Предпосылки создания изобретения
Область техники, к которой относится изобретение
Данное изобретение относится к способам и промежуточным соединениям для получения соединений бифенилимидазола, которые применимы при получении соединений, обладающих активностью антагониста рецептора типа 1 ангиотензина II и ингибирующей неприлизин активностью.
Уровень техники
Публикации заявок США № 2008/0269305 и 2009/0023228, обе Allegretti et al., поданные 23 апреля 2008, раскрывают новые соединения, которые обладают активностью антагониста рецептора AT1 и ингибирующей фермент неприлизин (NEP) активностью, содержание обеих публикаций включено в данное описание посредством ссылки. В одном варианте осуществления указанные заявки раскрывают новые соединения, такие как 4'-{2-этокси-4-этил-5-[((S)-2-меркапто-4-метилпентаноиламино)метил]имидазол-1-илметил)-3'-фторбифенил-2-карбоновая кислота.
При получении соединений для продолжительного хранения и при получении фармацевтических композиций и препаратов, часто желательно иметь кристаллическую форму терапевтического агента, которая не является ни гигроскопичной, ни расплывающейся. Выгодно также иметь кристаллическую форму, которая имеет относительно высокую температуру плавления, что позволяет перерабатывать материал без существенного разложения. Кристаллическая форма 4'-{2-этокси-4-этил-5-[((S)-2-меркапто-4-метилпентаноиламино)метил]имидазол-1-илметил}-3'-фторбифенил-2-карбоновой кислоты раскрыта в публикации заявки США № 2010/0081697, Chao et al., поданной 29 сентября 2009, содержание которой включено в данное описание посредством ссылки.
Соединения, раскрытые в указанных публикациях и заявках, получают по технологиям, которые обычно требуют, чтобы одно или несколько промежуточных соединений бифенилимидазола были очищены хроматографией. Существует несколько преимуществ в разработке способов, где такие стадии очистки не являются необходимыми. Данное изобретение направлено на удовлетворение этой потребности.
Сущность изобретения
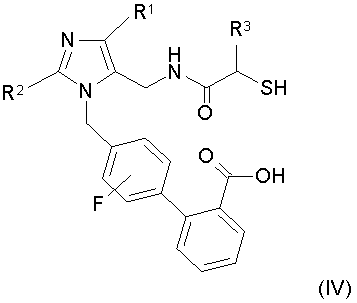
Данное изобретение относится к новым промежуточным соединениям и усовершенствованным способам получения промежуточных соединений, применимых для получения соединений формулы IV:
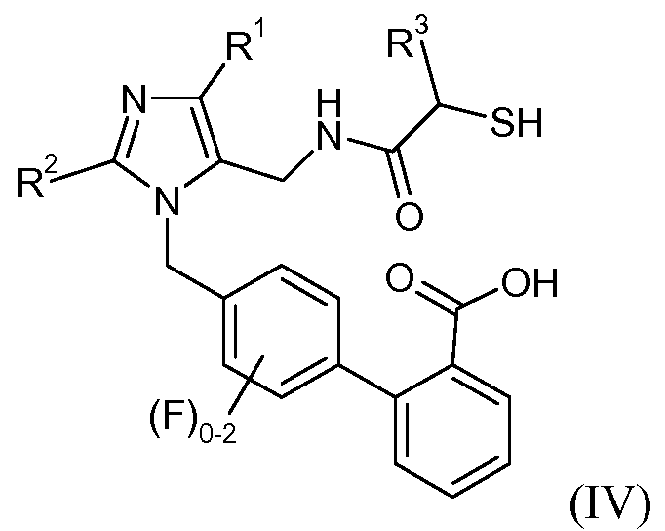
или его соли, где R1 означает -С1-6алкил, R2 означает -О-С1-5алкил и R3 означает -С1-6алкил, -С0-3алкиленарил, -С0-3алкиленгетероарил или -С0-3алкилен-С3-7циклоалкил. В одном конкретном варианте осуществления изобретение относится к способам получения промежуточных соединений, применимых для получения 4'-{2-этокси-4-этил-5-[((S)-2-меркапто-4-метилпентаноиламино)метил]имидазол-1-илметил)-3'-фторбифенил-2-карбоновой кислоты.
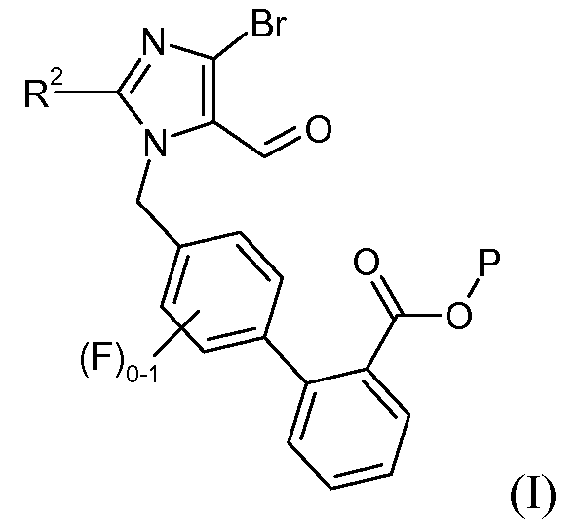
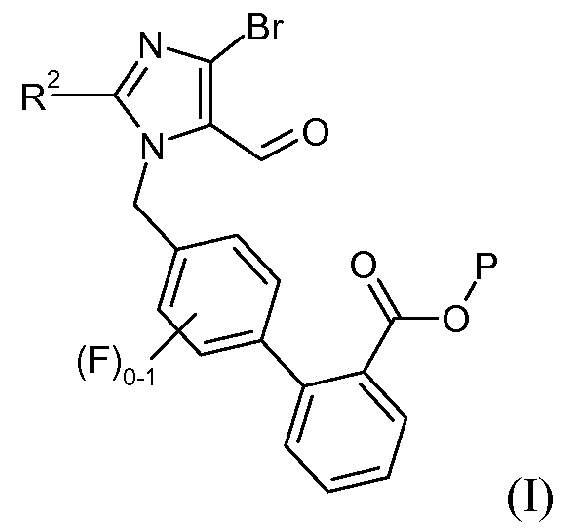
Один аспект изобретения относится к способу получения соединения формулы I:
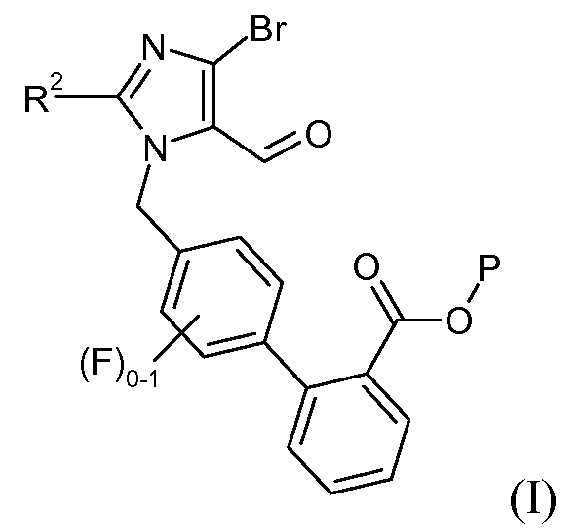
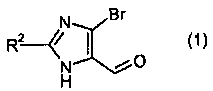
где R2 означает -О-С1-5алкил, и P означает защитную группу карбоновой кислоты, включающему стадию взаимодействия соединения формулы 1:
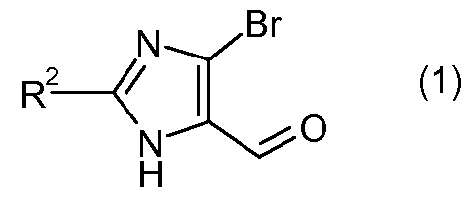
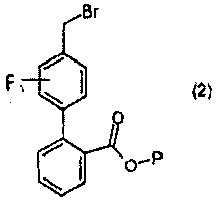
с соединением формулы 2:
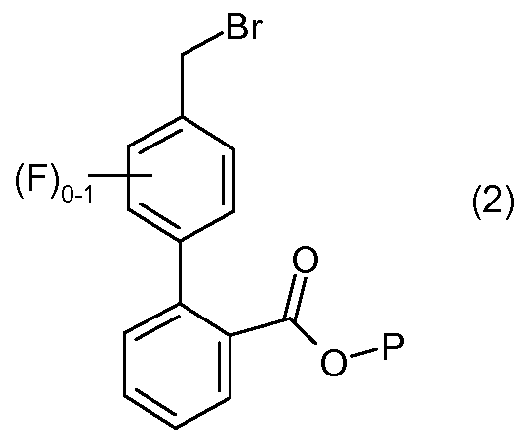
в органическом разбавителе и водном разбавителе основного характера в присутствии межфазного катализатора, где разбавители являются по существу несмешивающимися, с образованием соединения формулы I.
В одном варианте осуществления этот способ дополнительно включает стадию получения кристаллической формы соединения формулы I. Один аспект изобретения относится к кристаллическому трет-бутиловому эфиру 4'-(4-бром-2-этокси-5-формилимидазол-1-илметил)-3'-фторбифенил-2-карбоновой кислоты.
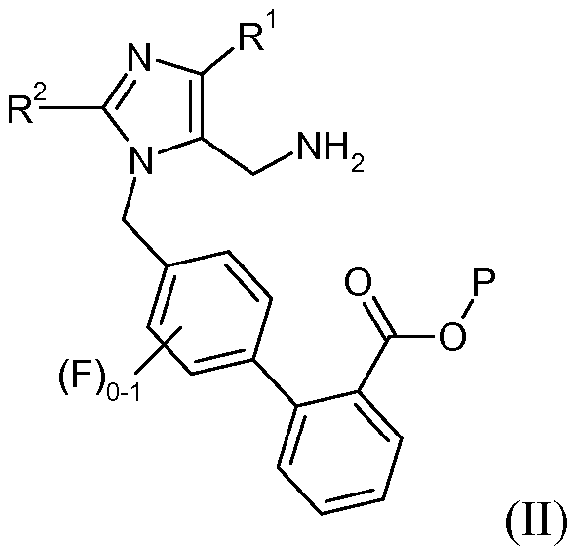
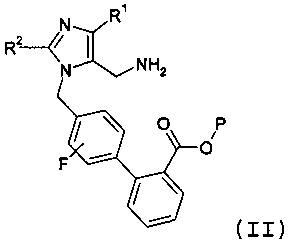
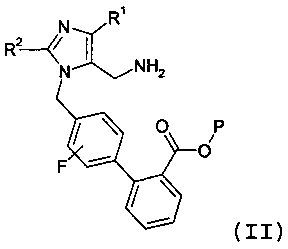
Другой аспект изобретения относится к способу получения соединения формулы II:
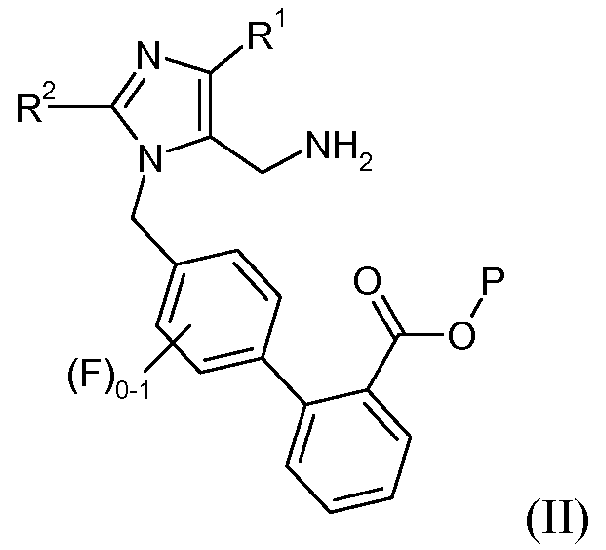
или его соли, где R1 представляет -С1-6алкил, R2 означает -O-С1-5алкил, и Р означает защитную группу карбоновой кислоты, включающему стадии:
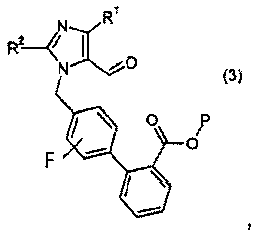
(a) взаимодействия соединения формулы I:
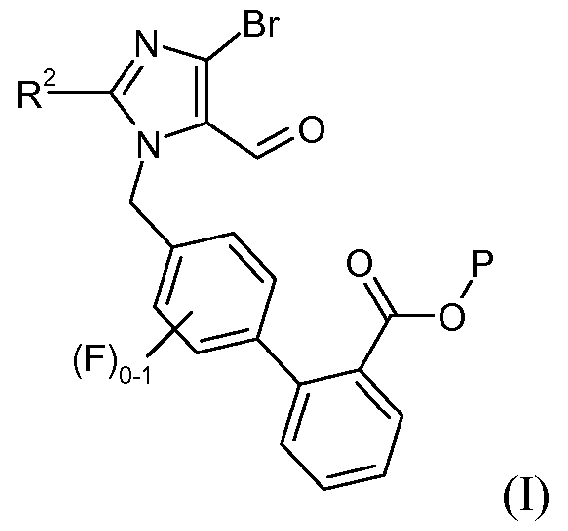
с реагентом калий-С1-6алкилтрифторборатом в присутствии палладий-фосфинового катализатора, с образованием соединения формулы 3:
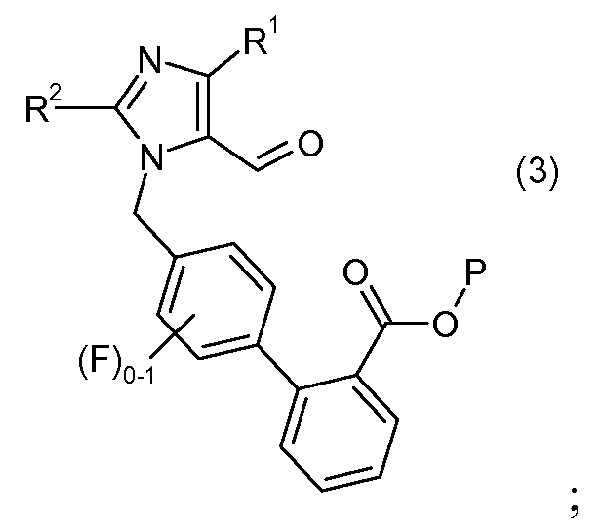
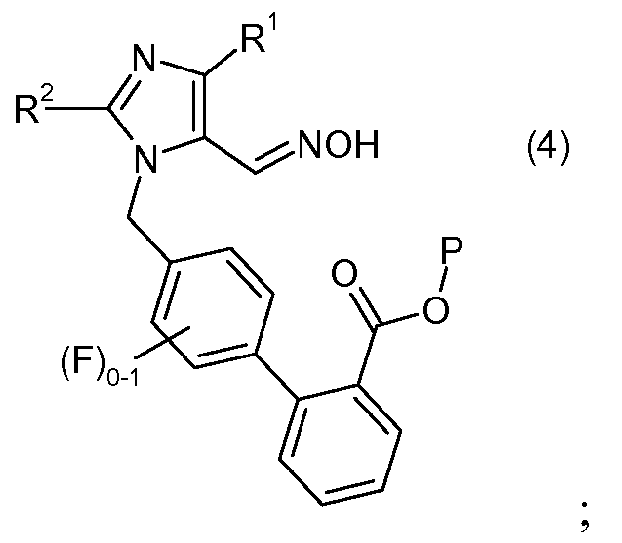
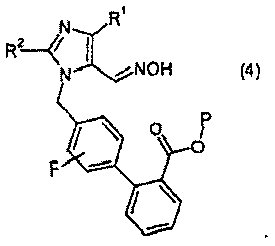
(b) взаимодействия соединения формулы 3 с гидроксиламином или его солью, с образованием соединения формулы 4:
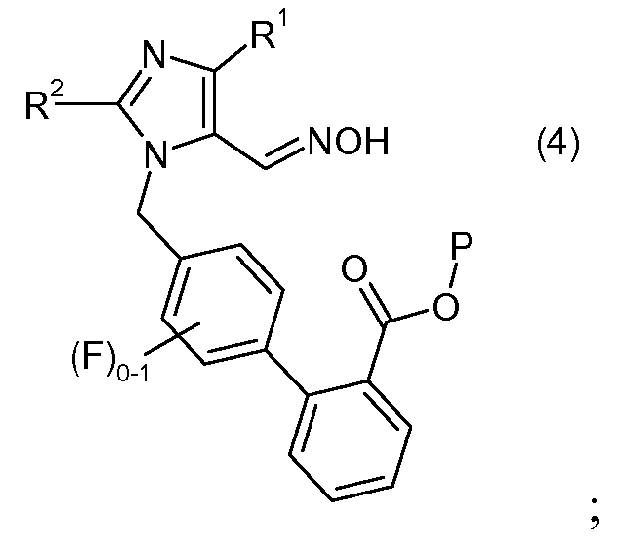
и
(с) взаимодействия соединения формулы 4 с восстановителем, с образованием соединения формулы II или его соли.
В одном варианте осуществления этот способ дополнительно включает стадию получения кристаллической формы соединения формулы II. Один аспект изобретения относится к кристаллическому трет-бутиловому эфиру 4'-(5-аминометил-2-этокси-4-этилимидазол-1-илметил)-3'-фторбифенил-2-карбоновой кислоты.
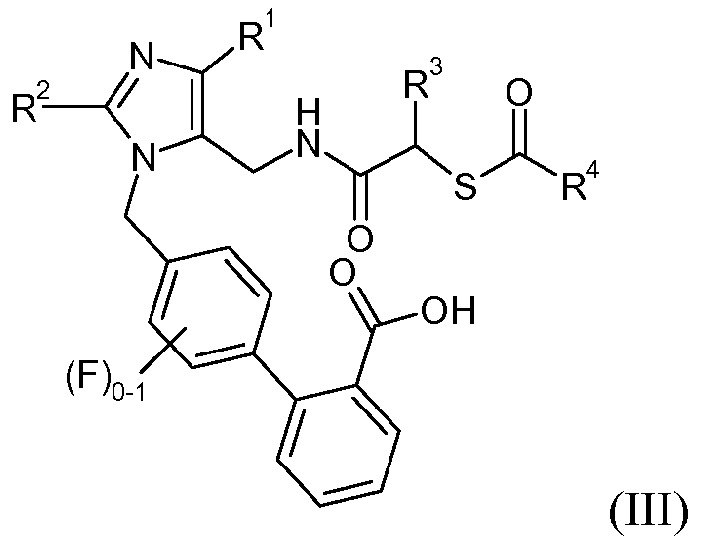
Еще один аспект изобретения относится к способу получения соединения формулы III:
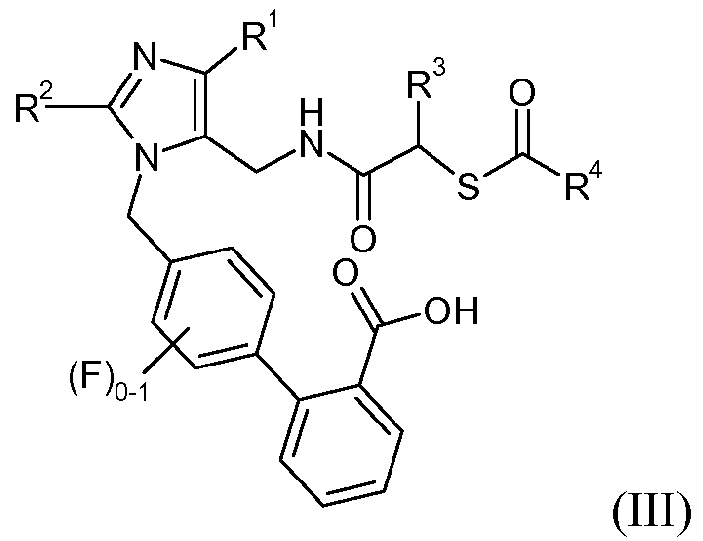
или его соли, где R1 означает -С1-6алкил, R2 означает -O-С1-5алкил, R3 означает -C1-6алкил, -C0-3алкиленарил, -C0-3алкиленгетероарил или -C0-3алкилен-C3-7циклоалкил, и R4 означает -С1-6алкил, -С0-6алкилен-C3-7циклоалкил, -С0-6алкиленарил или -С0-6алкиленморфолин; включающему стадии:
(a) взаимодействия соединения формулы I:
с реагентом калий-С1-6алкилтрифторборатом в присутствии палладий-фосфинового катализатора, с образованием соединения формулы 3:
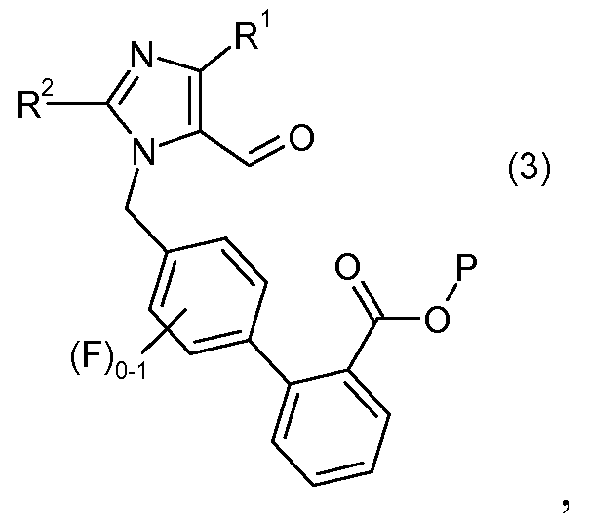
где P означает защитную группу карбоновой кислоты;
(b) взаимодействия соединения формулы 3 с гидроксиламином или его солью, с образованием соединения формулы 4:
(c) взаимодействия соединения формулы 4 с восстановителем с образованием соединения формулы II или его соли.
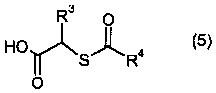
(d) взаимодействия соединения формулы II или его соли с соединением формулы 5:
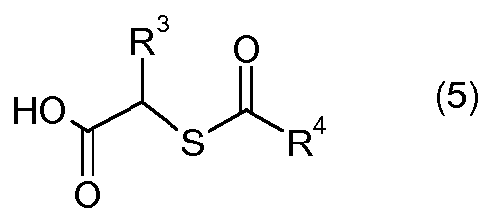
или его солью в присутствии реагента сочетания амин-карбоновая кислота, с образованием соединения формулы 6:
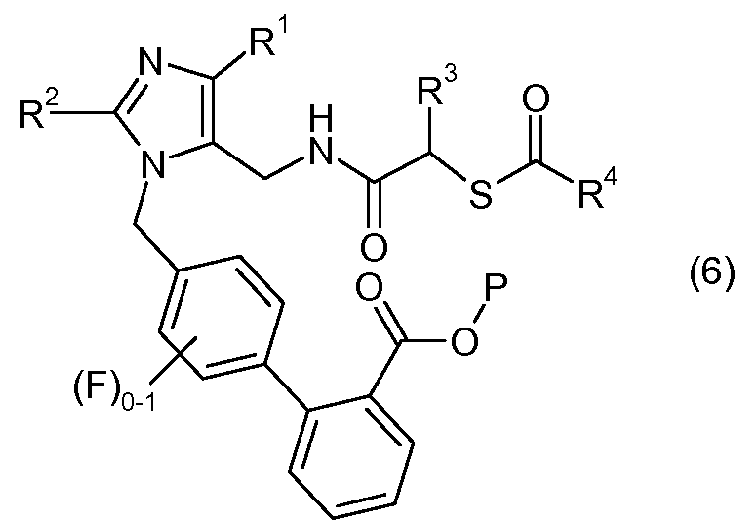
или его соли, и
(e) удаления защитной группы карбоновой кислоты, P, из соединения формулы 6 или его соли, с получением соединения формулы III или его соли.
В одном варианте осуществления способ дополнительно включает стадию получения кристаллической формы соединения формулы III. Один аспект изобретения относится к кристаллической 4'-{5-[((S)-2-ацетилсульфанил-4-метилпентаноиламино)метил]-2-этокси-4-этилимидазол-1-илметил)-3'-фторбифенил-2-карбоновой кислоте.
Другой аспект изобретения относится к новым промежуточным соединениям, применимым в способах по изобретению. В одном таком аспекте изобретения новые промежуточные соединения имеют формулу 3 или 4.
Краткое описание фигур
Различные аспекты данного изобретения поясняются ссылкой на сопровождающие фигуры.
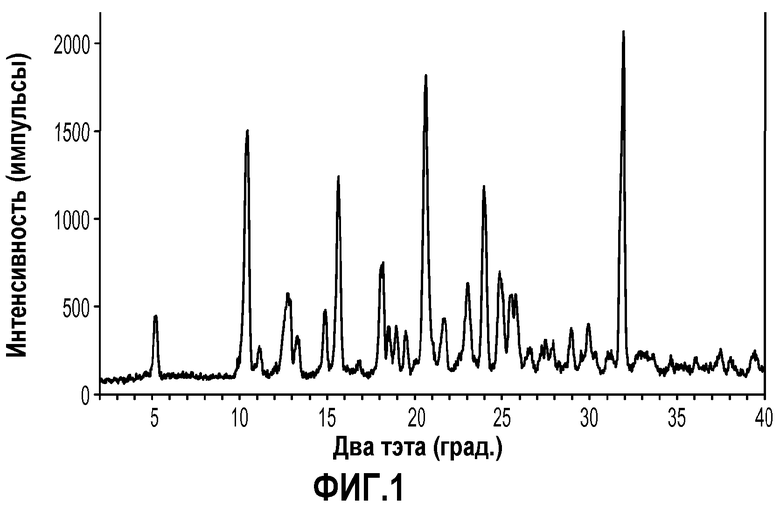
На фиг.1 показана порошковая рентгенограмма (PXRD) кристаллической формы трет-бутилового эфира 4'-(5-аминометил-2-этокси-4-этилимидазол-1-илметил)-3'-фторбифенил-2-карбоновой кислоты (формула IIa).
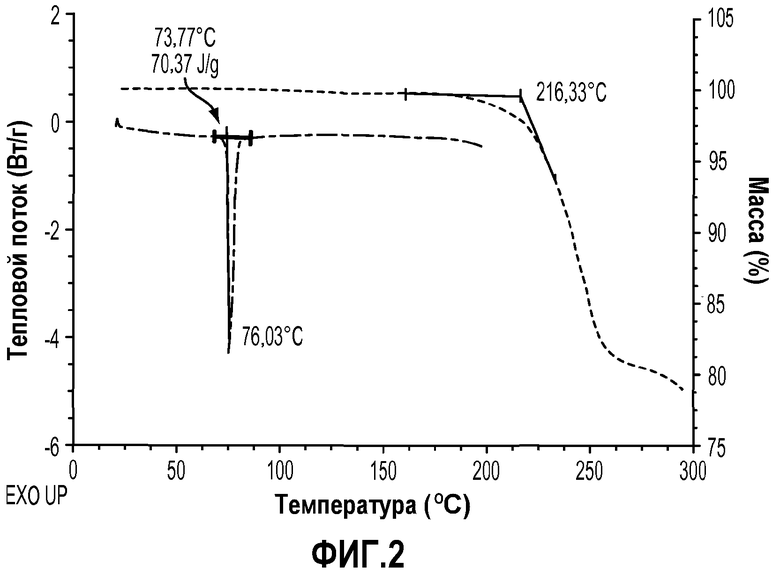
На фиг.2 показана термограмма дифференциальной сканирующей калориметрии (DSC) и термический гравиметрический анализ (TGA) указанного кристаллического соединения.
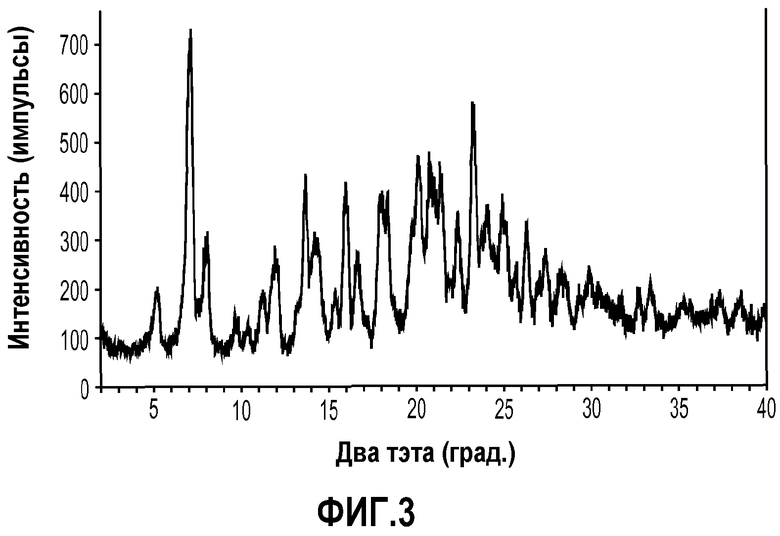
На фиг.3 показана порошковая рентгенограмма (PXRD) кристаллической формы 4'-{5-[((S)-2-ацетилсульфанил-4-метилпентаноиламино)метил]-2-этокси-4-этилимидазол-1-илметил}-3'-фторбифенил-2-карбоновой кислоты (формула IIIa).
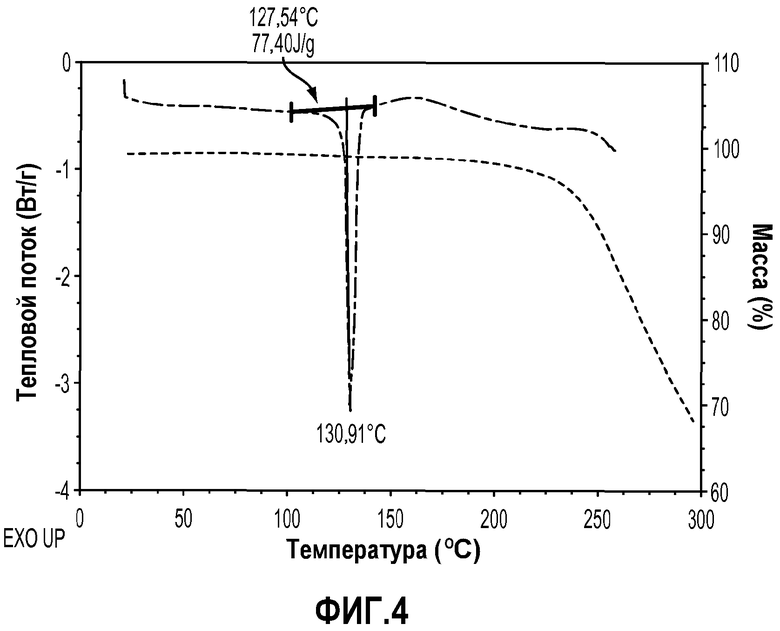
На фиг.4 показана DSC термограмма и TGA указанного кристаллического соединения.
Подробное описание изобретения
Изобретение относится к новым способам получения соединений формулы I:
и соединений формулы II:
и соединений формулы III:
или их солей.
Группа R1 представляет -С1-6алкил, примеры которого включают -CH3 и -CH2CH3. В одном конкретном варианте осуществления R1 означает -CH2CH3.
Группа R2 представляет -O-С1-5алкил, примеры которого включают -OCH3, -OCH2CH3, -OCH(CH3)2, -O(CH2)2CH3, -O(CH2)3CH3 и -OCH2CH(CH3)2. В одном конкретном варианте осуществления R2 означает -O-CH2CH3.
Группа R3 выбрана из -С1-6алкила, -C0-3алкиленарила, -C0-3алкиленгетероарила и -C0-3алкилен-C3-7циклоалкила. Примеры -С1-6алкила включают -CH3, -CH2CH3, -CH(CH3)2, -(CH2)2CH3, -(CH2)3CH3, -CH(CH3)CH2CH3, -CH2CH(CH3)2, -CH2C(CH3)3, -(CH2)2CH(CH3)2 и -(CH2)4CH3. В одном конкретном варианте осуществления R3 представляет -CH2CH(CH3)2. Примеры -C0-3алкиленарила включают фенил, бензил, -CH2-бифенил, -(CH2)2-фенил и -CH2-нафталин-1-ил. Примеры -C0-3алкиленгетероарила включают -CH2-пиридил, -CH2-фуранил, -CH2-тиенил и -CH2-тиофенил. Примеры -C0-3алкилен-C3-7циклоалкила включают -CH2-циклопропил, циклопентил, -CH2-циклопентил, -циклогексил и -CH2-циклогексил.
Группа R4 выбрана из -С1-6алкила, -C0-6алкилен-C3-7циклоалкила, -C0-6алкиленарила и -C0-6алкиленморфолина. Примеры -С1-6алкила включают -CH3, -CH2CH3, -CH(CH3)2, -C(CH3)3 и -CH2CH(CH3)2. В одном конкретном варианте осуществления R4 представляет -CH3. Примеры -C0-6алкилен-C3-7циклоалкила включают -циклопентил, -циклогексил и -CH2-циклопентил. Примеры -C0-6алкиленарила включают фенил. Примеры -C0-6алкиленморфолина включают -CH2-морфолин и -(CH2)2-морфолин.
Группа P представляет "защитную группу карбоновой кислоты", где термин используют в данном описании для обозначения группы, ковалентно связанной с карбоксильной функциональной группой, которая защищает функциональную группу от нежелательных реакций, но дает возможность регенерировать функциональную группу (т.е. удалить защиту или разблокировать) путем обработки защитной группы соответствующим реагентом. Примеры защитных групп карбоновой кислоты включают, но, не ограничиваясь ими, метил, этил, трет-бутил, бензил (Bn), п-метоксибензил (PMB), 9-флуоренилметил (Fm), триметилсилил (TMS), трет-бутилдиметилсилил (TBDMS), дифенилметил (бензгидрил, DPM) и тому подобное. В одном конкретном варианте осуществления P представляет собой трет-бутил. Другие примеры защитных групп карбоновой кислоты описаны, например, в T. W. Greene and G. M. Wuts, Protecting Groups in Organic Synthesis, Third Edition, Wiley, New York, 1999.
Определения
При описании соединений и способов по изобретению следующие термины имеют следующие значения, если не указано иное. Кроме того, используемые в данном описании формы единственного числа включают соответствующие формы множественного числа, если контекст не указывает явно на иное. Термины "содержащий", "включающий" и "имеющий" являются инклюзивными и означают, что могут быть дополнительные элементы помимо перечисленных. Все числа, обозначающие в данном описании количество ингредиентов, параметры характерных особенностей, таких как молекулярная масса, условия реакции и так далее, следует понимать как модифицируемые во всех случаях термином "около", если не указано иное. Соответственно, приведенные в данном описании числа являются аппроксимациями, которые могут изменяться в зависимости от желаемых свойств, которые хотят достигнуть данным изобретением. По меньшей мере и не как попытка ограничить применение системы эквивалентов к объему притязаний, каждое число по меньшей мере следует толковать в свете сообщаемых значащих цифр и путем применения обычных способов округления.
Названия описанных в данном описании соединений обычно даны с применением характеристики AutoNom, коммерчески доступного программного обеспечения MDL® ISIS/Draw (Symyx, Santa Clara, California).
Используемое в данном описании выражение "имеющее формулу" или "имеющее структуру" не предназначено для ограничения и используется таким же образом, как обычно используемый термин "содержащее".
Термин "алкил" означает одновалентную насыщенную углеводородную группу, которая может быть линейной или разветвленной. Если не дано другого определения, такие алкильные группы обычно содержат от 1 до 10 атомов углерода и включают, например, -С1-5алкил и -С1-6алкил. Примеры алкильных групп включают, например, метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, изобутил, трет-бутил, н-пентил, н-гексил, н-гептил, н-октил, н-нонил, н-децил и тому подобное.
Когда определенное число атомов углерода предназначается для конкретно используемого в данном описании термина, число атомов углерода указывают перед термином как нижний индекс. Например, термин "-С1-6алкил" означает алкильную группу, имеющую от 1 до 6 атомов углерода, и термин "-С3-7циклоалкил" означает циклоалкильную группу, имеющую от 3 до 7 атомов углерода, где атомы углерода находятся в любой приемлемой конфигурации.
Термин "алкилен" означает двухвалентную насыщенную углеводородную группу, которая может быть линейной или разветвленной. Если не указано иное, такие алкиленовые группы обычно содержат от 0 до 10 атомов углерода и включают, например, -С0-3алкилен- и -С0-6алкилен-. Примеры алкиленовых групп включают, например, метилен, этан-1,2-диил ("этилен"), пропан-1,2-диил, пропан-1,3-диил, бутан-1,4-диил, пентан-1,5-диил и тому подобное. Понятно, что, когда термин «алкилен» включает ноль атомов углерода, такой как -С0-3алкилен-, такие термины предназначаются для того, чтобы включать простую связь.
Термин "арил" означает одновалентный ароматический углеводород, имеющий одно кольцо (например, фенил) или конденсированные кольца. Конденсированные кольцевые системы включают такие, которые являются полностью ненасыщенными (например, нафталин), а также такие, которые являются частично ненасыщенными (например, 1,2,3,4-тетрагидронафталин). Если не указано иное, такие арильные группы обычно содержат от 6 до 10 атомов углерода кольца и включают, например, -С6-10арил. Примеры арильных групп включают, например, фенил и нафталин-1-ил, нафталин-2-ил и тому подобное.
Термин "циклоалкил" означает одновалентную насыщенную карбоциклическую углеводородную группу. Если не указано иное, такие циклоалкильные группы обычно содержат от 3 до 10 атомов углерода и включают, например, -С3-6циклоалкил и -С3-7циклоалкил. Примеры циклоалкильных групп включают, например, циклопропил, циклобутил, циклопентил, циклогексил и тому подобное.
Термин "гетероарил" означает одновалентную ароматическую группу, имеющую одно кольцо или два конденсированных кольца и содержащую в кольце (кольцах) по меньшей мере один гетероатом (обычно 1-3), выбранный из азота, кислорода и серы. Если не указано иное, такие гетероарильные группы обычно содержат в целом от 5 до 10 атомов кольца и включают, например, -С2-9гетероарил. Примеры гетероарильных групп включают, например, одновалентные радикалы пиррола, имидазола, тиазола, оксазола, фурана, тиофена, триазола, пиразола, изоксазола, изотиазола, пиридина, пиразина, пиридазина, пиримидина, триазина, индола, бензофурана, бензотиофена, бензимидазола, бензтиазола, хинолина, изохинолина, хиназолина, хиноксалина и тому подобное, где точка присоединения находится при любом доступном атоме углерода или азота кольца.
Используемый в данном описании термин "температура плавления" означает температуру, при которой наблюдается максимальный эндотермический тепловой поток при дифференциальной сканирующей калориметрии, для теплового перехода, который соответствует превращению фазы твердого вещества в жидкость.
Термин "соль", когда его используют в связи с соединением, означает соль соединения, полученную из неорганического или органического основания или из неорганической или органической кислоты. Соли, полученные из неорганических оснований, включают соли алюминия, аммония, кальция, меди, железа (III), железа (II), лития, магния, марганца (III), марганца (II), калия, натрия, цинка и тому подобное. Особенно предпочтительны соли аммония, кальция, магния, калия и натрия. Соли, полученные из органических оснований, включают соли первичных, вторичных и третичных аминов, включая замещенные амины, циклические амины, встречающиеся в природе амины и тому подобное, такие как аргинин, бетаин, кофеин, холин, N,N'-дибензилэтилендиамин, диэтиламин, 2-диэтиламиноэтанол, 2-диметиламиноэтанол, этаноламин, этилендиамин, N-этилморфолин, N-этилпиперидин, глюкамин, глюкозамин, гистидин, гидрабамин, изопропиламин, лизин, метилглюкамин, морфолин, пиперазин, пиперидин, полиаминосмолы, прокаин, пурины, теобромин, триэтиламин, триметиламин, трипропиламин, трометамин и тому подобное. Соли, полученные из кислот, включают соли уксусной, аскорбиновой, бензолсульфоновой, бензойной, камфорсульфоновой, лимонной, этансульфоновой, фумаровой, глюконовой, глюкуроновой, глутаминовой, гиппуровой, бромистоводородной, хлористоводородной, изетионовой, молочной, лактобионовой, малеиновой, яблочной, миндальной, метансульфоновой, слизевой, нафталинсульфоновой, никотиновой, азотной, памовой, пантотеновой, фосфорной, янтарной, серной, винной, п-толуолсульфоновой кислот и тому подобное. Особенно предпочтительны лимонная, бромистоводородная, хлористоводородная, малеиновая, фосфорная, серная и винная кислоты. Кроме того, когда соединение имеет в своем составе и часть основного характера, такую как амин или имидазол, и часть кислотного характера, такую как карбоновая кислота, могут образовываться цивиттерионы, и они включены в термин "соль", используемый в данном описании. Термин "фармацевтически приемлемая соль" означает соль, полученную из основания или кислоты, которые приемлемы для введения пациенту, такому как млекопитающее (например, соли, имеющие приемлемую для млекопитающего безопасность при заданном режиме дозирования). Однако понятно, что соли, охватываемые изобретением, необязательно должны быть фармацевтически приемлемыми солями, такие как соли промежуточных соединений, которые не предназначаются для введения пациенту.
Условия способа
Подходящие инертные разбавители для использования в способе по изобретению включают, для пояснения и не для ограничения, органические разбавители, такие как уксусная кислота, тетрагидрофуран (ТГФ), ацетонитрил (MeCN), N,N-диметилформамид (ДМФА), N,N-диметилацетамид, N-метилпирролидинон, диметилсульфоксид (ДМСО), толуол, дихлорметан (DCM), ацетон, этилацетат, изопропилацетат, простой метил-трет-бутиловый эфир, хлороформ (CHCl3), четыреххлористый углерод (CCl4), 1,4-диоксан, метанол, этанол, пропанол, изопропанол, бутанол, этиленгликоль и тому подобное. Водные разбавители также могут быть использованы, к ним относятся вода, а также водные разбавители основного и кислотного характера. Имеются в виду также комбинации любых из указанных разбавителей.
Подходящие полярные протонные растворители для использования в способе по изобретению включают, для пояснения и не для ограничения, метанол, этанол, пропанол, изопропанол, бутанол, этиленгликоль, воду, уксусную кислоту и тому подобное.
Существует много оснований, которые являются подходящими для использования в способе по изобретению. Примеры органических оснований включают, для пояснения и не для ограничения: амины, включая первичные алкиламины (например, метиламин, этаноламин, буферный агент трис и тому подобное), вторичные алкиламины (например, диметиламин, метилэтаноламин, N,N-диизопропилэтиламин (DIPEA) и тому подобное), третичные амины (например, триметиламин, триэтиламин и тому подобное); соединения аммония, такие как гидроксид аммония и гидразин; гидроксиды щелочных металлов, такие как гидроксид натрия, метоксид натрия, гидроксид калия, трет-бутоксид калия и тому подобное; гидриды металлов и карбоксилатные соли щелочных металлов, такие как ацетат натрия и тому подобное). Примеры неорганических оснований включают, для пояснения и не для ограничения: карбонаты щелочных металлов, такие как карбонат лития, карбонат калия, карбонат цезия, карбонат натрия, гидрокарбонат натрия и тому подобное; другие карбонаты, такие как гидрокарбонат кальция и тому подобное и фосфаты щелочных металлов, такие как фосфат калия и тому подобное).
Существует множество кислот, которые являются подходящими для использования в способе по изобретению, и к ним относятся, для пояснения и не для ограничения, борная, угольная, азотная (HNO3), фосфорная (Н3РО4), сульфаминовая и серная (H2SO4) кислоты, а также галоидводородные кислоты, такие как бромистоводородная (HBr), хлористоводородная (HCl), фтористоводородная (HF) и йодистоводородная (HI) кислота.
После завершения любых стадий способа, полученная смесь или продукт реакции могут быть дополнительно обработаны, с получением желаемого продукта. Например, полученная смесь или продукт реакции могут быть подвергнуты одной или нескольким из следующих процедур: концентрирование или распределение (например, между EtOAc и водой или между 5% ТГФ в EtOAc и 1M фосфорной кислотой); экстрагирование (например, EtOAc, CHCl3, DCM, HCl); промывание (например, этанолом, гептанами, насыщенным водным раствором NaCl, насыщенным раствором NaHCO3, Na2CO3 (5%), CHCl3 или 1M NaOH); перегонка; сушка (например, над MgSO4, над Na2SO4, в атмосфере азота или при пониженном давлении); осаждение; фильтрование; кристаллизация (например, из этанола, гептанов или изопропилацетата) и/или концентрирование (например, в вакууме).
После завершения любой из стадий кристаллизации, кристаллическое соединение может быть выделено из реакционной смеси обычными методами, такими как осаждение, концентрирование, центрифугирование, сушка (например, при комнатной температуре) и тому подобное.
Способ получения соединения формулы I представляет собой одностадийную реакцию алкилирования, которая предусматривает объединение соединения имидазола формулы 1 с соединением бифенила формулы 2, с образованием соединения формулы I. Соединения формулы 1 и 2 могут быть получены традиционными методами с использованием коммерчески доступных исходных материалов и традиционных реагентов. Например, см. описанные в данном описании примеры приготовления, а также публикации заявок США № 2008/0269305 и 2009/0023228, обе Allegretti et al.
В одном варианте осуществления используют небольшой избыток соединения имидазола формулы 1 в расчете на количество соединения бифенила формулы 2. В одном варианте осуществления используют от около 1 до около 2 эквивалентов имидазола, и в другом варианте осуществления используют от около 1 до 1,5 эквивалента.
Обычно, соединения формулы 1 и 2 объединяют в органическом разбавителе и водном разбавителе основного характера в присутствии межфазного катализатора. В одном варианте осуществления используют небольшой избыток водного разбавителя основного характера в расчете на количество соединения имидазола формулы 1. В одном варианте осуществления используют от около 1 до около 2 эквивалентов водного разбавителя основного характера, и в другом варианте осуществления используют от около 1 до 1,5 эквивалента.
Примеры межфазных катализаторов включают четвертичные аммониевые соли, такие как тетрабутиламмонийбромид (Bu4NBr), дидецилдиметиламмонийбромид (DDAB), метилтрифенилфосфонийбромид, метилтридециламмонийхлорид и тому подобное; и в одном варианте осуществления используют тетрабутиламмонийбромид. В одном варианте осуществления используют от около 0,01 до около 1,0 эквивалента межфазного катализатора в расчете на количество соединения бифенила формулы 2; и в другом варианте осуществления используют от около 0,03 до около 0,07 эквивалента.
Органический разбавитель и водный разбавитель основного характера являются по существу несмешивающимися, что означает, что два разбавителя не смешиваются с образованием раствора, т.е. они по существу нерастворимы друг в друге и обычно присутствуют в двух раздельных фазах, когда их смешивают, однако, следует отметить, что потенциально возможно небольшое смешивание разбавителей на поверхности их раздела. В одном варианте осуществления органическим разбавителем является толуол, и водным разбавителем основного характера является NaOH.
Получение соединения формулы I обычно проводят при температуре в пределах от около 20°C до около 40°C, и в одном варианте осуществления при температуре в пределах от около 25°C до около 35°C в течение около 24 до около 72 часов, и в одном варианте осуществления в течение от около 48 до 60 часов, или до тех пор, пока образование соединения формулы I по существу ни завершится.
Когда образование соединения формулы I по существу завершено, полученный продукт выделяют и очищают традиционными методами. Соединение формулы I необязательно кристаллизуют путем обработки этанолом до полного растворения, охлаждают для содействия кристаллизации, и выделяют полученное твердое вещество, с получением кристаллического вещества. Обычно растворение проводят при температуре в пределах от около 40°C до около 70°C и в одном варианте осуществления при температуре в пределах от около 50°C до 60°C. Стадию охлаждения осуществляют при температуре в пределах от около 0°C до около 10°C и в одном варианте осуществления при температуре в пределах от около 2°C до 6°C в течение от около 2 до 6 часов или пока ни завершится формирование кристаллов. После завершения стадии кристаллизации, кристаллическое соединение формулы I может быть выделено из реакционной смеси любыми традиционными способами.
Предшествующие способы получения соединений формулы I часто имели результатом получение высокого процентного содержания побочных продуктов формулы 1, часто настолько высокого, как 15%. Применение органического разбавителя и водного разбавителя основного характера в сочетании с межфазным катализатором, как в данном способе, снижает количество побочного продукта до менее чем 2%, обеспечивая реакцию с улучшенной селективностью, чем в известных ранее способах.
Способ получения соединения формулы II или его соли проводят в три стадии. Первой стадией способа является реакция сочетания Сузуки, которая предусматривает объединение одного эквивалента альдегида формулы I с одним или несколькими эквивалентами реагента калий-С1-6алкилтрифторбората в присутствии палладий-фосфинового катализатора, с образованием соединения формулы 3.
Альдегиды формулы I, используемые в способе по изобретению, могут быть получены описанными в данном описании способами или могут быть получены традиционными методами с использованием коммерчески доступных исходных веществ и традиционных реагентов. Например, см. описанные в данном описании приготовления, а также публикации США № 2008/0269305 и 2009/0023228, обе Allegretti et al., в которых описаны различные способы получения таких соединений.
Обычно альдегид формулы I и реагент калий-С1-6алкилтрифторборат объединяют с палладий-фосфиновым катализатором в инертном разбавителе в присутствии избыточного количества подходящего основания, с получением реакционной смеси. В одном варианте осуществления используют от около 1 до около 2 эквивалентов реагента калий-С1-6алкилтрифторбората в расчете на количество альдегида, и в другом варианте осуществления используют от около 1,4 до около 1,5 эквивалента.
Реагент калий-С1-6алкилтрифторборат выбирают на основании желаемой группы R1. Например, чтобы получить соединение формулы 3, где R1 означает этил, подходящим реагентом калий-С1-6алкилтрифторборатом является калий-этилтрифторборат.
Палладий-фосфиновый катализатор может быть единственным катализатором, содержащим палладий и фосфин, таким как бис(трифенилфосфин)палладий(II),
тетракис(трифенилфосфин)палладий(0) (Pd(PPh3)4),
[1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий(II),
бис[1,2-бис(дифенилфосфино)пропан]палладий(0) и тому подобное. Альтернативно, палладий-фосфиновый катализатор может быть комбинацией палладиевого катализатора и источника фосфина. Примеры палладиевых катализаторов включают ацетат палладия(II) (Pd(OAc)2), хлорид палладия(II) (PdCl2) и тому подобное. Соответствующие источники фосфина включают ди(1-адамантил)-н-бутилфосфин, трифенилфосфин, этилдифенилфосфин, дициклогексилфенилфосфин, 2-пиридилдифенилфосфин, бис(6-метил-2-пиридил)фенилфосфин, три-п-хлорфенилфосфин, три-п-метоксифенилфосфин и тому подобное. В одном варианте осуществления палладиевым катализатором является ацетат палладия(II) и источником фосфина является ди(1-адамантил)-н-бутилфосфин.
В одном варианте осуществления используют от около 0,01 до около 0,04 эквивалента палладиевого катализатора и от около 0,02 до около 0,06 эквивалента источника фосфина в расчете на количество альдегида, и в другом варианте осуществления используют от около 0,02 до около 0,03 эквивалента палладиевого катализатора и от около 0,03 до около 0,05 эквивалента источника фосфина. В другом варианте осуществления используют от около 0,03 до около 0,1 эквивалента палладий-фосфинового катализатора в расчете на количество альдегида, и в другом варианте осуществления используют от около 0,05 до около 0,08 эквивалента.
Используемое избыточное количество основания обычно составляет от около 3,0 до около 6,0 эквивалентов в расчете на количество альдегида и в одном варианте осуществления - от около 3,0 до около 4,0 эквивалентов. В одном варианте осуществления инертным разбавителем является смесь толуола и воды. В другом варианте осуществления основанием является карбонат щелочного металла, такой как карбонат цезия.
Получение соединения формулы 3 обычно осуществляют при температуре в пределах от около 80°C до около 100°C и в одном варианте осуществления при температуре в пределах от около 85°C до около 95°C в течение около 12 до около 20 часов, и в одном варианте осуществления в течение около 14 до 18 часов или пока по существу ни завершится образование соединения формулы 3. Когда образование соединения формулы 3 по существу завершено, полученный продукт выделяют и очищают традиционными методами. В одном варианте осуществления соединение формулы 3 получают в растворе.
Второй стадией способа является стадия образования оксима, которая включает объединение одного эквивалента альдегида формулы 3 с одним или несколькими эквивалентами гидроксиламина или его соли, с образованием оксима формулы 4.
Обычно соединение формулы 3 и гидроксиламин или его соль объединяют в присутствии избыточного количества подходящего основания для получения реакционной смеси. В одном варианте осуществления используют от около 1 до около 2 эквивалентов гидроксиламина или его соли в расчете на количество соединения формулы 3, и в другом варианте осуществления используют от около 1,4 до около 1,5 эквивалента.
Используемое избыточное количество основания обычно составляет от около 3,0 до около 6,0 эквивалентов в расчете на количество соединения формулы 3, и в одном варианте осуществления от около 3,0 до около 4,0 эквивалентов. В одном варианте осуществления основанием является карбонат щелочного металла, такой как гидрокарбонат натрия.
Оксим формулы 4 обычно получают при температуре в пределах от около 20°C до около 60°C, и в одном варианте осуществления при температуре в пределах от около 30°C до около 50°C в течение около 20 до около 28 часов, и в одном варианте осуществления в течение около 22 до 26 часов, или пока по существу ни завершится образование оксима. Когда образование оксима по существу завершено, полученный продукт выделяют и очищают традиционными методами.
Третьей стадией способа является восстановление оксима до первичного амина и взаимодействие оксима формулы 4 с восстановителем, с образованием амина формулы II или его соли.
Примерами восстановителей являются такие, которые пригодны для восстановления оксима до амина, и к ним относятся водород/никель Ренея, палладий на углероде (Pd/C) и Zn-HCl. Обычно оксим формулы 4 и восстановитель объединяют в полярном протонном растворителе и аминном основании для получения реакционной смеси. Получение амина обычно осуществляют при температуре окружающей среды в течение от 1 до около 5 часов, и в одном варианте осуществления в течение от около 2 до 4 часов, или пока по существу не завершится образование амина. В одном варианте осуществления аминным основанием является гидроксид аммония, и растворителем является этанол.
Когда образование амина по существу завершено, полученный продукт выделяют и очищают традиционными методами. Амин необязательно кристаллизуют путем обработки гептанами до полного растворения, охлаждения для содействия кристаллизации и выделения полученного твердого вещества, с получением кристаллического вещества. Обычно стадию охлаждения проводят при температуре в пределах от около 0°C до около 10°C, и в одном варианте осуществления при температуре в пределах от около 2°C до 6°C, в течение около 22 до 26 часов или пока не завершится образование кристаллов. После завершения стадии кристаллизации кристаллическое соединение формулы II или его соль может быть выделена из реакционной смеси любыми традиционными способами.
Способ получения соединения формулы III или его соли проводят в пять стадий. Первая, вторая и третья стадии описаны выше со ссылкой на способ получения соединения формулы II.
Четвертая стадия способа является стадией сочетания, которая предусматривает взаимодействие одного эквивалента амина формулы II или его соли с одним или несколькими эквивалентами карбоновой кислоты формулы 5 или ее соли в присутствии одного или нескольких эквивалентов реагента сочетания амин-карбоновая кислота, с получением соединения формулы 6 или его соли.
Карбоновые кислоты формулы 5, используемые в способе по изобретению, известны в данной области либо коммерчески доступны, либо могут быть получены традиционными методами с использованием коммерчески доступных исходных веществ и традиционных реагентов. Например, см. описанные в данном описании приготовления, а также публикации заявок США № 2008/0269305 и 2009/0023228, обе Allegretti et al., которые описывают различные способы получения таких соединений.
Обычно амин или его соль и карбоновую кислоту или ее соль объединяют в инертном разбавителе в присутствии реагента сочетания с образованием реакционной смеси. В одном варианте осуществления используют от около 1 до около 2 эквивалентов карбоновой кислоты в расчете на количество амина, и в другом варианте осуществления используют от около 1,1 до около 1,3 эквивалентов. В одном варианте осуществления используют от около 1 до около 2 эквивалентов реагента сочетания в расчете на количество амина, и в другом варианте осуществления используют от около 1,1 до около 1,3 эквивалента. Примеры инертных разбавителей включают дихлорметан и изопропилацетат.
Подходящие реагенты сочетания амин-карбоновая кислота включают (гексафторфосфат 2-(6-хлор-1H-бензотриазол-1-ил)-1,1,3,3-тетраметиламиния) (HCTU), гексафторфосфат бензотриазол-1-илокситрис(диметиламино)фосфония (BOP), гексафторфосфат бензотриазол-1-илокситрипирролидинофосфония (PyBOP), гексафторфосфат О-(7-азабензотриазол-1-ил-N,N,N',N'-тетраметилурония (HATU), дициклогексилкарбодиимид (DCC), N-(3-диметиламинопропил)-N'-этилкарбодиимид (EDC), карбонилдиимидазол (CDI) и тому подобное, и в одном конкретном варианте осуществления реагентом сочетания является HCTU.
Реакцию сочетания обычно проводят при температуре в пределах от около -5°C до около 5°C, и в одном варианте осуществления при температуре в пределах от около -1°C до около 3°C, в течение от около 5 до около 15 часов или пока образование соединения формулы 6 по существу не завершится. pH реакционной смеси доводят до около 5 добавлением подходящей кислоты, такой как 1н хлористоводородная кислота. Когда образование соединения формулы 6 по существу завершено, полученный продукт выделяют и очищают традиционными методами.
Пятой стадией способа является стадия удаления защиты, которая предусматривает удаление защитной группы карбоновой кислоты, P, из соединения формулы 6 или его соли, с получением соединения формулы III или его соли.
Для удаления группы Р используют стандартные приемы удаления защиты и реагенты, и они могут изменяться в зависимости от группы, которую используют. Например, NaOH используют, как правило, когда P представляет метил, кислоты, такие как TFA или HCl используют, как правило, когда P представляет трет-бутил, и условия каталитического гидрирования, такие как H2 (1 атм.) и 10% Pd/C в спиртовом растворителе ("H2/Pd/C") могут быть использованы, когда P представляет бензил. В одном конкретном варианте осуществления используют TFA.
Обычно соединение формулы 6 или его соль и реагент снятия защиты объединяют в инертном разбавителе. Используют избыточное количество реагента, в одном варианте осуществления используют от около 10 до около 30 эквивалентов реагента в расчете на количество соединения формулы 6. В одном варианте осуществления инертный разбавитель является безводным, такой как тетрагидрофуран, дихлорметан, N,N'-диметилформамид и 1,4-диоксан.
Стадию удаления защиты обычно проводят при температуре в пределах от около 10°C до около 30°C, и в одном варианте осуществления при температуре в пределах от около 15°C до около 25°C в течение от около 12 до около 20 часов, и в одном варианте осуществления в течение от около 14 до 18 часов, или пока реакция по существу не завершится. pH реакционной смеси затем доводят до от около 6 до около 7 добавлением подходящего основания, такого как водный карбонат калия.
Когда образование соединения формулы III по существу завершается, полученный продукт выделяют и очищают традиционными методами. Соединение формулы III необязательно кристаллизуют путем обработки изопропилацетатом до полного растворения, путем охлаждения для содействия кристаллизации и выделением полученного твердого вещества, с получением кристаллического вещества. Обычно стадию охлаждения проводят при температуре в пределах от около 0°C до около 10°C, и в одном варианте осуществления при температуре в пределах от около 2°C до 6°C в течение от около 14 до 18 часов или до тех пор, пока не образуются кристаллы. После завершения стадии кристаллизации кристаллическое соединение формулы III может быть выделено из реакционной смеси любыми традиционными способами.
Соединение формулы III может быть затем использовано для получения соединения формулы IV преобразованием группы сложного тиоэфира, -SC(O)-R4, в тиол, -SH. Это может быть осуществлено традиционными способами, такими как обработка основаниями, такими как гидроксид натрия, метоксид натрия, первичные алкиламины и гидразин.
Кристаллические свойства
Одним примером соединения формулы I является трет-бутиловый эфир 4'-(4-бром-2-этокси-5-формилимидазол-1-илметил)-3'-фторбифенил-2-карбоновой кислоты, который представлен формулой Ia:
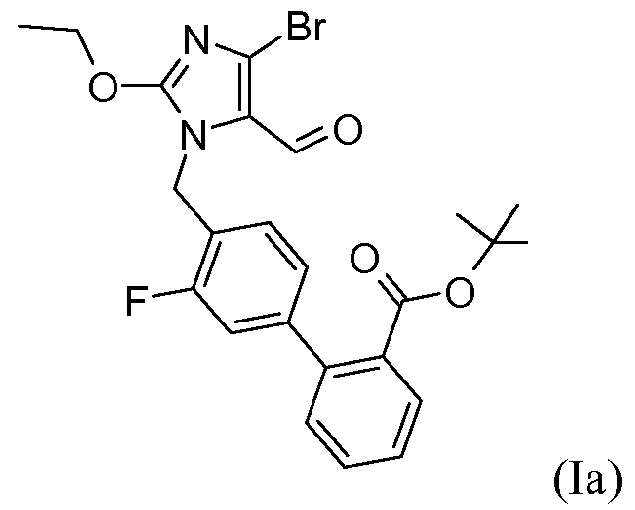
В одном варианте осуществления соединение формулы Ia находится в кристаллической форме.
Одним примером соединения формулы II является трет-бутиловый эфир 4'-(5-аминометил-2-этокси-4-этилимидазол-1-илметил)-3'-фторбифенил-2-карбоновой кислоты, который представлен формулой IIa:
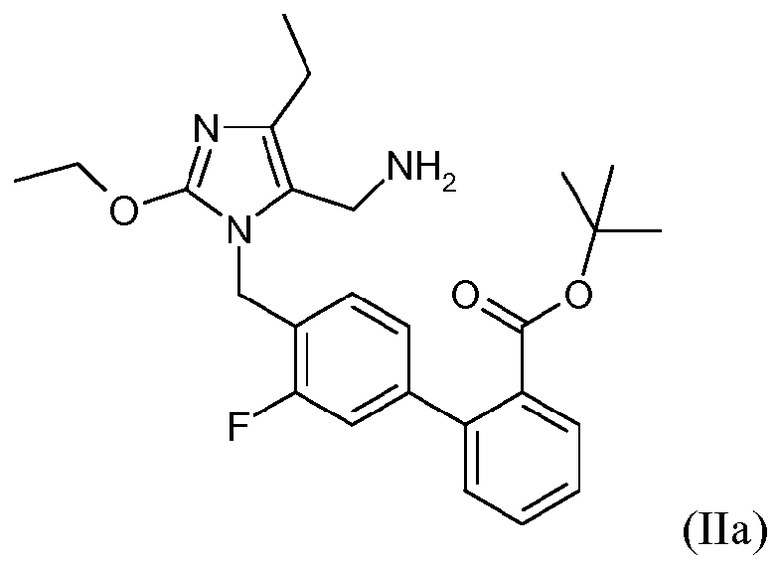
В одном варианте осуществления соединение формулы IIa находится в кристаллической форме. В другом варианте осуществления кристаллическая форма не ассоциирована с какими-либо противоионами и представлена как кристаллическая форма свободного основания.
Одним примером соединения формулы III является 4'-{5-[((S)-2-ацетилсульфанил-4-метилпентаноиламино)метил]-2-этокси-4-этилимидазол-1-илметил}-3'-фторбифенил-2-карбоновая кислота, которая представлена формулой IIIa:
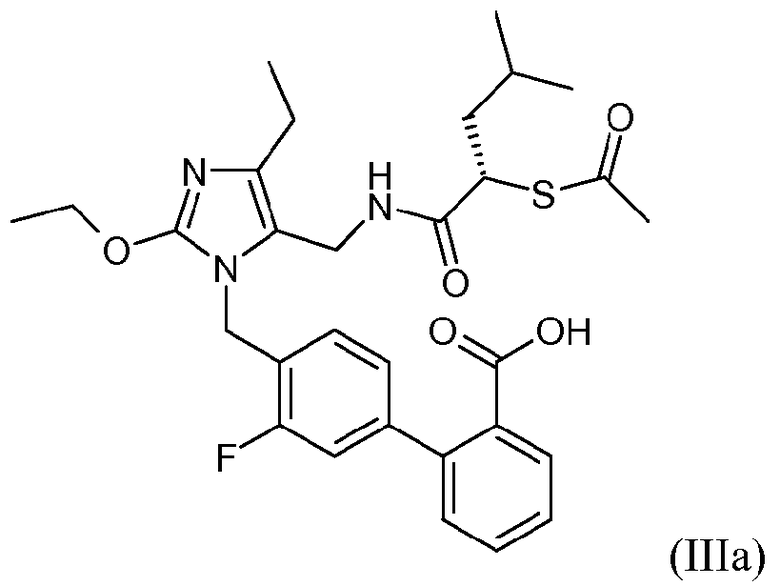
В одном варианте осуществления соединение формулы IIIa находится в кристаллической форме. В другом варианте осуществления кристаллическая форма является цвиттерионом.
Как хорошо известно в области порошковой рентгеновской дифракции, относительная высота пиков спектра PXRD зависит от некоторых факторов, относящихся к приготовлению образца и геометрии инструмента, тогда как положения пиков относительно нечувствительны к экспериментальным деталям. Рентгенограмму PXRD получали, как описано в примере 4. Таким образом, в одном варианте осуществления кристаллические соединения по изобретению характеризуются рентгенограммой PXRD, имеющей конкретные положения пиков.
Кристаллическая форма трет-бутилового сложного эфира 4'-(5-аминометил-2-этокси-4-этилимидазол-1-илметил)-3'-фторбифенил-2-карбоновой кислоты (формула IIa) характеризуется рентгенограммой PXRD, на которой положения пиков по существу находятся в соответствии положениями, которые показаны на фиг.1. Эти пики перечислены ниже в порядке снижения относительной интенсивности.
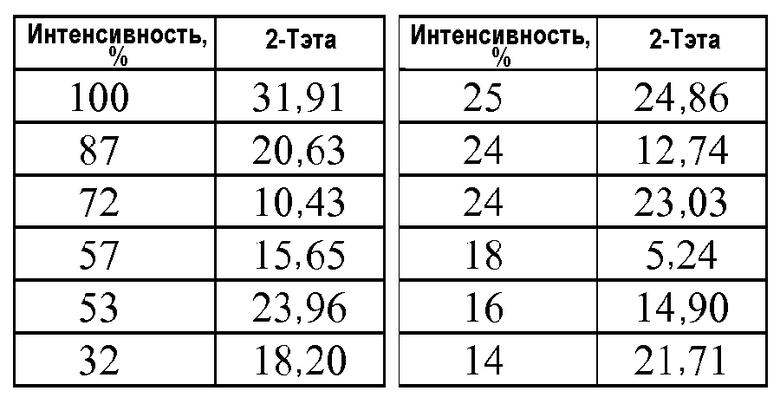
Таким образом, в одном варианте осуществления кристаллическая форма формулы IIa характеризуется порошковой рентгенограммой (PXRD), содержащей дифракционные пики при величинах 2θ 5,24±0,2, 10,43±0,2, 15,65±0,2, 20,63±0,2 и 31,91±0,2, и дополнительно характеризуется наличием одного или нескольких дополнительных дифракционных пиков при величинах 2θ, выбранных из 12,74±0,2, 14,90±0,2, 18,20±0,2, 21,71±0,2, 23,03±0,2, 23,96±0,2 и 24,86±0,2.
Кристаллическая форма 4'-{5-[((S)-2-ацетилсульфанил-4-метилпентаноиламино)метил]-2-этокси-4-этилимидазол-1-илметил}-3'-фторбифенил-2-карбоновой кислоты (формула IIIa) характеризуется рентгенограммой PXRD, на которой положения пиков по существу находятся в соответствии с показанными на фиг.3. Эти пики перечислены ниже в порядке снижения относительной интенсивности.
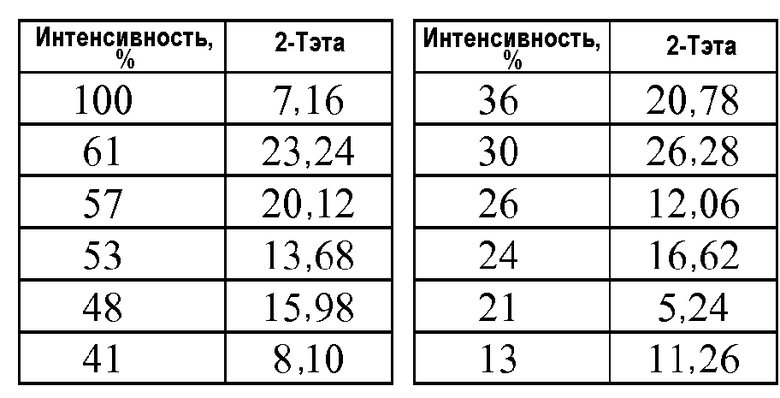
Так, в одном варианте осуществления кристаллическая форма формулы IIIa характеризуется порошковой рентгенограммой (PXRD), содержащей дифракционные пики при величинах 2θ 5,24±0,2, 7,16±0,2, 13,68±0,2 и 15,98±0,2, и дополнительно характеризуется наличием одного или нескольких дополнительных дифракционных пиков при величинах 2θ, выбранных из 8,10±0,2, 11,26±0,2, 12,06±0,2, 16,62±0,2, 20,12±0,2, 20,78±0,2, 23,24±0,2 и 26,28±0,2.
Кривые дифференциальной сканирующей калориметрии (DSC) получали, как изложено в примере 5. Таким образом, в одном варианте осуществления кристаллические соединения по изобретению характеризуются их термограммами DSC. В одном варианте осуществления кристаллическая форма формулы IIa характеризуется термограммой DSC, которая показывает температуру плавления около 76,0°C при отсутствии существенного термического разложения ниже 150,0°C, как видно на фиг.2. В одном варианте осуществления кристаллическая форма формулы IIIa характеризуется термограммой DSC, которая показывает температуру плавления около 130,9°C при отсутствии существенного термического разложения ниже 150,0°C, как видно на фиг.4.
Термогравиметрический анализ (TGA) проводили на кристаллических соединениях по изобретению, как описано в примере 5. Таким образом, в одном варианте осуществления кристаллические соединения по изобретению характеризуются их кривыми TGA. В одном варианте осуществления кристаллическая форма формулы IIa характеризуется кривой TGA, которая показывает потерю растворителей и/или воды (<0,5%) при температурах ниже 150°C (которые значительно выше, чем температура плавления), как видно на фиг.2. В одном варианте осуществления кристаллическая форма формулы IIIa характеризуется кривой TGA, которая показывает потерю растворителей и/или воды (<0,5%) при температурах ниже 150°C (которые значительно выше, чем температура плавления), как видно на фиг.4.
Эти свойства кристаллических соединений по изобретению дополнительно поясняются в примерах ниже.
Примеры
Следующие приготовления и примеры приведены для пояснения конкретных вариантов осуществления данного изобретения. Эти конкретные варианты осуществления, однако, не предназначены для ограничения объема настоящего изобретения каким-либо образом, если особо не указано иное.
Следующие сокращения имеют следующие значения, если не указано иное, и любые другие используемые в данном описании сокращения, которым не дано определение, имеют свои стандартные общепринятые значения:
Если не указано иное, все материалы, такие как реагенты, исходные вещества и растворители, приобретали от коммерческих поставщиков (таких как Sigma-Aldrich, Fluka Riedel-de Haen, Strem Chemicals, Inc. и тому подобное) и использовали без дополнительной очистки.
Реакции проводили в атмосфере азота, если не указано иное. Течение реакций отслеживали тонкослойной хроматографией (ТСХ), аналитической высокоэффективной жидкостной хроматографией (аналитическая. ВЭЖХ) и масс-спектрометрией, подробности которых даны в конкретных примерах. Растворителями, используемыми в аналитической ВЭЖХ, были следующие: растворитель A - 98% воды/2% MeCN/1,0 мл/л TFA, растворитель B - 90% MeCN/10% воды/1,0 мл/л TFA.
Реакционные смеси обрабатывали, как конкретно описано в каждом приготовлении или примере, как правило, реакционные смеси очищали экстракцией и другими способами очистки, такими как зависимая от температуры и растворителя кристаллизация и осаждение. В дополнение, реакционные смеси обычно очищали препаративной ВЭЖХ, обычно с использованием набивок колонок Microsorb CIS и Microsorb BDS и традиционных элюентов. Продукты реакции обычно характеризовали с помощью масс-спектрометрии и 1Н-ЯМР. Для измерения ЯМР образцы растворяли в дейтерированном растворителе (CD3OD, CDCl3 или ДМСО-d6), и спектр 1Н-ЯМР получали с использованием прибора Varian Gemini 2000 (400 МГц) в условиях стандартного наблюдения. Масс-спектрометрическую идентификацию соединений обычно проводили, используя метод электрораспылительной ионизации (ESMS) на приборе Applied Biosystems (Foster City, CA) model API 150 EX instrument или прибором Agilent (Palo Alto, CA) model 1200 LC/MSD.
Приготовление 1
5-Бром-2-этокси-3H-имидазол-4-карбальдегид
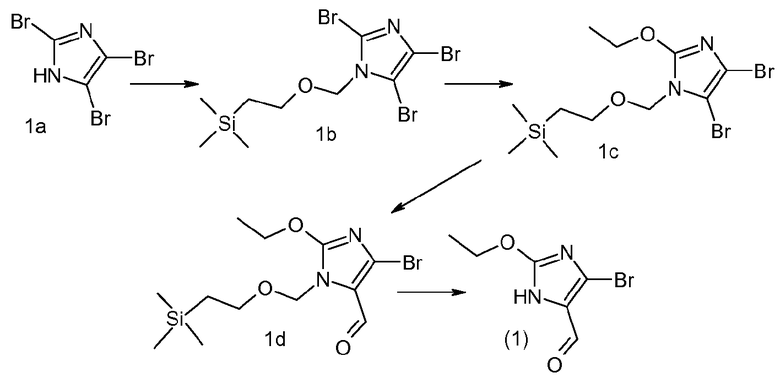
2,4,5-Трибром-1H-имидазол (la) (98,7 г, 324 ммоль, 1,0 экв.) растворяли в 1,20 л DCM и охлаждали до 0°C. К этому добавляли DIPEA (62 мл, 360 ммоль, 1,1 экв.) с последующим медленным добавлением [β-(триметилсилил)этокси]метилхлорида (60,2 мл, 340 ммоль, 1,05 экв.). Раствор медленно нагревали до комнатной температуры. Через 2 часа смесь промывали смесью 1M H3PO4/насыщенный водный NaCl (1:10; 2×600 мл). Органический слой сушили над MgSO4 и выпаривали досуха, с получением промежуточного соединения (1b) в виде бледно-желтой жидкости, которая отверждалась при стоянии (137 г).
Промежуточное соединение (1b) (130 г, 290 ммоль, 1,0 экв.) растворяли в безводном EtOH (650 мл). К этому медленно добавляли трет-бутоксид калия (98,6 г, 879 ммоль, 3,0 экв.) и смесь нагревали при кипячении с обратным холодильником в течение 16 часов. Смесь затем охлаждали до комнатной температуры, фильтровали и концентрировали. Полученное масло растворяли в EtOAc (800 мл) и промывали насыщенным раствором NaHCO3 (400 мл). Слои разделяли, и органический слой промывали насыщенным водным раствором NaCl, сушили над MgSO4, фильтровали и концентрировали, с получением промежуточного соединения (1c) в виде бурого масла (115,3 г). MC m/z: [M+H+] рассчитано для C11H20Br2N2O2Si, 401,9, найдено 401,2.
Промежуточное соединение (1c) (69,5 г, 174 ммоль, 1,0 экв.) растворяли в безводном ТГФ (600 мл) и охлаждали до -78°C в атмосфере азота. Добавляли по каплям 2,5M раствор н-бутиллития в гексанах (72,9 мл, 180 ммоль, 1,05 экв.) и смесь перемешивали при -78°C в течение 10 минут. Затем добавляли ДМФА (40 мл, 520 ммоль, 3,0 экв.), смесь перемешивали при -78°C в течение 15 минут и затем нагревали до комнатной температуры. Реакцию гасили водой (10 мл), разбавляли EtOAc (600 мл) и промывали водой (100 мл), насыщенным водным раствором NaCl, сушили над MgSO4 и концентрировали при пониженном давлении. Выделенное вещество очищали хроматографией на силикагеле (15-30% EtOAc:гексаны), с получением промежуточного соединения (1d) в виде бледно-желтого масла (45 г).
Промежуточное соединение (1d) (105,8 г, 303 ммоль, 1,0 экв.) охлаждали при 0°C во льду. TFA (300 мл) добавляли и смесь перемешивали при 0°C в течение 15 минут, затем нагревали до комнатной температуры. Через 90 минут смесь концентрировали при пониженном давлении и повторно растворяли в EtOAc (700 мл). Органическую фазу промывали насыщенным раствором гидрокарбоната (2×600 мл), насыщенным водным раствором NaCl, сушили над MgSO4 и концентрировали при пониженном давлении, с получением желтого твердого вещества. Полученное вещество суспендировали в гексанах (300 мл) и перемешивали при 0°C в течение 30 минут. Полученные вещества фильтровали и твердое вещество промывали холодными гексанами (150 мл), с получением указанного в заголовке соединения (1) в виде не совсем белого твердого вещества (61,2 г).
1Н ЯМР (CDCl3) δ (м.д.): 1,4 (м, 3Н), 4,5 (м, 2Н), 5,2 (с, 1Н), 9,2 (д, 1Н).
Приготовление 2
трет-Бутиловый эфир 4'-бромметил-3'-фторбифенил-2-карбоновой кислоты
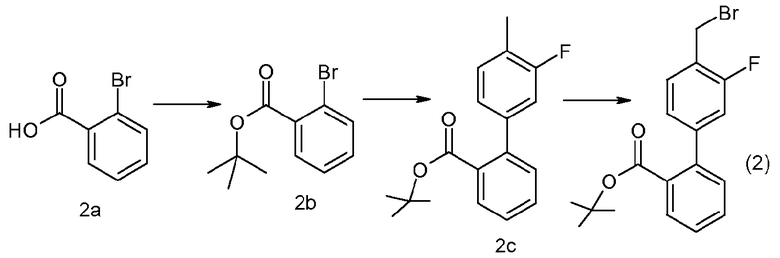
К раствору 1,0 M DCC в DCM (800 мл, 800 моль), охлажденному до 0°C, добавляли 2-бромбензойную кислоту (2a) (161 г, 800 ммоль) с последующим добавлением DMAP (9,0 г, 740 ммоль) и трет-бутилового спирта (82,4 мл, 880 ммоль). Смесь перемешивали при комнатной температуре в течение 10 минут, затем нагревали до комнатной температуры и перемешивали. Затем, спустя 16 часов, смесь фильтровали. Органическую фазу промывали насыщенным раствором NaHCO3 (400 мл), насыщенным водным раствором NaCl, сушили над MgSO4, фильтровали и концентрировали при пониженном давлении, с получением неочищенного промежуточного соединения (2b) виде масла (228,8 г).
Неочищенное промежуточное соединение (2b) (109,6 г, 426 ммоль) и 3-фтор-4-метилфенилбороновую кислоту (72,2 г, 449 ммоль) суспендировали в изопропиловом спирте (360 мл, 4,7 ммоль). Добавляли 2,0M раствор карбоната натрия в воде (360 мл, 720 ммоль) и смесь дегазировали в атмосфере азота. Тетракис(трифенилфосфин)палладий(0) (4,9 г, 4,3 ммоль) затем добавляли и смесь перемешивали при 90°C в течение 46 часов. Смесь охлаждали до комнатной температуры, разбавляли EtOAc (800 мл) и слои разделяли. Органическую фазу промывали насыщенным водным раствором NaCl и концентрировали при пониженном давлении. Выделенное масло очищали хроматографией на силикагеле (3× 4-6% EtOAc:гексаны), с получением промежуточного соединения (2c) в виде светлого масла (93,3 г).
Промежуточное соединение (2c) (89,8 г, 314 ммоль, 1,0 экв.) растворяли в CCl4 (620 мл, 6,4 моль) и дегазировали в атмосфере азота. Добавляли NBS (55,8 г, 314 ммоль) с последующим добавлением бензоилпероксида (1,5 г, 6,3 ммоль) и смесь нагревали при 90°C в атмосфере азота в течение 7 часов. Реакционную смесь охлаждали на бане со льдом, фильтровали и концентрировали при пониженном давлении. Выделенное масло растирали с 150 мл смеси 3% EtOAc:гексаны. Раствор охлаждали при -20°C в течение 2 часов, затем фильтровали и промывали холодным раствором 3% EtOAc:гексаны (200 мл), с получением указанного в заголовке соединения (2) в виде не совсем белого твердого вещества (88,9 г).
1Н ЯМР (CDCl3) δ (м.д.): 1,3 (м, 9Н), 4,6 (с, 2Н), 7,0-7,1 (м, 2Н), 7,2 (дд, 1Н), 7,4 (м, 1Н), 7,5 (м, 1Н), 7,8 (дд, 1Н).
Пример 1
Кристаллический трет-бутиловый эфир 4'-(5-аминометил-2-этокси-4-этилимидазол-1-илметил)-3'-фторбифенил-2-карбоновой кислоты
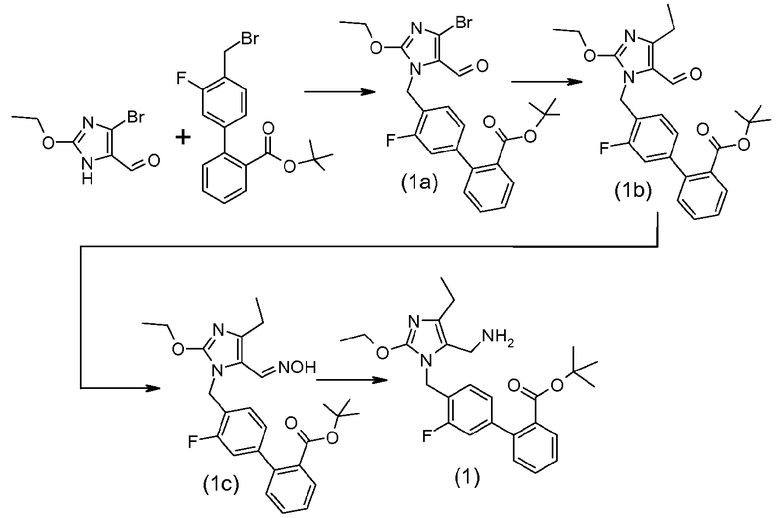
5-Бром-2-этокси-3H-имидазол-4-карбальдегид (22,0 г, 100 ммоль, 1,1 экв.), трет-бутиловый эфир 4'-бромметил-3'-фторбифенил-2-карбоновой кислоты (33,0 г, 90 ммоль, 1 экв.) и Bu4NBr (1,6 г, 5 ммоль, 0,05 экв.) растворяли в толуоле (400 мл) и 1н NaOH (120 мл, 120 ммоль, 1,2 экв.). Полученную смесь перемешивали при 27°C в течение 48-60 часов. Слой толуола отделяли, промывали водой (2×200 мл), затем удаляли перегонкой. К остатку добавляли EtOH (350 мл) и смесь нагревали при 50-60°C до тех пор, пока не растворялись твердые вещества. Смесь охлаждали до комнатной температуры более 4 часов, затем охлаждали до 4°C и перемешивали при 4°C в течение 4 часов. Твердые вещества отфильтровывали, промывали холодным EtOH (60 мл) и сушили при комнатной температуре в вакууме в течение 24 часов, с получением промежуточного соединения (1a) (~39 г).
Промежуточное соединение (1a) (20,0 г, 40 ммоль, 1 экв.), этилтрифторборат калия (7,1 г, 52 ммоль, 1,3 экв.), ацетат палладия(II) (224 мг, 1 ммоль, 0,025 экв.), cataCXium®A (бутилди-1-адамантилфосфин; CAS# 321921-71-5; 538 мг, 1,45 ммоль, 0,04 экв.) и Cs2CO3 (45 г, 138 ммоль, 3,45 экв.) растворяли в толуоле (240 мл) и воде (80 мл). Смесь промывали током азота (3×) в вакууме, затем нагревали при 90°C в течение 16 часов. Смесь затем охлаждали до комнатной температуры и слои разделяли. Органический слой промывали водой (2×200 мл), затем перегоняли при пониженном давлении, с получением масла. Масло растворяли в EtOH (240 мл). Добавляли воду (80 мл) и смесь перемешивали в течение 30 минут. Смесь фильтровали, для удаления твердых веществ, твердые вещества промывали 75% EtOH (130 мл) и фильтрат собирали, с получением промежуточное соединение (1b) в растворе EtOH, которое использовали непосредственно на следующей стадии.
Раствор в EtOH промежуточного соединения (Ib) (10 ммоль, 1 экв.) объединяли с гидрохлоридом гидроксиламина (27,2 г, 52 ммоль, 1,3 экв.) и NaHCO3 (35,2 г, 3,45 экв.). Смесь перемешивали при 40°C в течение 24 часов, затем охлаждали до комнатной температуры. Осадок отфильтровывали, промывали 75% EtOH (100 мл) и 50% EtOH (200 мл), затем сушили при пониженном давлении при 30°C в течение 24 часов, с получением промежуточного соединения (1c) (15 г).
Промежуточное соединение (1c) (5 г) объединяли с EtOH (100 мл), NH4OH (28%, 6 мл) и никелем Ренея (влажный 10 г) до образования суспензии. Смесь дегазировали в атмосфере азота (3×), дегазировали в атмосфере водорода (3×), затем перемешивали в атмосфере водорода (1 атм.) в течение 3 часов. Смесь фильтровали, для удаления катализатора, и твердые вещества промывали EtOH (20 мл). Фильтрат затем обрабатывали древесным углем (0,5 г) и снова фильтровали. Фильтрат затем перегоняли в вакууме, с получением масла. Добавляли гептаны (50 мл) и смесь перегоняли до масла (2×). Оставшееся масло растворяли в гептанах (60 мл) при нагревании смеси и перемешивании при 4°C в течение 24 часов. Твердые вещества затем отфильтровывали, промывали холодными гептанам (10 мл) и сушили при комнатной температуре в течение 24 часов, с получением указанного в заголовке соединения в виде кристаллического вещества (3,8 г).
Приготовление 3
(S)-2-Ацетилсульфанил-4-метилпентановая кислота
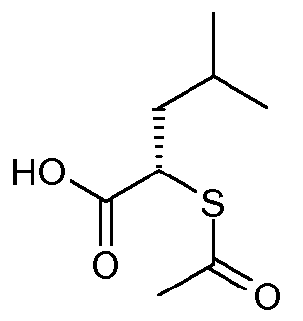
D-Лейцин (8,2 г, 62,7 ммоль) растворяли в 3,0M HBr в воде (99 мл, 0,3 моль) и охлаждали до 0°C. Раствор NaNO2 (6,9 г, 100 ммоль) в воде (11,3 мл, 627 ммоль) медленно добавляли в течение 20 минут. Смесь перемешивали при 0°C в течение 3 часов и затем экстрагировали дважды простым этиловым эфиром, промывали водой, затем насыщенным водным раствором NaCl, сушили над MgSO4, фильтровали и концентрировали, с получением (R)-2-бром-4-метилпентановой кислоты (11,5 г) в виде желтоватого масла. Его использовали на следующей стадии без дополнительной очистки.
Тиоуксусную кислоту (4,2 г, 54,4 ммоль) и ДМФА (100 мл, 1,0 моль) объединяли и смесь охлаждали на бане со льдом. Добавляли карбонат натрия (5,8 г, 54,4 ммоль). Спустя 30 минут, (R)-2-бром-4-метилпентановую кислоту (10,1 г, 51,8 ммоль) в ДМФА (20 мл) добавляли по каплям и смесь перемешивали при температуре от 0°C до комнатной температуры в течение 6 часов. Смесь разбавляли 100 мл EtOAc и экстрагировали 100 мл смеси 1:1 1н HCl:насыщенный водный раствор NaCl. Слои разделяли и водную фазу экстрагировали дополнительным EtOAc (100 мл). Органические вещества объединяли, промывали насыщенным водным раствором NaCl, сушили над MgSO4, фильтровали и концентрировали при пониженном давлении. Выделенное масло растворяли в простом диизопропиловом эфире (45 мл, 320 ммоль) и охлаждали до 0°C. Дициклогексиламин (10,1 мл, 50,7 ммоль) добавляли по каплям и твердому веществу давали возможность осаждения из раствора. После перемешивания в течение дополнительных 30 минут материал фильтровали и промывали 75 мл холодного простого диизопропилового эфира. Выделенное твердое вещество (14 г) суспендировали в 100 мл EtOAc. Добавляли 150 мл 5% KHSO4 и слои разделяли. Органическую фазу промывали насыщенным водным раствором NaCl, сушили над MgSO4, фильтровали и концентрировали при пониженном давлении. Выделенное масло затем подвергали азеотропной обработке (3×25 мл толуола), с получением указанного в заголовке соединения (6,1 г) в виде соли дициклогексиламина.
Пример 2
Кристаллическая 4'-(5-[((S)-2-ацетилсульфанил-4-метилпентаноиламино)метил]-2-этокси-4-этилимидазол-1-илметил)-3'-фторбифенил-2-карбоновая кислота
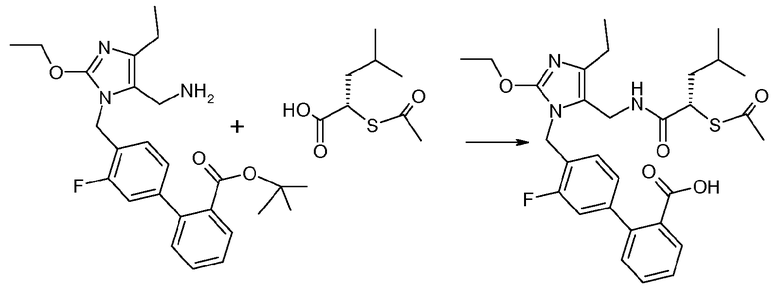
Кристаллический трет-бутиловый эфир 4'-(5-аминометил-2-этокси-4-этилимидазол-1-илметил)-3'-фторбифенил-2-карбоновой кислоты (соль дициклогексиламина, 18 г, 40 ммоль, 1 экв.), (S)-2-ацетилсульфанил-4-метилпентановую кислоту (18 г, 48 ммоль, 1,2 экв.) и HCTU (19 г, 48 ммоль, 1,2 экв.) объединяли в предварительно охлажденном сосуде (0°C в течение 10 минут) и добавляли холодный DCM (240 мл). Смесь перемешивали при 1±2°C в течение 5-15 часов. Добавляли 4% NaHCO3 (200 мл) и смесь перемешивали в течение 15 минут. Слой DCM отделяли и перегоняли до ~100 мл. Добавляли IPAc (150 мл) и перегоняли до 150 мл. Дополнительное количество IPAc (200 мл) добавляли и смесь промывали 4% NaHCO3 (2×200 мл) и водой (200 мл). Раствор перемешивали с 15% NH4Cl (300 мл) в течение 15 минут, pH доводили до 5,5 1н HCl и затем перемешивали в течение 1 часа. Твердые вещества отфильтровывали. Фильтрат промывали IPAc (50 мл) и слой IPAc отделяли. Слой IPAc перемешивали с 15% NH4Cl (200 мл) в течение 3 часов и любые твердые вещества отфильтровывали. Фильтрат промывали насыщенным водным раствором NaCl (150 мл) и перегоняли в вакууме до ~60 мл. Добавляли DCM (50 мл) и отгоняли. Добавляли DCM (200 мл) и смесь охлаждали до 0-5°C. TFA (70 мл) добавляли медленно (легкая экзотермичность) при температуре ниже 15°C и смесь перемешивали при 20°C в течение 16 часов. Смесь концентрировали до ~150 мл и добавляли IPAc (150 мл). Смесь перегоняли до ~150 мл. Дополнительное количество IPAc (150 мл) добавляли и снова перегоняли до ~150 мл. Добавляли IPAc (200 мл) и полученный раствор медленно добавляли к предварительно охлажденному K2CO3 (52 г) в воде (250 мл) при температуре ниже 10°C (умеренная экзотермичность, pH>7 возможно >6 во время гашения) свыше 15 минут. pH отслеживали во время перехода и дополнительное количество основания (8 г) добавляли, когда pH падал ниже 6. Слой IPAc отделяли и промывали насыщенным водным раствором NaCl (150 мл). Раствор IPAc перегоняли до ~50 мл. MTBE (100 мл) добавляли и смесь перегоняли до ~50 мл. Дополнительно добавляли MTBE (100 мл) и смесь перемешивали при комнатной температуре в течение 3 часов, с получением суспензии, которую затем перемешивали при 4°C в течение 16 часов. Твердые вещества отфильтровывали и промывали смесью MTBE/диизопропиловый эфир (1:1, 100 мл). Твердые вещества затем сушили при комнатной температуре в течение 60 часов в атмосфере азота, с получением указанного в заголовке соединения в виде кристаллического вещества (18,2 г).
Пример 3
Кристаллическая 4'-{2-этокси-4-этил-5-[((S)-2-меркапто-4-метилпентаноиламино)метил]имидазол-1-илметил}-3'-фторбифенил-2-карбоновая кислота
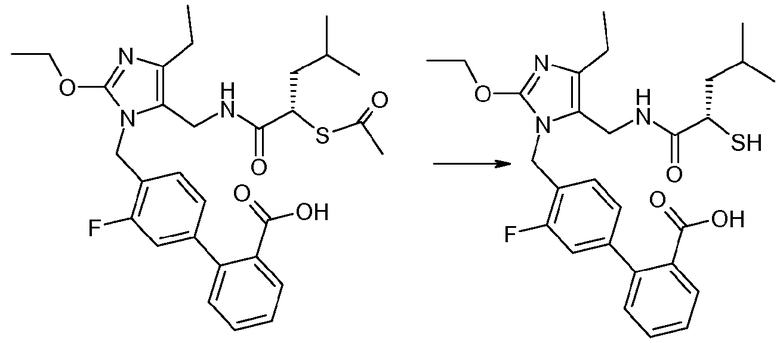
Кристаллическую 4'-{5-[((S)-2-ацетилсульфанил-4-метилпентаноиламино)метил]-2-этокси-4-этилимидазол-1-илметил}-3'-фторбифенил-2-карбоновую кислоту (2,3 г, 4 ммоль, 1 экв.) и DTT (62 мг, 0,4 ммоль, 0,1 экв.) растворяли в MeOH (30 мл). Полученный раствор дегазировали азотом (3 раза) и охлаждали при 0°C. NaOMe (25% в MeOH, 1,7 мл) добавляли и смесь перемешивали при 0°C в течение 30 минут. Добавляли AcOH (3 г, 50 ммоль, 4 экв.), для резкого охлаждения реакционной смеси до 0°C. Смесь нагревали до 20°C. Медленно добавляли деионизированную воду (10 мл). Смесь перемешивали при 20°C в течение 3 часов и затем перемешивали при 4°C в течение 1 часа, пока не образовывался осадок. Твердые вещества отфильтровывали и промывали MeOH/Н2О (2:1, 30 мл), затем сушили в атмосфере азота при 20°C в течение 48 часов, с получением указанного в заголовке кристаллического соединения (1,2 г).
Пример 4
Порошковая дифракция рентгеновских лучей
Порошковые рентгенограммы получали с помощью дифрактометра Rigaku Miniflex PXRD с применением излучения Cu Kα (30,0 кВ, 15,0 мА). Анализ проводили с гониометром, работающим в режиме непрерывного сканирования 2° (2θ) в минуту с размером шага 0,03° в диапазоне от 2 до 40° угла два тэта. Образцы на кварцевых держателях образцов готовили в виде тонкого слоя порошкообразного вещества. Прибор калибровали со стандартом кремний-металл в пределах ±0,02° угла два тэта. Конкретная рентгенограмма PXRD для образца кристаллического соединения примера 1 показана на фиг.1. Конкретная рентгенограмма PXRD для образца кристаллического соединения примера 2 показана на фиг.3. Множественные интенсивные пики порошковой дифракции и относительно плоская базовая линия, отображенные на фиг.1 и 3, убедительно показывают, что кристаллические соединения формулы IIa и IIIa обладают хорошей кристалличностью.
Пример 5
Термический анализ
Дифференциальную сканирующую калориметрию (DSC) проводили, используя инструмент TA Instruments Model Q-100 module с устройством управления Thermal Analyst controller. Данные собирали и анализировали, используя программное обеспечение TA Instruments Thermal Solutions. Образец 2,05 мг кристаллического соединения примера 1 точно взвешивали в покрытом алюминием лотке. После 5-минутного периода изотермического уравновешивания при 22°C образец нагревали с линейной скоростью нагрева 10°C/мин от 22°C до 250°C. Конкретная термограмма DSC показана на фиг.2.
Термограмма DSC показывает, что данное кристаллическое соединение обладает превосходной термостойкостью с температурой плавления около 76,0°C и не подвержено термическому разложению ниже 150,0°C. Некомплексный термический профиль не показывает какого-либо нежелательного эндотермического или экзотермического пика перед эндотермой плавления при 76,0°C, которая подтверждает, что данное кристаллическое твердое вещество является, наиболее вероятно, безводной кристаллической формой.
Конкретная кривая TGA показана на фиг.2, и она свидетельствует о том, что образец кристаллического соединения примера 1 потерял небольшое количество (<0,5%) массы в интервале температур от комнатной до 150,0°C, которое согласуется с потерей остаточной влаги или растворителя.
Образец 1,12 мг кристаллического соединения примера 2 оценивали подобным образом. Конкретная термограмма DSC показана на фиг.4. Термограмма DSC демонстрирует, что это кристаллическое соединение обладает превосходной термостойкостью с температурой плавления около 130,9°C и не подвержено термическому разложению ниже 150,0°C. Некомплексный термический профиль не показывает какого-либо нежелательного эндотермического или экзотермического пика перед эндотермой плавления при 130,9°C, которая подтверждает, что данное кристаллическое твердое вещество является, наиболее вероятно, безводной кристаллической формой.
Конкретная кривая TGA показана на фиг.4, и она свидетельствует о том, что образец кристаллического соединения примера 1 потерял небольшое количество (<0,5%) массы в интервале температур от комнатной до 150,0°C, которое согласуется с потерей остаточной влаги или растворителя.
Хотя данное изобретение описано со ссылкой на конкретные аспекты или варианты его осуществления специалисту в данной области будет понятно, что могут быть сделаны различные изменения или использованы эквивалентные решения, не отходя от сущности и объема изобретения. Дополнительно, в пределах, допустимых действующим патентным законодательством и нормативными актами, все публикации, патенты и патентные заявки, цитируемые в данном описании, включены посредством ссылки во всей их полноте в той же степени, как если бы каждый документ отдельно был включен в данное описание посредством ссылки.
| название | год | авторы | номер документа |
|---|---|---|---|
| АНТИГИПЕРТЕНЗИВНЫЕ СОЕДИНЕНИЯ ДВОЙНОГО ДЕЙСТВИЯ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ НА ИХ ОСНОВЕ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ | 2008 |
|
RU2476427C2 |
| ИНГИБИТОРЫ НЕПРИЛИЗИНА | 2011 |
|
RU2605557C2 |
| ЗАМЕЩЕННЫЕ АМИНОМАСЛЯНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ НЕПРИЛИЗИНА | 2012 |
|
RU2604522C2 |
| ИНГИБИТОРЫ НЕПРИЛИЗИНА | 2011 |
|
RU2622288C2 |
| ИНГИБИТОРЫ MAGL | 2017 |
|
RU2754536C1 |
| (2S,4R)-5-(5'-ХЛОР-2'-ФТОРБИФЕНИЛ-4-ИЛ)-4-(ЭТОКСИОКСАЛИЛАМИНО)-2-ГИДРОКСИМЕТИЛ-2-МЕТИЛПЕНТАНОВАЯ КИСЛОТА | 2016 |
|
RU2726623C2 |
| ИНГИБИТОРЫ НЕПРИЛИЗИНА | 2013 |
|
RU2650114C2 |
| ИНГИБИТОРЫ НЕПРИЛИЗИНА | 2012 |
|
RU2629930C2 |
| ИНГИБИТОРЫ MAGL НА ОСНОВЕ ПИРАЗОЛА | 2018 |
|
RU2789157C2 |
| ПРОИЗВОДНЫЕ 4-ПИПЕРИДИНИЛАЛКИЛАМИНА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ АНТАГОНИСТИЧЕСКИМ ДЕЙСТВИЕМ В ОТНОШЕНИИ МУСКАРИНОВЫХ РЕЦЕПТОРОВ | 2002 |
|
RU2300524C2 |
Изобретение относится к области органической химии, а именно к способам получения промежуточных соединений формул (I), (II) и (III) для получения соединений формулы (IV), где R1 означает -С1-6алкил; R2 означает -O-С1-5алкил, R3 означает -С1-6алкил. Также изобретение относится к кристаллическим формам промежуточных соединений формул (II) и (III). Технический результат: разработаны способы получения новых промежуточных соединений, полезных при синтезе соединения (IV), необходимого для синтезирования биологически активных соединений. 8 н. и 19 з.п. ф-лы, 4 ил., 5 пр.
 ,
,  ,
,  ,
, 
1. Способ получения соединения формулы I:
где R2 означает -O-С1-5алкил, и Р представляет защитную группу карбоновой кислоты, выбранную из метила, этила, трет-бутила, бензила, п-метоксибензила, 9-флуоренилметила, триметилсилила, трет-бутилдиметилсилила и дифенилметила, включающий стадию взаимодействия соединения формулы 1:
с соединением формулы 2:
в органическом разбавителе и водном разбавителе основного характера в присутствии межфазного катализатора, где разбавители являются по существу несмешивающимися, с образованием соединения формулы I.
2. Способ по п.1, где органическим разбавителем является толуол.
3. Способ по п.1, где водным разбавителем основного характера является NaOH.
4. Способ по п.1, где межфазным катализатором является тетрабутиламмонийбромид.
5. Способ получения соединения формулы II:
или его соли; где R1 представляет -С1-6алкил, R2 представляет -OC1-5алкил, и Р представляет защитную группу карбоновой кислоты, выбранную из метила, этила, трет-бутила, бензила, п-метоксибензила, 9-флуоренилметила, триметилсилила, трет-бутилдиметилсилила и дифенилметила; включающий стадии:
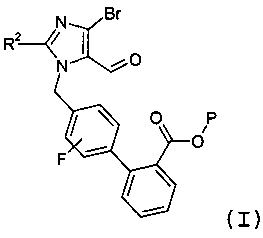
(а) взаимодействия соединения формулы I:
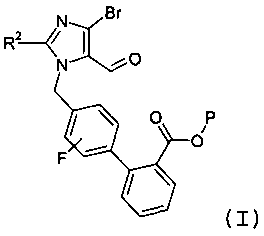
с реагентом калий-С1-6алкилтрифторборатом в присутствии палладий-фосфинового катализатора, с образованием соединения формулы 3:
(b) взаимодействия соединения формулы 3 с гидроксиламином или его солью, с образованием соединения формулы 4:
и
(c) взаимодействия соединения формулы 4 с восстановителем, с образованием соединения формулы II или его соли.
6. Способ по п.5, где палладий-фосфиновым катализатором является комбинация палладиевого катализатора и источника фосфина.
7. Способ по п.6, где палладиевым катализатором является ацетат палладия(II), и источником фосфина является ди(1-адамантил)-н-бутилфосфин.
8. Способ по п.5, где восстановителем является водород/никель Ренея или Pd/C.
9. Способ по п.5, дополнительно включающий стадию получения кристаллической формы соединения формулы II.
10. Способ получения соединения формулы III:
или его соли; где R1 означает -С1-6алкил; R2 означает -O-С1-5алкил, R3 означает -С1-6алкил, и R4 означает -С1-6алкил, включающий стадии:
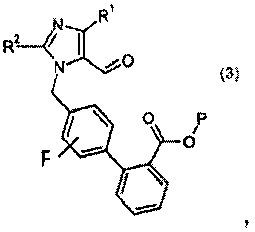
(a) взаимодействия соединения формулы I:
с реагентом калий-С1-6алкилтрифторборатом в присутствии палладий-фосфинового катализатора, с получением соединения формулы 3:
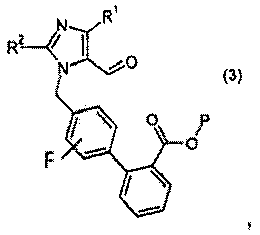
где Р означает защитную группу карбоновой кислоты, выбранную из метила, этила, трет-бутила, бензила, п-метоксибензила, 9-флуоренилметила, триметилсилила, трет-бутилдиметилсилила и дифенилметила;
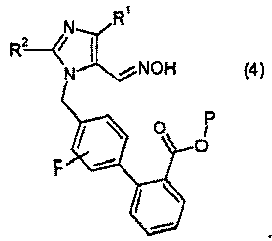
(b) взаимодействия соединения формулы 3 с гидроксиламином или его солью, с образованием соединения формулы 4:
(c) взаимодействия соединения формулы 4 с восстановителем, с образованием соединения формулы II:
или его соли;
(d) взаимодействия соединения формулы II или его соли с соединением формулы 5:
или его солью в присутствии реагента сочетания амин-карбоновая кислота, с образованием соединения формулы 6:
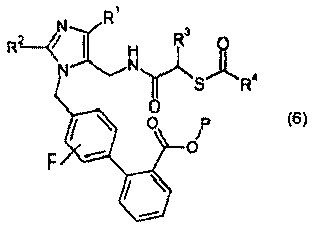
или его соли; и
(e) удаления защитной группы карбоновой кислоты, Р, из соединения формулы 6 или его соли, с образованием соединения формулы III или его соли.
11. Способ по п.10, где палладий-фосфиновым катализатором на стадии (а) является комбинация палладиевого катализатора и источника фосфина.
12. Способ по п.11, где палладиевым катализатором является ацетат палладия(II), и источником фосфина является ди(1-адамантил)-н-бутилфосфин.
13. Способ по п.10, где восстановителем на стадии (с) является водород/никель Ренея.
14. Способ по п.10, дополнительно включающий стадию получения кристаллической формы соединения формулы III.
15. Соединение формулы 3:
где R1 означает -С1-6алкил, R2 означает -О-С1-5алкил, и Р означает защитную группу карбоновой кислоты, представляющую собой трет-бутил.
16. Кристаллический трет-бутиловый эфир 4' -(5-аминометил-2-этокси-4-этилимидазол-1-илметил)-3'-фторбифенил-2-карбоновой кислоты, характеризующийся порошковой рентгенограммой, включающей дифракционные пики при величинах 2θ 5,24±0,2, 10,43±0,2, 15,65±0,2, 20,63±0,2 и 31,91±0,2.
17. Соединение по п.16, где соединение дополнительно характеризуется наличием одного или нескольких дополнительных дифракционных пиков при величинах 2θ, выбранных из 12,74±0,2, 14,90±0,2, 18,20±0,2, 21,71±0,2, 23,03±0,2, 23,96±0,2 и 24,86±0,2.
18. Соединение по п.16, где соединение характеризуется порошковой рентгенограммой, на которой положения пиков по существу согласуются с положениями пиков рентгенограммы, показанной на фиг.1.
19. Соединение по п.16, где соединение характеризуется кривой дифференциальной сканирующей калориметрии, которое имеет температуру плавления около 76,0ºC.
20. Соединение по п.16, где соединение характеризуется кривой дифференциальной сканирующей калориметрии по существу в соответствии с показанной на фиг.2.
21. Способ получения кристаллического соединения по п.16, включающий:
a) обработку трет-бутилового эфира 4'-(5-аминометил-2-этокси-4-этилимидазол-1-илметил)-3'-фторбифенил-2-карбоновой кислоты гептанами до полного растворения;
b) охлаждение для содействия кристаллизации и
c) выделение полученного твердого вещества, с получением кристаллического соединения по п.16.
22. Кристаллическая 4'-{5-[((S)-2-ацетилсульфанил-4-метилпентаноиламино)метил]-2-этокси-4-этилимидазол-1-илметил}-3'-фторбифенил-2-карбоновая кислота, характеризующаяся порошковой рентгенограммой, включающей дифракционные пики при величинах 2θ 5,24±0,2, 7,16±0,2, 13,68±0,2 и 15,98±0,2.
23. Соединение по п.22, где соединение дополнительно характеризуется наличием одного или нескольких дополнительных дифракционных пиков при величинах 2θ, выбранных из 8,10±0,2, 11,26±0,2, 12,06±0,2, 16,62±0,2, 20,12±0,2, 20,78±0,2, 23,24±0,2 и 26,28±0,2.
24. Соединение по п.22, где соединение характеризуется порошковой рентгенограммой, на которой положения пиков по существу согласуются с положениями пиков рентгенограммы, показанной на фиг.3.
25. Соединение по п.22, где соединение характеризуется кривой дифференциальной сканирующей калориметрии, которое имеет температуру плавления около 130,9ºC.
26. Соединение по п.22, где соединение характеризуется кривой дифференциальной сканирующей калориметрии по существу в соответствии с показанной на фиг.4.
27. Способ получения кристаллического соединения по п.22, включающий:
a) обработку 4'-{5-[((S)-2-ацетилсульфанил-4-метилпентаноиламино)метил]-2-этокси-4-этилимидазол-1-илметил}-3'-фторбифенил-2-карбоновой кислоты изопропилацетатом до полного растворения;
b) охлаждение для содействия кристаллизации и
c) выделение полученных твердых веществ, с получением кристаллического соединения по п.22.
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |
| ПРОИЗВОДНЫЕ ИМИДАЗОЛА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ПОНИЖАЮЩАЯ КРОВЯНОЕ ДАВЛЕНИЕ | 1992 |
|
RU2104272C1 |
| ПРОИЗВОДНЫЕ ИМИДАЗОЛА | 1992 |
|
RU2017733C1 |

