Область техники, к которой относится изобретение
Данное изобретение относится к химии органических соединений, фармакологии и медицине и касается терапии онкологических, хронических воспалительных и прочих заболеваний с помощью новых семейств химических соединений, обладающих повышенной эффективностью в ингибировании терапевтически значимых киназ, в частности, ALK-киназы и ее мутантных форм, а также повышенной селективностью и биодоступностью.
Уровень техники
Протеинкиназы являются важным семейством белков, участвующим в регуляции ключевых клеточных процессов, нарушение активности которых может приводить к онкологическим заболеваниям, хроническим воспалительным заболеваниям, заболеваниям центральной нервной системы и т.д. Список киназ, терапевтическая значимость которых к настоящему времени имеет доклиническую или клиническую валидацию, включает: ABL1, ALK, AKT, AKT2, AURKA, BRAF, BCR-ABL, BLK, BRK, C-KIT, C-MET, C-SRC, CAMK2B, CDK1, CDK2, CDK3, CDK4, CDK5, CDK6, CDK7, CDK8, CDK9, CRAF1, CHEK1, CHEK2, CLK1, CLK3, CSF1R, CSK, CSNK1G2, CSNK1G3, CSNK2A1, DAPK1, DAPK2, DAPK3, EGFR, EPHA2, EPHA3, EPHA5, ERBB2, ERBB3, ERBB4, ERK, ERK2, ERK3, FES, FGFR1, FGFR2, FGFR3, FGFR4, FGFR5, FGR, FLT-1, FYN, GSK3B, HCK, IGF1R, INSR, ITK, JAK1, JAK2, JAK3, JNK1, JNK2, JNK3, KIT, LCK, LOK, MAP3K5, MAPKAPK2, MARK1, MEK1, MEK2, MET, MKNK2, MST1, NEK2, P38α, P38δ, P38γ, PAK1, PAK4, PAK6, PAK7, PDPK1, PDGFR, PIK3CG, PIM1, PIM2, РКС, PLK1, PLK4, PRKCQ, PRKR, PTK2, PTK2B, RET, ROCK1, ROS1, RPS6KA1, SLK, SRC, SRPK1, STK16, SYK, TAK1, TGFBR1, TIE, TIE2, TNK2, TRK, VEGFR2, WEE1, ZAP70 (Karaman, M.W. et al., Nat Biotechnol, 2008, 26, 127-32; Bhagwat, S.S., Purinergic Signal, 2009, 5, 107-15). С появлением новых экспериментальных данных этот список постоянно растет.
Перспективным подходом для терапии заболеваний, ассоциированных с нарушенной активностью протеинкиназ, является применение низкомолекулярных химических соединений для ингибирования их активности. Примерами таких ингибиторов, одобренных для применения в клинической практике, являются: Иматиниб (Imatinib), Нилотиниб (Nilotinib), Дазатитниб (Dasatinib), Сунитиниб (Sunitinib), Сорафениб (Sorafenib), Лапатиниб (Lapatinib), Гефитиниб (Gefitinib), Эрлотиниб (Erlotinib), Кризотиниб (Crizotinib). Большое количество лекарственных кандидатов, ингибиторов киназ, находятся в настоящее время на стадии клинических испытаний и в предклинической разработке.
Киназа анапластической лимфомы (Anaplastic Lymphoma Kinase, ALK) - трансмембранная рецепторная тирозинкиназа, принадлежащая к семейству инсулиновых рецепторов. Наболее интенсивно ALK киназа экспрессируется в мозге новорожденных, что свидетельствует о возможной роли ALK в развитии мозга (Duyster, J. et al., Oncogene, 2001, 20, 5623-37).
Аберрантная активность киназы анапластической лимфомы является причиной многих онкологических заболеваний. Например, причиной 3-6% случаев немелкоклеточного рака легких (НМКРЛ) является хромосомная транслокация, вызывающая образование химерного белка, состоящего из белка EML4 и внутриклеточного домена ALK (Pao, W. et al., Lancet Oncol, 2011, 72, 175-80; Shaw, А.Т. et al., Clin Cancer Res, 2011, 17, 2081-6). Другая хромосомная транслокация приводит к образованию химерного белка NPM-ALK, и является причиной около 60% случаев анапластической крупноклеточной лимфомы (АККЛ) (Kutok, J.L. et al., J Clin Oncol, 2002, 20, 3691-702). Конститутивная тирозинкиназная активность химерных белков, EML4-ALK в случае НМКРЛ, или NPM-ALK в случае АККЛ, вызывает активацию нижестоящих сигнальных путей, ответственных за клеточное деление и защиту от апоптоза и в итоге ведущих к онкотрансформации клетки (Falini, В. et al., Blood, 1999, 94, 3509-15; Morris, S.W. et al., Br J Haematol, 2001, 113, 275-95; Bai, R.Y. et al., Blood, 2000, 96, 4319-27). ALK-положительные раковые опухоли являются онкоген-зависимыми: блокирование ферментативной активности с помощью ингибиторов ALK ведет к аресту клеточного цикла и апоптозу раковых клеток (Christensen, J.G. et al., Mol Cancer Ther, 2007, 6, 3314-22).
Ингибирование ALK является перспективной стратегией борьбы с ALK-положительными формами немелкоклеточного рака легких, анапластической крупноклеточной лимфомы и другими онкологическими заболеваниями, причиной которых является конститутивная активность ALK. Клинические испытания ингибитора ALK Кризотиниба на пациентах с поздней стадией НМКРЛ показали, что средняя продолжительность жизни пациентов увеличивается на 9 и более месяцев (Di Maio, M. et al., J Clin Oncol, 2009, 27, 1836-43) до 2-х лет (Kwak, E.L. et al., N Engl J Med, 2010, 363, 1693-703). К настоящему времени известны многочисленные ингибиторы ALK, включающие индазолоизохинолины (WO 2005/0093 89), тиазольные и оксазольные амиды (WO 2005/097765), пирролопиримидины (WO 2005/080393), пиримидиндиамины (WO 2005/016894), аминопиридины и аминопиразины (WO 2004/076412; WO 2007/066187), пиридопиразины (WO 2007/130468).
Применение низкомолекулярных ингибиторов ALK в терапевтической практике выявило несколько серьезных проблем, связанных с их эффективностью. Во-первых, эти проблемы связаны с низкой активностью ингибиторов по отношению к мутированным формам ALK, которые могут со временем появляться у пациентов. Например, известно, что киназный домен продукта гена EML4-ALK мишени немелкоклеточного рака легких подвержен появлению мутаций, которые обусловливают резистентность к Кризотинибу (мутации L1196M, C1156Y, G1269A и F1174L) (Choi, Y.L. et al., N Engl J Med, 2010, 363, 1734-9; Sasaki, T. et al., Cancer Res, 2010, 70, 10038-43). Частота подобных мутаций достигает 30% (Doebele, R.С. et al., Clin Cancer Res, 2012). Во-вторых, увеличение продолжительности жизни пациентов повышает вероятность образования метастазов в мозг: в среднем за 2 года лечения метастазы появляются у 50% пациентов (Shaw, А.Т. et al., Lancet Oncol, 2011, 12, 1004-12). Кризотиниб практически не проникает через гематоэнцефалический барьер и поэтому не воздействует на метастазы в мозге несмотря на то, что первичная опухоль в легких у того же пациента может продолжать сокращаться (Costa, D.В. et al., J Clin Oncol, 2011, 29, e443-5). Таким образом, создание новых соединений, способных ингибировать мутантные формы киназ и проникать через гематоэнцефалический барьер, является практически важной задачей.
Данное изобретение касается новых семейств химических соединений, обладающих повышенной эффективностью в ингибировании протеинкиназ и их мутантных форм и перспективных для применения в терапии онкологических, хронических воспалительных и прочих заболеваний.
Раскрытие изобретения
Задачей (техническим результатом) настоящего изобретения является получение новых химических соединений, обладающих повышенной эффективностью в ингибировании протеинкиназ, в частности ALK-киназы, и их мутантных форм, а также повышенной селективностью и биодоступностью, и перспективных для применения в терапии онкологических, хронических воспалительных и прочих заболеваний, в частности, опухолей центральной нервной системы - за счет способности новых соединений проникать через гематоэнцефалический барьер.
1. Общее описание соединений изобретения
1.1. Предметом настоящего изобретения являются соединения общей формулы I,
их таутомеры, изомеры или энантиомеры или их фармацевтически приемлемые соли, сольваты или гидраты, где:
LA представляет собой CH2 или CH(СН3);
LB представляет собой ковалентную химическую связь, OC0-3-алкил, SC0-3-алкил, NHC(O)C0-3-алкил, C(O)NHC0-3-алкил, С(O)C0-3-алкил, NHC0-3-алкил, CH2O, CH2S, CH2C(O)NH или CH2NH;
цикл A представляет собой фенил или 5-6-членный гетероарил, содержащий 0-3 атома N и 0-1 атом O или S, и опционально замещенный 1-4 группами RA;
RA выбирается независимо и представляет собой галоген, частично или полностью галогенированные C1-5-алкил, C2-5-алкенил, C2-5-алкинил, (CH2)mO(CH2)nH, (CH2)mNH(CH2)nH, (CH2)mC(O)O(CH2)nH, (CH2)mOC(O)(CH2)nH, (CH2)mC(O)NH(CH2)nH, (CH2)mNHC(O)(CH2)nH, CN, P(O)(RF)2, P(O)2(RF), Р(O)2OH, SRE, S(O)RE, S(O)2RE или S(O)2OH;
цикл B представляет собой фенил, C3-8 циклоалкил, 4-8-членный насыщенный или частично насыщенный гетероцикл, содержащий 0-3 атома N, и 0-1 атом О или S; или 5-6-членный гетероарильный цикл, содержащий 0-3 атома N, и 0-1 атом О или S; цикл В опционально содержит 1-5 заместителей RB;
RB выбирается независимо и представляет собой LC-RC, LC-H, галоген, частично или полностью галогенированные C1-5-алкил, С2-5-алкенил, C2-5-алкинил или CN; альтернативно две соседние группы RB, совместно с атомами, к которым они присоединены, могут образовывать 5-, 6- или 7-членный насыщенный, частично насыщенный или ненасыщенный цикл, содержащий 0-3 гетероатомов, выбранных из N, О, S, S(O), S(O)2, и опционально замещенный 1-4 заместителями RC или RD;
LC представляет собой ковалентную химическую связь, C1-3-алкил, (CH2)mO(CH2)n, (CH2)mNH(CH2)n, (CH2)mC(O)(CH2)n, (CH2)mC(O)O(CH2)n, (CH2)mOC(O)(CH2)n, (CH2)mC(O)NH(CH2)n или (CH2)mNHC(O)(CH2)n;
RC выбирается независимо и представляет собой фенил, C1-6-алкил, C3-7 циклоалкил или 3-7-членный гетероалициклил, содержащий 0-3 атома N, 0-2 атома О и 0-2 атома S; Rc опционально содержит 1-5 заместителей RD или CH2RD;
RD выбирается независимо и представляет собой галоген, (CH2)mCH3, (CH2)mO(CH2)nH, (CH2)mC(O)NH(CH2)nH, (CH2)mC(O)(CH2)nH, (CH2)mNH2, NHRF, N(RF)2 или 3-7-членный гетероалицикл, содержащий 0-3 атома N, 0-2 атома О, 0-2 атома S и опционально содержащий 1-3 заместителя RF;
RE выбирается независимо и представляет собой C1-3-алкил, NHC1-3-алкил или N(C1-3-алкил)2;
RF выбирается независимо и представляет собой C1-3-алкил;
m и n независимо выбираются из 0, 1, 2, 3;
2. Предпочтительные варианты воплощения изобретения
Отдельный подкласс соединений, представляющих интерес, включает соединения формулы I, в которых:
LA представляет собой CH2 или CH(CH3);
LB представляет собой ковалентную химическую связь, OC0-3-алкил, SC0-3-алкил, NHC(O)C0-3-алкил, С(O)NHC0-3-алкил, С(O)C0-3-алкил, NHC0-3-алкил, CH2O, CH2S, CH2C(O)NH или CH2NH;
цикл А представляет собой фенил, опционально замещенный 1-3 группами RA;
RA представляет собой галоген, частично или полностью галогенированные - C1-3-алкил, OC1-3-алкил, S(O)C1-3-алкил, S(O)2C1-3-алкил, S(O)NHC1-3-алкил, S(O)2NHC1-3-алкил, S(O)N(C1-3-алкил)2, S(O)2N(C1-3-алкил)2 или P(O)(C1-3-алкил)2;
цикл B представляет собой фенил; C3-7 циклоалкил; 4-6-членный насыщенный или частично насыщенный гетероцикл, содержащий 0-3 атома N, и 0-1 атом О или S; или 5-6-членный гетероарильный цикл, содержащий 0-3 атома N, и 0-1 атом О или S; цикл B опционально содержит 1-5 заместителей RB;
RB выбирается независимо и представляет собой LC-RC, LC-H, галоген или частично или полностью галогенированный C1-3-алкил; альтернативно две соседние группы RB, совместно с атомами, к которым они присоединены, могут образовывать 5-, 6- или 7-членный насыщенный, частично насыщенный или ненасыщенный цикл, содержащий 0-3 гетероатомов, выбирающихся из N, О, S, и опционально замещенный 1-4 заместителями RC или RD;
LC представляет собой ковалентную химическую связь, C1-3-алкил, (CH2)mC(O)(CH2)n, (CH2)mC(O)NH(CH2)n или (CH2)mO(CH2)n;
RC выбирается независимо и представляет собой фенил, C1-6-алкил или 4-6 членный гетероалициклил, содержащий 0-2 атома N и 0-1 атом О; RC опционально содержит 1-5 заместителей RD или CH2RD;
RD выбирается независимо и представляет собой (CH2)mCH3, (CH2)mO(CH2)nH, (CH2)mC(O)NH(CH2)nH, (CH2)mC(O)(CH2)nH, (CH2)mNH2, N(RF)2 или 4-6-членный гетероалицикл, содержащий 0-2 атома N, 0-1 атом О; RD опционально содержит 1-3 заместителя C1-3-алкил;
m и n независимо выбираются из 0, 1, 2, 3.
Другой подкласс соединений, представляющих интерес, включает соединения формулы I, в которых:
LA представляет собой CH2 или CH(CH3);
LB представляет собой ковалентную химическую связь, C(O)NH или NH;
цикл А представляет собой фенил, опционально замещенный 1-3 группами RA;
RA представляет собой Cl, F, CF3 или OCH3;
цикл B представляет собой фенил; 5-членный гетероарильный цикл, содержащий 1-3 атома N; 5-членный гетероарильный цикл, содержащий 1-2 атома N и 1 атом О или 6-членный гетероарильный цикл, содержащий 1-3 атома N; цикл B опционально содержит 1-3 заместителя RB;
RB выбирается независимо и представляет собой LC-RC или LC-H;
LC представляет собой ковалентную химическую связь, CH2, C(O), C(O)NH, CH2C(O)NH, C(O)NHCH2, C(O)NH(CH2)2 или OCH2;
RC выбирается независимо и представляет собой фенил, C1-3-алкил или 4-6-членный гетероалициклил, содержащий 0-2 атома N и 0-1 атом О; RC опционально содержит 1-3 заместителей RD или CH2RD;
RD выбирается независимо и представляет собой CH3, OCH3, OH, CH2C(O)NH2, C(O)CH3, N(RF)2 или 4-6 членный гетероалицикл, содержащий 0-2 атома N, 0-1 атом О, и RD опционально содержит 1 -3 заместителя RF;
RF представляет собой CH3.
Следующий подкласс соединений, представляющих интерес, включает соединения формулы I, в которых LB представляет собой ковалентную химическую связь, NH или C(O)NH.
Кроме того, подкласс соединений, представляющих интерес, включает также соединения формулы I, в которых цикл A является фенильным.
2.1. Отдельный подкласс соединений, представляющих интерес, включает соединения формулы I, в которых цикл В представляет собой фенил.
Другой подкласс соединений, представляющих интерес, включает соединения формулы I, в которых цикл A представляет собой C3-7 циклоалкил.
Другой подкласс соединений, представляющих интерес, включает соединения формулы I, в которых цикл B представляет собой 4-6-членный насыщенный или частично насыщенный гетероцикл, содержащий 0-3 атома N, и 0-1 атом О или S.
Другой подкласс соединений, представляющий интерес, включает соединения формулы I, в которых цикл B представляет собой 5-6-членный гетероарильный цикл, содержащий 0-3 атома N, и 0-1 атом O или S.
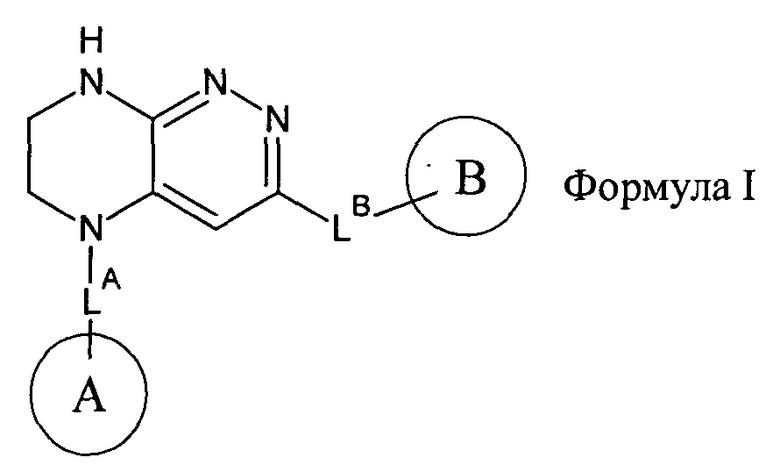
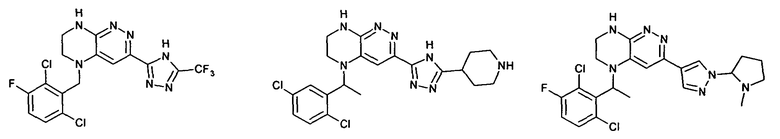
2.2. Отдельный класс соединений, представляющих интерес, включает соединения по формуле I, в которых линкер LB представляет NHC0-3-алкил или CH2NH. Иллюстративными примерами этого класса являются следующие соединения:
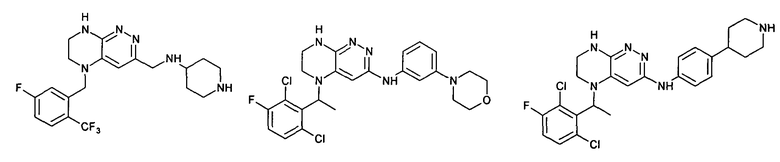
2.3. Другой класс соединений, представляющих интерес, включает соединения по формуле I, в которых линкер LB представляет C(O)C0-3-алкил. Иллюстративными примерами этого класса являются следующие соединения:
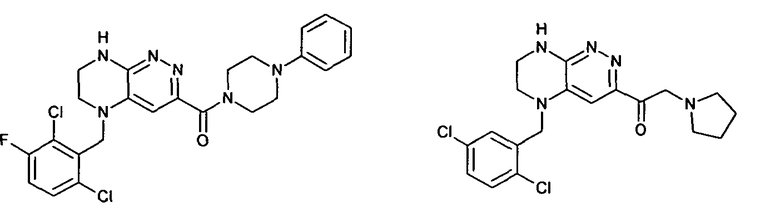
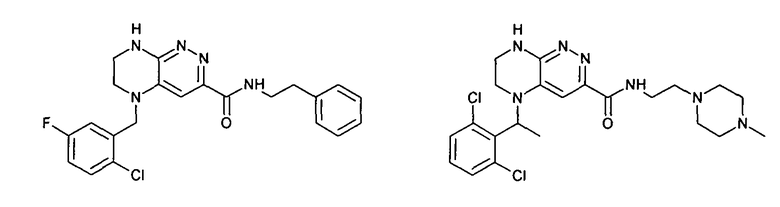
2.4. Другой класс соединений, представляющих интерес, включает соединения по формуле I, в которых линкер LB представляет C(O)NHC0-3-алкил. Иллюстративными примерами этого класса являются следующие соединения:
2.5. Еще один класс соединений, представляющих интерес, включает соединения по формуле I, в которых линкер LB представляет OC0-3-алкил, SC0-3-алкил, CH2O или CH2S. Иллюстративными примерами этого класса являются следующие соединения:
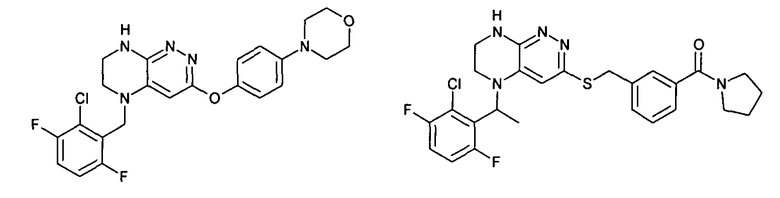
2.6. Еще один класс соединений, представляющих интерес, включает соединения по формуле I, в которых линкер LB представляет NHC(O)C0-3-алкил или CH2C(O)NH. Иллюстративными примерами этого класса являются следующие соединения:
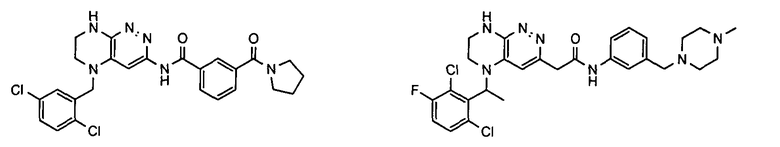
2.7. Другой предпочтительный вариант воплощения изобретения представляют соединения по формуле I, в которых LB представляет ковалентную химическую связь. Этот подкласс соединений иллюстрируется формулой IA:
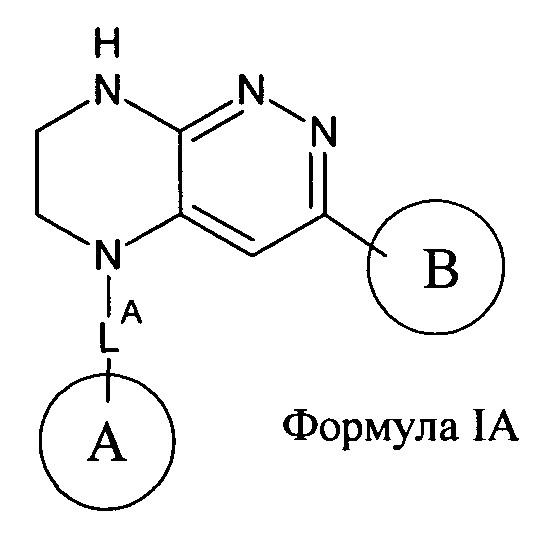
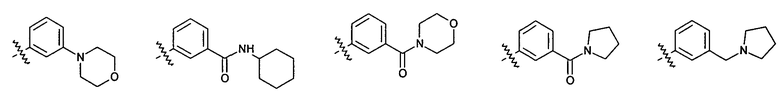
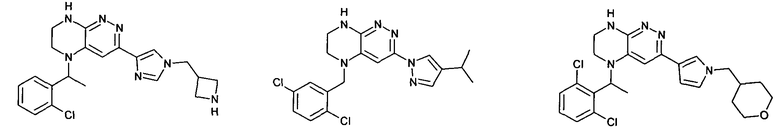
2.8. Одним из предпочтительных вариантов воплощения изобретения являются соединения по формуле I, IA и другие классы и подклассы изобретения, в которых цикл В представляет фенил, опционально замещенный 1-5 заместителями RB.
Иллюстративные примеры фенильной группы с заместителями RB представлены ниже:
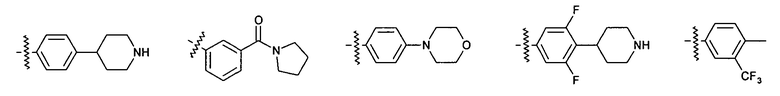
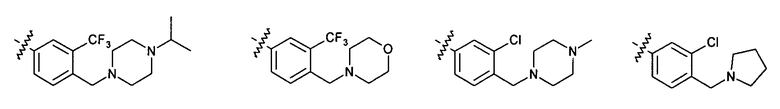
2.9. Другой вариант воплощения изобретения представляют соединения по формуле I, IA и другие классы и подклассы изобретения, в которых цикл B представляет C3-7 циклоалкил, опционально замещенный 1-5 заместителями RB. Нелимитирующими примерами этого класса являются следующие соединения:
2.10. Другой вариант воплощения изобретения представляют соединения по формулам I и IA, в которых цикл B представляет 4-6-членный насыщенный или частично насыщенный гетероцикл, содержащий атомы C, 0-3 атома N и 0-1 атом О или S, опционально замещенный 1-5 заместителями RB. Нелимитирующими примерами данного класса являются следующие соединения:
2.11. Отдельный предпочтительный вариант воплощения изобретения составляют соединения по формуле I, IA и другие классы и подклассы изобретения, в которых цикл B представляет 5- или 6-членный гетероарильный цикл, содержащий 0-3 атома N, и 0-1 атом О или S, и опционально замещенный 1-4 заместителями RB.
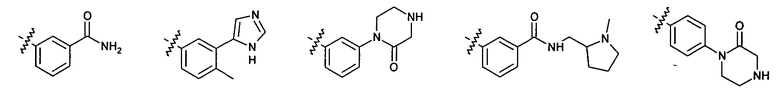
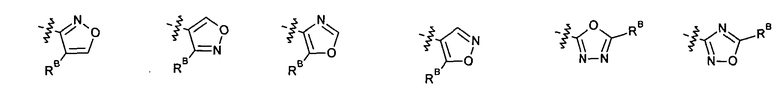
2.12. Особый интерес представляет вариант предыдущего предпочтительного воплощения изобретения, включающий соединения по формуле I, IA или другие подклассы, в которых цикл B представляет 5-членный гетероарильный цикл, состоящий из атомов C и 1-3 атомов азота, и опционально замещенный 1-3 заместителями RB. Нелимитирующими примерами этого класса являются соединения со следующими циклами B:
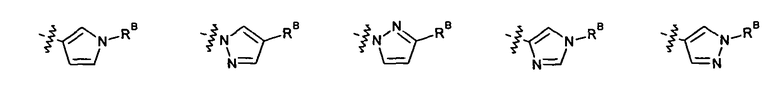
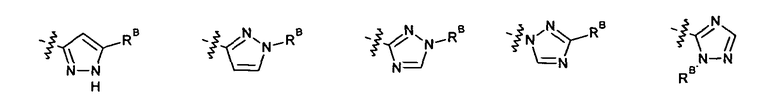
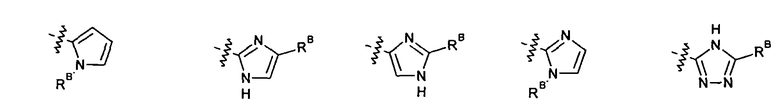
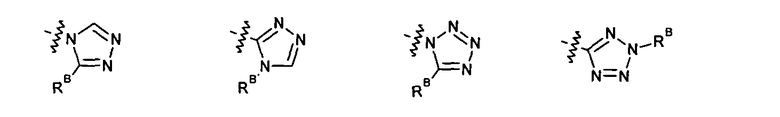
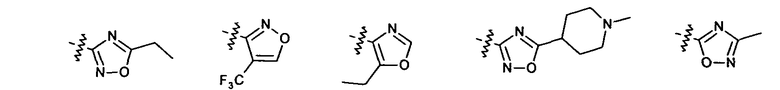
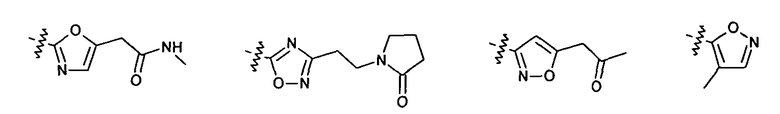
Нелимитирующие иллюстративные примеры вышеуказанных вариантов цикла B, замещенных RB, имеют следующий вид:
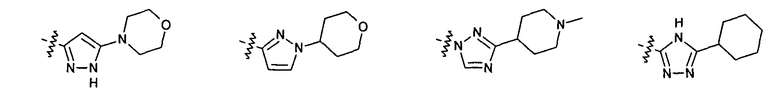
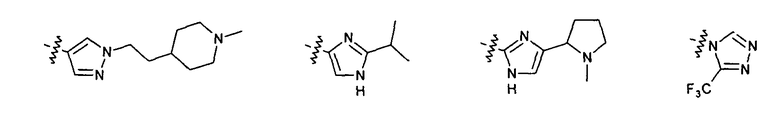
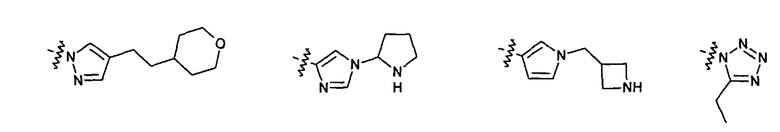
Нелимитирующие иллюстративные примеры этого класса соединений имеют следующие формулы:
2.13. Другой подкласс предыдущего воплощения, имеющий особый интерес, представляют соединения по формуле I, IA или другие классы и подклассы изобретения, в которых цикл В представляет 5-членный гетероарильный цикл, состоящий из атомов C, 1-2 атомов азота и 1 атом O, опционально замещенный 1-3 заместителями RB. Нелимитирующими примерами этого класса являются соединения, содержащие следующие циклы B:

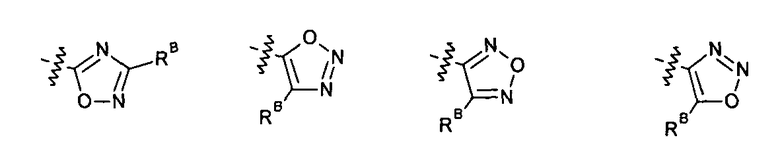
Нелимитирующие иллюстративные примеры вышеуказанных вариантов цикла B, замещенных RB, имеют следующий вид:
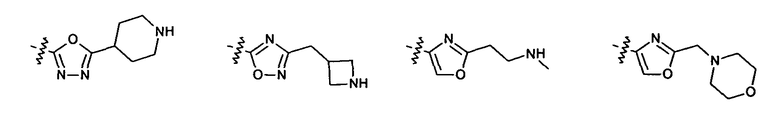
Нелимитирующие иллюстративные примеры этого класса соединений имеют следующие формулы:
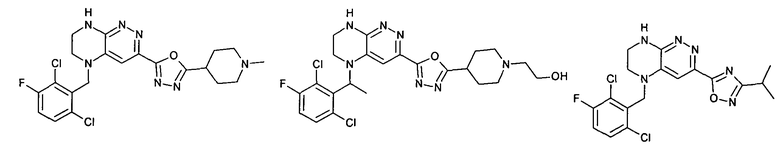
2.14. Отдельный представляющий интерес вариант предыдущего воплощения изобретения включает соединения по формуле I, IA и другие классы и подклассы изобретения, в которых цикл B представляет 6-членный гетероарильный цикл, содержащий 1-3 атома N, и опционально замещенный 1-3 заместителями RB. Нелимитирующими примерами этого класса являются соединения, содержащие следующие структуры цикла B:
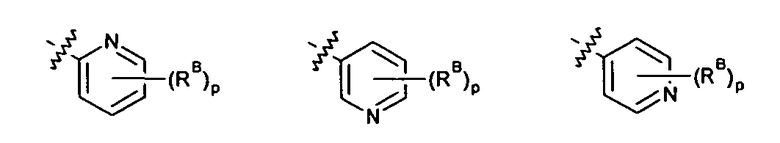
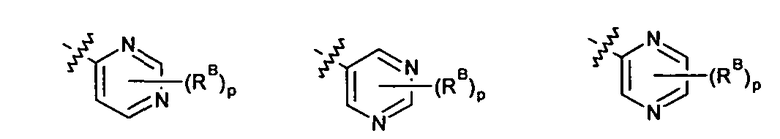
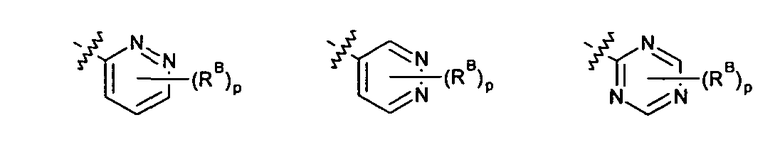
где p выбирается из 0, 1, 2, 3.
Нелимитирующие иллюстративные примеры вышеуказанных вариантов цикла B, замещенных RB, имеют следующий вид:
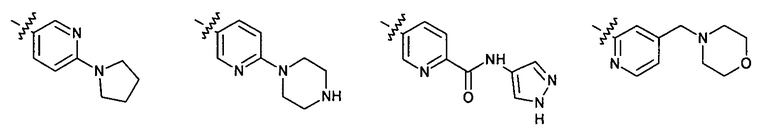
Нелимитирующие иллюстративные примеры этого класса соединений имеют следующие формулы:
2.15. Одним из вариантов воплощением изобретения являются соединения по формулам I и IA, в которых цикл A представляет фенил, опционально замещенный 1-3 группами RA.
2.16. Особый интерес представляет подкласс соединений по формулам I и IA или принадлежащих к другим приведенным выше подклассам, в которых группа RA представляет галоген, частично или полностью галогенированные -C1-3-алкил, -O-C1-3-алкил, S(O)C1-3-алкил, S(O)2C1-3-алкил, S(O)NHC1-3-алкил, S(O)2NHC1-3-алкил, S(O)N(C1-3-алкил)2, S(O)2N(C1-3-алкил)2 или P(O)(C1-3-алкил)2.
2.17. Отдельный особый интерес представляет подкласс соединений по формулам I и IA или принадлежащих к другим приведенным выше подклассам, в которых в группа RA представляет Cl, F, CF3 или OCH3.
2.18. Особый интерес представляет подкласс соединений по формулам I, IA, в которых цикл A представляет фенил, и цикл В представляет 5-6-членный гетероарил. Иллюстративные нелимитирующие примеры подобных соединений включают соединения по формулам, приведенным ниже:
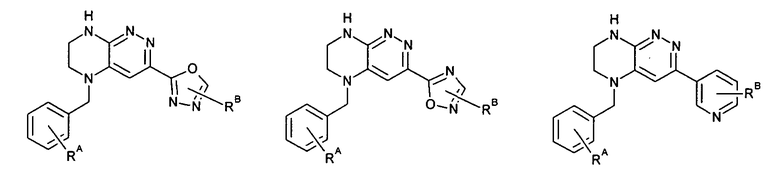
2.19. Также особый интерес представляет подкласс соединений по формуле I, в которых цикл A представляет фенил, линкер LB представляет C(O)NH, и цикл B представляет фенил.
2.20. Отдельный особый интерес представляет подкласс соединений по формуле I, в которых цикл A представляет фенил, линкер LB представляет NH, и цикл B представляет фенил.
Соединения настоящего изобретения, представляющие особый интерес, обладают одной или несколькими из следующих характеристик:
- молекулярная масса менее 1000, предпочтительно менее 750, и наиболее предпочтительно менее 650 г/моль (не включая массу каких-либо сольватирующих или совместно кристаллизующихся веществ, а также противоионов в случае соли); или
- ингибиторная активность по отношению к нативным или мутантным (особенно клинически значимым мутантным) киназам, особенно к киназам ALK, MET, ROS1, EGFR или другим киназам, представляющим интерес, со значением IC50 1 мкМ или менее (полученного с помощью любого научно обоснованного эксперимента по определению ингибирования киназ), предпочтительно с IC50 500 нМ или ниже, и оптимально с IC50 250 нМ или ниже; или
- ингибиторная активность по отношению к данной киназе с IC50 как минимум в 100 раз меньшим, чем соответствующие значения IC50 для других киназ, представляющих интерес; или
- цитотоксический или цитостатический эффект в отношении опухолевых клеток, определенный in vitro, или в исследованиях на животных, с использованием научно приемлемой модели (предпочтительны соединения, ингибирующие рост культуры клеток Ba/F3 NPM-ALK, Ba/F3 EML4-ALK, Karpas 299, SU-DHL-1, NCI-H3122 или NCI-H2228 с эффективностью, превышающей эффективность Кризотиниба, предпочтительно с эффективностью как минимум вдвое лучшей, чем у Кризотиниба, и наиболее предпочтительно с эффективностью как минимум в 10 раз лучшей, чем у Кризотиниба).
Настоящее изобретение также относится к применению соединений, являющихся предметом изобретения, для получения фармацевтической композиции для лечения и/или предотвращения заболевания, связанного с аберрантной активностью протеинкиназ.
В частности, такое заболевание может представлять собой рак легкого, кости, поджелудочной железы, кожи, шеи и головы, кожную или внутриглазную меланому, рак матки, яичника, прямой кишки, анального канала, рак желудка, почек, молочной железы, карциному фаллопиевых труб, слизистой оболочки и шейки матки, вагины, вульвы, ходжкинскую лимфому, рак пищевода, тонкого кишечника, эндокринной системы, щитовидной железы, паращитовидной железы, надпочечников, саркому мягких тканей, рак мочеиспускательного канала, пениса, простаты, хронический или острый миелолейкоз, лимфоцитарную лимфому, рак мочевого пузыря, почки или мочеточника, карциному почечного эпителия, карциному почечной лоханки, рабдомиосаркому, неопластические образования в центральной нервной системе, первичную лимфому ЦНС, опухоли спинного мозга, глиома мозгового ствола, аденома гипофиза и их комбинации.
Такое заболевание также может представлять собой немелкоклеточный рак легкого, анапластическую крупноклеточную лимфому, диффузную B-клеточную лимфому, воспалительную миофибробластическую опухоль, нейробластому, рабдомиосаркому, анапластический рак щитовидной железы, мультиформную глиобластому, холангиокарциному, аденокарциному желудка, хроническую миеломоноцитарную лейкемию, саркому Юинга, воспалительный рак груди, карциному папиллярного почечного эпителия, плоскоклеточную карциному.
Кроме того, изобретением предусматриваются фармацевтические композиции, содержащие как минимум одно соединение, являющееся предметом изобретения, или соль, гидрат или другой сольват такового, и как минимум один фармацевтически приемлемый носитель, адъювант, растворитель и/или наполнитель. Такие композиции предназначены для лечения и/или предотвращения заболевания, связанного с аберрантной активностью протеинкиназ, и могут быть введены объекту, нуждающемуся, в частности, в ингибировании роста, развития или метастазов раковой опухоли, включая солидные опухоли (например, рак простаты, толстой кишки, поджелудочной железы, яичников, молочной железы, пищевода, немелкоклеточный рак легкого (НМКРЛ), опухолевые заболевания головного мозга, в том числе глиобластома и нейробластома; раковые заболевания мягких тканей, в том числе рабдомиосаркома и др.), различные формы лимфомы, как например неходжкинская лимфома (НХЛ) известная как анапластическая крупноклеточная лимфома (АККЛ), различные формы лейкемии и другие формы рака, в том числе устойчивые к лечению Кризотинибом или другими киназными ингибиторами, а также в целом для лечения и профилактики заболеваний или неблагоприятных состояний организма, вызванных одной или несколькими киназами, которые ингибируются соединениями изобретения.
Настоящее изобретение также относится к способу лечения рака, который, согласно настоящему изобретению, включает в себя введение (в качестве монотерапии или в комбинации с одним или несколькими противораковыми агентами, одним или несколькими агентами для смягчения побочных эффектов, облучением и т.п.) терапевтически эффективного количества соединения, являющегося предметом изобретения, в организм человека или животного, нуждающегося в остановке, замедлении или обращении роста, развития или распространения рака, включая солидные опухоли или другие формы рака, такие как лейкемия. Такое введение представляет собой метод лечения или профилактики заболеваний, вызываемых одной или несколькими киназами, ингибируемыми одним из раскрываемых соединений или их фармакологически приемлемых производных. «Введение» в организм соединения настоящего изобретения включает доставку к реципиенту соединения, описанного в настоящем изобретении, пролекарства, или другого фармакологически приемлемого производного такого соединения, используя любые допустимые препараты или пути введения в организм, как описано в настоящем документе. Обычно соединение вводится в организм пациента один или несколько раз в неделю, например, ежедневно, через день, 5 дней в неделю и т.п. Пероральное и внутривенное введение представляет особый интерес.
Изобретение также включает получение соединений по любой из формул I, IA, или любых других соединений настоящего изобретения.
Кроме того, изобретение также включает использование соединения изобретения или его фармакологически приемлемого производного в производстве лекарственного средства для лечения как острой или хронической формы рака (включая лимфому и солидные опухоли, первичные или метастатические, включая уже отмеченные в настоящем документе типы рака, и включая резистентные или устойчивые к одному или нескольким видам лечения виды рака). Соединения, составляющие суть настоящего изобретения, могут быть использованы в производстве противораковых препаратов. Соединения настоящего изобретения также могут быть использованы в производстве лекарственных препаратов для ослабления или предотвращения различных расстройств путем ингибирования одной или нескольких киназ, включающих, но не ограничивающихся такими киназами, как как ALK, EGFR, MET, ROS1.
Изобретение также охватывает композиции, содержащие соединения настоящего изобретения, включая соединения любого из описанных классов или подклассов, в том числе любой из формул, описанных выше, помимо прочего, предпочтительно в терапевтически эффективном количестве, в соединении с по крайней мере одним терапевтически допустимым носителем, адъювантом или растворителем.
Соединения настоящего изобретения также могут быть использованы в качестве стандартов и реагентов для характеристики различных киназ, в особенности, но не ограничиваясь, киназами ALK, EGFR, MET, ROS1, также как и для изучения роли таких киназ в биологических и патологических явлениях; для изучения внутриклеточных путей передачи сигнала, осуществляемого с помощью таких киназ, для сравнительной оценки новых киназных ингибиторов; а также для изучения различных видов рака в моделях линий клеток и животных.
3. Определения
Следующие определения применяются в данном документе, если иное не указано явно. Кроме того, если не указано иное, все вхождения функциональных групп выбираются независимо, на что может указывать использование косого штриха для определения, что два вхождения могут быть как одинаковыми, так и разными (например, R, R', R''; Y, Y', Y'' и т.п.).
3.1. Термин «алифатический» в настоящем документе означает как насыщенную, так и ненасыщенную (но не ароматическую) прямую (т.е. неразветвленную), разветвленную, циклическую или полициклическую неароматическую углеводородную цепочку - остаток, который может быть опционально замещен одной или более функциональной группой. Если иное не указано явно, алкил, другие алифатические, алкокси и ацильные группы обычно содержат 1-8 (т.е. C1-8), а в большинстве случаев 1-6 (C1-6) смежных алифатических атомов углерода. В качестве примера такие алифатические группы включают метил, этил, изопропил, циклопропил, метилен, метилциклопропил, циклобутилметил, циклопентил производные и т.п., которые могут содержать один или несколько заместителей. Термин «алифатический», таким образом, подразумевает включение алкил, алкенил, алкинил, циклоалкил и циклоалкенил фрагментов.
Термин «алкил» в настоящем документе означает как неразветвленные, так и разветвленные, циклические или полициклические алкильные группы. Аналогичные условности применяются и к другим общим терминам, таким как «алкенил», «алкинил» и т.п. Кроме того, «алкил», «алкенил», «алкинил» и подобные группы могут быть как замещенными, так и незамещенными.
3.2. Термин «алкил» в настоящем документе относится к группам, обычно имеющим от одного до восьми, предпочтительно от одного до шести атомов углерода. Например, «алкил» может означать метил, этил, н-пропил, изопропил, изогексил, циктогексил и т.д. В качестве иллюстрации, замещенные алкильные группы включают, но не ограничиваются, следующими группами: фторметил, дифторметил, трифторметил, бензил, замещенный бензил, фенетил, замещенный фенетил и т.д. Термин C1-6 алкил означает алкил, содержащий от 1 до 6 атомов углерода, и включает C1, C2, C3, C4, C5 и C6-алкильные группы.
3.3. Термин «алкокси» относится к алкильным группам, соответствующим определению, приведенному выше, и которые присоединяются к молекуле посредством мостикового атома кислорода. Например, термин «алкокси» означает -O-алкил, где алкильная группа содержит от 1 до 8 атомов углерода в виде линейной (неразветвленной) или разветвленной цепи или в виде цикла. В качестве иллюстрации алкокси группы включают, но не ограничиваются, следующими группами: метокси, этокси, н-пропокси, н-бутокси, трет-бутокси и т.д.
3.4. Термин «галогеналкил» включает разветвленные и линейные насыщенные углеводородные цепи, в которых один или несколько атомов водорода замещены на галоген. Примеры галогеналкильных групп включают, но не ограничиваются, следующие группы: трифторметил, трихлорметил, пентафторэтил, -C(CF3)2CH3 и т.п.
3.5. Термин «алкенил» относится к группам, обычно имеющим от двух до восьми, чаще от двух до шести атомов углерода, и включающим линейные и разветвленные углеводородные цепи либо циклы, и имеющие одну или более двойных углерод-углеродных связей и расположенных в любой стабильной точке цикла или цепи. Например, «алкенил» может означать, но не ограничивается, следующими группами: проп-2-енил, бут-2-енил, бут-3-енил и т.п. Термин «алкинил» относится к группам, обычно имеющим от двух до восьми, чаще от двух до шести атомов углерода, и включающим линейные и разветвленные углеводородные цепи либо циклы, и имещие одну или более тройных углерод-углеродных связей. Например, «алкинил» может означать, но не ограничивается, следующими группами: проп-2-инил, бут-2-инил, бут-3-инил, гекс-2-инил, гекс-5-инил и т.д.
3.6. Термин «циклоалкил» относится к группам, имеющим от трех до 12, обычно от трех до десяти атомов углерода в моно-, ди- или полициклической (т.е. кольцевой) структуре. В качестве иллюстрации, циклоалкилы включают, но не ограничиваются, следующими радикалами: циклопропил, циклобутил, циклопентил и т.п., которые, как и в случае других алифатических или гетероалифатических или гетероциклических заместителей, могут быть замещенными. Термины «циклоалкил» и «карбоцикл» являются эквивалентными.
Термин «циклоалкенил» относится к алкенильным группам, содержащим от трех до 13, обычно от 5 до 8 атомов углерода в моно- или полициклической структуре, содержащей одну или более ненасыщенную двойную углерод-углеродную связь. Например, «циклоалкенил» может означать, но не ограничивается следующими группами: циклопентенил, циклогексенил и т.д.
3.7. Термин «гетероалифатический» в настоящем документе означает алифатические заместители, которые содержат атом кислорода, серы, азота, фосфора или кремния на месте одного или нескольких атомов углерода. Гетероалифатические заместители могут быть неразветвленными, разветвленными или циклическими, а также включать ациклические фрагменты, такие как CH3OCH2CH2O-, также как и гетероциклы, такие как морфолино, пирролидинил и т.д.
3.8. Термин «гетероцикл», «гетероциклил» или «гетероциклический» означает здесь неароматические циклические системы, имеющие от пяти до четырнадцати, как правило от пяти до десяти кольцевых атомов углерода, в которых существует один или несколько углеродных циклов, как правило от одного до четырех, в которых присутствует замещение гетероатомом, таким как N, O или S. Примеры гетероциклических колец включают, но не ограничиваются, следующими: тетрагидрофуран-2-ил, тетрагидрофуран-3-ил, тетрагидротиофен-2-ил, морфолин-2-ил, тиоморфолин-4-ил, пиперазин-1-ил, пиперазин-2-ил, фталимидин-1-ил, бензоксоланил и т.д.. Кроме того, в рамках термина «гетероциклил» или «гетероциклический», в том смысле как они использованы здесь, находятся группы, в которых неароматический цикл, содержащий гетероатом, соединен с одним или несколькими ароматическими или неароматическими циклами, такие как индолинил, хроманил, и т.д., в которых радикал-атом или место присоединения лежит в неароматическом цикле, содержащем гетероатом. Термин «гетероцикл», «гетероциклил» или «гетероциклический», также относятся к циклам, насыщенным или частично ненасыщенным, которые могут быть замещенными.
3.9. Термин «арил», используемый самостоятельно, или как часть большего фрагмента, такого как «аралкил», «аралкокси» или «арилоксиалкил», означает группы, содержащие ароматический цикл, или полициклические ароматические системы, имеющие от шести до четырнадцати атомов углерода. Примеры используемых арильных циклических групп включают в себя, но не ограничиваются такими группами, как фенил, нафтил, фенантрил, антрил, фенантро и т.п., так же как и нафт-1-ил, нафт-2-ил, антрац-1-ил и антрац-2-ил. Кроме того, в значение термина «арил», так, как оно используется здесь, входят группы, в которых ароматический цикл соединен с одним или более неароматическими циклами, такие как инданил, фенантридинил или тетрагидронафтил, в которых радикальный атом или место соединения принадлежит ароматическому циклу.
3.10. Термин «гетероарил» как он используется здесь, означает стабильный гетероциклический и полигетероциклический ароматический фрагмент, имеющий 5-14 атомов в цикле. Гетероарильная группа может быть замещенной или незамещенной и может содержать одно или несколько колец. Возможные заместители включают, помимо прочего, любой из ранее упомянутых заместителей. Примерами типичных гетероарильных циклов являются пяти- и шестичленные моноциклические группы, такие как тиенил, пирролил, имидазолил, пиразолил, пиридил, пиримидинил, пиридазинил, триазинил и т.п.; а также полициклические гетероциклические группы, такие как бензо[b]тиенил, нафто[2,3-b]тиенил, тиантренил, изобензофуранил, хроменил, изоиндолил, бензимидазол, птеридинил, и т.п. (см. A.R Katritzky, Handbook of Heterocyclic Chemistry). Кроме того, гетероарильные группы содержат группы, в которых гетероароматическое кольцо соединено с одним или несколькими ароматическими или неароматическими циклами, и радикальный атом или место присоединения принадлежит гетероароматическому кольцу. Примеры включают тетрагидрохинолинил, тетрагидроизохинолинил и пиридо[3,4-d]пиримидинил. Термин «гетероарил» также включает кольца с возможными заместителями. Термин «гетероарил» может использоваться эквивалентно с терминами «гетероарильный цикл» или «гетероароматический».
3.11. Арильная группа (включая арильную часть аралкил, аралкокси или арилоксиалкил-групп и т.п.) или гетероарильная группа (включая гетероарильную часть гетероаралкилов или гетероаралкокси фрагментов и т.п.) могут содержать один или несколько заместителей. Примеры подходящего заместителя на ненасыщенном атоме углерода арильной или гетероарильной группы включают галоген (F, Cl, Br или I), алкил, алкенил, алкинил, -CN, -R, -OR, -S(O)pR (где p выбирается из 0, 1, 2), -SO2NRR', -NRR', -(CO)YR, -O(CO)YR, -NR(CO)YR', -S(CO)YR, где каждое вхождение Y представляет собой независимо -O-, -S-, -NR- или ковалентную химическую связь; таким образом -YR включает -R, -OR, -SR и -NRR', a -(CO)YR включает -C(=O)R, -C(=O)OR, и -C(=O)NRR'. Дополнительные заместители включают -YC(=NR)NR'R'', -COCOR, -COMCOR (где M -алифатическая группа, содержащая 1-4 атома углерода), -YP(=O)(Y'R)(Y''R'), -NO2, -NRSO2R' и -NRSO2NR'R''. Для дальнейшей иллюстрации, заместители, в которой Y является -NR, таким образом, включают, среди прочих, -NRC(=O)R', -NRC(=O)NR'R'', -NRC(=O)OR' и -NRC(=NH)NR'R''.
Заместители R, R' и R'' включают водород, алкил, алкенил, алкинил, циклоалкил, циклоалкенил, арил, гетероарил, гетероциклил. Следует отметить, что заместители R могут в свою очередь быть замещенными или незамещенными. Так, заместитель R включает, но не ограничивается, галогеналкильными и галогенарильными группами (такими как хлорметил, трихлорметил или галофенил); алкоксиалкильными и алкоксиарильными группами (такими как метоксиэтил, моно-, ди- и триалкоксифенил; метилендиоксифенил или этилендиоксифенил); алкиламиногруппами. Кроме того, иллюстративные примеры включают 1,2-метилен-диокси, 1,2-этилендиокси, защищенный ОН (например, ацилокси), фенил, замещенный фенил, -O-фенил, -О-(замещенный) фенил, -бензил, -замещенный бензил, -O-фенэтил, -О-(замещенный) фенэтил и т.д. Сверх того, примеры заместителей включают амино, алкиламино, диалкиламино группы, аминокарбонил, галоген, алкил, алкиламинокарбонил, алкиламинокарбонилокси, диалкиламинокарбонилокси, алкокси, нитро, циано, карбокси, алкоксикарбонил, алкилкарбонил, гидрокси, галогеналкилокси и галогеналкильные группы.
Алифатическая, гетероалифатическая или неароматическая гетероциклическая группа может также содержать один или несколько заместителей. Примеры подходящих заместителей на таких группах включают в себя все вышеперечисленные заместители для атомов углерода арильной или гетероарильной группы, а в дополнение включают следующие заместители для насыщенного атома углерода: =О, =S, -NR, =NNRR', =NNHC(O)R, =NNHCOR, или =NNHSO2R, где R представляет водород, алкил, алкенил, алкинил, циклоалкил, арил, гетероарил, гетероциклил. Наглядные примеры заместителей на алифатической, гетероалифатической или гетероциклической группе включают амино, алкамино, диалкамино, аминокарбонил, галоген, алкил, алкиламинокарбонил, диалкиламинокарбонил, алкиламинокарбонилокси, алкокси, нитро, циано, карбокси, алкоксикарбонил, алкилкарбонил, гидрокси, галогеналкокси или галогеналкил группы.
Иллюстративные примеры заместителей на атоме азота ароматического или неароматического гетероцикла включают R, -NRR', -C(=O)R, -C(=O)OR, -C(=O)NRR', -C(=NR)NR'R'', -COCOR, -COMCOR (где M - алифатическая группа, содержащая 1-4 атома углерода), -CN, -NRSO2R' и -NRSO2NR'R', где R представляет водород, алкил, алкенил, алкинин, циклоалкил, арил, гетероарил и гетероциклил.
3.12. Данное изобретение содержит только такие комбинации заместителей и производных, которые образуют стабильное или химически возможное соединение. Стабильным или химически возможным соединением называется такое соединение, стабильности которого достаточно для его синтеза и аналитического детектирования. Предпочтительные соединения данного изобретения являются достаточно стабильными и не разлагаются при температуре до 40°С в отсутствие химически активных условий, в течение по крайней мере одной недели.
3.13. Некоторые соединения данного изобретения могут существовать в таутомерных формах, и это изобретение включает в себя все такие таутомерные формы таких соединений, если не указано иное.
3.14. Если не указано иначе, изображенные здесь структуры также подразумевают и все стереоизомеры, то есть R- и S- изомеры для каждого асимметричного центра. Кроме того, отдельные стереохимические изомеры, равно как и энантиомеры и диастереомерные смеси настоящих соединений, также являются предметом данного изобретения. Таким образом, данное изобретение охватывает каждый диастереомер или энантиомер, свободный в значительной степени от других изомеров (>90%, а предпочтительно >95% мольной чистоты), так же как и смесь таких изомеров.
Конкретный оптический изомер может быть получен разделением рацемической смеси в соответствии со стандартной процедурой, например путем получения диастереоизомерных солей путем обработки оптически активной кислотой или основанием с последующим разделением смеси диастереомеров кристаллизацией с последующим выделением оптически активных оснований из этих солей. Примерами соответствующих кислот являются винная, диацетилвинная, дибензоилвинная, дитолуолвинная и камфорсульфоновая кислота. Другая методика разделения оптических изомеров заключается в использовании хиральной хроматографической колонки. Кроме того, другой метод разделения включает синтез ковалентных диастереомерных молекул путем реакции соединений изобретения с оптически чистой кислоты в активированной форме или оптически чистым изоцианатом. Полученные диастереомеры можно разделить обычными способами, например, хроматографией, дистилляцией, кристаллизацией или сублимацией, а затем гидролизовать для получения энантиомерно чистого соединения.
Оптически активные соединения данного изобретения могут быть получены с использованием оптически активных исходных материалов. Такие изомеры могут находиться в форме свободной кислоты, свободного основания, эфира или соли.
3.15. Соединения, составляющие суть данного изобретения, могут существовать в меченной радиоизотопом форме, т.е. указанные соединения могут содержать один или несколько атомов, чья атомная масса или массовое число отличается от атомной массы или массового числа наиболее распространенных природных изотопов. Радиоизотопы водорода, углерода, фосфора, хлора включают 3H, 14C, 32P, 35S и 36Cl, соответственно. Соединения данного изобретения, которые содержат такие радиоизотопы и/или другие радиоизотопы других атомов, находятся в сфере настоящего изобретения. Тритиевые, т.е. 3H и углеродные, т.е. 14С радиоизотопы являются особенно предпочтительными благодаря простоте приготовления и обнаружения.
Соединения настоящего изобретения, меченные радиоактивными изотопами, могут быть получены с помощью методов, хорошо известных специалистам в данной области. Меченые соединения могут быть получены с помощью процедур, описанных здесь, простой заменой немеченых реагентов соответствующими мечеными реагентами.
Осуществление изобретения
4. Обзор методов получения соединений изобретения
Соединения, являющиеся предметом настоящего изобретения, могут быть получены с использованием описанных ниже синтетических методов. Перечисленные методы не являются исчерпывающими и допускают введение разумных модификаций. Указанные реакции должны проводиться с использованием подходящих растворителей и материалов. При реализации данных общих методик для синтеза конкретных веществ необходимо учитывать присутствующие в веществах функциональные группы и их влияние на протекание реакции. Для получения некоторых веществ необходимо изменить порядок стадий либо отдать предпочтение одной из нескольких альтернативных схем синтеза.
Защитная группа - функциональная группа, вводящаяся в молекулу химического соединения для обеспечения хемоселективности протекания необходимой химической реакции. Защитные группы играют важную роль в органическом синтезе. Некоторые используемые в органическом синтезе реагенты могут взаимодействовать сразу со многими функциональными группами преобразуемой молекулы. В том случае, если необходимо провести реакцию только с одним типом функциональных групп, не задевая остальные, последние модифицируют («защищают») при помощи защитных групп. Примером защитной группы может быть трет-бутоксикарбонильная группа (Boc).
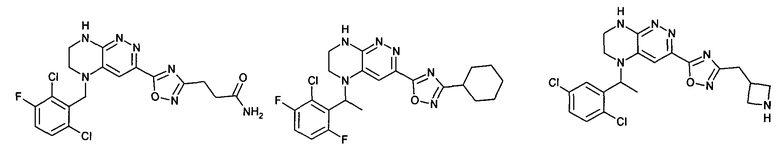
Синтез соединений, являющихся предметом данного изобретения, может быть осуществлен в соответствии со схемами I-XIII по стандартным методикам.
4.1. Промежуточные продукты для получения большинства соединений по формулам I и IA - соединения I-1, I-2, I-3 и I-4 - могут быть получены по схеме I.
На первой стадии происходит синтез моно-Boc-замещенного этилендиамина, из которого на второй стадии путем восстановительного аминирования получается интермедиат I-1. Взаимодействие данного интермедиата с 3,4,6-трихлорпиридазином в присутствии основания приводит к замещению атома хлора в 4 положении пиридазинового кольца. Взаимодействие полученного соединения с трифторуксусной кислотой приводит к снятию защитной Boc-группы. На следующем этапе происходит реакция внутримолекулярного нуклеофильного замещения, приводящая к получению бициклического продукта, взаимодействие которого с Boc-ангидридом приводит к интермедиату I-2. Карбонилирование этого интермедиата в метаноле в присутствии палладиевого катализатора приводит к метиловому эфиру I-3, гидролиз которого гидроксидом лития с последующим подкислением приводит к кислоте I-4.
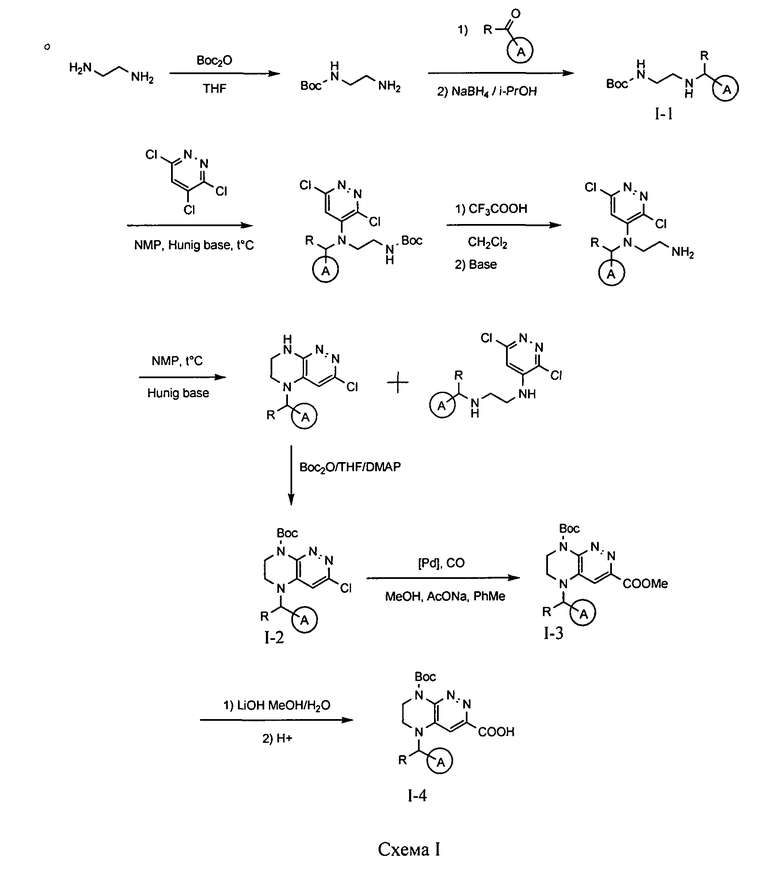
На схеме I цикл A определен по п.1.1. описания изобретения, а заместитель R представляет -H или -CH3.
4.2. Соединения по формуле IA, в которых цикл А определен по п.1.1 описания изобретения, заместитель R представляет -H, -CH3, цикл B представляет 1,2,4-оксадиазол, LB представляет ковалентную химическую связь, а заместитель RB присоединен к позиции 3 и определен по п.1.1 описания изобретения, могут быть получены из промежуточного продукта I-4 по схеме IIa.
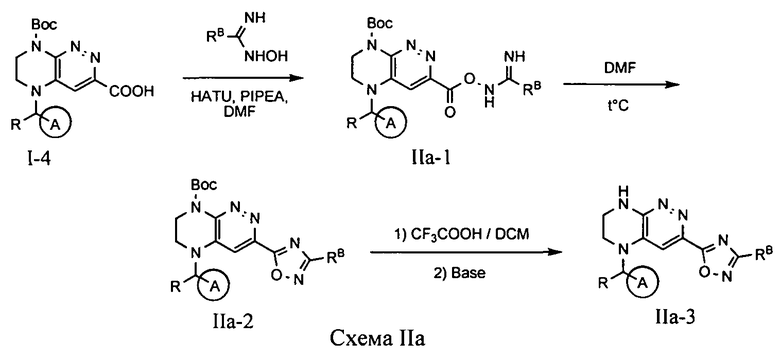
На первой стадии при реакции, активированной при помощи HATU кислоты I-4 с амидоксимом, происходит образование O-ациламидоксима, который затем циклизуется при нагревании в ДМФА с образованием соответствующего оксадиазола. На последней стадии происходит снятие защитной группы Boc.
Соединения по формуле IA, в которых цикл A определен по п.1.1 описания изобретения, заместитель R представляет -H, -CH3, цикл B представляет 1,2,4-оксадиазол, LB представляет ковалентную химическую связь, а заместитель RB определенный по п.1.1 описания изобретения, присоединен к позиции 5, могут быть получены из промежуточного продукта I-2 по схеме IIb.
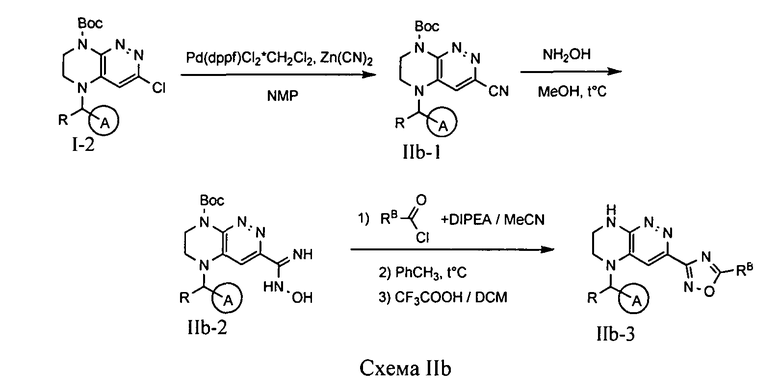
На первой стадии происходит палладий-катализируемое нуклеофильное замещение атома хлора на нитрильную группу под действием цианида цинка. Полученный нитрил обрабатывают гидроксиламином в метаноле, что приводит к амидоксиму IIb-2. Ацилирование полученного амидоксима хлорангидридом соответствующей кислоты в присутствии основания, нагревание продукта в толуоле и удаление защитной Boc-группы при обработке трифторуксусной кислотой приводят к искомому соединению.
4.3. Соединения по формуле IA, в которых цикл A определен по п.1.1 описания изобретения, заместитель R представляет -H, -CH3, цикл B представляет 1,2,3-оксадиазол, a LB представляет ковалентную химическую связь, могут быть получены из промежуточного продукта I-3 по схеме III.
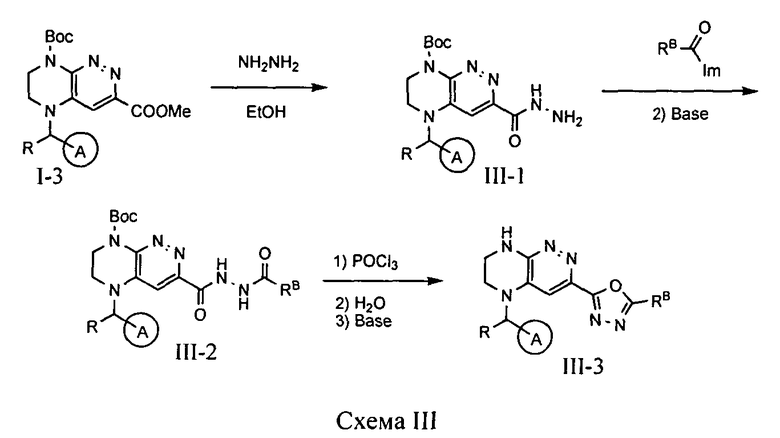
На первой стадии при реакции метилового эфира I-3 с гидразином происходит образование соответствующего гидразида, который затем подвергается ацилированию имидазолидом карбоновой кислоты с образованием диацилгидразина. Интермедиат III-2 подвергают взаимодействию с оксихлоридом фосфора, при котором происходит циклизация с образованием искомого соединения.
4.4. Соединения по формуле IA, в которых цикл A определен по п.1.1 описания изобретения, заместитель R представляет -H, -CH3, цикл B представляет 1,2,4-триазол, a LB представляет ковалентную химическую связь, могут быть получены из промежуточного продукта I-2 по схеме IV.
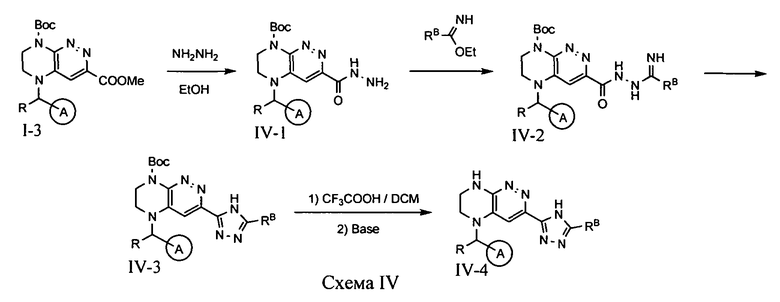
На первой стадии при реакции метилового эфира I-3 с гидразином происходит образование соответствующего гидразида, который затем подвергается взаимодействию с иминоэфиром. Нагревание полученного интермедиата приводит к образованию триазола, с которого на последней стадии снимается защитная Boc-группа.
4.5. Для синтеза соединений по формуле IA, в которых цикл A определен по п.1.1 описания изобретения, заместитель R представляет -H, -CH3, LB представляет ковалентную химическую связь, а цикл B определен по п.1.1 описания изобретения, может быть использована схема Va
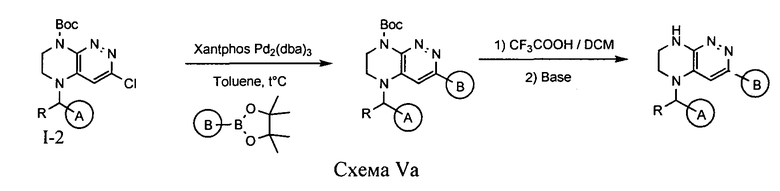
Палладий-катализируемая реакция интермедиата I-2 с пинаколбораном по реакции Сузуки приводит к образованию интермедиата, при удалении защитной группы с которого образуется искомое соединение.
Альтернативно, для синтеза соединений по формуле IA, в которых LB представляет ковалентную химическую связь, может быть использована схема Vb:
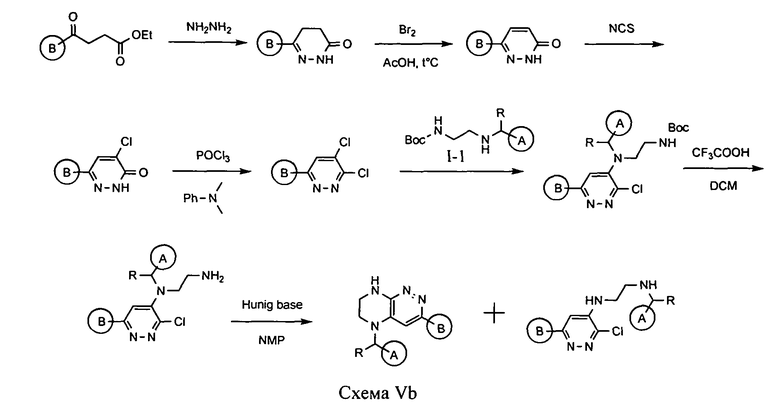
На первом этапе при взаимодействии с гидразином происходит образование циклического гидразида, который затем окисляется под действием брома в уксусной кислоте. Полученный интермедиат хлорируют N-хлорсукцинимидом. Продукт этой реакции обрабатывают оксихлоридом фосфора. Взаимодействие полученного интермедиата с Boc-замещенным производным этилендиамина I-1, удаление защитной Boc-группы под действием трифторуксусной кислоты и последующая циклизация приводят к искомому продукту.
4.6. Для синтеза соединений по формуле I, в которых цикл A определен по п.1.1 описания изобретения, заместитель R представляет -H, -CH3, LB представляет C(O)NHC0-3-алкил, а цикл B определен по п.1.1 описания изобретения, может быть использована схема VIa или VIb.
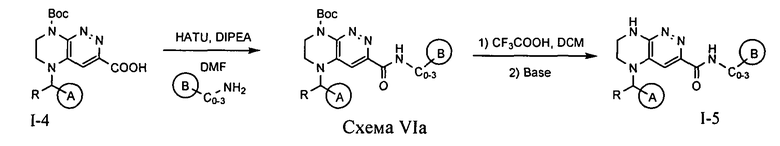
На первой стадии происходит активация карбоновой кислоты при помощи HATU и реакция с аминосоединением, приводящая к амиду. Снятие защитной Boc-группы под действием трифторуксусной кислоты приводит к искомому соединению.
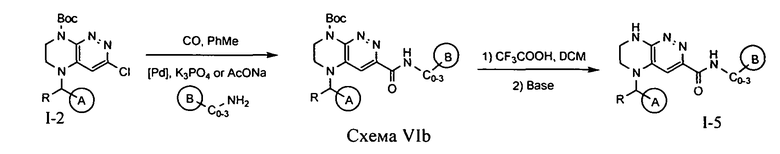
Палладий-катализируемое карбонилирование соединения 1-2 монооксидом углерода в присутствии аминосоединения приводит к амиду, снятие с которого защитной Boc-группы приводит к искомому соединению.
Синтез соединений по формуле I, в которых цикл A определен по п.1.1 описания изобретения, заместитель R представляет -H, -CH3, LB представляет CH2NH, а цикл B определен по п.1.1 описания изобретения, осуществляется по схеме VIc из соединения I-5, полученного по схемам VIa или VIb.
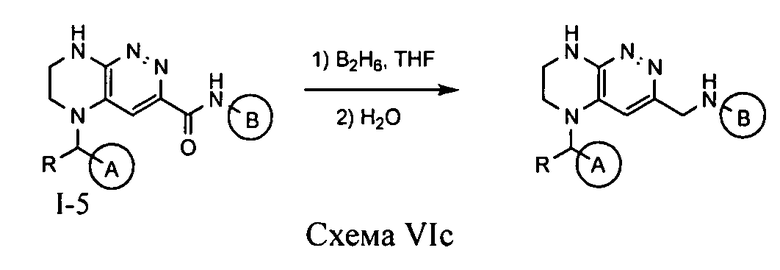
Восстановление соединения I-5 дибораном приводит к искомому соединению.
4.7. Для синтеза соединений по формуле I, в которых цикл А определен по п.1.1 описания изобретения, заместитель R представляет -H, -CH3, LB представляет NH, а цикл В определен по п.1.1 описания изобретения, могут быть использованы схемы VIIa или VIIb.
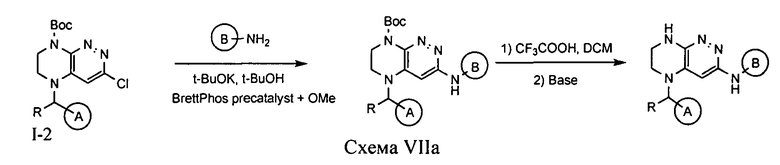
Палладий-катализируемое нуклеофильное замещение атома хлора под действием аминосоединения с последующим удалением защитной Boc-группы приводит к искомому соединению.
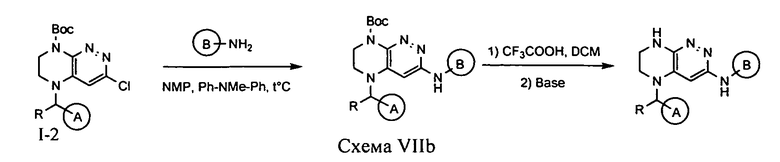
Нуклеофильное замещение атома хлора под действием аминосоединения с последующим удалением защитной Boc-группы приводит к искомому соединению.
4.8. Соединения по формуле I, в которых цикл A определен по п.1.1 описания изобретения, заместитель R представляет -H, -CH3, LB представляет -X-, цикл B определен по п.1.1 описания изобретения, а X представляет -O- или -S-, могут быть синтезированы по схеме VIIIa.
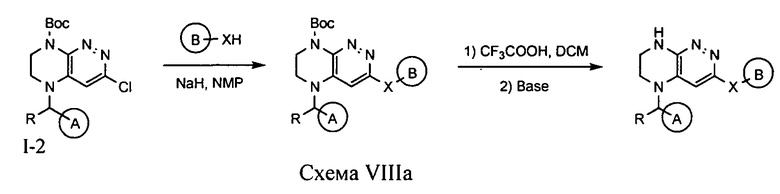
Нуклеофильное замещение атома хлора под действием алкоголят- или тиолят-аниона (полученного при взаимодействии гидрида натрия с соответствующим спиртом) с последующим удалением защитной Boc-группы приводит к искомому соединению.
Синтез соединений по формуле I, в которых цикл A определен по п.1.1 описания изобретения, заместитель R представляет -H, -CH3, LB представляет -X-C1-3-алкил, цикл B представляет собой фенил, опционально содержащий 1-5 заместителей RB, а X представляет O или S, осуществляется по схеме VIIIb.
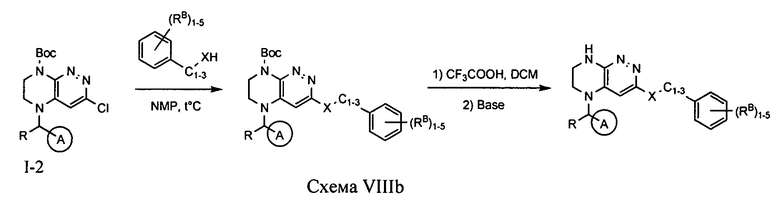
Нуклеофильное замещение атома хлора в исходном соединении под действием алкоголята или тиолята с последующим удалением защитной Boc-группы приводит к искомому соединению.
4.9. Соединения по формуле I, в которых цикл A определен по п.1.1 описания изобретения, заместитель R представляет -H, -CH3, LB представляет -NHC(O)C0-3-алкил, а цикл В определен по п.1.1 описания изобретения, могут быть синтезированы по схеме IX.
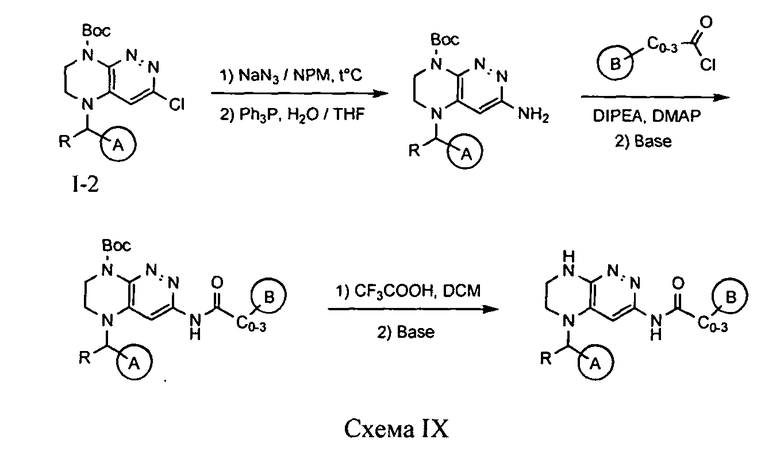
Нуклеофильное замещение атома хлора на азидогруппу с последующим ее расщеплением под действием трифенилфосфина приводит к аминосоединению, ацилирование которого хлорангидридом соответствующей карбоновой кислоты с последующим снятием защитной группы приводит к целевому соединению.
4.10. Соединения по формуле I, в которых цикл A определен по п.1.1 описания изобретения, заместитель R представляет -H, -CH3, LB представляет -CH2X-, цикл B определен по п.1.1 описания изобретения, а X представляет O или S, могут быть синтезированы по схеме X.
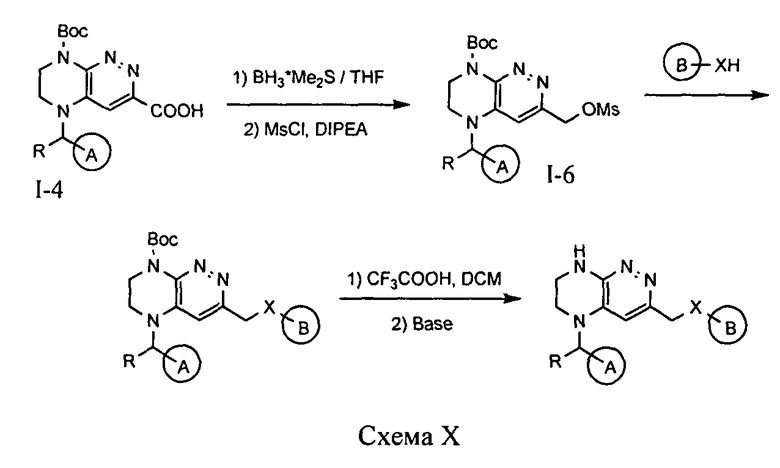
На первой стадии происходит восстановление кислоты I-4 до бензилового спирта под действием комплекса боран-диметилсульфида. Мезилирование полученного спирта, последующее нуклеофильное замещение под действием соответствующего алкоголята или тиоалкоголята и снятие защитной группы приводит к целевому соединению.
4.11. Соединения по формуле I, в которых цикл A определен по п.1.1 описания изобретения, заместитель R представляет -H, -CH3, LB представляет -CH2C(O)NH, цикл B определен по п.1.1 изобретения, а X представляет O или S, могут быть синтезированы по схеме XI.
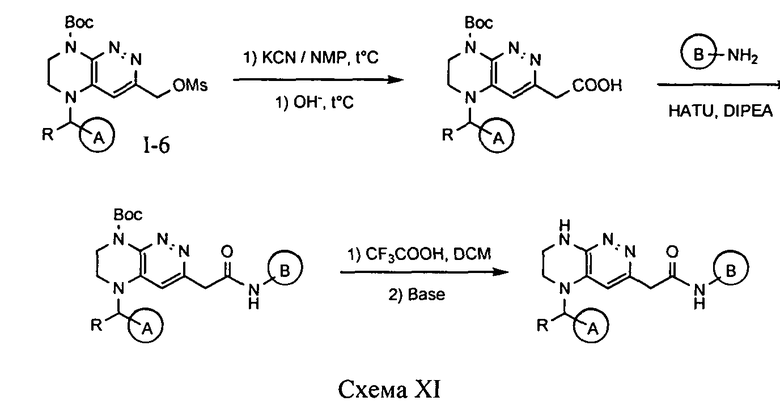
Нуклеофильное замещение мезилата I-6 под действием цианида калия и гидролиз полученного нитрила в щелочной среде приводит к арилуксусной кислоте. Ацилирование соответствующего амина полученной арилуксусной кислотой и снятие защитной группы приводит к целевому соединению.
4.12. Соединения по формуле I, в которых цикл А определен по п.1.1 описания изобретения, заместитель R представляет -H, -CH3, LB представляет -NHC0-3-алкил, цикл B определен по п.1.1 изобретения, могут быть синтезированы по схеме XII.
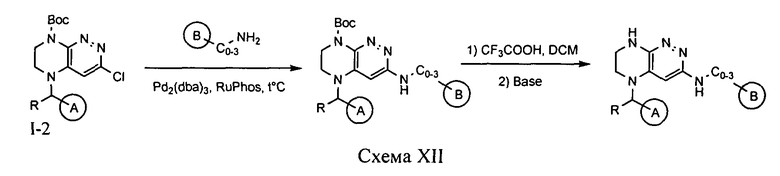
Палладий-катализируемое замещение атома хлора соединения 1-2 на аминогруппу под действуем соответствующего аминосоединения и последующее снятие защитной группы приводит к целевому продукту.
4.13. Соединения по формуле I, в которых цикл A определен по п.1.1 описания изобретения, заместитель R представляет -H, -CH3, LB представляет -C(O)C0-3-алкил, цикл В определен по п.1.1 изобретения, могут быть синтезированы по схеме XIII.
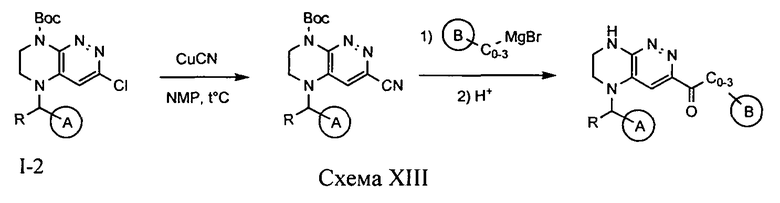
На первой стадии происходит замещение атома хлора на цианогруппу под действием цианида меди (I). Воздействие соответствующим магнийорганическим соединением на полученный нитрил приводит к целевому соединению.
Все соединения, являющиеся предметом данного изобретения, могут быть получены на основании вышеизложенных синтетических подходов, примеров экспериментальных методик и общеизвестных методик и материалов.
5. Применение химических соединений изобретения
5.1. Применение соединений по медицинским показаниям
Соединения, описанные в данном изобретении, могут применяться для терапии заболеваний, в патогенезе которых участвуют протеинкиназы. В частности, соединения, описанные в данном изобретении, способны ингибировать тирозинкиназы ALK, ROS1, MET, EGFR, которые участвуют в росте, развитии и метастазировании раковых опухолей. Кроме того, показано, что ряд соединений, составляющих настоящее изобретение, обладает антипролиферативной активностью in vitro по отношению к раковым клеточным линиям, таким, как например, Karpas-299, SU-DHL-1, NCI-H3122 или NCI-H2228. Такие соединения представляют интерес для лечения различных видов рака, включающих как солидные опухоли, так и лимфомы, и в особенности для лечения разновидностей рака, резистентных к другим способам терапии.
К разновидностям рака, для лечения которых могут применяться соединения данного изобретения, относятся солидные опухоли (например, рак простаты, толстой кишки, поджелудочной железы, яичников, молочной железы, пищевода, немелкоклеточный рак легких (НМКРЛ), опухолевые заболевания головного мозга, в том числе глиобластома и нейробластома; раковые заболевания мягких тканей, в том числе рабдомиосаркома и др.), различные формы лимфомы, как например неходжкинская лимфома (НХЛ), известная как анапластическая крупноклеточная лимфома (АККЛ), различные формы лейкемии и другие формы рака, патогенез которых связан с активностью ALK, MET, EGFR, ROS1.
Поскольку аберрантная активность ALK-киназы является причиной многих онкологических заболеваний, мы предполагаем, что использование ингибитора ALK как препарата для монотерапии или в сочетании с текущими средствами химиотерапии против НМКРЛ, АККЛ и других перечисленных выше онкологических заболеваний, позволит достигнуть их существенной и длительной ремиссии; ингибитор ALK также может быть использован как средство поддерживающей терапии, предназначенное для предотвращения возможных рецидивов у пациентов, нуждающихся в таком лечении.
5.2. Способ терапевтического применения соединений
Предмет данного изобретения также включает введение субъекту, нуждающемуся в соответствующем лечении, терапевтически эффективного количества соединения общей формулы I.
«Терапевтически эффективным количеством» называется такое количество соединения, которое необходимо для детектируемого уничтожения раковых клеток или ингибирования их роста или скорости распространения по организму, размера или количества опухолей, или других характеристик ракового заболевания. Точное требуемое количество может меняться от субъекта к субъекту в зависимости от вида, возраста и общего состояния пациента, тяжести заболевания, особенностей противоракового агента, методики введения препарата, комбинированного лечения с другими препаратами и т.п.
Вещество, или фармацевтическая композиция, содержащая вещество, может быть введена в организм пациента в любом количестве и любым путем введения, эффективным для уничтожения раковых клеток или ингибирования их роста.
Разовые дозы противораковых соединений изобретения предпочтительно формулируются в виде, удобном для введения в организм пациента. Выражение «разовая доза» в терминах настоящего изобретения означает порцию противоопухолевого агента, подходящую для лечения пациента. Согласно существующей практике, совокупная дневная доза соединений и композиций, описанных в настоящем изобретении, назначается лечащим врачом с опорой на тщательное медицинское заключение. Конкретный терапевтически эффективный уровень дозировки для каждого конкретного пациента или организма зависит от ряда факторов, включая тип расстройства, тяжесть заболевания, активность конкретного используемого препарата, особенности фармацевтической композиции, возраст, массу тела, общее состояние здоровья, пол и диету пациента, способ и график введения, скорость метаболизма и/или выведения соединения, продолжительность лечения, лекарственные препараты, используемые в комбинации или совместно с введением соединения из изобретения, и тому подобные факторы, хорошо известные в медицине.
После смешения лекарственного препарата с конкретным подходящим фармацевтически допустимым носителем в желаемой дозировке, композиции, составляющие суть изобретения, могут быть введены в организм человека или других животных перорально, ректально, парентерально, интрацистернально, интравагинально, интраперитонально, местно (с помощью кожных пластырей, порошков, мазей или капель), сублингвально, буккально, в виде спрея для рта или носа и т.п.
Эффективная системная дозировка соединения, вводимая разово или в виде нескольких отдельных доз, как правило, лежит в диапазоне от 0.01 до 500 мг соединения на кг массы тела пациента, предпочтительно от 0.1 до 125 мг/кг. Обычно соединение вводится пациенту, нуждающемуся в таком лечении, в дневной дозировке ориентировочно от 50 до 2000 мг на пациента. Введение может осуществляться как разово, так и несколько раз в день, неделю (или любой другой временной интервал), или время от времени. Например, соединение может быть введено в организм пациента один или несколько раз в день на недельной основе (например, каждый понедельник) в течение неопределенного времени или в течение нескольких недель (например, 4-10 недель). Кроме того, соединение может вводиться в организм пациента ежедневно в течение определенного периода дней (например, 2-10 дней), а затем следует период без приема вещества (например, 1-30 дней). Такой цикл может повторяться неопределенное время или в течение заданного числа циклов, например 4-10 циклов. В качестве примера, соединение настоящего изобретения может вводиться в организм пациента ежедневно в течение 5 дней, затем следует перерыв на 9 дней, и так далее, повторяя цикл неопределенное число раз, или в течение 4-10 циклов.
Количество соединения, которое будет эффективным в лечении или профилактике конкретного расстройства или состояния, зависит, в частности, от хорошо известных факторов, влияющих на эффективную дозировку препаратов. Кроме того, опционально могут применяться измерения in vitro или in vivo для определения оптимального дозового диапазона. Грубым путем определения эффективной дозы может стать экстраполяция кривых доза - отклик, которые будут зависеть от модели тестирования in vitro или на животных. Точный уровень дозировки, определяемый лечащим врачом, зависит от хорошо известных факторов, включающих способ введения препарата, а также возраста, массы тела, пола и общего состояния здоровья пациента; характера, тяжести и клинического состояния заболевания; использования (или неиспользования) сопутствующей терапии; а также характера и степени генетических изменений в клетках пациента.
При приеме внутрь для лечения или подавления конкретного состояния заболевания или расстройства, эффективная дозировка соединения данного изобретения может изменяться в зависимости от конкретного применяемого соединения, пути введения препарата в организм, условий и тяжести такого введения; состояния болезни, а также различного числа физических факторов, связанных с пациентом, проходящим лечение. В большинстве случаев удовлетворительный результат может быть достигнут при введении пациенту соединения в дневной дозировке от около 0.01 мг/кг до 500 мг/кг, обычно между 0.1 и 125 мг/кг. Предполагаемая дневная дозировка, как ожидается, может изменяться в зависимости от способа введения в организм пациента. Так, уровень дозировки при парентеральном введении часто составляет от 10 до 20% уровня пероральной дозировки.
В том случае, когда соединение данного изобретения используется как часть режима комбинированной терапии, доза каждого из компонентов комбинированной терапии вводится в течение требуемого периода лечения. Соединения, составляющие комбинированную терапию, могут вводиться в организм пациента как единовременно, в виде дозировки, содержащей все компоненты, так и в виде индивидуальных дозировок компонентов; кроме того, соединения комбинации могут быть введены в организм пациента в разное время в течение периода лечения, или одно из них может быть введено в качестве предварительной терапии для другого.
5.3. Фармацевтически приемлемые производные соединений
Соединения данного изобретения могут существовать в свободной форме в процессе обработки, или, если требуется, в виде фармацевтически приемлемой соли или другого производного. Используемый здесь термин «фармацевтически приемлемая соль» относится к таким солям, которые, в рамках проведенного медицинского заключения, пригодны для использования в контакте с тканями человека и животных без излишней токсичности, раздражения, аллергической реакции и т.д., и отвечают разумному соотношению пользы и риска. Фармацевтически приемлемые соли аминов, карбоновых кислот, фосфонатов и другие типы соединений хорошо известны в медицине. Подробное описание свойств таких солей дано Berge S.M., et al., в "Pharmaceutical Salts" J. Pharmaceutical Science, 66: 1-19 (1977), приведенном здесь в качестве ссылки. Соли могут быть получены in situ в процессе выделения или очистки соединений изобретения, а также могут быть получены отдельно, путем взаимодействия свободной кислоты или свободного основания соединения изобретения с подходящим основанием или кислотой, соответственно. Примером фармацевтически приемлемых, нетоксичных солей кислот могут служить соли аминогруппы, образованные неорганическими кислотами, такими как соляная, бромоводородная, фосфорная, серная и хлорная кислоты, или органическими кислотами, такими как уксусная, щавелевая, малеиновая, винная, янтарная или малоновая кислоты, или полученные другими методами, используемыми в данной области, например, с помощью ионного обмена. К другим фармацевтически приемлемым солям относятся адипинат, альгинат, аскорбат, аспартат, бензолсульфонат, бензоат, бисульфат, борат, бутират, камфорат, камфорсульфонат, цитрат, циклопентанпропионат, диглюконат, додецилсульфат, этансульфонат, формиат, фумарат, глюкогептонат, глицерофосфат, глюконат, гемисульфат, гептанат, гексанат, гидройодид, 2-гидрокси-этансульфонат, лактобионат, лактат, лаурат, лаурил сульфат, малат, малеат, малонат, метансульфонат, 2-нафталинсульфонат, никотинат, нитрат, олеат, оксалат, пальмитат, памоат, пектинат, персульфат, 3-фенилпропионат, фосфат, пикрат, пивалат, пропионат, стеарат, сукцинат, сульфат, тартрат, тиоцианат, п-толуолсульфонат, ундеканат, валериат и подобные. Типичные соли щелочных и щелочноземельных металлов содержат натрий, литий, калий, кальций, магний и другие. Кроме того, фармацевтически приемлемые соли могут содержать, если требуется, нетоксичные катионы аммония, четвертичного аммония и амина, полученные с использованием таких противоионов, как галогениды, гидроксиды, карбоксилаты, сульфаты, фосфаты, нитраты, низшие алкил сульфонаты и арил сульфонаты.
Cоединения, заявляемые в данном изобретении, могут образовывать сольваты - ассоциации или комплексы из одной или нескольких молекул растворителя и соединения по изобретению. Примеры растворителей, образующих сольваты, включают, но ими не ограничиваются, воду, изопропанол, этанол, метанол, ДМСО, этилацетат, уксусную кислоту и этаноламин. Комплексы, в которых молекулами растворителя является вода, относятся к гидратам.
Кроме того, термин «фармацевтически приемлемый сложный эфир», как он используется здесь, обозначает гидролизующийся in vivo сложный эфир, который легко разлагается в теле человека до исходных соединений или их солей. Подходящая эфирная группа включает, например, производные фармацевтически приемлемых алифатических карбоновых кислот, в частности алкановых, алкеновых, циклоалкановых и алкандиеновых кислот, в которых каждый алкильный или алкенильный компонент обычно имеет не более 6 углеродных атомов. Примерами конкретных эфиров могут служить производные формиатов, ацетатов, пропионатов, бутиратов, акрилатов и этилсукцинатов. Очевидно, что эфиры также могут быть образованы гидроксильной группой или группой карбоновой кислоты соединения изобретения.
Термин «фармацевтически приемлемая пролекарственная форма», в контексте данного изобретения, означает такие пролекарства из числа соединений, составляющих суть данного изобретения, которые пригодны для использования человеком и животными без излишней токсичности, раздражения, аллергической реакции и т.д., отвечают разумному соотношению пользы и риска. Термин «пролекарства» означает соединения, которые трансформируются in vivo с образованием исходного соединения указанной выше формулы, например, при гидролизе в крови.
5.4. Фармацевтические композиции
Изобретение также относится с фармацевтическим композициям, которые содержат по меньшей мере одно из описанных здесь соединений (или пролекарственную форму, фармацевтически приемлемую соль или другое фармацевтически приемлемое производное) и один или несколько фармацевтически приемлемых носителей, растворителей и/или наполнителей. Данные композиции также могут содержать один или несколько дополнительных терапевтических агентов. С другой стороны, соединение данного изобретения может быть введено пациенту, нуждающемуся в соответствующей терапии, в комбинации с одним или более других терапевтических режимов (например, совместно с Кризотинибом или другими ингибиторами киназ, интерфероном, трансплантацией костного мозга, ингибиторами фарнезилтрансферазы, бисфосфонатами, талидомидом, противоопухолевыми вакцинами, гормональной терапией, антителами, облучением и т.д.). К примеру, дополнительными терапевтическими агентами для совместного введения или включения в фармацевтическую композицию с соединениями данного изобретения могут быть один или несколько противоопухолевых агентов.
Фармацевтические композиции, заявляемые в данном изобретении, содержат соединения данного изобретения совместно с фармацевтически приемлемыми носителями, которые могут включать в себя любые растворители, разбавители, дисперсии или суспензии, поверхностно-активные вещества, изотонические агенты, загустители и эмульгаторы, консерванты, вяжущие вещества, смазочные материалы и т.д., подходящие для конкретной формы дозирования. Remington's pharmaceutical sciences (15th edition, E.W. Martin, Mack Publishing Co., Easton, Pa., 1975) раскрывает различные носители, использованные при разработке фармацевтических композиций и известные методы их приготовления. За исключением таких случаев, когда среда обычных носителей несовместима с соединением изобретения, например, при появлении любых нежелательных биологических эффектов и иных нежелательных взаимодействий с любым другим компонентом (компонентами) фармацевтической композиции, использование таких композиций находится в рамках данного изобретения. Материалы, которые могут служить фармацевтически приемлемыми носителями, включают, но не ограничиваются, моно- и олигосахаридами, а также их производными; солодом, желатином; тальком; эксципиентами, такими как: какао-масло и воск для суппозиториев; масла, такие как арахисовое, хлопковое, сафроловое, кунжутное, оливковое, кукурузное и соевое масло; гликоли, такие как пропиленгликоль; сложные эфиры, такие как этилолеат и этиллаурат; агар; буферные вещества, такие как гидроксид магния и гидроксид алюминия; альгиновая кислота; апирогенная вода; изотонический раствор, раствор Рингера; этиловый спирт и фосфатные буферные растворы. Также в составе композиции могут быть другие нетоксичные совместимые смазочные вещества, такие как лаурилсульфат натрия и стеарат магния, а также красители, разделительные жидкости, пленкообразователи, подсластители, вкусовые добавки и ароматизаторы, консерванты и антиоксиданты.
5.5. Лекарственные формы
Предметом данного изобретения являются также лекарственные формы - класс фармацевтических композиций, состав которых оптимизирован для определенного пути введения в организм в терапевтически эффективной дозе. Лекарственные композиции данного изобретения могут быть введены в организм орально, местно, ректально, внутриглазным способом, пульмональным, например, в виде ингаляционного спрея, или внутрисосудистым способом, интраназально, интраперитонеально, подкожно, внутримышечно, интрастернально, а также инфузионным способом, в рекомендованных дозировках.
Лекарственная форма данного изобретения может содержать соединение описанной здесь формулы или его фармацевтически приемлемую соль, и дополнительный препарат, например, выбранный из числа следующих: ингибитор киназы, антидепрессант, противоопухолевый препарат, противовирусный препарат, противовоспалительный препарат, противогрибковый препарат или соединение против сосудистой гиперпролиферации, и любой фармацевтически приемлемый носитель, адъювант или растворитель. Термин «фармацевтически допустимый носитель или адъювант» означает носитель или адъювант, который может быть введен в организм пациента совместно с соединением, составляющем суть данного изобретения, и который не разрушает фармакологической активности этого соединения, и является нетоксичным при введении в дозах, достаточных для доставки терапевтического количества соединения.
Лекарственные формы данного изобретения могут содержать составы, полученные методами использования липосом или микрокапсуляционные методы, методами приготовления наноформ препарата, и прочие примеры, известные в фармацевтике.
5.6. Применение соединений в комбинированной терапии
Несмотря на то, что соединения данного изобретения могут вводиться в качестве индивидуального активного фармацевтического средства, их также можно использовать в сочетании с одним или несколькими соединениями изобретения, или одним или несколькими другими агентами. При совместном приеме внутрь терапевтические агенты могут представлять собой разные лекарственные формы, которые вводятся одновременно или последовательно в разное время, либо терапевтические агенты могут быть объединены в одну лекарственную форму.
Фраза «комбинированная терапия» в отношении соединений данного изобретения в сочетании с другими фармацевтическими агентами, означает одновременный или последовательный прием всех агентов, который так или иначе обеспечит благоприятное воздействие сочетания лекарств. Совместное введение подразумевает, в частности, совместную доставку, например, в одной таблетке, капсуле, инъекции или в другой форме, имеющий фиксированное соотношение активных веществ, также как и одновременную доставку в нескольких, отдельных лекарственных формах для каждого соединения соответственно.
Таким образом, введение соединений данного изобретения может быть осуществлено в сочетании с дополнительными методами лечения, известными специалистам в области профилактики и лечения опухолевых заболеваний, включающими лучевую терапию, применение цитостатических и цитотоксических препаратов, других противоопухолевых средств и препаратов для подавления симптомов или побочных эффектов одного из лекарств.
Если лекарственная форма представляет собой фиксированную дозу, такая комбинация использует соединения данного изобретения в приемлемом дозовом диапазоне. Вещества данного изобретения также могут быть введены в организм пациента последовательно с другими противоопухолевыми или цитотоксическими агентами, в том случае, когда комбинация этих препаратов невозможна. Изобретение не ограничено последовательностью введения; соединения данного изобретения могут быть введены в организм пациента совместно, до или после введения другого противоопухолевого или цитотоксического препарата.
Примеры
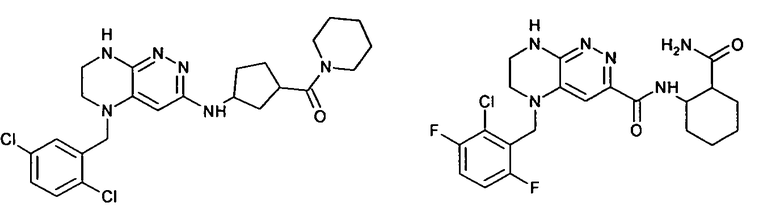
1. Получение 3-хлоро-5-(2-хлоро-3,6-дифторобензил)-5,6,7,8-тетрагидропиразино [2,3-с] пиридазина.
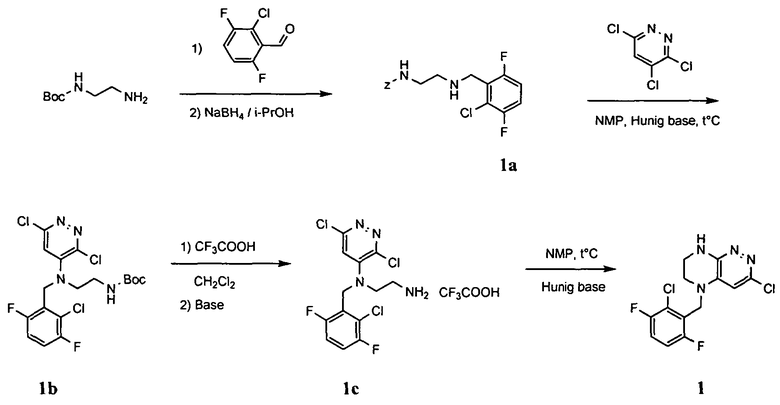
1). 17.7 г (100 ммоль) 2-хлор-3,6-дифторбензальдегида растворяют в 150 мл безводного метанола, прибавляют 0.1 мл ледяной уксусной кислоты и 16.0 г (100 ммоль) трет-бутил-2-аминоэтилкарбамата. Реакционную смесь перемешивают 4 часа при комнатной температуре, затем охлаждают до 0°C и прибавляют порциями 13.3 г (350 ммоль) боргидрида натрия, поддерживая заданную температуру (0°C), и оставляют на ночь при комнатной температуре. Растворитель удаляют, к остатку прибавляют 2.5 М раствор гидроксида натрия (~80 мл) до pH~10 и экстрагируют дихлорметаном (3×200 мл), объединенные экстракты промывают насыщенным раствором NaHCO3 (2×200 мл), а затем водой до нейтральной реакции, сушат, растворитель удаляют, остаток разделяют хроматографически. Получают: 24.9 г (78%) 1a.
2). К раствору 10.1 г (55 ммоль) 3,4,6-трихлорпиридазина в 80 мл безводного N-метилпирролидона прибавляют 16.0 г (50 ммоль) 1а и 9.6 мл (55 ммоль) основания Хунига. Реакционную смесь перемешивают в атмосфере аргона при 70°C в течение 90 часов (ТСХ-контроль). N-метилпирролидон удаляют в вакууме, остаток растворяют в 300 мл дихлорметана и промывают насыщенным раствором NaHCO3 (2×150 ml), а затем водой до нейтральной реакции, сушат, растворитель удаляют в вакууме, остаток перекристаллизовывают из диэтилового эфира. Получают: 15.7 г (67%) 1b.
3). Раствор 9.4 г (20 ммоль) 1b в 100 мл безводного дихлорметана охлаждают до -5°С и прибавляют 25 мл 20% (v/v) раствора трифторуксусной кислоты в безводном дихлорметане. Реакционную смесь перемешивают при комнатной температуре 18 часов, растворители удаляют, остаток промывают безводным эфиром (3×75 мл) и сушат. Получают: 9.2 г (96%) 1c.
4). К перемешиваемому раствору 9.6 г (20 ммоль) 1с в 100 мл безводного N-метилпирролидона прибавляют 8.7 мл (0.05 моль) основания Хунига и выдерживают полученную смесь 10 часов при 100°С (ТСХ-контроль), охлаждают, растворитель удаляют в вакууме, к остатку прибавляют 200 мл насыщенного раствора NaHCO3 и отфильтровывают образовавшийся осадок, который промывают водой (3×50 мл), сушат и разделяют хроматографически. Получают: 1.7 г (25%) соединения 1, m/z=330.03.
2. Получение метил 3-(3-(5-(2,6-дихлоро-3-фторобензил)-5,6,7,8-тетрагидропиразино[2,3-с]пиридазин-3-ил)-1,2,4-оксадиазол-5-ил) пропаната.
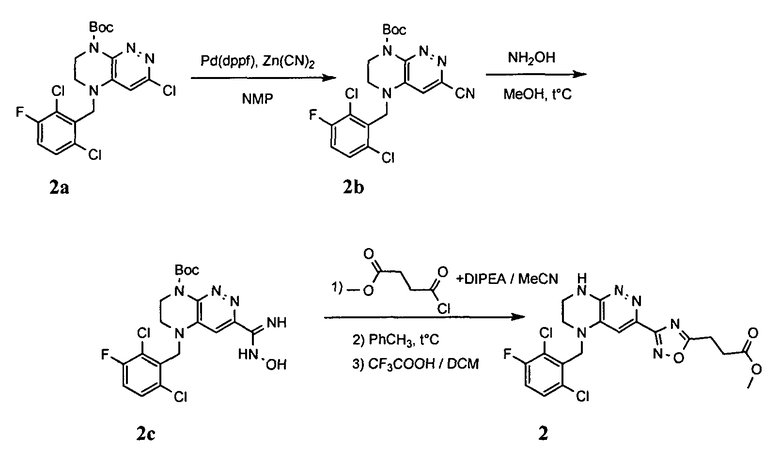
1). К раствору 4.48 г (10 ммоль) 2a в 40 мл безводного N-метилпирролидона добавляют 1.4 г (12 ммоль) цианида цинка и 0.37 г (0.5 ммоль, 5 моль %) комплекса Pd(dppf) с дихлорметаном и перемешивают в атмосфере аргона при 100°C в течение 12 часов (ТСХ-контроль). Полученную смесь вливают в 200 мл 1М раствора цианида калия. Полученный раствор экстрагируют дихлорметаном (3×100 мл). Органические фазы объединяют, промывают водой до нейтральной реакции, сушат, растворитель удаляют в вакууме. Остаток разделяют хроматографически. Получают 27% 2b.
2). 2.2 г (5 ммоль) 2b растворяют в 30 мл метанола и прибавляют 0,7 мл (10 ммоль) 50% водного раствора гидроксиламина. Полученную смесь перемешивают 6 часов при 45°C. Растворитель удаляют. Получают 0,94 г (95%) амидоксима 2c, который используется без дополнительной очистки.
3). 0,94 г (2 ммоль) амидоксима 2с растворяют в 30 мл безводного ацетонитрила и прибавляют 0,38 мл (2.2 ммоль) основания Хунига и 0,31 г (2.1 ммоль) хлорангидрида монометилового эфира янтарной кислоты. Реакционную смесь перемешивают 4 часа при комнатной температуре, растворитель удаляют, остаток промывают на фильтре водой (2×5 мл), сушат, прибавляют к 20 мл толуола и кипятят 2 часа (ТСХ-контроль). Толуол отгоняют, остаток растворяют в дихлорметане (30 мл) и прибавляют 1 мл 20% трифторуксусной кислоты в дихлорметане. Реакционную смесь оставляют на ночь. К реакционной смеси прибавляют 30 мл насыщенного раствора NaHCO3, промывают водой до нейтральной реакции и сушат. Растворитель удаляют в вакууме, остаток разделяют хроматографически. Получают: 42% соединения 2, m/z=446.07.
3. Получение 5-(5-(1-(2,6-дихлоро-3-фторобензил)этил)-5,6,7,8-тетрагидропиразино [2,3-с] пиридазин-3-ил)-3-циклогексил-1,2,4-оксадиазола.
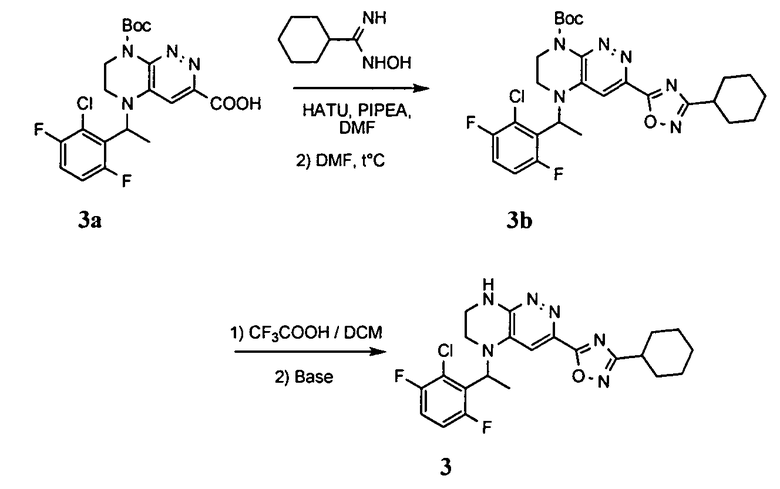
1). Стадия 1. 9 мг (20 ммоль) 3a суспендируют в 20 мл ацетонитрила и прибавляют 3,5 г (22 ммоль) карбонилдиимидазола и перемешивают 1 час. К реакционной смеси прибавляют 3,1 г (22 ммоль) циклогексиламидоксима (полученного по стандартной методике из нитрила циклогексанкарбоновой кислоты). Реакционную смесь перемешивают 2 часа, после чего растворитель удаляют. К остатку прибавляют 30 мл безводного ДМФА и перемешивают в атмосфере аргона 6 ч при 100°C. Растворитель удаляют в вакууме, остаток растворяют в дихлорметане, промывают водой, сушат, растворитель удаляют, остаток разделяют хроматографически. Получают: 42% 3b.
2). 3,4 г (6 ммоль) 3b растворяют в 10 мл безводного дихлорметана и прибавляют 5 мл 20% раствора трифторуксусной кислоты в дихлорметане. Реакционную смесь перемешивают 8 часов, растворители удаляют, к остатку прибавляют 20 мл насыщенного водного раствора NaHCO3, образовавшийся осадок отфильтровывают, промывают водой и сушат. Получают: 94% соединения 3, m/z=460.16.
4). Получение 4-((5-(5-(2-хлоро-3,6-дифторобензил)-5,6,7,8-тетрагидропиразино[2,3-с]пиридазин-3-ил)-1,3,4-оксадиазол-2-ил)метил)морфолина.
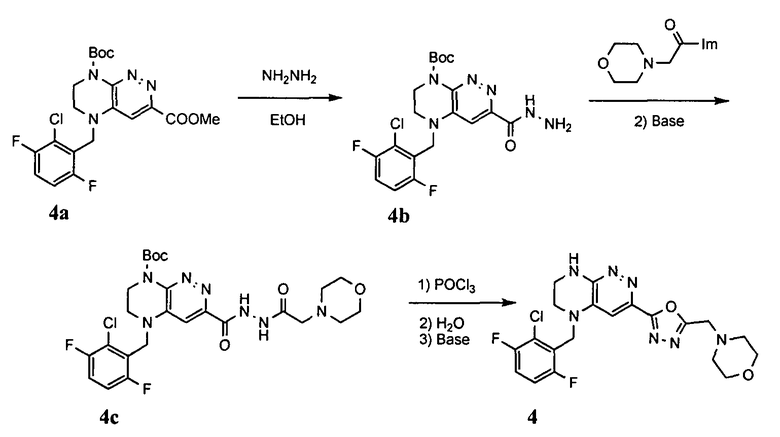
1). 9 г (20 ммоль) 4а растворяют в 20 мл метанола и прибавляют 2.2 мл (45 ммоль) гидразин-гидрата. Реакционную смесь перемешивают 4 часа. Растворитель удаляют, остаток растирают с 5 мл воды, отфильтровывают, промывают водой (2×10 мл) и сушат. Получают: 97% 4b.
2). 8.2 г (18 ммоль) 4b растворяют в 10 мл безводного ацетонитрила, прибавляют 2,8 мл (20 ммоль) триэтиламина, после чего по каплям прибавляют раствор 3,5 г (18 ммоль) имидазолида морфолинуксусной кислоты (полученного по стандартной методике из морфолинуксусной кислоты и карбонилдиимидазола) в 20 мл ацетонитрила. Реакционную смесь перемешивают 2 часа при комнатной температуре. Растворитель удаляют, остаток промывают водой (2×5 мл), сушат и используют в следующей стадии без дополнительной очистки. Получают: 83% 4c.
3). 7 г (12 ммоль) диацилгидразина 4с растворяют в 10 мл POCl3 и перемешивают реакционную смесь при 50°C 4 часа (ТСХ-контроль), охлаждают и выливают в лед и подщелачивают NaHCO3. Полученную смесь экстрагируют дихлорметаном (2×100 мл), объединенные органические фазы промывают водой до нейтральной реакции, растворитель удаляют, остаток разделяют хроматографически. Получают: 31% соединения 4, m/z=463.13.
5. Получение 2-(5-(5-хлоро-2-метоксибензил)-5,6,7,8-тетрагидрапиразино[2,3-с]пиридазин-3-ил)-5-(пиперидин-4-ил)-1,3,4-оксадиазола.
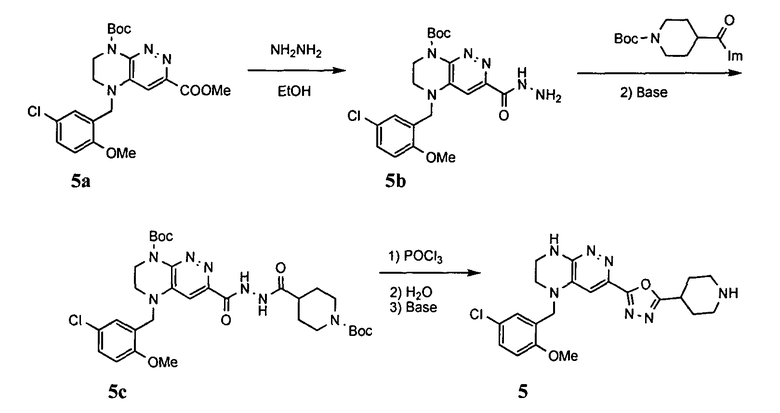
1). 9 г (20 ммоль) 5a растворяют в 20 мл метанола и прибавляют 2.2 мл (45 ммоль) гидразин-гидрата. Реакционную смесь перемешивают 4 часа. Растворитель удаляют, остаток растирают с 5 мл воды, отфильтровывают, промывают водой (2×10 мл) и сушат. Получают: 97% 5b.
2). Гидразид 5b 8,1 г (18 ммоль) растворяют в 10 мл безводного ацетонитрила, прибавляют 2.8 мл (20 ммоль) триэтиламина, после чего по каплям прибавляют раствор 5 г (18 ммоль) имидазолида Boc-пиникотиновой кислоты (полученного по стандартной методике из пиникотиновой кислоты и карбонилдиимидазола) в 20 мл ацетонитрила. Реакционную смесь перемешивают 2 часа при комнатной температуре. Растворитель удаляют, остаток промывают водой (2×5 мл), сушат и используют в следующей стадии без дополнительной очистки. Получают: 78% 5c.
3). Диацилгидразин 5c 9,9 г (15 ммоль) растворяют в 10 мл POCl3 и перемешивают реакционную смесь при 50°C 4 часа (ТСХ-контроль), охлаждают и выливают в лед и подщелачивают NaHCO3. Полученную смесь экстрагируют дихлорметаном (2×100 мл), объединенные органические фазы промывают водой до нейтральной реакции, растворитель удаляют, остаток разделяют хроматографически. Получают: 24% соединения 5, m/z=441.17.
6. Получение 3-[5-(1-метилэтил)-1,3,4-оксадиазол-2-ил]-5-[1-(2,5-дихлорфенил)этил]-5,6,7,8-тетрагидрапиразино[2,3-с]пиридазина.
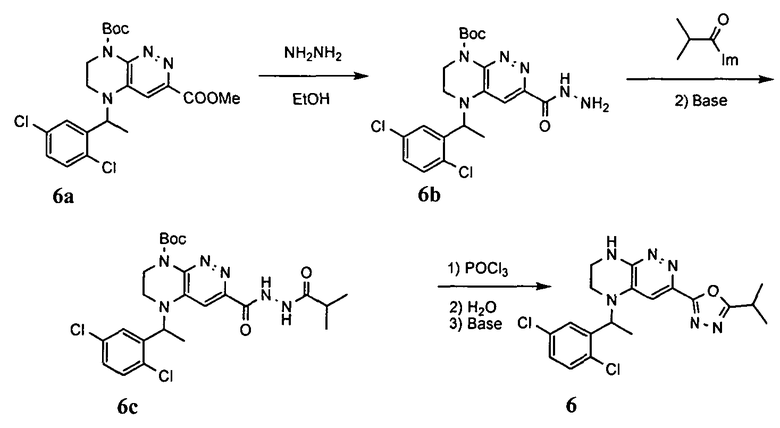
1). 9.3 г (20 ммоль) 6a растворяют в 20 мл метанола и прибавляют 2,2 мл (45 ммоль) гидразин-гидрата. Реакционную смесь перемешивают 4 часа. Растворитель удаляют, остаток растирают с 5 мл воды, отфильтровывают, промывают водой (2×10 мл) и сушат. Получают: 95% 6b.
2). Гидразид 6b 7.9 г (18 ммоль) растворяют в 10 мл безводного ацетонитрила, прибавляют 2.8 мл (20 ммоль) триэтиламина, после чего по каплям прибавляют раствор 2.5 г (18 ммоль) имидазолида изобутановой кислоты (полученного по стандартной методике из изобутановой кислоты и карбонилдиимидазола) в 20 мл ацетонитрила. Реакционную смесь перемешивают 2 часа при комнатной температуре. Растворитель удаляют, остаток промывают водой (2×5 мл), сушат и используют в следующей стадии без дополнительной очистки. Получают: 83% 6c.
3). Диацилгидразин 6с 8 г (15 ммоль) растворяют в 10 мл POCl3 и перемешивают реакционную смесь при 50°C 4 часа (ТСХ-контроль), охлаждают и выливают в лед и подщелачивают NaHCO3. Полученную смесь экстрагируют дихлорметаном (2×100 мл), объединенные органические фазы промывают водой до нейтральной реакции, растворитель удаляют, остаток разделяют хроматографически. Получают: 31% соединения 6, m/z=418.11.
7). Получение 2-(4-(5-(5-(2,6-дихлоро-3-фторбензил)-5,6,7,8-тетрагидропиразино[2,3-c]пиридазин-3-ил)-4Н-1,2,4-триазол-3-ил)пиперидин-1-ил)этанола.
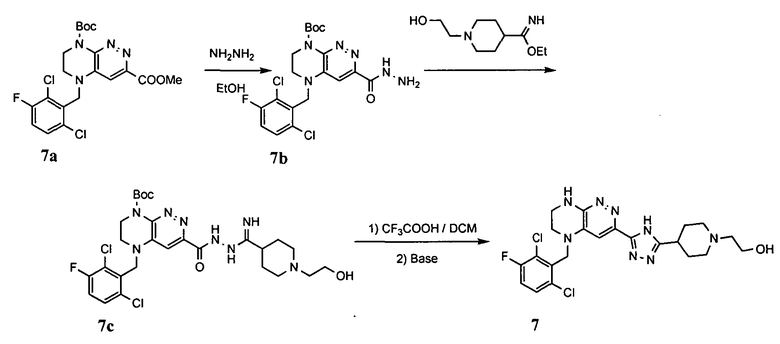
1). 9.4 г (20 ммоль) 7а растворяют в 20 мл метанола и прибавляют 2,2 мл (45 ммоль) гидразин-гидрата. Реакционную смесь перемешивают 4 часа. Растворитель удаляют, остаток растирают с 5 мл воды, отфильтровывают, промывают водой (2×10 мл) и сушат. Получают: 95% 7b.
2). К раствору 2.8 г (20 ммоль) 4-циано-1-этилпиперидина в 50 мл безводного диэтилового эфира прибавляют 50 мл безводного этанола. Реакционную смесь охлаждают до 0°C и прибавляют раствор 45 ммоль хлористого водорода в 100 мл эфира. Реакционную смесь перемешивают 8 часов, растворители удаляют, остаток растворяют в 200 мл безводного этанола и прибавляют порциями 2.8 г (40 ммоль) этилата натрия, поддерживая температуру не выше +10°C. Реакционную смесь перемешивают 30 минут, выпавший осадок отфильтровывают, к фильтрату прибавляют 8,5 г (18 ммоль) гидразида 7b и перемешивают 2 часа. Растворители удаляют, к остатку прибавляют 50 мл безводного толуола и кипятят полученную смесь 8 часов. Растворитель удаляют, остаток разделяют хроматографически. Получают: 28% 7c.
3). К раствору 3.1 г (5 ммоль) триазина 7с в 50 мл безводного хлористого метилена прибавляют 20 мл 20% раствора трифторуксусной кислоты в хлористом метилене. Реакционную смесь перемешивают 8 часов, растворители удаляют, к остатку прибавляют 20 мл насыщенного водного раствора NaHCO3. Образовавшийся осадок отфильтровывают, промывают водой и сушат. Получают: 98% соединения 7, m/z=506.15.
8. Получение 5-(2,5-дихлоробензил)-3-(5-(трифторометил)-4Н-1,2,4-триазол-3-ил)-6,7-дигидропиразино [2,3-с]пиридазина.
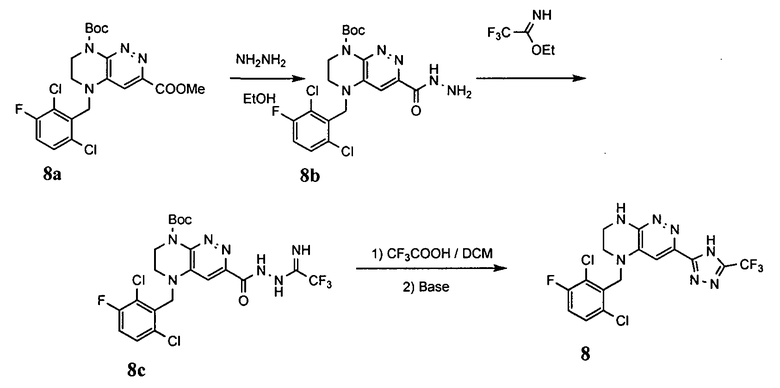
1). 9.4 г (20 ммоль) 8а растворяют в 20 мл метанола и прибавляют 2.2 мл (45 ммоль) гидразин-гидрата. Реакционную смесь перемешивают 4 часа. Растворитель удаляют, остаток растирают с 5 мл воды, отфильтровывают, промывают водой (2×10 мл) и сушат. Получают: 95% 8b.
2). К раствору 2.8 г (20 ммоль) трифторацетамидина в 50 мл безводного диоксана прибавляют 8.5 г (18 ммоль) гидразида 8b и перемешивают 2 часа, затем кипятят 8 часов. Растворитель удаляют, остаток разделяют хроматографически. Получают: 31% 8c.
3). К раствору 2.8 г (5 ммоль) триазина 8с в 50 мл безводного хлористого метилена прибавляют 20 мл 20% раствора трифторуксусной кислоты в хлористом метилене. Реакционную смесь перемешивают 8 часов, растворители удаляют, к остатку прибавляют 20 мл насыщенного водного раствора NaHCO3. Образовавшийся осадок отфильтровывают, промывают водой и сушат. Получают: 92% соединения 8, m/z=447.04.
9. Получение 3-(5-азетидин-3-илметил)-4H-1,2,4-триазол-3-ил)-5-(1-(2,5-дихлорофенил) этил)-6,7-дигидропиразино[2,3-с] пиридазина.

1). 9.3 г (20 ммоль) 9а растворяют в 20 мл метанола и прибавляют 2.2 мл (45 ммоль) гидразин-гидрата. Реакционную смесь перемешивают 4 часа. Растворитель удаляют, остаток растирают с 5 мл воды, отфильтровывают, промывают водой (2×10 мл) и сушат. Получают: 95% 9b.
2). К раствору 4.3 г (20 ммоль) 1-параметоксибензил-3-цианометилазета в 100 мл эфира прибавляют 40 ммоль абсолютного этанола. Реакционную смесь охлаждают до 0°C и прибавляют раствор 45 ммоль хлористого водорода в 100 мл эфира. Реакционную смесь перемешивают 8 часов, растворители удаляют, остаток растворяют в 200 мл безводного этанола и прибавляют порциями 2.7 г (40 ммоль) этилата натрия, поддерживая температуру не выше +10°C. Реакционную смесь перемешивают 30 минут, выпавший осадок отфильтровывают, к фильтрату прибавляют 8.4 г (18 ммоль) гидразида 9b, полученного на предыдущей стадии, и перемешивают 2 часа. Растворители удаляют, к остатку прибавляют 50 мл безводного толуола и кипятят полученную смесь 8 часов. Растворитель удаляют, остаток разделяют хроматографически. Получают: 14% 9c.
3). Раствор 1.1 г (2 ммоль) триазина 9с в 20 мл безводной трифторуксусной кислоты перемешивают 8 часов, растворители удаляют, к остатку прибавляют 10 мл насыщенного водного раствора NaHCO3. Образовавшийся осадок отфильтровывают, промывают водой и сушат. Получают: 53% соединения 9, m/z=444.13.
10. Получение 5-(1-(2,6-дихлоро-3-фторофенил)этил)-N-(4-(пирролидин-1-карбонил)фенил)-5,6,7,8-тетрагидропиразино [2,3-с] пиридазин-3-карбоксамида.
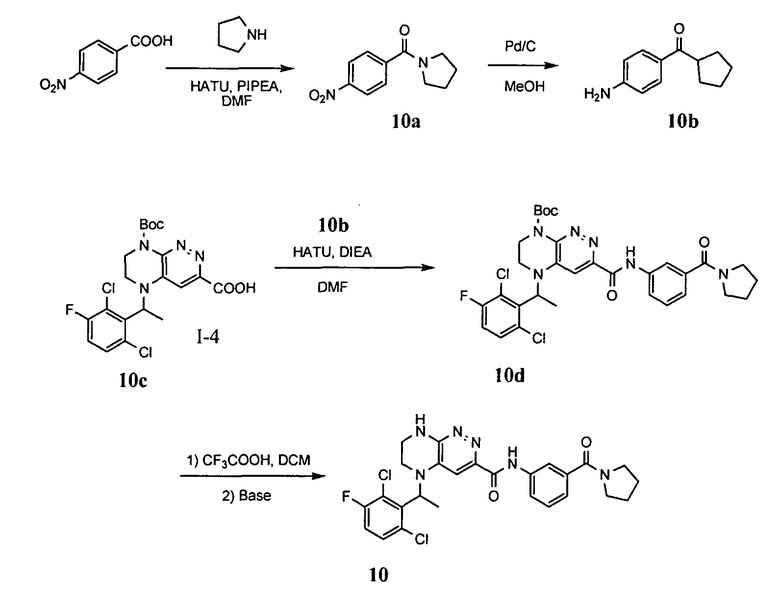
1). К раствору 4-нитробензойной кислоты (500 мг, 3 ммоль), HATU (1.71 г, 4.5 ммоль) и основания Хунига (1.16 г, 9 ммоль) в ДМФ добавляют пирролидин (320 мг, 4.5 ммоль). Реакционную смесь перемешивают в течение ночи при комнатной температуре. Растворитель удаляют, остаток разделяют хроматографически. Получают: 0.52 г (79%) 10a.
2). К раствору 1.7 мг 10а в метаноле добавляются 10% Pd/C (200 мг). Смесь гидрируют при комнатной температуре в течение 1 часа. Реакционную смесь фильтруют, из фильтрата удаляют растворитель. Получают: 0.28 мг (87%) 10b.
3). К раствору 10с (174 мг, 0.37 ммоль) в 10 мл ДМФ прибавляют сначала HATU (0.2 мг, 55 ммоль), затем основание Хунига (95 мг, 0.73 ммоль). Смесь перемешивают при комнатной температуре в течение 30 минут, и добавляют 10b (104 мг, 0.55 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 1.5 часов. Растворитель удаляют, остаток разделяют хроматографически. Получают 10d (169 мг, 71%).
4). К раствору 10d (169 мг, 0.26 ммоль) в 3 мл хлористого метилена добавляют 1 мл безводной трифторуксусной кислоты. Реакционную смесь перемешивают при комнатной температуре в течение 1 часа, растворители удаляют, к остатку прибавляют 10 мл насыщенного водного раствора NaHCO3 до pH 9. Полученную смесь экстрагируют дихлорметаном (4×5 мл). Объединенные органические фазы высушивают, растворитель удаляют, остаток разделяют хроматографически. Получают: 41% соединения 10 (58 мг), m/z=542.14.
11. Получение 5-(5-хлоро-2-(трифторметил)бензил)-N-(4-(4-(диметиламино)пиперидин-1-ил)-2-метоксифенил)-5,6,7,8-тетрагидропиразино[2,3-с]пиридазин-3-амин.
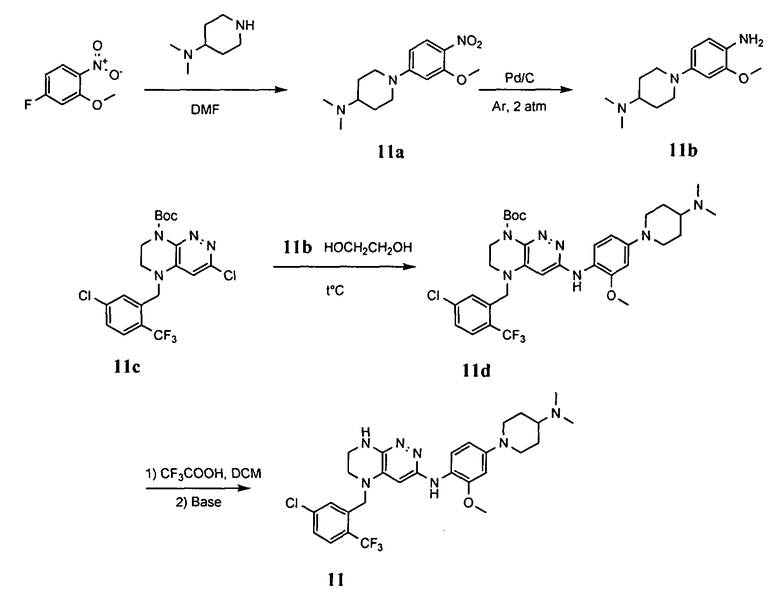
1). К раствору 5-фторо-2-нитроанизола (500 мг, 2.92 ммоль) в 3 мл DMF добавляют N,N-диметилпиперидин-4-амин (374 мг, 2.92 ммоль) и K2CO3 (0.808 г, 5.84 ммоль). Реакционную смесь перемешивают при 120°C в течение 18 ч. К реакционной смеси добавляют насыщенный раствор NaHCO3, полученную смесь экстрагируют этилацетатом. Органическую фазу очищают хроматографически. Получают: 88% (0.712 г) 11a.
2). Соединение 11a (250 мг, 0.9 ммоль) растворяют в 10 мл этанола в атмосфере аргона, добавляют 10% Pd/C (0.06 г). Гидрирование проводится в течение 4 часов при давлении 2 атм. Реакционную смесь фильтруют на диатомитовом фильтре (Celite) и добавляют раствор хлористого водорода в этаноле. Фильтрат концентрируют, получают 11b (200 мг, 88%).
3). К раствору соединения 11с (74 мг, 0.16 ммоль) в 1 мл 2-метоксиэтанола добавляют 40 мг (0.16 ммоль) 11b. Реакционная смесь перемешивается в течение 18 часов при 110°C. К смеси прибавляют насыщенный раствор Na2CO3, раствор экстрагируют этилацетатом и разделяют хроматографически. Получают: 23% (25 мг) соединения 11d.
4). К раствору 11d (175 мг, 0.26 ммоль) в 3 мл хлористого метилена добавляют 1 мл безводной трифторуксусной кислоты. Реакционную смесь перемешивают при комнатной температуре в течение 1 часа, растворители удаляют, к остатку прибавляют 10 мл насыщенного водного раствора NaHCO3 до pH 9. Полученную смесь экстрагируют дихлорметаном (4×5 мл). Объединенные органические фазы высушивают, растворитель удаляют, остаток разделяют хроматографически. Получают: 45% соединения 11 (67 мг), m/z=575.24.
12. Получение 5-(1-(2,6-дихлоро-3-фторофенил)этил)-3-(6-(пирролидин-1-ил)пиридин-3-ил)-5,6,7,8-тетрагидропиразино[2,3-с]пиридазина
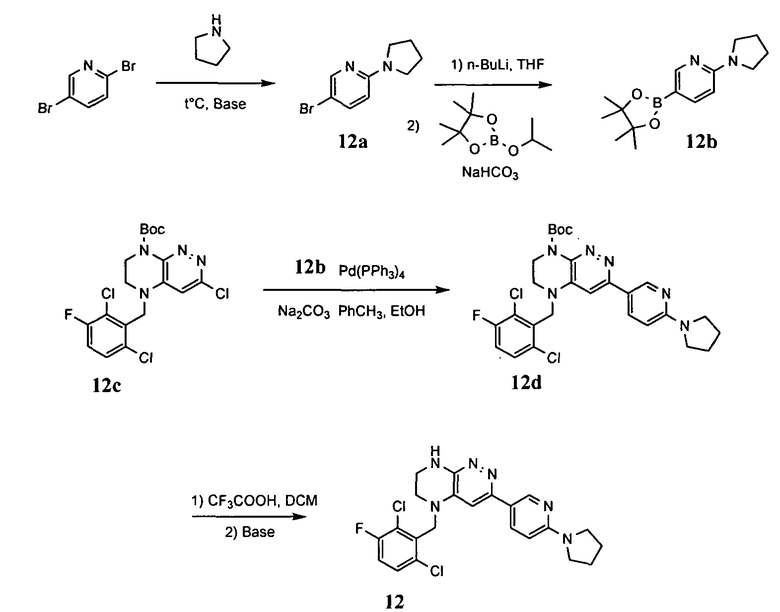
1). Смесь 1 г (4.2 ммоль) 2,5-дибромопиридина и пирролидина (1.5 мл) нагревают до 110°C в течение 90 минут. Избыток пирролидина отгоняют под вакуумом, остаток добавляют в водный раствор NaHCO3. Полученную смесь экстрагируют этилацетатом. Растворитель удаляют, получают: 55% (0.52 г) 12a.
2). 0.52 г (2.3 ммоль) 12a в 15 мл ТГФ охлаждают до -78°C и по каплям добавляют 1.7 мл 1.6М раствора н-бутиллития в гексане. Реакционную смесь перемешивают в течение 30 минут, после чего добавляют 2-изопропокси-4,4,5,5-тетраметил-1,3,2-диоксаборолан (511 мг, 2.7 ммоль). В течение 2 часов смесь оставляют нагреваться до комнатной температуры. Реакцию останавливают добавлением водного раствора NaHCO3. Смесь экстрагируют этилацетатом, высушивают с помощью Na2SO4 и удаляют растворитель. Полученный боронат используют в неочищенном виде для дальнейших реакций. Получают: 190 мг (30%) 12b.
3). 223 мг (0.5 ммоль) 12с и 165 мг (0.6 ммоль) 12b растворяют в смеси 6:1 толуола и этанола. К раствору добавляют 25 мг Pd(PPh3)4 и 1 мл 2М раствора Na2CO3. Полученную смесь греют при 80°C в течение 8 ч. Реакционную смесь разбавляют CH2Cl2, высушивают над MgSO4, отфильтровывают, концентрируют и очищают гель-хроматографией. Получают: 28% (78 мг) 12c.
4). К раствору 12d (145 мг, 0.26 ммоль) в 3 мл хлористого метилена добавляют 1 мл безводной трифторуксусной кислоты. Реакционную смесь перемешивают при комнатной температуре в течение 1 часа, растворители удаляют, к остатку прибавляют 10 мл насыщенного водного раствора NaHCO3 до pH 9. Полученную смесь экстрагируют дихлорметаном (4×5 мл). Объединенные органические фазы высушивают, растворитель удаляют, остаток разделяют хроматографически. Получают: 45% соединения 12 (54 мг), m/z=472.13.
13. Получение (R)-3-хлоро-5-(1-(2,6-дихлоро-3-фторофенил)этил)-5,6,7,8-тетрагидропиразино[2,3-с] пиридазина
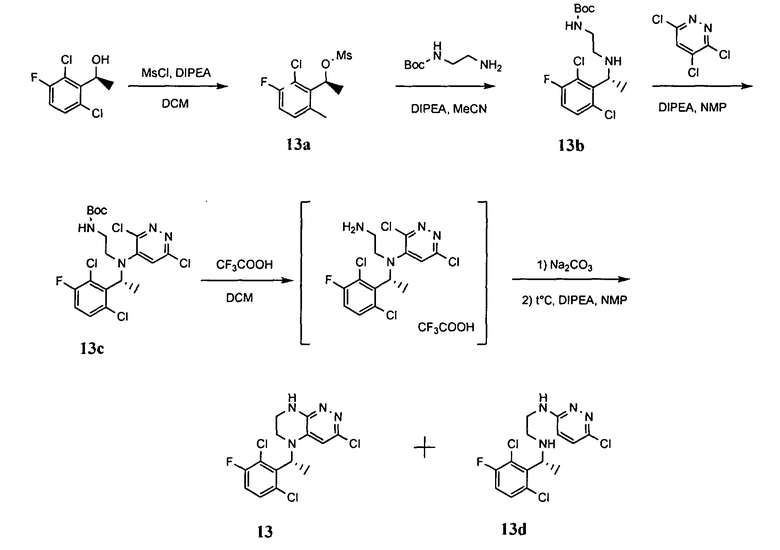
1). К раствору 4.18 г (0.02 моль) (S)-1-(2,6-дихлор-3-фторфенил)этанола в 50 мл безводного дихлорметана прибавляют 4.3 мл (~0.025 моль) N,N-диизопропилэтиламина, смесь охлаждают до -5°C и прибавляют по каплям, поддерживая заданную температуру 1.6 мл (0.021 моль) метансульфохлорида в 10 мл безводного дихлорметана. Реакционную смесь перемешивают 2 часов при 0°C, а затем еще 4 часа при комнатной температуре, затем к реакционной смеси прибавляют 10 мл насыщенного раствора NaHCO3, органическую фазу отделяют и промывают водой до нейтральной реакции, раствор сушат, растворитель удаляют. Получают: 5.3 г (93%) 13a, который используется в следующей стадии без дополнительной очистки.
2). К раствору 5.17 г (0.018 моль) 13а в 25 мл безводного ацетонитрила, при 0°C прибавляют раствор смеси 3.20 г (0.02 моль) трет-бутил 2-аминоэтилкарбамата и 3.5 мл (0.02 моль) N,N-диизопропилэтиламина в 20 мл безводного ацетонитрила. Реакционную смесь перемешивают 18 часов при комнатной температуре, растворитель удаляют, остаток растворяют в 200 мл эфира и промывают насыщенным раствором NaHCO3 (2×50 мл), а затем водой до нейтральной реакции. Раствор сушат, растворитель удаляют, остаток сушат в вакууме и разделяют хроматографически (CH2Cl2->CH2Cl2:MeOH (4:1)). Получают: 4.86 (77%) 13b.
3). К раствору 3.51 (0.01 моль) 13b в 10 мл безводного N-метилпирролидона прибавляют 2.1 мл (0.012 моль) N,N-диизопропилэтиламина и 2.20 (0.012 моль) 3,4,6-трихлорпиридазина; реакционную смесь перемешивают в атмосфере аргона без доступа влаги при 85°C в течение 200 часов (ТСХ-контроль). Растворитель удаляют в вакууме, остаток растворяют в 300 мл дихлорметана и промывают насыщенным раствором NaHCO3 (1×50 мл), а затем водой до нейтральной реакции. Раствор сушат, растворитель удаляют, остаток сушат в вакууме и разделяют хроматографически (CH2Cl2→CH2Cl2:MeOH (4:1)). Получают: 2.84 г (57%) 13c.
4). К раствору 2.50 г (5 ммоль) 13с в 100 мл дихлорметана прибавляют 20 мл 20% раствора трифторуксусной кислоты в дихлорметане. Реакционную смесь перемешивают при комнатной температуре 14 часов, прибавляют 200 мл дихлорметана и 150 мл насыщенного раствора NaHCO3, органическую фазу отделяют, промывают водой до нейтральной реакции, сушат, растворитель удаляют, остаток растворяют в 10 мл безводного N-метилпирролидона, прибавляют 1.2 мл (~0.07 моль) N,N-диизопропилэтиламина и перемешивают реакционную смесь в атмосфере аргона без доступа влаги при 100°C в течение 60 часов (ТСХ-контроль). Растворители удаляют в вакууме, остаток растворяют в 300 мл дихлорметана и промывают насыщенным раствором NaHCO3 (2×100 мл), а затем водой до нейтральной реакции, сушат, растворитель удаляют, остаток сушат в вакууме и разделяют хроматографически (Гексан : Этилацетат 1:1→CH2Cl2→CH2Cl2:MeOH (9:1)). Получают: 415 мг (23%) соединения 13, m/z=360.01 и 418 мг соединения 13d.
14. Примеры синтеза нехиральных 5-замещенных производных 3-хлоро-5,6,7,8-тетрагидропиразино[2,3-с]пиридазина
Нехиральные 5-замещенные производные 3-хлоро-5,6,7,8-тетрагидропиразино[2,3-c]пиридазина синтезируются аналогично соединению 1, методика синтеза которого приведена выше. Примеры полученных соединений представлены в таблице.
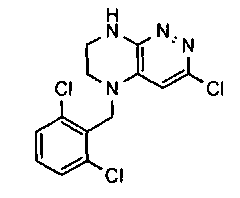
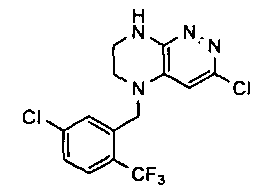
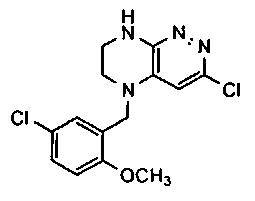
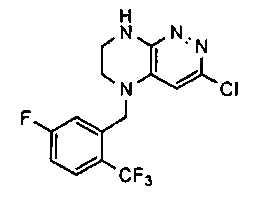
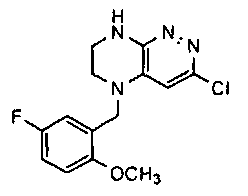
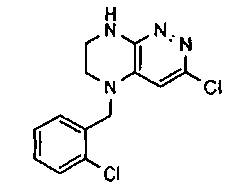
15. Примеры синтеза смеси энатниомеров хиральных 5-замещенных производных 3-хлоро-5,6,7,8-тетрагидропиразино [2,3-с]пиридазина
Смесь этантиомеров хиральных 5-замещенных производных 3-хлоро-5,6,7,8-тетрагидропиразино [2,3-с]пиридазина синтезируются аналогично соединению 1, методика синтеза которого приведена выше. Примеры полученных соединений представлены в таблице.
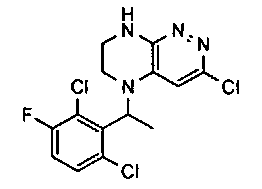
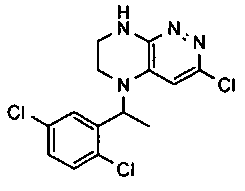
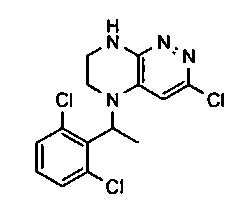
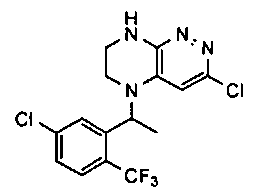
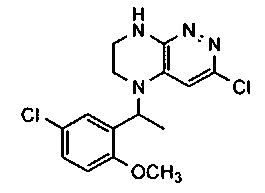
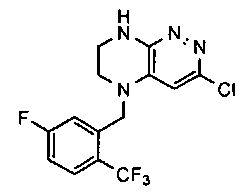
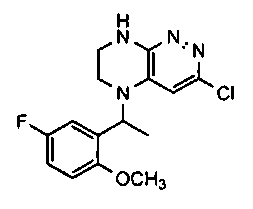
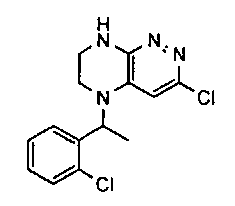
16. Примеры синтеза хиральных 5-замещенных производных 3-хлоро-5,6,7,8-тетрагидропиразино [2,3-с]пиридазина
Хиральные 5-замещенные производные 3-хлоро-5,6,7,8-тетрагидропиразино [2,3-с]пиридазина синтезируются аналогично соединению 13, методика синтеза которого приведена выше. Примеры полученных соединений представлены ниже.
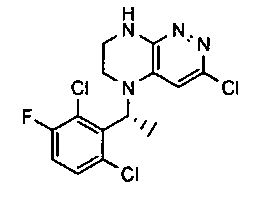
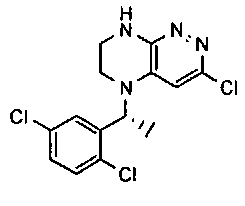
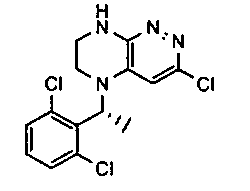
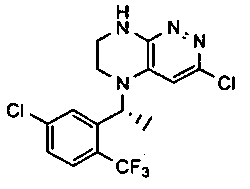
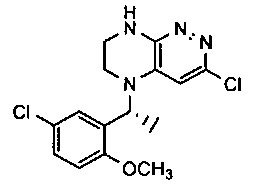
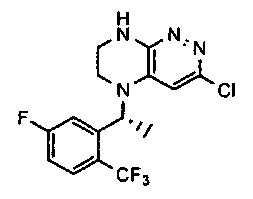
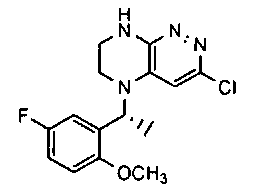
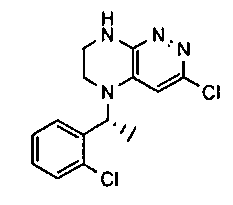
17. Примеры синтеза 5-замещенных производных 3-(5-бензил-5,6,7,8-тетрагидропиразино [2,3-с]пиридазин-3-ил)-1,2,4-оксадиазола
5-замещенные производные 3-(5-замещенного-5,6,7,8-тетрагидропиразино [2,3-с]пиридазин-3-ил)-1,2,4-оксадиазола синтезируются из соответствующих Boc-защищенных 5-замещенных производных 3-хлоро-5,6,7,8-тетрагидропиразино [2,3-с]пиридазина аналогично соединению 2, методика получения которого приведена выше. Примеры полученных соединений представлены ниже.
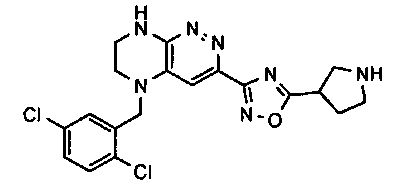
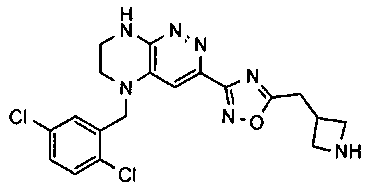
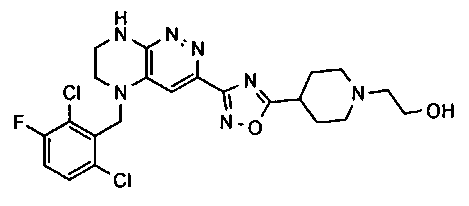
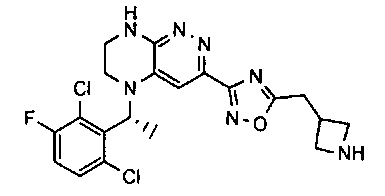
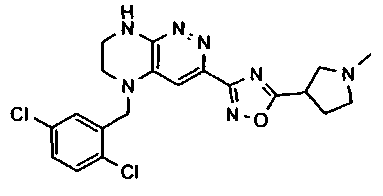
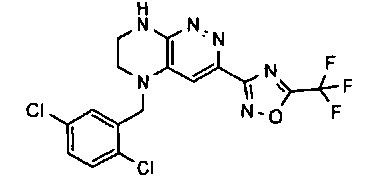
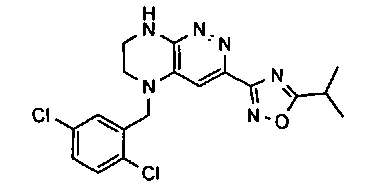
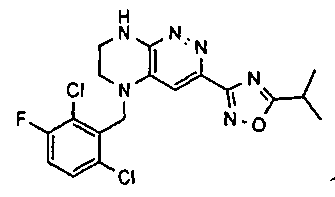

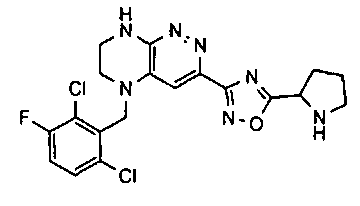
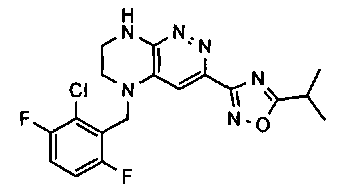
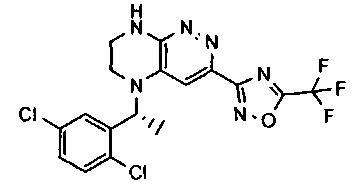
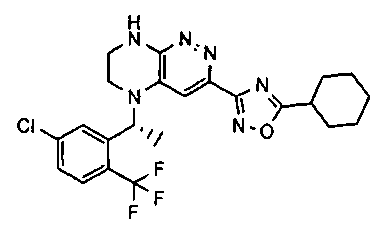
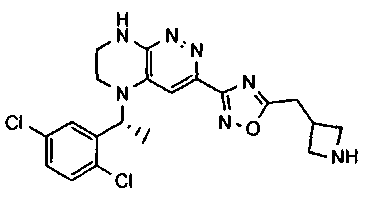
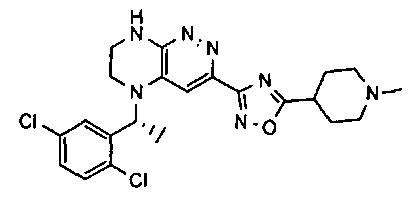
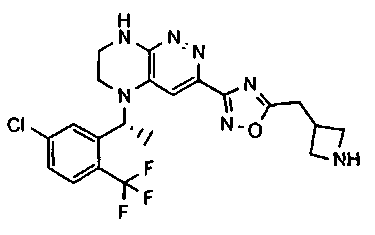
18. Примеры синтеза 5-замещенных производных 3-(5-бензил-5,6,7,8-тетрагидропиразино [2,3-с]пиридазин-3-ил)-1,2,4-оксадиазола
3-замещенные производные 5-(5-замещенного-5,6,7,8-тетрагидропиразино [2,3-с]пиридазин-3-ил)-1,2,4-оксадиазола синтезируются из соответствующих Boc-защищенных 5-замещенных производных 3-хлоро-5,6,7,8-тетрагидропиразино [2,3-с]пиридазина аналогично соединению 3, методика синтеза которого приведена выше. Примеры полученных соединений представлены в таблице.
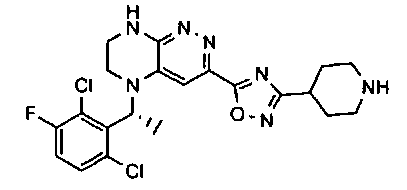
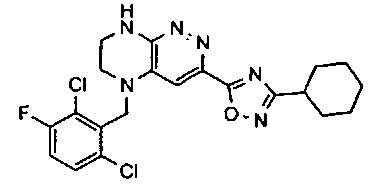
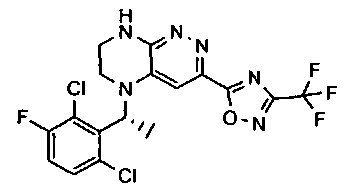
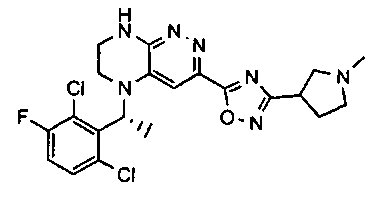
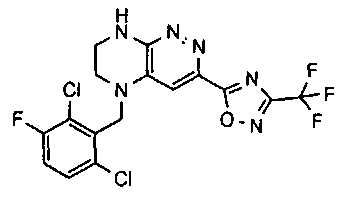
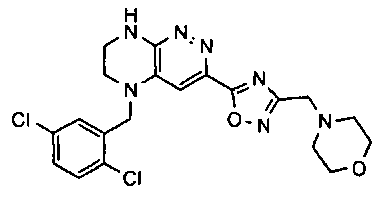
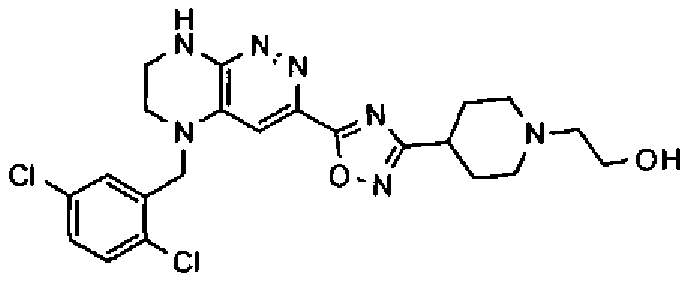
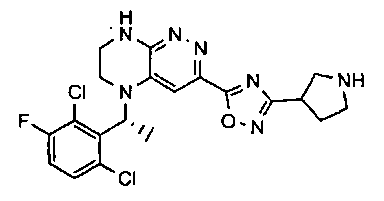
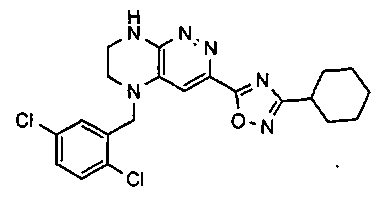
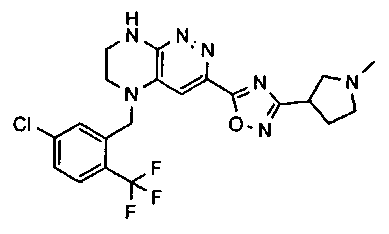
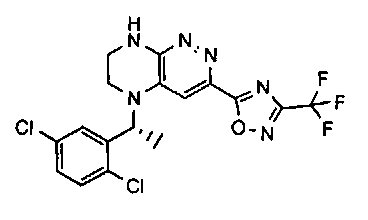
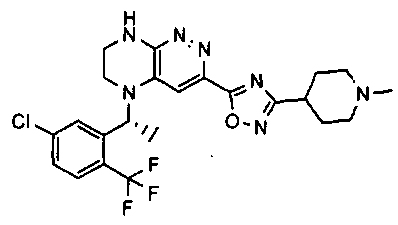
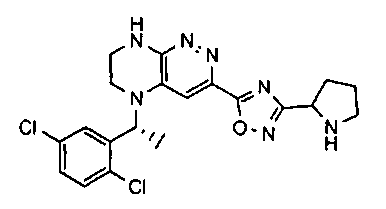
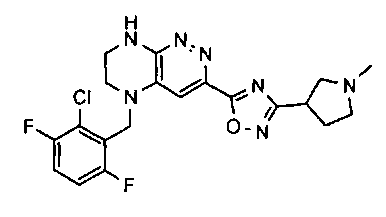
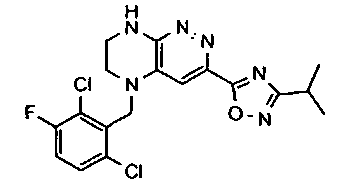
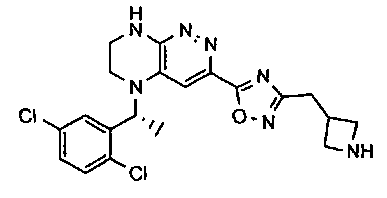
19. Примеры синтеза 5-замещенных производных 2-(5-замещенного-5,6,7,8-тетрагидропиразино [2,3-с]пиридазин-3-ил)-1,3,4-оксадиазола
2-замещенные производные 5-(5-замещенного-5,6,7,8-тетрагидропиразино [2,3-с]пиридазин-3-ил)-1,3,4-оксадиазола синтезируются из соответствующих Boc-защищенных 5-замещенных производных 3-хлоро-5,6,7,8-тетрагидропиразино [2,3-с]пиридазина аналогично соединению 4, методика синтеза которого приведена выше. Примеры полученных соединений представлены ниже.
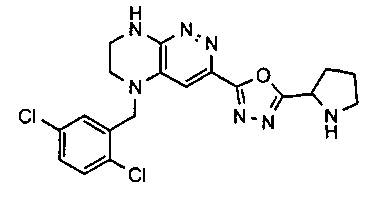
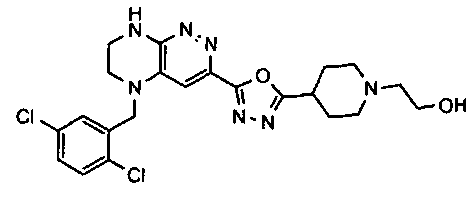
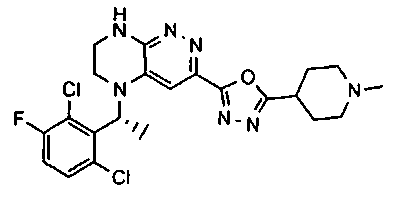
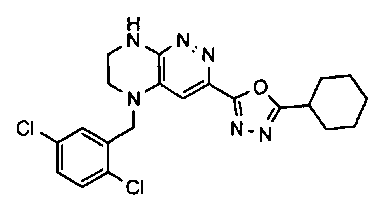
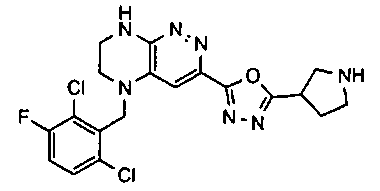
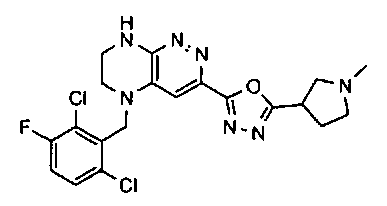
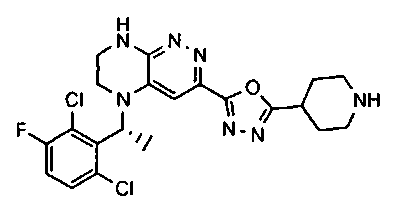
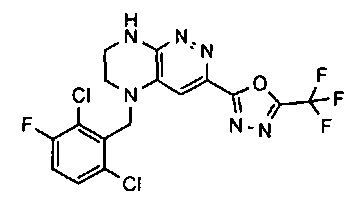
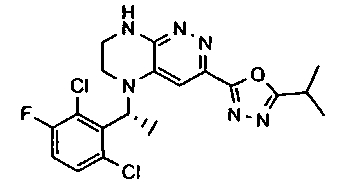
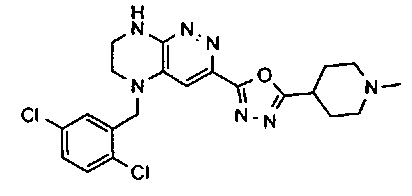
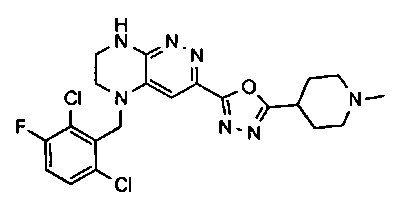
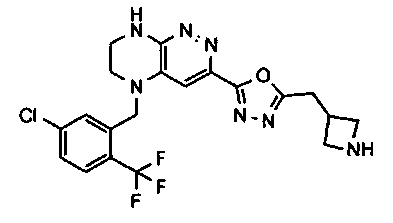
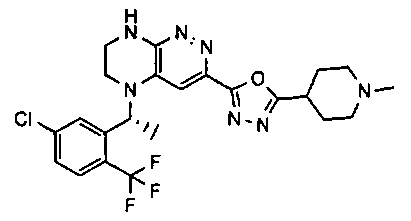
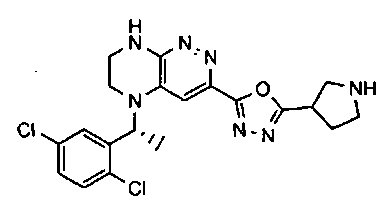
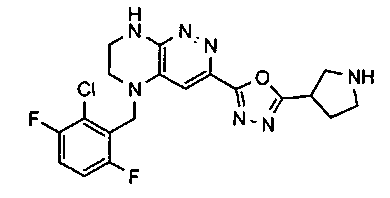
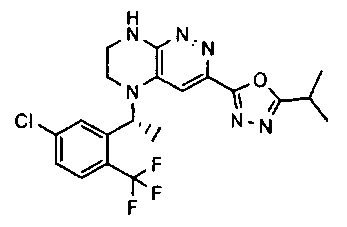
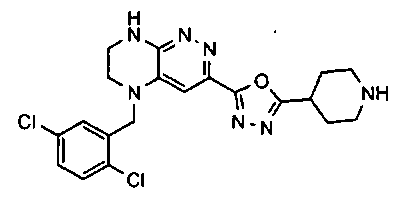
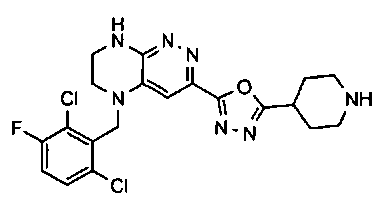
20. Примеры синтеза 5-замещенных производных 3-(5-замещенного-5,6,7,8-тетрагидропиразино [2,3-с]пиридазин-3-ил)-1,2,4-триазола
5-замещенные производные 3-(5-замещенного-5,6,7,8-тетрагидропиразино [2,3-с]пиридазин-3-ил)-1,2,4-триазола синтезируются из соответствующих Boc-защищенных 5-замещенных производных 3-хлоро-5,6,7,8-тетрагидропиразино [2,3-с]пиридазина аналогично соединению 7, методика синтеза которого приведена выше. Примеры полученных соединений представлены в таблице.
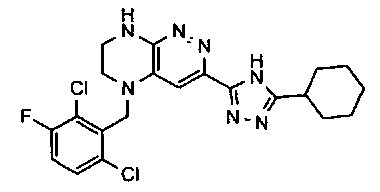
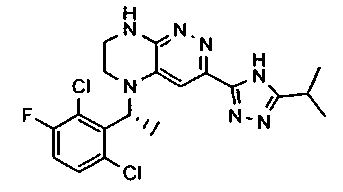
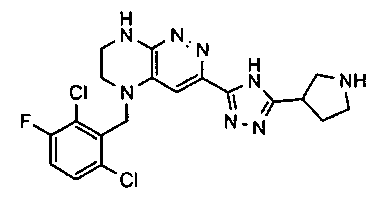
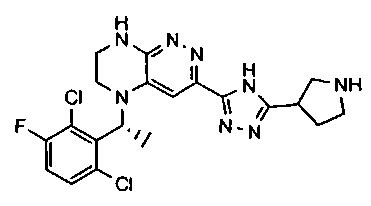
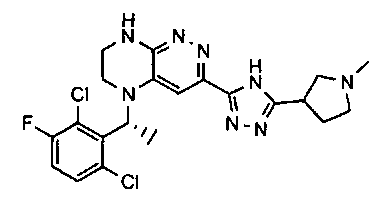
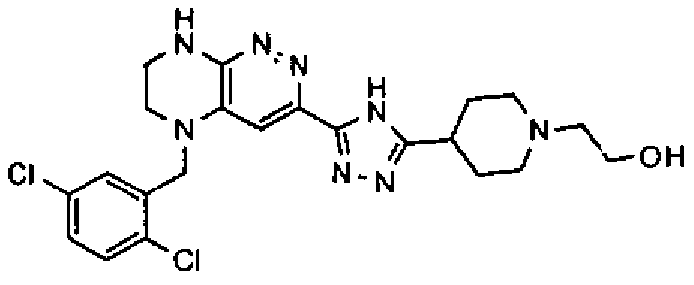
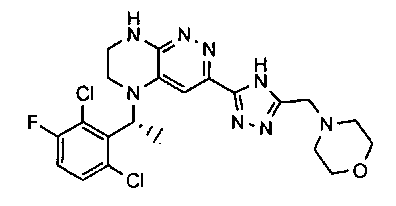
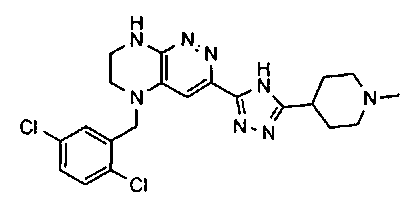
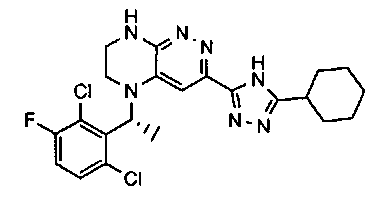
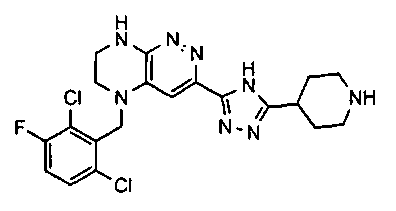
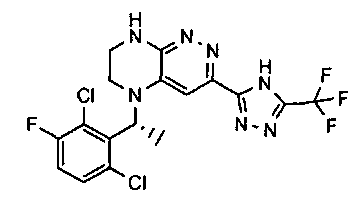
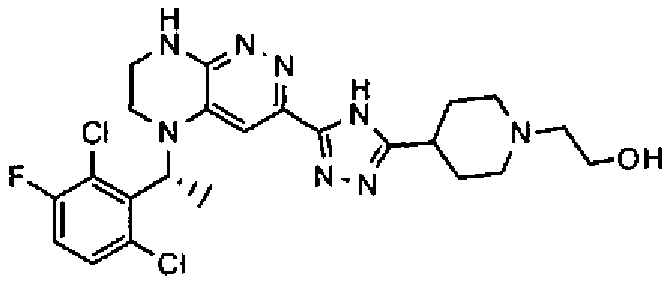
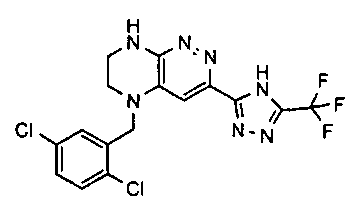
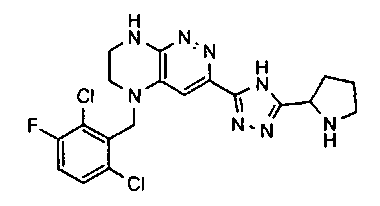
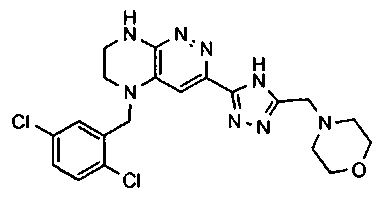
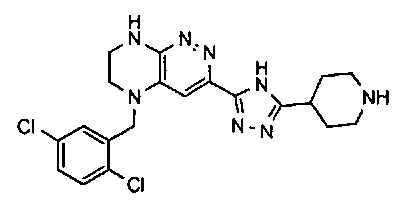
21. Примеры синтеза замещенных производных 5-замещенного-N-фенил-5,6,7,8-тетрагидропиразино [2,3-с]пиридазино-3-карбоксамида
Замещенные производные 5-замещенного-N-фенил-5,6,7,8-тетрагидропиразино [2,3-с]пиридазино-3-карбоксамида синтезируются из соответствующих Boc-защищенных 5-замещенных производных 3-хлоро-5,6,7,8-тетрагидропиразино [2,3-с]пиридазина аналогично соединению 10, методика синтеза которого приведена выше. Примеры полученных соединений представлены в таблице.
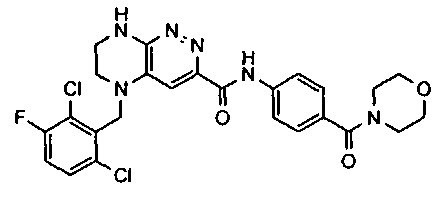
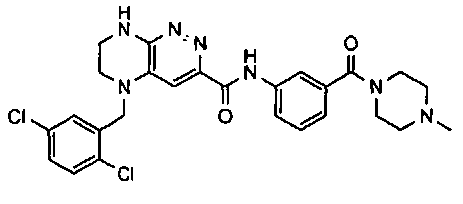
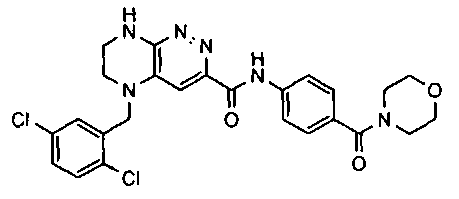
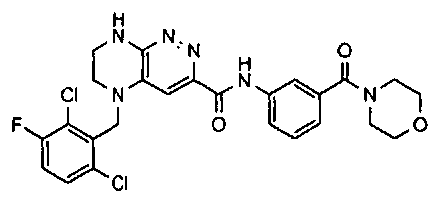
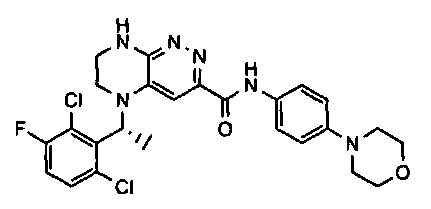
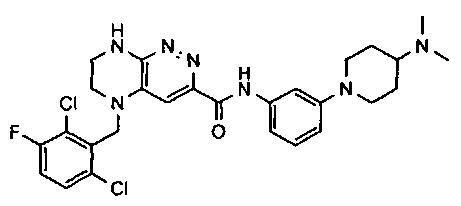
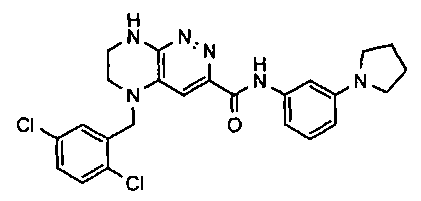
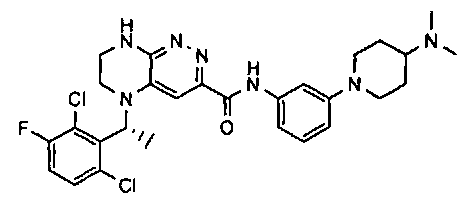
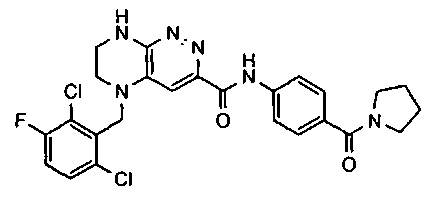
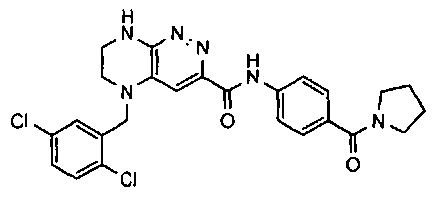
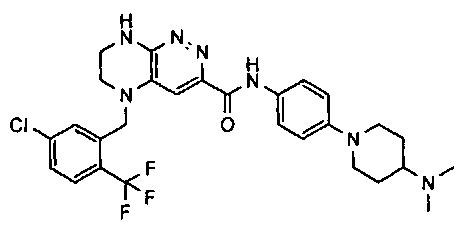
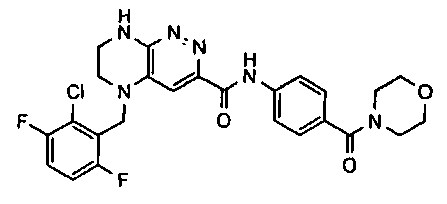
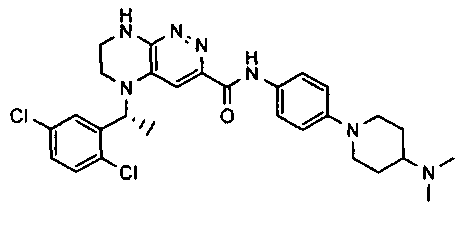
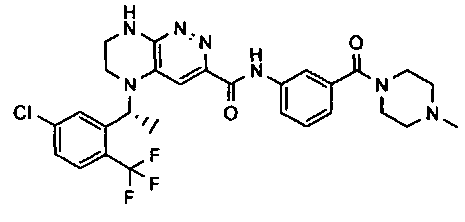
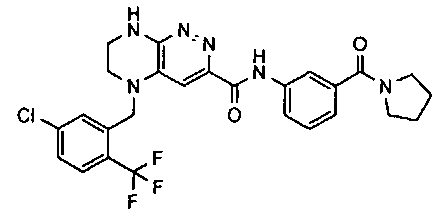
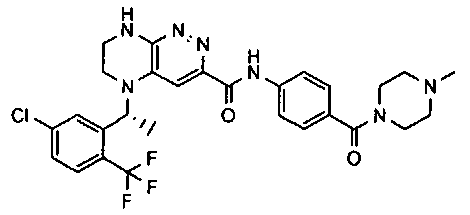
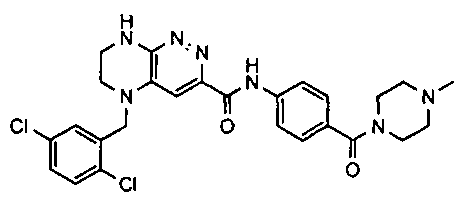
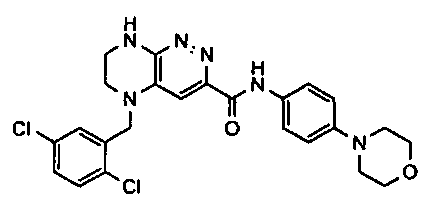
22. Примеры синтеза замещенных производных 5-замещенного-N-фенил-5,6,7,8-тетрагидропиразино [2,3-с] пиридазино-3-амида
Замещенные производные 5-замещенного-N-фенил-5,6,7,8-тетрагидропиразино [2,3-с]пиридазино-3-амида синтезируются из соответствующих Boc-защищенных 5-замещенных производных 3-хлоро-5,6,7,8-тетрагидропиразино [2,3-с]пиридазина аналогично соединению 11, методика синтеза которого приведена выше. Примеры полученных соединений представлены в таблице.
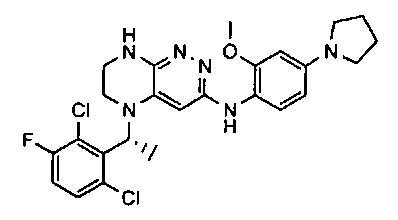
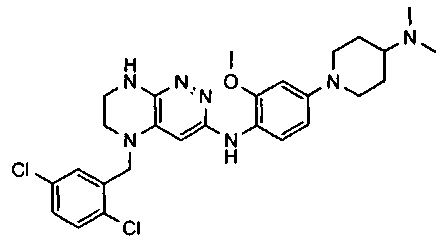
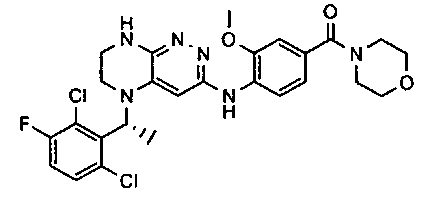
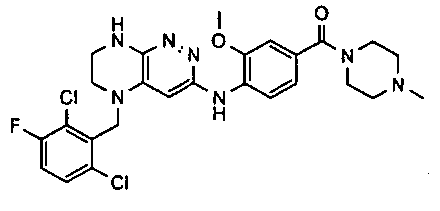
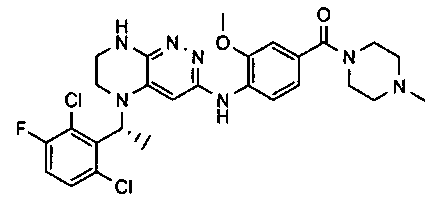
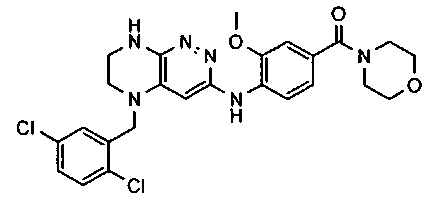
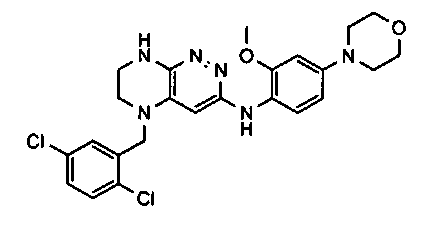
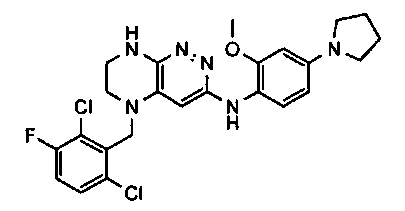
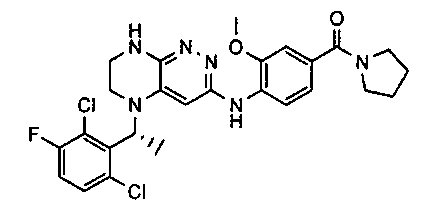
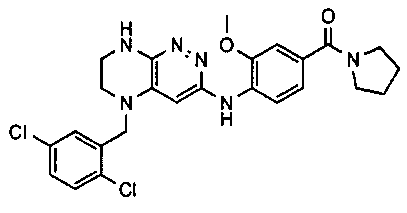
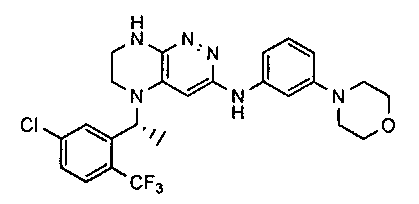
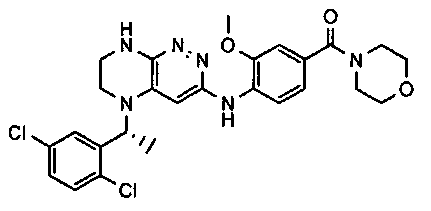
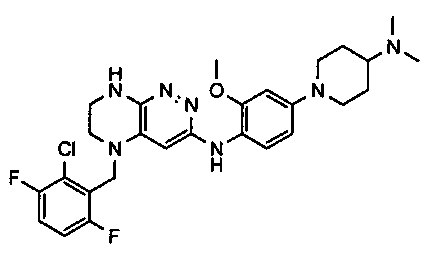
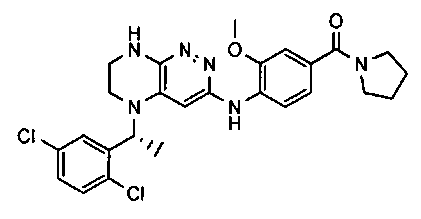
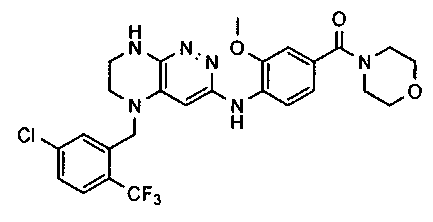
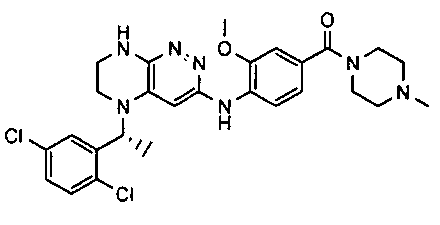
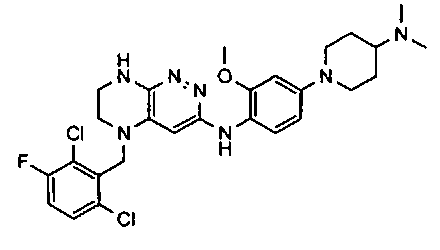
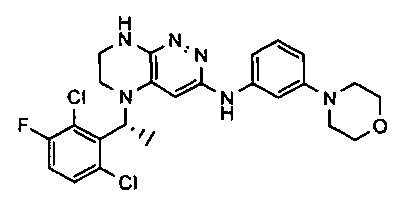
23. Примеры синтеза замещенных производных 5-замещенного-3-фенил-5,6,7,8-тетрагидропиразино [2,3-с]пиридазина
Замещенные производные 5-замещенного-3-фенил-5,6,7,8-тетрагидропиразино [2,3-с]пиридазина синтезируются из соответствующих Boc-защищенных 5-замещенных производных 3-хлоро-5,6,7,8-тетрагидропиразино [2,3-с]пиридазина аналогично соединению 12, методика синтеза которого приведена выше. Примеры полученных соединений представлены в таблице.
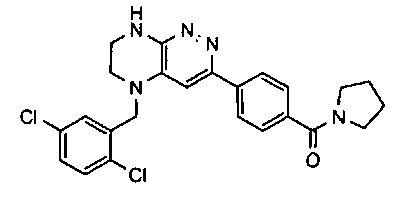
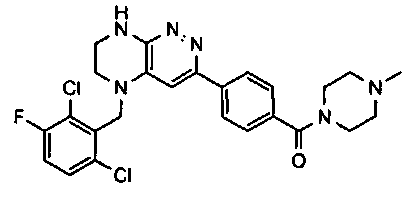
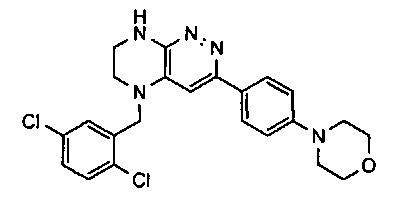
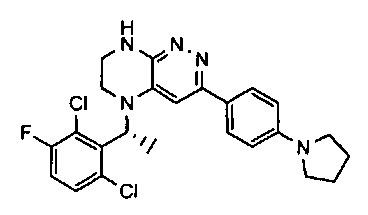
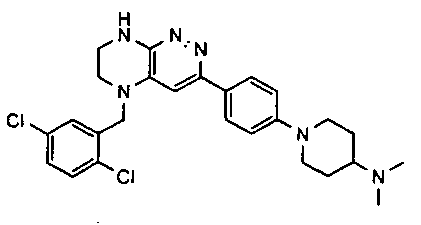
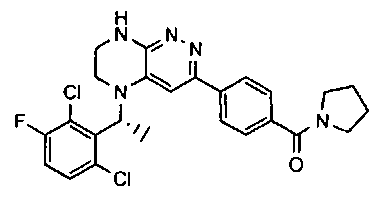
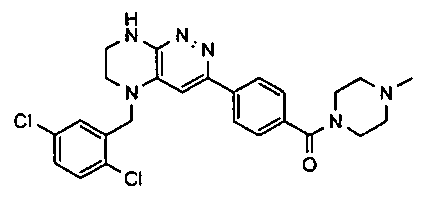
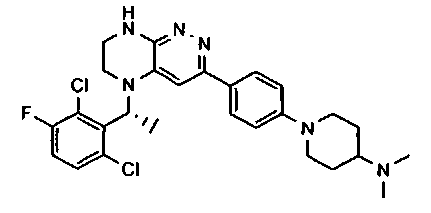
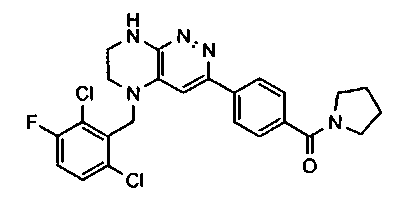
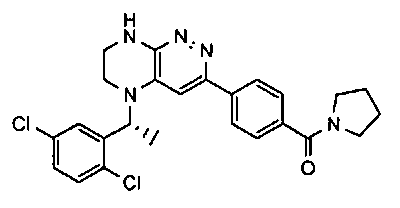
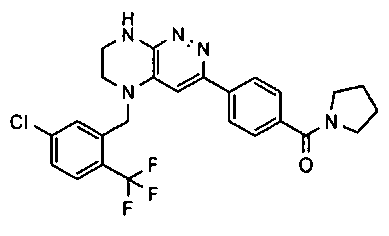
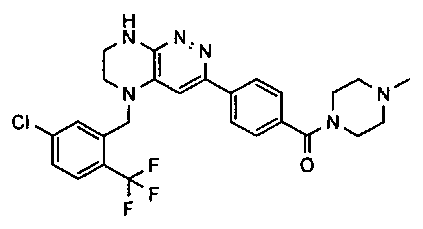
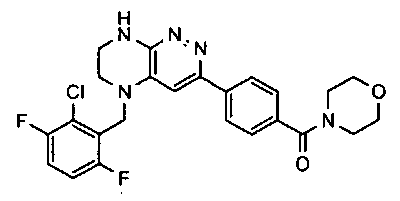
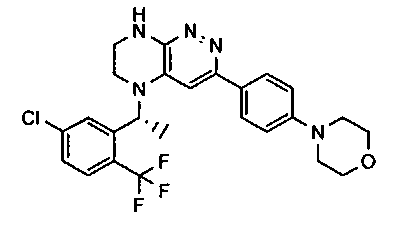
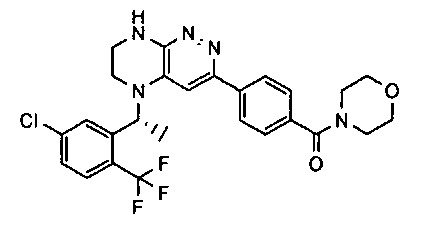
23.1. Другие примеры соединений по изобретению
Соединение 158 может быть синтезировано сочетанием Вос-защищенного 3-хлоро-5-(2-хлоро-3,6-дифторобензил]-5,6,7,8-тетрагидропиразино[2,3-с]пиридазина и 4-морфолинофенола согласно схеме VIIIa. Соединение 159 может быть синтезировано сочетанием Вос-защищенного 3-хлоро-5-(1-(2-хлоро-3,6-дифторофенил)этил)-5,6,7,8-тетрагидропиразино[2,3-с]пиридазина и (3-(меркаптометил)фенил)(пирролидин-1-ил)метанона согласно схеме VIIIb.
Для синтеза соединения 160 Вос-защищенный 3-хлоро-5-(2,5-дихлоробензил)-5,6,7,8-тетрагидропиразино[2,3-с]пиридазин превращается в Вос-защищенный 5-(2,5-дихлоробензил)-5,6,7,8-тетрагидропиразино[2,3-с]пиридазин-3-амин замещением атома хлора на азидогруппу с последующим ее расщеплением под действием трифенилфосфина. Сочетание аминопроизводного с 3-(пирролидин-1-карбонил)бензоил хлоридом проводят согласно схеме IX.
Соединение 161 получается из Вос-защищенной 5-(2,6-дихлоро-3-фторобензил)-5,6,7,8-тетрагидропиразино[2,3-с]пиридазин-3-карбоновой кислоты аналогично соединению 3.
Для синтеза соединения 162 Вос-защищенный 3-хлоро-5-(1-(2,6-дихлоро-3-фторфенил)этил)-5,6,7,8- тетрагидропиразино[2,3-с]пиридазин карбонилируется до метилового эфира соответствующей кислоты. После гидролиза кислоту восстанавливают до бензилового спирта под действием комплекса борандиметилсульфид. Нуклеофильное замещение мезилата под действием цианида калия и гидролиз полученного нитрила в щелочной среде приводит к арилуксусной кислоте, сочетание которой с 3-((4-метилпиперазин-1-ил)метил)анилином и последющее снятие защиты в кислой среде приводят к соединению 162.
Соединение 163 может быть получено из Вос-защищенного 3-хлоро-5-(2,5-дихлоробензил]-5,6,7,8-тетрагидропиразино[2,3-с)пиридазина и (3-аминоциклопентил) (пиперидин-1-ил)метанона аналогично соединению 11.
Соединение 164 получают палладий-катализируемым карбонилированием Вос-защищенного 3-хлоро-5-(1-(2,6-дихлорофенил)этил)-5,6,7,8-тетрагидропиразино[2,3-с]пиридазина в присутсвии 2-морфолиноэтанамина и последующим снятием Вос-защиты в кислой среде.
Палладий-катализируемое карбонилирование Вос-защищенного 3-хлоро-5-(5-фторо-2-(трифторметил)бензил]-тетрагидропиразино[2,3-с]пиридазина в присутствии третбутил 4-аминопиперидин-1-карбоксилата и восстановление продукта дибораном приводит к соединению 165.
Соединение 166 может быть получено из Вос-защищенной 5-(2,5-дихлоробензил)-5,6,7,8-тетрагидропиразино[2,3-с]пиридазин-3-карбоновой кислоты и пирролидина аналогично соединению 10.
Соединения 167 и 168 могут быть получены из соответствующих Вос-защищенных производных 3-хлоро-5,6,7,8-тетрагидропиразино[2,3-с]пиридазина по методике, аналогичной методике получения соединения 12.
Соединения 169-179 могут быть получены из соответствующих Вос-защищенных производных 3-хлоро-5,6,7,8-тетрагидропиразино[2,3-с]пиридазина по методике, аналогичной методике получения соединения 10.
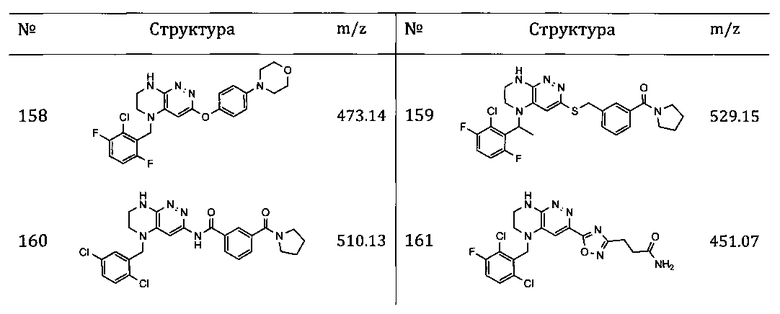
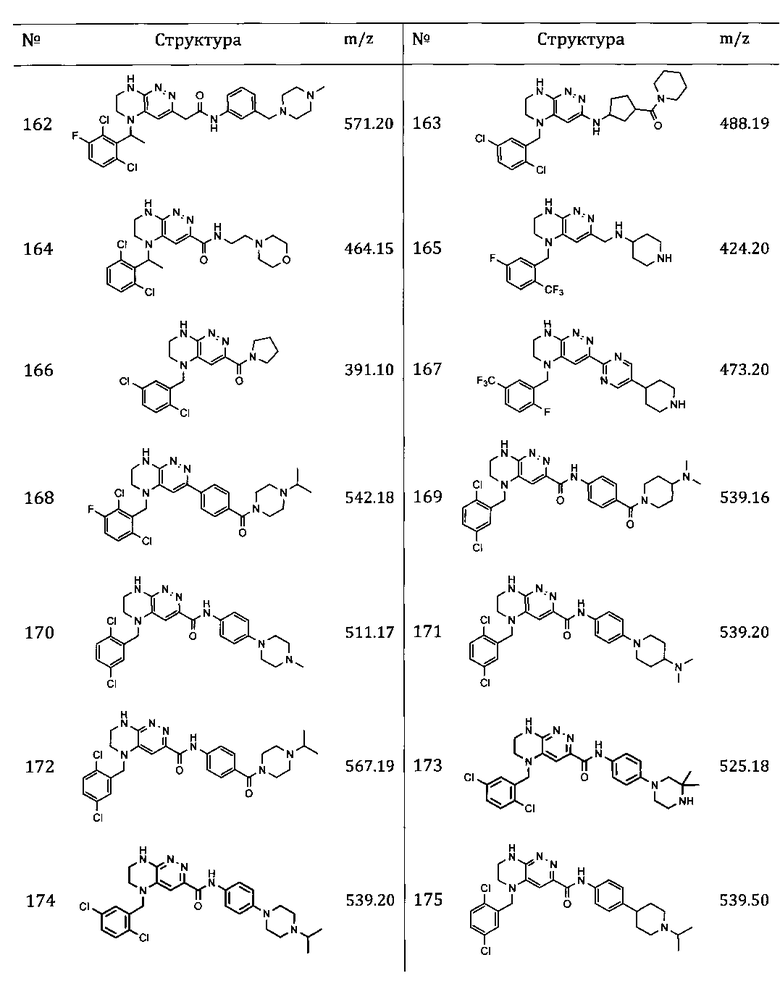
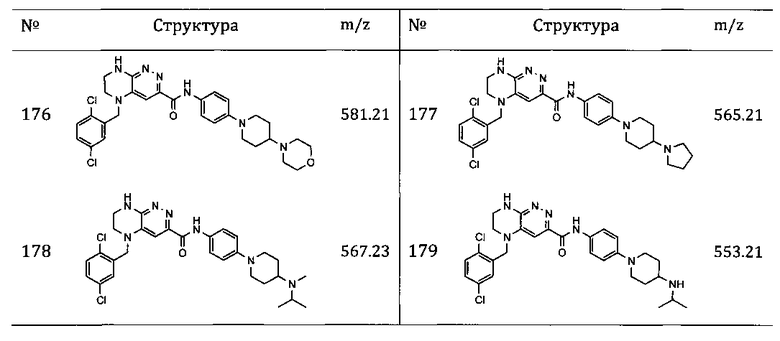
24. Характеристика биологической активности соединений
Биологическая активность соединений, являющихся предметом настоящего изобретения, была изучена различными методами. Например, было исследовано ингибирование киназной активности данными соединениями. Некоторые соединения показали существенную ингибиторную активность в наномолярной концентрации по отношению к киназе ALK, ALKL1196M, EGFR, ROS1, МЕТ. Также некоторые соединения показали существенную антипролиферативную активность на клетках Karpas-299, SU-DHL-1, NCI-H2228, NCI-H3122 в концентрациях 1-1000 нМ.
Иллюстративные примеры соединений, обладающих ингибиторной и антипролиферативной активностью, представлены ниже.
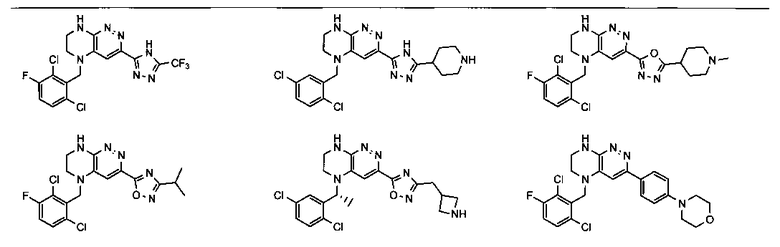
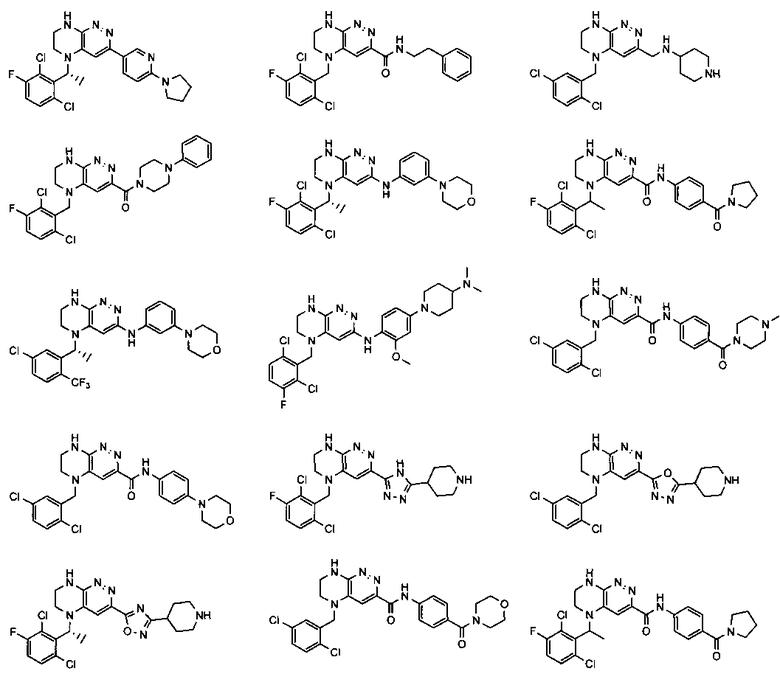
Далее изобретение будет проиллюстрировано следующими неограничивающими примерами.
24.1. Ингибирование киназ
Для соединений, являющихся предметом данного изобретения, была изучена способность ингибировать киназы, представляющие интерес для терапии онкологических, хронических воспалительных и прочих заболеваний. Список киназ, ингибирование которых изучалось в соответствии с описанной методикой, включал, (но принципиально не ограничен) киназами ALK, JAK2, BRAF, MET, TIE-2, FLT3, ABL, LCK, LYN, SRC, FYN, SYK, ZAP70, ITK, ТЕС, ВТК, EGFR, ERB2, PDGFRa, PDGFRb, KFR, IGF-1R, FLT1, ТЕК, АКТ, ROS1, EPHA1, а также их мутантными формами, в том числе придающими резистентность к существующим методам терапии онкологических заболеваний, например, мутантных форм ALK L1196M, C1156Y, F1174L, G1269A (Choi, Y. L., М. Soda,N Engl J Med, 2010, 363, 1734-9; Sasaki, Т., К. Okuda, Cancer Res, 2010, 70,10038-43; Doebele, R. С, A. B. Pilling, Clin Cancer Res, 2012).
Киназы, в виде киназного домена или полноразмерного белка, соединенного с глутатион-S-трансферазой (GST) или поли-гистидиновым фрагментов экспрессировались в зараженных бакуловирусом клетках насекомых (например, Sf21) или в клетках E.Coli. После выделения из клеток белки очищались до практически полной гомогенности с помощью афинной хроматографии по известным методикам. В некоторых случаях киназы ко-экспрессировались или смешивались с очищенными или частично очищенными регуляторными полипептидами до измерения активности.
Активность и ингибирование киназ определялось в соответствии с известными протоколами. Мерой ферментативной активности служила скорость переноса меченого 33PO4 с АТФ на синтетические субстраты поли(Clu, Tyr) 4:1 или поли(Arg, Ser) 3:1, присоединенные к биоактивной поверхности микротитрационной подложки. По окончании периода инкубации (120 мин) подложку промывали 0,5% фосфорной кислотой, добавляли жидкий сцинтиллянт, и на основании подсчета количества сцинтилляций на жидкостном сцинтилляционном детекторе определяли количество перенесенного фосфата. IC50 соответствовало концентрации вещества, уменьшающей на 50% количество 33Р, перенесенного на субстрат, связанный с подложкой.
Для определения ингибирования киназ могут использоваться и другие методики, основанные на измерении степени переноса фосфата на пептид или полипептид, содержащий тирозин, серин или треонин и присутствующий в растворенном или иммобилизованном виде.
Соединения, описанные в данном изобретении, имеют наномолярные значения IC50 в отношении различных киназ, в том числе ALK, MET, EGFR и ROS1. Также соединения, описанные в данном изобретении, являются селективными и в концентрации до 1000 нМ значимо не ингибируют такие киназы, как ABL, АКТ2, AURA, AURC, AXL, CDK2, СТК, FAK, IGF1R, IR, IRR, ITK, mTOR, MUSK, РКА, РКС9, RON, SYK, SRC, TYR03, ZAP70.
Ниже приведен список соединений, ингибирующих ALK киназу со значением IC50 менее 100 нМ
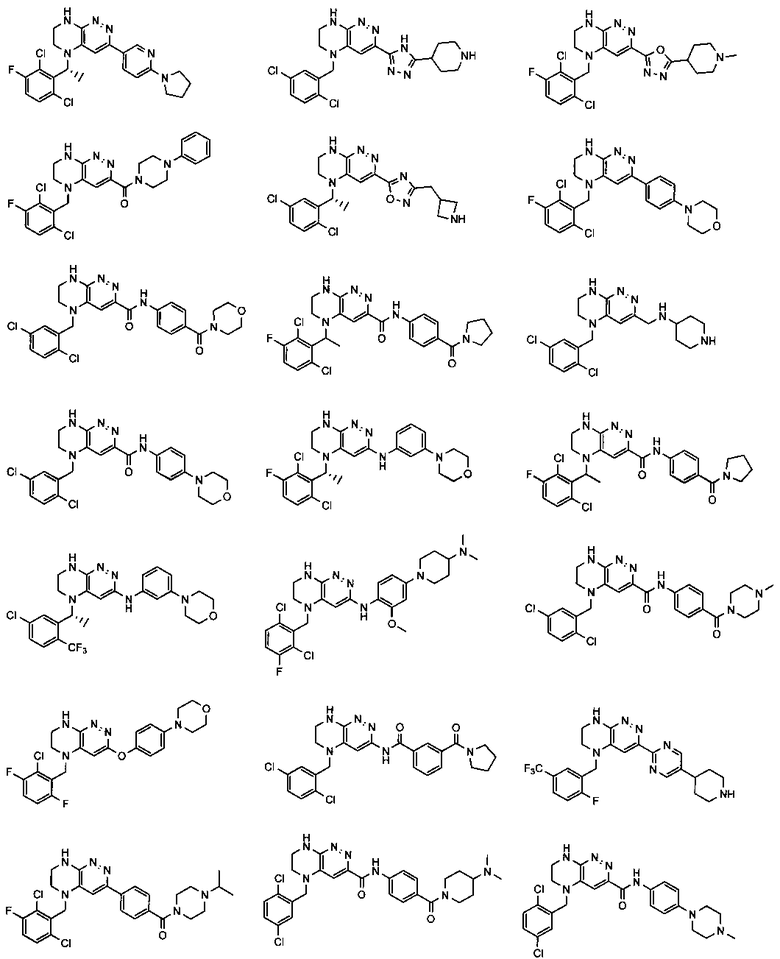
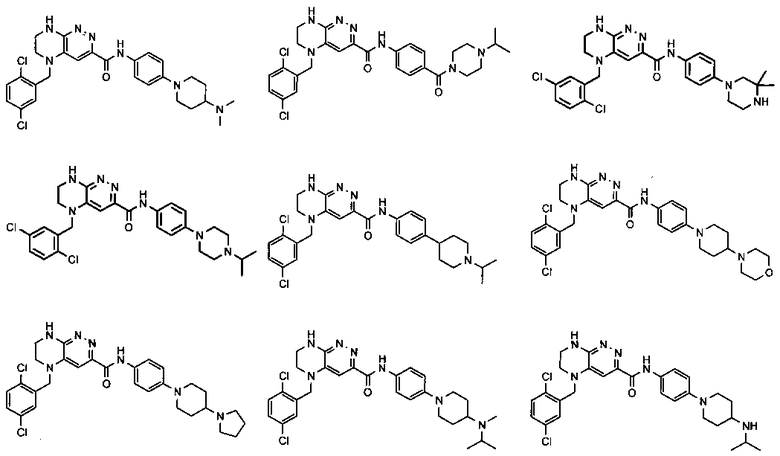
Список соединений, ингибирующих мутантную форму киназы ALKL1196M со значения IC50 менее 100 нМ
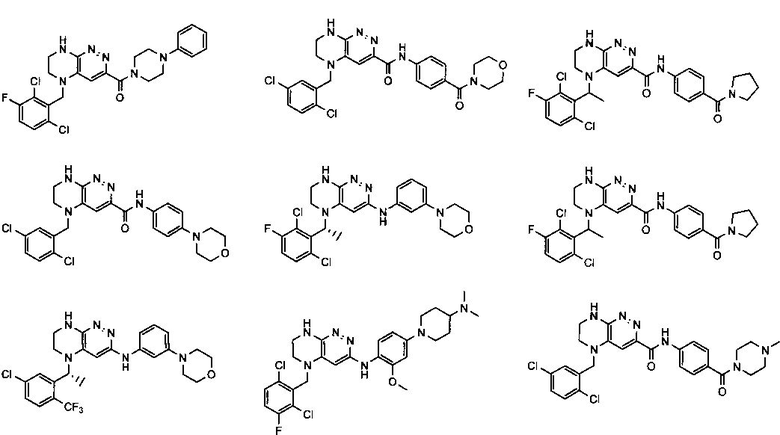
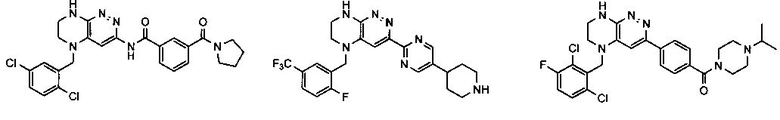
Список соединений, ингибирующих киназу ROS1 со значения IC50 менее 1 мкМ
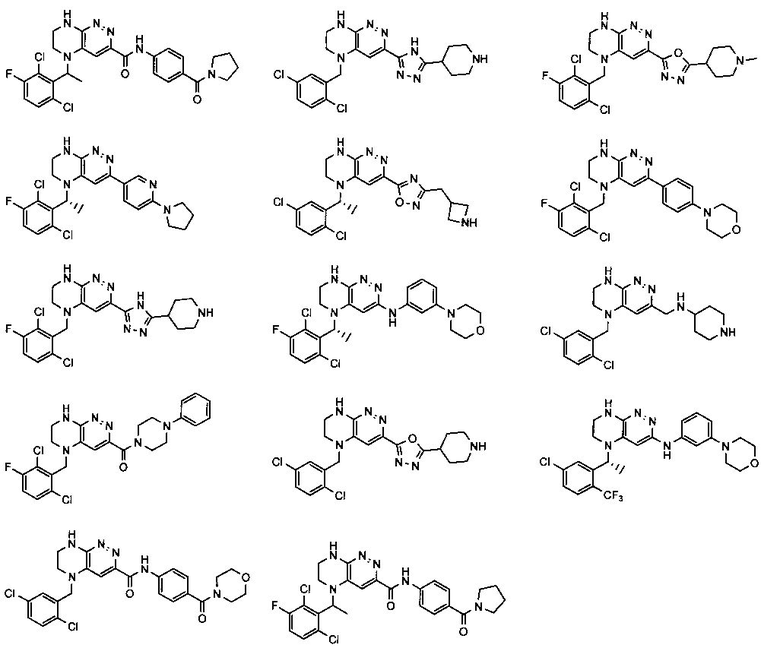
Список соединений, ингибирующих мутантную форму киназы EGFR (d746-750) со значения IC50 менее 10 мкМ
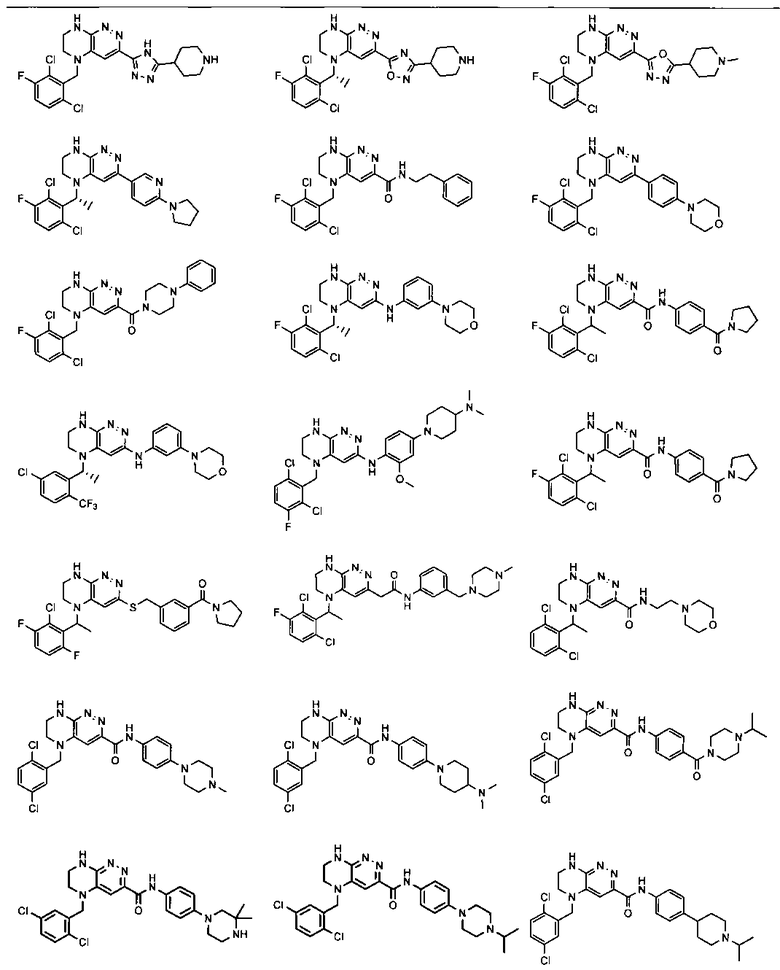
Список соединений, ингибирующих киназу МЕТ со значения IC50 менее 10 мкМ
24.2. Эксперименты с клеточными культурами
Некоторые соединения, являющиеся предметом данного изобретения, проявляют цитотоксичность или ингибируют рост опухолевых и других раковых клеточных линий и, таким образом, могут быть применены для лечения рака и других пролиферативных заболеваний.
Клеточные методы определения противоопухолевой активности хорошо известны и могут быть использованы для сравнительной характеристики соединений, описанных в данном изобретении. В целом, эксперименты по определению пролиферации клеток и количества жизнеспособных клеток дают детектируемый сигнал, пропорциональный количеству метаболически активных клеток. Противоопухолевая активность соединений может быть определена с использованием любой характеристики, отражающей уменьшение метаболической активности клеток после воздействия соединения. Традиционно используются методы, в которых в качестве меры жизнеспособности клетки выступают целостность мембраны (например, анализ элиминирования трипанового голубого) и синтез ДНК (например, определение включения BrdU или 3Н-тимидина).
В некоторых методах по определению пролиферации клеток используются реагенты, которые превращаются в детектируемые соединения в ходе пролиферации клеток. Предпочтительными реагентами для такого определения являются соли тетразолия, включающие, например, МТТ (3-(4,5-диметилтиазол-2-ил)-2,5-дифенилтетразолий бромид), MTS (3-(4,5-диметилтиазол-2-ил)-5-(3-карбоксиметоксифенил)-2-(4-сульфофенил)-2Н-тетразолий), ХТТ (2,3-бис(2-метокси-4-нитро-5-сульфофенил)-2Н-тетразолий-5-карбоксанилид), INT (2-(4-йодофенил)-3-(4-нитрофенил)-5-фенил тетразолий), NBT (2Н-тетразолий, 2,2′-(3,3′-диметокси[1,1′-бифенил]-4,4′-диил1)бис[3-(4-нитрофенил)-5-фенил, дихлорид) (Bernas, Т. et al., Biochim Biophys Acta, 1999, 1451, 73-81). Для определения пролиферации клеток проводят измерение количества продукта ферментативного превращения солей тетразолия в синие производные формазана, которые легко детектируются спектроскопическими методами.
В целом, предпочтительные методы определения пролиферации клеток включают инкубацию клеток в выбранной для роста среде в присутствии тестируемого вещества или без него. Условия роста для различных прокариотических и эукариотических клеток подробно описаны (Ausubel et al. Current Protocols in Molecular Biology. Wiley and Sons. 1999; Bonifacio et al. Current Protocols in Cell Biology. Wiley and Sons. 1999). Для определения пролиферации клеток после инкубации к ним добавляется соль тетразолия и затем определяется количество образовавшегося формазана. Количество образовавшихся производных формазана определяется по оптической плотности обработанных клеток.
Для определения антипролиферативной активности соединений могут использоваться раковые клеточные линии, например COLO 205, DLD-1, НСТ-15, НТ29 (рак толстой кишки); HEP G2 (гепатома); К-562 (лейкемия); А549, NCI-H249, NCI-H2228, NCI-H3122 (рак легкого); Karpas-299, SU-DHL-1 (лимфома); MCF7, MDA-MB-231 (рак молочной железы); SAOS-2 (остеосаркома); OVCAR-3 (рак яичника); PANC-1 (рак поджелудочной железы); DU-145, РС-3 (рак простаты); ACHN, CAKI-1 (рак почек); MG-63 (саркома).
Хотя для определения антипролиферативной активности соединений предпочтительно используются клетки млекопитающих, для этой цели также могут быть использованы клетки низших эукариотов, например дрожжей. Предпочтительно использование клеточных линий человека, крыс, мышей, кроликов, низших обезьян, хомяков и морских свинок, поскольку клеточные линии из этих организмов наиболее хорошо изучены и полно охарактеризованы.
Ниже приведен пример определения активности соединения на клетках. В данном эксперименте использовалась клеточная линия Ba/F3 (мышиные про-В-клетки), стабильно трансфецированная вектором pClneo™ (Promega Corp., Madison WI), кодирующим химерный белок NPM-ALK, и прошедшие селекцию по сопротивляемости к G418. Для выживания клеток контрольной линии, в которых трансфекция не проводилась, требовался интерлейкин IL-3. В то же время Ba/F3 клетки, экспрессирующие NPM-ALK (Ba/F3-NPM-ALK), были жизнеспособны в отсутствие IL-3, поскольку киназная активность NPM-ALK приводила к активации сигнальных путей, отвечающих за пролиферацию. Предполагаемое ингибирование NPM-ALK киназы таким образом блокирует сигналы клеточного роста, что проявляется в антипролиферативной активности соединений. Антипролиферативная активность, тем не менее, может быть преодолена добавлением избыточного количества IL-3, вызывающего клеточный рост по независимому от ALK механизму. Пример аналогичных экспериментов с использованием FLT3 киназы изложен в (Weisberg, Е. et. al., Cancer Cell, 2002,1,433-43).
Ингибирующая способность соединений определялась следующим образом: клетки Ba/F3-NPM-ALK переносились в дубликатах на 96-луночную плашку (15000 на одну лунку). Исследуемые вещества растворялись в ДМСО, затем раствор разбавлялся ДМСО и питательной средой до необходимой концентрации так, чтобы конечная концентрация ДМСО не превышала 1% по объему, и переносился в лунку с клетками. Финальная концентрация веществ составляла от 0,5 нМ до 10 µМ. В качестве контроля использовался ДМСО в том же количестве, что и при добавлении растворов веществ. После инкубирования клеток с соединениями в течение 3 дней определялось количество жизнеспособных клеток. Для этого к ним добавлялся раствор МТТ, проводилась инкубация и определялась оптическая плотность при 540 и 620 нм (количество жизнеспособных клеток пропорционально отношению оптических плотностей при этих длинах волн). IC50 определялось из подобранных с помощью компьютерной обработки кривых, наиболее адекватно описывающих экспериментальные данные.
Для определения антипролиферативной активности могут быть использованы клеточные линии, в которых активность рекомбинантной киназы необходима для выживания, и ингибирование киназной активности приводит к гибели клеток, которую можно фиксировать по изменению концентрации АТФ. Антипролиферативная активность соединений определяется по следующей методике: клетки, экспрессирующие рецепторную тирозинкиназу, например ALK, культурируются в питательной среде RPMI-1640, содержащей 10% эмбриональной телячьей сыворотки и антибиотики. Пересев клеток осуществляется в логарифмической фазе роста. Клетки переносятся в дубликатах в 384-луночную культуральную плашку (5000 клеток на лунку) в 50 µл питательной среды. В каждую ячейку добавляется 50 нл раствора исследуемого соединения, и клетки инкубируются в течение 48 часов при 37°C в увлажненной атмосфере, содержащей 5% СО2.Количество выживших клеток определяется добавлением 15 µл реагента CellTiter-Glo и измерением люменисценции. IC50 определяется из подобранных с помощью компьютерной обработки кривых, наиболее адекватно описывающих экспериментальные данные.
Кроме того, антипролиферативная активность соединений, являющихся предметом данного изобретения, может быть изучена на клеточных линиях KARPAS-299 анапластической крупноклеточной лимфомы по следующей методике. Клетки KARPAS-299, культивировавшихся в питательной среде RPMI-1640, переносятся в дубликатах в 96-луночную культуральную плашку (10000 клеток на лунку) и к ним добавляется раствор исследуемых веществ в питательной среде в различных концентрациях (конечный объем в одной лунке - 100 µл). Твердые вещества растворялись в ДМСО, затем раствор разбавлялся ДМСО до необходимой концентрации, смешивался с равным по объему количеством питательной среды и переносился в лунку с клетками. Финальная концентрация веществ составляла от 0,5 нМ до 10 µМ. В качестве контроля использовался ДМСО (в том же количестве, что и при добавлении растворов веществ). После инкубирования клеток с соединениями в течение 72 часов определялось количество жизнеспособных клеток. Для этого старая среда удалялась, в каждую лунку добавлялось 100 µл свежей среды и 40 µл раствора MTS, содержащего 5 мг/мл PBS. Плашку инкубировали в течение 2 ч при 37°C, затем добавляли 100 µл ДМСО в каждую лунку и перемешивали 1 мин. Затем определялась оптическая плотность при 490 нм и рассчитывался процент ингибирования пролиферации по сравнению с контролем (не содержащим исследуемых веществ).
Иллюстративные примеры соединений, обладающих антипролиферативной активностью, представлены ниже:
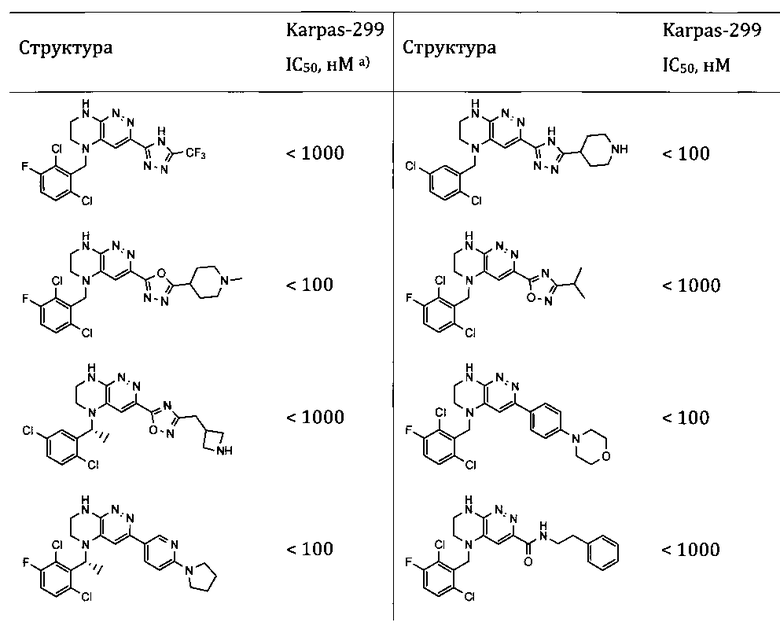
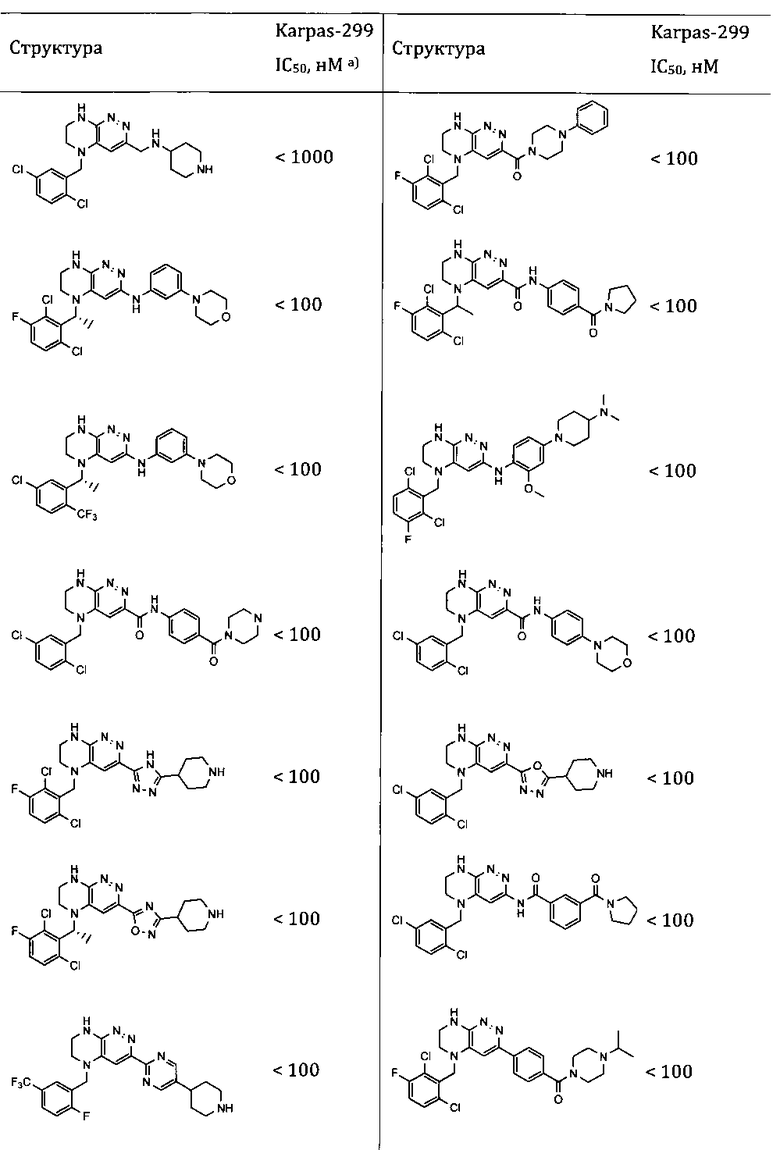
а) - концентрация соединения, при которой количество живых клеток в условиях эксперимента снижается в 2 раза по сравнению с отсутствием соединения.
24.3. Эксперименты с животными
Соединения, показавшие антипролиферативную активность в клеточных экспериментах, затем исследуются in vivo на организмах млекопитающих. Чаще всего эксперименты in vivo проводятся на грызунах, таких как мыши и крысы. Как правило, опухоль трансплантируется в мышь со сниженным иммунитетом для того, чтобы снизить вероятность отторжения. К таким мышам относятся, например, бестимусные голые мыши и SCID-мыши (мыши с тяжелым комбинированным иммунодефицитом).
Обычно опухоль имплантируется подкожно, и исследуемому животному в некоторой дозе и по некоторому режиму вводится лекарственный кандидат. Эффективность исследуемого соединения определяется по сокращению размера опухоли, измерение которой проводится с определенной периодичностью. Некоторые опухоли имплантируются в другие точки организма (например, внутрибрюшинно), и критерием эффективности служит среднее время выживания организма. Как правило, в ходе исследований на животных сравнивают разные модели опухолей, способы введения, дозировки и режимы приема лекарственных кандидатов.
Эффективность в животной модели немелкоклеточного рака легких
Для исследований использовались самцы бестимусных мышей. Клетки NCI-H3122 в количестве 1×107 вводились в составе 0,2 мл раствора Матригеля (BD Pharmingen) в левую ногу мыши под кетамин-ксилазиновой анестезией. Через неделю после введения клеток мыши были разделены на терапевтическую и контрольную группу и рандомизированы по размеру опухоли. Объем опухоли рассчитывали по формуле V=0,5×W2×L. Животные контрольной группы ежедневно получали 0,3 мл 0,5% раствора метилцеллюлозы, животные терапевтической группы - 0,3 мл 0,5% суспензии метилцеллюлозы, содержащей препарат в исследуемой дозе. Растворы вводились с помощью желудочного зонда. Лечение продолжали в течение 20 дней. Для определения эффективности ингибирования роста опухоли по окончании лечения рассчитывалось отношение среднего объема для терапавтической/контрольной групп (% Т/С). Для полученных данных был проведен анализ статистической достоверности с помощью теста Даннета. Эффективности исследованных соединений в дозе 30 мг/кг приведены ниже.
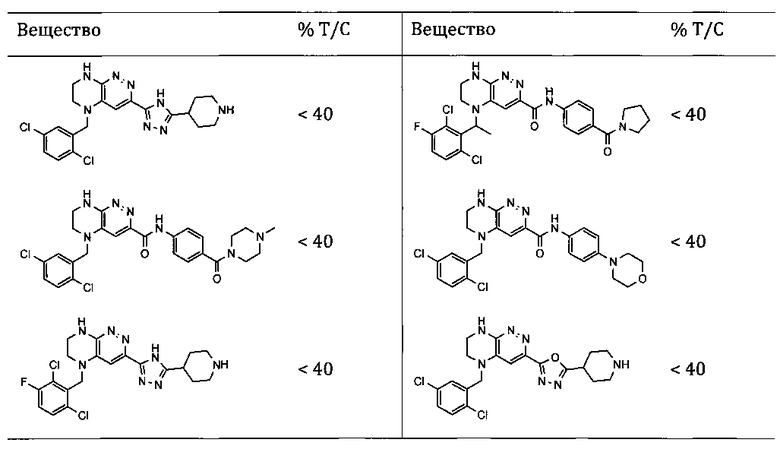
Эффективность в животной модели анапластической крупноклеточной лимфомы
Для исследований использовались SCID-мыши с опухолями, развившимися вследствие введения клеток Karpas-299. При достижении размера опухоли 220 мм3 животные были разделены на терапевтическую и контрольную группу и рандомизированы по размеру опухоли. Животные контрольной группы ежедневно получали 0,3 мл 0,5% раствора метилцеллюлозы, животные терапевтической группы - 0,3 мл 0,5% суспензии метилцеллюлозы, содержащей препарат в исследуемой дозе. Растворы вводились с помощью желудочного зонда. Лечение продолжали в течение 23 дней. Для определения эффективности ингибирования роста опухоли по окончании лечения рассчитывалось отношение среднего объема для терапавтической/контрольной групп (% Т/С). Для полученных данных был проведен анализ статистической достоверности с помощью теста Даннета. Эффективность исследованных соединений в дозе 30 мг/кг приведена ниже.
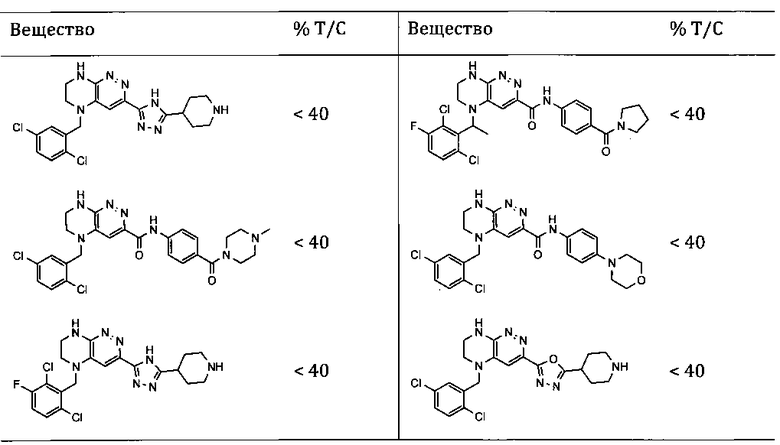
25. Примеры фармацевтической композиции
Вещества, описанные в данном изобретении, могут быть использованы для профилактики и лечения болезней человека, например, в виде следующих составов (под «Веществом» понимается активный ингредиент):
Данные составы могут быть приготовлены в соответствии со стандартными фармацевтическими методиками. Таблетки (1)-(3) могут быть покрыты кишечнорастворимой оболочкой с использованием, например, фталата ацетата целлюлозы. Аэрозольный состав (8) могут быть использован в сочетании со стандартными диспенсерами. В качестве суспендирующего агента вместо триолеата сорбитана и соевого лецитина может быть использован моноолеат сорбитана, полуолеат сорбитана, полисорбат 80, олеат полиглицерина или олеиновая кислота.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЙ СПОСОБ СИНТЕЗА ПРОИЗВОДНЫХ ТЕТРАГИДРОПИРАЗИНО[2,3-c]ПИРИДАЗИНА | 2016 |
|
RU2631430C1 |
| ЗАМЕЩЕННЫЕ ПИРИДИНЫ И ПИРИДАЗИНЫ, ОБЛАДАЮЩИЕ СПОСОБНОСТЬЮ ИНГИБИРОВАТЬ АНГИОГЕНЕЗ | 2000 |
|
RU2260008C2 |
| ПРОИЗВОДНЫЕ ИМИДАЗО [1,2-b]ПИРИДАЗИНА И ПИРАЗОЛО[1,5-a]ПИРИМИДИНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРА ПРОТЕИНКИНАЗ | 2007 |
|
RU2487875C2 |
| МАКРОЦИКЛЫ ПИРИДАЗИНОНА В КАЧЕСТВЕ ИНГИБИТОРОВ IRAK-КИНАЗЫ И ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2708066C2 |
| ЗАМЕЩЕННЫЕ ПИРИДАЗИН-КАРБОКСАМИДНЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ СОЕДИНЕНИЙ, ИНГИБИРУЮЩИХ КИНАЗЫ | 2009 |
|
RU2526618C2 |
| ИНГИБИТОРЫ mTOR КИНАЗЫ ДЛЯ ОНКОЛОГИЧЕСКИХ ПОКАЗАНИЙ И ЗАБОЛЕВАНИЙ, СВЯЗАННЫХ С mTOR/PI3K/AKT ПУТЕМ МЕТАБОЛИЗМА | 2009 |
|
RU2546658C2 |
| КОНДЕНСИРОВАННОЕ АЗА-ГЕТЕРОЦИКЛИЧЕСКОЕ АМИДНОЕ СОЕДИНЕНИЕ И ЕГО ПРИМЕНЕНИЕ | 2021 |
|
RU2811975C1 |
| СОЕДИНЕНИЯ И СПОСОБЫ МОДУЛИРОВАНИЯ КИНАЗЫ И ПОКАЗАНИЯ К ИХ ПРИМЕНЕНИЮ | 2013 |
|
RU2666146C2 |
| БИ- И ПОЛИЦИКЛИЧЕСКИЕ ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ ИЗОХИНОЛИНА И ИЗОХИНОЛИНОНА, ПОЛЕЗНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ RHO-КИНАЗЫ | 2009 |
|
RU2532481C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ 3-ФЕНИЛПРОПИОНОВОЙ КИСЛОТЫ ДЛЯ ЛЕЧЕНИЯ ДИАБЕТА | 2005 |
|
RU2360906C2 |
Изобретение относится к новым химическим соединениям общей формулы I, в которых LA, LB, LC, цикл A, цикл B, RA, RB, RC, RD, RE и RF имеют значения, указанные в формуле изобретения. Соединения формулы (I) являются ингибиторами протеинкиназ. Изобретение также относится к фармацевтическим композициям, содержащим указанные соединения, а также к применению вышеуказанных соединений для лечения и/или предотвращения заболеваний, связанных с аберрантной активностью протеинкиназ, в частности раковых заболеваний. 3 н. и 7 з.п. ф-лы, 14 табл., 25 пр.
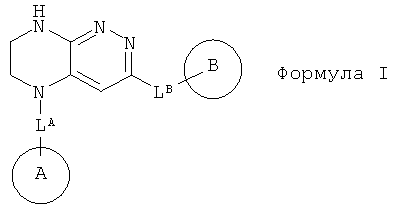
1. Соединение общей формулы I
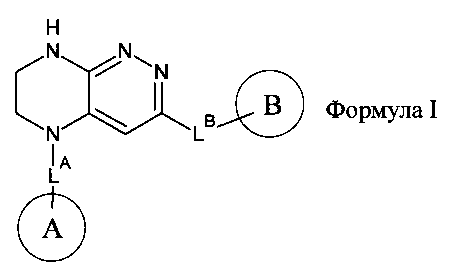
или его энантиомер или фармацевтически приемлемая соль, сольват или гидрат, где:
LA представляет собой СН2 или СН(СН3);
LB представляет собой ковалентную химическую связь, ОС0-3-алкил, SC0-3-алкил, NHC(O)С0-3-алкил, С(О)NHC0-3-алкил, С(О)С0-3-алкил, NHC0-3-алкил, CH2C(O)NH или CH2NH;
цикл А представляет собой фенил, опционально замещенный 1-3 группами RA;
RA выбирается независимо и представляет собой галоген, частично или полностью галогенированные C1-5-алкил, OC1-3-алкил;
цикл В представляет собой фенил; С5-6 циклоалкил; 5-6-членный насыщенный гетероцикл, содержащий 1-2 атома N и 0-1 атом О; 5-членный гетероарильный цикл, содержащий 1-3 атома N; 5-членный гетероарильный цикл, содержащий 2 атома N и 1 атом О; 6-членный гетероарильный цикл, содержащий 1-2 атома N; цикл В необязательно содержит 1-3 заместителя RB;
RB выбирается независимо и представляет собой LC-RC, LC-H, галоген, частично или полностью галогенированный C1-5-алкил;
LC представляет собой ковалентную химическую связь, C1-3-алкил, O(СН2)n, С(О), (СН2)mC(O)O(СН2)n или (CH2)mC(O)NH(CH2)n;
RC выбирается независимо и представляет собой фенил, C1-6-алкил, С3-7 циклоалкил, 4-6-членный гетероалициклил, содержащий 1-2 атома N и 0-1 атом О; RC необязательно содержит 1-3 заместителя RD;
RD выбирается независимо и представляет собой галоген, (СН2)mCH3, (СН2)mOH, NHRF, N(RF)2 или 4-6-членный гетероалицикл, содержащий 0-1 атом N и 0-1 атом О; RD необязательно содержит 1-3 заместителя C1-3-алкил;
RF выбирается независимо и представляет собой C1-3-алкил;
m и n независимо выбираются из 0, 1, 2, 3.
2. Соединение по п. 1, в котором:
LA представляет собой СН2 или СН(СН3);
LB представляет собой ковалентную химическую связь, ОС0-3-алкил, SC0-3-алкил, NНС(O)С0-3-алкил, С(O)NHC0-3-алкил, С(O)С0-3-алкил, NHC0-3-алкил, CH2C(O)NH или CH2NH;
цикл А представляет собой фенил, опционально замещенный 1-3 группами RA;
RA представляет собой галоген, частично или полностью галогенированные C1-3-алкил, OC1-3-алкил;
цикл В представляет собой фенил; C5-6 циклоалкил; 5-6-членный насыщенный гетероцикл, содержащий 1-2 атома N и 0-1 атом О; 5-членный гетероарильный цикл, содержащий 1-3 атома N; 5-членный гетероарильный цикл, содержащий 2 атома N и 1 атом О; 6-членный гетероарильный цикл, содержащий 1-2 атома N; цикл В необязательно содержит 1-3 заместителя RB;
RB выбирается независимо и представляет собой LC-RC, LC-H, галоген, частично или полностью галогенированный C1-3-алкил;
LC представляет собой ковалентную химическую связь, C1-3-алкил, С(О), (CH2)mC(O)NH(CH2)n или O(СН2)n;
RC выбирается независимо и представляет собой фенил, C1-6-алкил или 4-6-членный гетероалициклил, содержащий 1-2 атома N и 0-1 атом О; RC необязательно содержит 1-3 заместителя RD;
RD выбирается независимо и представляет собой (СН2)mCH3, (СН2]mOH, N(RF)2 или 4-6 членный гетероалицикл, содержащий 0-1 атом N и 0-1 атом 0; RD опционально содержит 1-3 заместителя С1-3-алкил; m и n независимо выбираются из 0, 1, 2, 3.
3. Соединение по п. 1, в котором:
LA представляет собой СН2 или СН(СН3);
LB представляет собой ковалентную химическую связь или C(O)NH, NH;
цикл А представляет собой фенил, опционально замещенный 1-3 группами RA;
RA представляет собой Cl, F, CF3 или ОСН3;
цикл В представляет собой фенил; 5-членный гетероарильный цикл, содержащий 1-3 атома N; 5-членный гетероарильный цикл, содержащий 2 атома N и 1 атом О, или 6-членный гетероарильный цикл, содержащий 1-2 атома N; цикл В необязательно содержит 1-3 заместителя RB;
RB выбирается независимо и представляет собой LC-RC или LC-H;
LC представляет собой ковалентную химическую связь, СН2, С(O), C(O)NH, CH2C(O)NH, C(O)NHCH2, C(O)NH(CH2)2 или OCH2;
RC выбирается независимо и представляет собой фенил, C1-3-алкил или 4-6-членный гетероалициклил, содержащий 1-2 атома N и 0-1 атом О; RC необязательно содержит 1-3 заместителей RD;
RD выбирается независимо и представляет собой СН3, ОН, N(RF)2 или 4-6-членный гетероалицикл, содержащий 0-1 атом N и 0-1 атом О; RD необязательно содержит 1-2 заместителя RF;
RF представляет собой СН3.
4. Соединение по п. 1, в котором LB представляет собой ковалентную химическую связь, NH или C(O)NH.
5. Соединение по п. 1, представляющее собой:
6. Применение соединения по любому из пп. 1-5 для получения фармацевтической композиции для лечения и/или предотвращения заболевания, связанного с аберрантной активностью протеинкиназ.
7. Применение по п. 6, в котором заболевание представляет собой рак легкого, кости, поджелудочной железы, кожи, шеи и головы, кожную или внутриглазную меланому, рак матки, яичника, прямой кишки, анального канала, рак желудка, почек, молочной железы, карциному фаллопиевых труб, слизистой оболочки и шейки матки, вагины, вульвы, ходжкинскую лимфому, рак пищевода, тонкого кишечника, эндокринной системы, щитовидной железы, паращитовидной железы, надпочечников, саркому мягких тканей, рак мочеиспускательного канала, пениса, простаты, хронический или острый миелолейкоз, лимфоцитарную лимфому, рак мочевого пузыря, почки или мочеточника, карциному почечного эпителия, карциному почечной лоханки, рабдомиосаркому, неопластические образования в центральной нервной системе, первичную лимфому ЦНС, опухоли спинного мозга, глиому мозгового ствола, аденому гипофиза или их комбинации.
8. Применение по п. 7, в котором заболевание представляет собой немелкоклеточный рак легкого, анапластическую крупноклеточную лимфому, диффузную В-клеточную лимфому, воспалительную миофибробластическую опухоль, нейробластому, рабдомиосаркому, анапластический рак щитовидной железы, мультиформную глиобластому, холангиокарциному, аденокарциному желудка, хроническую миеломоноцитарную лейкемию, саркому Юинга, воспалительный рак груди, рак почки, карциному папиллярного почечного эпителия или плоскоклеточную карциному.
9. Применение по п. 7, в котором заболевание представляет собой немелкоклеточный рак легкого или анапластическую крупноклеточную лимфому.
10. Фармацевтическая композиция для лечения и/или предотвращения заболевания, связанного с аберрантной активностью протеинкиназ, характеризующаяся тем, что содержит эффективное количество соединения по любому из пп. 1-5 и фармацевтически приемлемый носитель, растворитель и/или наполнитель.
| Изложница с суживающимся книзу сечением и с вертикально перемещающимся днищем | 1924 |
|
SU2012A1 |
| US 2942001 A, 21.06.1960, | |||
| ЦИКЛИЧЕСКИЕ БИОИЗОСТЕРЫ ПРОИЗВОДНЫХ ПУРИНОВОЙ СИСТЕМЫ И ИХ ПРИМЕНЕНИЕ В ТЕРАПИИ | 2003 |
|
RU2374248C9 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |

