Настоящее изобретение относится к способу получения алкил(мет)-акрилатов.
Сложные эфиры акриловой кислоты и метакриловой кислоты, ниженазываемые алкил(мет)-акрилатами, нашли основную область применения в получении полимеров и сополимеров с другими способными полимеризоваться соединениями.
Кроме того, сложный эфир метакриловой кислоты, например, такой как метилметакрилат, является важным мономером для разных особых сложных эфиров на основе метакриловой кислоты, которые получают переэтерификацией соответствующим спиртом.
Метилметакрилат и метакриловую кислоту в настоящее время преимущественно получают из синильной кислоты и ацетона через образующийся в качестве основного промежуточного соединения ацетонциангидрин (АЦГ).
В соответствующей патентной литературе описаны и реализованы в промышленном масштабе другие способы получения метилметакрилата и метакриловой кислоты, предусматривающие использование иных исходных соединений вместо АЦГ. В связи с этим, сырье на основе углеводородов с четырьмя атомами углерода, например, таких как изобутилен или трет-бутанол, в настоящее время используют в качестве реагентов, которые превращают в целевые производные метакриловой кислоты посредством многостадийного способа.
Дополнительным объектом интенсивного исследования явилось использование пропилена в качестве основного сырья, причем ступенчатое гидрокарбонилирование (с образованием изомасляной кислоты) и окислительное дегидрирование приводит к получению метакриловой кислоты с умеренными выходами.
Известно, что в качестве основного сырья можно использовать пропаналь или пропионовую кислоту, которые могут быть получены в технически реализуемых процессах из этилена и структур с одним атомом углерода, таких как монооксид углерода. В подобных процессах образующееся по реакции альдольной конденсации с формальдегидом β-гидроксикарбонильное соединение дегидратацией in situ превращают в соответствующее α,β-ненасыщенное соединение. Обзор общепринятых способов получения метакриловой кислоты и ее сложных эфиров приведен, например, в Weissermel, Агре „Industrielle organische Chemie", VCH, Вейнгейм 1994, 4-е издание, страница 305 и следующие, а также в Kirk Othmer „Encyclopedia of Chemical Technology", 3-е издание, том 15, страница 357.
Общеизвестно, что первую реакционную стадию технических способов, основанных на использовании АЦГ (так называемое амидирование АЦГ), осуществляют высококонцентрированной серной кислотой (около 100% мас. H2SO4) при температуре от 80 до почти 110°С.
Предвтавительным для такого процесса является, например, патент US 4529816, в соответствии с которым амидирование АЦГ осуществляют при температуре около 100°С и молярном отношении АЦГ к H2SO4 в примерном интервале от 1:1,5 до 1:1,8. Основными стадиями процесса по этому способу являются а) амидирование, b) конверсия и с) этерификация.
На стадии амидирования в качестве основных продуктов взаимодействия получают амид сульфокси-α-гидроксиизомасляной кислоты-водород сульфат (СИМА) и амид метакриловой кислоты-водородсульфат (МАКА×H2SO4) в виде раствора в избыточной серной кислоте. Кроме того, типичный раствор стадии амидирования содержит амид α-гидроксиизомасляной кислоты-водород-сульфат (ГИМА×H2SO4), выход которого в пересчете на АЦГ составляет менее 5%. При более или менее полном превращении АЦГ подобный весьма селективный процесс амидирования протекает с суммарным выходом указанных промежуточных соединений примерно от 96 до 97%.
Однако на стадии амидирования в качестве побочных продуктов образуются незначительные количества монооксида углерода, ацетона, продуктов сульфирования ацетона и продуктов циклоконденсации ацетона с различными промежуточными соединениями.
В зависимости от содержания воды в используемой серной кислоте образующаяся на стадии амидирования смесь, наряду с СИМА, содержит также ГИМА. Так, например, при использовании серной кислоты с концентрацией 97% мас. (1,5 эквивалента H2SO4 относительно АЦГ) образуется уже около 25% мас. ГИМА, которую не удается селективно и полностью конвертировать в МАКА. Таким образом, относительно высокое содержание воды на стадии осуществляемого при температуре от 90 до 110°С амидирования обусловливает относительно высокое содержание ГИМА, который может быть превращен обычной конверсией в целевой промежуточный продукт МАКА×H2SO4 лишь с относительно невысокой селективностью.
Целью конверсии является максимально полное превращение СИМА и ГИМА в МАКА, протекающее благодаря β-отщеплению серной кислоты (в используемой в качестве растворителя избыточной серной кислоте).
Таким образом, на стадии конверсии при повышенных температурах (от 140 до 160°С) и незначительном времени контакта реагентов (около 10 минут или менее) осуществляют превращение содержащихся в сернокислотном (безводном) растворе ГИМА, СИМА и МАКА (в виде соответствующих гидросульфатов).
Реакционная смесь стадии конверсии характеризуется значительным избытком серной кислоты и примерным содержанием основного продукта МАКА×H2SO4 в растворе, составляющем от 30 до 35% мас. (в зависимости от используемого избытка серной кислоты).
При более или менее полном превращении СИМА×H2SO4 на стадии конверсии примерный выход МАКА×H2SO4 составляет от 94 до 95%. Таким образом, с учетом потерь при амидировании, обусловленных указанными выше побочными реакциями, для последующей этерификации в целевой продукт - метилметакрилат (ММА) в распоряжении имеется лишь от 90 до 92% МАКА (в пересчете на АЦГ).
Побочными продуктами, образующимися в жестких реакционных условиях стадии конверсии, являются значительные количества продуктов взаимной конденсации и присоединения промежуточных соединений.
Цель этерификации состоит в максимально полном превращении полученного на стадии конверсии МАКА × H2SO4 в ММА. Этерификацию осуществляют добавлением к сернокислотному раствору МАКА смеси воды с метанолом, и она, по меньшей мере, частично протекает с образованием метакриловой кислоты (МАК) в качестве промежуточного продукта. Этерификацию можно осуществлять под давлением или без давления.
В результате омыления/этерификации подвергнутого конверсии раствора при температуре от 90 до 140°С и времени реакции от одного до нескольких часов обычно получают сернокислотный раствор ММА, МАК и образующегося гидросульфата аммония.
Селективность превращения метанола на стадии этерификации вследствие специфических условий реакции и присутствия свободной серной кислоты составляет лишь около 90% или менее, причем побочным продуктом является образующийся вследствие конденсации метанола диметиловый эфир.
При более или менее полном превращении МАКА × H2SO4 примерный выход ММА на стадии этерификации составляет от 98 до 99% в пересчете на используемый МАКА (суммарная селективность образования МАК+ММА). Таким образом, с учетом потерь на стадиях амидирования и конверсии, обусловленных указанными выше побочными реакциями, максимальный выход ММА в результате осуществления включающего все стадии общего технологического процесса при оптимальном осуществлении реакций составляет 90% в пересчете на АЦГ.
Наряду с неудовлетворительным общим выходом целевых продуктов, недостатком рассмотренного выше процесса, реализация которого, особенно в производственном масштабе, связана с образованием значительных количеств побочных продуктов и газообразных отходов, состоит в необходимости использования сверхстехиометрических количеств серной кислоты.
Кроме того, при переработке содержащей гидросульфат аммония и серную кислоту технологической кислоты на установке для регенерации контактной серной кислоты выделяются дегтеподобные твердые продукты конденсации, которые препятствуют надежному транспортированию технологической кислоты и устранение которых связано со значительными затратами.
В связи со значительными потерями выхода целевых продуктов, характерными для предложенного в патенте US 4529816 способа, предлагалось осуществлять амидирование и гидролиз АЦГ в присутствии воды при сохранении, по меньшей мере, на первых стадиях превращения присоединенных к молекулам гидроксильных функциональных групп.
Подобные альтернативные варианты реализуемого в присутствии воды амидирования в зависимости от того, осуществляют их в присутствии метанола или без него, сопровождаются образованием либо сложного метилового эфира 2-гидроксиизомасляной кислоты (ГИМКМ), либо 2-гидроксиизомасляной кислоты (ГИМК).
2-Гидроксиизомасляная кислота является основным промежуточным продуктом получения метакриловой кислоты и соответствующих сложных эфиров метакриловой кислоты, особенно метилметакрилата.
Другой альтернативный вариант получения из АЦГ сложных эфиров 2-гидроксиизо-масляной кислоты, особенно сложного метилового эфира 2-гидроксиизомасляной кислоты, предложен в японском патенте JP Hei 4-193845. В соответствии с подобным вариантом АЦГ сначала амидируют серной кислотой, используемой в количестве от 0,8 до 1,25 эквивалента, в присутствии менее 0,8 эквивалента воды при температуре ниже 60°С, а затем с целью превращения продуктов амидирования в ГИМКМ или соответствующие сложные эфиры подвергают взаимодействию при температуре выше 55°С со спиртом, особенно метанолом, используемым в количестве более 1,2 эквивалента. Сведения о присутствии снижающих вязкость сред, стабильных по отношению к реакционной матрице, в указанной публикации отсутствуют.
Недостатки и проблемы технической реализации указанного выше способа обусловлены чрезвычайно сильным повышением вязкости на конечной стадии реакции.
В патентной литературе приведены некоторые составы, предназначенные для утилизации и преобразования ГИМКМ дегидратацией в метилметакрилат.
Так, например, согласно европейскому патенту ЕР 0429800 ГИМКМ или смесь ГИМКМ с соответствующим альфа- или бета-алкоксилированным сложным эфиром подвергают превращению в газовой фазе в присутствии метанола в качестве совместно используемого исходного компонента на гетерогенном катализаторе, состоящем из кристаллического алюмосиликата и смешанной примеси, состоящей из щелочного металла, с одной стороны, и благородного металла, с другой стороны. Хотя, по меньшей мере, в начале реакции катализатор обеспечивает вполне удовлетворительные степени превращения и селективность, однако по мере продолжения реакции катализатор весьма резко утрачивает свою активность, что сопровождается снижением выходов.
Аналогичный состав предложен в европейском патенте ЕР 0941984, в котором описана газофазная дегидратация ГИМКМ в виде отдельной стадии синтеза ММА, реализуемой в присутствии гетерогенного катализатора, состоящего из соли щелочного металла и фосфорной кислоты на диоксиде кремния. Однако соответствующая многостадийная технология в целом весьма сложна, отдельные стадии подлежат реализации при повышенном давлении, а следовательно, с использованием дорогостоящего оборудования, и она не обеспечивает удовлетворительных выходов.
Наряду с указанными выше публикациями, касающимися дегидратации ГИМКМ и родственных сложных эфиров в газовой фазе с их превращением в соответствующие альфа-бета-ненасыщенные производные метакриловой кислоты, дегидратацию предлагается осуществлять также в жидкой фазе.
Получение МАК из 2-гидроксиизомасляной кислоты описано, например, в патенте US 3487101, в котором предложен жидкофазный синтез различных производных метакриловой кислоты, прежде всего, метакриловой кислота и ее сложных эфиров, из 2-гидроксиизомасляной кислоты, отличающийся тем, что превращение ГИМК в метакриловую кислоту осуществляют в присутствии растворенного щелочного катализатора при повышенной температуре, составляющей от 180 до 320°С, в присутствии высококипящих сложных эфиров (например, диметилфталата) и внутренних ангидридов (например, фталевого ангидрида). Согласно указанному патенту селективность образования МАК при степенях превращения ГИМК, превышающих 90%, достигает 98%. Сведения, касающиеся долговременной стабильности жидкого раствора катализатора, прежде всего истощения используемого ангидрида, отсутствуют.
Наряду с этим в японском патенте JP 184047/1985 рассматривается дегидратация ГИМКМ в присутствии высококонцентрированной серной кислоты (концентрация от 90 до 100% мас.). Недостатком подобного метода является повышенный расход серной кислоты и выход больших количеств водной серной кислоты, образующейся вследствие выделения воды из ГИМКМ в результате дегидратации. В связи с образованием больших количеств отработанной серной кислоты указанный метод не имеет экономического значения.
Немецкая заявка на патент DE-OS 1191367 относится к получению метакриловой кислоты из 2-гидроксиизомасляной кислоты в жидкой фазе, отличающемуся тем, что превращение ГИМК в метакриловую кислоту осуществляют в присутствии ингибиторов полимеризации (например, таких как порошок меди) и смеси катализаторов, состоящей из галогенидов металлов и галогенидов щелочных металлов, при повышенной температуре, составляющей от 180 до 220°С. Согласно указанной заявке селективность образования МАК при степенях превращения ГИМК, превышающих 90%, составляет более 99%. Наилучших результатов достигают благодаря использованию смесей катализаторов на основе бромида цинка и бромида лития. Однако общеизвестно, что использование содержащих галогениды катализаторов при высоких температурах предъявляет жесткие требования к применяемым конструкционным материалам, причем проблемы, обусловленные присутствием в дистилляте уносимых галогенированных побочных продуктов, относятся также к последующим узлам технологического оборудования.
Из европейского патента ЕР 0487853 известно получение метакриловой кислоты из ацетонциангидрина, отличающееся тем, что на первой стадии АЦГ подвергают взаимодействию с водой при умеренных температурах в присутствии гетерогенного катализатора гидролиза, на второй стадии осуществляют взаимодействие амида 2-гидроксиизомасляной кислоты с метилформиатом или метанолом/монооксидом углерода, приводящее к образованию формамида и сложного метилового эфира гидроксиизомасляной кислоты, на третьей стадии ГИМКМ в присутствии гетерогенного ионита омыляют водой до гидроксиизомасляной кислоты, а на четвертой стадии осуществляют дегидратацию ГИМК в жидкой фазе при высоких температурах в присутствии растворенной соли щелочного металла. Метакриловую кислоту получают из ГИМК с высокими степенями превращения (99%) и более или менее количественной селективностью. Однако в связи с необходимостью осуществления большого числа реакционных стадий и промежуточного выделения индивидуальных промежуточных соединений, а также в особенности в связи с необходимостью реализации отдельных технологических операций при повышенном давлении подобная технология является слишком сложной, а следовательно, в конечном итоге нерентабельной. Кроме того, приходится синтезировать формамид, который часто рассматривают в качестве нежелательного побочного продукта, подлежащего дорогостоящей утилизации.
В немецкой заявке на патент DE-OS 1768253 описан способ получения метакриловой кислоты дегидратацией α-гидроксиизомасляной кислоты, отличающийся тем, что ГИМК подвергают жидкофазному превращению при температуре, составляющей по меньшей мере 160°С, в присутствии катализатора дегидратации, представляющего собой металлическую соль α-гидроксиизомасляной кислоты. Особенно пригодными в данном случае являются соли ГИМК со щелочными и щелочноземельными металлами, которые могут быть получены в расплаве ГИМК in situ в результате превращения пригодной соли металла. Согласно указанной публикации выход МАК из ГИМК достигает 95%, причем исходная реакционная смесь при непрерывном осуществлении способа содержит ГИМК и около 1,5% мас. соли ГИМК со щелочным металлом.
Из патента RU 89631 известен способ получения метакриловой кислоты из 2-гидроксиизомасляной кислоты отщеплением воды в жидкой фазе, отличающийся тем, что реакцию осуществляют в отсутствие катализатора в водном растворе ГИМК (до 62% мас. ГИМК в воде) под давлением и при повышенных температурах, составляющих от 200 до 240°С.
Кроме того, известно о получении 2-гидроксиизомасляной кислоты из ацетонциангидрина, которое может быть реализовано омылением нитрильных функциональных групп в присутствии минеральных кислот (смотри J. Brit. Chem. Soc. (1930) и Спет.Вег.72 (1939), 800).
В соответствии с процессом подобного типа, известным, например, из японского патента JP Sho 63-61932, 2-гидроксиизомасляную кислоту (ГИМК) получают двухстадийным омылением АЦГ. При этом сначала АЦГ в присутствии от 0,2 до 1,0 моля воды и от 0,5 до 2 эквивалентов серной кислоты превращают в соответствующие соли амида. Уже на этой стадии, предусматривающей использование небольших количеств воды и серной кислоты, необходимых для обеспечения высоких выходов, сокращения времени реакции и образования незначительных количеств отходов технологической кислоты, сталкиваются с серьезной проблемой пригодности амидируемой смеси для перемешивания, что обусловлено ее высокой вязкостью, в особенности на конечной стадии реакции.
Повышение количества воды с целью снижения вязкости приводит к резкому замедлению амидирования и побочным реакциям, прежде всего фрагментированию АЦГ на эдукты (ацетон и синильную кислоту), которые в реакционных условиях образуют продукты последовательно протекающих реакций. Хотя повышение температуры в соответствии с японским патентом JP SHO 63-61932 и позволяет регулировать вязкость реакционной смеси, снижение которой обеспечивает возможность перемешивания реакционных композиций, однако в этом случае уже при умеренных температурах также резко усиливается протекание побочных реакций, что в конечном итоге проявляется в снижение выхода целевого продукта (смотри сравнительные примеры).
При осуществлении амидирования при пониженных температурах (ниже 50°С), то есть в условиях, которые могли бы обеспечить более высокую селективность, к моменту завершения реакции вследствие повышения концентрации трудно растворимых в реакционных условиях солей амида сначала происходит образование трудно перемешиваемой суспензии, а затем полное затвердевание реакционной системы.
На второй стадии предложенного в японском патенте JP SHO 63-61932 способа к полученному в результате амидирования раствору добавляют воду и осуществляют гидролиз при аналогичной амидированию повышенной температуре, причем из полученных при амидировании солей образуется 2-гидроксиизомасляная кислота (ГИМК) с одновременным выделением свободного гидросульфата аммония.
Для обеспечения рентабельности технического процесса, наряду с селективным образованием ГИМК в качестве целевого продукта реакции, большое значение имеет также ее выделение из реакционной матрицы, соответственно отделение ГИМК от остаточной технологической кислоты.
В соответствии с используемым в японском патенте JP Sho 57-131736 методом выделения α-оксиизомасляной кислоты (ГИМК) указанная проблема решается благодаря обработке содержащего α-гидроксиизомасляную кислоту и кислый гидросульфат аммония реакционного раствора, полученного в результате гидролитического расщепления ацетонциангидрина при его взаимодействии с серной кислотой и водой, экстрагирующим агентом, причем 2-гидроксиизомасляная кислота переходит в экстрагирующий агент, а кислый сульфат аммония остается в водной фазе.
В соответствии с указанной технологией с целью повышения степени экстракции ГИМК органическим экстрагентом содержащуюся в реакционной системе свободную серную кислоту перед экстракцией нейтрализуют благодаря обработке щелочной средой. Нейтрализация серной кислоты связана со значительными дополнительными расходами на аминное или минеральное основание и образованием значительного количества отходов в виде соответствующих солей, которые невозможно утилизировать без отрицательного воздействия на экологию и экономичность процесса.
Таким образом, недостатки предложенного в японском патенте JP Sho 57-131736 способа получения ММА через амид метакриловой кислоты - гидросульфат (реакционная последовательность: амидирование - конверсия - гидролитическая этерификация) состоят в следующем.
a) Использование значительного молярного избытка серной кислоты по отношению к АЦГ (в технически реализуемом процессе примерно от 1,5 до 2 эквивалентов серной кислоты на 1 эквивалент АЦГ).
b) Значительные потери выхода на стадии амидирования (примерно от 3 до 4%) и стадии конверсии (примерно от 5 до 6%), что в конечном итоге негативно отражается на максимальном выходе амидосульфата метакриловой кислота, составляющем около 91%.
c) Значительные потоки отходов в виде водной серной кислоты с растворенными в ней гидросульфатом аммония и органическими побочными продуктами. Выделение дегтеподобных остатков неопределенного состава из подобной отработанной технологической кислоты требует осуществления дополнительной обработки, соответственно затратоемкой утилизации.
Недостатки предложенного в японском патенте JP Sho 57-131736 способа получения ММА через гидроксиизомасляную кислоту в качестве основного промежуточного соединения (реакционная последовательность: амидирование - гидролитический синтез ГИМК- синтез МАК - гидролитическая этерификация) можно резюмировать следующим образом.
а) Использование серной кислоты даже при ее незначительном молярном избытке по отношению к АЦГ (не более 1,0 эквивалента серной кислоты на один эквивалент АЦГ) приводит к серьезным проблемам, обусловленным повышением вязкости и недостаточной пригодностью амидируемой среды для перемешивания, вплоть до полного затвердевания реакционных составов; предлагаемое разбавление спиртами (метанолом) или различными сложными эфирами в используемых для амидирования реакционных условиях приводит к неполному превращению АЦГ, резкому усилению протекания побочных реакций или химической деструкции разбавителей.
b) Значительные потери выхода на стадии амидирования (примерно от 5 до 6%) и дорогостоящая экстракция органическим растворителем, сопровождаемая образованием содержащей воду и ГИМК фазы экстрагирующего агента, выделение ГИМК из которой методом дистилляции связано с повышенным расходом энергии. На каждый килограмм ГИМК приходится около двух килограммов образующейся отработанной технологической кислоты, содержащей около 34% мас. воды и 66% мас. гидросульфата аммония (смотри пример 4 в японском патенте JP Sho 57-131736). Регенерация отработанного солевого раствора с высоким содержанием воды на установке для получения контактной серной кислоты требует значительных затрат энергии, что существенно ограничивает производственную мощность подобной установки.
Общей особенностью всех рассмотренных выше способов является чрезвычайно дорогостоящее выделение ГИМК из содержащей гидросульфат аммония водной реакционной матрицы. Присутствие слишком большого количества воды в содержащей ГИМК экстракционной фазе обусловливает также унос гидросульфата аммония на последующую стадию синтеза МАК, непрерывная реализация которой в техническом масштабе в течение допустимого периода времени не представляется возможной. Кроме того, предлагаемые способы вследствие высоких энергетических затрат на регенерацию как содержащей воду высококонцентрированной технологической кислоты, так и экстракционных потоков, являются нерентабельными и не могут служить подлинной альтернативой хотя и неселективной, но надежной технологии, приводящей к конечной цели благодаря реализации небольшого количества несложных технологических операций.
С учетом рассмотренного выше уровня техники в основу настоящего изобретения была положена задача предложить способ получения алкил(мет)акрилатов, отличающийся простотой и экономичностью осуществления.
Другая задача настоящего изобретения состояла в разработке способа, позволяющего получать алкил(мет)акрилаты с чрезвычайно высокой селективностью.
Кроме того, в основу настоящего изобретения была положена задача предложить способ получения алкил(мет)акрилатов, реализация которого сопровождалась бы образованием лишь незначительного количества побочных продуктов. При этом целевой продукт необходимо получать с максимально высокими выходами при незначительном суммарном потреблении энергии.
Другая задача настоящего изобретения состояла в том, чтобы предложить способ получения алкил(мет)акрилатов, которой можно было бы осуществлять особенно просто и экономично.
Указанные выше задачи, а также другие задачи изобретения, не сформулированные в явном виде, однако безусловно вытекающие из контекста соответствующего описания, решаются благодаря способу, отличительные признаки которого сформулированы в пункте 1 формулы изобретения. Целесообразные варианты предлагаемого в изобретении способа представлены в соответствующих зависимых пунктах формулы изобретения.
Таким образом, объектом настоящего изобретения является способ получения алкил(мет)акрилатов, включающий стадию переэтерификации сложного алкилового эфира α-гидроксикарбоновой кислоты (мет)акриловой кислотой, сопровождаемую образованием алкил(мет)акрилатов и α-гидроксикарбоновой кислоты, и стадию дегидратации α-гидроксикарбоновой кислоты, сопровождаемую образованием (мет)акриловой кислоты.
Благодаря реализации предлагаемых в изобретении операций могут быть достигнуты, в частности, следующие преимущества.
Способ позволяет отказаться от использования больших количеств серной кислоты в качестве реагента. Следовательно, в соответствии с предлагаемым в изобретении способом отсутствует образование больших количеств отходов гидросульфата аммония.
Алкил(мет)акрилаты, получаемые предлагаемым в изобретении способом, образуются с высокими выходами. Прежде всего, это становится очевидным из сравнения предлагаемого в изобретении способа со способом, описанным в европейской заявке на патент ЕР-А-0941984, в соответствии с которым сложные алкиловые эфиры α-гидроксикарбоновой кислоты подвергают непосредственной дегидратации в алкил(мет)акрилаты. Неожиданно было установлено, что, благодаря дополнительной реакционной стадии переэтерификации сложного алкилового эфира α-гидроксикарбоновой кислоты (мет)акриловой кислотой, в конечном итоге достигают более высоких значений селективности.
При этом образуются весьма незначительные количества побочных продуктов. Кроме того, прежде всего, с учетом высокой селективности, достигают высоких степеней превращения.
Предлагаемый в изобретении способ приводит к образованию незначительных количеств побочных продуктов.
Предлагаемый в изобретении способ может быть реализован рентабельно и, прежде всего, при незначительном потреблении энергии. При этом используемые для дегидратации и переэтерификации катализаторы можно эксплуатировать в течение продолжительного времени без снижения селективности или активности.
Предлагаемый в изобретении способ может быть реализован в промышленном масштабе.
Согласно изобретению сложный алкиловый эфир α-гидроксикарбоновой кислоты подвергают взаимодействию с (мет)акриловой кислотой. Используемые при этом (мет)акриловые кислоты известны, и являются коммерчески доступными продуктами. Наряду с акриловой кислотой (пропеновой кислотой) и метакриловой кислотой (2-метилпропеновой кислотой) к подобным кислотам, прежде всего, относятся соответствующие замещенные производные. Пригодными заместителями, прежде всего, являются галогены, такие как хлор, фтор и бром, а также алкильные группы, которые предпочтительно могут содержать от 1 до 10, особенно предпочтительно от 1 до 4 атомов углерода. К пригодным кислотам относятся, в частности, β-метилакриловая кислота (бутеновая кислота), α,β-диметилакриловая кислота, β-этилакриловая кислота, а также β,β-диметилакриловая кислота. Предпочтительными кислотами являются акриловая кислота (пропеновая кислота) и метакриловая кислота (2-метилпропеновая кислота), причем особенно предпочтительной является метакриловая кислота.
Подвергаемые переэтерификации сложные алкиловые эфиры α-гидроксикарбоновой кислоты являются известными эфирами, спиртовый остаток которых предпочтительно содержит от 1 до 20, прежде всего, от 1 до 10, особенно предпочтительно от 1 до 5 атомов углерода. Предпочтительные спиртовые остатки, прежде всего, происходят от метанола, этанола, пропанола, бутанола, в особенности н-бутанола и 2-метил-1-пропанола, пентанола, гексанола и 2-этилгексанола, причем особенно предпочтительные спиртовые остатки происходят от метанола и этанола.
Кислотный остаток подвергаемых переэтерификации сложных алкиловых эфиров α-гидроксикарбоновой кислоты предпочтительно происходит от (мет)акриловой кислоты, которая может быть получена дегидратацией α-гидроксикарбоновой кислоты. Так, например, если используют метакриловую кислоту, то переэтерификации подвергают сложный эфир α-гидроксиизомасляной кислоты. В случае использования акриловой кислоты переэтерификации предпочтительно подвергают сложный эфир α-гидроксиизопропионовой кислоты.
Предпочтительно используемыми сложными алкиловыми эфирами α-гидроксикарбоновой кислоты являются сложный метиловый эфир α-гидроксипропионовой кислоты, сложный этиловый эфир α-гидроксипропионовой кислоты, сложный метиловый эфир α-гидроксиизомасляной кислоты и сложный этиловый эфир α-гидроксиизомасляной кислоты.
Подобные сложные алкиловые эфиры α-гидроксикарбоновой кислоты часто с оптимальными затратами получают из соответствующих циангидринов. При этом степень чистоты циангидрина не является критическим параметром. Следовательно, для осуществления гидролиза можно использовать очищенный или неочищенный циангидрин. В соответствии с этим подлежащие использованию согласно изобретению сложные алкиловые эфиры α-гидроксикарбоновой кислоты могут быть синтезированы из кетонов и альдегидов, а также синильной кислоты и соответствующего спирта.
На первой стадии подобного синтеза карбонильное соединение, например, кетон, прежде всего, ацетон, или альдегид, например, ацетальдегид, пропаналь или бутаналь, превращают в соответствующий циангидрин взаимодействием с синильной кислотой. При этом превращению, в типичном случае осуществляемому с использованием в качестве катализатора незначительного количества щелочи или амина, особенно предпочтительно подвергают ацетон и/или ацетальдегид.
Полученный циангидрин на следующей стадии подвергают взаимодействию с водой, сопровождаемому образованием амида гидроксикарбоновой кислоты.
Подобное взаимодействие в типичном случае осуществляют в присутствии катализатора. Для этой цели, прежде всего, пригодны катализаторы на основе оксида марганца, описанные, например, в европейских заявках на патент ЕР-А-0945429, ЕР-А-0561614 и ЕР-А-0545697. При этом оксид марганца можно использовать в форме диоксида марганца, получаемого осуществляемой в кислой среде обработкой сульфата марганца перманганатом калия (смотри Biochem.J., 50, страница 43 (1951) и J.Chem.Soc., 1953, страница 2189, 1953) или электролитическим окислением сульфата марганца в водном растворе. В общем случае катализатор часто используют в виде порошка или гранулята с частицами подходящего размера. Кроме того, катализатор может быть нанесен на носитель. При этом, прежде всего, можно использовать также реакторы со взвешенным или неподвижным слоем катализатора, в частности, описанные в европейской заявке на патент ЕР-А-956 898.
Кроме того, реакцию гидролиза циангидрина можно катализировать ферментами. Пригодными ферментами являются, в частности, нитрилгидратазы. Подобная реакция приведена, например, в "Screening, Characterization and Application of Cyanide-resistant Nitrile Hydratases" Eng. Life. Sci. 2004, 4, №6.
Наряду с этим реакцию гидролиза циангидрина можно катализировать кислотами, в особенности серной кислотой. Кислотный катализ предлагается использовать, в частности, в японском патенте JP Hei 4-193845.
Воду, необходимую для осуществления гидролиза циангидрина, часто можно использовать в качестве растворителя. Молярное соотношение воды к циангидрину предпочтительно составляет, по меньшей мере, 1:1 и особенно предпочтительно находится в интервале от 0,5:1 до 25:1, еще более предпочтительно в интервале от 1:1 до 10:1.
Используемая для гидролиза вода может обладать высокой степенью чистоты. Однако это условие не является обязательным. Так, например, наряду со свежей водой, можно использовать также производственную или технологическую воду, содержащую более или менее значительные количества примесей. Следовательно, для гидролиза можно использовать также оборотную воду.
Кроме того, в используемой для гидролиза циангидрина реакционной смеси могут присутствовать и другие компоненты. К подобным компонентам относятся, в частности, альдегиды и кетоны, прежде всего, те из них, которые были использованы для синтеза циангидрина. Так, например, реакционная смесь может содержать ацетон и/или ацетальдегид. Подобная реакционная смесь приведена, например, в патенте US 4018829-A. Степень чистоты добавляемых альдегидов и/или кетонов в общем случае не является особенно критическим параметром. Следовательно, указанные вещества могут содержать примеси, прежде всего, спирты, например, метанол, воду и/или сложный метиловый эфир α-гидроксиизомасляной кислоты (ГИМКМ). Количество используемых в реакционной смеси карбонильных соединений, прежде всего, ацетона и/или ацетальдегида, можно варьировать в широких пределах. Предпочтительное количество используемого карбонильного соединения составляет от 0,1 до 6 молей, предпочтительно от 0,1 до 2 молей в расчете на моль циангидрина.
Температура, при которой осуществляют реакцию гидролиза, в общем случае может находиться в интервале от 10 до 150°С, предпочтительно от 20 до 100°С и особенно предпочтительно от 30 до 80°С.
Гидролиз циангидрина можно осуществлять, например, в реакторе со стационарным или взвешенным слоем катализатора.
Полученная указанным образом реакционная смесь, наряду с целевым амидом гидроксикислоты, в общем случае содержит другие компоненты, прежде всего, непревращенный циангидрин, а также, при необходимости, используемый ацетон и/или ацетальдегид. В соответствии с этим реакционная смесь может быть подвергнута очистке, при которой непревращенный циангидрин может быть расщеплен до ацетона и синильной кислоты, которые повторно используют для получения циангидрина. То же самое относится и к выделенному ацетону и/или ацетальдегиду.
Кроме того, очищенная реакционная смесь, содержащая амид гидроксикислоты, может быть освобождена от других компонентов, благодаря пропусканию через колонны с ионообменными смолами.
В качестве ионообменных смол, прежде всего, можно использовать катиониты и аниониты. Пригодные для этого иониты являются известными продуктами. Так, например, пригодные катиониты могут быть получены сульфированием сополимеров стирола с дивинилбензолом. Щелочные аниониты содержат четвертичные аммониевые группы, ковалентно соединенные с сополимерами стирола с дивинилбензолом.
Стадии получения амидов α-гидроксикарбоновой кислоты подробно рассмотрены, в частности, в европейской заявке на патент ЕР-А-0686623.
Полученный рассмотренным выше образом амид α-гидроксикарбоновой кислоты на следующей стадии может быть превращен в сложный алкиловый эфир α-гидроксикарбоновой кислоты. Соответствующая реакция может быть реализована, например, при использовании алкилформиатов. Пригодным, прежде всего, является метилформиат или смесь метанола с монооксидом углерода (пример подобной реакции приведен в европейской заявке на патент ЕР-А-0407811).
Амид α-гидроксикарбоновой кислоты предпочтительно подвергают алкоголизу спиртом предпочтительно с 1-10, особенно предпочтительно с 1-5 атомами углерода. Предпочтительными спиртами являются, в частности, метанол, этанол, пропанол, бутанол, прежде всего, н-бутанол и 2-метил-1-пропанол, пентанол, гексанол, гептанол, 2-этилгексанол, октанол, нонанол и деканол. Особенно предпочтительным спиртом является метанол и/или этанол, еще более предпочтительным спиртом является метанол. Взаимодействие амидов карбоновой кислоты со спиртами с целью получения сложных эфиров карбоновой кислоты является общеизвестной реакцией. Указанную реакцию можно ускорить, используя, например, щелочные катализаторы. Катализаторы могут быть как гомогенными, так и гетерогенными.
К гомогенным катализаторам относятся алкоголяты щелочных металлов и металлорганические соединения, содержащие титан, олово или алюминий. В качестве катализатора предпочтительно используют алкоголят титана или алкоголят олова, например, тетраизопропилоксид титана или тетрабутилоксид олова. К гетерогенным катализаторам относятся, в частности, оксид магния, оксид кальция, а также указанные выше щелочные иониты.
Молярное соотношение амида α-гидроксикарбоновой кислоты к спирту, например, амида α-гидроксиизомасляной кислоты к метанолу, не является критическим параметром и предпочтительно находится в интервале от 2:1 до 1:20.
Температуру реакции также можно варьировать в широких пределах, причем скорость реакции по мере повышения температуры в общем случае возрастает. Верхний предел возможного температурного диапазона в общем случае соответствует температуре кипения используемого спирта. Температура реакции предпочтительно составляет от 40 до 300°С, особенно предпочтительно от 160 до 240°С. В зависимости от температуры реакцию можно осуществлять при разрежении или избыточном давлении. Рассматриваемую реакцию осуществляют при давлении, предпочтительно находящемся в интервале от 0,5 до 35 бар, особенно предпочтительно от 5 до 30 бар.
Образующийся аммиак обычно выводят из реакционной системы, причем реакцию часто осуществляют при температуре кипения.
Выделяющийся при алкоголизе аммиак можно несложным методом рециркулировать в общий технологический процесс. Так, например, аммиак можно подвергать взаимодействию с метанолом, приводящему к образованию синильной кислоты. Подобное взаимодействие предложено, например, в европейской заявке на патент ЕР-А-0941984. Кроме того, аммиак и метан можно использовать для синтеза синильной кислоты методом фирмы ВМА или методом Андрусова, приведенными в Ullmann's Encyclopedia of Industrial Chemistry, 5-е издание (в записи на CD-ROM), раздел „Inorganic Cyano Compounds".
На следующей стадии сложный алкиловый эфир α-гидроксикарбоновой кислоты подвергают взаимодействию с (мет)акриловой кислотой, приводящему к образованию алкил(мет)акрилата, а также α-гидроксикарбоновой кислоты.
Наряду с указанными реагентами, реакционная смесь может содержать другие компоненты, например, такие как растворители, катализаторы, ингибиторы полимеризации и воду. Взаимодействие сложного эфира алкилгидроксикарбоновой кислоты с (мет)акриловой кислотой можно катализировать, по меньшей мере, одной кислотой или, по меньшей мере, одним основанием. При этом возможно использование как гомогенных, так и гетерогенных катализаторов. Особенно пригодными кислотными катализаторами, прежде всего, являются неорганические кислоты, например, серная кислота или соляная кислота, а также органические кислоты, например, сульфокислоты, прежде всего, п-толуолсульфокислота, а также кислый катионит.
Особенно пригодными катионообменными смолами, прежде всего, являются сополимеры стирола с дивинилбензолом, содержащие сульфокислотные группы. Особенно пригодные катионообменные смолы выпускает фирма Rohm&Haas под торговым названием Amberlyst® и фирма Вауеr под торговым названием Lewatit®.
Концентрация катализатора предпочтительно составляет от 1 до 30% мас., особенно предпочтительно от 5 до 15% мас. в пересчете на сумму исходных реагентов: сложного эфира α-алкилгидроксикарбоновой кислоты и (мет)акриловой кислоты.
К предпочтительно используемым ингибиторам полимеризации относятся, в частности, фенотиазин, трет-бутилкатехол, монометиловый эфир гидрохинона, гидрохинон, 4-гидрокси-2,2,6,6-тетраметилпиперидинооксил или их смеси, причем эффективность указанных ингибиторов в определенной степени можно повысить благодаря использованию кислорода. Ингибиторы полимеризации можно использовать в концентрации от 0,001 до 2,0% мас., особенно предпочтительно от 0,01 до 0,2% мас. в пересчете на сумму исходных реагентов: сложного эфира α-алкилгидроксикарбоновой кислоты и (мет)акриловой кислоты.
Переэтерификацию предпочтительно осуществляют в температурном интервале от 50 до 200°С, особенно предпочтительно от 70 до 130°С, прежде всего, от 80 до 120°С. еще более предпочтительно от 90 до 110°С.
В зависимости от температуры переэтерификацию можно осуществлять при разрежении или избыточном давлении. Переэтерификацию предпочтительно осуществляют под давлением, составляющим от 0,02 до 5 бар, прежде всего от 0,2 до 3 бар и особенно предпочтительно от 0,3 до 0,5 бар.
Молярное соотношение (мет)акриловой кислоты к сложному алкиловому эфиру α-гидроксикарбоновой кислоты предпочтительно находится в интервале от 4:1 до 1:4, прежде всего, от 3:1 до 1:3 и особенно предпочтительно от 2:1 до 1:2.
Селективность предпочтительно составляет, по меньшей мере, 90%, особенно предпочтительно 98%. Селективность определяют как отношение суммарного количества образующихся алкил(мет)акрилатов и α-гидроксикарбоновых кислот к суммарному количеству превращенных исходных реагентов: сложного алкилового эфира α-гидроксикарбоновой кислоты и (мет)акриловой кислоты.
Согласно особому варианту осуществления настоящего изобретения переэтерификацию можно осуществлять в присутствии воды. Содержание воды предпочтительно составляет от 0,1 до 50% мас., особенно предпочтительно от 0,5 до 20% мас. и еще более предпочтительно от 1 до 10% мас. в пересчете на массу используемого сложного алкилового эфира α-гидроксикарбоновой кислоты.
Неожиданно было обнаружено, что добавление незначительных количеств воды позволяет повысить селективность переэтерификации. Несмотря на добавление воды количество образующегося при этом метанола неожиданно удается поддерживать на невысоком уровне. При концентрации воды, составляющей от 10 до 15% мас. в пересчете на массу используемого сложного алкилового эфира α-гидроксикарбоновой кислоты, температуре реакции 120°С и ее длительности, соответственно времени пребывания реагентов, составляющей от 5 до 180 минут, предпочтительно образуется менее 5% мас. метанола.
Переэтерификацию можно осуществлять в периодическом или непрерывном режиме, причем предпочтительной является непрерывная переэтерификация.
Продолжительность переэтерификации зависит от молекулярной массы исходных соединений, а также от температуры реакции, причем указанные параметры можно варьировать в широком диапазоне. Время реакции переэтерификации сложного алкилового эфира α-гидроксикарбоновой кислоты (мет)акриловой кислотой предпочтительно составляет от 30 секунд до 15 часов, особенно предпочтительно от 5 минут до 5 часов и еще более предпочтительно от 15 минут до 3 часов.
При осуществлении переэтерификации в непрерывном режиме время пребывания реагентов предпочтительно составляет от 30 секунд до 15 часов, особенно предпочтительно от 5 минут до 5 часов и еще более предпочтительно от 15 минут до 3 часов.
При получении метилметакрилата из сложного метилового эфира α-гидроксиизомасляной кислоты температура предпочтительно составляет от 60 до 130°С, особенно предпочтительно от 80 до 120°С и еще более предпочтительно от 90 до 110°С. Давление предпочтительно находится в интервале от 50 до 1000 мбар, особенно предпочтительно в интервале от 300 до 800 мбар. Молярному соотношению метакриловой кислоты к сложному метиловому эфиру α-гидроксиизомасляной кислоты предпочтительно соответствует интервал от 2:1 до 1:2, прежде всего, от 1,5:1 до 1:1,5.
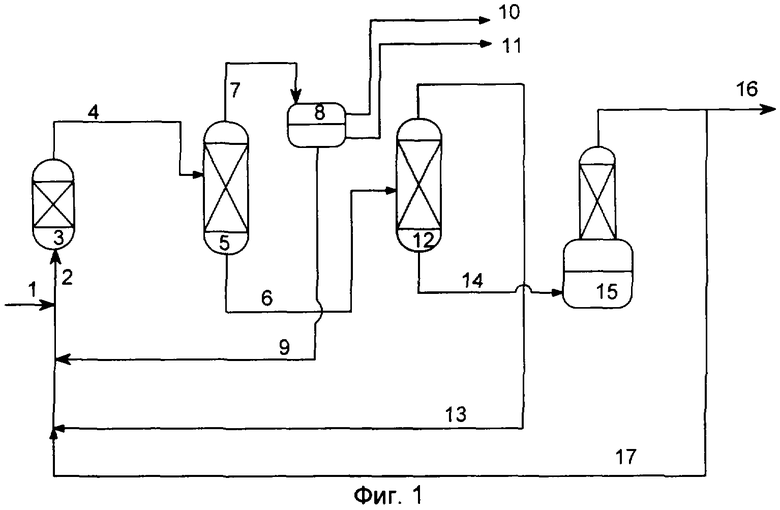
Переэтерификацию можно осуществлять, например, на установке, показанной на Фиг.1. Сложный эфир гидроксикарбоновой кислоты, например, сложный метиловый эфир гидроксиизомасляной кислоты, по трубопроводу (1) поступает в реактор (3) со стационарным слоем катионообменной смолы. (Мет)акриловую кислоту, например, 2-метилпропеновую кислоту, вводят в реактор (3) со стационарным слоем по трубопроводу (2) или трубопроводу (17). Для сокращения количества линий питания реактора трубопровод (2) может быть соединен с другими трубопроводами, например, с трубопроводом (9) и трубопроводом (13). Однако трубопроводы (9), (13) и/или (17) могут быть также непосредственно соединены с реактором (3) со стационарным слоем. В указанных выше реакционных условиях образуется реакционная смесь, которая наряду с метанолом, а также непревращенным сложным метиловым эфиром гидроксиизомасляной кислоты и метакриловой кислотой содержит продукты реакции: гидроксиизомасляную кислоту, а также метилметакрилат. Указанную реакционную смесь по трубопроводу (4) направляют в дистилляционную колонну (5). В дистилляционной колонне (5) в качестве головного продукта получают воду, метилметакрилат, а также метанол, которые по трубопроводу (7) направляют в разделитель фаз (8). Верхнюю фазу, состоящую из метилметакрилата, а также метанола, выводят из системы по трубопроводу (10). Нижняя фаза разделителя фаз (8) прежде всего содержит воду, которая может быть удалена из системы по трубопроводу (11) или направлена по трубопроводу (9) в реактор (3) со стационарным слоем.
В кубе дистилляционной колонны (5) могут присутствовать сложный метиловый эфир гидроксиизомасляной кислоты, гидроксиизомасляная кислота, а также метакриловая кислота, которые по трубопроводу (6) могут быть направлены во вторую дистилляционную колонну (12). В колонне (12) осуществляют отгонку сложного метилового эфира гидроксиизомасляной кислоты, а также метакриловой кислоты, которые по трубопроводу (13) возвращают на переэтерификацию. Содержащуюся в кубе дистилляционной колонны (12) гидроксиизомасляную кислоту по трубопроводу (14) направляют в реактор (15) для дегидратации. Полученная в результате дегидратации метакриловая кислота по трубопроводу (17) может быть направлена на переэтерификацию или выведена из системы по трубопроводу (16).
В соответствии с особенно предпочтительным вариантом осуществления изобретения переэтерификацию можно осуществлять в дистилляционной колонне. При этом катализатор может находиться в любой зоне дистилляционной колонны. Так, например, он может находиться в зоне куба или в самой колонне. Однако при этом должна быть обеспечена возможность приведения эдуктов в контакт с катализатором. Кроме того, катализатор может находиться в отдельной зоне дистилляционной колонны, соединенной с другими ее зонами, например, кубом и/или самой колонной. Подобное отдельное расположение зоны катализатора является предпочтительным.
Благодаря указанному выше предпочтительному варианту неожиданно удается повысить селективность переэтерификации. В этой связи необходимо констатировать, что давление реакции переэтерификации можно регулировать независимо от внутреннего давления в дистилляционной колонне. Это позволяет поддерживать низкую температуру кипения без увеличения времени реакции, соответственно времени пребывания реагентов. Кроме того, температуру реакции можно варьировать в широких пределах. Благодаря этому удается сократить ее длительность. Наряду с этим предоставляется возможность произвольного выбора объема катализатора вне зависимости от геометрических параметров дистилляционной колонны. Кроме того, можно, например, добавлять дополнительный реагент. Все указанные выше технические мероприятия способствуют повышению селективности и производительности при неожиданно обнаруженном достижении синергических эффектов.
В соответствии с рассматриваемым вариантом сложный алкиловый эфир α-гидроксикарбоновой кислоты, например, сложный метиловый эфир α-гидроксиизомасляной кислоты вводят в дистилляционную колонну. Кроме того, в колонну вводят (мет)акриловую кислоту, например, метакриловую кислоту. Дистилляцию предпочтительно осуществляют в таких условиях, чтобы отгонке подлежал строго один продукт, в то время как другой продукт оставался в кубе и подвергался непрерывному удалению из куба. При использовании спиртов с небольшим числом атомов углерода, прежде всего, этанола или метанола, из реакционной смеси предпочтительно отгоняют алкил(мет)акрилат. Эдукты периодически пропускают через зону катализатора. Вследствие этого непрерывно образуется алкил(мет)акрилат, а также α-гидроксикарбоновая кислота.
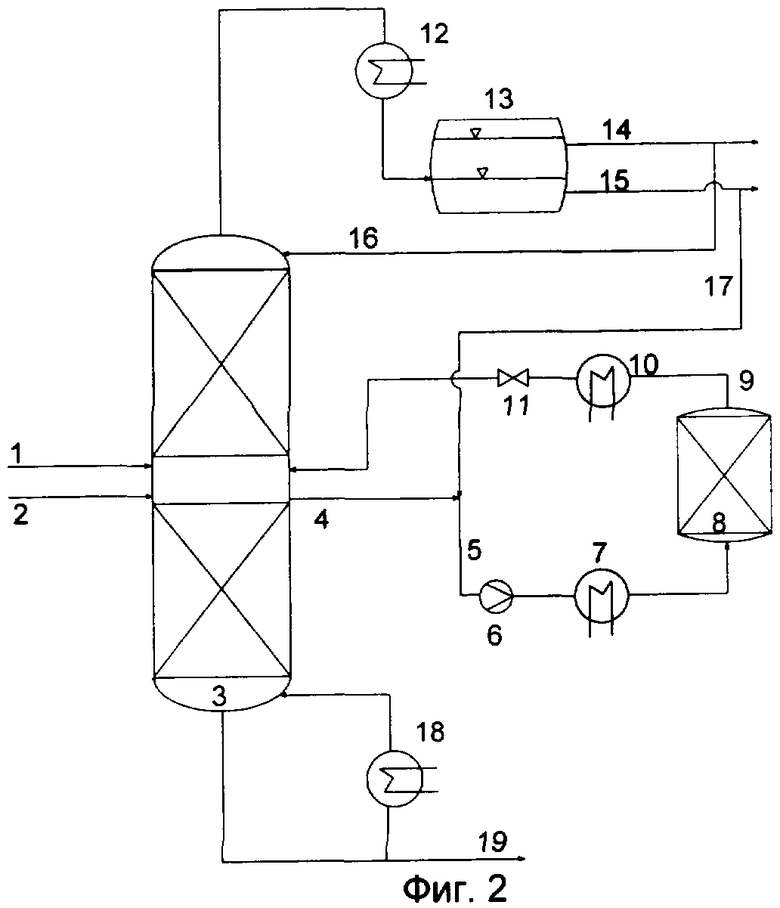
Предпочтительный вариант осуществления реакционной дистилляции схематически показан на Фиг.2. Эдукты можно подавать в дистилляционную колонну (3) по общему трубопроводу (1) или по двум отдельным трубопроводам (1) и (2). Подачу эдуктов предпочтительно осуществляют по отдельным трубопроводам. При этом эдукты можно вводить на одну и ту же ступень разделения или в любую точку колонны.
При этом температуру реагентов на входе в колонну можно регулировать посредством теплообменника (необходимое для этого оборудование не показано на Фиг.2). В предпочтительном варианте эдукты подают в колонну по отдельности, причем легкокипящий компонент вводят ниже точки подачи высококипящего компонента. В подобном случае легкокипящий компонент предпочтительно вводят в газообразном состоянии.
Для реализации настоящего изобретения можно использовать любую многоступенчатую дистилляционную колонну (3) с двумя или более ступенями разделения. Количеством ступеней разделения согласно настоящему изобретению обозначают число тарелок тарельчатой колонны или число теоретических ступеней разделения насадочной колонны или колонны с элементами насадки.
Примерами тарельчатых многоступенчатых дистилляционных колонн могут служить колонны с колпачковыми, сетчатыми, туннельными, клапанными, щелевыми, сетчатощелевыми, сетчатоколпачковыми, дюзовыми или центробежными тарелками, примерами многоступенчатых дистилляционных колонн с элементами насадки являются колонны с кольцами Рашига, кольцами Лессинга, кольцами Паля, седлами Берля или седлами Intalox, примерами насадочных многоступенчатых дистилляционных колонн являются колонны с насадкой типа Mellapak (фирма Sulzer), Rombopak (фирма Kühni), Montz-Pak (фирма Montz) и насадкой с гнездами для катализатора, например, Kata-Pak.
Кроме того, можно использовать также комбинированную дистилляционную колонну, включающую зоны тарелок, зоны с элементами насадки или зоны с насадками.
Колонна (3) может быть оснащена дополнительными узлами. Колонна предпочтительно включает конденсатор (12) для конденсации паров и кубовый испаритель (18).
Дистилляционная установка предпочтительно обладает, по меньшей мере, одной зоной, ниже называемой реактором, который заполнен, по меньшей мере, одним катализатором. Подобный реактор может находиться внутри дистилляционной колонны. Однако подобный реактор предпочтительно располагается в отдельной зоне вне колонны (3), причем один из таких предпочтительных вариантов конструктивного исполнения подробно показан на Фиг.2.
Для осуществления переэтерификации в отдельном реакторе (8) часть стекающей вниз внутри колонны жидкой фазы можно улавливать посредством накопителя и выводить из колонны в виде отдельного потока (4). Положение накопителя определяется концентрационным профилем отдельных компонентов в колонне. При этом концентрационный профиль можно регулировать варьированием температуры и/или флегмы. Накопитель предпочтительно располагают таким образом, чтобы выводимый из колонны поток содержал оба реагента, особенно предпочтительно в достаточно высокой концентрации и еще более предпочтительно при молярном соотношении кислоты к сложному эфиру, находящемся в интервале от 1,5:1 до 1:1,5. Кроме того, может быть предусмотрено несколько находящихся в разных местах дистилляционной колонны накопителей, причем молярное соотношение можно регулировать количеством отбираемых реагентов.
Кроме того, для регулирования соотношения кислоты к сложному эфиру в продуктах перекрестной переэтерификации или повышения ее селективности к выводимому из колонны потоку можно добавлять дополнительный реагент, например воду. Добавляемую воду можно вводить снаружи по соответствующему трубопроводу (на Фиг.1 он не показан) или отбирать ее из разделителя фаз (13). Давление обогащенного водой потока (5) может быть затем повышено посредством устройства (6) для повышения давления, например, насоса.
Повышение давления потока (5) позволяет уменьшить, соответственно предотвратить образование пара в реакторе, например, в реакторе со стационарным слоем катализатора. Благодаря этому можно обеспечить равномерное пропускание потока через реактор и смачивание частиц катализатора. Поток может быть пропущен через теплообменник (7) с целью регулирования необходимой температуры переэтерификации. При этом в зависимости от потребности поток может быть подвергнут нагреванию или охлаждению. Кроме того, варьирование температуры переэтерификации позволяет регулировать соотношение сложного эфира к кислоте в продуктах реакции.
В реакторе (8) расположен стационарный слой катализатора реакции переэтерификации. Реакционную смесь можно пропускать через реактор сверху вниз или снизу вверх. Выходящий из реактора поток (9) с определенным содержанием продуктов реакции и непревращенных эдуктов, зависящим от времени пребывания реагентов в реакторе, массы катализатора, температуры реакции и соотношения эдуктов, а также от количества добавляемой воды, сначала пропускают через теплообменник (10), чтобы отрегулировать температуру потока, предпочтительную для его последующего введения в дистилляционную колонну. Температуру потока предпочтительно регулируют таким образом, чтобы она соответствовала температуре дистилляционной колонны в точке введения потока.
При этом точка ввода в колонну рециркулируемого из реактора потока может располагаться выше или ниже точки отбора потока, направляемого из колонны в реактор, однако точка ввода указанного потока предпочтительно располагается выше точки отбора. Рециркуляцируемый в колонну поток можно подвергать дросселированию посредством клапана (11), причем давление дросселированного потока регулируют таким образом, чтобы оно было предпочтительно аналогично давлению в колонне. В предпочтительном варианте давление в дистилляционной колонне ниже давления дросселированного потока. Преимуществом подобного варианта является снижение температуры кипения подлежащих разделению компонентов, благодаря чему дистилляцию можно выполнять при более низких температурах, то есть в условиях, более благоприятных с точки зрения расхода энергии и термического воздействия на реагенты.
В дистилляционной колонне (3) осуществляют разделение смеси продуктов реакции. Низкокипящие компоненты, которыми предпочтительно являются образующиеся в результате переэтерификации сложные эфиры, отгоняют через верхнюю часть колонны. Дистилляционную колонну предпочтительно эксплуатируют в режиме, позволяющем отгонять добавляемую перед реактором со стационарным слоем воду также в виде головного продукта. Выходящий из верхней части колонны парообразный поток конденсируют в конденсаторе (12), и полученный конденсат подвергают разделению в отстойнике (13) на водную фазу и фазу, содержащую целевой эфир. Водную фазу можно направить по трубопроводу (15) на переработку или по трубопроводу (17) полностью или частично рециркулировать на переэтерификацию. Часть потока содержащей эфир фазы может быть возвращена в колонну по трубопроводу (14) в качестве флегмы (16) или выведена из дистилляционной колонны. Высококипящие компоненты (предпочтительно образующуюся в результате перекрестной переэтерификации кислоту) выводят из колонны (19) в виде кубового потока.
Использование рассмотренного выше предпочтительного варианта неожиданно позволяет повысить селективность переэтерификации. В этой связи необходимо подчеркнуть, что давление реакции переэтерификации можно регулировать независимо от внутреннего давления в дистилляционной колонне. Это позволяет поддерживать низкую температуру кипения без увеличения длительности реакции, соответственно времени пребывания реагентов. Кроме того, температуру переэтерификации можно варьировать в широких пределах. Благодаря этому удается сократить ее продолжительность. Наряду с этим предоставляется возможность произвольного выбора объема катализатора вне зависимости от геометрических параметров дистилляционной колонны. Кроме того, можно, например, добавлять дополнительный реагент.
Полученную в результате переэтерификации α-гидроксикарбоновую кислоту, например, гидроксиизомасляную кислоту, можно дегидратировать известными методами. В общем случае α-гидроксикарбоновую кислоту, например, α-гидроксиизомасляную кислоту, нагревают в присутствии, по меньшей мере, одной соли металла, например, соли щелочного и/или щелочноземельного металла, при температуре от 160 до 300°С, особенно предпочтительно от 200 до 240°С, причем в общем случае получают (мет)акриловую кислоту, а также воду. Пригодными солями металла являются, в частности, гидроксид натрия, гидроксид калия, гидроксид кальция, гидроксид бария, гидроксид магния, сульфит натрия, карбонат натрия, карбонат калия, карбонат стронция, карбонат магния, бикарбонат натрия, ацетат натрия, ацетат калий и дигидрофосфат натрия.
Дегидратацию α-гидроксикарбоновой кислоты предпочтительно можно осуществлять под давлением от 0,05 до 2,5 бар, особенно предпочтительно от 0,1 до 1 бар.
Согласно особому варианту осуществления настоящего изобретения дегидратацию осуществляют под давлением, которое практически аналогично давлению, используемому для реализации рассмотренной выше переэтерификации сложного алкилового эфира α-гидроксикарбоновой кислоты (мет)акриловой кислотой, однако подобное условие не следует рассматривать как ограничение объекта изобретения. Разность давлений при переэтерификации и дегидратации предпочтительно составляет менее 0,1 бар, особенно предпочтительно менее 0,05 бар. Согласно особому варианту осуществления настоящего изобретения полученная газообразная (мет)акриловая кислота может быть направлена на переэтерификацию без конденсации и повторного испарения.
Дегидратация α-гидроксикарбоновых кислот описана, например, в немецкой заявке на патент DE-A-1768253.
Полученную при дегидратации (мет)акриловую кислоту можно вновь использовать для синтеза алкил(мет)акрилатов. Кроме того, (мет)акриловая кислота является товарным продуктом. Таким образом, неожиданно выяснилось, что установку для получения алкил(мет)акрилатов можно использовать также для синтеза (мет)акриловой кислоты, причем соотношение между получаемыми алкил(мет)акрилатами и (мет)акриловой кислотой можно легко регулировать варьированием концентрации воды и/или температуры переэтерификации сложного алкилового эфира α-гидрокси-карбоновой кислоты.
Таким образом, алкил(мет)акрилаты можно получать из карбонильных соединений, синильной кислоты и спиртов простым и экономичным способом, включающим следующие стадии:
A) взаимодействие, по меньшей мере, одного карбонильного соединения с синильной кислотой с образованием, по меньшей мере, одного циангидрина,
B) гидролиз циангидрина, соответственно циангидринов, с образованием, по меньшей мере, одного амида α-гидроксикарбоновой кислоты,
C) алкоголиз амида α-гидроксикарбоновой кислоты, соответственно амидов α-гидроксикарбоновой кислоты, с образованием, по меньшей мере, одного сложного алкилового эфира α-гидроксикарбоновой кислоты,
D) переэтерификация сложного алкилового эфира α-гидроксикарбоновой кислоты, соответственно сложных алкиловых эфиров α-гидроксикарбоновой кислоты, (мет)акриловой кислотой с образованием, по меньшей мере, одного алкил(мет)акрилата и, по меньшей мере, одной α-гидроксикарбоновой кислоты,
Е) дегидратация α-гидроксикарбоновой кислоты, соответственно α-гидроксикарбоновых кислот, с образованием (мет)акриловой кислоты.
Настоящее изобретение более подробно поясняется на приведенных ниже примерах, включая сравнительный пример.
Пример 1
В показанную на Фиг.2 реакционную дистилляционную колонну в течение 48 часов вводили 4619 г сложного метилового эфира α-гидроксиизомасляной кислоты (ГИМКМ), а также 3516 г метакриловой кислоты (МАК). Взаимодействие вводимых реагентов осуществляли при температуре 120°С и давлении 250 мбар. Образующуюся α-гидроксиизомасляную кислоту выводили из куба колонны. Метилметакрилат (ММА) отгоняли через верхнюю часть колонны. Реакцию осуществляли в присутствии 16% мас. воды в пересчете на массу сложного метилового эфира α-гидроксиизомасляной кислоты. Переэтерификации осуществляли с использованием кислого катализатора (катионита типа Lewatit® K2431 фирмы Вауеr).
Селективность, определяемая отношением количества образовавшихся метилметакрилата (ММА) и α-гидроксиизомасляной кислоты (ГИМК) к количеству превращенных ГИМКМ и МАК, составляла 99%.
Выделенную α-гидроксиизомасляную кислоту подвергали дегидратации в соответствии с немецкой заявкой на патент DE-OS 1768253.
Итоговая селективность, определяемая отношением количества образовавшегося ММА к количеству превращенной ГИМКМ, составляла 98,5%.
Сравнительный пример 1
Метилметакрилат получали дегидратацией сложного метилового эфира α-гидроксиизомасляной кислоты. Дегидратацию осуществляли в соответствии с европейской заявкой на патент ЕР-А-0941984. Смесь 20 г дигидросульфата натрия и 80 г воды добавляли к 60 г силикагеля. При пониженном давлении из смеси удаляли воду. С целью получения катализатора остаток сушили в течение ночи при 150°С. 10 г полученного катализатора помещали в снабженную испарителем кварцевую трубку. Кварцевую трубку нагревали в печи, причем температура слоя катализатора составляла около 400°С. Смесь метанола со сложным метиловым эфиром α-гидроксиизомасляной кислоты (2:1) непрерывно испаряли со скоростью 10 г в час и пропускали через слой катализатора. Селективность реакции, определяемую отношением количества образовавшегося ММА к количеству превращенного ГИМКМ, составляла 88%.
Примеры 2-18
По существу повторяли пример 1, однако воду к реакционной смеси не добавляли. Переэтерификацию осуществляли, используя указанные в таблице 1 условия, прежде всего, температуру, время пребывания и молярное соотношение эдуктов. В таблице 1 приведена также селективность переэтерификации, определяемая отношением количества образующихся ММА и ГИМК к количеству превращенных ГИМКМ и МАК.
Примеры 19-38
По существу повторяли пример 1, однако переэтерификацию осуществляли, используя указанные в таблице 2 условия, прежде всего, температуру и время пребывания реагентов. Молярное соотношение ГИМКМ к МАК составляло 1:1. Кроме того, количество добавляемой воды варьировали в соответствии с приведенными в таблице 2 данными. Кроме того, в таблица 2 указана селективность переэтерификации, определяемая отношением количества образующихся ММА и ГИМК к количеству превращенных ГИМКМ и МАК, а также молярное отношение ГИМК к ММА.
Приведенные выше примеры показывают, что образование алкил(мет)-акрилатов в соответствии с настоящим изобретением может происходить с чрезвычайно высокой селективностью, причем отношение алкил(мет)-акрилатов к α-гидроксикарбоновой кислоте близко к единице и при относительно высоких концентрациях воды. В соответствии с этим образуются относительно небольшие количества метанола. При этом молярное отношение алкил(мет)акрилатов к α-гидроксикарбоновой кислоте можно регулировать также варьированием температуры.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ АМИДА КАРБОНОВОЙ КИСЛОТЫ ИЗ КАРБОНИЛЬНОГО СОЕДИНЕНИЯ И ЦИАНИСТОВОДОРОДНОЙ КИСЛОТЫ | 2009 |
|
RU2552619C9 |
| СПОСОБ ПЕРЕЭТЕРИФИКАЦИИ | 2006 |
|
RU2452725C2 |
| СПОСОБ ПОЛУЧЕНИЯ АЛЬФА-ГИДРОКСИКАРБОНОВЫХ КИСЛОТ | 2007 |
|
RU2454399C2 |
| СПОСОБ ПОЛУЧЕНИЯ СЛОЖНЫХ ЭФИРОВ МЕТАКРИЛОВОЙ КИСЛОТЫ | 1994 |
|
RU2131867C1 |
| СПОСОБ ПОЛУЧЕНИЯ АЦЕТОНЦИАНГИДРИНА | 2007 |
|
RU2497805C2 |
| СПОСОБ ПОЛУЧЕНИЯ МЕТАКРИЛОВОЙ КИСЛОТЫ | 2012 |
|
RU2602080C2 |
| СПОСОБ НЕПРЕРЫВНОГО ПОЛУЧЕНИЯ АЛКИЛАМИНО(МЕТ)АКРИЛАМИДОВ | 2004 |
|
RU2374221C2 |
| СПОСОБ ПОЛУЧЕНИЯ СЛОЖНЫХ АЛКИЛОВЫХ ЭФИРОВ МЕТАКРИЛОВОЙ КИСЛОТЫ АЗЕОТРОПНОЙ ДИСТИЛЛЯЦИЕЙ | 2007 |
|
RU2472770C2 |
| СПОСОБ ПОЛУЧЕНИЯ АЦЕТОНЦИАНГИДРИНА И ЕГО ПРОИЗВОДНЫХ ПРОДУКТОВ ПУТЕМ ЦЕЛЕНАПРАВЛЕННОГО ОХЛАЖДЕНИЯ | 2007 |
|
RU2491272C2 |
| СПОСОБ АДСОРБЦИОННОЙ ОЧИСТКИ СЛОЖНЫХ АЛКИЛОВЫХ ЭФИРОВ МЕТАКРИЛОВОЙ КИСЛОТЫ | 2007 |
|
RU2460718C2 |
Изобретение относится к усовершенствованному способу получения алкил(мет)акрилатов, применяющихся в получении полимеров и сополимеров с другими способными полимеризоваться соединениями, включающему стадию переэтерификации сложного алкилового эфира α-гидроксикарбоновой кислоты (мет)акриловой кислотой, сопровождаемую образованием алкил(мет)акрилатов и α-гидроксикарбоновой кислоты, и стадию дегидратации α-гидроксикарбоновой кислоты, сопровождаемую образованием (мет)акриловой кислоты. Способ позволяет получать алкил(мет)акрилаты с высокой селективностью. 21 з.п. ф-лы, 2 табл., 2 ил.
1. Способ получения алкил(мет)акрилатов, включающий стадию переэтерификации сложного алкилового эфира α-гидроксикарбоновой кислоты (мет)акриловой кислотой, сопровождаемую образованием алкил(мет)акрилатов и α-гидроксикарбоновой кислоты, и стадию дегидратации α-гидроксикарбоновой кислоты, сопровождаемую образованием (мет)акриловой кислоты.
2. Способ по п.1, отличающийся тем, что сложный алкиловый эфир α-гидроксикарбоновой кислоты получают алкоголизом амида гидроксикарбоновой кислоты.
3. Способ по п.2, отличающийся тем, что амид гидроксикарбоновой кислоты получают гидролизом циангидрина.
4. Способ по п.3, отличающийся тем, что циангидрином является ацетонциангидрин.
5. Способ по п.3, отличающийся тем, что для гидролиза используют катализатор.
6. Способ по п.5, отличающийся тем, что катализатор содержит оксид марганца, серную кислоту или фермент.
7. Способ по п.2, отличающийся тем, что спирт, используемый для алкоголиза амида гидроксикарбоновой кислоты, содержит от 1 до 10 атомов углерода.
8. Способ по п.7, отличающийся тем, что спиртом является метанол и/или этанол.
9. Способ по п.2, отличающийся тем, что алкоголиз осуществляют при температуре от 160 до 240°С.
10. Способ по п.2, отличающийся тем, что алкоголиз осуществляют под давлением от 5 до 30 бар.
11. Способ по п.2, отличающийся тем, что для алкоголиза используют, по меньшей мере, один щелочной катализатор.
12. Способ по п.1, отличающийся тем, что переэтерификацию сложного алкилового эфира α-гидроксикарбоновой кислоты (мет)акриловой кислотой катализируют кислотой.
13. Способ по п.12, отличающийся тем, что кислотой является ионит.
14. Способ по п.12, отличающийся тем, что переэтерификацию осуществляют в дистилляционной колонне.
15. Способ по п.1, отличающийся тем, что переэтерификацию сложного алкилового эфира α-гидроксикарбоновой кислоты (мет)акриловой кислотой осуществляют под давлением от 100 мбар до 3 бар.
16. Способ по п.1, отличающийся тем, что переэтерификацию сложного алкилового эфира α-гидроксикарбоновой кислоты (мет)акриловой кислотой осуществляют при температуре от 70 до 130°С.
17. Способ по п.1, отличающийся тем, что переэтерификацию сложного алкилового эфира α-гидроксикарбоновой кислоты (мет)акриловой кислотой осуществляют в присутствии воды.
18. Способ по п.17, отличающийся тем, что концентрация воды составляет от 0,1 до 50 мас.% в пересчете на массу сложного алкилового эфира α-гидроксикарбоновой кислоты.
19. Способ по п.1, отличающийся тем, что молярное соотношение сложного алкилового эфира α-гидроксикарбоновой кислоты к (мет)акриловой кислоте при его переэтерификации (мет)акриловой кислотой составляет от 3:1 до 1:3.
20. Способ по п.1, отличающийся тем, что время реакции при переэтерификации сложного алкилового эфира α-гидроксикарбоновой кислоты (мет)акриловой кислотой составляет от 5 мин до 5 ч.
21. Способ по одному из пп.1-20, отличающийся тем, что дегидратацию α-гидроксикарбоновой кислоты и переэтерификацию сложного алкилового эфира α-гидроксикарбоновой кислоты (мет)акриловой кислотой осуществляют при одинаковом давлении.
22. Способ по одному из пп.1-20, отличающийся тем, что газообразную (мет)акриловую кислоту, получаемую дегидратацией α-гидроксикарбоновой кислоты, направляют на переэтерификацию без конденсации и повторного испарения.
| US 3487101 А, 13.12.1969 | |||
| УСТРОЙСТВО ДЛЯ ИЗМЕЛЬЧЕНИЯ ПИЩЕВЫХ ПРОДУКТОВ | 1972 |
|
SU429800A1 |
| СПОСОБ ПОЛУЧЕНИЯ ВЫСШИХ АЛКИЛОВЫХ ЭФИРОВ МЕТАКРИЛОВОЙ КИСЛОТЫ | 0 |
|
SU330160A1 |

