Область техники
Изобретение относится к области фармакологии и медицины и касается способа синтеза производных тетрагидропиразино[2,3-с]пиридазина, которые могут быть использованы для лечения онкологических, хронических воспалительных и прочих заболеваний.
Уровень техники
Онкологические заболевания являются одной из основных причин заболеваемости и смертности во всем мире и смертности во всем мире - по данным Всемирной Организации Здравоохранения, в 2012 году произошло около 14 миллионов новых случаев заболевания и 8,2 миллиона случаев смерти, связанных с раком. Немелкоклеточный рак легкого (НМКРЛ) - остаточно распространенное (14% всех онкологических пациентов) и первое по смертности (28% всех онкологических смертей) среди онкологических заболеваний. Заболевание характеризуется поздней стадией диагностирования и высоким риском мета-стазирования в мозг, что осложняет хирургическое вмешательство и снижает эффективность химиотерапии. Дополнительным осложняющим фактором является высокая гетерогенность заболевания: порядка 15% случаев обусловлены аберрантной активностью (в следствие мутаций) ГТФазы KRAS, 10% - киназы EGFR, 5% - киназы ALK, при этом остальные идентифицированные группы заболеваний имеют еще меньший размер, а порядка 50% случаев не имеют однозначно установленной мишени заболевания.
Перспективным подходом для терапии онкологических заболеваний, вызванных нарушением активности протеинкиназ является применение низкомолекулярных химических соединений для ингибирования их активности. Примерами Перспективным подходом для терапии заболеваний, ассоциированных с нарушенной активностью протеинкиназ, является применение низкомолекулярных химических соединений для ингибирования их активности. Примерами таких ингибиторов, одобренных для применения в клинической практике, являются: иматиниб, нилотиниб, дазатитниб, лапатиниб, гефитиниб, эрлотиниб, кризотиниб и др.
Эффективными ингибиторами киназы ALK являются производные тетрагидропира-зино[2,3-с]пиридазина, которые характеризуются высокой активностью и селективностью по сравнению с другими препаратами. Способ синтеза производных тетрагидропиразино[2,3-с]пиридазина описан в международной публикации WO 2015047133. Общая схема синтеза ключевых интермедиатов конечных соединений выглядит следующим образом:
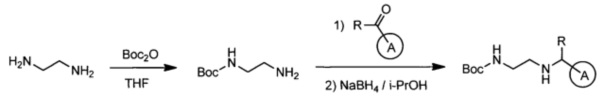
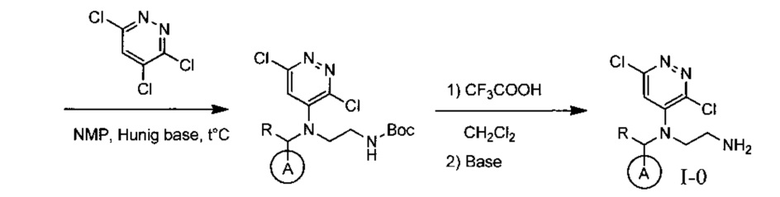
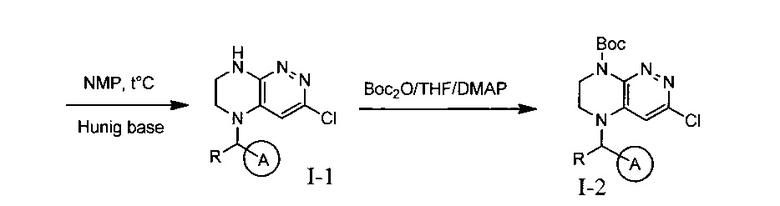
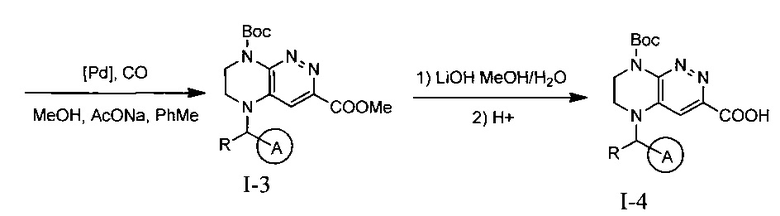
На данной схеме цикл А представляет фенил, опционально замещенный 1-3 группами RA, R представляет Н или СН3, RA выбирается независимо и представляет собой галоген, частично или полностью галогенированный С1-5 алкил, ОС1-3-алкил.
Конечные продукты - ингибиторы ALK получают интермедиата 1-4
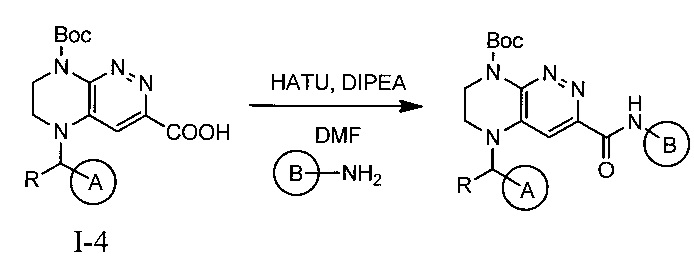
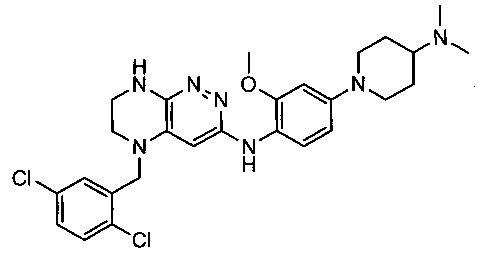
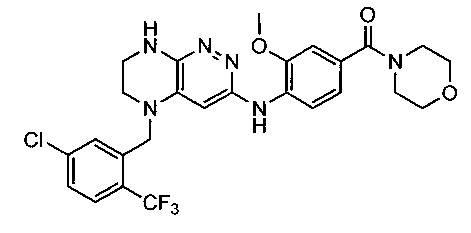
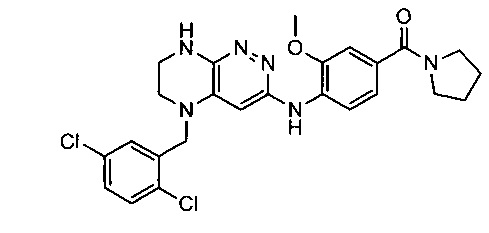
Не ограничивающие примеры ингибиторов ALK, которые можно получить по данной схеме, приведены ниже
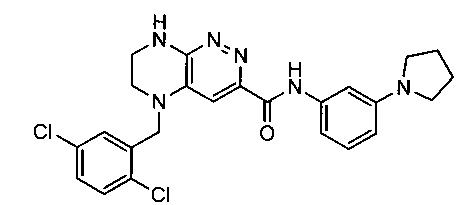
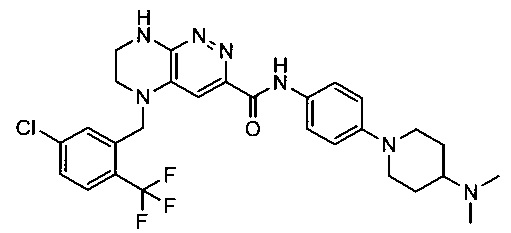
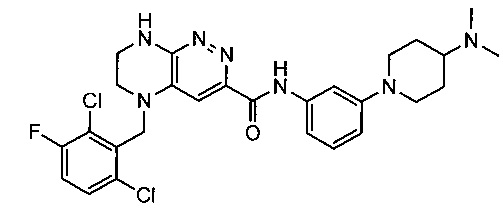
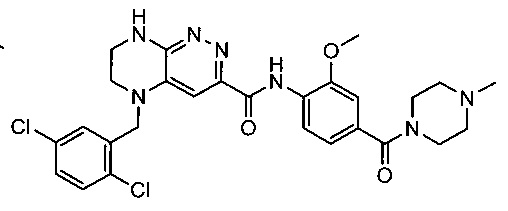
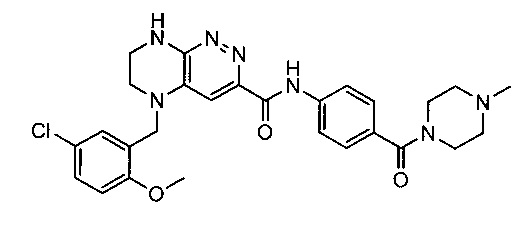
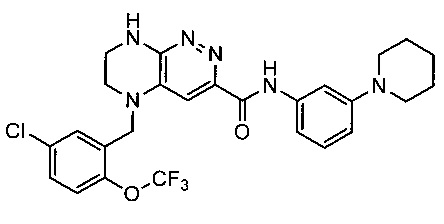
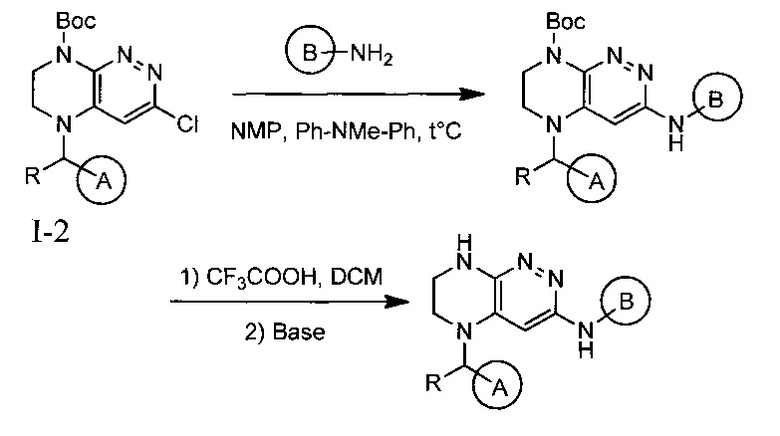
Паладий-катализируемое нуклеофильное замещение соединения 1-2 с последующим удалением Вос-группы приводит к другому классу ингибиторов ALK
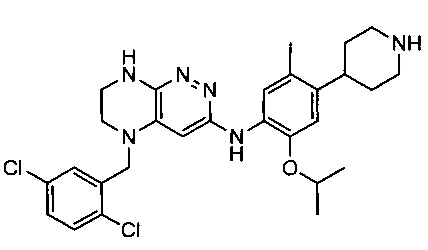
Не ограничивающие примеры ингибиторов ALK, которые можно получить по данной схеме, приведены ниже
Одной из ключевых стадий указанного способа получения указанных ингибиторов киназы ALK является стадия циклизации солевых производных I-0 с образованием интер-медиата I-1. Для этого к перемешиваемому раствору 9.6 г (20 ммоль) интермедиата I-0 в 100 мл безводного N-метилпирролидона прибавляют 8.7 мл (0.05 моль) основания Хунига и выдерживают полученную смесь 10 часов при 100°С (ТСХ-контроль), охлаждают, растворитель удаляют в вакууме, к остатку прибавляют 200 мл насыщенного раствора NaHCO3 и отфильтровывают образовавшийся осадок, который промывают водой (3×50 мл), сушат и разделяют хроматографически. Получают: 1.7 г (25%) соединения I-1.
Необходимость хроматографического продукта I-1 связана с тем, что данная реакция протекает даже при комнатной температуре с образованием двух продуктов, а именно продукта атаки концевого атома азота по атому углерода в положении 3 с образованием целевого соединения I-1, а также продукта ипсо-атаки концевого азота по углероду в положении 4 (известной как перегруппировка Смайлса), с образованием побочного продукта I-1B
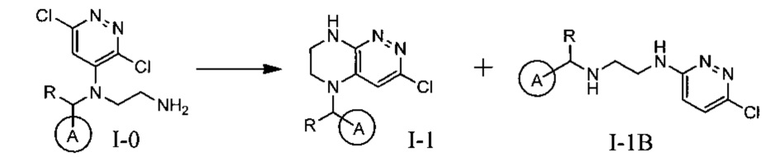
Побочный продукт I-1В по своим свойствам очень похож на продукт I-1, поэтому разделение перекристаллизацией приводит к большим потерям целевого соединения. Использование хроматографического разделения позволяет добиться увеличения выхода до 25%, однако существенно удорожает синтез продукта в промышленном масштабе.
Раскрытие изобретения
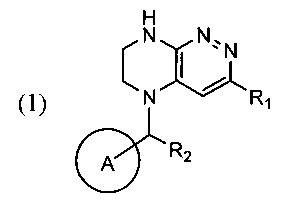
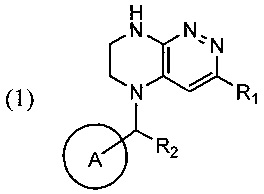
Задачей изобретения является создание нового способа синтеза производных тетра-гидропиразино[2,3-с]пиридазина, свободного от образования трудноотделимого побочного продукта, где указанные производные тетрагидропиразино[2,3-с]пиридазина в совокупности представлены общей формулой (1)
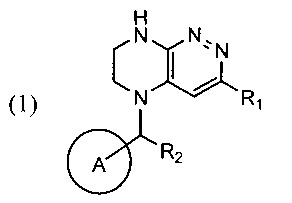
где цикл А представляет собой фенил, опционально замещенный 1-3 группами RA;
RA выбирается независимо и представляет собой галоген, частично или полностью галогенированные C1-5-алкил, ОС1-3-алкил;
R1 представляет собой галоген,
R2 представляет собой водород или -СН3.
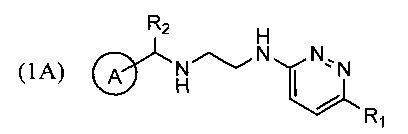
Техническим результатом, достигаемым при использовании изобретения, является повышение выхода реакции циклизации с образованием производных тетрагидропиразино[2,3-с]пиридазина (1), повышение чистоты образующихся продуктов и устранение стадии хроматографического разделения продуктов реакции за счет снижения количества образующегося побочного продукта перегруппировки - соединений формулы 1А
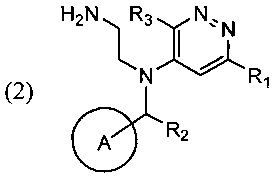
Поставленная задача и требуемый технический результат достигаются за счет нового способа синтеза указанного соединения, который включает циклизацию соли соединения общей формулы (2)
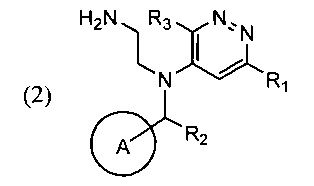
в ходе которой соль соединения (2) смешивают с подходящим растворителем и при повышенной температуре добавляют к раствору или суспензии основания в соответствующим растворителе.
В качестве соли соединения (2) может быть использован хлорид, бромид, иодид, ацетат или трифторацетат соединения (2), в качестве растворителя для реакции циклизации могут быть использованы диметилформамид, диметилацетамид, N-метилпирролидон, диметилсульфоксид; в качестве основания может быть использован карбонат калия, карбонат цезия, безводный ацетат натрия, триэтиламин, трибутиламин, тетраметилгуанидин, N-этилморфолин. Циклизацию осуществляют при температуре 130-180°С, раствор соединения (2) добавляют в реакционную смесь по каплям или вводят под слой раствора или суспензии основания в подходящем растворителе.
Существенное отличие предложенной методики от известной из уровня техники заключается в том, что раствор соединения (2) добавляют в реакционную смесь (раствору или суспензии основания в подходящем растворителе) при повышенной температуре. Нами было установлено, что образование целевой продукт (1) является термодинамическим продуктом, а побочный продукт (1А) - кинетическим продуктом. Изменение последовательности добавления реагентов, а также введение солей (2) в нагретый раствор или суспензию основания с образованием свободного основания (2) при высоких температурах позволяет практически полностью исключить образование побочного продукта в ходе реакции.
Краткое описание чертежей
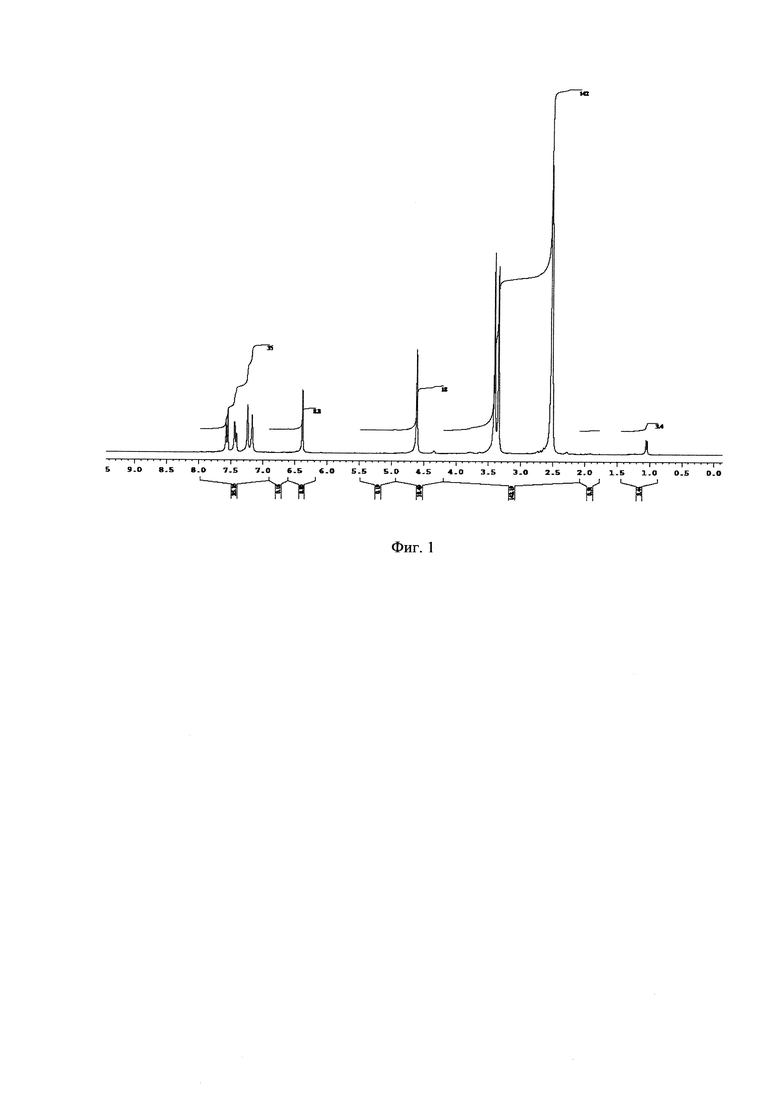
На Фиг. 1 - изображен 1Н-ЯМР спектр основного продукта 5-(2,5-дихлоробензил)-3-хлоро-5,6,7,8-тетрагидропиразино [2,3-с]пиридазина.
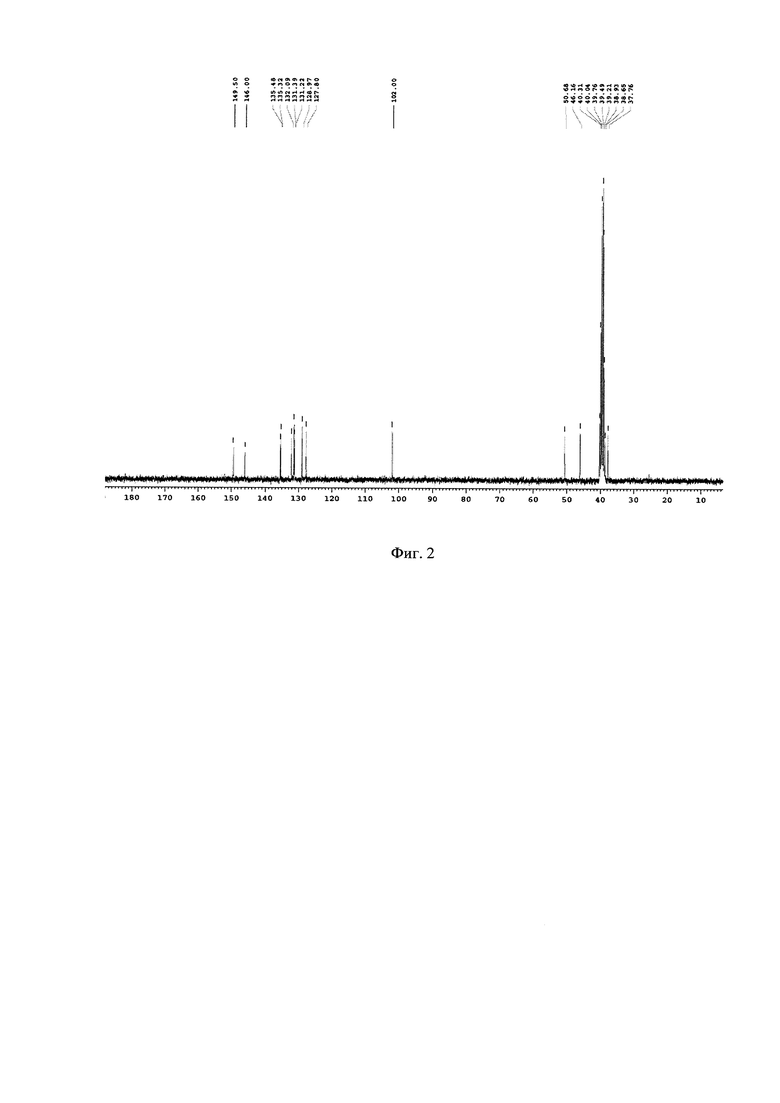
На Фиг. 2 - изображен 13С-ЯМР спектр основного продукта 5-(2,5-дихлоробензил)-3-хлоро-5,6,7,8-тетрагидропиразино[2,3-с]пиридазина.
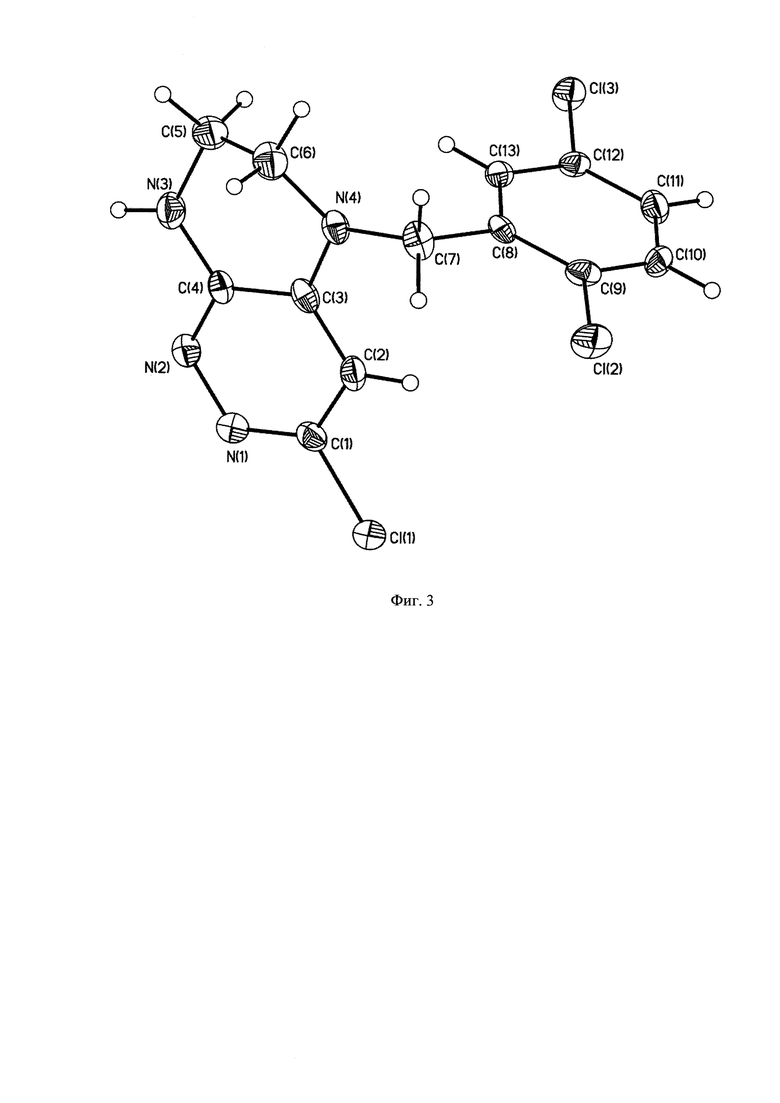
На Фиг. 3 - изображена структура основного продукта 5-(2,5-дихлоробензил)-3-хлоро-5,6,7,8-тетрагидропиразино [2,3-с]пиридазина.
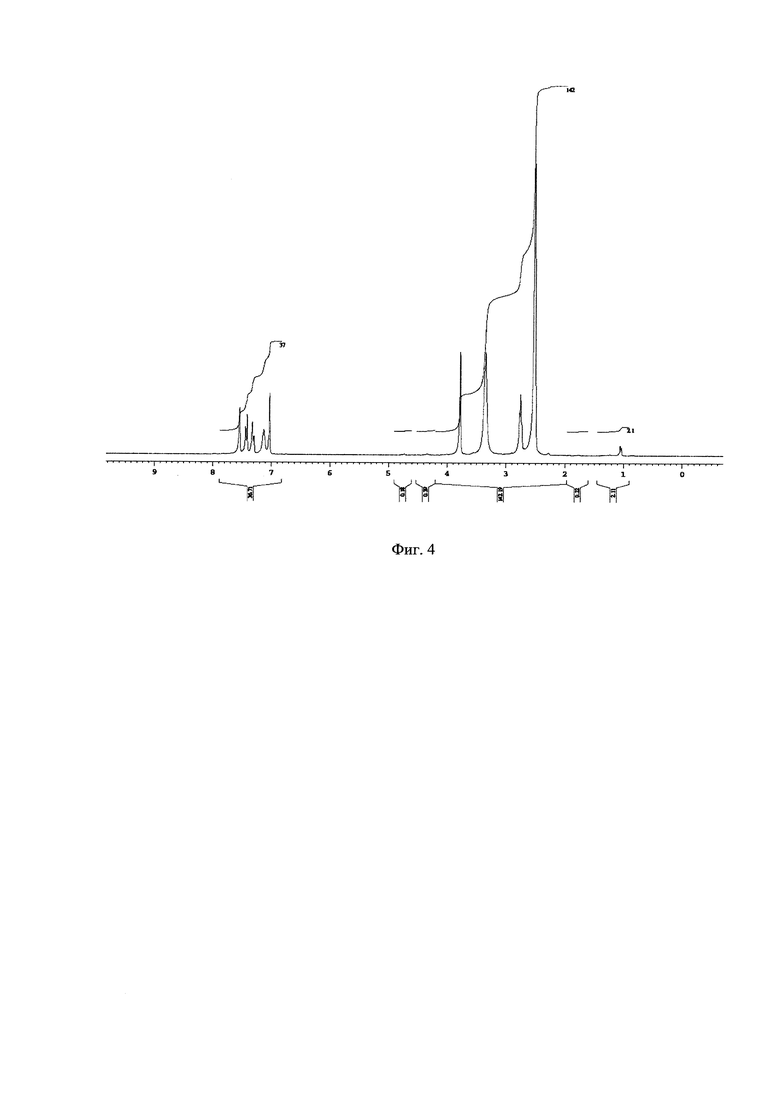
На Фиг. 4 - изображен 1Н-ЯМР спектр побочного продукта перегруппировки N1-(2,5-дихлоробензил)-N6-(3,6-дихлоропиридазин-4-ил)этан-1,2-диамина.
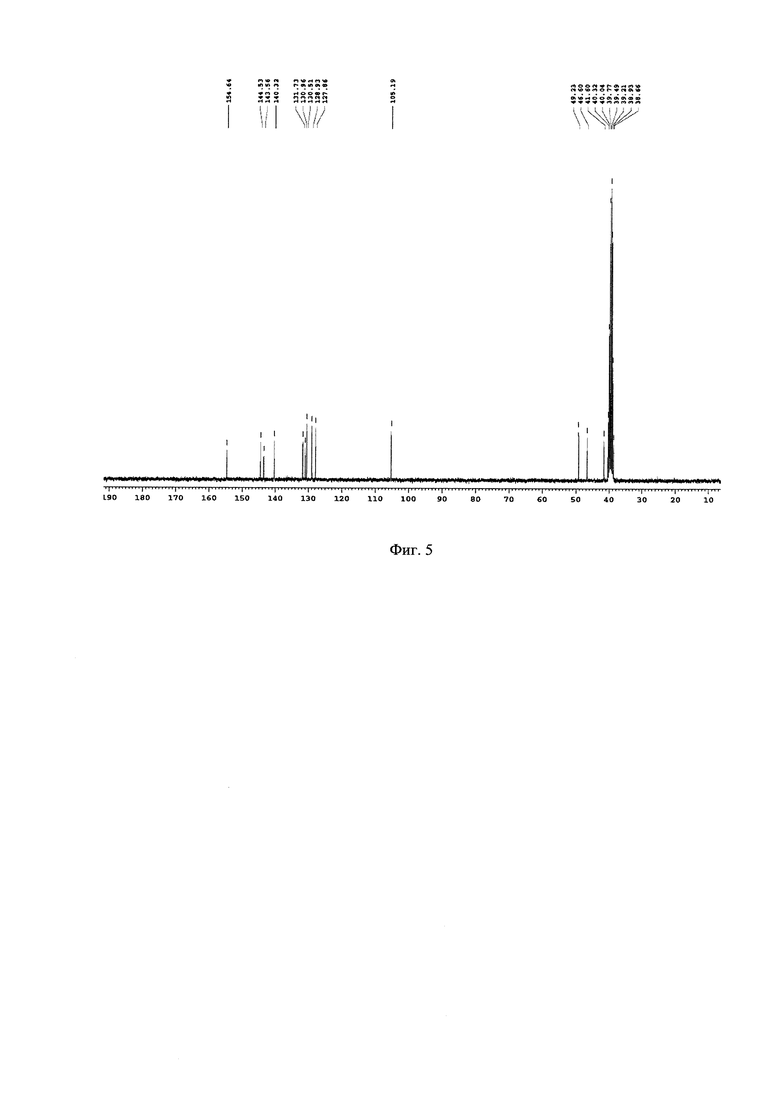
На Фиг. 5 - изображен 13С-ЯМР спектр побочного продукта перегруппировки N1-(2,5-дихлоробензил)-N2-(3,6-дихлоропиридазин-4-ил)этан-1,2-диамина.
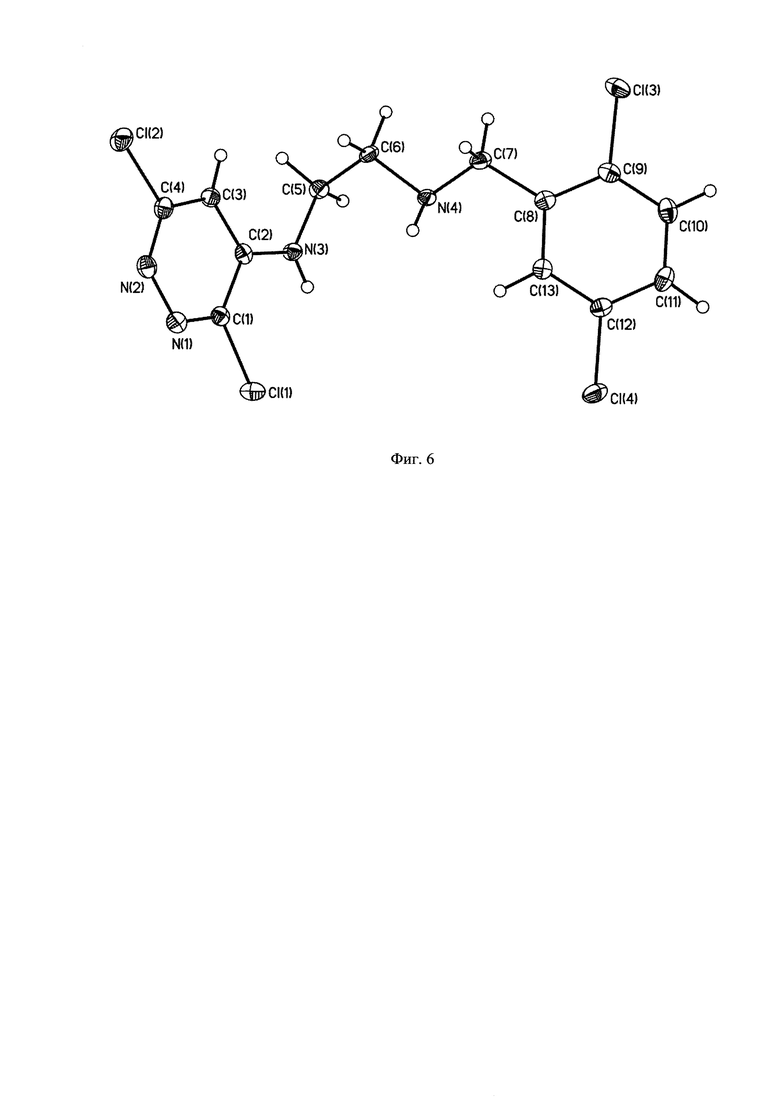
На Фиг. 6 - изображена структура побочного продукта перегруппировки N1-(2,5-дихлоробензил)-N2-(3,6-дихлоропиридазин-4-ил)этан-1,2-диамина.
Осуществление изобретения
Ниже с целью иллюстрации отдельных аспектов осуществления изобретения приведены примеры осуществления предлагаемого способа. Приведенные ниже примеры не предназначены для того, чтобы каким-либо образом ограничивать объем настоящего изобретения.
Пример 1. Способ синтеза 5-(2,5-дихлоробензил)-3-хлоро-5,6,7,8-тетрагидропиразино[2,3-с]пиридазина
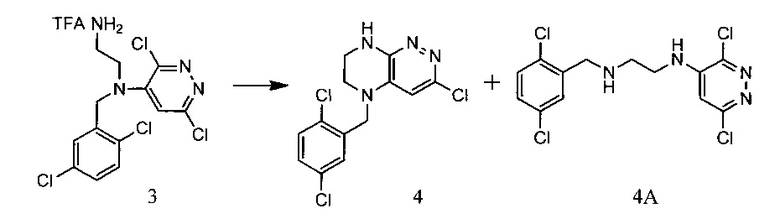
45,4 г (~0,5 моль) трифторацетата диамина 3 смешивают с 200 мл безводного диметилацетамида, и по каплям, поддерживая температуру около 150°С и прибавляют к суспензии 80 г безводного поташа в 500 мл безводного диметилацетамида. По окончании прибавления реакционную смесь перемешивают при заданной температуре еще 30 минут, охлаждают, растворитель удаляют в вакууме, к остатку прибавляют 600 мл этилацетата и 500 мл воды, органический слой отделяют, промывают водой 3×300 мл, сушат над сульфатом натрия, растворитель удаляют в вакууме и сушат. Остаток перекристаллизовывают из изопропанола. Получают: 7,8 г (48%) целевого 5-(2,5-дихлоробензил)-3-хлоро-5,6,7,8-тетрагидропиразино[2,3-с]пиридазина.
1Н-ЯМР, 13С-ЯМР спектры, а также структура целевого продукта реакции приведены на Фиг. 1, 2 и 3 соответственно.
Подбор оптимальных условий циклизации был осуществлен для реакции образования 5-(2,5-дихлоробензил)-3-хлоро-5,6,7,8-тетрагидропиразино[2,3-с]пиридазина (4) на загрузках 5-10 г. Зависимость соотношения продуктов 4:4А от условий протекания реакции представлена в таблице. 1Н-ЯМР, 13С-ЯМР спектры, а также структура побочного продукта перегруппировки приведены на Фиг. 4, 5 и 6 соответственно.
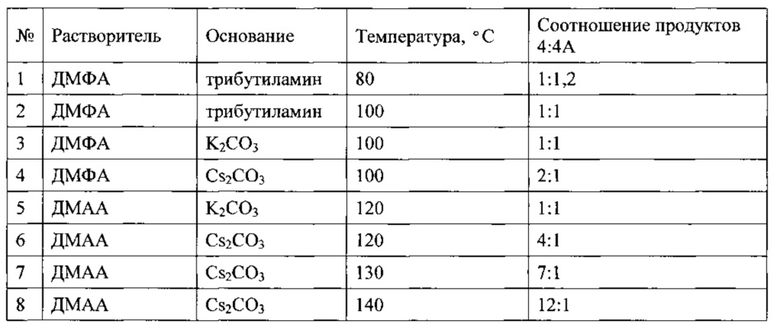
В результате подбора условий нами было достигнуто соотношение продуктов 12:1 в пользу целевого соединения. Дальнейшее повышение температуры приводило к существенному снижению выхода целевого соединения 4 за счет осмоления реакционной смеси.
При увеличении загрузок до 50-100 г, за счет увеличения продолжительности прибавления раствора исходного соединения наблюдалось значительное осмоление реакционной смеси. Мы предполагаем, что данный процесс связан с высокой основностью карбоната цезия, а умеренная растворимость Cs2CO3 в диметилацетамиде (DMAA) в сочетании с высокой насыпной плотностью приводят к сложностям с перемешиванием, и как следствие, возникновению диффузионного фактора в процессе протекания реакции, и к неравномерностям нагревания реакционной смеси. Совокупность этих процессов при масштабировании не только приводит к заметному осмолению, но так же является причиной плохой воспроизводимости получаемых результатов. В результате дополнительной оптимизации условий реакции циклизации, мы перешли к использованию N-метилпироллидона (NMP) растворимость неорганических карбонатов в котором значительно выше, а так же предприняли попытку использовать гомогенные высококипящие органические основания, такие как тетраметилгуанидин (TMG), трибутиламин (Bu3N) и N-этилморфолин. Результаты оптимизации представлены ниже
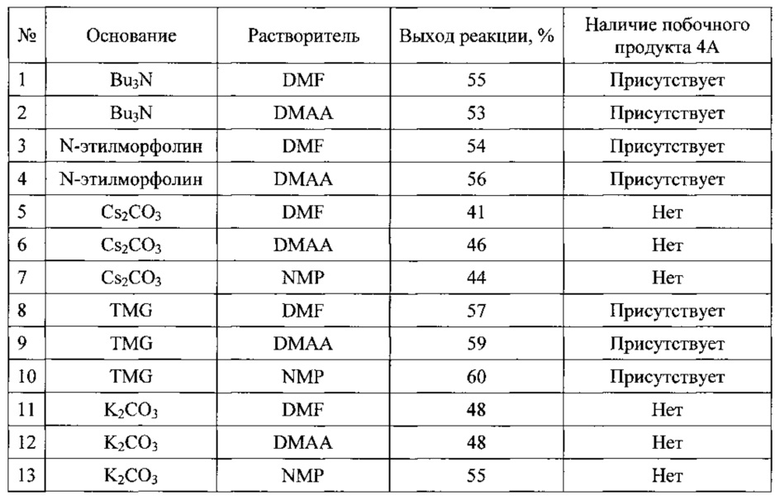
Как видно из приведенных данных, использование органических оснований - трибу-тиламина, N-этилморфолина и тетраметилгуанидина приводит к образованию побочного продукта, что, по видимому, связано с наличием основания в парах над реакционной смесью. Это приводит к образованию свободного амина в растворе соли при температуре ниже температуры реакционной смеси и как следствие к образованию продукта побочного продукта (продукта перегруппировки Смайлса). Использования в качестве основания карбоната калия, имеющего по сравнению с карбонатом цезия значительно более низкую насыпную плотность, позволило с одной стороны, избежать появления основания в газовой фазе над реакционной смесью, а с другой, позволяло осуществлять энергичное перемешивания с распределением карбоната калия по всему объему реакционной смеси. Дополнительным преимущество этого карбоната калия является дешевизна реагента.
Среди использованных нами растворителей N-метилпирролидон обладает самой высокой температурой кипения (202-204°С), что позволяет сильнее нагревать реакционную смесь и таким образом повышать выход реакции. Однако, как уже было отмечено выше, оборотной стороной высокой температуры проведения реакции является начинающееся осмоление реакционной смеси, а так же сложность удаления растворителя. Диметилфор-мамид (DMF) при этих температурах в присутствии оснований значительно разлагается с выделением нуклеофильного диметиламина, что приводит к образованию побочных продуктов (данные продукты были зафиксированы нами по данным ВЭЖХ/МС-анализа реакционной смеси). Таким образом, диметилацетамид, является оптимальным растворителем для проведения данной реакции, так как позволяет достичь приемлемых выходов и может быть отогнан из реакционной смеси в вакууме водоструйного насоса при температуре ниже 100°С.
Пример 2. 5-(2,6-дихлоро-3-фторобензил)-3-хлоро-5,6,7,8-тетрагидропиразино[2,3-с]пиридазина
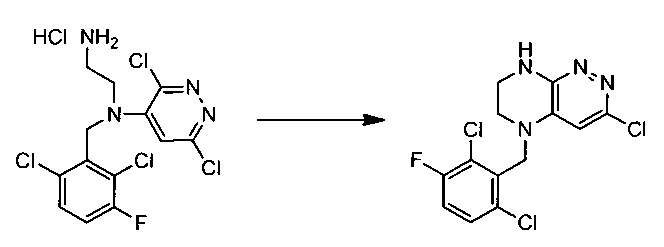
38,4 г (~0,1 моль) хлорида N1-(2,6-дихлоро-3-фторобензил)-N1-(3,6-дихлоропиридазин-4-ил)этан-1,2-диамина смешивают с 100 мл безводного диметилацетамида, и по каплям, поддерживая температуру около 150°С прибавляют к суспензии 80 г безводного поташа в 500 мл безводного диметилацетамида. По окончании прибавления реакционную смесь перемешивают при заданной температуре еще 45 минут, охлаждают, растворитель удаляют в вакууме, к остатку прибавляют 600 мл этилацетата и 500 мл воды, органический слой отделяют, промывают водой 3×300 мл, сушат над сульфатом натрия, растворитель удаляют в вакууме и сушат. Получают: 15,3 г (44%) целевого продукта 5-(2,6-дихлоро-3-фторобензил)-3-хлоро-5,6,7,8-тетрагидропиразино [2,3-с]пиридазина.
Пример 3. Синтез 5-(2,6-дихлоро-3-фторобензил)-3-хлоро-5,6,7,8-тетрагидропиразино[2,3-с]пиридазина
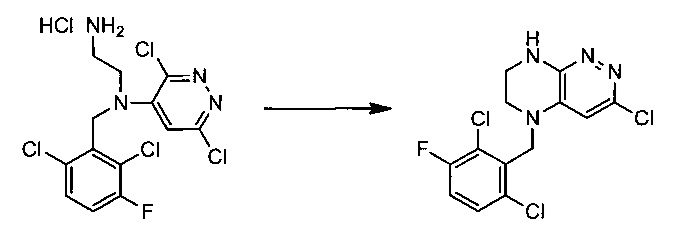
38,4 г (~0,1 моль) хлорида N1-(2,6-дихлоро-3-фторобензил)-N1-(3,6-дихлоропиридазин-4-ил)этан-1,2-диамина смешивают с 100 мл безводного диметилацетамида, и по каплям, поддерживая температуру около 150°С прибавляют к суспензии 80 г безводного поташа в 500 мл безводного диметилацетамида. По окончании прибавления реакционную смесь перемешивают при заданной температуре еще 45 минут, охлаждают, растворитель удаляют в вакууме, к остатку прибавляют 600 мл этилацетата и 500 мл воды, органический слой отделяют, промывают водой 3×300 мл, сушат над сульфатом натрия, растворитель удаляют в вакууме и сушат. Получают: 15,3 г (44%) целевого продукта 5-(2,6-дихлоро-3-фторобензил)-3-хлоро-5,6,7,8-тетрагидропиразино[2,3-с]пиридазина.
Пример 3. Синтез 3-хлоро-5-(5-хлоро-2-(трифторметил)бензил)-5,6,7,8-тетрагидропиридазин-4-ил)этан-1,2-диамина
49,6 г (~0,1 моль) трифторацетата N1-(2-хлоро-5-(трифторметил)бензил)-N1-(3,6-дихлоропиридазин-4-ил)этан-1,2-диамина смешивают с 100 мл безводного диметилацетамида, и по каплям, поддерживая температуру около 140°С прибавляют к суспензии 80 г безводного поташа в 500 мл безводного диметилацетамида. По окончании прибавления реакционную смесь перемешивают при заданной температуре еще 45 минут, охлаждают, растворитель удаляют в вакууме, к остатку прибавляют 600 мл этилацетата и 500 мл воды, органический слой отделяют, промывают водой 3×300 мл, сушат над сульфатом натрия, растворитель удаляют в вакууме и сушат. Получают: 13,8 г (38%) целевого продукта 3-хлоро-5-(5-хлоро-2-(трифторметил)бензил)-5,6,7,8-тетрагидропиридазин-4-ил)этан-1,2-диамина.
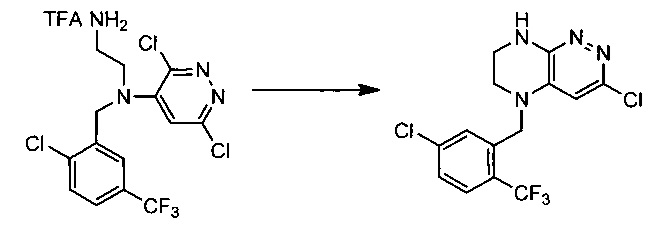
Пример 4. Синтез 3-бромо-5-(1-(5-хлоро-2-(трифторметил)фенил)этил)-5,6,7,8-тетрагидропиразино[2,3-с]пиридазина
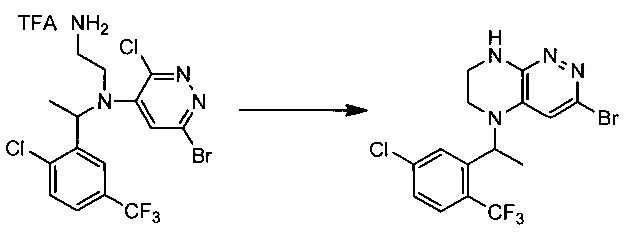
55,5 г (~0,1 моль) трифторацетата N1-(6-бромо-3-хлоропиридазин-4-ил)-N1-(1-(2-хлоро-5-(трифторметил)фенил)этил)этан-1,2-диамина смешивают с 100 мл безводного диметилацетамида, и по каплям, поддерживая температуру около 160°С прибавляют к суспензии 120 г безводного карбоната цезия в 500 мл безводного диметилацетамида. По окончании прибавления реакционную смесь перемешивают при заданной температуре еще 60 минут, охлаждают, растворитель удаляют в вакууме, к остатку прибавляют 600 мл этилацетата и 500 мл воды, органический слой отделяют, промывают водой 3×300 мл, сушат над сульфатом натрия, растворитель удаляют в вакууме и сушат. Получают: 22,3 г (53%) целевого продукта 3-бромо-5-(1-(5-хлоро-2-(трифторметил)фенил)этил)-5,6,7,8-тетрагидропиразино [2,3-с] пиридазина.
Пример 5. Синтез 3-хлоро-5-(1-(5-хлоро-2-метоксифенил)этил)-5,6,7,8-тетрагидропиразино[2,3-с]пиридазина
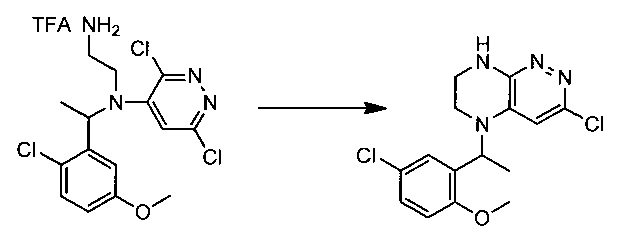
47,3 г (~0,1 моль) трифторацетата N1-(1-(2-хлоро-5-метоксифенил)этил)-N1-(3,6-дихлоропиридазин-4-ил)этан-1,2-диамина смешивают с 100 мл безводного диметилацетамида, и по каплям, поддерживая температуру около 150°С прибавляют к раствору 32 мл (~0.25 моль) тетраметилгуанидина в 500 мл безводного диметилацетамида. По окончании прибавления реакционную смесь перемешивают при заданной температуре еще 45 минут, охлаждают, растворитель удаляют в вакууме, к остатку прибавляют 600 мл этилацетата и 500 мл воды, органический слой отделяют, промывают водой 3×300 мл, сушат над сульфатом натрия, растворитель удаляют в вакууме и сушат. Получают: 17,3 г (51%) целевого продукта 3-хлоро-5-(1-(5-хлоро-2-метоксифенил)этил)-5,6,7,8-тетрагидропиразино[2,3-с]пиридазина.
Предложенное изобретение не ограничено описанными выше вариантами осуществления, а наоборот оно охватывает различные модификации и варианты в рамках сущности и объема предлагаемой формулы изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ ХИМИЧЕСКИЕ СОЕДИНЕНИЯ (ВАРИАНТЫ) И ИХ ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ ОНКОЛОГИЧЕСКИХ ЗАБОЛЕВАНИЙ | 2013 |
|
RU2550346C2 |
| СПОСОБ ПОЛУЧЕНИЯ АНТРА[2,3-b]ФУРАН-3-КАРБОНОВОЙ КИСЛОТЫ | 2014 |
|
RU2554937C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ 5-АМИНО-2,4,6-ТРИИОДО- 1,3-БЕНЗОЛКАРБОНОВОЙ КИСЛОТЫ | 1990 |
|
RU2046795C1 |
| Способ получения 8-алкил-5-оксо-5,8дигидропиридо/2,3- /пиримидин-6-карбоновых кислот | 1973 |
|
SU691091A3 |
| ПРОИЗВОДНЫЕ 3-АМИНОПИРИДАЗИНОВ ИЛИ ИХ КИСЛОТНО-АДДИТИВНЫЕ ФАРМАЦЕВТИЧЕСКИЕ ПРИЕМЛЕМЫЕ СОЛИ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ, СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ 3-АМИНОПИРИДАЗИНОВ ИЛИ ИХ КИСЛОТНО-АДДИТИВНЫХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫХ СОЛЕЙ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1992 |
|
RU2088577C1 |
| ОДНОРЕАКТОРНЫЙ СПОСОБ ПОЛУЧЕНИЯ ПОЛУПРОДУКТА ДЛЯ СИНТЕЗА ЛИНЕЗОЛИДА | 2020 |
|
RU2760198C1 |
| СПОСОБ СИНТЕЗА ФТОРКЛОЗАПИНА И ЕГО ПРОИЗВОДНЫХ | 2014 |
|
RU2557241C1 |
| СПОСОБ ПОЛУЧЕНИЯ 1R,3S-2,2-ДИМЕТИЛ-3-(2-ОКСОПРОПИЛ)-ЦИКЛОПРОПАНАЦЕТОНИТРИЛА | 1989 |
|
RU1689376C |
| ПРОИЗВОДНЫЕ МЕТИЛПИПЕРАЗИНАЗЕПИНА ИЛИ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ ЭТИХ ПРОИЗВОДНЫХ | 1991 |
|
RU2111966C1 |
| Способ получения 2-(2-хлоралкокси)-2-оксо-1,4,2-диоксафосфепанов | 1982 |
|
SU1033497A1 |
Изобретение относится к способe синтеза производных тетрагидропиразино[2,3-с]пиридазина формулы (1)
которые могут быть использованы для изготовления лекарственных средств для лечения онкологических, хронических воспалительных и прочих заболеваний. Технический результат: новый способ синтез позволяет получить соединения общей формулы (1), обладающие повышенной степенью чистоты. 6 з.п. ф-лы, 6 ил., 5 пр.
1. Способ получения соединения формулы (1)
цикл А представляет собой фенил, опционально замещенный 1-3 группами RA;
RA выбирается независимо и представляет собой галоген, частично или полностью галогенированные С1-5-алкил, OC1-3-алкил;
R1 представляет собой галоген OSO2R,
R2 представляет собой водород или -СН3,
R представляет собой частично или полностью галогенированный C1-3-алкил или фенил, опционально 1-3 группами СН3 или атомами галогена,
который включает циклизацию соли соединения соли (2)
с образованием соединения формулы (1), в ходе которой соль соединения (2) смешивают с подходящим растворителем и при повышенной температуре добавляют к раствору или суспензии основания в соответствующим растворителе.
2. Способ по п. 1, при котором используют хлорид, бромид, иодид, ацетат, сульфат, тозилат, мезилат, трифторацетат соединения (2).
3. Способ по п. 1, при котором в качестве растворителя используют диметилформамид, диметилацетамид, N-метилпирролидон.
4. Способ по п. 1, при котором в качестве основания используют карбонат калия, карбонат цезия, безводный ацетат натрия, триэтиламин, трибутиламин, тетраметилгуанидин, N-этилморфолин.
5. Способ по п. 1, при котором добавление соли (2) осуществляют при температуре от 130 до 180°C.
6. Способ по п. 1, при котором соль (2) добавляют к раствору или суспензии основания.
7. Способ по п. 1, при котором соль (2) вводят под слой раствора или суспензии основания.
| НОВЫЕ ХИМИЧЕСКИЕ СОЕДИНЕНИЯ (ВАРИАНТЫ) И ИХ ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ ОНКОЛОГИЧЕСКИХ ЗАБОЛЕВАНИЙ | 2013 |
|
RU2550346C2 |
| WO 2012048259 A2, 12.04.2012. | |||

