5,7 Гц, 1H), 7.89 (d, J=8,4 Гц, 1H), 7.75-7.79 (m, 1H), 7.62 (d, J=5,7 Гц, 1H), 7.39 (s, 1H), 6.42 (s, 1H), 3.43-3.73 (m, 4H), 2.87 (s, 2H), 1.64-2.01 (m, 4H), 1.61 (s, 9H).
Стадия 2: 2'-трет-бутил-1-(1-(диметиламино)изохинолин-7-карбонил)-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(2'Н)-он
Раствор диметиламина в метаноле (1,75 мл; 3,50 ммоль; 2 M) добавляли к 2'-трет-бутил-1-(1-хлоризохинолин-7-карбонил)-4',6'-дигидроспиро-[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(2'Н)-ону (158 мг; 0,350 ммоль). Реакционный сосуд герметично закрывали, смесь нагревали до 60°C и перемешивали в течение 65 часов. Реакционную смесь охлаждали до комнатной температуры и концентрировали. После очистки колоночной флэш-хроматографией (1-15% метанол/дихлорметан) получали указанное в заголовке соединение (99 мг, 61%) в виде белого твердого вещества. +APCl (M+H) 461,4; 1H ЯМР (400 МГц, CDCl3, δ): 8.16-8.20 (m, 1H), 8.12 (d, J=5,9 Гц, 1H), 7.75 (d, J=8,2 Гц, 1H), 7.58-7.64 (m, 1H), 7.37 (s, 1H), 7.14 (d, J=5,9 Гц, 1H), 6.00 (br. s., 1H), 3.40-3.71 (m, 4H), 3.10-3.28 (m, 6H), 2.85 (s, 2H), 1.64-1.99 (m, 4H), 1.60 (s, 9H).
Пример 104: 2'-трет-бутил-1-(2-хлорхинолин-7-карбонил)-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(2'Н)-он
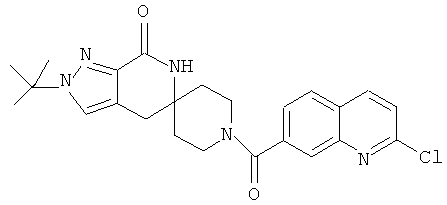
Указанное в заголовке соединение получали способом, аналогичным описанному для Примера 3, используя гидрохлоридную соль 2'-трет-бутил-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(2'Н)-она (промежуточное соединение 4) и 2-хлорхинолин-7-карбоновую кислоту (промежуточное соединение 44). +ESI (M+H) 452,3; 1H ЯМР (400 МГц, CDCl3, δ): 8.12 (d, J=8,2 Гц, 1H), 7.98 (br. s., 1H), 7.88 (dd, J=8,4 Гц, 1H), 7.61 (dd, J=8,4, 1,6 Гц, 1H), 7.44 (d, J=8,6 Гц, 1H), 7.39 (s, 1H), 5.91 (br. s., 1H), 4.06-4.22 (m, 1H), 3.38-3.64 (m, 3H), 2.85 (br. s., 2H), 1.67-1.97 (m, 4H), 1.61 (s, 9H).
Пример 105: 2'-трет-бутил-1-(2-(диметиламино)хинолин-7-карбонил)-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(2'Н)-он
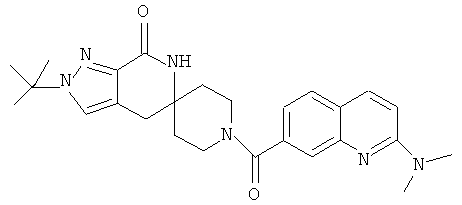
Раствор диметиламина в тетрагидрофуране (2,2 мл; 4,4 ммоль; 2,0 M) добавляли к 2'-трет-бутил-1-(2-хлорхинолин-7-карбонил)-4',6'-дигидро-спиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(2'Н)-ону (100 мг; 0,2 ммоль). Реакционный сосуд герметично закрывали и смесь нагревали до 70°C в течение 15 часов. Реакционную смесь охлаждали до комнатной температуры и концентрировали. После очистки обращенно-фазовой HPLC получали указанное в заголовке соединение (25 мг, 25%). +ESI (M+H) 461,2; время удерживания при HPLC 1,96 минуты (метод А).
Соединения, приведенные ниже в Таблице 3, получали, используя методики, аналогичные описанным выше для синтеза соединения из Примера 105, с применением соответствующих исходных веществ, имеющихся в продаже, полученных с использованием способов, хорошо известных специалистам в данной области, или полученных аналогично путям, описанным выше для других промежуточных соединений. Приведенные ниже соединения сначала были выделены в виде свободного основания, и для тестирования их можно превратить в фармацевтически приемлемую соль.
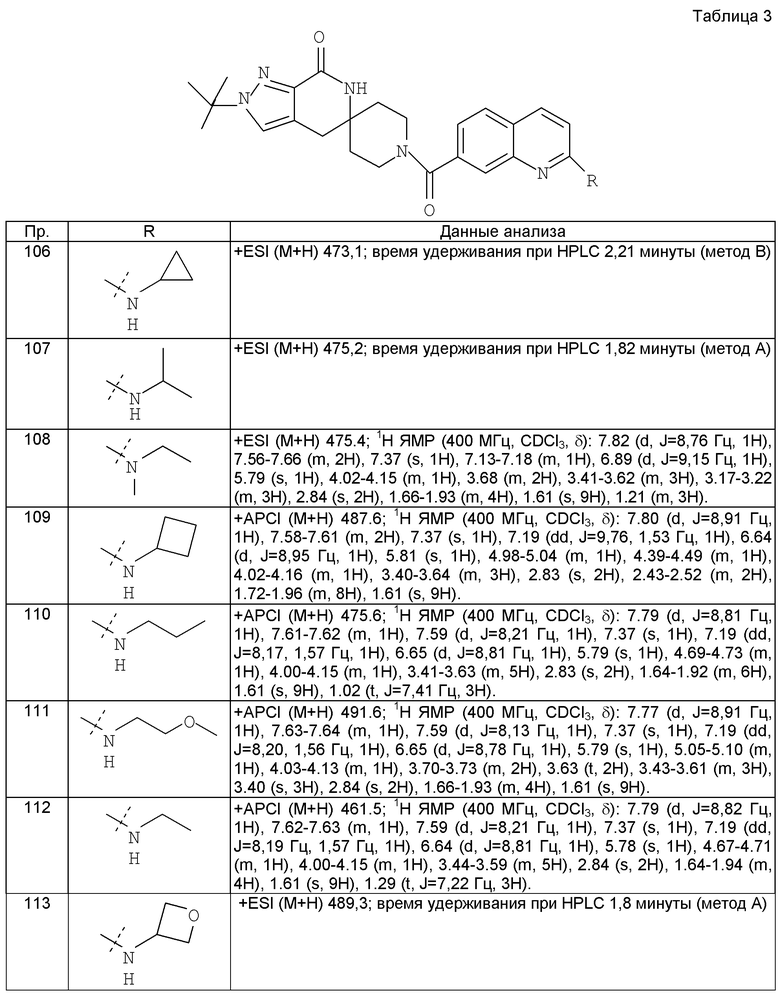
Соединения, приведенные ниже в Таблице 4, получали, используя методики, аналогичные описанным выше для синтеза соединения из Примеров 1-3, с применением соответствующих исходных веществ, имеющихся в продаже, полученных с использованием способов, хорошо известных специалистам в данной области, или полученных аналогично путям, описанным выше для других промежуточных соединений. Приведенные ниже соединения сначала были выделены в виде свободного основания, и для тестирования их можно превратить в фармацевтически приемлемую соль.
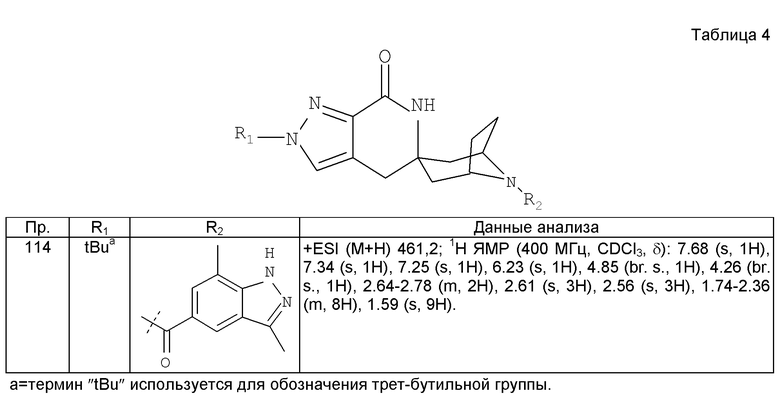
Соединения, приведенные ниже в Таблице 5, получали, используя методики, аналогичные описанным выше для синтеза соединения из Примера 103, с применением соответствующих исходных веществ, имеющихся в продаже, полученных с использованием способов, хорошо известных специалистам в данной области, или полученных аналогично путям, описанным выше для других промежуточных соединений. Приведенные ниже соединения сначала были выделены в виде свободного основания, и для тестирования их можно превратить в фармацевтически приемлемую соль.
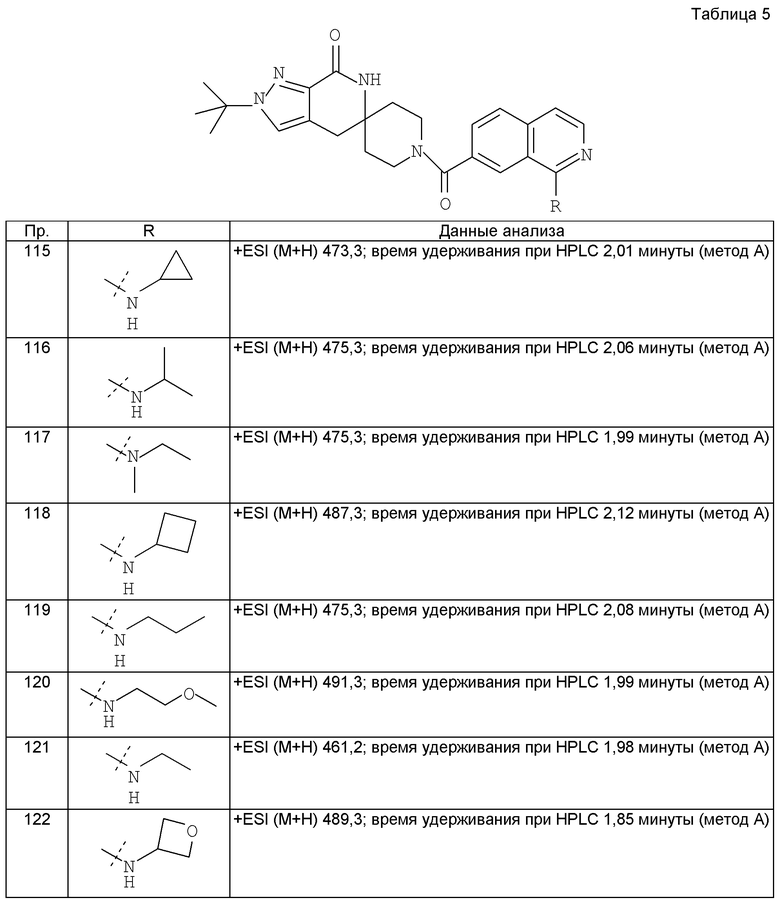
Пример 123: 2'-(трет-бутил)-1-(1-(трет-бутиламино)изохинолин-7-карбонил)-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(2'Н)-он
Стадия 1: 1-хлоризохинолин-7-карбоновая кислота
К суспензии 7-бром-1-хлоризохинолина (2,000 г; 8,247 ммоль) в THF (12 мл) и диэтилового эфира (12 мл), охлажденной до -78°C, добавляли н-BuLi (н-бутиллитий) (3,96 мл; 9,9 ммоль; 2,5 М в гексанах). Перемешивали в течение пяти минут и барботировали в течение приблизительно одной минуты диоксидом углерода, выпускаемым через иглу. Реакционную смесь нагревали до 0°C и добавляли 15 мл 1 н. водного гидроксида натрия. Смесь разбавляли диэтиловым эфиром и перемешивали в течение 18 ч. Органические и водные слои разделяли и органические слои промывали 1 н. водным гидроксидом натрия и водой. Водные фракции объединяли и подкисляли до рН 4, используя 1 н. водную соляную кислоту. Полученные твердые вещества собирали фильтрацией и сушили, получая указанное в заголовке соединение (1,252 г; 73%). +ESI (М+Н) 208,1; 1H ЯМР (400 МГц, DMSO-d6) δ ppm 13.58 (br. s., 1H), 8.86 (m, 1Н), 8.43 (d, J=5,67 Гц, 1H), 8.33 (dd, J=8,61, 1,57 Гц, 1H), 8.19 (d, J=8,41 Гц, 1H), 8.01 (dd, 1H).
Стадия 2: 2'-(трет-бутил)-1-(1-(трет-бутиламино)изохинолин-7-карбонил)-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(2'Н)-он
К суспензии 1-хлоризохинолин-7-карбоновой кислоты (100 мг; 0,482 ммоль), RuPhos (2-дициклогексилфосфино-2',6'-диизопропоксибифенил) (6,5 мг; 0,014 ммоль), BrettPhos (2-(дициклогексилфосфино)-3,6-диметокси-2',4',6'-тризопропил-1,1'-бифенил) (11,2 мг; 0,014 ммоль) и трет-бутилата натрия (70,2 мг; 0,723 ммоль) в диоксане (0,5 мл) добавляли трет-бутиламин (0,254 мл; 2,41 ммоль). Сосуд герметично закрывали, смесь нагревали до 110°С и перемешивали в течение ночи. Реакционную смесь охлаждали до комнатной температуры и добавляли бистриметилсилиламид лития (0,136 мл; 0,723 ммоль). Реакционную смесь нагревали до 110°С и оставляли перемешиваться в течение ночи. Реакционную смесь охлаждали до комнатной температуры, фильтровали через целит и промывали метанолом. Фильтрат концентрировали при пониженном давлении и добавляли 1 н. водный гидроксид натрия (1 мл). Проводили распределение между этилацетатом и смесью воды и 1 н. водного гидроксида натрия. Слои разделяли и водный слой подкисляли до рН 4. Водный слой экстрагировали в этилацетат. Экстракты сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении, получая 1-(трет-бутиламино)изохинолин-7-карбоновую кислоту.
К суспензии 1-(трет-бутиламино)изохинолин-7-карбоновой кислоты (24,7 мг; 0,101 ммоль) и гидрохлоридной соли 2'-трет-бутил-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(2'Н)-она (33,9 мг; 0,101 ммоль) в N,N-диметилформамиде (1 мл) добавляли триэтиламин (0,07 мл; 0,50 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 10 минут. Затем добавляли циклический ангидрид 1-пропанфосфоновой кислоты (0,07 мл; 0,12 ммоль; 50%-ный раствор в этилацетате) и реакционную смесь перемешивали при комнатной температуре в течение ночи. N,N-диметилформамид удаляли при пониженном давлении и остаток очищали обращенно-фазовой HPLC, получая 2'-(трет-бутил)-1-(1-(трет-бутиламино)изохинолин-7-карбонил)-4',6'-дигидроспиро[пиперидин-4,5'-пиразоло[3,4-с]пиридин]-7'(2'Н)-он (6,1 мг; 24%). +ESI (m+H) 489,3; время удерживания при HPLC 2,94 минуты (метод В).
Соединения, приведенные ниже в Таблице 6, получали, используя методики, аналогичные описанным выше для синтеза соединений из Примеров 1-3, с применением соответствующих исходных веществ, имеющихся в продаже, полученных с использованием способов, хорошо известных специалистам в данной области, или полученных аналогично путям, описанным выше для других промежуточных соединений. Приведенные ниже соединения сначала были выделены в виде свободного основания, и для тестирования их можно превратить в фармацевтически приемлемую соль.
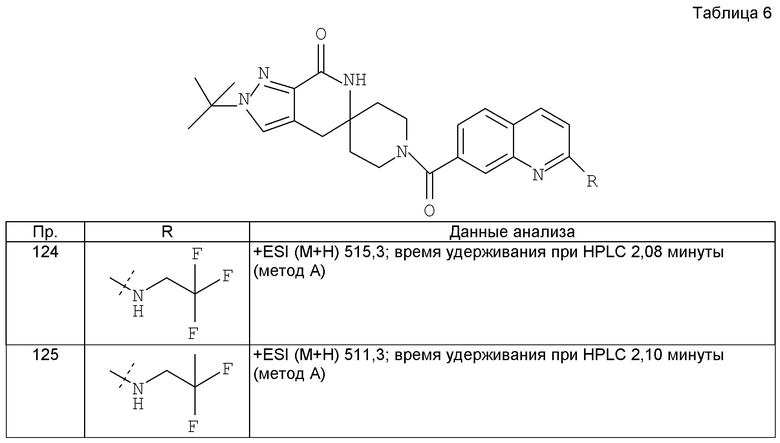
ФАРМАКОЛОГИЧЕСКИЕ ДАННЫЕ
Биологические протоколы
Полезность соединений по настоящему изобретению в лечении заболеваний (как например, подробно описанных в настоящем изобретении) у животных, в частности млекопитающих (например, людей), можно продемонстрировать по их активности в традиционных анализах, известных среднему специалисту в данной области, включая анализы in vitro и in vivo, описанные ниже. Посредством таких анализов также можно провести сравнение активностей соединения по настоящему изобретению с активностями других известных соединений.
Прямое ингибирование активности АСС1 и АСС2
АСС-ингибирующую активность соединения по настоящему изобретению демонстрировали, используя способы, основанные на стандартных методиках. Например, прямое ингибирование активности АСС для соединения формулы (I) определяли, используя препараты рекомбинантной АСС1 человека (rhACC1) и рекомбинантной АСС2 человека (rhACC2). Репрезентативные последовательности рекомбинантных АСС1 и АСС2 человека, которые могут быть использованы в данном анализе, приведены на Фиг.1 (SEQ ID NO.1) и Фиг.2 (SEQ. ID NO.2), соответственно.
[1] Получение rhACC1. Два литра клеток SF9, инфицированных рекомбинантным бакуловирусом, содержащим полноразмерную кДНК АСС1 человека, суспендировали в охлажденном во льду лизирующем буфере (25 мМ Трис, рН 7,5; 150 мМ NaCl; 10% глицерина; 5 мМ имидазол (EMD Bioscience; Gibbstown, NJ); 2 мМ ТСЕР (трис(2-карбоксиэтил)фосфин) (BioVectra; Charlottetown, Canada); нуклеаза бензоназа (10000 ед./100 г клеточной массы; Novagen; Madison, WI); не содержащая EDTA (этилендиаминтетрауксусная кислота) смесь ингибиторов протеаз (1 таб./50 мл; Roche Diagnostics; Mannheim, Germany). Клетки лизировали, используя 3 цикла замораживания-оттаивания, и центрифугировали при 40000×g в течение 40 минут (4°C). Супернатант наносили непосредственно на колонку с крупнозернистым носителем HisTrap FF (GE Healthcare; Piscataway, NJ) и элюировали градиентом имидазола до 0,5 М включительно, используя 20 объемов колонки (CV). Собирали АСС1-содержащие фракции, разбавляли 1:5, используя 25 мМ Трис, pH 7,5, 2 мМ ТСЕР, 10%-ный глицерин, наносили непосредственно на колонку CaptoQ (GE Healthcare) и элюировали градиентом NaCl до 1 М включительно, используя 20 CV. Отщепление фосфатных групп в очищенной АСС1 проводили посредством инкубации с лямбда-фосфатазой (100 ед./10 мкМ целевого белка; New EngIand BioIabs; Beveriy, MA) в течение 14 часов при 4°C; для ингибирования фосфатазы добавляли окадаиковую кислоту (конечная концентрация 1 мкМ; Roche Diagnostics). Выполняли замену буфера в очищенной АСС1 на 25 мМ Трис, рН 7,5, 2 мМ ТСЕР, 10%-ный глицерин, 0,5 М NaCl, используя диализ при 4°C в течение 6 часов. Готовили аликвоты и замораживали при -80°C.
[2] Измерение ингибирования rhACC1. Анализ с использованием hACC1 проводили в 384-луночном планшете Costar №3676 (Costar, Cambridge, MA) с применением набора Transcreener ADP detection FP assay kit (набор для детекции АДФ (аденозиндифосфат) посредством анализа FP (поляризация флуоресценции)) (Bellbrook Labs, Madison, Wisconsin), следуя рекомендованным производителем условиям для реакции с 50 мкМ АТФ (аденозинтрифосфат). Для данного анализа использовали следующие конечные условия: 50 мМ HEPES (N-2-гидроксиэтил-пиперазин-N-2-этансульфоновая кислота), рН 7,2; 10 мМ MgCl2, 7,5 мМ трикалийцитрат, 2 мМ DTT (дитиотреит); 0,1 мг/мл BSA (бычий сывороточный альбумин); 30 мкМ ацетил-КоА; 50 мкМ АТФ и 10 мМ КНСО3. Обычно реакцию в объеме 10 мкл проводили в течение 120 мин при 25°C, добавляли 10 мкл буфера Transcreener Stop and Detect buffer (буфер для остановки реакции и детекции) и эту смесь инкубировали при комнатной температуре в течение еще 1 часа. Данные получали с использованием флуоресцентного ридера Envision Fluorescence reader (PerkinElmer), применяя обычное двойное зеркало (general dual mirror) Су5 FP возбуждения 620, фильтр Су5 FP возбуждения 620, фильтр эмиссии 688 (S) и эмиссии 688 (Р).
[3] Получение rhACC2. Ингибирование АСС2 человека измеряли, используя очищенную рекомбинантную АСС2 человека (rhACC2). Кратко, полноразмерную клонированную АСС2 (Cytomax) приобретали у Cambridge Bioscience Limited, секвенировали и субклонировали в PCDNA5 FRT ТО-ТОРО (Invitrogen, Carlsbad, CA). АСС2 экспрессировали в клетках СНО (яичники китайского хомячка) путем индукции тетрациклином и получали в 5 литрах DMEM/F12 (модифицированная Дульбекко среда Игла с питательной смесью Хэма F12), содержащей глутамин, биотин, гигромицин и бластицидин с 1 мкг/мл тетрациклина (Invitrogen, Carlsbad, CA). Кондиционированную среду, содержащую АСС2, затем наносили на колонку Softlink Soft Release Avidin column (колонка с иммобилизованным авидином с мягким высвобождением) (Promega, Madison, Wisconsin) и элюировали 5 мМ биотином. Элюировали 4 мг АСС2 в концентрации 0,05 мг/мл (определенной по А280) с чистотой приблизительно 95% (определенной по А280). Очищенную АСС2 диализовали против буфера, содержащего 50 мМ Трис, 200 мМ NaCl, 4 мМ DTT, 2 мМ EDTA и 5% глицерина. Собранный белок замораживали и хранили при -80°С без какой-либо потери активности при размораживании. Для измерения активности АСС2 и оценки ингибирования АСС2 тестируемые соединения растворяли в DMSO (диметилсульфоксид) и добавляли к ферменту rhACC2 в виде 5-кратного концентрированного раствора с получением конечной концентрации DMSO 1%.
[4] Измерение ингибирования АСС2 человека. Анализ с использованием hACC2 проводили в 384-луночном планшете Costar №3676 (Costar, Cambridge, MA) с применением набора Transcreener ADP detection FP assay kit (BeIIbrook Labs, Madison, Wisconsin), следуя рекомендованным производителем условиям для реакции с 50 мкМ АТФ. Для данного анализа использовали следующие конечные условия: 50 мМ HEPES, рН 7,2; 5 мМ MgCl2, 5 мМ трикалийцитрат, 2 мМ DTT; 0,1 мг/мл BSA; 30 мкМ ацетил-КоА; 50 мкМ АТФ и 8 мМ КНСО3. Обычно реакцию в объеме 10 мкл проводили в течение 50 мин при 25°C, добавляли 10 мкл буфера Transcreener Stop and Detect buffer и эту смесь инкубировали при комнатной температуре в течение еще 1 часа. Данные получали с использованием флуоресцентного ридера Envision Fluorescence reader (PerkinElmer), применяя обычное двойное зеркало (general dual mirror) Су5 FP возбуждения 620, фильтр Су5 FP возбуждения 620, фильтр эмиссии 688 (S) и эмиссии 688 (Р).
Результаты анализов рекомбинантной hACC1 и рекомбинантной hACC2 с применением Transcreener суммированы ниже в таблице для соединений формулы (I), приведенных выше в разделе Примеры.
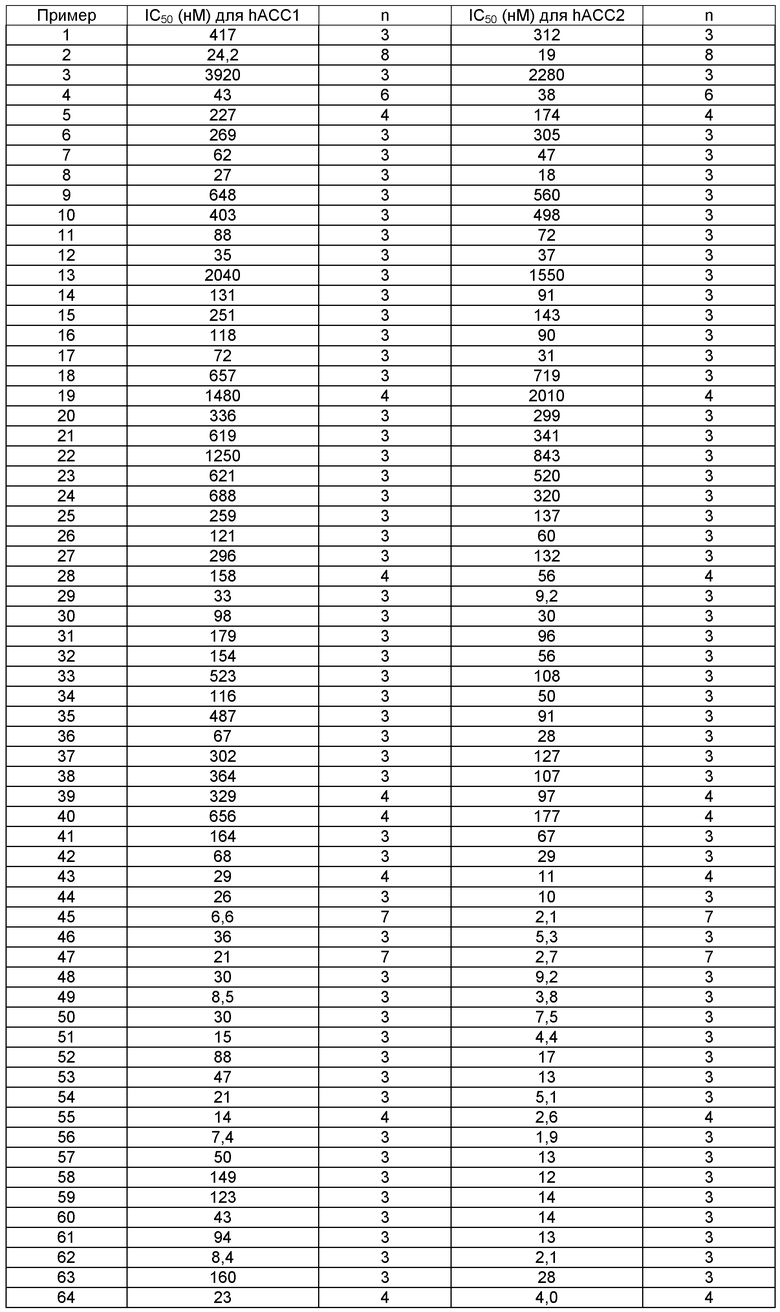
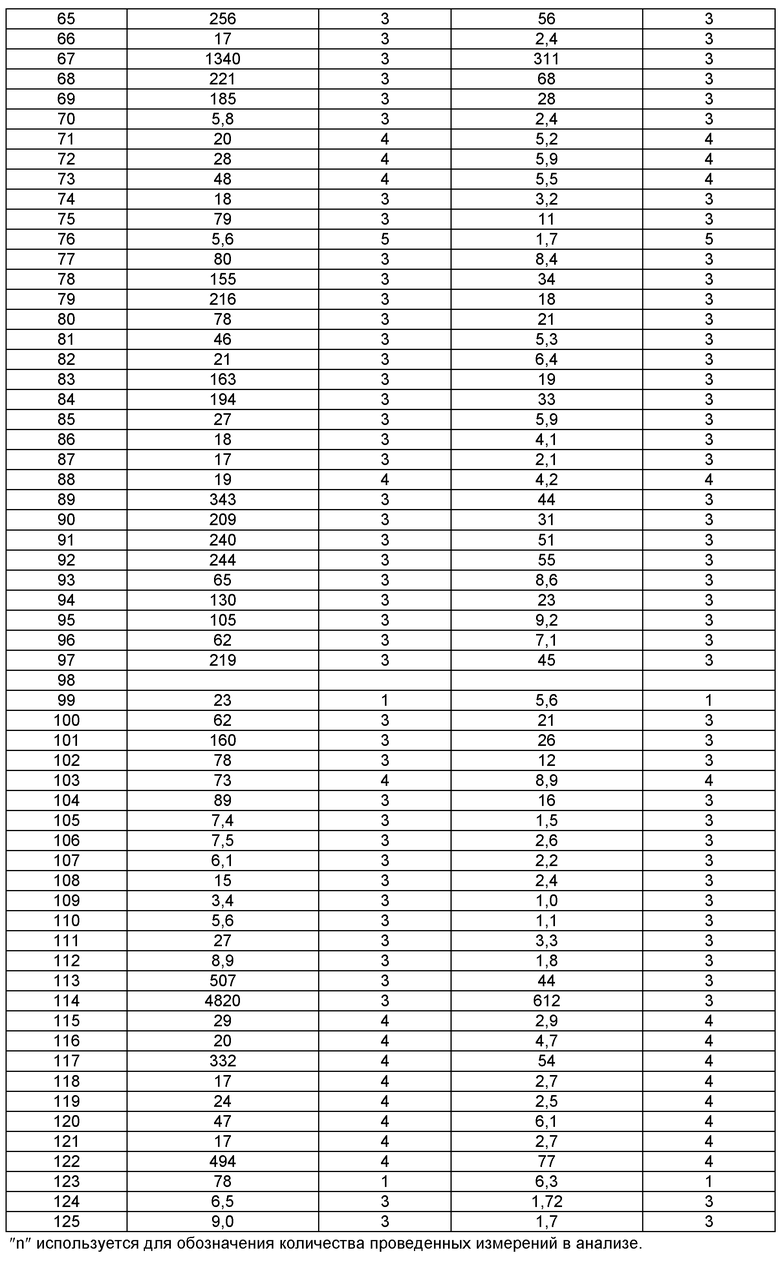
В Перечне последовательностей под номером 1 представлена последовательность рекомбинантной АСС1 человека (SEQ. ID NO.1), которая может быть использована в анализе in vitro с применением Transcreener.
В Перечне последовательностей под номером 2 представлена последовательность рекомбинантной АСС2 человека (SEQ. ID NO.2), которая может быть использована в анализе in vitro с применением Transcreener.
Оценка ингибирования АСС in vivo в остром опыте на экспериментальных животных
АСС-ингибирующая активность соединений по настоящему изобретению может быть подтверждена in vivo посредством оценки их способности снижать уровни малонил-КоА в печени и мышечной ткани у получавших лечение животных.
Измерение ингибирования образования малонил-КоА у экспериментальных животных может быть проведено с использованием приведенной далее методологии.
При применении этого метода самцов крыс Sprague-Dawley (225-275 г), содержащихся на стандартном корме и с доступом к воде без ограничения, случайным образом распределяли по группам перед исследованием. Одних животных кормили, другим не давали корма в течение 18 часов перед началом эксперимента. Через два часа после начала светового цикла животным перорально вводили дозу 0,5%-ной метилцеллюлозы (разбавителя) или соответствующего соединения (приготовленного в разбавителе) в объеме 5 мл/кг. Контрольные крысы, получавшие корм и разбавитель, были включены с целью определения фоновых уровней малонил-КоА в тканях, в то время как животные, не получавшие корма, были включены для определения того, как влияло голодание на уровни малонил-КоА. Через один час после введения соединений у животных вызывали асфиксию, используя CO2, и ткани извлекали. Конкретно, кровь отбирали путем сердечной пункции и помещали в пробирки BD Microtainer, содержащие EDTA (BD Biosciences, NJ), перемешивали и помещали на лед. Для определения воздействия лекарственного средства использовали плазму крови. Печень и четырехглавую мышцу извлекали, незамедлительно фиксировали замораживанием, заворачивали в фольгу и хранили в жидком азоте.
Ткани измельчали под жидким N2 для обеспечения однородности при приготовлении образцов. Из ткани (150-200 мг) экстрагировали малонил-КоА, используя 5 объемов 10%-ной трикарбоновой кислоты в Lysing Matrix A (лизирующей матрице А) (МР Biomedicals, PN 6910) на приборе FastPrep FP120 (Thermo Scientific, скорость=5,5; в течение 45 секунд). Супернатант, содержащий малонил-КоА, отделяли от клеточного дебриса после центрифугирования при 15000×g в течение 30 минут (Eppendorf Centrifuge 5402). Образцы надежно замораживали при -80°C до завершения анализа.
Оценка уровней малонил-КоА в печени и мышечной ткани может быть проведена в анализах с использованием приведенной далее методологии.
В этом методе были использованы следующие вещества: тетралитиевая соль малонил-КоА и трилитиевая соль малонил-13С3-КоА, которые были приобретены у Isotec (Miamisburg, ОН, USA), перхлорат натрия (Sigma, номер по каталогу 410241), трихлоруксусная кислота (ACROS, номер по каталогу 42145), фосфорная кислота (J.T.Baker, номер по каталогу 0260-01), формиат аммония (Пика, номер по каталогу 17843), метанол (квалификация ″для HPLC″, J.T. Baker, номер по каталогу 9093-33) и вода (квалификация ″для HPLC″, J.T. Baker, 4218-03), которые были использованы для приготовления необходимых подвижных фаз. Колонки для твердофазной экстракции (SPE) в режиме он-лайн Strata-X 25 мкм; 20 мм × 2,0 мм в.д. (внутренний диаметр) (номер по каталогу 00M-S033-B0-CB) были получены от Phenomenex (Torrance, СА, USA). Колонки для обращенно-фазовой хроматографии SunFire C18 (3,5 мкм; 100 мм × 3,0 мм в.д. (номер по каталогу 186002543) были приобретены у Waters Corporation (Milford, MA, USA).
Этот метод выполняли с применением следующего оборудования. Для двойной хроматографии использовали бинарный насос Agilent 1100, насос для четырехкомпонентных смесей Agilent 1100 и два 6-канальных двухпозиционных клапана Vaico Cheminert. Образцы вводили, применяя автосэмплер НТС PAL от LEAP с охлажденным посредством использования эффекта Пельтье штативом, поддерживаемым при 10°C, и петлей для образцов (20 мкл). Для промывки иглы автосэмплера использовали следующие растворы: 10%-ную трихлоруксусную кислоту в воде (масс./об.) для промывки 1 и смесь 90:10 метанол:вода для промывки 2. Температуру в аналитической колонке (Sunfire) поддерживали при 35°C, используя термостат для микроколонок для LC (жидкостная хроматография) от MicroTech Scientific (Micro-LC Column Over). Элюат анализировали на тройном квадрупольном масс-спектрометре API3000 от ABI Sciex с ионизатором Turbo Ion Spray.
Двойную хроматографию проводили, параллельно используя различные условия градиентного элюирования для он-лайн твердофазной экстракции и обращенно-фазовой хроматографии. Общая схема этого метода состояла в том, что первую стадию использовали для очистки образца и захвата представляющего интерес аналита, с последующим кратковременным соединением обеих стадий для элюирования с первой стадии во вторую стадию. Затем эти стадии разъединяли, чтобы осуществить градиентное элюирование данного аналита со второй стадии с целью количественного определения, одновременно готовя первую стадию для следующего образца в последовательности. В момент кратковременного соединения обеих стадий друг с другом, движение потока подвижной фазы в первой стадии меняли на противоположное для элюирования аналита во вторую стадию, что позволяло получить оптимальную ширину пика, форму пика и время элюирования.
В первой стадии в HPLC-системе использовали колонку для твердофазной экстракции в режиме он-лайн Strata-X от Phenomenex и подвижную фазу, составленную из смеси 100 мМ перхлорат натрия/0,1% (об./об.) фосфорной кислоты для растворителя А и метанола для растворителя В.
Во второй стадии в HPLC-системе использовали колонку для обращенно-фазовой хроматографии SunFire C18 от Waters и подвижную фазу, составленную из 100 мМ формиата аммония для растворителя А и метанола для растворителя В. Начальное состояние градиента выдерживали в течение 2 минут, и в течение этого периода времени происходил перенос аналита в аналитическую колонку. Важно, чтобы элюирующая сила в начальном состоянии была достаточной для осуществления элюирования аналита с колонки для он-лайн SPE с одновременным удержанием его на аналитической колонке. Сразу после этого градиент линейно увеличивали до 74,5% А в течение 4,5 минуты, после чего выполняли стадию промывки и повторного уравновешивания.
Масс-спектрометрия в сочетании с HPLC может представлять собой высокоселективный и чувствительный метод количественного определения аналитов в образцах сложного состава, однако на результаты по-прежнему оказывают влияние хроматографическая интерференция и подавление. В результате сочетания двухстадийной HPLC и масс-спектрометрии эта хроматографическая интерференция была значительно снижена. Кроме того, благодаря использованию в тройном квадрупольном масс-спектрометре режима мониторинга множественных реакций (MRM) было значительно улучшено соотношение сигнала к шуму.
Что касается этого анализа, то работу на масс-спектрометре осуществляли в режиме положительных ионов с напряжением на ионизаторе TurbolonSpray составляющем 2250 В. Распыляющий газ нагревали до 450°C. Потенциал декластеризации (DP), фокусирующий потенциал (FP) и энергию соударений (СЕ) устанавливали на 60, 340 и 42 В, соответственно. Для квадруполя 1 (Q1) устанавливали единичное разрешение, для квадруполя 3 (Q3) устанавливали низкое разрешение. Заданное значение в отношении газа для CAD (столкновительно активируемая диссоциация) составляло 8. Регистрировали следующие MRM-переходы: для малонил-КоА 854,1→347,0 m/z (L. Gao et al. (2007) J. Chromatogr. В 853, 303-313); и для малонил-13С3-СоА 857,1→350,0 m/z с временами измерения 200 мс. Элюент направляли в масс-спектрометр в момент, близкий к ожидаемому времени элюирования аналита, в остальное время его направляли в слив для сохранения источника и улучшения эксплутационной надежности прибора. Интегрирование полученных хроматофамм выполняли, используя программное обеспечение Analyst (Applied Biosystems). Концентрации малонил-КоА в тканях рассчитывали, используя калибровочную кривую, полученную с применением 10%-ного раствора трихлоруксусной кислоты в воде.
Образцы для построения калибровочной кривой с целью количественного определения малонил-КоА в экстрактах тканей готовили в 10%-ной (масс./об.) трихлоруксусной кислоте (ТСА) и в диапазоне от 0,01 до 1 пмоль/мкл. В качестве внутреннего стандарта в каждый компонент калибровочной кривой и образец добавляли малонил-13С3-КоА (в конечной концентрации 0,4 пмоль/мкл).
Готовили шесть внутрианалитических качественных контролей: три из объединенного экстракта, полученного от животных, не получавших корма, и три из объединенного экстракта, полученного от животных, получавших корм. Их анализировали в качестве независимых образцов, дополненных 12С-малонил-КоА в концентраци 0,0,1 или 0,3 пмоль/мкл, а также малонил-13С3-КоА (0,4 пмоль/мкл). Каждый из внутрианалитических качественных контролей содержал 85% водного экстракта ткани, а остальной вклад вносил внутренний стандарт (0,4 пмоль/мкл) и 12С-малонил-КоА. При проведении каждого измерения включали межаналитические контроли; они состоят из объединенных образцов четырехглавых мышц, по одному образцу от животных, не получавших корма и получавших корм, и/или из объединенных образцов печени, по одному образцу от животных, не получавших корма и получавших корм. Все подобные контроли дополнены малонил-13С3-КоА (0,4 пмоль/мкл).
Все публикации, включая, но не ограничиваясь этим, выданные патенты, патентные заявки и журнальные статьи, приведенные в этой заявке, включены по отдельности в настоящее описание посредством ссылки во всей своей полноте.
Хотя данное изобретение изложено выше со ссылкой на описанные воплощения, специалистам в данной области техники будет несложно понять, что подробно описанные конкретные эксперименты являются только иллюстрациями данного изобретения. Следует понимать, что могут быть выполнены различные модификации без отступления от сущности изобретения. Соответственно, данное изобретение ограничено только приведенной далее формулой изобретения.
ПЕРЕЧЕНЬ ПОСЛЕДОВАТЕЛЬНОСТЕЙ
<110> Пфайзер Инк.
БэГЛИ Скотт Уилльям
ДОУ Роберт Ли
ГРИФФИТ Дэвид Эндрю
СМИТ Аарон Кристофер
<120> N1/N2-Лактамные ингибиторы ацетил-КоA-карбоксилаз
<130> PC71687A
<150> 61/531,744
<151> 2011-09-07
<150> 61/408,127
<151> 2010-10-29
<160> 2
<170> PatentIn version 3.5
<210> 1
<211> 2356
<212> ПРТ
<213> Homo sapiens
<400> 1
Met Ala His His His His His His Asp Glu Val Asp Asp Glu Pro Ser
1 5 10 15
Pro Leu Ala Gln Pro Leu Glu Leu Asn Gln His Ser Arg Phe Ile Ile
20 25 30
Gly Ser Val Ser Glu Asp Asn Ser Glu Asp Glu Ile Ser Asn Leu Val
35 40 45
Lys Leu Asp Leu Leu Glu Lys Glu Gly Ser Leu Ser Pro Ala Ser Val
50 55 60
Gly Ser Asp Thr Leu Ser Asp Leu Gly Ile Ser Ser Leu Gln Asp Gly
65 70 75 80
Leu Ala Leu His Ile Arg Ser Ser Met Ser Gly Leu His Leu Val Lys
85 90 95
Gln Gly Arg Asp Arg Lys Lys Ile Asp Ser Gln Arg Asp Phe Thr Val
100 105 110
Ala Ser Pro Ala Glu Phe Val Thr Arg Phe Gly Gly Asn Lys Val Ile
115 120 125
Glu Lys Val Leu Ile Ala Asn Asn Gly Ile Ala Ala Val Lys Cys Met
130 135 140
Arg Ser Ile Arg Arg Trp Ser Tyr Glu Met Phe Arg Asn Glu Arg Ala
145 150 155 160
Ile Arg Phe Val Val Met Val Thr Pro Glu Asp Leu Lys Ala Asn Ala
165 170 175
Glu Tyr Ile Lys Met Ala Asp His Tyr Val Pro Val Pro Gly Gly Pro
180 185 190
Asn Asn Asn Asn Tyr Ala Asn Val Glu Leu Ile Leu Asp Ile Ala Lys
195 200 205
Arg Ile Pro Val Gln Ala Val Trp Ala Gly Trp Gly His Ala Ser Glu
210 215 220
Asn Pro Lys Leu Pro Glu Leu Leu Leu Lys Asn Gly Ile Ala Phe Met
225 230 235 240
Gly Pro Pro Ser Gln Ala Met Trp Ala Leu Gly Asp Lys Ile Ala Ser
245 250 255
Ser Ile Val Ala Gln Thr Ala Gly Ile Pro Thr Leu Pro Trp Ser Gly
260 265 270
Ser Gly Leu Arg Val Asp Trp Gln Glu Asn Asp Phe Ser Lys Arg Ile
275 280 285
Leu Asn Val Pro Gln Glu Leu Tyr Glu Lys Gly Tyr Val Lys Asp Val
290 295 300
Asp Asp Gly Leu Gln Ala Ala Glu Glu Val Gly Tyr Pro Val Met Ile
305 310 315 320
Lys Ala Ser Glu Gly Gly Gly Gly Lys Gly Ile Arg Lys Val Asn Asn
325 330 335
Ala Asp Asp Phe Pro Asn Leu Phe Arg Gln Val Gln Ala Glu Val Pro
340 345 350
Gly Ser Pro Ile Phe Val Met Arg Leu Ala Lys Gln Ser Arg His Leu
355 360 365
Glu Val Gln Ile Leu Ala Asp Gln Tyr Gly Asn Ala Ile Ser Leu Phe
370 375 380
Gly Arg Asp Cys Ser Val Gln Arg Arg His Gln Lys Ile Ile Glu Glu
385 390 395 400
Ala Pro Ala Thr Ile Ala Thr Pro Ala Val Phe Glu His Met Glu Gln
405 410 415
Cys Ala Val Lys Leu Ala Lys Met Val Gly Tyr Val Ser Ala Gly Thr
420 425 430
Val Glu Tyr Leu Tyr Ser Gln Asp Gly Ser Phe Tyr Phe Leu Glu Leu
435 440 445
Asn Pro Arg Leu Gln Val Glu His Pro Cys Thr Glu Met Val Ala Asp
450 455 460
Val Asn Leu Pro Ala Ala Gln Leu Gln Ile Ala Met Gly Ile Pro Leu
465 470 475 480
Tyr Arg Ile Lys Asp Ile Arg Met Met Tyr Gly Val Ser Pro Trp Gly
485 490 495
Asp Ser Pro Ile Asp Phe Glu Asp Ser Ala His Val Pro Cys Pro Arg
500 505 510
Gly His Val Ile Ala Ala Arg Ile Thr Ser Glu Asn Pro Asp Glu Gly
515 520 525
Phe Lys Pro Ser Ser Gly Thr Val Gln Glu Leu Asn Phe Arg Ser Asn
530 535 540
Lys Asn Val Trp Gly Tyr Phe Ser Val Ala Ala Ala Gly Gly Leu His
545 550 555 560
Glu Phe Ala Asp Ser Gln Phe Gly His Cys Phe Ser Trp Gly Glu Asn
565 570 575
Arg Glu Glu Ala Ile Ser Asn Met Val Val Ala Leu Lys Glu Leu Ser
580 585 590
Ile Arg Gly Asp Phe Arg Thr Thr Val Glu Tyr Leu Ile Lys Leu Leu
595 600 605
Glu Thr Glu Ser Phe Gln Met Asn Arg Ile Asp Thr Gly Trp Leu Asp
610 615 620
Arg Leu Ile Ala Glu Lys Val Gln Ala Glu Arg Pro Asp Thr Met Leu
625 630 635 640
Gly Val Val Cys Gly Ala Leu His Val Ala Asp Val Ser Leu Arg Asn
645 650 655
Ser Val Ser Asn Phe Leu His Ser Leu Glu Arg Gly Gln Val Leu Pro
660 665 670
Ala His Thr Leu Leu Asn Thr Val Asp Val Glu Leu Ile Tyr Glu Gly
675 680 685
Val Lys Tyr Val Leu Lys Val Thr Arg Gln Ser Pro Asn Ser Tyr Val
690 695 700
Val Ile Met Asn Gly Ser Cys Val Glu Val Asp Val His Arg Leu Ser
705 710 715 720
Asp Gly Gly Leu Leu Leu Ser Tyr Asp Gly Ser Ser Tyr Thr Thr Tyr
725 730 735
Met Lys Glu Glu Val Asp Arg Tyr Arg Ile Thr Ile Gly Asn Lys Thr
740 745 750
Cys Val Phe Glu Lys Glu Asn Asp Pro Ser Val Met Arg Ser Pro Ser
755 760 765
Ala Gly Lys Leu Ile Gln Tyr Ile Val Glu Asp Gly Gly His Val Phe
770 775 780
Ala Gly Gln Cys Tyr Ala Glu Ile Glu Val Met Lys Met Val Met Thr
785 790 795 800
Leu Thr Ala Val Glu Ser Gly Cys Ile His Tyr Val Lys Arg Pro Gly
805 810 815
Ala Ala Leu Asp Pro Gly Cys Val Leu Ala Lys Met Gln Leu Asp Asn
820 825 830
Pro Ser Lys Val Gln Gln Ala Glu Leu His Thr Gly Ser Leu Pro Arg
835 840 845
Ile Gln Ser Thr Ala Leu Arg Gly Glu Lys Leu His Arg Val Phe His
850 855 860
Tyr Val Leu Asp Asn Leu Val Asn Val Met Asn Gly Tyr Cys Leu Pro
865 870 875 880
Asp Pro Phe Phe Ser Ser Lys Val Lys Asp Trp Val Glu Arg Leu Met
885 890 895
Lys Thr Leu Arg Asp Pro Ser Leu Pro Leu Leu Glu Leu Gln Asp Ile
900 905 910
Met Thr Ser Val Ser Gly Arg Ile Pro Pro Asn Val Glu Lys Ser Ile
915 920 925
Lys Lys Glu Met Ala Gln Tyr Ala Ser Asn Ile Thr Ser Val Leu Cys
930 935 940
Gln Phe Pro Ser Gln Gln Ile Ala Asn Ile Leu Asp Ser His Ala Ala
945 950 955 960
Thr Leu Asn Arg Lys Ser Glu Arg Glu Val Phe Phe Met Asn Thr Gln
965 970 975
Ser Ile Val Gln Leu Val Gln Arg Tyr Arg Ser Gly Ile Arg Gly His
980 985 990
Met Lys Ala Val Val Met Asp Leu Leu Arg Gln Tyr Leu Arg Val Glu
995 1000 1005
Thr Gln Phe Gln Asn Gly His Tyr Asp Lys Cys Val Phe Ala Leu
1010 1015 1020
Arg Glu Glu Asn Lys Ser Asp Met Asn Thr Val Leu Asn Tyr Ile
1025 1030 1035
Phe Ser His Ala Gln Val Thr Lys Lys Asn Leu Leu Val Thr Met
1040 1045 1050
Leu Ile Asp Gln Leu Cys Gly Arg Asp Pro Thr Leu Thr Asp Glu
1055 1060 1065
Leu Leu Asn Ile Leu Thr Glu Leu Thr Gln Leu Ser Lys Thr Thr
1070 1075 1080
Asn Ala Lys Val Ala Leu Arg Ala Arg Gln Val Leu Ile Ala Ser
1085 1090 1095
His Leu Pro Ser Tyr Glu Leu Arg His Asn Gln Val Glu Ser Ile
1100 1105 1110
Phe Leu Ser Ala Ile Asp Met Tyr Gly His Gln Phe Cys Ile Glu
1115 1120 1125
Asn Leu Gln Lys Leu Ile Leu Ser Glu Thr Ser Ile Phe Asp Val
1130 1135 1140
Leu Pro Asn Phe Phe Tyr His Ser Asn Gln Val Val Arg Met Ala
1145 1150 1155
Ala Leu Glu Val Tyr Val Arg Arg Ala Tyr Ile Ala Tyr Glu Leu
1160 1165 1170
Asn Ser Val Gln His Arg Gln Leu Lys Asp Asn Thr Cys Val Val
1175 1180 1185
Glu Phe Gln Phe Met Leu Pro Thr Ser His Pro Asn Arg Gly Asn
1190 1195 1200
Ile Pro Thr Leu Asn Arg Met Ser Phe Ser Ser Asn Leu Asn His
1205 1210 1215
Tyr Gly Met Thr His Val Ala Ser Val Ser Asp Val Leu Leu Asp
1220 1225 1230
Asn Ser Phe Thr Pro Pro Cys Gln Arg Met Gly Gly Met Val Ser
1235 1240 1245
Phe Arg Thr Phe Glu Asp Phe Val Arg Ile Phe Asp Glu Val Met
1250 1255 1260
Gly Cys Phe Ser Asp Ser Pro Pro Gln Ser Pro Thr Phe Pro Glu
1265 1270 1275
Ala Gly His Thr Ser Leu Tyr Asp Glu Asp Lys Val Pro Arg Asp
1280 1285 1290
Glu Pro Ile His Ile Leu Asn Val Ala Ile Lys Thr Asp Cys Asp
1295 1300 1305
Ile Glu Asp Asp Arg Leu Ala Ala Met Phe Arg Glu Phe Thr Gln
1310 1315 1320
Gln Asn Lys Ala Thr Leu Val Asp His Gly Ile Arg Arg Leu Thr
1325 1330 1335
Phe Leu Val Ala Gln Lys Asp Phe Arg Lys Gln Val Asn Tyr Glu
1340 1345 1350
Val Asp Arg Arg Phe His Arg Glu Phe Pro Lys Phe Phe Thr Phe
1355 1360 1365
Arg Ala Arg Asp Lys Phe Glu Glu Asp Arg Ile Tyr Arg His Leu
1370 1375 1380
Glu Pro Ala Leu Ala Phe Gln Leu Glu Leu Asn Arg Met Arg Asn
1385 1390 1395
Phe Asp Leu Thr Ala Ile Pro Cys Ala Asn His Lys Met His Leu
1400 1405 1410
Tyr Leu Gly Ala Ala Lys Val Glu Val Gly Thr Glu Val Thr Asp
1415 1420 1425
Tyr Arg Phe Phe Val Arg Ala Ile Ile Arg His Ser Asp Leu Val
1430 1435 1440
Thr Lys Glu Ala Ser Phe Glu Tyr Leu Gln Asn Glu Gly Glu Arg
1445 1450 1455
Leu Leu Leu Glu Ala Met Asp Glu Leu Glu Val Ala Phe Asn Asn
1460 1465 1470
Thr Asn Val Arg Thr Asp Cys Asn His Ile Phe Leu Asn Phe Val
1475 1480 1485
Pro Thr Val Ile Met Asp Pro Ser Lys Ile Glu Glu Ser Val Arg
1490 1495 1500
Ser Met Val Met Arg Tyr Gly Ser Arg Leu Trp Lys Leu Arg Val
1505 1510 1515
Leu Gln Ala Glu Leu Lys Ile Asn Ile Arg Leu Thr Pro Thr Gly
1520 1525 1530
Lys Ala Ile Pro Ile Arg Leu Phe Leu Thr Asn Glu Ser Gly Tyr
1535 1540 1545
Tyr Leu Asp Ile Ser Leu Tyr Lys Glu Val Thr Asp Ser Arg Thr
1550 1555 1560
Ala Gln Ile Met Phe Gln Ala Tyr Gly Asp Lys Gln Gly Pro Leu
1565 1570 1575
His Gly Met Leu Ile Asn Thr Pro Tyr Val Thr Lys Asp Leu Leu
1580 1585 1590
Gln Ser Lys Arg Phe Gln Ala Gln Ser Leu Gly Thr Thr Tyr Ile
1595 1600 1605
Tyr Asp Ile Pro Glu Met Phe Arg Gln Ser Leu Ile Lys Leu Trp
1610 1615 1620
Glu Ser Met Ser Thr Gln Ala Phe Leu Pro Ser Pro Pro Leu Pro
1625 1630 1635
Ser Asp Met Leu Thr Tyr Thr Glu Leu Val Leu Asp Asp Gln Gly
1640 1645 1650
Gln Leu Val His Met Asn Arg Leu Pro Gly Gly Asn Glu Ile Gly
1655 1660 1665
Met Val Ala Trp Lys Met Thr Phe Lys Ser Pro Glu Tyr Pro Glu
1670 1675 1680
Gly Arg Asp Ile Ile Val Ile Gly Asn Asp Ile Thr Tyr Arg Ile
1685 1690 1695
Gly Ser Phe Gly Pro Gln Glu Asp Leu Leu Phe Leu Arg Ala Ser
1700 1705 1710
Glu Leu Ala Arg Ala Glu Gly Ile Pro Arg Ile Tyr Val Ser Ala
1715 1720 1725
Asn Ser Gly Ala Arg Ile Gly Leu Ala Glu Glu Ile Arg His Met
1730 1735 1740
Phe His Val Ala Trp Val Asp Pro Glu Asp Pro Tyr Lys Gly Tyr
1745 1750 1755
Arg Tyr Leu Tyr Leu Thr Pro Gln Asp Tyr Lys Arg Val Ser Ala
1760 1765 1770
Leu Asn Ser Val His Cys Glu His Val Glu Asp Glu Gly Glu Ser
1775 1780 1785
Arg Tyr Lys Ile Thr Asp Ile Ile Gly Lys Glu Glu Gly Ile Gly
1790 1795 1800
Pro Glu Asn Leu Arg Gly Ser Gly Met Ile Ala Gly Glu Ser Ser
1805 1810 1815
Leu Ala Tyr Asn Glu Ile Ile Thr Ile Ser Leu Val Thr Cys Arg
1820 1825 1830
Ala Ile Gly Ile Gly Ala Tyr Leu Val Arg Leu Gly Gln Arg Thr
1835 1840 1845
Ile Gln Val Glu Asn Ser His Leu Ile Leu Thr Gly Ala Gly Ala
1850 1855 1860
Leu Asn Lys Val Leu Gly Arg Glu Val Tyr Thr Ser Asn Asn Gln
1865 1870 1875
Leu Gly Gly Ile Gln Ile Met His Asn Asn Gly Val Thr His Cys
1880 1885 1890
Thr Val Cys Asp Asp Phe Glu Gly Val Phe Thr Val Leu His Trp
1895 1900 1905
Leu Ser Tyr Met Pro Lys Ser Val His Ser Ser Val Pro Leu Leu
1910 1915 1920
Asn Ser Lys Asp Pro Ile Asp Arg Ile Ile Glu Phe Val Pro Thr
1925 1930 1935
Lys Thr Pro Tyr Asp Pro Arg Trp Met Leu Ala Gly Arg Pro His
1940 1945 1950
Pro Thr Gln Lys Gly Gln Trp Leu Ser Gly Phe Phe Asp Tyr Gly
1955 1960 1965
Ser Phe Ser Glu Ile Met Gln Pro Trp Ala Gln Thr Val Val Val
1970 1975 1980
Gly Arg Ala Arg Leu Gly Gly Ile Pro Val Gly Val Val Ala Val
1985 1990 1995
Glu Thr Arg Thr Val Glu Leu Ser Ile Pro Ala Asp Pro Ala Asn
2000 2005 2010
Leu Asp Ser Glu Ala Lys Ile Ile Gln Gln Ala Gly Gln Val Trp
2015 2020 2025
Phe Pro Asp Ser Ala Phe Lys Thr Tyr Gln Ala Ile Lys Asp Phe
2030 2035 2040
Asn Arg Glu Gly Leu Pro Leu Met Val Phe Ala Asn Trp Arg Gly
2045 2050 2055
Phe Ser Gly Gly Met Lys Asp Met Tyr Asp Gln Val Leu Lys Phe
2060 2065 2070
Gly Ala Tyr Ile Val Asp Gly Leu Arg Glu Cys Cys Gln Pro Val
2075 2080 2085
Leu Val Tyr Ile Pro Pro Gln Ala Glu Leu Arg Gly Gly Ser Trp
2090 2095 2100
Val Val Ile Asp Ser Ser Ile Asn Pro Arg His Met Glu Met Tyr
2105 2110 2115
Ala Asp Arg Glu Ser Arg Gly Ser Val Leu Glu Pro Glu Gly Thr
2120 2125 2130
Val Glu Ile Lys Phe Arg Arg Lys Asp Leu Val Lys Thr Met Arg
2135 2140 2145
Arg Val Asp Pro Val Tyr Ile His Leu Ala Glu Arg Leu Gly Thr
2150 2155 2160
Pro Glu Leu Ser Thr Ala Glu Arg Lys Glu Leu Glu Asn Lys Leu
2165 2170 2175
Lys Glu Arg Glu Glu Phe Leu Ile Pro Ile Tyr His Gln Val Ala
2180 2185 2190
Val Gln Phe Ala Asp Leu His Asp Thr Pro Gly Arg Met Gln Glu
2195 2200 2205
Lys Gly Val Ile Ser Asp Ile Leu Asp Trp Lys Thr Ser Arg Thr
2210 2215 2220
Phe Phe Tyr Trp Arg Leu Arg Arg Leu Leu Leu Glu Asp Leu Val
2225 2230 2235
Lys Lys Lys Ile His Asn Ala Asn Pro Glu Leu Thr Asp Gly Gln
2240 2245 2250
Ile Gln Ala Met Leu Arg Arg Trp Phe Val Glu Val Glu Gly Thr
2255 2260 2265
Val Lys Ala Tyr Val Trp Asp Asn Asn Lys Asp Leu Ala Glu Trp
2270 2275 2280
Leu Glu Lys Gln Leu Thr Glu Glu Asp Gly Val His Ser Val Ile
2285 2290 2295
Glu Glu Asn Ile Lys Cys Ile Ser Arg Asp Tyr Val Leu Lys Gln
2300 2305 2310
Ile Arg Ser Leu Val Gln Ala Asn Pro Glu Val Ala Met Asp Ser
2315 2320 2325
Ile Ile His Met Thr Gln His Ile Ser Pro Thr Gln Arg Ala Glu
2330 2335 2340
Val Ile Arg Ile Leu Ser Thr Met Asp Ser Pro Ser Thr
2345 2350 2355
<210> 2
<211> 2458
<212> ПРТ
<213> Homo sapiens
<400> 2
Met Val Leu Leu Leu Cys Leu Ser Cys Leu Ile Phe Ser Cys Leu Thr
1 5 10 15
Phe Ser Trp Leu Lys Ile Trp Gly Lys Met Thr Asp Ser Lys Pro Ile
20 25 30
Thr Lys Ser Lys Ser Glu Ala Asn Leu Ile Pro Ser Gln Glu Pro Phe
35 40 45
Pro Ala Ser Asp Asn Ser Gly Glu Thr Pro Gln Arg Asn Gly Glu Gly
50 55 60
His Thr Leu Pro Lys Thr Pro Ser Gln Ala Glu Pro Ala Ser His Lys
65 70 75 80
Gly Pro Lys Asp Ala Gly Arg Arg Arg Asn Ser Leu Pro Pro Ser His
85 90 95
Gln Lys Pro Pro Arg Asn Pro Leu Ser Ser Ser Asp Ala Ala Pro Ser
100 105 110
Pro Glu Leu Gln Ala Asn Gly Thr Gly Thr Gln Gly Leu Glu Ala Thr
115 120 125
Asp Thr Asn Gly Leu Ser Ser Ser Ala Arg Pro Gln Gly Gln Gln Ala
130 135 140
Gly Ser Pro Ser Lys Glu Asp Lys Lys Gln Ala Asn Ile Lys Arg Gln
145 150 155 160
Leu Met Thr Asn Phe Ile Leu Gly Ser Phe Asp Asp Tyr Ser Ser Asp
165 170 175
Glu Asp Ser Val Ala Gly Ser Ser Arg Glu Ser Thr Arg Lys Gly Ser
180 185 190
Arg Ala Ser Leu Gly Ala Leu Ser Leu Glu Ala Tyr Leu Thr Thr Gly
195 200 205
Glu Ala Glu Thr Arg Val Pro Thr Met Arg Pro Ser Met Ser Gly Leu
210 215 220
His Leu Val Lys Arg Gly Arg Glu His Lys Lys Leu Asp Leu His Arg
225 230 235 240
Asp Phe Thr Val Ala Ser Pro Ala Glu Phe Val Thr Arg Phe Gly Gly
245 250 255
Asp Arg Val Ile Glu Lys Val Leu Ile Ala Asn Asn Gly Ile Ala Ala
260 265 270
Val Lys Cys Met Arg Ser Ile Arg Arg Trp Ala Tyr Glu Met Phe Arg
275 280 285
Asn Glu Arg Ala Ile Arg Phe Val Val Met Val Thr Pro Glu Asp Leu
290 295 300
Lys Ala Asn Ala Glu Tyr Ile Lys Met Ala Asp His Tyr Val Pro Val
305 310 315 320
Pro Gly Gly Pro Asn Asn Asn Asn Tyr Ala Asn Val Glu Leu Ile Val
325 330 335
Asp Ile Ala Lys Arg Ile Pro Val Gln Ala Val Trp Ala Gly Trp Gly
340 345 350
His Ala Ser Glu Asn Pro Lys Leu Pro Glu Leu Leu Cys Lys Asn Gly
355 360 365
Val Ala Phe Leu Gly Pro Pro Ser Glu Ala Met Trp Ala Leu Gly Asp
370 375 380
Lys Ile Ala Ser Thr Val Val Ala Gln Thr Leu Gln Val Pro Thr Leu
385 390 395 400
Pro Trp Ser Gly Ser Gly Leu Thr Val Glu Trp Thr Glu Asp Asp Leu
405 410 415
Gln Gln Gly Lys Arg Ile Ser Val Pro Glu Asp Val Tyr Asp Lys Gly
420 425 430
Cys Val Lys Asp Val Asp Glu Gly Leu Glu Ala Ala Glu Arg Ile Gly
435 440 445
Phe Pro Leu Met Ile Lys Ala Ser Glu Gly Gly Gly Gly Lys Gly Ile
450 455 460
Arg Lys Ala Glu Ser Ala Glu Asp Phe Pro Ile Leu Phe Arg Gln Val
465 470 475 480
Gln Ser Glu Ile Pro Gly Ser Pro Ile Phe Leu Met Lys Leu Ala Gln
485 490 495
His Ala Arg His Leu Glu Val Gln Ile Leu Ala Asp Gln Tyr Gly Asn
500 505 510
Ala Val Ser Leu Phe Gly Arg Asp Cys Ser Ile Gln Arg Arg His Gln
515 520 525
Lys Ile Val Glu Glu Ala Pro Ala Thr Ile Ala Pro Leu Ala Ile Phe
530 535 540
Glu Phe Met Glu Gln Cys Ala Ile Arg Leu Ala Lys Thr Val Gly Tyr
545 550 555 560
Val Ser Ala Gly Thr Val Glu Tyr Leu Tyr Ser Gln Asp Gly Ser Phe
565 570 575
His Phe Leu Glu Leu Asn Pro Arg Leu Gln Val Glu His Pro Cys Thr
580 585 590
Glu Met Ile Ala Asp Val Asn Leu Pro Ala Ala Gln Leu Gln Ile Ala
595 600 605
Met Gly Val Pro Leu His Arg Leu Lys Asp Ile Arg Leu Leu Tyr Gly
610 615 620
Glu Ser Pro Trp Gly Val Thr Pro Ile Ser Phe Glu Thr Pro Ser Asn
625 630 635 640
Pro Pro Leu Ala Arg Gly His Val Ile Ala Ala Arg Ile Thr Ser Glu
645 650 655
Asn Pro Asp Glu Gly Phe Lys Pro Ser Ser Gly Thr Val Gln Glu Leu
660 665 670
Asn Phe Arg Ser Ser Lys Asn Val Trp Gly Tyr Phe Ser Val Ala Ala
675 680 685
Thr Gly Gly Leu His Glu Phe Ala Asp Ser Gln Phe Gly His Cys Phe
690 695 700
Ser Trp Gly Glu Asn Arg Glu Glu Ala Ile Ser Asn Met Val Val Ala
705 710 715 720
Leu Lys Glu Leu Ser Ile Arg Gly Asp Phe Arg Thr Thr Val Glu Tyr
725 730 735
Leu Ile Asn Leu Leu Glu Thr Glu Ser Phe Gln Asn Asn Asp Ile Asp
740 745 750
Thr Gly Trp Leu Asp Tyr Leu Ile Ala Glu Lys Val Gln Ala Glu Lys
755 760 765
Pro Asp Ile Met Leu Gly Val Val Cys Gly Ala Leu Asn Val Ala Asp
770 775 780
Ala Met Phe Arg Thr Cys Met Thr Asp Phe Leu His Ser Leu Glu Arg
785 790 795 800
Gly Gln Val Leu Pro Ala Asp Ser Leu Leu Asn Leu Val Asp Val Glu
805 810 815
Leu Ile Tyr Gly Gly Val Lys Tyr Ile Leu Lys Val Ala Arg Gln Ser
820 825 830
Leu Thr Met Phe Val Leu Ile Met Asn Gly Cys His Ile Glu Ile Asp
835 840 845
Ala His Arg Leu Asn Asp Gly Gly Leu Leu Leu Ser Tyr Asn Gly Asn
850 855 860
Ser Tyr Thr Thr Tyr Met Lys Glu Glu Val Asp Ser Tyr Arg Ile Thr
865 870 875 880
Ile Gly Asn Lys Thr Cys Val Phe Glu Lys Glu Asn Asp Pro Thr Val
885 890 895
Leu Arg Ser Pro Ser Ala Gly Lys Leu Thr Gln Tyr Thr Val Glu Asp
900 905 910
Gly Gly His Val Glu Ala Gly Ser Ser Tyr Ala Glu Met Glu Val Met
915 920 925
Lys Met Ile Met Thr Leu Asn Val Gln Glu Arg Gly Arg Val Lys Tyr
930 935 940
Ile Lys Arg Pro Gly Ala Val Leu Glu Ala Gly Cys Val Val Ala Arg
945 950 955 960
Leu Glu Leu Asp Asp Pro Ser Lys Val His Pro Ala Glu Pro Phe Thr
965 970 975
Gly Glu Leu Pro Ala Gln Gln Thr Leu Pro Ile Leu Gly Glu Lys Leu
980 985 990
His Gln Val Phe His Ser Val Leu Glu Asn Leu Thr Asn Val Met Ser
995 1000 1005
Gly Phe Cys Leu Pro Glu Pro Val Phe Ser Ile Lys Leu Lys Glu
1010 1015 1020
Trp Val Gln Lys Leu Met Met Thr Leu Arg His Pro Ser Leu Pro
1025 1030 1035
Leu Leu Glu Leu Gln Glu Ile Met Thr Ser Val Ala Gly Arg Ile
1040 1045 1050
Pro Ala Pro Val Glu Lys Ser Val Arg Arg Val Met Ala Gln Tyr
1055 1060 1065
Ala Ser Asn Ile Thr Ser Val Leu Cys Gln Phe Pro Ser Gln Gln
1070 1075 1080
Ile Ala Thr Ile Leu Asp Cys His Ala Ala Thr Leu Gln Arg Lys
1085 1090 1095
Ala Asp Arg Glu Val Phe Phe Ile Asn Thr Gln Ser Ile Val Gln
1100 1105 1110
Leu Val Gln Arg Tyr Arg Ser Gly Ile Arg Gly Tyr Met Lys Thr
1115 1120 1125
Val Val Leu Asp Leu Leu Arg Arg Tyr Leu Arg Val Glu His His
1130 1135 1140
Phe Gln Gln Ala His Tyr Asp Lys Cys Val Ile Asn Leu Arg Glu
1145 1150 1155
Gln Phe Lys Pro Asp Met Ser Gln Val Leu Asp Cys Ile Phe Ser
1160 1165 1170
His Ala Gln Val Ala Lys Lys Asn Gln Leu Val Ile Met Leu Ile
1175 1180 1185
Asp Glu Leu Cys Gly Pro Asp Pro Ser Leu Ser Asp Glu Leu Ile
1190 1195 1200
Ser Ile Leu Asn Glu Leu Thr Gln Leu Ser Lys Ser Glu His Cys
1205 1210 1215
Lys Val Ala Leu Arg Ala Arg Gln Ile Leu Ile Ala Ser His Leu
1220 1225 1230
Pro Ser Tyr Glu Leu Arg His Asn Gln Val Glu Ser Ile Phe Leu
1235 1240 1245
Ser Ala Ile Asp Met Tyr Gly His Gln Phe Cys Pro Glu Asn Leu
1250 1255 1260
Lys Lys Leu Ile Leu Ser Glu Thr Thr Ile Phe Asp Val Leu Pro
1265 1270 1275
Thr Phe Phe Tyr His Ala Asn Lys Val Val Cys Met Ala Ser Leu
1280 1285 1290
Glu Val Tyr Val Arg Arg Gly Tyr Ile Ala Tyr Glu Leu Asn Ser
1295 1300 1305
Leu Gln His Arg Gln Leu Pro Asp Gly Thr Cys Val Val Glu Phe
1310 1315 1320
Gln Phe Met Leu Pro Ser Ser His Pro Asn Arg Met Thr Val Pro
1325 1330 1335
Ile Ser Ile Thr Asn Pro Asp Leu Leu Arg His Ser Thr Glu Leu
1340 1345 1350
Phe Met Asp Ser Gly Phe Ser Pro Leu Cys Gln Arg Met Gly Ala
1355 1360 1365
Met Val Ala Phe Arg Arg Phe Glu Asp Phe Thr Arg Asn Phe Asp
1370 1375 1380
Glu Val Ile Ser Cys Phe Ala Asn Val Pro Lys Asp Thr Pro Leu
1385 1390 1395
Phe Ser Glu Ala Arg Thr Ser Leu Tyr Ser Glu Asp Asp Cys Lys
1400 1405 1410
Ser Leu Arg Glu Glu Pro Ile His Ile Leu Asn Val Ser Ile Gln
1415 1420 1425
Cys Ala Asp His Leu Glu Asp Glu Ala Leu Val Pro Ile Leu Arg
1430 1435 1440
Thr Phe Val Gln Ser Lys Lys Asn Ile Leu Val Asp Tyr Gly Leu
1445 1450 1455
Arg Arg Ile Thr Phe Leu Ile Ala Gln Glu Lys Glu Phe Pro Lys
1460 1465 1470
Phe Phe Thr Phe Arg Ala Arg Asp Glu Phe Ala Glu Asp Arg Ile
1475 1480 1485
Tyr Arg His Leu Glu Pro Ala Leu Ala Phe Gln Leu Glu Leu Asn
1490 1495 1500
Arg Met Arg Asn Phe Asp Leu Thr Ala Val Pro Cys Ala Asn His
1505 1510 1515
Lys Met His Leu Tyr Leu Gly Ala Ala Lys Val Lys Glu Gly Val
1520 1525 1530
Glu Val Thr Asp His Arg Phe Phe Ile Arg Ala Ile Ile Arg His
1535 1540 1545
Ser Asp Leu Ile Thr Lys Glu Ala Ser Phe Glu Tyr Leu Gln Asn
1550 1555 1560
Glu Gly Glu Arg Leu Leu Leu Glu Ala Met Asp Glu Leu Glu Val
1565 1570 1575
Ala Phe Asn Asn Thr Ser Val Arg Thr Asp Cys Asn His Ile Phe
1580 1585 1590
Leu Asn Phe Val Pro Thr Val Ile Met Asp Pro Phe Lys Ile Glu
1595 1600 1605
Glu Ser Val Arg Tyr Met Val Met Arg Tyr Gly Ser Arg Leu Trp
1610 1615 1620
Lys Leu Arg Val Leu Gln Ala Glu Val Lys Ile Asn Ile Arg Gln
1625 1630 1635
Thr Thr Thr Gly Ser Ala Val Pro Ile Arg Leu Phe Ile Thr Asn
1640 1645 1650
Glu Ser Gly Tyr Tyr Leu Asp Ile Ser Leu Tyr Lys Glu Val Thr
1655 1660 1665
Asp Ser Arg Ser Gly Asn Ile Met Phe His Ser Phe Gly Asn Lys
1670 1675 1680
Gln Gly Pro Gln His Gly Met Leu Ile Asn Thr Pro Tyr Val Thr
1685 1690 1695
Lys Asp Leu Leu Gln Ala Lys Arg Phe Gln Ala Gln Thr Leu Gly
1700 1705 1710
Thr Thr Tyr Ile Tyr Asp Phe Pro Glu Met Phe Arg Gln Ala Leu
1715 1720 1725
Phe Lys Leu Trp Gly Ser Pro Asp Lys Tyr Pro Lys Asp Ile Leu
1730 1735 1740
Thr Tyr Thr Glu Leu Val Leu Asp Ser Gln Gly Gln Leu Val Glu
1745 1750 1755
Met Asn Arg Leu Pro Gly Gly Asn Glu Val Gly Met Val Ala Phe
1760 1765 1770
Lys Met Arg Phe Lys Thr Gln Glu Tyr Pro Glu Gly Arg Asp Val
1775 1780 1785
Ile Val Ile Gly Asn Asp Ile Thr Phe Arg Ile Gly Ser Phe Gly
1790 1795 1800
Pro Gly Glu Asp Leu Leu Tyr Leu Arg Ala Ser Glu Met Ala Arg
1805 1810 1815
Ala Glu Gly Ile Pro Lys Ile Tyr Val Ala Ala Asn Ser Gly Ala
1820 1825 1830
Arg Ile Gly Met Ala Glu Glu Ile Lys His Met Phe His Val Ala
1835 1840 1845
Trp Val Asp Pro Glu Asp Pro His Lys Gly Phe Lys Tyr Leu Tyr
1850 1855 1860
Leu Thr Pro Gln Asp Tyr Thr Arg Ile Ser Ser Leu Asn Ser Val
1865 1870 1875
His Cys Lys His Ile Glu Glu Gly Gly Glu Ser Arg Tyr Met Ile
1880 1885 1890
Thr Asp Ile Ile Gly Lys Asp Asp Gly Leu Gly Val Glu Asn Leu
1895 1900 1905
Arg Gly Ser Gly Met Ile Ala Gly Glu Ser Ser Leu Ala Tyr Glu
1910 1915 1920
Glu Ile Val Thr Ile Ser Leu Val Thr Cys Arg Ala Ile Gly Ile
1925 1930 1935
Gly Ala Tyr Leu Val Arg Leu Gly Gln Arg Val Ile Gln Val Glu
1940 1945 1950
Asn Ser His Ile Ile Leu Thr Gly Ala Ser Ala Leu Asn Lys Val
1955 1960 1965
Leu Gly Arg Glu Val Tyr Thr Ser Asn Asn Gln Leu Gly Gly Val
1970 1975 1980
Gln Ile Met His Tyr Asn Gly Val Ser His Ile Thr Val Pro Asp
1985 1990 1995
Asp Phe Glu Gly Val Tyr Thr Ile Leu Glu Trp Leu Ser Tyr Met
2000 2005 2010
Pro Lys Asp Asn His Ser Pro Val Pro Ile Ile Thr Pro Thr Asp
2015 2020 2025
Pro Ile Asp Arg Glu Ile Glu Phe Leu Pro Ser Arg Ala Pro Tyr
2030 2035 2040
Asp Pro Arg Trp Met Leu Ala Gly Arg Pro His Pro Thr Leu Lys
2045 2050 2055
Gly Thr Trp Gln Ser Gly Phe Phe Asp His Gly Ser Phe Lys Glu
2060 2065 2070
Ile Met Ala Pro Trp Ala Gln Thr Val Val Thr Gly Arg Ala Arg
2075 2080 2085
Leu Gly Gly Ile Pro Val Gly Val Ile Ala Val Glu Thr Arg Thr
2090 2095 2100
Val Glu Val Ala Val Pro Ala Asp Pro Ala Asn Leu Asp Ser Glu
2105 2110 2115
Ala Lys Ile Ile Gln Gln Ala Gly Gln Val Trp Phe Pro Asp Ser
2120 2125 2130
Ala Tyr Lys Thr Ala Gln Ala Ile Lys Asp Phe Asn Arg Glu Lys
2135 2140 2145
Leu Pro Leu Met Ile Phe Ala Asn Trp Arg Gly Phe Ser Gly Gly
2150 2155 2160
Met Lys Asp Met Tyr Asp Gln Val Leu Lys Phe Gly Ala Tyr Ile
2165 2170 2175
Val Asp Gly Leu Arg Gln Tyr Lys Gln Pro Ile Leu Ile Tyr Ile
2180 2185 2190
Pro Pro Tyr Ala Glu Leu Arg Gly Gly Ser Trp Val Val Ile Asp
2195 2200 2205
Ala Thr Ile Asn Pro Leu Cys Ile Glu Met Tyr Ala Asp Lys Glu
2210 2215 2220
Ser Arg Gly Gly Val Leu Glu Pro Glu Gly Thr Val Glu Ile Lys
2225 2230 2235
Phe Arg Lys Lys Asp Leu Ile Lys Ser Met Arg Arg Ile Asp Pro
2240 2245 2250
Ala Tyr Lys Lys Leu Met Glu Gln Leu Gly Glu Pro Asp Leu Ser
2255 2260 2265
Asp Lys Asp Arg Lys Asp Leu Glu Gly Arg Leu Lys Ala Arg Glu
2270 2275 2280
Asp Leu Leu Leu Pro Ile Tyr His Gln Val Ala Val Gln Phe Ala
2285 2290 2295
Asp Phe His Asp Thr Pro Gly Arg Met Leu Glu Lys Gly Val Ile
2300 2305 2310
Ser Asp Ile Leu Glu Trp Lys Thr Ala Arg Thr Phe Leu Tyr Trp
2315 2320 2325
Arg Leu Arg Arg Leu Leu Leu Glu Asp Gln Val Lys Gln Glu Ile
2330 2335 2340
Leu Gln Ala Ser Gly Glu Leu Ser His Val His Ile Gln Ser Met
2345 2350 2355
Leu Arg Arg Trp Phe Val Glu Thr Glu Gly Ala Val Lys Ala Tyr
2360 2365 2370
Leu Trp Asp Asn Asn Gln Val Val Val Gln Trp Leu Glu Gln His
2375 2380 2385
Trp Gln Ala Gly Asp Gly Pro Arg Ser Thr Ile Arg Glu Asn Ile
2390 2395 2400
Thr Tyr Leu Lys His Asp Ser Val Leu Lys Thr Ile Arg Gly Leu
2405 2410 2415
Val Glu Glu Asn Pro Glu Val Ala Val Asp Cys Val Ile Tyr Leu
2420 2425 2430
Ser Gln His Ile Ser Pro Ala Glu Arg Ala Gln Val Val His Leu
2435 2440 2445
Leu Ser Thr Met Asp Ser Pro Ala Ser Thr
2450 2455
| название | год | авторы | номер документа |
|---|---|---|---|
| ЛИНКЕРЫ НА ОСНОВЕ СУЛЬФОМАЛЕИМИДА И СООТВЕТСТВУЮЩИЕ КОНЪЮГАТЫ | 2019 |
|
RU2815199C2 |
| КОНЪЮГАТЫ АНТИТЕЛА И ЛЕКАРСТВЕННОГО СРЕДСТВА (ADC) И КОНЪЮГАТЫ АНТИТЕЛА И ПРОЛЕКАРСТВА (APDC), СОДЕРЖАЩИЕ ФЕРМЕНТАТИВНО РАСЩЕПЛЯЕМЫЕ ГРУППЫ | 2016 |
|
RU2751512C2 |
| 1-(4-АМИНО-5-БРОМ-6-(1H-ПИРАЗОЛ-1-ИЛ)ПИРИМИДИН-2-ИЛ)-1H-ПИРАЗОЛ-4-ОЛ И ЕГО ПРИМЕНЕНИЕ В ЛЕЧЕНИИ РАКА | 2018 |
|
RU2791531C2 |
| ПРОИЗВОДНЫЕ 3-(5-АМИНО-1-ОКСОИЗОИНДОЛИН-2-ИЛ)ПИПЕРИДИН-2,6-ДИОНА И ИХ ПРИМЕНЕНИЕ В ЛЕЧЕНИИ ЗАБОЛЕВАНИЙ, СВЯЗАННЫХ С БЕЛКОМ С "ЦИНКОВЫМИ ПАЛЬЦАМИ" 2 СЕМЕЙСТВА IKAROS (IKZF2) | 2019 |
|
RU2815714C2 |
| ПРОИЗВОДНЫЕ МАЙТАНЗИНОИДА С САМОРАСЩЕПЛЯЮЩИМИСЯ ПЕПТИДНЫМИ ЛИНКЕРАМИ И ИХ КОНЪЮГАТЫ | 2018 |
|
RU2765098C2 |
| КОМПОЗИЦИЯ ДЛЯ ЛЕЧЕНИЯ РАКА, ЭКСПРЕССИРУЮЩЕГО IGF-1R | 2016 |
|
RU2728568C2 |
| ПРОИЗВОДНЫЕ 3-(5-ГИДРОКСИ-1-ОКСОИЗОИНДОЛИН-2-ИЛ) ПИПЕРИДИН-2,6-ДИОНА И ИХ ПРИМЕНЕНИЕ В ЛЕЧЕНИИ ЗАБОЛЕВАНИЙ, СВЯЗАННЫХ С БЕЛКОМ С "ЦИНКОВЫМИ ПАЛЬЦАМИ" 2 (IKZF2) СЕМЕЙСТВА IKAROS | 2019 |
|
RU2797559C2 |
| СПОСОБ ПРИМЕНЕНИЯ ВОССТАНОВЛЕНИЯ ДЛЯ ПОЛУЧЕНИЯ (S)-НИКОТИНА | 2022 |
|
RU2833281C2 |
| Соединения бензотиа(ди)азепина и их применение в качестве модуляторов желчных кислот | 2020 |
|
RU2838220C1 |
| КОНЪЮГАТЫ АНТИТЕЛА И ЛЕКАРСТВЕННОГО СРЕДСТВА ДЛЯ РАЗРУШЕНИЯ ГЕМОПОЭТИЧЕСКИХ СТВОЛОВЫХ КЛЕТОК | 2017 |
|
RU2781444C2 |










Изобретение относится к соединению формулы (I) или его фармацевтически приемлемой соли, где G представляет собой группу формулы (II) или (III), R1 представляет собой изопропил или трет-бутил; R2 представляет собой нафтил, хинолинил или изохинолинил, R2 возможно замещен независимо одной-двумя группами метил, метокси или хлоро; R3 представляет собой водород. Также изобретение относится к фармацевтической композиции, содержащей соединение формулы (I). Технический результат - соединения формулы (I) в качестве ингибиторов ацетил-КоА-карбоксилаз(ы). 2 н. и 4 з.п. ф-лы, 10 ил., 7 табл., 125 пр.
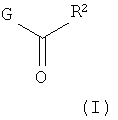
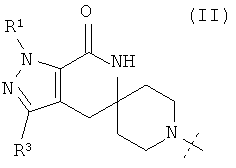
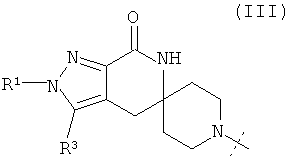
1. Соединение формулы (I)
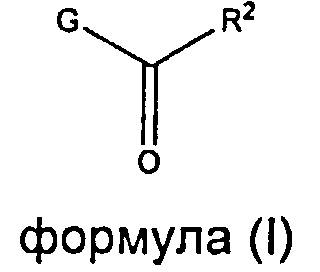
или его фармацевтически приемлемая соль, где
G представляет собой
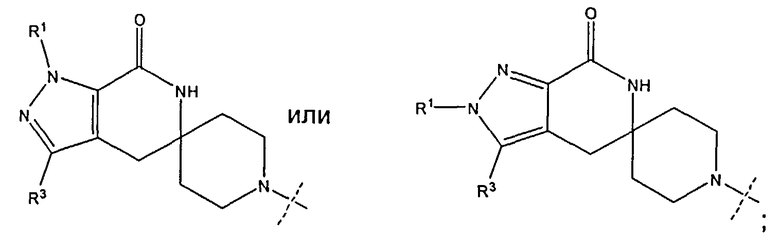
R1 представляет собой изопропил или трет-бутил;
R2 представляет собой нафтил, хинолинил или изохинолинил, причем указанный R2 возможно замещен независимо одной-двумя группами метил, метокси или хлоро; и
R3 представляет собой водород.
2. Соединение по п. 1, имеющее структуру
или его фармацевтически приемлемая соль.
3. Соединение по п. 1, имеющее структуру
или его фармацевтически приемлемая соль.
4. Соединение по п. 1, имеющее структуру
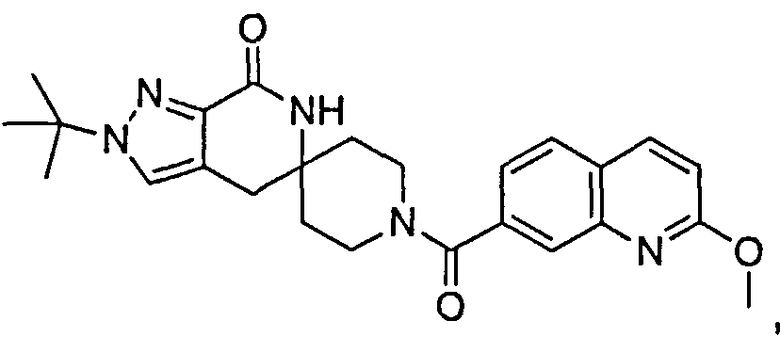
или его фармацевтически приемлемая соль.
5. Соединение по п. 1, имеющее структуру
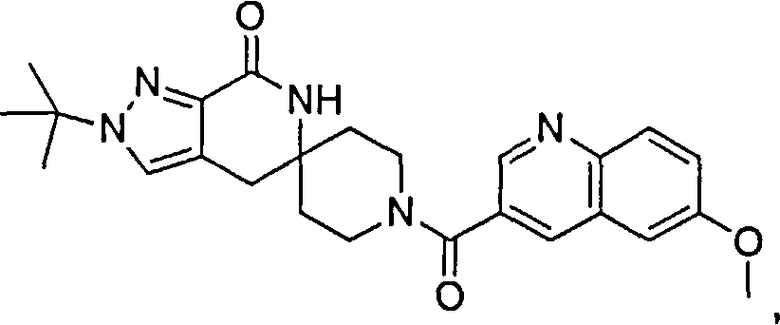
или его фармацевтически приемлемая соль.
6. Фармацевтическая композиция, обладающая ингибирующей активностью в отношении ацетил-КоА-карбоксилаз(ы), содержащая терапевтически эффективное количество соединения по любому из п.п. 1-5 или его фармацевтически приемлемой соли и фармацевтически приемлемый эксципиент, разбавитель или носитель.
| ТЕПЛООБМЕННЫЙ ЭЛЕМЕНТ | 1994 |
|
RU2123652C1 |
| Колосоуборка | 1923 |
|
SU2009A1 |
| ЕА 200900613 А1, 30.10.2009 | |||

