Настоящее изобретение относится к способу получения эфиров (мет)акриловой кислоты.
Эфиры (мет)акриловой кислоты представляют собой широко известные и часто используемые мономеры. В соответствии с этим известны многочисленные способы получения этих соединений. Для получения специальных (мет)акрилатов чаще всего проводят реакции переэтерификации, в ходе которых метил(мет)акрилат реагирует с соответствующим спиртом. До настоящего времени для повышения выхода и улучшения селективности этой реакции использовались различные катализаторы.
Так, например, в патенте ФРГ №2805702 описано получение эфиров ненасыщенных карбоновых кислот. Для катализа представленных в ней реакций могут быть использованы, в частности, соединения, которые содержат цирконий и/или кальций. К числу катализаторов, которые подходят лучше всего, принадлежит, в частности, ацетилацетонат циркония. Реакции протекают с высокими выходами, составляющими около 97% из расчета на используемый спирт.
Кроме того, для ускорения реакции переэтерификации могут быть использованы кислоты или основания. Такие реакции представлены, например, в патенте Китая №1355161, в патенте ФРГ №3423443 или в заявке на Европейский патент № А 0534666. К катализаторам с основными свойствами относится, в частности, амид лития, как это представлено в материалах патента ФРГ №3423443, патента Канады №795814 и патента США №6194530.
Кроме того, было предложено улучшить экономические показатели производства путем прибавления исходных соединений по ходу реакции. Так, например, в материалах патента США №5072027 описано прибавление спирта и метилметакрилата после достижения высокой степени превращения использовавшихся на начальной стадии реакции исходных соединений.
Представленные ранее превращения уже приводят к высокой степени превращения и к получению чистых продуктов. Однако ввиду важного экономического значения специальных (мет)акрилатов существует постоянное стремление к улучшению способов их получения.
Недостаток представленных ранее способов состоит также в том, что катализаторы должны быть выделены из реакционных смесей. При таком разделении многие из представленных ранее катализаторов подвергаются необратимому превращению и поэтому не могут быть снова использованы.
В соответствии с уровнем техники задача настоящего изобретения состояла в разработке способа получения (мет)акрилатов, в соответствии с которым целевой продукт может быть получен с очень хорошими экономическими показателями. Кроме того, получаемый эфир (мет)акриловой кислоты должен содержать лишь незначительные количества побочных продуктов и остатков катализатора.
Еще одна задача изобретения состояла в том, чтобы разработать способ, в соответствии с которым можно получать (мет)акрилаты с очень высокой селективностью.
Кроме того, задача настоящего изобретения состояла в разработке такого способа получения (мет)акрилатов, который может быть реализован легко и с небольшими затратами. При этом продукция должна получаться с максимально возможными выходами и с небольшими суммарными расходами энергии.
Кроме того, существовала потребность в разработке способа, который может быть реализован без трудоемкого отделения катализатора.
Эти задачи, а также другие, не выделенные специально, но легко выводящиеся из обсуждавшихся в вводной части взаимосвязей или следующие из них задачи, решаются способом со всеми признаками п.1 формулы изобретения. Практически значимые варианты соответствующего изобретению способа защищаются ссылающимися на п.1 зависимыми пунктами формулы.
В соответствии с этим, объектом настоящего изобретения является способ получения (мет)акрилатов, включающий переэтерификацию низкокипящего эфира (мет)акриловой кислоты используемым в качестве исходного продукта спиртом в присутствии катализаторов, при этом способ отличается тем, что переэтерификация катализируется основным ионообменным веществом.
Благодаря этому неожиданно удалось разработать способ получения (мет)акрилатов, в соответствии с которым получение продукции осуществляется очень экономично. Удивительно, что получаемая продукция содержит лишь очень незначительные количества побочных продуктов и остатков катализатора. В соответствии с изобретением применяемые катализаторы можно отделять особенно просто и без остатков. При этом катализатор можно без значительных издержек снова возвращать в процесс.
Кроме того, соответствующий изобретению способ представляет возможность исключительно селективного получения (мет)акрилатов.
Кроме того, соответствующий изобретению способ может быть реализован просто и без больших затрат, причем продукция может быть получена с высокими выходами и в целом с уменьшенным расходом энергии. В дополнение к этому соответствующий изобретению способ можно исключительно легко осуществлять по непрерывной схеме. Благодаря этому достигается высокое качество продукции при очень низких затратах.
В соответствии с изобретением получают (мет)акрилаты, при этом понятие (мет)акрилатов относится к эфирам метакриловой кислоты, эфирам акриловой кислоты и к их смесям.
(Мет)акрилаты широко известны. К этим соединениям относятся наряду с другими (мет)акрилаты, которые являются производными насыщенных спиртов, например, гексил(мет)акрилат, 2-этилгексил(мет)акрилат, гептил(мет)акрилат, 2-(трет-бутил)-аминоэтил(мет)акрилат, октил(мет)акрилат, 3-изопропилгептил(мет)акрилат, нонил-(мет)акрилат, децил(мет)акрилат, ундецил(мет)акрилат, 5-метилундецил(мет)акрилат, додецил(мет)акрилат, 2-метилдодецил(мет)акрилат, тридецил(мет)акрилат, 5-метил-тридецил(мет)акрилат, тетрадецил(мет)акрилат, пентадецил(мет)акрилат, гексадецил-(мет)акрилат, 2-метилгексадецил(мет)акрилат, гептадецил(мет)акрилат, 5-изопропил-гептадецил(мет)акрилат, 4-трет-бутилоктадецил(мет)акрилат, 5-этилоктадецил-(мет)акрилат, 3-изопропилоктадецил(мет)акрилат, октадецил(мет)акрилат, нонадецил-(мет)акрилат, эйкозил(мет)акрилат, цетилэйкозил(мет)акрилат, стеарилэйкозил-(мет)акрилат, докозил(мет)акрилат и/или эйкозилтетратриаконтил(мет)акрилат;
(мет)акрилаты, которые являются производными ненасыщенных спиртов, например, 2-пропинил(мет)акрилат, аллил(мет)акрилат, винил(мет)акрилат, олеил(мет)акрилат;
такие циклоалкил(мет)акрилаты, как циклопентил(мет)акрилат, 3-винилциклогексил-(мет)акрилат, циклогексил(мет)акрилат, борнил(мет)акрилат;
(мет)акрилаты, которые содержат две (мет)акрилатные группы или несколько (мет)акрилатных групп, такие ди(мет)акрилаты гликолей, как ди(мет)акрилат этиленгликоля, ди(мет)акрилат диэтиленгликоля, ди(мет)акрилат триэтиленгликоля, ди(мет)акрилаты тетра- и полиэтиленгликоля, ди(мет)акрилат 1,3-бутандиола, ди(мет)акрилат 1,4-бутандиола, ди(мет)акрилат 1,6-гександиола, ди(мет)акрилат глицерина и диметакрилаты этоксилированного бисфенола A;
такие (мет)акрилаты с тремя или с несколькими двойными связями, как, например, три(мет)акрилат глицерина, три(мет)акрилат триметилолпропана, тетра(мет)акрилат пентаэритрита и пента(мет)акрилат дипентаэритрита.
В число особо предпочтительных (мет)акрилатов входят, в частности, 2-этилгексил-(мет)акрилат, ди(мет)акрилат этиленгликоля (1,2-этандииловый диэфир 2-метил-пропеновой кислоты, CAS №97-905), диметакрилат этоксилированного бисфенола A, диметакрилат 1,3-бутандиола (CAS №1189-08-8), диметакрилат 1,4-бутандиола (CAS №2082-81-7) и/или триметакрилат триметилолпропана.
В соответствии с изобретением для получения (мет)акрилатов используют спирт, который в данных материалах называют также исходным спиртом. При этом свойства спирта определяются тем, какое соединение должно быть получено. В соответствии с этим могут быть, в частности, использованы спирты с числом атомов углерода от пяти и более, ненасыщенные спирты и/или многоатомные спирты. К предпочтительным спиртам с числом атомов углерода от пяти и более относятся, например, пентанол, гексанол, 3-метилбутанол, гептанол, 2-этилгексиловый спирт, 2-трет-бутилгептанол, октанол, 3-изопропилгептанол, нонанол, деканол, ундеканол, 5-метилундеканол, додеканол, 2-метилдодеканол, тридеканол, 5-метилтридеканол, тетрадеканол, пентадеканол, гексадеканол, 2-метилгексадеканол, гептадеканол, 5-изопропил-гептадеканол, 4-трет-бутилоктадеканол, 5-этилоктадеканол, 3-изопропилоктадеканол, октадеканол, нонадеканол, эйкозанол, цетилэйкозанол, стеарилэйкозанол, докозанол и/или эйкозилтетратриаконтанол. К предпочтительным ненасыщенным спиртам относятся, в частности, 2-пропин-1-ол, аллиловый спирт и виниловый спирт и/или олеиловый спирт. В соответствии с особым аспектом настоящего изобретения могут быть использованы одноатомные спирты, в составе которых есть только одна гидроксильная группа.
В особо предпочтительном случае используют многоатомные спирты. Многоатомные спирты представляют собой органические соединения с двумя, тремя, четырьмя или с несколькими гидроксильными группами. В число таких соединений входят, в частности, этиленгликоль, триметилолпропан, 1,2-пропандиол, 1,3-пропандиол, 1,3-бутандиол, 1,4-бутандиол, этоксилированный бисфенол А, 1,6-гександиол, пентаэритрит, полиэтиленгликоль, в частности, полиэтиленгликоль 400, и/или глицерин, при этом особое предпочтение отдается триметилолпропану. Эти соединения могут быть приобретены коммерческим путем, поскольку они производятся, например, фирмами BASF AG, Celanese AG или Clariant GmbH.
В соответствии с настоящим изобретением проводят взаимодействие спирта с низкокипящим эфиром (мет)акриловой кислоты. Понятие «низкокипящий эфир (мет)акриловой кислоты» означает, что в качестве исходного соединения используется сложный эфир с более низкой температурой кипения, чем температура кипения образующегося в результате переэтерификации сложного эфира. В предпочтительном случае при давлении 10 мбар разница в температурах кипения в предпочтительном случае составляет по крайней мере 2°C, в особо предпочтительном случае по крайней мере 10°C и в самом предпочтительном случае по крайней мере 20°C. В частности, более всего для этого подходят (мет)акриловые эфиры, образующиеся из спиртов с числом атомов углерода от одного до четырех. К этим спиртам относятся прежде всего метанол, этанол, н-пропанол, изопропанол, н-бутанол и трет-бутанол. В частности, особое предпочтение отдается использованию этил(мет)акрилата или метил(мет)акрилата, причем наиболее предпочтительным является метилметакрилат.
Отношение массы исходного спирта по отношению к низкокипящему эфиру (мет)акриловой кислоты лежит в предпочтительном случае в пределах от 2:1 до 1:20, в особо предпочтительном случае от 1:2 до 1:15, в самом предпочтительном случае 1:3 до 1:10. Отношение массы исходного спирта к массе низкокипящего эфира (мет)акриловой кислоты может также лежать в пределах от 2:1 до 1:10.
Соответствующая переэтерификация катализируется основным ионообменным веществом. Такие основные ионообменные вещества широко известны, например, они достаточно подробно описаны в Ullmanns Encyclopedia of Industrial Chemistry (6-е издание).
Основные ионообменные вещества имеют в предпочтительном случае полимерное строение, которое модифицировано группами с основными свойствами. К предпочтительным полимерным структурам относятся, в частности, полимеры на основе стирола и поли(мет)акрилатов, при этом в особо предпочтительном случае эти полимеры имеют сетчатую структуру. Такие поперечные сшивки могут быть получены, например, с дивинилстиролом. Кроме того, могут быть использованы фенол-формальдегидные смолы или полиалкиламинные смолы. Предпочтительные основные группы представлены, в частности, группами, в состав которых входит азот, при этом они могут быть алифатическими или ароматическими.
В соответствии с этим основные ионообменные вещества в активной форме могут содержать, например, аммонийные и/или пиридиниевые группы. В предпочтительном случае аминогруппы соответствуют общей формуле 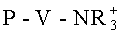
В предпочтительном случае реакцию переэтерификации можно проводить с ионообменным веществом, представляющим собой сильное основание. Сильно основные ионообменные вещества содержат группу с четвертичным атомом азота, то есть атом азота в этом случае не имеет ни одной связи с атомом водорода. В соответствии с этим сильно основные ионообменные вещества содержат, например, алкилированные по атому азота пиридиниевые группы или четвертичные аммонийные группы, при этом, например, в формуле 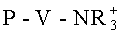
При этом ионообменные вещества могут также включать различные азотсодержащие группы. В соответствии с этим могут быть использованы ионообменные вещества, которые содержат первичные, вторичные, третичные и/или четвертичные аминные группы. Такие вещества проявляют свойства как сильноосновных, так и слабоосновных ионообменных веществ.
Для проведения реакции ионообменные вещества переводят в общем случае в форму, которая при взаимодействии с водой приводит к повышению значения pH, то есть для этого ионообменное вещество обрабатывают основным анионом так, чтобы получилось основное ионообменное вещество. В число предпочтительных основных анионов входят, в частности, гидроксидные ионы и карбонатные ионы.
Предпочтительно, когда ионообменные вещества в сухом виде имеют определяемый с помощью сит размер частиц в пределах от 0,3 до 0,8 мм, в особо предпочтительном случае в пределах от 0,5 до 0,7 мм.
Ионообменивающая способность ионообменных веществ в общем случае не имеет решающего значения, при этом предпочтительно, когда она лежит в пределах от 0,5 экв/л до 1,8 экв/л, в особо предпочтительном случае в пределах от 0,7 экв/л до 1,5 экв/л (в сухом виде).
Если исходить из стоимости и из ионообменивающей способности, то предпочтение отдается прежде всего содержащим четвертичные аммонийные группы основным ионообменным веществам на основе полистирола.
Описанные выше анионообменные вещества могут быть приобретены коммерческим путем у компании Bayer AG под торговой маркой ®Lewatit, у компании Rohm & Haas Comp./USA под торговой маркой ®Amberlite, ®Ambersep, а также у компании ®Amberlyst и у компании The Dow Chemical Com./USA под торговой маркой ®Dowex, а также у других компаний. В особо предпочтительном случае для переэтерификации могут быть использованы, в частности, ®Ambersep 900 и ®Amberlyst A26.
Количество используемого катализатора может изменяться в широких пределах. Однако особый интерес представляют способы, в соответствии с которыми количество катализатора из расчета на массу загруженных участников реакции лежит в пределах от 0,1 до 30 мас.%, в предпочтительном случае в пределах от 1 до 20 мас.% и в особо предпочтительном случае в пределах от 5 до 15 мас.%. Эта масса представляет собой сухую массу используемого ионообменного вещества.
Взаимодействие можно проводить при повышенном или при пониженном давлении. В соответствии с особо предпочтительным вариантом настоящего изобретения переэтерификацию можно проводить при давлении в пределах от 200 до 2000 мбар, в особо предпочтительном случае в пределах от 500 до 1300 мбар.
Температура реакции также может изменяться в широких пределах, в частности, в зависимости от давления. В соответствии с предпочтительным вариантом реализации настоящего изобретения переэтерификацию целесообразно проводить при температурах в пределах от 50°C до 140°C, в особо предпочтительном случае в пределах от 70°C до 120°C и в наиболее предпочтительном случае в пределах от 80 до 110°C.
Неожиданно оказалось, что особые преимущества достигаются в том случае, когда при проведении реакции температуру повышают по мере протекания взаимодействия. В соответствии с таким предпочтительным вариантом соответствующего изобретению способа предпочтительно, когда температура в начале взаимодействия, в частности, до достижения степени превращения 80%, в предпочтительном случае до достижения степени превращения 70% из расчета на массу используемого спирта, лежит в пределах от 50°C до 90°C, а в конце взаимодействия, в частности, после достижения степени превращения 80%, в предпочтительном случае после достижения степени превращения 90% из расчета на массу используемого спирта, лежит в пределах от 95°C до 130°С.
Неожиданно оказалось также, что при названных выше температурах проведения реакции не происходит заметного дезактивирования ионообменного вещества. В частности, для находящихся в OH-форме используемых в предпочтительном случае сильноосновных ионообменных веществ рекомендуемая производителями максимальная рабочая температура лежит значительно ниже той температуры, которая используется для проведения реакции переэтерификации. Так, например, максимальная рабочая температура по спецификации, представляемой для ®Amberlyst A 26 OH и ®Ambersep 900 OH составляет 60°C. Сообщается, что выше этой температуры происходит разложение структур четвертичных аммонийных солей путем отщепления аминов.
Благодаря тому, что дезактивирование катализатора оказалось неожиданно незначительным, основное ионообменное вещество, которое уже использовалось в реакции переэтерификации, может быть использовано в качестве катализатора в другой реакции. Предпочтительно, когда использовавшееся основное ионообменное вещество направляют на регенерацию с помощью обработки водным раствором гидроксида или карбоната щелочного металла.
Соответствующий изобретению способ может быть реализован на самих веществах, то есть без использования еще и растворителя. В случае необходимости можно также использовать инертный растворитель. К ним наряду с другими относятся бензол, толуол, н-гексан, циклогексан и метилизобутилкетон, а также метилэтилкетон.
В предпочтительном случае переэтерификацию проводят в безводных условиях, при этом чаще всего допускается присутствие небольших количеств воды. В соответствии с таким аспектом настоящего изобретения в реакционной смеси содержится в предпочтительном случае не более 0,5 мас.% воды, в особо предпочтительном случае не более 0,05 мас.% воды.
В наиболее целесообразном варианте проведения соответствующей изобретению переэтерификации смешивают все компоненты, например, спирт, эфир метакриловой кислоты и катализатор, после этого образующуюся реакционную смесь нагревают до кипения. При этом можно удалять из реакционной смеси выделяющийся спирт, например, метанол или этанол, дистиллятивным путем, в случае необходимости в виде азеотропной смеси с метилметакрилатом или этилметакрилатом.
Время реакции наряду с другими факторами определяется выбранными параметрами, например, давлением и температурой. Однако в общем случае это время лежит в пределах от 1 до 24 часов, в предпочтительном случае от 3 до 20 часов и в наиболее предпочтительном случае от 4 до 12 часов. При реализации способа по непрерывной схеме время пребывания в общем случае лежит в пределах от 0,5 до 24 часов, в предпочтительном случае от 1 до 12 часов и в наиболее предпочтительном случае от 2 до 4 часов. Другие данные, относящиеся к продолжительности протекания реакции, специалист может определить на основании приведенных примеров.
В предпочтительном случае реакция может протекать при перемешивании, при этом в особо предпочтительном случае скорость перемешивания может лежать в пределах от 50 до 2000 об/мин, в наиболее предпочтительном случае от 100 до 500 об/мин.
Значение pH может находиться в широких пределах. В целесообразном случае взаимодействие можно проводить при значении pH в пределах от 8 до 14, в предпочтительном случае от 9 до 13. Для определения значения pH можно внести часть реакционной смеси в избыточное количество воды (например, в десятикратное количество по массе). После этого определяют значение pH водной фазы обычным способом при 25°C.
Для предотвращения нежелательной полимеризации метакрилатов в ходе реакции можно использовать ингибиторы полимеризации. Такие соединения хорошо известны специалисту, например, это гидрохиноны, такие простые эфиры гидрохинона, как монометиловый эфир гидрохинона, или же это ди-трет-бутилпирокатехин, фенотиазин, N,N′-дифенил-n-фенилендиамин, 4-гидрокси-2,2,6,6-тетраметилпиперидин-1-оксил, n-фенилендиамин, Метиленовый синий или стерически затрудненные фенолы. Эти соединения, которые могут быть использованы в качестве индивидуальных веществ или в виде смесей, в общем случае могут быть приобретены коммерческим путем. Действие стабилизаторов основано чаще всего на том, что они выступают в качестве акцепторов радикалов для образующихся в процессе полимеризации свободных радикалов. Дополнительные сведения можно получить из обычных специальных литературных источников, в частности, это Römpp-Lexikon Chemie; Издатель: J. Falbe, M. Regitz; Штуттгарт, Нью-Йорк; 10-е Издание (1996); ключевое слово «Antioxidantien (антиоксиданты)», а также из цитированных в этом справочнике литературных источников.
В частности, в предпочтительном случае в качестве ингибиторов полимеризации используют фенолы. Наиболее неожиданные преимущества достигаются при использовании монометилового эфира гидрохинона. Из расчета на массу всей реакционной смеси содержание ингибиторов в чистом виде или в виде смеси может составлять от 0,01 до 0,5% (отношение масс).
Эти ингибиторы полимеризации могут быть добавлены к реакционной смеси до начала реакции или в ее начале. Кроме того, можно также добавлять порции используемых ингибиторов полимеризации по ходу переэтерификации. Особый интерес при этом представляют, в частности, способы, при реализации которых часть ингибитора полимеризации прибавляют путем введения ее в возвращающуюся из колонны жидкую фазу. Наиболее целесообразно, когда эти смеси состоят из метилметакрилата, монометилового эфира гидрохинона и 4-гидрокси-2,2,6,6-тетраметилпиперидин-1-оксила. Благодаря этому можно исключить нежелательную полимеризацию внутри дистилляционной колонны.
Для ингибирования может быть также использован кислород. Он может присутствовать, например, в виде воздуха, при этом его следует подавать в таком количестве, чтобы его содержание в газовой фазе над реакционной смесью не превышало нижней границы взрывоопасности. Особое предпочтение при этом отдается количеству воздуха в пределах от 0,05 до 0,5 л в час на моль спирта. При работе в периодическом режиме это количество может относиться к начальной загрузке спирта. При работе по непрерывной схеме это количество может относиться к подаваемому количеству спирта. Можно также использовать смеси инертного газа и кислорода, например, смеси азота и кислорода или смеси аргона и кислорода.
В соответствии со специальным вариантом реализации настоящего изобретения для ингибирования можно использовать сочетание кислорода и не менее чем одного фенола, в предпочтительном случае метилового эфира гидрохинона.
Выделяющийся из используемого (мет)акрилата спирт, например, метанол и/или этанол, в предпочтительном случае может быть отделен с помощью дистилляции. При этом в предпочтительном случае можно, например, отделять смесь, содержащую метилметакрилат и метанол. Неожиданно оказалось, что часть отделенной смеси можно с успехом возвращать в новую загрузку. В соответствии с этим вариантом возвращаемую часть отделенной смеси получают в конце реакции, в частности, после достижения степени превращения 80%, в предпочтительном случае после достижения степени превращения 90% по используемому спирту. Так, например, количество возвращенной в процесс смеси в начале превращения следующей партии может составлять от 10 до 50% из расчета на общее количество загружаемого на переэтерификацию эфира метакриловой кислоты.
Переэтерификацию можно проводить как по непрерывной, так и по периодической схеме. Превращение по непрерывной схеме можно в предпочтительном случае проводить в установках с несколькими реакторами, при этом можно также менять температуру реакции и выделять из реакционной системы спирт, который образуется из низкокипящего эфира (мет)акриловой кислоты.
Особый интерес представляют, в частности, полупериодические способы, в соответствии с которыми загружают часть реакционной смеси. На следующих операциях или при работе по непрерывной схеме можно в начале реакции прибавлять в реакционную смесь низкокипящий эфир (мет)акриловой кислоты.
В соответствии со специальным вариантом настоящего изобретения присутствующее в реакционной смеси молярное отношение низкокипящего эфира (мет)акриловой кислоты к исходному спирту может быть повышено во время взаимодействия путем прибавления низкокипящего эфира (мет)акриловой кислоты. Выражение «прибавление низкокипящего эфира (мет)акриловой кислоты» означает, что это соединение добавляют из внешнего источника. В соответствии с этим простое возвращение в виде сконденсировавшейся фазы, которое происходит в дистилляционной колонне, не относится к его прибавлению. Особый интерес представляют, в частности, способы, в соответствии с которыми присутствующее в реакционной смеси мольное отношение содержания низкокипящего эфира (мет)акриловой кислоты к содержанию используемого для переэтерификации спирта повышается во время превращения путем прибавления низкокипящего эфира (мет)акриловой кислоты не менее чем на 10%, в особо предпочтительном случае не менее чем на 40% и в наиболее предпочтительном случае не менее чем на 100%. Так, например, мольное отношение количества прибавляемого во время превращения низкокипящего сложного эфира к количеству загруженного в начале реакции низкокипящего сложного эфира может лежать в пределах от 1:5 до 2:1, в особо предпочтительном случае от 1:3 до 1:1,5. В оптимальном случае количественное отношение исходного спирта к низкокипящему эфиру (мет)акриловой кислоты в начале реакции может лежать в предпочтительных пределах от 1:2 до 1:8, в особо предпочтительном случае оно составляет от 1:2,5 до 1:6 и в самом предпочтительном случае в пределах от 1:3 до 1:4. Путем прибавления низкокипящего сложного эфира во время реакции это отношение можно повышать, например, до значений от 1:3 до 1:20, в предпочтительном случае от 1:4 до 1:15 и в наиболее предпочтительном случае от 1:6 до 1:10. В каждом отдельном случае эти значения относятся к содержащемуся в реакционной смеси исходному спирту. Чаще всего количество уже превращенного в целевой сложный эфир исходного спирта можно определять по количеству отогнанного спирта, который выделился в результате реакции. Кроме того, находящееся в реакционной смеси количество исходного спирта можно определять с помощью газовой хроматографии.
В соответствии со специальным вариантом реализации количество добавляемых низкокипящих эфиров (мет)акриловой кислоты можно регулировать по количеству выделившегося в результате реакции и отогнанного спирта. Для такого регулирования можно, например, использовать температуру, которая устанавливается в колонне на определенной высоте. Можно также по температуре устанавливать флегмовое число во время дистиллятивного удаления выделяющегося из исходного продукта спирта. Если, например, из реакционной массы удаляется смесь, содержащая метанол и метилметакрилат, то тогда можно на длительное время установить температуру от примерно 75 до 85°C, выше которой дистиллят не отбирается. Соответствующее количество смеси отбирается только ниже этой температуры. В результате этого достигается поддерживаемое в течение длительного времени сравнительно высокое отношение низкокипящего (мет)акрилата к исходному спирту и тогда отпадает необходимость в добавлении к реакционной смеси слишком больших количеств низкокипящего (мет)акрилата.
Неожиданно оказалось, что путем прибавления во время переэтерификации низкокипящего сложного эфира достигается особенно высокое значение объемной производительности реактора и, благодаря этому, можно производить значительные количества специальных (мет)акрилатов на сравнительно небольших установках. Кроме того, может быть увеличено количество специального (мет)акрилата, которое получается с одной загрузки, и тогда можно получить дополнительные преимущества, поскольку снижаются затраты на проведение реакции из расчета на полученное количество продукции.
Прибавление низкокипящего эфира (мет)акриловой кислоты целесообразно проводить в течение времени, которое составляет по крайней мере 30%, в предпочтительном случае по крайней мере 50% и в самом предпочтительном случае по крайней мере 70% от времени реакции. При этом прибавление в течение этого времени проводят отдельными порциями, когда для первого прибавления устанавливают начало этого периода времени и для последнего прибавления устанавливают окончание этого времени. В предпочтительном случае прибавление проводят по крайней мере в три этапа, в особо предпочтительном случае по крайней мере в пять этапов и в наиболее предпочтительном случае по крайней мере в десять этапов. Кроме того, это прибавление можно также проводить в непрерывном режиме.
Особый интерес представляют также периодические способы проведения процесса, в соответствии с которыми метил(мет)акрилат прибавляют во время переэтерификации. Такому варианту отдается предпочтение, например, в том случае, когда метил(мет)акрилат отгоняется из реакционной смеси вместе с метанолом. В предпочтительном случае отношение масс прибавляемого во время переэтерификации количества метил(мет)акрилата к количеству отделяемой смеси метанола и метил(мет)акрилата может лежать в пределах от 2:1 до 1:2, в особо предпочтительном случае от 1,5:1 до 1:1,5.
При периодическом способе проведения процесса в конце реакции можно отделять избыточный исходный продукт, в частности, непревращенный эфир (мет)акриловой кислоты, с помощью дистилляции. Он может быть также снова использован для следующей загрузки без дополнительной очистки.
Полученный в начале превращения дистиллят, который, например, может содержать значительные количества метанола или этанола, может быть также направлен на рецикл, например, путем использования его в функционирующей параллельно установке для получения поступающего на переэтерификацию эфира (мет)акриловой кислоты.
Подходящая установка для. проведения соответствующей изобретению переэтерификации может, например, состоять из обогреваемого паром реактора с мешалкой, дистилляционной колонны и конденсатора. Такие часто используемые установки описаны, например, в Ullmanns Encyclopedia of Industrial Chemistry (6-e издание), издательство Wiley-VCH, Вейнгейм 2003, Том 10, страница 647. Масштаб установки зависит от объема производства (мет)акрилата, при этом соответствующий изобретению способ может быть реализован как в лабораторном, так и в промышленном масштабе. В соответствии с таким специальным аспектом реактор с мешалкой может соответственно иметь объем от 1 м3 до 30 м3, в предпочтительном случае от 3 м3 до 20 м3. Перемешивающее устройство реактора с мешалкой может представлять собой, в частности, якорную мешалку, пропеллерную мешалку, лопастную мешалку или многоступенчатую импульсную противоточную мешалку (MIG-impeller, multistage impulse countercurrent).
Роль дистилляционной колонны состоит в том, чтобы обеспечить вывод азеотропной смеси, обогащенной метанолом или, соответственно, этанолом, и свести таким образом к минимуму потери обязательно уходящего вместе с ними исходного эфира. Дистилляционная колонна может иметь одну ступень разделения, две или несколько ступеней разделения. Число ступеней разделения в тарельчатой колонне равно числу ее тарелок или же оно равно числу теоретических тарелок в насадочной колонне или в колонне, заполненной насадкой. Примером многоступенчатой дистилляционной тарельчатой колоны служит колонна с колпачковыми тарелками, с ситчатыми тарелками, туннельными тарелками, клапанными тарелками, провальными тарелками, ситчатыми и пропровальными тарелками, ситчатыми и колпачковыми тарелками, дюзовыми тарелками, центрифугальными тарелками; для многоступенчатой дистилляционной колонны с насадкой используются кольца Рашига, кольца Лессинга, кольца Паля, седла Берля, седла Инталокс, а для заполненной насадкой многоступенчатой дистилляционной колонны используются насадки типа Меллапак (Sulzer), Ромбопак (Kühni), Монтц-Пак (Montz). При регуляции зависящего от степени превращения значения флегмового числа можно, например, при использовании метилметакрилата устанавливать в дистилляте содержание метанола более 60% для самых разных значений степени превращения.
Подходящие конденсаторы, которые могут быть элементом установки для проведения соответствующей изобретению переэтерификации, представлены пластинчатыми и кожухотрубными теплообменниками.
После окончания взаимодействия полученный (мет)акрилат уже соответствует представленным выше высоким требованиям, вследствие чего дальнейшая его очистка чаще всего оказывается излишней. Для дополнительного повышения качества и, в частности, для отделения катализатора можно очистить полученную смесь известными способами.
В еще одном варианте реализации соответствующего изобретению способа можно проводить очистку полученной смеси продуктов с помощью фильтрования. Такие способы известны из уровня техники (W. Gösele, Chr. Alt в Ullmann′s Encyclopedia of Industrial Chemistry, (6-е издание), издательство Wiley-VCH, Вейнгейм 2003, Том 13, страницы 731 и 746), при этом могут быть использованы такие обычные вспомогательные средства для фильтрации, как, например, отбеливающая глина и/или силикат алюминия (перлит). Так, например, наряду с другими могут быть использованы фильтры непрерывного действия для намывной фильтрации или патронные фильтры.
Дополнительное повышение качества продукции может быть достигнуто, например, путем дистилляции полученного фильтрата. Из-за склонности мономера к полимеризации рекомендуются способы дистилляции, при которых термическое воздействие на перегоняемое вещество сведено к минимуму. Лучше всего подходят установки, в которых идет постоянное испарение мономера из тонкой пленки, например пленочные испарители с падающим слоем и испарители с вращающимся распределителем. Могут быть также использованы установки молекулярной разгонки. Такие устройства известны (Ullmanns Encyclopedia of Industrial Chemistry (6-е издание), издательство Wiley-VCH, Вейнгейм 2003, Том 36, страница 505). Так, например, можно использовать соединенный с колонной испаритель непрерывного действия с вращающимся распределителем. Дистилляцию можно проводить, например, при давлении в пределах от 1 до 40 мбар и при температуре в испарителе от 120°С до 150°С.
Далее настоящее изобретение иллюстрируется несколькими примерами, которые не могут быть использованы для ограничения объема притязаний.
Пример 1
В четырехгорлой круглодонной колбе объемом 1000 мл, снабженной пропущенной через втулку для мешалки сабельной мешалкой, мотором для мешалки, трубкой для ввода воздуха, погруженным в жидкость термометром и дистилляционной колонной с насадкой для отбора флегмы и приспособлением для возвращения в колбу конденсата, смешивают 170 г (1,3 моль) 2-этилгексилового спирта, 455 г (4,55 моль) метилового эфира метакриловой кислоты, 0,129 г монометилового эфира гидрохинона и 0,003 г 4-гидрокси-2,2,6,6-тетраметилпиперидин-1-оксила (ингибиторы), а также в качестве катализатора 62,5 г (10 мас.%) ®Amberlyst A26 (Rohm & Haas Comp./США) в гидроксидной форме. После этого реакционную смесь 7 часов перемешивают при температуре в жидкости от 100°C до 123°C, пропуская воздух, при этом идет отгонка метанола и метилметакрилата. После окончания реакции реакционную смесь фильтруют через складчатый фильтр для отделения ее от катализатора и освобождают от метилметакрилата при давлении 10 мбар.
Выход полученного 2-этилгексилметакрилата составляет 117 г (45% из расчета на исходный 2-этилгексанол). Чистота продукта составляет 92,3% (по результатам анализа с помощью газовой хроматографии).
Часть продукта адсорбционно связана с катализатором, который целесообразно использовать для получения следующей партии продукта.
Пример 2
В четырехгорлой круглодонной колбе объемом 1000 мл, снабженной пропущенной через втулку для мешалки сабельной мешалкой, мотором для мешалки, трубкой для ввода воздуха, погруженным в жидкость термометром и дистилляционной колонной с насадкой для отбора флегмы и приспособлением для возвращения в колбу конденсата, смешивают 606 г (6,06 моль) метилового эфира метакриловой кислоты, 0,124 г монометилового эфира гидрохинона и 0,0025 г 4-гидрокси-2,2,6,б-тетраметилпиперидин-1-оксила (ингибиторы), а также в качестве катализатора 70,5 г (10 мас.%) ®Amberlyst A26 (Rohm & Haas Comp./США) в гидроксидной форме. Исходные продукты нагревают до кипения при пропускании воздуха и обезвоживают реакционную массу, отгоняя азеотропную смесь до достижения в жидкости температуры 98°C. После этого прибавляют 99 г (1,1 моль) 1,4-бутандиола и метилметакрилат в количестве, которое соответствует отогнанному количеству метилметакрилата в виде его смеси с водой (240 г). Затем реакционную смесь 5 часов перемешивают при температуре в жидкости от 100°C до 113°C, пропуская воздух, при этом идет отгонка метанола и метилметакрилата. После окончания реакции реакционную смесь фильтруют через складчатый фильтр для отделения ее от катализатора и освобождают от метилметакрилата при давлении 2 мбар.
Выход полученного диметакрилата 1,4-бутандиола составляет 133 г (53% из расчета на исходный спирт). Чистота продукта составляет 87,5% (по результатам анализа с помощью газовой хроматографии). Продукт содержит 10% полимеризующихся высококипящих соединений, вследствие чего содержание реакционноспособных целевых сложных эфиров составляет 97,5%.
Часть продукта адсорбционно связана с катализатором, который целесообразно использовать для получения следующей партии продукта.
Пример 3
В четырехгорлой круглодонной колбе объемом 1000 мл, снабженной пропущенной через втулку для мешалки сабельной мешалкой, мотором для мешалки, трубкой для ввода воздуха, погруженным в жидкость термометром и дистилляционной колонной с насадкой для отбора флегмы и приспособлением для возвращения в колбу конденсата, смешивают 74,5 г (1,2 моль) этиленгликоля, 600 г (6 молей) метилового эфира метакриловой кислоты, 0,119 г монометилового эфира гидрохинона и 0,0024 г 4-гидрокси-2,2,6,6-тетраметилпиперидин-1-оксила (ингибиторы), а также в качестве катализатора 67,5 г (10 мас.%) ®Amberlyst A26 (Rohm & Haas Comp./США) в гидроксидной форме. После этого реакционную смесь 5 часов перемешивают при температуре в жидкости от 100°C до 121°C, пропуская воздух, при этом идет отгонка метанола и метилметакрилата. После окончания реакции реакционную смесь фильтруют через складчатый фильтр для отделения ее от катализатора, катализатор экстрагируют метилметакрилатом (100 г) и освобождают от метилметакрилата реакционную смесь вместе с экстрактом при давлении 10 мбар.
Выход полученного диметакрилата этиленгликоля составляет 178 г (74,8% из расчета на исходный этиленгиколь). Чистота продукта составляет 89,8% (по результатам анализа с помощью газовой хроматографии). Продукт содержит 5,7% полимеризующихся высококипящих соединений, а также 0,47% гидроксиэтилметакрилата, что соответствует содержанию реакционноспособных целевых сложных эфиров 96%. Показатель цветности по платино-кобальтовой шкале равен 44.
Часть продукта адсорбционно связана с катализатором, который целесообразно использовать для получения следующей партии продукта.
Пример 4
В четырехгорлой круглодонной колбе объемом 1000 мл, снабженной пропущенной через втулку для мешалки сабельной мешалкой, мотором для мешалки, трубкой для ввода воздуха, погруженным в жидкость термометром и дистилляционной колонной с дефлегматором и приспособлением для возвращения в колбу конденсата, смешивают 495 г (4,95 моль) метилового эфира метакриловой кислоты, 0,06 г фенотиазина и 0,0045 г 4-гидрокси-2,2,6,6-тетраметилпиперидин-1-оксила (ингибиторы), а также в качестве катализатора 78,4 г (10 мас.%) ®Amberlyst A26 (Rohm & Haas Comp./США) в гидроксидной форме. Исходные продукты нагревают до кипения при пропускании воздуха и обезвоживают реакционную массу, отгоняя азеотропную смесь до достижения в жидкости температуры 98°С. После этого прибавляют 285 г (0,9 моль) Dianol® 220 (дважды этоксилированный бисфенол А фирмы Seppic, Франция) и метилметакрилат в количестве, которое соответствует отогнанному количеству метилметакрилата в виде его смеси с водой (310 г). Затем реакционную смесь 8,5 часов перемешивают, поддерживая температуру в жидкости от 100°C до 120°C и пропуская воздух, при этом идет отгонка метанола и метилметакрилата. После окончания реакции реакционную смесь фильтруют через складчатый фильтр для отделения ее от катализатора, экстрагируют его метилметакрилатом (100 г) и освобождают от метилметакрилата смесь фильтрата и экстракта при давлении 3 мбар.
Выход полученного диметакрилата этоксилированного бисфенола А составляет 338 г (83% из расчета на исходное соединение). Чистота продукта составляет 83,4% (по результатам анализа с помощью газовой хроматографии). Продукт содержит 13,9% полимеризующихся высококипящих соединений, что соответствует содержанию реакционноспособных целевых сложных эфиров 97,3%. Показатель цветности по платино-кобальтовой шкале равен 30.
Часть продукта адсорбционно связана с катализатором, который целесообразно использовать для получения следующей партии продукта.
Пример 5
В четырехгорлой круглодонной колбе объемом 1000 мл, снабженной пропущенной через втулку для мешалки сабельной мешалкой, мотором для мешалки, трубкой для ввода воздуха, погруженным в жидкость термометром и дистилляционной колонной с дефлегматором и устройством для возвращения в колбу конденсата, смешивают 285 г (0,9 моль) Newpol® BPE 20-F (дважды этоксилированный бисфенол А фирмы Sanyo, Япония), 495 г (4,95 моль) метилового эфира метакриловой кислоты, 0,06 г фенотиазина и 0,0045 г 4-гидрокси-2,2,6,6-тетраметилпиперидин-1-оксила (ингибиторы), а в качестве катализатора 78,4 г (10 мас.%) использованного в примере 4 катализатора. Затем реакционную смесь 8 часов перемешивают, поддерживая температуру в жидкости от 100°C до 120°C и пропуская воздух, при этом идет отгонка метанола и метилметакрилата. После окончания реакции фильтруют реакционную смесь через складчатый фильтр для отделения ее от катализатора, экстрагируют его метилметакрилатом (100 г) и освобождают от метилметакрилата смесь фильтрата и экстракта при давлении 3 мбар.
Выход полученного диметакрилата этоксилированного бисфенола А составляет 350 г (86% из расчета на исходное соединение). Чистота продукта составляет 70,8% (по результатам анализа с помощью газовой хроматографии). Продукт содержит 24,5% полимеризующихся высококипящих соединений, что соответствует содержанию реакционноспособных целевых сложных эфиров 95,3%. Показатель цветности по платино-кобальтовой шкале равен 25.
Часть продукта адсорбционно связана с катализатором, который целесообразно использовать для получения следующей партии продукта.
Пример 6
В четырехгорлой круглодонной колбе объемом 1000 мл, снабженной пропущенной через втулку для мешалки сабельной мешалкой, мотором для мешалки, трубкой для ввода воздуха, погруженным в жидкость термометром и дистилляционной колонной с дефлегматором и устройством для возвращения в колбу конденсата, смешивают 170 г (1,3 моль) 2-этилгексилового спирта. 455 г (4,55 моль) метилового эфира метакриловой кислоты, 0,129 г монометилового эфира гидрохинона и 0,003 г 4-гидрокси-2,2,6,6-тетраметилпиперидин-1-оксила (ингибиторы) и в качестве катализатора - 80 г (10 мас.%) использованного в примере 1 катализатора. Затем реакционную смесь 8 часов перемешивают, поддерживая температуру в жидкости от 100°C до 123°C и пропуская воздух, при этом идет отгонка метанола и метилметакрилата. После окончания реакции фильтруют реакционную смесь через складчатый фильтр для отделения ее от катализатора и освобождают от метилметакрилата при давлении 10 мбар.
Выход полученного 2-этилгексилметакрилата составляет 156 г (60% из расчета на исходный 2-этилгексиловый спирт). Чистота продукта составляет 91,8% (по результатам анализа с помощью газовой хроматографии).
Часть продукта адсорбционно связана с катализатором, который целесообразно использовать для получения следующей партии продукта.
Пример 7
Опыт по примеру 7 проводят в основном по аналогии с примером 1, но при этом используют 62,5 г Ambersep® 900 (Rohm & Haas Comp./США) в гидроксидной форме.
Выход полученного 2-этилгексилметакрилата составляет 120 г (46% из расчета на исходный 2-этилгексиловый спирт). Чистота продукта составляет 93% (по результатам анализа с помощью газовой хроматографии).
Часть продукта адсорбционно связана с катализатором, который целесообразно использовать для получения следующей партии продукта.
Пример 8
В хроматографическую колонку с помощью полностью обессоленной воды переносят 100 г ®Amberlyst A26 (Rohm & Haas Comp./США) в хлоридной форме и обрабатывают смолу 10%-ным раствором карбоната натрия до полного отсутствия в выходящем элюате ионов хлора. После этого промывают полностью обессоленной водой до нейтральной реакции выходящего элюата. Затем промывают метанолом и сушат полученный ®Amberlyst A26 в карбонатной форме на воздухе.
В четырехгорлой круглодонной колбе объемом 1000 мл, снабженной пропущенной через втулку для мешалки сабельной мешалкой, мотором для мешалки, трубкой для ввода воздуха, погруженным в жидкость термометром и дистилляционной колонной с дефлегматором и устройством для возвращения в колбу конденсата, смешивают 170 г (1,3 моль) 2-этилгексилового спирта, 455 г (4,55 моль) метилового эфира метакриловой кислоты, 0,129 г монометилового эфира гидрохинона и 0,003 г 4-гидрокси-2,2,6,6-тетраметилпиперидин-1-оксила (ингибиторы) и в качестве катализатора 62,5 г (10 мас.%) ®Amberlyst A26 (Rohm & Haas Comp./США) в карбонатной форме. Затем реакционную смесь 8,5 часов перемешивают, поддерживая температуру в жидкости от 100°C до 123°C и пропуская воздух, при этом идет отгонка метанола и метилметакрилата. После окончания реакции фильтруют реакционную смесь через складчатый фильтр для отделения ее от катализатора и освобождают от метилметакрилата при давлении 9 мбар.
Выход полученного 2-этилгексилметакрилата составляет 125 г (48% из расчета на исходный 2-этилгексиловый спирт). Чистота продукта составляет 94% (по результатам анализа с помощью газовой хроматографии).
Часть продукта адсорбционно связана с катализатором, который целесообразно использовать для получения следующей партии продукта.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ НЕПРЕРЫВНОГО ПОЛУЧЕНИЯ АЛКИЛАМИНО(МЕТ)АКРИЛАМИДОВ | 2004 |
|
RU2374221C2 |
| СПОСОБ НЕПРЕРЫВНОГО ПОЛУЧЕНИЯ АЛКИЛАМИНО(МЕТ)АКРИЛАМИДОВ | 2010 |
|
RU2546670C2 |
| СПОСОБ ПОЛУЧЕНИЯ АЛКИЛ(МЕТ)АКРИЛАТОВ | 2006 |
|
RU2409552C2 |
| СПОСОБ ПОЛУЧЕНИЯ АМИДА КАРБОНОВОЙ КИСЛОТЫ ИЗ КАРБОНИЛЬНОГО СОЕДИНЕНИЯ И ЦИАНИСТОВОДОРОДНОЙ КИСЛОТЫ | 2009 |
|
RU2552619C9 |
| МАКРОМОНОМЕРЫ, СОДЕРЖАЩИЕ ПОЛИИЗОБУТЕНОВЫЕ ГРУППЫ, И ИХ ГОМО- И СОПОЛИМЕРЫ | 2017 |
|
RU2745788C2 |
| СПОСОБ ПОЛУЧЕНИЯ ДИ(МЕТ)АКРИЛАТА ИЗОСОРБИДА | 2016 |
|
RU2703461C2 |
| СПОСОБ ГЕТЕРОГЕННО КАТАЛИЗИРУЕМОГО ПОЛУЧЕНИЯ (МЕТ)АКРИЛАТОВ N-ГИДРОКСИАЛКИЛИРОВАННЫХ ЛАКТАМОВ | 2006 |
|
RU2430084C2 |
| УСОВЕРШЕНСТВОВАННЫЙ СПОСОБ НЕПРЕРЫВНОГО ПОЛУЧЕНИЯ АЛКИЛ(МЕТ)АКРИЛАТОВ С МНОГОКРАТНОЙ РЕЦИРКУЛЯЦИЕЙ КАТАЛИЗАТОРА | 2003 |
|
RU2407733C2 |
| СПОСОБ ПЕРЕЭТЕРИФИКАЦИИ | 2006 |
|
RU2452725C2 |
| КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ОКСИД ДИАЛКИЛОЛОВА, И ЕЕ ПРИМЕНЕНИЕ В КАЧЕСТВЕ КАТАЛИЗАТОРА ПЕРЕЭТЕРИФИКАЦИИ ПРИ СИНТЕЗЕ СЛОЖНЫХ (МЕТ)АКРИЛОВЫХ ЭФИРОВ | 2010 |
|
RU2529863C2 |
Изобретение относится к усовершенствованному способу получения эфиров (мет)акриловой кислоты, включающему переэтерификацию низкокипящего эфира (мет)акриловой кислоты, температура кипения которого ниже, чем температура кипения образующегося в результате переэтерификации сложного эфира, исходным спиртом в присутствии основного ионообменного вещества, в качестве катализатора, и ингибитора полимеризации, причем переэтерификацию проводят при температуре в пределах от 50°C до 140°C. Получаемый продукт содержит незначительные количества побочных продуктов и остатков катализатора. Способ реализуется без трудоемкого отделения катализатора. 19 з.п. ф-лы, 8 пр.
1. Способ получения эфиров (мет)акриловой кислоты, включающий переэтерификацию низкокипящего эфира (мет)акриловой кислоты, температура кипения которого ниже, чем температура кипения образующегося в результате переэтерификации сложного эфира, исходным спиртом в присутствии основного ионообменного вещества, в качестве катализатора, и ингибитора полимеризации, причем переэтерификацию проводят при температуре в пределах от 50°C до 140°C.
2. Способ по п.1, отличающийся тем, что применяют ионообменное вещество со свойствами сильного основания.
3. Способ по п.1, отличающийся тем, что в качестве низкокипящего эфира (мет)акриловой кислоты используют метил(мет)акрилат и/или этил(мет)акрилат.
4. Способ по п.1, отличающийся тем, что спирт, выделяющийся из низкокипящего эфира (мет)акриловой кислоты, отделяют с помощью дистилляции.
5. Способ по п.1, отличающийся тем, что в качестве исходного спирта используют одноатомный спирт.
6. Способ по п.5, отличающийся тем, что в качестве исходного спирта используют бутанол, гексанол, 3-метилбутанол, гептанол, 2-этилгексиловый спирт, 2-трет-бутилгептанол, октанол, 3-изопропилгептанол, нонанол, деканол, ундеканол, 5-метилундеканол, додеканол, 2-метилдодеканол, тридеканол, 5-метил-тридеканол, тетрадеканол, пентадеканол, гексадеканол, 2-метилгексадеканол, гептадеканол, 5-изопропилгептадеканол, 4-трет-бутилоктадеканол, 5-этил-октадеканол, 3-изопропилоктадеканол, октадеканол, нонадеканол, эйкозанол, цетилэйкозанол, стеарилэйкозанол, докозанол и/или эйкозилтетратриаконтанол.
7. Способ по п.1, отличающийся тем, что в качестве исходного спирта используют многоатомный спирт.
8. Способ по п.7, отличающийся тем, что в качестве исходного спирта используют этиленгликоль, триметилолпропан, 1,2-пропандиол, 1,3-пропандиол, 1,3-бутандиол, 1,4-бутандиол, этоксилированный бисфенол A, 1,6-гександиол и/или пентаэритрит.
9. Способ по п.1, отличающийся тем, что реакционная смесь содержит не более 0,5 мас.% воды.
10. Способ по п.9, отличающийся тем, что реакционная смесь содержит не более 0,05 мас.% воды.
11. Способ по п.1, отличающийся тем, что присутствующее в реакционной смеси мольное отношение низкокипящего эфира (мет)акриловой кислоты к исходному спирту повышают во время превращения по крайней мере на 40% путем прибавления низкокипящего эфира (мет)акриловой кислоты.
12. Способ по п.1, отличающийся тем, что отделяют смесь, содержащую метилметакрилат и метанол.
13. Способ по п.1, отличающийся тем, что во время переэтерификации прибавляют метилметакрилат.
14. Способ по п.1, отличающийся тем, что время реакции лежит в пределах от 3 до 20 часов.
15. Способ по п.1, отличающийся тем, что основное ионообменное вещество содержит гидроксидные и/или карбонатные ионы.
16. Способ по п.1, отличающийся тем, что отношение массы исходного спирта к массе низкокипящего эфира (мет)акриловой кислоты лежит в пределах от 2:1 до 1:10.
17. Способ по п.1, отличающийся тем, что взаимодействие проводят при давлении в пределах от 200 до 2000 мбар.
18. Способ по п.1, отличающийся тем, что способ реализуют по непрерывной схеме.
19. Способ по одному из пп.1-18, отличающийся тем, что в качестве катализатора используют основное ионообменное вещество, которое уже использовалось в реакции переэтерификации.
20. Способ по одному из пп.1-18, отличающийся тем, что использованное основное ионообменное вещество регенерируют путем обработки водным раствором гидроксида или карбоната щелочного металла.
| Способ очистки ионогенных поверхностно-активных веществ | 1986 |
|
SU1401843A1 |
| Кулачок | 1982 |
|
SU1094998A1 |
| СПОСОБ СТРОИТЕЛЬСТВА МНОГОЭТАЖНЫХ ЗДАНИЙ ИЗ ОБЪЕМНЫХ БЛОКОВ | 1992 |
|
RU2037608C1 |
| US 6977310 B2, 20.12.2005 | |||
| US 7294240 B2, 13.11.2007 | |||
| СПОСОБ ПОЛУЧЕНИЯ СЛОЖНОГО ЭФИРА РЕАКЦИЕЙ ЭТЕРИФИКАЦИИ | 1997 |
|
RU2178783C2 |

