Изобретение относится в общем к ингибированию протеинов сгустков крови и особенно к специфическим ингибиторам фактора Xa свертывающей системы крови.
Способность образовывать сгустки крови необходима для выживания. При некоторых болезнях, однако, образование сгустков крови в системе кровообращения само по себе является источником заболеваемости. Таким образом иногда желательно предотвращать образование сгустков крови. Однако полное ингибирование свертывающей системы крови нежелательно, так как последует кровотечение, угрожающее жизни.
Для того, чтобы снизить внутрисосудистое образование сгустков крови, специалисты разработали эффективный ингибитор протромбиназы или фактора Xa, который вводится в комплекс протромбиназы, в котором он активизирует тромбин во время образования сгустков.
Соответствующие концентрации ингибитора фактора Xa повысят уровень агентов, образующих протромбиназу, требуемых для инициирования образования сгустков, но не будут вызывать ненужного пролонгирования процесса свертывания крови, как только достигается пороговая концентрация тромбина. Однако несмотря на давнее признание желательности создания такого ингибитора, в настоящее время в клинической практике не существует эффективного специфического ингибитора фактора Xa.
Во многих клинических областях существует необходимость в лечении противосвертывающими средствами.
Имеющиеся в настоящее время лекарственные средства во многих клинических областях являются неудовлетворительными. Например, почти у 50% пациентов, которые подверглись полной замене тазобедренного сустава, развивается глубокий венечный тромбоз (ГВТ).
В принятой терапии применяют фиксированную дозу низкомолекулярного гепарина (НМГ), и изменяющуюся дозу гепарина. Даже при таких схемах лечения у 10-20% пациентов развивается ГВТ и у 5-10% развиваются осложнения с кровотечением.
Другой клинической ситуацией, когда требуются более эффективные антикоагулянты, являются пластические операции на коронарных сосудах с повышенным риском инфаркта миокарда или стенокардия.
Современная общепринятая терапия, которая включает применение гепарина и аспирина, связана с 6-8% случаев закупорки сосудов в течение 24-часовой процедуры. Показатель осложнений, связанных с кровотечением, требующих переливания крови из-за применения гепарина, также составляет примерно 7%.
Более того, хотя довольно значительное количество случаев обходится без закупорки сосудов, применение гепарина после окончания процедур малоэффективно и может быть неблагоприятным.
Наиболее широко применяемыми ингибиторами свертывания крови являются гепарин и родственные сульфосодержащие полисахариды, НМГ и гепаринсульфат. Молекулы этих соединений проявляют противосвертывающие свойства путем промотирования связывания природного регулятора процесса свертывания, анти-тромбина III, с тромбином и фактором Xa. Ингибирующая активность гепарина первоначально направлена в отношении тромбина, который инактивируется примерно в 100 раз быстрее, чем фактор Ха. Хотя в сравнении с гепарином гепаринсульфат и НМГ являются более эффективными ингибиторами Xa, чем в случае тромбина, разница in vitro небольшая - (в 3-30 раз) и действие in vivo может быть непостоянным. Гирудин и гирулог являются также антикоагулянтами, специфическими в отношении тромбина, используемыми в современной клинике. Однако эти антикоагулянты, которые ингибируют тромбин, также могут вызывать кровотечение.
Доклинические испытания на бабуинах и собаках показали, что специфические ингибиторы фактора Xa предотвращают образование сгустков, не вызывая побочных кровотечений, наблюдаемых при использовании прямых ингибиторов тромбина. Такие ингибиторы фактора Xa включают, например, 2.7-бис-(4-амидинобензилиден)циклогептанон и метиловый эфир Na-тозилглицил-3-амидинофенилаланина ("TENSTOP"), эффективные ингибирующие концентрации (Ki's) которых составляют 20 нм и 800 нм соответственно, (+)-(2S)-2-(4({(3S)-1-ацетимидоил-3-пирролидинил} окси)фенил)-3-(7-амидино-2-нафтил)пропановая кислота также является представителем класса ингибиторов фактора Xa (Katakura et al., Biochem. Biophys. Res.Comm. 197:965-972 (1993)). Однако эти соединения не применяются в клинической практике.
Специфические протеиновые ингибиторы фактора Xa также были обнаружены и включают, например, антистазин (ATC) и антикоагулирующий пептид, выделенный из клеща (КАП). ATC, который выделен из пиявки Haementerin officinalis, содержит 119 аминокислот и имеет Ki для фактора Xa, равную 0,05 нм. КАП, выделенный из клеща Ornithodoros moubata, содержит 60 аминокислот, для него Ki для фактора Xa составляет примерно 0,5 нм.
Эффективность полученных рекомбинантно АТС и КАП изучалась на ряде животных. Оба ингибитора уменьшают время кровотечения по сравнению с другими антикоагулянтами и предотвращают свертывание крови при испытании на модели венечного тромбоза, вызванного тромбопластином, в лигированных яремных венах.
Результаты, полученные на этой модели, коррелируются с результатами, полученными при использовании распространенного в настоящее время лекарства - гепарина.
Было также обнаружено, что подкожное введение АТС является эффективным при испытании на модели рассеянной внутрисосудистой коагуляции (РВК), вызванной тромбопластином. КАП эффективно предотвращает артериальный тромбоз и "пониженный кровоток", вызванный хирургическим введением полиэфирного ("DACRON") имплантата, в дозах, обеспечивающих клинически приемлемое пролонгирование времени активированного частичного тромбопластина (аЧТП), т.е. менее чем в два раза. Для сравнения стандартный гепарин, даже в дозах, вызывающих пятикратное увеличение аЧТП, не предотвращает тромбоз и пониженный кровоток внутри имплантата. АЧТП является клиническим методом испытания коагуляции, который особенно чувствителен к ингибиторам тромбина.
АТС и КАП не нашли применение в клинике. Один важный недостаток этих двух ингибиторов состоит в том, что введение требуемых повторяющихся доз вызывает образование нейтрализующих антител, ограничивая тем самым их клиническое использование.
Более того, размеры КАП и АТС делают оральное введение невозможным, ограничивая число пациентов, способных получить пользу от этих агентов.
Специфический ингибитор фактора Xa будет иметь практическое значение в медицине. В частности ингибитор фактора Xa будет эффективен в обстоятельствах, когда применяемые гепарин и сульфосодержащие полисахариды являются неэффективными или слабоэффективными. Таким образом существует необходимость в создании специфического низкомолекулярного ингибитора фактора Xa, предотвращающего образование сгустков крови, который является эффективным, но не вызывает нежелательных побочных эффектов.
Данное изобретение удовлетворяет эту потребность и обеспечивает нужные преимущества.
Данное изобретение относится к соединениям, которые специфически ингибируют активность фактора Xa.
Соединение по изобретению имеет структуру X1-Y-I-R-X2, где X1 означает водород (Н), ацил, алкил или арилалкил, или одну или несколько аминокислот, и X2 означает модифицированную С-концевую последовательность, одну или несколько групп, защищающих карбоксигруппу (см. ниже), одну или несколько аминокислот или другие заместители, и Y, I и R относятся к аминокислотам - тирозину, изолейцину и аргинину, соответственно, и к пептидомиметическим или органическим структурам, обладающим такой же функциональной активностью, что и тирозин, изолейцин и аргинин, соответственно. Кроме того соединение по изобретению имеет структуру A1-A2-(A3)m-B, как указано в этом описании.
Соединение по изобретению может быть линейным или циклическим, содержащим от примерно 2 до примерно 43 остатков по длине молекулы и модифицированным по N-содержащим и/или С-содержащим концевым последовательностям. Такие соединения проявляют специфическое ингибирование активности фактора Xa с Ki<100 мкМ, предпочтительно с Ki< 2 нМ и практически не ингибируют активность других протеаз, участвующих в каскадной циркуляции. Конкретные примеры таких соединений включают Ac-Tyr-Ile-Arq-Leu-Ala-NH2; Ac-Tyr-Ile-Arg-Leu-Pro-NH2; Ac-(iBu)-Tyr-Ile-Arg-Leu-Pro-NH2; Ac-Tyr-Ile-Arg-N(CH3)O(CH3); Ac-Tyr-{ Φ (CH2NH)} -Ile-Arg-Leu-Pro-NH2 где Φ означает псевдопептидную связь, которая, например, может быть восстанавливающей связью, как указанная "CH2NH)"; псевдопептидные связи указаны символом "Ф", заключенным в скобки, "{Φ}"; Ac-Tyr-Ile-Arg-NH-CH2(4-пиpидил); Ac-Tyr-Ile-{ Φ (CH2NH)} -Arg-Leu-Pro-NH2; Ac-Tyr-Chg-Arg(NO2)-{ Φ(CH2NH)} -Leu-NH2; Ac-Tyr-Ile-Arg-{Φ(COCH2)}- Gly-Pro-NH2; Ac-Tyr-Ile-Dab(Nγ- C3H7N)-Leu-Ala-NH2; Ac-Tyr-Ile-PalMe(3)-NH2; Tyr-Ile-Arg-NH2; (D)-Tyr-Ile-Arg-Leu-Pro-NH2; Ac-(Bzl)Gly-(CHx)Gly-(3-гуанидопропил)Cly-NH2; Cyclo(Gly-Tyr-Ile-Arg-Gly); Tfa-(iBu)Tyr-Chg-Arg-Leu-Pro-NH2; Ac-pAph-Chg-Arg-Leu-Pro-NH2; Ac-Nal(2)-Chg-Arg-Leu-Pro-NH2; Ac-pAph-Chg-Palme(3)-NH2; и их фармацевтически приемлемые соли и производные с концевыми C-содержащими группами, например, амиды, сложные эфиры, спирты и альдегиды (см. также таблицу 5).
Изобретение предусматривает методы специфического ингибирования активности фактора Xa и ингибирования свертываемости крови у отдельной особи. Предусмотрены также методы определения уровней фактора Xa или активности.
На фиг. 1 представлена схематическая диаграмма каскада коагуляции крови.
На фиг. 2 приведен пример структуры соединения по изобретению.
На фиг. 3 А, Б показаны схемы реакций получения некоторых соединений по изобретению.
Коагуляция крови является сложным процессом, включающим ряд прогрессивно-распространяющихся реакций активизации ферментов, в которых зимогены плазмы последовательно активируются ограниченным протеолизом. Механически каскадная циркуляция крови разделена на внутренний и внешний циклы, которые сходятся при активации фактора Xа; последующее образование тромбина протекает по одному общему циклу (см. фиг. 1).
Настоящее исследование предполагает, что внутренний цикл играет важную роль в поддержании и росте образования фибрина, в то время как внешний цикл является критическим в фазе инициирования коагуляции крови. Общепринято считать, что коагуляция крови физически инициируется после образования комплекса тканевый фактор/фактор VIIa. Сразу же после образования этот комплекс быстро инициирует коагуляцию путем активирования факторов IX и X. Вновь образовавшийся фактор Xa затем образует 1:1 комплекс с фактором Va и фосфолипидами с образованием комплекса протромбиназы, который является ответственным за превращение растворимого фибриногена в нерастворимый фибрин. По мере прохождения времени активность комплекса фактор VIIa/тканевый фактор (внешний цикл) подавляется протеином ингибитора протеазы типа Kunitz, TFPI, который, когда образует комплекс с фактором Xa, может непосредственно ингибировать протеолитическую активность комплекса фактор Vlla/тканевый фактор.
Для того, чтобы поддержать процесс коагуляции в присутствии ингибированной внешней системы, продуцируется дополнительно фактор Xa благодаря активности внутреннего цикла, опосредствованный через тромбин. Таким образом тромбин играет двойную автокаталитическую роль, осуществляя свое собственное продуцирование и превращение фибриногена в фибрин.
Автокаталитическая природа образования тромбина является важной гарантией против нерегулируемого кровотечения и обеспечивает то, что, по достижении данной пороговой концентрации протромбиназы, коагуляция крови будет происходить до конца, приводя, например, к окончанию кровотечения.
Таким образом наиболее желательно создание агентов, которые ингибируют коагуляцию без непосредственного ингибирования тромбина.
Данное изобретение обеспечивает получение YIR пептидов, которые являются соединениями, ингибирующими активность фактора Xa, и практически не ингибирующими активность других протеаз, участвующих в процессе коагуляции крови.
Используемый здесь термин "соединение" или "YIR пептид" относится к неприродным Tyr-Ile-Arg (YIR) пептиду и его аналогам и миметикам, которые могут ингибировать активность фактора Xa.
YIR последовательность сама называется в данном описании "YIR фрагментом" и состоит из трипептида тирозин-изолеицин-аргинин или его функционального эквивалента, например, pAph-Chg-PalMe(3), pAph-Chg-PalMe(3)-NH2 и pAph-Chg-AMP(4) (см. сокращения в таблице 1).
Такие соединения по изобретению содержат, по меньшей мере, один YIR фрагмент или его функциональный эквивалент и способны специфически ингибировать активность фактора Xa. Для удобства термин "соединение" и "YIR пептид" используются здесь широко для обозначения пептидов по изобретению, включая функциональные эквиваленты, такие как аналоги пептидов, миметические пептиды и синтетические органические соединения. Функциональный эквивалент YIR пептида по изобретению может быть охарактеризован частично как имеющий структуру, описанную в данном описании, и величиной Ki 100 мкМ для ингибирования активности фактора Xa (см. пример XXXVII).
Пептидные аналоги YIR пептида согласно изобретению включают, например, пептиды, содержащие неприродные аминокислоты или химически модифицированные аминокислоты, при условии, что соединение сохраняет ингибирующую активность в отношении фактора Xa (например, см. таблицу 2). Точно также миметические пептиды представляют неаминокислотные химические структуры, которые имитируют структуру YIR пептида по изобретению и сохраняют ингибирующую активность в отношении фактора Xa. Такие миметики в общем характеризуются как проявляющие похожие физические свойства, такие как размер, заряд или гидрофобность, которые представлены в соответствующей пространственной ориентации, как найдено в противоионе обычного YIR пептида. Конкретным примером миметического пептида является соединение, в котором амидная связь между одной или несколькими аминокислотами замещена, например, связью углерод-углерод или другой связью, как это хорошо известно из уровня техники (см., например. Sawyer in Peptide Based Drug Design p.p. 387-422 (ACS, Washington DC 1995). Таким образом, изобретение обеспечивает получение соединений, ингибирующих фактор Xa, имеющих структуру A1-A2-(A3)m-B, где m = 0 или 1, описанных ниже. Примеры таких пептидов, которые могут быть миметическими, приведены в данном описании.
Термин "аминокислота" используется в самом широком смысле для обозначения двадцати природных аминокислот, которые транслированы из генетического кода и состоят из блоков протеинов, включая, если иначе не оговаривается, L-аминокислоты и D-аминокислоты, а также химически модифицированные аминокислоты, такие как аминокислотные аналоги, природные аминокислоты, которые обычно не вводятся в протеины, например, норлейцин и химические синтетические соединения, имеющие свойства, характерные для аминокислот. Например, аналоги или миметики фенилаланина или пролина, которые допускают то же конформационное ограничение пептидов, как природные Phe или Pro, включены в определение "аминокислоты" и хорошо известны специалистам.
Такие аналоги и миметики называются в данном описании "функциональными эквивалентами" аминокислоты. Другие примеры аминокислоты и их аналогов приведены Roberts и Vollaccio (The Peptides: Analysis, Sunthesis, Biology, Eds Grass and Meienhofer, Vol.5, p.341, Academic Press, Inc., N.Y. 1983). Сокращения аминокислот, аналогов аминокислот и миметиков приведены в таблице 1.
Используемый в данном описании термин "активность фактора Xa" относится к способности фактора Xa, самого по себе или в совокупности подъединиц, известный как комплекс протромбиназы, катализировать превращение протромбина в тромбин. При использовании в отношении активности фактора Xa термин "ингибирование" означает как прямое, так и косвенное ингибирование активности фактора Xa. Прямое ингибирование активности фактора Xa можно осуществить, например, путем связывания YIR пептида по изобретению с фактором Xa или протромбиназой с тем, чтобы предотвратить связывание протромбина активным центром комплекса протромбиназы. Косвенное ингибирование активности фактора Xa можно осуществить, например, путем связывания соединения по изобретению с растворимым фактором Xa для того, чтобы предотвратить его вхождение в протромбиназный комплекс.
Используемый в отношении ингибирования активности фактора Xa термин "специфическое" означает, что YIR пептид может ингибировать активность фактора Xa, не оказывая ингибирующего действия на активность других указанных протеаз, включая плазмин и тромбин (при той же концентрации ингибитора). Такие протеазы участвуют в процессе коагуляции крови и каскадном фибринолизе (см. таблицу 2; см. также пример XXVII).
Результаты, приведенные в таблице 2, показывают, что YIR пептиды по изобретению пригодны в качестве ингибиторов фактора Xa, но практически не ингибируют активность других сериновых протеаз, таких как тромбин или плазмин, которые участвуют в процессе коагуляции крови и фибринолиза.
Используемый здесь термин "заместитель" относится к любой из различных химических групп, которая введена в основную цепь пептида или боковую цепь пептида, аналога пептида, миметика или органического соединения, описанных в данном описании. Заместитель может быть любым, известным специалистам (см., например, Giannis and Kolter, Angew. Chem. Int. Ed. Engl. 32: 1244-1267 (1993)). В данном описании приведены многочисленные примеры, показывающие различные заместители, включая, например, введение pNH2 заместителем в фенилаланин с получением F(pNH2) и введение галогена в тирозин с получением, например, Y(3-1) или Y(3,5-1). Кроме того заместитель может быть, например, гетероатомом, таким как азот (N; см., например. Pal), кислород (O; см., например, O-метилтирозин) или сера (S; см., например, Тyr(SO3H)), которые могут содержать заместитель. Таким образом, N-, S-, или O-содержащий фрагмент, такой как -SO3H, рассматривается как заместитель.
Кроме того, заместитель может быть группой, защищающей амногруппу или карбоксильную группу.
Используемый здесь термин "алкил" применяется в самом широком смысле для обозначения насыщенных или ненасыщенных, линейных, разветвленных или циклических цепей с примерно 1-13 атомами C. Так, термин "алкил" включает, например, метил, этил, н-пропил, изопропил, втор.бутил, 1-метилбутил, 2,2-диметилбутил, 2-метилпентил, 2,2-диметилпропил, н-пентил и н-гексил, алкиленовые группы, карбоциклические группы, такие как циклогексил и циклопентил, а также комбинации линейных или разветвленных цепей и карбоциклических цепей, такие как метил-циклогексил или циклопропил-метилен. Кроме того следует иметь в виду, что алкил может содержать заместитель. Точно так же термин "ацил" используется в самом широком смысле для обозначения насыщенных или ненасыщенных, линейных, разветвленных или циклических цепей с примерно 1-13 атомами углерода, которые содержат карбоксильную группу. Так, термин "ацил" охватывает, например, такие группы, как формил, ацетил, бензоил и т.п.
Термин "арил" относится к ароматическим группам, содержащим 5-13 атомов углерода и, по меньшей мере, одно "кольцо", имеющее сопряженную систему пи-электронов. Примерами арилов являются, например, гетероциклические арильные группы, диарильные группы, их аналоги и производные, которые могут содержать один или несколько заместителей.
Термин "арилалкил" относится к алкилу, определенному выше, замещенному арильной группой. Подходящие арилалкильные группы включают бензил, пиколил и т.п., причем все они могут быть замещенными.
Термин "гетероалкил", "гетероарилалкил" и "гетероарил" также используются в данном описании и относятся к алкилу, арилалкилу и арилу, соответственно, который замещен одним или несколькими гетероатомами, такими как атомы N, О или S. Кроме того термин "гетероциклический" используется в отношении циклической алкильной или арильной группы, которая замещена одним или несколькими гетероатомами. Многочисленные примеры гетероалкилов, гетероарилалкилов, гетероарилов и гетероциклов приведены, например, в таблицах 1 и 3, а также известны из уровня техники.
Пептиды по изобретению могут быть модифицированы по концевым N- или C-содержащим последовательностям с использованием группы, замещающей аминогруппу или карбоксильную группу, соответственно.
Многочисленные примеры таких модификаций приведены в данном описании (см., например. таблицу 3). N-содержащая концевая группа пептида или пептидного аналога может быть химически модифицирована таким образом, что концевая аминогруппа замещена, например, ацетильной, циклопентилкарбоксильной, изохинолилкарбоксильной, фурильной, тозильной, пиразинкарбоксильной или другой подобной группой, которая может быть замещена заместителем, как описано выше. Концевая аминогруппа может быть также замещена, например, обратимой амидной связью. Следует иметь в виду, что термин "аминогруппа" применяется здесь в широком смысле для обозначения любой свободной аминогруппы, включая первичную, вторичную или третичную аминогруппу, имеющуюся в пептиде. По сравнению с этим "N-концевая" последовательность означает альфа-аминогруппу первой аминокислоты, содержащейся в пептиде, формула которого написана общепринятым образом.
N-концевая группа пептида по изобретению может быть защищена присоединением к ней группы, защищающей аминогруппу. Термин "защищающая аминогруппу" используется здесь в широком смысле для обозначения химической группы, которая может реагировать со свободной аминогруппой, включая, например, альфа-аминогруппу, находящуюся на конце молекулы пептида по изобретению. Взаимодействуя с последней, защищающая группа защищает реакционноспособную в других условиях аминогруппу от нежелательных реакций, которые могут происходить, например, в процессе синтеза или вследствие активности экзопептидазы у конечного соединения. Модификация аминогруппы также может обеспечить дополнительные преимущества, включая, например, увеличение растворимости или активности соединения. В данном описании описаны различные группы, защищающие аминогруппу (см. таблицу 3), а также они известны из уровня техники и включают, например, ацильные группы, такие как ацетил, пиколил, терт. бутилацетил, терт. бутилоксикарбонил, бензилоксикарбонил, бензоил, включая, например, бензилоксим, такой как 2-арил-2-O-бензилоксим (см. пример XVI), а также аминоацильный остаток, который сам может быть модифицирован группой, защищающей аминогруппу. Другие группы, защищающие аминогруппу, описаны, например, в The Peptides, eds.Gross and Meinhofer, Vol.3 (Academic Press, Inc. , N. Y. , 1981); и Green and Wuts, "Protective Groups in organic Synthesis, 2-nd ed., p.p. 309-405 (John Wiley and Sons, New York (1991)). Продукт любой такой модификации концевой аминогруппы пептида или аналога пептида по изобретению называется в данном описании "N-концевое производное".
Подобным образом карбоксильная группа, такая, как содержащая в качестве C-концевой в пептиде, может быть химически модифицирована с использованием группы, защищающей карбоксигруппу. Термин "карбоксильная группа" и "C-концевая" используются по аналогиии с терминами "аминогруппа" и "N-концевая", указанными выше.
Карбоксильная группа, такая как содержащаяся в C-концевой группе пептида, может быть модифицирована восстановлением C-концевой карбоксигруппы до спирта или альдегида или путем образования стоматического сложного эфира или замещением карбоксигруппы заместителем, таким как тиазолил, циклогексил, или другой группой. Стоматические эфиры хорошо известны и включают, например, алкоксиметильные группы, такие как метоксиметил, этоксиметил, изопропоксиметил и т.п., альфа-(C1-C4)алкоксиэтилы, такие как метоксиэтил, этоксиэтил, пропоксиэтил, изопропоксиэтил и т. п.; 2-оксо-1,3-диоксилен-4-илметильные группы, такие как 5-метил-2-оксо-1,3-диоксилен-4-илметил и т.п.; C1-C3-алкилтиометилы, такие как метилтиометил, этилтиометил, изопропилтиометил и т.п.; ацилоксиметильные группы, такие как пивалилоксиметил, альфа-ацетоксиметил и т. п.; этоксикарбонил-1-метильную группу; альфа-ацилокси-альфа-замещенные метильные группы, такие как альфа-ацетоксиэтил, 3-фталидил или 5,6-диметилфталидил, 1-(C1-C4-алкилоксикарбонилокси)эт-1-ильные группы, такие как 1-(этоксикарбонилокси)эт-1-ил; и 1-(C1-C4-алкиламинокарбонилокси)эт-1-ильную группу, такую как 1-(метиламинокарбонилокси)эт-1-ил.
Пептид по изобретению может быть модифицирован присоединением группы, защищающей карбоксильную. Защищающие карбоксильные группы хорошо известны и, будучи присоединены к пептиду, защищают карбоксигруппу против нежелательных реакций (см., например, Greene and Wuts, supra, p.p. 224-276 (1991)).
Специалисту известно, что такие модификации, как описаны выше, которые могут осуществляться с N-концевой аминогруппой или C-концевой карбоксильной группой пептида, подобным образом можно осуществлять с любой имеющейся реакционноспособной аминогруппой или карбоксильной группой, например, в боковой цепи аминокислоты или аналога аминокислоты в пептиде по изобретению. Методы осуществления таких модификаций описаны ниже, а также известны из уровня техники.
Данное изобретение обеспечивает получение соединений, которые специфически ингибируют активность фактора Xa. Соединение по изобретению имеет общую структуру X1-YIR-X2 или является ее функциональным эквивалентом, где X1 означает H, ацил, алкил, арилалкил или одну или несколько аминокислот, и X2 означает модифицированную C-концевую группу, одну или несколько групп, защищающих карбоксигруппу, или одну или несколько аминокислот или другой заместитель, такой как группа, защищающая аминогруппу.
Соединение по изобретению полезно в качестве антикоагулянта в терапевтическом лечении различных клинических заболеваний. Соединение по изобретению также пригодно для различных лабораторных анализов для предотвращения свертывания образцов крови.
Изобретение также предусматривает соединение, которое специфически ингибирует активность фактора Xa и имеет общую формулу A1-A2-(A3)m-B, где m = 0 или 1 и A1 означает R1-R2-R3, A2 означает R4-R5-R6 и A3 означает R7-R8-R9; где R1 выбран из группы, состоящей из 1-20 аминокислот;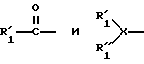
X выбран из группы, состоящей N, CH и NC(O), и R'1, R''1 выбраны из группы, состоящей из алкила, ацила, арила, арилалкила и групп, защищающих аминогруппу и где R1 может быть замещенным; R2 означает -CR99R100, где R99 и R100 независимо выбраны из группы, включающей H, алкил, арилалкил, гетероарилалкил и гетероарил, и где R99 и R100 независимо могут быть замещенными; R3 выбран из группы, включающей -С(О)-, -CH2-, -CHR99-C(O)- и -С(О)-NR35-CH2-C(О)-, где R35 означает CHR55 мостиковой группы - C(O)-CR55-; R4 выбран из группы, включающей -CH2- и -NR50-, где R50 выбран из группы, включающей H, алкил, арилалкил, и гетероцикл; R5 означает = CR201R202, где R201 и R202 независимо выбраны из группы, включающей H, алкил, арил и арилалкил, и где R201 и R202 независимо могут быть замещенными; R6 означает заместитель, выбранный из группы, включающей -С(O), -CH2- и -CHR99-C(O)-; R7 выбран из группы, включающей -CH2- и -NR51, где R51 означает H, алкил, арилалкил, гетероалкил и гетероарилалкил, и любой из этих радикалов замещен заместителем, выбранным из группы, включающей Q и -(CH2)n-Q, где n = 1-5 и Q выбран из группы, включающей аминную, амидиновую, имидазольную и гуанидиновую группы, которые могут быть замещенными, и моно-, ди-, три- или тетраалкиламмоний фармацевтически приемлемой соли, ее изоуреид или изотиоуреид; R8 означает -CR210R211-, где R210 и R211 независимо выбраны из группы, включающей H, алкил, алкиларил и гетероцикл, причем любой из этих радикалов может быть замещен заместителем, выбранным из группы, включающей Q или -(CH2)n-Q, где n = 1-5 и Q выбран из группы, включающей аминную, амидиновую, имидазольную и гуанидиновую группы, которые могут быть замещенными, и моно-, ди-, три- или тетраалкиламмоний фармацевтически приемлемой соли, ее изоуреид или изотиоуреид; R9 выбран из группы, состоящей из -С(O)-, -CH2- и -CHR99-C(O); и где, когда m = 1, B выбран из группы, включающей 1-20 аминокислот, -NHR52, -NR60R61-, OR70 и -CHR60R61, где R52 выбран из группы, включающей H, алкил, арилалкил, гетероарилалкил и гетероарил; где R60 и R61 независимо выбраны из группы, включающей H, алкил, арилалкил, арил, гетероарилалкил, и где, когда m = 0, B выбран из группы, включающей 1-20 аминокислот, -OR70, -NHR52 и NR60R61, который присоединен к R6 амидной связью или сложноэфирной связью; где В может быть замещен заместителем, при условии, что когда R3 означает -CH2- или -CHR99-C(О)-, R4 означает NR50; когда R4 означает -CH2-, R3 означает -С(О)- или - CHR99-C(O)-, R4 означает NR50; когда R4 означает -CH2-, R3 означает -С(О)- или -CHR99-C(O)-; когда R6 означает -CH2-, R7 означает -NHR51-; когда R7 означает -CH2-, R6 означает -С(О)- или -CHR99-C(O)-; когда R4 означает -NR50- и R1 означает
R50 и R'1 вместе образуют мостиковую группу формулы -C(O)-CHR55-, где CHR55 является R50 и карбонильная группа является R'1, и R''1 и R55 независимо означают H, C1-C6-алкил или арилалкил; и когда R3 означает -С(О)-NR35-CH2-C(О)-, тогда R4 означает -NR50-, R1 означает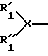
R35 и R'1 вместе образуют мостиковую группу формулы -С(О)CHR55-, где С(O) является R'1 и CHR55 является R35; R''1 и R55 независимо означают H или C1-C6-алкил (см., например, фиг. 2).
Соединение по изобретению может содержать циклическую N-концевую группу, образованную R1, R2, R3 и, если это желательно, R4. Такое соединение обозначается, например, структурой A1-A2-(A3)m-B, как описано выше, где R4 означает -NR50-, R1 означает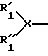
R50 и R'1 вместе образуют мостиковую группу формулы -C(O)-CHR55, где R55 означает H; R1 означает H или метил; R99 и R100 независимо выбраны из группы, включающей H, арилалкил, алкил и гетероалкил или 1-3 атома углерода, и где R99 и R100 далее могут быть соединены с фрагментом, выбранным из группы, включающей фенил, тиенил, тиазолил, пиридил, нафтил, тионафтил, индолил или насыщенный алкил, алкокси, моноалкиламино, диалкиламино, тетраалкиламмоний, арилалкиламино, аиноалкиларил, карбокси, галоид, гидрокси, амино, амидо, амидино, гуанидино, триазолил и сульфонил, и R3 выбран из группы, состоящей из -С(О)- и -C(O)-NR35-CH2-C(O)-.
Кроме того, в соединении A1-A2-(A3)m-B R'1 и R''1 могут содержать до шести заместителей, включая, например, алкил, и могут быть связаны группой, такой как -OCH2-, -SCH2-, =N-CH2-, =NC(О)-, -CO- или -NY-CO-NZ, где Y и Z могут означать H, алкил, арилалкил или гетероаралкил. Более того, R99 и R100 независимо могут быть замещены заместителем, таким как фенил, тиенил, тиазолил, пиридил, нафтил, тионафтил или индолил, или насыщенная группа, который может содержать до пяти групп, выбранных из алкила, алкокси, моно-, ди- или триалкиламина, тетраалкиламмония, арилалкиламина, аминоалкиларила, карбокси, галогенов, гидрокси, амино, амидо, амидино, гуанидино, триазолила или сульфонила.
Предпочтительным соединением с заместителями в R2-положении является соединение, где R100 означает H и R99 означает или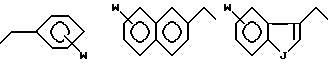
где W в замещенном соединении может быть, например, галогеном, гидроксилом, амино- или амидиновой группой, и J может означать, например, O, S или -NR, где R означает H или алкил, арил, или арилалкил.
Соединение по изобретению, которое содержит заместитель, замещенный у A2 и проявляет ингибирующую активность в отношении фактора Xa, может иметь, например, замещение у R50, R201 или R202 одним или несколькими гетероатомами, такими как N, О или S. R202 также может быть замещен заместителем, выбранным из: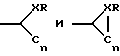
когда X означает C, N или S, R отсутствует или может означать H или алкил, который может быть замещен гетероатомом и n = 1-5.
Соединение по изобретению, которое содержит замесититель у A3 и проявляет ингибирующую активность в отношении фактора Xa, может включать, например, замещение R51 одним или несколькими заместителями, такими, как H, алкил, арилалкил или гетероцикл, возможно также замещенными гетероатомом, таким как N, O или S.
R210 и R211 могут означать, например, заместитель -(CH2)n-Q, где n равен примерно 1-5 и где Q означает амино, амидино, мочевину, имидазол, гуанидин, моно-, ди-, три- или тетраалкиламмоний фармацевтически приемлемой соли, ее изоуреид или изотиоуреид. Кроме того, R210 или R211 могут означать, например, алкил, арил или алкиларил. Эти группы так же могут быть замещены заместителем, таким как гидроксил или C1-C4-алкокси.
Соединение по изобретению может иметь альтернативное расположение заместителей, составляющих фрагмент В. Такое альтернативное расположение заместителей может включать, например, замещение R52 азотом, кислородом или серой или замещение R60, R61 или R70 одним или несколькими гетероатомами или алкильными группами.
Общие структуры, описанные здесь, представляют различные соединения по изобретению, которые сохраняют ингибирующую активность по отношению к фактору Xa, такую как присущую трипептиду YIR. К структурам, описанным здесь, относятся также соединения, содержащие неприродные аминокислоты, аминокислотные миметики и другие органические структуры и заместители, проявляющие похожую функцию.
Такие функциональные эквиваленты обеспечивают соответствующие пространственные группировки желаемых зарядов и сил, которые придают эффективную ингибирующую активность по отношению к фактору Xa.
Конкретные примеры соединений по изобретению включают, например,
Ac-Tyr-Ile-Arg-Leu-Ala-NH2; Ac-Tyr-Ile-Arg-Leu-Pro-NH2; Ac-(iBu)Tyr-Ile-Arg-Leu-Ala-NH2; Ac-Tyr-Ilе-Arg-Leu-N(CH3)O(CH3); Ac-Tyr-{Y(CH2NH)}-Ile-Arg-Leu-Pro-NH2; Ac-Tyr-Ile-Arg-NH-CH2(4-Pyridil); Ac-Tyr-Ile-{Y(CH2NH)} -Arg-Leu-Pro-NH2; Ac-Tyr-Chg-Arg(NO2)-{Y(CH2NH)}-Leu-NH2; Ac-Tyr-Ile-Arg-{ Y(COCH2)} -Gly-Pro-NH2; Ac-Tyr-Ile-Dab(Ny-C3H7N)-Leu-Ala-NH2; Ac-Tyr-Ile-PaIMe(3)-NH2; Tyr-Ile-Arg-NH2; D-Tyr-Ile-Arg-Leu-Pro-NH2, Ac-(Bzl)Gly-(Chx)Gly-(3-гуанидопропил)Gly-NH2; Цикло(Gly-Tyr-Ile-Arg-Gly); Tfa-(iBu)Tyr-Chg-Arg-Leu-Pro-NH2; Ac-pAph-Chg-Arg-Leu-Pro-NH2; и Ac-Nal(2)-Chg-Arg-Leu-Pro-NH2. Другие YIR пептиды по изобретению показаны, например, в таблицах 3 и 5.
Данное изобретение также относится к соединению со структурой A1-A2-(A3)m-B, где R1 означает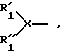
R'1 выбран из группы, включающей H, -CO-Ra, -SO2-Ra, группу, защищающую аминогруппу, 1-6 аминокислот, которые могут быть замещенными и где N-концевая группа указанных 1-6 аминокислот замещена заместителем, выбранным из группы, включающей H, -CO-Ra, -SO2-Ra и группу, защищающую аминогруппу; и где Ra выбран из группы, включающей алкил, арил и гетероалкил; R''1 выбран из группы, включающей H, ацил и алкил; X означает N; R2 означает -CHR99-, где R99 выбран из группы, включающей алкил, арил, арилалкил, гетероалкил и гетероарил, которые могут быть замещены заместителем, выбранным из группы, включающей 1-6 атомов фтора, хлора, брома, йода, 1-6 аминогрупп, нитрогрупп, амидиновых групп, амидогрупп, карбоксигрупп, сложноэфирных групп, групп простого эфира и гидроксильных групп; R3 означает -С(О)-; R4 означает -NH-; R5 означает -CHR201-, где R201 является алкилом; R6 означает -С(О)-; R7 означает -NH-; R8 означает -CHR210-; где R210 является гетероалкилом, имеющим, по меньшей мере, один формальный положительный заряд, где гетероатомом являются 1-6 атомов азота; R9 означает -С(О)-; и B выбран из группы, включающей -ORb и -N-RcRd, где Rb выбран из группы, состоящей из H, алкила и группы, защищающей карбоксигруппу, Rc выбран из группы, включающей H и алкил, и Rd выбран из группы, включающей алкил, гетероалкил и 1-20 аминокислот, которые могут быть замещены заместителем, и где C-концевая группа может быть модифицирована группой, защищающей карбоксигруппу, группой первичного амида или частью циклического пептида в качестве вторичной или третичной амидной группы, образовавшейся с аминогруппой радикала R1. Такое соединение может содержать одну или несколько групп, защищающих аминогруппы.
Например, соединение по изобретению содержит A1, выбранный из группы, включающей Tyr, F(pNH2), mAph, pAph и Nal(2), который содержит 0 или 1 групп, защищающих аминогруппы; A2 выбран из группы, состоящей из Arg, PalMe(3), Dab(Nγ-C3H7N), Dap(Nβ-C3H7N) и Orn(Nδ-C3H7N); и B выбран из группы, включающей H, ОН, NH2, одну-пять аминокислот или их функциональных эквивалентов и группу, защищающую карбоксигруппу. Примеры таких соединений включают Ac-pAph-Chg-PaIMe(3)-NH-CH2-CHx; Ac-pAph-Chg-PalMe(3)-NH-Chx; Bzf-pAph-Chg-PaIMe(3)-NH2; циклопентил-CO-pAph-Chg-PalMe(3)- NH2; 3Iqc-pAph-Chg-PaIMe(3)-NH2; 2-фуроил-pAph-Chg-PalMe(3)-NH2; 5-Ме-тиенил-CO-pAph-Chg-PalMe(3)-NH2; и Ac-pAph-Chg-PalMe (3)-ол (см. также таблицу 5).
Изобретение далее относится к соединению, имеющему структуру A1-A2-B, а именно A1-A2-(A3)m-B, где m = 0.
В таком соединении B может означать гетероарилалкил, такой как (4-(N-метилпиридиний))метил; 2-(3-(N-метилпиридиний))метил; 2-(3-(N-метилпиридиний))метил; 2-(3-(N-метилпиридиний)эт-1-ил; 1-(4-(N-метилпиридиний))-эт-1-ил; (n-амидин)бензил; 2-(4-(N-метилпиридиний))проп-1-ил; и 2-(4-(N-метилпиридиний))эт-1-ил. Ac-pAph-Chg-AMP(4) и Ac-pAph-Chg-AEMP(4) являются примерами таких соединений.
Выбор включения L- или D-аминокислоты в соединение по данному изобретению может зависеть, частично, от желаемых характеристик пептида. Например, включение одной или нескольких D-аминокислот может придать повышенную стабильность соединению in vitro или in vivo. Включение одной или нескольких D-аминокислот также может повысить или понизить фармакологическую активность соединения. В некоторых случаях может быть желательным дать соединению остаться активным только в течение короткого промежутка времени.
В таких случаях включение одной или нескольких L-аминокислот в соединение может позволить эндогенным пептидазам в организме человека усвоить соединение in vivo, тем самым ограничив действие активного соединения на организм.
Специалист может определить требуемые желаемые характеристики соединения по изобретению, принимая во внимание, например, возраст и общее состояние здоровья пациента.
Соединение по изобретению может быть синтезировано с использованием, например, автоматического синтезатора (см. пример 1).
Избирательная модификация реакционноспособной группы, такой, как группа, имеющаяся в боковой цепи аминокислоты, или N-концевая или C-концевая реакционноспособная группа в пептиде, может придать соединению желаемые характеристики, такие как повышенная растворимость или улучшенная ингибирующая активность.
Если используются методы твердофазного синтеза, можно манипулировать химическим составом соединения, когда пептид присоединяется к смоле или после того как пептид отщепился от смолы, с получением, например, производного с N-концевой группой, такого как ацилированное соединение с N-концевой группой.
Подобные модификации могут быть также осуществимы с карбоксильной группой соединения, включая C-концевую карбоксильную группу, которая может быть, например, амидирована.
Специалист может также синтезировать YIR пептид, используя метод синтеза в среде растворителя. Синтезированное соединение может быть очищено с использованием хорошо известных методов, таких как обратимая жидкостная хроматография высокого разрешения (ЖХВР, см. пример 1) или другие методы выделения, основанные, например, на использовании размера, заряда или гидрофобности соединения. Для характеристики структуры соединения по изобретению можно также использовать хорошо известные методы, такие как секвенирование аминокислот или масс-спектрометрия (MS).
YIR пептиды по изобретению могут быть линейными или циклическими (см., например. таблицу 3 ниже). Циклизацию можно проводить путем образования мостика между двумя несмежными остатками, фрагментами или заместителями, которые могут быть в составе или вне YIR фрагмента.
Циклизацию также можно проводить, например, путем образования мостика между одним из остатков в составе YIR фрагмента и несмежным остатком, фрагментом и заместителем вне YIR последовательности.
Например, пептиды или пептидомиметики могут быть циклизованы путем образования связей S-S, -CH2-S-, -CH2-О-CH-, лактамной или сложноэфирной связей или известными, ранее описанными методами (см. Hruby, Life Sci. 31: 189-199 (1982); Toniolo, Int.J.Pept.Prot.Res. 35:287-300 (1990); Kates et al., Tetr.Lett. 34:1549-1552 (1993)).
Используемое выражение "вне YIR фрагмента" означает не включающий тирозин, изолейцин или аргинин остаток YIR последовательности или его эквивалента, находящийся в YIR пептиде по изобретению.
В противоположность этому выражение "в составе YIR фрагмента" означает включающие, по меньшей мере, один остаток тирозина, изолейцина и аргинина YIR последовательность или ее эквивалент. Термин "мостик" в отношении циклического соединения означает связь, образовавшуюся между двумя несмежными аминокислотами, находящимися в YIR пептиде по изобретению.
Циклизация может быть достигнута путем образования, например, дисульфидной связи или лактамной связи между X1 и X2. Остатки, способные образовать дисульфидную связь, включают, например, Cys, Pen, Mpr и Mpp и его эквиваленты, содержащие 2-аминогруппы.
Остатки, способные образовать лактамный мостик, включают, например, Asp, Glu, Lys, Orn, альфа, бета-диаминопропионовую кислоту, альфа-аминоадипиновую кислоту, альфа, гамма-диаминомасляную кислоту, диаминоуксусную кислоту, аминобензойную кислоту и меркаптобензойную кислоту. Соединения, описанные в данном описании, могут быть циклизованы, например, через лактамную связь, которая может использовать группу в боковой цепи одного смежного остатка с образованием ковалентного присоединения к N-концевой аминогруппе X1 или Y. Альтернативные мостиковые структуры также можно использовать для циклизации соединений по изобретению, включая, например, пептиды и пептидомиметики, которые могут быть циклизованы через связи S-S, -CH2-S-, -CH2-O-CH2-, лактамные, сложноэфирные или другие связи (см., например, Hruby, Supra, 1982; Toniolo, Supra, 1990; Kates et al. Supra, 1993).
Соединение согласно данному изобретению можно получить как гомогенное или в виде смеси соединений, содержащих различные комбинации заместителей.
Большой выбор заместителей позволяет регулировать физико-химические свойства аналогов пептидного соединения.
Выбор заместителя также влияет на связывающее средство соединения (см. примеры).
Различные соединения, содержащие различные расположения заместителей, проявляют различные уровни ингибирующей активности в отношении фактора Xa. Эти соединения были синтезированы согласно методикам, описанным в примерах. Испытания пептидов на ингибирующую активность осуществляли с использованием методов, описанных в примерах XXXVII и XXXVIII. Используя такие методы, специалист может синтезировать соединение, описанное здесь, включая его модификацию, и определить ингибирующую активность по отношению к фактору Xa.
Изобретение предусматривает соединения, которые специфически ингибируют активность фактора Xa.
Такие соединения имеют Ki 100 мкм, предпочтительно, 2 нм по отношению к активности фактора Xa и практически не ингибируют активность других протеаз, участвующих в коагуляции и фибринолизе (см. таблицу 2). Такие другие протеазы включают, например, тромбин и плазмин. Специфичность соединений по изобретению показана в примере XXXVII ниже (см. также таблицу 2).
Соединение по изобретению можно применять преимущественно в качестве антикоагулянта, который может контактировать с образцом крови для предотвращения коагуляции. Например, эффективное количество соединения по изобретению может контактировать со свежевзятым образцом крови для предотвращения ее коагуляции. Используемый термин "эффективное количество", применяемый в отношении соединения по изобретению, означает количество соединения, которое ингибирует активность фактора Xa. Специалист хорошо знает, что эффективное количество соединения по изобретению можно определить, используя описанные здесь методы (см. примеры XXXVII и XXXVIII), а также известные методы.
Имея в виду описанную полезность соединения по изобретению, специалист понимает, что агент, такой как гепарин, может быть заменен соединением по изобретению. Такое применение соединения по изобретению может привести, например, к экономии средств по сравнению с другими антикоагулянтами.
Кроме того, соединение по изобретению может применяться для лечения различных клинических заболеваний, включая, например, лечение сердечно-сосудистых заболеваний и осложнений, связанных, например, с инфекцией или хирургическими операциями.
Примеры осложнений после хирургического вмешательства включают хронический и проксимальный тромбофлебит, которые могут развиться после операций. Таким образом соединение по изобретению пригодно в качестве лекарства для снижения или предотвращения нежелательной коагуляции крови у пациента.
Поскольку YIR пептид по изобретению может ингибировать активность фактора Xa, такое соединение может быть пригодным для уменьшения или ингибирования свертывания крови у индивидуума. Используемый здесь термин "индивидуум" означает позвоночное, включая млекопитающее, такое как человек, у которого фактор Xa участвует в каскадном свертывании.
Свертывание крови у индивидуума можно уменьшить или предотвратить введением индивидууму терапевтически эффективного количества YIR пептида по изобретению. Используемый термин "терапевтически эффективное количество" означает дозу YIR пептида, которую нужно ввести индивидууму для ингибирования активности фактора Xa у индивидуума. Более конкретно терапевтически эффективное количество соединения по изобретению ингибирует каталитическую активность фактора Xa или непосредственно в комплексе протромбиназы или в виде растворимой производной единицы, или косвенно путем ингибирования внедрения фактора Xa в комплекс протромбиназы. В частности такие соединения могут ингибировать активность фактора Xa при Ki< 100 мкМ и, предпочтительно, при K<2 нМ. Терапевтически эффективное количество можно определить, используя методы, описанные, например, в примерах XXXVII и XXXVIII, и другие, известные из уровня техники.
В практике терапевтического лечения по настоящему изобретению конкретная доза для получения терапевтически эффективного количества вводимой индивидууму фармацевтической композиции будет зависеть от множества факторов, включая, например, природу или степень заболевания, схему введения и возраст и физические данные индивидуума. Соответствующую дозу можно определить клиническими методами, хорошо известными в медицине. Таким образом изобретение предусматривает способ специфического ингибирования активности фактора Xa путем контактирования фактора Xa с соединением, имеющим последовательность X1-YIR-X2 или A1-A2-(A3)m-B, где m = 0 или 1, или его функциональным эквивалентам.
Изобретение далее предусматривает способ уменьшения или ингибирования образования сгустка крови у индивидуума путем введения терапевтически эффективного количества соединения по изобретению.
Соединение по изобретению обычно вводится индивидууму в виде композиции, содержащей соединение и фармацевтически приемлемый носитель.
Термин "фармацевтически приемлемый носитель" означает среду или состав, которые являются нетоксичными для индивидуума или имеют допустимую токсичность, соответствующую установленным требованиям.
Термин "фармацевтически приемлемый носитель" охватывает любой из стандартных фармацевтических носителей, таких как физиологический раствор с фосфатным буфером, вода, эмульсия типа масло/вода или вода/масло или различные типы смачивающих агентов. Подходящие фармацевтические носители и их состав описаны Martin (в Remington's Pharmaceutical Sciences, 15th ed. (Mack Publishing Co., Easton 1975).
Такие композиции в общем содержат терапевтически эффективное количество соединения по изобретению вместе с подходящим количеством носителя, чтобы получить соответствующую дозу для введения индивидууму.
Таким образом, заявленные соединения могут быть полезны как лекарства для ингибирования активности фактора Xa и свертывания крови у индивидуума.
Фармацевтически приемлемые носители могут также включать, например, другие среды, соединения или модификаторы ингибитора фактора Xa, которые улучшают его фармакологические функции. Фармацевтически приемлемая среда может включать, например, кислую соль, такую как соль, полученную с неорганической кислотой, например, хлористоводородной, бромистоводородной, фосфорной кислотой, серной кислотой или перхлорной кислотой, или с органической кислотой, например, уксусной, щавелевой, малеиновой, винной, лимонной, янтарной или малоновой кислотой. Другие фармацевтически приемлемые соли включают, например, неорганические нитрат, сульфат, ацетат, малеат, формиат, лактат, тартрат, сукцинат, цитрат, п-толуолсульфонат и т.п., включающие, не ограничивая изобретение, катионы щелочных и щелочноземельных металлов, например, натрия, лития, калия, кальция или магния, а также в качестве нетоксичного аммония четвертичный аммоний и катионы аминов, например, аммоний, метиламмоний, диметиламмоний, триметиламмоний, тетраметиламмоний, этиламмоний, триэтиламмоний и тетраэтиламмоний.
Примеры модификаций, которые усиливают фармакологическую функцию соединения, включают, например, этерификацию, например, образование C1-C6-алкиловых эфиров, предпочтительно, C1-C4-алкиловых эфиров, где алкил является линейным или разветвленным. Другие приемлемые эфиры включают, например, C5-C7-циклоалкиловые эфиры и арилалкиловые эфиры, например, бензиловые. Такие эфиры могут быть получены из соединений, описанных в данном описании с использованием обычных методов, хорошо известных в химии пептидов.
Фармацевтические приемлемые модификации могут включать, например, образование амидов пептида. Такие амидные модификации, которые можно проводить с соединениями по изобретению, включают, например, полученные с использованием аммония, первичных C1-C6-диалкиламинов, в которых алкилы являются линейными или разветвленными, или ариламинов, имеющих различные заместители. В случае вторичных аминов амин может быть в виде 5- или 6-членного гетероцикла, содержащего, например, атом азота. Способы получения таких амидов хорошо известны.
Согласно другому аспекту изобретения YIR пептид может быть использован при исследованиях для идентификации наличия фактора Xa или для выделения фактора Xa в чистом виде.
Предпочтительно, соединение по изобретению получает метку при помощи, например, радиоизотопа, и меченое соединение обнаруживают с использованием обычного метода, пригодного для обнаружения конкретной метки. Кроме того YIR пептид можно использовать в качестве пробы для обнаружения места расположения или количества активности фактора Xa in vivo, in vitro или ex vivo.
Следует иметь в виду, что модификации, которые практически не влияют на различные аспекты данного изобретения, также входят в объем этого изобретения. Соответственно нижеследующие примеры только иллюстрируют изобретение, не ограничивая его.
Пример I.
Методики синтеза пептида
Исходные материалы, используемые при синтезе, получены от химических фирм, таких как Aldrich, Sigma, Fluka, Nova Biochem и Advance Chemtech.
Во время синтеза этих соединений функциональные группы производных аминокислот были защищены блокирующими группами для предотвращения побочных реакций на стадиях сочетания. Примеры подходящих блокирующих групп и их применение описаны в The Peptides, Supra, 1981, и в Vol. 9, Udenfriend and Meienhofer, ed. 1987.
Для получения соединений по изобретению используют общий метод синтеза пептидов в твердой фазе. Такие методы описаны, например. Steward and Young (Solid Phase Peptide Synthesis (Freeman and Co, San Francisko, 1969).
Если не указано иначе, пептиды синтезируют на полистироле, сшитом 1% дивинилбензола. Для связывания с твердой подложкой используют чувствительный к кислоте линкер (линкер Rink) (Rink, Tetr Lett. 28:3787 (1987); Sieber, Tetr.Lett. 28: 2107 (1987)).
Связывание осуществляют, используя N,N'-диизопропилкарбодиимид (ДИК) в присутствии эквивалентного количества HOBt. Все реакции связывания осуществляют или в N,N-диметилформамиде (ДМФ), или 1:1 смеси ДМФ:дихлорметан при комнатной температуре (КТ) в течение 40 мин. Завершение реакции связывания определяют по пробе с нингидрином.
Деблокировку Fmoc проводят, используя 50% пиперидина в ДМФ в течение 10 мин. Количество выделившегося Fmoc определяют по абсорбции при 300 нМ раствора после деблокировки, объему промывочных растворов и весу смолы, используемой при синтезе. Второе (двойное) связывание осуществляют, пока не завершится связывание в первом случае.
При этом используют следующие цикл связывания и методы (см. табл. А).
После завершения связывания пептида со смолой осуществляют конечную деблокировку Fmoc, сопровождаемую обычными циклами промывки и определением количества Fmoc, выделившегося при деблокировке. В некоторых случаях Nальфа-незащищенный пептид ацетилируют при встряхивании пептида с 20-кратным избытком смеси уксусный ангидрид/пиридин (1:1) в течение 15 мин.
Пептид промывают последовательно ДХМ, ДМФ и ДХМ, затем сушат под вакуумом. Пептид суспендируют в реагенте K (King et al., Int. J.Pept.Prot. Res. 36: 255-266 (1990), публикация введена в качестве ссылки) (5 мл/г пептида) в течение 180 мин при комнатной температуре, затем расщепляющую смесь отфильтровывают в безводный диэтиловый эфир и отделяют твердый остаток центрифугированием и сушат в вакууме над твердыми гранулами КОН. Высушенный пептид подвергают очистке при помощи ЖХВР, используя соответствующий градиент 0,1% ТФК в воде и ацетонитриле (АЦН).
После сбора пика, содержащего целевой продукт, раствор модифицируют и подвергают пептид методам анализа, позволяющим его идентификацию, эти методы включают масс-спектрометрию при электроразбрызгивании и анализ аминокислот для определения состава синтезированного соединения.
Для очистки пептида образец технического лиофилизированного пептида растворяют в смеси 0,1% водной ТФК, содержащей 10-50% АЦН. Раствор пептида обычно отфильтровывают через шприц, соединенный с нейлоновым 0,45 мкм "ACRODISC" 13 (GELMAN Sciences; Ann Arbor MI) фильтром.
Соответствующий объем отфильтрованного раствора пептида впрыскивают в полупрепаративную колонку C18 (Vygac Protein and Peptide C18, 218TP1010; The Separation Group; Hesperia CA). Скорость прохождения градиента или изократической смеси 0,1% ТФК буфера и АЦН (ЖХВР) в качестве элюента поддерживается при помощи Beckman "SYSTEM GOLD" ЖХВР. Элюирование пептида регулируют при помощи УФ при 230 нм (Beckman System Gold, Programmable Solvent Module 126 и Programmable Detector Module 166, управляемый программным обеспечением "SYSTEM GOLD"). После определения пика, соответствующего синтезируемому соединению, с использованием MS, соединение собирают, лиофилизируют и подвергают биологическому испытанию. MS (масс-спектрометрию) проводят, используя прибор SCIEX API III+. Кроме того осуществляют ЯМР, используя прибор General Electric (300 МГц). В случае ЯМР образцы обычно измеряют в гексадейтеродиметилсульфоксиде или дейтерохлороформе (CDCl3; Aldrich).
Альдегиды аминокислот получают, используя хорошо известные методы. Аминокислоты и альдегиды пептидов были описаны, например, Fehrentz and Castro, Sysnthesis 676 (1983); Bajusz et al., J.Med.Chem. 33:1729, (1990); Kawamura et al., Chem.Pharm.Bull. 17:1902 (1969), and Someno et al., Chem. Pharm, Bull., 34:1748 (1986). Получение восстановленных пептидных связей осуществляют на уровне дипептида в растворе (например, Tyr-{Φ(CH2NH)}-Ile), затем соответствующим образом замещенный дипептид связывают с остатком пептида на смоле, используя метод синтеза пептидов в твердой фазе. По другому варианту альдегид защищенной аминокислоты связывают с пептидом на смоле, используя методы, описанные Но et al. (Pent Res. 6:10-12 (1993)).
Пример II. Синтез Ac-Tyr-Ile-Arg-Leu-Ala-NH2
Для синтеза Ac-Tyr-Ile-Arg-Leu-Ala-NH2 1 г смолы Rink'a (0,6 ммоль NH2/г смолы) используют в способе, описанном выше. Полученный пептид анализируют методом масс-спектрометрии. (М+Н)+): найдено 659,4, вычислено (выч.) 659,9.
Пример III. Синтез Ac-Tyr-Ile-Arg-Leu-Pro-NH2
Для синтеза Ac-Tyr-Ile-Arg-Leu-Pro-NH2 1 г смолы Rink'a (0,6 ммоль NH2/г смолы) используют в способе, описанном в примере 1. Полученный пептид характеризуется: (М + Н)+: найдено 685,4, выч. 685,9.
Пример IV. Синтез Ac-(iBu)Tyr-Ile-Arg-Leu-Pro-NH2
Используют 1 г смолы Rink'a (0,6 ммоль NH2/г смолы).
Применяют общий метод синтеза в твердой фазе. После деблокировки Tyr и соответствующей промывки пептида добавляют 50 экв изобутиральдегида в ДМФ, содержащем 2% ледяной уксусной кислоты. Полученную смесь встряхивают в течение 4 час при комнатной температуре. После промывки пептида ДМФ, содержащим 2% уксусной кислоты (2 x 8 мл), добавляют 1 г NaBH3CN в 10 мл ДМФ, содержащего 2% уксусной кислоты. Пептидную смолу встряхивают в течение 30 мин, затем отфильтровывают и добавляют свежую смесь NaBH3CN в смеси ДМФ/уксусная кислота, реакцию продолжают еще 30 мин.
Пептидную смолу затем промывают смесью ДМФ/2% уксусной кислоты (2 x 8 мл) и ДМФ (2 x 8 мл). Полученную моноалкилированную пептидную смолу ацетилируют смесью уксусного ангидрида с триэтиламином в ДМФ (30 экв, 6 час).
После соответствующей промывки пептида, пептид подвергают расщеплению и деблокировке, как описано в примере I.
Очищенный ЖХВР пептид анализируют методом масс-спектрометрии. (М + Н)+: найдено 758,4, выч. 758,5.
Пример V. Синтез Tfa-(iBu)Tyr-Ile-Arg-Leu-Pro-NH2
Ту же процедуру, которая описана в примере IV, используют для получения (iBu)Tyr-Ile-Arg-Leu-Pro-Rink смола. Окончательное трифторацетилирование осуществляют путем обработки пептида-смолы 0,7 М трифторуксусного ангидрида в присутствии диизопропилэтиламина (ДИЭА) и N-метилимидазола (NМИ) (1:3:0,3 экв) в течение 45 мин.
Расщепление пептида от смолы и выделение пептида осуществляют как описано в примере IV. Очищенный пептид идентифицируют методом МС. (М + Н)+: найдено 812,4, выч. 812,5.
Пример VI. Синтез Ac-Tyr-Ile-Arg-N(CH3)O(CH3).
Синтез Boc-Arg(Ng-Tos)-N(CH3)O(CH3) проводят в соответствии с описанным в литературе методом (Fehrentz and Castro, Supra, 1983). Boc-Arg(N-Tos)-N(CH3)O(CH3) (200 мг) смешивают с 5 мл трифторуксусной кислоты (ТФК) при комнатной температуре и перемешивают в течение 20 мин. Исчезновение исходного соединения контролируют методом тонкослойной хроматографии (ТСХ), используя CHC13 : MeOH : CH3COOH (90:9:1) и обнаруживают визуально при помощи нингидринового разбрызгивания и УФ-света. Выпаривание оставшейся ТФК под вакуумом и сушка в вакууме над гранулами КОН приводит к твердому материалу, имеющему соответствующую массу. (М + H)+ найдено 371,2, выч. 371,4.
В первой колбе в 1 мл ДМФ растворяют 150 мг материала, полученного выше, затем добавляют 57 мкл триэтиламина и охлаждают смесь до 0oC. Во второй колбе в безводном тетрагидрофуране (ТГФ) растворяют 171 мг Z-Tyr-Ile-OH (Biochem Bioscience Inc.; Philadelphia PA) и охлаждают до -10oC, затем добавляют 44 мкл NNM мкл изобутилхлорформиата в атмосфере азота и смесь перемешивают 15 мин. Ранее полученный раствор Arg(Tos)N(CH3(OCH3) в ДМФ добавляют к смешанному ангидриду Z-Tyr-Ile-OH дипептида и смесь перемешивают при -10oC в течение 30 мин, а затем в течение ночи при комнатной температуре.
После обработки реакционной смеси, как описано в примере I, пептид сушат под вакуумом и небольшую часть очищают ЖХВР и анализируют методом MS; пептид имеет ожидавшийся молекулярный вес (781). Полученный пептид Z-Tyr-Ile-Arg(Tos)-N(CH3))OCH3 смешивают с 500 мкл анизола и подвергают деблокировке с HF обычным методом.
После обработки выделяют 169 мг Tyr-Ile-Arg-N(CH3)O(CH3) и идентифицируют методом МС (найдено 493,6, выч. 494).
Остаточный пептид затем растворяют в 1 мл 1N HCl и подвергают лиофилизации.
Tyr-Ile-Arg-N(CH3)OCH3•2HCl (76 мг) растворяют в ацетонитриле, охлаждают до 0oC и добавляют 13 мкл пиридина, а затем 15 мкл уксусного ангидрида. Смесь перемешивают при 0oC в течение 3 час и окончание реакции устанавливают по нингидриновой пробе. После перемешивания при комнатной температуре в течение 8 час реакционную смесь обрабатывают и полученный продукт Ac-Tyr-Ile-Arg-N(CH3)OCH3 анализируют методом МС (найдено 535,6, выч. 535,3).
Пример VII. Синтез Ac-Tyr-{Φ CH2NH)}-Ile-Arg-Leu-Pro-NH2
а) Синтез Fmoc-Tyr(But)-H
4,6 г (10,0 ммол) Fmoc-Tyr(But)-ОН, 2,1 г (10,1 ммол) дициклогексилкарбодиимида (ДЦК), 1,26 г (10,1 ммол) бензилмеркаптана и 0,12 г DMAP реагируют в дихлорметане, как: описано Но и Ngu (J.Org.Chem. 58:2315(1993)).
После обработки выделяют Fmoc-Tyr-(But)-S-CH2C6H125 и после восстановления тиоэфира путем перемешивания с триэтилсиланом в присутствии 10% Pd на угле и очистки методом хроматографии в тонком слое получают Fmoc-Tyr(But)-H с выходом 81%. Данные ЯМР и выход продукта соответствуют ожидаемому.
б) Синтез Fmoc-Tyr(But)-{ Φ (CH2NH)}-Ile-(O-Аллил)
0,73 г (1,66 ммол) Fmoc-Tyr(But)-ОН и 0,209 г (3,32 ммол) NaBH3CN в 20 мл 1% AcOH в ДМФ добавляют к раствору 0,516 г (1,82 ммол) ТФК•Ile-(O-Аллил) в 2 мл ДМФ. Через 2 часа реакционную смесь обрабатывают и конечный продукт очищают хроматографией в тонком слое (этилацетат: гексан, 35: 65) с получением масла, характеризующегося соответствующими данными ЯМР и МС. (М + Н): найдено 599, выч. 598,7.
в) Синтез Fmoc-Tyr(But)-{Φ(CH2NH)}Ile-OH
К 0,467 г (0,78 ммол) Fmoc-Tyr(But)-{ Φ (CH2NH}-Ile-O-Аллил в 10 мл дихлорметана добавляют 89 мкл (1,56 ммол) HOAc, 20 мкл триэтиламина (ИЭА) и 0,02 г комплекса PdCl2(Ph3)2.
Добавляют сразу 231 мкл (8,86 нмол) Bu3SnH и перемешивают смесь в течение 1 час при комнатной температуре. После соответствующей обработки реакционной смеси продукт очищают хроматографией в тонком слое (CHCl3 : MeOH, 20 : 1) с получением пептида с выходом 69% (0,319 г). (М+H+): найдено 559, выч. 558.
Fmoc-Tyr(But)-{ Φ(CH2NH)}Ile-OH затем связывают со смолой Arg(Pmc)-Leu-Pro-Rink, используя общий метод синтеза в твердой фазе, как описано в примере I. Целевую пептидную смолу Ac-Tyr(But)-{Φ(CH3NH)}-Ile-Arg(Pmc)-Leu-Pro-Rink деблокируют и расщепляют, как описано в примере I и очищают методом ЖХВР на колонке C18.
Пример VIII. Синтез Ac-Tyr-Ile-Arg-NH-CH2(4-пиридил)а
Оксимную смолу (DeGrado и Kaiser, J.Org. Chem. 45: 1295 (1980) (0,862 г, 0,6 ммол/г) связывают в течение ночи с Boc-Arg(Tos)-OH в присутствии ДИК/HOBt. Смолу промывают ДМФ, затем ДХМ и ацетилируют смесью уксусный ангидрид/ДИЭА (1: 1 экв) в ДХМ.
После промывки смолы ДХМ, ДМФ и ДХМ ее деблокируют 25% ТФК в ДХМ в течение 30 мин. Полученную смолу промывают ДХМ, изопропанолом и ДХМ. С ТФК. Arg(Tos)-OxmR связывают Boc-Ile-OH в симметричной ангидридной форме (3 экв) в присутствии 1,5 экв ДИЭА в ДХМ. Цикл промывки, ацетилирования и деблокировки, описанный выше, повторяют. После деблокировки добавляют связыванием, как Ile, Boc-Tyr(2-BrZ)OH, затем пептидную смолу Boc-Tyr(2-BrZ)-Ile-Arg(Tos)-OxmR деблокируют и ацетилируют с получением Av-Tyr(2-BrZ)-Ile-Arg(Tos)-OxmR. Пептидную смолу сушат под вакуумом с получением прироста веса 0,216 г.
К 1/3 этой смолы добавляют 100 мкл (800 мкм) 4-(диметиламино)пиридина в присутствии 60 мкл ледяной уксусной кислоты и 120 мкл ДИЭА в 6 мл ДХМ.
Смолу встряхивают в течение ночи при комнатной температуре. После фильтрования раствора полученную смолу промывают 3 мл ДМФ и промывные воды соединяют с ДХМ фильтратом. После выпаривания растворителя полученный пептид деблокируют при помощи HF/анизол и обрабатывают как обычно для получения целевого пептида. Осуществляют электрораспылительную МС. (М + Н)+: найдено 582,3, выч. 582.
Пример IX. Синтез Ac-Tyr-Ile-{ Φ (CH2NH)}-Arg-Leu-Pro-NH2
a) Синтез Boc-Ile-H
Альдегид синтезируют из 1 г Boc-Ile-N(Me)OMe, как описано Fehrentz и Castro (Supra, 1983). Альдегид идентифицируют методом тонкослойной хроматографии и ЯМР, как описано в указанной ссылке.
б) Синтез Arg(Tos)-Leu-Pro-MBHA
Синтез трипептидной смолы осуществляют в твердой фазе, как описано в примере I.
в) Синтез Boc-Ile-{Φ(CH2NH)}-Arg(Tos)-Leu-Pro-MBHA
Boc-Ile-H связывают с трипептидной смолой Arg(Tos)-Leu-Pro-МВМА путем восстановительного аминирования с использованием NaBH3CN в ДМФ, содержащем 1% уксусной кислоты. Boc-группа была отщеплена как обычно и Ac-Tyr-OH связывают с использованием ДИК/HoBt.
Конечную пептидную смолу (0,7 г) деблокируют и пептид отщепляют от смолы, используя смесь HF/тиоанизол. 19 г сырого Ac-Tyr-Ile-{Φ(CH2NH)}-Arg-Leu-Pro-NH2 очищают методом ЖХВР на колонке C18 с получением примерно 5 мг > 90% чистого целевого пептида. (М + H+): найдено 688,4, выч. 687,9.
Пример Х. Синтез Ac-Tyr-Ile-Dab(Nγ- C3H7N)-Leu-Ala-NH2
0,2 г SCAL-TG (0,2 ммол NH2/г) (Patek and Lebl, Tetr.Lett. 32: 3891-3894 (1991)) связывают с Fmoc-Ala-OH и Fmoc-Leu-OH, Fmoc-Dab(Воc)-ОН, Fmoc-Ile-OH и Fmoc-Tyr(But)-ОН, используя методы, описанные в примере I.
После ацетилирования N-концевой группы и деблокирования боковой цепи ТФК, промывают пептидную смолу Ac-Tyr-Ile-Dab-Leu-Ala-SCAL-TG, нейтрализуют и обрабатывают 0,3 М PyBroP/NМИ в ДМФ в течение 2 час. Полученный пептид отщепляют от смолы, используя 1М смеси трифенилфосфин/(CH3)3SiCl в ДХМ (3 x 1 час), затем 100% ТФК (1 час). После выделения сырого пептида путем осаждения диэтиловым эфиром пептид лиофилизируют из 0,1% водного раствора ТФК. Пептид Ac-Tyr-Ile-Dab(Nγ-C3H7N)-Leu-Ala-NH2 очищают ЖХВР и проводят МС. (М + H+): найдено 676,4, выч. 676,4.
Пример XI. Синтез Ac-Tyr-Ile-PalMe(3)-NH2
С 1,0 г смолы Rink (0,48 ммол NH2/г связывают Fmoc-Pal(3)-ОН, Fmoc-Ile-OH и Fmoc-Tyr(But)-ОН, используя методы, описанные в примере I. К 0,25 г полученной пептидной смолы Fmoc-Tyr(But)-Ile-Pal(3)-Rink добавляют 500 мел метилиодида (МИ) в ДХМ и встряхивают в течение 6 час. Полученную пептидную смолу Fmoc-Tyr (But)-Ile-PalMe(3)-Rink деблокируют и ацетилируют и отщепляют, как описано в примере I. Часть сырого пептида очищают методом ЖХВР и проводят МС.
Пример XII. Синтез Ac-Cyclo(Glu-Tyr-Ile-Arg-Leu-Lys)-NH2
1 г SCAL-TG (0,29 ммол NH2/г) (см. пример X) связывают с Fmoc-Lys(Воc)-ОН, Fmoc-Leu-OH, Fmoc-Arg(Pmc)-ОН, Fmoc-Ile-OH, Fmoc-Tyr(But)-ОН и Fmoc-Glu(OtBu)-OH, используя методы, описанные в примере I.
После удаления Fmoc пептидную смолу ацетилируют и промывают ДМФ, затем ДХМ. Пептидную смолу Ac-Glu(OtBu)-Tyr(But)-Ile-Arg(Pmc)-Leu-Lys(Boc)-SCAL-Tg деблокируют реагентом K, промывают, нейтрализуют и циклизуют, используя BOP/HOBt/ДИЭА (5: 5: 5 экв), в ДМФ в течение 2 час. Окончание связывания контролируют пробой с применением нингидрина, как описано Kaiser (Kaiser et al. , Anal. Biochem. 34:595 (1970)). После циклизации пептид отщепляют от смолы, очищают методом ЖХВР и проводят МС. (М+Н)+: найдено, 844,5, выч. 844,5.
Пример XIII. Синтез Цикло(Cly-Tyr-Ile-Arg-Gly)
1 г оксимной смолы (см. пример VIII) (0,6 ммол NH2/г) связывают в течение ночи с Boc-Gly-OH в присутствии ДИК/HOBt. После промывки и деблокировки смолы сочетают Boc-Arg(Tos)-OH, Boc-Ile-OH и Boc-Tyr(2BrZ)-OH, используя методы, описанные в примере VIII.
1/3 пептидной смолы, Boc-Tyr-(2BrZ)-lle-Arg(Tos)-Gly-Oxime деблокируют и связывают с Boc-Gly при помощи ДИК/HOBt. Пептидную смолу деблокируют, нейтрализуют и циклизуют в течение ночи в ДМФ, содержащем 1% уксусной кислоты. Смолу отфильтровывают и промывают (ДМФ), соединяют фильтраты и удаляют органический растворитель испарением в вакууме.
Пептид деблокируют (HF/анизол), лиофилизируют, очищают методом ЖХВР и осуществляют МС. (М + Н)+: найдено 547,8, выч. 547,8.
Пример XIV. Синтез N-замещенных глициновых соединений: синтез Ac-(Bzl)Gly-(Chx)Gly-(3-гуанидопропил)Gly-NH2
Для синтеза N-замещенных глицинов применяют методику Zuckermann'a и др. (J. Am. Chem. Soc. 114:10646 (1992), ссылка), 1 г SCAL-TG (0,29 ммол NH2/г) (см. пример X) связывают с бромуксусной кислотой через симметричный ангидрид в ДХМ/ДМФ. Каждую реакцию сочетания повторяют дважды. К смоле Br-CH2CO-SCAL-TG добавляют Вос-NH-CH2CH2CH2NH2 в ДМСО и встряхивают смолу в течение 2 час.
После деблокировки процесс повторяют, чередуя связывание Br-CH2COOH со смолой и реакцию смолы, содержащей бромуксусную кислоту, и соответствующего амина.
Смолу (Bzl)Gly-(Chx)Gly-(Boc-NH-(CH2)3)Gly-SCAL-TG ацетилируют смесью уксусный ангидрид/ДИЭА/NMU (1:1:0,25) в ДМФ в течение ночи. После деблокировки группы Вос смолу Ac-(Bzl)Gly-(Chx) Gly-(3-аминопропил)Cly-SCAL-TG обрабатывают 1,8 М карбоксиамидинопиразола•HCI (Bernatowicz et al., J.Org.Chem. 57:2497-2502 (1992)) в присутствии ДИЭА (1:1) в ДМФ в течение 3 час при комнатной температуре.
Окончание гуанилирования определяют пробой по Kaiser'у. Отщепление и обработку полученного пептида проводят, как описано в примере X, и осуществляют МС. (М + Н)+: найдено 502,3, выч. 502,3.
Пример XV. Синтез дикетопиперазиновых соединений: синтез цикло(Ser-Ida)-Ile-Arg-Leu-Ala-NH2
Исходный защищенный тетрапептид, Fmoc-Ile-Arg(Pmc)-Leu-Ala-Rink, получают через стратегию Fmoc со смолой Rink (см. пример I). После деблокировки Fmoc пептидной смолы Fmoc-Ida(OMe)-OH (3 экв; ДИК, HOBt) и Fmoc-Ser(tBu)-OH (7 экв; симметричный ангидрид) связывают последовательно. Окончательное деблокирование и спонтанное закрытие кольца осуществляют одновременно путем выдержки в 50% пиперидина/ДМФ в течение 1 час. После промывки в несколько стадий конечный пептид отщепляют и деблокируют, используя смесь ТФК/тиоанизол/H2O (95:2,5:2,5).
Полученный пептид обрабатывают, как описано выше, и анализируют методом ЖХВР (> 95%) и методом МС. (М + Н)+: найдено 655,4, выч. 655,38.
Пример XVI. Синтез Ph-C(NOCH2Ph)-CO-I-R-NH2
0,2 г смолы Rink'а связывают с Fmoc-Arg(Pmoc)-OH, Fmoc-Ile-OH с последующим удалением защиты Fmoc (см. пример I).
С пептидной смолой, Ile-Arg(Pmoc)-Rink, связывают Ph-C(NOCH2Ph)-COOH, используя ДИК/HOBt, как описано выше. Пептидную смолу, Ph-C(NOCH2Ph)-CO-Ile-Arg(Pmc)-Rink, обрабатывают, как описано в примере I, и осуществляют МС. (М + Н)+: найдено 524,3, выч. 524,6.
Пример XVII. Синтез Ac-pAph-Ile-Arg-Leu-Pro-NH2
Синтез осуществляют со 100 мг смолы Rink'a (0,48 ммол/г) согласно методу, описанному в примере I, используя следующие производные аминокислот: Fmoc-Pro-OH, Fmoc-Leu-OH, Fmoc-Arg(Pmc)-ОН, Fmoc-Ile-OH и Fmoc-pAph-(Fmoc)-OH (рацемическую смесь).
Отщепление и выделение пептида осуществляют, как описано в примере I. Оба диастереомерных пептида выделяются методом хроматографии с обращенной фазой - ЖХВР и идентифицируются методом МС. (М + Н)+: найдено 754,4, выч. 754,5.
Пример XVIII. Синтез Ac-Tyr-Chg-Arg-ol
Пептидную последовательность получают с 0,25 г Fmoc-Arg(Pmc)-Sasrin смолы (0,5 ммол NH2/г смолы; Bachem Bioscience), используя способ, описанный в примере I. После деблокировки N-концевой Fmoc и ацетилирования защищенный пептид отщепляют от смолы восстановительным отщеплением в виде C-концевого спирта (Mergler et al. Peptides p.p. 177-178 (eds. Schneider and Eberle; Leiden 1993), включенная в качестве ссылки). Пептидную смолу встряхивают с раствором NaBH4 (4 экв) в 2 мл THF:EtOH (6:1) в течение 24 час. После проведения реакции отщепления смолу промывают ДХМ, затем расщепляющим раствором, и промывные воды соединяют и лиофилизируют. Лиофилизированный пептид деблокируют обработкой ТФК/вода/тиоанизол (90: 5:5) в течение 2 час и выделяют осаждением, очищенный методом ЖХВР пептид анализируют методом МС. (М + Н)+: найдено 505,3, выч. 505,3.
Пример XIX. Синтез Ac-Tyr-Chg-Arg-ол.ацетат
Защищенный пептидный спирт получают как описано в примере XVIII.
10 мг сырого продукта растворяют в смеси ДХМ/АЦН и обрабатывают уксусным ангидридом (2 ммол) в присутствии ТЭА (2,4 ммол) в течение 20 мин. Раствор фильтруют, выпаривают и пептид деблокируют, как описано выше. Пептид очищают методом ЖХВР и анализируют методом МС. (М + Н)+: найдено 547,3, выч. 547,3.
Пример XX. Синтез Ac-Phe(pNH2)-Chg-Orn(C(NH)CH3)-Leu-Pro-NH2
1 г "TENTAGEL'S" NH2 смолы (0,28 ммол NH2/г смолы; Rapp Polymer; Tubingen Germany) функционализируют линкером SCAL, как описано в примере X и связывают следующие аминокислоты: Fmoc-Pro-OH; Fmoc-Leu-OH; Fmoc-Orn-(Boc)-ОН и Fmoc-Chg-OH. Пептидную смолу Fmoc-Chg-Orn (Boc)-Leu-Pro-SCAL-TG обрабатывают 50% ТФК в ДХМ (1 промывка в течение 1 мин, затем 1 промывка в течение 30 мин), промывают 3 раза ДХМ, нейтрализуют 5% ДИЭА в ДХМ (2 x 30 с) и 2 раза ДХМ. К пептидной смоле добавляют раствор 1,5 г гидрохлорида этилацетимидата (Aldrich) в 4 мл смеси пиридин: ДИЭА и 3 мл ДМФ и продолжают связывание в течение ночи при комнатной температуре.
Пептидную смолу, Fmoc-Chg-Orn(С(NH)-CH3)-Leu-Pro-SCAL-TG, деблокируют 2% пиперидина в ДМФ, 4 раза ДХМ и связывают при помощи ДИК/HoBt в ДМФ. Деблокирование Fmoc и ацетилирование смесью уксусный ангидрид: пиридин (1:1) в течение 20 мин приводит к получению пептидной смолы Ac-Phe-(pNH-BOC)-Chg-Orn-(С(NH)CH3)- Leu-Pro-SCAL-TG. Восстановление линкера SCAL и отщепление пептида, сопровождаемое очисткой сырого продукта методом ЖХВР, приводит к получению ожидаемого продукта. (М + Н)+: найдено 740,2, выч. 740,48.
Пример XXI. Синтез Ac-Phe(pNH2)-Chg-Dap(Nβ-C6H11N)-Leu-Pro-NH2
0,5 г SCAL-TG (0,32 ммол NH2/г) связывают с Fmoc-Pro-OH, Fmoc-Pro-OH, Fmoc-Leu-OH, Fmoc-Dap(Boc)-ОН и Fmoc-Chg-OH. Группу Boc из боковой цепи удаляют, используя 50% ТФК, в течение 20 мин, и пептидную смолу нейтрализуют промывкой 10% ДИЭА/ДХМ. Свободную аминогруппу в боковой цепи превращают в диметиламидиниевую группу обработкой пептидной смолы 0,3 М PyBroP/NMI в ДМФ в течение 20 мин. Деблокировка группы Fmoc смесью 50% пиперидина/ДМФ в течение 60 мин приводит к замене диметиламидиниевой группы пиперидиниевой в боковой цепи Dap. Последовательность заканчивают связыванием с Fmoc-Phe(Boc)-ОН и деблокировкой группы Fmoc. Пептид ацетилируют и отщепляют, как описано в примере X.
Пептид, очищенный методом ЖХВР, анализируют методом МС. (М + Н)+: найдено 752,4, выч. 752,4.
Пример XXII. Синтез Ac-pAph-Chg-PalMe(3)-NH2
Рацемический H-Phe(pCN)-OH синтезируют методом с применением ацетамидомалоната (Wagner et al. Патент ГДР 155954). Рацемический Ac-pAph-OH синтезируют превращением цианогруппы аммонолизом соответствующего метилтиоимидата (реакция цианогруппы с сернистым водородом) и последующим метилированием MeI.
1 г смолы "TENTAGEL" (замещение = 0,21 ммол NH2/г смолы) и линкер Knorr (Bernatowicz et al. , Tetr. Lett. 30: 4645 (1989)) используют для синтеза пептида.
Дипептид, Fmoc-Chg-Pal-Knorr-TG, получают как описано в примере I.
3-Пиридилаланин последовательно метилируют 1 мл MeI в ДХМ в течение ночи. После деблокировки Fmoc Ac-pAph-OH связывают методом с использованием ДИК/HOBt, и пептид обрабатывают как описано в примере I. (М+Н)+: найдено 550,3, выч. 550,31.
Пример XXIII. Синтез Ac-Tyr-Chg-pAph-Leu-Pro-NH2
Сборку пентапептида Ac-Tyr(But)-Chg-Phe-(pCN)-Leu-Pro-Knorr-TG, проводят на 0,4 г "TENTAGEL" (замещение = 0,2 ммол NH2/г смолы), как описано в примере I. Смолу обрабатывают в течение ночи в закрытом шприце 8 мл смеси пиридин/триэтиламин (75: 25), насыщенной H2S. Связанный со смолой тиоамид метилируют, используя 0,5 г MeI в 8 мл ацетона в течение 30 мин при 50oC, затем промывают ацетоном и метанолом.
Осуществляют реакцию метилтиоимида с ацетатом аммония в метаноле в течение 3 час при 55oC для получения целевого продукта, который отщепляют от смолы и очищают, как описано выше. (М + Н)+: найдено 761,4, выч. 760,43.
Пример XXIV. Синтез Ac-Phe(pCH2NH2)-Chg-Arg-Leu-Pro-NH2
Ac-DL-Phe(pCN)-Chg-Arg-Leu-Pro-NH2 (сырой пептид) синтезируют на 1 г смолы Rink'а (0,6 ммол NH2/г смолы) как описано в примере I. 125 мг сырого пептида растворяют в 50 мл MeOH и добавляют 0,5 мл суспензии Ni Ренея (Aldrich). Смесь пептида и катализатора гидрируют при давлении 35 ф/дюйм2 (241,3 кПа) в течение 4 час при комнатной температуре. Катализатор отфильтровывают, а раствор выпаривают досуха. Остаток лиофилизируют из 0,1% водной ТФК, содержащей 30% АЦЕ.
Высушенный технический продукт очищают методом ЖХВР и анализируют методом МС. (М + Н)+: найдено 741,4, выч. 741,7.
Пример XXV. Синтез Ac-Phe(pC(NOH)NH2)-Chg-Arg- Leu-Pro-NH2
21,1 мг Технического пептида, полученного как описано в примере XXIV, смешивают с 60,3 мг NH2OH•HCI (Aldrich) в 1,5 мл MeOH, 0,7 мл пиридина и 0,5 мл ТЭА. Смесь перемешивают 72 час при комнатной температуре и затем в вакууме выпаривают растворитель и летучие вещества. Пептид очищают методом ЖХВР и анализируют методом МС. (М + Н)+: найдено 770,4, выч. 770,3.
Пример XXVI. Синтез соединений A1-A2-B
A1-A2-B, т. е. соединения A1-A2-(A3)m-B, где m = 0, получают, как показано на фиг. 3. Вкратце, связывание рацемического N-ацетил-4-цианфенилаланина с метиловым эфиром L-циклогексилглицина (H-Chg-ОМе) приводит к образованию смеси двух диастереомерных дипептидов, которые разделяют хроматографией. Рацемический n-ацетил-4-цианфенилаланин частично снова растворяется при получении соли с метиловым эфиром L-циклогексилглицина. Менее растворимая D,L-соль легко кристаллизуется, и последующее сочетание приводит к получению сырого Ac-F(pCN)-Chg-OMe, который далее очищают хроматографией над силикагелем. Эти эфиры дипептида гидролизуют до соответствующих кислот, применяя гидроокись лития в смеси метанол/вода при комнатной температуре. Обе дипептидные кислоты превращают в соответствующие амиды обычным связыванием с подходящими аминами RNH2. Амины, не являющиеся товарными продуктами, получают стандартными методами.
Превращение цианогрупп в соответствующие амидины осуществляют обычными методами или через тиоамид и метилтиоимидат, или гидрированием соответствующего амидоксима (пример XXV).
Последний получают реакцией нитрила с гидроксиламином. Примеры, описанные ниже, иллюстрируют получение названных соединений этими методами. Соединения по изобретению могут быть получены и другими методами; выбранные способы, описанные в примерах, используются для удобства.
Пример XXVII. Синтез Ac-pAph-Chg-NHCH2-(4-метилпиридиния)
Синтез Ac-pAph-Chg-NHCH2-(4-метилпиридиния) осуществляют превращением Ac-F(pCN)-Chg-NH-CH2-(4-пиридил)а, используя методы, описанные в примере XXII. Конечный продукт очищают методом ЖХВР, как описано в примере I. МС анализ: (М + Н)+: найдено 493,3, выч. 493,29.
Исходное вещество получают следующим образом:
a) 2,32 г (10 ммол) Ac-(D,L)-F(pCN) растворяют в 75 мл этанола при нагревании. Добавляют метиловый эфир L-циклогексилглицина (1,75 г, 10 ммол) и перемешивают смесь 2 часа при комнатной температуре. Осажденные кристаллы отфильтровывают и сушат с получением 1,55 г D,L-соли. Фильтрат частично выпаривают и разбавляют эфиром.
Выделенные кристаллы собирают и высушивают, остается 2,1 г L,L-соли, загрязненной D,L-солью. Сырую L,L-соль соединяют с 20 мл ДМФ, 0,71 г HOBt и 1,18 г ДЦК. Смесь перемешивают 24 часа при комнатной температуре. Мочевину отфильтровывают, и фильтрат выпаривают.
Остаток растворяют в метиленхлориде, и раствор промывают 1 N HCl и насыщенным водным раствором бикарбоната натрия. Органический слой сушат и выпаривают, осадок хроматографируют над 60 г силикагеля, используя 20%(об.) раствор ацетона в метиленхлориде для элюирования. Кристаллизация соединенных чистых фракций из смеси метиленхлорид/эфир/гексан приводит к получению 1,6 г Ac-F(pCN)-Chg-ОМе в виде бесцветных кристаллов с точкой плавления (т.пл.), равной 178-180oC.
б) Смесь 1,93 г (5 ммол) Ac-F(pCN)-Chg-OMe (по примеру XXVII.а выше), 100 мл метанола, 10 мл воды и 0,75 г гидрата гидроокиси лития перемешивают в атмосфере азота в течение 24 час при комнатной температуре.
Добавляют 2 мл уксусной кислоты, выпаривают растворители, и остаток разделяют между метиленхлоридом, содержащим 20% изопропанола, и 1 N HCl. Органический слой высушивают и выпаривают, а остаток кристаллизуют из смеси метиленхлорид/эфир/гексан, получают 1,6 г Ac-F(pCN)Chg-OH в виде бесцветных кристаллов с т.пл. 216-218oC.
в) Смесь 150 мг (0,4 ммол) Ac-F-(pCN)-Chg-OH (см. выше), 65 мг (0,6 ммол) 4-аминометилпиридина, 124 мг (0,6 ммол) ДЦК, 60 мг (0,44 ммол) HOBt и 5 мл ДМФ перемешивают в течение 20 час при комнатной температуре. Мочевину удаляют фильтрованием, фильтрат выпаривают. Остаток суспендируют в метаноле и собирают нерастворимый продукт, оставляя 140 мг бесцветного Ac-F-(pCN)-Chg-NHCH2(4-пиридил)а.
Хроматографией над силикагелем с использованием смеси ацетон:метиленхлорид: метанол (4:5:1) получают аналитический образец. Кристаллическое твердое вещество имеет т.пл. > 250oC.
Пример XXVIII. Ac-F(4-амидин)-Chg-NHCH2(4-метилпиридиний)
Это соединение получают взаимодействием 150 мг Ac-F(pCN)-Chg-NHCH2(4-пиридил)а (см. выше) с сернистым водородом, затем с метилйодидом и ацетатом аммония.
Продукт выделяют ЖХВР в виде гомогенного вещества. Данные МС: (М + Н)+ найдено 493,3, выч. 493,29.
Исходное вещество получают следующим образом:
а) Смесь 2,8 г Ac-F(pCN), метилового эфира (L)-циклогексилглицина, 940 мг HOBt, 1,57 г ДЦК и 30 мл ДМФ перемешивают 2 дня при комнатной температуре. Фильтрованием удаляют мочевину и выпаривают фильтрат. Остаток растворяют в метиленхлориде и промывают раствором 1 N HCl и 10% водным раствором карбоната натрия. Органическую фазу сушат и выпаривают. Кристаллизацией остатка из смеси метиленхлорид/эфир/гексан получают 2,05 г бесцветного Ac-F(pCN)-Chg-OMe с т.пл. 181-183oC.
б) Гидролиз 1,93 г Ac-F(pCN)-Chg-OMe (см. выше) при помощи 0,75 г моногидрата гидроокиси лития в 100 мл метанола и 10 мл воды проводят, как описано для L,L-изомера в примере XXVII выше, кристаллизация из смеси метиленхлорид/эфир приводит к получению 1,65 г Ac-F(pCN)-Chg-OH с т.пл. 180-182oC.
в) Смесь 225 мг Ac-F(pCN)-Chg-OH (см.выше), 100 мг 4-аминометилпиридина, 90 мг HOBt, 180 мг ДЦК и 6 мл ДМФ перемешивают в течение уикэнда при комнатной температуре. Отфильтровывают мочевину и выпаривают фильтрат, остаток перемешивают с метанолом, удаляют твердый остаток фильтрованием, при этом остается 190 мг кристаллического Ac-F(pCN)-Chg-NHCH2(4-пиридил)а с т.пл. > 250oC.
Пример XXIX. Ac-pAph-Chg-NHCH2CH2 (3-метилпиридиний)
Смесь 125 мг Ac-F(pCN)-Chg-NHCH2CH2(3-пиридил)а, 2 мл ДМСО, 10 мл пиридина и 3 мл триэтиламина насыщают сернистым водородом при охлаждении смесью лед/вода.
После перемешивания в герметичной пробирке в течение ночи при комнатной температуре растворители выпаривают, остаток растворяют в смеси ацетон/эфир и высушивают с получением 125 мг тиоамида. Этот продукт соединяют с 2 мл ДМСО, 5 мл ацетона и 0,75 мл метилиодида, и смесь перемешивают в герметичной пробирке в течение ночи при комнатной температуре. После разбавления толуолом растворители выпаривают, и остаток перемешивают вместе с эфиром. Эфир декантируют, заменяют свежим эфиром и продолжают перемешивание в течение ночи, пока не затвердеет резиноподобное вещество, затем оставшийся эфир отфильтровывают, и остаток сушат.
Оставшийся остаток растворяют в 20 мл метанола и обрабатывают 0,3 мл уксусной кислоты и 0,4 г ацетата аммония.
Смесь нагревают до 55-60oC в течение 2,5 час, затем растворители выпаривают.
Остаток растворяют в смеси вода/ацн/ТФК и лиофилизируют. Сырой продукт очищают ЖХВР. Данные МС: (М + Н)+ найдено 507,3, выч. 507,31.
Исходное вещество получают следующим образом. Смесь 150 мг (0,4 ммол) Ac-F(pCN)-Chg-OH, 120 мг (0,6 ммол) 2-(3-пиридил)этиламина гидрохлорида, 125 мг ДЦК, 60 мг HOBt, 0,5 мл диизопропилэтиламина и 10 мл ДМФ перемешивают 24 час при комнатной температуре. После выпаривания растворителя остаток перемешивают с метанолом, нерастворимый продукт собирают путем фильтрации и промывают метанолом и эфиром, получая бесцветные кристаллы. Фильтрат выпаривают, и остаток растворяют в смеси метиленхлорид/изопропанол.
Этот раствор промывают 10%-ным водным раствором карбоната натрия, сушат и выпаривают. Остаток хроматографируют над 14 г силикагеля, используя смесь метиленхлорид: ацетон: метанол (5: 4:1), с получением 40 мг Ac-F(pCN)-Chg-NHCH2CH2(3-пиpидил)a с т.пл. 265-268oC.
б) Дигидрохлорид 2-(3-пиридил)этиламина получают следующим образом. Смесь 1,3 г 3-пиридилацетонитрила, примерно 3 г никеля Ренея и 30 мл метанола, содержащего 10% об. аммиака, гидрируют при давлении 35 ф/дюйм2 (241,3 кПа) в течение 20 час, используя гидрогенатор Parr. Катализатор отфильтровывают над целитом, и фильтрат выпаривают. Остаток растворяют в метиленхлориде, сушат над сульфатом магния, фильтруют и выпаривают. Продукт превращают в дигидрохлорид, используя хлористый водород в диоксане. Кристаллизацией из смеси метанол/эфир получают 1,4 г бесцветных кристаллов с т.пл. 145-148oC.
Пример XXX. Ac-pAph-Chg-NHCH2CH2(4-метилпиридиний)
Это соединение получают методами, описанными выше, взаимодействием Ac-F(pCN)-Chg-NHCH2CH2-(4-пиридил)а с сернистым водородом с последующим метилированием метилиодидом и реакцией с ацетатом аммония.
Сырой продукт очищают ЖХВР. Данные МС: (М + Н)+ найдено 507,3, выч. 507,31.
Исходное вещество получают связыванием Ac-F(pCN)-Chq-OH с дигидрохлоридом 2-(4-пиридил)этиламина, как описано в примере XXIX выше.
Дигидрохлорид 2-(4-пиридил)этиламина получают как описано в случае дигидрохлорида 2-(3-пиридил)этиламина (см.выше) гидрированием пиридил-4-ацетонитрила над никелем Ренея в присутствии аммиака. Дигидрохлорид имеет т.пл. 220oC.
Пример XXXI. Ac-pAph-Chg-NHCH2(4-амидинофенил)
Это соединение получают, используя методы, похожие на описанный выше для Ac-F(pCN)-Chg-NHCH2(4-цианфенил)а, путем обработки сернистым водородом в ДМСО, пиридине и триэтиламине. Полученный бис-тиоамид метилируют метилиодидом в смеси ДМСО/ацетон, затем проводят реакцию с ацетатом аммония, как описано выше. Сырой продукт очищают ЖХВР. Данные МС: (М + Н)+ найдено 520,3, выч. 520,3.
Исходное вещество получают следующим образом. Смесь 75 мг (0,2 ммол) Ac-F(pCN)-Chg-OH, 50 мг (0,3 ммол) гидрохлорида (4-цианфенил)метиламина, 62 мг ДЦК, 30 мг HOBt, 0,2 мл ДИЭА и 2 мл ДМФ перемешивают 24 час при комнатной температуре. После фильтрования растворитель выпаривают и растворяют остаток в метиленхлориде, содержащем 20% изопропанола. Раствор промывают 1N HCl и 10% водным раствором карбоната натрия, затем сушат и выпаривают растворитель.
Остаток перемешивают в небольшом количестве смеси метанол/вода, отделившееся твердое вещество сушат, получая 80 мг Ac-F(pCN)-Chg-NHCH2(4-цианфенил)а.
Гидрохлорид (4-цианфенил)метиламина получают следующим образом. Смесь 2 г (10 ммол) α-бром-п-толуолнитрила, 2 г (10,8 ммол) фталимида калия и 30 мл ДМФ нагревают с обратным холодильником 1 мин.
После охлаждения смесь подкисляют уксусной кислотой и разбавляют водой, чтобы вызвать кристаллизацию продукта. Кристаллы отфильтровывают, промывают водой и сушат, получают 2,24 г бесцветного N-(4-цианфенил)метилфталимид с т. пл. 182-184oC.
1,5 г N-(4-цианфенил)метилфталимида суспендируют в 50 мл кипящего метанола и обрабатывают 1 мл гидразингидрата. Через 5 мин получается прозрачный раствор. Выпаривают метанол и обрабатывают остаток 2N HCI. Суспензию нагревают до кипения и затем охлаждают на льду. Твердые вещества отфильтровывают и выпаривают фильтрат. Остаток растворяют в воде. Полученный раствор опять нагревают до кипения, охлаждают и фильтруют. Фильтрат делают щелочным добавлением гидроокиси натрия и экстрагируют метиленхлоридом, содержащим изопропанол. Органическую фазу сушат, растворитель выпаривают, а остаток превращают в гидрохлорид, кристаллизуют из смеси изопропанол/эфир, получая 0,43 г бесцветных кристаллов с т.пл. > 260oC.
Гидрохлорид (3-цианфенил) метиламина получают взаимодействием (α-бром-м-толуолнитрила с фталимидом калия с получением N-(3-цианфенил)метилфталимида с т. пл. = 147-148oC. Реакция этого соединения с гидразингидратом и превращение в гидрохлорид, как описано выше, позволяет получить 3-цианфенилметиламин с т.пл. 223-226oC.
Пример XXXII. Ac-pAph-Chg-NHCH2(3-амидинофенил)
Это соединение получают, используя описанные выше методы. Ac-F(pCN)-Chg-NHCH2(3-цианфенил) обрабатывают сернистым водородом в ДМСО, пиридине и триэтиламине. Полученный бис-тиоамид метилируют метилиодидом в смеси ДМСО/ацетон, затем проводят реакцию с ацетатом аммония, как описано выше. Сырой продукт очищают ЖХВР. Данные МС: (М + Н)+ найдено 520,3, выч. 520,3.
Исходное вещество получают следующим образом. Смесь 300 мг (0,8 ммол) Ac-F(pCN)-Chg-OH, 200 мг (1,2 ммол) (3-цианфенил)метиламина гидрохлорида, 250 мг ДЦК, 120 мг HOBt, 0,8 мл ДИЭА и 10 мл ДМФ перемешивают 24 часа при комнатной температуре. После фильтрования выпаривают растворитель и растворяют осадок в большом объеме метиленхлорида, содержащего 20% изопропанола.
Раствор промывают 1 N НCI и 10% водным раствором карбоната натрия, сушат и выпаривают растворитель. Остаток перемешивают со смесью изопропанол/эфир, выделившееся твердое вещество отделяют, сушат и получают 400 мг Ac-F(pCN)-Chg-NHCH2(3-цианфенил)а.
Пример XXXIII. Ac-pAph-Chg-NHCH(Me)(4-метилпиридиний)
Смесь диастереомеров названного соединения получают взаимодействием смеси двух диастереомеров Ac-F(pCN)-Chg- NНCH(Ме)(4пиридил)а с сернистым водородом, затем с MeI и ацетатом аммония.
Диастереомеры разделяют ЖХВР. Данные МС: (М + Н)+ найдено 507,3, выч. 507,31.
Исходный материал получают следующим образом. Смесь 150 мг (0,4 ммол) Ac-F(pCN)-Chg-OH, 120 мг (0,6 ммол) рацемического 1-(4-пиридил)этиламина гидрохлорида, 125 мг ДЦК, 60 мг HOBt, 0,5 мл ДИЭА и 10 мл ДМФ перемешивают 24 часа при комнатной температуре. После фильтрации выпаривают растворитель и растворяют остаток в большом объеме метиленхлорида, содержащего 20% изопропанола.
Раствор промывают 10% водным раствором карбоната натрия, сушат и упаривают. Остаток перемешивают со смесью изопропанол/эфир, собирают выделившееся твердое вещество и сушат, получая 125 мг Ac-F(pCN)-Chg-NHCH(Ме)(4-пиридил)а в виде смеси двух диастереомеров.
Рацемический гидрохлорид 1-(4-пиридил)этиламина получают следующим образом.
Смесь 1 г 4-ацетилпиридин-N-окиси, 2 г никеля Ренея и 30 мл метанола, содержащего 20% аммиака (об/об) гидрируют 24 часа при давлении 30 ф/дюйм2 (206,8 кПа).
Катализатор удаляют фильтрованием над целитом, а фильтрат упаривают.
Остаток растворяют в метиленхлориде, фильтруют раствор и упаривают.
Остаток растворяют в изопропаноле и обрабатывают хлористым водородом в среде эфира. Собирают осажденные кристаллы и сушат их, получая 0,9 г продукта с т.пл. 198-200oC.
Пример XXXIV. Синтез ДИПА(м)pAph-Chg-Arg-Leu-Pro-NH2
а) Синтез N, N-Диизопропиламида (p-цианбензил) малоновой кислоты (ДИПА(м)Phe(pCN))-ОН
Синтез 2-(п-цианбензил)малоновой кислоты осуществлять по измененной методике (см. Pinori et al. Патент США N 506191). К раствору 3,8 г 2,2-диметил-1,3-диоксан-4,6-диона (Meldrum's acid; Aldrich) и 1,12 г NaCNBH3 (Aldrich) в 25 мл ДМФ добавляют 2,3 г п-цианбензальдегида (Aldrich) и перемешивают смесь в течение 2 час при комнатной температуре. К реакционной смеси добавляют 400 мл воды и охлаждают раствор на ледяной бане, pH доводят до 3,8-4 путем добавления по каплям 20% водного раствора HCl. Белый осадок собирают на стеклянной воронке Бюхнера и промывают холодной водой. Осадок сушат в вакууме над CaCl2 в течение 24 час. Данные ЯМР твердого вещества в CDCl3 показывают наличие 2,2-диметил-5-(п-циан)бензил-1,3-диоксан-4,6-диона (ДЦБД) с т.пл. 135-142oC и Rt 0,45 (CHCl3 : MeOH : уксусная кислота; 95:4: 1).
К 1,5 мл диизопропиламина в 45 мл ДХМ добавляют 3 мл N,O-Бис(триметилсилил)ацетамида (БСА) и нагревают раствор с обратным холодильником в реакционной колбе, снабженной магнитной мешалкой и конденсатором, содержащим трубку с CaCl2, в течение 7 час. После охлаждения раствора до комнатной температуры добавляют 0,8 г ДЦБД и реакционную смесь нагревают с обратным холодильником в течение 3 час (пока не закончится превращение в целевой продукт, что показывает ТСХ). После охлаждения реакционной смеси аккуратно добавляют 5-8 мл 20% водного раствора соляной кислоты. После разделения слоев органический слой промывают водой, сушат (MgSO4) и выпаривают досуха с получением чистого продукта, который используют на следующей стадии без дальнейшей очистки.
Идентификация соединений осуществляется методом ЯМР в CDCl3 и МС.
б) Синтез DIPA(m)pAph-Chg-Arg-Leu-Pro-NH2
Пептидную смолу DIPA(m)Phe(pCN)-Chg-Arg(PMc)-Leu-Pro-Rink синтезируют методом, описанным в примере I. Полученную смолу обрабатывают солянокислым гидроксиламином, как описано в примере I. Полученную смолу обрабатывают солянокислым гидроксиламином, как описано в примере XXV с получением DIPA(m)Phe (pC(NOH)NH2)-Chg-Arg(PMC)-Leu-Pro-Rink. После отщепления пептида от смолы и лиофилизации сырой продукт (120 мг) растворяют в 80 мл MeOH и 10 мл насыщенного раствора MH3 в MeOH. К реакционной смеси добавляют 0,25 мл суспензии никеля Ренея (Aldrich) и гидрируют смесь в течение 24 час под давлением 45 ф/дюйм2 (310,2 кПа). Катализатор отфильтровывают, растворитель выпаривают досуха и осадок лиофилизируют из 1:1 водного раствора 0,1% ТФК и АЦН. Сырой пептид очищают методом ЖХВР и идентифицируют соединение МС. (М + Н)+ найдено 824,2, выч. 824,5.
Пример XXXV
Соединения со многими заместителями, оказавшиеся эффективными ингибиторами фактора Xa:
Соединение - Вычисл. (Найдено)
Ac-(2-CF3Bzl)-Y-I-R-L-P-NH2 - 860.5 (860.3)
Ac-(CH3CH2CH2CH(CH3)CH2)-Y-I-R-L-P-NH2 - 786.5 (786.5)
CH3OCO-Y-I-R-L-P-NH2 - 742.4 (742.4)
Ac-Y-Chg-R-NH2 - 518.2 (518.2)
Nal(2)-Cha-R-D(O-Allyl)-NH2 - 679.4 (679.4)
y-Tle-R-Nle-P-NH2 - 660.4 (660.4)
Phe(pF)-I-R-L-P-NH - 662.3 (662.3)
Ac-(D)Tic(ОН)-I-R-L-P-NH2 - 714.4 (714.4)
Ac-Phe(pCN)-I-R-L-P-NH2 - 711.4 (711.4)
Ac-Phe(pCONH2)-Chg-R-L-P-NH2 - 755.4 (755.4)
y-Chg-R-NH2 - 476.2 (476.2)
Ac-W-Chg-R-L-P-NH2 - 751.3 (751.3)
Ac-Y-I-R-NH-CH(CH3)-(CH2)2-CH3 - 562.3 (562.3)
Ac-Y-Pgl-R-L-P-NH2 - 722.2 (722.2)
Ac-Y-Chg-R-Ina-NH2 - 629.4 (629.4)
Ac-Tza-Chg-R-NH2 - 509.3 (509.3)
Ac-Y-Chg-R-Pip-NH2 - 629.4 (629.4)
Ac-Phe(pNH2)-Chg-R-NH2 - 517.2 (517.2)
Ac-(Bzl)G-(Chx)Gly-(3-guanidinopropyl)G-NH2 - 502.3 (502.3)
Ac-Y-Chg-R-ol.acetate - 547.3 (547.3)
Ac-Y-Chg-R-ОCH3 - 533.3 (533.3)
Ac-Y-Chg-R-OH - 519.3 (519.3)
Bz-Y-Chg-R-NH2 - 532.2 (532.2)
Пример XXXVI. Сочетания химических изменений, улучшающие активность, когда отдельные изменения могут не улучшать активность.
Измеряют ингибирование активности фактора Xa. Однако любое релевантное измерение биологической активности, такое как действие YIR пептида по изобретению на процесс коагуляции, активность in vivo, период полувыведения in vivo, оральная биодоступность, оральная доступность или период полувыведения, может служить мерой активности пептида по изобретению.
Описаны многие специфические изменения. В качестве примера осуществляют два изменения для того, чтобы показать, что дальнейшее повышение активности получают при сочетании изменений, даже тогда, когда отдельные изменения незначительно повышают активность.
Единичные химические изменения приводят к получению Ac-Y-I-R-L-P с Ki = 0,49 мкМ и (iBu)Y-I-R-L-P с Ki = 2,6 мкМ по сравнению с родительским соединением Y-I-R-L-P (Ki = 5,3 мкМ). Сочетание этих двух изменений приводит к получению Ac-(iBu)Y-l-R-L-P-NH2 с Ki = 0,04 мкМ. Таким образом эти результаты показывают, что пептид по изобретению с сочетанием двух химических изменений может обладать значительно повышенной ингибирующей активностью в отношении фактора Xa по сравнению с соответствующими аналогами с единичными изменениями даже тогда, когда одно родительское соединение, такое как (iBu)Y-I-R-L-P-NH2 не обладает значительно повышенной активностью в сравнении с родительским соединением Y-I-R-L-P-NH2.
В таблице 3 приведены примеры конкретных химических модификаций, которые обеспечивают получение соединений, имеющих значения Ki между 100 мкМ и 1 пМ в отношении ингибирования фактора Xa.
Пример XXXVII. Ингибирование in vitro выбранных очищенных коагуляционных ферментов и других сериновых протеаз.
Способность соединения по изобретению ингибировать фактор Xa, тромбин, плазмин, эластазу и трипсин оценивается путем определения концентрации YIR пептида, которая приводит к ингибированию активности фермента на 50 % (ИК50). Очищенные ферменты используют в хромогенных методах. Для определения константы ингибирования корректируют величину ИК50 для сравнения с субстратом, используя формулу:
Ki = ИК50•(1/{1 + ((концентрация субстрата))/Km субстрата)}) (Chen and Prusoff, Biochem.Pharmacol. 22:3099- 3018(1973)).
а. Опыт с фактором Xa
Для этого метода используют буфер TBS-P (50 мМ Tris-Cl, pH 7,8, 200 мМ NaCI, 0,05% (вес/об) NaN3). ИК50 определяют путем соединения в соответствующих колодцах титрационного микропланшета Costar 25 мкл человеческого фактора Xa (Enzyme Research Laboratories, Inc.; South Bend, IN) в TBS-P; 40 мкл 10% (об/об) ДМСО в TBS-P (неингибированные контрольные образцы или различных концентраций испытуемого пептида, разведенного в 10% (об/об) ДМСО в TBS-P; и субстрата S-2765 (Na-бензил-оксикарбонил-D-Arg-Cly-L-Arg-п-нитроанимид); Kabi Pharmacia, Inc; Franklin, ОН) в TBS-P.
Определение осуществляют путем прекультивирования пептидного ингибитора плюс фермент в течение 10 мин, затем начинают опыт путем добавления субстрата до получения конечного объема 100 мкл. Первоначальную скорость гидролиза хромогенного субстрата определяют по изменению абсорбции при 405 нИ, используя кинетический планшет-ридер Bio-tek Instruments (Ceres UV900HDi) при 25oC в линейной части отрезка времени (обычно 1-5 мин после добавления субстрата). Концентрация ингибитора, вызывающая 50%-ное снижение скорости гидролиза субстрата, предопределяется линейной регрессией после построения зависимости относительных скоростей гидролиза (в сравнении с неингибированными контрольными образцами) от log концентрации пептида. Концентрация фермента равна 0,5 нМ, а концентрация субстрата - 140 мкМ.
б) Опыт с тромбином
Для этого метода используют буфер TBS-P. ИК50 определяют, как описано в примере XXXVII. а, за исключением того, что субстратом является S-2366 (L-PyroGlu-L-Pro-L-Arg-п-нитроанимид; Kabi) и ферментом служит человеческий тромбин (Enzyme Research Laboratories, Inc.; South Bend.IN). Концентрация фермента равна 1 нМ, и концентрация субстрата - 175 мкМ.
в) Опыт с плазмином
Для этого метода используют буфер TBS-P. ИК50 определяют, как в примере XXXVII. а, но в качестве субстрата используют S-2251 ((D)-Val-L-Leu-L-Lуs-п-нитpoaнимид; Kabi), а в качестве фермента - человеческий плазмин (Kabi). Концентрация фермента равна 5 нМ, и концентрация субстрата - 300 уМ.
г) Опыт с трипсином
Для этого опыта используют TBS-P, содержащий 10 мМ CaCl2. ИК50 определяют, как описано в примере XXXVII.а, но субстратом является BAPNA (Бензоил-L-Arg-п-нитроанимид; Sigma Chemical Со; St. Louis, Mo, а ферментом является бычий панкреатический трипсин (Тип XIII, обработанный ТРСК; Sigma). Концентрация фермента равна 50 нМ, а концентрация субстрата - 300 мкМ.
д) Опыт с эластазой
Для этого опыта применяют буфер Tris-Cl pH 7,4, 300 мМ, 300 мМ NaCI, 2% (об/об) N-метилпирролидона, 0,01% (вес/об) NaN3.
ИК50 определяют, как описано в примере XXXVII.а, но субстратом является сукцинил-Ala-Ala-п-нитроанимид (Calbiochem-Nova Biochem Corp. ; San Diego CA), а ферментом - эластаза человеческого нейтрофила (Athens GA). Концентрация фермента составляет 75 нМ, концентрация субстрата - 600 мкм.
Величина Ki для испытуемых соединений в сравнении с контрольным соединением "TENSTOP" (N-альфа-тозил-Cly-п-амидинофенилаланина метиловый эфир; American Diagnostica, Inc., Greenwich CT), которое является обратимым ингибитором фактора Xa (Sturzebecher et al., Thromb.Res. 54:245-252 (1989); Hauptmann et al., Thromb Haerm. 63:220-223 (1990)) приведены в таблице 2. Данные показывают, что YIR пептиды по изобретению могут ингибировать активность фактора Xa, но практически не ингибируют активность других сериновых протеаз, включая тромбин и плазмин, которые участвуют в процессе коагуляции крови и фибринолиза.
Пример XXXVIII. Опыт по определению ингибирования коагуляции.
Соединения по изобретению оценивают по их способности ингибировать активность фактора Xa. Эффективность различных соединений оценивают по времени протромбина (ПВ) in vitro, используя плазму группового донора.
Используют также опыт ex vivo, в котором собирают плазму в разное время после внутривенного (вв) введения соединения крысам или кроликам или интрадуоденального введения крысам и, применяя определение ПВ, определяют период полураспада плазмы, определение ПВ инициируют разбавлением тромбопластина для того, чтобы получить протяженную и легко воспроизводимую конечную точку коагуляции, это называется "определение ПА с разбавлением", как описано ниже. Эффективность различных соединений также определяют, используя in vivo шунтирующую модель тромба у крыс.
а) Определение in vitro времени разбавленного протромбина
10 мкл подогретой (37oC) бедной тромбоцитами плазмы групповой человеческой крови (БТП) добавляют в чашку фиброметра. (Baxter Diagnostics, Inc.; McGaw Park Il).
Добавляют 50 мкл испытуемого соединения в различных концентрациях в TBS-BSA с кальцием (50 мМ Tris-Cl, 100 мМ NaCl, 0,1% (вес/об) альбумина бычьей сыворотки, 20 мМ CaCl2). В контрольных опытах для измерения времени неингибированной коагуляции добавляют TBS-BSA с кальцием, но без испытуемого соединения.
В чашку фиброметра добавляют 150 мкл разбавленного подогретого тромбопластина кролика и запускают таймер фиброметра. До испытания соединения получают кривую разбавления тромбопластина кролика и используют ее для выбора степени разбавления тромбопластина, которая дает ПВ, равное примерно 30 с для неингибированных контрольных образцов. Опытная концентрация, обеспечивающая 50% ингибирование коагуляции (ИК50) испытуемым соединением, (см. таблица 4) рассчитывается по временным параметрам кривой разбавления.
По другому варианту определение времени разбавленного протромбина осуществляют, используя "исследовательский метод" на автоматическом приборе ACL3000-plus, Instrumentation Laboratories (IL; Milan, Italy). Тромбопластин разбавляют, пока не достигнут времени свертывания, равного 30-35 с. Это время свертывания принимают за 100%. Строят стандартную кривую калибровки путем серийного 2-х-разового разбавления разбавленного тромбопластина. (Тромбопластин мозга кролика IL-группы). Во время опыта образец, 50 мкл, (плазма, отделенная центрифугированием) смешивают со 100 мкл тромбопластина и через 169 с снимают нефелометрические показатели. Время коагуляции определяют из максимальной скорости изменения светорассеяния, рассчитанной на приборе.
Ингибирование выражают как активность в процентах, определенную путем сравнения с калибровочной кривой.
б) Ex vivo определение времени разбавленного протромбина
Испытуемое соединение вводят внутривенно или через хвостовую вену (крыса) или ушную вену (кролик) по одобренной методике.
Отбирают образцы крови по 1 мл через измеренные промежутки времени после введения испытуемого соединения из канюлированной сонной артерии (крыса) или ушной артерии (кролик). После центрифугирования для получения БТП плазму немедленно помещают для хранения на лед или замораживают.
Для определения времени разбавленного протромбина плазму подогревают и анализируют, как указано выше.
Процент ингибирования рассчитывают из кривой разбавления тромбопластина, которая строилась для каждой серии образцов и использовалась для определения времени, при котором примерно 50% от первоначальной антикоагулирующей активности остается у плазмы (T 1/2). Результаты этого опыта показывают, что YIR пептиды по изобретению могут ингибировать коагуляцию крови in vitro и после введения in vivo (см. таблицу 4).
Различные соединения испытывают также на антикоагулирующую активность, используя ex vivo определение времени разбавленного протромбина, применяя внутривенное болюсное введение различных доз крысам.
Все соединения, перечисленные в таблице 5, показывают, по меньшей мере, 30%-ное ингибирование через 10 мин после введения <2 мг/кг указанного соединения. Эти результаты показывают, что различные представители YIR пептидов по изобретению обладают значительной антикоагулирующей активностью. Структуры всех соединений, перечисленных в таблице 5, были подтверждены ИС и данными анализа аминокислот.
Таблица 5.
1. Ac-pAph-Chg-PalMe(3)-NH-CH2-Chx
2. Ac-pAph-Chg-PalMe(3)-NH-2CMT
3. Ac-pAph-Chg-PalMe(3)-NH-Chx
4. Ac-F(pNH2)-Chg-Dab(Ny-C3NH7)L-P-NH2
5. Bz-F(pNH2)-Chg-R-L-P-NH2
6. Tos-F(pNH2)-Chg-R-L-P-NH2
7. Ac-Y(3-I)-Chg-R-L-P-NH2
8. Ac-pAph-Chg-AMP(4)
9. y-Chg-R-L-NH2
10. Ac-F(pNH2)-Chg-R-ol
11. Cyclopentyl-CO-pAph-Chg-PalMe(3)-NH2
12. 2-Iqc-pAph-Chg-PalMe(3)-NH2
13. Bzf-pAph-Chg-PalMe(3)-NH2
14. 3-Iqc-F(pNH2)-Chg-R-L-P-NH2
15. Ac-F(pNH2)-Chg-R-Thiazolyl
16. 2-Furoyl-pAph-Chg-PalMe(3)-NH2
17. 5-Me-thienyl-CO-pAph-Chg-PalMe(3)-NH2
18. Ac-Nal(2)-Chg-R-Thiazolyl
19. 2-Bzf-f(pNH2)-Chg-R-L-P-NH2
20. Ac-pAph-Chg-Dab(Ny-C3NH7)-L-P-NH2
21. Ac-Orn-Nal(2)-Chg-PaiMe(3)-Sar-E-NH2
22. Ac-Phe(2-1,4-NH2)-Chg-R-L-P-NH2
23. Ac-(iBu)pAph-Chg-R-L-P-NH2
24. Ac-pAph-Chg-R-Gla-P-NH2
25. Ac-pAph-Chg-R-Pen (CH2COOH)-P-NH2
26. Ac-pAph-Ghg-R-L-P-NH2
27. AC-F(pNH2)-Chg-R-(Me)L-P-NH2
28. Ac-F(pNH2)-Chg-R-OEt
29. Ac-F(pNH2)-Chg-Orn(Nδ- C3H7N)-L-P-NH2
30. Ac-F(pNH2)-Chg-R-L-P-NH2
31. Ac-Nal(2)-Chg-R-L-P-NH2
32. Ac-pAph-Chg-Dab(Ny-C3H7N)
33. Ac-pAph-Chg-PalMe(3)-NH2
34. Ac-pAph-Chg-PaiMe(3)-L-P-NH2
35. Ac-pAph-Chg-R-NH2
36. Ac-pAph-Chg-R-OH
37. Ac-pAph-Chg-R-NH-Nip-NH2
38. Ac-K-Nal(2)-Chg-R-Hyp-E-NH2
39. DIPA-pAph-Chg-R-L-P-NH2
40. DIPA-mF(pNH2)-Chg-R-L-P-NH2
41. Isn-F(pNH2)-Chg-R-L-NH2
42. Pza-F(pNH2)-Chg-R-L-P-NH2
43. Tfa-(iBu)F(pNH2)-Chg-R-L-P-NH2
44. Tfa-(iBu)Y-Chg-R-L-P-NH2
45. Tfa-(iBu)Y-I-Orn(Nδ- C3H7N)-L-P-NH2
В некоторых опытах испытуемые соединения вводились крысам интрадуоденально. Мужские особи крыс Spraque-Dawley подвергают анестезии при помощи сочетания кетамин/ксилазин, вводимых подкожно. Правую сонную артерию канюлируют для взятия проб крови. Осуществляют лапаротомию и канюлирование двенадцатиперстной кишки иглой с шарообразным кончиком и прижимают ее, чтобы обеспечить периферическое расположение шва относительно точки введения. Дополнительная растяжка помещается проксимально к точке введения для предотвращения просачивания содержимого желудка. Эффективность шва в предотвращении достижения соединением участка введения испытывают, применяя давление в конце каждого опыта.
Точка введения располагается на расстоянии примерно 4 см от соединения двенадцатиперстная кишка-желудок. Соединения вводят в 1 мл физиологического раствора. Образец крови 0,7 мл отбирают до введения испытуемого соединения и через 15, 30, 60, 90 и 120 мин после введения. Плазму отделяют цетрифугированием и анализируют на ингибирование коагуляции с использованием метода определения времени разбавленного протромбина.
Следующие соединения проявляют, по меньшей мере, 30% ингибирование при определении времени разбавленного протромбина с последующим интрадуоденальным введением 50 мг/кг соединения: Ac-pAph-Chg-PalMe(3)-NH-CHz-Chx; Ac-pAph-Chg-PalMe(3)-NH-Chx; Bzf-pAph-Chg-PalMe(3)-NH2; Ac-F(pNH2)-Chg-R-L-P-NH2; Ac-pAph-Chg-PalMe(3) -L-P-NH2; Ac-pAph-Chg-PalMe(3)-NH2; Ac-Aph-Chg-AMP(4); Cyclopentyl-CO-pAph-Chg-PaIMe(3)-NH2; 3-Iqc-pAph-Chg-PalMe(3)-NH2; 2-Furoyl-pAph-Chg-PalMe(3)-NH2; 5-Me-thienyl-CO-pAph-Chg-PalMe (3)-NH2, Ac-Y(3-I)-Chg-R-L-P-NH2, Ac-F(pNH2)-Chg-R-ol и Ac-pAph-Chg-PalMe(3)-ol.
в) Модель артериовенозного анастомоза тромбоза у крыс
Антитромботическую эффективность различных соединений по изобретению оценивают, используя экстракорпоральное артериовенозное (АВ) шунтирование.
Контур АВ шунтирования (анастомоза) состоит из трубки длиной 20 см из полиэтилена (ПЭ) 60, вставленной в правую сонную артерию, трубки длиной 6 см из ПЭ160, содержащей нить из мерсеризованного хлопка длиной 6,5 см (отрезок длиной 5 см соприкасается с потоком крови), и второй трубки из ПЭ60 длиной 20 см, заканчивающей контур в левой яремной вене.
Весь контур заполнен физиологическим раствором до введения соединений.
Испытуемые соединения вводят путем непрерывного вливания в хвостовую вену, используя поршневой насос и катетер (объем вливания 1,02 мл/час). Соединение вводят в течение 30 мин, затем открывают шунт и дают крови течь в течение 15 мин (всего 45 мин вливания). В конце 15-минутного периода шунт зажимают, нить аккуратно удаляют и взвешивают на аналитических весах.
Процент ингибирования образования тромба определяют по весу тромба, образовавшегося у контрольных крыс, которым вливали физиологический раствор.
Следующие соединения ингибируют рост тромба, по меньшей мере, на 30% после вливания 33 мкг/кг/мин: Ac-pAph-Chg-PalMe(3)-NH-CH2-Chx; Ac-pAph-Chg-PalMe(3) -NH-Chx; Bzf-pAph-Chg-PaIMe(3)-NH2; Ac-pAph-Chg-PalMe(3) -L-P-NH2; Ac-pAph-Chg-PalMe(3)-NH2; Ac-pAph-Chg-AMP (4); Cyclopentyl-CO-pAph-Chg-PaIMe(3)-NH2; 3-Iqc-pAph-Chg-PalMe(3)-NH2; 2-Furoyl-pAph-Chg-PalMe(3)-NH2; 5-Me-thienyl-CO-pAph-Chg-PalMe(3)-NH2, Ac-pAph-Chg-PalMe (3)-ol и Tos-F(pNH2)-Chg-R-L-P-NH2.
Хотя изобретение было раскрыто с описанием различных вариантов, специалистам ясно, что подробные конкретные примеры только иллюстрируют изобретение. Следует иметь в виду, что, не выходя за рамки изобретения, можно сделать различные изменения.
Соответственно изобретение ограничено только нижеследующей формулой изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ МЕТАСТИНА И ИХ ПРИМЕНЕНИЕ | 2007 |
|
RU2454425C2 |
| ПЕПТИДЫ, ОБЛАДАЮЩИЕ АГОНИСТИЧЕСКОЙ АКТИВНОСТЬЮ В ОТНОШЕНИИ РЕЦЕПТОРА НЕЙРОПЕПТИДА-2-(Y2R) | 2006 |
|
RU2383553C2 |
| ПРОЛЕКАРСТВЕННЫЕ КОМПОЗИЦИИ С ВЫСОКОЙ СТЕПЕНЬЮ ПРОНИКНОВЕНИЯ НА ОСНОВЕ ПЕПТИДОВ И РОДСТВЕННЫХ ПЕПТИДАМ СОЕДИНЕНИЙ | 2010 |
|
RU2627065C2 |
| ПРОИЗВОДНЫЕ МЕТАСТИНА И ИХ ПРИМЕНЕНИЕ | 2006 |
|
RU2430107C2 |
| ПЕПТИДНЫЕ ВЕКТОРЫ | 2004 |
|
RU2361876C2 |
| БЛОКАТОРЫ Kv1.3 | 2020 |
|
RU2825633C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ПЕПТИДАМИДОВ ИЛИ ИХ ФИЗИОЛОГИЧЕСКИ СОВМЕСТИМЫХ АЦЕТАТОВ ИЛИ ГИДРОХЛОРИДОВ | 1991 |
|
RU2036200C1 |
| ПЕПТИДНЫЕ КОМПОЗИЦИИ | 2014 |
|
RU2725150C2 |
| АНАЛОГИ ПЕПТИДА ЛГ-РФ, ИХ ПРИМЕНЕНИЕ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 1998 |
|
RU2212247C2 |
| Ингибиторы фактора VIIa | 1999 |
|
RU2223967C2 |





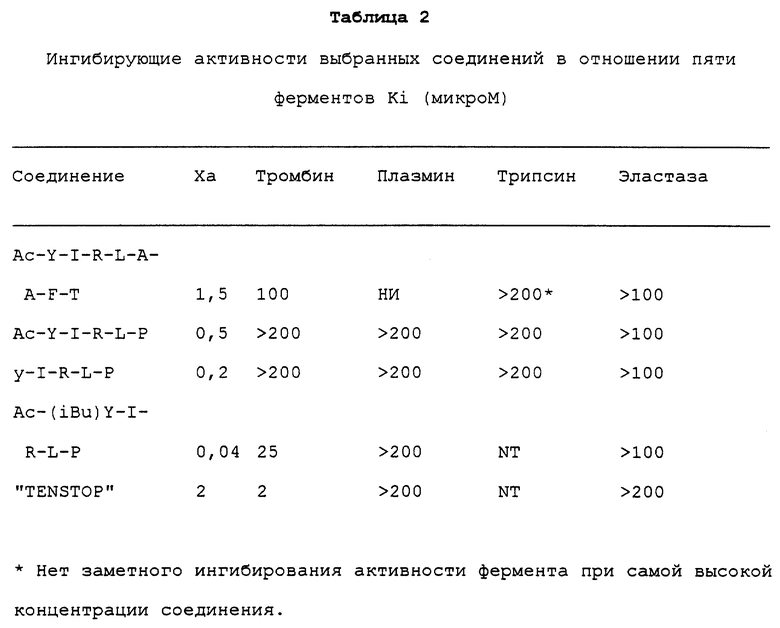


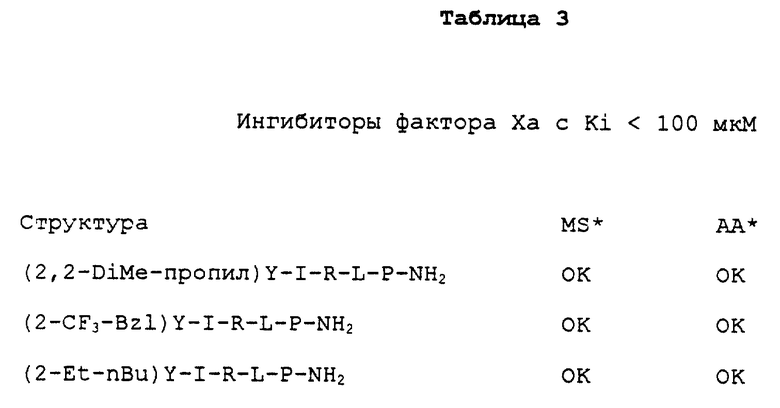











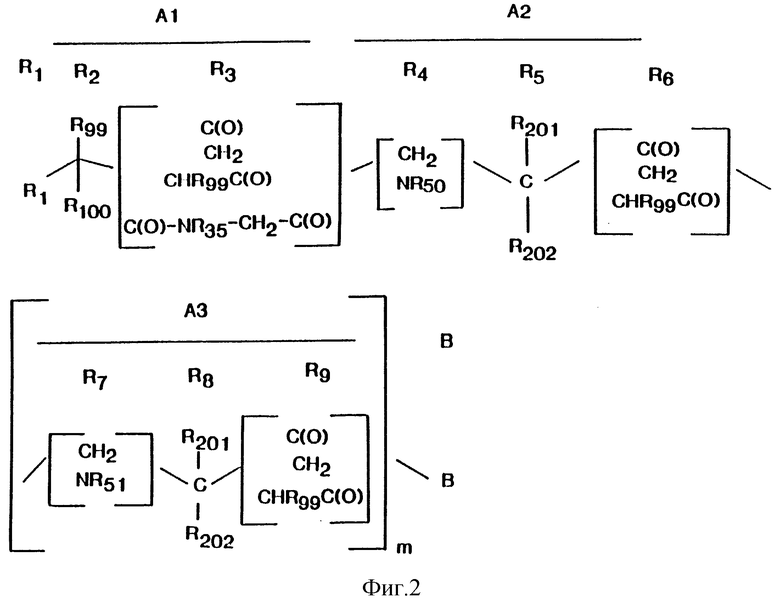

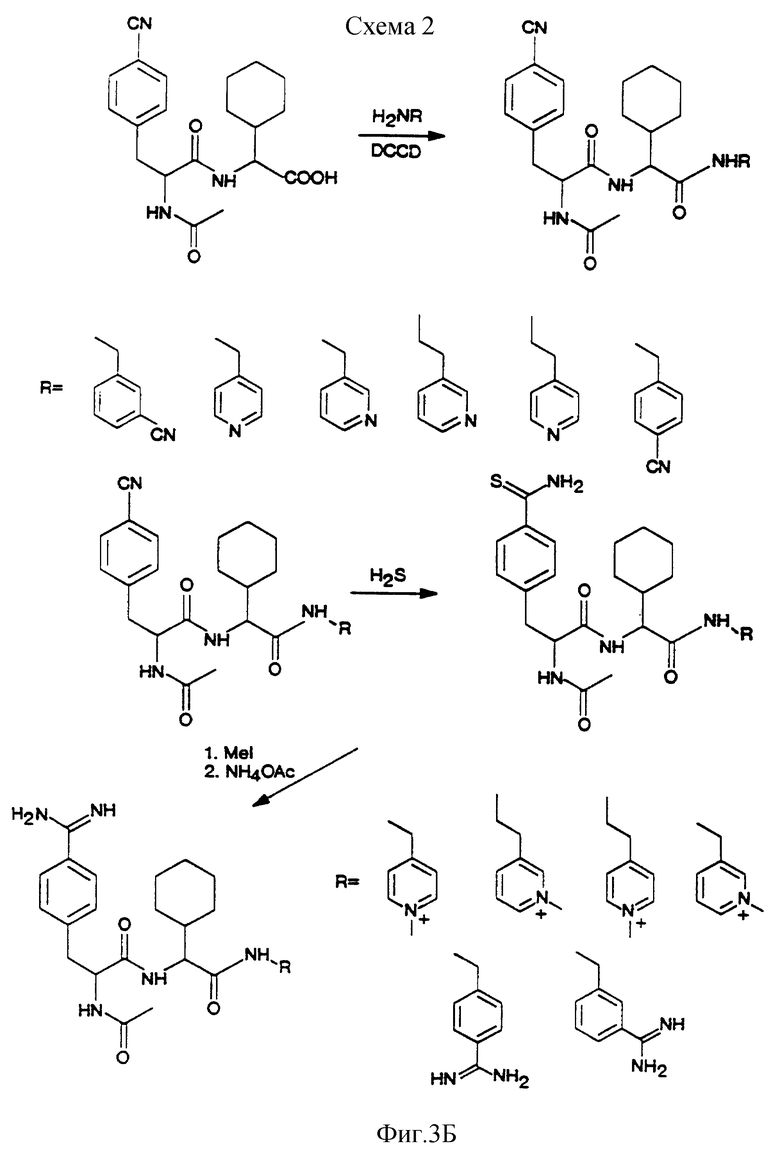
Описывается соединение неприродного происхождения, специфически ингибирующее активность фактора Ха, имеющее общую формулу (I) А1-А2-(А3)m-В, где m = 0 или 1; А1 - R1-R2-R3; А3 - R7-R8-R9; R1 выбран из группы, состоящей из R'1-X-R1", где X - N и R'1 и R''1 независимо выбраны из группы, состоящей из Н, алкила, ацила арила, арилалкила и групп, защищающих аминогруппу; R2 - -CR99R100, где R99 и R100 независимо выбраны из группы, включающей Н, алкил, арилалкил, гетероарилалкил и гетероарил; R3 - -С(O)- или -СН2-; R4 - -NR50-, где R50 - Н; R5 - -СR201R202, где R201 и R202 независимо выбраны из группы, включающей Н, алкил, арил и арилалкил; R6 - -С(O) или -СН2-; R7 - -NR51, где R51 - Н; R8 - -CR210R211-, где R210 и R211 независимо выбраны из группы, включающей Н, алкил, арилалкил и гетероцикл, причем алкил, алкиларил и гетероцикл может быть замещен Q или -(CH2)n-Q, где n = 1-5 и Q выбран из группы, включающей аминную, амидиновую, имидазольную и гуанидиновую группы, которые могут быть замещенными, и моно-, ди-, три- или тетраалкиламмоний фармацевтически приемлемой соли, ее изоуреид или изотиоуреид; R9 - -С(O)- или -СН2-; В - -NHR52, где R52 выбран из группы, включающей Н, алкил, арилалкил, гетероарилалкил, гетероарил, одну или две аминокислоты, или его фармацевтически приемлемую соль, амид, эфир, спирт или альдегид. Соединение формулы (I) предотвращает образование сгустков крови, являясь специфическим низкомолекулярным ингибитором фактора Ха, эффективен, но не вызывает нежелательных побочных эффектов. 8 с. и 20 з.п. ф-лы, 5 табл., 4 ил.
A1-A2-(A3)m-B,
где m = 0 или 1;
A1 - R1-R2-R3;
A2 - R4-R5-R6;
A3 - R7-R8-R9;
R1 выбран из группы, состоящей из
где X - N и R'1 и R''1 независимо выбраны из группы, состоящей из H, алкила, ацила, арила, арилалкила и групп, защищающих аминогруппу,
R2 - CR99R100, где R99 и R100, независимо выбраны из группы, включающей H, алкил, арилалкил, гетероарилалкил и гетероарил;
R3 - -C(O)- или -CH2-;
R4 - -NR50-, где R50 означает H;
R5 - CR201R202, где R201 и R202 независимо выбраны из группы, включающей H, алкил, арил и арилалкил;
R6 - -C(O) или -CH2-;
R7 - -NR51, где R51 - H;
R6 - -CR210, R211-, где R210, R211, независимо выбраны из группы, включающей H, алкил, арилалкил и гетероцикл, причем алкил, алкиларил и гетероцикл могут быть замещены Q или -(CH2)n-Q, где n = 1 - 5 и Q выбран из группы, включающей аминную, амидиновую, имидазольную и гуанидиновую группы, которые могут быть замещенными, и моно-, ди-, три- или тетраалкиламмоний фармацевтически приемлемой соли, ее изоуреид или изотиогуреид;
R9 - -C(O)- или -СH2-;
B - -NHR52, где R52выбран из группы, включающей H, алкил, арилалкил, гетероарилалкил, гетероарил, одну аминокислоту или две аминокислоты,
или его фармацевтически приемлемую соль, амид, эфир, спирт или альдегид.
Ac-pAph-Chg-PalMe(3)-NH-CH2-Chx;
Ac-pAph-Chg-PalMe(3)-NH-2CMT;
Ac-pAph-Chg-PalMe(3)-NH-Chx;
Ac-F(pNH2)-Chg-Dab(Nγ-C3NH7)-L-P-NH2(SEQ ID NO : 263);
Bz-F(pNH2)-Chg-R-L-P-NH2(SEQ ID NO : 264);
Tos-F(pNH2)-Chg-R-L-P-NH2(SEQ ID NO : 265);
Ac-Y(3 - 1)-Chg-R-L-P-NH2(SEQ ID NO : 266);
y-Chg-R-L-NH2;
Ac-F(pNH2)-Chg-R-ol;
Cyclopentyl-CO-pAph-Chg-PalMe(3)-NH2;
3-Igc-pAph-Chg-PalMe(3)-NH2;
Bzf-pAph-Chg-PalMe(3)-NH2;
3-Igc-F(pNH2)-Chg-R-L-P-NH2;
Ac-F(pNH2)-Chg-R-NH-2-thiazolyl;
2-Furoyl-pAph-Chg-PalMe(3)-NH2;
5-Me-2-thienyl-CO-pAph-Chg-PalMe(3)-NH2;
Ac-Nal(2)-Chg-R-NH-2-thiazolyl;
2-Bzf-F(pNH2)-Chg-R-L-P-NH2;
Ac-pAph-Chg-Dab(Nγ-C3H7N)-L-P-NH2(SEQ ID NO : 268);
Ac-(iBu)pAph-Chg-R-L-P-NH2(SEQ ID NO : 271);
Ac-pAph-Chg-R-Gla-P-NH2(SEQ ID NO : 272);
Ac-pAph-Chg-R-Pen(CH2COOH)-P-NH2(SEQ ID NO : 273);
Ac-pAph-Chg-R-L-P-NH2(SEQ ID NO : 274);
Ac-F(pNH2)-Chg-R-(Me)L-P-NH2(SEQ ID NO : 275);
Ac-F(pNH2)-Chg-R-OEt;
Ac-F(pNH2)-Chg-Orn(Nδ-C3H7N)-L-P-NH2(SEQ ID NO : 276);
Ac-F(pNH2)-Chg-R-L-P-NH2(SEQ ID NO : 277);
Ac-Nal(2)-Chg-R-L-P-NH2(SEQ ID NO : 278);
Ac-pAph-Chg-Dab(Nγ-C3H7N)-NH2;
Ac-pAph-Chg-PalMe(3)-NH2;
Ac-pAph-Chg-PalMe(3)-L-P-NH2(SEQ ID NO : 279);
Ac-pAph-Chg-R-NH2;
Ac-pAph-Chg-R-OH;
Ac-pAph-Chg-R-ol;
DIPA-(m)pAph-Chg-R-L-P-NH2(SEQ ID NO : 36);
DIPA-(m)F(pNH2)-Chg-R-L-P-NH2(SEQ ID NO : 289);
Isn-F(pNH2)-Chg-R-L-P-NH2(SEQ ID NO : 282);
Pza-F(pNH2)-Chg-R-L-P-NH2(SEQ ID NO : 283);
Tfa-(iBu)Y-Chg-R-L-P-NH2(SEQ ID NO : 285);
Tfa-(iBu)Y-1-Orn(Nδ-C3H7N)-L-P-NH2(SEQ ID NO : 286);
CF3C(O)-(iBu)Phe(NH2)-Chg-Arg-Leu-Pro-NH2;
Ac-pAph-Ile-Arg-Leu-Pro-NH2;
CF3C(O)-(iBu)Nal(2)-Chg-Arg-Leu-Pro-NH2;
Ac-Phe-(31,4NH2)-Chg-Arg-Leu-Pro-NH2;
CF3C(O)-Tyr-Chg-Arg-Leu-Pro-NH2;
(5-blnzimidazoyl)-Phe(NH2)-Chg-Arg-Leu-Pro-NH2;
CF3C(O)-(iBu)Tyr-Ile-Arg-Leu-Pro-NH2;
AC-(Chx-CH2)Tyr-Ile-Arg-Leu-Pro-NH2;
D-Tyr-Chg-Arg-Leu-Pro-NH2;
Ac-Trp-Chg-Arg-Leu-Pro-NH2;
23. Соединение неприродного происхождения, выбранное из группы, включающей
Ac-pAph-Chg-PalMe(3)-NH-CH2-Chx;
Ac-pAph-Chg-PalMe(3)-NH2;
Brf-pAph-Chg-PalMe(3)-NH2;
Ac-pAph-Chg-PalMe(3)-L-P-NH2(SEQ ID NO : 279);
Ac-pAph-Chg-PalMe(3)-NH2;
Cyclopentyl-CO-pAph-Chg-PalMe(3)-NH2;
3-Igc-pAph-Chg-PalMe(3)-NH2;
2-Furoyl-pAph-Chg-PalMe(3)-NH2;
5-Me-thienyl-CO-pAph-Chg-PalMe(3)-NH2; и
Ac-pAph-Chg-PalMe(4)-ol.
Ac-Tyr-Ile-Arg-Leu-Ala-NH2(SEQ ID NO : 2);
Ac-Tyr-Ile-Arg-Leu-NH2(SEQ ID NO : 3);
Ac-(iBu)Tyr-Ile-Arg-Leu-Pro-NH2(SEQ ID NO : 4);
Ac-Tyr-Ile-Arg-N(CH3)O(CH3);
Ac-Tyr-{ψ(CH2NH)}Ile-Arg-Leu-Pro-NH2(SEQ ID NO : 5);
AC-Tyr-Ile-Arg-NH-CH2(4-Pyridyl);
Ac-Tyr-Ile-{ψ(CH2NH)}-Arg-Leu-Pro-NH2(SEQ ID NO : 6);
Ac-Tyr-Chg-Arg(NO2)-{ψ(CH2NH)}-Leu-NH2(SEQ ID NO : 7);
Ac-Tyr-Ile-Arg-{ψ(COCH2)}-Gly-Pro-NH2(SEQ ID NO : 8);
Ac-Tyr-Ile-Dab(Nγ-C3H7N)Leu-Ala-NH2(SEQ ID NO : 9);
Ac-Tyr-Ile-PalMe(3)-NH2;
Tyr-Ile-Arg-NH2;
D-Tyr-Ile-Arg-Leu-Pro-NH2;
Ac-(Bzl)Gly-(Chx)Gly-(3-guanidopropyl)Gly-NH2;
Cyclo(Gly-Tyr-Ile-Arg-Gly)(SEQ ID NO : 10);
Tfa-(iBu)Tyr-Chg-Arg-Leu-Pro-NH2(SEQ ID NO : 11);
Ac-pAph-Chg-Arg-Leu-Pro-NH2(SEQ ID NO : 12);
Ac-Nal(2)-Chg-Arg-Leu-Pro-NH2(SEQ ID NO : 13);
Ac-pAph-Chg-PalMe-NH2.
Ac-Y-I-R-L-A-NH2(SEQ ID NO : 290);
Ac-Y-I-R-L-P-NH2(SEQ ID NO : 149);
Ac-(iBu)-Y-I-R-L-P-NH2(SEQ ID NO : 94);
Ac-Y-I-R-N-(CH3)O(CH3);
Ac-Y-{ψ(CH2NH)}-I-R-L-P-NH2(SEQ ID NO : 135);
AC-Y-I-R-NH-CH2(4-Pyridyl);
Ac-Y-I-{ψ(CH2NH)}-R-L-P-NH2(SEQ ID NO : 141);
Ac-Y-Ghg-R(NO2){ψ(CH2NH)}-L-NH2(SEQ ID NO : 131);
Ac-Y-I-R-{ψ(COCH2)}-G-P-NH2(SEQ ID NO : 136);
Ac-Y-I-Dab(Nγ-C3H7N)-L-A-NH2(SEQ ID NO : 137);
Ac-Y-I-PalMe(3)-NH2;
Y-I-R-NH2;
D-Y-I-R-L-P-NH2;
Ac-(Bzl)Gly-(Chx)Gly-(3-guanidopropyl)Gly-NH2;
Cyclo(G-Y-I-R-G)(SEQ ID NO : 183);
Tfa-(iBu)Y-Chg-R-L-P-NH2(SEQ ID NO : 221);
Ac-pAph-Chg-R-L-P-NH2(SEQ ID NO : 122) и
Ac-Nal(2)-Chg-R-L-P-NH2(SEQ ID NO : 119).

