Изобретение относится к катализатору, способу его получения и его использованию в области гидрообработки.
Катализатор гидрообработки углеводородных фракций традиционно предназначен для удаления содержащихся в них сернистых или азотистых соединений с целью, например, доведения качества нефтепродуктов до характеристик, требуемых (по содержанию серы, содержанию ароматических соединений и т.д.) для заданного применения (автомобильное горючее, бензин или дизельное топливо, мазут, реактивное топливо). Речь может идти также о предварительной обработке такого сырья с целью удаления примесей перед осуществлением с ним различных процессов преобразования для изменения физико-химических свойств, таких как, например, процессы конверсии, гидрокрекинга вакуумных дистиллятов, каталитического крекинга, гидроконверсии кубовых остатков атмосферной или вакуумной перегонки. Состав и использование катализаторов гидрообработки особенно хорошо описаны в статье B.S. Clausen, H.T. Topsøe и F.E. Massoth, вышедшей в сборнике Catalysis Science and Technology, volume 11 (1996), Springer-Verlag. После сульфидирования на носителе присутствует много участков поверхности, которые совсем не обладают хорошими эксплуатационными характеристиками для требуемых реакций. Эти участки особенно хорошо описаны в статье Topsøe и соавт., опубликованной в номере 26 Catalysis Review Science and Engineering, 1984, pages 395-420.
Ужесточение стандартов по загрязнению окружающей среды автомобилями в Европейском сообществе (Journal Officiel de l'Union européenne, L76, 22 mars 2003, Directive 2003/70/CE, pages L76/10-L76/19) принудило специалистов по нефтепереработке очень сильно уменьшить содержание серы в дизельном топливе и бензине (не более 10 массовых частей на миллион (млн-1) серы по состоянию на 1 января 2009 года против 50 млн-1 по состоянию на 1 января 2005 года). В то же время, специалисты по нефтепереработке оказываются вынужденными использовать сырье, все более стойкое к процессам гидрообработки, потому что, с одной стороны, сырая нефть становится все более тяжелой и, следовательно, содержит все больше примесей, а с другой стороны, в силу увеличения числа процессов конверсии на нефтеочистительных установках. На практике, в таких процессах образуются смеси, поддающиеся гидроочистке труднее, чем фракции, непосредственно образующиеся при атмосферной перегонке. В качестве примера можно упомянуть фракцию газойля, образующегося при каталитическом крекинге и называемого также LCO (Light Cycle Oil (легкий рецикловый газойль)), в связи с высоким содержанием в ней ароматических соединений. Эти фракции обрабатывают вместе с фракцией газойля, образующейся при атмосферной перегонке; они требуют применения катализаторов, обладающих функцией гидрообессеривания и гидрирования, существенно улучшенной по сравнению с традиционными катализаторами, так чтобы уменьшать содержание ароматических соединений для получения плотности и цетанового числа согласно техническим условиям.
Кроме того, в процессах конверсии, таких как каталитический крекинг или гидрокрекинг, используют катализаторы, имеющие кислотную группу, что делает их особенно чувствительными к присутствию азотистых примесей и, в частности, азотистых соединений основного характера. Таким образом, необходимо использовать катализаторы для предварительной обработки этих исходных смесей с целью удаления таких соединений. Эти катализаторы гидрообработки должны обладать также улучшенной функцией гидрирования в тех случаях, когда первая стадия гидродеазотирования принята в качестве стадии гидрирования ароматических циклов, примыкающих к связи C-N.
Таким образом, из этого следует перспективность нахождения способов получения катализаторов гидрообработки с целью получения новых катализаторов с улучшенными эксплуатационными характеристиками.
Добавка органического соединения к катализаторам гидрообработки для улучшения их активности хорошо известна в настоящее время специалистам в данной области техники. Использование различных групп органических соединений, таких как моно-, ди- или полиспирты, этерифицированные при необходимости, защищено многими патентами (WO 96/41848, WO 01/76741, US 4012340, US 3954673, EP 601722). Катализаторы, модифицированные сложными моноэфирами C2-C14, описаны в заявках EP 466568 и EP 1046424, однако эти модификации по-прежнему не обеспечивают повышения в достаточной степени эксплуатационных характеристик катализатора, чтобы удовлетворять характеристикам по содержанию серы в топливе, которые становятся все более стесняющими специалистов по нефтепереработке.
Для исправления такой ситуации в патенте WO 2006/077326 компании Total предложено использование катализатора, содержащего металлы групп VIB и VIII, тугоплавкий оксид в качестве носителя и органическое соединение, имеющее по меньшей мере 2 функциональные группировки сложного эфира карбоновой кислоты, формулы R1-O-CO-R2-CO-O-R1 или R1-CO-O-R2-O-CO-R1, в которой каждый R1 независимо представляет собой алкил C1-C18, алкенил C2-C18, арил C6-C18, циклоалкил C3-C8, алкиларил или арилалкил C7-C20 или 2 группы R1 совместно образуют двухвалентную группу C2-C18, а R2 представляет собой алкилен C1-C18, арилен C6-C18, циклоалкилен C3-C7 или их комбинацию, причем углеродная цепь углеводородных групп, представляемых R1 и R2, может содержать или быть связанной с одним или несколькими гетероатомами, выбранными из N, S и O, а каждый из R1 и R2 может иметь один или несколько заместителей формулы -C(=O)O-R1 или -O-C(=O)-R1, где R1 имеет указанные ранее значения. В предпочтительном варианте используют диалкил(C1-C4)сукцинат, в частности диметилсукцинат, приведенный в качестве примера. Эти соединения могут быть введены в присутствии растворителя (перечень важных растворителей приведен) или карбоновой кислоты. Среди трех десятков поименно упомянутых кислот приведена уксусная кислота, не упомянутая в числе десятка предпочтительных кислот. Следует отметить, что лимонная кислота является предпочтительной.
Способ получения катализатора, соответственно описанному в патенте WO 2006/077326, включает в себя стадии созревания и термической обработки, которые могут длиться в течение нескольких дней, например от 49 до 115 дней, что очень сильно ограничивает производство этих катализаторов и, следовательно, требует внесения улучшений.
В других патентах предшествующего уровня техники описано повышение активности, связанное с комбинированным использованием органической кислоты или спирта в катализаторе гидрообработки. Так, например, в заявке JP 1995-136523, опубликованной компанией KK Japan Energy, предложено решение, состоящее том, что:
- согласно первому предпочтительному варианту изобретения получают раствор, содержащий носитель катализатора, один или несколько металлов групп VI и VIII периодической системы элементов и органическую кислоту. Согласно второму предпочтительному варианту изобретения этот раствор содержит также предшественник фосфора;
- осуществляют термическую обработку в интервале от 200 до 400°C;
- осуществляют пропитку полученного ранее катализатора органической кислотой или спиртом в соотношении от 0,1 до 2 на моль металлов.
Один из предпочтительных вариантов изобретения включает в себя затем стадию сушки при температуре ниже 200°C, в то время как второй предпочтительный вариант включает в себя финишную термическую обработку при температуре, большей или равной 400°C.
Было установлено, что данные катализаторы не обладают активностью, достаточной для удовлетворения новым экологическим стандартам в случае все более обедненного водородом сырья, находящегося в распоряжении специалистов по нефтепереработке.
Аналогичным образом, в патенте WO 2005/035691 заявлен способ активации, позволяющий схематично уменьшить содержание кристаллической фазы типа CoMoO4, присутствующей в регенерируемых катализаторах, содержащих оксиды металлов групп VIII и VIB, причем способ включает в себя приведение в контакт регенерируемого катализатора с кислотой и органической добавкой. С этой целью в большом числе примеров было реализовано применение в регенерируемом катализаторе комбинации лимонной кислоты (CA) и полиэтиленгликоля (PEG). Настоящее изобретение относится к катализатору и способу его получения, причем катализатор является приемлемым для использования при гидрообработке и имеет улучшенные каталитические качества (в частности, каталитическую активность) по сравнению с катализаторами предшествующего уровня техники. На практике, было выявлено, что использование пары диалкил(C1-C4)сукцинат, в частности диметилсукцинат, и уксусной кислоты в высушенном предшественнике катализатора неожиданным образом ведет к каталитической активности, четко улучшенной по сравнению с любым из соединений пары.
Более точно изобретение относится к катализатору, содержащему аморфный носитель на основе оксида алюминия, фосфор, по меньшей мере один диалкил(C1-C4)сукцинат, уксусную кислоту и функциональную группу с гидрирующей/дегидрирующей способностью, содержащую по меньшей мере один элемент группы VIII и по меньшей мере один элемент группы VIB, при этом спектр комбинационного рассеяния этого катализатора содержит характеристические полосы по меньшей мере одного гетерополианиона Кеггина в области 990 и/или 974 см-1, характеристические полосы упомянутого сукцината и основную характеристическую полосу уксусной кислоты в области 896 см-1. Функциональная группа с гидрирующей/дегидрирующей способностью предпочтительно образована из кобальта и молибдена. Она может содержать также по меньшей мере один элемент группы VIII и по меньшей мере один элемент группы VIB за исключением случая функциональной группы с гидрирующей/дегидрирующей способностью, образованной из кобальта и молибдена.
Полученный катализатор имеет спектр комбинационного рассеяния, сгруппированный по характеристическим полосам:
1) характеристические полосы одного или нескольких гетерополианионов типа Кеггина PXY11O40 x- и/или PXY12O40 x-, где Y означает металл группы VIB, а X означает металл группы VIII.
Согласно Griboval, Blanchard, Payen, Fournier, Dubois, Catalysis Today, 45 (1998), 277, fig. 3e), основные полосы структуры PCoMo11O40 x- в высушенном катализаторе соответствуют 232, 366, 943, 974 см-1, а согласно M.T. Pope в "Heteropoly and Isopoly oxometalates", Springer Verlag, p. 8, эти полосы не являются характеристическими соответственно природе атома X или Y, а соответствуют структуре HPA. Наиболее интенсивная характеристическая полоса этого типа HPA лакунарной структуры Кеггина расположена в области 974 см-1.
Согласно Griboval, Blanchard, Gengembre, Payen, Fournier, Dubois, Bernard, Journal of Catalysis, 188 (1999), 102, fig. 1a), основные полосы PMo12O40 x- расположены в случае HPA в состоянии плотной массы, например, с кобальтом в качестве противоиона, в области 251, 603, 902, 970, 990 см-1. Наиболее интенсивная характеристическая полоса этого HPA Кеггина расположена в области 990 см-1. M.T. Pope в "Heteropoly and Isopoly oxometalates", Springer Verlag, p. 8, сообщает также, что эти полосы не являются характеристическими соответственно природе атома X или Y, а соответствуют HPA полной, лакунарной или замещенной структуры Кеггина;
2) характеристические полосы одного или нескольких используемых диалкилсукцинатов. Спектр комбинационного рассеяния диметилсукцината образует конфигурацию, являющуюся однозначной для молекулы этого соединения. В спектральной области 300-1800 см-1 этот спектр характеризуется серией следующих полос (приведены только наиболее интенсивные полосы в см-1): 391, 853 (наиболее интенсивная полоса), 924, 964, 1739 см-1. Спектр диэтилсукцината содержит в рассмотренной спектральной области следующие основные полосы: 861 (наиболее интенсивная полоса), 1101, 1117 см-1. Соответственно для дибутилсукцината: 843, 1123, 1303, 1439, 1463 см-1 и для диизопропилсукцината: 833, 876, 1149, 1185, 1469 (наиболее интенсивная полоса), 1733 см-1;
3) основные характеристические полосы уксусной кислоты: 448, 623, 896 cм-1. Наиболее интенсивная полоса расположена в области 896 см-1.
Точное положение полос, их форма и относительная интенсивность могут варьировать в некоторой мере в зависимости от условий записи спектра, оставаясь при этом характеристическими для данной молекулы. С другой стороны, спектры комбинационного рассеяния органических соединений хорошо документированы как в базах данных по спектрам комбинационного рассеяния (см., например, Spectral Database for Organic Compounds, http://riodb01.ibase.aist.go.jp/sdbs/cgi-bin/direct_frame_top.cgi), так и поставщиками реактивов (см., например, www.sigmaaldrich.com).
Спектры комбинационного рассеяния получали на спектрометре комбинационного рассеяния, оснащенном ионным аргоновым лазером (514 нм). Пучок лазерного излучения фокусировали на образце посредством микроскопа, оснащенного объективом с 50-кратным увеличением и длинным фокусным расстоянием. Мощность лазерного излучения на уровне образца составляла около 1 мВт. Сигнал комбинационного рассеяния, испускаемый образцом, улавливали тем же объективом, диспергировали посредством сетки при 1800 об/мин и затем улавливали детектором CCD. Полученное разрешение спектра составило около 0,5 см-1. Зарегистрированная область спектра находилась в интервале от 300 до 1800 см-1. Продолжительность записи устанавливали равной 120 с для каждого записанного спектра комбинационного рассеяния.
Используемый диалкилсукцинат предпочтительно представляет собой диметилсукцинат, а катализатор имеет в своем спектре комбинационного рассеяния основные характеристические полосы одного или нескольких гетерополианионов Кеггина в области 990 и/или 974 см-1, диметилсукцината в области 853 см-1 и уксусной кислоты в области 896 см-1.
Катализатор по настоящему изобретению предпочтительно содержит носитель, образованный из оксида алюминия или смеси "диоксид кремния-оксид алюминия".
Катализатор по настоящему изобретению может содержать также бор и/или фтор, и/или кремний.
В тексте заявки описан также способ получения катализатора по настоящему изобретению, который включает в себя по меньшей мере одну стадию пропитки при температуре ниже 180°C высушенного предшественника катализатора, содержащего по меньшей мере фосфор и функциональную группу с гидрирующей/дегидрирующей способностью, а также аморфный носитель, пропитанный раствором, содержащим комбинацию уксусной кислоты и диалкил(C1-C4)сукцината, последующую стадию созревания пропитанного предшественника катализатора и затем стадию сушки при температуре ниже 180°C без последующей стадии прокаливания (термической обработки в атмосфере воздуха); полученный катализатор предпочтительно подвергают сульфидированию.
Функциональная группа с гидрирующей/дегидрирующей способностью содержит по меньшей мере один элемент группы VIII и по меньшей мере один элемент группы VIB. Функциональная группа с гидрирующей/дегидрирующей способностью предпочтительно образована из кобальта и молибдена.
Таким образом, простой и быстрый способ получения с отдельными стадиями, не длящимися более нескольких часов, обеспечивает более хорошую производительность в промышленном масштабе, чем способы предшествующего уровня техники.
Более точно способ получения катализатора гидрообработки по настоящему изобретению включает в себя следующие последовательные стадии, подробно описанные далее:
a) по меньшей мере одну стадию пропитки аморфного носителя на основе оксида алюминия по меньшей мере одним раствором, содержащим элементы функциональной группы с гидрирующей/дегидрирующей способностью и фосфор; полученный продукт можно назвать "предшественник катализатора";
b) стадию сушки при температуре ниже 180°C без последующего прокаливания; полученный продукт можно назвать "высушенный предшественник катализатора";
c) по меньшей мере одну стадию пропитки пропиточным раствором, содержащим по меньшей мере один диалкил(C1-C4)сукцинат, уксусную кислоту и по меньшей мере одно соединение фосфора, если последний не был введен полностью на стадии a); полученный продукт можно назвать "пропитанный высушенный предшественник катализатора";
d) стадию созревания;
e) стадию сушки при температуре ниже 180°C без стадии последующего прокаливания; полученный продукт можно назвать "катализатор".
Продукт, полученный на выходе стадии e), предпочтительно подвергают сульфидированию на стадии f).
Таким образом, соответственно описанному далее, способ по настоящему изобретению осуществляют предпочтительно в вариантах, осуществляемых индивидуально или в комбинации: из оксида алюминия или смеси "диоксид кремния-оксид алюминия" образуют носитель; все количество функциональной группы с гидрирующей способностью вводят на стадии a); все количество фосфора вводят на стадии a); диалкилсукцинат представляет собой диметилсукцинат; стадию c) осуществляют в отсутствие растворителя; стадию d) осуществляют при температуре от 17 до 50°C; стадию e) осуществляют при температуре в интервале от 80 до 160°C.
Наиболее предпочтительно способ по настоящему изобретению включает в себя следующие последовательные стадии:
a) по меньшей мере одну стадию пропитки носителя в сухом состоянии раствором, содержащим все количество элементов функциональной группы с гидрирующей/дегидрирующей способностью и все количество фосфора;
b) стадию сушки при температуре в интервале от 75 до 130°C без последующего прокаливания;
c) по меньшей мере одну стадию пропитки в сухом состоянии пропиточным раствором, содержащим диметилсукцинат и уксусную кислоту;
d) стадию созревания при 17-50°C;
e) стадию сушки предпочтительно в атмосфере азота при температуре в интервале от 80 до 160°C без стадии последующего прокаливания.
Предшественник катализатора, содержащий функциональную группу с гидрирующей/дегидрирующей способностью, аморфный носитель на основе оксида алюминия, а также способ его получения описаны далее.
Предшественник катализатора, полученный на выходе стадии a) способа по настоящему изобретению, может быть получен в большинстве случаев любыми способами, хорошо известными специалистам в данной области техники.
Предшественник катализатора содержит функциональную группу с гидрирующей/дегидрирующей способностью и фосфор и/или бор, и/или фтор в качестве легирующего элемента, а также аморфный носитель. Функциональная группа с гидрирующей/дегидрирующей способностью содержит по меньшей мере один элемент группы VIB и по меньшей мере один элемент группы VIII. Функциональная группа с гидрирующей/дегидрирующей способностью предпочтительно образована из кобальта и молибдена.
Аморфный носитель предшественника катализатора представляет собой продукт на основе оксида алюминия, то есть он содержит больше 50% оксида алюминия - и в общем случае он содержит только оксид алюминия или смесь "диоксид кремния-оксид алюминия", определенную далее, - и при необходимости один или несколько металлов и/или один или несколько легирующих элементов, вводимых не на стадии пропитки (вводимых, например, во время получения - при перемешивании, пластификации и т.д. - носителя или его формования). Носитель получают формованием (например, экструзией) и прокаливанием в общем случае в интервале 300-600°C.
Носитель предпочтительно образован из оксида алюминия и предпочтительно из экструдированного оксида алюминия. Оксид алюминия предпочтительно представляет собой гамма-форму оксида алюминия, а аморфный носитель предпочтительно образован из гамма-формы оксида алюминия.
В другом предпочтительном варианте используют смесь "диоксид кремния-оксид алюминия", содержащую меньшей мере 50% оксида алюминия. Содержание диоксида кремния в носителе составляет не более 50% масс., более часто меньше или равно 45% масс. и предпочтительно меньше или равно 40%.
Источники кремния хорошо известны специалистам в данной области техники. В качестве примеров можно упомянуть кремниевую кислоту, диоксид кремния в виде порошка или в коллоидном состоянии (золь диоксида кремния), тетраэтилортосиликат Si(OEt)4.
Функциональная группа с гидрирующей/дегидрирующей способностью предшественника катализатора содержит по меньшей мере один элемент группы VIB и по меньшей мере один элемент группы VIII. Пара, образованная из кобальта и молибдена, является предпочтительной. Общее содержание элементов с гидрирующей/дегидрирующей способностью предпочтительно составляет больше 6% масс. оксида по отношению к общей массе катализатора. Предпочтительными элементами группы VIB являются молибден и вольфрам и в общем случае молибден. Предпочтительными элементами группы VIII являются неблагородные металлы, в частности кобальт и никель.
Функциональную группу с гидрирующей способностью преимущественно выбирают из группы, в которую входят комбинации элементов "кобальт-молибден", "никель-молибден" или "никель-кобальт-молибден", или "никель-молибден-вольфрам".
В случае, когда требуется значительная активность при гидрообессеривании или при гидродеазотировании и гидрировании ароматических соединений, функциональная группа с гидрирующей/дегидрирующей способностью предпочтительно содержит ассоциацию никеля и молибдена; ассоциация никеля и вольфрама в присутствии молибдена также может быть предпочтительной. В случае сырья типа вакуумных дистиллятов или более тяжелого сырья преимущественно могут быть использованы комбинации типа "кобальт-никель-молибден".
Предшественники молибдена, которые могут быть использованы, также хорошо известны специалистам в данной области техники. Например, в качестве источников молибдена можно использовать оксиды и гидроксиды, молибденовые кислоты и их соли, предпочтительно соли аммония, такие как молибдат аммония, гептамолибдат аммония, фосфорномолибденовую кислоту (H3PMo12O40) и ее соли и при необходимости кремниймолибденовую кислоту (H4SiMo12O40) и ее соли. Источниками молибдена могут быть также любые гетерополисоединения, например, типа Кеггина, лакунарной структуры Кеггина, замещенной структуры Кеггина, Доусона, Андерсона, Страндберга. Предпочтительно используют триоксид молибдена и гетерополисоединения (гетерополианионы) типа Страндберга, Кеггина, лакунарной структуры Кеггина или замещенной структуры Кеггина.
Предшественники вольфрама, которые могут быть использованы, также хорошо известны специалистам в данной области техники. Например, в качестве источников вольфрама можно использовать оксиды и гидроксиды, вольфрамовые кислоты и их соли, предпочтительно соли аммония, такие как вольфрамат аммония, метавольфрамат аммония, фосфорновольфрамовую кислоту и ее соли и при необходимости кремнийвольфрамовую кислоту (H4SiW12O40) и ее соли. Источниками вольфрама могут быть также любые гетерополисоединения, например, типа Кеггина, лакунарной структуры Кеггина, замещенной структуры Кеггина, Доусона. Предпочтительно используют оксиды и соли аммония, такие как метавольфрамат аммония, или гетерополианионы типа Кеггина, лакунарной структуры Кеггина, замещенной структуры Кеггина.
Количество одного или нескольких предшественников одного или нескольких элементов группы VIB предпочтительно находится в интервале от 5 до 40% масс., предпочтительно в интервале от 8 до 35% масс. и более предпочтительно в интервале от 10 до 30% масс. оксидов группы VIB по отношению к общей массе предшественника катализатора.
Предшественники одного или нескольких элементов группы VIII, которые могут быть использованы, предпочтительно выбраны из оксидов, гидроксидов, гидроксикарбонатов, карбонатов и нитратов, например, предпочтительно используют гидроксикарбонат никеля, карбонат кобальта или гидроксид кобальта.
Количество одного или нескольких предшественников одного или нескольких элементов группы VIII предпочтительно находится в интервале от 1 до 10% масс., предпочтительно в интервале от 1,5 до 9% масс. и более предпочтительно в интервале от 2 до 8% масс. оксидов группы VIII по отношению к общей массе предшественника катализатора.
Функциональная группа с гидрирующей/дегидрирующей способностью предшественника катализатора может быть введена в катализатор на разных стадиях получения и разными способами. Функциональную группу с гидрирующей/дегидрирующей способностью всегда вводят по меньшей мере частично и предпочтительно полностью при пропитке сформованного носителя. Она также может быть введена частично во время формования аморфного носителя.
В случае, когда функциональную группу с гидрирующей/дегидрирующей способностью вводят частично во время формования аморфного носителя, она может быть введена частично (например, до 10% одного или нескольких элементов группы VIB может быть введено при перемешивании) только во время перемешивания с гелем оксида алюминия, выбранного в качестве матрицы, причем остаток одного или нескольких элементов с гидрирующей способностью вводят в последующее время. В случае, когда функциональную группу с гидрирующей/дегидрирующей способностью вводят частично во время перемешивания, содержание одного или нескольких элементов группы VIB, вводимых в ходе этой стадии, предпочтительно составляет меньше 5% от общего количества одного или нескольких элементов группы VIB, вводимых в конечный катализатор. По меньшей мере один элемент (или все элементы) группы VIB предпочтительно вводят в то же самое время, что и по меньшей мере один элемент (или все элементы) группы VIII, независимо от способа введения. Эти способы и количества, относящиеся к введению элементов, используют предпочтительно в случае, когда функциональная группа с гидрирующей/дегидрирующей способностью образована из Co и Mo.
В случае, когда функциональную группу с гидрирующей/дегидрирующей способностью вводят по меньшей мере частично и предпочтительно полностью после формования аморфного носителя, введение функциональной группы с гидрирующей/дегидрирующей способностью в аморфный носитель может быть преимущественно осуществлено посредством одной или нескольких пропиток сформованного и прокаленного носителя избытком раствора или предпочтительно посредством одной или нескольких пропиток в сухом состоянии и предпочтительно посредством одной пропитки сформованного и прокаленного носителя в сухом состоянии растворами, содержащими соли-предшественники металлов. Более предпочтительно функциональную группу с гидрирующей/дегидрирующей способностью вводят полностью после формования аморфного носителя посредством пропитки носителя в сухом состоянии пропиточным раствором, содержащим соли-предшественники металлов. Введение функциональной группы с гидрирующей/дегидрирующей способностью также может быть преимущественно осуществлено посредством одной или нескольких пропиток сформованного и прокаленного носителя раствором одного или нескольких предшественников активной фазы. В случае, когда элементы вводят посредством нескольких пропиток соответствующими солями предшественников, промежуточную стадию сушки катализатора в общем случае осуществляют при температуре в интервале от 50 до 180°C, более предпочтительно в интервале от 60 до 150°C и наиболее предпочтительно в интервале от 75 до 130°C.
В катализатор вводят также фосфор. В катализатор также может быть введена другая легирующая добавка, выбранная из бора и фтора, используемых по отдельности или в смеси. Легирующая добавка представляет собой прибавляемый элемент, который сам по себе не имеет каких-либо каталитических свойств, но увеличивает каталитическую активность одного или нескольких металлов.
Легирующая добавка может быть преимущественно введена отдельно или в смеси по меньшей мере с одним из элементов функциональной группы с гидрирующей/дегидрирующей способностью.
Она также может быть введена во время синтеза носителя.
Она также может быть введена непосредственно перед или после пластификации выбранной матрицы, представляющей собой, например и предпочтительно, оксигидроксид алюминия (бемит), являющийся предшественником оксида алюминия.
Легирующая добавка также может быть преимущественно введена полностью или частично в смеси с одним или несколькими предшественниками функциональной группы с гидрирующей/дегидрирующей способностью в сформованный аморфный носитель, предпочтительно из оксида алюминия или смеси "диоксид кремния-оксид алюминия" в экструдированном виде, посредством пропитки аморфного носителя в сухом состоянии раствором, содержащим соли-предшественники металлов и один или несколько предшественников одной или нескольких легирующих добавок.
Источник бора может представлять собой борную кислоту, предпочтительно ортоборную кислоту H3BO3, диборат или пентаборат аммония, оксид бора, сложные борные эфиры. Бор может быть введен, например, посредством раствора борной кислоты в смеси "вода/спирт" или также в смеси "вода/этаноламин".
Предпочтительный источник фосфора представляет собой ортофосфорную кислоту H3PO4, при этом ее сложные эфиры и соли, такие как фосфаты аммония, являются также приемлемыми. Фосфор также может быть введен в то же самое время, что и один или несколько элементов группы VIB в виде гетерополианионов Кеггина, лакунарной структуры Кеггина, замещенной структуры Кеггина или типа Страндберга.
Предшественники фтора, которые могут быть использованы, хорошо известны специалистам в данной области техники. Например, фторидные анионы могут быть введены в виде фтороводородной кислоты или ее солей. Эти соли образованы щелочными металлами, аммонием или органическими соединениями. В последнем случае соль предпочтительно образуется в реакционной смеси при реакции между органическим соединением и фтороводородной кислотой. Фтор может быть введен, например, посредством пропитки водным раствором фтороводородной кислоты или фторида аммония, или также дифторида аммония.
Легирующую добавку предпочтительно вводят в предшественник катализатора в виде оксида легирующего элемента в количестве по отношению к катализатору:
- в интервале от 0 до 40%, предпочтительно в интервале от 0 до 30%, более предпочтительно в интервале от 0 до 20%, предпочтительно в интервале от 0 и 15% и наиболее предпочтительно в интервале от 0 до 10% в случае, когда легирующая добавка представляет собой бор; в случае, когда содержится бор, его минимальное количество предпочтительно составляет 0,1% масс.;
- в интервале от 0,1 до 20%, предпочтительно в интервале от 0,1 до 15% и наиболее предпочтительно в интервале от 0,1 до 10% масс., в случае, когда легирующая добавка представляет собой фосфор;
- в интервале от 0 до 20%, предпочтительно в интервале от 0 до 15% и наиболее предпочтительно в интервале от 0 до 10% в случае, когда легирующая добавка представляет собой фтор; в случае, когда содержится фтор, его минимальное количество предпочтительно составляет 0,1% масс.
Фосфор содержится во всех вариантах. Его вводят по меньшей мере частично (и предпочтительно полностью) посредством пропитки предшественника катализатора на стадии a) и при необходимости в высушенный предшественник катализатора на стадии c). Аналогичным образом, данное положение предпочтительно относится к другим легирующим добавкам. Тем не менее, как было отмечено ранее, легирующие добавки могут быть введены частично или полностью (за исключением фосфора) во время получения носителя (включая формование).
Введение функциональной группы с гидрирующей/дегидрирующей способностью и при необходимости легирующей добавки в носитель или на носитель, прошедший формование и прокаливание, затем преимущественно продолжают на стадии сушки b), в ходе которой растворитель солей-предшественников металлов одного или нескольких оксидов одного или нескольких металлов (в общем случае растворитель представляет собой воду) удаляют при температуре в интервале от 50 до 180°C, более предпочтительно в интервале от 60 до 150°C или также в интервале от 65 до 145°C и наиболее предпочтительно в интервале от 70 до 140°C, или также в интервале от 75 до 130°C. После стадии сушки "высушенного предшественника катализатора", полученного таким образом, никогда не следует стадия прокаливания в атмосфере воздуха при температуре выше 200°C. Преимущественно действуют в этих диапазонах температур до температуры не более 150°C и без последующего прокаливания при температуре выше 180°C.
На стадии a) способа по настоящему изобретению "предшественник катализатора" предпочтительно получают посредством пропитки в сухом состоянии раствором, содержащим один или несколько предшественников функциональной группы с гидрирующей/дегидрирующей способностью и фосфор, сформованного и прокаленного аморфного носителя на основе оксида алюминия с последующей сушкой при температуре ниже 180°C, предпочтительно в интервале от 50 до 180°C, более предпочтительно в интервале от 60 до 150°C и наиболее предпочтительно в интервале от 75 до 130°C.
Таким образом, на выходе стадии b) получают "высушенный предшественник катализатора".
На стадии a) способа по настоящему изобретению возможно получать пропиточный раствор, содержащий по меньшей мере одну легирующую добавку, выбранную из бора и фтора, используемых по отдельности или в смеси.
Еще более предпочтительно "предшественник катализатора" на стадии a) способа по настоящему изобретению получают с пропиточным раствором, содержащим по меньшей мере один предшественник каждого элемента функциональной группы с гидрирующей/дегидрирующей способностью в присутствии предшественника фосфора, причем аморфный носитель образуют из оксида алюминия или смеси "диоксид кремния-оксид алюминия".
Соответственно стадии c) способа по настоящему изобретению высушенный предшественник катализатора пропитывают пропиточным раствором, содержащим по меньшей мере один диалкил(C1-C4)сукцинат (предпочтительно диметилсукцинат) и уксусную кислоту.
Упомянутые соединения преимущественно вводят в пропиточный раствор стадии c) способа по настоящему изобретению в количестве, соответствующем:
- молярному отношению диалкилсукцината (например, диметилсукцината) к одному или нескольким элементам группы VIB, вводимым в предшественник катализатора, в интервале от 0,15 до 2 моль/моль, предпочтительно в интервале от 0,3 до 1,8 моль/моль, более предпочтительно в интервале от 0,5 до 1,5 моль/моль и наиболее предпочтительно в интервале от 0,8 до 1,2 моль/моль;
- молярному отношению уксусной кислоты к одному или нескольким элементам группы VIB, вводимым в предшественник катализатора, в интервале от 0,1 до 5 моль/моль, предпочтительно в интервале от 0,5 до 4 моль/моль, более предпочтительно в интервале от 1,3 до 3 моль/моль и наиболее предпочтительно в интервале от 1,5 до 2,5 моль/моль. Это указание предпочтительно относится к случаю, когда функциональная группа с гидрирующей/дегидрирующей способностью образована из Co и Mo.
Соответственно стадии c) способа по настоящему изобретению комбинацию диалкилсукцината и уксусной кислоты вводят в высушенный предшественник катализатора посредством по меньшей мере одной стадии пропитки и предпочтительно посредством только стадии пропитки пропиточным раствором данного высушенного предшественника катализатора.
Упомянутая комбинация преимущественно может быть нанесена на одной или нескольких стадиях пропиткой суспензией или пропиткой избытком, или пропиткой в сухом состоянии, или любым другим способом, известным специалистам в данной области техники.
В предпочтительном варианте осуществления стадии c) способа получения по настоящему изобретению стадия c) представляет собой только стадию пропитки в сухом состоянии.
Соответственно стадии c) способа по настоящему изобретению пропиточный раствор стадии c) содержит по меньшей мере одну комбинацию диалкил(C1-C4)сукцината (в частности, диметилсукцината) и уксусной кислоты.
Пропиточный раствор, используемый на стадии c) способа по настоящему изобретению, может быть дополнен любым апротонным растворителем, известным специалистам в данной области техники и содержащим, в частности, толуол, ксилол.
Пропиточный раствор, используемый на стадии c) способа по настоящему изобретению, может быть дополнен любым полярным растворителем, известным специалистам в данной области техники. Используемый полярный растворитель предпочтительно выбирают из группы, в которую входят метанол, этанол, вода, фенол, циклогексанол, используемые по отдельности или в смеси. Полярный растворитель, используемый на стадии c) способа по настоящему изобретению, также может быть предпочтительно выбран из группы, в которую входят пропиленкарбонат, ДМСО (диметилсульфоксид) или сульфолан, используемые по отдельности или в смеси. Предпочтительно используют полярный протонный растворитель. Перечень традиционных полярных растворителей, а также их диэлектрические постоянные могут быть найдены в книге "Solvents and Solvent Effects in Organic Chemistry", C. Reichardt, Wiley-VCH, 3eme édition, 2003, pages 472-474. Используемый растворитель более предпочтительно представляет собой этанол.
Предпочтительно растворитель в пропиточном растворе, используемом на стадии c) способа по настоящему изобретению, отсутствует, что облегчает реализацию в промышленном масштабе. Предпочтительно он содержит только диалкилсукцинат и уксусную кислоту.
Используемый диалкилсукцинат предпочтительно выбирают из группы, в которую входят диметилсукцинат, диэтилсукцинат, дипропилсукцинат, диизопропилсукцинат и дибутилсукцинат. Используемый диалкил(C1-C4)сукцинат предпочтительно представляет собой диметилсукцинат или диэтилсукцинат. Используемый диалкил(C1-C4)сукцинат более предпочтительно представляет собой диметилсукцинат. Используют по меньшей мере один диалкил(C1-C4)сукцинат, предпочтительно только один диалкилсукцинат и предпочтительно диметилсукцинат.
Соответственно стадии d) способа получения по настоящему изобретению пропитанный предшественник катализатора, выходящий со стадии c), направляют на стадию созревания. Созревание предпочтительно осуществляют при атмосферном давлении и при температуре в интервале от 17 до 50°C, в общем случае продолжительность созревания в интервале от десяти минут до сорока восьми часов и предпочтительно в интервале от тридцати минут до пяти часов является достаточной. Более долгая продолжительность не исключается. Простое средство регулировать продолжительность созревания состоит в контроле образования гетерополианионов Кеггина спектроскопией комбинационного рассеяния в пропитанном высушенном предшественнике катализатора, выходящем со стадии c) способа по настоящему изобретению. Для увеличения производительности без изменения количества преобразованных гетерополианионов продолжительность созревания более предпочтительно находится в интервале от тридцати минут до четырех часов. Еще больше предпочтительно продолжительность созревания находится в интервале от тридцати минут до трех часов.
Соответственно стадии e) способа получения по настоящему изобретению предшественник катализатора, выходящий со стадии d), подвергают сушке при температуре ниже 180°C без стадии последующего прокаливания при температуре выше 200°C.
Цель этой стадии состоит в получении катализатора, приемлемого для транспортировки, хранения и осуществления манипуляций, в частности для загрузки в установку гидрообработки. Преимущественно речь идет, в зависимости от выбранного варианта осуществления настоящего изобретения, об удалении всего количества или части необходимого растворителя, обеспечившего введение комбинации диалкил(C1-C4)сукцината (в частности, диметилсукцината) и уксусной кислоты. Во всех случаях, в частности в случае, когда используют только комбинацию диалкил(C1-C4)сукцината (в частности диметилсукцината) и уксусной кислоты, речь идет о переводе катализатора в сухое состояние, чтобы избежать склеивания экструдированных элементов между собой во время транспортировки, хранения, манипуляций или загрузки.
Стадию сушки e) способа по настоящему изобретению предпочтительно осуществляют по любой технологии, известной специалистам в данной области техники. Эту стадию предпочтительно осуществляют при атмосферном или пониженном давлении. Предпочтительно сушку осуществляют при атмосферном давлении.
Стадию e) предпочтительно осуществляют при температуре в интервале от 50°C до 180°C (не выше), предпочтительно в интервале от 60 до 170°C и более предпочтительно в интервале от 80 до 160°C. Преимущественно действуют в этих диапазонах температур до температуры не более 160°C (предпочтительный интервал составляет 80-180°C) и без последующего прокаливания при температуре выше 180°C.
Эту стадию предпочтительно осуществляют в проницаемом слое, используя горячий воздух или любой другой газ. В случае, когда сушку осуществляют в неподвижном слое, используемый газ предпочтительно представляет собой воздух или инертный газ, такой как аргон или азот. Более предпочтительно сушку осуществляют в проницаемом слое в атмосфере азота.
Продолжительность этой стадии предпочтительно находится в интервале от 30 минут до 4 часов и предпочтительно в интервале от 1 часа до 3 часов.
На выходе стадии e) способа по настоящему изобретению получают высушенный катализатор, который не направляют на какую-либо дальнейшую стадию прокаливания в атмосфере воздуха, например, при температуре выше 200°C.
Катализатор, полученный на выходе стадии d) или стадии e), имеет спектр комбинационного рассеяния, содержащий наиболее интенсивные полосы в области 990 и 974 см-1 (гетерополианионы типа Кеггина), полосы, соответствующие сукцинату (в случае диметилсукцината наиболее интенсивная полоса расположена в области 853 см-1), и характеристические полосы уксусной кислоты, наиболее интенсивная из которых находится в области 896 см-1.
Перед использованием высушенный или прокаленный катализатор предпочтительно преобразуют в сульфидированный катализатор для формирования его активного состояния. Эту стадию активации или сульфидирования осуществляют способами, хорошо известными специалистам в данной области техники, и преимущественно в атмосфере, редуцирующей серу, в присутствии водорода и сероводорода.
После стадии e) способа по настоящему изобретению высушенный катализатор, полученный таким образом, преимущественно подвергают сульфидированию на стадии f) без стадии промежуточного прокаливания.
Высушенный катализатор предпочтительно сульфидируют вне места применения или по месту применения. Сульфидирующие агенты представляют собой газообразный H2S или любые другие соединения, содержащие серу и используемые для активации углеводородного сырья с целью сульфидирования катализатора. Соединения, содержащие серу, предпочтительно выбирают из алкилдисульфидов, таких как, например, диметилдисульфид (DMDS), алкилсульфидов, таких как, например, диметилсульфид, н-бутилмеркаптан, полисульфидных соединений типа трет-нонилполисульфида, такого как, например, TPS-37 или TPS-54, реализуемые компанией ARKEMA, или любых других соединений, известных специалистам в данной области техники и позволяющих получить хорошее качество сульфидирования катализатора. Катализатор предпочтительно сульфидируют по месту применения в присутствии сульфидирующего агента и исходной углеводородной смеси. Более предпочтительно катализатор сульфидируют по месту применения посредством исходной углеводородной смеси с добавкой диметилдисульфида.
В заключение, другой целью настоящего изобретения является использование катализатора по настоящему изобретению в процессах гидрообработки, предпочтительно в процессах гидрообессеривания, гидродеазотирования, гидродеметаллизации, гидрирования ароматических соединений и гидроконверсии нефтяных фракций.
Высушенные катализаторы, полученные способом по настоящему изобретению и предпочтительно предварительно обработанные на стадии сульфидирования f), преимущественно используют для реакций гидрообработки углеводородного сырья, такого как нефтяные фракции, фракции, получаемые из угля, или углеводороды, получаемые из природного газа, и более предпочтительно для реакций гидрирования, гидродеазотирования, гидродеароматизации, гидрообессеривания, гидродеметаллизации или гидроконверсии углеводородного сырья.
В этих случаях использования катализаторы, полученные способом по настоящему изобретению и предпочтительно предварительно обработанные на стадии сульфидирования f), имеют активность, улучшенную по сравнению с катализаторами предшествующего уровня техники. Данные катализаторы также предпочтительно могут быть использованы при предварительной обработке сырья для каталитического крекинга или гидрообессеривания кубовых остатков или для глубокого гидрообессеривания дизельного топлива (ULSD Ultra Low Sulfur Diesel (сверхмалосернистое дизельное топливо)).
Исходные смеси, используемые в процессах гидрообработки, представляют собой, например, бензиновые фракции, фракции дизельного топлива, вакуумные газойли, кубовые остатки атмосферной перегонки, кубовые остатки вакуумной перегонки, атмосферные дистилляты, вакуумные дистилляты, тяжелое топливо, масла, воски и парафины, масла, бывшие в употреблении, деасфальтированные кубовые остатки или сырая нефть, фракции, образующиеся при процессах термической или каталитической конверсии, используемые по отдельности или в смеси. Обрабатываемые смеси, в частности смеси, упомянутые ранее, содержат в общем случае гетероатомы, такие как атомы серы, кислорода и азота, а в случае тяжелых смесей они очень часто содержат также и металлы.
Рабочие условия, устанавливаемые в процессах, в которых используются реакции гидрообработки углеводородных смесей, описанных ранее, в общем случае следующие: температура преимущественно находится в интервале от 180 до 450°C и предпочтительно в интервале от 250 до 440°C, давление преимущественно находится в интервале от 0,5 до 30 МПа и предпочтительно в интервале от 1 до 18 МПа, часовая объемная скорость преимущественно находится в интервале от 0,1 до 20 ч-1 и предпочтительно в интервале от 0,2 до 5 ч-1, а соотношение "водород/исходная смесь", выраженное в пересчете на объем водорода, измеренный в нормальных условиях по температуре и давлению, к объему жидкой исходной смеси находится предпочтительно в интервале от 50 до 2000 л/л.
В приведенных далее примерах показано значительное повышение активности катализаторов, полученных способом по настоящему изобретению, по сравнению с катализаторами предшествующего уровня техники и описаны подробности изобретения, однако без ограничения объема патентной охраны.
Пример 1. Получение прокаленного катализатора (NiMoP/оксид алюминия) C1A, высушенного катализатора (NiMoP/оксид алюминия) с добавкой лимонной кислоты (CA) и полиэтиленгликоля (PEG) C1E (C1A и C1E не соответствуют изобретению), а также высушенного катализатора (NiMoP/оксид алюминия) с добавкой уксусной кислоты и диметилсукцината C1B и C1F (соответствуют изобретению)
Использовали матрицу, состоящую из ультрадисперсного слоистого бемита или геля оксида алюминия, реализуемого компанией Condea Chemie GmbH. Этот гель смешали с водным раствором, содержавшим азотную кислоту с концентрацией 66% (7% масс. кислоты на грамм сухого геля), и затем перемешивали в течение 15 минут. Полученную после перемешивания пасту пропускали через фильеру с круглыми отверстиями диаметром 1,6 мм. Затем экструдированные элементы сушили в течение ночи при 120°C и далее прокаливали при 600°C в течение 2 часов в атмосфере влажного воздуха, содержавшей 50 г воды на кг сухого воздуха. Таким образом, получали экструдированные элементы носителя, имеющие удельную поверхность 300 м2/г. Рентгеноструктурным анализом обнаружено, что носитель состоит только из гамма-формы кубического оксида алюминия со слабой кристалличностью.
К носителю из оксида алюминия, описанному ранее и находящемуся в "экструдированном" виде, прибавляют никель, молибден и фосфор. Пропиточный раствор готовят растворением при нагревании оксида молибдена и гидроксикарбоната никеля в водном растворе фосфорной кислоты с целью получения состава с соотношением приблизительно 4/22,5/4, выраженным в пересчете на содержание в % масс. оксидов никеля, молибдена и фосфорного ангидрида по отношению к количеству сухого вещества конечного катализатора. После пропитки в сухом состоянии экструдированные элементы оставляют созревать в атмосфере, насыщенной водой, в течение 12 ч и затем сушат в течение ночи при 90°C. Полученный таким образом высушенный предшественник катализатора обозначен как C1. Прокаливание C1 при 450°C в течение 2 часов дает прокаленный катализатор C1A. Катализаторы C1 и C1A имеют следующий конечный состав, выраженный в пересчете на оксиды: MoO3=22,4±0,2% масс., NiO=4,1±0,1% масс. и P2O5=4,0±0,1% масс.
Катализатор C1E получают пропиткой высушенного предшественника катализатора C1 раствором, содержащим лимонную кислоту (CA) и полиэтиленгликоль (PEG) в виде раствора в этаноле, так чтобы получить объем пропиточного раствора, равный объему пор высушенного предшественника катализатора C1. Принятое содержание лимонной кислоты (CA) и полиэтиленгликоля (PEG) составляет по 10% масс.
Катализатор C1B, соответствующий изобретению, получают из высушенного предшественника катализатора C1 посредством пропитки в сухом состоянии раствором, содержащим смесь диметилсукцината и уксусной кислоты в этаноле, для получения в конечном катализаторе также 10% масс. уксусной кислоты и 10% масс. диметилсукцината.
Катализатор C1F, соответствующий изобретению, получают аналогично, но в отсутствие этанола. Конечное содержание уксусной кислоты принимают равным 13% масс., а конечное содержание диметилсукцината - 20% масс.
Далее катализаторы направляют на стадию созревания в течение 3 ч при 20°C в атмосфере воздуха и затем на термическую обработку при 110°C в течение 3 ч в печи с проницаемым слоем.
Пример 2. Оценка катализатора NiMoP/оксид алюминия C1A (не соответствующего изобретению), катализатора C1E (не соответствующего изобретению), катализатора C1B (соответствующего изобретению) при гидрообработке дистиллятного газойля
Сульфидирование катализатора (30 см3 катализатора в виде экструдированных элементов, смешанных с 10 см3 SiC крупностью 0,8 мм) осуществляют при 50 бар, VVH=2 ч-1, с соотношением (объемной подачей) H2/HC на входе = 400 нл/л. Исходную смесь, используемую при сульфидировании (газойль с добавкой 2% DMDS Evolution компании Arkema), вводят в реактор с атмосферой H2 по достижении 150°C. После выдерживания в течение часа при 150°C температуру повышают со скоростью 25°C/ч до 220°C и затем со скоростью 12°C/ч до достижения плато 350°C, выдерживаемого в течение 12 часов.
После сульфидирования температуру понижают до 330°C и вводят испытуемую исходную смесь. Испытание по катализу осуществляют при общем давлении 50 бар, сбросе водорода, VVH=2 ч-1, с соотношением H2/HC на входе 400 нл/л (подача H2=24 нл·ч-1, подача исходной смеси = 60 см3·ч-1) при 330, 340 и 350°C.
Для того чтобы иметь возможность оценивать эксплуатационные характеристики катализаторов при HDS и для освобождения от присутствия H2S в приемниках, приемный сосуд продувают азотом из расчета 10 л·ч-1.
Используемый в данном случае газойль получен из неочищенной тяжелой арабской нефти. Он содержит 0,89% масс. серы, 100 млн-1 масс. азота, его TMP [(T5+2T50+4T95)/7] равна 324°C, а плотность составляет 0,848 г/см3.
Активность при HDS определяют исходя из конверсии при HDS по формуле:
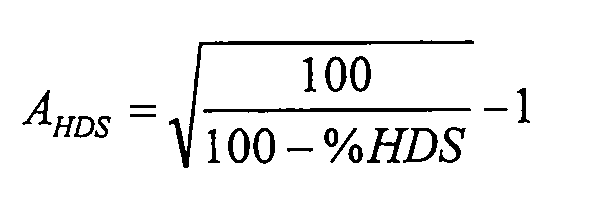
где конверсия при HDS (%HDS) определена как:

В ходе испытания плотность уходящих жидких потоков, полученных при каждой температуре, измеряли при 15°C. Изменение плотности показано на фигуре 1. Данный график позволяет определить температуру, при которой необходимо действовать с целью получения заданной плотности, причем специалисты по нефтепереработке очень заинтересованы в использовании катализатора, который обеспечивал бы этот показатель при наиболее низкой температуре. На фигуре 1 показано, что катализатор по настоящему изобретению позволяет при равной плотности уходящего жидкого потока уменьшить рабочую температуру приблизительно на 15°C по сравнению с катализатором C1A предшествующего уровня техники. Результаты, полученные при гидрообессеривании в ходе этого испытания, представлены в приведенной далее таблице.
Полученные результаты показывают, что при гидрообработке газойля, как при гидрообессеривании, так и при гидродеароматизации, представляет интерес (что выражается в изменении плотности уходящих жидких потоков) введение в катализатор добавки диметилсукцината в комбинации с уксусной кислотой способом по настоящему изобретению. На практике, как показано в предыдущей таблице, полученная активность при HDS равна 126 при высокой температуре (соответствует уровню ULSD, то есть содержание серы близко к 10 млн-1 масс.) для катализатора по настоящему изобретению, тогда как для прокаленного катализатора значение равно 100 (сравнительное значение), а для катализатора предшествующего уровня техники C1E значение равно 106.
Пример 3. Оценка катализатора NiMoP/оксид алюминия C1A (не соответствующего изобретению) и катализатора C1F (соответствующего изобретению) при гидродеазотировании вакуумного дистиллята (DSV) для применения типа предварительной обработки при гидрокрекинге
Основные характеристики используемого вакуумного дистиллята приведены далее:
плотность при 20°C: 0,9365;
сера: 2,92% масс.;
общий азот: 1400 млн-1 масс.;
модельная перегонка:
PI: 361°C;
10%: 430°C;
50%: 492°C;
90%: 567°C;
PF: 598°C.
Испытание проводят в экспериментальном изотермическом реакторе с неподвижным проницаемым слоем, причем жидкие потоки проходят снизу вверх. После сульфидирования по месту применения при 350°C на установке, работающей под давлением, посредством дистиллятного газойля прямой перегонки, к которому прибавлено 2% масс. диметилдисульфида, испытание по гидрообработке проводили в следующих рабочих условиях:
общее давление: 12 МПа;
объем катализатора: 40 см3;
температура: 380°C;
подача водорода: 40 л/ч;
подача исходной смеси: 40 см3/ч.
Эксплуатационные каталитические характеристики испытуемых катализаторов представлены в приведенной далее таблице. Характеристики выражены в относительной активности, при этом активность катализатора C1A принята равной 100, а соотношение активностей предположительно составляет около 1,5. Зависимость, связывающая активность и конверсию при гидрообессеривании (%HDS), выражается следующей формулой:

Такая же зависимость применима для гидродеазотирования (%HDN и AHDN).
Кроме того, оценивают также общую конверсию фракции, имеющей температуру кипения меньше 380°C и полученной с каждым катализатором. Ее выражают, исходя из результатов модельной перегонки (методика ASTM D86), по зависимости:
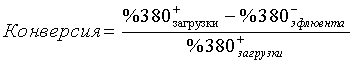
В приведенной далее таблице представлены результаты испытаний, полученные для трех катализаторов.
(%)
Полученные результаты по катализу показывают, что в случае применения типа предварительной обработки при гидрокрекинге катализатор по настоящему изобретению более эффективен, чем прокаленный катализатор NiMoP, в той мере, в какой катализатор по настоящему изобретению обеспечивает преимущество по гидрообессериванию, а также по гидродеазотированию и, что более неожиданно, по конверсии.
Пример 4. Получение прокаленного катализатора NiMoP на основе смеси "диоксид кремния-оксид алюминия" C2A (не соответствующего изобретению) и высушенного катализатора NiMoP на основе смеси "диоксид кремния-оксид алюминия" с добавкой уксусной кислоты и диметилсукцината C2B (соответствующего изобретению)
Два катализатора NiMoP получали с составом 3,6/18/1,6 на основе смеси "диоксид кремния-оксид алюминия" типа SIRALOX, реализуемой компанией SASOL, с содержанием диоксида кремния 25%. Высушенный предшественник катализатора типа NiMoP/"диоксид кремния-оксид алюминия" получали исходя из предшественников MoO3 и Ni(OH)2, солюбилизированных посредством H3PO4, и синтезировали при нагревании с обратным холодильником в течение 2 ч при 90°C. Затем прозрачный раствор сгущали выпариванием воды для достижения пропиточного объема и далее им пропитывали при комнатной температуре смесь "диоксид кремния-оксид алюминия". Экструдированные элементы носителя, пропитанные таким образом, направляли на стадию созревания в закрытой камере, насыщенной водой, выдерживая в течение ночи, и далее сушили в сушильном шкафу при 120°C в течение 24 ч. Затем этот предшественник катализатора делили на две части:
первую часть прокаливали при 450°C в течение 2 ч в атмосфере воздуха в неподвижном проницаемом слое для получения катализатора C2A (не соответствующего изобретению);
вторую часть использовали соответственно методике по настоящему изобретению, пропитывая в капельном режиме раствором, содержавшим уксусную кислоту и диметилсукцинат с молярным соотношением "диметилсукцинат/уксусная кислота" 0,58, до появления выступающей влаги, указывающего на то, что все поры заполнены. Затем катализатор оставляли созревать в течение 3 ч и подвергали термической обработке при 125°C в течение 2 ч для получения катализатора C2B, соответствующего изобретению.
Пример 5. Оценка при гидрировании толуола в присутствии анилина и при мягком гидрокрекинге DSV прокаленного катализатора NiMoP на основе смеси "диоксид кремния-оксид алюминия" C2A (не соответствующего изобретению) и высушенного катализатора NiMoP на основе смеси "диоксид кремния-оксид алюминия" с добавкой уксусной кислоты и диметилсукцината C2B (соответствующего изобретению)
Испытание по гидрированию толуола в присутствии анилина (испытание "HTA") имеет целью оценку гидрирующей активности (HYD) нанесенных или цельнотельных сульфидированных катализаторов в присутствии H2S и под давлением водорода. Изомеризация и крекинг, которые характеризуют кислотную функцию катализатора, нанесенного на смесь "диоксид кремния-оксид алюминия", ингибируются в присутствии NH3 (вследствие разложения анилина). Таким образом, анилин и/или NH3 будут реагировать по принципу реакции нейтрализации кислоты основанием в местах нахождения кислотных групп на носителе. Все испытания осуществляли на установке, содержавшей несколько параллельно соединенных микрореакторов. Во время испытания "HTA" используют одну и ту же исходную смесь для сульфидирования катализатора и собственно на стадии испытания по катализу. Перед загрузкой катализатор кондиционируют: его измельчают и сортируют, так чтобы крупность образца находилась в интервале от 2 до 4 мм. В реакторы загружают по 4 см3 измельченного катализатора, смешанного с 4 см3 карборунда (SiC, 500 мкм).
В этом испытании используют следующую исходную смесь:
толуол: 20% масс.;
циклогексан: 73,62% масс.;
DMDS (диметилдисульфид): 5,88% масс. (3,8% масс. S);
анилин: 0,5% масс. (750 млн-1 N).
В реактор загружают катализатор в его сухой, неактивной форме. Активацию (сульфидирование) осуществляют на установке с одной и той же исходной смесью. Именно H2S, образующийся вследствие разложения DMDS, сульфидирует оксидную фазу. Количество анилина, присутствующего в исходной смеси, выбирали для получения, после разложения, содержания приблизительно 750 млн-1 NH3.
Во время испытания по гидрированию толуола приняты следующие рабочие условия:
P=6 МПа;
VVH=2 ч-1 (подача исходной смеси = 8 см3/ч);
H2/HC=450 нл/л (подача H2=3,6 нл/л);
T=350°C.
Оценивают процентную долю превращенного толуола и, принимая гипотезу реакции 1-го порядка, вычисляют активность по следующей формуле:
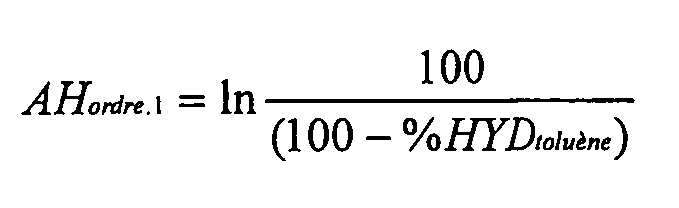
где %HYD toluène = процентная доля превращенного толуола.
Катализатор C2A (не соответствующий изобретению) имеет активность 0,52, а активность катализатора C2B (соответствующего изобретению) составляет 0,93, что представляет собой заметное повышение и показывает перспективность комбинации уксусной кислоты и диметилсукцината для увеличения гидрирующей активности катализаторов типа NiMoP на основе смеси "диоксид кремния-оксид алюминия" при мягком гидрокрекинге. С целью количественного определения повышения эффективности по конверсии и гидрообессериванию осуществляли испытание по гидрообработке исходной смеси типа вакуумного дистиллята ("DSV").
Используемая исходная смесь представляет собой смесь типа "DSV", основные характеристики которой представлены в приведенной далее таблице.
 где Tx% означает температуру кипения x%-ной жидкой фракции.
где Tx% означает температуру кипения x%-ной жидкой фракции.
Испытывают фракцию экструдированных элементов длиной от 2 до 4 мм. В реактор загружают 4 см3 катализатора в его сухой неактивной форме. Активацию (сульфидирование) осуществляют на установке перед началом испытания смесью, называемой сульфидирующей (дистиллятный газойль прямой перегонки + 2% масс. DMDS). Именно H2S, образующийся вследствие разложения DMDS, сульфидирует катализатор.
Во время испытания приняты следующие рабочие условия:
P=6 МПа;
VVH=0,6 ч-1;
H2/HC на выходе = 480 нл/л;
T=380°C.
Благодаря этому испытанию классификацию катализаторов осуществляют по оценке общей конверсии фракции 370+ во фракцию 370-: общая конверсия во фракцию 370- = % масс. фракции 370°C- в уходящих жидких потоках.
Результаты катализа представлены в приведенной далее таблице. Катализатор C2B, соответствующий изобретению, обеспечивает повышение конверсии на 5% и, главным образом, в отношении HDS по сравнению с катализатором C2A (не соответствующим изобретению), так как содержание S в уходящих жидких потоках изменяется с 60 до 32 млн-1 в случае, когда в используемый катализатор внесена добавка диметилсукцината в комбинации с уксусной кислотой согласно методике по настоящему изобретению.
Полученные результаты показывают, что кроме преимущества при гидрировании катализатор по настоящему изобретению может позволить получить интересные преимущества при мягком гидрокрекинге по сравнению со стандартным прокаленным катализатором, имеющим подобный состав.
Пример 6. Получение прокаленного катализатора CoMoNiP/оксид алюминия C3A (не соответствующего изобретению) и высушенного катализатора CoMoNiP/оксид алюминия с добавкой диметилсукцината и уксусной кислоты C3B (соответствующего изобретению), а также высушенного катализатора CoMoNiP/оксид алюминия с добавкой диметилсукцината C3C (не соответствующего изобретению)
Оксид алюминия, использованный в примере 1, был использован также в этом примере для получения "высушенного предшественника катализатора" с составом "NiCoMoP/оксид алюминия". Используемые предшественники представляют собой триоксид молибдена, карбонат кобальта, гидроксикарбонат никеля и фосфорную кислоту. Пропиточный раствор получают за одну стадию при нагревании с обратным холодильником этих предшественников. Целью является получение содержания, выраженного в пересчете на содержание в % масс. оксидов по отношению к сухому катализатору (за вычетом потерь после прокаливании при 550°C): NiO/CoO/MoO3/P2O5=1/2,3/15/4,4. После пропитки экструдированные элементы оставляют созревать в течение ночи в атмосфере, насыщенной водой, и затем помещают на 2 ч в печь при 120°C. При этом получают высушенный предшественник катализатора, который, как и в примере 4, делят на три части:
первую часть прокаливают при 450°C в течение 3 ч для получения катализатора C3A (не соответствующего изобретению);
вторую часть пропитывают раствором, содержащим уксусную кислоту и диметилсукцинат, согласно методике по настоящему изобретению: соотношение "диметилсукцинат/уксусная кислота" в растворе составляет 0,58, высушенный предшественник катализатора пропитывают этим раствором до появления выступающей влаги, указывающего на то, что поры предшественника катализатора заполнены раствором, содержащим диметилсукцинат и уксусную кислоту. Затем осуществляют стадию созревания в течение 2 ч с последующей термической обработкой при 140°C в течение 1 ч. Полученный таким образом катализатор представляет собой катализатор C3B, соответствующий изобретению;
третью часть пропитывают диметилсукцинатом до появления выступающей влаги, указывающего на то, что поры предшественника катализатора заполнены раствором диметилсукцината. Затем осуществляют стадию созревания в течение 2 ч с последующей термической обработкой при 140°C в течение 1 ч. Полученный таким образом катализатор представляет собой катализатор C3C, не соответствующий изобретению.
Пример 7. Оценка прокаленного катализатора CoMoNiP/оксид алюминия C3A (не соответствующего изобретению), высушенного катализатора CoMoNiP/оксид алюминия с добавкой диметилсукцината и уксусной кислоты C3B (соответствующего изобретению) и высушенного катализатора CoMoNiP/оксид алюминия с добавкой диметилсукцината C3C (не соответствующего изобретению) в испытании с молекулярной моделью гидрирования толуола
В таких применениях, как гидрообработка вакуумных дистиллятов и кубовых остатков, функциональная группа с гидрирующей/дегидрирующей способностью играет критическую роль с учетом значительного содержания ароматических соединений в таком сырье. Таким образом, испытание по гидрированию толуола использовали для изучения перспективности катализаторов, предназначенных для таких применений, как предварительная обработка при каталитическом крекинге или гидрообессеривание кубовых остатков.
Катализаторы, описанные ранее в примере 6, сульфидируют по месту применения в динамическом режиме в трубчатом реакторе с неподвижным проницаемым слоем на экспериментальной установке типа Microcat (разработчик: компании Vinci), причем жидкие потоки проходят сверху вниз. Определение гидрирующей активности осуществляют немедленно после сульфидирования под давлением и без выгрузки из реактора на воздух с использованием углеводородной смеси, служившей для сульфидирования катализаторов.
Сульфидирующая и испытуемая смесь состоит из 5,8% диметилдисульфида (DMDS), 20% толуола и 74,2% циклогексана (по массе).
Сульфидирование осуществляют при температуре от комнатной до 350°C со скоростью подъема температуры 2°C/мин, VVH=4 ч-1 и H2/HC=450 нл/л. Испытание по катализу осуществляют при 350°C с VVH=2 ч-1 и H2/HC, равном значению при сульфидировании, с отбором не менее 4 проб, анализируемых способом газофазной хроматографии.
Таким образом, определяют стабилизированную каталитическую активность равных объемов катализаторов в реакции гидрирования толуола.
Приняты следующие конкретные условия определения активности:
общее давление: 6,0 МПа;
давление толуола: 0,37 МПа;
давление циклогексана: 1,42 МПа;
давление метана: 0,22 МПа;
давление водорода: 3,68 МПа;
давление H2S: 0,22 МПа;
объем катализатора: 4 см3 (экструдированные элементы длиной от 2 до 4 мм);
часовая объемная скорость: 2 ч-1;
температура при сульфидировании и испытании: 350°C.
Пробы, отбираемые из уходящего жидкого потока, анализируют способом газофазной хроматографии. Определение молярных концентраций непревращенного толуола (T) и концентраций продуктов его гидрирования (метилциклогексан (MCC6), этилциклопентан (EtCC5) и диметилциклопентаны (DMCC5)) позволяет рассчитывать степень гидрирования толуола XHYD, определяемую по формуле:
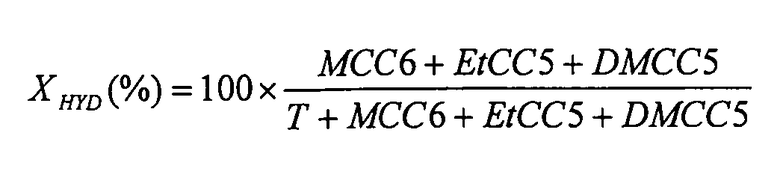
Поскольку реакция гидрирования толуола в условиях осуществляемого испытания имеет 1-й порядок, а реактор соответствует реактору идеального вытеснения, гидрирующую активность AHYD катализаторов рассчитывают, используя формулу:
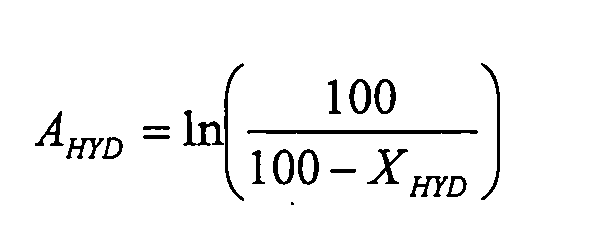
Приведенная далее таблица позволяет сравнить относительные гидрирующие активности катализаторов, полученных в примере 6.
кислоты
кислоты (% масс.
по отношению к конечному катализатору)
добавки
органической
добавки
(% масс. по отношению
к конечному катализатору)
Полученные результаты по катализу показывают особый эффект в отношении гидрирующей активности комбинации уксусной кислоты (AA) и диметилсукцината (DMSU) в высушенном предшественнике катализатора CoMoNiP/оксид алюминия (соответствующем изобретению) по сравнению с прокаленным катализатором CoMoNiP/оксид алюминия предшествующего уровня техники. Это повышение гидрирующей активности является особенно предпочтительным для применения в отношении тяжелых исходных смесей, например, при предварительной обработке при каталитическом крекинге или также при гидрообессеривании кубовых остатков.
Спектры комбинационного рассеяния получали на спектрометре комбинационного рассеяния, оснащенном ионным аргоновым лазером (514 нм). Пучок лазерного излучения фокусировали на образце посредством микроскопа, оснащенного объективом с 50-кратным увеличением и длинным фокусным расстоянием. Мощность лазерного излучения на уровне образца составляла около 1 мВт. Сигнал комбинационного рассеяния, испускаемый образцом, улавливали тем же объективом, диспергировали посредством сетки при 1800 об/мин и затем улавливали детектором CCD. Полученное разрешение спектра составило около 0,5 см-1. Зарегистрированная область спектра находилась в интервале от 300 до 1800 см-1. Продолжительность записи устанавливали равной 120 с для каждого записанного спектра комбинационного рассеяния.
Спектроскопия комбинационного рассеяния, осуществленная с катализаторами C16-C19, позволила показать для катализаторов, соответствующих изобретению, присутствие в спектре комбинационного рассеяния наиболее интенсивных характеристических полос HPA Кеггина, диметилсукцината и уксусной кислоты. Точное положение полос, их форма и относительная интенсивность могут варьировать в некоторой мере в зависимости от условий записи спектра, оставаясь при этом характеристическими для данной молекулы. С другой стороны, спектры комбинационного рассеяния органических соединений хорошо документированы как в базах данных по спектрам комбинационного рассеяния (см., например, Spectral Database for Organic Compounds, http://riodb01.ibase.aist.go.jp/sdbs/cgi-bin/direct_frame_top.cgi), так и поставщиками реактивов (см., например, www.sigmaaldrich.com).
Спектры комбинационного рассеяния, записанные для катализаторов C3B (DMSU+AA) и C3C (только DMSU), представлены на фигуре 2. Каждое определение повторяли в 3 разных зонах экструдированных элементов. В случае катализатора C3B установлено присутствие двух характеристических полос гетерополианионов Кеггина в области 990 и 971 см-1. Менее интенсивные полосы, также приписываемые этим образцам, представляют собой полосы гетерополианиона Кеггина в области 970, 902 и 602 см-1. В двух этих спектрах также установлено присутствие интенсивной полосы диметилсукцината в области 851 см-1. Зато полоса в области 896 см-1 присутствует только в случае катализатора по настоящему изобретению.
Таким образом, в спектре комбинационного рассеяния катализатора C3B по настоящему изобретению присутствуют характеристические полосы гетерополианионов Кеггина, диметилсукцината и уксусной кислоты, в то время как в случае катализатора C3C присутствуют только характеристические полосы гетерополианионов Кеггина и диметилсукцината.
Пример 8. Получение катализаторов CoMoP на основе оксида алюминия C1, C2, C3, C4 (не соответствующих изобретению)
Использовали матрицу, состоящую из ультрадисперсного слоистого бемита или геля оксида алюминия, реализуемого компанией Condea Chemie GmbH. Этот гель смешали с водным раствором, содержавшим азотную кислоту с концентрацией 66% (7% масс. кислоты на грамм сухого геля), и затем перемешивали в течение 15 минут. Полученную после перемешивания пасту пропускали через фильеру с круглыми отверстиями диаметром 1,6 мм. Затем экструдированные элементы сушили в течение ночи при 120°C и далее прокаливали при 600°C в течение 2 часов в атмосфере влажного воздуха, содержавшей 50 г воды на кг сухого воздуха. Таким образом, получали экструдированные элементы носителя, имеющие удельную поверхность 300 м2/г. Рентгеноструктурным анализом обнаружено, что носитель состоит только из гамма-формы кубического оксида алюминия со слабой кристалличностью.
К носителю из оксида алюминия, описанному ранее и находящемуся в "экструдированном" виде, прибавляют кобальт, молибден и фосфор. Пропиточный раствор готовят растворением при нагревании оксида молибдена (24,34 г) и гидроксида кобальта (5,34 г) в растворе фосфорной кислоты (7,47 г) в виде водного раствора. После пропитки в сухом состоянии экструдированные элементы оставляют созревать в атмосфере, насыщенной водой, в течение 12 ч и затем сушат в течение ночи при 90°C. Полученный таким образом высушенный предшественник катализатора обозначен как C1. Прокаливание предшественника катализатора C1 при 450°C в течение 2 часов дает прокаленный катализатор C2. Катализаторы C1 и C2 имеют следующий конечный состав, выраженный в пересчете на оксиды: MoO3=22,5±0,2% масс., CoO=4,1±0,1% масс. и P2O5=4,0+0,1% масс.
Прокаленный катализатор C2 загружают в установку с проницаемым слоем и сульфидируют дистиллятным газойлем прямой перегонки с добавкой 2% масс. диметилдисульфида. Затем в течение 300 ч проводят испытание по HDS со смесью дистиллятного газойля прямой перегонки и газойля, полученного при каталитическом крекинге. После испытания использованный катализатор выгружают, собирают и промывают толуолом при нагревании с обратным холодильником и далее делят на две части. Первую часть регенерируют в печи с контролируемым горением, вводя на каждом плато температуры возрастающие количества кислорода, что позволяет ограничивать экзотермические эффекты, связанные со сгоранием кокса. Конечное плато регенерации находится при 450°C. Отрегенерированный таким образом катализатор анализируют способом DRX. Отмечено отсутствие характеристической спектральной линии при 26°, связанной с наличием кристаллического CoMoO4. Этот катализатор в дальнейшем обозначен как C3. Вторую часть промытого использованного катализатора регенерируют в муфельной печи при 400°C без контроля экзотермических эффектов сгорания кокса. Анализ способом DRX, осуществленный после регенерации, показывает присутствие тонкой спектральной линии при 26°, характерной для наличия кристаллического CoMoO4. Кроме того, этот катализатор, обозначаемый далее как C4, имеет сильно выраженный яркий синий цвет.
Пример 9. Получение катализаторов CoMoP на основе оксида алюминия с добавками C5, C6, C7, C8, C9, C10, C11, C12 (не соответствующих изобретению)
Катализатор C5 получают пропиткой высушенного предшественника катализатора C1 раствором, содержащим лимонную кислоту (CA) и полиэтиленгликоль (PEG) в виде раствора в этаноле, так чтобы получить объем пропиточного раствора, равный объему пор высушенного предшественника катализатора C1. Принятое содержание лимонной кислоты (CA) и полиэтиленгликоля (PEG) составляет по 10% масс.
Катализатор C6 получают пропиткой высушенного предшественника катализатора C1 раствором, содержащим лимонную кислоту (CA) и полиэтиленгликоль (PEG) в виде раствора в этаноле. Принятое содержание лимонной кислоты (CA) и полиэтиленгликоля (PEG) составляет 4 и 10% масс. соответственно.
Катализатор C7 получают пропиткой прокаленного катализатора C2 раствором, содержащим лимонную кислоту и полиэтиленгликоль в виде раствора в этаноле, так чтобы получить объем пропиточного раствора, равный объему пор прокаленного катализатора C2. Принятое содержание лимонной кислоты (CA) и полиэтиленгликоля (PEG) составляет по 10% масс.
Катализатор C8 получают пропиткой прокаленного катализатора C2 раствором, содержащим лимонную кислоту и полиэтиленгликоль в виде раствора в этаноле. Принятое содержание лимонной кислоты (CA) и полиэтиленгликоля (PEG) составляет 4 и 10% масс. соответственно.
Катализатор C9 получают пропиткой прокаленного катализатора C2 раствором, содержащим уксусную кислоту (AA) и диметилсукцинат (DMSU) в виде раствора в этаноле. Принятое содержание уксусной кислоты (AA) и диметилсукцината (DMSU) составляет 4 и 10% масс. соответственно.
Катализатор C10 получают пропиткой регенерированного катализатора C3, не содержащего тугоплавкую фазу типа CoMoO4, раствором, содержащим лимонную кислоту (CA) и полиэтиленгликоль (PEG) в этаноле, так чтобы получить объем пропиточного раствора, равный объему пор катализатора C3. Принятое содержание лимонной кислоты (CA) и полиэтиленгликоля (PEG) составляет по 10% масс.
Катализатор C11 получают пропиткой регенерированного катализатора C3, не содержащего тугоплавкую фазу типа CoMoO4, раствором, содержащим лимонную кислоту (CA) и полиэтиленгликоль (PEG) в этаноле. Принятое содержание лимонной кислоты (CA) и полиэтиленгликоля (PEG) составляет 4 и 10% масс. соответственно.
Катализатор C12 получают пропиткой регенерированного катализатора C3, не содержащего тугоплавкую фазу типа CoMoO4, раствором, содержащим уксусную кислоту и диметилсукцинат в этаноле. Принятое содержание уксусной кислоты и диметилсукцината составляет 4 и 10% масс. соответственно.
Катализатор C13 получают пропиткой регенерированного катализатора C4, содержащего CoMoO4, раствором, содержащим лимонную кислоту и полиэтиленгликоль в этаноле, так чтобы получить объем пропиточного раствора, равный объему пор катализатора C4. Принятое содержание лимонной кислоты и полиэтиленгликоля составляет по 10% масс.
Катализатор C14 получают пропиткой регенерированного катализатора C4, содержащего CoMoO4, раствором, содержащим лимонную кислоту и полиэтиленгликоль в этаноле. Принятое содержание лимонной кислоты и полиэтиленгликоля составляет 4 и 10% масс. соответственно.
Катализатор C15 получают пропиткой регенерированного катализатора C4, содержащего CoMoO4, раствором, содержащим уксусную кислоту и диметилсукцинат в этаноле. Принятое содержание уксусной кислоты и диметилсукцината составляет 4 и 10% масс. соответственно.
Затем катализаторы C5-C15 направляют на стадию созревания в течение 3 ч с последующей стадией термической обработки (сушки) в течение 1 ч при 140°C в атмосфере азота.
Пример 10. Получение катализатора CoMoP с добавкой C16 (соответствующего изобретению)
Катализатор C16 получают пропиткой высушенного предшественника катализатора C1 раствором, содержащим уксусную кислоту, диметилсукцинат и этанол. Принятое содержание уксусной кислоты и диметилсукцината составляет 4 и 10% масс. соответственно. Затем катализатор направляют на стадию созревания в течение 3 ч в атмосфере воздуха при комнатной температуре с последующей стадией термической обработки (сушки) при 140°C в течение 1 ч в атмосфере азота.
Пример 11. Сравнительное испытание катализаторов C1-C16 при гидрировании толуола в циклогексане под давлением в присутствии сероводорода
Описанные ранее катализаторы сульфидируют по месту применения в динамическом режиме в трубчатом реакторе с неподвижным проницаемым слоем на экспериментальной установке типа Microcat (разработчик: компании Vinci), причем жидкие потоки проходят сверху вниз. Определение гидрирующей активности осуществляют немедленно после сульфидирования под давлением и без выгрузки из реактора на воздух с использованием углеводородной смеси, служившей для сульфидирования катализаторов.
Сульфидирующая и испытуемая смесь состоит из 5,8% диметилдисульфида (DMDS), 20% толуола и 74,2% циклогексана (по массе).
Сульфидирование осуществляют при температуре от комнатной до 350°C со скоростью подъема температуры 2°C/мин, VVH=4 ч-1 и H2/HC=450 нл/л. Испытание по катализу осуществляют при 350°C с VVH=2 ч-1 и H2/HC, равном значению при сульфидировании, с отбором не менее 4 проб, анализируемых способом газофазной хроматографии.
Таким образом, определяют стабилизированную каталитическую активность равных объемов катализаторов в реакции гидрирования толуола.
Приняты следующие конкретные условия определения активности:
- общее давление: 6,0 МПа;
- давление толуола: 0,37 МПа;
- давление циклогексана: 1,42 МПа;
- давление метана: 0,22 МПа;
- давление водорода: 3,68 МПа;
- давление H2S: 0,22 МПа;
- объем катализатора: 4 см3 (экструдированные элементы длиной от 2 до 4 мм);
- часовая объемная скорость: 2 ч-1;
- температура при сульфидировании и испытании: 350°C.
Пробы, отбираемые из уходящего жидкого потока, анализируют способом газофазной хроматографии. Определение молярных концентраций непревращенного толуола (T) и концентраций продуктов его гидрирования (метилциклогексан (MCC6), этилциклопентан (EtCC5) и диметилциклопентаны (DMCC5)) позволяет рассчитывать степень гидрирования толуола XHYD, определяемую по формуле:
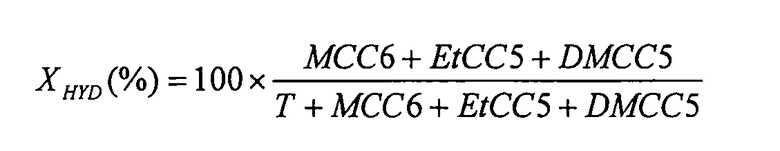
Поскольку реакция гидрирования толуола в условиях осуществляемого испытания имеет 1-й порядок, а реактор соответствует реактору идеального вытеснения, гидрирующую активность AHYD катализаторов рассчитывают, используя формулу:

В таблице 1 приведено сравнение относительных гидрирующих активностей катализаторов с добавками, полученными исходя из высушенного предшественника катализатора C1 (не соответствующего изобретению), равных по сравнению с активностью катализатора с добавкой, с активностью исходного прокаленного катализатора C2 (не соответствующего изобретению), взятого в качестве сравнительного образца (100%-я активность).
Относительные активности по сравнению с прокаленным катализатором C2 (не соответствующим изобретению) при гидрировании толуола с катализаторами C5 и C6 с добавками (не соответствующими изобретению) и C16 (соответствующим изобретению), полученными исходя из высушенного катализатора C1 (не соответствующего изобретению)
кислоты
В таблице 1 показано, что высушенный предшественник катализатора C1 (не соответствующий изобретению) обладает активностью, меньшей, чем прокаленный катализатор C2 (не соответствующий изобретению). Катализатор C5 с добавкой (не соответствующий изобретению), полученный прибавлением 10% масс. лимонной кислоты (CA) и 10% полиэтиленгликоля (PEG) к высушенному катализатору C1, не мог быть испытан, поскольку экструдированные элементы склеились в одну массу после сушки, что показывает, что избыток кислоты и добавки не подходит для случая, когда исходный катализатор представляет собой высушенный катализатор. Катализатор C6 (не соответствующий изобретению), полученный прибавлением 4% лимонной кислоты (CA) и 10% полиэтиленгликоля (PEG) к высушенному катализатору C1, имеет активность, улучшенную по сравнению с исходным высушенным катализатором на 40%. Тем не менее, так как высушенный исходный катализатор C1 имеет активность, уменьшенную на 25% по сравнению со стандартным прокаленным катализатором C2, относительная гидрирующая активность катализатора C6 по сравнению с катализатором C2 повышена только на 5%, что соответствует допускаемой в этом испытании ошибке. Таким образом, внесение добавки в высушенный катализатор комбинации "PEG+CA" не представляет интереса. Наконец, катализатор C16 (соответствующий изобретению), полученный прибавлением 4% уксусной кислоты (AA) и 10% диметилсукцината (DMSU), имеет повышение на 84% по сравнению с исходным высушенным катализатором C1 (не соответствующим изобретению). По сравнению с прокаленным катализатором C2 (не соответствующим изобретению), используемым традиционно, активность катализатора C16 (соответствующего изобретению) повышена на 38%, что выше, чем у нового поколения катализаторов (повышение от 25 до 30%). Полученные результаты по катализу показывают особый и неожиданный эффект комбинации уксусной кислоты (AA) и диметилсукцината (DMSU) в высушенном катализаторе (соответствует изобретению) по сравнению с комбинацией лимонной кислоты (CA) и полиэтиленгликоля (PEG) (не соответствует изобретению).
Аналогичным образом, в таблице 2 приведено сравнение относительных гидрирующих активностей катализаторов с добавками, полученных исходя из высушенного катализатора C1 (не соответствующего изобретению), равных по сравнению с активностью катализатора с добавкой, с активностью исходного прокаленного катализатора C2 (не соответствующего изобретению), взятого в качестве сравнительного образца (100%-я активность).
Относительные активности по сравнению с прокаленным катализатором C2 (не соответствующим изобретению) при гидрировании толуола с катализаторами C7, C8 и C9 с добавками (не соответствующими изобретению), полученными исходя из прокаленного катализатора C2 (не соответствующего изобретению)
В таблице 2 показано, что неожиданным образом катализатор C7 с добавкой (не соответствующий изобретению), полученный прибавлением 10% масс. лимонной кислоты (AC) и 10% полиэтиленгликоля (PEG) к прокаленному катализатору C2, имеет активность, близкую, даже эквивалентную, к активности исходного прокаленного катализатора C2 (не соответствующего изобретению), что показывает, что избыток кислоты и добавки, который не подходит в случае, когда исходный катализатор представлял собой высушенный катализатор, является малополезным в случае прокаленного катализатора. Катализатор C8 (не соответствующий изобретению), полученный прибавлением 4% лимонной кислоты (CA) и 10% полиэтиленгликоля (PEG) к прокаленному катализатору C2, имеет активность, улучшенную по сравнению с исходным прокаленным катализатором на 14%. Наконец, катализатор C9 (не соответствующий изобретению), полученный прибавлением 4% уксусной кислоты (AA) и 10% диметилсукцината (DMSU), имеет активность, близкую (повышение на 5%) к активности исходного прокаленного катализатора C2 (не соответствующего изобретению). Полученные результаты по катализу хорошо показывают особую перспективность комбинации уксусной кислоты (AA) и диметилсукцината (DMSU) только в случае высушенного предшественника катализатора C1 (комбинация, соответствующая изобретению), а не в случае прокаленного катализатора C2 (комбинация, не соответствующая изобретению).
Аналогичным образом, в таблице 3 приведено сравнение относительных гидрирующих активностей катализаторов с добавками (не соответствующих изобретению), полученных исходя из регенерированного катализатора C3, не содержащего тугоплавкую фазу типа CoMoO4.
Относительные активности по сравнению с прокаленным катализатором C2 (не соответствующим изобретению) при гидрировании толуола с катализаторами C10, C11 и C12 с добавками (не соответствующими изобретению), полученными исходя из регенерированного катализатора C3 (не соответствующего изобретению), не содержащего кристаллическую фазу CoMoO4
добавки
масс. по отношению к катализатору)
В таблице 3 показано, что катализатор C10 с добавкой (не соответствующий изобретению), полученный прибавлением 10% масс. лимонной кислоты (CA) и 10% полиэтиленгликоля (PEG) к регенерированному катализатору C3, не содержащему тугоплавкую фазу типа CoMoO4, имеет активность, близкую, даже эквивалентную, к активности исходного прокаленного катализатора C2 (не соответствующего изобретению), что показывает, что избыток кислоты и добавки является также малополезным в случае регенерированного катализатора, не содержащего кристаллическую фазу CoMoO4. Катализатор C11 (не соответствующий изобретению), полученный прибавлением 4% лимонной кислоты (CA) и 10% полиэтиленгликоля (PEG) к регенерированному катализатору C3, не содержащему тугоплавкую фазу типа CoMoO4, имеет активность, улучшенную по сравнению с исходным катализатором C3 (не соответствующим изобретению) на 12%, что соответствует активности, улучшенной на 9% по сравнению со свежим прокаленным катализатором C2. Наконец, катализатор C12 (не соответствующий изобретению), полученный прибавлением 4% уксусной кислоты (AA) и 10% диметилсукцината (DMSU), имеет активность, близкую к активности прокаленного катализатора C2 (не соответствующего изобретению). Полученные результаты по катализу хорошо показывают особую перспективность комбинации уксусной кислоты (AA) и диметилсукцината (DMSU) в высушенном катализаторе C1 (соответствующем изобретению) по сравнению с такой же комбинацией в регенерированном катализаторе C3, не содержащем кристаллическую фазу типа CoMoO4 (не соответствующем изобретению).
Аналогичным образом, в таблице 4 приведено сравнение относительных гидрирующих активностей катализаторов с добавками, полученных исходя из регенерированного катализатора C4 (не соответствующего изобретению), содержащего CoMoO4. Присутствие CoMoO4 подтверждено анализом способом DRX.
Относительные активности по сравнению с прокаленным катализатором C2 (не соответствующим изобретению) при гидрировании толуола с катализаторами C4, C13, C14 и C14 с добавками (не соответствующими изобретению), полученными исходя из регенерированного катализатора C4 (не соответствующего изобретению), содержащего кристаллические фазы типа CoMoO4
органической добавки
масс. по отношению к катализатору)
В таблице 4 показано, что катализатор C13 с добавкой (не соответствующий изобретению), полученный прибавлением 10% масс. лимонной кислоты (CA) и 10% полиэтиленгликоля (PEG) к регенерированному катализатору C4, содержащему CoMoO4, имеет активность, близкую, даже эквивалентную, к активности исходного прокаленного катализатора C2 (не соответствующего изобретению), что показывает, что избыток кислоты и добавки является полезным в случае регенерированного катализатора, содержащего кристаллические фазы CoMoO4. На практике, катализатор C14 (не соответствующий изобретению), полученный прибавлением 4% лимонной кислоты (CA) и 10% полиэтиленгликоля (PEG) к регенерированному катализатору C4, содержащему кристаллические фазы типа CoMoO4 (не соответствующему изобретению), имеет активность, недостаточно улучшенную по сравнению с исходным катализатором C4 (не соответствующим изобретению), так как его остаточная активность меньше активности свежего прокаленного катализатора C2. Наконец, катализатор C15 (не соответствующий изобретению), полученный прибавлением 4% уксусной кислоты (AA) и 10% диметилсукцината (DMSU), имеет активность, близкую (повышение на 3%) к активности катализатора C4 (не соответствующего изобретению) и значительно более низкую по сравнению со свежим прокаленным катализатором C2. Полученные результаты по катализу хорошо показывают особую перспективность комбинации уксусной кислоты (AA) и диметилсукцината (DMSU) в высушенном катализаторе C1 (соответствующем изобретению) по сравнению с такой же комбинацией в регенерированном катализаторе C4, содержащем кристаллические фазы типа CoMoO4 (не соответствующем изобретению). Эта комбинация является особенно неэффективной в катализаторах, которые содержат огнеупорные кристаллические фазы типа CoMoO4, в противоположность комбинации лимонной кислоты (CA) и полиэтиленгликоля (PEG).
Пример 12. Получение катализаторов C17 и C18 (не соответствующих изобретению) и C19 (соответствующего изобретению) и сравнение при HDS газойля катализаторов C2 (не соответствующего изобретению), C17 и C18 (не соответствующих изобретению), C16 и C19 (соответствующих изобретению)
Катализатор C17 получают пропиткой предшественника катализатора C1 только диметилсукцинатом (DMSU). При этом в конечном катализаторе предусматривают содержание 30% масс. диметилсукцината. Далее катализатор направляют на стадию созревания в течение 3 ч при 20°C и затем на термическую обработку при 140°C в атмосфере воздуха в течение 1 ч в печи с проницаемым слоем. Катализатор, полученный после этой термической обработки, обозначен как C17. Этот катализатор не соответствует настоящему изобретению, поскольку он не содержит уксусную кислоту в комбинации с диметилсукцинатом.
Катализатор C18 получают аналогично, но заполняя поры предшественника катализатора C1 уксусной кислотой. Получают 31% масс. по отношению к массе катализатора. Стадии созревания/термической обработки подобны случаю катализатора C17.
Катализатор C19 получают пропиткой предшественников катализаторов C1 раствором, содержащим только смесь диметилсукцината и уксусной кислоты с молярным соотношением диметилсукцината к молибдену 1,1. При этом предусматривают содержание по отношению к конечному катализатору 25 и 18% масс. для диметилсукцината и уксусной кислоты соответственно. Далее катализатор направляют на стадию созревания в течение 3 ч при комнатной температуре и затем на термическую обработку при 140°C в атмосфере воздуха в течение 1 ч в печи с проницаемым слоем. Катализатор, полученный после этой термической обработки, обозначен как C19. Этот катализатор соответствует настоящему изобретению.
Катализаторы C2 (не соответствующий изобретению), C16 (соответствующий изобретению), C17 (не соответствующий изобретению) и C19 (соответствующий изобретению) испытывали при HDS газойля.
Характеристики использованного газойля:
- плотность при 15°C: 0,8522;
- сера: 1,44% масс.;
- модельная перегонка:
PI: 155°C;
10%: 247°C;
50%: 315°C;
90%: 392°C;
PF: 444°C.
Испытание проводят в экспериментальном изотермическом реакторе с неподвижным проницаемым слоем, причем жидкие потоки проходят снизу вверх. После сульфидирования по месту применения при 350°C на установке, работающей под давлением, посредством испытуемого газойля, к которому прибавлено 2% масс. диметилдисульфида, проводили испытание по гидрообессериванию в следующих рабочих условиях:
- общее давление: 7 МПа;
- объем катализатора: 30 см3;
- температура: 340°C;
- подача водорода: 24 л/ч;
- подача исходной смеси: 60 см3/ч.
Эксплуатационные каталитические характеристики испытуемых катализаторов приведены в таблице 3. Характеристики выражены в относительной активности, при этом активность прокаленного катализатора C2 принята равной 100, а соотношение активностей предположительно составляет около 1,5. Зависимость, связывающая активность и конверсию при гидрообессеривании (обозначено как %HDS), выражается следующей формулой:

Полученные результаты приведены в таблице 5.
Относительная активность при равных объемах при гидрообессеривании газойля с катализаторами C16 (соответствующим изобретению), C17 (не соответствующим изобретению) и C18 (соответствующим изобретению) по сравнению с прокаленным катализатором C2 (не соответствующим изобретению)
относительно
C2 (%)
853 (DMSU)
896 (AA)
853 (DMSU)
896 (AA)
851 (DMSU)
895 (AA)
В таблице 5 ясно показаны синергическое действие и особая перспективность комбинации уксусной кислоты и диметилсукцината в высушенном предшественнике катализатора. На практике, катализаторы C17 и C18 (не соответствующие изобретению) имеют активности меньше активностей, полученных для катализаторов C16 и C19, соответствующих изобретению. Более интересно отметить, что в случае катализатора C19 веденное количество добавки DMSU больше, чем в случае катализатора C16, однако повышение активности происходит в противоположность тому, что было выявлено в случае PEG и лимонной кислоты в примере 1.
Спектры комбинационного рассеяния получали на спектрометре комбинационного рассеяния, оснащенном ионным аргоновым лазером (514 нм). Пучок лазерного излучения фокусировали на образце посредством микроскопа, оснащенного объективом с 50-кратным увеличением и длинным фокусным расстоянием. Мощность лазерного излучения на уровне образца составляла около 1 мВт. Сигнал комбинационного рассеяния, испускаемый образцом, улавливали тем же объективом, диспергировали посредством сетки при 1800 об/мин и затем улавливали детектором CCD. Полученное разрешение спектра составило около 0,5 см-1. Зарегистрированная область спектра находилась в интервале от 300 до 1800 см-1. Продолжительность записи устанавливали равной 120 с для каждого записанного спектра комбинационного рассеяния.
Спектроскопия комбинационного рассеяния, осуществленная с катализаторами C16-C19, позволила показать для катализаторов, соответствующих изобретению, присутствие в спектре комбинационного рассеяния наиболее интенсивных характеристических полос HPA Кеггина, диметилсукцината и уксусной кислоты. Точное положение полос, их форма и относительная интенсивность могут варьировать в некоторой мере в зависимости от условий записи спектра, оставаясь при этом характеристическими для данной молекулы. С другой стороны, спектры комбинационного рассеяния органических соединений хорошо документированы как в базах данных по спектрам комбинационного рассеяния (см., например, Spectral Database for Organic Compounds, http://riodb01.ibase.aist.go.jp/sdbs/cgi-bin/direct_frame_top.cgi), так и поставщиками реактивов (см., например, www.sigmaaldrich.com).
Без привязки к какой-либо теории было установлено, что некомплексообразующие органические добавки позволяют преобразовывать гетерополианионы в растворе в порах катализатора. Типичен именно этот феномен, выявленный с появлением полос гетерополианионов после пропитки раствором, содержащим смесь диалкилсукцината и уксусной кислоты. Этот эффект был квалифицирован как "преобразование гетерополианионов", поскольку они изначально содержатся в пропиточном растворе, но не содержатся в свежепропитанном и высушенном предшественнике катализатора и, напротив, преобразуются во время стадии созревания после пропитки добавкой. В случае PEG в присутствии лимонной кислоты комплексообразующее действие обеспечивает исчезновение кристаллических фаз, легко выявляемых способом DRX, но при этом никакой гетерополианион не преобразуется.

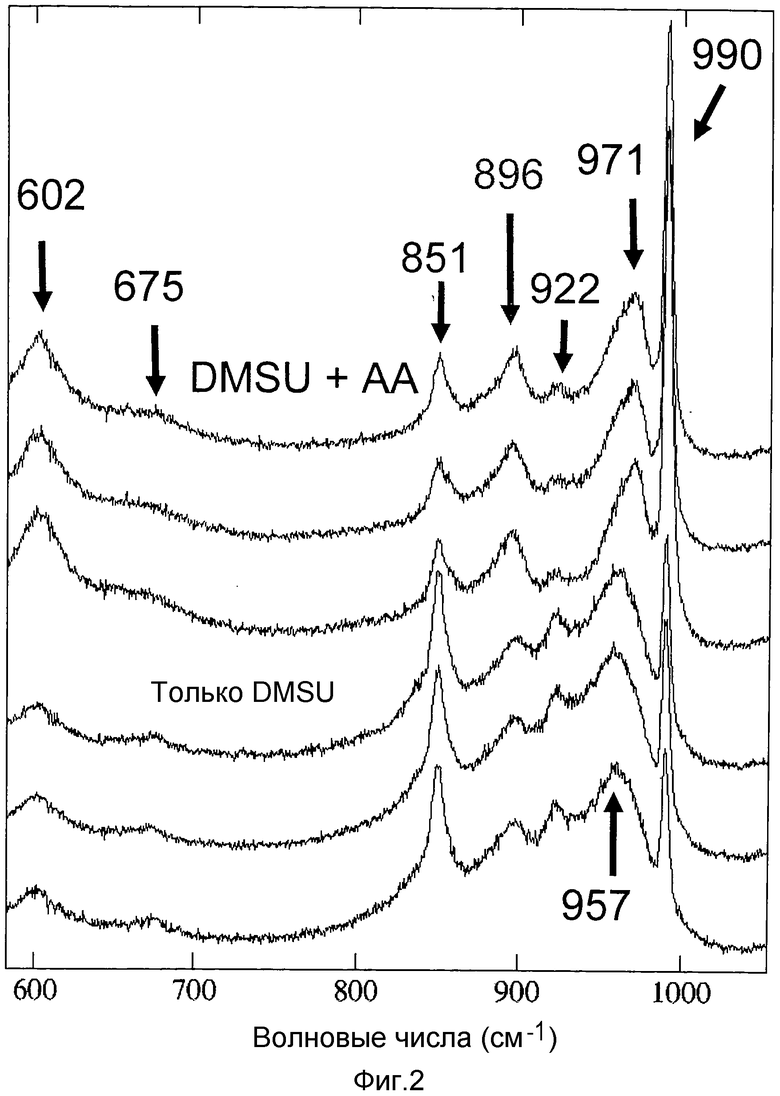
Изобретение относится к катализатору гидрообработки углеводородного сырья. Данный катализатор содержит аморфный носитель на основе оксида алюминия, фосфор, по меньшей мере один диалкил(C1-C4)сукцинат, уксусную кислоту и функциональную группу с гидрирующей/дегидрирующей способностью, содержащую по меньшей мере один элемент группы VIB и по меньшей мере один элемент группы VIII, выбранный из кобальта и/или никеля. При этом в спектре комбинационного рассеяния указанного катализатора присутствуют характеристические полосы по меньшей мере одного гетерополианиона Кеггина в области 990 и/или 974 см-1, характеристические полосы указанного сукцината и основная характеристическая полоса уксусной кислоты в области 896 см-1. Предлагаемый катализатор обладает повышенной активностью в процессах гидрообработки за счет синергического действия комбинации уксусной кислоты и диметилсукцината. Изобретение также относится к способу получения такого катализатора и катализатору, полученному этим способом, а также способу гидрообработки углеводородного сырья в присутствии такого катализатора. 4 н. и 20 з.п. ф-лы, 2 ил., 5 табл., 12 пр.
1. Катализатор гидрообработки углеводородного сырья, содержащий аморфный носитель на основе оксида алюминия, фосфор, по меньшей мере один диалкил(C1-C4)сукцинат, уксусную кислоту и функциональную группу с гидрирующей/дегидрирующей способностью, содержащую по меньшей мере один элемент группы VIB и по меньшей мере один элемент группы VIII, выбранный из кобальта и/или никеля, причем в спектре комбинационного рассеяния указанного катализатора присутствуют характеристические полосы по меньшей мере одного гетерополианиона Кеггина в области 990 и/или 974 см-1, характеристические полосы указанного сукцината и основная характеристическая полоса уксусной кислоты в области 896 см-1.
2. Катализатор по п.1, в котором диалкилсукцинат представляет собой диметилсукцинат, причем в спектре комбинационного рассеяния указанного катализатора присутствуют основные характеристические полосы гетерополианионов Кеггина в области 990 и/или 974 см-1, характеристическая полоса диметилсукцината в области 853 см-1 и характеристическая полоса уксусной кислоты в области 896 см-1.
3. Катализатор по п.1, в котором диалкилсукцинат представляет собой диэтилсукцинат, дибутилсукцинат или диизопропилсукцинат.
4. Катализатор по любому из предыдущих пунктов, содержащий носитель, образованный из оксида алюминия.
5. Катализатор по любому из пп.1-3, содержащий носитель, образованный из смеси "диоксид кремния-оксид алюминия".
6. Катализатор по любому из пп.1-3, содержащий также бор и/или фтор.
7. Катализатор по любому из пп.1, 2, в котором функциональная группа с гидрирующей/дегидрирующей способностью образована из кобальта и молибдена, а носитель образован из оксида алюминия.
8. Катализатор по любому из пп.1-3, в котором функциональная группа с гидрирующей способностью выбрана из группы, в которую входят комбинации элементов "никель-молибден" или "никель-кобальт-молибден", или "никель-молибден-вольфрам".
9. Катализатор по любому из пп.1, 2, подвергнутый сульфидированию.
10. Способ получения катализатора по любому из предыдущих пунктов, причем указанный способ включает в себя следующие последовательные стадии:
a) по меньшей мере одну стадию пропитки аморфного носителя на основе оксида алюминия по меньшей мере одним раствором, содержащим элементы функциональной группы с гидрирующей/дегидрирующей способностью и фосфор;
b) стадию сушки при температуре ниже 180°C без последующего прокаливания;
c) по меньшей мере одну стадию пропитки пропиточным раствором, содержащим по меньшей мере один диалкил(C1-C4)сукцинат, уксусную кислоту и по меньшей мере одно соединение фосфора, если последний не был введен полностью на стадии a);
d) стадию созревания;
e) стадию сушки при температуре ниже 180°C без стадии последующего прокаливания.
11. Способ по п.10, в котором все количество функциональной группы с гидрирующей способностью вводят на стадии a).
12. Способ по любому из пп.10, 11, в котором стадию c) осуществляют в отсутствие растворителей.
13. Способ по любому из пп.10, 11, в котором стадию c) осуществляют в присутствии растворителя, выбранного из группы, в которую входят метанол, этанол, вода, фенол, циклогексанол, используемые по отдельности или в смеси.
14. Способ по п.10, в котором диалкилсукцинат и уксусную кислоту вводят в пропиточный раствор на стадии c) в количестве, соответствующем молярному отношению диалкилсукцината к одному или нескольким элементам группы VIB, вводимым в предшественник катализатора, в интервале от 0,15 до 2 моль/моль, и молярному отношению уксусной кислоты к одному или нескольким элементам группы VIB, вводимым в предшественник катализатора, в интервале от 0,1 до 5 моль/моль.
15. Способ по п.10 или 14, в котором стадию d) осуществляют при температуре от 17 до 50°C.
16. Способ по п.10 или 14, в котором стадию e) осуществляют при температуре от 80 до 160°C без последующего прокаливания при температуре выше 180°C.
17. Способ по п.10 или 14, причем указанный способ включает в себя следующие последовательные стадии:
a) по меньшей мере одну стадию пропитки носителя в сухом состоянии раствором, содержащим все количество элементов функциональной группы с гидрирующей/дегидрирующей способностью и фосфор;
b) стадию сушки при температуре в интервале от 75 до 130°C без последующего прокаливания;
c) по меньшей мере одну стадию пропитки в сухом состоянии пропиточным раствором, содержащим диметилсукцинат и уксусную кислоту;
d) стадию созревания при 17-50°C;
e) стадию сушки при температуре в интервале от 80 до 160°C без стадии последующего прокаливания.
18. Способ по п.10 или 14, в котором фосфор вводят на стадии a) или на стадии c), если он не был введен полностью на стадии a), причем количество, вводимое на стадии a) и выраженное в пересчете на количество оксидов по отношению к катализатору, находится в интервале от 0,1 до 20%, предпочтительно в интервале от 0,1 до 15% и наиболее предпочтительно в интервале от 0,1 до 10 мас.%.
19. Способ по п.10 или 14, в котором стадию e) осуществляют в атмосфере азота.
20. Способ по п.10 или 14, в котором продукт, полученный на выходе стадии e), подвергают сульфидированию.
21. Способ гидрообработки углеводородного сырья в присутствии катализатора, заявленного в любом из пп.1-9 или полученного способом по любому из пп.10-20.
22. Способ по п.21, в котором гидрообработка представляет собой гидрообессеривание, гидродеазотирование, гидродеметаллизацию, гидрирование ароматических соединений или гидроконверсию.
23. Способ по п.22, в котором гидрообработка представляет собой глубокое гидрообессеривание газойлей.
24. Катализатор, полученный в соответствии со способом по любому из пп.10-20.
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| КАТАЛИТИЧЕСКАЯ СИСТЕМА ДЛЯ КАТАЛИТИЧЕСКИХ ГИДРОДЕСУЛЬФУРИЗАЦИИ, ГИДРОДЕНИТРОГЕНИЗАЦИИ, РЕФОРМИНГА, ГИДРИРОВАНИЯ-ДЕГИДРИРОВАНИЯ И ИЗОМЕРИЗАЦИИ УГЛЕВОДОРОДНОГО СЫРЬЯ, СПОСОБ ЕЕ ПОЛУЧЕНИЯ, АКТИВАЦИИ, РЕГЕНЕРАЦИИ И ИСПОЛЬЗОВАНИЯ | 1995 |
|
RU2146171C1 |

