Изобретение относится к синтезу пептидов, в частности к получению трипептида (I), используемого в качестве промежуточного соединения (1-3 фрагмент LH-RH) в синтезе биологически активных синтетических агонистов гонадотропин-рилизинг-гормона LH-RH (ЛГ-РГ):
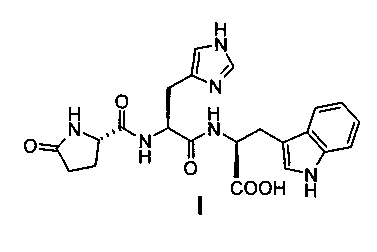
Гонадотропин-рилизинг-гормон (ГнРГ, гонадолиберин), известный также как лютеинизирующий-рилизинг-гормон (ЛГ-РГ), содержит последовательность из 10 аминокислот и является одним из представителей рилизинг-гормонов гипотоламуса:
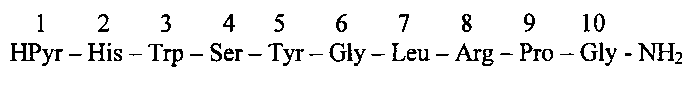
Ввиду быстрого разрушения ГнРГ в организме эндопептидазами (период полувыведения эндогенного ГнРГ составляет 2-8 мин) в фармацевтической практике получили широкое распространение синтетические модифицированные агонисты ГнРГ, обладающие гораздо более длительным периодом действия при сохранении (или усилении) специфической биологической активности. Как правило, модификация молекулы гонадолиберина происходит путем замещения аминокислот в позициях 6 и 10, что позволяет при сохранении аффинитета к гипофизарным рецепторам замедлить ее расщепление эндопептидазами. При этом было обнаружено, что кроме повышенной устойчивости к энзимам агонисты ГнРГ значительно превосходят эндогенный гонадолиберин по биологической активности. Первоначально агонисты ГнРГ разрабатывались для лечения бесплодия на почве гипогонадотропного гипогонадизма, в соответствии с физиологическим значением гонадолиберина. Однако впоследствии оказалось, что повышение секреции гонадотропинов происходит только при применении препарата ГнРГ в импульсном режиме, а длительное применение агонистов ГнРГ приводит к транзиторной стимуляции секреции гонадотропинов и, как следствие, к понижению уровня секреции половых стероидов. В связи с этим показания к применения агонистов ГнРГ существенно расширились. В настоящее время их применяют при прогрессирующем раке предстательной железы, раке молочной железы, фибромиомы матки, эндометриозе, а также при экстракорпоральном оплодотворении (ЭКО).
В настоящее время наиболее широко применяются 4 синтетических агониста ГнРГ: бусерелин, гозерелин. лейпрорелин и трипторелин, в производстве которых трипептид I является ключевым полупродуктом [1, 2, 3] при получении по жидкофазному методу.
В частности известен способ получения нонапептида лейпрорелина [1]:
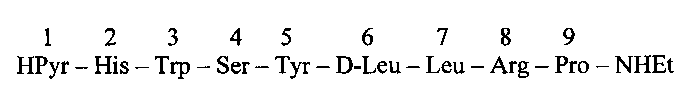
заключающийся в конденсации трипептида I и гексапептида HSerTyr-D-LeuLeuArgProNHEt.
Способ получения декапептида трипторелина [2]:
по схеме 3+7, где в качестве трипептида используют соединение I.
В RU №2444525, 10.03.2012, описан способ получения нонапептида буселерина [3]:
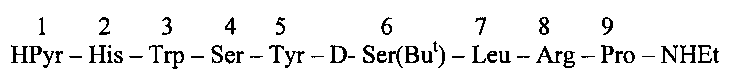
по схеме 3+6 с использованием в качестве промежуточного соединения гидрохлорида трипептида I.
Из US №4024248, 17.03.1977, известен способ получения аналога LH-RH (нонапептидэтиламида) конденсацией пептидных фрагментов по схеме 3+6 азидным методом, в котором в качестве промежуточного соединения используют трипептид формулы HPyr-His-Trp-NH-NH2.
Наиболее близким по технической сущности является известный из DD №226895, 04.09.1985, способ получения трипептида I (HPyr-His-TrpOH), включающий 7 стадий:
- постановка бензилоксикарбонильной защиты на L-пироглутаминовую кислоту;
- получение метилового эфира гистидина;
- конденсация защищенной L-пироглутаминовой кислоты и метилового эфира L-гистидина в присутствии дициклогексилкарбодиимида (ДЦК) с образованием метилового эфира защищенного дипептида - Z-PyrHisOMe;
- снятие Z-защиты с использованием палладия на угле;
- гудразинолиз метилового эфира дипептида с образованием Н-PyrHisNHNH2;
- получение этилового эфира целевого трипептида Н-PyrGluHisTrpOEt реакцией H-PyrHisNHNH2 и гидрохлорида этилового эфира L-триптофана с использованием стандартного протокола конденсации азидным методом;
- гидролиз H-PyrGluHisTrpOEt гидроксидом натрия в метаноле с образованием трипептида.
Недостатками этого метода являются:
- относительно большое количество стадий (за счет стадий постановки и снятия защитных групп),
- высокая себестоимость процесса,
- использование токсичных реагентов (бензилоксикарбонилхлорид, гидразин гидрат, трет-бутил нитрит),
- жесткие условия проведения процесса (при проведении конденсации азидным методом необходимо охлаждение до -25 - -30°С).
Задачей данного изобретения является:
- упрощение процесса получения,
- исключение реагентов на постановку и снятие защитных групп,
- снижение себестоимости производства I,
- снижение токсичности процесса.
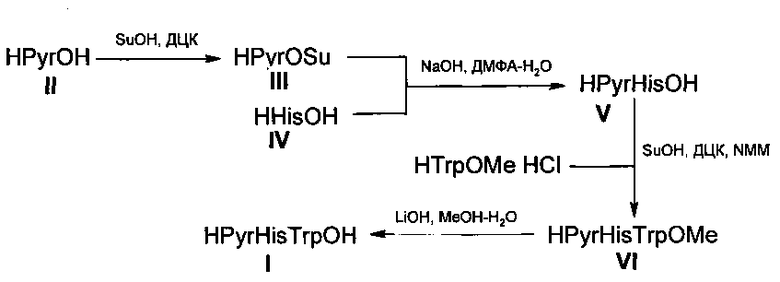
Поставленная техническая задача и достигаемый технический результат достигается способом получения трипептида формулы (I) HPyr-His-TrpOH методом жидкофазного пептидного синтеза без использования стадий постановки и снятия защитных групп, включающим следующие стадии:
1. Конденсация L-пироглутаминовой кислоты (II) HPyrOH и N-гидроксисукцинимида (SuOH) в присутствии дициклогексилкарбодиимида (ДЦК) в тетрагидрофуране с образованием активированного эфира (III);
2. Взаимодействие полученного эфира III с натриевой солью L-гистидина (IV) HHisOH в смеси растворителей диметилформамид-вода с образованием дипептида HPyrHisOH (V) ;
3. Реакция активированного сукцинимидового эфира, полученного без выделения конденсацией дипептида (V) и SuOH, с метиловым эфиром L-триптофана в диметилформамиде с образованием метилового эфира трипептида - HPyrHisTrpOMe (VI) ;
4. Гидролиз метилового эфира формулы (VI) в водно-метанольном растворе с использованием гидроксида лития и дальнейшее осаждение ионов лития эквимольным количеством ортофосфорной кислоты дает целевой трипептид I.
Разработанный метод синтеза I, позволяющий решить поставленную задачу 4-стадийном синтезом, выполнен по следующей схеме:
Ниже представлен в общем виде способ, заявленный в качестве изобретения, раскрывающий сущность данного способа.
Конденсацию L-пироглутаминовой кислоты (II) и N-гидроксисукцинимида (SuOH) в присутствии ДЦК проводят в тетрагидрофуране в течение 48 ч. Выпавшую дициклогексилмочевину отфильтровывают и удаляют растворитель в вакууме. Остаток растворяют в хлористом метилене и оставляют кристаллизоваться при 5°С. После фильтрования получают кристаллический активированный эфир III с выходом 69%.
Реакция активированного эфира III и натриевой соли L-гистидина, полученной без выделения нейтрализацией IV водным раствором гидроксида натрия, дает дипептид V. Сырец V, полученный после отгонки в вакууме растворителей, растворяют в метаноле, доводят рН концентрированной соляной кислотой до 3 и высаживают тетрагидрофураном при температуре 5°С. Выход V (чистотой 96%) составляет 82%.
Конденсация дипептида V и метилового эфира L-триптофана с использованием SuOH и ДЦК в диметилформамиде в течение 24 ч приводит к метиловому эфиру трипептида VI. N-Метилморфолин применяют для перевода гидрохлорида в свободное основание. Продукт реакции, полученный после удаления диметилформамида в вакууме, растворяют в 2-фазной системе этилацетат-вода. Соединение VI высаживают из водного слоя раствором бикарбоната натрия при рН=8. Дополнительную очистку производят промыванием сырца VI этилацетатом. Выход VI (чистотой 96.6%) составляет 65.2%.
Целевой трипептид I получают гидролизом VI водно-метанольным раствором гидроксида лития в течение 1.5 ч. После окончания реакции Li+ связывают эквимолярным количеством ортофосфорной кислоты, удаляют метанол в вакууме, а водный раствор I лиофилизуют. Получают I с выходом 95% и чистотой 98.5% (ВЭЖХ).
Чистота полученного трипептида HPyr-His-TrpOH и его полупродуктов подтверждена ВЭЖХ и ЯМР спектроскопией. Аналитическую ВЭЖХ проводили на хроматографе фирмы Shimadzu. Колонка: Grom-Sil 12J ODS-4HE, 5 мкм, 250×4,6 мм. Условия: градиент 20%В (0 мин) 60%В (10 мин) 70%В (15 мин) 70%В (30 мин). А - фосфатный буфер рН 3 (20,4 г KH2PO4 растворяли в 3 л дистиллированной воды и доводили рН до 3 добавлением концентрированной фосфорной кислоты), В - ацетонитрил. Спектры ПМР получены на приборе Bruker АМ-360 (рабочая частота 360.13 МГц). Спектры ЯМР 13С получены на приборе Bruker АМ-360 (рабочая частота 90.56 МГц). Угол удельного вращения измеряли на автоматическом поляриметре "OTAGO" АР-300. Спектры ESI-MS регистрировали на приборе "Agilent LC/MS 1200" при ионизации пробы электрораспылением в режиме регистрации положительных ионов. Пробы готовили в системе ацетонитрил/вода 1/1, концентрация 2 мг/мл. Условия анализа: поток 1 мл/мин, давление на нибулайзере 20 psi, температура 360°C, скорость потока осушающего газа 9 л/мин, напряжение 3500 В, целевая масса от 100 до 2000.
Описываемый способ иллюстрируется следующим примером, отражающим подробное осуществление каждой стадии синтеза.
A. H-Pyr-OSu (III).
Суспендируют 30 г (0.23 моль) пироглутаминовой кислоты (II) в 0.5 л тетрагидрофурана, присыпают 26.7 г (0.23 моль) N-гидроксисукцинимида. Охлаждают суспензию до 0°С и присыпают 47.3 г (0.23 моль) дициклогексилкарбодиимида. Отогревают реакционную массу до комнатной температуры и перемешивают в течение 48 ч. Отфильтровывают выпавшую мочевину, фильтрат упаривают в вакууме до окончания отгонки (50°С, 12 мм рт.ст.). Растворяют маслообразный остаток в 200 мл хлористого метилена, вносят затравку конечного продукта и оставляют при 5°С на 2 ч. Продукт отфильтровывают, промывают 50 мл холодного (5°С) хлористого метилена и сушат в вакууме (40°С, 12 мм рт.ст.) до постоянного веса. Получают 36 г (69.2%) белого кристаллического III. Т.пл. 136-138°С. 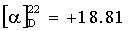
Б. H-Pyr-His-OH (V).
Растворяют 18.9 г (0.084 моль) III в 35 мл диметилформамида. К полученному раствору при 0°С прикапывают раствор 9.3 г (0.06 моль) гистидина (IV) и 2.4 г (0.06 моль) NaOH в 30 мл воды в течение 20 мин. Перемешивают реакционную массу 30 мин при 10°С и затем 2 ч при комнатной температуре до окончания реакции. Упаривают реакционную массу в вакууме до окончания отгонки (50°С, 2 мм рт.ст.). Маслообразный остаток растворяют в 50 мл метилового спирта, подкисляют конц. соляной кислотой до рН=3, добавляют 100 мл тетрагидрофурана и оставляют при 5°С в течение 12 ч. Осадок фильтруют, промывают 50 мл тетрагидрофурана и сушат в вакууме до постоянного веса (50°С, 2 мм рт.ст.). Получают 13.1 г (82%) белого аморфного V. Содержание основного вещества 96% (ВЭЖХ). 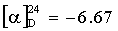
B. H-Pyr-His-TrpOMe (VI).
Растворяют 6.42 г (0.024 моль) V в 55 мл диметилформамида. К полученному раствору присыпают 6.11 г (0.024 моль) метилового эфира L-триптофана, 3.3 г (0.029 моль) N-гидроксисукцинимида и 3.3 г (0.032 моль) N-метилморфолина. После полного растворения реакционную массу охлаждают до 10°С и при этой температуре в течение 1 ч прикапывают раствор 6.5 г (0.032 моль) дициклогексилкарбодиимида в 20 мл диметилформамида. Отогревают до комнатной температуры и перемешивают в течение 24 ч. Осадок дициклогексилмочевины отфильтровывают, а фильтрат упаривают в вакууме до окончания отгонки (50°С, 2 мм рт.ст.). Оставшееся масло растворяют в 50 мл воды, добавляют 30 мл этилацетата, фильтруют и отделяют водный слой. К водному слою прикапывают насыщенный раствор гидрокарбоната натрия до рН 8, приливают 50 мл этилацетата и оставляют при 5°С на 3 ч. Сырец VI фильтруют, перемешивают в смеси этилацетат-вода 30 мл воды и 30 мл этилацетата, фильтруют, промывают на фильтре 20 мл этилацетата и сушат в вакууме до постоянного веса. Получают 7.3 г (65.2%) белого порошка VI. Содержание основного вещества 96.6% (ВЭЖХ). 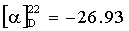
Г. H-Pyr-His-TrpOH (I).
Суспендируют 6.3 г (0.0135 моль) VI в 47 мл метилового спирта. К полученной суспензии при комнатной температуре приливают раствор 0.44 г гидроксида лития (0.0184 моль) в 20 мл воды. Перемешивают реакционную массу 1.5 ч до окончания реакции (ТСХ). Прикапывают ортофосфорную кислоту (85%) до рН 7-7.5 (эквимолярное количество). Фильтруют выпавший фосфат лития, а фильтрат упаривают до объема 15 мл (отгонка метанола) и лиофилизуют остаток. Получают 5.8 г (94.9%) белого порошка I. Содержание основного вещества 98.5% (ВЭЖХ). 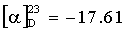
Как видно из вышеприведенного примера, описанный способ существенно упрощает процесс получения целевого соединения за счет уменьшения количества стадий синтеза, делает его более экономически эффективным за счет отсутствия стадий постановки и снятия защитных групп и позволяет получать трипептид I с чистотой более 98%.
Источники информации
1. Химико-фармацевтический журнал. 2014, 48(3), 54-56.
2. Российский биотерапевтический журнал 2013, 12(4), 55-58.
3. RU №2444525. Способ получения нонапептидэтиламида и промежуточные соединения для его получения, 10.03.2012.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ДИАЦЕТАТА ТРИПЕПТИДА | 2014 |
|
RU2551276C1 |
| СПОСОБ ПОЛУЧЕНИЯ НОНАПЕПТИДЭТИЛАМИДА И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДЛЯ ЕГО ПОЛУЧЕНИЯ | 2010 |
|
RU2444525C2 |
| СПОСОБ ПОЛУЧЕНИЯ АНАЛОГОВ АДЕНОКОРТИКОТРОПНОГО ГОРМОНА (АКТГ), ПОСЛЕДОВАТЕЛЬНОСТИ (4-10), ОБЛАДАЮЩИХ НЕЙРОТРОПНОЙ АКТИВНОСТЬЮ, И ТЕТРАПЕПТИД ДЛЯ ЕГО ПОЛУЧЕНИЯ | 2006 |
|
RU2315057C2 |
| СПОСОБ ПОЛУЧЕНИЯ ТРИПЕПТИДОВ | 2006 |
|
RU2303602C2 |
| СПОСОБ УСКОРЕННОГО ПЕПТИДНОГО СИНТЕЗА В РАСТВОРЕ | 2002 |
|
RU2237673C2 |
| ТРИПЕПТИД, ОБЛАДАЮЩИЙ НЕЙРОПСИХОТРОПНОЙ АКТИВНОСТЬЮ | 2023 |
|
RU2823022C1 |
| ДИПЕПТИДЫ В КАЧЕСТВЕ КОРМОВЫХ ДОБАВОК | 2010 |
|
RU2536467C2 |
| СПОСОБ ПОЛУЧЕНИЯ ДОДЕКАПЕПТИДА И ТРИПЕПТИД ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ | 2007 |
|
RU2340626C1 |
| Дипептидные лиганды TSPO, обладающие нейропсихотропной активностью | 2018 |
|
RU2756772C2 |
| СПОСОБ ПОЛУЧЕНИЯ БУСЕРЕЛИНА И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДЛЯ ЕГО ПОЛУЧЕНИЯ | 2010 |
|
RU2442791C1 |
Изобретение относится к области пептидной и фармацевтической химии, конкретно к способу получения HPyr-His-TrpOH (I), используемого в качестве ключевого полупродукта (1-3 фрагмента) при синтезе синтетических агонистов гонадотропин-рилизинг-гормона, LH-RH (ЛГ-РГ). Трипептид (I) получают методом жидкофазного пептидного синтеза без использования стадий постановки и снятия защитных групп. Способ позволяет уменьшить количество стадий синтеза, является более экономически эффективным за счет отсутствия стадий постановки и снятия защитных групп и позволяет получать трипептид (I) с чистотой более 98%. 1 пр.
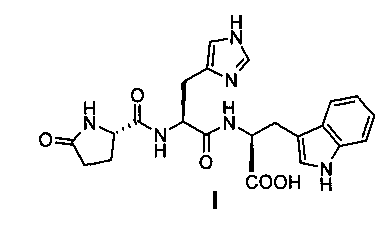
Способ получения трипептида общей формулы (I) HPyr-His-TrpOH, используемого в качестве промежуточного соединения (1-3 фрагмента) в синтезе синтетических агонистов гонадотропинрилизинг-гормона (LH-RH), методом жидкофазного пептидного синтеза без постановки и снятий защитных групп, включающий следующие стадии:
а) конденсацию L-пироглутаминовой кислоты (II) HPyrOH и N-гидроксисукцинимида SuOH в присутствии дициклогексилкарбодиимида (ДЦК) в тетрагидрофуране;
б) взаимодействие образовавшегося на стадии (а) активированного эфира (III) HPyrOSu с натриевой солью L-гистидина HHisOH (IV) в смеси растворителей диметилформамид-вода с образованием дипептида HPyrHisOH (V);
в) реакцию активированного сукцинимидного эфира, полученного без выделения конденсацией дипептида (V) и SuOH, с метиловым эфиром L-триптофана в диметилформамиде с образованием метилового эфира трипептида (VI) HPyr-His-TrpOMe;
г) гидролиз метилового эфира трипептида (VI) в водно-метанольном растворе с использованием гидроксида лития;
д) осаждение ионов лития эквимольным количеством ортофосфорной кислоты после гидролиза метилового эфира трипептида (VI);
е) удаление метанола в вакууме и лиофилизация водного раствора целевого продукта трипептида формулы (I).
| СПОСОБ ПОЛУЧЕНИЯ НОНАПЕПТИДЭТИЛАМИДА И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДЛЯ ЕГО ПОЛУЧЕНИЯ | 2010 |
|
RU2444525C2 |
| Способ определения октатиона | 1981 |
|
SU1008656A1 |
| DD 226895 A1, 04.09.1985 | |||
| US 3953416, 27.04.1976 | |||
| US 3947569, 30.03.1976. | |||

