Настоящее изобретение относится к способу получения амида карбоновой кислоты из карбонильного соединения и цианистоводородной кислоты. Кроме того, настоящее изобретение относится к способу получения алкил(мет)акрилатов и полимеров, а также к способу изготовления формовочных масс и полимерных формованных изделий.
Из уровня техники давно известно о получении амидов карбоновых кислот путем гидролиза нитрилов карбоновой кислоты в присутствии содержащего диоксид марганца катализатора. Амиды карбоновой кислоты часто используют в технике в качестве полупродуктов. Так, например, амид α-гидроксиизомасляной кислоты используют для получения метакриловой кислоты или сложных эфиров метакриловой кислоты, в частности метилметакрилата.
Особенно предпочтительный способ получения амидов карбоновых кислот описан в международной заявке WO 2008/061822 А1.
Способ синтеза, предложенный в указанной выше международной заявке, обладает относительной экономичностью, однако существует настоятельная необходимость в его усовершенствовании. Циангидрин, образующийся в результате взаимодействия цианистоводородной кислоты с карбонильным соединением, в частности ацетоном, обычно стабилизируют путем добавления кислоты. Перед превращением циангидрина в амид карбоновой кислоты указанную стабилизацию необходимо устранять, для чего обычно осуществляют дистилляцию циангидрина. В соответствии с цитируемой заявкой смесь, образующуюся в результате взаимодействия ацетона с цианистоводородной кислотой, можно использовать для гидролиза. Однако вопрос о необходимости выполнения ее очистки остается открытым. Подобная очистка в общем случае включает двухстадийную дистилляцию, на первой стадии который выделяют непревращенные исходные продукты. На второй стадии из циангидрина обычно удаляют используемую для его стабилизации кислоту. Отсутствие очистки, например, дистилляции, является причиной относительно существенного сокращения срока службы катализатора. Использование дистиллированной реакционной смеси позволяет значительно увеличить срок службы катализатора, однако вследствие повышения энергопотребления снижается общая эффективность соответствующего процесса.
С учетом уровня техники в основу настоящего изобретения была положена задача предложить особенно простой и экономичный способ получения амидов карбоновых кислот с высоким выходом. Особая проблема при этом, в частности, состояла в том, чтобы способ позволял получать амид карбоновой кислоты с высокой скоростью, низким расходом энергии и незначительными потерями выхода при особенно длительном сроке службы катализатора и длительном периоде эксплуатации производственной установки.
Указанная задача, а также другие конкретно не указанные, но вытекающие из контекста нижеследующего описания задачи, решаются благодаря способу, отличительные признаки которого приведены в пункте 1 формулы изобретения. Целесообразные варианты осуществления предлагаемого способа представлены в соответствующих зависимых пунктах. Решение задач, относящихся к получению алкил(мет)акрилатов, полимеров, формовочных масс и полимерных формованных изделий, приведено в пунктах 22, 24, 26 и 27 формулы изобретения.
В соответствии с этим объектом настоящего изобретения является способ получения амида карбоновой кислоты из карбонильного соединения и цианистоводородной кислоты, который включает следующие стадии:
A) взаимодействие карбонильного соединения с цианистоводородной кислотой с целью получения нитрила гидроксикарбоновой кислоты,
B) гидролиз полученного на стадии А) нитрила гидроксикарбоновой кислоты в присутствии содержащего диоксид марганца катализатора,
и отличается тем, что для взаимодействия карбонильного соединения с цианистоводородной кислотой на стадии А) используют молярный избыток карбонильного соединения по отношению к цианистоводородной кислоте, причем полученную на стадии А) реакционную смесь перед выполняемым на стадии В) гидролизом не подвергают дистилляционной очистке. Благодаря этому предложенный способ неожиданно отличается особенно высокой энергоэффективностью и позволяет продлить срок службы катализатора.
Предлагаемый в изобретении способ позволяет достичь также ряд других преимуществ. К ним относится, в частности, неожиданная возможность значительного увеличения продолжительности эксплуатации установки для получения амида карбоновой кислоты. Таким образом, предлагаемый в изобретении способ можно осуществлять особенно эффективно, экономично, с высокой скоростью, низким расходом энергии и незначительными потерями выхода целевого продукта.
Предлагаемый в изобретении способ позволяет эффективно получать амиды карбоновых кислот. При этом, в частности, используют карбонильные соединения, которые в общем случае содержат группы формулы -СО-. Амиды карбоновой кислоты содержат по меньшей мере одну группу формулы -CONH2. Указанные соединения известны специалистам и описаны, например, в справочнике Chemie Lexikon (2-е издание в записи на CD-ROM).
В качестве исходных продуктов, в частности, можно использовать алифатические или циклоалифатические, насыщенные или ненасыщенные, ароматические или гетероароматические карбонильные соединения. Карбонильные соединения, подлежащие использованию в качестве исходных продуктов, могут содержать одну, две или более карбонильных групп. Кроме того, можно использовать также карбонильные соединения, ароматический или алифатический остаток которых содержит гетероатомы, в частности атомы галогенов, таких как хлор, бром или фтор, атом кислорода, серы и/или азота. Особенно пригодные карбонильные соединения предпочтительно содержат 1-100 атомов углерода, предпочтительно 2-20 атомов углерода, еще более предпочтительно 2-5 атомов углерода.
К особенно предпочтительным карбонильным соединениям относятся алифатические или гетероалифатические кетоны с 3-5 атомами углерода, например, такие как ацетон, и алифатические или гетероалифатические альдегиды с 2-5 атомами углерода, например, такие как 3-метилмеркапто-пропиональдегид или ацетальдегид. При этом особенно предпочтительным исходным продуктом является ацетон.
Указанные соединения можно подвергать взаимодействию с цианистоводородной кислотой до α-нитрилов гидроксикарбоновой кислоты (циангидринов), например, таких как α-гидрокси-γ-метилтиобутиронитрил (2-гидрокси-4-метил-тиобутиронитрил), 2-гидроксипропионитрил (лактонитрил) или 2-гидрокси-2-метилпропионитрил (ацетонциангидрин), по отдельности или в виде смеси, причем особенно предпочтительным продуктом подобного взаимодействия является ацетонциангидрин.
Карбонильное соединение используют в молярном избытке по отношению к цианистоводородной кислоте. Молярное отношение карбонильного соединения к цианистоводородной кислоте предпочтительно находится в интервале от 1,1:1 до 7:1, предпочтительно от 1,5:1 до 5:1, еще более предпочтительно от 2:1 до 3:1.
Превращение карбонильного соединения с цианистоводородной кислотой на стадии А) предпочтительно осуществляют в присутствии основания. В качестве основания можно использовать анионообменные смолы. Предпочтительному использованию подлежат гидроксиды или оксиды, причем особенно предпочтительными являются гидроксиды или оксиды щелочноземельных или щелочных металлов. К ним относятся, в частности, Са(ОН)2, Мg(ОН)2, МgО, CaO, NaOH, КОН, LiOH и Li2O. В качестве основания еще более предпочтительно используют LiOH или Li2O. Для превращения карбонильного соединения с цианистоводородной кислотой к реакционной смеси предпочтительно добавляют от 0,001 до 10% масс., особенно предпочтительно от 0,01% масс. до 2% масс. гидроксида и/или оксида. В соответствии с особым вариантом осуществления настоящего изобретения количество добавляемого гидроксида и/или оксида следует выбирать таким образом, чтобы отсутствовала необходимость добавления другого основания на последующей стадии гидролиза В) с целью регулирования показателя рН.
Для регулирования показателя рН теоретически можно использовать также растворимые амины. Однако обнаружено, что подобные амины могут оказывать негативное влияние на срок службы используемого на стадии гидролиза В) катализатора. Помимо органических соединений с одним атомом азота к аминам относится также аммиак (NН3). Таким образом, содержание растворимых аминов в реакционной смеси предпочтительно составляет не более 0,1% масс., особенно предпочтительно не более 0,01% масс. и еще более предпочтительно не более 0,001% масс. В соответствии с особым вариантом к реакционной смеси можно не добавлять существенных количеств указанных аминов с целью регулирования показателя рН.
Температура, при которой реализуют взаимодействие карбонильного соединения с цианистоводородной кислотой, в общем случае находится в интервале от -30 до 70°С, предпочтительно от -20 до 60°С, в частности от -10 до 50°С и особенно предпочтительно от -5 до 40°С.
В зависимости от температуры превращения карбонильного соединения с цианистоводородной кислотой до нитрила гидроксикарбоновой кислоты стадию А) осуществляют при пониженном или повышенном давлении. Указанное превращение предпочтительно осуществляют при давлении, составляющем от 0,5 до 10 бар, особенно предпочтительно от 0,8 до 3 бар.
Длительность синтеза нитрила гидроксикарбоновой кислоты на стадии А) зависит, в частности, от исходного карбонильного соединения, активности катализатора и температуры реакции, причем указанные параметры можно варьировать в широких пределах. Длительность превращения карбонильного соединения с цианистоводородной кислотой до нитрила гидроксикарбоновой кислоты предпочтительно составляет от 30 секунд до 15 часов, особенно предпочтительно от 10 минут до 5 часов и еще более предпочтительно от 30 минут до 3 часов.
При осуществлении превращения на стадии А) в непрерывном режиме время контакта реагентов предпочтительно составляет от 30 секунд до 15 часов, особенно предпочтительно от 10 минут до 5 часов и еще более предпочтительно от 30 минут до 3 часов.
В отличие от способов уровня техники реакционную смесь, образующуюся в результате превращения реагентов на стадии А), перед выполняемым на стадии В) гидролизом не подвергают дистилляционной очистке. При этом под дистилляцией подразумевают разделение реакционной смеси на компоненты, обладающие разными точками кипения. Отказ от подобной очистки позволяет значительно повысить эффективность предлагаемого в изобретении способа. Согласно предпочтительному варианту реализуемого на стадии А) превращения необходимость в очистке продуктов этого превращения отсутствует. Полученную в результате превращения на стадии А) смесь непосредственно направляют на стадию гидролиза В).
Согласно изобретению гидролиз полученного на стадии А) нитрила карбоновой кислоты осуществляют в присутствии катализатора, содержащего диоксид марганца. Стехиометрическому составу природного, а также синтетического диоксида марганца благодаря присутствию в его кристаллической решетке марганца с другими степенями валентности предпочтительно может соответствовать диапазон от MnO1,7 до МnO2,0. Диоксид марганца существует в нескольких аллотропных модификациях. Эти модификации сильно отличаются друг от друга по каталитическим свойствам. Наиболее высокой степенью кристалличности обладает пиролюзит, представляющий собой бета-модификацию диоксида марганца, которая обладает наибольшей стабильностью. Другие модификации диоксида марганца, к которым, в частности, относятся альфа-модификация и дельта-модификация, обладают менее выраженной кристалличностью или являются аморфными продуктами. Модификации диоксида марганца могут быть установлены на основании дифракции рентгеновских лучей. Химически и каталитически особенно активные формы диоксида марганца могут быть частично гидратированы и могут дополнительно содержать гидроксильные группы.
В состав содержащего диоксид марганца катализатора могут входить другие соединения или ионы. К последним, в частности, относятся ионы щелочных и/или щелочноземельных металлов, которые внедряются в кристаллическую решету или концентрируются на поверхности катализатора при его получении. К предпочтительным ионам щелочных металлов, в частности, относятся ионы лития, натрия и/или калия. К предпочтительным ионам щелочноземельных металлов, в частности, относятся ионы кальция и/или магния. Количество атомов щелочного и/или щелочноземельного металла в расчете на атом марганца предпочтительно может составлять менее 0,6. Отношение количества атомов щелочного и/или щелочноземельного металла к количеству атомов марганца предпочтительно находится в интервале от 0,01:1 до 0,5:1, особенно предпочтительно в интервале от 0,05:1 до 0,4:1.
Кроме того, в состав содержащего диоксид марганца катализатора могут входить промоторы, которые также могут быть внедрены в кристаллическую решетку или могут концентрироваться на поверхности катализатора. К предпочтительным промоторам относятся, в частности, титан, цирконий, ванадий, ниобий, тантал, хром, молибден, вольфрам, цинк, галлий, индий, германий, олово и платина. Количество атомов промотора в расчете на атом марганца предпочтительно может составлять менее 0,3. Отношение количества атомов промотора к количеству атомов марганца предпочтительно находится в интервале от 0,001:1 до 0,2:1, особенно предпочтительно от 0,005:1 до 0,1:1. В состав содержащего диоксид марганца катализатора предпочтительно может входить от 0,01 до 10% масс., особенно предпочтительно от 0,1 до 5% масс. промоторов (в пересчете на массу соответствующего металла или иона металла).
С целью повышения механической стабильности в состав пригодных катализаторов может быть включен также диоксид кремния или дополнительные связующие (смотри, например, европейскую заявку на патент ЕР-А-0956898).
Особенно предпочтительные катализаторы содержат, например, следующие компоненты:
от 0,0 до 25% масс., в частности от 0,1 до 2% масс.SiO2,
от 0,1 до 10% масс., в частности от 2 до 7% масс. K2О,
от 0,0 до 5% масс., в частности от 0,2 до 4% масс. ZrO2 и
от 75 до 99% масс., в частности от 85 до 98% масс. МnO2.
Катализаторы могут содержать также другие указанные выше элементы. Состав катализаторов может быть определен путем полуколичественного рентгенофлуоресцентного анализа.
На рентгеновских спектрах содержащих диоксид марганца предпочтительных порошкообразных катализаторов, которые снимают методом дифракционного рентгеновского анализа, присутствует по меньшей мере один рефлекс в диапазоне дифракционных углов от 32,0 до 42,0°. Для дифракционного рентгеновского анализа можно использовать, например, прибор Xpert фирмы Panalytical. Проявляющийся в диапазоне от 32,0° до 42,0° рефлекс в его максимуме особенно предпочтительно обладает наибольшей интенсивностью в сравнении с другими рефлексами в диапазоне от 20° до 65°. Особенно предпочтительные катализаторы обладают низкой кристалличностью, о степени которой можно судить, в частности, по рентгеновскому спектру. Согласно банку данных Международного центра дифракционных характеристик (ICDD) особенно предпочтительные катализаторы обладают структурой номер 44-0141 или номер 72-1982, причем особенно предпочтительными являются катализаторы, которые обладают структурой номер 44-0141.
Ионы щелочных и/или щелочноземельных металлов, а также промоторы при получении катализаторов можно добавлять, например, в виде солей. Под солями, в частности, подразумевают галогениды, нитраты, сульфаты, карбонаты, фосфаты и гидроксиды указанных выше веществ, причем предпочтительно используют растворимые в воде соединения.
В содержащем диоксид марганца катализаторе предпочтительно может присутствовать по меньшей мере 50% масс., особенно предпочтительно по меньшей мере 80% масс. диоксида марганца с брутто-формулой МnОх, в которой индексу x соответствует интервал от 1,7 до 2,0.
Согласно особому варианту осуществления настоящего изобретения содержащий диоксид марганца катализатор обладает удельной поверхностью по БЭТ (определяемой согласно DIN 66131) в интервале от 50 до 1000 м2/г, особенно предпочтительно от 100 до 300 м2/г, еще более предпочтительно от 150 до 250 м2/г.
В зависимости от типа реактора катализатор можно использовать, например, в виде порошка или гранулята, размер частиц которого чаще всего зависит от используемого реакционного объема.
Получение рассмотренных выше содержащих диоксид марганца катализаторов известно (смотри, например, европейские заявки на патент ЕР-А-0379111, ЕР-А-0956898, ЕР-А-0545697 и ЕР-А-0433611). Содержащие диоксид марганца катализаторы, подлежащие использованию согласно изобретению, предпочтительном можно получать путем окисления содержащих Mn2+ солей, например, MnSO4, перманганатами, например, перманганатом калия (смотри Biochem.J., 43 (1951), с.50, а также J.Chem. Soc., с.2189, 1953). Кроме того, диоксид марганца, пригодный для использования в составе катализатора, может быть получен путем электролитического окисления сульфата марганца в водном растворе.
Катализаторы со структурой номер 44-0141 можно получать, например, путем медленного добавления водного раствора, содержащего 0,71 моля Mn(II)SO4 (суммарное содержание Mn2+ в растворе 15% масс.), 0,043 моля Zr(IV)(SO4)2, 0,488 моля концентрированной серной кислоты и 13,24 молей воды, к раствору 1,09 моля КМnО4 в 64,5 молях воды, при 70°С. Надосадочный раствор вместе с образовавшимся осадком можно термостатировать в течение 3 часов при 90°С. Затем осадок может быть отделен фильтрованием, четырежды промыт литром воды и подвергнут 12-часовой сушке при 110°С.
Показатель рН реакционной смеси, добавляемой к содержащему диоксид марганца катализатору, преимущественно находится в интервале от 6,0 до 11,0, предпочтительно от 6,5 до 10,0, еще более предпочтительно от 8,5 до 9,5. Показатель рН в данном случае равен отрицательному десятичному логарифму активности ионов оксония (Н3О+). Данная характеристика зависит, в частности, от температуры, причем она коррелирует с температурой реакции. Для целей настоящего изобретения часто оказывается достаточным выполнять определение показателя рН электрическим измерительным прибором (рН-метром), причем для многих целей подобное определение можно осуществлять не при температуре реакции, а при комнатной температуре. Регулирование показателя рН предпочтительно следует осуществлять уже при синтезе нитрила гидроксикарбоновой кислоты на стадии А), причем в предпочтительном варианте для этой цели можно использовать указанные выше оксиды и гидроксиды.
При этом следует констатировать, что содержащий диоксид марганца катализатор часто обладает амфотерными свойствами, в связи с чем показатель рН реакционной смеси при взаимодействии сильно зависит от типа и количества катализатора. Определение «реакционная смесь, добавляемая к содержащему диоксид марганца катализатору», используют, чтобы подчеркнуть то обстоятельство, что показатель рН измеряют в отсутствие катализатора. Другими компонентами реакционной смеси являются, например, растворитель, вода, нитрил карбоновой кислоты и так далее.
Неожиданно было установлено, что выполнение гидролиза в присутствии ионов лития способствует существенному продлению срока службы содержащего диоксид марганца катализатора. В соответствии с этим с целью дополнительной оптимизации предлагаемого в изобретении способа к реакционной смеси можно добавлять соединения лития, в частности водорастворимые соли лития, например LiCl, LiBr, Li2SO4, LiOH и/или Li2О. Концентрация соединений лития в реакционной смеси предпочтительно составляет от 0,001 до 5% масс., особенно предпочтительно от 0,01% масс. до 1% масс. Соединения лития можно добавлять во время гидролиза или перед гидролизом.
Гидролиз нитрила карбоновой кислоты до амида карбоновой кислоты предпочтительно осуществляют в присутствии окисляющего средства. Пригодные окисляющие средства хорошо известны специалистам. К ним относятся, в частности, кислородсодержащие газы, пероксиды, например, пероксид водорода (H2O2), пероксид натрия, пероксид калия, пероксид магния, пероксид кальция, пероксид бария, пероксид бензоила и дипероксид ацетила, пероксикислоты или соли пероксикислот, например, надмуравьиная кислота, надуксусная кислота, персульфат натрия, персульфат аммония и персульфат калия, оксокислоты или соли оксокислот, например, надйодная кислота, перйодат калия, перйодат натрия, перхлорная кислота, перхлорат калия, перхлорат натрия, хлорат калия, хлорат натрия, бромат калия, йодат натрия, йодная кислота, гипохлорит натрия, перманганаты, например, перманганат калия, перманганат натрия и перманганат лития, а также соли хромовой кислоты, например, хромат калия, хромат натрия и хромат аммония.
Количество используемого окисляющего средства можно варьировать в широких пределах, причем реагенты и продукты синтеза не должны подвергаться окислению окисляющим средством. В соответствии с этим количество последнего может быть ограничено чувствительностью реагентов и продуктов синтеза к окислению. Нижний предел количества окисляющего средства определяется тем, до какой степени необходимо оптимизировать стабильность катализатора. Молярное отношение окисляющего средства к нитрилу карбоновой кислоты предпочтительно находится в интервале от 0,001:1 до 2:1, особенно предпочтительно от 0,01:1 до 1,5:1.
Указанные окисляющие средства можно добавлять к реакционной смеси, например, в виде раствора и/или газа. В качестве окисляющих средств особенно предпочтительно используют газы, которые содержат кислород. При этом газы могут содержать молекулярный кислород (O2) или озон (О3). Кроме того, используемый в качестве окисляющего средства газ может содержать другие газы, в частности инертные газы, такие как азот или благородные газы. В соответствии с особым вариантом используемый в качестве окисляющего средства газ предпочтительно может содержать от 50 до 98% об. инертного газа и от 2 до 50% об. молекулярного кислорода (O2). К предпочтительным газам, используемым в качестве окисляющего средства, в частности относится воздух. Кроме того, в качестве окисляющего средства можно использовать газ, содержащий менее 20% об., в частности менее 10% об. молекулярного кислорода, причем подобные газы в общем случае содержат по меньшей мере 1% об., предпочтительно по меньшей мере 2% об. кислорода.
Расход содержащего кислород газа, пропускаемого через реакционную смесь, предпочтительно составляет от 1 до 5000 литр/час, особенно предпочтительно от 10 до 1000 литр/час (в расчете на 1 кг содержащего диоксид марганца катализатора).
Воду, необходимую для осуществления гидролиза нитрила карбоновой кислоты, часто можно использовать также в качестве растворителя. Молярное отношение воды к нитрилу карбоновой кислоты предпочтительно составляет по меньшей мере 1:1 и особенно предпочтительно находится в интервале от 0,5:1 до 25:1, еще более предпочтительно в интервале от 1:1 до 10:1.
Используемая для гидролиза вода может обладать высокой степенью чистоты. Однако это условие вовсе не является обязательным. Так, например, наряду со свежей водой можно использовать также промышленную или технологическую воду, которая может обладать более или менее высоким содержанием примесей. Таким образом, для гидролиза можно использовать также вторичную воду.
Кроме того, в реакционной смеси, используемой для гидролиза нитрила карбоновой кислоты, могут присутствовать другие компоненты. К ним относятся, в частности, карбонильные соединения, такие как альдегиды и кетоны, в частности аналогичные используемым для синтеза циангидринов, подлежащих предпочтительному превращению в качестве исходного нитрила карбоновой кислоты. Так, например, в исходной реакционной смеси может присутствовать ацетон и/или ацетальдегид. Подобный вариант описан, например, в патенте США US 4018829-A. Степень чистоты добавляемых к реакционной смеси альдегидов и/или кетонов в общем случае не является особенно критическим параметром. Таким образом, указанные вещества могут содержать примеси, в частности спирты, например метанол, воду и/или сложный метиловый эфир α-гидроксиизомасляной кислоты. Количество добавляемых к исходной реакционной смеси карбонильных соединений, в частности ацетона и/или ацетальдегида, можно варьировать в широких пределах. Карбонильное соединение преимущественно добавляют в количестве от 0,1 до 6 молей, предпочтительно от 0,1 до 2 молей на моль нитрила карбоновой кислоты. В соответствии с особым вариантом осуществления настоящего изобретения указанное карбонильное соединение можно полностью добавлять на стадии А), а соответствующий избыток рециркулировать.
Температура, при которой осуществляют реакцию гидролиза, в общем случае находится в интервале от 10 до 150°С, предпочтительно от 20 до 100°С, особенно предпочтительно от 30 до 80°С.
Реакцию гидролиза в зависимости от температуры можно осуществлять при пониженном или повышенном давлении. Давление, при котором осуществляют реакцию гидролиза, предпочтительно находится в интервале от 0,1 до 10 бар, особенно предпочтительно в интервале от 0,5 до 5 бар.
Длительность реакции гидролиза зависит, в частности, от используемых нитрилов карбоновой кислоты, активности катализатора и температуры реакции, причем указанные параметры можно варьировать в широких пределах. Длительность реакции гидролиза предпочтительно составляет от 30 секунд до 15 часов, особенно предпочтительно от 15 минут до 10 часов и еще более предпочтительно от 60 минут до 5 часов.
При осуществлении гидролиза в непрерывном режиме время контакта соответствующих реагентов предпочтительно составляет от 30 секунд до 15 часов, особенно предпочтительно от 15 минут до 10 часов и еще более предпочтительно от 60 минут до 5 часов.
Расход нитрила карбоновой кислоты в расчете на катализатор можно варьировать в широких пределах. Предпочтительно используют от 0,01 до 2,0 г, особенно предпочтительно от 0,05 до 1,0 г, еще более предпочтительно от 0,1 до 0,4 г нитрила карбоновой кислоты на грамм катализатора в час.
Гидролиз на стадии В) можно осуществлять, например, в реакторе со стационарным слоем катализатора или в суспензионном реакторе. При использовании в качестве окисляющего средства газа гидролиз, в частности, можно осуществлять в так называемом реакторе с орошаемым слоем, в котором может быть обеспечен оптимальный контакт между газом, твердым веществом и жидкостью. Катализатор в реакторе с орошаемым слоем находится в виде стационарного слоя. При этом указанный реактор можно эксплуатировать в режиме прямотока или противотока.
Образующаяся на стадии В) реакционная смесь в общем случае помимо целевого амида карбоновой кислоты содержит также другие компоненты, в частности непревращенный нитрил карбоновой кислоты или цианистоводородную кислоту, используемое в избытке карбонильное соединение, в частности ацетон и/или ацетальдегид, а также используемую в избытке воду.
В соответствии с этим в общем случае осуществляют разделение реакционной смеси стадии В), целью которого является повторное превращение содержащихся в ней исходных продуктов. В соответствии с одним из вариантов осуществления настоящего изобретения полученная на стадии В) реакционная смесь может быть подвергнута очистке путем двухступенчатой дистилляции.
Образующаяся на стадии В) реакционная смесь в общем случае содержит непревращенный нитрил гидроксикарбоновой кислоты. Температура его кипения превышает точку кипения воды. Обусловленные этим проблемы могут быть легко решены путем расщепления непревращенного нитрила гидроксикарбоновой кислоты на карбонильное соединение и цианистоводородную кислоту. Подобное расщепление можно катализировать, например, основанием, предпочтительно анионообменной смолой, присутствие которой (или соответственно которых) может быть предусмотрено, например, в кубе дистилляционной колонны.
Согласно первой модификации указанного предпочтительного варианта полученный на стадии В) амид карбоновой кислоты на первой ступени а) дистилляции может быть отделен от смеси, которая содержит воду, карбонильное соединение, нитрил гидроксикарбоновой кислоты и/или цианистоводородную кислоту. При этом можно осуществлять расщепление нитрила гидроксикарбоновой кислоты предпочтительно на карбонильное соединение и цианистоводородную кислоту. Указанную смесь можно подвергать очистке на второй ступени b) дистилляции, причем карбонильное соединение и цианистоводородную кислоту отбирают в верхней части второй дистилляционной колонны, в то время как воду отбирают из ее куба.
Первую ступень а) дистилляции, целью которой является разделение полученной на стадии В) реакционной смеси на амид карбоновой кислоты и смесь, содержащую воду, карбонильное соединение, нитрил гидроксикарбоновой кислоты и/или цианистоводородную кислоту, предпочтительно можно осуществлять при температуре от 110 до 260°С, особенно предпочтительно от 140 до 230°С. При этом давление предпочтительно находится в интервале от 0,002 до 1 бар, особенно предпочтительно от 10 до 500 мбар. Под указанными выше температурами дистилляции, в частности, подразумевают температуру в кубе дистилляционной колонны.
Температура, при которой воду отделяют от карбонильного соединения и цианистоводородной кислоты на ступени b) дистилляции, в общем случае может находиться в интервале от 50 до 150°С, предпочтительно от 70 до 120°С, особенно предпочтительно от 90 до 110°С. Вторую ступень b) дистилляции предпочтительно осуществляют при давлении от 0,2 до 5 бар, особенно предпочтительно от 0,7 до 1,5 бар.
В соответствии с особенно предпочтительным вариантом на первой ступени а') дистилляции из полученной на стадии В) реакционной смеси в верхней части дистилляционной колонны сначала можно выделить карбонильное соединение и цианистоводородную кислоту. При этом нитрил гидроксикарбоновой кислоты сначала расщепляют на карбонильное соединение и цианистоводородную кислоту, которые также выделяют из смеси в верхней части дистилляционной колонны. Выделенные указанным образом цианистоводородную кислоту и карбонильное соединение можно использовать для получения нитрила гидроксикарбоновой кислоты на стадии А). Остающаяся в кубе дистилляционной колонны смесь содержит воду и амид карбоновой кислоты. Эту смесь разделяют на второй ступени b'), причем амид карбоновой кислоты может быть отделен от воды предпочтительно путем многоступенчатого выпаривания. При этом произведенный за счет первичной энергии выпар после снижения давления используют на второй ступени дистилляции в качестве средства нагревания жидкой фазы. С целью экономии энергии принцип выпаривания может быть реализован на нескольких ступенях. Многоступенчатое выпаривание предпочтительно включает от двух до четырех ступеней. Принцип многоступенчатого выпаривания подробно рассматривается, в частности, в руководстве H.G. Hirschberg, Handbuch der Verfahrens-technik und des Aniagenbau, издательство Springer 1999, которое следует считать ссылкой, способствующей раскрытию сущности настоящего изобретения. Указанный вариант осуществления изобретения неожиданно позволяет выполнять очистку полученной на стадии В) реакционной смеси с особенно высокой экономией энергии. Выделенную при этом воду можно использовать для осуществления гидролиза на стадии В).
Первую ступень а') дистилляции, реализуемой с целью разделения полученной на стадии В) реакционной смеси на амид карбоновой кислоты и воду, а также смесь, содержащую карбонильное соединение и цианисто-водородную кислоту, предпочтительно можно осуществлять при температуре от 50 до 170°С, особенно предпочтительно от 90 до 120°С. Давление на ступени а') дистилляции предпочтительно находится в интервале от 0,4 до 5 бар, особенно предпочтительно от 0,7 до 2 бар. Под указанной выше температурой дистилляции, в частности, подразумевают температуру в кубе дистилляционной колонны.
Температура, при которой на стадии b') выделяют воду и амид карбоновой кислоты, в общем случае может находиться в интервале от 90 до 260°С, предпочтительно от 100 до 180°С. Вторую ступень дистилляции b') предпочтительно можно осуществлять при давлении от 10 мбар до 20 бар, особенно предпочтительно от 100 мбар до 10 бар. Высокие давления, в частности, используют на первых ступенях многоступенчатого выпаривания.
Неожиданных преимуществ в отношении продолжительности эксплуатации установки и срока службы катализатора можно достичь, в частности, благодаря тому, что в дистилляционную колонну, используемую для выделения воды и цианистоводородной кислоты, через флегму вводят смесь, которая содержит используемое на стадии А) карбонильное соединение, причем указанная смесь обладает более низким содержанием цианистоводородной кислоты, чем смесь, отбираемая в верхней части дистилляционной колонны. Массовое количество цианистоводородной кислоты, содержащейся в смеси, вводимой в дистилляционную колонну с флегмой, предпочтительно составляет не более 60%, особенно предпочтительно не более 40% и еще более предпочтительно не более 10% от массового количества цианистоводородной кислоты, содержащейся в отбираемой в верхней части колонны смеси. В особенно предпочтительном варианте цианистоводородная кислота в смеси, вводимой в дистилляционную колонну с флегмой, в основном отсутствует. В этой связи необходимо констатировать, что из выделяемых карбонильного соединения и цианистоводородной кислоты может образоваться нитрил гидроксикарбоновой кислоты, который также не подлежит возвращению в дистилляционную колонну. В соответствии с этим приведенные выше данные, касающиеся количества возвращаемой в дистилляционную колонну цианистоводородной кислоты, относятся к суммарному количеству свободной цианистоводородной кислоты и цианистоводородной кислоты, химически связанной в виде нитрила гидроксикарбоновой кислоты. Количество химически связанной цианистоводородной кислоты может быть определено путем деструкции нитрила гидроксикарбоновой кислоты. В соответствии с этим смесь, вводимая в дистилляционную колонну с флегмой, преимущественно не содержит также нитрила гидроксикарбоновой кислоты.
Согласно предпочтительному варианту осуществления настоящего изобретения выделенное при разделении воды и цианистоводородной кислоты карбонильное соединение используют для синтеза нитрила гидроксикарбоновой кислоты на стадии А). В соответствии с этим головной продукт соответствующей ступени дистилляции предпочтительно используют для получения нитрила гидроксикарбоновой кислоты, подлежащего превращению на стадии В). Выделенную воду соответственно можно использовать для гидролиза нитрила гидроксикарбоновой кислоты на стадии В).
Дополнительное продление срока службы катализатора может быть достигнуто, в частности, благодаря использованию дистилляционной колонны с высокой разделяющей способностью, что в первую очередь относится к разделению воды и цианистоводородной кислоты. В соответствии с этим для указанной цели предпочтительно используют дистилляционную колонну, которая обладает двумя или более ступенями разделения. В соответствии с настоящим изобретением под числом ступеней разделения подразумевают количество тарелок в случае использования тарельчатой колонны или количество теоретических ступеней разделения в случае использования колонны с насадочным материалом или колонны с насадочными телами.
Примерами пригодных многоступенчатых тарельчатых дистилляционных колонн являются дистилляционные колонны, оснащенные колпачковыми, сетчатыми, туннельными, клапанными, щелевыми, комбинированными сетчатыми/щелевыми, комбинированными сетчатыми/колпачковыми, дюзовыми или центробежными тарелками, примерами пригодных многоступенчатых дистилляционных колонн с насадочными телами дистилляционные колонны, заполненные кольцами Рашига, кольцами Лессинга, кольцами Палля, седлами Берля или седлами Intalox, примерами пригодных многоступенчатых дистилляционных колонн с насадочным материалом являются дистилляционные колонны с насадками типа Mellapak (фирма Sulzer), Rombopak (фирма Kuhni), Montz-pak (фирма Montz) и насадками с гнездами для катализатора, например, Kata-pak.
Можно использовать также комбинированную дистилляционную колонну, включающую секции с тарелками, секции с насадочными телами и секции с насадочным материалом.
Благодаря использованию дистилляционной колонны, которая обладает высокой разделяющей способностью, а также флегмы с низким содержанием цианистоводородной кислоты, содержание цианистоводородной кислоты в возвращаемой на стадию гидролиза водной фазе можно поддерживать на чрезвычайно низком уровне. На стадию гидролиза предпочтительно возвращают водную фазу, которая содержит весьма незначительное количество цианистоводородной кислоты, которое может составлять менее 1% масс., особенно предпочтительно менее 0,5% масс., особенно предпочтительно менее 0,1% масс. в пересчете на возвращаемую водную фазу.
Неожиданных преимуществ можно достичь, в частности, благодаря тому, что количество карбонильного соединения, подаваемого на ступень дистилляции, реализуемой с целью разделения воды и цианистоводородной кислоты, выбирают таким образом, чтобы его было достаточно для получения на стадии А) предусматриваемого количества нитрила гидроксикарбоновой кислоты. В соответствии с этим необходимое для взаимодействия с цианистоводородной кислотой количество карбонильного соединения предпочтительно полностью подают с флегмой в колонну, используемую для дистилляции смеси воды, цианистоводородной кислоты и превращаемого на стадии А) карбонильного соединения. В зависимости от исполнения это можно осуществлять на первой или второй ступени дистилляции.
Кроме того, содержащую очищенный амид карбоновой кислоты реакционную смесь можно подвергать очистке от других компонентов на колоннах с ионообменной смолой.
Для этой цели, в частности, можно использовать катионообменные или анионообменные смолы. Пригодные ионообменные смолы являются известными продуктами. Пригодные катионообменные смолы могут быть получены, например, путем сульфирования сополимеров стирола с дивинилбензолом. Анионообменные смолы со щелочным характером содержат группы четвертичного аммония, ковалентно присоединенные к сополимеру стирола с дивинилбензолом.
Очистка амидов α-гидроксикарбоновой кислоты подробно описана, в частности, в европейской заявке на патент ЕР-А-0686623.
Согласно настоящему изобретению реакцию гидролиза, в частности, можно осуществлять в виде промежуточной ступени способа получения (мет)акриловых мономеров. При этом под (мет)акриловыми мономерами подразумевают метакриловые мономеры, акриловые мономеры и смеси мономеров обоих указанных типов. К (мет)акриловым мономерам, в частности, относятся (мет)акриловые кислоты, в частности акриловая кислота (пропеновая кислота) и метакриловая кислота (2-метилпропеновая кислота), а также сложные эфиры указанных кислот, называемые также (мет)акрилатами. Таким образом, объектом настоящего изобретения является также способ получения алкил(мет)акрилатов, в частности метилметакрилата, который включает стадию гидролиза, реализуемую в соответствии с предлагаемым в настоящем изобретении способом. Способы, которые могут включать стадию гидролиза циангидринов, реализуемую с целью получения (мет)акриловой кислоты и/или алкил-(мет)акрилатов, описаны, в частности, в европейских заявках на патент ЕР-А-0406676, ЕР-А-0407811, ЕР-А-0686623 и ЕР-А-0941984.
Согласно особенно предпочтительному варианту осуществления настоящего изобретения алкил(мет)акрилаты можно получать из карбонильных соединений, цианистоводородной кислоты и спиртов, используя простой и экономичный способ, который включает следующие стадии:
A) получение по меньшей мере одного циангидрина путем взаимодействия по меньшей мере одного карбонильного соединения с цианистоводородной кислотой,
B) гидролиз циангидрина, соответственно циангидринов, с образованием по меньшей мере одного амида α-гидроксикарбоновой кислоты,
C) алкоголиз амида α-гидроксикарбоновой кислоты, соответственно амидов α-гидроксикарбоновых кислот, с образованием по меньшей мере одного сложного алкилового эфира α-гидроксикарбоновой кислоты,
D) переэтерификацию сложного алкилового эфира α-гидроксикарбоновой кислоты, соответственно сложных алкиловых эфиров α-гидроксикарбоновых кислот, (мет)акриловой кислотой с образованием по меньшей мере одного алкил(мет)акрилата и по меньшей мере одной α-гидроксикарбоновой кислоты,
Е) дегидратацию α-гидроксикарбоновой кислоты, соответственно α-гидроксикарбоновых кислот, с образованием (мет)акриловой кислоты.
Стадии А) и В) подробно рассмотрены выше. Полученный на этих стадиях амид α-гидроксикарбоновой кислоты на следующей стадии С) может быть превращен в сложный алкиловый эфир α-гидроксикарбоновой кислоты. Превращение на стадии С) можно осуществлять, например, с использованием алкилформиатов. Пригодным, в частности, является метилформиат или смесь метанола с монооксидом углерода, причем пример осуществления соответствующей реакции приведен в европейской заявке на патент ЕР-А-0407811.
Превращение амида α-гидроксикарбоновой кислоты путем алкоголиза предпочтительно осуществляют с использованием спирта предпочтительно с 1-10 атомами углерода, особенно предпочтительно с 1-5 атомами углерода. Предпочтительными спиртами, в частности, являются метанол, этанол, пропанол, бутанол (в частности, н-бутанол и 2-метил-1-пропанол), пентанол, гексанол, гептанол, 2-этилгексанол, октанол, нонанол и деканол. В качестве спирта особенно предпочтительно используют метанол и/или этанол, еще более предпочтительно метанол. Превращение амидов карбоновых кислот посредством спиртов с целью получения сложных эфиров карбоновых кислот в общем случае осуществляют известными методами.
Молярное отношение амида α-гидроксикарбоновой кислоты к спирту, например амида α-гидроксиизомасляной кислоты к метанолу, не является критическим параметром и предпочтительно составляет от 3:1 до 1:20. Еще более целесообразно указанное отношение находится в интервале от 2:1 до 1:15, особенно предпочтительно от 1:1 до 1:10.
Температуру алкоголиза также можно варьировать в широких пределах, причем по мере ее повышения скорость алкоголиза в общем случае возрастает. Верхний температурный предел этой реакции в общем случае определяется точкой кипения используемого спирта. Алкоголиз предпочтительно осуществляют в температурном интервале от 40 до 300°С, особенно предпочтительно от 160 до 240°С. В зависимости от температуры реакции ее можно осуществлять при пониженном или избыточном давлении. Реакцию предпочтительно осуществляют в интервале давлений от 0,5 до 200 бар, особенно целесообразно от 1 до 100 бар и особенно предпочтительно от 5 до 30 бар.
Согласно особому варианту осуществления настоящего изобретения взаимодействие амида альфа-гидроксикарбоновой кислоты со спиртом осуществляют в функционирующем под давлением реакторе. Под подобным реактором в принципе подразумевают реакционный объем, в котором во время превращения можно поддерживать избыточное давление. В этой связи избыточным считают давление, которое превышает атмосферное давление, то есть, в частности, составляет более 1 бар. Предпочтительному давлению соответствует интервал от 1 до 100 бар. В соответствии с этим давление как во время превращения/алкоголиза амида альфа-гидроксикарбоновой кислоты, так и во время выделения/удаления аммиака из смеси продуктов превращения/алкоголиза может превышать атмосферное давление, то есть может составлять более 1 бар. Таким образом, образующийся аммиак можно удалять из смеси продуктов превращения путем отгонки под давлением выше 1 бар, причем при дистилляционном удалении аммиака можно полностью отказаться от использования вспомогательных средств, таких как отпаривающий газ.
В смеси продуктов алкоголиза можно уменьшать содержание не только аммиака, но и непревращенного спирта. В частности, если для алкоголиза используют метанол, то образуется смесь продуктов реакции, содержащая в принципе очень трудно разделимые аммиак и метанол. В наиболее простом случае с целью обеднения подобной смеси продуктов реакции аммиаком и спиртом последние удаляют из нее непосредственно в виде смеси аммиака со спиртом. Последнюю затем подвергают разделению, выполняемому, например, путем ректификации. Кроме того, оба указанных компонента, то есть спирт (метанол) и аммиак, можно совместно выделять из смеси продуктов реакции и одновременно отделять друг от друга.
Алкоголиз и удаление аммиака/спирта из смеси продуктов реакции можно осуществлять в соответствующих агрегатах, которые пространственно отделены друг от друга. Для этой цели можно предусмотреть, например, один или несколько функционирующих под давлением реакторов, соединенных с функционирующей под давлением дистилляционной колонной. Речь при этом идет об одном или нескольких реакторах, упорядоченных в отдельной зоне вне дистилляционной колонны.
В предпочтительном варианте можно использовать непрерывный способ получения сложных эфиров альфа-гидроксикарбоновых кислот, в соответствии с которым в присутствии катализатора осуществляют превращение амида альфа-гидроксикарбоновой кислоты со спиртом, приводящее к образованию смеси продуктов реакции, которая содержит сложный эфир альфа-гидроксикарбоновой кислоты, аммиак, непревращенный амид альфа-гидроксикарбоновой кислоты, а также спирт и катализатор, причем:
а') в функционирующий под давлением реактор вводят потоки используемых в качестве исходных продуктов амида альфа-гидроксикарбоновой кислоты, спирта и катализатора,
b') в функционирующем под давлением реакторе реализуют взаимодействие указанных потоков при давлении от 1 до 100 бар,
с') полученную на стадии b') смесь продуктов реакции, содержащую сложный эфир альфа-гидроксикарбоновой кислоты, непревращенный амид альфа-гидроксикарбоновой кислоты и катализатор, выгружают из функционирующего под давлением реактора, и
d') снижают содержание спирта и аммиака в указанной смеси продуктов реакции, причем аммиак отгоняют под давлением, которое поддерживают на постоянном уровне, составляющем более 1 бар.
При этом в соответствии с особенно целесообразным вариантом:
b1) в функционирующем под давлением реакторе реализуют взаимодействие исходных продуктов при давлении от 5 до 70 бар,
b2) давление полученной на стадии b1) смеси продуктов реакции снижают до величины, которой соответствует интервал от 1 бар до давления в функционирующем под давлением реакторе,
с1) смесь продуктов реакции со сниженным на стадии b2) давлением направляют в дистилляционную колонну,
с2) из верхней части дистилляционной колонны, давление в которой составляет от 1 до 10 бар, отбирают аммиак и спирт, и
d1) из дистилляционной колонны отбирают обедненную аммиаком и спиртом смесь полученных на стадии с2) продуктов реакции, содержащую сложный эфир альфа-гидроксикарбоновой кислоты, непревращенный амид альфа-гидроксикарбоновой кислоты и катализатор.
В соответствии с указанным предпочтительным вариантом осуществления предлагаемого в изобретении способа превращение исходных продуктов и выделение аммиака/спирта осуществляют в двух пространственно отделенных друг от друга агрегатах. Другими словами, реактор/реакционный объем и агрегат для выделения аммиака/спирта из смеси продуктов алкоголиза отделены друг от друга. Преимущество подобного варианта состоит в возможности осуществления реакции/превращения исходных продуктов и последующего выделения аммиака/спирта в разных диапазонах давления. Деление способа на стадию превращения в функционирующем под давлением реакторе и стадию разделения в функционирующей при более высоком давлении дистилляционной колонне, причем обе стадии осуществляют под избыточным давлением, составляющим более 1 бар, неожиданно позволяет дополнительно существенно повысить эффективность разделения продуктов алкоголиза и выделения смеси аммиака со спиртом.
Дополнительного улучшения указанных качественных параметров можно достичь благодаря однократному или многократному повторению в функционирующем под давлением реакторе превращения смеси продуктов реакции, обедненной аммиаком и спиртом в кубе разделительной колонны (функционирующей под давлением дистилляционной колонне), причем стадию превращения осуществляют в нескольких функционирующих под давлением реакторах, последовательно соединенных друг с другом.
Таким образом, еще более предпочтительным является вариант осуществления способа, который отличается тем, что:
e) выгружаемую со стадии d1) смесь продуктов реакции сжимают до давления, составляющего от 5 до 70 бар,
f) сжатую на стадии е) смесь направляют в другой функционирующий под давлением реактор, в котором вновь осуществляют взаимодействие, и
g) повторяют указанные выше стадии b2), c1), c2) и d1).
Таким образом, особый интерес представляет вариант осуществления предлагаемого в изобретении способа, в соответствии с которым обедненную аммиаком и спиртом смесь отбирают с тарелки, находящейся выше куба первой дистилляционной колонны, сжимают ее до давления, превышающего давление в дистилляционной колонне, и направляют во второй функционирующий под давлением реактор, в котором осуществляют повторное взаимодействие при повышенном давлении и повышенной температуре, затем давление полученной в результате двукратного превращения смеси продуктов реакции уменьшают до величины, более низкой по сравнению с давлением во втором функционирующем под давлением реакторе, но превышающей 1 бар, и возвращают в первую дистилляционную колонну ниже тарелки, с которой был осуществлен отбор во второй функционирующий под давлением реактор, но выше куба этой колонны, и из ее верхней части вновь отбирают аммиак и спирт, получая двукратно обедненную аммиаком и спиртом смесь продуктов реакции.
Указанный технологический процесс можно повторять неоднократно, причем в особенно благоприятном варианте его повторяют, например, три или четыре раза. Таким образом, предпочтительным является способ, в соответствии с которым несколько раз повторяют взаимодействие в функционирующем под давлением реакторе, снижение давления полученной в результате превращения смеси, подачу указанной смеси в первую дистилляционную колонну, ее обеднение аммиаком и спиртом в первой дистилляционной колонне, отбор обедненной аммиаком и спиртом смеси, ее сжатие и последующую подачу в другой функционирующий под давлением реактор, причем в соответствии с числом n последовательно соединенных, функционирующих под давлением реакторов в функционирующей под давлением дистилляционной колонне получают смесь продуктов реакции, n-кратно обедненную аммиаком и спиртом. При этом n может означать положительное целое число больше ноля. Числу n предпочтительно соответствует интервал от 2 до 10.
В целесообразном варианте осуществления способа предусматривают многократное повторение указанных выше стадий от е) до g).
В соответствии с особым вариантом осуществления способа реализуют четырехкратное превращение и обеднение, используя четыре функционирующие под давлением реактора, последовательно соединенные друг с другом, и получая четырежды обедненную аммиаком и спиртом смесь продуктов реакции. Следовательно, подобный вариант осуществления способа отличается тем, что стадии от е) до g) повторяют по меньшей мере дважды, то есть превращение осуществляют в общей сложности по меньшей мере в четырех функционирующих под давлением реакторах, последовательно соединенных друг с другом.
Для осуществления указанного варианта особенно целесообразным является использование разных температурных интервалов в дистилляционной колонне и реакторе.
Так, например, температура в функционирующей под давлением дистилляционной колонне в общем случае предпочтительно находится в примерном интервале от 50 до 160°С. Точная температура в типичных случаях определяется точкой кипения системы, которая зависит от преобладающего в дистилляционной колонне давления.
Температура в реакторе предпочтительно находится в примерном интервале от 120 до 240°С. При этом еще более целесообразным является снижение температуры в реакторе по сравнению с указанными значениями, например, на величину, составляющую от 3 до 15°С, предпочтительно от 4 до 10°С, еще более целесообразно на 5°С. Подобное снижение температуры оказывает положительное влияние на селективность превращения.
Другое техническое мероприятие, которое способствует повышению селективности, состоит в уменьшении реакционного объема при переходе от одного реактора к другому. По мере уменьшения объема реактора наряду с повышением селективности достигают повышения степени превращения.
Как указано выше, благоприятным является отбор смеси продуктов реакции из определенной точки функционирующей под давлением дистилляционной колонны. При этом для определения относительного местоположения точки отбора в качестве ориентира используют расстояние от этой точки до куба дистилляционной колонны. В соответствии с настоящим изобретением особенно целесообразным является вариант, в соответствии с которым смесь продуктов реакции со сниженным давлением после очередного взаимодействия в функционирующем под давлением реакторе вводят на стадии с1) в дистилляционную колонну в точке, которая находится на меньшем расстоянии от нижней части этой колонны (то есть от куба) по сравнению с точкой подачи смеси продуктов реакции, осуществляемой после предыдущего превращения.
Наряду с рассмотренным выше вариантом, согласно которому взаимодействие амида альфа-гидроксикарбоновой кислоты со спиртом и удаление образующегося при этом, в частности, аммиака осуществляют в двух пространственно отделенных друг от друга, но взаимосвязанных агрегатах, может оказаться предпочтительным другой вариант, в соответствии с которым стадию взаимодействия указанных реагентов и стадию удаления аммиака реализуют в едином агрегате. При этом функционирующий под давлением реактор и функционирующая под давлением дистилляционная колонна в известной степени объединены в единый агрегат.
Давление, подлежащее предпочтительному соблюдению в реакционной дистилляционной колонне, которую в соответствии с указанным выше предлагаемым в изобретении вариантом используют в качестве реактора, можно варьировать в широких пределах. При этом согласно предпочтительному варианту осуществления изобретения стадии от а) до с) одновременно реализуют в реакционной дистилляционной колонне, функционирующей под давлением от 5 до 40 бар. Особенно целесообразным является способ, в соответствии с которым стадии от а) до с) одновременно реализуют в реакционной дистилляционной колонне при давлении от 10 до 30 бар.
В соответствии с предпочтительным вариантом осуществления способа превращение исходных продуктов реализуют в реакционной дистилляционной колонне, сконструированной в виде функционирующей под давлением колонны, причем образующийся в процессе превращения аммиак непрерывно выводят из ее верхней части. Благодаря этому достигают неожиданного эффекта, который состоит в возможности чрезвычайно простого выделения и повторного использования обладающего высокой степенью чистоты аммиака без необходимости снижения давления. Особый интерес при этом представляет также вариант, в соответствии с которым аммиак под давлением выводят из верхней части колонны, в то время как спирт отбирают из куба колонны или выводят из нее в виде бокового потока. Таким образом, благодаря надлежащему расчету эффективности разделения в подобной реакционной дистилляционной колонне обеспечивают непосредственное разделение аммиака и спирта.
Согласно указанному варианту осуществления настоящего изобретения можно использовать любую выдерживающую давление реакционную дистилляционную колонну, которая предпочтительно обладает двумя или более ступенями разделения. Подобные реакционные дистилляционные колонны более подробно описаны ниже при рассмотрении стадии D), причем их можно использовать также для превращения амида карбоновой кислоты со спиртом.
Обедненная аммиаком смесь продуктов реакции, в частности, содержит целевой сложный эфир альфа-гидроксикарбоновой кислоты. В целесообразном варианте осуществления способа с целью дополнительного выделения и очистки сложного эфира обедненную аммиаком смесь продуктов реакции можно отбирать из куба реакционной дистилляционной колонны и направлять в другую (вторую) дистилляционную колонну, в которой спирт выводят из верхней части и предпочтительно возвращают в реактор, получая при этом смесь продуктов, обедненную как аммиаком, так и спиртом.
С целью дополнительного выделения сложного эфира альфа-гидроксикарбоновой кислоты из обедненной аммиаком и спиртом смеси и его очистки обедненную аммиаком и спиртом смесь в предпочтительном варианте осуществления способа выводят из куба дополнительной дистилляционной колонны и направляют в другую дистилляционную колонну, причем эфир альфа-гидроксикарбоновой кислоты отбирают из верхней части этой колонны, а обедненную аммиаком, спиртом и эфиром альфа-гидроксикарбоновой кислоты смесь при необходимости после переработки на дополнительных стадиях очистки возвращают в реактор. Отбираемый из верхней части этой колонны сложный эфир альфа-гидроксикарбоновой кислоты обладает высокой степенью чистоты и в особенно предпочтительном варианте может быть направлен, например, на другие реакционные стадии с целью получения алкил(мет)акрилатов.
Как указано выше, дистилляционная система предпочтительно включает по меньшей мере одну зону, называемую реактором, в которой предусмотрен по меньшей мере один катализатор. Подобный реактор, как указано выше, предпочтительно находится внутри дистилляционной колонны.
Согласно изобретению предпочтительным является удаление из реакционной системы через газовую фазу не более 10% масс., предпочтительно не более 5% масс., особенно предпочтительно не более 1% масс. находящегося в реакционной фазе спирта. Подобное мероприятие способствует особенно экономичному осуществлению алкоголиза.
Для ускорения указанной реакции можно использовать, например, катализаторы с основным характером. Речь при этом идет как о гомогенных, так и о гетерогенных катализаторах.
К гомогенным катализаторам относятся алкоголяты щелочных металлов и металлоорганические соединения титана, олова или алюминия. В качестве гомогенного катализатора предпочтительно используют алкоголят титана или алкоголят олово, например тетраизопропилоксид титана или тетрабутилоксид олова. К гетерогенным катализаторам, относятся, в частности, оксид магния, оксид кальция, а также указанные выше ионообменные смолы с основным характером.
В качестве катализаторов, используемых для осуществления предлагаемого в изобретении способа, особый интерес представляют водостойкие соединения лантаноидов. Использование гомогенных катализаторов указанного типа приводит к неожиданным благоприятным результатам. Определение подобного катализатора «водостойкий» означает, что он сохраняет свою каталитическую способность в присутствии воды. В соответствии с этим предлагаемое в изобретении превращение можно осуществлять в присутствии до 2% масс. воды без существенного негативного воздействия последней на каталитическую активность катализатора. При этом определение «существенное» означает, что скорость и/или селективность реакции снижается не более чем на 50% по сравнению с превращением, осуществляемым в отсутствие воды.
К соединениям лантаноидов относятся соединения лантана, цезия, празеодима, неодима, прометия, самария, европия, гадолиния, тербия, диспрозия, гольмия, эрбия, тулия, иттербия и/или лютеция. Предпочтительному использованию подлежат соединения лантаноидов, которые содержат лантан. Растворимость соединений лантаноидов в воде при 25°С преимущественно составляет по меньшей мере 1 г/л, предпочтительно по меньшей мере 10 г/л. Предпочтительными соединениями лантаноидов являются соли, которые предпочтительно находятся в степени окисления 3. К особенно предпочтительным водостойким соединениям лантаноидов относятся La(NO3)3 и/или LaCl3. Указанные соединения можно добавлять к реакционной смеси в виде солей или они образуются in situ.
Особый вариант осуществления способа предусматривает использование в качестве катализатора растворимого комплекса, содержащего титан и/или олово, а также амид альфа-гидроксикарбоновой кислоты.
Другой особый вариант осуществления предлагаемого в изобретении способа предусматривает использование в качестве катализатора трифторметансульфоната металла. При этом предпочтительно используют трифторметансульфонат металла, выбранного из группы, включающей элементы 1, 2, 3, 4, 11, 12, 13 и 14 групп периодической системы элементов. Предпочтительными являются трифторметансульфонаты одного или нескольких металлов, относящихся к лантаноидам.
Наряду с предпочтительными вариантами гомогенного катализа в известных условиях целесообразным является также осуществление способа, предусматривающее использование гетерогенных катализаторов. К хорошо пригодным гетерогенным катализаторам относятся, в частности, оксид магния, оксид кальция, а также ионообменные вещества с основным характером и другие вещества. Так, например, может быть предпочтительным способ, в соответствии с которым в качестве катализатора используют нерастворимый оксид по меньшей мере одного металла, выбранного из группы, включающей сурьму, скандий, ванадий, лантан, церий, титан, цирконий, гафний, ниобий, тантал, хром, молибден, вольфрам, технеций, рений, железо, кобальт, никель, медь, алюминий, кремний, олово, свинец и висмут. В другом варианте предпочтительным может быть способ, в соответствии с которым в качестве катализатора используют нерастворимый металл, выбранный из группы, включающей титан, цирконий, гафний, ванадий, ниобий, тантал, хром, молибден, вольфрам, железо, кобальт, никель, медь, галлий, индий, висмут и теллур.
Образующийся аммиак обычно выводят из реакционной системы, причем превращение часто осуществляют при температуре кипения.
Выделяющийся в процессе алкоголиза свободный аммиак может быть легко возвращен в общий технологический процесс. Так, например, аммиак может быть подвергнут превращению с метанолом до цианистоводородной кислоты. Подобное превращение описано, например, в европейской заявке на патент ЕР-А-0941984. Кроме того, цианистоводородную кислоту можно получать из аммиака и метана согласно методу фирмы ВМА или по реакции Андрусова, причем указанные методы получения HCN приведены в Ullmann's Encyclopedia of Industrial Chemistry (5-е издание на CD-ROM, ключевое слово "Inorganic Cyano Compounds").
Предпочтительный вариант осуществления алкоголиза амида α-гидроксикарбоновой кислоты на стадии С) приведен в международной заявке WO 2007/131829, зарегистрированной 28.03.2007 Европейским патентным ведомством под номером РСТ/ЕР2007/052951, причем описанный в ней вариант превращения гидроксиамида карбоновой кислоты со спиртом посредством данной ссылки, включен в описание настоящего изобретения.
На следующей стадии D) сложный алкиловый эфир α-гидроксикарбоновой кислоты подвергают превращению с (мет)акриловой кислотой, в результате которого получают алкил(мет)акрилат, а также α-гидроксикарбоновую кислоту.
Согласно другому аспекту настоящего изобретения сложный алкиловый эфир α-гидроксикарбоновой кислоты можно подвергать превращению с (мет)акриловой кислотой. Используемые при этом (мет)акриловые кислоты являются коммерчески доступными известными продуктами. Помимо акриловой (пропеновой) кислоты и метакриловой (2-метилпропеновой) кислоты к пригодным (мет)акриловым кислотам, в частности, относятся соответствующие производные, которые содержат заместители. К пригодным заместителям, в частности, относятся галогены, такие как хлор, фтор и бром, а также алкильные группы предпочтительно с 1-10 атомами углерода, особенно предпочтительно с 1-4 атомами углерода. К соответствующим производным относятся, в частности, β-метилакриловая (бутеновая) кислота, α,β-диметилакриловая кислота, β-этилакриловая кислота, а также β,β-диметилакриловая кислота. Предпочтительными являются акриловая (пропеновая) кислота и метакриловая (2-метилпропеновая) кислота, причем особенно предпочтительной является метакриловая кислота.
Используемые сложные алкиловые эфиры α-гидроксикарбоновой кислоты являются известными соединениями, спиртовой остаток которых предпочтительно содержит 1-20 атомов углерода, в частности 1-10 атомов углерода, особенно предпочтительно 1-5 атомов углерода. Предпочтительные спиртовые остатки, в частности, являются производными метанола, этанола, пропанола, бутанола (в частности, н-бутанола и 2-метил-1-пропанола), пентанола, гексанола или 2-этилгексанола, причем особенно предпочтительными являются остатки метанола и этанола.
Кислотный остаток используемых для переэтерификации сложных алкиловых эфиров α-гидроксикарбоновой кислоты предпочтительно является производным (мет)акриловой кислоты, которая может быть получена путем дегидратации α-гидроксикарбоновой кислоты. В случае метакриловой кислоты используют, например, сложный эфир α-гидроксиизомасляной кислоты. В случае акриловой кислоты предпочтительно используют, например, α-гидроксиизопропионовую кислоту.
К предпочтительно используемым сложным алкиловым эфирам α-гидроксикарбоновой кислоты относится метиловый эфир α-гидроксипропионовой кислоты, этиловый эфир α-гидроксипропионовой кислоты, метиловый эфир α-гидроксиизомасляной кислоты и этиловый эфир α-гидроксиизомасляной кислоты.
Реакционная смесь помимо реагентов может содержать также другие компоненты, например, такие как растворители, катализаторы, ингибиторы полимеризации и воду.
Превращение сложного алкилового эфира α-гидроксикарбоновой кислоты можно катализировать по меньшей мере одной кислотой или по меньшей мере одним основанием. При этом можно использовать как гомогенные, так и гетерогенные катализаторы. В качестве кислотных катализаторов особенно пригодны, в частности, неорганические кислоты, например, серная кислота или соляная кислота, органические кислоты, например, сульфокислоты, в частности п-толуолсульфокислота, а также кислотные катионообменные смолы.
К особенно пригодным катионообменным смолам, в частности, относятся сополимеры стирола с дивинилбензолом, содержащие сульфокислотные группы. Особенно пригодными катионообменными смолами являются продукты, поставляемые фирмой Rohm&Haas под торговым названием Amberlyst®, а также фирмой Lanxess под торговым названием Lewatit®.
Концентрация катализатора предпочтительно составляет от 1 до 30% масс., особенно предпочтительно от 5 до 15% масс. в пересчете на суммарное количество используемого сложного эфира α-алкилгидроксикарбоновой кислоты и используемой (мет)акриловой кислоты.
К предпочтительно используемым ингибиторам полимеризации относятся, в частности, фенотиазин, трет-бутилпирокатехин, монометиловый эфир гидрохинона, гидрохинон, 4-гидрокси-2,2,6,6-тетраметилпиперидинооксил (TEMPOL) или их смеси, причем эффективность указанных ингибиторов может быть в определенной степени повышена благодаря использованию кислорода. Ингибиторы полимеризации можно использовать в концентрации от 0,001 до 2,0% масс., особенно предпочтительно от 0,01 до 0,2% масс., в пересчете на суммарное количество используемого сложного эфира α-алкилгидроксикарбоновой кислоты и используемой (мет)акриловой кислоты.
Переэтерификацию предпочтительно осуществляют в температурном интервале от 50 до 200°С, особенно предпочтительно от 70 до 130°С, в частности от 80 до 120°С и еще более предпочтительно от 90 до 110°С.
В зависимости от температуры переэтерификацию можно осуществлять при пониженном или избыточном давлении. Переэтерификацию предпочтительно осуществляют в интервале давлений от 0,02 до 5 бар, в частности от 0,2 до 3 бар, особенно предпочтительно от 0,3 до 0,5 бар.
Молярное отношение (мет)акриловой кислоты к сложному алкиловому эфиру α-гидроксикарбоновой кислоты предпочтительно находится в интервале от 4:1 до 1:4, в частности от 3:1 до 1:3, особенно предпочтительно от 2:1 до 1:2.
Селективность переэтерификации предпочтительно составляет по меньшей мере 90%, особенно предпочтительно 98%. Селективность переэтерификации определяется отношением суммарного количества образующихся алкил(мет)акрилатов и α-гидроксикарбоновых кислот к суммарному количеству превращенных исходных соединений, то есть алкилового эфира α-гидроксикарбоновой кислоты и (мет)акриловой кислоты.
Согласно особому варианту осуществления настоящего изобретения переэтерификацию можно осуществлять в присутствии воды. Содержание воды предпочтительно составляет от 0,1 до 50% масс., особенно предпочтительно от 0,5 до 20% масс., еще более предпочтительно от 1 до 10% масс. в пересчете на массу используемого сложного алкилового эфира α-гидроксикарбоновой кислоты.
Добавление небольших количеств воды неожиданно позволяет повысить селективность переэтерификации. При этом несмотря на добавление воды неожиданно удается поддерживать количество образующегося метанола на низком уровне. При концентрации воды от 10 до 15% масс. (в пересчете на массу используемого сложного алкилового эфира α-гидроксикарбоновой кислоты), температуре переэтерификации 120°С и ее длительности (соответственно времени контакта реагентов) от 5 до 180 минут предпочтительно образуется менее 5% масс. метанола.
Переэтерификацию можно осуществлять в периодическом или непрерывном режиме, причем предпочтительным является непрерывный режим. Продукты переэтерификации с целью смещения равновесия предпочтительно можно отделять от соответствующих исходных продуктов.
Длительность переэтерификации зависит от молекулярной массы реагентов и температуры, причем указанные параметры можно варьировать в широких пределах. Длительность переэтерификации сложного алкилового эфира α-гидроксикарбоновой кислоты (мет)акриловой кислотой предпочтительно составляет от 30 секунд до 15 часов, особенно предпочтительно от 5 минут до 5 часов и еще более предпочтительно от 15 минут до 3 часов.
При осуществлении переэтерификации в непрерывном режиме время контакта реагентов предпочтительно составляет от 30 секунд до 15 часов, особенно предпочтительно от 5 минут до 5 часов и еще более предпочтительно от 15 минут до 3 часов.
При получении метилметакрилата путем переэтерификации сложного метилового эфира α-гидроксиизомасляной кислоты температура предпочтительно составляет от 60 до 130°С, особенно предпочтительно от 80 до 120°С, еще более предпочтительно от 90 до 110°С. При этом давление предпочтительно находится в интервале от 50 до 1000 мбар, особенно предпочтительно от 300 до 800 мбар. Молярное отношение метакриловой кислоты к сложному метиловому эфиру α-гидроксиизомасляной кислоты предпочтительно составляет от 2:1 до 1:2, в частности от 1,5:1 до 1:1,5.
В особенно предпочтительном варианте переэтерификацию можно осуществлять в дистилляционной колонне. При этом катализатор можно вводить в любую зону колонны. Так, например, катализатор можно вводить в зону куба или в зону колонны. Однако при этом должен быть реализован контакт исходные продуктов с катализатором. Кроме того, катализатор можно вводить в отдельную зону дистилляционной колонны, соединенную с другими ее зонами, например, кубом и/или колонной. Подобное отдельное расположение зоны подачи катализатора является предпочтительным.
Осуществление переэтерификации в соответствии с указанным предпочтительным вариантом неожиданно позволяет повысить ее селективность. В этой связи следует констатировать, что давление реакции можно регулировать независимо от давления внутри дистилляционных колонн. Благодаря этому можно поддерживать температуру кипения на низком уровне без необходимости соответствующего увеличения длительности реакции, соответственно времени контакта реагентов. Температуру переэтерификации также можно варьировать в широких пределах. Тем самым можно сократить длительность этой реакции. Кроме того, можно использовать любой объем катализатора без необходимости учета геометрических параметров колонны. Наряду с этим можно добавлять, например, другой реагент. Все указанные мероприятия могут способствовать повышению селективности и производительности, причем неожиданно достигают синергических эффектов.
При этом в дистилляционную колонну подают сложный алкиловый эфир α-гидроксикарбоновой кислоты, например, метиловый эфир α-гидроксиизомасляной кислоты. В дистилляционную колонну подают также (мет)акриловую кислоту, например, метакриловую кислоту. Дистилляцию предпочтительно осуществляют в таких условиях, чтобы можно было отгонять только один продукт, а второй продукт оставлять в кубе и непрерывно отбирать. В случае использования спиртов с небольшим числом атомов углерода, в частности этанола или метанола, в дистилляционной колонне из реакционной смеси предпочтительно отгоняют алкил(мет)акрилат. Исходные продукты в циклическом режиме пропускают через зону катализатора. При этом непрерывно образуется алкил(мет)акрилат, а также α-гидроксикарбоновая кислота.
Предпочтительный вариант конструктивного исполнения дистилляционной колонны схематически показан на фиг.1. Исходные продукты можно подавать в дистилляционную колонну (3) по общему трубопроводу (1) или по двум отдельным трубопроводам (1) и (2). Подачу исходных продуктов предпочтительно осуществляют по двум отдельным трубопроводам. При этом исходные продукты можно подавать на одну и ту же ступень или в любое место дистилляционной колонны (3).
При этом температуру реагентов можно регулировать путем их пропускания через теплообменник, не показанный на фиг.1. В предпочтительном варианте исходные продукты подают в дистилляционную колонну по отдельности, причем более легкокипящий компонент вводят в колонну ниже места подачи более высококипящего компонента. В подобном случае более легкокипящий компонент подают в дистилляционную колонну предпочтительно в парообразном состоянии.
Для осуществления настоящего изобретения можно использовать любую многоступенчатую дистилляционную колонну (3), которая обладает двумя или более ступенями разделения. В соответствии с настоящим изобретением под числом ступеней разделения подразумевают количество тарелок в случае использования тарельчатой колонны или число теоретических ступеней разделения в случае использования колонны с насадочным материалом или колонны с насадочными телами.
Примерами пригодных многоступенчатых тарельчатых дистилляционных колонн являются дистилляционные колонны, оснащенные колпачковыми, сетчатыми, туннельными, клапанными, щелевыми, комбинированными сетчатыми/щелевыми, комбинированными сетчатыми/колпачковыми, дюзовыми или центробежными тарелками, примерами пригодных многоступенчатых дистилляционных колонн с насадочными телами дистилляционные колонны, заполненные кольцами Рашига, кольцами Лессинга, кольцами Палля, седлами Берля или седлами Intalox, примерами пригодных многоступенчатых дистилляционных колонн с насадочным материалом являются дистилляционные колонны с насадками типа Mellapak (фирма Sulzer), Rombopak (фирма Kuhni), Montz-pak (фирма Montz) и насадками с гнездами для катализатора, например, Kata-pak.
Можно использовать также комбинированную дистилляционную колонну, включающую секции с тарелками, секции с насадочными телами и секции с насадочным материалом.
Дистилляционная колонна (3) может быть снабжена внутренними устройствами. Колонна (3) предпочтительно оснащена конденсатором (12) для конденсации паров и кубовым испарителем (18).
Используемая для дистилляции аппаратура предпочтительно включает по меньшей мере одну зону, в дальнейшем называемую реактором, в которой предусмотрен по меньшей мере один катализатор. Подобный реактор может находиться внутри дистилляционной колонны. Однако его предпочтительно устанавливают в отдельной зоне вне дистилляционной колонны (3), причем этот предпочтительный вариант более подробно показан на фиг.1.
В случае осуществления переэтерификации в отдельном реакторе (8) часть стекающей в дистилляционной колонне жидкой фазы можно направлять в сборник и выводить из дистилляционной колонны в виде частичного потока (4). Положение сборника определяется профилем концентрации отдельных компонентов в колонне. При этом профиль концентраций можно регулировать путем варьирования температуры и/или флегмового числа. Сборник предпочтительно позиционируют таким образом, чтобы подаваемый в него из дистилляционной колонны поток содержал оба реагента, особенно предпочтительно реагенты в достаточно высокой концентрации, еще более предпочтительно при молярном отношении кислоты к сложному эфиру от 1,5:1 до 1:1,5. Кроме того, можно предусмотреть несколько сборников в разных местах дистилляционной колонны, причем молярное отношение можно регулировать путем варьирования расхода отбираемых реагентов.
Для установления необходимого отношения кислоты к сложному эфиру при перекрестной переэтерификации или для повышения селективности этой реакции к выводимому из дистилляционной колонны потоку можно добавлять дополнительный реагент, например, воду. Воду можно добавлять по трубопроводу (на фиг.1 не показан) из внешнего источника или из аппарата для разделения фаз (13). Напор обогащенного водой потока (5) можно повышать посредством устройства для нагнетания (6), например, насоса.
Повышение давления указанного потока позволяет уменьшать, соответственно предотвращать образование пара в реакторе, например, в реакторе со стационарным слоем катализатора. Благодаря этому можно обеспечивать равномерное пропускание реагентов через реактор и смачивание частиц катализатора. Обогащенный водой поток можно пропускать через теплообменник (7), посредством которого можно регулировать температуру переэтерификации. При этом указанный поток в зависимости от потребности можно нагревать или охлаждать. Кроме того, варьирование температуры реакции позволяет управлять соотношением между сложным эфиром и кислотой.
В реакторе (8) находится стационарный слой катализатора переэтерификации. Реагенты можно пропускать через слой катализатора сверху вниз или снизу вверх. Выходящий из реактора поток (9), содержащий определенное количество продуктов переэтерификации и непревращенные исходные продукты, причем содержание указанных компонентов в потоке зависит от времени пребывания, массы катализатора, температуры реакции и соотношения исходных продуктов, а также от количества добавляемой воды, сначала пропускают через теплообменник (10), в котором устанавливают температуру, предпочтительную для подачи потока в дистилляционную колонну. При этом устанавливают температуру, предпочтительно соответствующую температуре в месте ввода потока в дистилляционную колонну.
При этом место подачи возвращаемого из реактора в дистилляционную колонну потока может располагаться выше или ниже места отбора возвращаемого в реактор потока из дистилляционной колонны, предпочтительно выше этого места. Перед возвращением в дистилляционную колонну поток можно подвергать разряжению путем пропускания через клапан (11), причем давление потока предпочтительно устанавливают на уровне, аналогичном давлению в дистилляционной колонне. При этом давление в дистилляционной колонне предпочтительно ниже давления в реакторе. Преимуществом подобного варианта является снижение точки кипения подлежащих разделению компонентов, что позволяет осуществлять дистилляцию на более низком температурном уровне, а, следовательно, в энергосберегающем и термически благоприятном режиме.
В дистилляционной колонне (3) осуществляют разделение смеси продуктов переэтерификации. Продукт с более низкой температурой кипения, которым предпочтительно является образующийся в результате переэтерификации сложный эфир, выделяют в верхней части дистилляционной колонны (3). Дистилляционную колонну предпочтительно эксплуатируют в таком режиме, чтобы в качестве головного продукта можно было выделять также воду, добавляемую перед реактором со стационарным слоем катализатора. Отбираемый в верхней части дистилляционной колонны парообразный поток конденсируется в конденсаторе (12), и полученный конденсат разделяют в декантаторе (13) на водную фазу и фазу, содержащую сложный эфир. Водную фазу по трубопроводу (15) можно направлять на переработку или полностью или частично возвращать на переэтерификацию по трубопроводу (17). Часть содержащей сложный эфир фазы можно возвращать по трубопроводу (14) в дистилляционную колонну в качестве флегмы (16) или частично выводить из дистиллятора. Высококипящий продукт, которым предпочтительно является образующаяся в результате перекрестной переэтерификации кислота, отбирают из дистилляционной колонны в качестве кубового потока (19).
Образующуюся в результате переэтерификации α-гидроксикарбоновую кислоту, например гидроксиизомасляную кислоту, можно подвергать дегидратации, осуществляемой известными методами на последующей стадии Е). В общем случае α-гидроксикарбоновую кислоту, например α-гидроксиизомасляную кислоту, нагревают в присутствии по меньшей мере одной соли металла, например, соли щелочного металла и/или соли щелочноземельного металла, до температуры от 160 до 300°С, особенно предпочтительно от 200 до 240°С, причем в общем случае получают (мет)акриловую кислоту, а также воду. К пригодным солям металлов относятся, в частности, гидроксид натрия, гидроксид калия, гидроксид кальция, гидроксид бария, гидроксид магния, сульфит натрия, карбонат натрия, карбонат калия, карбонат стронция, карбонат магния, бикарбонат натрия, ацетат натрия, ацетат калия и дигидрофосфат натрия.
Дегидратацию α-гидроксикарбоновой кислоты можно осуществлять при давлении, предпочтительно находящемся в интервале от 0,05 до 2,5 бар, особенно предпочтительно в интервале от 0,1 до 1 бар.
Дегидратация α-гидроксикарбоновых кислот описана, например, в немецкой заявке на патент DE-A-1768253.
Полученную, как указано выше, (мет)акриловую кислоту, в свою очередь, можно использовать для получения алкил(мет)акрилатов. Кроме того, (мет)акриловая кислота является товарным продуктом. Таким образом, способ синтеза алкил(мет)акрилатов неожиданно можно использовать также для получения (мет)акриловой кислоты, причем соотношение между алкил(мет)акрилатами и (мет)акриловой кислотой легко можно установить путем варьирования концентрации воды при переэтерификации сложного алкилового эфира α-гидроксикарбоновой кислоты и/или температуры этой реакции.
Предлагаемый в изобретении способ можно рассматривать также в качестве ступени способа получения полимеров, формовочных масс и полимерных формованных изделий, который реализуют с использованием настоящего изобретения и который также является новым способом, обладающим признаками изобретения.
(Мет)акрилаты, которые могут быть получены согласно изобретению, в частности подлежащий предпочтительному получению метилметакрилат, можно превращать в полимеры путем радикальной полимеризации.
Соответствующие полимеры в общем случае получают путем радикальной полимеризации смесей, которые содержат метилметакрилат. Подобные смеси в общем случае содержат по меньшей мере 40% масс., предпочтительно по меньшей мере 60% масс. и особенно предпочтительно по меньшей мере 80% масс. метилметакрилата в пересчете на массу мономеров.
Помимо метилметакрилата подобные смеси могут содержать другие (мет)акрилаты, способные сополимеризоваться с метилметакрилатом. При этом под (мет)акрилатами согласно изобретению подразумевают метакрилаты и акрилаты, а также их смеси.
Указанные мономеры являются хорошо известными соединениями, к которым, в частности, относятся:
(мет)акрилаты, которые являются производными насыщенных спиртов, например, такие как метилакрилат, этил(мет)акрилат, пропил(мет)акрилат, н-бутил(мет)акрилат, трет-бутил(мет)акрилат, пентил(мет)акрилат или 2-этилгексил(мет)акрилат,
(мет)акрилаты, которые являются производными ненасыщенных спиртов, например, такие как олеил(мет)акрилат, 2-пропинил(мет)акрилат, аллил-(мет)акрилат или винил(мет)акрилат,
арил(мет)акрилаты, например, такие как бензил(мет)акрилат или фенил-(мет)акрилат, причем соответствующие арильные остатки могут быть незамещенными или могут содержать до четырех заместителей,
циклоалкил(мет)акрилаты, например, такие как 3-винилциклогексил(мет)-акрилат или борнил(мет)акрилат,
гидроксилалкил(мет)акрилаты, например, такие как 3-гидроксипропил-(мет)акрилат, 3,4-дигидроксибутил(мет)акрилат, 2-гидроксиэтил(мет)акрилат или 2 гидроксипропил(мет)акрилат,
гликольди(мет)акрилаты, например, такие как 1,4-бутандиол(мет)акрилат,
(мет)акрилаты на основе неполных эфиров многоатомных спиртов, например, такие как тетрагидрофурфурил(мет)акрилат или винилоксиэтоксиэтил(мет)акрилат,
амиды и нитрилы (мет)акриловой кислоты, например, такие как N-(3-ди-метиламинопропил)(мет)акриламид, N-(диэтилфосфоно)(мет)акриламид или 1-метакрилоиламидо-2-метил-2-пропанол,
серосодержащие метакрилаты, такие как этилсульфинилэтил(мет)акрилат, 4-тиоцианатобутил(мет)акрилат, этилсульфонилэтил(мет)акрилат, тио-цианатометил(мет)акрилат, метилсульфинилметил(мет)акрилат или бис((мет)акрилоилоксиэтил)сульфид,
многоатомные (мет)акрилаты, такие как триметилолпропантри-(мет)акрилат.
Помимо указанных выше (мет)акрилатов подвергаемые полимеризации смеси могут содержать также другие ненасыщенные мономеры, способные сополимеризоваться с метилметакрилатом и указанными выше (мет)акрилатами, к которым, в частности, относятся:
1-алкены, например, такие как гексен-1 или гептен-1,
разветвленные алкены, например, такие как винилциклогексан, 3,3-диметил-1-пропен, 3-метил-1-диизобутилен или 4-метилпентен-1,
акрилонитрил,
сложные виниловые эфиры, например, такие как винилацетат,
стирол, замещенные стиролы с алкильным заместителем в боковой цепи, например, такие как α-метилстирол или α-этилстирол, замещенные стиролы с алкильным заместителем в кольце, например, такие как винилтолуол или п-метилстирол, а также галогенированные стиролы, например, такие как монохлорстиролы, дихлорстиролы, трибромстиролы или тетрабромстиролы,
гетероциклические виниловые соединения, например, такие как 2-винил-пиридин, 3-винилпиридин, 2-метил-5-винилпиридин, 3-этил-4-винил-пиридин, 2,3-диметил-6-винилпиридин, винилпиримидин, винилпиперидин, 9-винилкарбазол, 3-винилкарбазол, 4-винилкарбазол, 1-винилимидазол, 2-метил-1-винилимидазол, N-винилпирролидон, 2-винилпирролидон, N-винилпирролидин, 3-винилпирролидин, N-винилкапролактам, N-винил-бутиролактам, винилоксолан, винилфуран, винилтиофен, винилтиолан, винилтиазолы, гидрированные винилтиазолы, винилоксазолы и гидрированные винилоксазолы,
простые виниловые и изопрениловые эфиры,
производные малеиновой кислоты, например, такие как малеиновый ангидрид, метилмалеиновый ангидрид, малеинимид или имид метилмалеиновой кислоты,
диены, например, такие как дивинилбензол.
В общем случае указанные сомономеры используют в количестве от 0 до 60% масс., предпочтительно от 0 до 40% масс., особенно предпочтительно от 0 до 20% масс. в пересчете на массу мономеров, причем их можно использовать по отдельности или в виде смеси.
В общем случае полимеризацию инициируют посредством радикальных инициаторов. К предпочтительным радикальным инициаторам относятся, в частности, хорошо известные специалистам азоинициаторы, например, такие как азобисизобутиронитрил или 1,1-азобисциклогексанкарбонитрил, а также пероксидные соединения, например, такие как пероксид метил-этилкетона, пероксид ацетилацетона, пероксид дилаурила, трет-бутилпер-2-этилгексаноат, пероксид кетона, пероксид метилизобутилкетона, пероксид циклогексанона, пероксид дибензоила, трет-бутил-пероксибензоат, трет-бутилпероксиизопропилкарбонат, 2,5-бис(2-этил-гексаноилперокси)-2,5-диметилгексан, трет-бутилперокси-2-этилгексаноат, трет-бутилперокси-3,5,5-триметил-гексаноат, пероксид дикумила, 1,1-бис(трет-бутилперокси)циклогексан, 1,1-бис(трет-бутилперокси)-3,3,5-триметилциклогексан, гидропероксид кумила, гидропероксид трет-бутила, бис(4-трет-бутилциклогексил)-пероксидикарбонат, смеси двух или более указанных выше соединений друг с другом, а также их смеси с неуказанными соединениями, которые также могут образовывать радикалы.
Указанные инициаторы чаще всего используют в количестве от 0,01 до 10% масс., предпочтительно от 0,5 до 3% масс., в пересчете на массу мономеров.
Полимеризацию предпочтительно можно осуществлять в температурном интервале от 20 до 120°С.
Получение гомополимеров и/или сополимеров (мет)акрилатов различными методами радикальной полимеризации известно. Указанную полимеризацию можно осуществлять в массе, растворе, суспензии или эмульсии. Полимеризация в массе описана, например, в Houben-Weyl, том Е20, часть 2 (1987), cc. 1145 и следующие. Ценную информацию относительно полимеризации в растворе можно найти на с.1156 и следующей указанного справочника, тогда как информация по поводу техники суспензионной полимеризации приведена на с.1149 и следующей, а по поводу техники эмульсионной полимеризации на с.1150 и следующей.
Указанные выше полимеры, в частности, можно использовать для изготовления формовочных масс, которые помимо полимеров обычно могут содержать добавки, например, такие как красители, пигменты, в частности, пигменты типа металлик, а также УФ-стабилизаторы и наполнители. Содержание указанных добавок определяется назначением конкретной формовочной массы, причем его можно варьировать в широких пределах. В случае присутствия указанных добавок их содержание предпочтительно может составлять от 0 до 30% масс., особенно предпочтительно от 0,1 до 5% масс.
Предпочтительные формовочные массы, соответственно полимеры, можно перерабатывать в формованные изделия обычными методами формования, например, методом литья под давлением или экструзии, причем объектом настоящего изобретения является также способ изготовления формованных изделий с использование полимеров, которые могут быть получены предлагаемым в изобретении способом.
Кроме того, алкил(мет)акрилаты, которые могут быть получены предлагаемым в изобретении способом, в частности метилметакрилат, можно использовать для изготовления литых стекол. Подобные формованные изделия, получаемые методом камерного литья, характеризуются особенно высокими показателями молекулярной массы, в связи с чем в отличие от полимеров, пригодных для переработки в термопластичном состоянии, они обладают иными механическими свойствами. Способ изготовления подобных формованных изделий, предусматривающий использование (мет)акрилатов, которые могут быть получены предлагаемым в изобретении способом, также является объектом настоящего изобретения.
Приведенный ниже пример служит для более подробного пояснения настоящего изобретения.
Пример 1
Готовят смесь, содержащую 1 моль цианистоводородной кислоты, 2,5 моля ацетона и 200 м.д. Li2O, которую при 20°С подвергают превращению до установления равновесия. Полученная реакционная смесь содержит 47,7% масс. ацетонциангидрина и 51,5% масс. ацетона. Содержание цианистоводородной кислоты в ней составляет менее 8000 м.д.
Указанную реакционную смесь непрерывно подают в реактор для гидролиза, в который добавляют 38,6% масс. воды. При этом содержание цианистоводородной кислоты в основном остается неизменным.
Часть воды добавляют из возврата, получаемого в кубе дистилляционной колонны при очистке реакционной смеси после гидролиза ацетонциангидрина. Гидролиз ацетонциангидрина осуществляют с использованием в качестве катализатора МnO2, который более подробно описан, в частности, в примерах к международной заявке WO 2008/061822. Аналогично приведенному в указанной заявке примеру 1 для стабилизации катализатора используют воздух. Показатель рН составляет 9,0.
Из реакционной смеси, полученной в результате гидролиза ацетонциангидрина, в качестве кубового продукта первой ступени дистилляции выделяют амид карбоновой кислоты, причем дистилляцию осуществляют при температуре куба 175°С и давлении 0,4 бар.
Головной продукт, содержащий 52% масс. воды, 43% масс. ацетона, 4,8% масс. ацетонциангидрина и 0,2% масс. цианистоводородной кислоты, подвергают очистке в дистилляционной колонне с внутренними устройствами, которую осуществляют в непрерывном режиме, причем подача продукта в колонну составляет 1,2 кг/ч. В качестве флегмы в указанную дистилляционную колонну подают 0,3 кг/ч ацетона, благодаря чему обеспечивают высокую эффективность разделения. Дистилляционную установку можно эксплуатировать в течение неограниченного времени: например, в течение 60 дней не было обнаружено снижения эффективности разделения. Цианистоводородная кислота в кубовом продукте указанной дистилляционной колонны отсутствует. Головной продукт содержит 86% масс. ацетона, 5,8% масс. ацетонциангидрина, 0,2% масс. цианистоводородной кислоты и 8% масс. воды.
Срок службы катализатора (промежуток времени, в течение которого степень превращения снижается, достигая 50% от первоначального значения) составляет более 60 дней.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ АЛКИЛ(МЕТ)АКРИЛАТОВ | 2006 |
|
RU2409552C2 |
| СПОСОБ ПОЛУЧЕНИЯ АЛЬФА-ГИДРОКСИКАРБОНОВЫХ КИСЛОТ | 2007 |
|
RU2454399C2 |
| СПОСОБ ПОЛУЧЕНИЯ МЕТАКРИЛОВОЙ КИСЛОТЫ | 2012 |
|
RU2602080C2 |
| СПОСОБ НЕПРЕРЫВНОГО ПОЛУЧЕНИЯ АЛКИЛАМИНО(МЕТ)АКРИЛАМИДОВ | 2010 |
|
RU2546670C2 |
| СПОСОБ ПОЛУЧЕНИЯ СЛОЖНЫХ АЛКИЛОВЫХ ЭФИРОВ МЕТАКРИЛОВОЙ КИСЛОТЫ АЗЕОТРОПНОЙ ДИСТИЛЛЯЦИЕЙ | 2007 |
|
RU2472770C2 |
| СПОСОБ ПЕРЕЭТЕРИФИКАЦИИ | 2006 |
|
RU2452725C2 |
| СПОСОБ ПОЛУЧЕНИЯ ЭФИРОВ (МЕТ)АКРИЛОВОЙ КИСЛОТЫ | 2009 |
|
RU2515985C2 |
| СПОСОБ НЕПРЕРЫВНОГО ПОЛУЧЕНИЯ АЛКИЛАМИНО(МЕТ)АКРИЛАМИДОВ | 2004 |
|
RU2374221C2 |
| КАТАЛИЗАТОР ДЛЯ ПРЕВРАЩЕНИЯ НИТРИЛОВ КАРБОНОВЫХ КИСЛОТ | 2009 |
|
RU2496575C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ КАРБОНОВЫХ КИСЛОТ | 2007 |
|
RU2448949C2 |
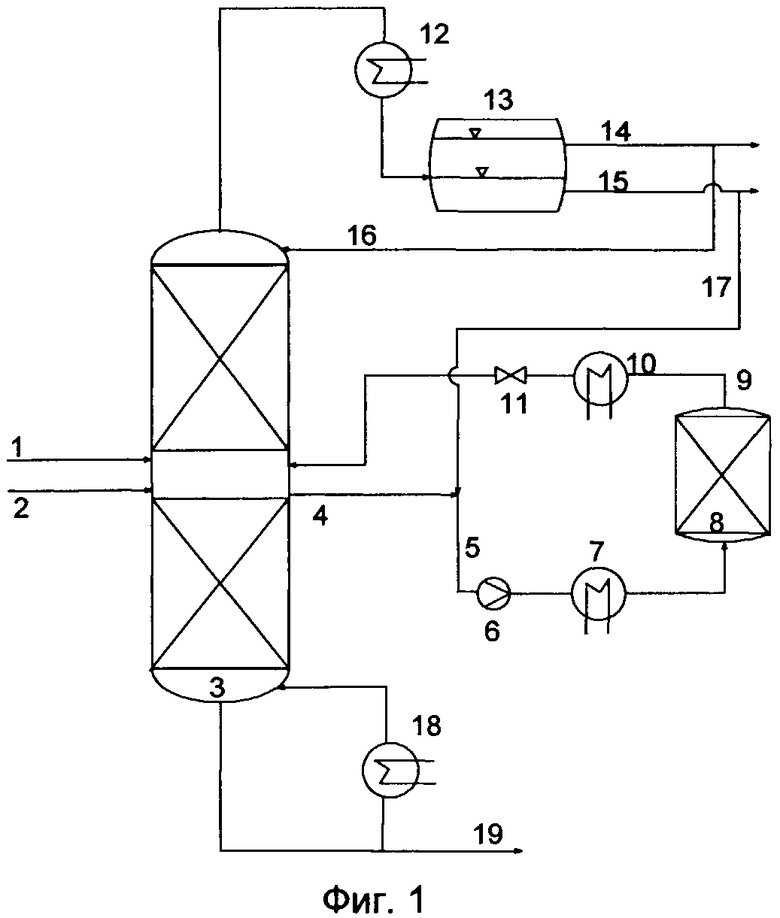
Изобретение относится к способу получения амида карбоновой кислоты из алифатического кетона с 3-5 атомами углерода и цианистоводородной кислоты. Способ включает стадии: А) взаимодействие кетона, взятого в молярном избытке, с цианистоводородной кислотой с получением нитрила соответствующей гидроксикарбоновой кислоты, Б) гидролиз полученного нитрила гидроксикарбоновой кислоты в присутствии содержащего диоксид марганца катализатора, В) переработку реакционной смеси, полученной после стадии Б), путем дистилляции. Дистилляцию осуществляют в две стадии, причем на первой стадии полученный амид карбоновой кислоты отделяют от смеси, содержащей воду, указанный кетон, нитрил гидроксикарбоновой кислоты и/или цианистоводородную кислоту, а на второй стадии указанную смесь разделяют с отбором указанного кетона и цианистоводородной кислоты в качестве головного продукта, а воды в качестве кубового продукта, при этом в дистилляционную колонну, используемую для разделения воды и цианистоводородной кислоты, с флегмой вводят смесь, которая содержит используемый на стадии А) кетон и обладает более низким содержанием цианистоводородной кислоты, чем смесь, отбираемая в верхней части этой колонны. Технический результат - усовершенствованный способ получения амида карбоновый кислоты, позволяющий увеличить период эксплуатации производственной установки при одновременном повышении срока службы катализатора гидролиза. 10 з.п. ф-лы, 1 ил., 1 пр.
1. Способ получения амида карбоновой кислоты из алифатического кетона с 3-5 атомами углерода и цианистоводородной кислоты, включающий следующие стадии:
A) взаимодействие указанного кетона с цианистоводородной кислотой с целью получения нитрила соответствующей гидроксикарбоновой кислоты, при этом указанный кетон используют в молярном избытке по отношению к цианистоводородной кислоте,
Б) гидролиз полученного на стадии А) нитрила гидроксикарбоновой кислоты в присутствии содержащего диоксид марганца катализатора,
B) переработку реакционной смеси, полученной после стадии Б) путем дистилляции,
отличающийся тем, что дистилляцию осуществляют в две стадии, причем на первой стадии полученный амид карбоновой кислоты отделяют от смеси, содержащей воду, указанный кетон, нитрил гидроксикарбоновой кислоты и/или цианистоводородную кислоту, а на второй стадии указанную смесь разделяют с отбором указанного кетона и цианистоводородной кислоты в качестве головного продукта, а воды в качестве кубового продукта, при этом в дистилляционную колонну, используемую для разделения воды и цианистоводородной кислоты, с флегмой вводят смесь, которая содержит используемый на стадии А) кетон и обладает более низким содержанием цианистоводородной кислоты, чем смесь, отбираемая в верхней части этой колонны.
2. Способ по п. 1, отличающийся тем, что молярное отношение указанного кетона к цианистоводородной кислоте составляет от 1,1:1 до 7:1.
3. Способ по п. 2, отличающийся тем, что молярное отношение указанного кетона к цианистоводородной кислоте составляет от 1,5:1 до 5:1.
4. Способ по п. 1, отличающийся тем, что количество добавляемого кетона выбирают таким образом, чтобы его было достаточно для получения на стадии А) предусматриваемого количества нитрила гидроксикарбоновой кислоты.
5. Способ по 1, отличающийся тем, что полученный в результате переработки реакционной смеси кетон используют для получения нитрила гидроксикарбоновой кислоты на стадии А).
6. Способ по п. 1, отличающийся тем, что в качестве указанного кетона используют ацетон.
7. Способ по п. 1, отличающийся тем, что стадию А) осуществляют в температурном интервале от -10 до 50оС.
8. Способ по п. 1, отличающийся тем, что стадию А) осуществляют при давлении от 0,3 до 3 бар.
9. Способ по п. 1, отличающийся тем, что молярное отношение воды к нитрилу гидроксикарбоновой кислоты при гидролизе на стадии Б) составляет от 0,5:1 до 25:1.
10. Способ по п. 1, отличающийся тем, что гидролиз на стадии Б) осуществляют в температурном интервале от 10 до 150оС.
11. Способ по одному из пп. 1-10, отличающийся тем, что гидролиз на стадии Б) осуществляют при давлении от 0,1 до 10 бар.
| ПЕРЕНОСНОЙ УНОР | 0 |
|
SU407811A1 |
| Приспособление для точного наложения листов бумаги при снятии оттисков | 1922 |
|
SU6A1 |
| DE 102006055430 A1, 29.05.2008 | |||
| JP 2002371046 A, 26.12.2002 | |||
| Способ получения ацетонциангидрина | 1970 |
|
SU486506A3 |
| СПОСОБ ПОЛУЧЕНИЯ МЕТАКРИЛАМИДА | 0 |
|
SU267511A1 |

