Изобретение относится к области гидрометаллургии платиновых металлов, а именно, к осадительному способу выделения рутения в форме малорастворимых комплексных соединений, и может быть использовано в процессах аффинажного производства, а также при переработке и захоронении отработанного топлива АЭС, что особенно важно, так как содержание осколочных платиновых металлов, в том числе и рутения, в них во много раз превышает содержание этих металлов в используемом ныне сырье.
Образующиеся в процессах аффинажа платиновых металлов и при переработке отработанного топлива АЭС производственные растворы содержат рутений, в основном в виде нитрозонитро-, нитрозохлоро-, хлоро- и амминокомплексов рутения. Эти соединения устойчивы и хорошо растворимы в воде, что весьма осложняет процесс их извлечения из раствора с целью дальнейшей переработки для выделения рутения (Ru).
Известен способ выделения рутения и родия из растворов продуктов деления ядерного топлива (пат. GB №941985, кл. C 01g(G21), 1960) посредством осаждения сульфидов из солянокислого раствора действием сероводорода. Способ применим при отсутствии окислителей в обрабатываемом растворе, что достигается многократным упариванием исходных азотнокислых растворов с соляной кислотой. Высокая токсичность сероводорода является существенным недостатком процесса.
Известен способ выделения рутения, родия и палладия из водных растворов хлоридных солей этих металлов действием монооксида углерода на слабокислые водные растворы (пат. US №4081271, кл. C22B 011/04, 1978. Ugo R. Process for separating and recovering rhodium and iridium from their mixtures with other precious metals). Недостатком способа является высокая токсичность монооксида углерода.
Развитие вышеупомянутого способа изложено в (пат. US №4163664, кл. C22B 011/04, 1979. Ugo R. Process for precipitating precious metals from solutions which contain them). Недостаток способа сохранился без изменений.
Известен способ группового выделения осколочных платиноидов из азотнокислых растворов отработанного ядерного топлива, включающий отгонку воды и азотной кислоты, получение твердой шихты с добавлением соединений бора (нитрида бора, карбида бора или борогидрида натрия), восстановительную плавку шихты при 2000°C и отделение сплава платиновых металлов от расплавленной оксидной фазы (пат. US №5082603, кл. G21F 009/00, 1992. Horie М., Fukumoto М., Yoneya М. Method of treatment of high-level radioactive waste). Недостатками способа являются потери рутения при отгонке азотной кислоты, высокая температура проведения процесса и сложность отделения металлической фазы от оксидной при этой температуре.
Известен способ получения концентрата платиновых металлов путем их осаждения из царсководочного раствора тиосульфатом натрия (заявка РФ №97111284, кл. C22B 11/00, 1999. Лолейт С.И., Калмыков Ю.М., Давыдова В.Я. и др. «Способ извлечения платиновых металлов из содержащего их материала»). Недостатком способа является большой расход тиосульфата натрия, поскольку большей частью он идет на удаление окислителя в царсководочном растворе. Кроме того, поведение редких платиновых металлов в этом процессе не описано.
Известен способ выделения рутения, родия и палладия из растворов отработанного ядерного топлива (пат. US №5393322, кл. C22B 011/00, 1995. Ugo R. "Process for recovering noble metals from solutions deriving from the treatment of nuclear fuels"), основанный на осаждении платиновых металлов путем восстановительного карбонилирования азотнокислых растворов при pH 2-4 монооксидом углерода при давлении 1 атм и температуре от 20 до 100°C в течение 6-100 часов. Основным недостатком метода является предварительное удаление основной массы азотной кислоты из раствора с помощью формальдегида. Такой прием, как было показано (King R.B., Bhattacharyya N.K. // Environ. Sci. Technol. 1997. V. 31. №4. P.984), приводит к частичному восстановлению азотной кислоты до аммиака с последующим образованием аммиачных комплексов платиновых металлов, которые практически не поддаются восстановлению. При проведении процесса в области pH 2-4 для предотвращения гидролиза и выпадения его продуктов в твердую фазу требуется введение в систему маскирующего комплексообразователя - Трилона Б (ЭДТА), что усложняет процесс. Использование высокотоксичного газообразного монооксида углерода в качестве восстановителя предъявляет повышенные требования к оборудованию и технике безопасности.
Известен способ выделения рутения из нитратно-цитратных растворов (пат. RU №2230034, кл. C01G 55/00, 2004. Корнилов А.С., Филимонов В.Т. «Способ выделения рутения-106 из рафинатов производства трансплутониевых элементов»), заключающийся в следующем. Рафинаты производства трансплутониевых элементов, содержащие 0,2-0,7 моль/л нитрат-ионов и 0,3-0,5 моль/л цитрат ионов, подвергаются обработке раствором серы в сульфиде натрия до концентрации серы 0-0,5 моль/л, сульфида натрия до 0,3-0,5 моль/л. Проводят сорбцию при pH 8-12 на анионите высокоосновном (АВ-17) в нитратной форме. Промывают анионит водой. Десорбцию рутения проводят раствором аммиака концентрацией не менее 1 моль/л, содержащим перекись водорода в количестве не менее стехиометрического по отношению к сорбированным сульфид- и полисульфид-ионам. Недостатком метода является низкая степень извлечения рутения (до 78%).
Известен способ выделения рутения, родия и палладия из азотнокислых растворов (пат. RU №2239666, кл. C22B 11/00, C22B 3/44, 2004. Беляев А.В., Корда Т.М., Храненко С.П., Емельянов В.А. «Способ получения концентрата родия, палладия и рутения из азотнокислых растворов»), заключающийся в том, что в раствор с концентрацией азотной кислоты 2-3 моль/л вводится в качестве осадителя твердый тиоцианат щелочного металла, с избытком, составляющим 1/3 от количества азотной кислоты в растворе, в интервале температур 18-80°C. Последующее прокаливание полученного осадка ведут при 750-800°C. Недостатком способа является неполное количественное извлечение рутения (94-96%), а также продолжительное время проведения процесса (не менее 14 ч). Способ ограничивается использованием азотнокислых растворов рутения.
Известен способ сорбционного удаления рутения из урансодержащих растворов (выбран нами в качестве прототипа) (пат. RU №2086508, кл. C01G 43/00, C10G 55/00, 1997. Флореансинг А., Никола Ф. «Способ извлечения рутения, содержащегося в урансодержащем растворе»). В раствор с кислотностью 0,01 н и 0,5 н, содержащий рутений, вносят нитриты щелочных металлов либо аммония до концентраций 0,1-50 г/л и рутений связывают в результате пропускания через смолу на основе четвертичного аммония или третичного амина. При необходимости элюирование проводят азотной кислотой в качестве сильной кислоты, предпочтительно, в виде концентрированного раствора. Способ позволяет удалять рутений из указанных растворов, содержащих от нескольких миллионных долей до многих г/л рутения. Недостатком способа является хранение рутения в сорбированном виде, что ведет к удорожанию процесса. В модификации способа удаления рутения, включающей десорбцию рутения, образуются азотнокислые растворы рутения, обладающие высокой коррозионной активностью.
Технической задачей данного изобретения является полное количественное (100%) извлечение рутения в форме малорастворимого комплексного соединения из хлоридных, нитритных, нитратных, аммиачных растворов рутения.
В настоящее время нет способа, который позволял бы выделять рутений (Ru) в форме чистого, устойчивого, нерастворимого в воде соединения с количественным (100%) выходом, тем самым решая проблему минимизации объемов производственных растворов. Кроме того, предлагаемый нами способ позволяет выделять в твердую фазу комплекс рутения (Ru) и из растворов, получающихся при переработке отходов ядерного топлива, наличие в которых платиновых металлов препятствует обязательной при захоронении жидких радиоактивных отходов операции «остекловывания».
Поставленная задача решается тем, что в способе выделения рутения, включающем взаимодействие комплексных соединений рутения с нитритом щелочного металла в качестве исходного соединения используют аммино-, хлоро-, нитрозохлоро- или нитрозонитрокомплексы рутения либо их кислые растворы и проводят взаимодействие с насыщенным водным раствором нитрита калия при температуре кипения реакционной смеси.
Отличительными от прототипа признаками являются:
- насыщенный раствор нитрита калия (KNO2);
- взаимодействие реагентов при температуре кипения водного раствора.
Продолжительность процесса составляет 30 минут.
Способы выделения рутения с такими отличительными признаками авторам не известны.
Сущность предлагаемого способа заключается в следующем. Процесс количественного (100%) выделения рутения в форме малорастворимого K4[Ru(NO2)6]·0,5H2O проводят в насыщенном водном растворе нитрита калия 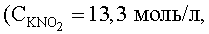
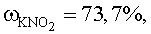
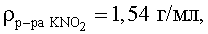
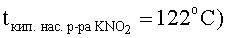
При использовании в качестве стартовых форм рутения твердых комплексных соединений (нитро-, хлоро-, нитрато-, аммино-, нитрозокомплексы рутения, а также комплексы, смешанные по этим лигандам) навеску комплекса вносят в насыщенный при комнатной температуре водный раствор нитрита калия из расчета 1,0·10-3÷2,6·10-1 моль рутения на 1 л насыщенного водного раствора KNO2.
В случае использования экстрактов рутения в органических растворителях соотношения соблюдаются те же.
При использовании кислых растворов комплексов рутения нитрит калия берут с 1,5-избытком от количества кислоты в растворе, при этом соотношение рутения к нитриту калия также приводят к 1,0·10-3-2,6-10-1 моль рутения на 1 л насыщенного водного раствора KNO2; нитрит калия вносят либо в твердом виде, либо в виде насыщенного раствора.
Проведение процесса в течение 30 минут позволяет получить 100% выход целевого продукта (Таблица 1) и именно нитрит калия позволяет получить нерастворимую соль. При этом получаемый гексанитрорутенат (II) калия кристаллизуется с 0,5 молекулы воды и полностью переходит в твердую фазу. Продукт отделяют от маточного раствора фильтрованием, промывают разбавленным водным раствором нитрита калия, затем 96% спиртом и диэтиловым эфиром.
Инфракрасный спектр и дифрактограмма получаемого желтого порошка идентичны приведенным в работах [Alomer S.S., Askar К.А., Habboush D.A. // Termochimica Acta. 1981. V. 45. № 1. P.1, Емельянов B.A., Беляев A.B., Федотов M.A. и др. // Журн. Неорган. химии. Т.37. №11. С.2515], что подтверждает получение именно гексанитросоединения Ru.
При необходимости затем проводят термическое разложение смеси K4[Ru(NO2)6]·0,5H2O с NH4Cl (1:1 по массе) на воздухе при 400°C в течение 1 часа, образующийся RuO2 отмывают водой от KCl.
Понижение температуры проведения процесса приводит к увеличению времени процесса и снижению выхода продукта. При меньших концентрациях KNO2 в растворе понижается температура реакционной смеси, что также увеличивает время проведения процесса и снижает выход целевого продукта. Проведение процесса в открытой системе с упариванием досуха и повторным растворением твердой фазы в воде также ведет к увеличению продолжительности процесса и снижению выхода продукта (Таблица 2).
Промышленная применимость способа иллюстрируется следующими примерами.
Пример 1. В круглодонную стеклянную колбу объемом 10 мл, снабженную обратным холодильником, помещают 0,039 г K2[Ru(NO)Cl5] (νRu=1,0·10-4 моль) и добавляют 5 мл насыщенного водного раствора KNO2 (5,7 г KNO2 в 2,0 мл H2O). Затем раствор нагревают до кипения и кипятят в течение 25 минут. После охлаждения реакционную смесь фильтруют, осадок промывают водой, сушат и взвешивают. Выход целевого продукта K4[Ru(NO2)6]·0,5H2O - 99,93%.
Пример 2. Реакцию проводят аналогично примеру 1. Навеска K2[Ru(NO)Cl5] - 0,3755 г (νRu=9,715·10-4 моль), объем насыщенного водного раствора KNO2 - 5 мл (5,7 г KNO2 в 2,0 мл H2O). Смесь кипятят 30 мин. Выход целевого продукта 99,995%.
Пример 3. Реакцию проводят аналогично примеру 1, но в качестве исходного соединения берут K2[Ru(NO)(NO2)4(OH)]. Навеска - 0,0394 г (νRu=9,60·10-5 моль), объем насыщенного водного раствора KNO2 - 5 мл (5,7 г KNO2 в 2,0 мл H2O). Смесь кипятят 20 минут. Выход целевого продукта - 100%.
Пример 4. В круглодонную колбу объемом 10 мл помещают 5 мл 0,02 моль/л раствора RuCl3 (νRu=1·10-4 моль) в 0,1 моль/л HCl, вводят 15 г KNO2 (0,2 моль) и кипятят 30 минут, затем проводят все операции, как описано в примере 1. Выход целевого продукта 99,95%.
Пример 5. Реакцию проводят аналогично примеру 1. Навеска [Ru(NH3)5Cl]Cl2 - 0,381 г (νRu=1,30·10-3 моль), объем насыщенного раствора KNO2 - 5 мл (5,7 г KNO2 в 2,0 мл H2O). Смесь кипятят 30 мин. Выход целевого продукта 100,0%.
Пример 6. В круглодонную колбу объемом 20 мл помещают 10 мл раствора нитрозонитроаквакомплексов рутения в трис-н-бутилфосфате (ТБФ) (cRu=0,795 г/л) и добавляют 5 мл насыщенного водного раствора KNO2 (5,7 г KNO2 в 2,0 мл H2O). Двухфазную систему (органический раствор и водный раствор) нагревают до температуры кипения водной фазы и кипятят 30 мин. После охлаждения отделяют органическую фазу (органический растворитель может быть возвращен в реакционный цикл). Водная фаза с осадком обрабатывается, как в примере 1. Выход целевого продукта 99,98%.
Пример 7. В круглодонную колбу объемом 25 мл помещают 0,5 мл раствора нитрозонитроаквакомплексов рутения в 3 М HNO3 (cRu=20,26 г/л, νRu=1,016·10-4 моль), 4,5 мл, и добавляют 15,2 г KNO2(0,2 моль). Систему нагревают до температуры кипения и кипятят 30 мин. После охлаждения проводят все операции, как в примере 1. Выход целевого продукта 100%.
Результаты химического анализа показывают, что получаемое соединение соответствует химической формуле K4[Ru(NO2)6]·0,5H2O и продукт не загрязнен другими компонентами реакционной смеси.
В результате химического анализа полученных желтых порошков найдено, %: K - 28,2±0,5, Ru - 18,6±0,3, Н - 0,21, N - 15,1±0,5. Для K4[Ru(NO2)6]·0,5H2O вычислено, %: K - 28,8, Ru - 18,6, H - 0,18, N - 15,5.
Помимо элементного анализа, индивидуальность получаемых соединений подтверждается данными ИК-спектроскопического и рентгенофазового исследований, показывающими идентичность ИК-спектров и дифрактограмм синтезированных комплексов приведенными в работах [Alomer S.S., Askar К.А., Habboush D.A. // Termochimica Acta. 1981. V. 45. №1. P.1, Емельянов B.A., Беляев A.B., Федотов M.A. и др. // Журн. неорган, химии. Т.37. №11. С.2515].
Предложенный способ выделения рутения в форме гексанитрорутената(II) калия, позволяющий полностью извлечь рутений в форме чистого продукта, может быть использован в аффинаже платиновых металлов и при переработке отходов отработанного топлива АЭС.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ КОНЦЕНТРАТА РОДИЯ, ПАЛЛАДИЯ И РУТЕНИЯ ИЗ АЗОТНОКИСЛЫХ РАСТВОРОВ | 2003 |
|
RU2239666C1 |
| СПОСОБ КОНЦЕНТРИРОВАНИЯ РУТЕНИЯ | 2002 |
|
RU2223918C1 |
| СПОСОБ СОРБЦИОННОГО ИЗВЛЕЧЕНИЯ И РАЗДЕЛЕНИЯ РОДИЯ И РУТЕНИЯ | 2014 |
|
RU2573853C2 |
| Способ извлечения родия, рутения и палладия из азотнокислых растворов | 2020 |
|
RU2762694C1 |
| Способ извлечения рутения из кислых растворов | 1990 |
|
SU1791752A1 |
| Способ выделения рутения из концентратов, содержащих благородные металлы | 2021 |
|
RU2758957C1 |
| СПОСОБ ИЗВЛЕЧЕНИЯ МЕТАЛЛОВ ПЛАТИНОВОЙ ГРУППЫ ИЗ ПРОДУКТА КИСЛОТНОГО РАСТВОРЕНИЯ ВОЛОКСИДИРОВАННОГО ОТРАБОТАВШЕГО ЯДЕРНОГО ТОПЛИВА | 2015 |
|
RU2607644C2 |
| СПОСОБ ИЗВЛЕЧЕНИЯ РУТЕНИЯ | 2013 |
|
RU2540163C1 |
| СПОСОБ ВЫДЕЛЕНИЯ РУТЕНИЯ ИЗ НЕРАСТВОРИМЫХ ОСТАТКОВ ОТ ПЕРЕРАБОТКИ ОБЛУЧЕННОГО ЯДЕРНОГО ТОПЛИВА | 2004 |
|
RU2289636C2 |
| СПОСОБ ОЧИСТКИ ТЕХНОЛОГИЧЕСКИХ УРАНОВЫХ ПРОДУКТОВ ПЕРЕРАБОТКИ ОТРАБОТАВШЕГО ЯДЕРНОГО ТОПЛИВА ОТ РУТЕНИЯ | 2014 |
|
RU2576530C1 |
Изобретение относится к области гидрометаллургии платиновых металлов, а именно к способам выделения рутения в форме гексанитрорутената (II) калия. Изобретение может быть использовано в процессах аффинажного производства, а также при переработке и захоронении отработанного топлива АЭС. Рутений количественно осаждают в форме гексанитрорутената (II) калия из растворов, содержащих хлоро-, аммино-, нитрозохлоро-, нитрозонитрато- или нитрозонитрокомплексы рутения в насыщенном растворе нитрита калия при температуре кипения реакционной смеси. Техническим результатом является количественное (100%) извлечение рутения в форме малорастворимого комплексного соединения. 6 з.п. ф-лы, 2 табл., 7 пр.
1. Способ извлечения рутения из комплексных соединений рутения, отличающийся тем, что в качестве исходных соединений используют соединения, выбранные из твердых хлоро-, аммино-, нитрозо-, нитрато-, нитрокомплексов рутения, комплексов, смешанных по этим лигандам, комплексов, взятых в виде кислых водных растворов этих комплексов, и комплексов, взятых в виде растворов соединений рутения в органических растворителях, при этом обработку исходного сырья осуществляют нитритом калия (KNO2), взятым в твердом виде или в виде насыщенного водного раствора нитрита калия при не менее чем 200 кратном мольном избытке нитрита калия по отношению к рутению и температуре кипения реакционной смеси с последующим отделением продукта в нерастворимой форме.
2. Способ по п.1, отличающийся тем, что обработку исходного сырья проводят при температуре кипения насыщенного водного раствора нитрита калия.
3. Способ по п.1, отличающийся тем, что его проводят в замкнутой системе с возвратом органического растворителя.
4. Способ по п.1, отличающийся тем, что обработку проводят в течение 30 мин.
5. Способ по п.1, отличающийся тем, что при использовании в качестве исходных соединений твердых комплексных соединений нитро-, хлоро-, нитрато-, аммино-, нитрозокомплексов рутения и комплексов, смешанных по этим лигандам, навеску комплекса вносят в насыщенный при комнатной температуре водный раствор нитрита калия из расчета 1,0·10-3÷2,6·10-1 моль рутения на 1 л насыщенного водного раствора KNO2.
6. Способ по п.1, отличающийся тем, что при использовании в качестве исходных соединений комплексов в виде растворов в органических растворителях, насыщенный водный раствор нитрита калия берут из расчета 1,0·10-3÷2,6·10-1 моль рутения на 1 л насыщенного водного раствора KNO2.
7. Способ по п.1, отличающийся тем, что при использовании в качестве исходных соединений кислых растворов комплексов рутения нитрит калия берут с 1,5-избытком от количества кислоты в растворе, после чего отношение рутения к нитриту калия доводят до значения 1,0·10-3÷2,6·10-1 моль рутения на 1 л насыщенного водного раствора KNO2.
| СПОСОБ ИЗВЛЕЧЕНИЯ РУТЕНИЯ, СОДЕРЖАЩЕГОСЯ В УРАНСОДЕРЖАЩЕМ РАСТВОРЕ | 1992 |
|
RU2086508C1 |
| СПОСОБ КОНЦЕНТРИРОВАНИЯ РУТЕНИЯ | 2002 |
|
RU2223918C1 |
| СПОСОБ ВЫДЕЛЕНИЯ РУТЕНИЯ-106 ИЗ РАФИНАТОВ ПРОИЗВОДСТВА ТРАНСПЛУТОНИЕВЫХ ЭЛЕМЕНТОВ | 2002 |
|
RU2230034C2 |
| US 4282112 A, 04.08.1981 | |||
| JP 9203792 А, 05.08.1997 | |||
| JP 2001098335 A, 10.04.2001 | |||
| Прибор, замыкающий сигнальную цепь при повышении температуры | 1918 |
|
SU99A1 |
| EP 0756013 А1, 29.01.1997 | |||

